Compositions And Methods For Treating The Vertebral Column
Hart; Charles ; et al.
U.S. patent application number 17/373330 was filed with the patent office on 2022-04-07 for compositions and methods for treating the vertebral column. The applicant listed for this patent is BioMimetic Therapeutics, LLC. Invention is credited to Charles Hart, Samuel E. Lynch, Dan Perrien, Conan Young.
| Application Number | 20220105246 17/373330 |
| Document ID | / |
| Family ID | 1000006038558 |
| Filed Date | 2022-04-07 |


| United States Patent Application | 20220105246 |
| Kind Code | A1 |
| Hart; Charles ; et al. | April 7, 2022 |
COMPOSITIONS AND METHODS FOR TREATING THE VERTEBRAL COLUMN
Abstract
The present invention relates to compositions and methods useful for treating structures of the vertebral column, including vertebral bodies. In one embodiment, a method for promoting bone formation in a vertebral body comprising providing a composition comprising a PDGF solution and a biocompatible matrix and applying the composition to at least one vertebral body. Promoting bone formation in a vertebral body, according to some embodiments, can increase bone volume, mass, and/or density leading to an increase in mechanical strength of the vertebral body treated with a composition of the present invention.
| Inventors: | Hart; Charles; (Brentwood, TN) ; Lynch; Samuel E.; (Franklin, TN) ; Young; Conan; (Franklin, TN) ; Perrien; Dan; (Murfreesboro, TN) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 1000006038558 | ||||||||||
| Appl. No.: | 17/373330 | ||||||||||
| Filed: | July 12, 2021 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 14853901 | Sep 14, 2015 | 11058801 | ||
| 17373330 | ||||
| 12631731 | Dec 4, 2009 | 9161967 | ||
| 14853901 | ||||
| PCT/US2008/065666 | Jun 3, 2008 | |||
| 12631731 | ||||
| PCT/US2007/003582 | Feb 9, 2007 | |||
| 12631731 | ||||
| 11704685 | Feb 9, 2007 | 7799754 | ||
| 12631731 | ||||
| 61026835 | Feb 7, 2008 | |||
| 60933202 | Jun 4, 2007 | |||
| 60817988 | Jun 30, 2006 | |||
| 60859809 | Nov 17, 2006 | |||
| 60817988 | Jun 30, 2006 | |||
| 60859809 | Nov 17, 2006 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61K 49/0452 20130101; A61L 27/48 20130101; A61K 47/02 20130101; A61K 9/14 20130101; A61K 47/42 20130101; A61L 2430/38 20130101; A61L 27/227 20130101; A61L 27/425 20130101; A61L 2300/414 20130101; A61L 2300/252 20130101; A61K 9/0024 20130101; A61L 2300/202 20130101; A61K 49/0433 20130101; A61L 27/54 20130101; A61L 2300/44 20130101; A61L 2400/06 20130101; A61L 2300/45 20130101; A61L 27/40 20130101; A61K 38/1858 20130101 |
| International Class: | A61L 27/48 20060101 A61L027/48; A61K 9/00 20060101 A61K009/00; A61K 49/04 20060101 A61K049/04; A61L 27/54 20060101 A61L027/54; A61K 47/02 20060101 A61K047/02; A61K 47/42 20060101 A61K047/42; A61L 27/40 20060101 A61L027/40; A61K 9/14 20060101 A61K009/14; A61K 38/18 20060101 A61K038/18; A61L 27/22 20060101 A61L027/22; A61L 27/42 20060101 A61L027/42 |
Claims
1-14. (canceled)
15. A method for increasing bone density in at least one vertebral body of a patient comprising: applying to the vertebral body a composition comprising: a) a biocompatible matrix having incorporated therein a solution of platelet derived growth factor (PDGF) at a concentration in a range of about 0.1 mg/ml to about 1.0 mg/ml in a buffer, wherein the biocompatible matrix comprises (i) particles of a porous calcium phosphate in a range of about 100 .mu.m to about 3 mm in size or (ii) (a) particles of a porous calcium phosphate in a range of about 100 .mu.m to about 3 mm in size and (b) collagen, wherein the calcium phosphate comprises interconnected pores and a porosity greater than 50%, and b) b) a contrast agent.
16. The method of claim 15, wherein the biocompatible matrix comprises the particles of porous calcium phosphate and the collagen.
17. The method of claim 16, wherein the collagen comprises Type I collagen.
18. The method of claim 16, wherein the weight ratio of calcium phosphate:collagen is about 80:20.
19. The method of claim 15, wherein the PDGF is at a concentration of about 0.3 mg/ml.
20. The method of claim 15, wherein the PDGF comprises PDGF-BB.
21. The method of claim 20, wherein the PDGF-BB comprises recombinant human PDGF-BB (rhPDGF-BB) or a fragment thereof.
22. The method of claim 21, wherein the rhPDGF-BB comprises at least 65% of intact rhPDGF-BB.
23. The method of claim 21, wherein the fragment of rhPDGF-BB is selected from the group consisting of amino acid sequences 1-31, 1-32, 33-108, 33-109 and 1-108 of the entire B chain.
24. The method of claim 15, wherein the calcium phosphate comprises particles in a range of about 100 .mu.m to about 300 .mu.m in size.
25. The method of claim 15, wherein the calcium phosphate comprises particles in a range of about 1000 .mu.m to about 2000 .mu.m in size.
26. The method of claim 15, wherein the calcium phosphate comprises particles in a range of about 250 .mu.m to about 1000 .mu.m in size.
27. The method of claim 15, where the calcium phosphate comprises .beta.-tricalcium phosphate.
28. The method of claim 16, wherein the composition is flowable.
29. The method of claim 28, wherein applying the composition to the vertebral body comprises injecting the composition into the vertebral body.
30. The method of claim 15, wherein the method increases bone volume and/or mass in the vertebral body.
31. The method of claim 15, wherein the contrast agent is chosen from a cationic contrast agent, an anionic contrast agent, a nonionic contrast agents, or a mixture thereof.
32. The method of claim 15, wherein the contrast agent is a radiopaque contrast agent.
33. The method of claim 32, wherein the radiopaque contrast agent is (S)--N,N'-bis[2-hydroxy-1-(hydroxymethyl)-ethyl]-2,4,6-triiodo-5-lactamid- oisophthalamide or a derivative thereof.
34. The method of claim 15, further comprising locally or systemically administering to the patient a vitamin or an osteoclast inhibitor.
Description
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application is a continuation-in-part of International Application No. PCT/US2008/065666, filed on Jun. 3, 2008, which claims priority of U.S. Provisional Patent Application Ser. Nos. 60/933,202, filed Jun. 4, 2007, and 61/026,835 filed Feb. 7, 2008, all of which are incorporated herein by reference in their entirety. This application is also a continuation-in-part of International Application No. PCT/US2007/003582 and U.S. patent application Ser. No. 11/704,685, both of which were filed on Feb. 9, 2007, and both of which claim priority of U.S. Provisional Patent Application Ser. Nos. 60/817,988, filed Jun. 30, 2006, and 60/859,809, filed Nov. 17, 2006, all of which are incorporated herein by reference in their entirety.
FIELD OF THE INVENTION
[0002] The present invention relates to compositions and methods useful for treating structures of the vertebral column, including vertebral bodies.
BACKGROUND OF THE INVENTION
[0003] Musculoskeletal problems are pervasive throughout the population in all age groups and in both sexes. Half of Americans will need services for fractures at some point in their lifetimes according to a widely published article presented at the 2003 annual meeting of the American Academy of Orthopedic Surgeons (AAOS). More than $10 billion per year is spent in the U.S. on hospital care associated with fracture treatment according to this report.
[0004] Vertebral compression fractures (VCFs) are the most common osteoporotic fractures, occurring in about 20% of post-menopausal women (Eastell et al., J Bone Miner Res 1991; 6:207-215). It is estimated that 700,000 VCFs occur annually, and only 250,000 of these are diagnosed and treated. Because these fractures are left untreated, osteoporosis may remain untreated and progress rapidly. Post-menopausal women have a 5-fold increased risk of sustaining another vertebral fracture within the coming year and 2-fold increased risk of other fragility fractures, including hip fractures (Klotzbuecher et al, J Bone Miner Res, 2000; 15:721-739).
[0005] VCFs occur when there is a break in one or both of the vertebral body end plates, usually due to trauma, causing failure of the anterior column and weakening the vertebrae from supporting the body during activities of daily living. Vertebral compression fractures caused by osteoporosis can cause debilitating back pain, spinal deformity, and height loss. Both symptomatic and asymptomatic vertebral fractures are associated with increased morbidity and mortality. With the number of aged people at risk for osteoporosis is expected to increase dramatically in the coming decades, accurate identification of VCFs and treatment intervention is necessary to reduce the enormous potential impact of this disease on patients and health care systems.
[0006] Traditionally, VCFs caused by osteoporosis have been treated with bed rest, narcotic analgesics, braces, and physical therapy. Bed rest, however, leads to accelerated bone loss and physical deconditioning, further aggravating the patient as well as contributing to the problem of osteoporosis. Moreover, the use of narcotics can worsen the mood and mentation problem that may already be prevalent in the elderly. Additionally, brace wear is not well-tolerated by the elderly. Although the current treatments of osteoporosis such as hormone replacement, bisphosphonates, calcitonin, and parathyroid hormone (PTH) analogs deal with long-term issues, except for calcitonin, they provide no immediate benefit in terms of pain control once a fracture occurs (Kapuscinski et al., Master Med. Pol. 1996; 28:83-86).
[0007] Recently, minimally invasive treatments for vertebral body compression fractures, vertebroplasty and kyphoplasty, have been developed to address the issues of pain and fracture stabilization. Vertebroplasty is the filling of a fractured vertebral body with the goals of stabilizing the bone, preventing further collapse, and eliminating acute fracture pain. Vertebroplasty, however, does not attempt to restore vertebral height and/or sagittal alignment. In addition, because there is no void in the bone, vertebral filling is performed under less control with less viscous cement and, as a consequence, filler leaks are common.
[0008] Kyphoplasty is a minimally invasive surgical procedure with the goal of safety, improving vertebral height and stabilizing VCF. Guided by x-ray images, an inflatable bone tamp is inflated in the fractured vertebral body. This compacts the inner cancellous bone as it pushes the fractured cortices back toward their normal position. Fixation can then be done by filling the void with a biomaterial under volume control with a more viscous cement. Although kyphoplasty is considered a safe and effective treatment of vertebral compression fractures, biomechanical studies demonstrate that cement augmentation places additional stress on adjacent levels. In fact, this increased stiffness can decrease the ultimate load to failure of adjacent vertebrae by 8 to 30% and provoke subsequent fractures (Berlemann et al., J Bone Joint Surgery BR, 2002; 84:748-52). Compression fracture of one or more vertebral bodies subsequent to vertebroplasty or kyphoplasty is referred to herein as a "secondary vertebral compression fracture."
[0009] In a recent clinical study, a higher rate of secondary vertebral compression fracture was observed after kyphoplasty compared with historical data for untreated fractures. Most of these occurred at an adjacent level within 2 months of the index procedure. After this two-month period, there were only occasional secondary vertebral compression fractures which occurred at remote levels. This study confirmed biomechanical studies showing that cement augmentation places additional stress on adjacent level. (Fribourg et al., Incidence of subsequent vertebral fracture after kyphoplasty, Spine, 2004; 20; 2270-76).
[0010] Given the increased incidence of the use of minimally invasive surgical techniques for the treatment of vertebral compression fractures, and the predisposition of adjacent vertebrae to undergo secondary compression fracture, an unmet clinical need exists to prophylactically treat and prevent secondary VCFs.
SUMMARY OF THE INVENTION
[0011] The present invention provides compositions and methods useful for treating structures of the vertebral column, including vertebral bodies. In some embodiments of the present invention, compositions are provided for promoting bone formation in a vertebral body. In other embodiments, compositions and methods are provided for preventing or decreasing the likelihood of vertebral compression fractures. In another embodiment, methods and compositions are provided for preventing or decreasing the likelihood of secondary vertebral compression fractures associated with vertebroplasty and/or kyphoplasty. The present compositions and methods can be useful in treating vertebral bodies of compromised patients, such as those with osteoporosis, diabetes, or other diseases or conditions.
[0012] In one aspect, a composition for promoting bone formation in a vertebral body comprises a solution comprising platelet derived growth factor (PDGF) and biocompatible matrix, wherein the solution is disposed or incorporated in the biocompatible matrix. In some embodiments, the PDGF is absorbed by the biocompatible matrix. In other embodiments, the PDGF is adsorbed onto one or more surfaces of the biocompatible matrix. In a further embodiment, the PDGF is absorbed by the biocompatible matrix and adsorbed onto one or more surfaces of the biocompatible matrix.
[0013] In some embodiments, PDGF is present in the solution in a concentration ranging from about 0.01 mg/ml to about 10 mg/ml, from about 0.05 mg/ml to about 5 mg/ml, from about 0.1 mg/ml to about 1.0 mg/ml, or from about 0.2 mg/ml to about 0.4 mg/mi. The concentration of PDGF within the solution may be within any of the concentration ranges stated above.
[0014] In some embodiments of the present invention, PDGF comprises PDGF homodimers and heterodimers, including PDGF-AA, PDGF-BB, PDGF-AB, PDGF-CC, PDGF-DD, and mixtures and derivatives thereof. In one embodiment, PDGF comprises PDGF-BB. In another embodiment PDGF comprises a recombinant human (rh) PDGF such as recombinant human PDGF-BB (rhPDGF-BB).
[0015] In some embodiments of the present invention, PDGF comprises PDGF fragments. In one embodiment rhPDGF-B comprises the following fragments: amino acid sequences 1-31, 1-32, 33-108, 33-109, and/or 1-108 of the entire B chain. The complete amino acid sequence (1-109) of the B chain of PDGF is provided in FIG. 15 of U.S. Pat. No. 5,516,896. It is to be understood that the rhPDGF compositions of the present invention may comprise a combination of intact rhPDGF-B (1-109) and fragments thereof. Other fragments of PDGF may be employed such as those disclosed in U.S. Pat. No. 5,516,896. In some embodiments, rhPDGF-BB comprises at least 65% of intact rhPDGF-B (1-109).
