Murine, chimeric, humanized or human anti-TNF-alpha antibodies
Goldenberg , et al. A
U.S. patent number 10,385,139 [Application Number 15/897,847] was granted by the patent office on 2019-08-20 for murine, chimeric, humanized or human anti-tnf-alpha antibodies. This patent grant is currently assigned to IBC Pharmaceuticals, Inc.. The grantee listed for this patent is IBC Pharmaceuticals, Inc.. Invention is credited to Chien-Hsing Chang, David M. Goldenberg, Rongxiu Li.










View All Diagrams
| United States Patent | 10,385,139 |
| Goldenberg , et al. | August 20, 2019 |
Murine, chimeric, humanized or human anti-TNF-alpha antibodies
Abstract
The present invention concerns compositions and methods of use of bispecific antibodies comprising at least one anti-TNF-.alpha. antibody or antigen-binding fragment thereof and at least one anti-IL-6 antibody or antigen-binding fragment thereof. Preferably, the bispecific antibody is in the form of a DNL.RTM. complex. The anti-TNF-.alpha. or anti-IL-6 antibodies may comprise specific CDR sequences disclosed herein. The compositions and methods are of use to treat autoimmune disease, immune system dysfunction or inflammatory disease, as disclosed herein.
| Inventors: | Goldenberg; David M. (Mendham, NJ), Li; Rongxiu (Parsippany, NJ), Chang; Chien-Hsing (Downingtown, PA) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Applicant: |
|
||||||||||
| Assignee: | IBC Pharmaceuticals, Inc.
(Morris Plains, NJ) |
||||||||||
| Family ID: | 53005010 | ||||||||||
| Appl. No.: | 15/897,847 | ||||||||||
| Filed: | February 15, 2018 |
Prior Publication Data
| Document Identifier | Publication Date | |
|---|---|---|
| US 20180186898 A1 | Jul 5, 2018 | |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | Issue Date | ||
|---|---|---|---|---|---|
| 15620126 | Jun 12, 2017 | 9932413 | |||
| 15206571 | Jul 11, 2016 | ||||
| 14525690 | Aug 16, 2016 | 9416197 | |||
| 61898798 | Nov 1, 2013 | ||||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | C07K 14/46 (20130101); A61K 47/6803 (20170801); A61K 47/6845 (20170801); C07K 16/248 (20130101); C07K 16/468 (20130101); A61K 45/06 (20130101); C07K 16/241 (20130101); A61K 47/6889 (20170801); A61K 47/6801 (20170801); C07K 2317/565 (20130101); C07K 2317/76 (20130101); C07K 2317/92 (20130101); C07K 2319/70 (20130101); C07K 2317/33 (20130101); C07K 2317/60 (20130101); C07K 2317/24 (20130101); A61K 39/3955 (20130101); C07K 2317/31 (20130101); C07K 2317/55 (20130101); C07K 2319/00 (20130101); A61K 2039/505 (20130101) |
| Current International Class: | C07K 16/46 (20060101); A61K 47/68 (20170101); C07K 16/24 (20060101); C07K 14/46 (20060101); A61K 45/06 (20060101); A61K 39/00 (20060101); A61K 39/395 (20060101) |
References Cited [Referenced By]
U.S. Patent Documents
| 4046722 | September 1977 | Rowland |
| 4699784 | October 1987 | Shih et al. |
| 4868109 | September 1989 | Lansdorp et al. |
| 5770198 | June 1998 | Coller et al. |
| 5871945 | February 1999 | Lockerbie et al. |
| 6261537 | July 2001 | Klaveness et al. |
| 6306393 | October 2001 | Goldenberg et al. |
| 6524854 | February 2003 | Monia et al. |
| 7060506 | June 2006 | Craig |
| 7521056 | April 2009 | Chang et al. |
| 7527787 | May 2009 | Chang et al. |
| 7534866 | May 2009 | Chang et al. |
| 7550143 | June 2009 | Chang et al. |
| 8003111 | August 2011 | Chang et al. |
| 8034352 | October 2011 | Chang et al. |
| 8158129 | April 2012 | Chang et al. |
| 8163291 | April 2012 | Chang et al. |
| 8211440 | July 2012 | Chang et al. |
| 8246960 | August 2012 | Chang et al. |
| 8277817 | October 2012 | Chang et al. |
| 8282934 | October 2012 | Chang et al. |
| 8349332 | January 2013 | Chang et al. |
| 8435540 | May 2013 | Chang et al. |
| 8475794 | July 2013 | Chang et al. |
| 8481041 | July 2013 | Chang et al. |
| 8491914 | July 2013 | Chang et al. |
| 8551480 | October 2013 | Chang et al. |
| 8562988 | October 2013 | Chang et al. |
| 8597659 | December 2013 | Chang et al. |
| 2003/0198956 | October 2003 | Makowski et al. |
| 2003/0232420 | December 2003 | Braun et al. |
| 2004/0018587 | January 2004 | Makowski et al. |
| 2004/0126361 | July 2004 | Saifer et al. |
| 2005/0003403 | January 2005 | Rossi et al. |
| 2006/0210475 | September 2006 | Goldenberg et al. |
| 2007/0264265 | November 2007 | Goldenberg et al. |
| 2009/0111143 | April 2009 | Goldenberg et al. |
| 2011/0020273 | January 2011 | Chang et al. |
| 2011/0064754 | March 2011 | Taylor et al. |
| 2011/0143417 | June 2011 | Chang et al. |
| 2011/0158905 | June 2011 | Goldenberg et al. |
| 2011/0189083 | August 2011 | Chang et al. |
| 2012/0196346 | August 2012 | Chang et al. |
| 2012/0276100 | November 2012 | Chang et al. |
| 2012/0276608 | November 2012 | Chang et al. |
| 2013/0078183 | March 2013 | Chang et al. |
| 2013/0109073 | May 2013 | Chang et al. |
| 2013/0136718 | May 2013 | Chang et al. |
| 2013/0177532 | July 2013 | Chang et al. |
| 2013/0217091 | August 2013 | Chang et al. |
| 2013/0295005 | November 2013 | Chang et al. |
| 1544466 | Nov 2004 | CN | |||
| 2000/068248 | Nov 2000 | WO | |||
| 2005005638 | Jan 2005 | WO | |||
| 2006063150 | Jun 2006 | WO | |||
| 2006/107617 | Oct 2006 | WO | |||
| 2006/107786 | Oct 2006 | WO | |||
| 2007/046893 | Apr 2007 | WO | |||
| 20071075270 | Jul 2007 | WO | |||
| 2009149189 | Dec 2009 | WO | |||
| 2010003101 | Jan 2010 | WO | |||
| 2010003108 | Jan 2010 | WO | |||
| 2011084714 | Jul 2011 | WO | |||
| 2012163521 | Dec 2012 | WO | |||
Other References
|
Remicade European Medicines Agency, 2007. cited by examiner . Freise et al, Molecular Immunology 2015; vol. 67, pp. 142-152. cited by examiner . Smith, UNM college of pharmacy, vol. 17, lesson 1, introduction to diagnostic and therapeutic antibodies, 2012,. cited by examiner . Abbas et al., Cellular and Molecular Immunology, W.B. Saunders Comp. 1991, p. 43. cited by applicant . Alto et al., "Bioinformatic design of A-kinase anchoring protein-in silico: a potent and selective peptide antagonist of type II protein kinase A anchoring" Proc. Natl. Acad. Sci USA Apr. 15, 2003; 100(8):4445-50. cited by applicant . Backer et al., "Self-Assembled "Dock and Lock" System for Linking Payloads to Targeting Proteins" Bioconjugate Chem., 2006, 17(4):912-919. cited by applicant . Baillie et al., "Compartmentalisation of phospodiesterases and protein kinase A: opposites attract" FEBS Letters 2005; 579:3264-3270. cited by applicant . Banky et al., "Dimerization/Docking Domain of the Type I.alpha. Regulatory Subunit of cAMP-dependent Protein Kinase" J. Biol. Chem. 273:35048-55, 1998. cited by applicant . Basu et al., "Structure-Function Engineering of Interferon-.beta.-1b for Improving Stability, Solubility, Potency, Immunogenicity, and Pharmacokinetic Properties by Site-Selective Mono-PEGylation" Bioconjugate Chem. 2006; 17:618-630. cited by applicant . Belardelli et al., "Interferon-alpha in tumor immunity and immunotherapy" Cytokine Growth Factor Rev. 13(2):119-134(2002). cited by applicant . Belardelli et al., "International Meeting on Cancer Vaccines: How Can We Enhance Efficacy of Therapeutic Vaccines?" Cancer Res. 64:6827-6830 (2004). cited by applicant . Belardelli et al., "The neglected role of type I interferon in the T-cell response: implications for its clinical use" Immunol. Today 17(8):369-72 (1996). cited by applicant . Biron et al., "Natural killer cells in antiviral defense: function and regulation by innate cytokines" Annu. Rev. Immunol. 17:189-220 (1999). cited by applicant . Brunda et al., "Modulation of Murine Natural Killer Cell Activity in Vitro and in Vivo by Recombinant Human Interferons" Cancer Res. 44:597-601 (1984). cited by applicant . Burns-Hamuro et al., "Distinct interaction modes of an AKAP bound to two regulatory subunit isoforms of protein kinase A revealed by amide hydrogen/deuterium exchange" Protein Science (2005), 14:2982-2992. cited by applicant . Carr et al., "Interaction of the Regulatory Subunit (RII) of cAMP-dependent Protein Kinase with RII-anchoring Proteins Occurs through an Amphipathic Helix Binding Motif" J. Biol. Chem. 266:14188-92 (1991). cited by applicant . Carr et al., "Identification of Sperm-specific Proteins That Interact with A-kinase Anchoring Proteins in a Manner Similar to the Type II Regulatory Subunit of PKA" J. Biol. Chem. 276(20):17332-17338 (2001). cited by applicant . Carrero et al., "Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes" J. Exp. Med. 203(4):933-940 (2006). cited by applicant . Chang et al., "The Dock and Lock Method: A Novel Platform Technology for Building Multivalent, Multifunctional Structures of Defined Composition with Retained Bioactivity" Clin. Cancer Res. Sep. 15, 2007;13(18 Suppl), pp. 5586-5591. cited by applicant . Chmura et al., "Antibodies with infinite affinity" Proc. Natl. Acad. Sci. USA 98(15):8480-8484 (2001). cited by applicant . Colledge et al., "AKAPs: from structure to function" Trends Cell Biol. 6:216-21 (1999). cited by applicant . Corbin et al., "Regulation of Adenosine 3',5'-Monophosphate-dependent Protein Kinase" J. Biol. Chem. 248:1813-21 (1973). cited by applicant . Dhalluin et al., "Structural and Biophysical Characterization of the 40 kDa PEG-Interferon-.alpha.2a and Its Individual Positional Isomers" Bioconjugate Chem. 2005;16:504-517. cited by applicant . Dodart et al., "Immunotherapy for Alzheimer's Disease: will vaccination work?" Trends Mol. Med. 9(3):85-87 (2003). cited by applicant . Doherty et al., "Site-Specific PEGylation of Engineered Cysteine Analogues of Recombinant Human Granulocyte-Macrophage Colony-Stimulating Factor" Bioconjugate Chem. 2005;16:1291-1298. cited by applicant . Ferrantini et al., "IFN-.alpha.1 Gene Expression into a Metastatic Murine Adenocarcinoma (TS/A) Results in CD8+ T Cell-Mediated Tumor Rejection and Development of Antitumor Immunity" J. Immunol. 153:4604-15 (1994). cited by applicant . Ferrantini et al., "Interferon-.alpha. and cancer: Mechanisms of action and new perspectives of clinical use" Biochimie 89: 884-893 (2007). cited by applicant . Foser et al., "Improved biological and transcriptional activity of monopegylated interferon-.alpha.-2a isomers" The Pharmacogenomics J 3:312-319 (2003). cited by applicant . Gillies et al., "High-level expression of chimeric antibodies using adapted cDNA variable region cassettes" J. Immunol. Methods 125 (1989) 191-202. cited by applicant . Glennie et al., "Mechanisms of killing by anti-CD20 monoclonal antibodies" Mol. Immunol. 44:3823-3837 (2007). cited by applicant . Gold et al., "A Novel Bispecific, Trivalent Antibody Construct for Targeting Pancreatic Carcinoma", Cancer Res. 68:4819-26, 2008. cited by applicant . Gold et al., "Molecular Basis of AKAP Specificity for PKA Regulatory Subunits" Mol. Cell Nov. 3, 2006;24(3):383-95. cited by applicant . Goldenberg et al., "Multifunctional Antibodies by the Dock-and-Lock Method for Improved Cancer Imaging and Therapy by Pretargeting" J. Nucl. Med. 49:158-63, 2008. cited by applicant . Goldenberg et al., "Properties and structure-function relationships of veltuzumab (hA20), a humanized anti-CD20 monoclonal antibody" Blood 113:1062-70 (2009). cited by applicant . Goodson et al., "Site-Directed PEGylation of Recombinant Interleukin-2 at its Glycosylation Site" Nat. Biotechnology Apr. 1990;8(4):343-6. cited by applicant . Grace et al., "Site of Pegylation and Polyethylene Glycol Molecule Size Attenuate Interferon-.alpha. Antiviral and Antiproliferative Activities through the JAK/STAT Signaling Pathway" J. Biol. Chem. 2005;280(8):6327-6336. cited by applicant . Grimley et al., "Prolonged STAT1 Activation Related to the Growth Arrest of Malignant Lymphoma Cells by Interferon-.alpha." Blood 91(8):3017-27 (1998). cited by applicant . Gutterman et al., "Leukocyte Interferon-Induced Tumor Regression in Human Metastatic Breast Cancer, Multiple Myeloma, and Malignant Lymphoma" Ann. Intern. Med. 93(3):399-406 (1980). cited by applicant . Gutterman et al., "Cytokine therapeutics: Lessons from interferon .alpha." Proc. Natl. Acad. Sci. USA 91:1198-205 (1994). cited by applicant . Harris et al., "Effect of pegylation on pharmaceuticals" Nat. Rev. Drug. Discov. 2:214-221 (2003). cited by applicant . Hausken et al., "Mutational Analysis of the A-Kinase Anchoring Protein (AKAP)-binding Site on RII" J. Biol. Chem. 271:29016-22 (1996). cited by applicant . Hodneland et al., Selective immobilization of proteins to self-assembled monolayers presenting active site-directed capture ligands, Proc. Natl. Acad. Sci. USA 2002; 99:5048-5052. cited by applicant . Huang et al., "Targeting IFN-.alpha. to B Cell Lymphoma by a Tumor-Specific Antibody Elicits Potent Antitumor Activities" J. Immunol. 179:6881-88 (2007). cited by applicant . Hundsrucker et al., "High-affinity AKAP7.delta.-protein kinase A interaction yields novel protein kinase A-anchoring disruptor peptides" Biochem. J. (2006) 396, 297-306. cited by applicant . Kimby et al., "Long-term molecular remissions in patients with indolent lymphoma treated with rituximab as a single agent or in combination with interferon alpha-2a: A randomized phase II study from the Nordic Lymphoma Group" Leuk. Lymphoma 49(1)102-112 (2008). cited by applicant . Kinderman et al., "A Dynamic Mechanism for AKAP Binding to RII Isoforms of cAMP-Dependent Protein Kinase" Mol. Cell 24(3):397-408 (2006). cited by applicant . Kinstler et al., "Characterization and Stability of N-terminally PEGylated rhG-CSF" Pharm. Res. 1996;13(7):996-1002. cited by applicant . Kramer et al., "Cell and virus sensitivity studies with recombinant human alpha interferons" J. Interferon. Res. 3(4):425-35 (1983). cited by applicant . Le Bon et al., "Type I Interferons Potently Enhance Humoral Immunity and Can Promote Isotype Switching by Stimulating Dendritic Cells In Vivo" Immunity 14:461-470 (2001). cited by applicant . Lee et al., "Solid-Phase PEGylation of Recombinant Interferon .alpha.-2a for Site-Specific Modification: Process Performance, Characterization, and in Vitro Bioactivity" Bioconjugate Chem. 2007; 18:1728-34. cited by applicant . Lohmann et al., "High-affinity binding of the regulatory subunit (RII) of cAMP-dependent protein kinase to microtubule-associated and other cellular proteins" Proc. Natl. Acad. Sci. USA 81:6723-27 (1984). cited by applicant . Luft et al., "Type I IFNs Enhance the Terminal Differentiation of Dendritic Cells" J. Immunol. 161:1947-1953 (1998). cited by applicant . Mason, Anthony J., "Functional Analysis of the Cysteine Residues of Activin A" Mol. Endocrinol. 8:325-32 (1994). cited by applicant . Matarrese et al., "Type I Interferon Gene Transfer Sensitizes Melanoma Cells to Apoptosis via a Target Activity on Mitochondrial Function" Am. J. Pathol. 2002, 160(4):1507-1520. cited by applicant . Mecchia et al., "Type I consensus interferon (CIFN) gene transfer into human melanoma cells up-regulates p53 and enhances cisplatin-induced apoptosis: implications for new therapeutic strategies with IFN-alpha" Gene Ther. (2000) 7, 167-179. cited by applicant . Newlon et al., "A Novel Mechanism of PKA Anchoring Revealed by Solution Structures of Anchoring Complexes" EMBO J. 2001; 20:1651-1662. cited by applicant . Newlon et al., "The molecular basis for protein kinase A anchoring revealed by solution NMR" Nature Struct. Biol. 1999; 3:222-227. cited by applicant . Ngo et al., "Computational Complexity, Protein Structure Prediction, and the Levinthal Paradox", The Protein Folding Problem and Tertiary Structure Prediction, Ch. 14, pp. 492-495, (Mertz & Le Grand, Eds.), Birkhauser Boston, 1994. cited by applicant . Osborn et al., "Pharmacokinetic and Pharmacodynamic Studies of a Human Serum Albumin-Interferon-.alpha. Fusion Protein in Cynomolgus Monkeys" J. Pharmacol. Exp. Ther. 303(2):540-548 (2002). cited by applicant . Oyen et al., "Human testis cDNA for the regulatory subunit RII.alpha. of cAMP-dependent protein kinase encodes an alternate amino-terminal region" FEBS Letters 246:57-64, 1989. cited by applicant . Ozzello et al., "Conjugation of interferon alpha to a humanized monoclonal antibody (HuBrE-3vI) enhances the selective localization and antitumor effects of interferon in breast cancer xenografts" Breast Cancer Res. Treat. 48: 135-147 (1998). cited by applicant . Paquette et al., "Interferon-.alpha. and granulocyte-macrophage colony-stimulating factor differentiate peripheral blood monocytes into potent antigen-presenting cells" J. Leukoc. Biol. 64:358-367; 1998. cited by applicant . Pelham et al., "Interferon-.alpha. conjugation to human osteogenic sarcoma monoclonal antibody 791T/36" Cancer Immunol. Immuother 1983;15(3):210-216. cited by applicant . Pepinsky et al., "Improved Pharmacokinetic Properties of a Polyethylene Glycol-Modified Form of Interferon-.beta.-1a with Preserved in Vitro Bioactivity" Pharmacol. Exp. Ther. 2001; 297(3):1059-1066. cited by applicant . Pilling et al., "Interferon-.beta. mediates stromal cell rescue of T cells from apoptosis" Eur. J. Immunol. 29:1041-1050 (1999). cited by applicant . Rabjohn et al., "Molecular Cloning and Epitope Analysis of the Peanut Allergen Ara h 3" J. Clinical Investigation 103(4):535-542 (1999). cited by applicant . Raefsky et al., "Studies of Interferon as a regulator of hematopoietic cells proliferation" J. Immunol. 135(4):2507-2512 (1985). cited by applicant . Rose et al., "Structural basis of dimerization, coactivator recognition and MODY3 mutations in HNF-1.alpha." Nature Struct. Biol. 2000; 7:744-748. cited by applicant . Rosendahl et al., "A Long-Acting, Highly Potent Interferon .alpha.-2 Conjugate Created Using Site-Specific PEGylation" Bioconjugate Chem. 2005;16:200-207. cited by applicant . Rossi et al., "Novel Designs of Multivalent Anti-CD20 Humanized Antibodies as Improved Lymphoma Therapeutics" Cancer Res. 68:8384-92 (2008). cited by applicant . Rossi et al., "Stably tethered multifunctional structures of defined composition made by the dock and lock method for use in cancer targeting" Proc. Nal Acad. Sci. Epub Apr. 24, 2006, vol. 103, No. 18, pp. 6841-6846. cited by applicant . Rustandi et al., "The Ca2+-Dependent Interaction of S100B(.beta..beta.) with a Peptide Derived from p53", Biochemistry 1998; 37: 1951-1960. cited by applicant . Sabaawy et al., "Enhancement of 5-fluorouracil cytotoxicity on human colon cancer cells by retrovirus-mediated interferon-.alpha. gene transfer" Int. J. Oncol. Jun. 1999; 14(6):1143-51. cited by applicant . Salles et al., "Rituximab combined with chemotherapy and interferon in follicular lymphoma patients: results of the GELA-GOELAMS FL2000 study" Blood 2008; 112:4824-4831. cited by applicant . Santini et al., "Type I Interferon as a Powerful Adjuvant for Monocyte-derived Dendritic Cell Development and Activity In Vivo and in Hu-PBL-SCID Mice" J. Exp. Med. 191(10):1777-1788 (2000). cited by applicant . Scott et al., "Type II Regulatory Subunit Dimerization Determines the Subcellular Localization of the cAMP-dependent Protein Kinase" J. Biol. Chem. 265:21561-66 (1990). cited by applicant . Scott et al., "Cyclic nucleotide-dependent protein kinases" Pharmacol. Ther. 1991;50(1):123-45. cited by applicant . Seffernick et al., "Melamine Deaminase and Atrazine Chlorohydrolase: 98 Percent Identical but Functionally Different" J. Bacteriol. 183(8):2405-2410 (2001). cited by applicant . Sharkey et al., "Improved Therapeutic Results by Pretargeted Radioimmunotherapy of Non-Hodgkin's Lymphoma with a New Recombinant, Trivalent, Anti-CD20, Bispecific Antibody" Cancer Res. 68:5282-90 (2008). cited by applicant . Sharkey et al., "Metastatic Human Colonic Carcinoma: Molecular Imaging with Pretargeted SPECT and PET in a Mouse Model" Radiology 246:497-507 (2008). cited by applicant . Sidky et al., "Inhibition of Angiogenesis by Interferons: Effects on Tumor- and Lymphocyte-induced Vascular Responses" Cancer Res. 47:5155-5161, Oct. 1, 1987. cited by applicant . Stein et al., "Characterization of a New Humanized Anti-CD20 Monoclonal Antibody, IMMU-106, and Its Use in Combination with the Humanized Anti-CD22 Antibody, Epratuzumab, for the Therapy of Non-Hodgkin's Lymphoma" Clin. Cancer Res. vol. 10, 2868-2878, Apr. 15, 2004. cited by applicant . Stein et al., "Characterization of a humanized IgG4 anti-HLA-DR monoclonal antibody that lacks effector cell functions but retains direct antilymphoma activity and increases the potency of rituximab" Blood 2006;108:2736-2744. cited by applicant . Stokka et al., "Characterization of A-kinase-anchoring disruption using a solution-based assay" Biochem. J. (2006) 400, 493-499. cited by applicant . Stryer et al., "Levels of Structure in Protein Architecture", Biochemistry, 3rd Ed., pp. 31-33, W.H. Freeman & Co., New York, 1988. cited by applicant . Takaoka et al., "Integration of interferon-.alpha./.beta. signalling to p53 responses in tumour suppression and antiviral defence" Nature Jul. 31, 2003;424(6948):516-23. cited by applicant . Taylor, S., "cAMP-dependent Protein Kinase" J. Biol. Chem. 1989;264(15):8443-8446. cited by applicant . Walsh et al., "An Adenosine 3', 5'-Monophosphate-dependant Protein Kinase from Rabbit Skeletal Muscle" J. Biol. Chem. 243(13):3763-3774 (1968). cited by applicant . Weck et al., "Comparison of the Antiviral Activities of Various Cloned Human Interferon-.alpha. Subtypes in Mammalian Cell Cultures" J. Gen. Virol. (1981), 57, 233-237. cited by applicant . Winkler et al., "Changing the Antigen Binding Specificity by Single Point Mutations of an Anti-p24 (HIV-1) Antibody" J. Immunol. 165:4505-14 (2000). cited by applicant . Witkowski et al., "Conversion of a .beta.-Ketoacyl Synthase to a Malonyl Decarboxylase by Replacement of the Active-Site Cysteine with Glutamine" Biochemistry 38(36):11643-50 (1999). cited by applicant . Wong et al., "AKAP Signalling Complexes: Focal Points in Space and Time" Nat. Rev. Mol. Cell Biol. 12:959-70 (2004). cited by applicant . Zhu et al., "Inhibition of tumor growth and metastasis by targeting tumor-associated angiogenesis with antagonists to the receptors of vascular endothelial growth factor" Invest. New Drugs 17:195-212, 1999. cited by applicant . Allard et al., "Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs", Clin Cancer Res. Oct. 15, 2013;19(20):5626-35. cited by applicant . Almoallim et al., "Anti-Tumor Necrosis Factor-.alpha. Induced Systemic Lupus Erythematosus", Open Rheumatol J. 2012;6:315-9. cited by applicant . Baeuerle et al., "Bispecific T-cell engaging antibodies for cancer therapy", Cancer Res. Jun. 15, 2009;69(12):4941-4. cited by applicant . Cardillo et al., "Targeting both IGF-1R and mTOR synergistically inhibits growth of renal cell carcinoma in vitro", BMC Cancer. Apr. 1, 2013;13:170. cited by applicant . Callahan et al., "At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy", J Leukoc Biol. Jul. 2013;94(1):41-53. cited by applicant . Chang et al., "A new method to produce monoPEGylated dimeric cytokines shown with human interferon-.alpha.2b", Bioconjug Chem. Oct. 21, 2009;20(10):1899-907. cited by applicant . Chang et al., "A novel class of anti-HIV agents with multiple copies of enfuvirtide enhances inhibition of viral replication and cellular transmission in vitro", PLoS One. 2012;7(7):e41235. cited by applicant . Chang et al., "Evaluation of a novel hexavalent humanized anti-IGF-1R antibody and its bivalent parental IgG in diverse cancer cell lines", PLoS One. 2012;7(8):e44235. cited by applicant . Fischer et al., "Combined inhibition of tumor necrosis factor .alpha. and interleukin-17 as a therapeutic opportunity in rheumatoid arthritis: development and characterization of a novel bispecific antibody", Arthritis Rheumatol. Jan. 2015;67(1):51-62. cited by applicant . Flieger et al., "A bispecific single-chain antibody directed against EpCAM/CD3 in combination with the cytokines interferon alpha and interleukin-2 efficiently retargets T and CD3+CD56+ natural-killer-like T lymphocytes to EpCAM-expressing tumor cells", Cancer Immunol Immunother. Oct. 2000;49(8):441-8. cited by applicant . Goldenberg, DM., "Targeted therapy of cancer with radiolabeled antibodies", J Nucl Med. May 2002;43(5):693-713. cited by applicant . Gleason et al., "Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production", Mol Cancer Ther. Dec. 2012;11(12):2674-84. cited by applicant . Hennigan et al., "Interleukin-6 inhibitors in the treatment of rheumatoid arthritis", Ther Clin Risk Manag. Aug. 2008;4(4):767-75. cited by applicant . Hirabara et al., "Clinical efficacy of abatacept, tocilizumab, and etanercept in Japanese rheumatoid arthritis patients with inadequate response to anti-TNF monoclonal antibodies", Clin Rheumatol. Sep. 2014;33(9):1247-54. cited by applicant . Intlekofer et al., "At the bench: preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy", J Leukoc Biol. Jul. 2013;94(1):25-39. cited by applicant . Kipriyanov et al., "Synergistic antitumor effect of bispecific CD19.times.CD3 and CD19.times.CD16 diabodies in a preclinical model of non-Hodgkin's lymphoma", J Immunol. Jul. 1, 2002;169(1):137-44. cited by applicant . Kyi et al., "Checkpoint blocking antibodies in cancer immunotherapy", FEBS Lett. Jan. 21, 2014;588(2):368-76. cited by applicant . Liu et al., "Trop-2-targeting tetrakis-ranpirnase has potent antitumor activity against triple-negative breast cancer", Mol Cancer. Mar. 10, 2014;13:53. cited by applicant . Lum et al., "Targeted T-cell Therapy in Stage IV Breast Cancer: A Phase I Clinical Trial", Clin Cancer Res. May 15, 2015;21(10):2305-14. cited by applicant . Morales-Kastresana et al., "Combined immunostimulatory monoclonal antibodies extend survival in an aggressive transgenic hepatocellular carcinoma mouse model", Clin Cancer Res. Nov. 15, 2013;19(22):6151-62. cited by applicant . Mori et al., "IL-1.beta. and TNF.alpha.-initiated IL-6-STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis", Int Immunol. Nov. 2011;23(11):701-12. cited by applicant . Oberst et al., "CEA/CD3 bispecific antibody MEDI-565/AMG 211 activation of T cells and subsequent killing of human tumors is independent of mutations commonly found in colorectal adenocarcinomas", MAbs. 2014;6(6):1571-84. cited by applicant . Ofei et al., "Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM", Diabetes. Jul. 1996;45(7):881-5. cited by applicant . Peng et al., "The CEA/CD3-bispecific antibody MEDI-565 (MT111) binds a nonlinear epitope in the full-length but not a short splice variant of CEA", PLoS One. 2012;7(5):e36412. cited by applicant . Podojil et al., "Targeting the B7 family of co-stimulatory molecules: successes and challenges", BioDrugs. Feb. 2013;27(1):1-13. cited by applicant . Rossi et al., "Hexavalent bispecific antibodies represent a new class of anticancer therapeutics: 1. Properties of anti-CD20/CD22 antibodies in lymphoma", Blood. Jun. 11, 2009;113(24):6161-71. cited by applicant . Rossi et al., "The dock-and-lock method combines recombinant engineering with site-specific covalent conjugation to generate multifunctional structures", Bioconjug Chem. Mar. 21, 2012;23(3):309-23. cited by applicant . Rossi et al., "Complex and defined biostructures with the dock-and-lock method", Trends Pharmacol Sci. Sep. 2012;33(9):474-81. cited by applicant . Rossi et al., "Optimization of multivalent bispecific antibodies and immunocytokines with improved in vivo properties", Bioconjug Chem. Jan. 16, 2013;24(1):63-71. cited by applicant . Rossi et al., "A new class of bispecific antibodies to redirect T cells for cancer immunotherapy", MAbs. Mar.-Apr. 2014;6(2):381-91. cited by applicant . Rossi et al., "A New Platform for Trivalent Bispecific Antibodies Used for T-Cell Redirected Killing of B-Cell Malignancies", Nov. 15, 2013; Blood: 122 (21). cited by applicant . Rossi et al., "A novel Trop-2/CD3 trivalent bispecific antibody effectively redirects T cells to kill target human pancreatic and gastric cancer cells", Proceedings of the 105th Annual Meeting of the American Association for Cancer Research; Apr. 5-9, 2014; San Diego, CA; Cancer Res 2014;74(19 Suppl):Abstract # 2655. cited by applicant . Rossi et al., Novel T-cell redirecting trivalent bispecific antibodies, Proceedings of the 104th Annual Meeting of the American Association for Cancer Research; Apr. 6-10, 2013; Washington, DC; Cancer Res 2013;73(8 Suppl):Abstract # 4747. cited by applicant . Rossi et al., "Redirected T-cell killing of solid cancers targeted with an anti-CD3/Trop-2-bispecific antibody is enhanced in combination with interferon-.alpha.", Mol Cancer Ther. Oct. 2014;13(10):2341-51. cited by applicant . Sharkey et al., "Improved cancer therapy and molecular imaging with multivalent, multispecific antibodies", Cancer Biother Radiopharm. Feb. 2010;25(1):1-12. cited by applicant . Shubert et al., "A recombinant triplebody with specificity for CD19 and HLA-DR mediates preferential binding to antigen double-positive cells by dual-targeting", MAbs. Jan.-Feb. 2012;4(1):45-56. cited by applicant . Stedman'S Online Medical Dictionary, Feb. 3, 2017; p. 1. cited by applicant . Tanaka et al., "Targeting interleukin-6: all the way to treat autoimmune and inflammatory diseases", Int J Biol Sci. 2012;8(9)1227-36. cited by applicant . Topalian et al., "Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity", Curr Opin Immunol. Apr. 2012;24(2):207-12. cited by applicant . Vallera et al., "A bispecific recombinant immunotoxin, DT2219, targeting human CD19 and CD22 receptors in a mouse xenograft model of B-cell leukemia/lymphoma", Clin Cancer Res. May 15, 2005;11(10):3879-88. cited by applicant . Wu et al., "Molecular construction and optimization of anti-human IL-1alpha/beta dual variable domain immunoglobulin (DVD-Ig) molecules", MAbs. Jul.-Aug. 2009;1(4):339-47. cited by applicant. |
Primary Examiner: Bunner; Bridget E
Assistant Examiner: Hamud; Fozia
Attorney, Agent or Firm: Nakashima; Richard A.
