Separation Moieties And Methods Of Use Thereof
WINSTON; William ; et al.
U.S. patent application number 17/082955 was filed with the patent office on 2021-04-22 for separation moieties and methods of use thereof. The applicant listed for this patent is Werewolf Therapeutics, Inc.. Invention is credited to Vinay BHASKAR, Heather BRODKIN, Luke EVNIN, Daniel HICKLIN, Giselle KNUDSEN, Jose Andres SALMERON GARCIA, Cynthia SEIDEL-DUGAN, William WINSTON.
| Application Number | 20210115102 17/082955 |
| Document ID | / |
| Family ID | 1000005326084 |
| Filed Date | 2021-04-22 |








View All Diagrams
| United States Patent Application | 20210115102 |
| Kind Code | A1 |
| WINSTON; William ; et al. | April 22, 2021 |
SEPARATION MOIETIES AND METHODS OF USE THEREOF
Abstract
Provided herein are separation moieties that are suitable for use in conjunction with a variety of therapeutic payloads. The separation moieties serve to generate conditionally active macromolecules whereby the macromolecules have reduced or minimal biological activity until the separation moieties are modified under specific conditions.
| Inventors: | WINSTON; William; (West Newton, MA) ; EVNIN; Luke; (San Francisco, CA) ; BHASKAR; Vinay; (San Francisco, CA) ; KNUDSEN; Giselle; (San Anselmo, CA) ; HICKLIN; Daniel; (Montclair, NJ) ; SEIDEL-DUGAN; Cynthia; (Belmont, MA) ; SALMERON GARCIA; Jose Andres; (Westminster, MA) ; BRODKIN; Heather; (West Newton, MA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 1000005326084 | ||||||||||
| Appl. No.: | 17/082955 | ||||||||||
| Filed: | October 28, 2020 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| PCT/US2020/032988 | May 14, 2020 | |||
| 17082955 | ||||
| 62938786 | Nov 21, 2019 | |||
| 62847914 | May 14, 2019 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | C07K 2319/02 20130101; C07K 2319/30 20130101; C07K 2317/92 20130101; C07K 2317/73 20130101; C07K 2317/622 20130101; C07K 2319/03 20130101; C07K 14/57 20130101; C07K 14/7155 20130101; C07K 2319/50 20130101; C07K 2319/31 20130101; C07K 16/2887 20130101; C07K 14/55 20130101; A61K 2039/505 20130101 |
| International Class: | C07K 14/55 20060101 C07K014/55; C07K 14/715 20060101 C07K014/715; C07K 14/57 20060101 C07K014/57; C07K 16/28 20060101 C07K016/28 |
Claims
1-74. (canceled)
75. A polypeptide comprising Formula I: [D1]-[L1]-[D2], wherein D1 is a first domain of interest; L1 is a separation moiety that connects or links D1 to D2, wherein the separation moiety comprises an amino acid sequence selected from SEQ ID NOs: 195-220 or an amino acid sequence that has at least about 75% identity to SEQ ID NOs:195-220; and D2 is a second domain of interest.
76. The polypeptide of claim 1, wherein the separation moiety comprises an amino acid sequence that is a substrate for at least one protease present in a tumor microenvironment of a human tumor.
77. The polypeptide of claim 1, comprising two or more separation moieties, wherein the two or more separation moieties are each a substrate for a protease and, wherein each separation moiety independently comprises an amino acid sequence selected from SEQ ID NOs: 195-220 or an amino acid sequence that has at least about 75% identity to SEQ ID NOs:195-220.
78. The polypeptide of claim 1, further comprising a non-cleavable linker sequence.
79. The polypeptide of claim 1, wherein [D1] or [D2] comprises a cytokine, chemokine, growth factor, a soluble receptor, or a fragment thereof, or any combination thereof.
80. The polypeptide of claim 1, wherein the polypeptide comprises at least one of an extracellular domain, a transmembrane domain, and an intracellular domain.
81. The polypeptide of claim 1, wherein [D1] or [D2] comprises a cell surface receptor, a chimeric antigen receptor (CAR), or a T Cell Receptor (TCR) subunit, or a fragment thereof.
82. The polypeptide of claim 1, wherein [D1] or [D2] comprises an antigen-binding polypeptide, an antibody or an antigen-binding portion thereof.
83. The polypeptide of claim 1, wherein [D1] or [D2] comprises a half-life extension domain.
84. The polypeptide of claim 1, wherein the cleavable moiety is cleaved with either (a) greater catalytic efficiency, (b) greater specificity, or (c) both (a) and (b), by one or more proteases than a reference polypeptide sequence.
85. The polypeptide of claim 1, wherein the cleavable moiety is cleaved with reduced catalytic efficiency by one or more proteases than a reference polypeptide sequence.
86. The polypeptide of claim 1, wherein the cleavable moiety is cleaved with reduced catalytic efficiency by one or more serum proteases, one or more hepatic proteases, or a protease present in a normal healthy tissue.
87. A recombinant pro-protein comprising: a. a recombinant polypeptide comprising a cleavable moiety that is a substrate for a protease, wherein the cleavable moiety comprises an amino acid sequence selected from the group consisting of SEQ ID NOs: 195-220 or an amino acid sequence at least at least 75% identical to an amino acid sequence selected from the group consisting of SEQ ID NOs: 195-220; and b. a polypeptide with biological activity, wherein the pro-protein has attenuated biological activity, and wherein cleavage of the cleavable moiety by the protease produces a polypeptide with biological activity that is not attenuated.
88. The recombinant pro-protein of claim 87, wherein the polypeptide with biological activity comprises a cytokine, chemokine, growth factor, a soluble receptor or a combination thereof.
89. The recombinant pro-protein of claim 87, wherein the polypeptide with biological activity comprises at least one of an extracellular domain, a transmembrane domain, and an intracellular domain.
90. The recombinant pro-protein of claim 87, wherein the polypeptide with biological activity comprises a cell surface receptor, a chimeric antigen receptor (CAR), or a T Cell Receptor (TCR) subunit.
91. The recombinant pro-protein of claim 87, wherein the polypeptide with biological activity comprises an antigen-binding polypeptide, an antibody or an antigen-binding portion thereof.
92. A fusion protein comprising: a. a signaling protein or molecule; b. a blocking moiety selected from a steric blocking moiety, a specific blocking moiety, and the combination thereof; and c. a peptide linker that comprises a cleavable moiety that is a substrate for a protease, wherein the cleavable moiety comprises an amino acid sequence selected from the group consisting of SEQ ID NOs: 195-220 or an amino acid sequence at least at least 75% identical to an amino acid sequence selected from the group consisting of SEQ ID NOs: 195-220, wherein (a) and (b) are operably linked by (c).
93. The fusion protein of claim 92, wherein the steric blocking moiety comprises human serum albumin (HSA), an anti-HSA antibody, an immunoglobulin Fc, or a fragment thereof.
Description
[0001] The present application claims the benefit of U.S. Provisional Application No. 62/847,914 filed on May 14, 2019 and U.S. Provisional Application No. 62/938,786 filed on Nov. 21, 2019, each of which are incorporated herein by reference in their entireties.
1. SEQUENCE LISTING
[0002] The instant application contains a Sequence Listing which has been submitted electronically in ASCII format and is hereby incorporated by reference in its entirety. Said ASCII copy, created on May 14, 2020, is named 761146_000140_SL.txt and is 896,815 bytes in size.
2. BACKGROUND
[0003] Recombinant fusion proteins containing two or more functional polypeptides have utility in many fields including, for use in protein purification, imaging, as therapeutics, and drug delivery. For example, protein drugs can be fused to Fc domains of antibodies or to carrier proteins (i.e., human serum albumin) for targeting, to extend their plasma half-lives and/or to achieve therapeutic effects. Chen et al., (2013), Adv Drug Deliv Rev. 65(10):1357-1369.
[0004] Direct fusion of functional polypeptide or domains without a linker may lead to many undesirable outcomes, including misfolding of the fusion proteins (Zhao et al., (2008), Protein Expr. Purif., 61:73-77), low yield in protein production (Amet et al., (2009), Pharm. Res. 26:523-528), or impaired bioactivity (Bai et al., (2006) Proc. Nat Acad. Sci. USA, 102:7292-7296). One approach to overcome these difficulties is to use linker sequences between the component polypeptides or domains in a fusion protein. However, the selection of a suitable linker to join protein domains together can be complicated and is often neglected in the design of fusion proteins. Chen et al., (2013), Adv Drug Deliv Rev. 65(10):1357-1369. The properties of linker sequences, such as length, hydrophobicity, amino acid composition, secondary structures, and the overall folding can affect linker suitability and need to be taken into consideration in designing and selecting an appropriate linker. In addition, linkers that can be cleaved under selected conditions or in selected biological locations (e.g., in tumor microenvironment) to deliver active therapeutic agents (e.g., therapeutic polypeptides) can provide targeted pharmacological activity of the therapeutic agents and reduce unwanted systemic effects. This introduces further complexity into designing suitable linkers.
[0005] There is a need for improved liner sequences that can be used to prepare stable fusion proteins, including linkers that can be cleaved under selected conditions. Accordingly, novel separation moieties or linkers are disclosed herein. The separation moieties or linkers disclosed herein can be utilized, for example to specifically deliver prodrugs such as conditionally active and/or targeted cytokines to target sites where the linkers are processed to activate bioactivity.
3. SUMMARY
[0006] Provided herein are compositions and methods to generate and use high efficiency separation moieties and/or linkers. The linkers can confer site-selectivity with regards to biological activity of the attached payload or payloads. In some embodiments, the separation moieties and/or linkers are used in conjugation with therapeutic proteins to treat a disease or disorder, such as proliferative disease, a tumorous disease, an inflammatory disease, an immunological disorder, an autoimmune disease, an infectious disease, a viral disease, an allergic reaction, a parasitic reaction, graft-versus-host disease and the like.
[0007] Disclosed herein are recombinant polypeptides comprising a separation moiety, wherein the separation moiety comprises an amino acid sequence is a substrate for an enzyme, specifically a protease. The protease can be selected from the group consisting of Fibroblast activation protein alpha (FAP.alpha., also known as prolyl endopeptidase FAP), Cathepsin L (CTSL1), an ADAM selected from ADAM 8, ADAM 9, ADAM 10, ADAM12 ADAM17, and ADAMTS1, and an MMP selected from MMP1, MMP2, MMP9 or MMP14. The cleavable moiety can also be a substrate for a cathepsin, such as cathepsin B, cathepsin C, cathepsin D, cathepsin E, cathepsin G, cathepsin K and/or cathepsin L. Preferably, the cathepsin is Cathepsin L. Preferably, the protease is MMP14 or cathepsin L. The cleavable moiety can comprise an amino acid sequence that is a substrate for at least two proteases. In another embodiment, the separation moiety comprises two or more cleavable moieties, each of which is a substrate for a protease. The separation moiety can comprise a first cleavable moiety comprising a first amino acid sequence that is a substrate for a first protease and a second cleavable moiety comprising a second amino acid sequence that is a substrate for a second protease. In embodiments, the disclosure related to recombinant polypeptides that comprise a separation moiety that contains a protease cleavage motif as disclosed herein. The recombinant polypeptide can comprise a separation moiety that comprises an amino acid sequence selected from the group consisting of SEQ ID NOs: 195-220, or an amino acid sequence that has at least 90% identity to SEQ ID NOs:195-220. Preferred separation moieties comprise the sequence GPAGLYAQ (SEQ ID NO: 195) or ALFKSSFP (SEQ ID NO: 198). The disclosure also relates to functional variants of separation moieties comprising SEQ ID NOs: 195-220. The functional variants of SEQ ID NO: 195 can comprise any of SEQ ID NOS: 258-331. The functional variants of SEQ ID NO: 198 can comprise of SEQ ID NO:198 comprises SEQ ID NOS: 199 or any of SEQ ID NOS: 332-408. The separation moieties disclosed herein can comprise Formula I: [D1]-[L1]-[D2]. D1 is a first domain of interest. L1 is a separation moiety that connects or links D1 to D2, wherein the separation moiety comprises an amino acid sequence selected from SEQ ID NOs: 195-220 or an amino acid sequence that has at least about 90% identity to SEQ ID NOs:195-220. D2 is a second domain of interest.
[0008] In one embodiment, the recombinant polypeptide can further comprise a non-cleavable linker sequence. The recombinant polypeptide can include a therapeutic protein, such as a cytokine, chemokine, growth factor, soluble receptor, antigen-binding portion of an antibody (e.g., scFV, dAb) and the like. In another embodiment, the recombinant polypeptide comprises a cytokine, chemokine, growth factor, a soluble receptor, or any combination thereof. In another embodiment, the recombinant polypeptide comprises at least one of an extracellular domain, a transmembrane domain, and an intracellular domain. In one embodiment, the recombinant polypeptide comprises a cell surface receptor, a chimeric antigen receptor (CAR), or a T Cell Receptor (TCR) subunit. In one embodiment, the recombinant polypeptide comprises an antigen-binding polypeptide, an antibody or an antigen-binding portion thereof.
[0009] In one embodiment, the cleavable moiety is cleaved with either (a) greater catalytic efficiency or (b) greater specificity or (c) both (a) and (b), by one or more proteases than a reference polypeptide sequence. In another embodiment, the one or more proteases are selected from the group consisting of FAP.alpha., CTSL1, an ADAM selected from ADAM 8, ADAM 9, ADAM 10, ADAM12 ADAM17, and ADAMTS1, and an MMP selected from MMP1, MMP2, MMP9 and MMP14. The one or more proteases can also include a protease selected from cathepsins, such as cathepsin B, cathepsin C, cathepsin D, cathepsin E, cathepsin G, cathepsin K and/or cathepsin L. In another embodiment, the reference polypeptide sequence is present in a naturally occurring polypeptide substrate for FAP.alpha., CTSL1, an ADAM selected from ADAM 8, ADAM 9, ADAM 10, ADAM12 ADAM17, and ADAMTS1, and an MMP selected from MMP1, MMP2, MMP9 and MMP14, a cathepsin, such as cathepsin B, cathepsin C, cathepsin D, cathepsin E, cathepsin G, cathepsin K and/or cathepsin L or a combination thereof. In another embodiment, the cleavable moiety is cleaved with reduced catalytic efficiency by one or more proteases than a reference polypeptide sequence. In another embodiment the cleavable moiety is cleaved with reduced catalytic efficiency by one or more serum proteases. In another embodiment, the cleavable moiety is cleaved with reduced catalytic efficiency by one or more hepatic proteases. In another embodiment, the cleavable moiety is cleaved with reduced catalytic efficiency by one or more Factor Xa, hepsin, or thrombin.
[0010] In one embodiment, the recombinant polypeptide comprises two or more separation moieties. In one embodiment, the recombinant polypeptide is operably linked to a moiety selected from the group consisting of a polypeptide moiety, a lipid moiety, a nucleic acid moiety, a detectable moiety, and a small molecule.
[0011] Provided herein is a recombinant pro-protein comprising: a recombinant polypeptide comprising a cleavable moiety that is a substrate for a protease, wherein the cleavable moiety comprises an amino acid sequence selected from the group consisting of SEQ ID NOs: 195-220; and a polypeptide with biological activity, wherein the pro-protein has attenuated biological activity and wherein cleavage of the cleavable moiety by the protease produces a polypeptide with biological activity that is not attenuated. In one embodiment, the polypeptide with biological activity comprises a cytokine, chemokine, growth factor, a soluble receptor or a combination thereof. In some preferred aspects, the linkers are components of fusion proteins with therapeutic utility, and the linkers are not cleaved or are cleaved with low efficiency in the peripheral circulation but are cleaved with higher efficiency at a desired location in the body, such as a tumor microenvironment or site of inflammation. In another embodiment, the polypeptide with biological activity comprises at least one of an extracellular domain, a transmembrane domain, and an intracellular domain. In another embodiment, the polypeptide with biological activity comprises a cell surface receptor, a chimeric antigen receptor (CAR), or a T Cell Receptor (TCR) subunit. In another embodiment, the polypeptide with biological activity comprises an antigen-binding polypeptide, an antibody or an antigen-binding portion thereof.
[0012] The disclosure also relates to a fusion protein comprising: a. a signaling protein or molecule; and b. a blocking moiety selected from a steric blocking moiety, a specific blocking moiety, and the combination thereof; and c. a peptide linker that comprises a cleavable moiety, e.g., a cleavable moiety disclosed herein, having at least one protease-cleavable sequence. In one embodiment, the fusion protein, wherein (a) and (b) are operably linked by (c). In another embodiment, the fusion protein, wherein the peptide linker comprises two or more copies of the same cleavable moiety. In another embodiment, the fusion protein wherein the steric blocking moiety comprises human serum albumin (HSA) or an anti-HSA antibody. In another embodiment, the fusion protein wherein the signaling protein is an interleukin-2 amino acid sequence comprising: (i) a non-native N terminus and/or (ii) a non-native C terminus. In another embodiment, the fusion protein wherein the signaling protein is an interleukin-2 amino acid sequence. In another embodiment, the fusion protein further comprising one or more half-life extension domains that are not also a specific blocker. In another embodiment, the fusion protein wherein the non-native N and/or C termini are generated by circular permutation.
[0013] The fusion polypeptide provided herein can a first polypeptide fusion partner linked to a ligand by a protease cleavable linker, wherein the cleavable linker has been optimized for catalytic efficiency, and wherein the ligand has been optionally modified, wherein the first polypeptide fusion partner is a blocking moiety which prevents binding of the modified ligand to a target receptor or a subunit of a target receptor until cleavage of the protease cleavable linker.
[0014] The fusion polypeptide provided herein can also a fusion polypeptide comprising a first polypeptide fusion partner linked to a ligand by a protease cleavable linker, wherein the cleavable linker has been optimized for catalytic efficiency, and wherein the ligand has been optionally modified, including by circularly permutation to create a non-native N-terminus and a new C-terminus as compared to a native ligand, and wherein at least one of the new N-terminus or the new C-terminus of the modified ligand is operably linked to a first polypeptide fusion partner to form a fusion polypeptide wherein the first polypeptide fusion partner is a blocking moiety which prevents binding of the modified ligand to a target receptor or a subunit of a target receptor until cleavage of the protease cleavable linker.
[0015] In one embodiment, the first polypeptide fusion partner is selected from the group consisting of an antibody, an antibody fragment, and an albumin molecule. In another embodiment, the first polypeptide fusion partner further comprising a second polypeptide fusion partner comprising a second blocking moiety. In another embodiment, the second polypeptide fusion partner is a different kind of blocking moiety than the first polypeptide fusion partner. In another embodiment, the first polypeptide fusion partner is albumin and the second polypeptide fusion partner is a domain comprising a complementary amino acid sequence that blocks activity of the cytokine. In another embodiment, the first polypeptide fusion partner is a steric blocker, such as albumin, and the second polypeptide is a specific blocker, such as a cytokine receptor, portion of a cytokine receptor, a de novo affinity peptide specific for the cytokine, or an antibody or antibody fragment that specifically binds the cytokine of the fusion polypeptide. In another embodiment, the second polypeptide fusion partner is the same kind of blocking moiety as the first polypeptide fusion partner.
[0016] In one embodiment, the fusion protein further comprises a tumor antigen binding component. In another embodiment, the fusion protein further comprises a serum half-life extension domain. In another embodiment, the ligand is selected from the group consisting of helix bundle proteins and cytokines (including, but not limited to, growth hormone, IL-2, IL-4, IL-5, IL-6, IL-10, IL-22, IL-23p19, IL-11, IL-13, IL-15, IL-12p35, IL-21, IL-30 (IL27p28), IL-34, IL-35, IL-35p35, IFN-.alpha., IFN-.beta., IFN.gamma., LIF, CNTF, oncostatin M, CLCF-1, GCSF, GM-CSF, EPO, ferritin, leptin, placental lactogen, prolactin, apolipoprotein e), b-trefoil proteins (including, but not limited to, IL-1.alpha., IL-1.beta., IL-1Ra, IL18, IL-33, IL-36Ra, IL-36a, IL-36b, IL-36g, IL-37, IL-38, IL1Hy2, FGF-1, FGF-2, FGF-3, FGF-4, FGF-5, FGF-6, FGF-7, FGF-8a, FGF-8b, FGF-8e, FGF-8f, FGF-9, FGF-10, FGF-11, FGF-12, FGF-13, FGF-14, FGF-16, FGF-17, FGF-18, FGF-19, FGF-20, FGF-21, FGF-22, FGF-23), .alpha./.beta. (TIM) barrel proteins (including, but not limited to, triosephosphate isomerase), beta sandwich proteins (including, but not limited to, galectin-1, galectin-3, TNF-beta, seven .beta.-propeller proteins, class 1 MHC .alpha.1.alpha.2 domain, integrin I domain, GYF domain, C1 domain, C2 domain (for example, from cPLA2, PKC, synaptotagmin), PDZ domains, C3d, C5a. In one embodiment, wherein the ligand comprises IL-2 polypeptide or a fragment or fragments thereof. In another embodiment, the protease-cleavable linker polypeptide comprises a sequence that is capable of being cleaved by at least one protease selected from the group consisting of a kallikrein, thrombin, chymase, carboxypeptidase A, cathepsin G, an elastase, a FAP, an ADAM selected from ADAM 8, ADAM 9, ADAM 10, ADAM12 ADAM17, and ADAMTS1, PR-3, granzyme M, a calpain, a matrix metalloproteinase (MMP), a plasminogen activator, a cathepsin, a caspase, a tryptase, and a tumor cell surface protease. In another embodiment, the cytokine or fragment or mutein thereof is substantially dissociated from the cytokine blocking moiety after the protease-cleavable polypeptide linker is cleaved by a protease.
[0017] Disclosed herein are fusion polypeptide comprising at least one of each of: a cytokine polypeptide or functional fragment or mutein thereof [A]; a cytokine blocking moiety [B]; and an optimized protease-cleavable polypeptide linker [L]; wherein the blocking moiety is selected from the group consisting of an antibody, an antibody fragment, and an albumin, and wherein the cytokine comprises a circularly permuted cytokine. In some embodiments, the fusion protein further comprises a tumor antigen binding component and/or a serum half-life extension domain. In some embodiment, the fusion polypeptide wherein the cytokine peptide or functional fragment or mutein thereof is selected from the group consisting of helix bundle proteins and cytokines (including, but not limited to, growth hormone, IL-2, IL-4, IL-5, IL-6, IL-10, IL-22, IL-23p19, IL-11, IL-13, IL-15, IL-12p35, IL-21, IL-30 (IL27p28), IL-34, IL-35, IL-35p35, IFN-.beta., IFN.gamma., LIF, CNTF, oncostatin M, CLCF-1, GCSF, GM-CSF, EPO, ferritin, leptin, placental lactogen, prolactin, apolipoprotein e), a FAP (e.g., Fap.alpha.), an ADAM selected from ADAM 8, ADAM 9, ADAM 10, ADAM12 ADAM17, and ADAMTS1, b-trefoil proteins (including, but not limited to, IL-1.alpha., IL-1.beta., IL-1Ra, IL18, IL-33, IL-36Ra, IL-36a, IL-36b, IL-36g, IL-37, IL-38, IL1Hy2, FGF-1, FGF-2, FGF-3, FGF-4, FGF-5, FGF-6, FGF-7, FGF-8a, FGF-8b, FGF-8e, FGF-8f, FGF-9, FGF-10, FGF-11, FGF-12, FGF-13, FGF-14, FGF-16, FGF-17, FGF-18, FGF-19, FGF-20, FGF-21, FGF-22, FGF-23), .alpha./.beta. (TIM) barrel proteins (including, but not limited to, triosephosphate isomerase), beta sandwich proteins (including, but not limited to, galectin-1, galectin-3, TNF-.beta., seven .beta.-propeller proteins, class 1 MHC .alpha.1.alpha.2 domain, integrin I domain, GYF domain, C1 domain, C2 domain (for example, from cPLA2, PKC, synaptotagmin), PDZ domains, C3d, C5a. In one embodiment, the cytokine peptide or functional fragment or mutein thereof comprises IL-2. In another embodiment, the cytokine blocking moiety comprises a ligand binding domain or fragment or mutein of a cognate receptor for the cytokine, a single domain antibody or scFv that binds the cytokine polypeptide or functional fragment or mutein thereof, or an antibody or antibody fragment that binds a receptor of the cytokine. In another embodiment, antibody is a single domain antibody or scFv. In another embodiment, the blocking moiety extends the serum half-life of the cytokine or fragment thereof.
[0018] Disclosed herein are fusion polypeptides comprising a protease cleavable moiety, wherein the sequence is catalytically optimized for cleavage by certain proteases and wherein protease cleavage renders the composition inducible in a tumor microenvironment. In one embodiment, the fusion protein further comprises a biologically inactive polypeptide, wherein cleavage of the cleavable moiety by the protease converts the biologically inactive polypeptide to a biologically active polypeptide. In one embodiment, the biologically inactive polypeptide comprises a cytokine, chemokine, growth factor, or soluble receptor. In another embodiment, the biologically inactive polypeptide comprises at least one of an extracellular domain, a transmembrane domain, and an intracellular domain. In another embodiment, the biologically inactive polypeptide comprises a cell surface receptor, a chimeric antigen receptor (CAR), or a T Cell Receptor (TCR) subunit. In another embodiment, the biologically inactive polypeptide comprises an antigen-binding polypeptide, an antibody or an antigen-binding portion thereof.
[0019] The disclosure further relates to a nucleic acid encoding any of the polypeptides disclosed herein, a vector comprising any of the nucleic acids encoding any of the polypeptides disclosed herein, and a host cell comprising said vector.
[0020] Methods of making a pharmaceutical composition, comprising culturing the host cell comprising a vector comprising a nucleic acid encoding any of the polypeptides disclosed herein, under suitable conditions for expression and collection of desired polypeptides are provided herein. Method of using any of the polypeptides disclosed herein comprising administering an effective amount of a pharmaceutical composition comprising such polypeptides to a subject in need thereof are described. For example, use for treating a subject with a disease or disorder disclosed herein.
[0021] Provided herein are pharmaceutical compositions comprising an effective amount of any of the recombinant polypeptides, any of the pro-proteins, any of the fusion proteins, any of the fusion polypeptides, any of the nucleic acids, any of the vectors, or any of the host cells comprising such vectors disclosed herein. For example, a pharmaceutical composition for treating a subject with a disease or disorder disclosed herein.
[0022] Also disclosed is the use of the recombinant polypeptides, pro-proteins, fusion proteins, fusion polypeptides, nucleic acids, vectors, or host cells comprising such vectors disclosed herein for the manufacture of a medicament for treating a disease or disorder disclosed herein.
4. BRIEF DESCRIPTION OF THE DRAWINGS
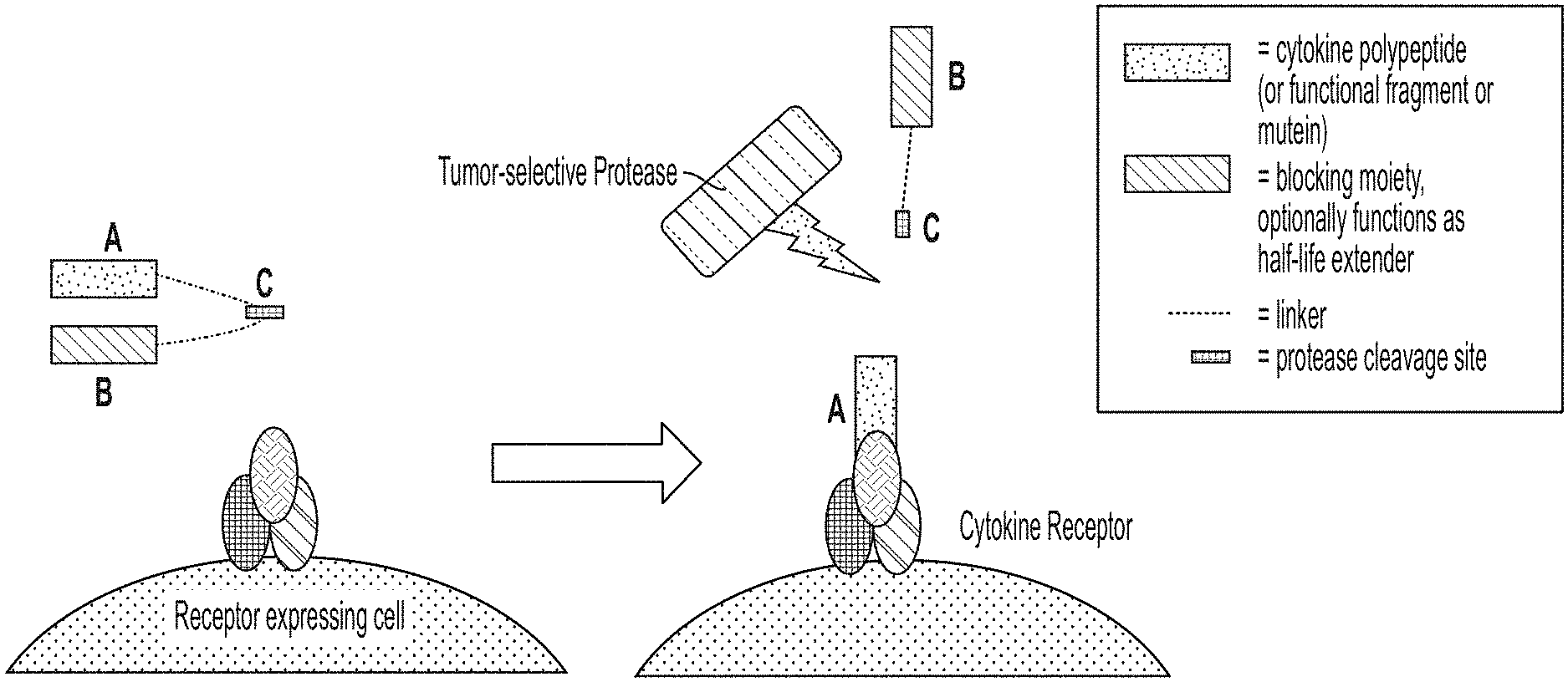
[0023] FIG. 1A is a schematic illustrating a protease-activated cytokine or chemokine that includes a blocking moiety. The blocking moiety may optionally function as a serum half-life extending domain. To the left of the arrow the drawing shows that a cytokine is connected to a blocking moiety via a protease-cleavable linker, thus blocking its ability to bind to its receptor. To the right of the arrow the drawing shows that in an inflammatory or tumor environment a protease cleaves at a protease-cleavage site on the linker, releasing the blocking moiety and allowing the cytokine to bind to its receptor.
[0024] FIG. 1B is a schematic illustrating a protease-activated cytokine or chemokine wherein HSA (blocking moiety) is directly bound to the cytokine or chemokine of interest, with a protease cleavage site between the HSA and a cytokine or chemokine of interest. To the left of the arrow the drawing shows that a cytokine is connected to a blocking moiety via a protease-cleavable linker, thus blocking its ability to bind to its receptor. To the right of the arrow the drawing shows that in an inflammatory or tumor environment, the protease cleaves at a protease-cleavage site on linker, releasing the blocking moiety and allowing the cytokine to bind to its receptor.
[0025] FIG. 1C is a schematic illustrating a protease-activated cytokine or chemokine wherein more than one HSA (blocking moiety) is bound directly to the molecule of interest. If desired, one or more of the HSA can be bonded to the cytokine or chemokine through a linker, such as a linker that contains a protease cleavage site. To the left of the arrow the drawing shows that a cytokine is connected to a blocking moiety via a protease-cleavable linker, thus blocking its ability to bind to its receptor. To the right of the arrow the drawing shows that in an inflammatory or tumor environment, protease cleaves at protease-cleavage site on linker, releasing the blocking moiety and allowing cytokine to bind receptor. The cytokine now has similar pK properties as compared to the native cytokine (e.g., has a short half-life).
[0026] FIG. 1D is a schematic illustrating a protease-activated cytokine or chemokine comprising more than one cytokine, of the same type or different type, each of which is bonded to a binding domain through a protease-cleavable linker. To the left of the arrow the drawing shows that a cytokine is connected to a blocking moiety via a protease-cleavable linker, thus blocking its ability to bind to its receptor. To the right of the arrow the drawing shows that in an inflammatory or tumor environment a protease cleaves at a protease cleavage site on linker, releasing the blocking moiety and allowing the cytokine to bind to its receptor.
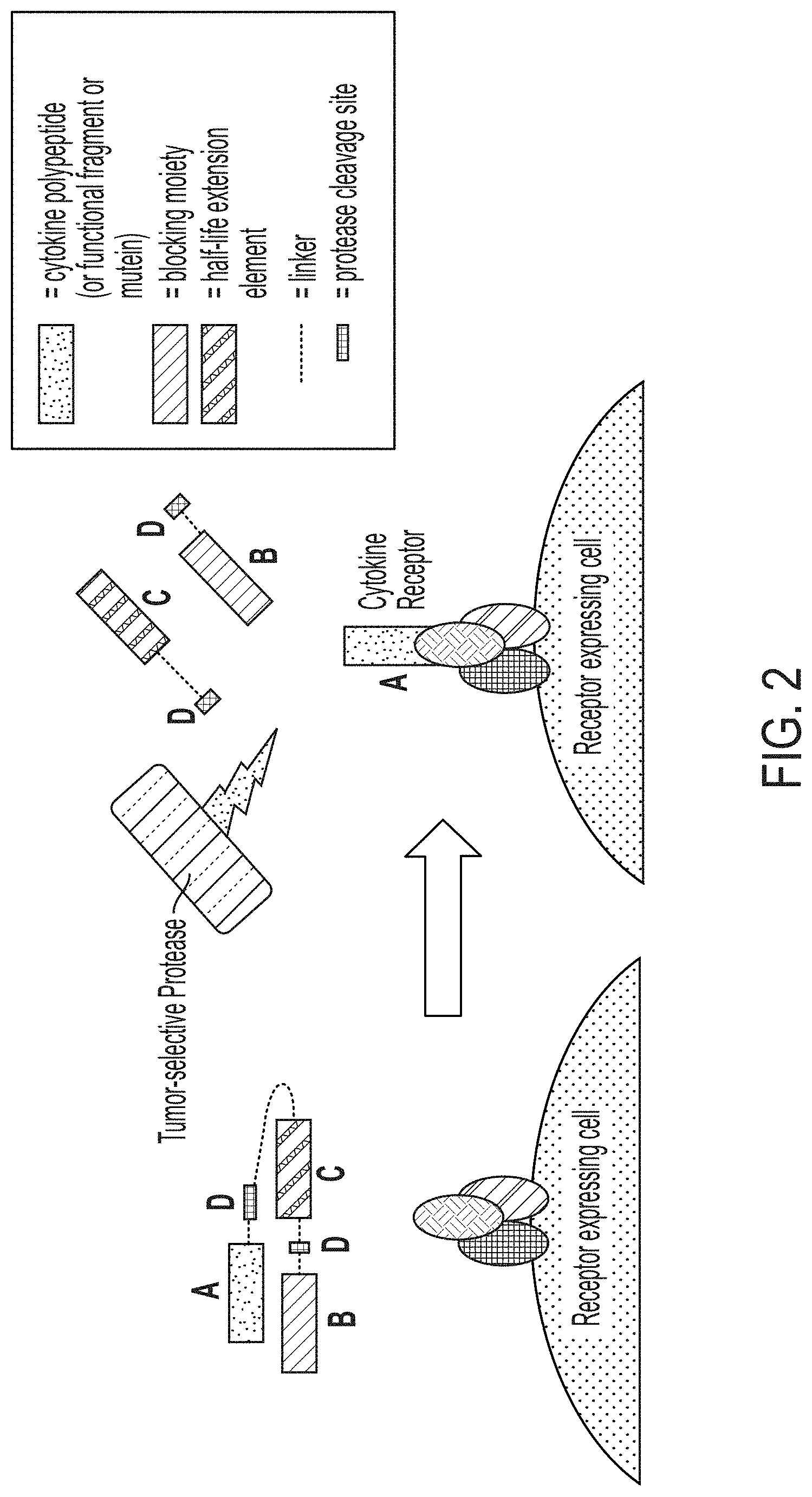
[0027] FIG. 2 is a schematic illustrating a protease-activated cytokine or chemokine comprising a cytokine or chemokine polypeptide, a blocking moiety, and a serum half-life extending domain connected by at least one protease-cleavable linker. To the left of the arrow the drawing shows that a cytokine is connected to a blocking moiety via protease-cleavable linkers, thus blocking its ability to bind to its receptor. It is also bound to a separate half-life extension element, which extends half-life in serum. To the right of the arrow the drawing shows that in an inflammatory or tumor environment a protease cleaves at a protease-cleavage site on linker, thus releasing the serum half-life extension element and the blocking moiety and allowing the cytokine to bind to its receptor. The cytokine now has similar pK properties as compared to the native cytokine (e.g., a short half-life).
[0028] FIG. 3 is a schematic illustrating a protease-activated cytokine or chemokine comprising a cytokine or chemokine polypeptide, a blocking moiety, and a targeting domain connected by at least one protease-cleavable linker. To the left of the arrow the drawing shows that a cytokine is connected to a blocking moiety and a targeting domain via a protease-cleavable linker, thus blocking its ability to bind to its receptor. To the right of the arrow the drawing shows that in an inflammatory or tumor microenvironment a protease cleaves at the protease cleavage site in the linker, releasing the targeting domain and the blocking moiety and allowing the cytokine to bind to its receptor.
[0029] FIG. 4A is a schematic illustrating a protease-activated cytokine or chemokine comprising a cytokine or chemokine polypeptide, a blocking moiety, a targeting domain, and a serum half-life extending domain connected by at least one protease-cleavable linker, wherein the cytokine polypeptide and the targeting domain are connected by a protease-cleavable linker. To the left of the arrow, the drawing shows that a cytokine or chemokine is connected to targeting domain, blocking moiety, and half-life extension element via protease-cleavable linker(s), thus blocking its ability to bind to its receptor. To the right of the arrow the drawing shows that in an inflammatory or tumor environment, the protease cleaves at a protease-cleavage site on linker(s), releasing the half-life extension element, the targeting domain, and the blocking moiety, and allowing the cytokine to bind to its receptor. The cytokine now has similar pK properties as compared to the native cytokine (e.g., short half-life).
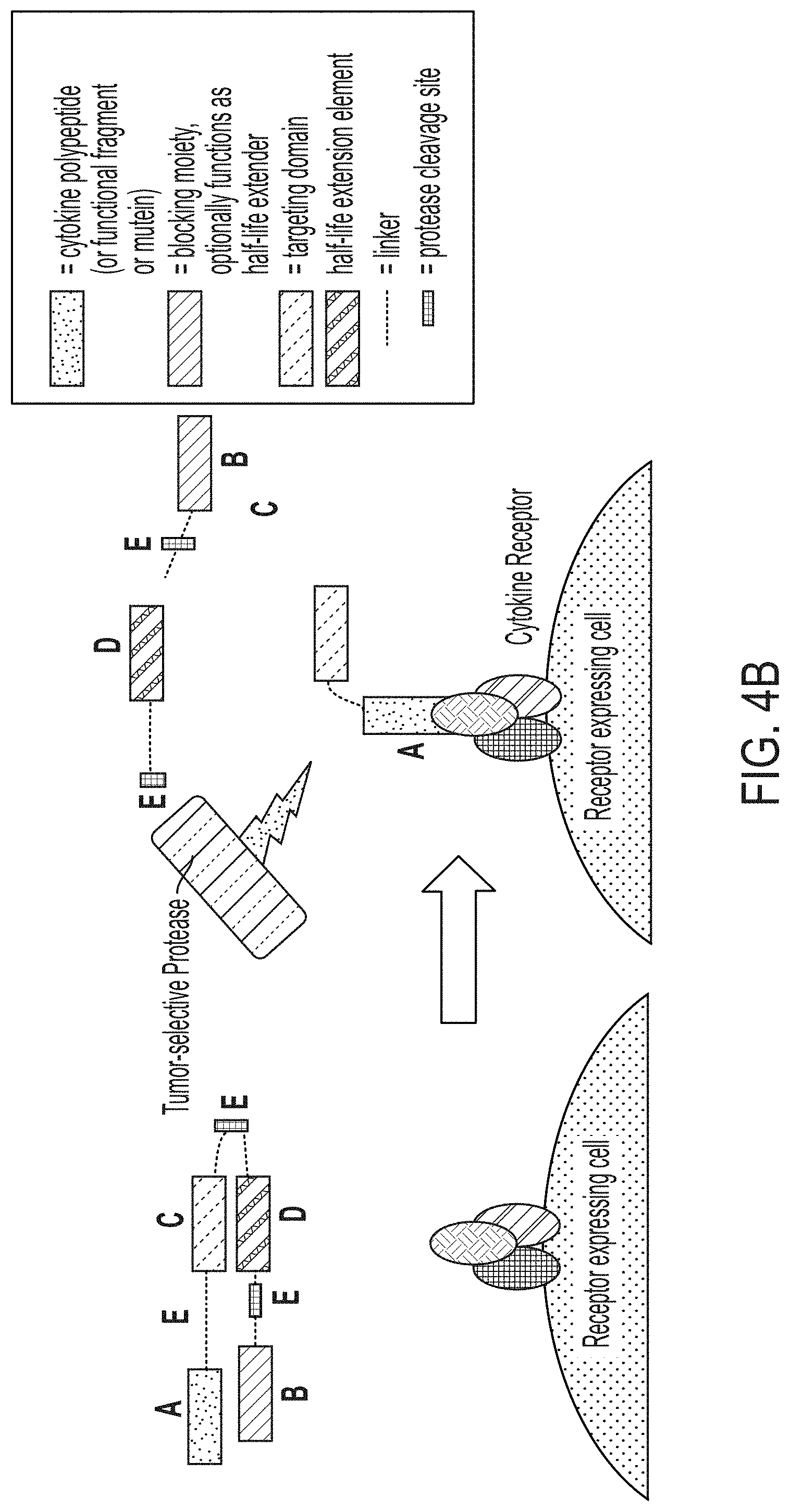
[0030] FIG. 4B is a schematic illustrating a protease-activated cytokine or chemokine comprising a cytokine or chemokine polypeptide, a blocking moiety, a targeting domain, and a serum half-life extending domain connected by at least one protease-cleavable linker. To the left of the arrow, the drawing shows that a cytokine is connected to targeting domain, a blocking moiety, and a half-life extension element via protease-cleavable linker(s), thus blocking its ability to bind to its receptor. To the right of the arrow the drawing shows that in an inflammatory or tumor environment, the protease cleaves at a protease-cleavage site on linker(s), releasing the half-life extension element and the blocking moiety and allowing the cytokine to bind to the receptor. The targeting moiety remains bound, keeping the cytokine in the tumor microenvironment. The cytokine now has similar pK properties as compared to the native cytokine (e.g., a short half-life).
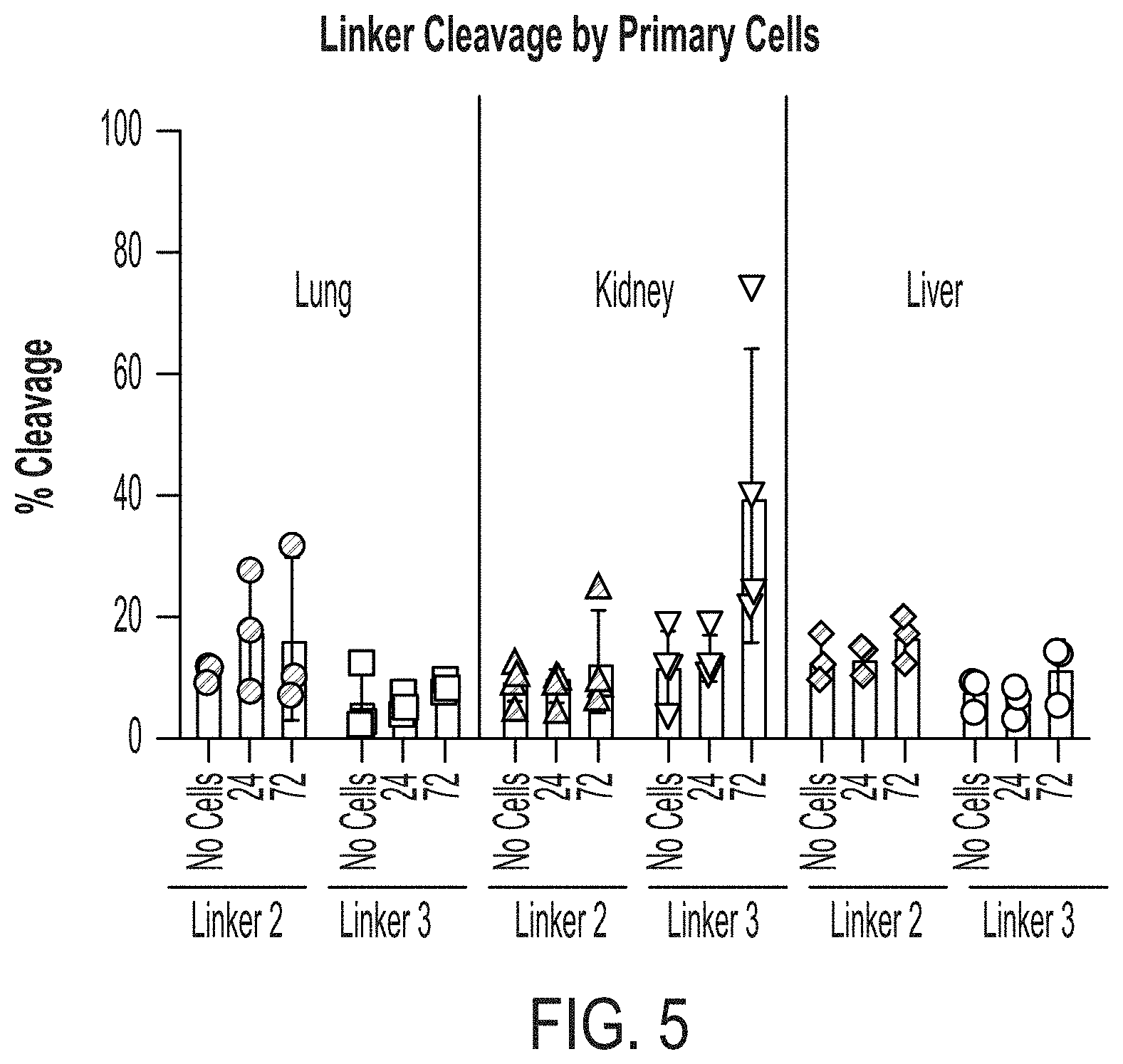
[0031] FIG. 5 depicts a graph showing that Linkers-2 (GPAGLYAQ, SEQ ID NO: 195) and Linkers-3 (ALFKSSFP, SEQ ID NO: 198) are minimally cleaved in lung, kidney, and livery cells.
[0032] FIGS. 6A-6B show graphs that polypeptides containing recombinant human IL-2 and the sequence for Linker-1 (GPAGMKGL, SEQ ID NO: 196), Linker-2 (GPAGLYAQ, SEQ ID NO: 195), or Linker-3 (ALFKSSFP, SEQ ID NO: 198) are not processed by healthy lung fibroblasts.
[0033] FIGS. 7A-7H is a series of graphs showing activity of exemplary IL-2 fusion proteins in IL-2 dependent cytotoxic T lymphocyte cell line CTLL-2. Each graph shows results of the IL-2 proliferation assay as quantified by CellTiter-Glo.RTM. (Promega) luminescence-based cell viability assay. Each proliferation assay was performed with HSA (FIGS. 7B, 7D, 7F, 7H) or without (FIGS. 7A, 7C, 7E, 7G). Each fusion protein comprises an anti-HSA binder, and both uncleaved and MMP9 protease cleaved versions of the fusion protein were used in each assay.
[0034] FIGS. 8A-8F is a series of graphs showing activity of exemplary IL-2 fusion proteins in IL-2 dependent cytotoxic T lymphocyte cell line CTLL-2. Each graph shows results of the IL-2 proliferation assay as quantified by CellTiter-Glo (Promega) luminescence-based cell viability assay. Both uncleaved and MMP9 protease cleaved versions of the fusion protein were used in each assay.
[0035] FIGS. 9A-9Z is a series of graphs showing activity of exemplary IL-2 fusion proteins in IL-2 dependent cytotoxic T lymphocyte cell line CTLL-2. Each graph shows results of the IL-2 proliferation assay as quantified by CellTiter-Glo (Promega) luminescence-based cell viability assay. Both uncleaved and MMP9 protease cleaved versions of the fusion protein were used in each assay.
[0036] FIG. 10 shows results of protein cleavage assay. Fusion protein ACP16 was run on an SDS-PAGE gel in both cleaved and uncleaved form. As can be seen in the gel, cleavage was complete.
[0037] FIGS. 11A-11B are a series of graphs depicting results from a HEK-Blue IL-12 reporter assay performed on human p40/murine p35 IL12 fusion proteins and recombinant human IL12 (Rec hIL-12). Analysis was performed based on quantification of Secreted Alkaline Phosphatase (SEAP) activity using the reagent QUANTI-Blue.RTM. (InvivoGen). Results confirm that IL12 protein fusion proteins are active.
[0038] FIGS. 12A-12F show a series of graphs depicting the results of HEK-blue assay of four IL-12 fusion proteins, before and after cleavage by MMP9. Analysis was performed based on quantification of Secreted Alkaline Phosphatase (SEAP) activity using the reagent QUANTI-Blue (InvivoGen). The data show greater activity in the cleaved IL12 than in the full fusion protein. Constructs tested were ACP06 (FIG. 12A), ACP07 (FIG. 12C), ACP08 (FIG. 12B), ACP09 (FIG. 12D), ACP10 (FIG. 12E), ACP11 (FIG. 12F)
[0039] FIG. 13 shows results of protein cleavage assay. Fusion protein ACP11 was run on an SDS-PAGE gel in both cleaved and uncleaved form. As can be seen in the gel, cleavage was complete.
[0040] FIG. 14 is a schematic which depicts a non-limiting example of an inducible cytokine protein, wherein the construct is activated upon protease cleavage of a linker attached between two subunits of the cytokine.
[0041] FIGS. 15A-15D are graphs depicting results from a HEK-Blue assay performed on human p40/murine p35 IL12 fusion proteins and recombinant human IL12 (Rec hIL-12). Results confirm that IL12 protein fusion proteins are active. Each proliferation assay was performed with HSA or without HSA.
[0042] FIGS. 16A-16F are a series of graphs showing activity of exemplary IFN.gamma. fusion proteins compared to activity of mouse IFN.gamma. control using WEHI 279 cell survival assay. Each assay was performed with medium containing HSA (+HSA) or not containing HSA (-HSA). Each fusion protein comprises an anti-HSA binder, and both uncleaved and MMP9 protease cleaved versions of the fusion protein were used in each assay.
[0043] FIGS. 17A-17F are a series of graphs showing activity of exemplary IFN.gamma. fusion proteins compared to activity of mouse IFN.gamma. control using B16 reporter assay. Each assay was performed with medium containing HSA (+HSA) or not containing HSA (-HSA). Each fusion protein comprises an anti-HSA binder, and both uncleaved and MMP9 protease cleaved versions of the fusion protein were used in each assay.
[0044] FIGS. 18A-18B shows results of protein cleavage assay. Two constructs, ACP31 (IFN-a fusion protein; FIG. 18A) and ACP55 (IFN-.gamma. fusion protein; 18B), were run on an SDS-PAGE gel in both cleaved and uncleaved form. As can be seen in the gel, cleavage was complete.
[0045] FIGS. 19A-19B are a series of graphs showing activity of exemplary IFN fusion proteins compared to activity of mouse IFN.gamma. control using B16 reporter assay. Each assay was performed with culture medium containing HSA, and each fusion protein comprises an anti-HSA binder. Both uncleaved and MMP9 protease cleaved versions of the fusion protein were used in each assay.
[0046] FIGS. 20A-20B are a series of graphs showing activity of exemplary IFN.alpha. fusion proteins compared to activity of mouse IFNalphaA control using a B16 reporter assay. Each assay was performed with medium containing HSA, and each fusion protein comprises an anti-HSA binder. Both uncleaved and MMP9 protease cleaved versions of the fusion protein were used in each assay.
[0047] FIGS. 21A-21D are a series of graphs depicting the results of tumor growth studies using the MC38 cell line. FIG. 21A-21C show the effect of IFN.gamma. and IFN.gamma. fusion proteins on tumor growth when injected intraperitoneally (IP) using different dosing levels and schedules (.mu.g=micrograms, BID=twice daily, BIW=twice weekly, QW=weekly). FIG. 21D shows the effect of intratumoral (IT) injection of IFN.gamma. and IL-2 on tumor growth.
[0048] FIGS. 22A-22B are a series of graphs showing activity of exemplary IFN.gamma. fusion proteins (ACP51 and ACP52) cleaved by MMP9 protease compared to activity of uncleaved fusion proteins using B16 reporter assay. Each fusion protein comprises an anti-HSA binder and a tumor targeting domain.
[0049] FIGS. 23A-23B are a series of graphs showing activity of exemplary IFN.gamma. fusion proteins (ACP53 and ACP54) cleaved by MMP9 protease compared to activity of uncleaved fusion proteins using B16 reporter assay. Each fusion protein comprises IFN.gamma. directly fused to albumin.
[0050] FIGS. 24A-24B are two graphs showing the stability of IL-2 fusion proteins containing Linker-1 (GPAGMKGL, SEQ ID NO: 196), Linker-2 (GPAGLYAQ, SEQ ID NO: 195), or Linker-3 (ALFKSSFP, SEQ ID NO: 198) in human serum in normal patient and a cancer patient. FIG. 24A depicts the stability of the IL-2 fusion proteins containing Linker-1 (GPAGMKGL, SEQ ID NO: 196), Linker-2 (GPAGLYAQ, SEQ ID NO: 195), or Linker-3 (ALFKSSFP, SEQ ID NO: 198) at 24 hours. FIG. 24B depicts the stability of the IL-2 fusion proteins containing Linker-1 (GPAGMKGL, SEQ ID NO: 196), Linker-2 (GPAGLYAQ, SEQ ID NO: 195), or Linker-3 (ALFKSSFP, SEQ ID NO: 198) at 72 hours.
[0051] FIGS. 25A and 25B are two graphs showing analysis of ACP16 (FIG. 25A) and ACP124 (FIG. 25B) in a HEKBlue IL-2 reporter assay in the presence of HSA. Circles depict the activity of the uncut polypeptide, squares depict activity of the cut polypeptide, and triangles depict IL-2 alone as a control. FIG. 25C is a graph showing results of a CTLL-2 proliferation assay. CTLL2 cells (ATCC) were plated in suspension at a concentration of 500,000 cells/well in culture media with or without 40 mg/ml human serum albumin (HSA) and stimulated with a dilution series of recombinant hIL2 or activatable hIL2 for 72 hours at 37.degree. C. and 5% CO.sub.2. Activity of uncleaved and cleaved activatable ACP16 was tested. Cleaved activatable hIL2 was generated by incubation with active MMP9 Cell activity was assessed using a CellTiter-Glo (Promega) luminescence-based cell viability assay. Triangles show wile-type cytokine, circles depict intact fusion protein, and squares depict protease-cleaved fusion protein.
[0052] FIGS. 26A-26C are a series of graphs showing activity of fusion proteins in an HEKBlue IL-12 reporter assay. FIG. 26A is a graph showing activity of cut and uncut ACP11 (a human p40/murine p35 IL12 fusion protein). FIG. 26B is a graph showing analysis of ACP91 (a chimeric IL-12 fusion protein). Squares depict activity of the uncut ACP91 polypeptide, and triangles depict the activity of the cut polypeptide (ACP91+MMP9). EC50 values for each are shown in the table. FIG. 26C is a graph showing analysis of ACP136 (a chimeric IL-12 fusion protein). Squares depict activity of the uncut ACP136 polypeptide, and triangles depict the activity of the cut polypeptide (ACP136+MMP9). EC50 values for each are shown in the table insert.
[0053] FIGS. 27A-27F are a series of graphs showing that cleaved IL-12 polypeptides are active in a HEKBlue IL2 reporter assay. Fusion proteins are evaluated both uncut (circles) and cut (squares) form, and wild type IL2 is used as a control +HSA for FIGS. 27A-C; ACP131 is used as a control (triangles) for FIGS. 27D-27F. Shown are data for APC31+HSA (FIG. 27A), ACP125+HSA (FIG. 27B), ACP126+HSA (FIG. 27C), ACP127 (FIG. 27D), ACP128 (FIG. 27E), and ACP129 (FIG. 27F). The EC50 values for each are shown in the table below each graph.
[0054] FIGS. 28A-28N are a series of graphs depicting the activity of APC56 (FIG. 28A), APC57 (FIG. 28B) APC58 (FIG. 28C), APC59 (FIG. 28D), APC60 (FIG. 28E), APC61+HSA (FIG. 28F), ACP30+HSA (FIG. 28G), ACP73 (FIG. 28H), ACP70+HSA (FIG. 28I), ACP71 (FIG. 28J), ACP72 (FIG. 28K), ACP 73 (FIG. 28L), ACP74 (FIG. 28M), and ACP75 (FIG. 28N) in a HEKBlue IFN.alpha. reporter assay. Each fusion was tested for its activity when cut (squares) and uncut (circles). Analysis of murine IFN.gamma. is included in each graph as a comparator.
[0055] FIGS. 29A-29B is two graphs showing results of analyzing ACP31 (mouse IFN.alpha.1 fusion protein) and ACP11 (a human p40/murine p35 IL12 fusion protein) in a tumor xenograft model. FIG. 29A shows tumor volume over time in mice treated with 33 .mu.g ACP31 (circles), 110 .mu.g ACP31 (triangles), 330 .mu.g ACP31 (diamonds), and as controls 1 .mu.g murine wild type IFN.alpha.1 (dashed line, squares) and 10 .mu.g mIFN.alpha.1 (dashed line, small circles). Vehicle alone is indicated by large open circles. The data show tumor volume decreasing over time in a dose-dependent manner in mice treated with ACP31. FIG. 29B shows tumor volume over time in mice treated with 17.5 .mu.g ACP11 (squares), 175 .mu.g ACP31 (triangles), 525 .mu.g ACP31 (circles), and as controls 2 .mu.g ACP04 (dashed line, triangles) and 10 .mu.g ACP04 (dashed line, diamonds). Vehicle alone is indicated by large open circles. The data show tumor volume decreasing over time in a dose-dependent manner in mice treated with both ACP11 and ACP04 (a human p40/murine p35 IL12 fusion protein).
[0056] FIGS. 30A-30F are a series of spaghetti plots showing tumor volume over time in a mouse xenograft tumor model in mice each treated with vehicle alone (FIG. 30A), 2 .mu.g ACP04 (FIG. 30B), 10 .mu.g ACP04 (FIG. 30C), 17.5 .mu.g ACP11 (FIG. 30D), 175 .mu.g ACP11 (FIG. 30E), and 525 .mu.g ACP11 (FIG. 30F). Each line represents a single mouse.
[0057] FIGS. 31A-31C depicts three graphs showing results of analyzing ACP16 and ACP124 in a tumor xenograft model. FIG. 31A shows tumor volume over time in mice treated with 4.4 .mu.g ACP16 (squares), 17 .mu.g ACP16 (triangles), 70 .mu.g ACP16 (downward triangles), 232 .mu.g ACP16 (dark circles), and as a comparator 12 .mu.g wild type IL-2 (dashed line, triangles) and 36 .mu.g wild type IL-2 (dashed line, diamonds. Vehicle alone is indicated by large open circles. The data show tumor volume decreasing over time in a dose-dependent manner in mice treated with ACP16 at higher concentrations. FIG. 31B shows tumor volume over time in mice treated with 17 .mu.g ACP124 (squares), 70 .mu.g ACP124 (triangles), 230 .mu.g ACP124 (downward triangles), and 700 .mu.g ACP124. Vehicle alone is indicated by large open circles. FIG. 31C shows tumor volume over time in mice treated with 17 .mu.g ACP16 (triangles), 70 .mu.g ACP16 (circles), 232 .mu.g ACP16 (dark circles), and as a comparator 17 .mu.g ACP124 (dashed line, triangles) 70 .mu.g ACP124 (dashed line, diamonds), 230 .mu.g ACP124 (dashed line, diamonds). Vehicle alone is indicated by dark downward triangles. The data show tumor volume decreasing over time in a dose-dependent manner in mice treated with ACP16, but not ACP124.
[0058] FIGS. 32A-32C are a series of spaghetti plots showing activity of fusion proteins in an MC38 mouse xenograft model. Each line in the plots is a single mouse.
[0059] FIG. 33 is a graph showing tumor volume over time in a mouse xenograft model showing tumor growth in control mice (open circles) and AP16-treated mice (squares).
[0060] FIGS. 34A-34D are a series of survival plots showing survival of mice over time after treatment with cleavable fusion proteins. FIG. 34A shows data for mice treated with vehicle alone (gray line), 17 .mu.g ACP16 (dark line), and 11 .mu.g ACP124 (dashed line). FIG. 34B shows data for mice treated with vehicle alone (gray line), 70 .mu.g ACP16 (dark line), and 70 .mu.g ACP124 (dashed line). FIG. 34C shows data for mice treated with vehicle alone (gray line), 232 .mu.g ACP16 (dark line), and 230 .mu.g ACP124 (dashed line). FIG. 34D shows data for mice treated with vehicle alone (gray line), 232 .mu.g ACP16 (dark line), and 700 .mu.g ACP124 (dashed line).
[0061] FIG. 35 a series of spaghetti plots showing activity of fusion proteins in an MC38 mouse xenograft model. All mouse groups were given four doses total except for the highest three doses of APC132, wherein fatal toxicity was detected after 1 week/2 doses. Shown are vehicle alone (top), 17, 55, 70, and 230 .mu.g ACP16 (top full row), 9, 28, 36, and 119 .mu.g ACP132 (middle full row), and 13, 42, 54, and 177 .mu.g ACP21 (bottom full row). Each line in the plots represents an individual animal.
[0062] FIG. 36 is a schematic illustrating a substrate cleavage activity in conditioned complete (+FBS) media by FRET endpoint assay across four cell lines. The ratio of tumor vs control activity was approximated by averaging the three tumor cell lines and comparing to the control myofibroblast cell line where signal was detectable. FIG. 36 discloses SEQ ID NOs: 201, 198, 197, 196 and 195, respectively, in order of appearance.
[0063] FIG. 37 is a schematic illustrating ADAM17_2 substrate kinetics in cell culture conditioned media. FIG. 37 discloses SEQ ID NO: 235.
[0064] FIG. 38 is a schematic illustrating FAP.alpha._1 substrate kinetics in conditioned media. FIG. 38 discloses SEQ ID NO: 197.
[0065] FIG. 39 is a schematic illustrating FAP.alpha._1 substrate kinetics in cell lysates. FIG. 39 discloses SEQ ID NO: 197.
[0066] FIG. 40 is a schematic illustrating MMP9_1 substrate kinetics in cell lysates. FIG. 40 discloses SEQ ID NO: 196.
[0067] FIG. 41 is a schematic illustrating a substrate cleavage activity in cell lysates by FRET endpoint assay. FIG. 41 discloses SEQ ID NOS 198 and 197, respectively, in order of appearance.
[0068] FIG. 42 is a schematic illustrating CTSL1_1 substrate kinetics in cell lysates. FIG. 42 discloses SEQ ID NO: 198.
[0069] FIG. 43 is a schematic illustrating MMP14_1 substrate kinetics in cell lysates. FIG. 43 discloses SEQ ID NO: 195.
[0070] FIG. 44 is a schematic illustrating calculated concentration of enzyme equivalents per cell culture-derived sample. FIG. 44 discloses SEQ ID NOS 201, 198, 197, 197, 196 and 195, respectively, in order of appearance.
[0071] FIG. 45 is a schematic illustrating an enzyme progress curve for CTSL1 cleavage of CSTL1_2 vs CTSL1_1. FIG. 45 discloses SEQ ID NOS 198, 199 and 236, respectively, in order of appearance.
[0072] FIG. 46 is a schematic illustrating 30-mer cleavage of CTSL1_1 (ALFKSSFP, SEQ ID NO: 198) vs CTSL1_2 (ALFFSSPP, SEQ ID NO: 199).
[0073] FIG. 47 is a schematic illustrating susceptibility of the CTSL1 FRET substrates to CTSK cleavage. Rates of product formation were measured as a specific activity in units pmol min.sup.-1 .mu.g.sup.-1. The threshold value for the reference substrate, Z-LR-AMC is shown as a dashed line. FIG. 47 discloses SEQ ID NOS 198 and 199, respectively, in order of appearance.
[0074] FIG. 48 is a schematic illustrating 30-mer Substrate Degradation by MMP9. Ranking of the substrates by relative rates of degradation are shown with "+"; uncleaved substrates are indicated as "-". FIG. 48 discloses SEQ ID NOS 204, 205, 214, 216, 202, 217, 203, 211, 219, 207, 215, 212, 213, 206, 208, 209, 210, 218 and 220, respectively, in order of appearance.
[0075] FIG. 49 is a schematic illustrating tandem MMP14_1 Motif degradation by MMP9. Top: substrate degradation traces, modeled with first-order kinetics. Bottom: product formation traces showing complex kinetics. FIG. 49 discloses SEQ ID NOS 202-205, respectively, in order of appearance.
[0076] FIG. 50 is a schematic illustrating 30-mer Substrate Degradation by FAP.alpha.. Ranking of the substrates by relative rates of degradation are shown with "+"; uncleaved substrates are indicated as "-". FIG. 50 discloses SEQ ID NOS 205, 204, 206, 217, 203, 218, 219, 213, 216, 207, 214, 210, 202, 211, 208, 209, 212, 215 and 220, respectively, in order of appearance.
[0077] FIG. 51 is a schematic illustrating 30-mer Substrate Degradation by CTSL1. Ranking of the substrates by relative rates of degradation are shown with "+"; uncleaved substrates are indicated as "-". FIG. 51 discloses SEQ ID NOS 207, 208, 202, 218, 219, 212, 215, 217, 211, 209, 214, 206, 213, 210, 216, 203, 204, 205 and 220, respectively, in order of appearance.
[0078] FIG. 52 is a schematic illustrating 30-mer Substrate Degradation by ADAM17. Ranking of the substrates by relative rates of degradation are shown with "+"; uncleaved substrates are indicated as "-". FIG. 52 discloses SEQ ID NOS 208, 209, 211, 214, 217, 219, 213, 218, 215, 210, 212, 216, 207, 206, 202, 203, 204, 205 and 220, respectively, in order of appearance.
[0079] FIG. 53 is a schematic illustrating 30-mer Substrate Degradation by Factor Xa. Ranking of the substrates by relative rates of degradation are shown with "+"; uncleaved substrates are indicated as "-". FIG. 53 discloses SEQ ID NOS 220, 206, 202, 214, 208, 209, 215, 210, 218, 217, 207, 213, 216, 211, 212, 219, 203, 204 and 205, respectively, in order of appearance.
[0080] FIG. 54 is a schematic illustrating 30-mer Substrate Degradation by Thrombin. Ranking of the substrates by relative rates of degradation are shown with "+"; uncleaved substrates are indicated as "-". FIG. 54 discloses SEQ ID NOS 220, 204, 202, 207, 205, 211, 212, 215, 209, 218, 219, 217, 210, 213, 216, 214, 208, 206 and 203, respectively, in order of appearance.
[0081] FIG. 55 is a schematic illustrating 30-mer Substrate Degradation by hepsin. Ranking of the substrates by relative rates of degradation are shown with "+"; uncleaved substrates are indicated as "-". FIG. 55 discloses SEQ ID NOS 220, 209, 216, 215, 210, 213, 206, 214, 212, 207, 217, 208, 211, 218, 219, 202, 203, 204 and 205, respectively, in order of appearance.
[0082] FIGS. 56A-56C show western blots probed with an IL-2 antibody demonstrating the stability of ACP16 (FIG. 56A), ACP153 (FIG. 56B), and ACP157 (FIG. 56C) in 90% serum. Serum was pooled from three human donors. Constructs of interest were incubated with PBS, Serum, or MMP9 protease and cleavage was assessed at T=0 hours and at T=24 hours.
[0083] FIG. 57A-57B show western blots probed with an IL-2 antibody demonstrating the stability of ACP153, ACP155, ACP156, ACP16, and ACP372 in 90% serum. Serum was pooled from three human donors. Constructs of interest were incubated with PBS, Serum, or MMP9 protease and cleavage was assessed at T=24 hours and at T=72 hours. FIG. 57A shows the result using serum from a human donor and FIG. 57B shows the result using serum from a mouse donor.
[0084] FIGS. 58A-58D show a series of spaghetti plots showing activity of fusion proteins in an MC38 mouse xenograft model. Shown are vehicle alone (FIG. 58A, top), 17, 55, and 230 .mu.g ACP16 (FIG. 58A), 55 and 230 .mu.g ACP153 (FIG. 58B), 55 and 230 .mu.g ACP155 (FIG. 58C), and 55 and 230 .mu.g ACP156 (FIG. 58D). Each line in the plots represents an individual animal.
[0085] FIG. 59 shows a graph depicting results from a STAT activation reporter assay performed on IL-2 fusion proteins and recombinant human IL2 (Rec hIL-2). Analysis was performed based on quantification of Secreted Alkaline Phosphatase (SEAP) activity using the reagent QUANTI-Blue (InvivoGen).
[0086] FIG. 60 shows a graph depicting results from a STAT activation reporter assay performed on IL-2 fusion proteins and recombinant human IL2 (Rec hIL-2). Analysis was performed based on quantification of Secreted Alkaline Phosphatase (SEAP) activity using the reagent QUANTI-Blue (InvivoGen).
[0087] FIG. 61 shows a graph depicting results from a STAT activation reporter assay performed on IL-2 fusion proteins and recombinant human IL2 (Rec hIL-2). Analysis was performed based on quantification of Secreted Alkaline Phosphatase (SEAP) activity using the reagent QUANTI-Blue (InvivoGen).
[0088] FIG. 62 shows a graph depicting results from a STAT activation reporter assay performed on IL-2 fusion proteins and recombinant human IL2 (Rec hIL-2). Analysis was performed based on quantification of Secreted Alkaline Phosphatase (SEAP) activity using the reagent QUANTI-Blue (InvivoGen).
[0089] FIG. 63 shows a table reporting engineered cleavage substrates described herein and the extent of cleavage observed using relevant proteases. Flanking sequences are lower case, the first cleavable sequence is underlined, the second is in bold font, and the third cleavable sequence is in italics. In some cases, there is overlap between cleavable sequences, which are indicated accordingly. FIG. 63 discloses SEQ ID NOS 202-220, respectively, in order of appearance.
[0090] FIG. 64 is a schematic of an inducible tetravalent antibody format.
[0091] FIG. 65A-65B show that multivalent 4-1BB antibodies are able to inducibly agonize 4-1BB.
5. DETAILED DESCRIPTION
[0092] This disclosure relates to novel separation moieties or linkers and to polypeptides, such as fusion proteins, that contain the linkers. The linkers are preferably protease cleavable and link a first amino acid sequence of interest (e.g. a first domain of interest) to a second amino acid sequence of interest (e.g. a second domain of interest).
[0093] The disclosed separation moieties confer site-selectivity with regard to the action of the attached payload or payloads. The payload can be a therapeutic agent, a half-life extender, a blocking agent and the like, or any combination thereof. The separation moieties may be used to attach any payload of interest, including e.g. cytokines, antibodies, cell-based therapies, etc. The separation moieties may be used individually or be used in tandem, triplicate, quadruplicate, and so forth, as long as the separation moiety is smaller than about 100 amino acids. Individual separation moieties may be directly joined to each other, or may be interspersed with non-cleavable linkers, whichever promotes high efficiency and site-specificity.
[0094] The various embodiments of the present disclosure are further described in detail in the paragraphs below.
[0095] Unless otherwise defined, all terms of art, notations and other scientific terminology used herein are intended to have the meanings commonly understood by those of skill in the art to which this invention pertains. In some cases, terms with commonly understood meanings are defined herein for clarity and/or for ready reference, and the inclusion of such definitions herein should not necessarily be construed to represent a difference over what is generally understood in the art. The techniques and procedures described or referenced herein are generally well understood and commonly employed using conventional methodologies by those skilled in the art, such as, for example, the widely utilized molecular cloning methodologies described in Sambrook et al., Molecular Cloning: A Laboratory Manual 4th ed. (2012) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. As appropriate, procedures involving the use of commercially available kits and reagents are generally carried out in accordance with manufacturer-defined protocols and conditions unless otherwise noted.
[0096] "Cytokine" is a well-known term of art that refers to any of a class of immunoregulatory proteins (such as interleukin or interferon) that are secreted by cells especially of the immune system and that are modulators of the immune system. Cytokine polypeptides that can be used in the fusion proteins disclosed herein include, but are not limited to transforming growth factors, such as TGF-.alpha. and TGF-.beta. (e.g., TGFbeta1, TGFbeta2, TGFbeta3); interferons, such as interferon-.alpha., interferon-.beta., interferon-.gamma., interferon-kappa and interferon-omega; interleukins, such as IL-1, IL-1.alpha., IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-11, IL-12, IL-13, IL-14, IL-15, IL-16, IL-17, IL-18, IL-21 and IL-25; tumor necrosis factors, such as tumor necrosis factor alpha and lymphotoxin; transforming growth factor beta (TGFbeta) family proteins, chemokines (e.g., C-X-C motif chemokine 10 (CXCL10), CCL19, CCL20, CCL21), and granulocyte macrophage-colony stimulating factor (GM-CS), as well as fragments of such polypeptides that active the cognate receptors for the cytokine (i.e., functional fragments of the foregoing). "Chemokine" is a term of art that refers to any of a family of small cytokines with the ability to induce directed chemotaxis in nearby responsive cells.
[0097] Cytokines are well-known to have short serum half-lives that frequently are only a few minutes. Even forms of cytokines that have altered amino acid sequences intended to extend the serum half-life yet retain receptor agonist activity typically also have short serum half-lives. As used herein, a "short-half-life cytokine" refers to a cytokine that has a substantially brief half-life circulating in the serum of a subject, such as a serum half-life that is less than 10, less than 15, less than 30, less than 60, less than 90, less than 120, less than 240, or less than 480 minutes. As used herein, a short half-life cytokine includes cytokines which have not been modified in their sequence to achieve a longer than usual half-life in the body of a subject and polypeptides that have altered amino acid sequences intended to extend the serum half-life yet retain receptor agonist activity. This latter case is not meant to include the addition of heterologous protein domains, such as a bona fide half-life extension element, such as serum albumin.
[0098] A "conservative" amino acid substitution, as used herein, generally refers to substitution of one amino acid residue with another amino acid residue from within a recognized group which can change the structure of the peptide but biological activity of the peptide is substantially retained. Conservative substitutions of amino acids are known to those skilled in the art. Conservative substitutions of amino acids can include, but not limited to, substitutions made amongst amino acids within the following groups: (a) M, I, L, V; (b) F, Y, W; (c) K, R, H; (d) A, G; (e) S, T; (f) Q, N; and (g) E, D. For instance, a person of ordinary skill in the art reasonably expect that an isolated replacement of a leucine with an isoleucine or valine, an aspartate with a glutamate, a threonine with a serine, or a similar replacement of an amino acid with a structurally related amino acid will not have a major effect on the biological activity of the resulting molecule.
[0099] "Sortases" are transpeptidases that modify proteins by recognizing and cleaving a carboxyl-terminal sorting signal embedded in or terminally attached to a target protein or peptide. Sortase A catalyzes the cleavage of the LPXTG motif (where X is any standard amino acid) (SEQ ID NO: 237) between the Thr and Gly residue on the target protein, with transient attachment of the Thr residue to the active site Cys residue on the enzyme, forming an enzyme-thioacyl intermediate. To complete transpeptidation and create the peptide-monomer conjugate, a biomolecule with an N-terminal nucleophilic group, typically an oligoglycine motif, attacks the intermediate, displacing Sortase A and joining the two molecules.
[0100] As used herein, the term "steric blocker" refers to a polypeptide or polypeptide moiety that can be covalently bonded to a cytokine polypeptide directly or indirectly through other moieties such as linkers, for example in the form of a chimeric polypeptide (fusion protein), but otherwise does not covalently bond to the cytokine polypeptide. A steric blocker can non-covalently bond to the cytokine polypeptide, for example though electrostatic, hydrophobic, ionic or hydrogen bonding. A steric blocker typically inhibits or blocks the activity of the cytokine moiety due to its proximity to the cytokine moiety and comparative size.
[0101] As used and described herein, a "half-life extension element" is apart of the chimeric polypeptide that increases the serum half-life and improve pK, for example, by altering its size (e.g., to be above the kidney filtration cutoff), shape, hydrodynamic radius, charge, or parameters of absorption, biodistribution, metabolism, and elimination.
[0102] The term "separation moiety" or "linker" as used herein refers to an amino acid sequence typically less than about 100 amino acids that connects or links a first amino acid sequence of interest (e.g., an amino acid sequence that folds to form a first protein domain) to a second amino acid sequence of interest (e.g., an amino acid sequence that folds to form a second protein domain) in a contiguous polypeptide chain. The separation moiety or linker typically include one or more protease cleavage sites and thus is protease cleavable. A "tandem linker" refers to a linker that comprises two or more protease cleavages sites which can be cleaved by the same or different proteases, and which can be arranged in any desired orientation, such as one cleavage site adjacent to another cleavage site, one cleavage site overlapping another cleavage site, one cleavage site following by another cleavage site with intervening amino acids between the two cleavage sites.
[0103] As used herein, the terms "activatable," "activate," "induce," and "inducible" refer to the ability of a protein, i.e. a cytokine, that is part of a conjugate, to bind its receptor and effectuate activity upon cleavage of additional elements from the conjugate.
[0104] As used herein, "plasmids" or "viral vectors" are agents that transport the disclosed nucleic acids into the cell without degradation and include a promoter yielding expression of the nucleic acid molecule and/or polypeptide in the cells into which it is delivered.
[0105] As used herein, the terms "peptide", "polypeptide", or "protein" are used broadly to mean two or more amino acids linked by a peptide bond. Protein, peptide, and polypeptide are also used herein interchangeably to refer to amino acid sequences. It should be recognized that the term polypeptide is not used herein to suggest a particular size or number of amino acids comprising the molecule and that a peptide of the invention can contain up to several amino acid residues or more.
[0106] As used throughout, "subject" can be a vertebrate, more specifically a mammal (e.g. a human, horse, cat, dog, cow, pig, sheep, goat, mouse, rabbit, rat, and guinea pig), birds, reptiles, amphibians, fish, and any other animal. The term does not denote a particular age or sex. Tus, adult and newborn subjects, whether male or female, are intended to be covered.
[0107] As used herein, "patient" or "subject" may be used interchangeably and can refer to a subject with a disease or disorder (e.g. cancer). The term patient or subject includes human and veterinary subjects.
[0108] As used herein the terms "treatment", "treat", "treating," or grammatically related terms refer to a method of reducing the effects of a disease or condition or symptom of the disease or condition. Thus, in the disclosed method, treatment can refer to at least about 10%, at least about 20%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, or substantially complete reduction in the severity of an established disease or condition or symptom of the disease or condition. For example, a method for treating a disease is considered to be a treatment if there is a 10% reduction in one or more symptoms of the disease in a subject as compared to a control. Thus, the reduction can be a 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, or any percent reduction in between 10% and 100% as compared to native or control levels. It is well understood in the art that treatment does not necessarily refer to a cure or complete ablation of the disease, condition, or symptoms of the disease or condition. Desirable effects of treatment include, but are not limited to, preventing occurrence or recurrence of disease, alleviation of symptoms, diminishment of any direct or indirect pathological consequences of the disease, preventing metastasis, decreasing the rate of disease progression, amelioration or palliation of the disease state, and remission or improved prognosis.
[0109] As used herein, the terms "prevent", "preventing", and "prevention" of a disease or disorder refers to an action, for example, administration of the chimeric polypeptide or nucleic acid sequence encoding the chimeric polypeptide, that occurs before or at about the same time a subject begins to show one or more symptoms of the disease or disorder, which inhibits or delays onset or exacerbation of one or more symptoms of the disease or disorder.
[0110] As used herein, references to "decreasing", "reducing", or "inhibiting" include a change of at least about 10%, of at least about 20%, of at least about 30%, of at least about 40%, of at least about 50%, of at least about 60%, of at least about 70%, of at least about 80%, of at least about 90% or greater as compared to a suitable control level. Such terms can include but do not necessarily include complete elimination of a function or property, such as agonist activity.
[0111] An "attenuated cytokine receptor agonist" is a cytokine receptor agonist that has decreased receptor agonist activity as compared to the cytokine receptor's naturally occurring agonist. An attenuated cytokine agonist may have at least about 10.times., at least about 50.times., at least about 100.times., at least about 250.times., at least about 500.times., at least about 1000.times. or less agonist activity as compared to the receptor's naturally occurring agonist. When a fusion protein that contains a cytokine polypeptide as described herein is described as "attenuated" or having "attenuated activity", it is meant that the fusion protein is an attenuated cytokine receptor agonist.
[0112] An "intact fusion protein" is a fusion protein in which no domain has been removed from the fusion protein, for example by protease cleavage. A domain may be removable by protease cleavage or other enzymatic activity, but when the fusion protein is "intact", this has not occurred.
[0113] As used herein "moiety" refers to a portion of a molecule that has a distinct function within that molecule, and that function may be performed by that moiety in the context of another molecule. A moiety may be a chemical entity with a particular function, or a portion of a biological molecule with a particular function. For example, a "blocking moiety" within a fusion protein is a portion of the fusion protein which is capable of blocking the activity of some or all of the fusion polypeptide. his may be a protein domain, such as serum albumin.
[0114] A. Separation Moiety or Linker
[0115] The disclosure relates to novel protease cleavable separation moieties. As described herein, the protease cleavable separation moieties were designed so that the separation moieties are cleaved with high efficiency by proteases at a desired location (e.g., proteases that are selectively expressed or expressed at high levels in the tumor microenvironment) but are stable and not cleaved or cleaved with low efficiency in other locations (e.g., in the periphery, for example healthy tissue or serum).
