Methods And Materials For Activating An Internal Ribosome Entry Site In Exon 5 Of The Dmd Gene
Flanigan; Kevin ; et al.
U.S. patent application number 17/335251 was filed with the patent office on 2022-04-28 for methods and materials for activating an internal ribosome entry site in exon 5 of the dmd gene. The applicant listed for this patent is RESEARCH INSTITUTE AT NATIONWIDE CHILDREN'S HOSPITAL, UNIVERSITY OF WESTERN AUSTRALIA. Invention is credited to Kevin Flanigan, Nicolas Sebastien Wein, Stephen Wilton.
| Application Number | 20220127607 17/335251 |
| Document ID | / |
| Family ID | 1000006075436 |
| Filed Date | 2022-04-28 |








View All Diagrams
| United States Patent Application | 20220127607 |
| Kind Code | A1 |
| Flanigan; Kevin ; et al. | April 28, 2022 |
METHODS AND MATERIALS FOR ACTIVATING AN INTERNAL RIBOSOME ENTRY SITE IN EXON 5 OF THE DMD GENE
Abstract
The present invention relates to the delivery of oligomers for treating patients with a 5' mutation in their DMD gene other than a DMD exon 2 duplication. The invention provides methods and materials for activating an internal ribosome entry site in exon 5 of the DMD gene resulting in translation of a functional truncated isoform of dystrophin. The methods and materials can be used for the treatment of muscular dystrophies arising from 5' mutations in the DMD gene such as Duchenne Muscular Dystrophy or Becker Muscular Dystrophy.
| Inventors: | Flanigan; Kevin; (Columbus, OH) ; Wein; Nicolas Sebastien; (Columbus, OH) ; Wilton; Stephen; (Perth, AU) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 1000006075436 | ||||||||||
| Appl. No.: | 17/335251 | ||||||||||
| Filed: | June 1, 2021 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 15502702 | Feb 8, 2017 | 11053494 | ||
| PCT/US15/44366 | Aug 7, 2015 | |||
| 17335251 | ||||
| 62035395 | Aug 9, 2014 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61K 9/0019 20130101; C12N 2750/14143 20130101; C12N 2840/203 20130101; C12N 2310/3513 20130101; C12N 15/113 20130101; C12N 2310/315 20130101; C12N 2310/3519 20130101; C12N 2320/33 20130101; C12N 2320/31 20130101; C12N 2310/346 20130101; C12N 2310/11 20130101; C12N 2310/3231 20130101; A61K 31/58 20130101; C12N 2330/51 20130101; A61K 31/573 20130101; A61K 31/712 20130101; C12N 2310/3233 20130101 |
| International Class: | C12N 15/113 20060101 C12N015/113; A61K 9/00 20060101 A61K009/00; A61K 31/573 20060101 A61K031/573; A61K 31/58 20060101 A61K031/58; A61K 31/712 20060101 A61K031/712 |
Goverment Interests
STATEMENT OF GOVERNMENT INTEREST
[0002] This invention was made with Government support under NS043264 awarded by the National Institutes of Health. The government has certain rights in the invention.
Claims
1-23. (canceled)
24. A method of ameliorating Duchenne Muscular Dystrophy or Becker Muscular Dystrophy in a patient with a 5' mutation in a DMD gene but without a DMD exon 2 duplication, the method comprising administering to the patient a recombinant adeno-associated virus (rAAV) comprising a DMD exon 5 internal ribosome entry site (IRES)-activating oligomer construct comprising: (a) the nucleotide sequence set forth in SEQ ID NO: 5, 7, or 8; or (b) a nucleotide sequence that expresses an RNA transcript comprising the nucleotide sequence set forth in SEQ ID NO: 9, 11, or 12.
25. The method of claim 24, wherein the progression of a dystrophic pathology is inhibited in the patient following administration of the rAAV to the patient.
26. The method of claim 24, wherein muscle function is improved in the patient following administration of the rAAV to the patient.
27. The method of claim 26, wherein the improvement in muscle function is an improvement in muscle strength or an improvement in stability in standing and walking.
28. The method of claim 24, wherein the genome of the rAAV lacks adeno-associated virus rep and cap DNA.
29. The method of claim 24, wherein the genome of the rAAV is a self-complementary genome.
30. The method of claim 24, wherein the genome of the rAAV is a single-stranded genome.
31. The method of claim 24, wherein the rAAV comprises one or more capsid proteins from AAV1, AAV2, AAV3, AAV4, AAV5, AAV6, AAV7, AAV8, AAV9, AAV10, AAV11, or AAVrh74.
32. The method of claim 31, wherein the rAAV comprises one or more capsid proteins from AAV1, AAV6, AAV8, AAV9, or AAVrh74.
33. The method of claim 24, wherein the DMD exon 5 IRES-activating oligomer construct comprises the nucleotide sequence set forth in SEQ ID NO: 5.
34. The method of claim 24, wherein the DMD exon 5 IRES-activating oligomer construct comprises the nucleotide sequence set forth in SEQ ID NO: 7.
35. The method of claim 24, wherein the DMD exon 5 IRES-activating oligomer construct comprises the nucleotide sequence set forth in SEQ ID NO: 8.
36. The method of claim 24, wherein the DMD exon 5 IRES-activating oligomer construct comprises a nucleotide sequence that expresses an RNA comprising the nucleotide sequence set forth in SEQ ID NO: 9.
37. The method of claim 24, wherein the DMD exon 5 IRES-activating oligomer construct comprises a nucleotide sequence that expresses an RNA comprising the nucleotide sequence set forth in SEQ ID NO: 11.
38. The method of claim 24, wherein the DMD exon 5 IRES-activating oligomer construct comprises a nucleotide sequence that expresses an RNA comprising the nucleotide sequence set forth in SEQ ID NO: 12.
39. The method of claim 24, further comprising administering a glucocorticoid to the patient.
Description
[0001] This application is a continuation of U.S. application Ser. No. 15/502,702, now U.S. Pat. No. 11,053,494, issued Jul. 6, 2021, which is a U.S. national stage application of PCT/US2015/044366, filed Aug. 7, 2015, which claims the benefit of the filing date of U.S. Provisional Patent Application No. 62/035,395 filed Aug. 9, 2014, which is incorporated by reference herein in its entirety.
INCORPORATION BY REFERENCE OF THE DEQUENCE LISTING
[0003] This application contains, as a separate part of disclosure, a Sequence Listing in computer-readable form (filename: 48873A_Seqlisting.txt; 20,279 bytes--ASCII text file; created May 25, 2021) which is incorporated by reference herein in its entirety.
FIELD OF THE INVENTION
[0004] The present invention relates to the delivery of oligomers for treating patients with a 5' mutation in their DMD gene other than a DMD exon 2 duplication. The invention provides methods and materials for activating an internal ribosome entry site in exon 5 of the DMD gene resulting in a functional truncated isoform of dystrophin. The methods and materials can be used for the treatment of muscular dystrophies arising from 5' mutations in the DMD gene such as Duchenne Muscular Dystrophy or Becker Muscular Dystrophy.
BACKGROUND
[0005] Muscular dystrophies (MDs) are a group of genetic diseases. The group is characterized by progressive weakness and degeneration of the skeletal muscles that control movement. Some forms of MD develop in infancy or childhood, while others may not appear until middle age or later. The disorders differ in terms of the distribution and extent of muscle weakness (some forms of MD also affect cardiac muscle), the age of onset, the rate of progression, and the pattern of inheritance.
[0006] One form of MD is Duchenne Muscular Dystrophy (DMD). It is the most common severe childhood form of muscular dystrophy affecting 1 in 5000 newborn males. DMD is caused by mutations in the DMD gene leading to absence of dystrophin protein (427 KDa) in skeletal and cardiac muscles, as well as GI tract and retina. Dystrophin not only protects the sarcolemma from eccentric contractions, but also anchors a number of signaling proteins in close proximity to sarcolemma. Many clinical cases of DMD are linked to deletion mutations in the DMD gene. Despite many lines of research following the identification of the DMD gene, treatment options are limited. Corticosteroids are clearly beneficial but even with added years of ambulation the benefits are offset by long-term side effects. The original controlled, randomized, double-blind study reported more than 20 years ago showed benefits using prednisone [Mendell et al., N. Engl. J. Med., 320: 1592-1597 (1989)]. Subsequent reports showed equal efficacy using deflazacort, a sodium-sparing steroid [Biggar et al., J. Pediatr., 138: 45-50 (2001)]. Recent studies also demonstrate efficacy by exon skipping, prolonging walking distance on the 6MWT. Thus far, published clinical studies have reported benefit for only mutations where the reading frame is restored by skipping exon 51 [Cirak et al., Lancet, 378: 595-605 (2011) and Goemans et al., New Engl. J. Med. 364: 1513-1522 (2011)]. In the only report of a double blind, randomized treatment trial promising results were demonstrated with eteplirsen, a phosphorodiamidate morpholino oligomer (PMO) [Mendell et al., Annals Neurology, 74(5): 637-647 (2013)]. In all of these exon-skipping trials, the common denominator of findings has been a plateau in walking ability after an initial modest improvement. Another exon-skipping article is Greer et al., Molecular Therapy--Nucleic Acids, 3: 3155 (2014).
[0007] See also, U.S. Patent Application Publication Nos. 2012/0077860 published Mar. 29, 2012; 2013/0072541 published Mar. 21, 2013; and 2013/0045538 published Feb. 21, 2013.
[0008] In contrast to the deletion mutations, DMD exon duplications account for around 5% of disease-causing mutations in unbiased samples of dystrophinopathy patients [Dent et al., Am. J. Med. Genet., 134(3): 295-298 (2005)], although in some catalogues of mutations the number of duplications is higher [including that published by the United Dystrophinopathy Project in Flanigan et al., Hum. Mutat., 30(12): 1657-1666 (2009), in which it was 11%].
[0009] Mutations in the DMD gene result in either the more severe DMD or the milder Becker muscular dystrophy (BMD). The phenotype generally depends upon whether the mutation results in the complete absence of the protein product dystrophin (in DMD) or preserves a reading frame that allows translation of a partially functional dystrophin protein (in BMD) [Monaco, Trends in Biochemical Sciences, 14: 412-415 (1989)]. We previously identified a particular BMD founder allele (c.9T>G; p.Trp3X) that did not follow this reading frame rule [Flanigan et al., Neuromuscular Disorders: NMD, 19: 743-748 (2009) and Flanigan et al., Human Mutation, 30: 1657-1666 (2009)]. Although this nonsense mutation is predicted to result in no protein translation, muscle biopsy revealed significant amounts (.about.21%) of dystrophin expression of minimally decreased size and the clinical phenotype is one of a very mild dystrophinopathy [Flanigan et al., Neuromuscular Disorders: NMD, 19: 743-748 (2009)]. In cellulo and in vitro translation studies demonstrated that in p.Trp3X patients translation is initiated from AUGs in exon 6, suggesting alternate translation initiation as a mechanism of phenotypic amelioration [Gurvich et al., Human Mutation, 30: 633-640 (2009)]. Noting that most truncating mutations reported in 5' exons were in fact associated with BMD rather than DMD, we proposed that altered translation initiation may be a general mechanism of phenotypic rescue for 5' mutations in this gene, a prediction supported by subsequent reports [Witting and Vissing, Neuromuscular Disorders: NMD, 23: 25-28 (2013) and Flanigan et al., Neuromuscular Disorders: NMD, 23: 192 (2013)]. The canonical actin-binding domain 1 (ABD1) was previously proposed to be essential for protein function [Gimona et al., FEBS Letters, 513: 98-106 (2002).
[0010] Translation initiation is commonly understood to occur by cap-dependent initiation. Internal ribosome entry sites (IRESs) are RNA regulatory sequences that govern cap-independent translation initiation in eukaryotic cells, which is activated when cap-dependent translation is compromised (e.g., during cell stress). Ribosomes are recruited directly to these IRESs on the mRNA and can then continue scanning in a 5' to 3' direction for alternative initiation codons. They were first described in viruses, and among the earliest characterized was the encephalomyocarditis virus (EMCV) IRES. Almost 85 cellular IRESs have been described to date and are mainly located in 5'UTR regions; for example, the 5'UTR of utrophin A, an autosomal homologue of dystrophin, contains an IRES that is both particularly active in regenerating muscle and inducible by exposure to glucocorticoid (the mainstay of therapy for DMD) [Miura et al., J. Biol. Chem., 280: 32997-33005 (2005) and Miura et al., PloS One, 3: e2309 (2008)]. However, other eukaryotic IRESs have been described within coding sequences, and some have also been implicated in the modulation of pathology. These include an IRES in the APC gene linked to a mild version of familial adenomatous polyposis coli in which patients with certain 5' mutations still produce a partially functional protein through the use of a downstream initiation codon.
[0011] Adeno-associated virus (AAV) is a replication-deficient parvovirus, the single-stranded DNA genome of which is about 4.7 kb in length including 145 nucleotide inverted terminal repeat (ITRs). There are multiple serotypes of AAV. The nucleotide sequences of the genomes of the AAV serotypes are known. For example, the complete genome of AAV-1 is provided in GenBank Accession No. NC_002077; the complete genome of AAV-2 is provided in GenBank Accession No. NC_001401 and Srivastava et al., J. Virol., 45: 555-564 (1983); the complete genome of AAV-3 is provided in GenBank Accession No. NC_1829; the complete genome of AAV-4 is provided in GenBank Accession No. NC_001829; the AAV-5 genome is provided in GenBank Accession No. AF085716; the complete genome of AAV-6 is provided in GenBank Accession No. NC_00 1862; at least portions of AAV-7 and AAV-8 genomes are provided in GenBank Accession Nos. AX753246 and AX753249, respectively (see also U.S. Pat. Nos. 7,282,199 and 7,790,449 relating to AAV-8); the AAV-9 genome is provided in Gao et al., J. Virol., 78: 6381-6388 (2004); the AAV-10 genome is provided in Mol. Ther., 13(1): 67-76 (2006); and the AAV-11 genome is provided in Virology, 330(2): 375-383 (2004). The sequence of the AAV rh.74 genome is provided herein. Cis-acting sequences directing viral DNA replication (rep), encapsidation/packaging and host cell chromosome integration are contained within the AAV ITRs. Three AAV promoters (named p5, p19, and p40 for their relative map locations) drive the expression of the two AAV internal open reading frames encoding rep and cap genes. The two rep promoters (p5 and p19), coupled with the differential splicing of the single AAV intron (at nucleotides 2107 and 2227), result in the production of four rep proteins (rep 78, rep 68, rep 52, and rep 40) from the rep gene. Rep proteins possess multiple enzymatic properties that are ultimately responsible for replicating the viral genome. The cap gene is expressed from the p40 promoter and it encodes the three capsid proteins VP1, VP2, and VP3. Alternative splicing and non-consensus translational start sites are responsible for the production of the three related capsid proteins. A single consensus polyadenylation site is located at map position 95 of the AAV genome. The life cycle and genetics of AAV are reviewed in Muzyczka, Current Topics in Microbiology and Immunology, 158: 97-129 (1992).
[0012] AAV possesses unique features that make it attractive as a vector for delivering foreign DNA to cells, for example, in gene therapy. AAV infection of cells in culture is noncytopathic, and natural infection of humans and other animals is silent and asymptomatic. Moreover, AAV infects many mammalian cells allowing the possibility of targeting many different tissues in vivo. Moreover, AAV transduces slowly dividing and non-dividing cells, and can persist essentially for the lifetime of those cells as a transcriptionally active nuclear episome (extrachromosomal element). The AAV proviral genome is infectious as cloned DNA in plasmids which makes construction of recombinant genomes feasible. Furthermore, because the signals directing AAV replication, genome encapsidation and integration are contained within the ITRs of the AAV genome, some or all of the internal approximately 4.3 kb of the genome (encoding replication and structural capsid proteins, rep-cap) may be replaced with foreign DNA. The rep and cap proteins may be provided in trans. Another significant feature of AAV is that it is an extremely stable and hearty virus. It easily withstands the conditions used to inactivate adenovirus (56o to 65.degree. C. for several hours), making cold preservation of AAV less critical. AAV may even be lyophilized. Finally, AAV-infected cells are not resistant to superinfection.
[0013] There remains a need in the art for treatments for muscular dystrophies including DMD and BMD.
SUMMARY
[0014] The present disclosure contemplates methods and products for preventing disease, delaying the progression of disease, and/or treating patients with one or more 5' mutations of the DMD gene. The methods are based on the identification of a glucocorticoid-inducible IRES in exon 5 of the DMD gene, the activation of which can generate a functional N-terminally truncated dystrophin isoform
[0015] The disclosure contemplates methods of ameliorating Duchenne Muscular Dystrophy or Becker Muscular Dystrophy in a patient with a 5' mutation in the DMD gene comprising the step of administering a DMD exon 5 IRES-activating oligomer construct to the patient, wherein the patient does not have a DMD exon 2 duplication.
[0016] In some embodiments of the methods, the DMD exon 5 IRES-activating oligomer construct targets one of the following portions of exon 2 of the DMD gene:
TABLE-US-00001 (SEQ ID NO: 1) 5' TCAAAAGAAAACATTCACAAAATGGGTA 3', (SEQ ID NO: 2) 5' GCACAATTTTCTAAGGTAAGAAT 3', (SEQ ID NO: 3) 5' TAGATGAAAGAGAAGATGTTCAAAAGAAAAC 3', or (SEQ ID NO: 4) 5' TAGATGAAAGAGAAGATGTTC 3'.
[0017] In some embodiments of the methods, the DMD exon 5 IRES-activating oligomer construct is a U7snRNA polynucleotide construct in the genome of a recombinant adeno-associated virus. In some of these embodiments, the genome of the recombinant adeno-associated virus lacks adeno-associated virus rep and cap DNA. In some of these embodiments, the virus genome is a self-complementary genome. In some of these embodiments the recombinant adeno-associated virus is a recombinant AAV1 virus, a recombinant AAV6 virus, a recombinant AAV9 virus or a recombinant AAV rh74 virus. In some embodiments, the U7snRNA polynucleotide construct comprises: the U7-B antisense polynucleotide TACCCATTTTGCGAATGTTTTCTTTTGA (SEQ ID NO: 5), the U7-C antisense polynucleotide ATTCTTACCTTAGAAAATTGTGC (SEQ ID NO: 6), the U7-AL antisense polynucleotide GTTTTCTTTTGAAGATCTTCTCTTTCATCTA (SEQ ID NO: 7), or the U7-AS antisense polynucleotide GAAGATCTTCTCTTTCATCTA (SEQ ID NO: 8).
[0018] In some embodiments of the methods, the DMD exon 5 IRES-activating oligomer construct is an antisense oligomer. In some embodiments, the antisense oligomer is an exon 2-targeting antisense oligomer: B antisense oligomer UACCCAUUUUGCGAAUGUUUUCUUUUGA (SEQ ID NO: 9), C antisense oligomer AUUCUUACCUUAGAAAAUUGUGC (SEQ ID NO: 10), AL antisense oligomer GUUUUCUUUUGAACAUCUUCUCUUUCAUCUA (SEQ ID NO: 11) or AS antisense oligomer GAACAUCUUCUCUUUCAUCUA (SEQ ID NO:12). In some of these embodiments, the exon 2-targeting antisense oligomer: is a phosphorodiamidate morpholino oligomer (PMO), is a cell penetrating peptide-conjugated PMO (PPMO), is a PMO internalizing peptide (PIP), comprises tricyclo-DNA (tcDNA) or comprises 2'O-methyl-phosphorothioate modifications.
[0019] In some embodiments of the methods, the progression of a dystrophic pathology is inhibited in the patient.
[0020] In some embodiments of the methods, muscle function is improved in the patient. The improvement in muscle function can be an improvement in muscle strength or an improvement in stability in standing and walking.
[0021] In some embodiments, the contemplated methods further comprise administering a glucocorticoid to the patient.
[0022] The disclosure contemplates a recombinant adeno-associated virus (AAV) comprising a DMD exon 5 IRES-activating oligomer construct, wherein the DMD exon 5 IRES-activating oligomer construct is a U7snRNA polynucleotide construct comprising:
TABLE-US-00002 U7-B antisense sequence (SEQ ID NO: 5) TACCCATTTTGCGAATGTTTTCTTTTGA, U7-C antisense sequence (SEQ ID NO: 6) ATTCTTACCTTAGAAAATTGTGC, U7-AL antisense polynucleotide (SEQ ID NO: 7) GTTTTCTTTTGAAGATCTTCTCTTTCATCTA, or U7-AS antisense polynucleotide (SEQ ID NO: 8) GAAGATCTTCTCTTTCATCTA.
In some embodiments, the genome of the recombinant AAV lacks AAV rep and cap DNA. In some embodiments, the recombinant AAV genome is a self-complementary genome. In some embodiments, the recombinant adeno-associated virus is a recombinant AAV1 virus, a recombinant AAV6 virus, a recombinant AAV9 virus or a recombinant AAV rh74 virus. In some embodiments, the self-complementary genome comprises the DMD exon 5 IRES-activating U7 snRNA polynucleotide construct U7_ACCA (FIG. 15A shows the genome insert 3' to 5' while FIG. 15B shows the reverse complement of the sequence of FIG. 15A).
[0023] The disclosure contemplates a DMD exon 5 IRES-activating oligomer construct, wherein the DMD exon 5 IRES-activating oligomer construct is an exon 2-targeting antisense oligomer: B antisense oligomer UACCCAUUUUGCGAAUGUUUUCUUUUGA (SEQ ID NO: 9), C antisense oligomer AUUCUUACCUUAGAAAAUUGUGC (SEQ ID NO: 10), AL antisense oligomer GUUUUCUUUUGAACAUCUUCUCUUUCAUCUA (SEQ ID NO: 11) or AS antisense oligomer GAACAUCUUCUCUUUCAUCUA (SEQ ID NO:12). In some embodiments, the exon 2-targeting antisense oligomer: is a phosphorodiamidate morpholino oligomer (PMO), is a cell penetrating peptide-conjugated PMO (PPMO), is a PMO internalizing peptide (PIP), comprises tricyclo-DNA (tcDNA) or comprises 2'O-methyl-phosphorothioate modifications.
BRIEF DESCRIPTION OF THE DRAWING
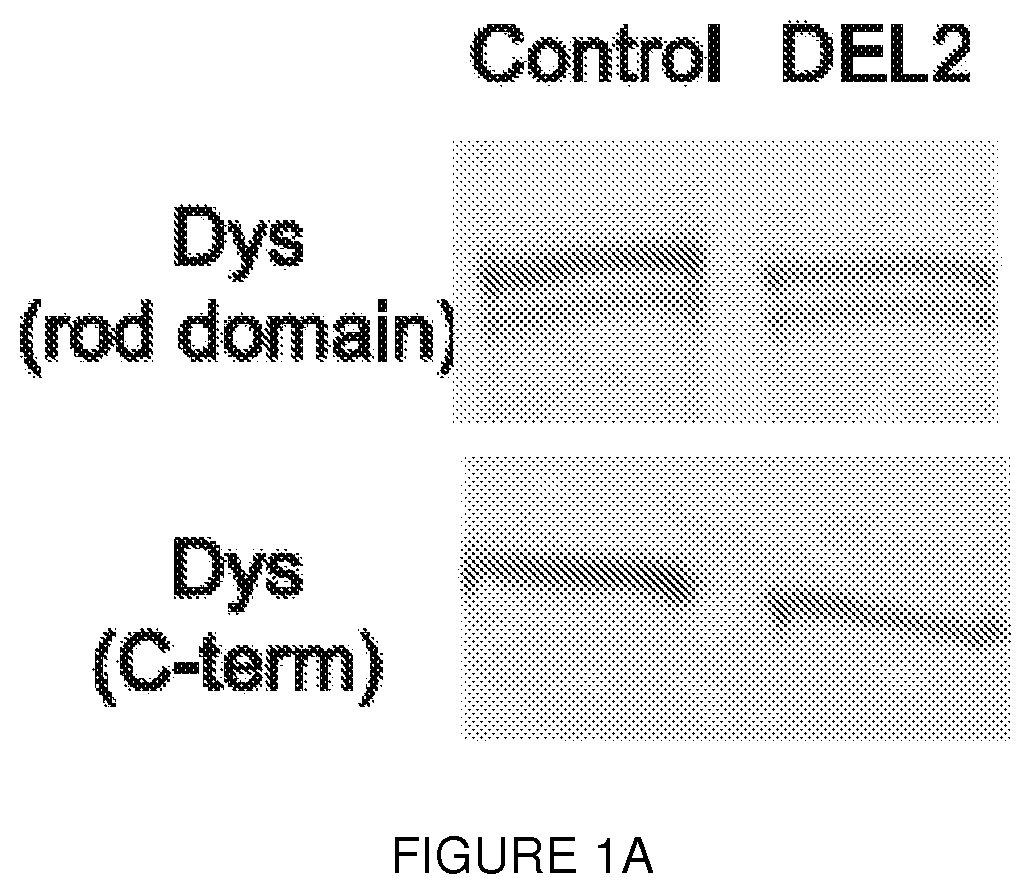
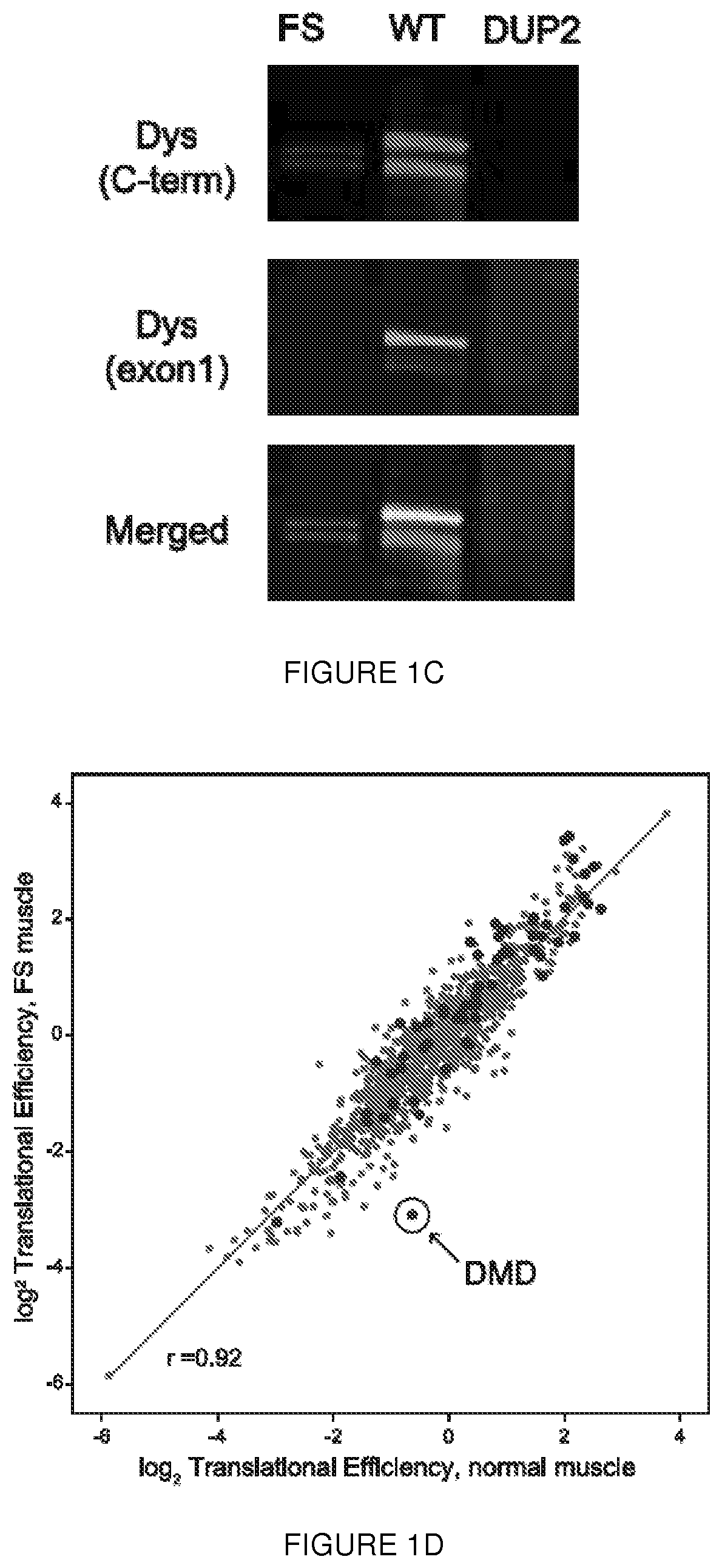
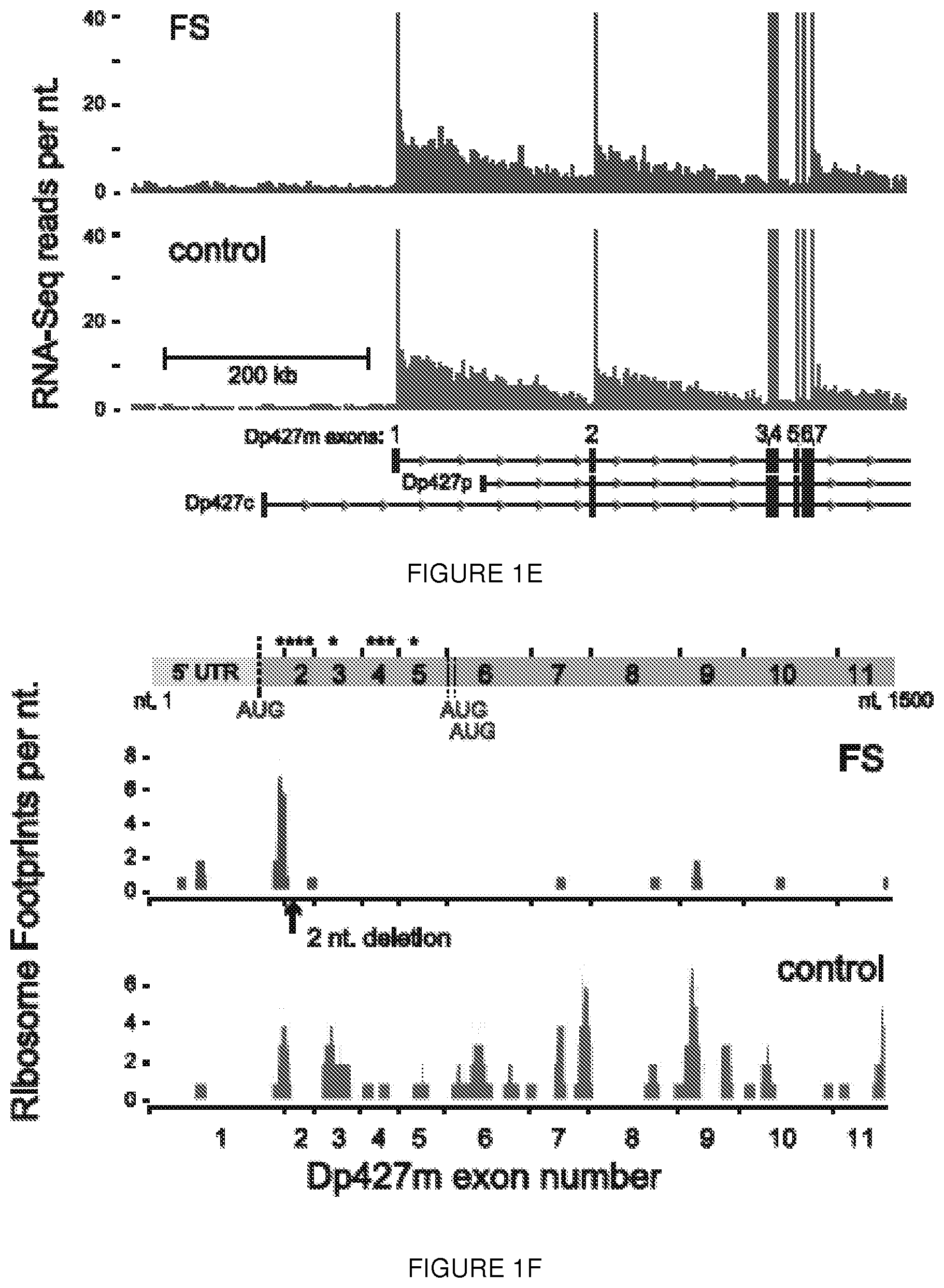
[0024] FIGS. 1A-1F. Human biopsy samples corroborate translation from exon 6. FIG. 1A shows immunoblot analysis of muscle from an asymptomatic individual with a deletion of exon 2 (DEL2) resulting in a frameshift and premature stop codon (p.Tyr11PhefsX7) demonstrates expression of dystrophin of minimally decreased size. Antibodies: NCL-DYS1 (rod domain), NCL-DYS2 (C-terminal). FIG. 1B shows sequences from mass spectrometric analysis of dystrophin peptides from muscle biopsy of the deletion exon 2 individual (SEQ ID NO: 28) results in the identification of no peptides encoded prior to M124 (in exon 6), whereas peptides encoded within exon 2, 3, 4 were readily identified in control muscle (Control) (SEQ ID NO: 29). The control peptides of FIG. 1B are found at positions 24-61, 83-104, 125-141, and 196-226 of the dystrophin reference sequence, i.e., UniProt accession number P11532, set out in SEQ ID NO: 27. FIG. 1C shows immunoblot analysis of dystrophin expression of muscle from a BMD patient with a truncating frameshift (FS) mutation in exon 2 (c.40_41del), from a normal control (WT), and from a DMD patient with a duplication of exon 2 (DUP2). In the presence of the premature stop codon induced by the frameshifting mutation, a dystrophin protein of diminished size and amount can be detected using a C-terminal antibody (PA1-21011, Thermo, Inc.; red) but not using an antibody detected to epitopes encoded within exon 1 (Manex1A, green). In contrast, dystrophin is entirely absent in the Dup2 patient. FIG. 1D shows ribosome profiling data was used to compute a translation efficiency (TE) metric for each of the 1000 most abundant transcripts (by mRNA mass) from patient FS (c.40_41del) and normal control muscle. TE value for each gene was calculated from the normalized number of ribosome footprint sequence reads divided by the number of RNA-Seq reads mapped within the coding (CDS) sequence. The rank transcript abundance of the top 1000 genes was computed from the total number of mapped reads per transcript. The subset of genes classified as `sarcomeric` by Gene Ontology annotation are colored red and the location of the DMD gene is circled. FIG. 1E shows RNA-Seq read depth from muscle total RNA mapped to the 5' region of the DMD gene (hg19, chrX:32,737,599-33,487,390). Read depth for Dp427m exons 1 through 7 was truncated at 40 reads per nucleotide; the exonic read depth ranged from 67 to 91 (FS, c.40_41del) and 58 to 89 (normal) reads per nucleotide. FIG. 1F shows ribosome footprints mapped to the 5' region (nt. 1 to 1500) of the Dp427m (NM_004006.2) transcript. The locations of the exon 1 Dp427m start codon and the c.40_41del mutations are shown, with the short ORF (p.Glu14Argfs*17) as the first CDS segment (green) separated from the remainder of the CDS (green) beginning at the exon 6 alternate AUG (green) initiation codons. Asterisks show the locations of the 9 out-of-frame AUG codons in exons 1 through 5.
[0025] FIGS. 2A-2F. Dystrophin exon 5 can induce cap-independent translation. (FIG. 2A) Induction of translation of the downstream (FLuc) cistron in an in vitro transcription/translation system (rabbit reticulocyte lysate [RRL], left) and following transfection into C2C12 cells (right) in a dicistronic dual luciferase reporter. Results are expressed as the ratio of Firefly:Renilla luciferase (F/L), and normalized to the empty vector (set as 1). (FIG. 2B) Formaldehyde electrophoresis of the T7 transcription products used in the RRL assay confirms RNA integrity. (FIG. 2C) Mapping of the exon 5 IRES: dicistronic mapping constructs (left) used to map cap-independent translation activity. In each case, numbering is based upon the Dp427m cDNA sequence; the full-length construct pRdEF+4+369 (exon 1 to 6) begins at the +4 position to exclude the native AUG initiation codon. Exon 6 was preserved, and AUG2 (M124) and AUG3 (M128) were cloned in-frame with the downstream FLuc reporter. FLuc luminescence (cap-independent) is expressed as a percentage of RLuc luminescence (cap-dependent) after transfection of the dicistronic constructs in C2C12 cells (right). All results were normalized to the exon 6 alone vector, the FLuc:RLuc ratio of which was set at a value of 1. Statistical analysis was performed using a Kruskal-Wallis test, comparing the results for each construct versus the exon 6 alone vector, which resulted in levels of expression comparable to an entirely empty vector (p>0.99). Significantly increased translation of the downstream reporter was demonstrated with the exon 1 to 6 (p<0.0001), exon 2 to 6 (p=0.0175), exon 3 to 6 (p=0.0009), exon 3* to 6 (p=0.0078), exon 4 to 6 (p=0.0078), or exon 5 to 6 (p=0.0019). In contrast, deletion of exon 5 (either in whole or in part) resulted in no significant difference for all three in comparison to exon 6 alone. (FIG. 2D) RT-PCR products amplified from RNA derived from transfected C2C12 cells, using primers located as depicted as arrows on the scheme in panel (FIG. 2C), shows no evidence of altered splicing. (FIG. 2E) Northern blot analysis of C2C12 transfected cells using a P.sup.32 radiolabeled probe targeting the FLuc cistron shows no evidence of RNA strand breakage to explain the increased signal in the presence of dystrophin exons 1-5. (A non-specific band of approximately 3 kb is detected in every transfection condition, including the empty vector, and is therefore unrelated to the increase in FLuc or EMCV signals compared to empty vector. (FIG. 2F) IRES activity is abrogated by the presence of a duplicated exon 2, but not by a deletion of exon 2. Error bars represent s.d.