Parent Case Text
RELATED APPLICATIONS
This application is a divisional of U.S. patent application Ser. No. 15/620,126, filed Jun. 12, 2017, which was a divisional of U.S. patent application Ser. No. 15/206,571 (now abandoned), filed Jul. 11, 2016, which was a divisional of U.S. patent application Ser. No. 14/525,690 (now issued U.S. Pat. No. 9,416,197), filed Oct. 28, 2014, which claimed the benefit under 35 U.S.C. 119(e) of provisional U.S. Patent Application Ser. No. 61/898,798, filed Nov. 1, 2013.
Claims
What is claimed is:
1. A murine, chimeric, humanized or human anti-TNF-.alpha. antibody or antigen-binding fragment thereof comprising the heavy chain CDR sequences CDR1 (GFWN, SEQ ID NO:113), CDR2 (YISYSGRTYYNPSLKS, SEQ ID NO:114) and CDR3 (DANYVLDY, SEQ ID NO:115) and the light chain CDR sequences CDR1 (KSSQSLLNSSTQKNYLA, SEQ ID NO:116), CDR2 (FASARES, SEQ ID NO:117) and CDR3 (QQHYRTPFT, SEQ ID NO:118).
2. The anti-TNF-.alpha. antibody or fragment thereof of claim 1, wherein the antibody allotype is selected from the group consisting of nG1m1, G1m3, nG1m1,2 and Km3.
3. The anti-TNF-.alpha. antibody or fragment thereof of claim 1, wherein the antibody or fragment is a naked antibody or fragment.
4. The anti-TNF-.alpha. antibody or fragment thereof of claim 1, wherein the antibody is conjugated to an agent selected from the group consisting of a drug, an anti-angiogenic agent, a pro-apoptotic agent, an antibiotic, a hormone, a hormone antagonist, an immunomodulator, a cytokine, a chemokine, a prodrug, and an enzyme.
5. The anti-TNF-.alpha. antibody or fragment thereof of claim 4, wherein the drug possesses a pharmaceutical property selected from the group consisting of antimitotic, antikinase, anti-tyrosine kinase, alkylating, antimetabolite, antibiotic, alkaloid, anti-angiogenic, pro-apoptotic agent, and immune modulator.
6. The anti-TNF-.alpha. antibody or fragment thereof of claim 4, wherein the drug is selected from the group consisting of 5-fluorouracil, aplidin, azaribine, anastrozole, anthracyclines, bendamustine, bleomycin, bortezomib, bryostatin-1, busulfan, calicheamycin, camptothecin, carboplatin, 10-hydroxycamptothecin, carmustine, celecoxib, chlorambucil, cisplatinum, Cox-2 inhibitors, irinotecan (CPT-11), SN-38, carboplatin, cladribine, camptothecans, cyclophosphamide, cytarabine, dacarbazine, docetaxel, dactinomycin, daunorubicin, doxorubicin, 2-pyrrolinodoxorubicine (2P-DOX), pro-2P-DOX, cyano-morpholino doxorubicin, doxorubicin glucuronide, epirubicin glucuronide, estramustine, epipodophyllotoxin, estrogen receptor binding agents, etoposide (VP16), etoposide glucuronide, etoposide phosphate, floxuridine (FUdR), 3',5'-O-dioleoyl-FudR (FUdR-dO), fludarabine, flutamide, farnesyl-protein transferase inhibitors, gemcitabine, hydroxyurea, idarubicin, ifosfamide, L-asparaginase, lenolidamide, leucovorin, lomustine, mechlorethamine, melphalan, mercaptopurine, 6-mercaptopurine, methotrexate, mitoxantrone, mithramycin, mitomycin, mitotane, navelbine, nitrosourea, plicomycin, procarbazine, paclitaxel, pentostatin, PSI-341, raloxifene, semustine, streptozocin, tamoxifen, temazolomide, transplatinum, thalidomide, thioguanine, thiotepa, teniposide, topotecan, uracil mustard, vinorelbine, vinblastine, vincristine and vinca alkaloids.
7. The anti-TNF-.alpha. antibody or fragment thereof of claim 4, wherein the chemokine is selected from the group consisting of RANTES, MCAF, MIP1-alpha, MIP1-Beta and IP-10.
8. The anti-TNF-.alpha. antibody or fragment thereof of claim 4, wherein the anti-angiogenic agent is selected from the group consisting of angiostatin, baculostatin, canstatin, maspin, anti-VEGF antibody, anti-PlGF peptide, anti-vascular growth factor antibody, anti-Flk-1 antibody, anti-Flt-1 antibody, anti-Kras antibody, anti-cMET antibody, anti-MIF (macrophage migration-inhibitory factor) antibody, laminin peptide, fibronectin peptide, plasminogen activator inhibitor, tissue metalloproteinase inhibitor, interferon, interleukin-12, IP-10, Gro- , thrombospondin, 2-methoxyoestradiol, proliferin-related protein, carboxiamidotriazole, CM101, Marimastat, pentosan polysulphate, angiopoietin-2, interferon-alpha, herbimycin A, PNU145156E, 16K prolactin fragment, Linomide (roquinimex), thalidomide, pentoxifylline, genistein, TNP-470, endostatin, paclitaxel, accutin, angiostatin, cidofovir, vincristine, bleomycin, AGM-1470, platelet factor 4 and minocycline.
9. The anti-TNF-.alpha. antibody or fragment thereof of claim 4, wherein the immunomodulator is selected from the group consisting of a cytokine, a stem cell growth factor, a lymphotoxin, a hematopoietic factor, a colony stimulating factor (CSF), an interferon (IFN), erythropoietin, and thrombopoietin.
10. The anti-TNF-.alpha. antibody or fragment thereof of claim 9, wherein the cytokine is selected from the group consisting of human growth hormone, N-methionyl human growth hormone, bovine growth hormone, parathyroid hormone, thyroxine, insulin, proinsulin, relaxin, prorelaxin, follicle stimulating hormone (FSH), thyroid stimulating hormone (TSH), luteinizing hormone (LH), hepatic growth factor, prostaglandin, fibroblast growth factor, prolactin, placental lactogen, OB protein, tumor necrosis factor-.alpha., tumor necrosis factor- , mullerian-inhibiting substance, mouse gonadotropin-associated peptide, inhibin, activin, vascular endothelial growth factor, integrin, thrombopoietin (TPO), NGF- , platelet-growth factor, transforming growth factor-.alpha. (TGF-.alpha.), TGF- , insulin-like growth factor-I, insulin-like growth factor-II, interferon-.alpha., interferon-.beta., interferon-.gamma., interferon-.lamda., macrophage-CSF, interleukin-1 (IL-1), IL-la, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-11, IL-12, IL-13, IL-14, IL-15, IL-16, IL-17, IL-18, IL-21, IL-25, LIF, FLT-3, angiostatin, thrombospondin, endostatin, and LT (lymphotoxin).
Description
SEQUENCE LISTING
The instant application contains a Sequence Listing which has been submitted electronically in ASCII format and is hereby incorporated by reference in its entirety. Said ASCII copy, created on Oct. 27, 2014, is named IBC139US1 SL.txt and is 58,035 bytes in size.
FIELD OF THE INVENTION
The present invention relates to compositions and methods of use of complexes comprising at least one anti-TNF-.alpha. antibody or antigen-binding fragment thereof and at least one anti-IL-6 antibody or antigen-binding fragment thereof. The complex may be a bispecific or multispecific antibody or fragment thereof. Preferably, the complex is a DOCK-AND-LOCK.RTM. (DNL.RTM.) complex, in which the components are joined using the binding affinity between a DDD (dimerization and docking domain) moiety of human protein kinase A (PKA) regulatory subunit RI.alpha., RI.beta., RII.alpha. or RII.beta., and an AD (anchoring domain) moiety of an A-kinase anchoring protein (AKAP), wherein a pair of DDD moieties forms a dimer that binds to a complementary sequence on the AD moiety. Although the basic DNL.RTM. complex is trimeric, complexes with other stoichiometries are possible, such as tetrameric, pentameric or hexameric. The subject complexes are of use to treat autoimmune disease, inflammatory disease or other conditions in which TNF-.alpha. and IL-6 play a pathogenic role. In particularly preferred embodiments, the disease or condition is selected from the group consisting of systemic lupus erythematosus (SLE), rheumatoid arthritis, inflammatory bowel disease, type II diabetes, obesity, atherosclerosis and cachexia related to cancer.
BACKGROUND OF THE INVENTION
TNF-.alpha. and IL-6 are proinflammatory cytokines involved in the pathogenesis of various autoimmune diseases, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), inflammatory bowel disease, and type 2 diabetes. Blocking the biological activities of TNF-.alpha. has demonstrated clinical benefits in patients with RA and Crohn's disease, as exemplified by five antibody- or receptor-based therapeutics currently on the market. The promise of IL-6 blockade was also reinforced by the regulatory approval of one anti-IL-6R antibody for treating RA and juvenile idiopathic arthritis, with additional antibodies targeting either IL-6R or IL-6 in advanced clinical trials. As reported by Mori et al. (Int Immunol 2011; 23: 701-12), IL-6 directly activates STAT3, whereas TNF-.alpha. indirectly activates STAT3 via stimulating the expression of IL-6, which then activates STAT3 and triggers a cytokine amplification loop of IL-6, resulting in sustained STAT3 activation and chronic inflammation.
Numerous antibodies against TNF-.alpha. are commercially available and/or publicly known, including infliximab (Jansenn Biotech, Inc.), adalimumab (Abbvie, Inc.), certolizumab pegol (UCB, Inc.) and golimumab (Centocor). Although these therapeutic agents have significantly improved the treatment of certain autoimmune diseases, such as rheumatoid arthritis (RA), it has been reported that about 30% of RA patients treated with TNF inhibitors (including anti-TNF.alpha. antibodies) show little to no effect of the therapy, with about two thirds demonstrating moderate to high disease activity at 1 year after treatment (Hirabara et al., 2014, Clin Rheumatol 33:1247-54). Further, loss of therapeutic efficacy is frequently observed with anti-TNF monoclonal antibodies (adalimumab, infliximab) in patients receiving concomitant low-dose methotrexate, due to immunogenicity-related issues (Hirabara et al, 2014). A need exists for more effective compositions and methods for use of anti-TNF antibodies in treating diseases and conditions related to TNF-.alpha..
Dysregulated IL-6 production has been demonstrated to play a pathological role in various autoimmune and chronic inflammatory diseases. Therapies against IL-6 pathways have commonly targeted the IL-6 receptor (IL-6R), including the anti-IL-6R antibodies tocilizumab, and sarilumab. Antibodies targeted directly against IL-6 have also been developed, such as olokizumab (UCB), siltuximab (Janssen), BMS-943429 (Bristol-Myers Squibb) and sirukumab (Centocor). The latter have been used against various autoimmune diseases and cancers. Following regulatory approval of tocilizumab for rheumatoid arthritis, Castleman's disease and systemic juvenile idiopathic arthritis, favorable results of off-label use have been reported in systemic lupus erythematosus, systemic sclerosis, polymyositis, vasculitis syndrome including giant cell arteritis, Takayasu arteritis, cryoglobulinemia, glomerulonephritis and rheumatoid vasculitis (see, e.g., Tanaka & Kishimoto, 2012, Int J Biol Sci 8:1227-36). While these results are promising, no antibodies against IL-6 (as opposed to IL-6R) have yet been approved for human use in any indication.
A need exists in the field for more effective, well-tolerated therapeutic agents targeted against TNF and IL-6.
SUMMARY OF THE INVENTION
The present invention concerns compositions and methods of use of bispecific or multispecific antibodies comprising at least one anti-TNF-.alpha. antibody or antigen-binding fragment thereof and at least one anti-IL-6 antibody or antigen-binding fragment thereof. Preferably, the bispecific or multispecific antibody is in the form of a DNL.RTM. complex, comprising AD and DDD moiety binding pairs as described below.
The antibodies may be chimeric, humanized or human antibodies. In certain preferred embodiments, the antibodies are humanized, comprising the CDR sequences of, e.g., a murine anti-IL-6 or anti-TNF-.alpha. antibody and the framework (FR) and constant region sequences from one or more human antibodies. Methods of antibody humanization are well known in the art, as discussed in detail below. The antibody can be of various isotypes, preferably human IgG1, IgG2, IgG3 or IgG4, more preferably comprising human IgG1 hinge and constant region sequences. More preferably, the antibody or fragment thereof may be designed or selected to comprise human constant region sequences that belong to specific allotypes, which may result in reduced immunogenicity. Preferred allotypes for administration include a non-G1m1 allotype (nG1m1), such as G1m3, G1m3,1, G1m3,2 or G1m3,1,2. More preferably, the allotype is selected from the group consisting of the nG1m1, G1m3, nG1m1,2 and Km3 allotypes.
Numerous anti-TNF-.alpha. antibodies are commercially available and/or publicly known, including but not limited to CDP571 (Ofei et al., 2011, Diabetes 45:881-85); MTNFAI, M2TNFAI, M3TNFAI, M3TNFABI, M302B and M303 (Thermo Scientific); 3H15L1, D13H3, TN3, 17H1L4, MP9-20A4, and 68B6A3 L1 (Life Technologies); NBP1-19532, NB600-587, NBP2-27223, and NBP2-27224, (NOVUS BIOLOGICALS.RTM.); ab9635, (ABCAM.RTM.); certolizumab pegol (UCB, Brussels, Belgium); adalimumab (Abbvie); infliximab and golimumab (Centocor). These and many other known anti-TNF-.alpha. antibodies may be used in the claimed methods and compositions.
Numerous anti-IL-6 antibodies are commercially available and/or publicly known, including but not limited to 5IL6, 4HCLC, 4H16L21, 677B6A2, and 20F3 (Thermo Scientific); NBP1-47810, NBP2025275, NBP1047355, and NBP2021624 (NOVUS BIOLOGICALS.RTM.); olokizumab (UCB); siltuximab (Janssen); BMS-943429 (Bristol-Myers Squibb); and sirukumab (Centocor). These and many other known anti-IL-6 antibodies may be used in the claimed methods and compositions.
The subject antibodies may be co-administered with one or more other therapeutic agents. The therapeutic agents may be conjugated to the antibodies or administered separately, either before, concomitantly with or after the antibody. Therapeutic agents of use for treating immune or inflammatory diseases are preferably selected from drugs, anti-angiogenic agents, pro-apoptotic agents, antibiotics, hormones, hormone antagonists, chemokines, prodrugs, enzymes, immunomodulators, cytokines or other known agents of use for immune or inflammatory diseases.
Drugs of use may possess a pharmaceutical property selected from the group consisting of antimitotic, antikinase (e.g., anti-tyrosine kinase), alkylating, antimetabolite, antibiotic, alkaloid, anti-angiogenic, pro-apoptotic agents, immune modulators, and combinations thereof.
Exemplary drugs of use may include 5-fluorouracil, aplidin, azaribine, anastrozole, anthracyclines, bendamustine, bleomycin, bortezomib, bryostatin-1, busulfan, calicheamycin, camptothecin, carboplatin, 10-hydroxycamptothecin, carmustine, celecoxib, chlorambucil, cisplatin (CDDP), Cox-2 inhibitors, irinotecan (CPT-11), SN-38, carboplatin, cladribine, camptothecans, cyclophosphamide, cytarabine, dacarbazine, docetaxel, dactinomycin, daunorubicin, doxorubicin, 2-pyrrolinodoxorubicine (2P-DOX), cyano-morpholino doxorubicin, doxorubicin glucuronide, epirubicin glucuronide, estramustine, epipodophyllotoxin, estrogen receptor binding agents, etoposide (VP16), etoposide glucuronide, etoposide phosphate, floxuridine (FUdR), 3',5'-O-dioleoyl-FudR (FUdR-dO), fludarabine, flutamide, farnesyl-protein transferase inhibitors, gemcitabine, hydroxyurea, idarubicin, ifosfamide, L-asparaginase, lenolidamide, leucovorin, lomustine, mechlorethamine, melphalan, mercaptopurine, 6-mercaptopurine, methotrexate, mitoxantrone, mithramycin, mitomycin, mitotane, navelbine, nitrosourea, plicomycin, procarbazine, paclitaxel, pentostatin, PSI-341, raloxifene, semustine, streptozocin, tamoxifen, temazolomide (an aqueous form of DTIC), transplatinum, thalidomide, thioguanine, thiotepa, teniposide, topotecan, uracil mustard, vinorelbine, vinblastine, vincristine and vinca alkaloids.
Chemokines of use may include RANTES, MCAF, MIP1-alpha, MIP1-Beta and IP-10.
In certain embodiments, anti-angiogenic agents, such as angiostatin, baculostatin, canstatin, maspin, anti-VEGF antibodies, anti-PlGF peptides and antibodies, anti-vascular growth factor antibodies, anti-Flk-1 antibodies, anti-Flt-1 antibodies and peptides, anti-Kras antibodies, anti-cMET antibodies, anti-MIF (macrophage migration-inhibitory factor) antibodies, laminin peptides, fibronectin peptides, plasminogen activator inhibitors, tissue metalloproteinase inhibitors, interferons, interleukin-12, IP-10, Gro- , thrombospondin, 2-methoxyoestradiol, proliferin-related protein, carboxiamidotriazole, CM101, Marimastat, pentosan polysulphate, angiopoietin-2, interferon-alpha, herbimycin A, PNU145156E, 16K prolactin fragment, Linomide (roquinimex), thalidomide, pentoxifylline, genistein, TNP-470, endostatin, paclitaxel, accutin, angiostatin, cidofovir, vincristine, bleomycin, AGM-1470, platelet factor 4 or minocycline may be of use.
Immunomodulators of use may be selected from a cytokine, a stem cell growth factor, a lymphotoxin, a hematopoietic factor, a colony stimulating factor (CSF), an interferon (IFN), erythropoietin, thrombopoietin and a combination thereof. Specifically useful are lymphotoxins such as tumor necrosis factor (TNF), hematopoietic factors, such as interleukin (IL), colony stimulating factor, such as granulocyte-colony stimulating factor (G-CSF) or granulocyte macrophage-colony stimulating factor (GM-CSF), interferon, such as interferons-.alpha., -.beta. or -.gamma., and stem cell growth factor, such as that designated "S1 factor". Included among the cytokines are growth hormones such as human growth hormone, N-methionyl human growth hormone, and bovine growth hormone; parathyroid hormone; thyroxine; insulin; proinsulin; relaxin; prorelaxin; glycoprotein hormones such as follicle stimulating hormone (FSH), thyroid stimulating hormone (TSH), and luteinizing hormone (LH); hepatic growth factor; prostaglandin, fibroblast growth factor; prolactin; placental lactogen, OB protein; tumor necrosis factor-.alpha. and - ; mullerian-inhibiting substance; mouse gonadotropin-associated peptide; inhibin; activin; vascular endothelial growth factor; integrin; thrombopoietin (TPO); nerve growth factors such as NGF- ; platelet-growth factor; transforming growth factors (TGFs) such as TGF-.alpha. and TGF- ; insulin-like growth factor-I and -II; erythropoietin (EPO); osteoinductive factors; interferons such as interferon-.alpha., -.beta., and -.gamma.; colony stimulating factors (CSFs) such as macrophage-CSF (M-CSF); interleukins (ILs) such as IL-1, IL-1.alpha., IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-11, IL-12; IL-13, IL-14, IL-15, IL-16, IL-17, IL-18, IL-21, IL-25, LIF, kit-ligand or FLT-3, angiostatin, thrombospondin, endostatin, tumor necrosis factor and LT. Lenolidamide is yet another immunomodulator that has shown activity in controlling certain cancers, such as multiple myeloma and hematopoietic tumors.
The antibodies or complexes may be used to treat a variety of diseases or conditions in which TNF-.alpha. and IL-6 play a pathogenic role, such as autoimmune, immune dysfunction or inflammatory diseases. Exemplary diseases or conditions may be selected from the group consisting of rheumatoid arthritis (RA), systemic lupus erythematosus, type 2 diabetes, Crohn's disease, Castleman's disease, juvenile idiopathic arthritis, systemic sclerosis, polymyositis, vasculitis syndrome, Takayasu arteritis, cryoglobulinemia, glomerulonephritis, rheumatoid vasculitis, arthritis, sepsis, septic shock, inflammation, non-septic hyperinflammatory disorder, nephritis, inflammatory bowel disease, inflammatory liver injury, acute pancreatitis, acute respiratory distress syndrome, ischemia-reperfusion injury, ischemic stroke, graft-vs.-host disease and cachexia related to cancer.
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. 1. Assay for neutralizing anti-IL-6 antibodies. Supernatants from clones were incubated with human IL-6 at 37.degree. C. for 1 hour, prior to incubation with HT-29 cells. The cells were incubated with rhIL-6 alone or in combination with serum for 15 min at 37.degree. C. and phosphorylation of STAT3 was detected by Western blotting.
FIG. 2A. Titration of neutralizing anti-IL-6 antibodies. The ability to block IL-6 induced phosphorylation of STAT3 was determined by Western blot analysis using the indicated concentrations of the 2-3B2 anti-IL-6 antibody. A substantial inhibition of IL-6 dependent phosphorylation was seen as low as 0.067 nM antibody.
FIG. 2B. Titration of neutralizing anti-IL-6 antibodies. The ability to block IL-6 induced phosphorylation of STAT3 was determined by Western blot analysis using the indicated concentrations of the 4-4E6 anti-IL-6 antibody. Approximately equivalent effects on phosphorylation were observed at 0.67 nM 4-4E6 vs. 0.0067 nM 2-34B2 antibody (FIG. 2A).