[0116] The protease cleavable separation moieties were designed using a process that included prioritizing proteases that would be suitable for cleaving the separation moieties based on expression in target indications, such as, expression in particular types of tumors (e.g., colon, lung, breast, melanoma). Multiple data sources for increased expression or specific expression of proteases in the target indications were used, including mRNA, proteomics and tissue staining data. The proteases were also prioritized based on their specific activity as well as intrinsic specificity, with high specific activity and high intrinsic activity preferred. Stability in the serum is an important design consideration, and to avoid potential off-target cleavage of the separation moieties by serum proteases, proteases that are not dependent on arginine in their substrate were selected, since many off-target enzymes are active towards arginine residues.
[0117] Starting sequences for the design process were selected using a diverse peptide library as substrates for proteases with mass spectrometric detection of protease cleaved products to identify preferred sequence motifs for each candidate protease. For selected initial motifs, a new peptide library that was tailored to the preferred sequence motif for the candidate protease was designed, created and analyzed. The peptide motifs were also counter-screened for cleavage by the serum proteases thrombin and Factor Xa as well as the liver/kidney protease hepsin. his process yielded peptides containing sequence motifs that are cleaved by certain tumor-associated proteases (e.g., Matrix Metaloprotease 9 (MMP9), MMP14 and/or Cathepsin L) with high efficiency, but are stable (not cleaved or cleaved with low efficiency) in the serum or normal healthy tissue (e.g., by thrombin, Factor Xa, hepsin and the like). The separation moieties disclosed herein are efficient cleavage by human tumors and minimal cleavage by normal tissues or serum.
[0118] This disclosure relates to separation moieties or linkers that connect a first amino acid sequence of interest (e.g. a first domain of interest) to a second amino acid sequence of interest (e.g. a second domain of interest). Typically, the first amino acid sequence of interest and the second amino acid sequence of interest are not found together in a naturally occurring protein. For example, the separation moiety can connect or link a first domain of interest to a second domain of interest in a fusion protein. The separation moiety is an amino acid sequence that can be of any suitable length, and preferably can be cleaved by a protease.
[0119] The separation moieties disclosed herein can confer functionality, including flexibility as well as the ability to be cleaved. Flexible linkers are usually applied when joined domains requires a certain degree of movement or interaction. Cleavable linkers are introduced to release free and functional domains in vivo at a target site. The separation moieties disclosed herein serve to connect at least two domains of interest. The separation moieties can maintain cooperative inter-domain interactions or preserving biological activity. The separation moieties can join functional domains (e.g., a payload and half-life extension element) that are released from the separation moiety at a target site (e.g. a tumor microenvironment).
[0120] In a preferred embodiment, the separation moiety is cleavable by a cleaving agent, e.g., an enzyme. Preferably, the separation moiety comprises a protease cleavage site. In some cases, the separation moiety comprises one or more cleavage sites. The separation moiety can comprise a single protease cleavage site. The separation moiety can also comprise 2 or more protease cleavage sites. For example, 2 cleavage sites, 3 cleavage sites, 4, cleavage sites, 5 cleavage sites, or more. In cases the separation moiety comprises 2 or more protease cleavage sites, the cleavage sites can be cleaved by the same protease or different proteases. A separation moiety comprising two or more cleavage sites is referred to as a "tandem linker." The two or more cleavage sites can be arranged in any desired orientation, including, but not limited tom one cleavage site adjacent to another cleavage site, one cleavage site overlapping another cleavage site, or one cleavage site following by another cleavage site with intervening amino acids between the two cleavage sites.
[0121] Of particular interest in the present invention are disease specific protease-cleavable linkers. Also preferred are protease-cleavable linkers that are preferentially cleaved at a desired location in the body, such as the tumor microenvironment, relative to the peripheral circulation. For example, the rate at which the protease-cleavable linker is cleaved in the tumor microenvironment can be at least about 10 times, at least about 100 times, at least about 1000 times or at least about 10,000 times faster in the desired location in the body, e.g., the tumor microenvironment, in comparison to in the peripheral circulation (e.g., in plasma).
[0122] Proteases known to be associated with diseased cells or tissues include but are not limited to serine proteases, cysteine proteases, aspartate proteases, threonine proteases, glutamic acid proteases, metalloproteases, asparagine peptide lyases, serum proteases, cathepsins, Cathepsin B, Cathepsin C, Cathepsin D, Cathepsin E, Cathepsin G, Cathepsin K, Cathepsin L, kallikreins, hK1, hK10, hK15, plasmin, collagenase, Type IV collagenase, stromelysin, Factor Xa, chymotrypsin-like protease, trypsin-like protease, elastase-like protease, subtilisin-like protease, actinidain, bromelain, calpain, caspases, caspase-3, Mirl-CP, papain, HIV-1 protease, HSV protease, CMV protease, chymosin, renin, pepsin, matriptase, legumain, plasmepsin, nepenthesin, metalloexopeptidases, metalloendopeptidases, matrix metalloproteases (MMP), MMP1, MMP2, MMP3, MMP8, MMP9, MMP13, MMP11, MMP14, urokinase plasminogen activator (uPA), enterokinase, prostate-specific antigen (PSA, hK3), interleukin-1.beta. converting enzyme, thrombin, FAP (FAP.alpha.), dipeptidyl peptidase, meprins, granzymes and dipeptidyl peptidase IV (DPPIV/CD26). Proteases capable of cleaving linker amino acid sequences (which can be encoded by the chimeric nucleic acid sequences provided herein) can, for example, be selected from the group consisting of a prostate specific antigen (PSA), a matrix metalloproteinase (MMP), an A Disintigrin and a Metalloproteinase (ADAM), a plasminogen activator, a cathepsin, a caspase, a tumor cell surface protease, and an elastase. The MMP can, for example, be matrix metalloproteinase 2 (MMP2), matrix metalloproteinase 9 (MMP9), matrix metalloproteinase 14 (MMP14). In addition, or alternatively, the linker can be cleaved by a cathepsin, such as, Cathepsin B, Cathepsin C, Cathepsin D, Cathepsin E, Cathepsin G, Cathepsin K and/or Cathepsin L. Preferably, the linker can be cleaved by MMP14 or Cathepsin L.
[0123] Proteases useful for cleavage of linkers and for use in the methods disclosed herein are presented in Table 1, and exemplary proteases and their cleavage site are presented in Table 1a:
TABLE-US-00001 TABLE 1 Proteases relevant to inflammation and cancer Protease Specificity Other aspects Secreted by killer T cells: Granzyme B (grB) Cleaves after Asp Type of serine protease; strongly residues (asp-ase) implicated in inducing perforin-dependent target cell apoptosis Granzyme A (grA) trypsin-like, cleaves after Type of serine protease; basic residues Granzyme H (grH) Unknown substrate Type of serine protease; specificity Other granzymes are also secreted by killer T cells, but not all are present in humans Caspase-8 Cleaves after Asp Type of cysteine protease; plays essential residues role in TCR-induced cellular expansion- exact molecular role unclear Mucosa-associated Cleaves after arginine Type of cysteine protease; likely acts both lymphoid tissue residues as a scaffold and proteolytically active (MALT1) enzyme in the CBM-dependent signaling pathway Tryptase Targets: angiotensin I, Type of mast cell-specific serine protease; fibrinogen, prourokinase, trypsin-like; resistant to inhibition by TGF.beta.; preferentially macromolecular protease inhibitors cleaves proteins after expressed in mammals due to their lysine or arginine tetrameric structure, with all sites facing residues narrow central pore; also associated with inflammation Associated with inflammation: Thrombin Targets: FGF-2, Type of serine protease; modulates HB-EGF, Osteo-pontin, activity of vascular growth factors, PDGF, VEGF chemokines and extracellular proteins; strengthens VEGF-induced proliferation; induces cell migration; angiogenic factor; regulates hemostasis Chymase Exhibit chymotrypsin- Type of mast cell-specific serine protease like specificity, cleaving proteins after aromatic amino acid residues Carboxypeptidase A Cleaves amino acid Type of zinc-dependent metalloproteinase (MC-CPA) residues from C-terminal end of peptides and proteins Kallikreins Targets: high molecular Type of serine protease; modulate weight relaxation response; contribute to kininogen, pro-urokinase inflammatory response; fibrin degradation Elastase Targets: E-cadherin, GM- Type of neutrophil serine protease; CSF, IL-1, IL-2, IL-6, degrades ECM components; regulates IL8, p38.sup.MAPK, TNF.alpha., VE- inflammatory response; activates pro- cadherin apoptotic signaling Cathepsin G Targets: EGF, ENA-78, Type of serine protease; degrades ECM IL-8, MCP-1, MMP-2, components; chemo-attractant of MT1-MMP, leukocytes; regulates inflammatory PAI-1, RANTES, TGF.beta., response; promotes apoptosis TNF.alpha. PR-3 Targets: ENA-78, IL-8, Type of serine protease; promotes IL-18, JNK, p38.sup.MAPK, inflammatory response; activates pro- TNF.alpha. apoptotic signaling Granzyme M (grM) Cleaves after Met and Type of serine protease; only expressed in other long, unbranched NK cells hydrophobic residues Calpains Cleave between Arg and Family of cysteine proteases; calcium- Gly dependent; activation is involved in the process of numerous inflammation- associated diseases
TABLE-US-00002 TABLE 1a Exemplary Proteases and Protease Recognition Sequences SEQ ID Protease Cleavage Domain Sequence NO: MMP7 KRALGLPG 3 MMP7 (DE).sub.8RPLALWRS(DR).sub.8 4 MMP9 PR(S/T)(L/I)(S/T) 5 MMP9 LEATA 6 MMP11 GGAANLVRGG 7 MMP14 SGRIGFLRTA 8 MMP PLGLAG 9 MMP PLGLAX 10 MMP PLGC(me)AG 11 MMP ESPAYYTA 12 MMP RLQLKL 13 MMP RLQLKAC 14 MMP2, MMP9, MMP14 EP(Cit)G(Hof)YL 15 Urokinase plasminogen SGRSA 16 activator (uPA) Urokinase plasminogen DAFK 17 activator (uPA) Urokinase plasminogen GGGRR 18 activator (uPA) Lysosomal Enzyme GFLG 19 Lysosomal Enzyme ALAL 20 Lysosomal Enzyme FK 21 Cathepsin B NLL 22 Cathepsin D PIC(Et)FF 23 Cathepsin K GGPRGLPG 24 Prostate Specific Antigen HSSKLQ 25 Prostate Specific Antigen HSSKLQL 26 Prostate Specific Antigen HSSKLQEDA 27 Herpes Simplex Virus Protease LVLASSSFGY 28 HIV Protease GVSQNYPIVG 29 CMV Protease GVVQASCRLA 30 Thrombin F(Pip)RS 31 Thrombin DPRSFL 32 Thrombin PPRSFL 33 Caspase-3 DEVD 34 Caspase-3 DEVDP 35 Caspase-3 KGSGDVEG 36 Interleukin 1.beta. converting enzyme GWEHDG 37 Enterokinase EDDDDKA 38 FAP KQEQNPGST 39 Kallikrein 2 GKAFRR 40 Plasmin DAFK 41 Plasmin DVLK 42 Plasmin DAFK 43 TOP ALLLALL 44 GPLGVRG 221 IPVSLRSG 222 VPLSLYSG 223 SGESPAYYTA 224
[0124] Exemplary protease linkers include, but are not limited to kallikrein cleavable linkers, thrombin cleavable linkers, chymase cleavable linkers, carboxypeptidase A cleavable linkers, cathepsin cleavable linkers, elastase cleavable linkers, FAP cleavable linkers, ADAM cleavable linkers, PR-3 cleavable linkers, granzyme M cleavable linkers, a calpain cleavable linkers, a matrix metalloproteinase (MMP) cleavable linkers, a plasminogen activator cleavable linkers, a caspase cleavable linkers, a tryptase cleavable linkers, or a tumor cell surface protease. Specifically, MMP9 cleavable linkers, ADAM cleavable linkers, CTSL1 cleavable linkers, FAP.alpha. cleavable linkers, and cathepsin cleavable linkers. Some preferred protease-cleavable linkers are cleaved by a MMP and/or a cathepsin.
[0125] The separation moieties disclosed herein are typically less than 100 amino acids. Such separation moieties can be of different lengths, such as from 1 amino acid (e.g., Gly) to 30 amino acids, from 1 amino acid to 40 amino acids, from 1 amino acid to 50 amino acids, from 1 amino acid to 60 amino acids, from 1 to 70 amino acids, from 1 to 80 amino acids, from 1 to 90 amino acids, and from 1 to 100 amino acids. In some embodiments, the linker is at least about 1, about 2, about 3, about 4, about 5, about 10, about 15, about 20, about 25, about 30, about 35, about 40, about 45, about 50, about 55, about 60, about 65, about 70, about 75, about 80, about 85, about 90, about 95, or about 100 amino acids in length. Preferred linkers are typically from about 5 amino acids to about 30 amino acids.
[0126] Preferably the lengths of linkers vary from 2 to 30 amino acids, optimized for each condition so that the linker does not impose any constraints on the conformation or interactions of the linked domains.
TABLE-US-00003 In some embodiments, the separation moiety comprises the sequence (SEQ ID NO: 195) GPAGLYAQ; (SEQ ID NO: 196) GPAGMKGL; (SEQ ID NO: 197) PGGPAGIG; (SEQ ID NO: 198) ALFKSSFP; (SEQ ID NO: 199) ALFFSSPP; (SEQ ID NO: 200) LAQRLRSS; (SEQ ID NO; 201) LAQKLKSS; (SEQ ID NO: 202) GALFKSSFPSGGGPAGLYAQGGSGKGGSGK; (SEQ ID NO: 203) RGSGGGPAGLYAQGSGGGPAGLYAQGGSGK; (SEQ ID NO: 204) KGGGPAGLYAQGPAGLYAQGPAGLYAQGSR; (SEQ ID NO: 205) RGGPAGLYAQGGPAGLYAQGGGPAGLYAQK; (SEQ ID NO: 206) KGGALFKSSFPGGPAGIGPLAQKLKSSGGS; (SEQ ID NO: 207) SGGPGGPAGIGALFKSSFPLAQKLKSSGGG; (SEQ ID NO: 208) RGPLAQKLKSSALFKSSFPGGPAGIGGGGK; (SEQ ID NO: 209) GGGALFKSSFPLAQKLKSSPGGPAGIGGGR; (SEQ ID NO: 210) RGPGGPAGIGPLAQKLKSSALFKSSFPGGG; (SEQ ID NO: 211) RGGPLAQKLKSSPGGPAGIGALFKSSFPGK; (SEQ ID NO: 212) RSGGPAGLYAQALFKSSFPLAQKLKSSGGG; (SEQ ID NO: 213) GGPLAQKLKSSALFKSSFPGPAGLYAQGGR; (SEQ ID NO: 214) GGALFKSSFPGPAGLYAQPLAQKLKSSGGK; (SEQ ID NO: 215) RGGALFKSSFPLAQKLKSSGPAGLYAQGGK; (SEQ ID NO: 216) RGGGPAGLYAQPLAQKLKSSALFKSSFPGG; (SEQ ID NO: 217) SGPLAQKLKSSGPAGLYAQALFKSSFPGSK; (SEQ ID NO: 218) KGGPGGPAGIGPLAQRLRSSALFKSSFPGR; (SEQ ID NO: 219) KSGPGGPAGIGALFFSSPPLAQKLKSSGGR; or (SEQ ID NO: 220) SGGFPRSGGSFNPRTFGSKRKRRGSRGGGG
[0127] Certain preferred separation moieties comprises the sequence GPAGLYAQ (SEQ ID NO: 195) or ALFKSSFP (SEQ ID NO: 198). The separation moieties disclosed herein can comprise one or more cleavage motif or functional variants that are the same or different. The separation moieties can comprise 1, 2, 3, 4, 5, or more cleavage motifs or functional variants. Separation moieties comprising 30 amino acids can contain 2 cleavage motifs or functional variants, 3 cleavage motifs or functional variants or more. A "functional variant" of a separation moiety retains the ability to be cleaved with high efficiency at a target site (e.g., a tumor microenvironment that expresses high levels of the protease) and are not cleaved or cleaved with low efficiency in the periphery (e.g., serum). For example, the functional variants retain at least about 50%, about 55%, about 60%, about 70%, about 80%, about 85%, about 95% or more of the cleavage efficiency of a separation moiety comprising any one of SEQ ID NOs. 195-220.
[0128] The separation moieties comprising more than one cleavage motif can be selected from SEQ ID NOs: 195-201 and combinations thereof. Preferred separation moieties comprising more than one cleavage motif comprise the amino acids selected from SEQ ID NO: 202-220.
[0129] The separation moiety can comprise both ALFKSSFP (SEQ ID NO: 198) and GPAGLYAQ (SEQ ID NO: 195). The separation moiety can comprise two cleavage motifs that each have the sequence GPAGLYAQ (SEQ ID NO: 195). Alternatively or additionally, the separation moiety can comprise two cleavage motifs that each have the sequence ALFKSSFP (SEQ ID NO: 198). The separation moiety can comprise a third cleavage motif that is the same or different.
[0130] In some embodiments, the separation moiety comprises an amino acid sequence that is at least about 90%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, or at least 99% identical to SEQ ID NOs: 195 to SEQ ID NO: 220 over the full length of SEQ ID NO: 195-220.
[0131] The disclosure also relates to functional variants of separation moieties comprising SEQ ID NOs. 195-220. The functional variants of separation moieties comprising SEQ ID NOs: 195-220 generally differ from SEQ ID NOs. 195-220 by one or a few amino acids (including substitutions, deletions, insertions, or any combination thereof), and substantially retain their ability to be cleaved by a protease.
[0132] The functional variants can contain at least one or more amino acid substitutions, deletions, or insertions relative to the separation moieties comprising SEQ ID NOs. 195-220. The functional variant can comprise 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 amino acid alterations comparted to the separation moieties comprising SEQ ID NOs. 195-220. In some preferred embodiments, the functional variant differs from the separation moiety comprising SEQ ID NOs. 195-220 by less than 10, less, than 8, less than 5, less than 4, less than 3, less than 2, or one amino acid alterations, e.g., amino acid substitutions or deletions. In other embodiments, the functional variant may comprise 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 amino acid substitutions compared to SEQ ID NOs. 195-220. The amino acid substitution can be a conservative substitution or a non-conservative substitution, but preferably is a conservative substitution.
[0133] In other embodiments, the functional variants of the separation moieties may comprise 1, 2, 3, 4, or 5 or more non-conservative amino acid substitutions compared the separation moieties comprising SEQ ID NOs: 195-220. Non-conservative amino acid substitutions could be recognized by one of skill in the art. The functional variant of the separation moiety preferably contains no more than 1, 2, 3, 4, or 5 amino acid deletions.
[0134] The amino acid sequences disclosed in the separation moieties can be described by the relative linear position in the separation moiety with respect to the sissile bond. As will be well-understood by persons skilled in the art, separation moieties comprising 8 amino acid protease substrates (e.g., SEQ ID Nos: 195-201) contain amino acid at positions P4, P3, P2, P1, P1', P2', P3', P4', wherein the sissile bond is between P1 and P1'. For example, amino acid positions for the separation moiety comprising the sequence GPAGLYAQ (SEQ ID NO: 195) can be described as follows:
TABLE-US-00004 G P A G L Y A Q P4 P3 P2 P1 P1` P2` P3` P4`
[0135] Amino acids positions for the separation moiety comprising the sequence ALFKSSFP (SEQ ID NO: 198) can be described as follows:
TABLE-US-00005 A L F K S S F P P4 P3 P2 P1 P1` P2` P3` P4`
[0136] Preferably, the amino acids surrounding the cleavage site (e.g., positions P1 and P1' for SEQ ID NOs: 195-201) are not substituted.
[0137] In embodiments, the separation moiety comprises the sequence GPAGLYAQ (SEQ ID NO: 195) or ALFKSSFP (SEQ ID NO: 198) or a functional variant of SEQ ID NO: 195 or a function variant of SEQ ID NO: 198. As described herein, a functional variant of PAGLYAQ (SEQ ID NO: 195) or ALFKSSFP (SEQ ID NO: 198) can comprise one or more amino acid substitutions, and substantially retain their ability to be cleaved by a protease. Specifically, the functional variants of GPAGLYAQ (SEQ ID NO: 195) is cleaved by MMP14, and the functional variant of ALFKSSFP (SEQ ID NO: 198) is cleaved by Capthepsin L (CTSL1). The functional variants also retain their ability to be cleaved with high efficiency at a target site (e.g., a tumor microenvironment that expresses high levels of the protease). For example, the functional variants of GPAGLYAQ (SEQ ID NO: 195) or ALFKSSFP (SEQ ID NO: 198) retain at least about 50%, about 55%, about 60%, about 70%, about 80%, about 85%, about 95% or more of the cleavage efficiency of a separation moiety comprising amino acid sequence GPAGLYAQ (SEQ ID NO: 195) or ALFKSSFP (SEQ ID NO: 198), respectively.
[0138] Preferably, the functional variant of GPAGLYAQ (SEQ ID NO: 195) or ALFKSSFP (SEQ ID NO: 198) comprise no more than 1, 2, 3, 4, or 5 conservative amino acid substitutions compared to GPAGLYAQ (SEQ ID NO: 195) or ALFKSSFP (SEQ ID NO: 198). Preferably, the amino acids at position P1 and P1' are not substituted. The amino acids at positions P1 and P1' in SEQ ID NO: 195 are G and L, and the amino acids at positions P1 and P1' in SEQ ID NO: 198 are K and S.
[0139] The functional variant of GPAGLYAQ (SEQ ID NO: 195) can preferably comprise one or more of the following: a) an arginine amino acid substitution at position P4, b) a leucine, valine, asparagine, or proline amino acid substitution at position P3, c) a asparagine amino acid substitution at position P2, d) a histidine, asparagine, or glycine amino acid substitution at position P1, e) a asparagine, isoleucine, or leucine amino acid substitution at position P1', f) a tyrosine or arginine amino acid substitution at position P2', g) a glycine, arginine, or alanine amino acid substitution at position P3', h) or a serine, glutamine, or lysine amino acid substitution at position P4'. The following amino acid substitutions are disfavored in functional variants of GPAGLYAQ (SEQ ID NO: 195): a) arginine or isoleucine at position P3, b) alanine at position P2, c) valine at position P1, d) arginine, glycine, asparagine, or threonine at position P1', e) aspartic acid or glutamic acid at position P2', f) isoleucine at position P3', g) valine at position P4'. In some embodiments, the functional variant of GPAGLYAQ (SEQ ID NO: 195) does not comprise an amino acid substitution at position P1 and/or P1'.
[0140] The amino acid substitution of the functional variant of GPAGLYAQ (SEQ ID NO: 195) preferably comprises an amino acid substitution at position P4 and/or P4'. For example, the functional variant of GPAGLYAQ (SEQ ID NO: 195) can comprises a leucine at position P4, or serine, glutamine, lysine, or phenylalanine at position P4. Alternatively or additionally, the functional variant of GPAGLYAQ (SEQ ID NO: 195) can comprises a glycine, phenylalanine, or a proline at position P4'.
[0141] In some embodiments, the amino acid substitutions at position P2 or P2' of GPAGLYAQ (SEQ ID NO: 195) are not preferred.
[0142] In some embodiments, the functional variant of GPAGLYAQ (SEQ ID NO: 195) comprises the amino acid sequence selected from SEQ ID NOs: 258-331. Specific functional variants of GPAGLYAQ (SEQ ID NO: 195) include GPLGLYAQ (SEQ ID NO: 295), and GPAGLKGA (SEQ ID NO: 285).
[0143] The functional variants of LFKSSFP (SEQ ID NO: 198) preferably comprises hydrophobic amino acid substitutions. The functional variant of LFKSSFP (SEQ ID NO: 198) can preferably comprise one or more of the following: (a) lysine, histidine, serine, glutamine, leucine, proline, or phenylalanine at position P4; (b) lysine, histidine, glycine, proline, asparagine, phenylalanine at position P3; (c) arginine, leucine, alanine, glutamine, or histatine at position P2; (d) phenylalanine, histidine, threonine, alanine, or glutamine at position P1; (e) histidine, leucine, lysine, alanine, isoleucine, arginine, phenylalanine, asparagine, glutamic acid, or glycine at position P1', (f) phenylalanine, leucine, isoleucine, lysine, alanine, glutamine, or proline at position P2'; (g) phenylalanine, leucine, glycine, serine, valine, histidine, alanine, or asparagine at position P3'; and phenylalanine, histidine, glycine, alanine, serine, valine, glutamine, lysine, or leucine.
[0144] The inclusion of aspartic acid and/or glutamic acid in functional variants of SEQ ID NO:198 are generally disfavored and avoided. The following amino acid substitutions are also disfavored in functional variants of LFKSSFP (SEQ ID NO: 198): (a) alanine, serine, or glutamic acid at position P3; (b) proline, threonine, glycine, or aspartic acid at position P2; (c) proline at position P1; (d) proline at position P1'; (e) glycine at position P2'; (f) lysine or glutamic acid at position P3'; (g) aspartic acid at position P4'.
[0145] The amino acid substitution of the functional variant of LFKSSFP (SEQ ID NO: 198) preferably comprises an amino acid substitution at position P4 and/or P1. In some embodiments, an amino acid substitution of the functional variant of LFKSSFP (SEQ ID NO: 198) at position P4' is not preferred.
[0146] In some embodiments, the functional variant of LFKSSFP (SEQ ID NO: 198) comprises the amino acid sequence selected from SEQ ID NOs: 332-408. Specific functional variants of LFKSSFP (SEQ ID NO: 198) include ALFFSSPP (SEQ ID NO: 199), ALFKSFPP (SEQ ID NO: 381), ALFKSLPP (SEQ ID NO: 382); ALFKHSPP (SEQ ID NO: 370); ALFKSIPP (SEQ ID NO: 383); ALFKSSLP (SEQ ID NO: 390); or SPFRSSRQ (SEQ ID NO: 333).
[0147] The separation moieties disclosed herein can form a stable complex under physiological conditions with the amino acid sequences (e.g. domains) that they link, while being capable of being cleaved by a protease. For example, the separation moiety is stable (e.g., not cleaved or cleaved with low efficiency) in the circulation and cleaved with higher efficiency at a target site (i.e. a tumor microenvironment). Accordingly, fusion polypeptides that include the linkers disclosed herein can, if desired, have a prolonged circulation half-life and/or lower biological activity in the circulation in comparison to the components of the fusion polypeptide as separate molecular entities. Yet, when in the desired location (e.g., tumor microenvironment) the linkers can be efficiently cleaved to release the components that are joined together by the linker and restoring or nearly restoring the half-life and biological activity of the components as separate molecular entities.
[0148] The separation moiety desirably remains stable in the circulation for at least 2 hours, at least 5, hours, at least 10 hours, at least 15 hours, at least 20 hours, at least 24 hours, at least 30 hours, at least 35 hours, at least 40 hours, at least 45 hours, at least 50 hours, at least 60 hours, at least 65 hours, at least 70 hours, at least 80 hours, at least 90 hours, or longer.
[0149] In some embodiments, the separation moiety is cleaved by less than 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 20%, 5%, or 1% in the circulation as compared to the target location. The separation moiety is also stable in the absence of an enzyme capable of cleaving the linker. However, upon expose to a suitable enzyme (i.e., a protease), the separation moiety is cleaved resulting in separation of the linked domain.
[0150] B. Polypeptides and Compositions Comprising a Separation Moiety
[0151] The separation moieties disclosed herein can be used in a wide range of applications. Without limitation, they are suitable for us in fusion proteins. As further described herein, the separation moieties are particularly useful for preparing therapeutic fusion proteins in which the therapeutic of biological activity of the fusion protein is attenuated and the attenuation is removed upon cleavage of the separation moiety. The separation moieties can also be used to conjugate a variety of payloads such as therapeutic and/or diagnostic agents to carriers or targeting agents (e.g., antibodies and fragments of antibodies, nanoparticles). Suitable methods for prepare such conjugates are well-known in the art, see for example, Bioconjugate Techniques, Third Ed., G. T. Hermanson (Ed.) Academic Press 2013. Exemplary payloads include, but are not limited to cytokines, antibodies, cell based-therapies, antibiotics, cytotoxic drugs, or other recombinant polypeptide complexes. Of specific interest are separation moieties that are suitable for use in conjugation with payloads that target or are targeted to tumor microenvironments.
[0152] This disclosure relates recombinant polypeptides in which a separation moiety as disclosed herein links a first amino acid sequence of interest (e.g. a first domain of interest) to a second amino acid sequence of interest (e.g. a second domain of interest). Typically, the first amino acid sequence of interest and the second amino acid sequence of interest are not found together in a naturally occurring protein. Preferred linkers are SEQ ID NOS:195-220. In embodiments, at least one of the first amino acid sequence of interest and the second amino acid sequence of interest is the amino acid sequence of a therapeutic polypeptide. In some embodiments in which at least one of the first amino acid sequence of interest and the second amino acid sequence of interest is the amino acid sequence of a therapeutic polypeptide, the other amino acid sequence of interest can be the amino acid sequence of a targeting polypeptide, a half-life extending polypeptide and/or a blocking polypeptide.
[0153] The polypeptides that contain a separation moiety can be represented by Formula I: [D1]-[L1]-[D2], wherein D1 is a first amino acid sequence of interest (e.g., a domain of interest), L1 is a separation moiety that connects or links D1 to D2; and D2 is a second amino acid sequence of interest (e.g., a second domain of interest). Preferably, L1 is a protease-cleavable separation moiety, and more preferably L1 comprises or consists of any of SEQ ID NOS: 195-220.
[0154] The polypeptides can also be represented by Formula II: [D1]-[L1]-[D2]-[L2]-[D3], wherein D1 is a first amino acid of interest (e.g., a domain of interest), L1 and L2 are each, independently, a linker; D2 is a second amino acid sequence of interest (e.g., a domain of interest), and D3 is a third amino acid of interest (e.g., a domain of interest), wherein at least one of L1 and L2 is a protease-cleavable separation moiety, and preferably at least one of L1 and L2 comprises or consists of any of SEQ ID NOS: 195-220.
[0155] The polypeptide can also be represented by Formula III: [D1]-[L1]-[D2]-[L2]-[D3]-[L3]-[D4], wherein D1 is a first amino acid of interest (e.g., a domain of interest), L1, L2 and L3 are each, independently, a linker; D2 is a second amino acid sequence of interest (e.g., a domain of interest), D3 is a third amino acid of interest (e.g., a domain of interest); and wherein D4 is a fourth amino acid of interest (e.g., a domain of interest), wherein at least one of L1, L2 and L3 is a protease-cleavable separation moiety, and preferably at least one of L1, L2 and L3 comprises or consists of any of SEQ ID NOS: 195-220.
[0156] Additional specific applications of the separation moieties are described in more detail herein.
[0157] i. Delivery of Payloads
[0158] The separation moieties as described herein can be used to attach a therapeutic drug-moiety. In this approach, a therapeutic drug moiety is attached to the separation moiety to make a therapeutic drug moiety complex. The separate drug moiety complex can be a prodrug that is inactive until the target protease cleaves the prodrug, releasing the free drug.
[0159] ii. Antibody-Drug Conjugates
[0160] Another example of use of the separation moiety is in the field of antibody-drug conjugates (ADCs) that are mainly directed toward the treatment of cancer. ADCs typically are an antibody that is linked to a cytotoxic moiety, such as a cytotoxic drug. The ADCs discriminate between the healthy and diseased cells and provide targeted delivery of drug (e.g. cytotoxic drug) to diseased cells. ADCs typically comprise an antibody that targets a tumor marker that is specific to tumor cells, whereupon the antibody attaches itself to the tumor cell, causing the ADC to be absorbed into the cell, which enables the cytotoxic component to be released to kill the tumor cell. A key aspect of ADCs is the provision of a stable linker between the antibody component and the cytotoxic agent. In such applications, linkers may be cleavable or non-cleavable. For non-cleavable linkers, the antibody, linker and cytotoxic unit is incorporated into the tumor cell. The nature of the linker typically determines the release profile of the cytotoxic agent. For example, cleavable linkers between the antibody and the cytotoxic agent are typically catalyzed by enzymes in the tumor cell or in the tumor microenvironment, wherein the antibody and the cytotoxic agents are cleaved to release the cytotoxic agent.
[0161] In certain embodiments, the separation moiety disclosed herein links or connects a drug moiety to an antibody moiety.
[0162] iii. Peptide-Drug Conjugates
[0163] The separation moieties disclosed herein are suitable for use in peptide-drug conjugates. These compounds typically comprise a cytotoxic payload and linker; however, instead of antibodies, peptide-drug conjugates are equipped with peptides that have the ability to penetrate tumors, thus allowing the cargo to be delivered inside the tumor. In some embodiments, the separation moiety links or connects a cytotoxic payload with a peptide. The peptide-drug conjugate remains stable and has no biological activity until the target protease cleaves the separation moiety.
[0164] iv. Inducible Adoptive Cell Therapy
[0165] The separation moieties disclosed herein are suitable for use in constructs engineered for use in adoptive cell transfer (ACT) therapies. The field of adoptive cell transfer (ACT) is currently comprised of chimeric antigen receptor (CAR) engineered T cells (and next generation therapies) that target T cells to cell surface expressed targets (e.g., tumor cells expressing surface targets), and T cell receptor (TCR) engineered T cells that can target intracellular antigens.
[0166] In one embodiment, the separation moiety is used to tether a targeting moiety to a CAR construct. A CAR replaces the endogenous TCR complex with a new receptor that uses a fragment of a human or mouse antibody to bind to targets outside of a cancer cell. The antibody fragment is linked to various signaling proteins inside T-cell which mediate receptor activation when the CAR binds to its target. Porter et al., (2011) NEJM, 365:725-733; Grupp et al., (2013) NEJM, 368:1509-1518; U.S. Ser. No. 10/221,245/WO/2014/153270, Treatment of cancer using humanized anti-CD19 chimeric antigen receptor.
[0167] In another embodiment, the separation moiety is used to tether a targeting moiety to a TCR construct. A TCR is based on the gene for the protein receptor that is already naturally present in T-cells. The gene for a desired TCR can be discovered in a single patient, e.g., a patient that is able to mount an effective immune response against a type of cancer. This gene can then be introduced into other patients by incorporating it into TCR T cell constructs or reengineered to improve the binding interaction with its MHC target. Guy et al., (2013) Nat Immunol., 14(3):262-70; Kuhns et al., (2012) Front Immunol., 25; 3:159; Fesnak et al., (2016) Nat Rev Cancer, 16(9): 566-581.
[0168] These types of engineered T cells, whether autologous or allogenic, comprise engineered T cell receptor components that comprise a targeting agent such as an isolated human or humanized antibody. In CAR-T and TCR-T cells, the binding affinity of the targeting moiety may be affected by the steric, chemical, or flexibility properties of the separation moiety tethering the targeting moiety to the rest of the construct and the T cell. The separation moieties disclosed herein are suitable for use with engineered constructs for making CAR T and TCR T cells.
[0169] v. Antigen-Binding Proteins
[0170] The separation moieties disclosed herein are suitable for use in antigen-binding proteins. An "antigen-binding protein" (ABP) is a protein comprising one or more antigen-binding domains that specifically bind to an antigen or epitope. In some embodiments, the antigen-binding domain binds the antigen or epitope with specificity and affinity similar to that of naturally occurring antibodies. Typically, the separation moieties link a polypeptide that blocks the antigen-binding site of the ABP from binding to its cognate antigen. But when the separation moiety is cleaved the blocking polypeptide can diffuse away from the ABP antigen binding site and the ABP can bind to its cognate antigen. Exemplary binding polypeptides that can block the antigen binding site of an ABP include steric blocker such as human serum albumin, and peptides that interact with one or more of the complementarity determining regions (CDRs) in the antigen binding site of the ABP. Such blocking peptides can be obtained by screening libraries or by screening peptide fragments of the cognate antigen of an ABP of interest. Typically, when the ABP contains and an antigen binding site of an antibody, the separation moiety and blocker will be bonded to the amino terminus of the antibody light chain, or the amino terminus of the antibody heavy chain, so that the blocker is tethered close to and will readily block the antigen binding site. Suitable variations of this approach are used when the ABP contains an alternative scaffold for the binding site. Similarly, when single chain antibody binding sites are use, such as scFV of dAb, the blocker-separation moiety is typically bonded to the amino terminus near the antigen binding site. In certain embodiments, the ABP comprises an antibody binding site that comprises a VH and a VL, and the blocker-separation moiety is bonded to the amino terminus of the VL.
[0171] The ABP can be an antibody (e.g., the first and second antigen binding domains are in the form of an antibody). Preferably, at least one antigen binding domain of the ABP is in the form of an antibody. In another preferred embodiment, a first or second antigen binding domain is in the form of an antibody, and a first or second antigen binding domain is in the form of an antigen-binding fragment (e.g., the first antigen binding domain is an antibody and the second antigen binding domain is an antigen-binding fragment. Alternatively, the first antigen binding domain is an antigen binding fragment and the second antigen binding domain is an antibody).
[0172] In some embodiments, the ABP consists of an antibody. In some embodiments, the ABP consists essentially of an antibody. In some embodiments, the ABP comprises an alternative scaffold. In some embodiments, the ABP consists of an alternative scaffold. In some embodiments, the ABP consists essentially of an alternative scaffold. In some embodiments, the ABP comprises an antibody fragment. In some embodiments, the ABP consists of an antibody fragment. In some embodiments, the ABP consists essentially of an antibody fragment.
[0173] In some embodiments, the separation moieties disclosed herein are suitable for use in conjunction with an antibody. The term "antibody" is used herein in its broadest sense and includes certain types of immunoglobulin molecules comprising one or more antigen-binding domains that specifically bind to an antigen or epitope. An antibody specifically includes intact antibodies (e.g., intact immunoglobulins), antibody fragments, and multi-specific antibodies. An antibody is one type of ABP.
[0174] In some embodiments, the separation moieties disclosed herein are suitable for use in conjunction with an antigen binding protein comprising an alternative scaffold. The term "alternative scaffold" refers to a molecule in which one or more regions may be diversified to produce one or more antigen-binding domains that specifically bind to an antigen or epitope.
[0175] In some embodiments, the antigen-binding domain binds the antigen or epitope with specificity and affinity similar to that of an antibody. Exemplary alternative scaffolds include those derived from fibronectin (e.g., Adnectins.TM.), the .beta.-sandwich (e.g., iMab), lipocalin (e.g., Anticalins.RTM.), EETI-II/AGRP, BPTI/LACI-D1/ITI-D2 (e.g., Kunitz domains), thioredoxin peptide aptamers, protein A (e.g., Affibody.RTM.), ankyrin repeats (e.g., DARPins), gamma-B-crystallin/ubiquitin (e.g., Affilins), CTLD3 (e.g., Tetranectins), Fynomers, and (LDLR-A module) (e.g., Avimers). Additional information on alternative scaffolds is provided in Binz et al., Nat. Biotechnol., 2005 23:1257-1268; Skerra, Current Opin. in Biotech., 2007 18:295-304; and Silacci et al., J. Biol. Chem., 2014, 289:14392-14398; each of which is incorporated by reference in its entirety. An alternative scaffold is one type of ABP.
[0176] In some embodiments, the separation moieties disclosed herein are suitable for use in conjunction with an antibody fragment. An "antibody fragment" comprises a portion of an intact antibody, such as the antigen-binding or variable region of an intact antibody. Antibody fragments include, for example, Fv fragments, Fab fragments, F(ab')2 fragments, Fab' fragments, scFv (sFv) fragments, and scFv-Fc fragments.
[0177] In some embodiments, the separation moieties disclosed herein are suitable for use in conjunction with one or more Fv, Fab, or F(ab')2 fragments. "Fv" fragments comprise a non-covalently-linked dimer of one heavy chain variable domain and one light chain variable domain. "Fab" fragments comprise, in addition to the heavy and light chain variable domains, the constant domain of the light chain and the first constant domain (CH1) of the heavy chain. Fab fragments may be generated, for example, by recombinant methods or by papain digestion of a full-length antibody. "F(ab')2" fragments contain two Fab' fragments joined, near the hinge region, by disulfide bonds. F(ab')2 fragments may be generated, for example, by recombinant methods or by pepsin digestion of an intact antibody. The F(ab') fragments can be dissociated, for example, by treatment with 1-mercaptoethanol.
[0178] In some embodiments, the separation moieties disclosed herein are suitable for use in conjunction with an scFv or scFv-Fc. "Single-chain Fv" or "sFv" or "scFv" antibody fragments comprise a VH domain and a VL domain in a single polypeptide chain. The VH and VL are generally linked by a peptide linker. See Pluckthun A. (1994). Any suitable linker may be used.
[0179] In some embodiments, the linker is a (GGGGS)n (SEQ ID NO: 231). In some embodiments, n=1, 2, 3, 4, 5, or 6. See Antibodies from Escherichia coli. In Rosenberg M. & Moore G. P. (Eds.), The Pharmacology of Monoclonal Antibodies vol. 113 (pp. 269-315). Springer-Verlag, New York, incorporated by reference in its entirety. "scFv-Fc" fragments comprise an scFv attached to an Fc domain. For example, an Fc domain may be attached to the C-terminal of the scFv. The Fc domain may follow the VH or VL, depending on the orientation of the variable domains in the scFv (i.e., VH-VL or VL-VH). Any suitable Fc domain known in the art or described herein may be used. In some cases, the Fc domain comprises an IgG4 Fc domain.
[0180] In some embodiments, the separation moieties disclosed herein are suitable for use in conjunction with a single domain antibody. The term "single domain antibody" refers to a molecule in which one variable domain of an antibody specifically binds to an antigen without the presence of another variable domain. Single domain antibodies, and fragments thereof, are described in Arabi Ghahroudi et al., FEBS Letters, 1998, 414:521-526 and Muyldermans et al., Trends in Biochem. Sci., 2001, 26:230-245, each of which is incorporated by reference in its entirety. Single domain antibodies are also known as sdAbs or nanobodies.
[0181] In some embodiments, the separation moieties disclosed herein are suitable for use in conjunction with a monospecific ABP. A "monospecific ABP" is an ABP that comprises one or more binding sites that specifically bind to the same epitope. An example of a monospecific ABP is a naturally occurring IgG molecule which, while divalent (i.e., having two antigen-binding domains), recognizes the same epitope at each of the two antigen-binding domains. The binding specificity may be present in any suitable valency.
[0182] In some embodiments, the separation moieties disclosed herein are suitable for use in conjunction with a monoclonal antibody. The term "monoclonal antibody" refers to an antibody from a population of substantially homogeneous antibodies. A population of substantially homogeneous antibodies comprises antibodies that are substantially similar and that bind the same epitope(s), except for variants that may normally arise during production of the monoclonal antibody. Such variants are generally present in only minor amounts. A monoclonal antibody is typically obtained by a process that includes the selection of a single antibody from a plurality of antibodies. For example, the selection process can be the selection of a unique clone from a plurality of clones, such as a pool of hybridoma clones, phage clones, yeast clones, bacterial clones, or other recombinant DNA clones. The selected antibody can be further altered, for example, to improve affinity for the TNFR superfamily member proteins ("affinity maturation"), to humanize the antibody, to improve its production in cell culture, and/or to reduce its immunogenicity in a subject.
[0183] In some embodiments, the separation moieties disclosed herein are suitable for use in conjunction with a chimeric antibody. The term "chimeric antibody" refers to an antibody in which a portion of the heavy and/or light chain is derived from a particular source or species, while the remainder of the heavy and/or light chain is derived from a different source or species.
[0184] In some embodiments, the separation moieties disclosed herein are suitable for use in conjunction with a humanized antibody. "Humanized" forms of non-human antibodies are chimeric antibodies that contain minimal sequence derived from the non-human antibody. A humanized antibody is generally a human antibody (recipient antibody) in which residues from one or more CDRs are replaced by residues from one or more CDRs of a non-human antibody (donor antibody). The donor antibody can be any suitable non-human antibody, such as a mouse, rat, rabbit, chicken, or non-human primate antibody having a desired specificity, affinity, or biological effect. In some instances, selected framework region residues of the recipient antibody are replaced by the corresponding framework region residues from the donor antibody. Humanized antibodies may also comprise residues that are not found in either the recipient antibody or the donor antibody. Such modifications may be made to further refine antibody function. For further details, see Jones et al., Nature, 1986, 321:522-525; Riechmann et al., Nature, 1988, 332:323-329; and Presta, Curr. Op. Struct. Biol., 1992, 2:593-596, each of which is incorporated by reference in its entirety.
[0185] In some embodiments, the separation moieties disclosed herein are suitable for use in conjunction with a human antibody. A "human antibody" is one which possesses an amino acid sequence corresponding to that of an antibody produced by a human or a human cell, or derived from a non-human source that utilizes a human antibody repertoire or human antibody-encoding sequences (e.g., obtained from human sources or designed de novo). Human antibodies specifically exclude humanized antibodies.
[0186] In some embodiments, the ABPs provided herein specifically bind to the extracellular domain of a TNFR Superfamily Protein. In some embodiments, the TNFR superfamily protein is CD27, CD137, CD40, GITR, LT-betaR, CD30, HVEM, TNFR1, TNFR2, or OX-40. The TNFR superfamily protein may be expressed on the surface of any suitable target cell. In some embodiments, the target cell is a T cell. In some embodiments, the target cell is an effector T cell. In some embodiments, the target cell is a regulatory T cell. In some embodiments, the target cell is a natural killer (NK) cell. In some embodiments, the target cell is a natural killer T (NKT) cell. In some embodiments, the target cell is a B cell. In some embodiments, the target cell is a myeloid derived cell. In some embodiments, the target cell is a myeloid derived suppressor cell. In some embodiments, the target cell is a dendritic cell.
[0187] In some embodiments, an ABP provided herein is an antibody. In some embodiments, an ABP provided herein is an antibody fragment. In some embodiments, an ABP provided herein is an alternative scaffold.
[0188] In some embodiments, the ABPs provided herein comprise an immunoglobulin molecule. In some embodiments, the ABPs provided herein consist of an immunoglobulin molecule. In some embodiments, the ABPs provided herein consist essentially of an immunoglobulin molecule. In some aspects, the immunoglobulin molecule comprises an antibody. In some aspects, the immunoglobulin molecule consists of an antibody. In some aspects, the immunoglobulin molecule consists essentially of an antibody.
[0189] In some embodiments, the ABPs provided herein comprise alight chain. In some aspects, the light chain is a kappa light chain. In some aspects, the light chain is a lambda light chain.
[0190] In some embodiments, the ABPs provided herein comprise a heavy chain. In some aspects, the heavy chain is an IgA. In some aspects, the heavy chain is an IgD. In some aspects, the heavy chain is an IgE. In some aspects, the heavy chain is an IgG. In some aspects, the heavy chain is an IgM. In some aspects, the heavy chain is an IgG1. In some aspects, the heavy chain is an IgG2. In some aspects, the heavy chain is an IgG3. In some aspects, the heavy chain is an IgG4. In some aspects, the heavy chain is an IgA1. In some aspects, the heavy chain is an IgA2.
[0191] In some embodiments, the separation moieties connect to a further blocking moiety.
[0192] In some embodiments, the separation moieties are positioned such that movement of the antigen-binding protein subunits is restricted.
[0193] a. Multispecific and Monospecific Multivalent Antigen Binding Proteins
[0194] In some embodiments, the separation moieties disclosed herein are suitable for use in conjunction with a multi-specific antigen binding protein (ABP). Multispecific ABP's provided herein bind more than one antigen. For instance, a multispecific antibody can bind 2 antigens, 3 antigens, binds 4 antigens, 5 antigens, or more. Alternatively, the multispecific ABP can bind 2 or more different epitopes. When the ABP binds two or more epitopes, the two or more different epitopes may be epitopes on the same antigen (e.g., a single TNFR superfamily protein molecule expressed by a cell) or on different antigens (e.g., different TNFR superfamily member protein molecules expressed by the same cell, or a TNFR superfamily member protein molecule and a non-TNFR Superfamily member protein molecule). In some aspects, a multi-specific ABP binds two different epitopes (i.e., a "bispecific ABP"). In some aspects, a multi-specific ABP binds three different epitopes (i.e., a "trispecific ABP"). In some aspects, a multi-specific ABP binds four different epitopes (i.e., a "quadspecific ABP"). In some aspects, a multi-specific ABP binds five different epitopes (i.e., a "quintspecific ABP"). In some aspects, a multi-specific ABP binds 6, 7, 8, or more different epitopes. Each binding specificity may be present in any suitable valency.
[0195] In various embodiments, the antigen binding protein comprises a blocking domain. The separation moiety disclosed herein links the blocking domain to a first antigen binding domain and consequently the blocking domain inhibits (a) binding affinity or avidity of the antigen binding protein on the epitope and/or (b) agonist or antagonist activity of the antigen binding protein on the epitope. Preferably the separation moiety comprises a cleavage site. Upon cleavage of the separation moiety by a disease-specific enzyme (i.e., a protease) (a) the binding affinity or avidity of the antigen binding protein on the epitope and/or (b) agonist or antagonist activity of the antigen binding protein on the epitope is increased. In some embodiments, the antigen binding protein comprises an additional separation moiety that links or connects an additional blocking domain to an antigen binding domain. The additional separation moiety comprises a cleavage site.
[0196] In various embodiments, the antigen binding protein comprises an additional blocking domain (e.g., a second, third, or fourth blocking domain) that is/are operably linked to an antigen binding domain (e.g., the second, third, or fourth antigen binding domain) by a cleavable separation moiety as disclosed herein. The blocking domain can be a steric blocker or a specific blocker. In some embodiments, the steric blocker is independently selected from the group consisting of a fragment of the extracellular portion of the antibody binding protein, serum albumin, a fragment of serum albumin, and an antibody or antigen binding fragment thereof that binds serum albumin. In some embodiments, the specific blocker is independently selected from a fragment of an antibody or antigen-binding fragment thereof that binds to the first, second, third or fourth antigen binding domain of the antigen binding protein described herein. Preferably, the blocking domain is serum albumin or a fragment thereof, or an antibody or antigen binding fragment thereof that binds serum albumin.
[0197] The antigen binding domain may further comprise a third antigen binding domain and a fourth antigen binding domain each with binding specificity for a target antigen. TNFR superfamily target antigens are well known in the art and are encompassed by this disclosure. Exemplary TNFR superfamily members include, but are not limited to CD27, CD30, CD137 (4-1BB), TNFR1 (CD120a), TNFR2 (CD120b), CD40, CD95 (Fas/Apo-1), HVEM, LT-betaR, GITR, nerve growth factor receptor or OX-40 (CD34). Preferred TNFR superfamily target antigens are CD27, CD137, and OX40.
[0198] The antigen binding domains with specificity for an antigen can have binding specificity for the same epitope or for a different epitope on the same antigen. For example, the first, second and third antigen binding domains can have binding specificity for the same epitope, for a different epitope, two of the antigen binding domains may binding specificity for the same epitope, or three of the antigen binding domains may have binding specificity for the same epitope. In some embodiments, the antigen binding protein may further comprise a fifth antigen binding domain with specificity for a tumor antigen.
[0199] In some embodiments, the antigen binding polypeptide comprises an Fc region. In one format, two antigen binding domains can be located on each end of the Fc region. Another format comprises two antibody arms attached to the N terminus of the Fc region, each arm comprising 2 antigen binding domains. The antigen binding domain can also comprise a half-life extension domain. The half-life extension domain can be albumin, an antigen binding domain that recruits albumin, or an immunoglobulin Fc, or a fragment thereof. In some embodiments, the half-life extension domain is operably linked to the antigen binding polypeptide by a cleavable linker.
[0200] The disclosure also relates to an antigen binding protein comprising at least a first polypeptide and a second polypeptide. The first polypeptide comprises at least a portion of an antibody heavy chain constant region and an antibody heavy chain variable region (VH). The second polypeptide comprises at least a portion of an antibody light chain constant region and an antibody light chain variable region (VL). At least one of the first and second polypeptide further comprises a blocking domain that is operably linked to the VH or VL through a protease cleavable linker. The first polypeptide associates with the second polypeptide, and VH and VL form an antigen binding site with binding specificity for a target antigen (e.g., 4-1BB or CS27). The blocking domain inhibits binding of the antigen binding site to the target antigen (e.g., 4-1BB or CD27). In some embodiments, the first polypeptide further comprises a second VH, and the first polypeptide associates with two of said second polypeptides to form two VH/VL antigen binding sites that each have specificity for human 4-1BB. In some embodiments, the antigen binding protein is a dimer of the first polypeptide with the associated second polypeptides. The antigen binding protein can further comprise a third, fourth, fifth, or a sixth polypeptide.
[0201] In some embodiments the ABP is monospecific multivalent ABP. Such formats can have a variety of structures and be prepared using suitable antibody engineering techniques. For example, a dual Fab antibody that contains a heavy chain with the structure VH-CH1-noncleavable liner-VH-CH1-CH2-CH3 can be prepare. Such a heavy chain can be expressed and can pair with two light chains. The heavy chain can dimerize through conventional interchain disulfide bonds to form an antibody format that contains 4 Fab antigen binding sites. In such formats, the separation moieties described herein can be bonded to the amino terminus of the light chain polypeptides to link a blocker to each of the Fab antigen binding sites. Other suitable monospecific, multivalent ABP formats can be readily envisioned by those of skill in the art. In one such example a heavy chain is prepared that has the structure VH-CH1-CH2-CH3-VH-CH1. Two such heavy chains and dimerize and associate with four light chains to form a monospecific tetravalent ABP format. In such formats, the separation moieties described herein can be bonded to the amino terminus of the light chain polypeptides to link a blocker to each of the Fab antigen binding sites. The binding activity of such monospecific tetravalent ABPs is masked by the blocking domain, and the mask is removed upon cleavage of the separation domain (e.g., in a tumor microenvironment) and the ABP is able to bind its cognate antigen.
[0202] The monospecific multivalent ABP formats can have binding specificity for any desired antigen. In some embodiments, the monospecific multivalent ABP formats specifically bind to the extracellular domain of a TNFR Superfamily Protein, such as CD27, CD30, CD137 (4-1BB), TNFR1 (CD120a), TNFR2 (CD120b), CD40, CD95 (Fas/Apo-1), HVEM, LT-betaR, GITR, nerve growth factor receptor or OX-40 (CD34).
[0203] In embodiments, the multivalent antigen binding protein can comprise a first antigen binding site with specificity for a target antigen (e.g., CD27 or TNFR1), second antigen binding site with specificity for the target antigen, a blocking polypeptide, at least one protease cleavable linker, and an optional half-life extension element. In such embodiments, a first blocking polypeptide is operably linked to the first antigen binding site by a protease cleavable linker and, optionally, a second blocking polypeptide is operably linked to the second antigen binding site by a protease cleavable linker. Preferably, the blocking polypeptide is operably linked to the second antigen binding site by a protease cleavable linker. The blocking polypeptide inhibits (a) binding affinity or avidity of the antigen binding protein on the target antigen and/or (b) agonist or antagonist activity of the antigen binding protein on the target antigen and upon cleavage of the protease cleavable linker (a) the binding affinity or avidity of the antigen binding protein on the target antigen and/or (b) agonist activity of the antigen binding protein on the target antigen is increased. The optional half-life extension element can be operably linked to the first antigen binding site and/or second antigen binding site through a optionally protease cleavable linker. In such embodiments, the first and second binding sites can have specificity for the same epitope or different epitopes.
[0204] In embodiments, the antigen binding protein further comprises a third antigen binding site with specificity for the same target antigen. The antigen binding protein can further comprise a fourth antigen binding site with specificity for the same target antigen as the first and second and third antigen binding sites. The third and fourth antigen binding sites can each further comprise a blocking polypeptide that is operable linked to the antigen-binding sight through a protease cleavable linker.
[0205] This disclosure also relates to a tetravalent antigen binding protein that can comprise a first polypeptide that comprises at least a portion of an antibody heavy chain constant region and an antibody heavy chain variable region (VH) and a second polypeptide that comprises at least a portion of an antibody light chain constant region, an antibody light chain variable region (VL). The first polypeptide associates with the second polypeptide and the VH and VL form an antigen binding site with binding specificity for a target antigen. The tetravalent antigen binding protein further comprises a blocking polypeptide that is operably linked to the VH or VL through a protease cleavable linker. The blocking polypeptide inhibits binding of the antigen binding site to the target antigen. In some embodiments, the first polypeptide can further comprise a second VH. The first polypeptide associates with two of the second polypeptides to form two VH/VL antigen binding site that each have specificity for the target antigen.
[0206] The tetravalent antigen binding protein can further comprise a third polypeptide, a fourth polypeptide, a fifth polypeptide, a sixth polypeptide, a seventh polypeptide, and an eight polypeptide. The third polypeptide can comprise at least a portion of an antibody heavy chain constant region and an antibody heavy chain variable region (VH). The fourth polypeptide can comprise at least a portion of an antibody light chain constant region, an antibody light chain variable region (VL). The firth polypeptide can comprise at least portion of an antibody heavy chain constant region and an antibody heavy chain variable region (VH). The sixth polypeptide can comprise at least a portion of an antibody light chain constant region, an antibody light chain variable region (VL). The seventh polypeptide can comprise at least portion of an antibody heavy chain constant region and an antibody heavy chain variable region (VH). The eighth polypeptide can comprise at least a portion of an antibody light chain constant region, an antibody light chain variable region (VL).
[0207] In some embodiments at least one of the third and fourth polypeptide further comprises a blocking polypeptide that is operably linked to the VH or VL through a protease cleavable linker. The third polypeptide associates with the fourth polypeptide and VH and VL form an antigen binding site with binding specificity for a target antigen, and the blocking domain inhibits binding of the antigen binding site to the target antigen.
[0208] In some embodiments, the fifth and sixth polypeptide further comprises a blocking polypeptide that is operably linked to the VH or VL through a protease cleavable linker. The fifth polypeptide associates with the sixth polypeptide and VH and VL form an antigen binding site with binding specificity for a target antigen, and the blocking domain inhibits binding of the antigen binding site to the target antigen.
[0209] In some embodiments, the seventh and eighth polypeptide further comprises a blocking polypeptide that is operably linked to the VH or VL through a protease cleavable linker. The seventh polypeptide associates with the eighth polypeptide and VH and VL form an antigen binding site with binding specificity for a target antigen, and the blocking domain inhibits binding of the antigen binding site to the target antigen.
[0210] vi. Inducible Cytokines
[0211] Disclosed herein are methods and compositions to engineer and use constructs comprising inducible cytokines. Cytokines are potent immune agonists, which lead to them being considered promising therapeutic agents for oncology. However, cytokines have a very narrow therapeutic window. Cytokines have short serum half-lives and are also considered to be highly potent. Consequently, therapeutic administration of cytokines produces undesirable systemic effects and toxicities. These were exacerbated by the need to administer large quantities of cytokine in order to achieve the desired levels of cytokine at the intended site of cytokine action (e.g., a tumor). Unfortunately, due to the biology of cytokines and inability to effectively target and control their activity, cytokines have not achieved the hoped-for clinical advantages in the treatment of tumors.
[0212] Disclosed herein are fusion proteins that overcome the toxicity and short half-life problems that have severely limited the clinical use of cytokines in oncology. The fusion proteins contain cytokine polypeptides that have receptor agonist activity. But in the context of the fusion protein, the cytokine receptor agonist activity is attenuated, and the circulating half-life is extended. The fusion proteins include protease cleave sites, which are cleaved by proteases that are associated with a desired site of cytokine activity (e.g., a tumor), and are typically enriched or selectively present at the site of desired activity. Thus, the fusion proteins are preferentially (or selectively) and efficiently cleaved at the desired site of activity to limit cytokine activity substantially to the desired site of activity, such as the tumor microenvironment. Protease cleavage at the desired site of activity, such as in a tumor microenvironment, releases a form of the cytokine from the fusion protein that is much more active as a cytokine receptor agonist than the fusion protein (typically at least about 100.times. more active than the fusion protein). The form of the cytokine that is released upon cleavage of the fusion protein typically has a short half-life, which is often substantially similar to the half-life of the naturally occurring cytokine, further restricting cytokine activity to the tumor microenvironment. Even though the half-life of the fusion protein is extended, toxicity is dramatically reduced or eliminated because the circulating fusion protein is attenuated, and active cytokine is targeted to the tumor microenvironment. The fusion proteins described herein, for the first time, enable the administration of an effective therapeutic dose of a cytokine to treat tumors with the activity of the cytokine substantially limited to the tumor microenvironment, and dramatically reduces or eliminates unwanted systemic effects and toxicity of the cytokine.
[0213] In general, the therapeutic use of cytokines is strongly limited by their systemic toxicity. TNF, for example, was originally discovered for its capacity of inducing the hemorrhagic necrosis of some tumors, and for its in vitro cytotoxic effect on different tumoral lines, but it subsequently proved to have strong pro-inflammatory activity, which can, in case of overproduction conditions, dangerously affect the human body. As the systemic toxicity is a fundamental problem with the use of pharmacologically active amounts of cytokines in humans, novel derivatives and therapeutic strategies are now under evaluation, aimed at reducing the toxic effects of this class of biological effectors while keeping their therapeutic efficacy.
[0214] IL-2 exerts both stimulatory and regulatory functions in the immune system and is, along with other members of the common .gamma. chain (.gamma.c) cytokine family, central to immune homeostasis. IL-2 mediates its action by binding to IL-2 receptors (IL-2R), consisting of either trimeric receptors made of IL-2R.alpha. (CD25), IL-2R.beta. (CD122), and IL-2R.gamma. (.gamma.c, CD132) chains or dimeric .beta..gamma. IL-2Rs (1, 3). Both IL-2R variants are able to transmit signal upon IL-2 binding. However, trimeric .alpha..beta..gamma. IL-2Rs have a roughly 10-100 times higher affinity for IL-2 than dimeric .beta..gamma. IL-2Rs (3), implicating that CD25 confers high-affinity binding of IL-2 to its receptor but is not crucial for signal transduction. Trimeric IL-2Rs are found on activated T cells and CD4+ forkhead box P3 (FoxP3)+ T regulatory cells (Treg), which are sensitive to IL-2 in vitro and in vivo. Conversely, antigen-experienced (memory) CD8+, CD44 high memory-phenotype (MP) CD8+, and natural killer (NK) cells are endowed with high levels of dimeric .beta..gamma. IL-2Rs, and these cells also respond vigorously to IL-2 in vitro and in vivo.
[0215] Expression of the high-affinity IL-2R is critical for endowing T cells to respond to low concentrations of IL-2 that is transiently available in vivo. IL-2R.alpha. expression is absent on naive and memory T cells but is induced after antigen activation. IL-2R.beta. is constitutively expressed by NK, NKT, and memory CD8+ T cells but is also induced on naive T cells after antigen activation. .gamma.c is much less stringently regulated and is constitutively expressed by all lymphoid cells. Once the high-affinity IL-2R is induced by antigen, IL-2R signaling upregulates the expression of IL-2Ra in part through Stat5-dependent regulation of Il2ra transcription (Kim et al., 2001). This process represents a mechanism to maintain expression of the high-affinity IL-2R and sustain IL-2 signaling while there remains a source of IL-2.
[0216] IL-2 is captured by IL-2R.alpha. through a large hydrophobic binding surface surrounded by a polar periphery that results in a relatively weak interaction (Kd 10-8 M) with rapid on-off binding kinetics. However, the IL-2R.alpha.-IL-2 binary complex leads to a very small conformational change in IL-2 that promotes association with IL-2R.beta. through a distinct polar interaction between IL-2 and IL-2R.beta.. The pseudo-high affinity of the IL2/.alpha./.beta. trimeric complex (i.e. Kd.about.300 pM) clearly indicates that the trimeric complex is more stable than either IL2 bound to the .alpha. chain alone (Kd=10 nM) or to the .beta. chain alone (Kd=450 nM). In any event, the IL2/.alpha./.beta. trimer then recruits the .gamma. chain into the quaternary complex capable of signaling, which is facilitated by the large composite binding site on the IL2-bound .beta. chain for the .gamma. chain.
[0217] In other words, the ternary IL-2R.alpha.-IL-2R.beta.-IL-2 complex then recruits .gamma.c through a weak interaction with IL-2 and a stronger interaction with IL-2R.beta. to produce a stable quaternary high-affinity IL-2R (Kd 10-11 M which is 10 pM). The formation of the high-affinity quaternary IL-2-IL-2R complex leads to signal transduction through the tyrosine kinases Jak1 and Jak3, which are associated with IL-2R.beta. and .gamma.c, respectively (Nelson and Willerford, 1998). The quaternary IL-2-IL-2R complex is rapidly internalized, where IL-2, IL-2R.beta., and .gamma.c are rapidly degraded, but IL-2R.alpha. is recycled to the cell surface (Hemar et al., 1995; Yu and Malek, 2001). Thus, those functional activities that require sustained IL-2R signaling require a continued source of IL-2 to engage IL-2R.alpha. and form additional IL-2-IL-2R signaling complexes.