[0016] A biocompatible matrix, according to some embodiments of the present invention, comprises a bone substituting agent (also called a scaffolding material herein) and optionally a biocompatible binder. Bone substituting agents, in some embodiments, comprise calcium phosphate including amorphous calcium phosphate, monocalcium phosphate monohydrate (MCPM), monocalcium phosphate anhydrous (MCPA), dicalcium phosphate dihydrate (DCPD), dicalcium phosphate anhydrous (DCPA), octacalcium phosphate (OCP), .alpha.-tricalcium phosphate, .beta.-TCP, hydroxyapatite (OHAp), poorly crystalline hydroxapatite, tetracalcium phosphate (TTCP), heptacalcium decaphosphate, calcium metaphosphate, calcium pyrophosphate dihydrate, calcium pyrophosphate, carbonated calcium phosphate, hydroxyapatite, or derivatives or mixtures thereof. In some embodiments, bone substituting agents comprise calcium sulfate or demineralized bone such as dried cortical or cancellous bone.
[0017] In another aspect, the present invention provides a composition for promoting bone formation in a vertebral body comprising a PDGF solution disposed in a biocompatible matrix, wherein the biocompatible matrix comprises a bone scaffolding material and a biocompatible binder. The PDGF solution may have a concentration of PDGF as described above. A bone scaffolding material, in some embodiments, comprises calcium phosphate. In an embodiment, calcium phosphate comprises .beta.-TCP. In one aspect, biocompatible matrices may include calcium phosphate particles with or without biocompatible binders or bone allograft such as demineralized freeze dried bone allograft (DFDBA), mineralized freeze dried bone allograft (FDBA), or particulate demineralized bone matrix (DBM). In another aspect, biocompatible matrices may include bone allograft such as DFDBA, DBM, or other bone allograft materials including cortical bone shapes, such as blocks, wedges, cylinders, or particles, or cancellous bone particles of various shapes and sizes.
[0018] Moreover, a biocompatible binder, according to some embodiments of the present invention, comprises proteins, polysaccharides, nucleic acids, carbohydrates, synthetic polymers, or mixtures thereof. In one embodiment, a biocompatible binder comprises collagen. In another embodiment, a biocompatible binder comprises hyaluronic acid.
[0019] In another aspect the present invention provides a composition for preventing or decreasing the likelihood of vertebral compression fractures, including secondary vertebral compression fractures. In some embodiments, a composition for preventing or decreasing the likelihood of vertebral compression fractures comprises a solution comprising PDGF and a biocompatible matrix wherein the solution is disposed in the biocompatible matrix. In other embodiments, a composition for preventing or decreasing the likelihood of vertebral compression fractures comprises a PDGF solution disposed in a biocompatible matrix, wherein the biocompatible matrix comprises a bone scaffolding material and a biocompatible binder. In embodiments of a composition for preventing or decreasing the likelihood of vertebral compression fractures, a PDGF solution may have a concentration of PDGF as described above. Moreover, a bone scaffolding material, in some embodiments, comprises calcium phosphate. In an embodiment, calcium phosphate comprises .beta.-tricalcium phosphate. A biocompatible binder, according to some embodiments of the present invention, comprises proteins, polysaccharides, nucleic acids, carbohydrates, synthetic polymers, or mixtures thereof. In one embodiment, a biocompatible binder comprises collagen. In another embodiment, a biocompatible binder comprises collagen, such as bovine collagen.
[0020] In some embodiments of the present invention, compositions for promoting bone formation in vertebral bodies and compositions for preventing or reducing the likelihood of vertebral compression fractures further comprise at least one contrast agent. Contrast agents, according to embodiments of the present invention, are substances operable to at least partially provide differentiation of two or more bodily tissues when imaged. Contrast agents, according to some embodiments, comprise cationic contrast agents, anionic contrast agents, nonionic contrast agents, or mixtures thereof. In some embodiments, contrast agents comprise radiopaque contrast agents. Radiopaque contrast agents, in some embodiments, comprise iodo-compounds including (S)--N,N'-bis[2-hydroxy-1-(hydroxymethyl)-ethyl]-2,4,6-triiodo-5-lactamid- oisophthalamide (Iopamidol) and derivatives thereof.
[0021] In another aspect, the present invention provides a kit comprising a biocompatible matrix in a first package and a solution comprising PDGF in a second package. In some embodiments, the biocompatible matrix comprises a scaffolding material, a scaffolding material and a biocompatible binder, and/or bone allograft such as DFDBA or particulate DBM. In one embodiment, the scaffolding material comprises a calcium phosphate, such as .beta.-TCP. Moreover, in some embodiments, the solution comprises a predetermined concentration of PDGF. The concentration of the PDGF can be predetermined according to the surgical procedure being performed, such as promoting or accelerating bone growth in a vertebral body or preventing or decreasing the likelihood of secondary vertebral compression fractures. Moreover, in some embodiments, the biocompatible matrix can be present in the kit in a predetermined amount. The amount of biocompatible matrix provided by a kit can be dependent on the surgical procedure being performed. In some embodiments, the second package containing the PDGF solution comprises a syringe. A syringe can facilitate disposition of the PDGF solution in the biocompatible matrix. Once the PDGF solution has been disposed in the biocompatible matrix, in some embodiments, the resulting composition can placed in a second syringe and/or cannula and delivered to a vertebral body.
[0022] The present invention also provides methods of producing compositions for promoting bone formation in vertebral bodies and preventing or decreasing the likelihood of compression fractures of vertebral bodies, including secondary vertebral compression fractures. In one embodiment, a method for producing such compositions comprises providing a solution comprising PDGF, providing a biocompatible matrix, and disposing the solution in the biocompatible matrix. In some embodiments, a method of producing compositions for promoting bone formation in a vertebral body and preventing or decreasing the likelihood of compression fracture in a vertebral body further comprises providing a contrast agent and disposing the contrast agent in the biocompatible matrix.
[0023] In another aspect, the present invention provides methods for promoting or accelerating bone formation in a vertebral body comprising providing a composition comprising a PDGF solution disposed in a biocompatible matrix and applying an effective amount of the composition to at least one vertebral body. Applying the composition to at least one vertebral body, in some embodiments, comprises injecting the composition into the at least one vertebral body.
[0024] In another aspect, the present invention provides methods comprising preventing or decreasing the likelihood of vertebral compression fractures, including secondary vertebral compression fractures. Preventing or decreasing the likelihood of vertebral compression fractures, according to embodiments of the present invention comprises providing a composition comprising a PDGF solution disposed in a biocompatible matrix and applying an effective amount of the composition to at least one vertebral body. In some embodiments, applying the composition to at least one vertebral body comprises injecting the composition into the at least one vertebral body. In one embodiment, the composition is applied to a second vertebral body, in some instances an adjacent vertebral body, subsequent to a vertebroplasty or kyphoplasty of a first vertebral body. In some embodiments, a composition comprising a PDGF solution disposed in a biocompatible matrix is applied to at least one high risk vertebral body. "High risk vertebral bodies" (HVB), as used herein, refer to vertebral bodies of vertebrae T5 through T12 as well as L1 through L4, which are at the greatest risk of undergoing secondary vertebral compression fracture.
[0025] In some embodiments of methods of the present invention, the biocompatible matrix comprises a bone scaffolding material. In some embodiments, the biocompatible matrix comprises a bone scaffolding material and a biocompatible binder.
[0026] In some embodiments, methods for promoting bone formation in vertebral bodies and preventing or decreasing the likelihood of compression fractures of vertebral bodies further comprise providing at least one pharmaceutical composition in addition to the composition comprising a PDGF solution disposed in a biocompatible matrix and administering the at least one pharmaceutical composition locally and/or systemically. The at least one pharmaceutical composition, in some embodiments, comprises vitamins, calcium supplements, or any osteoclast inhibitor known to one of skill in the art, including bisphosphonates. In some embodiments, the at least one pharmaceutical composition is administered locally. In such embodiments, the at least one pharmaceutical composition can be incorporated into the biocompatible matrix or otherwise disposed in and around a vertebral body. In other embodiments, the at least one pharmaceutical composition is administered systemically to a patient. In one embodiment, for example, the at least one pharmaceutical composition is administered orally to a patient. In another embodiment, the at least one pharmaceutical composition is administered intravenously to a patient.
[0027] Accordingly, it is an object of the present invention to provide a composition comprising PDGF useful in promoting bone formation in vertebral bodies.
[0028] It is another object of the present invention to provide a composition comprising PDGF useful in strengthening vertebral bodies.
[0029] It is another object of the present invention to provide a composition comprising PDGF useful in strengthening vertebral bodies of patients with osteoporosis.
[0030] It is another object of the present invention to provide a composition comprising PDGF useful in preventing or decreasing the likelihood of vertebral compression fractures, including secondary vertebral compression fractures.
[0031] Another object of the present invention is to provide methods for promoting bone formation in vertebral bodies using compositions comprising PDGF.
[0032] A further object of the present invention is to provide methods of preventing or decreasing the likelihood of vertebral compression fractures, including secondary vertebral compression fractures, using compositions comprising PDGF.
[0033] These and other embodiments of the present invention are described in greater detail in the detailed description which follows. These and other objects, features and advantages of the present invention will become apparent after a review of the following detailed description of the disclosed embodiments and claims.
BRIEF DESCRIPTION OF THE FIGURES
[0034] FIG. 1 illustrates a syringe and related apparatus penetrating tissue overlaying a vertebral body to deliver a composition of the present invention to the vertebral body according to an embodiment of the present invention.
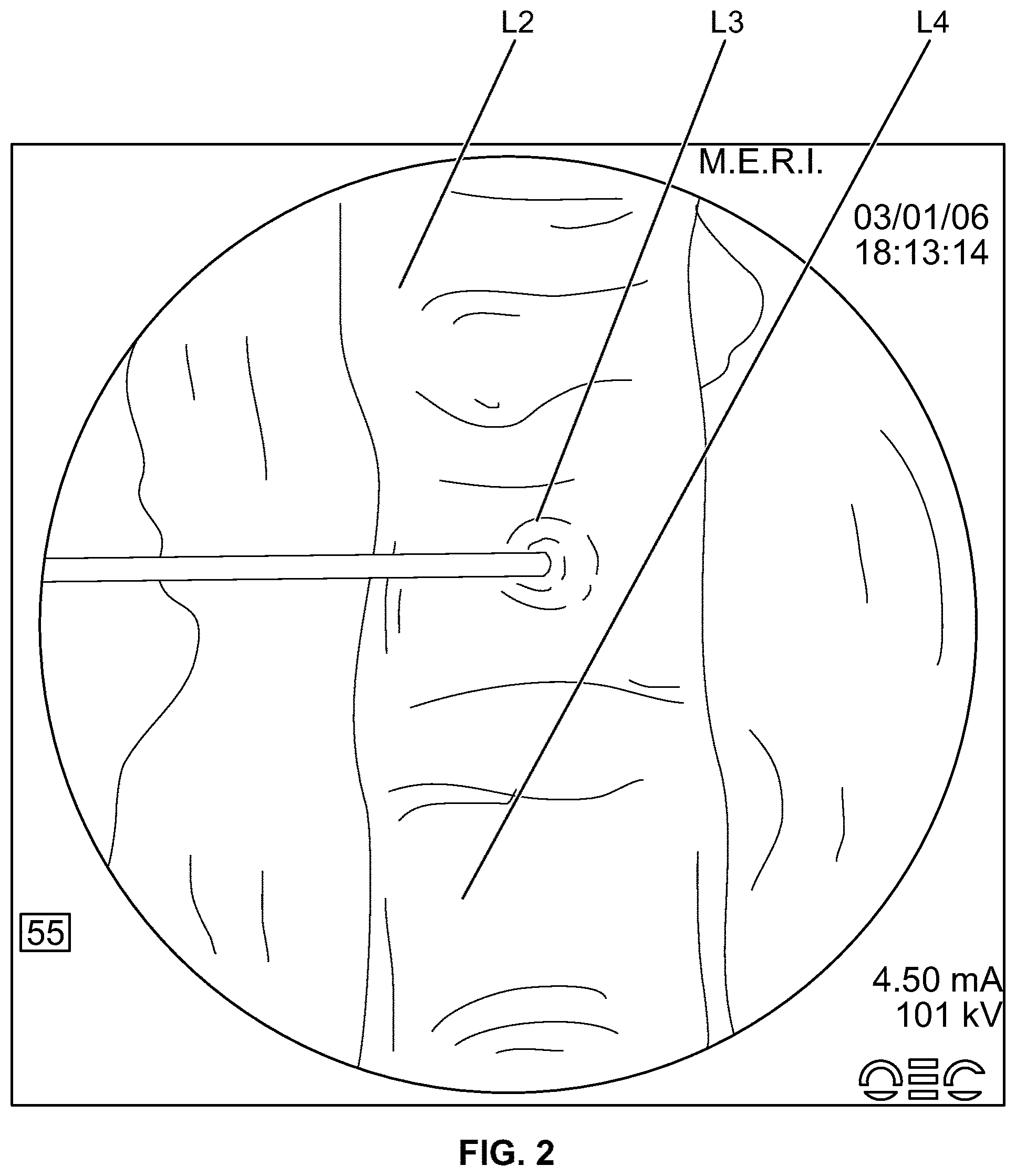
[0035] FIG. 2 is a radiograph illustrating injection of a composition into a vertebral body according to an embodiment of the present invention.