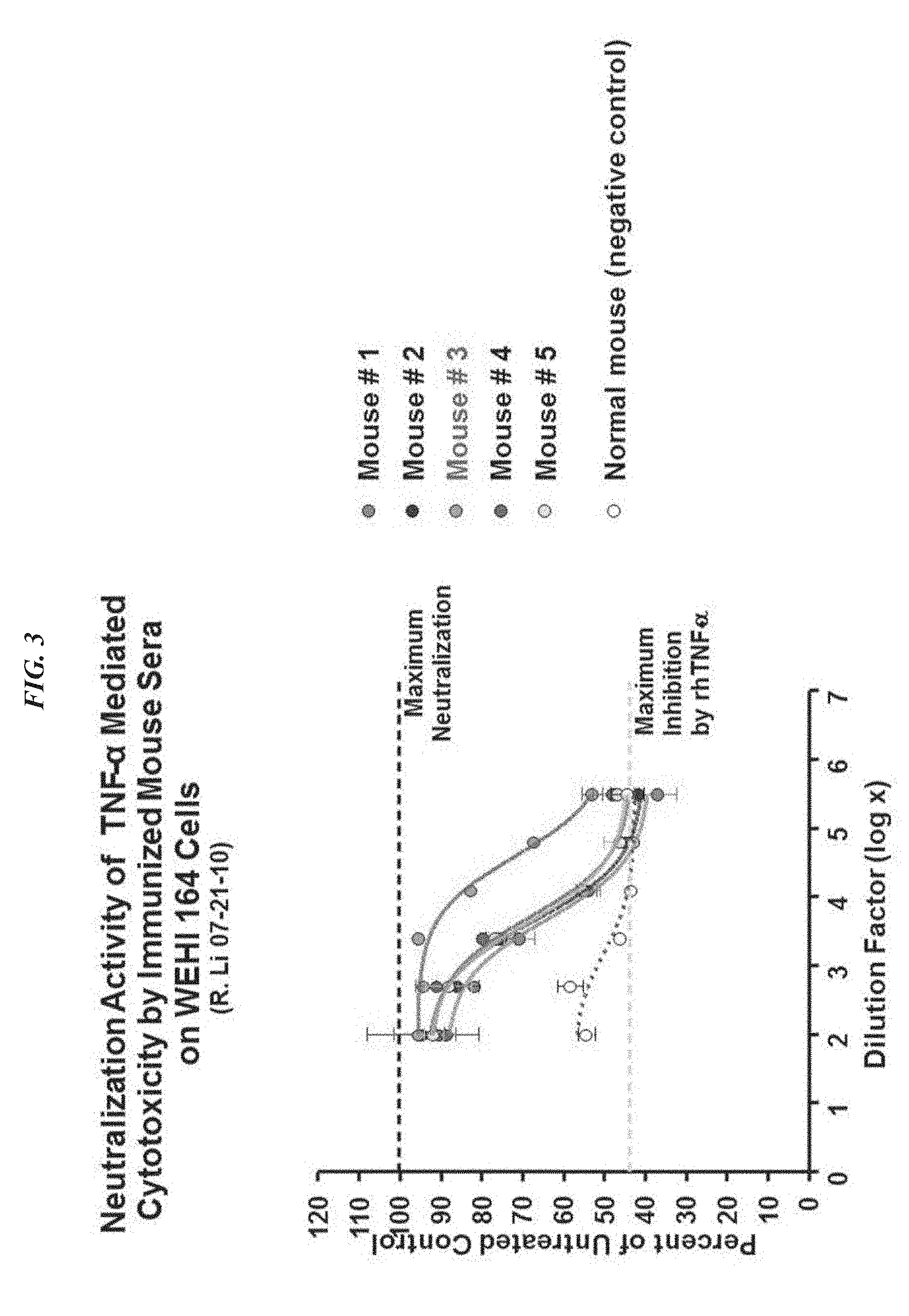
FIG. 3. Neutralization activity of TNF-.alpha. mediated cytotoxicity by immunized mouse sera on WEHI 164 cells. Serum from mouse #3 was the most effective at inhibiting TNF-.alpha. mediated cytotoxicity.
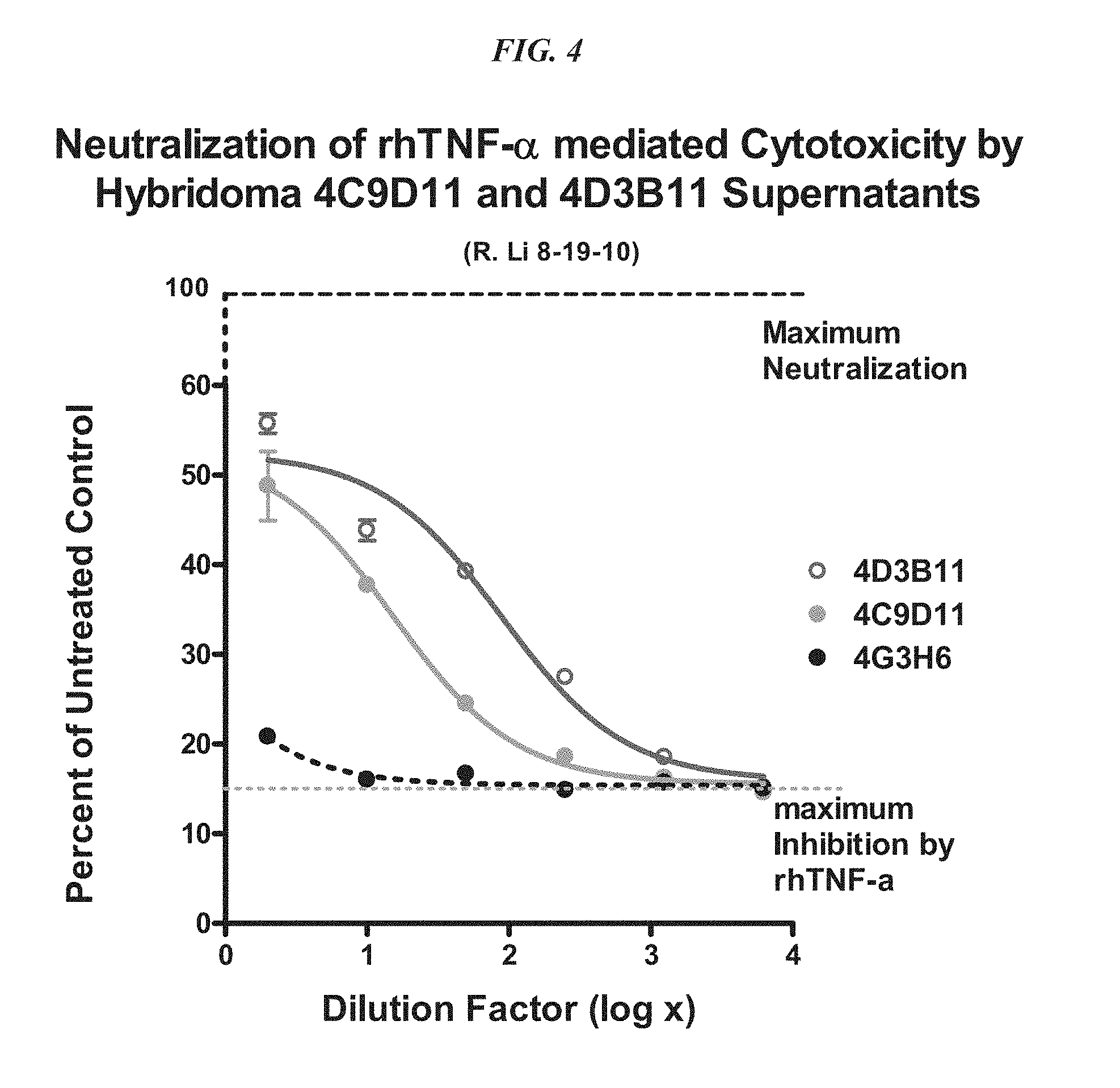
FIG. 4. Neutralization activity of TNF-.alpha. mediated cytotoxicity by antibodies from clones 4C9D11 and 4D3B11 in WEHI 164 cells.
FIG. 5. Neutralization activity of TNF-.alpha. mediated cytotoxicity by antibodies from clones 4C9D11G11 and 4D3B11C4 in L929 cells.
FIG. 6. Antibody-based neutralization of rhTNF-.alpha.-induced cell surface expression of ICAM-1 in ECV-304 cells (a derivative of T24 bladder cancer cell line).
FIG. 7. Amino acid sequence of the anti-IL-6 antibody (2-3B2) heavy chain (VH) sequence (SEQ ID NO:94). The sequence of a homologous heavy chain of the B34781 antibody (SEQ ID NO:95), obtained from the NCBI protein sequence database, is shown for comparison. Putative CDR sequences (underlined) were identified by comparison with the known sequence of the homologous B34781 antibody.
FIG. 8. Amino acid sequence of the anti-IL-6 antibody (2-3B2) light chain (VK) sequence (SEQ ID NO:96). The sequence of a homologous light chain of AAB53778.1 (SEQ ID NO:97), obtained from the NCBI protein sequence database, is shown for comparison. Putative CDR sequences (underlined) were identified by comparison with the known sequence of the homologous AAB53778.1.
FIG. 9. Activity of cIL6/TNF.alpha. DVD construct for neutralizing IL-6 induced phosphorylation of STAT3 in HT-29 cells, compared to parent 2-3B2 anti-IL-6 antibody.
FIG. 10. Amino acid sequence of the anti-TNF-.alpha. antibody (4C9) heavy chain (VH) sequence (SEQ ID NO:98). The sequence of a homologous heavy chain of the AAS66033.1 antibody (SEQ ID NO:99), obtained from the NCBI protein sequence database, is shown for comparison. Putative CDR sequences (underlined) were identified by comparison with the known sequence of the homologous AAS66033.1 antibody.
FIG. 11. Amino acid sequence of the anti-IL-6 antibody (4C9) light chain (VK) sequence (SEQ ID NO:100). The sequence of a homologous heavy chain of AAS66032.1 (SEQ ID NO:101), obtained from the NCBI protein sequence database, is shown for comparison. Putative CDR sequences (underlined) were identified by comparison with the known sequence of the homologous AAS66032.1.
FIG. 12. Schematic illustration of the synthesis of C.sub.K-AD2-cIgG-anti-TNF-.alpha.-pdHL2.
FIG. 13. Inhibition of IL-6 induced phosphorylation of STAT3 by cT*-(c6)-(c6) complex compared to Fab-DDD2-cIL-6 protein.
FIG. 14. Inhibition of natural IL-6 induced phosphorylation of STAT3 by cT*-(c6)-(c6) complex compared to Fab-DDD2-cIL-6 protein.
FIG. 15. Inhibition of rhTNF-.alpha. induced cell death in L929 cells by anti-TNF-.alpha. antibody constructs.
FIG. 16. Inhibition of cell death induced by natural TNF-.alpha. in L929 cells by anti-TNF-.alpha. antibody constructs.
FIG. 17. Relative affinities of cT*-(c6)-(c6), c-anti-TNF-.alpha. and c-anti-IL-6 for IL-6 and TNF-.alpha. from different species.
FIG. 18A. Role of STAT3 in IL-6 and TNF-.alpha. mediated pathways.
FIG. 18B. Role of STAT3 in IL-6 and TNF-.alpha. mediated disease processes.
DEFINITIONS
Unless otherwise specified, "a" or "an" means "one or more".
As used herein, the terms "and" and "or" may be used to mean either the conjunctive or disjunctive. That is, both terms should be understood as equivalent to "and/or" unless otherwise stated.
A "therapeutic agent" is an atom, molecule, or compound that is useful in the treatment of a disease. Examples of therapeutic agents include antibodies, antibody fragments, peptides, drugs, toxins, enzymes, nucleases, hormones, immunomodulators, antisense oligonucleotides, small interfering RNA (siRNA), chelators, boron compounds, photoactive agents, dyes, and radioisotopes.
A "diagnostic agent" is an atom, molecule, or compound that is useful in diagnosing a disease. Useful diagnostic agents include, but are not limited to, radioisotopes, dyes (such as with the biotin-streptavidin complex), contrast agents, fluorescent compounds or molecules, and enhancing agents (e.g., paramagnetic ions) for magnetic resonance imaging (MM).
An "antibody" as used herein refers to a full-length (i.e., naturally occurring or formed by normal immunoglobulin gene fragment recombinatorial processes) immunoglobulin molecule (e.g., an IgG antibody) or an immunologically active (i.e., specifically binding) portion of an immunoglobulin molecule, like an antibody fragment. An "antibody" includes monoclonal, polyclonal, bispecific, multispecific, murine, chimeric, humanized and human antibodies.
A "naked antibody" is an antibody or antigen binding fragment thereof that is not attached to a therapeutic or diagnostic agent. The Fc portion of an intact naked antibody can provide effector functions, such as complement fixation and ADCC (see, e.g., Markrides, Pharmacol Rev 50:59-87, 1998). Other mechanisms by which naked antibodies induce cell death may include apoptosis. (Vaswani and Hamilton, Ann Allergy Asthma Immunol 81: 105-119, 1998.)
An "antibody fragment" is a portion of an intact antibody such as F(ab').sub.2, F(ab).sub.2, Fab', Fab, Fv, sFv, scFv, dAb and the like. Regardless of structure, an antibody fragment binds with the same antigen that is recognized by the full-length antibody. For example, antibody fragments include isolated fragments consisting of the variable regions, such as the "Fv" fragments consisting of the variable regions of the heavy and light chains or recombinant single chain polypeptide molecules in which light and heavy variable regions are connected by a peptide linker ("scFv proteins"). "Single-chain antibodies", often abbreviated as "scFv" consist of a polypeptide chain that comprises both a V.sub.H and a V.sub.L domain which interact to form an antigen-binding site. The V.sub.H and V.sub.L domains are usually linked by a peptide of 1 to 25 amino acid residues. Antibody fragments also include diabodies, triabodies and single domain antibodies (dAb).
An antibody or antibody complex preparation, or a composition described herein, is said to be administered in a "therapeutically effective amount" if the amount administered is physiologically significant. An agent is physiologically significant if its presence results in a detectable change in the physiology of a recipient subject. In particular embodiments, an antibody preparation is physiologically significant if its presence invokes an antitumor response or mitigates the signs and symptoms of an autoimmune disease state. A physiologically significant effect could also be the evocation of a humoral and/or cellular immune response in the recipient subject leading to growth inhibition or death of target cells.
DOCK-AND-LOCK.RTM. (DNL.RTM.)
In preferred embodiments, a bivalent or multivalent antibody is formed as a DOCK-AND-LOCK.RTM. (DNL.RTM.) complex (see, e.g., U.S. Pat. Nos. 7,521,056; 7,527,787; 7,534,866; 7,550,143 and 7,666,400, the Examples section of each of which is incorporated herein by reference.) Generally, the technique takes advantage of the specific and high-affinity binding interactions that occur between a dimerization and docking domain (DDD) sequence of the regulatory (R) subunits of cAMP-dependent protein kinase (PKA) and an anchor domain (AD) sequence derived from any of a variety of AKAP proteins (Baillie et al., FEBS Letters. 2005; 579: 3264. Wong and Scott, Nat. Rev. Mol. Cell Biol. 2004; 5: 959). The DDD and AD peptides may be attached to any protein, peptide or other molecule. Because the DDD sequences spontaneously dimerize and bind to the AD sequence, the technique allows the formation of complexes between any selected molecules that may be attached to DDD or AD sequences.
Although the standard DNL.RTM. complex comprises a trimer with two DDD-linked molecules attached to one AD-linked molecule, variations in complex structure allow the formation of dimers, trimers, tetramers, pentamers, hexamers and other multimers. In some embodiments, the DNL.RTM. complex may comprise two or more antibodies, antibody fragments or fusion proteins which bind to the same antigenic determinant or to two or more different antigens. The DNL.RTM. complex may also comprise one or more other effectors, such as proteins, peptides, immunomodulators, cytokines, interleukins, interferons, binding proteins, peptide ligands, carrier proteins, toxins, ribonucleases such as onconase, inhibitory oligonucleotides such as siRNA, antigens or xenoantigens, polymers such as PEG, enzymes, therapeutic agents, hormones, cytotoxic agents, anti-angiogenic agents, pro-apoptotic agents or any other molecule or aggregate.
PKA, which plays a central role in one of the best studied signal transduction pathways triggered by the binding of the second messenger cAMP to the R subunits, was first isolated from rabbit skeletal muscle in 1968 (Walsh et al., J. Biol. Chem. 1968; 243:3763). The structure of the holoenzyme consists of two catalytic subunits held in an inactive form by the R subunits (Taylor, J. Biol. Chem. 1989; 264:8443). Isozymes of PKA are found with two types of R subunits (RI and RII), and each type has .alpha. and .beta. isoforms (Scott, Pharmacol. Ther. 1991; 50:123). Thus, the four isoforms of PKA regulatory subunits are RI.alpha., RI.beta., RII.alpha. and RII.beta.. The R subunits have been isolated only as stable dimers and the dimerization domain has been shown to consist of the first 44 amino-terminal residues of RII.alpha. (Newlon et al., Nat. Struct. Biol. 1999; 6:222). As discussed below, similar portions of the amino acid sequences of other regulatory subunits are involved in dimerization and docking, each located near the N-terminal end of the regulatory subunit. Binding of cAMP to the R subunits leads to the release of active catalytic subunits for a broad spectrum of serine/threonine kinase activities, which are oriented toward selected substrates through the compartmentalization of PKA via its docking with AKAPs (Scott et al., J. Biol. Chem. 1990; 265; 21561)
Since the first AKAP, microtubule-associated protein-2, was characterized in 1984 (Lohmann et al., Proc. Natl. Acad. Sci USA. 1984; 81:6723), more than 50 AKAPs that localize to various sub-cellular sites, including plasma membrane, actin cytoskeleton, nucleus, mitochondria, and endoplasmic reticulum, have been identified with diverse structures in species ranging from yeast to humans (Wong and Scott, Nat. Rev. Mol. Cell Biol. 2004; 5:959). The AD of AKAPs for PKA is an amphipathic helix of 14-18 residues (Carr et al., J. Biol. Chem. 1991; 266:14188). The amino acid sequences of the AD are quite varied among individual AKAPs, with the binding affinities reported for RII dimers ranging from 2 to 90 nM (Alto et al., Proc. Natl. Acad. Sci. USA. 2003; 100:4445). AKAPs will only bind to dimeric R subunits. For human RII.alpha., the AD binds to a hydrophobic surface formed by the 23 amino-terminal residues (Colledge and Scott, Trends Cell Biol. 1999; 6:216). Thus, the dimerization domain and AKAP binding domain of human RII.alpha. are both located within the same N-terminal 44 amino acid sequence (Newlon et al., Nat. Struct. Biol. 1999; 6:222; Newlon et al., EMBO J. 2001; 20:1651), which is termed the DDD herein.
We have developed a platform technology to utilize the DDD of human PKA regulatory subunit RI.alpha., RI.beta., RII.alpha. or RII.beta. and the AD of AKAP as an excellent pair of linker modules for docking any two entities, referred to hereafter as A and B, into a noncovalent complex, which could be further locked into a DNL.RTM. complex through the introduction of cysteine residues into both the DDD and AD at strategic positions to facilitate the formation of disulfide bonds. The general methodology of the approach is as follows. Entity A is constructed by linking a DDD sequence to a precursor of A, resulting in a first component hereafter referred to as a. Because the DDD sequence would effect the spontaneous formation of a dimer, A would thus be composed of a.sub.2. Entity B is constructed by linking an AD sequence to a precursor of B, resulting in a second component hereafter referred to as b. The dimeric motif of DDD contained in a.sub.2 will create a docking site for binding to the AD sequence contained in b, thus facilitating a ready association of a.sub.2 and b to form a binary, trimeric complex composed of a.sub.2b. This binding event is made irreversible with a subsequent reaction to covalently secure the two entities via disulfide bridges, which occurs very efficiently based on the principle of effective local concentration because the initial binding interactions should bring the reactive thiol groups placed onto both the DDD and AD into proximity (Chmura et al., Proc. Natl. Acad. Sci. USA. 2001; 98:8480) to ligate site-specifically. Using various combinations of linkers, adaptor modules and precursors, a wide variety of DNL.RTM. constructs of different stoichiometry may be produced and used (see, e.g., U.S. Pat. Nos. 7,550,143; 7,521,056; 7,534,866; 7,527,787 and 7,666,400.)
By attaching the DDD and AD away from the functional groups of the two precursors, such site-specific ligations are also expected to preserve the original activities of the two precursors. This approach is modular in nature and potentially can be applied to link, site-specifically and covalently, a wide range of substances, including peptides, proteins, antibodies, antibody fragments, and other effector moieties with a wide range of activities. Utilizing the fusion protein method of constructing AD and DDD conjugated effectors described in the Examples below, virtually any protein or peptide may be incorporated into a DNL.RTM. construct. However, the technique is not limiting and other methods of conjugation may be utilized.
A variety of methods are known for making fusion proteins, including nucleic acid synthesis, hybridization and/or amplification to produce a synthetic double-stranded nucleic acid encoding a fusion protein of interest. Such double-stranded nucleic acids may be inserted into expression vectors for fusion protein production by standard molecular biology techniques (see, e.g. Sambrook et al., Molecular Cloning, A laboratory manual, 2.sup.nd Ed, 1989). In such preferred embodiments, the AD and/or DDD moiety may be attached to either the N-terminal or C-terminal end of an effector protein or peptide. However, the skilled artisan will realize that the site of attachment of an AD or DDD moiety to an effector moiety may vary, depending on the chemical nature of the effector moiety and the part(s) of the effector moiety involved in its physiological activity. Site-specific attachment of a variety of effector moieties may be performed using techniques known in the art, such as the use of bivalent cross-linking reagents and/or other chemical conjugation techniques.
Structure-Function Relationships in AD and DDD Moieties
For different types of DNL.RTM. constructs, different AD or DDD sequences may be utilized. Exemplary DDD and AD sequences are provided below.
TABLE-US-00001 DDD1 (SEQ ID NO: 1) SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA DDD2 (SEQ ID NO: 2) CGHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA AD1 (SEQ ID NO: 3) QIEYLAKQIVDNAIQQA AD2 (SEQ ID NO: 4) CGQIEYLAKQIVDNAIQQAGC
The skilled artisan will realize that DDD1 and DDD2 are based on the DDD sequence of the human RII.alpha. isoform of protein kinase A. However, in alternative embodiments, the DDD and AD moieties may be based on the DDD sequence of the human RI.alpha. form of protein kinase A and a corresponding AKAP sequence, as exemplified in DDD3, DDD3C and AD3 below.
TABLE-US-00002 DDD3 (SEQ ID NO: 5) SLRECELYVQKHNIQALLKDSIVQLCTARPERPMAFLREYFERLEKEEAK DDD3C (SEQ ID NO: 6) MSCGGSLRECELYVQKHNIQALLKDSIVQLCTARPERPMAFLREYFERLE KEEAK AD3 (SEQ ID NO: 7) CGFEELAWKIAKMIWSDVFQQGC
In other alternative embodiments, other sequence variants of AD and/or DDD moieties may be utilized in construction of the DNL.RTM. complexes. For example, there are only four variants of human PKA DDD sequences, corresponding to the DDD moieties of PKA RI.alpha., RII.alpha., RI.beta. and RII.beta.. The RII.alpha. DDD sequence is the basis of DDD1 and DDD2 disclosed above. The four human PKA DDD sequences are shown below. The DDD sequence represents residues 1-44 of RII.alpha., 1-44 of RII.beta., 12-61 of RI.alpha. and 13-66 of RI.beta.. (Note that the sequence of DDD1 is modified slightly from the human PKA RII.alpha. DDD moiety.)
TABLE-US-00003 PKA RI.alpha. (SEQ ID NO: 8) SLRECELYVQKHNIQALLKDVSIVQLCTARPERPMAFLREYFEK LEKEEAK PKA RI.beta. (SEQ ID NO: 9) SLKGCELYVQLHGIQQVLKDCIVHLCISKPERPMKFLREHFEKL EKEENRQILA PKA RII.alpha. (SEQ ID NO: 10) SHIQIPPGLTELLQGYTVEVGQQPPDLVDFAVEYFTRLREARRQ PKA RII.beta. (SEQ ID NO: 11) SIEIPAGLTELLQGFTVEVLRHQPADLLEFALQHFTRLQQENER
The structure-function relationships of the AD and DDD domains have been the subject of investigation. (See, e.g., Burns-Hamuro et al., 2005, Protein Sci 14:2982-92; Carr et al., 2001, J Biol Chem 276:17332-38; Alto et al., 2003, Proc Natl Acad Sci USA 100:4445-50; Hundsrucker et al., 2006, Biochem J 396:297-306; Stokka et al., 2006, Biochem J 400:493-99; Gold et al., 2006, Mol Cell 24:383-95; Kinderman et al., 2006, Mol Cell 24:397-408, the entire text of each of which is incorporated herein by reference.)
For example, Kinderman et al. (2006, Mol Cell 24:397-408) examined the crystal structure of the AD-DDD binding interaction and concluded that the human DDD sequence contained a number of conserved amino acid residues that were important in either dimer formation or AKAP binding, underlined in SEQ ID NO:1 below. (See FIG. 1 of Kinderman et al., 2006, incorporated herein by reference.) The skilled artisan will realize that in designing sequence variants of the DDD sequence, one would desirably avoid changing any of the underlined residues, while conservative amino acid substitutions might be made for residues that are less critical for dimerization and AKAP binding.
TABLE-US-00004 (SEQ ID NO: 1) SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA
As discussed in more detail below, conservative amino acid substitutions have been characterized for each of the twenty common L-amino acids. Thus, based on the data of Kinderman (2006) and conservative amino acid substitutions, potential alternative DDD sequences based on SEQ ID NO:1 are shown in Table 1. In devising Table 1, only highly conservative amino acid substitutions were considered. For example, charged residues were only substituted for residues of the same charge, residues with small side chains were substituted with residues of similar size, hydroxyl side chains were only substituted with other hydroxyls, etc. Because of the unique effect of proline on amino acid secondary structure, no other residues were substituted for proline. A limited number of such potential alternative DDD moiety sequences are shown in SEQ ID NO:12 to SEQ ID NO:31 below. The skilled artisan will realize that an almost unlimited number of alternative species within the genus of DDD moieties can be constructed by standard techniques, for example using a commercial peptide synthesizer or well known site-directed mutagenesis techniques. The effect of the amino acid substitutions on AD moiety binding may also be readily determined by standard binding assays, for example as disclosed in Alto et al. (2003, Proc Natl Acad Sci USA 100:4445-50).
TABLE-US-00005 TABLE 1 Conservative Amino Acid Substitutions in DDD1 (SEQ ID NO: 1). Consensus sequence disclosed as SEQ ID NO: 87. S H I Q I P P G L T E L L Q G Y T V E V L R T K N A S D N A S D K R Q Q P P D L V E F A V E Y F T R L R E A R A N N E D L D S K K D L K L I I I V V V THIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 12) SKIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 13) SRIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 14) SHINIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 15) SHIQIPPALTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 16) SHIQIPPGLSELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 17) SHIQIPPGLTDLLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 18) SHIQIPPGLTELLNGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 19) SHIQIPPGLTELLQAYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 20) SHIQIPPGLTELLQGYSVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 21) SHIQIPPGLTELLQGYTVDVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 22) SHIQIPPGLTELLQGYTVEVLKQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 23) SHIQIPPGLTELLQGYTVEVLRNQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 24) SHIQIPPGLTELLQGYTVEVLRQNPPDLVEFAVEYFTRLREARA (SEQ ID NO: 25) SHIQIPPGLTELLQGYTVEVLRQQPPELVEFAVEYFTRLREARA (SEQ ID NO: 26) SHIQIPPGLTELLQGYTVEVLRQQPPDLVDFAVEYFTRLREARA (SEQ ID NO: 27) SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFLVEYFTRLREARA (SEQ ID NO: 28) SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFIVEYFTRLREARA (SEQ ID NO: 29) SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFVVEYFTRLREARA (SEQ ID NO: 30) SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVDYFTRLREARA (SEQ ID NO: 31)
Alto et al. (2003, Proc Natl Acad Sci USA 100:4445-50) performed a bioinformatic analysis of the AD sequence of various AKAP proteins to design an RII selective AD sequence called AKAP-IS (SEQ ID NO:3), with a binding constant for DDD of 0.4 nM. The AKAP-IS sequence was designed as a peptide antagonist of AKAP binding to PKA. Residues in the AKAP-IS sequence where substitutions tended to decrease binding to DDD are underlined in SEQ ID NO:3 below. The skilled artisan will realize that in designing sequence variants of the AD sequence, one would desirably avoid changing any of the underlined residues, while conservative amino acid substitutions might be made for residues that are less critical for DDD binding. Table 2 shows potential conservative amino acid substitutions in the sequence of AKAP-IS (AD1, SEQ ID NO:3), similar to that shown for DDD1 (SEQ ID NO:1) in Table 1 above.
A limited number of such potential alternative AD moiety sequences are shown in SEQ ID NO:32 to SEQ ID NO:49 below. Again, a very large number of species within the genus of possible AD moiety sequences could be made, tested and used by the skilled artisan, based on the data of Alto et al. (2003). It is noted that FIG. 2 of Alto (2003) shows an even large number of potential amino acid substitutions that may be made, while retaining binding activity to DDD moieties, based on actual binding experiments.