[0218] Interleukin-15 (IL-15), another member of the 4-alpha-helix bundle family of cytokines, has also emerged as an immunomodulator for the treatment of cancer. IL-15 is initially captured via IL-15R.alpha., which is expressed on antigen-presenting dendritic cells, monocytes and macrophages. IL-15 exhibits broad activity and induces the differentiation and proliferation of T, B and natural killer (NK) cells via signaling through the IL-15/IL-2-R-.beta. (CD122) and the common .gamma. chain (CD132). It also enhances cytolytic activity of CD8.sup.+ T cells and induces long-lasting antigen-experienced CD8.sup.+CD44 memory T cells. IL-15 stimulates differentiation and immunoglobulin synthesis by B cells and induces maturation of dendritic cells. It does not stimulate immunosuppressive T regulatory cells (Tregs). Thus, boosting IL-15 activity selectively in the tumor micro-environment could enhance innate and specific immunity and fight tumors (Waldmann et al., 2012). IL-15 was initially identified for its ability to stimulate T cell proliferation in an IL-2-like manner through common receptor components (IL-2R/15R.beta.-.gamma.c) and signaling through JAK1/JAK3 and STAT3/STAT5. Like IL-2, IL-15 has been shown to stimulate proliferation of activated CD4-CD8-, CD4+CD8+, CD4+ and CD8+ T cells as well as facilitate the induction of cytotoxic T-lymphocytes, and the generation, proliferation and activation of NK cells (Waldmann et al., 1999). However, unlike IL-2 which is required to maintain forkhead box P3 (FOXP3)-expressing CD4+CD25+ Treg cells and for the retention of these cells in the periphery, IL-15 has little effect on Tregs (Berger et al., 2009). This is important as FOXP3-expressing CD4+CD25+ Tregs inhibit effector T cells, thereby inhibiting immune responses including those directed against the tumor. IL-2 also has a crucial role in initiating activation induced cell death (AICD), a process that leads to the elimination of self-reactive T cells, whereas IL-15 is an anti-apoptotic factor for T cells (Marks-Konczalik et al., 2000). IL-15 co-delivered with HIV peptide vaccines has been shown to overcome CD4+ T cell deficiency by promoting longevity of antigen-specific CD8+ T cells and blocking TRAIL-mediated apoptosis (Oh et al., 2008). Furthermore, IL-15 promotes the long-term maintenance of CD8+CD44hi memory T cells (Kanegane et al., 1996).
[0219] The importance of IL-15 and IL-15R.alpha. to T and NK cell development is further highlighted by the phenotype of IL-15R.alpha..sup.-/- and IL-15.sup.-/- mice. Knockout mice demonstrate decreased numbers of total CD8+ T cells, and are deficient in memory-phenotype CD8+ T cells, NK cells, NK/T cells and some subsets of intestinal intraepithelial lymphocytes, indicating that IL-15 provides essential positive homeostatic functions for these subsets of cells (Lodolce et al., 1996; Kennedy et al., 1998). The similarities in the phenotypes of these two strains of knockout mice suggest the importance of IL-15R.alpha. in maintaining physiologically relevant IL-15 signals.
[0220] IL-15 is presented in trans by the IL-15 receptor alpha-chain to the IL-15R.beta..gamma.c complex displayed on the surface of T cells and natural killer (NK) cells (Han et al., 2011). The IL-15Ra-chain plays a role of chaperone protein, stabilizes, and increases IL-15 activity (Desbois et al., 2016). It has been shown that exogenous IL-15 may have a limited impact on patients with cancer due to its dependency on IL-15Ra frequently downregulated in cancer patients. Therefore, the fusion protein RLI, composed of the sushi+ domain of IL15Ra coupled via a linker to IL-15, has been suggested as an alternative approach to IL15 therapy (Bessard et al., 2009). It was found that administration of soluble IL-15/IL-15R.alpha. complexes greatly enhanced IL-15 serum half-life and bioavailability in vivo (Stoklasek et al., 2010).
[0221] In addition to the effects on T and NK cells, IL-15 also has several effects on other components of the immune system. IL-15 protects neutrophils from apoptosis, modulates phagocytosis and stimulates the secretion of IL-8 and IL-1R antagonist. It functions through the activation of JAK2, p38 and ERK1/2 MAPK, Syk kinase and the NF-kB transcriptional factor (Pelletier et al., 2002). In mast cells, IL-15 can act as a growth factor and an inhibitor of apoptosis. In these cells IL-15 activates the JAK2/STAT5 pathway without the requirement of .gamma.c binding (Tagaya et al., 1996). IL-15 also induces B lymphocyte proliferation and differentiation, and increases immunoglobulin secretion (Armitage et al., 1995). It also prevents Fas-mediated apoptosis and allows induction of antibody responses partially independent of CD4-help (Demerci et al., 2004; Steel et al., 2010). Monocytes, macrophages and dendritic cells effectively transcribe and translate IL-15. They also respond to IL-15 stimulation. Macrophages respond by increasing phagocytosis, inducing IL-8, IL-12 and MCP-1 expression, and secreting IL-6, IL-8 and TNF.alpha. (Budagian et al., 2006). Dendritic cells incubated with IL-15 demonstrate maturation with increased CD83, CD86, CD40, and MHC class II expression, are also resistant to apoptosis, and show enhanced interferon-.gamma. secretion (Anguille et al., 2009).
[0222] IL-15 has also been shown to have effects on non-hematological cells including myocytes, adipocytes, endothelial and neural cells. IL-15 has an anabolic effect on muscle and may support muscle cell differentiation (Quinn et al., 1995). It stimulates myocytes and muscle fibers to accumulate contractile protein and is able to slow muscle wasting in rats with cancer-related cachexia (Figueras et al., 2004). IL-15 has also been shown to stimulate angiogenesis (Angiolillo et al., 1997) and induce microglial growth and survival (Hanisch et al., 1997).
[0223] Interleukin-7 (IL-7), also of the IL-2/IL-15 family, is a well-characterized pleiotropic cytokine, and is expressed by stromal cells, epithelial cells, endothelial cells, fibroblasts, smooth muscle cells and keratinocytes, and following activation, by dendritic cells (Alpdogan et al., 2005). Although it was originally described as a growth and differentiation factor for precursor B lymphocytes, subsequent studies have shown that IL-7 is critically involved in T-lymphocyte development and differentiation. Interleukin-7 signaling is essential for optimal CD8 T-cell function, homeostasis and establishment of memory (Schluns et al., 2000); it is required for the survival of most T-cell subsets, and its expression has been proposed to be important for regulating T-cell numbers.
[0224] IL-7 binds to a dimeric receptor, including IL-7R.alpha. and .gamma..sub.c to form a ternary complex that plays fundamental roles in extracellular matrix remodeling, development, and homeostasis of T and B cells (Mazzucchelli and Durum, 2007). IL-7R.alpha. also cross-reacts to form a ternary complex with thymic stromal lymphopoietin (TSLP) and its receptor (TSLPR), and activates the TSLP pathway, resulting in T and dendritic cell proliferation in humans and further B cell development in mice (Leonard, 2002). Tight regulation of the signaling cascades activated by the complexes are therefore crucial to normal cellular function. Under-stimulation of the IL-7 pathway caused by mutations in the IL-7Ra ectodomain inhibits T and B cell development, resulting in patients with a form of severe combined immunodeficiency (SCID) (Giliani et al., 2005; Puel et al., 1998).
[0225] IL-7 has a potential role in enhancing immune reconstitution in cancer patients following cytotoxic chemotherapy. IL-7 therapy enhances immune reconstitution and can augment even limited thymic function by facilitating peripheral expansion of even small numbers of recent thymic emigrants. Therefore, IL-7 therapy could potentially repair the immune system of patients who have been depleted by cytotoxic chemotherapy (Capitini et al., 2010).
[0226] Interleukin-12 (IL-12) is a disulfide-linked heterodimer of two separately encoded subunits (p35 and p40), which are linked covalently to give rise to the so-called bioactive heterodimeric (p70) molecule (Lieschke et al., 1997; Jana et al., 2014). Apart from forming heterodimers (IL-12 and IL-23), the p40 subunit is also secreted as a monomer (p40) and a homodimer (p40.sub.2). It is known in the art that synthesis of the heterodimer as a single chain with a linker connecting the p35 to the p40 subunit preserves the full biological activity of the heterodimer. IL-12 plays a critical role in the early inflammatory response to infection and in the generation of Th1 cells, which favor cell-mediated immunity. It has been found that overproduction of IL-12 can be dangerous to the host because it is involved in the pathogenesis of a number of autoimmune inflammatory diseases (e.g. MS, arthritis, type 1 diabetes).
[0227] The IL-12 receptor (IL-12R) is a heterodimeric complex consisting of IL-12R.beta.1 and IL-12R.beta.2 chains expressed on the surface of activated T-cells and natural killer cells (Trinchieri et al., 2003). The IL-12R.beta.1 chain binds to the IL-12p40 subunit, whereas IL-12p35 in association with IL-12R.beta.2 confers an intracellular signaling ability (Benson et al., 2011). Signal transduction through IL-12R induces phosphorylation of Janus kinase (Jak2) and tyrosine kinase (Tyk2), that phosphorylate and activate signal transducer and activator of transcription (STAT)1, STAT3, STAT4, and STAT5. The specific cellular effects of IL-12 are due mainly to activation of STAT4. IL-12 induces natural killer and T-cells to produce cytokines, in particular interferon (IFN).gamma., that mediate many of the proinflammatory activities of IL-12, including CD4+ T-cell differentiation toward the Th1 phenotype (Montepaone et al., 2014).
[0228] Treg cells actively suppress activation of the immune system and prevent pathological self-reactivity and consequent autoimmune disease. Developing drugs and methods to selectively activate regulatory T cells for the treatment of autoimmune disease is the subject of intense research and, until the development of the present invention, which can selectively deliver active interleukins at the site of inflammation, has been largely unsuccessful. Treg are a class of CD4+CD25+ T cells that suppress the activity of other immune cells. Treg are central to immune system homeostasis and play a major role in maintaining tolerance to self-antigens and in modulating the immune response to foreign antigens. Multiple autoimmune and inflammatory diseases, including Type 1 Diabetes (TD), Systemic Lupus Erythematosus (SLE), and Graft-versus-Host Disease (GVHD) have been shown to have a deficiency of Treg cell numbers or Treg function.
[0229] Consequently, there is great interest in the development of therapies that boost the numbers and/or function of Treg cells. One treatment approach for autoimmune diseases being investigated is the transplantation of autologous, ex vivo-expanded Treg cells (Tang, Q., et al, 2013, Cold Spring Harb. Perspect. Med., 3:1-15). While this approach has shown promise in treating animal models of disease and in several early stage human clinical trials, it requires personalized treatment with the patient's own T cells, is invasive, and is technically complex. Another approach is treatment with low dose Interleukin-2 (IL-2). Treg cells characteristically express high constitutive levels of the high affinity IL-2 receptor, IL2R.alpha..beta..gamma., which is composed of the subunits IL2R.alpha. (CD25), IL2R.beta. (CD122), and IL2R.gamma. (CD132), and Treg cell growth has been shown to be dependent on IL-2 (Malek, T. R., et al., 2010, Immunity, 33:153-65).
[0230] Conversely, immune activation has also been achieved using IL-2, and recombinant IL-2 (Proleukin.RTM.) has been approved to treat certain cancers. High-dose IL-2 is used for the treatment of patients with metastatic melanoma and metastatic renal cell carcinoma with a long-term impact on overall survival.
[0231] Clinical trials of low-dose IL-2 treatment of chronic GVHD (Koreth, J., et al., 2011, N Engl J Med., 365:2055-66) and HCV-associated autoimmune vasculitis patients (Saadoun, D., et al., 2011, N Engl J Med., 365:2067-77) have demonstrated increased Treg levels and signs of clinical efficacy. New clinical trials investigating the efficacy of IL-2 in multiple other autoimmune and inflammatory diseases have been initiated. The rationale for using so-called low dose IL-2 was to exploit the high IL-2 affinity of the trimeric IL-2 receptor which is constitutively expressed on Tregs while leaving other T cells which do not express the high affinity receptor in the inactivated state. Aldesleukin (marketed as Proleukin.RTM. by Prometheus Laboratories, San Diego, Calif.), the recombinant form of IL-2 used in these trials, is associated with high toxicity. Aldesleukin is approved for the treatment of metastatic melanoma and metastatic renal cancer, but its side effects are so severe that its use is only recommended in a hospital setting with access to intensive care (Web address: www.proleukin.com/assets/pdf/proleukin.pdf).
[0232] The clinical trials of IL-2 in autoimmune diseases have employed lower doses of IL-2 in order to target Treg cells, because Treg cells respond to lower concentrations of IL-2 than many other immune cell types due to their expression of IL2R alpha (Klatzmann D, 2015 Nat Rev Immunol. 15:283-94). However, even these lower doses resulted in safety and tolerability issues, and the treatments used have employed daily subcutaneous injections, either chronically or in intermittent 5-day treatment courses. Therefore, there is a need for an autoimmune disease therapy that potentiates Treg cell numbers and function, that targets Treg cells more specifically than IL-2, that is safer and more tolerable, and that is administered less frequently.
[0233] One approach that has been suggested for improving the therapeutic index of IL-2-based therapy is to use variants of IL-2 that are selective for Treg cells relative to other immune cells. IL-2 receptors are expressed on a variety of different immune cell types, including T cells, NK cells, eosinophils, and monocytes, and this broad expression pattern likely contributes to its pleiotropic effect on the immune system and high systemic toxicity. In particular, activated T effector cells express IL2R.alpha..beta..gamma., as do pulmonary epithelial cells. But, activating T effector cells runs directly counter to the goal of down-modulating and controlling an immune response, and activating pulmonary epithelial cells leads to known dose-limiting side effects of IL-2 including pulmonary edema. In fact, the major side effect of high-dose IL-2 immunotherapy is vascular leak syndrome (VLS), which leads to accumulation of intravascular fluid in organs such as lungs and liver with subsequent pulmonary edema and liver cell damage. There is no treatment of VLS other than withdrawal of IL-2. Low-dose IL-2 regimens have been tested in patients to avoid VLS, however, at the expense of suboptimal therapeutic results.
[0234] According to the literature, VLS is believed to be caused by the release of proinflammatory cytokines from IL-2-activated NK cells. However, there is strong evidence that pulmonary edema results from direct binding of IL-2 to lung endothelial cells, which expressed low to intermediate levels of functional .alpha..beta..gamma. IL-2Rs. And, the pulmonary edema associated with interaction of IL-2 with lung endothelial cells was abrogated by blocking binding to CD25 with an anti-CD25 monoclonal antibody (mAb), in CD25-deficient host mice, or by the use of CD122-specific IL-2/anti-IL-2 mAb (IL-2/mAb) complexes, thus preventing VLS.
[0235] Treatment with interleukin cytokines other than IL-2 has been more limited. IL-15 displays immune cell stimulatory activity similar to that of IL-2 but without the same inhibitory effects, thus making it a promising immunotherapeutic candidate. Clinical trials of recombinant human IL-15 for the treatment of metastatic malignant melanoma or renal cell cancer demonstrated appreciable changes in immune cell distribution, proliferation, and activation and suggested potential antitumor activity (Conlon et. al., 2014). IL-15 is currently in clinical trials to treat various forms of cancer. However, IL-15 therapy is known to be associated with undesired and toxic effects, such as exacerbating certain leukemias, graft-versus-host disease, hypotension, thrombocytopenia, and liver injury. (Mishra A., et al., Cancer Cell, 2012, 22(5):645-55; Alpdogan O. et al., Blood, 2005, 105(2):866-73; Conlon K C et al., J Clin Oncol, 2015, 33(1):74-82.)
[0236] IL-7 promotes lymphocyte development in the thymus and maintains survival of naive and memory T cell homeostasis in the periphery. Moreover, it is important for the organogenesis of lymph nodes (LN) and for the maintenance of activated T cells recruited into the secondary lymphoid organs (SLOs) (Gao et. al., 2015). In clinical trials of IL-7, patients receiving IL-7 showed increases in both CD4+ and CD8+ T cells, with no significant increase in regulatory T cell numbers as monitored by FoxP3 expression (Sportes et al., 2008). In clinical trials reported in 2006, 2008 and 2010, patients with different kinds of cancers such as metastatic melanoma or sarcoma were injected subcutaneously with different doses of IL-7. Little toxicity was seen except for transient fevers and mild erythema. Circulating levels of both CD4+ and CD8+ T cells increased significantly and the number of Treg reduced. TCR repertoire diversity increased after IL-7 therapy. However, the anti-tumor activity of IL-7 was not well evaluated (Gao et. al., 2015). Results suggest that IL-7 therapy could enhance and broaden immune responses.
[0237] IL-12 is a pleiotropic cytokine, the actions of which create an interconnection between the innate and adaptive immunity. IL-12 was first described as a factor secreted from PMA-induced EBV-transformed B-cell lines. Based on its actions, IL-12 has been designated as cytotoxic lymphocyte maturation factor and natural killer cell stimulatory factor. Due to bridging the innate and adaptive immunity and potently stimulating the production of IFN.gamma., a cytokine coordinating natural mechanisms of anticancer defense, IL-12 seemed ideal candidate for tumor immunotherapy in humans. However, severe side effects associated with systemic administration of IL-12 in clinical investigations and the very narrow therapeutic index of this cytokine markedly tempered enthusiasm for the use of this cytokine in cancer patients (Lasek et. al., 2014). Approaches to IL-12 therapy in which delivery of the cytokine is tumor-targeted, which may diminish some of the previous issues with IL-12 therapy, are currently in clinical trials for cancers.
[0238] The direct use of IL-2 as an agonist to bind the IL-2R and modulate immune responses therapeutically has been problematic due its well-documented therapeutic risks, e.g., its short serum half-life and high toxicity. These risks have also limited the therapeutic development and use of other cytokines. New forms of cytokines that reduce these risks are needed. Disclosed herein are compositions and methods comprising IL-2 and IL-15 and other cytokines, functional fragments and muteins of cytokines as well as conditionally active cytokines designed to address these risks and provide needed immunomodulatory therapeutics.
[0239] The present invention is designed to address the shortcomings of direct IL-2 therapy and therapy using other cytokines, for example using cytokine blocking moieties, e.g. steric blocking polypeptides, serum half-life extending polypeptides, targeting polypeptides, linking polypeptides, including protease cleavable linkers, and combinations thereof. Cytokines, including interleukins (e.g., IL-2, IL-7, IL-12, IL-15, IL-18, IL-21 IL-23), interferons (IFNs, including IFN.alpha., IFN.beta. and IFN.gamma.), tumor necrosis factors (e.g., TNF.alpha., lymphotoxin), transforming growth factors (e.g., TGF.beta.1, TGF.beta.2, TGF.beta.3), chemokines (C-X-C motif chemokine 10 (CXCL10), CCL19, CCL20, CCL21), and granulocyte macrophage-colony stimulating factor (GM-CS) are highly potent when administered to patients. As used herein, "chemokine" means a family of small cytokines with the ability to induce directed chemotaxis in nearby responsive cells Cytokines can provide powerful therapy but are accompanied by undesired effects that are difficult to control clinically and which have limited the clinical use of cytokines. This disclosure relates to new forms of cytokines that can be used in patients with reduced or eliminated undesired effects. In particular, this disclosure relates to pharmaceutical compositions including chimeric polypeptides (fusion proteins), nucleic acids encoding fusion proteins and pharmaceutical formulations of the foregoing that contain cytokines or active fragments or muteins of cytokines that have decreased cytokine receptor activating activity in comparison to the corresponding cytokine. However, under selected conditions or in a selected biological environment the chimeric polypeptides activate their cognate receptors, often with the same or higher potency as the corresponding naturally occurring cytokine. As described herein, this is typically achieved using a cytokine blocking moiety that blocks or inhibits the receptor activating function of the cytokine, active fragment or mutein thereof under general conditions but not under selected conditions, such as those present at the desired site of cytokine activity (e.g., an inflammatory site or a tumor).
[0240] The chimeric polypeptides and nucleic acids encoding the chimeric polypeptides can be made using any suitable method. For example, nucleic acids encoding a chimeric polypeptide can be made using recombinant DNA techniques, synthetic chemistry or combinations of these techniques, and expressed in a suitable expression system, such as in CHO cells. Chimeric polypeptides can similarly be made, for example by expression of a suitable nucleic acid, using synthetic or semi-synthetic chemical techniques, and the like. In some embodiments, the blocking moiety can be attached to the cytokine polypeptide via sortase-mediated conjugation. "Sortases" are transpeptidases that modify proteins by recognizing and cleaving a carboxyl-terminal sorting signal embedded in or terminally attached to a target protein or peptide. Sortase A catalyzes the cleavage of the LPXTG motif (where X is any standard amino acid) (SEQ ID NO: 237) between the Thr and Gly residue on the target protein, with transient attachment of the Thr residue to the active site Cys residue on the enzyme, forming an enzyme-thioacyl intermediate. To complete transpeptidation and create the peptide-monomer conjugate, a biomolecule with an N-terminal nucleophilic group, typically an oligoglycine motif, attacks the intermediate, displacing Sortase A and joining the two molecules.
[0241] To form the cytokine-blocking moiety conjugate, the cytokine polypeptide is first tagged at the N-terminus with a polyglycine sequence, or alternatively, with at the C-terminus with a LPXTG (SEQ ID NO: 237) motif. The blocking moiety or other element has respective peptides attached that serve as acceptor sites for the tagged polypeptides. For conjugation to domains carrying a LPXTG (SEQ ID NO: 237) acceptor peptide attached via its N-terminus, the polypeptide will be tagged with an N-terminal poly-glycine stretch. For conjugation to domain carrying a poly-glycine peptide attached via its C-terminus, the polypeptide will be tagged at its C-terminus with a LPXTG (SEQ ID NO: 237) sortase recognition sequence. Recognizing poly-glycine and LPXTG (SEQ ID NO: 237) sequences, sortase will form a peptide bond between polymer-peptide and tagged polypeptides. The sortase reaction cleaves off glycine residues as intermediates and occurs at room temperature.
[0242] A variety of mechanisms can be exploited to remove or reduce the inhibition caused by the blocking moiety. For example, the pharmaceutical compositions can include a cytokine moiety and a blocking moiety, e.g. a steric blocking moiety, with a protease cleavable linker comprising a protease cleavage site located between the cytokine and cytokine blocking moiety or within the cytokine blocking moiety. When the protease cleavage site is cleaved, the blocking moiety can dissociate from cytokine, and the cytokine can then activate cytokine receptor.
[0243] Any suitable linker can be used. For example, the linker can comprise glycine-glycine, a sortase-recognition motif, or a sortase-recognition motif and a peptide sequence (Gly.sub.4Ser).sub.n (SEQ ID NO: 238) or (Gly.sub.3Ser).sub.n (SEQ ID NO: 239), wherein n is 1, 2, 3, 4 or 5. Typically, the sortase-recognition motif comprises a peptide sequence LPXTG (SEQ ID NO: 237), where X is any amino acid. In some embodiments, the covalent linkage is between a reactive lysine residue attached to the C-terminal of the cytokine polypeptide and a reactive aspartic acid attached to the N-terminal of the blocker or other domain. In other embodiments, the covalent linkage is between a reactive aspartic acid residue attached to the N-terminal of the cytokine polypeptide and a reactive lysine residue attached to the C-terminal of the blocker or another domain.
[0244] Accordingly, as described in detail herein, the cytokine blocking moieties used can be steric blockers. As used herein, a "steric blocker" refers to a polypeptide or polypeptide moiety that can be covalently bonded to a cytokine polypeptide directly or indirectly through other moieties such as linkers, for example in the form of a chimeric polypeptide (fusion protein), but otherwise does not covalently bond to the cytokine polypeptide. A steric blocker can non-covalently bond to the cytokine polypeptide, for example though electrostatic, hydrophobic, ionic or hydrogen bonding. A steric blocker typically inhibits or blocks the activity of the cytokine moiety due to its proximity to the cytokine moiety and comparative size. The steric inhibition of the cytokine moiety can be removed by spatially separating the cytokine moiety from the steric blocker, such as by enzymatically cleaving a fusion protein that contains a steric blocker and a cytokine polypeptide at a site between the steric blocker and the cytokine polypeptide.
[0245] As described in greater detail herein, the blocking function can be combined with or due to the presence of additional functional components in the pharmaceutical composition, such as a targeting domain, a serum half-life extension element, and protease-cleavable linking polypeptides. For example, a serum half-life extending polypeptide can also be a steric blocker.
[0246] In the interest of presenting a concise disclosure of the full scope of the invention, aspects of the invention are described in detail using the cytokine IL-2 as an exemplary cytokine. However, the invention and this disclosure are not limited to IL-2. It will be clear to a person of skill in the art that this disclosure, including the disclosed methods, polypeptides and nucleic acids, adequately describes and enables the use of other cytokines, fragments and muteins, such as IL-2, IL-7, IL-12, IL-15, IL-18, IL-21 IL-23, IFN.alpha., IFN.beta., IFN.gamma., TNF.alpha., lymphotoxin, TGF-.beta.1, TGF.beta.2, TGF.beta.3, GM-CSF, CXCL10, CCL19, CCL20, CCL21 and functional fragments or muteins of any of the foregoing.
[0247] Various elements ensure the delivery and activity of IL-2 preferentially at the site of desired IL-2 activity and to severely limit systemic exposure to the interleukin via a blocking and/or a targeting strategy preferentially linked to a serum half-life extension strategy. In this serum half-life extension strategy, the blocked version of interleukin circulates for extended times (preferentially 1-2 or more weeks) but the activated version has the typical serum half-life of the interleukin.
[0248] By comparison to a serum half-life extended version, the serum half-life of IL-2 administered intravenously is only .about.10 minutes due to distribution into the total body extracellular space, which is large, .about.15 L in an average sized adult. Subsequently, IL-2 is metabolized by the kidneys with a half-life of .about.2.5 hours. (Smith, K. "Interleukin 2 immunotherapy." Therapeutic Immunology 240 (2001)). By other measurements, IL-2 has a very short plasma half-life of 85 minutes for intravenous administration and 3.3 hours subcutaneous administration (Kirchner, G. I., et al., 1998, Br J Clin Pharmacol. 46:5-10). In some embodiments of this invention, the half-life extension element is linked to the interleukin via a linker which is cleaved at the site of action (e.g. by inflammation-specific proteases) releasing the interleukin's full activity at the desired site and also separating it from the half-life extension of the uncleaved version. In such embodiments, the fully active and free interleukin would have very different pharmacokinetic (pK) properties--a half-life of hours instead of weeks. In addition, exposure to active cytokine is limited to the site of desired cytokine activity (e.g., an inflammatory site or tumor) and systemic exposure to active cytokine, and associated toxicity and side effects, are reduced.
[0249] Other cytokines envisioned in this invention have similar pharmacology (e.g. IL-15 as reported by Blood 2011 117:4787-4795; doi: doi.org/10.1182/blood-2010-10-311456) as IL-2 and accordingly, the designs of this invention address the shortcomings of using these agents directly, and provide chimeric polypeptides that can have extended half-life and/or be targeted to a site of desired activity (e.g., a site of inflammation or a tumor).
[0250] If desired, IL-2 can be engineered to bind the IL-2R complex generally or one of the three IL-2R subunits specifically with an affinity that differs from that of the corresponding wild-type IL-2, for example to selectively activate Tregs or Teff (Effector T Cell). For example, IL-2 polypeptides that are said to have higher affinity for the trimeric form of the IL-2 receptor relative to the dimeric beta/gamma form of the I1-2 receptor in comparison to wild type IL-2 can have an amino acid sequence that includes one of the following sets of mutations with respect to SEQ ID NO: 1 (a mature IL-2 protein comprising amino acids 21-153 of human IL-2 having the UniProt Accession No. P60568-1): (a) K64R, V69A, and Q74P; (b) V69A, Q74P, and T101A; (c) V69A, Q74P, and I128T; (d) N30D, V69A, Q74P, and F103S; (e) K49E, V69A, A73V, and K76E; (f) V69A, Q74P, T101A, and T133N; (g) N30S, V69A, Q74P, and I128A; (h) V69A, Q74P, N88D, and S99P; (i) N30S, V69A, Q74P, and I128T; (j) K9T, Q11R, K35R, V69A, and Q74P; (k) A1T, M46L, K49R, E61D, V69A, and H79R; (l) K48E, E68D, N71T, N90H, F103S, and I114V; (m) S4P, T10A, Q11R, V69A, Q74P, N88D, and T133A; (n) E15K, N30S Y31H, K35R, K48E, V69A, Q74P, and I92T; (o) N30S, E68D, V69A, N71A, Q74P, S75P, K76R, and N90H; (p) N30S, Y31C, T37A, V69A, A73V, Q74P, H79R, and I128T; (q) N26D, N29S, N30S, K54R, E67G, V69A, Q74P, and I92T; (r) K8R, Q13R, N26D, N30T, K35R, T37R, V69A, Q74P, and 192T; and (s) N29S, Y31H, K35R, T37A, K48E, V69A, N71R, Q74P, N88D, and I89V. This approach can also be applied to prepare muteins of other cytokines including interleukins (e.g., IL-2, IL-7, IL-12, IL-15, IL-18, IL-23), interferons (IFNs, including IFNalpha, IFNbeta and IFNgamma), tumor necrosis factors (e.g., TNFalpha, lymphotoxin), transforming growth factors (e.g., TGFbeta1, TGFbeta2, TGFbeta3) and granulocyte macrophage-colony stimulating factor (GM-CS). For example, muteins can be prepared that have desired binding affinity for a cognate receptor.
[0251] As noted above, any of the mutant IL-2 polypeptides disclosed herein can include the sequences described; they can also be limited to the sequences described and otherwise identical to SEQ ID NO:1. Moreover, any of the mutant IL-2 polypeptides disclosed herein can optionally include a substitution of the cysteine residue at position 125 with another residue (e.g., serine) and/or can optionally include a deletion of the alanine residue at position 1 of SEQ ID NO:1.
[0252] Another approach to improving the therapeutic index of an IL-2 based therapy is to optimize the pharmacokinetics of the molecule to maximally activate Treg cells. Early studies of IL-2 action demonstrated that IL-2 stimulation of human T cell proliferation in vitro required a minimum of 5-6 hours exposure to effective concentrations of IL-2 (Cantrell, D. A., et. al., 1984, Science, 224: 1312-1316). When administered to human patients, IL-2 has a very short plasma half-life of 85 minutes for intravenous administration and 3.3 hours subcutaneous administration (Kirchner, G. I., et al., 1998, Br J Clin Pharmacol. 46:5-10). Because of its short half-life, maintaining circulating IL-2 at or above the level necessary to stimulate T cell proliferation for the necessary duration necessitates high doses that result in peak IL-2 levels significantly above the EC50 for Treg cells or will require frequent administration. These high IL-2 peak levels can activate IL2R.beta..gamma. receptors and have other unintended or adverse effects, for example VLS as noted above. An IL-2 analog, or a multifunctional protein with IL-2 attached to a domain that enables binding to the FcRn receptor, with a longer circulating half-life than IL-2 can achieve a target drug concentration for a specified period of time at a lower dose than IL-2, and with lower peak levels. Such an IL-2 analog will therefore require either lower doses or less frequent administration than IL-2 to effectively stimulate Treg cells. Less frequent subcutaneous administration of an IL-2 drug will also be more tolerable for patients. A therapeutic with these characteristics will translate clinically into improved pharmacological efficacy, reduced toxicity, and improved patient compliance with therapy. Alternatively, IL-2 or muteins of IL-2 (herein, "IL-2*") can be selectively targeted to the intended site of action (e.g. sites of inflammation). This targeting can be achieved by one of several strategies, including the addition of domains to the administered agent that comprise blockers of the IL-2 (or muteins) that are cleaved away or by targeting domains or a combination of the two.
[0253] In some embodiments, IL-2* partial agonists can be tailored to bind with higher or lower affinity depending on the desired target; for example, an IL-2* can be engineered to bind with enhanced affinity to one of the receptor subunits and not the others. These types of partial agonists, unlike full agonists or complete antagonists, offer the ability to tune the signaling properties to an amplitude that elicits desired functional properties while not meeting thresholds for undesired properties. Given the differential activities of the partial agonists, a repertoire of IL-2 variants could be engineered to exhibit an even finer degree of distinctive signaling activities, ranging from almost full to partial agonism to complete antagonism.
[0254] In some embodiments, the IL-2* has altered affinity for IL-2R.alpha.. In some embodiments, the IL-2* has a higher affinity for IL-2R.alpha. than wild-type IL-2. In other embodiments, the IL-2* has altered affinity for IL-2R.beta.. In one embodiment, IL-2* has enhanced binding affinity for IL-2R.beta., e.g., the N-terminus of IL-2R.beta., that eliminates the functional requirement for IL-2R.alpha.. In another embodiment, an IL-2* is generated that has increased binding affinity for IL-2R.beta. but that exhibited decreased binding to IL-2R.gamma., and thereby is defective IL-2R.gamma..beta. heterodimerization and signaling.
[0255] Blocking moieties, described in further detail below, can also be used to favor binding to or activation of one or more receptors. In one embodiment, blocking moieties are added such that IL-2R.beta..gamma. binding or activation is blocked but IL-2R.alpha. binding or activation is not changed. In another embodiment, blocking moieties are added such that IL-2R.alpha. binding or activation is diminished. In another embodiment, blocking moieties are added such that binding to and or activation of all three receptors is inhibited. This blocking may be relievable by removal of the blocking moieties in a particular environment, for example by proteolytic cleavage of a linker linking one or more blocking moieties to the cytokine.
[0256] A similar approach can be applied to improve other cytokines, particularly for use as immunostimulatory agents, for example for treating cancer. For example, in this aspect, the pharmacokinetics and/or pharmacodynamics of the cytokine (e.g., IL-2, IL-7, IL-12, IL-15, IL-18, IL-21 IL-23, IFN.alpha., IFN.beta. and IFN.gamma., TNF.alpha., lymphotoxin, TGF-.beta.1, TGF.beta.2, TGF.beta.3, GM-CSF, CXCL10, CCL19, CCL20, and CCL21 can be tailored to maximally activate effector cells (e.g., effect T cells, NK cells) and/or cytotoxic immune response promoting cells (e.g., induce dendritic cell maturation) at a site of desired activity, such as in a tumor, but preferably not systemically.
[0257] Thus, provided herein are pharmaceutical compositions comprising at least one cytokine polypeptide, such as interleukins (e.g., IL-2, IL-7, IL-12, IL-15, IL-18, IL-21, IL-23), interferons (IFNs, including IFN.alpha., IFN.beta. and IFN.gamma.), tumor necrosis factors (e.g., TNF.alpha., lymphotoxin), transforming growth factors (e.g., TGF-.beta.1, TGF.beta.2, TGF.beta.3), chemokines (e.g. CXCL10, CCL19, CCL20, CCL21) and granulocyte macrophage-colony stimulating factor (GM-CS) or a functional fragment or mutein of any of the foregoing. The polypeptide typically also includes at least one linker amino acid sequence, wherein the amino acid sequence is in certain embodiments capable of being cleaved by an endogenous protease. In one embodiment, the linker comprises an amino acid sequence comprising HSSKLQ (SEQ ID NO: 25), GPLGVRG (SEQ ID NO: 221), IPVSLRSG (SEQ ID NO: 222), VPLSLYSG (SEQ ID NO: 223), or SGESPAYYTA (SEQ ID NO: 224). In other embodiments, the chimeric polypeptide further contains a blocking moiety, e.g. a steric blocking polypeptide moiety, capable of blocking the activity of the interleukin polypeptide. The blocking moiety, for example, can comprise a human serum albumin (HSA) binding domain or an optionally branched or multi-armed polyethylene glycol (PEG). Alternatively, the pharmaceutical composition comprises a first cytokine polypeptide or a fragment thereof, and blocking moiety, e.g. a steric blocking polypeptide moiety, wherein the blocking moiety blocks the activity of the cytokine polypeptide on the cytokine receptor, and wherein the blocking moiety in certain embodiments comprises a protease cleavable domain. In some embodiments, blockade and reduction of cytokine activity is achieved simply by attaching additional domains with very short linkers to the N or C terminus of the interleukin domain. In such embodiments, it is anticipated the blockade is relieved by protease digestion of the blocking moiety or of the short linker that tethers the blocker to the interleukin. Once the domain is clipped or is released, it will no longer be able to achieve blockade of cytokine activity.
[0258] The pharmaceutical composition e.g., chimeric polypeptide can comprise two or more cytokines, which can be the same cytokine polypeptide or different cytokine polypeptides. For example, the two or more different types of cytokines have complementary functions. In some examples, a first cytokine is IL-2 and a second cytokine is IL-12. In some embodiments, each of the two or more different types of cytokine polypeptides have activities that modulate the activity of the other cytokine polypeptides. In some examples of chimeric polypeptides that contain two cytokine polypeptides, a first cytokine polypeptide is T-cell activating, and a second cytokine polypeptide is non-T-cell-activating. In some examples of chimeric polypeptides that contain two cytokine polypeptides, a first cytokine is a chemoattractant, e.g., CXCL10, and a second cytokine is an immune cell activator.
[0259] Preferably, the cytokine polypeptides (including functional fragments) that are included in the fusion proteins disclosed herein are not mutated or engineered to alter the properties of the naturally occurring cytokine, including receptor binding affinity and specificity or serum half-life. However, changes in amino acid sequence from naturally occurring (including wild type) cytokine are acceptable to facilitate cloning and to achieve desired expression levels, for example.
[0260] a. Blocking Moiety
[0261] The blocking moiety can be any moiety that inhibits the ability of the cytokine to bind and/or activate its receptor. The blocking moiety can inhibit the ability of the cytokine to bind and/or activate its receptor sterically blocking and/or by noncovalently binding to the cytokine. Examples of suitable blocking moieties include the full length or a cytokine-binding fragment or mutein of the cognate receptor of the cytokine. Antibodies and fragments thereof including, a polyclonal antibody, a recombinant antibody, a human antibody, a humanized antibody a single chain variable fragment (scFv), single-domain antibody such as a heavy chain variable domain (VH), a light chain variable domain (VL) and a variable domain of camelid-type nanobody (VHH), a sdAb and the like that bind the cytokine can also be used. Other suitable antigen-binding domain that bind the cytokine can also be used, include non-immunoglobulin proteins that mimic antibody binding and/or structure such as, anticalins, affilins, affibody molecules, affimers, affitins, alphabodies, avimers, DARPins, fynomers, kunitz domain peptides, monobodies, and binding domains based on other engineered scaffolds such as SpA, GroEL, fibronectin, lipocalin and CTLA4 scaffolds. Further examples of suitable blocking polypeptides include polypeptides that sterically inhibit or block binding of the cytokine to its cognate receptor. Advantageously, such moieties can also function as half-life extending elements. For example, a peptide that is modified by conjugation to a water-soluble polymer, such as PEG, can sterically inhibit or prevent binding of the cytokine to its receptor. Polypeptides, or fragments thereof, that have long serum half-lives can also be used, such as serum albumin (human serum albumin), immunoglobulin Fc, transferring and the like, as well as fragments and muteins of such polypeptides.
[0262] Antibodies and antigen-binding domains that bind to, for example, a protein with a long serum half-life such as HSA, immunoglobulin or transferrin, or to a receptor that is recycled to the plasma membrane, such as FcRn or transferrin receptor, can also inhibit the cytokine, particularly when bound to their antigen. Examples of such antigen-binding polypeptides include a single chain variable fragment (scFv), single-domain antibody such as a heavy chain variable domain (VH), a light chain variable domain (VL) and a variable domain of camelid-type nanobody (VHH), a sdAb and the like. Other suitable antigen-binding domain that bind the cytokine can also be used, include non-immunoglobulin proteins that mimic antibody binding and/or structure such as, anticalins, affilins, affibody molecules, affimers, affitins, alphabodies, avimers, DARPins, fynomers, kunitz domain peptides, monobodies, and binding domains based on other engineered scaffolds such as SpA, GroEL, fibronectin, lipocalin and CTLA4 scaffolds.
[0263] In illustrative examples, when IL-2 is the cytokine in the chimeric polypeptide, the blocking moiety can be the full length or fragment or mutein of the alpha chain of IL-2 receptor (IL-2R.alpha.) or beta (IL-2R.beta.) or gamma chain of IL-2 receptor (IL-2R.gamma.), an anti-IL-2 single-domain antibody (sdAb) or scFv, an anti-CD25 antibody or fragment thereof, and anti-HSA sdAb or scFv, and the like.
[0264] b. In Vivo Half-Life Extension Elements
[0265] Preferably, the chimeric polypeptides comprise an in vivo half-life extension element. Increasing the in vivo half-life of therapeutic molecules with naturally short half-lives allows for a more acceptable and manageable dosing regimen without sacrificing effectiveness. As used herein, a "half-life extension element" is a part of the chimeric polypeptide that increases the in vivo half-life and improve pK, for example, by altering its size (e.g., to be above the kidney filtration cutoff), shape, hydrodynamic radius, charge, or parameters of absorption, biodistribution, metabolism, and elimination. An exemplary way to improve the pK of a polypeptide is by expression of an element in the polypeptide chain that binds to receptors that are recycled to the plasma membrane of cells rather than degraded in the lysosomes, such as the FcRn receptor on endothelial cells and transferrin receptor. Three types of proteins, e.g., human IgGs, HSA (or fragments), and transferrin, persist for much longer in human serum than would be predicted just by their size, which is a function of their ability to bind to receptors that are recycled rather than degraded in the lysosome. These proteins, or fragments of them that retain the FcRn binding are routinely linked to other polypeptides to extend their serum half-life. In one embodiment, the half-life extension element is a human serum albumin (HSA) binding domain. HSA (SEQ ID NO:2) may also be directly bound to the pharmaceutical compositions or bound via a short linker. Fragments of HSA may also be used. HSA and fragments thereof can function as both a blocking moiety and a half-life extension element. Human IgGs can also carry out a similar function.
[0266] The serum half-life extension element can also be antigen-binding polypeptide that binds to a protein with a long serum half-life such as serum albumin, transferrin and the like. Examples of such polypeptides include antibodies and fragments thereof including, a polyclonal antibody, a recombinant antibody, a human antibody, a humanized antibody a single chain variable fragment (scFv), single-domain antibody such as a heavy chain variable domain (VH), a light chain variable domain (VL) and a variable domain of camelid-type nanobody (VHH), a sdAb and the like. Other suitable antigen-binding domain include non-immunoglobulin proteins that mimic antibody binding and/or structure such as, anticalins, affilins, affibody molecules, affimers, affitins, alphabodies, avimers, DARPins, fynomers, kunitz domain peptides, monobodies, and binding domains based on other engineered scaffolds such as SpA, GroEL, fibronectin, lipocalin and CTLA4 scaffolds. Further examples of antigen-binding polypeptides include a ligand for a desired receptor, a ligand-binding portion of a receptor, a lectin, and peptides that binds to or associates with one or more target antigens.
[0267] Some preferred serum half-life extension elements are polypeptides that comprise complementarity determining regions (CDRs), and optionally non-CDR loops. Advantageously, such serum half-life extension elements can extend the serum half-life of the cytokine, and also function as inhibitors of the cytokine (e.g., via steric blocking, non-covalent interaction or combination thereof) and/or as targeting domains. In some instances, the serum half-life extension elements are domains derived from an immunoglobulin molecule (Ig molecule) or engineered protein scaffolds that mimic antibody structure and/or binding activity. The Ig may be of any class or subclass (IgG1, IgG2, IgG3, IgG4, IgA, IgE, IgM etc.). A polypeptide chain of an Ig molecule folds into a series of parallel beta strands linked by loops. In the variable region, three of the loops constitute the "complementarity determining regions" (CDRs) which determine the antigen binding specificity of the molecule. An IgG molecule comprises at least two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds, or an antigen binding fragment thereof. Each heavy chain is comprised of a heavy chain variable region (abbreviated herein as VH) and a heavy chain constant region. The heavy chain constant region is comprised of three domains, CH1, CH2 and CH3. Each light chain is comprised of a light chain variable region (abbreviated herein as VL) and a light chain constant region. The light chain constant region is comprised of one domain, CL. The VH and VL regions can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDRs) with are hypervariable in sequence and/or involved in antigen recognition and/or usually form structurally defined loops, interspersed with regions that are more conserved, termed framework regions (FR). Each VH and VL is composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. In some embodiments of this disclosure, at least some or all of the amino acid sequences of FR1, FR2, FR3, and FR4 are part of the "non-CDR loop" of the binding moieties described herein. A variable domain of an immunoglobulin molecule has several beta strands that are arranged in two sheets. The variable domains of both light and heavy immunoglobulin chains contain three hypervariable loops, or complementarity-determining regions (CDRs). e three CDRs of a V domain (CDR1, CDR2, CDR3) cluster at one end of the beta barrel. The CDRs are the loops that connect beta strands B-C, C'-C'', and F-G of the immunoglobulin fold, whereas the bottom loops that connect beta strands AB, CC', C''-D and E-F of the immunoglobulin fold, and the top loop that connects the D-E strands of the immunoglobulin fold are the non-CDR loops. In some embodiments of this disclosure, at least some amino acid residues of a constant domain, CH1, CH2, or CH3, are part of the "non-CDR loop" of the binding moieties described herein. Non-CDR loops comprise, in some embodiments, one or more of AB, CD, EF, and DE loops of a C1-set domain of an Ig or an Ig-like molecule; AB, CC', EF, FG, BC, and EC' loops of a C2-set domain of an Ig or an Ig-like molecule; DE, BD, GF, A(A1A2)B, and EF loops of I(Intermediate)-set domain of an Ig or Ig-like molecule.
[0268] Within the variable domain, the CDRs are believed to be responsible for antigen recognition and binding, while the FR residues are considered a scaffold for the CDRs. However, in certain cases, some of the FR residues play an important role in antigen recognition and binding. Framework region residues that affect Ag binding are divided into two categories. The first are FR residues that contact the antigen, thus are part of the binding-site, and some of these residues are close in sequence to the CDRs. Other residues are those that are far from the CDRs in sequence but are in close proximity to it in the 3-D structure of the molecule, e.g., a loop in heavy chain.
[0269] The binding moieties are any kinds of polypeptides. For example, in certain instances the binding moieties are natural peptides, synthetic peptides, or fibronectin scaffolds, or engineered bulk serum proteins. The bulk serum protein comprises, for example, albumin, fibrinogen, or a globulin. In some embodiments, the binding moieties are engineered scaffolds. Engineered scaffolds comprise, for example, a sdAb, a scFv, a Fab, a VHH, a fibronectin type III domain, immunoglobulin-like scaffold (as suggested in Halaby et al., 1999. Prot Eng 12(7):563-571), DARPin, cysteine knot peptide, lipocalin, three-helix bundle scaffold, protein G-related albumin-binding module, or a DNA or RNA aptamer scaffold.
[0270] In some cases, the serum half-life extending element comprises a binding site for a bulk serum protein. In some embodiments, the CDRs provide the binding site for the bulk serum protein. The bulk serum protein is, in some examples, a globulin, albumin, transferrin, IgG1, IgG2, IgG4, IgG3, IgA monomer, Factor XIII, Fibrinogen, IgE, or pentameric IgM. In some embodiments, the CDR form a binding site for an immunoglobulin light chain, such as an Ig.kappa. free light chain or an Ig.lamda. free light chain.
[0271] The serum half-life extension element can be any type of binding domain, including but not limited to, domains from a monoclonal antibody, a polyclonal antibody, a recombinant antibody, a human antibody, a humanized antibody. In some embodiments, the binding moiety is a single chain variable fragment (scFv), single-domain antibody such as a heavy chain variable domain (VH), a light chain variable domain (VL) and a variable domain (VHH) of camelid derived nanobody. In other embodiments, the binding moieties are non-Ig binding domains, i.e., antibody mimetic, such as anticalins, affilins, affibody molecules, affimers, affitins, alphabodies, avimers, DARPins, fynomers, kunitz domain peptides, and monobodies.
[0272] In other embodiments, the serum half-life extension element can be a water-soluble polymer or a peptide that is conjugated to a water-soluble polymer, such as PEG. "PEG," "polyethylene glycol" and "poly(ethylene glycol)" as used herein, are interchangeable and encompass any nonpeptidic water-soluble poly(ethylene oxide). The term "PEG" also means a polymer that contains a majority, that is to say, greater than 50%, of --OCH.sub.2CH.sub.2-- repeating subunits. With respect to specific forms, the PEG can take any number of a variety of molecular weights, as well as structures or geometries such as "branched," "linear," "forked," "multifunctional," and the like, to be described in greater detail below. The PEG is not limited to a particular structure and can be linear (e.g., an end capped, e.g., alkoxy PEG or a bifunctional PEG), branched or multi-armed (e.g., forked PEG or PEG attached to a polyol core), a dendritic (or star) architecture, each with or without one or more degradable linkages. Moreover, the internal structure of the PEG can be organized in any number of different repeat patterns and can be selected from the group consisting of homopolymer, alternating copolymer, random copolymer, block copolymer, alternating tripolymer, random tripolymer, and block tripolymer. PEGs can be conjugated to polypeptide and peptides through any suitable method. Typically a reactive PEG derivative, such as N-hydroxysuccinimidyl ester PEG, is reacted with a peptide or polypeptide that includes amino acids with a side chain that contains an amine, sulfhydryl, carboxylic acid or hydroxyl functional group, such as cysteine, lysine, asparagine, glutamine, threonine, tyrosine, serine, aspartic acid, and glutamic acid.
[0273] c. Targeting and Retention Domains
[0274] For certain applications, it may be desirable to maximize the amount of time the construct is present in its desired location in the body. This can be achieved by including one further domain in the chimeric polypeptide (fusion protein) to influence its movements within the body. For example, the chimeric nucleic acids can encode a domain that directs the polypeptide to a location in the body, e.g., tumor cells or a site of inflammation; this domain is termed a "targeting domain" and/or encode a domain that retains the polypeptide in a location in the body, e.g., tumor cells or a site of inflammation; this domain is termed a "retention domain". In some embodiments a domain can function as both a targeting and a retention domain. In some embodiments, the targeting domain and/or retention domain are specific to a protease-rich environment. In some embodiments, the encoded targeting domain and/or retention domain are specific for regulatory T cells (Tregs), for example targeting the CCR4 or CD39 receptors. Other suitable targeting and/or retention domains comprise those that have a cognate ligand that is overexpressed in inflamed tissues, e.g., the IL-1 receptor, or the IL-6 receptor. In other embodiments, the suitable targeting and/or retention domains comprise those who have a cognate ligand that is overexpressed in tumor tissue, e.g., Epcam, CEA or mesothelin. In some embodiments, the targeting domain is linked to the interleukin via a linker which is cleaved at the site of action (e.g. by inflammation or cancer specific proteases) releasing the interleukin full activity at the desired site. In some embodiments, the targeting and/or retention domain is linked to the interleukin via a linker which is not cleaved at the site of action (e.g. by inflammation or cancer specific proteases), causing the cytokine to remain at the desired site.
[0275] Antigens of choice, in some cases, are expressed on the surface of a diseased cell or tissue, for example a tumor or a cancer cell. Antigens useful for tumor targeting and retention include but are not limited to EpCAM, EGFR, HER-2, HER-3, c-Met, FoIR, and CEA. Pharmaceutical compositions disclosed herein, also include proteins comprising two targeting and/or retention domains that bind to two different target antigens known to be expressed on a diseased cell or tissue. Exemplary pairs of antigen binding domains include but are not limited to EGFR/CEA, EpCAM/CEA, and HER-2/HER-3.
[0276] Suitable targeting and/or retention domains include antigen-binding domains, such as antibodies and fragments thereof including, a polyclonal antibody, a recombinant antibody, a human antibody, a humanized antibody a single chain variable fragment (scFv), single-domain antibody such as a heavy chain variable domain (VH), a light chain variable domain (VL) and a variable domain of camelid-type nanobody (VHH), a sdAb and the like. Other suitable antigen-binding domain include non-immunoglobulin proteins that mimic antibody binding and/or structure such as, anticalins, affilins, affibody molecules, affimers, affitins, alphabodies, avimers, DARPins, fynomers, kunitz domain peptides, monobodies, and binding domains based on other engineered scaffolds such as SpA, GroEL, fibronectin, lipocalin and CTLA4 scaffolds. Further examples of antigen-binding polypeptides include a ligand for a desired receptor, a ligand-binding portion of a receptor, a lectin, and peptides that binds to or associates with one or more target antigens.
[0277] In some embodiments, the targeting and/or retention domains specifically bind to a cell surface molecule. In some embodiments, the targeting and/or retention domains specifically bind to a tumor antigen. In some embodiments, the targeting polypeptides specifically and independently bind to a tumor antigen selected from at least one of EpCAM, EGFR, HER-2, HER-3, cMet, CEA, and FoIR. In some embodiments, the targeting polypeptides specifically and independently bind to two different antigens, wherein at least one of the antigens is a tumor antigen selected from EpCAM, EGFR, HER-2, HER-3, cMet, CEA, and FoIR.
[0278] The targeting and/or retention antigen can be a tumor antigen expressed on a tumor cell. Tumor antigens are well known in the art and include, for example, EpCAM, EGFR, HER-2, HER-3, c-Met, FolR, PSMA, CD38, BCMA, and CEA. 5T4, AFP, B7-H3, Cadherin-6, CAIX, CD117, CD123, CD138, CD166, CD19, CD20, CD205, CD22, CD30, CD33, CD352, CD37, CD44, CD52, CD56, CD70, CD71, CD74, CD79b, DLL3, EphA2, FAP, FGFR2, FGFR3, GPC3, gpA33, FLT-3, gpNMB, HPV-16 E6, HPV-16 E7, ITGA2, ITGA3, SLC39A6, MAGE, mesothelin, Muc1, Muc16, NaPi2b, Nectin-4, P-cadherin, NY-ESO-1, PRLR, PSCA, PTK7, ROR1, SLC44A4, SLTRK5, SLTRK6, STEAP1, TIM1, Trop2, WT1.
[0279] The targeting and/or retention antigen can be an immune checkpoint protein. Examples of immune checkpoint proteins include but are not limited to CD27, CD137, 2B4, TIGIT, CD155, ICOS, HVEM, CD40L, LIGHT, TIM-1, OX40, DNAM-1, PD-L1, PD1, PD-L2, CTLA-4, CD8, CD40, CEACAM1, CD48, CD70, A2AR, CD39, CD73, B7-H3, B7-H4, BTLA, IDO1, IDO2, TDO, KIR, LAG-3, TIM-3, or VISTA.
[0280] The targeting and/or retention antigen can be a cell surface molecule such as a protein, lipid or polysaccharide. In some embodiments, a targeting and/or retention antigen is a on a tumor cell, virally infected cell, bacterially infected cell, damaged red blood cell, arterial plaque cell, inflamed or fibrotic tissue cell. The targeting and/or retention antigen can comprise an immune response modulator. Examples of immune response modulator include but are not limited to granulocyte-macrophage colony stimulating factor (GM-CSF), macrophage colony stimulating factor (M-CSF), granulocyte colony stimulating factor (G-CSF), interleukin 2 (IL-2), interleukin 3 (IL-3), interleukin 12 (IL-12), interleukin 15 (IL-15), B7-1 (CD80), B7-2 (CD86), GITRL, CD3, or GITR.
[0281] The targeting and/or retention antigen can be a cytokine receptor. Examples, of cytokine receptors include but are not limited to Type I cytokine receptors, such as GM-CSF receptor, G-CSF receptor, Type I IL receptors, Epo receptor, LIF receptor, CNTF receptor, TPO receptor; Type II Cytokine receptors, such as IFN-alpha receptor (IFNAR1, IFNAR2), IFB-beta receptor, IFN-gamma receptor (IFNGR1, IFNGR2), Type II IL receptors; chemokine receptors, such as CC chemokine receptors, CXC chemokine receptors, CX3C chemokine receptors, XC chemokine receptors; tumor necrosis receptor superfamily receptors, such as TNFRSF5/CD40, TNFRSF8/CD30, TNFRSF7/CD27, TNFRSF1A/TNFR1/CD120a, TNFRSF1B/TNFR2/CD120b; TGF-beta receptors, such as TGF-beta receptor 1, TGF-beta receptor 2; Ig super family receptors, such as IL-1 receptors, CSF-R, PDGFR (PDGFRA, PDGFRB), SCFR.
[0282] d. Linkers
[0283] As stated above, the pharmaceutical compositions comprise one or more linker sequences. A linker sequence serves to provide flexibility between polypeptides, such that, for example, the blocking moiety is capable of inhibiting the activity of the cytokine polypeptide. The linker sequence can be located between any or all of the cytokine polypeptide, the serum half-life extension element, and/or the blocking moiety. As described herein, at least one of the linkers is protease cleavable, and contains a (one or more) cleavage site for a (one or more) desired protease. Preferably, the desired protease is enriched or selectively expressed at the desired site of cytokine activity (e.g., the tumor microenvironment). Thus, the fusion protein is preferentially or selectively cleaved at the site of desired cytokine activity.
[0284] The orientation of the components of the pharmaceutical composition, are largely a matter of design choice and it is recognized that multiple orientations are possible, and all are intended to be encompassed by this disclosure. For example, a blocking moiety can be located C-terminally or N-terminally to a cytokine polypeptide.
[0285] Provided herein are pharmaceutical compositions comprising polypeptide sequences. As with all peptides, polypeptides, and proteins, including fragments thereof, it is understood that additional modifications in the amino acid sequence of the chimeric polypeptides (amino acid sequence variants) can occur that do not alter the nature or function of the peptides, polypeptides, or proteins. Such modifications include conservative amino acid substitutions and are discussed in greater detail below.
[0286] The compositions provided herein have a desired function. The compositions are comprised of at least a cytokine polypeptide, such as IL-2, IL-7, IL-12, IL-15, IL-18, IL-21, or IFN.gamma., or a chemokine, such as CXCL10, CCL19, CCL20, CCL21, a blocking moiety, e.g. a steric blocking polypeptide, and an optional serum half-life extension element, and an optional targeting polypeptide, with one or more linkers connecting each polypeptide in the composition. The first polypeptide, e.g., an IL-2 mutein, is provided to be an active agent. The blocking moiety is provided to block the activity of the interleukin. The linker polypeptide, e.g., a protease cleavable polypeptide, is provided to be cleaved by a protease that is specifically expressed at the intended target of the active agent. Optionally, the blocking moiety blocks the activity of the first polypeptide by binding the interleukin polypeptide. In some embodiments, the blocking moiety, e.g. a steric blocking peptide, is linked to the interleukin via a protease-cleavable linker which is cleaved at the site of action (e.g. by inflammation specific proteases) releasing the cytokine full activity at the desired site.
[0287] In some embodiments, the linker comprises glycine-glycine, a sortase-recognition motif, or a sortase-recognition motif and a peptide sequence (Gly.sub.4Ser).sub.n (SEQ ID NO: 238) or (Gly.sub.3Ser).sub.n (SEQ ID NO: 239), wherein n is 1, 2, 3, 4 or 5. In one embodiment, the sortase-recognition motif comprises a peptide sequence LPXTG, where X is any amino acid (SEQ ID NO: 237). In one embodiment, the covalent linkage is between a reactive lysine residue attached to the C-terminal of the cytokine polypeptide and a reactive aspartic acid attached to the N-terminal of the blocking or other moiety. In one embodiment, the covalent linkage is between a reactive aspartic acid residue attached to the N-terminal of the cytokine polypeptide and a reactive lysine residue attached to the C-terminal of the blocking or other moiety.
[0288] e. Cleavage and Inducibility
[0289] As described herein, the activity of the cytokine polypeptide the context of the fusion protein is attenuated, and protease cleavage at the desired site of activity, such as in a tumor microenvironment, releases a form of the cytokine from the fusion protein that is much more active as a cytokine receptor agonist than the fusion protein. For example, the cytokine-receptor activating (agonist) activity of the fusion polypeptide can be at least about 10.times., at least about 50.times., at least about 100.times., at least about 250.times., at least about 500.times., or at least about 1000.times. less than the cytokine receptor activating activity of the cytokine polypeptide as a separate molecular entity. The cytokine polypeptide that is part of the fusion protein exists as a separate molecular entity when it contains an amino acid that is substantially identical to the cytokine polypeptide and does not substantially include additional amino acids and is not associated (by covalent or non-covalent bonds) with other molecules. If necessary, a cytokine polypeptide as a separate molecular entity may include some additional amino acid sequences, such as a tag or short sequence to aid in expression and/or purification.
[0290] In other examples, the cytokine-receptor activating (agonist) activity of the fusion polypeptide is at least about 10.times., at least about 50.times., at least about 100.times., at least about 250.times., at least about 500.times., or about 1000.times. less than the cytokine receptor activating activity of the polypeptide that contains the cytokine polypeptide that is produced by cleavage of the protease cleavable linker in the fusion protein. In other words, the cytokine receptor activating (agonist) activity of the polypeptide that contains the cytokine polypeptide that is produced by cleavage of the protease cleavable linker in the fusion protein is at least about 10.times., at least about 50.times., at least about 100.times., at least about 250.times., at least about 500.times., or at least about 1000.times. greater than the cytokine receptor activating activity of the fusion protein. In other examples, a recombinant polypeptide is in conjugation with a cleavable moiety wherein the cleavable moiety is cleaved with reduced catalytic efficiency by one or more proteases than a reference polypeptide sequence.
[0291] In some embodiments, the cleavable moiety is resistant to proteolytic cleavage by one or more proteases. A cleavable moiety is resistant to a protease if the sequence comprises a binding site that is altered from the canonical cleavable motif sequence for the specific protease. In some embodiments, a binding site is altered compared to a reference sequence by making one or more substitutions in the protease cleavage motif sequence. For example, the protease cathepsin S cleaves the sequence GAVVRGA (SEQ ID NO: 240); a sequence is made to substitution can be made to substitute the arginine (R) with a glutamine (Q), thus changing a charged residue to a shorter, polar residue, reducing the ability of cathepsin S to bind and cleave the sequence. Such semi-conservative amino acid substitutions in protease target sequence motifs can lead to reduced binding ability, and such altered sequence motifs are therefore protease resistant. An uncleavable moiety can be made by inserting a disruptive amino acid into the protease target sequence motif, such as a proline (causes curves in the secondary structure of the peptide) or histidine (causes steric interference with other amino acid side chains). As used herein, a "protease-resistant" peptide linker is one with reduced or undetectable cleavage by one or more specified proteases. Exemplary protease-resistant peptide linkers can be tested, e.g., in vitro by incubation with a specific protease followed by analysis of the digestion products by western blot.
[0292] f. Polypeptide Substitutions
[0293] The polypeptides described herein can include components (e.g., the cytokine, the blocking moiety) that have the same amino acid sequence of the corresponding naturally occurring protein (e.g., IL-2, IL-15, HSA) or can have an amino acid sequence that differs from the naturally occurring protein so long as the desired function is maintained. It is understood that one way to define any known modifications and derivatives or those that might arise, of the disclosed proteins and nucleic acids that encode them is through defining the sequence variants in terms of identity to specific known reference sequences. Specifically disclosed are polypeptides and nucleic acids which have at least, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 percent identity to the chimeric polypeptides provided herein. For example, provided are polypeptides or nucleic acids that have at least, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 percent identity to the sequence of any of the nucleic acids or polypeptides described herein. This includes polypeptides or nucleic acids that have at least, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 percent identity to the sequence of the cleavable linkers provided herein. This also includes variants of the linker or inducible polypeptides that comprise 1, 2, 3, 4, 5, or 6 variants from the cleavage domain sequences. Those of skill in the art readily understand how to determine the identity of two polypeptides or two nucleic acids. For example, the identity can be calculated after aligning the two sequences so that the identity is at its highest level.
[0294] Another way of calculating identity can be performed by published algorithms. Optimal alignment of sequences for comparison may be conducted by the local identity algorithm of Smith and Waterman Adv. Appl. Math. 2:482 (1981), by the identity alignment algorithm of Needleman and Wunsch, J. Mol. Biol. 48:443 (1970), by the search for similarity method of Pearson and Lipman, Proc. Natl. Acad. Sci. USA 85:2444 (1988), by computerized implementations of these algorithms (GAP, BESTFIT, FASTA, and TFASTA in the Wisconsin Genetics Software Package, Genetics Computer Group, 575 Science Dr., Madison, Wis.), or by inspection.
[0295] The same types of identity can be obtained for nucleic acids by, for example, the algorithms disclosed in Zuker, Science 244:48-52 (1989); Jaeger et al., Proc. Natl. Acad. Sci. USA 86:7706-7710 (1989); Jaeger et al., Methods Enzymol. 183:281-306 (1989), which are herein incorporated by reference for at least material related to nucleic acid alignment. It is understood that any of the methods typically can be used and that in certain instances the results of these various methods may differ, but the skilled artisan understands if identity is found with at least one of these methods, the sequences would be said to have the stated identity, and be disclosed herein.