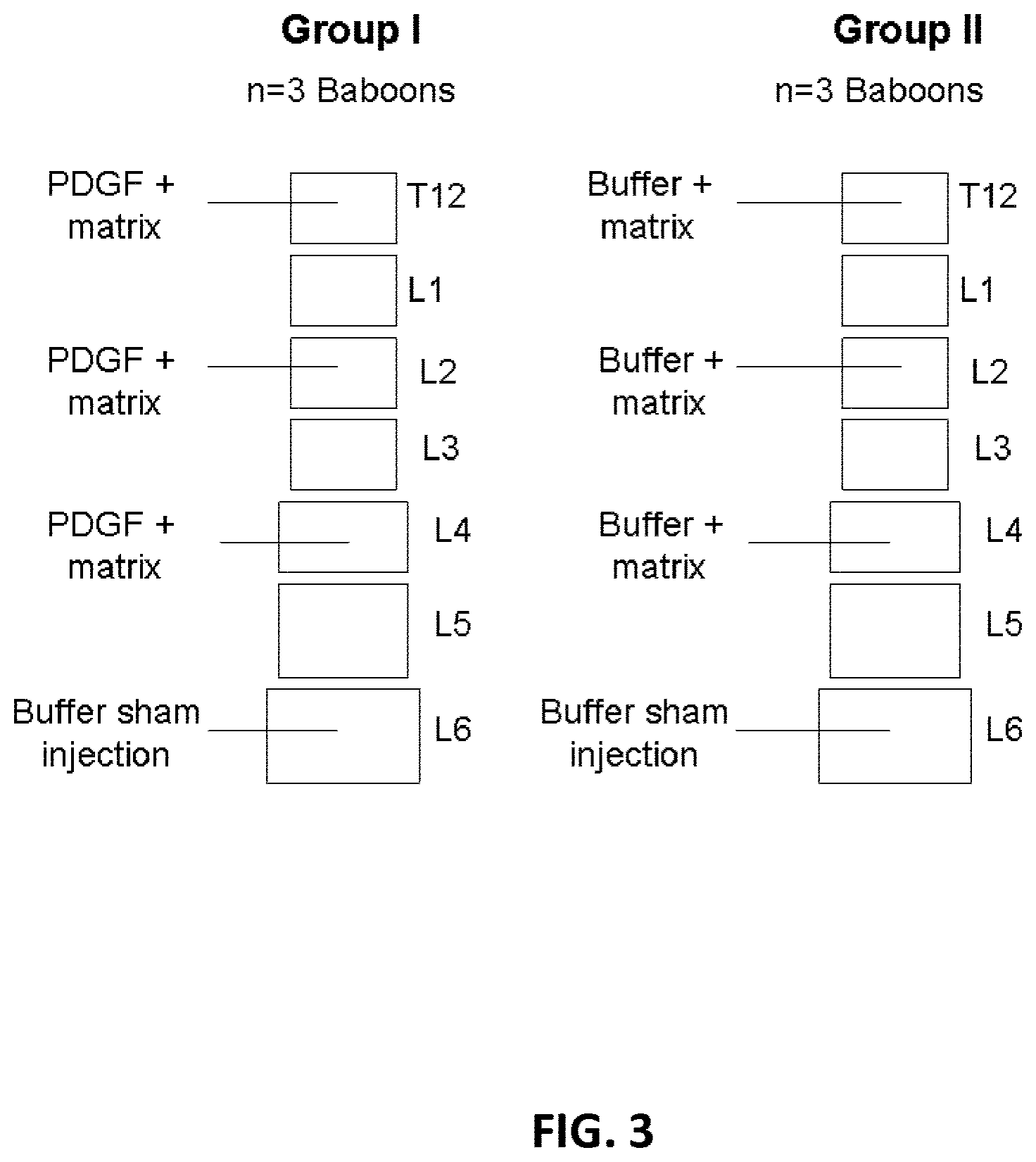
[0036] FIG. 3 illustrates vertebrae receiving compositions of the present invention according to one embodiment of the present invention.
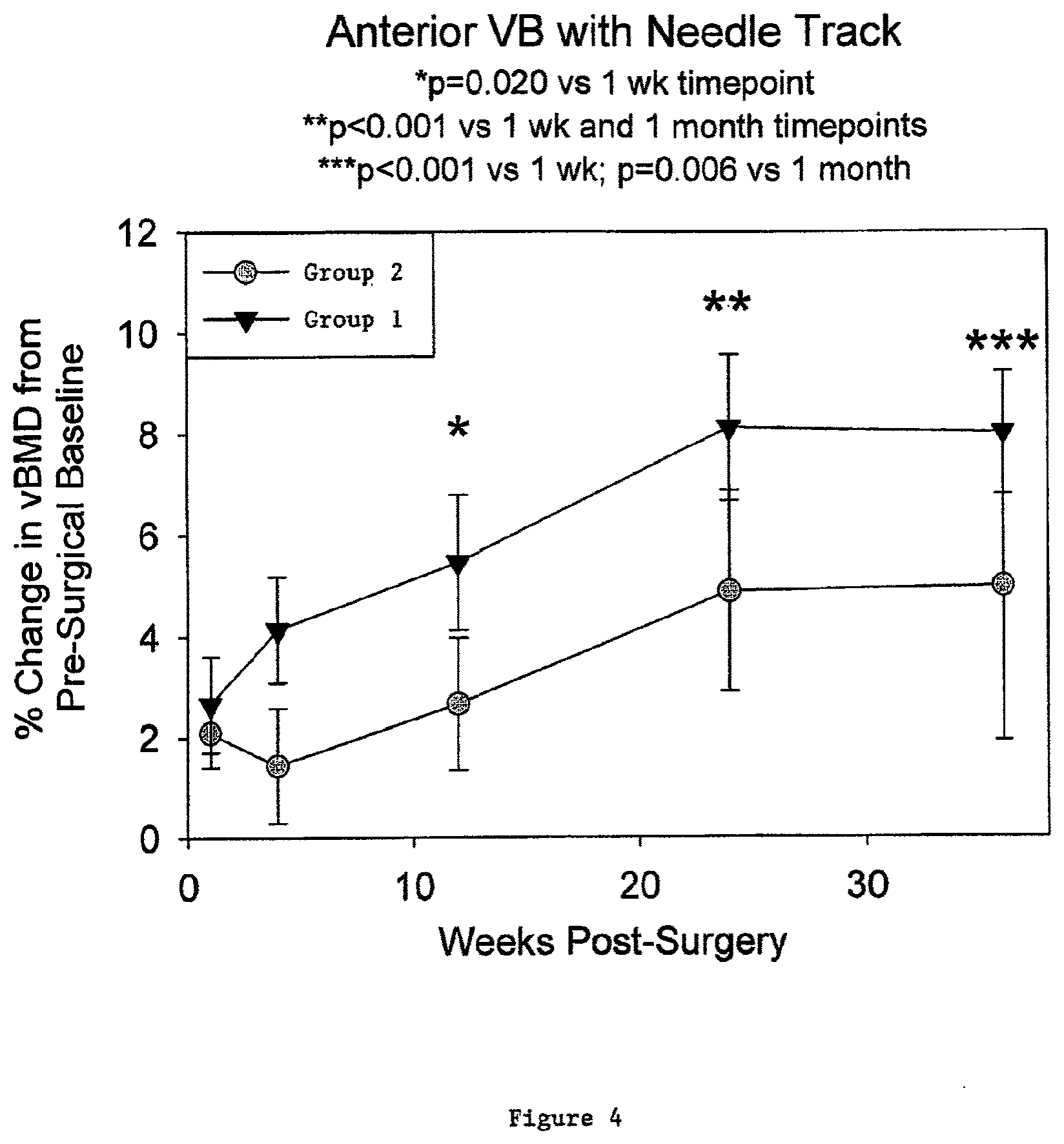
[0037] FIG. 4 illustrates percent change in volumetric bone mineral density for vertebral bodies receiving a composition comprising 1.0 mg/ml of rhPDGF-BB disposed in a .beta.-TCP/collagen matrix in comparison with vertebral bodies receiving a composition comprising 20 mM sodium acetate buffer disposed in a .beta.-TCP/collagen matrix according to one embodiment of the present invention.
[0038] FIG. 5 illustrates percent change in volumetric bone mineral density for vertebral bodies receiving a composition comprising 1.0 mg/ml of rhPDGF-BB disposed in a .beta.-TCP/collagen matrix in comparison with vertebral bodies receiving a composition comprising 20 mM sodium acetate buffer disposed in a .beta.-TCP/collagen matrix according to one embodiment of the present invention.
DETAILED DESCRIPTION
[0039] The present invention provides compositions and methods useful for treating structures of the vertebral column, including vertebral bodies. According to embodiments described herein, the present invention provides compositions for promoting bone formation in a vertebral body and compositions for preventing or decreasing the likelihood of vertebral compression fractures, including secondary vertebral compression fractures. In one embodiment, the compositions comprise a solution comprising PDGF and a biocompatible matrix, wherein the solution is disposed in the biocompatible matrix. In another embodiment, the compositions comprise a PDGF solution disposed in a biocompatible matrix, wherein the biocompatible matrix comprises a bone scaffolding material and a biocompatible binder. In one aspect, biocompatible matrices include calcium phosphate particles with or without biocompatible binders or bone allograft such as DFDBA or particulate DBM. In another aspect, biocompatible matrices may include DFDBA or DBM.
[0040] Turning now to components that can be included in various embodiments of the present invention, compositions of the present invention comprise a solution comprising PDGF.
PDGF Solutions
[0041] PDGF plays an important role in regulating cell growth and migration. PDGF, as with other growth factors, binds with the extracellular domains of receptor tyrosine kinases. The binding of PDGF to these transmembrane proteins activate the kinase activity of their catalytic domains located on the cytosolic side of the membrane. By phosphorylating tyrosine residues of target proteins, the kinases induce a variety of cellular processes that include cell growth and extracellular matrix production.
[0042] In one aspect, a composition provided by the present invention comprises a solution comprising PDGF and a biocompatible matrix, wherein the solution is disposed or incorporated in the biocompatible matrix. In some embodiments, PDGF is present in the solution in a concentration ranging from about 0.01 mg/ml to about 10 mg/ml, from about 0.05 mg/ml to about 5 mg/ml, or from about 0.1 mg/ml to about 1.0 mg/ml. PDGF may be present in the solution at any concentration within these stated ranges including the upper limit and lower limit of each range. In other embodiments, PDGF is present in the solution at any one of the following concentrations: about 0.05 mg/ml; about 0.1 mg/ml; about 0.15 mg/ml; about 0.2 mg/ml; about 0.25 mg/ml; about 0.3 mg/ml; about 0.35 mg/ml; about 0.4 mg/ml; about 0.45 mg/ml; about 0.5 mg/ml, about 0.55 mg/ml, about 0.6 mg/ml, about 0.65 mg/ml, about 0.7 mg/ml; about 0.75 mg/ml; about 0.8 mg/ml; about 0.85 mg/ml; about 0.9 mg/ml; about 0.95 mg/ml; or about 1.0 mg/ml. In some embodiments, PDGF is present in the solution in a concentration ranging from about 0.2 mg/ml to about 2 mg/ml, from about 0.3 mg/ml to about 3 mg/ml, from about 0.4 mg/ml to about 4 mg/ml, or from about 0.5 mg/ml to about 5 mg/ml. It is to be understood that these concentrations are simply examples of particular embodiments, and that the concentration of PDGF may be within any of the concentration ranges stated above including the upper limit and the lower limit of each range.
[0043] Various amounts of PDGF may be used in the compositions of the present invention. Amounts of PDGF that could be used include amounts in the following ranges: about 1 .mu.g to about 50 mg, about 10 .mu.g to about 25 mg, about 100 .mu.g to about 10 mg, and about 250 .mu.g to about 5 mg.
[0044] The concentration of PDGF or other growth factors in embodiments of the present invention can be determined by using an enzyme-linked immunoassay as described in U.S. Pat. Nos. 6,221,625, 5,747,273, and 5,290,708, or any other assay known in the art for determining PDGF concentration. When provided herein, the molar concentration of PDGF is determined based on the molecular weight (MW) of PDGF dimer (e.g., PDGF-BB; MW about 25 kDa).
[0045] In some embodiments of the present invention, PDGF comprises PDGF homodimers and heterodimers, including PDGF-AA, PDGF-BB, PDGF-AB, PDGF-CC, PDGF-DD, and mixtures and derivatives thereof. In one embodiment, for example, PDGF comprises PDGF-BB. In another embodiment PDGF comprises a recombinant human PDGF, such as rhPDGF-BB. In some embodiments, PDGF comprises mixtures of the various homodimers and/or heterodimers. Embodiments of the present invention contemplate any combination of PDGF-AA, PDGF-BB, PDGF-AB, PDGF-CC, and/or PDGF-DD.
[0046] PDGF, in some embodiments, can be obtained from natural sources. In other embodiments, PDGF can be produced by recombinant DNA techniques. In other embodiments, PDGF or fragments thereof may be produced using peptide synthesis techniques known to one of ordinary skill in the art, such as solid phase peptide synthesis. When obtained from natural sources, PDGF can be derived from biological fluids. Biological fluids, according to some embodiments, can comprise any treated or untreated fluid associated with living organisms including blood
[0047] Biological fluids, in another embodiment, can also comprise blood components including platelet concentrate (PC), apheresed platelets, platelet-rich plasma (PRP), plasma, serum, fresh frozen plasma (FFP), and buffy coat (BC). Biological fluids, in a further embodiment, can comprise platelets separated from plasma and resuspended in a physiological fluid.
[0048] When produced by recombinant DNA techniques, a DNA sequence encoding a single monomer (e.g., PDGF B-chain or A-chain), in some embodiments, can be inserted into cultured prokaryotic or eukaryotic cells for expression to subsequently produce the homodimer (e.g. PDGF-BB or PDGF-AA). In other embodiments, a PDGF heterodimer can be generated by inserting DNA sequences encoding for both monomeric units of the heterodimer into cultured prokaryotic or eukaryotic cells and allowing the translated monomeric units to be processed by the cells to produce the heterodimer (e.g. PDGF-AB). Commercially available GMP recombinant PDGF-BB can be obtained commercially from Novartis Corporation (Emeryville, Calif.). Research grade rhPDGF-BB can be obtained from multiple sources including R&D Systems, Inc. (Minneapolis, Minn.), BD Biosciences (San Jose, Calif.), and Chemicon, International (Temecula, Calif.). In some embodiments, monomeric units can be produced in prokaryotic cells in a denatured form, wherein the denatured form is subsequently refolded into an active molecule.
[0049] In embodiments of the present invention, PDGF comprises PDGF fragments. In one embodiment rhPDGF-B comprises the following fragments: amino acid sequences 1-31, 1-32, 33-108, 33-109, and/or 1-108 of the entire B chain. The complete amino acid sequence (1-109) of the B chain of PDGF is provided in FIG. 15 of U.S. Pat. No. 5,516,896. It is to be understood that the rhPDGF compositions of the present invention may comprise a combination of intact rhPDGF-B (1-109) and fragments thereof. Other fragments of PDGF may be employed such as those disclosed in U.S. Pat. No. 5,516,896. In accordance with one embodiment, the rhPDGF-BB comprises at least 60% of intact rhPDGF-B (1-109). In another embodiment, the rhPDGF-BB comprises at least 65%, 75%, 80%, 85%, 90%, 95%, or 99% of intact rhPDGF-B (1-109).