TABLE-US-00006 AKAP-IS (SEQ ID NO: 3) QIEYLAKQIVDNAIQQA
TABLE-US-00007 TABLE 2 Conservative Amino Acid Substitutions in AD1 (SEQ ID NO: 3). Consensus sequence disclosed as SEQ ID NO: 88. Q I E Y L A K Q I V D N A I Q Q A N L D F I R N E Q N N L V T V I S V NIEYLAKQIVDNAIQQA (SEQ ID NO: 32) QLEYLAKQIVDNAIQQA (SEQ ID NO: 33) QVEYLAKQIVDNAIQQA (SEQ ID NO: 34) QIDYLAKQIVDNAIQQA (SEQ ID NO: 35) QIEFLAKQIVDNAIQQA (SEQ ID NO: 36) QIETLAKQIVDNAIQQA (SEQ ID NO: 37) QIESLAKQIVDNAIQQA (SEQ ID NO: 38) QIEYIAKQIVDNAIQQA (SEQ ID NO: 39) QIEYVAKQIVDNAIQQA (SEQ ID NO: 40) QIEYLARQIVDNAIQQA (SEQ ID NO: 41) QIEYLAKNIVDNAIQQA (SEQ ID NO: 42) QIEYLAKQIVENAIQQA (SEQ ID NO: 43) QIEYLAKQIVDQAIQQA (SEQ ID NO: 44) QIEYLAKQIVDNAINQA (SEQ ID NO: 45) QIEYLAKQIVDNAIQNA (SEQ ID NO: 46) QIEYLAKQIVDNAIQQL (SEQ ID NO: 47) QIEYLAKQIVDNAIQQI (SEQ ID NO: 48) QIEYLAKQIVDNAIQQV (SEQ ID NO: 49)
Gold et al. (2006, Mol Cell 24:383-95) utilized crystallography and peptide screening to develop a SuperAKAP-IS sequence (SEQ ID NO:50), exhibiting a five order of magnitude higher selectivity for the RII isoform of PKA compared with the RI isoform. Underlined residues indicate the positions of amino acid substitutions, relative to the AKAP-IS sequence, which increased binding to the DDD moiety of RII.alpha.. In this sequence, the N-terminal Q residue is numbered as residue number 4 and the C-terminal A residue is residue number 20. Residues where substitutions could be made to affect the affinity for RII.alpha. were residues 8, 11, 15, 16, 18, 19 and 20 (Gold et al., 2006). It is contemplated that in certain alternative embodiments, the SuperAKAP-IS sequence may be substituted for the AKAP-IS AD moiety sequence to prepare DNL.RTM. constructs. Other alternative sequences that might be substituted for the AKAP-IS AD sequence are shown in SEQ ID NO:51-53. Substitutions relative to the AKAP-IS sequence are underlined. It is anticipated that, as with the AD2 sequence shown in SEQ ID NO:4, the AD moiety may also include the additional N-terminal residues cysteine and glycine and C-terminal residues glycine and cysteine.
TABLE-US-00008 SuperAKAP-IS (SEQ ID NO: 50) QIEYVAKQIVDYAIHQA Alternative AKAP sequences (SEQ ID NO: 51) QIEYKAKQIVDHAIHQA (SEQ ID NO: 52) QIEYHAKQIVDHAIHQA (SEQ ID NO: 53) QIEYVAKQIVDHAIHQA
FIG. 2 of Gold et al. disclosed additional DDD-binding sequences from a variety of AKAP proteins, shown below.
TABLE-US-00009 RII-Specific AKAPs AKAP-KL (SEQ ID NO: 54) PLEYQAGLLVQNAIQQAI AKAP79 (SEQ ID NO: 55) LLIETASSLVKNAIQLSI AKAP-Lbc (SEQ ID NO: 56) LIEEAASRIVDAVIEQVK RI-Specific AKAPs AKAPce (SEQ ID NO: 57) ALYQFADRFSELVISEAL RIAD (SEQ ID NO: 58) LEQVANQLADQIIKEAT PV38 (SEQ ID NO: 59) FEELAWKIAKMIWSDVF Dual-Specificity AKAPs AKAP7 (SEQ ID NO: 60) ELVRLSKRLVENAVLKAV MAP2D (SEQ ID NO: 61) TAEEVSARIVQVVTAEAV DAKAP1 (SEQ ID NO: 62) QIKQAAFQLISQVILEAT DAKAP2 (SEQ ID NO: 63) LAWKIAKMIVSDVMQQ
Stokka et al. (2006, Biochem J 400:493-99) also developed peptide competitors of AKAP binding to PKA, shown in SEQ ID NO:64-66. The peptide antagonists were designated as Ht31 (SEQ ID NO:64), RIAD (SEQ ID NO:65) and PV-38 (SEQ ID NO:66). The Ht-31 peptide exhibited a greater affinity for the RII isoform of PKA, while the RIAD and PV-38 showed higher affinity for RI.
TABLE-US-00010 Ht31 (SEQ ID NO: 64) DLIEEAASRIVDAVIEQVKAAGAY RIAD (SEQ ID NO: 65) LEQYANQLADQIIKEATE PV-38 (SEQ ID NO: 66) FEELAWKIAKMIWSDVFQQC
Hundsrucker et al. (2006, Biochem J 396:297-306) developed still other peptide competitors for AKAP binding to PKA, with a binding constant as low as 0.4 nM to the DDD of the RII form of PKA. The sequences of various AKAP antagonistic peptides are provided in Table 1 of Hundsrucker et al., reproduced in Table 3 below. AKAPIS represents a synthetic RII subunit-binding peptide. All other peptides are derived from the RII-binding domains of the indicated AKAPs.
TABLE-US-00011 TABLE 3 AKAP Peptide sequences Peptide Sequence AKAPIS QIEYLAKQIVDNAIQQA (SEQ ID NO: 3) AKAPIS-P QIEYLAKQIPDNAIQQA (SEQ ID NO: 67) Ht31 KGADLIEEAASRIVDAVIEQVKAAG (SEQ ID NO: 68) Ht31-P KGADLIEEAASRIPDAPIEQVKAAG (SEQ ID NO: 69) AKAP7.delta.-wt-pep PEDAELVRLSKRLVENAVLKAVQQY (SEQ ID NO: 70) AKAP7.delta.-L304T-pep PEDAELVRTSKRLVENAVLKAVQQY (SEQ ID NO: 71) AKAP7.delta.-L308D-pep PEDAELVRLSKRDVENAVLKAVQQY (SEQ ID NO: 72) AKAP7.delta.-P-pep PEDAELVRLSKRLPENAVLKAVQQY (SEQ ID NO: 73) AKAP7.delta.-PP-pep PEDAELVRLSKRLPENAPLKAVQQY (SEQ ID NO: 74) AKAP7.delta.-L314E-pep PEDAELVRLSKRLVENAVEKAVQQY (SEQ ID NO: 75) AKAP1-pep EEGLDRNEEIKRAAFQIISQVISEA (SEQ ID NO: 76) AKAP2-pep LVDDPLEYQAGLLVQNAIQQAIAEQ (SEQ ID NO: 77) AKAP5-pep QYETLLIETASSLVKNAIQLSIEQL (SEQ ID NO: 78) AKAP9-pep LEKQYQEQLEEEVAKVIVSMSIAFA (SEQ ID NO: 79) AKAP10-pep NTDEAQEELAWKIAKMIVSDIMQQA (SEQ ID NO: 80) AKAP11-pep VNLDKKAVLAEKIVAEAIEKAEREL (SEQ ID NO: 81) AKAP12-pep NGILELETKSSKLVQNIIQTAVDQF (SEQ ID NO: 82) AKAP14-pep TQDKNYEDELTQVALALVEDVINYA (SEQ ID NO: 83) Rab32-pep ETSAKDNINIEEAARFLVEKILVNH (SEQ ID NO: 84)
Residues that were highly conserved among the AD domains of different AKAP proteins are indicated below by underlining with reference to the AKAP IS sequence (SEQ ID NO:3). The residues are the same as observed by Alto et al. (2003), with the addition of the C-terminal alanine residue. (See FIG. 4 of Hundsrucker et al. (2006), incorporated herein by reference.) The sequences of peptide antagonists with particularly high affinities for the RII DDD sequence were those of AKAP-IS, AKAP7.delta.-wt-pep, AKAP7.delta.-L304T-pep and AKAP7.delta.-L308D-pep.
TABLE-US-00012 AKAP-IS (SEQ ID NO: 3) QIEYLAKQIVDNAIQQA
Carr et al. (2001, J Biol Chem 276:17332-38) examined the degree of sequence homology between different AKAP-binding DDD sequences from human and non-human proteins and identified residues in the DDD sequences that appeared to be the most highly conserved among different DDD moieties. These are indicated below by underlining with reference to the human PKA RII.alpha. DDD sequence of SEQ ID NO:1. Residues that were particularly conserved are further indicated by italics. The residues overlap with, but are not identical to those suggested by Kinderman et al. (2006) to be important for binding to AKAP proteins. The skilled artisan will realize that in designing sequence variants of DDD, it would be most preferred to avoid changing the most conserved residues (italicized), and it would be preferred to also avoid changing the conserved residues (underlined), while conservative amino acid substitutions may be considered for residues that are neither underlined nor italicized.
TABLE-US-00013 (SEQ ID NO: 1) SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA
A modified set of conservative amino acid substitutions for the DDD1 (SEQ ID NO:1) sequence, based on the data of Carr et al. (2001) is shown in Table 4. Even with this reduced set of substituted sequences, there are over 65,000 possible alternative DDD moiety sequences that may be produced, tested and used by the skilled artisan without undue experimentation. The skilled artisan could readily derive such alternative DDD amino acid sequences as disclosed above for Table 1 and Table 2.
TABLE-US-00014 TABLE 4 Conservative Amino Acid Substitutions in DDD1 (SEQ ID NO: 1). Consensus sequence disclosed as SEQ ID NO: 89. S H I Q I P P G L T E L L Q G Y T V E V L R T N S I L A Q Q P P D L V E F A V E Y F T R L R E A R A N L D S K K L L I I I V V V
The skilled artisan will realize that these and other amino acid substitutions in the DDD or AD amino acid sequences may be utilized to produce alternative species within the genus of AD or DDD moieties, using techniques that are standard in the field and only routine experimentation.
Amino Acid Substitutions
In alternative embodiments, the disclosed methods and compositions may involve production and use of proteins or peptides with one or more substituted amino acid residues. For example, the DDD and/or AD sequences used to make DNL.RTM. constructs may be modified as discussed above.
The skilled artisan will be aware that, in general, amino acid substitutions typically involve the replacement of an amino acid with another amino acid of relatively similar properties (i.e., conservative amino acid substitutions). The properties of the various amino acids and effect of amino acid substitution on protein structure and function have been the subject of extensive study and knowledge in the art.
For example, the hydropathic index of amino acids may be considered (Kyte & Doolittle, 1982, J. Mol. Biol., 157:105-132). The relative hydropathic character of the amino acid contributes to the secondary structure of the resultant protein, which in turn defines the interaction of the protein with other molecules. Each amino acid has been assigned a hydropathic index on the basis of its hydrophobicity and charge characteristics (Kyte & Doolittle, 1982), these are: isoleucine (+4.5); valine (+4.2); leucine (+3.8); phenylalanine (+2.8); cysteine/cystine (+2.5); methionine (+1.9); alanine (+1.8); glycine (-0.4); threonine (-0.7); serine (-0.8); tryptophan (-0.9); tyrosine (-1.3); proline (-1.6); histidine (-3.2); glutamate (-3.5); glutamine (-3.5); aspartate (-3.5); asparagine (-3.5); lysine (-3.9); and arginine (-4.5). In making conservative substitutions, the use of amino acids whose hydropathic indices are within .+-.2 is preferred, within .+-.1 are more preferred, and within .+-.0.5 are even more preferred.
Amino acid substitution may also take into account the hydrophilicity of the amino acid residue (e.g., U.S. Pat. No. 4,554,101). Hydrophilicity values have been assigned to amino acid residues: arginine (+3.0); lysine (+3.0); aspartate (+3.0); glutamate (+3.0); serine (+0.3); asparagine (+0.2); glutamine (+0.2); glycine (0); threonine (-0.4); proline (-0.5.+-0.1); alanine (-0.5); histidine (-0.5); cysteine (-1.0); methionine (-1.3); valine (-1.5); leucine (-1.8); isoleucine (-1.8); tyrosine (-2.3); phenylalanine (-2.5); tryptophan (-3.4). Replacement of amino acids with others of similar hydrophilicity is preferred.
Other considerations include the size of the amino acid side chain. For example, it would generally not be preferred to replace an amino acid with a compact side chain, such as glycine or serine, with an amino acid with a bulky side chain, e.g., tryptophan or tyrosine. The effect of various amino acid residues on protein secondary structure is also a consideration. Through empirical study, the effect of different amino acid residues on the tendency of protein domains to adopt an alpha-helical, beta-sheet or reverse turn secondary structure has been determined and is known in the art (see, e.g., Chou & Fasman, 1974, Biochemistry, 13:222-245; 1978, Ann. Rev. Biochem., 47: 251-276; 1979, Biophys. J., 26:367-384).
Based on such considerations and extensive empirical study, tables of conservative amino acid substitutions have been constructed and are known in the art. For example: arginine and lysine; glutamate and aspartate; serine and threonine; glutamine and asparagine; and valine, leucine and isoleucine. Alternatively: Ala (A) leu, ile, val; Arg (R) gln, asn, lys; Asn (N) his, asp, lys, arg, gln; Asp (D) asn, glu; Cys (C) ala, ser; Gln (Q) glu, asn; Glu (E) gln, asp; Gly (G) ala; His (H) asn, gln, lys, arg; Ile (I) val, met, ala, phe, leu; Leu (L) val, met, ala, phe, ile; Lys (K) gln, asn, arg; Met (M) phe, ile, leu; Phe (F) leu, val, ile, ala, tyr; Pro (P) ala; Ser (S), thr; Thr (T) ser; Trp (W) phe, tyr; Tyr (Y) trp, phe, thr, ser; Val (V) ile, leu, met, phe, ala.
Other considerations for amino acid substitutions include whether or not the residue is located in the interior of a protein or is solvent exposed. For interior residues, conservative substitutions would include: Asp and Asn; Ser and Thr; Ser and Ala; Thr and Ala; Ala and Gly; Ile and Val; Val and Leu; Leu and Ile; Leu and Met; Phe and Tyr; Tyr and Trp. (See, e.g., PROWL website at rockefeller.edu) For solvent exposed residues, conservative substitutions would include: Asp and Asn; Asp and Glu; Glu and Gln; Glu and Ala; Gly and Asn; Ala and Pro; Ala and Gly; Ala and Ser; Ala and Lys; Ser and Thr; Lys and Arg; Val and Leu; Leu and Ile; Ile and Val; Phe and Tyr. (Id.) Various matrices have been constructed to assist in selection of amino acid substitutions, such as the PAM250 scoring matrix, Dayhoff matrix, Grantham matrix, McLachlan matrix, Doolittle matrix, Henikoff matrix, Miyata matrix, Fitch matrix, Jones matrix, Rao matrix, Levin matrix and Risler matrix (Idem.)
In determining amino acid substitutions, one may also consider the existence of intermolecular or intramolecular bonds, such as formation of ionic bonds (salt bridges) between positively charged residues (e.g., His, Arg, Lys) and negatively charged residues (e.g., Asp, Glu) or disulfide bonds between nearby cysteine residues.
Methods of substituting any amino acid for any other amino acid in an encoded protein sequence are well known and a matter of routine experimentation for the skilled artisan, for example by the technique of site-directed mutagenesis or by synthesis and assembly of oligonucleotides encoding an amino acid substitution and splicing into an expression vector construct.
Antibodies and Antibody Fragments
Techniques for preparing monoclonal antibodies against virtually any target antigen, such as IL-6 or TNF-.alpha., are well known in the art. See, for example, Kohler and Milstein, Nature 256: 495 (1975), and Coligan et al. (eds.), CURRENT PROTOCOLS IN IMMUNOLOGY, VOL. 1, pages 2.5.1-2.6.7 (John Wiley & Sons 1991). Briefly, monoclonal antibodies can be obtained by injecting mice with a composition comprising an antigen, removing the spleen to obtain B-lymphocytes, fusing the B-lymphocytes with myeloma cells to produce hybridomas, cloning the hybridomas, selecting positive clones which produce antibodies to the antigen, culturing the clones that produce antibodies to the antigen, and isolating the antibodies from the hybridoma cultures.
MAbs can be isolated and purified from hybridoma cultures by a variety of well-established techniques. Such isolation techniques include affinity chromatography with Protein-A SEPHAROSE.RTM., size-exclusion chromatography, and ion-exchange chromatography. See, for example, Coligan at pages 2.7.1-2.7.12 and pages 2.9.1-2.9.3. Also, see Baines et al., "Purification of Immunoglobulin G (IgG)," in METHODS IN MOLECULAR BIOLOGY, VOL. 10, pages 79-104 (The Humana Press, Inc. 1992).
After the initial raising of antibodies to the immunogen, the antibodies can be sequenced and subsequently prepared by recombinant techniques. Humanization and chimerization of murine antibodies and antibody fragments are well known to those skilled in the art. The use of antibody components derived from humanized, chimeric or human antibodies obviates potential problems associated with the immunogenicity of murine constant regions.
Chimeric Antibodies
A chimeric antibody is a recombinant protein in which the variable regions of a human antibody have been replaced by the variable regions of, for example, a mouse antibody, including the complementarity-determining regions (CDRs) of the mouse antibody. Chimeric antibodies exhibit decreased immunogenicity and increased stability when administered to a subject. General techniques for cloning murine immunoglobulin variable domains are disclosed, for example, in Orlandi et al., Proc. Nat'l Acad. Sci. USA 86: 3833 (1989). Techniques for constructing chimeric antibodies are well known to those of skill in the art. As an example, Leung et al., Hybridoma 13:469 (1994), produced an LL2 chimera by combining DNA sequences encoding the V.sub..kappa. and V.sub.H domains of murine LL2, an anti-CD22 monoclonal antibody, with respective human .kappa. and IgG.sub.1 constant region domains.
Humanized Antibodies
Techniques for producing humanized MAbs are well known in the art (see, e.g., Jones et al., Nature 321: 522 (1986), Riechmann et al., Nature 332: 323 (1988), Verhoeyen et al., Science 239: 1534 (1988), Carter et al., Proc. Nat'l Acad. Sci. USA 89: 4285 (1992), Sandhu, Crit. Rev. Biotech. 12: 437 (1992), and Singer et al., J. Immun. 150: 2844 (1993)). A chimeric or murine monoclonal antibody may be humanized by transferring the mouse CDRs from the heavy and light variable chains of the mouse immunoglobulin into the corresponding variable domains of a human antibody. The mouse framework regions (FR) in the chimeric monoclonal antibody are also replaced with human FR sequences. As simply transferring mouse CDRs into human FRs often results in a reduction or even loss of antibody affinity, additional modification might be required in order to restore the original affinity of the murine antibody. This can be accomplished by the replacement of one or more human residues in the FR regions with their murine counterparts to obtain an antibody that possesses good binding affinity to its epitope. See, for example, Tempest et al., Biotechnology 9:266 (1991) and Verhoeyen et al., Science 239: 1534 (1988). Generally, those human FR amino acid residues that differ from their murine counterparts and are located close to or touching one or more CDR amino acid residues would be candidates for substitution.
Human Antibodies
Methods for producing fully human antibodies using either combinatorial approaches or transgenic animals transformed with human immunoglobulin loci are known in the art (e.g., Mancini et al., 2004, New Microbiol. 27:315-28; Conrad and Scheller, 2005, Comb. Chem. High Throughput Screen. 8:117-26; Brekke and Loset, 2003, Curr. Opin. Phamacol. 3:544-50). A fully human antibody also can be constructed by genetic or chromosomal transfection methods, as well as phage display technology, all of which are known in the art. See for example, McCafferty et al., Nature 348:552-553 (1990). Such fully human antibodies are expected to exhibit even fewer side effects than chimeric or humanized antibodies and to function in vivo as essentially endogenous human antibodies. In certain embodiments, the claimed methods and procedures may utilize human antibodies produced by such techniques.
In one alternative, the phage display technique may be used to generate human antibodies (e.g., Dantas-Barbosa et al., 2005, Genet. Mol. Res. 4:126-40). Human antibodies may be generated from normal humans or from humans that exhibit a particular disease state, such as cancer (Dantas-Barbosa et al., 2005). The advantage to constructing human antibodies from a diseased individual is that the circulating antibody repertoire may be biased towards antibodies against disease-associated antigens.
In one non-limiting example of this methodology, Dantas-Barbosa et al. (2005) constructed a phage display library of human Fab antibody fragments from osteosarcoma patients. Generally, total RNA was obtained from circulating blood lymphocytes (Id.). Recombinant Fab were cloned from the .mu., .gamma. and .kappa. chain antibody repertoires and inserted into a phage display library (Id.). RNAs were converted to cDNAs and used to make Fab cDNA libraries using specific primers against the heavy and light chain immunoglobulin sequences (Marks et al., 1991, J. Mol. Biol. 222:581-97). Library construction was performed according to Andris-Widhopf et al. (2000, In: Phage Display Laboratory Manual, Barbas et al. (eds), 1.sup.st edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. pp. 9.1 to 9.22). The final Fab fragments were digested with restriction endonucleases and inserted into the bacteriophage genome to make the phage display library. Such libraries may be screened by standard phage display methods, as known in the art (see, e.g., Pasqualini and Ruoslahti, 1996, Nature 380:364-366; Pasqualini, 1999, The Quart. J. Nucl. Med. 43:159-162).
Phage display can be performed in a variety of formats, for their review, see e.g. Johnson and Chiswell, Current Opinion in Structural Biology 3:5564-571 (1993). Human antibodies may also be generated by in vitro activated B cells. See U.S. Pat. Nos. 5,567,610 and 5,229,275, incorporated herein by reference in their entirety. The skilled artisan will realize that these techniques are exemplary and any known method for making and screening human antibodies or antibody fragments may be utilized.
In another alternative, transgenic animals that have been genetically engineered to produce human antibodies may be used to generate antibodies against essentially any immunogenic target, using standard immunization protocols. Methods for obtaining human antibodies from transgenic mice are disclosed by Green et al., Nature Genet. 7:13 (1994), Lonberg et al., Nature 368:856 (1994), and Taylor et al., Int. Immun. 6:579 (1994). A non-limiting example of such a system is the XENOMOUSE.RTM. (e.g., Green et al., 1999, J. Immunol. Methods 231:11-23) from Abgenix (Fremont, Calif.). In the XENOMOUSE.RTM. and similar animals, the mouse antibody genes have been inactivated and replaced by functional human antibody genes, while the remainder of the mouse immune system remains intact.
The XENOMOUSE.RTM. was transformed with germline-configured YACs (yeast artificial chromosomes) that contained portions of the human IgH and Igkappa loci, including the majority of the variable region sequences, along accessory genes and regulatory sequences. The human variable region repertoire may be used to generate antibody producing B cells, which may be processed into hybridomas by known techniques. A XENOMOUSE.RTM. immunized with a target antigen will produce human antibodies by the normal immune response, which may be harvested and/or produced by standard techniques discussed above. A variety of strains of XENOMOUSE.RTM. are available, each of which is capable of producing a different class of antibody. Transgenically produced human antibodies have been shown to have therapeutic potential, while retaining the pharmacokinetic properties of normal human antibodies (Green et al., 1999). The skilled artisan will realize that the claimed compositions and methods are not limited to use of the XENOMOUSE.RTM. system but may utilize any transgenic animal that has been genetically engineered to produce human antibodies.
Antibody Fragments
Antibody fragments which recognize specific epitopes can be generated by known techniques. Antibody fragments are antigen binding portions of an antibody, such as F(ab').sub.2, Fab', F(ab).sub.2, Fab, Fv, sFv and the like. F(ab').sub.2 fragments can be produced by pepsin digestion of the antibody molecule and Fab' fragments can be generated by reducing disulfide bridges of the F(ab').sub.2 fragments. Alternatively, Fab' expression libraries can be constructed (Huse et al., 1989, Science, 246:1274-1281) to allow rapid and easy identification of monoclonal Fab' fragments with the desired specificity. F(ab).sub.2 fragments may be generated by papain digestion of an antibody.
A single chain Fv molecule (scFv) comprises a VL domain and a VH domain. The VL and VH domains associate to form a target binding site. These two domains are further covalently linked by a peptide linker (L). Methods for making scFv molecules and designing suitable peptide linkers are described in U.S. Pat. Nos. 4,704,692, 4,946,778, R. Raag and M. Whitlow, "Single Chain Fvs." FASEB Vol 9:73-80 (1995) and R. E. Bird and B. W. Walker, "Single Chain Antibody Variable Regions," TIBTECH, Vol 9: 132-137 (1991).
Techniques for producing single domain antibodies are also known in the art, as disclosed for example in Cossins et al. (2006, Prot Express Purif 51:253-259), incorporated herein by reference. Single domain antibodies (VHH) may be obtained, for example, from camels, alpacas or llamas by standard immunization techniques. (See, e.g., Muyldermans et al., TIBS 26:230-235, 2001; Yau et al., J Immunol Methods 281:161-75, 2003; Maass et al., J Immunol Methods 324:13-25, 2007). The VHH may have potent antigen-binding capacity and can interact with novel epitopes that are inaccessible to conventional VH-VL pairs. (Muyldermans et al., 2001). Alpaca serum IgG contains about 50% camelid heavy chain only IgG antibodies (HCAbs) (Maass et al., 2007). Alpacas may be immunized with known antigens, such as TNF-.alpha., and VHHs can be isolated that bind to and neutralize the target antigen (Maass et al., 2007). PCR primers that amplify virtually all alpaca VHH coding sequences have been identified and may be used to construct alpaca VHH phage display libraries, which can be used for antibody fragment isolation by standard biopanning techniques well known in the art (Maass et al., 2007). In certain embodiments, anti-pancreatic cancer VHH antibody fragments may be utilized in the claimed compositions and methods.
An antibody fragment can be prepared by proteolytic hydrolysis of the full length antibody or by expression in E. coli or another host of the DNA coding for the fragment. An antibody fragment can be obtained by pepsin or papain digestion of full length antibodies by conventional methods. These methods are described, for example, by Goldenberg, U.S. Pat. Nos. 4,036,945 and 4,331,647 and references contained therein. Also, see Nisonoff et al., Arch Biochem. Biophys. 89: 230 (1960); Porter, Biochem. J. 73: 119 (1959), Edelman et al., in METHODS IN ENZYMOLOGY VOL. 1, page 422 (Academic Press 1967), and Coligan at pages 2.8.1-2.8.10 and 2.10.-2.10.4.
Known Antibodies
Various embodiments, for example in combination therapy, may involve the use of antibodies binding to target antigens besides IL-6 or TNF-.alpha.. A variety of antibodies are commercially available and/or known in the art. Antibodies of use may be commercially obtained, for example, from the American Type Culture Collection (ATCC, Manassas, Va.). A large number of antibodies against various disease targets, including but not limited to tumor-associated antigens, have been deposited at the ATCC and/or have published variable region sequences and are available for use in the claimed methods and compositions. See, e.g., U.S. Pat. Nos. 7,312,318; 7,282,567; 7,151,164; 7,074,403; 7,060,802; 7,056,509; 7,049,060; 7,045,132; 7,041,803; 7,041,802; 7,041,293; 7,038,018; 7,037,498; 7,012,133; 7,001,598; 6,998,468; 6,994,976; 6,994,852; 6,989,241; 6,974,863; 6,965,018; 6,964,854; 6,962,981; 6,962,813; 6,956,107; 6,951,924; 6,949,244; 6,946,129; 6,943,020; 6,939,547; 6,921,645; 6,921,645; 6,921,533; 6,919,433; 6,919,078; 6,916,475; 6,905,681; 6,899,879; 6,893,625; 6,887,468; 6,887,466; 6,884,594; 6,881,405; 6,878,812; 6,875,580; 6,872,568; 6,867,006; 6,864,062; 6,861,511; 6,861,227; 6,861,226; 6,838,282; 6,835,549; 6,835,370; 6,824,780; 6,824,778; 6,812,206; 6,793,924; 6,783,758; 6,770,450; 6,767,711; 6,764,688; 6,764,681; 6,764,679; 6,743,898; 6,733,981; 6,730,307; 6,720,155; 6,716,966; 6,709,653; 6,693,176; 6,692,908; 6,689,607; 6,689,362; 6,689,355; 6,682,737; 6,682,736; 6,682,734; 6,673,344; 6,653,104; 6,652,852; 6,635,482; 6,630,144; 6,610,833; 6,610,294; 6,605,441; 6,605,279; 6,596,852; 6,592,868; 6,576,745; 6,572,856; 6,566,076; 6,562,618; 6,545,130; 6,544,749; 6,534,058; 6,528,625; 6,528,269; 6,521,227; 6,518,404; 6,511,665; 6,491,915; 6,488,930; 6,482,598; 6,482,408; 6,479,247; 6,468,531; 6,468,529; 6,465,173; 6,461,823; 6,458,356; 6,455,044; 6,455,040, 6,451,310; 6,444,206; 6,441,143; 6,432,404; 6,432,402; 6,419,928; 6,413,726; 6,406,694; 6,403,770; 6,403,091; 6,395,276; 6,395,274; 6,387,350; 6,383,759; 6,383,484; 6,376,654; 6,372,215; 6,359,126; 6,355,481; 6,355,444; 6,355,245; 6,355,244; 6,346,246; 6,344,198; 6,340,571; 6,340,459; 6,331,175; 6,306,393; 6,254,868; 6,187,287; 6,183,744; 6,129,914; 6,120,767; 6,096,289; 6,077,499; 5,922,302; 5,874,540; 5,814,440; 5,798,229; 5,789,554; 5,776,456; 5,736,119; 5,716,595; 5,677,136; 5,587,459; 5,443,953, 5,525,338, the Examples section of each of which is incorporated herein by reference. These are exemplary only and a wide variety of other antibodies and their hybridomas are known in the art. The skilled artisan will realize that antibody sequences or antibody-secreting hybridomas against almost any disease-associated antigen may be obtained by a simple search of the ATCC, NCBI and/or USPTO databases for antibodies against a selected disease-associated target of interest. The antigen binding domains of the cloned antibodies may be amplified, excised, ligated into an expression vector, transfected into an adapted host cell and used for protein production, using standard techniques well known in the art (see, e.g., U.S. Pat. Nos. 7,531,327; 7,537,930; 7,608,425 and 7,785,880, the Examples section of each of which is incorporated herein by reference).