[0296] Protein modifications include amino acid sequence modifications. Modifications in amino acid sequence may arise naturally as allelic variations (e.g., due to genetic polymorphism), may arise due to environmental influence (e.g., by exposure to ultraviolet light), or may be produced by human intervention (e.g., by mutagenesis of cloned DNA sequences), such as induced point, deletion, insertion and substitution mutants. These modifications can result in changes in the amino acid sequence, provide silent mutations, modify a restriction site, or provide other specific mutations. Amino acid sequence modifications typically fall into one or more of three classes: substitutional, insertional or deletional modifications. Insertions include amino and/or carboxyl terminal fusions as well as intrasequence insertions of single or multiple amino acid residues. Insertions ordinarily will be smaller insertions than those of amino or carboxyl terminal fusions, for example, on the order of one to four residues. Deletions are characterized by the removal of one or more amino acid residues from the protein sequence. Typically, no more than about from 2 to 6 residues are deleted at any one site within the protein molecule. Amino acid substitutions are typically of single residues but can occur at a number of different locations at once; insertions usually will be on the order of about from 1 to 10 amino acid residues; and deletions will range about from 1 to 30 residues. Deletions or insertions preferably are made in adjacent pairs, i.e. a deletion of 2 residues or insertion of 2 residues. Substitutions, deletions, insertions or any combination thereof may be combined to arrive at a final construct. The mutations must not place the sequence out of reading frame and preferably will not create complementary regions that could produce secondary mRNA structure. Substitutional modifications are those in which at least one residue has been removed and a different residue inserted in its place. Such substitutions generally are made in accordance with the following Table 2 and are referred to as conservative substitutions.
TABLE-US-00006 TABLE 2 Exemplary amino acid substitutions Amino Acid Exemplary Substitutions Ala Ser, Gly, Cys Arg Lys, Gln, Met, Ile Asn Gln, His, Glu, Asp Asp Glu, Asn, Gln Cys Ser, Met, Thr Gln Asn, Lys, Glu, Asp Glu Asp, Asn, Gln Gly Pro, Ala His Asn, Gln Ile Leu, Val, Met Leu Ile, Val, Met Lys Arg, Gln, Met, Ile Met Leu, Ile, Val Phe Met, Leu, Tyr, Trp, His Ser Thr, Met, Cys Thr Ser, Met, Val Trp Tyr, Phe Tyr Trp, Phe, His Val Ile, Leu, Met
[0297] Modifications, including the specific amino acid substitutions, are made by known methods. For example, modifications are made by site specific mutagenesis of nucleotides in the DNA encoding the polypeptide, thereby producing DNA encoding the modification, and thereafter expressing the DNA in recombinant cell culture. Techniques for making substitution mutations at predetermined sites in DNA having a known sequence are well known, for example M13 primer mutagenesis and PCR mutagenesis.
[0298] Modifications can be selected to optimize binding. For example, affinity maturation techniques can be used to alter binding of the scFv by introducing random mutations inside the complementarity determining regions (CDRs). Such random mutations can be introduced using a variety of techniques, including radiation, chemical mutagens or error-prone PCR. Multiple rounds of mutation and selection can be performed using, for example, phage display.
[0299] The disclosure also relates to nucleic acids that encode the chimeric polypeptides described herein, and to the use of such nucleic acids to produce the chimeric polypeptides and for therapeutic purposes. For example, the invention includes DNA and RNA molecules (e.g., mRNA, self-replicating RNA) that encode a chimeric polypeptide and to the therapeutic use of such DNA and RNA molecules.
[0300] g. Exemplary Compositions
[0301] Exemplary fusion proteins of the invention combine the above described elements in a variety of orientations. The orientations described in this section are meant as examples and are not to be considered limiting.
[0302] In some embodiments, the fusion protein comprises a cytokine, a blocking moiety and a half-life extension element. In some embodiments, the cytokine is positioned between the half-life extension element and the blocking moiety. In some embodiments, the cytokine is N-terminal to the blocking moiety and the half-life extension element. In some such embodiments, the cytokine is proximal to the blocking moiety; in some such embodiments, the cytokine is proximal to the half-life extension element. At least one protease-cleavable linker must be included in all embodiments, such that the cytokine may be active upon cleavage. In some embodiments, the cytokine is C-terminal to the blocking moiety and the half-life extension element. Additional elements may be attached to one another by a cleavable linker, a non-cleavable linker, or by direct fusion.
[0303] In some embodiments, the blocking domains used are capable of extending half-life, and the cytokine is positioned between two such blocking domains. In some embodiments, the cytokine is positioned between two blocking domains, one of which is capable of extending half-life.
[0304] In some embodiments, two cytokines are included in the same construct. In some embodiments, the cytokines are connected to two blocking domains each (three in total in one molecule), with a blocking domain between the two cytokine domains. In some embodiments, one or more additional half-life extension domains may be included to optimize pharmacokinetic properties.
[0305] In some embodiments, three cytokines are included in the same construct. In some embodiments, the third cytokine may function to block the other two in place of a blocking domain between the two cytokines.
[0306] Preferred half-life extension elements for use in the fusion proteins are human serum albumin (HSA), an antibody or antibody fragment (e.g., scFV, dAb) which binds serum albumin, a human or humanized IgG, or a fragment of any of the foregoing. In some preferred embodiments, the blocking moiety is human serum albumin (HSA), or an antibody or antibody fragment which binds serum albumin, an antibody which binds the cytokine and prevents activation of binding or activation of the cytokine receptor, another cytokine, or a fragment of any of the foregoing. In preferred embodiments comprising an additional targeting domain, the targeting domain is an antibody which binds a cell surface protein which is enriched on the surface of cancer cells, such as EpCAM, FOLR1, and Fibronectin.
[0307] vii. Other Uses
[0308] The separation moieties disclosed herein can be used for antibody-antibiotic conjugates. The separation moiety disclosed herein connects or links an antimicrobial antibiotic to an antibody specific for a bacterial strain (e.g., Staphylococcus aureus Ab). The antibody-antibiotic conjugate does not display antibacterial activity when the antibody is linked to the antibiotic. However, upon internalization into host cells, the separation moiety is cleaved by proteases releasing the free antibiotic. The free antibiotic kills the intracellular bacteria.
[0309] The separation moieties disclosed herein can be used for applications in chemical probes use for detection and isolation of proteins. Chemical probes are designed based on small molecule interaction with proteins. The probes typically comprise a covalent binding motif in order for the probe to interact with the target protein, a detection/purification tag for visualization/purification of the protein target and a linker group. The separation moieties described herein can be incorporated to enable the target protein to be detected and isolated.
[0310] C. Methods of Treatment and Pharmaceutical Compositions
[0311] Further provided are methods of treating a subject with or at risk of developing an of a disease or disorder, such as proliferative disease, a tumorous disease, an inflammatory disease, an immunological disorder, an autoimmune disease, an infectious disease, a viral disease, an allergic reaction, a parasitic reaction, or graft-versus-host disease. The methods administering to a subject in need thereof an effective amount of a fusion protein as disclosed herein that is typically administered as a pharmaceutical composition. In some embodiments, the method further comprises selecting a subject with or at risk of developing such a disease or disorder. The pharmaceutical composition preferably comprises a blocked cytokine, fragment or mutein thereof that is activated at a site of inflammation. In one embodiment, the chimeric polypeptide comprises a cytokine polypeptide, fragment or mutein thereof and a serum half-life extension element. In another embodiment, the chimeric polypeptide comprises a cytokine polypeptide, fragment or mutein thereof and a blocking moiety, e.g. a steric blocking polypeptide, wherein the steric blocking polypeptide is capable of sterically blocking the activity of the cytokine polypeptide, fragment or mutein thereof. In another embodiment, the chimeric polypeptide comprises a cytokine polypeptide, fragment or mutein thereof, a blocking moiety, and a serum half-life extension element.
[0312] Inflammation is part of the complex biological response of body tissues to harmful stimuli, such as pathogens, damaged cells, or irritants, and is a protective response involving immune cells, blood vessels, and molecular mediators. The function of inflammation is to eliminate the initial cause of cell injury, clear out necrotic cells and tissues damaged from the original insult and the inflammatory process, and to initiate tissue repair. Inflammation can occur from infection, as a symptom or a disease, e.g., cancer, atherosclerosis, allergies, myopathies, HIV, obesity, or an autoimmune disease. An autoimmune disease is a chronic condition arising from an abnormal immune response to a self-antigen. Autoimmune diseases that may be treated with the polypeptides disclosed herein include but are not limited to lupus, celiac disease, diabetes mellitus type 1, Graves' disease, inflammatory bowel disease, multiple sclerosis, psoriasis, rheumatoid arthritis, and systemic lupus erythematosus.
[0313] The pharmaceutical composition can comprise one or more protease-cleavable linker sequences. The linker sequence serves to provide flexibility between polypeptides, such that each polypeptide is capable of inhibiting the activity of the first polypeptide. The linker sequence can be located between any or all of the cytokine polypeptide, fragment or mutein thereof, the blocking moiety, and serum half-life extension element. Optionally, the composition comprises, two, three, four, or five linker sequences. The linker sequence, two, three, or four linker sequences can be the same or different linker sequences. In one embodiment, the linker sequence comprises GGGGS (SEQ ID NO: 232), GSGSGS (SEQ ID NO: 233), or G(SGGG).sub.2SGGT (SEQ ID NO: 234). In another embodiment, the linker comprises a protease-cleavable sequence selected from group consisting of HSSKLQ (SEQ ID NO: 25), GPLGVRG (SEQ ID NO:221), IPVSLRSG (SEQ ID NO: 222), VPLSLYSG (SEQ ID NO: 223), or SGESPAYYTA (SEQ ID NO: 224). In some embodiments, the linker is cleaved by a protease selected from the group consisting of a kallikrein, thrombin, chymase, carboxypeptidase A, cathepsin G, an elastase, PR-3, granzyme M, a calpain, a matrix metalloproteinase (MMP), a plasminogen activator, a cathepsin, a caspase, a tryptase, or a tumor cell surface protease.
[0314] Further provided are methods of treating a subject with or at risk of developing cancer. The methods comprise administering to the subject in need thereof an effective amount of a chimeric polypeptide (a fusion protein) as disclosed herein that is typically administered as a pharmaceutical composition. In some embodiments, the method further comprises selecting a subject with or at risk of developing cancer. The pharmaceutical composition preferably comprises a blocked cytokine, fragment or mutein thereof that is activated at a tumor site. Preferably, the tumor is a solid tumor. The cancer may be a colon cancer, a lung cancer, a melanoma, a sarcoma, a renal cell carcinoma, a breast cancer,
[0315] The method can further involve the administration of one or more additional agents to treat cancer, such as chemotherapeutic agents (e.g., Adriamycin, Cerubidine, Bleomycin, Alkeran, Velban, Oncovin, Fluorouracil, Thiotepa, Methotrexate, Bisantrene, Noantrone, Thiguanine, Cytaribine, Procarabizine), immuno-oncology agents (e.g., anti-PD-L1, anti-CTLA4, anti-PD-1, anti-CD47, anti-GD2), cellular therapies (e.g, CAR-T, T-cell therapy), oncolytic viruses and the like.
[0316] Provided herein are pharmaceutical formulations or compositions containing the chimeric polypeptides and a pharmaceutically acceptable carrier. The herein provided compositions are suitable for administration in vitro or in vivo. By pharmaceutically acceptable carrier is meant a material that is not biologically or otherwise undesirable, i.e., the material is administered to a subject without causing undesirable biological effects or interacting in a deleterious manner with the other components of the pharmaceutical formulation or composition in which it is contained. The carrier is selected to minimize degradation of the active ingredient and to minimize adverse side effects in the subject.
[0317] Suitable carriers and their formulations are described in Remington: The Science and Practice ofPharmacy 21.sup.st Edition, David B. Troy, ed., Lippicott Williams & Wilkins (2005). Typically, an appropriate amount of a pharmaceutically acceptable salt is used in the formulation to render the formulation isotonic, although the formulate can be hypertonic or hypotonic if desired. Examples of the pharmaceutically acceptable carriers include, but are not limited to, sterile water, saline, buffered solutions like Ringer's solution, and dextrose solution. The pH of the solution is generally about 5 to about 8 or from about 7 to 7.5. Other carriers include sustained release preparations such as semipermeable matrices of solid hydrophobic polymers containing the immunogenic polypeptides. Matrices are in the form of shaped articles, e.g., films, liposomes, or microparticles. Certain carriers may be more preferable depending upon, for instance, the route of administration and concentration of composition being administered. Carriers are those suitable for administration of the chimeric polypeptides or nucleic acid sequences encoding the chimeric polypeptides to humans or other subjects.
[0318] The pharmaceutical formulations or compositions are administered in a number of ways depending on whether local or systemic treatment is desired and, on the area to be treated. The compositions are administered via any of several routes of administration, including topically, orally, parenterally, intravenously, intra-articularly, intraperitoneally, intramuscularly, subcutaneously, intracavity, transdermally, intrahepatically, intracranially, nebulization/inhalation, or by installation via bronchoscopy. In some embodiments, the compositions are administered locally (non-systemically), including intratumorally, intra-articularly, intrathecally, etc.
[0319] Preparations for parenteral administration include sterile aqueous or non-aqueous solutions, suspensions, and emulsions. Examples of non-aqueous solvents are propylene glycol, polyethylene glycol, vegetable oils such as olive oil, and injectable organic esters such as ethyl oleate. Aqueous carriers include water, alcoholic/aqueous solutions, emulsions or suspensions, including saline and buffered media. Parenteral vehicles include sodium chloride solution, Ringer's dextrose, dextrose and sodium chloride, lactated Ringer's, or fixed oils. Intravenous vehicles include fluid and nutrient replenishers, electrolyte replenishers (such as those based on Ringer's dextrose), and the like. Preservatives and other additives are optionally present such as, for example, antimicrobials, antioxidants, chelating agents, and inert gases and the like.
[0320] Formulations for topical administration include ointments, lotions, creams, gels, drops, suppositories, sprays, liquids, and powders. Conventional pharmaceutical carriers, aqueous, powder, or oily bases, thickeners and the like are optionally necessary or desirable.
[0321] Compositions for oral administration include powders or granules, suspension or solutions in water or non-aqueous media, capsules, sachets, or tables. Thickeners, flavorings, diluents, emulsifiers, dispersing aids or binders are optionally desirable.
[0322] Optionally, the chimeric polypeptides or nucleic acid sequences encoding the chimeric polypeptides are administered by a vector. There are a number of compositions and methods which can be used to deliver the nucleic acid molecules and/or polypeptides to cells, either in vitro or in vivo via, for example, expression vectors. These methods and compositions can largely be broken down into two classes: viral based delivery systems and non-viral based delivery systems. Such methods are well known in the art and readily adaptable for use with the compositions and methods described herein. Such compositions and methods can be used to transfect or transduce cells in vitro or in vivo, for example, to produce cell lines that express and preferably secrete the encoded chimeric polypeptide or to therapeutically deliver nucleic acids to a subject. The components of the chimeric nucleic acids disclosed herein typically are operably linked in frame to encode a fusion protein.
[0323] As used herein, plasmid or viral vectors are agents that transport the disclosed nucleic acids into the cell without degradation and include a promoter yielding expression of the nucleic acid molecule and/or polypeptide in the cells into which it is delivered. Viral vectors are, for example, Adenovirus, Adeno-associated virus, herpes virus, Vaccinia virus, Polio virus, Sindbis, and other RNA viruses, including these viruses with the HIV backbone. Also preferred are any viral families which share the properties of these viruses which make them suitable for use as vectors. Retroviral vectors, in general are described by Coffin et al., Retroviruses, Cold Spring Harbor Laboratory Press (1997), which is incorporated by reference herein for the vectors and methods of making them. The construction of replication-defective adenoviruses has been described (Berkner et al., J. Virol. 61:1213-20 (1987); Massie et al., Mol. Cell. Biol. 6:2872-83 (1986); Haj-Ahmad et al., J. Virol. 57:267-74 (1986); Davidson et al., J. Virol. 61:1226-39 (1987); Zhang et al., BioTechniques 15:868-72 (1993)). The benefit and the use of these viruses as vectors is that they are limited in the extent to which they can spread to other cell types, since they can replicate within an initial infected cell, but are unable to form new infectious viral particles. Recombinant adenoviruses have been shown to achieve high efficiency after direct, in vivo delivery to airway epithelium, hepatocytes, vascular endothelium, CNS parenchyma, and a number of other tissue sites. Other useful systems include, for example, replicating and host-restricted non-replicating vaccinia virus vectors.
[0324] The provided polypeptides and/or nucleic acid molecules can be delivered via virus like particles. Virus like particles (VLPs) consist of viral protein(s) derived from the structural proteins of a virus. Methods for making and using virus like particles are described in, for example, Garcea and Gissmann, Current Opinion in Biotechnology 15:513-7 (2004).
[0325] The provided polypeptides can be delivered by subviral dense bodies (DBs). DBs transport proteins into target cells by membrane fusion. Methods for making and using DBs are described in, for example, Pepperl-Klindworth et al., Gene Therapy 10:278-84 (2003).
[0326] The provided polypeptides can be delivered by tegument aggregates. Methods for making and using tegument aggregates are described in International Publication No. WO 2006/110728.
[0327] Non-viral based delivery methods can include expression vectors comprising nucleic acid molecules and nucleic acid sequences encoding polypeptides, wherein the nucleic acids are operably linked to an expression control sequence. Suitable vector backbones include, for example, those routinely used in the art such as plasmids, artificial chromosomes, BACs, YACs, or PACs. Numerous vectors and expression systems are commercially available from such corporations as Novagen (Madison, Wis.), Clonetech (Pal Alto, Calif.), Stratagene (La Jolla, Calif.), and Invitrogen/Life Technologies (Carlsbad, Calif.). Vectors typically contain one or more regulatory regions. Regulatory regions include, without limitation, promoter sequences, enhancer sequences, response elements, protein recognition sites, inducible elements, protein binding sequences, 5' and 3' untranslated regions (UTRs), transcriptional start sites, termination sequences, polyadenylation sequences, and introns. Such vectors can also be used to make the chimeric polypeptides by expression is a suitable host cell, such as CHO cells.
[0328] Preferred promoters controlling transcription from vectors in mammalian host cells may be obtained from various sources, for example, the genomes of viruses such as polyoma, Simian Virus 40 (SV40), adenovirus, retroviruses, hepatitis B virus, and most preferably cytomegalovirus (CMV), or from heterologous mammalian promoters, e.g. .beta.-actin promoter or EF1.alpha. promoter, or from hybrid or chimeric promoters (e.g., CMV promoter fused to the .beta.-actin promoter). Of course, promoters from the host cell or related species are also useful herein.
[0329] Enhancer generally refers to a sequence of DNA that functions at no fixed distance from the transcription start site and can be either 5' or 3' to the transcription unit. Furthermore, enhancers can be within an intron as well as within the coding sequence itself. hey are usually between 10 and 300 base pairs (bp) in length, and they function in cis. Enhancers usually function to increase transcription from nearby promoters. Enhancers can also contain response elements that mediate the regulation of transcription. While many enhancer sequences are known from mammalian genes (globin, elastase, albumin, fetoprotein, and insulin), typically one will use an enhancer from a eukaryotic cell virus for general expression. Preferred examples are the SV40 enhancer on the late side of the replication origin, the cytomegalovirus early promoter enhancer, the polyoma enhancer on the late side of the replication origin, and adenovirus enhancers.
[0330] The promoter and/or the enhancer can be inducible (e.g. chemically or physically regulated). A chemically regulated promoter and/or enhancer can, for example, be regulated by the presence of alcohol, tetracycline, a steroid, or a metal. A physically regulated promoter and/or enhancer can, for example, be regulated by environmental factors, such as temperature and light. Optionally, the promoter and/or enhancer region can act as a constitutive promoter and/or enhancer to maximize the expression of the region of the transcription unit to be transcribed. In certain vectors, the promoter and/or enhancer region can be active in a cell type specific manner. Optionally, in certain vectors, the promoter and/or enhancer region can be active in all eukaryotic cells, independent of cell type. Preferred promoters of this type are the CMV promoter, the SV40 promoter, the .beta.-actin promoter, the EF1.alpha. promoter, and the retroviral long terminal repeat (LTR).
[0331] The vectors also can include, for example, origins of replication and/or markers. A marker gene can confer a selectable phenotype, e.g., antibiotic resistance, on a cell. The marker product is used to determine if the vector has been delivered to the cell and once delivered is being expressed. Examples of selectable markers for mammalian cells are dihydrofolate reductase (DHFR), thymidine kinase, neomycin, neomycin analog G418, hygromycin, puromycin, and blasticidin. When such selectable markers are successfully transferred into a mammalian host cell, the transformed mammalian host cell can survive if placed under selective pressure. Examples of other markers include, for example, the E. coli lacZ gene, green fluorescent protein (GFP), and luciferase. In addition, an expression vector can include a tag sequence designed to facilitate manipulation or detection (e.g., purification or localization) of the expressed polypeptide. Tag sequences, such as GFP, glutathione S-transferase (GST), polyhistidine, c-myc, hemagglutinin, or FLAG.TM. tag (Kodak; New Haven, Conn.) sequences typically are expressed as a fusion with the encoded polypeptide. Such tags can be inserted anywhere within the polypeptide including at either the carboxyl or amino terminus.
[0332] As used herein, the terms peptide, polypeptide, or protein are used broadly to mean two or more amino acids linked by a peptide bond. Protein, peptide, and polypeptide are also used herein interchangeably to refer to amino acid sequences. It should be recognized that the term polypeptide is not used herein to suggest a particular size or number of amino acids comprising the molecule and that a peptide of the invention can contain up to several amino acid residues or more. As used throughout, subject can be a vertebrate, more specifically a mammal (e.g. a human, horse, cat, dog, cow, pig, sheep, goat, mouse, rabbit, rat, and guinea pig), birds, reptiles, amphibians, fish, and any other animal. The term does not denote a particular age or sex. Thus, adult and newborn subjects, whether male or female, are intended to be covered. As used herein, patient or subject may be used interchangeably and can refer to a subject with a disease or disorder (e.g. cancer). The term patient or subject includes human and veterinary subjects.
[0333] A subject at risk of developing a disease or disorder can be genetically predisposed to the disease or disorder, e.g., have a family history or have a mutation in a gene that causes the disease or disorder, or show early signs or symptoms of the disease or disorder. A subject currently with a disease or disorder has one or more than one symptom of the disease or disorder and may have been diagnosed with the disease or disorder.
[0334] The methods and agents as described herein are useful for both prophylactic and therapeutic treatment. For prophylactic use, a therapeutically effective amount of the chimeric polypeptides or chimeric nucleic acid sequences encoding the chimeric polypeptides described herein are administered to a subject prior to onset (e.g., before obvious signs of cancer or inflammation) or during early onset (e.g., upon initial signs and symptoms of cancer or inflammation). Prophylactic administration can occur for several days to years prior to the manifestation of symptoms of cancer or inflammation. Prophylactic administration can be used, for example, in the preventative treatment of subjects diagnosed with a genetic predisposition to cancer. Therapeutic treatment involves administering to a subject a therapeutically effective amount of the chimeric polypeptides or nucleic acid sequences encoding the chimeric polypeptides described herein after diagnosis or development of cancer or inflammation (e.g., an autoimmune disease). Prophylactic use may also apply when a patient is undergoing a treatment, e.g., a chemotherapy, in which inflammation is expected.
[0335] According to the methods taught herein, the subject is administered an effective amount of the agent (e.g., a chimeric polypeptide). The terms effective amount and effective dosage are used interchangeably. The term effective amount is defined as any amount necessary to produce a desired physiologic response. Effective amounts and schedules for administering the agent may be determined empirically, and making such determinations is within the skill in the art. The dosage ranges for administration are those large enough to produce the desired effect in which one or more symptoms of the disease or disorder are affected (e.g., reduced or delayed). The dosage should not be so large as to cause substantial adverse side effects, such as unwanted cross-reactions, anaphylactic reactions, and the like. Generally, the dosage will vary with the age, condition, sex, type of disease, the extent of the disease or disorder, route of administration, or whether other drugs are included in the regimen, and can be determined by one of skill in the art. The dosage can be adjusted by the individual physician in the event of any contraindications. Dosages can vary and can be administered in one or more dose administrations daily, for one or several days. Guidance can be found in the literature for appropriate dosages for given classes of pharmaceutical products.
[0336] As used herein the terms treatment, treat, or treating refers to a method of reducing the effects of a disease or condition or symptom of the disease or condition. Thus, in the disclosed method, treatment can refer to a 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, or 100% reduction in the severity of an established disease or condition or symptom of the disease or condition. For example, a method for treating a disease is considered to be a treatment if there is a 10% reduction in one or more symptoms of the disease in a subject as compared to a control. Thus, the reduction can be a 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, or any percent reduction in between 10% and 100% as compared to native or control levels. It is understood that treatment does not necessarily refer to a cure or complete ablation of the disease, condition, or symptoms of the disease or condition.
[0337] As used herein, the terms prevent, preventing, and prevention of a disease or disorder refers to an action, for example, administration of the chimeric polypeptide or nucleic acid sequence encoding the chimeric polypeptide, that occurs before or at about the same time a subject begins to show one or more symptoms of the disease or disorder, which inhibits or delays onset or exacerbation of one or more symptoms of the disease or disorder. As used herein, references to decreasing, reducing, or inhibiting include a change of 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% or greater as compared to a control level. Such terms can include but do not necessarily include complete elimination.
[0338] IL-2 variants have been developed that are selective for IL2R.alpha..beta..gamma. relative to IL2R.beta..gamma. (Shanafelt, A. B., et al., 2000, Nat Biotechnol. 18:1197-202; Cassell, D. J., et. al., 2002, Curr Pharm Des., 8:2171-83). These variants have amino acid substitutions which reduce their affinity for IL2RB. Because IL-2 has undetectable affinity for IL2RG, these variants consequently have reduced affinity for the IL2R.beta..gamma. receptor complex and reduced ability to activate IL2R.beta..gamma.-expressing cells but retain the ability to bind IL2RA and the ability to bind and activate the IL2R.alpha..beta..gamma. receptor complex.
[0339] One of these variants, IL2/N88R (Bay 50-4798), was clinically tested as a low-toxicity version of IL-2 as an immune system stimulator, based on the hypothesis that IL2R.beta..gamma.-expressing NK cells are a major contributor to toxicity. Bay 50-4798 was shown to selectively stimulate the proliferation of activated T cells relative to NK cells, and was evaluated in phase I/I clinical trials in cancer patients (Margolin, K., et. al., 2007, Clin Cancer Res., 13:3312-9) and HIV patients (Davey, R. T., et. al., 2008, J Interferon Cytokine Res., 28:89-100). These clinical trials showed that Bay 50-4798 was considerably safer and more tolerable than aldesleukin, and also showed that it increased the levels of CD4+CD25+ T cells, a cell population enriched in Treg cells. Subsequent to these trials, research in the field more fully established the identity of Treg cells and demonstrated that Treg cells selectively express IL2R.alpha..beta..gamma. (reviewed in Malek, T. R., et al., 2010, Immunity, 33:153-65). Based on this new research, it can now be understood that IL2R.alpha..beta..gamma. selective agonists should be selective for Treg cells.
[0340] In addition, mutants can be made that selectively alter the affinity for the CD25 chain relative to native I1-2.
[0341] IL-2 can be engineered to produce mutants that bind the IL-2R complex generally or the IL-2Ra subunit specifically with an affinity that differs from that of the corresponding wild-type IL-2 or of a presently available mutant (referred to as C125S, as the cysteine residue at position 125 is replaced with a serine residue).
[0342] Accordingly, the present invention features mutant interleukin-2 (IL-2*) polypeptides that include an amino acid sequence that is at least 80% identical to wild-type IL-2 (e.g., 85, 87, 90, 95, 97, 98, or 99% identical) and that bind, as compared to WT IL-2, with higher to the IL-2 trimeric receptor relative to the dimeric IL-2 receptor. Typically, the muteins will also bind an IL-2 receptor .alpha. subunit (IL-2R.alpha.) with an affinity that is greater than the affinity with which wild type IL-2 binds the IL-2R.alpha.. The amino acid sequence within mutant IL-2 polypeptides can vary from SEQ ID NO:1 (UniProtKB accession number P60568) by virtue of containing (or only containing) one or more amino acid substitutions, which may be considered conservative or non-conservative substitutions. Non-naturally occurring amino acids can also be incorporated. Alternatively, or in addition, the amino acid sequence can vary from SEQ ID NO:1 (which may be considered the "reference" sequence) by virtue of containing and addition and/or deletion of one or more amino acid residues. More specifically, the amino acid sequence can differ from that of SEQ ID NO:1 by virtue of a mutation at least one of the following positions of SEQ ID NO:1: 1, 4, 8, 9, 10, 11, 13, 15, 26, 29, 30, 31, 35, 37, 46, 48, 49, 54, 61, 64, 67, 68, 69, 71, 73, 74, 75, 76, 79, 88, 89, 90, 92, 99, 101, 103, 114, 125, 128, or 133 (or combinations thereof). As noted, as few as one of these positions may be altered, as may two, three, four, five, six, seven, eight, nine, ten, or 11 or more (including up to all) of the positions. For example, the amino acid sequence can differ from SEQ ID NO:1 at positions 69 and 74 and further at one or more of positions 30, 35, and 128. The amino acid sequence can also differ from SEQ ID NO:2 (as disclosed in U.S. Pat. No. 7,569,215, incorporated herein by reference) at one of the following sets of positions: (a) positions 64, 69, and 74; (b) positions 69, 74, and 101; (c) positions 69, 74, and 128; (d) positions 30, 69, 74, and 103; (e) positions 49, 69, 73, and 76; (f) positions 69, 74, 101, and 133; (g) positions 30, 69, 74, and 128; (h) positions 69, 74, 88, and 99; (i) positions 30, 69, 74, and 128; (j) positions 9, 11, 35, 69, and 74; (k) positions 1, 46, 49, 61, 69, and 79; (l) positions 48, 68, 71, 90, 103, and 114; (m) positions 4, 10, 11, 69, 74, 88, and 133; (n) positions 15, 30 31, 35, 48, 69, 74, and 92; (O) positions 30, 68, 69, 71, 74, 75, 76, and 90; (p) positions 30, 31, 37, 69, 73, 74, 79, and 128; (q) positions 26, 29, 30, 54, 67, 69, 74, and 92; (r) positions 8, 13, 26, 30, 35, 37, 69, 74, and 92; and (s) positions 29, 31, 35, 37, 48, 69, 71, 74, 88, and 89. Aside from mutations at these positions, the amino acid sequence of the mutant IL-2 polypeptide can otherwise be identical to SEQ ID NO:1. With respect to specific substitutions, the amino acid sequence can differ from SEQ ID NO:1 by virtue of having one or more of the following mutations: A1T, S4P, K8R, K9T, T10A, Q11R, Q13R, E15K, N26D, N29S, N30S, N30D, N30T, Y31H, Y31C, K35R, T37A, T37R, M46L, K48E, K49R, K49E, K54R, E61D, K64R, E67G, E68D, V69A, N71T, N71A, N71R, A73V, Q74P, S75P, K76E, K76R, H79R, N88D, I89V, N90H, I92T, S99P, T101A, F103S, I114V, I128T, I128A, T133A, or T133N. Our nomenclature is consistent with that of the scientific literature, where the single letter code of the amino acid in the wild-type or reference sequence is followed by its position within the sequence and then by the single letter code of the amino acid with which it is replaced. Thus, A1T designates a substitution of the alanine residue a position 1 with threonine. Other mutant polypeptides within the scope of the invention include those that include a mutant of SEQ ID NO:2 having substitutions at V69 (e.g. A) and Q74 (e.g., P). For example, the amino acid sequence can include one of the following sets of mutations with respect to SEQ ID NO:2: (a) K64R, V69A, and Q74P; (b) V69A, Q74P, and T101A; (c) V69A, Q74P, and I128T; (d) N30D, V69A, Q74P, and F103S; (e) K49E, V69A, A73V, and K76E; (f) V69A, Q74P, T101A, and T133N; (g) N30S, V69A, Q74P, and I128A; (h) V69A, Q74P, N88D, and S99P; (i) N30S, V69A, Q74P, and I128T; (j) K9T, Q11R, K35R, V69A, and Q74P; (k) A1T, M46L, K49R, E61D, V69A, and H79R; (l) K48E, E68D, N71T, N90H, F103S, and I114V; (m) S4P, T10A, Q1R, V69A, Q74P, N88D, and T133A; (n) E15K, N30S Y31H, K35R, K48E, V69A, Q74P, and I92T; (o) N30S, E68D, V69A, N71A, Q74P, S75P, K76R, and N90H; (p) N30S, Y31C, T37A, V69A, A73V, Q74P, H79R, and I128T; (q) N26D, N29S, N30S, K54R, E67G, V69A, Q74P, and I92T; (r) K8R, Q13R, N26D, N30T, K35R, T37R, V69A, Q74P, and I92T; and (s) N29S, Y31H, K35R, T37A, K48E, V69A, N71R, Q74P, N88D, and I89V. SEQ ID NO:2 is disclosed in U.S. Pat. No. 7,569,215, which is incorporated herein by reference as an exemplary IL-2 polypeptide sequence that can be used in the invention.
[0343] As noted above, any of the mutant IL-2 polypeptides disclosed herein can include the sequences described; they can also be limited to the sequences described and otherwise identical to SEQ ID NO:1. Moreover, any of the mutant IL-2 polypeptides described herein can optionally include a substitution of the cysteine residue at position 125 with another residue (e.g., serine) and/or can optionally include a deletion of the alanine residue at position 1 of SEQ ID NO:1.
[0344] The mutant IL-2 polypeptides disclosed herein can bind to the IL-2R.alpha. subunit with a K.sub.d of less than about 28 nM (e.g., less than about 25 nM; less than about 5 nM; about 1 nM; less than about 500 pM; or less than about 100 pM). More specifically, a mutant IL-2 polypeptide can have an affinity equilibrium constant less than 1.0 nM (e.g., about 0.8, 0.6, 0.4, or 0.2 nM). Affinity can also be expressed as a relative rate of dissociation from an IL-2R.alpha. subunit or from an IL-2 receptor complex (e.g., a complex expressed on the surface of a cell or otherwise membrane bound). For example, the mutant IL-2 polypeptides can dissociate from, e.g., IL-2R.alpha., at a decreased rate relative to a wild-type polypeptide or to an IL-2 based therapeutic, e.g., IL-2*. Alternatively, affinity can be characterized as the time, or average time, an IL-2* polypeptide persists on, for example, the surface of a cell expressing an IL-2R. For example, an IL-2*polypeptide can persist on the receptor for at least about 2, 5, 10, 50, 100, or 250 times (or more).
[0345] Disclosed are materials, compositions, and components that can be used for, can be used in conjunction with, can be used in preparation for, or are products of the disclosed methods and compositions. These and other materials are disclosed herein, and it is understood that when combinations, subsets, interactions, groups, etc. of these materials are disclosed that while specific reference of each various individual and collective combinations and permutations of these compounds may not be explicitly disclosed, each is specifically contemplated and described herein. For example, if a method is disclosed and discussed and a number of modifications that can be made to a number of molecules including the method are discussed, each and every combination and permutation of the method, and the modifications that are possible are specifically contemplated unless specifically indicated to the contrary. Likewise, any subset or combination of these is also specifically contemplated and disclosed. his concept applies to all aspects of this disclosure including, but not limited to, steps in methods using the disclosed compositions. Thus, if there are a variety of additional steps that can be performed, it is understood that each of these additional steps can be performed with any specific method steps or combination of method steps of the disclosed methods, and that each such combination or subset of combinations is specifically contemplated and should be considered disclosed.
[0346] Publications cited herein and the material for which they are cited are hereby specifically incorporated by reference in their entireties.
6. INCORPORATION BY REFERENCE
[0347] All publications, patents, and patent applications mentioned in this specification are herein incorporated by reference to the same extent as if each individual publication, patent, or patent application was specifically and individually indicated to be incorporated by reference. However, the citation of a reference herein should not be construed as an acknowledgement that such reference is prior art to the present invention. To the extent that any of the definitions or terms provided in the references incorporated by reference differ from the terms and discussion provided herein, the present terms and definitions control.
7. EXAMPLES
[0348] The following are examples of methods and compositions of the invention. It is understood that various other embodiments may be practiced, given the general description provided herein.
Example 1. Detection of IL-2, IL-2 Mutein, IL-2R.alpha. and IL-2R.gamma. in Fusion Proteins by ELISA
[0349] IL-2 mutein is detected with a commercially available antibody, e.g., the anti-IL-2 monoclonal (JES6-1A12) (BD Pharmingen; San Jose, Calif.). A positive control is used to show whether the monoclonal antibody recognizes the cytokine or mutein. Antibodies against IL-2R.alpha. and IL-2R.gamma. chain are also used. Wells of a 96-well plate are coated with an antibody (2.5 .mu.g/ml) in PBS. Wells are blocked with 5% non-fat milk in PBS with 0.2% Tween.RTM. 20 (PBS-M-Tw) and fusion proteins are added for 1-2 hours at 37.degree. C. After washing, an anti-IL-2 biotin-labeled antibody, e.g., JES5H4 (BD Pharmingen) is added and binding is detected using Streptavidin HRP (Southern Biotechnology Associates; Birmingham, Ala.). The ELISA plate is developed by adding 50 .mu.l O-phenylenediamine (OPD) (Sigma-Aldrich) in 0.1M Citrate pH 4.5 and 0.04% H.sub.2O.sub.2, stopped by adding 50 .mu.l/well 2N H.sub.2SO.sub.4 and the absorbance was read at 490 nm.
Example 2: Protease Cleavage of Fusion Protein by MMP9 Protease
[0350] One of skill in the art would be familiar with methods of setting up protein cleavage assay. 100 .mu.g of protein in 1.times.PBS pH 7.4 were cleaved with 1 .mu.g active MMP9 (Sigma catalog #SAE0078-50 or Enzo catalog BML-SE360) and incubated at room temperature for up to 16 hours. Digested protein is subsequently used in functional assays or stored at -80.degree. C. prior to testing. Extent of cleavage was monitored by SDS PAGE using methods well known in the art. As shown in FIGS. 10, 13, 18A, 18b, and 27A full cleavage of the fusion proteins by MMP9 protease is seen.
Example 3: CTLL-2 Assay
[0351] CTLL2 cells (ATCC) were plated in suspension at a concentration of 500,000 cells/well in culture media with or without 40 mg/ml human serum albumin (HSA) and stimulated with a dilution series of recombinant hIL2 or activatable hIL2 for 72 hours at 37.degree. C. and 5% CO.sub.2. Activity of uncleaved and cleaved activatable hIL2 was tested. Cleaved activatable hIL2 was generated by incubation with active MMP9. Cell activity was assessed using a CellTiter-Glo.RTM. (Promega) luminescence-based cell viability assay. Results are shown in FIGS. 8A-8F, FIGS. 9A-9Z, FIG. 25C.
Example 4: Protease Cleavage of the IL-2/IL-2R.alpha./IL-2R.gamma. Chimeric Polypeptide Results in Increased Accessibility to Antibodies and Biologically Active IL-2 Mutein
[0352] The IL-2 mutein fusion proteins are biochemically characterized before and after cleavage with a protease, e.g., PSA. Immunoblot analyses will show that the fusion proteins can be cleaved by PSA and that there is an increase in intensity of the predicted low molecular weight cleavage product of approximately 20 kDa reactive with an anti-IL-2 antibody after treatment of the samples with PSA. The degree of cleavage is dependent upon the amount of PSA as well as the time of incubation. Interestingly, when the fusion protein is analyzed before and after PSA treatment by ELISA, it was found that the apparent amount of IL-2 is increased after PSA cleavage. In this experiment, there is an approximately 2 or 4-fold increase in the apparent amount of IL-2 detected using this sandwich ELISA depending on the construct, suggesting that the antibody binding is partially hindered in the intact fusion protein. Aliquots of the same samples are also analyzed after PSA treatment using the CTLL-2 cell line that requires IL-2 for growth and survival and the viability of cells can be ascertained using the colorimetric MTT assay. In this assay, the more a supernatant can be diluted, the more biologically active IL-2 it contains, and there is an increase in the amount of biologically active IL-2 after PSA cleavage. The amount of IL-2 mutein increase will suggest that after PSA cleavage there is an increase in the predicted low molecular weight cleavage fragment of approximately 20 kDa reactive with an anti-IL-2 antibody, an increase in antibody accessibility, and most importantly, an increase in the amount of biologically active IL-2 mutein.
Example 5. In Vivo Delivery of a Protease Activated Fusion Protein Results in Decreased Tumor Growth
[0353] The chimeric polypeptide is examined to determine if it could have biological effects in vivo. For these experiments a system is used in which tumor cells injected intraperitoneally rapidly and preferentially attach and grow initially on the milky spots, a series of organized immune aggregates found on the omentum (Gerber et al., Am. J. Pathol. 169:1739-52 (2006)). This system offers a convenient way to examine the effects of fusion protein treatment on tumor growth since fusion proteins can be delivered intraperitoneally multiple times and tumor growth can be analyzed by examining the dissociated omental cells. For these experiments, the Colon 38 cell line, a rapidly growing tumor cell line that expresses both MMP2 and MMP9 in vitro, may be used. The omental tissue normally expresses a relatively small amount of MMP2 and MMP9, but, when Colon 38 tumor is present on the omentum, MMP levels increase. Using this tumor model, the ability of IL-2 mutein fusion proteins to affect tumor growth is examined. Colon 38 cells are injected intraperitoneally, allowed to attach and grow for 1 day, and then treated daily with fusion protein intraperitoneally. At day 7, the animals are sacrificed and the omenta examined for tumor growth using flow cytometry and by a colony-forming assay.
Example 6: Determination of Antigen Affinity by Flow Cytometry
[0354] Activatable interleukin proteins are tested for their binding affinities to human CD20.sup.+ cells and cynomolgus CD20.sup.+ cells.
[0355] CD20.sup.+ cells are incubated with 100 .mu.L of serial dilutions of the activatable interleukin proteins and at least one protease. After washing three times with FACS buffer the cells are incubated with 0.1 mL of 10 .mu.g/mL mouse monoclonal anti-idiotype antibody in the same buffer for 45 min on ice. After a second washing cycle, the cells are incubated with 0.1 mL of 15 g/mL FITC-conjugated goat anti-mouse IgG antibodies under the same conditions as before. As a control, cells are incubated with the anti-His IgG followed by the FITC-conjugated goat anti-mouse IgG antibodies without the activatable IL2 proteins. The cells were then washed again and resuspended in 0.2 mL of FACS buffer containing 2 .mu.g/mL propidium iodide (PI) in order to exclude dead cells. The fluorescence of 1.times.10.sup.4 living cells is measured using a Beckman-Coulter FC500 MPL flow cytometer using the MXP software (Beckman-Coulter, Krefeld, Germany) or a Millipore Guava EasyCyte flow cytometer using the Incyte software (Merck Millipore, Schwalbach, Germany). Mean fluorescence intensities of the cell samples are calculated using CXP software (Beckman-Coulter, Krefeld, Germany) or Incyte software (Merck Millipore, Schwalbach, Germany). After subtracting the fluorescence intensity values of the cells stained with the secondary and tertiary reagents alone the values are then used for calculation of the K.sub.D values with the equation for one-site binding (hyperbola) of the GraphPad Prism (version 6.00 for Windows, GraphPad Software, La Jolla Calif. USA).
[0356] CD20 binding and crossreactivity are assessed on the human CD20.sup.+ tumor cell lines. The K.sub.D ratio of crossreactivity is calculated using the K.sub.D values determined on the CHO cell lines expressing either recombinant human or recombinant cynomolgus antigens.
Example 7: Cytotoxicity Assay
[0357] Activatable interleukin protein is evaluated in vitro on its mediation of immune response to CD20.sup.+ target cells.
[0358] Fluorescence labeled CD20.sup.+ REC-1 cells (a Mantle cell lymphoma cell line, ATCC CRL-3004) are incubated with isolated PBMC of random donors or CB15 T-cells (standardized T-cell line) as effector cells in the presence of the activatable IL2 protein and at least one protease. After incubation for 4 h at 37.degree. C. in a humidified incubator, the release of the fluorescent dye from the target cells into the supernatant is determined in a spectro-fluorimeter. Target cells incubated without the activatable IL2 protein and target cells totally lysed by the addition of saponin at the end of the incubation serve as negative and positive controls, respectively.
[0359] Based on the measured remaining living target cells, the percentage of specific cell lysis is calculated according to the following formula: [1-(number of living targets.sub.(sample)/number of living targets.sub.(spontaneous)].times.100%. Sigmoidal dose response curves and EC.sub.50 values are calculated by non-linear regression/4-parameter logistic fit using the GraphPad Software. The lysis values obtained for a given antibody concentration are used to calculate sigmoidal dose-response curves by 4 parameter logistic fit analysis using the Prism.RTM. GraphPad.RTM. software.
Example 8: Pharmacokinetics of Activatable Interleukin Proteins
[0360] Activatable interleukin protein is evaluated for half-time elimination in animal studies.
[0361] The activatable IL2 protein is administered to cynomolgus monkeys as a 0.5 mg/kg bolus injection into the saphenous vein. Another cynomolgus monkey group receives a comparable IL2 construct in size but lacking a serum half-life extension element. A third and fourth group receive an IL2 construct with serum half-life extension element and a cytokine with CD20 and serum half-life extension elements respectively, and both comparable in size to the activatable interleukin protein. Each test group consists of 5 monkeys. Serum samples are taken at indicated time points, serially diluted, and the concentration of the proteins is determined using a binding ELISA to CD20.
[0362] Pharmacokinetic analysis is performed using the test article plasma concentrations. Group mean plasma data for each test article conforms to a multi-exponential profile when plotted against the time post-dosing. The data are fit by a standard two-compartment model with bolus input and first-order rate constants for distribution and elimination phases. The general equation for the best fit of the data for i.v. administration is: c(t)=Ae.sup.-.alpha.t+Be.sup.-.beta.t, where c(t) is the plasma concentration at time t, A and B are intercepts on the Y-axis, and .alpha. and .beta. are the apparent first-order rate constants for the distribution and elimination phases, respectively. The .alpha.-phase is the initial phase of the clearance and reflects distribution of the protein into all extracellular fluid of the animal, whereas the second or .beta.-phase portion of the decay curve represents true plasma clearance. Methods for fitting such equations are well known in the art. For example, A=D/V(.alpha.-k21)/(.alpha.-.beta.), B=D/V(.beta.-k21)/(.alpha.-.beta.), and .alpha. and .beta. (for .alpha.>.beta.) are roots of the quadratic equation: r.sup.2+(k12+k21+k10)r+k21k10=0 using estimated parameters of V=volume of distribution, k10=elimination rate, k12=transfer rate from compartment 1 to compartment 2 and k21=transfer rate from compartment 2 to compartment 1, and D=the administered dose.
[0363] Data analysis: Graphs of concentration versus time profiles are made using KaleidaGraph (KaleidaGraph.TM. V. 3.09 Copyright 1986-1997. Synergy Software. Reading, Pa.). Values reported as less than reportable (LTR) are not included in the PK analysis and are not represented graphically. Pharmacokinetic parameters are determined by compartmental analysis using WinNonlin software (WinNonlin.RTM. Professional V. 3.1 WinNonlin.RTM. Copyright 1998-1999. Pharsight Corporation. Mountain View, Calif.). Pharmacokinetic parameters are computed as described in Ritschel W A and Kearns G L, 1999, IN: Handbook of Basic Pharmacokinetics Including Clinical Applications. 5th edition, American Pharmaceutical Assoc., Washington, D.C.
[0364] It is expected that the activatable interleukin protein has improved pharmacokinetic parameters such as an increase in elimination half-time as compared to proteins lacking a serum half-life extension element.
Example 9: Xenograft Tumor Model
[0365] Activatable IL2 protein is evaluated in a xenograft model.
[0366] Female immune-deficient NOD/scid mice are sub-lethally irradiated (2 Gy) and subcutaneously inoculated with 4.times.10.sup.6 Ramos RA1 cells into the right dorsal flank. When tumors reach 100 to 200 mm.sup.3, animals are allocated into 3 treatment groups. Groups 2 and 3 (8 animals each) are intraperitoneally injected with 1.5.times.10.sup.7 activated human T-cells. Three days later, animals from Group 3 are subsequently treated with a total of 9 intravenous doses of 50 .mu.g activatable interleukin protein. Groups 1 and 2 are only treated with vehicle. Body weight and tumor volume are determined for 30 days.
[0367] It is expected that animals treated with the activatable interleukin protein have a statistically significant delay in tumor growth in comparison to the respective vehicle-treated control group.
Example 10: Mouse IFN.gamma. WEHI Cell Survival Assay
[0368] WEHI279 cells (ATCC) were plated in suspension at a concentration of 25,000 cells/well in culture media with or without 1.5% human serum albumin (HSA) and stimulated with a dilution series of recombinant mIFN.gamma. or inducible mIFN.gamma. for 72 hours at 37.degree. C. and 5% CO.sub.2. Activity of uncleaved and cleaved inducible mIFN.gamma. was tested. Cleaved inducible mIFN.gamma. was generated by incubation with active MMP9. Cell survival was assessed using a CellTiter-Glo (Promega) luminescence-based cell viability assay. The EC50 values for cleaved inducible mIFN.gamma. molecules were at least 100.times. more potent than un-cleaved inducible mIFN.gamma. molecules. As shown in FIGS. 16A-16, greater inducibility was seen in assays wherein the culture medium contained human serum albumin.
Example 11: Reserved
Example 12: Mouse IFN.gamma. B16 Reporter Cell Assay
[0369] B16-Blue IFN.gamma. cells (InvivoGen) were plated at a concentration of 75,000 cells/well in culture media with or without 1.5% human serum albumin (HSA) and stimulated with a dilution series of recombinant mIFN.gamma. or inducible mIFN.gamma. for 24 hours at 37.degree. C. and 5% CO.sub.2. Activity of uncleaved and cleaved inducible mIFN.gamma. was tested. Cleaved inducible mIFN.gamma. was generated by incubation with active MMP9. Supernatants were harvested, and SEAP activation was assessed by adding QUANTI-Blue Reagent (InvivoGen), incubating at 37.degree. C. for 2 hours, and measuring absorbance at 620 nm. The EC50 values for cleaved inducible mIFN.gamma. molecules were at least 100.times. more potent than un-cleaved inducible mIFN.gamma. molecules. Results are shown in e.g., FIG. 19A-19B, FIGS. 22A-22B, FIGS. 23A-23B. This experiment was repeated with for IFN.alpha. conjugates using B16-Blue IFN.alpha./.beta. cells. The EC50 values for cleaved inducible mIFN.alpha. molecules were at least 100.times. more potent than un-cleaved inducible mIFN.alpha. molecules. See FIGS. 20A-20B.
Example 13. In Vivo Delivery of a Protease Activated Fusion Protein Results in Decreased Tumor Growth
[0370] The chimeric polypeptide is examined to determine if it could have biological effects in vivo. For these experiments a system is used in which tumor cells injected intraperitoneally rapidly and preferentially attach and grow initially on the milky spots, a series of organized immune aggregates found on the omentum (Gerber et al., Am. J. Pathol. 169:1739-52 (2006)). This system offers a convenient way to examine the effects of fusion protein treatment on tumor growth since fusion proteins can be delivered intraperitoneally multiple times and tumor growth can be analyzed by examining the dissociated omental cells. For these experiments, the Colon 38 cell line, a rapidly growing tumor cell line that expresses both MMP2 and MMP9 in vitro, may be used. The omental tissue normally expresses a relatively small amount of MMP2 and MMP9, but, when Colon 38 tumor is present on the omentum, MMP levels increase. Using this tumor model, the ability of IFN fusion proteins to affect tumor growth is examined. Colon 38 cells are injected intraperitoneally, allowed to attach and grow for 1 day, and then treated daily with fusion protein intraperitoneally. At day 7, the animals are sacrificed and the omenta examined for tumor growth using flow cytometry and by a colony-forming assay.
Example 14: The Chimeric Polypeptide was Examined to Determine its Biological Effects in Vivo
[0371] The MC38 cell line, a rapidly growing colon adenocarcinoma cell line that expresses MMP9 in vitro, was used. Using this tumor model, the ability of IFN.gamma. fusion proteins to affect tumor growth was examined. MC38 cells were injected subcutaneously, allowed to grow for 10-14 days, and then treated with fusion protein twice weekly intraperitoneally for a total of four doses. As a comparator, wild type mIFN.gamma. was administered at the dose levels indicated, twice daily for 2 weeks on a 5 day on/2 day off schedule (10 total doses). Tumor growth and body weight were monitored approximately twice per week for two weeks.
Example 15: Construction of an Exemplary IFN.gamma. Protein Targeting CD20
[0372] 15.1 Generation of an Activatable Cytokine Domain
[0373] An IFN.gamma. polypeptide capable of binding to CD20 polypeptide present in a tumor or on a tumor cell is produced as follows. A nucleic acid is produced that contains nucleic acid sequences: (1) encoding an IFN.gamma. polypeptide sequence and (2) one or more polypeptide linkers. Activatable IFN.gamma. plasmid constructs can have optional Flag, His or other affinity tags, and are electroporated into HEK293 or other suitable human or mammalian cell lines and purified. Validation assays include T cell activation assays using T cells responsive to IFN.gamma. stimulation in the presence of a protease.
[0374] 15.2 Generation of a scFv CD20 Binding Domain
[0375] CD20 is one of the cell surface proteins present on B-lymphocytes. CD20 antigen is found in normal and malignant pre-B and mature B lymphocytes, including those in over 90% of B-cell non-Hodgkin's lymphomas (NHL). The antigen is absent in hematopoietic stem cells, activated B lymphocytes (plasma cells) and normal tissue. As such, several antibodies mostly of murine origin have been described: 1F5, 2B8/C2B8, 2H7, and 1H4.
[0376] Human or humanized anti-CD20 antibodies are therefore used to generate scFv sequences for CD20 binding domains of an activatable IFN.gamma. protein. DNA sequences coding for human or humanized VL and VH domains are obtained, and the codons for the constructs are, optionally, optimized for expression in cells from Homo sapiens. The order in which the VL and VH domains appear in the scFv is varied (i.e., VL-VH, or VH-VL orientation), and three copies of the "G4S" (SEQ ID NO: 241) or "G.sub.4S" (SEQ ID NO: 241) subunit (G.sub.4S).sub.3 (SEQ ID NO: 242) connect the variable domains to create the scFv domain. Anti-CD20 scFv plasmid constructs can have optional Flag, His or other affinity tags, and are electroporated into HEK293 or other suitable human or mammalian cell lines and purified. Validation assays include binding analysis by FACS, kinetic analysis using Proteon, and staining of CD20-expressing cells.
[0377] 15.3 Cloning of DNA Expression Constructs Encoding the Activatable IFN.gamma. Protein
[0378] The activatable IFN.gamma. construct with protease cleavage site domains is used to construct an activatable IFN.gamma. protein in combination with an anti-CD20 scFv domain and a serum half-life extension element (e.g., a HSA binding peptide or VH domain). For expression of an activatable IFN.gamma. protein in CHO cells, coding sequences of all protein domains are cloned into a mammalian expression vector system. In brief, gene sequences encoding the activatable IFN.gamma. domain, serum half-life extension element, and CD20 binding domain along with peptide linkers L1 and L2 are separately synthesized and subcloned. The resulting constructs are then ligated together in the order of CD20 binding domain-L1-IFN.gamma. subunit 1-L2-protease cleavage domain-L3-IFN.gamma. subunit2-L4-anti-CD20 scFv-L5-serum half-life extension element to yield a final construct. All expression constructs are designed to contain coding sequences for an N-terminal signal peptide and a C-terminal hexahistidine (6.times.His)-tag (SEQ ID NO: 243) to facilitate protein secretion and purification, respectively.
[0379] 15.4 Expression of Activatable IFN.gamma. Proteins in Stably Transfected CHO Cells
[0380] A CHO cell expression system (Flp-In.RTM., Life Technologies), a derivative of CHO-K1 Chinese Hamster ovary cells (ATCC, CCL-61) (Kao and Puck, Proc. Natl. Acad. Sci. USA 1968; 60(4):1275-81), is used. Adherent cells are subcultured according to standard cell culture protocols provided by Life Technologies.
[0381] For adaption to growth in suspension, cells are detached from tissue culture flasks and placed in serum-free medium. Suspension-adapted cells are cryopreserved in medium with 10% DMSO.
[0382] Recombinant CHO cell lines stably expressing secreted activatable IFN.gamma. proteins are generated by transfection of suspension-adapted cells. During selection with the antibiotic Hygromycin B viable cell densities are measured twice a week, and cells are centrifuged and resuspended in fresh selection medium at a maximal density of 0.1.times.10.sup.6 viable cells/mL. Cell pools stably expressing activatable IFN.gamma. proteins are recovered after 2-3 weeks of selection at which point cells are transferred to standard culture medium in shake flasks. Expression of recombinant secreted proteins is confirmed by performing protein gel electrophoresis or flow cytometry. Stable cell pools are cryopreserved in DMSO containing medium.
[0383] Activatable IFN.gamma. proteins are produced in 10-day fed-batch cultures of stably transfected CHO cell lines by secretion into the cell culture supernatant. Cell culture supernatants are harvested after 10 days at culture viabilities of typically >75%. Samples are collected from the production cultures every other day and cell density and viability are assessed. On day of harvest, cell culture supernatants are cleared by centrifugation and vacuum filtration before further use.
[0384] Protein expression titers and product integrity in cell culture supernatants are analyzed by SDS-PAGE.
[0385] 15.5 Purification of Activatable IFN.gamma. Proteins
[0386] Activatable IFN.gamma. proteins are purified from CHO cell culture supernatants in a two-step procedure. The constructs are subjected to affinity chromatography in a first step followed by preparative size exclusion chromatography (SEC) on Superdex 200 in a second step. Samples are buffer-exchanged and concentrated by ultrafiltration to a typical concentration of >1 mg/mL. Purity and homogeneity (typically >90%) of final samples are assessed by SDS PAGE under reducing and non-reducing conditions, followed by immunoblotting using an anti-HSA or anti idiotype antibody as well as by analytical SEC, respectively. Purified proteins are stored at aliquots at -80.degree. C. until use.
Example 16: Determination of Antigen Affinity by Flow Cytometry
[0387] The activatable IFN.gamma. proteins are tested for their binding affinities to human CD20.sup.+ cells and cynomolgus CD20.sup.+ cells.
[0388] CD20.sup.+ cells are incubated with 100 .mu.L of serial dilutions of the activatable IFN.gamma. proteins and at least one protease. After washing three times with FACS buffer the cells are incubated with 0.1 mL of 10 .mu.g/mL mouse monoclonal anti-idiotype antibody in the same buffer for 45 min on ice. After a second washing cycle, the cells are incubated with 0.1 mL of 15 .mu.g/mL FITC-conjugated goat anti-mouse IgG antibodies under the same conditions as before. As a control, cells are incubated with the anti-His IgG followed by the FITC-conjugated goat anti-mouse IgG antibodies without the activatable IFN.gamma. proteins. The cells were then washed again and resuspended in 0.2 mL of FACS buffer containing 2 .mu.g/mL propidium iodide (PI) in order to exclude dead cells. The fluorescence of 1.times.10.sup.4 living cells is measured using a Beckman-Coulter FC500 MPL flow cytometer using the MXP software (Beckman-Coulter, Krefeld, Germany) or a Millipore Guava EasyCyte flow cytometer using the Incyte software (Merck Millipore, Schwalbach, Germany). Mean fluorescence intensities of the cell samples are calculated using CXP software (Beckman-Coulter, Krefeld, Germany) or Incyte software (Merck Millipore, Schwalbach, Germany). After subtracting the fluorescence intensity values of the cells stained with the secondary and tertiary reagents alone the values are then used for calculation of the K.sub.D values with the equation for one-site binding (hyperbola) of the GraphPad Prism (version 6.00 for Windows, GraphPad Software, La Jolla Calif. USA).
[0389] CD20 binding and cross-reactivity are assessed on the human CD20.sup.+ tumor cell lines. The K.sub.D ratio of cross-reactivity is calculated using the K.sub.D values determined on the CHO cell lines expressing either recombinant human or recombinant cynomolgus antigens.
Example 17: Cytotoxicity Assay
[0390] The activatable IFN.gamma. protein is evaluated in vitro on its mediation of immune response to CD20.sup.+ target cells.
[0391] Fluorescence labeled CD20.sup.+ REC-1 cells (a Mantle cell lymphoma cell line, ATCC CRL-3004) are incubated with isolated PBMC of random donors or CB15 T-cells (standardized T-cell line) as effector cells in the presence of the activatable IFN.gamma. protein and at least one protease. After incubation for 4 h at 37.degree. C. in a humidified incubator, the release of the fluorescent dye from the target cells into the supernatant is determined in a spectrofluorometer. Target cells incubated without the activatable IFN.gamma. protein and target cells totally lysed by the addition of saponin at the end of the incubation serve as negative and positive controls, respectively.
[0392] Based on the measured remaining living target cells, the percentage of specific cell lysis is calculated according to the following formula: [1-(number of living targets.sub.(sample)/number of living targets.sub.(spontaneous)].times.100%. Sigmoidal dose response curves and EC.sub.50 values are calculated by non-linear regression/4-parameter logistic fit using the GraphPad Software. The lysis values obtained for a given antibody concentration are used to calculate sigmoidal dose-response curves by 4 parameter logistic fit analysis using the Prism software.
Example 18: Pharmacokinetics of Activatable IFN.gamma. Proteins
[0393] The activatable IFN.gamma. protein is evaluated for half-time elimination in animal studies.
[0394] The activatable IFN.gamma. protein is administered to cynomolgus monkeys as a 0.5 mg/kg bolus injection into the saphenous vein. Another cynomolgus monkey group receives a comparable cytokine in size but lacking a serum half-life extension element. A third and fourth group receive a cytokine with serum half-life extension elements and a cytokine with CD20 and serum half-life extension elements respectively, and both comparable in size to the activatable IFN.gamma. protein. Each test group consists of 5 monkeys. Serum samples are taken at indicated time points, serially diluted, and the concentration of the proteins is determined using a binding ELISA to CD20.
[0395] Pharmacokinetic analysis is performed using the test article plasma concentrations. Group mean plasma data for each test article conforms to a multi-exponential profile when plotted against the time post-dosing. The data are fit by a standard two-compartment model with bolus input and first-order rate constants for distribution and elimination phases. The general equation for the best fit of the data for i.v. administration is: c(t)=Ae.sup.-.alpha.t+Be.sup.-.beta.t, where c(t) is the plasma concentration at time t, A and B are intercepts on the Y-axis, and .alpha. and .beta. are the apparent first-order rate constants for the distribution and elimination phases, respectively. The .alpha.-phase is the initial phase of the clearance and reflects distribution of the protein into all extracellular fluid of the animal, whereas the second or .beta.-phase portion of the decay curve represents true plasma clearance. Methods for fitting such equations are well known in the art. For example, A=D/V(.alpha.-k21)/(.alpha.-.beta.), B=D/V(.beta.-k21)/(.alpha.-.beta.), and .alpha. and .beta. (for .alpha.>.beta.) are roots of the quadratic equation: r.sup.2+(k12+k21+k10)r+k21k10=0 using estimated parameters of V=volume of distribution, k10=elimination rate, k12=transfer rate from compartment 1 to compartment 2 and k21=transfer rate from compartment 2 to compartment 1, and D=the administered dose.
[0396] Data analysis: Graphs of concentration versus time profiles are made using KaleidaGraph (KaleidaGraph.TM. V. 3.09 Copyright 1986-1997. Synergy Software. Reading, Pa.). Values reported as less than reportable (LTR) are not included in the PK analysis and are not represented graphically. Pharmacokinetic parameters are determined by compartmental analysis using WinNonlin software (WinNonlin.RTM. Professional V. 3.1 WinNonlin.TM. Copyright 1998-1999. Pharsight Corporation. Mountain View, Calif.). Pharmacokinetic parameters are computed as described in Ritschel W A and Kearns G L, 1999, IN: Handbook of Basic Pharmacokinetics Including Clinical Applications. 5th edition, American Pharmaceutical Assoc., Washington, D.C.