[0050] In some embodiments of the present invention, PDGF can be purified. Purified PDGF, as used herein, comprises compositions having greater than about 95% by weight PDGF prior to incorporation in solutions of the present invention. The solution may be any pharmaceutically acceptable solution. In other embodiments, the PDGF can be substantially purified. Substantially purified PDGF, as used herein, comprises compositions having about 5% to about 95% by weight PDGF prior to incorporation into solutions of the present invention. In one embodiment, substantially purified PDGF comprises compositions having about 65% to about 95% by weight PDGF prior to incorporation into solutions of the present invention. In other embodiments, substantially purified PDGF comprises compositions having about 70% to about 95%, about 75% to about 95%, about 80% to about 95%, about 85% to about 95%, or about 90% to about 95%, by weight PDGF, prior to incorporation into solutions of the present invention. Purified PDGF and substantially purified PDGF may be incorporated into scaffolds and binders.
[0051] In a further embodiment, PDGF can be partially purified. Partially purified PDGF, as used herein, comprises compositions having PDGF in the context of platelet rich plasma (PRP), fresh frozen plasma (FFP), or any other blood product that requires collection and separation to produce PDGF. Embodiments of the present invention contemplate that any of the PDGF isoforms provided herein, including homodimers and heterodimers, can be purified or partially purified. Compositions of the present invention containing PDGF mixtures may contain PDGF isoforms or PDGF fragments in partially purified proportions. Partially purified and purified PDGF, in some embodiments, can be prepared as described in U.S. patent application Ser. No. 11/159,533 (Publication No: 20060084602).
[0052] In some embodiments, solutions comprising PDGF are formed by solubilizing PDGF in one or more buffers. Buffers suitable for use in PDGF solutions of the present invention can comprise, but are not limited to, carbonates, phosphates (e.g. phosphate buffered saline), histidine, acetates (e.g. sodium acetate), acidic buffers such as acetic acid and HCl, and organic buffers such as lysine, Tris buffers (e.g. tris(hydroxymethyl)aminoethane), N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES), and 3-(N-morpholino) propanesulfonic acid (MOPS). Buffers can be selected based on biocompatibility with PDGF and the buffer's ability to impede undesirable protein modification. Buffers can additionally be selected based on compatibility with host tissues. In one embodiment, sodium acetate buffer is used. The buffers may be employed at different molarities, for example, about 0.1 mM to about 100 mM, about 1 mM to about 50 mM, about 5 mM to about 40 mM, about 10 mM to about 30 mM, or about 15 mM to about 25 mM, or any molarity within these ranges. In some embodiments, an acetate buffer is employed at a molarity of about 20 mM.
[0053] In another embodiment, solutions comprising PDGF are formed by solubilizing lyophilized PDGF in water, wherein prior to solubilization the PDGF is lyophilized from an appropriate buffer.
[0054] Solutions comprising PDGF, according to embodiments of the present invention, can have a pH ranging from about 3.0 to about 8.0. In one embodiment, a solution comprising PDGF has a pH ranging from about 5.0 to about 8.0, from about 5.5 to about 7.0, or from about 5.5 to about 6.5, or any value within these ranges. The pH of solutions comprising PDGF, in some embodiments, can be compatible with the prolonged stability and efficacy of PDGF or any other desired biologically active agent. PDGF is more stable in an acidic environment. Therefore, in accordance with one embodiment the present invention comprises an acidic storage formulation of a PDGF solution. In accordance with this embodiment, the PDGF solution preferably has a pH from about 3.0 to about 7.0 or from about 4.0 to about 6.5. The biological activity of PDGF, however, can be optimized in a solution having a neutral pH range. Therefore, in a further embodiment, the present invention comprises a neutral pH formulation of a PDGF solution. In accordance with this embodiment, the PDGF solution preferably has a pH from about 5.0 to about 8.0, from about 5.5 to about 7.0, or from about 5.5 to about 6.5. In accordance with a method of the present invention, an acidic PDGF solution is reformulated to a neutral pH composition, wherein such composition is then used to treat bone and promote bone growth and/or healing. In accordance with some embodiments of the present invention, the PDGF utilized in the solutions is rh-PDGF-BB. In a further embodiment, the pH of the PDGF containing solution may be altered to optimize the binding kinetics of PDGF to a matrix substrate or linker. If desired, the pH of the material equilibrates to adjacent material, the bound PDGF may become labile.
[0055] The pH of solutions comprising PDGF, in some embodiments, can be controlled by the buffers recited herein. Various proteins demonstrate different pH ranges in which they are stable. Protein stabilities are primarily reflected by isoelectric points and charges on the proteins. The pH range can affect the conformational structure of a protein and the susceptibility of a protein to proteolytic degradation, hydrolysis, oxidation, and other processes that can result in modification to the structure and/or biological activity of the protein.
[0056] In some embodiments, solutions comprising PDGF can further comprise additional components, such as other biologically active agents. In other embodiments, solutions comprising PDGF can further comprise cell culture media, other stabilizing proteins such as albumin, antibacterial agents, protease inhibitors [e.g., ethylenediaminetetraacetic acid (EDTA), ethylene glycol-bis(beta-aminoethylether)-N, N,N',N'-tetraacetic acid (EGTA), aprotinin, .epsilon.-aminocaproic acid (EACA), etc.] and/or other growth factors such as fibroblast growth factors (FGFs), epidermal growth factors (EGFs), transforming growth factors (TGFs), keratinocyte growth factors (KGFs), insulin-like growth factors (IGFs), hepatocyte growth factors (HGFs), bone morphogenetic proteins (BMPs), or other PDGFs including compositions of PDGF-AA, PDGF-BB, PDGF-AB, PDGF-CC and/or PDGF-DD.
[0057] In addition to solutions comprising PDGF, compositions of the present invention also comprise a biocompatible matrix in which to dispose the PDGF solutions and may also comprise a biocompatible binder either with or without addition of a biocompatible matrix.
Biocompatible Matrix
[0058] Bone Scaffolding Material
[0059] A biocompatible matrix, according to embodiments of the present invention, comprises a bone scaffolding material. It is to be understood that the terms bone scaffolding material and bone substituting agent are used interchangeably in the present application. The bone scaffolding material provides the framework or scaffold for new bone and tissue growth to occur. In some embodiments, a bone scaffolding material has multidirectional and interconnected pores of varying diameters. In some embodiments, a bone scaffolding material comprises a plurality of pockets and non-interconnected pores in addition to the interconnected pores. A bone scaffolding material, in some embodiments, is one that can permanently or temporarily replace bone. Following implantation, the bone scaffolding material can be retained by the body or it can be resorbed by the body and replaced by bone.
[0060] A bone scaffolding material, in some embodiments, comprises at least one calcium phosphate. In other embodiments, a bone scaffolding material can comprise a plurality of calcium phosphates. Calcium phosphates suitable for use as a bone scaffolding material, in embodiments of the present invention, have a calcium to phosphorus atomic ratio ranging from 0.5 to 2.0. In some embodiments, a bone scaffolding material comprises an allograft such as DFDBA, FDBA, or particulate DBM. In some embodiments, a bone scaffolding material comprises mineralized bone allograft, mineralized bone, mineralized deproteinized xenograft, or demineralized bone.
[0061] Non-limiting examples of calcium phosphates suitable for use as bone scaffolding materials comprise amorphous calcium phosphate, monocalcium phosphate monohydrate (MCPM), monocalcium phosphate anhydrous (MCPA), dicalcium phosphate dihydrate (DCPD), dicalcium phosphate anhydrous (DCPA), octacalcium phosphate (OCP), .alpha.-tricalcium phosphate, .beta.-TCP, hydroxyapatite (OHAp), poorly crystalline hydroxapatite, tetracalcium phosphate (TTCP), heptacalcium decaphosphate, calcium metaphosphate, calcium pyrophosphate dihydrate, calcium pyrophosphate, carbonated calcium phosphate, hydroxyapatite, or derivatives or mixtures thereof.
[0062] In some embodiments, a bone scaffolding material comprises a polymeric material. A polymeric scaffold, in some embodiments, comprises collagen, polylactic acid, poly(L-lactide), poly(D,L-lactide), polyglycolic acid, poly(L-lactide-co-glycolide), poly(L-lactide-co-D,L-lactide), polyacrylate, polymethacrylate, polymethylmethacrylate, chitosan, or combinations or derivatives thereof.
[0063] In some embodiments, a bone scaffolding material comprises porous structure. Porosity is a desirable characteristic as it facilitates cell migration and infiltration into the scaffolding material so that the infiltrating cells can secrete extracellular bone matrix. Porosity also provides access for vascularization. Porosity also provides a high surface area for enhanced resorption and release of active substances as well as increased cell-matrix interaction. A bone scaffolding material, in some embodiments, can be sized and shaped prior to use. In some embodiments. the bone scaffolding material can be provided in a shape suitable for implantation.
[0064] Porous bone scaffolding materials, according to some embodiments, can comprise pores having diameters ranging from about 1 .mu.m to about 1 mm. In one embodiment, a bone scaffolding material comprises macropores having diameters ranging from about 100 .mu.m to about 1 mm or greater. In another embodiment, a bone scaffolding material comprises mesopores having diameters ranging from about 10 .mu.m to about 100 .mu.m. In a further embodiment, a bone scaffolding material comprises micropores having diameters less than about 10 .mu.m. Embodiments of the present invention contemplate bone scaffolding materials comprising macropores, mesopores, and micropores or any combination thereof.
[0065] A porous bone scaffolding material, in one embodiment, has a porosity greater than about 25% or greater than about 40%. In another embodiment, a porous bone scaffolding material has a porosity greater than about 50%, greater than about 60%, greater than about 65%, greater than about 70%, greater than about 80%, or greater than about 85%. In a further embodiment, a porous bone scaffolding material has a porosity greater than about 90%. In some embodiments, a porous bone scaffolding material comprises a porosity that facilitates cell migration into the scaffolding material.
[0066] In some embodiments, a bone scaffolding material comprises a plurality of particles. A bone scaffolding material, for example, can comprise a plurality of calcium phosphate particles. Particles of a bone scaffolding material, in some embodiments, can individually demonstrate any of the pore diameters and porosities provided here for the bone scaffolding material. In other embodiments, particles of a bone scaffolding material can form an association to produce a matrix having any of the pore diameters or porosities provided herein for the bone scaffolding material.
[0067] Bone scaffolding particles may be mm, .mu.m, or submicron (nm) in size. Bone scaffolding particles, in one embodiment, have an average diameter ranging from about 1 .mu.m to about 5 mm. In other embodiments, particles have an average diameter ranging from about 1 mm to about 2 mm, from about 1 mm to about 3 mm, or from about 250 .mu.m to about 750 .mu.m. Bone scaffolding particles, in another embodiment, have an average diameter ranging from about 100 .mu.m to about 300 .mu.m. In a further embodiment, bone scaffolding particles have an average diameter ranging from about 75 .mu.m to about 300 .mu.m. In additional embodiments, bone scaffolding particles have an average diameter less than about 25 .mu.m, less than about 1 .mu.m, or less than about 1 mm. In some embodiments, scaffolding particles have an average diameter ranging from about 100 .mu.m to about 5 mm or from about 100 .mu.m to about 3 mm. In other embodiments, bone scaffolding particles have an average diameter ranging from about 250 .mu.m to about 2 mm, from about 250 .mu.m to about 1 mm, or from about 200 .mu.m to about 3 mm. Particles may also be in the range of about 1 nm to about 1 .mu.m, less than about 500 nm, or less than about 250 nm.
[0068] Bone scaffolding materials, according to some embodiments, can be provided in a shape suitable for implantation (e.g., a sphere, a cylinder, or a block). In other embodiments, bone scaffolding materials are moldable, extrudable and/or injectable. Moldable, extrudable, and injectable bone scaffolding materials can facilitate efficient placement of compositions of the present invention in and around vertebral bodies. In some embodiments, bone scaffolding materials are flowable. Flowable bone scaffolding materials, in some embodiments, can be applied vertebral bodies through a syringe and needle or cannula. In some embodiments, bone scaffolding materials harden in vivo.
[0069] In some embodiments, bone scaffolding materials are bioresorbable. A bone scaffolding material, in one embodiment, can be at least 30%, 40%, 50%, 60%, 70%, 75%, or 90% resorbed within one year subsequent to in vivo implantation. In another embodiment, a bone scaffolding material can be resorbed at least 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 75%, or 90% within 1, 3, 6, 9, 12, or 18 months of in vivo implantation. In some embodiments, a bone scaffolding material is greater than 90% resorbed within 1, 3, 6, 9, 12, or 18 months of in vivo implantation. Bioresorbability will be dependent on: (1) the nature of the matrix material (i.e., its chemical make up, physical structure and size); (2) the location within the body in which the matrix is placed; (3) the amount of matrix material that is used; (4) the metabolic state of the patient (diabetic/non-diabetic, osteoporotic, smoker, old age, steroid use, etc.); (5) the extent and/or type of injury treated; and (6) the use of other materials in addition to the matrix such as other bone anabolic, catabolic and anti-catabolic factors.
Bone Scaffolding Comprising .beta.-Tricalcium Phosphate (.beta.-TCP)
[0070] A bone scaffolding material for use as a biocompatible matrix can comprise .beta.-TCP. .beta.-TCP, according to some embodiments, can comprise a porous structure having multidirectional and interconnected pores of varying diameters. In some embodiments, .beta.-TCP comprises a plurality of pockets and non-interconnected pores of various diameters in addition to the interconnected pores. The porous structure of .beta.-TCP, in one embodiment, comprises macropores having diameters ranging from about 100 .mu.m to about 1 min or greater, mesopores having diameters ranging from about 10 .mu.m to about 100 .mu.m, and micropores having diameters less than about 10 .mu.m. Macropores and micropores of the .beta.-TCP can facilitate osteoinduction and osteoconduction while macropores, mesopores and micropores can permit fluid communication and nutrient transport to support bone regrowth throughout the .beta.-TCP biocompatible matrix.