Particular antibodies that may be of use for therapy of cancer within the scope of the claimed methods and compositions include, but are not limited to, LL1 (anti-CD74), LL2 and RFB4 (anti-CD22), RS7 (anti-epithelial glycoprotein-1 (EGP-1)), PAM4 and KC4 (both anti-mucin), MN-14 (anti-carcinoembryonic antigen (CEA, also known as CD66e), Mu-9 (anti-colon-specific antigen-p), Immu 31 (an anti-alpha-fetoprotein), TAG-72 (e.g., CC49), Tn, J591 or HuJ591 (anti-PSMA (prostate-specific membrane antigen)), AB-PG1-XG1-026 (anti-PSMA dimer), D2/B (anti-PSMA), G250 (anti-carbonic anhydrase IX), hL243 (anti-HLA-DR), alemtuzumab (anti-CD52), bevacizumab (anti-VEGF), cetuximab (anti-EGFR), gemtuzumab (anti-CD33), ibritumomab tiuxetan (anti-CD20); panitumumab (anti-EGFR); rituximab (anti-CD20); tositumomab (anti-CD20); GA101 (anti-CD20); and trastuzumab (anti-ErbB2). Such antibodies are known in the art (e.g., U.S. Pat. Nos. 5,686,072; 5,874,540; 6,107,090; 6,183,744; 6,306,393; 6,653,104; 6,730.300; 6,899,864; 6,926,893; 6,962,702; 7,074,403; 7,230,084; 7,238,785; 7,238,786; 7,256,004; 7,282,567; 7,300,655; 7,312,318; 7,585,491; 7,612,180; 7,642,239; and U.S. Patent Application Publ. No. 20040202666 (now abandoned); 20050271671; and 20060193865; the Examples section of each incorporated herein by reference.) Specific known antibodies of use include hPAM4 (U.S. Pat. No. 7,282,567), hA20 (U.S. Pat. No. 7,251,164), hA19 (U.S. Pat. No. 7,109,304), hIMMU31 (U.S. Pat. No. 7,300,655), hLL1 (U.S. Pat. No. 7,312,318,), hLL2 (U.S. Pat. No. 7,074,403), hMu-9 (U.S. Pat. No. 7,387,773), hL243 (U.S. Pat. No. 7,612,180), hMN-14 (U.S. Pat. No. 6,676,924), hMN-15 (U.S. Pat. No. 7,541,440), hR1 (U.S. patent application Ser. No. 12/772,645), hRS7 (U.S. Pat. No. 7,238,785), hMN-3 (U.S. Pat. No. 7,541,440), AB-PG1-XG1-026 (U.S. patent application Ser. No. 11/983,372, deposited as ATCC PTA-4405 and PTA-4406) and D2/B (WO 2009/130575) the text of each recited patent or application is incorporated herein by reference with respect to the Figures and Examples sections.
Anti-TNF-.alpha. antibodies are known in the art and may be of use to treat immune diseases, such as autoimmune disease, immune dysfunction (e.g., graft-versus-host disease, organ transplant rejection) or diabetes. Known antibodies against TNF-.alpha. include the human antibody CDP571 (Ofei et al., 2011, Diabetes 45:881-85); murine antibodies MTNFAI, M2TNFAI, M3TNFAI, M3TNFABI, M302B and M303 (Thermo Scientific, Rockford, Ill.); infliximab (Centocor, Malvern, Pa.); certolizumab pegol (UCB, Brussels, Belgium); and adalimumab (Abbott, Abbott Park, Ill.). These and many other known anti-TNF-.alpha. antibodies may be used in the claimed methods and compositions. Other antibodies of use for therapy of immune dysregulatory or autoimmune disease include, but are not limited to, anti-B-cell antibodies such as veltuzumab, epratuzumab, milatuzumab or hL243; tocilizumab (anti-IL-6 receptor); basiliximab (anti-CD25); daclizumab (anti-CD25); efalizumab (anti-CD11a); muromonab-CD3 (anti-CD3 receptor); anti-CD40L (UCB, Brussels, Belgium); natalizumab (anti-.alpha.4 integrin) and omalizumab (anti-IgE).
Type-2 diabetes may be treated using known antibodies against B-cell antigens, such as CD22 (epratuzumab), CD74 (milatuzumab), CD19 (hA19), CD20 (veltuzumab) or HLA-DR (hL243) (see, e.g., Winer et al., 2011, Nature Med 17:610-18). Anti-CD3 antibodies also have been proposed for therapy of type 1 diabetes (Cernea et al., 2010, Diabetes Metab Rev 26:602-05).
Macrophage migration inhibitory factor (MIF) is an important regulator of innate and adaptive immunity and apoptosis. It has been reported that CD74 is the endogenous receptor for MIF (Leng et al., 2003, J Exp Med 197:1467-76). The therapeutic effect of antagonistic anti-CD74 antibodies on MIF-mediated intracellular pathways may be of use for treatment of a broad range of disease states, such as cancers of the bladder, prostate, breast, lung, colon and chronic lymphocytic leukemia (e.g., Meyer-Siegler et al., 2004, BMC Cancer 12:34; Shachar & Haran, 2011, Leuk Lymphoma 52:1446-54); autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus (Morand & Leech, 2005, Front Biosci 10:12-22; Shachar & Haran, 2011, Leuk Lymphoma 52:1446-54); kidney diseases such as renal allograft rejection (Lan, 2008, Nephron Exp Nephrol. 109:e79-83); and numerous inflammatory diseases (Meyer-Siegler et al., 2009, Mediators Inflamm epub Mar. 22, 2009; Takahashi et al., 2009, Respir Res 10:33; Milatuzumab (hLL1) is an exemplary anti-CD74 antibody of therapeutic use for treatment of MIF-mediated diseases.
Anti-CD3 antibodies have been reported to reduce development and progression of atherosclerosis (Steffens et al., 2006, Circulation 114:1977-84). Antibodies against oxidized LDL induced a regression of established atherosclerosis in a mouse model (Ginsberg, 2007, J Am Coll Cardiol 52:2319-21). Anti-ICAM-1 antibody was shown to reduce ischemic cell damage after cerebral artery occlusion in rats (Zhang et al., 1994, Neurology 44:1747-51).
Antibody Allotypes
Immunogenicity of therapeutic antibodies is associated with increased risk of infusion reactions and decreased duration of therapeutic response (Baert et al., 2003, N Engl J Med 348:602-08). The extent to which therapeutic antibodies induce an immune response in the host may be determined in part by the allotype of the antibody (Stickler et al., 2011, Genes and Immunity 12:213-21). Antibody allotype is related to amino acid sequence variations at specific locations in the constant region sequences of the antibody. The allotypes of IgG antibodies containing a heavy chain .gamma.-type constant region are designated as Gm allotypes (1976, J Immunol 117:1056-59).
For the common IgG1 human antibodies, the most prevalent allotype is G1m1 (Stickler et al., 2011, Genes and Immunity 12:213-21). However, the G1m3 allotype also occurs frequently in Caucasians (Id.). It has been reported that G1m1 antibodies contain allotypic sequences that tend to induce an immune response when administered to non-G1m1 (nG1m1) recipients, such as G1m3 patients (Id.). Non-G1m1 allotype antibodies are not as immunogenic when administered to G1m1 patients (Id.).
The human G1m1 allotype comprises the amino acids aspartic acid at Kabat position 356 and leucine at Kabat position 358 in the CH3 sequence of the heavy chain IgG1. The nG1m1 allotype comprises the amino acids glutamic acid at Kabat position 356 and methionine at Kabat position 358. Both G1m1 and nG1m1 allotypes comprise a glutamic acid residue at Kabat position 357 and the allotypes are sometimes referred to as DEL and EEM allotypes. A non-limiting example of the heavy chain constant region sequences for G1m1 and nG1m1 allotype antibodies is shown for the exemplary antibodies rituximab (SEQ ID NO:85) and veltuzumab (SEQ ID NO:86).
TABLE-US-00015 Rituximab heavy chain variable region sequence (SEQ ID NO: 85) ASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGV HTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKAEP KSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVS HEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGK EYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTC LVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRW QQGNVFSCSVMHEALHNHYTQKSLSLSPGK Veltuzumab heavy chain variable region (SEQ ID NO: 86) ASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGV HTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEP KSCDKTHTCPPCPAPELLGGPSVFLEPPKPKDTLMISRTPEVTCVVVDVS HEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGK EYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTC LVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRW QQGNVFSCSVMHEALHNHYTQKSLSLSPGK
Jefferis and Lefranc (2009, mAbs 1:1-7) reviewed sequence variations characteristic of IgG allotypes and their effect on immunogenicity. They reported that the G1m3 allotype is characterized by an arginine residue at Kabat position 214, compared to a lysine residue at Kabat 214 in the G1m17 allotype. The nG1m1,2 allotype was characterized by glutamic acid at Kabat position 356, methionine at Kabat position 358 and alanine at Kabat position 431. The G1m1,2 allotype was characterized by aspartic acid at Kabat position 356, leucine at Kabat position 358 and glycine at Kabat position 431. In addition to heavy chain constant region sequence variants, Jefferis and Lefranc (2009) reported allotypic variants in the kappa light chain constant region, with the Km1 allotype characterized by valine at Kabat position 153 and leucine at Kabat position 191, the Km1,2 allotype by alanine at Kabat position 153 and leucine at Kabat position 191, and the Km3 allotype characterized by alanine at Kabat position 153 and valine at Kabat position 191.
With regard to therapeutic antibodies, veltuzumab and rituximab are, respectively, humanized and chimeric IgG1 antibodies against CD20, of use for therapy of a wide variety of hematological malignancies and/or autoimmune diseases. Table 5 compares the allotype sequences of rituximab vs. veltuzumab. As shown in Table 5, rituximab (G1m17,1) is a DEL allotype IgG1, with an additional sequence variation at Kabat position 214 (heavy chain CH1) of lysine in rituximab vs. arginine in veltuzumab. It has been reported that veltuzumab is less immunogenic in subjects than rituximab (see, e.g., Morchhauser et al., 2009, J Clin Oncol 27:3346-53; Goldenberg et al., 2009, Blood 113:1062-70; Robak & Robak, 2011, BioDrugs 25:13-25), an effect that has been attributed to the difference between humanized and chimeric antibodies. However, the difference in allotypes between the EEM and DEL allotypes likely also accounts for the lower immunogenicity of veltuzumab.
TABLE-US-00016 TABLE 5 Allotypes of Rituximab vs. Veltuzumab Heavy chain position and associated allotypes Complete 214 356/358 431 allotype (allotype) (allotype) (allotype) Rituximab G1m17, 1 K 17 D/L 1 A -- Veltuzumab G1m3 R 3 E/M -- A --
In order to reduce the immunogenicity of therapeutic antibodies in individuals of nG1m1 genotype, it is desirable to select the allotype of the antibody to correspond to the G1m3 allotype, characterized by arginine at Kabat 214, and the nG1m1,2 null-allotype, characterized by glutamic acid at Kabat position 356, methionine at Kabat position 358 and alanine at Kabat position 431. Surprisingly, it was found that repeated subcutaneous administration of G1m3 antibodies over a long period of time did not result in a significant immune response. In alternative embodiments, the human IgG4 heavy chain in common with the G1m3 allotype has arginine at Kabat 214, glutamic acid at Kabat 356, methionine at Kabat 359 and alanine at Kabat 431. Since immunogenicity appears to relate at least in part to the residues at those locations, use of the human IgG4 heavy chain constant region sequence for therapeutic antibodies is also a preferred embodiment. Combinations of G1m3 IgG1 antibodies with IgG4 antibodies may also be of use for therapeutic administration.
Immunoconjugates
In certain embodiments, the antibodies or complexes may be conjugated to one or more therapeutic or diagnostic agents. The therapeutic agents do not need to be the same but can be different, e.g. a drug and a radioisotope. For example, .sup.131I can be incorporated into a tyrosine of an antibody or fusion protein and a drug attached to an epsilon amino group of a lysine residue. Therapeutic and diagnostic agents also can be attached, for example to reduced SH groups and/or to carbohydrate side chains. Many methods for making covalent or non-covalent conjugates of therapeutic or diagnostic agents with antibodies or fusion proteins are known in the art and any such known method may be utilized.
A therapeutic or diagnostic agent can be attached at the hinge region of a reduced antibody component via disulfide bond formation. Alternatively, such agents can be attached using a heterobifunctional cross-linker, such as N-succinyl 3-(2-pyridyldithio)propionate (SPDP). Yu et al., Int. J. Cancer 56: 244 (1994). General techniques for such conjugation are well-known in the art. See, for example, Wong, CHEMISTRY OF PROTEIN CONJUGATION AND CROSS-LINKING (CRC Press 1991); Upeslacis et al., "Modification of Antibodies by Chemical Methods," in MONOCLONAL ANTIBODIES: PRINCIPLES AND APPLICATIONS, Birch et al. (eds.), pages 187-230 (Wiley-Liss, Inc. 1995); Price, "Production and Characterization of Synthetic Peptide-Derived Antibodies," in MONOCLONAL ANTIBODIES: PRODUCTION, ENGINEERING AND CLINICAL APPLICATION, Ritter et al. (eds.), pages 60-84 (Cambridge University Press 1995). Alternatively, the therapeutic or diagnostic agent can be conjugated via a carbohydrate moiety in the Fc region of the antibody. The carbohydrate group can be used to increase the loading of the same agent that is bound to a thiol group, or the carbohydrate moiety can be used to bind a different therapeutic or diagnostic agent.
Methods for conjugating peptides to antibody components via an antibody carbohydrate moiety are well-known to those of skill in the art. See, for example, Shih et al., Int. J. Cancer 41: 832 (1988); Shih et al., Int. J. Cancer 46: 1101 (1990); and Shih et al., U.S. Pat. No. 5,057,313, incorporated herein in their entirety by reference. The general method involves reacting an antibody component having an oxidized carbohydrate portion with a carrier polymer that has at least one free amine function. This reaction results in an initial Schiff base (imine) linkage, which can be stabilized by reduction to a secondary amine to form the final conjugate.
The Fc region may be absent if the antibody used as the antibody component is an antibody fragment. However, it is possible to introduce a carbohydrate moiety into the light chain variable region of a full length antibody or antibody fragment. See, for example, Leung et al., J. Immunol. 154: 5919 (1995); Hansen et al., U.S. Pat. No. 5,443,953 (1995), Leung et al., U.S. Pat. No. 6,254,868, incorporated herein by reference in their entirety. The engineered carbohydrate moiety is used to attach the therapeutic or diagnostic agent.
In some embodiments, a chelating agent may be attached to an antibody, antibody fragment or fusion protein and used to chelate a therapeutic or diagnostic agent, such as a radionuclide. Exemplary chelators include but are not limited to DTPA (such as Mx-DTPA), DOTA, TETA, NETA or NOTA. Methods of conjugation and use of chelating agents to attach metals or other ligands to proteins are well known in the art (see, e.g., U.S. Pat. No. 7,563,433, the Examples section of which is incorporated herein by reference).
In certain embodiments, radioactive metals or paramagnetic ions may be attached to proteins or peptides by reaction with a reagent having a long tail, to which may be attached a multiplicity of chelating groups for binding ions. Such a tail can be a polymer such as a polylysine, polysaccharide, or other derivatized or derivatizable chains having pendant groups to which can be bound chelating groups such as, e.g., ethylenediaminetetraacetic acid (EDTA), diethylenetriaminepentaacetic acid (DTPA), porphyrins, polyamines, crown ethers, bis-thiosemicarbazones, polyoximes, and like groups known to be useful for this purpose.
Chelates may be directly linked to antibodies or peptides, for example as disclosed in U.S. Pat. No. 4,824,659, incorporated herein in its entirety by reference. Particularly useful metal-chelate combinations include 2-benzyl-DTPA and its monomethyl and cyclohexyl analogs, used with diagnostic isotopes in the general energy range of 60 to 4,000 keV, such as .sup.125I, .sup.131I, .sup.123I, .sup.124I, .sup.62Cu, .sup.64Cu, .sup.18F, .sup.111In, .sup.67Ga, .sup.68Ga, .sup.99mTe, .sup.94mTe, .sup.11C, .sup.13N, .sup.15O, .sup.76Br, for radioimaging. The same chelates, when complexed with non-radioactive metals, such as manganese, iron and gadolinium are useful for MRI. Macrocyclic chelates such as NOTA, DOTA, and TETA are of use with a variety of metals and radiometals, most particularly with radionuclides of gallium, yttrium and copper, respectively. Such metal-chelate complexes can be made very stable by tailoring the ring size to the metal of interest. Other ring-type chelates such as macrocyclic polyethers, which are of interest for stably binding nuclides, such as .sup.223Ra for RAIT are encompassed.
More recently, methods of .sup.18F-labeling of use in PET scanning techniques have been disclosed, for example by reaction of F-18 with a metal or other atom, such as aluminum. The .sup.18F--Al conjugate may be complexed with chelating groups, such as DOTA, NOTA or NETA that are attached directly to antibodies or used to label targetable constructs in pre-targeting methods. Such F-18 labeling techniques are disclosed in U.S. Pat. No. 7,563,433, the Examples section of which is incorporated herein by reference.
Therapeutic Agents
In alternative embodiments, therapeutic agents such as cytotoxic agents, anti-angiogenic agents, pro-apoptotic agents, antibiotics, hormones, hormone antagonists, chemokines, drugs, prodrugs, toxins, enzymes or other agents may be used, either conjugated to the subject antibody complexes or separately administered before, simultaneously with, or after the antibody complex. Drugs of use may possess a pharmaceutical property selected from the group consisting of antimitotic, kinase inhibitor, Bruton kinase inhibitor, alkylating, antimetabolite, antibiotic, alkaloid, anti-angiogenic, pro-apoptotic agents and combinations thereof.
Exemplary drugs of use include, but are not limited to, 5-fluorouracil, afatinib, aplidin, azaribine, anastrozole, anthracyclines, axitinib, AVL-101, AVL-291, bendamustine, bleomycin, bortezomib, bosutinib, bryostatin-1, busulfan, calicheamycin, camptothecin, carboplatin, 10-hydroxycamptothecin, carmustine, celecoxib, chlorambucil, cisplatin (CDDP), Cox-2 inhibitors, irinotecan (CPT-11), SN-38, carboplatin, cladribine, camptothecans, crizotinib, cyclophosphamide, cytarabine, dacarbazine, dasatinib, dinaciclib, docetaxel, dactinomycin, daunorubicin, doxorubicin, 2-pyrrolinodoxorubicine (2P-DOX), cyano-morpholino doxorubicin, doxorubicin glucuronide, epirubicin glucuronide, erlotinib, estramustine, epidophyllotoxin, entinostat, estrogen receptor binding agents, etoposide (VP16), etoposide glucuronide, etoposide phosphate, exemestane, fingolimod, floxuridine (FUdR), 3',5'-O-dioleoyl-FudR (FUdR-dO), fludarabine, flutamide, farnesyl-protein transferase inhibitors, flavopiridol, fostamatinib, ganetespib, GDC-0834, GS-1101, gefitinib, gemcitabine, hydroxyurea, ibrutinib, idarubicin, idelalisib, ifosfamide, imatinib, L-asparaginase, lapatinib, lenolidamide, leucovorin, LFM-A13, lomustine, mechlorethamine, melphalan, mercaptopurine, 6-mercaptopurine, methotrexate, mitoxantrone, mithramycin, mitomycin, mitotane, navelbine, neratinib, nilotinib, nitrosurea, olaparib, plicomycin, procarbazine, paclitaxel, PCI-32765, pentostatin, PSI-341, raloxifene, semustine, sorafenib, streptozocin, SU11248, sunitinib, tamoxifen, temazolomide (an aqueous form of DTIC), transplatinum, thalidomide, thioguanine, thiotepa, teniposide, topotecan, uracil mustard, vatalanib, vinorelbine, vinblastine, vincristine, vinca alkaloids and ZD1839.
Toxins of use may include ricin, abrin, alpha toxin, saporin, ribonuclease (RNase), e.g., onconase, DNase I, Staphylococcal enterotoxin-A, pokeweed antiviral protein, gelonin, diphtheria toxin, Pseudomonas exotoxin, and Pseudomonas endotoxin.
Chemokines of use may include RANTES, MCAF, MIP1-alpha, MIP1-Beta and IP-10.
In certain embodiments, anti-angiogenic agents, such as angiostatin, baculostatin, canstatin, maspin, anti-VEGF antibodies, anti-PlGF peptides and antibodies, anti-vascular growth factor antibodies, anti-Flk-1 antibodies, anti-Flt-1 antibodies and peptides, anti-Kras antibodies, anti-cMET antibodies, anti-MIF (macrophage migration-inhibitory factor) antibodies, laminin peptides, fibronectin peptides, plasminogen activator inhibitors, tissue metalloproteinase inhibitors, interferons, interleukin-12, IP-10, Gro- , thrombospondin, 2-methoxyoestradiol, proliferin-related protein, carboxiamidotriazole, CM101, Marimastat, pentosan polysulphate, angiopoietin-2, interferon-alpha, herbimycin A, PNU145156E, 16K prolactin fragment, Linomide (roquinimex), thalidomide, pentoxifylline, genistein, TNP-470, endostatin, paclitaxel, accutin, angiostatin, cidofovir, vincristine, bleomycin, AGM-1470, platelet factor 4 or minocycline may be of use.
Immunomodulators of use may be selected from a cytokine, a stem cell growth factor, a lymphotoxin, a hematopoietic factor, a colony stimulating factor (CSF), an interferon (IFN), erythropoietin, thrombopoietin and a combination thereof. Specifically useful are lymphotoxins such as tumor necrosis factor (TNF), hematopoietic factors, such as interleukin (IL), colony stimulating factor, such as granulocyte-colony stimulating factor (G-CSF) or granulocyte macrophage-colony stimulating factor (GM-CSF), interferon, such as interferons-.alpha., -.beta. or -.gamma., and stem cell growth factor, such as that designated "S1 factor". Included among the cytokines are growth hormones such as human growth hormone, N-methionyl human growth hormone, and bovine growth hormone; parathyroid hormone; thyroxine; insulin; proinsulin; relaxin; prorelaxin; glycoprotein hormones such as follicle stimulating hormone (FSH), thyroid stimulating hormone (TSH), and luteinizing hormone (LH); hepatic growth factor; prostaglandin, fibroblast growth factor; prolactin; placental lactogen, OB protein; tumor necrosis factor-.alpha. and - ; mullerian-inhibiting substance; mouse gonadotropin-associated peptide; inhibin; activin; vascular endothelial growth factor; integrin; thrombopoietin (TPO); nerve growth factors such as NGF- ; platelet-growth factor; transforming growth factors (TGFs) such as TGF-.alpha. and TGF- ; insulin-like growth factor-I and -II; erythropoietin (EPO); osteoinductive factors; interferons such as interferon-.alpha., -.beta., and -.gamma.; colony stimulating factors (CSFs) such as macrophage-CSF (M-CSF); interleukins (ILs) such as IL-1, IL-1.alpha., IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-11, IL-12; IL-13, IL-14, IL-15, IL-16, IL-17, IL-18, IL-21, IL-25, LIF, kit-ligand or FLT-3, angiostatin, thrombospondin, endostatin, tumor necrosis factor and LT.
Radionuclides of use include, but are not limited to--.sup.111In, .sup.177Lu, .sup.212Bi, .sup.213Bi, .sup.211At, .sup.62Cu, .sup.67Cu, .sup.90Y, .sup.125I, .sup.131I, .sup.32P, .sup.33P, .sup.47Sc, .sup.111Ag, .sup.67Ga, .sup.142Pr, .sup.153Sm, .sup.161Tb, .sup.166Dy, .sup.166Ho, .sup.186Re, .sup.188Re, .sup.189Re, .sup.212Pb, .sup.223Ra, .sup.225Ac, .sup.59Fe, .sup.75Se, .sup.77As, .sup.89Sr, .sup.99Mo, .sup.105Rh, .sup.109Pd, .sup.143Pr, .sup.149Pm, .sup.169Er, .sup.194Ir, .sup.198Au, .sup.199Au, .sup.227Th and .sup.211Pb. The therapeutic radionuclide preferably has a decay-energy in the range of 20 to 6,000 keV, preferably in the ranges 60 to 200 keV for an Auger emitter, 100-2,500 keV for a beta emitter, and 4,000-6,000 keV for an alpha emitter. Maximum decay energies of useful beta-particle-emitting nuclides are preferably 20-5,000 keV, more preferably 100-4,000 keV, and most preferably 500-2,500 keV. Also preferred are radionuclides that substantially decay with Auger-emitting particles. For example, Co-58, Ga-67, Br-80m, Tc-99m, Rh-103m, Pt-109, In-111, Sb-119, 1-125, Ho-161, Os-189m and Ir-192. Decay energies of useful beta-particle-emitting nuclides are preferably <1,000 keV, more preferably <100 keV, and most preferably <70 keV. Also preferred are radionuclides that substantially decay with generation of alpha-particles. Such radionuclides include, but are not limited to: Dy-152, At-211, Bi-212, Ra-223, Rn-219, Po-215, Bi-211, Ac-225, Fr-221, At-217, Bi-213, Th-227 and Fm-255. Decay energies of useful alpha-particle-emitting radionuclides are preferably 2,000-10,000 keV, more preferably 3,000-8,000 keV, and most preferably 4,000-7,000 keV. Additional potential radioisotopes of use include .sup.11C, .sup.13N, .sup.15O, .sup.75Br, .sup.198Au, .sup.224Ac, .sup.126I, .sup.133I, .sup.77Br, .sup.113mIn, .sup.95Ru, .sup.97Ru, .sup.103Ru, .sup.105Ru, .sup.107Hg, .sup.203Hg, .sup.121mTe, .sup.122mTe, .sup.125mTe, .sup.165Tm, .sup.167Tm, .sup.168Tm, .sup.197Pt, .sup.109Pd, .sup.105Rh, .sup.142Pr, .sup.143Pr, .sup.161Tb, .sup.166Ho, .sup.199Au, .sup.57Co, .sup.58Co, .sup.51Cr, .sup.59Fe, .sup.75Se, .sup.201Tl, .sup.225Ac, .sup.76Br, .sup.169Yb, and the like. Some useful diagnostic nuclides may include .sup.18F, .sup.52Fe, .sup.62Cu, .sup.64Cu, .sup.67Cu, .sup.67Ga, .sup.68Ga, .sup.86Y, .sup.89Zr, .sup.94Tc, .sup.94mTc, .sup.99mTc, or In. Radionuclides and other metals may be delivered, for example, using chelating groups attached to an antibody or conjugate. Macrocyclic chelates such as NOTA, DOTA, and TETA are of use with a variety of metals and radiometals, most particularly with radionuclides of gallium, yttrium and copper, respectively. Such metal-chelate complexes can be made very stable by tailoring the ring size to the metal of interest. Other ring-type chelates, such as macrocyclic polyethers for complexing .sup.223Ra, may be used.