[0397] It is expected that the activatable IFN protein has improved pharmacokinetic parameters such as an increase in elimination half-time as compared to proteins lacking a serum half-life extension element.
Example 19: Xenograft Tumor Model
[0398] The activatable IFN.gamma. protein is evaluated in a xenograft model.
[0399] Female immune-deficient NOD/scid mice are sub-lethally irradiated (2 Gy) and subcutaneously inoculated with 4.times.10.sup.6 Ramos RA1 cells into the right dorsal flank. When tumors reach 100 to 200 mm.sup.3, animals are allocated into 3 treatment groups. Groups 2 and 3 (8 animals each) are intraperitoneally injected with 1.5.times.10.sup.7 activated human T-cells. Three days later, animals from Group 3 are subsequently treated with a total of 9 intravenous doses of 50 .mu.g activatable IFN.gamma. protein. Groups 1 and 2 are only treated with vehicle. Body weight and tumor volume are determined for 30 days.
[0400] It is expected that animals treated with the activatable IFN.gamma. protein have a statistically significant delay in tumor growth in comparison to the respective vehicle-treated control group.
[0401] While preferred embodiments of the present invention have been shown and described herein, it will be obvious to those skilled in the art that such embodiments are provided by way of example only. Numerous variations, changes, and substitutions will now occur to those skilled in the art without departing from the invention. It should be understood that various alternatives to the embodiments of the invention described herein may be employed in practicing the invention. It is intended that the following claims define the scope of the invention and that methods and structures within the scope of these claims and their equivalents be covered thereby.
Example 20: HEK Blue Assay
[0402] HEK-Blue IL12 cells (InvivoGen) were plated in suspension at a concentration of 250,000 cells/well in culture media with or without 40 mg/ml human serum albumin (HSA) and stimulated with a dilution series of recombinant hIL12, chimeric IL12 (mouse p35/human p40) or activatable hIL12 for 24 hours at 37.degree. C. and 5% CO.sub.2. Activity of uncleaved and cleaved activatable hIL12 was tested. Cleaved inducible hIL12 was generated by incubation with active MMP9. IL12 activity was assessed by quantification of Secreted Alkaline Phosphatase (SEAP) activity using the reagent QUANTI-Blue (InvivoGen), a colorimetric based assay.
[0403] HEK-Blue IL2 cells (InvivoGen) were plated in suspension at a concentration of 50,000 cells/well in culture media with or without 15-40 mg/ml human serum albumin (HSA) and stimulated with a dilution series of recombinant hIL2 or activatable hIL2 for 24 hours at 37.degree. C. and 5% CO.sub.2. Activity of uncleaved and cleaved activatable hIL2 was tested. Cleaved inducible hIL2 was generated by incubation with active MMP9 or another protease. IL2 activity was assessed by quantification of Secreted Alkaline Phosphatase (SEAP) activity using the reagent QUANTI-Blue (InvivoGen), a colorimetric based assay. Results are shown in FIGS. 59-62.
Example 21: Splenocyte T-Blast Assay
[0404] T-Blasts were induced from murine splenocytes with a 6-day incubation with PHA and a 24 hr incubation with recombinant hIL12. T-blasts were then plated in suspension at a concentration of 200,000 cells/well in culture media with or without 40 mg/ml human serum albumin (HSA) and stimulated with a dilution series of recombinant hIL12 or chimeric IL12 (mouse p35/human p40) or mouse IL12 for 72 hours at 37.degree. C. and 5% CO2. Activity of uncleaved and cleaved IL12 was tested. Cleaved inducible hIL12 was generated by incubation with active MMP9. IL12 activity was assessed by downstream quantification of IFN.gamma. production using a mIFN.gamma. alphaLISA.
Example 22: In Vivo Delivery of a Protease Activated Fusion Protein Results in Decreased Tumor Growth
[0405] The chimeric polypeptide is examined to determine if it could have biological effects in vivo. For these experiments a system is used in which tumor cells injected intraperitoneally rapidly and preferentially attach and grow initially on the milky spots, a series of organized immune aggregates found on the omentum (Gerber et al., Am. J. Pathol. 169:1739-52 (2006)). This system offers a convenient way to examine the effects of fusion protein treatment on tumor growth since fusion proteins can be delivered intraperitoneally multiple times and tumor growth can be analyzed by examining the dissociated omental cells. For these experiments, the Colon 38 cell line, a rapidly growing tumor cell line that expresses both MMP2 and MMP9 in vitro, may be used. The omental tissue normally expresses a relatively small amount of MMP2 and MMP9, but, when Colon 38 tumor is present on the omentum, MMP levels increase. Using this tumor model, the ability of IL-2 mutein fusion proteins to affect tumor growth is examined. Colon 38 cells are injected intraperitoneally, allowed to attach and grow for 1 day, and then treated daily with fusion protein intraperitoneally. At day 7, the animals are sacrificed and the omenta examined for tumor growth using flow cytometry and by a colony-forming assay.
Example 23A: Construction of an Exemplary Activatable Interleukin Protein Targeting CD20
[0406] 23.1 Generation of an Activatable Interleukin Domain
[0407] The human IL-12p35 chain canonical sequence is UniProt Accession No. P29459. The human IL-12p40 chain canonical sequence is UniProt Accession No. P29460. IL-12p35 and IL-12p40 are cloned into an expression construct. A protease cleavage site is included between the IL-12p35 and IL-12p40 domains. An IL-12 polypeptide capable of binding to CD20 polypeptide present in a tumor or on a tumor cell is produced as follows. A nucleic acid is produced that contains nucleic acid sequences: (1) encoding an IFN.gamma. polypeptide sequence and (2) one or more polypeptide linkers. Activatable interleukin plasmid constructs can have optional Flag, His or other affinity tags, and are electroporated into HEK293 or other suitable human or mammalian cell lines and purified. Validation assays include T cell activation assays using T cells responsive to IL-12 stimulation in the presence of a protease.
[0408] 23.2 Generation of a scFv CD20 Binding Domain
[0409] CD20 is one of the cell surface proteins present on B-lymphocytes. CD20 antigen is found in normal and malignant pre-B and mature B lymphocytes, including those in over 90/of B-cell non-Hodgkin's lymphomas (NHL). The antigen is absent in hematopoietic stem cells, activated B lymphocytes (plasma cells) and normal tissue. As such, several antibodies mostly of murine origin have been described: 1F5, 2B8/C2B8, 2H7, and 1H4.
[0410] Human or humanized anti-CD20 antibodies are therefore used to generate scFv sequences for CD20 binding domains of an activatable interleukin protein. DNA sequences coding for human or humanized VL and VH domains are obtained, and the codons for the constructs are, optionally, optimized for expression in cells from Homo sapiens. The order in which the VL and VH domains appear in the scFv is varied (i.e., VL-VH, or VH-VL orientation), and three copies of the "G4S" (SEQ ID NO: 241) or "G.sub.4S" (SEQ ID NO: 241) subunit (G.sub.4S).sub.3 (SEQ ID NO: 242) connect the variable domains to create the scFv domain. Anti-CD20 scFv plasmid constructs can have optional Flag, His or other affinity tags, and are electroporated into HEK293 or other suitable human or mammalian cell lines and purified. Validation assays include binding analysis by FACS, kinetic analysis using Proteon, and staining of CD20-expressing cells.
[0411] 23.3 Cloning of DNA Expression Constructs Encoding the Activatable Interleukin Protein
[0412] The activatable interleukin construct with protease cleavage site domains are used to construct an activatable interleukin protein in combination with an anti-CD20 scFv domain and a serum half-life extension element (e.g., a HSA binding peptide or VH domain). For expression of an activatable interleukin protein in CHO cells, coding sequences of all protein domains are cloned into a mammalian expression vector system. In brief, gene sequences encoding the activatable interleukin domain, serum half-life extension element, and CD20 binding domain along with peptide linkers L1 and L2 are separately synthesized and subcloned. The resulting constructs are then ligated together in the order of CD20 binding domain-L1-IL-12p35-L2-protease cleavage domain-L3-IL-12p40-L4-anti-CD20 scFv-L5-serum half-life extension element to yield a final construct. All expression constructs are designed to contain coding sequences for an N-terminal signal peptide and a C-terminal hexahistidine (6.times.His)-tag (SEQ ID NO: 243) to facilitate protein secretion and purification, respectively.
[0413] 23.4 Expression of Activatable Interleukin Proteins in Stably Transfected CHO Cells
[0414] A CHO cell expression system (Flp-In.RTM., Life Technologies), a derivative of CHO-K1 Chinese Hamster ovary cells (ATCC, CCL-61) (Kao and Puck, Proc. Nat. Acad Sci USA 1968; 60(4):1275-81), is used. Adherent cells are subcultured according to standard cell culture protocols provided by Life Technologies.
[0415] For adaption to growth in suspension, cells are detached from tissue culture flasks and placed in serum-free medium. Suspension-adapted cells are cryopreserved in medium with 10% DMSO.
[0416] Recombinant CHO cell lines stably expressing secreted activatable interleukin proteins are generated by transfection of suspension-adapted cells. During selection with the antibiotic Hygromycin B viable cell densities are measured twice a week, and cells are centrifuged and resuspended in fresh selection medium at a maximal density of 0.1.times.10.sup.6 viable cells/mL. Cell pools stably expressing activatable interleukin proteins are recovered after 2-3 weeks of selection at which point cells are transferred to standard culture medium in shake flasks. Expression of recombinant secreted proteins is confirmed by performing protein gel electrophoresis or flow cytometry. Stable cell pools are cryopreserved in DMSO containing medium.
[0417] Activatable interleukin proteins are produced in 10-day fed-batch cultures of stably transfected CHO cell lines by secretion into the cell culture supernatant. Cell culture supernatants are harvested after 10 days at culture viabilities of typically >75%. Samples are collected from the production cultures every other day and cell density and viability are assessed. On day of harvest, cell culture supernatants are cleared by centrifugation and vacuum filtration before further use.
[0418] Protein expression titers and product integrity in cell culture supernatants are analyzed by SDS-PAGE.
[0419] 23.5 Purification of Activatable Interleukin Proteins
[0420] Activatable interleukin proteins are purified from CHO cell culture supernatants in a two-step procedure. The constructs are subjected to affinity chromatography in a first step followed by preparative size exclusion chromatography (SEC) on Superdex 200 in a second step. Samples are buffer-exchanged and concentrated by ultrafiltration to a typical concentration of >1 mg/mL. Purity and homogeneity (typically >90%) of final samples are assessed by SDS PAGE under reducing and non-reducing conditions, followed by immunoblotting using an anti-HSA or anti idiotype antibody as well as by analytical SEC, respectively. Purified proteins are stored at aliquots at -80.degree. C. until use.
Example 23B: Determination of Antigen Affinity by Flow Cytometry
[0421] Activatable interleukin proteins are tested for their binding affinities to human CD20.sup.+ cells and cynomolgus CD20.sup.+ cells.
[0422] CD20.sup.+ cells are incubated with 100 .mu.L of serial dilutions of the activatable interleukin proteins and at least one protease. After washing three times with FACS buffer the cells are incubated with 0.1 mL of 10 .mu.g/mL mouse monoclonal anti-idiotype antibody in the same buffer for 45 min on ice. After a second washing cycle, the cells are incubated with 0.1 mL of 15 .mu.g/mL FITC-conjugated goat anti-mouse IgG antibodies under the same conditions as before. As a control, cells are incubated with the anti-His IgG followed by the FITC-conjugated goat anti-mouse IgG antibodies without the activatable interleukin proteins. The cells were then washed again and resuspended in 0.2 mL of FACS buffer containing 2 .mu.g/mL propidium iodide (PI) in order to exclude dead cells. The fluorescence of 1.times.10.sup.4 living cells is measured using a Beckman-Coulter FC500 MPL flow cytometer using the MXP software (Beckman-Coulter, Krefeld, Germany) or a Millipore Guava EasyCyte flow cytometer using the Incyte software (Merck Millipore, Schwalbach, Germany). Mean fluorescence intensities of the cell samples are calculated using CXP software (Beckman-Coulter, Krefeld, Germany) or Incyte software (Merck Millipore, Schwalbach, Germany). After subtracting the fluorescence intensity values of the cells stained with the secondary and tertiary reagents alone the values are then used for calculation of the K.sub.D values with the equation for one-site binding (hyperbola) of the GraphPad Prism (version 6.00 for Windows, GraphPad Software, La Jolla Calif. USA).
[0423] CD20 binding and cross-reactivity are assessed on the human CD20.sup.+ tumor cell lines. The K.sub.D ratio of cross-reactivity is calculated using the K.sub.D values determined on the CHO cell lines expressing either recombinant human or recombinant cynomolgus antigens.
Example 24: Cytotoxicity Assay
[0424] The activatable interleukin protein is evaluated in vitro on its mediation of immune response to CD20.sup.+ target cells.
[0425] Fluorescence labeled CD20.sup.+ REC-1 cells (a Mantle cell lymphoma cell line, ATCC CRL-3004) are incubated with isolated PBMC of random donors or CB15 T-cells (standardized T-cell line) as effector cells in the presence of the activatable interleukin protein and at least one protease. After incubation for 4 h at 37.degree. C. in a humidified incubator, the release of the fluorescent dye from the target cells into the supernatant is determined in a spectrofluorometer. Target cells incubated without the activatable interleukin protein and target cells totally lysed by the addition of saponin at the end of the incubation serve as negative and positive controls, respectively.
[0426] Based on the measured remaining living target cells, the percentage of specific cell lysis is calculated according to the following formula: [1-(number of living targets.sub.(sample)/number of living targets.sub.(spontaneous))].times.100%. Sigmoidal dose response curves and EC.sub.50 values are calculated by non-linear regression/4-parameter logistic fit using the GraphPad Software. The lysis values obtained for a given antibody concentration are used to calculate sigmoidal dose-response curves by 4 parameter logistic fit analysis using the Prism software.
Example 25: Pharmacokinetics of Activatable Interleukin Proteins
[0427] The activatable interleukin protein is evaluated for half-time elimination in animal studies.
[0428] The activatable interleukin protein is administered to cynomolgus monkeys as a 0.5 mg/kg bolus injection into the saphenous vein. Another cynomolgus monkey group receives a comparable cytokine in size but lacking a serum half-life extension element. A third and fourth group receive a cytokine with serum half-life extension elements and a cytokine with CD20 and serum half-life extension elements respectively, and both comparable in size to the activatable interleukin protein. Each test group consists of 5 monkeys. Serum samples are taken at indicated time points, serially diluted, and the concentration of the proteins is determined using a binding ELISA to CD20.
[0429] Pharmacokinetic analysis is performed using the test article plasma concentrations. Group mean plasma data for each test article conforms to a multi-exponential profile when plotted against the time post-dosing. The data are fit by a standard two-compartment model with bolus input and first-order rate constants for distribution and elimination phases. The general equation for the best fit of the data for i.v. administration is: c(t)=Ae.sup.-.alpha.t+Be.sup.-.beta.t, where c(t) is the plasma concentration at time t, A and B are intercepts on the Y-axis, and .alpha. and .beta. are the apparent first-order rate constants for the distribution and elimination phases, respectively. The .alpha.-phase is the initial phase of the clearance and reflects distribution of the protein into all extracellular fluid of the animal, whereas the second or .beta.-phase portion of the decay curve represents true plasma clearance. Methods for fitting such equations are well known in the art. For example, A=D/V(.alpha.-k21)/(.alpha.-.beta.), B=D/V(.beta.-k21)/(.alpha.-.beta.), and .alpha. and .beta. (for .alpha.>.beta.) are roots of the quadratic equation: r.sup.2+(k12+k21+k10)r+k21k10=0 using estimated parameters of V=volume of distribution, k10=elimination rate, k12=transfer rate from compartment 1 to compartment 2 and k21=transfer rate from compartment 2 to compartment 1, and D=the administered dose.
[0430] Data analysis: Graphs of concentration versus time profiles are made using KaleidaGraph (KaleidaGraph.TM. V. 3.09 Copyright 1986-1997. Synergy Software. Reading, Pa.). Values reported as less than reportable (LTR) are not included in the PK analysis and are not represented graphically. Pharmacokinetic parameters are determined by compartmental analysis using WinNonlin software (WinNonlin.RTM. Professional V. 3.1 WinNonlin.TM. Copyright 1998-1999. Pharsight Corporation. Mountain View, Calif.). Pharmacokinetic parameters are computed as described in Ritschel W A and Kearns G L, 1999, IN: Handbook of Basic Pharmacokinetics Including Clinical Applications. 5th edition, American Pharmaceutical Assoc., Washington, D.C.
[0431] It is expected that the activatable interleukin protein has improved pharmacokinetic parameters such as an increase in elimination half-time as compared to proteins lacking a serum half-life extension element.
Example 26: Xenograft Tumor Model
[0432] Activatable interleukin protein is evaluated in a xenograft model.
[0433] Female immune-deficient NOD/scid mice are sub-lethally irradiated (2 Gy) and subcutaneously inoculated with 4.times.10.sup.6 Ramos RA1 cells into the right dorsal flank. When tumors reach 100 to 200 mm.sup.3, animals are allocated into 3 treatment groups. Groups 2 and 3 (8 animals each) are intraperitoneally injected with 1.5.times.10.sup.7 activated human T-cells. Three days later, animals from Group 3 are subsequently treated with a total of 9 intravenous doses of 50 .mu.g activatable interleukin protein. Groups 1 and 2 are only treated with vehicle. Body weight and tumor volume are determined for 30 days.
TABLE-US-00007 TABLE 3 Summary of the treatment modes Gr. N Agent Formulation dose Route Schedule .sup. 1.sup.# 10 Vehicle -- ip biwk x 3 2 7 ACP16 700 .mu.g/animal ip biwk x 3 3 7 ACP16 230 .mu.g/animal ip biwk x 3 4 7 ACP16 70 .mu.g/animal ip biwk x 3 5 7 ACP16 55 ug/animal ip biwk x 3 6 7 ACP16 17 .mu.g/animal ip biwk x 3 7 7 ACP132 361 .mu.g/animal ip biwk x 3 8 7 ACP132 119 .mu.g/animal ip biwk x 3 9 7 ACP132 36 .mu.g/animal ip biwk x 3 10 7 ACP132 28 .mu.g/animal ip biwk x 3 11 7 ACP132 9 .mu.g/animal ip biwk x 3 12 7 ACP21 540 .mu.g/animal ip biwk x 3 13 7 ACP21 177 .mu.g/animal ip biwk x 3 14 7 ACP21 54 .mu.g/animal ip biwk x 3 15 7 ACP21 42 .mu.g/animal ip biwk x 3 16 7 ACP21 13 .mu.g/animal ip biwk x 3 17 7 ACP133 210 .mu.g/animal ip bid x 5 then 2-day pause then bid x 5 then 2-day pause 18 7 ACP133 105 .mu.g/animal ip bid x 5 then 2-day pause then bid x 5 then 2-day pause 19 7 ACP133 40 .mu.g/animal ip bid x 5 then 2-day pause then bid x 5 then 2-day pause 20 7 ACP133 3 .mu.g/animal ip bid x 5 then 2-day pause then bid x 5 then 2-day pause .sup.#Control Group
[0434] It is expected that animals treated with the activatable interleukin protein have a statistically significant delay in tumor growth in comparison to the respective vehicle-treated control group.
[0435] While preferred embodiments of the present invention have been shown and described herein, it will be obvious to those skilled in the art that such embodiments are provided by way of example only. Numerous variations, changes, and substitutions will now occur to those skilled in the art without departing from the invention. It should be understood that various alternatives to the embodiments of the invention described herein may be employed in practicing the invention. It is intended that the following claims define the scope of the invention and that methods and structures within the scope of these claims and their equivalents be covered thereby.
[0436] The MC38 cell line, a rapidly growing colon adenocarcinoma cell line that expresses MMP9 in vitro, was used. Using this tumor model, the ability of fusion proteins to affect tumor growth was examined.
Example 27A: MC38 IL-2POC
[0437] 27A.1 Agents and Treatment
[0438] Additional studies were carried out in non-tumor bearing animals as described below.
TABLE-US-00008 TABLE 4 Summarizes the treatment regime. Gr. N Agent Formulation dose Route Schedule 1 8 Vehicle -- ip biwk x 4 2 8 ACP16 700 .mu.g/animal ip biwk x 4 3 8 ACP16 230 .mu.g/animal ip biwk x 4 4 8 ACP16 70 .mu.g/animal ip biwk x 4 8 8 ACP153 700 .mu.g/animal ip biwk x 4 9 8 ACP153 230 .mu.g/animal ip biwk x 4 10 8 ACP153 70 .mu.g/animal ip biwk x 4 11 8 ACP154 700 .mu.g/animal ip biwk x 4 12 8 ACP154 230 .mu.g/animal ip biwk x 4 13 8 ACP154 70 .mu.g/animal ip biwk x 4 14 8 ACP155 700 .mu.g/animal ip biwk x 4 15 8 ACP155 230 .mu.g/animal ip biwk x 4 16 8 ACP155 70 .mu.g/animal ip biwk x 4 17 8 ACP156 700 .mu.g/animal ip biwk x 4 18 8 ACP156 230 .mu.g/animal ip biwk x 4 19 8 ACP156 70 .mu.g/animal ip biwk x 4 20 8 ACP157 700 .mu.g/animal ip biwk x 4 21 8 ACP157 230 .mu.g/animal ip biwk x 4 22 8 ACP157 70 .mu.g/animal ip biwk x 4
TABLE-US-00009 TABLE 5 Describes the constructs used in the MC38 IL-2POC animal study. Construct Name Description MW ACP16 IL2-X-HSA-LX-blocker Fusion protein- 58256 6xHis ACP133 IL-2 with C term 6x His 16462 ACP132 IL2-L-HSA 29996 ACP21 IL2-XL-blocker Fusion protein-6xHis 44843
Example 27: MC3IL-2
TABLE-US-00010 [0439] TABLE 6 Summarizes the treatment regime. Gr. N Agent Formulation dose Route Schedule .sup. 1.sup.# 12 Vehicle -- ip biwk x 2 2 8 ACP16 4.4 .mu.g/animal ip biwk x 2 3 8 ACP16 17 .mu.g/animal ip biwk x 2 4 8 ACP16 70 .mu.g/animal ip biwk x 2 5 8 ACP16 232 .mu.g/animal ip biwk x 2 6 8 ACP130 19 .mu.g/animal ip biwk x 2 7 8 ACP130 45 .mu.g/animal ip biwk x 2 8 8 ACP130 180 .mu.g/animal ip biwk x 2 9 8 ACP130 600 .mu.g/animal ip biwk x 1 12 8 ACP124 17 .mu.g/animal ip biwk x 2 13 8 ACP124 70 .mu.g/animal ip biwk x 2 14 8 ACP124 230 .mu.g/animal ip biwk x 2 15 8 ACP124 700 .mu.g/animal ip biwk x 2 16 8 IL-2- 12 .mu.g/animal ip bid x 5 then 2-day WTI pause then bid x 5 then 2-day pause 17 8 IL-2- 36 .mu.g/animal ip bid x 5 then 2-day WTI pause then bid x 5 then 2-day pause .sup.#Control Group
[0440] 27B.1 Procedure
[0441] Mice were anaesthetized with isoflurane for implant of cells to reduce the ulcerations. 308 CR female C57BL/6 mice were set up with 5.times.105 MC38 tumor cells in 0% Matrigel sc in flank. Cell Injection Volume was 0.1 mL/mouse. Mouse age at start date was 8 to 12 weeks. Pair matches were performed when tumors reach an average size of 100-150 mm.sup.3 and begin treatment. Body weights were taken at initiation and then biweekly to the end. Caliper measurements were taken biweekly to the end. Any adverse reactions were to be reported immediately. Any individual animal with a single observation of >than 30% body weight loss or three consecutive measurements of >25% body weight loss was euthanized. Any group with a mean body weight loss of >20% or >10% mortality stopped dosing; the group was not euthanized and recovery is allowed. Within a group with >20% weight loss, individuals hitting the individual body weight loss endpoint were euthanized. If the group treatment related body weight loss is recovered to within 10% of the original weights, dosing resumed at a lower dose or less frequent dosing schedule. Exceptions to non-treatment body weight % recovery were allowed on a case-by-case basis. Endpoint was tumor growth delay (TGD). Animals were monitored individually. The endpoint of the experiment was a tumor volume of 1500 mm3 or 45 days, whichever comes first. Responders were followed longer. When the endpoint was reached, the animals are to be euthanized.
[0442] 27B.2 Dosing Instructions
[0443] No compounds in salt form were used. The amount needed per week was calculated, aliquoted accordingly, and stored at -20 C. For each week of dosing, one aliquot was thawed, stored at 4 C, and diluted in the required amount with PBS right before each injection. IL-2-WTI required protection from light; pre-formulation stored at -4.degree. C., post-formulation--stored at -20.degree. C. Lyophilized material was reconstituted as directed by instructions, similar to above. ACP16, ACP130, ACP124, and IL-2 WTI were prepared for dosing in PBS. IL-2-WTI indicates Proleukin (aldesleukin) in PBS; the vehicle was PBS.
[0444] Dosing volume was 0.2 mL/mouse for IL-2-WTI; 0.3 mL for ACP16, ACP130; 0.5 mL for ACP124. Do not adjust for body weight.
[0445] 27B.3 Special Instructions
[0446] ACP16: current amount of required compound--13.45 mg
[0447] ACP130: current amount of required compound--25.83 mg
[0448] 2 ACP124: current amount of required compound--42.31 mg
[0449] 3 IL-2-WTI: current amount of required compound--9.98 mg
[0450] Necropsy was to be performed in case of unexpected toxicity
Example 27C: MC38 IFN.alpha. and IL-12
[0451] 27C.1 Agents and Treatment:
TABLE-US-00011 TABLE 7 Summarizes the treatment regime. Gr. N Agent Formulation dose Route Schedule .sup. 1.sup.# 12 Vehicle -- ip biwk x 3 2 8 ACP11 17.5 .mu.g/animal ip biwk x 3 3 8 ACP11 175 .mu.g/animal ip biwk x 3 4 8 ACP11 525 .mu.g/animal ip biwk x 3 5 8 ACP31 33 .mu.g/animal ip biwk x 3 6 8 ACP31 110 .mu.g/animal ip biwk x 3 7 8 ACP31 330 .mu.g/animal ip biwk x 3 8 8 ACP131 1 .mu.g/animal ip bid x 5 then 2-day pause then bid x 5 then 2-day pause 9 8 ACP131 10 .mu.g/animal ip bid x 5 then 2-day pause then bid x 5 then 2-day pause 10 8 ACP131 30 .mu.g/animal ip bid x 5 then 2-day pause then bid x 5 then 2-day pause 11 8 mIFNa1- 1 .mu.g/animal ip bid x 5 then 2-day WTI pause then bid x 5 then 2-day pause 12 8 mIFNa1- 10 .mu.g/animal ip bid x 5 then 2-day WTI pause then bid x 5 then 2-day pause 13 8 IL-12-HM- 2 .mu.g/animal ip bid x 5 then 2-day WTI pause then bid x 5 then 2-day pause 14 8 IL-12-HM- 10 .mu.g/animal ip bid x 5 then 2-day WTI pause then bid x 5 then 2-day pause 15 8 ACP131 5 .mu.g/animal itu bid x 5 then 2-day pause then bid x 5 then 2-day pause .sup.#Control Group
[0452] 27C.2 Procedures
[0453] Mice were anaesthetized with isoflurane for implant of cells to reduce the ulcerations. 308 CR female C57BL/6 mice were set up with 5.times.10.sup.5 MC38 tumor cells in 0% Matrigel sc in flank. Cell Injection Volume was 0.1 mL/mouse. Mouse age at start date was 8 to 12 weeks. Pair matches were performed when tumors reach an average size of 100-150 mm.sup.3 and begin treatment. Body weights were taken at initiation and then biweekly to the end. Caliper measurements were taken biweekly to the end. Any adverse reactions were to be reported immediately. Any individual animal with a single observation of >than 30% body weight loss or three consecutive measurements of >25% body weight loss was euthanized. Any group with a mean body weight loss of >20% or >10% mortality stopped dosing; the group was not euthanized and recovery is allowed. Within a group with >20% weight loss, individuals hitting the individual body weight loss endpoint were euthanized. If the group treatment related body weight loss is recovered to within 10% of the original weights, dosing resumed at a lower dose or less frequent dosing schedule. Exceptions to non-treatment body weight % recovery were allowed on a case-by-case basis. Endpoint was tumor growth delay (TGD). Animals were monitored individually. The endpoint of the experiment was a tumor volume of 1500 mm.sup.3 or 45 days, whichever comes first. Responders were followed longer. When the endpoint was reached, the animals are to be euthanized.
[0454] 27C.3 Dosing Instructions
[0455] No compounds in salt form were used. Prepared dosing solutions were as follows: IL-12-HM-WTI was stored to provide protection from light; pre-formulation at -4.degree. C., post-formulation at -20.degree. C. mIFNa1-WTI was stored at -20.degree. C., protected from light; pre-formulation stored at -20.degree. C., post-formulation stored at -4.degree. C. Lyophilized material was reconstituted as directed in instructions. The amount needed per week was calculated, aliquoted accordingly, and stored at -20.degree. C. For each week of dosing, one aliquot was thawed and stored at 4 C, the required amount was diluted with PBS right before each injection.
[0456] For ACP11, ACP31, ACP131, the amount needed per week was calculated, aliquoted accordingly, and stored at -20 C. For each week of dosing, one aliquot was thawed and stored at 4 C, the required amount was diluted with PBS right before each injection. PBS was used as the vehicle for all tests.
[0457] Intraperitoneal (ip) dosing volume ACP131, mIFNa1-WTI, IL-12-HM-WTI=0.2 mL/mouse. Dosing volume for ACP11=0.4 mL (before Day 18, 2/26/19) and 0.55 mL (starting on Day 18, 2/26/19).
[0458] Dosing volume for ACP31=0.3 mL. Dosage was not adjusted for body weight. Intratumoral (itu) dosing volume for Gr.16 mIFNa1-WTI and Gr.17 ACP131=0.05 mL/mouse. Dosage was not adjusted for body weight.
Example 27D: MC38 Rechallenge
[0459] 27D.1 Agents and Treatment:
TABLE-US-00012 TABLE 8 Summarizes the treatment regime. Gr. N Agent Formulation dose Route Schedule .sup. 1.sup.# 33 No -- -- -- Treatment 2 7 ACP16 70 .mu.g/animal ip MC38-e415 Gr 4 An 1-4, 6-8 (ACP16 biwkx2) 3 8 ACP16 232 .mu.g/animal ip MC38-e415 Gr 5 An 1-8 (ACP16 biwkx2) 5 5 IL-2- 12 .mu.g/animal ip MC38-e415 WTI Gr 16 An 1, 3-6 (IL-2-WTI bid x 5 then 2-day pause then bid x 5 then 2-day pause) 6 7 IL-2- 36 .mu.g/animal ip MC38-e415 WTI Gr 17 An 1-7 (IL-2-WTI bid x 5 then 2-day pause then bid x 5 then 2-day pause) .sup.#Control Group
[0460] 27D.2 Procedures
[0461] Mice were anaesthetized with isoflurane for implant of cells to reduce the ulcerations. This portion of the study began on the day of implant (Day 1). Group 1 consisted of 33 CR female C57BL/6 mice set up with 5.times.105 MC38 tumor cells in 0% Matrigel subcutaneously in the flank. Groups 2-6 consisted of 33 CR female C57BL/6 mice set up with 5.times.105 MC38 tumor cells in 0%/Matrigel sc in the left flank. The tumors from the previous MC38 experiment (example 25.times.) were implanted in the right flank of each animal. Cell Injection Volume was 0.1 mL/mouse. Age of control mice at initiation was 14 to 17 weeks. These mice were age matched to mice from the previous MC38 experiment (example 25.times.). No dosing of active agent occurred during rechallenge. Body Weights were taken biweekly until end, as were caliper measurements. Any adverse reactions or death were reported immediately. Any individual animal with a single observation of >than 30% body weight loss or three consecutive measurements of >25% body weight loss was euthanized. Endpoint was tumor growth delay (TGD). Animals were monitored individually. The endpoint of the experiment was a tumor volume of 1000 mm3 or 45 days, whichever comes first. Responders were followed longer when possible. When the endpoint is reached, the animals were euthanized.
Example 27E: Treatment with ACP16, APC153, ACP155, and ACP156 (See FIG. 58)
[0462] 27E.1 Agents and Treatment:
TABLE-US-00013 TABLE 9 Summarizes the treatment regime. Gr. N Agent Formulation dose Route Schedule .sup. 1.sup.# 12 Vehicle -- ip biwk x 2 2 8 ACP16 17 .mu.g/animal ip biwk x 2 3 8 ACP16 55 .mu.g/animal ip biwk x 2 4 8 ACP16 230 .mu.g/animal ip biwk x 2 5 8 ACP155 55 .mu.g/animal ip biwk x 2 6 8 ACP155 230 .mu.g/animal ip biwk x 2 7 8 ACP153 55 .mu.g/animal ip biwk x 2 8 8 ACP153 230 .mu.g/animal ip biwk x 2 9 8 ACP156 55 .mu.g/animal ip biwk x 2 10 8 ACP156 230 .mu.g/animal ip biwk x 2
[0463] 27E.2 Procedures:
[0464] Mice were anaesthetized with isoflurane for implant of cells to reduce the ulcerations. CR female C57BL/6 mice were set up with 5.times.10.sup.5 MC38 tumor cells in 0% Matrigel sc in flank. Cell Injection Volume was 0.1 mL/mouse. Mouse age at start date was 8 to 12 weeks. Pair matches were performed when tumors reach an average size of 100-150 mm.sup.3 and begin treatment. ACP16 was dosed at 17, 55 or 230 g/animal; ACP153, ACP155 and ACP156 were dosed at 55 or 230 .mu.g/animal. Body weights were taken at initiation and then biweekly to the end. Caliper measurements were taken biweekly to the end. Any adverse reactions were to be reported immediately. Any individual animal with a single observation of >than 30% body weight loss or three consecutive measurements of >25% body weight loss was euthanized. Any group with a mean body weight loss of >20% or >10% mortality stopped dosing; the group was not euthanized and recovery is allowed. Within a group with >20% weight loss, individuals hitting the individual body weight loss endpoint were euthanized. If the group treatment related body weight loss is recovered to within 10% of the original weights, dosing resumed at a lower dose or less frequent dosing schedule. Exceptions to non-treatment body weight % recovery were allowed on a case-by-case basis. Endpoint was tumor growth delay (TGD). Animals were monitored individually. The endpoint of the experiment was a tumor volume of 1500 mm.sup.3 or 45 days, whichever comes first. Responders were followed longer. When the endpoint was reached, the animals are to be euthanized. Results are shown in FIG. 58A-58D.
Example 28: FRET Screens of Conditioned Media
[0465] Proteolytic activity was screened in the conditioned media samples by fluorescent resonance energy transfer (FRET) assays with the substrates listed in Table 10.
TABLE-US-00014 TABLE 10 Substrate motif sequences. Experimentally Tested Substrate SEQ ID in vitro Conditioned 30-mer Name P4-P4' Sequence NO: kinetics media MSP-MS MMP14_1 GPAGLYAQ 195 MMP9_1 GPAGMKGL 196 FAP.alpha._1 PGGPAGIG 197 CTSL1_1 ALFKSSFP 198 CTSL1_2 ALFFSSPP 199 ADAM17_1 LAQRLRSS 200 ADAM17_2 LAQKLKSS 201
[0466] 28.1 Reaction Conditions
[0467] Protease specificity screening was performed using Multiplexed Substrate Profiling by Mass Spectrometry (MSP-MS), the method described in, e.g. O'Donoghue A. J. et al., Nat Methods. 2012; 9(11): 1095-1100. This method employs a physico-chemically diverse peptide library as substrates for proteases, and reactions are monitored over time with mass spectrometric detection of cleaved products. The resulting cleavages are assessed for specific cleavage in the enzyme-treated sample by comparison with results from a no-enzyme control incubation. Sequence logos for motif analysis are generated with iceLogo software (v.1.2) (iomics.ugent.be/icelogoserver/).
[0468] Recombinant human enzymes were sourced from R&D Systems (Bio-techne): CTSL1 (#952-CY), ADAM17 (a.k.a. TACE, TNF-alpha Converting Enzyme) (#930-ADB), FAP-alpha (#930-ADB-010), MMP-14 (#918-MP-010), furin (#1503-SE-010), MMP-9 (#911-MP-010), thrombin (#2196-SE-200), thermolysin (#3097-ZN), Factor Xa (#1063-SE-010), hepsin (#4776-SE) and ADAM-TS1 (#2197-AD). Enzymes were activated and assayed following the manufacturer's recommendations.
[0469] For CTSL1, enzyme was activated by pre-incubation in assay buffer (50 mM MES, 5 mM DTT, 1 mM EDTA, pH=6) for 15 min. MSP-MS reactions were then started with the mixing of enzyme and substrate, at final CTSL1 concentration of 0.04 ng/.mu.l (0.77 nM), and peptide substrate concentration at 500 nM.
[0470] ADAM17 activity was monitored following manufacturer's recommended conditions using the FRET substrate Mca-PLAQAV-Dpa-RSSSR-NH.sub.2(SEQ ID NO: 244), in the recommended assay buffer: 25 mM TRIS, 2.5 .mu.M ZnCl.sub.2 at the optimal pH 9.0, and also at physiological pH 7.4. ADAM17 enzyme specificity was profiled in the MSP-MS experiment at pH 9.0 using 10 nM enzyme, and at pH 7.4 using 50 nM enzyme.
[0471] MMP-14 was activated following manufacturer's recommended conditions by pre-incubation with the enzyme furin, at a molar ratio of 1:100 furin to MMP-14, at 37.degree. C. and pH 9.0 for 1.5 hours. FRET assays were performed using 20 nM MMP-14 at 37.degree. C. in activation buffer: 50 mM TRIS HCl, 3 mM CaCl.sub.2, 1 .mu.M ZnCl.sub.2 at pH 8.5.
[0472] MMP9 was activated by incubation with 1 mM p-amino phenyl mercuric acetate (prepared from a stock at 100 mM in DMSO) for 24h at 37.degree. C. in activation buffer: 50 mM TRIS HCl (pH 7.5), 10 mM CaCl.sub.2, 150 mM NaCl, 0.05% Brij35. The final enzyme concentration used for fluorescence assays was 2.5 nM.
[0473] FAP.alpha. activity was tested with fluorescence detection using a generic Z-GP-AMC substrate. For FRET assays, the final enzyme concentration was 5 nM and the buffer was 50 mM TRIS HCl, 1 M NaCl, 1 mg/mL BSA, pH 7.5.
[0474] Thrombin was activated following the manufacturer's recommended conditions by pre-incubation with the enzyme with thermolysin for 15 min, and then the thermolysin was quenched with 1,10 phenanthroline treatment. Activated thrombin was assayed at 1.2 nM in activation buffer: 50 mM TRIS HCl, 10 mM CaCl.sub.2, 150 mM NaCl, 0.05% (w/v) Brij-35, pH 7.5.
[0475] Factor Xa was assayed at 4.7 nM enzyme, at 37.degree. C. in the manufacturer recommended buffer: 50 mM TRIS, 10 mM CaCl.sub.2, 150 mM NaCl, pH 7.5.
[0476] Hepsin was pre-activated overnight at 37.degree. C. in the manufacturer recommended buffer: 100 mM TRIS, 10 mM CaCl.sub.2, 150 mM NaCl, 0.05% Brij-35, pH 8.0. Activity was measured with 2.4 or 24 nM enzyme in the buffer: 50 mM TRIS HCl at pH 7.4.
[0477] ADAM-TS1 was assayed at 5 .mu.M enzyme concentration in the manufacturer recommended buffer: 50 mM TRIS, 10 mM CaCl.sub.2), 150 mM NaCl at pH 7.5.
[0478] 28.2 Samples
[0479] Conditioned media and cell lysate samples were received from Charles River Laboratories for the murine colon cancer cell lines: MC38 (epithelial), CT26 (fibroblast), and CT26 transduced with a lentivirus for doxycyclin-inducible expression of MMP9 (CT26 pLVX MMP9-5T4+dox). An immortalized mouse intestinal myofibroblast cell line (ABM Good #T0565) was used as a control stromal cell line, grown under manufacturer recommended conditions. Conditioned media and cell lysate samples were also received for screening purposes from ABM Good for the immortalized mouse colonic epithelial cell line YAMC (ABM Good #T0567).
[0480] Conditioned media samples were prepared according to the standard protocol using logarithmic phase cells. Briefly, cells were grown for 16h in either serum-free media or in complete media containing 10% fetal bovine serum (FBS). After conditioned media collection, the adherent cells were washed then lysed on plate with non-denaturing lysis buffer to produce a lysed "cell pellet" to capture any activity that remained associated with the cells. All cell culture-derived conditioned media samples, cell lysates from Charles River Laboratories, and cell lysates derived internally were processed with identical methods, to allow comparison between samples.
[0481] The resulting conditioned media was buffer exchanged with PBS and concentrated to 10.times. the original titer as a stock solution.
[0482] 28.3 Results
[0483] Method development experiments with these samples showed that conditioned media containing fetal bovine serum (FBS) were amenable to FRET screening, although a small amount of background fluorescence for each substrate was obtained in the presence of FBS. FBS background controls were therefore used for baseline subtraction at each concentration of substrate throughout the FRET experiments.
[0484] In the end-point screening assays, a fixed titer of 10.times. concentrated sample and a substrate concentration of 10 .mu.M was used. Values reported in the end-point experiments are initial velocity measurements, in relative fluorescence units (RFU) per second. End-point measurements are a starting point for a screen, but the rate of the reaction is non-linearly related to substrate concentration. Therefore, a more accurate representation of activity would be given by steady-state kinetic measurements covering a range of substrate concentrations. To compare activity across cell lines, the data were treated with a Michaelis-Menten model, and the concentration of equivalent enzyme units [E.sub.0] was solved.
[0485] Accordingly, initial velocity was measured as a function of substrate concentration, using a 2-fold dilution series from 250 to 1 .mu.M. Data were fitted using non-linear least squares fitting with GraphPad Prism software v 8.0 to the Michaelis-Menten equation:
velocity = k cat [ E 0 ] [ S ] ( K M + [ S ] ) ##EQU00001##
where k.sub.cat is the rate of product formation under steady-state conditions in units (s.sup.-1), and K.sub.M is the Michaelis constant that gives the substrate concentration in molar units (M) at which half maximum velocity of the reaction is produced for a given enzyme, [E.sub.0] is the enzyme concentration, and [S] the substrate concentration in (M).
[0486] In the assays where purified recombinant enzyme is used, the Michaelis-Menten parameters are as classically defined above. For conditioned media samples, the effective titer of conditioned media per assay was adjusted to approximately a 2.times. concentrate of the original unprocessed media samples to assure that the detection range matched the signal produced. The enzyme component in the calculation is redefined as a mixture of possible proteases that contribute to the cumulative observed activity, each with intrinsic reactivity toward a substrate. In this case, it is more appropriate to consider k.sub.cat and K.sub.M as macroscopic constants that represent the overall efficiency of catalysis, and E.sub.0 is an enzyme equivalent, expressed in units of concentration. To solve the data fit, k.sub.cat/K.sub.M can be fixed at that of the recombinant enzyme and enzyme equivalents per cell culture volume can be output. The effective titer of enzyme equivalents in this case can be used to compare the activity produced by the different cell lines.
[0487] The ratios of enzyme equivalents between tumor and control cell lines may be a contributing factor to therapeutic index in a pro-drug employing protease-cleavable linkers.
[0488] For all six motif substrates, greater activity was detected in the conditioned media from the tumor cell lines than from the control myofibroblast cell line (FIG. 36).
[0489] The ADAM17 substrate with the sequence LAQKLKSS (ADAM17_2) (SEQ ID NO: 201), based on the results in the end-point screen experiments, had approximately 3-fold greater activity in the conditioned media produced from the three tumor cell lines, compared to the myofibroblast cell line (FIG. 36, black bars). For reference, the activity of recombinant ADAM17 at 12 nM is shown in the same assay format. The steady state kinetic curves for ADAM17 substrate are shown in FIG. 37. There was essentially undetectable ADAM17_2 background activity in the myofibroblast sample.
[0490] The CTSL1 substrate, ALFKSSFP (SEQ ID NO: 198) (CTSL1_1), based on the results in the end-point screen experiments, had undetectable activity in the control myofibroblast cell line, as well had low activity in the MC38 and CT26 parental cell lines, and had undetectable activity in the CT26-MMP9+ cell line (FIG. 36). This assay was benchmarked with CTSL1 at 1.5 nM, showing that the extracellular concentration of secreted CTSL1 activity was below this effective concentration across the four cell lines. Re-analysis of conditioned media with the CTSL1_1 substrate using the steady-state kinetics analysis yielded no measurable activity above background.
[0491] The secreted activity for FAP.alpha., based on the results in the end-point screen experiments, was very low, but was still detected in all three tumor cell lines (FIG. 36, blue bars). The FAP.alpha._1 activity was benchmarked with 5 nM FAP.alpha. enzyme. This enzyme is also found in a plasma membrane-associated form, therefore FAP.alpha. activity was also tested in the cell pellet fraction that was collected after the media conditioning procedure. These results will be discussed below.
[0492] As observed in the end-point screen, re-analysis of the conditioned media for activity toward the FAP.alpha._1 substrate showed lower activity than that associated with the cell pellet, measured in cell lysates (FIG. 38 and FIG. 39). Myofibroblasts had no measurable secreted activity but did have cell-associated activity (FIG. 38 and FIG. 39).
[0493] The MMP substrates MMP9_1 and MMP14_1 all had high activity in the end point screen, and the activity was significantly higher in the tumor cell lines than in the myofibroblast control cells. MMP9_1 (second column from right, FIG. 38) had approximately 4-fold greater activity in the tumor lines than the myofibroblast control; and MMP14_1 had approximately 57-fold greater activity in the tumor lines. The MMP14_1 substrate had the lowest activity in the myofibroblast cells, thus contributing to the higher ratio of tumor vs control in this assay. Secreted activity was measured for the MMP9_1 substrate in the conditioned media for each of the tumor cell lines using the steady-state kinetics analysis (FIG. 36). The myofibroblast cell line had an artefact in analysis of MMP9_1 secreted activity (FIG. 36); this will be addressed with a repeat analysis at higher enzyme titer. As with the MMP9 substrates, the MMP14_1 substrate had significant measurable activity in the conditioned media from the tumor cells but not in the myofibroblast cell line.
[0494] Taken together, the results of the FRET screens of conditioned media demonstrated tumor-specific activity for the enzymes ADAM17, CTSL1, MMP9 and MMP14. Soluble FAP-alpha activity was low.
Example 29: Tumor-Specific Activity Toward the FAP.alpha. Substrate in the Cell Lysates
[0495] To test for cell pellet-associated FAP.alpha. activity, the cell lysates were clarified by ultracentrifugation and then assayed neat in the FRET assay. As shown in FIG. 41, FAP.alpha._1 cleavage activity was detected in all four cell lines (blue bars, right hand bar of each pair). A modest 2-fold greater activity was detected in the three tumor cell lines over the myofibroblast cell line. For comparison, CTSL1 activity was also tested in the cell lysates. CTSL1 is a lysosomal enzyme; therefore, activity toward CTSL1_1 was expected to be similarly abundant in all four cell lines. The ratio of CTSL1_1 cleavage in tumor vs control cell lines in this screening format was .about.1.times. (grey bars, left-hand bar of each pair, FIG. 41). FAP.alpha. (fibroblast activating protein-alpha) is a marker for fibroblast (CT26) and myofibroblast cell lines. Therefore, an additional non-fibroblast, epithelial cell line was obtained as an alternate negative control cell line, comparable to the MC38 epithelial cell line.
Example 30: Cell-Associated Activity Toward CTSL1 Substrate
[0496] Cell-associated activity toward CTSL1 substrate was detected in all of the cell lysates, indicating cell-associated activity for this enzyme. The activity was significantly greater in MC38 cells over the myofibroblast, CT26 parental, and CT26-MMP9+ cells (FIG. 42).
Example 31: Secreted Activity in Conditioned Media from Immortalized Myofibroblast Cell Line
[0497] Low background-corrected fluorescence was observed in all the myofibroblast reactions. Thus, it can be concluded that the immortalized myofibroblast cell line had low conditioned media activity.
Example 32: Screening of an Additional Control "Normal" Epithelial Cell Line
[0498] An immortalized mouse colonic epithelial cell line (YAMC) was used to test activity in the presence of additional control "normal" epithelial cells. YAMC's are given the calculated enzyme equivalents (nM) resulting from all of these steady state experiments, to allow for comparison of the amount of equivalent activity per sample. For example, the YAMC control epithelial cell line produced activity with the ADAM17_2 substrate equivalent to that of 0.52.+-.0.03 nM of ADAM17 recombinant enzyme. This cell line compares directly to MC38, also of epithelial origin, which had activity equivalent to that of 0.32.+-.0.03 nM of ADAM17 recombinant enzyme. The results of the screening show that the YAMC cell line generally had more background activity than did the myofibroblast cell line with all of the substrates. This cell line required non-standard cell culture conditions, including growth with murine IFN.gamma., and at low temperature to maintain, and may not be fully representative of epithelial cell lines.
Example 33: Cleavage of CTSL1_2 Motif in the Context of an 8-Mer FRET Peptide and in the Context of an Extended Tandem Linker
[0499] Additional in vitro kinetic experiments were performed to characterize a new candidate motif for CTSL1 enzyme; the CTSL1_2 motif has the sequence ALFFSSPP (SEQ ID NO: 199). CTSL1 cleaves the FRET substrate bearing the CTSL1_1 motif highly efficiently (FIG. 46). Yet, initial experiments with the CTSL1_2 FRET substrate were inconclusive, possibly due to artifacts in the FRET assay; therefore, a new FRET peptide construct was designed with the 9-mer sequence ALFFSSPPS (SEQ ID NO: 236). This 9-mer FRET substrate bearing the CTSL1_2 motif produced comparable catalytic efficiency to the CTSL1_1 motif.
[0500] All of the FRET substrates used in these studies have a methoxycoumarin (Mca) fluorophore and dinitrophenyl-lysyl (Dnp) quencher pair at the N- and C-termini of the sequences, respectively. The motif ALFFSSPP (SEQ ID NO: 199) was efficiently cleaved in previous experiments at ALFF|SSPP (SEQ ID NO: 199) (where | indicates the scissile bond) within 14-mer peptide constructs that were used in the MSP-MS tailored library assay. It was cleaved also at the ALF|FSSPP (SEQ ID NO: 199) and ALFFS|SPP (SEQ ID NO: 199) sites within 14-mer peptides, indicating that the enzyme recognition in the CTSL1_2 motif may be shifted up- or down-sequence by +/-1 site in the 14-mer peptide experiments. This also suggests that the 8-mer design using Mca-ALFFSSPPK-Dnp (SEQ ID NO: 245) may have unfavorable interactions (steric or otherwise) with the fluor/quencher pair in the shifted P4 or P4' positions that were alleviated by the redesign as a 9-mer substrate using Mca-ALFFSSPPSK-Dnp (SEQ ID NO: 246). This FRET experiment indicated that cleavage of this sequence most likely requires binding recognition at the +/-1 positionally-shifted site and that the fluor or quencher may interfere with this binding.
[0501] This hypothesis was tested in the context of the hybrid linker design experiment. Shown in FIG. 46 are the results of cleavage of two matched 30-mer peptides with the following sequences:
TABLE-US-00015 (CTSL1_1) (SEQ ID NO: 207) SGGPGGPAGIGALFKSSFPLAQKLKSSGGG (CTSL1_2) (SEQ ID NO: 219) KSGPGGPAGIGALFFSSPPLAQKLKSSGGR
[0502] Both peptides showed rapid disappearance of substrate (squares) and product formation from cleavage at the target scissile bond (site 0, blue triangles, FIG. 46). Where the peptides differed is in the permissive cleavage at the -1 and +1 sites. CTSL1_1 had a -1 site product at ALF|KSSFP (SEQ ID NO: 198) (downward triangles) but CTSL1_2 did not, and the +1 site product was more rapidly formed from the CTSL1_2 substrate (ALFFS|SPP (SEQ ID NO: 199)) than with the CTSL1_1 substrate (ALFKS|SFP) (SEQ ID NO: 198).
[0503] Counter-screening of CTSL1_1 and CTSL1_2 was performed with the enzyme Cathepsin K (CTSK). The substrate CTSL1_2, although lacking a basic P1 residue (ALFFSSPP (SEQ ID NO: 199)) was measurably cleaved by CTSK but at a 15.times. lower level than the CTSL1_1 substrate (ALFKSSFP (SEQ ID NO: 198)) FIG. 47. For comparison, the specific activity for reference substrates such as Z-LR-AMC and Z-FR-AMC is 15,000-25,000 pmol/min/.mu.g, with the average value indicated by a dashed line in FIG. 47.
[0504] Thus, these results show that the new CTSL1_2 motif was not cleaved in the context of an 8-mer FRET peptide, but it was cleaved as a 9-mer FRET peptide and it was rapidly cleaved in the context of an extended tandem linker.
Example 34: Tandem Linker Analysis with Mass Spectrometric Detection
[0505] 34.1 Substrate Profiling by Mass Spectrometry
[0506] To analyze the catalytic efficiency of candidate tandem linker designs, a tailored library approach using MSP-MS was applied. For the tailored library, a set of nineteen synthetic peptides, 30 amino acids in length (30-mers), were designed to test whether sequence context affects the efficiency of cleavage. Peptides were assayed in a multiplex format together as a substrate library with individual recombinant enzymes, using MSP-MS. For quantitative comparisons, kinetic analysis was performed over a time course, and results are reported either as a catalytic efficiency k.sub.cat/K.sub.M for well-behaved first-order kinetics, or as observed rates as k.sub.obs for more complex kinetic behavior.
[0507] 34.2 the Library of Tandem Linker Sequences
[0508] The library of tandem linker sequences was designed to incorporate three individual protease motifs within the context of a longer, 30-mer peptide sequence. Table 11 lists the sequences and motif arrangement of the 30-mer peptides.
TABLE-US-00016 TABLE 11 30-mer peptide sequences. Substrate SEQ ID Name Sequence NO: ALU30-1 GALFKSSFPSGGGPAGLYAQGGSGKGGSGK 202 ALU30-2 RGSGGGPAGLYAQGSGGGPAGLYAQGGSGK 203 ALU30-3 KGGGPAGLYAQGPAGLYAQGPAGLYAQGSR 204 ALU30-4 RGGPAGLYAQGGPAGLYAQGGGPAGLYAQK 205 ALU30-5 KGGALFKSSFPGGPAGIGPLAQKLKSSGGS 206 ALU30-6 SGGPGGPAGIGALFKSSFPLAQKLKSSGGG 207 ALU30-7 RGPLAQKLKSSALFKSSFPGGPAGIGGGGK 208 ALU30-8 GGGALFKSSFPLAQKLKSSPGGPAGIGGGR 209 ALU30-9 RGPGGPAGIGPLAQKLKSSALFKSSFPGGG 210 ALU30-10 RGGPLAQKLKSSPGGPAGIGALFKSSFPGK 211 ALU30-11 RSGGPAGLYAQALFKSSFPLAQKLKSSGGG 212 ALU30-12 GGPLAQKLKSSALFKSSFPGPAGLYAQGGR 213 ALU30-13 GGALFKSSFPGPAGLYAQPLAQKLKSSGGK 214 ALU30-14 RGGALFKSSFPLAQKLKSSGPAGLYAQGGK 215 ALU30-15 RGGGPAGLYAQPLAQKLKSSALFKSSFPGG 216 ALU30-16 SGPLAQKLKSSGPAGLYAQALFKSSFPGSK 217 ALU30-17 KGGPGGPAGIGPLAQRLRSSALFKSSFPGR 218 ALU30-18 KSGPGGPAGIGALFFSSPPLAQKLKSSGGR 219 ALU30-19 SGGFPRSGGSFNPRTFGSKRKRRGSRGGGG 220
[0509] To test for additive effects of having one, two, or three repeat MMP14_1 motifs with small differences in spacing between the motifs, a set of four tandem linkers were designed (ALU30-1 to -4).
[0510] To test whether neighboring effects from adjacent sites could alter the catalytic efficiency of a given protease toward its target motif, the three motifs in a set were arranged in all permutations. For example, the combination of motifs A, B, and C could be arranged as: A-B-C, C-A-B, B-C-A, B-A-C, A-C-B, and C-B-A. These permutations were tested for the tandem combinations of MMP14_1, ADAM17_2, and CTSL1_1 as a broad set of motifs susceptible to matrix metalloprotease (MMP) activity (ALU30-11 to -16), and for FAP.alpha._1 with ADAM17_2, and CTSL1_1 as a set with lower MMP sensitivity (ALU30-5 to -10).
[0511] Also tested within the library were two alternate substrates for ADAM17 and CTSL1. Two additional peptides were designed, swapping the ADAM17_2 motif with ADAM17_1 (in ALU30-17), or CTSL1_1 with CTSL1_2 (in ALU30-18), demonstrating a strategy for testing further tandem linker designs with new individual protease motifs.
[0512] 34.3 Counter-Screening
[0513] Counter-screening was performed against the enzymes Thrombin, Factor Xa and Hepsin using this library as well, and for calibration of activity, a positive-control peptide bearing favorable motifs for these enzymes was also designed (ALU30-19).
[0514] Outside of these motifs, minor variations were made to the bracketing sequences at the N- or C-terminus of each tandem linker peptide to generate unique sequences that allow for differentiation of peptides using mass spectrometric detection.
[0515] Catalytic efficiency (k.sub.cat/K.sub.M) estimations were used to rank the top cleaved substrates in the MSP-MS reaction. Peptides were quantified by mass spectrometric label-free quantitation from the MS1 precursor ion peak areas for each peptide. Enzyme progress curves were modeled from six-time point measurements in GraphPad Prism. Data were fitted using non-linear least squares fitting to the first order kinetics equation:
Y = e ( - k cat K M [ E 0 ] t ) ##EQU00002##
where Y=percent product formation or substrate consumption, and t=time. The observed rate is a function of the enzyme concentration [E.sub.0], and an observed catalytic efficiency (k.sub.cat/K.sub.M) in units M.sup.-1s.sup.-1
[0516] 34.4 Results
[0517] Analyses with the 30-mer tandem linker library showed specific cleavages with each of the recombinant enzymes. In this experiment, the rates of substrate degradation and of product formation were both useful comparisons for understanding the efficiency of linker cleavage.
[0518] For example, the substrate degradation traces for the 30-mer library peptides are shown from MMP9-treatment in FIG. 55. The most efficient MMP9 substrates were those that bore three-repeats of the MMP14_1 motif. The next most efficient substrates contained the pair of motifs where MMP14_1 was followed directly by ADAM17_2. The single or double MMP14_1 motif-bearing peptides were next in the order. An unanticipated result was that the FAP.alpha._1 motif was also cleaved within the peptides bearing FAP.alpha._1 directly followed by the CTSL1_1 motif at PGGPAG|GALF (SEQ ID NO: 247); these were lower efficiency cleavages. The FAP.alpha._1 motif was not cleaved by MMP9, however, when followed either by the ADAM17_2 motif or the spacing residues GG. The slowest peptides bear the MMP14_1 motif near the C-terminus of the 30-mer peptide. From this experiment, a trend emerged that MMP9 has additional sequence preferences in the downstream prime-side positions, and that the combination of MMP14_1 upstream of ADAM17_2 is most efficient.
[0519] To better understand the intrinsic rates of product formation with MMP9 treatment, it is also possible to monitor the cleavage products at a specific peptide bond in these experiments. A comparison of peptides bearing one, two or three MMP14_1 tandem motifs is shown in FIG. 50. The most rapidly degraded peptides were those bearing three repeat motifs of MMP14_1 (top panel). Overall, the peptides bearing one or two MMP14_1 motifs were degraded at approximately the same rate (top panel). However, considering intrinsic rates of bond cleavage at individual sites, the cleavage product of Alu30-2 at bond 9 appeared more rapidly than the cleavage product of Alu30-1 at bond 16 or of Alu30-2 at bond 21 (bottom panel). Thus, the specific cleavage of the Alu30-2 peptide was more efficient than the Alu30-1 peptide. These individual bond cleavage events are monitored by tracking unique peptide fragments, and although too complex for data fitting, the ranking of products is possible.
[0520] FAP.alpha. was able to cleave both FAP.alpha._1 and MMP14_1 motifs, at PGGP|AGIG (SEQ ID NO: 197) and GP|AGLYAQ (SEQ ID NO: 195) respectively. Degradation of the 30-mer peptides by FAP.alpha. showed highest cleavage activity toward the peptides bearing tandem MMP14_1 motifs (FIG. 50, Alu30-4 or Alu30-3). The peptide bearing two MMP14_1 motifs (Alu30-2) was also efficiently cleaved. FAP.alpha._1 motifs were cleaved more efficiently when followed by the ADAM17_1 or ADAM17_2 motifs than when followed by CTSL1_1 or CTSL1_2. Among these peptides, the Lys-bearing ADAM17_2 or CTSL1_1 motifs were lower efficiency than the new motifs tested in this experiment, ADAM17_1 and CTSL1_2.
[0521] Another trend was a preference for cleavage of the FAP.alpha._1 motif in the first or second position relative to the N-terminus of these peptides. When the FAP.alpha._1 motif was in the third position closest to the C-terminus, these peptides were not cleaved (Alu30-7 or Alu30-8). The MMP14_1 motif was readily cleaved in the third position closest to the C-terminus, but this was most evident in the tandem MMP14_1 motif-bearing peptides, where N-terminal cleavages assist in shortening the 30-mer. Thus, it may be that FAP.alpha. could serve as a secondary-cleaving enzyme after activation with a first enzyme elsewhere in a hybrid linker design.
[0522] CTSL1 treatment of the 30-mer peptide library produced cleavages in all peptides bearing a CTSL1_1 or CTSL1_2 motif (FIG. 51). The most efficiently cleaved peptides were those containing the FAPa_1, CTSL1_1 and ADAM17_2 motifs, in multiple orders. Peptides bearing the MMP14_1 motif with CTSL1_1 and ADAM17_2 were slightly lower efficiency. CTSL1 motifs in the middle of the 30-mer peptides were also favorable, thus CTSL1 may be able to serve as a first cleavage in the hybrid motifs.
[0523] ADAM17 produced cleavages in all peptides bearing an ADAM17_1 or ADAM17_2 motif as well (FIG. 52). The most efficient peptide cleavages occurred in the peptides containing ADAM17_2, CTSL1_1 and FAP.alpha._1 motifs. Slightly lower efficiency cleavages were obtained in peptides containing the MMP14_1 with ADAM17_2 and CTSL1_1. Peptides bearing only the MMP14_1 motif were not cleaved. Peptides Alu30-9 and Alu30-17, containing the ADAM17_2 and ADAM17_1 motifs respectively, had similar cleavage efficiencies.
[0524] Counter-screening with Thrombin, Factor Xa and Hepsin was also performed following the same analysis. The library peptide Alu30-19 was designed to include authentic sites for each enzyme, for comparison of overall cleavage efficiency as well as cleavage site specificity within each motif. The substrate designed for Factor Xa has the motif FNPR|TFGS (SEQ ID NO: 248), derived from the Factor Xa cleavage site in Thrombin. The substrate motif for Thrombin was FPR|, a common tool substrate motif. The substrate motif for hepsin was RKRR|GSRG (SEQ ID NO: 249) from filaggrin.
[0525] Shown in FIG. 53 are the results of Factor Xa cleavage. The authentic cleavage site motif was Peptide Alu30-19 with this motif was completely degraded before the 5 min time point. Products of the Alu30-19 cleavage were formed from all three sites in this peptide. In comparison, the only library peptide with significant cleavage was Alu30-5, cleaved at the CTSL1_1 motif ALFKS|SFP (SEQ ID NO: 198), upstream of a FAP.alpha._1 motif.
[0526] Otherwise, all other peptides bearing a CTSL1_1 motif were cleaved by Factor Xa at either ALF|KSSFP (SEQ ID NO: 198) or ALFKS|SFP (SEQ ID NO: 198). The ADAM17 motifs were also cleaved at KLK|SSGP (SEQ ID NO: 250), KLKSS|ALF (SEQ ID NO: 251), or RLR|SSALF SEQ ID NO: 252), but these cleavages were all lower efficiency. The peptide with the motif CTSL1_2 was cleaved at the lowest efficiency.
[0527] Thrombin showed higher activity than Factor Xa, and it cleaved each of the 30-mer peptides. The MMP14_1 motifs were cleaved at GPAG|LYAQ (SEQ ID NO: 195), the CTSL1_1 motifs at ALFK|SSFP (SEQ ID NO: 198), and the ADAM17_2 motifs at LAQK|LKSS (SEQ ID NO: 201) or LAQKLK|SS (SEQ ID NO: 201) (or LAQR|LRSS (SEQ ID NO: 200) and LAQRL|SS (SEQ ID NO: 253) in ADAM17_1). The two alternate ADAM17 motifs were cleaved with similar efficiency (peptides Alu30-17 vs Alu30-9). However, the CTSL1_2 motif had lower cleavage activity than did the CTSL1_1 motif (peptides Alu30-18 vs Alu30-6). The peptides Alu30-1 and Alu30-3 were the most rapidly cleaved in the library, and Alu30-4 was also an efficient substrate, showing the susceptibility of MMP14_1 motifs in this experiment. The peptide Alu30-2, bearing two MMP14_1 motifs, was less susceptible to thrombin, however, potentially due to altered spacing with additional glycine residues between motifs. Also, the peptides Alu30-12, Alu30-13 and Alu30-15, with MMP14_1 motifs in combination with ADAM17_2 and CTSL1_1, were cleaved at lower efficiency by thrombin. Thus, increased spacing between the motifs or specific arrangements may rescue a thrombin-susceptible linker design (FIG. 54).
[0528] The favored arrangements for reducing thrombin susceptibility include using ADAM17_2 upstream of CTSL1_1, and CTSL1_1 upstream of FAP.alpha._1.
[0529] Finally, hepsin treatment of the 30-mer library produced lower efficiency cleavages overall (FIG. 55). The kinetics of these peptide degradation reactions were complex due to the formation of multiple cleavage produces; for example, Alu30-19 was cleaved at all three motifs within the first 15 min of the reaction at RKRR| (SEQ ID NO: 254), FPR| and FNPR| (SEQ ID NO: 255). The literature value for the catalytic efficiency of a FRET peptide bearing this motif was 3.5.times.10.sup.5 M.sup.-1s.sup.-1. The authentic peptide Alu30-19, as well as the top cleaved peptides Alu30-8,-9, -12, and -14 all had apparent catalytic efficiencies on the same order of 10.sup.5 M.sup.-1s.sup.-1. The features that made peptides more susceptible to hepsin appear to be placement of an ADAM17_2 motif in the second motif position of the peptide, or in the combination of ADAM17_2 followed by CTSL1_1. In general, the library peptides were all cleaved at P1 Lys sites, QKLK| (SEQ ID NO: 256) or ALFK| (SEQ ID NO: 257). The ADAM17_1 motif was less efficiently cleaved than ADAM17_2, and the alternate CTSL1_2 motif was also cleaved at a much slower rate than CTSL1_1.
[0530] To conclude, the tandem linker analysis with mass spectrometric detection revealed motif order preferences and unexpected side-reactions.
Example 35: 30-Mer Tandem Linker Design
[0531] To generate the most efficient tandem linker, the cleavage efficiencies for each of the targeted enzymes toward the 30-mer library peptides can be compared (FIG. 64).
[0532] The tandem linker with the arrangement of ADAM17_2-MMP14_1-CTSL1_1 in the peptide Alu30-16 had generally high activity toward the full set of five targeted enzymes, as well as the lowest susceptibility to thrombin, Factor Xa, and hepsin. The next best configuration was MMP14_1-ADAM17_2-CTSL1_1 in Alu30-15 which, although it had slightly higher hepsin susceptibility and lower CTSL1 activity may be rescued by the replacement with the CTSL1_2 motif. Replacement of the ADAM17_2 motif with ADAM17_1 may also enhance activity toward FAP-alpha and CTSL1, even if ADAM17 activity enhancement is minor, and hepsin susceptibility would be predicted to be reduced.
Example 36. Stability in Serum and Plasma
[0533] The stability of constructs of interest was measured in human or mouse serum. Each construct (approximately 30 mg/mL) was combined with serum (1:9 ratio of construct to serum), MMP9, or PBS. The mixture was incubated at 37.degree. C. for 24 or 72 hours. Samples were taken at T=0 hours for comparison. After incubation, samples were diluted 1:500 (in human serum, FIG. 56) or 1:200 (in mouse serum, FIG. 57) in PBS and run on an SDS PAGE gel for western blot analysis. A polyclonal anti-IL-2 antibody (R&D Systems) was used to probe the blots. Results are shown in FIG. 56 and FIG. 57.
Example 37. IL-2 Serum Stability
[0534] The stability of IL2 fusion proteins in human serum (normal and cancer patient) was measured using capillary electrophoresis-based immunoassays (Jess instrument, Protein Simple). Fusion proteins were concentrated to 10 mg/ml and incubated with serum (90%). A time zero sample was immediately stored on ice while 24 hour and 72 hour samples were placed at 37.degree. C. Post incubation, samples were diluted 1:1000 with 0.1.times. sample buffer (Protein Simple) and loaded on the Jess cartridge per the manufacturer's protocol. The primary antibody was a monoclonal human IL2 antibody (R&D Systems, cat #AF-202-NA, stock concentration 0.2 mg/ml, working concentration 1:100), and the secondary antibody was a peroxidase-conjugated AffiniPure Bovine Anti Goat IgG (H+L) (Jackson Immuno Research, cat #805-035-18, reconstituted at 0.8 mg/ml, working concentration 1:5000 dilution). All antibodies were diluted in milk free diluent (Protein Simple). Quantitation of IL2 containing species was quantitated using Compass software (Protein Simple) to determine the extent of cleavage (i.e. amount of IL2 containing species smaller than the input fusion protein). Results are shown in FIGS. 24A-24B.
Example 38: Tissue Stability
[0535] Primary human lung epithelial cells and renal epithelial cells were obtained from ATCC. Primary hepatocytes were obtained from Lonza and Sigma. Cells were thawed, counted, and plated at 1e4 cells per well in a 96 well round bottom plate in their respective growth medias. Polypeptides containing recombinant human IL-2 and the sequence for Linker-2 and Linker-3 were incubated with cells or media alone for 24 and 72 hours at a concentration 5 .mu.g/mL. Cell culture supernatants were collected, and cells were discarded. Protein cleavage was measured by western blot for IL-2 using the protein simple JESS system and Compass software. Results are shown in FIG. 5.
[0536] Primary human lung fibroblasts were obtained from ATCC. Prior to the processing assay, human PBMCs were stimulated with 5 ug/mL PHA for 72 hours to generate T blasts, which were then frozen and used to measure polypeptide processing. Primary human fibroblasts were thawed, counted, and plated at 1e5 cells per well in a 96 well round bottom plate in X-Vivo 15 media. Polypeptides containing recombinant human IL-2 and the sequence for Linker-1 (GPAGMKGL, SEQ ID NO: 196), Linker-2 (GPAGLYAQ, SEQ ID NO: 195), or Linker-3 (ALFKSSFP, SEQ ID NO: 198), or a non-cleavable sequence were incubated with or without fibroblasts cells for 48 hours at a concentration of either A) 3.3 nM or B) 0.33 nM. As a positive control, some wells were also incubated with a polypeptide that was pre-cut in vitro overnight with 1 .mu.g pre-activated MMP9 enzyme at 37.degree. C. After 48 hours, cell culture supernatants were collected and healthy fibroblasts were discarded. Polypeptide processing was measured either by A) western blot for IL-2 using the protein simple JESS system, or by B) measuring the capacity of the cell culture supernatants to stimulate T blast proliferation. Briefly, T blasts were incubated for 72 hours with cell culture supernatants containing polypeptides previously exposed to healthy human fibroblasts, before T blast proliferation was measured by Cell Titer Glow analysis. Results are shown in FIG. 6A-6B.
Example 39: Linker-2 is Efficiently Cleaved in Human Tumor Cells
[0537] Cleavage efficiency of Linker-2 (GPAGLYAQ, SEQ ID NO: 195) was evaluated in tumor tissue derived from human samples. A total of 66 samples were analyzed from seven solid tumor types. The tumor types are shown in Table 5 below.
[0538] Briefly, frozen dissociated tumor cells (DTC) were thawed, counted and plated in X-Vivo media. Cells or "No Cell Control" wells were incubated with an inducible IL-2 cytokine containing Linker 1 (a cleavable linker that was not designed using the processes described herein) or Linker-2 (GPAGLYAQ, SEQ ID NO: 195). These inducible IL-2 proteins have no or minimal IL-2 biological activity when the linker is intact, and IL-2 biological activity is induced when the linker is cleaved. Cell culture supernatants were collected and frozen before analysis for IL-2 biological activity using T-Blast proliferation and Jess protein assays.
