Inhibitors Of Cyclin-dependent Kinases
Gray; Nathanael S. ; et al.
U.S. patent application number 15/758982 was filed with the patent office on 2020-06-04 for inhibitors of cyclin-dependent kinases. This patent application is currently assigned to Dana-Farber Cancer Institute, Inc.. The applicant listed for this patent is Dana-Farber Cancer Institute, Inc.. Invention is credited to Nathanael S. Gray, Mingfeng Hao, Nicholas Paul Kwiatkowski, Tinghu Zhandg.
| Application Number | 20200172499 15/758982 |
| Document ID | / |
| Family ID | 58240329 |
| Filed Date | 2020-06-04 |









View All Diagrams
| United States Patent Application | 20200172499 |
| Kind Code | A9 |
| Gray; Nathanael S. ; et al. | June 4, 2020 |
INHIBITORS OF CYCLIN-DEPENDENT KINASES
Abstract
The present invention provides novel compounds of Formulae (I'), (I), (II'), and (II), and pharmaceutically acceptable salts, solvates, hydrates, polymorphs, co-crystals, tautomers, stereoisomers, isotopically labeled derivatives, prodrugs, and compositions thereof. Also provided are methods and kits involving the inventive compounds or compositions for treating and/or preventing proliferative diseases (e.g., cancers (e.g., leukemia, acute lymphoblastic leukemia, lymphoma, Burkitt's lymphoma, melanoma, multiple myeloma, breast cancer, Ewing's sarcoma, osteosarcoma, brain cancer, ovarian cancer, neuroblastoma, lung cancer, colorectal cancer), benign neoplasms, diseases associated with angiogenesis, inflammatory diseases, autoinflammatory diseases, and autoimmune diseases) in a subject. Treatment of a subject with a proliferative disease using a compound or composition of the invention may inhibit the aberrant activity of a kinase, such as a cyclin-dependent kinase (CDK) (e.g., CDK7, CDK12, or CDK13), and therefore, induce cellular apoptosis and/or inhibit transcription in the subject.
| Inventors: | Gray; Nathanael S.; (Boston,, MA) ; Zhandg; Tinghu; (Brookline, MA) ; Kwiatkowski; Nicholas Paul; (Brookline, MA) ; Hao; Mingfeng; (Boston, MA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Assignee: | Dana-Farber Cancer Institute,
Inc. Boston MA |
||||||||||
| Prior Publication: |
|
||||||||||
| Family ID: | 58240329 | ||||||||||
| Appl. No.: | 15/758982 | ||||||||||
| Filed: | September 9, 2016 | ||||||||||
| PCT Filed: | September 9, 2016 | ||||||||||
| PCT NO: | PCT/US16/51118 PCKC 00 | ||||||||||
| 371 Date: | March 9, 2018 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 62216271 | Sep 9, 2015 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61P 35/00 20180101; A61K 31/497 20130101; C07D 417/04 20130101; C07D 417/12 20130101; A61K 31/497 20130101; C07D 277/46 20130101; A61P 25/00 20180101; A61K 2300/00 20130101; C07D 417/14 20130101 |
| International Class: | C07D 277/46 20060101 C07D277/46; C07D 417/04 20060101 C07D417/04; C07D 417/14 20060101 C07D417/14; C07D 417/12 20060101 C07D417/12 |
Goverment Interests
GOVERNMENT SUPPORT
[0002] This invention was made with government support under grant number 1 R01 CA179483-01A1 awarded by the National Institutes of Health. The government has certain rights in the invention.
Claims
1. A compound of Formula (I'): ##STR00370## or a pharmaceutically acceptable salt thereof, wherein: R.sup.1 is hydrogen, halogen, or optionally substituted alkyl; M is O, S, or NR.sup.M; R.sup.M is hydrogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted acyl, or a nitrogen protecting group; Ring A is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl; Ring B is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl; Ring C is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted monocyclic or bicyclic aryl, or optionally substituted monocyclic or bicyclic heteroaryl; each instance of R.sup.A, R.sup.B, and R.sup.C is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --OR.sup.a, --N(R.sup.a).sub.2, --SR.sup.a, --CN, --SCN, --NO.sub.2, --N.sub.3, or optionally substituted acyl; each instance of R.sup.a is independently hydrogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted acyl, a nitrogen protecting group when attached to a nitrogen atom, an oxygen protecting group when attached to an oxygen atom, or a sulfur protecting group when attached to a sulfur atom, or two instances of R.sup.a are joined to form a substituted or unsubstituted, heterocyclic ring, or substituted or unsubstituted, heteroaryl ring; each of a1, b1, and c1 is independently 0, 1, 2, 3, 4, 5, or 6, as valency permits; L.sup.1 is --CH.sub.2--, .sup.lc--S(.dbd.O).sub.2--.sup.lb, --O--, --S--, --NR.sup.L1--, --C(.dbd.O)--, .sup.lc--NR.sup.L1C(.dbd.O)--.sup.lb, .sup.lc--C(.dbd.O)NR.sup.L1--.sup.lb, .sup.lc--OC(.dbd.O).sup.lb, or .sup.lc--C(.dbd.O)O--.sup.lb; wherein .sup.lb indicates the point of attachment is to Ring B; and .sup.lc indicates the point of attachment is to Ring C; L.sup.2 is --O--, --S--, --NR.sup.L2--, .sup.lb--NR.sup.L2C(.dbd.O)--.sup.lm, .sup.lb--C(.dbd.O)NR.sup.L2--.sup.lm; wherein .sup.lb indicates the point of attachment is to Ring B; and .sup.lm indicates the point of attachment is to the heteroaryl ring with M; X is .sup.xm--CH.sub.2CH.sub.2--.sup.xa, .sup.xm--CH.dbd.CH--.sup.xa, .sup.xm--CH.sub.2--NR.sup.LXxa, .sup.xm--CH.sub.2--O--CH.sub.2--.sup.xa, .sup.xm--CH.sub.2--NR.sup.LX--CH.sub.2--.sup.xa, --O--, --S--, --NR.sup.LX--, .sup.xm--O--CH.sub.2--.sup.xa, .sup.xm--S--CH.sub.2--.sup.xa, .sup.xm--S--C(.dbd.O)CH.sub.2--.sup.xa, or .sup.xm--NR.sup.LX--CH.sub.2--.sup.xa; wherein .sup.xa indicates the point of attachment is to Ring A; and .sup.xm indicates the point of attachment is to the heteroaryl ring with M; each of R.sup.L1, R.sup.L2, and R.sup.LX is independently hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group; R.sup.2 is any of Formulae (i-1)-(i-41): ##STR00371## ##STR00372## ##STR00373## ##STR00374## ##STR00375## ##STR00376## wherein: L.sup.3 is a bond or an optionally substituted C.sub.1-4 hydrocarbon chain, optionally wherein one or more carbon units of the hydrocarbon chain are independently replaced with --C.dbd.O--, --O--, --S--, --NR.sup.L3a--, --NR.sup.L3aC(.dbd.O)--, --C(.dbd.O)NR.sup.L3a--, --SC(.dbd.O)--, --C(.dbd.O)S--, --OC(.dbd.O)--, --C(.dbd.O)O--, --NR.sup.L3aC(.dbd.S)--, --C(.dbd.S)NR.sup.L3a--, trans-CR.sup.L3b.dbd.CR.sup.L3b--, cis-CR.sup.L3b.dbd.CR.sup.L3b--, --C.ident.C--, --S(.dbd.O)--, --S(.dbd.O)O--, --OS(.dbd.O)--, --S(.dbd.O)NR.sup.L3a--, --NR.sup.L3aS(.dbd.O)--, --S(.dbd.O).sub.2--, --S(.dbd.O).sub.2O--, --OS(.dbd.O).sub.2--, --S(.dbd.O).sub.2NR.sup.L3a--, or --NR.sup.L3aS(.dbd.O).sub.2--, wherein R.sup.L3a is hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group, and wherein each occurrence of R.sup.L3b is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl, or two R.sup.L3b groups are joined to form an optionally substituted carbocyclic or optionally substituted heterocyclic ring; L.sup.4 is a bond or an optionally substituted, branched or unbranched C.sub.1-6 hydrocarbon chain; each of R.sup.E1, R.sup.E2, and R.sup.E3 is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --CN, --CH.sub.2OR.sup.EE, --CH.sub.2N(R.sup.EE).sub.2, --CH.sub.2SR.sup.EE, --OR.sup.EE, --N(R.sup.EE).sub.2, --Si(R.sup.EE).sub.3, and --SR.sup.EE, wherein each occurrence of R.sup.EE is independently hydrogen, optionally substituted alkyl, optionally substituted alkoxy, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl, or two R.sup.EE groups are joined to form an optionally substituted heterocyclic ring; or R.sup.E1 and R.sup.E3, or R.sup.E2 and R.sup.E3, or R.sup.E1 and R.sup.E2 are joined to form an optionally substituted carbocyclic or optionally substituted heterocyclic ring; R.sup.E4 is a leaving group; R.sup.E5 is halogen; R.sup.E6 is hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group; each instance of Y is independently O, S, or NR.sup.E7, wherein R.sup.E7 is hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group; a is 1 or 2; and each instance of z is independently 0, 1, 2, 3, 4, 5, or 6, as valency permits.
2. (canceled)
3. The compound of claim 1, wherein the compound is of the formula: ##STR00377## or a pharmaceutically acceptable salt thereof.
4. The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein the Ring B is optionally substituted monocyclic heterocyclyl.
5. The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein the Ring B is optionally substituted piperidinyl.
6. The compound of claim 5, wherein the compound is of Formula (I-i), (I-ii), (I-ii-a), (I-ii-b), (I-ii-c), (I-iii), or (I'-i): ##STR00378## ##STR00379## or a pharmaceutically acceptable salt thereof.
7-11. (canceled)
12. The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein Ring B is optionally substituted monocyclic carbocyclyl.
13. (canceled)
14. The compound of claim 1, wherein the compound is of Formula (I-iv), (I-iv-a), or (I-iv-b): ##STR00380## or a pharmaceutically acceptable salt thereof.
15-17. (canceled)
18. The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein Ring C is optionally substituted.bicyclic aryl or heteroaryl.
19-20. (canceled)
21. A compound of Formula (II'): ##STR00381## or a pharmaceutically acceptable salt thereof, wherein: R.sup.1 is hydrogen, halogen, or optionally substituted alkyl; M is O, S, or NR.sup.M; R.sup.M is hydrogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted acyl, or a nitrogen protecting group; Ring A is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl; Ring B is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl; each instance of R.sup.A and R.sup.B is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --OR.sup.a, --N(R.sup.a).sub.2, --SR.sup.a, --CN, --SCN, --C(.dbd.NR.sup.a)R.sup.a, --C(.dbd.NR.sup.a)OR.sup.a, --C(.dbd.NR.sup.a)N(R.sup.a).sub.2, --C(.dbd.O)R.sup.a, --C(.dbd.O)OR.sup.a, --C(.dbd.O)N(R.sup.a).sub.2, --NO.sub.2, --NR.sup.aC(.dbd.O)R.sup.a, --NR.sup.aC(.dbd.O)OR.sup.a, --NR.sup.aC(.dbd.O)N(R.sup.a).sub.2, --OC(.dbd.O)R.sup.a, --OC(.dbd.O)OR.sup.a, or --OC(.dbd.O)N(R.sup.a).sub.2; each instance of R.sup.a is independently hydrogen, optionally substituted acyl, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, a nitrogen protecting group when attached to a nitrogen atom, an oxygen protecting group when attached to an oxygen atom, or a sulfur protecting group when attached to a sulfur atom, or two instances of R.sup.a are joined to form a substituted or unsubstituted, heterocyclic ring, or substituted or unsubstituted, heteroaryl ring; each of a1 and b1 is independently 0, 1, 2, 3, 4, 5, or 6, as valency permits; L.sup.2 is --O--, --S--, --NR.sup.L2--, .sup.lb--NR.sup.L2C(.dbd.O)--.sup.lm, .sup.lb--C(.dbd.O)NR.sup.L2--.sup.lm; wherein .sup.lb indicates the point of attachment is to Ring B; and .sup.lm indicates the point of attachment is to the heteroaryl ring with M; X is a bond, --O--, --S--, --NR.sup.LX--, .sup.xm--O--CH.sub.2--.sup.xa, .sup.xm--S--CH.sub.2--.sup.xa, or .sup.xm--NR.sup.LX--CH.sub.2--.sup.xa; wherein .sup.xa indicates the point of attachment is to Ring A; and .sup.xm indicates the point of attachment is to the heteroaryl ring with M; each of R.sup.L2 and R.sup.LX is independently hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group; R.sup.2 is any of Formulae (i-1)-(i-41): ##STR00382## ##STR00383## ##STR00384## ##STR00385## ##STR00386## ##STR00387## wherein: L.sup.3 is a bond or an optionally substituted C.sub.1-4 hydrocarbon chain, optionally wherein one or more carbon units of the hydrocarbon chain are independently replaced with --C.dbd.O--, --O--, --S--, --NR.sup.L3a--, --NR.sup.L3aC(.dbd.O)--, --C(.dbd.O)NR.sup.L3a--, --SC(.dbd.O)--, --C(.dbd.O)S--, --OC(.dbd.O)--, --C(.dbd.O)O--, --NR.sup.L3aC(.dbd.S)--, --C(.dbd.S)NR.sup.L3a--, trans-CR.sup.L3b.dbd.CR.sup.L3b--, cis-CR.sup.L3b.dbd.CR.sup.L3b--, --C.ident.C--, --S(.dbd.O)--, --S(.dbd.O)O--, --OS(--O)--, --S(--O)NR.sup.L3a--, --NR.sup.L3aS(.dbd.O)--, --S(.dbd.O).sub.2--, --S(.dbd.O).sub.2O--, --OS(.dbd.O).sub.2--, --S(.dbd.O).sub.2NR.sup.L3a--, or --NR.sup.L3aS(.dbd.O).sub.2--, wherein R.sup.L3a is hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group, and wherein each occurrence of R.sup.L3b is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl, or two R.sup.L3b groups are joined to form an optionally substituted carbocyclic or optionally substituted heterocyclic ring; L.sup.4 is a bond or an optionally substituted, branched or unbranched C.sub.1-6 hydrocarbon chain; each of R.sup.E1, R.sup.E2, and R.sup.E3 is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --CN, --CH.sub.2OR.sup.EE, --CH.sub.2N(R.sup.EE).sub.2, --CH.sub.2SR.sup.EE, --OR.sup.EE, --N(R.sup.EE).sub.2, --Si(R.sup.EE).sub.3, and --SR.sup.EE, wherein each occurrence of R.sup.EE is independently hydrogen, optionally substituted alkyl, optionally substituted alkoxy, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl, or two R.sup.EE groups are joined to form an optionally substituted heterocyclic ring; or R.sup.E1 and R.sup.E3, or R.sup.E2 and R.sup.E3, or R.sup.E1 and R.sup.E2 are joined to form an optionally substituted carbocyclic or optionally substituted heterocyclic ring; R.sup.E4 is a leaving group; R.sup.E5 is halogen; R.sup.E6 is hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group; each instance of Y is independently O, S, or NR.sup.E7, wherein R.sup.E7 is hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group; a is 1 or 2; and each instance of z is independently 0, 1, 2, 3, 4, 5, or 6, as valency permits.
22-30. (canceled)
31. The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein Ring A is optionally substituted monocyclic heteroaryl.
32-39. (canceled)
40. The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein Ring A is optionally substituted phenyl.
41-46. (canceled)
47. The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein Ring A is optionally substituted monocyclic carbocyclyl.
48. (canceled)
49. The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein Ring A is optionally substituted monocyclic heterocyclyl.
50-54. (canceled)
55. The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein Ring A is optionally substituted 6-membered heteroaryl.
56. (canceled)
57. The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein L.sup.1 is --CH.sub.2-- or .sup.lc--S(.dbd.O).sub.2--.sup.lb.
58-82. (canceled)
83. The compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein R.sup.2 is of Formula (i-1): ##STR00388##
84-90. (canceled)
91. The compound of claim 1, wherein the compound is of one of the following formulae: ##STR00389## ##STR00390## ##STR00391## ##STR00392## ##STR00393## or a pharmaceutically acceptable salt thereof.
92. The compound of claim 1, wherein the compound is of one of the following formulae: ##STR00394## ##STR00395## ##STR00396## ##STR00397## ##STR00398## ##STR00399## ##STR00400## ##STR00401## ##STR00402## ##STR00403## ##STR00404## ##STR00405## ##STR00406## ##STR00407## ##STR00408## ##STR00409## ##STR00410## or a pharmaceutically acceptable salt thereof.
93. The compound of claim 1, wherein the compound is of one of the following formulae: ##STR00411## ##STR00412## ##STR00413## ##STR00414## ##STR00415## ##STR00416## ##STR00417## or a pharmaceutically acceptable salt thereof.
94-95. (canceled)
96. A pharmaceutical composition comprising a compound of claim 1, or a pharmaceutically acceptable salt thereof, and optionally a pharmaceutically acceptable excipient.
97. (canceled)
98. A method of treating a proliferative disease in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of a compound of claim 1, or a pharmaceutically acceptable salt thereof.
99-131. (canceled)
132. A method of inhibiting the activity of a cyclin-dependent kinase (CDK) in a biological sample or subject, the method comprising administering to the subject or contacting the biological sample with a therapeutically effective amount of a compound of claim 1, or a pharmaceutically acceptable salt thereof.
133-153. (canceled)
Description
RELATED APPLICATIONS
[0001] This application claims priority under 35 U.S.C. .sctn. 119(e) to U.S. Provisional application, U.S. Ser. No. 62/216,271, filed Sep. 9, 2015, which is incorporated herein by reference.
BACKGROUND OF THE INVENTION
[0003] The members of the cyclin-dependent kinase (CDK) family play critical regulatory roles in cell proliferation. There are currently twenty known mammalian CDKs. While CDK7 to CDK13 have been linked to transcription, CDK1, 2, 4, and 6 show demonstrable association with the cell cycle.
[0004] Unique among the mammalian CDKs, CDK7 has consolidated kinase activities, regulating both the cell cycle and transcription. In the cytosol, CDK7 exists as a heterotrimeric complex and is believed to function as a CDK1/2-activating kinase (CAK), whereby phosphorylation of conserved residues in CDK1/2 by CDK7 is required for full catalytic CDK activity and cell cycle progression (Desai et al., "Effects of phosphorylation by CAK on cyclin binding by CDC2 and CDK2." Mol. Cell Biol. 15, 345-350 (1995); Kaldis et al., "Analysis of CAK activities from human cells." Eur. J. Biochem. 267, 4213-4221 (2000); Larochelle et al., "Requirements for CDK7 in the assembly of CDK1/cyclin B and activation of CDK2 revealed by chemical genetics in human cells." Mol. Cell 25, 839-850 (2007)). In the nucleus, CDK7 forms the kinase core of the RNA polymerase (RNAP) II general transcription factor complex and is charged with phosphorylating the C-terminal domain (CTD) of RNAP II, a requisite step in gene transcriptional initiation (Serizawa. et al., "Association of CDK-activating kinase subunits with transcription factor TFIIH." Nature 374, 280-282 (1995); Shiekhattar et al., "CDK-activating kinase complex is a component of human transcription factor TFIIH." Nature 374, 283-287 (1995); Drapkin et al., "Human cyclin-dependent kinase-activating kinase exists in three distinct complexes." Proc. Natl. Acad. Sci. U.S.A. 93, 6488-6493 (1996); Liu. et al., "Two cyclin-dependent kinases promote RNA polymerase II transcription and formation of the scaffold complex." Mol. Cell Biol. 24, 1721-1735 (2004); Akhtar et al., "TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II." Mol. Cell 34, 387-393 (2009); Glover-Cutter et al., "TFIIH-associated CDK7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II." Mol. Cell Biol. 29, 5455-5464 (2009)). Together, the two functions of CDK7, i.e., CAK and CTD phosphorylation, support critical facets of cellular proliferation, cell cycling, and transcription.
[0005] CDK12 and CDK13 were identified in cDNA screens for cell cycle regulators. Because their cyclin partners were not yet known, they were initially named CRKRS and CDC2L5 (Ko et al., J. Cell Sci., 2001, 114, 2591-2603; Marques et al., Biochem Biophys Res Commun., 2000, 279(3):832-837), respectively. They were found to be 1490- and 1512-amino acid proteins, respectively, with a conserved central CTD kinase domain and degenerate RS domains identified in their N- and C-terminal regions (Even et al., J Cell Biochem., 2006, 99(3), 890-904).
[0006] Evidence has shown CDK12 and CDK13 play an important role in cancer development. A comprehensive genomic approach identified CDK12 to be one of the most frequently somatically mutated genes in high-grade serous ovarian cancer, the most fatal form of the disease (Erratum, Nature, 2011, 474(7353), 609-615). Several identified point mutations in the kinase domain point to the critical importance of the kinase activity of CDK12 for the development/progression of this disease. CDK12 has also been found to contribute to the development of breast cancer. Notably, CDK12 is located on chromosome 17, within the 17q21 locus that contains several candidate genes for breast cancer susceptibility (Kauraniemi et al., Cancer Res., 2001, 61(22), 8235-8240), and it is co-amplified with the tyrosine kinase receptor ERBB2, a protein amplified and overexpressed in about 20% of breast tumors. Gene fusion between CDK12 and ERBB2 was also detected in gastric cancer (Zang et al., Cancer Res., 2011, 71(1), 29-39). CDK12 is also implicated in the modification of tamoxifen sensitivity in estrogen-positive breast cancer via the modulation of the mitogen-activated protein kinase pathway (Iorns et al., Carcinogenesis, 2009, 30(10):1696-1701).
[0007] Due to the important regulatory functions of kinases, such as CDK7, CDK12, and CDK13, in cell cycle control, cell proliferation, differentiation, and apoptosis, it is important to develop modulators of the activities of these kinases, including selective modulators, for use as research tools as well as therapeutic agents in the treatment of diseases.
SUMMARY OF THE INVENTION
[0008] Cyclin dependent kinases (CDKs), e.g., CDK7, CDK12, and CDK13, are key regulators of the cell cycle. Their successive activation and inactivation drives the cycle forward. The activity of CDKs is regulated by multiple mechanisms such as positive and negative phosphorylation, binding of regulatory proteins like cyclins, and CDK inhibitors. Most CDKs require the phosphorylation of a threonine residue located in the T-loop to achieve full kinase activity. This threonine residue is conserved in all CDKs that function in cell cycle regulation. The enzyme responsible for this phosphorylation is therefore termed CDK-activating-kinase or CAK. CAK complexes have been found to be composed of CDK7, CDK12, CDK13, cyclin H, and MAT1. Besides its CAK function, CDK7, CDK12, and CDK13 also play a role in transcription and possibly in DNA repair. This suggests that the CDK7, CDK12, and CDK13 enzyme complexes are involved in multiple functions in the cell, e.g., cell cycle control, apoptosis, transcription regulation, and DNA repair.
[0009] The present invention provides compounds of Formulae (I'), (II'), (I), (II), and pharmaceutically acceptable salts, solvates, hydrates, polymorphs, co-crystals, tautomers, stereoisomers, isotopically labeled derivatives, prodrugs, and compositions thereof. The compounds of Formulae (I'), (II'), (I), (II), and pharmaceutically acceptable salts, solvates, hydrates, polymorphs, co-crystals, tautomers, stereoisomers, isotopically labeled derivatives, prodrugs, and compositions thereof, may inhibit the activity of a kinase. The compounds described herein may in certain embodiments selectively inhibit specific CDK subtypes, for example, CDK7, CDK12, or CDK13. In certain embodiments, the compounds of Formulae (I'), (II'), (I), and (II) are selective for CDK7 compared to other kinases. In certain embodiments, the compounds of Formulae (I'), (II'), (I), and (II) are selective for CDK12 and/or CDK13 compared to other kinases. The present invention also provides methods of using the inventive compounds, and pharmaceutically acceptable salts, solvates, hydrates, polymorphs, co-crystals, tautomers, stereoisomers, isotopically labeled derivatives, prodrugs, and compositions thereof, to study the inhibition of a kinase (e.g., CDK7, CDK12, and/or CDK13) and as therapeutics for the prevention and/or treatment of diseases associated with the overexpression and/or aberrant activity of a kinase (e.g., CDK7, CDK12, and/or CDK13). In certain embodiments, the inventive compounds are used for the prevention and/or treatment of proliferative diseases (e.g., cancers (e.g., leukemia, acute lymphoblastic leukemia, lymphoma, Burkitt's lymphoma, melanoma, multiple myeloma, breast cancer, Ewing's sarcoma, osteosarcoma, brain cancer, neuroblastoma, lung cancer, colorectal cancer), benign neoplasms, diseases associated with angiogenesis, inflammatory diseases, autoinflammatory diseases, and autoimmune diseases) in a subject.
[0010] Since the discovery of selective inhibitors of CDK7, CDK12, and CDK13 has been hampered by the high sequence and structural similarities of the kinase domain of CDK family members, the development of selective inhibitors of the transcriptional cyclin-dependent kinases (tCDKs) will allow dissection of their individual contributions to the regulation of transcription and evaluation of their therapeutic potential. Without wishing to be bound by any particular theory, the inventive compounds' selectivity for CDK7, CDK12, and/or CDK13 may be due to the compounds' ability to covalently modify a specific cysteine residue of these kinases (e.g., Cys312 of CDK7, Cys1039 of CDK12, Cys1017 of CDK13).
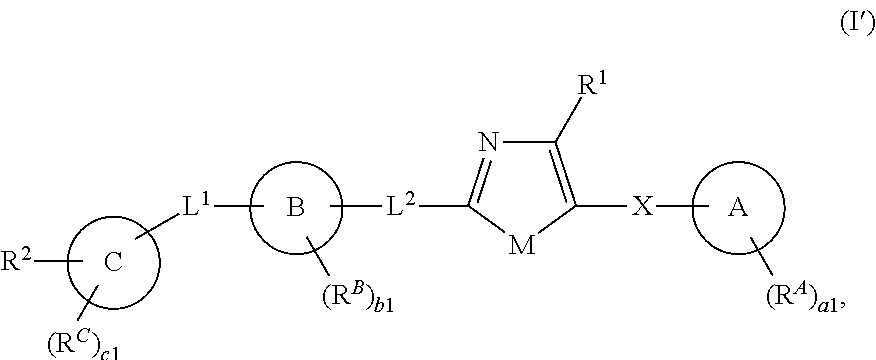
[0011] In one aspect, the present invention provides compounds of Formula (I'):
##STR00001##
and pharmaceutically acceptable salts, solvates, hydrates, polymorphs, co-crystals, tautomers, stereoisomers, isotopically labeled derivatives, and prodrugs thereof, wherein R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, Ring A, Ring B, Ring C, L.sup.1, L.sup.2, X, a1, b1, and c1 are as defined herein.
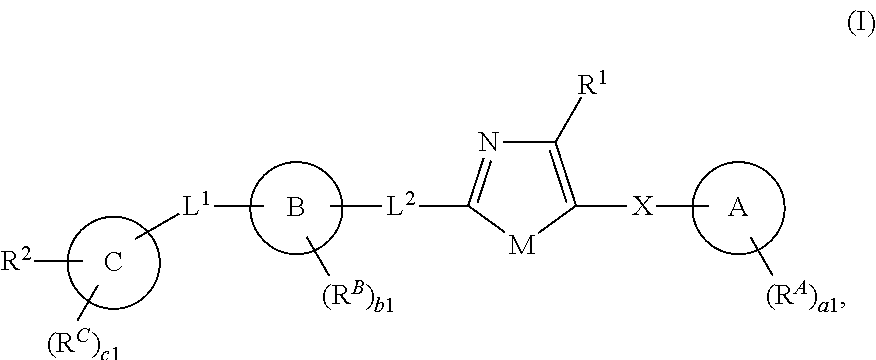
[0012] In one aspect, the present invention provides compounds of Formula (I):
##STR00002##
and pharmaceutically acceptable salts, solvates, hydrates, polymorphs, co-crystals, tautomers, stereoisomers, isotopically labeled derivatives, and prodrugs thereof, wherein R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, Ring A, Ring B, Ring C, L.sup.1, L.sup.2, X, a1, b1, and c1 are as defined herein.
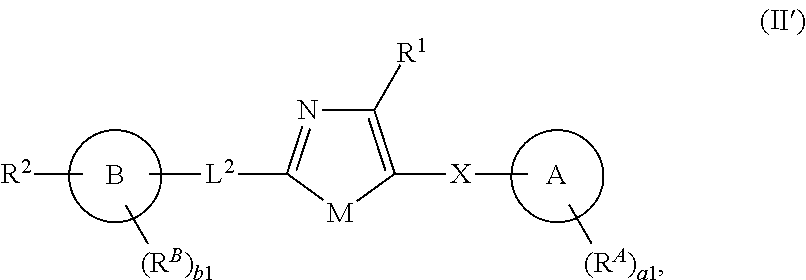
[0013] In one aspect, the present invention provides compounds of Formula (II'):
##STR00003##
and pharmaceutically acceptable salts, solvates, hydrates, polymorphs, co-crystals, tautomers, stereoisomers, isotopically labeled derivatives, and prodrugs thereof, wherein R.sup.1, R.sup.2, R.sup.A, R.sup.B, Ring A, Ring B, L.sup.2, X, a1, and b1 are as defined herein.
[0014] In one aspect, the present invention provides compounds of Formula (II):
##STR00004##
and pharmaceutically acceptable salts, solvates, hydrates, polymorphs, co-crystals, tautomers, stereoisomers, isotopically labeled derivatives, and prodrugs thereof, wherein R.sup.1, R.sup.2, R.sup.A, R.sup.B, Ring A, Ring B, L.sup.2, X, a1, and b1 are as defined herein.
[0015] In another aspect, the present disclosure provides pharmaceutical compositions including a compound described herein, and optionally a pharmaceutically acceptable excipient. In certain embodiments, the pharmaceutical compositions described herein include a therapeutically or prophylactically effective amount of a compound described herein. The pharmaceutical composition may be useful for treating a proliferative disease in a subject in need thereof, preventing a proliferative disease in a subject in need thereof, inhibiting the activity of a protein kinase in a subject, biological sample, tissue, or cell, and/or inducing apoptosis in a cell. In certain embodiments, the proliferative disease is an inflammatory disease. In certain embodiments, the inflammatory disease is rheumatoid arthritis, Crohn's disease, or fibrosis.
[0016] In another aspect, the present invention provides methods for treating and/or preventing a proliferative disease. Exemplary proliferative diseases which may be treated include cancer, benign neoplasms, diseases associated with angiogenesis, inflammatory diseases, autoinflammatory diseases, and autoimmune diseases. In certain embodiments, the cancer is selected from the group consisting of pancreatic cancer, lung cancer (e.g., small cell lung cancer (SCLC), and non-small cell lung cancer), prostate cancer, breast cancer, ovarian cancer, kidney cancer, liver cancer, Ewing's sarcoma, osteosarcoma, brain cancer, neuroblastoma, and colorectal cancer.
[0017] Another aspect of the invention relates to methods of inhibiting the activity of a kinase (e.g., CDK (e.g., CDK7, CDK12, CDK13)) using a compound described herein in a biological sample or subject. In certain embodiments, the method involves the selective inhibition of CDK7. In certain embodiments, the method involves the selective inhibition of CDK12. In certain embodiments, the method involves the selective inhibition of CDK13.
[0018] Also provided by the present invention are methods of inhibiting the transcription of one or more genes in the cell of a biological sample or subject using a compound described herein. The transcription of genes affected by the activity of CDK7 may be inhibited by a compound of the invention. In certain embodiments, these genes are one or more selected from the group consisting of MYC, RUNX1, MYB, TAL1, GATA3, KLF2, HNRPDL, p21, ASCL1, MYCN, INSM1, NEUROD1, NEUROG1, FOXG1, FOXA1, SOX2, SOX4, BCL11A, OTX2, GAT2, PHOX2B, PLK2, TAF1, CTGF, WEE1, SDIM, JUN, PIM1, IL8, and FOS1. The transcription of genes affected by the activity of CDK12 may be inhibited by a compound of the invention. In certain embodiments, these genes are one or more selected from the group consisting of BRCA1, FANCI, ATR, FANCD2, APEX1, NEK9, CHEK1, CHEK2, ATM, RAD51C, RAD51D, ORC3L, MDC1, TERF2, ERCC4, FANCF, PARP9, RUNX1, MYB, TAL1, MCL1, MYC, BCL2, ETS1, and EWS-FLI. The transcription of genes affected by the activity of CDK13 may be inhibited by a compound of the invention. In certain embodiments, the gene is SNORA38.
[0019] The present invention also provides methods of inhibiting cell growth in a biological sample or subject. In still another aspect, the present invention provides methods of inducing apoptosis of a cell in a biological sample or subject.
[0020] The present invention provides methods for administering to a subject in need thereof an effective amount of a compound, or pharmaceutical composition thereof, as described herein. Also described are methods for contacting a cell with an effective amount of a compound, or pharmaceutical composition thereof, as described herein. In certain embodiments, a method described herein further includes administering to the subject an additional pharmaceutical agent. In certain embodiments, a method described herein further includes contacting the cell with an additional pharmaceutical agent. The methods described herein may further include performing radiotherapy, immunotherapy, and/or transplantation on the subject.
[0021] In yet another aspect, the present invention provides compounds of Formulae (I'), (II'), (I), (II), and pharmaceutically acceptable salts, solvates, hydrates, polymorphs, co-crystals, tautomers, stereoisomers, isotopically labeled derivatives, prodrugs, and compositions thereof, for use in the treatment of a disease (e.g., a proliferative disease such as cancer) in a subject.
[0022] Another aspect of the present disclosure relates to kits comprising a container with a compound, or pharmaceutical composition thereof, as described herein. The kits described herein may include a single dose or multiple doses of the compound or pharmaceutical composition. The kits may be useful in a method of the disclosure. In certain embodiments, the kit further includes instructions for using the compound or pharmaceutical composition. A kit described herein may also include information (e.g. prescribing information) as required by a regulatory agency such as the U.S. Food and Drug Administration (FDA).
[0023] The details of one or more embodiments of the invention are set forth herein. Other features, objects, and advantages of the invention will be apparent from the Detailed Description, Examples, Figures, and Claims.
BRIEF DESCRIPTION OF THE DRAWINGS
[0024] The accompanying drawing, which are incorporated in and constitute a part of this specification, illustrate several embodiments of the invention and together with the description, serve to explain the principles of the invention.
[0025] FIG. 1 shows the chemical structures of exemplary compounds described herein and the IC.sub.50 values of the exemplary compounds in inhibiting select cyclin-dependent kinases.
[0026] FIG. 2A shows that compound B12 exhibits binding of intracellular CDK12 and CDK13 at concentrations between 62.5 nM to 1 .mu.M treatment. Jurkat cells treated with compound B12 for 6 hours show decreased pulldown of CDK12 and CDK13-associated cyclin K by biotin-THZ1 relative to DMSO-treated cells. These data indicate that B12 cellular treatment blocks biotin-THZ1 from binding CDK12 and CDK13-associated cyclin K complexes by successful binding of these complexes in cells at these concentrations. Cyclin H (Cyc H) pulldown was not affected, indicating that CDK7 binding is not affected. FIG. 2B shows the structure of compound B12.
[0027] FIG. 3 shows the binding of compounds B12, B15, and B16 (relative to DMSO control) to CDK12 and CDK13-associated cyclin K at concentrations of 100 nM, 50 nM, 25 nM, and 12.5 nM. Jurkat cells were treated with each compound (or DMSO) for 6 hours, followed by lysis and pulldown with biotin-THZ1, and subsequent western blotting for cyclin K (Cyc K) and cyclin H (Cyc H). Compound B12 exhibits binding to CDK12 and CDK13-associated cyclin K, while compounds B15 and B16 do not. Compound B12 is able to block pulldown of CDK12 and CDK13-associated cyclin K down to 25 nM treatment, while compounds B15 and B16 show very little effect on cyclin K pulldown. These results indicate that at concentrations as low as 25 nM B12 successfully targets intracellular CDK12 and CDK13-associated cyclin K complexes.
[0028] FIG. 4 shows binding of intracellular CDK12 and CDK13-associated cyclin K complexes by exemplified compounds. Jurkat cells were treated with each compound at a concentration of 500 nM for 6 hours, followed by lysing and pulldown with biotin-THZ1, and subsequent western blotting for cyclin K (Cyc K) and cyclin H (Cyc H). Compounds B1, B3, B4, B5, B6, B8, B9, and B12 show a loss in CDK12 and CDK13-associated cyclin K pulldown, indicating that these compounds successfully bind CDK12 and CDK13-associated cyclin K complexes in cells thus blocking biotin-THZ1 pull down, while compounds B5, B8, and B9 show a more pronounced loss of cyclin H pulldown, indicating that these compounds successfully target CDK7-cyclin H complexes in cells and interfere with biotin-THZ1 pull down.
[0029] FIG. 5 shows exemplary results of growth assays of select compounds described herein. Jurkat cells were plated at 30,000 cells/well and treated with a titration of compounds indicated. Cells were allowed to grow for 72 hours. Cells were assayed using CELLTITER GLO (Promega) to determine cell viability by measuring the amount of ATP present, which is an indicator of cell metabolic activity. Top panel: luminescent values (y-axis); concentration in .mu.M (uM) (x-axis); the curves are generated using PRISM. Bottom panel: IC.sub.50 values in .mu.M. Error bars indicate +/- standard deviation. All compounds tested showed anti-proliferative effects to varying extents.
[0030] FIG. 6 shows exemplary mass spectrum labeling of CDK12 with compound B12. Compound B12 is able to label CDK12 once treated with a 5-fold excess of compound B12 for 1 hour at 4.degree. C.
[0031] FIG. 7 shows the binding of intracellular CDK12 and CDK13-associated cyclin K complexes by exemplified compounds. Jurkat cells were treated with each compound at a concentration of 500 nM for 6 hours, followed by lysing and pull down with biotin-THZ1, and subsequent western blotting for cyclin K (Cyc K) and cyclin H (Cyc H). Compounds that decrease the pull down efficiency of CDK12 and CDK13-associated cyclin K complexes indicate those compounds successfully targeted intracellular CDK12 and CDK13-associated cyclin K complexes and therefore were able to block biotin-THZ1 pull down of these complexes. In FIG. 7A, compounds MFH 2-90-1 and MFH 2-102-1, show a loss in cyclin K pulldown by biotin-THZ1, indicating that these compounds were able to bind intracellular CDK12 and CDK13-associated cyclin K complexes and block pull down by biotin-THZ1. In FIG. 7B, compounds MFH 2-90-1, MFH 3-103-1, and MFH 3-151-1, show a loss in cyclin K pulldown by biotin-THZ1, indicating that these compounds were able to bind intracellular CDK12 and CDK13-associated cyclin K complexes and thus block pull down by biotin-THZ1.
[0032] FIG. 8 shows the binding of intracellular CDK12 and CDK13-associated cyclin K complexes by exemplified compounds. Jurkat cells were treated with each compound at a concentration of 500 nM for 6 hours, followed by lysing and pull down with biotin-THZ1, and subsequent western blotting for cyclin K (Cyc K) and cyclin H (Cyc H). Compounds that decrease the pull down efficiency of CDK12 and CDK13-associated cyclin K complexes indicate those compounds successfully targeted intracellular CDK12 and CDK13-associated cyclin K complexes and therefore were able to block biotin-THZ1 pull down of these complexes. In FIG. 8A, compound THZ 5-31 and MFH 2-90-1 show a loss in cyclin K pulldown by biotin-THZ1, indicating that these compounds successfully targeted CDK12 and CDK13-associated complexes in cells and block biotin-THZ1 binding. In FIG. 8B, compounds THZ 5-31, MFH 2-90-1, THZ-CE B-15, and THZ-CE B-16, show a loss in cyclin K pull down by biotin-THZ1, indicating that these compounds successfully targeted CDK12 and CDK13-associated complexes in cells and block biotin-THZ1 binding.
[0033] FIG. 9 shows the binding of intracellular CDK12 and CDK13-associated cyclin K complexes by exemplified compounds. Jurkat cells were treated with each compound at a concentration of 500 nM for 4 hours, followed by lysing and pull down with biotin-THZ1, and subsequent western blotting for cyclin K (Cyc K) and cyclin H (Cyc H). Compounds that decrease the pull down efficiency of CDK12 and CDK13-associated cyclin K complexes indicate those compounds successfully targeted intracellular CDK12 and CDK13-associated cyclin K complexes and therefore were able to block biotin-THZ1 pull down of these complexes. In FIG. 9A, compounds THZ 5-31, MFH 2-90-1, MFH 3-75-1, and MFH 3-81-1 show a loss in cyclin K pull down by biotin-THZ1, indicating that these compounds successfully targeted CDK12 and CDK13-associated complexes in cells and block biotin-THZ1 binding. In FIG. 9B, compounds THZ 5-31, MFH 4-70-1, and MFH 4-70-1 show a loss in cyclin K pull down, by biotin-THZ1, indicating that these compounds successfully targeted CDK12 and CDK13-associated complexes in cells and block biotin-THZ1 binding.
DEFINITIONS
[0034] Definitions of specific functional groups and chemical terms are described in more detail below. The chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 75.sup.th Ed., inside cover, and specific functional groups are generally defined as described therein.
[0035] Additionally, general principles of organic chemistry, as well as specific functional moieties and reactivity, are described in Thomas Sorrell, Organic Chemistry, University Science Books, Sausalito, 1999; Smith and March, March's Advanced Organic Chemistry, 5.sup.th Edition, John Wiley & Sons, Inc., New York, 2001; Larock, Comprehensive Organic Transformations, VCH Publishers, Inc., New York, 1989; and Carruthers, Some Modern Methods of Organic Synthesis, 3.sup.rd Edition, Cambridge University Press, Cambridge, 1987. The disclosure is not intended to be limited in any manner by the exemplary listing of substituents described herein.
[0036] Compounds described herein can comprise one or more asymmetric centers, and thus can exist in various isomeric forms, e.g., enantiomers and/or diastereomers. For example, the compounds described herein can be in the form of an individual enantiomer, diastereomer or geometric isomer, or can be in the form of a mixture of stereoisomers, including racemic mixtures and mixtures enriched in one or more stereoisomer. Isomers can be isolated from mixtures by methods known to those skilled in the art, including chiral high pressure liquid chromatography (HPLC) and the formation and crystallization of chiral salts; or preferred isomers can be prepared by asymmetric syntheses. See, for example, Jacques et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen et al., Tetrahedron 33:2725 (1977); Eliel, Stereochemistry of Carbon Compounds (McGraw-Hill, N Y, 1962); and Wilen, Tables of Resolving Agents and Optical Resolutions p. 268 (E. L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, Ind. 1972). The disclosure additionally encompasses compounds described herein as individual isomers substantially free of other isomers, and alternatively, as mixtures of various isomers.
[0037] When a range of values is listed, it is intended to encompass each value and sub-range within the range. For example "C.sub.1-6" is intended to encompass, C.sub.1, C.sub.2, C.sub.3, C.sub.4, C.sub.5, C.sub.6, C.sub.1-6, C.sub.1-5, C.sub.1-4, C.sub.1-3, C.sub.1-2, C.sub.2-6, C.sub.2-5, C.sub.2-4, C.sub.2-3, C.sub.3-6, C.sub.3-5, C.sub.3-4, C.sub.4-6, C.sub.4-5, and C.sub.5-6.
[0038] The term "aliphatic" includes both saturated and unsaturated, straight chain (i.e., unbranched), branched, acyclic, cyclic, or polycyclic aliphatic hydrocarbons, which are optionally substituted with one or more functional groups. As will be appreciated by one of ordinary skill in the art, "aliphatic" is intended herein to include, but is not limited to, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, and cycloalkynyl moieties. Thus, the term "alkyl" includes straight, branched and cyclic alkyl groups. An analogous convention applies to other generic terms such as "alkenyl", "alkynyl", and the like. Furthermore, the terms "alkyl", "alkenyl", "alkynyl", and the like encompass both substituted and unsubstituted groups. In certain embodiments, "lower alkyl" is used to indicate those alkyl groups (cyclic, acyclic, substituted, unsubstituted, branched or unbranched) having 1-6 carbon atoms.
[0039] In certain embodiments, the alkyl, alkenyl, and alkynyl groups employed in the disclosure contain 1-20 aliphatic carbon atoms. In certain other embodiments, the alkyl, alkenyl, and alkynyl groups employed in the disclosure contain 1-10 aliphatic carbon atoms. In yet other embodiments, the alkyl, alkenyl, and alkynyl groups employed in the disclosure contain 1-8 aliphatic carbon atoms. In still other embodiments, the alkyl, alkenyl, and alkynyl groups employed in the disclosure contain 1-6 aliphatic carbon atoms. In yet other embodiments, the alkyl, alkenyl, and alkynyl groups employed in the disclosure contain 1-4 carbon atoms. Illustrative aliphatic groups thus include, but are not limited to, for example, methyl, ethyl, n-propyl, isopropyl, cyclopropyl, --CH.sub.2-cyclopropyl, vinyl, allyl, n-butyl, sec-butyl, isobutyl, tert-butyl, cyclobutyl, --CH.sub.2-cyclobutyl, n-pentyl, sec-pentyl, isopentyl, tert-pentyl, cyclopentyl, --CH.sub.2-cyclopentyl, n-hexyl, sec-hexyl, cyclohexyl, --CH.sub.2-cyclohexyl moieties and the like, which again, may bear one or more substituents. Alkenyl groups include, but are not limited to, for example, ethenyl, propenyl, butenyl, 1-methyl-2-buten-1-yl, and the like. Representative alkynyl groups include, but are not limited to, ethynyl, 2-propynyl (propargyl), 1-propynyl, and the like.
[0040] The term "alkyl" refers to a radical of a straight-chain or branched saturated hydrocarbon group having from 1 to 10 carbon atoms ("C.sub.1-10 alkyl"). In some embodiments, an alkyl group has 1 to 9 carbon atoms ("C.sub.1-9 alkyl"). In some embodiments, an alkyl group has 1 to 8 carbon atoms ("C.sub.1-8 alkyl"). In some embodiments, an alkyl group has 1 to 7 carbon atoms ("C.sub.1-7 alkyl"). In some embodiments, an alkyl group has 1 to 6 carbon atoms ("C.sub.1-6 alkyl"). In some embodiments, an alkyl group has 1 to 5 carbon atoms ("C.sub.1-5 alkyl"). In some embodiments, an alkyl group has 1 to 4 carbon atoms ("C.sub.1-4 alkyl"). In some embodiments, an alkyl group has 1 to 3 carbon atoms ("C.sub.1-3 alkyl"). In some embodiments, an alkyl group has 1 to 2 carbon atoms ("C.sub.1-2 alkyl"). In some embodiments, an alkyl group has 1 carbon atom ("C.sub.1 alkyl"). In some embodiments, an alkyl group has 2 to 6 carbon atoms ("C.sub.2-6 alkyl"). Examples of C.sub.1-6 alkyl groups include methyl (C.sub.1), ethyl (C.sub.2), propyl (C.sub.3) (e.g., n-propyl, isopropyl), butyl (C.sub.4) (e.g., n-butyl, tert-butyl, sec-butyl, iso-butyl), pentyl (C.sub.5) (e.g., n-pentyl, 3-pentanyl, amyl, neopentyl, 3-methyl-2-butanyl, tertiary amyl), and hexyl (C.sub.6) (e.g., n-hexyl). Additional examples of alkyl groups include n-heptyl (C.sub.7), n-octyl (C.sub.8), and the like. Unless otherwise specified, each instance of an alkyl group is independently unsubstituted (an "unsubstituted alkyl") or substituted (a "substituted alkyl") with one or more substituents (e.g., halogen, such as F). In certain embodiments, the alkyl group is an unsubstituted C.sub.1-10 alkyl (such as unsubstituted C.sub.1-6 alkyl, e.g., --CH.sub.3 (Me), unsubstituted ethyl (Et), unsubstituted propyl (Pr, e.g., unsubstituted n-propyl (n-Pr), unsubstituted isopropyl (i-Pr)), unsubstituted butyl (Bu, e.g., unsubstituted n-butyl (n-Bu), unsubstituted tert-butyl (tert-Bu or t-Bu), unsubstituted sec-butyl (sec-Bu), unsubstituted isobutyl (i-Bu)). In certain embodiments, the alkyl group is a substituted C.sub.1-10 alkyl (such as substituted C.sub.1-6 alkyl, e.g., --CF.sub.3, Bn).
[0041] "Alkenyl" refers to a radical of a straight-chain or branched hydrocarbon group having from 2 to 20 carbon atoms, one or more carbon-carbon double bonds, and no triple bonds ("C.sub.2-20 alkenyl"). In some embodiments, an alkenyl group has 2 to 10 carbon atoms ("C.sub.2-10 alkenyl"). In some embodiments, an alkenyl group has 2 to 9 carbon atoms ("C.sub.2-9 alkenyl"). In some embodiments, an alkenyl group has 2 to 8 carbon atoms ("C.sub.2-8 alkenyl"). In some embodiments, an alkenyl group has 2 to 7 carbon atoms ("C.sub.2-7 alkenyl"). In some embodiments, an alkenyl group has 2 to 6 carbon atoms ("C.sub.2-6 alkenyl"). In some embodiments, an alkenyl group has 2 to 5 carbon atoms ("C.sub.2-5 alkenyl"). In some embodiments, an alkenyl group has 2 to 4 carbon atoms ("C.sub.2-4 alkenyl"). In some embodiments, an alkenyl group has 2 to 3 carbon atoms ("C.sub.2-3 alkenyl"). In some embodiments, an alkenyl group has 2 carbon atoms ("C.sub.2 alkenyl"). The one or more carbon-carbon double bonds can be internal (such as in 2-butenyl) or terminal (such as in 1-butenyl). Examples of C.sub.2-4 alkenyl groups include ethenyl (C.sub.2), 1-propenyl (C.sub.3), 2-propenyl (C.sub.3), 1-butenyl (C.sub.4), 2-butenyl (C.sub.4), butadienyl (C.sub.4), and the like. Examples of C.sub.2-6 alkenyl groups include the aforementioned C.sub.2-4 alkenyl groups as well as pentenyl (C.sub.5), pentadienyl (C.sub.5), hexenyl (C.sub.6), and the like. Additional examples of alkenyl include heptenyl (C.sub.7), octenyl (C.sub.8), octatrienyl (C.sub.8), and the like. Unless otherwise specified, each instance of an alkenyl group is independently optionally substituted, i.e., unsubstituted (an "unsubstituted alkenyl") or substituted (a "substituted alkenyl") with one or more substituents. In certain embodiments, the alkenyl group is unsubstituted C.sub.2-10 alkenyl. In certain embodiments, the alkenyl group is substituted C.sub.2-10 alkenyl. In an alkenyl group, a C.dbd.C double bond for which the stereochemistry is not specified (e.g., --CH.dbd.CHCH.sub.3 or
##STR00005##
may be an (E)- or (Z)-double bond.
[0042] "Alkynyl" refers to a radical of a straight-chain or branched hydrocarbon group having from 2 to 20 carbon atoms, one or more carbon-carbon triple bonds, and optionally one or more double bonds ("C.sub.2-20 alkynyl"). In some embodiments, an alkynyl group has 2 to 10 carbon atoms ("C.sub.2-10 alkynyl"). In some embodiments, an alkynyl group has 2 to 9 carbon atoms ("C.sub.2-9 alkynyl"). In some embodiments, an alkynyl group has 2 to 8 carbon atoms ("C.sub.2-8 alkynyl"). In some embodiments, an alkynyl group has 2 to 7 carbon atoms ("C.sub.2-7 alkynyl"). In some embodiments, an alkynyl group has 2 to 6 carbon atoms ("C.sub.2-6 alkynyl"). In some embodiments, an alkynyl group has 2 to 5 carbon atoms ("C.sub.2-5 alkynyl"). In some embodiments, an alkynyl group has 2 to 4 carbon atoms ("C.sub.2-4 alkynyl"). In some embodiments, an alkynyl group has 2 to 3 carbon atoms ("C.sub.2-3 alkynyl"). In some embodiments, an alkynyl group has 2 carbon atoms ("C.sub.2 alkynyl"). The one or more carbon-carbon triple bonds can be internal (such as in 2-butynyl) or terminal (such as in 1-butynyl). Examples of C.sub.2-4 alkynyl groups include, without limitation, ethynyl (C.sub.2), 1-propynyl (C.sub.3), 2-propynyl (C.sub.3), 1-butynyl (C.sub.4), 2-butynyl (C.sub.4), and the like. Examples of C.sub.2-6 alkenyl groups include the aforementioned C.sub.2-4 alkynyl groups as well as pentynyl (C.sub.5), hexynyl (C.sub.6), and the like. Additional examples of alkynyl include heptynyl (C.sub.7), octynyl (C.sub.8), and the like. Unless otherwise specified, each instance of an alkynyl group is independently optionally substituted, i.e., unsubstituted (an "unsubstituted alkynyl") or substituted (a "substituted alkynyl") with one or more substituents. In certain embodiments, the alkynyl group is unsubstituted C.sub.2-10 alkynyl. In certain embodiments, the alkynyl group is substituted C.sub.2-10 alkynyl.
[0043] "Carbocyclyl" or "carbocyclic" refers to a radical of a non-aromatic cyclic hydrocarbon group having from 3 to 10 ring carbon atoms ("C.sub.3-10 carbocyclyl") and zero heteroatoms in the non-aromatic ring system. In some embodiments, a carbocyclyl group has 3 to 8 ring carbon atoms ("C.sub.3-8 carbocyclyl"). In some embodiments, a carbocyclyl group has 3 to 6 ring carbon atoms ("C.sub.3-6 carbocyclyl"). In some embodiments, a carbocyclyl group has 3 to 6 ring carbon atoms ("C.sub.3-6 carbocyclyl"). In some embodiments, a carbocyclyl group has 5 to 10 ring carbon atoms ("C.sub.5-10 carbocyclyl"). Exemplary C.sub.3-6 carbocyclyl groups include, without limitation, cyclopropyl (C.sub.3), cyclopropenyl (C.sub.3), cyclobutyl (C.sub.4), cyclobutenyl (C.sub.4), cyclopentyl (C.sub.5), cyclopentenyl (C.sub.5), cyclohexyl (C.sub.6), cyclohexenyl (C.sub.6), cyclohexadienyl (C.sub.6), and the like. Exemplary C.sub.3-8 carbocyclyl groups include, without limitation, the aforementioned C.sub.3-6 carbocyclyl groups as well as cycloheptyl (C.sub.7), cycloheptenyl (C.sub.7), cycloheptadienyl (C.sub.7), cycloheptatrienyl (C.sub.7), cyclooctyl (C.sub.8), cyclooctenyl (C.sub.8), bicyclo[2.2.1]heptanyl (C.sub.7), bicyclo[2.2.2]octanyl (C.sub.8), and the like. Exemplary C.sub.3-10 carbocyclyl groups include, without limitation, the aforementioned C.sub.3-8 carbocyclyl groups as well as cyclononyl (C.sub.9), cyclononenyl (C.sub.9), cyclodecyl (C.sub.10), cyclodecenyl (C.sub.10), octahydro-1H-indenyl (C.sub.9), decahydronaphthalenyl (C.sub.10), spiro[4.5]decanyl (C.sub.10), and the like. As the foregoing examples illustrate, in certain embodiments, the carbocyclyl group is either monocyclic ("monocyclic carbocyclyl") or contain a fused, bridged or spiro ring system such as a bicyclic system ("bicyclic carbocyclyl") and can be saturated or can be partially unsaturated. "Carbocyclyl" also includes ring systems wherein the carbocyclic ring, as defined above, is fused with one or more aryl or heteroaryl groups wherein the point of attachment is on the carbocyclic ring, and in such instances, the number of carbons continue to designate the number of carbons in the carbocyclic ring system. Unless otherwise specified, each instance of a carbocyclyl group is independently optionally substituted, i.e., unsubstituted (an "unsubstituted carbocyclyl") or substituted (a "substituted carbocyclyl") with one or more substituents. In certain embodiments, the carbocyclyl group is unsubstituted C.sub.3-10 carbocyclyl. In certain embodiments, the carbocyclyl group is substituted C.sub.3-10 carbocyclyl.
[0044] In some embodiments, "carbocyclyl" is a monocyclic, saturated carbocyclyl group having from 3 to 10 ring carbon atoms ("C.sub.3-10 cycloalkyl"). In some embodiments, a cycloalkyl group has 3 to 8 ring carbon atoms ("C.sub.3-8 cycloalkyl"). In some embodiments, a cycloalkyl group has 3 to 6 ring carbon atoms ("C.sub.3-6 cycloalkyl"). In some embodiments, a cycloalkyl group has 5 to 6 ring carbon atoms ("C.sub.5-6 cycloalkyl"). In some embodiments, a cycloalkyl group has 5 to 10 ring carbon atoms ("C.sub.5-10 cycloalkyl"). Examples of C.sub.5-6 cycloalkyl groups include cyclopentyl (C.sub.5) and cyclohexyl (C.sub.5). Examples of C.sub.3-6 cycloalkyl groups include the aforementioned C.sub.5-6 cycloalkyl groups as well as cyclopropyl (C.sub.3) and cyclobutyl (C.sub.4). Examples of C.sub.3-8 cycloalkyl groups include the aforementioned C.sub.3-6 cycloalkyl groups as well as cycloheptyl (C.sub.7) and cyclooctyl (C.sub.8). Unless otherwise specified, each instance of a cycloalkyl group is independently unsubstituted (an "unsubstituted cycloalkyl") or substituted (a "substituted cycloalkyl") with one or more substituents. In certain embodiments, the cycloalkyl group is unsubstituted C.sub.3-10 cycloalkyl. In certain embodiments, the cycloalkyl group is substituted C.sub.3-10 cycloalkyl.
[0045] "Heterocyclyl" or "heterocyclic" refers to a radical of a 3- to 10-membered non-aromatic ring system having ring carbon atoms and 1 to 4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, sulfur, boron, phosphorus, and silicon ("3-10 membered heterocyclyl"). In heterocyclyl groups that contain one or more nitrogen atoms, the point of attachment can be a carbon or nitrogen atom, as valency permits. A heterocyclyl group can either be monocyclic ("monocyclic heterocyclyl") or a fused, bridged, or spiro ring system, such as a bicyclic system ("bicyclic heterocyclyl"), and can be saturated or can be partially unsaturated. Heterocyclyl bicyclic ring systems can include one or more heteroatoms in one or both rings. "Heterocyclyl" also includes ring systems wherein the heterocyclic ring, as defined above, is fused with one or more carbocyclyl groups wherein the point of attachment is either on the carbocyclyl or heterocyclic ring, or ring systems wherein the heterocyclic ring, as defined above, is fused with one or more aryl or heteroaryl groups, wherein the point of attachment is on the heterocyclic ring, and in such instances, the number of ring members continue to designate the number of ring members in the heterocyclic ring system. Unless otherwise specified, each instance of heterocyclyl is independently optionally substituted, i.e., unsubstituted (an "unsubstituted heterocyclyl") or substituted (a "substituted heterocyclyl") with one or more substituents. In certain embodiments, the heterocyclyl group is unsubstituted 3-10 membered heterocyclyl. In certain embodiments, the heterocyclyl group is substituted 3-10 membered heterocyclyl.
[0046] In some embodiments, a heterocyclyl group is a 5-10 membered, non-aromatic ring system having ring carbon atoms and 1-4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, sulfur, boron, phosphorus, and silicon ("5-10 membered heterocyclyl"). In some embodiments, a heterocyclyl group is a 5-8 membered non-aromatic ring system having ring carbon atoms and 1-4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur ("5-8 membered heterocyclyl"). In some embodiments, a heterocyclyl group is a 5-6 membered non-aromatic ring system having ring carbon atoms and 1-4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur ("5-6 membered heterocyclyl"). In some embodiments, the 5-6 membered heterocyclyl has 1-3 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In some embodiments, the 5-6 membered heterocyclyl has 1-2 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In some embodiments, the 5-6 membered heterocyclyl has one ring heteroatom selected from nitrogen, oxygen, and sulfur.
[0047] Exemplary 3-membered heterocyclyl groups containing one heteroatom include, without limitation, azirdinyl, oxiranyl, thiiranyl. Exemplary 4-membered heterocyclyl groups containing one heteroatom include, without limitation, azetidinyl, oxetanyl and thietanyl. Exemplary 5-membered heterocyclyl groups containing one heteroatom include, without limitation, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothiophenyl, dihydrothiophenyl, pyrrolidinyl, dihydropyrrolyl, and pyrrolyl-2,5-dione. Exemplary 5-membered heterocyclyl groups containing two heteroatoms include, without limitation, dioxolanyl, oxasulfuranyl, disulfuranyl, and oxazolidin-2-one. Exemplary 5-membered heterocyclyl groups containing three heteroatoms include, without limitation, triazolinyl, oxadiazolinyl, and thiadiazolinyl. Exemplary 6-membered heterocyclyl groups containing one heteroatom include, without limitation, piperidinyl, tetrahydropyranyl, dihydropyridinyl, and thianyl. Exemplary 6-membered heterocyclyl groups containing two heteroatoms include, without limitation, piperazinyl, morpholinyl, dithianyl, and dioxanyl. Exemplary 6-membered heterocyclyl groups containing two heteroatoms include, without limitation, triazinanyl. Exemplary 7-membered heterocyclyl groups containing one heteroatom include, without limitation, azepanyl, oxepanyl and thiepanyl. Exemplary 8-membered heterocyclyl groups containing one heteroatom include, without limitation, azocanyl, oxecanyl and thiocanyl. Exemplary 5-membered heterocyclyl groups fused to a C.sub.6 aryl ring (also referred to herein as a 5,6-bicyclic heterocyclic ring) include, without limitation, indolinyl, isoindolinyl, dihydrobenzofuranyl, dihydrobenzothienyl, benzoxazolinonyl, and the like. Exemplary 6-membered heterocyclyl groups fused to an aryl ring (also referred to herein as a 6,6-bicyclic heterocyclic ring) include, without limitation, tetrahydroquinolinyl, tetrahydroisoquinolinyl, and the like.
[0048] "Aryl" refers to a radical of a monocyclic or polycyclic (e.g., bicyclic or tricyclic) 4n+2 aromatic ring system (e.g., having 6, 10, or 14 pi electrons shared in a cyclic array) having 6-14 ring carbon atoms and zero heteroatoms provided in the aromatic ring system ("C.sub.6-14 aryl"). In some embodiments, an aryl group has six ring carbon atoms ("C.sub.6 aryl"; e.g., phenyl). In some embodiments, an aryl group has ten ring carbon atoms ("C.sub.10 aryl"; e.g., naphthyl such as 1-naphthyl and 2-naphthyl). In some embodiments, an aryl group has fourteen ring carbon atoms ("C.sub.14 aryl"; e.g., anthracyl). "Aryl" also includes ring systems wherein the aryl ring, as defined above, is fused with one or more carbocyclyl or heterocyclyl groups, wherein the radical or point of attachment is on the aryl ring, and in such instances, the number of carbon atoms continue to designate the number of carbon atoms in the aryl ring system. Unless otherwise specified, each instance of an aryl group is independently optionally substituted, i.e., unsubstituted (an "unsubstituted aryl") or substituted (a "substituted aryl") with one or more substituents. In certain embodiments, the aryl group is unsubstituted C.sub.6-14 aryl. In certain embodiments, the aryl group is substituted C.sub.6-14 aryl.
[0049] "Aralkyl" refers to an optionally substituted alkyl group substituted by an optionally substituted aryl group. In certain embodiments, the aralkyl is optionally substituted benzyl. In certain embodiments, the aralkyl is benzyl. In certain embodiments, the aralkyl is optionally substituted phenethyl. In certain embodiments, the aralkyl is phenethyl.
[0050] "Heteroaryl" refers to a radical of a 5-10 membered, monocyclic or bicyclic 4n+2 aromatic ring system (e.g., having 6 or 10 pi electrons shared in a cyclic array) having ring carbon atoms and 1-4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen and sulfur ("5-10 membered heteroaryl"). In heteroaryl groups that contain one or more nitrogen atoms, the point of attachment can be a carbon or nitrogen atom, as valency permits. Heteroaryl bicyclic ring systems can include one or more heteroatoms in one or both rings. "Heteroaryl" includes ring systems wherein the heteroaryl ring, as defined above, is fused with one or more carbocyclyl or heterocyclyl groups wherein the point of attachment is on the heteroaryl ring, and in such instances, the number of ring members continue to designate the number of ring members in the heteroaryl ring system. "Heteroaryl" also includes ring systems wherein the heteroaryl ring, as defined above, is fused with one or more aryl groups wherein the point of attachment is either on the aryl or heteroaryl ring, and in such instances, the number of ring members designates the number of ring members in the fused (aryl/heteroaryl) ring system. Bicyclic heteroaryl groups wherein one ring does not contain a heteroatom (e.g., indolyl, quinolinyl, carbazolyl, and the like) the point of attachment can be on either ring, i.e., either the ring bearing a heteroatom (e.g., 2-indolyl) or the ring that does not contain a heteroatom (e.g., 5-indolyl).
[0051] In some embodiments, a heteroaryl group is a 5-10 membered aromatic ring system having ring carbon atoms and 1-4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur ("5-10 membered heteroaryl"). In some embodiments, a heteroaryl group is a 5-8 membered aromatic ring system having ring carbon atoms and 1-4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur ("5-8 membered heteroaryl"). In some embodiments, a heteroaryl group is a 5-6 membered aromatic ring system having ring carbon atoms and 1-4 ring heteroatoms provided in the aromatic ring system, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur ("5-6 membered heteroaryl"). In some embodiments, the 5-6 membered heteroaryl has 1-3 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In some embodiments, the 5-6 membered heteroaryl has 1-2 ring heteroatoms selected from nitrogen, oxygen, and sulfur. In some embodiments, the 5-6 membered heteroaryl has 1 ring heteroatom selected from nitrogen, oxygen, and sulfur. Unless otherwise specified, each instance of a heteroaryl group is independently optionally substituted, i.e., unsubstituted (an "unsubstituted heteroaryl") or substituted (a "substituted heteroaryl") with one or more substituents. In certain embodiments, the heteroaryl group is unsubstituted 5-14 membered heteroaryl. In certain embodiments, the heteroaryl group is substituted 5-14 membered heteroaryl.
[0052] Exemplary 5-membered heteroaryl groups containing one heteroatom include, without limitation, pyrrolyl, furanyl, and thiophenyl. Exemplary 5-membered heteroaryl groups containing two heteroatoms include, without limitation, imidazolyl, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, and isothiazolyl. Exemplary 5-membered heteroaryl groups containing three heteroatoms include, without limitation, triazolyl, oxadiazolyl, and thiadiazolyl. Exemplary 5-membered heteroaryl groups containing four heteroatoms include, without limitation, tetrazolyl. Exemplary 6-membered heteroaryl groups containing one heteroatom include, without limitation, pyridinyl. Exemplary 6-membered heteroaryl groups containing two heteroatoms include, without limitation, pyridazinyl, pyrimidinyl, and pyrazinyl. Exemplary 6-membered heteroaryl groups containing three or four heteroatoms include, without limitation, triazinyl and tetrazinyl, respectively. Exemplary 7-membered heteroaryl groups containing one heteroatom include, without limitation, azepinyl, oxepinyl, and thiepinyl. Exemplary 5,6-bicyclic heteroaryl groups include, without limitation, indolyl, isoindolyl, indazolyl, benzotriazolyl, benzothiophenyl, isobenzothiophenyl, benzofuranyl, benzoisofuranyl, benzimidazolyl, benzoxazolyl, benzisoxazolyl, benzoxadiazolyl, benzthiazolyl, benzisothiazolyl, benzthiadiazolyl, indolizinyl, and purinyl. Exemplary 6,6-bicyclic heteroaryl groups include, without limitation, naphthyridinyl, pteridinyl, quinolinyl, isoquinolinyl, cinnolinyl, quinoxalinyl, phthalazinyl, and quinazolinyl.
[0053] "Heteroaralkyl" is a subset of alkyl and heteroaryl and refers to an optionally substituted alkyl group substituted by an optionally substituted heteroaryl group.
[0054] "Unsaturated" or "partially unsaturated" refers to a group that includes at least one double or triple bond. A "partially unsaturated" ring system is further intended to encompass rings having multiple sites of unsaturation, but is not intended to include aromatic groups (e.g., aryl or heteroaryl groups). Likewise, "saturated" refers to a group that does not contain a double or triple bond, i.e., contains all single bonds.
[0055] Alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl groups, which are divalent linking groups, are further referred to using the suffix-ene, e.g., alkylene, alkenylene, alkynylene, carbocyclylene, heterocyclylene, arylene, and heteroarylene.
[0056] An atom, moiety, or group described herein may be unsubstituted or substituted, as valency permits, unless otherwise provided expressly. The term "optionally substituted" refers to substituted or unsubstituted.
[0057] A group is optionally substituted unless expressly provided otherwise. The term "optionally substituted" refers to being substituted or unsubstituted. In certain embodiments, alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl groups are optionally substituted (e.g., "substituted" or "unsubstituted" alkyl, "substituted" or "unsubstituted" alkenyl, "substituted" or "unsubstituted" alkynyl, "substituted" or "unsubstituted" carbocyclyl, "substituted" or "unsubstituted" heterocyclyl, "substituted" or "unsubstituted" aryl or "substituted" or "unsubstituted" heteroaryl group). In general, the term "substituted", whether preceded by the term "optionally" or not, means that at least one hydrogen present on a group (e.g., a carbon or nitrogen atom) is replaced with a permissible substituent, e.g., a substituent which upon substitution results in a stable compound, e.g., a compound which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, or other reaction. Unless otherwise indicated, a "substituted" group has a substituent at one or more substitutable positions of the group, and when more than one position in any given structure is substituted, the substituent is either the same or different at each position. The term "substituted" is contemplated to include substitution with all permissible substituents of organic compounds, any of the substituents described herein that results in the formation of a stable compound. The present disclosure contemplates any and all such combinations in order to arrive at a stable compound. For purposes of this disclosure, heteroatoms such as nitrogen may have hydrogen substituents and/or any suitable substituent as described herein which satisfy the valencies of the heteroatoms and results in the formation of a stable moiety. In certain embodiments, the substituent is a carbon atom substituent. In certain embodiments, the substituent is a nitrogen atom substituent. In certain embodiments, the substituent is an oxygen atom substituent. In certain embodiments, the substituent is a sulfur atom substituent.
[0058] Exemplary carbon atom substituents include, but are not limited to, halogen, --CN, --NO.sub.2, --N.sub.3, --SO.sub.2H, --SO.sub.3H, --OH, --OR.sup.aa, --ON(R.sup.bb).sub.2, --N(R.sup.bb).sub.2, --N(R.sup.bb).sub.3.sup.+X.sup.-, --N(OR.sup.cc)R.sup.bb, --SH, --SR.sup.aa, --SSR.sup.cc, --C(.dbd.O)R.sup.aa, --CO.sub.2H, --CHO, --C(OR.sup.cc).sub.2, --CO.sub.2R.sup.aa, --OC(.dbd.O)R.sup.aa, --OCO.sub.2R.sup.aa, --C(.dbd.O)N(R.sup.bb).sub.2, --OC(.dbd.O)N(R.sup.bb).sub.2, --NR.sup.bbC(.dbd.O)R.sup.aa, --NR.sup.bbCO.sub.2R.sup.aa, --NR.sup.bbC(.dbd.O)N(R.sup.bb).sub.2, --C(.dbd.NR.sup.bb)R.sup.aa, --C(.dbd.NR.sup.bb)OR.sup.aa, --OC(.dbd.NR.sup.bb)R.sup.aa, --OC(.dbd.NR.sup.bb)OR.sup.aa, --C(.dbd.NR.sup.bb)N(R.sup.bb).sub.2, --OC(.dbd.NR.sup.bb)N(R.sup.bb).sub.2, --NR.sup.bbC(.dbd.NR.sup.bb)N(R.sup.bb).sub.2, --C(.dbd.O)NR.sup.bbSO.sub.2R.sup.aa, --NR.sup.bbSO.sub.2R.sup.aa, --SO.sub.2N(R.sup.bb).sub.2, --SO.sub.2R.sup.aa, --SO.sub.2OR.sup.aa, --OSO.sub.2R.sup.aa, --S(.dbd.O)R.sup.aa, --OS(.dbd.O)R.sup.aa, --Si(R.sup.aa).sub.3, --OSi(R.sup.aa).sub.3--C(.dbd.S)N(R.sup.bb).sub.2, --C(.dbd.O)SR.sup.aa, --C(.dbd.S)SR.sup.aa, --SC(.dbd.S)SR.sup.aa, --SC(.dbd.O)SR.sup.aa, --OC(.dbd.O)SR.sup.aa, --SC(.dbd.O)OR.sup.aa, --SC(.dbd.O)R.sup.aa, --P(.dbd.O)(R.sup.aa).sub.2, --P(.dbd.O)(OR.sup.cc).sub.2, --OP(.dbd.O)(R.sup.aa).sub.2, --OP(.dbd.O)(OR.sup.cc).sub.2, --P(.dbd.O)(N(R.sup.bb).sub.2).sub.2, --OP(.dbd.O)(N(R.sup.bb).sub.2).sub.2, --NR.sup.bbP(.dbd.O)(R.sup.aa).sub.2, --NR.sup.bbP(.dbd.O)(OR.sup.cc).sub.2, --NR.sup.bbP(.dbd.O)(N(R.sup.bb).sub.2).sub.2, --P(R.sup.cc).sub.2, --P(OR.sup.cc).sub.2, --P(R.sup.cc).sub.3.sup.+X.sup.-, --P(OR.sup.cc).sub.3.sup.+X.sup.-, --P(R.sup.cc).sub.4, --P(OR.sup.cc).sub.4, --OP(R.sup.cc).sub.2, --OP(R.sup.cc).sub.3.sup.+X.sup.-, --OP(OR.sup.cc).sub.2, --OP(OR.sup.cc).sub.3.sup.+X.sup.-, --OP(R.sup.cc).sub.4, --OP(OR.sup.cc).sub.4, --B(R.sup.aa).sub.2, --B(OR.sup.cc).sub.2, --BR.sup.aa(OR.sup.cc), C.sub.1-10 alkyl, C.sub.1-10 perhaloalkyl, C.sub.2-10 alkenyl, C.sub.2-10 alkynyl, heteroC.sub.1-10 alkyl, heteroC.sub.2-10 alkenyl, heteroC.sub.2-10 alkynyl, C.sub.3-10 carbocyclyl, 3-14 membered heterocyclyl, C.sub.6-14 aryl, and 5-14 membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R.sup.dd groups; wherein X.sup.- is a counterion;
[0059] or two geminal hydrogens on a carbon atom are replaced with the group .dbd.O, .dbd.S, .dbd.NN(R.sup.bb).sub.2, .dbd.NNR.sup.bbC(.dbd.O)R.sup.aa, .dbd.NNR.sup.bbC(.dbd.O)OR.sup.aa, .dbd.NNR.sup.bbS(.dbd.O).sub.2R.sup.aa, .dbd.NR.sup.bb, or .dbd.NOR.sup.cc;
[0060] each instance of R.sup.aa is, independently, selected from C.sub.1-10 alkyl, C.sub.1-10 perhaloalkyl, C.sub.2-10 alkenyl, C.sub.2-10 alkynyl, C.sub.3-10 carbocyclyl, 3-14 membered heterocyclyl, C.sub.6-14 aryl, and 5-14 membered heteroaryl, or two R.sup.aa groups are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R.sup.dd groups;
[0061] each instance of R.sup.bb is, independently, selected from hydrogen, --OH, --OR.sup.aa, --N(R.sup.cc).sub.2, --CN, --C(.dbd.O)R.sup.aa, --C(.dbd.O)N(R.sup.cc).sub.2, --CO.sub.2R.sup.aa, --SO.sub.2R.sup.aa, --C(.dbd.NR.sup.cc)OR.sup.aa, --C(.dbd.NR.sup.cc)N(R.sup.cc).sub.2, --SO.sub.2N(R.sup.cc).sub.2, --SO.sub.2R.sup.cc, --SO.sub.2OR.sup.cc, --SOR.sup.aa, --C(.dbd.S)N(R.sup.cc).sub.2, --C(.dbd.O)SR.sup.cc, --C(.dbd.S)SR.sup.cc, --P(.dbd.O)(R.sup.aa).sub.2, --P(.dbd.O)(OR.sup.cc).sub.2, --P(.dbd.O)(N(R.sup.cc).sub.2).sub.2, C.sub.1-10 alkyl, C.sub.1-10 perhaloalkyl, C.sub.2-10 alkenyl, C.sub.2-10 alkynyl, heteroC.sub.1-10alkyl, heteroC.sub.2-10alkenyl, heteroC.sub.2-10alkynyl, C.sub.3-10 carbocyclyl, 3-14 membered heterocyclyl, C.sub.6-14 aryl, and 5-14 membered heteroaryl, or two R.sup.bb groups are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R.sup.dd groups; wherein X.sup.- is a counterion;
[0062] each instance of R.sup.cc is, independently, selected from hydrogen, C.sub.1-10 alkyl, C.sub.1-10 perhaloalkyl, C.sub.2-10 alkenyl, C.sub.2-10 alkynyl, C.sub.3-10 carbocyclyl, 3-14 membered heterocyclyl, C.sub.6-14 aryl, and 5-14 membered heteroaryl, or two R.sup.cc groups are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R.sup.dd groups;
[0063] each instance of R.sup.dd is, independently, selected from halogen, --CN, --NO.sub.2, --N.sub.3, --SO.sub.2H, --SO.sub.3H, --OH, --OR.sup.ee, --ON(R.sup.ff).sub.2, --N(R.sup.ff).sub.2, --N(R.sup.ff).sub.3.sup.+X.sup.-, --N(OR.sup.ee)R.sup.ff, --SH, --SR.sup.ee, --SSR.sup.ee, --C(.dbd.O)R.sup.ee, --CO.sub.2H, --CO.sub.2R.sup.ee, --OC(.dbd.O)R.sup.ee, --OCO.sub.2R.sup.ee, --C(.dbd.O)N(R.sup.ff).sub.2, --OC(.dbd.O)N(R.sup.ff).sub.2, --NR.sup.ffC(.dbd.O)R.sup.ee, --NR.sup.ffCO.sub.2R.sup.ee, --NR.sup.ffC(.dbd.O)N(R.sup.ff).sub.2, --C(.dbd.NR.sup.ff)OR.sup.ee, --OC(.dbd.NR.sup.ff)R.sup.ee, --OC(.dbd.NR.sup.ff)OR.sup.ee, --C(.dbd.NR.sup.ff)N(R.sup.ff).sub.2, --OC(.dbd.NR.sup.ff)N(R.sup.ff).sub.2, --NR.sup.ffC(.dbd.NR.sup.ff)N(R.sup.ff).sub.2, --NR.sup.ffSO.sub.2R.sup.ee, --SO.sub.2N(R.sup.ff).sub.2, --SO.sub.2R.sup.ee, --SO.sub.2OR.sup.ee, --OSO.sub.2R.sup.ee, --S(.dbd.O)R.sup.ee, --Si(R.sup.ee).sub.3, --OSi(R.sup.ee).sub.3, --C(.dbd.S)N(R.sup.ff).sub.2, --C(.dbd.O)SR.sup.ee, --C(.dbd.S)SR.sup.ee, --SC(.dbd.S)SR.sup.ee, --P(.dbd.O)(OR.sup.ee).sub.2, --P(.dbd.O)(R.sup.ee).sub.2, --OP(.dbd.O)(R.sup.ee).sub.2, --OP(.dbd.O)(OR.sup.ee).sub.2, C.sub.1-6 alkyl, C.sub.1-6 perhaloalkyl, C.sub.2-6 alkenyl, C.sub.2-6 alkynyl, heteroC.sub.1-6alkyl, heteroC.sub.2-6alkenyl, heteroC.sub.2-6alkynyl, C.sub.3-10 carbocyclyl, 3-10 membered heterocyclyl, C.sub.6-10 aryl, 5-10 membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R.sup.gg groups, or two geminal R.sup.dd substituents can be joined to form .dbd.O or .dbd.S; wherein X.sup.- is a counterion;
[0064] each instance of R.sup.ee is, independently, selected from C.sub.1-6 alkyl, C.sub.1-6 perhaloalkyl, C.sub.2-6 alkenyl, C.sub.2-6 alkynyl, C.sub.3-10 carbocyclyl, C.sub.6-10 aryl, 3-10 membered heterocyclyl, and 3-10 membered heteroaryl, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R.sup.gg groups;
[0065] each instance of R.sup.ff is, independently, selected from hydrogen, C.sub.1-6 alkyl, C.sub.1-6 perhaloalkyl, C.sub.2-6 alkenyl, C.sub.2-6 alkynyl, C.sub.3-10 carbocyclyl, 3-10 membered heterocyclyl, C.sub.6-10 aryl and 5-10 membered heteroaryl, or two R.sup.ff groups are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R.sup.gg groups; and
[0066] each instance of R.sup.gg is, independently, halogen, --CN, --NO.sub.2, --N.sub.3, --SO.sub.2H, --SO.sub.3H, --OH, --OC.sub.1-6 alkyl, --ON(C.sub.1-6 alkyl).sub.2, --N(C.sub.1-6 alkyl).sub.2, --N(C.sub.1-6 alkyl).sub.3.sup.+X.sup.-, --NH(C.sub.1-6 alkyl).sub.2.sup.+X.sup.-, --NH.sub.2(C.sub.1-6 alkyl).sup.+X.sup.-, --NH.sub.3.sup.+X.sup.-, --N(OC.sub.1-6 alkyl)(C.sub.1-6 alkyl), --N(OH)(C.sub.1-6 alkyl), --NH(OH), --SH, --SC.sub.1-6 alkyl, --SS(C.sub.1-6 alkyl), --C(.dbd.O)(C.sub.1-6 alkyl), --CO.sub.2H, --CO.sub.2(C.sub.1-6 alkyl), --OC(.dbd.O)(C.sub.1-6 alkyl), --OCO.sub.2(C.sub.1-6 alkyl), --C(.dbd.O)NH.sub.2, --C(.dbd.O)N(C.sub.1-6 alkyl).sub.2, --OC(.dbd.O)NH(C.sub.1-6 alkyl), --NHC(.dbd.O)(C.sub.1-6 alkyl), --N(C.sub.1-6 alkyl)C(.dbd.O)(C.sub.1-6 alkyl), --NHCO.sub.2(C.sub.1-6 alkyl), --NHC(.dbd.O)N(C.sub.1-6 alkyl).sub.2, --NHC(.dbd.O)NH(C.sub.1-6 alkyl), --NHC(.dbd.O)NH.sub.2, --C(.dbd.NH)O(C.sub.1-6 alkyl), --OC(.dbd.NH)(C.sub.1-6 alkyl), --OC(.dbd.NH)OC.sub.1-6 alkyl, --C(.dbd.NH)N(C.sub.1-6 alkyl).sub.2, --C(.dbd.NH)NH(C.sub.1-6 alkyl), --C(.dbd.NH)NH.sub.2, --OC(.dbd.NH)N(C.sub.1-6 alkyl).sub.2, --OC(NH)NH(C.sub.1-6 alkyl), --OC(NH)NH.sub.2, --NHC(NH)N(C.sub.1-6 alkyl).sub.2, --NHC(.dbd.NH)NH.sub.2, --NHSO.sub.2(C.sub.1-6 alkyl), --SO.sub.2N(C.sub.1-6 alkyl).sub.2, --SO.sub.2NH(C.sub.1-6 alkyl), --SO.sub.2NH.sub.2, --SO.sub.2C.sub.1-6 alkyl, --SO.sub.2OC.sub.1-6 alkyl, --OSO.sub.2C.sub.1-6 alkyl, --SOC.sub.1-6 alkyl, --Si(C.sub.1-6 alkyl).sub.3, --OSi(C.sub.1-6 alkyl).sub.3-C(.dbd.S)N(C.sub.1-6 alkyl).sub.2, C(.dbd.S)NH(C.sub.1-6 alkyl), C(.dbd.S)NH.sub.2, --C(.dbd.O)S(C.sub.1-6 alkyl), --C(.dbd.S)SC.sub.1-6 alkyl, --SC(.dbd.S)SC.sub.1-6 alkyl, --P(.dbd.O)(OC.sub.1-6 alkyl).sub.2, --P(.dbd.O)(C.sub.1-6 alkyl).sub.2, --OP(.dbd.O)(C.sub.1-6 alkyl).sub.2, --OP(.dbd.O)(OC.sub.1-6 alkyl).sub.2, C.sub.1-6 alkyl, C.sub.1-6 perhaloalkyl, C.sub.2-6 alkenyl, C.sub.2-6 alkynyl, heteroC.sub.1-6alkyl, heteroC.sub.2-6alkenyl, heteroC.sub.2-6alkynyl, C.sub.3-10 carbocyclyl, C.sub.6-10 aryl, 3-10 membered heterocyclyl, 5-10 membered heteroaryl; or two geminal R.sup.gg substituents can be joined to form .dbd.O or .dbd.S; wherein X is a counterion.
[0067] A "counterion" or "anionic counterion" is a negatively charged group associated with a positively charged group in order to maintain electronic neutrality. An anionic counterion may be monovalent (i.e., including one formal negative charge). An anionic counterion may also be multivalent (i.e., including more than one formal negative charge), such as divalent or trivalent. Exemplary counterions include halide ions (e.g., F.sup.-, Cl.sup.-, Br.sup.-, I.sup.-), NO.sub.3.sup.-, ClO.sub.4.sup.-, OH.sup.-, H.sub.2PO.sub.4.sup.-, HCO.sub.3.sup.-, HSO.sub.4.sup.-, sulfonate ions (e.g., methansulfonate, trifluoromethanesulfonate, p-toluenesulfonate, benzenesulfonate, 10-camphor sulfonate, naphthalene-2-sulfonate, naphthalene-1-sulfonic acid-5-sulfonate, ethan-1-sulfonic acid-2-sulfonate, and the like), carboxylate ions (e.g., acetate, propanoate, benzoate, glycerate, lactate, tartrate, glycolate, gluconate, and the like), BF.sub.4.sup.-, PF.sub.4.sup.-, PF.sub.6.sup.-, AsF.sub.6.sup.-, SbF.sub.6.sup.-, B[3,5-(CF.sub.3).sub.2C.sub.6H.sub.3].sub.4].sup.-, B(C.sub.6F.sub.5).sub.4.sup.-, BPh.sub.4.sup.-, Al(OC(CF.sub.3).sub.3).sub.4.sup.-, and carborane anions (e.g., CB.sub.11H.sub.12.sup.- or (HCB.sub.11Me.sub.5Br.sub.6).sup.-). Exemplary counterions which may be multivalent include CO.sub.3.sup.2-, HPO.sub.4.sup.2-, PO.sub.4.sup.3-, B.sub.4O.sub.7.sup.2-, SO.sub.4.sup.2-, S.sub.2O.sub.3.sup.2-, carboxylate anions (e.g., tartrate, citrate, fumarate, maleate, malate, malonate, gluconate, succinate, glutarate, adipate, pimelate, suberate, azelate, sebacate, salicylate, phthalates, aspartate, glutamate, and the like), and carboranes.
[0068] "Halo" or "halogen" refers to fluorine (fluoro, --F), chlorine (chloro, --Cl), bromine (bromo, --Br), or iodine (iodo, --I).
[0069] The term "hydroxyl" or "hydroxy" refers to the group --OH. The term "substituted hydroxyl" or "substituted hydroxyl," by extension, refers to a hydroxyl group wherein the oxygen atom directly attached to the parent molecule is substituted with a group other than hydrogen, and includes groups selected from --OR.sup.aa, --ON(R.sup.bb).sub.2, --OC(.dbd.O)SR.sup.aa, --OC(.dbd.O)R.sup.aa, --OCO.sub.2R.sup.aa, --OC(.dbd.O)N(R.sup.bb).sub.2, --OC(.dbd.NR.sup.bb)R.sup.aa, --OC(.dbd.NR.sup.bb)OR.sup.aa, --OC(.dbd.NR.sup.bb)N(R.sup.bb).sub.2, --OS(.dbd.O)R.sup.aa, --OSO.sub.2R.sup.aa, --OSi(R.sup.aa).sub.3, --OP(R.sup.cc).sub.2, --OP(R.sup.cc).sub.3.sup.+X.sup.-, --OP(OR.sup.cc).sub.2, --OP(OR.sup.cc).sub.3.sup.+X.sup.-, --OP(.dbd.O)(R.sup.aa).sub.2, --OP(.dbd.O)(OR.sup.cc).sub.2, and --OP(.dbd.O)(N(R.sup.bb)).sub.2, wherein X.sup.-, R.sup.aa, R.sup.bb, and R.sup.cc are as defined herein.
[0070] "Acyl" refers to a moiety selected from the group consisting of --C(.dbd.O)R.sup.aa, --CHO, --CO.sub.2R.sup.aa, --C(.dbd.O)N(R.sup.bb).sub.2, --C(.dbd.NR.sup.bb)R.sup.aa, --C(.dbd.NR.sup.bb)OR.sup.aa, --C(.dbd.NR.sup.bb)N(R.sup.bb).sub.2, --C(.dbd.O)NR.sup.bbSO.sub.2R.sup.aa, --C(.dbd.S)N(R.sup.bb).sub.2, --C(.dbd.O)SR.sup.aa, or --C(.dbd.S)SR.sup.aa, wherein R.sup.aa and R.sup.bb are as defined herein.
[0071] The term "amino" refers to the group --NH.sub.2. The term "substituted amino," by extension, refers to a monosubstituted amino, a disubstituted amino, or a trisubstituted amino. In certain embodiments, the "substituted amino" is a monosubstituted amino or a disubstituted amino group.