[0026] FIGS. 3A-3D. Out-of-frame exon-skipping stimulates IRES activity in patient-derived cell lines. (FIG. 3A) Schematic representation of the human DMD exon 1-10 reading frame (blue) and 5'UTR (red). Blue numbers above each exon indicate cDNA positions; red numbers at the base of each exon indicate the amino acid position. The canonical actin binding domain 1 is represented, along with the predicted (via ScanProsite) CH and ABS domains. (FIG. 3B) Schematic representation of exon 2 (SEQ ID NO: 30). The selected targeted sequences are indicated below, affecting either splice acceptor (S.A.), splice donor (S.D.), or exon splice enhancer (E.S.E.) sequences (as predicted using Human Splicing Finder or ESE finder 3.0). (FIG. 3C) Two copies each of U7-C and U7-AL were cloned into the same AAV plasmid, as they were the most efficient in skipping exon 2 (see FIGS. 10A-10B). The resulting construct is referred to as U7-ACCA. RT-PCR results after infection of U7-ACCA vector (1E11 vg) or H2A antisense oligonucleotide (AON H2A) into either wt or duplicated exon 2 FibroMyoD (FM) cells. These are derived from patient fibroblast lines stably infected with hTERT and a tet-inducible MyoD lentivirus; treatment with doxycycline results in transdifferentiation into a myogenic lineage, with subsequent dystrophin mRNA expression. (FIG. 3D) Immunoblot performed 14 days after infection of FM cells with U7-ACCA shows expression of the smaller N-truncated dystrophin protein (arrow). Antibody: C-terminal dystrophin (PA1-21011, Thermo, Inc.). A smaller band of approximately 390 kDa is detected in every lane, but is non-specific (as seen in the untreated sample) and does not correspond to the IRES-driven isoform. (The image was assembled for clarity; complete images are included as FIGS. 11A-11B).
[0027] FIGS. 4A-4F. Intramuscular delivery of U7-ACCA results in significant N-truncated dystrophin expression in Dup2 mice, restoring localization of dystrophin-associated proteins. (FIGS. 4A-4B) RT-PCR results performed 4 weeks after TA intramuscular injection of 1e11vg U7-ACCA show nearly complete skipping of both copies of exon 2 in both (FIG. 4A) Dup2 and (FIG. 4B) control BI6 mice (PDN: methylprednisolone 1 mg/kg/day intraperitoneal). In Dup2 animals (FIG. 4A), quantification revealed the Dup2 transcript to be 5.1% of total, whereas the wild type was 8.6% and the Del2 transcript was 86.3%. In wild type BI6 animals (FIG. 4B), the wild type transcript was 14.2% and Del2 transcript was 85.8%. (FIG. 4C) RNA-Seq read depth using a tibialis anterior muscle total RNA library from Dup2 U7-ACCA treated (upper) and Dup2 untreated (lower) mice, mapped to the 5' region of the mouse Dmd gene (mm9, chrX:80,150,000-81,050,000). (FIG. 4D) Immunoblot performed a month after infection shows significant expression of the N-truncated isoform (asterisk) in both Dup2 and control BI6 mice. The protein induced in BI6 males injected with U7-ACCA is of the same size as that expressed in the Dup2 treated animals, confirming the size difference between this protein and the full-length isoform. (C-terminal antibody: PA1-21011,Thermo, Inc). Coomassie staining of the same samples demonstrates no difference in migration behavior. (FIG. 4E) Immunofluorescent staining of dystrophin (C-terminal antibody: PA1-21011,Thermo, Inc), .beta.-dystroglycan (Beta-DG; MANDAG2); and neuronal nitric oxide synthetase (nNOS; sc-648; Santa Cruz). (FIG. 4F) Evans blue (EBD) protection assay in Dup2 mice one month after intramuscular injection with 1e11vg shows stabilization of muscle membranes. Evans blue uptake (red) is seen only in fibers without positive dystrophin expression (green, C-terminal antibody: PA1-21011,Thermo, Inc). (Dup2 mice used for these panel; n=5)
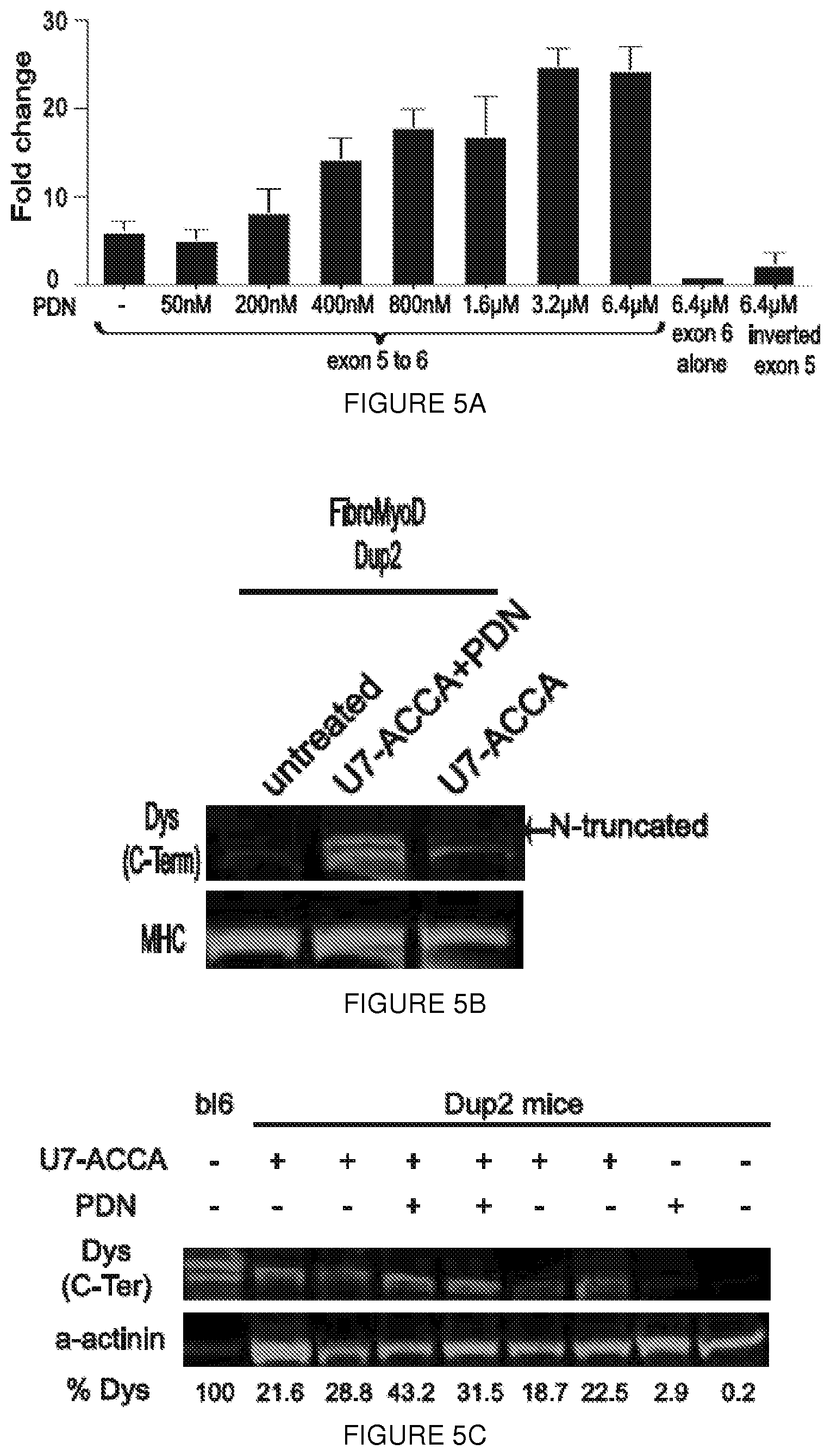
[0028] FIGS. 5A-5F. Glucorticoid activation of the dystrophin IRES. (FIG. 5A) Dual luciferase assay performed on lysates from C2C12 cells transfected with the pRDEF vector carrying the exon 5-6 IRES construct. Methylprednisolone (PDN) increases IRES activity in a dose-dependent fashion. Error bars representate s.d. (FIG. 5B) Dup2 FM cells treated with both U7-ACCA and PDN (6.4 .mu.M) show increased dystrophin expression. The image intensity for the wild-type lane was lowered to allow identification of bands. MHC=myosin heavy chain (loading control). (FIG. 5C) Representative immunoblot demonstrates increased expression of dystrophin in Dup2 mice injected with 1e11vg U7-ACCA after treatment with PDN (1 mg/kg/day). % Dys: intensity ratio of dystrophin:.alpha.-actinin, normalized to control muscle. (FIG. 5D) Quantification of the dystrophin/.alpha.-actinin signal in U7-ACCA treated muscles in the presence or absence of PDN. Five animals treated with U7-ACCA in the tibialis anterior muscles were injected with either PBS or PDN (1 mg/kg/day). Immunoblot was performed on each muscle in duplicate, and the signals for both dystrophin and .alpha.-actinin from the resulting 5 lanes were quantified using ImageJ. Significantly more dystrophin was present in muscles from PDN-treated animals (P=0.0159, two tailed Mann-Whitney test, error bars represent s.d.). (FIG. 5E) Representative western blot demonstrates an increased level of utrophin in Dup2 compared to BI6 mouse. Treatment with PDN (1 mg/kg/day) does not increase expression of utrophin. (FIG. 5F) Quantification of the utrophin/a-actinin signal in treated muscles in the presence or absence of U7-ACCA and PDN. Five animals treated with U7-ACCA in the tibialis anterior muscles were injected with either PBS or PDN (1 mg/kg/day). The signals for both utrophin and .alpha.-actinin from the resulting 5 lanes were quantified using ImageJ. No significance was detected between the four (Kruskal-Wallis, error bars represent s.d.).
[0029] FIGS. 6A-6E. Expression of the IRES-driven isoform improves muscle membrane integrity and protects Dup2 muscle from contraction-induced damage. Dup2 tibialis anterior muscles were treated by intramuscular injection of 5e11vg U7-ACCA alone or with methylprednisolone (PDN: 1 mg/kg/day intraperitoneal) and analyzed at 4 weeks post-injection. (FIG. 6A) Central nucleation in untreated Dup2 animals (73.0.+-.1.6% of myofibers) was significantly reduced by treatment with U7-ACCA alone (65.2.+-.2.2%, ***p=0.0002). No significant difference was observed between Dup2 and Dup2+PDN. (FIG. 6B) The percentage of Evans blue dye (EBD)-positive fibers in untreated Dup2 muscle (14.7.+-.6.6%; one outlier is represented as a dot) is reduced by treatment with U7-ACCA alone (2.8.+-.1.8%, *p=0.0310) or in combination with prednisone (0.65.+-.0.5%, ***p=0.0005). No significant difference was observed betweenDup2 and Dup2+PDN. EBD-positive fibers were quantified as a percent out of a total of 5,000 fibers counted per animal. (FIG. 6C) Normalized maximum hindlimb (Norm max HL) grip strength in untreated Dup2 mice (2.22.+-.0.26 kg force/kg mass of animal, or kgf/kg) is significantly lower than B16 (3.36.+-.0.37 kgf/kg, ***p<0.0001). Significantly improved strength follows treatment with either U7-ACCA alone (3.35.+-.0.32 kgf/kg, ***p<0.0001) or in combination with prednisone (3.17.+-.0.28 kgf/kg, ***p=0.0002), both of which restore strength to a level not significantly different from that seen in BI6. No significant difference was observed between Dup2 and Dup2+PDN. (FIG. 6D) Normalized specific force following tetanic contraction in untreated Dup2 animals (170.9.+-.14.3 mN/mm.sup.2) is significantly less than in BI6 (274.0.+-.12.1 mN/mm.sup.2,**p=0.0061). Significantly increased force follows treatment with U7-ACCA alone (236.04.+-.19.4 mN/mm.sup.2, *p=0.0350) or with prednisone (251.2.+-.10.4 mN/mm.sup.2, **p=0.0025), both of which restore specific force to a level not significantly different from that seen in BI6. No significant difference was observed between Dup2 and Dup2+PDN. (FIG. 6E) Treatment significantly protects Dup2 muscle from loss of force following repetitive eccentric contractions. Two-way analysis of variance demonstrates significant improvement in decay curves versus untreated Dup2 (*p<0.05 and ***p<0.001), and Bonferroni post-hoc analysis demonstrates that the combination of both treatment showed no significant difference from control BI6 in force retention following contractions #3 to #10 (*p<0.05 and ***p<0.001). No significant difference was observed between Dup2 and Dup2+PDN (p<0.99). Two way ANOVA demonstrates significant difference between Dup2+U7 and Dup2+U7+PDN (*p<0.05) (FIGS. 6A-6C) n=4 animals studied for each condition and when applied 2000 fibers count/mouse, two tailed Kruskal-Wallis, error bar as s.d.); (FIGS. 6D-6E) n=5 muscles from at least 3 animals, error bar as s.e.m.
[0030] FIGS. 7A-7G. Mutational analysis in the deletion exon 2 asymptomatic boy. (FIG. 7A) H&E-stained muscle section from the patient with deletion of exon 2 (DEL2) reveals an absence of dystrophic features. (FIG. 7B) Immunohistochemical staining of muscle sections from the same patient using NCL-DYS3 antibody (exons 10-12). Manex1A staining (exon 1 specific) was not performed at that time, and tissue is no longer available. (FIG. 7C) CGH profile of the genomic context (top panel) and of the entire X chromosome (bottom panel) of the 12.983 bp deletion including exon 2 (shown in the overlay track at the bottom of the top panel). (FIG. 7D) Alignment of the sequenced junction with the reference genome sequence (NCBI hg18) (SEQ ID NOs: 31-33, respectively). Proximal and distal reference sequences are colored differently and the junction is in black. Vertical bars between the sequences represent sequence homology. A microhomology of 5 bp (CTGTG, shown a box) is found at the junction between the distal and proximal sequences, characteristic for non-homologous end joining. (FIG. 7E) Genomic sequences of the breakpoint with the microhomology sequence underlined in blue (SEQ ID NO: 34). (FIGS. 7F-7G) RT-PCR and sequencing results confirm the deletion of exon 2 at RNA level.
[0031] FIGS. 8A-8D. Immunofluorescent analysis of muscle from the frameshift (c.40_41del) patient. (FIG. 8A) Immunostaining using a dystrophin antibody (Abcam 15277, C-terminal) shows dystrophin at the sarcolemmal membrane in both control and patient muscle biopsies whereas Manex1A staining is absent in the patient sample, confirming the lack of expression of the epitope encoded by exon 1. (FIG. 8B) Ribosome Profiling of the DMD muscle-isoform transcript. The normalized average reads (read depth per nt. versus the average read depth on NM_004006) for RNA-Seq reads are plotted every 25 nucleotides using an averaged normalized average read depth per nucleotide calculated from a 500 bp. sliding window. Reads from patient FS(c.40_41del) are shown in red and from control muscle in grey, with regression lines shown for each set of averages. (FIG. 8C) as in (FIG. 8B) except using RPF-Seq reads, with the linear regression line calculated for the CDS region only. (FIG. 8D) The exon structure of the NM_004006 transcript is drawn to the same scale as the x-axis from (FIG. 8B) and (FIG. 8C). The arrow indicates the location of the alternate translation initiation sites in exon 6. Since the experiment used total RNA, the RNA-Seq reads mapping to NM_004006 are derived from both nascent and mature transcripts. The 5' to 3' gradient of RNA-Seq reads shown in (FIG. 8B) agrees with an original estimate from human skeletal muscle of the relative excess of 5' exons in nascent RNA due to the transit time (.about.16 hrs.) for RNA polymerase to transcribe across the .about.2.2 Mb of chr. X region containing the 79 DMD exons of the muscle isoform. Regression analysis of the RPF-Seq reads does not indicate a 5' to 3' gradient, inferring that ribosomes are equally distributed across the length of the mature transcript.
[0032] FIGS. 9A-9C. The dystrophin RES is not ubiquitously active. (FIG. 9A) Dual luciferase assays demonstrate activation in two myogenic cell lines (C2C12, and a commercial human skeletal muscle myoblast line [hSKMM]), but not in HEK209K cells, suggesting preferential activation in cells of a myogenic lineage. (FIG. 9B) Northern blot from transfected C2C12 and 293k using a probe against Firefly luciferase demonstrates the presence of the transcript as well as the previously described (FIGS. 2A-2F) nonspecific band. Notably, this band is present in all conditions, including following transfection with the exon 6 alone construct, and therefore is unrelated to the the fold change seen with exon 5 containing constructs. (FIG. 9C) RT-PCR products amplified from RNA derived from transfected 293k cells shows no evidence of altered splicing. Error bars represent s.d.
[0033] FIGS. 10A-10B. Optimization of AAV mediated U7 exon-skipping. (FIG. 10A)
Four different target sequences (AS, AL, B or C) were cloned into AAV under the control of U7. Infection of these AAV either alone (FIG. 10A) or in combination (FIG. 10B) were performed in both control and duplicated exon 2 patient derived-FibroMyoD. 3 days post AAV infection, RT-PCR results demonstrated that in U7-C is able to induce exon-skipping in both control and duplicated exon 2 patient FibroMyoD whereas U7-AL is only able to induce skipping in the patient cell lines. Two copies constructs U7-C and U7-AL were cloned into a same AAV plasmid (U7-ACCA). (c) Transfection of a Dup2 patient's cultured MyoD transformed fibroblasts and primary myoblasts, using an AON (AONH2A) which targets an internal exon 2 sequence, gave similar results, but at lower efficiency than U7-mediated skipping.
[0034] FIGS. 11A-11B. Western blot from patient-derived cell lines. (FIG. 11A) The original western blot from FIG. 3d seen in two different imaging intensities, low (upper panel) and high (lower panel). Lane 1 from the upper panel and lanes 2 and 4 from the lower panel were used to assemble FIG. 3d. FM=FibroMyoD derived control cell lines; FM Dup=FibroMyoD patient-derived cell lines from an exon 2 duplicated patient; FS=protein from muscle biopsy of c.40_41del. (FIG. 11B) Coomassie staining of the same samples as seen in FIG. 4c demonstrates no significant difference in migration behavior.
[0035] FIGS. 12A-12B. Glucocorticoid increases IRES activity but cannot force its activation. (FIG. 12A) Dual luciferase assay results after transfection of 3 constructs in 293k treated with glucocorticoid demonstrate that IRES activity cannot be induced by this compound. Error bars represent s.d. (FIG. 12B) Genomic qPCR of AAV copy number confirm that increase of dystrophin level detected by western blot in PDN treated mice is not due an increased number of AAV vector in the PDN treated animals. N=4 animals per group. Error bars represent s.d.
[0036] FIG. 13 is the rh74 genome sequence (SEQ ID NO: 14) wherein nucleotides 210-2147 are the Rep 78 gene open reading frame, 882-208 are the Rep52 open reading frame, 2079-2081 are the Rep78 stop, 2145-2147 are the Rep78 stop, 1797-1800 are a splice donor site, 2094-2097 are a splice acceptor site, 2121-2124 are a splice acceptor site, 174-181 are the p5 promoter+1 predicted, 145-151 are the p5 TATA box, 758-761 are the p19 promoter+1 predicted, 732-738 are the p19 TATA box, 1711-1716 are the p40 TATA box, 2098-4314 are the VP1 Cap gene open reading frame, 2509-2511 are the VP2 start, 2707-2709 are the VP3 start and 4328-4333 are a polyA signal.
[0037] FIG. 14 shows a map of a plasmid with an AAV genome insert of an exemplary exon 2-targeted U7snRNA.
[0038] FIG. 15A shows the AAV genome insert (3' to 5') (SEQ ID NO: 15 is the same sequence in the 5' to 3' direction) of the plasmid of FIG. 14. FIG. 15B shows the reverse complement (SEQ ID NO: 26) of the sequence in FIG. 15A.
[0039] U7 encode for a U7snRNP that share some features with spliceosomal snRNPs. Although it is not involved in pre-mRNA splicing, it processes the 3' ends of histone mRNA (Muer and Schumperli 1997; Dominski and Marzluff 1999). Nucleotides 1-113 of SEQ ID NO: 15 correspond to the 3' ITR, nucleotides 114-220 of SEQ ID NO: 15 correspond to the 3' untranslated region (UTR) (reverse orientation sequence). Nucleotides 221-251 of SEQ ID NO: 15 correspond to SmOPT (reverse orientation sequence). SmOPT is a modification of the original Sm-binding site of U7 snRNA with a consensus sequence derived from spliceosomal snRNAs (Grimm et al. 1993; Stefanovic et al. 1995a). Nucleotides 252-262: of SEQ ID NO: 15 correspond to a loop (reverse orientation sequence). Nucleotides 263-295 correspond to U7-Along (reverse orientation sequence), which is an antisense sequence that targets the acceptor site of exon 2. Nucleotides 296-551 of SEQ ID NO: 15 correspond to U7 (reverse orientation sequence), nucleotides 558-664 of SEQ ID NO: 15 correspond to 3' UTR (reverse orientation sequence), nucleotides 665-695 of SEQ ID NO: 15 correspond to SmOPT (reverse orientation sequence), and nucleotides 696-706 of SEQ ID NO: 15 correspond to a loop (reverse orientation sequence). Nucleotides 707-731 of SEQ ID NO: 15 correspond to U7-C (reverse orientation sequence), which is an antisense sequence that targets the donor site of exon 2. Nucleotides 732-987 of SEQ ID NO: 15 correspond to U7 (reverse orientation sequence), nucleotides 994-1100 of SEQ ID NO: 15 correspond to 3' UTR (reverse orientation sequence), nucleotides 1111-1131 of SEQ ID NO: 15 correspond to SmOPT (reverse orientation sequence), nucleotides 1132-1142 of SEQ ID NO: 15 correspond to a loop (reverse orientation sequence), nucleotides 1143-1167 of SEQ ID NO: 15 correspond to U7-C (reverse orientation sequence), nucleotides 1168-1423 of SEQ ID NO: 15 correspond to U7 (reverse orientation sequence), nucleotides 1430-1536 of SEQ ID NO: 15 correspond to 3' UTR (reverse orientation sequence), nucleotides 1537-1567 of SEQ ID NO: 15 correspond to SmOPT (reverse orientation sequence), nucleotides 1568-1578 of SEQ ID NO: 15 correspond to a loop (reverse orientation sequence), nucleotides 1579-1611 of SEQ ID NO: 15 correspond to U7-Along (reverse orientation sequence), nucleotides 1612-1867 of SEQ ID NO: 15 correspond to U7 (reverse orientation sequence) and nucleotides 1920-2052 of of SEQ ID NO: 15 correspond to the ITR.
[0040] FIG. 16 shows a schematic of a vector used in creation of a mdx.sup.dup2 (Dup2) mouse.
[0041] FIGS. 17A-17F shows (FIG. 17A) RT-PCR performed on 5 different Dup2 mouse muscles one month after tail vein injection of AAV9.U7-ACCA (3.3E12 vg/kg). As demonstrated by the presence of multiple transcripts (here labeled Dup2, wt, and Del2), U7-ACCA treatment is able to force skipping of one or both copies of exon 2 in all muscles tested. (TA: tibialis anterior; Gas: gastrocnemius; : heart; Tri: triceps; dia: diaphragm.) (FIG. 17B) Western blot performed on 5 different muscles one month after injection demonstrates the presence of dystrophin in all tested muscles. (FIG. 17C) Immunostaining of dystrophin on the same samples confirms dystrophin expression and its proper localization at the sarcolemma. (FIG. 17D) Evaluation of both forelimb and hindlimb grip strength demonstrates a complete correction of grip strength in Dup2 animals treated with AAV9.U7-ACCA. (FIG. 17E) Normalized specific and total forces following tetanic contraction show improvement in muscle force in comparison to untreated Dup2 animals. (FIG. 17F) Cardiac papillary muscles demonstrate improvements in length-dependent force generation in treated animals.
[0042] FIGS. 18A-18G. (FIG. 18A) IM study design. Escalating doses of the AAV9.U7snRNA-ACCA vector were delivered to the tibialis anterior muscle at 2 months, and muscle analyzed at 3 months by mRNA, protein, and electrophysiology studies. (FIG. 18B) Quantification of mRNA by RT-PCR at ascending dose levels of IM injection. Transcripts contain either two (Dup2), one (WT), or zero copies (.DELTA.2) of exon 2. Expression of the N-truncated dystrophin following ascending dose levels of IM injection. Protein expression by (FIG. 18C) immunofluorescence or (FIG. 18D) immunoblot demonstrates a dose response. (FIG. 18E) Quantification of the immunoblot suggests maximal protein expression at 3.1E11 vg. Amelioration of deficits in absolute force (FIG. 18F), specific force (FIG. 18G), in response to eccentric contraction following IM injection into the tibialis anterior muscle of 3.1 E11 vg.
[0043] FIGS. 19A-19E. (FIG. 19A) IV study design. Escalating doses of the AAV9.U7snRNA-ACCA vector were delivered systemically at 2 months, and muscle analyzed at 3 months by mRNA, protein, and electrophysiology studies. (FIG. 19B) Quantification of mRNA by RT-PCR at ascending dose levels of IV injection. Transcripts contain either two (Dup2), one (WT), or zero copies (.DELTA.2) of no exon 2. (FIG. 19C) Quantification of dystrophin by immunoblot following IV injection. Expression follows a dose response, with expression in triceps lagging that in heart and diaphragm. (FIG. 19D) Immunostaining of dystrophin from BI6 and Dup2. (FIG. 19E) expression following IV injection. A dose response is seen, with significant dystrophin expression in the heart and diaphragm at higher doses.
[0044] FIG. 20. Early injection of AAV9.U7-ACCA prevents the muscle pathology in the Dup2 mouse. Immunostaining of dystrophin demonstrates production and localization of N-terminally truncated dystrophin at the plasma membrane. No centronucleation was observed following hematoxylin and eosin staining. By 6 months of age, untreated Dup2 mice typically demonstrate 60% of their fibers with central nuclei (data not shown).
[0045] FIG. 21. Generation of alternative N-terminally truncated dystrophins in human cell lines derived from patients carrying mutations within the first nine exons. RT-PCR results after skipping of exon 2 using either AAV1.U7-ACCA vector (1.times.10E11 vector genomes) or H2A antisense oligonucleotide (AON H2A) in various patient cell lines carrying mutation within exon 1 to 4. This results in approximately 90% of transcript lacking exon 2 (quantification not shown). FM=FibroMyoD cells derived from healthy human subject. Immunoblot performed 14 d after infection of FibroMyod cells with AAV1.U7-ACCA shows expression of the N-terminally trucated dystrophin protein. A smaller band of approximately 390 kDa is detected in every lane but is nonspecific (as seen in the untreated sample) and does not correspond to the IRES-driven isoform. (The image was assembled for clarity, with wild-type contrast altered to clearly show bands.)
[0046] FIGS. 22A-22C. Expression of the N-truncated dystrophin following treatment with PPMO antisense oligonucleotide. (FIG. 22A) transfection in C2C12 mouse myoblasts or (FIG. 22B) intramuscular injection into Dup2 mouse tibialis anterior muscles of AL-PPMO. RT-PCR results from treated cells or muscles demonstrate an efficient skipping of exon 2. (FIG. 22C) immunofluorescence of dystrophin shows expression of a plasma membrane protein following intramuscular injection of AL-PPMO antisense oligonucleotide.
DESCRIPTION
[0047] As noted above, the present disclosure contemplates methods and products for preventing, delaying the progression of, and/or treating patients with one or more 5' mutations of the DMD gene that are based on the activation of a glucocorticoid-inducible IRES in exon 5 of the DMD gene. The activation of the inducible IRES in exon 5 of the DMD gene generates a functional N-terminally truncated dystrophin isoform.
[0048] As used herein, a "5' mutation of the DMD gene" is a mutation within or affecting exon 1, 2, 3 or 4 of the DMD gene. In the methods of the invention, the patients treated do not have a DMD exon 2 duplication, but a "mutation affecting exon 1, 2, 3 or 4" as contemplated herein can be a duplication other than a DMD exon 2 duplication.
[0049] In one aspect, the methods involve using an "DMD exon 5 IRES-activating oligomer construct." As used herein, a DMD exon 5 IRES-activating oligomer construct targets exon 2 to induce altered splicing that results in the exclusion of exon 2 from the mature RNA causing a frameshift in the DMD gene reading frame and inducing utilization of the IRES in exon 5 for translational initiation.
[0050] In some embodiments, the DMD exon 5 IRES-activating oligomer construct targets one of the following portions (shown 5' to 3') of exon 2 of the DMD gene.
TABLE-US-00003 B: (SEQ ID NO: 1) TCAAAAGAAAACATTCACAAAATGGGTA (+17 + 44) C: (SEQ ID NO: 2) GCACAATTTTCTAAGGTAAGAAT (+48 - 8) AL: (SEQ ID NO: 3) TAGATGAAAGAGAAGATGTTCAAAAGAAAAC (-3 + 28) AS: (SEQ ID NO: 4) TAGATGAAAGAGAAGATGTTC (-3 + 18)
[0051] In some embodiments, a rAAV is used to deliver a U7 small nuclear RNA polynucleotide construct that is targeted to DMD exon 2 by an antisense polynucleotide. In some embodiments, the U7 small nuclear RNA is a human U7 small nuclear RNA. In some embodiments, the polynucleotide construct is inserted in the genome of a rAAV9, the genome of a rAAV6 or the genome of a rAAVrh74. In some embodiments, the U7 small nucleotide RNA construct comprises exemplary targeting antisense polynucleotides including, but not limited to the following where, for example, the "U7-AL antisense polynucleotide" is respectively complementary to and targets the "AL" exon 2 sequence in the preceding paragraph.
TABLE-US-00004 U7-B antisense polynucleotide: (SEQ ID NO: 5) TACCCATTTTGCGAATGTTTTCTTTTGA U7-C antisense polynucleotide: (SEQ ID NO: 6) ATTCTTACCTTAGAAAATTGTGC U7-AL antisense polynucleotide: (SEQ ID NO: 7) GTTTTCTTTTGAAGATCTTCTCTTTCATCTA U7-AS antisense polynucleotide: (SEQ ID NO: 8) GAAGATCTTCTCTTTCATCTA
[0052] In some embodiments, the DMD exon 5 IRES-activating oligomer construct is an exon 2-targeting antisense oligomer. In some embodiments, the antisense oligomers are contemplated to include modifications compared to the native phosphodiester oligodeoxynucleotide polymer to limit their nuclease sensitivity. Contemplated modifications include, but are not limited to, phosphorodiamidate morpholino oligomers (PPOs), cell penetrating peptide-conjugated PMOs (PPMOs), PMO internalizing peptides (PIP) [(Betts et al., Sci. Rep., 5: 8986 (2015)], tricyclo-DNA (tcDNA) [Goyenvalle et al., Nat. Med., 21: 270-275 (2015)] and 2'O-methyl-phosphorothioate modifications. Exemplary DMD exon 5 IRES-activating oligomer constructs that are exon 2-targeting antisense oligomers include, but are not limited to, the following antisense oligomers (shown 5' to 3') where, for example, the "B antisense oligomer" respectively targets the "B" exon 2 target in paragraph [0032].
TABLE-US-00005 B antisense oligomer: (SEQ ID NO: 9) UACCCAUUUUGCGAAUGUUUUCUUUUGA C antisense oligomer: (SEQ ID NO: 10) AUUCUUACCUUAGAAAAUUGUGC AL antisense oligomer: (SEQ ID NO: 11) GUUUUCUUUUGAACAUCUUCUCUUUCAUCUA AS antisense oligomer: (SEQ ID NO: 12) GAACAUCUUCUCUUUCAUCUA H2A (+12 + 41): (SEQ ID NO: 13) CCAUUUUGUGAAUGUUUUCUUUUGAACAUC
[0053] In another aspect, a method of ameliorating a muscular dystrophy (such as DMD or BMD) in a patient with a 5' mutation of the DMD gene is provided. In some embodiments, the method comprises the step of administering a rAAV to the patient, wherein the genome of the rAAV comprises a DMD exon 5 IRES-activating oligomer construct. In some embodiments, the method comprises the step of administering a DMD exon 5 IRES-activating oligomer construct that is an exon 2-targeting antisense oligomer. In some embodiments, the patient is also treated with a glucocorticoid.
[0054] In yet another aspect, the invention provides a method of inhibiting the progression of dystrophic pathology associated with a muscular dystrophy (such as DMD or BMD). In some embodiments, the method comprises the step of administering a rAAV to a patient with a 5' mutation of the DMD gene, wherein the genome of the rAAV comprises a DMD exon 5 IRES-activating oligomer construct. In some embodiments, the method comprises the step of administering a DMD exon 5 IRES-activating oligomer construct that is an exon 2-targeting antisense oligomer. In some embodiments, the patient is also treated with a glucocorticoid.