[0539] The minimum fold change in IL-2 activity is 1.00, and the maximum is 2.50. A 1.3 fold change is indicative of a significant increase in biological activity relative to the uncleaved control. Table 5 shows the average fold change for Linker-2 (GPAGLYAQ, SEQ ID NO: 195) in comparison to an uncleaved control, a cleaved control, and Linker-1. In Table 12 the symbol "-" indicate essentially no increase in activity; "+/-" indicates some increase in activity but not significant; "+," "++," and "+++" indicate the relative significant increase in activity compared to the uncleavable control. Linker-2 is sufficiently cleaved in human tumor types to induce IL-2 biological activity.
TABLE-US-00017 TABLE 12 Cleavage efficiency of Linker-2 in human tumor types Average Fold Change Over Non-Cleavable Diagnosis Uncleavable Linker 1 Linker 2 Pre-cut Melanoma - - +/- +++ (n = 8) Kidney Cancer, - - + +++ Renal Cell Carcinoma (n = 11) Head and Neck Cancer, - +/- +++ +++ Squamous Cell (n = 6) Colorectal Cancer, - +/- + +++ Adenocarcinoma (n = 10) Lung Cancer, - - ++ +++ Squamous Cell Carcinoma (n = 13) Lung Cancer, - - + +++ Adenocarcinoma (n = 10) Breast Cancer - - +/- +++ (n = 5)
Example 40: Linker-3 is Efficiently Cleaved in Human Tumor Cells
[0540] Cleavage efficiency of Linker-3 (ALFKSSFP, SEQ ID NO: 198) was evaluated in tumor tissue derived from human samples. A total of 66 samples were analyzed from seven solid tumor types. The tumor types are shown in Table 6 below.
[0541] Briefly, frozen DTC cells were thawed, counted and plated in X-Vivo media. Cells or "No Cell Control" wells were incubated with an inducible IL-2 cytokine containing Linker 1 (a cleavable linker that was not designed using the processes described herein) or Linker-2 (GPAGLYAQ, SEQ ID NO: 195). These inducible IL-2 proteins have no or minimal IL-2 biological activity when the linker is intact, and IL-2 biological activity is induced when the linker is cleaved. Cell culture supernatants were collected and frozen before analysis for IL-2 biological activity using T-Blast proliferation and Jess protein assays.
[0542] The minimum fold change in IL-2 activity is 1.00, and the maximum is 2.50. A 1.3 fold change is indicative of a significant increase in biological activity relative to the uncleaved control. Table 6 shows the average fold change for Linker-3 (ALFKSSFP, SEQ ID NO: 198) in comparison to an uncleaved control, a cleaved control, and Linker 1. In Table 13 the symbol "-" indicate essentially no increase in activity; "+/-" indicates some increase in activity but not significant; "+," "++," and "+++" indicate the relative significant increase in activity compared to the uncleavable control. Linker-3 (ALFKSSFP, SEQ ID NO: 198) is sufficiently cleaved in human tumor types to induce IL-2 biological activity.
TABLE-US-00018 TABLE 13 Cleavage efficiency of Linker-3 in human tumor types Average Fold Change Over Non-Cleavable Diagnosis Uncleavable Linker 1 Linker 3 Pre-cut Melanoma - - + +++ (n = 8) Kidney Cancer, - - + +++ Renal Cell Carcinoma (n = 11) Head and Neck Cancer, - +/- +++ +++ Squamous Cell (n = 6) Colorectal Cancer, - +/- +++ +++ Adenocarcinoma (n = 10) Lung Cancer, - - +++ +++ Squamous Cell Carcinoma (n = 13) Lung Cancer, - - +++ +++ Adenocarcinoma (n = 10) Breast Cancer - - + +++ (n = 5)
Example 41: Measuring Inducibility of Agonist Anti-4-1BB Antibodies PGP-41
[0543] A stable HT-1080 cell line expressing human 4-1BB was established. Agonism of human-4-1BB in these cells resulted in increased secretion of IL-8. Tetravalent monospecific antibodies, inducible format tetravalent monospecific antibodies (protease cleaved or uncleaved), or trimeric ligands were tested for ability to agonize 4-1BB.
[0544] Before addition to the cultured cells, some samples of the antibody to be tested were incubated with an appropriate protease under suitable conditions for proteolysis. Some samples were maintained in an uncleaved state. The extent of inducibility was determined by comparison of uncleaved (uninduced) inducible format tetravalent monospecific antibody with the corresponding protease cleaved inducible format tetravalent monospecific antibody.
[0545] After incubation at 37.degree. C. and 5% CO2 for 6 hours, the agonistic activities of the cleaved and uncleaved antibodies were evaluated by the quantification of IL-8 production using an IL-8 AlphaLISA or ELISA. The EC50s and maximum IL-8 levels were compared. Results are shown in FIG. 65A-65B.
Example 42: Protease Cleavage of Anti-4-1BB Antibodies by MMP9
[0546] One of skill in the art would be familiar with methods of setting up protein cleavage assay. 100 .mu.g of protein in 1.times.PBS pH 7.4 were cleaved with 1 .mu.g active MMP9 (Sigma catalog #SAE0078-50 or Enzo catalog BML-SE360) and incubated at room temperature for up to 16 hours. Digested protein is subsequently used in functional assays or stored at -80.degree. C. prior to testing. The extent of cleavage was monitored by SDS PAGE using methods well known in the art.
Example 43: Protease Cleavage of Anti-4-1BB Antibodies by MMP14 or CTSL1
[0547] One of skill in an be replaced with Linker-3. the art would be familiar with methods of setting up protein cleavage assay. 100 .mu.g of protein in 1.times.PBS pH 7.4 are cleaved with 1 .mu.g active MMP14 (R&D Systems Catalog #9518-MP-010) or CTSL1 (R&D Systems Catalog #952-CY) and incubated at room temperature for up to 16 hours. Digested protein are subsequently used in functional assays or stored at -80.degree. C. prior to testing. The extent of cleavage is monitored by SDS PAGE using methods well known in the art.
8. OTHER EMBODIMENTS
[0548] The disclosure set forth above may encompass multiple distinct inventions with independent utility. Although each of these inventions has been disclosed in its preferred form(s), the specific embodiments thereof as disclosed and illustrated herein are not to be considered in a limiting sense, because numerous variations are possible. The subject matter of the inventions includes all novel and nonobvious combinations and subcombinations of the various elements, features, functions, and/or properties disclosed herein. The following claims particularly point out certain combinations and subcombinations regarded as novel and nonobvious. Inventions embodied in other combinations and subcombinations of features, functions, elements, and/or properties may be claimed in this application, in applications claiming priority from this application, or in related applications. Such claims, whether directed to a different invention or to the same invention, and whether broader, narrower, equal, or different in scope in comparison to the original claims, also are regarded as included within the subject matter of the inventions of the present disclosure.
[0549] Exemplary polypeptide constructs are detailed herein in Appendix A. While the exemplary polypeptides that contain Linker-2 (GPAGLYAQ, SEQ ID NO: 195), or Linker-3 (ALFKSSFP, SEQ ID NO: 198) or other cleavable linkers are disclosed in Appendix A, for each construct, the disclosed linker can be replaced with either Linker-2 (GPAGLYAQ, SEQ ID NO: 195), or Linker-3 (ALFKSSFP, SEQ ID NO: 198) or other cleavable linkers disclosed herein. For example construct
ACP355 (IgG4_Fc(S228P)-X-CD25ecd_C213S-LX-IL2-LX Blocker_(Blocker=VHVL.F2.high.A02_Vh\Vl_A46S;X=MMP14-1) can contain Linker-2, however it can be replaced with Linker-3. Alternatively, construct ACPT464 (IgG4_Fc(S228P)-X-IL2-LX-blocker_(Blocker=VHVL.F2.high.A02_Vh/Vl_VH105-VL- 43_disulfidel;X=CTSL1-1) can contain Linker-3, however, it can be replaced with Linker-2.
[0550] The elements of the polypeptide constructs provided in Appendix A contain the abbreviations as follows: "L," "X," "LX," and "XL" each refer to a linker. "X" refers to a cleavable linker. "L" refers a linker that is optionally cleavable. When L is the only linker in a polypeptide, L is cleavable. "LX" or "XL" each refer to a cleavable linker with an extended non-cleavable sequence adjacent to it. Cleav. Lin. Also refers to a cleavable linker. Other abbreviations used include: "mIFNg" for mouse interferon gamma (IFNg); (5) "hAlbumin" for human serum albumin (HSA); and (6) "mAlbumin" indicates mouse serum albumin.
TABLE-US-00019 APPENDIX A CONSTRUCT PERMUTATION TABLE Construct Name Construct Description ACP01 (anti-HSA)-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-(anti-HSA)-6xHis ACP02 (anti-HSA)-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-(anti- HSA)-6xHis ACP03 (anti-HSA)-(cleav. link.)-mouse IFN.gamma.-mouse IFN.gamma.-(cleav. link.)-(anti-HSA)-6xHis ACP50 (anti-EpCAM)-(anti-HSA)-(cleav. link.)-mouse IFN.gamma.-mouse IFN.gamma.-(cleav. link.)-(anti- HSA)-6xHis ACP51 (anti-EpCAM)-Linker-(anti-HSA)-(cleav. link.)-mIFN.gamma.-(cleav. link.)-(anti-HSA)- 6xHis ACP52 (anti-HSA)-(cleav. link.)-mIFN.gamma.-(cleav. link.)-(anti-HSA)-Linker-(anti-EpCAM)- 6xHis ACP53 mAlbumin-(cleav. link.)-mIFN.gamma.-(cleav. link.)-mAlbumin-6xHis ACP54 mAlbumin-(cleav. link.)-mIFN.gamma.-Linker-mIFN.gamma.-(cleav. link.)-mAlbumin-6xHis ACP30 (anti-HSA)-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-(anti-HSA)-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-(anti-HSA)-6xHis ACP55 (anti-HSA)-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-(anti-HSA)-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-(anti-HSA)-6xHis-C-tag ACP56 (anti-FOLR1)-Linker-(anti-HSA)-(cleav. link.)-mIFN.gamma.-(cleav. link.)-(anti-HSA)-6xHis ACP57 (anti-HSA)-(cleav. link.)-mIFN.gamma.-(cleav. link.)-(anti-HSA)-Linker-(anti-FOLR1)-6xHis ACP58 (anti-HSA)-(cleav. link.)-mIFN.gamma.-(cleav. link.)-mIFN.gamma.-(cleav. link.)-(anti-HSA)- Linker-(anti-EpCAM)-6xHis ACP59 (anti-FOLR1)-Linker-(anti-HSA)-(cleav. link.)-mIFN.gamma.-(cleav. link.)-mIFN.gamma.-(cleav. link.)-(anti-HSA)-6xHis ACP60 (anti-HSA)-(cleav. link.)-mIFN.gamma.-(cleav. link.)-mIFN.gamma.-(cleav. link.)-(anti-HSA)- Linker-(anti-FOLR1)-6xHis ACP61 (anti-HSA)-(cleav. link.)-mIFN.gamma.-(cleav. link.)-mIFN.gamma.-(cleav. link.)-(anti-HSA)- Linker-FN(CGS-2)-6xHis ACP63 anti-FN CGS-2 scFv (Vh/Vl)-6xHis ACP69 (anti-HSA)-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-(anti-HSA)-(cleav. link.)-mouse IFN.gamma. ACP70 mouse IFN.gamma.-(cleav. link.)-(anti-HSA)-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-(anti- HSA) ACP71 mouse IFN.gamma.-(cleav. link.)-mAlbumin-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)- mAlbumin ACP72 mAlbumin-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-mAlbumin-(cleav. link.)-mouse IFN.gamma. ACP73 mAlbumin-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-mAlbumin-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-mAlbumin ACP74 mAlbumin-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-5mer linker-mAlbumin-5mer linker-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-mAlbumin ACP75 mAlbumin-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-10mer linker-mAlbumin-10mer linker-(cleav. link.)-mouse IFN.gamma.-(cleav. link.)-mAlbumin ACP78 (anti-HSA)-Linker-mouse_IFN.gamma.-Linker-(anti-HSA)-Linker-mouse_IF- N.gamma.-Linker-(anti- HSA)_(non-cleavable_control) ACP134 Anti-HSA-X-mouse_IFN.gamma.-X-anti-HSA-X-mouse_IFN.gamma.-X-anti-HS- A-L-anti-FOLR1 ACP135 Anti-FOLR1-L-HSA-X-mouse_IFN.gamma.-X-HSA-X-mouse_IFN.gamma.-X-HSA ACP04 human p40-murine p35-6xHis ACP05 human p40-human p35-6xHis ACP34 mouse p35-(Cleavable Linker)-mouse p40-6xHis ACP35 mouse p35-GS-(Cleavable Linker)-GS-mouse p40-6xHis ACP36 (anti-HSA)-(Cleav. Linker)-mouse p40-mouse p35-(Cleav. Linker)-(anti-HSA)-6xHis ACP37 (anti-EpCAM)-(anti-HSA)-(Cleav. Linker)-mouse p40-mouse p35-(Cleav. Linker)- (anti-HSA)-6xHis ACP79 (anti-EpCAM)-Linker-(anti-HSA)-(Cleavable Linker)-mIL12-(Cleavable Linker)- (Anti-HSA)-6xHis ACP80 (anti-HSA)-(Cleavable Linker)-mIL12-(Cleavable Linker)-(anti-HSA)-Linker-(anti- EpCAM)-6xHis ACP06 Blocker12-Linker-(Cleavable Linker)-human p40-Linker-mouse p35-(Cleavable Linker)-(anti-HSA)-6xHis ACP07 Blocker12-Linker-(Cleavable Linker)-human p40-Linker-mouse p35-(Cleavable Linker)-(anti-HSA)-Linker-(anti-FOLR1)-6xHis ACP08 (anti-FOLR1)-Linker-Blocker12-Linker-(Cleavable Linker)-human p40-Linker-mouse p35-(Cleavable Linker)-(anti-HSA)-6xHis ACP09 (anti-HSA)-Linker-Blocker12-Linker-(Cleavable Linker)-human p40-Linker-mouse p35-6xHis ACP10 (anti-HSA)-(Cleavable Linker)-human p40-L-mouse p35-(Cleavable Linker)-Linker- Blocker12-6xHis ACP11 hp40-Linker-mp35-(Cleavable Linker)-Linker-Blocker12-Linker-(anti-HSA)-6xHis ACP91 human_p40-Linker-mouse_p35-Linker-Linker-Blocker-Linker-(anti-HSA) (non- cleavable_control) ACP136 human p40-L-mouse p35-XL-Blocker ACP138 human_p40-L-mouse_p35-XL-Blocker-L-HSA-L-FOLR1 ACP139 FOLR1-L-human_p40-L-mouse_p35-XL-Blocker-L-HSA ACP140 FOLR1-X-human_p40-L-mouse_p35-XL-Blocker-L-HSA ACP12 (anti-EpCAM)-IL2-(cleav. link.)-(anti-HSA)-blocker-6xHis ACP13 (anti-EpCAM)-Blocker2-(anti-HSA)-(cleav. link.)-IL2-6xHis ACP14 Blocker2-Linker-(cleav. link.)-IL2- (cleav. link.)-(anti-HSA)-6xHis ACP15 Blocker2-Linker-(anti-HSA)-Linker-(cleav. link.)- IL2 -6xHis ACP16 IL2-(cleav. link.)-(anti-HSA)-Linker-(cleav. link.)-Blocker2-6xHis ACP17 (anti-EpCAM)-Linker-IL2-(cleav. link.)-(anti-HSA)-Linker-(cleav. link.)-Blocker2- 6xHis ACP18 (anti-EpCAM)-Linker-IL2-(cleav. link.)-(anti-HSA)-Linker-vh(cleav. link.)vl-6xHis ACP19 IL2-(cleav. link.)-Linker-Blocker2-Linker-(anti-HSA)-Linker-(anti-EpCAM) -6xHis ACP20 IL2-(cleav. link.)-Blocker2-6xHis ACP21 IL2-(cleav. link.)-Linker-Blocker2-6xHis ACP22 IL2-(cleav. link.)-Linker-blocker-(cleav. link.)-(anti-HSA)-Linker-(anti-EpCAM)- 6xHis ACP23 (anti-FOLR1)-(cleav. link.)-Blocker2-Linker-(cleav. link.)-(anti-HSA)-(cleav. link.)- IL2-6xHis ACP24 (Blocker2)-(cleav. link.)-(IL2)-6xHis ACP25 Blocker2-Linker-(cleav. link.)-IL2-6xHis ACP26 (anti-EpCAM)-Linker-IL2-(cleav. link.)-(anti-HSA)-Linker-blocker(NARA1 Vh/Vl) ACP27 (anti-EpCAM)-Linker-IL2-(cleav. link.)-(anti-HSA)-Linker-blocker(NARA1 Vl/Vh) ACP28 IL2-(cleav. link.)-Linker-Blocker2-(NARA1 Vh/Vl)-Linker-(anti-HSA)-Linker-(anti- EpCAM) ACP29 IL2-(cleav. link.)-Linker-Blocker2-(NARA1 Vl/Vh)-Linker-(anti-HSA)-Linker-(anti- EpCAM) ACP38 IL2-(cleav. link.)-blocker-(anti-HSA)-(anti-EpCAM)-6xHis ACP39 (anti-EpCAM)-(cleav. link.)-(anti-HSA)-(cleav. link.)-Blocker2-(cleav. link.)-IL-2- 6xHis ACP40 CD25ecd-Linker-(cleav. link.)-IL2-6xHis ACP41 IL2-(cleav. link.)-Linker-CD25ecd-6xHis ACP42 (anti-HSA)-Linker-CD25ecd-Linker-(cleav. link.)-IL2-6xHis ACP43 IL2-(cleav. link.)-Linker-CD25ecd-Linker-(anti-HSA)-6xHis ACP44 IL2-(cleav. link.)-Linker-CD25ecd-(cleav. link.)-(anti-HSA)-6xHis ACP45 (anti-HSA)-(cleav. link.)-Blocker2-Linker-(cleav. link.)-IL2-6xHis ACP46 IL2-(cleav. link.)-linkerL-vh(cleav. link.)vl-Linker-(anti-HSA)-L-(anti-EpCAM)- 6xHis ACP47 (anti-EpCAM)-Linker-IL2-(Cleavable Linker)-(anti-HSA)-Linker-Blocker2-6xHis ACP48 IL2-(cleav. link.)-Blocker2-Linker-(anti-HSA)-6xHis ACP49 IL2-(cleav. link.)-Linker-Blocker2-Linker-(anti-HSA)-6xHis ACP92 (anti-HSA)-(16mer Cleav. Link.)-IL2-(16mer Cleav. Link.)-(anti-HSA)-6XHis ACP93 (anti-EpCAM)-(anti-HSA)-(anti-EpCAM)-Blocker2-(cleav. link.)-IL2-6xHis ACP94 (anti-EpCAM)-(anti-HSA)-Blocker2-(cleav. link.)-IL2-6xHis ACP95 (anti-EpCAM)-(anti-HSA)-(cleav. link.)-IL2-6xHis ACP96 (anti-EpCAM)-(16mer cleav. link.)-IL2-(16mer cleav. link.)-(anti-HSA) ACP38 IL2-(cleav. link.)-blocker-(anti-HSA)-(anti-EpCAM)-6xHis ACP97 (anti-EpCAM)-(anti-HSA)-(cleav. link.)-IL2-(cleav. link.)-(anti-HSA)-6xHis ACP99 (anti-EpCAM)-Linker-IL2-(cleav. link.)-(anti-HSA)-6xHis ACP100 (anti-EpCAM)-Linker-IL2-6xHis ACP101 IL2-(cleav. link.)-(anti-HSA)-6xHis ACP102 (anti-EpCAM)-(cleav. link.)-IL2-(cleav. link.)-(anti-HSA)-Linker-blocker-6xHis ACP103 IL2-(cleav. link.)-Linker-Blocker2-Linker-(anti-HSA)-Linker-(antiI-FOLR1)-6xHis ACP104 (anti-FOLR1)-IL2-(cleav. link.)-(anti-HSA)-Linker-Blocker2-6xHis ACP105 Blocker2-Linker-(cleav. link.)-IL2-(cleav. link.)-(anti-HSA)-Linker-(anti-FOLR1)- 6xHis ACP106 (anti-FOLR1)-Linker-(anti-HSA)-(cleav. link.)-blocker-Linker-(cleav. link.)-IL2 - 6xHis ACP107 Blocker2-Linker-(anti-HSA)-(cleav. link.)-IL2-Linker-(anti-FOLR1)-6xHis ACP108 (anti-EpCAM)-IL2-(Dually cleav. link.)-(anti-HSA)-Linker-blocker-6xHis ACP117 anti-FN CGS-2 scFv (Vh/Vl)-6xHis ACP118 NARA1 Vh/Vl non-cleavable ACP119 NARA1 Vh/Vl cleavable ACP120 NARA1 Vl/Vh non-cleavable ACP121 NARA1 Vl/Vh cleavable ACP124 IL2-Linker-(anti-HSA)-Linker-Linker-blocker_(non-cleavable_control) ACP132 IL2-L-HSA ACP141 IL2-L-hAlb ACP142 IL2-X-hAlb ACP144 IL2-X-HSA-LX-blocker-L-FOLR1 ACP145 FOLR1-L-IL2-X-HSA-LX-blocker ACP146 FOLR1-X-IL2-X-HSA-LX-blocker ACP133 IL-2-6x His ACP147 IL2-X-HSA-LX-blocker-L-TAA ACP148 TAA-L-IL2-X-HSA-LX-blocker ACP149 TAA-X-IL2-X-HSA-LX-blocker ACP31 (anti-HSA)-(cleav. link.)-mIFNa1-(cleav. link.)-(anti-HSA) ACP32 (anti-HSA)-(cleav. link.)-mIFNa1(N + C trunc)-(cleav. link.)-(anti-HSA) ACP33 (anti-HSA)-(cleav. link.)-mIFNa1(C trunc)-(cleav. link.)-(anti-HSA) ACP131 mIFNa1 ACP125 HSA-X-mIFNa1 ACP126 mIFNa1-X-HSA ACP127 mAlb-X-mIFNa1-X-mAlb ACP128 mAlb-X-mIFNa1 ACP129 mIFNa1-X-mAlb ACP150 FOLR1-L-HSA-X-mIFNa1-X-HSA ACP151 FOLR1-L-HSA-X-mIFNa1-X-HSA-L-FOLR1 ACP152 HSA-L-mIFNa1-L-HSA_(non-cleavable_control) ACP203 HSA-X-mIFNa1-X-HSA_(X = MMP14-1) ACP204 HSA-X-mIFNa1-X-HSA_(X = CTSL1-1) ACP205 HSA-X-mIFNa1-X-HSA_(X = ADAM17-2) ACP206 HSA-X-Human_IFNA2b-X-HSA_(X = MMP14-1) ACP207 HSA-X-Human_IFNA2b-X-HSA _(X = CTSL1-1) ACP208 HSA-X-Human_IFNA2b-X-HSA _(X = ADAM17-2) ACP336 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh-X- Vl_A46S; X = MMP14-1) ACP337 IL2-X-anti-HSA-LX- blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_A46S; X = MMP14-1) ACP338 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh-X-Vl; X = MMP14-1) ACP339 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl;X = MMP14-1) ACP340 IL2-X-anti-HSA-LX-blocker_(Blocker = Hu2TOW91_B; X = MMP14-1) ACP341 IL2-X-anti-HSA-LX-blocker_(Blocker = Hu3TOW85_A; X = MMP14-1) ACP342 CD25ecd_C213S-LX-IL2-X-anti-HSA-LX- blocker_(Blocker = VHVL.F2.high.A02_Vh-X-Vl_A46S; X = MMP14-1) ACP343 CD25ecd_C213 S-LX-IL2-X-anti-HS A-LX- blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_A46S; X = MMP14-1) ACP344 CD25ecd_C213S-LX-IL2-X-anti-HSA-LX- blocker_(Blocker = VHVL.F2.high.F03_Vh-X-Vl; X = MMP14-1) ACP345 CD25ecd_C213S-LX-IL2-X-anti-HSA-LX- blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl; X = MMP14-1) ACP346 CD25ecd_C213S-LX-IL2-X-anti-HSA-LX- blocker_(Blocker = Hu2TOW91_B; X = MMP14-1) ACP347 CD25ecd_C213S-LX-IL2-X-anti-HSA-LX- blocker_(Blocker = Hu3TOW85_A; X = MMP14-1) ACP348 IgG4_Fc(S228P)-X-IL2-LX-Blocker_(Blocker = VHVL.F2.high.A02_Vh-X-
Vl_A46S; X = MMP14-1) ACP349 IgG4_Fc(S228P)-X-IL2-LX- Blocker_(Blocker = VHVL.F2.high.A02_Vh\Vl_A46S; X = MMP14-1) ACP350 IgG4_Fc(S228P)-X-IL2-LX-Blocker_(Blocker = VHVL.F2.high.F03_Vh-X- V1; X = MMP14-1) ACP351 IgG4_Fc(S228P)-X-IL2-LX- Blocker_(Blocker = VHVL.F2.high.F03_Vh\Vl; X = MMP14-1) ACP352 IgG4_Fc(S228P)-X-IL2-LX-Blocker_(Blocker = Hu2TOW91_B; X = MMP14-1) ACP353 IgG4_Fc(S228P)-X-IL2-LX-Blocker_(Blocker = Hu3TOW85_A; X = MMP14-1) ACP354 IgG4_Fc(S228P)-X-CD25ecd_C213S-LX-IL2-LX- Blocker_(Blocker = VHVL.F2.high.A02_Vh-X-Vl_A46S; X = MMP14-1) ACP355 IgG4_Fc(S228P)-X-CD25ecd_C213S-LX-IL2-LX- Blocker_(Blocker = VHVL.F2.high.A02_Vh\Vl_A46S; X = MMP14-1) ACP356 IgG4_Fc(S228P)-X-CD25ecd_C213S-LX-IL2-LX- Blocker_(Blocker = VHVL.F2.high.F03_Vh-X-Vl; X = MMP14-1) ACP357 IgG4_Fc(S228P)-X-CD25ecd_C213S-LX-IL2-LX- Blocker_(Blocker = VHVL.F2.high.F03_Vh\Vl; X = MMP14-1) ACP358 IgG4_Fc(S228P)-X-CD25ecd_C213S-LX-IL2-LX- Blocker_(Blocker = Hu2TOW91_B; X = MMP14-1) ACP359 IgG4_Fc(S228P)-X-CD25ecd_C213S-LX-IL2-LX- Blocker_(Blocker = Hu3TOW85_A; X = MMP14-1) ACP360 MT204_Vh/Vl_3xG4S_A46S ACP361 MT204_Vh-X-Vl_X = MMP14-1 ACP362 MT204_Vh-X-Vl_X = MMP14-1,_A46S ACP365 VHVL.F2.high.A02_Vh-X-Vl_X = MMP14-1 ACP366 VHVL.F2.high.A02_Vh-X-Vl_X = MMP14-1,_A46S ACP368 VHVL.F2.high.F03_Vh-X-Vl_X = MMP14-1 ACP371 IL2-X-anti-HSA-LX-blocker_(Blocker = MT204_Vh/Vl_VH44- VL100_disulfide; X = MMP14-1) ACP372 IL2-X-anti-HSA-LX-blocker_(Blocker = MT204_Vh/Vl_VH105- VL43_disulfide; X = MMP14-1) ACP373 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_VH44- VL100_disulfide; X = MMP14-1) ACP374 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_VH105- VL43_disulfide; X = MMP14-1) ACP375 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl_VH44- VL100_disulfide; X = MMP14-1) ACP376 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl_VH105- VL43_disulfideX = MMP14-1) ACP377 IL2-X-anti-HSA-LX-blocker_(Blocker = Hu2TOW91_A; X = MMP14-1) ACP378 IL2-X-anti-HSA-LX-Heavy_blocker_Fab_(Blocker = MT204_VH-CH1; X = MMP14-1) ACP379 IgG4_Fc(S228P)-X-IL2-LX-Heavy_blocker_Fab_(Blocker = MT204_VH- CH1; X = MMP14-1) ACP383 IgG4_Fc(S228P)-X-IL2-LX-blocker_(Blocker = MT204_Vh/Vl_VH44- VL100_disulfide; X = MMP14-1) ACP384 IgG4_Fc(S228P)-X-IL2-LX-blocker_(Blocker = MT204_Vh/Vl_VH105- VL43_disulfide; X = MMP14-1) ACP385 IgG4_Fc(S228P)-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_VH44- VL100_disulfide; X = MMP14-1) ACP386 IgG4_Fc(S228P)-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_VH105- VL43_disulfide; X = MMP14-1) ACP387 IgG4_Fc(S228P)-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl_VH44- VL100_disulfide; X = MMP14-1) ACP388 IgG4_Fc(S228P)-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl_VH105- VL43_disulfide; X = MMP14-1) ACP389 IgG4_Fc(S228P)-X-IL2-LX-blocker_(Blocker = Hu2TOW91_A; X = MMP14-1) ACP390 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_A46S_VH44- VL100_disulfide; X = MMP14-1) ACP391 IgG4_Fc(S228P)-X-IL2-LX- blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_A46S_VH44- VL100_disulfide; X = MMP14-1) ACP392 IL2-XL-CD25ecd_C213S-X-HSA-LX- blocker_(Blocker = VHVL.F2.high.A02_Vh_G44C_Vl_A46S_G100C; X = MMP14-1) ACP393 IL2-XL-CD25ecd_C213S-X-HSA-LX- blocker_(Blocker = VHVL.F2.high.A02_Vh_Q105CV1A43C; X = MMP14-1) ACP394 IL2-XL-CD25ecd_C213S-X-HSA-LX- blocker_(Blocker = VHVL.F2.high.F03_Vh_G44C_Vl_G100C; X = MMP14-1) ACP395 IL2-XL-CD25ecd_C213S-X-HSA-LX- blocker_(Blocker = VHVL.F2.high.F03_Vh_Q105C_Vl_A43C; X = MMP14-1) ACP396 IL2-XL-CD25ecd_C213S-X-HSA-LX-blocker_(Blocker = Hu2TOW91_A; X = MMP14- 1) ACP397 IL2-XL-CD25ecd_C213S-X-HSA-LX-blocker_(Blocker = Hu2TOW91_B; X = MMP14- 1) ACP398 IL2-XL-CD25ecd_C213S-X-HSA-LX-Heavy_blocker_Fab_(Blocker = MT204_VH- CH1; X = MMP14-1) ACP399 Blocker-XL-HSA-X-IL2(Nterm-41)-X- HSA_(Blocker = VHVL.F2.high.A02_Vh_G44C_Vl_A46S_G100C; X = MMP14-1) ACP400 Blocker-XL-HSA-X-IL2(Nterm-41)-X- HSA_(Blocker = VHVL.F2.high.A02_Vh_Q105CV1A43C; X = MMP14-1) ACP401 Blocker-XL-HSA-X-IL2(Nterm-41)-X- HSA_(Blocker = VHVL.F2.high.F03_Vh_G44C_Vl_G100C; X = MMP14-1) ACP402 Blocker-XL-HSA-X-IL2(Nterm-41)-X- HSA_(Blocker = VHVL.F2.high.F03_Vh_Q105C_Vl_A43C; X = MMP14-1) ACP403 Blocker-XL-HSA-X-IL2(Nterm-41)-X-HSA_(Blocker = Hu2TOW91_A; X = MMP14-1) ACP404 Blocker-XL-HSA-X-IL2(Nterm-41)-X-HSA_(Blocker = Hu2TOW91_B; X = MMP14-1) ACP405 Heavy_Blocker_Fab-XL-HSA-X-IL2(Nterm-41)-X-HSA_(Blocker = MT204_VH- CH1; X = MMP14-1) ACP406 mIgG1_Fc(S228P)-X-IL2-LX-Heavy_blocker_Fab_(Blocker = MT204_VH- CH1; X = MMP14-1) ACP407 mIgG1_Fc(S228P)-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_VH44- VL100_disulfide; X = MMP14-1) ACP408 mIgG1_Fc(S228P)-X-IL2-LX- blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_A46S_VH44- VL100_disulfide; X = MMP14-1) ACP409 mIgG1_Fc(S228P)-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_VH105- VL43_disulfidel; X = MMP14-1) ACP410 mIgG1_Fc(S228P)-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl_VH44- VL100_disulfidel; X = MMP14-1) ACP411 mIgG1_Fc(S228P)-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl_VH105- VL43_disulfidel; X = MMP14-1) ACP412 mIgG1_Fc(S228P)-X-IL2-LX-blocker_(Blocker = Hu2TOW91_A; X = MMP14-1) ACP415 IL2-XL-blocker-L-CD25_213S-X- HSA_Blocker = VHVL.F2.high.A02_Vh_G44C_Vl_A46S_G100C; X = MMP14-1) ACP416 IL2-XL-blocker-L-CD25_213S-X- HSA_(Blocker = VHVL.F2.high.A02_Vh_Q105C_Vl_A43C; X = MMP14-1) ACP417 IL2-XL-blocker-L-CD25_213 S-X- HSA_(Blocker = VHVL.F2.high.F03_Vh_G44C_Vl_G100C; X = MMP14-1) ACP418 IL2-XL-blocker-L-CD25_213S-X- HSA_(Blocker = VHVL.F2.high.F03_Vh_Q105C_Vl_A43C; X = MMP14-1) ACP419 IL2-XL-blocker-L-CD25_213 S-X-HSA_(Blocker = Hu2TOW91_A; X = MMP14-1) ACP420 IL2-XL-blocker-L-CD25_213S-X-HSA_(Blocker = Hu2TOW91_B; X = MMP14-1) ACP421 HSA-X-blocker-L-CD25_213S-LX- IL2_(Blocker = VHVL.F2.high.A02_Vh_G44C_Vl_A46S_G100C; X = MMP14-1) ACP422 HSA-X-blocker-L-CD25_213S-LX- IL2_(Blocker = VHVL.F2.high.A02_Vh_Q105C_Vl_A43C; X = MMP14-1) ACP423 HSA-X-blocker-L-CD25_213S-LX- IL2_(Blocker = VHVL.F2.high.F03_Vh_G44C_Vl_G100C; X = MMP14-1) ACP424 HSA-X-blocker-L-CD25_213S-LX- IL2_(Blocker = VHVL.F2.high.F03_Vh_Q105CV1A43C; X = MMP14-1) ACP425 HSA-X-blocker-L-CD25_213S-LX-IL2_(Blocker = Hu2TOW91_A; X = MMP14-1) ACP426 HSA-X-blocker-L-CD25_213S-LX-IL2_(Blocker = Hu2TOW91B; X = MMP14-1) ACP427 IL2-X-anti-HSA-LX-Blocker1-L- Blocker2_(Blocker1 = VHVL.F2.high.A02_Vh_G44C_Vl_A46S_G100C, Blocker2 = Hu2TOW91_A; X = MMP14-1) ACP428 IL2-X-anti-HSA-LX-Blocker1-L- Blocker2_(Blocker1 = VHVL.F2.high.A02_Vh_Q105C_Vl_A43C, Blocker2 = Hu2TOW91_A; X = MMP14-1) ACP429 IL2-X-anti-HSA-LX-Blocker1-L- Blocker2_(Blocker1 = VHVL.F2.high.F03_Vh_G44C_Vl_G100C, Blocker2 = Hu2TOW91_A; X = MMP14-1) ACP430 IL2-X-anti-HSA-LX-Blocker1-L- Blocker2_(Blocker1 = VHVL.F2.high.F03_Vh_Q105C_Vl_A43C, Blocker2 = Hu2TOW91_A; X = MMP14-1) ACP431 IL2-X-anti-HSA-LX-Blocker1-L- Blocker2_(Blocker1 = VHVL.F2.high.A02_Vh_G44C_Vl_A46S_G100C, Blocker2 = Hu2TOW91_B; X = MMP14-1) ACP432 IL2-X-anti-HSA-LX-Blocker1-L- Blocker2_(Blocker1 = VHVL.F2.high.A02_Vh_Q105C_Vl_A43C, Blocker2 = Hu2TOW91_B; X = MMP14-1) ACP433 IL2-X-anti-HSA-LX-Blocker1-L- Blocker2_(Blocker1 = VHVL.F2.high.F03_Vh_G44C_Vl_G100C, Blocker2 = Hu2TOW91_B; X = MMP14-1) ACP434 IL2-X-anti-HSA-LX-Blocker1-L- Blocker2_(Blocker1 = VHVL.F2.high.F03_Vh_Q105C_Vl_A43C, Blocker2 = Hu2TOW91_B; X = MMP14-1) ACP439 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.C07_Vh/Vl; X = MMP14-1) ACP440 IL2-X-anti-HSA-LX- blocker_(Blocker = VHVL.F2.high.C07_Vh/Vl_A46S; X = MMP14-1) ACP441 IL2-X-anti-HSA-LX- blocker_(Blocker = VHVL.F2.high.C07_Vh/Vl_A46L; X = MMP14-1) ACP442 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.C07_Vh/Vl_A46S_VH44- VL100_disulfide; X = MMP14-1) ACP443 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.C07_Vh/Vl_A46L_VH44- VL100_disulfide; X = MMP14-1) ACP444 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.C07_Vh/Vl_VH105- VL43_disulfide; X = MMP14-1) ACP445 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh-X- V1_A46L; X = MMP14-1) ACP446 IL2-X-anti-HSA-LX- blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_A46L; X = MMP14-1) ACP447 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_A46L_VH44- VL100_disulfide; X = MMP14-1) ACP451 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_A46S; X = CTSL1- 1) ACP452 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl; X = CTSL1-1) ACP453 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_A46S_VH44- VL100_disulfide; X = CTSL1-1) ACP454 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_VH105- VL43_disulfidel; X = CTSL1-1) ACP455 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl_VH44- VL100_disulfide; X = CTSL1-1) ACP456 IL2-X-anti-HSA-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl_VH105- VL43_disulfideX = CTSL1-1) ACP457 IL2-X-anti-HSA-LX-Heavy_blocker_Fab_(Blocker = MT204_VH-CH1; X = CTSL1-1) ACP458 IgG4_Fc(S228P)-X-IL2-LX-Heavy_blocker_Fab_(Blocker = MT204_VH- CH1; X = CTSL1-1) ACP459 IgG4_Fc(S228P)-X-IL2-LX- Blocker_(Blocker = VHVL.F2.high.A02_Vh\Vl_A46S; X = CTSL1-1) ACP460 IgG4_Fc(S228P)-X-IL2-LX- Blocker_(Blocker = VHVL.F2.high.F03_Vh\Vl; X = CTSL1-1) ACP461 IgG4_Fc(S228P)-X-IL2-LX- blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_A46S_VH44- VL100_disulfide; X = CTSL1-1) ACP462 IgG4_Fc(S228P)-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_VH105- VL43_disulfidel; X = CTSL1-1) ACP463 IgG4_Fc(S228P)-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl_VH44- VL100_disulfidel; X = CTSL1-1) ACP464 IgG4_Fc(S228P)-X-IL2-LX-blocker_(Blocker=VHVL.F2.high.F03_Vh/Vl_VH1- 05- VL43_disulfidel; X = CTSL1-1) ACP465 mIgG1_Fc-X-IL2-LX- Blocker_(Blocker = VHVL.F2.high.A02_Vh\Vl_A46S; X = CTSL1-1) ACP466 mIgG1_Fc-X-IL2-LX-Blocker_(Blocker = VHVL.F2.high.F03_Vh\Vl; X = CTSL1-1) ACP467 mIgGl_Fc-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_A46S_VH44- VL100_disulfide; X = CTSL1-1) ACP468 mIgG1_Fc-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.A02_Vh/Vl_VH105- VL43_disulfidel; X = CTSL1-1) ACP469 mIgG1_Fc-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl_VH44- VL100_disulfidel; X = CTSL1-1) ACP470 mIgG1_Fc-X-IL2-LX-blocker_(Blocker = VHVL.F2.high.F03_Vh/Vl_VH105- VL43_disulfidel; X = CTSL1-1) ACP471 mIgG1_Fc-X-IL2-LX-Heavy_blocker_Fab_(Blocker = MT204_VH-CH1; X = CTSL1-1)
TABLE-US-00020 APPENDIX B Sequences SEQ ID NO. Name Sequence 1 Human IL-2 MYRMQLLSCI ALSLALVTNS APTSSSTKKT QLQLEHLLLD LQMILNGINN YKNPKLTRML TFKFYMPKKA TELKHLQCLE EELKPLEEVL NLAQSKNFHL RPRDLISNIN VIVLELKGSE TTFMCEYADE TATIVEFLNR WITFCQSIISTLT 2 Human MKWVTFISLL FLFSSAYSRG VFRRDAHKSE VAHRFKDLGE serum ENFKALVLIA FAQYLQQCPF EDHVKLVNEV TEFAKTCVAD albumin ESAENCDKSL HTLFGDKLCT VATLRETYGE MADCCAKQEP ERNECFLQHK DDNPNLPRLV RPEVDVMCTA FHDNEETFLK KYLYEIARRH PYFYAPELLF FAKRYKAAFT ECCQAADKAA CLLPKLDELR DEGKASSAKQ GLKCASLQKF GERAFKAWAV ARLSQRFPKA EFAEVSKLVT DLTKVHTECC HGDLLECADD RADLAKYICE NQDSISSILK ECCEKPLLEK SHCIAEVEND EMPADLPSLA ADFVGSKDVC KNYAEAKDVF LGMFLYEYAR RHPDYSVVLL LRLAKTYETT LEKCCAAADP HECYAKVFDE FKPLVEEPQN LIKQNCELFE QLGEYKFQNA LLVRYTKKVP QVSTPILVEV SRNLGKVGSK CCKHPEAKRM PCAEDCLSVF LNQLCVLHEK TPVSDRYTKC CTESLVNGRPCFSALEVDETYVPKEFNAETFTFHADICTLSEKERQIK KQTALV ELVKHK PKATKEQLKAVMDDFAAFVEKCCKADDKET CFAEEGKKLVAASQAALGL 45 ACP12 (IL2 QVQLQESGGGLVQAGGSLRLSCAASGRIFSIDIMSWYRQAPGKQREL conjugate) VARITRGGTISYDDSVKGRFTISRDNAKNTVYLQMNSLKPEDTGVYY CNALYGTDYWGKGTQVTVSSggggsggggsggggsaptssstkktqlqlehllldlqmil nginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelk- g settfmceyadetativeflnrwitfcqsiistltSGGPGPAGMKGLPGSEVQLVESGGG LVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGR DTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLS VSSQGTLVTVSSggggsggggsggggsEVQLVESGGGLVQPGGSLRLSCAA SGFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRFTISR DNAKNSLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTTVTV SSGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQNVG TNVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLTISSL QPEDFATYYCQQYYTYPYTFGGGTKVEIKHHHHHH 46 ACP13 (IL2 QVQLQESGGGLVQAGGSLRLSCAASGRIFSIDIMSWYRQAPGKQREL conjugate) VARITRGGTISYDDSVKGRFTISRDNAKNTVYLQMNSLKPEDTGVYY CNALYGTDYWGKGTQVTVSSggggsggggsggggsEVQLVESGGGLVQP GGSLRLSCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPD TVRGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNWDALDY WGQGTTVTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVT ITCKASQNVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGS GTDFTLTISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKggggsggggs ggggsEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPG KGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPE DTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGSaptssstkkt qlqlehlllqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrpr dlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltHHHHHH 47 ACP14 (IL2 EVQLVESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPGKGLE conjugate) WVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAEDTAV YYCARDSNWDALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDIQM TQSPSSLSASVGDRVTITCKASQNVGTNVGWYQQKPGKAPKALIYS ASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPYTF GGGTKVEIKggggsggggsggggsggggsggggsggggsSGGPGPAGMKGLPGSa ptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnla- q sknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGMK GLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAP GKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRP EDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHH 48 ACP15 (IL2 EVQLVESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPGKGLE conjugate) WVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAEDTAV YYCARDSNWDALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDIQM TQSPSSLSASVGDRVTITCKASQNVGTNVGWYQQKPGKAPKALIYS ASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPYTF GGGTKVEIKggggsggggsggggsggggsggggsggggsEVQLVESGGGLVQPG NSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAE SVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGT LVTVSSggggsggggsggggsSGGPGPAGMKGLPGSaptssstkktqlqlehllldlqmil nginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelk- g settfmceyadetativeflnrwitfcqsiistltHHHHHH 49 ACP16 (IL2 aptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnla conjugate) qsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGMK GLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAP GKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRP EDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggsggggsggggsggg gsSGGPGPAGMKGLPGSEVQLVESGGGLVQPGGSLRLSCAASGFTFS SYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRFTISRDNAKN SLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTTVTVSSGGG GSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQNVGTNVG WYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQPED FATYYCQQYYTYPYTFGGGTKVEIKHHHHHH 50 ACP17 (IL2 QVQLQESGGGLVQAGGSLRLSCAASGRIFSIDIMSWYRQAPGKQREL conjugate) VARITRGGTISYDDSVKGRFTISRDNAKNTVYLQMNSLKPEDTGVYY CNALYGTDYWGKGTQVTVSSggggsggggsggggsaptssstkktqlqlehllldlqmil nginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelk- g settfmceyadetativeflnrwitfcqsiistltSGGPGPAGMKGLPGSEVQLVESGGG LVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGR DTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLS VSSQGTLVTVSSggggsggggsggggsggggsggggsggggsSGGPGPAGMKGLP GSEVQLVESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPGKG LEWVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAEDTA VYYCARDSNWDALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDIQ MTQSPSSLSASVGDRVTITCKASQNVGTNVGWYQQKPGKAPKALIY SASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPYT FGGGTKVEIKHHHHHH 51 ACP18 (IL2 QVQLQESGGGLVQAGGSLRLSCAASGRIFSIDIMSWYRQAPGKQREL conjugate) VARITRGGTISYDDSVKGRFTISRDNAKNTVYLQMNSLKPEDTGVYY CNALYGTDYWGKGTQVTVSSggggsggggsggggsaptssstkktqlqlehllldlqmil nginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelk- g settmceyadetativeflnrwitfcqsiistltSGGPGPAGMKGLPGSEVQLVESGGG LVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGR DTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLS VSSQGTLVTVSSggggsggggsggggsggggsggggsggggsEVQLVESGGGLVQ PGGSLRLSCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSP DTVRGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNWDALD YWGQGTTVTVSSsggpgpagmkglpgsDIQMTQSPSSLSASVGDRVTITCK ASQNVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTD FTLTISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKHHHHHH 52 ACP19 (IL2 aptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnla conjugate) qsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGMK GLPGSggggsggggsggggsggggsggggsggggsEVQLVESGGGLVQPGGSLRL SCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRF TISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTT VTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQ NVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLT ISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKggggsggggsggggsEVQ LVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVS SISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYC TIGGSLSVSSQGTLVTVSSggggsggggsggggsQVQLQESGGGLVQAGGS LRLSCAASGRIFSIDIMSWYRQAPGKQRELVARITRGGTISYDDSVKG RFTISRDNAKNTVYLQMNSLKPEDTGVYYCNALYGTDYWGKGTQV TVSSHHHHHH** 53 ACP20 (IL2 aptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnla conjugate) qsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGMK GLPGSEVQLVESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPG KGLEWVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAED TAVYYCARDSNWDALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDI QMTQSPSSLSASVGDRVTITCKASQNVGTNVGWYQQKPGKAPKALI YSASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPY TFGGGTKVEIKHHHHHH 54 ACP21 (IL2 aptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnla conjugate) qsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGMK GLPGSggggsggggsggggsggggsggggsggggsEVQLVESGGGLVQPGGSLRL SCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRF TISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTT VTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQ NVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLT ISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKHHHHHH 55 ACP22 (IL2 aptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnla conjugate) qsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGMK GLPGSggggsggggsggggsggggsggggsggggsEVQLVESGGGLVQPGGSLRL SCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRF TISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTT VTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQ NVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLT ISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKSGGPGPAGMKGLPG SEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggsQVQLQESGGGLVQ AGGSLRLSCAASGRIFSIDIMSWYRQAPGKQRELVARITRGGTISYDD SVKGRFTISRDNAKNTVYLQMNSLKPEDTGVYYCNALYGTDYWGK GTQVTVSSHHHHHH 56 ACP23 (IL2 QVQLQESGGGLAQAGGSLSLSCAASGFTVSNSVMAWYRQTPGKQR conjugate) EFVAIINSVGSTNYADSVKGRFTISRDNAKNTVYLQMNNLKPEDTAV YVCNRNFDRIYWGQGTQVTVSSSGGPGPAGMKGLPGSEVQLVESGG GLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSS YTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNW DALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASV GDRVTITCKASQNVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSR FSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKggg gsggggsggggsggggsggggsggggsSGGPGPAGMKGLPGSEVQLVESGGGL VQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRD TLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSV SSQGTLVTVSSSGGPGPAGMKGLPGSaptssstkktqlqlehllldlqmilnginnykn pkltrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmc- e yadetativeflnrwitfcqsiistltHHEIHRH 57 ACP24 (IL2 EVQLVESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPGKGLE conjugate) WVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAEDTAV YYCARDSNWDALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDIQM TQSPSSLSASVGDRVTITCKASQNVGTNVGWYQQKPGKAPKALIYS ASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPYTF GGGTKVEIKSGGPGPAGMKGLPGSaptssstkktqlqlehllldlqmilnginnyknpkl trmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceya- d etativeflnrwitfcqsiistltHHHHHH 58 ACP25 EVQLVESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPGKGLE (IL2 WVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAEDTAV conjugate) YYCARDSNWDALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDIQM TQSPSSLSASVGDRVTITCKASQNVGTNVGWYQQKPGKAPKALIYS ASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPYTF GGGTKVEIKggggsggggsggggsggggsggggsggggsSGGPGPAGMKGLPGSa ptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnla- q sknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltHHHHHH 59 ACP26 (IL2 QVQLQESGGGLVQAGGSLRLSCAASGRIFSIDIMSWYRQAPGKQREL conjugate) VARITRGGTISYDDSVKGRFTISRDNAKNTVYLQMNSLKPEDTGVYY CNALYGTDYWGKGTQVTVSSggggsggggsggggsaptssstkktqlqlehllldlqmil nginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelk- g settfmceyadetativeflnrwitfcqsiistltSGGPGPAGMKGLPGSEVQLVESGGG LVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGR DTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLS VSSQGTLVTVSSggggsggggsggggsggggsQVQLQQSGAELVRPGTSVKV SCKASGYAFTNYLIEWVKQRPGQGLEWIGVINPGSGGTNYNEKFKG KATLTADKSSSTAYMQLSSLTSDDSAVYFCARWRGDGYYAYFDVW GAGTTVTVSSggggsggggsggggsDIVLTQSPASLAVSLGQRATISCKASQ SVDYDGDSYMNWYQQKPGQPPKLLIYAASNLESGIPARFSGSGSGT DFTLNIHPVEEEDAATYYCQQSNEDPYTFGGGTKLEIKHHHHHHEPE A 60 ACP27 (IL2 QVQLQESGGGLVQAGGSLRLSCAASGRIFSIDIMSWYRQAPGKQREL conjugate) VARITRGGTISYDDSVKGRFTISRDNAKNTVYLQMNSLKPEDTGVYY CNALYGTDYWGKGTQVTVSSggggsggggsggggsaptssstkktqlqlehllldlqmil nginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelk- g settfmceyadetativeflnrwitfcqsiistltSGGPGPAGMKGLPGSEVQLVESGGG LVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGR DTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLS
VSSQGTLVTVSSggggsggggsggggsggggsDIVLTQSPASLAVSLGQRATIS CKASQSVDYDGDSYMNWYQQKPGQPPKLLIYAASNLESGIPARFSG SGSGTDFTLNIHPVEEEDAATYYCQQSNEDPYTFGGGTKLEIKggggsg gggsggggsQVQLQQSGAELVRPGTSVKVSCKASGYAFTNYLIEWVKQ RPGQGLEWIGVINPGSGGTNYNEKFKGKATLTADKSSSTAYMQLSSL TSDDSAVYFCARWRGDGYYAYFDVWGAGTTVTVSSHEIREIHHEPE A 61 ACP28 (IL2 aptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnla conjugate) qsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGMK GLPGSggggsggggsggggsggggsggggsQVQLQQSGAELVRPGTSVKVSCK ASGYAFTNYLIEWVKQRPGQGLEWIGVINPGSGGTNYNEKFKGKAT LTADKSSSTAYMQLSSLTSDDSAVYFCARWRGDGYYAYFDVWGAG TTVTVSSggggsggggsggggsDIVLTQSPASLAVSLGQRATISCKASQSVD YDGDSYMNWYQQKPGQPPKLLIYAASNLESGIPARFSGSGSGTDFTL NIHPVEEEDAATYYCQQSNEDPYTFGGGTKLEIKggggsggggsggggsEV QLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEW VSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVY YCTIGGSLSVSSQGTLVTVSSggggsggggsggggsQVQLQESGGGLVQAG GSLRLSCAASGRIFSIDIMSWYRQAPGKQRELVARITRGGTISYDDSV KGRFTISRDNAKNTVYLQMNSLKPEDTGVYYCNALYGTDYWGKGT QVTVSSHHEIHRHEPEA 62 ACP29 (IL2 aptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnla conjugate) qsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGMK GLPGSggggsggggsggggsggggsggggsDIVLTQSPASLAVSLGQRATISCKA SQSVDYDGDSYMNWYQQKPGQPPKLLIYAASNLESGIPARFSGSGSG0 TDFTLNIHPVEEEDAATYYCQQSNEDPYTFGGGTKLEIKggggsggggsg gggsQVQLQQSGAELVRPGTSVKVSCKASGYAFTNYLIEWVKQRPGQ GLEWIGVINPGSGGTNYNEKFKGKATLTADKSSSTAYMQLSSLTSDD SAVYFCARWRGDGYYAYFDVWGAGTTVTVSSggggsggggsggggsEVQ LVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVS SISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYC TIGGSLSVSSQGTLVTVSSggggsggggsggggsQVQLQESGGGLVQAGGS LRLSCAASGRIFSIDIMSWYRQAPGKQRELVARITRGGTISYDDSVKG RFTISRDNAKNTVYLQMNSLKPEDTGVYYCNALYGTDYWGKGTQV TVSSHHHHHHEPEA 63 IL2Ra 10 20 30 40 50 MDSYLLMWGL LTFIMVPGCQ AELCDDDPPE IPHATFKAMA YKEGTMLNCE 60 70 80 90 100 CKRGFRRIKS GSLYMLCTGN SSHSSWDNQC QCTSSATRNT TKQVTPQPEE 110 120 130 140 150 QKERKTTEMQ SPMQPVDQAS LPGHCREPPP WENEATERIY HFVVGQMVYY 160 170 180 190 200 QCVQGYRALH RGPAESVCKM THGKTRWTQP QLICTGEMET SQFPGEEKPQ 210 220 230 240 250 ASPEGRPESE TSCLVTTTDF QIQTEMAATM ETSIFTTEYQ VAVAGCVFLL 260 270 ISVLLLSGLT WQRRQRKSRR TI 64 IL2Rb 10 20 30 40 50 MAAPALSWRL PLLILLLPLA TSWASAAVNG TSQFTCFYNS RANISCVWSQ 60 70 80 90 100 DGALQDTSCQ VHAWPDRRRW NQTCELLPVS QASWACNLIL GAPDSQKLTT 110 120 130 140 150 VDIVTLRVLC REGVRWRVMA IQDFKPFENL RLMAPISLQV VHVETHRCNI 160 170 180 190 200 SWEISQASHY FERHLEFEAR TLSPGHTWEE APLLTLTQKQ EWICLETLTP 210 220 230 240 250 DTQYEFQVRV KPLQGEFTTW SPWSQPLAFR TKPAALGKDT IPWLGHLLVG 260 270 280 290 300 LSGAFGFIIL VYLLINCRNT GPWLKKVLKC NTPDPSKFFS QLSSEHGGDV 310 320 330 340 350 QKWLSSPFPS SSFSPGGLAP EISPLEVLER DKVTQLLLQQ DKVPEPASLS 360 370 380 390 400 SNHSLTSCFT NQGYFFFHLP DALEIEACQV YFTYDPYSEE DPDEGVAGAP 410 420 430 440 450 TGSSPQPLQP LSGEDDAYCT FPSRDDLLLF SPSLLGGPSP PSTAPGGSGA 460 470 480 490 500 GEERMPPSLQ ERVPRDWDPQ PLGPPTPGVP DLVDFQPPPE LVLREAGEEV 510 520 530 540 550 PDAGPREGVS FPWSRPPGQG EFRALNARLP LNTDAYLSLQ ELQGQDPTHL V 65 IL2Rg 10 20 30 40 50 MLKPSLPFTS LLFLQLPLLG VGLNTTILTP NGNEDTTADF FLTTMPTDSL 60 70 80 90 100 SVSTLPLPEV QCFVFNVEYM NCTWNSSSEP QPTNLTLHYW YKNSDNDKVQ 110 120 130 140 150 KCSHYLFSEE ITSGCQLQKK EIHLYQTFVV QLQDPRFPRR QATQMLKLQN 160 170 180 190 200 LVIPWAPENL TLHKLSESQL ELNWNNRFLN HCLEHLVQYR TDWDHSWTEQ 210 220 230 240 250 SVDYRHKFSL PSVDGQKRYT FRVRSRFNPL CGSAQHWSEW SHPIHWGSNT 260 270 280 290 300 SKENPFLFAL EAVVISVGSM GLIISLLCVY FWLERTMPRI PTLKNLEDLV 310 320 330 340 350 TEYHGNFSAW SGVSKGLAES LQPDYSERLC LVSEIPPKGG ALGEGPGASP 360 CNQHSPYWAP PCYTLKPET 66 ACP04 iwelkkdvyvveldwypdapgemvvltcdtpeedgitwtldqssevlgsgktltiqvkefgdagq- ytch (human kggevlshsllllhkkedgiwstdilkdqkepknktflrceaknysgrftcwwlttistdltfsvk- ssrgssd p40/murine pqgvtcgaatlsaervrgdnkeyeysvecqedsacpaaeeslpievmvdavhklkyenytssffirdiik p35 IL12 pdppknlqlkplknsrqvevsweypdtwstphsyfsltfcvqvqgkskrekkdrvftdktsatv- icrkna conjugate) sisvraqdryyssswsewasvpcsggggsggggsggggsrvipvsgparclsqsrnllkaddmvktar eklkhysctaedidheditrdqtstlktclplelhknesclatretssttrgsclppqktslmmtlclgsiy- edlk myqtefqainaalqnhnhqqiildkgmlvaidelmqslnhngetlrqkppvgeadpyrvkmklcillha fstrvvtinrvmgylssaHHHHHH 67 ACP05 iwelkkdvyvveldwypdapgemvvltcdtpeedgitwtldqssevlgsgktltiqvkefgdagq- ytch (human kggevlshsllllhkkedgiwstdilkdqkepknktflrceaknysgrftcwwlttistdltfsvk- ssrgssd p40/murine pqgvtcgaatlsaervrgdnkeyeysvecqedsacpaaeeslpievmvdavhklkyenytssffirdiik p35 IL12 pdppknlqlkplknsrqvevsweypdtwstphsyfsltfcvqvqgkskrekkdrvftdktsatv- icrkna conjugate) sisvraqdryyssswsewasvpcsggggsggggsggggsrnlpvatpdpgmfpclhhsqnllravsn mlqkarqtlefypctseeidheditkdktstveaclpleltknesclnsretsfitngsclasrktsfmmal- cls siyedlkmyqvefktmnakllmdpkrqifldqnmlavidelmqalnfnsetvpqkssleepdfyktkik lcillhafriravtidrvmsylnasHHHHHH 68 ACP06 QSVLTQPPSVSGAPGQRVTISCSGSRSNIGSNTVKWYQQLPGTAPKLL (human IYYNDQRPSGVPDRFSGSKSGTSASLAITGLQAEDEADYYCQSYDRY p40/murine THPALLFGTGTKVTVLggggsggggsggggsQVQLVESGGGVVQPGRSLR p35 IL12 LSCAASGFTFSSYGMHWVRQAPGKGLEWVAFIRYDGSNKYYADSV conjugate) KGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCKTHGSHDNWGQGT MVTVSSggggsggggsggggsggggsggggsggggsSGGPGPAGMKGLPGSiwelk kdvyvveldwypdapgemvvltcdtpeedgitwtldqssevlgsgktltiqvkefgdagqytchkgge vlshsllllhkkedgiwstdilkdqkepknktflrceaknysgrftcwwlttistdltfsvkssrgssdpqg- vt cgaatlsaervrgdnkeyeysvecqedsacpaaeeslpievmvdavhklkyenytssffirdiikpdppk nlqlkplknsrqvevsweypdtwstphsyfsltfcvqvqgkskrekkdrvftdktsatvicrknasisvra qdryyssswsewasvpcsggggsggggsggggsrvipvsgparclsqsrnllkttddmvktareklkhy sctaedidheditrdqtstlktclplelhknesclatretssttrgsclppqktslmmtlclgsiyedlkmy- qte fqainaalqnhnhqqiildkgmlvaidelmqslnhngetlrqkppvgeadpyrvkmklcillhafstrvv tinrvmgylssaSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCA ASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTI SRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHH FIRHHEPEA 69 ACP07 QSVLTQPPSVSGAPGQRVTISCSGSRSNIGSNTVKWYQQLPGTAPKLL (human IYYNDQRPSGVPDRFSGSKSGTSASLAITGLQAEDEADYYCQSYDRY p40/murine THPALLFGTGTKVTVLggggsggggsggggsQVQLVESGGGVVQPGRSLR p35 IL12 LSCAASGFTFSSYGMHWVRQAPGKGLEWVAFIRYDGSNKYYADSV conjugate) KGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCKTHGSHDNWGQGT MVTVSSggggsggggsggggsggggsggggsggggsSGGPGPAGMKGLPGSiwelk kdvyvveldwypdapgemvvltcdtpeedgitwtldqssevlgsgktltiqvkefgdagqytchkgge vlshsllllhkkedgiwstdilkdqkepknktflrceaknysgrftcwwlttistdltfsvkssrgssdpqg- vt cgaatlsaervrgdnkeyeysvecqedsacpaaeeslpievmvdavhklkyenytssffirdiikpdppk nlqlkplknsrqvevsweypdtwstphsyfsltfcvqvqgkskrekkdrvftdktsatvicrknasisvra qdryyssswsewasvpcsggggsggggsggggsrvipvsgparclsqsrnllkttddmvktareklkhy sctaedidheditrdqtstlktclplelhknesclatretssttrgsclppqktslmmtlclgsiyedlkmy- qte fqainaalqnhnhqqiildkgmlvaidelmqslnhngetlrqkppvgeadpyrvkmklcillhafstrvv tinrvmgylssaSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCA ASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTI SRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggg gsggggsggggsQVQLQESGGGLAQAGGSLSLSCAASGFTVSNSVMAWY RQTPGKQREFVAIINSVGSTNYADSVKGRFTISRDNAKNTVYLQMNN LKPEDTAVYVCNRNFDRIYWGQGTQVTVSSHHHHHHEPEA 70 ACP08 QVQLQESGGGLAQAGGSLSLSCAASGFTVSNSVMAWYRQTPGKQR (human EFVAIINSVGSTNYADSVKGRFTISRDNAKNTVYLQMNNLKPEDTAV p40/murine YVCNRNFDRIYWGQGTQVTVSSggggsggggsggggsQSVLTQPPSVSGAP p35 IL12 GQRVTISCSGSRSNIGSNTVKWYQQLPGTAPKLLIYYNDQRPSGVPD conjugate) RFSGSKSGTSASLAITGLQAEDEADYYCQSYDRYTHPALLFGTGTKV TVLggggsggggsggggsQVQLVESGGGVVQPGRSLRLSCAASGFTFSSYG MHWVRQAPGKGLEWVAFIRYDGSNKYYADSVKGRFTISRDNSKNT LYLQMNSLRAEDTAVYYCKTHGSHDNWGQGTMVTVSSggggsggggsg gggsggggsggggsggggsSGGPGPAGMKGLPGSiwelkkdvyvveldwypdapgem vvltcdtpeedgitwtldqssevlgsgktltiqvkefgdagqytchkggevlshsllllhkkedgiwstdil- k dqkepknktflrceaknysgrftcwwlttistdltfsvkssrgssdpqgvtcgaatlsaervrgdnkeyeys vecqedsacpaaeeslpievmvdavhklkyenytssffirdiikpdppknlqlkplknsrqvevsweyp dtwstphsyfsltfcvqvqgkskrekkdrvftdktsatvicrknasisvraqdryyssswsewasvpcsgg ggsggggsggggsrvipvsgparclsqsrnllkttddmvktareklkhysctaedidheditrdqtstlktc- l plelhknesclatretssttrgsclppqktslmmtlclgsiyedlkmyqtefqainaalqnhnhqqiildkg mlvaidelmqslnhngetlrqkppvgeadpyrvkmklcillhafstrvvtinrvmgylssaSGGPGP AGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWV RQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMN SLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHHEPEA 71 ACP09 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLE (human WVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAV p40/murine YYCTIGGSLSVSSQGTLVTVSSggggsggggsggggsQSVLTQPPSVSGAPG p35 IL12 QRVTISCSGSRSNIGSNTVKWYQQLPGTAPKLLIYYNDQRPSGVPDR conjugate) FSGSKSGTSASLAITGLQAEDEADYYCQSYDRYTHPALLFGTGTKVT VLggggsggggsggggsQVQLVESGGGVVQPGRSLRLSCAASGFTFSSYG MHWVRQAPGKGLEWVAFIRYDGSNKYYADSVKGRFTISRDNSKNT LYLQMNSLRAEDTAVYYCKTHGSHDNWGQGTMVTVSSggggsggggsg gggsggggsggggsggggsSGGPGPAGMKGLPGSiwelkkdvyvveldwypdapgem vvltcdtpeedgitwtldqssevlgsgktltiqvkefgdagqytchkggevlshsllllhkkedgiwstdil- k dqkepknktflrceaknysgrftcwwlttistdltfsvkssrgssdpqgvtcgaatlsaervrgdnkeyeys vecqedsacpaaeeslpievmvdavhklkyenytssffirdiikpdppknlqlkplknsrqvevsweyp dtwstphsyfsltfcvqvqgkskrekkdrvftdktsatvicrknasisvraqdryyssswsewasvpcsgg ggsggggsggggsrvipvsgparclsqsrnllkaddmvktareklkhysctaedidheditrdqtstlktcl plelhknesclatretssttrgsclppqktslmmtlclgsiyedlkmyqtefqainaalqnhnhqqiildkg mlvaidelmqslnhngeftrqkppvgeadpyrvkmklcillhafstrvvtinrvmgylssaHHHHH HEPEA 72 ACP10 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLE (human WVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAV p40/murine YYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGSiwelkkdvyvveld p35 IL12 wypdapgemvvltcdtpeedgitwtldqssevlgsgktltiqvkefgdagqytchkggevlshs- llllhk conjugate) kedgiwstdilkdqkepknktflrceaknysgrftcwwlttistdltfsvkssrgssdpqgvtcgaatlsaer vrgdnkeyeysvecqedsacpaaeeslpievmvdavhklkyenytssffirdiikpdppknlqlkplkn srqvevsweypdtwstphsyfsltfcvqvqgkskrekkdrvftdktsatvicrknasisvraqdryysssw sewasvpcsggggsggggsggggsrvipvsgparclsqsrnllkttddmvktareklkhysctaedidhe ditrdqtstlktclplelhknesclatretssttrgsclppqktslmmtlclgsiyedlkmyqtefqainaa- lqn hnhqqiildkgmlvaidelmqslnhngetlrqkppvgeadpyrvkmklcillhafstrvvtinrvmgyls saSGGPGPAGMKGLPGSggggsggggsggggsggggsggggsggggsQSVLTQPPS VSGAPGQRVTISCSGSRSNIGSNTVKWYQQLPGTAPKLLIYYNDQRP SGVPDRFSGSKSGTSASLAITGLQAEDEADYYCQSYDRYTHPALLFG TGTKVTVLggggsggggsggggsQVQLVESGGGVVQPGRSLRLSCAASGF TFSSYGMHWVRQAPGKGLEWVAFIRYDGSNKYYADSVKGRFTISRD NSKNTLYLQMNSLRAEDTAVYYCKTHGSHDNWGQGTMVTVSSHH HHHHEPEA 73 ACP11 iwelkkdvyvveldwypdapgemvvltcdtpeedgitwtldqssevlgsgktltiqvkefgdagq- ytch (human kggevlshsllllhkkedgiwstdilkdqkepknktflrceaknysgrftcwwlttistdltfsvk- ssrgssd p40/murine pqgvtcgaatlsaervrgdnkeyeysvecqedsacpaaeeslpievmvdavhklkyenytssffirdiik p35 IL12 pdppknlqlkplknsrqvevsweypdtwstphsyfsltfcvqvqgkskrekkdrvftdktsatv- icrkna conjugate) sisvraqdryyssswsewasvpcsggggsggggsggggsrvipvsgparclsqsrnllkttddmvktar eklkhysctaedidheditrdqtstlktclplelhknesclatretssttrgsclppqktslmmtlclgsiy- edlk myqtefqainaalqnhnhqqiildkgmlvaidelmqslnhngetlrqkppvgeadpyrvkinklcillha
fstrvvtinrvmgylssaSGGPGPAGMKGLPGSggggsggggsggggsggggsggggsgg ggsQSVLTQPPSVSGAPGQRVTISCSGSRSNIGSNTVKWYQQLPGTAP KLLIYYNDQRPSGVPDRFSGSKSGTSASLAITGLQAEDEADYYCQSY DRYTHPALLFGTGTKVTVLggggsggggsggggsQVQLVESGGGVVQPGR SLRLSCAASGFTFSSYGMHWVRQAPGKGLEWVAFIRYDGSNKYYA DSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCKTHGSHDNWG QGTMVTVSSggggsggggsggggsEVQLVESGGGLVQPGNSLRLSCAASG FTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRD NAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHH HHEPEA 74 IL12 p40 10 20 30 40 50 human MCHQQLVISW FSLVFLASPL VAIWELKKDV YVVELDWYPD APGEMVVLTC (Uniprot 60 70 80 90 100 Accession DTPEEDGITW TLDQSSEVLG SGKTLTIQVK EFGDAGQYTC HKGGEVLSHS No. P29460) 110 120 130 140 150 LLLLHKKEDG IWSTDILKDQ KEPKNKTFLR CEAKNYSGRF TCWWLTTSIT 160 170 180 190 200 DLTFSVKSSR GSSDPQGVTC GAATLSAERV RGDNKEYEYS VECQEDSACP 210 220 230 240 250 AAEESLPIEV MVDAVHKLKY ENYTSSFFIR DIIKPDPPKN LQLKPLKNSR 260 270 280 290 300 QVEVSWEYPD TWSTPHSYFS LTFCVQVQGK SKREKKDRVF TDKTSATVIC 310 320 RKNASISVRA QDRYYSSSWS EWASVPCS 75 IL1 p35 10 20 30 40 50 mouse MCQSRYLLFL ATLALLNHLS LARVIPVSGP ARCLSQSRNL LKTTDDMVKT (Uniprot 60 70 80 90 100 Accession AREKLKHYSC TAEDIDHEDI TRDQTSTLKT CLPLELHKNE SCLATRETSS No. P43431) 110 120 130 140 150 TTRGSCLPPQ KTSLMMTLCL GSIYEDLKMY QTEFQAINAA LQNHNHQQII 160 170 180 190 200 LDKGMLVAID ELMQSLNHNG ETLRQKPPVG EAEPYRVKMK LCILLHAFST 210 RVVTINRVMG YLSSA 76 IL12Rb-2 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 310 320 330 340 350 360 370 380 390 400 410 420 430 440 450 460 470 480 490 500 510 520 530 540 550 560 570 580 590 600 610 620 630 640 650 660 670 680 690 700 710 720 730 740 750 760 770 780 790 800 810 820 830 840 850 860 ML 77 IL12Rb-1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 310 320 330 340 350 360 370 380 390 400 410 420 430 440 450 460 470 480 490 500 510 520 530 540 550 560 570 580 590 600 610 620 630 640 650 660 KM 79 IL-12 p35 10 20 30 40 50 human MCHQQLVISW FSLVFLASPL VAIWELKKDV YVVELDWYPD APGEMVVLTC (Uniprot 60 70 80 90 100 accession DTPEEDGITW TLDQSSEVLG SGKTLTIQVK EFGDAGQYTC HKGGEVLSHS no. P29459) 110 120 130 140 150 LLLLHKKEDG IWSTDILKDQ KEPKNKTFLR CEAKNYSGRF TCWWLTTIST 160 170 180 190 200 DLTFSVKSSR GSSDPQGVTC GAATLSAERV RGDNKEYEYS VECQEDSACP 210 220 230 240 250 AAEESLPIEV MVDAVHKLKY ENYTSSFFIR KIIKPDPPKN LQLKPLKNSR 260 270 280 290 300 QVEVSWEYPD TWSTPHSYFS LTFCVQVQGK SKREKKDRVF TDKTSATVIC 310 320 RKNASISVRA QDRYYSSSWS EWASVPCS 80 IL-12 p40 10 20 30 40 50 mouse MCPQKLTISW FAIVLLVSPL MAMWELEKDV YVVEVDWTPD APGETVNLTC (Uniprot 60 70 80 90 100 accession DTPEEDDITW TSDQRHGVIG SGKTLTITVK EFLDAGQYTC HKGGETLSHS no. P43432) 110 120 130 140 150 HLLLHKKENG IWSTEILKNF KNKTFLKCEA PNYSGRFTCS WLVQRNMDLK 160 170 180 190 200 FNIKSSSSSP DSRAVTCGMA SLSAEKVTLD QRDYEKYSVS CQEDVTCPTA 210 220 230 240 250 EETLPIELAL EARQQNKYEN YSTSFFIRDI IKPDPPKNLQ MKPLKNSQVE 260 270 280 290 300 VSWEYPDSES TPHSYFSLKF FVRIQRKKEK MKETEECGNQ KGAFLVEKTS 310 320 330 TEVQCKGGNV CVQAQDRYYN SSCSKWACVP CRVRS 81 ACP01 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLE (mouse IFN.gamma. WVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAV conjugate) YYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGShgtviesleslnnyfn ssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskakk- d afmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGPGPAGMKGLPGSEVQ LVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVS SISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYC TIGGSLSVSSQGTLVTVSSHHHHHH 82 ACP02 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLE (mouse IFN.gamma. WVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAV conjugate) YYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGShgtviesleslnnyfn ssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskakk- d afmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGPGPAGMKGLPGShgtvi esleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshli- tt ffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGPGPAGMKG LPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPG KGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPE DTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHH 83 ACP03 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLE (mouse IFN.gamma. WVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAV conjugate) YYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGShgtviesleslnnyfn ssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskakk- d afmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcggggsggggsggggshgtvieslesln nyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsns- k akkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGPGPAGMKGLPGS EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLE WVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAV YYCTIGGSLSVSSQGTLVTVSSHHHHHH 84 Human IFN- 10 20 30 40 50 g (Uniprot MKYTSYILAF QLCIVLGSLG CYCQDPVVKE AENLKKYFNA GHSDVADNGT Accession 60 70 80 90 100 No. P01579) LFLGILKNWK EESDRKIMQA QIVSFYFKLF KNFKDDQSIG KSVETIKEHM 110 120 130 140 150 NVKFFNSNKK KRDDFEKLTN YSVTDLNVQR KAIHELIQVM AELSPAAKTG 160 KRKSQMLFR GRRASQ 85 Mouse IFN- 10 20 30 40 50 g (Uniprot MNATHCILAL QLFLMAVSGC YCHGTVIESL ESLNNYFNSS GIDVEEKSLF Accession 60 70 80 90 100 No. P01580) LDIWRNWQKD GDMKILQSQI ISFYLRLFEV LKDNQAISNN ISVIESHLIT 110 120 130 140 150 TFFSNSKAKK DAFMSIAKFE VNNPQVQRQA FNELIRVVHQ LLPESSLRKR KRSRC 86 ACP30 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF (mouse IFN.gamma. GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate) LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqai snnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGP GPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMS WVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYL QMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLP GShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnis vieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGPGPA GMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVR QAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNS LRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHH 87 ACP31 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLE (mouse WVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAV INFa1 YYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGScdlpqthnlrnkraltl conjugate) lvqmrrlsplsclkdrkdfgfpqekvdaqqikkaqaipvlseltqqilniftskdssaawnttlldsfcndlh qqlndlqgclmqqvgvqefpltqedallavrkyfhritvylrekkhspcawevvraevwralsssanvlg rlreekSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFT FSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNA KTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHHE PEA 88 ACP32 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLE (mouse WVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAV INFa1 YYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGScdlpqthnlrnkraltl conjugate) lvqmrrlsplsclkdrkdfgfpqekvdaqqikkaqaipvlseltqqilniftskdssaawnttlldsfcndlh qqlndlqgclmqqvgvqefpltqedallavrkyfhritvylrekkhspcawevvraevwralsssanS GGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHHEPEA 89 IFN.gamma.R1 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 310 320 330 340 350 360 370 380 390 400 410 420 430 440 450 460 470 480 90 IFN.gamma.R2 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150
160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 310 320 330 91 ACP51 QVQLQESGGGLVQAGGSLRLSCAASGRIFSIDIMSWYRQAPGKQREL Mouse IFG VARITRGGTISYDDSVKGRFTISRDNAKNTVYLQMNSLKPEDTGVYY conjugate CNALYGTDYWGKGTQVTVSSggggsggggsggggsEVQLVESGGGLVQP GNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLY AESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQ GTLVTVSSSGGPGPAGMKGLPGShgtviesleslnnyfnssgidveekslfldiwrnwq kdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqa- f nelirvvhqllpesslrkrkrsrcSGGPGPAGMKGLPGSEVQLVESGGGLVQPGN SLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAES VKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTL VTVSSHHHHHH 92 ACP52 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLE Mouse IFG WVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAV conjugate YYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGShgtviesleslnnyfn ssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskakk- d afmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGPGPAGMKGLPGSEVQ LVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVS SISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYC TIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGSEVQLVESGGGLVQ PGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLY AESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQ GTLVTVSSggggsggggsggggsQVQLQESGGGLVQAGGSLRLSCAASGRI FSIDIMSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKN TVYLQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSHHHHHH 93 ACP53 eahkseiahryndlgeqhfkglvliafsqylqkcsydehaklvqevtdfaktcvadesaancdks- lhtlfg Mouse IFG dklcaipnlrenygeladcctkqepernecflqhkddnpslppferpeaeamctsfkenpttf- mghylhe conjugate varrhpyfyapellyyaeqyneiltqccaeadkescltpkidgykekalvssvrqrmkcssmq- kfgeraf kawavarlsqtfpnadfaeitklatdltkvnkecchgdllecaddraelakymcenqatissklqtccdkpl lkkahclsevehdtmpadlpaiaadfvedqeycknyaeakdvflgtflyeysahpdysyslllrlakkye atlekccaeanppacygtvlaefqplveepknlvktncdlyeklgeygfqnailvrytqkapqvstptlve aarnlgrvgtkcctlpedqrlpcvedylsailnrvcllhektpysehvtkccsgslverrpcfsaltvdety- vp kefkaetftfhsdictlpekekqikkqtalaelvkhkpkataeqlktvmddfaqfldtcckaadkdtcfste gpnlvtrckdalaSGGPGPAGMKGLPGShgtviesleslnnyfnssgidveekslfldiwrnw qkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrq afnelirvvhqllpesslrkrkrsrcSGGPGPAGMKGLPGSeahkseiahryndlgeqhfkglvl iafsqylqkcsydehaklvqevtdfaktcvadesaancdkslhtlfgdklcaipnlrenygeladcctkqep ernecflqhkddnpslppferpeaeamctsfkenpttfmghylhevarrhpyfyapellyyaeqyneilt qccaeadkescltpkldgvkekalvssvrqrmkcssmqkfgerafkawavarlsqtfpnadfaeitklat dltkvnkecchgdllecaddraelakymcenqatissklqtccdkpllkkahclsevehdtmpadlpaia adfvedqevcknyaeakdvflgtflyeysrrhpdysvslllrlakkyeatlekccaeanppacygtvlaefq plveepknlvktncdlyeklgeygfqnailvrytqkapqvstptlyeaarnlgrvgtkcctlpedqrlpcve dylsailnrvcllhektpvsehvtkccsgslverrpcfsaltvdetyvpkefkaetftfhsdictlpekekq- ik kqtalaelvkhkpkataeqlktvmddfaqfldtcckaadkdtcfstegpnlvtrckdalaHHHHHH 94 ACP54 eahkseiahryndlgeqhfkglvliafsqylqkcsydehaklvqevtdfaktcvadesaancdks- lhtlfg Mouse IFG dklcaipnlrenygeladcctkqepernecflqhkddnpslppferpeaeamctsfkenpttf- mghylhe conjugate varrhpyfyapellyyaeqyneiltqccaeadkescltpkldgvkekalvssvrqrmkcssmq- kfgeraf kawavarlsqtfpnadfaeitklatdltkvnkecchgdllecaddraelakymcenqatissklqtccdkpl lkkahclsevehdtmpadlpaiaadfvedqevcknyaeakdvflgtflyeysrrhpdysvslllrlakkye atlekccaeanppacygtvlaefqplveepknlvktncdlyeklgeygfqnailvrytqkapqvstptlve aarnlgrvgtkcctlpedqrlpcvedylsailnrvcllhektpvsehvtkccsgslverrpcfsaltvdety- vp kefkaetftfhsdictlpekekqikkqtalaelvkhkpkataeqlktvmddfaqfldtcckaadkdtcfste gpnlvtrckdalaSGGPGPAGMKGLPGShgtviesleslnnyfnssgidveekslfldiwrnw qkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrq afnelirvvhqllpesslrkrkrsrcggggsggggsggggshgtviesleslnnyfnssgidveekslfldi- w rnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqv qrqafnelirvvhqllpesslrkrkrsrcSGGPGPAGMKGLPGSeahkseiahryndlgeqhfk glyliafsqylqkcsydehaklvqevtdfaktcvadesaancdkslhtlfgdklcaipnlrenygeladcct kqepernecflqhkddnpslppferpeaeamctsfkenpttfmghylhevarrhpyfyapellyyaeqy neiltqccaeadkescltpkldgykekalvssvrqrmkcssmqkfgerafkawavarlsqtfpnadfaeit klatdltkvnkecchgdllecaddraelakymcenqatissklqtccdkpllkkahclsevehdtmpadlp aiaadfvedqevcknyaeakdvflgtflyeysrrhpdysyslllrlakkyeatlekccaeanppacygtvla efqplveepknlvktncdlyeklgeygfqnailvrytqkapqvstptlveaarnlgrvgtkcctlpedqrlp cvedylsailnrvcllhektpvsehvtkccsgslverrpcfsaltvdetyvpkefkaetftfhsdictlpek- ek qikkqtalaelvkhkpkataeqlktvmddfaqfldtcckaadkdtcfstegpnlvtrckdalaHHHHH H 95 ACP50 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI Mouse IFG MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSggggsggggsgggg sEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGShgtviesleslnnyf nssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskak- k dafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcggggsggggsggggshgtvieslesl nnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsn- s kakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGPGPAGMKGLPG SEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSHHHHHH 96 ACP55 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IFG GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqai snnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGP GPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMS WVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYL QMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLP GShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnis vieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGPGPA GMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVR QAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNS LRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHH 97 ACP56 mdmrvpaqllgllllwlrgarcQVQLQESGGGLAQAGGSLSLSCAASGFTVSNS Mouse IFG VMAWYRQTPGKQREFVAIINSVGSTNYADSVKGRFTISRDNAKNTV conjugate YLQMNNLKPEDTAVYVCNRNFDRIYWGQGTQVTVSSggggsggggsggg gsEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGShgtviesleslnnyf nssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskak- k dafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGPGPAGMKGLPGSEV QLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEW VSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVY YCTIGGSLSVSSQGTLVTVSSHEIREIHHEPEA 98 ACP57 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IFG GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqai snnisvieshlittffsnskakkdainsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGP GPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMS WVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYL QMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggsQV QLQESGGGLAQAGGSLSLSCAASGFTVSNSVMAWYRQTPGKQREFV AIINSVGSTNYADSVKGRFTISRDNAKNTVYLQMNNLKPEDTAVYV CNRNFDRIYWGQGTQVTVSSHHHHHHEPEA 99 ACP58 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IFG GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqai snnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGP GPAGMKGLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrl fevlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrk- r krsrcSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFT FSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNA KTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggs ggggsQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDIMSWYRQAPGK QRELVARITRGGTISYDDSVKGRFTISRDNAKNTVYLQMNSLKPEDT GVYYCNALYGTDYWGKGTQVTVSSHHHHHHEPEA 100 ACP59 mdmrvpaqllgllllwlrgarcQVQLQESGGGLAQAGGSLSLSCAASGFTVSNS Mouse IFG VMAWYRQTPGKQREFVAIINSVGSTNYADSVKGRFTISRDNAKNTV conjugate YLQMNNLKPEDTAVYVCNRNFDRIYWGQGTQVTVSSggggsggggsggg gsEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGShgtviesleslnnyf nssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskak- k dafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGPGPAGMKGLPGShgt viesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisviesh- l ittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGPGPAGMK GLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAP GKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRP EDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHHEPEA 101 ACP60 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IFG GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqai snnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGP GPAGMKGLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrl fevlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrk- r krsrcSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFT FSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNA KTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggs ggggsQVQLQESGGGLAQAGGSLSLSCAASGFTVSNSVMAWYRQTPG KQREFVAIINSVGSTNYADSVKGRFTISRDNAKNTVYLQMNNLKPED TAVYVCNRNFDRIYWGQGTQVTVSSHHHHHHEPEA 102 ACP61 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IFG GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqai snnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGP GPAGMKGLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrl fevlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrk- r krsrcSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFT FSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNA KTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggs ggggsEVQLVESGGGLVQPGGSLRLSCAASGFTFSSYAMSWVRQAPG KGLEWVSAISGSGGSTYYADSVKGRFTISRDNSKNTLYLQMNSLRAE DTAVYYCARGVGAFRPYRKHEWGQGTLVTVSRggggsggggsggggsSSE LTQDPAVSVALGQTVRITCQGDSLRSYYASWYQQKPGQAPVLVIYG KNNRPSGIPDRFSGSSSGNTASLTTTGAQAEDEADYYCNSSPFEHNL VVFGGGTKLTVLHHHHHHEPEA 103 ACP63 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGGSLRLSCAASGFTFSSY Anti-FN AMSWVRQAPGKGLEWVSAISGSGGSTYYADSVKGRFTISRDNSKNT CGS-2 scFv LYLQMNSLRAEDTAVYYCARGVGAFRPYRKHEWGQGTLVTVSRgg ggsggggsggggsSSELTQDPAVSVALGQTVRITCQGDSLRSYYASWYQQ KPGQAPVLVIYGKNNRPSGIPDRFSGSSSGNTASLTTTGAQAEDEAD YYCNSSPFEHNLVVFGGGTKLTVLHHHHHHEPEA 104 ACP69 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IFG GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqai snnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcSGGP GPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMS WVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYL QMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLP GShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnis vieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcHHHHHH EPEA