[0072] Nitrogen atoms can be substituted or unsubstituted as valency permits, and include primary, secondary, tertiary, and quaternary nitrogen atoms. Exemplary nitrogen atom substituents include, but are not limited to, hydrogen, --OH, --OR.sup.aa, --N(R.sup.cc).sub.2, --CN, --C(.dbd.O)R.sup.aa, --C(.dbd.O)N(R.sup.cc).sub.2, --CO.sub.2R.sup.aa, --SO.sub.2R.sup.aa, --C(.dbd.NR.sup.bb)R.sup.aa, --C(.dbd.NR.sup.cc)OR.sup.aa, --C(.dbd.NR.sup.cc)N(R.sup.cc).sub.2, --SO.sub.2N(R.sup.cc).sub.2, --SO.sub.2R.sup.cc, --SO.sub.2OR.sup.cc, --SOR.sup.aa, --C(.dbd.S)N(R.sup.cc).sub.2, --C(.dbd.O)SR.sup.cc, --C(.dbd.S)SR.sup.cc, --P(.dbd.O)(OR.sup.cc).sub.2, --P(.dbd.O)(R.sup.aa).sub.2, --P(.dbd.O)(N(R.sup.cc).sub.2).sub.2, C.sub.1-10 alkyl, C.sub.1-10 perhaloalkyl, C.sub.2-10 alkenyl, C.sub.2-10 alkynyl, heteroC.sub.1-10alkyl, heteroC.sub.2-10alkenyl, heteroC.sub.2-10alkynyl, C.sub.3-10 carbocyclyl, 3-14 membered heterocyclyl, C.sub.6-14 aryl, and 5-14 membered heteroaryl, or two R.sup.cc groups attached to an N atom are joined to form a 3-14 membered heterocyclyl or 5-14 membered heteroaryl ring, wherein each alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, carbocyclyl, heterocyclyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R.sup.dd groups, and wherein R.sup.aa, R.sup.bb, R.sup.cc and R.sup.dd are as defined above.
[0073] In certain embodiments, the substituent present on a nitrogen atom is a nitrogen protecting group (also referred to as an amino protecting group). Nitrogen protecting groups include, but are not limited to, --OH, --OR.sup.aa, --N(R.sup.cc).sub.2, --C(.dbd.O)R.sup.aa, --C(.dbd.O)N(R.sup.cc).sub.2, --CO.sub.2R.sup.aa, --SO.sub.2R.sup.aa, --C(.dbd.NR.sup.cc)R.sup.aa, --C(.dbd.NR.sup.cc)OR.sup.aa, --C(.dbd.NR.sup.cc)N(R.sup.cc).sub.2, --SO.sub.2N(R.sup.cc).sub.2, --SO.sub.2R.sup.cc, --SO.sub.2OR.sup.cc, --SOR.sup.aa, --C(.dbd.S)N(R.sup.cc).sub.2, --C(.dbd.O)SR.sup.cc, --C(.dbd.S)SR.sup.cc, C.sub.1-10 alkyl (e.g., aralkyl, heteroaralkyl), C.sub.2-10 alkenyl, C.sub.2-10 alkynyl, C.sub.3-10 carbocyclyl, 3-14 membered heterocyclyl, C.sub.6-14 aryl, and 5-14 membered heteroaryl groups, wherein each alkyl, alkenyl, alkynyl, carbocyclyl, heterocyclyl, aralkyl, aryl, and heteroaryl is independently substituted with 0, 1, 2, 3, 4, or 5 R.sup.dd groups, and wherein R.sup.aa, R.sup.bb, R.sup.cc and R.sup.dd are as defined herein. Nitrogen protecting groups are well known in the art and include those described in detail in Protecting Groups in Organic Synthesis, T. W. Greene and P. G. M. Wuts, 3.sup.rd edition, John Wiley & Sons, 1999, incorporated herein by reference.
[0074] For example, nitrogen protecting groups such as amide groups (e.g., --C(.dbd.O)R.sup.aa) include, but are not limited to, formamide, acetamide, chloroacetamide, trichloroacetamide, trifluoroacetamide, phenylacetamide, 3-phenylpropanamide, picolinamide, 3-pyridylcarboxamide, N-benzoylphenylalanyl derivative, benzamide, p-phenylbenzamide, o-nitrophenylacetamide, o-nitrophenoxyacetamide, acetoacetamide, (N'-dithiobenzyloxyacylamino)acetamide, 3-(p-hydroxyphenyl)propanamide, 3-(o-nitrophenyl)propanamide, 2-methyl-2-(o-nitrophenoxy)propanamide, 2-methyl-2-(o-phenylazophenoxy)propanamide, 4-chlorobutanamide, 3-methyl-3-nitrobutanamide, o-nitrocinnamide, N-acetylmethionine derivative, o-nitrobenzamide, and o-(benzoyloxymethyl)benzamide.
[0075] Nitrogen protecting groups such as carbamate groups (e.g., --C(.dbd.O)OR.sup.aa) include, but are not limited to, methyl carbamate, ethyl carbamate, 9-fluorenylmethyl carbamate (Fmoc), 9-(2-sulfo)fluorenylmethyl carbamate, 9-(2,7-dibromo)fluorenylmethyl carbamate, 2,7-di-t-butyl-[9-(10,10-dioxo-10,10,10,10-tetrahydrothioxanthyl)]methyl carbamate (DBD-Tmoc), 4-methoxyphenacyl carbamate (Phenoc), 2,2,2-trichloroethyl carbamate (Troc), 2-trimethylsilylethyl carbamate (Teoc), 2-phenylethyl carbamate (hZ), 1-(1-adamantyl)-1-methylethyl carbamate (Adpoc), 1,1-dimethyl-2-haloethyl carbamate, 1,1-dimethyl-2,2-dibromoethyl carbamate (DB-t-BOC), 1,1-dimethyl-2,2,2-trichloroethyl carbamate (TCBOC), 1-methyl-1-(4-biphenylyl)ethyl carbamate (Bpoc), 1-(3,5-di-t-butylphenyl)-1-methylethyl carbamate (t-Bumeoc), 2-(2'- and 4'-pyridyl)ethyl carbamate (Pyoc), 2-(N,N-dicyclohexylcarboxamido)ethyl carbamate, t-butyl carbamate (BOC or Boc), 1-adamantyl carbamate (Adoc), vinyl carbamate (Voc), allyl carbamate (Alloc), 1-isopropylallyl carbamate (Ipaoc), cinnamyl carbamate (Coc), 4-nitrocinnamyl carbamate (Noc), 8-quinolyl carbamate, N-hydroxypiperidinyl carbamate, alkyldithio carbamate, benzyl carbamate (Cbz), p-methoxybenzyl carbamate (Moz), p-nitrobenzyl carbamate, p-bromobenzyl carbamate, p-chlorobenzyl carbamate, 2,4-dichlorobenzyl carbamate, 4-methylsulfinylbenzyl carbamate (Msz), 9-anthrylmethyl carbamate, diphenylmethyl carbamate, 2-methylthioethyl carbamate, 2-methylsulfonylethyl carbamate, 2-(p-toluenesulfonyl)ethyl carbamate, [2-(1,3-dithianyl)]methyl carbamate (Dmoc), 4-methylthiophenyl carbamate (Mtpc), 2,4-dimethylthiophenyl carbamate (Bmpc), 2-phosphonioethyl carbamate (Peoc), 2-triphenylphosphonioisopropyl carbamate (Ppoc), 1,1-dimethyl-2-cyanoethyl carbamate, m-chloro-p-acyloxybenzyl carbamate, p-(dihydroxyboryl)benzyl carbamate, 5-benzisoxazolylmethyl carbamate, 2-(trifluoromethyl)-6-chromonylmethyl carbamate (Tcroc), m-nitrophenyl carbamate, 3,5-dimethoxybenzyl carbamate, o-nitrobenzyl carbamate, 3,4-dimethoxy-6-nitrobenzyl carbamate, phenyl(o-nitrophenyl)methyl carbamate, t-amyl carbamate, S-benzyl thiocarbamate, p-cyanobenzyl carbamate, cyclobutyl carbamate, cyclohexyl carbamate, cyclopentyl carbamate, cyclopropylmethyl carbamate, p-decyloxybenzyl carbamate, 2,2-dimethoxyacylvinyl carbamate, o-(N,N-dimethylcarboxamido)benzyl carbamate, 1,1-dimethyl-3-(N,N-dimethylcarboxamido)propyl carbamate, 1,1-dimethylpropynyl carbamate, di(2-pyridyl)methyl carbamate, 2-furanylmethyl carbamate, 2-iodoethyl carbamate, isoborynl carbamate, isobutyl carbamate, isonicotinyl carbamate, p-(p'-methoxyphenylazo)benzyl carbamate, 1-methylcyclobutyl carbamate, 1-methylcyclohexyl carbamate, 1-methyl-1-cyclopropylmethyl carbamate, 1-methyl-1-(3,5-dimethoxyphenyl)ethyl carbamate, i-methyl-1-(p-phenylazophenyl)ethyl carbamate, 1-methyl-1-phenylethyl carbamate, 1-methyl-1-(4-pyridyl)ethyl carbamate, phenyl carbamate, p-(phenylazo)benzyl carbamate, 2,4,6-tri-t-butylphenyl carbamate, 4-(trimethylammonium)benzyl carbamate, and 2,4,6-trimethylbenzyl carbamate.
[0076] Nitrogen protecting groups such as sulfonamide groups (e.g., --S(.dbd.O).sub.2R.sup.aa) include, but are not limited to, p-toluenesulfonamide (Ts), benzenesulfonamide, 2,3,6,-trimethyl-4-methoxybenzenesulfonamide (Mtr), 2,4,6-trimethoxybenzenesulfonamide (Mtb), 2,6-dimethyl-4-methoxybenzenesulfonamide (Pme), 2,3,5,6-tetramethyl-4-methoxybenzenesulfonamide (Mte), 4-methoxybenzenesulfonamide (Mbs), 2,4,6-trimethylbenzenesulfonamide (Mts), 2,6-dimethoxy-4-methylbenzenesulfonamide (iMds), 2,2,5,7,8-pentamethylchroman-6-sulfonamide (Pmc), methanesulfonamide (Ms), .beta.-trimethylsilylethanesulfonamide (SES), 9-anthracenesulfonamide, 4-(4',8'-dimethoxynaphthylmethyl)benzenesulfonamide (DNMBS), benzylsulfonamide, trifluoromethylsulfonamide, and phenacylsulfonamide.
[0077] Other nitrogen protecting groups include, but are not limited to, phenothiazinyl-(10)-acyl derivative, N'-p-toluenesulfonylaminoacyl derivative, N'-phenylaminothioacyl derivative, N-benzoylphenylalanyl derivative, N-acetylmethionine derivative, 4,5-diphenyl-3-oxazolin-2-one, N-phthalimide, N-dithiasuccinimide (Dts), N-2,3-diphenylmaleimide, N-2,5-dimethylpyrrole, N-1,1,4,4-tetramethyldisilylazacyclopentane adduct (STABASE), 5-substituted 1,3-dimethyl-1,3,5-triazacyclohexan-2-one, 5-substituted 1,3-dibenzyl-1,3,5-triazacyclohexan-2-one, 1-substituted 3,5-dinitro-4-pyridone, N-methylamine, N-allylamine, N-[2-(trimethylsilyl)ethoxy]methylamine (SEM), N-3-acetoxypropylamine, N-(1-isopropyl-4-nitro-2-oxo-3-pyroolin-3-yl)amine, quaternary ammonium salts, N-benzylamine, N-di(4-methoxyphenyl)methylamine, N-5-dibenzosuberylamine, N-triphenylmethylamine (Tr), N-[(4-methoxyphenyl)diphenylmethyl]amine (MMTr), N-9-phenylfluorenylamine (PhF), N-2,7-dichloro-9-fluorenylmethyleneamine, N-ferrocenylmethylamino (Fcm), N-2-picolylamino N-oxide, N-1,1-dimethylthiomethyleneamine, N-benzylideneamine, N-p-methoxybenzylideneamine, N-diphenylmethyleneamine, N-[(2-pyridyl)mesityl]methyleneamine, N--(N',N'-dimethylaminomethylene)amine, N,N'-isopropylidenediamine, N-p-nitrobenzylideneamine, N-salicylideneamine, N-5-chlorosalicylideneamine, N-(5-chloro-2-hydroxyphenyl)phenylmethyleneamine, N-cyclohexylideneamine, N-(5,5-dimethyl-3-oxo-1-cyclohexenyl)amine, N-borane derivative, N-diphenylborinic acid derivative, N-[phenyl(pentaacylchromium- or tungsten)acyl]amine, N-copper chelate, N-zinc chelate, N-nitroamine, N-nitrosoamine, amine N-oxide, diphenylphosphinamide (Dpp), dimethylthiophosphinamide (Mpt), diphenylthiophosphinamide (Ppt), dialkyl phosphoramidates, dibenzyl phosphoramidate, diphenyl phosphoramidate, benzenesulfenamide, o-nitrobenzenesulfenamide (Nps), 2,4-dinitrobenzenesulfenamide, pentachlorobenzenesulfenamide, 2-nitro-4-methoxybenzenesulfenamide, triphenylmethylsulfenamide, and 3-nitropyridinesulfenamide (Npys).
[0078] Exemplary oxygen atom substituents include, but are not limited to, --R.sup.aa, --N(R.sup.bb).sub.2, --C(.dbd.O)SR.sup.aa, --C(.dbd.O)R.sup.aa, --CO.sub.2R.sup.aa, --C(.dbd.O)N(R.sup.bb).sub.22, --C(.dbd.NR.sup.bb)R.sup.aa, --C(.dbd.NR.sup.bb)OR.sup.aa, --C(.dbd.NR.sup.bb)N(R.sup.bb), --S(.dbd.O)R.sup.aa, --SO.sub.2R.sup.aa, --Si(R.sup.aa).sub.3, --P(R.sup.cc).sub.2, --P(R.sup.cc).sub.3.sup.+X.sup.-, --P(OR.sup.cc).sub.2, --P(OR.sup.cc).sub.3.sup.+X.sup.-, --P(.dbd.O)(R.sup.aa).sub.2, --P(.dbd.O)(OR.sup.cc).sub.2, and --P(.dbd.O)(N(R.sup.bb).sub.2).sub.2, wherein X.sup.-, R.sup.aa, R.sup.bb, and R.sup.cc are as defined herein. In certain embodiments, the oxygen atom substituent present on an oxygen atom is an oxygen protecting group (also referred to as a hydroxyl protecting group). Oxygen protecting groups are well known in the art and include those described in detail in Protecting Groups in Organic Synthesis, T. W. Greene and P. G. M. Wuts, 3.sup.rd edition, John Wiley & Sons, 1999, incorporated herein by reference. Exemplary oxygen protecting groups include, but are not limited to, methyl, t-butyloxycarbonyl (BOC or Boc), methoxylmethyl (MOM), methylthiomethyl (MTM), t-butylthiomethyl, (phenyldimethylsilyl)methoxymethyl (SMOM), benzyloxymethyl (BOM), p-methoxybenzyloxymethyl (PMBM), (4-methoxyphenoxy)methyl (p-AOM), guaiacolmethyl (GUM), t-butoxymethyl, 4-pentenyloxymethyl (POM), siloxymethyl, 2-methoxyethoxymethyl (MEM), 2,2,2-trichloroethoxymethyl, bis(2-chloroethoxy)methyl, 2-(trimethylsilyl)ethoxymethyl (SEMOR), tetrahydropyranyl (THP), 3-bromotetrahydropyranyl, tetrahydrothiopyranyl, 1-methoxycyclohexyl, 4-methoxytetrahydropyranyl (MTHP), 4-methoxytetrahydrothiopyranyl, 4-methoxytetrahydrothiopyranyl S,S-dioxide, 1-[(2-chloro-4-methyl)phenyl]-4-methoxypiperidin-4-yl (CTMP), 1,4-dioxan-2-yl, tetrahydrofuranyl, tetrahydrothiofuranyl, 2,3,3a,4,5,6,7,7a-octahydro-7,8,8-trimethyl-4,7-methanobenzofuran-2-yl, 1-ethoxyethyl, 1-(2-chloroethoxy)ethyl, 1-methyl-1-methoxyethyl, 1-methyl-1-benzyloxyethyl, 1-methyl-1-benzyloxy-2-fluoroethyl, 2,2,2-trichloroethyl, 2-trimethylsilylethyl, 2-(phenylselenyl)ethyl, t-butyl, allyl, p-chlorophenyl, p-methoxyphenyl, 2,4-dinitrophenyl, benzyl (Bn), p-methoxybenzyl, 3,4-dimethoxybenzyl, o-nitrobenzyl, p-nitrobenzyl, p-halobenzyl, 2,6-dichlorobenzyl, p-cyanobenzyl, p-phenylbenzyl, 2-picolyl, 4-picolyl, 3-methyl-2-picolyl N-oxido, diphenylmethyl, p,p'-dinitrobenzhydryl, 5-dibenzosuberyl, triphenylmethyl, .alpha.-naphthyldiphenylmethyl, p-methoxyphenyldiphenylmethyl, di(p-methoxyphenyl)phenylmethyl, tri(p-methoxyphenyl)methyl, 4-(4'-bromophenacyloxyphenyl)diphenylmethyl, 4,4',4''-tris(4,5-dichlorophthalimidophenyl)methyl, 4,4',4''-tris(levulinoyloxyphenyl)methyl, 4,4',4''-tris(benzoyloxyphenyl)methyl, 3-(imidazol-1-yl)bis(4',4''-dimethoxyphenyl)methyl, 1,1-bis(4-methoxyphenyl)-1'-pyrenylmethyl, 9-anthryl, 9-(9-phenyl)xanthenyl, 9-(9-phenyl-10-oxo)anthryl, 1,3-benzodisulfuran-2-yl, benzisothiazolyl S,S-dioxido, trimethylsilyl (TMS), triethylsilyl (TES), triisopropylsilyl (TIPS), dimethylisopropylsilyl (IPDMS), diethylisopropylsilyl (DEIPS), dimethylthexylsilyl, I-butyldimethylsilyl (TBDMS), t-butyldiphenylsilyl (TBDPS), tribenzylsilyl, tri-p-xylylsilyl, triphenylsilyl, diphenylmethylsilyl (DPMS), t-butylmethoxyphenylsilyl (TBMPS), formate, benzoylformate, acetate, chloroacetate, dichloroacetate, trichloroacetate, trifluoroacetate, methoxyacetate, triphenylmethoxyacetate, phenoxyacetate, p-chlorophenoxyacetate, 3-phenylpropionate, 4-oxopentanoate (levulinate), 4,4-(ethylenedithio)pentanoate (levulinoyldithioacetal), pivaloate, adamantoate, crotonate, 4-methoxycrotonate, benzoate, p-phenylbenzoate, 2,4,6-trimethylbenzoate (mesitoate), alkyl methyl carbonate, 9-fluorenylmethyl carbonate (Fmoc), alkyl ethyl carbonate, alkyl 2,2,2-trichloroethyl carbonate (Troc), 2-(trimethylsilyl)ethyl carbonate (TMSEC), 2-(phenylsulfonyl) ethyl carbonate (Psec), 2-(triphenylphosphonio) ethyl carbonate (Peoc), alkyl isobutyl carbonate, alkyl vinyl carbonate alkyl allyl carbonate, alkyl p-nitrophenyl carbonate, alkyl benzyl carbonate, alkyl p-methoxybenzyl carbonate, alkyl 3,4-dimethoxybenzyl carbonate, alkyl o-nitrobenzyl carbonate, alkyl p-nitrobenzyl carbonate, alkyl S-benzyl thiocarbonate, 4-ethoxy-1-naphthyl carbonate, methyl dithiocarbonate, 2-iodobenzoate, 4-azidobutyrate, 4-nitro-4-methylpentanoate, o-(dibromomethyl)benzoate, 2-formylbenzenesulfonate, 2-(methylthiomethoxy)ethyl, 4-(methylthiomethoxy)butyrate, 2-(methylthiomethoxymethyl)benzoate, 2,6-dichloro-4-methylphenoxyacetate, 2,6-dichloro-4-(1,1,3,3-tetramethylbutyl)phenoxyacetate, 2,4-bis(1,1-dimethylpropyl)phenoxyacetate, chlorodiphenylacetate, isobutyrate, monosuccinoate, (E)-2-methyl-2-butenoate, o-(methoxyacyl)benzoate, .alpha.-naphthoate, nitrate, alkyl N,N,N',N'-tetramethylphosphorodiamidate, alkyl N-phenylcarbamate, borate, dimethylphosphinothioyl, alkyl 2,4-dinitrophenylsulfenate, sulfate, methanesulfonate (mesylate), benzylsulfonate, and tosylate (Ts).
[0079] Exemplary sulfur atom substituents include, but are not limited to, --R.sup.aa, --N(R.sup.bb).sub.2, --C(.dbd.O)SR.sup.aa, --C(.dbd.O)R.sup.aa, --CO.sub.2R.sup.aa, --C(.dbd.O)N(R.sup.bb).sub.2, --C(.dbd.NR.sup.bb)R.sup.aa, --C(.dbd.NR.sup.bb)OR.sup.aa, --C(.dbd.NR.sup.bb)N(R.sup.bb).sub.2, --S(.dbd.O)R.sup.aa, --SO.sub.2R.sup.aa, --Si(R.sup.aa).sub.3, --P(R.sup.cc).sub.2, --P(R.sup.cc).sub.3.sup.+X.sup.-, --P(OR.sup.cc).sub.2, --P(OR.sup.cc).sub.3.sup.+X.sup.-, --P(.dbd.O)(R.sup.aa).sub.2, --P(.dbd.O)(OR.sup.cc).sub.2, and --P(.dbd.O)(N(R.sup.bb).sub.2).sub.2, wherein R.sup.aa, R.sup.bb, and R.sup.cc are as defined herein. In certain embodiments, the sulfur atom substituent present on a sulfur atom is a sulfur protecting group (also referred to as a thiol protecting group). Sulfur protecting groups are well known in the art and include those described in detail in Protecting Groups in Organic Synthesis, T. W. Greene and P. G. M. Wuts, 3.sup.rd edition, John Wiley & Sons, 1999, incorporated herein by reference.
[0080] A "hydrocarbon chain" refers to a substituted or unsubstituted divalent alkyl, alkenyl, or alkynyl group. A hydrocarbon chain includes (1) one or more chains of carbon atoms immediately between the two radicals of the hydrocarbon chain; (2) optionally one or more hydrogen atoms on the chain(s) of carbon atoms; and (3) optionally one or more substituents ("non-chain substituents," which are not hydrogen) on the chain(s) of carbon atoms. A chain of carbon atoms consists of consecutively connected carbon atoms ("chain atoms" or "carbon units") and does not include hydrogen atoms or heteroatoms. However, a non-chain substituent of a hydrocarbon chain may include any atoms, including hydrogen atoms, carbon atoms, and heteroatoms. For example, hydrocarbon chain --C.sup.AH(C.sup.BH.sub.2C.sup.CH.sub.3)-- includes one chain atom C.sup.A, one hydrogen atom on C.sup.A, and non-chain substituent --(C.sup.BH.sub.2C.sup.CH.sub.3). The term "C.sub.x hydrocarbon chain," wherein x is a positive integer, refers to a hydrocarbon chain that includes x number of chain atom(s) between the two radicals of the hydrocarbon chain. If there is more than one possible value of x, the smallest possible value of x is used for the definition of the hydrocarbon chain. For example, --CH(C.sub.2H.sub.5)-- is a C.sub.1 hydrocarbon chain, and
##STR00006##
is a C.sub.3 hydrocarbon chain. When a range of values is used, the meaning of the range is as described herein. For example, a C.sub.3-10 hydrocarbon chain refers to a hydrocarbon chain where the number of chain atoms of the shortest chain of carbon atoms immediately between the two radicals of the hydrocarbon chain is 3, 4, 5, 6, 7, 8, 9, or 10. A hydrocarbon chain may be saturated (e.g., --(CH.sub.2).sub.4--). A hydrocarbon chain may also be unsaturated and include one or more C.dbd.C and/or C.ident.C bonds anywhere in the hydrocarbon chain. For instance, --CH.dbd.CH--(CH.sub.2).sub.2--, --CH.sub.2--C.ident.C--CH.sub.2--, and --C.ident.C--CH.dbd.CH-- are all examples of a unsubstituted and unsaturated hydrocarbon chain. In certain embodiments, the hydrocarbon chain is unsubstituted (e.g., --C.ident.C-- or --(CH.sub.2).sub.4--). In certain embodiments, the hydrocarbon chain is substituted (e.g., --CH(C.sub.2H.sub.5)-- and --CF.sub.2--). Any two substituents on the hydrocarbon chain may be joined to form an optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl ring. For instance,
##STR00007##
are all examples of a hydrocarbon chain. In contrast, in certain embodiments,
##STR00008##
are not within the scope of the hydrocarbon chains described herein. When a chain atom of a C.sub.x hydrocarbon chain is replaced with a heteroatom, the resulting group is referred to as a C.sub.x hydrocarbon chain wherein a chain atom is replaced with a heteroatom, as opposed to a C.sub.x-1 hydrocarbon chain. For example,
##STR00009##
is a C.sub.3 hydrocarbon chain wherein one chain atom is replaced with an oxygen atom.
[0081] The term "leaving group" is given its ordinary meaning in the art of synthetic organic chemistry and refers to an atom or a group capable of being displaced by a nucleophile. Examples of suitable leaving groups include, but are not limited to, halogen (such as F, Cl, Br, or I (iodine)), alkoxycarbonyloxy, aryloxycarbonyloxy, alkanesulfonyloxy, arenesulfonyloxy, alkyl-carbonyloxy (e.g., acetoxy), arylcarbonyloxy, aryloxy, methoxy, N,O-dimethylhydroxylamino, pixel, and haloformates. In some cases, the leaving group is a sulfonic acid ester, such as toluenesulfonate (tosylate, --OTs), methanesulfonate (mesylate, --OMs), p-bromobenzenesulfonyloxy (brosylate, --OBs), --OS(.dbd.O).sub.2(CF.sub.2).sub.3CF.sub.3 (nonaflate, --ONf), or trifluoromethanesulfonate (triflate, -OTf). In some cases, the leaving group is a brosylate, such as p-bromobenzenesulfonyloxy. In some cases, the leaving group is a nosylate, such as 2-nitrobenzenesulfonyloxy. The leaving group may also be a phosphineoxide (e.g., formed during a Mitsunobu reaction) or an internal leaving group such as an epoxide or cyclic sulfate. Other non-limiting examples of leaving groups are water, ammonia, alcohols, ether moieties, thioether moieties, zinc halides, magnesium moieties, diazonium salts, and copper moieties. Exemplary leaving groups include, but are not limited to, halo (e.g., chloro, bromo, iodo) and activated substituted hydroxyl groups (e.g., --OC(.dbd.O)SR.sup.aa, --OC(.dbd.O)R.sup.aa, --OCO.sub.2R.sup.aa, --OC(.dbd.O)N(R.sup.bb).sub.2, --OC(.dbd.NR.sup.bb)R.sup.aa, --OC(.dbd.NR.sup.bb)OR.sup.aa, --OC(.dbd.NR.sup.bb)N(R.sup.bb).sub.2, --OS(.dbd.O)R.sup.aa, --OSO.sub.2R.sup.aa, --OP(R.sup.cc).sub.2, --OP(R.sup.cc).sub.3, --OP(.dbd.O).sub.2R.sup.aa, --OP(.dbd.O)(R.sup.aa).sub.2, --OP(.dbd.O)(OR.sup.cc).sub.2, --OP(.dbd.O).sub.2N(R.sup.bb).sub.2, and --OP(.dbd.O)(NR.sup.bb).sub.2, wherein R.sup.aa, R.sup.bb, and R.sup.cc are as defined herein).
[0082] The term "pharmaceutically acceptable salt" refers to those salts which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response, and the like, and are commensurate with a reasonable benefit/risk ratio. Pharmaceutically acceptable salts are well known in the art. For example, Berge et al. describe pharmaceutically acceptable salts in detail in J. Pharmaceutical Sciences, 1977, 66, 1-19, incorporated herein by reference. Pharmaceutically acceptable salts of the compounds described herein include those derived from suitable inorganic and organic acids and bases. Examples of pharmaceutically acceptable, non-toxic acid addition salts are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid, and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid, or malonic acid or by using other methods known in the art such as ion exchange. Other pharmaceutically acceptable salts include adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate, undecanoate, valerate salts, and the like. Salts derived from appropriate bases include alkali metal, alkaline earth metal, ammonium and N.sup.+(C.sub.1-4 alkyl).sub.4.sup.- salts. Representative alkali or alkaline earth metal salts include sodium, lithium, potassium, calcium, magnesium, and the like. Further pharmaceutically acceptable salts include, when appropriate, nontoxic ammonium, quaternary ammonium, and amine cations formed using counterions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, lower alkyl sulfonate, and aryl sulfonate.
[0083] The term "solvate" refers to forms of the compound that are associated with a solvent, usually by a solvolysis reaction. This physical association may include hydrogen bonding. Conventional solvents include water, methanol, ethanol, acetic acid, DMSO, THF, diethyl ether, and the like. The compounds described herein may be prepared, e.g., in crystalline form, and may be solvated. Suitable solvates include pharmaceutically acceptable solvates and further include both stoichiometric solvates and non-stoichiometric solvates. In certain instances, the solvate will be capable of isolation, for example, when one or more solvent molecules are incorporated in the crystal lattice of a crystalline solid. "Solvate" encompasses both solution-phase and isolatable solvates. Representative solvates include hydrates, ethanolates, and methanolates.
[0084] The term "hydrate" refers to a compound that is associated with water. Typically, the number of the water molecules contained in a hydrate of a compound is in a definite ratio to the number of the compound molecules in the hydrate. Therefore, a hydrate of a compound may be represented, for example, by the general formula R.x H.sub.2O, wherein R is the compound, and x is a number greater than 0. A given compound may form more than one type of hydrate, including, e.g., monohydrates (x is 1), lower hydrates (x is a number greater than 0 and smaller than 1, e.g., hemihydrates (R.0.5H.sub.2O)), and polyhydrates (x is a number greater than 1, e.g., dihydrates (R.2H.sub.2O) and hexahydrates (R.6H.sub.2O)).
[0085] The term "tautomers" or "tautomeric" refers to two or more interconvertible compounds resulting from at least one formal migration of a hydrogen atom and at least one change in valency (e.g., a single bond to a double bond, a triple bond to a single bond, or vice versa). The exact ratio of the tautomers depends on several factors, including temperature, solvent, and pH. Tautomerizations (i.e., the reaction providing a tautomeric pair) may catalyzed by acid or base. Exemplary tautomerizations include keto-to-enol, amide-to-imide, lactam-to-lactim, enamine-to-imine, and enamine-to-(a different enamine) tautomerizations.
[0086] It is also to be understood that compounds that have the same molecular formula but differ in the nature or sequence of bonding of their atoms or the arrangement of their atoms in space are termed "isomers". Isomers that differ in the arrangement of their atoms in space are termed "stereoisomers."
[0087] Stereoisomers that are not mirror images of one another are termed "diastereomers" and those that are non-superimposable mirror images of each other are termed "enantiomers." When a compound has an asymmetric center, for example, it is bonded to four different groups, a pair of enantiomers is possible. An enantiomer can be characterized by the absolute configuration of its asymmetric center and is described by the R- and S-sequencing rules of Cahn and Prelog, or by the manner in which the molecule rotates the plane of polarized light and designated as dextrorotatory or levorotatory (i.e., as (+) or (-)-isomers respectively). A chiral compound can exist as either individual enantiomer or as a mixture thereof. A mixture containing equal proportions of the enantiomers is called a "racemic mixture."
[0088] The term "polymorphs" refers to a crystalline form of a compound (or a salt, hydrate, or solvate thereof) in a particular crystal packing arrangement. All polymorphs have the same elemental composition. Different crystalline forms usually have different X-ray diffraction patterns, infrared spectra, melting points, density, hardness, crystal shape, optical and electrical properties, stability, and solubility. Recrystallization solvent, rate of crystallization, storage temperature, and other factors may cause one crystal form to dominate. Various polymorphs of a compound can be prepared by crystallization under different conditions.
[0089] The term "co-crystal" refers to a crystalline structure composed of at least two components. In certain embodiments, a co-crystal may contain a compound of the present invention and one or more other component, including but not limited to, atoms, ions, molecules, or solvent molecules. In certain embodiments, a co-crystal may contain a compound of the present invention and one or more components related to said compound, including not limited to, an isomer, tautomer, salt, solvate, hydrate, synthetic precursor, synthetic derivative, fragment or impurity of said compound.
[0090] The term "isotopically labeled derivative" or "isotopically labeled" refers to a compound wherein one or more atoms in the compound (or in an associated ion or molecule of a salt, hydrate, or solvate) has been replaced with an isotope of the same element. For the given element or position in the molecule the isotope will be enriched, or present in a higher percentage of all atoms of the element or of all atoms at the position in the molecule in a sample, relative to an unlabeled variant. In certain embodiments, the enriched isotope will be a stable isotope. In certain embodiments, the enriched isotope will be an unstable or radioactive isotope (e.g., a radionuclide). In certain embodiments, the enriched isotope may be detected by a measurement technique, including but not limited to nuclear magnetic resonance, mass spectrometry, infrared spectroscopy, or a technique that measures radioactive decay.
[0091] The term "prodrugs" refers to compounds that have cleavable groups and become by solvolysis or under physiological conditions the compounds described herein, which are pharmaceutically active in vivo. Such examples include, but are not limited to, choline ester derivatives and the like, N-alkylmorpholine esters and the like. Other derivatives of the compounds described herein have activity in both their acid and acid derivative forms, but in the acid sensitive form often offer advantages of solubility, tissue compatibility, or delayed release in the mammalian organism (see, Bundgard, H., Design of Prodrugs, pp. 7-9, 21-24, Elsevier, Amsterdam 1985). Prodrugs include acid derivatives well known to practitioners of the art, such as, for example, esters prepared by reaction of the parent acid with a suitable alcohol, or amides prepared by reaction of the parent acid compound with a substituted or unsubstituted amine, or acid anhydrides, or mixed anhydrides. Simple aliphatic or aromatic esters, amides, and anhydrides derived from acidic groups pendant on the compounds described herein are particular prodrugs. In some cases it is desirable to prepare double ester type prodrugs such as (acyloxy)alkyl esters or ((alkoxycarbonyl)oxy)alkylesters. C.sub.1-C.sub.8 alkyl, C.sub.2-C.sub.8 alkenyl, C.sub.2-C.sub.8 alkynyl, aryl, C.sub.7-C.sub.12 substituted aryl, and C.sub.7-C.sub.12 arylalkyl esters of the compounds described herein may be preferred.
[0092] The term "inhibition", "inhibiting", "inhibit," or "inhibitor" refer to the ability of a compound to reduce, slow, halt or prevent activity of a particular biological process (e.g., CDK kinase activity)) in a cell relative to vehicle.
[0093] When a compound, pharmaceutical composition, method, use, or kit is referred to as "selectively," "specifically," or "competitively" binding a first protein kinase, the compound, pharmaceutical composition, method, use, or kit binds the first protein kinase (e.g., CDK) with a higher binding affinity (e.g., not less than about 2-fold, not less than about 5-fold, not less than about 10-fold, not less than about 30-fold, not less than about 100-fold, not less than about 1,000-fold, or not less than about 10,000-fold) than binding a second protein or second chromatin that is different from the first protein and the first chromatin.
[0094] When a compound, pharmaceutical composition, method, use, or kit is referred to as "selectively," "specifically," or "competitively" modulating (e.g., increasing or inhibiting) the activity of a first protein kinase, the compound, pharmaceutical composition, method, use, or kit modulates the activity of the first protein kinase (e.g., CDK) to a greater extent (e.g., not less than about 2-fold, not less than about 5-fold, not less than about 10-fold, not less than about 30-fold, not less than about 100-fold, not less than about 1,000-fold, or not less than about 10,000-fold) than the activity of a second protein kinase that is different from the first protein kinase.
[0095] The term "aberrant activity" refers to activity deviating from normal activity, that is, abnormal activity. The term "increased activity" refers to activity higher than normal activity.
[0096] The terms "composition" and "formulation" are used interchangeably.
[0097] A "subject" to which administration is contemplated refers to a human (i.e., male or female of any age group, e.g., pediatric subject (e.g., infant, child, or adolescent) or adult subject (e.g., young adult, middle-aged adult, or senior adult)) or non-human animal. In certain embodiments, the non-human animal is a mammal (e.g., primate (e.g., cynomolgus monkey or rhesus monkey), commercially relevant mammal (e.g., cattle, pig, horse, sheep, goat, cat, or dog), or bird (e.g., commercially relevant bird, such as chicken, duck, goose, or turkey)). In certain embodiments, the non-human animal is a fish, reptile, or amphibian. The non-human animal may be a male or female at any stage of development. The non-human animal may be a transgenic animal or genetically engineered animal. A "patient" refers to a human subject in need of treatment of a disease.
[0098] The term "biological sample" refers to any sample including tissue samples (such as tissue sections and needle biopsies of a tissue); cell samples (e.g., cytological smears (such as Pap or blood smears) or samples of cells obtained by microdissection); samples of whole organisms (such as samples of yeasts or bacteria); or cell fractions, fragments or organelles (such as obtained by lysing cells and separating the components thereof by centrifugation or otherwise). Other examples of biological samples include blood, serum, urine, semen, fecal matter, cerebrospinal fluid, interstitial fluid, mucous, tears, sweat, pus, biopsied tissue (e.g., obtained by a surgical biopsy or needle biopsy), nipple aspirates, milk, vaginal fluid, saliva, swabs (such as buccal swabs), or any material containing biomolecules that is derived from another biological sample.
[0099] The terms "administer," "administering," or "administration" refers to implanting, absorbing, ingesting, injecting, inhaling, or otherwise introducing a compound described herein, or a composition thereof, into, in, or on a subject.
[0100] The terms "treatment," "treat," and "treating" refer to reversing, alleviating, delaying the onset of or inhibiting the progress of a disease described herein. In some embodiments, treatment may be administered after one or more signs or symptoms of the disease have developed or have been observed. In other embodiments, treatment may be administered in the absence of signs or symptoms of the disease. For example, treatment may be administered to a susceptible subject prior to the onset of symptoms (e.g., in light of a history of symptoms and/or in light of exposure to a pathogen). Treatment may also be continued after symptoms have resolved, for example, to delay or prevent recurrence.
[0101] The terms "condition," "disease," and "disorder" are used interchangeably.
[0102] An "effective amount" of a compound described herein refers to an amount sufficient to elicit the desired biological response, i.e., treating the condition. As will be appreciated by those of ordinary skill in this art, the effective amount of a compound described herein may vary depending on such factors as the desired biological endpoint, the pharmacokinetics of the compound, the condition being treated, the mode of administration, and the age and health of the subject. In certain embodiments, an effective amount is a therapeutically effective amount. In certain embodiments, an effective amount is a prophylactic treatment. In certain embodiments, an effective amount is the amount of a compound described herein in a single dose. In certain embodiments, an effective amount is the combined amounts of a compound described herein in multiple doses.
[0103] A "therapeutically effective amount" of a compound described herein is an amount sufficient to provide a therapeutic benefit in the treatment of a condition or to delay or minimize one or more symptoms associated with the condition. A therapeutically effective amount of a compound means an amount of therapeutic agent, alone or in combination with other therapies, which provides a therapeutic benefit in the treatment of the condition. The term "therapeutically effective amount" can encompass an amount that improves overall therapy, reduces or avoids symptoms, signs, or causes of the condition, and/or enhances the therapeutic efficacy of another therapeutic agent.
[0104] A "prophylactically effective amount" of a compound described herein is an amount sufficient to prevent a condition, or one or more symptoms associated with the condition or prevent its recurrence. A prophylactically effective amount of a compound means an amount of a therapeutic agent, alone or in combination with other agents, which provides a prophylactic benefit in the prevention of the condition. The term "prophylactically effective amount" can encompass an amount that improves overall prophylaxis or enhances the prophylactic efficacy of another prophylactic agent.
[0105] A "proliferative disease" refers to a disease that occurs due to abnormal growth or extension by the multiplication of cells (Walker, Cambridge Dictionary of Biology; Cambridge University Press: Cambridge, UK, 1990). A proliferative disease may be associated with: 1) the pathological proliferation of normally quiescent cells; 2) the pathological migration of cells from their normal location (e.g., metastasis of neoplastic cells); 3) the pathological expression of proteolytic enzymes such as the matrix metalloproteinases (e.g., collagenases, gelatinases, and elastases); or 4) the pathological angiogenesis as in proliferative retinopathy and tumor metastasis. Exemplary proliferative diseases include cancers (i.e., "malignant neoplasms"), benign neoplasms, diseases associated with angiogenesis, inflammatory diseases, and autoimmune diseases.
[0106] The term "angiogenesis" refers to the physiological process through which new blood vessels form from pre-existing vessels. Angiogenesis is distinct from vasculogenesis, which is the de novo formation of endothelial cells from mesoderm cell precursors. The first vessels in a developing embryo form through vasculogenesis, after which angiogenesis is responsible for most blood vessel growth during normal or abnormal development. Angiogenesis is a vital process in growth and development, as well as in wound healing and in the formation of granulation tissue. However, angiogenesis is also a fundamental step in the transition of tumors from a benign state to a malignant one, leading to the use of angiogenesis inhibitors in the treatment of cancer. Angiogenesis may be chemically stimulated by angiogenic proteins, such as growth factors (e.g., VEGF). "Pathological angiogenesis" refers to abnormal (e.g., excessive or insufficient) angiogenesis that amounts to and/or is associated with a disease.
[0107] The terms "neoplasm" and "tumor" are used herein interchangeably and refer to an abnormal mass of tissue wherein the growth of the mass surpasses and is not coordinated as in the growth of normal tissue. A neoplasm or tumor may be "benign" or "malignant," depending on the following characteristics: degree of cellular differentiation (including morphology and functionality), rate of growth, local invasion, and metastasis. A "benign neoplasm" is generally well differentiated, has characteristically slower growth than a malignant neoplasm, and remains localized to the site of origin. In addition, a benign neoplasm does not have the capacity to infiltrate, invade, or metastasize to distant sites. Exemplary benign neoplasms include, but are not limited to, lipoma, chondroma, adenomas, acrochordon, senile angiomas, seborrheic keratoses, lentigos, and sebaceous hyperplasias. In some cases, certain "benign" tumors may later give rise to malignant neoplasms, which may result from additional genetic changes in a subpopulation of the tumor's neoplastic cells, and these tumors are referred to as "pre-malignant neoplasms." An exemplary pre-malignant neoplasm is a teratoma. In contrast, a "malignant neoplasm" is generally poorly differentiated (anaplasia) and has characteristically rapid growth accompanied by progressive infiltration, invasion, and destruction of the surrounding tissue. Furthermore, a malignant neoplasm generally has the capacity to metastasize to distant sites. The term "metastasis," "metastatic," or "metastasize" refers to the spread or migration of cancerous cells from a primary or original tumor to another organ or tissue and is typically identifiable by the presence of a "secondary tumor" or "secondary cell mass" of the tissue type of the primary or original tumor and not of that of the organ or tissue in which the secondary (metastatic) tumor is located. For example, a prostate cancer that has migrated to bone is said to be metastasized prostate cancer and includes cancerous prostate cancer cells growing in bone tissue.
[0108] The term "cancer" refers to a class of diseases characterized by the development of abnormal cells that proliferate uncontrollably and have the ability to infiltrate and destroy normal body tissues. See, e.g., Stedman's Medical Dictionary, 25th ed.; Hensyl ed.; Williams & Wilkins: Philadelphia, 1990. Exemplary cancers include, but are not limited to, hematological malignancies. Additional exemplary cancers include, but are not limited to, acoustic neuroma; adenocarcinoma; adrenal gland cancer, anal cancer; angiosarcoma (e.g., lymphangiosarcoma, lymphangioendotheliosarcoma, hemangiosarcoma); appendix cancer; benign monoclonal gammopathy; biliary cancer (e.g., cholangiocarcinoma); bladder cancer; breast cancer (e.g., adenocarcinoma of the breast, papillary carcinoma of the breast, mammary cancer, medullary carcinoma of the breast, triple negative breast cancer (TNBC)); brain cancer (e.g., meningioma, glioblastomas, glioma (e.g., astrocytoma, oligodendroglioma), medulloblastoma); bronchus cancer; carcinoid tumor; cervical cancer (e.g., cervical adenocarcinoma); choriocarcinoma; chordoma; craniopharyngioma; colorectal cancer (e.g., colon cancer, rectal cancer, colorectal adenocarcinoma); connective tissue cancer; epithelial carcinoma; ependymoma; endotheliosarcoma (e.g., Kaposi's sarcoma, multiple idiopathic hemorrhagic sarcoma); endometrial cancer (e.g., uterine cancer, uterine sarcoma); esophageal cancer (e.g., adenocarcinoma of the esophagus, Barrett's adenocarcinoma); Ewing's sarcoma; ocular cancer (e.g., intraocular melanoma, retinoblastoma); familiar hypereosinophilia; gall bladder cancer; gastric cancer (e.g., stomach adenocarcinoma); gastrointestinal stromal tumor (GIST); germ cell cancer; head and neck cancer (e.g., head and neck squamous cell carcinoma, oral cancer (e.g., oral squamous cell carcinoma), throat cancer (e.g., laryngeal cancer, pharyngeal cancer, nasopharyngeal cancer, oropharyngeal cancer)); heavy chain disease (e.g., alpha chain disease, gamma chain disease, mu chain disease; hemangioblastoma; hypopharynx cancer; inflammatory myofibroblastic tumors; immunocytic amyloidosis; kidney cancer (e.g., nephroblastoma a.k.a. Wilms' tumor, renal cell carcinoma); liver cancer (e.g., hepatocellular cancer (HCC), malignant hepatoma); lung cancer (e.g., bronchogenic carcinoma, small cell lung cancer (SCLC), non-small cell lung cancer (NSCLC), adenocarcinoma of the lung); leiomyosarcoma (LMS); mastocytosis (e.g., systemic mastocytosis); muscle cancer; myelodysplastic syndrome (MDS); mesothelioma; myeloproliferative disorder (MPD) (e.g., polycythemia vera (PV), essential thrombocytosis (ET), agnogenic myeloid metaplasia (AMM) a.k.a. myelofibrosis (MF), chronic idiopathic myelofibrosis, chronic myelocytic leukemia (CML), chronic neutrophilic leukemia (CNL), hypereosinophilic syndrome (HES)); neuroblastoma; neurofibroma (e.g., neurofibromatosis (NF) type 1 or type 2, schwannomatosis); neuroendocrine cancer (e.g., gastroenteropancreatic neuroendocrine tumor (GEP-NET), carcinoid tumor); osteosarcoma (e.g., bone cancer); ovarian cancer (e.g., cystadenocarcinoma, ovarian embryonal carcinoma, ovarian adenocarcinoma); papillary adenocarcinoma; pancreatic cancer (e.g., pancreatic andenocarcinoma, intraductal papillary mucinous neoplasm (IPMN), Islet cell tumors); penile cancer (e.g., Paget's disease of the penis and scrotum); pinealoma; primitive neuroectodermal tumor (PNT); plasma cell neoplasia; paraneoplastic syndromes; intraepithelial neoplasms; prostate cancer (e.g., prostate adenocarcinoma); rectal cancer; rhabdomyosarcoma; salivary gland cancer; skin cancer (e.g., squamous cell carcinoma (SCC), keratoacanthoma (KA), melanoma, basal cell carcinoma (BCC)); small bowel cancer (e.g., appendix cancer); soft tissue sarcoma (e.g., malignant fibrous histiocytoma (MFH), liposarcoma, malignant peripheral nerve sheath tumor (MPNST), chondrosarcoma, fibrosarcoma, myxosarcoma); sebaceous gland carcinoma; small intestine cancer; sweat gland carcinoma; synovioma; testicular cancer (e.g., seminoma, testicular embryonal carcinoma); thyroid cancer (e.g., papillary carcinoma of the thyroid, papillary thyroid carcinoma (PTC), medullary thyroid cancer); urethral cancer; vaginal cancer; and vulvar cancer (e.g., Paget's disease of the vulva).
[0109] The term "hematological malignancy" refers to tumors that affect blood, bone marrow, and/or lymph nodes. Exemplary hematological malignancies include, but are not limited to, leukemia, such as acute lymphoblastic leukemia (ALL) (e.g., B-cell ALL, T-cell ALL), acute myelocytic leukemia (AML) (e.g., B-cell AML, T-cell AML), chronic myelocytic leukemia (CML) (e.g., B-cell CML, T-cell CML), and chronic lymphocytic leukemia (CLL) (e.g., B-cell CLL, T-cell CLL)); lymphoma, such as Hodgkin lymphoma (HL) (e.g., B-cell HL, T-cell HL) and non-Hodgkin lymphoma (NHL) (e.g., B-cell NHL, such as diffuse large cell lymphoma (DLCL) (e.g., diffuse large B-cell lymphoma (DLBCL, e.g., activated B-cell (ABC) DLBCL (ABC-DLBCL))), follicular lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), mantle cell lymphoma (MCL), marginal zone B-cell lymphoma (e.g., mucosa-associated lymphoid tissue (MALT) lymphoma, nodal marginal zone B-cell lymphoma, splenic marginal zone B-cell lymphoma), primary mediastinal B-cell lymphoma, Burkitt's lymphoma, Waldenstrom's macroglobulinemia (WM, lymphoplasmacytic lymphoma), hairy cell leukemia (HCL), immunoblastic large cell lymphoma, precursor B-lymphoblastic lymphoma, central nervous system (CNS) lymphoma (e.g., primary CNS lymphoma and secondary CNS lymphoma); and T-cell NHL, such as precursor T-lymphoblastic lymphoma/leukemia, peripheral T-cell lymphoma (PTCL) (e.g., cutaneous T-cell lymphoma (CTCL) (e.g., mycosis fungoides, Sezary syndrome), angioimmunoblastic T-cell lymphoma, extranodal natural killer T-cell lymphoma, enteropathy type T-cell lymphoma, subcutaneous panniculitis-like T-cell lymphoma, and anaplastic large cell lymphoma); lymphoma of an immune privileged site (e.g., cerebral lymphoma, ocular lymphoma, lymphoma of the placenta, lymphoma of the fetus, testicular lymphoma); a mixture of one or more leukemia/lymphoma as described above; myelodysplasia; and multiple myeloma (MM).
[0110] The term "inflammatory disease" refers to a disease caused by, resulting from, or resulting in inflammation. The term "inflammatory disease" may also refer to a dysregulated inflammatory reaction that causes an exaggerated response by macrophages, granulocytes, and/or T-lymphocytes leading to abnormal tissue damage and/or cell death. An inflammatory disease can be either an acute or chronic inflammatory condition and can result from infections or non-infectious causes. Inflammatory diseases include, without limitation, atherosclerosis, arteriosclerosis, autoimmune disorders, multiple sclerosis, systemic lupus erythematosus, polymyalgia rheumatica (PMR), gouty arthritis, degenerative arthritis, tendonitis, bursitis, psoriasis, cystic fibrosis, arthrosteitis, rheumatoid arthritis, inflammatory arthritis, Sjogren's syndrome, giant cell arteritis, progressive systemic sclerosis (scleroderma), ankylosing spondylitis, polymyositis, dermatomyositis, pemphigus, pemphigoid, diabetes (e.g., Type I), myasthenia gravis, Hashimoto's thyroiditis, Graves' disease, Goodpasture's disease, mixed connective tissue disease, sclerosing cholangitis, inflammatory bowel disease, Crohn's disease, ulcerative colitis, pernicious anemia, inflammatory dermatoses, usual interstitial pneumonitis (UIP), asbestosis, silicosis, bronchiectasis, berylliosis, talcosis, pneumoconiosis, sarcoidosis, desquamative interstitial pneumonia, lymphoid interstitial pneumonia, giant cell interstitial pneumonia, cellular interstitial pneumonia, extrinsic allergic alveolitis, Wegener's granulomatosis and related forms of angiitis (temporal arteritis and polyarteritis nodosa), inflammatory dermatoses, hepatitis, delayed-type hypersensitivity reactions (e.g., poison ivy dermatitis), pneumonia, respiratory tract inflammation, Adult Respiratory Distress Syndrome (ARDS), encephalitis, immediate hypersensitivity reactions, asthma, hay fever, allergies, acute anaphylaxis, rheumatic fever, glomerulonephritis, pyelonephritis, cellulitis, cystitis, chronic cholecystitis, ischemia (ischemic injury), reperfusion injury, allograft rejection, host-versus-graft rejection, appendicitis, arteritis, blepharitis, bronchiolitis, bronchitis, cervicitis, cholangitis, chorioamnionitis, conjunctivitis, dacryoadenitis, dermatomyositis, endocarditis, endometritis, enteritis, enterocolitis, epicondylitis, epididymitis, fasciitis, fibrositis, gastritis, gastroenteritis, gingivitis, ileitis, iritis, laryngitis, myelitis, myocarditis, nephritis, omphalitis, oophoritis, orchitis, osteitis, otitis, pancreatitis, parotitis, pericarditis, pharyngitis, pleuritis, phlebitis, pneumonitis, proctitis, prostatitis, rhinitis, salpingitis, sinusitis, stomatitis, synovitis, testitis, tonsillitis, urethritis, urocystitis, uveitis, vaginitis, vasculitis, vulvitis, vulvovaginitis, angitis, chronic bronchitis, osteomyelitis, optic neuritis, temporal arteritis, transverse myelitis, necrotizing fasciitis, and necrotizing enterocolitis.
[0111] An "autoimmune disease" refers to a disease arising from an inappropriate immune response of the body of a subject against substances and tissues normally present in the body. In other words, the immune system mistakes some part of the body as a pathogen and attacks its own cells. This may be restricted to certain organs (e.g., in autoimmune thyroiditis) or involve a particular tissue in different places (e.g., Goodpasture's disease which may affect the basement membrane in both the lung and kidney). The treatment of autoimmune diseases is typically with immunosuppression, e.g., medications which decrease the immune response. Exemplary autoimmune diseases include, but are not limited to, glomerulonephritis, Goodpasture's syndrome, necrotizing vasculitis, lymphadenitis, peri-arteritis nodosa, systemic lupus erythematosis, rheumatoid arthritis, psoriatic arthritis, systemic lupus erythematosis, psoriasis, ulcerative colitis, systemic sclerosis, dermatomyositis/polymyositis, anti-phospholipid antibody syndrome, scleroderma, pemphigus vulgaris, ANCA-associated vasculitis (e.g., Wegener's granulomatosis, microscopic polyangiitis), uveitis, Sjogren's syndrome, Crohn's disease, Reiter's syndrome, ankylosing spondylitis, Lyme disease, Guillain-Barre syndrome, Hashimoto's thyroiditis, and cardiomyopathy.
[0112] The term "kinase" is a class of enzyme that transfers a phosphate group from a high energy donor molecules, such as ATP, to a specific substrate, referred to as phosphorylation. Kinases are part of the larger family of phosphotransferases. One of the largest groups of kinases are protein kinases, which act on and modify the activity of specific proteins. Kinases are used extensively to transmit signals and control complex processes in cells. Various other kinases act on small molecules such as lipids, carbohydrates, amino acids, and nucleotides, either for signaling or to prime them for metabolic pathways. Kinases are often named after their substrates. More than 500 different protein kinases have been identified in humans. Exemplary human protein kinases include, but are not limited to, AAK1, ABL, ACK, ACTR2, ACTR2B, AKT1, AKT2, AKT3, ALK, ALK1, ALK2, ALK4, ALK7, AMPKa1, AMPKa2, ANKRD3, ANPa, ANPb, ARAF, ARAFps, ARG, AurA, AurAps1, AurAps2, AurB, AurBps1, AurC, AXL, BARK1, BARK2, BIKE, BLK, BMPR1A, BMPR1Aps1, BMPR1Aps2, BMPR1B, BMPR2, BMX, BRAF, BRAFps, BRK, BRSK1, BRSK2, BTK, BUB1, BUBR1, CaMK1a, CaMK1b, CaMK1d, CaMK1g, CaMK2a, CaMK2b, CaMK2d, CaMK2g, CaMK4, CaMKK1, CaMKK2, caMLCK, CASK, CCK4, CCRK, CDC2, CDC7, CDK10, CDK11, CDK2, CDK3, CDK4, CDK4ps, CDK5, CDK5ps, CDK6, CDK7, CDK7ps, CDK8, CDK8ps, CDK9, CDKL1, CDKL2, CDKL3, CDKL4, CDKL5, CGDps, CHED, CHK1, CHK2, CHK2ps1, CHK2ps2, CK1a, CK1a2, CK1aps1, CK1aps2, CK1aps3, CK1d, CK1e, CK1g1, CK1g2, CK1g2ps, CK1g3, CK2a1, CK2a1-rs, CK2a2, CLIK1, CLIK1L, CLK1, CLK2, CLK2ps, CLK3, CLK3ps, CLK4, COT, CRIK, CRK7, CSK, CTK, CYGD, CYGF, DAPK1, DAPK2, DAPK3, DCAMKL1, DCAMKL2, DCAMKL3, DDR1, DDR2, DLK, DMPK1, DMPK2, DRAK1, DRAK2, DYRK1A, DYRK1B, DYRK2, DYRK3, DYRK4, EGFR, EphA1, EphA10, EphA2, EphA3, EphA4, EphA5, EphA6, EphA7, EphA8, EphB1, EphB2, EphB3, EphB4, EphB6, Erk1, Erk2, Erk3, Erk3ps1, Erk3ps2, Erk3ps3, Erk3ps4, Erk4, Erk5, Erk7, FAK, FER, FERps, FES, FGFR1, FGFR2, FGFR3, FGFR4, FGR, FLT1, FLT1ps, FLT3, FLT4, FMS, FRK, Fused, FYN, GAK, GCK, GCN2, GCN22, GPRK4, GPRK5, GPRK6, GPRK6ps, GPRK7, GSK3A, GSK3B, Haspin, HCK, HER2/ErbB2, HER3/ErbB3, HER4/ErbB4, HH498, HIPK1, HIPK2, HIPK3, HIPK4, HPK1, HRI, HRIps, HSER, HUNK, ICK, IGF1R, IKKa, IKKb, IKKe, ILK, INSR, IRAK1, IRAK2, IRAK3, IRAK4, IRE1, IRE2, IRR, ITK, JAK1, JAK2, JAK3, JNK1, JNK2, JNK3, KDR, KHS1, KHS2, KIS, KIT, KSGCps, KSR1, KSR2, LATS1, LATS2, LCK, LIMK1, LIMK2, LIMK2ps, LKB1, LMR1, LMR2, LMR3, LOK, LRRK1, LRRK2, LTK, LYN, LZK, MAK, MAP2K1, MAP2K1ps, MAP2K2, MAP2K2ps, MAP2K3, MAP2K4, MAP2K5, MAP2K6, MAP2K7, MAP3K1, MAP3K2, MAP3K3, MAP3K4, MAP3K5, MAP3K6, MAP3K7, MAP3K8, MAPKAPK2, MAPKAPK3, MAPKAPK5, MAPKAPKps1, MARK1, MARK2, MARK3, MARK4, MARKps01, MARKps02, MARKps03, MARKps04, MARKps05, MARKps07, MARKps08, MARKps09, MARKps10, MARKps11, MARKps12, MARKps13, MARKps15, MARKps16, MARKps17, MARKps18, MARKps19, MARKps20, MARKps21, MARKps22, MARKps23, MARKps24, MARKps25, MARKps26, MARKps27, MARKps28, MARKps29, MARKps30, MAST1, MAST2, MAST3, MAST4, MASTL, MELK, MER, MET, MISR2, MLK1, MLK2, MLK3, MLK4, MLKL, MNK1, MNK1ps, MNK2, MOK, MOS, MPSK1, MPSK1ps, MRCKa, MRCKb, MRCKps, MSK1, MSK12, MSK2, MSK22, MSSK1, MST1, MST2, MST3, MST3ps, MST4, MUSK, MYO3A, MYO3B, MYT1, NDR1, NDR2, NEK1, NEK10, NEK11, NEK2, NEK2ps1, NEK2ps2, NEK2ps3, NEK3, NEK4, NEK4ps, NEK5, NEK6, NEK7, NEK8, NEK9, NIK, NIM1, NLK, NRBP1, NRBP2, NuaK1, NuaK2, Obscn, Obscn2, OSR1, p38a, p38b, p38d, p38g, p70S6K, p70S6Kb, p70S6Kps1, p70S6Kps2, PAK1, PAK2, PAK2ps, PAK3, PAK4, PAK5, PAK6, PASK, PBK, PCTAIRE1, PCTAIRE2, PCTAIRE3, PDGFRa, PDGFRb, PDK1, PEK, PFTAIRE1, PFTAIRE2, PHKg1, PHKg1ps1, PHKg1ps2, PHKg1ps3, PHKg2, PIK3R4, PIM1, PIM2, PIM3, PINK1, PITSLRE, PKACa, PKACb, PKACg, PKCa, PKCb, PKCd, PKCe, PKCg, PKCh, PKCi, PKCips, PKCt, PKCz, PKD1, PKD2, PKD3, PKG1, PKG2, PKN1, PKN2, PKN3, PKR, PLK1, PLK1ps1, PLK1ps2, PLK2, PLK3, PLK4, PRKX, PRKXps, PRKY, PRP4, PRP4ps, PRPK, PSKH1, PSKH1ps, PSKH2, PYK2, QIK, QSK, RAF1, RAF1ps, RET, RHOK, RIPK1, RIPK2, RIPK3, RNAseL, ROCK1, ROCK2, RON, ROR1, ROR2, ROS, RSK1, RSK12, RSK2, RSK22, RSK3, RSK32, RSK4, RSK42, RSKL1, RSKL2, RYK, RYKps, SAKps, SBK, SCYL1, SCYL2, SCYL2ps, SCYL3, SGK, SgK050ps, SgK069, SgK071, SgK085, SgK110, SgK196, SGK2, SgK223, SgK269, SgK288, SGK3, SgK307, SgK384ps, SgK396, SgK424, SgK493, SgK494, SgK495, SgK496, SIK (e.g., SIK1, SIK2), skMLCK, SLK, Slob, smMLCK, SNRK, SPEG, SPEG2, SRC, SRM, SRPK1, SRPK2, SRPK2ps, SSTK, STK33, STK33ps, STLK3, STLK5, STLK6, STLK6ps1, STLK6-rs, SuRTK106, SYK, TAK1, TAO1, TAO2, TAO3, TBCK, TBK1, TEC, TESK1, TESK2, TGFbR1, TGFbR2, TIE1, TIE2, TLK1, TLK1ps, TLK2, TLK2ps1, TLK2ps2, TNK1, Trad, Trb1, Trb2, Trb3, Trio, TRKA, TRKB, TRKC, TSSK1, TSSK2, TSSK3, TSSK4, TSSKps1, TSSKps2, TTBK1, TTBK2, TTK, TTN, TXK, TYK2, TYK22, TYRO3, TYRO3ps, ULK1, ULK2, ULK3, ULK4, VACAMKL, VRK1, VRK2, VRK3, VRK3ps, Wee1, Wee1B, Wee1Bps, Wee1ps1, Wee1ps2, Wnk1, Wnk2, Wnk3, Wnk4, YANK1, YANK2, YANK3, YES, YESps, YSK1, ZAK, ZAP70, ZC1/HGK, ZC2/TNIK, ZC3/MINK, and ZC4/NRK.
[0113] The term "SRC family kinase" refers to a family of non-receptor tyrosine protein kinases that includes nine members: SRCA subfamily that includes c-SRC (proto-oncogene tyrosine-protein kinase SRC), YES (proto-oncogene tyrosine-protein kinase Yes), FYN (proto-oncogene tyrosine-protein kinase FYN), and FGR (Gardner-Rasheed feline sarcoma viral (v-FGR) oncogene homolog); SRCB subfamily that includes LCK (lymphocyte-specific protein tyrosine kinase), HCK (tyrosine-protein kinase HCK, hemopoietic cell kinase), BLK (tyrosine-protein kinase BLK), and LYN (tyrosine-protein kinase LYN); and FRK (Fyn-related kinase).
[0114] The term "CDK" refers to a cyclin-dependent kinase. A CDK binds a cyclin (e.g., Cyclin H), which is a regulatory protein. CDKs phosphorylate their substrates at serines and threonines. The consensus sequence for the phosphorylation site in the amino acid sequence of a CDK substrate is [S/T*]PX[K/R], where S/T* is the phosphorylated serine or threonine, P is proline, X is any amino acid, K is lysine, and R is arginine. CDKs include CDK1, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8, CDK9, CDK10, CDK11, CDK12, CDK13, CDK14, CDK15, CDK16, CDK17, CDK18, CDK19. and CDK20.
[0115] CDK7, cyclin-dependent kinase 7, is a CDK, wherein the substrate is Cyclin H, MAT1 (e.g., MNAT1), or Cyclin H and MAT1. CDK7 is alternatively referred to as CAK1, HCAK, MO15, STK1, CDKN7, and p39MO15. Non-limiting examples of the nucleotide and protein sequences for human CDK7 are described in GenBank Accession Number: NP_001790, incorporated herein by reference. The amino acid sequence of this CDK7 is as follows:
TABLE-US-00001 (SEQ ID NO: 1) MALDVKSRAKRYEKLDFLGEGQFATVYKARDKNTNQIVAIKKIKLGHRSE AKDGINRTALREIKLLQELSHPNIIGLLDAFGHKSNISLVEDFMETDLEV IIKDNSLVLTPSHIKAYMLMTLQGLEYLHQHWILHRDLKPNNLLLDENGV LKLADFGLAKSFGSPNRAYTHQVVTRWYRAPELLFGARMYGVGVDMWAVG CILAELLLRVPFLPGDSDLDQLTRIFETLGTPTEEQWPDMCSLPDYVTFK SFPGIPLHHIFSAAGDDLLDLIQGLFLFNPCARITATQALKMKYFSNRPG PTPGCQLPRPNCPVETLKEQSNPALAIKRKRTEALEQGGLPKKLIF
[0116] CDK12, cyclin-dependent kinase 12, is a CDK, wherein the substrate is Cyclin K or flavopiridol. CDK12 is alternatively referred to as Cdc2-related kinase, CDC2-related protein kinase 7, Cell division cycle 2-related protein kinase 7, Cell division protein kinase 12, CRK7, CRKR, CRKRS, cyclin-dependent kinase 12, or KIAA0904. Non-limiting examples of the nucleotide and protein sequences for human CDK12 are described in Uniprot Number: Q9NYV4, which is incorporated herein by reference. The amino acid sequence of this CDK12 is as follows:
TABLE-US-00002 (SEQ ID NO 2) MPNSERHGGKKDGSGGASGTLQPSSGGGSSNSRERHRLVSKHKRHKSKHS KDMGLVTPEAASLGTVIKPLVEYDDISSDSDTFSDDMAFKLDRRENDERR GSDRSDRLHKHRHHQHRRSRDLLKAKQTEKEKSQEVSSKSGSMKDRISGS SKRSNEETDDYGKAQVAKSSSKESRSSKLHKEKTRKERELKSGHKDRSKS HRKRETPKSYKTVDSPKRRSRSPHRKWSDSSKQDDSPSGASYGQDYDLSP SRSHTSSNYDSYKKSPGSTSRRQSVSPPYKEPSAYQSSTRSPSPYSKRQR SVSPYSRRRSSSYERSGSYSGRSPSPYGRRRSSSPFLSKRSLSRSPLPSR KSMKSRSRSPAYSRHSSSHSKKKRSSSRSRHSSISPVRLPLNSSLGAELS RKKKERAAAAAAAKMDGKESKGSPVFLPRKENSSVEAKDSGLESKKLPRS VKLEKSAPDTELVNVTHLNTEVKNSSDTGKVKLDENSEKHLVKDLKAQGT RDSKPIALKEEIVTPKETETSEKETPPPLPTIASPPPPLPTTTPPPQTPP LPPLPPIPALPQQPPLPPSQPAFSQVPASSTSTLPPSTHSKTSAVSSQAN SQPPVQVSVKTQVSVTAAIPHLKTSTLPPLPLPPLLPGDDDMDSPKETLP SKPVKKEKEQRTRHLLTDLPLPPELPGGDLSPPDSPEPKAITPPQQPYKK RPKICCPRYGERRQTESDWGKRCVDKFDIIGIIGEGTYGQVYKAKDKDTG ELVALKKVRLDNEKEGFPITAIREIKILRQLIHRSVVNMKEIVTDKQDAL DFKKDKGAFYLVFEYMDHDLMGLLESGLVHFSEDHIKSFMKQLMEGLEYC HKKNFLHRDIKCSNILLNNSGQIKLADEGLARLYNSEESRPYTNKVITLW YRPPELLLGEERYTPAIDVWSCGCILGELFTKKPIFQANLELAQLELISR LCGSPCPAVWPDVIKLPYFNTMKPKKQTRRRLREEFSFIPSAALDLLDHM LTLDPSKRCTAEQTLQSDFLKDVELSKMAPPDLPHWQDCHELWSKKRRRQ RQSGVVVEEPPPSKTSPKETTSGTSTEPVKNSSPAPPQPAPGKVESGAGD AIGLADITQQLNQSELAVLLNLLQSQTDLSIPQMAQLLNIHSNPEMQQQL EALNQSISALTEATSQQQDSETMAPEESLKEAPSAPVILPSAEQTTLEAS STPADMQNILAVLLSQLMKTQEPAGSLEENNSDKNSGPQGPRRTPTMPQE EAAACPPHILPPEKRPPEPPGPPPPPPPPPLVEGDLSSAPQELNPAVTAA LLQLLSQPEAEPPGHLPHEHQALRPMEYSTRPRPNRTYGNTDGPETGESA IDTDERNSGPALTESLVQTLVKNRTFSGSLSHLGESSSYQGTGSVQFPGD QDLRFARVPLALHPVVGQPFLKAEGSSNSVVHAETKLQNYGELGPGTTGA SSSGAGLHWGGPTQSSAYGKLYRGPTRVPPRGGRGRGVPY
[0117] CDK13, cyclin-dependent kinase 13, is a CDK, wherein the relevant cyclin is cyclin K and a reference inhibitor is the pan-CDK inhibitor flavopiridol and the c-terminal domain (CTD) of RNA-polymerase II is a physiological substrate. CDK13 is alternatively referred to as CHED; CDC2L; CDC2L5; or hCDK13. Non-limiting examples of the nucleotide and protein sequences for human CDK12 are described in GenBank Accession Number M80629, which is incorporated herein by reference. The amino acid sequence of this CDK13 is as follows:
TABLE-US-00003 (SEQ ID NO: 3) MPSSSDTALGGGGGLSWAEKKLEERRKRRRFLSPQQPPLLLPLLQPQLLQ PPPPPPPLLFLAAPGTAAAAAAAAAASSSCFSPGPPLEVKRLARGKRRAG GRQKRRRGPRAGQEAEKRRVFSLPQPQQDGGGGASSGGGVTPLVEYEDVS SQSEQGLLLGGASAATAATAAGGTGGSGGSPASSSGTQRRGEGSERRPRR DRRSSSGRSKERHREHRRRDGQRGGSEASKSRSRESHSGEERAEVAKSGS SSSSGGRRKSASATSSSSSSRKDRDSKAHRSRTKSSKEPPSAYKEPPKAY REDKTEPKAYRRRRSLSPLGGRDDSPVSHRASQSLRSRKSPSPAGGGSSP YSRRLPRSPSPYSRRRSPSYSRHSSYERGGDVSPSPYSSSSWRRSRSPYS PVLRRSGKSRSRSPYSSRHSRSRSRHRLSRSRSRHSSISPSTLTLKSSLA AELNKNKKARAAEAARAAEAAKAAEATKAAEAAAKAAKASNTSTPTKGNT ETSASASQTNHVKDVKKIKIEHAPSPSSGGTLKNDKAKTKPPLQVTKVEN NLIVDKATKYAVIVGKESKSAATKEESVSLKEKTKPLTPSIGAKEKEQHV ALVTSTLPPLPLPPMLPEDKEADSLRGNISVKAVKKEVEKKLRCLLADLP LPPELPGGDDLSKSPEEKKTATQLHSKRRPKICGPPYGETKEKDIDWGKR CVDKFDISGIIGEGTYGQVYKARDKDTGEMVALKKVRLDNEKEGFPITAI REIKILRQLTHQSIINMKEIVTDKEDALDFKKDKGAFYLVFEYMDHDLMG LLESGLVHFNENHIKSFMRQLMEGLDYCHKKNFLHRDIKCSNILLNNRGQ IKLADFGLARLYSSEESRPYTNKVITLWYRPPELLLGEERYTPAIDVWSC GCILGELFTKKPIFQANQELAQLELISRICGSPCPAVWPDVIKLPYFNTM KPKKQYRRKLREEFVFIPAAALDLFDYMLALDPSKRCTAEQALQCEFLRD VEPSKMPPPDLPLWQDCHELWSKKRRRQKQMGMTDDVSTIKAPRKDLSLG LDDSRTNTPQGVLPSSQLKSQGSSNVAPVKTGPGQHLNHSELAILLNLLQ SKTSVNMADFVQVLNIKVNSETQQQLNKINLPAGILATGEKQTDPSTPQQ ESSKPLGGIQPSSQTIQPKVETDAAQAAVQSAFAVLLTQLIKAQQSKQKD VLLEERENGSGHEASLQLRPPPEPSTPVSGQDDLIQHQDMRILELTPEPD RPRILPPDQRPPEPPEPPPVTEEDLDYRTENQHVPTTSSSLTDPHAGVKA ALLQLLAQHQPQDDPKREGGIDYQAGDTYVSTSDYKDNFGSSSFSSAPYV SNDGLGSSSAPPLERRSFIGNSDIQSLDNYSTASSHSGGPPQPSAFSESF PSSVAGYGDIYLNAGPMLFSGDKDHRFEYSHGPIAVLANSSDPSTGPEST HPLPAKMHNYNYGGNLQENPSGPSLMHGQTWTSPAQGPGYSQGYRGHIST STGRGRGRGLPY
DETAILED DESCRIPTION OF CERTAIN EMBODIMENTS OF THE INVENTION
[0118] Cyclin dependent kinases (CDKs) are key regulators of the cell cycle. Their successive activation and inactivation drives the cycle forward. The activity of CDKs is regulated by multiple mechanisms such as positive and negative phosphorylation, binding of regulatory proteins like cyclins, and CDK inhibitors. CDK7 plays a critical role in the regulation of RNA polymerase II-mediated transcription of protein-encoding genes. Disruption of CDK7, CDK12, and/or CDK13 signaling causes defects in transcription. However, a complete understanding of how these disruptions affect global transcription is lacking. Furthermore, the absence of selective inhibitors of CDK7, CDK12, and CDK13 has hindered investigation of the transcriptional and functional consequences of acute and long-term inhibition of these kinases under normal and pathological conditions. The present invention provides selective CDK7, CDK12, and/or CDK13 inhibitors, which covalently modify a cysteine residue located outside of the canonical kinase domain (i.e., Cys312 of CDK7, Cys1039 of CDK12, and Cys1017 of CDK13). Selective covalent inhibitors of these kinases may be useful in the treatment of various proliferative diseases including cancer.
[0119] The present invention provides compounds, which inhibit the activity of a kinase, for the prevention and/or treatment of a subject with a proliferative disease. In certain embodiments, the inventive compounds inhibit the activity of a cyclin-dependent kinase (CDK). In certain embodiments, the inventive compounds inhibit the activity of a cyclin-dependent kinase 7 (CDK7). In certain embodiments, the inventive compounds inhibit the activity of a cyclin-dependent kinase 12 (CDK12). In certain embodiments, the inventive compounds inhibit the activity of a cyclin-dependent kinase 13 (CDK13). The present invention also provides methods of using the compounds described herein, e.g., as biological probes to study the inhibition of the activity of a kinase (e.g., CDK (e.g. CDK7, CDK12, and/or CDK13)), and as therapeutics, e.g., in the prevention and/or treatment of diseases associated with the overexpression and/or aberrant activity of a kinase (e.g., CDK (e.g. CDK7, CDK12, and/or CDK13)). In certain embodiments, the diseases are proliferative diseases. The proliferative diseases that may be treated and/or prevented include, but are not limited to, cancers (e.g., breast cancer, leukemia, melanoma, multiple myeloma), benign neoplasms, diseases associated with angiogenesis, inflammatory diseases, autoinflammatory diseases, and autoimmune diseases. Also provided by the present disclosure are pharmaceutical compositions, kits, methods, and uses including a compound of Formulae (I'), (II'), (I), and (II) as described herein.
Compounds
[0120] Aspects of the present disclosure relate to the compounds described herein. The compounds described herein may be useful in treating and/or preventing proliferative diseases in a subject, inhibiting the activity of a protein kinase (e.g., CDK) in a subject or biological sample, and inducing apoptosis of a cell. In certain embodiments, a compound described herein is a compound of any one of Formulae (I'), (II'), (I), (II), or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof. In certain embodiments, a compound described herein is a compound of Formula (I'), or a pharmaceutically acceptable salt thereof. In certain embodiments, a compound described herein is a compound of Formula (I), or a pharmaceutically acceptable salt thereof. In certain embodiments, a compound described herein is a compound of Formula (II'), or a pharmaceutically acceptable salt thereof. In certain embodiments, a compound described herein is a compound of Formula (II), or a pharmaceutically acceptable salt thereof.
[0121] In certain embodiments, a compound described herein is of Formula (I'):
##STR00010##
or a pharmaceutically acceptable salt thereof, wherein:
[0122] R.sup.1 is hydrogen, halogen, or optionally substituted alkyl;
[0123] M is O, S, or NR.sup.M;
[0124] R.sup.M is hydrogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted acyl, or a nitrogen protecting group;
[0125] Ring A is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl;
[0126] Ring B is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl;
[0127] Ring C is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted monocyclic or bicyclic aryl, or optionally substituted monocyclic or bicyclic heteroaryl;
[0128] each instance of R.sup.A, R.sup.B, and R.sup.C is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --OR.sup.a, --N(R.sup.a).sub.2, --SR.sup.a, --CN, --SCN, --NO.sub.2, --N.sub.3, or optionally substituted acyl;
[0129] each instance of R.sup.a is independently hydrogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted acyl, a nitrogen protecting group when attached to a nitrogen atom, an oxygen protecting group when attached to an oxygen atom, or a sulfur protecting group when attached to a sulfur atom, or two instances of R.sup.a are joined to form a substituted or unsubstituted, heterocyclic ring, or substituted or unsubstituted, heteroaryl ring;
[0130] each of a1, b1, and c1 is independently 0, 1, 2, 3, 4, 5, or 6, as valency permits;
[0131] L.sup.1 is --CH.sub.2--, .sup.lc--S(.dbd.O).sub.2--.sup.lb, --O--, --S--, --NR.sup.L1--, --C(.dbd.O)--, .sup.lc--NR.sup.L1C(.dbd.O)--.sup.lb, .sup.lc--C(.dbd.O)NR.sup.L1--.sup.lb, .sup.lc--OC(.dbd.O)--.sup.lb, or .sup.lc--C(.dbd.O)O--.sup.lb; wherein .sup.lb indicates the point of attachment is to Ring B; and .sup.lc indicates the point of attachment is to Ring C;
[0132] L.sup.2 is --O--, --S--, --NR.sup.L2--, .sup.lb--NR.sup.L2C(.dbd.O)--.sup.lm, .sup.lb--C(.dbd.O)NR.sup.L2--.sup.lm; wherein .sup.lb indicates the point of attachment is to Ring B; and .sup.lm indicates the point of attachment is to the heteroaryl ring with M;
[0133] X is .sup.xm--CH.sub.2CH.sub.2--.sup.xa, .sup.xm--CH.dbd.CH--.sup.xa, .sup.xm--CH.sub.2--NR.sup.LXxa, .sup.xm--CH.sub.2--O--CH.sub.2--.sup.xa, .sup.xm--CH.sub.2--NR.sup.LX--CH.sub.2--.sup.xa, --O--, --S--, --NR.sup.LX--, .sup.xm--O--CH.sub.2--.sup.xa, .sup.xm--S--CH.sub.2--.sup.xa, .sup.xm--S--C(.dbd.O)CH.sub.2--.sup.xa, or .sup.xm--NR.sup.LX--CH.sub.2--.sup.xa; wherein .sup.xa indicates the point of attachment is to Ring A; and .sup.xm indicates the point of attachment is to the heteroaryl ring with M;
[0134] each of R.sup.L1, R.sup.L2, and R.sup.LX is independently hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group;
[0135] R.sup.2 is any of Formulae (i-1)-(i-41):
##STR00011## ##STR00012## ##STR00013## ##STR00014## ##STR00015##
wherein: [0136] L.sup.3 is a bond or an optionally substituted C.sub.14 hydrocarbon chain, optionally wherein one or more carbon units of the hydrocarbon chain are independently replaced with --C.dbd.O--, --O--, --S--, --NR.sup.L3a--, --NR.sup.L3aC(.dbd.O)--, --C(.dbd.O)NR.sup.L3a--, --SC(.dbd.O)--, --C(.dbd.O)S--, --OC(.dbd.O)--, --C(.dbd.O)O--, --NR.sup.L3aC(.dbd.S)--, --C(.dbd.S)NR.sup.L3a--, trans-CR.sup.L3b.dbd.CR.sup.L3b--, cis-CR.sup.L3b.dbd.CR.sup.L3b--, --C.dbd.C--, --S(.dbd.O)--, --S(.dbd.O)O--, --OS(.dbd.O)--, --S(.dbd.O)NR.sup.L3a--, --NR.sup.L3aS(.dbd.O)--, --S(.dbd.O).sub.2--, --S(.dbd.O).sub.2O--, --OS(.dbd.O).sub.2--, --S(.dbd.O).sub.2NR.sup.L3a--, or --NR.sup.L3aS(.dbd.O).sub.2--, wherein R.sup.L3a is hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group, and wherein each occurrence of R.sup.L3b is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl, or two R.sup.L3b groups are joined to form an optionally substituted carbocyclic or optionally substituted heterocyclic ring; [0137] L.sup.4 is a bond or an optionally substituted, branched or unbranched C.sub.1-6 hydrocarbon chain; [0138] each of R.sup.E1, R.sup.E2, and R.sup.E3 is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --CN, --CH.sub.2OR.sup.EE, --CH.sub.2N(R.sup.EE).sub.2, --CH.sub.2SR.sup.EE, --OR.sup.EE, --N(R.sup.EE).sub.2, --Si(R.sup.EE).sub.3, and --SR.sup.EE, wherein each occurrence of R.sup.EE is independently hydrogen, optionally substituted alkyl, optionally substituted alkoxy, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl, or two R.sup.EE groups are joined to form an optionally substituted heterocyclic ring; [0139] or R.sup.E1 and R.sup.E3, or R.sup.E2 and R.sup.E3, or R.sup.E1 and R.sup.E2 are joined to form an optionally substituted carbocyclic or optionally substituted heterocyclic ring; [0140] R.sup.E4 is a leaving group; [0141] R.sup.E5 is halogen; [0142] R.sup.E6 is hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group; [0143] each instance of Y is independently O, S, or NR.sup.E7, wherein R.sup.E7 is hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group; [0144] a is 1 or 2; and [0145] each instance of z is independently 0, 1, 2, 3, 4, 5, or 6, as valency permits.
[0146] In certain embodiments, a compound described herein is of Formula (I):
##STR00016##
or a pharmaceutically acceptable salt thereof, wherein:
[0147] R.sup.1 is hydrogen, halogen, or optionally substituted alkyl;
[0148] M is O, S, or NR.sup.M;
[0149] R.sup.M is hydrogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted acyl, or a nitrogen protecting group;
[0150] Ring A is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl;
[0151] Ring B is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl;
[0152] Ring C is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl;
[0153] each instance of R.sup.A, R.sup.B, and R.sup.C is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --OR.sup.a, --N(R.sup.a).sub.2, --SR.sup.a, --CN, --SCN, --NO.sub.2, --N.sub.3, or optionally substituted acyl;
[0154] each instance of R.sup.a is independently hydrogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted acyl, a nitrogen protecting group when attached to a nitrogen atom, an oxygen protecting group when attached to an oxygen atom, or a sulfur protecting group when attached to a sulfur atom, or two instances of R.sup.a are joined to form a substituted or unsubstituted, heterocyclic ring, or substituted or unsubstituted, heteroaryl ring;
[0155] each of a1, b1, and c1 is independently 0, 1, 2, 3, 4, 5, or 6, as valency permits;
[0156] L.sup.1 is --O--, --S--, --NR.sup.L1--, --C(.dbd.O)--, .sup.lc--NR.sup.L1C(.dbd.O)--.sup.lb, .sup.lc--C(.dbd.O)NR.sup.L1--.sup.lb, .sup.lc--OC(.dbd.O)--.sup.lb, or .sup.lc--C(.dbd.O)O--.sup.lb; wherein .sup.lb indicates the point of attachment is to Ring B; and .sup.lc indicates the point of attachment is to Ring C;
[0157] L.sup.2 is --O--, --S--, --NR.sup.L2--, .sup.lb--NR.sup.L2C(.dbd.O)--.sup.lm, .sup.lb--C(.dbd.O)NR.sup.L2--.sup.lm; wherein .sup.lb indicates the point of attachment is to Ring B; and .sup.lm indicates the point of attachment is to the heteroaryl ring with M;
[0158] X is --O--, --S--, --NR.sup.LX--, .sup.xm--O--CH.sub.2--.sup.xa, .sup.xm--S--CH.sub.2--.sup.xa, or .sup.xm--NR.sup.LX--CH.sub.2--.sup.xa; wherein .sup.xa indicates the point of attachment is to Ring A; and .sup.xm indicates the point of attachment is to the heteroaryl ring with M;
[0159] each of R.sup.L1, R.sup.L2, and R.sup.LX is independently hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group;
[0160] R.sup.2 is any of Formulae (i-1)-(i-46):
##STR00017## ##STR00018## ##STR00019## ##STR00020## ##STR00021##
[0161] L.sup.3 is a bond or an optionally substituted C.sub.1-4 hydrocarbon chain, optionally wherein one or more carbon units of the hydrocarbon chain are independently replaced with --C.dbd.O--, --O--, --S--, --NR.sup.L3a--, --NR.sup.L3aC(.dbd.O)--, --C(.dbd.O)NR.sup.L3a--, --SC(.dbd.O)--, --C(.dbd.O)S--, --OC(.dbd.O)--, --C(.dbd.O)O--, --NR.sup.L3aC(.dbd.S)--, --C(.dbd.S)NR.sup.L3a--, trans-CR.sup.L3b.dbd.CR.sup.L3b--, cis-CR.sup.L3b.dbd.CR.sup.L3b, --C.ident.C--, --S(.dbd.O)--, --S(.dbd.O)O--, --OS(.dbd.O)--, --S(.dbd.O)NR.sup.L3a--, --NR.sup.L3aS(.dbd.O)--, --S(.dbd.O).sub.2--, --S(.dbd.O).sub.2O--, --OS(.dbd.O).sub.2--, --S(.dbd.O).sub.2NR.sup.L3a, or --NR.sup.L3aS(.dbd.O).sub.2--, wherein R.sup.L3a is hydrogen, substituted or unsubstituted C.sub.1-6 alkyl, or a nitrogen protecting group, and wherein each occurrence of R.sup.L3b is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl, or two R.sup.L3b groups are joined to form an optionally substituted carbocyclic or optionally substituted heterocyclic ring;
[0162] L.sup.4 is a bond or an optionally substituted, branched or unbranched C.sub.1 hydrocarbon chain;
[0163] each of R.sup.E1, R.sup.E2, and R.sup.E3 is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --CN, --CH.sub.2OR.sup.EE, --CH.sub.2N(R.sup.EE).sub.2, --CH.sub.2SR.sup.EE, --OR.sup.EE, --N(R.sup.EE).sub.2, --Si(R.sup.EE).sub.3, and --SR.sup.EE, wherein each occurrence of R.sup.EE is independently hydrogen, optionally substituted alkyl, optionally substituted alkoxy, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl, or two R.sup.EE groups are joined to form an optionally substituted heterocyclic ring; or R.sup.E1 and R.sup.E3, or R.sup.E2 and R.sup.E3, or R.sup.E1 and R.sup.E2 are joined to form an optionally substituted carbocyclic or optionally substituted heterocyclic ring;
[0164] R.sup.E4 is a leaving group;
[0165] R.sup.E5 is halogen;
[0166] R.sup.E6 is hydrogen, substituted or unsubstituted C.sub.1-6 alkyl, or a nitrogen protecting group;
[0167] each instance of Y is independently O, S, or NR.sup.E7, wherein R.sup.E7 is hydrogen, substituted or unsubstituted C.sub.1-6 alkyl, or a nitrogen protecting group;
[0168] a is 1 or 2; and
[0169] each instance of z is independently 0, 1, 2, 3, 4, 5, or 6, as valency permits.
[0170] In certain embodiments, a compound described herein is of Formula (II'):
##STR00022##
[0171] R.sup.1 is hydrogen, halogen, or optionally substituted alkyl;
[0172] M is O, S, or NR.sup.M;
[0173] R.sup.M is hydrogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted acyl, or a nitrogen protecting group;
[0174] Ring A is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl;
[0175] Ring B is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl;
[0176] each instance of R.sup.A and R.sup.B is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --OR.sup.a, --N(R.sup.a).sub.2, --SR.sup.a, --CN, --SCN, --C(.dbd.NR.sup.a)R.sup.a, --C(.dbd.NR.sup.a)OR.sup.a, --C(.dbd.NR.sup.a)N(R.sup.a).sub.2, --C(.dbd.O)R.sup.a, --C(.dbd.O)OR.sup.a, --C(.dbd.O)N(R.sup.a).sub.2, --NO.sub.2, --NR.sup.aC(.dbd.O)R.sup.a, --NR.sup.aC(.dbd.O)OR.sup.a, --NR.sup.aC(.dbd.O)N(R.sup.a).sub.2, --OC(.dbd.O)R.sup.a, --OC(.dbd.O)OR.sup.a, or --OC(.dbd.O)N(R.sup.a).sub.2;
[0177] each instance of R.sup.a is independently hydrogen, optionally substituted acyl, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, a nitrogen protecting group when attached to a nitrogen atom, an oxygen protecting group when attached to an oxygen atom, or a sulfur protecting group when attached to a sulfur atom, or two instances of R.sup.a are joined to form a substituted or unsubstituted, heterocyclic ring, or substituted or unsubstituted, heteroaryl ring;
[0178] each of a1 and b1 is independently 0, 1, 2, 3, 4, 5, or 6, as valency permits;
[0179] L.sup.2 is --O--, --S--, --NR.sup.L2--, .sup.lb--NR.sup.L2C(.dbd.O)--.sup.lm, .sup.lb--C(.dbd.O)NR.sup.L2--.sup.lm; wherein .sup.lb indicates the point of attachment is to Ring B; and .sup.lm indicates the point of attachment is to the heteroaryl ring with M;
[0180] X is a bond, --O--, --S--, --NR.sup.LX--, .sup.xm--O--CH.sub.2--.sup.xa, .sup.xm--S--CH.sub.2--.sup.xa, or .sup.xm--NR.sup.LX--CH.sub.2--.sup.xa; wherein .sup.xa indicates the point of attachment is to Ring A; and .sup.xm indicates the point of attachment is to the heteroaryl ring with M;
[0181] each of R.sup.L2 and R.sup.LX is independently hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group;
[0182] R.sup.2 is any of Formulae (i-1)-(i-41):
##STR00023## ##STR00024## ##STR00025## ##STR00026## ##STR00027##
wherein: [0183] L.sup.3 is a bond or an optionally substituted C.sub.1-4 hydrocarbon chain, optionally wherein one or more carbon units of the hydrocarbon chain are independently replaced with --C.dbd.O--, --O--, --S--, --NR.sup.L3a--, --NR.sup.L3aC(.dbd.O)--, --C(.dbd.O)NR.sup.L3a--, --SC(.dbd.O)--, --C(.dbd.O)S--, --OC(.dbd.O)--, --C(.dbd.O)O--, --NR.sup.L3aC(.dbd.S)--, --C(.dbd.S)NR.sup.L3a--, trans-CR.sup.L3b.dbd.CR.sup.L3b--, cis-CR.sup.L3bCR.sup.L3b--, --C.dbd.C--, --S(.dbd.O)--, --S(.dbd.O)O--, --OS(.dbd.O)--, --S(.dbd.O)NR.sup.L3a, --NR.sup.L3aS(.dbd.O)--, --S(.dbd.O).sub.2--, --S(.dbd.O).sub.2O--, --OS(.dbd.O).sub.2--, --S(.dbd.O).sub.2NR.sup.L3a--, or --NR.sup.L3aS(.dbd.O).sub.2--, wherein R.sup.L3a is hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group, and wherein each occurrence of R.sup.L3b is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl, or two R.sup.L3b groups are joined to form an optionally substituted carbocyclic or optionally substituted heterocyclic ring; [0184] L.sup.4 is a bond or an optionally substituted, branched or unbranched C.sub.1-6 hydrocarbon chain; [0185] each of R.sup.E1, R.sup.E2, and R.sup.E3 is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --CN, --CH.sub.2OR.sup.EE, --CH.sub.2N(R.sup.EE).sub.2, --CH.sub.2SR.sup.EE, --OR.sup.EE, --N(R.sup.EE).sub.2, --Si(R.sup.EE).sub.3, and --SR.sup.EE, wherein each occurrence of R.sup.EE is independently hydrogen, optionally substituted alkyl, optionally substituted alkoxy, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl, or two R.sup.EE groups are joined to form an optionally substituted heterocyclic ring; [0186] or R.sup.E1 and R.sup.E3, or R.sup.E2 and R.sup.E3, or R.sup.E1 and R.sup.E2 are joined to form an optionally substituted carbocyclic or optionally substituted heterocyclic ring; [0187] R.sup.E4 is a leaving group; [0188] R.sup.E5 is halogen; [0189] R.sup.E6 is hydrogen, optionally substituted C.sub.14 alkyl, or a nitrogen protecting group; each instance of Y is independently O, S, or NR.sup.E7, wherein R.sup.E7 is hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group; [0190] a is 1 or 2; and [0191] each instance of z is independently 0, 1, 2, 3, 4, 5, or 6, as valency permits.
[0192] In certain embodiments, a compound described herein is of Formula (I):
##STR00028##
or a pharmaceutically acceptable salt thereof, wherein:
[0193] R.sup.1 is hydrogen, halogen, or optionally substituted alkyl;
[0194] M is O, S, or NR.sup.M;
[0195] R.sup.M is hydrogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted acyl, or a nitrogen protecting group;
[0196] Ring A is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl;