[0071] In comprising a porous structure, .beta.-TCP, in some embodiments, can have a porosity greater than 25% or greater than about 40%. In other embodiments, .beta.-TCP can have a porosity greater than 50%, greater than about 60%, greater than about 65%, greater than about 70%, greater than about 75%, greater than about 80%, or greater than about 85%. In a further embodiment, .beta.-TCP can have a porosity greater than 90%. In some embodiments, .beta.-TCP can have a porosity that facilitates cell migration into the .beta.-TCP.
[0072] In some embodiments, a .beta.-TCP bone scaffolding material comprises .beta.-TCP particles. .beta.-TCP particles, in some embodiments, can individually demonstrate any of the pore diameters, pore structures, and porosities provided herein for scaffolding materials.
[0073] .beta.-TCP particles, in one embodiment have an average diameter ranging from about 1 .mu.m to about 5 mm. In other embodiments, .beta.-TCP particles have an average diameter ranging from about 1 mm to about 2 mm, from about 1 mm to about 3 mm, from about 100 .mu.m to about 5 mm, from about 100 .mu.m to about 3 mm, from about 250 .mu.m to about 2 mm, from about 250 .mu.m to about 750 .mu.m, from about 250 .mu.m to about 1 mm, from about 250 .mu.m to about 2 mm, or from about 200 .mu.m to about 3 mm. In another embodiment, .beta.-TCP particles have an average diameter ranging from about 100 .mu.m to about 300 .mu.m. In some embodiments, .beta.-TCP particles have an average diameter ranging from about 75 .mu.m to about 300 .mu.m. In some embodiments, .beta.-TCP particles have an average diameter of less than about 25 .mu.m, less than about 1 .mu.m, or less than about 1 mm. In some embodiments, .beta.-TCP particles have an average diameter ranging from about 1 nm to about 1 .mu.m. In a further embodiment, .beta.-TCP particles have an average diameter less than about 500 nm or less than about 250 nm.
[0074] A biocompatible matrix comprising a .beta.-TCP bone scaffolding material, in some embodiments, can be provided in a shape suitable for implantation (e.g., a sphere, a cylinder, or a block). In other embodiments, a .beta.-TCP bone scaffolding material can be moldable, extrudable, and/or flowable thereby facilitating application of the matrix to vertebral bodies. Flowable matrices may be applied through syringes, tubes, cannulas, or spatulas.
[0075] A .beta.-TCP bone scaffolding material, according to some embodiments, is bioresorbable. In one embodiment, a .beta.-TCP bone scaffolding material can be at least 30%, 40%, 50%, 60%, 65%, 70%, 75%, 80%, or 85% resorbed one year subsequent to in vivo implantation. In another embodiment, a .beta.-TCP bone scaffolding material can be greater than 90% resorbed one year subsequent to in viva implantation.
Bone Scaffolding Material and Biocompatible Binder
[0076] In another embodiment, a biocompatible matrix comprises a bone scaffolding material and a biocompatible binder. Bone scaffolding materials in embodiments of a biocompatible matrix further comprising a biocompatible binder are consistent with those provided hereinabove.
[0077] Biocompatible binders, according to some embodiments, can comprise materials operable to promote cohesion between combined substances. A biocompatible binder, for example, can promote adhesion between particles of a bone scaffolding material in the formation of a biocompatible matrix. In certain embodiments, the same material may serve as both a scaffolding material. In some embodiments, for example, polymeric materials described herein such as collagen and chitosan may serve as both scaffolding material and a binder.
[0078] Biocompatible binders, in some embodiments, can comprise collagen, polysaccharides, nucleic acids, carbohydrates, proteins, polypeptides, poly(.alpha.-hydroxy acids), poly(lactones), poly(amino acids), poly(anhydrides), polyurethanes, poly(orthoesters), poly(anhydride-co-imides), poly(orthocarbonates), poly(.alpha.-hydroxy alkanoates), poly(dioxanones), poly(phosphoesters), polylactic acid, poly(L-lactide) (PLLA), poly(D,L-lactide) (PDLLA), polyglycolide (PGA), poly(lactide-co-glycolide (PLGA), poly(L-lactide-co-D,L-lactide), poly(D,L-lactide-co-trimethylene carbonate), polyglycolic acid, polyhydroxybutyrate (PHB), poly(.epsilon.-caprolactone), poly(.delta.-valerolactone), poly(.gamma.-butyrolactone), poly(caprolactone), polyacrylic acid, polycarboxylic acid, poly(allylamine hydrochloride), poly(diallyldimethylammonium chloride), poly(ethyleneimine), polypropylene fumarate, polyvinyl alcohol, polyvinylpyrrolidone, polyethylene, polymethylmethacrylate, carbon fibers, poly(ethylene glycol), poly(ethylene oxide), poly(vinyl alcohol), poly(vinylpyrrolidone), poly(ethyloxazoline), poly(ethylene oxide)-co-poly(propylene oxide) block copolymers, poly(ethylene terephthalate)polyamide, and copolymers and mixtures thereof.
[0079] Biocompatible binders, in other embodiments, can comprise alginic acid, arabic gum, guar gum, xantham gum, gelatin, chitin, chitosan, chitosan acetate, chitosan lactate, chondroitin sulfate, N,O-carboxymethyl chitosan, a dextran (e.g., .alpha.-cyclodextrin, .beta.-cyclodextrin, .gamma.-cyclodextrin, or sodium dextran sulfate), fibrin glue, lecithin, phosphatidylcholine derivatives, glycerol, hyaluronic acid, sodium hyaluronate, a cellulose (e.g., methylcellulose, carboxymethylcellulose, hydroxypropyl methylcellulose, or hydroxyethyl cellulose), a glucosamine, a proteoglycan, a starch (e.g., hydroxyethyl starch or starch soluble), lactic acid, pluronic acids, sodium glycerophosphate, glycogen, a keratin, silk, and derivatives and mixtures thereof.
[0080] In some embodiments, a biocompatible binder is water-soluble. A water-soluble binder can dissolve from the biocompatible matrix shortly after its implantation, thereby introducing macroporosity into the biocompatible matrix. Macroporosity, as discussed herein, can increase the osteoconductivity of the implant material by enhancing the access and, consequently, the remodeling activity of the osteoclasts and osteoblasts at the implant site.
[0081] In some embodiments, a biocompatible binder can be present in a biocompatible matrix in an amount ranging from about 5 weight percent to about 50 weight percent of the matrix. In other embodiments, a biocompatible binder can be present in an amount ranging from about 10 weight percent to about 40 weight percent of the biocompatible matrix. In another embodiment, a biocompatible binder can be present in an amount ranging from about 15 weight percent to about 35 weight percent of the biocompatible matrix. In a further embodiment, a biocompatible binder can be present in an amount of about 20 weight percent of the biocompatible matrix. In another embodiment, a biocompatible binder can be present in a biocompatible matrix in an amount greater than about 50 weight percent or 60 weight percent of the matrix. In one embodiment, a biocompatible binder can be present in a biocompatible matrix in an amount up to about 99 weight percent of the matrix.
[0082] A biocompatible matrix comprising a bone scaffolding material and a biocompatible binder, according to some embodiments, can be flowable, moldable, and/or extrudable. In such embodiments, a biocompatible matrix can be in the form of a paste or putty. A biocompatible matrix in the form of a paste or putty, in one embodiment, can comprise particles of a bone scaffolding material adhered to one another by a biocompatible binder.
[0083] A biocompatible matrix in paste or putty form can be molded into the desired implant shape or can be molded to the contours of the implantation site. In one embodiment, a biocompatible matrix in paste or putty form can be injected into an implantation site with a syringe or cannula.
[0084] In some embodiments, a biocompatible matrix in paste or putty form does not harden and retains a flowable and moldable form subsequent to implantation. In other embodiments, a paste or putty can harden subsequent to implantation, thereby reducing matrix flowability and moldability.
[0085] A biocompatible matrix comprising a bone scaffolding material and a biocompatible binder, in some embodiments, can also be provided in a predetermined shape including a block, sphere, or cylinder or any desired shape, for example a shape defined by a mold or a site of application.
[0086] A biocompatible matrix comprising a bone scaffolding material and a biocompatible binder, in some embodiments, is bioresorbable. A biocompatible matrix, in such embodiments, can be resorbed within one year of in vivo implantation. In another embodiment, a biocompatible matrix comprising a bone scaffolding material and a biocompatible binder can be resorbed within 1, 3, 6, or 9 months of in vivo implantation. In some embodiments, a biocompatible matrix comprising a scaffolding material and a biocompatible binder can be resorbed within 1, 3, or six years of in vivo implantation. Bioresorbablity will be dependent on: (1) the nature of the matrix material (i.e., its chemical make up, physical structure and size); (2) the location within the body in which the matrix is placed; (3) the amount of matrix material that is used; (4) the metabolic state of the patient (diabetic/non-diabetic, osteoporotic, smoker, old age, steroid use, etc.); (5) the extent and/or type of injury treated; and (6) the use of other materials in addition to the matrix such as other bone anabolic, catabolic and anti-catabolic factors.
Biocompatible Matrix Comprising .beta.-TCP and Collagen
[0087] In some embodiments, a biocompatible matrix can comprise a .beta.-TCP bone scaffolding material and a biocompatible collagen binder. .beta.-TCP bone scaffolding materials suitable for combination with a collagen binder are consistent with those provided hereinabove.
[0088] A collagen binder, in some embodiments, can comprise any type of collagen, including Type I, Type II, and Type III collagens. In one embodiment, a collagen binder comprises a mixture of collagens, such as a mixture of Type I and Type II collagen. In other embodiments, a collagen binder is soluble under physiological conditions. Other types of collagen present in bone or musculoskeletal tissues may be employed. Recombinant, synthetic and naturally occurring forms of collagen may be used in the present invention.
[0089] A biocompatible matrix, according to some embodiments, can comprise a plurality of .beta.-TCP particles adhered to one another with a collagen binder. In some embodiments, .beta.-TCP particles for combination with a collagen binder have an average diameter ranging from about 1 .mu.m to about 5 mm. In other embodiments, .beta.-TCP particles have an average diameter ranging from about 1 mm to about 2 mm, from about 1 mm to about 3 mm, from about 100 .mu.m to about 5 mm, from about 100 .mu.m to about 3 mm, from about 250 .mu.m to about 2 mm, from about 250 .mu.m to about 750 .mu.m, from about 250 .mu.m to about 1 mm, from about 250 .mu.m to about 2 mm, or from about 200 .mu.m to about 3 mm. In another embodiment, .beta.-TCP particles have an average diameter ranging from about 100 .mu.m to about 300 .mu.m. In some embodiments, .beta.-TCP particles have an average diameter ranging from about 75 .mu.m, to about 300 .mu.m. In some embodiments, .beta.-TCP particles have an average diameter of less than about 25 .mu.m, less than about 1 .mu.m, or less than about 1 mm. In some embodiments, .beta.-TCP particles have an average diameter ranging from about 1 nm to about 1 .mu.m. In a further embodiment, .beta.-TCP particles have an average diameter less than about 500 nm or less than about 250 nm.
[0090] .beta.-TCP particles, in some embodiments, can be adhered to one another by the collagen binder so as to produce a biocompatible matrix having a porous structure. In some embodiments, the porous structure of a biocompatible matrix comprising .beta.-TCP particles and a collagen binder demonstrates multidirectional and interconnected pores of varying diameters. In some embodiments, a the biocompatible matrix comprises a plurality of pockets and non-interconnected pores of various diameters in addition to the interconnected pores.
[0091] In some embodiments, a biocompatible matrix comprising .beta.-TCP particles and a collagen binder can comprise pores having diameters ranging from about 1 .mu.m to about 1 mm. A biocompatible matrix comprising .beta.-TCP particles and a collagen binder can comprise macropores having diameters ranging from about 100 .mu.m to about 1 mm or greater, mesopores having diameters ranging from about 10 .mu.m to 100 .mu.m, and micropores having diameters less than about 10 .mu.m.
[0092] A biocompatible matrix comprising .beta.-TCP particles and a collagen binder can have a porosity greater than about 25% or greater than about 40%. In another embodiment, the biocompatible matrix can have a porosity greater than about 50%, greater than about 65%, greater than about 70%, greater than about 75%, greater than about 80%, or greater than about 85%. In a further embodiment, the biocompatible matrix can have a porosity greater than about 90%. In some embodiments, the biocompatible matrix can have a porosity that facilitates cell migration into the matrix.
[0093] In some embodiments, the .beta.-TCP particles can individually demonstrate any of the pore diameters, pore structures, and porosities provided herein for a biocompatible matrix comprising the .beta.-TCP and collagen binder.
[0094] A biocompatible matrix comprising .beta.-TCP particles, in some embodiments, can comprise a collagen binder in an amount ranging from about 5 weight percent to about 50 weight percent of the matrix. In other embodiments, a collagen binder can be present in an amount ranging from about 10 weight percent to about 40 weight percent of the biocompatible matrix. In another embodiment, a collagen binder can be present in an amount ranging from about 15 weight percent to about 35 weight percent of the biocompatible matrix. In a further embodiment, a collagen binder can be present in an amount of about 20 weight percent of the biocompatible matrix.
[0095] A biocompatible matrix comprising .beta.-TCP particles and a collagen binder, according to some embodiments, can be flowable, moldable, and/or extrudable. In such embodiments, the biocompatible matrix can be in the form of a paste or putty. A paste or putty can be molded into the desired implant shape or can be molded to the contours of the implantation site. In one embodiment, a biocompatible matrix in paste or putty form comprising .beta.-TCP particles and a collagen binder can be injected into an implantation site with a syringe or cannula.
[0096] In some embodiments, a biocompatible matrix in paste or putty form comprising .beta.-TCP particles and a collagen binder can retain a flowable and moldable form when implanted. In other embodiments, the paste or putty can harden subsequent to implantation, thereby reducing matrix flowability and moldability.
[0097] A biocompatible matrix comprising .beta.-TCP particles and a collagen binder, in some embodiments, can be provided in a predetermined shape such as a block, sphere, or cylinder.
[0098] A biocompatible matrix comprising .beta.-TCP particles and a collagen binder can be resorbable. In one embodiment, a biocompatible matrix comprising .beta.-TCP particles and a collagen binder can be at least 75% resorbed one year subsequent to in vivo implantation. In another embodiment, a biocompatible matrix comprising .beta.-TCP particles and a collagen binder can be greater than 90% resorbed one year subsequent to in vivo implantation.