105 ACP 70 mdmrvpaqllgllllwlrgarchgtviesleslnnyfnssgidveekslfldiwrnwqkdgdm- kilqsqii Mouse IFG sfylrlfevlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnel- irvvhqllpes conjugate slrkrkrsrcSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAAS GFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISR DNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGP GPAGMKGLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrl fevlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrk- r krsrcSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFT FSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNA KTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHHE PEA 106 ACP 71 mdmrvpaqllgllllwlrgarchgtviesleslnnyfnssgidveekslfldiwrnwqkdgdm- kilqsqii Mouse IFG sfylrlfevlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnel- irvvhqllpes conjugate slrkrkrsrcSGGPGPAGMKGLPGSEAHKSEIAHRYNDLGEQHFKGLVLIA FSQYLQKCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHTLFGD KLCAIPNLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPFERPE AEAMCTSFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQYNEIL TQCCAEADKESCLTPKLDGVKEKALVSSVRQRMKCSSMQKFGERAF KAWAVARLSQTFPNADFAEITKLATDLTKVNKECCHGDLLECADDR AELAKYMCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMPADLP AIAADFVEDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSVSLLLRL AKKYEATLEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKTNCDLY EKLGEYGFQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTKCCTLPE DQRLPCVEDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVERRPCFS ALTVDETYVPKEFKAETFTFHSDICTLPEKEKQIKKQTALAELVKHKP KATAEQLKTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVTRCKDA LASGGPGPAGMKGLPGShgtviesleslnnyfnssgidveeksifidiwrnwqkdgdmkil qsqiisfylrlfevlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvv- hq llpesslrkrkrsrcSGGPGPAGMKGLPGSEAHKSEIAHRYNDLGEQHFKGL VLIAFSQYLQKCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHT LFGDKLCAIPNLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPF ERPEAEAMCTSFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQY NEILTQCCAEADKESCLTPKLDGVKEKALVSSVRQRMKCSSMQKFG ERAFKAWAVARLSQTFPNADFAEITKLATDLTKVNKECCHGDLLEC ADDRAELAKYMCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMP ADLPAIAADFVEDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSVSL LLRLAKKYEATLEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKTN CDLYEKLGEYGFQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTKC CTLPEDQRLPCVEDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVER RPCFSALTVDETYVPKEFKAETFTFHSDICTLPEKEKQIKKQTALAEL VKHKPKATAEQLKTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVT RCKDALAHHHHHHEPEA 107 ACP72 mdmrvpaqllgllllwlrgarcEAHKSEIAHRYNDLGEQHFKGLVLIAFSQYLQ Mouse IFG KCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHTLFGDKLCAIP conjugate NLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPFERPEAEAMCT SFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQYNEILTQCCAEA DKESCLTPKLDGVKEKALVSSVRQRMKCSSMQKFGERAFKAWAVA RLSQTFPNADFAEITKLATDLTKVNKECCHGDLLECADDRAELAKY MCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMPADLPAIAADFV EDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSVSLLLRLAKKYEAT LEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKTNCDLYEKLGEYG FQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTKCCTLPEDQRLPCV EDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVERRPCFSALTVDETY VPKEFKAETFTFHSDICTLPEKEKQIKKQTALAELVKHKPKATAEQL KTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVTRCKDALASGGPGP AGMKGLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfev lkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkr- sr cSGGPGPAGMKGLPGSEAHKSEIAHRYNDLGEQHFKGLVLIAFSQYL QKCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHTLFGDKLCAI PNLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPFERPEAEAMC TSFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQYNEILTQCCAE ADKESCLTPKLDGVKEKALVSSVRQRMKCSSMQKFGERAFKAWAV ARLSQTFPNADFAEITKLATDLTKVNKECCHGDLLECADDRAELAK YMCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMPADLPAIAADF VEDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSVSLLLRLAKKYEA TLEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKTNCDLYEKLGEY GFQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTKCCTLPEDQRLPC VEDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVERRPCFSALTVDET YVPKEFKAETFTFHSDICTLPEKEKQIKKQTALAELVKHKPKATAEQ LKTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVTRCKDALASGGPG PAGMKGLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfe vlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrk- r srcHHHHHHEPEA 108 ACP 73 mdmrvpaqllgllllwlrgarcEAHKSEIAHRYNDLGEQHFKGLVLIAFSQYLQ Mouse IFG KCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHTLFGDKLCAIP conjugate NLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPFERPEAEAMCT SFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQYNEILTQCCAEA DKESCLTPKLDGVKEKALVSSVRQRMKCSSMQKFGERAFKAWAVA RLSQTFPNADFAEITKLATDLTKVNKECCHGDLLECADDRAELAKY MCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMPADLPAIAADFV EDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSVSLLLRLAKKYEAT LEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKTNCDLYEKLGEYG FQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTKCCTLPEDQRLPCV EDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVERRPCFSALTVDETY VPKEFKAETFTFHSDICTLPEKEKQIKKQTALAELVKHKPKATAEQL KTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVTRCKDALASGGPGP AGMKGLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfev lkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkr- sr cSGGPGPAGMKGLPGSEAHKSEIAHRYNDLGEQHFKGLVLIAFSQYL QKCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHTLFGDKLCAI PNLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPFERPEAEAMC TSFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQYNEILTQCCAE ADKESCLTPKLDGVKEKALVSSVRQRMKCSSMQKFGERAFKAWAV ARLSQTFPNADFAEITKLATDLTKVNKECCHGDLLECADDRAELAK YMCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMPADLPAIAADF VEDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSVSLLLRLAKKYEA TLEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKTNCDLYEKLGEY GFQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTKCCTLPEDQRLPC VEDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVERRPCFSALTVDET YVPKEFKAETFTFHSDICTLPEKEKQIKKQTALAELVKHKPKATAEQ LKTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVTRCKDALASGGPG PAGMKGLPGShgtviesleslnnyfnssgidveeksifidiwrnwqkdgdmkilqsqiisfylrlfe vlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrk- r srcSGGPGPAGMKGLPGSEAHKSEIAHRYNDLGEQHFKGLVLIAFSQY LQKCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHTLFGDKLC AIPNLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPFERPEAEA MCTSFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQYNEILTQC CAEADKESCLTPKLDGVKEKALVSSVRQRMKCSSMQKFGERAFKA WAVARLSQTFPNADFAEITKLATDLTKVNKECCHGDLLECADDRAE LAKYMCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMPADLPAI AADFVEDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSVSLLLRLAK KYEATLEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKTNCDLYEK LGEYGFQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTKCCTLPED QRLPCVEDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVERRPCFSAL TVDETYVPKEFKAETFTFHSDICTLPEKEKQIKKQTALAELVKHKPK ATAEQLKTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVTRCKDAL AHHHHHHEPEA 109 ACP74 mdmrvpaqllgllllwlrgarcEAHKSEIAHRYNDLGEQHFKGLVLIAFSQYLQ Mouse IFG KCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHTLFGDKLCAIP conjugate NLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPFERPEAEAMCT SFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQYNEILTQCCAEA DKESCLTPKLDGVKEKALVSSVRQRMKCSSMQKFGERAFKAWAVA RLSQTFPNADFAEITKLATDLTKVNKECCHGDLLECADDRAELAKY MCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMPADLPAIAADFV EDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSVSLLLRLAKKYEAT LEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKTNCDLYEKLGEYG FQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTKCCTLPEDQRLPCV EDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVERRPCFSALTVDETY VPKEFKAETFTFHSDICTLPEKEKQIKKQTALAELVKHKPKATAEQL KTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVTRCKDALASGGPGP AGMKGLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfev lkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkr- sr cSGGPGPAGMKGLPGSggggsEAHKSEIAHRYNDLGEQHFKGLVLIAF SQYLQKCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHTLFGD KLCAIPNLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPFERPE AEAMCTSFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQYNEIL TQCCAEADKESCLTPKLDGVKEKALVSSVRQRMKCSSMQKFGERAF KAWAVARLSQTFPNADFAEITKLATDLTKVNKECCHGDLLECADDR AELAKYMCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMPADLP AIAADFVEDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSVSLLLRL AKKYEATLEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKTNCDLY EKLGEYGFQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTKCCTLPE DQRLPCVEDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVERRPCFS ALTVDETYVPKEFKAETFTFHSDICTLPEKEKQIKKQTALAELVKHKP KATAEQLKTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVTRCKDA LAggggsSGGPGPAGMKGLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdg dmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafne- li rvvhqllpesslrkrkrsrcSGGPGPAGMKGLPGSEAHKSEIAHRYNDLGEQHF KGLVLIAFSQYLQKCSYDEHAKLVQEVTDFAKTCVADESAANCDKS LHTLFGDKLCAIPNLRENYGELADCCTKQEPERNECFLQHKDDNPSL PPFERPEAEAMCTSFKENPTTFMGHYLHEVARRHPYFYAPELLYYAE QYNEILTQCCAEADKESCLTPKLDGVKEKALVSSVRQRMKCSSMQK FGERAFKAWAVARLSQTFPNADFAEITKLATDLTKVNKECCHGDLL ECADDRAELAKYMCENQATISSKLQTCCDKPLLKKAHCLSEVEHDT MPADLPAIAADFVEDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSV SLLLRLAKKYEATLEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKT NCDLYEKLGEYGFQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTK CCTLPEDQRLPCVEDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVE RRPCFSALTVDETYVPKEFKAETFTFHSDICTLPEKEKQIKKQTALAE LVKHKPKATAEQLKTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVT RCKDALAHHHHHHEPEA 110 ACP75 mdmrvpaqllgllllwlrgarcEAHKSEIAHRYNDLGEQHFKGLVLIAFSQYLQ Mouse IFG KCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHTLFGDKLCAIP conjugate NLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPFERPEAEAMCT SFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQYNEILTQCCAEA DKESCLTPKLDGVKEKALVSSVRQRMKCSSMQKFGERAFKAWAVA RLSQTFPNADFAEITKLATDLTKVNKECCHGDLLECADDRAELAKY MCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMPADLPAIAADFV EDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSVSLLLRLAKKYEAT LEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKTNCDLYEKLGEYG FQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTKCCTLPEDQRLPCV EDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVERRPCFSALTVDETY VPKEFKAETFTFHSDICTLPEKEKQIKKQTALAELVKHKPKATAEQL KTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVTRCKDALASGGPGP AGMKGLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfev lkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkr- sr cSGGPGPAGMKGLPGSggggsggggsEAHKSEIAHRYNDLGEQHFKGLV LIAFSQYLQKCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHTL FGDKLCAIPNLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPFE RPEAEAMCTSFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQYN EILTQCCAEADKESCLTPKLDGVKEKALVSSVRQRMKCSSMQKFGE RAFKAWAVARLSQTFPNADFAEITKLATDLTKVNKECCHGDLLECA DDRAELAKYMCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMPA DLPAIAADFVEDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSVSLLL RLAKKYEATLEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKTNCD LYEKLGEYGFQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTKCCT LPEDQRLPCVEDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVERRPC FSALTVDETYVPKEFKAETFTFHSDICTLPEKEKQIKKQTALAELVKH KPKATAEQLKTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVTRCKD ALAggggsggggsSGGPGPAGMKGLPGShgtviesleslnnyfnssgidveekslfldiwr nwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskakkdafmsiakfevnnpqv qrqafnelirvvhqllpesslrkrkrsrcSGGPGPAGMKGLPGSEAHKSEIAHRYNDL GEQHFKGLVLIAFSQYLQKCSYDEHAKLVQEVTDFAKTCVADESAA NCDKSLHTLFGDKLCAIPNLRENYGELADCCTKQEPERNECFLQHKD DNPSLPPFERPEAEAMCTSFKENPTTFMGHYLHEVARRHPYFYAPEL LYYAEQYNEILTQCCAEADKESCLTPKLDGVKEKALVSSVRQRMKC SSMQKFGERAFKAWAVARLSQTFPNADFAEITKLATDLTKVNKECC HGDLLECADDRAELAKYMCENQATISSKLQTCCDKPLLKKAHCLSE VEHDTMPADLPAIAADFVEDQEVCKNYAEAKDVFLGTFLYEYSRRH PDYSVSLLLRLAKKYEATLEKCCAEANPPACYGTVLAEFQPLVEEPK NLVKTNCDLYEKLGEYGFQNAILVRYTQKAPQVSTPTLVEAARNLG RVGTKCCTLPEDQRLPCVEDYLSAILNRVCLLHEKTPVSEHVTKCCS GSLVERRPCFSALTVDETYVPKEFKAETFTFHSDICTLPEKEKQIKKQ TALAELVKHKPKATAEQLKTVMDDFAQFLDTCCKAADKDTCFSTE GPNLVTRCKDALAHHHHHHEPEA 111 ACP78 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IFG GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsgggg shgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisv- i eshlittffsnskakkdafmsiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcggggsggggsg gggsEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGK GLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPED TAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggshgtviesleslnnyfnss gidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskakkda- f msiakfevnnpqvqrqafnelirvvhqllpesslrkrkrsrcggggsggggsggggsEVQLVESG GGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGS GRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGS LSVSSQGTLVTVSSHHHHHHEPEA 112 ACP134 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IFG GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqai snnisvieshlittffsnskakkdafmsiakfevnnpqvqrqafnelftyvhqllpesslrkrkrsrcSGGP GPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMS WVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYL QMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLP GShgtviesleslnnyfnssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnis
vieshlittffsnskakkdafmsiakfevnnpqvqrqafneliryvhqllpesslrkrkrsrcSGGPGPA GMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVR QAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNS LRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggsQVQLQE SGGGLAQAGGSLSLSCAASGFTVSNSVMAWYRQTPGKQREFVAIINS VGSTNYADSVKGRFTISRDNAKNTVYLQMNNLKPEDTAVYVCNRN FDRIYWGQGTQVTVSSHHHHHHEPEA 113 ACP135 mdmrypaqllgllllwlrgarcQVQLQESGGGLAQAGGSLSLSCAASGFTVSNS Mouse IFG VMAWYRQTPGKQREFVAIINSVGSTNYADSVKGRFTISRDNAKNTV conjugate YLQMNNLKPEDTAVYVCNRNFDRIYWGQGTQVTVSSggggsggggsggg gsEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGShgtviesleslnnyf nssgidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskak- k dafmsiakfevnnpqvqrqafneliryvhqllpesslrkrkrsrcSGGPGPAGMKGLPGSEV QLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEW VSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVY YCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGShgtviesleslnnyfnss gidveekslfldiwrnwqkdgdmkilqsqiisfylrlfevlkdnqaisnnisvieshlittffsnskakkda- f msiakfevnnpqvqrqafneliryvhqllpesslrkrkrsrcSGGPGPAGMKGLPGSEVQL VESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSS ISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTI GGSLSVSSQGTLVTVSSHHEIHRHEPEA 114 ACP34 mdmrvpaqllgllllwlrgarcrvipvsgparclsqsrnllkttddmvktareklkhysctaed- idheditr Mouse IL-12 dqtstlktclplelhknesclatretssttrgsclppqktslmmtlclgsiyedlkmyqtefqainaalqnhn- h conjugate qqnldkgmlvaidelmqslnhngetlrqkppygeadpyrykmklcillhafstrvvtinrymg- ylssaS GGPGPAGMKGLPGSmwelekdvyvveydwtpdapgetvnltcdtpeedditwtsdqrhgv igsgktltitvkefldagqytchkggetlshshlllhkkengiwsteilknfknktflkceapnysgrftcs- wl vqrnmdlkfnikssssspdsravtcgmaslsaekvtldqrdyekysvscqedvtcptaeetlpielalearq qnkyenystsffirdiikpdppknlqmkplknsqveysweypdswstphsyfslkffyriqrkkekmk eteegcnqkgaflvektstevqckggnvcvqaqdryynsscskwacvpcrvrsHHHHHH 115 ACP35 mdmrvpaqllgllllwlrgarcrvipvsgparclsqsrnllkttddmvktareklkhysctaed- idheditr Mouse IL-12 dqtstlktclplelhknesclatretssttrgsclppqktslmmtlclgsiyedlkmyqtefqainaalqnhn- h conjugate qqnldkgmlvaidelmqslnhngetlrqkppygeadpyrvkmklcillhafstrvvtinrvmg- ylssag gggsggggsggggsSGGPGPAGMKGLPGSggggsggggsggggsmwelekdvyvvey dwtpdapgetvnltcdtpeedditwtsdqrhgvigsgktltitvkefldagqytchkggetlshshlllhkk engiwsteilknfknktflkceapnysgrftcswlvqrnmdlkfnikssssspdsravtcgmaslsaekvtl dqrdyekysvscqedvtcptaeetlpielalearqqnkyenystsffirdiikpdppknlqmkplknsqve vsweypdswstphsyfslkffvriqrkkekmketeegcnqkgaflvektstevqckggnvcvqaqdry ynsscskwacvpcrvrsHHHHHH 116 ACP36 mdmrypaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IL-12 GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGSmwelekdvyvvevdwtpdapgetvnltcdtpeedditwtsdqrhgvigsgktltitvkefld agqytchkggetlshshlllhkkengiwsteilknfknktflkceapnysgrftcswlvqrnmdlkfnikss ssspdsravtcgmaslsaekvtldqrdyekysyscqedvtcptaeetlpielalearqqnkyenystsffir- d iikpdppknlqmkplknsqvevsweypdswstphsyfslkffvriqrkkekmketeegcnqkgaflve ktstevqckggnvcvqaqdryynsscskwacvpcrvrsggggsggggsggggsrvipvsgparclsqs rnllkttddmvktareklkhysctaedidheditrdqtstlktclplelhknesclatretssttrgsclpp- qktsl mmtlclgsiyedlkmyqtefqainaalqnhnhqqiildkgmlvaidelmqslnhngefirqkppvgea dpyrvkmklcillhafstrvvtinrvmgylssaSGGPGPAGMKGLPGSEVQLVESGGG LVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGR DTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLS VSSQGTLVTVSSHHHHHH 117 ACP37 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI Mouse IL-12 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSggggsggggsgggg sEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGSmwelekdvyvve vdwtpdapgetvnltcdtpeedditwtsdqrhgvigsgktltitvkefldagqytchkggetlshshlllhk kengiwsteilknfknktflkceapnysgrftcswlvqrnmdlkfnikssssspdsravtcgmaslsaekv tldqrdyekysvscqedvtcptaeetlpielalearqqnkyenystsffirdiikpdppknlqmkplknsqv evsweypdswstphsyfslkffvriqrkkekmketeegcnqkgaflvektstevqckggnvcvqaqdr yynsscskwacvpcrvrsggggsggggsggggsrvipvsgparclsqsrnllkttddmvktareklkhy sctaedidheditrdqtstlktclplelhknesclatretssttrgsclppqktslmmtlclgsiyedlkmy- qte fqainaalqnhnhqqiildkgmlvaidelmqslnhngetlrqkppvgeadpyrvkmklcillhafstrvv tinrvmgylssaSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCA ASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTI SRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHH HHHH 118 ACP79 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI Mouse IL-12 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSggggsggggsgggg sEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGSmwelekdvyvve vdwtpdapgetvnltcdtpeedditwtsdqrhgvigsgktltitvkefldagqytchkggetlshshlllhk kengiwsteilknfknktflkceapnysgrftcswlvqrnmdlkfnikssssspdsravtcgmaslsaekv tldqrdyekysvscqedvtcptaeetlpielalearqqnkyenystsffirdiikpdppknlqmkplknsqv evsweypdswstphsyfslkffvriqrkkekmketeegcnqkgaflvektstevqckggnvcvqaqdr yynsscskwacvpcrvrsggggsggggsggggsrvipvsgparclsqsrnllkttddmvktareklkhy sctaedidheditrdqtstlktclplelhknesclatretssttrgsclppqktslmmtlclgsiyedlkmy- qte fqainaalqnhnhqqiildkgmlvaidelmqslnhngetlrqkppvgeadpyrvkmklcillhafstrvv tinrvmgylssaSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCA ASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTI SRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSShh HHHH 119 ACP 80 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IL-12 GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGSmwelekdvyvvevdwtpdapgetvnltcdtpeedditwtsdqrhgvigsgktltitvkefld agqytchkggetlshshlllhkkengiwsteilknfknktflkceapnysgrftcswlvqrnmdlkfnikss ssspdsravtcgmaslsaekvtldqrdyekysvscqedvtcptaeetlpielalearqqnkyenystsffir- d iikpdppknlqmkplknsqvevsweypdswstphsyfslkffvriqrkkekmketeegcnqkgaflve ktstevqckggnvcvqaqdryynsscskwacvpcrvrsggggsggggsggggsrvipvsgparclsqs rnllkttddmvktareklkhysctaedidheditrdqtstlktclplelhknesclatretssttrgsclpp- qktsl mmtlclgsiyedlkmyqtefqainaalqnhnhqqiildkgmlvaidelmqslnhngefirqkppvgea dpyrvkmklcillhafstrvvtinrvmgylssaSGGPGPAGMKGLPGSEVQLVESGGG LVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGR DTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLS VSSQGTLVTVSSggggsggggsggggsQVQLQESGGGLVQAGGSLRLSCA ASGRIFSIDIMSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISR DNAKNTVYLQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSH HHHHH 120 ACP91 mdmrvpaqllgllllwlrgarciwelkkdvyvveldwypdapgemvvltcdtpeedgitwtldq- ssevl Chimeric IL- gsgktltiqvkefgdagqytchkggevlshsllllhkkedgiwstdilkdqkepknktflrceaknysgrft 12 conjugate cwwlttistdltfsvkssrgssdpqgvtcgaatlsaervrgdnkeyeysvecqedsacpaaeeslpievmv davhklkyenytssffirdiikpdppknlqlkplknsrqvevsweypdtwstphsyfsltfcvqvqgksk rekkdrvftdktsatvicrknasisvraqdryyssswsewasvpcsggggsggggsggggsrvipvsgp arclsqsrnllkttddmvktareklkhysctaedidheditrdqtstlktclplelhknesclatretsstt- rgscl ppqktslmmtlclgsiyedlkmyqtefqainaalqnhnhqqiildkgmlvaidelmqslnhngefirqk ppvgeadpyrvkmklcillhafstrvvtinrvmgylssaggggsggggsggggsggggsggggsggg gsggggsggggsggggsQSVLTQPPSVSGAPGQRVTISCSGSRSNIGSNTVK WYQQLPGTAPKLLIYYNDQRPSGVPDRFSGSKSGTSASLAITGLQAE DEADYYCQSYDRYTHPALLFGTGTKVTVLggggsggggsggggsQVQLVE SGGGVVQPGRSLRLSCAASGFTFSSYGMHWVRQAPGKGLEWVAFIR YDGSNKYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCKT HGSHDNWGQGTMVTVSSggggsggggsggggsEVQLVESGGGLVQPGNS LRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESV KGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLV TVSSHHHHHHEPEA 121 ACP136 mdmrvpaqllgllllwlrgarciwelkkdvyvveldwypdapgemvvltcdtpeedgitwtld- qssevl Chimeric IL- gsgktltiqvkefgdagqytchkggevlshsllllhkkedgiwstdilkdqkepknktflrceaknysgrft 12 conjugate cwwlttistdltfsvkssrgssdpqgvtcgaatlsaervrgdnkeyeysvecqedsacpaaeeslpievmv davhklkyenytssffirdiikpdppknlqlkplknsrqvevsweypdtwstphsyfsltfcvqvqgksk rekkdrvftdktsatvicrknasisvraqdryyssswsewasvpcsggggsggggsggggsrvipvsgp arclsqsrnllkttddmvktareklkhysctaedidheditrdqtstlktclplelhknesclatretsstt- rgscl ppqktslmmtlclgsiyedlkmyqtefqainaalqnhnhqqiildkgmlvaidelmqslnhngefirqk ppvgeadpyrvkmklcillhafstrvvtinrvmgylssaSGGPGPAGMKGLPGSggggsgg ggsggggsggggsggggsggggsQSVLTQPPSVSGAPGQRVTISCSGSRSNIGS NTVKWYQQLPGTAPKLLIYYNDQRPSGVPDRFSGSKSGTSASLAITG LQAEDEADYYCQSYDRYTHPALLFGTGTKVTVLggggsggggsggggsQV QLVESGGGVVQPGRSLRLSCAASGFTFSSYGMHWVRQAPGKGLEW VAFIRYDGSNKYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAV YYCKTHGSHDNWGQGTMVTVSSHHHHHHEPEA 122 ACP138 mdmrvpaqllgllllwlrgarciwelkkdvyvveldwypdapgemvvltcdtpeedgitwtld- qssevl Chimeric IL- gsgktltiqvkefgdagqytchkggevlshsllllhkkedgiwstdilkdqkepknktflrceaknysgrft 12 conjugate cwwlttistdltfsvkssrgssdpqgvtcgaatlsaervrgdnkeyeysvecqedsacpaaeeslpievmv davhklkyenytssffirdiikpdppknlqlkplknsrqvevsweypdtwstphsyfsltfcvqvqgksk rekkdrvftdktsatvicrknasisvraqdryyssswsewasvpcsggggsggggsggggsrvipvsgp arclsqsrnllkttddmvktareklkhysctaedidheditrdqtstlktclplelhknesclatretsstt- rgscl ppqktslmmticlgsiyedlkmyqtefqainaalqnhnhqqiildkgmlvaidelmqslnhngefirqk ppvgeadpyrvkmklcillhafstrvvtinrvmgylssaSGGPGPAGMKGLPGSggggsgg ggsggggsggggsggggsggggsQSVLTQPPSVSGAPGQRVTISCSGSRSNIGS NTVKWYQQLPGTAPKLLIYYNDQRPSGVPDRFSGSKSGTSASLAITG LQAEDEADYYCQSYDRYTHPALLFGTGTKVTVLggggsggggsggggsQV QLVESGGGVVQPGRSLRLSCAASGFTFSSYGMHWVRQAPGKGLEW VAFIRYDGSNKYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAV YYCKTHGSHDNWGQGTMVTVSSggggsggggsggggsEVQLVESGGGLV QPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTL YAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSS QGTLVTVSSggggsggggsggggsQVQLQESGGGLAQAGGSLSLSCAASGF TVSNSVMAWYRQTPGKQREFVAIINSVGSTNYADSVKGRFTISRDNA KNTVYLQMNNLKPEDTAVYVCNRNFDRIYWGQGTQVTVSSHHHHH HEPEA 123 ACP139 mdmrvpaqllgllllwlrgarcQVQLQESGGGLAQAGGSLSLSCAASGFTVSNS Chimeric IL- VMAWYRQTPGKQREFVAIINSVGSTNYADSVKGRFTISRDNAKNTV 12 conjugate YLQMNNLKPEDTAVYVCNRNFDRIYWGQGTQVTVSSggggsggggsggg gsiwelkkdvyvveldwypdapgemvvltcdtpeedgitwtldqssevlgsgktltiqvkefgdagqyt chkggevlshsllllhkkedgiwstdilkdqkepknktfIrceaknysgrftcwwlttistdltfsvkssrg- ss dpqgvtcgaatlsaervrgdnkeyeysvecqedsacpaaeeslpievmvdavhklkyenytssffirdii kpdppknlqlkplknsrqvevsweypdtwstphsyfsltfcvqvqgkskrekkdrvftdktsatvicrkn asisvraqdryyssswsewasvpcsggggsggggsggggsrvipvsgparclsqsrnllkttddmvkta reklkhysctaedidheditrdqtstlktclplelhknesclatretssttrgsclppqktslmmticlgsi- yedl kmyqtefqainaalqnhnhqqiildkgmlvaidelmqslnhngetlrqkppvgeadpyrvkmklcillh afstrvvtinrvmgylssaSGGPGPAGMKGLPGSggggsggggsggggsggggsggggsg gggsQSVLTQPPSVSGAPGQRVTISCSGSRSNIGSNTVKWYQQLPGTAP KLLIYYNDQRPSGVPDRFSGSKSGTSASLAITGLQAEDEADYYCQSY DRYTHPALLFGTGTKVTVLggggsggggsggggsQVQLVESGGGVVQPGR SLRLSCAASGFTFSSYGMHWVRQAPGKGLEWVAFIRYDGSNKYYA DSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCKTHGSHDNWG QGTMVTVSSggggsggggsggggsEVQLVESGGGLVQPGNSLRLSCAASG FTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRD NAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHH HHEPEA 124 ACP140 mdmrvpaqllgllllwlrgarcQVQLQESGGGLAQAGGSLSLSCAASGFTVSNS Chimeric IL- VMAWYRQTPGKQREFVAIINSVGSTNYADSVKGRFTISRDNAKNTV 12 conjugate YLQMNNLKPEDTAVYVCNRNFDRIYWGQGTQVTVSSSGGPGPAGM KGLPGSiwelkkdvyvveldwypdapgemvvltcdtpeedgitwtldqssevlgsgktltiqvkef gdagqytchkggevlshsllllhkkedgiwstdilkdqkepknktfirceaknysgrftcwwlttistdltf- s vkssrgssdpqgvtcgaatlsaervrgdnkeyeysvecqedsacpaaeeslpievmvdavhklkyenyt ssffirdiikpdppknlqlkplknsrqvevsweypdtwstphsyfsltfcvqvqgkskrekkdrvftdkts atvicrknasisvraqdryyssswsewasvpcsggggsggggsggggsrvipvsgparclsqsrnllkttd dmvktareklkhysctaedidheditrdqtstlktclplelhknesclatretssttrgsclppqktslmmt- icl gsiyedlkmyqtefqainaalqnhnhqqiildkgmlvaidelmqslnhngetlrqkppvgeadpyrvk mklcillhafstrvvtinrvmgylssaSGGPGPAGMKGLPGSggggsggggsggggsggggs ggggsggggsQSVLTQPPSVSGAPGQRVTISCSGSRSNIGSNTVKWYQQL PGTAPKLLIYYNDQRPSGVPDRFSGSKSGTSASLAITGLQAEDEADY YCQSYDRYTHPALLFGTGTKVTVLggggsggggsggggsQVQLVESGGGV VQPGRSLRLSCAASGFTFSSYGMHWVRQAPGKGLEWVAFIRYDGSN KYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCKTHGSHD NWGQGTMVTVSSggggsggggsggggsEVQLVESGGGLVQPGNSLRLSCA ASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTI SRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHH HHHHEPEA
125 ACP38 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmltf- kfympkka IL-2 telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativefl- nrwitfc conjugate qsiistltSGGPGPAGMKGLPGSEVQLVESGGGLVQPGGSLRLSCAASGF TFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRFTISRDNA KNSLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTTVTVSSG GGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQNVGTN VGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQP EDFATYYCQQYYTYPYTFGGGTKVEIKggggsggggsggggsEVQLVESG GGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGS GRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGS LSVSSQGTLVTVSSggggsggggsggggsQVQLQESGGGLVQAGGSLRLSC AASGRIFSIDIMSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTIS RDNAKNTVYLQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSS HHHHHH 126 ACP39 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI IL-2 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSSGGPGPAGM KGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQA PGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLR PEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGSEVQL VESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPGKGLEWVAA IDSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCAR DSNWDALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDIQMTQSPSS LSASVGDRVTITCKASQNVGTNVGWYQQKPGKAPKALIYSASFRYS GVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPYTFGGGTKV EIKSGGPGPAGMKGLPGSaptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfy mpkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativef- ln rwitfcqsiistltHHHHHH** 127 ACP40 mdmrvpaqllgllllwlrgarcelcdddppeiphatfkamaykegtmlnceckrgfrriksgsl- ymlctg IL-2 nsshsswdnqcqctssatrnttkqvtpqpeeqkerkttemqspmqpvdqaslpghcrepppweneate conjugate riyhfvvgqmvyyqcvqgyralhrgpaesyckmthgktrwtqpqlictgemetsqfpgeekpq- aspe grpesetsclvtadfqiqtemaatmetsiftteyqggggsggggsggggsggggsggggsggggsSG GPGPAGMKGLPGSaptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkate khlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfc- qsi istltHHHHHH 128 ACP41 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmltf- kfympkka IL-2 telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativefl- nrwitfc conjugate qsiistltSGGPGPAGMKGLPGSggggsggggsggggsggggsggggsggggselcdddpp eiphatfkamaykegtmlnceckrgfrriksgslymlctgnsshsswdnqcqctssatrnttkqvtpqpe eqkerkttemqspmqpvdqaslpghcrepppweneateriyhfvvgqmvyyqcvqgyralhrgpae svckmthgktrwtqpqlictgemetsqfpgeekpqaspegrpesetsclvtadfqiqtemaatmetsiftt eyqHHHHHH 129 ACP42 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF IL-2 GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsgggg selcdddppeiphatfkamaykegtmlnceckrgfrriksgslymlctgnsshsswdnqcqctssatrntt kqvtpqpeeqkerkttemqspmqpvdqaslpghcrepppweneateriyhfvvgqmvyyqcvqgy ralhrgpaesyckmthgktrwtqpqlictgemetsqfpgeekpqaspegrpesetsclvtttdfqiqtema atmetsiftteyqggggsggggsggggsggggsggggsggggsSGGPGPAGMKGLPGSapt ssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleeylnlaqs- k nfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltHHHHHH 130 ACP43 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmltf- kfympkka IL-2 telkhlqcleeelkpleeylnlaqsknfhlrprdlisninvivlelkgsettfmceyadetatiyefl- nrwitfc conjugate qsiistltSGGPGPAGMKGLPGSggggsggggsggggsggggsggggsggggselcdddpp eiphatfkamaykegtmlnceckrgfrriksgslymlctgnsshsswdnqcqctssatrnttkqvtpqpe eqkerkttemqspmqpydqaslpghcrepppweneateriyhfvygqmyyyqcyqgyralhrgpae svckmthgktrwtqpqlictgemetsqfpgeekpqaspegrpesetsclvtttdfqiqtemaatmetsiftt eyqggggsggggsggggsEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFG MSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTL YLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHH 131 ACP44 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmltf- kfympkka IL-2 telkhlqcleeelkpleeylnlaqsknfhlrprdlisninvivlelkgsettfmceyadetatiyefl- nrwitfc conjugate qsiistltSGGPGPAGMKGLPGSggggsggggsggggsggggsggggsggggselcdddpp eiphatfkamaykegtmlnceckrgfrriksgslymlctgnsshsswdnqcqctssatrnttkqvtpqpe eqkerkttemqspmqpydqaslpghcrepppweneateriyhfvygqmyyyqcyqgyralhrgpae svckmthgktrwtqpqlictgemetsqfpgeekpqaspegrpesetsclytadfqiqtemaatmetsiftt eyqSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTF SKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNA KTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHH 132 ACP45 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF IL-2 GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGSEVQLVESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPG KGLEWVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAED TAVYYCARDSNWDALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDI QMTQSPSSLSASVGDRVTITCKASQNVGTNVGWYQQKPGKAPKALI YSASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPY TFGGGTKVEIKggggsggggsggggsggggsggggsggggsSGGPGPAGMKGLPG Saptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleeyln- l aqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltHHHHHH 133 ACP46 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmltf- kfympkka IL-2 telkhlqcleeelkpleeylnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativefl- nrwitfc conjugate qsiistltSGGPGPAGMKGLPGSggggsggggsggggsggggsggggsggggsEVQLV ESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPGKGLEWVAAI DSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCAR DSNWDALDYWGQGTTVTVSSsggpgpagmkglpgsDIQMTQSPSSLSASV GDRVTITCKASQNVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSR FSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKggg gsggggsggggsEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVR QAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNS LRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggsQVQLQE SGGGLVQAGGSLRLSCAASGRIFSIDIMSWYRQAPGKQRELVARITR GGTISYDDSVKGRFTISRDNAKNTVYLQMNSLKPEDTGVYYCNALY GTDYWGKGTQVTVSSHHHHHH 134 ACP47 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI IL-2 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conujgate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSggggsggggsgggg saptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleeyln- l aqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGM KGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQA PGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLR PEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggsggggsggggsgg ggsEVQLVESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPGKG LEWVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAEDTA VYYCARDSNWDALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDIQ MTQSPSSLSASVGDRVTITCKASQNVGTNVGWYQQKPGKAPKALIY SASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPYT FGGGTKVEIKHHHHHH 135 ACP48 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmltf- ldympkka IL-2 telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativefl- nrwitfc conjugate qsiistltSGGPGPAGMKGLPGSEVQLVESGGGLVQPGGSLRLSCAASGF TFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRFTISRDNA KNSLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTTVTVSSG GGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQNVGTN VGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQP EDFATYYCQQYYTYPYTFGGGTKVEIKggggsggggsggggsEVQLVESG GGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGS GRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGS LSVSSQGTLVTVSSHHHHHH 136 ACP49 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmltf- kfympkka IL-2 telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativefl- nrwitfc conjugate qsiistltSGGPGPAGMKGLPGSggggsggggsggggsggggsggggsggggsEVQLV ESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPGKGLEWVAAI DSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCAR DSNWDALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDIQMTQSPSS LSASVGDRVTITCKASQNVGTNVGWYQQKPGKAPKALIYSASFRYS GVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPYTFGGGTKV EIKggggsggggsggggsEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFG MSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTL YLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHEIREIREI 137 ACP92 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF IL-2 GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGSaptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpl eevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPG- P AGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWV RQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMN SLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHH 138 ACP93 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI IL-2 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSgsgsgsgsgsgsgsg sEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSgsgsgsgsgsgsgsgsQVQLQESGGGLVQ AGGSLRLSCAASGRIFSIDIMSWYRQAPGKQRELVARITRGGTISYDD SVKGRFTISRDNAKNTVYLQMNSLKPEDTGVYYCNALYGTDYWGK GTQVTVSSgsgsgsgsgsgsgsgsEVQLVESGGGLVQPGGSLRLSCAASGFT FSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRFTISRDNAK NSLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTTVTVSSGG GGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQNVGTNV GWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQPE DFATYYCQQYYTYPYTFGGGTKVEIKSGGPGPAGMKGLPGSaptssstk ktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhl- r prdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltHHHHHH 139 ACP94 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI IL-2 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSgsgsgsgsgsgsgsg sEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSgsgsgsgsgsgsgsgsEVQLVESGGGLVQ PGGSLRLSCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSP DTVRGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNWDALD YWGQGTTVTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRV TITCKASQNVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSG SGTDFTLTISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKSGGPGPA GMKGLPGSaptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcle eelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistl- tHH HHHH 140 ACP95 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI IL-2 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSgsgsgsgsgsgsgsg sEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGSaptssstkktqlqle hllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlis- n invivlelkgsettfmceyadetativeflnrwitfcqsiistltHHHHHH 141 ACP96 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI IL-2 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSSGGPGPAGM KGLPGSaptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelk pleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGG- PG PAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSW VRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQM NSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHH 142 ACP97 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI IL-2 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSggggsggggsgggg sEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGSaptssstkktqlqle hllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlis- n invivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGMKGLPGSEVQL VESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSS ISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTI GGSLSVSSQGTLVTVSSHHHHHH 143 ACP99 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI IL-2 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSggggsggggsgggg saptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevln- l aqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGM KGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQA PGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLR
PEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHH 144 ACP100 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI IL-2 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSggggsggggsgggg saptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevln- l aqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltHHHHHH 145 A CP101 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkka IL-2 telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativefl- nrwitfc conjugate qsiistltSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGF TFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDN AKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHH H 146 ACP102 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI IL-2 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSSGGPGPAGM KGLPGSaptssstkktqlqlehllldlqmilnginnyknpkhrmltfkfympkkatelkhlqcleeelk pleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGG- PG PAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSW VRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQM NSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggsggggsg gggsggggsEVQLVESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQA PGKGLEWVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRA EDTAVYYCARDSNWDALDYWGQGTTVTVSSGGGGSGGGGSGGGG SDIQMTQSPSSLSASVGDRVTITCKASQNVGTNVGWYQQKPGKAPK ALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYT YPYTFGGGTKVEIKHHHHHH 147 A CP103 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkka IL-2 telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativefl- nrwitfc conjugate qsiistltSGGPGPAGMKGLPGSggggsggggsggggsggggsggggsggggsEVQLV ESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPGKGLEWVAAI DSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCAR DSNWDALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDIQMTQSPSS LSASVGDRVTITCKASQNVGTNVGWYQQKPGKAPKALIYSASFRYS GVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPYTFGGGTKV EIKggggsggggsggggsEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFG MSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTL YLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggs QVQLQESGGGLAQAGGSLSLSCAASGFTVSNSVMAWYRQTPGKQR EFVAIINSVGSTNYADSVKGRFTISRDNAKNTVYLQMNNLKPEDTAV YVCNRNFDRIYWGQGTQVTVSSHHHHHH 148 ACP104 mdmrvpaqllgllllwlrgarcQVQLQESGGGLAQAGGSLSLSCAASGFTVSNS IL-2 VMAWYRQTPGKQREFVAIINSVGSTNYADSVKGRFTISRDNAKNTV conjugate YLQMNNLKPEDTAVYVCNRNFDRIYWGQGTQVTVSSaptssstkktqlqle hllldlqmilnginnyknpkhrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlisn invivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGMKGLPGSEVQL VESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSS ISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTI GGSLSVSSQGTLVTVSSggggsggggsggggsggggsggggsggggsEVQLVESG GGLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSS SYTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCARDSN WDALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSA SVGDRVTITCKASQNVGTNVGWYQQKPGKAPKALIYSASFRYSGVP SRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIK HHHHHH 149 ACP105 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGGSLRLSCAASGFTFSSYT IL-2 LAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLY conjugate LQMNSLRAEDTAVYYCARDSNWDALDYWGQGTTVTVSSGGGGSG GGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQNVGTNVGWYQ QKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATY YCQQYYTYPYTFGGGTKVEIKggggsggggsggggsggggsggggsggggsSGGP GPAGMKGLPGSaptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkh lqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqs- iistl tSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSK FGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKT TLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggg gsQVQLQESGGGLAQAGGSLSLSCAASGFTVSNSVMAWYRQTPGKQ REFVAIINSVGSTNYADSVKGRFTISRDNAKNTVYLQMNNLKPEDTA VYVCNRNFDRIYWGQGTQVTVSSHHHHHH 150 ACP106 mdmrvpaqllgllllwlrgarcQVQLQESGGGLAQAGGSLSLSCAASGFTVSNS IL-2 VMAWYRQTPGKQREFVAIINSVGSTNYADSVKGRFTISRDNAKNTV conjugate YLQMNNLKPEDTAVYVCNRNFDRIYWGQGTQVTVSSggggsggggsggg gsEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGSEVQLVESGG GLVQPGGSLRLSCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSS YTYSPDTVRGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNW DALDYWGQGTTVTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASV GDRVTITCKASQNVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSR FSGSGSGTDFTLTISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKggg gsggggsggggsggggsggggsggggsSGGPGPAGMKGLPGSaptssstkktqlqlehllld lqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlisninvi vlelkgsettfmceyadetativeflnrwitfcqsiistltHHHHHH 151 ACP107 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGGSLRLSCAASGFTFSSYT IL-2 LAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLY conjugate LQMNSLRAEDTAVYYCARDSNWDALDYWGQGTTVTVSSGGGGSG GGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQNVGTNVGWYQ QKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATY YCQQYYTYPYTFGGGTKVEIKggggsggggsggggsggggsggggsggggsEVQ LVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVS SISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYC TIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGSaptssstkktqlqlehllldlq milnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivl- e lkgsettfmceyadetativeflnrwitfcqsiistltggggsggggsggggsQVQLQESGGGLA QAGGSLSLSCAASGFTVSNSVMAWYRQTPGKQREFVAIINSVGSTNY ADSVKGRFTISRDNAKNTVYLQMNNLKPEDTAVYVCNRNFDRIYW GQGTQVTVSSHHHHHH 152 ACP108 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI IL-2 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSggggsggggsgggg saptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevln- l aqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGM KGLPGSrgetgpaaPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFG MSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTL YLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggs ggggsggggsggggsEVQLVESGGGLVQPGGSLRLSCAASGFTFSSYTLAW VRQAPGKGLEWVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLYLQM NSLRAEDTAVYYCARDSNWDALDYWGQGTTVTVSSGGGGSGGGG SGGGGSDIQMTQSPSSLSASVGDRVTITCKASQNVGTNVGWYQQKP GKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQ QYYTYPYTFGGGTKVEIKHEIHHHH 153 ACP117 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGGSLRLSCAASGFTFSSY Anti-FN AMSWVRQAPGKGLEWVSAISGSGGSTYYADSVKGRFTISRDNSKNT CGS-2 scFv LYLQMNSLRAEDTAVYYCARGVGAFRPYRKHEWGQGTLVTVSRgg ggsggggsggggsSSELTQDPAVSVALGQTVRITCQGDSLRSYYASWYQQ KPGQAPVLVIYGKNNRPSGIPDRFSGSSSGNTASLTTTGAQAEDEAD YYCNSSPFEHNLVVFGGGTKLTVLHHHHHHEPEA 154 ACP118 mdmrvpaqllgllllwlrgarcQVQLQQSGAELVRPGTSVKVSCKASGYAFTN NARA1 YLIEWVKQRPGQGLEWIGVINPGSGGTNYNEKFKGKATLTADKSSST Vh/Vl non- AYMQLSSLTSDDSAVYFCARWRGDGYYAYFDVWGAGTTVTVSSgg cleavable ggsggggsggggsDIVLTQSPASLAVSLGQRATISCKASQSVDYDGDSYM NWYQQKPGQPPKLLIYAASNLESGIPARFSGSGSGTDFTLNIHPVEEE DAATYYCQQSNEDPYTFGGGTKLEIKHHHHHHEPEA 155 ACP119 mdmrvpaqllgllllwlrgarcQVQLQQSGAELVRPGTSVKVSCKASGYAFTN NARA1 YLIEWVKQRPGQGLEWIGVINPGSGGTNYNEKFKGKATLTADKSSST Vh/Vl AYMQLSSLTSDDSAVYFCARWRGDGYYAYFDVWGAGTTVTVSSSG cleavable GPGPAGMKGLPGSDIVLTQSPASLAVSLGQRATISCKASQSVDYDGD SYMNWYQQKPGQPPKLLIYAASNLESGIPARFSGSGSGTDFTLNIHPV EEEDAATYYCQQSNEDPYTFGGGTKLEIKHHHHHHEPEA 156 ACP120 mdmrvpaqllgllllwlrgarcDIVLTQSPASLAVSLGQRATISCKASQSVDYDG NARA1 DSYMNWYQQKPGQPPKLLIYAASNLESGIPARFSGSGSGTDFTLNIHP Vl/Vh non- VEEEDAATYYCQQSNEDPYTFGGGTKLEIKggggsggggsggggsQVQLQ cleavable QSGAELVRPGTSVKVSCKASGYAFTNYLIEWVKQRPGQGLEWIGVI NPGSGGTNYNEKFKGKATLTADKSSSTAYMQLSSLTSDDSAVYFCA RWRGDGYYAYFDVWGAGTTVTVSSHHHHHHEPEA 157 ACP121 mdmrvpaqllgllllwlrgarcDIVLTQSPASLAVSLGQRATISCKASQSVDYDG NARA1 DSYMNWYQQKPGQPPKLLIYAASNLESGIPARFSGSGSGTDFTLNIHP Vl/Vh VEEEDAATYYCQQSNEDPYTFGGGTKLEIKSGGPGPAGMKGLPGSQ cleavable VQLQQSGAELVRPGTSVKVSCKASGYAFTNYLIEWVKQRPGQGLE WIGVINPGSGGTNYNEKFKGKATLTADKSSSTAYMQLSSLTSDDSAV YFCARWRGDGYYAYFDVWGAGTTVTVSSHHHHHHEPEA 158 ACP124 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmlt- fkfympkka IL-2 telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativefl- nrwitfc conjugate qsiistltggggsggggsggggsEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHHHEPEA 159 ACP132 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkttrmlt- fkfympkka IL-2 telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativefl- nrwitfc conjugate qsiistltggggsggggsggggsdahksevahrfkdlgeenfkalvliafaqylqqcpfedhv- klvnevte faktcvadesaencdkslhtlfgdklctvatlretygemadccakqepernecflqhkddnpnlprlvrpe vdvmctafhdneetflkkylyeiarrhpyfyapellffakrykaafteccqaadkaacllpkldelrdegka ssakqrlkcaslqkfgerafkawavarlsqrfpkaefaevsklvtdltkvhtecchgdllecaddradlaky- i cenqdsissklkeccekpllekshciaevendempadlpslaadfveskdvcknyaeakdvflgmflye yarrhpdysvvlllrlaktyettlekccaaadphecyakvfdefkplveepqnlikqncelfeqlgeykfqn allvrytkkvpqvstptivevsrnlgkvgskcckhpeakrmpcaedylsvvinqlcvlhektpvsdrytk ccteslvnapcfsalevdetyvpkefnaetftfhadictlsekerqikkqtalvelvkhkpkatkeqlkavm ddfaafvekcckaddketcfaeegkklvaasqaalglHHHHHHEPEA 160 ACP141 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmlt- fkfympkka IL-2 telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativefl- nrwitfc conjugate qsiistltggggsggggsggggsdahksevahrfkdlgeenfkalvliafaqylqqcpfedhv- klvnevte faktcvadesaencdkslhtlfgdklctvatlretygemadccakqepernecflqhkddnpnlprlvrpe vdvmctafhdneetflkkylyeiarrhpyfyapellffakrykaafteccqaadkaacllpkldelrdegka ssakqrlkcaslqkfgerafkawavarlsqrfpkaefaevsklvtdltkvhtecchgdllecaddradlaky- i cenqdsissklkeccekpllekshciaevendempadlpslaadfveskdvcknyaeakdvflgmflye yarrhpdysvvlllrlaktyettlekccaaadphecyakvfdefkplveepqnlikqncelfeqlgeykfqn allvrytkkvpqvstptivevsrnlgkvgskcckhpeakrmpcaedylsvvinqlcvlhektpvsdrytk ccteslvnapcfsalevdetyvpkefnaetftfhadictlsekerqikkqtalvelvkhkpkatkeqlkavm ddfaafvekcckaddketcfaeegkklvaasqaalglHHHHHHEPEA 161 ACP142 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmlt- fkfympkka IL-2 telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativefl- nrwitfc conjugate qsiistltSGGPGPAGMKGLPGSdahksevahrfkdlgeenfkalvliafaqylqqcpfedhv- kl vnevtefaktcvadesaencdkslhtlfgdklctvaftretygemadccakqepernecflqhkddnpnlp rlvrpevdvmctafhdneetflkkylyeiarrhpyfyapellffakrykaafteccqaadkaacllpkldel- r degkassakqrlkcaslqkfgerafkawavarlsqrfpkaefaevsklvtdltkvhtecchgdllecaddra dlakyicenqdsissklkeccekpllekshciaevendempadlpslaadfveskdvcknyaeakdvflg mflyeyarrhpdysvvlllrlaktyettlekccaaadphecyakvfdefkplveepqnlikqncelfeqlge ykfqnallvrytkkvpqvstptivevsrnlgkvgskcckhpeakrmpcaedylsvvinqlcvlhektpvs drvtkccteslvnapcfsalevdetyvpkefnaetftfhadictlsekerqikkqtalvelvkhkpkatkeq- l kavmddfaafvekcckaddketcfaeegkklvaasqaalglHHHHHHEPEA 162 ACP144 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmlt- fkfympkka IL-2 telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativefl- nrwitfc conjugate qsiistltSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGF TFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDN AKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggg gsggggsggggsggggsggggsSGGPGPAGMKGLPGSEVQLVESGGGLVQPG GSLRLSCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDT VRGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNWDALDYW GQGTTVTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTIT CKASQNVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSG TDFTLTISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKggggsggggsgg ggsQVQLQESGGGLAQAGGSLSLSCAASGFTVSNSVMAWYRQTPGK QREFVAIINSVGSTNYADSVKGRFTISRDNAKNTVYLQMNNLKPEDT AVYVCNRNFDRIYWGQGTQVTVSSHHEIREIHEPEA 163 ACP145 mdmrvpaqllgllllwlrgarcQVQLQESGGGLAQAGGSLSLSCAASGFTVSNS IL-2 VMAWYRQTPGKQREFVAIINSVGSTNYADSVKGRFTISRDNAKNTV conjugate YLQMNNLKPEDTAVYVCNRNFDRIYWGQGTQVTVSSggggsggggsggg
gsaptssstkktqlqlehllldlqmilnginnyknpkhrmltfkfympkkatelkhlqcleeelkpleevln laqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGM KGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQA PGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLR PEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggsggggsggggsgg ggsSGGPGPAGMKGLPGSEVQLVESGGGLVQPGGSLRLSCAASGFTFS SYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRFTISRDNAKN SLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTTVTVSSGGG GSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQNVGTNVG WYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQPED FATYYCQQYYTYPYTFGGGTKVEIKHHHHHHEPEA 164 ACP146 mdmrvpaqllgllllwlrgarcQVQLQESGGGLAQAGGSLSLSCAASGFTVSNS IL-2 VMAWYRQTPGKQREFVAIINSVGSTNYADSVKGRFTISRDNAKNTV conjugate YLQMNNLKPEDTAVYVCNRNFDRIYWGQGTQVTVSSSGGPGPAGM KGLPGSaptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelk pleevinlaqsknfhlrprdlisninvivlelkgsettfinceyadetativeflnrwitfcqsiistltSG- GPG PAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSW VRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQM NSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggsggggsg gggsggggsSGGPGPAGMKGLPGSEVQLVESGGGLVQPGGSLRLSCAAS GFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRFTISRD NAKNSLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTTVTVS SGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQNVGT NVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQ PEDFATYYCQQYYTYPYTFGGGTKVEIKHHHHHHEPEA 165 ACP133 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmlt- fkfympkka IL-2-6xHis telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwit- fc qsnstltHHHHHH 166 ACP147 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmlt- fkfympkka IL-2 telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativefl- nrwitfc conjugate qsiistltSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGF TFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDN AKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggg gsggggsggggsggggsggggsSGGPGPAGMKGLPGSEVQLVESGGGLVQPG GSLRLSCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDT VRGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNWDALDYW GQGTTVTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTIT CKASQNVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSG TDFTLTISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKggggsggggsgg ggsQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDIMSWYRQAPGKQ RELVARITRGGTISYDDSVKGRFTISRDNAKNTVYLQMNSLKPEDTG VYYCNALYGTDYWGKGTQVTVSSHHHHHHEPEA 167 ACP148 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI IL-2 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSggggsggggsgggg saptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelkpleevln- l aqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGGPGPAGM KGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQA PGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLR PEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggsggggsggggsgg ggsSGGPGPAGMKGLPGSEVQLVESGGGLVQPGGSLRLSCAASGFTFS SYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRFTISRDNAKN SLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTTVTVSSGGG GSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQNVGTNVG WYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQPED FATYYCQQYYTYPYTFGGGTKVEIKHHHHHHEPEA 168 ACP149 mdmrvpaqllgllllwlrgarcQVQLQESGGGLVQAGGSLRLSCAASGRIFSIDI IL-2 MSWYRQAPGKQRELVARITRGGTISYDDSVKGRFTISRDNAKNTVY conjugate LQMNSLKPEDTGVYYCNALYGTDYWGKGTQVTVSSSGGPGPAGM KGLPGSaptssstkktqlqlehllldlqmilnginnyknpkltrmltfkfympkkatelkhlqcleeelk pleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativeflnrwitfcqsiistltSGG- PG PAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSW VRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQM NSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsggggsggggsg gggsggggsSGGPGPAGMKGLPGSEVQLVESGGGLVQPGGSLRLSCAAS GFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRFTISRD NAKNSLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTTVTVS SGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQNVGT NVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQ PEDFATYYCQQYYTYPYTFGGGTKVEIKHHHHHHEPEA 169 ACP33 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IFNa- GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGScdlpqthnlrnkraltllvqmalsplsclkdrkdfgfpqekvdaqqikkagaipvlseltqqiln iftskdssaawnttldsfcndlhqqlndlqgclmqqvgvqefpltqedallavrkyfhritvylrekkhsp cawevvraevwralsssanvSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNS LRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESV KGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLV TVSSHHHHHHEPEA 170 A CP131 mdmrvpaqllgllllwlrgarccdlpqthnlrnkraltllvqmalsplsclkdrkdfgfpqekvdaqqikk Mouse IFNa aqaipvlseltqqflniftskdssaawnttlldsfcndlhqqlndlqgclmqqvgvqefpltqedallavrky fhritvylrekkhspcawevvraevwralsssanvlgrlreekHHHHHHEPEA 171 ACP125 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IFNa- GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGScdlpqthnlrnkraltllvqmrrlsplsclkdrkdfgfpqekvdaqqikkagaipvlseltqqiln iftskdssaawnttlldsfcndlhqqlndlqgclmqqvgvqefpltqedallavrkyfhritvylrekkhsp cawevvraevwralsssanvlgrlreekHHHHHHEPEA 172 ACP126 mdmrvpaqllgllllwlrgarccdlpqthnlrnkraltllvqmrrlsplsclkdrkdfgfpqe- kvdaqqikk Mouse IFNa- aqaipvlseltqqflniftskdssaawnttlldsfcndlhqqlndlqgclmqqvgvqefpltqedallavrky conjugate fhritvylrekkhspcawevvraevwralsssanvlgrlreekSGGPGPAGMKGLPGSEVQ LVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVS SISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYC TIGGSLSVSSQGTLVTVSSHHHHHHEPEA 173 ACP127 mdmrvpaqllgllllwlrgarcEAHKSEIAHRYNDLGEQHFKGLVLIAFSQYLQ Mouse IFNa- KCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHTLFGDKLCAIP conjugate NLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPFERPEAEAMCT SFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQYNEILTQCCAEA DKESCLTPKLDGVKEKALVSSVRQRMKCSSMQKFGERAFKAWAVA RLSQTFPNADFAEITKLATDLTKVNKECCHGDLLECADDRAELAKY MCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMPADLPAIAADFV EDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSVSLLLRLAKKYEAT LEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKTNCDLYEKLGEYG FQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTKCCTLPEDQRLPCV EDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVERRPCFSALTVDETY VPKEFKAETFTFHSDICTLPEKEKQIKKQTALAELVKHKPKATAEQL KTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVTRCKDALASGGPGP AGMKGLPGScdlpqthnlrnkraltllvqmalsplsclkdrkdfgfpqekvdaqqikkaqaipvls eltqqilniftskdssaawnttlldsfcndlhqqlndlqgclmqqvgvqefpltqedallavrkyfhrityy- lr ekkhspcawevvraevwralsssanvlgrlreekSGGPGPAGMKGLPGSEAHKSEIAH RYNDLGEQHFKGLVLIAFSQYLQKCSYDEHAKLVQEVTDFAKTCVA DESAANCDKSLHTLFGDKLCAIPNLRENYGELADCCTKQEPERNECF LQHKDDNPSLPPFERPEAEAMCTSFKENPTTFMGHYLHEVARRHPYF YAPELLYYAEQYNEILTQCCAEADKESCLTPKLDGVKEKALVSSVR QRMKCSSMQKFGERAFKAWAVARLSQTFPNADFAEITKLATDLTKV NKECCHGDLLECADDRAELAKYMCENQATISSKLQTCCDKPLLKKA HCLSEVEHDTMPADLPAIAADFVEDQEVCKNYAEAKDVFLGTFLYE YSRRHPDYSVSLLLRLAKKYEATLEKCCAEANPPACYGTVLAEFQPL VEEPKNLVKTNCDLYEKLGEYGFQNAILVRYTQKAPQVSTPTLVEA ARNLGRVGTKCCTLPEDQRLPCVEDYLSAILNRVCLLHEKTPVSEHV TKCCSGSLVERRPCFSALTVDETYVPKEFKAETFTFHSDICTLPEKEK QIKKQTALAELVKHKPKATAEQLKTVMDDFAQFLDTCCKAADKDT CFSTEGPNLVTRCKDALAHHHHHHEPEA 174 ACP128 mdmrvpaqllgllllwlrgarcEAHKSEIAHRYNDLGEQHFKGLVLIAFSQYLQ Mouse IFNa- KCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHTLFGDKLCAIP conjugate NLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPFERPEAEAMCT SFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQYNEILTQCCAEA DKESCLTPKLDGVKEKALVSSVRQRMKCSSMQKFGERAFKAWAVA RLSQTFPNADFAEITKLATDLTKVNKECCHGDLLECADDRAELAKY MCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMPADLPAIAADFV EDQEVCKNYAEAKDVFLGTFLYEYSRRHPDYSVSLLLRLAKKYEAT LEKCCAEANPPACYGTVLAEFQPLVEEPKNLVKTNCDLYEKLGEYG FQNAILVRYTQKAPQVSTPTLVEAARNLGRVGTKCCTLPEDQRLPCV EDYLSAILNRVCLLHEKTPVSEHVTKCCSGSLVERRPCFSALTVDETY VPKEFKAETFTFHSDICTLPEKEKQIKKQTALAELVKHKPKATAEQL KTVMDDFAQFLDTCCKAADKDTCFSTEGPNLVTRCKDALASGGPGP AGMKGLPGScdlpqthnlrnkraltllvqmalsplsclkdrkdfgfpqekvdaqqikkaqaipvls eltqqilniftskdssaawnttlldsfcndlhqqlndlqgclmqqvgvqefpltqedallavrkyfhrityy- lr ekkhspcawevvraevwralsssanvlgrlreekHHHHHHEPEA 175 ACP129 mdmrvpaqllgllllwlrgarccdlpqthnlrnkraltllvqmalsplsclkdrkdfgfpqek- vdaqqikk Mouse IFNa- aqaipvlseltqqilniftskdssaawnttlldsfendlhqqlndlqgclmqqvgvqefpltqedallavrky conjugate fhritvylrekkhspcawevvraevwralsssanvlgrlreekSGGPGPAGMKGLPGSEAH KSEIAHRYNDLGEQHFKGLVLIAFSQYLQKCSYDEHAKLVQEVTDF AKTCVADESAANCDKSLHTLFGDKLCAIPNLRENYGELADCCTKQE PERNECFLQHKDDNPSLPPFERPEAEAMCTSFKENPTTFMGHYLHEV ARRHPYFYAPELLYYAEQYNEILTQCCAEADKESCLTPKLDGVKEK ALVSSVRQRMKCSSMQKFGERAFKAWAVARLSQTFPNADFAEITKL ATDLTKVNKECCHGDLLECADDRAELAKYMCENQATISSKLQTCCD KPLLKKAHCLSEVEHDTMPADLPAIAADFVEDQEVCKNYAEAKDVF LGTFLYEYSRRHPDYSVSLLLRLAKKYEATLEKCCAEANPPACYGTV LAEFQPLVEEPKNLVKTNCDLYEKLGEYGFQNAILVRYTQKAPQVST PTLVEAARNLGRVGTKCCTLPEDQRLPCVEDYLSAILNRVCLLHEKT PVSEHVTKCCSGSLVERRPCFSALTVDETYVPKEFKAETFTFHSDICT LPEKEKQIKKQTALAELVKHKPKATAEQLKTVMDDFAQFLDTCCKA ADKDTCFSTEGPNLVTRCKDALAHHHHHHEPEA 176 ACP150 mdmrvpaqllgllllwlrgarcQVQLQESGGGLAQAGGSLSLSCAASGFTVSNS Mouse IFNa- VMAWYRQTPGKQREFVAIINSVGSTNYADSVKGRFTISRDNAKNTV conjugate YLQMNNLKPEDTAVYVCNRNFDRIYWGQGTQVTVSSggggsggggsggg gsEVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGL EWVSSISGSGRDTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTA VYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMKGLPGScdlpqthnlrnkral tllvqmalsplsclkdrkdfgfpqekvdaqqikkaqaipvlseltqqilniftskdssaawnttlldsfcnd- l hqqlndlqgclmqqvgvqefpltqedallavrkyfhritvylrekkhspcawevvraevwralsssanvl grlreekSGGPGPAGMKGLPGSEVQLVESGGGLVQPGNSLRLSCAASGF TFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDN AKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSHHHHH HEPEA 177 ACP151 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IFNa- GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSSGGPGPAGMK GLPGScdlpqthnlrnkraltllvqmalsplsclkdrkdfgfpqekvdaqqikkaqaipvlseltqqiln iftskdssaawnttlldsfcndlhqqlndlqgclmqqvgvqefpltqedallavrkyfhritvylrekkhsp cawevvraevwralsssanvlgrlreekSGGPGPAGMKGLPGSEVQLVESGGGLV QPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTL YAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSS QGTLVTVSSggggsggggsggggsQVQLQESGGGLAQAGGSLSLSCAASGF TVSNSVMAWYRQTPGKQREFVAIINSVGSTNYADSVKGRFTISRDNA KNTVYLQMNNLKPEDTAVYVCNRNFDRIYWGQGTQVTVSSHHHHH HEPEA 178 ACP152 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGNSLRLSCAASGFTFSKF Mouse IFNa- GMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAKTT conjugate LYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsgggg scdlpqthnlrnkraltllvqmalsplsclkdrkdfgfpqekvdaqqikkaqaipvlseltqqilniftskd- s saawnttlldsfcndlhqqlndlqgclmqqvgvqefpltqedallavrkyfhritvylrekkhspcawevv raevwralsssanvlgrlreekggggsggggsggggsEVQLVESGGGLVQPGNSLRLSC AASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRF TISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS HHHHHHEPEA 179 ACP153 mdmrvpaqllgllllwlrgarcaptssstkktqlqlehllldlqmilnginnyknpkltrmlt- fkfympkka (IL-2 telkhlqcleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmceyadetativef- lnrwitfc Conjugate) qsiistltsggpGPAGLYAQpgsEVQLVESGGGLVQPGNSLRLSCAASGFTFS KFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAK TTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsg gggsggggsggggsggggssggpGPAGLYAQpgsEVQLVESGGGLVQPGGSLR LSCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGR FTISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGT TVTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKAS QNVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFT LTISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKHHHHHHEPEA 180 ACP154 mdmrvpaqllgllllwlrgareaptssstkktqlqlehllldlqmilnginnyknpkltrmlt- fkfympkka (IL-2 telkhlqeleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmeeyadetativef- lnrwitfe Conjugate) qsiistltsggpPGGPAGIGpgsEVQLVESGGGLVQPGNSLRLSCAASGFTFS KFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAK TTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsg gggsggggsggggsggggssggpPGGPAGIGpgsEVQLVESGGGLVQPGGSLRL
SCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRF TISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTT VTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQ NVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLT ISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKHHHHHHEPEA 181 ACP155 mdmrvpaqllgllllwlrgareaptssstkktqlqlehllldlqmilnginnyknpkltrmlt- fkfympkka (IL-2 telkhlqeleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmeeyadetativef- lnrwitfe Conjugate) qsiistltsggpALFKSSFPpgsEVQLVESGGGLVQPGNSLRLSCAASGFTFS KFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNAK TTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggsg gggsggggsggggsggggssggpALFKSSFPpgsEVQLVESGGGLVQPGGSLRL SCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRF TISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQGTT VTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKASQ NVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFTLT ISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKHHHHHHEPEA 182 ACP156 mdmrvpaqllgllllwlrgareaptssstkktqlqlehllldlqmilnginnyknpkltrmlt- fkfympkka (IL-2 telkhlqeleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmeeyadetativef- lnrwitfe Conjugate) qsiistltsggpPLAQKLKSSpgsEVQLVESGGGLVQPGNSLRLSCAASGFTF SKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRDNA KTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSSggggsggggs ggggsggggsggggsggggssggpPLAQKLKSSpgsEVQLVESGGGLVQPGGSL RLSCAASGFTFSSYTLAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRG RFTISRDNAKNSLYLQMNSLRAEDTAVYYCARDSNWDALDYWGQG TTVTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCKA SQNVGTNVGWYQQKPGKAPKALIYSASFRYSGVPSRFSGSGSGTDFT LTISSLQPEDFATYYCQQYYTYPYTFGGGTKVEIKHHHHHHEPEA 183 ACP157 mdmrvpaqllgllllwlrgareaptssstkktqlqlehllldlqmilnginnyknpkltrmlt- fkfympkka (IL-2 telkhlqeleeelkpleevlnlaqsknfhlrprdlisninvivlelkgsettfmeeyadetativef- lnrwitfe Conjugate) qsiistltsggpPGGPAGIGalfkssfpPLAQKLKSSpgsEVQLVESGGGLVQPGN SLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAES VKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTL VTVSSggggsggggsggggsggggsggggsggggssggpPGGPAGIGalfkssfpPLAQ KLKSSpgsEVQLVESGGGLVQPGGSLRLSCAASGFTFSSYTLAWVRQ APGKGLEWVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLYLQMNSLR AEDTAVYYCARDSNWDALDYWGQGTTVTVSSGGGGSGGGGSGGG GSDIQMTQSPSSLSASVGDRVTITCKASQNVGTNVGWYQQKPGKAP KALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYY TYPYTFGGGTKVEIKHHHHHHEPEA 184 Place hold 185 Place hold 186 Place hold 187 Place hold 188 Place hold 189 Place hold 190 Place hold 191 Place hold 192 Blocker 2 mdmrvpaqllgllllwlrgarcEVQLVESGGGLVQPGGSLRLSCAASGFTFSSYT (IL2 blocker) LAWVRQAPGKGLEWVAAIDSSSYTYSPDTVRGRFTISRDNAKNSLY LQMNSLRAEDTAVYYCARDSNWDALDYWGQGTTVTVSSggggsgggg sggggsDIQMTQSPSSLSASVGDRVTITCKASQNVGTNVGWYQQKPGK APKALIYSASFRYSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQY YTYPYTFGGGTKVEIKHHHHHH 193 Blocker 12 mdmrvpaqllgllllwlrgarcQSVLTQPPSVSGAPGQRVTISCSGSRSNIGSNTV (IL-12 KWYQQLPGTAPKLLIYYNDQRPSGVPDRFSGSKSGTSASLAITGLQA blocker) EDEADYYCQSYDRYTHPALLFGTGTKVTVLggggsggggsggggsQVQLV ESGGGVVQPGRSLRLSCAASGFTFSSYGMHWVRQAPGKGLEWVAFI RYDGSNKYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYC KTHGSHDNWGQGTMVTVSSFIRREIHH 194 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQGGGGGLD GNEEPGGLEWVSSISGSGRDTLYADSVKGRFTISRDNAKTTLYLQMN SLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS 225 ACP203 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGR DTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS sggpGPAGLYAQpgscdlpqthnlrnkraltllvqmrrlsplsclkdrkdfgfpqekvdaqqikkaqai pvlseltqqilniftskdssaawnttlldsfcndlhqqlndlqgclmqqvgvqefpltqedallavrkyfhr- it vylrekkhspcawevvraeywralsssanylgrlreeksggpGPAGLYAQpgsEVQLVESGGGLV QPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFT ISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS 226 ACP204 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGR DTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS sggpALFKSSFPpgscdlpqthnlrnkraltllvqmrrlsplsclkdrkdfgfpqekvdaqqikkaqaipv lseltqqilniftskdssaawnttlldsfcndlhqqlndlqgclmqqvgvqefpltqedallavrkyfhrit- yyl rekkhspcawevvraeywralsssanylgrlreeksggpALFKSSFPpgsEVQLVESGGGLVQP GNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTIS RDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS 227 ACP205 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGR DTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS sggpPLAQKLKSSpgscdlpqthnlrnkraltllvqmrrlsplsclkdrkdfgfpqekvdaqqikkaqai pvlseltqqilniftskdssaawnttlldsfcndlhqqlndlqgclmqqvgvqefpltqedallavrkyfhr- it vylrekkhspcawevvraeywralsssanylgrlreeksggpPLAQKLKSSpgsEVQLVESGGGL VQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGR FTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS 228 ACP206 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGR DTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS sggpGPAGLYAQpgscdlpqthslgsrrtlmllaqmrrislfsclkdrhdfgfpqeefgnqfqkaetipvl hemiqqifnlfstkdssaawdetlldkfytelyqqlndleacviqgvgytetplmkedsilayrkyfqritl- yl kekkyspcawevvraeimrsfslstnlqeslrskesggpGPAGLYAQpgsEVQLVESGGGLVQP GNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTIS RDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS 229 ACP207 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGR DTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS sggpALFKSSFPpgscdlpqthslgsrrtlmllaqmrrislfsclkdrhdfgfpqeefgnqfqkaetipylh emiqqifnlfstkdssaawdetlldkfytelyqqlndleacviqgvgytetplmkedsilayrkyfqritly- lk ekkyspcawevvraeimrsfslstnlqeslrskesggpALFKSSFPpgsEVQLVESGGGLVQPGN SLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTISRD NAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS 230 ACP208 EVQLVESGGGLVQPGNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGR DTLYAESVKGRFTISRDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS sggpPLAQKLKSSpgscdlpqthslgsrrtlmllaqmrrislfsclkdrhdfgfpqeefgnqfqkaetipv lhemiqqifnlfstkdssaawdetlldkfytelyqqlndleacviqgvgytetplmkedsilayrkyfqrit- ly lkekkyspcawevvraeimrsfslstnlqeslrskesggpPLAQKLKSSpgsEVQLVESGGGLVQP GNSLRLSCAASGFTFSKFGMSWVRQAPGKGLEWVSSISGSGRDTLYAESVKGRFTIS RDNAKTTLYLQMNSLRPEDTAVYYCTIGGSLSVSSQGTLVTVSS 258 MMP14 GPLGLKAQ substrate motif sequence 259 MMP14 LPLGLKAQ substrate motif sequence 260 MMP14 SPLGLKAQ substrate motif sequence 261 MMP14 QPLGLKAQ substrate motif sequence 262 MMP14 KPLGLKAQ substrate motif sequence 263 MMP14 FPLGLKAQ substrate motif sequence 264 MMP14 HPLGLKAQ substrate motif sequence 265 MMP14 PPLGLKAQ substrate motif sequence 266 MMP14 APLGLKAQ substrate motif sequence 267 MMP14 DPLGLKAQ substrate motif sequence 268 MMP14 GPHGLKAQ substrate motif sequence 269 MMP14 GPSGLKAQ substrate motif sequence 270 MMP14 GpQGLKAQ substrate motif sequence 271 MMP14 GPPGLKAQ substrate motif sequence 272 MMP14 GPEGLKAQ substrate motif sequence 273 MMP14 GPFGLKAQ substrate motif sequence 274 MMP14 GPRGLKAQ substrate motif sequence 275 MMP14 GPGGLKAQ substrate motif sequence 276 MMP14 GPAGLKAQ substrate motif sequence 277 MMP14 LPAGLKGA substrate motif sequence 195 MMP14 GPAGLYAQ substrate motif sequence 278 MMP14 GPANLVAQ substrate motif sequence 279 MMP14 GPAALVGA substrate motif
sequence 280 MMP14 GPANLRAQ substrate motif sequence 281 MMP14 GPAGLRAQ substrate motif sequence 282 MMP14 GPAGLVAQ substrate motif sequence 283 MMP14 GPAGLRGA substrate motif sequence 284 MMP14 LPAGLVGA substrate motif sequence 285 MMP14 GPAGLKGA substrate motif sequence 286 MMP14 GPLALKAQ substrate motif sequence 287 MMP14 GPLNLKAQ substrate motif sequence 288 MMP14 GPLHLKAQ substrate motif sequence 289 MMP14 GPLYLKAQ substrate motif sequence 290 MMP14 GPLPLKAQ substrate motif sequence 291 MMP14 GPLELKAQ substrate motif sequence 292 MMP14 GPLRLKAQ substrate motif sequence 293 MMP14 GPLLLKAQ substrate motif sequence 294 MMP14 GPLSLKAQ substrate motif sequence 295 MMP14 GPLGLYAQ substrate motif sequence 296 MMP14 GPLGLFAQ substrate motif sequence 297 MMP14 GPLGLLAQ substrate motif sequence 298 MMP14 GPLGLHAQ substrate motif sequence 299 MMP14 GPLGLRAQ substrate motif sequence 300 MMP14 GPLGLAAQ substrate motif sequence 301 MMP14 GPLGLEAQ substrate motif sequence 302 MMP14 GPLGLGAQ substrate motif sequence 303 MMP14 GPLGLPAQ substrate motif sequence 304 MMP14 GPLGLQAQ substrate motif sequence 305 MMP14 GPLGLSAQ substrate motif sequence 306 MMP14 GPLGLVAQ substrate motif sequence 307 MMP14 GPLGLKLQ substrate motif sequence 308 MMP14 GPLGLKFQ substrate motif sequence 309 MMP14 GPLGLKEQ substrate motif sequence 310 MMP14 GPLGLKKQ substrate motif sequence 311 MMP14 GPLGLKQQ substrate motif sequence 312 MMP14 GPLGLKSQ substrate motif sequence 313 MMP14 GPLGLKGQ substrate motif sequence 314 MMP14 GPLGLKHQ substrate motif sequence 315 MMP14 GPLGLKPQ substrate motif sequence 316 MMP14 GPLGLKAG substrate motif sequence 317 MMP14 GPLGLKAF substrate motif sequence 318 MMP14 GPLGLKAP substrate motif sequence 319 MMP14 GPLGLKAL substrate motif sequence 320 MMP14 GPLGLKAE substrate motif sequence 321 MMP14 GPLGLKAA substrate motif sequence 322 MMP14 GPLGLKAH substrate motif sequence 323 MMP14 GPLGLKAK substrate motif sequence 324 MMP14 GPLGLKAS substrate motif sequence 325 MMP14 GPLGLFGA substrate motif sequence 326 MMP14 GPLGLQGA substrate motif sequence 327 MMP14 GPLGLVGA substrate motif sequence 328 MMP14 GPLGLAGA substrate motif sequence 329 MMP14 GPLGLLGA substrate motif sequence
330 MMP14 GPLGLRGA substrate motif sequence 331 MMP14 GPLGLYGA substrate motif sequence 332 CTSL1 ALFKSSPP substrate motif sequence 333 CTSL1 SPFRSSRQ substrate motif sequence 334 CTSL1 KLFKSSPP substrate motif sequence 335 CTSL1 HLFKSSPP substrate motif sequence 336 CTSL1 SLFKSSPP substrate motif sequence 337 CTSL1 QLFKSSPP substrate motif sequence 338 CTSL1 LLFKSSPP substrate motif sequence 339 CTSL1 PLFKSSPP substrate motif sequence 340 CTSL1 FLFKSSPP substrate motif sequence 341 CTSL1 GLFKSSPP substrate motif sequence 342 CTSL1 VLFKSSPP substrate motif sequence 343 CTSL1 ELFKSSPP substrate motif sequence 344 CTSL1 AKFKSSPP substrate motif sequence 345 CTSL1 AHFKSSPP substrate motif sequence 346 CTSL1 AGFKSSPP substrate motif sequence 347 CTSL1 APFKSSPP substrate motif sequence 348 CTSL1 ANFKSSPP substrate motif sequence 349 CTSL1 AFFKSSPP substrate motif sequence 350 CTSL1 AAFKSSPP substrate motif sequence 351 CTSL1 ASFKSSPP substrate motif sequence 352 CTSL1 AEFKSSPP substrate motif sequence 353 CTSL1 ALRKSSPP substrate motif sequence 354 CTSL1 ALLKSSPP substrate motif sequence 355 CTSL1 ALAKSSPP substrate motif sequence 356 CTSL1 ALQKSSPP substrate motif sequence 357 CTSL1 ALHKSSPP substrate motif sequence 358 CTSL1 ALPKSSPP substrate motif sequence 359 CTSL1 ALTKSSPP substrate motif sequence 360 CTSL1 ALGKSSPP substrate motif sequence 361 CTSL1 ALDKSSPP substrate motif sequence 199 CTSL1 ALFFSSPP substrate motif sequence 362 CTSL1 ALFHSSPP substrate motif sequence 363 CTSL1 ALFTSSPP substrate motif sequence 364 CTSL1 ALFASSPP substrate motif sequence 365 CTSL1 ALFQSSPP substrate motif sequence 366 CTSL1 ALFLSSPP substrate motif sequence 367 CTSL1 ALFGSSPP substrate motif sequence 368 CTSL1 ALFESSPP substrate motif sequence 369 CTSL1 ALFPSSPP substrate motif sequence 370 CTSL1 ALFKHSPP substrate motif sequence 371 CTSL1 ALFKLSPP substrate motif sequence 372 CTSL1 ALFKKSPP substrate motif sequence 373 CTSL1 ALFKASPP substrate motif sequence 374 CTSL1 substrate ALFKISPP motif sequence 375 CTSL1 ALFKGSPP substrate motif sequence 376 CTSL1 ALFKNSPP substrate motif sequence 377 CTSL1 ALFKRSPP substrate motif sequence 378 CTSL1 ALFKESPP substrate motif sequence
379 CTSL1 ALFKFSPP substrate motif sequence 380 CTSL1 ALFKPSPP substrate motif sequence 381 CTSL1 ALFKSFPP substrate motif sequence 382 CTSL1 ALFKSLPP substrate motif sequence 383 CTSL1 ALFKSIPP substrate motif sequence 384 CTSL1 ALFKSKPP substrate motif sequence 385 CTSL1 ALFKSAPP substrate motif sequence 386 CTSL1 ALFKSQPP substrate motif sequence 387 CTSL1 ALFKSPPP substrate motif sequence 388 CTSL1 ALFKSEPP substrate motif sequence 389 CTSL1 ALFKSGPP substrate motif sequence 198 CTSL1 ALFKSSFP substrate motif sequence 390 CTSL1 ALFKSSLP substrate motif sequence 391 CTSL1 substrate ALFKSSGP motif sequence 392 CTSL1 ALFKSSSP substrate motif sequence 393 CTSL1 ALFKSSVP substrate motif sequence 394 CTSL1 ALFKSSHP substrate motif sequence 395 CTSL1 ALFKSSAP substrate motif sequence 396 CTSL1 ALFKSSNP substrate motif sequence 397 CTSL1 ALFKSSKP substrate motif sequence 398 CTSL1 ALFKSSEP substrate motif sequence 399 CTSL1 ALFKSSPF substrate motif sequence 400 CTSL1 ALFKSSPH substrate motif sequence 401 CTSL1 ALFKSSPG substrate motif sequence 402 CTSL1 ALFKSSPA substrate motif sequence 403 CTSL1 ALFKSSPS substrate motif sequence 404 CTSL1 ALFKSSPV substrate motif sequence 405 CTSL1 ALFKSSPQ substrate motif sequence 406 CTSL1 ALFKSSPK substrate motif sequence 407 CTSL1 ALFKSSPL substrate motif sequence 408 CTSL1 ALFKSSPD substrate motif sequence indicates data missing or illegible when filed
Sequence CWU 0 SQTB SEQUENCE LISTING The patent application
contains a lengthy "Sequence Listing" section. A copy of the
"Sequence Listing" is available in electronic form from the USPTO
web site
(https://seqdata.uspto.gov/?pageRequest=docDetail&DocID=US20210115102A1).
An electronic copy of the "Sequence Listing" will also be available
from the USPTO upon request and payment of the fee set forth in 37
CFR 1.19(b)(3).
0 SQTB SEQUENCE LISTING The patent application contains a lengthy
"Sequence Listing" section. A copy of the "Sequence Listing" is
available in electronic form from the USPTO web site
(https://seqdata.uspto.gov/?pageRequest=docDetail&DocID=US20210115102A1).
An electronic copy of the "Sequence Listing" will also be available
from the USPTO upon request and payment of the fee set forth in 37
CFR 1.19(b)(3).
References
D00000
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
D00012