[0055] In still another aspect, a method of improving muscle function in a patient with a 5' mutation of the DMD gene is provided. In some embodiments, the method comprises the step of administering a rAAV to the patient, wherein the genome of the rAAV comprises a DMD exon 5 IRES-activating oligomer construct. In some embodiments, the method comprises the step of administering a DMD exon 5 IRES-activating oligomer construct that is an exon 2-targeting antisense oligomer. In some embodiments, the improvement in muscle function is an improvement in muscle strength. The improvement in muscle strength is determined by techniques known in the art such as the maximal voluntary isometric contraction testing (MVICT). In some instances, the improvement in muscle function is an improvement in stability in standing and walking. The improvement in stability strength is determined by techniques known in the art such as the 6-minute walk test (6MWT) or timed stair climb. In some embodiments, the patient is also treated with a glucocorticoid.
[0056] In another aspect, the invention provides a method of delivering a DMD exon 5 IRES-activating oligomer construct to an animal (including, but not limited to, a human) with a 5' mutation of the DMD gene. In some embodiments, the method comprises the step of a rAAV to the patient, wherein the genome of the rAAV comprises a DMD exon 5 IRES-activating oligomer construct. In some embodiments, the method comprises the step of administering a DMD exon 5 IRES-activating oligomer construct that is an exon 2-targeting antisense oligomer. In some embodiments, the animal is also treated with a glucocorticoid.
[0057] Cell transduction efficiencies of the methods of the invention described herein may be at least about 60, about 65, about 70, about 75, about 80, about 85, about 90 or about 95 percent.
[0058] In some embodiments of the foregoing methods of the invention, the virus genome is a self-complementary genome. In some embodiments of the methods, the genome of the rAAV lacks AAV rep and cap DNA. In some embodiments of the methods, the rAAV is a SC rAAV U7_ACCA comprising the exemplary genome set out in FIG. 15. In some embodiments, the rAAV is a rAAV6. In some embodiments, the rAAV is a rAAV9. In some embodiments the rAAV is a rAAV rh74 (FIG. 13).
[0059] In yet another aspect, the invention provides a rAAV comprising the AAV rAAV9 capsid and a genome comprising the exemplary DMD exon 5 IRES-activating U7 snRNA polynucleotide construct U7_ACCA. In some embodiments, the genome of the rAAV lacks AAV rep and cap DNA. In some embodiments, the rAAV comprises a self-complementary genome. In some embodiments of the methods, the rAAV is a SC rAAV U7_ACCA comprising the exemplary genome is set out in FIGS. 15A-15B. In some embodiments, the rAAV is a rAAV6. In some embodiments, the rAAV is a rAAV9. In some embodiments the rAAV is a rAAV rh74 (FIG. 13).
[0060] Recombinant AAV genomes of the invention comprise one or more AAV ITRs flanking at least one DMD exon 5 IRES-activating U7 snRNA polynucleotide construct. Genomes with DMD exon 5 IRES-activating U7 snRNA polynucleotide constructs comprising each of the targeting antisense sequences set out in paragraph [0033] are specifically contemplated, as well as genomes with DMD exon 5 IRES-activating U7 snRNA polynucleotide constructs comprising each possible combination of two or more of the targeting antisense sequences set out in paragraph [0033]. In some embodiments, including the exemplified embodiments, the U7 snRNA polynucleotide includes its own promoter. AAV DNA in the rAAV genomes may be from any AAV serotype for which a recombinant virus can be derived including, but not limited to, AAV serotypes AAV-1, AAV-2, AAV-3, AAV-4, AAV-5, AAV-6, AAV-7, AAV-8, AAV-9, AAV-10, AAV-11 and AAV rh.74. As noted in the Background section above, the nucleotide sequences of the genomes of various AAV serotypes are known in the art. In some embodiments of the invention, the promoter DNAs are muscle-specific control elements, including, but not limited to, those derived from the actin and myosin gene families, such as from the myoD gene family [See Weintraub et al., Science, 251: 761-766 (1991)], the myocyte-specific enhancer binding factor MEF-2 [Cserjesi and Olson, Mol. Cell. Biol., 11: 4854-4862 (1991)], control elements derived from the human skeletal actin gene [Muscat et al., Mol. Cell. Biol., 7: 4089-4099 (1987)], the cardiac actin gene, muscle creatine kinase sequence elements [Johnson et al., Mol. Cell. Biol., 9:3393-3399 (1989)] and the murine creatine kinase enhancer (MCK) element, desmin promoter, control elements derived from the skeletal fast-twitch troponin C gene, the slow-twitch cardiac troponin C gene and the slow-twitch troponin I gene: hypoxia-inducible nuclear factors [Semenza et al., Proc. Natl. Acad. Sci. USA, 88: 5680-5684 (1991)], steroid-inducible elements and promoters including the glucocorticoid response element (GRE) [See Mader and White, Proc. Natl. Acad. Sci. USA, 90: 5603-5607 (1993)], and other control elements.
[0061] DNA plasmids of the invention comprise rAAV genomes of the invention. The DNA plasmids are transferred to cells permissible for infection with a helper virus of AAV (e.g., adenovirus, E1-deleted adenovirus or herpesvirus) for assembly of the rAAV genome into infectious viral particles. Techniques to produce rAAV particles, in which an AAV genome to be packaged, rep and cap genes, and helper virus functions are provided to a cell are standard in the art. Production of rAAV requires that the following components are present within a single cell (denoted herein as a packaging cell): a rAAV genome, AAV rep and cap genes separate from (i.e., not in) the rAAV genome, and helper virus functions. The AAV rep genes may be from any AAV serotype for which recombinant virus can be derived and may be from a different AAV serotype than the rAAV genome ITRs, including, but not limited to, AAV serotypes AAV-1, AAV-2, AAV-3, AAV-4, AAV-5, AAV-6, AAV-7, AAV-8, AAV-9, AAV-10, AAV-11 and AAV rh74. Use of cognate components is specifically contemplated. Production of pseudotyped rAAV is disclosed in, for example, WO 01/83692 which is incorporated by reference herein in its entirety.
[0062] A method of generating a packaging cell is to create a cell line that stably expresses all the necessary components for AAV particle production. For example, a plasmid (or multiple plasmids) comprising a rAAV genome lacking AAV rep and cap genes, AAV rep and cap genes separate from the rAAV genome, and a selectable marker, such as a neomycin resistance gene, are integrated into the genome of a cell. AAV genomes have been introduced into bacterial plasmids by procedures such as GC tailing [Samulski et al., Proc. Natl. Acad. S6. USA, 79:2077-2081 (1982)], addition of synthetic linkers containing restriction endonuclease cleavage sites [Laughlin et al., Gene, 23:65-73 (1983)] or by direct, blunt-end ligation [Senapathy & Carter, J. Biol. Chem., 259:4661-4666 (1984)]. The packaging cell line is then infected with a helper virus such as adenovirus. The advantages of this method are that the cells are selectable and are suitable for large-scale production of rAAV. Other examples of suitable methods employ adenovirus or baculovirus rather than plasmids to introduce rAAV genomes and/or rep and cap genes into packaging cells.
[0063] General principles of rAAV production are reviewed in, for example, Carter, Current Opinions in Biotechnology, 1533-1539 (1992); and Muzyczka, Curr. Topics in Microbial. and Immunol., 158:97-129 (1992). Various approaches are described in Ratschin et al., Mol. Cell. Biol., 4:2072 (1984); Hermonat et al., Proc. Natl. Acad. Sci. USA, 81:6466 (1984); Tratschin et al., Mol. Cell. Biol. 5:3251 (1985); McLaughlin et al., J. Virol., 62:1963 (1988); and Lebkowski et al., Mol. Cell. Biol., 7:349 (1988). Samulski et al., J. Virol., 63:3822-3828 (1989); U.S. Pat. No. 5,173,414; WO 95/13365 and corresponding U.S. Pat. No. 5,658.776; WO 95/13392; WO 96/17947; PCT/US98/18600; WO 97/09441 (PCT/US96/14423); WO 97/08298 (PCT/US96/13872); WO 97/21825 (PCT/US96/20777); WO 97/06243 (PCT/FR96/01064); WO 99/11764; Perrin et al., Vaccine, 13:1244-1250 (1995); Paul et al., Human Gene Therapy, 4:609-615 (1993); Clark et al., Gene Therapy, 3:1124-1132 (1996); U.S. Pat. Nos. 5,786,211; 5,871,982; and 6,258,595. The foregoing documents are hereby incorporated by reference in their entirety herein, with particular emphasis on those sections of the documents relating to rAAV production.
[0064] The invention thus provides packaging cells that produce infectious rAAV. In one embodiment packaging cells may be stably transformed cancer cells such as HeLa cells, 293 cells and PerC.6 cells (a cognate 293 line). In another embodiment, packaging cells are cells that are not transformed cancer cells, such as low passage 293 cells (human fetal kidney cells transformed with E1 of adenovirus), MRC-5 cells (human fetal fibroblasts), WI-38 cells (human fetal fibroblasts), Vero cells (monkey kidney cells) and FRhL-2 cells (rhesus fetal lung cells).
[0065] The rAAV may be purified by methods standard in the art such as by column chromatography or cesium chloride gradients. Methods for purifying rAAV vectors from helper virus are known in the art and include methods disclosed in, for example, Clark et al., Hum. Gene Ther., 10(6): 1031-1039 (1999); Schenpp and Clark, Methods Mol. Med., 69:427-443 (2002); U.S. Pat. No. 6,566,118 and WO 98/09657.
[0066] In another embodiment, the invention contemplates compositions comprising a DMD exon 5 IRES-activating oligomer construct of the present invention in a viral delivery vector or other delivery vehicle. Compositions of the invention comprise a pharmaceutically acceptable carrier. The compositions may also comprise other ingredients such as diluents. Acceptable carriers and diluents are nontoxic to recipients and are preferably inert at the dosages and concentrations employed, and include buffers such as phosphate, citrate, or other organic acids; antioxidants such as ascorbic acid; low molecular weight polypeptides; proteins, such as serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine, arginine or lysine; monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrins; chelating agents such as EDTA; sugar alcohols such as mannitol or sorbitol; salt-formig counterions such as sodium; and/or nonionic surfactants such as Tween, pluronics or polyethylene glycol (PEG).
[0067] Sterile injectable solutions are prepared by incorporating the active ingredient in the required amount in the appropriate solvent with various other ingredients enumerated above, as required, followed by filter sterilization. Generally, dispersions are prepared by incorporating the sterilized active ingredient into a sterile vehicle which contains the basic dispersion medium and the required other ingredients from those enumerated above. In the case of sterile powders for the preparation of sterile injectable solutions, the preferred methods of preparation are vacuum drying and the freeze drying technique that yield a powder of the active ingredient plus any additional desired ingredient from the previously sterile-filtered solution thereof.
[0068] Titers of rAAV to be administered in methods of the invention will vary depending, for example, on the particular rAAV, the mode of administration, the treatment goal, the individual, and the cell type(s) being targeted, and may be determined by methods standard in the art. Titers of rAAV may range from about 1.times.10.sup.6, about 1.times.10.sup.7, about 1.times.10.sup.8, about 1.times.10.sup.9, about 1.times.10.sup.10, about 1.times.10.sup.11, about 1.times.10.sup.12, about 1.times.10.sup.13 to about 1.times.10.sup.14 or more DNase resistant particles (DRP) per ml. Dosages may also be expressed in units of viral genomes (vg) (i.e., 1.times.10.sup.1 vg, 1.times.10.sup.8 vg, 1.times.10.sup.9 vg, 1.times.10.sup.10 vg, 1.times.10.sup.11 vg, 1.times.10.sup.12 vg, 1.times.10.sup.13 vg, 1.times.10.sup.14 vg, respectively).
[0069] Methods of transducing a target cell (e.g., a skeletal muscle) of a patient with a 5' mutation of the DMD gene with a rAAV of the invention, in vivo or in vitro, are contemplated herein. The methods comprise the step of administering an effective dose, or effective multiple doses, of a composition comprising a rAAV of the invention to an animal (including a human being) with a 5' mutation of the DMD gene. If the dose is administered prior to development of DMD, the administration is prophylactic. If the dose is administered after the development of DMD, the administration is therapeutic. In embodiments of the invention, an "effective dose" is a dose that alleviates (eliminates or reduces) at least one symptom associated with DMD being treated, that slows or prevents progression to DMD, that slows or prevents progression of a disorder/disease state, that diminishes the extent of disease, that results in remission (partial or total) of disease, and/or that prolongs survival.
[0070] Administration of an effective dose of the compositions may be by routes standard in the art including, but not limited to, intramuscular, parenteral, intravenous, oral, buccal, nasal, pulmonary, intracranial, intraosseous, intraocular, rectal, or vaginal. Route(s) of administration and serotype(s) of AAV components of rAAV (in particular, the AAV ITRs and capsid protein) of the invention may be chosen and/or matched by those skilled in the art taking into account the infection and/or disease state being treated and the target cells/tissue(s). In some embodiments, the route of administration is intramuscular. In some embodiments, the route of administration is intravenous.
[0071] Combination therapies are also contemplated by the invention. Combination therapy as used herein includes simultaneous treatment or sequential treatments. Combinations of methods of the invention with standard medical treatments (e.g., corticosteroids and/or immunosuppressive drugs) are specifically contemplated, as are combinations with other therapies such as those mentioned in the Background section above. In some embodiments, the corticosteroid is a glucocorticoid such as prednisone, deflazacort or Medrol (6-methyl-prednisolone; PDN).
EXAMPLES
[0072] Aspects and embodiments of the invention are illustrated by the following examples.
[0073] Most mutations that truncate the reading frame of the DMD gene cause loss of dystrophin expression and lead to DMD. However, amelioration of disease severity can result from alternate translation initiation beginning in DMD exon 6 that leads to expression of a highly functional N-truncated dystrophin. This novel isoform results from usage of an IRES within exon 5 that is glucocorticoid-inducible. IRES activity was confirmed in patient muscle by both peptide sequencing and ribosome profiling as described below. Generation of a truncated reading frame upstream of the IRES by exon skipping led to synthesis of a functional N-truncated isoform in both patient-derived cell lines and in a DMD mouse model, where expression protects muscle from contraction-induced injury and corrects muscle force to the same level as control mice. These results support a novel therapeutic approach for patients with mutations within the 5' exons of the DMD gene. See also, Wein et al., Abstracts/Neuromuscular Disorders, 23: 738-852 (2013).
Example 1
Evidence for IRES-Induced Translation from Human Muscle Samples
[0074] We previously published that nonsense and frameshifting mutations leading to a stop codon within at least the first two DMD exons should result in the mild BMD phenotype via exon 6 translation initiation [Gurvich et al., Human Mutation, 30: 633-640 (2009)]. However, duplication of exon 2--which is the most common single exon duplication and results in a premature stop codon within the duplicated exon 2 sequence--would seem to be an exception to this prediction, as it is usually associated with DMD [White et al., Human Mutation, 27: 938-945 (2006)]. However, a deletion of exon 2, which also results in a premature stop codon, has not been described, either in our large cohort [Flanigan et al., Human Mutation, 30: 1657-1666 (2009)] or in other large publicly available catalogues (www.dmd.nl). We interpreted this lack of reported cases to mean that the clinical features in patients with exon 2 deletions are either asymptomatic or exceedingly mild due to expression of the N-truncated isoform.
[0075] This interpretation was confirmed by the detection of a deletion of exon 2 (DEL2) in an Italian boy who first presented at age 6 years for evaluation of an incidentally detected elevation of serum creatine kinase (550 iu/l; normal value<200 iu/l). Normal early motor milestones were reported and no muscle dystrophy was ever reported in the family. His neurological examination was entirely normal at 15 years of age. Muscle biopsy showed slight fiber size variability (FIG. 7A), and in some sections an increased number of central nuclei along with some densely stained hypercontracted fibers. Immunofluorescent analysis using a C-terminal antibody showed the presence of dystrophin at the membrane (FIG. 7B). Interestingly, western blot revealed that the detected dystrophin had a smaller molecular weight (.about.410 kDa) (FIG. 1a), and mutational analysis revealed a deletion of exon 2 (FIGS. 7C-G). Subsequent peptide sequencing using tandem mass spectrometry (LC-MS/MS).sup.20 confirmed the absence of any residues encoded by exons 1 through 5 among the 99 unique peptides detected and matched to dystrophin, consistent with translation initiation within exon 6 (FIG. 1B and Table 1).
TABLE-US-00006 TABLE 1 Peptide spectrum match in human muscle. Dystrophin peptides encoded in exons 1-10. (N) represents the number of times a peptide sequence was detected in normal control muscle or in muscle from the patient with a deletion of exon 2. Dystrophin Peptide Spectrum Match (N) Peptide Normal SEQ se- MW control Del2 ID quence [Da] Exon muscle Muscle NO WVN 9,795,009 2 1 0 16 AQF SK QHI 16,718,084 3 1 0 17 ENL FSD LQD GR LLD 12,997,520 4 1 0 18 LLE GLT GQK VLQ 24,782,521 4-5 2 0 19 NNN VDL VNI GST DIV DGN HK NLM 14,496,990 6 3 0 20 AGL QQT NSE K LEH 10,705,737 7 1 0 21 AFN IAR YQL 8,504,665 7 1 1 22 GIE K LLD 17,488,606 7-8 2 3 23 PED VDT TYP DKK SYA 18,948,820 9 3 2 24 YTQ AAY VTT SDP TR SPF 15,817,538 9-10 3 1 25 PSQ HLE APE DK
[0076] In a complementary approach, we examined DMD translation efficiency, promoter usage, and alternate splicing using muscle RNA isolated from a mild BMD patient with an exon 2 frameshift mutation (c.40_41del [p.Glu14ArgfsX17], referred to as FS) whose western blot also revealed expression of the same smaller molecular weight dystrophin (.about.410 kDa) which lacked the N-terminal epitope (FIG. 1C; FIG. 8A). To confirm our western blot results, muscle homogenate from the same FS patient was used to construct RNA-Seq libraries for ribosome-protected fragments (i.e., ribosome footprints isolated after RNase digestion) and for total RNA. We compared the mRNA translation efficiency in normal versus patient muscle using the ratio of reads from ribosome-protected fragments (RPFs) to reads from RNA-Seq. Among the top 1000 most abundant muscle mRNAs, DMD displayed the greatest change in translation efficiency (FIG. 1D), indicating a .about.5-fold reduction in the amount of ribosomes translating the DMD muscle transcript in the frameshifted patient FS. This decreased amount of translation is consistent with both the expected reduction in dystrophin level given the patient's mild BMD phenotype, and with the amount of dystrophin seen in p.Trp3X patients.sup.4 and other 5' mutation alleles (FIG. 1C).
[0077] The saw-tooth RNA-Seq pattern observed in DMD introns 1 through 8 (FIG. 1E) confirmed that the major transcription start was located at the dystrophin muscle-specific promoter (Dp427m) and that DMD exons 1 through 7 underwent efficient co-transcriptional splicing [Ameur et al., Nature Structural & Molecular Biology, 18: 1435-1440 (2011)] in both the control and FS patient samples. Two alternate 427 kD isoforms of dystrophin (Dp427p and Dp427c) are expressed primarily in the central nervous system, and differ from Dp427m only in the use of alternate exon 1 sequences. The lack of a strong nascent RNA signal from either the Dp427p or Dp427c promoters confirmed that up-regulation of alternate promoters does not contribute to alternate AUG usage in exon 6 (FIG. 1E). In both samples, RNA-Seq reads spanning exon-exon junctions mapped exclusively to the known junctions between Dp427m exon 1 and exon 11, indicating that splicing of novel 5' UTRs from alternate promoters did not contribute to exon 6 AUG usage. The distribution of ribosome footprints mapped on Dp427m exons 1 through 11 revealed normal levels of exon 1 AUG initiation, followed by premature termination in exon 2 and resumption of translation following the exon 6 in-frame AUG codons (FIG. 1F) that continued into the body of the DMD transcript (FIGS. 8B-8D), consistent with efficient alternate translation initiation.
Example 2
In Vitro Transcription/Translation Studies
[0078] Having demonstrated new evidence for efficient alternate translation initiation using both ribosome profiling and protein analysis directly in patient muscle, we sought to characterize the elements contributing to the high translation efficiency. To determine whether exons 1 through 5 of DMD contain an IRES, we cloned the 5' portion of the cDNA encompassing exons 1 through part of exon 6, beginning at the +4 position to exclude the native AUG initiation codon (c.4_c.369, referred as exon 1 to 6), into the dicistronic dual luciferase reporter vector pRDEF. This vector contains an upstream cap-dependent renilla luciferase (RLuc) open reading frame (ORF) under control of an SV40 promoter and a downstream cap-independent firefly luciferase (FLuc) ORF under the control of the sequences of interest, with the two ORFs separated by a secondary structure element (dEMCV) that prevents ribosomal scanning (FIG. 2C). We used the EMCV IRES sequence as a positive control, and normalized all values to the empty vector. In each case we included 49 nucleotides from exon 6 that placed the exon 6 AUGs in-frame with the downstream FLuc reporter. This sequence corresponds to the first 39 nt, inclusive of the two in-frame AUGs (M124 and M128), and 10 additional nucleotides used for cloning purposes. T7 mediated RNA were generated from the different constructs and were used to perform rabbit reticulocyte lysate (RRL) translation assays (FIG. 2A, left panel). Size and integrity of the corresponding RNAs were checked using a formaldehyde agarose gel (FIG. 2B). Cap-independent translation activity (represented as the ratio of downstream FLuc to the RLuc luminescence) of the exons 1-5 of DMD results in a 1.5-1.7 fold increase in FLuc signal, less than the 3.4-3.8 increase seen with the control EMCV IRES but consistent with IRES activity (FIG. 2A, left panel).
Example 3
IRES Activity in Cell Cultures
[0079] RRL-based translation may underestimate IRES activity of either viral or eukaryotic cellular IRESs, possibly due to the limiting amounts of RNA binding proteins in this specialized extract or due to the lack of tissue-specific IRES trans-acting factors (ITAFs). Therefore, the assay was performed in C2C12 myoblasts which express dystrophin, and we observed that the presence of the exon 1 to 6 construct leads to .about.8 fold higher FLuc expression relative to exon 6 alone vector (FIG. 2A, right panel). This represents .about.50% of the activity of the control EMCV IRES, suggesting the presence of a relatively strong IRES within exons 1-5. To map the position of the IRES, deletion constructs consisting of the 5' portion of the DMD gene (exons 1-5) or appropriate controls were cloned into pRDEF (FIG. 2C). Deletion of the first 300 nucleotides (nt) of this sequence did not significantly change the FLuc expression, whereas removal or inversion of the last 71 nt (representing nearly all of exon 5) completely abrogates expression of the FLuc reporter, and further deletions within exon 5 result in greatly reduced FLuc expression. To test the hypothesis that the putative IRES required muscle specific factors, we repeated the experiments in HEK293K cells, which do not endogenously express dystrophin, and in a commercial human myoblast cell line (hSKMM). Unlike the ECMV IRES, the putative DMD IRES did not stimulate FLuc expression in 293K cells whereas the level of stimulation in hSKMM cells replicated the C2C12 results (FIG. 9A), suggesting that the IRES is preferentially active in muscle.
[0080] Control experiments were performed to exclude the possibility of aberrant splicing events, cryptic promoter activities, or other potential artifacts leading to misinterpretation of the dicistronic assay. We removed the upstream SV40 promoter to generate a promoterless version of the pRDEF vector containing the exon 1 to 6 (c.4_c.369) DMD sequence. Transfection of this construct into C2C12 myoblasts showed only minimal background luminescence from both RLuc and FLuc, strongly arguing against any cryptic promoter activity in the DMD coding sequence (data not shown). No aberrant splicing was detected by RT-PCR (FIG. 2D and FIG. 9C), and RNA integrity was confirmed by a northern blot (FIGS. 2e and 9b).
[0081] Notably, although either duplication or deletion of exon 2 results in an interrupted reading frame, the disparate associated clinical phenotypes led to the hypothesis that IRES activity may be diminished in the presence of an exon 2 duplication. We tested this hypothesis in C2C12 cells and showed that IRES activation was equivalent between the full length (exons 1-6) and deletion 2 cDNAs, but was markedly reduced in the presence of an exon 2 duplication (FIG. 2F) confirming that duplication but not deletion of exon 2 ablates IRES activity.
Example 4
Out-of-Frame Exon-Skipping is Able to Drive an IRES Mediated Dystrophin In Vitro
[0082] In considering skipping of exons prior to the exon 5 IRES, only the removal of exon 2 will disrupt the reading frame and result in a premature stop codon (FIG. 3A). We contemplated that deletion of this exon could be used therapeutically to increase activation of the IRES, whether by use of antisense oligonucleotides (AONs) [Wood et al., Brain: A Journal of Neurology, 133: 957-972 (2010); van Deutekom et al., New England Journal of Medicine, 357: 2677-2686 (2007) and Kinali et al., Lancet Neurology, 8: 918-928 (2009)] or by use of AAV-U7 mediated antisense delivery [Goyenvalle et al., Science, 306: 1796-1799 (2004) and Vulin et al., Molecular Therapy: Journal of the American Society of Gene Therapy, 20: 2120-2133 (2012)]. We selected four different sequences (respectively labeled "B", "AL", "AS" and "C" in FIG. 3B) for U7snRNA targeting and cloned each into AAV1 to assess exon-skipping efficiency in myoblasts generated from either a wild type or an exon 2 duplication fibroblast cell lines that expresses a doxycycline-inducible MyoD (referred as FibroMyoD) [Chaouch et al., Human Gene Therapy, 20: 784-790 (2009)]. All constructs were able to skip either one or two copies of exon 2 (FIGS. 10A-10B). Subsequently, in order to increase skipping efficiency, two copies of each of the U7-C and U7-AL targeting antisense sequences were cloned into the single self-complementary (sc) AAV1 vector (and designated AAV1.U7-ACCA), the genome of which is shown in FIGS. 15A-15B in the 3' to 5' orientation. U7-C and U7-AL were used to avoid any possible overlap in the antisense sequence between AL and B. A known antisense sequence (AON H2A) was used as a positive control of skipping [Tennyson and Worton, Nucleic Acids Res., 24: 3059-3064 (1996)]. Infection of FibroMyoD cells resulted in 88.6% of the DMD transcript with complete skipping of exon 2 leading to the production of N-terminally truncated dystrophin (FIGS. 3C-3D and FIG. 12A).