[0099] A solution comprising PDGF can be disposed in a biocompatible matrix to produce a composition for treating structures of the vertebral column according to embodiments described herein.
[0100] In some embodiments, compositions comprising a PDGF solution disposed in a biocompatible matrix for promoting bone formation in a vertebral body and preventing or reducing the likelihood of vertebral compression fractures, as described herein, further comprise at least one contrast agent. Contrast agents, according to some embodiments, comprise cationic contrast agents, anionic contrast agents, nonionic contrast agents or mixtures thereof. In some embodiments, contrast agents comprise radiopaque contrast agents. Radiopaque contrast agents, in some embodiments, comprise iodo-compounds including (S)--N,N'-bis[2-hydroxy-1-(hydroxymethyl)-ethyl]-2,4,6-triiodo-5-lactamid- oisophthalamide (Iopamidol) and derivatives thereof.
Disposing PDGF Solution in a Biocompatible Matrix
[0101] The present invention provides methods for producing compositions for promoting bone formation in a vertebral body and preventing or reducing the likelihood of compression fractures of vertebral bodies, including secondary vertebral fractures. In one embodiment, a method for producing such compositions comprises providing a solution comprising PDGF, providing a biocompatible matrix, and disposing the solution in the biocompatible matrix. PDGF solutions and biocompatible matrices suitable for combination are consistent with those described hereinabove.
[0102] In some embodiments, a PDGF solution can be disposed in a biocompatible matrix by soaking the biocompatible matrix in the PDGF solution. A PDGF solution. in another embodiment, can be disposed in a biocompatible matrix by injecting the biocompatible matrix with the PDGF solution. In some embodiments, injecting a PDGF solution can comprise disposing the PDGF solution in a syringe and expelling the PDGF solution into the biocompatible matrix to saturate the biocompatible matrix.
[0103] In some embodiments, the PDGF is absorbed into the pores of the biocompatible matrix. In some embodiments, the PDGF is adsorbed onto one or more surfaces of the biocompatible matrix, including surfaces within pores of the biocompatible matrix.
[0104] The biocompatible matrix, according to some embodiments, can be in a predetermined shape, such as a brick or cylinder, prior to receiving a PDGF solution. Subsequent to receiving a PDGF solution, the biocompatible matrix can have a paste or putty form that is flowable, extrudable, and/or injectable. In other embodiments, the biocompatible matrix can already demonstrate a flowable paste or putty form prior to receiving a solution comprising PDGF. Flowable, extrudable, and/or injectable forms of compositions comprising a PDGF solution disposed in a biocompatible matrix are advantageous for use in methods of the present application as they can applied to vertebral bodies with syringes and/or cannulas.
[0105] In some embodiments, methods of producing compositions for promoting bone formation in vertebral bodies and preventing or decreasing the likelihood of compression fractures in vertebral bodies further comprise providing at least one contrast agent and disposing the at least one contrast agent in the biocompatible matrix. In some embodiments, disposing at least one contrast agent in a biocompatible matrix comprises combining the at least one contrast agent with a PDGF solution and injecting the biocompatible matrix with the PDGF/contrast agent solution.
[0106] In another embodiment, disposing at least one contrast agent in a biocompatible matrix comprises combining the at least one contrast agent with a PDGF solution and soaking the biocompatible matrix in the PDGF/contrast agent solution. Alternatively, in some embodiments, a contrast agent is disposed in a biocompatible matrix independent of the PDGF solution.
[0107] Contrast agents, according to some embodiments of the present invention, facilitate placement or application of compositions of the present invention in and around vertebral bodies. Contrast agents, according to some embodiments, comprise cationic contrast agents, anionic contrast agents, nonionic contrast agents, or mixtures thereof. In some embodiments, contrast agents comprise radiopaque contrast agents. Radiopaque contrast agents, in some embodiments, comprise iodo-compounds including (S)--N,N'-bis[2-hydroxy-1-(hydroxymethyl)-ethyl]-2,4,6-triiodo-- 5-lactamidoisophthalamide (Iopamidol) and derivatives thereof.
Compositions Further Comprising Biologically Active Agents
[0108] Compositions of the present invention, according to some embodiments, can further comprise one or more biologically active agents in addition to PDGF. Biologically active agents that can be incorporated into compositions of the present invention, in addition to PDGF, can comprise organic molecules, inorganic materials, proteins, peptides, nucleic acids (e.g., genes, gene fragments, small interfering ribonucleic acids [si-RNAs], gene regulatory sequences, nuclear transcriptional factors, and antisense molecules), nucleoproteins, polysaccharides (e.g., heparin), glycoproteins, and lipoproteins. Non-limiting examples of biologically active compounds that can be incorporated into compositions of the present invention, including, e.g., anti-cancer agents, antibiotics, analgesics, anti-inflammatory agents, immunosuppressants, enzyme inhibitors, antihistamines, hormones, muscle relaxants, prostaglandins, trophic factors, osteoinductive proteins, growth factors, and vaccines, are disclosed in U.S. patent application Ser. No. 11/159,533 (Publication No: 20060084602). In some embodiments, biologically active compounds that can be incorporated into compositions of the present invention include osteostimulatory factors such as insulin-like growth factors, fibroblast growth factors, or other PDGFs. In accordance with other embodiments, biologically active compounds that can be incorporated into compositions of the present invention preferably include osteoinductive and osteostimulatory factors such as bone morphogenetic proteins (BMPs), BMP mimetics, calcitonin, or calcitonin mimetics, statins, statin derivatives, fibroblast growth factors, insulin-like growth factors, growth differentiating factors, small molecule or antibody blockers of Wnt antagonists (e.g. sclerostin, DKK, soluble Wnt receptors), and/or parathyroid hormone. In some embodiments, factors also include protease inhibitors, as well as osteoporotic treatments that decrease bone resorption including bisphosphonates, teriparadide, and antibodies to the activator receptor of the NF-kB ligand (RANK) ligand.
[0109] Standard protocols and regimens for delivery of additional biologically active agents are known in the art. Additional biologically active agents can introduced into compositions of the present invention in amounts that allow delivery of an appropriate dosage of the agent to the implant site. In most cases, dosages are determined using guidelines known to practitioners and applicable to the particular agent in question. The amount of an additional biologically active agent to be included in a composition of the present invention can depend on such variables as the type and extent of the condition, the overall health status of the particular patient, the formulation of the biologically active agent, release kinetics, and the bioresorbability of the biocompatible matrix. Standard clinical trials may be used to optimize the dose and dosing frequency for any particular additional biologically active agent.
[0110] A composition of the present invention, according to some embodiments, can further comprise the addition of additional bone grafting materials with PDGF including autologous bone marrow, autologous platelet extracts, allografts, synthetic bone matrix materials, xenografts, and derivatives thereof.
Methods of Treating Vertebral Bodies
[0111] In some embodiments, the present invention provides methods for promoting bone formation in a vertebral body comprising providing a composition comprising a PDGF solution disposed in a biocompatible matrix and applying the composition to at least one vertebral body. In some embodiments, the composition can be applied to a plurality of vertebral bodies. Applying the composition, in some embodiments, comprises injecting at least one vertebral body with the composition. Compositions of the present invention, in some embodiments, are injected into the cancellous bone of a vertebral body. Vertebral bodies, in some embodiments, comprise thoracic vertebral bodies, lumbar vertebral bodies, or combinations thereof. Vertebral bodies, in some embodiments, comprise cervical vertebral bodies, coccygeal vertebral bodies, the sacrum, or combinations thereof.
[0112] In another aspect, the present invention provides methods for preventing or decreasing the likelihood of vertebral compression fractures, including secondary vertebral compression fractures by strengthening vertebrae. Preventing or decreasing the likelihood of vertebral compression fractures, according to embodiments of the present invention, comprises providing a composition comprising a PDGF solution disposed in a biocompatible matrix and applying the composition to at least one vertebral body. In some embodiments, applying the composition to at least one vertebral body comprises injecting the composition into the at least one vertebral body.
[0113] In some embodiments, a composition of the present invention is applied to a second vertebral body subsequent to vertebroplasty or kyphoplasty of a first vertebral body. In some embodiments, the second vertebral body is adjacent to the first vertebral body. In other embodiments, the second vertebral body is not adjacent to the first vertebral body. In a further embodiment, a composition of the present invention is applied to a third vertebral body subsequent to vertebroplasty or kyphoplasty of a first vertebral body. In some embodiments, the third vertebral body is adjacent to the first vertebral body. In other embodiments, the third vertebral body is not adjacent to the first vertebral body. Embodiments of the present invention additionally contemplate application of compositions provided herein to a plurality of vertebral bodies, including high risk vertebral bodies, subsequent to vertebroplasty or kyphoplasty of a first vertebral body. It is to be understood that first, second, and third vertebral bodies, as used herein, do not refer to any specific position in the vertebral column as methods for inhibiting vertebral compression fractures, including secondary compression fractures, can be applied to all types of vertebral bodies including thoracic vertebral bodies, lumbar vertebral bodies, cervical vertebral bodies, coccygeal vertebral bodies, and the sacrum.
[0114] In some embodiments, methods for promoting bone formation in vertebral bodies and preventing or decreasing the likelihood of compression fractures of vertebral bodies further comprise providing at least one pharmaceutical composition in addition to the composition comprising a PDGF solution disposed in a biocompatible matrix and administering the at least one pharmaceutical composition locally and/or systemically. The at least one pharmaceutical composition, in some embodiments, comprises vitamins, such as vitamin D3, calcium supplements, or any osteoclast inhibitor known to one of skill in the art, including bisphosphonates. In some embodiments, the at least one pharmaceutical composition is administered locally. In such embodiments, the at least one pharmaceutical composition can be incorporated into the biocompatible matrix or otherwise disposed in and around a vertebral body. In other embodiments, the at least one pharmaceutical composition is administered systemically to a patient. In one embodiment, for example, the at least one pharmaceutical composition is administered orally to a patient. In another embodiment, the at least one pharmaceutical composition is administered intravenously to a patient.
[0115] The following examples will serve to further illustrate the present invention without, at the same time, however, constituting any limitation thereof. On the contrary, it is to be clearly understood that resort may be had to various embodiments, modifications and equivalents thereof which, after reading the description herein, may suggest themselves to those skilled in the art without departing from the spirit of the invention.
Example 1
Preparation of a Composition Comprising a Solution of PDGF and a Biocompatible Matrix
[0116] A composition comprising a solution of PDGF and a biocompatible matrix was prepared according to the following procedure.
[0117] A pre-weighed block of biocompatible matrix comprising .beta.-TCP and collagen was obtained. The .beta.-TCP comprised .beta.-TCP particles having an average diameter ranging from about 100 .mu.m to about 300 .mu.m. The .beta.-TCP particles were formulated with about 20% weight percent soluble bovine type I collagen binder. Such a .beta.-TCP/collagen biocompatible matrix can be commercially obtained from Kensey Nash (Exton, Pa.).
[0118] A solution comprising rhPDGF-BB was obtained. rhPDGF-BB is commercially available from Novartis Corporation at a stock concentration of 10 mg/ml (i.e., Lot #QA2217) in a sodium acetate buffer. The rhPDGF-BB is produced in a yeast expression system by Chiron Corporation and is derived from the same production facility as the rhPDGF-BB that is utilized in the products REGRANEX, (Johnson & Johnson) and GEM 21S (BioMimetic Therapeutics) which have been approved for human use by the United States Food and Drug Administration. This rhPDGF-BB is also approved for human use in the European Union and Canada. The rhPDGF-BB solution was diluted to 0.3 mg/ml in the acetate buffer. The rhPDGF-BB solution can be diluted to any desired concentration according to embodiments of the present invention, including 1.0 mg/ml.
[0119] A ratio of about 3 ml of rhPDGF-BB solution to about 1 g dry weight of the .beta.-TCP/collagen biocompatible matrix was used to produce the composition. In the preparation of the composition, the rhPDGF-BB solution was expelled on the biocompatible matrix with a syringe, and the resulting composition was blended into a paste for placement into a syringe for subsequent injection into a vertebral body.
Example 2
Method of Inhibiting Secondary Vertebral Compression Fractures
Experimental Design and Overview
[0120] This prospective, randomized, controlled, single-center clinical trial is to evaluate the efficacy of compositions comprising a PDGF solution disposed in a tricalcium phosphate matrix for inhibiting secondary compression fractures in high risk vertebral bodies (HVBs) at the time of kyphoplasty of vertebral compression fractures. Comparisons are made between vertebral bodies treated with a .beta.-tricalcium phosphate+rhPDGF-BB composition and untreated vertebral bodies. The present study is a pilot, clinical trial to support the proof-or-principle of .beta.-TCP+rh-PDGF-BB to prevent or decrease the likelihood of secondary vertebral compression fractures by increased bone formation in HVBs.
[0121] The study is performed on up to a total of 10 patients requiring prophylactic treatment of HVBs at the time of kyphoplasty. Potential patients are screened to determine if they meet the inclusion and exclusion criteria If all entry criteria are achieved, the potential patients are invited to participate in the clinical trial. All patients considered for entry into the study are documented on the Screening Log and reasons for exclusion are recorded.
[0122] All patients have undergone kyphoplasty and do not have a symptomatic VCF adjacent to the two vertebral bodies treated in this study. The subject is not to be enrolled into the study if the surgeon determines intraoperatively that the fracture does not meet the fracture enrollment criteria or other fractures exist that would preclude treatment in this protocol.
[0123] A total of 10 patients are enrolled and treated in the present study. A first vertebral body adjacent to the kyphoplasty of each patient is injected with 3.0 ml of a composition of .beta.-TCP+0.3 mg/ml of rhPDGF-BB prepared in accordance with Example 1 herein. The second vertebral body adjacent to the kyphoplasty remains untreated and serves as a control. The treated vertebral body may be either cranial or caudal to the kyphoplasty and is determined randomly.
[0124] Patients are treated according to the standard protocols and follow-up for kyphoplasty/vertebroplasty. Each patient is examined by the surgeon at 7-14 days, and at 6, 12, 24, and 52 weeks for clinical, radiographic and quantitative computed tomography (QCT). All over-the-counter and prescribed medication usage is recorded. An independent radiologist, unaware of the patients' treatment group assignments, performs QCT analysis to assess bone density. These measurements are documented and analyzed.