[0197] Ring B is optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl;
[0198] each instance of R.sup.A and R.sup.B is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --OR.sup.a, --N(R.sup.a).sub.2, --SR.sup.a, --CN, --SCN, --C(.dbd.NR.sup.a)R.sup.a, --C(.dbd.NR.sup.a)OR.sup.a, --C(.dbd.NR.sup.a)N(R.sup.a).sub.2, --C(.dbd.O)R.sup.a, --C(.dbd.O)OR.sup.a, --C(.dbd.O)N(R.sup.a).sub.2, --NO.sub.2, --NR.sup.aC(.dbd.O)R.sup.a, --NR.sup.aC(.dbd.O)OR.sup.a, --NR.sup.aC(.dbd.O)N(R.sup.a).sub.2, --OC(.dbd.O)R.sup.a, --OC(.dbd.O)OR.sup.a, or --OC(.dbd.O)N(R.sup.a).sub.2;
[0199] each instance of R.sup.a is independently hydrogen, optionally substituted acyl, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, a nitrogen protecting group when attached to a nitrogen atom, an oxygen protecting group when attached to an oxygen atom, or a sulfur protecting group when attached to a sulfur atom, or two instances of R.sup.a are joined to form a substituted or unsubstituted, heterocyclic ring, or substituted or unsubstituted, heteroaryl ring;
[0200] each of a1 and b1 is independently 0, 1, 2, 3, 4, 5, or 6, as valency permits;
[0201] L.sup.2 is --O--, --S--, --NR.sup.L2--, .sup.lb--NR.sup.L2C(.dbd.O)--.sup.lm, .sup.lb--C(.dbd.O)NR.sup.L2--.sup.lm; wherein .sup.lb indicates the point of attachment is to Ring B; and .sup.lm indicates the point of attachment is to the heteroaryl ring with M;
[0202] X is --O--, --S--, --NR.sup.LX--, .sup.xm--O--CH.sub.2--.sup.xa, .sup.xm--S--CH.sub.2--.sup.xa, or .sup.xm--NR.sup.LX--CH.sub.2--.sup.xa; wherein .sup.xa indicates the point of attachment is to Ring A; and .sup.xm indicates the point of attachment is to the heteroaryl ring with M;
[0203] each of R.sup.L2 and R.sup.LX is independently hydrogen, optionally substituted C.sub.1-6 alkyl, or a nitrogen protecting group;
[0204] R.sup.2 is any of Formulae (i-1)-(i-46):
##STR00029## ##STR00030## ##STR00031## ##STR00032## ##STR00033##
wherein:
[0205] L.sup.3 is a bond or an optionally substituted C.sub.1-4 hydrocarbon chain, optionally wherein one or more carbon units of the hydrocarbon chain are independently replaced with --C.dbd.O--, --O--, --S--, --NR.sup.L3a--, --NR.sup.L3aC(.dbd.O)--, --C(.dbd.O)NR.sup.L3a--, --SC(.dbd.O)--, --C(.dbd.O)S--, --OC(.dbd.O)--, --C(.dbd.O)O--, --NR.sup.L3aC(.dbd.S)--, --C(.dbd.S)NR.sup.L3a, trans-CR.sup.L3b.dbd.CR.sup.L3b--, cis-CR.sup.L3b.dbd.CR.sup.L3b--, --C.ident.C--, --S(.dbd.O)--, --S(.dbd.O)O--, --OS(.dbd.O)--, --S(.dbd.O)NR.sup.L3a--, --NR.sup.L3aS(.dbd.O)--, --S(.dbd.O).sub.2--, --S(.dbd.O).sub.2O--, --OS(.dbd.O).sub.2--, --S(.dbd.O).sub.2NR.sup.L3a--, or --NR.sup.L3aS(.dbd.O).sub.2--, wherein R.sup.L3a is hydrogen, substituted or unsubstituted C.sub.1-6 alkyl, or a nitrogen protecting group, and wherein each occurrence of R.sup.L3b is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl, or two R.sup.L3b groups are joined to form an optionally substituted carbocyclic or optionally substituted heterocyclic ring;
[0206] L.sup.4 is a bond or an optionally substituted, branched or unbranched C.sub.1-6 hydrocarbon chain;
[0207] each of R.sup.E1, R.sup.E2, and R.sup.E3 is independently hydrogen, halogen, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --CN, --CH.sub.2OR.sup.EE, --CH.sub.2N(R.sup.EE).sub.2, --CH.sub.2SR.sup.EE, --OR.sup.EE, --N(R.sup.EE).sub.2, --Si(R.sup.EE).sub.3, and --SR.sup.EE, wherein each occurrence of R.sup.EE is independently hydrogen, optionally substituted alkyl, optionally substituted alkoxy, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl, or two R.sup.EE groups are joined to form an optionally substituted heterocyclic ring;
[0208] or R.sup.E1 and R.sup.E3, or R.sup.E2 and R.sup.E3, or R.sup.E1 and R.sup.E2 are joined to form an optionally substituted carbocyclic or optionally substituted heterocyclic ring;
[0209] R.sup.E4 is a leaving group;
[0210] R.sup.E5 is halogen;
[0211] R.sup.E6 is hydrogen, substituted or unsubstituted C.sub.1-6 alkyl, or a nitrogen protecting group;
[0212] each instance of Y is independently O, S, or NR.sup.E7, wherein R.sup.E7 is hydrogen, substituted or unsubstituted C.sub.1-6 alkyl, or a nitrogen protecting group;
[0213] a is 1 or 2; and
[0214] each instance of z is independently 0, 1, 2, 3, 4, 5, or 6, as valency permits.
[0215] As generally defined herein in Formulae (I'), (II'), (I), and (II), R.sup.1 is hydrogen, halogen, or optionally substituted alkyl. As generally defined herein in Formulae (I)-(II), R.sup.1 is hydrogen, halogen, or optionally substituted alkyl. In certain embodiments, R.sup.1 is hydrogen. In certain embodiments, R.sup.1 is halogen. In certain embodiments, R.sup.1 is F. In certain embodiments, R.sup.1 is Cl. In certain embodiments, R.sup.1 is Br. In certain embodiments, R.sup.1 is I. In certain embodiments, R.sup.1 is optionally substituted alkyl. In certain embodiments, R.sup.1 is unsubstituted alkyl. In certain embodiments, R.sup.1 is unsubstituted C.sub.1-6 alkyl. In certain embodiments, R.sup.1 is methyl. In certain embodiments, R.sup.1 is substituted alkyl. In certain embodiments, R.sup.1 is substituted C.sub.1-6alkyl.
[0216] In certain embodiments, M is O. In certain embodiments, M is S. In certain embodiments, M is NR.sup.M, wherein R.sup.M is as defined herein. In certain embodiments, M is NH. In certain embodiments, M is NR.sup.M, wherein R.sup.M is optionally substituted alkyl. In certain embodiments, M is NR.sup.M, wherein R.sup.M is unsubstituted alkyl. In certain embodiments, M is NCH.sub.3. In certain embodiments, M is NAc.
[0217] Compounds of any one of Formulae (I'), (II'), (I), and (II) include Ring A attached to linker X. Compounds of any one of Formulae (I)-(II) include Ring A attached to linker X. Ring A may be optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl. In certain embodiments, Ring A is optionally substituted monocyclic carbocyclyl. In certain embodiments, Ring A is optionally substituted cyclohexyl. In certain embodiments, Ring A is optionally substituted monocyclic heterocyclyl. In certain embodiments, Ring A is optionally substituted piperidinyl. In certain embodiments, Ring A is optionally substituted piperizinyl. In certain embodiments, Ring A is optionally substituted tetrahydropyranyl. In certain embodiments, Ring A is optionally substituted phenyl. In certain embodiments, Ring A is phenyl substituted with only X. In certain embodiments, Ring A is optionally substituted monocyclic heteroaryl. In certain embodiments, Ring A is optionally substituted 5-membered heteroaryl. In certain embodiments, Ring A is optionally substituted 6-membered heteroaryl. In certain embodiments, Ring A is optionally substituted pyridine. In certain embodiments, Ring A is optionally substituted pyrimidine.
[0218] In certain embodiments, Ring A is of Formula (x-i):
##STR00034##
wherein:
[0219] each of V.sup.10, V.sup.11, V.sup.12, V.sup.13, and V.sup.14 is independently O, S, N, NR.sup.A1, C, or CR.sup.A2, as valency permits;
[0220] each instance of R.sup.A1 is independently selected from the group consisting of hydrogen, substituted or unsubstituted acyl, substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted carbocyclyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, and a nitrogen protecting group;
[0221] each instance of R.sup.A2 is independently selected from the group consisting of hydrogen, halogen, substituted or unsubstituted acyl, substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted carbocyclyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, --CN, --OR.sup.A2a, --N(R.sup.A2a).sub.2, and --SR.sup.A2a; and
[0222] each occurrence of R.sup.A2a is independently selected from the group consisting of hydrogen, substituted or unsubstituted acyl, substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted carbocyclyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, a nitrogen protecting group when attached to a nitrogen atom, an oxygen protecting group when attached to an oxygen atom, and a sulfur protecting group when attached to a sulfur atom, or two R.sup.A2a groups are joined to form a substituted or unsubstituted heterocyclic ring.
[0223] In certain embodiments, only one of V.sup.10, V.sup.11, V.sup.12, V.sup.13, and V.sup.14 is selected from the group consisting of O, S, N, and NR.sup.A1. In certain embodiments, Ring A is of the formula:
##STR00035##
[0224] In certain embodiments, Ring A is of the formula:
##STR00036##
[0225] In certain embodiments, Ring A is of the formula:
##STR00037##
[0226] In certain embodiments, only two of V.sup.10, V.sup.11, V.sup.12, V.sup.13, and V.sup.14 are each independently selected from the group consisting of O, S, N, and NR.sup.A1. In certain embodiments, Ring A is of the formula:
##STR00038##
[0227] In certain embodiments, Ring A is of the formula:
##STR00039##
[0228] In certain embodiments, Ring A is of Formula (x-ii):
##STR00040##
[0229] In certain embodiments, Ring A is of the formula:
##STR00041##
[0230] In certain embodiments, Ring A is of the formula:
##STR00042##
[0231] In certain embodiments, Ring A is of the formula:
##STR00043##
[0232] In certain embodiments, Ring A is of the formula:
##STR00044##
In certain embodiments, Ring A is of the formula:
##STR00045##
In certain embodiments, Ring A is of the formula:
##STR00046##
[0233] In certain embodiments, only three of V.sup.10, V.sup.11, V.sup.12, V.sup.13, and V.sup.14 are each independently selected from the group consisting of O, S, N, and NR.sup.A1. In certain embodiments, Ring A is of the formula:
##STR00047##
[0234] In certain embodiments, Ring A is of the formula:
##STR00048##
[0235] In certain embodiments, Ring A is of the formula:
##STR00049##
[0236] In certain embodiments, only four of V.sup.10, V.sup.11, V.sup.12, V.sup.13, and V.sup.14 are each independently selected from the group consisting of N and NR.sup.A1. In certain embodiments, Ring A is of the formula:
##STR00050##
[0237] In certain embodiments, Ring A may also be a substituted or unsubstituted 6-membered heteroaryl ring. In certain embodiments, Ring A is of the formula:
##STR00051##
wherein each of V.sup.10, V.sup.11, V.sup.12, V.sup.13, V.sup.14, and V.sup.15 is independently N, C, or CR.sup.A2, as valency permits, wherein R.sup.A2 is as defined herein. In certain embodiments, only one of V.sup.10, V.sup.11, V.sup.12, V.sup.13, V.sup.14, and V.sup.15 is N. In certain embodiments, Ring A is of the formula:
##STR00052##
wherein k is 0, 1, 2, 3, or 4, and R.sup.A2 is as defined herein. In certain embodiments, Ring A is of the formula:
##STR00053##
[0238] In certain embodiments, only two of V.sup.10, V.sup.11, V.sup.12, V.sup.13, V.sup.14, and V.sup.15 are N. In certain embodiments, Ring A is of the formula:
##STR00054##
[0239] In certain embodiments, only three of V.sup.10, V.sup.11, V.sup.12, V.sup.13, V.sup.14, and V.sup.15 are N. In certain embodiments, Ring A is of the formula:
##STR00055##
[0240] In certain embodiments, Ring A is of the formula:
##STR00056##
wherein R.sup.A and a1 are as defined herein.
[0241] In certain embodiments, a1 is 1; and Ring A is one of the formulae:
##STR00057##
In certain embodiments, a1 is 2; and Ring A is one of the formulae:
##STR00058##
In certain embodiments, a2 is 3; and R.sub.61 is one of the formulae:
##STR00059##
In certain embodiments, a2 is 4; and R.sub.61 is one of the formulae:
##STR00060##
In certain embodiments, a2 is 5; and R.sub.61 is of the formula
##STR00061##
[0242] In certain embodiments, Ring A is of the formula:
##STR00062##
wherein R.sup.A and a1 are as defined herein.
[0243] In certain embodiments, a1 is 1; and Ring A is one of the formulae:
##STR00063##
In certain embodiments, a1 is 2; and Ring A is one of the formulae:
##STR00064##
In certain embodiments, a2 is 3; and R.sub.61 is one of the formulae:
##STR00065##
In certain embodiments, a2 is 4; and R.sub.61 is one of the formulae:
##STR00066##
In certain embodiments, a2 is 5; and R.sub.61 is of the formula
##STR00067##
[0244] In certain embodiments, Ring A is optionally substituted monocyclic heterocyclyl. In certain embodiments, Ring A is optionally substituted 5-membered heterocyclyl. In certain embodiments, Ring A is optionally substituted 6-membered heterocyclyl. In certain embodiments, Ring A is optionally substituted 6-membered heterocyclyl with one heteroatom selected from the group of S, O, and N.
[0245] In certain embodiments, Ring A is of the formula:
##STR00068##
wherein H.sup.A is S, O, or NR.sup.HA; R.sup.HA is hydrogen, optionally substituted alkyl, or a nitrogen protecting group; and R.sup.A and a1 are as defined herein.
[0246] In certain embodiments, Ring A is of the formula:
##STR00069##
wherein H.sup.A, R.sup.A and a1 are as defined herein.
[0247] In certain embodiments, Ring A is of the formula:
##STR00070##
wherein H.sup.A, R.sup.A and a1 are as defined herein.
[0248] In certain embodiments, H.sup.A is S. In certain embodiments, H.sup.A is O. In certain embodiments, H.sup.A is NR.sup.HA.
[0249] In certain embodiments, Ring A is of the formula:
##STR00071##
[0250] In certain embodiments, Ring A is of the formula:
##STR00072##
wherein R.sup.HA is hydrogen, optionally substituted alkyl, or a nitrogen protecting group; and R.sup.A and a1 are as defined herein. In certain embodiments, Ring A is of the formula:
##STR00073##
[0251] In certain embodiments, R.sup.A is hydrogen. In certain embodiments, R.sup.A is halogen. In certain embodiments, R.sup.A is F. In certain embodiments, R.sup.A is Cl. In certain embodiments, R.sup.A is Br. In certain embodiments, R.sup.A is I. In certain embodiments, R.sup.A is optionally substituted alkyl. In certain embodiments, R.sup.A is unsubstituted alkyl. In certain embodiments, R.sup.A is methyl, ethyl, n-propyl, i-propyl, n-butyl, s-butyl, t-butyl, n-pentyl, t-pentyl, neo-pentyl, i-pentyl, s-pentyl, or 3-pentyl. In certain embodiments, R.sup.A is t-butyl. In certain embodiments, R.sup.A is substituted alkyl. In certain embodiments, R.sup.A is haloalkyl. In certain embodiments, R.sup.A is --CF.sub.3, --CHF.sub.2, or --CH.sub.2F. In certain embodiments, R.sup.A is optionally substituted alkenyl. In certain embodiments, R.sup.A is optionally substituted alkynyl. In certain embodiments, R.sup.A is optionally carbocyclyl. In certain embodiments, R.sup.A is optionally substituted heterocyclyl. In certain embodiments, R.sup.A is optionally substituted aryl. In certain embodiments, R.sup.A is optionally substituted heteroaryl. In certain embodiments, R.sup.A is --OR.sup.a, wherein R.sup.a is as defined herein. In certain embodiments, R.sup.A is --OH. In certain embodiments, R.sup.A is --OR.sup.a, wherein R.sup.a is optionally substituted alkyl or an oxygen protecting group. In certain embodiments, R.sup.A is --OR.sup.a, wherein R.sup.a is unsubstituted alkyl. In certain embodiments, R.sup.A is --OCH.sub.3. In certain embodiments, R.sup.A is --N(R.sup.a).sub.2, wherein R.sup.a is as defined herein. In certain embodiments, R.sup.A is --NH.sub.2. In certain embodiments, R.sup.A is --N(R.sup.a).sub.2, wherein each instance of R.sup.a is optionally substituted alkyl or a nitrogen protecting group. In certain embodiments, R.sup.A is --N(CH.sub.3).sub.2. In certain embodiments, R.sup.A is --NHR.sup.a, wherein R.sup.a is optionally substituted alkyl or a nitrogen protecting group. In certain embodiments, R.sup.A is --NHCH.sub.3.
[0252] In certain embodiments, at least one instance of R.sup.A1 is H (hydrogen). In certain embodiments, at least one instance of R.sup.A1 is halogen. In certain embodiments, at least one instance of R.sup.A1 is F (fluorine). In certain embodiments, at least one instance of R.sup.A1 is Cl (chlorine). In certain embodiments, at least one instance of R.sup.A1 is Br (bromine). In certain embodiments, at least one instance of R.sup.A1 is I (iodine). In certain embodiments, at least one instance of R.sup.A1 is substituted acyl. In certain embodiments, at least one instance of R.sup.A1 is unsubstituted acyl. In certain embodiments, at least one instance of R.sup.A1 is acetyl. In certain embodiments, at least one instance of R.sup.A1 is substituted acetyl. In certain embodiments, at least one instance of R.sup.A1 is substituted alkyl. In certain embodiments, at least one instance of R.sup.A1 is unsubstituted alkyl. In certain embodiments, at least one instance of R.sup.A1 is C.sub.1-6 alkyl. In certain embodiments, at least one instance of R.sup.A1 is methyl. In certain embodiments, at least one instance of R.sup.A1 is ethyl. In certain embodiments, at least one instance of R.sup.A1 is propyl. In certain embodiments, at least one instance of R.sup.A1 is butyl. In certain embodiments, at least one instance of R.sup.A1 is substituted alkenyl. In certain embodiments, at least one instance of R.sup.A1 is unsubstituted alkenyl. In certain embodiments, at least one instance of R.sup.A1 is vinyl. In certain embodiments, at least one instance of R.sup.A1 is substituted alkynyl. In certain embodiments, at least one instance of R.sup.A1 is unsubstituted alkynyl. In certain embodiments, at least one instance of R.sup.A1 is ethynyl. In certain embodiments, at least one instance of R.sup.A1 is substituted carbocyclyl. In certain embodiments, at least one instance of R.sup.A1 is unsubstituted carbocyclyl. In certain embodiments, at least one instance of R.sup.A1 is substituted heterocyclyl. In certain embodiments, at least one instance of R.sup.A1 is unsubstituted heterocyclyl. In certain embodiments, at least one instance of R.sup.A1 is substituted aryl. In certain embodiments, at least one instance of R.sup.A1 is unsubstituted aryl. In certain embodiments, at least one instance of R.sup.A1 is substituted phenyl. In certain embodiments, at least one instance of R.sup.A1 is unsubstituted phenyl. In certain embodiments, at least one instance of R.sup.A1 is substituted heteroaryl. In certain embodiments, at least one instance of R.sup.A1 is unsubstituted heteroaryl. In certain embodiments, at least one instance of R.sup.A1 is substituted pyridyl. In certain embodiments, at least one instance of R.sup.A1 is unsubstituted pyridyl. In certain embodiments, at least one instance of R.sup.A1 is a nitrogen protecting group. In certain embodiments, at least one instance of R.sup.A1 is BOC.
[0253] In certain embodiments, at least one R.sup.A1 is hydrogen, substituted or unsubstituted C.sub.1-6 alkyl, or a nitrogen protecting group. In certain embodiments, all instances of R.sup.A1 are each independently hydrogen, substituted or unsubstituted C.sub.1-6 alkyl, or a nitrogen protecting group. In certain embodiments, all instances of R.sup.A1 are hydrogen.
[0254] In certain embodiments, at least one R.sup.A2 is H. In certain embodiments, at least one R.sup.A2 is halogen. In certain embodiments, at least one R.sup.A2 is F. In certain embodiments, at least one R.sup.A2 is Cl. In certain embodiments, at least one R.sup.A2 is Br. In certain embodiments, at least one R.sup.A2 is I (iodine). In certain embodiments, at least one R.sup.A2 is substituted acyl. In certain embodiments, at least one R.sup.A2 is unsubstituted acyl. In certain embodiments, at least one R.sup.A2 is acetyl. In certain embodiments, at least one R.sup.A2 is substituted acetyl. In certain embodiments, at least one R.sup.A2 is substituted alkyl. In certain embodiments, at least one R.sup.A2 is unsubstituted alkyl. In certain embodiments, at least one R.sup.A2 is C.sub.1-6 alkyl. In certain embodiments, at least one R.sup.A2 is methyl. In certain embodiments, at least one R.sup.A2 is ethyl. In certain embodiments, at least one R.sup.A2 is propyl. In certain embodiments, at least one R.sup.A2 is butyl. In certain embodiments, at least one R.sup.A2 is substituted alkenyl. In certain embodiments, at least one R.sup.A2 is unsubstituted alkenyl. In certain embodiments, at least one R.sup.A2 is vinyl. In certain embodiments, at least one R.sup.A2 is substituted alkynyl. In certain embodiments, at least one R.sup.A2 is unsubstituted alkynyl. In certain embodiments, at least one R.sup.A2 is ethynyl. In certain embodiments, at least one R.sup.A2 is substituted carbocyclyl. In certain embodiments, at least one R.sup.A2 is unsubstituted carbocyclyl. In certain embodiments, at least one R.sup.A2 is substituted heterocyclyl. In certain embodiments, at least one R.sup.A2 is unsubstituted heterocyclyl. In certain embodiments, at least one R.sup.A2 is substituted aryl. In certain embodiments, at least one R.sup.A2 is unsubstituted aryl. In certain embodiments, at least one R.sup.A2 is substituted phenyl. In certain embodiments, at least one R.sup.A2 is unsubstituted phenyl. In certain embodiments, at least one R.sup.A2 is substituted heteroaryl. In certain embodiments, at least one R.sup.A2 is unsubstituted heteroaryl. In certain embodiments, at least one R.sup.A2 is substituted pyridyl. In certain embodiments, at least one R.sup.A2 is unsubstituted pyridyl. In certain embodiments, at least one R.sup.A2 is --OR.sup.A2, wherein R.sup.A2a is as defined herein. In certain embodiments, at least one R.sup.A2 is --OR.sup.A2a, wherein R.sup.A2a is hydrogen. In certain embodiments, at least one R.sup.A2 is --OR.sup.A2a, wherein R.sup.A2a is substituted or unsubstituted C.sub.1-6 alkyl. In certain embodiments, at least one R.sup.A2 is --OR.sup.A2a, wherein R.sup.A2a is unsubstituted C.sub.1-6 alkyl. In certain embodiments, at least one R.sup.A2 is --OCH.sub.3. In certain embodiments, at least one R.sup.A2 is --N(R.sup.A2a).sub.2. In certain embodiments, at least one R.sup.A2 is --SR.sup.A2a. In certain embodiments, all instances of R.sup.A2 are hydrogen.
[0255] In certain embodiments, all R.sup.A1 and R.sup.A2 are hydrogen. In certain embodiments, R.sup.A1 is hydrogen; and at least one R.sup.A2 is substituted or unsubstituted alkyl. In certain embodiments, R.sup.A1 is hydrogen; and at least one R.sup.A2 is unsubstituted alkyl. In certain embodiments, R.sup.A1 is hydrogen; and at least one R.sup.A2 is methyl, ethyl, or n-propyl. In certain embodiments, R.sup.A1 is hydrogen; and at least one R.sup.A2 is --OR.sup.A2a, wherein R.sup.A2a is as defined herein. In certain embodiments, R.sup.A1 is hydrogen; and at least one R.sup.A2 is --OR.sup.A2a, wherein R.sup.A2a is substituted or unsubstituted C.sub.1-6 alkyl. In certain embodiments, R.sup.A1 is hydrogen; and at least one R.sup.A2 is --OR.sup.A2a, wherein R.sup.A2a is unsubstituted C.sub.1-6 alkyl. In certain embodiments, R.sup.A1 is hydrogen; and at least one R.sup.A2 is --OCH.sub.3.
[0256] In certain embodiments, Ring A is of the formula:
##STR00074##
[0257] In certain embodiments, Ring A is of the formula:
##STR00075##
[0258] In certain embodiments, Ring A is of the formula:
##STR00076##
In certain embodiments, Ring A is of one of the formula:
##STR00077##
[0259] In certain embodiments, Ring A is of the formula:
##STR00078##
[0260] Compounds of any one of Formulae (I'), (II'), (I), and (II) include Ring B between linker L.sup.1 and linker L.sup.2. Compounds of any one of Formulae (I)-(II) include Ring B between linker L.sup.1 and linker L.sup.2. Ring B may be optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl. In certain embodiments, Ring B is optionally substituted monocyclic carbocyclyl. In certain embodiments, Ring B is optionally substituted monocyclic heterocyclyl. In certain embodiments, Ring B is optionally substituted pyrrolidinyl. In certain embodiments, Ring B is optionally substituted phenyl. In certain embodiments, Ring B is optionally substituted monocyclic heteroaryl. In certain embodiments, Ring B is phenyl substituted with only L.sup.1 and L.sup.2. In certain embodiments, Ring B is optionally substituted cyclohexyl. In certain embodiments, Ring B is optionally substituted piperidinyl. In certain embodiments, Ring B is optionally substituted piperizinyl. In certain embodiments, Ring B is optionally substituted pyridinyl. In certain embodiments, Ring B is optionally substituted pyrimidinyl.
[0261] In certain embodiments of Formulae (I'), (II'), (I), and (II), Ring B is
##STR00079##
wherein each ring atom is optionally substituted. In certain embodiments, Ring B is
##STR00080##
wherein each ring atom is optionally substituted. In certain embodiments, Ring B is
##STR00081##
wherein each ring atom is optionally substituted. In certain embodiments, Ring B is
##STR00082##
wherein each ring atom is optionally substituted, and L.sup.1 and L.sup.2 may attach to Ring B at either indicated position. In certain embodiments, Ring B is
##STR00083##
wherein each ring atom is optionally substituted, and L.sup.2 and R.sup.2 may attach to Ring B at either indicated position. In certain embodiments, Ring B is
##STR00084##
wherein each ring atom is optionally substituted, and L.sup.2 and R.sup.2 is attached to Ring B at either position indicated. In certain embodiments, Ring B is
##STR00085##
wherein each ring atom is optionally substituted, and L.sup.1 and L.sup.2 may attach to Ring B at either indicated position. In certain embodiments, Ring B is
##STR00086##
wherein each ring atom is optionally substituted, and L.sup.1 and L.sup.2 may attach to Ring B at either indicated position.
[0262] Compounds of Formula (I') and (I) include Ring C between linker L.sup.1 and R.sup.2. Ring C may be optionally substituted monocyclic carbocyclyl, optionally substituted monocyclic heterocyclyl, optionally substituted phenyl, or optionally substituted monocyclic heteroaryl. In certain embodiments, Ring C is optionally substituted monocyclic carbocyclyl. In certain embodiments, Ring C is optionally substituted monocyclic heterocyclyl. In certain embodiments, Ring C is optionally substituted monocyclic aryl. In certain embodiments, Ring C is optionally substituted phenyl. In certain embodiments, Ring C is optionally substituted bicyclic aryl. In certain embodiments, Ring C is optionally substituted 2,3-dihydro-1H-indene. In certain embodiments, Ring C is optionally substituted naphthalene. In certain embodiments, Ring C is:
##STR00087##
In certain embodiments, Ring C is:
##STR00088##
In certain embodiments, Ring C is optionally substituted monocyclic heteroaryl. In certain embodiments, Ring C is phenyl substituted with only L.sup.1 and R.sup.2. In certain embodiments, Ring C is optionally substituted cyclohexyl. In certain embodiments, Ring C is optionally substituted pyridinone. In certain embodiments, Ring C is:
##STR00089##
In certain embodiments, Ring C is:
##STR00090##
In certain embodiments, Ring C is optionally substituted piperidinyl. In certain embodiments, Ring C is optionally substituted piperizinyl. In certain embodiments, Ring C is optionally substituted pyridinyl. In certain embodiments, Ring C is optionally substituted pyrimidinyl. In certain embodiments, Ring C is optionally substituted bicyclic heteroaryl. In certain embodiments, Ring C is optionally substituted indolyl. In certain embodiments, Ring C is optionally substituted indolinyl. In certain embodiments, Ring C is:
##STR00091##
In certain embodiments, Ring C is:
##STR00092##
[0263] In certain embodiments, for Formula (I'), L.sup.1 is --CH.sub.2--. In certain embodiments, for Formula (I'), L.sup.1 is .sup.lc--S(.dbd.O).sub.2--.sup.lb. In certain embodiments, L.sup.1 is --O--. In certain embodiments, L.sup.1 is --S--. In certain embodiments, L.sup.1 is --NR.sup.L1--. In certain embodiments, L.sup.1 is --C(.dbd.O)--. In certain embodiments, L.sup.1 is --NR.sup.L1--. In certain embodiments, L.sup.1 is .sup.lc--OC(.dbd.O)--.sup.lb. In certain embodiments, L.sup.1 is .sup.lc--C(.dbd.O)O--.sup.lb. In certain embodiments, L.sup.1 is .sup.lc--NR.sup.L1C(.dbd.O)--.sup.lb. In certain embodiments, L.sup.1 is .sup.lc--C(O)NR.sup.L1--.sup.lb. As used herein, .sup.lb indicates the point of attachment is to Ring B; and .sup.lc indicates the point of attachment is to Ring C.
[0264] In certain embodiments, R.sup.L1 is hydrogen. In certain embodiments, R.sup.L1 is optionally substituted C.sub.1-6 alkyl. In certain embodiments, R.sup.L1 is unsubstituted C.sub.1-6 alkyl. In certain embodiments, R.sup.L1 is methyl. In certain embodiments, R.sup.L1 is substituted C.sub.1-6 alkyl. In certain embodiments, R.sup.L1 is a nitrogen protecting group.
[0265] In certain embodiments, L.sup.2 is --O--. In certain embodiments, L.sup.2 is --S--. In certain embodiments, L.sup.2 is --NR.sup.L2--. In certain embodiments, L.sup.2 is .sup.lb--NR.sup.L2C(.dbd.O)--.sup.lm. In certain embodiments, L.sup.2 is .sup.lb--C(.dbd.O)NR.sup.L2--.sup.lm. As used herein, .sup.lb indicates the point of attachment is to Ring B; and .sup.lm indicates the point of attachment is to the heteroaryl ring with M.
[0266] In certain embodiments, R.sup.L2 is hydrogen. In certain embodiments, R.sup.L2 is optionally substituted C.sub.1-6 alkyl. In certain embodiments, R.sup.L2 is unsubstituted C.sub.1-6 alkyl. In certain embodiments, R.sup.L2 is methyl. In certain embodiments, R.sub.L2 is substituted C.sub.1-6 alkyl. In certain embodiments, R.sup.L2 is a nitrogen protecting group.
[0267] In certain embodiments, for Formula (II'), X is a bond. In certain embodiments, for Formula (I'), X is .sup.xm--CH.sub.2CH.sub.2--.sup.xa. In certain embodiments, for Formula (I'), X is .sup.xm--CH.dbd.CH--.sup.xa. In certain embodiments, for Formula (I'), X is .sup.xm--CH.sub.2--NR.sup.LX--.sup.xa. In certain embodiments, for Formula (I'), X is .sup.xm--CH.sub.2--NH--.sup.xa. In certain embodiments, for Formula (I'), X is .sup.xm--CH.sub.2--O--CH.sub.2--.sup.xa. In certain embodiments, for Formula (I'), X is .sup.xa--CH.sub.2--NH--CH.sub.2--.sup.xa. In certain embodiments, X is --O--. In certain embodiments, X is --S--. In certain embodiments, X is .sup.xm--S--C(.dbd.O)CH.sub.2--.sup.xa. In certain embodiments, X is --NR.sup.LX--. In certain embodiments, X is --O--CH.sub.2--. In certain embodiments, X is --S--CH.sub.2--. In certain embodiments, X is --NR.sup.LX--CH.sub.2--. In certain embodiments, X is --NH--CH.sub.2--.
[0268] In certain embodiments, a1 is 1. In certain embodiments, a1 is 2. In certain embodiments, a1 is 3.
[0269] In certain embodiments, b1 is 1. In certain embodiments, b1 is 2. In certain embodiments, b1 is 3.
[0270] In certain embodiments, c1 is 1. In certain embodiments, c1 is 2. In certain embodiments, c1 is 3.
[0271] In certain embodiments, R.sup.B is optionally substituted alkyl. In certain embodiments, R.sup.B is optionally substituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.B is unsubstituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.B is methyl or ethyl. In certain embodiments, R.sup.B is substituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.B is hydroxy C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.B is --CH.sub.2OH. In certain embodiments, R.sup.B is --CH.sub.2CH.sub.2OH. In certain embodiments, R.sup.B is --N(R.sup.a).sub.2, wherein R.sup.a is as defined herein. In certain embodiments, R.sup.B is --NHR.sup.a, wherein R.sup.a is as defined herein. In certain embodiments, R.sup.B is --NHR.sup.a, wherein R.sup.a is hydrogen or optionally substituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.B is --NH.sub.2. In certain embodiments, R.sup.B is --NHR.sup.a, wherein R.sup.a is optionally substituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.B is --NHR.sup.a, wherein R.sup.a is unsubstituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.B is --NHR.sup.a, wherein R.sup.a is methyl or ethyl. In certain embodiments, R.sup.B is --NHCH.sub.3. In certain embodiments, R.sup.B is --NHR.sup.a, wherein R.sup.a is a nitrogen protecting group. In certain embodiments, R.sup.B is --N(CH.sub.3)R.sup.a, wherein R.sup.a is optionally substituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.B is --N(CH.sub.3)R.sup.a, wherein R.sup.a is unsubstituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.B is --N(CH.sub.3)R.sup.a, wherein R.sup.a is methyl or ethyl. In certain embodiments, R.sup.B is --N(CH.sub.3).sub.2. In certain embodiments, R.sup.B is --N(CH.sub.3)R.sup.a, wherein R.sup.a is a nitrogen protecting group.
[0272] In certain embodiments, R.sup.C is optionally substituted alkyl. In certain embodiments, R.sup.C is optionally substituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.C is unsubstituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.C is methyl or ethyl. In certain embodiments, R.sup.C is substituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.C is hydroxy C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.C is --CH.sub.2OH. In certain embodiments, R.sup.C is --CH.sub.2CH.sub.2OH. In certain embodiments, R.sup.C is --N(R.sup.a).sub.2, wherein R.sup.a is as defined herein. In certain embodiments, R.sup.C is --NHR.sup.a, wherein R.sup.a is as defined herein. In certain embodiments, R.sup.C is --NHR.sup.a, wherein R.sup.a is hydrogen or optionally substituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.C is --NH.sub.2. In certain embodiments, R.sup.C is --NHR.sup.a, wherein R.sup.a is optionally substituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.C is --NHR.sup.a, wherein R.sup.a is unsubstituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.C is --NHR.sup.a, wherein R.sup.a is methyl or ethyl. In certain embodiments, R.sup.C is --NHCH.sub.3. In certain embodiments, R.sup.C is --NHR.sup.a, wherein R.sup.a is a nitrogen protecting group. In certain embodiments, R.sup.C is --N(CH.sub.3)R.sup.a, wherein R.sup.a is optionally substituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.C is --N(CH.sub.3)R.sup.a, wherein R.sup.a is unsubstituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.C is --N(CH.sub.3)R.sup.a, wherein R.sup.a is methyl or ethyl. In certain embodiments, R.sup.C is --N(CH.sub.3).sub.2. In certain embodiments, R.sup.C is --N(CH.sub.3)R.sup.a, wherein R.sup.a is a nitrogen protecting group.
[0273] Compounds of Formula (I') include R.sup.2 attached to Ring C. Compounds of Formula (I) include R.sup.2 attached to Ring C. Compounds of Formula (I) include R.sup.2 attached to Ring B. Compounds of Formula (I') include R.sup.2 attached to Ring B. In certain embodiments, R.sup.2 comprises an electrophilic moiety. In certain embodiments, R.sup.2 comprises a Michael acceptor moiety. The electrophilic moiety (e.g., Michael acceptor moiety) may react with a cysteine residue of a kinase (e.g., CDK (e.g., CDK7)) to allow for covalent attachment of the compound to the kinase. In certain embodiments, the electrophilic moiety (e.g., Michael acceptor moiety) may react with a cysteine residue of a kinase (e.g., CDK (e.g., CDK7)). In certain embodiments, the electrophilic moiety (e.g., Michael acceptor moiety) may react with the Cys312 residue of CDK7. In certain embodiments, the covalent attachment is irreversible. In certain embodiments, the covalent attachment is reversible.
[0274] As generally defined herein in Formulae (I'), (II'), (I), and (II), R.sup.2 may be any one of Formulae (i-1)-(i-41). In certain embodiments, R.sup.2 is of Formula (i-1):
##STR00093##
In certain embodiments, R.sup.2 is of Formula (i-2):
##STR00094##
In certain embodiments, R.sup.2 is of Formula (i-3):
##STR00095##
In certain embodiments, R.sup.2 is of Formula (i-4):
##STR00096##
In certain embodiments, R.sup.2 is of Formula (i-5):
##STR00097##
In certain embodiments, R.sup.2 is of Formula (i-6):
##STR00098##
In certain embodiments, R.sup.2 is of Formula (i-7):
##STR00099##
In certain embodiments, R.sup.2 is of Formula (i-8):
##STR00100##
In certain embodiments, R.sup.2 is of Formula (i-9):
##STR00101##
In certain embodiments, R.sup.2 is of Formula (i-10):
##STR00102##
In certain embodiments, R.sup.2 is of Formula (i-11):
##STR00103##
In certain embodiments, R.sup.2 is of Formula (i-12):
##STR00104##
In certain embodiments, R.sup.2 is of Formula (i-13):
##STR00105##
In certain embodiments, R.sup.2 is of Formula (i-14):
##STR00106##
In certain embodiments, R.sup.2 is of Formula (i-15):
##STR00107##
In certain embodiments, R.sup.2 is of Formula (i-16):
##STR00108##
In certain embodiments, R.sup.2 is of Formula (i-17):
##STR00109##
In certain embodiments, R.sup.2 is of Formula (i-18):
##STR00110##
In certain embodiments, R.sup.2 is of Formula (i-19):
##STR00111##
In certain embodiments, R.sup.2 is of Formula (i-20):
##STR00112##
In certain embodiments, R.sup.2 is of Formula (i-21):
##STR00113##
In certain embodiments, R.sup.2 is of Formula (i-22):
##STR00114##
In certain embodiments, R.sup.2 is of Formula (i-23):
##STR00115##
In certain embodiments, R.sup.2 is of Formula (i-24):
##STR00116##
In certain embodiments, R.sup.2 is of Formula (i-25):
##STR00117##
In certain embodiments, R.sup.2 is of Formula (i-26):
##STR00118##
In certain embodiments, R.sup.2 is of Formula (i-27):
##STR00119##
In certain embodiments, R.sup.2 is of Formula (i-28):
##STR00120##
In certain embodiments, R.sup.2 is of Formula (i-29):
##STR00121##
In certain embodiments, R.sup.2 is of Formula (i-30):
##STR00122##
In certain embodiments, R.sup.2 is of Formula (i-31):
##STR00123##
In certain embodiments, R.sup.2 is of Formula (i-32):
##STR00124##
In certain embodiments, R.sup.2 is of Formula (i-33):
##STR00125##
In certain embodiments, R.sup.2 is of Formula (i-34):
##STR00126##
In certain embodiments, R.sup.2 is of Formula (i-35):
##STR00127##
In certain embodiments, R.sup.2 is of Formula (i-36):
##STR00128##
In certain embodiments, R.sup.2 is of Formula (i-37):
##STR00129##
In certain embodiments, R.sup.2 is of Formula (i-38):
##STR00130##
In certain embodiments, R.sup.2 is of Formula (i-39)
##STR00131##
In certain embodiments, R.sup.2 is of Formula (i-40):
##STR00132##
In certain embodiments, R.sup.2 is of Formula (i-41):
##STR00133##
[0275] In certain embodiments, R.sup.2 is of Formula (i-1a):
##STR00134##
In certain embodiments, R.sup.2 is of Formula (i-1b):
##STR00135##
In certain embodiments, R.sup.2 is of Formula (i-1c):
##STR00136##
In certain embodiments, R.sup.2 is of Formula (i-1d):
##STR00137##
In certain embodiments, R.sup.2 is of Formula (i-1e):
##STR00138##
In certain embodiments, R.sup.2 is of Formula (i-1f):
##STR00139##
In certain embodiments, R.sup.2 is of Formula (i-1g)
##STR00140##
In certain embodiments, R.sup.2 is
##STR00141##
In certain embodiments, R.sup.2 is
##STR00142##
In certain embodiments, R.sup.2 is
##STR00143##
In certain embodiments, R.sup.2 is of Formula (i-1h):
##STR00144##
In certain embodiments, R.sup.2 is
##STR00145##
[0276] In certain embodiments, R.sup.2 is of Formula (i-1a):
##STR00146##
In certain embodiments, R.sup.2 is of Formula (i-1b):
##STR00147##
In certain embodiments, R.sup.2 is of Formula (i-1c):
##STR00148##
[0277] In certain embodiments, R.sup.2 is of Formula (i-18a):
##STR00149##
In certain embodiments, R.sup.2 is of Formula (i-18b):
##STR00150##
In certain embodiments, R.sup.2 is of Formula (i-18c):
##STR00151##
[0278] In certain embodiments, R.sup.2 is of Formula (i-15a):
##STR00152##
In certain embodiments, R.sup.2 is of Formula (i-15b):
##STR00153##
In certain embodiments, R.sup.2 is of Formula (i-15c):
##STR00154##
[0279] R.sup.2 may contain linker L.sup.3 or L.sup.4. In certain embodiments, L.sup.3 is a bond. L.sup.3 is an optionally substituted C.sub.1-4 hydrocarbon chain. In certain embodiments, L.sup.3 is an optionally substituted C.sub.1-4 hydrocarbon chain, wherein one or more carbon units of the hydrocarbon chain are independently replaced with --C(.dbd.O)--, --O--, --S--, --NR.sup.L3a--, --NR.sup.L3aC(.dbd.O)--, --C(.dbd.O)NR.sup.L3a--, --SC(.dbd.O)--, --C(.dbd.O)S--, --OC(.dbd.O)--, --C(.dbd.O)O--, --NR.sup.L3aC(.dbd.S)--, --C(--S)NR.sup.L3a--, trans-CR.sup.L3b.dbd.CR.sup.L3b--, cis-CR.sup.L3b.dbd.CR.sup.L3b--, --C.ident.C--, --S(.dbd.O)--, --S(.dbd.O)O--, --OS(.dbd.O)--, --S(.dbd.O)NR.sup.L3a--, --NR.sup.L3aS(.dbd.O)--, --S(.dbd.O).sub.2--, --S(.dbd.O).sub.2O--, --OS(.dbd.O).sub.2--, --S(.dbd.O).sub.2NR.sup.L3a--, or --NR.sup.L3aS(.dbd.O).sub.2--. In certain embodiments, L.sup.3 is an optionally substituted C.sub.1-4 hydrocarbon chain, wherein one carbon unit of the hydrocarbon chain is replaced with --NR.sup.L3a-- (e.g., --NH--). In certain embodiments, L.sup.3 is of the formula: --(CH.sub.2).sub.1-4--NR.sup.L3a-- (e.g., --(CH.sub.2).sub.1-4--NH--) or --NR.sup.L3a--CH.sub.2).sub.1-4--(e.g., --NH--CH.sub.2).sub.1-4--). In certain embodiments, L.sup.3 is --NR.sup.L3a. In certain embodiments, L.sup.3 is --NR.sup.L3a(C.dbd.O)--. In certain embodiments, L.sup.3 is --(C.dbd.O)NR.sup.L3a--. In certain embodiments, L.sup.3 is --NH--. In certain embodiments, L.sup.3 is --(C.dbd.O)--. In certain embodiments, L.sup.3 is --NH(C.dbd.O)--. In certain embodiments, L.sup.3 is --(C.dbd.O)NH--. In certain embodiments, L.sup.3 is --O--. In certain embodiments, L.sup.3 is --S--. In certain embodiments, L.sup.4 is a bond. In certain embodiments, L.sup.4 is an optionally substituted C.sub.1-4 hydrocarbon chain.
[0280] Linker L.sup.3 may contain groups R.sup.L3a or R.sup.L3b. In certain embodiments, R.sup.L3a is hydrogen. In certain embodiments, at least one instance of R.sup.L3b is hydrogen. In certain embodiments, each instance of R.sup.L3b is hydrogen. In certain embodiments, at least one instance of R.sup.L3b is --Cl, --Br, or --I. In certain embodiments, each instance of R.sup.L3b is --Cl, --Br, or --I. In certain embodiments, at least one instance of R.sup.L3b is --F. In certain embodiments, each instance of R.sup.L3b is --F. In certain embodiments, at least one instance of R.sup.L3b is optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl. In certain embodiments, two R.sup.L3b groups are joined to form an optionally substituted carbocyclic or optionally substituted heterocyclic ring.
[0281] R.sup.2 may contain groups R.sup.E1, R.sup.E2, and/or R.sup.E3. In certain embodiments, R.sup.E1 is hydrogen. In certain embodiments, R.sup.E2 is hydrogen. In certain embodiments, R.sup.E3 is hydrogen. In certain embodiments, R.sup.E1 is --Cl, --Br, or --I. In certain embodiments, R.sup.E2 is --Cl, --Br, or --I. In certain embodiments, R.sup.E3 is --Cl, --Br, or --I. In certain embodiments, R.sup.E1 is --F. In certain embodiments, R.sup.E2 is --F. In certain embodiments, R.sup.E3 is --F. In certain embodiments, R.sup.E1 is optionally substituted alkyl (e.g., substituted or unsubstituted C.sub.1-6 alkyl). In certain embodiments, R.sup.E2 is optionally substituted alkyl (e.g., substituted or unsubstituted C.sub.1-6 alkyl). In certain embodiments, R.sup.E3 is optionally substituted alkyl (e.g., substituted or unsubstituted C.sub.1-6 alkyl). In certain embodiments, R.sup.E1 is optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --CN, --CH.sub.2OR.sup.EE, --CH.sub.2N(R.sup.EE).sub.2, --CH.sub.2SR.sup.EE, --OR.sup.EE, --N(R.sup.EE).sub.2, --Si(R.sup.EE).sub.3, or --SR.sup.EE. In certain embodiments, R.sup.E2 is optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --CN, --CH.sub.2OR.sup.EE, --CH.sub.2N(R.sup.EE).sub.2, --CH.sub.2SR.sup.EE, --OR.sup.EE, --N(R.sup.EE).sub.2, --Si(R.sup.EE).sub.3, or --SR.sup.EE. In certain embodiments, R.sup.E3 is optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted heteroaryl, --CN, --CH.sub.2OR.sup.EE, --CH.sub.2N(R.sup.EE).sub.2, --CH.sub.2SR.sup.EE, --OR.sup.EE, --N(R.sup.EE).sub.2, --Si(R.sup.EE).sub.3, or --SR.sup.EE. In certain embodiments, R.sup.E1 is --N(R.sup.EE).sub.2. In certain embodiments, R.sup.E2 is --N(R.sup.EE).sub.2. In certain embodiments, R.sup.E3 is --N(R.sup.EE).sub.2. In certain embodiments, R.sup.E1 is --N(CH.sub.3).sub.2. In certain embodiments, R.sup.E2 is --N(CH.sub.3).sub.2. In certain embodiments, R.sup.E3 is --N(CH.sub.3).sub.2. In certain embodiments, R.sup.E1 is --CH.sub.2N(R.sup.EE).sub.2. In certain embodiments, R.sup.E2 is --CH.sub.2N(R.sup.EE).sub.2. In certain embodiments, R.sup.E3 is --CH.sub.2N(R.sup.EE).sub.2. In certain embodiments, R.sup.E1 is --CH.sub.2N(CH.sub.3).sub.2. In certain embodiments, R.sup.E2 is --CH.sub.2N(CH.sub.3).sub.2. In certain embodiments, R.sup.E3 is --CH.sub.2N(CH.sub.3).sub.2. In certain embodiments, R.sup.E1 is --CN. In certain embodiments, R.sup.E2 is --CN. In certain embodiments, R.sup.E3 is --CN.
[0282] In certain embodiments, R.sup.E1 and R.sup.E3 are joined to form an optionally substituted carbocyclic ring. In certain embodiments, R.sup.E1 and R.sup.E3 are joined to form an optionally substituted heterocyclic ring. In certain embodiments, R.sup.E2 and R.sup.E3 are joined to form an optionally substituted carbocyclic ring. In certain embodiments, R.sup.E2 and R.sup.E3 are joined to form an optionally substituted heterocyclic ring. In certain embodiments, R.sup.E1 and R.sup.E2 are joined to form an optionally substituted carbocyclic ring. In certain embodiments, R.sup.E1 and R.sup.E2 are joined to form an optionally substituted heterocyclic ring.
[0283] R.sup.2 may contain group R.sup.E4, where R.sup.E4 is a leaving group. In certain embodiments, R.sup.E4 is --Cl, --Br, or --I. In certain embodiments, R.sup.E4 is --F. In certain embodiments, R.sup.E4 is --OS(.dbd.O)R.sup.E4a or --OS(.dbd.O).sub.2R.sup.E4a, wherein R.sup.E4a is substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted carbocyclyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, or substituted or unsubstituted heteroaryl. In certain embodiments, R.sup.E4 is --OR.sup.E4a. In certain embodiments, R.sup.E4 is --OMs, --OTf --OTs, --OBs, or 2-nitrobenzenesulfonyloxy. In certain embodiments, R.sup.E4 is --OR.sup.E4a. In certain embodiments, R.sup.E4 is --OMe, --OCF.sub.3, or --OPh. In certain embodiments, R.sup.E4 is --OC(.dbd.O)R.sup.E4a. In certain embodiments, R.sup.E4 is --OC(.dbd.O)Me, --OC(.dbd.O)CF.sub.3, --OC(.dbd.O)Ph, or --OC(.dbd.O)Cl. In certain embodiments, R.sup.E4 is --OC(O)OR.sup.E4a. In certain embodiments, R.sup.E4 is --OC(.dbd.O)OMe or --OC(.dbd.O)O(t-Bu).
[0284] R.sup.2 may contain group R.sup.E5, where R.sup.E5 is a halogen. In certain embodiments, R.sup.E5 is --Cl, --Br, or --I. In certain embodiments, R.sup.E5 is --F.
[0285] R.sup.2 may contain group R.sup.E6. In certain embodiments, R.sup.E6 is hydrogen. In certain embodiments, R.sup.E6 is substituted or unsubstituted C.sub.1-C.sub.6 alkyl. In certain embodiments, R.sup.E6 is a nitrogen protecting group.
[0286] In certain embodiments, a is 1. In certain embodiments, a is 2.
[0287] In certain embodiments, z is 0. In certain embodiments, z is 1. In certain embodiments, z is 2. In certain embodiments, z is 3, 4, 5, or 6.
[0288] R.sup.2 may contain group Y. In certain embodiments, Y is O. In certain embodiments, Y is S. In certain embodiments, Y is NR.sup.E7. In certain embodiments, Y is NH. In certain embodiments, a compound of Formula (I') is of Formula (I).
[0289] In certain embodiments, the compound of Formula (I) is the formula:
##STR00155##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0290] In certain embodiments, the compound of Formula (I) is the formula:
##STR00156##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0291] In certain embodiments, the compound of Formula (I) is the formula:
##STR00157##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0292] In certain embodiments, a compound of Formula (I) is of Formula (I-i):
##STR00158##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, Ring A, Ring C, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0293] In certain embodiments, a compound of Formula (I) is of Formula (I-ii):
##STR00159##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof wherein L.sup.1, L.sup.2, X, Ring A, Ring C, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0294] In certain embodiments, a compound of Formula (I) is of Formula (I-ii-a):
##STR00160##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0295] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00161##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0296] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00162##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0297] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00163##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0298] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00164##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0299] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00165##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0300] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00166##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0301] In certain embodiments, a compound of Formula (I) is of Formula (I-ii-b):
##STR00167##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0302] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00168##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0303] In certain embodiments, a compound of Formula (I) is of Formula (I-ii-c):
##STR00169##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0304] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00170##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0305] In certain embodiments, a compound of Formula (I) is of Formula (I-ii-A):
##STR00171##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, b1, and c1 are as defined herein.
[0306] In certain embodiments, a compound of Formula (I) is of Formula (I-ii):
##STR00172##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof; wherein L.sup.1, L.sup.2, X, Ring A, Ring C, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0307] In certain embodiments, a compound of Formula (I) is of Formula (I-iv):
##STR00173##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, Ring A, Ring C, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0308] In certain embodiments, a compound of Formula (I) is of Formula (I-iv-a):
##STR00174##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0309] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00175##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0310] In certain embodiments, a compound of Formula (I) is of Formula (I-iv-b):
##STR00176##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0311] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00177##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0312] In certain embodiments, a compound of Formula (I) is of Formula (I-v):
##STR00178##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof wherein L.sup.1, L.sup.2, X, Ring A, Ring C, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0313] In certain embodiments, a compound of Formula (I) is of Formula (I-v-a):
##STR00179##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0314] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00180##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0315] In certain embodiments, a compound of Formula (I) is of Formula (I-v-b):
##STR00181##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof wherein L.sup.1, L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0316] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00182##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0317] In certain embodiments, a compound of Formula (I') is of Formula (I'-A):
##STR00183##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0318] In certain embodiments, a compound of Formula (I') is of the formula:
##STR00184##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, b1, and c1 are as defined herein.
[0319] In certain embodiments, a compound of Formula (I') is of Formula (I'-i):
##STR00185##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof; wherein L.sup.1, L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0320] In certain embodiments, a compound of Formula (I') is of Formula (I'-ii):
##STR00186##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof; wherein L.sup.1, L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0321] In certain embodiments, a compound of Formula (I') is of Formula (I'-iii):
##STR00187##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof; wherein L.sup.1, L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, a1, b1, and c1 are as defined herein.
[0322] In certain embodiments, a compound of Formula (I') is of the formula:
##STR00188##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.1, L.sup.2, X, R.sup.1, R.sup.2, R.sup.A, R.sup.B, R.sup.C, b1, and c1 are as defined herein.
[0323] In certain embodiments, a compound of Formula (II') is of Formula (II).
[0324] In certain embodiments, the compound of Formula (II) is the formula:
##STR00189##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0325] In certain embodiments, the compound of Formula (II) is the formula:
##STR00190##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0326] In certain embodiments, the compound of Formula (II) is the formula:
##STR00191##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0327] In certain embodiments, a compound of Formula (II) is of Formula (II-i):
##STR00192##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, a1, and b1 are as defined herein.
[0328] In certain embodiments, a compound of Formula (II) is of the formula:
##STR00193##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0329] In certain embodiments, a compound of Formula (II) is of the formula:
##STR00194##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0330] In certain embodiments, a compound of Formula (II') is of Formula (II'-i):
##STR00195##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, wherein L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, and b1 are as defined herein.
[0331] In certain embodiments, the compound of Formula (II') is of the formula:
##STR00196##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0332] In certain embodiments, a compound of Formula (II) is of Formula (II-ii):
##STR00197##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof; wherein L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, a1, and b1 are as defined herein.
[0333] In certain embodiments, a compound of Formula (II) is of the formula:
##STR00198##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0334] In certain embodiments, a compound of Formula (II) is of the formula:
##STR00199##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0335] In certain embodiments, a compound of Formula (II) is of Formula (II-iii):
##STR00200##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof wherein L.sup.2, X, Ring A, R.sup.1, R.sup.2, R.sup.A, R.sup.B, a1, and b1 are as defined herein.
[0336] In certain embodiments, a compound of Formula (II) is of the formula:
##STR00201##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0337] In certain embodiments, a compound of Formula (II) is of the formula:
##STR00202##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0338] In certain embodiments, a compound of Formula (I') is of the formula:
##STR00203## ##STR00204## ##STR00205## ##STR00206## ##STR00207## ##STR00208## ##STR00209## ##STR00210##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0339] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00211## ##STR00212## ##STR00213## ##STR00214## ##STR00215##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0340] In certain embodiments, a compound of Formula (I) is of the formula:
##STR00216## ##STR00217## ##STR00218## ##STR00219## ##STR00220## ##STR00221## ##STR00222## ##STR00223## ##STR00224## ##STR00225## ##STR00226## ##STR00227## ##STR00228## ##STR00229## ##STR00230## ##STR00231## ##STR00232## ##STR00233## ##STR00234## ##STR00235## ##STR00236##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0341] In certain embodiments, a compound of Formula (II') is of the formula:
##STR00237##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
[0342] In certain embodiments, a compound of Formula (II) is of the formula:
##STR00238## ##STR00239##
or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof.
Pharmaceutical Compositions and Administration
[0343] The pharmaceutical compositions described herein may be useful in treating and/or preventing proliferative diseases (e.g., cancers (e.g., leukemia, acute lymphoblastic leukemia, lymphoma, Burkitt's lymphoma, melanoma, multiple myeloma, breast cancer, Ewing's sarcoma, osteosarcoma, brain cancer, neuroblastoma, lung cancer, colorectal cancer), benign neoplasms, diseases associated with angiogenesis, inflammatory diseases, autoinflammatory diseases, and autoimmune diseases) in a subject. The compositions described herein may also be useful for inhibiting the activity of a protein kinase (e.g., CDK (e.g., CDK7, CDK12, and/or CDK13)) in a subject, biological sample, tissue, or cell. The compositions described herein may also be useful for inducing apoptosis in a cell.
[0344] The present disclosure provides pharmaceutical compositions comprising a compound described herein (e.g., a compound of any one of Formulae (I'), (II'), (I), (II), or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, and optionally a pharmaceutically acceptable excipient. In certain embodiments, the pharmaceutical composition of the invention comprises a compound described herein, or a pharmaceutically acceptable salt thereof, and optionally a pharmaceutically acceptable excipient. In certain embodiments, a pharmaceutical composition described herein comprises a compound described herein, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient. In certain embodiments, the compound described herein, or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, is provided in an effective amount in the pharmaceutical composition.
[0345] In certain embodiments, the effective amount is a therapeutically effective amount (e.g., amount effective for treating a proliferative disease in a subject in need thereof). In certain embodiments, the effective amount is an amount effective for inhibiting the activity of a protein kinase (e.g., CDK (e.g., CDK7, CDK12, and/or CDK13)) in a subject in need thereof. In certain embodiments, the effective amount is an amount effective for inhibiting the activity of a protein kinase (e.g., CDK (e.g., CDK7, CDK12, and/or CDK13)) in a cell. In certain embodiments, the effective amount is an amount effective for inducing apoptosis in a cell. In certain embodiments, the effective amount is a prophylactically effective amount (e.g., amount effective for preventing a proliferative disease in a subject in need thereof and/or for keeping a subject in need thereof in remission of a proliferative disease).
[0346] In certain embodiments, the protein kinase being inhibited is a CDK. In certain embodiments, the protein kinase being inhibited is CDK1, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8, CDK9, CDK10, CDK11, CDK12, CDK13, CDK14, CDK15, CDK16, CDK17, CDK18, CDK19, or CDK20. In certain embodiments, the protein kinase being inhibited is CDK7. In certain embodiments, the protein kinase being inhibited is CDK12. In certain embodiments, the protein kinase being inhibited is CDK13. In certain embodiments, the protein kinase being inhibited is a Src family kinase. In certain embodiments, the protein kinase being inhibited is SRC. In certain embodiments, the protein kinase being inhibited is FGR. In certain embodiments, the protein kinase being inhibited is BUB1B. In certain embodiments, the protein kinase being inhibited is CHEK2. In certain embodiments, the protein kinase being inhibited is HIPK4. In certain embodiments, the protein kinase being inhibited is PRKCQ. In certain embodiments, the protein kinase being inhibited is R.sup.EET. In certain embodiments, the protein kinase being inhibited is MELK. In certain embodiments, the protein kinase being inhibited is IRAK1, IRAK4, BMX, or PI3K. In certain embodiments, the protein kinase being inhibited is ABL, ARG, BLK, CSK, EphB1, EphB2, FGR, FRK, FYN, SRC, YES, LCK, LYN, MAP2K5, NLK, p38a, SNRK, or TEC. In certain embodiments, the protein kinase being inhibited is ABL1(H396P)-phosphorylated, ABL1-phosphorylated, BLK, EPHA4, EPHB2, EPHB3, EPHB4, FGR, JAK3(JH1domain-catalytic), KIT, KIT(L576P), KIT(V559D), PDGFRB, SRC, YES, ABL1(H396P)-nonphosphorylated, ABL1 (Y253F)-phosphorylated, ABL1-nonphosphorylated, FRK, LYN, ABL1(Q252H)-nonphosphorylated, DDR1, EPHB1, ERBB4, p38-alpha, ABL2, ABL1(Q252H)-phosphorylated, SIK, EPHA8, MEK5, ABL1(E255K)-phosphorylated, ABL1(F317L)-nonphosphorylated, FYN, LCK, EPHA2, ABL1(M351T)-phosphorylated, TXK, EGFR(L858R), EGFR(L861Q), ERBB2, ERBB3, EPHA5, ABL1(F317I)-nonphosphorylated, EGFR(L747-E749del, A750P), CSK, EPHA1, ABL1(F317L)-phosphorylated, BRAF(V600E), EGFR, KIT-autoinhibited, or EGFR(E746-A750del). In certain embodiments, the protein kinase being inhibited is ABL1(F317L)-nonphosphorylated, ABL1 (H396P)-nonphosphorylated, ABL1 (H396P)-phosphorylated, ABL1-phosphorylated, BLK, EPHA4, EPHB2, EPHB3, EPHB4, JAK3(JH1domain-catalytic), KIT, KIT(L576P), KIT(V559D), LYN, PDGFRB, SRC, YES, ABL1-nonphosphorylated, ABL1(Y253F)-phosphorylated, ERBB3, FGR, FRK, p38-alpha, ABL1(F317I)-nonphosphorylated, DDR1, EPHA2, ABL1 (Q252H)-phosphorylated, MEK5, ABL1 (Q252H)-nonphosphorylated, ABL2, FYN, EPHB1, ABL1(E255K)-phosphorylated, ABL1 (F317L)-phosphorylated, EPHA1, ABL1(M351T)-phosphorylated, ERBB4, TXK, LCK, EPHA8, SIK, EPHA5, EGFR(L861Q), CSF1R-autoinhibited, BRAF(V600E), BRK, CSK, KIT(D816V), KIT-autoinhibited, EGFR(L747-T751del,Sins), EGFR(L858R), EGFR(L747-E749del, A750P), or CSF1R.
[0347] In certain embodiments, the effective amount is an amount effective for inhibiting the activity of a protein kinase (e.g., CDK (e.g., CDK7, CDK12, and/or CDK13)) by at least about 10%, at least about 20%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, at least about 95%, or at least about 98%. In certain embodiments, the effective amount is an amount effective for inhibiting the activity of a protein kinase (e.g., CDK (e.g., CDK7, CDK12, and/or CDK13)) by not more than 10%, not more than 20%, not more than 30%, not more than 40%, not more than 50%, not more than 60%, not more than 70%, not more than 80%, not more than 90%, not more than 95%, or not more than 98%. In certain embodiments, the effective amount is an amount effective for inhibiting the activity of a protein kinase (e.g., CDK (e.g., CDK7, CDK12, and/or CDK13)) by a range between a percentage described in this paragraph and another percentage described in this paragraph, inclusive.
[0348] Pharmaceutical compositions described herein can be prepared by any method known in the art of pharmacology. In general, such preparatory methods include bringing the compound described herein (i.e., the "active ingredient") into association with a carrier or excipient, and/or one or more other accessory ingredients, and then, if necessary and/or desirable, shaping, and/or packaging the product into a desired single- or multi-dose unit.
[0349] Pharmaceutical compositions can be prepared, packaged, and/or sold in bulk, as a single unit dose, and/or as a plurality of single unit doses. A "unit dose" is a discrete amount of the pharmaceutical composition comprising a predetermined amount of the active ingredient. The amount of the active ingredient is generally equal to the dosage of the active ingredient which would be administered to a subject and/or a convenient fraction of such a dosage, such as one-half or one-third of such a dosage.
[0350] Relative amounts of the active ingredient, the pharmaceutically acceptable excipient, and/or any additional ingredients in a pharmaceutical composition described herein will vary, depending upon the identity, size, and/or condition of the subject treated and further depending upon the route by which the composition is to be administered. The composition may comprise between about 0.1% and about 100% (w/w) of the active ingredient.
[0351] Pharmaceutically acceptable excipients used in the manufacture of provided pharmaceutical compositions include inert diluents, dispersing and/or granulating agents, surface active agents and/or emulsifiers, disintegrating agents, binding agents, preservatives, buffering agents, lubricating agents, and/or oils. Excipients such as cocoa butter and suppository waxes, coloring agents, coating agents, sweetening, flavoring, and perfuming agents may also be present in the composition.
[0352] Exemplary diluents include calcium carbonate, sodium carbonate, calcium phosphate, dicalcium phosphate, calcium sulfate, calcium hydrogen phosphate, sodium phosphate lactose, sucrose, cellulose, microcrystalline cellulose, kaolin, mannitol, sorbitol, inositol, sodium chloride, dry starch, cornstarch, powdered sugar, and mixtures thereof.
[0353] Exemplary granulating and/or dispersing agents include potato starch, corn starch, tapioca starch, sodium starch glycolate, clays, alginic acid, guar gum, citrus pulp, agar, bentonite, cellulose, and wood products, natural sponge, cation-exchange resins, calcium carbonate, silicates, sodium carbonate, cross-linked poly(vinyl-pyrrolidone) (crospovidone), sodium carboxymethyl starch (sodium starch glycolate), carboxymethyl cellulose, cross-linked sodium carboxymethyl cellulose (croscarmellose), methylcellulose, pregelatinized starch (starch 1500), microcrystalline starch, water insoluble starch, calcium carboxymethyl cellulose, magnesium aluminum silicate (Veegum), sodium lauryl sulfate, quaternary ammonium compounds, and mixtures thereof.
[0354] Exemplary surface active agents and/or emulsifiers include natural emulsifiers (e.g., acacia, agar, alginic acid, sodium alginate, tragacanth, chondrux, cholesterol, xanthan, pectin, gelatin, egg yolk, casein, wool fat, cholesterol, wax, and lecithin), colloidal clays (e.g., bentonite (aluminum silicate) and Veegum (magnesium aluminum silicate)), long chain amino acid derivatives, high molecular weight alcohols (e.g., stearyl alcohol, cetyl alcohol, oleyl alcohol, triacetin monostearate, ethylene glycol distearate, glyceryl monostearate, and propylene glycol monostearate, polyvinyl alcohol), carbomers (e.g., carboxy polymethylene, polyacrylic acid, acrylic acid polymer, and carboxyvinyl polymer), carrageenan, cellulosic derivatives (e.g., carboxymethylcellulose sodium, powdered cellulose, hydroxymethyl cellulose, hydroxypropyl cellulose, hydroxypropyl methylcellulose, methylcellulose), sorbitan fatty acid esters (e.g., polyoxyethylene sorbitan monolaurate (Tween.RTM. 20), polyoxyethylene sorbitan (Tween.RTM. 60), polyoxyethylene sorbitan monooleate (Tween.RTM. 80), sorbitan monopalmitate (Span.RTM. 40), sorbitan monostearate (Span.RTM. 60), sorbitan tristearate (Span.RTM.65), glyceryl monooleate, sorbitan monooleate (Spana 80), polyoxyethylene esters (e.g., polyoxyethylene monostearate (Myrj.RTM.45), polyoxyethylene hydrogenated castor oil, polyethoxylated castor oil, polyoxymethylene stearate, and Solutol.RTM.), sucrose fatty acid esters, polyethylene glycol fatty acid esters (e.g., Cremophor.RTM.), polyoxyethylene ethers, (e.g., polyoxyethylene lauryl ether (Brij.RTM. 30)), poly(vinyl-pyrrolidone), diethylene glycol monolaurate, triethanolamine oleate, sodium oleate, potassium oleate, ethyl oleate, oleic acid, ethyl laurate, sodium lauryl sulfate, Pluronic.RTM. F-68, poloxamer P-188, cetrimonium bromide, cetylpyridinium chloride, benzalkonium chloride, docusate sodium, and/or mixtures thereof.
[0355] Exemplary binding agents include starch (e.g., cornstarch and starch paste), gelatin, sugars (e.g., sucrose, glucose, dextrose, dextrin, molasses, lactose, lactitol, mannitol, etc.), natural and synthetic gums (e.g., acacia, sodium alginate, extract of Irish moss, panwar gum, ghatti gum, mucilage of isapol husks, carboxymethylcellulose, methylcellulose, ethylcellulose, hydroxyethylcellulose, hydroxypropyl cellulose, hydroxypropyl methylcellulose, microcrystalline cellulose, cellulose acetate, poly(vinyl-pyrrolidone), magnesium aluminum silicate (Veegum.RTM.), and larch arabogalactan), alginates, polyethylene oxide, polyethylene glycol, inorganic calcium salts, silicic acid, polymethacrylates, waxes, water, alcohol, and/or mixtures thereof.
[0356] Exemplary preservatives include antioxidants, chelating agents, antimicrobial preservatives, antifungal preservatives, antiprotozoan preservatives, alcohol preservatives, acidic preservatives, and other preservatives. In certain embodiments, the preservative is an antioxidant. In other embodiments, the preservative is a chelating agent.
[0357] Exemplary antioxidants include alpha tocopherol, ascorbic acid, ascorbyl palmitate, butylated hydroxyanisole, butylated hydroxytoluene, monothioglycerol, potassium metabisulfite, propionic acid, propyl gallate, sodium ascorbate, sodium bisulfite, sodium metabisulfite, and sodium sulfite.
[0358] Exemplary chelating agents include ethylenediaminetetraacetic acid (EDTA) and salts and hydrates thereof (e.g., sodium edetate, disodium edetate, trisodium edetate, calcium disodium edetate, dipotassium edetate, and the like), citric acid and salts and hydrates thereof (e.g., citric acid monohydrate), fumaric acid and salts and hydrates thereof, malic acid and salts and hydrates thereof, phosphoric acid and salts and hydrates thereof, and tartaric acid and salts and hydrates thereof. Exemplary antimicrobial preservatives include benzalkonium chloride, benzethonium chloride, benzyl alcohol, bronopol, cetrimide, cetylpyridinium chloride, chlorhexidine, chlorobutanol, chlorocresol, chloroxylenol, cresol, ethyl alcohol, glycerin, hexetidine, imidurea, phenol, phenoxyethanol, phenylethyl alcohol, phenylmercuric nitrate, propylene glycol, and thimerosal.
[0359] Exemplary antifungal preservatives include butyl paraben, methyl paraben, ethyl paraben, propyl paraben, benzoic acid, hydroxybenzoic acid, potassium benzoate, potassium sorbate, sodium benzoate, sodium propionate, and sorbic acid.
[0360] Exemplary alcohol preservatives include ethanol, polyethylene glycol, phenol, phenolic compounds, bisphenol, chlorobutanol, hydroxybenzoate, and phenylethyl alcohol.
[0361] Exemplary acidic preservatives include vitamin A, vitamin C, vitamin E, beta-carotene, citric acid, acetic acid, dehydroacetic acid, ascorbic acid, sorbic acid, and phytic acid.
[0362] Other preservatives include tocopherol, tocopherol acetate, deteroxime mesylate, cetrimide, butylated hydroxyanisol (BHA), butylated hydroxytoluened (BHT), ethylenediamine, sodium lauryl sulfate (SLS), sodium lauryl ether sulfate (SLES), sodium bisulfite, sodium metabisulfite, potassium sulfite, potassium metabisulfite, Glydant.RTM. Plus, Phenonip.RTM., methylparaben, Germall.RTM. 115, Germaben.RTM. II, Neolone.RTM., Kathon.RTM., and Euxyl.RTM..
[0363] Exemplary buffering agents include citrate buffer solutions, acetate buffer solutions, phosphate buffer solutions, ammonium chloride, calcium carbonate, calcium chloride, calcium citrate, calcium glubionate, calcium gluceptate, calcium gluconate, D-gluconic acid, calcium glycerophosphate, calcium lactate, propanoic acid, calcium levulinate, pentanoic acid, dibasic calcium phosphate, phosphoric acid, tribasic calcium phosphate, calcium hydroxide phosphate, potassium acetate, potassium chloride, potassium gluconate, potassium mixtures, dibasic potassium phosphate, monobasic potassium phosphate, potassium phosphate mixtures, sodium acetate, sodium bicarbonate, sodium chloride, sodium citrate, sodium lactate, dibasic sodium phosphate, monobasic sodium phosphate, sodium phosphate mixtures, tromethamine, magnesium hydroxide, aluminum hydroxide, alginic acid, pyrogen-free water, isotonic saline, Ringer's solution, ethyl alcohol, and mixtures thereof.
[0364] Exemplary lubricating agents include magnesium stearate, calcium stearate, stearic acid, silica, talc, malt, glyceryl behanate, hydrogenated vegetable oils, polyethylene glycol, sodium benzoate, sodium acetate, sodium chloride, leucine, magnesium lauryl sulfate, sodium lauryl sulfate, and mixtures thereof.
[0365] Exemplary natural oils include almond, apricot kernel, avocado, babassu, bergamot, black current seed, borage, cade, camomile, canola, caraway, carnauba, castor, cinnamon, cocoa butter, coconut, cod liver, coffee, corn, cotton seed, emu, eucalyptus, evening primrose, fish, flaxseed, geraniol, gourd, grape seed, hazel nut, hyssop, isopropyl myristate, jojoba, kukui nut, lavandin, lavender, lemon, litsea cubeba, macademia nut, mallow, mango seed, meadowfoam seed, mink, nutmeg, olive, orange, orange roughy, palm, palm kernel, peach kernel, peanut, poppy seed, pumpkin seed, rapeseed, rice bran, rosemary, safflower, sandalwood, sasquana, savoury, sea buckthorn, sesame, shea butter, silicone, soybean, sunflower, tea tree, thistle, tsubaki, vetiver, walnut, and wheat germ oils. Exemplary synthetic oils include, but are not limited to, butyl stearate, caprylic triglyceride, capric triglyceride, cyclomethicone, diethyl sebacate, dimethicone 360, isopropyl myristate, mineral oil, octyldodecanol, oleyl alcohol, silicone oil, and mixtures thereof.
[0366] Liquid dosage forms for oral and parenteral administration include pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs. In addition to the active ingredients, the liquid dosage forms may comprise inert diluents commonly used in the art such as, for example, water or other solvents, solubilizing agents and emulsifiers such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, dimethylformamide, oils (e.g., cottonseed, groundnut, corn, germ, olive, castor, and sesame oils), glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, and mixtures thereof. Besides inert diluents, the oral compositions can include adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, and perfuming agents. In certain embodiments for parenteral administration, the conjugates described herein are mixed with solubilizing agents such as Cremophor.RTM., alcohols, oils, modified oils, glycols, polysorbates, cyclodextrins, polymers, and mixtures thereof.
[0367] Injectable preparations, for example, sterile injectable aqueous or oleaginous suspensions can be formulated according to the known art using suitable dispersing or wetting agents and suspending agents. The sterile injectable preparation can be a sterile injectable solution, suspension, or emulsion in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol. Among the acceptable vehicles and solvents that can be employed are water, Ringer's solution, U.S.P., and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose any bland fixed oil can be employed including synthetic mono- or di-glycerides. In addition, fatty acids such as oleic acid are used in the preparation of injectables.
[0368] The injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium prior to use.
[0369] In order to prolong the effect of a drug, it is often desirable to slow the absorption of the drug from subcutaneous or intramuscular injection. This can be accomplished by the use of a liquid suspension of crystalline or amorphous material with poor water solubility. The rate of absorption of the drug then depends upon its rate of dissolution, which, in turn, may depend upon crystal size and crystalline form. Alternatively, delayed absorption of a parenterally administered drug form may be accomplished by dissolving or suspending the drug in an oil vehicle.
[0370] Compositions for rectal or vaginal administration are typically suppositories which can be prepared by mixing the conjugates described herein with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol, or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active ingredient.
[0371] Solid dosage forms for oral administration include capsules, tablets, pills, powders, and granules. In such solid dosage forms, the active ingredient is mixed with at least one inert, pharmaceutically acceptable excipient or carrier such as sodium citrate or dicalcium phosphate and/or (a) fillers or extenders such as starches, lactose, sucrose, glucose, mannitol, and silicic acid, (b) binders such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidinone, sucrose, and acacia, (c) humectants such as glycerol, (d) disintegrating agents such as agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate, (e) solution retarding agents such as paraffin, (f) absorption accelerators such as quaternary ammonium compounds, (g) wetting agents such as, for example, cetyl alcohol and glycerol monostearate, (h) absorbents such as kaolin and bentonite clay, and (i) lubricants such as talc, calcium stearate, magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate, and mixtures thereof. In the case of capsules, tablets, and pills, the dosage form may include a buffering agent.
[0372] Solid compositions of a similar type can be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethylene glycols and the like. The solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings and other coatings well known in the art of pharmacology. They may optionally comprise opacifying agents and can be of a composition that they release the active ingredient(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner. Examples of encapsulating compositions which can be used include polymeric substances and waxes. Solid compositions of a similar type can be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethylene glycols and the like.
[0373] The active ingredient can be in a micro-encapsulated form with one or more excipients as noted above. The solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings, release controlling coatings, and other coatings well known in the pharmaceutical formulating art. In such solid dosage forms the active ingredient can be admixed with at least one inert diluent such as sucrose, lactose, or starch. Such dosage forms may comprise, as is normal practice, additional substances other than inert diluents, e.g., tableting lubricants and other tableting aids such a magnesium stearate and microcrystalline cellulose. In the case of capsules, tablets and pills, the dosage forms may comprise buffering agents. They may optionally comprise opacifying agents and can be of a composition that they release the active ingredient(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner. Examples of encapsulating agents which can be used include polymeric substances and waxes.
[0374] Dosage forms for topical and/or transdermal administration of a compound described herein may include ointments, pastes, creams, lotions, gels, powders, solutions, sprays, inhalants, and/or patches. Generally, the active ingredient is admixed under sterile conditions with a pharmaceutically acceptable carrier or excipient and/or any needed preservatives and/or buffers as can be required. Additionally, the present disclosure contemplates the use of transdermal patches, which often have the added advantage of providing controlled delivery of an active ingredient to the body. Such dosage forms can be prepared, for example, by dissolving and/or dispensing the active ingredient in the proper medium. Alternatively or additionally, the rate can be controlled by either providing a rate controlling membrane and/or by dispersing the active ingredient in a polymer matrix and/or gel.