Example 5
IRES Driven N-Truncated Dystrophin is Expressed After Out-of-Frame Exon-Skipping in a Novel Mouse Model Harboring a Duplication of Exon 2
[0083] We tested the ability of the U7-ACCA vector to skip exon 2 in vivo in a mouse model carrying a duplication of exon 2 on a C57BL/6 background (the Dup2 mouse; described in Example 8 below). The resulting DMD mRNA contains two copies of exon 2, disrupting the reading frame and resulting in nearly complete absence of dystrophin expression. AAV1.U7-ACCA (1e11vg) was injected directly into the tibialis anterior muscle in six to eight week-old Dup2 mice (n=5) or BI6 control mice. Four weeks later, RT-PCR analysis from injected muscles demonstrates nearly complete exon-skipping of exon 2 in Dup2 or BI6 (FIGS. 4A-4B). Consistent with the RT-PCR results, the saw-tooth RNA-Seq pattern observed in Dmd introns 1 and 2 confirmed the suppression of co-transcriptional splicing of the duplicated exon 2 as well as the high-efficiency of co-transcriptional splicing of exon 1 to exon 3 in the treated mice (FIG. 4C). Western blot and immunostaining demonstrate expression of the N-truncated protein. Sarcolemmal staining is restored for .beta.-dystroglycan and nNOS (FIGS. 4D-4E), suggesting the presence of a functional dystroglycan complex.
[0084] We also performed a dose escalation study using intramuscular injection (IM) into the tibialis anterior (TA) of Dup2 mice in order to assess the degree of dose response for exon skipping and protein expression. IM escalating doses are set out in FIG. 18A. As seen in FIG. 18B, the degree of skipped transcript shows an expected dose response. FIG. 18B shows a similar expected dose response in protein expression, maximal at 3.1E11 vg per injection, with significant correction of physiologic force defects (FIG. 18C).
Example 6
Glucocorticoid Increases Activation of the Dystrophin IRES
[0085] We examined the effect of glucocorticoid exposure on IRES activity as a muscle-specific IRES found in the 5' UTR of utrophin, an analog of dystrophin, was found to be glucocorticoid-activated [Miura et al., PloS One, 3: e2309 (2008)]. Furthermore, treatment with the glucocorticoids prednisone and deflazacort are standard treatment for DMD. We assayed exon 5 IRES activity using the exon 5 to 6 construct in C2C12 cells in the presence of increasing concentrations of 6-methyl-prednisolone (PDN) and found that downstream FLuc activity increased in a dose-dependent fashion from around 7 fold change in the absence of PDN to over 20 fold at 6.4 .mu.M PDN (FIG. 5A).This glucocorticoid activation was not seen after transfection of the exon 6 alone or the inverted exon 5 control constructs or in 293K (FIG. 5A). An increase in dystrophin expression was seen in Dup2 FibroMyoD cells treated with 6.4 .mu.M PDN (FIG. 5B) and co-treatment of Dup2 mice (n=5) with both U7-ACCA and PDN resulted in an increase in dystrophin expression over U7-ACCA alone (FIGS. 5C-5D), consistent with glucocorticoid inducibility. An increase to less than 3% compared to untreated Dup2 was seen with PDN alone in rare samples (represented in FIG. 5C), suggesting some leakiness of the IRES in the Dup2 model. In all cases, this increase of dystrophin expression was not due to a difference in the AAV vector genome copy number (data not shown). Because utrophin translation may be regulated by corticosteroids and overexpression can compensate for absent dystrophin, we assessed utrophin levels in the same injected muscles (FIG. 5E). In untreated Dup2 animals, utrophin levels were increased in comparison to B16, similar to what has been reported in mdx, the standard dystrophinopathy mouse model. Comparison of the four groups reveals no statistically significant difference in utrophin levels between PDN treated and untreated animals (FIG. 5F), excluding utrophin upregulation as a cause of functional rescue following PDN treatment.
Example 7
Local IRES Driven N-Truncated Dystrophin Expression Stabilizes Muscle Membrane and Corrects Force Deficits in Dup2 Mouse Muscle
[0086] We examined whether expression of the IRES-driven isoform improved muscle integrity and physiology in the Dup2 mouse. Similar to the case in mdx mice, dystrophic changes in Dup2 mice are quantifiable at 4 weeks of age as widespread muscle regeneration characterized by centralized nuclei (Vulin et al., manuscript in press). One month after intramuscular injection of AAV1.U7-ACCA into the tibialis anterior muscle of 4-week old Dup2 mice, expression of the IRES driven isoform results in a significant reduction of centralized nuclei (FIG. 6A). To demonstrate that this isoform restores membrane integrity, treated and untreated Dup2 mice were subjected to a downhill running protocol and injected with Evans blue dye (EBD), which enters skeletal muscle fibers that have been permeabilized by membrane damage. Following intraperitoneal injection of EBD, uptake is found only in fibers without dystrophin staining, suggesting the N-truncated protein stabilizes the sarcolemma and provides further evidence for the functionality of this protein in vivo (FIG. 4F). Quantification of the number of EBD positive fiber confirms that expression of the IRES driven isoform results in protection of muscle fibers in these mice (FIG. 6B). Importantly, this membrane protection is associated with restoration of hindlimb grip strength (FIG. 6C) and muscle specific force (FIG. 6D) to the levels seen in BI6 control mice. Dup2 muscles injected with U7-ACCA with or without prednisone were significantly more resistant to contraction-induced injury than untreated Dup2 muscle, and the combination of both treatments showed no significant difference from BI6 controls (FIG. 6E), Despite the minimal (<3%) expression of dystrophin seen in some Dup2 muscles by PDN (FIG. 5C), treatment of the Dup2 muscles by PDN alone does not result in a significant amelioration of the muscle physiology (FIGS. 6A-6E).
Example 8
DMD Models
[0087] Examples of models of the DMD exon 2 duplication include in vivo and in vitro models as follows.
mdx.sup.dup2 Mouse Model
[0088] Mice carrying a duplication of exon 2 within the DMD locus were developed. The exon 2 duplication mutation is the most common human duplication mutation and results in relatively severe DMD.
[0089] A map of the insertion vector is shown in FIG. 16 In the map, the numbers indicate the relative positions of cloning sites and exons and restriction sites. The neo cassette is in the same direction of the gene and the insertion point is precisely at 32207/32208 bp in the intron2. At least 150 bp extra intronic sequences are kept on each side of inserted exon 2, E2 region is 1775-2195 bp. Sizes of exon 2 and intron 2 are 62 bp and 209572 bp respectively.
[0090] Male C57BL/6 ES cells were transfected with the vector (FIG. 16) carrying an exon2 construct and then insertion was checked by PCR. One good clone was found, amplified and injected in dozens of albino BL/6 blastocysts. Injected blastocysts were implanted into recipient mice. The dystrophin gene from chimeric males was checked by PCR and then by RT-PCR. The colony was expanded and includes some female mice bred to homozygosity. Dystrophin expression in muscles from a 4 week old hemizygous mdxdup2 mouse was essentially absent.
Immortalized and Conditionally Inducible FibroMyoD Cell Lines
[0091] Expression of the MyoD gene in mammalian fibroblasts results in transdifferentiation of cells into the myogenic lineage. Such cells can be further differentiated into myotubes, and they express muscle genes, including the DMD gene.
[0092] Immortalized cell lines that conditionally express MyoD under the control of a tetracycline-inducible promoter were generated. This is achieved by stable transfection of the primary fibroblast lines of a lentivirus the tet-inducible MyoD and containing the human telomerase gene (TER). The resultant stable line allows MyoD expression to be initiated by treatment with doxycycline. Such cell lines were generated from patients with DMD who carry a duplication of exon 2.
[0093] Using the line, duplication skipping using 2'-O-methyl antisense oligomers (AONs) provided by Dr. Steve Wilton (Perth, Australia) was demonstrated. Multiple cell lines were tested.
Transiently MyoD-Transfected Primary Cell Lines
[0094] Proof-of-principle experiments using primary fibroblast lines transiently transfected with adenovirus-MyoD were conducted. The adenovirus constructs were not integrated in the cell genomes, yet MyoD was transiently expressed. The resulting DMD expression was sufficient to perform exon skipping experiments (although reproducibility favors the stably transfected lines.)
Example 9
Intravenous Injection of AAV9-U7_ACCA in the Dup2 Mouse Model Results in Significant Expression of the N-Truncated Isoform and Correction of Strength Deficit
[0095] We tested the ability of an AAV9-U7-ACCA genome to skip exon 2 in vivo in Dup2 mice upon intravenous injection. The U7-ACCA genome was cloned into a rAAV9 vector (designated AAV9-U7_ACCA herein) for administration to the mice. AAV9-U7_ACCA was injected into the tail vein (3.3E12 vg/kg) of five Dup2 mice. One month after injection, treated animals were examined.
[0096] Results of the experiment are shown in FIGS. 17A-17F.
[0097] We also carried out dose escalation studies of intravenous dosing (FIG. 19A). As seen in FIG. 19B, the degree of skipped transcript shows an expected dose response, as was seen in the IM studies. At the highest level, the majority of transcript consists of either wild-type transcript, which is translated into full-length dystrophin, or exon 2-deleted transcript, which is translated into the N-truncated isoform; importantly, either isoform provides a functional benefit to the mouse (as to humans). FIG. 19C shows a similar expected dose response in protein expression. Quite importantly, in terms of clinical utility, at the higher doses there is unquestionable and abundant expression of dystrophin in the diaphragm and heart muscles. Quantification of protein expression on immunoblot (FIG. 19D) confirms the dose escalation response.
[0098] Newborn screening (NBS) for DMD in human newborns is now feasible, therefore we tested the benefits of early expression of the N-trucated isoform by delivery of AAV9.U7-ACCA vector (8.times.10.sup.11 vg) results at postnatal day 1 (P1) in Dup2 mice. This single injection results in widespread expression of the N-truncated isoform in all muscles, with sustained protection of muscle fibers through one and six months post treatment (FIG. 20).
Example 10
[0099] PPMOs having following sequences (shown 5' to 3') are administered to Dup2 mice.
TABLE-US-00007 C antisense oligomer: (SEQ ID NO: 10) AUUCUUACCUUAGAAAAUUGUGC AL antisense oligomer: (SEQ ID NO: 11) GUUUUCUUUUGAACAUCUUCUCUUUCAUCUA
[0100] We transfected the AL-PPMO into wild type C2C12 mouse myoblasts (FIG. 22). Three days following transfection, an RT-PCR was performed and demonstrated an efficient exon 2 skipping (FIG. 22A). A similar experiment was performed in the Dup2 mouse model. Intramuscular injection of the AL-PPMO into the tibialis anterior (TA) of Dup2 mice was performed in order to assess the degree of exon 2 skipping and protein expression. As seen in FIG. 22B, exon 2 skipping was achieved efficiently. FIG. 22C was obtained using the same treated TA muscles. Immunostaining of dystrophin was carried out and of dystophrin he results demonstrated efficient production and localization to the plasma membrane protein.
[0101] In another experiment, systemic injections are given in the tail vein of another cohort of mice of three doses weekly at 12 mg/kg. We will evaluate skipping and dystrophin restoration at 4 weeks after the first injection.
Example 11
[0102] Patients harboring a nonsense mutation within exon 1 or 2 still express the highly functional N-terminally truncated dystrophin isoform. This is due to the presence of IRES in exon 5 that allow re-entry of the ribosome and translation from exon 6. Therefore we hypothesize that creation of a nonsense mutation should force activation of the IRES in human patient cell lines carrying either missense mutation or in frame deletion duplication, within exon 1 to 4. Only removal of exon 2 generates a stop codon in exon 3. Therefore complete skipping of exon 2 in patient carrying the above mentioned mutation, would induce a stop codon in exon 3, and thereby production of the IRES-mediated N-terminally truncated isoform.
[0103] We collected cells from human patients carrying mutation in these exons. The cells were then infected with a lentivirus expressing an inducible MyoD that forces conversion of fibroblasts to myoblasts which can then be further differentiated into myotubes, the cell type that expresses dystrophin (referred to hereafter as "myofibroblasts"). Despite aiming to collect cells from patients harboring missense mutation or in frame deletion or duplication within exon 1 to 4, only cells from patient carrying a nonsense mutation were available. These cells were derived from BMD patients, and as they carry a nonsense mutation they already naturally expressed the N-terminally truncated dystrophin isoform. However, treatment with AAV1.U7-ACCA at differentiation resulted in higher expression of the IRES-initiated isoform by day 14 (FIG. 21).
Discussion of Results in the Examples
[0104] We have demonstrated the presence of a glucocorticoid-responsive IRES within DMD exon 5 that can drive the expression of an N-truncated but functional dystrophin. Ribosome profiling from a BMD patient with an exon 2 frameshifting mutation demonstrated a mild reduction in dystrophin translation efficiency and a ribosome footprint pattern consistent with ribosome loading beginning in exons 5 and 6. The relevance of this IRES-induced isoform to the amelioration of disease severity, which we first described in patients with exon 1 nonsense mutations [Flanigan et al., Neuromuscular Disorders: NMD, 19: 743-748 (2009)], is also confirmed by the mass spectrometric data from the first ever reported case of an exon 2 deletion, found in an entirely asymptomatic subject. Finally, in a novel therapeutic approach, we have induced out-of-frame exon-skipping to generate a premature stop codon and consequently force activation of the IRES in both patient-derived cell lines and in a novel DMD mouse model, in which we restored components of the dystrophin complex and corrected the pathologic and physiologic features of muscle injury.
[0105] Most eukaryotic mRNAs are monocistronic and possess a specialized cap structure at their 5' terminus, which is required for translation initiation as this is where scanning by the 40S ribosomal subunit begins. Despite clear evidence for the cap-dependent 5.fwdarw.3' scanning model of initiation, bioinformatic analysis has suggested that .about.50% of human transcripts contain 5'UTR short upstream open reading frames (uORFs) that may mediate transcript-specific translation efficiency and control. uORFs may function by modulating either leaky scanning or termination-dependent reinitiation, although uORFs can also dynamically regulate access to IRES elements as shown for the mammalian cationic amino acid transporter 1 gene, CAT1/SLC7A1. Recognizing the cautions raised regarding IRES identification via reporter assays, all control experiments performed in this study--including assessment of RNA integrity by RT-PCR and Northern blot, use of a promoterless plasmid, and use of an appropriate positive IRES control - were consistent with cap-independent initiation due to IRES activity. We mapped a minimal region harboring a DMD IRES activity to 71 nt, of a small length compared to EMCV (588 nt) but similar in size to that identified in the c-myc 5'UTR (50 nt). This is an important feature as such small IRESs can be used in dicistronic vectors, where space is limited when packaged into viral vectors such as AAV.
[0106] Although the precise molecular mechanism by which cellular IRESs modulate translation has not been defined in the literature, the requirement of ITAFs has been strongly suggested. These cellular proteins act in trans to augment IRES activity. Almost all ITAFs have been shown to harbor RNA binding domains and have been hypothesized to act as RNA chaperones, helping the IRES primary sequence attain appropriate conformational state intrinsic to its activity. This is likely relevant to the loss of dystrophin IRES activity in the presence of an exon 2 duplication, which may ablate IRES function by formation of a complex secondary structure or cause the formation of an inhibitory uORF that interferes with ITAF access to the exon 5 IRES.
[0107] Our results provide a molecular explanation for the rescue of 5' truncating mutations via a heretofore undescribed mechanism of post-transcriptional regulation of dystrophin expression. The identification of this new cellular IRES and the resultant dystrophin isoform has significant implications for understanding the basic biology of muscle and dystrophin. We note that exon 5 of DMD is highly conserved, with 87% identity to human found in the dog, mouse, horse, and chicken DMD genes, and 67% among 39 species including D. rerio and X. tropicalis. The presence of an IRES within such a highly conserved region strongly suggests selective pressure favoring a programmed role for alternate translation initiation. The role of the IRES under normal conditions is unclear, but ongoing efforts to understand the relevant cell lineage-specific and/or conditional activation signals will shed light on underlying mechanisms of IRES control and elucidate potentially novel functions of dystrophin.
[0108] An intriguing question is how the N-truncated isoform remains functional. A key cellular role for dystrophin is presumed to be transmitting the force of contraction across the sarcolemma to extracellular structures by serving as an important architectural bridge role between the F-actin cytoskeleton and the muscle plasma membrane. Two regions within dystrophin are responsible for F-actin binding: ABD1 (actin binding domain, spanning residues 15-237) and ABD2 (spanning residues 1468-2208). A number of studies have shown a lack of stability of dystrophin in the setting of deletions within the ABD1 domain. However, we note that most of these studies were performed with microdystrophin constructs lacking the ABD2 domain, which has been shown to enhance the interaction between ABD1 and actin. Such miniproteins bind actin and modify actin dynamics in a different manner compared to the full length version. Although results with such constructs show that absence of ABD2 does not completely abrogate binding of dystrophin to actin, it is unlikely that absence of ABD1 completely disrupts the interaction between dystrophin and actin. Expression of transgenes deleted for ABD1 lessens the mdx phenotype and restores the costameric pattern of the M band and Z lines, suggesting that the link between dystrophin and the subsarcolemmal cytoskeleton involves more than an interaction with ABD1. In agreement with this, other members of the cytoskeleton have been shown to interact with the dystrophin spectrin-repeat.
[0109] Although some series suggest that BMD due to mutations affecting ABD1 is more severe [Beggs et al., American Journal of Human Genetics, 49: 54-67 (1991)], our clinical and experimental observations--as well reports of other BMD patients lacking part or all of the ABD1 domain [Winnard et al., Human Molecular Genetics, 2: 737-744 (1993); Winnard et al., American Journal of Human Genetics, 56: 158-166 (1995) and Heald et al., Neurology, 44: 2388-2390 (1994)]--clearly indicate the significant functionality of the IRES-driven N-truncated isoform despite lacking the first half of the canonical ABD1 (FIG. 3a). This is of particular interest since forcing expression of this isoform by generating an out-of-frame transcript in order to induce IRES activity holds substantial therapeutic potential. This novel out-of-frame strategy could be combined with glucocorticoid treatment, a drug already used in DMD/BMD patients, which should increase IRES activation. Significantly, rather than being a personalized exon-skipping approach for patients with exon 2 duplications (who represent nearly 2% of DMD patients in one large series), out-of-frame skipping of exon 2 to induce expression of such a protein is contemplated for treatment of all patients who harbor mutations at the 5' end of the DMD gene (up to 6% in the same cohort) [Flanigan et al., Neuromuscular Disorders: NMD, 19: 743-748 (2009)].
[0110] While the present invention has been described in terms of specific embodiments, it is understood that variations and modifications will occur to those skilled in the art. Accordingly, only such limitations as appear in the claims should be placed on the invention.
[0111] All documents referred to in this application are hereby incorporated by reference in their entirety with particular attention to the content for which they are referred.
Sequence CWU 1 SEQUENCE LISTING <160> NUMBER OF SEQ ID
NOS: 34 <210> SEQ ID NO 1 <211> LENGTH: 28 <212>
TYPE: DNA <213> ORGANISM: Homo sapiens <400> SEQUENCE:
1 tcaaaagaaa acattcacaa aatgggta 28 <210> SEQ ID NO 2
<211> LENGTH: 23 <212> TYPE: DNA <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 2 gcacaatttt ctaaggtaag aat 23
<210> SEQ ID NO 3 <211> LENGTH: 31 <212> TYPE:
DNA <213> ORGANISM: Homo sapiens <400> SEQUENCE: 3
tagatgaaag agaagatgtt caaaagaaaa c 31 <210> SEQ ID NO 4
<211> LENGTH: 21 <212> TYPE: DNA <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 4 tagatgaaag agaagatgtt c 21
<210> SEQ ID NO 5 <211> LENGTH: 28 <212> TYPE:
DNA <213> ORGANISM: Adeno-associated virus <400>
SEQUENCE: 5 tacccatttt gcgaatgttt tcttttga 28 <210> SEQ ID NO
6 <211> LENGTH: 23 <212> TYPE: DNA <213>
ORGANISM: Adeno-associated virus <400> SEQUENCE: 6 attcttacct
tagaaaattg tgc 23 <210> SEQ ID NO 7 <211> LENGTH: 31
<212> TYPE: DNA <213> ORGANISM: Adeno-associated virus
<400> SEQUENCE: 7 gttttctttt gaagatcttc tctttcatct a 31
<210> SEQ ID NO 8 <211> LENGTH: 21 <212> TYPE:
DNA <213> ORGANISM: Adeno-associated virus <400>
SEQUENCE: 8 gaagatcttc tctttcatct a 21 <210> SEQ ID NO 9
<211> LENGTH: 28 <212> TYPE: RNA <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 9 uacccauuuu gcgaauguuu ucuuuuga
28 <210> SEQ ID NO 10 <211> LENGTH: 23 <212>
TYPE: RNA <213> ORGANISM: Homo sapiens <400> SEQUENCE:
10 auucuuaccu uagaaaauug ugc 23 <210> SEQ ID NO 11
<211> LENGTH: 31 <212> TYPE: RNA <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 11 guuuucuuuu gaacaucuuc
ucuuucaucu a 31 <210> SEQ ID NO 12 <211> LENGTH: 21
<212> TYPE: RNA <213> ORGANISM: Homo sapiens
<400> SEQUENCE: 12 gaacaucuuc ucuuucaucu a 21 <210> SEQ
ID NO 13 <211> LENGTH: 30 <212> TYPE: RNA <213>
ORGANISM: Homo sapiens <400> SEQUENCE: 13 ccauuuugug
aauguuuucu uuugaacauc 30 <210> SEQ ID NO 14 <211>
LENGTH: 4472 <212> TYPE: DNA <213> ORGANISM:
Adeno-associated virus rh74 <400> SEQUENCE: 14 ctccatcact
aggggtaacc gcgaagcgcc tcccacgctg ccgcgtcagc gctgacgtaa 60
attacgtcat aggggagtgg tcctgtatta gctgtcacgt gagtgctttt gcgacatttt
120 gcgacaccac gtggccattc atggtatata tggccgagtg agcgagcagg
atctccattt 180 tgaccgcgaa atttgaacga gcagcagcca tgccgggctt
ctacgagatc gtgcttaagg 240 tgccgagcga cctggacgag cacctgccgg
gcatttctga ctcgtttgtg aactgggtgg 300 cagagaagga atgggagctg
cccccggatt ctgacatgga tcggaatctg attgagcagg 360 cacccctgac
cgtggccgag aagctacagc gcgacttcct ggtccaatgg cgccgcgtga 420
gtaaggcccc ggaggccctc ttctttgttc agttcgagaa gggcgagtcc tacttccacc
480 tccatattct ggtagagacc acgggggtca aatccatggt gctgggccgc
ttcctgagtc 540 agattcggga caagctggtg cagaccatct accgcgggat
cgagccgacc ctgcccaact 600 ggttcgcggt gacaaagacg cgtaatggcg
ccggaggggg gaacaaggtg gtggacgagt 660 gctacatccc caactacctg
ctgcccaaga ctcagcccga gctgcagtgg gcgtggacta 720 acatggagga
gtatataagc gcgtgcttga acctggccga gcgcaaacgg ctcgtggcgc 780
agcacctgac ccacgtcagc cagacccagg agcagaacaa ggagaatctg aacccgaatt
840 ctgacgcgcc tgtcatccgg tcaaaaacct ccgcgcgcta catggagctg
gtcgggtggc 900 tggtggaccg gggcatcacc tccgagaagc agtggatcca
ggaggaccag gcctcgtaca 960 tctccttcaa cgccgcctcc aactcgcggt
ctcagatcaa ggccgcgctg gacaatgccg 1020 gcaagatcat ggcgctgacc
aaatccgcgc ccgactacct ggtaggcccc gctctgcccg 1080 cggacattaa
atccaaccgc atctaccgca tcctggagct gaatggctac gaccctgcct 1140
acgccggttc cgtctttctc ggctgggccc agaaaaagtt tggcaaaagg aacaccatct
1200 ggctgtttgg gccggccacc acgggcaaga ccaacatcgc ggaagccatc
gcccacgccg 1260 tgcccttcta cggctgcgtc aactggacca atgagaactt
tcccttcaac gattgcgtcg 1320 acaagatggt gatctggtgg gaggagggca
agatgacggc caaggtcgtg gagtccgcca 1380 aggccattct cggcggcagc
aaggtgcgcg tggaccaaaa gtgcaagtcg tccgcccaga 1440 tcgatcccac
ccccgtgatc gtcacctcca acaccaacat gtgcgccgtg attgacggga 1500
acagcaccac cttcgagcac cagcagccgt tgcaggaccg gatgttcaaa tttgaactta
1560 cccgccgtct ggagcacgac tttggcaagg tgacaaagca ggaagtcaaa
gagttcttcc 1620 gctgggcgca ggatcacgtg accgaggtgg cgcatgagtt
ctacgtcaga aagggtggag 1680 ctaacaaaag acccgccccc gatgacgcgg
atataagcga gcccaagcgg gcctgcccct 1740 cagtcgcgga tccatcgacg
tcagacgcgg aaggagctcc ggtggacttt gccgacaggt 1800 accaaaacaa
atgttctcgt cacgcgggca tgcttcagat gctgtttccc tgcaaaacat 1860
gcgagagaat gaatcagaat ttcaacattt gcttcacgca cgggaccaga gactgttcag
1920 aatgtttccc tggcgtgtca gaatctcaac cggtcgtcag aaaaaagacg
tatcggaaac 1980 tctgtgcgat tcatcatctg ctggggcggg cacccgagat
tgcttgctcg gcctgcgacc 2040 tggtcaacgt ggacctggat gactgtgttt
ctgagcaata aatgacttaa accaggtatg 2100 gctgccgatg gttatcttcc
agattggctc gaggacaacc tctctgaggg cattcgcgag 2160 tggtgggacc
tgaaacctgg agccccgaaa cccaaagcca accagcaaaa gcaggacaac 2220
ggccggggtc tggtgcttcc tggctacaag tacctcggac ccttcaacgg actcgacaag
2280 ggggagcccg tcaacgcggc ggacgcagcg gccctcgagc acgacaaggc
ctacgaccag 2340 cagctccaag cgggtgacaa tccgtacctg cggtataatc
acgccgacgc cgagtttcag 2400 gagcgtctgc aagaagatac gtcttttggg
ggcaacctcg ggcgcgcagt cttccaggcc 2460 aaaaagcggg ttctcgaacc
tctgggcctg gttgaatcgc cggttaagac ggctcctgga 2520 aagaagagac
cggtagagcc atcaccccag cgctctccag actcctctac gggcatcggc 2580
aagaaaggcc agcagcccgc aaaaaagaga ctcaattttg ggcagactgg cgactcagag
2640 tcagtccccg accctcaacc aatcggagaa ccaccagcag gcccctctgg
tctgggatct 2700 ggtacaatgg ctgcaggcgg tggcgctcca atggcagaca
ataacgaagg cgccgacgga 2760 gtgggtagtt cctcaggaaa ttggcattgc
gattccacat ggctgggcga cagagtcatc 2820 accaccagca cccgcacctg
ggccctgccc acctacaaca accacctcta caagcaaatc 2880 tccaacggga
cctcgggagg aagcaccaac gacaacacct acttcggcta cagcaccccc 2940
tgggggtatt ttgacttcaa cagattccac tgccactttt caccacgtga ctggcagcga
3000 ctcatcaaca acaactgggg attccggccc aagaggctca acttcaagct
cttcaacatc 3060 caagtcaagg aggtcacgca gaatgaaggc accaagacca
tcgccaataa ccttaccagc 3120 acgattcagg tctttacgga ctcggaatac
cagctcccgt acgtgctcgg ctcggcgcac 3180 cagggctgcc tgcctccgtt
cccggcggac gtcttcatga ttcctcagta cgggtacctg 3240 actctgaaca
atggcagtca ggctgtgggc cggtcgtcct tctactgcct ggagtacttt 3300
ccttctcaaa tgctgagaac gggcaacaac tttgaattca gctacaactt cgaggacgtg
3360 cccttccaca gcagctacgc gcacagccag agcctggacc ggctgatgaa
ccctctcatc 3420 gaccagtact tgtactacct gtcccggact caaagcacgg
gcggtactgc aggaactcag 3480 cagttgctat tttctcaggc cgggcctaac
aacatgtcgg ctcaggccaa gaactggcta 3540 cccggtccct gctaccggca
gcaacgcgtc tccacgacac tgtcgcagaa caacaacagc 3600 aactttgcct
ggacgggtgc caccaagtat catctgaatg gcagagactc tctggtgaat 3660
cctggcgttg ccatggctac ccacaaggac gacgaagagc gattttttcc atccagcgga