[0125] All postoperative complications and device-related adverse events are recorded on the appropriate case report form. If a subject experiences a subsequent VCF during the study period or another surgical procedure for a serious adverse event or the investigational device is removed, the subject is monitored for safety until the end of the study. Those subjects who are re-operated and/or have the fracture fixation hardware removed are requested to give permission to examine the explants for histological purposes. All patients are monitored during the 12-month trial and any subject who requests study withdrawal or is withdrawn by the investigator is requested to provide a reason for study discontinuance. Table 1 provides a timeline summary for the present study.
TABLE-US-00001 TABLE 1 Study Timeline Survey Visit 1 Visit 2 Screening Surgical Visit Visit .dwnarw. .dwnarw. Visit 3 Visit 4 Visit 5 Visit 6 Visit 7 Within 21 Within 21 Post Tx Post Tx Post Tx Post Tx Post Tx Days of Days of Follow Up Follow Up Follow Up Follow Up Follow Up Surgery Screening .dwnarw. .dwnarw. .dwnarw. .dwnarw. .dwnarw. Day 0 Day 7-14 Week 6 Week 12 Week 24 Week 52 .+-.3 days .+-.7 days .+-.7 days .+-.14 days
[0126] The primary endpoint is the bone density at 12 weeks post-operatively measured by QCT scans. Secondary endpoints include subject pain and quality of life assessments.
Surgical Protocol
[0127] After patients have been enrolled in the study, satisfying both the inclusion and exclusion criteria, the following surgical protocol is undertaken.
[0128] Patients are brought into an operating room (OR) in the standard fashion, and standard methods are used to perform the kyphoplasty procedure with methyl methacrylate cement augmentation of the fractured vertebral body. Standard radiographs are taken of the vertebral bodies treated with kyphoplasty and with preventative bone augmentation treatment.
[0129] Following the kyphoplasty treatment, the investigator identifies and qualifies the two levels to be treated with prophylactic bone augmentation. If two (2) qualified vertebral bodies are not available for treatment, as determined at the time of surgery, the patient is considered a screen failure and not enrolled into the study.
[0130] Upon identification of the two HVBs, the investigator requests that the randomization code be opened to determine the study treatment administered. The randomization code specifies treatment with the .beta.-TCP+rhPDGF composition either proximally or distally in relation to the level treated with kyphoplasty. The other HVB remains untreated.
[0131] The .beta.-TCP+rhPDGF composition is mixed according to the procedure provided in Example 1. Once mixed, the paste is loaded into a syringe for injection using aseptic technique. Once the .beta.-TCP+rhPDGF composition is mixed, the clinician waits about 10 minutes prior to implantation. A new sterile mixing device (spatula) is used for each mix. The investigator directs the assistant who performs the mixing to record the cumulative amount of implanted composition, as well as the residual amount of composition not implanted. The amount of composition is calculated and documented using qualitative relative measurements (1/3, 2/3, All).
[0132] An 8 to 16 gauge JAMSHIDI.RTM. needle available from Cardinal Health of Dublin, Ohio is inserted through an extrapedicular approach into the vertebral bodies requiring prophylactic treatment. The wire is passed through the JAMSHIDI.RTM. needle and the JAMSHIDI.RTM. needle through the stylet over the wire The appropriate mixed preparation is injected into the subject vertebral body. Care should be taken to minimize leakage of the paste outside of the vertebral body.
[0133] Contrast agents, according to embodiments of the present invention, can assist in identifying the leakage of the paste outside the vertebral body. FIG. 1 illustrates a syringe and related apparatus penetrating tissue overlaying a vertebral body to deliver a composition of the present invention to the vertebral body. FIG. 2 is a radiograph illustrating injection of a composition of the present invention into the vertebral body of the L3 vertebra according to one embodiment.
[0134] The instrumentation is removed. Thorough irrigation and standard wound closure techniques are employed.
Follow-up Evaluations
[0135] Patients are seen for post-operative evaluations at days 7-14, and at 6 (.+-.3 days), 12 (.+-.7 days), 24 (.+-.7 days), and 52 (.+-.14 days) weeks post-surgery. Routine evaluations and procedures are performed during the follow-up period, as specified in the study flowchart of Table 2 below.
TABLE-US-00002 TABLE 2 Study Flow Chart and Follow-up Assessments Post-Treatment Follow-Up Evaluations Surgery Visit Visit Visit Visit Screening Visit Visit 4 5 6 7 Visit 2 3 Week 6 Week 12 Week 24 Week 52 Procedure 1 Day 0 Day 7-14 .+-.3 Days .+-.7 Days .+-.7 Days .+-.14 Days Informed Consent X.sup.1 Screening Log X Medical History X Physical Examination of Spine X X X X X X X Subject Eligibility X X Criteria Verification Identification of High- X Risk Vertebral Bodies Randomization X Kyphoplasty and X Preventative Bone Augmentation Volume of Graft Material X Placed Qualitative CT X X X X Assessments.sup.2 Plain Radiographic X X X X X X Assessments Adverse X X X X X X Events/Complications Concomitant Medications X X X X X X X Review .sup.1Must occur prior to any study-specific procedures. .sup.2Quantitative Computed Tomography (QCT) is performed according to standard protocol to obtain BMD data which is determined by the designated musculoskeletal radiologist.
Assessment of Effectiveness
[0136] Outcome data is collected from this study on findings derived from radiographs, QCTs, and from direct examination of function. The schedule of these measurements is provided in Table 3.
TABLE-US-00003 TABLE 3 Frequency of Radiographic and Functional Assessments Study Parameters Plain film Qualitative Timepoint radiographs CT Scans Pain Function Prior to X X X X Treatment Immediately X Post-Treatment Day 7-14 X Week 6 X X X X Week 12 X X X X Week 24 X X X X Week 52 X X X X
[0137] Vertebral bodies injected with a .beta.-TCP+rhPDGF composition are expected to display increased bone mineral density (BMD) in comparison to untreated vertebral bodies. Increased bone mineral density in a vertebral body can render the vertebral body less susceptible to fractures including secondary fractures induced by kyphoplasty/vertebroplasty operations.
Example 3
Method of Inhibiting Vertebral Compression Fractures in Osteoporotic Individuals
[0138] A method of inhibiting vertebral compression fractures in osteoporotic individuals comprises promoting bone formation in vertebral bodies through treatment with compositions comprising a PDGF solution disposed in a biocompatible matrix such as .beta.-tricalcium phosphate.
[0139] Compositions of the present invention are mixed in accordance with that provided in Example 1. The concentration of PDGF in the PDGF solutions ranges from 0.3 mg/ml to 1.0 mg/ml. Once mixed, the composition is loaded into a syringe for injection using aseptic technique. The surgeon waits about 10 minutes prior to implantation. A new sterile mixing device (spatula) is used for each mix.
[0140] The JAMSHIDI.RTM. needle is inserted through an extrapedicular approach into the vertebral bodies requiring prophylactic treatment. Vertebral bodies requiring prophylactic treatment, in some embodiments, comprise high risk vertebral bodies including vertebral bodies T5 through T12 and L1 through L4. The wire is passed through the JAMSHIDI.RTM. needle and the JAMSHIDI.RTM. needle through the stylet over the wire The mixed composition is injected into the subject vertebral body. Care is taken to minimize leakage of the paste outside of the vertebral body. A plurality of vertebral bodies are treated according to the present example. Osteoporotic patients receiving this treatment have a lower incidence of vertebral compression fractures than untreated osteoporotic patients.
Example 4
Evaluation of the Chronic Safety of rh-PDGF-BB Combined with Collagen/.beta.-tricalcium Phosphate Matrix in a Rabbit Paravertebral Implant Model
Experimental Design and Overview
[0141] This study evaluated the safety of implanting injectable rhPDGF-BB/collagen/.beta.-TCP material in a paravertebral intramuscular site adjacent to the spine of rabbits. The animals were observed for signs of neurotoxicity, and the implant sites with adjacent vertebral bodies and spinal cord were examined histologically to document tissue-specific responses to the material.
[0142] The study protocol and animal care was approved by the local IACUC and conducted according to AAALAC guidelines. Twelve (12) naive, female, albino New Zealand rabbits weighing .gtoreq.2.5 kg were assigned to one of 4 groups: 0.3 mg/ml PDGF; 1.0 mg/ml PDGF; rubber; or acetate buffer. PDGF treated rabbits received 0.2 cc implants of appropriately concentrated rhPDGF-BB in matrix injected into a 1 cm pocket in the right paravertebral muscle adjacent to the L4-L5 vertebral bodies while high density polyethylene (HDPE) was implanted in a similar incision in the left paravertebral muscles near L2-L3 of the same animals. Rabbits in the sodium acetate buffer group received sodium acetate buffer in place of the PDGF+matrix implant, while those in the rubber group received only rubber in the right paravertebral muscle. One rabbit in each group was sacrificed at 29, 90, and 180 days post-surgery.
[0143] Body weights were measured prior to surgery and biweekly following surgery for the duration of the study. Radiographs were taken prior to surgery, immediately following surgery, and immediately prior to sacrifice. Digital photography of the surgical sites was performed during surgery and at the study end points. Weekly clinical observations of the implant sites were recorded for signs of erythema, edema, and inflammation and for signs of neurotoxicity, such as ambulatory changes. At necropsy, each implant site along with the adjacent vertebral body and spinal cord were harvested en bloc, fixed in formalin, and prepared for decalcified, paraffin embedded histopathological analysis.
Materials
[0144] The dosages of rhPDGF-BB tested in this study included 0.3 mg/ml and 1.0 mg/ml in 20 mM sodium acetate buffer, pH 6.0+/-0.5. The matrix material consisted of 20% lyophilized bovine type I collagen and 80% .beta.-TCP with a particle size of 100-300 .mu.m (Kensey Nash Corporation). Negative control material consisted of high-density polyethylene (HDPE) and positive control material consisted of black rubber. Immediately prior to surgery, the rhPDGF-BB and control solutions were mixed with matrix material in a 3:1 liquid to mass ratio.
[0145] Briefly, the PDGF solution was allowed to saturate the material for about 2 minutes then was manually mixed for about 3 minutes to generate a paste-like consistency. The homogeneous distribution of rhPDGF-BB throughout the mixed material using this mixing technique was confirmed by eluting the PDGF from samples of similar mass and then quantifying the PDGF by ELISA (R&D Systems).
Results
[0146] Following manual mixing of 0.3 mg/ml rhPDGF-BB with the collagen/.beta.-TCP matrix, the homogeneity of rhPDGF-BB throughout the mixed material was confirmed within +/-4% error across samples.
[0147] All animals recovered from surgery, and at the time of this writing, all clinical observations were reported to be normal with no signs of neurotoxicity or abnormal wound healing at the surgical sites. Two animals treated with sodium acetate buffer and matrix control exhibited minor scabbing at the surgical wounds which healed completely. One animal that received 0.3 mg/mi rhPDGF-BB exhibited slight erythema at the surgical site 3-4 days after surgery and then returned to normal appearance. A histopathological analysis of test article implant sites 29 days post-surgery indicated a mild amount of tissue in-growth into the implanted test materials and a mild inflammatory response. No ectopic or abnormal bone formation was observed in the vertebral bodies adjacent to the implant sites. These findings are summarized in Table 4 and compared with ratings for negative control HDPE implant sites.
TABLE-US-00004 TABLE 4 Summary of Histopathology Findings at Implant Sites 29 Days After Surgery [PDGF-BB] Tissue In- Ectopic (mg/ml) Macrophages MGCs growth Bone Exostosis 0.3 3, 1(NC) 2, 0(NC) 2, 0(NC) 0, 0(NC) 0, 0(NC) 1.0 2, 2(NC) 2, 0(NC) 2, 0(NC) 0, 0(NC) 0, 0(NC) NC = Negative Control; MGC = multinucleated giant cells; Bioreactivity scale: 0 = Absent, 1 = Minimal/Slight, 2 = Mild, 3 = Moderate, 4 = Marked/Severe
[0148] Preliminary evidence from this study based on clinical observations, suggests that collagen/.beta.-tricalcium phosphate combined with either 1.0 mg/ml, 0.3 mg/ml rhPDGF-BB, or sodium acetate buffer does not elicit any acute or chronic neurotoxic effects. Histopathological assessment of the implant sites 29 days post-surgery indicated a normal and expected mild amount of tissue in-growth into the implanted material and a mild inflammatory response. No ectopic bone formation, exostosis, or abnormal bone resorption was observed at any of the implant sites. Based on observations of the animals treated in this study, collagen/.beta.-tricalcium phosphate combined with either 1.0 mg/ml, 0.3 mg/ml rhPDGF-BB is safe to use when injected in close proximity to the spinal column.
Example 5
Evaluation of the Safety of PDGF-BB Combined with a Bovine Type I Collagen/.beta.-TCP Matrix for Vertebral Therapy
[0149] This study evaluated the safety of a composition comprising rhPDGF-BB combined with a biocompatible matrix comprising .beta.-tricalcium phosphate and type I collagen for bone augmentation following injection of the composition into vertebral bodies of baboons. Experimental Design
[0150] A total of 6 female baboons (Papio anubis) of 18 to 21 years of age were studied, each baboon being assigned to one of two treatment groups as provided in Table 5. During the study, the animals were imaged and analyzed using radiography, quantitative computed tomography (QCT), magnetic resonance imaging (MRI) techniques, terminal histology and non-GLP microcomputed tomography (microCT).
[0151] Four vertebral levels (T12, L2, L4 and L6) were investigated in each animal. Each animal of Group 1 received an injection of about 0.5 cc of a 1.0 mg/ml rhPDGF-BB+collagen/.beta.-TCP (matrix) composition into each of the T12, L2 and L4 vertebral bodies. The 1.0 mg/ml rhPDGF-BB+collagen/.beta.-TCP (matrix) compositions were prepared as set forth in Example 1 hereinabove. Each animal of Group 11 received an injection of about 0.5 cc of a sodium acetate buffer+collagen/.beta.-TCP (matrix) composition into each of the T12, L2, and L4 vertebral bodies. Animals of each group I and II additionally received an injection of about 0.5 cc of a sodium acetate buffer into the L6 vertebral bodies. Therefore, a total of four (4) vertebral bodies per animal received an injection. FIG. 3 summarizes the injection strategy of the present study. Documentation of the treatments in each animal was recorded on study forms.