[0375] Suitable devices for use in delivering intradermal pharmaceutical compositions described herein include short needle devices. Intradermal compositions can be administered by devices which limit the effective penetration length of a needle into the skin. Alternatively or additionally, conventional syringes can be used in the classical mantoux method of intradermal administration. Jet injection devices which deliver liquid formulations to the dermis via a liquid jet injector and/or via a needle which pierces the stratum corneum and produces a jet which reaches the dermis are suitable. Ballistic powder/particle delivery devices which use compressed gas to accelerate the compound in powder form through the outer layers of the skin to the dermis are suitable.
[0376] Formulations suitable for topical administration include, but are not limited to, liquid and/or semi-liquid preparations such as liniments, lotions, oil-in-water and/or water-in-oil emulsions such as creams, ointments, and/or pastes, and/or solutions and/or suspensions. Topically administrable formulations may, for example, comprise from about 1% to about 10% (w/w) active ingredient, although the concentration of the active ingredient can be as high as the solubility limit of the active ingredient in the solvent. Formulations for topical administration may further comprise one or more of the additional ingredients described herein.
[0377] A pharmaceutical composition described herein can be prepared, packaged, and/or sold in a formulation suitable for pulmonary administration via the buccal cavity. Such a formulation may comprise dry particles which comprise the active ingredient and which have a diameter in the range from about 0.5 to about 7 nanometers, or from about 1 to about 6 nanometers. Such compositions are conveniently in the form of dry powders for administration using a device comprising a dry powder reservoir to which a stream of propellant can be directed to disperse the powder and/or using a self-propelling solvent/powder dispensing container such as a device comprising the active ingredient dissolved and/or suspended in a low-boiling propellant in a sealed container. Such powders comprise particles wherein at least 98% of the particles by weight have a diameter greater than 0.5 nanometers and at least 95% of the particles by number have a diameter less than 7 nanometers. Alternatively, at least 95% of the particles by weight have a diameter greater than 1 nanometer and at least 90% of the particles by number have a diameter less than 6 nanometers. Dry powder compositions may include a solid fine powder diluent such as sugar and are conveniently provided in a unit dose form.
[0378] Low boiling propellants generally include liquid propellants having a boiling point of below 65.degree. F. at atmospheric pressure. Generally the propellant may constitute 50 to 99.9% (w/w) of the composition, and the active ingredient may constitute 0.1 to 20% (w/w) of the composition. The propellant may further comprise additional ingredients such as a liquid non-ionic and/or solid anionic surfactant and/or a solid diluent (which may have a particle size of the same order as particles comprising the active ingredient).
[0379] Pharmaceutical compositions described herein formulated for pulmonary delivery may provide the active ingredient in the form of droplets of a solution and/or suspension. Such formulations can be prepared, packaged, and/or sold as aqueous and/or dilute alcoholic solutions and/or suspensions, optionally sterile, comprising the active ingredient, and may conveniently be administered using any nebulization and/or atomization device. Such formulations may further comprise one or more additional ingredients including, but not limited to, a flavoring agent such as saccharin sodium, a volatile oil, a buffering agent, a surface active agent, and/or a preservative such as methylhydroxybenzoate. The droplets provided by this route of administration may have an average diameter in the range from about 0.1 to about 200 nanometers.
[0380] Formulations described herein as being useful for pulmonary delivery are useful for intranasal delivery of a pharmaceutical composition described herein. Another formulation suitable for intranasal administration is a coarse powder comprising the active ingredient and having an average particle from about 0.2 to 500 micrometers. Such a formulation is administered by rapid inhalation through the nasal passage from a container of the powder held close to the nares.
[0381] Formulations for nasal administration may, for example, comprise from about as little as 0.1% (w/w) to as much as 100% (w/w) of the active ingredient, and may comprise one or more of the additional ingredients described herein. A pharmaceutical composition described herein can be prepared, packaged, and/or sold in a formulation for buccal administration. Such formulations may, for example, be in the form of tablets and/or lozenges made using conventional methods, and may contain, for example, 0.1 to 20% (w/w) active ingredient, the balance comprising an orally dissolvable and/or degradable composition and, optionally, one or more of the additional ingredients described herein. Alternately, formulations for buccal administration may comprise a powder and/or an aerosolized and/or atomized solution and/or suspension comprising the active ingredient. Such powdered, aerosolized, and/or aerosolized formulations, when dispersed, may have an average particle and/or droplet size in the range from about 0.1 to about 200 nanometers, and may further comprise one or more of the additional ingredients described herein.
[0382] A pharmaceutical composition described herein can be prepared, packaged, and/or sold in a formulation for ophthalmic administration. Such formulations may, for example, be in the form of eye drops including, for example, a 0.1-1.0% (w/w) solution and/or suspension of the active ingredient in an aqueous or oily liquid carrier or excipient. Such drops may further comprise buffering agents, salts, and/or one or more other of the additional ingredients described herein. Other opthalmically-administrable formulations which are useful include those which comprise the active ingredient in microcrystalline form and/or in a liposomal preparation. Ear drops and/or eye drops are also contemplated as being within the scope of this disclosure.
[0383] Although the descriptions of pharmaceutical compositions provided herein are principally directed to pharmaceutical compositions which are suitable for administration to humans, such compositions are generally suitable for administration to animals of all sorts. Modification of pharmaceutical compositions suitable for administration to humans in order to render the compositions suitable for administration to various animals is well understood, and the ordinarily skilled veterinary pharmacologist can design and/or perform such modification with ordinary experimentation.
[0384] The compounds provided herein are typically formulated in dosage unit form for ease of administration and uniformity of dosage. It will be understood, however, that the total daily usage of the compositions described herein will be decided by a physician within the scope of sound medical judgment. The specific therapeutically effective dose level for any particular subject or organism will depend upon a variety of factors including the disease being treated and the severity of the disorder; the activity of the specific active ingredient employed; the specific composition employed; the age, body weight, general health, sex, and diet of the subject; the time of administration, route of administration, and rate of excretion of the specific active ingredient employed; the duration of the treatment; drugs used in combination or coincidental with the specific active ingredient employed; and like factors well known in the medical arts.
[0385] The compounds and compositions provided herein can be administered by any route, including enteral (e.g., oral), parenteral, intravenous, intramuscular, intra-arterial, intramedullary, intrathecal, subcutaneous, intraventricular, transdermal, interdermal, rectal, intravaginal, intraperitoneal, topical (as by powders, ointments, creams, and/or drops), mucosal, nasal, bucal, sublingual; by intratracheal instillation, bronchial instillation, and/or inhalation; and/or as an oral spray, nasal spray, and/or aerosol. Specifically contemplated routes are oral administration, intravenous administration (e.g., systemic intravenous injection), regional administration via blood and/or lymph supply, and/or direct administration to an affected site. In general, the most appropriate route of administration will depend upon a variety of factors including the nature of the agent (e.g., its stability in the environment of the gastrointestinal tract), and/or the condition of the subject (e.g., whether the subject is able to tolerate oral administration). In certain embodiments, the compound or pharmaceutical composition described herein is suitable for topical administration to the eye of a subject.
[0386] The exact amount of a compound required to achieve an effective amount will vary from subject to subject, depending, for example, on species, age, and general condition of a subject, severity of the side effects or disorder, identity of the particular compound, mode of administration, and the like. An effective amount may be included in a single dose (e.g., single oral dose) or multiple doses (e.g., multiple oral doses). In certain embodiments, when multiple doses are administered to a subject or applied to a biological sample, tissue, or cell, any two doses of the multiple doses include different or substantially the same amounts of a compound described herein. In certain embodiments, when multiple doses are administered to a subject or applied to a biological sample, tissue, or cell, the frequency of administering the multiple doses to the subject or applying the multiple doses to the tissue or cell is three doses a day, two doses a day, one dose a day, one dose every other day, one dose every third day, one dose every week, one dose every two weeks, one dose every three weeks, or one dose every four weeks. In certain embodiments, the frequency of administering the multiple doses to the subject or applying the multiple doses to the tissue or cell is one dose per day. In certain embodiments, the frequency of administering the multiple doses to the subject or applying the multiple doses to the tissue or cell is two doses per day. In certain embodiments, the frequency of administering the multiple doses to the subject or applying the multiple doses to the tissue or cell is three doses per day. In certain embodiments, when multiple doses are administered to a subject or applied to a biological sample, tissue, or cell, the duration between the first dose and last dose of the multiple doses is one day, two days, four days, one week, two weeks, three weeks, one month, two months, three months, four months, six months, nine months, one year, two years, three years, four years, five years, seven years, ten years, fifteen years, twenty years, or the lifetime of the subject, biological sample, tissue, or cell. In certain embodiments, the duration between the first dose and last dose of the multiple doses is three months, six months, or one year. In certain embodiments, the duration between the first dose and last dose of the multiple doses is the lifetime of the subject, biological sample, tissue, or cell. In certain embodiments, a dose (e.g., a single dose, or any dose of multiple doses) described herein includes independently between 0.1 .mu.g and 1 .mu.g, between 0.001 mg and 0.01 mg, between 0.01 mg and 0.1 mg, between 0.1 mg and 1 mg, between 1 mg and 3 mg, between 3 mg and 10 mg, between 10 mg and 30 mg, between 30 mg and 100 mg, between 100 mg and 300 mg, between 300 mg and 1,000 mg, or between 1 g and 10 g, inclusive, of a compound described herein. In certain embodiments, a dose described herein includes independently between 1 mg and 3 mg, inclusive, of a compound described herein. In certain embodiments, a dose described herein includes independently between 3 mg and 10 mg, inclusive, of a compound described herein. In certain embodiments, a dose described herein includes independently between 10 mg and 30 mg, inclusive, of a compound described herein. In certain embodiments, a dose described herein includes independently between 30 mg and 100 mg, inclusive, of a compound described herein.
[0387] Dose ranges as described herein provide guidance for the administration of provided pharmaceutical compositions to an adult. The amount to be administered to, for example, a child or an adolescent can be determined by a medical practitioner or person skilled in the art and can be lower or the same as that administered to an adult. In certain embodiments, a dose described herein is a dose for an adult human whose body weight is approximately 70 kg.
[0388] A compound or composition, as described herein, may be administered in combination with one or more additional pharmaceutical agents (e.g., therapeutically and/or prophylactically active agents) useful in treating and/or preventing a proliferative disease. The compounds or compositions can be administered in combination with additional pharmaceutical agents that improve their activity (e.g., activity (e.g., potency and/or efficacy) in treating a proliferative disease in a subject in need thereof, in preventing a proliferative disease in a subject in need thereof, and/or in inhibiting the activity of a protein kinase (e.g., CDK (e.g., CDK7, CDK12, and/or CDK13)) in a subject, biological sample, tissue, or cell), improve bioavailability, improve safety, reduce drug resistance, reduce and/or modify metabolism, inhibit excretion, and/or modify distribution in a subject, biological sample, tissue, or cell. It will also be appreciated that the therapy employed may achieve a desired effect for the same disorder, and/or it may achieve different effects. In certain embodiments, a pharmaceutical composition described herein including a compound described herein and an additional pharmaceutical agent shows a synergistic effect that is absent in a pharmaceutical composition including one of the compound and the additional pharmaceutical agent, but not both.
[0389] The compound or composition can be administered concurrently with, prior to, or subsequent to one or more additional pharmaceutical agents, which are different from the compound or composition and may be useful as, e.g., combination therapies in treating and/or preventing a proliferative disease. Pharmaceutical agents include therapeutically active agents. Pharmaceutical agents also include prophylactically active agents. Pharmaceutical agents include small organic molecules such as drug compounds (e.g., compounds approved for human or veterinary use by the U.S. Food and Drug Administration as provided in the Code of Federal Regulations (CFR)), peptides, proteins, carbohydrates, monosaccharides, oligosaccharides, polysaccharides, nucleoproteins, mucoproteins, lipoproteins, synthetic polypeptides or proteins, small molecules linked to proteins, glycoproteins, steroids, nucleic acids, DNAs, RNAs, nucleotides, nucleosides, oligonucleotides, antisense oligonucleotides, lipids, hormones, vitamins, and cells. In certain embodiments, the additional pharmaceutical agent is a pharmaceutical agent useful in treating a proliferative disease. In certain embodiments, the additional pharmaceutical agent is a pharmaceutical agent useful in preventing a proliferative disease. In certain embodiments, the additional pharmaceutical agent is a pharmaceutical agent useful in inhibiting the activity of a protein kinase (e.g., CDK (e.g., CDK7, CDK12, and/or CDK13)) in a subject, biological sample, tissue, or cell. In certain embodiments, the additional pharmaceutical agent is a pharmaceutical agent useful in inducing apoptosis in a cell. In certain embodiments, the additional pharmaceutical agent is a pharmaceutical agent approved by a regulatory agency (e.g., the US FDA) for treating and/or preventing a proliferative disease. Each additional pharmaceutical agent may be administered at a dose and/or on a time schedule determined for that pharmaceutical agent. The additional pharmaceutical agent(s) may also be administered together with each other and/or with the compound or composition described herein in a single dose or administered separately in different doses. The particular combination to employ in a regimen will take into account compatibility of the compound described herein with the additional pharmaceutical agent(s) and/or the desired therapeutic and/or prophylactic effect to be achieved. In general, it is expected that the additional pharmaceutical agent(s) in combination be utilized at levels that do not exceed the levels at which they are utilized individually. In some embodiments, the levels utilized in combination will be lower than those utilized individually.
[0390] In certain embodiments, the additional pharmaceutical agent is a cytotoxic agent. In certain embodiments, the additional pharmaceutical agent is an anti-proliferative agent (e.g., anti-cancer agent). In certain embodiments, the additional pharmaceutical agent is an anti-leukemia agent. In certain embodiments, the additional pharmaceutical agent is ABITREXATE (methotrexate), ADE, Adriamycin RDF (doxorubicin hydrochloride), Ambochlorin (chlorambucil), ARRANON (nelarabine), ARZERRA (ofatumumab), BOSULIF (bosutinib), BUSULFEX (busulfan), CAMPATH (alemtuzumab), CERUBIDINE (daunorubicin hydrochloride), CLAFEN (cyclophosphamide), CLOFAREX (clofarabine), CLOLAR (clofarabine), CVP, CYTOSAR-U (cytarabine), CYTOXAN (cyclophosphamide), ERWINAZE (Asparaginase Erwinia Chrysanthemi), FLUDARA (fludarabine phosphate), FOLEX (methotrexate), FOLEX PFS (methotrexate), GAZYVA (obinutuzumab), GLEEVEC (imatinib mesylate), Hyper-CVAD, ICLUSIG (ponatinib hydrochloride), IMBRUVICA (ibrutinib), LEUKERAN (chlorambucil), LINFOLIZIN (chlorambucil), MARQIBO (vincristine sulfate liposome), METHOTREXATE LPF (methorexate), MEXATE (methotrexate), MEXATE-AQ (methotrexate), mitoxantrone hydrochloride, MUSTARGEN (mechlorethamine hydrochloride), MYLERAN (busulfan), NEOSAR (cyclophosphamide), ONCASPAR (Pegaspargase), PURINETHOL (mercaptopurine), PURIXAN (mercaptopurine), Rubidomycin (daunorubicin hydrochloride), SPRYCEL (dasatinib), SYNRIBO (omacetaxine mepesuccinate), TARABINE PFS (cytarabine), TASIGNA (nilotinib), TREANDA (bendamustine hydrochloride), TRISENOX (arsenic trioxide), VINCASAR PFS (vincristine sulfate), ZYDELIG (idelalisib), or a combination thereof. In certain embodiments, the additional pharmaceutical agent is an anti-lymphoma agent. In certain embodiments, the additional pharmaceutical agent is ABITREXATE (methotrexate), ABVD, ABVE, ABVE-PC, ADCETRIS (brentuximab vedotin), ADRIAMYCIN PFS (doxorubicin hydrochloride), ADRIAMYCIN RDF (doxorubicin hydrochloride), AMBOCHLORIN (chlorambucil), AMBOCLORIN (chlorambucil), ARRANON (nelarabine), BEACOPP, BECENUM (carmustine), BELEODAQ (belinostat), BEXXAR (tositumomab and iodine I 131 tositumomab), BICNU (carmustine), BLENOXANE (bleomycin), CARMUBRIS (carmustine), CHOP, CLAFEN (cyclophosphamide), COPP, COPP-ABV, CVP, CYTOXAN (cyclophosphamide), DEPOCYT (liposomal cytarabine), DTIC-DOME (dacarbazine), EPOCH, FOLEX (methotrexate), FOLEX PFS (methotrexate), FOLOTYN (pralatrexate), HYPER-CVAD, ICE, IMBRUVICA (ibrutinib), INTRON A (recombinant interferon alfa-2b), ISTODAX (romidepsin), LEUKERAN (chlorambucil), LINFOLIZIN (chlorambucil), Lomustine, MATULANE (procarbazine hydrochloride), METHOTREXATE LPF (methotrexate), MEXATE (methotrexate), MEXATE-AQ (methotrexate), MOPP, MOZOBIL (plerixafor), MUSTARGEN (mechlorethamine hydrochloride), NEOSAR (cyclophosphamide), OEPA, ONTAK (denileukin diftitox), OPPA, R-CHOP, REVLIMID (lenalidomide), RITUXAN (rituximab), STANFORD V, TREANDA (bendamustine hydrochloride), VAMP, VELBAN (vinblastine sulfate), VELCADE (bortezomib), VELSAR (vinblastine sulfate), VINCASAR PFS (vincristine sulfate), ZEVALIN (ibritumomab tiuxetan), ZOLINZA (vorinostat), ZYDELIG (idelalisib), or a combination thereof. In certain embodiments, the additional pharmaceutical agent is ABITREXATE (methotrexate), ABRAXANE (paclitaxel albumin-stabilized nanoparticle formulation), AC, AC-T, ADE, ADRIAMYCIN PFS (doxorubicin hydrochloride), ADRUCIL (fluorouracil), AFINITOR (everolimus), AFINITOR DISPERZ (everolimus), ALDARA (imiquimod), ALIMTA (pemetrexed disodium), AREDIA (pamidronate disodium), ARIMIDEX (anastrozole), AROMASIN (exemestane), AVASTIN (bevacizumab), BECENUM (carmustine), BEP, BICNU (carmustine), BLENOXANE (bleomycin), CAF, CAMPTOSAR (irinotecan hydrochloride), CAPOX, CAPRELSA (vandetanib), CARBOPLATIN-TAXOL, CARMUBRIS (carmustine), CASODEX (bicalutamide), CEENU (lomustine), CERUBIDINE (daunorubicin hydrochloride), CERVARIX (recombinant HPV bivalent vaccine), CLAFEN (cyclophosphamide), CMF, COMETRIQ (cabozantinib-s-malate), COSMEGEN (dactinomycin), CYFOS (ifosfamide), CYRAMZA (ramucirumab), CYTOSAR-U (cytarabine), CYTOXAN (cyclophosphamide), DACOGEN (decitabine), DEGARELIX, DOXIL (doxorubicin hydrochloride liposome), DOXORUBICIN HYDROCHLORIDE, DOX-SL (doxorubicin hydrochloride liposome), DTIC-DOME (dacarbazine), EFUDEX (fluorouracil), ELLENCE (epirubicin hydrochloride), ELOXATIN (oxaliplatin), ERBITUX (cetuximab), ERIVEDGE (vismodegib), ETOPOPHOS (etoposide phosphate), EVACET (doxorubicin hydrochloride liposome), FARESTON (toremifene), FASLODEX (fulvestrant), FEC, FEMARA (letrozole), FLUOROPLEX (fluorouracil), FOLEX (methotrexate), FOLEX PFS (methotrexate), FOLFIRI, FOLFIRI-BEVACIZUMAB, FOLFIRI-CETUXIMAB, FOLFIRINOX, FOLFOX, FU-LV, GARDASIL (recombinant human papillomavirus (HPV) quadrivalent vaccine), GEMCITABINE-CISPLATIN, GEMCITABINE-OXALIPLATIN, GEMZAR (gemcitabine hydrochloride), GILOTRIF (afatinib dimaleate), GLEEVEC (imatinib mesylate), GLIADEL (carmustine implant), GLIADEL WAFER (carmustine implant), HERCEPTIN (trastuzumab), HYCAMTIN (topotecan hydrochloride), IFEX (ifosfamide), IFOSFAMIDUM (ifosfamide), INLYTA (axitinib), INTRON A (recombinant interferon alfa-2b), IRESSA (gefitinib), IXEMPRA (ixabepilone), JAKAFI (ruxolitinib phosphate), JEVTANA (cabazitaxel), KADCYLA (ado-trastuzumab emtansine), KEYTRUDA (pembrolizumab), KYPROLIS (carfilzomib), LIPODOX (doxorubicin hydrochloride liposome), LUPRON (leuprolide acetate), LUPRON DEPOT (leuprolide acetate), LUPRON DEPOT-3 MONTH (leuprolide acetate), LUPRON DEPOT-4 MONTH (leuprolide acetate), LUPRON DEPOT-PED (leuprolide acetate), MEGACE (megestrol acetate), MEKINIST (trametinib), METHAZOLASTONE (temozolomide), METHOTREXATE LPF (methotrexate), MEXATE (methotrexate), MEXATE-AQ (methotrexate), MITOXANTRONE HYDROCHLORIDE, MITOZYTREX (mitomycin c), MOZOBIL (plerixafor), MUSTARGEN (mechlorethamine hydrochloride), MUTAMYCIN (mitomycin c), MYLOSAR (azacitidine), NAVELBINE (vinorelbine tartrate), NEOSAR (cyclophosphamide), NEXAVAR (sorafenib tosylate), NOLVADEX (tamoxifen citrate), NOVALDEX (tamoxifen citrate), OFF, PAD, PARAPLAT (carboplatin), PARAPLATIN (carboplatin), PEG-INTRON (peginterferon alfa-2b), PEMETREXED DISODIUM, PERJETA (pertuzumab), PLATINOL (cisplatin), PLATINOL-AQ (cisplatin), POMALYST (pomalidomide), prednisone, PROLEUKIN (aldesleukin), PROLIA (denosumab), PROVENGE (sipuleucel-t), REVLIMID (lenalidomide), RUBIDOMYCIN (daunorubicin hydrochloride), SPRYCEL (dasatinib), STIVARGA (regorafenib), SUTENT (sunitinib malate), SYLATRON (peginterferon alfa-2b), SYLVANT (siltuximab), SYNOVIR (thalidomide), TAC, TAFINLAR (dabrafenib), TARABINE PFS (cytarabine), TARCEVA (erlotinib hydrochloride), TASIGNA (nilotinib), TAXOL (paclitaxel), TAXOTERE (docetaxel), TEMODAR (temozolomide), THALOMID (thalidomide), TOPOSAR (etoposide), TORISEL (temsirolimus), TPF, TRISENOX (arsenic trioxide), TYKERB (lapatinib ditosylate), VECTIBIX (panitumumab), VEIP, VELBAN (vinblastine sulfate), VELCADE (bortezomib), VELSAR (vinblastine sulfate), VEPESID (etoposide), VIADUR (leuprolide acetate), VIDAZA (azacitidine), VINCASAR PFS (vincristine sulfate), VOTRIENT (pazopanib hydrochloride), WELLCOVORIN (leucovorin calcium), XALKORI (crizotinib), XELODA (capecitabine), XELOX, XGEVA (denosumab), XOFIGO (radium 223 dichloride), XTANDI (enzalutamide), YERVOY (ipilimumab), ZALTRAP (ziv-aflibercept), ZELBORAF (vemurafenib), ZOLADEX (goserelin acetate), ZOMETA (zoledronic acid), ZYKADIA (ceritinib), ZYTIGA (abiraterone acetate), or a combination thereof. In certain embodiments, the additional pharmaceutical agent is a kinase inhibitor. In certain embodiments, the additional pharmaceutical agent is a protein kinase inhibitor (e.g., tyrosine protein kinase inhibitor). In certain embodiments, the additional pharmaceutical agent is an inhibitor of a Src family kinase. In certain embodiments, the additional pharmaceutical agent is a CDK inhibitor. In certain embodiments, the additional pharmaceutical agent is a CDK7 inhibitor. In certain embodiments, the additional pharmaceutical agent is a CDK12 inhibitor. In certain embodiments, the additional pharmaceutical agent is a CDK13 inhibitor. In certain embodiments, the additional pharmaceutical agent is an inhibitor of one or more protein kinases selected from the group consisting of IRAK1, IRAK4, BMX, and PI3K. In certain embodiments, the additional pharmaceutical agent is an inhibitor of one or more protein kinases selected from the group consisting of BUB1B, CDK2, CDK9, CHEK2, FGR, HIPK4, PRKCQ, RET, SRC, and MELK. In certain embodiments, the additional pharmaceutical agent is an inhibitor of one or more protein kinases selected from the group consisting of ABL, ARG, BLK, CSK, EphB1, EphB2, FOR, FRK, FYN, SRC, YES, LCK, LYN, MAP2K5, NLK, p38a, SNRK, and TEC. In certain embodiments, the additional pharmaceutical agent is an inhibitor of one or more protein kinases selected from the group consisting of ABL1(H396P)-phosphorylated, ABL1-phosphorylated, BLK, EPHA4, EPHB2, EPHB3, EPHB4, FOR, JAK3(JH1domain-catalytic), KIT, KIT(L576P), KIT(V559D), PDGFRB, SRC, YES, ABL1(H396P)-nonphosphorylated, ABL1(Y253F)-phosphorylated, ABL1-nonphosphorylated, FRK, LYN, ABL1(Q252H)-nonphosphorylated, DDR1, EPHB1, ERBB4, p38-alpha, ABL2, ABL1(Q252H)-phosphorylated, SIK, EPHA8, MEK5, ABL1(E255K)-phosphorylated, ABL1(F317L)-nonphosphorylated, FYN, LCK, EPHA2, ABL1(M351T)-phosphorylated, TXK, EGFR(L858R), EGFR(L861Q), ERBB2, ERBB3, EPHA5, ABL1(F317I)-nonphosphorylated, EGFR(L747-E749del, A750P), CSK, EPHA1, ABL1(F317L)-phosphorylated, BRAF(V600E), EGFR, KIT-autoinhibited, and EGFR(E746-A750del). In certain embodiments, the additional pharmaceutical agent is an inhibitor of one or more protein kinases selected from the group consisting of ABL1 (F317L)-nonphosphorylated, ABL1 (H396P)-nonphosphorylated, ABL1 (H396P)-phosphorylated, ABL1-phosphorylated, BLK, EPHA4, EPHB2, EPHB3, EPHB4, JAK3(JH1domain-catalytic), KIT, KIT(L576P), KIT(V559D), LYN, PDGFRB, SRC, YES, ABL1-nonphosphorylated, ABL1(Y253F)-phosphorylated, ERBB3, FOR, FRK, p38-alpha, ABL1(F317I)-nonphosphorylated, DDR1, EPHA2, ABL1(Q252H)-phosphorylated, MEK5, ABL1(Q252H)-nonphosphorylated, ABL2, FYN, EPHB1, ABL1 (E255K)-phosphorylated, ABL1(F317L)-phosphorylated, EPHA1, ABL1(M351T)-phosphorylated, ERBB4, TXK, LCK, EPHA8, SIK, EPHA5, EGFR(L861Q), CSF1R-autoinhibited, BRAF(V600E), BRK, CSK, KIT(D816V), KIT-autoinhibited, EGFR(L747-T751del,Sins), EGFR(L858R), EGFR(L747-E749del, A750P), and CSF1R. In certain embodiments, the additional pharmaceutical agent is an anti-angiogenesis agent, anti-inflammatory agent, immunosuppressant, anti-bacterial agent, anti-viral agent, cardiovascular agent, cholesterol-lowering agent, anti-diabetic agent, anti-allergic agent, pain-relieving agent, or a combination thereof. In certain embodiments, the compounds described herein or pharmaceutical compositions can be administered or used in combination with an anti-cancer therapy including, but not limited to, transplantation (e.g., bone marrow transplantation, stem cell transplantation), surgery, radiation therapy, immunotherapy, and chemotherapy.
[0391] Also encompassed by the disclosure are kits (e.g., pharmaceutical packs). The kits provided may comprise a pharmaceutical composition or compound described herein and a container (e.g., a vial, ampule, bottle, syringe, and/or dispenser package, or other suitable container). In some embodiments, provided kits may optionally further include a second container comprising a pharmaceutical excipient for dilution or suspension of a pharmaceutical composition or compound described herein. In some embodiments, the pharmaceutical composition or compound described herein provided in the first container and the second container are combined to form one unit dosage form.
Methods of Treatment and Uses
[0392] The present invention also provides methods for the treatment or prevention of a proliferative disease (e.g., cancers (e.g., leukemia, acute lymphoblastic leukemia, lymphoma, Burkitt's lymphoma, melanoma, multiple myeloma, breast cancer, Ewing's sarcoma, osteosarcoma, brain cancer, neuroblastoma, lung cancer, colorectal cancer), benign neoplasms, diseases associated with angiogenesis, inflammatory diseases, autoinflammatory diseases, and autoimmune diseases).
[0393] The compounds described herein may exhibit kinase inhibitory activity; the ability to inhibit cyclin-dependent kinase (CDK); the ability to inhibit cyclin-dependent kinase 7 (CDK7); the ability to inhibit cyclin-dependent kinase 7 (CDK7), without inhibiting another cyclin-dependent kinase (CDK); the ability to inhibit cyclin-dependent kinase 12 (CDK12); the ability to inhibit cyclin-dependent kinase 12 (CDK12), without inhibiting another cyclin-dependent kinase (CDK); the ability to inhibit cyclin-dependent kinase 13 (CDK13); the ability to inhibit cyclin-dependent kinase 13 (CDK13), without inhibiting another cyclin-dependent kinase (CDK); the ability to inhibit cyclin-dependent kinases 12 and 13 (CDK12 and CDK13); the ability to inhibit cyclin-dependent kinases 12 and 13 (CDK12 and CDK13), without inhibiting another cyclin-dependent kinase (CDK); a therapeutic effect and/or preventative effect in the treatment of cancers; a therapeutic effect and/or preventative effect in the treatment of Myc-dependent cancers; and/or a therapeutic profile (e.g., optimum safety and curative effect) that is superior to existing chemotherapeutic agents.
[0394] Without wishing to be bound by any particular theory, the compounds described herein are able to bind (e.g., covalently modify) the protein kinase being inhibited. In certain embodiments, the R.sup.2 group of a compound described herein is able to bind (e.g., covalently modify) to the protein kinase. In certain embodiments, the R.sup.2 group of a compound described herein is able to covalently bind a cysteine residue of the protein kinase. In certain embodiments, the compound is capable of covalently modifying CDK7 (e.g., Cys312 of CDK7). In certain embodiments, the R.sup.2 group of a compound described herein is able to covalently modify residue Cys312 of CDK7. In certain embodiments, the compound is capable of covalently modifying CDK12 (e.g., Cys1039 of CDK12). In certain embodiments, the R.sup.2 group of a compound described herein is able to covalently modify residue Cys1039 of CDK12. In certain embodiments, the compound is capable of covalently modifying CDK13 (e.g., Cys1017 of CDK13). In certain embodiments, the R.sup.2 group of a compound described herein is able to covalently modify residue Cys1017 of CDK13.
[0395] In another aspect, the present disclosure provides methods of inhibiting the activity of a protein kinase in a subject, the methods comprising administering to the subject an effective amount (e.g., therapeutically effective amount) of a compound, or pharmaceutical composition thereof, as described herein.
[0396] In another aspect, the present disclosure provides methods of inhibiting the activity of a protein kinase in a biological sample, the methods comprising contacting the biological sample with an effective amount of a compound, or pharmaceutical composition thereof, as described herein.
[0397] In another aspect, the present disclosure provides methods of inhibiting the activity of a protein kinase in a tissue, the methods comprising contacting the tissue with an effective amount of a compound, or pharmaceutical composition thereof, as described herein.
[0398] In another aspect, the present disclosure provides methods of inhibiting the activity of a protein kinase in a cell, the methods comprising contacting the cell with an effective amount of a compound, or pharmaceutical composition thereof, as described herein.
[0399] In certain embodiments, the subject being treated is a mammal. In certain embodiments, the subject is a human. In certain embodiments, the subject is a domesticated animal, such as a dog, cat, cow, pig, horse, sheep, or goat. In certain embodiments, the subject is a companion animal such as a dog or cat. In certain embodiments, the subject is a livestock animal such as a cow, pig, horse, sheep, or goat. In certain embodiments, the subject is a zoo animal. In another embodiment, the subject is a research animal such as a rodent, dog, or non-human primate. In certain embodiments, the subject is a non-human transgenic animal such as a transgenic mouse or transgenic pig.
[0400] In certain embodiments, the biological sample being contacted with the compound or composition is breast tissue, bone marrow, lymph node, lymph tissue, spleen, or blood.
[0401] In certain embodiments, the cell being contacted with the compound or composition is in vitro. In certain embodiments, the cell being contacted with the compound or composition is in vivo. In certain embodiments, the cell being contacted with the compound or composition is ex vivo. In certain embodiments, the cell being contacted with the compound or composition is a malignant cell (e.g., malignant blood cell). In certain embodiments, the cell being contacted with the compound or composition is a malignant hematopoietic stem cell (e.g., malignant myeloid cell or malignant lymphoid cell). In certain embodiments, the cell being contacted with the compound or composition is a malignant lymphocyte (e.g., malignant T-cell or malignant B-cell). In certain embodiments, the cell being contacted with the compound or composition is a malignant red blood cell, malignant white blood cell, or malignant platelet. In certain embodiments, the cell being contacted with the compound or composition is a malignant neutrophil, malignant macrophage, or malignant plasma cell. In certain embodiments, the cell being contacted with the compound or composition is a carcinoma cell. In certain embodiments, the cell being contacted with the compound or composition is a carcinoma breast cell. In certain embodiments, the cell being contacted with the compound or composition is a sarcoma cell. In certain embodiments, the cell being contacted with the compound or composition is a sarcoma cell from breast tissue.
[0402] The proliferative disease to be treated or prevented using the compounds described herein may be associated with overexpression of a kinase, such as cyclin-dependent kinase (CDK). The process of eukaryotic cell division may be broadly divided into a series of sequential phases termed G1, S, G2, and M. Correct progression through the various phases of the cell cycle has been shown to be critically dependent upon the spatial and temporal regulation of a family of proteins known as cyclin dependent kinases (CDKs) and a diverse set of their cognate protein partners termed cyclins. CDKs are CDC2 (also known as CDK1) homologous serine-threonine kinase proteins that are able to utilize ATP as a substrate in the phosphorylation of diverse polypeptides in a sequence-dependent context. Cyclins are a family of proteins characterized by a homology region, containing approximately 100 amino acids, termed the "cyclin box" which is used in binding to, and defining selectivity for, specific CDK partner proteins.
[0403] Modulation of the expression levels, degradation rates, protein levels, and activity levels of various CDKs and cyclins throughout the cell cycle leads to the cyclical formation of a series of CDK/cyclin complexes, in which the CDKs are enzymatically active. The formation of these complexes controls passage through discrete cell cycle checkpoints and thereby enables the process of cell division to continue. Failure to satisfy the prerequisite biochemical criteria at a given cell cycle checkpoint, i.e., failure to form a required CDK/cyclin complex, can lead to cell cycle arrest and/or cellular apoptosis. Aberrant cellular proliferation can often be attributed to loss of correct cell cycle control. Inhibition of CDK enzymatic activity therefore provides a means by which abnormally dividing cells can have their division arrested and/or be killed. The diversity of CDKs, and CDK complexes, and their critical roles in mediating the cell cycle, provides a broad spectrum of potential therapeutic targets selected on the basis of a defined biochemical rationale.
[0404] CDK7, a member of the CDK family, was originally isolated as the catalytic subunit of the trimeric CDK-activating kinase (CAK) complex. This complex, consisting of CDK7, cyclin H, and MAT1, is responsible for activation of the mitotic promoting factor in vitro. The discovery that CDK7 was also a component of the basal transcription repair factor IIH (TFIIH) implicated a dual role for CDK7 in transcription as part of TFIIH and in the control of the cell cycle as the trimeric CAK complex. TFIIH is a multi-subunit protein complex identified as a factor required for RNA polymerase II (RNAP 1)-catalyzed transcription, and subsequently this complex was found to play a key role in nucleotide excision repair. CDK7 is a component of at least three complexes, i.e., the trimeric CAK complex, the quaternary complex with the XPD (or ERCC2, a protein involved in transcription-coupled nucleotide excision repair), and the nine-subunit TFIIH complex. The two functions of CDK7 in CAK and CTD phosphorylation support critical facets of cellular proliferation, cell cycling, and transcription. Overexpression of CDK7 may inhibit apoptosis, promote transcription and cell proliferation, and/or disrupt DNA repair, and therefore, cause proliferative diseases. In certain embodiments, the proliferative disease to be treated or prevented using the compounds described herein may be associated with overexpression of a CDK (e.g., CDK7).
[0405] Cdk12 and Cdk13 are Cdc2-related proteins that share 92% identity in their kinase domains (Chen et al., Exp. Neurol., 2014, 261, 10-21). CDK12 plays a critical role in cell processes, for example, regulating transcription and splicing machinery by stabilizing the RNAPII and DNA interaction, and regulating DNA damage response (DDR) and maintenance of genomic stability by modulating the expression of DDR genes. Overexpression of CDK12 has been found to correlate, both at the transcriptional and protein level, with pathological parameters of breast cancer disease.
[0406] A proliferative disease may be associated with aberrant activity of a CDK (e.g., CDK7, CDK12, and/or CDK13). Aberrant activity of a CDK (e.g., CDK7, CDK12, and/or CDK13) may be an elevated and/or an inappropriate activity of the CDK. Deregulation of cell cycle progression is a characteristic of a proliferative disease, and a majority of proliferative diseases have abnormalities in some component of CDK (e.g., CDK7, CDK12, and/or CDK13) activity, frequently through elevated and/or inappropriate CDK activation. Inhibition of the catalytic activity of CDK7, CDK12, and/or CDK13 would be expected to inhibit cell cycle progression by blocking the phosphorylation of cell cycle CDKs, and would additionally inhibit transcription of effectors of cell division. In certain embodiments, CDK7 is not overexpressed, and the activity of CDK7 is elevated and/or inappropriate. In certain other embodiments, CDK7 is overexpressed, and the activity of CDK7 is elevated and/or inappropriate. In certain embodiments, CDK12 is not overexpressed, and the activity of CDK12 is elevated and/or inappropriate. In certain embodiments, CDK12 is overexpressed, and the activity of CDK12 is elevated and/or inappropriate. In certain other embodiments, CDK13 is not overexpressed, and the activity of CDK13 is elevated and/or inappropriate. In certain other embodiments, CDK13 is overexpressed, and the activity of CDK13 is elevated and/or inappropriate. The compounds described herein, and pharmaceutically acceptable salts, solvates, hydrates, polymorphs, co-crystals, tautomers, stereoisomers, isotopically labeled derivatives, prodrugs, and compositions thereof, may inhibit the activity of CDK7 and be useful in treating and/or preventing proliferative diseases. The compounds described herein, and pharmaceutically acceptable salts, solvates, hydrates, polymorphs, co-crystals, tautomers, stereoisomers, isotopically labeled derivatives, prodrugs, and compositions thereof, may inhibit the activity of CDK12 and/or CDK13 and be useful in treating and/or preventing proliferative diseases.
[0407] A proliferative disease may also be associated with inhibition of apoptosis of a cell in a biological sample or subject. All types of biological samples described herein or known in the art are contemplated as being within the scope of the invention. Apoptosis is the process of programmed cell death. Inhibition of apoptosis may result in uncontrolled cell proliferation and, therefore, may cause proliferative diseases. The cell cycle CDKs (CDK1, 2, 4, and 6) are activated by phosphorylation by CDK7/cyclin H (also called CAK). Inhibition of CDK7 would therefore result in cell-cycle arrest at multiple points in the cell cycle due to failure to activate the cell cycle CDKs. CDK7 activates transcription by phosphorylating the CTD of RNAP II. Inhibition of CTD phosphorylation has been shown to inhibit transcription and reduce expression of short lived proteins, including those involved in apoptosis regulation. It is appreciated in the art that stalling of RNA polymerase may activate p53 (also known as protein 53 or tumor protein 53, a tumor suppressor protein that is encoded in humans by the TP53 gene), leading to apoptosis. Thus, inhibition of the activity of CDK7 are expected to cause cytotoxicity by inducing apoptosis. The compounds described herein, and pharmaceutically acceptable salts, solvates, hydrates, polymorphs, co-crystals, tautomers, stereoisomers, isotopically labeled derivatives, prodrugs, and compositions thereof, may induce apoptosis, and therefore, be useful in treating and/or preventing proliferative diseases.
[0408] The CycK/Cdk12 complex regulates phosphorylation of Ser2 in the C-terminal domain of RNA polymerase II and expression of a small subset of human genes, as revealed in expression microarrays. Through regulation of expression of DNA damage response genes (i.e. oncogenes), CycK/Cdk12 protects cells from genomic instability. In certain embodiments, the DNA damage response genes are BRCA1, BRCA2, HER1, HER2, ATR, FANCI, or FANCD2. In certain embodiments, the DNA damage response genes are BRCA1, HER2, ATR, FANCI, and FANCD2. In certain embodiments, the DNA damage response genes are BRCA1. In certain embodiments, the DNA damage response genes are HER2.
[0409] In certain embodiments, the proliferative disease to be treated or prevented using the compounds described herein is cancer. All types of cancers disclosed herein or known in the art are contemplated as being within the scope of the invention. In certain embodiments, the proliferative disease is a cancer associated with BCL-2 anti-apoptotic proteins (e.g., MCL-1 and/or XIAP) (e.g., cancer associated with dependence on BCL-2 anti-apoptotic proteins). In certain embodiments, the proliferative disease is a cancer associated with overexpression of MYC (a gene that codes for a transcription factor). In certain embodiments, the cancer is a MYC-dependent cancer. In certain embodiments, the proliferative disease is a cancer associated with amplification of BRCA1. In certain embodiments, the proliferative disease is a cancer associated with amplification of HER2. In certain embodiments, the proliferative disease is a hematological malignancy. In certain embodiments, the proliferative disease is a blood cancer. In certain embodiments, the proliferative disease is a hematological malignancy. In certain embodiments, the proliferative disease is leukemia. In certain embodiments, the proliferative disease is chronic lymphocytic leukemia (CLL). In certain embodiments, the proliferative disease is acute lymphoblastic leukemia (ALL). In certain embodiments, the proliferative disease is T-cell acute lymphoblastic leukemia (T-ALL). In certain embodiments, the proliferative disease is chronic myelogenous leukemia (CML). In certain embodiments, the proliferative disease is acute myelogenous leukemia (AML). In certain embodiments, the proliferative disease is acute monocytic leukemia (AMoL). In certain embodiments, the proliferative disease is lymphoma. In some embodiments, the proliferative disease is Burkitt's lymphoma. In certain embodiments, the proliferative disease is a Hodgkin's lymphoma. In certain embodiments, the proliferative disease is a non-Hodgkin's lymphoma. In certain embodiments, the proliferative disease is multiple myeloma. In certain embodiments, the proliferative disease is melanoma. In certain embodiments, the proliferative disease is colorectal cancer. In certain embodiments, the proliferative disease is breast cancer. In certain embodiments, the proliferative disease is recurring breast cancer. In certain embodiments, the proliferative disease is mutant breast cancer. In certain embodiments, the proliferative disease is HER2+ breast cancer. In certain embodiments, the proliferative disease is HER2-breast cancer. In certain embodiments, the proliferative disease is triple-negative breast cancer (TNBC). In certain embodiments, the proliferative disease is a bone cancer. In certain embodiments, the proliferative disease is osteosarcoma. In certain embodiments, the proliferative disease is Ewing's sarcoma. In some embodiments, the proliferative disease is a brain cancer. In some embodiments, the proliferative disease is neuroblastoma. In some embodiments, the proliferative disease is a lung cancer. In some embodiments, the proliferative disease is small cell lung cancer (SCLC). In some embodiments, the proliferative disease is non-small cell lung cancer. In some embodiments, the proliferative disease is a benign neoplasm. All types of benign neoplasms disclosed herein or known in the art are contemplated as being within the scope of the invention. In some embodiments, the proliferative disease is associated with angiogenesis. All types of angiogenesis disclosed herein or known in the art are contemplated as being within the scope of the invention. In certain embodiments, the proliferative disease is an inflammatory disease. All types of inflammatory diseases disclosed herein or known in the art are contemplated as being within the scope of the invention. In certain embodiments, the inflammatory disease is rheumatoid arthritis.
In certain embodiments, the proliferative disease is an acute inflammatory disease. In certain embodiments, the acute inflammatory disease is rheumatoid arthritis, chron's disease, or fibrosis. In some embodiments, the proliferative disease is an autoinflammatory disease. All types of autoinflammatory diseases disclosed herein or known in the art are contemplated as being within the scope of the invention. In some embodiments, the proliferative disease is an autoimmune disease. All types of autoimmune diseases disclosed herein or known in the art are contemplated as being within the scope of the invention.
[0410] Another aspect of the invention relates to methods of inhibiting the activity of a kinase in a biological sample, tissue, cell, or subject. In certain embodiments, the kinase is a CDK. In certain embodiments, the kinase is CDK7. In certain embodiments, the kinase is CDK12. In certain embodiments, the kinase is CDK13. In certain embodiments, the activity of the kinase is aberrant activity of the kinase. In certain embodiments, the activity of the kinase is increased activity of the kinase. In certain embodiments, the inhibition of the activity of the kinase is irreversible. In other embodiments, the inhibition of the activity of the kinase is reversible. In certain embodiments, the methods of inhibiting the activity of the kinase include attaching a compound described herein to the kinase.
[0411] Also provided in the present invention are methods of inhibiting transcription of genes in a biological sample or subject. In certain embodiments, the transcription of genes affected by the activity of CDK7 may be inhibited by a compound of the invention. In certain embodiments, the genes which may have their transcription inhibited by the activity of CDK7 are one or more selected from the group consisting of MYC, RUNX1, MYB, TAL1, GATA3, KLF2, HNRPDL, p21, ASCL1, MYCN, INSM1, NEUROD1, NEUROG1, FOXG1, FOXA1, SOX2, SOX4, BCL11A, OTX2, GAT2, PHOX2B, PLK2, TAF1, CTGF, WEE1, SDIM, JUN, PIM1, IL8, and FOS1. In certain embodiments, the genes which may have their transcription inhibited by the activity of CDK7 include MYC, KLF2, E2F2, CDK6, CCND3, E2F3, HNRPDL, TET1, IL7R, BRCA1, BRCA2, HER1, and HER2. In certain embodiments, the transcription of genes affected by the activity of CDK12 may be inhibited by a compound of the invention. In certain embodiments, the genes which may have their transcription inhibited by the activity of CDK12 are one or more selected from the group consisting of BRCA1, FANCI, ATR, FANCD2, APEX1, NEK9, CHEK1, CHEK2, ATM, RAD51C, RAD51D, ORC3L, MDC1, TERF2, ERCC4, FANCF, PARP9, RUNX1, MYB, TAL1, MCL1, MYC, BCL2, ETS1, and EWS-FLI. In certain embodiments, the transcription of genes affected by the activity of CDK13 may be inhibited by a compound of the invention. In certain embodiments, the gene is SNORA38.
[0412] The present invention also provides methods of inhibiting cell growth in a biological sample, tissue, cell, or subject.
[0413] In still another aspect, the present invention provides methods of inducing apoptosis of a cell in a biological sample, tissue, cell, or subject.
[0414] In certain embodiments, the methods described herein include administering to a subject or contacting a biological sample with an effective amount of a compound described herein, or a pharmaceutically acceptable salt, solvate, hydrate, polymorph, co-crystal, tautomer, stereoisomer, isotopically labeled derivative, or prodrug thereof, or a pharmaceutical composition thereof. In certain embodiments, the methods described herein include administering to a subject or contacting a biological sample with an effective amount of a compound described herein, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof. In certain embodiments, the compound is contacted with a biological sample. In certain embodiments, the compound is administered to a subject. In certain embodiments, the compound is administered in combination with one or more additional pharmaceutical agents described herein. The additional pharmaceutical agent may be an anti-proliferative agent. In certain embodiments, the additional pharmaceutical agent is an anti-cancer agent. The additional pharmaceutical agent may also be a kinase inhibitor. In certain embodiments, the additional pharmaceutical agent is an inhibitor of a CDK. In certain embodiments, the additional pharmaceutical agent is an inhibitor of CDK7. In certain embodiments, the additional pharmaceutical agent is a selective inhibitor of CDK7. In certain embodiments, the additional pharmaceutical agent is a nonselective inhibitor of CDK7. In certain embodiments, the additional pharmaceutical agent is an inhibitor of CDK12. In certain embodiments, the additional pharmaceutical agent is a selective inhibitor of CDK12. In certain embodiments, the additional pharmaceutical agent is a nonselective inhibitor of CDK12. In certain embodiments, the additional pharmaceutical agent is an inhibitor of CDK13. In certain embodiments, the additional pharmaceutical agent is a selective inhibitor of CDK13. In certain embodiments, the additional pharmaceutical agent is a nonselective inhibitor of CDK13. In certain embodiments, the additional pharmaceutical agent is an inhibitor of another CDK. In certain embodiments, the additional pharmaceutical agent is a selective inhibitor of another CDK. In certain embodiments, the additional pharmaceutical agent is a nonselective inhibitor of another CDK. In certain embodiments, the additional pharmaceutical agent is flavopiridol, triptolide, SNS-032 (BMS-387032), PHA-767491, PHA-793887, BS-181, (S)--CR8, (R)--CR8, or NU6140. In certain embodiments, the additional pharmaceutical agent is an inhibitor of a mitogen-activated protein kinase (MAPK). In certain embodiments, the additional pharmaceutical agent is an inhibitor of a glycogen synthase kinase 3 (GSK3). In certain embodiments, the additional pharmaceutical agent is an inhibitor of an AGC kinase. In certain embodiments, the additional pharmaceutical agent is an inhibitor of a calmodulin-dependent kinase (CaM Kinase). In certain embodiments, the additional pharmaceutical agent is an inhibitor of a casein kinase 1. In certain embodiments, the additional pharmaceutical agent is an inhibitor of a STE kinase. In certain embodiments, the additional pharmaceutical agent is an inhibitor of a tyrosine kinase.
[0415] In some embodiments, the additional pharmaceutical agent is a topoisomerase inhibitor, a MCL1 inhibitor, a BCL-2 inhibitor, a BCL-xL inhibitor, a BRD4 inhibitor, a BRCA1 inhibitor, BRCA2 inhibitor, HER1 inhibitor, HER2 inhibitor, a CDK9 inhibitor, a Jumonji histone demethylase inhibitor, or a DNA damage inducer. In some embodiments, the additional pharmaceutical agent is etoposide, obatoclax, navitoclax, JQ1, 4-(((5'-chloro-2'-(((1R,4R)-4-(((R)-1-methoxypropan-2-yl)amino)cyclohexyl- )amino)-[2,4'-bipyridin]-6-yl)amino)methyl)tetrahydro-2H-pyran-4-carbonitr- ile, JIB04, or cisplatin. In some embodiments, the additional pharmaceutical agent is etoposide, obatoclax, or navitoclax, and the disease to be treated is breast cancer, e.g., triple-negative breast cancer, HER2 positive breast cancer, HER2 negative breast cancer, ER-positive breast cancer, ER-negative breast cancer, or ER/PR-positive breast cancer. In some embodiments, the additional pharmaceutical agent is etoposide, JIB04, or cisplatin, and the disease to be treated is Ewing's sarcoma. In some embodiments, the additional pharmaceutical agent is JQ1 or NVP2, and the disease to be treated is leukemia, e.g., acute myelogenous leukemia, myeloblastic leukemia, promyelocytic leukemia, myelomonocytic leukemia, monocytic leukemia, monoblastic leukemia, or megakaryoblastic leukemia. In certain embodiments, a pharmaceutical composition described herein further comprises a combination of the additional pharmaceutical agents described herein.
[0416] The inventive compounds or compositions may synergistically augment inhibition of CDK7 induced by the additional pharmaceutical agent(s) in the biological sample or subject. The inventive compounds or compositions may synergistically augment inhibition of CDK12 induced by the additional pharmaceutical agent(s) in the biological sample or subject. The inventive compounds or compositions may synergistically augment inhibition of CDK12 and/or CDK13 induced by the additional pharmaceutical agent(s) in the biological sample or subject. Thus, the combination of the inventive compounds or compositions and the additional pharmaceutical agent(s) may be useful in treating proliferative diseases resistant to a treatment using the additional pharmaceutical agent(s) without the inventive compounds or compositions.
[0417] In some embodiments, the activity of a protein kinase is non-selectively inhibited by the compounds or pharmaceutical compositions described herein. In some embodiments, the activity of the protein kinase being inhibited is selectively inhibited by the compounds or pharmaceutical compositions described herein, compared to the activity of a different protein (e.g., a different protein kinase). In certain embodiments, the activity of CDK (e.g., CDK7, CDK12, or CDK13) is selectively inhibited by a compound or pharmaceutical composition described herein, compared to the activity of a different protein. In certain embodiments, the activity of CDK7 is selectively inhibited by a compound or pharmaceutical composition described herein, compared to the activity of a different CDK protein. In certain embodiments, the activity of CDK7 is selectively inhibited by a compound or pharmaceutical composition described herein, compared to the activity of CDK12. In certain embodiments, the activity of CDK7 is selectively inhibited by a compound or pharmaceutical composition described herein, compared to the activity of CDK13. In certain embodiments, the activity of CDK7 is selectively inhibited by a compound or pharmaceutical composition described herein, compared to the activity of CDK12 and the activity of CDK13. In certain embodiments, the activity of CDK12 is selectively inhibited by a compound or pharmaceutical composition described herein, compared to the activity of CDK7. In certain embodiments, the activity of CDK13 is selectively inhibited by a compound or pharmaceutical composition described herein, compared to the activity of CDK7. In certain embodiments, the activity of CDK12 and the activity of CDK13 are selectively inhibited by a compound or pharmaceutical composition described herein, compared to the activity of CDK7.
[0418] The selectivity of a compound or pharmaceutical composition described herein in inhibiting the activity of a protein kinase over a different protein (e.g., a different protein kinase) may be measured by the quotient of the IC.sub.50value of the compound or pharmaceutical composition in inhibiting the activity of the different protein over the IC.sub.50 value of the compound or pharmaceutical composition in inhibiting the activity of the protein kinase. The selectivity of a compound or pharmaceutical composition described herein for a protein kinase over a different protein may also be measured by the quotient of the K.sub.d value of an adduct of the compound or pharmaceutical composition and the different protein over the K.sub.d value of an adduct of the compound or pharmaceutical composition and the protein kinase. In certain embodiments, the selectivity is at least 2-fold, at least 3-fold, at least 5-fold, at least 10-fold, at least 30-fold, at least 100-fold, at least 300-fold, at least 1,000-fold, at least 3,000-fold, at least 10,000-fold, at least 30,000-fold, or at least 100,000-fold. In certain embodiments, the selectivity is not more than 100,000-fold, not more than 10,000-fold, not more than 1,000-fold, not more than 100-fold, not more than 10-fold, or not more than 2-fold. Combinations of the above-referenced ranges (e.g., at least 2-fold and not more than 10,000-fold) are also within the scope of the disclosure.
[0419] In certain embodiments, a kit described herein includes a first container comprising a compound or pharmaceutical composition described herein. In certain embodiments, a kit described herein is useful in treating a proliferative disease (e.g., cancers (e.g., leukemia, acute lymphoblastic leukemia, lymphoma, Burkitt's lymphoma, melanoma, multiple myeloma, breast cancer, Ewing's sarcoma, osteosarcoma, brain cancer, neuroblastoma, lung cancer, colorectal cancer), benign neoplasms, diseases associated with angiogenesis, inflammatory diseases, autoinflammatory diseases, and autoimmune diseases) in a subject in need thereof, preventing a proliferative disease in a subject in need thereof, inhibiting the activity of a protein kinase (e.g., CDK (e.g., CDK7, CDK12, or CDK13)) in a subject, biological sample, tissue, or cell, and/or inducing apoptosis in a cell.
[0420] In certain embodiments, a kit described herein further includes instructions for using the compound or pharmaceutical composition included in the kit. A kit described herein may also include information as required by a regulatory agency such as the U.S. Food and Drug Administration (FDA). In certain embodiments, the information included in the kits is prescribing information. In certain embodiments, the kits and instructions provide for treating a proliferative disease in a subject in need thereof, preventing a proliferative disease in a subject in need thereof, inhibiting the activity of a protein kinase (e.g., CDK (e.g., CDK7, CDK12, or CDK13)) in a subject, biological sample, tissue, or cell, and/or inducing apoptosis in a cell. A kit described herein may include one or more additional pharmaceutical agents described herein as a separate composition.
EXAMPLES
[0421] In order that the invention described herein may be more fully understood, the following examples are set forth. The synthetic and biological examples described in this application are offered to illustrate the compounds, pharmaceutical compositions, and methods provided herein and are not to be construed in any way as limiting their scope.
Pulldown Assay (FIGS. 2-4 and 7-9)
[0422] Jurkat cells were treated with DMSO or concentration of compound indicated. 6 hours after treatment, cells were washed and harvested by resuspending in lysis buffer (50 mM Hepes pH 7.4, 150 mM NaCl, 1% NP-40, 5 mM EDTA, protease and phosphatase inhibitors) and lysing on ice 30 minutes. Lysates were cleared by centrifugation at 15,000 rpm 30 minutes. Biotin-labeled THZ1 was added to 1 .mu.M to lysates and rotated at 4.degree. C. overnight. Streptavidin-agarose beads were washed and 30 .mu.L slurry was added to each lysate and rotated for 1 hour at 4.degree. C. Beads were washed 5 times with lysis buffer and 50 .mu.L 2.times.LDS buffer was added to each sample. Samples were boiled and equal volume of protein was loaded onto gel. Gel was transferred to nitrocellulose and blotted for Cyclin K and Cyclin H.
Interpretation of Results
[0423] We conclude that pre-treatment of cells with several of the compounds, but not DMSO, blocks biotin-THZ1 from being able to bind to CDK12, which blocks the pulldown of CDK12 and CDK13-associated Cyclin K. This indicates that active compounds are able to engage CDK12 and CDK13-associated cyclin K complexes in cells and block binding of these complexes by bio-THZ1. We do not see a similar loss of pulldown of Cyclin H, indicating that these compounds are not able to bind to CDK7-associated cyclin H complexes and block its association with biotin-THZ1.
Growth Assay (FIG. 5)
[0424] Jurkat cells were plated at 30,000 cells/well and treated with a titration of compounds indicated. Cells were allowed to grow for 72 hours. Cells were assayed using CELLTITER GLO (Promega) to determine cell viability by measuring the amount of ATP present, which is an indicator of cell metabolic activity. Results are graphed as luminescent values. Curves were generated using PRISM and an IC.sub.50 value was determined.
Interpretation of Results
[0425] We can conclude that several of the compounds generated and tested are more potent against cell growth than the parent SNS-032 compound in Jurkat cells.
Synthesis of the Compounds
[0426] The compounds provided herein can be prepared from readily available starting materials using the following general methods and procedures. Reactions were monitored by thin layer chromatography (TLC) with 0.25 mm E. Merck pre-coated silica gel plates (60 F.sub.2 4) and Waters LCMS system (Waters 2489 UV/Visible Detector, Waters 3100 Mass, Waters 515 HPLC pump, Waters 2545 Binary Gradient Module, Waters Reagent Manager, Waters 2767 Sample Manager) using SunFire.TM. C18 column (4.6.times.50 mm, 5 m particle size): solvent gradient=95% A at 0 min, 0% A at 5 min; solvent A=0.5% TFA in Water; solvent B=Methanol; flow rate: 1.5 mL/min. Purification of reaction products was carried out by flash chromatography using CombiFlash.RTM. Rf with Teledyne Isco RediSep.RTM. Rf High Performance Gold or Silicycle SiliaSep.TM. High Performance columns (4 g, 12 g, 24 g, 40 g, 80 g or 120 g) or by Waters preparative HPLC system with a C18 column: solvent gradient=100% A at 0 min, 0% A at 15 min; solvent A=0.5% TFA in Water; solvent B=Methanol; flow rate: 20 mL/min. The purity of all compounds was over 95% and was analyzed with Waters LCMS system. .sup.1H NMR and .sup.13C NMR spectra were obtained using a Varian Inova-600 or 400 MHz spectrometer. Chemical shifts are reported relative to chloroform (.delta.=7.24) for .sup.1H NMR or dimethyl sulfoxide (.delta.=2.50) for .sup.1H NMR and dimethyl sulfoxide (.delta.=39.51) for .sup.13C NMR. Data are reported as (br=broad, s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet).
##STR00240## ##STR00241##
5-Thiocyanatothiazol-2-amine (MFH-2-76-1)
[0427] A mixture of 2-amino-5-bromothiazole hydrobromide (6.0 g, 23.08 mmol) and potassium thiocyanate (9.0 g, 92.32 mmol) in methanol (150 mL) was stirred at room temperature for 20 h. Methanol was evaporated and water (20 ml) was added. The pH of the aqueous solution was adjusted to pH=12 with 10% NaOH and then a precipitate was formed. The solid was collected by filtration to yield compound MFH-2-76-1 (1.5 g, 41%) as a solid. LCMS (m/z): 158 [M+H]+.
5-((5-tert-Butyloxazol-2-yl)methylthio)thiazol-2-amine (MFH-2-80-1)
[0428] To a solution of compound MFH-2-76-1 (315 mg, 2 mmol) in EtOH (5 ml) was added NaBH.sub.4 (151 mg, 4 mmol) at room temperature. The mixture was stirred for 1 h, and then acetone (3 ml) was slowly added. After 1 h, a solution of 5-tert-Butyl-2-chloromethyloxazole (348 mg, 2 mmol) in EtOH (3 ml) was added. The resulting dark reaction mixture was heated to reflux for 1 h, and was then cooled and concentrated in reduced pressure. The residue was then dissolved in ethyl acetate and washed with water. The organic phase was separated, dried (MgSO.sub.4) and concentrated in reduced pressure to give a crude product MFH-2-80-1 (310 mg, 57%) LCMS (m/z): 270 [M+H]+.
5-[[(5-t-buty2-oxazolyl)methyl]thio]-2-bromo (MFH-2-83-1)
[0429] To a solution of CuBr.sub.2 (1.8 g, 8 mmol) in acetonitrile (48 mL) at 0.degree. C. was added t-BuONO (827 mg, 8 mmol) followed by compound MFH-2-80-1 (1.8 g, 6.68 mmol). The mixture was stirred at 0.degree. C. for 1 h and then was warmed up to room temperature. Ethyl acetate was added and the organic mixture washed with hydrochloric acid (50 mL), dried over magnesium sulfate, filtered through a pad of silica gel, and concentrated in reduced pressure. The residue was purified by chromatographed on silica gel to give product. MFH-2-83-1 (1.5g, 67%). LCMS (m/z): 334 [M+H]+.
(R)-tert-butyl3-(5-((5-tert-butyloxazol-2-yl)methylthio)triazol-2-ylamino)- piperidine-1-carboxylate (MFH-2-84-1)
[0430] The mixture of MFH-2-83-1 (1.5g, 4.5 mmol), (R)-tert-butyl 3-aminopiperidine-1-carboxylate (1.6 g, 8.1 mmol) and DIEA (1.5 g, 11.25 mmol) in NMP (5 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-2-84-1 (1.36 g, yield 66.8%). LCMS (m/z): 453 [M+H].sup.+.
(R)-5-((5-tert-butyloxazol-2-yl)methylthio)-N-(piperidin-3-yl)thiazol-2-am- ine (MFH-2-85-1)
[0431] To a solution of MFH-2-85-1 (1.36 g, 3 mmol) in methanol (18 mL) was added 4N HCl/dioxane (18 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 353 [M+H].sup.+.
[0432] (R)-(3-(5-((5-tert-butyloxazol-2-yl)methylthi)triazol-2-ylamino)pip- eridin-1-yl)(4-nitrophenyl)methanone (MFH-2-86-1)
[0433] The mixture of MFH-2-85-1 (250 mg, 0.643 mmol), 4-nitrobenzoyl chloride (132 mg, 0.71 mmol) in pyridine (3 mL) was stirred for overnight at room temperature. Then the reaction mixture was concentrated under reduced pressure and the residue was directly used in the next step. LCMS (m/z): 502 [M+H].sup.+.
(R)-(4-aminophenyl)(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-yl- amino)piperidin-1-yl)methanone (MFH-2-89-1)
[0434] To a solution of MFH-2-86-1 (270 mg, 0.54 mmol) in ethyl acetate and methanol (1:1) were added Tin (11) chloride dehydrate (1.2 g, 5.4 mmol) and conc. HCl (0.2 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-2-89-1 (140 mg, yield 55%). LCMS (m/z): 472 [M+H]+.
(R)--N-(4-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)triazol-2-ylamino)pip- eridine-1-carbonyl)phenyl)acrylamide (MFH-2-90-1)
[0435] To a solution of MFH-2-89-1 (140 mg, 0.30 mmol) and DIPEA (0.2 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (35 mg, 0.39 mmol) in DCM (0.8 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1 h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-2-90-1 (24.7 mg, yield 16%). LCMS (m/z): 526 [M+H]+. 1H NMR (500 MHz, DMSO) .delta. 10.27 (s, 1H), 7.97 (s, 1H), 7.67 (s, 2H), 7.35 (d, J=6.7 Hz, 2H), 6.87 (d, J=26.3 Hz, 1H), 6.70 (s, 1H), 6.43 (dd, J=17.0, 10.1 Hz, 1H), 6.27 (dd, J=17.0, 1.8 Hz, 1H), 5.77 (dd, J=10.1, 1.8 Hz, 1H), 3.94 (s, 2H), 3.79 (s, 1H), 3.63 (d, J=23.9 Hz, 2H), 3.14 (d, J=65.1 Hz, 2H), 1.95 (s, 1H), 1.76 (s, 1H), 1.52 (d, J=7.8 Hz, 2H), 1.17 (s, 9H).
##STR00242## ##STR00243##
(S)-tert-butyl3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino- )piperline-1-carboxylate (MFH-2-82-1)
[0436] The mixture of MFH-2-83-1 (150 mg, 0.45 mmol), (S)-tert-butyl 3-aminopiperidine-1-carboxylate (160 mg, 0.81 mmol) and DIEA (150 mg, 1.13 mmol) in NMP (1 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-2-82-1 (136 mg, yield 67%). LCMS (m/z): 453 [M+H].sup.+.
(S)-5-((5-tert-butyloxazol-2-yl)methylthio)-N-(piperidin-3-yl)thiazol-2-am- ine (MFH-2-87-1)
[0437] To a solution of MFH-2-82-1 (136 mg, 0.3 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 353 [M+H].sup.+.
(S)-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)piperidin- -1-yl)(4-nitrophenyl)methanone (MFH-2-88-1)
[0438] The mixture of MFH-2-87-1 (250 mg, 0.643 mmol), 4-nitrobenzoyl chloride (132 mg, 0.71 mmol) in pyridine (3 mL) was stirred for overnight at room temperature. Then the reaction mixture was concentrated under reduced pressure and the residue was directly used in the next step. LCMS (m/z): 502 [M+H].sup.+.
(S)-(4-aminophenyl)(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-yl- amino)piperidin-1-yl)methanone (MFH-2-91-1)
[0439] To a solution of MFH-2-88-1 (270 mg, 0.54 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (1.2 g, 5.4 mmol) and cone. HCl (0.2 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol(4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-2-91-1 (140 mg, yield 55%). LCMS (m/z): 472 [M+H]+.
(S)--N-(4-(35-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)piper- idine-1-carbonyl)phenyl)acrylamide (MFH-2-92-1)
[0440] To a solution of MFH-2-91-1 (30 mg, 0.06 mmol) and DIPEA (0.2 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (8 mg, 0.08 mmol) in DCM (0.5 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-2-92-1 (11 mg, yield 33%). LCMS (m/z): 526 [M+H]+. 1H NMR (500 MHz, DMSO) .delta. 10.30 (s, 1H), 8.07 (s, 1H), 7.65 (s, 2H), 7.35 (d, J=6.7 Hz, 2H), 6.88 (d, J=37.7 Hz, 1H), 6.70 (s, 1H), 6.43 (dd, J=16.9, 10.1 Hz, 1H), 6.27 (dd, J=17.0, 1.6 Hz, 1H), 5.77 (dd, J=10.2, 1.7 Hz, 1H), 3.95 (s, 2H), 3.79 (s, 1H), 3.63 (d, J=23.9 Hz, 2H), 3.14 (d, J=65.1 Hz, 2H), 1.92 (s, J=18.7 Hz, 1H), 1.76 (s, 1H), 1.52 (d, J=6.6 Hz, 2H), 1.17 (s, 9H).
##STR00244##
(R)-tert-butyl4-3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylami- no)piperidine-1-carbonyl)cyclohexylcarbamate (MFH-2-93-1)
[0441] The mixture of MFH-2-85-1 (116 mg, 0.33 mmol), 4-(tert-butoxycarbonylamino)cyclohexanecarboxylic acid (120 mg, 0.5 mmol), HOBT (68 mg, 0.5 mmol) and EDCI (96 mg, 0.5 mmol) in DCM (10 ml) was stirred for overnight. The reaction mixture was diluted with DCM (25 ml). The organic phase was washed with saturated Na.sub.2CO.sub.3 and brine (20 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-2-93-1 (80 mg, yield 42%). LCMS (m/z): 578 [M+H]+.
(R)-(4-aminocyclohexyl)(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-- 2-ylamino)piperidin-1-yl)methanone (MFH-2-94-1)
[0442] To a solution of MFH-2-93-1 (80 mg, 0.14 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 478 [M+H].sup.+.
(R)--N-(4-(3-(5-(5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)pipe- ridine-1-carbonyl)cyclohexyl)acrylamide (MFH-2-95-1)
[0443] To a solution of MFH-2-94-1 (30 mg, 0.06 mmol) and DIPEA (0.2 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (8 mg, 0.08 mmol) in DCM (0.5 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-2-95-1 (15.9 mg, yield 48%). LCMS (m/z): 532 [M+H]+.
##STR00245##
(R)-tert-butyl4-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)triazol-2-ylam- ino)piperidine-1-carbonyl)benzylcarbamate (MFH-2-96-1)
[0444] The mixture of MFH-2-85-1 (88 mg, 0.25 mmol), 4-((tert-butoxycarbonylamino)methyl)benzoic acid (94 mg, 0.38 mmol), HOBT (51 mg, 0.38 mmol) and EDCI (72 mg, 0.38 mmol) in DCM (8 ml) was stirred for overnight. The reaction mixture was diluted with DCM (25 ml). The organic phase was washed with saturated Na.sub.2CO.sub.3 and brine (20 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-2-96-1 (125 mg, yield 85%). LCMS (m/z): 586 [M+H]+.
(R)-(4-(aminomethyl)phenyl)(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thia- zol-2-ylamino)piperidin-1-yl)methanone (MFH-2-97-1)
[0445] To a solution of MFH-2-96-1 (125 mg, 0.21 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 486 [M+H].sup.+.
(R)--N-(4-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)pip- eridine-1-carbonyl)benzyl)acrylamide (MFH-2-98-1)
[0446] To a solution of MFH-2-97-1 (50 mg, 0.1 mmol) and DIPEA (0.2 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (12 mg, 0.13 mmol) in DCM (0.5 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1 h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-2-98-1 (16.4 mg, yield 30%). LCMS (m/z): 540 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 8.62 (s, 1H), 8.08 (d, J=54.5 Hz, 1H), 7.28 (d, J=31.4 Hz, 4H), 6.91 (d, J=65.2 Hz, 1H), 6.72 (s, 1H), 6.28 (dd, J=17.1, 10.2 Hz, 1H), 6.13 (dd, J=17.1, 2.2 Hz, 1H), 5.62 (dd, J=10.2, 1.9 Hz, 1H), 4.36 (s, 2H), 3.95 (s, 2H), 3.67-3.62 (m, 2H), 3.33 (s, 1H), 3.14-2.83 (m, 2H), 1.94 (s, 1H), 1.74 (d, J=65.1 Hz, 1H), 1.53 (s, 2H), 1.20 (s, 9H).
##STR00246##
(R)-4-nitrophenyl3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylam- ino)piperidine-1-carboxylate (MFH-2-99-1)
[0447] To a solution of MFH-2-85-1 (210 mg, 0.6 mmol) and DIPEA (116 mg, 0.9 mmol) in DCM (8 mL) was added 4-nitrophenyl chloroformate (133 mg, 0.66 mmol) in DCM (1 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1 h. The solution was then concentrated under reduced pressure and the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-2-99-1 (283 mg, yield 91%). LCMS (m/z): 518 [M+H]+.
(R)-4-aminophenyl3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylami- no)piperidine-1-carboxylate (MFH-2-100-1)
[0448] To a solution of MFH-2-99-1 (106 mg, 0.21 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (462 mg, 2.1 mmol) and conc. HCl (0.1 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-2-100-1 (60 mg, yield 60%). LCMS (m/z): 488 [M+H]+.
(R)-4-acrylamidophenyl3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-- ylamino)piperidine-1-carboxylate (MFH-2-102-1)
[0449] To a solution of MFH-2-100-1 (60 mg, 0.12 mmol) and DIPEA (0.2 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (15 mg, 0.16 mmol) in DCM (0.5 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-2-102-1 (18.1 mg, yield 27%). LCMS (m/z): 542 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.17 (s, 1H), 8.13 (d, J=14.2 Hz, 1H), 7.64 (s, 2H), 7.08 (s, 1H), 7.01 (d, J=9.0 Hz, 1H), 6.95 (s, 1H), 6.72 (dd, J=11.3, 7.1 Hz, 1H), 6.42 (dd, J=16.9, 10.1 Hz, 1H), 6.25 (dd, J=17.0, 1.7 Hz, 1H), 5.82-5.68 (m, 1H), 3.94 (s, 2H), 3.84 (s, 1H), 3.74 (s, 1H), 3.26 (d, J=44.4 Hz, 2H), 2.93 (s, 1H), 1.92 (d, J=13.1 Hz, 1H), 1.79 (d, J=6.3 Hz, 1H), 1.52 (s, 2H), 1.21 (s, 9H).
##STR00247##
(R)-tert-butyl1-(3-(5-(5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylami- no)piperidine-1-carbonyl)piperidin-4-ylcarbamate (MFH-2-101-1)
[0450] The mixture of MFH-2-99-1 (177 mg, 0.34 mmol) and tert-butyl piperidin-4-ylcarbamate (89 mg, 0.44 mmol) in DMSO (3 mL) was stirred at 60.degree. C. overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-2-101-1 (150 mg, yield 76%). LCMS (m/z): 579 [M+H].sup.+.
(R)-(4-aminopiperidin-1-yl)(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thia- zol-2-ylamino)piperidin-1-yl)methanone (MFH-2-103-1)
[0451] To a solution of MFH-2-101-1 (150 mg, 0.26 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 479 [M+H].sup.+.
(R)--N-(1-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)pip- eridine-1-carbonyl)piperidin-4-yl)acrylamide (MFH-2-104-1)
[0452] To a solution of MFH-2-103-1 (30 mg, 0.06 mmol) and DIPEA (0.2 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (8 mg, 0.08 mmol) in DCM (0.5 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-2-104-1 (10.2 mg, yield 30%). LCMS (m/z): 533 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 8.13 (s, 1H), 8.07 (d, J=7.7 Hz, 1H), 6.97 (s, 1H), 6.70 (d, J=5.5 Hz, 1H), 6.18 (dd, J=17.1, 10.1 Hz, 1H), 6.07 (dd, J=17.1, 2.2 Hz, 1H), 5.57 (dd, J=10.1, 2.2 Hz, 1H), 3.94 (s, 2H), 3.81-3.71 (m, 2H), 3.48 (s, 2H), 3.34-3.24 (m, 2H), 2.79 (t, J=11.6 Hz, 3H), 2.70 (dd, J=13.2, 9.5 Hz, 1H), 1.89 (d, J=10.4 Hz, 1H), 1.71 (d, J=13.1 Hz, 3H), 1.43 (dd, J=16.4, 8.5 Hz, 2H), 1.32 (m, 2H), 1.18 (s, 9H).
##STR00248## ##STR00249##
5-thiocyanato-1,3,4-thiadiazol-2-amine (MFH-2-202-1)
[0453] A mixture of 5-bromo-1,3,4-thiadiazol-2-amine (1.5 g, 8.3 mmol) and potassium thiocyanate (3.2 g, 33.3 mmol) in methanol (30 mL) was stirred at room temperature for 20 h. Methanol was evaporated and water (10 ml) was added. The pH of the aqueous solution was adjusted to pH=12 with 10% NaOH and precipitate formed. The solid was collected by filtration to yield compound MFH-2-202-1 (600 mg, 46%) as solid. LCMS (m/z): 159 [M+H]+.
5-((5-tert-butyloxazol-2-yl)methylthio)-1,3,4-thiadiazol-2-amine (MFH-2-204-1)
[0454] To a solution of compound MFH-2-202-1 (315 mg, 2 mmol) in EtOH (5 ml) was added NaBH.sub.4 (151 mg, 4 mmol) at room temperature. The mixture was stirred for 1h, and then acetone (3 ml) was slowly added. After 1 h, a solution of 5-tert-Butyl-2-chloromethyloxazole (348 mg, 2 mmol) in EtOH (3 ml) was added. The resulting reaction mixture was heated to reflux for 1h and then was cooled followed by concentration in reduced pressure. The residue was dissolved in ethyl acetate. The organic phase was washed with brine (30 mL) and then the solvent was removed after drying with MgSO.sub.4. The crude product was then obtained MFH-2-204-1 (310 mg, 57%) as solid LCMS (m/z): 271 [M+H]+.
2-((5-bromo-1,3,4-thiadiazol-2-ylthio)methyl)-5-tert-butyloxazole (MFH-2-208-1)