3720 gtcttaatgt ttgggaaaca gggagctgga aaagacaacg tggactatag
cagcgtgatg 3780 ctaaccagcg aggaagaaat aaagaccacc aacccagtgg
ccacagaaca gtacggcgtg 3840 gtggccgata acctgcaaca gcaaaacgcc
gctcctattg taggggccgt caatagtcaa 3900 ggagccttac ctggcatggt
gtggcagaac cgggacgtgt acctgcaggg tcccatctgg 3960 gccaagattc
ctcatacgga cggcaacttt catccctcgc cgctgatggg aggctttgga 4020
ctgaagcatc cgcctcctca gatcctgatt aaaaacacac ctgttcccgc ggatcctccg
4080 accaccttca ctaaggccaa gctggcttct ttcatcacgc agtacagtac
cggccaggtc 4140 agcgtggaga tcgagtggga gctgcagaag gagaacagca
aacgctggaa cccagagatt 4200 cagtacactt ccaactacta caaatctaca
aatgtggact ttgctgtcaa tactgagggt 4260 acttattccg agcctcgccc
cattggcacc cgttacctca cccgtaatct gtaattacat 4320 gttaatcaat
aaaccggtta attcgtttca gttgaacttt ggtctcctgt ccttcttatc 4380
ttatcggtta ccatagaaac tggttactta ttaactgctt ggtgcgcttc gcgataaaag
4440 acttacgtca tcgggttacc cctagtgatg ga 4472 <210> SEQ ID NO
15 <211> LENGTH: 2052 <212> TYPE: DNA <213>
ORGANISM: Adeno-associated virus <400> SEQUENCE: 15
cgaccgcgcg agcgagcgag tgactccggc gggcccgttt cgggcccgca gcccgctgga
60 aaccagcggg ccggagtcac tcgctcgctc gcgcgtctct ccctcaccgg
ttgaggtagt 120 gatccccaag gaacatcaat tactaattgg gcggtacgat
gaatagatgc atcggtacga 180 gatctattgt tgtatcctcg acactaaccg
acaaaagtcg gttagtcgtg actgagtaaa 240 cgtatcggaa atgttcgcca
gtgtttgagt tctttgctcg ccaaaattat cagaaaatct 300 tataacaaat
agcttggctt attccttgac acgaaacact aagtgtatag tcacctcccc 360
acacctttac cgtggaacta gagtgggagt agctttcacc tcaactacag gaagggaccg
420 agcgatgtct gcgtgaaggc gttcaaaaga aaacttctag aagagaaagt
agatttaaaa 480 acctcgtcca aaagactgaa gccagccttt tggggagggt
taaagtgacc agatgttact 540 ttcgttttgt caagagaagg ggcgaggggc
cacacactct ccccgaaact aggaagagac 600 caaaggatcc tttgcgcata
caccgatcta ttgttgtatc ctcgacacta accgacaaaa 660 gtcggttagt
cgtgactgag taaacgtatc ggaaatgttc gccagtgttt gagttctttg 720
ctcgccaaaa ttatcagaaa atcttataac aaatagcttg gcttattcct tgacacgaaa
780 cactaagtgt atagtcacct ccccacacct ttaccgtgga actagagtgg
gagtagcttt 840 cacctcaact acaggaaggg accgagcgat gtctgcgtga
aggcgtttaa gaatggaatc 900 ttttaacacg ttaaaaacct cgtccaaaag
actgaagcca gccttttggg gagggttaaa 960 gtgaccagat gttactttcg
ttttgtcaag agaaggggcg aggggccaca cactctcccc 1020 gaaactagga
agagaccaaa ggatcctttg cgcatacacc gatctattgt tgtatcctcg 1080
acactaaccg acaaaagtcg gttagtcgtg actgagtaaa cgtatcggaa atgttcgcca
1140 gtgtttgagt tctttgctcg ccaaaattat cagaaaatct tataacaaat
agcttggctt 1200 attccttgac acgaaacact aagtgtatag tcacctcccc
acacctttac cgtggaacta 1260 gagtgggagt agctttcacc tcaactacag
gaagggaccg agcgatgtct gcgtgaaggc 1320 gtttaagaat ggaatctttt
aacacgttaa aaacctcgtc caaaagactg aagccagcct 1380 tttggggagg
gttaaagtga ccagatgtta ctttcgtttt gtcaagagaa ggggcgaggg 1440
gccacacact ctccccgaaa ctaggaagag accaaaggat cctttgcgca tacaccgatc
1500 tattgttgta tcctcgacac taaccgacaa aagtcggtta gtcgtgactg
agtaaacgta 1560 tcggaaatgt tcgccagtgt ttgagttctt tgctcgccaa
aattatcaga aaatcttata 1620 acaaatagct tggcttattc cttgacacga
aacactaagt gtatagtcac ctccccacac 1680 ctttaccgtg gaactagagt
gggagtagct ttcacctcaa ctacaggaag ggaccgagcg 1740 atgtctgcgt
gaaggcgttc aaaagaaaac ttctagaaga gaaagtagat ttaaaaacct 1800
cgtccaaaag actgaagcca gccttttggg gagggttaaa gtgaccagat gttactttcg
1860 ttttgtcaag agaaggggcg aggggccaca cactctcccc gaaactagga
agagaccaaa 1920 ggatcctttg cgcatacaca tgttggggtg agggagagac
gcgcgagcga gcgagtgact 1980 ccggcccgct ggtttccagc gggctgcggg
cccgaaacgg gcccgccgga gtcactcgct 2040 cgctcgcgcg tc 2052
<210> SEQ ID NO 16 <211> LENGTH: 8 <212> TYPE:
PRT <213> ORGANISM: Homo sapiens <400> SEQUENCE: 16 Trp
Val Asn Ala Gln Phe Ser Lys 1 5 <210> SEQ ID NO 17
<211> LENGTH: 14 <212> TYPE: PRT <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 17 Gln His Ile Glu Asn Leu Phe
Ser Asp Leu Gln Asp Gly Arg 1 5 10 <210> SEQ ID NO 18
<211> LENGTH: 12 <212> TYPE: PRT <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 18 Leu Leu Asp Leu Leu Glu Gly
Leu Thr Gly Gln Lys 1 5 10 <210> SEQ ID NO 19 <211>
LENGTH: 23 <212> TYPE: PRT <213> ORGANISM: Homo sapiens
<400> SEQUENCE: 19 Val Leu Gln Asn Asn Asn Val Asp Leu Val
Asn Ile Gly Ser Thr Asp 1 5 10 15 Ile Val Asp Gly Asn His Lys 20
<210> SEQ ID NO 20 <211> LENGTH: 13 <212> TYPE:
PRT <213> ORGANISM: Homo sapiens <400> SEQUENCE: 20 Asn
Leu Met Ala Gly Leu Gln Gln Thr Asn Ser Glu Lys 1 5 10 <210>
SEQ ID NO 21 <211> LENGTH: 9 <212> TYPE: PRT
<213> ORGANISM: Homo sapiens <400> SEQUENCE: 21 Leu Glu
His Ala Phe Asn Ile Ala Arg 1 5 <210> SEQ ID NO 22
<211> LENGTH: 7 <212> TYPE: PRT <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 22 Tyr Gln Leu Gly Ile Glu Lys 1
5 <210> SEQ ID NO 23 <211> LENGTH: 15 <212> TYPE:
PRT <213> ORGANISM: Homo sapiens <400> SEQUENCE: 23 Leu
Leu Asp Pro Glu Asp Val Asp Thr Thr Tyr Pro Asp Lys Lys 1 5 10 15
<210> SEQ ID NO 24 <211> LENGTH: 17 <212> TYPE:
PRT <213> ORGANISM: Homo sapiens <400> SEQUENCE: 24 Ser
Tyr Ala Tyr Thr Gln Ala Ala Tyr Val Thr Thr Ser Asp Pro Thr 1 5 10
15 Arg <210> SEQ ID NO 25 <211> LENGTH: 14 <212>
TYPE: PRT <213> ORGANISM: Homo sapiens <400> SEQUENCE:
25 Ser Pro Phe Pro Ser Gln His Leu Glu Ala Pro Glu Asp Lys 1 5 10
<210> SEQ ID NO 26 <211> LENGTH: 2052 <212> TYPE:
DNA <213> ORGANISM: Adeno-associated virus <400>
SEQUENCE: 26 gctggcgcgc tcgctcgctc actgaggccg cccgggcaaa gcccgggcgt
cgggcgacct 60 ttggtcgccc ggcctcagtg agcgagcgag cgcgcagaga
gggagtggcc aactccatca 120 ctaggggttc cttgtagtta atgattaacc
cgccatgcta cttatctacg tagccatgct 180 ctagataaca acataggagc
tgtgattggc tgttttcagc caatcagcac tgactcattt 240 gcatagcctt
tacaagcggt cacaaactca agaaacgagc ggttttaata gtcttttaga 300
atattgttta tcgaaccgaa taaggaactg tgctttgtga ttcacatatc agtggagggg
360 tgtggaaatg gcaccttgat ctcaccctca tcgaaagtgg agttgatgtc
cttccctggc 420 tcgctacaga cgcacttccg caagttttct tttgaagatc
ttctctttca tctaaatttt 480 tggagcaggt tttctgactt cggtcggaaa
acccctccca atttcactgg tctacaatga 540 aagcaaaaca gttctcttcc
ccgctccccg gtgtgtgaga ggggctttga tccttctctg 600 gtttcctagg
aaacgcgtat gtggctagat aacaacatag gagctgtgat tggctgtttt 660
cagccaatca gcactgactc atttgcatag cctttacaag cggtcacaaa ctcaagaaac
720 gagcggtttt aatagtcttt tagaatattg tttatcgaac cgaataagga
actgtgcttt 780 gtgattcaca tatcagtgga ggggtgtgga aatggcacct
tgatctcacc ctcatcgaaa 840 gtggagttga tgtccttccc tggctcgcta
cagacgcact tccgcaaatt cttaccttag 900 aaaattgtgc aatttttgga
gcaggttttc tgacttcggt cggaaaaccc ctcccaattt 960 cactggtcta
caatgaaagc aaaacagttc tcttccccgc tccccggtgt gtgagagggg 1020
ctttgatcct tctctggttt cctaggaaac gcgtatgtgg ctagataaca acataggagc
1080 tgtgattggc tgttttcagc caatcagcac tgactcattt gcatagcctt
tacaagcggt 1140 cacaaactca agaaacgagc ggttttaata gtcttttaga
atattgttta tcgaaccgaa 1200 taaggaactg tgctttgtga ttcacatatc
agtggagggg tgtggaaatg gcaccttgat 1260 ctcaccctca tcgaaagtgg
agttgatgtc cttccctggc tcgctacaga cgcacttccg 1320 caaattctta
ccttagaaaa ttgtgcaatt tttggagcag gttttctgac ttcggtcgga 1380
aaacccctcc caatttcact ggtctacaat gaaagcaaaa cagttctctt ccccgctccc
1440 cggtgtgtga gaggggcttt gatccttctc tggtttccta ggaaacgcgt
atgtggctag 1500 ataacaacat aggagctgtg attggctgtt ttcagccaat
cagcactgac tcatttgcat 1560 agcctttaca agcggtcaca aactcaagaa
acgagcggtt ttaatagtct tttagaatat 1620 tgtttatcga accgaataag
gaactgtgct ttgtgattca catatcagtg gaggggtgtg 1680 gaaatggcac
cttgatctca ccctcatcga aagtggagtt gatgtccttc cctggctcgc 1740
tacagacgca cttccgcaag ttttcttttg aagatcttct ctttcatcta aatttttgga
1800 gcaggttttc tgacttcggt cggaaaaccc ctcccaattt cactggtcta
caatgaaagc 1860 aaaacagttc tcttccccgc tccccggtgt gtgagagggg
ctttgatcct tctctggttt 1920 cctaggaaac gcgtatgtgt acaaccccac
tccctctctg cgcgctcgct cgctcactga 1980 ggccgggcga ccaaaggtcg
cccgacgccc gggctttgcc cgggcggcct cagtgagcga 2040 gcgagcgcgc ag 2052
<210> SEQ ID NO 27 <211> LENGTH: 240 <212> TYPE:
PRT <213> ORGANISM: Homo sapiens <300> PUBLICATION
INFORMATION: <308> DATABASE ACCESSION NUMBER:
UniProtKB/Swiss-Prot: P11532.3 <309> DATABASE ENTRY DATE:
2015-07-22 <313> RELEVANT RESIDUES IN SEQ ID NO: (1)..(240)
<400> SEQUENCE: 27 Met Leu Trp Trp Glu Glu Val Glu Asp Cys
Tyr Glu Arg Glu Asp Val 1 5 10 15 Gln Lys Lys Thr Phe Thr Lys Trp
Val Asn Ala Gln Phe Ser Lys Phe 20 25 30 Gly Lys Gln His Ile Glu
Asn Leu Phe Ser Asp Leu Gln Asp Gly Arg 35 40 45 Arg Leu Leu Asp
Leu Leu Glu Gly Leu Thr Gly Gln Lys Leu Pro Lys 50 55 60 Glu Lys
Gly Ser Thr Arg Val His Ala Leu Asn Asn Val Asn Lys Ala 65 70 75 80
Leu Arg Val Leu Gln Asn Asn Asn Val Asp Leu Val Asn Ile Gly Ser 85
90 95 Thr Asp Ile Val Asp Gly Asn His Lys Leu Thr Leu Gly Leu Ile
Trp 100 105 110 Asn Ile Ile Leu His Trp Gln Val Lys Asn Val Met Lys
Asn Ile Met 115 120 125 Ala Gly Leu Gln Gln Thr Asn Ser Glu Lys Ile
Leu Leu Ser Trp Val 130 135 140 Arg Gln Ser Thr Arg Asn Tyr Pro Gln
Val Asn Val Ile Asn Phe Thr 145 150 155 160 Thr Ser Trp Ser Asp Gly
Leu Ala Leu Asn Ala Leu Ile His Ser His 165 170 175 Arg Pro Asp Leu
Phe Asp Trp Asn Ser Val Val Cys Gln Gln Ser Ala 180 185 190 Thr Gln
Arg Leu Glu His Ala Phe Asn Ile Ala Arg Tyr Gln Leu Gly 195 200 205
Ile Glu Lys Leu Leu Asp Pro Glu Asp Val Asp Thr Thr Tyr Pro Asp 210
215 220 Lys Lys Ser Ile Leu Met Tyr Ile Thr Ser Leu Phe Gln Val Leu
Pro 225 230 235 240 <210> SEQ ID NO 28 <211> LENGTH: 22
<212> TYPE: PRT <213> ORGANISM: Homo sapiens
<400> SEQUENCE: 28 Tyr Gln Leu Gly Ile Glu Lys Leu Leu Asp
Pro Glu Asp Val Asp Thr 1 5 10 15 Thr Tyr Pro Asp Lys Lys 20
<210> SEQ ID NO 29 <211> LENGTH: 112 <212> TYPE:
PRT <213> ORGANISM: Homo sapiens <400> SEQUENCE: 29 Trp
Val Asn Ala Gln Phe Ser Lys Phe Gly Lys Gln His Ile Glu Asn 1 5 10
15 Leu Phe Ser Asp Leu Gln Asp Gly Arg Arg Leu Leu Asp Leu Leu Glu
20 25 30 Gly Leu Thr Gly Gln Lys Val Leu Gln Asn Asn Asn Val Asp
Leu Val 35 40 45 Asn Ile Gly Ser Thr Asp Ile Val Asp Gly Asn His
Lys Asn Ile Met 50 55 60 Ala Gly Leu Gln Gln Thr Asn Ser Glu Lys
Ile Leu Leu Leu Glu His 65 70 75 80 Ala Phe Asn Ile Ala Arg Tyr Gln
Leu Gly Ile Glu Lys Leu Leu Asp 85 90 95 Pro Glu Asp Val Asp Thr
Thr Tyr Pro Asp Lys Lys Ser Ile Leu Met 100 105 110 <210> SEQ
ID NO 30 <211> LENGTH: 75 <212> TYPE: DNA <213>
ORGANISM: Homo sapiens <400> SEQUENCE: 30 tagatgaaag
agaagatgtt caaaagaaaa cattcacaaa atgggtaaat gcacaatttt 60
ctaaggtaag aatgg 75 <210> SEQ ID NO 31 <211> LENGTH: 80
<212> TYPE: DNA <213> ORGANISM: Homo sapiens
<400> SEQUENCE: 31 aactccattt atcaattaac caaaaactcc
caaataactg tgtaggttcc atgagttcag 60 gagattccac ttcaactaat 80
<210> SEQ ID NO 32 <211> LENGTH: 80 <212> TYPE:
DNA <213> ORGANISM: Homo sapiens <400> SEQUENCE: 32
aactccattt atcaattaac caaaaactcc caaataactg tggtgtatat ttatctattt
60 ttatgggttg caaaatactt 80 <210> SEQ ID NO 33 <211>
LENGTH: 80 <212> TYPE: DNA <213> ORGANISM: Homo sapiens
<400> SEQUENCE: 33 caaattctcc cctgaaaatt taaaaaaata
cattgttctg tggtgtatat ttatctattt 60 ttatgggttg caaaatactt 80
<210> SEQ ID NO 34 <211> LENGTH: 55 <212> TYPE:
DNA <213> ORGANISM: Homo sapiens <400> SEQUENCE: 34
aattaaccaa aaactcccaa ataactgtgg tgtatattta tctattttta tgggt 55
1 SEQUENCE LISTING <160> NUMBER OF SEQ ID NOS: 34 <210>
SEQ ID NO 1 <211> LENGTH: 28 <212> TYPE: DNA
<213> ORGANISM: Homo sapiens <400> SEQUENCE: 1
tcaaaagaaa acattcacaa aatgggta 28 <210> SEQ ID NO 2
<211> LENGTH: 23 <212> TYPE: DNA <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 2 gcacaatttt ctaaggtaag aat 23
<210> SEQ ID NO 3 <211> LENGTH: 31 <212> TYPE:
DNA <213> ORGANISM: Homo sapiens <400> SEQUENCE: 3
tagatgaaag agaagatgtt caaaagaaaa c 31 <210> SEQ ID NO 4
<211> LENGTH: 21 <212> TYPE: DNA <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 4 tagatgaaag agaagatgtt c 21
<210> SEQ ID NO 5 <211> LENGTH: 28 <212> TYPE:
DNA <213> ORGANISM: Adeno-associated virus <400>
SEQUENCE: 5 tacccatttt gcgaatgttt tcttttga 28 <210> SEQ ID NO
6 <211> LENGTH: 23 <212> TYPE: DNA <213>
ORGANISM: Adeno-associated virus <400> SEQUENCE: 6 attcttacct
tagaaaattg tgc 23 <210> SEQ ID NO 7 <211> LENGTH: 31
<212> TYPE: DNA <213> ORGANISM: Adeno-associated virus
<400> SEQUENCE: 7 gttttctttt gaagatcttc tctttcatct a 31
<210> SEQ ID NO 8 <211> LENGTH: 21 <212> TYPE:
DNA <213> ORGANISM: Adeno-associated virus <400>
SEQUENCE: 8 gaagatcttc tctttcatct a 21 <210> SEQ ID NO 9
<211> LENGTH: 28 <212> TYPE: RNA <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 9 uacccauuuu gcgaauguuu ucuuuuga
28 <210> SEQ ID NO 10 <211> LENGTH: 23 <212>
TYPE: RNA <213> ORGANISM: Homo sapiens <400> SEQUENCE:
10 auucuuaccu uagaaaauug ugc 23 <210> SEQ ID NO 11
<211> LENGTH: 31 <212> TYPE: RNA <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 11 guuuucuuuu gaacaucuuc
ucuuucaucu a 31 <210> SEQ ID NO 12 <211> LENGTH: 21
<212> TYPE: RNA <213> ORGANISM: Homo sapiens
<400> SEQUENCE: 12 gaacaucuuc ucuuucaucu a 21 <210> SEQ
ID NO 13 <211> LENGTH: 30 <212> TYPE: RNA <213>
ORGANISM: Homo sapiens <400> SEQUENCE: 13 ccauuuugug
aauguuuucu uuugaacauc 30 <210> SEQ ID NO 14 <211>
LENGTH: 4472 <212> TYPE: DNA <213> ORGANISM:
Adeno-associated virus rh74 <400> SEQUENCE: 14 ctccatcact
aggggtaacc gcgaagcgcc tcccacgctg ccgcgtcagc gctgacgtaa 60
attacgtcat aggggagtgg tcctgtatta gctgtcacgt gagtgctttt gcgacatttt
120 gcgacaccac gtggccattc atggtatata tggccgagtg agcgagcagg
atctccattt 180 tgaccgcgaa atttgaacga gcagcagcca tgccgggctt
ctacgagatc gtgcttaagg 240 tgccgagcga cctggacgag cacctgccgg
gcatttctga ctcgtttgtg aactgggtgg 300 cagagaagga atgggagctg
cccccggatt ctgacatgga tcggaatctg attgagcagg 360 cacccctgac
cgtggccgag aagctacagc gcgacttcct ggtccaatgg cgccgcgtga 420
gtaaggcccc ggaggccctc ttctttgttc agttcgagaa gggcgagtcc tacttccacc
480 tccatattct ggtagagacc acgggggtca aatccatggt gctgggccgc
ttcctgagtc 540 agattcggga caagctggtg cagaccatct accgcgggat
cgagccgacc ctgcccaact 600 ggttcgcggt gacaaagacg cgtaatggcg
ccggaggggg gaacaaggtg gtggacgagt 660 gctacatccc caactacctg
ctgcccaaga ctcagcccga gctgcagtgg gcgtggacta 720 acatggagga
gtatataagc gcgtgcttga acctggccga gcgcaaacgg ctcgtggcgc 780
agcacctgac ccacgtcagc cagacccagg agcagaacaa ggagaatctg aacccgaatt
840 ctgacgcgcc tgtcatccgg tcaaaaacct ccgcgcgcta catggagctg
gtcgggtggc 900 tggtggaccg gggcatcacc tccgagaagc agtggatcca
ggaggaccag gcctcgtaca 960 tctccttcaa cgccgcctcc aactcgcggt
ctcagatcaa ggccgcgctg gacaatgccg 1020 gcaagatcat ggcgctgacc
aaatccgcgc ccgactacct ggtaggcccc gctctgcccg 1080 cggacattaa
atccaaccgc atctaccgca tcctggagct gaatggctac gaccctgcct 1140
acgccggttc cgtctttctc ggctgggccc agaaaaagtt tggcaaaagg aacaccatct
1200 ggctgtttgg gccggccacc acgggcaaga ccaacatcgc ggaagccatc
gcccacgccg 1260 tgcccttcta cggctgcgtc aactggacca atgagaactt
tcccttcaac gattgcgtcg 1320 acaagatggt gatctggtgg gaggagggca
agatgacggc caaggtcgtg gagtccgcca 1380 aggccattct cggcggcagc
aaggtgcgcg tggaccaaaa gtgcaagtcg tccgcccaga 1440 tcgatcccac
ccccgtgatc gtcacctcca acaccaacat gtgcgccgtg attgacggga 1500
acagcaccac cttcgagcac cagcagccgt tgcaggaccg gatgttcaaa tttgaactta
1560 cccgccgtct ggagcacgac tttggcaagg tgacaaagca ggaagtcaaa
gagttcttcc 1620 gctgggcgca ggatcacgtg accgaggtgg cgcatgagtt
ctacgtcaga aagggtggag 1680 ctaacaaaag acccgccccc gatgacgcgg
atataagcga gcccaagcgg gcctgcccct 1740 cagtcgcgga tccatcgacg
tcagacgcgg aaggagctcc ggtggacttt gccgacaggt 1800 accaaaacaa
atgttctcgt cacgcgggca tgcttcagat gctgtttccc tgcaaaacat 1860
gcgagagaat gaatcagaat ttcaacattt gcttcacgca cgggaccaga gactgttcag
1920 aatgtttccc tggcgtgtca gaatctcaac cggtcgtcag aaaaaagacg
tatcggaaac 1980 tctgtgcgat tcatcatctg ctggggcggg cacccgagat
tgcttgctcg gcctgcgacc 2040 tggtcaacgt ggacctggat gactgtgttt
ctgagcaata aatgacttaa accaggtatg 2100 gctgccgatg gttatcttcc
agattggctc gaggacaacc tctctgaggg cattcgcgag 2160 tggtgggacc
tgaaacctgg agccccgaaa cccaaagcca accagcaaaa gcaggacaac 2220
ggccggggtc tggtgcttcc tggctacaag tacctcggac ccttcaacgg actcgacaag
2280 ggggagcccg tcaacgcggc ggacgcagcg gccctcgagc acgacaaggc
ctacgaccag 2340 cagctccaag cgggtgacaa tccgtacctg cggtataatc
acgccgacgc cgagtttcag 2400 gagcgtctgc aagaagatac gtcttttggg
ggcaacctcg ggcgcgcagt cttccaggcc 2460 aaaaagcggg ttctcgaacc
tctgggcctg gttgaatcgc cggttaagac ggctcctgga 2520 aagaagagac
cggtagagcc atcaccccag cgctctccag actcctctac gggcatcggc 2580
aagaaaggcc agcagcccgc aaaaaagaga ctcaattttg ggcagactgg cgactcagag
2640 tcagtccccg accctcaacc aatcggagaa ccaccagcag gcccctctgg
tctgggatct 2700 ggtacaatgg ctgcaggcgg tggcgctcca atggcagaca
ataacgaagg cgccgacgga 2760 gtgggtagtt cctcaggaaa ttggcattgc
gattccacat ggctgggcga cagagtcatc 2820 accaccagca cccgcacctg
ggccctgccc acctacaaca accacctcta caagcaaatc 2880 tccaacggga
cctcgggagg aagcaccaac gacaacacct acttcggcta cagcaccccc 2940
tgggggtatt ttgacttcaa cagattccac tgccactttt caccacgtga ctggcagcga
3000 ctcatcaaca acaactgggg attccggccc aagaggctca acttcaagct
cttcaacatc 3060 caagtcaagg aggtcacgca gaatgaaggc accaagacca
tcgccaataa ccttaccagc 3120 acgattcagg tctttacgga ctcggaatac
cagctcccgt acgtgctcgg ctcggcgcac 3180 cagggctgcc tgcctccgtt
cccggcggac gtcttcatga ttcctcagta cgggtacctg 3240
actctgaaca atggcagtca ggctgtgggc cggtcgtcct tctactgcct ggagtacttt
3300 ccttctcaaa tgctgagaac gggcaacaac tttgaattca gctacaactt
cgaggacgtg 3360 cccttccaca gcagctacgc gcacagccag agcctggacc
ggctgatgaa ccctctcatc 3420 gaccagtact tgtactacct gtcccggact
caaagcacgg gcggtactgc aggaactcag 3480 cagttgctat tttctcaggc
cgggcctaac aacatgtcgg ctcaggccaa gaactggcta 3540 cccggtccct
gctaccggca gcaacgcgtc tccacgacac tgtcgcagaa caacaacagc 3600
aactttgcct ggacgggtgc caccaagtat catctgaatg gcagagactc tctggtgaat
3660 cctggcgttg ccatggctac ccacaaggac gacgaagagc gattttttcc
atccagcgga 3720 gtcttaatgt ttgggaaaca gggagctgga aaagacaacg
tggactatag cagcgtgatg 3780 ctaaccagcg aggaagaaat aaagaccacc
aacccagtgg ccacagaaca gtacggcgtg 3840 gtggccgata acctgcaaca
gcaaaacgcc gctcctattg taggggccgt caatagtcaa 3900 ggagccttac
ctggcatggt gtggcagaac cgggacgtgt acctgcaggg tcccatctgg 3960
gccaagattc ctcatacgga cggcaacttt catccctcgc cgctgatggg aggctttgga
4020 ctgaagcatc cgcctcctca gatcctgatt aaaaacacac ctgttcccgc
ggatcctccg 4080 accaccttca ctaaggccaa gctggcttct ttcatcacgc
agtacagtac cggccaggtc 4140 agcgtggaga tcgagtggga gctgcagaag
gagaacagca aacgctggaa cccagagatt 4200 cagtacactt ccaactacta
caaatctaca aatgtggact ttgctgtcaa tactgagggt 4260 acttattccg
agcctcgccc cattggcacc cgttacctca cccgtaatct gtaattacat 4320
gttaatcaat aaaccggtta attcgtttca gttgaacttt ggtctcctgt ccttcttatc
4380 ttatcggtta ccatagaaac tggttactta ttaactgctt ggtgcgcttc
gcgataaaag 4440 acttacgtca tcgggttacc cctagtgatg ga 4472
<210> SEQ ID NO 15 <211> LENGTH: 2052 <212> TYPE:
DNA <213> ORGANISM: Adeno-associated virus <400>
SEQUENCE: 15 cgaccgcgcg agcgagcgag tgactccggc gggcccgttt cgggcccgca
gcccgctgga 60 aaccagcggg ccggagtcac tcgctcgctc gcgcgtctct
ccctcaccgg ttgaggtagt 120 gatccccaag gaacatcaat tactaattgg
gcggtacgat gaatagatgc atcggtacga 180 gatctattgt tgtatcctcg
acactaaccg acaaaagtcg gttagtcgtg actgagtaaa 240 cgtatcggaa
atgttcgcca gtgtttgagt tctttgctcg ccaaaattat cagaaaatct 300
tataacaaat agcttggctt attccttgac acgaaacact aagtgtatag tcacctcccc
360 acacctttac cgtggaacta gagtgggagt agctttcacc tcaactacag
gaagggaccg 420 agcgatgtct gcgtgaaggc gttcaaaaga aaacttctag
aagagaaagt agatttaaaa 480 acctcgtcca aaagactgaa gccagccttt
tggggagggt taaagtgacc agatgttact 540 ttcgttttgt caagagaagg
ggcgaggggc cacacactct ccccgaaact aggaagagac 600 caaaggatcc
tttgcgcata caccgatcta ttgttgtatc ctcgacacta accgacaaaa 660
gtcggttagt cgtgactgag taaacgtatc ggaaatgttc gccagtgttt gagttctttg
720 ctcgccaaaa ttatcagaaa atcttataac aaatagcttg gcttattcct
tgacacgaaa 780 cactaagtgt atagtcacct ccccacacct ttaccgtgga
actagagtgg gagtagcttt 840 cacctcaact acaggaaggg accgagcgat
gtctgcgtga aggcgtttaa gaatggaatc 900 ttttaacacg ttaaaaacct
cgtccaaaag actgaagcca gccttttggg gagggttaaa 960 gtgaccagat
gttactttcg ttttgtcaag agaaggggcg aggggccaca cactctcccc 1020
gaaactagga agagaccaaa ggatcctttg cgcatacacc gatctattgt tgtatcctcg
1080 acactaaccg acaaaagtcg gttagtcgtg actgagtaaa cgtatcggaa
atgttcgcca 1140 gtgtttgagt tctttgctcg ccaaaattat cagaaaatct
tataacaaat agcttggctt 1200 attccttgac acgaaacact aagtgtatag
tcacctcccc acacctttac cgtggaacta 1260 gagtgggagt agctttcacc
tcaactacag gaagggaccg agcgatgtct gcgtgaaggc 1320 gtttaagaat
ggaatctttt aacacgttaa aaacctcgtc caaaagactg aagccagcct 1380
tttggggagg gttaaagtga ccagatgtta ctttcgtttt gtcaagagaa ggggcgaggg
1440 gccacacact ctccccgaaa ctaggaagag accaaaggat cctttgcgca
tacaccgatc 1500 tattgttgta tcctcgacac taaccgacaa aagtcggtta
gtcgtgactg agtaaacgta 1560 tcggaaatgt tcgccagtgt ttgagttctt
tgctcgccaa aattatcaga aaatcttata 1620 acaaatagct tggcttattc
cttgacacga aacactaagt gtatagtcac ctccccacac 1680 ctttaccgtg
gaactagagt gggagtagct ttcacctcaa ctacaggaag ggaccgagcg 1740
atgtctgcgt gaaggcgttc aaaagaaaac ttctagaaga gaaagtagat ttaaaaacct
1800 cgtccaaaag actgaagcca gccttttggg gagggttaaa gtgaccagat
gttactttcg 1860 ttttgtcaag agaaggggcg aggggccaca cactctcccc
gaaactagga agagaccaaa 1920 ggatcctttg cgcatacaca tgttggggtg
agggagagac gcgcgagcga gcgagtgact 1980 ccggcccgct ggtttccagc
gggctgcggg cccgaaacgg gcccgccgga gtcactcgct 2040 cgctcgcgcg tc 2052
<210> SEQ ID NO 16 <211> LENGTH: 8 <212> TYPE:
PRT <213> ORGANISM: Homo sapiens <400> SEQUENCE: 16 Trp
Val Asn Ala Gln Phe Ser Lys 1 5 <210> SEQ ID NO 17
<211> LENGTH: 14 <212> TYPE: PRT <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 17 Gln His Ile Glu Asn Leu Phe
Ser Asp Leu Gln Asp Gly Arg 1 5 10 <210> SEQ ID NO 18
<211> LENGTH: 12 <212> TYPE: PRT <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 18 Leu Leu Asp Leu Leu Glu Gly
Leu Thr Gly Gln Lys 1 5 10 <210> SEQ ID NO 19 <211>
LENGTH: 23 <212> TYPE: PRT <213> ORGANISM: Homo sapiens
<400> SEQUENCE: 19 Val Leu Gln Asn Asn Asn Val Asp Leu Val
Asn Ile Gly Ser Thr Asp 1 5 10 15 Ile Val Asp Gly Asn His Lys 20
<210> SEQ ID NO 20 <211> LENGTH: 13 <212> TYPE:
PRT <213> ORGANISM: Homo sapiens <400> SEQUENCE: 20 Asn
Leu Met Ala Gly Leu Gln Gln Thr Asn Ser Glu Lys 1 5 10 <210>
SEQ ID NO 21 <211> LENGTH: 9 <212> TYPE: PRT
<213> ORGANISM: Homo sapiens <400> SEQUENCE: 21 Leu Glu
His Ala Phe Asn Ile Ala Arg 1 5 <210> SEQ ID NO 22
<211> LENGTH: 7 <212> TYPE: PRT <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 22 Tyr Gln Leu Gly Ile Glu Lys 1
5 <210> SEQ ID NO 23 <211> LENGTH: 15 <212> TYPE:
PRT <213> ORGANISM: Homo sapiens <400> SEQUENCE: 23 Leu
Leu Asp Pro Glu Asp Val Asp Thr Thr Tyr Pro Asp Lys Lys 1 5 10 15
<210> SEQ ID NO 24 <211> LENGTH: 17 <212> TYPE:
PRT <213> ORGANISM: Homo sapiens <400> SEQUENCE: 24 Ser
Tyr Ala Tyr Thr Gln Ala Ala Tyr Val Thr Thr Ser Asp Pro Thr 1 5 10
15 Arg <210> SEQ ID NO 25 <211> LENGTH: 14 <212>
TYPE: PRT <213> ORGANISM: Homo sapiens <400> SEQUENCE:
25 Ser Pro Phe Pro Ser Gln His Leu Glu Ala Pro Glu Asp Lys 1 5 10
<210> SEQ ID NO 26 <211> LENGTH: 2052 <212> TYPE:
DNA <213> ORGANISM: Adeno-associated virus <400>
SEQUENCE: 26 gctggcgcgc tcgctcgctc actgaggccg cccgggcaaa gcccgggcgt
cgggcgacct 60 ttggtcgccc ggcctcagtg agcgagcgag cgcgcagaga
gggagtggcc aactccatca 120 ctaggggttc cttgtagtta atgattaacc
cgccatgcta cttatctacg tagccatgct 180 ctagataaca acataggagc
tgtgattggc tgttttcagc caatcagcac tgactcattt 240
gcatagcctt tacaagcggt cacaaactca agaaacgagc ggttttaata gtcttttaga
300 atattgttta tcgaaccgaa taaggaactg tgctttgtga ttcacatatc
agtggagggg 360 tgtggaaatg gcaccttgat ctcaccctca tcgaaagtgg
agttgatgtc cttccctggc 420 tcgctacaga cgcacttccg caagttttct
tttgaagatc ttctctttca tctaaatttt 480 tggagcaggt tttctgactt
cggtcggaaa acccctccca atttcactgg tctacaatga 540 aagcaaaaca
gttctcttcc ccgctccccg gtgtgtgaga ggggctttga tccttctctg 600
gtttcctagg aaacgcgtat gtggctagat aacaacatag gagctgtgat tggctgtttt
660 cagccaatca gcactgactc atttgcatag cctttacaag cggtcacaaa
ctcaagaaac 720 gagcggtttt aatagtcttt tagaatattg tttatcgaac
cgaataagga actgtgcttt 780 gtgattcaca tatcagtgga ggggtgtgga
aatggcacct tgatctcacc ctcatcgaaa 840 gtggagttga tgtccttccc
tggctcgcta cagacgcact tccgcaaatt cttaccttag 900 aaaattgtgc
aatttttgga gcaggttttc tgacttcggt cggaaaaccc ctcccaattt 960
cactggtcta caatgaaagc aaaacagttc tcttccccgc tccccggtgt gtgagagggg
1020 ctttgatcct tctctggttt cctaggaaac gcgtatgtgg ctagataaca
acataggagc 1080 tgtgattggc tgttttcagc caatcagcac tgactcattt
gcatagcctt tacaagcggt 1140 cacaaactca agaaacgagc ggttttaata
gtcttttaga atattgttta tcgaaccgaa 1200 taaggaactg tgctttgtga
ttcacatatc agtggagggg tgtggaaatg gcaccttgat 1260 ctcaccctca
tcgaaagtgg agttgatgtc cttccctggc tcgctacaga cgcacttccg 1320
caaattctta ccttagaaaa ttgtgcaatt tttggagcag gttttctgac ttcggtcgga
1380 aaacccctcc caatttcact ggtctacaat gaaagcaaaa cagttctctt
ccccgctccc 1440 cggtgtgtga gaggggcttt gatccttctc tggtttccta
ggaaacgcgt atgtggctag 1500 ataacaacat aggagctgtg attggctgtt
ttcagccaat cagcactgac tcatttgcat 1560 agcctttaca agcggtcaca
aactcaagaa acgagcggtt ttaatagtct tttagaatat 1620 tgtttatcga
accgaataag gaactgtgct ttgtgattca catatcagtg gaggggtgtg 1680
gaaatggcac cttgatctca ccctcatcga aagtggagtt gatgtccttc cctggctcgc
1740 tacagacgca cttccgcaag ttttcttttg aagatcttct ctttcatcta
aatttttgga 1800 gcaggttttc tgacttcggt cggaaaaccc ctcccaattt
cactggtcta caatgaaagc 1860 aaaacagttc tcttccccgc tccccggtgt
gtgagagggg ctttgatcct tctctggttt 1920 cctaggaaac gcgtatgtgt
acaaccccac tccctctctg cgcgctcgct cgctcactga 1980 ggccgggcga
ccaaaggtcg cccgacgccc gggctttgcc cgggcggcct cagtgagcga 2040
gcgagcgcgc ag 2052 <210> SEQ ID NO 27 <211> LENGTH: 240
<212> TYPE: PRT <213> ORGANISM: Homo sapiens
<300> PUBLICATION INFORMATION: <308> DATABASE ACCESSION
NUMBER: UniProtKB/Swiss-Prot: P11532.3 <309> DATABASE ENTRY
DATE: 2015-07-22 <313> RELEVANT RESIDUES IN SEQ ID NO:
(1)..(240) <400> SEQUENCE: 27 Met Leu Trp Trp Glu Glu Val Glu
Asp Cys Tyr Glu Arg Glu Asp Val 1 5 10 15 Gln Lys Lys Thr Phe Thr
Lys Trp Val Asn Ala Gln Phe Ser Lys Phe 20 25 30 Gly Lys Gln His
Ile Glu Asn Leu Phe Ser Asp Leu Gln Asp Gly Arg 35 40 45 Arg Leu
Leu Asp Leu Leu Glu Gly Leu Thr Gly Gln Lys Leu Pro Lys 50 55 60
Glu Lys Gly Ser Thr Arg Val His Ala Leu Asn Asn Val Asn Lys Ala 65
70 75 80 Leu Arg Val Leu Gln Asn Asn Asn Val Asp Leu Val Asn Ile
Gly Ser 85 90 95 Thr Asp Ile Val Asp Gly Asn His Lys Leu Thr Leu
Gly Leu Ile Trp 100 105 110 Asn Ile Ile Leu His Trp Gln Val Lys Asn
Val Met Lys Asn Ile Met 115 120 125 Ala Gly Leu Gln Gln Thr Asn Ser
Glu Lys Ile Leu Leu Ser Trp Val 130 135 140 Arg Gln Ser Thr Arg Asn
Tyr Pro Gln Val Asn Val Ile Asn Phe Thr 145 150 155 160 Thr Ser Trp
Ser Asp Gly Leu Ala Leu Asn Ala Leu Ile His Ser His 165 170 175 Arg
Pro Asp Leu Phe Asp Trp Asn Ser Val Val Cys Gln Gln Ser Ala 180 185
190 Thr Gln Arg Leu Glu His Ala Phe Asn Ile Ala Arg Tyr Gln Leu Gly
195 200 205 Ile Glu Lys Leu Leu Asp Pro Glu Asp Val Asp Thr Thr Tyr
Pro Asp 210 215 220 Lys Lys Ser Ile Leu Met Tyr Ile Thr Ser Leu Phe
Gln Val Leu Pro 225 230 235 240 <210> SEQ ID NO 28
<211> LENGTH: 22 <212> TYPE: PRT <213> ORGANISM:
Homo sapiens <400> SEQUENCE: 28 Tyr Gln Leu Gly Ile Glu Lys
Leu Leu Asp Pro Glu Asp Val Asp Thr 1 5 10 15 Thr Tyr Pro Asp Lys
Lys 20 <210> SEQ ID NO 29 <211> LENGTH: 112 <212>
TYPE: PRT <213> ORGANISM: Homo sapiens <400> SEQUENCE:
29 Trp Val Asn Ala Gln Phe Ser Lys Phe Gly Lys Gln His Ile Glu Asn
1 5 10 15 Leu Phe Ser Asp Leu Gln Asp Gly Arg Arg Leu Leu Asp Leu
Leu Glu 20 25 30 Gly Leu Thr Gly Gln Lys Val Leu Gln Asn Asn Asn
Val Asp Leu Val 35 40 45 Asn Ile Gly Ser Thr Asp Ile Val Asp Gly
Asn His Lys Asn Ile Met 50 55 60 Ala Gly Leu Gln Gln Thr Asn Ser
Glu Lys Ile Leu Leu Leu Glu His 65 70 75 80 Ala Phe Asn Ile Ala Arg
Tyr Gln Leu Gly Ile Glu Lys Leu Leu Asp 85 90 95 Pro Glu Asp Val
Asp Thr Thr Tyr Pro Asp Lys Lys Ser Ile Leu Met 100 105 110
<210> SEQ ID NO 30 <211> LENGTH: 75 <212> TYPE:
DNA <213> ORGANISM: Homo sapiens <400> SEQUENCE: 30
tagatgaaag agaagatgtt caaaagaaaa cattcacaaa atgggtaaat gcacaatttt
60 ctaaggtaag aatgg 75 <210> SEQ ID NO 31 <211> LENGTH:
80 <212> TYPE: DNA <213> ORGANISM: Homo sapiens
<400> SEQUENCE: 31 aactccattt atcaattaac caaaaactcc
caaataactg tgtaggttcc atgagttcag 60 gagattccac ttcaactaat 80
<210> SEQ ID NO 32 <211> LENGTH: 80 <212> TYPE:
DNA <213> ORGANISM: Homo sapiens <400> SEQUENCE: 32
aactccattt atcaattaac caaaaactcc caaataactg tggtgtatat ttatctattt
60 ttatgggttg caaaatactt 80 <210> SEQ ID NO 33 <211>
LENGTH: 80 <212> TYPE: DNA <213> ORGANISM: Homo sapiens
<400> SEQUENCE: 33 caaattctcc cctgaaaatt taaaaaaata
cattgttctg tggtgtatat ttatctattt 60 ttatgggttg caaaatactt 80
<210> SEQ ID NO 34 <211> LENGTH: 55 <212> TYPE:
DNA <213> ORGANISM: Homo sapiens <400> SEQUENCE: 34
aattaaccaa aaactcccaa ataactgtgg tgtatattta tctattttta tgggt 55
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
D00012
D00013