Therapeutic agents may include a photoactive agent or dye. Fluorescent compositions, such as fluorochrome, and other chromogens, or dyes, such as porphyrins sensitive to visible light, have been used to detect and to treat lesions by directing the suitable light to the lesion. In therapy, this has been termed photoradiation, phototherapy, or photodynamic therapy. See Joni et al. (eds.), PHOTODYNAMIC THERAPY OF TUMORS AND OTHER DISEASES (Libreria Progetto 1985); van den Bergh, Chem. Britain (1986), 22:430. Moreover, monoclonal antibodies have been coupled with photoactivated dyes for achieving phototherapy. See Mew et al., J. Immunol. (1983), 130:1473; idem., Cancer Res. (1985), 45:4380; Oseroff et al., Proc. Natl. Acad. Sci. USA (1986), 83:8744; idem., Photochem. Photobiol. (1987), 46:83; Hasan et al., Prog. Clin. Biol. Res. (1989), 288:471; Tatsuta et al., Lasers Surg. Med. (1989), 9:422; Pelegrin et al., Cancer (1991), 67:2529.
Other useful therapeutic agents may comprise oligonucleotides, especially antisense oligonucleotides that preferably are directed against oncogenes and oncogene products, such as bcl-2 or p53. A preferred form of therapeutic oligonucleotide is siRNA.
Diagnostic Agents
Diagnostic agents are preferably selected from the group consisting of a radionuclide, a radiological contrast agent, a paramagnetic ion, a metal, a fluorescent label, a chemiluminescent label, an ultrasound contrast agent and a photoactive agent. Such diagnostic agents are well known and any such known diagnostic agent may be used. Non-limiting examples of diagnostic agents may include a radionuclide such as .sup.110In, .sup.111In, .sup.177Lu, .sup.18F, .sup.52Fe, .sup.62Cu, .sup.64Cu, .sup.67Cu, .sup.67Ga, .sup.68Ga, .sup.86Y, .sup.90Y, .sup.89Zr, .sup.94mTc, .sup.94Tc, .sup.99mTc, .sup.120I, .sup.123I, .sup.124I, .sup.125I, .sup.131I, .sup.154-158Gd, .sup.32P, .sup.11C, .sup.13N, .sup.15O, .sup.186Re, .sup.188Re, .sup.51Mn, .sup.52mMn, .sup.55Co, .sup.72As, .sup.75Br, .sup.76Br, .sup.82mRb, .sup.83Sr, or other gamma-, beta-, or positron-emitters. Paramagnetic ions of use may include chromium (III), manganese (II), iron (III), iron (II), cobalt (II), nickel (II), copper (II), neodymium (III), samarium (III), ytterbium (III), gadolinium (III), vanadium (II), terbium (III), dysprosium (III), holmium (III) or erbium (III). Metal contrast agents may include lanthanum (III), gold (III), lead (II) or bismuth (III). Ultrasound contrast agents may comprise liposomes, such as gas filled liposomes. Radiopaque diagnostic agents may be selected from compounds, barium compounds, gallium compounds, and thallium compounds. A wide variety of fluorescent labels are known in the art, including but not limited to fluorescein isothiocyanate, rhodamine, phycoerytherin, phycocyanin, allophycocyanin, o-phthaldehyde and fluorescamine. Chemiluminescent labels of use may include luminol, isoluminol, an aromatic acridinium ester, an imidazole, an acridinium salt or an oxalate ester.
Therapeutic Use
In another aspect, the invention relates to a method of treating a subject, comprising administering a therapeutically effective amount of an antibody complex as described herein to a subject. Diseases that may be treated with the antibody complexes described herein include, but are not limited to immune diseases (e.g., SLE, RA, juvenile idiopathic arthritis, Crohn's disease, type 2 diabetes, Castleman's disease) or inflammatory diseases (e.g., sepsis, septic shock, inflammation, inflammatory bowel disease, inflammatory liver injury, acute pancreatitis). Such therapeutics can be given once or repeatedly, depending on the disease state and tolerability of the conjugate, and can also be used optimally in combination with other therapeutic modalities, such as immunomodulator therapy, immunotherapy, chemotherapy, antisense therapy, interference RNA therapy, gene therapy, and the like. Each combination will be adapted to patient condition and prior therapy, and other factors considered by the managing physician.
As used herein, the term "subject" refers to any animal (i.e., vertebrates and invertebrates) including, but not limited to mammals, including humans. It is not intended that the term be limited to a particular age or sex. Thus, adult and newborn subjects, as well as fetuses, whether male or female, are encompassed by the term.
In preferred embodiments, the antibodies that are used in the treatment of human disease are human or humanized (CDR-grafted) versions of antibodies; although murine and chimeric versions of antibodies can be used. Same species IgG molecules are mostly preferred to minimize immune responses. This is particularly important when considering repeat treatments. For humans, a human or humanized IgG antibody is less likely to generate an anti-IgG immune response from patients.
In another preferred embodiment, diseases that may be treated using the antibody complexes include, but are not limited to immune dysregulation disease and related autoimmune diseases, including Class III autoimmune diseases such as immune-mediated thrombocytopenias, such as acute idiopathic thrombocytopenic purpura and chronic idiopathic thrombocytopenic purpura, dermatomyositis, Sjogren's syndrome, multiple sclerosis, Sydenham's chorea, myasthenia gravis, systemic lupus erythematosus, lupus nephritis, rheumatic fever, polyglandular syndromes, bullous pemphigoid, diabetes mellitus, Henoch-Schonlein purpura, post-streptococcal nephritis, erythema nodosum, Takayasu's arteritis, Addison's disease, rheumatoid arthritis, sarcoidosis, ulcerative colitis, erythema multiforme, IgA nephropathy, polyarteritis nodosa, ankylosing spondylitis, Goodpasture's syndrome, thromboangitis obliterans, Sj gren's syndrome, primary biliary cirrhosis, Hashimoto's thyroiditis, thyrotoxicosis, scleroderma, chronic active hepatitis, rheumatoid arthritis, polymyositis/dermatomyositis, polychondritis, pemphigus vulgaris, Wegener's granulomatosis, membranous nephropathy, amyotrophic lateral sclerosis, tabes dorsalis, giant cell arteritis/polymyalgia, pernicious anemia, rapidly progressive glomerulonephritis and fibrosing alveolitis, and also juvenile diabetes, as disclosed in U.S. Provisional Application Ser. No. 60/360,259, filed Mar. 1, 2002 (now expired). Antibodies that may be of use for combination therapy in these diseases include, but are not limited to, those reactive with HLA-DR antigens, B-cell and plasma-cell antigens (e.g., CD19, CD20, CD21, CD22, CD23, CD4, CD5, CD8, CD14, CD15, CD19, CD20, CD21, CD22, CD23, CD25, CD33, CD37, CD38, CD40, CD40L, CD46, CD52, CD54, CD74, CD80, CD126, CD138, B7, MUC1, Ia, HM1.24, and HLA-DR), IL-6, IL-17. Since many of these autoimmune diseases are affected by autoantibodies made by aberrant B-cell populations, depletion of these B-cells is a preferred method of autoimmune disease therapy. In a preferred embodiment, the anti-B-cell, anti-T-cell, or anti-macrophage or other such antibodies of use in the co-treatment of patients with autoimmune diseases also can be conjugated to result in more effective therapeutics to control the host responses involved in said autoimmune diseases, and can be given alone or in combination with other therapeutic agents, such as TNF inhibitors or anti-IL-6R antibodies and the like.
In a preferred embodiment, a more effective therapeutic agent can be provided by using multivalent, multispecific antibodies. Exemplary bivalent and bispecific antibodies are found in U.S. Pat. Nos. 7,387,772; 7,300,655; 7,238,785; and 7,282,567, the Examples section of each of which is incorporated herein by reference. These multivalent or multispecific antibodies are particularly preferred in the targeting of disease associated cells which express multiple antigen targets and even multiple epitopes of the same antigen target, but which often evade antibody targeting and sufficient binding for immunotherapy because of insufficient expression or availability of a single antigen target on the cell. By targeting multiple antigens or epitopes, said antibodies show a higher binding and residence time on the target, thus affording a higher saturation with the drug being targeted in this invention.
Formulation and Administration
Suitable routes of administration of the conjugates include, without limitation, oral, parenteral, rectal, transmucosal, intestinal administration, intramuscular, subcutaneous, intramedullary, intrathecal, direct intraventricular, intravenous, intravitreal, intraperitoneal, intranasal, or intraocular injections. The preferred routes of administration are parenteral. Alternatively, one may administer the compound in a local rather than systemic manner, for example, via injection of the compound directly into a solid tumor.
Antibody complexes or immunoconjugates can be formulated according to known methods to prepare pharmaceutically useful compositions, whereby the antibody complex or immunoconjugate is combined in a mixture with a pharmaceutically suitable excipient. Sterile phosphate-buffered saline is one example of a pharmaceutically suitable excipient. Other suitable excipients are well-known to those in the art. See, for example, Ansel et al., PHARMACEUTICAL DOSAGE FORMS AND DRUG DELIVERY SYSTEMS, 5th Edition (Lea & Febiger 1990), and Gennaro (ed.), REMINGTON'S PHARMACEUTICAL SCIENCES, 18th Edition (Mack Publishing Company 1990), and revised editions thereof.
The antibody complex or immunoconjugate can be formulated for intravenous administration via, for example, bolus injection or continuous infusion. Preferably, the antibody of the present invention is infused over a period of less than about 4 hours, and more preferably, over a period of less than about 3 hours. For example, the first 25-50 mg could be infused within 30 minutes, preferably even 15 min, and the remainder infused over the next 2-3 hrs. Formulations for injection can be presented in unit dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative. The compositions can take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and can contain formulatory agents such as suspending, stabilizing and/or dispersing agents. Alternatively, the active ingredient can be in powder form for constitution with a suitable vehicle, e.g., sterile pyrogen-free water, before use.
Additional pharmaceutical methods may be employed to control the duration of action of the antibody complex. Control release preparations can be prepared through the use of polymers to complex or adsorb the antibody complex. For example, biocompatible polymers include matrices of poly(ethylene-co-vinyl acetate) and matrices of a polyanhydride copolymer of a stearic acid dimer and sebacic acid. Sherwood et al., Bio/Technology 10: 1446 (1992). The rate of release of an antibody complex or immunoconjugate from such a matrix depends upon the molecular weight, the amount of antibody complex or immunoconjugate within the matrix, and the size of dispersed particles. Saltzman et al., Biophys. J. 55: 163 (1989); Sherwood et al., supra. Other solid dosage forms are described in Ansel et al., PHARMACEUTICAL DOSAGE FORMS AND DRUG DELIVERY SYSTEMS, 5th Edition (Lea & Febiger 1990), and Gennaro (ed.), REMINGTON'S PHARMACEUTICAL SCIENCES, 18th Edition (Mack Publishing Company 1990), and revised editions thereof.
Generally, the dosage of an administered antibody complex or immunoconjugate for humans will vary depending upon such factors as the patient's age, weight, height, sex, general medical condition and previous medical history. It may be desirable to provide the recipient with a dosage that is in the range of from about 1 mg/kg to 25 mg/kg as a single intravenous infusion, although a lower or higher dosage also may be administered as circumstances dictate. A dosage of 1-20 mg/kg for a 70 kg patient, for example, is 70-1,400 mg, or 41-824 mg/m.sup.2 for a 1.7-m patient. The dosage may be repeated as needed, for example, once per week for 4-10 weeks, once per week for 8 weeks, or once per week for 4 weeks. It may also be given less frequently, such as every other week for several months, or monthly or quarterly for many months, as needed in a maintenance therapy.
Alternatively, an antibody complex or immunoconjugate may be administered as one dosage every 2 or 3 weeks, repeated for a total of at least 3 dosages. Or, twice per week for 4-6 weeks. If the dosage is lowered to approximately 200-300 mg/m.sup.2 (340 mg per dosage for a 1.7-m patient, or 4.9 mg/kg for a 70 kg patient), it may be administered once or even twice weekly for 4 to 10 weeks. Alternatively, the dosage schedule may be decreased, namely every 2 or 3 weeks for 2-3 months. It has been determined, however, that even higher doses, such as 20 mg/kg once weekly or once every 2-3 weeks can be administered by slow i.v. infusion, for repeated dosing cycles. The dosing schedule can optionally be repeated at other intervals and dosage may be given through various parenteral routes, with appropriate adjustment of the dose and schedule.
Expression Vectors
Still other embodiments may concern DNA sequences comprising a nucleic acid encoding an antibody, antibody fragment, toxin or constituent fusion protein of an antibody complex, such as a DNL.RTM. construct. Fusion proteins may comprise an antibody or fragment or toxin attached to, for example, an AD or DDD moiety.
Various embodiments relate to expression vectors comprising the coding DNA sequences. The vectors may contain sequences encoding the light and heavy chain constant regions and the hinge region of a human immunoglobulin to which may be attached chimeric, humanized or human variable region sequences. The vectors may additionally contain promoters that express the encoded protein(s) in a selected host cell, enhancers and signal or leader sequences. Vectors that are particularly useful are pdHL2 or GS. More preferably, the light and heavy chain constant regions and hinge region may be from a human EU myeloma immunoglobulin, where optionally at least one of the amino acid in the allotype positions is changed to that found in a different IgG1 allotype, and wherein optionally amino acid 253 of the heavy chain of EU based on the EU number system may be replaced with alanine. See Edelman et al., Proc. Natl. Acad. Sci USA 63: 78-85 (1969). In other embodiments, an IgG1 sequence may be converted to an IgG4 sequence.
The skilled artisan will realize that methods of genetically engineering expression constructs and insertion into host cells to express engineered proteins are well known in the art and a matter of routine experimentation. Host cells and methods of expression of cloned antibodies or fragments have been described, for example, in U.S. Pat. Nos. 7,531,327 and 7,537,930, the Examples section of each incorporated herein by reference.
Kits
Various embodiments may concern kits containing components suitable for treating or diagnosing diseased tissue in a patient. Exemplary kits may contain one or more antibody complexes as described herein. If the composition containing components for administration is not formulated for delivery via the alimentary canal, such as by oral delivery, a device capable of delivering the kit components through some other route may be included. One type of device, for applications such as parenteral delivery, is a syringe that is used to inject the composition into the body of a subject. Inhalation devices may also be used. In certain embodiments, a therapeutic agent may be provided in the form of a prefilled syringe or autoinjection pen containing a sterile, liquid formulation or lyophilized preparation.
The kit components may be packaged together or separated into two or more containers. In some embodiments, the containers may be vials that contain sterile, lyophilized formulations of a composition that are suitable for reconstitution. A kit may also contain one or more buffers suitable for reconstitution and/or dilution of other reagents. Other containers that may be used include, but are not limited to, a pouch, tray, box, tube, or the like. Kit components may be packaged and maintained sterilely within the containers. Another component that can be included is instructions to a person using a kit for its use.
EXAMPLES
In the working Examples below, the DOCK-AND-LOCK.RTM. (DNL) technology was used to generate the first bispecific antibody (bsAb) with high potency to neutralize both TNF-.alpha. and IL-6. This prototype DNL.RTM. construct, designated cT*-(c6)-(c6), comprises a chimeric anti-TNF-.alpha. IgG linked at the carboxyl terminus of each light chain to a pair of dimerized Fab's derived from a chimeric anti-IL-6 antibody, thus featuring a hexavalent bsAb capable of blocking 2 and 4 molecules of TNF-.alpha. and IL-6, respectively, as well as a fully functional Fc. As discussed below, the exemplary anti-TNF-.alpha./anti-IL-6 bispecific antibody showed potent activity in in vitro assays designed to test efficacy for immune diseases such as SLE or RA. However, the person of ordinary skill will realize that the subject complexes of use are not limited to the specific DNL.RTM. cT*-(c6)-(c6) complex discussed below, but more generally encompass bispecific antibodies and/or antigen-binding antibody fragments with at least one binding site for IL-6 an at least one binding site for TNF-.alpha..
Example 1. Generation of Neutralizing Mouse Anti-Human IL-6 Monoclonal Antibody
The 2-3B2 mouse monoclonal antibody against human IL-6 was produced using standard immunological techniques, discussed below, that may be used to make anti-human IL-6 antibodies in general.
Recombinant human IL-6 (rhIL-6) was obtained from ProSpec-Tany TechnoGene Ltd. (Rehovot, Israel). Multiple mice were initially immunized with 30 .mu.g rhIL-6 administered i.p., followed by booster injections of 30 or 10 .mu.g with or without adjuvant, according to a standard boosting schedule. Animals were tested for presence of anti-IL-6 antibodies by ELISA assay using rhIL-6 coated microtiter plates and serial dilutions of serum. Prior to fusion, the presence of neutralizing anti-IL-6 antibodies was detected by the ability to block IL-6 stimulated protein phosphorylation (of STAT3) using Western blotting (data not shown).
Cells secreting neutralizing anti-IL-6 antibodies were fused with the P3-X63.Ag8.653 myeloma cell line by PEG mediated cell fusion using standard techniques to generate antibody-secreting hybridomas cell lines. The 2-3B2, 4-4F5 and 4-4E6 anti-IL-6 clones were obtained by selection on HAT medium, cloning and subcloning. Supernatants from isolated clones containing neutralizing anti-IL-6 were detected by the ability to block IL-6 stimulated STAT3 protein phosphorylation, determined by Western blotting (FIG. 1). Clones 2-3B2, 4-4E6 and control anti-IL-6 MAb206, but not clone 4-4F5, were able to block rhIL-6 induced phosphorylation (FIG. 1). Size exclusion HPLC of antibodies purified by protein A column chromatography demonstrated the presence of homogeneous antibodies, which was confirmed by SDS-PAGE (data not shown). Isotyping using an SBA CLONETYPING.TM. system showed that the anti-IL-6 antibodies were IgG1/.kappa. murine isotypes.
Binding to human IL-6 was determined Western blotting against rhIL-6 (FIG. 2). Based on the intensity of labeling using identical concentrations of antibody, it was determined that the 2-3B2 clone (FIG. 2A) showed higher affinity for human IL-6 than the 4-4E6 clone (FIG. 2B) and 2-3B2 was selected for production of chimeric and humanized anti-IL-6 antibodies. Serial dilution demonstrated that the 2-3B2 antibody was about 100-fold more potent than 4-4E6 for inhibiting the IL-6 induced phosphorylation of STAT3 (FIG. 2A-B). Neither antibody bound to murine IL-6 (not shown).
Example 2. Generation of Neutralizing Mouse Anti-Human TNF-.alpha. Monoclonal Antibody
Monoclonal antibodies against human TNF-.alpha. were prepared using standard techniques, as discussed in Example 1 above for IL-6. Mice were immunized with recombinant human TNF-.alpha. obtained from ProSpec-Tany TechnoGene Ltd. (Rehovot, Israel). Testing of serum from immunized mice for anti-TNF-.alpha. antibodies was performed by ELISA (data not shown).
Neutralizing antibodies were also detected by cytotoxicity assay. Briefly, WEHI 164 cells (mouse fibrosarcoma) were cultured in RPMI complete media. Cells were plated at a density of 1.times.10.sup.4 cells/well in 75 .mu.L of medium in 96-well plates and kept in a 37.degree. C. incubator overnight before the assay. On the day of the assay, sera from the immunized mice were diluted 1:25, 1:125, 1:625, 1:3, 125, 1:15,625, and 1:78,125 in RPMI complete medium containing 8 .mu.g/mL of actinomycin-D and 0.4 ng/mL of rhTNF-.alpha.. Twenty-five .mu.L of the diluted sera were added to the cells in the corresponding wells. The addition of the sera to the cells made the final dilutions of the sera as 1:100, 1:500, 1:2,500, 1:12,500, 1:62,500, and 1:312,500. The final concentration of actinomycin-D was 2 .mu.g/mL and rhTNF-.alpha. was 0.1 ng/mL. Plates were incubated in a 37.degree. C./5% CO2 incubator for 20 hours. After this incubation, 20 .mu.L of MTS reagent was added to all the wells and the absorbance in each well determined in a plate reader at 490 nm after two hours. As a negative control, serum from a naive mouse (not immunized) was diluted in a like manner. One set of wells was incubated with only actinomycin-D and rhTNF-.alpha. to determine maximum growth inhibition. Another set of cells remained untreated (cells grown in media lacking actinomycin-D and rhTNF-.alpha.). Growth inhibition was measured as percent of untreated control cell growth.
The results of the cytotoxicity assay are shown in FIG. 3. Serum from each of the inoculated mice showed the ability to neutralize rhTNF-.alpha. mediated cytotoxicity. Serum from mouse #3 showed the greatest ability to inhibit rhTNF-.alpha. mediated cytotoxicity.
Hybridomas were produced from splenocytes of mice showing the presence of anti-TNF-.alpha. antibodies by PEG fusion, essentially as discussed above. Selection of fused hybridomas was performed using HAT medium. Neutralizing clones 4C9 and 4D3 were obtained from mouse #3. After further subcloning, antibodies were purified by chromatography on protein G columns. Purified antibodies were determined to be homogeneous by size separation HPLC and SDS-PAGE (data not shown). Isotype analysis, performed as discussed above, showed that 4C9 was IgG1/.kappa. while 4D3 was IgG2a/.kappa..
The ability of anti-TNF-.alpha. antibodies from clones 4C9D11 and 4D3B11 to neutralize TNF-.alpha.-mediated cytotoxicity was determined (FIG. 4). WEHI 164 cells were seeded at 1.times.10.sup.4 cells/well into 96-well plates and grown in 200 .mu.L of RPMI complete medium overnight. On the day of the assay, supernatants from clones were collected and diluted 1:2. A further 1:5 dilution was made thereafter. Each dilution was made in RPMI complete medium containing a final concentration of actinomycin-D at 2 .mu.g/mL and rhTNF-.alpha. at 0.1 ng/mL. Before addition of the diluted supernatant, the medium in the plate for WEHI 164 cells growth was removed, and replaced with the diluted supernatant in the corresponding wells, 100 .mu.L/well. The plate was incubated for 20 hours at 37.degree. C. in a 5% CO2 incubator. After this incubation, 20 .mu.L of MTS was added to all the wells and the absorbance in each well determined in a plate reader at 490 nm after two hours. As a negative control, supernatant from a clone which stopped producing antibody (ELISA negative) was diluted in a like manner. One set of wells was incubated with only actinomycin-D and rhTNF-.alpha. to determine maximum growth inhibition. Another set of cells remained untreated (cells grown in media lacking actinomycin-D and rhTNF-.alpha.). Growth inhibition was measured as percent of untreated control cell growth. The antibody from clone 4D3B11 was more effective at blocking TNF-.alpha. mediated cytotoxicity in this assay (FIG. 4).
Antibody binding specificity was determined by Western blotting against rhTNF-.alpha.. Under reducing conditions, 4D3B11C4 and the anti-TNF-.alpha. antibody REMICADE.RTM. (infliximab) showed no or weak binding to human TNF-.alpha., with no binding to human TNF-.beta. or murine TNF-.alpha. (not shown). Under the same reducing conditions, antibody 4C9D11G11 showed strong binding to human TNF-.alpha., with no binding to human TNF-.beta. or murine TNF-.alpha. (not shown).
Neutralization of rhTNF-.alpha. induced cytotoxicity by anti-TNF-.alpha. antibodies was determined in a different in vitro system (FIG. 5). L929 cells (mouse fibroblasts) were seeded at 2.times.10.sup.4 cells/well into 96-well plates and grown in 90 .mu.L of MEM medium overnight (10% horse serum complete medium). On the following day, the purified antibodies were diluted 1:5 in MEM medium (containing a final concentration of actinomycin-D at 20 .mu.g/mL and rhTNF-.alpha. at 1 ng/mL) for an antibody concentration range of 10,000 to 3.2 ng/mL. The antibodies were pre-incubated with rhTNF-.alpha. at RT for one hour. After this pre-incubation, 10 .mu.L of the diluted antibodies were then added to the 90 .mu.L cells in the corresponding wells, that made the final concentration of the antibodies from 1000 to 0.32 ng/mL, with a final concentration of actinomycin-D and rhTNF-.alpha. at 2 .mu.g/mL and 0.1 ng/mL, respectively. The plate was incubated for 20 hours at 37.degree. C. Following this incubation, 20 .mu.L of MTS was added to all the wells and the absorbance in each well determined in a plate reader at 490 nm after two hours. As a negative control, an anti-hTNF-.alpha. antibody, 4C3 (non-neutralizing), was diluted in a like manner. REMICADE.RTM., the commercial anti-TNF-.alpha. antibody was also diluted in a like manner as a positive control. One set of wells was incubated with only actinomycin-D and rhTNF-.alpha. to determine maximum growth inhibition.
Under these conditions, the 4C9D11G11 antibody (EC.sub.50 11.2 ng/mL) was more effective than 4D3B11C4 (EC.sub.50 22.1 ng/mL) at inhibiting TNF-.alpha.-induced cytotoxicity (FIG. 5). Neither monoclonal antibody was as effective as REMICADE.RTM. (EC.sub.50 3.6 ng/mL) (FIG. 5).
An assay was performed for antibody based neutralization of rhTNF-.alpha.-induced cell surface expression of ICAM-1 (FIG. 6). ECV-304 cells (a derivative of T24, bladder cancer cell line) were seeded at 2.times.10.sup.5 cells/well into 6-well plates, grown in 10% FBS Medium 199 for 6 hours for attaching. Varying doses of the mAbs or REMICADE.RTM. (positive control) were mixed with constant amounts of rhTNF-.alpha. (10 ng/mL). The mixture of the antibodies and rhTNF-.alpha. was pre-incubated at 37.degree. C. for two hours, and then pipetted into the appropriate corresponding wells in duplicate. Cells were then grown for 72 hours in a 37.degree. C. incubator. After this incubation, supernatant was removed and cells were trypsinized and transferred to 15 mL tubes. Cells were washed with cold PBS/0.5% BSA two times, supernatant was removed and the cell pellets were re-suspended in the residual wash buffer (.about.100 .mu.L). An aliquot of 25 .mu.L from the cell suspension from each sample was then transferred to 4 mL flow tubes. Cells were Fc-blocked by treatment with 1 .mu.g of human IgG for 15 min at RT and then incubated with fluorescent-conjugated anti-CD54 reagent for 45 min at 4.degree. C. Cells were then washed with 4 mL of PBS/5% BSA for two times and re-suspended in 400 .mu.L of PBS and then subjected to flow-cytometric analysis (FACS). One set of cells remained untreated as background fluorescent control. Another set of cells treated with only 10 ng/mL of rhTNF-.alpha. served as the positive control for obtaining maximum fluorescent (i.e. maximum ICAM-1 up-regulation).
The 4C9 clone again showed higher neutralizing activity than the 4D3 clone and 4C9 was selected for chimerization.
Example 3. Production of Chimeric Anti-IL-6 Antibody from 2-3B2 Hybridoma
Total RNA was extracted from hybridomas 2-3B2 cells by standard techniques and mRNA was separated from the total RNA fraction. The mRNA was used as a template for VH and VK cDNA synthesis, using a QIAGEN.RTM. OneStep RT-PCR kit. Primers used were as shown below (restriction sites are underlined).