[0455] To a solution of CuBr.sub.2 (594 mg, 2.66 mmol) in acetonitrile (8 mL) at 0.degree. C. was added t-BuONO (275 mg, 2.66 mmol) followed by compound MFH-2-204-1 (600 mg, 2.22 mmol). The mixture was stirred at 0.degree. C. for 1h and then was warmed up to room temperature. After stirring for 1 h, ethyl acetate was added and the organic mixture washed with hydrochloric acid (50 mL), dried over magnesium sulfate, filtered through a pad of silica gel, and concentrated in reduced pressure. The residue was chromatographed on silica gel to give the MFH-2-208-1 (700 mg, 94%). LCMS (m/z): 335 [M+H]+.
(R)-tert-butyl3-(5-((5-tert-butyloxazol-2-yl)methylthio)-1,3,4-thiadiazol-- 2-ylamino)piperidine-1-carboxylate (MFH-3-2-1)
[0456] The mixture of MFH-2-208-1 (250 mg, 0.75 mmol), (R)-tert-butyl 3-aminopiperidine-1-carboxylate (225 mg, 1.12 mmol) and DIEA (242 mg, 1.87 mmol) in NMP (1 mL) was stirred at 140.degree. C. overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-2-1 (180 mg, yield 53%). LCMS (m/z): 454 [M+H].sup.+.
(R)-5-((5-tert-butyloxazol-2-yl)methylthio)-N-(piperidin-3-yl)-1,3,4-thiad- iazol-2-amine (MFH-3-11-1)
[0457] To a solution of MFH-3-2-1 (250 mg, 0.55 mmol) in methanol (5 mL) was added 4N HCl/dioxane (5 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 354 [M+H].sup.+.
(R)-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)-1,3,4-thiadiazol-2-ylamino- )piperidin-1-yl)(4-nitrophenyl)methanone (MFH-3-14-1)
[0458] The mixture of MFH-3-11-1 (140 mg, 0.4 mmol), 4-nitrobenzoyl chloride (88 mg, 0.48 mmol) in pyridine (3 mL) was stirred overnight at room temperature. Then the reaction mixture was concentrated under reduced pressure and the residue was directly used in the next step. LCMS (m/z): 503 [M+H].sup.+.
(R)-(4-aminophenyl)(3-(5-((5-tert-butyloxazol-2-yl)methylthio)-1,3,4-thiad- iazol-2-ylamino)piperidin-1-yl)methanone (MFH-3-18-1)
[0459] To a solution of MFH-3-14-1 (200 mg, 0.4 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (904 mg, 4 mmol) and conc. HCl (0.2 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-3-18-1 (45 mg, yield 24%). LCMS (m/z): 473 [M+H]+.
(R)--N-(4-(3-((5-tert-butyloxazol-2-yl)methylthio)-1,3,4-thiadiazol-2-ylam- ino)piperidine-1-carbonyl)phenyl)acrylamide (MFH-3-25-1)
[0460] To a solution of MFH-3-18-1 (22 mg, 0.05 mmol) and DIPEA (0.2 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (6 mg, 0.06 mmol) in DCM (0.2 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1 h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-25-1 (8.8 mg, yield 36%). LCMS (m/z): 527 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.29 (s, 1H), 8.02 (d, J=5.0 Hz, 1H), 7.68 (s, 2H), 7.35 (s, 2H), 6.75 (s, 1H), 6.44 (dd, J=17.0, 10.1 Hz, 1H), 6.28 (d, J=16.9 Hz, 1H), 5.86-5.70 (m, 1H), 4.33 (s, 2H), 3.74 (s, 1H), 3.60 (s, 2H), 3.21 (d, J=32.1 Hz, 2H), 1.99 (s, 1H), 1.77 (s, 1H), 1.58 (dd, J=22.4, 11.7 Hz, 2H), 1.29 (s, 9H).
##STR00250## ##STR00251##
N1-(4-nitrophenyl)cyclohexane-1,3-diamine (MFH-3-26-1)
[0461] The mixture of 1-fluoro-4-nitrobenzene (200 mg, 1.42 mmol), cyclohexane-1,3-diamine (485 mg, 4.25 mmol) and K.sub.2CO.sub.3 (587 mg, 4.25 mmol) in DMF (3 mL) was stirred at 90.degree. C. for 6h. The solution was then diluted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-26-1 (230 mg, yield 69%). LCMS (m/z): 236 [M+H].sup.+.
(5-((5-tert-butyloxaxzol-2-yl)methylthio)thiazol-2-yl)-N3-(4-nitrophenyl)c- yclohexane-1,3-diamine (MFH-3-29-1)
[0462] The mixture of MFH-2-83-1 (272 mg, 0.82 mmol), MFH-3-26-1 (230 mg, 0.98 mmol) and DIEA (316 mg, 2.46 mmol) in NMP (2 mL) was stirred at 140.degree. C. overnight. The solution was diluted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-29-1 (80 mg, yield 20%). LCMS (m/z): 458 [M+H].sup.+.
N1-(3 ((5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)cyclohexy- l)benzene-1,4-diamine (MFH-3-33-1)
[0463] To a solution of MFH-3-29-1 (80 mg, 0.16 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (370 mg, 1.6 mmol) and conc. HCl (0.1 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-3-33-1 (20 mg, yield 27%). LCMS (m/z): 458 [M+H]+.
N-(4-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)cyclohex- ylamino)phenyl)acrylamide (MFH-3-35-1)
[0464] To a solution of MFH-3-33-1 (20 mg, 0.04 mmol) and DIPEA (0.1 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (4 mg, 0.04 mmol) in DCM (0.2 mL) dropwise. The mixture was then stirred at -40.degree. C. for 1h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-35-1 (9.3 mg, yield 41%). LCMS (m/z): 512 [M+H]+.
##STR00252##
(R)-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)piperidi- n-1-yl)(2-methyl-4-nitrophenyl)methanone (MFH-3-64-1)
[0465] The mixture of MFH-2-85-1 (44 mg, 0.13 mmol), 4-((tert-butoxycarbonylamino)methyl)benzoic acid (47 mg, 0.19 mmol), HOBT (25 mg, 0.19 mmol) and EDCI (36 mg, 0.19 mmol) in DCM (3 ml) was stirred for overnight. The reaction mixture was diluted with DCM (25 ml). The organic phase was washed with saturated Na.sub.2CO.sub.3 and brine (20 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-64-1 (63 mg, yield 94%). LCMS (m/z): 516 [M+H]+.
(R)-(4-amino-2-methylphenyl)(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thi- azol-2-ylamino)piperidin-1-yl)methanone (MFH-3-66-1)
[0466] To a solution of MFH-3-64-1 (63 mg, 0.12 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (138 mg, 0.61 mmol) and conc. HCl (0.1 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-3-66-1 (20 mg, yield 34%). LCMS (m/z): 486 [M+H]+.
(R)--N-(4-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)pip- eridine-1-carbonyl)-3-methylphenyl)acrylamide (MFH-3-75-1)
[0467] To a solution of MFH-3-66-1 (20 mg, 0.04 mmol) and DIPEA (0.1 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (4 mg, 0.04 mmol) in DCM (0.2 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1 h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-75-1 (9.4 mg, yield 41%). LCMS (m/z): 540 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.16 (d, J=35.4 Hz, 1H), 8.03 (d, J=59.1 Hz, 1H), 7.54 (d, J=9.3 Hz, 1H), 7.24-7.08 (m, 1H), 7.00 (d, J=9.0 Hz, 1H), 6.75 (d, J=10.5 Hz, 1H), 6.69 (s, 1H), 6.43 (dt, J=17.0, 10.5 Hz, 1H), 6.32-6.20 (m, 1H), 5.76 (t, J=11.0 Hz, 1H), 3.96 (s, J=11.8 Hz, 2H), 3.59 (d, J=52.3 Hz, 4H), 3.00 (s, 1H), 2.19 (s, 3H), 1.92 (dd, J=59.5, 33.2 Hz, 2H), 1.57 (d, J=57.5 Hz, 2H), 1.20 (s, 9H).
##STR00253##
(R)-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)piperidi- n-1-yl)(2-methoxy-4-nitrophenyl)methanone (MFH-3-65-1)
[0468] The mixture of MFH-2-85-1 (44 mg, 0.13 mmol), 4-((tert-butoxycarbonylamino)methyl)benzoic acid (47 mg, 0.19 mmol), HOBT (25 mg, 0.19 mmol) and EDCI (36 mg, 0.19 mmol) in DCM (3 ml) was stirred overnight. The reaction mixture was diluted with DCM (25 ml). The organic phase was washed with saturated Na.sub.2CO.sub.3 and brine (20 mL) and dried over Na.sub.2SO.sub.3. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-64-1 (71 mg, yield 100%). LCMS (m/z): 532 [M+H]+.
(R)-4-amino-2-methoxyphenyl)(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thi- azol-2-ylamino)piperidin-1-yl)methanone (MFH-3-67-1)
[0469] To a solution of MFH-3-64-1 (71 mg, 0.13 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (151 mg, 0.67 mmol) and conc. HCl (0.1 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-3-67-1 (30 mg, yield 45%). LCMS (m/z): 502 [M+H]+.
(R)--N-(4-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)pip- eridine-1-carbonyl)-3-methoxyphenyl)acrylamide (MFH-3-81-1)
[0470] To a solution of MFH-3-67-1 (30 mg, 0.06 mmol) and DIPEA (0.1 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (7 mg, 0.08 mmol) in DCM (0.2 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-81-1 (11.8 mg, yield 35%). LCMS (m/z): 556 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.36-10.15 (m, 1H), 8.16-7.83 (m, 1H), 7.49 (d, J=14.3 Hz, 1H), 7.15 (dd, J=19.9, 7.7 Hz, 1H), 7.04-6.94 (m, 1H), 6.74 (dd, J=28.4, 22.6 Hz, 2H), 6.43 (dt, J=18.7, 9.3 Hz, 1H), 6.27 (ddd, J=17.0, 9.5, 1.8 Hz, 1H), 5.83-5.72 (m, 1H), 3.91 (s, 2H), 3.78 (s, 3H), 3.73 (s, 1H), 3.45 (d, J=13.3 Hz, 1H), 3.24 (t, J=32.2 Hz, 1H), 2.88 (dd, J=34.4, 22.2 Hz, 2H), 1.97 (d, J=18.3 Hz, 1H), 1.78 (s, 1H), 1.46 (s, 2H), 1.20 (s, 9H).
##STR00254## ##STR00255##
(1R,3S)--3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)cycl- ohexanecarboxylic acid (MFH-3-79-1)
[0471] The mixture of MFH-2-83-1 (200 mg, 0.6 mmol), 3-aminocyclohexanecarboxylic acid (138 mg, 0.96 mmol) and DIEA (233 mg, 1.8 mmol) in NMP (1 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-79-1 (60 mg, yield 26%). LCMS (m/z): 396 [M+H].sup.+.
tert-butyl1-((1R,3S)--3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-- ylamino)cyclohexanecarbonyl)piperidin-4-ylcarbamate (MFH-3-83-1)
[0472] The mixture of MFH-3-79-1 (60 mg, 0.15 mmol), tert-butyl piperidin-4-ylcarbamate (46 mg, 0.23 mmol), HOBT (31 mg, 0.23 mmol) and EDCI (44 mg, 0.23 mmol) in DCM (6 ml) was stirred for overnight. The reaction mixture was diluted with DCM (25 ml). The organic phase was washed with saturated Na.sub.2CO.sub.3 and brine (20 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-83-1 (80 mg, yield 91%). LCMS (m/z): 578 [M+H]+.
(4-aminopiperidin-1-yl)((1R,3S)--3-(5-((5-tert-butyloxazol-2-yl)methylthio- )triazol-2-ylamino)cyclohexyl)methanone (MFH-3-85-1)
[0473] To a solution of MFH-3-83-1 (80 mg, 0.14 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 478 [M+H].sup.+.
N-(1-((1R,3S)--3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino- )cyclohexanecarbonyl)piperidin-4-yl)acrylamide (MFH-3-88-1)
[0474] To a solution of MFH-3-85-1 (20 mg, 0.04 mmol) and DIPEA (0.1 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (5 mg, 0.05 mmol) in DCM (0.1 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-88-1 (3.7 mg, yield 17%). LCMS (m/z): 532 [M+H]+.
##STR00256##
5-((5-tert-butyloxazol-2-yl)methylthio)-N-((1R,3R)-3-(4-nitrophenoxy)cycl- ohexyl)thiazol-2-amine (MFH-3-97-1)
[0475] The mixture of MFH-2-83-1 (180 mg, 0.54 mmol), (1R,3R)-3-(4-nitrophenoxy)cyclohexanamine (204 mg, 0.86 mmol) and DIEA (140 mg, 1.1 mmol) in NMP (1 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-97-1 (80 mg, yield 30%). LCMS (m/z): 489 [M+H].sup.+.
N-((1R,3R)-3-(4-aminophenoxy)cyclohexyl)-5-((5-tert-butyloxazol-2-yl)methy- lthio)thiazol-2-amine (MFH-3-102-1)
[0476] To a solution of MFH-3-97-1 (80 mg, 0.16 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (300 mg, 1.3 mmol) and cone. HCl (0.1 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol(4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-3-102-1 (40 mg, yield 54%). LCMS (m/z): 459 [M+H].sup.+.
N-(4-((1R,3R)-3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)- cyclohexyloxy)phenyl)acrylamide (MFH-3-103-1)
[0477] To a solution of MFH-3-102-1 (40 mg, 0.09 mmol) and DIPEA (0.1 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (10 mg, 0.11 mmol) in DCM (0.2 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1 h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-103-1 (9.6 mg, yield 21%). LCMS (m/z): 513 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.00 (s, 1H), 8.04 (d, J=7.3 Hz, 1H), 7.56 (d, J=9.0 Hz, 2H), 6.94 (s, 1H), 6.93-6.89 (m, 2H), 6.71 (s, 1H), 6.39 (dd, J=17.0, 10.1 Hz, 1H), 6.21 (dd, J=17.0, 2.0 Hz, 1H), 5.71 (dd, J=10.1, 2.0 Hz, 1H), 4.66 (s, 1H), 3.94 (s, 2H), 3.83 (s, 1H), 2.01 (d, J=13.3 Hz, 1H), 1.85 (s, 1H), 1.74-1.54 (m, 5H), 1.34 (dd, J=21.1, 11.1 Hz, 1H), 1.18 (s, 9H).
##STR00257## ##STR00258##
(R)-tert-butyl3-(5-cyanothiazol-2-ylamino)piperidine-1-carboxylate (MFH-3-72-1)
[0478] The mixture of 2-bromothiazole-5-carbonitrile (800 mg, 4.23 mmol), (R)-tert-butyl 3-aminopiperidine-1-carboxylate (1.1 g, 5.5 mmol) and DIEA (820 mg, 6.35 mmol) in THF (15 mL) was stirred at 80.degree. C. for 6h. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-72-1 (1.3 g, yield 100%). LCMS (m/z): 309 [M+H].sup.+.
(R)-tert-butyl 3-(5-(aminomethyl)thiazol-2-ylamino)piperidine-1-carboxylate (MFH-3-82-1)
[0479] To a solution of MFH-3-72-1 (300 mg, 1 mmol) and CoCl.sub.2 6H.sub.2O (232 mg, 1 mmol) in ethanol (8 mL) was added NaBH.sub.4 (110 mg, 3 mmol) at room temperature. The vial was sealed and stirred at room temperature for 3h and then was quenched with water. The obtained mixture was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-82-1 (89 g, yield 28%). LCMS (m/z): 313 [M+H].sup.+.
(R)-tert-butyl3-((5-chloro-4-trifluoromethyl)pyridin-2-ylamino)methyl)thia- zol-2-ylamino)piperidine-1-carboxylate (MFH-3-99-1)
[0480] The mixture of MFH-2-82-1 (175 mg, 0.56 mmol), 2-bromo-5-chloro-4-(trifluoromethyl)pyridine (146 mg, 0.56 mmol) and DIEA (108 mg, 0.84 mmol) in NMP (1 mL) was stirred at 80.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-99-1 (90 mg, yield 58%). LCMS (m/z): 492 [M+H].sup.+.
(R)-5-((5-chloro-4-(trifluoromethyl)pyridin-2-ylamino)methyl)-N-(piperidin- -3-yl)thiazol-2-amine (MFH-3-106-1)
[0481] To a solution of MFH-3-99-1 (90 mg, 0.18 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 392 [M+H].sup.+.
(R)--N-(4-(3-(5-((5-chloro-4-trifluoromethyl)pyridin-2-ylamino)methyl)thia- zol-2-ylamino)piperidine-1-carbonyl)phenyl)acrylamide (MFH-3-107-1)
[0482] The mixture of MFH-3-106-1 (20 mg, 0.05 mmol), 4-acrylamidobenzoic acid (10 mg, 0.05 mmol), HOBT (8 mg, 0.06 mmol) and EDCI (11 mg, 0.06 mmol) in DMF (0.5 ml). Then the reaction mixture was stirred for overnight. The reaction mixture was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-107-1 (3.8 mg, yield 13%). LCMS (m/z): 565 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.31 (s, 1H), 8.28 (s, 1H), 7.68 (s, 3H), 7.36 (d, J=7.2 Hz, 2H), 7.25-6.81 (m, 3H), 6.45 (dd, J=17.0, 10.2 Hz, 1H), 6.28 (dd, J=17.0, 1.9 Hz, 1H), 5.78 (dd, J=10.1, 1.9 Hz, 1H), 4.46 (s, 2H), 3.69 (s, 3H), 3.18 (d, J=11.7 Hz, 2H), 1.96 (s, 1H), 1.75 (s, 1H), 1.55 (m, 2H).
##STR00259## ##STR00260##
diethyl (5-tert-butyloxazol-2-yl)methylphosphonate (MFH-3-91-1)
[0483] A solution of 5-tert-butyl-2-(chloromethyl)oxazole (800 mg, 4.61 mmol) in triethyl phosphite (3.83 g, 23.04 mmol) was heated at 120.degree. C. for overnight. The solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 276 [M+H].sup.+.
(E)-tert-butyl5-(2-(5-tert-butyloxazol-2-yl)vinyl)thiazol-2-ylcarbamate (MFH-3-94-1)
[0484] To a solution of MFH-3-91-1 (500 mg, 1.82 mmol) in THF (5 mL) was added t-BuOK (370 mg, 3.3 mmol) at room temperature. The vial was sealed and stirred at room temperature for 10 min. Then, a solution of tert-butyl 5-formylthiazol-2-ylcarbamate (345 mg, 1.5 mmol) in THF (3 mL) was added. After completion, the reaction mixture was quenched with water. The obtained mixture was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-94-1 (440 mg, yield 84%). LCMS (m/z): 350 [M+H].sup.+.
(E)-5-(2-(5-tert-butyloxzol-2-yl)vinyl)thiazol-2-amine (MFH-3-98-1)
[0485] To a solution of MFH-3-94-1 (220 mg, 0.63 mmol) in methanol (5 mL) was added 4N HCl/dioxane (5 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 250 [M+H].sup.+.
(E)-2-(2-(2-bromothiazol-5-yl)vinyl)-5-tert-butyloxazole (MFH-3-105-1)
[0486] To a solution of CuBr.sub.2 (172 mg, 0.77 mmol) in acetonitrile (15 mL) at 0.degree. C. was added t-BuONO (79 mg, 0.77 mmol) followed by compound MFH-3-98-1 (160 mg, 0.64 mmol). The mixture was stirred at 0.degree. C. for one hour, then at room temperature for one hour, ethyl acetate was added and the organic mixture washed with hydrochloric acid (20 mL), dried over magnesium sulfate, filtered through a pad of silica gel, and concentrated under reduced pressure. The residue was chromatographed on silica gel to give the MFH-3-105-1 (50 mg, 25%). LCMS (m/z): 314 [M+H]+.
(R,E)-tert-butyl3-(5-(2-(5-tert-butyloxazol-2-yl))vinyl)thiazol-2-ylamino)- piperidine-1-carboxylate (MFH-3-108-1)
[0487] The mixture of MFH-3-105-1 (50 mg, 0.16 mmol), (R)-tert-butyl 3-aminopiperidine-1-carboxylate (51 mg, 0.26 mmol) and DIEA (41 mg, 0.32 mmol) in NMP (0.5 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (10 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-108-1 (40 mg, yield 58%). LCMS (m/z): 433 [M+H].sup.+.
(R,E)-5-(2-(5-tert-butyloxazol-2-yl)vinyl)-N-(piperidin-3-yl)thiazol-2-ami- ne (MFH-3-109-1)
[0488] To a solution of MFH-3-108-1 (40 g, 0.09 mmol) in methanol (2 mL) was added 4N HCl/dioxane (2 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 333 [M+H].sup.+.
(R,E)-N-(4-(3-(5-(2-(5-tert-butyloxazol-2-yl)vinyl)triazol-2-ylamino)piper- idine-1-carbonyl)phenyl)acrylamide (MFH-3-110-1)
[0489] The mixture of MFH-3-109-1 (35 mg, 0.1 mmol), 4-acrylamidobenzoic acid (30 mg, 0.16 mmol), HOBT (21 mg, 0.16 mmol) and EDCI (30 mg, 0.16 mmol) in DMF (0.5 ml). Then the reaction mixture was stirred for overnight. The reaction mixture was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-110-1 (5 mg, yield 10%). LCMS (m/z): 506 [M+H]+.
##STR00261## ##STR00262##
5-(2-(5-tert-butyloxazol-2-yl)ethyl)thiazol-2-amine (MFH-3-104-1)
[0490] A mixture of compound MFH-3-94-1 (250 mg, 1 mmol) and 10% Pd/C (20 mg) in MeOH (10 mL) was stirred for 5h at room temperature under H.sub.2 balloon. The mixture was filtered through celite. The filtrate was added 4N HCl/dioxane (5 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 252 [M+H].sup.+.
2-(2-(2-bromothiazol-5-yl)ethyl)-5-tert-butyloxazole (MFH-3-114-1)
[0491] To a solution of CuBr.sub.2 (53 mg, 0.24 mmol) in acetonitrile (2 mL) at 0.degree. C. was added t-BuONO (25 mg, 0.24 mmol) followed by compound MFH-3-104-1 (60 mg, 0.24 mmol). The mixture was stirred at 0.degree. C. for one hour, and then was warmed up to room temperature. Ethyl acetate was added and the organic mixture was washed with hydrochloric acid (20 mL), dried over magnesium sulfate, filtered through a pad of silica gel, and concentrated in vacuo. The residue was chromatographed on silica gel to give the MFH-3-114-1 (40 mg, 53%). LCMS (m/z): 316 [M+H]+.
(R)-tert-butyl3-(5-(2-(5-tert-butyloxazol-2-yl)ethyl)thiazol-2-ylamino)pip- eridin-1-carboxylate (MFH-3-115-1)
[0492] The mixture of MFH-3-114-1 (40 mg, 0.13 mmol), (R)-tert-butyl 3-aminopiperidine-1-carboxylate (41 mg, 0.2 mmol) and DIEA (33 mg, 0.25 mmol) in NMP (0.5 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (10 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-115-1 (35 mg, yield 63%). LCMS (m/z): 435 [M+H].sup.+.
(R)-5-(2-(5-tert-butyloxazol-2-yl)ethyl)-N-(piperidin-3-yl)thiazol-2-amine
[0493] To a solution of MFH-3-115-1 (35 mg, 0.08 mmol) in methanol (2 mL) was added 4N HCl/dioxane (2 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 335 [M+H].sup.+.
(R)--N-(4-(3-(5-(2-(5-tert-butyloxazol-2-yl)ethyl)thiazol-2-ylamino)piperi- dine-1-carbonyl)phenyl)acrylamide (MFH-3-116-1)
[0494] The mixture of (R)-5-(2-(5-tert-butyloxazol-2-yl)ethyl)-N-(piperidin-3-yl)thiazol-2-amin- e (23 mg, 0.07 mmol), 4-acrylamidobenzoic acid (15 mg, 0.08 mmol), HOBT (12 mg, 0.09 mmol) and EDCI (17 mg, 0.09 mmol) in DMF (0.5 ml) was stirred for overnight. The reaction mixture was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-116-1 (5.5 mg, yield 16%). LCMS (m/z): 508 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.38 (s, 1H), 8.29 (d, J=7.5 Hz, 1H), 7.82 (d, J=8.8 Hz, 2H), 7.74 (d, J=8.8 Hz, 2H), 6.96 (s, 1H), 6.68 (s, 1H), 6.45 (dd, J=17.0, 10.2 Hz, 1H), 6.29 (dd, J=17.0, 1.9 Hz, 1H), 5.79 (dd, J=10.1, 1.9 Hz, 1H), 3.87 (dd, J=12.3, 4.0 Hz, 1H), 3.75-3.63 (m, 2H), 3.13-2.99 (m, 4H), 2.96 (t, J=6.8 Hz, 2H), 1.88 (m, 2H), 1.63 (m, 2H), 1.22 (s, 9H).
##STR00263## ##STR00264##
2-bromo-5-((3-(trifluoromethyl)benzyloxy)methyl)thiazole (MFH-3-111-1)
[0495] The mixture of (2-bromothiazol-5-yl)methanol (180 mg, 0.93 mmol), 1-(bromomethyl)-3-(trifluoromethyl)benzene (288 mg, 1.2 mmol) and NaOH (67 mg, 1.67 mmol) in DMF (2 mL) was stirred at 120.degree. C. for 3h. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (PE/EA=0-50%) to obtain MFH-3-111-1 (160 mg, yield 49%). LCMS (m/z): 353 [M+H].sup.+.
(R)-tert-butyl35-((3-(trifluoromethyl)benzyloxy)methyl)thiazol-2-ylamino)p- iperidine-1-carboxylate (MFH-3-112-1)
[0496] The mixture of MFH-3-111-1 (160 mg, 0.34 mmol), (R)-tert-butyl 3-aminopiperidine-1-carboxylate (109 mg, 0.55 mmol) and DIEA (88 mg, 0.68 mmol) in NMP (1 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-112-1 (35 mg, yield 22%). LCMS (m/z): 472 [M+H].sup.+.
(R)--N-(piperidin-3-yl)-5-((3-(trifluoromethyl)benzyloxy)methyl)thiazol-2-- amine (MFH-3-119-1)
[0497] To a solution of MFH-3-112-1 (35 mg, 0.08 mmol) in methanol (2 mL) was added 4N HCl/dioxane (2 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 372 [M+H].sup.+.
(R)--N-(4-(3-(5-((3-(trifluoromethyl)benzyloxy)methyl)thiazol-2-ylamino)pi- peridine-1-carbonyl)phenyl)acrylamide (MFH-3-120-1)
[0498] The mixture of MFH-3-119-1 (23 mg, 0.06 mmol), 4-acrylamidobenzoic acid (15 mg, 0.08 mmol), HOBT (12 mg, 0.09 mmol) and EDCI (17 mg, 0.09 mmol) in DMF (0.5 ml). Then the reaction mixture was stirred for overnight. The reaction mixture was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-120-1 (8.7 mg, yield 26%). LCMS (m/z): 545 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.32 (s, 1H), 7.72-7.57 (m, 7H), 7.37 (d, J=5.1 Hz, 3H), 6.47-6.40 (m, 1H), 6.27 (dd, J=16.6, 6.9 Hz, 1H), 5.77 (t, J=8.3 Hz, 1H), 4.58 (d, J=8.2 Hz, 2H), 4.56 (s, 2H), 3.78 (s, 3H), 3.20 (d, J=6.7 Hz, 2H), 1.98 (s, 1H), 1.76 (s, 1H), 1.55 (m, 2H).
##STR00265## ##STR00266##
2-(((2-bromothiazol-5-yl)methoxy)methyl)-5-tert-butyloxazole (MFH-3-121-1)
[0499] The mixture of (2-bromothiazol-5-yl)methanol (220 mg, 1.1 mmol), 5-tert-butyl-2-(chloromethyl)oxazole (256 mg, 1.5 mmol) and NaOH (82 mg, 2.04 mmol) in DMF (2 mL) was stirred at 120.degree. C. for 3h. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (PE/EA=0-50%) to obtain MFH-3-111-1 (170 mg, yield 45%). LCMS (m/z): 332 [M+H].sup.+.
(R)-tert-butyl3-(5-(((5-tert-butyloxazol-2-yl)methoxy)methyl)triazol-2-yla- mino)piperidine-1-carboxylate (MFH-3-123-1)
[0500] The mixture of MFH-3-121-1 (170 mg, 0.51 mmol), (R)-tert-butyl 3-aminopiperidine-1-carboxylate (164 mg, 0.82 mmol) and DIEA (133 mg, 1 mmol) in NMP (1 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-123-1 (50 mg, yield 22%). LCMS (m/z): 451 [M+H].sup.+.
(R)-5-(((5-tert-butyloxazol-2-yl)methoxy)methyl)-N-(piperidin-3-yl)thiazol- -2-amine (MFH-3-126-1)
[0501] To a solution of MFH-3-123-1 (50 mg, 0.11 mmol) in methanol (2 mL) was added 4N HCl/dioxane (2 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 372 [M+H].sup.+.
(R)--N-(4-(3-(((5-tert-butyloxazol-2-yl)methoxy)methyl)thiazol-2-ylamino)p- iperidine-1-carbonyl)phenyl)acrylamide (MFH-3-128-1)
[0502] The mixture of MFH-3-126-1 (23 mg, 0.06 mmol), 4-acrylamidobenzoic acid (15 mg, 0.08 mmol), HOBT (12 mg, 0.09 mmol) and EDCI (17 mg, 0.09 mmol) in DMF (0.5 ml) was stirred for overnight. The reaction mixture was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-128-1 (7 mg, yield 22%). LCMS (m/z): 524 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.37 (s, 1H), 8.33 (d, J=38.9 Hz, 1H), 7.84 (d, J=8.5 Hz, 2H), 7.75 (d, J=8.4 Hz, 2H), 7.37 (d, J=8.3 Hz, 1H), 6.89-6.71 (m, 1H), 6.46 (dd, J=16.9, 10.1 Hz, 1H), 6.29 (d, J=17.0 Hz, 1H), 5.80 (d, J=10.0 Hz, 1H), 4.59-4.40 (m, 2H), 4.05 (s, 1H), 3.99-3.85 (m, 2H), 3.33 (dd, J=31.2, 10.4 Hz, 2H), 3.00 (m, 2H), 1.93 (d, J=15.1 Hz, 1H), 1.84 (s, 1H), 1.62 (s, 2H), 1.23 (s, 9H).
##STR00267## ##STR00268##
(R)-tert-butyl3-(5-(((5-tert-butyloxazol-2-yl)methylamino)methyl)thiazol-- 2-ylamino)piperidine-1-carboxylate (MFH-3-90-1)
[0503] The mixture of MFH-2-82-1 (160 mg, 0.51 mmol), 5-tert-butyl-2-(chloromethyl)oxazole (89 mg, 0.51 mmol) and DIEA (132 mg, 1 mmol) in NMP (1 mL) was stirred at 80.degree. C. for overnight. The solution was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-90-1 (133 mg, yield 58%). LCMS (m/z): 450 [M+H].sup.+.
(R)-5-(((5-tert-butyloxazol-2-yl)methylamino)methyl)-N-(piperidin-3-yl)thi- azol-2-amine (MFH-3-136-1)
[0504] To a solution of MFH-3-90-1 (60 mg, 0.13 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 350 [M+H]+.sup.+.
(R)--N-(4-(3-(5-((5-tert-butyloxazol-2-yl)methylamino)methyl)thiazol-2-yla- mino)piperidine-1-carbonyl)phenyl)acrylamide (MFH-3-137-1)
[0505] The mixture of MFH-3-136-1 (30 mg, 0.09 mmol), 4-acrylamidobenzoic acid (17 mg, 0.09 mmol) and TEA (9 mg, 0.09 mmol) in DMF (1 ml). The reaction mixture was cooled to -60.degree. C., and HATU (33 mg, 0.09 mmol) was added in the reaction mixture. The reaction mixture was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-137-1 (18 mg, yield 38%). LCMS (m/z): 523 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.31 (s, 1H), 8.79-8.59 (m, 2H), 8.10-7.97 (m, 1H), 7.69 (s, 1H), 7.37 (s, 1H), 7.13 (d, J=6.5 Hz, 1H), 6.96 (dd, J=9.8, 4.5 Hz, 1H), 6.78 (s, 1H), 6.43 (d, J=6.7 Hz, 1H), 6.28 (s, 1H), 5.79 (dd, J=12.2, 4.2 Hz, 1H), 4.31 (s, 2H), 3.86 (s, 2H), 3.72 (s, 2H), 3.43 (d, J=10.6 Hz, 1H), 2.93-2.75 (m, 2H), 2.00 (d, J=8.6 Hz, 1H), 1.89 (d, J=22.0 Hz, 1H), 1.59 (m, 2H), 1.23 (s, 9H).
##STR00269##
(R)-tert-butyl4-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)triazol-2-ylam- ino)piperidin-1-ylsulfonyl)phenylcarbamate (MFH-3-147-1)
[0506] The mixture of MFH-2-85-1 (60 mg, 0.17 mmol) and TEA (26 mg, 0.26 mmol) in DCM (1 ml) was cooled to 0.degree. C. and then a solution of tert-butyl 4-(chlorosulfonyl)phenylcarbamate (50 mg, 0.17 mmol) in DCM (1 mL) was added. The reaction mixture was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-147-1 (80 mg, yield 78%). LCMS (m/z): 608 [M+H].sup.+.
(R)--N-(1-(4-aminophenylsulfonyl)piperidin-3-yl)-5-((5-tert-butyloxazol-2-- yl)methylthio)thiazol-2-amine (MFH-3-148-1)
[0507] To a solution of MFH-3-147-1 (80 mg, 0.13 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 507 [M+H].sup.+.
(R)--N-(4-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)pip- eridin-1-ylsulfonyl)phenyl)acrylamide (MFH-3-151-1)
[0508] To a solution of MFH-3-148-1 (30 mg, 0.06 mmol) and DIPEA (0.2 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (7 mg, 0.08 mmol) in DCM (0.2 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1 h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-151-1 (8.5 mg, yield 25%). LCMS (m/z): 561 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.56 (d, J=22.3 Hz, 1H), 8.00 (d, J=7.3 Hz, 1H), 7.93-7.89 (m, 1H), 7.87 (dd, J=11.3, 4.4 Hz, 1H), 7.83-7.75 (m, 1H), 7.72-7.68 (m, 1H), 6.98 (d, J=2.7 Hz, 1H), 6.75-6.70 (m, 1H), 6.51-6.42 (m, 1H), 6.32 (ddd, J=17.0, 3.9, 1.9 Hz, 1H), 5.83 (ddd, J=10.1, 6.1, 1.9 Hz, 1H), 3.96 (d, J=3.8 Hz, 2H), 3.43-3.34 (m, 1H), 3.25 (d, J=11.8 Hz, 1H), 3.07-2.93 (m, 1H), 2.86 (dd, J=12.6, 9.7 Hz, 1H), 2.32-2.22 (m, 1H), 1.77 (d, J=6.7 Hz, 1H), 1.67 (d, J=3.5 Hz, 1H), 1.52 (d, J=10.2 Hz, 1H), 1.34 (dd, J=18.1, 10.1 Hz, 1H), 1.24-1.16 (m, 9H).
##STR00270## ##STR00271##
tert-butyl 5-(benzylthio)indoline-1-carboxylate (MFH-3-158-1)
[0509] A mixture of tert-butyl 5-bromoindoline-1-carboxylate (600 mg, 2.02 mmol), phenylmethanethiol (276 mg, 2.22 mmol), Pd.sub.2(dba).sub.3 (185 mg, 0.2 mmol), Xantphos (117 mg, 0.2 mmmol), and DIEA (783 mg, 6.06 mmol) in toluene (25 mL) was refluxed under N.sub.2 atmosphere for 5h. After cooling to room temperature, ethyl acetate (50 mL) and H.sub.2O (50 mL) were added to the mixture, and insoluble materials were removed by filtration. The filtrate was diluted with ethyl acetate (50 mL) and was washed with water, 0.2 M hydrochloric acid, aqueous NaHCO.sub.3 solution, and brine. The organic layer was dried over Na.sub.2SO.sub.4 and concentrated under reduced pressure. The residue was crystallized from AcOEt-i-Pr.sub.2O to give the title compound as a yellow solid (660 mg, 96%). LCMS (m/z): 342 [M+H]+.
tert-butyl 5-(chlorosulfonyl)indoline-1-carboxylate (MFH-3-159-1)
[0510] To a mixture of compound MFH-3-158-1 (660 g, 1.94 mmol), acetic acid (6 mL) in water (3 mL) was added N-chlorosuccinimide (1.1 g, 7.74 mol) at 0.degree. C. The mixture was stirred at room temperature for 4 h. Precipitated solid was collected by filtration, washed with water and dried to give the title compound as a pale yellow solid (400 mg, 65%). This compound was used for the next step without further purification.
(R)-tert-butyl5-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylami- no)piperidin-1-ylsulfonyl)indoline-1-carboxylate (MFH-3-162-1)
[0511] The mixture of MFH-2-85-1 (60 mg, 0.17 mmol) and TEA (26 mg, 0.26 mmol) in DCM (1 ml) was cooled to 0.degree. C. and a solution of MFH-3-159-1 (54 mg, 0.17 mmol) in DCM (1 mL) was added. And reaction completed, the reaction mixture was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-162-1 (82 mg, yield 76%). LCMS (m/z): 634 [M+H].sup.+.
(R)-5-((5-tert-butyloxazol-2-yl)methylthio)-N-(1-(indolin-5-ylsulfonyl)pip- eridin-3-yl)thiazol-2-amine (MFH-3-164-1)
[0512] To a solution of MFH-3-162-1 (82 mg, 0.13 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 534 [M+H].sup.+.
(R)-1-(5-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)pipe- ridin-1-ylsulfonyl)indolin-1-yl)prop-2-en-1-one (MFH-3-168-1)
[0513] To a solution of MFH-3-164-1 (20 mg, 0.04 mmol) and DIPEA (0.1 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (5 mg, 0.05 mmol) in DCM (0.1 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-168-1 (5 mg, yield 23%). LCMS (m/z): 588 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 8.29 (s, 1H), 7.94 (d, J=7.4 Hz, 1H), 7.57 (dd, J=12.2, 3.8 Hz, 2H), 6.96 (s, 1H), 6.81-6.74 (m, 1H), 6.74 (s, 1H), 6.35 (dd, J=16.7, 2.0 Hz, 1H), 5.89 (dd, J=10.3, 2.0 Hz, 1H), 4.29 (t, J=8.7 Hz, 2H), 3.96 (s, 2H), 3.69 (s, 1H), 3.55 (d, J=8.0 Hz, 1H), 3.25 (t, J=8.4 Hz, 3H), 2.31-2.24 (m, 1H), 2.18 (t, J=7.4 Hz, 1H), 1.77 (d, J=6.6 Hz, 2H), 1.49 (dd, J=17.4, 10.0 Hz, 2H), 1.24 (s, 9H).
##STR00272## ##STR00273##
5-(pyridin-4-ylmethylthio)thiazol-2-amine (MFH-3-146-1)
[0514] To a solution of compound MFH-2-76-1 (300 mg, 1.91 mmol) in EtOH (5 ml) was added NaBH.sub.4 (144 mg, 3.82 mmol) room temperature. The mixture was stirred for 1h, and then acetone (3 ml) was slowly introduced. After 1 h, a solution of 4-(bromomethyl)pyridine hydrobromide (483 mg, 1.91 mmol) in EtOH (3 ml) was added. The resulting dark reaction mixture was heated to reflux for 1h, and was then cooled and concentrated in vacuo. The residue was partitioned between EtOAc and brine. The organic phase was separated, dried (MgSO.sub.4), and concentrated in vacuo to give a crude solid which was triturated with diethyl ether/hexane to provide compound MFH-3-146-1 (240 mg, 56%) as solid LCMS (m/z): 224 [M+H]+.
2-bromo-5-(pyridin-4-ylmethylthio)thiazole (MFH-3-153-1)
[0515] To a solution of CuBr.sub.2 (288 mg, 1.29 mmol) in acetonitrile (8 mL) at 0.degree. C. was added t-BuONO (133 mg, 1.29 mmol) followed by compound MFH-3-146-1 (240 g, 1.08 mmol). The mixture was stirred at 0.degree. C. for one hour, then at room temperature for one hour. Ethyl acetate was added and the organic mixture washed with hydrochloric acid (20 mL), dried over magnesium sulfate, filtered through a pad of silica gel, and concentrated in vacuo. The residue was chromatographed on silica gel to give the MFH-3-153-1 (130 mg, 42%). LCMS (m/z): 288 [M+H]+.
(R)-tert-butyl3-(5-(pyridin-4-ylmethylthio)thiazol-2-ylamino)piperidine-1-- carboxylate (MFH-3-156-1)
[0516] The mixture of MFH-3-153-1 (130 mg, 0.45 mmol), (R)-tert-butyl 3-aminopiperidine-1-carboxylate (145 mg, 0.72 mmol) and DIEA (117 mg, 0.91 mmol) in NMP (1 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-156-1 (90 mg, yield 49%). LCMS (m/z): 407 [M+H].sup.+.
(R)--N-(piperidin-3-yl)-5-(pyridin-4-ylmethylthio)thiazol-2-amine (MFH-3-157-1)
[0517] To a solution of MFH-3-156-1 (90 mg, 0.22 mmol) in methanol (2 mL) was added 4N HCl/dioxane (2 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 307 [M+H].sup.+.
(R)-tert-butyl4-(3-(5-(pyridin-4-ylmethylthio)thiazol-2-ylamino)piperidine- -1-carbonyl)phenylcarbamate (MFH-3-177-1)
[0518] The mixture of MFH-3-157-1 (60 mg, 0.2 mmol), 4-(tert-butoxycarbonylamino)benzoic acid (15 mg, 0.08 mmol), HOBT (12 mg, 0.09 mmol) and EDCI (60 mg, 0.26 mmol) in DMF (1 ml) was stirred for overnight. The reaction mixture was diluted with chloroform and iso-propanol (4:1). The saturated Na.sub.2CO.sub.3 was added and the reaction mixture was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-3-177-1 (90 mg, yield 86%). LCMS (m/z): 526 [M+H]+.
(R)-(4-aminophenyl)(3-(5-(pyridin-4-ylmethylthio)thiazol-2-ylamino)piperid- in-1-yl)methanone (MFH-3-178-1)
[0519] To a solution of MFH-3-177-1 (90 mg, 0.17 mmol) in methanol (2 mL) was added 4N HCl/dioxane (2 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 426 [M+H].sup.+.
(R)--N-(4-(3-(5-(pyridin-4-ylmethylthio)thiazol-2-ylamino)piperidine-1-car- bonyl)phenyl)acrylamide (MFH-3-179-1)
[0520] To a solution of MFH-3-178-1 (40 mg, 0.09 mmol) and DIPEA (0.2 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (11 mg, 0.12 mmol) in DCM (0.1 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-179-1 (4.4 mg, yield 10%). LCMS (m/z): 480 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.30 (s, 1H), 8.06 (s, 1H), 7.85 (s, 2H), 7.68 (s, 2H), 7.36 (s, 2H), 7.25-7.02 (m, 2H), 6.80 (s, 1H), 6.49-6.37 (m, 1H), 6.27 (d, J=16.9 Hz, 1H), 5.77 (d, J=10.1 Hz, 1H), 4.12 (s, 1H), 3.99 (s, 2H), 3.43-3.31 (m, 2H), 3.14 (d, J=28.5 Hz, 2H), 1.95 (s, 1H), 1.76 (s, 1H), 1.55 (s, 2H).
##STR00274## ##STR00275##
5-(pyridin-3-ylmethylthio)thiazol-2-amine (MFH-3-171-1)
[0521] To a solution of compound MFH-2-76-1 (300 mg, 1.91 mmol) in EtOH (5 ml) was added NaBH.sub.4 (144 mg, 3.82 mmol) portionwise at room temperature. The mixture was stirred for 1h, and then acetone (3 ml) was slowly introduced. After 1 h, a solution of 3-(bromomethyl)pyridine hydrobromide (483 mg, 1.91 mmol) in EtOH (3 mL) was added. The resulting dark reaction mixture was heated to reflux for 1 h, and was then cooled and concentrated in vacuo. The residue was partitioned between EtOAc and brine. The organic phase was separated, dried (MgSO.sub.4), and concentrated in vacuo to give a crude solid which was triturated with diethyl ether/hexane to provide compound MFH-3-171-1 (310 mg, 73%) LCMS (m/z): 224 [M+H]+.
2-bromo-5-(pyridin-3-ylmethylthio)thiazole (MFH-3-180-1)
[0522] To a solution of CuBr.sub.2 (370 mg, 1.66 mmol) in acetonitrile (20 mL) at 0.degree. C. was added t-BuONO (172 mg, 1.66 mmol) followed by compound MFH-3-171-1 (310 mg, 1.39 mmol). The mixture was stirred at 0.degree. C. for one hour, then at room temperature for one hour, ethyl acetate was added and the organic mixture washed with hydrochloric acid (2.times.50 mL), dried over magnesium sulfate, filtered through a pad of silica gel, and concentrated in vacuo. The residue was chromatographed on silica gel to give the MFH-3-180-1 (288 mg, 72%). LCMS (m/z): 288 [M+H]+.
(R)-tert-butyl3-(5-(pyridin-3-ylmethylthio)thiazol-2-ylamino)piperidine-1-- carboxylate (MFH-3-182-1)
[0523] The mixture of MFH-3-180-1 (288 mg, 1 mmol), (R)-tert-butyl 3-aminopiperidine-1-carboxylate (320 mg, 1.6 mmol) and DIEA (194 mg, 1.5 mmol) in NMP (1 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-182-1 (180 mg, yield 44%). LCMS (m/z): 407 [M+H].sup.+.
(R)--N-(piperidin-3-yl-5-(pyridin-3-ylmethylthio)thiazol-2-amine (MFH-3-185-1)
[0524] To a solution of MFH-3-182-1 (180 mg, 0.44 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 307 [M+H].sup.+.
(R)-(4-nitrophenyl)(3-(5-(pyridin-3-ylmethylthio)thiazol-2-ylamino)piperid- in-1-yl)methanone (MFH-3-186-1)
[0525] The mixture of MFH-3-185-1 (120 mg, 0.39 mmol), 4-nitrobenzoyl chloride (72 mg, 0.39 mmol) in pyridine (2 mL) was stirred for overnight at room temperature. Then the reaction mixture was concentrated under reduced pressure and the residue was directly used in the next step. LCMS (m/z): 456 [M+H].sup.+.
(R)-(4-aminophenyl)(3-(5-(pyridin-3-ylmethylthio)thiazol-2-ylamino)piperid- in-1-yl)methanone (MFH-3-187-1)
[0526] To a solution of MFH-3-186-1 (178 mg, 0.39 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (881 mg, 3.9 mmol) and cone. HCl (0.2 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-3-187-1 (20 mg, yield 12%). LCMS (m/z): 426 [M+H]+.
(R)--N-(4-(3-(5-(pyridin-3-ylmethylthio)thiazol-2-ylamino)piperidine-1-car- bonyl)phenyl)acrylamide (MFH-3-191-1)
[0527] To a solution of MFH-3-187-1 (20 mg, 0.05 mmol) and DIPEA (0.1 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (6 mg, 0.06 mmol) in DCM (0.5 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-191-1 (6 mg, yield 26%). LCMS (m/z): 480 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.30 (s, 1H), 8.58 (d, J=53.1 Hz, 2H), 8.00 (d, J=44.0 Hz, 2H), 7.78-7.62 (m, 2H), 7.40 (d, J=29.7 Hz, 2H), 7.14 (dd, J=67.0, 35.1 Hz, 1H), 6.82 (s, 1H), 6.43 (dd, J=17.4, 10.0 Hz, 1H), 6.28 (t, J=12.8 Hz, 1H), 5.84-5.74 (m, 1H), 4.04 (s, 1H), 3.98 (s, 2H), 3.31 (s, 2H), 3.17 (s, 2H), 1.96 (s, 1H), 1.75 (s, 1H), 1.54 (s, 2H).
##STR00276## ##STR00277##
(1R,3R)-3-(4-nitro-2-(trifluoromethyl)phenoxy)cyclohexanamine (MFH-3-166-1)
[0528] To a solution of compound (1R,3R)-3-aminocyclohexanol hydrochloride (120 mg, 0.8 mmol) in absolute DMF (1 ml) was added NaH (130 mg, 3.3 mmol) portionwise at 0.degree. C. The mixture was stirred for 1 h, and then 1-fluoro-4-nitro-2-(trifluoromethyl)benzene (170 mg, 0.8 mmol) was slowly introduced. After 3h, the water was added slowly. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-166-1 (50 mg, yield 20%). LCMS (m/z): 305 [M+H].sup.+.
5-((5-tert-butyloxazol-2-yl)methylthio)-N-((1R,3R)-3-(4-nitro-2-(trifluoro- methyl)phenoxy)cyclohexyl)thiazol-2-amine (MFH-3-183-1)
[0529] The mixture of MFH-2-83-1 (55 mg, 0.16 mmol), MFH-3-166-1 (50 mg, 0.16 mmol) and DIEA (32 mg, 0.25 mmol) in NMP (0.5 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-183-1 (28 mg, yield 31%). LCMS (m/z): 557 [M+H].sup.+.
N-((1R,3R)-3-(4-amino-2-(trifluoromethyl)phenoxy)cyclohexyl)-5-((5-(tert-b- utyloxazol-2-yl)methylthio)thiazol-2-amine (MFH-3-188-1)
[0530] To a solution of MFH-3-183-1 (28 mg, 0.05 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (113 mg, 0.5 mmol) and conc. HCl (0.1 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-3-188-1 (20 mg, yield 75%). LCMS (m/z): 527 [M+H]+.
N-(4-((1R,3R)-3-(5-(((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino- )cyclohexyloxy)-3-(trifluoromethyl)phenyl)acrylamide (MFH-3-192-1)
[0531] To a solution of MFH-3-188-1 (20 mg, 0.04 mmol) and DIPEA (0.1 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (5 mg, 0.05 mmol) in DCM (0.1 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1 h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-192-1 (5.4 mg, yield 24%). LCMS (m/z): 581 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.27 (s, 1H), 8.04 (t, J=5.4 Hz, 2H), 7.82 (dd, J=9.0, 2.5 Hz, 1H), 7.25 (t, J=8.1 Hz, 1H), 6.95 (d, J=5.6 Hz, 1H), 6.70 (d, J=7.3 Hz, 1H), 6.39 (dd, J=17.0, 10.1 Hz, 1H), 6.26 (dd, J=17.0, 2.0 Hz, 1H), 5.77 (dd, J=10.1, 2.0 Hz, 1H), 4.92 (s, 1H), 3.93 (d, J=0.8 Hz, 2H), 2.10 (d, J=14.5 Hz, 1H), 1.93 (d, J=14.3 Hz, 1H), 1.79 (s, 1H), 1.65 (dd, J=22.7, 11.5 Hz, 2H), 1.59 (s, 2H), 1.32 (m, 1H), 1.23 (dd, J=11.2, 6.2 Hz, 1H), 1.17 (s, 9H).
##STR00278## ##STR00279##
5-((2-(trifluoromethyl)pyrimidin-5-yl)methylthio)thiazol-2-amine (MFH-3-184-1)
[0532] To a solution of compound MFH-2-76-1 (300 mg, 1.91 mmol) in absolute EtOH (5 ml) was added NaBH.sub.4 (144 mg, 3.82 mol) at room temperature. The mixture was stirred for 1 h, and then acetone (3 ml) was slowly introduced. After 1 h, a solution of 5-(chloromethyl)-2-(trifluoromethyl)pyrimidine (375 mg, 1.91 mmol) in EtOH (3 ml) was added. The resulting dark reaction mixture was heated to reflux for 1 h, and was then cooled and concentrated in vacuo. The residue was partitioned between EtOAc and brine. The organic phase was separated, dried (MgSO.sub.4), and concentrated in vacuo to give a crude solid which was triturated with diethyl ether/hexane to provide compound MFH-3-184-1 (500 mg, 90%) LCMS (m/z): 293 [M+H]+.
2-bromo-5-((2-(trifluoromethyl)pyrimidin-5-yl)methylthio)thiazole (MFH-3-189-1)
[0533] To a solution of CuBr.sub.2 (458 mg, 2.05 mmol) in acetonitrile (15 mL) at 0.degree. C. was added t-BuONO (211 mg, 2.05 mmol) followed by compound MFH-3-184-1 (500 mg, 1.71 mmol). The mixture was stirred at 0.degree. C. for one hour, then at room temperature for one hour. Ethyl acetate was added and the organic mixture was washed with hydrochloric acid (50 mL), dried over magnesium sulfate, filtered through a pad of silica gel, and concentrated in vacuo. The residue was chromatographed on silica gel to give the MFH-3-189-1 (430 mg, 70%). LCMS (m/z): 357 [M+H]+.
(R)-tert-butyl3-(5-((2-(trifluoromethyl)pyrimidin-5-yl)methylthio)thiazol-- 2-ylamino)piperidine-1-carboxylate (MFH-3-190-1)
[0534] The mixture of MFH-3-189-1 (430 mg, 1.21 mmol), (R)-tert-butyl 3-aminopiperidine-1-carboxylate (362 mg, 1.81 mmol) and DIEA (312 mg, 2.4 mmol) in NMP (1 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-190-1 (380 mg, yield 66%). LCMS (m/z): 476 [M+H].sup.+.
(R)--N-(piperidin-3-yl)-5-((2-(trifluoromethyl)pyrimidin-5-yl)methylthio)t- hiazol-2-amine (MFH-3-194-1)
[0535] To a solution of MFH-3-190-1 (380 mg, 0.8 mmol) in methanol (5 mL) was added 4N HCl/dioxane (5 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 376 [M+H].sup.+.
(R)-(4-nitrophenyl)(3-(5-((2-(trifluoromethyl)pyrimidin-5-yl)methylthio)tr- iazol-2-ylamino)piperidin-1-yl)methanone (MFH-3-195-1)
[0536] The mixture of MFH-3-194-1 (230 mg, 0.61 mmol), 4-nitrobenzoyl chloride (113 mg, 0.61 mmol) in pyridine (2 mL) was stirred for overnight at room temperature. Then the reaction mixture was concentrated under reduced pressure and the residue was directly used in the next step. LCMS (m/z): 525 [M+H].sup.+.
(R)-(4-aminophenyl)(3-(5-((2-(trifluoromethyl)pyrimidin-5-yl)methylthio)th- iazol-2-ylamino)piperidin-1-yl)methanone (MFH-3-198-1)
[0537] To a solution of MFH-3-195-1 (280 mg, 0.53 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (965 mg, 4.27 mmol) and cone. HCl (0.2 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-3-198-1 (200 mg, yield 76%). LCMS (m/z): 495 [M+H]+.
(R)--N-(4-(3-(5-((2-(trifluoromethyl)pyrimidin-5-yl)methylthio)thiazol-2-y- lamino)piperidine-1-carbonyl)phenyl)acrylamide (MFH-3-201-1)
[0538] To a solution of MFH-3-198-1 (40 mg, 0.08 mmol) and DIPEA (0.1 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (10 mg, 0.1 mmol) in DCM (0.2 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1 h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-201-1 (9.8 mg, yield 22%). LCMS (m/z): 549 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.26 (s, 1H), 8.81 (s, 2H), 8.11 (s, 1H), 7.79-7.59 (m, 2H), 7.38 (t, J=13.1 Hz, 2H), 6.99-6.73 (m, 1H), 6.50-6.35 (m, 1H), 6.34-6.17 (m, 1H), 5.80 (m, 1H), 4.01 (s, 2H), 3.81-3.73 (m, 3H), 3.45-3.20 (m, 2H), 1.94 (d, J=18.7 Hz, 1H), 1.78 (m, 1H), 1.54 (s, 2H).
##STR00280## ##STR00281##
5-(pyridin-2-ylmethylthio)thiazol-2-amine (MFH-3-170-1)
[0539] To a solution of compound MFH-2-7-1 (300 mg, 1.91 mmol) in absolute EtOH (5 ml) was added NaBH.sub.4 (144 mg, 3.82 mmol) portionwise at room temperature. The mixture was stirred for 1h, and then acetone (3 ml) was slowly introduced. After 1 h, a solution of 2-(chloromethyl)pyridine hydrochloride (313 mg, 1.91 mmol) in EtOH (3 ml) was added. The resulting dark reaction mixture was heated to reflux for 1 h, and was then cooled and concentrated in vacuo. The residue was partitioned between EtOAc and brine. The organic phase was separated, dried (MgSO.sub.4), and concentrated in vacuo to give a crude solid which was triturated with diethyl ether/hexane to provide compound MFH-3-170-1 (260 mg, 61%) LCMS (m/z): 224 [M+H]+.
2-bromo-5-(pyridin-2-ylmethylthio)thiazole (MFH-3-176-1)
[0540] To a solution of CuBr.sub.2 (313 mg, 1.4 mmol) in acetonitrile (20 mL) at 0.degree. C. was added t-BuONO (144 mg, 1.4 mmol) followed by compound MFH-3-170-1 (260 mg, 1.16 mmol). The mixture was stirred at 0.degree. C. for one hour, then at room temperature for one hour. Ethyl acetate was added and the organic mixture washed with hydrochloric acid (2.times.50 mL), dried over magnesium sulfate, filtered through a pad of silica gel, and concentrated in vacuo. The residue was chromatographed on silica gel to give the MFH-3-176-1 (90 mg, 27%). LCMS (m/z): 288 [M+H]+.
(R)-tert-butyl3-(5-(pyridin-2-ylmethylthio)thiazol-2-ylamino)piperidine-1-- carboxylate (MFH-3-196-1)
[0541] The mixture of MFH-3-176-1 (180 mg, 0.63 mmol), (R)-tert-butyl 3-aminopiperidine-1-carboxylate (200 mg, 1 mmol) and DIEA (162 mg, 1.3 mmol) in NMP (I mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-196-1 (120 mg, yield 47%). LCMS (m/z): 407 [M+H].sup.+.
(R)--N-(piperidin-3-yl)-5-(pyridin-2-ylmethylthio)thiazol-2-amine (MFH-3-197-1)
[0542] To a solution of MFH-3-196-1 (120 mg, 0.3 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 307 [M+H].sup.+.
(R)-(4-nitrophenyl)(3-(5-(pyridin-2-ylmethylthio)thiazol-2-ylamino)piperid- in-1-yl)methanone (MFH-3-199-1)
[0543] The mixture of MFH-3-197-1 (80 mg, 0.26 mmol), 4-nitrobenzoyl chloride (48 mg, 0.26 mmol) in pyridine (2 mL) was stirred for overnight at room temperature. Then the reaction mixture was concentrated under reduced pressure and the residue was directly used in the next step. LCMS (m/z): 456 [M+H].sup.+.
(R)-(4-aminophenyl)(3-(5-(pyridin-2-ylmethylthio)thiazol-2-ylamino)piperid- in-1-yl)methanone (MFH-3-200-1)
[0544] To a solution of MFH-3-199-1 (118 mg, 0.261 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (472 mg, 2.1 mmol) and conc. HCl (0.2 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-3-200-1 (60 mg, yield 54%). LCMS (m/z): 426 [M+H]+.
(R)--N-(4-(3-(5-(pyridin-3-ylmethylthio)thiazol-2-ylamino)piperidine-1-car- bonyl)phenyl)acrylamide (MFH-3-191-1)
[0545] To a solution of MFH-3-187-1 (40 mg, 0.1 mmol) and DIPEA (0.1 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (12 mg, 0.12 mmol) in DCM (0.2 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-3-203-1 (8.4 mg, yield 19%). LCMS (m/z): 480 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.30 (s, 1H), 8.62 (s, 1H), 8.06 (d, J=50.5 Hz, 2H), 7.72 (d, J=21.6 Hz, 2H), 7.51 (s, 2H), 7.37 (s, 2H), 6.86 (s, 1H), 6.44 (dd, J=17.1, 10.2 Hz, 1H), 6.27 (d, J=16.9 Hz, 1H), 5.85-5.74 (m, 1H). 4.04 (s, 1H), 3.98 (s, 2H), 3.31 (s, 2H), 3.17 (s, 2H), 1.96 (s, 1H), 1.75 (s, 1H), 1.54 (s, 2H).
##STR00282## ##STR00283##
5-((tetrahydro-2H-pyran-4-yl)methylthio)thiazol-2-amine (MFH-3-202-1)
[0546] To a solution of compound MFH-2-76-1 (194 mg, 1.23 mmol) in absolute EtOH (4 ml) was added NaBH.sub.4 (93 mg, 2.47 mmol) at room temperature. The mixture was stirred for 1h, and then acetone (2 ml) was slowly introduced. After 1h, a solution of 4-(bromomethyl)-tetrahydro-2H-pyran (221 mg, 1.23 mmol) in EtOH (2 ml) was added. The resulting dark reaction mixture was heated to reflux for 1 h, and was then cooled and concentrated in vacuo. The residue was partitioned between EtOAc and brine. The organic phase was separated, dried (MgSO.sub.4), and concentrated in vacuo to give a crude solid which was triturated with diethyl ether/hexane to provide compound MFH-3-202-1 (250 mg, 88%) LCMS (m/z): 231 [M+H]+.
2-bromo-5-((tetrahydro-2H-pyran-4-yl)methylthio)thiazole (MFH-3-204-1)
[0547] To a solution of CuBr.sub.2 (290 mg, 1.3 mmol) in acetonitrile (15 mL) at 0.degree. C. was added t-BuONO (175 mg, 1.3 mmol) followed by compound MFH-3-202-1 (250 mg, 1.1 mmol). The mixture was stirred at 0.degree. C. for one hour, then at room temperature for one hour, ethyl acetate was added and the organic mixture washed with hydrochloric acid (2.times.30 mL), dried over magnesium sulfate, filtered through a pad of silica gel, and concentrated in vacuo. The residue was chromatographed on silica gel to give the MFH-3-204-1 (160 mg, 50%). LCMS (m/z): 295 [M+H]+.
(R)-tert-butyl3-(5-((tetrahydro-2H-pyran-4-yl)methylthio)thiazol-2-ylamino- )piperidine-1-carboxylate (MFH-3-205-1)
[0548] The mixture of MFH-3-204-1 (160 mg, 0.54 mmol), (R)-tert-butyl 3-aminopiperidine-1-carboxylate (174 mg, 0.87 mmol) and DIEA (112 mg, 0.87 mmol) in NMP (0.5 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-3-205-1 (120 mg, yield 53%). LCMS (m/z): 414 [M+H].sup.+.
(R)--N-(piperidin-3-yl)-5-((tetrahydro-2H-pyran-4-yl)methylthio)thiazol-2-- amine (MFH-3-206-1)
[0549] To a solution of MFH-3-205-1 (120 mg, 0.3 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 314 [M+H].sup.+.
(R)-(4-nitrophenyl).sub.3-(5-((tetrahydro-2H-pyran-4-yl)methylthio)triazol- -2-ylamino)piperidin-1-yl)methanone (MFH-3-207-1)
[0550] The mixture of MFH-3-206-1 (80 mg, 0.26 mmol), 4-nitrobenzoyl chloride (48 mg, 0.26 mmol) in pyridine (2 mL) was stirred for overnight at room temperature. Then the reaction mixture was concentrated under reduced pressure and the residue was directly used in the next step. LCMS (m/z): 463 [M+H].sup.+.
(R)-(4-aminophenyl)(3-(5-((tetrahydro-2H-pyran-4-yl)methylthio)thiazol-2-y- lamino)piperidin-1-yl)methanone (MFH-4-2-1)
[0551] To a solution of MFH-3-207-1 (120 mg, 0.26 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (472 mg, 2.1 mmol) and conc. HCl (0.2 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-4-2-1 (60 mg, yield 54%). LCMS (m/z): 433 [M+H]+.
(R)--N-(4-(3-(5-((tetrahydro-2H-pyran-4-yl)methylthio)triazol-2-ylamino)pi- peridine-1-carbonyl)phenyl)acrylamide (MFH-4-4-1)
[0552] To a solution of MFH-4-2-1 (30 mg, 0.07 mmol) and DIPEA (0.1 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (8 mg, 0.09 mmol) in DCM (0.1 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-4-4-1 (4.6 mg, yield 13%). LCMS (m/z): 487 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.32 (d, J=28.3 Hz, 1H), 8.07 (s, 1H), 7.77-7.62 (m, 2H), 7.40 (dd, J=32.0, 7.9 Hz, 2H), 7.15-6.93 (m, 1H), 6.45 (dd, J=17.0, 10.1 Hz, 1H), 6.28 (dd, J=17.0, 1.9 Hz, 1H), 5.79 (dd, J=10.1, 1.9 Hz, 1H), 3.83 (d, J=9.4 Hz, 2H), 3.62 (s, 4H), 3.24 (t, J=11.1 Hz, 2H), 3.11 (s, 1H), 2.58 (d, J=6.4 Hz, 1H), 1.98 (d, J=12.6 Hz, 1H), 1.77 (s, 1H), 1.70 (d, J=11.6 Hz, 2H), 1.57 (m, 3H), 1.21 (m, 3H).
##STR00284## ##STR00285##
5-(pyrimidin-5-ylmethylthio)thiazol-2-amine (MFH-3-208-1)
[0553] To a solution of compound MFH-2-76-1 (230 mg, 1.46 mmol) in absolute EtOH (5 ml) was added NaBH.sub.4 (166 mg, 4.38 mmol) at room temperature. The mixture was stirred for 1h, and then acetone (3 ml) was slowly introduced. After 1 h, a solution of 5-(chloromethyl)pyrimidine (241 mg, 1.46 mmol) in EtOH (3 ml) was added. The resulting dark reaction mixture was heated to reflux for 1 h, and was then cooled and concentrated in vacuo. The residue was partitioned between EtOAc and brine. The organic phase was separated, dried (MgSO.sub.4), and concentrated in vacuo to give a crude solid which was triturated with diethyl ether/hexane to provide compound MFH-3-208-1 (180 mg, 55%) LCMS (m/z): 225 [M+H]+.
2-bromo-5-(pyrimidin-5-ylmethylthio)thiazole (MFH-4-1-1)
[0554] To a solution of CuBr.sub.2 (215 mg, 0.96 mmol) in acetonitrile (15 mL) at 0.degree. C. was added t-BuONO (99 mg, 0.96 mmol) followed by compound MFH-3-208-1 (180 mg, 0.8 mmol). The mixture was stirred at 0.degree. C. for one hour, then at room temperature for one hour, ethyl acetate was added and the organic mixture washed with hydrochloric acid (2.times.50 mL), dried over magnesium sulfate, filtered through a pad of silica gel, and concentrated in vacuo. The residue was chromatographed on silica gel to give the MFH-4-1-1 (90 mg, 39%). LCMS (m/z): 289 [M+H]+.
(R)-tert-butyl3-(5-(pyrimidin-5-ylmethylthio)triazol-2-ylamino)piperidine-- 1-carboxylate (MFH-4-3-1)
[0555] The mixture of MFH-4-1-1 (90 mg, 0.31 mmol), (R)-tert-butyl 3-aminopiperidine-1-carboxylate (100 mg, 0.5 mmol) and DIEA (65 mg, 0.5 mmol) in NMP (1 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (20 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-4-3-1 (66 mg, yield 52%). LCMS (m/z): 408 [M+H].sup.+.
(R)--N-(piperidin-3-yl)-5-(pyrimidin-5-ylmethylthio)thiazol-2-amine (MFH-4-5-1)
[0556] To a solution of MFH-4-3-1 (66 mg, 0.16 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 308 [M+H].sup.+.
(R)-(4-nitrophenyl)(3-(5-(pyrimidin-5-ylmethylthio)thiazol-2-ylamino)piper- idin-1-yl)methanone (MFH-4-6-1)
[0557] The mixture of MFH-4-5-1 (50 mg, 0.16 mmol), 4-nitrobenzoyl chloride (30 mg, 0.16 mmol) in pyridine (2 mL) was stirred for overnight at room temperature. Then the reaction mixture was concentrated under reduced pressure and the residue was directly used in the next step. LCMS (m/z): 457 [M+H].sup.+.
(R)-(4-aminophenyl)(3-(5-(pyrimidin-5-ylmethylthio)thiazol-2-ylamino)piper- idin-1-yl)methanone (MFH-4-7-1)
[0558] To a solution of MFH-4-6-1 (74 mg, 0.16 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (292 mg, 1.3 mmol) and cone. HCl (0.1 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-4-7-1 (15 mg, yield 22%). LCMS (m/z): 427 [M+H]+.
(R)--N-(4-(3-(5-(pyrimidin-5-ylmethylthio)thiazol-2-ylamino)piperidine-1-c- arbonyl)phenyl)acrylamide (MFH-4-10-1)
[0559] To a solution of MFH-4-7-1 (15 mg, 0.04 mmol) and DIPEA (0.1 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (4 mg, 0.05 mmol) in DCM (0.1 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1 h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-4-10-1 (2.2 mg, yield 13%). LCMS (m/z): 481 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.26 (s, 1H), 9.07 (s, 1H) 8.81 (s, 2H), 8.11 (s, 1H), 7.79-7.59 (m, 2H), 7.38 (t, J=13.1 Hz, 2H), 6.99-6.73 (m, 1H), 6.50-6.35 (m, 1H), 6.34-6.17 (m, 1H), 5.80 (ddd, J=23.3, 10.0, 2.6 Hz, 1H), 4.01 (s, 2H), 3.81-3.73 (m, 3H), 3.45-3.20 (m, 2H), 1.94 (d, J=18.7 Hz, 1H), 1.78 (m, 1H), 1.54 (s, 2H).
##STR00286##
(R,E)-N-(4-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)p- iperidine-1-carbonyl)phenyl)-4-(dimethylamino)but-2-enamide (MFH-4-13-1)
[0560] To a solution of (E)-4-bromobut-2-enoic acid (17 mg, 0.1 mmol) in SOCl.sub.2 (0.2 mL). The mixture was stirred at 70.degree. C. for 1h under N.sub.2 atmosphere. The mixture was cooled to room temperature and then was concentrated under reduced pressure. The residue was diluted with dichloromethane and the resulted solution was added dropwise to a solution of MFH-2-20-1 (37 mg, 0.08 mmol) and DIPEA (0.2 mL) in CH.sub.3CN (2 mL) at 0.degree. C. After stirring for 1 h at 0.degree. C., a solution of dimethylamine in THF (2 mol/L, 0.1 ml, 0.2 mmol) was added and the reaction mixture was stirred at room temperature for 1h. The removal of the solvent under reduced pressure provided the residue which was purified by HPLC (MeOH/H.sub.2O, 0.05% TFA) to obtain MFH-4-13-1 (18 mg, yield 39%). LCMS (m/z): 583 [M+H]+; .sup.1H NMR (500 MHz, DMSO) .delta. 10.18 (s, 1H), 7.96 (s, 1H), 7.66 (s, 2H), 7.34 (d, J=6.9 Hz, 2H), 6.84 (s, 1H), 6.80-6.65 (m, 2H), 6.27 (d, J=15.4 Hz, 1H), 3.94 (s, 2H), 3.79 (s, 1H), 3.65 (s, 2H), 3.14 (m, 2H), 3.06 (d, J=5.8, 2H), 2.18 (s, 6H), 1.95 (s, 1H), 1.75 (s, 1H), 1.51 (d, J=9.0 Hz, 2H), 1.17 (s, 9H).
##STR00287##
(R,E)-N-(4-(3-(5-((5-tert butyloxazol-2-yl)methylthio)thiazol-2-ylamino)piperidin-1-ylsulfonyl)phen- yl)-4-(dimethylamino)but-2-enamide (MFH-4-40-1)
[0561] To a solution of (E)-4-bromobut-2-enoic acid (12 mg, 0.07 mmol) in SOCl.sub.2 (0.2 mL). The mixture was stirred at 70.degree. C. for 1h under N.sub.2 atmosphere. The mixture was cooled to room temperature and then was concentrated under reduced pressure. The residue was diluted with dichloromethane and the resulted solution was added dropwise to a solution of MFH-4-40-1 (25 mg, 0.05 mmol) and DIPEA (0.2 mL) in CH.sub.3CN (2 mL) at 0.degree. C. After stirring for 1 h at 0.degree. C., a solution of dimethylamine in THF (2 mol/L, 0.05 ml, 0.1 mmol) was added and the reaction mixture was stirred at room temperature for 1h. The removal of the solvent under reduced pressure provided the residue which was purified by HPLC (MeOH/H.sub.2O, 0.05% TFA) to obtain MFH-4-40-1 (20 mg, yield 66%). LCMS (m/z): 619 [M+H].sup.+; .sup.1H NMR (500 MHz, DMSO) .delta. 10.76 (s, 1H), 7.98 (d, J=7.2 Hz, 1H), 7.90 (dd, J=6.5, 4.7 Hz, 2H), 7.70 (dd, J=6.6, 4.8 Hz, 2H), 6.97 (d, J=2.7 Hz, 1H), 6.83-6.75 (m, 1H), 6.72 (d, J=8.8 Hz, 1H), 6.48 (d, J=15.3 Hz, 1H), 3.96 (s, 2H), 3.71 (s, 2H), 3.53 (d, J=9.9 Hz, 2H), 3.24 (d, J=11.2 Hz, 1H), 3.17 (s, 2H), 2.81 (s, 6H), 2.34-2.22 (m, 1H), 1.84-1.70 (m, 2H), 1.52 (dd, J=10.1, 4.0 Hz, 1H), 1.22 (s, 9H).
##STR00288## ##STR00289##
(R)-tert-butyl3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino- )pyrrolidine-1-carboxylate (MFH-4-30-1)
[0562] The mixture of MFH-2-83-1 (150 mg, 0.45 mmol), (R)-tert-butyl 3-aminopyrrolidine-1-carboxylate (134 mg, 0.72 mmol) and DIEA (116 mg, 0.9 mmol) in NMP (1 mL) was stirred at 140.degree. C. for overnight. The residue was extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (30 mL.times.2) and dried over Na.sub.2SO.sub.4. After removal of the solvent, the residue was purified by silica gel (MeOH/DCM=0-20%) to obtain MFH-4-30-1 (110 mg, yield 56%). LCMS (m/z): 439 [M+H].sup.+.
(R)-5-((5-tert-butyloxazol-2-yl)methylthio)-N-(pyrrolidin-3-yl)thiazol-2-a- mine (MFH-4-39-1)
[0563] To a solution of MFH-4-30-1 (110 mg, 0.25 mmol) in methanol (3 mL) was added 4N HCl/dioxane (3 mL). The solution was then stirred for 3h at room temperature and the solvent was removed under reduced pressure to provide a crude which was directly used in the next step. LCMS (m/z): 339 [M+H].sup.+.
(R)-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)pyrrolidi- n-1-yl)(4-nitrophenyl)methanone (MFH-4-41-1)
[0564] The mixture of MFH-4-39-1 (80 mg, 0.24 mmol), 4-nitrobenzoyl chloride (53 mg, 0.28 mmol) in pyridine (2 mL) was stirred for overnight at room temperature. Then the reaction mixture was concentrated under reduced pressure and the residue was directly used in the next step. LCMS (m/z): 488 [M+H].sup.+.
(R)-(4-aminophenyl)(3-(5-((5-tert-butyloxazol-2-yl)methylthio)triazol-2-yl- amino)pyrrolidin-1-yl)methanone (MFH-4-44-1)
[0565] To a solution of MFH-4-41-1 (117 mg, 0.24 mmol) in ethyl acetate and methanol (1:1) were added Tin(II) chloride dehydrate (427 mg, 1.9 mmol) and conc. HCl (0.1 mL). After stirring for 3 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1), concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give MFH-4-44-1 (90 mg, yield 83%). LCMS (m/z): 458 [M+H]+.
(R)--N-(4-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)pyr- rolidine-1-carbonyl)phenyl)acrylamide (MFH-4-70-1)
[0566] To a solution of MFH-4-44-1 (22 mg, 0.05 mmol) and DIPEA (0.2 mL) in CH.sub.3CN (2 mL) was added acryloyl chloride (5 mg, 0.06 mmol) in DCM (0.1 mL) dropwise. The mixture was then stirred at 0.degree. C. for 1 h. The solution was then concentrated under reduced pressure and the residue was purified by prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to provide MFH-4-70-1 (13.4 mg, yield 54%). LCMS (m/z): 512 [M+H]+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.33 (s, 1H), 8.38-8.21 (m, 1H), 7.73 (d, J=8.3 Hz, 2H), 7.52 (t, J=8.6 Hz, 2H), 7.04-6.85 (m, 1H), 6.77-6.65 (m, 1H), 6.45 (dd, J=17.0, 10.1 Hz, 1H), 6.29 (d, J=16.9 Hz, 1H), 5.84-5.75 (m, 1H), 3.96 (d, J=24.0 Hz, 2H), 3.82-3.72 (m, 1H), 3.57 (ddd, J=25.0, 12.1, 7.0 Hz, 3H), 3.33 (d, J=7.5 Hz, 1H), 2.15 (dt, J=30.0, 14.1 Hz, 1H), 1.92 (m, 1H), 1.23-1.12 (m, 9H).
##STR00290##
(R,E)-N-(4-(3-(5-((5-tert-butyloxazol-2-yl)methylthio)thiazol-2-ylamino)p- yrrolidine-1-carbonyl)phenyl)-4-dimethylamino)but-2-enamide (MFH-4-73-1)
[0567] To a solution of (E)-4-bromobut-2-enoic acid (15 mg, 0.09 mmol) in SOCl.sub.2 (0.2 mL). The mixture was stirred at 70.degree. C. for 1h under N.sub.2 atmosphere. The mixture was cooled to room temperature and then was concentrated under reduced pressure. The residue was diluted with dichloromethane and the resulted solution was added dropwise to a solution of MFH-4-40-1 (30 mg, 0.07 mmol) and DIPEA (0.2 mL) in CH.sub.3CN (2 mL) at 0.degree. C. After stirring for 1 h at 0.degree. C., a solution of dimethylamine in THF (2 mol/L, 0.1 ml, 0.2 mmol) was added and the reaction mixture was stirred at room temperature for 1h. The removal of the solvent under reduced pressure provided the residue which was purified by HPLC (MeOH/H.sub.2O, 0.05% TFA) to obtain MFH-4-73-1 (23 mg, yield 61%). LCMS (m/z): 569 [M+H].sup.+; .sup.1H NMR (500 MHz, DMSO) .delta. 10.52 (s, 1H), 8.30-8.19 (m, 1H), 7.73 (d, J=8.3 Hz, 2H), 7.53 (t, J=8.4 Hz, 2H), 7.03-6.85 (m, 1H), 6.78 (dd, J=15.1, 7.4 Hz, 1H), 6.75-6.65 (m, 1H), 6.48 (d, J=15.3 Hz, 1H), 3.97 (s, 2H), 3.93 (s, 2H), 3.81-3.71 (m, 1H), 3.65-3.42 (m, 3H), 3.32 (dd, J=10.0, 2.5 Hz, 1H), 2.81 (d, J=2.5 Hz, 6H), 2.21-2.10 (m, 1H), 1.99-1.85 (m, 1H), 1.23-1.12 (m, 9H).
##STR00291##
(4-aminophenyl)((3R)-3-((5-((5-(tert-butyl)oxazol-2-yl)methyl)sulfinyl)th- iazol-2-yl)amino)piperidin-1-yl)methanone (YLIU-01-006-1)
[0568] To a solution of MFH-2-89-1 (30 mg, 0.064 mmol) in DCM (2 mL) was added m-CPBA (12 mg, 0.07 mmol), the reaction mixture was stirred at rt overnight. Then diluted with DCM, washed with NaHCO.sub.3 aq.(sat.), concentrated and purified with silica gel column (eluted with MeOH in DCM 0% to 15%) to give the title compound (20 mg, 64%) as a brown oil. LCMS (m/z): 488 [M+H]+.
N-(4-((3R)-3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)sulfinyl)thiazol-2-yl- )amino)piperidine-1-carbonyl)phenyl)acrylamide (YLIU-01-007-1)
[0569] To a solution of YLIU-01-006-1 (20 mg, 0.04 mmol) in MeCN (2 mL) was added DIPEA (25 mg, 0.2 mmol). At 0.degree. C., acryloyl chloride (4 mg, 0.044 mmol) was added dropwise. Monitored with LCMS, when the reaction was completed, Na.sub.2CO.sub.3 aq.(sat.) was added, the resulting mixture was extracted with DCM/i-PrOH (4/1) (20 mL). The combined organic layer was concentrated and purified with Prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to give the title compound (8 mg, 36.9%) as white solid. LCMS (m/z): 542 [M+H]+.
[0570] 1H NMR (500 MHz, DMSO) .delta. 10.18 (s, 1H), 8.45 (s, 1H), 7.71-7.48 (m, 2H), 7.39-7.10 (m, 3H), 6.68 (s, 1H), 6.42-6.24 (m, 1H), 6.23-6.06 (m, 1H), 5.77-5.57 (m, 1H), 4.62-4.44 (m, 2H), 4.42-4.20 (m, 2H), 3.56 (m, 3H), 2.02-1.68 (m, 2H), 1.57-1.32 (m, 2H), 0.99 (s, 9H).
##STR00292## ##STR00293##
5-Thiocyanatothiazol-2-amine (YLIU-01-004-1)
[0571] A mixture of 2-amino-5-bromothiazole hydrobromide (7.8 g, 30 mmol) and potassium thiocyanate (20.46 g, 210.93 mmol) in methanol (230 mL) was stirred at room temperature for 48 h. Methanol was evaporated and water (30 ml) was added. The pH of the aqueous solution was adjusted to pH=12 with 10% NaOH aq and precipitate formed. The solid was collected by filtration to yield compound YLIU-01-004-1 (3.3 g, 70%) as a brownish solid. LCMS (m/z): 158 [M+H]+.
5-((5-tert-Butyloxazol-2-yl)methylthio)thiazol-2-amine (YLIU-01-005-1)
[0572] To a solution of compound YLIU-01-004-1 (156 mg, 1 mmol) in absolute EtOH (3 ml) was added NaBH.sub.4 (76 mg, 2 mmol) portionwise at 0.degree. C. The mixture was stirred at rt for 1 h, and then acetone (2 ml) was slowly introduced. After 1 h, a solution of 2-(bromomethyl)-5-(tert-butyl)oxazole (240 mg, 1.1 mmol) in EtOH (2 ml) was added. The resulting dark reaction mixture was heated to 65.degree. C. for 1 h, and was then cooled and concentrated in vacuo. The residue was partitioned between EtOAc and brine. The organic phase was separated, dried (MgSO4), concentrated and purified with silica gel column (eluted with MeOH in DCM 0% to 10%) to provide the title compound (120 mg, 44%) as a brown solid. LCMS (m/z): 270 [M+H]+.
2-(((2-bromothiazol-5-yl)thio)methyl)-5-(tert-butyl)oxazole (YLIU-01-017-1)
[0573] To a solution of CuBr.sub.2 (2.32 g, 10 mmol) in acetonitrile (50 mL) at 0.degree. C. was added t-BuONO (1.1 g, 10 mmol) followed by compound YLIU-01-004-1 (1.66 g, 6.13 mmol). The mixture was stirred at 0.degree. C. for 1 h, then at room temperature for 1 h. Ethyl acetate was added and the organic layer was washed with hydrochloric acid (2.times.50 mL), dried over magnesium sulfate, filtered through a pad of silica gel, and concentrated in vacuo. The residue was purified on silica gel column to give the YLIU-01-017-1 (690 mg, 33%) as orange oil. LCMS (m/z): 333 [M+H]+.
tert-butyl (R)-3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl- )amino) piperidine-1-carboxylate (YLIU-01-038-1)
[0574] A mixture of YLIU-01-017-1 (150 mg, 0.45 mmol), tert-butyl (R)-3-aminopiperidine-1-carboxylate (180 mg, 0.9 mmol), DIPEA (0.24 mL, 1.35 mmol) in NMP (3 mL) was heated to 140.degree. C. overnight. The solution was diluted with water (20 mL) and extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated. The residue was purified by silica gel column (MeOH/DCM, 0-20%) to give YLIU-01-038-1 (150 mg, 73%). LCMS (m/z): 453 [M+H]+.
(R)-5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-N-(piperidin-3-yl)thiazol-- 22-amine (YLIU-01-047-1)
[0575] To a mixture of compound YLIU-01-038-1 (150 mg, 0.33 mmol) in methanol (2 mL) was added 4N HCl/dioxane (2 mL) and the resulted solution was stirred at rt for 3 h. The mixture was concentrated under reduced pressure to give the title compound as HCl salt, which was directly used in the next step. LCMS (m/z): 353 [M+H].sup.+.
(R)-(3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)amino)pip- eridin-1-yl)(5-nitropyridin-2-yl)methanone (YLIU-01-048-1)
[0576] To a mixture of YLIU-01-047-1 (50 mg, 0.12 mmol), 5-nitropicolinic acid (23 mg, 0.14 mmol) and DIPEA (78 mg, 0.6 mmol) in DCM (2 mL) was added T.sub.3P (50% solution in EA, 230 mg, 0.36 mmol) dropwise at rt. The reaction mixture was stirred at rt overnight. The solution was diluted with DCM (20 mL) and washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated. The residue was purified by silica gel column (MeOH/DCM, 0-20%) to give the title compound (46 mg, 76%) as yellow solid. LCMS (m/z): 503 [M+H].sup.+.
(R)-(5-aminopyridin-2-yl)(3-((5-((5-(tertbutyl)oxazol-2-yl)methyl)thio)thi- azol-2-yl)amino)piperidin-1-yl)methanone (YLIU-01-064-1)
[0577] To a solution of YLIU-01-048-1 (46 mg, 0.091 mmol) in ethyl acetate and methanol (2 mL, 1:1) was added Tin(II) chloride (138 mg, 0.73 mmol) at rt. After stirring for 2 h at 80.degree. C., the reaction mixture was diluted with chloroform and iso-propanol (4:1), neutralized with saturated NaHCO.sub.3 aq. and filtered. The filtrate was extracted with chloroform and iso-propanol (4:1). The combined organic layer was concentrated under reduced pressure and the resulting residue was purified by silica gel column chromatography (MeOH/DCM=0-20%) to give YLIU-01-064-1 (16 mg, 37%) as yellow solid. LCMS (m/z): 473 [M+H]+.
(R)--N-(6-(3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)ami- no)piperidine-1-carbonyl)pyridin-3-yl)acrylamide (YLIU-01-067-1)
[0578] To a solution of YLIU-01-064-1 (16 mg, 0.034 mmol) and DIPEA (13 mg, 0.1 mmol) in MeCN (2 mL) was added acryloyl chloride (4 mg, 0.044 mmol) dropwise at 0.degree. C. Monitored with LCMS, when the reaction was completed, Na.sub.2CO.sub.3 aq.(sat.) was added, the resulting mixture was extracted with DCM/i-PrOH (4/1) (20 mL.times.2). The combined organic layer was concentrated and purified with Prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to give the title compound (1.3 mg, 7.27%) as white solid. LCMS (m/z): 527 [M+H]+.
##STR00294## ##STR00295##
tert-butyl 4-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)amino)piperid- ine-1-carboxylate (YLIU-01-062-1)
[0579] A mixture of YLIU-01-017-1 (150 mg, 0.45 mmol), tert-butyl 4-aminopiperidine-1-carboxylate (180 mg, 0.9 mmol), DIPEA (0.24 mL, 1.35 mmol) in NMP (3 mL) was heated to 140.degree. C. overnight. The solution was diluted with water (20 mL) and extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated. The residue was purified by silica gel column (MeOH/DCM, 0-20%) to give YLIU-01-062-1 (150 mg, 73%). LCMS (m/z): 453 [M+H]+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-N-(piperidin-4-yl)thiazol-2-am- ine (YLIU-01-062A-1)
[0580] To a mixture of compound YLIU-01-062-1 (150 mg, 0.33 mmol) in methanol (2 mL) was added 4N HCl/dioxane (2 mL) and the resulted solution was stirred at rt for 3 h. The mixture was concentrated under reduced pressure to give the title compound as HCl salt, which was directly used in the next step. LCMS (m/z): 353 [M+H].sup.+.
(4-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)amino)piperid- in-1-yl)(3-nitrophenyl)methanone (YLIU-01-068-1)
[0581] The mixture of YLIU-01-062A-1 (330 mg, 0.89 mmol), 3-nitrobenzoyl chloride (330 mg, 1.78 mmol) in pyridine (2 mL) was stirred at rt overnight. Then the reaction mixture was concentrated under reduced pressure and the residue was purified by silica gel column (MeOH/DCM, 0-20%) to give YLIU-01-068-1 (320 mg, 72%) as yellow solid. LCMS (m/z): 502 [M+H]+.
(3-aminophenyl)(4-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-y- l)amino)piperidin-1-yl)methanone (YLIU-01-076-1)
[0582] To a solution of YLIU-01-068-1 (100 mg, 0.2 mmol) in MeOH (3 mL) was added Pd/C (10%, 20 mg) and N.sub.2H4-H.sub.2O (0.1 mL). The reaction mixture was stirred at 80.degree. C. overnight, then filtered through Celite. The filtrate was concentrated and purified by Prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to give the title compound (27 mg, 29%) as yellow oil. LCMS (m/z): 472 [M+H]+.
N-(3-(4-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)amino)pi- peridine-1-carbonyl)phenyl)acrylamide (YLIU-01-078-1)
[0583] To a solution of YLIU-01-076-1 (20 mg, 0.042 mmol) and DIPEA (17 mg, 0.13 mmol) in MeCN (2 mL) was added acryloyl chloride (4.2 mg, 0.046 mmol) dropwise at 0.degree. C. Monitored with LCMS, when the reaction was completed, Na.sub.2CO.sub.3 aq.(sat.) was added, the resulting mixture was extracted with DCM/i-PrOH (4/1) (20 mL.times.2). The combined organic layer was concentrated and purified with Prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to give the title compound (1.4 mg, 6.3%) as white solid. LCMS (m/z): 526 [M+H]+.
##STR00296## ##STR00297##
4-methyl-5-thiocyanatothiazol-2-amine (YLIU-01-056-1)
[0584] A mixture of 2-amino-5-bromothiazole hydrobromide (580 mg, 3 mmol) and potassium thiocyanate (2.9 g, 30 mmol) in methanol (20 mL) was stirred at room temperature for 48 h. Methanol was evaporated and water (3 ml) was added. The pH of the aqueous solution was adjusted to pH=12 with 10% NaOH aq and precipitate formed. The solid was collected by filtration to yield compound YLIU-01-056-1 (480 mg, 93%) as a brownish solid. LCMS (m/z): 172 [M+H]+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-4-methylthiazol-2-amine (YLIU-01-073-1)
[0585] To a solution of compound YLIU-01-056-1 (171 mg, 1 mmol) in absolute EtOH (3 ml) was added NaBH.sub.4 (76 mg, 2 mmol) portionwise at 0.degree. C. The mixture was stirred at rt for 1 h, and then acetone (2 ml) was slowly introduced. After 1 h, a solution of 2-(bromomethyl)-5-(tert-butyl)oxazole (240 mg, 1.1 mmol) in EtOH (2 ml) was added. The resulting dark reaction mixture was heated to 65.degree. C. for 1 h, and was then cooled and concentrated in vacuo. The residue was partitioned between EtOAc and brine. The organic phase was separated, dried (MgSO4), concentrated and purified with silica gel column (eluted with MeOH in DCM 0% to 10%) to provide the title compound (314 mg, >100%) as a brown oil. LCMS (m/z): 284 [M+H]+.
2-(((2-bromo-4-methylthiazol-5-yl)thio)methyl)-5-(tert-butyl)-4-methyloxaz- ole (YLIU-01-089-1)
[0586] To a solution of CuBr.sub.2 (423 mg, 1.9 mmol) in acetonitrile (10 mL) at 0.degree. C. was added t-BuONO (200 mg, 1.9 mmol) followed by compound YLIU-01-073-1 (314 mg, 1.1 mmol). The mixture was stirred at 0.degree. C. for 1 h, then at room temperature for 1 h. Ethyl acetate was added and the organic layer was washed with hydrochloric acid (2.times.50 mL), dried over magnesium sulfate, filtered through a pad of silica gel, and concentrated in vacuo. The residue was purified on silica gel column to give the YLIU-01-089-1 (140 mg, 37%) as orange oil. LCMS (m/z): 347 [M+H]+.
tert-butyl (R)-3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-4-methylthi- azol-2-yl) amino)piperidine-1-carboxylate (YLIU-01-092-1)
[0587] A mixture of YLIU-01-089-1 (140 mg, 0.4 mmol), tert-butyl (R)-3-aminopiperidine-1-carboxylate (160 mg, 0.8 mmol), DIPEA (0.22 mL, 1.2 mmol) in NMP (3 mL) was heated to 140.degree. C. overnight. The solution was diluted with water (20 mL) and extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated. The residue was purified by silica gel column (MeOH/DCM, 0-20%) to give YLIU-01-092-1 (220 mg, >100%). LCMS (m/z): 467 [M+H]+.
(R)-5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-4-methyl-N-(piperidin-3-yl- )thiazol-2-amine (YLIU-01-096-1)
[0588] To a mixture of compound YLIU-01-092-1 (220 mg, 0.47 mmol) in methanol (2 mL) was added 4N HCl/dioxane (2 mL) and the resulted solution was stirred at rt for 3 h. The mixture was concentrated and the residue was purified by Prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to give the title compound (88 mg, 51%) as yellow solid. LCMS (m/z): 367 [M+H]+.
(R)--N-(4-(3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-4-methylthiazol- -2-yl)amino)piperidine-1-carbonyl)phenyl)acrylamide (YLIU-01-099-1)
[0589] To a mixture of YLIU-01-096-1 (22 mg, 0.06 mmol), 4-acrylamidobenzoic acid (14 mg, 0.072 mmol) and DIPEA (39 mg, 0.3 mmol) in DCM (2 mL) was added T.sub.3P (50% solution in EA, 57 mg, 0.18 mmol) dropwise at rt. The reaction mixture was stirred at rt overnight. The solution was diluted with DCM (20 mL) and washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated. The residue was purified by Prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to give the title compound (17.1 mg, 53%) as yellow solid. LCMS (m/z): 540 [M+H].sup.+.
[0590] 1H NMR (500 MHz, DMSO) .delta. 10.28 (s, 1H), 8.44-7.89 (m, 1H), 7.81-7.57 (m, 2H), 7.36 (m, 2H), 6.81-6.60 (m, 1H), 6.51-6.36 (m, 1H), 6.32-6.20 (m, 1H), 5.88-5.67 (m, 1H), 3.87 (m, 3H), 3.63 (s, 2H), 3.12 (m, 2H), 1.90 (m, 2H), 1.63-1.39 (m, 5H), 1.19 (s, 9H).
##STR00298##
tert-butyl 5-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)amino)-3,3-di- fluoropiperidine-1-carboxylate (YLIU-01-101-1)
[0591] A mixture of YLIU-01-017-1 (130 mg, 0.4 mmol), tert-butyl 5-amino-3,3-difluoropiperidine-1-carboxylate (350 mg, 1.5 mmol), DIPEA (0.36 mL, 1.8 mmol) in NMP (3 mL) was heated to 140.degree. C. overnight. The solution was diluted with water (20 mL) and extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated. The residue was purified by silica gel column (MeOH/DCM, 0-20%) to give YLIU-01-101-1 (12 mg, 6%). LCMS (m/z): 489 [M+H]+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-N-(5,5-difluoropiperidin-3-yl)- triazol-2-amine (YLIU-01-111-1)
[0592] To a mixture of compound YLIU-01-101-1 (12 mg, 0.024 mmol) in methanol (2 mL) was added 4N HCl/dioxane (2 mL) and the resulted solution was stirred at rt for 3 h. The mixture was concentrated and the crude product was used into next step directly as HCl salt. LCMS (m/z): 389 [M+H]+.
N-(4-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)amino)-3,3-- difluoropiperidine-1-carbonyl)phenyl)acrylamide (YLIU-01-114-1)
[0593] To a mixture of YLIU-01-111-1 (10 mg, crude HCl salt), 4-acrylamidobenzoic acid (6 mg, 0.03 mmol) and DIPEA (16 mg, 0.12 mmol) in DCM (2 mL) was added T.sub.3P (50% solution in EA, 46 mg, 0.07 mmol) dropwise at rt. The reaction mixture was stirred at rt overnight. The solution was diluted with DCM (20 mL) and washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated. The residue was purified by Prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to give the title compound (2.5 mg, 19%) as yellow solid. LCMS (m/z): 562 [M+H]+.
[0594] 1H NMR (500 MHz, DMSO) .delta. 10.35 (s, 1H), 8.10 (s, 1H), 7.74 (d, J=8.3 Hz, 2H), 7.42 (d, J=8.6 Hz, 2H), 7.08-6.83 (m, 1H), 6.72 (m, 1H), 6.45 (m, 1H), 6.29 (m, 1H), 5.80 (m, 1H), 3.97 (s, 2H), 3.65-3.44 (m, 3H), 3.22-2.90 (m, 2H), 2.08 (m, 2H), 1.21 (s, 9H).
##STR00299##
Methyl 4-(N-acryloylacrylamido)-3-(trifluoromethyl)benzoate (YLIU-01-100-1)
[0595] To a solution of methyl 4-amino-3-(trifluoromethyl)benzoate (220 mg, 1 mmol) and DIPEA (390 mg, 3 mmol) in MeCN (5 mL) was added acryloyl chloride (108 mg, 1.2 mmol) dropwise at 0.degree. C. The reaction was stirred at rt for 1 h, then Na.sub.2CO.sub.3 aq.(sat.) was added, the resulting mixture was extracted with DCM (20 mL.times.2). The combined organic layer was concentrated and purified with SGC (Hexane/EA-4/1) to give YLIU-01-100-1 (114 mg, 35%) as white solid. LCMS (m/z): 328 [M+H]+.
4-acrylamido-3-(trifluoromethyl)benzoic acid (YLIU-01-115-1)
[0596] To a solution of YLIU-01-100-1 (114 mg, 0.35 mmol) in THF (4 mL) was added 1N LiOH aq. (4 mL). The reaction mixture was stirred at rt for 1h, then adjusted PH<7 with 4N HCl aq. The resulting mixture was extracted with EA (20 mL.times.3), washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated to give the desire product (110 mg, >100%) as a white solid. LCMS (m/z): 260 [M+H]+.
(R)--N-(4-(3-((4-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)ami- no)piperidine-1-carbonyl)-2-(trifluoromethyl)phenyl)acrylamide (YLIU-01-121-1)
[0597] To a mixture of YLIU-01-047-1 (20 mg, 0.057 mmol), YLIU-01-115-1 (18 mg, 0.068 mmol) and DIPEA (37 mg, 0.285 mmol) in DCM (2 mL) was added T.sub.3P (50% solution in EA, 108 mg, 0.17 mmol) dropwise at rt. The reaction mixture was stirred at rt overnight. The solution was diluted with DCM (20 mL) and washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated. The residue was purified by Prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to give the title compound (17.8 mg, 53%) as yellow solid. LCMS (m/z): 594 [M+H].sup.+. .sup.1H NMR (500 MHz, DMSO) .delta. 9.87 (m, 1H), 8.15 (m, 1H), 7.72 (m, 3H), 7.08-6.66 (m, 2H), 6.64-6.50 (m, 1H), 6.29 (m, 1H), 5.93-5.70 (m, 1H), 3.94 (m, 2H), 3.72 (m, 2H), 3.40 (m, 1H), 3.15 (m, 2H), 2.07-1.66 (m, 2H), 1.64-1.35 (m, 2H), 1.14 (s, 9H).
##STR00300##
methyl 4-acrylamido-3-fluorobenzoate (YLIU-01-116-1)
[0598] To a solution of methyl 4-amino-3-fluorobenzoate (500 mg, 3 mmol) and DIPEA (1.65 mL, 9 mmol) in MeCN (5 mL) was added acryloyl chloride (320 mg, 3.6 mmol) dropwise at 0.degree. C. The reaction was stirred at rt for 1 h, then Na.sub.2CO.sub.3 aq.(sat.) was added, the resulting mixture was extracted with DCM (20 mL.times.2). The combined organic layer was concentrated and recrystallized from EA to give YLIU-01-116-1 (190 mg, 28%) as white solid. LCMS (m/z): 224 [M+H]+.
4-acrylamido-3-fluorobenzoic acid (YLIU-01-119-1)
[0599] To a solution of YLIU-01-116-1 (190 mg, 0.85 mmol) in THF/water (4 mL/4 mL) was added LiOH (204 mg, 8.5 mmol). The reaction mixture was stirred at rt for 1h, then adjusted PH<7 with 4N HCl aq. The resulting mixture was extracted with EA (20 mL.times.3), washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated to give the desire product (110 mg, 61%) as a white solid. LCMS (m/z): 210 [M+H]+.
(R)--N-(4-(3-((4-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)ami- no)piperidine-1-carbonyl)-2-fluorophenyl)acrylamide (YLIU-01-123-1)
[0600] To a mixture of YLIU-01-047-1 (20 mg, 0.057 mmol), YLIU-01-115-1 (15 mg, 0.068 mmol) and DIPEA (37 mg, 0.285 mmol) in DCM (2 mL) was added T.sub.3P (50% solution in EA, 108 mg, 0.17 mmol) dropwise at rt. The reaction mixture was stirred at rt overnight. The solution was diluted with DCM (20 mL) and washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated. The residue was purified by Prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to give the title compound (23.8 mg, 77%) as yellow solid. LCMS (m/z): 544 [M+H].sup.+. .sup.1H NMR (500 MHz, DMSO) .delta. 10.06 (s, 1H), 8.05 (m, 2H), 7.44-6.97 (m, 2H), 6.74-6.53 (m, 3H), 6.36-6.21 (m, 1H), 5.80 (m, 1H), 3.69 (s, 2H), 3.53 (m, 3H), 3.31-3.01 (m, 2H), 1.86 (m, 2H), 1.51 (m, 2H), 1.16 (s, 9H).