D00014
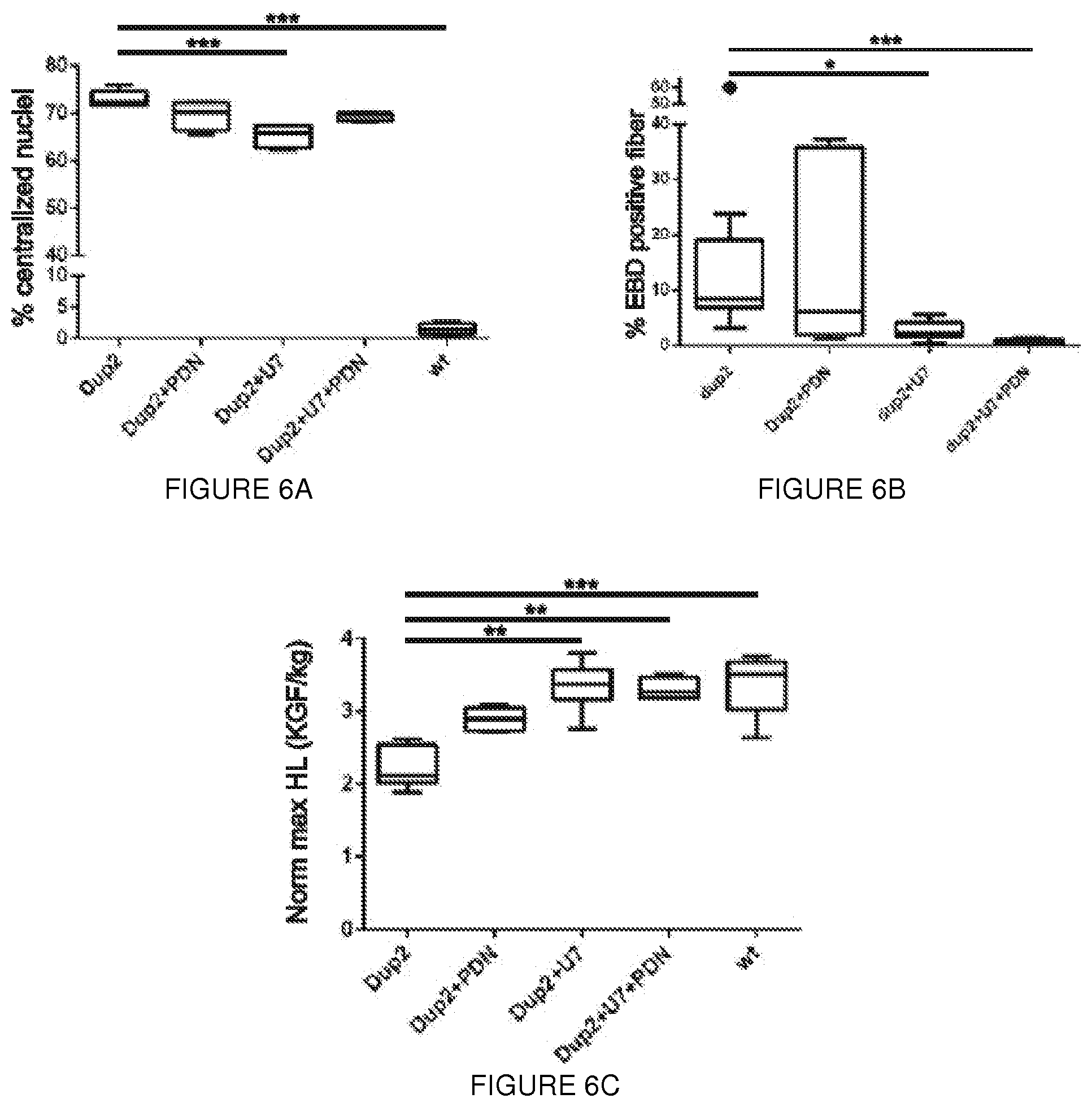
D00015
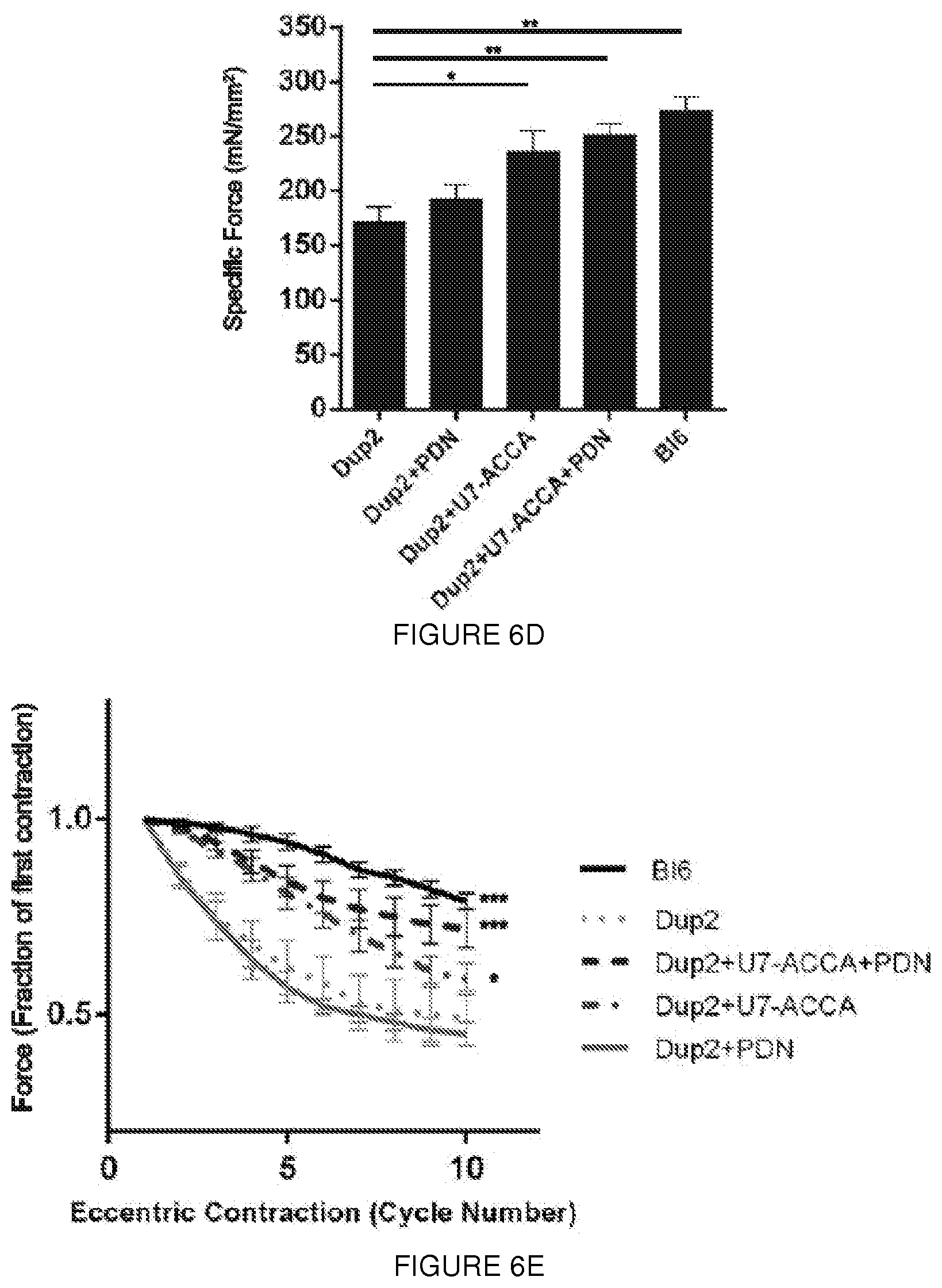
D00016