[0152] Surgery was conducted using a percutaneous, fluoroscopically guided approach. The procedure was performed similar to a vertebroplasty, except that an injectable 1.0 mg/ml rhPDGF-BB+collagen/.beta.-TCP matrix or appropriate control treatment was injected. About 0.5 cc of a rhPDGF-BB+collagen/.beta.-TCP material, control material, or buffer was injected into each vertebral body as described above. Each animal was provided anesthesia during the surgery.
TABLE-US-00005 TABLE 5 Summary of Treatments for Injection of rhPDGF-BB + Collagen/ .beta.-TCP Matrix in Vertebral Bodies of Baboons Group Dose Timepoints Analyses I 1.0 mg/ml PDGF + Pre- and post- QCT, MRI, Clinical collagen/.beta.-TCP surgery, 1 and Observations, Serum matrix. L6 received 3 months, Chemistry and Hematology sodium acetate (optionally (Pre- and post-surgery, buffer only 6 months) and at 1 month, 3 months, and 6 months post-surgery), Body Weights, Radiography, Histology, non-GLP microCT II 20 mM sodium Pre- and post- QCT, MRI, Clinical acetate buffer, pH surgery, 1 and Observations, Serum 6.0 + collagen/ 3 months, Chemistry and Hematology .beta.-TCP matrix. L6 (optionally (Pre- and post-surgery, received sodium 6 months) and at 1 month, 3 months, acetate buffer only and 6 months post-surgery), Body Weights, Radiography, Histology, non-GLP microCT
A. Assignment to Dose Group
[0153] Three animals were assigned to treatment groups by a manual scheme designed to achieve similar group mean body weights.
B. Assignment to Surgery Days
[0154] Animals were assigned to one of two surgery days (Day I or Day II). For each animal, a coin flip determined assignment into the Day I group or the Day 11 group. This process was continued until each of the days was filled with three animals. The animal numbers, their dosing group assignments, and surgery days were recorded. The animal treatment groups were known to the study monitors and study director. The radiologists and histopathologist were blind to the treatment groups.
C. In-Vivo Observations and Measurements
Clinical Observations
[0155] Animals were observed within their cages daily throughout the study. Recording of cageside observations was commenced after the pre-selection criteria was completed and is continued until the end of study. Each animal was observed for changes in general appearance and behavior, including changes in ambulation. Each animal was observed for evidence of menstrual cycling over the course of the study. The cycling readings were performed non-GLP and were recorded in the Southwest Foundation for Biomedical Research (SFBR) animal database.
[0156] Treatment of the animals was in accordance with SFBR standard operating procedures (SOPs), which adhere to the regulations outlined in the USDA Animal Welfare Act (9 CFR, Parts 1, 2 and 3) and the conditions specified in The Guide for Care and Use of Laboratory Animals (ILAR publication, 1996, National Academy Press). The study animals were observed and were recorded at least once daily for signs of illness or distress, including changes in ambulation, and any such observations were reported to the responsible veterinarian and study director.
Body Weight
[0157] Body weights were measured at the initial health check, prior to surgery, and prior to follow up radiographs. Food was withheld prior to sedation and subsequent body weight measurements.
Food Consumption
[0158] Except when animals were fasting for study procedures, food consumption was qualitatively assessed daily for each animal (as part of the cageside observations), beginning at least 7 days prior to surgery. Each animal was provided a full feeder of food once a day (non-sedation days) and the amount eaten was documented as per SFBR SOPs. On sedation days, the animals were fed once each day when they recovered from anesthesia.
D. Fluoroscopy, Photography, Radiography, MRI and QCT Imaging
[0159] Non-GLP digital photographs were taken of the injection sites pre-operatively, immediately post-operatively, and at 1, 3, 6, and 9 months post-operatively.
[0160] Anteroposterior and lateral radiographs were taken pretreatment and immediately following surgery as well as at about 1, 3, 6, and 9 months post-operatively. For anteroposterior radiographs, the animals were placed on their backs in supine position with their legs supported. For lateral radiographs, the animals were positioned lying on their left sides with arms and legs supported. Energy (kV) and intensity (mA) settings for each position and animal were recorded.
[0161] Non-GLP fluorographs were captured intraoperatively before, during, and after injection of the test and control articles into the vertebrae of the animals. Fluorographs were not assessed as an outcome of this study, but enable the surgeon to accurately insert the introduction needle into the vertebral bodies during surgery.
[0162] Magnetic resonance imaging (MRI) was performed to image the spine of each animal pre-operatively and within 4 to 10 days post-operatively. MRI was additionally performed to image the spine of each animal at about 1, 3, 6, and 9 months post-operatively. MRI sessions consisted of T1 and T2-weighted scans.
[0163] Quantitative computed tomography imaging (QCT) was performed to image the spine of each animal pre-operatively and within 4 to 10 days post-operatively. QCT was additionally performed to image the spine of each animal at about 1, 3, 6, and 9 months post-operatively. Scans consisted of a series of contiguous cross-sectional slices of the torso from the caudal endplate of the 11th thoracic vertebrae to the cranial endplate of the sacrum.
[0164] QCT was also used to obtain 3 mm thick cross sectional images of injected and intervening untreated vertebral bodies of each baboon 1 week prior to surgery (pre-surgical) and at 1, 4, and 12 weeks post surgery. A total of 5-8 slices were required to fully image each vertebral body. The DICOM (digital imaging and communications in medicine) format images produced by the QCT were transferred and converted to a file format for 3-dimensional volumetric analysis using software developed by Scanco AG (Bassersdorf, Switzerland). Volumetric bone mineral density (vBMD) of the anterior compartment of each vertebral body was determined by manually selecting a region of interest (ROI) that excluded the cortical shell in each slice. The evaluation software created a z-stack of the individual slices and ROI's before extracting the volumetric ROI and calculating volumetric density in arbitrary units derived from the gray-scale intensity in the images and a percent change from the baseline scans was calculated. One-way repeated measures ANOVA with Tukey's post-hoc test was used to determine the presence of any statistically significant changes in vBMD for animals of Group I and Group II from pre-surgical or 1 wk post-surgical to the conclusion of the study.
[0165] The radiographs, MRI, and QCT images were evaluated by one board certified clinical radiologist and one qualified associate to provide a consensus assessment of the neuropathological, osteopathological and surrounding soft tissue pathological outcomes resulting from the treatments of the vertebrae. The evaluation consisted of a qualitative examination of each image for abnormalities of the bone and of the neural tissues and adjacent surrounding soft tissues. The radiologist followed the Radiology Assessment Protocol to evaluate the radiology data.
E. Clinical Pathology Evaluation
Serum Chemistry
[0166] About 3 ml whole blood was collected into containers without anticoagulant pre-surgery and post-surgery. About 3 ml whole blood was also collected into containers without anticoagulant at about 1, 3, 6, and 9 months post-operatively. The animals were fasted overnight prior to blood collection for serum chemistry. Serum was analyzed for the following parameters set forth in Table 6.
TABLE-US-00006 TABLE 6 Serum Analysis Sodium Phosphorus Potassium Glucose Chloride Urea nitrogen (BUN) Total carbon dioxide (bicarbonate) Creatinine Total bilirubin Total protein Alkaline phosphatase (AP) Albumin Lactate dehydrogenase (LDH) Globulin Aspartate aminotransferase (AST) Albumin/globulin ratio Alanine aminotransferase (ALT) Cholesterol Gamma-glutamyltransferase (GGT) Triglycerides Calcium BUNICREAT Ratio Anion Gap Direct Bilirubin CPK
Hematology
[0167] About 3 ml of blood was collected in EDTA-containing tubes pre-surgery and post-surgery. About 3 ml of blood was also collected in EDTA-containing tubes at 1, 3, 6, and 9 months post-surgery. The whole blood samples were analyzed for the following parameters set forth in Table 7.
TABLE-US-00007 TABLE 7 Blood Analysis Red blood cell (RBC) counts Mean corpuscular hemoglobin (MCH) White blood cells (WBCs) Mean corpuscular hemoglobin (total and differential*) concentration (MCHC) Hemoglobin concentration Mean corpuscular volume (MCV) Hematocrit Platelet counts (Plt) RDW Abnormal blood cell morphology *Includes polysegmented neutrophils, band cells, lymphocytes, monocytes, basophils, eosinophils.
Serum Collection for Analysis by Sponsor or SFBR
[0168] About 14 ml of blood was collected into non-additive (i.e., "clot") tubes from all animals once pre-surgery and post-surgery. About 14 ml of blood was also collected into non-additive (i.e., "clot") tubes from all animals at 1, 3, 6, and 9 months post-surgery. The blood was centrifuged to obtain serum and divided into two aliquots. The serum is stored at -70.degree. C. or lower.
Anatomic Pathology
[0169] All animals are humanely sacrificed at the end of the study. A gross necropsy is conducted on each animal sacrificed in a moribund or diseased condition to determine the cause and/or nature of the moribund or diseased condition.
Necropsy
[0170] A complete necropsy is conducted under the supervision of the study pathologist on the sacrificed animals in a moribund or diseased condition during the study to determine the cause and/or nature of the moribund or diseased condition. A standard necropsy includes an examination of external surfaces and orifices, extremities, body cavities, and internal organ/tissues. All of the treated vertebrae are collected and are examined for abnormalities. A brief morphologic description of all macroscopic abnormalities is recorded on individual necropsy forms.
Tissue Collection and Preservation
[0171] The following tissues and organs are obtained at sacrifice and are preserved in 10% neutral-buffered formalin (except for the eyes, which were preserved in Bouin's Solution for optimum fixation). Each tissue or organ specimen is then embedded in paraffin for preservation purposes and is archived at a sponsor-approved site or used to help determine the cause of death.
[0172] For all baboons, all treated and adjacent untreated vertebrae (T12 thru L6) are individually harvested en bloc including the spinal cord and spinal canal and are appropriately identified as to the treatment received. The T12 vertebral body is identified by leaving a minimum of 2 cm of the ribs attached to the bone. All bone specimens are placed in formalin fixative in preparation for plastic embedding.
[0173] Each of the soft tissues or organ specimens is embedded in paraffin for preservation purposes and are archived. A summary of the tissue samples to be collected is provided in Table 8.
TABLE-US-00008 TABLE 8 Summary of Tissue Samples to be Collected at Necropsy Cardiovascular Urogenital Aorta Kidneys Heart Urinary Bladder Digestive Ovaries Salivary Gland (mandibular) Uterus Tongue Cervix Esophagus Vagina Stomach Skin/MusculoskeIetal Small Intestine Skin/Mammary Gland (males and females) Duodenum Bone (femoral head) Jejunum Bone (7th rib) Ileum Skeletal Muscle (thigh) Large Intestine Knee joint Cecum Shoulder joint Colon Mandible Rectum Right Foot Pancreas Left Ankle Liver Right Hand Gallbladder Left Wrist Respiratory Spine from T12 to L7/Sacrum-separated Trachea Nervous/Special Sense Lung (including bronchi) Eyes with optic nerve Lymphoid/Hematopoietic Sciatic Nerve Bone Marrow (sternum) Brain Thymus Optic chiasm Spleen Cerebrum Lymph Nodes Cerebellum Axillary Medulla Mandibular Pons Mesenteric Spinal Cord (thoracic) Endocrine Other Adrenals Animal Number Tattoo Pituitary Gross Lesions Thyroid/Parathyroids* Lacrimal glands *The occasional absence of the parathyroid gland from the routine tissue section will not require a recut of the section.
[0174] Vertebral bodies injected with a composition comprising a rhPDGF-BB solution disposed in a .beta.-TCP/collagen matrix displayed the formation of normal bone with no adverse neurotoxic effects. Moreover, soft tissues adjacent to vertebral bodies receiving a composition comprising a rhPDGF-BB solution disposed in a .beta.-TCP/collagen matrix did not demonstrate abnormalities resulting from the administration of the rh-PDGF/matrix composition.
[0175] Furthermore, vertebral bodies injected with a composition comprising a rhPDGF-BB solution disposed in a .beta.-TCP/collagen matrix additionally displayed increased volumetric bone mineral densities. FIG. 4 illustrates percent change in volumetric bone mineral density (vBMD) for vertebral bodies of animals of Group I and Group II. Each data point in FIG. 4 represents an average of all vertebral bodies treated within each group. For example, the first data point in FIG. 4 for Group I is the average of nine vertebral body measurements (T12, L2, and IA for each of three animals in Group I) taken after injection of the rhPDGF-BB matrix composition into the vertebral bodies. Similarly, the first data point in FIG. 4 for Group 2 is the average of nine vertebral body measurements (T12, L2 and L4 for each of three animals in Group II) taken after injection of a collagen/.beta.-TCP matrix into the vertebral bodies.
[0176] As illustrated in FIG. 4, vertebral bodies treated with a composition comprising a rhPDGF-BB solution disposed in a .beta.-TCP/collagen matrix (Group I) demonstrated a steady increase in vBMD over the course of the study, the increase in vBMD becoming statistically significant over the 1 week post-operative level by the third month [2.64%+/-1.16 (week 1) v. 5.93%+/-1.33 (week 12); p=0.023]. vBMD continued to increase through the sixth month of the study before reaching a plateau at the ninth month. Vertebral bodies treated with a composition comprising 20 mM sodium acetate buffer disposed in the .beta.-TCP/collagen matrix (Group II), however, did not demonstrate significant increases in vBMD over the course of the study.
[0177] Additionally, FIG. 5 illustrates percent change in vBMD for vertebral bodies of animals of Group I and Group II wherein the injected .beta.-TCP/collagen matrix is subtracted from the volumetric bone mineral density analysis. As in FIG. 4, each data point in FIG. 5 represents an average of all vertebral bodies treated within each group.
[0178] As illustrated in FIG. 5, vertebral bodies treated with a rhPDGF-BB matrix composition (Group I) demonstrated increases in vBMD. The subtraction of the .beta.-TCP/collagen matrix from the volumetric bone mineral density analysis provided a clear indication that vBMD increased throughout all regions of the vertebral bodies of Group I as opposed to regions local to the injection site of the rhPDGF-BB matrix composition.
[0179] All patents, publications and abstracts cited above are incorporated herein by reference in their entirety. It should be understood that the foregoing relates only to preferred embodiments of the present invention and that numerous modifications or alterations may be made therein without departing from the spirit and the scope of the present invention as defined in the following claims.
* * * * *
D00000
D00001
D00002
D00003
D00004
D00005
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.