##STR00301##
benzyl (2R,3R)-3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)triazol-2-y- l)amino)-2-methylpiperidine-1-carboxylate (YLIU-01-142-1)
[0601] A mixture of YLIU-01-017-1 (80 mg, 0.24 mmol), benzyl (2R,3R)-3-amino-2-methyl piperidine-1-carboxylate (300 mg, 1.2 mmol), DIPEA (0.42 mL, 2.4 mmol) in NMP (3 mL) was heated to 140.degree. C. overnight. The solution was diluted with water (20 mL) and extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated. The residue was purified by silica gel column (MeOH/DCM, 0-20%) to give YLIU-01-142-1 (70 mg, 58%) as yellow solid. LCMS (m/z): 501 [M+H]+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-N-((2R,3R)-2-methylpiperidin-3- -yl)triazol-2-amine (YLIU-01-150-1)
[0602] To a mixture of compound YLIU-01-142-1 (70 mg, 0.14 mmol) in MeCN (4 mL) was added TMSI (280 mg, 1.4 mmol) and the resulted solution was stirred at 0.degree. C. for 30 min. The reaction was quenched with water and the resulting solution was purified by Prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to give the title compound (34 mg, 66%) as yellow solid. LCMS (m/z): 367 [M+H]+.
N-(4-((2R,3R)-3-((5-((5-tert-butyl)oxazol-2-yl)methyl)thio)triazol-2-yl)am- ino)-2-methylpiperidine-1-carbonyl)phenyl)acrylamide (YLIU-01-155-1)
[0603] To a mixture of YLIU-01-150-1 (17 mg, 0.046 mmol), 4-acrylamidobenzoic acid (10 mg, 0.055 mmol) and DIPEA (0.04 mL, 0.23 mmol) in DMF (2 mL) was added HATU (35 mg, 0.093 mmol) at rt. The reaction mixture was stirred at rt overnight. The solution was diluted with DCM (20 mL) and washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated. The residue was purified by Prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to give the title compound (8.2 mg, 33%) as yellow solid. LCMS (m/z): 540 [M+H].sup.+.
[0604] 1H NMR (500 MHz, DMSO) .delta. 10.30 (s, 1H), 8.02 (s, 1H), 7.72 (d, J=8.6 Hz, 2H), 7.38 (d, J=8.3 Hz, 2H), 6.79 (m, 2H), 6.45 (dd, J=17.0, 10.2 Hz, 1H), 6.28 (dd, J=17.0, 1.9 Hz, 1H), 5.79 (dd, J=10.1, 1.9 Hz, 1H), 3.79 (m, 4H), 3.08 (m, 2H), 1.84-1.38 (m, 4H), 1.19 (s, 9H), 1.04 (s, 3H).
##STR00302##
benzyl (2S,5R)-5-((5-((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl- )amino)-2-methylpiperidine-1-carboxylate (YLIU-01-143-1)
[0605] A mixture of YLIU-01-017-1 (80 mg, 0.24 mmol), benzyl (2S,5R)-5-amino-2-methyl piperidine-1-carboxylate (300 mg, 1.2 mmol), DIPEA (0.42 mL, 2.4 mmol) in NMP (3 mL) was heated to 140.degree. C. overnight. The solution was diluted with water (20 mL) and extracted with chloroform and iso-propanol (4:1). The organic phase was washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated. The residue was purified by silica gel column (MeOH/DCM, 0-20%) to give YLIU-01-143-1 (50 mg, 42%) as yellow solid. LCMS (m/z): 501 [M+H]+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-N-((3R,6S)--6-methylpiperidin-- 3-yl)triazol-2-amine (YLIU-01-151-1)
[0606] To a mixture of compound YLIU-01-143-1 (50 mg, 0.1 mmol) in MeCN (4 mL) was added TMSI (200 mg, 1 mmol) and the resulted solution was stirred at 0.degree. C. for 30 min. The reaction was quenched with water and the resulting solution was purified by Prep-HPLC (MeOH/H.sub.2O, 0.05% TFA) to give the title compound (20 mg, 54%) as yellow solid. LCMS (m/z): 367 [M+H]+.
N-(4-((2,5R)-5-((((5-(tertbutyl)oxazol-2-yl)methyl)thio)triazol-2-yl)amino- )-2-methylpiperidine-1-carbonyl)phenyl)acrylamide (YLIU-01-156-1)
[0607] To a mixture of YLIU-01-151-1 (20 mg, 0.054 mmol), 4-acrylamidobenzoic acid (12.5 mg, 0.065 mmol) and DIPEA (0.05 mL, 0.27 mmol) in DMF (2 mL) was added HATU (41 mg, 0.108 mmol) at rt. The reaction mixture was stirred at rt overnight. The solution was diluted with DCM (20 mL) and washed with brine (50 mL.times.2) and dried over Na.sub.2SO.sub.4, filtered and concentrated. The residue was purified by Prep-HPLC (MeOH/H2O, 0.05% TFA) to give the title compound (11 mg, 38%) as yellow solid. LCMS (m/z): 540 [M+H]+. 1H NMR (500 MHz, DMSO) .delta. 10.30 (s, 1H), 8.03 (s, 1H), 7.73 (d, J=8.6 Hz, 2H), 7.39 (d, J=8.6 Hz, 2H), 6.97 (m, 1H), 6.71 (s, 1H), 6.45 (dd, J=17.0, 10.1 Hz, 1H), 6.28 (dd, J=17.0, 1.9 Hz, 1H), 5.79 (dd, J=10.1, 1.9 Hz, 1H), 3.95 (m, 2H), 3.57 (m, 2H), 3.34-3.02 (m, 1H), 2.68 (m, 1H), 1.71 (m, 4H), 1.35-0.88 (m, 12H).
Synthesis of B1
##STR00303## ##STR00304##
[0608] 5-thiocyanatothiazol-2-amine (1)
[0609] To a solution of 5-bromothiazol-2-amine hydrobromide (5.2 g, 20 mmol) in methanol (100 mL) was added KSCN (19.5 g, 200 mmol) at room temperature. The resulting mixture was stirred for 20 hours and then concentrated under vacuum. The residue was diluted with H.sub.2O (150 mL). The pH of the solution was adjusted to 10 with 10% Na.sub.2CO.sub.3. The precipitate was filtered and washed with water to obtain (1.8 g, 58%) of the title compound (1) as a brown solid. MS m/z 158.95 [M+H].sup.+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2)
[0610] To a solution of 5-thiocyanatothiazol-2-amine (1.8 g, 11.46 mmol) in absolute EtOH (50 mL) was added NaBH.sub.4 (0.87 g, 2.3 mmol) portionwise at room temperature. The resulting mixture was stirred for 1 hour, and then acetone (20 ml) was slowly introduced. After 1 hours, a solution of 5-(tert-butyl)-2-(chloromethyl)oxazole (2.19 g, 12.6 mmol) in EtOH (10 ml) was added, and the resulting mixture was cooled, concentrated in vacuo, and then partitioned between EtOAc and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (2.1 g, 68%) of the title compound (2) as a pale red-brown solid. MS m/z 270.06 [M+H].sup.+.
tert-butyl 4-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)car- bamoyl)piperidine-1-carboxylate (3)
[0611] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (186 mg, 0.69 mmol), 1-(tert-butoxycarbonyl)piperidine-4-carboxylic acid (229 mg, 0.94 mmol), DMAP (42.8 mg, 0.35 mmol), and triethylamine (0.2 ml, 1.4 mmol) in DMF (1 mL) and DCM (2 ml) was added EDAC (267 mg, 1.4 mmol) at room temperature. The reaction mixture was stirred for 1.5 hours, diluted with EtOAc, washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (226 mg, 83%) of the title compound (3) as a white solid. MS m/z 481.18[M+H]+.
N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)piperidine-4-ca- rboxamide hydrochloride (4)
[0612] To a solution of tert-butyl 4-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)carbamoyl)pip- eridine-1-carboxylate (117 mg, 0.24 mmol) in 4 M HCl EtOAc (5 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was concentrated under vacuum to afford (100 mg, 100%) of the title compound (4) as a white solid, which was used without further purification. MS m/z 381.14 [M+H].sup.+.
(E)-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(4-(dime- thylamino)but-2-enoyl)piperidine-4-carboxamide (B1)
[0613] To a solution of N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)piperidine-4-c- arboxamide hydrochloride (100 mg, 0.24 mmol) in DMF (1 mL) was added HATU (110 mg, 0.29 mmol), (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (45 mg, 0.27 mmol) and DIPEA (125 mg, 0.96 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (100 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (6 mg, 5.1%) of the title compound as a yellow solid. MS m/z 492.21 [M+H].sup.+. .sup.1H NMR (400 MHz, DMSO-d.sub.6) 12.42 (s, 1H), 10.66 (s, 2H), 7.77 (s, 2H), 7.40 (s, 3H), 6.83 (s, 1H), 6.72 (s, 1H), 6.51 (d, J=15.1 Hz, 1H), 4.52-4.17 (m, 1H), 4.07 (s, 2H), 3.63 (s, 1H), 3.08 (s, 2H), 2.77 (s, 6H), 2.02 (s, 2H), 1.72 (s, 3H), 1.46 (s, 2H), 1.19 (s, 9H).
Synthesis of B2
##STR00305## ##STR00306##
[0614] 5-thiocyanatothiazol-2-amine (1)
[0615] To a solution of 5-bromothiazol-2-amine hydrobromide (5.2 g, 20 mmol) in Methanol (100 mL) was added KSCN (19.5 g, 200 mmol) at room temperature. The resulting mixture was stirred for 20 hours and then concentrated under vacuum. The residue was diluted with H.sub.2O (150 mL). The pH of the solution was adjusted to 10 with 10% Na.sub.2CO.sub.3. The precipitate was filtered and washed with water to obtain (1.8 g, 58%) of the title compound (1) as a brown solid. MS m/z 158.95 [M+H].sup.+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2)
[0616] To a solution of 5-thiocyanatothiazol-2-amine (1.8 g, 11.46 mmol) in absolute EtOH (50 mL) was added NaBH.sub.4 (0.87 g, 2.3 mmol) portionwise at room temperature. The resulting mixture was stirred for 1 hour, and then acetone (20 ml) was slowly introduced. After 1 hour, a solution of 5-(tert-butyl)-2-(chloromethyl)oxazole (2.19 g, 12.6 mmol) in EtOH (10 ml) was added, and the resulting mixture was cooled, concentrated in vacuo, and then partitioned between EtOAc and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (2.1 g, 68%) of the title compound (2) as a pale red-brown solid. MS m/z 270.06 [M+H].sup.+.
tert-butyl 4-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)car- bamoyl)piperidine-1-carboxylate (3)
[0617] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (186 mg, 0.69 mmol), 1-(tert-butoxycarbonyl)piperidine-4-carboxylic acid (229 mg, 0.94 mmol), DMAP (42.8 mg, 0.35 mmol), and triethylamine (0.2 ml, 1.4 mmol) in DMF (1 mL) and DCM (2 ml) was added EDAC (267 mg, 1.4 mmol) at room temperature. The reaction mixture was stirred for 1.5 hours, diluted with EtOAc, washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (226 mg, 83%) of the title compound (3) as a white solid. MS m/z 481.18[M+H].sup.+.
N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)piperidine-4-ca- rboxamide hydrochloride (4)
[0618] To a solution of tert-butyl 4-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)carbamoyl)pip- eridine-1-carboxylate (20 mg, 0.047 mmol) in 4 M HCl EtOAc (3 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was concentrated under vacuum to afford (100 mg, 100%) of the title compound (4) as a white solid, which was used without further purification. MS m/z 381.14 [M+H]+.
1-acryloyl-N-(5-(((5-(tert-butyl)oxazol-2-yl)methylthio)thiazol-2-yl)piper- idine-4-carboxamide (B2)
[0619] To a solution of N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)piperidine-4-c- arboxamide hydrochloride (20 mg, 0.05 mmol) in THF (5 mL) was added DIPEA (17 mg, 0.15 mmol) and acryloyl chloride (43 mg, 0.05 mmol) at 0.degree. C. for 1 hour. The resulting mixture was stirred at room temperature for 2 hours. Then it was concentrated and purified by an silica gel column to afford (11 mg, 50.7%) of the title compound as a slightly white solid. MS m/z 435.15 [M+H].sup.+. .sup.1H NMR (400 MHz, CDCl.sub.3) .delta. 11.64 (s, 1H), 7.29 (s, 1H), 6.58 (q, J=11.0 Hz, 2H), 6.28 (d, J=16.7 Hz, 1H), 5.71 (d, J=10.6 Hz, 1H), 4.58 (d, J=12.1 Hz, 1H), 4.09 (dd, J=17.6, 10.2 Hz, 1H), 3.96 (s, 2H), 3.21 (s, 1H), 2.90 (s, 1H), 2.68 (t, J=10.4 Hz, 1H), 1.95 (d, J=12.0 Hz, 2H), 1.82 (s, 2H), 1.24 (s, 9H).
Synthesis of B3
##STR00307## ##STR00308##
[0620] 5-thiocyanatothiazol-2-amine (1)
[0621] To a solution of 5-bromothiazol-2-amine hydrobromide (5.2 g, 20 mmol) in methanol (100 mL) was added KSCN (19.5 g, 200 mmol) at room temperature. The resulting mixture was stirred for 20 hours and then concentrated under vacuum. The residue was diluted with H.sub.2O (150 mL). The pH of the solution was adjusted to 10 with 10% Na.sub.2CO.sub.3. The precipitate was filtered and washed with water to obtain (1.8 g, 58%) of the title compound (1) as a brown solid. MS m/z 158.95 [M+H].sup.+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2)
[0622] To a solution of 5-thiocyanatothiazol-2-amine (1.8 g, 11.46 mmol) in absolute EtOH (50 mL) was added NaBH.sub.4 (0.87 g, 2.3 mmol) portionwise at room temperature. The resulting mixture was stirred for 1 hour, and then acetone (20 m) was slowly introduced. After 1 hour, a solution of 5-(tert-butyl)-2-(chloromethyl)oxazole (2.19 g, 12.6 mmol) in EtOH (10 ml) was added, and the resulting mixture was cooled, concentrated in vacuo, and then partitioned between EtOAc and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (2.1 g, 68%) of the title compound (2) as a pale red-brown solid. MS m/z 270.06 [M+H].sup.+.
tert-butyl (3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)triazol-2-yl)ca- rbamoyl)cyclohexyl)carbamate (3)
[0623] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (160 mg, 0.6 mmol) in DMF (2 mL) was added HATU (456 mg, 1.2 mmol), 3-((tert-butoxycarbonyl)amino)cyclohexane-1-carboxylic acid (292 mg, 1.2 mmol), and DIPEA (143 mg, 2.4 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (150 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (210 mg, 70%) of the title compound (3) as a white solid. MS m/z 495.20[M+H].sup.+.
3-amino-N-(5-(((5-tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)cyclohex- ane-1-carboxamide hydrochloride (4)
[0624] To a solution of tert-butyl (3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)carbamoyl)cy- clohexyl)carbamate (210 mg, 0.424 mmol) in 4 M HCl EtOAc (5 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was concentrated under vacuum to afford (182 mg, 99%) of the title compound (4) as a white solid, which was used without further purification. MS m/z 395.15 [M+H].sup.+.
(E)-N-(5-((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-3-(4-(dimet- hylamino)but-2-enamido)cyclohexane-1-carboxamide (B3)
[0625] To a solution of 3-amino-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)cycloh- exane-1-carboxamide hydrochloride (130 mg, 0.3 mmol) in DMF (1 mL) was added HATU (228 mg, 0.6 mmol), (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (100 mg, 0.6 mmol), and DIPEA (0.214 ml, 1.2 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (100 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (48 mg, 33%) of the title compound as a white solid. MS m/z 506.26[M+H].sup.+. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 12.27 (s, 1H), 8.00 (d, J=8.0 Hz, 1H), 7.39 (s, 1H), 6.73 (s, 1H), 6.54 (d, J=16.2 Hz, 1H), 6.01 (d, J=15.7 Hz, 1H), 4.06 (s, 2H), 3.63 (d, J=26.0 Hz, 2H), 3.03 (s, 2H), 2.60 (s, 1H), 2.17 (s, 6H), 1.91 (d, J=11.2 Hz, 1H), 1.79 (s, 3H), 1.30 (s, 4H), 1.18 (s, 9H).
Synthesis of B4
##STR00309##
[0626] 5-thiocyanatothiazol-2-amine (1)
[0627] To a solution of 5-bromothiazol-2-amine hydrobromide (5.2 g, 20 mmol) in methanol (100 mL) was added KSCN (19.5 g, 200 mmol) at room temperature. The resulting mixture was stirred for 20 hours and then concentrated under vacuum. The residue was diluted with H.sub.2O (150 mL). The pH of the solution was adjusted to 10 with 10% Na.sub.2CO.sub.3. The precipitate was filtered and washed with water to obtain (1.8 g, 58%) of the title compound (1) as a brown solid. MS m/z 158.95 [M+H].sup.+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2)
[0628] To a solution of 5-thiocyanatothiazol-2-amine (1.8 g, 11.46 mmol) in absolute EtOH (50 mL) was added NaBH.sub.4 (0.87 g, 2.3 mmol) portionwise at room temperature. The resulting mixture was stirred for 1 hour, and then acetone (20 ml) was slowly introduced. After 1 hour, a solution of 5-(tert-butyl)-2-(chloromethyl)oxazole (2.19 g, 12.6 mmol) in EtOH (10 ml) was added, and the resulting mixture was cooled, concentrated in vacuo, and then partitioned between EtOAc and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (2.1 g, 68%) of the title compound (2) as a pale red-brown solid. MS m/z 270.06 [M+H].sup.+.
tert-butyl 3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)car- bamoyl)piperidine-1-carboxylate (3)
[0629] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (200 mg, 0.74 mmol) in DMF (2 mL) was added HATU (562 mg, 1.5 mmol), 1-(tert-butoxycarbonyl)piperidine-3-carboxylic acid (343 mg, 1.5 mmol), and DIPEA (0.54 ml, 3 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (150 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (260 mg, 73%) of the title compound (3) as a white solid. MS m/z 481.19[M+H].sup.+.
N-(5-(((5-tert-butyl)oxazol-2-yl)methyl)thio)triazol-2-yl)piperidine-3-car- boxamide hydrochloride (4)
[0630] To a solution of tert-butyl 3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)carbamoyl)pip- eridine-1-carboxylate (260 mg, 0.54 mmol) in 4 M HCl EtOAc (5 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was concentrated under vacuum to afford (180 mg, 80%) of the title compound (4) as a white solid, which was used without further purification. MS m/z 381.14 [M+H].sup.+.
tert-butyl (4-(3-((5-((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)- carbamoyl)piperidine-1-carbonyl)phenyl)carbamate (5)
[0631] To a solution of N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)piperidine-3-c- arboxamide hydrochloride (180 mg, 0.43 mmol) in DMF (2 mL) was added HATU (329 mg, 0.86 mmol), 4-((tert-butoxycarbonyl)amino)benzoic acid (205 mg, 0.86 mmol), and DIPEA (0.38 ml, 2.15 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (100 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (80 mg, 31%) of the title compound (5) as a white solid. MS m/z 600.23[M+H].sup.+.
1-(4-aminobenzoyl)-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2- -yl)piperidine-3-carboxamide hydrochloride (6)
[0632] To a solution of tert-butyl (4-(3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)carbamoyl- )piperidine-1-carbonyl)phenyl)carbamate (80 mg, 0.133 mmol) in 4 M HCl EtOAc (3 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was concentrated under vacuum to afford (65 mg, 91%) of the title compound (6) as a white solid, which was used without further purification. MS m/z 381.14 [M+H].sup.+.
(E)-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(4-(4-(d- imethylamino)but-2-enamido)benzoyl)piperidine-3-carboxamide (B4)
[0633] To a solution of 1-(4-aminobenzoyl)-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-- 2-yl)piperidine-3-carboxamide hydrochloride (0.60 g, 0.112 mmol) in DCM (5 mL) was added DIPEA (0.2 ml, 1.34 mmol) and (E)-4-bromobut-2-enoyl chloride (22.5 mg, 0.134 mmol) at 0.degree. C. for 3 min. Then it was added 4.0 M dimethylamine in THF (2 mL). The resulting mixture was stirred at room temperature for 2 hours. Then it was concentrated and purified by an silica gel column to afford (8 mg, 11.7%) of the title compound as a slightly white solid. MS m/z 611.24 [M+H].sup.+. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 12.42 (s, 1H), 10.66 (s, 2H), 7.77 (s, 2H), 7.40 (s, 3H), 6.83 (s, 1H), 6.72 (s, 1H), 6.51 (d, J=15.1 Hz, 1H), 4.52-4.17 (m, 1H), 4.07 (s, 2H), 3.63 (s, 1H), 3.08 (s, 2H), 2.77 (s, 6H), 2.02 (s, 2H), 1.72 (s, 3H), 1.46 (s, 2H), 1.19 (s, 9H).
Synthesis of B5
##STR00310## ##STR00311##
[0634] 5-thiocyanatothiazol-2-amine (1)
[0635] To a solution of 5-bromothiazol-2-amine hydrobromide (5.2 g, 20 mmol) in methanol (100 mL) was added KSCN (19.5 g, 200 mmol) at room temperature. The resulting mixture was stirred for 20 hours and then concentrated under vacuum. The residue was diluted with H.sub.2O (150 mL). The pH of the solution was adjusted to 10 with 10% Na.sub.2CO.sub.3. The precipitate was filtered and washed with water to obtain (1.8 g, 58%) of the title compound (1) as a brown solid. MS m/z 158.95 [M+H].sup.+.
5-((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2)
[0636] To a solution of 5-thiocyanatothiazol-2-amine (1.8 g, 11.46 mmol) in absolute EtOH (50 mL) was added NaBH.sub.4 (0.87 g, 2.3 mmol) portionwise at room temperature. The resulting mixture was stirred for 1 hour, and then acetone (20 ml) was slowly introduced. After 1 hour, a solution of 5-(tert-butyl)-2-(chloromethyl)oxazole (2.19 g, 12.6 mmol) in EtOH (10 ml) was added, and the resulting mixture was cooled, concentrated in vacuo, and then partitioned between EtOAc and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (2.1 g, 68%) of the title compound (2) as a pale red-brown solid. MS m/z 270.06 [M+H].sup.+.
tert-butyl 3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)car- bamoyl)piperidine-1-carboxylate (3)
[0637] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (65 mg, 0.24 mmol) in DMF (2 mL) was added HATU (183 mg, 0.48 mmol), 1-(tert-butoxycarbonyl)piperidine-3-carboxylic acid (83 mg, 0.36 mmol) and DIPEA (126 mg, 0.96 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (100 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (23 mg, 20%) of the title compound (3) as a white solid. MS m/z 481.19[M+H].sup.+.
N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)piperidine-3-ca- rboxamide hydrochloride (4)
[0638] To a solution of tert-butyl 3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)carbamoyl)pip- eridine-1-carboxylate (23 mg, 0.048 mmol) in 4 M HCl EtOAc (2 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was concentrated under vacuum to afford (16 mg, 80%) of the title compound (4) as a white solid, which was used without further purification. MS m/z 381.14 [M+H]+.
1-(4-acrylamidobenzoyl)-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thia- zol-2-yl)piperidine-3-carboxamide (B5)
[0639] To a solution of N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)piperidine-3-c- arboxamide hydrochloride (50 mg, 0.12 mmol) in DMF (1 mL) was added HATU (92 mg, 0.24 mmol), 4-acrylamidobenzoic acid (46 mg, 0.24 mmol), and DIPEA (0.22 ml, 1.2 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (100 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (8 mg, 12%) of the title compound as a white solid. MS m/z 554.21 [M+H].sup.+. .sup.1H NMR (400 MHz, DMSO-ds) .delta. 12.37 (s, 1H), 7.73 (d, J=7.8 Hz, 2H), 7.55-7.22 (m, 3H), 6.71 (s, 1H), 6.44 (dd, J=16.9, 10.3 Hz, 1H), 6.28 (d, J=16.8 Hz, 1H), 5.78 (d, J=9.9 Hz, 1H), 4.05 (s, 2H), 3.67 (s, 1H), 3.05 (s, 2H), 2.69 (s, 1H), 1.97 (s, 1H), 1.70 (d, J=12.1 Hz, 2H), 1.44 (s, 1H), 1.23 (s, 2H), 1.12 (s, 9H).
Synthesis of B6
##STR00312##
[0640] 5-thiocyanatothiazol-2-amine (1)
[0641] To a solution of 5-bromothiazol-2-amine hydrobromide (5.2 g, 20 mmol) in methanol (100 mL) was added KSCN (19.5 g, 200 mmol) at room temperature. The resulting mixture was stirred for 20 hours and then concentrated under vacuum. The residue was diluted with H.sub.2O (150 mL). The pH of the solution was adjusted to 10 with 10% Na.sub.2CO.sub.3. The precipitate was filtered and washed with water to obtain (1.8 g, 58%) of the title compound (1) as a brown solid. MS m/z 158.95 [M+H].sup.+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2)
[0642] To a solution of 5-thiocyanatothiazol-2-amine (1.8 g, 11.46 mmol) in absolute EtOH (50 mL) was added NaBH.sub.4 (0.87 g, 2.3 mmol) portionwise at room temperature. The resulting mixture was stirred for 1 hour, and then acetone (20 ml) was slowly introduced. After 1 hour, a solution of 5-(tert-butyl)-2-(chloromethyl)oxazole (2.19 g, 12.6 mmol) in EtOH (10 ml) was added, and the resulting mixture was cooled, concentrated in vacuo, and then partitioned between EtOAc and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (2.1 g, 68%) of the title compound (2) as a pale red-brown solid. MS m/z 270.06 [M+H].sup.+.
3-acrylamido-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)be- nzamide (B6)
[0643] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (54 mg, 0.2 mmol) in DMF (1 mL) was added HATU (160 mg, 0.42 mmol), 3-acrylamidobenzoic acid (60 mg, 0.31 mmol), and DIPEA (52 mg, 0.4 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (100 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (7 mg, 8%) of the title compound as a white solid. MS m/z 443.12[M+H].sup.+. .sup.1H NMR (400 MHz, CDCl.sub.3) .delta. 12.36 (s, 1H), 9.08 (s, 1H), 8.24-7.99 (m, 2H), 7.90 (s, 1H), 7.63 (d, J=7.6 Hz, 1H), 7.41 (t, J=7.9 Hz, 1H), 6.95 (s, 1H), 6.55 (s, 1H), 6.44 (d, J=16.8 Hz, 1H), 6.32 (dd, J=16.8, 10.0 Hz, 1H), 5.76 (d, J=10.0 Hz, 1H), 3.95 (s, 2H), 1.23 (s, 9H).
Synthesis of B7
##STR00313##
[0644] 5-thiocyanatothiazol-2-amine (1)
[0645] To a solution of 5-bromothiazol-2-amine hydrobromide (5.2 g, 20 mmol) in methanol (100 mL) was added KSCN (19.5 g, 200 mmol) at room temperature. The resulting mixture was stirred for 20 hours and then concentrated under vacuum. The residue was diluted with H.sub.2O (150 mL). The pH of the solution was adjusted to 10 with 10% Na.sub.2CO.sub.3. The precipitate was filtered and washed with water to obtain (1.8 g, 58%) of the title compound (1) as a brown solid. MS m/z 158.95 [M+H].sup.+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2)
[0646] To a solution of 5-thiocyanatothiazol-2-amine (1.8 g, 11.46 mmol) in absolute EtOH (50 mL) was added NaBH.sub.4 (0.87 g, 2.3 mmol) portionwise at room temperature. The resulting mixture was stirred for 1 hour, and then acetone (20 ml) was slowly introduced. After 1 hour, a solution of 5-(tert-butyl)-2-(chloromethyl)oxazole (2.19 g, 12.6 mmol) in EtOH (10 ml) was added, and the resulting mixture was cooled, concentrated in vacuo, and then partitioned between EtOAc and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (2.1 g, 68%) of the title compound (2) as a pale red-brown solid. MS m/z 270.06 [M+H].sup.+.
tert-butyl (3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)ca- rbamoyl)phenyl)carbamate (3)
[0647] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (404 mg, 1.5 mmol) in DMF (2 mL) was added HATU (1.14 g, 3 mmol), 3-((tert-butoxycarbonyl)amino)benzoic acid (573 mg, 3 mmol), and DIPEA (1.05 ml, 6 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (200 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (490 mg, 67%) of the title compound (3) as a white solid. MS m/z 489.15[M+H].sup.+.
3-amino-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)benzami- de hydrochloride (4)
[0648] To a solution of tert-butyl (3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)carbamoyl)ph- enyl)carbamate (245 mg, 0.5 mmol) in 4 M HCl EtOAc (5 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was concentrated under vacuum to afford (212 mg, 100%) of the title compound (4) as a white solid, which was used without further purification. MS m/z 389.11 [M+H].sup.+.
(E)-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-3-(4-(dime- thylamino)but-2-enamido)benzamide (B7)
[0649] To a solution of 3-amino-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)benzam- ide hydrochloride (83 mg, 0.195 mmol) in MeCN (5 mL) was added DIPEA (77.6 mg, 0.6 mmol), and (E)-4-bromobut-2-enoyl chloride (40.26 mg, 0.22 mmol) at 0.degree. C. for 3 min. Then it was added 4.0 M dimethylamine in THF (1 mL). The resulting mixture was stirred at room temperature for 2 hours. Then it was concentrated and purified by an silica gel column to afford (7 mg, 7.2%) of the title compound as a slightly yellow solid. MS m/z 500.18 [M+H].sup.+. .sup.1H NMR (400 MHz, CDCl.sub.3) .delta. 8.82 (s, 1H), 8.01 (d, J=16.7 Hz, 2H), 7.64 (s, 1H), 7.42 (s, 1H), 7.10 (s, 1H), 7.00 (s, 1H), 6.58 (s, 1H), 6.24 (d, J=13.7 Hz, 1H), 3.96 (s, 2H), 3.16 (s, 2H), 2.31 (s, 6H), 1.26 (s, 9H).
Synthesis of B8
##STR00314##
[0650] 5-thiocyanatothiazol-2-amine (1)
[0651] To a solution of 5-bromothiazol-2-amine hydrobromide (5.2 g, 20 mmol) in methanol (100 mL) was added KSCN (19.5 g, 200 mmol) at room temperature. The resulting mixture was stirred for 20 h and then concentrated under vacuum. The residue was diluted with H.sub.2O (150 mL). The pH of the solution was adjusted to 10 with 10% Na.sub.2CO.sub.3. The precipitate was filtered and washed with water to obtain (1.8 g, 58%) of the title compound (1) as a brown solid. MS m/z 158.95 [M+H]*.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2)
[0652] To a solution of 5-thiocyanatothiazol-2-amine (1.8 g, 11.46 mmol) in absolute EtOH (50 mL) was added NaBH.sub.4 (0.87 g, 2.3 mmol) portionwise at room temperature. The resulting mixture was stirred for 1 hour, and then acetone (20 mL) was slowly introduced. After 1 hour, a solution of 5-(tert-butyl)-2-(chloromethyl)oxazole (2.19 g, 12.6 mmol) in EtOH (10 mL) was added, and the resulting mixture was cooled, concentrated in vacuo, and then partitioned between EtOAc and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (2.1 g, 68%) of the title compound (2) as a pale red-brown solid. MS m/z 270.06 [M+H].sup.+.
tert-butyl (4-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)ca- rbamoyl)phenyl)carbamate (3)
[0653] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (220 mg, 0.82 mmol) in DMF (1 mL) was added HATU (623 mg, 1.64 mmol), 4-((tert-butoxycarbonyl)amino)benzoic acid (390 mg, 1.64 mmol) and DIPEA (0.6 ml, 3.28 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (150 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (200 mg, 50%) of the title compound (3) as a white solid. MS m/z 489.15[M+H].sup.+.
4-amino-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)benzami- de hydrochloride (4)
[0654] To a solution of tert-butyl (4-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)carbamoyl)ph- enyl)carbamate (200 mg, 0.41 mmol) in 4 M HCl EtOAc (5 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was concentrated under vacuum to afford (174 mg, 100%) of the title compound (4) as a white solid, which was used without further purification. MS m/z 389.11 [M+H].sup.+.
(E)-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-44-(dimeth- ylamino)but-2-enamido)benzamide (B8)
[0655] To a solution of 4-amino-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)benzam- ide hydrochloride (75 mg, 0.177 mmol) in MeCN (5 mL) was added DIPEA (0.3 ml, 2.6 mmol) and (E)-4-bromobut-2-enoyl chloride (46.85 mg, 0.256 mmol) at 0.degree. C. for 3 min. Then it was added 4.0 M dimethylamine in THF (1 mL). The resulting mixture was stirred at room temperature for 2 hours. Then it was concentrated and purified by an silica gel column to afford (7 mg, 8%) of the title compound as a slightly yellow solid. MS m/z 500.18 [M+H].sup.+. .sup.1H NMR (400 MHz, CDCl.sub.3) .delta. 8.57 (s, 1H), 7.90 (d, J=7.3 Hz, 2H), 7.76 (s, 2H), 7.19 (s, 1H), 7.05 (s, 1H), 6.61 (s, 1H), 6.23 (d, J=15.5 Hz, 1H), 3.98 (s, 2H), 3.18 (s, 2H), 2.34 (s, 6H), 1.27 (s, 9H).
Synthesis of B9
##STR00315##
[0656] 5-thiocyanatothiazol-2-amine (1)
[0657] To a solution of 5-bromothiazol-2-amine hydrobromide (5.2 g, 20 mmol) in methanol (100 mL) was added KSCN (19.5 g, 200 mmol) at room temperature. The resulting mixture was stirred for 20 h and then concentrated under vacuum. The residue was diluted with H.sub.2O (150 mL). The pH of the solution was adjusted to 10 with 10% Na.sub.2CO.sub.3. The precipitate was filtered and washed with water to obtain (1.8 g, 58%) of the title compound (1) as a brown solid. MS m/z 158.95 [M+H].sup.+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2)
[0658] To a solution of 5-thiocyanatothiazol-2-amine (1.8 g, 11.46 mmol) in absolute EtOH (50 mL) was added NaBH.sub.4 (0.87 g, 2.3 mmol) portionwise at room temperature. The resulting mixture was stirred for 1 hour, and then acetone (20 mL) was slowly introduced. After 1 hour, a solution of 5-(tert-butyl)-2-(chloromethyl)oxazole (2.19 g, 12.6 mmol) in EtOH (10 mL) was added, and the resulting mixture was cooled, concentrated in vacuo, and then partitioned between EtOAc and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (2.1 g, 68%) of the title compound (2) as a pale red-brown solid. MS m/z 270.06 [M+H].sup.+.
tert-butyl (3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl))thio)thiazol-2-yl)c- arbamoyl)phenyl)carbamate (3)
[0659] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (404 mg, 1.5 mmol) in DMF (2 mL) was added HATU (1.14 g, 3 mmol), 3-((tert-butoxycarbonyl)amino)benzoic acid (573 mg, 3 mmol), and DIPEA (1.05 ml, 6 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (200 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (490 mg, 67%) of the title compound (3) as a white solid. MS m/z 489.15 [M+H].sup.+.
3-amino-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)benzami- de hydrochloride (4)
[0660] To a solution of tert-butyl (3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)carbamoyl)ph- enyl)carbamate (245 mg, 0.5 mmol) in 4 M HCl EtOAc (5 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was concentrated under vacuum to afford (212 mg, 100%) of the title compound (4) as a white solid, which was used without further purification. MS m/z 389.11 [M+H].sup.+.
3-(4-acrylamidobenzamido)-N-(5-(((5-tert-butyl)oxazol-2-yl)methyl)thio)tri- azol-2-yl)benzamide (B9)
[0661] To a solution of 3-amino-N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)benzam- ide hydrochloride (212 mg, 0.5 mmol) in DMF (2 mL) was added HATU (380 mg, 1 mmol), 4-acrylamidobenzoic acid (191 mg, 1 mmol), and DIPEA (0.5 ml, 3 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (200 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (93 mg, 34%) of the title compound as a white solid. MS m/z 562.18[M+H].sup.+. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 12.83 (s, 1H), 10.47 (s, 1H), 10.40 (s, 1H), 8.46 (s, 1H), 8.00 (dd, J=15, 5.0 Hz, 3H), 7.83 (d, J=15.0 Hz, 3H), 7.52 (d, J=5.0 Hz, 2H), 6.75 (s, 1H), 6.46 (d, J=6.3 Hz, 1H), 6.31 (d, J=6.5 Hz, 1H), 5.82 (d, J=15.0 Hz, 1H), 4.12 (s, 2H), 1.22 (s, 9H).
Synthesis of B12
##STR00316##
[0662] 5-thiocyanatothiazol-2-amine (1)
[0663] To a solution of 5-bromothiazol-2-amine hydrobromide (26 g, 100 mmol) in methanol (500 mL) was added KSCN (50 g, 500 mmol) at room temperature. The resulting mixture was stirred for 20 hours and then concentrated under vacuum. The residue was diluted with H.sub.2O (300 mL). The pH of the solution was adjusted to 10 with 10% Na.sub.2CO.sub.3. The precipitate was filtered and washed with water to obtain (8 g, 51%) of the title compound (1) as a brown solid. MS m/z 158.95 [M+H].sup.+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2)
[0664] To a solution of 5-thiocyanatothiazol-2-amine (2 g, 13 mmol) in absolute EtOH (50 mL) was added NaBH.sub.4 (1 g, 26 mmol) portionwise at room temperature. The resulting mixture was stirred for 1 hour, and then acetone (20 mL) was slowly introduced. After 1 hour, a solution of 5-(tert-butyl)-2-(chloromethyl)oxazole (2.71 g, 15.6 mmol) in EtOH (10 mL) was added, and the resulting mixture was cooled, concentrated in vacuo, and then partitioned between EtOAc and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (2.8 g, 80%) of the title compound (2) as a pale red-brown solid. MS m/z 270.06 [M+H].sup.+.
2-(((2-bromothiazol-5-yl)thio)methyl)-5-(tert-butyl)oxazole (3)
[0665] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (404 mg, 1.5 mmol) in MeCN (10 mL) was added tert-butyl nitrite (239 mg, 2.25 mmol) at 0.degree. C. The resulting mixture was stirred for 10 min, and then CuBr.sub.2 (402 mg, 1.8 mmol) was introduced. The resulting mixture was stirred at room temperature for 3 hours, the resulting mixture was concentrated in vacuo. The residue was purified by an silica gel column to afford (143 mg, 29%) of the title compound (3) as a pale yellow oil. MS m/z 333.96 [M+H].sup.+.
tert-butyl 3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)ami- no)piperidine-1-carboxylate (4)
[0666] To a solution of 2-(((2-bromothiazol-5-yl)thio)methyl)-5-(tert-butyl)oxazole (80 mg, 0.22 mmol) in NMP (0.1 mL) was added tert-butyl 3-aminopiperidine-1-carboxylate (80 mg, 0.4 mmol) and DIPEA (0.084 ml, 0.48 mmol). The resulting mixture was heated at 140.degree. C. for 8 hours. Then it was diluted with EtOAc (150 mL) at room temperature, washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (30 mg, 28%) of the title compound (4) as a slightly yellow solid. MS m/z 453.20 [M+H].sup.+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-N-(piperidin-3-yl)thiazol-2-am- ine hydrochloride (5)
[0667] To a solution of tert-butyl 3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)amino)piperid- ine-1-carboxylate (38 mg, 0.084 mmol) in 4 M HCl EtOAc (5 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was concentrated under vacuum to afford (35 mg, 98%) of the title compound (5) as a white solid, which was used without further purification. MS m/z 353.14 [M+H].sup.+.
N-(4-(3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)amino)pi- peridine-1-carbonyl)phenyl)acrylamide (B12)
[0668] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-N-(piperidin-3-yl)thiazol-2-a- mine hydrochloride (35 mg, 0.082 mmol) in DMF (0.5 mL) was added HATU (62.4 mg, 0.164 mmol), 4-acrylamidobenzoic acid (31.5 mg, 0.164 mmol), and DIPEA (42.6 mg, 0.33 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (70 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (5 mg, 12%) of the title compound as a white solid. MS m/z 526.21[M+H].sup.+. .sup.1H NMR (400 MHz, CDCl.sub.3) .delta. 9.83 (s, 1H), 7.59 (d, J=7.2 Hz, 2H), 7.29 (s, 2H), 6.86 (s, 1H), 6.66 (s, 1H), 6.56 (s, 1H), 6.44 (d, J=16.9 Hz, 1H), 6.26 (s, 1H), 5.72 (s, 1H), 4.03 (s, 1H), 3.84 (s, 2H), 3.55 (s, 1H), 3.46 (d, J=31.2 Hz, 2H), 2.03 (s, 3H), 1.84 (s, 2H), 1.32 (s, 9H).
Synthesis of B15
##STR00317##
[0669] 5-thiocyanatothiazol-2-amine (1)
[0670] To a solution of 5-bromothiazol-2-amine hydrobromide (26 g, 100 mmol) in methanol (500 mL) was added KSCN (50 g, 500 mmol) at room temperature. The resulting mixture was stirred for 20 h and then concentrated under vacuum. The residue was diluted with H.sub.2O (300 mL). The pH of the solution was adjusted to 10 with 10% Na.sub.2CO.sub.3. The precipitate was filtered and washed with water to obtain (8 g, 51%) of the title compound (1) as a brown solid. MS m/z 158.95 [M+H].sup.+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2)
[0671] To a solution of 5-thiocyanatothiazol-2-amine (2 g, 13 mmol) in absolute EtOH (50 mL) was added NaBH.sub.4 (1 g, 26 mmol) portionwise at room temperature. The resulting mixture was stirred for 1 hour, and then acetone (20 mL) was slowly introduced. After 1 hour, a solution of 5-(tert-butyl)-2-(chloromethyl)oxazole (2.71 g, 15.6 mmol) in EtOH (10 mL) was added, and the resulting mixture was cooled, concentrated in vacuo, and then partitioned between EtOAc and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (2.8 g, 80%) of the title compound (2) as a pale red-brown solid. MS m/z 270.06 [M+H].sup.+.
2-(((2-bromothiazol-5-yl)thio)methyl)-5-(tert-butyl)oxazole (3)
[0672] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2.02 g, 7.5 mmol) in MeCN (50 mL) was added tert-butyl nitrite (1.2 g, 11.7 mmol) at 0.degree. C. The resulting mixture was stirred for 10 min, and then CuBr.sub.2 (2.01 mg, 9 mmol) was introduced. The resulting mixture was stirred at room temperature for 3 hours. the resulting mixture was concentrated in vacuo. The residue was purified by an silica gel column to afford (0.6 g, 24%) of the title compound (3) as a pale yellow oil. MS m/z 333.96 [M+H].sup.+.
tert-butyl 3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)ami- no)piperidine-1-carboxylate (4)
[0673] To a solution of 2-(((2-bromothiazol-5-yl)thio)methyl)-5-(tert-butyl)oxazole (0.6 g, 1.8 mmol) in NMP (0.6 mL) was added tert-butyl 3-aminopiperidine-1-carboxylate (0.72 mg, 3.6 mmol) and DIPEA (0.65 ml, 3.6 mmol). The resulting mixture was heated at 140.degree. C. for 8 hours. Then it was diluted with EtOAc (200 mL) at room temperature, washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (150 mg, 18.5%) of the title compound (4) as a slightly yellow solid. MS m/z 453.20 [M+H].sup.+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-N-(piperidin-3-yl)triazol-2-am- ine hydrochloride (5)
[0674] To a solution of tert-butyl 3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)amino)piperid- ine-1-carboxylate (150 mg, 0.33 mmol) in 4 M HCl EtOAc (5 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was concentrated under vacuum to afford (130 mg, 93%) of the title compound (5) as a white solid, which was used without further purification. MS m/z 353.14 [M+H].sup.+.
N-(3-(3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)triazol-2-yl)amino)pi- peridine-1-carbonyl)phenyl)acrylamide (B12)
[0675] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-N-(piperidin-3-yl)thiazol-2-a- mine hydrochloride (55 mg, 0.13 mmol) in DMF (0.5 mL) was added HATU (99 mg, 0.26 mmol), 3-acrylamidobenzoic acid (49.7 mg, 0.26 mmol), and DIPEA (67 mg, 0.52 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (70 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (12 mg, 18%) of the title compound as a white solid. MS m/z 526.21 [M+H].sup.+. .sup.1H NMR (400 MHz, CDCl.sub.3) .delta. 9.41 (s, 1H), 7.85 (s, 1H), 7.57 (s, 1H), 7.07 (s, 1H), 6.86 (s, 1H), 6.64 (s, 1H), 6.45 (d, J=17.1 Hz, 1H), 6.41-6.20 (m, 1H), 5.73 (s, 1H), 5.55 (s, 1H), 4.03-3.80 (m, 2H), 3.71 (s, 1H), 3.56 (s, 2H), 2.04 (s, 2H), 1.85 (s, 4H), 1.29 (s, 9H).
Synthesis of B16
##STR00318##
[0676] 5-thiocyanatothiazol-2-amine (1)
[0677] To a solution of 5-bromothiazol-2-amine hydrobromide (26 g, 100 mmol) in methanol (500 mL) was added KSCN (50 g, 500 mmol) at room temperature. The resulting mixture was stirred for 20 hours and then concentrated under vacuum. The residue was diluted with H.sub.2O (300 mL). The pH of the solution was adjusted to 10 with 10% Na.sub.2CO.sub.3. The precipitate was filtered and washed with water to obtain (8 g, 51%) of the title compound (1) as a brown solid. MS m/z 158.95 [M+H].sup.+.
5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2)
[0678] To a solution of 5-thiocyanatothiazol-2-amine (2 g, 13 mmol) in absolute EtOH (50 mL) was added NaBH.sub.4 (1 g, 26 mmol) portionwise at room temperature. The resulting mixture was stirred for 1 hour, and then acetone (20 mL) was slowly introduced. After 1 hour, a solution of 5-(tert-butyl)-2-(chloromethyl)oxazole (2.71 g, 15.6 mmol) in EtOH (10 mL) was added, and the resulting mixture was cooled, concentrated in vacuo, and then partitioned between EtOAc and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (2.8 g, 80%) of the title compound (2) as a pale red-brown solid. MS m/z 270.06 [M+H].sup.+.
2-(((2-bromothiazol-5-yl)thio)methyl)-5-(tert-butyl)oxazole (3)
[0679] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-amine (2.02 g, 7.5 mmol) in MeCN (50 mL) was added tert-butyl nitrite (1.2 g, 11.7 mmol) at 0.degree. C. The resulting mixture was stirred for 10 min, and then CuBr.sub.2 (2.01 mg, 9 mmol) was introduced. The resulting mixture was stirred at room temperature for 3 hours. the resulting mixture was concentrated in vacuo. The residue was purified by an silica gel column to afford (0.6 g, 24%) of the title compound (3) as a pale yellow oil. MS m/z 333.96 [M+H].sup.+.
tert-butyl 3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)ami- no)piperidine-1-carboxylate (4)
[0680] To a solution of 2-(((2-bromothiazol-5-yl)thio)methyl)-5-(tert-butyl)oxazole (0.6 g, 1.8 mmol) in NMP (0.6 mL) was added tert-butyl 3-aminopiperidine-1-carboxylate (0.72 mg, 3.6 mmol) and DIPEA (0.65 ml, 3.6 mmol). The resulting mixture was heated at 140.degree. C. for 8 hours. Then it was diluted with EtOAc (200 mL) at room temperature, washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (150 mg, 18.5%) of the title compound (4) as a slightly yellow solid. MS m/z 453.20 [M+H].sup.+.
5-((5-(tert-butyl)oxazol-2-yl)methyl)thio)-N-(piperidin-3-yl)thiazol-2-ami- ne hydrochloride (5)
[0681] To a solution of tert-butyl 3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)amino)piperid- ine-1-carboxylate (150 mg, 0.33 mmol) in 4 M HCl EtOAc (5 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was concentrated under vacuum to afford (130 mg, 93%) of the title compound (5) as a white solid, which was used without further purification. MS m/z 353.14 [M+H].sup.+.
tert-butyl (4-(3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl- )amino)piperidine-1-carbonyl)phenyl)carbamate (6)
[0682] To a solution of 5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)-N-(piperidin-3-yl)thiazol-2-a- mine hydrochloride (65 mg, 0.153 mmol) in DMF (0.5 mL) was added HATU (118 mg, 0.31 mmol), 4-((tert-butoxycarbonyl)amino)benzoic acid (72 mg, 0.31 mmol), and DIPEA (79 mg, 0.612 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (70 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (40 mg, 43%) of the title compound (6) as a white solid. MS m/z 572.22[M+H].sup.+.
(4-aminophenyl)(3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-y- l)amino)piperidin-1-yl)methanone hydrochloride (7)
[0683] To a solution of tert-butyl (4-(3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)amino)pip- eridine-1-carbonyl)phenyl)carbamate (40 mg, 0.07 mmol) in 4 M HCl EtOAc (1 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was concentrated under vacuum to afford (35 mg, 93%) of the title compound (7) as a white solid, which was used without further purification. MS m/z 472.18 [M+H].sup.+.
(E)-N-(4-(3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)amin- o)piperidine-1-carbonyl)phenyl)-4-dimethylamino)but-2-enamide (B16)
[0684] To a solution of (4-aminophenyl)(3-((5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-- yl)amino)piperidin-1-yl)methanone hydrochloride (50 mg, 0.1 mmol) in MeCN (5 mL) was added DIPEA (65 mg, 0.5 mmol) and (E)-4-bromobut-2-enoyl chloride (22 mg, 0.12 mmol) at 0.degree. C. for 3 min. Then it was added 4.0 M dimethylamine in THF (1 mL). The resulting mixture was stirred at room temperature for 2 hours. Then it was concentrated and purified by an silica gel column to afford (10 mg, 17%) of the title compound as a white solid. MS m/z 500.18 [M+H].sup.+. .sup.1H NMR (400 MHz, MeOD) .delta. 7.72 (s, 2H), 7.41 (s, 2H), 7.01-6.88 (m, 1H), 6.74 (d, J=22.4 Hz, 2H), 3.94 (s, 2H), 3.86-3.68 (m, 4H), 3.50 (s, 1H), 3.27 (d, J=6.6 Hz, 2H), 2.44 (s, 6H), 2.23 (d, J=21.3 Hz, 1H), 2.13 (d, J=20.0 Hz, 1H), 1.98 (s, 2H), 1.72 (s, 2H), 1.45-1.40 (m, 9H).
Synthesis of B17
##STR00319##
[0685] 5-morpholinothiazol-2-amine (1)
[0686] To a mixture of 5-bromothiazol-2-amine hydrobromide (2.6 g, 10 mmol) and powdered potassium carbonate (2.77 g, 20 mmol) in NMP (5 mL) was added morpholine (1.73 mL, 20 mmol) under argon atmosphere and heated at 60.degree. C. for 3 hours. Reaction mixture was cooled to room temperature and poured over ice cold water (100 mL), extracted with EtOAc (3.times.100 mL), washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by an silica gel column to afford (420 mg, 45.4%) of the title compound (1) as a brown solid. MS m/z 186.07[M+H].sup.+.
3-acrylamido-N-(5-morpholinothiazol-2-yl)benzamide (B17)
[0687] To a solution of 5-morpholinothiazol-2-amine (90 mg, 0.5 mmol) in DMF (0.5 mL) was added HATU (380 mg, 1 mmol), 3-acrylamidobenzoic acid (191 mg, 1 mmol), and DIPEA (129 mg, 1 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (100 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (36 mg, 21%) of the title compound as a yellow solid. MS m/z 359.14 [M+H].sup.+. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 12.30 (s, 1H), 10.37 (s, 1H), 8.29 (s, 1H), 7.91 (d, J=7.2 Hz, 1H), 7.78 (d, J=7.0 Hz, 1H), 7.49 (d, J=6.9 Hz, 1H), 6.78 (s, 1H), 6.47 (dd, J=15.8, 10.4 Hz, 1H), 6.31 (d, J=16.8 Hz, 1H), 5.81 (d, J=9.8 Hz, 1H), 3.75 (s, 4H), 3.05 (s, 4H).
Synthesis of B18
##STR00320##
[0688] 5-(4-methylpiperazin-1-yl)thiazol-2-amine (1)
[0689] To a mixture of 5-bromothiazol-2-amine hydrobromide (2.6 g, 10 mmol) and powdered potassium carbonate (2.77 g, 20 mmol) in NMP (5 mL) was added 1-methylpiperazine (2.25 mL, 20 mmol) under argon atmosphere and heated at 60.degree. C. for 3 hours. Reaction mixture was filtered and concentrated under reduced pressure. The residue was purified by an silica gel column to afford (340 mg, 34.3%) of the title compound (1) as a brown solid. MS m/z 199.10 [M+H].sup.+.
3-acrylamido-N-(5-(4-methylpiperazin-1-yl)thiazol-2-yl)benzamide (B18)
[0690] To a solution of 5-(4-methylpiperazin-1-yl)thiazol-2-amine (99 mg, 0.5 mmol) in DMF (0.5 mL) was added HATU (380 mg, 1 mmol), 3-acrylamidobenzoic acid (191 mg, 1 mmol), and DIPEA (129 mg, 1 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was purified by an silica gel column to afford (41 mg, 22%) of the title compound as a yellow solid. MS m/z 372.17 [M+H].sup.+. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 12.22 (s, 1H), 10.35 (s, 1H), 8.28 (s, 1H), 7.91 (d, J=6.9 Hz, 1H), 7.78 (d, J=6.7 Hz, 1H), 7.47 (t, J=7.7 Hz, 1H), 6.71 (s, 1H), 6.47 (dd, J=16.8, 10.1 Hz, 1H), 6.31 (d, J=17.0 Hz, 1H), 5.88-5.68 (m, 1H), 3.06 (s, 4H), 2.47 (s, 4H), 2.22 (s, 3H).
Synthesis of B19
##STR00321##
[0691] 5-(4-ethylpiperazin-1-yl)thiazol-2-amine (1)
[0692] To a mixture of 5-bromothiazol-2-amine hydrobromide (2.6 g, 10 mmol) and powdered potassium carbonate (2.77 g, 20 mmol) in NMP (5 mL) was added 1-ethylpiperazine (2.28 mL, 20 mmol) under argon atmosphere and heated at 60.degree. C. for 3 hours. Reaction mixture was filtered and concentrated under reduced pressure. The residue was purified by an silica gel column to afford (382 mg, 36%) of the title compound (1) as a brown solid. MS m/z 213.11 [M+H].sup.+.
3-acrylamido-N-(5-(4-methylpiperazin-1-yl)thiazol-2-yl)benzamide (B19)
[0693] To a solution of 5-(4-ethylpiperazin-1-yl)thiazol-2-amine (106 mg, 0.5 mmol) in DMF (0.5 mL) was added HATU (380 mg, 1 mmol), 3-acrylamidobenzoic acid (191 mg, 1 mmol) and DIPEA (129 mg, 1 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was purified by an silica gel column to afford (20 mg, 10.3%) of the title compound as a yellow solid. MS m/z 386.20 [M+H].sup.+. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 12.23 (s, 1H), 10.35 (s, 1H), 8.29 (s, 1H), 7.91 (d, J=7.0 Hz, 1H), 7.79 (d, J=6.8 Hz, 1H), 7.49 (d, J=7.3 Hz, 1H), 6.72 (s, 1H), 6.58-6.43 (m, 1H), 6.31 (d, J=17.2 Hz, 1H), 5.81 (d, J=10.0 Hz, 1H), 3.07 (s, 4H), 2.52 (s, 4H), 2.38 (d, J=6.3 Hz, 2H), 1.03 (s, 3H).
Synthesis of B20
##STR00322##
[0694] tert-butyl 4-(2-aminothiazol-5-yl)piperazine-1-carboxylate (1)
[0695] To a mixture of 5-bromothiazol-2-amine hydrobromide (2.6 g, 10 mmol) and powdered potassium carbonate (2.77 g, 20 mmol) in NMP (5 mL) was added tert-butyl piperazine-1-carboxylate (3.72 g, 20 mmol) under argon atmosphere and heated at 60.degree. C. for 3 hours. Reaction mixture was cooled to room temperature and poured over ice cold water (100 mL), extracted with EtOAc (3.times.100 mL), washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by an silica gel column to afford (600 mg, 42.3%) of the title compound (1) as a brown solid. MS m/z 285.13[M+H].sup.+.
tert-butyl 4-(2-(3-acrylamidobenzamido)triazol-5-yl)piperazine-1-carboxyla- te (B20)
[0696] To a solution of tert-butyl 4-(2-aminothiazol-5-yl)piperazine-1-carboxylate (142 mg, 0.5 mmol) in DMF (0.5 mL) was added HATU (380 mg, 1 mmol), 3-acrylamidobenzoic acid (191 mg, 1 mmol), and DIPEA (129 mg, 1 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was diluted with EtOAc (100 mL), washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated. The residue was purified by an silica gel column to afford (31 mg, 13%) of the title compound as a yellow solid. MS m/z 458.25 [M+H].sup.+. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 12.29 (s, 1H), 10.35 (s, 1H), 8.29 (s, 1H), 7.91 (d, J=7.5 Hz, 1H), 7.79 (d, J=7.4 Hz, 1H), 7.48 (t, J=7.9 Hz, 1H), 6.79 (s, 1H), 6.57-6.40 (m, 1H), 6.31 (d, J=16.6 Hz, 1H), 5.85-5.68 (m, 2H), 3.49 (s, 4H), 3.03 (s, 4H), 1.43 (s, 9H).
Synthesis of B22
##STR00323##
[0697] tert-butyl 1-(2-aminothiazol-5-yl)piperidin-4-ylcarbamate (1)
[0698] To a mixture of 5-bromothiazol-2-amine hydrobromide (2.6 g, 10 mmol) and powdered potassium carbonate (2.77 g, 20 mmol) in NMP (5 mL) was added tert-butyl piperidin-4-ylcarbamate (4.0 g, 20 mmol) under argon atmosphere and heated at 60.degree. C. for 3 hours. Reaction mixture was cooled to room temperature and poured over ice cold water (100 mL), extracted with EtOAc (3.times.100 mL), washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by an silica gel column to afford (400 mg, 13.4%) of the title compound (1) as a brown solid. MS m/z 299.15[M+H].sup.+.
5-(4-aminopiperidin-1-yl)thiazol-2-amine (2)
[0699] To a solution of tert-butyl 1-(2-aminothiazol-5-yl)piperidin-4-ylcarbamate (200 mg, 0.67 mmol) in 4 M HCl EtOAc (4 mL). The resulting mixture was stirred at room temperature for 30 min. Then it was diluted with EtOAc (100 mL). The pH of the solution was adjusted to 7 with 1 M NaOH. The organics washed with water and brine, dried (Na.sub.2SO.sub.4), and concentrated to afford (120 mg, 90%) of the title compound (2) as a slightly brown solid. MS m/z 199.10 [M+H].sup.+.
5-(4-(dimethylamino)piperidin-1-yl)thiazol-2-amine (3)
[0700] To a solution of 5-(4-aminopiperidin-1-yl)thiazol-2-amine (120 mg, 0.61 mmol) in MeOH (5 mL) was added paraformaldehyde (90 mg, 3 mmol), AcOH (1 drop), and NaBH.sub.3CN (251 mg, 4 mmol). The resulting mixture was stirred at room temperature for 15 hours. Then it was concentrated under reduced pressure. The residue was purified by an silica gel column to afford (9 mg, 5.8%) of the title compound (3) as a brown solid. MS m/z 227.15[M+H].sup.+.
3-acrylamido-N-(5-(4-(dimethylamino)piperidin-1-yl)triazol-2-yl)benzamide (B22)
[0701] To a solution of 5-(4-(dimethylamino)piperidin-1-yl)thiazol-2-amine (100 mg, 0.442 mmol) in DMF (0.5 mL) was added HATU (335 mg, 0.884 mmol), 3-acrylamidobenzoic acid (169 mg, 0.884 mmol), and DIPEA (228 mg, 1.768 mmol). The resulting mixture was stirred at room temperature for 1 hour. Then it was purified by an silica gel column to afford (35 mg, 17.5%) of the title compound as a yellow solid. MS m/z 400.20 [M+H]+. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 12.21 (s, 1H), 10.36 (s, 1H), 8.27 (s, 1H), 7.89 (s, 1H), 7.76 (s, 1H), 7.46 (s, 1H), 6.69 (s, 1H), 6.52-6.40 (m, 1H), 6.30 (d, J=17.1 Hz, 1H), 5.79 (d, J=9.6 Hz, 1H), 3.42 (d, J=9.5 Hz, 2H), 2.74 (t, J=11.1 Hz, 2H), 2.19 (s, 7H), 1.82 (d, J=11.2 Hz, 2H), 1.54 (d, J=10.9 Hz, 2H).
Structure-Activity Analyses for Selected Compounds
[0702] Select compounds described herein were evaluated for structure-activity analyses. Exemplary results are shown in Table 1.
TABLE-US-00004 TABLE 1 IC.sub.50 values of exemplary compounds described herein. WT CDK12/ HAP1 CDK13 CDK2 CDK7 CDK9 IC.sub.50 CS HAP1 Compound (nM) (nM) (nM) (nM) IC.sub.50 (nM) ##STR00324## >10,000 >10,000 >10,000 >10,000 >10,000 ##STR00325## >10,000 >10,000 >10,000 >10,000 >10,000 ##STR00326## >10,000 >10,000 >10,000 >10,000 >10,000 ##STR00327## >10,000 >10,000 >10,000 >10,000 >10,000 ##STR00328## >10,000 >10,000 >10,000 >10,000 >10,000 ##STR00329## 120 6020 114 8 308 ##STR00330## 185 5550 22 ##STR00331## 174 6170 182 ##STR00332## >10,000 >10,000 >10,000 ##STR00333## 24.6 362 20A ##STR00334## 267 9230 212 ##STR00335## >10,000 >10,000 >10,000 >10,000 >10,000 ##STR00336## 15.7 557 30.9 ##STR00337## ##STR00338## 126 >10,000 164 ##STR00339## 93.5 4400 199 ##STR00340## 334 1280 66.5 119 591 ##STR00341## >1,000 >10,000 >10,000 >10,000 >10,000 ##STR00342## 22.9 3400 15.1 ##STR00343## >10,000 >10,000 >10,000 >10,000 >10,000 ##STR00344## >1,000 >10,000 >10,000 >10,000 >10,000 ##STR00345## >1,000 >10,000 >10,000 >10,000 >10,000 ##STR00346## 3170 8870 7140 >10,000 >10,000 ##STR00347## 264 224 197 197 1096 ##STR00348## >1,000 >1,000 >1,000 ##STR00349## >1,000 >1,000 >1,000 ##STR00350## >370 2870 1000 ##STR00351## >1,000 >1,000 >1,000 ##STR00352## 124 447 1570 ##STR00353## 251 498 298 ##STR00354## >1,000 >1,000 >1,000 ##STR00355## >1,000 >1,000 7280 ##STR00356## 92.3 5000 65 ##STR00357## 689 158 474 ##STR00358## 9.86 242 33 ##STR00359## 11.9 230 54.5 ##STR00360## 10,000 10,000 ##STR00361## 122 2111 ##STR00362## 1539 3331 ##STR00363## 7480 10,000 ##STR00364## 1180 4960 ##STR00365## 412 944 ##STR00366## 61 618 ##STR00367## ##STR00368## ##STR00369##
Genome Editing:
[0703] With regard to the exemplary results for cellular assays shown in Table 1 above, genome editing was performed as follows. The CRISPR/Cas9 system was used to mutate the endogenous CDK12 and CDK13 loci to encode for CDK12 C1039S and CDK13 C1017S, both are putative CDK12/13 inhibitor-refractory mutants. Target-specific oligonucleotides were cloned into pX330, which carries a codon-optimized version of Cas9 and was further modified to express GFP for identifying transfectants. Cells were co-transfected (X-tremeGENE 9 (Roche)) with 1) pX330 expressing Cas9 and CDK12-targeting guide RNAs and 2) a pUC57-AMP construct bearing 1500 bp of modified CDK12 reference genome that is centered around the CRISPR targeting site in CDK12. Two days after transfection, cells were sorted using GFP as a marker of transfected cells and cells were re-plated for five days. Cells were then re-plated at low density to facilitate the isolation of individual clones. Individual clones were isolated, expanded, and PCR genotyped using mutant specific PCR primers. Following initial PCR screening, individual clones were Sanger sequenced to confirm the presence of the desired mutation. Western blot confirmed the presence of intact CDK12 kinase. The process was sequentially repeated this time with Cas9/sgRNA constructs to target and replace the CDK13 genetic loci. Subsequent experiments were conducted using a CDK12 C1039S/CDK13 C1017S clone and a WT control clone that was carried through the entirety of the CRISPR protocol but that was verified by Sanger sequencing to be WT for CDK12 and CDK13. The genomic sequence complementary to the CDK12-directed guide RNA that was cloned into pX330 and used in the genome editing experiments is: GGCAGGATTGCCATGAGTTG. The genomic sequence complementary to the CDK13-directed guide RNA that was cloned into pX330 and used in the genome editing experiments is: GGCAAGATTGTCATGAGTTA. The reference genome sequence used as a repair template for CDK12 and CDK13 CRISPR was edited to 1) introduce DNA coding for serine, 2) introduce mutations to either remove the PAM site (NGG) targeted by CRISPR/Cas9 or introduce sufficient wobble mutations such that the guide RNA could not recognize the repair template and thus could not be cut by CRISPR/Cas9, and 3) introduce mutations that could allow for mutant and WT-specific PCR amplification.
HAP1 Cell Proliferation Assay:
[0704] With regard to the exemplary results shown in Table 1 above, the HAP1 cell proliferation assay was performed as follows. HAP1 WT and double mutants cells were seeded at a density of 12,000 cells/well in 96-well plates. Twenty-four hours cells were then treated with the indicated compounds in a 10-pt dose escalation format from 1 nM to 10 .mu.M or DMSO control for 72 hrs. After 72 hrs, cells were assayed using CellTiter-Glo Luminescent Cell Viability Assay (Promega) to determine cell viability by measuring the amount of ATP present in each sample cell population, which is an indicator of cell metabolic activity. Results are graphed as fraction of the DMSO control at 72 hrs. All data points were performed in biological triplicate.
[0705] HAP1 cells expressing putative inhibitor-refractory mutations in CDK12 (C1039S) and CDK13 (C1017S) show up to 20-fold less sensitivity to exemplary compounds as compared to control WT HAP1 cells. This result indicates that a substantial portion of intracellular compound activity comes from covalent inhibition of CDK12 and/or CDK13 and mutation of the targeted cysteine (C1039 in CDK12 and C1017 in CDK13) to less nucleophilic serines is sufficient to rescue a substantial portion of anti-proliferative activity.
EQUIVALENTS AND SCOPE
[0706] In the claims articles such as "a," "an," and "the" may mean one or more than one unless indicated to the contrary or otherwise evident from the context. Claims or descriptions that include "or" between one or more members of a group are considered satisfied if one, more than one, or all of the group members are present in, employed in, or otherwise relevant to a given product or process unless indicated to the contrary or otherwise evident from the context. The invention includes embodiments in which exactly one member of the group is present in, employed in, or otherwise relevant to a given product or process. The invention includes embodiments in which more than one, or all of the group members are present in, employed in, or otherwise relevant to a given product or process.
[0707] Furthermore, the invention encompasses all variations, combinations, and permutations in which one or more limitations, elements, clauses, and descriptive terms from one or more of the listed claims is introduced into another claim. For example, any claim that is dependent on another claim can be modified to include one or more limitations found in any other claim that is dependent on the same base claim. Where elements are presented as lists, e.g., in Markush group format, each subgroup of the elements is also disclosed, and any element(s) can be removed from the group. It should it be understood that, in general, where the invention, or aspects of the invention, is/are referred to as comprising particular elements and/or features, certain embodiments of the invention or aspects of the invention consist, or consist essentially of, such elements and/or features. For purposes of simplicity, those embodiments have not been specifically set forth in haec verba herein. It is also noted that the terms "comprising" and "containing" are intended to be open and permits the inclusion of additional elements or steps. Where ranges are given, endpoints are included. Furthermore, unless otherwise indicated or otherwise evident from the context and understanding of one of ordinary skill in the art, values that are expressed as ranges can assume any specific value or sub-range within the stated ranges in different embodiments of the invention, to the tenth of the unit of the lower limit of the range, unless the context clearly dictates otherwise.
[0708] This application refers to various issued patents, published patent applications, journal articles, and other publications, all of which are incorporated herein by reference. If there is a conflict between any of the incorporated references and the instant specification, the specification shall control. In addition, any particular embodiment of the present invention that falls within the prior art may be explicitly excluded from any one or more of the claims. Because such embodiments are deemed to be known to one of ordinary skill in the art, they may be excluded even if the exclusion is not set forth explicitly herein. Any particular embodiment of the invention can be excluded from any claim, for any reason, whether or not related to the existence of prior art.
[0709] Those skilled in the art will recognize or be able to ascertain using no more than routine experimentation many equivalents to the specific embodiments described herein. The scope of the present embodiments described herein is not intended to be limited to the above Description, but rather is as set forth in the appended claims. Those of ordinary skill in the art will appreciate that various changes and modifications to this description may be made without departing from the spirit or scope of the present invention, as defined in the following claims.
Sequence CWU 1
1
51346PRTHomo sapiens 1Met Ala Leu Asp Val Lys Ser Arg Ala Lys Arg
Tyr Glu Lys Leu Asp 1 5 10 15 Phe Leu Gly Glu Gly Gln Phe Ala Thr
Val Tyr Lys Ala Arg Asp Lys 20 25 30 Asn Thr Asn Gln Ile Val Ala
Ile Lys Lys Ile Lys Leu Gly His Arg 35 40 45 Ser Glu Ala Lys Asp
Gly Ile Asn Arg Thr Ala Leu Arg Glu Ile Lys 50 55 60 Leu Leu Gln
Glu Leu Ser His Pro Asn Ile Ile Gly Leu Leu Asp Ala 65 70 75 80 Phe
Gly His Lys Ser Asn Ile Ser Leu Val Phe Asp Phe Met Glu Thr 85 90
95 Asp Leu Glu Val Ile Ile Lys Asp Asn Ser Leu Val Leu Thr Pro Ser
100 105 110 His Ile Lys Ala Tyr Met Leu Met Thr Leu Gln Gly Leu Glu
Tyr Leu 115 120 125 His Gln His Trp Ile Leu His Arg Asp Leu Lys Pro
Asn Asn Leu Leu 130 135 140 Leu Asp Glu Asn Gly Val Leu Lys Leu Ala
Asp Phe Gly Leu Ala Lys 145 150 155 160 Ser Phe Gly Ser Pro Asn Arg
Ala Tyr Thr His Gln Val Val Thr Arg 165 170 175 Trp Tyr Arg Ala Pro
Glu Leu Leu Phe Gly Ala Arg Met Tyr Gly Val 180 185 190 Gly Val Asp
Met Trp Ala Val Gly Cys Ile Leu Ala Glu Leu Leu Leu 195 200 205 Arg
Val Pro Phe Leu Pro Gly Asp Ser Asp Leu Asp Gln Leu Thr Arg 210 215
220 Ile Phe Glu Thr Leu Gly Thr Pro Thr Glu Glu Gln Trp Pro Asp Met
225 230 235 240 Cys Ser Leu Pro Asp Tyr Val Thr Phe Lys Ser Phe Pro
Gly Ile Pro 245 250 255 Leu His His Ile Phe Ser Ala Ala Gly Asp Asp
Leu Leu Asp Leu Ile 260 265 270 Gln Gly Leu Phe Leu Phe Asn Pro Cys
Ala Arg Ile Thr Ala Thr Gln 275 280 285 Ala Leu Lys Met Lys Tyr Phe
Ser Asn Arg Pro Gly Pro Thr Pro Gly 290 295 300 Cys Gln Leu Pro Arg
Pro Asn Cys Pro Val Glu Thr Leu Lys Glu Gln 305 310 315 320 Ser Asn
Pro Ala Leu Ala Ile Lys Arg Lys Arg Thr Glu Ala Leu Glu 325 330 335
Gln Gly Gly Leu Pro Lys Lys Leu Ile Phe 340 345 21490PRTHomo
sapiens 2Met Pro Asn Ser Glu Arg His Gly Gly Lys Lys Asp Gly Ser
Gly Gly 1 5 10 15 Ala Ser Gly Thr Leu Gln Pro Ser Ser Gly Gly Gly
Ser Ser Asn Ser 20 25 30 Arg Glu Arg His Arg Leu Val Ser Lys His
Lys Arg His Lys Ser Lys 35 40 45 His Ser Lys Asp Met Gly Leu Val
Thr Pro Glu Ala Ala Ser Leu Gly 50 55 60 Thr Val Ile Lys Pro Leu
Val Glu Tyr Asp Asp Ile Ser Ser Asp Ser 65 70 75 80 Asp Thr Phe Ser
Asp Asp Met Ala Phe Lys Leu Asp Arg Arg Glu Asn 85 90 95 Asp Glu
Arg Arg Gly Ser Asp Arg Ser Asp Arg Leu His Lys His Arg 100 105 110
His His Gln His Arg Arg Ser Arg Asp Leu Leu Lys Ala Lys Gln Thr 115
120 125 Glu Lys Glu Lys Ser Gln Glu Val Ser Ser Lys Ser Gly Ser Met
Lys 130 135 140 Asp Arg Ile Ser Gly Ser Ser Lys Arg Ser Asn Glu Glu
Thr Asp Asp 145 150 155 160 Tyr Gly Lys Ala Gln Val Ala Lys Ser Ser
Ser Lys Glu Ser Arg Ser 165 170 175 Ser Lys Leu His Lys Glu Lys Thr
Arg Lys Glu Arg Glu Leu Lys Ser 180 185 190 Gly His Lys Asp Arg Ser
Lys Ser His Arg Lys Arg Glu Thr Pro Lys 195 200 205 Ser Tyr Lys Thr
Val Asp Ser Pro Lys Arg Arg Ser Arg Ser Pro His 210 215 220 Arg Lys
Trp Ser Asp Ser Ser Lys Gln Asp Asp Ser Pro Ser Gly Ala 225 230 235
240 Ser Tyr Gly Gln Asp Tyr Asp Leu Ser Pro Ser Arg Ser His Thr Ser
245 250 255 Ser Asn Tyr Asp Ser Tyr Lys Lys Ser Pro Gly Ser Thr Ser
Arg Arg 260 265 270 Gln Ser Val Ser Pro Pro Tyr Lys Glu Pro Ser Ala
Tyr Gln Ser Ser 275 280 285 Thr Arg Ser Pro Ser Pro Tyr Ser Arg Arg
Gln Arg Ser Val Ser Pro 290 295 300 Tyr Ser Arg Arg Arg Ser Ser Ser
Tyr Glu Arg Ser Gly Ser Tyr Ser 305 310 315 320 Gly Arg Ser Pro Ser
Pro Tyr Gly Arg Arg Arg Ser Ser Ser Pro Phe 325 330 335 Leu Ser Lys
Arg Ser Leu Ser Arg Ser Pro Leu Pro Ser Arg Lys Ser 340 345 350 Met
Lys Ser Arg Ser Arg Ser Pro Ala Tyr Ser Arg His Ser Ser Ser 355 360
365 His Ser Lys Lys Lys Arg Ser Ser Ser Arg Ser Arg His Ser Ser Ile
370 375 380 Ser Pro Val Arg Leu Pro Leu Asn Ser Ser Leu Gly Ala Glu
Leu Ser 385 390 395 400 Arg Lys Lys Lys Glu Arg Ala Ala Ala Ala Ala
Ala Ala Lys Met Asp 405 410 415 Gly Lys Glu Ser Lys Gly Ser Pro Val
Phe Leu Pro Arg Lys Glu Asn 420 425 430 Ser Ser Val Glu Ala Lys Asp
Ser Gly Leu Glu Ser Lys Lys Leu Pro 435 440 445 Arg Ser Val Lys Leu
Glu Lys Ser Ala Pro Asp Thr Glu Leu Val Asn 450 455 460 Val Thr His
Leu Asn Thr Glu Val Lys Asn Ser Ser Asp Thr Gly Lys 465 470 475 480
Val Lys Leu Asp Glu Asn Ser Glu Lys His Leu Val Lys Asp Leu Lys 485
490 495 Ala Gln Gly Thr Arg Asp Ser Lys Pro Ile Ala Leu Lys Glu Glu
Ile 500 505 510 Val Thr Pro Lys Glu Thr Glu Thr Ser Glu Lys Glu Thr
Pro Pro Pro 515 520 525 Leu Pro Thr Ile Ala Ser Pro Pro Pro Pro Leu
Pro Thr Thr Thr Pro 530 535 540 Pro Pro Gln Thr Pro Pro Leu Pro Pro
Leu Pro Pro Ile Pro Ala Leu 545 550 555 560 Pro Gln Gln Pro Pro Leu
Pro Pro Ser Gln Pro Ala Phe Ser Gln Val 565 570 575 Pro Ala Ser Ser
Thr Ser Thr Leu Pro Pro Ser Thr His Ser Lys Thr 580 585 590 Ser Ala
Val Ser Ser Gln Ala Asn Ser Gln Pro Pro Val Gln Val Ser 595 600 605
Val Lys Thr Gln Val Ser Val Thr Ala Ala Ile Pro His Leu Lys Thr 610
615 620 Ser Thr Leu Pro Pro Leu Pro Leu Pro Pro Leu Leu Pro Gly Asp
Asp 625 630 635 640 Asp Met Asp Ser Pro Lys Glu Thr Leu Pro Ser Lys
Pro Val Lys Lys 645 650 655 Glu Lys Glu Gln Arg Thr Arg His Leu Leu
Thr Asp Leu Pro Leu Pro 660 665 670 Pro Glu Leu Pro Gly Gly Asp Leu
Ser Pro Pro Asp Ser Pro Glu Pro 675 680 685 Lys Ala Ile Thr Pro Pro
Gln Gln Pro Tyr Lys Lys Arg Pro Lys Ile 690 695 700 Cys Cys Pro Arg
Tyr Gly Glu Arg Arg Gln Thr Glu Ser Asp Trp Gly 705 710 715 720 Lys
Arg Cys Val Asp Lys Phe Asp Ile Ile Gly Ile Ile Gly Glu Gly 725 730
735 Thr Tyr Gly Gln Val Tyr Lys Ala Lys Asp Lys Asp Thr Gly Glu Leu
740 745 750 Val Ala Leu Lys Lys Val Arg Leu Asp Asn Glu Lys Glu Gly
Phe Pro 755 760 765 Ile Thr Ala Ile Arg Glu Ile Lys Ile Leu Arg Gln
Leu Ile His Arg 770 775 780 Ser Val Val Asn Met Lys Glu Ile Val Thr
Asp Lys Gln Asp Ala Leu 785 790 795 800 Asp Phe Lys Lys Asp Lys Gly
Ala Phe Tyr Leu Val Phe Glu Tyr Met 805 810 815 Asp His Asp Leu Met
Gly Leu Leu Glu Ser Gly Leu Val His Phe Ser 820 825 830 Glu Asp His
Ile Lys Ser Phe Met Lys Gln Leu Met Glu Gly Leu Glu 835 840 845 Tyr
Cys His Lys Lys Asn Phe Leu His Arg Asp Ile Lys Cys Ser Asn 850 855
860 Ile Leu Leu Asn Asn Ser Gly Gln Ile Lys Leu Ala Asp Phe Gly Leu
865 870 875 880 Ala Arg Leu Tyr Asn Ser Glu Glu Ser Arg Pro Tyr Thr
Asn Lys Val 885 890 895 Ile Thr Leu Trp Tyr Arg Pro Pro Glu Leu Leu
Leu Gly Glu Glu Arg 900 905 910 Tyr Thr Pro Ala Ile Asp Val Trp Ser
Cys Gly Cys Ile Leu Gly Glu 915 920 925 Leu Phe Thr Lys Lys Pro Ile
Phe Gln Ala Asn Leu Glu Leu Ala Gln 930 935 940 Leu Glu Leu Ile Ser
Arg Leu Cys Gly Ser Pro Cys Pro Ala Val Trp 945 950 955 960 Pro Asp
Val Ile Lys Leu Pro Tyr Phe Asn Thr Met Lys Pro Lys Lys 965 970 975
Gln Tyr Arg Arg Arg Leu Arg Glu Glu Phe Ser Phe Ile Pro Ser Ala 980
985 990 Ala Leu Asp Leu Leu Asp His Met Leu Thr Leu Asp Pro Ser Lys
Arg 995 1000 1005 Cys Thr Ala Glu Gln Thr Leu Gln Ser Asp Phe Leu
Lys Asp Val 1010 1015 1020 Glu Leu Ser Lys Met Ala Pro Pro Asp Leu
Pro His Trp Gln Asp 1025 1030 1035 Cys His Glu Leu Trp Ser Lys Lys
Arg Arg Arg Gln Arg Gln Ser 1040 1045 1050 Gly Val Val Val Glu Glu
Pro Pro Pro Ser Lys Thr Ser Arg Lys 1055 1060 1065 Glu Thr Thr Ser
Gly Thr Ser Thr Glu Pro Val Lys Asn Ser Ser 1070 1075 1080 Pro Ala
Pro Pro Gln Pro Ala Pro Gly Lys Val Glu Ser Gly Ala 1085 1090 1095
Gly Asp Ala Ile Gly Leu Ala Asp Ile Thr Gln Gln Leu Asn Gln 1100
1105 1110 Ser Glu Leu Ala Val Leu Leu Asn Leu Leu Gln Ser Gln Thr
Asp 1115 1120 1125 Leu Ser Ile Pro Gln Met Ala Gln Leu Leu Asn Ile
His Ser Asn 1130 1135 1140 Pro Glu Met Gln Gln Gln Leu Glu Ala Leu
Asn Gln Ser Ile Ser 1145 1150 1155 Ala Leu Thr Glu Ala Thr Ser Gln
Gln Gln Asp Ser Glu Thr Met 1160 1165 1170 Ala Pro Glu Glu Ser Leu
Lys Glu Ala Pro Ser Ala Pro Val Ile 1175 1180 1185 Leu Pro Ser Ala
Glu Gln Thr Thr Leu Glu Ala Ser Ser Thr Pro 1190 1195 1200 Ala Asp
Met Gln Asn Ile Leu Ala Val Leu Leu Ser Gln Leu Met 1205 1210 1215
Lys Thr Gln Glu Pro Ala Gly Ser Leu Glu Glu Asn Asn Ser Asp 1220
1225 1230 Lys Asn Ser Gly Pro Gln Gly Pro Arg Arg Thr Pro Thr Met
Pro 1235 1240 1245 Gln Glu Glu Ala Ala Ala Cys Pro Pro His Ile Leu
Pro Pro Glu 1250 1255 1260 Lys Arg Pro Pro Glu Pro Pro Gly Pro Pro
Pro Pro Pro Pro Pro 1265 1270 1275 Pro Pro Leu Val Glu Gly Asp Leu
Ser Ser Ala Pro Gln Glu Leu 1280 1285 1290 Asn Pro Ala Val Thr Ala
Ala Leu Leu Gln Leu Leu Ser Gln Pro 1295 1300 1305 Glu Ala Glu Pro
Pro Gly His Leu Pro His Glu His Gln Ala Leu 1310 1315 1320 Arg Pro
Met Glu Tyr Ser Thr Arg Pro Arg Pro Asn Arg Thr Tyr 1325 1330 1335
Gly Asn Thr Asp Gly Pro Glu Thr Gly Phe Ser Ala Ile Asp Thr 1340
1345 1350 Asp Glu Arg Asn Ser Gly Pro Ala Leu Thr Glu Ser Leu Val
Gln 1355 1360 1365 Thr Leu Val Lys Asn Arg Thr Phe Ser Gly Ser Leu
Ser His Leu 1370 1375 1380 Gly Glu Ser Ser Ser Tyr Gln Gly Thr Gly
Ser Val Gln Phe Pro 1385 1390 1395 Gly Asp Gln Asp Leu Arg Phe Ala
Arg Val Pro Leu Ala Leu His 1400 1405 1410 Pro Val Val Gly Gln Pro
Phe Leu Lys Ala Glu Gly Ser Ser Asn 1415 1420 1425 Ser Val Val His
Ala Glu Thr Lys Leu Gln Asn Tyr Gly Glu Leu 1430 1435 1440 Gly Pro
Gly Thr Thr Gly Ala Ser Ser Ser Gly Ala Gly Leu His 1445 1450 1455
Trp Gly Gly Pro Thr Gln Ser Ser Ala Tyr Gly Lys Leu Tyr Arg 1460
1465 1470 Gly Pro Thr Arg Val Pro Pro Arg Gly Gly Arg Gly Arg Gly
Val 1475 1480 1485 Pro Tyr 1490 31512PRTHomo sapiens 3Met Pro Ser
Ser Ser Asp Thr Ala Leu Gly Gly Gly Gly Gly Leu Ser 1 5 10 15 Trp
Ala Glu Lys Lys Leu Glu Glu Arg Arg Lys Arg Arg Arg Phe Leu 20 25
30 Ser Pro Gln Gln Pro Pro Leu Leu Leu Pro Leu Leu Gln Pro Gln Leu
35 40 45 Leu Gln Pro Pro Pro Pro Pro Pro Pro Leu Leu Phe Leu Ala
Ala Pro 50 55 60 Gly Thr Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala
Ser Ser Ser Cys 65 70 75 80 Phe Ser Pro Gly Pro Pro Leu Glu Val Lys
Arg Leu Ala Arg Gly Lys 85 90 95 Arg Arg Ala Gly Gly Arg Gln Lys
Arg Arg Arg Gly Pro Arg Ala Gly 100 105 110 Gln Glu Ala Glu Lys Arg
Arg Val Phe Ser Leu Pro Gln Pro Gln Gln 115 120 125 Asp Gly Gly Gly
Gly Ala Ser Ser Gly Gly Gly Val Thr Pro Leu Val 130 135 140 Glu Tyr
Glu Asp Val Ser Ser Gln Ser Glu Gln Gly Leu Leu Leu Gly 145 150 155
160 Gly Ala Ser Ala Ala Thr Ala Ala Thr Ala Ala Gly Gly Thr Gly Gly
165 170 175 Ser Gly Gly Ser Pro Ala Ser Ser Ser Gly Thr Gln Arg Arg
Gly Glu 180 185 190 Gly Ser Glu Arg Arg Pro Arg Arg Asp Arg Arg Ser
Ser Ser Gly Arg 195 200 205 Ser Lys Glu Arg His Arg Glu His Arg Arg
Arg Asp Gly Gln Arg Gly 210 215 220 Gly Ser Glu Ala Ser Lys Ser Arg
Ser Arg His Ser His Ser Gly Glu 225 230 235 240 Glu Arg Ala Glu Val
Ala Lys Ser Gly Ser Ser Ser Ser Ser Gly Gly 245 250 255 Arg Arg Lys
Ser Ala Ser Ala Thr Ser Ser Ser Ser Ser Ser Arg Lys 260 265 270 Asp
Arg Asp Ser Lys Ala His Arg Ser Arg Thr Lys Ser Ser Lys Glu 275 280
285 Pro Pro Ser Ala Tyr Lys Glu Pro Pro Lys Ala Tyr Arg Glu Asp Lys
290 295 300 Thr Glu Pro Lys Ala Tyr Arg Arg Arg Arg Ser Leu Ser Pro
Leu Gly 305 310 315 320 Gly Arg Asp Asp Ser Pro Val Ser His Arg Ala
Ser Gln Ser Leu Arg 325 330 335 Ser Arg Lys Ser Pro Ser Pro Ala Gly
Gly Gly Ser Ser Pro Tyr Ser 340 345 350 Arg Arg Leu Pro Arg Ser Pro
Ser Pro Tyr Ser Arg Arg Arg Ser Pro 355 360 365 Ser Tyr Ser Arg His
Ser Ser Tyr Glu Arg Gly Gly Asp Val Ser Pro 370 375 380 Ser Pro Tyr
Ser Ser Ser Ser Trp Arg Arg Ser Arg Ser Pro Tyr Ser 385 390 395 400
Pro Val Leu Arg Arg Ser Gly Lys Ser Arg Ser Arg Ser Pro Tyr Ser 405
410 415 Ser Arg His Ser Arg Ser Arg Ser Arg His Arg Leu Ser Arg Ser
Arg 420 425 430 Ser Arg His Ser Ser Ile Ser Pro Ser Thr Leu Thr
Leu
Lys Ser Ser 435 440 445 Leu Ala Ala Glu Leu Asn Lys Asn Lys Lys Ala
Arg Ala Ala Glu Ala 450 455 460 Ala Arg Ala Ala Glu Ala Ala Lys Ala
Ala Glu Ala Thr Lys Ala Ala 465 470 475 480 Glu Ala Ala Ala Lys Ala
Ala Lys Ala Ser Asn Thr Ser Thr Pro Thr 485 490 495 Lys Gly Asn Thr
Glu Thr Ser Ala Ser Ala Ser Gln Thr Asn His Val 500 505 510 Lys Asp
Val Lys Lys Ile Lys Ile Glu His Ala Pro Ser Pro Ser Ser 515 520 525
Gly Gly Thr Leu Lys Asn Asp Lys Ala Lys Thr Lys Pro Pro Leu Gln 530
535 540 Val Thr Lys Val Glu Asn Asn Leu Ile Val Asp Lys Ala Thr Lys
Lys 545 550 555 560 Ala Val Ile Val Gly Lys Glu Ser Lys Ser Ala Ala
Thr Lys Glu Glu 565 570 575 Ser Val Ser Leu Lys Glu Lys Thr Lys Pro
Leu Thr Pro Ser Ile Gly 580 585 590 Ala Lys Glu Lys Glu Gln His Val
Ala Leu Val Thr Ser Thr Leu Pro 595 600 605 Pro Leu Pro Leu Pro Pro
Met Leu Pro Glu Asp Lys Glu Ala Asp Ser 610 615 620 Leu Arg Gly Asn
Ile Ser Val Lys Ala Val Lys Lys Glu Val Glu Lys 625 630 635 640 Lys
Leu Arg Cys Leu Leu Ala Asp Leu Pro Leu Pro Pro Glu Leu Pro 645 650
655 Gly Gly Asp Asp Leu Ser Lys Ser Pro Glu Glu Lys Lys Thr Ala Thr
660 665 670 Gln Leu His Ser Lys Arg Arg Pro Lys Ile Cys Gly Pro Arg
Tyr Gly 675 680 685 Glu Thr Lys Glu Lys Asp Ile Asp Trp Gly Lys Arg
Cys Val Asp Lys 690 695 700 Phe Asp Ile Ile Gly Ile Ile Gly Glu Gly
Thr Tyr Gly Gln Val Tyr 705 710 715 720 Lys Ala Arg Asp Lys Asp Thr
Gly Glu Met Val Ala Leu Lys Lys Val 725 730 735 Arg Leu Asp Asn Glu
Lys Glu Gly Phe Pro Ile Thr Ala Ile Arg Glu 740 745 750 Ile Lys Ile
Leu Arg Gln Leu Thr His Gln Ser Ile Ile Asn Met Lys 755 760 765 Glu
Ile Val Thr Asp Lys Glu Asp Ala Leu Asp Phe Lys Lys Asp Lys 770 775
780 Gly Ala Phe Tyr Leu Val Phe Glu Tyr Met Asp His Asp Leu Met Gly
785 790 795 800 Leu Leu Glu Ser Gly Leu Val His Phe Asn Glu Asn His
Ile Lys Ser 805 810 815 Phe Met Arg Gln Leu Met Glu Gly Leu Asp Tyr
Cys His Lys Lys Asn 820 825 830 Phe Leu His Arg Asp Ile Lys Cys Ser
Asn Ile Leu Leu Asn Asn Arg 835 840 845 Gly Gln Ile Lys Leu Ala Asp
Phe Gly Leu Ala Arg Leu Tyr Ser Ser 850 855 860 Glu Glu Ser Arg Pro
Tyr Thr Asn Lys Val Ile Thr Leu Trp Tyr Arg 865 870 875 880 Pro Pro
Glu Leu Leu Leu Gly Glu Glu Arg Tyr Thr Pro Ala Ile Asp 885 890 895
Val Trp Ser Cys Gly Cys Ile Leu Gly Glu Leu Phe Thr Lys Lys Pro 900
905 910 Ile Phe Gln Ala Asn Gln Glu Leu Ala Gln Leu Glu Leu Ile Ser
Arg 915 920 925 Ile Cys Gly Ser Pro Cys Pro Ala Val Trp Pro Asp Val
Ile Lys Leu 930 935 940 Pro Tyr Phe Asn Thr Met Lys Pro Lys Lys Gln
Tyr Arg Arg Lys Leu 945 950 955 960 Arg Glu Glu Phe Val Phe Ile Pro
Ala Ala Ala Leu Asp Leu Phe Asp 965 970 975 Tyr Met Leu Ala Leu Asp
Pro Ser Lys Arg Cys Thr Ala Glu Gln Ala 980 985 990 Leu Gln Cys Glu
Phe Leu Arg Asp Val Glu Pro Ser Lys Met Pro Pro 995 1000 1005 Pro
Asp Leu Pro Leu Trp Gln Asp Cys His Glu Leu Trp Ser Lys 1010 1015
1020 Lys Arg Arg Arg Gln Lys Gln Met Gly Met Thr Asp Asp Val Ser
1025 1030 1035 Thr Ile Lys Ala Pro Arg Lys Asp Leu Ser Leu Gly Leu
Asp Asp 1040 1045 1050 Ser Arg Thr Asn Thr Pro Gln Gly Val Leu Pro
Ser Ser Gln Leu 1055 1060 1065 Lys Ser Gln Gly Ser Ser Asn Val Ala
Pro Val Lys Thr Gly Pro 1070 1075 1080 Gly Gln His Leu Asn His Ser
Glu Leu Ala Ile Leu Leu Asn Leu 1085 1090 1095 Leu Gln Ser Lys Thr
Ser Val Asn Met Ala Asp Phe Val Gln Val 1100 1105 1110 Leu Asn Ile
Lys Val Asn Ser Glu Thr Gln Gln Gln Leu Asn Lys 1115 1120 1125 Ile
Asn Leu Pro Ala Gly Ile Leu Ala Thr Gly Glu Lys Gln Thr 1130 1135
1140 Asp Pro Ser Thr Pro Gln Gln Glu Ser Ser Lys Pro Leu Gly Gly
1145 1150 1155 Ile Gln Pro Ser Ser Gln Thr Ile Gln Pro Lys Val Glu
Thr Asp 1160 1165 1170 Ala Ala Gln Ala Ala Val Gln Ser Ala Phe Ala
Val Leu Leu Thr 1175 1180 1185 Gln Leu Ile Lys Ala Gln Gln Ser Lys
Gln Lys Asp Val Leu Leu 1190 1195 1200 Glu Glu Arg Glu Asn Gly Ser
Gly His Glu Ala Ser Leu Gln Leu 1205 1210 1215 Arg Pro Pro Pro Glu
Pro Ser Thr Pro Val Ser Gly Gln Asp Asp 1220 1225 1230 Leu Ile Gln
His Gln Asp Met Arg Ile Leu Glu Leu Thr Pro Glu 1235 1240 1245 Pro
Asp Arg Pro Arg Ile Leu Pro Pro Asp Gln Arg Pro Pro Glu 1250 1255
1260 Pro Pro Glu Pro Pro Pro Val Thr Glu Glu Asp Leu Asp Tyr Arg
1265 1270 1275 Thr Glu Asn Gln His Val Pro Thr Thr Ser Ser Ser Leu
Thr Asp 1280 1285 1290 Pro His Ala Gly Val Lys Ala Ala Leu Leu Gln
Leu Leu Ala Gln 1295 1300 1305 His Gln Pro Gln Asp Asp Pro Lys Arg
Glu Gly Gly Ile Asp Tyr 1310 1315 1320 Gln Ala Gly Asp Thr Tyr Val
Ser Thr Ser Asp Tyr Lys Asp Asn 1325 1330 1335 Phe Gly Ser Ser Ser
Phe Ser Ser Ala Pro Tyr Val Ser Asn Asp 1340 1345 1350 Gly Leu Gly
Ser Ser Ser Ala Pro Pro Leu Glu Arg Arg Ser Phe 1355 1360 1365 Ile
Gly Asn Ser Asp Ile Gln Ser Leu Asp Asn Tyr Ser Thr Ala 1370 1375
1380 Ser Ser His Ser Gly Gly Pro Pro Gln Pro Ser Ala Phe Ser Glu
1385 1390 1395 Ser Phe Pro Ser Ser Val Ala Gly Tyr Gly Asp Ile Tyr
Leu Asn 1400 1405 1410 Ala Gly Pro Met Leu Phe Ser Gly Asp Lys Asp
His Arg Phe Glu 1415 1420 1425 Tyr Ser His Gly Pro Ile Ala Val Leu
Ala Asn Ser Ser Asp Pro 1430 1435 1440 Ser Thr Gly Pro Glu Ser Thr
His Pro Leu Pro Ala Lys Met His 1445 1450 1455 Asn Tyr Asn Tyr Gly
Gly Asn Leu Gln Glu Asn Pro Ser Gly Pro 1460 1465 1470 Ser Leu Met
His Gly Gln Thr Trp Thr Ser Pro Ala Gln Gly Pro 1475 1480 1485 Gly
Tyr Ser Gln Gly Tyr Arg Gly His Ile Ser Thr Ser Thr Gly 1490 1495
1500 Arg Gly Arg Gly Arg Gly Leu Pro Tyr 1505 1510 420DNAArtificial
SequenceSynthetic Polynucleotide 4ggcaggattg ccatgagttg
20520DNAArtificial SequenceSynthetic Polynucleotide 5ggcaagattg
tcatgagtta 20