D00017
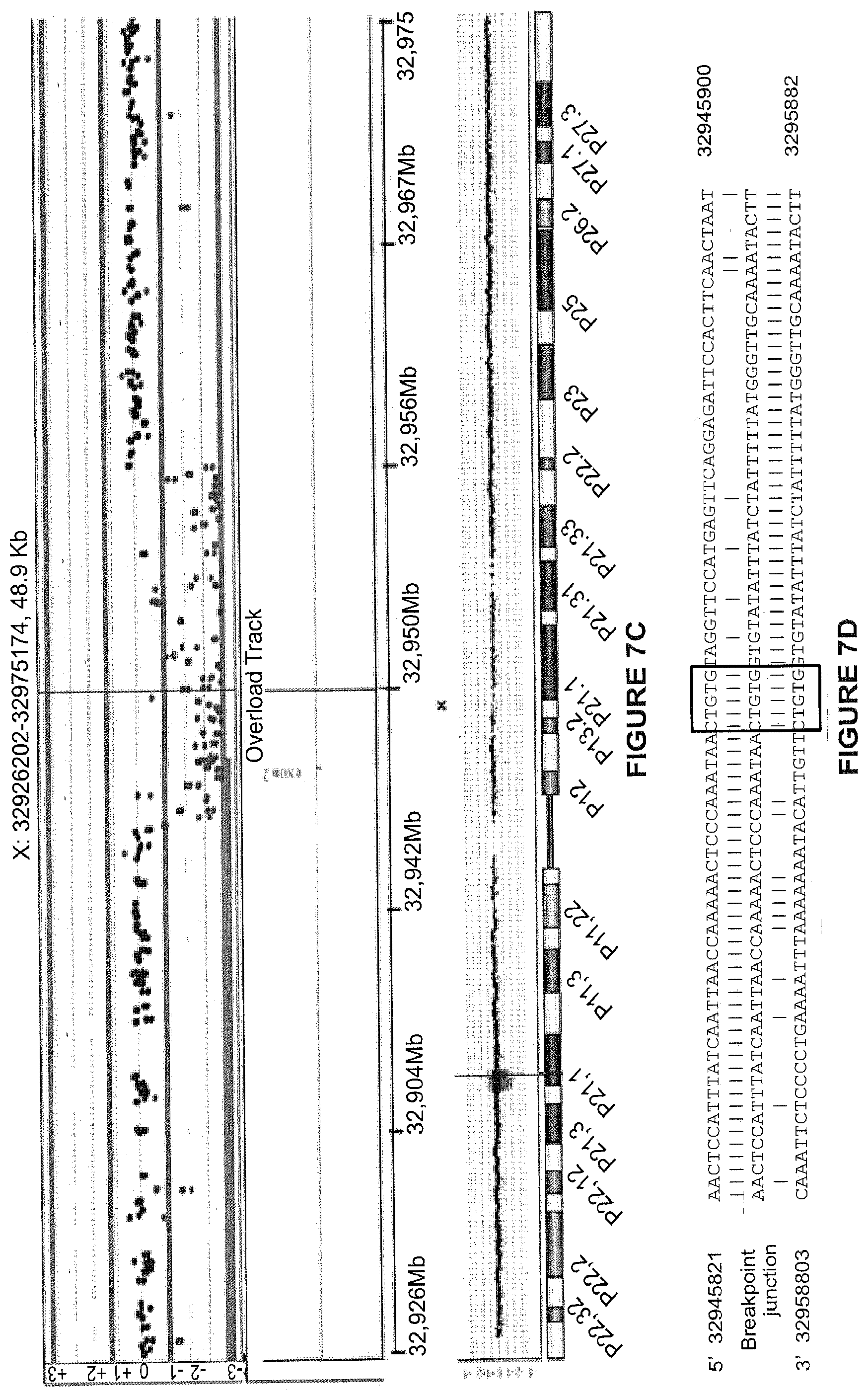
D00018

D00019

D00020
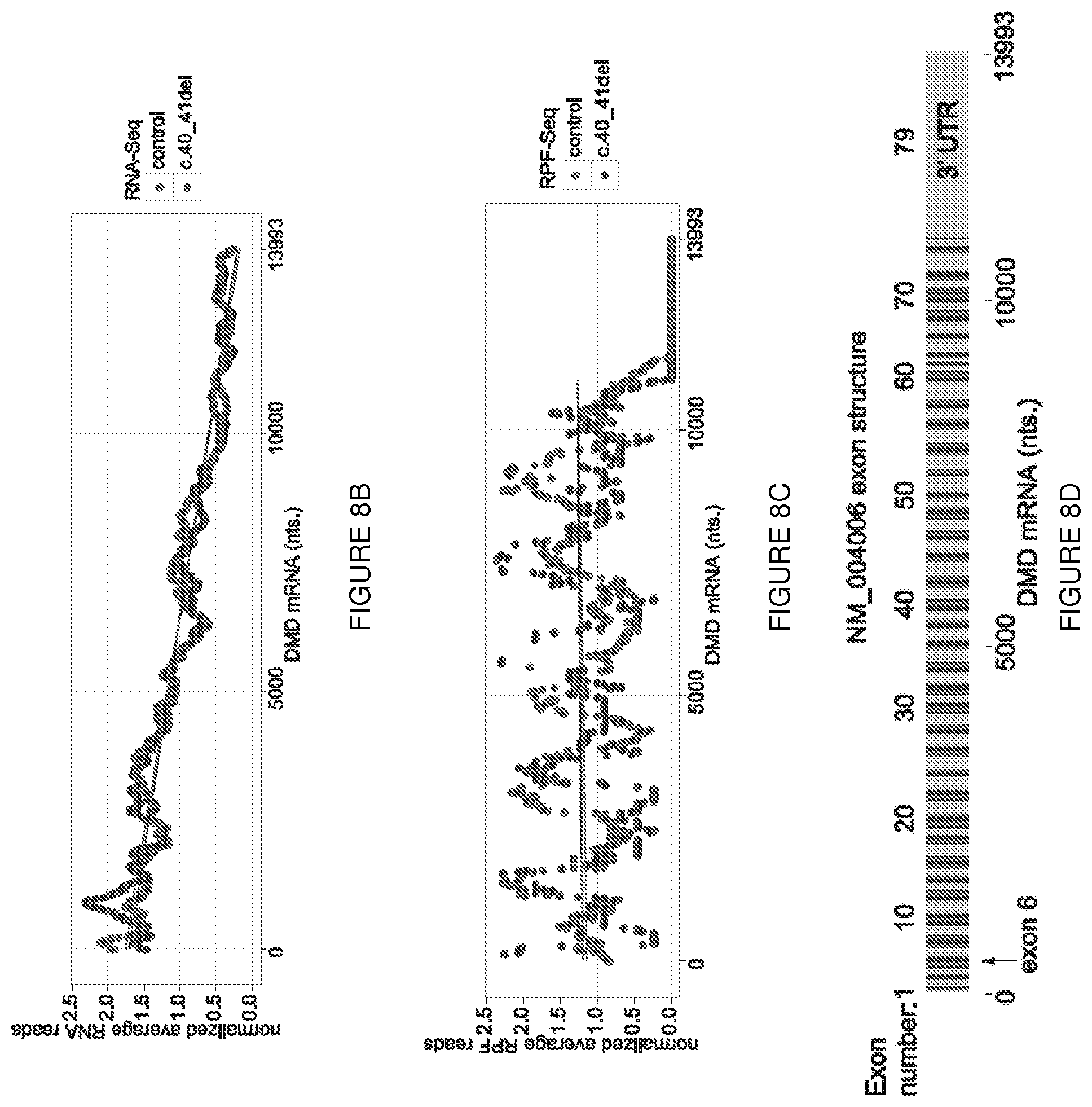
D00021
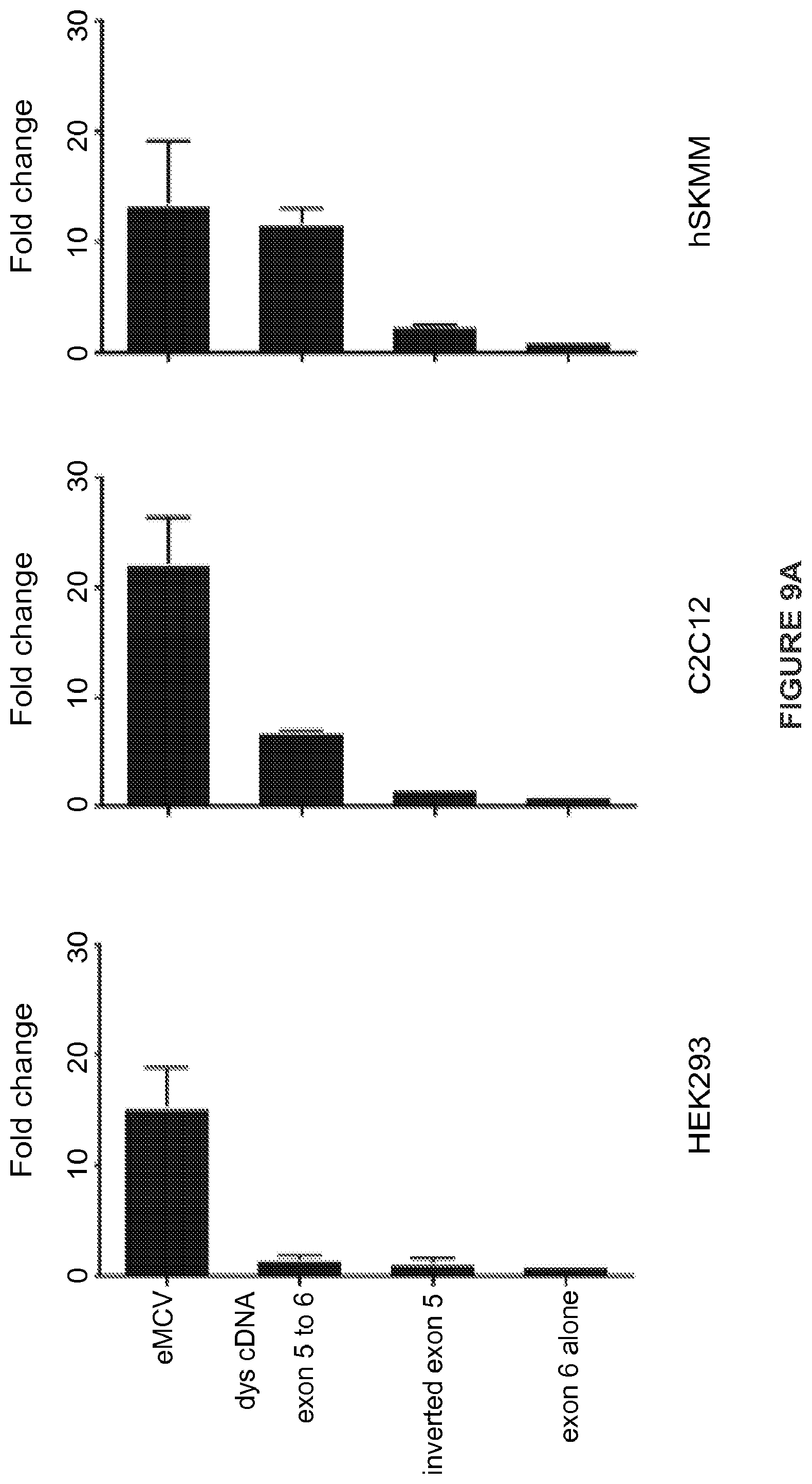
D00022
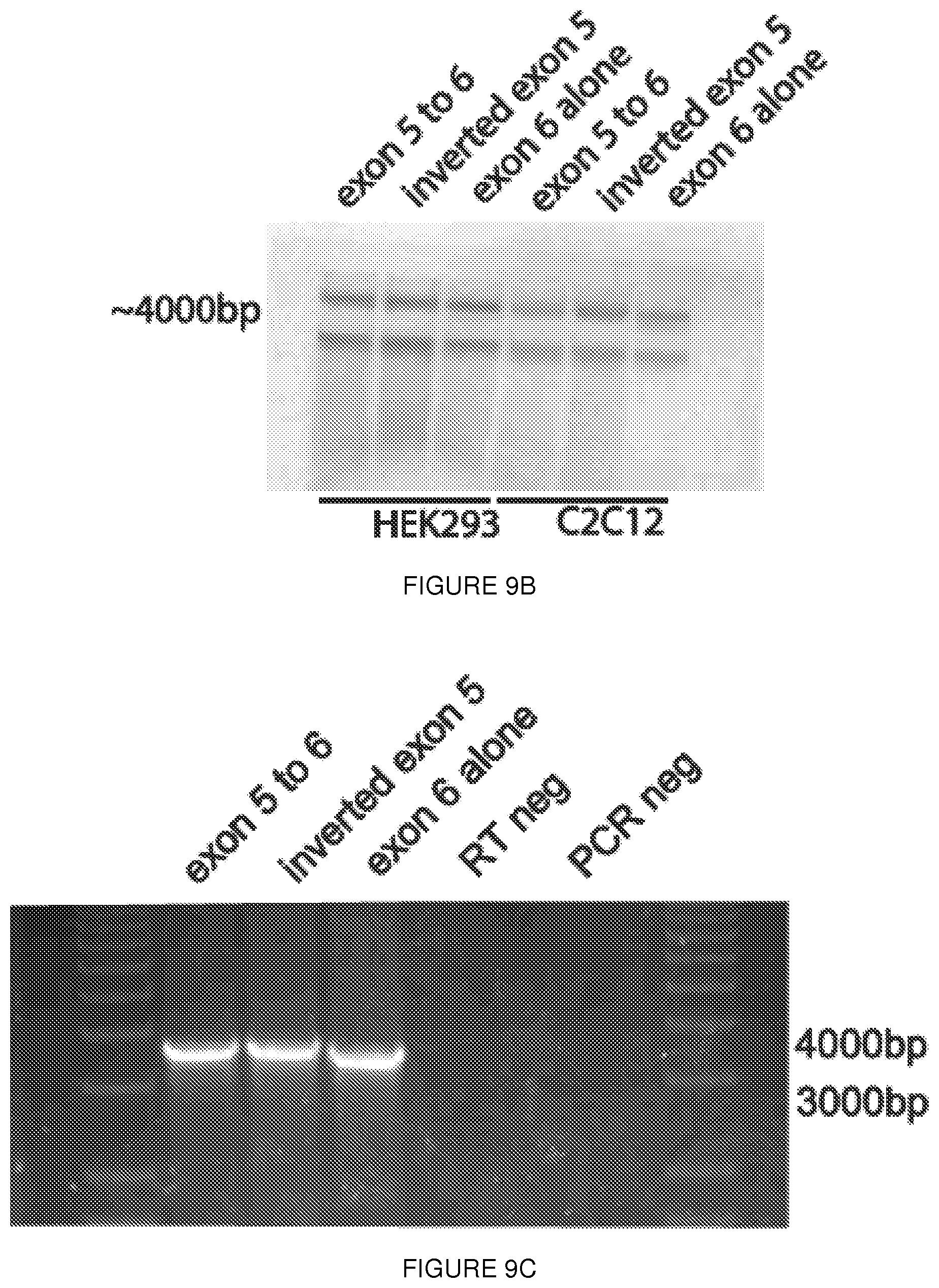
D00023
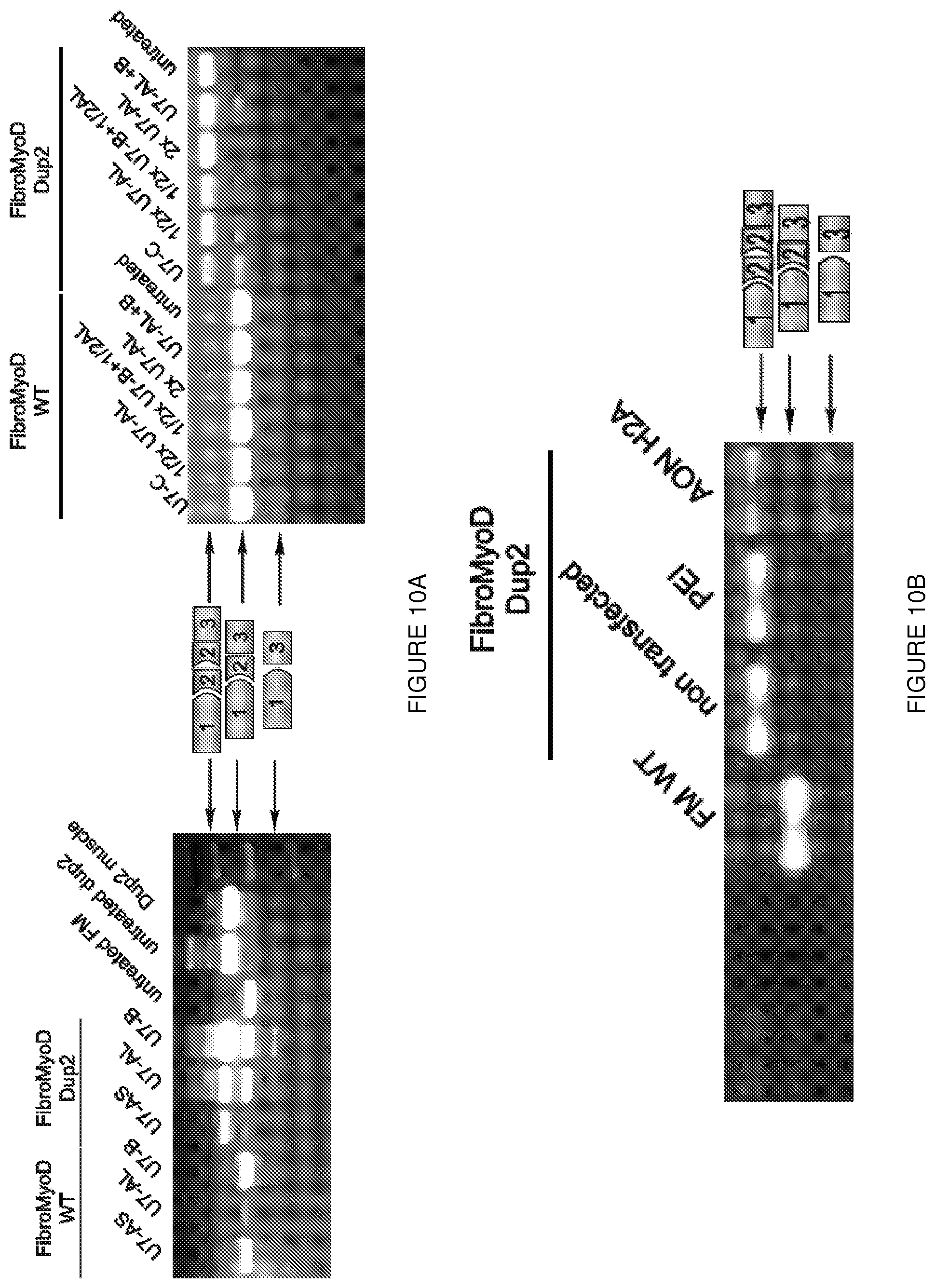
D00024
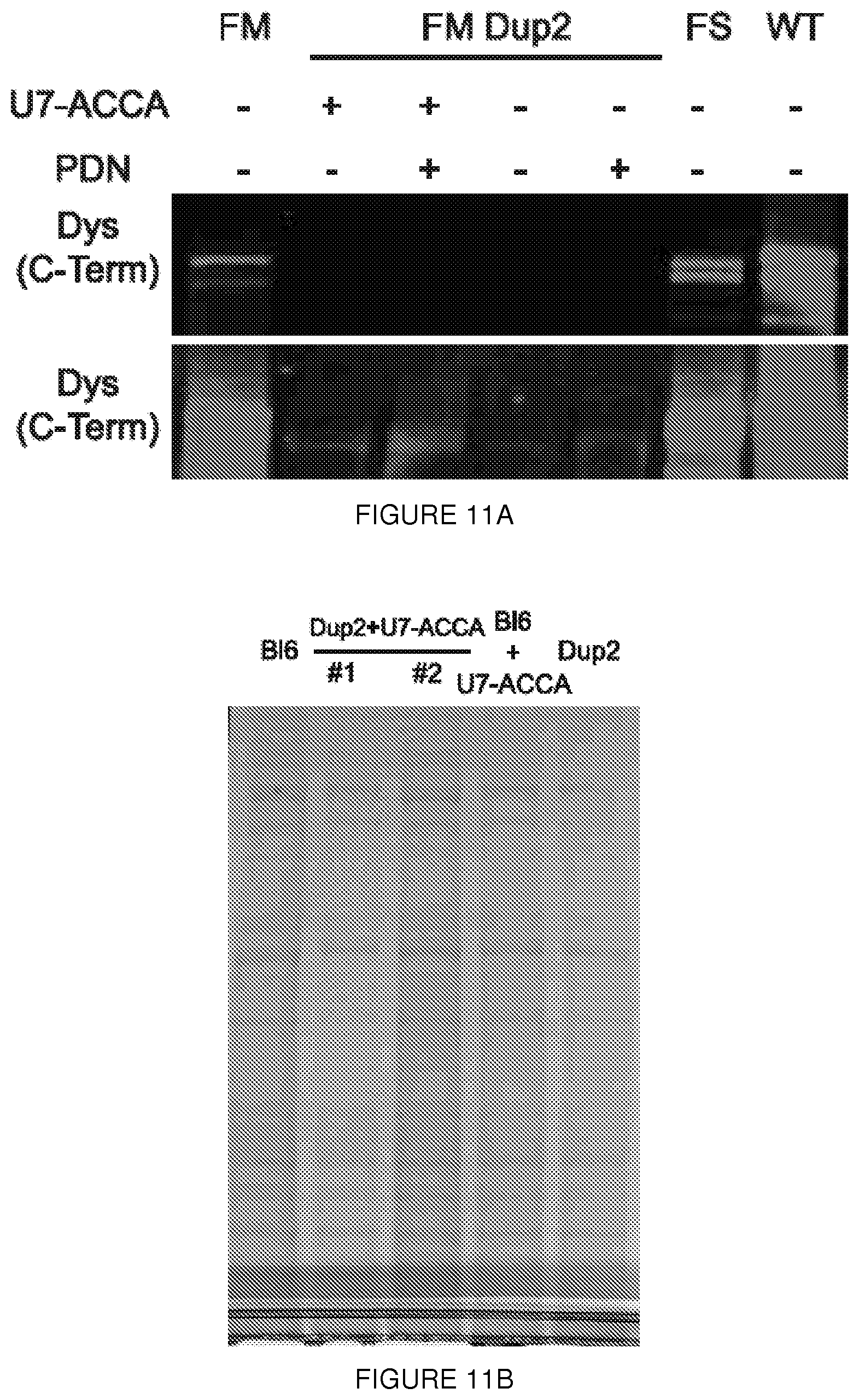
D00025
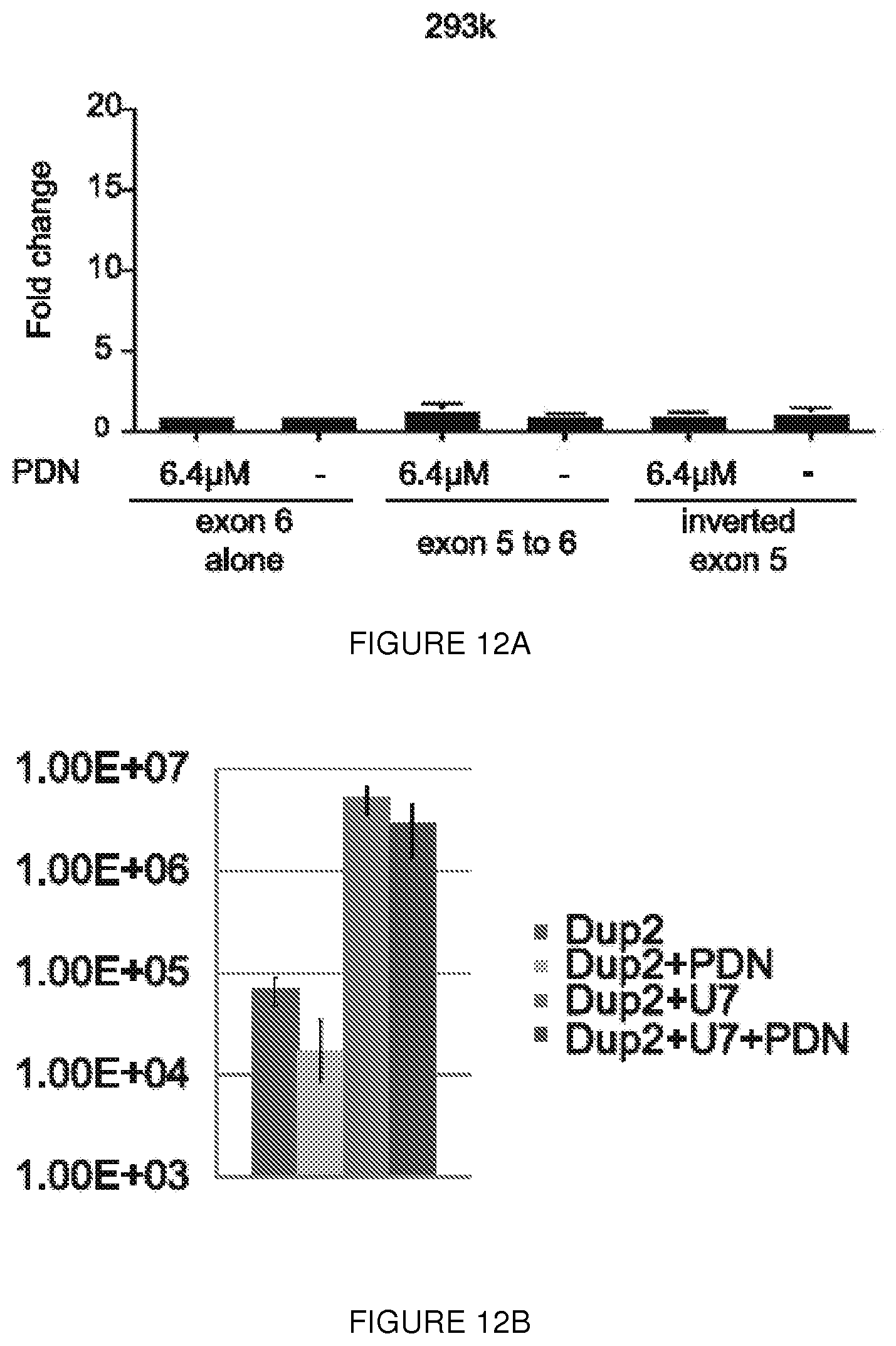
D00026