TABLE-US-00017 Vk1 BACK (PNAS 86:3833-3837,1989) (SEQ ID NO: 90) GACATTCAGCTGACCCAGTCTCCA CK3'-BH: (Biotechniques 15:286-291, 1993) (SEQ ID NO: 91) GCCGGATCCTCACTGGATGGTGGGAAGATGGATACA VH1 BACK: (PNAS 86:3833-3837, 1989) AGGTSMARCTGCAGSAGTCWGG (SEQ ID NO: 92, S = C/G, M = A/C, R = A/G, W = A/T) CH1-C: (Clinical Cancer Res 5:3095s-3100s, 1999) (SEQ ID NO: 93) AGCTGGGAAGGTGTGCAC
The VH and Vk cDNA sequences were cloned into the pGEMT vector for sequencing by the Sanger dideoxy technique, using an automated DNA sequencer. The putative VH (SEQ ID NO:94) and Vk (SEQ ID NO:96) murine amino sequences are shown in FIG. 7 and FIG. 8. The locations of the heavy and light chain CDR sequences are proposed, based on homology with the known heavy and light chain antibody sequences of B34871 (SEQ ID NO:95) and AAB53778.1 (SEQ ID NO:97), respectively, from the NCBI protein sequence database. The indicated 2-3B2 heavy chain CDR sequences are CDR1 (GFTFSRFGMH, SEQ ID NO:107), CDR2 (YIGRGSSTIYYADTVKG, SEQ ID NO:108) and CDR3 (SNWDGAMDY, SEQ ID NO:109). The 2-3B2 light chain CDR sequences are CDR1 (RASGNIHNFLA, SEQ ID NO:110), CDR2 (NAETLAD, SEQ ID NO:111) and CDR3 (QHFWSTPWT, SEQ ID NO:112).
The VH and VK sequences from the 2-3B2 anti-IL-6 antibody and the VH and VK sequences from the 4C9 anti-TNF-.alpha. antibody were used to make a cIL6/TNF.alpha. DVD (dual variable domain) antibody construct. (See, e.g., Wu et al., 2009, MAbs 1:339-47.) The resulting bispecific DVD construct was compared with the parent 2-3B2 anti-IL-6 antibody for the ability to inhibit IL-6 induced phosphorylation of STAT3 on HT-29 cells (FIG. 9). As shown in FIG. 9, the DVD construct showed the same efficacy as the parent anti-IL-6 antibody for inhibition of IL-6 mediated phosphorylation.
The sequences for restriction sites and leader peptides for cloning into vector pdHL2 were added to the VH and VK sequences of 2-3B2. The complete sequences were synthesized commercially (GenScript, Piscataway, N.J.). The 2-3B2-VH-pUC57 and 2-3B2-VK-pUC57 vectors were produced by incorporating the VH sequence as a XhoI-HindIII insert and the VK sequence as a XbaI-BamH1 insert into corresponding sites in pUC57. A vector expressing chimeric 2-3B2 antibody was produced starting with the hA20-pdHL2-IgG vector (see, e.g., Goldenberg et al., 2002, Blood 100:11 Abstract 2260). The hA20-VH sequence was replaced with cIL6-VH and the hA20-VK sequence was replaced with cIL6-VK by restriction enzyme digestion and ligation. The resulting chimeric 2-3B2 antibody comprised the murine VH and VK sequences of 2-3B2, attached to human antibody constant region sequences. After transfection, screening and antibody purification on a protein A column, a chimeric anti-IL6 clone 1B5 (c-IL6-1B5) was obtained as a homogeneous antibody preparation, as confirmed by HPLC and SDS-PAGE (not shown). The final clone is identified as 1B5A9.
Example 4. Production of Chimeric Anti-TNF-.alpha. Antibody from 4C9 Hybridoma
Total RNA was extracted from hybridomas 4C9 cells by standard techniques and mRNA was separated from the total RNA fraction. The mRNA was used as a template for VH and VK cDNA synthesis, using a PHUSION.RTM. High Fidelity PCR kit (Thermo Scientific, Pittsburgh, Pa.). Primers used were as disclosed in Example 3 above.
The VH and Vk cDNA sequences were cloned into the pGEMT vector for sequencing by the Sanger dideoxy technique, using an automated DNA sequencer. The putative VH (SEQ ID NO:98) and Vk (SEQ ID NO:100) murine amino sequences are shown in FIG. 10 and FIG. 11. The locations of the heavy and light chain CDR sequences are proposed, based on homology with the known heavy and light chain antibody sequences of AAS66033.1 (SEQ ID NO:99) and AAS66032.1 (SEQ ID NO:101), respectively, from the NCBI protein sequence database. The indicated 4C9 heavy chain CDR sequences are CDR1 (GFWN, SEQ ID NO:113), CDR2 (YISYSGRTYYNPSLKS, SEQ ID NO:114) and CDR3 (DANYVLDY, SEQ ID NO:115). The 4C9 light chain CDR sequences are CDR1 (KSSQSLLNSSTQKNYLA, SEQ ID NO:116), CDR2 (FASARES, SEQ ID NO:117) and CDR3 (QQHYRTPFT, SEQ ID NO:118).
An optimized DNA sequence encoding the TNF-.alpha. VH, also comprising a 5' leader sequence and 3' flanking sequence, was designed as shown in SEQ ID NO:102 below and cloned into pdHL2. The optimized 4C9-VH sequence is underlined. The DNA sequence was synthesized by GenScript (Piscataway, N.J.).
TABLE-US-00018 (SEQ ID NO: 102) CTCGAGCACACAGGACCTCACCATGGGATGGAGCTGTATCATCCTCTTCT TGGTAGCAACAGCTACAGGTAAGGGGCTCACAGTAGCAGGCTTGAGGTCT GGACATATATATGGGTGACAATGACATCCACTTTGCCTTTCTCTCCACAG GTGTCCACTCCGTGCAGCTGCAGGAGAGCGGACCCTCCCTGGTGAAGCCT AGTCAGACCCTGAGCCTGACATGCTCCGTGACTGGGGACTCTATCACCAG TGGCTTCTGGAACTGGATTCGGAAGTTCCCAGGAAACAAGTTTGAATACA TGGGATATATCTCTTACAGTGGGCGCACATACTATAACCCCAGCCTGAAG TCCAGGCTGTCTATTACAAGAGACACTTCTAAAAACCAGTTTTATCTGCA GCTGAACAGCGTGACTGCCGAGGATACTGCTACCTACTATTGTGCCAGGG ACGCTAATTATGTGCTGGATTACTGGGGCCAGGGAACCACACTGACCGTG AGCTCCGGTGAGTCCTTACAACCTCTCTCTTCTATTCAGCTTAAATAGAT TTTACTGCATTTGTTGGGGGGGAAATGTGTGTATCTGAATTTCAGGTCAT GAAGGACTAGGGACACCTTGGGAGTCAGAAAGGGTCATTGGGAAGCTT
An optimized DNA sequence encoding the TNF-.alpha. VK, also comprising a 5' leader sequence and 3' flanking sequence, was designed as shown in SEQ ID NO:103 below and cloned into pdHL2. The optimized 4C9-VK sequence is underlined. The DNA sequence was synthesized by GenScript (Piscataway, N.J.).
TABLE-US-00019 (SEQ ID NO: 103) TCTAGACACAGGACCTCACCATGGGATGGAGCTGTATCATCCTCTTCTTG GTAGCAACAGCTACAGGTAAGGGGCTCACAGTAGCAGGCTTGAGGTCTGG ACATATATATGGGTGACAATGACATCCACTTTGCCTTTCTCTCCACAGGT GTCCACTCCGACATCCAGCTGACCCAGAGCCCCAGCTCCCTGGCTATGTC CGTGGGACAGAAGGTGACAATGAACTGCAAATCTAGTCAGTCTCTGCTGA ACAGCTCCACTCAGAAGAATTACCTGGCTTGGTTCCAGCAGAAGCCCGGG CAGAGTCCTAAACTGCTGGTGTATTTTGCCTCTGCTAGGGAGAGTGGCGT GCCAGACAGATTCATCGGCAGCGGCAGCGGGACCGATTTTACCCTGACAA TTTCTAGTGTGCAGGCCGAGGACCTGGCTGATTACTTCTGTCAGCAGCAC TATCGGACTCCCTTCACCTTTGGCTCCGGAACAAAGCTGGAGATCAAGCG TGAGTAGAATTTAAACTTTGCTTCCTCAGTTGGATCC
The VH and VK coding sequences were inserted into pUC57 and then pdHL2 for expression of the chimeric 4C9 antibody as discussed in Example 3 above. The chimeric 4C9 was produced by transfection of pdHL2, screening for transfectants and antibody purification on a protein A column. The selected clone was designated 6A9. The purified antibody was determined to be homogeneous by HPLC and SDS-PAGE (not shown). A binding affinity assay for chimeric anti-TNF-.alpha. showed a dissociation constant (K.sub.D) of 4.13 e.sup.-11 (not shown).
Example 5. Construction of CH1-DDD2-cFab-anti-IL-6-pGSHL
The hLL2-Fab-DDD2-pGSHL#2 plasmid (see, e.g., WO2013181087A2; Rossi et al., 2009, Blood 113:6161-71; U.S. Patent Publ. Nos. 20130323204, 20140212425) was used as a starting material for production of a DDD2 conjugated Fab anti-IL-6 antibody fragment. The hLL2-DDD2 plasmid was digested with XbaI/XhoI and the 6577 bp vector was isolated. cIL6-pdHL2 (Example 3) was digested with XbaI/XhoI and the 2604 bp cIL6 coding insert was isolated. The two were ligated to form the 9182 bp Vk-cIL6-Fab-DDD2-pGSHL vector. After screening by PstI digestion and electrophoresis, the 9182 bp vector was digested with XhoI/Hind3/Alkaline phosphatase and an 8536 vector was isolated. The cIL6-pdHL2 vector, comprising a 648 bp cIL6-VH coding insert was digested with XhoI/Hind3. The 648 bp VH encoding insert was ligated with the 8536 bp vector and VK insert to generate C.sub.H1-DDD2-cFab-anti-IL-6-pGSHL. The final construct was then transfected, clones were picked and purified by Kappa-select (GE Healthcare Life Sciences, Piscataway, N.J.). The purified antibody product of C.sub.H1-DDD2-cFab-anti-IL-6 appeared homogeneous on HPLC and SDS-PAGE (not shown). The DDD2-derivatized cIL6-Fab showed equivalent activity to the underivatized cIL6 or an hR1-(IL6).sub.4 construct when assayed for inhibition of IL-6 induced STAT3 phosphorylation (not shown).
Example 6. Construction of C.sub.K-AD2-cIgG-anti-TNF-.alpha.-pdHL2
The Ck-AD2-IgG-hA20-pdHL2 plasmid (see, e.g., WO201262583A1; Chang et al., 2012, PLoS ONE 7(8): e44235; U.S. Patent Publ. Nos. 20130323204, 20070140966) was used as a starting material for production of an AD2 conjugated IgG anti-TNF-.alpha. antibody. The Ck-AD2-hA20 plasmid was digested with BamHI/XhoI to obtain the Ck-AD2 coding portion. Plasmid cIgG-anti-TNF-.alpha.-pdHL2 (Example 4) was digested with BamHI/XhoI to obtain .DELTA.C.sub.K-cIgG-anti-TNF-.alpha.-pdHL2. The two were ligated to form Ck-AD2-cIgG-anti-TNF-.alpha.-pdHL2 (see FIG. 12). The C.sub.K-AD2-cIgG-anti-TNF-.alpha.-pdHL2 vector was used to transform DHF.alpha. competent cells. Colonies were picked and purified by mini-Prep. Plasmid DNA was analyzed by restriction endonuclease digestion and agarose gel electrophoresis (not shown). The plasmid DNA was purified by Maxi-Prep and the insert was DNA sequenced. The DNA sequences encoding cTNF-.alpha.-VH, AD2 and cTNF-.alpha.-VK are shown in SEQ ID NOs 104-106 below.
TABLE-US-00020 cTNF-.alpha.-VH (SEQ ID NO: 104) GTGCAGCTGCAGGAGAGCGGACCCTCCCTGGTGAAGCCTAGTCAGACCCT GAGCCTGACATGCTCCGTGACTGGGGACTCTATCACCAGTGGCTTCTGGA ACTGGATTCGGAAGTTCCCAGGAAACAAGTTTGAATACATGGGATATATC TCTTACAGTGGGCGCACATACTATAACCCCAGCCTGAAGTCCAGGCTGTC TATTACAAGAGACACTTCTAAAAACCAGTTTTATCTGCAGCTGAACAGCG TGACTGCCGAGGATACTGCTACCTACTATTGTGCCAGGGACGCTAATTAT GTGCTGGATTACTGGGGCCAGGGAACCACACTGACCGTGAGCTCC AD2 (SEQ ID NO: 105) TGTGGCCAGATCGAGTACCTGGCCAAGCAGATCGTGGACAACGCCATCCA GCAGGCCGGGTGC cTNF-.alpha.-VK (SEQ ID NO: 106) GACATCCAGCTGACCCAGAGCCCCAGCTCCCTGGCTATGTCCGTGGGACA GAAGGTGACAATGAACTGCAAATCTAGTCAGTCTCTGCTGAACAGCTCCA CTCAGAAGAATTACCTGGCTTGGTTCCAGCAGAAGCCCGGGCAGAGTCCT AAACTGCTGGTGTATTTTGCCTCTGCTAGGGAGAGTGGCGTGCCAGACAG ATTCATCGGCAGCGGCAGCGGGACCGATTTTACCCTGACAATTTCTAGTG TGCAGGCCGAGGACCTGGCTGATTACTTCTGTCAGCAGCACTATCGGACT CCCTTCACCTTTGGCTCCGGAACAAAGCTGGAGATCAAGCGTGAGTAGAA TTTAAACTTTGCT
After transfection, screening, expression and antibody purification, clone 4A5 encoding C.sub.K-AD2-cIgG-4A5 was obtained.
Example 7. Construction of cT*-(c6)-(c6) Anti-IL-6/Anti-TNF-.alpha. Bispecific DNL.RTM. Complex
The Ck-AD2-cIgG-4A5 and C.sub.H1-DDD2-cFab-anti-IL-6 fusion proteins were used to make a DOCK-AND-LOCK.RTM. (DNL).RTM. complex, using techniques disclosed herein and in issued U.S. Pat. Nos. 7,550,143; 7,521,056; 7,534,866; 7,527,787; 7,666,400; 7,858,070; 7,871,622; 7,906,121; 7,906,118; 8,163,291; 7,901,680; 7,981,398; 8,003,111; 8,034,352; 8,562,988; 8,211,440; 8,491,914; 8,282,934; 8,246,960; 8,349,332; 8,277,817; 8,158,129; 8,475,794; 8,597,659; 8,481,041; 8,435,540 and 8,551,480, the Examples section of each incorporated herein by reference.
The intact DNL.RTM. complex was formed by mixing the AD2 and DDD2 components together under reducing conditions and allowing the complementary sequences on the DDD moiety to form a dimer that binds to the AD moiety. Twenty five mg of C.sub.K-AD2-cIgG-4A5 was mixed with 50 mg of C.sub.H1-DDD2-cFab-anti-IL-6. A 1/10 volume of 1M Tris, pH 7.5, 1 mM EDTA, 2 mM reduced glutathione was added to the reaction and the proteins were reduced overnight at room temperature. The complexes were then oxidized with 4 mM oxidized glutathione at room temperature for 3 hours to form disulfide bonds between the AD2 and DDD2 moieties to stabilize the complex.
Chromatography of the complex on a MABSELECT.TM. column was performed. After loading, the column was washed with 0.04M PBS, pH 7.4+1 mM oxidized glutathione, followed by a PBS wash and elution with 0.1M citrate (pH 3.5). The elution volume was 25 ml (2.5 ml of 3M Tris, pH 8.6+22.5 ml of eluate). The concentration measured by OD280 was 2.3 mg/ml (57.5 mg total).
The product was dialyzed against two 5-L changes of 0.04M PBS, pH 7.4. The final concentration by OD280 was 1.8 mg/ml (52.5 mg total). Purified complex was analyzed by SE-HPLC, which confirmed the presence of cT*-(c6)-(c6) as an apparently homogeneous peak (not shown). The results were confirmed by SDS-PAGE. The activity of the purified cT*-(c6)-(c6) bispecific antibody complex was then examined.
The cT*-(c6)-(c6) complex showed greater activity than the Fab-DDD2-cIL-6 protein for inhibiting IL-6 induced phosphorylation of STAT3 (FIG. 13). HT-29 cells were seeded at 2.times.10.sup.6 cells/well in 6-well plates, grown overnight. The indicated antibodies were pre-incubated with hIL6 at 37.degree. C. for 1 hour. Then media containing rhIL-6 alone or in combination with antibodies was added to the HT-29 cells for 30 min at 37.degree. C. After the incubation, the supernatant was removed, cells were washed and lysed.
Two SDS-PAGE gels were run, transferred to nitrocellulose membranes. Membranes were cut at 60 KDa, the upper portions was probed with either anti-p-STAT3 or anti-t-STAT3 (FIG. 13). The lower portions were probed with b-actin for loading control (FIG. 13). This assay showed that TNF-(IL6)-(IL6) neutralized IL-6 with similar or greater potency compared to anti-IL-6 Fab-DDD2.
FIG. 14 shows that the cT*-(c6)-(c6) complex was able to neutralize natural IL-6 induced phosphorylation of STAT3 in HT-29 cells. HT-29 cells were seeded at 2.times.10.sup.6 cells/well in 6-well plates and grown overnight. TNF-(IL6)-(IL6) or chimeric anti-IL6 1B5A9 was pre-incubated with the supernatant containing 10 ng/mL of natural IL6 released from collagen Type II stimulated RA patient PBMCs at 37.degree. C. for 1 hour. At the end of the incubation, the supernatant containing natural IL-6 alone or in combination with antibodies was added to the HT-29 cells for 30 min at 37.degree. C. After incubation, the supernatant was removed and cells were washed and lysed. Two SDS-PAGE gels were run, transferred to nitrocellulose membranes. Membranes were cut at 60 KDa, the upper portion was probed with either anti-p-STAT3 or anti-t-STAT3 (FIG. 14). The lower portions were probed with b-actin for loading control (FIG. 14).
The ability to neutralize TNF-.alpha. induced cell death was also examined for cT*-(c6)-(c6) compared to other anti-TNF-.alpha. antibody constructs (FIG. 15). In the presence of 2 .mu.g/mL of actinomycin-D, recombinant human TNF-.alpha. at 0.1 ng/mL induced about 70% cell death in L929 cells. As shown, the TNF-.alpha.-IL6-IL6, chimeric anti-TNF-.alpha. clone 6A9 and Ck-AD2-cTNF-.alpha.-IgG clone 4A5 were able to neutralize the activity of rhTNF-.alpha., and inhibit cell death in a dose-response manner (FIG. 15), just like their parent antibody 4C9.
TNF-.alpha.-(IL6)-(IL6) and Ck-AD2-cTNF-.alpha. were also able to neutralize cell death of L929 cells induced by natural human TNF-.alpha. (released from RA PBMCs) (FIG. 16). Upon stimulation by type II collagen for 5 days, natural human TNF-.alpha. is released from the cultured PBMCs isolated from a rheumatoid arthritis patient (S22). In the presence of 2 .mu.g/mL of actinomycin-D, TNF-.alpha. at 0.1 ng/mL induced about 76% cell death. As shown in FIG. 16, TNF-.alpha.-IL6-IL6 and Ck-AD2-cTNF-.alpha.-IgG clone 4A5 were able to neutralize the activity of the natural human TNF-.alpha., and inhibit cell death in a dose-response manner.
The ability of cT*-(c6)-(c6) to bind to IL-6 or TNF-.alpha. from rat, monkey or human was determined by ELISA. The results are summarized in FIG. 17, which shows that the affinity of cT*-(c6)-(c6) for IL-6 or TNF-.alpha. from different species was approximately the same as the individual antibodies, and that the antibodies showed approximately similar dissociation constants for human, Cynomolgus monkey and canine antigens.
As can be seen in FIG. 18, STAT3 plays a central role in both TNF-.alpha. and IL-6 mediated pathways and disease processes and inhibition of STAT3 phosphorylation induced by TNF-.alpha. or IL-6 is a reasonable surrogate to determine the efficacy of anti-TNF-.alpha. or anti-IL-6 antibody complexes as moderators of such disease processes. Because TNF-.alpha. and IL-6 play pathogenic roles in the development of a variety of autoimmune, immune dysfunction or inflammatory diseases, including but not limited to systemic lupus erythematosus, rheumatoid arthritis, inflammatory bowel disease, type II diabetes, obesity, atherosclerosis and cachexia related to cancer, the presence results show that bispecific anti-IL-6/anti-TNF-.alpha. antibodies, such as cT*-(c6)-(c6), are of use for treatment of such TNF-.alpha./IL-6 mediated diseases or conditions.