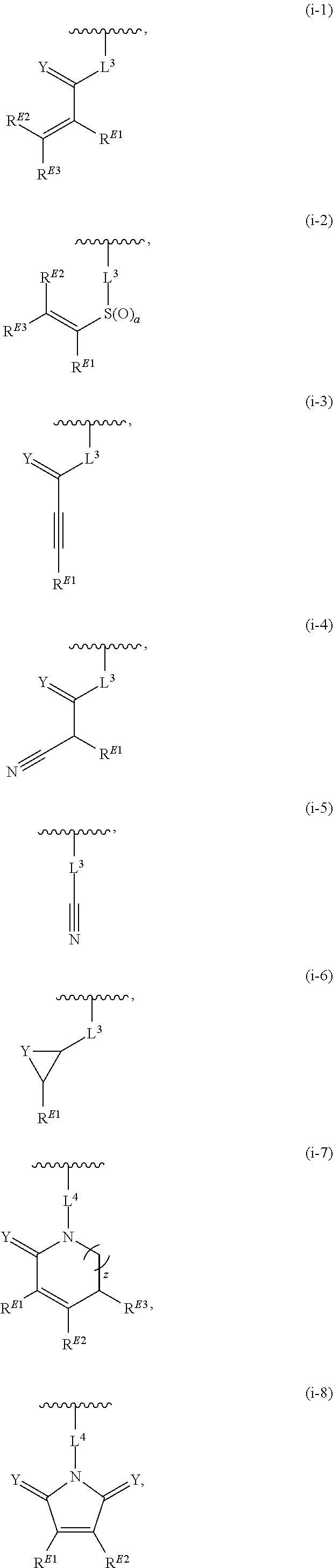








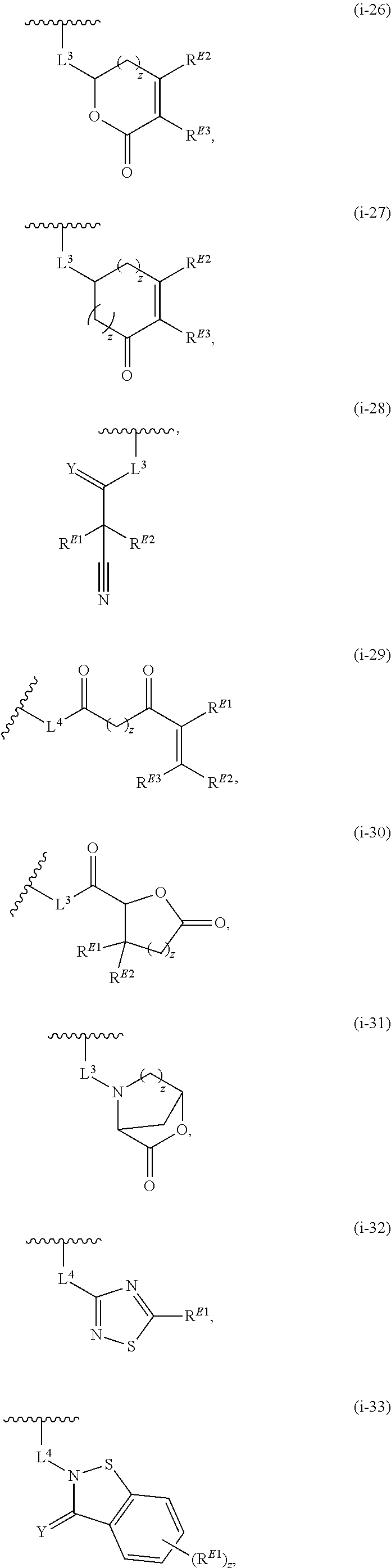














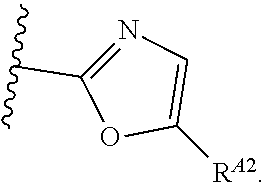










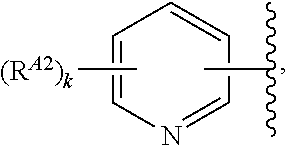

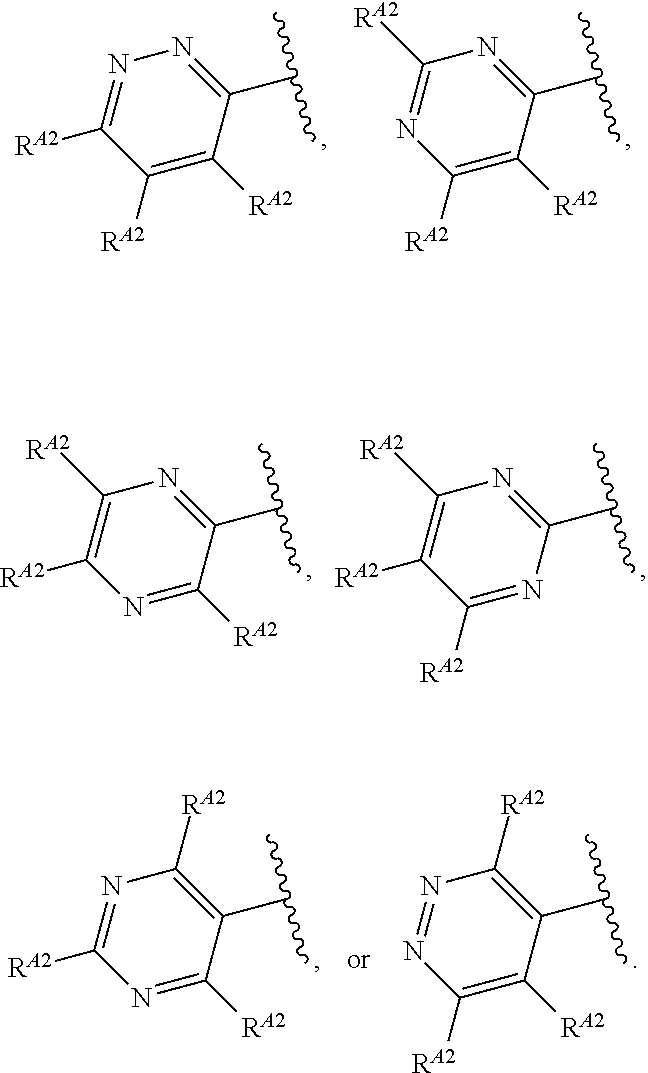
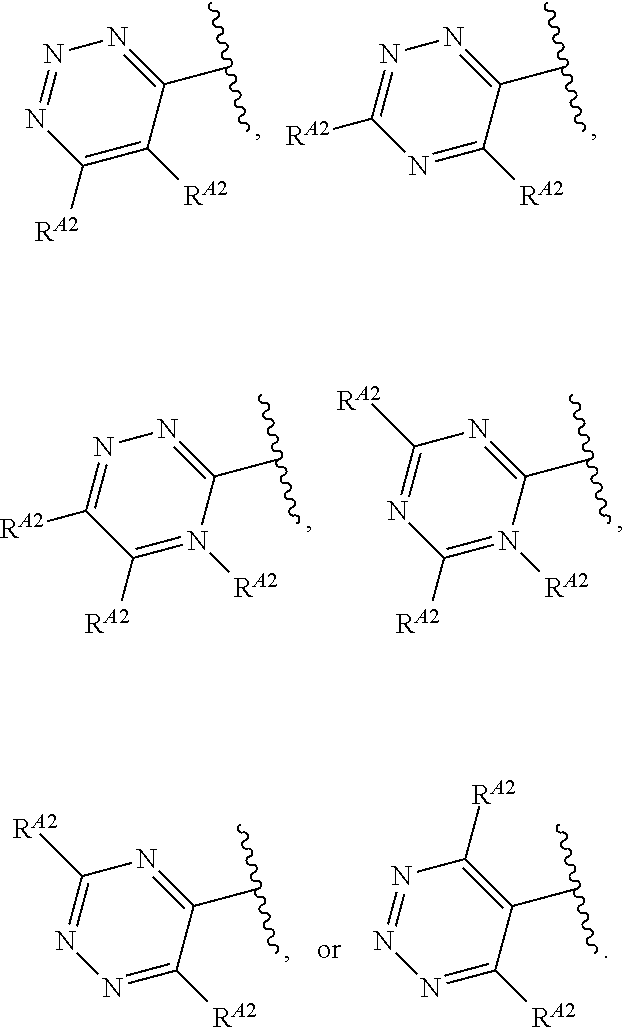








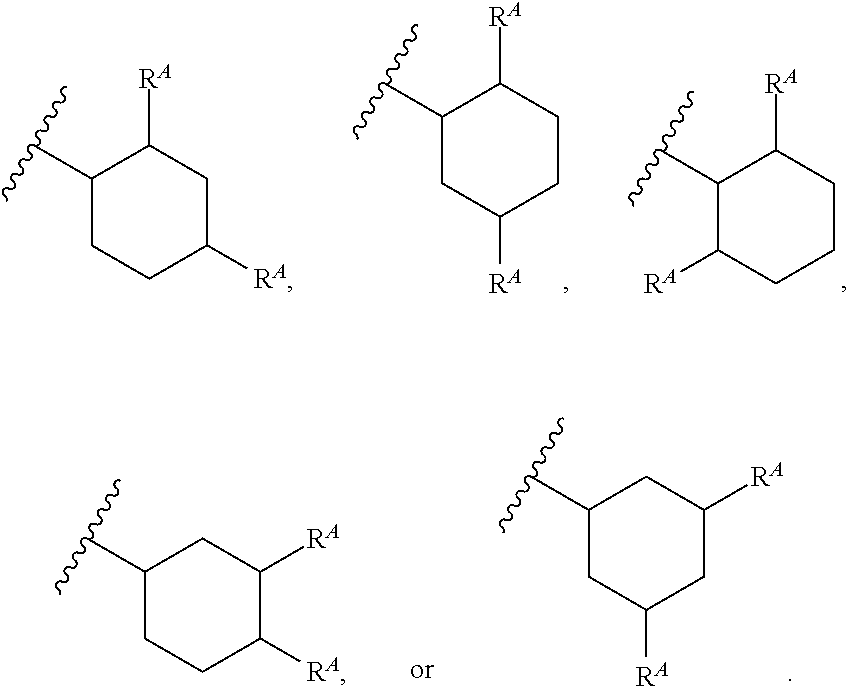

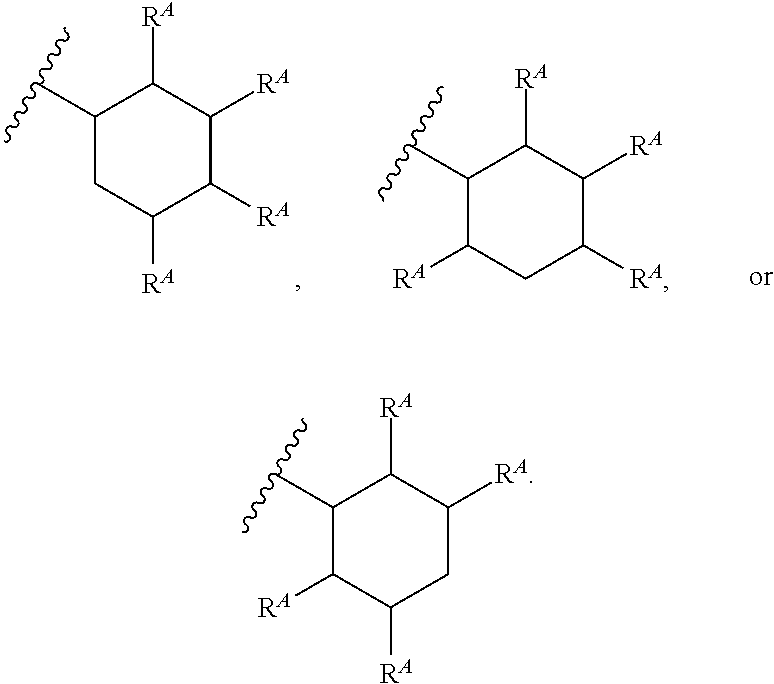


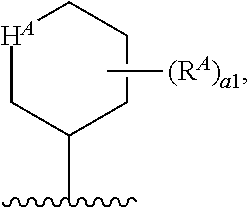


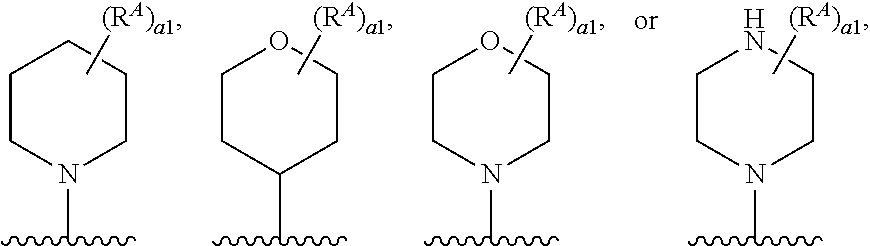



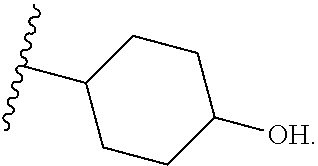















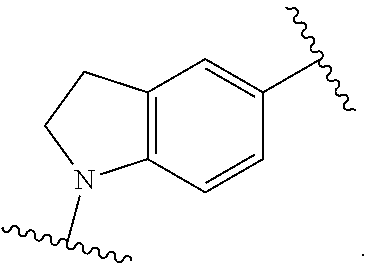












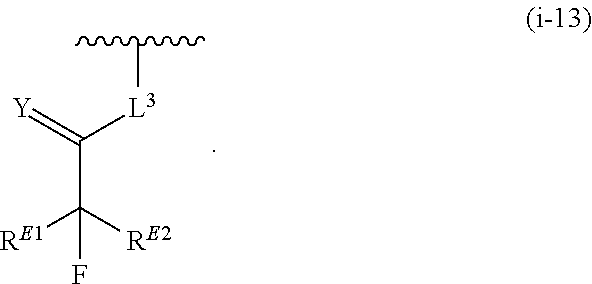





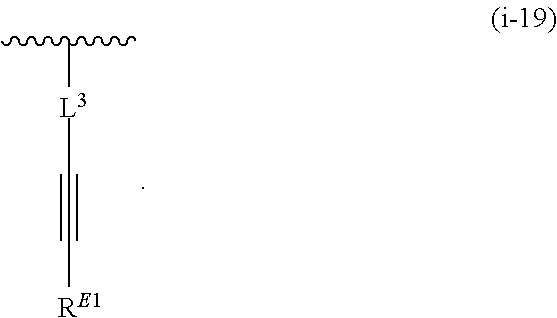



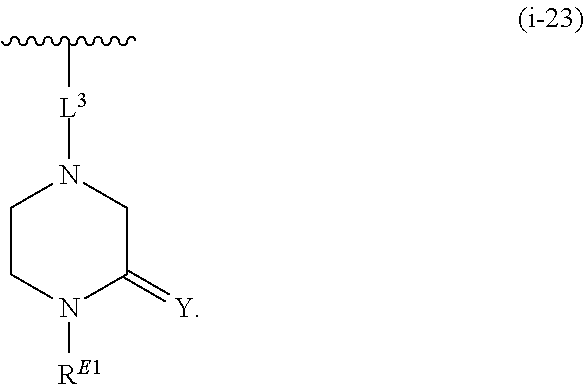








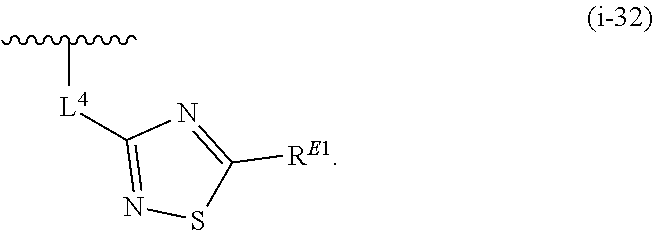







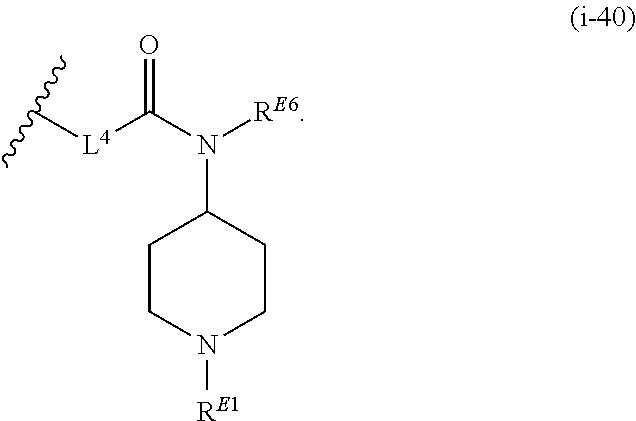


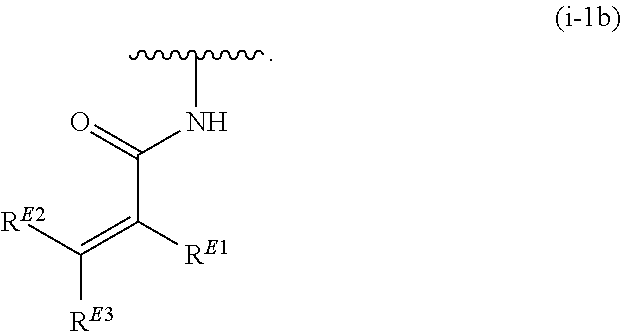





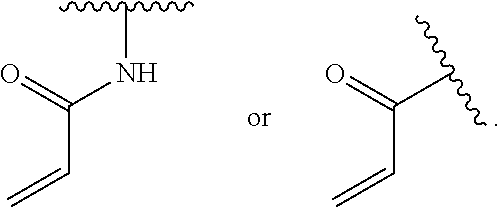



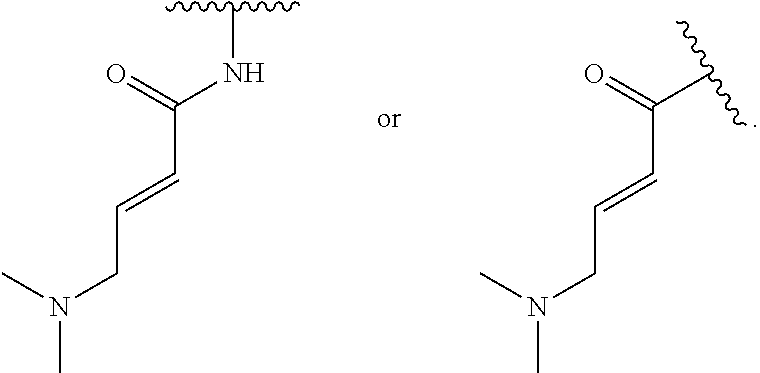




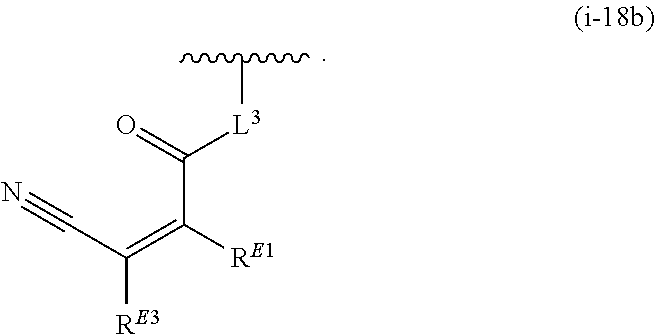


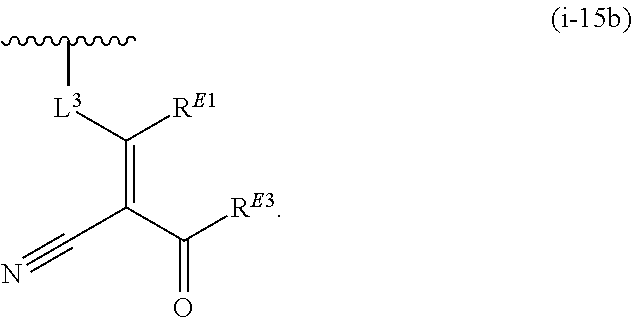
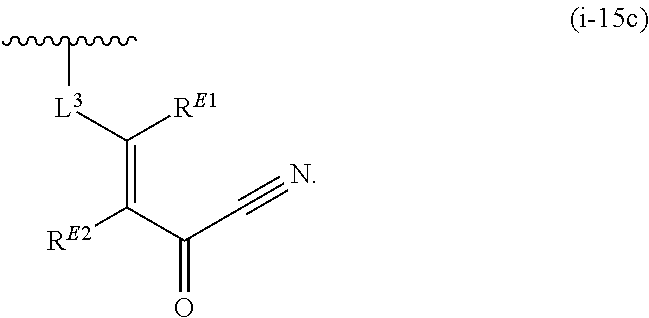