D00027

D00028

D00029

D00030
D00031

D00032

D00033

D00034

D00035

D00036

D00037
D00038

D00039

D00040

D00041
D00042
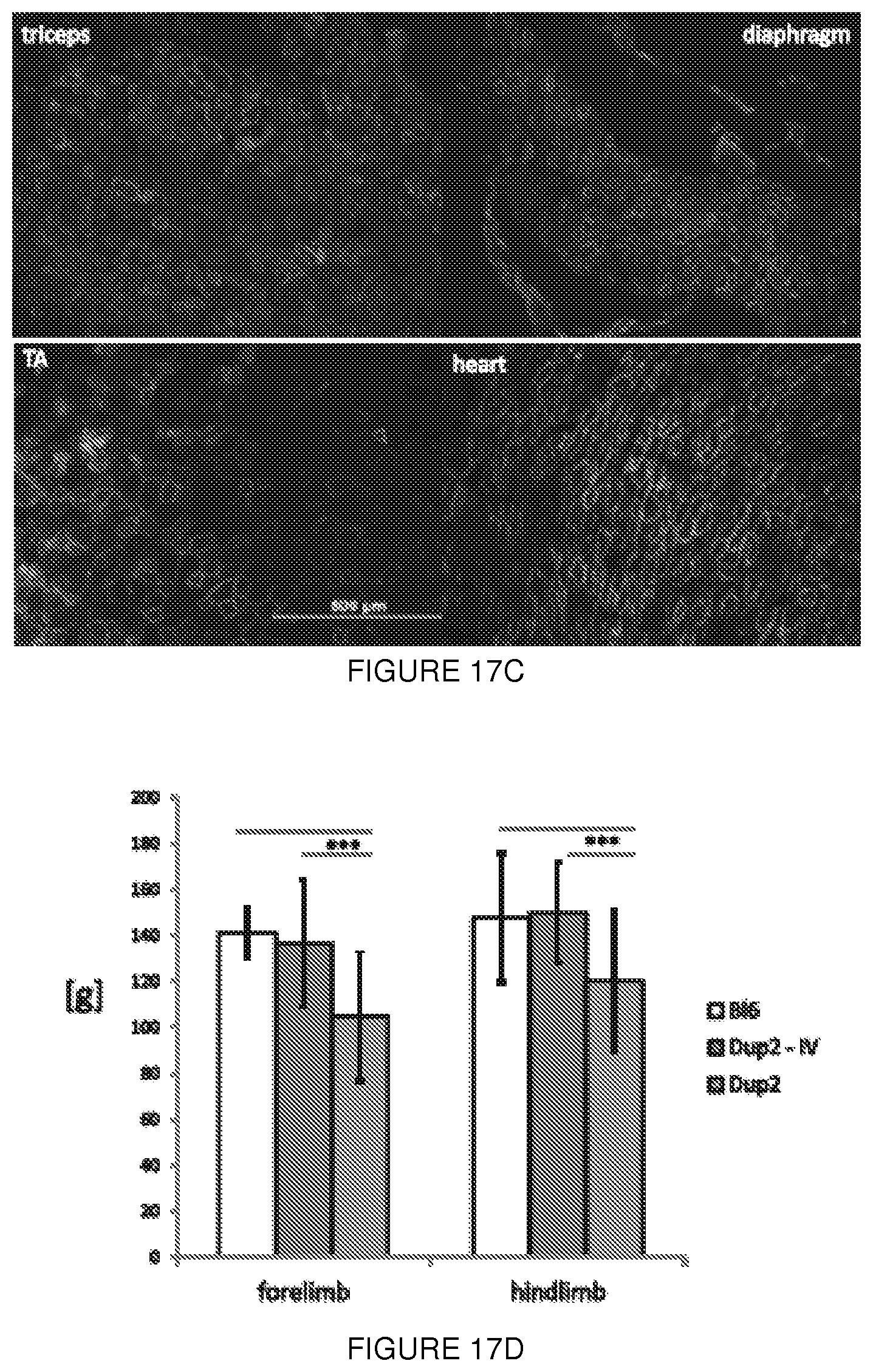
D00043
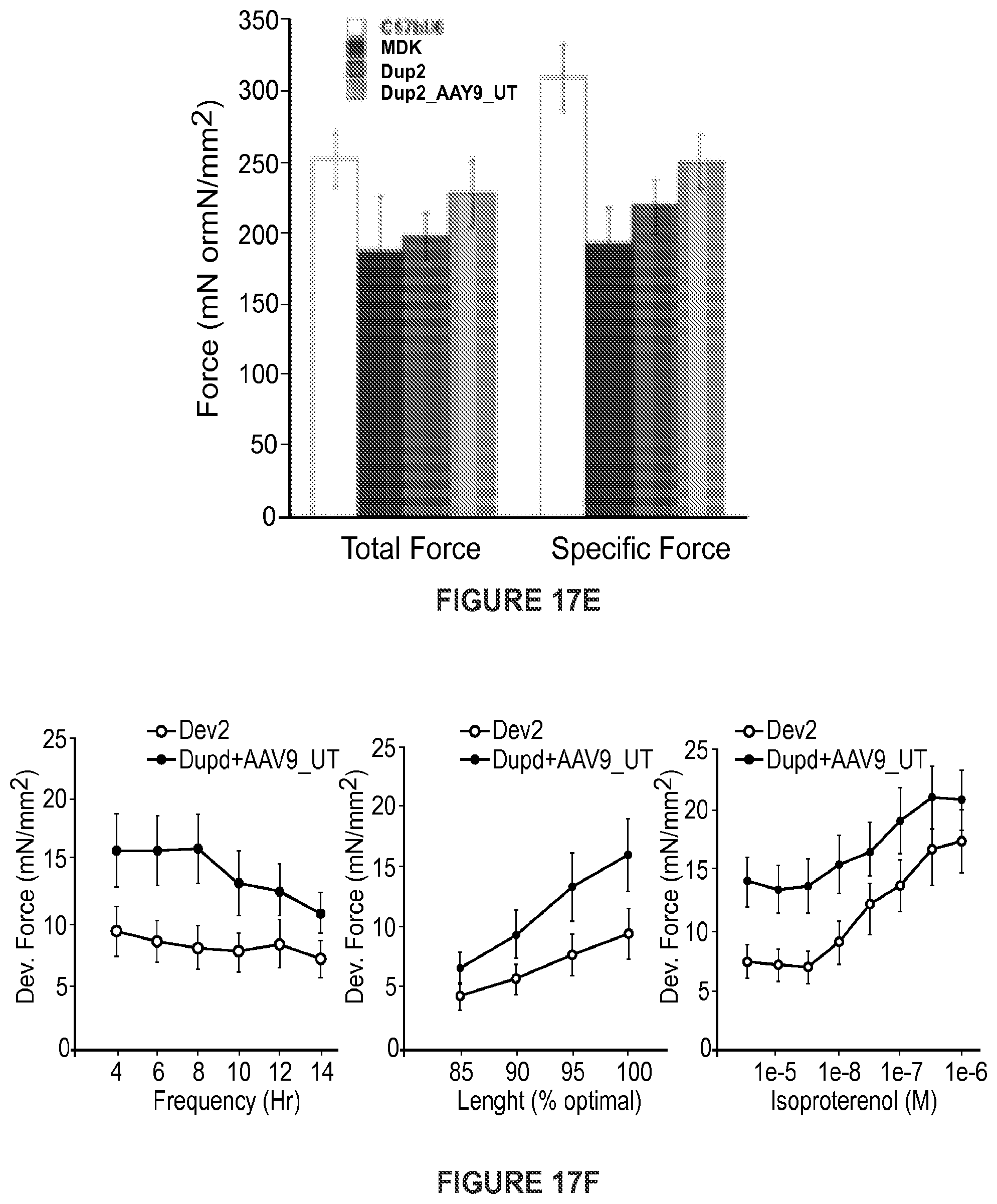
D00044
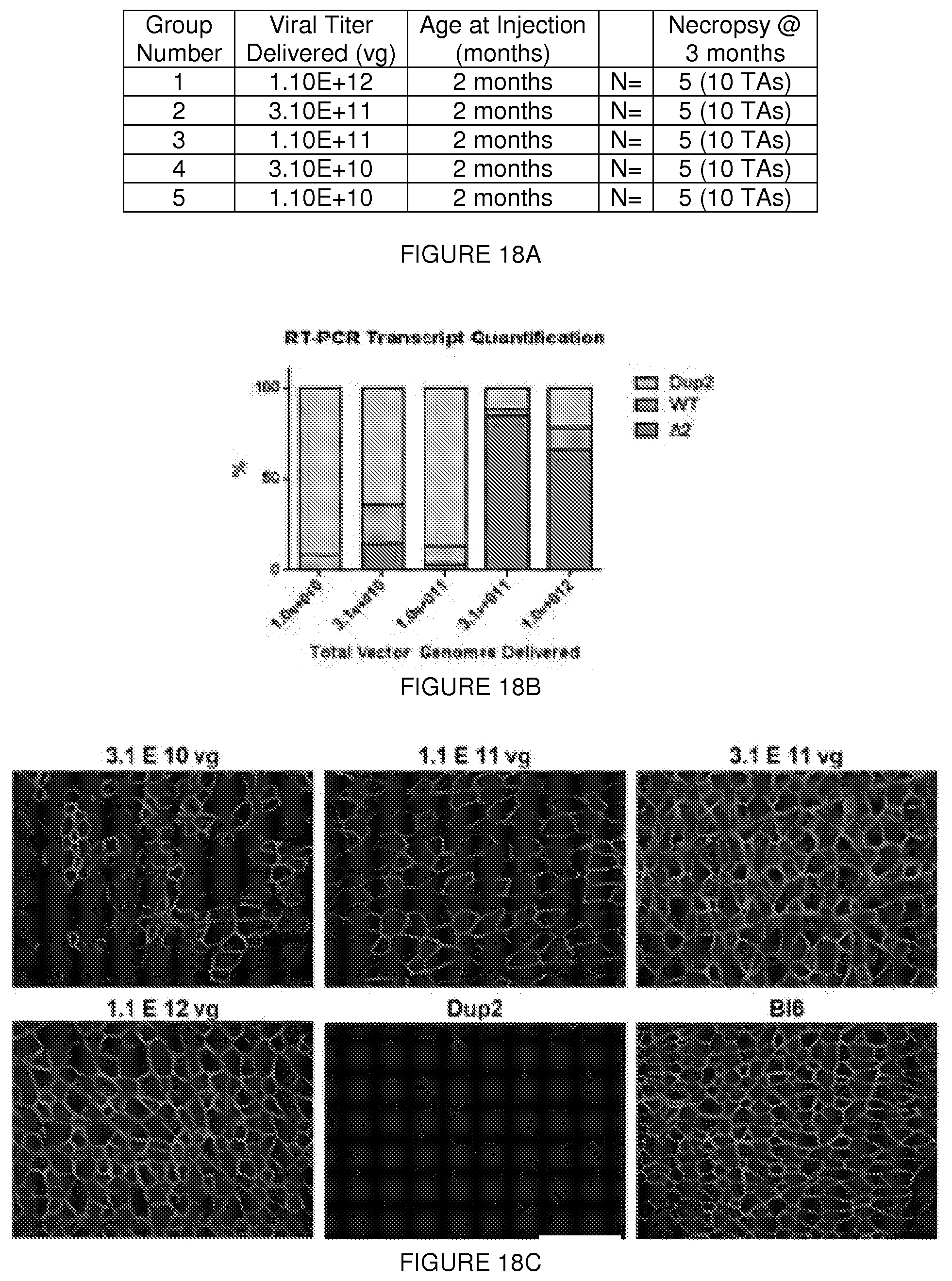
D00045

D00046
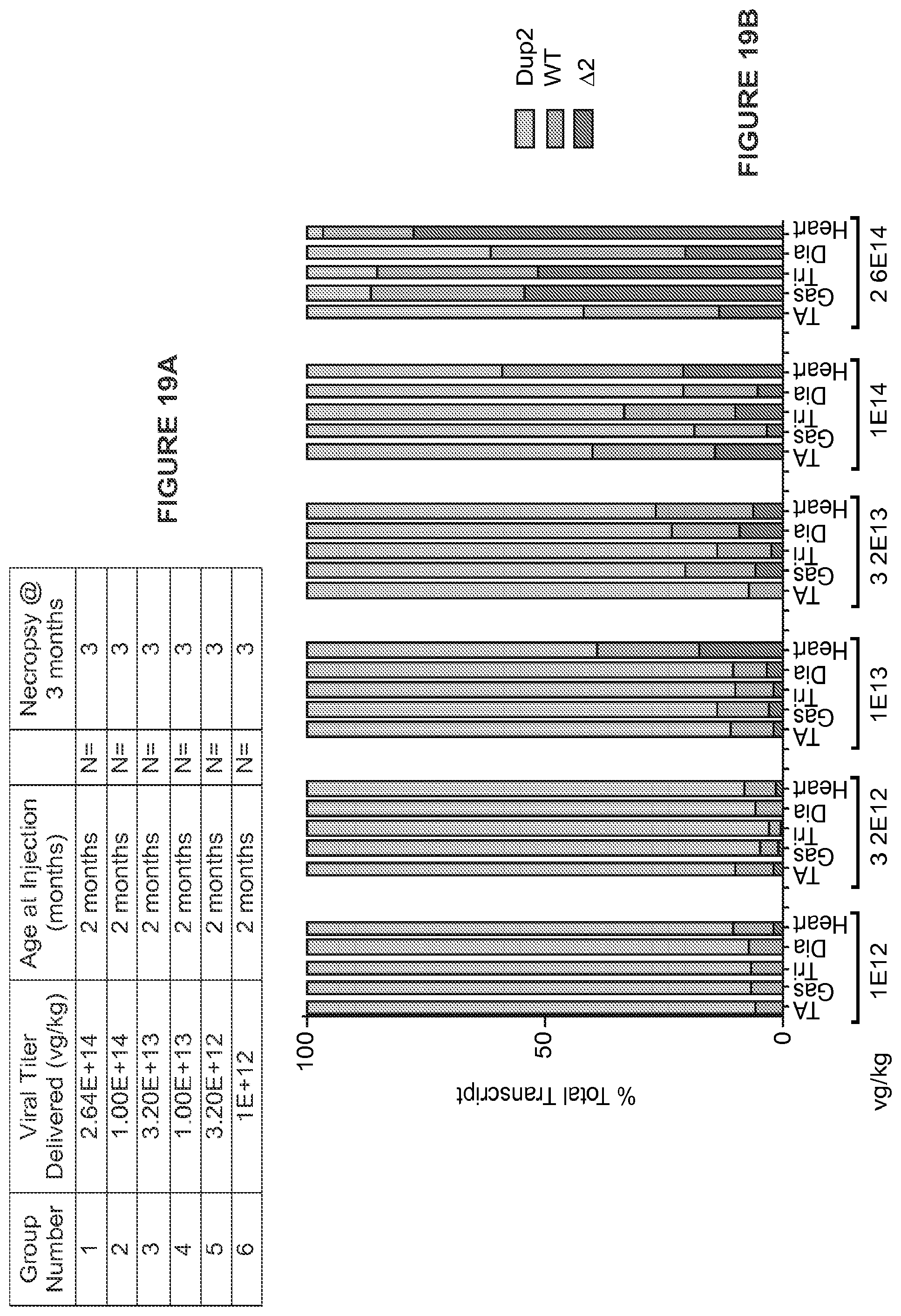
D00047
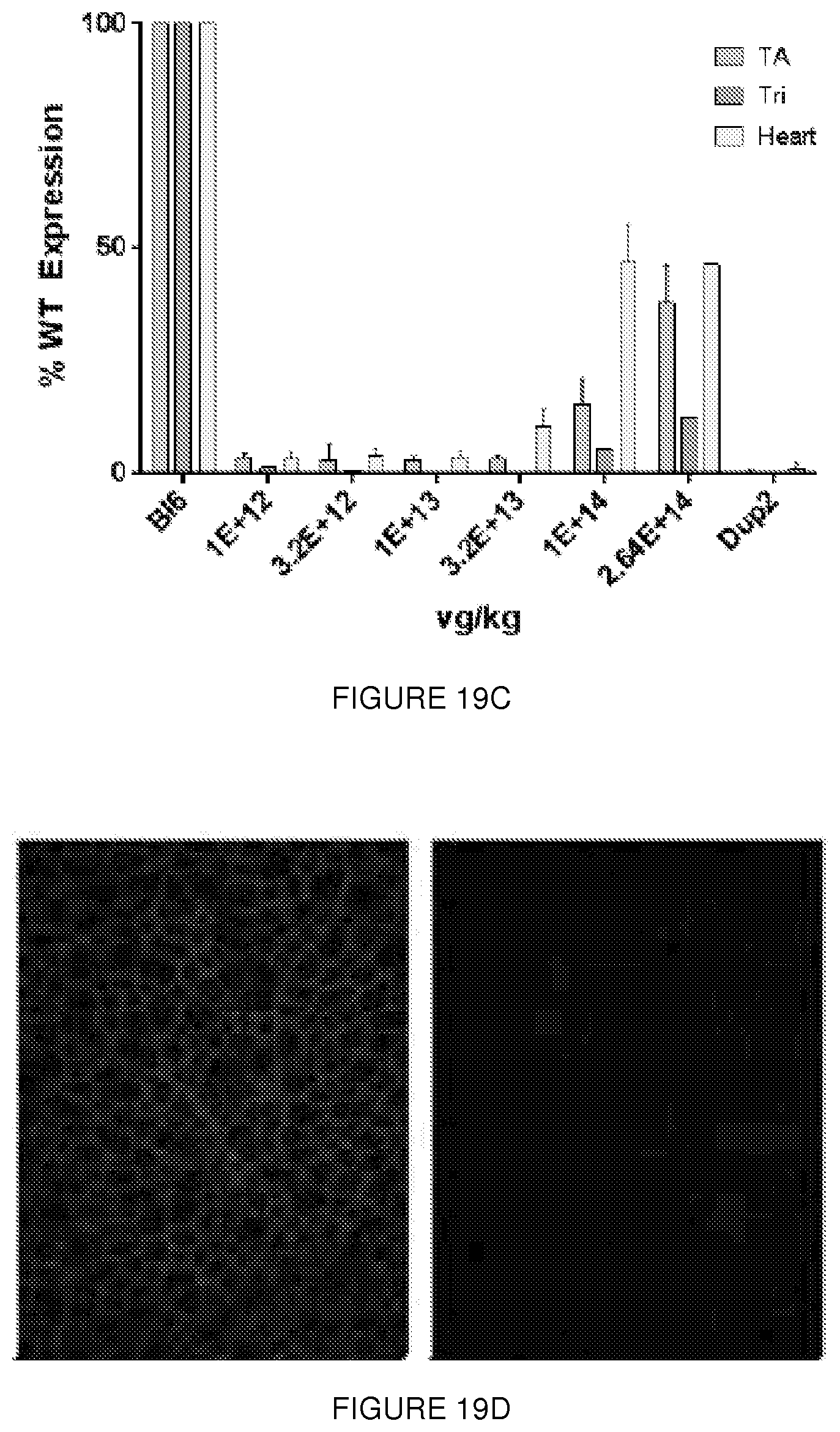
D00048
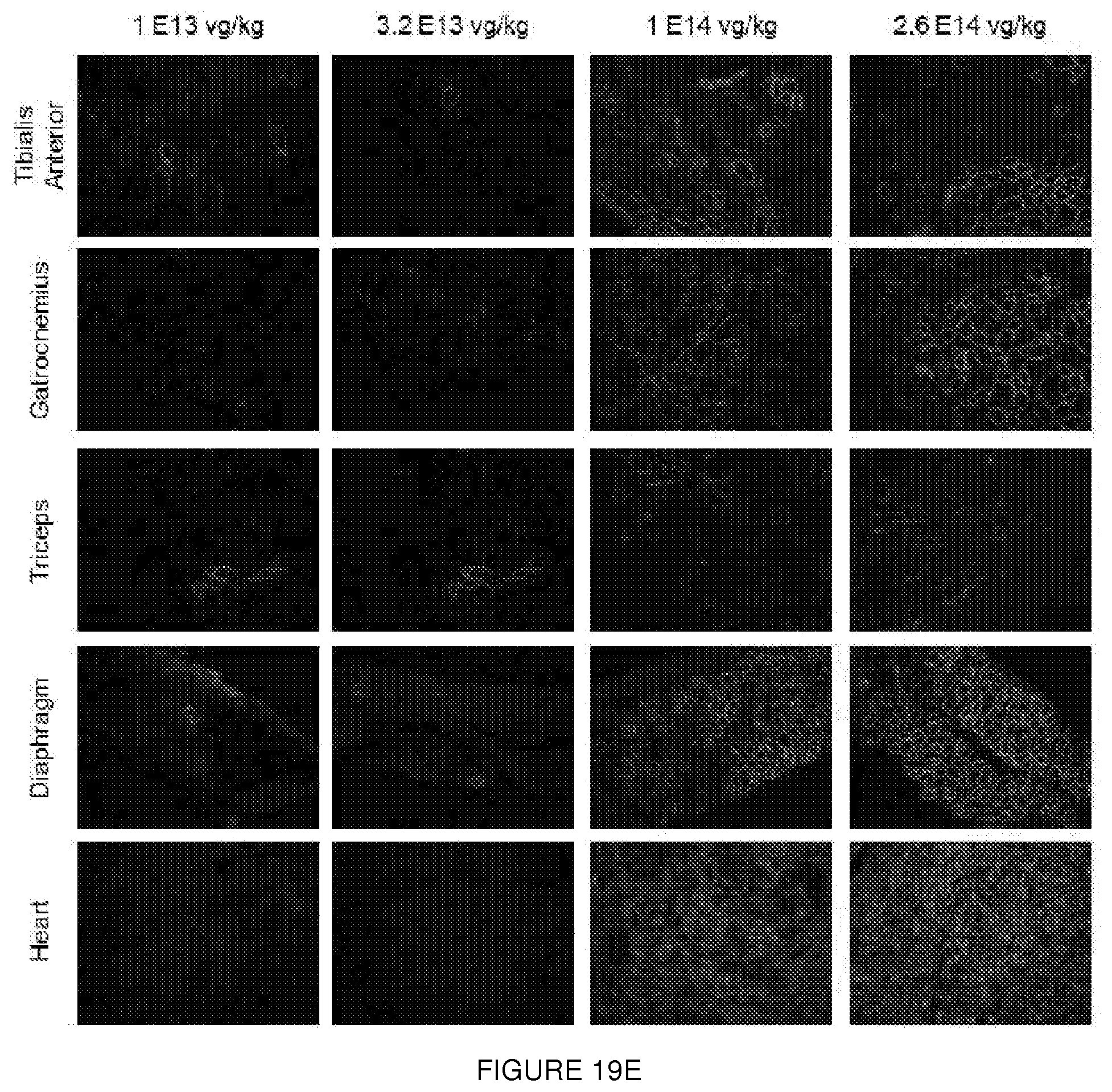
D00049

D00050
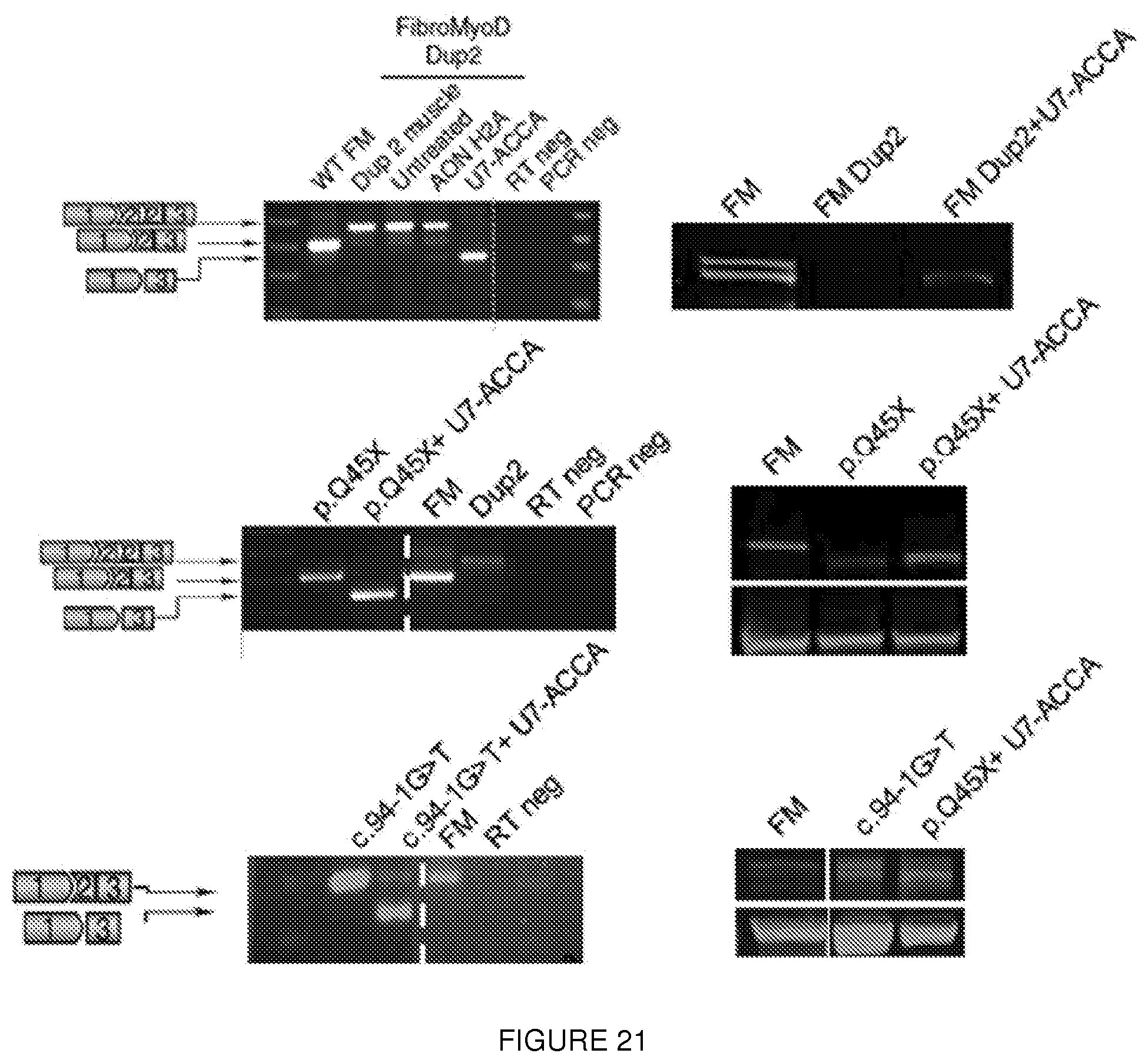
D00051
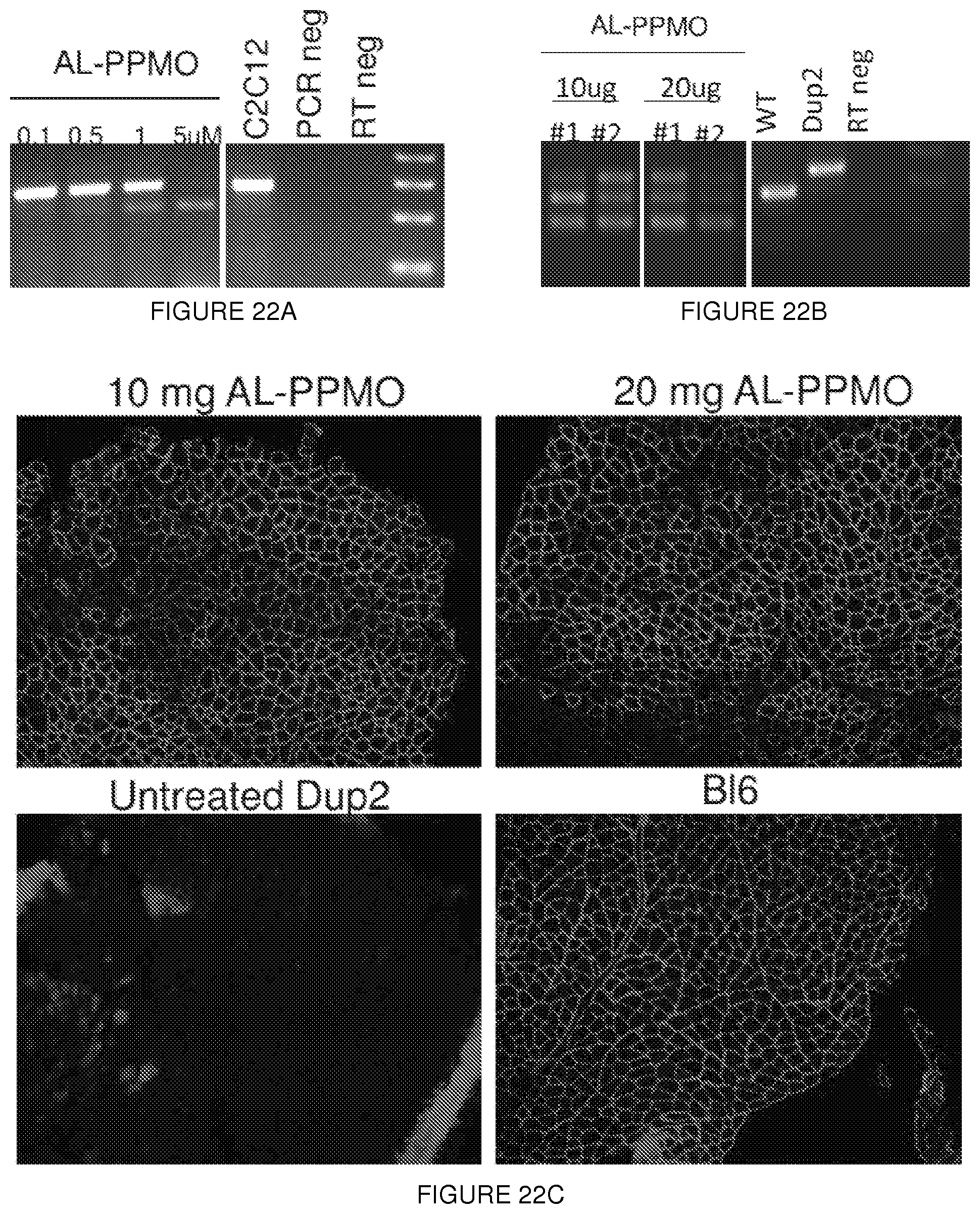
S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.