SEQUENCE LISTINGS
1
118144PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 1Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 40245PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 2Cys Gly His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly1 5 10 15Tyr Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe 20 25 30Ala Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 40 45317PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 3Gln Ile Glu Tyr Leu Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Ala421PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 4Cys Gly Gln Ile Glu Tyr Leu Ala Lys Gln Ile Val Asp Asn Ala Ile1 5 10 15Gln Gln Ala Gly Cys 20550PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 5Ser Leu Arg Glu Cys Glu Leu Tyr Val Gln Lys His Asn Ile Gln Ala1 5 10 15Leu Leu Lys Asp Ser Ile Val Gln Leu Cys Thr Ala Arg Pro Glu Arg 20 25 30Pro Met Ala Phe Leu Arg Glu Tyr Phe Glu Arg Leu Glu Lys Glu Glu 35 40 45Ala Lys 50655PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 6Met Ser Cys Gly Gly Ser Leu Arg Glu Cys Glu Leu Tyr Val Gln Lys1 5 10 15His Asn Ile Gln Ala Leu Leu Lys Asp Ser Ile Val Gln Leu Cys Thr 20 25 30Ala Arg Pro Glu Arg Pro Met Ala Phe Leu Arg Glu Tyr Phe Glu Arg 35 40 45Leu Glu Lys Glu Glu Ala Lys 50 55723PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 7Cys Gly Phe Glu Glu Leu Ala Trp Lys Ile Ala Lys Met Ile Trp Ser1 5 10 15Asp Val Phe Gln Gln Gly Cys 20851PRTHomo sapiens 8Ser Leu Arg Glu Cys Glu Leu Tyr Val Gln Lys His Asn Ile Gln Ala1 5 10 15Leu Leu Lys Asp Val Ser Ile Val Gln Leu Cys Thr Ala Arg Pro Glu 20 25 30Arg Pro Met Ala Phe Leu Arg Glu Tyr Phe Glu Lys Leu Glu Lys Glu 35 40 45Glu Ala Lys 50954PRTHomo sapiens 9Ser Leu Lys Gly Cys Glu Leu Tyr Val Gln Leu His Gly Ile Gln Gln1 5 10 15Val Leu Lys Asp Cys Ile Val His Leu Cys Ile Ser Lys Pro Glu Arg 20 25 30Pro Met Lys Phe Leu Arg Glu His Phe Glu Lys Leu Glu Lys Glu Glu 35 40 45Asn Arg Gln Ile Leu Ala 501044PRTHomo sapiens 10Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Gly Gln Gln Pro Pro Asp Leu Val Asp Phe Ala Val 20 25 30Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Arg Gln 35 401144PRTHomo sapiens 11Ser Ile Glu Ile Pro Ala Gly Leu Thr Glu Leu Leu Gln Gly Phe Thr1 5 10 15Val Glu Val Leu Arg His Gln Pro Ala Asp Leu Leu Glu Phe Ala Leu 20 25 30Gln His Phe Thr Arg Leu Gln Gln Glu Asn Glu Arg 35 401244PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 12Thr His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 401344PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 13Ser Lys Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 401444PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 14Ser Arg Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 401544PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 15Ser His Ile Asn Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 401644PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 16Ser His Ile Gln Ile Pro Pro Ala Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 401744PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 17Ser His Ile Gln Ile Pro Pro Gly Leu Ser Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 401844PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 18Ser His Ile Gln Ile Pro Pro Gly Leu Thr Asp Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 401944PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 19Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Asn Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 402044PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 20Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Ala Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 402144PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 21Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Ser Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 402244PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 22Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Asp Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 402344PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 23Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Lys Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 402444PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 24Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Asn Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 402544PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 25Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Asn Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 402644PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 26Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Glu Leu Val Glu Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 402744PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 27Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Asp Phe Ala 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 402844PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 28Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Leu 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 402944PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 29Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ile 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 403044PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 30Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Val 20 25 30Val Glu Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 403144PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 31Ser His Ile Gln Ile Pro Pro Gly Leu Thr Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Val Glu Val Leu Arg Gln Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Val Asp Tyr Phe Thr Arg Leu Arg Glu Ala Arg Ala 35 403217PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 32Asn Ile Glu Tyr Leu Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Ala3317PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 33Gln Leu Glu Tyr Leu Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Ala3417PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 34Gln Val Glu Tyr Leu Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Ala3517PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 35Gln Ile Asp Tyr Leu Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Ala3617PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 36Gln Ile Glu Phe Leu Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Ala3717PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 37Gln Ile Glu Thr Leu Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Ala3817PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 38Gln Ile Glu Ser Leu Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Ala3917PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 39Gln Ile Glu Tyr Ile Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Ala4017PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 40Gln Ile Glu Tyr Val Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Ala4117PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 41Gln Ile Glu Tyr Leu Ala Arg Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Ala4217PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 42Gln Ile Glu Tyr Leu Ala Lys Asn Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Ala4317PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 43Gln Ile Glu Tyr Leu Ala Lys Gln Ile Val Glu Asn Ala Ile Gln Gln1 5 10 15Ala4417PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 44Gln Ile Glu Tyr Leu Ala Lys Gln Ile Val Asp Gln Ala Ile Gln Gln1 5 10 15Ala4517PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 45Gln Ile Glu Tyr Leu Ala Lys Gln Ile Val Asp Asn Ala Ile Asn Gln1 5 10 15Ala4617PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 46Gln Ile Glu Tyr Leu Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Asn1 5 10 15Ala4717PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 47Gln Ile Glu Tyr Leu Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Leu4817PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 48Gln Ile Glu Tyr Leu Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Ile4917PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 49Gln Ile Glu Tyr Leu Ala Lys Gln Ile Val Asp Asn Ala Ile Gln Gln1 5 10 15Val5017PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 50Gln Ile Glu Tyr Val Ala Lys Gln Ile Val Asp Tyr Ala Ile His Gln1 5 10 15Ala5117PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 51Gln Ile Glu Tyr Lys Ala Lys Gln Ile Val Asp His Ala Ile His Gln1 5 10 15Ala5217PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 52Gln Ile Glu Tyr His Ala Lys Gln Ile Val Asp His Ala Ile His Gln1 5 10 15Ala5317PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 53Gln Ile Glu Tyr Val Ala Lys Gln Ile Val Asp His Ala Ile His Gln1 5 10 15Ala5418PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 54Pro Leu Glu Tyr Gln Ala Gly Leu Leu Val Gln Asn Ala Ile Gln Gln1 5 10 15Ala Ile5518PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 55Leu Leu Ile Glu Thr Ala Ser Ser Leu Val Lys Asn Ala Ile Gln Leu1 5 10 15Ser Ile5618PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 56Leu Ile Glu Glu Ala Ala Ser Arg Ile Val Asp Ala Val Ile Glu Gln1 5 10 15Val Lys5718PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 57Ala Leu Tyr Gln Phe Ala Asp Arg Phe Ser Glu Leu Val Ile Ser Glu1 5 10 15Ala Leu5817PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 58Leu Glu Gln Val Ala Asn Gln Leu Ala Asp Gln Ile Ile Lys Glu Ala1 5 10 15Thr5917PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 59Phe Glu Glu Leu Ala Trp Lys Ile Ala Lys Met Ile
Trp Ser Asp Val1 5 10 15Phe6018PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 60Glu Leu Val Arg Leu Ser Lys Arg Leu Val Glu Asn Ala Val Leu Lys1 5 10 15Ala Val6118PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 61Thr Ala Glu Glu Val Ser Ala Arg Ile Val Gln Val Val Thr Ala Glu1 5 10 15Ala Val6218PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 62Gln Ile Lys Gln Ala Ala Phe Gln Leu Ile Ser Gln Val Ile Leu Glu1 5 10 15Ala Thr6316PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 63Leu Ala Trp Lys Ile Ala Lys Met Ile Val Ser Asp Val Met Gln Gln1 5 10 156424PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 64Asp Leu Ile Glu Glu Ala Ala Ser Arg Ile Val Asp Ala Val Ile Glu1 5 10 15Gln Val Lys Ala Ala Gly Ala Tyr 206518PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 65Leu Glu Gln Tyr Ala Asn Gln Leu Ala Asp Gln Ile Ile Lys Glu Ala1 5 10 15Thr Glu6620PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 66Phe Glu Glu Leu Ala Trp Lys Ile Ala Lys Met Ile Trp Ser Asp Val1 5 10 15Phe Gln Gln Cys 206717PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 67Gln Ile Glu Tyr Leu Ala Lys Gln Ile Pro Asp Asn Ala Ile Gln Gln1 5 10 15Ala6825PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 68Lys Gly Ala Asp Leu Ile Glu Glu Ala Ala Ser Arg Ile Val Asp Ala1 5 10 15Val Ile Glu Gln Val Lys Ala Ala Gly 20 256925PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 69Lys Gly Ala Asp Leu Ile Glu Glu Ala Ala Ser Arg Ile Pro Asp Ala1 5 10 15Pro Ile Glu Gln Val Lys Ala Ala Gly 20 257025PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 70Pro Glu Asp Ala Glu Leu Val Arg Leu Ser Lys Arg Leu Val Glu Asn1 5 10 15Ala Val Leu Lys Ala Val Gln Gln Tyr 20 257125PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 71Pro Glu Asp Ala Glu Leu Val Arg Thr Ser Lys Arg Leu Val Glu Asn1 5 10 15Ala Val Leu Lys Ala Val Gln Gln Tyr 20 257225PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 72Pro Glu Asp Ala Glu Leu Val Arg Leu Ser Lys Arg Asp Val Glu Asn1 5 10 15Ala Val Leu Lys Ala Val Gln Gln Tyr 20 257325PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 73Pro Glu Asp Ala Glu Leu Val Arg Leu Ser Lys Arg Leu Pro Glu Asn1 5 10 15Ala Val Leu Lys Ala Val Gln Gln Tyr 20 257425PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 74Pro Glu Asp Ala Glu Leu Val Arg Leu Ser Lys Arg Leu Pro Glu Asn1 5 10 15Ala Pro Leu Lys Ala Val Gln Gln Tyr 20 257525PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 75Pro Glu Asp Ala Glu Leu Val Arg Leu Ser Lys Arg Leu Val Glu Asn1 5 10 15Ala Val Glu Lys Ala Val Gln Gln Tyr 20 257625PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 76Glu Glu Gly Leu Asp Arg Asn Glu Glu Ile Lys Arg Ala Ala Phe Gln1 5 10 15Ile Ile Ser Gln Val Ile Ser Glu Ala 20 257725PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 77Leu Val Asp Asp Pro Leu Glu Tyr Gln Ala Gly Leu Leu Val Gln Asn1 5 10 15Ala Ile Gln Gln Ala Ile Ala Glu Gln 20 257825PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 78Gln Tyr Glu Thr Leu Leu Ile Glu Thr Ala Ser Ser Leu Val Lys Asn1 5 10 15Ala Ile Gln Leu Ser Ile Glu Gln Leu 20 257925PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 79Leu Glu Lys Gln Tyr Gln Glu Gln Leu Glu Glu Glu Val Ala Lys Val1 5 10 15Ile Val Ser Met Ser Ile Ala Phe Ala 20 258025PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 80Asn Thr Asp Glu Ala Gln Glu Glu Leu Ala Trp Lys Ile Ala Lys Met1 5 10 15Ile Val Ser Asp Ile Met Gln Gln Ala 20 258125PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 81Val Asn Leu Asp Lys Lys Ala Val Leu Ala Glu Lys Ile Val Ala Glu1 5 10 15Ala Ile Glu Lys Ala Glu Arg Glu Leu 20 258225PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 82Asn Gly Ile Leu Glu Leu Glu Thr Lys Ser Ser Lys Leu Val Gln Asn1 5 10 15Ile Ile Gln Thr Ala Val Asp Gln Phe 20 258325PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 83Thr Gln Asp Lys Asn Tyr Glu Asp Glu Leu Thr Gln Val Ala Leu Ala1 5 10 15Leu Val Glu Asp Val Ile Asn Tyr Ala 20 258425PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptide 84Glu Thr Ser Ala Lys Asp Asn Ile Asn Ile Glu Glu Ala Ala Arg Phe1 5 10 15Leu Val Glu Lys Ile Leu Val Asn His 20 2585330PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 85Ala Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro Ser Ser Lys1 5 10 15Ser Thr Ser Gly Gly Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr 20 25 30Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser 35 40 45Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser 50 55 60Leu Ser Ser Val Val Thr Val Pro Ser Ser Ser Leu Gly Thr Gln Thr65 70 75 80Tyr Ile Cys Asn Val Asn His Lys Pro Ser Asn Thr Lys Val Asp Lys 85 90 95Lys Ala Glu Pro Lys Ser Cys Asp Lys Thr His Thr Cys Pro Pro Cys 100 105 110Pro Ala Pro Glu Leu Leu Gly Gly Pro Ser Val Phe Leu Phe Pro Pro 115 120 125Lys Pro Lys Asp Thr Leu Met Ile Ser Arg Thr Pro Glu Val Thr Cys 130 135 140Val Val Val Asp Val Ser His Glu Asp Pro Glu Val Lys Phe Asn Trp145 150 155 160Tyr Val Asp Gly Val Glu Val His Asn Ala Lys Thr Lys Pro Arg Glu 165 170 175Glu Gln Tyr Asn Ser Thr Tyr Arg Val Val Ser Val Leu Thr Val Leu 180 185 190His Gln Asp Trp Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val Ser Asn 195 200 205Lys Ala Leu Pro Ala Pro Ile Glu Lys Thr Ile Ser Lys Ala Lys Gly 210 215 220Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu Pro Pro Ser Arg Asp Glu225 230 235 240Leu Thr Lys Asn Gln Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr 245 250 255Pro Ser Asp Ile Ala Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn 260 265 270Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp Ser Asp Gly Ser Phe Phe 275 280 285Leu Tyr Ser Lys Leu Thr Val Asp Lys Ser Arg Trp Gln Gln Gly Asn 290 295 300Val Phe Ser Cys Ser Val Met His Glu Ala Leu His Asn His Tyr Thr305 310 315 320Gln Lys Ser Leu Ser Leu Ser Pro Gly Lys 325 33086330PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptide 86Ala Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro Ser Ser Lys1 5 10 15Ser Thr Ser Gly Gly Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr 20 25 30Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser 35 40 45Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser 50 55 60Leu Ser Ser Val Val Thr Val Pro Ser Ser Ser Leu Gly Thr Gln Thr65 70 75 80Tyr Ile Cys Asn Val Asn His Lys Pro Ser Asn Thr Lys Val Asp Lys 85 90 95Arg Val Glu Pro Lys Ser Cys Asp Lys Thr His Thr Cys Pro Pro Cys 100 105 110Pro Ala Pro Glu Leu Leu Gly Gly Pro Ser Val Phe Leu Phe Pro Pro 115 120 125Lys Pro Lys Asp Thr Leu Met Ile Ser Arg Thr Pro Glu Val Thr Cys 130 135 140Val Val Val Asp Val Ser His Glu Asp Pro Glu Val Lys Phe Asn Trp145 150 155 160Tyr Val Asp Gly Val Glu Val His Asn Ala Lys Thr Lys Pro Arg Glu 165 170 175Glu Gln Tyr Asn Ser Thr Tyr Arg Val Val Ser Val Leu Thr Val Leu 180 185 190His Gln Asp Trp Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val Ser Asn 195 200 205Lys Ala Leu Pro Ala Pro Ile Glu Lys Thr Ile Ser Lys Ala Lys Gly 210 215 220Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu Pro Pro Ser Arg Glu Glu225 230 235 240Met Thr Lys Asn Gln Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr 245 250 255Pro Ser Asp Ile Ala Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn 260 265 270Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp Ser Asp Gly Ser Phe Phe 275 280 285Leu Tyr Ser Lys Leu Thr Val Asp Lys Ser Arg Trp Gln Gln Gly Asn 290 295 300Val Phe Ser Cys Ser Val Met His Glu Ala Leu His Asn His Tyr Thr305 310 315 320Gln Lys Ser Leu Ser Leu Ser Pro Gly Lys 325 3308744PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptideMOD_RES(1)..(1)Ser or ThrMOD_RES(2)..(2)His, Lys or ArgMOD_RES(4)..(4)Gln or AsnMOD_RES(8)..(8)Gly or AlaMOD_RES(10)..(10)Thr or SerMOD_RES(11)..(11)Glu or AspMOD_RES(14)..(14)Gln or AsnMOD_RES(15)..(15)Gly or AlaMOD_RES(17)..(17)Thr or SerMOD_RES(19)..(19)Glu or AspMOD_RES(22)..(22)Arg or LysMOD_RES(23)..(24)Gln or AsnMOD_RES(27)..(27)Asp or GluMOD_RES(30)..(30)Glu or AspMOD_RES(32)..(32)Ala, Leu, Ile or ValMOD_RES(34)..(34)Glu or AspMOD_RES(37)..(37)Thr or SerMOD_RES(38)..(38)Arg or LysMOD_RES(40)..(40)Arg or LysMOD_RES(41)..(41)Glu or AspMOD_RES(42)..(42)Ala, Leu, Ile or ValMOD_RES(43)..(43)Arg or LysMOD_RES(44)..(44)Ala, Leu, Ile or Val 87Xaa Xaa Ile Xaa Ile Pro Pro Xaa Leu Xaa Xaa Leu Leu Xaa Xaa Tyr1 5 10 15Xaa Val Xaa Val Leu Xaa Xaa Xaa Pro Pro Xaa Leu Val Xaa Phe Xaa 20 25 30Val Xaa Tyr Phe Xaa Xaa Leu Xaa Xaa Xaa Xaa Xaa 35 408817PRTArtificial SequenceDescription of Artificial Sequence Synthetic peptideMOD_RES(1)..(1)Gln or AsnMOD_RES(2)..(2)Ile, Leu or ValMOD_RES(3)..(3)Glu or AspMOD_RES(4)..(4)Tyr, Phe, Thr or SerMOD_RES(5)..(5)Leu, Ile or ValMOD_RES(7)..(7)Lys or ArgMOD_RES(8)..(8)Gln or AsnMOD_RES(11)..(11)Asp or GluMOD_RES(12)..(12)Asn or GlnMOD_RES(15)..(16)Gln or AsnMOD_RES(17)..(17)Ala, Leu, Ile or Val 88Xaa Xaa Xaa Xaa Xaa Ala Xaa Xaa Ile Val Xaa Xaa Ala Ile Xaa Xaa1 5 10 15Xaa8944PRTArtificial SequenceDescription of Artificial Sequence Synthetic polypeptideMOD_RES(1)..(1)Ser or ThrMOD_RES(4)..(4)Gln or AsnMOD_RES(10)..(10)Thr or SerMOD_RES(18)..(18)Val, Ile, Leu or AlaMOD_RES(23)..(23)Gln or AsnMOD_RES(33)..(33)Val, Ile, Leu or AlaMOD_RES(34)..(34)Glu or AspMOD_RES(37)..(37)Thr or SerMOD_RES(38)..(38)Arg or LysMOD_RES(40)..(40)Arg or LysMOD_RES(42)..(42)Ala, Leu, Ile or ValMOD_RES(44)..(44)Ala, Leu, Ile or Val 89Xaa His Ile Xaa Ile Pro Pro Gly Leu Xaa Glu Leu Leu Gln Gly Tyr1 5 10 15Thr Xaa Glu Val Leu Arg Xaa Gln Pro Pro Asp Leu Val Glu Phe Ala 20 25 30Xaa Xaa Tyr Phe Xaa Xaa Leu Xaa Glu Xaa Arg Xaa 35 409024DNAArtificial SequenceDescription of Artificial Sequence Synthetic primer 90gacattcagc tgacccagtc tcca 249136DNAArtificial SequenceDescription of Artificial Sequence Synthetic primer 91gccggatcct cactggatgg tgggaagatg gataca 369222DNAArtificial SequenceDescription of Artificial Sequence Synthetic primer 92aggtsmarct gcagsagtcw gg 229318DNAArtificial SequenceDescription of Artificial Sequence Synthetic primer 93agctgggaag gtgtgcac 1894117PRTMus musculus 94Val Lys Leu Gln Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly Ser1 5 10 15Arg Lys Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Ser Arg Phe Gly 20 25 30Met His Trp Val Arg Gln Ala Pro Glu Lys Gly Leu Glu Trp Val Ala 35 40 45Tyr Ile Gly Arg Gly Ser Ser Thr Ile Tyr Tyr Ala Asp Thr Val Lys 50 55 60Gly Arg Phe Thr Ile Ser Arg Asp Asn Pro Lys Asn Thr Leu Phe Leu65 70 75 80Gln Met Thr Ser Leu Arg Ser Glu Asp Thr Ala Met Tyr Tyr Cys Ala 85 90 95Arg Ser Asn Trp Asp Gly Ala Met Asp Tyr Trp Gly Gln Gly Thr Ser 100 105 110Val Thr Val Ser Ser 11595120PRTMus musculus 95Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly Ser1 5 10 15Arg Gln Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Ser Ser Phe Gly 20 25 30Met His Trp Val Arg Gln Ala Pro Glu Lys Gly Leu Glu Trp Val Ala 35 40 45Tyr Ile Ser Arg Gly Gly Asn Thr Ile Tyr Tyr Ala Asn Thr Val Lys 50 55 60Gly Arg Phe Thr Ile Ser Arg Asp Asn Pro Lys Asn Thr Leu Phe Leu65 70 75 80Gln Met Thr Ser Leu Arg Ser Asp Asp Thr Ala Met Tyr Tyr Cys Ala 85 90 95Arg Ser His Tyr Tyr Gly Tyr Phe Tyr Ala Met Asp Tyr Trp Gly Gln 100 105 110Gly Thr Thr Leu Thr Val Ser Ser 115 12096123PRTMus musculus 96Asp Ile Gln Leu Thr Gln Ser Pro Ala Ser Leu Ser Ala Ser Val Gly1 5 10 15Glu Thr Val Thr Ile Thr Cys Arg Ala Ser Gly Asn Ile His Asn Phe 20 25 30Leu Ala Trp Tyr Gln Gln Lys Gln Gly Lys Ser Pro Gln Leu Leu Val 35 40 45Tyr Asn Ala Glu Thr Leu Ala Asp Gly Val Pro Ser Arg Phe Ser Gly 50 55 60Ser Gly Ser Gly Thr Gln Tyr Ser Leu Lys Ile Asn Ser Leu Gln Pro65 70 75 80Glu Asp Phe Gly Ser Tyr Tyr Cys Gln His Phe Trp Ser Thr Pro Trp 85 90 95Thr Phe Gly Gly Gly Thr Lys Leu Glu Ile Lys Arg Ala Asp Ala Ala 100 105 110Pro Thr Val Ser Ile Phe Pro Pro Ser Ser Glu 115 12097123PRTMus musculus 97Asp Ile Gln Met Thr Gln Ser Pro Ala Ser Leu Ser Ala Ser Val Gly1 5 10 15Glu Thr Val Thr Ile Thr Cys Arg Ala Ser Gly Asn Ile His Asn Tyr 20 25 30Leu Ala Trp Tyr Gln Gln Lys Gln Gly Lys Ser Pro Gln Leu Leu Val 35 40
45Tyr Asn Ala Lys Thr Leu Ala Asp Gly Val Pro Ser Arg Phe Ser Gly 50 55 60Ser Gly Ser Gly Thr Gln Tyr Ser Leu Lys Ile Asn Ser Leu Gln Pro65 70 75 80Glu Asp Phe Gly Thr Tyr Tyr Cys His His Phe Trp Ser Thr Pro Trp 85 90 95Thr Phe Gly Gly Gly Thr Lys Leu Glu Val Lys Arg Ala Asp Ala Ala 100 105 110Pro Thr Val Ser Ile Leu Pro Pro Ser Ser Glu 115 12098115PRTMus musculus 98Val Gln Leu Gln Glu Ser Gly Pro Ser Leu Val Lys Pro Ser Gln Thr1 5 10 15Leu Ser Leu Thr Cys Ser Val Thr Gly Asp Ser Ile Thr Ser Gly Phe 20 25 30Trp Asn Trp Ile Arg Lys Phe Pro Gly Asn Lys Phe Glu Tyr Met Gly 35 40 45Tyr Ile Ser Tyr Ser Gly Arg Thr Tyr Tyr Asn Pro Ser Leu Lys Ser 50 55 60Arg Leu Ser Ile Thr Arg Asp Thr Ser Lys Asn Gln Phe Tyr Leu Gln65 70 75 80Leu Asn Ser Val Thr Ala Glu Asp Thr Ala Thr Tyr Tyr Cys Ala Arg 85 90 95Asp Ala Asn Tyr Val Leu Asp Tyr Trp Gly Gln Gly Thr Thr Leu Thr 100 105 110Val Ser Ser 11599114PRTMus musculus 99Leu Gln Glu Ser Gly Pro Gly Leu Val Lys Pro Ser Gln Ser Leu Ser1 5 10 15Leu Thr Cys Ser Val Ser Gly Tyr Ser Ile Thr Ser Gly Tyr Phe Trp 20 25 30Asn Trp Ile Arg Gln Phe Ser Gly Asn Lys Leu Glu Trp Met Gly Tyr 35 40 45Ile Ser Tyr Asp Gly Ser Asn Asn Tyr Asn Pro Ser Leu Lys Asn Arg 50 55 60Ile Ser Ile Thr Arg Asp Thr Ser Lys Asn Gln Phe Phe Leu Lys Leu65 70 75 80Asn Ser Val Thr Pro Glu Asp Thr Ala Thr Tyr Tyr Cys Ala Arg Asp 85 90 95Gly Asp Tyr Tyr Phe Asp Tyr Trp Gly Gln Gly Thr Thr Val Thr Val 100 105 110Ser Ser100114PRTMus musculus 100Asp Ile Gln Leu Thr Gln Ser Pro Ser Ser Leu Ala Met Ser Val Gly1 5 10 15Gln Lys Val Thr Met Asn Cys Lys Ser Ser Gln Ser Leu Leu Asn Ser 20 25 30Ser Thr Gln Lys Asn Tyr Leu Ala Trp Phe Gln Gln Lys Pro Gly Gln 35 40 45Ser Pro Lys Leu Leu Val Tyr Phe Ala Ser Ala Arg Glu Ser Gly Val 50 55 60Pro Asp Arg Phe Ile Gly Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr65 70 75 80Ile Ser Ser Val Gln Ala Glu Asp Leu Ala Asp Tyr Phe Cys Gln Gln 85 90 95His Tyr Arg Thr Pro Phe Thr Phe Gly Ser Gly Thr Lys Leu Glu Ile 100 105 110Lys Arg101109PRTMus musculus 101Leu Thr Gln Ser Pro Ser Ser Leu Ala Met Ser Val Gly Gln Lys Val1 5 10 15Thr Met Asn Cys Lys Ser Ser Gln Ser Leu Leu Asn Ser Tyr Thr Gln 20 25 30Lys Asn Tyr Leu Ala Trp Tyr Gln Gln Lys Pro Gly Gln Ser Pro Lys 35 40 45Leu Leu Val Tyr Phe Ala Ser Thr Arg Glu Ser Gly Val Pro Asp Arg 50 55 60Phe Met Gly Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser65 70 75 80Val Gln Thr Glu Asp Leu Ala Asp Tyr Phe Cys Gln Gln His Tyr Arg 85 90 95Ile Pro Phe Thr Phe Gly Ser Gly Thr Lys Leu Glu Ile 100 105102648DNAArtificial SequenceDescription of Artificial Sequence Synthetic polynucleotide 102ctcgagcaca caggacctca ccatgggatg gagctgtatc atcctcttct tggtagcaac 60agctacaggt aaggggctca cagtagcagg cttgaggtct ggacatatat atgggtgaca 120atgacatcca ctttgccttt ctctccacag gtgtccactc cgtgcagctg caggagagcg 180gaccctccct ggtgaagcct agtcagaccc tgagcctgac atgctccgtg actggggact 240ctatcaccag tggcttctgg aactggattc ggaagttccc aggaaacaag tttgaataca 300tgggatatat ctcttacagt gggcgcacat actataaccc cagcctgaag tccaggctgt 360ctattacaag agacacttct aaaaaccagt tttatctgca gctgaacagc gtgactgccg 420aggatactgc tacctactat tgtgccaggg acgctaatta tgtgctggat tactggggcc 480agggaaccac actgaccgtg agctccggtg agtccttaca acctctctct tctattcagc 540ttaaatagat tttactgcat ttgttggggg ggaaatgtgt gtatctgaat ttcaggtcat 600gaaggactag ggacaccttg ggagtcagaa agggtcattg ggaagctt 648103537DNAArtificial SequenceDescription of Artificial Sequence Synthetic polynucleotide 103tctagacaca ggacctcacc atgggatgga gctgtatcat cctcttcttg gtagcaacag 60ctacaggtaa ggggctcaca gtagcaggct tgaggtctgg acatatatat gggtgacaat 120gacatccact ttgcctttct ctccacaggt gtccactccg acatccagct gacccagagc 180cccagctccc tggctatgtc cgtgggacag aaggtgacaa tgaactgcaa atctagtcag 240tctctgctga acagctccac tcagaagaat tacctggctt ggttccagca gaagcccggg 300cagagtccta aactgctggt gtattttgcc tctgctaggg agagtggcgt gccagacaga 360ttcatcggca gcggcagcgg gaccgatttt accctgacaa tttctagtgt gcaggccgag 420gacctggctg attacttctg tcagcagcac tatcggactc ccttcacctt tggctccgga 480acaaagctgg agatcaagcg tgagtagaat ttaaactttg cttcctcagt tggatcc 537104345DNAArtificial SequenceDescription of Artificial Sequence Synthetic polynucleotide 104gtgcagctgc aggagagcgg accctccctg gtgaagccta gtcagaccct gagcctgaca 60tgctccgtga ctggggactc tatcaccagt ggcttctgga actggattcg gaagttccca 120ggaaacaagt ttgaatacat gggatatatc tcttacagtg ggcgcacata ctataacccc 180agcctgaagt ccaggctgtc tattacaaga gacacttcta aaaaccagtt ttatctgcag 240ctgaacagcg tgactgccga ggatactgct acctactatt gtgccaggga cgctaattat 300gtgctggatt actggggcca gggaaccaca ctgaccgtga gctcc 34510563DNAArtificial SequenceDescription of Artificial Sequence Synthetic oligonucleotide 105tgtggccaga tcgagtacct ggccaagcag atcgtggaca acgccatcca gcaggccggg 60tgc 63106363DNAArtificial SequenceDescription of Artificial Sequence Synthetic polynucleotide 106gacatccagc tgacccagag ccccagctcc ctggctatgt ccgtgggaca gaaggtgaca 60atgaactgca aatctagtca gtctctgctg aacagctcca ctcagaagaa ttacctggct 120tggttccagc agaagcccgg gcagagtcct aaactgctgg tgtattttgc ctctgctagg 180gagagtggcg tgccagacag attcatcggc agcggcagcg ggaccgattt taccctgaca 240atttctagtg tgcaggccga ggacctggct gattacttct gtcagcagca ctatcggact 300cccttcacct ttggctccgg aacaaagctg gagatcaagc gtgagtagaa tttaaacttt 360gct 36310710PRTMus musculus 107Gly Phe Thr Phe Ser Arg Phe Gly Met His1 5 1010817PRTMus musculus 108Tyr Ile Gly Arg Gly Ser Ser Thr Ile Tyr Tyr Ala Asp Thr Val Lys1 5 10 15Gly1099PRTMus musculus 109Ser Asn Trp Asp Gly Ala Met Asp Tyr1 511011PRTMus musculus 110Arg Ala Ser Gly Asn Ile His Asn Phe Leu Ala1 5 101117PRTMus musculus 111Asn Ala Glu Thr Leu Ala Asp1 51129PRTMus musculus 112Gln His Phe Trp Ser Thr Pro Trp Thr1 51134PRTMus musculus 113Gly Phe Trp Asn111416PRTMus musculus 114Tyr Ile Ser Tyr Ser Gly Arg Thr Tyr Tyr Asn Pro Ser Leu Lys Ser1 5 10 151158PRTMus musculus 115Asp Ala Asn Tyr Val Leu Asp Tyr1 511617PRTMus musculus 116Lys Ser Ser Gln Ser Leu Leu Asn Ser Ser Thr Gln Lys Asn Tyr Leu1 5 10 15Ala1177PRTMus musculus 117Phe Ala Ser Ala Arg Glu Ser1 51189PRTMus musculus 118Gln Gln His Tyr Arg Thr Pro Phe Thr1 5
* * * * *
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
D00012
D00013

D00014

D00015
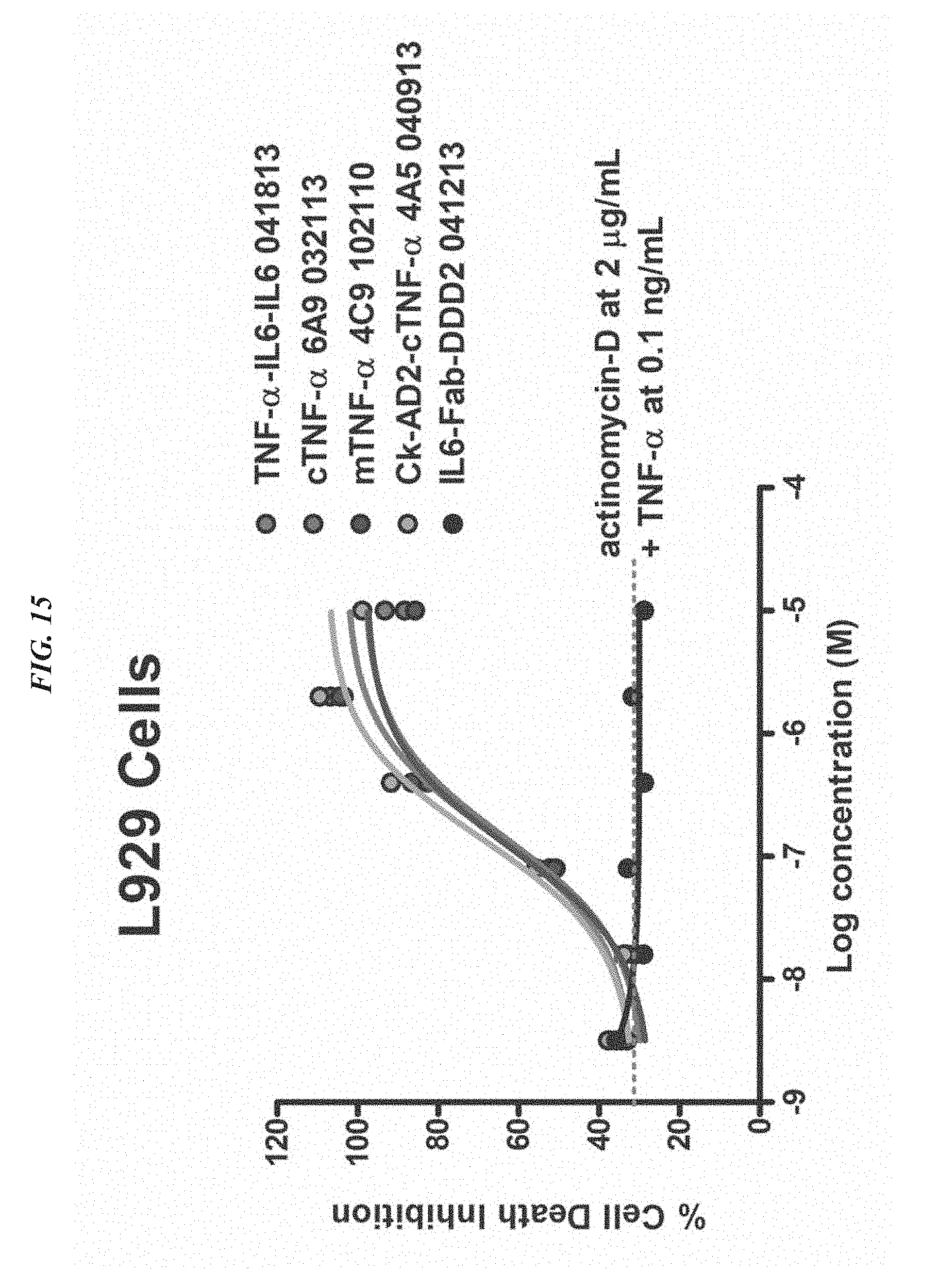
D00016

D00017

D00018

S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.