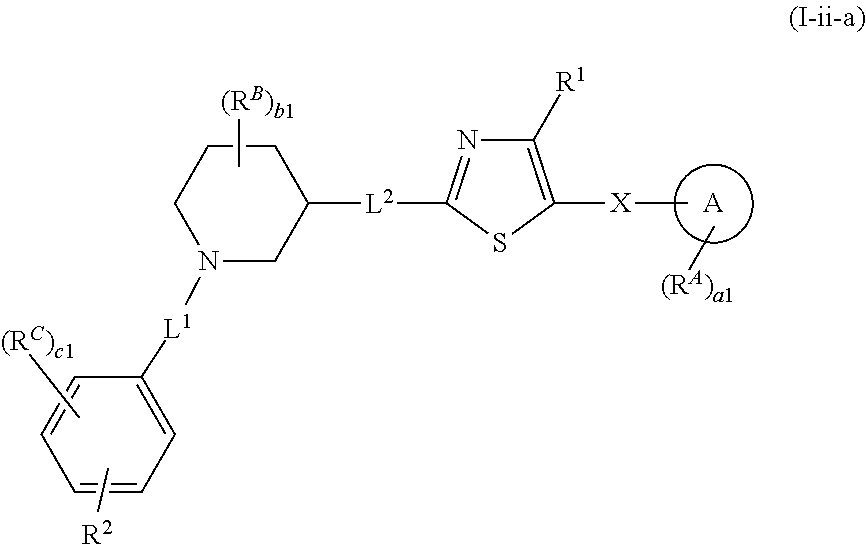

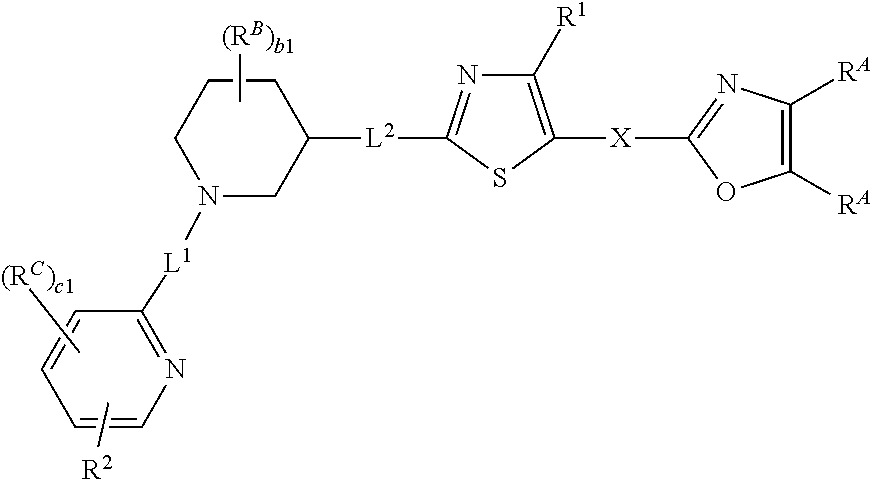

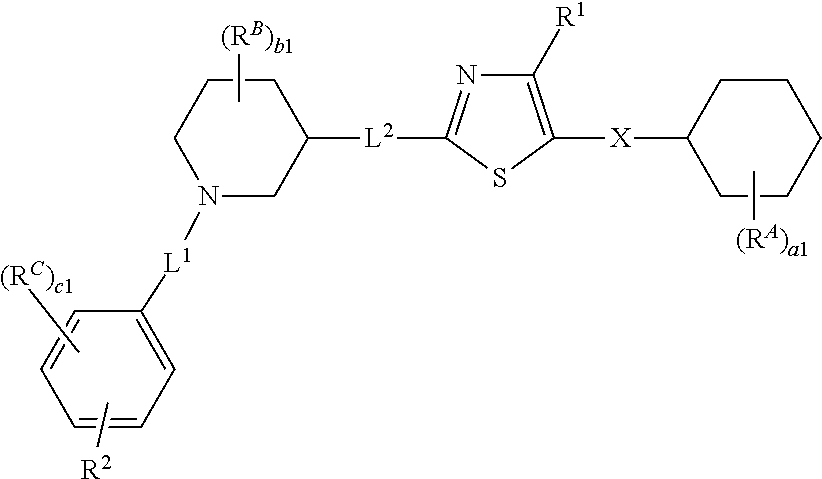





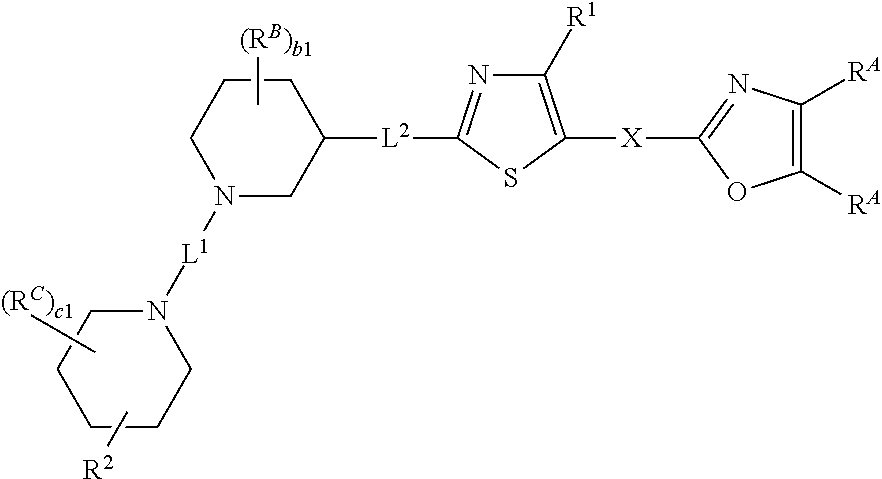

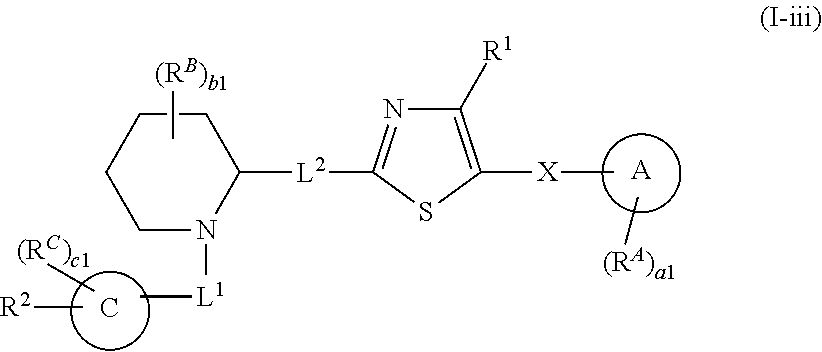


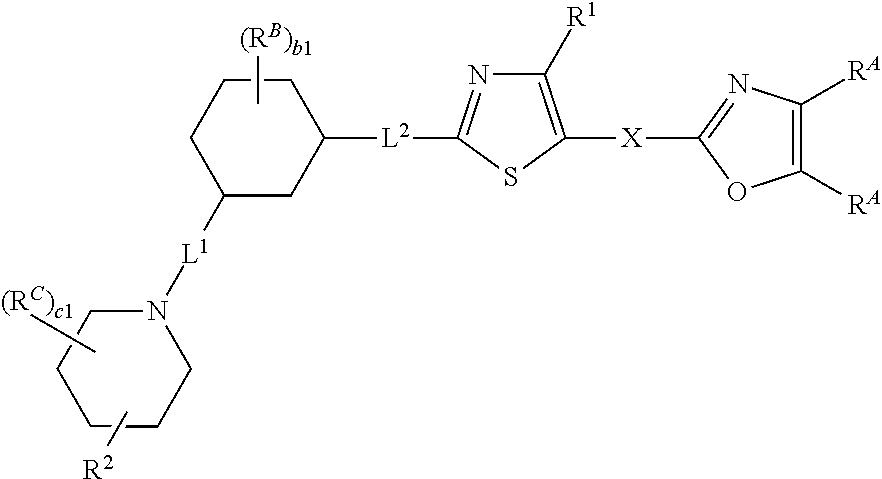



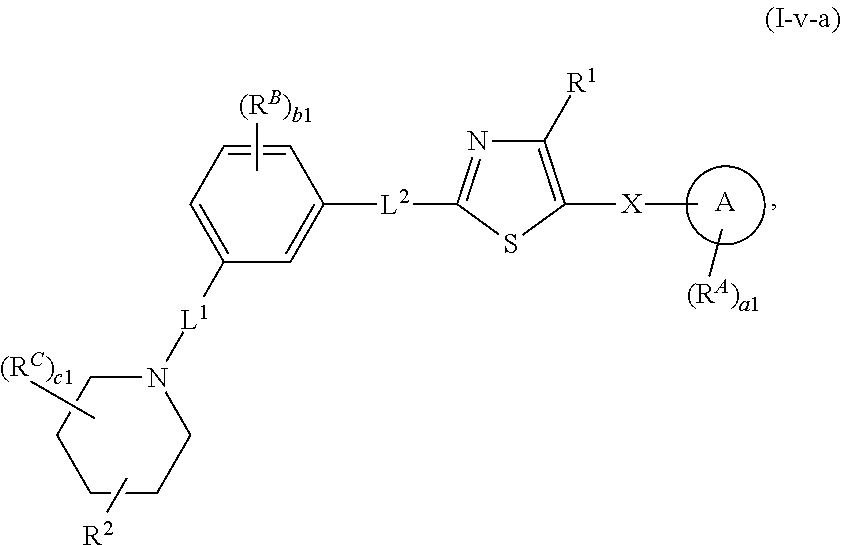





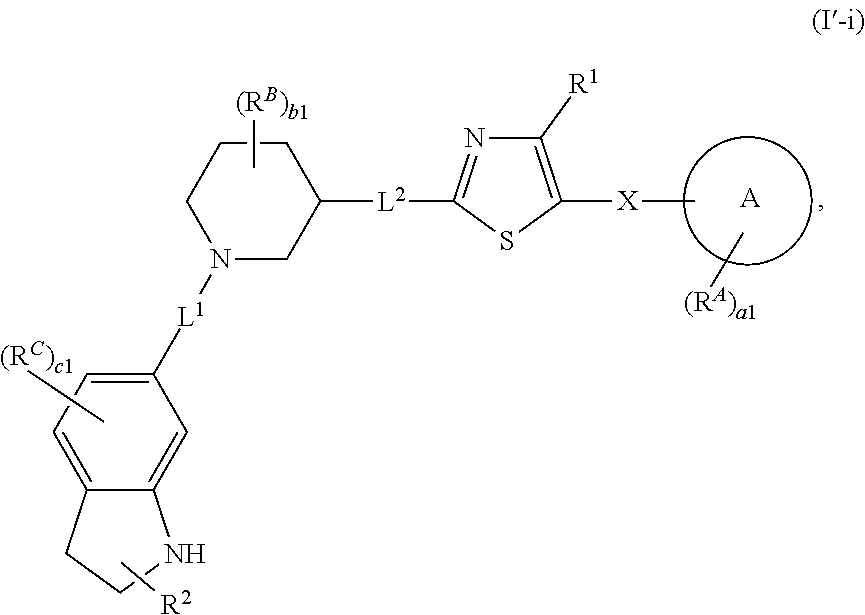













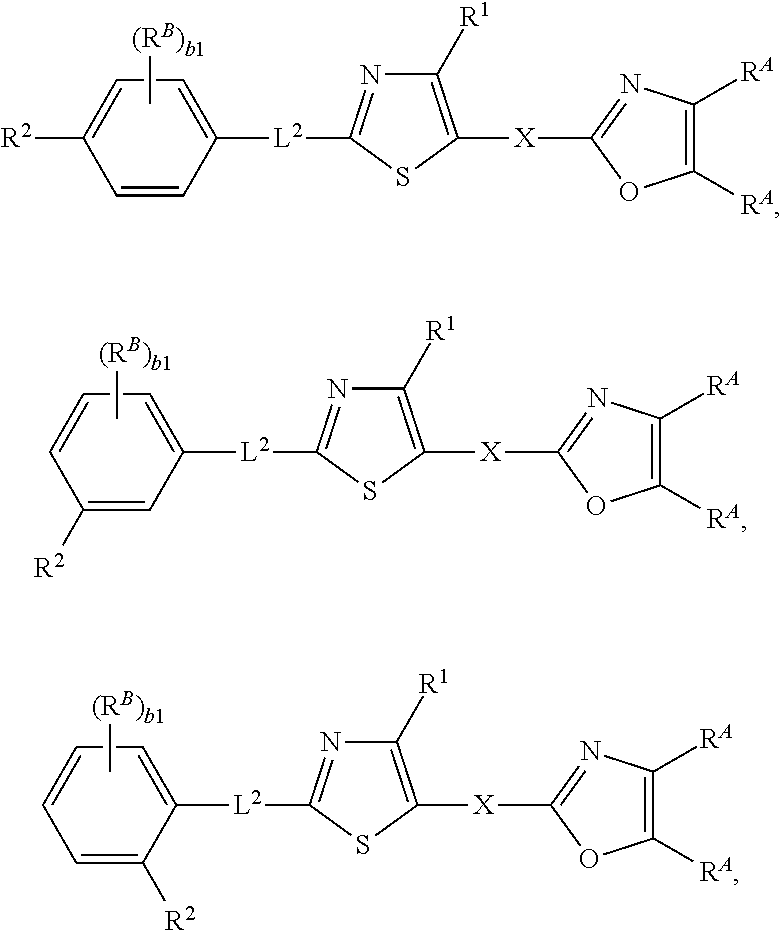


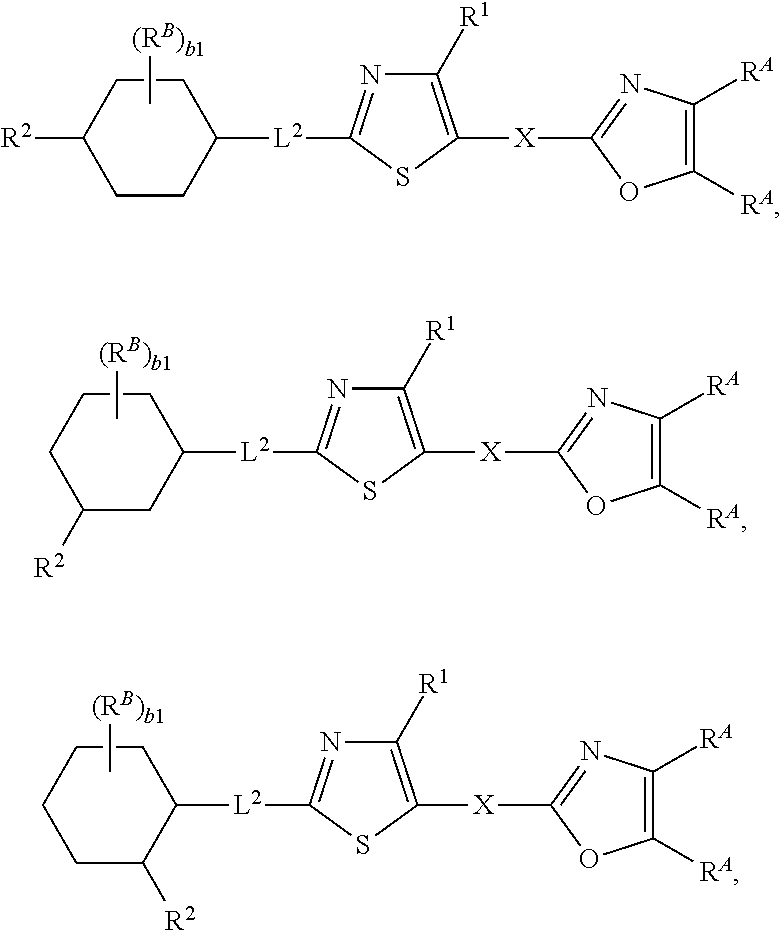


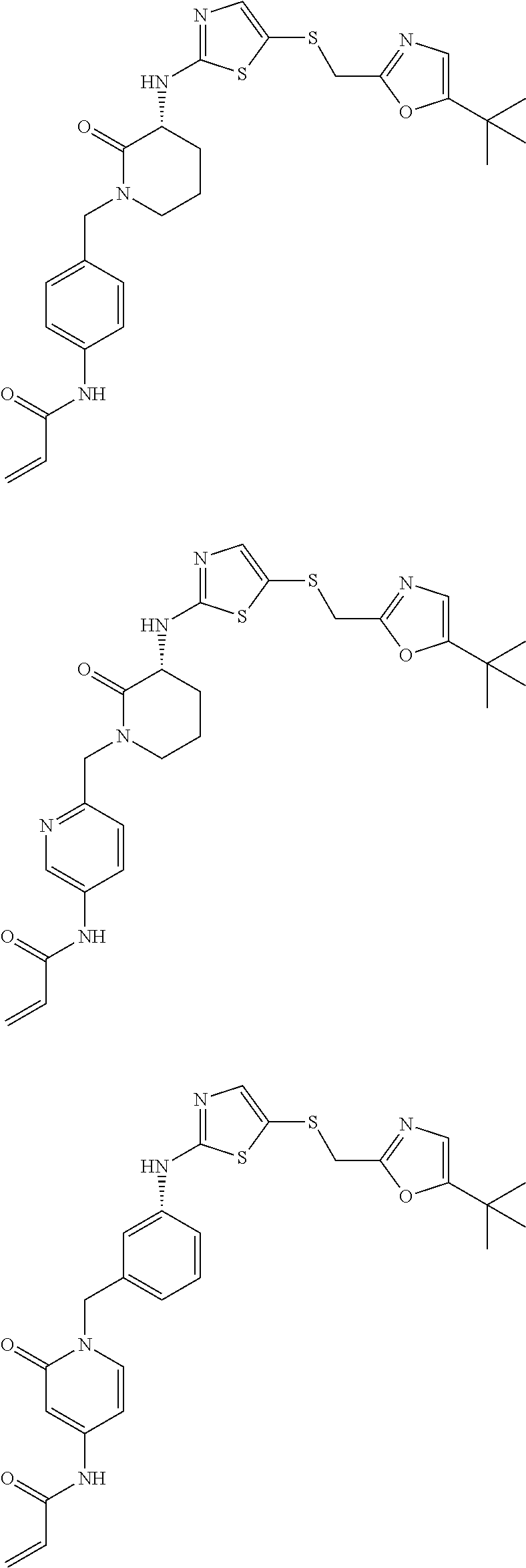

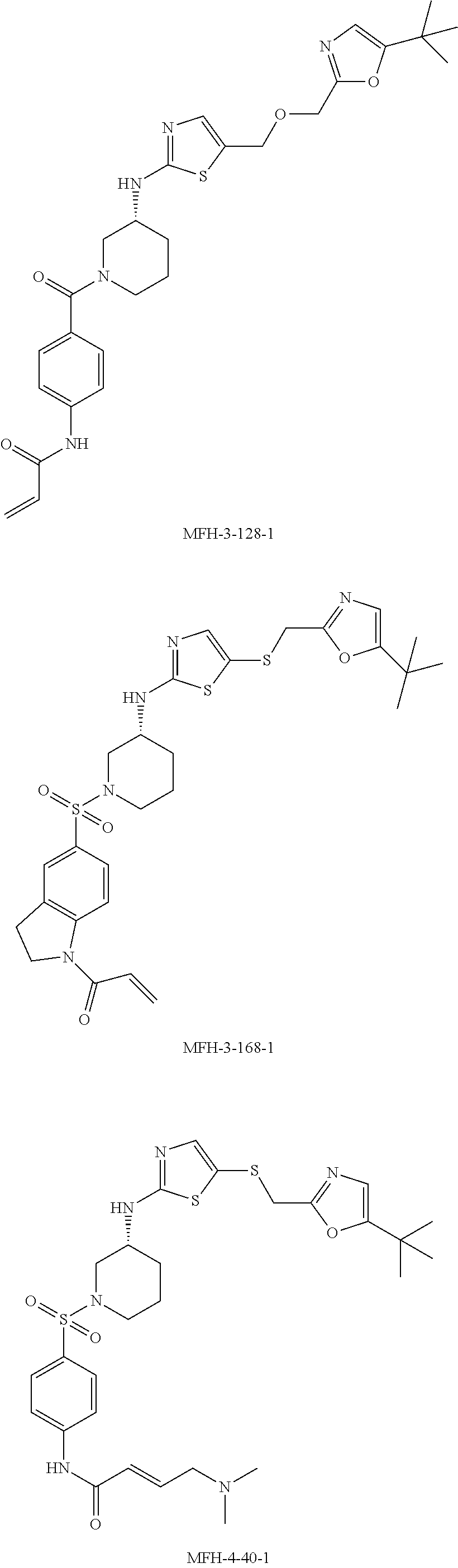





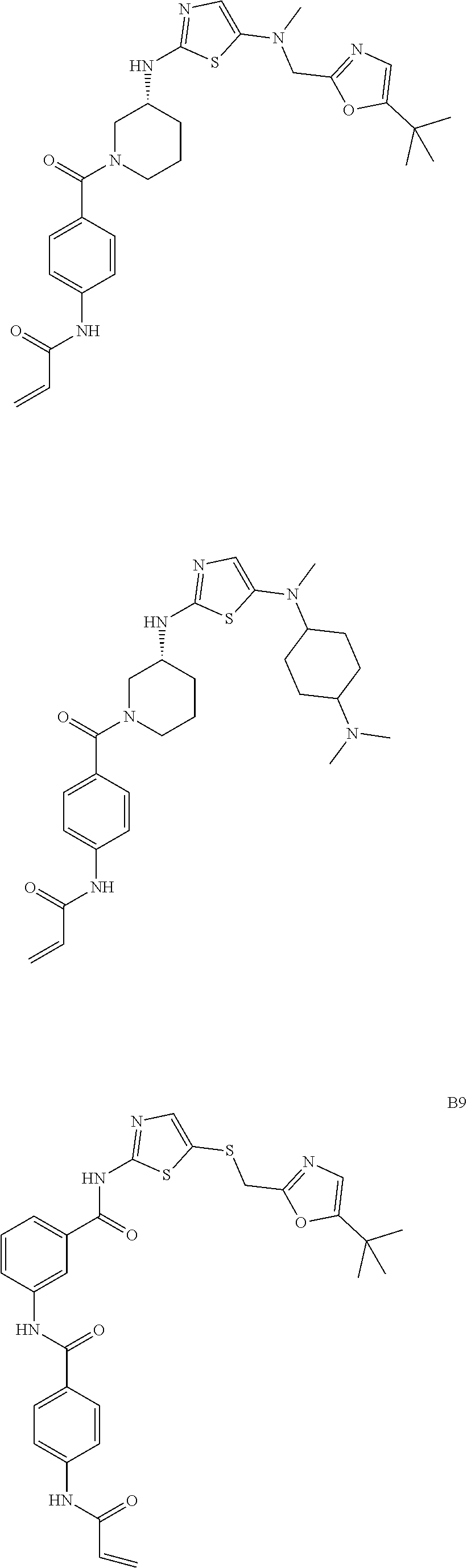

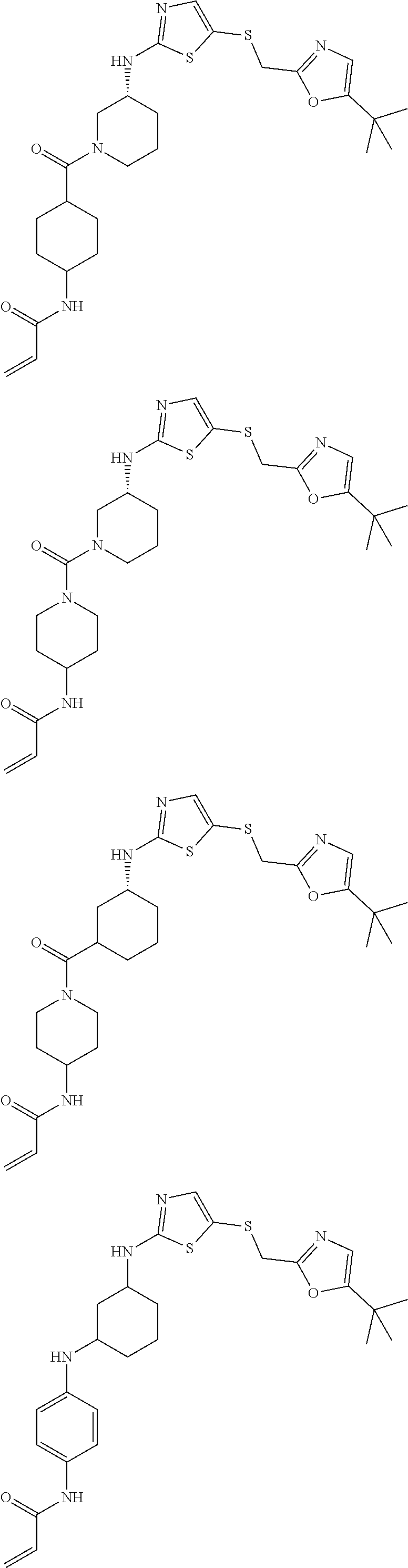


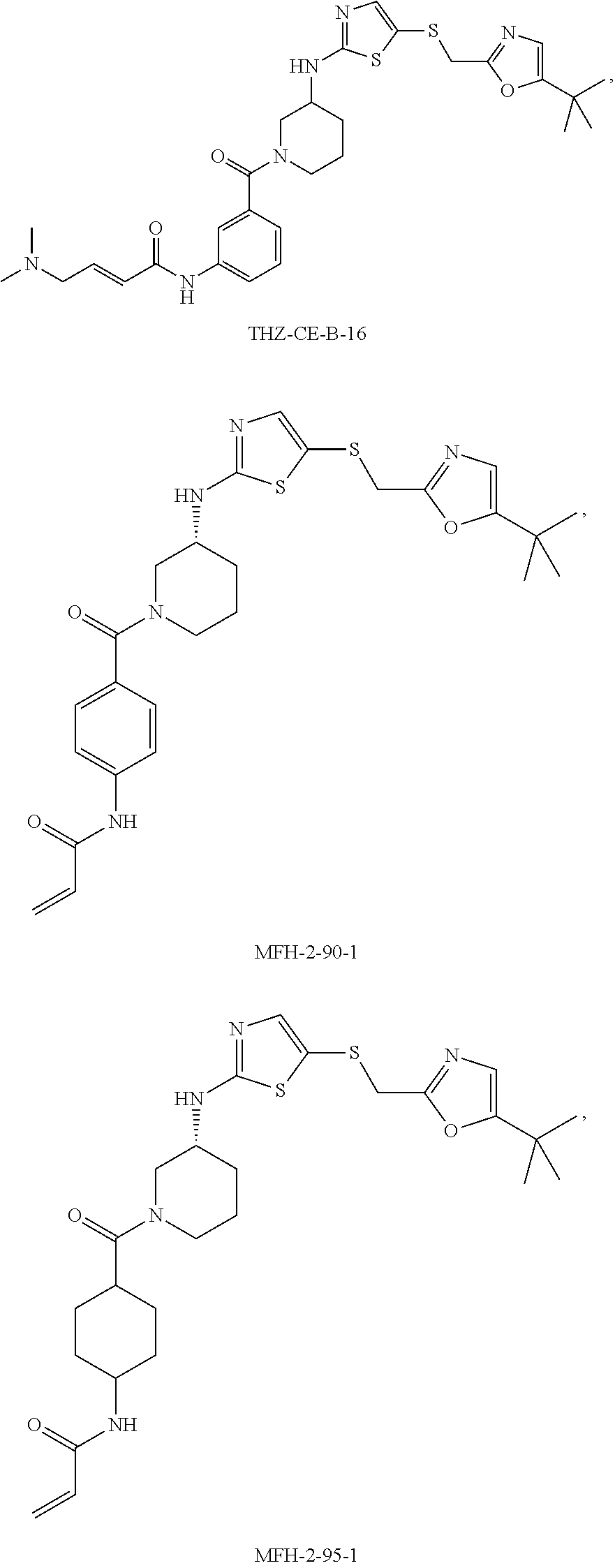
















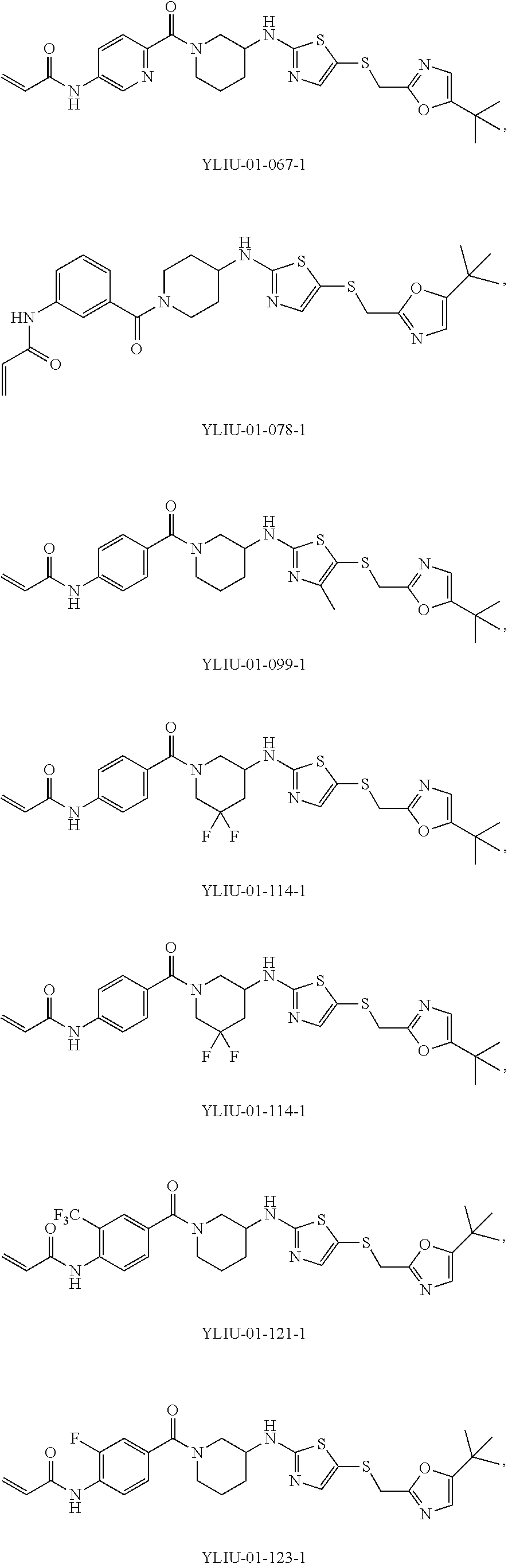


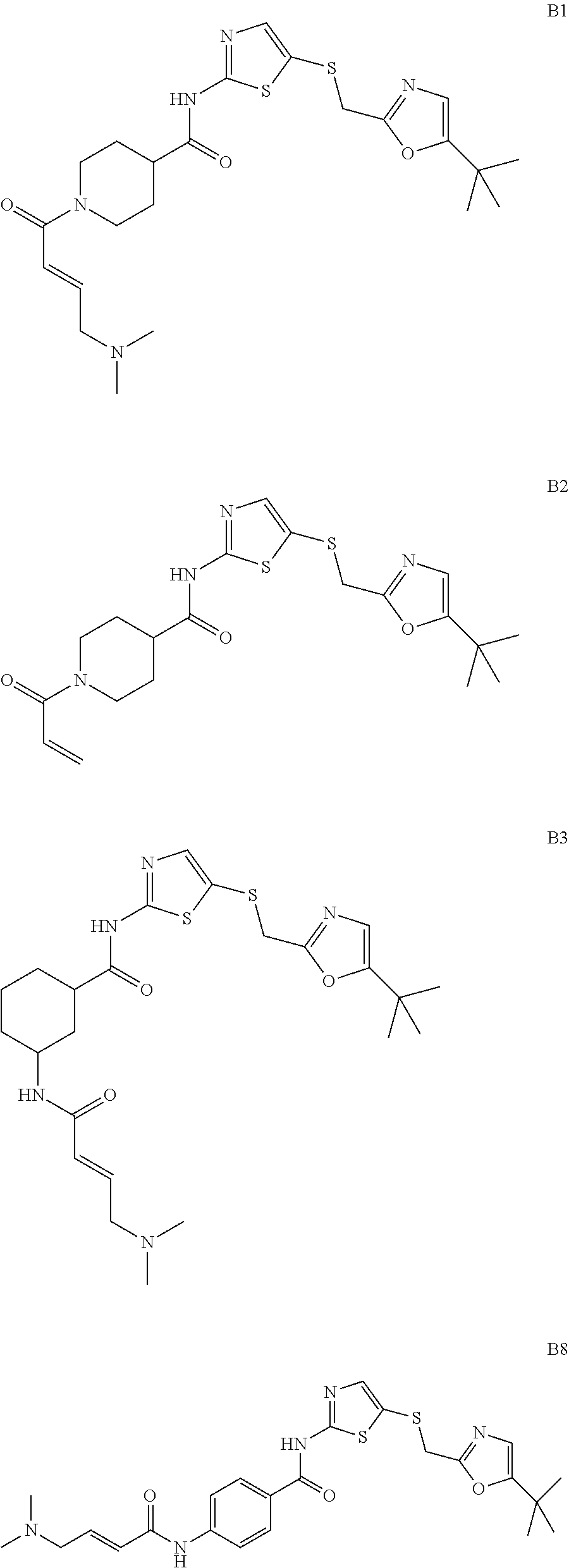


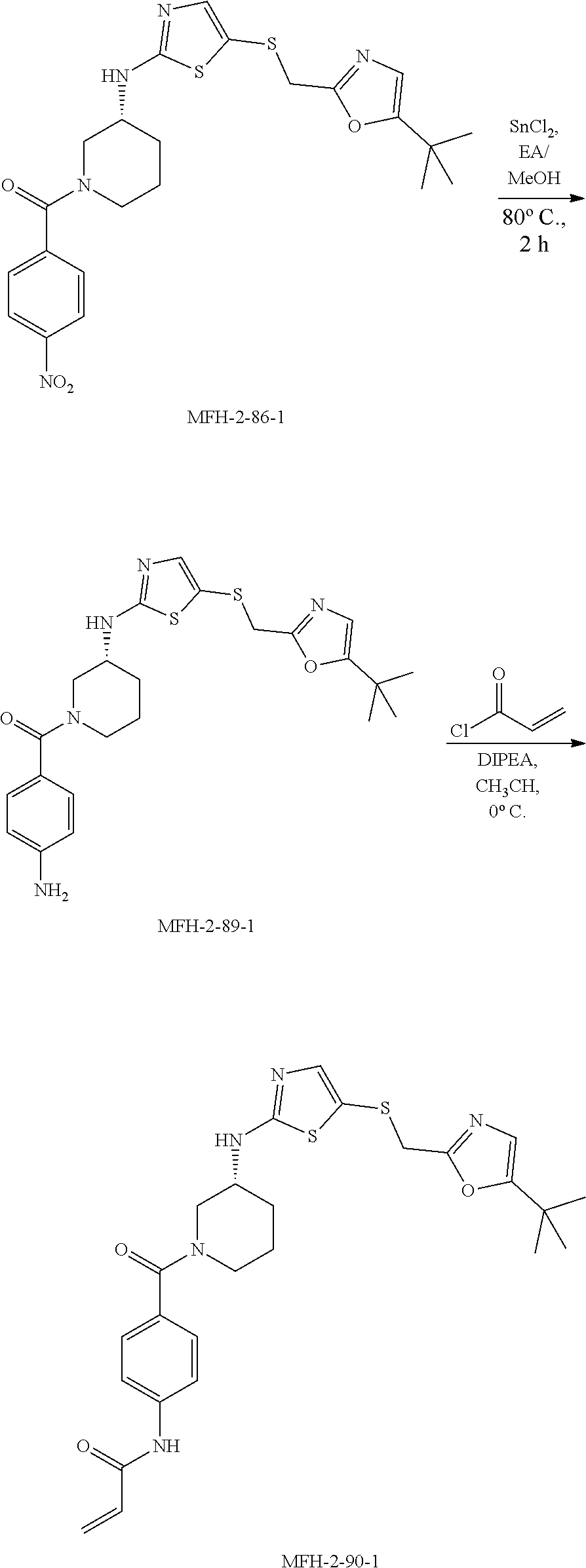




















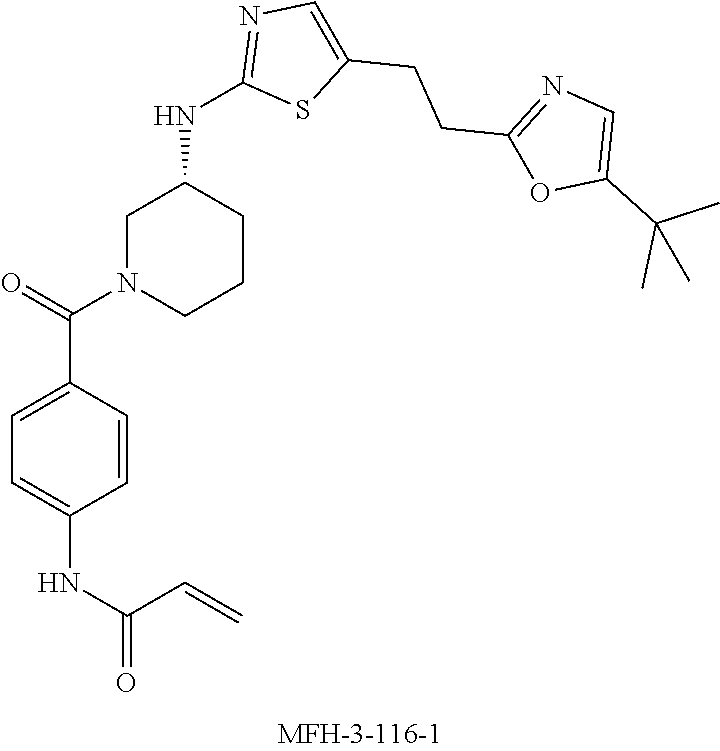


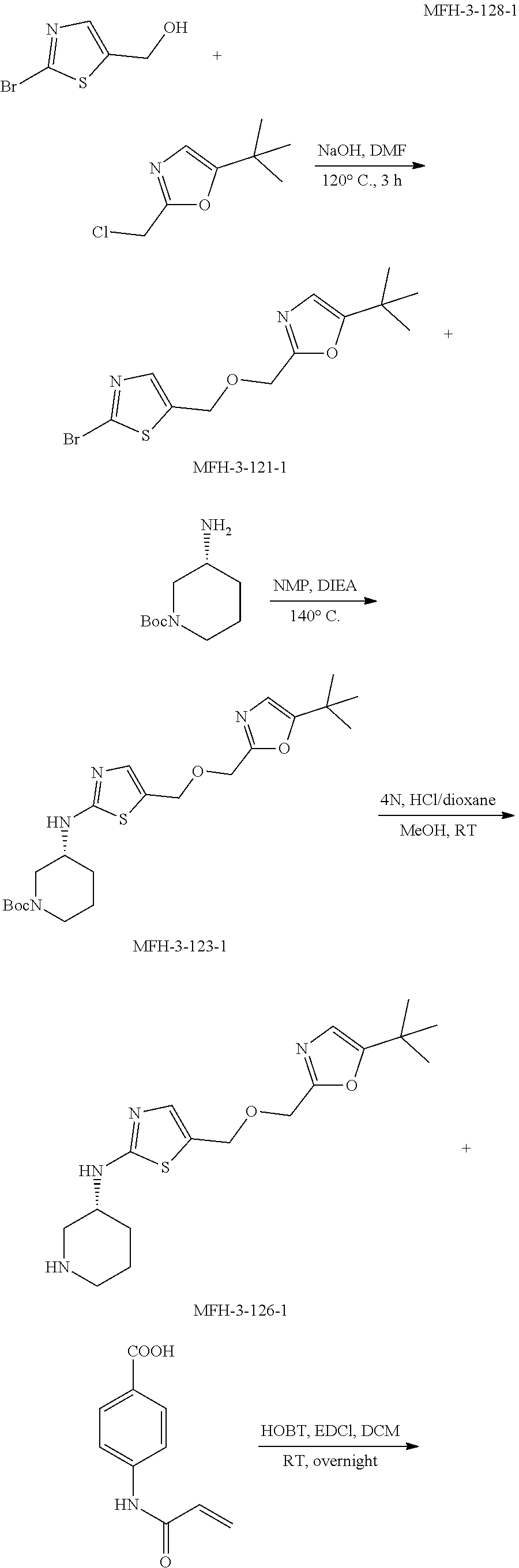







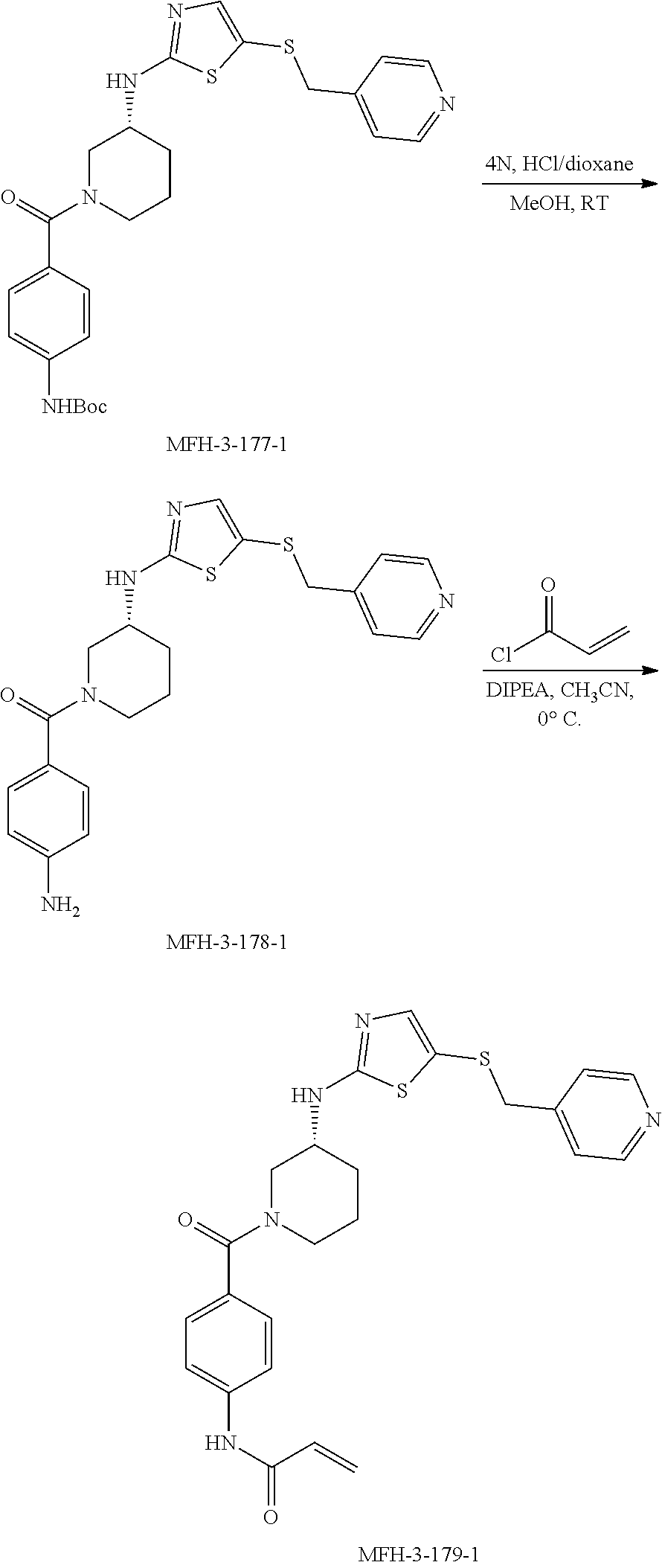

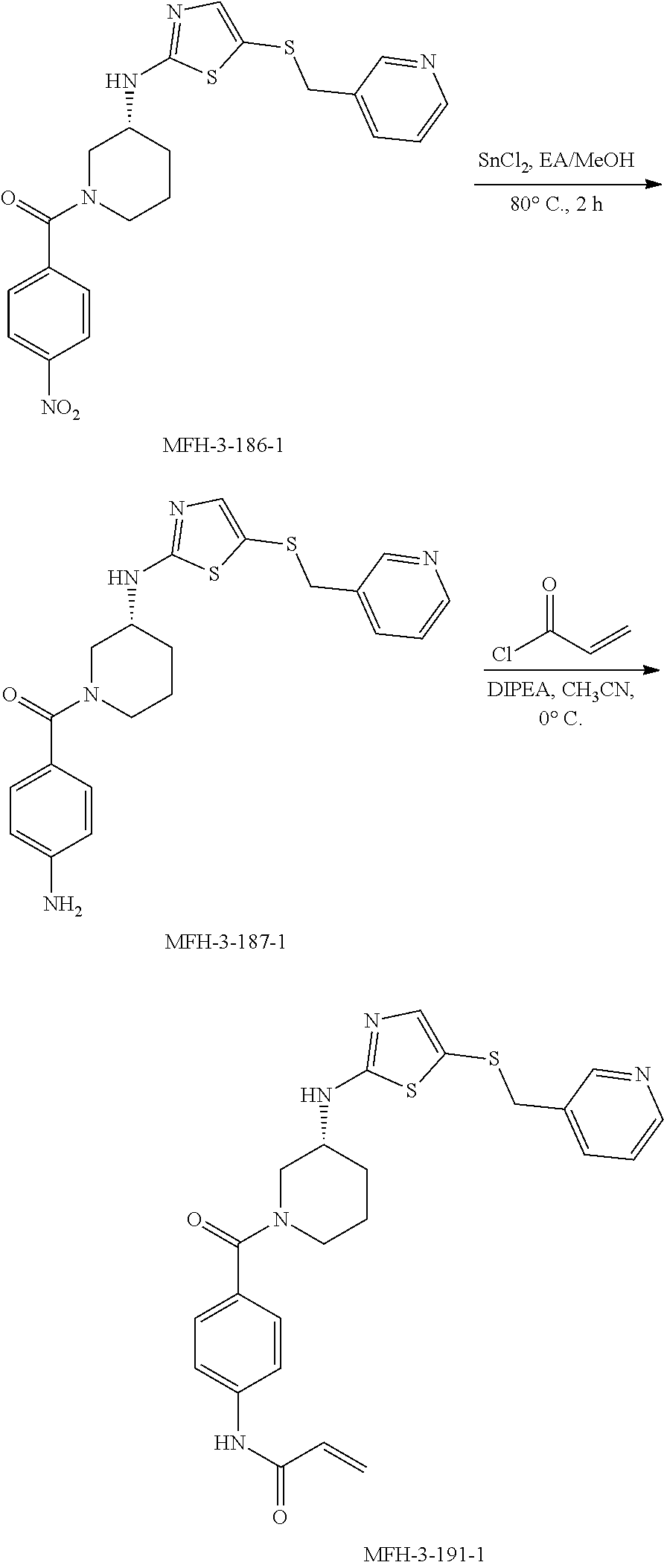
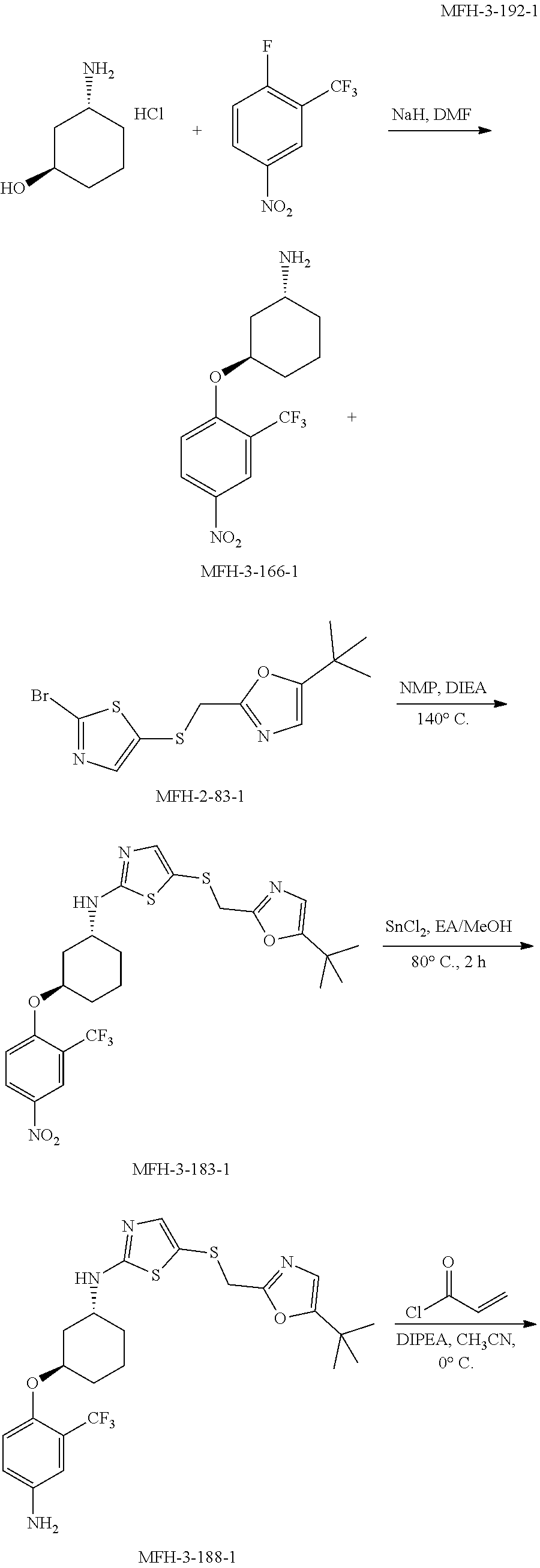







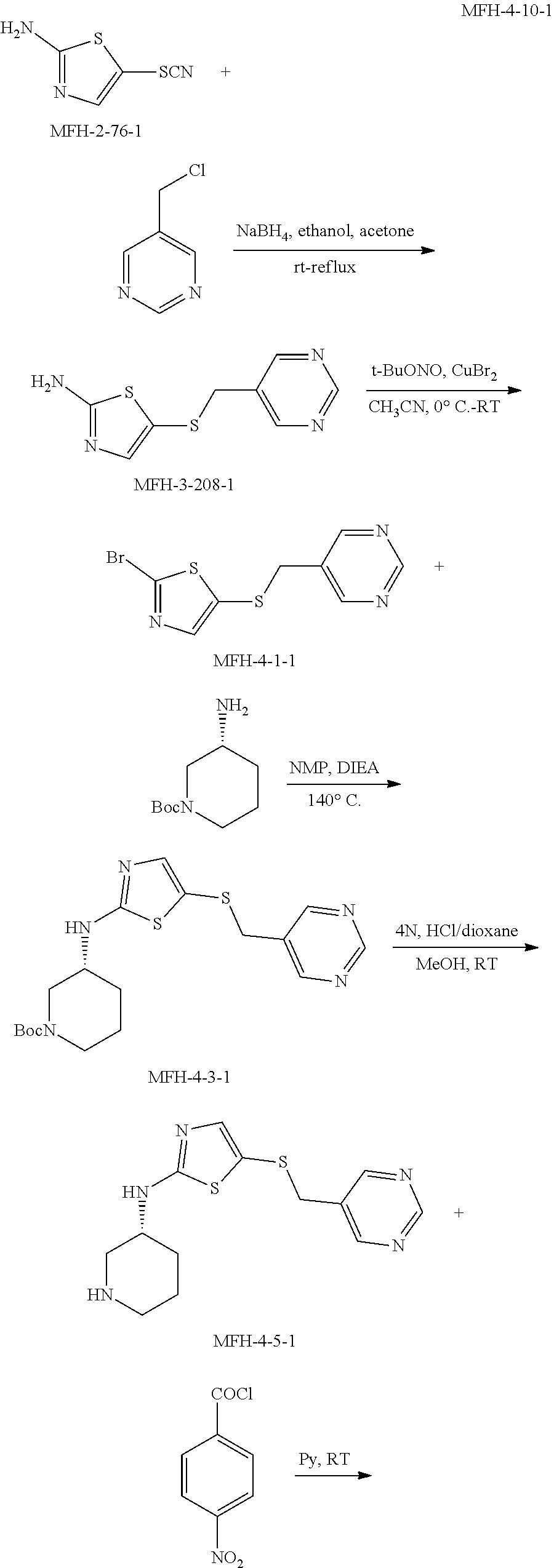




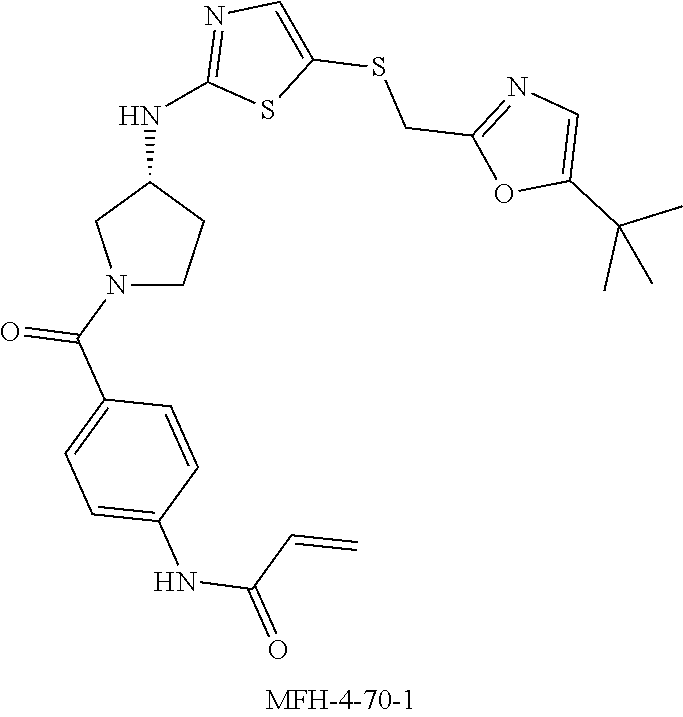
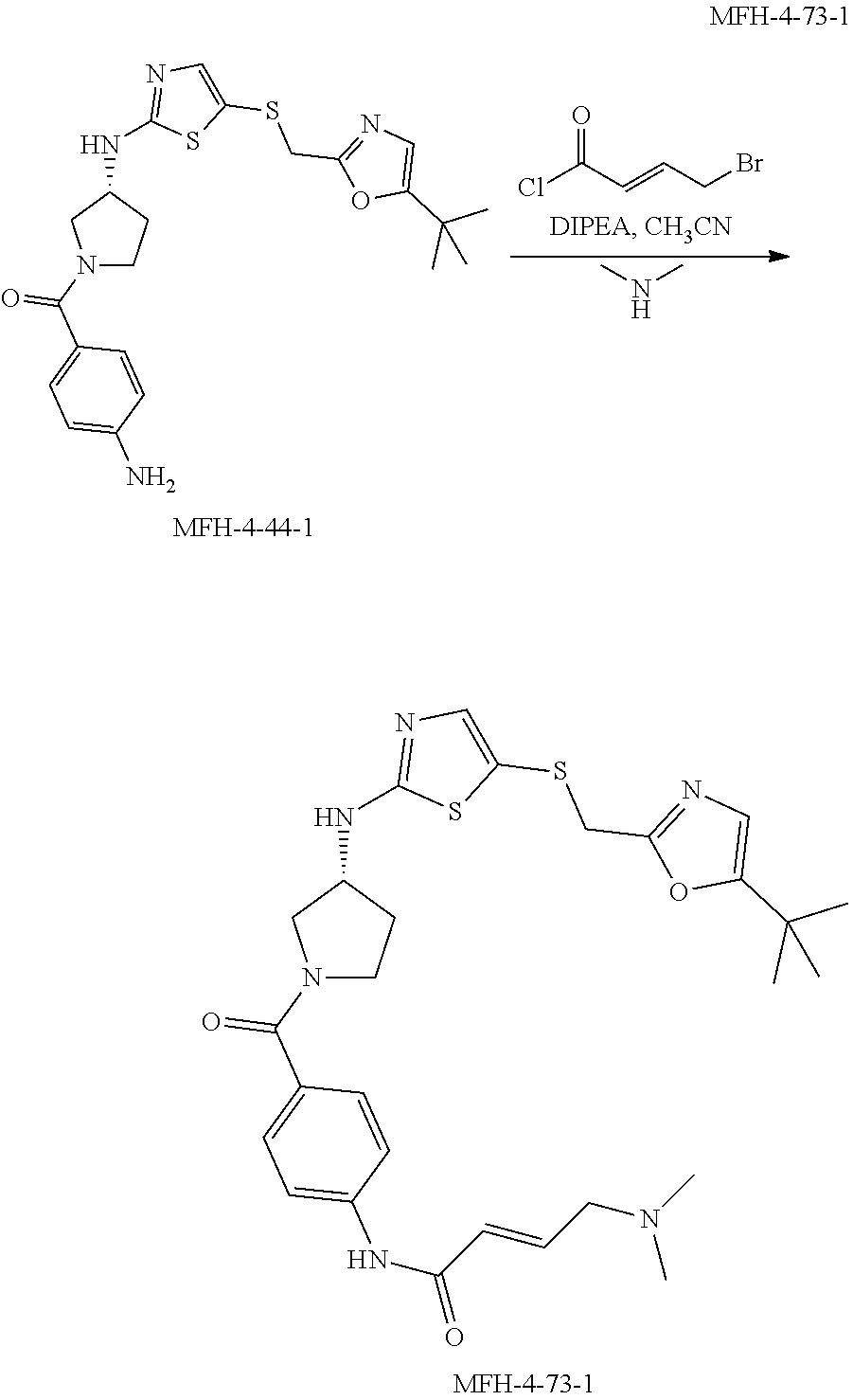


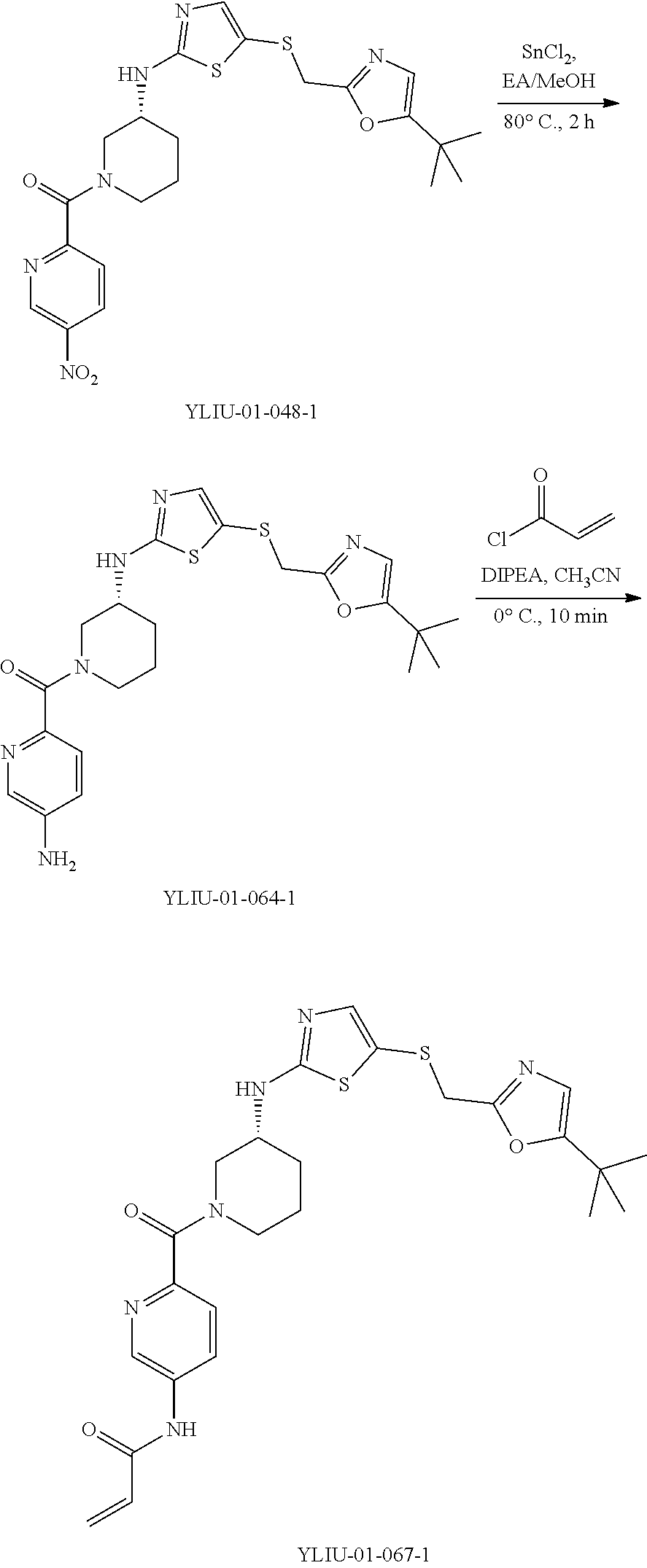





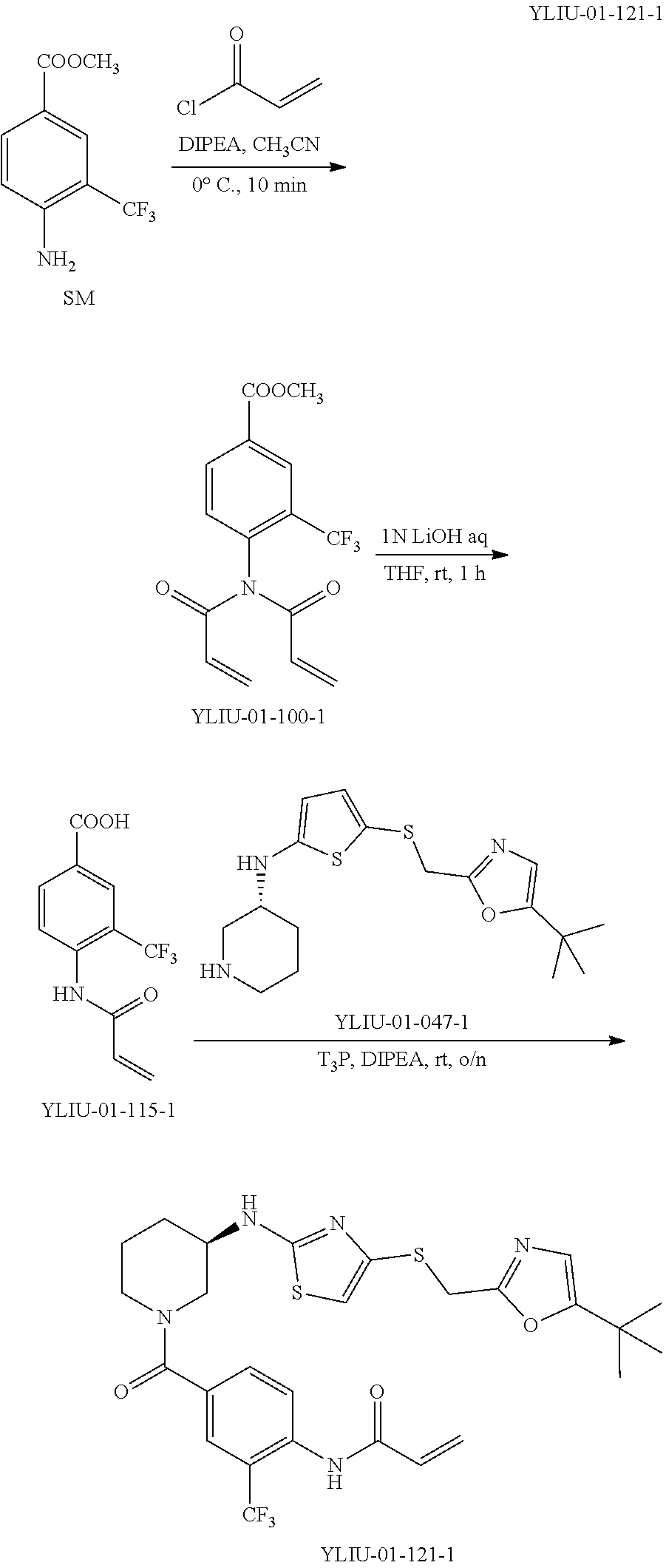
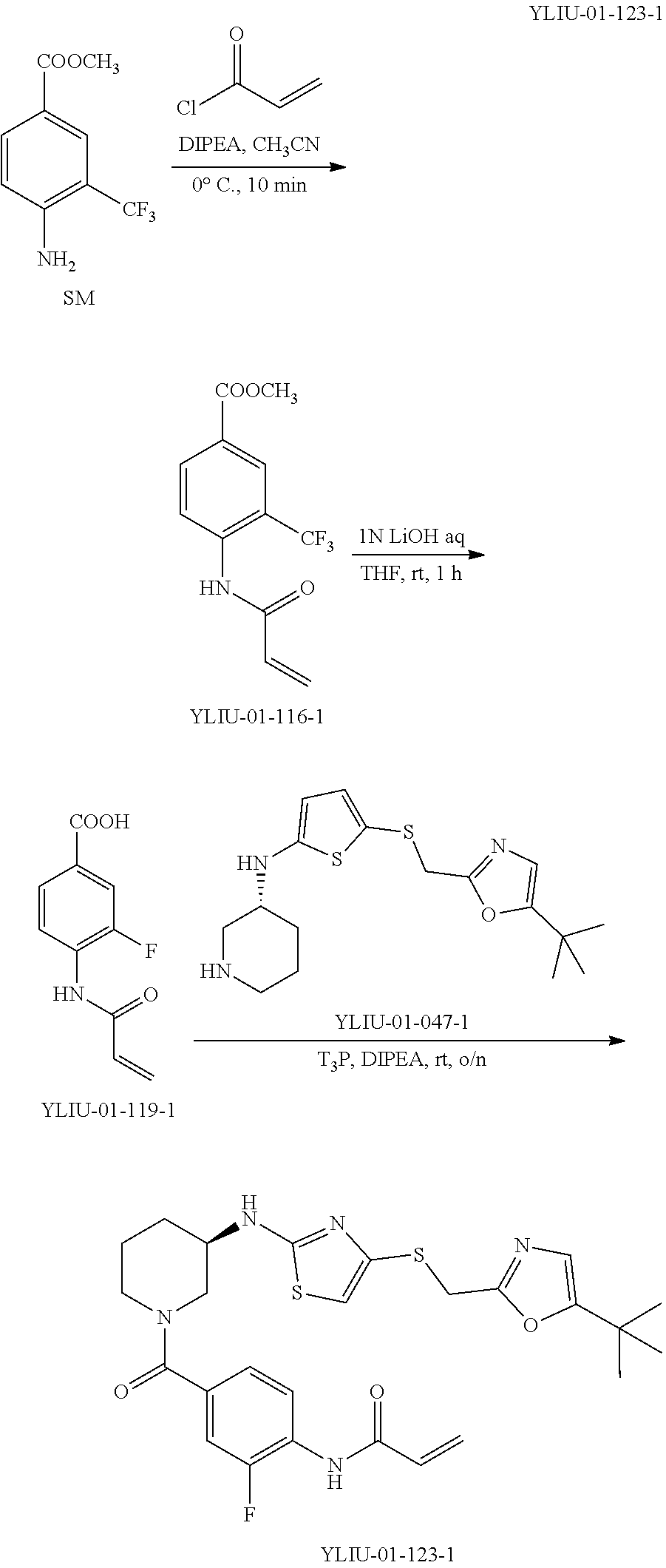

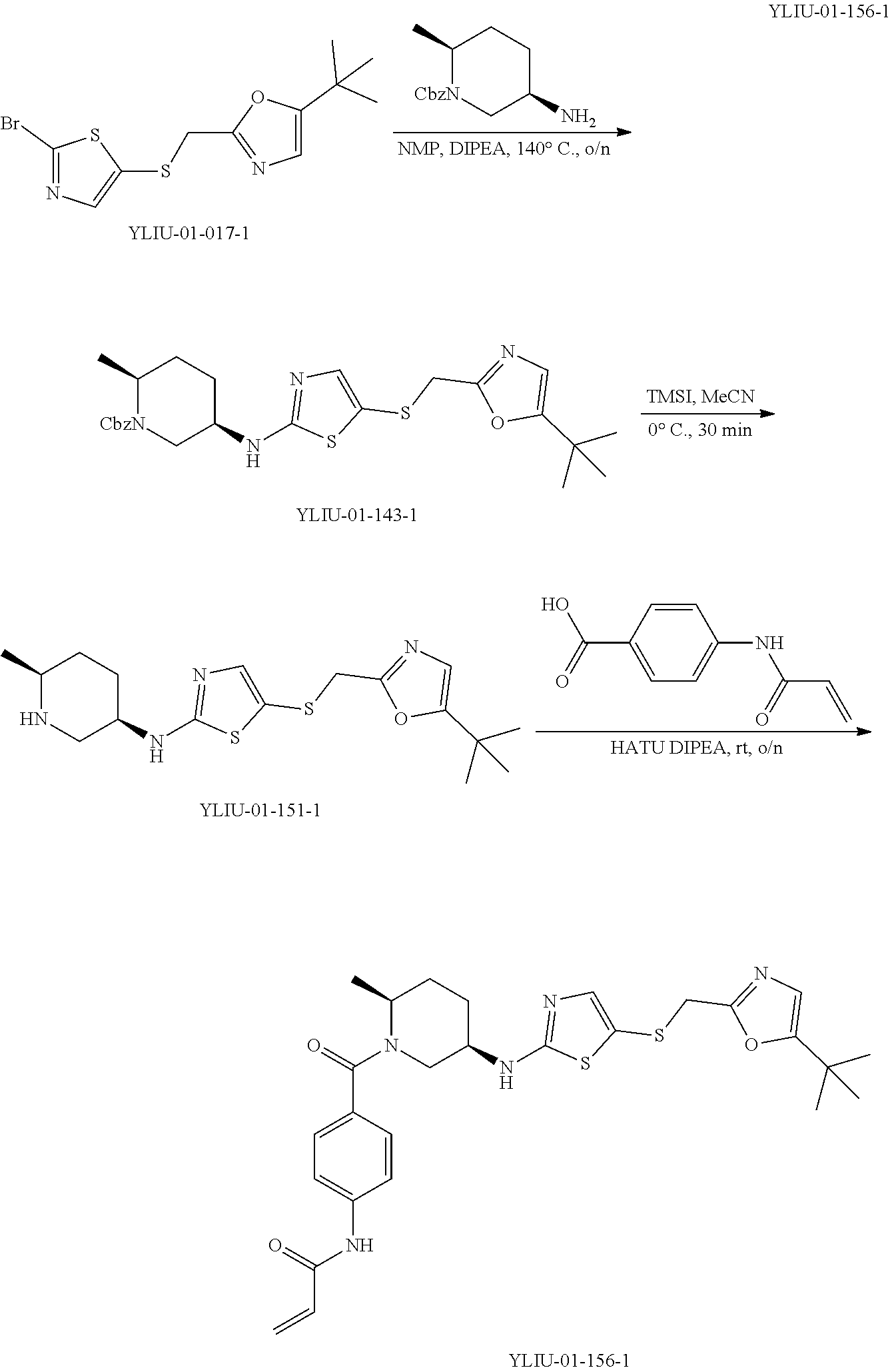




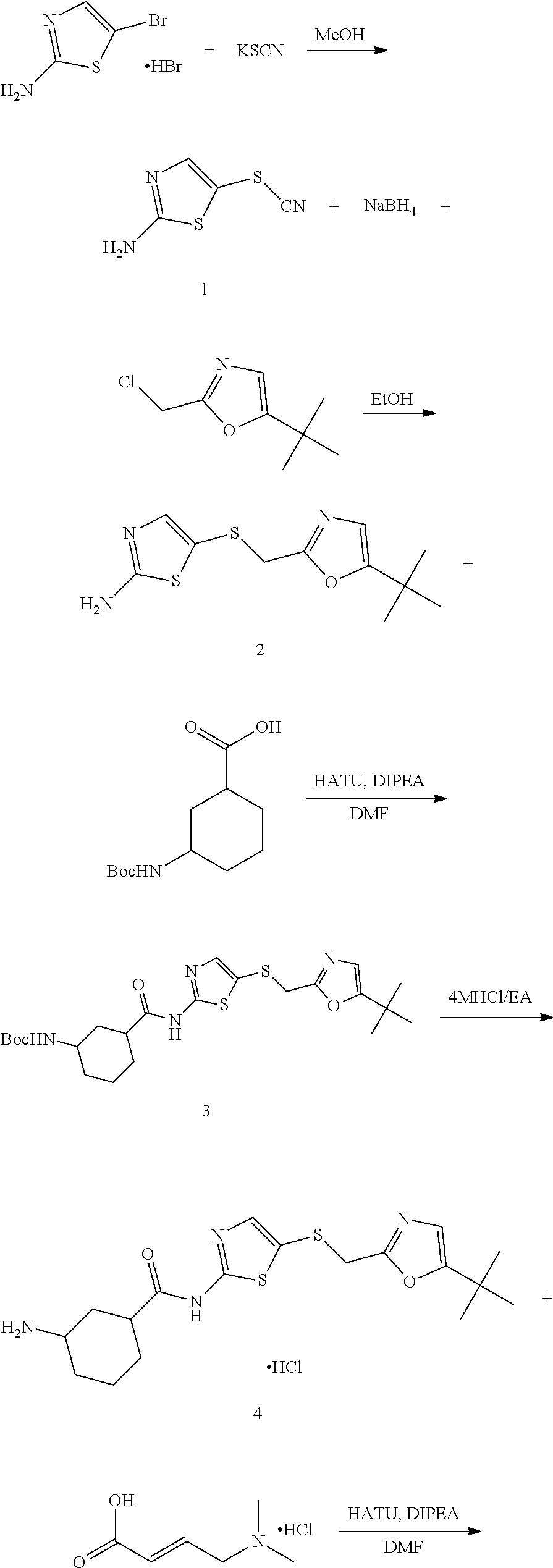






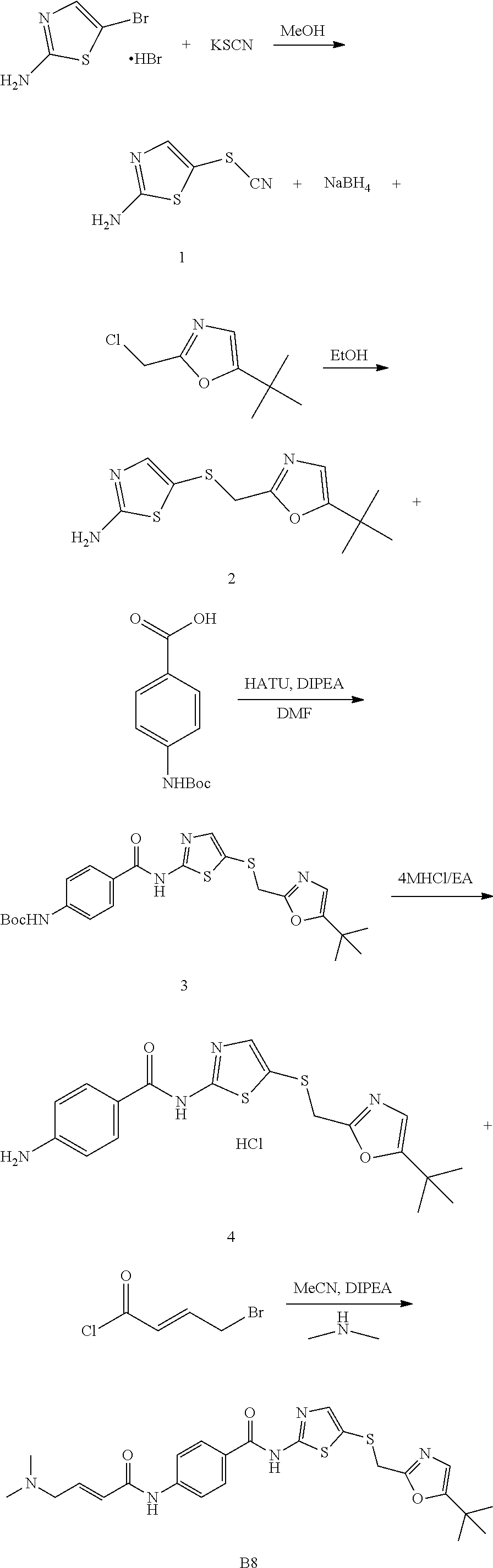









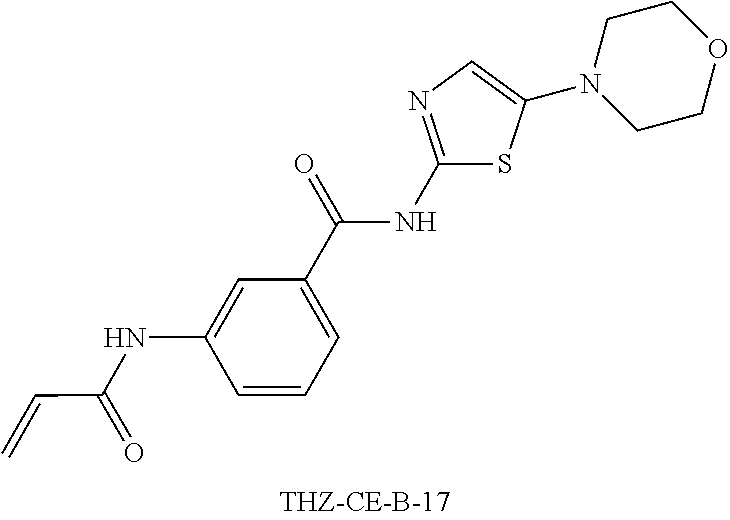










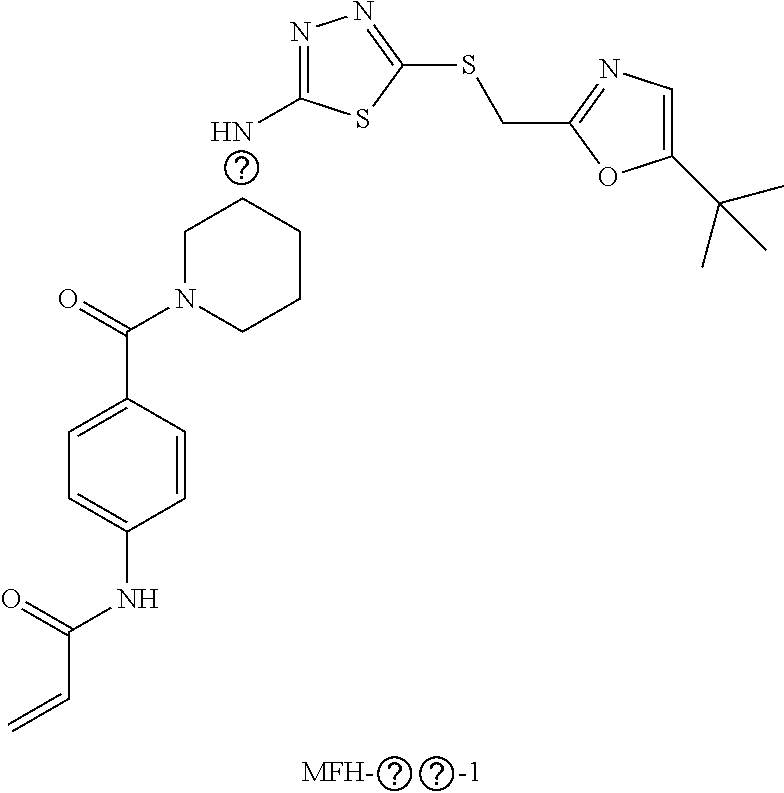







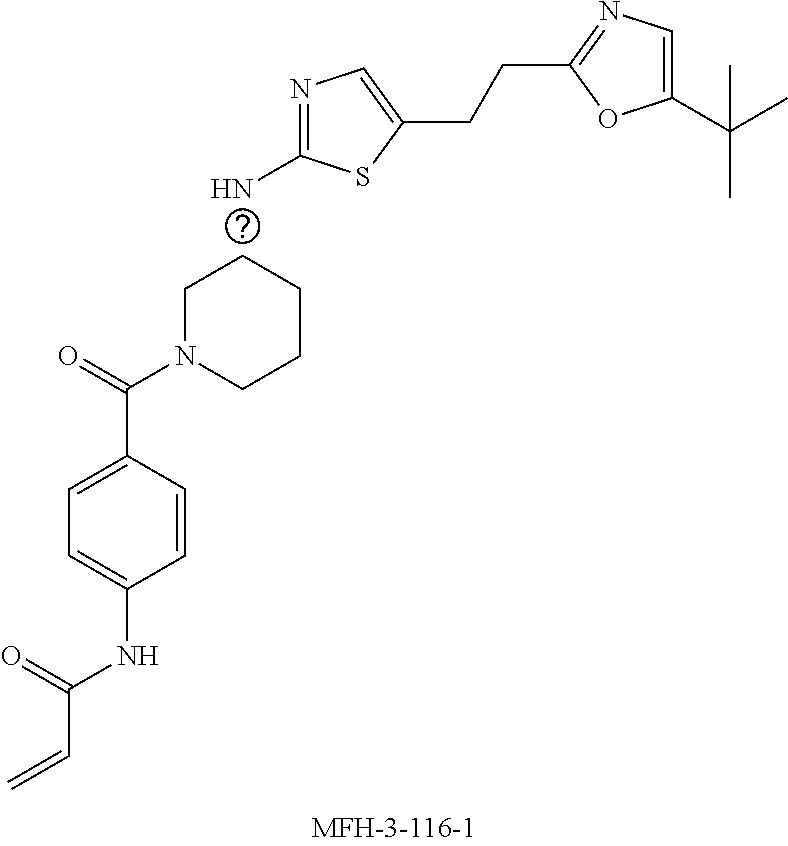











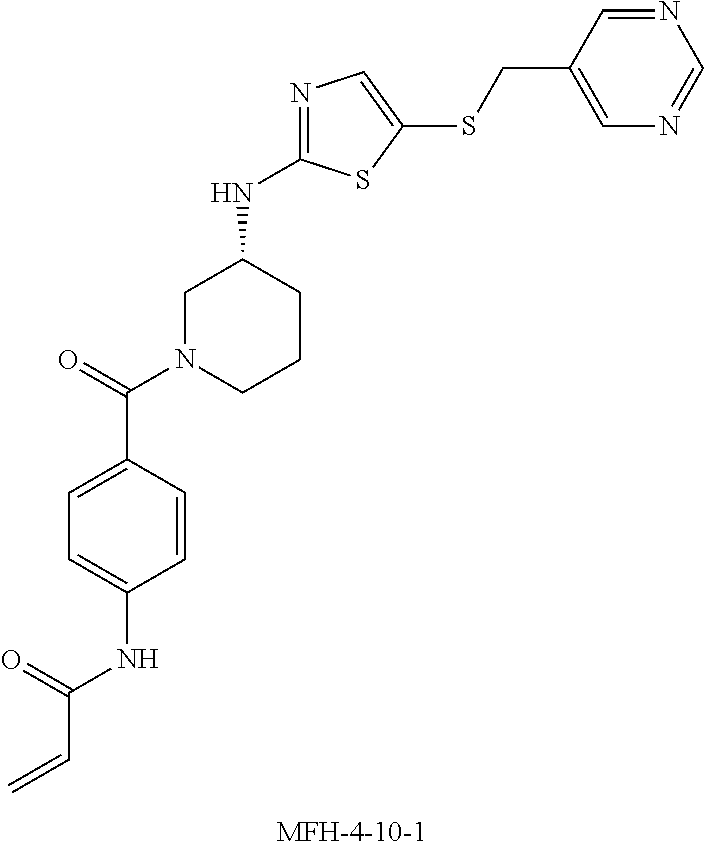

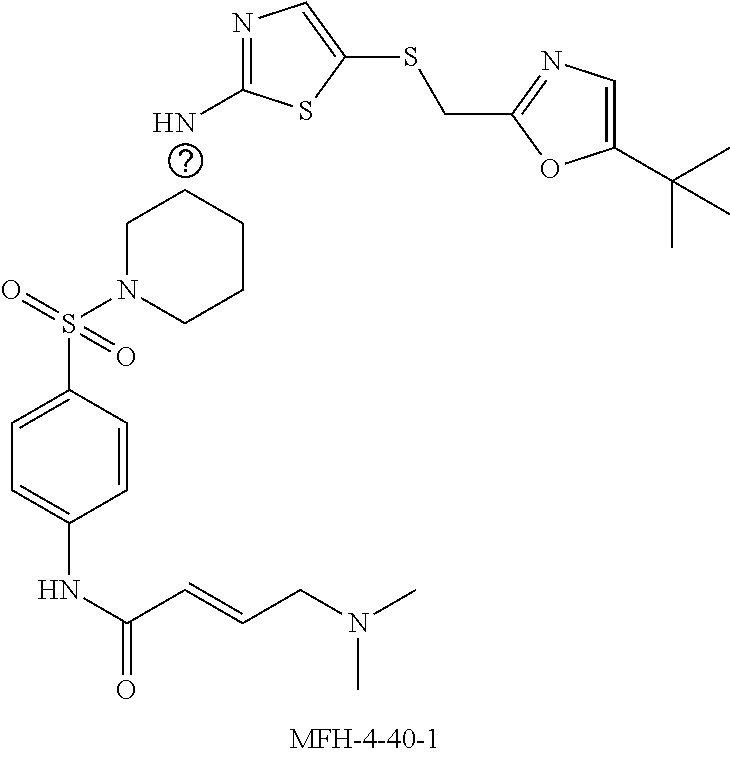






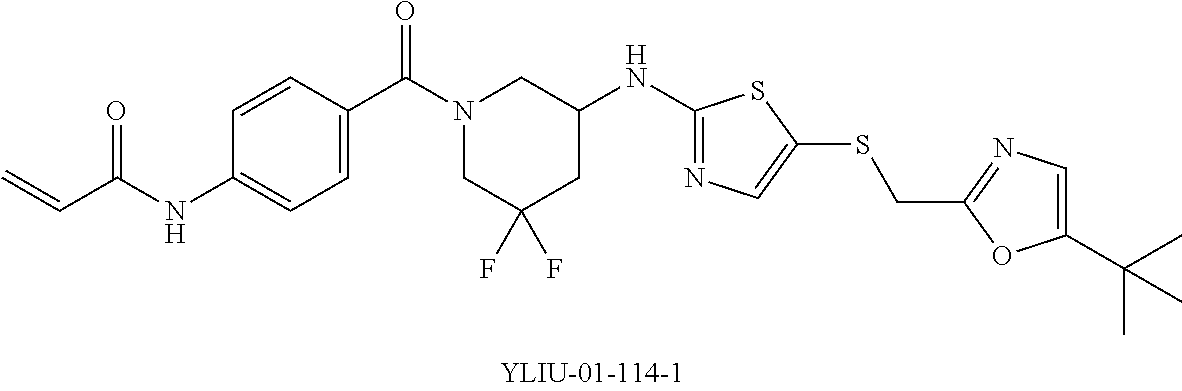
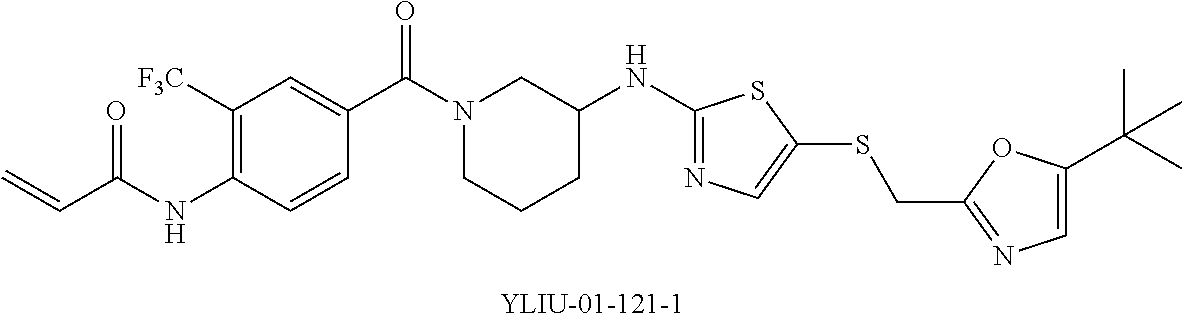




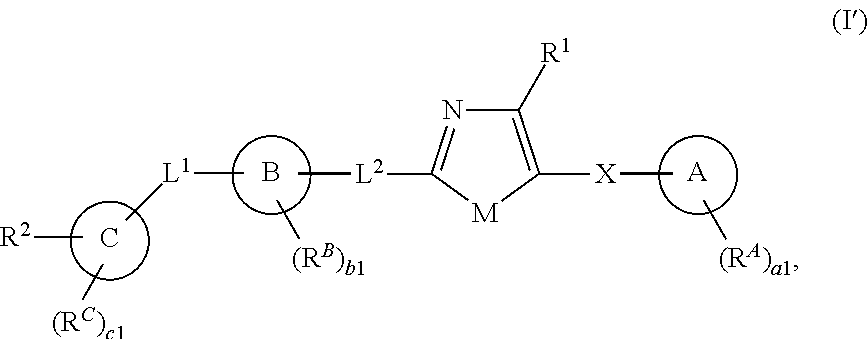






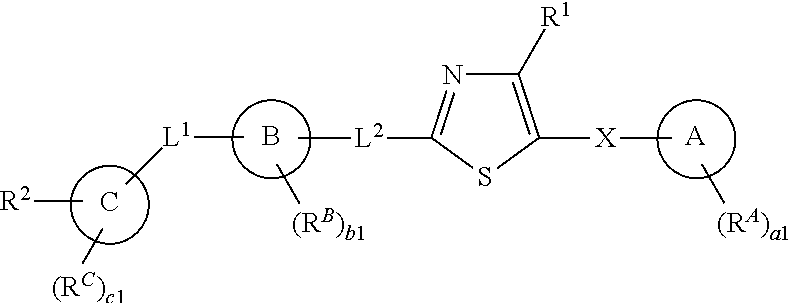

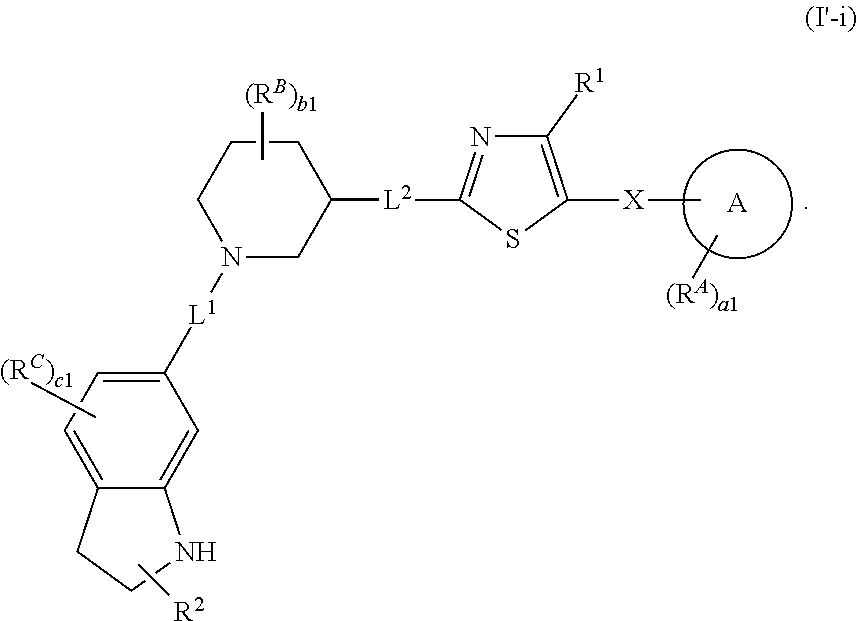
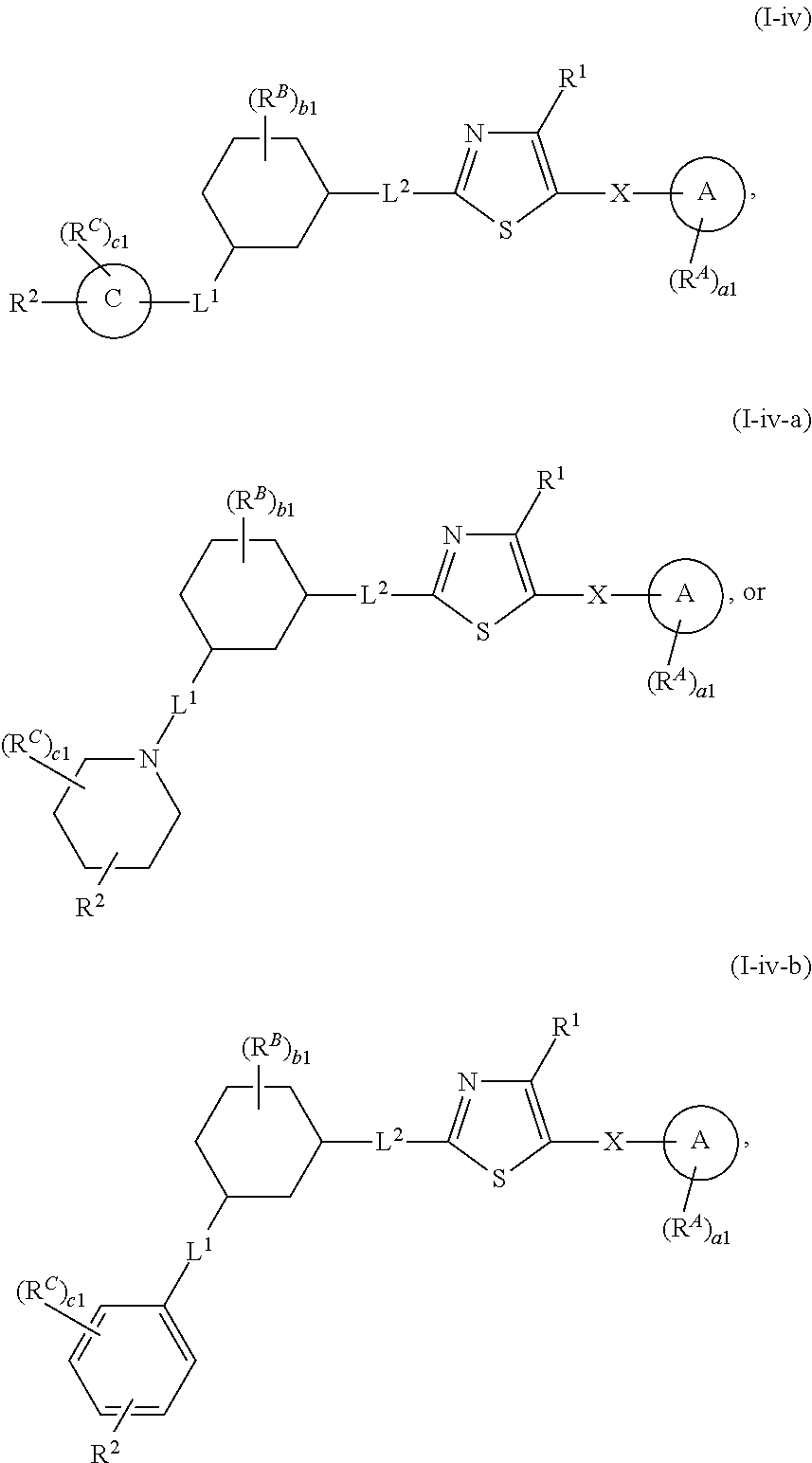


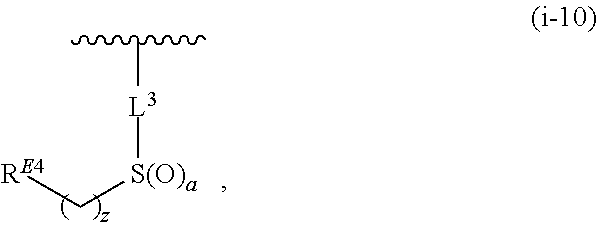









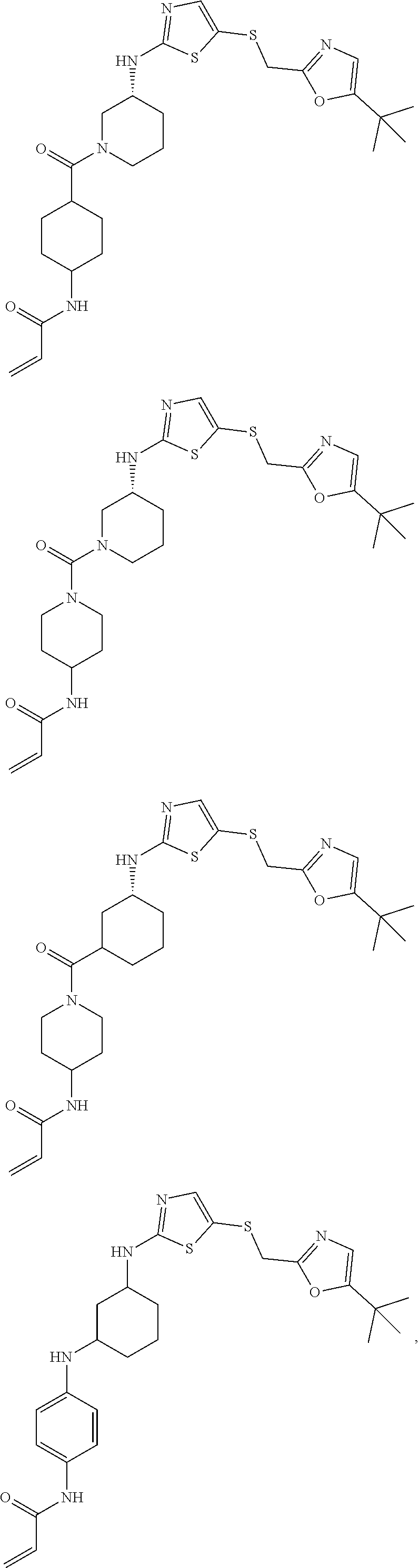

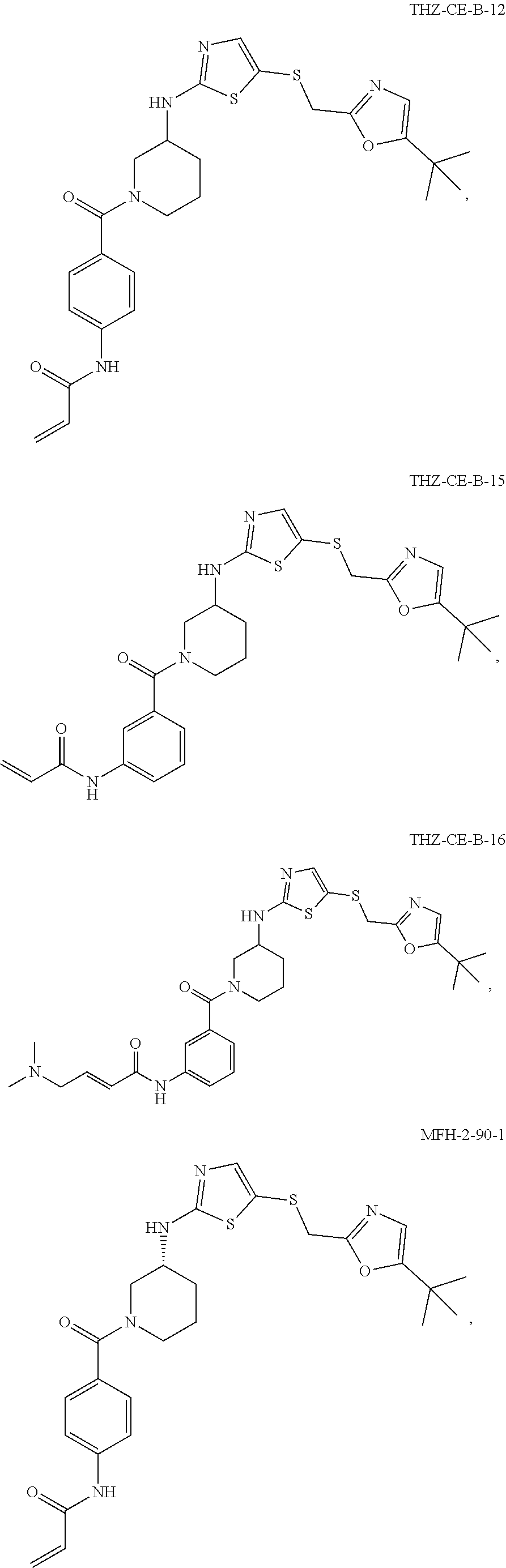


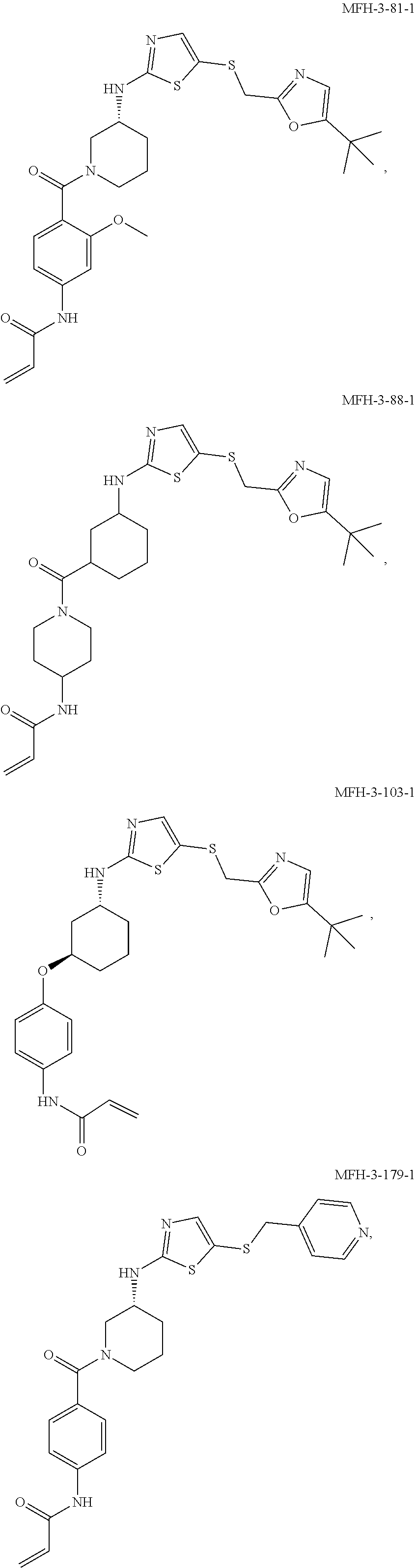




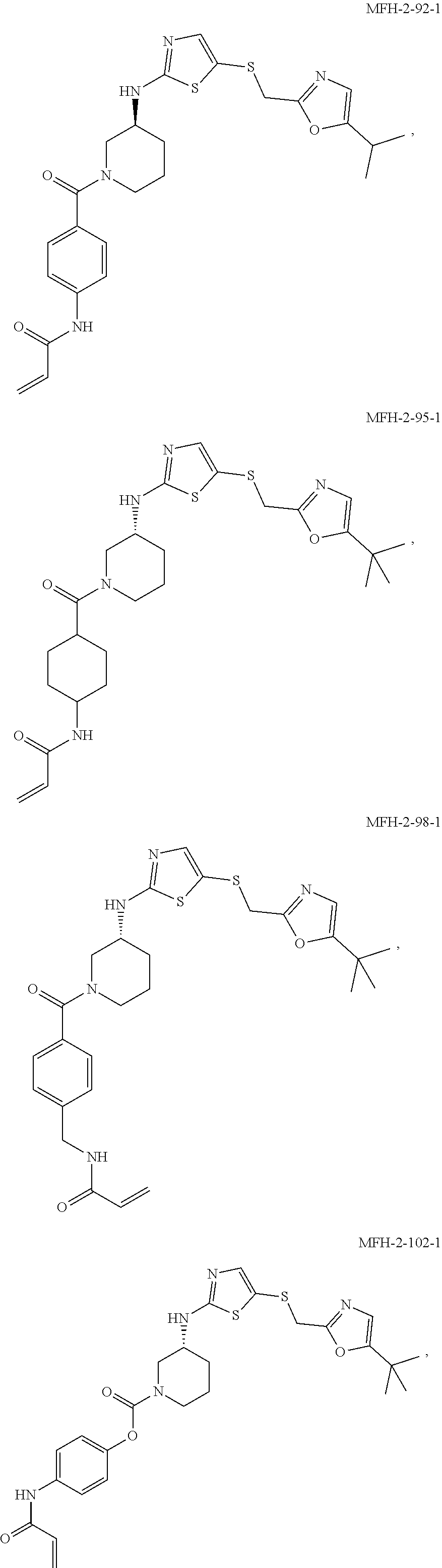



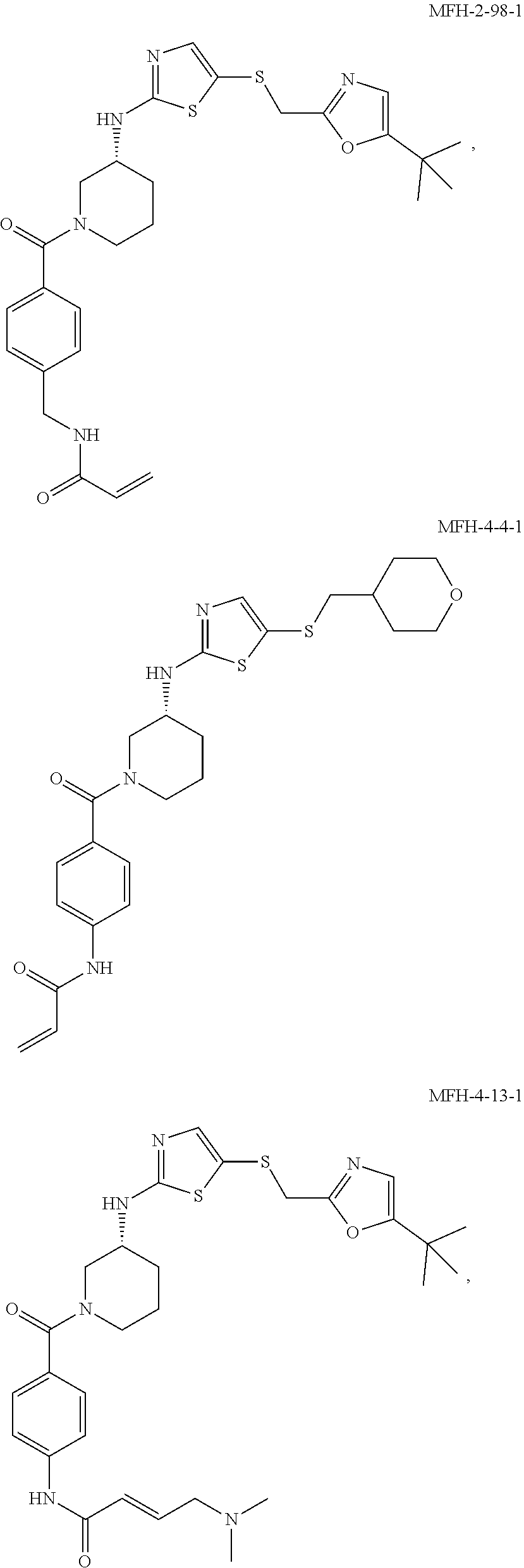
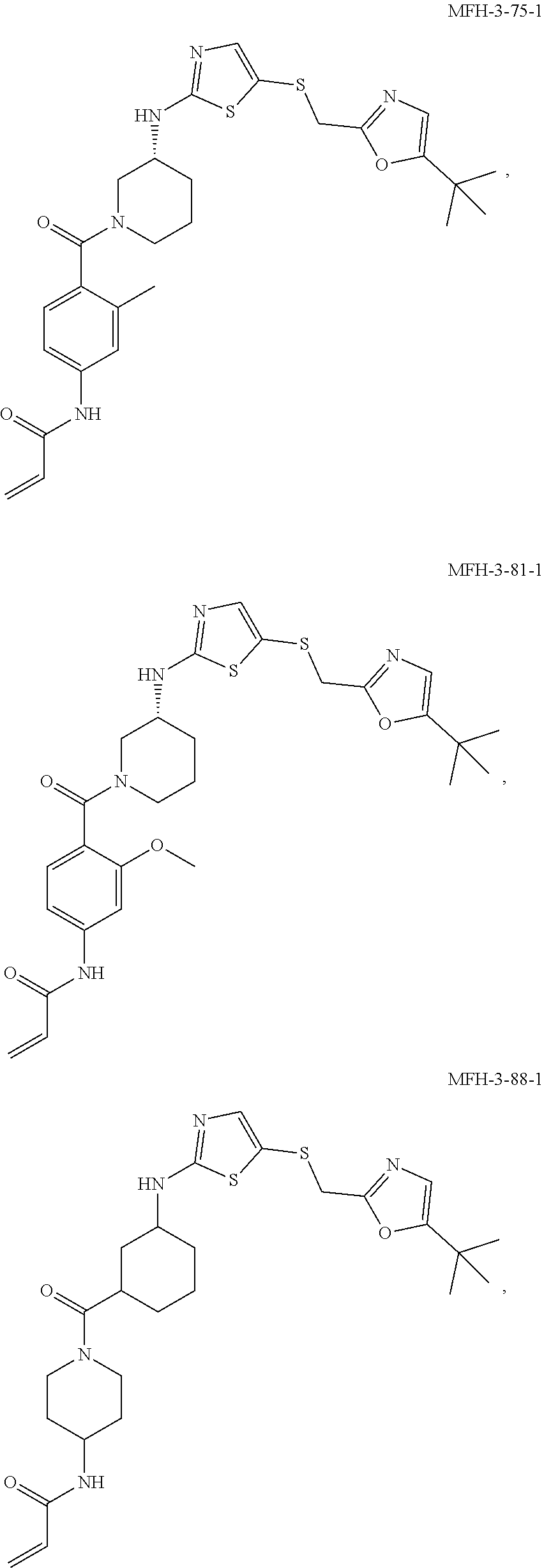









D00001
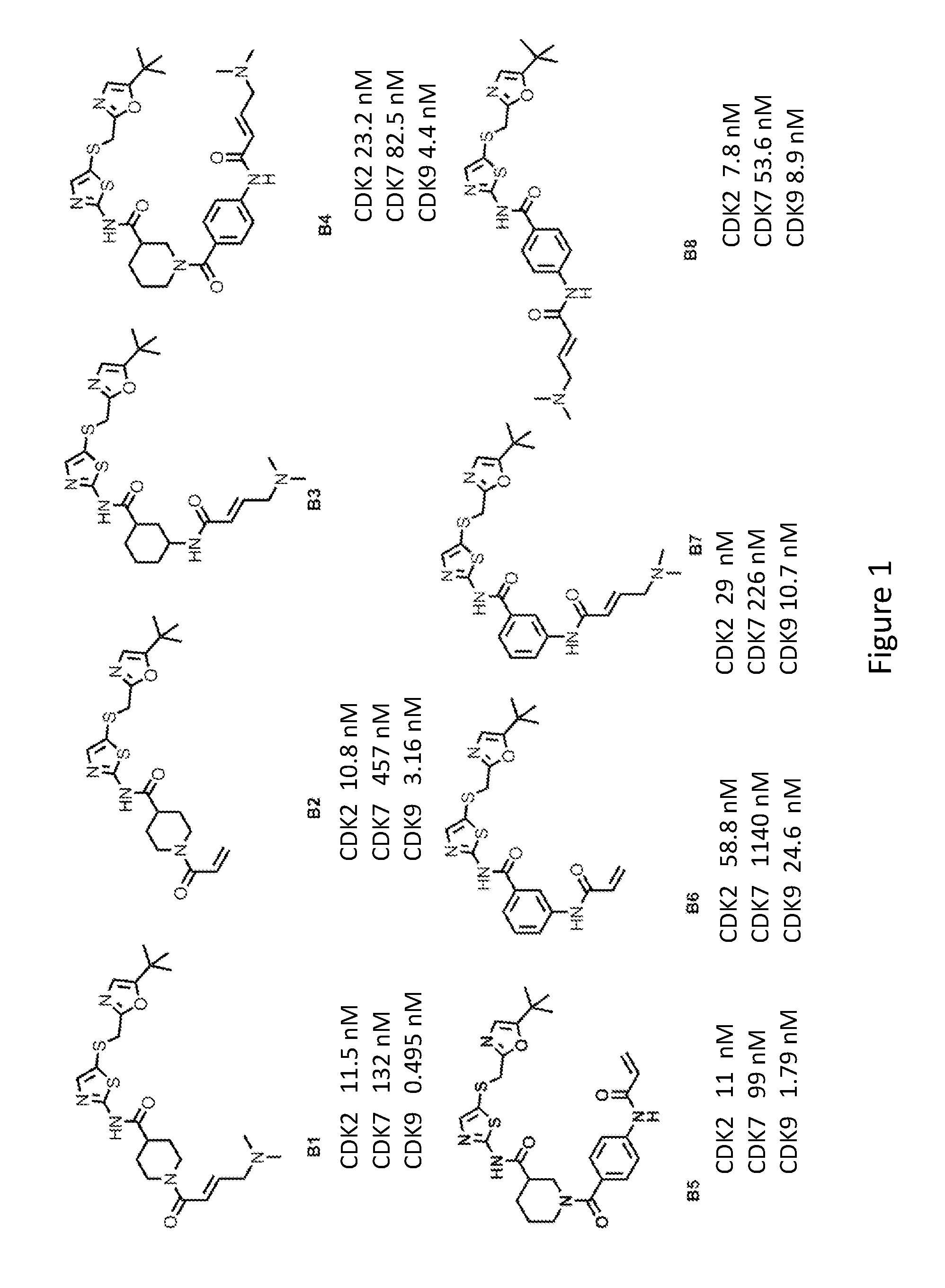
D00002

D00003

D00004

D00005

D00006

D00007

D00008

D00009

D00010
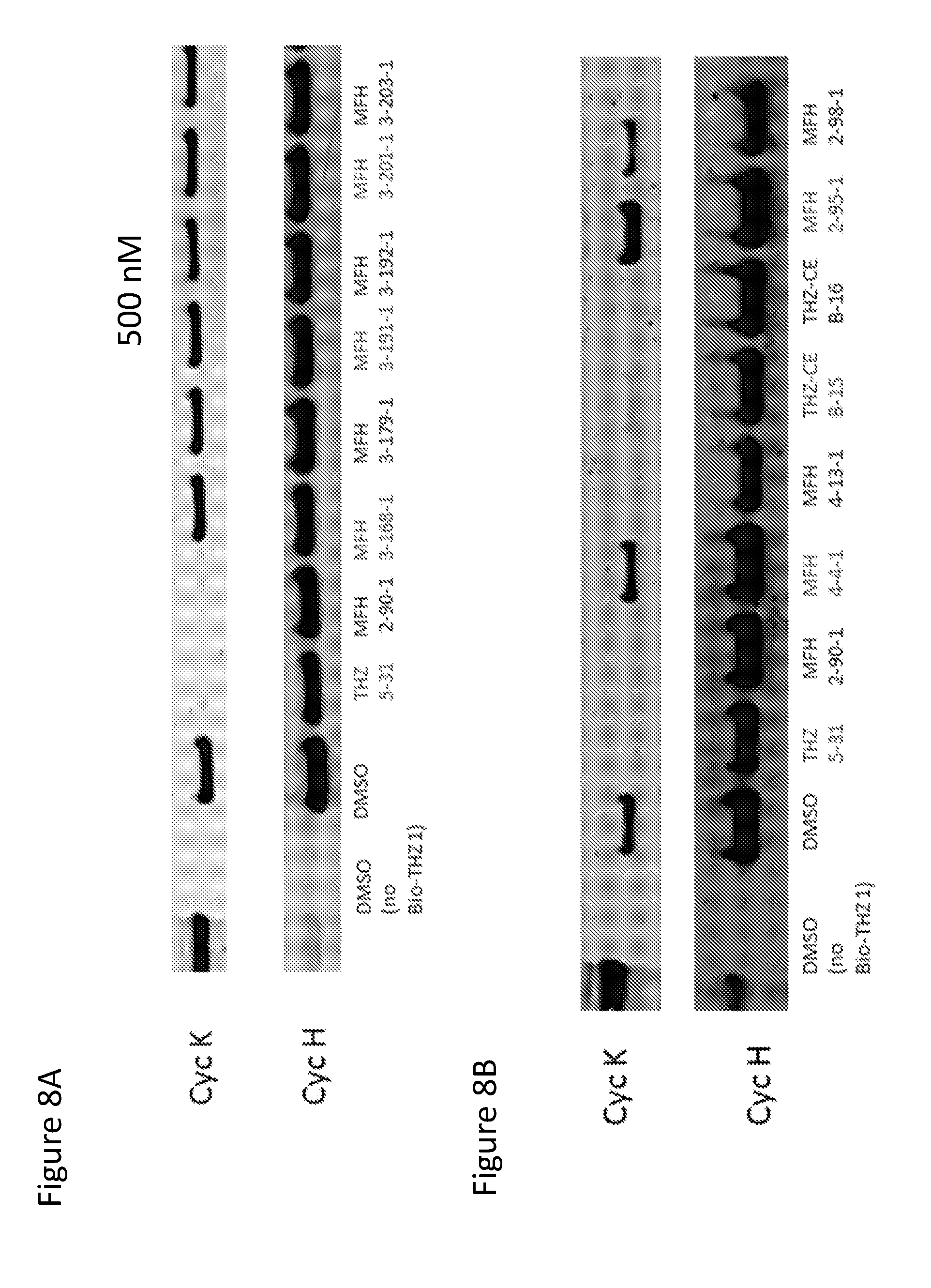
D00011

S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.