D00013

D00014

D00015

D00016
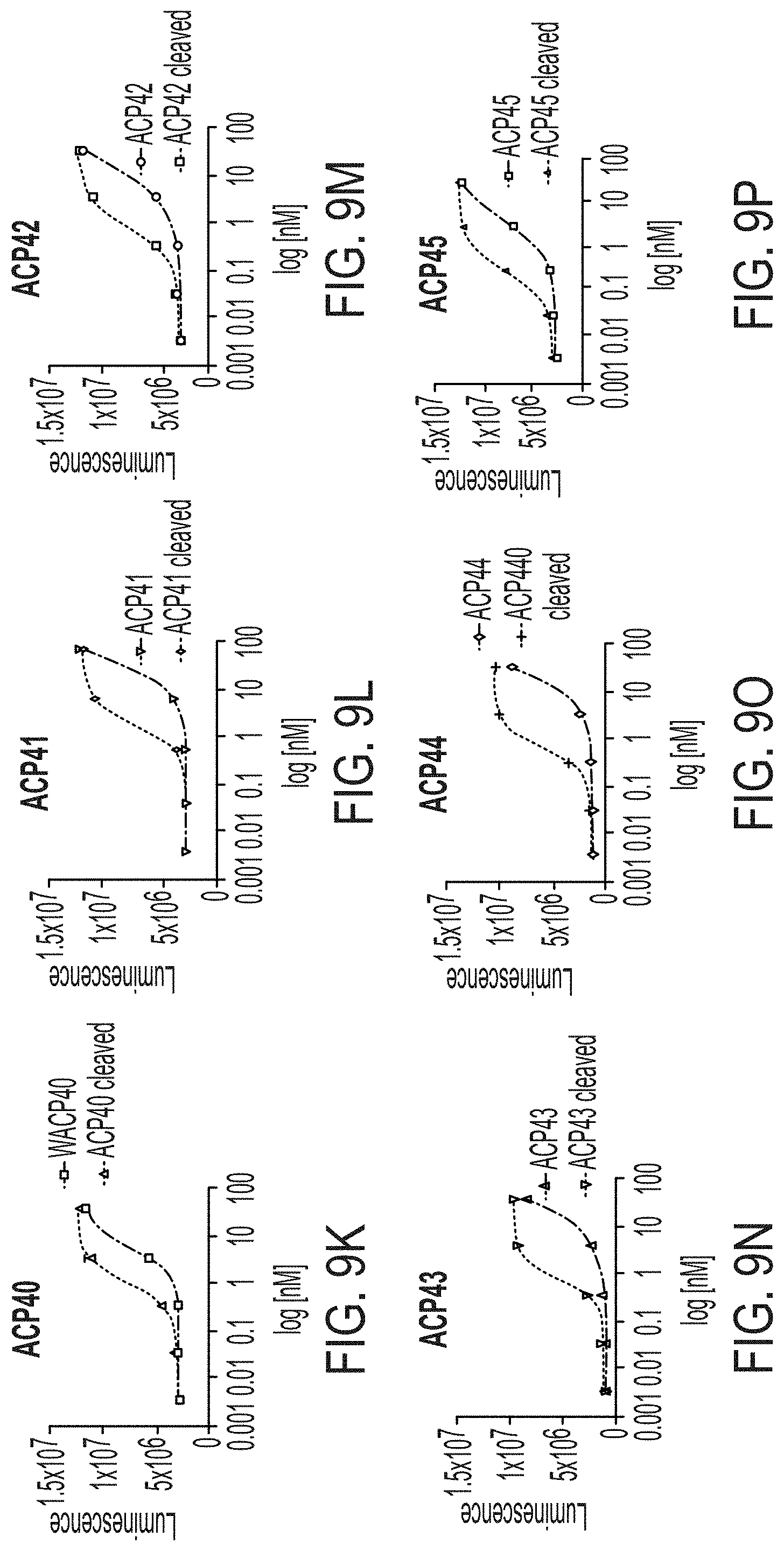
D00017

D00018

D00019

D00020

D00021

D00022

D00023

D00024

D00025

D00026
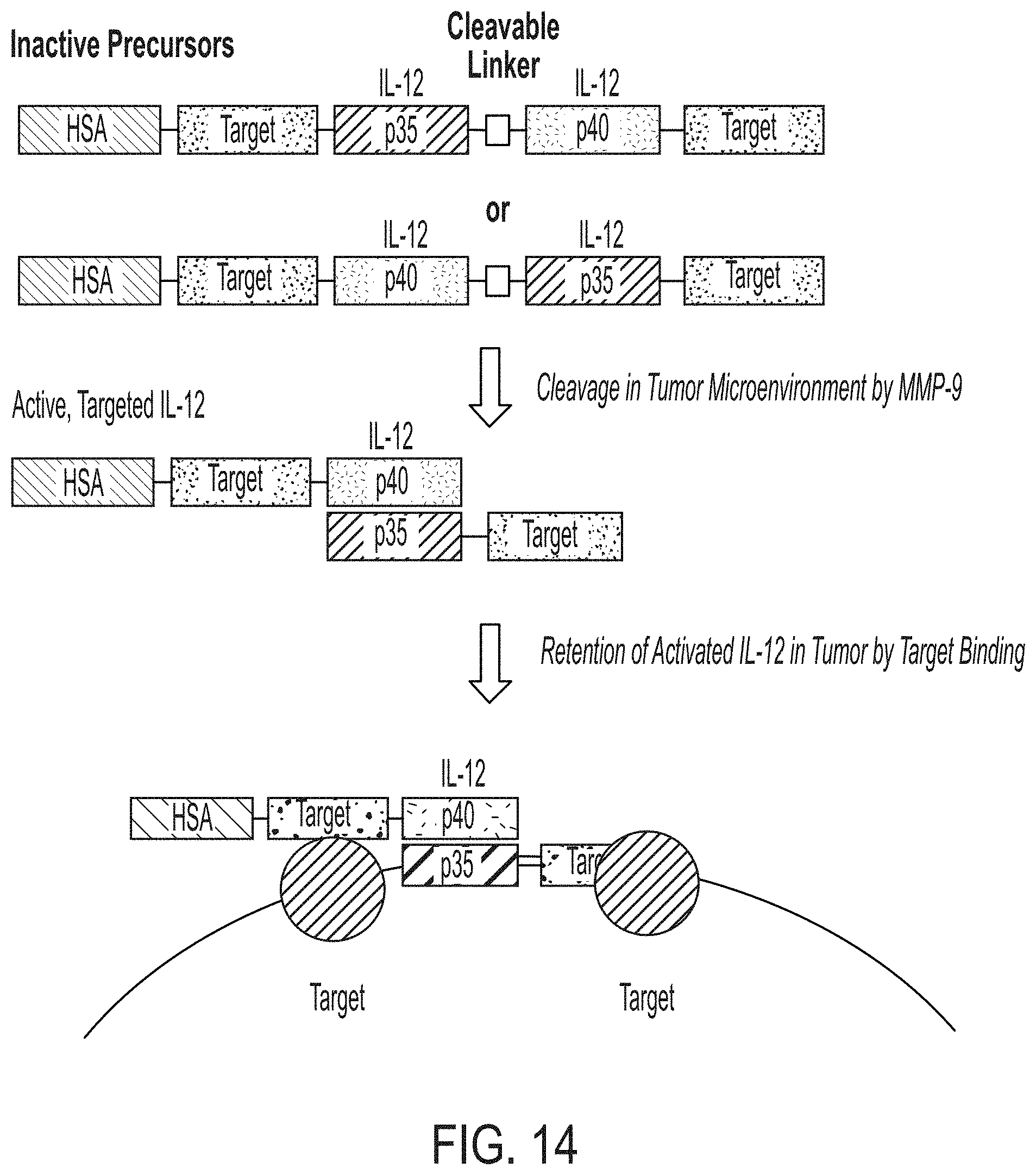
D00027

D00028

D00029

D00030
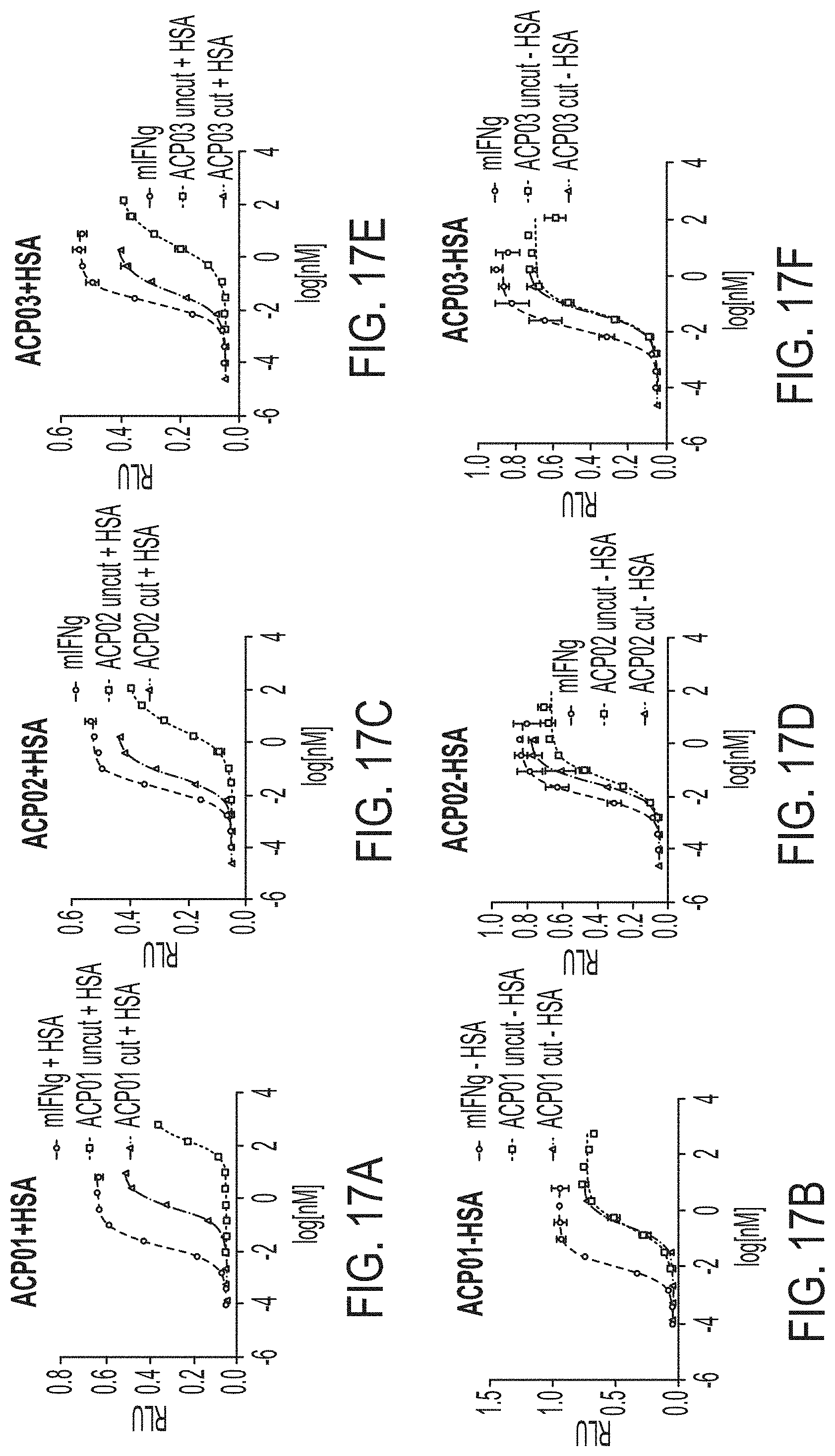
D00031
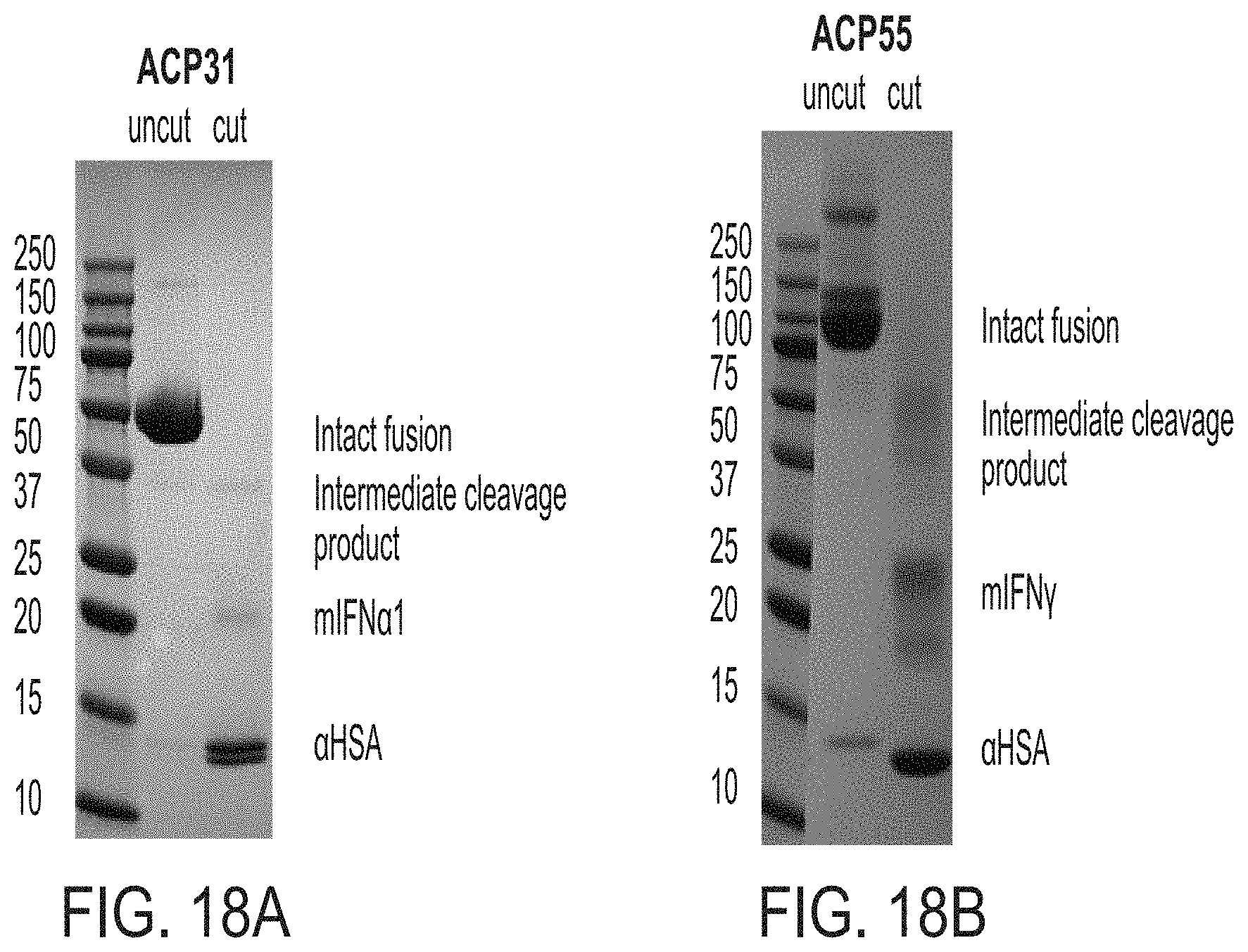
D00032

D00033
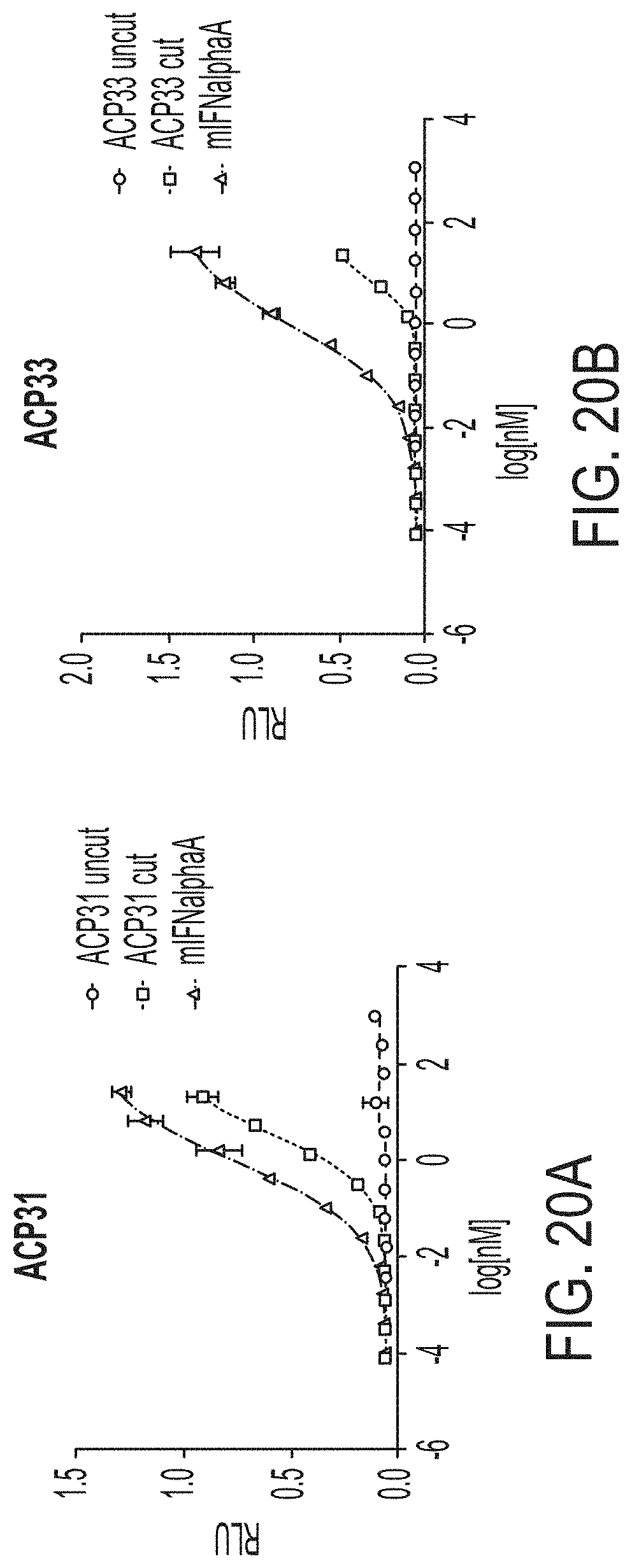
D00034

D00035

D00036
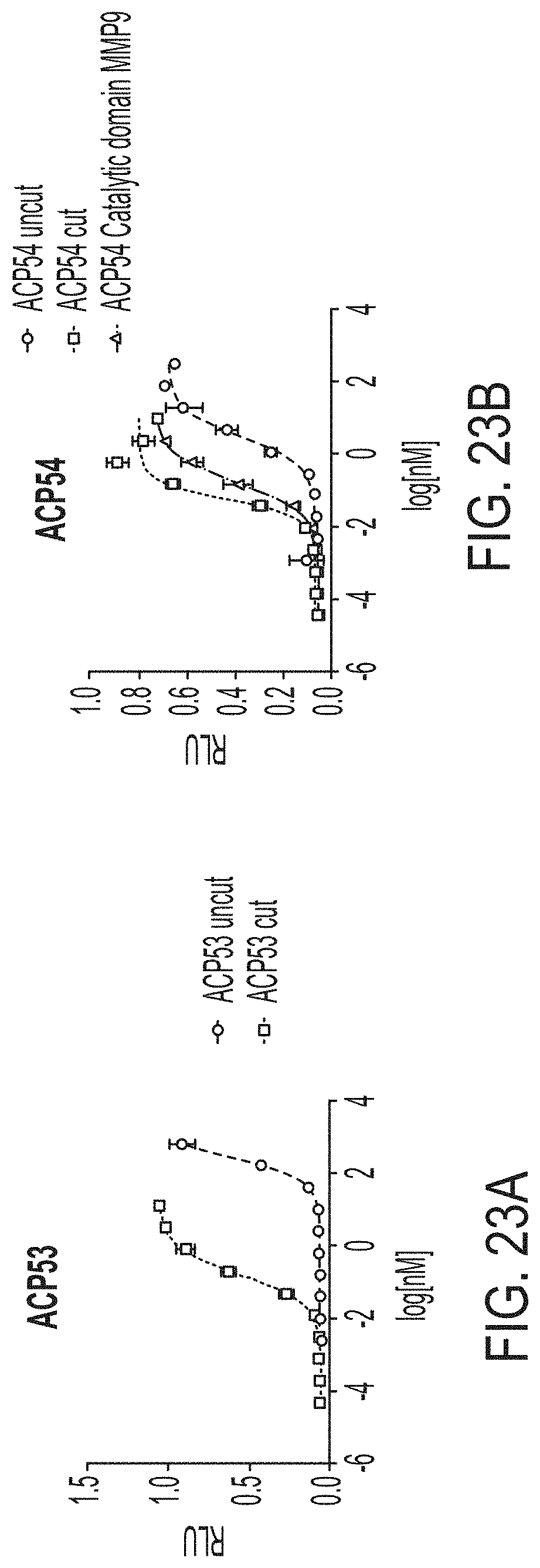
D00037

D00038

D00039

D00040

D00041

D00042

D00043

D00044

D00045

D00046

D00047
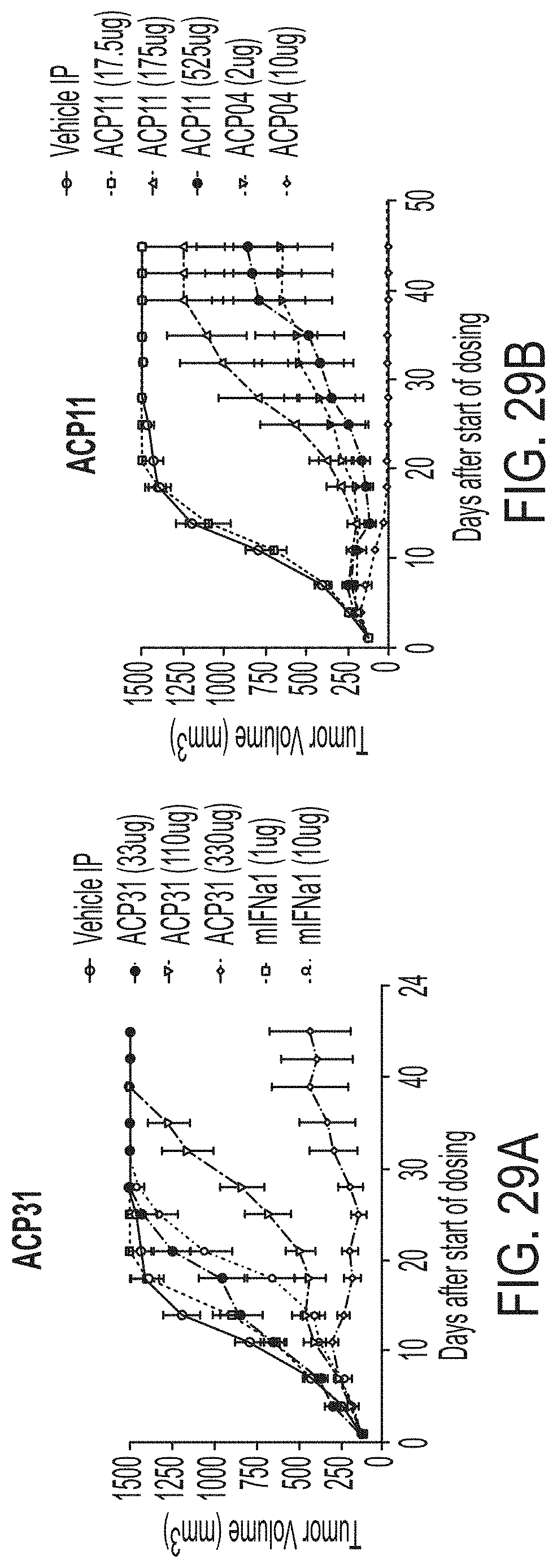
D00048

D00049

D00050
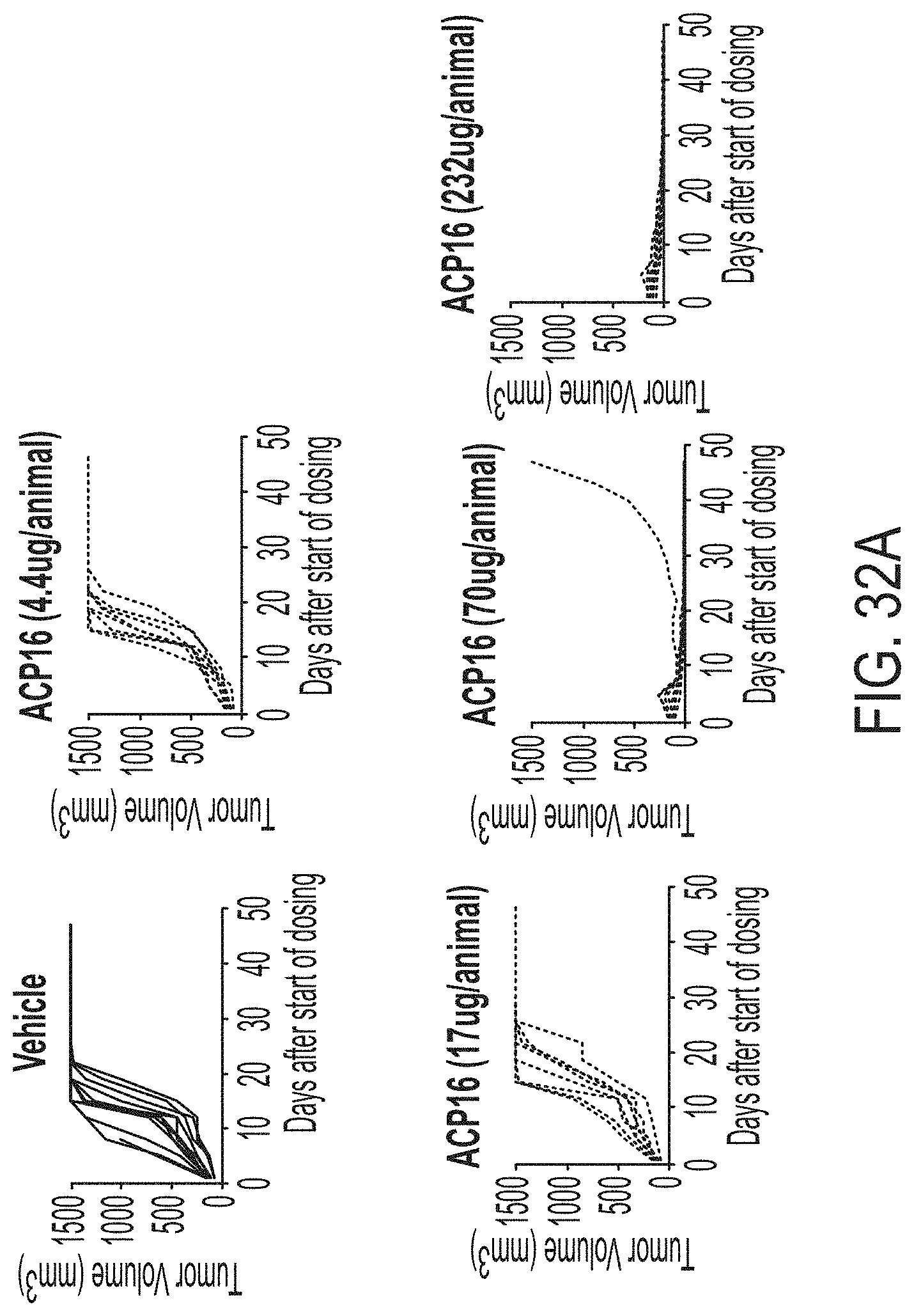
D00051

D00052

D00053
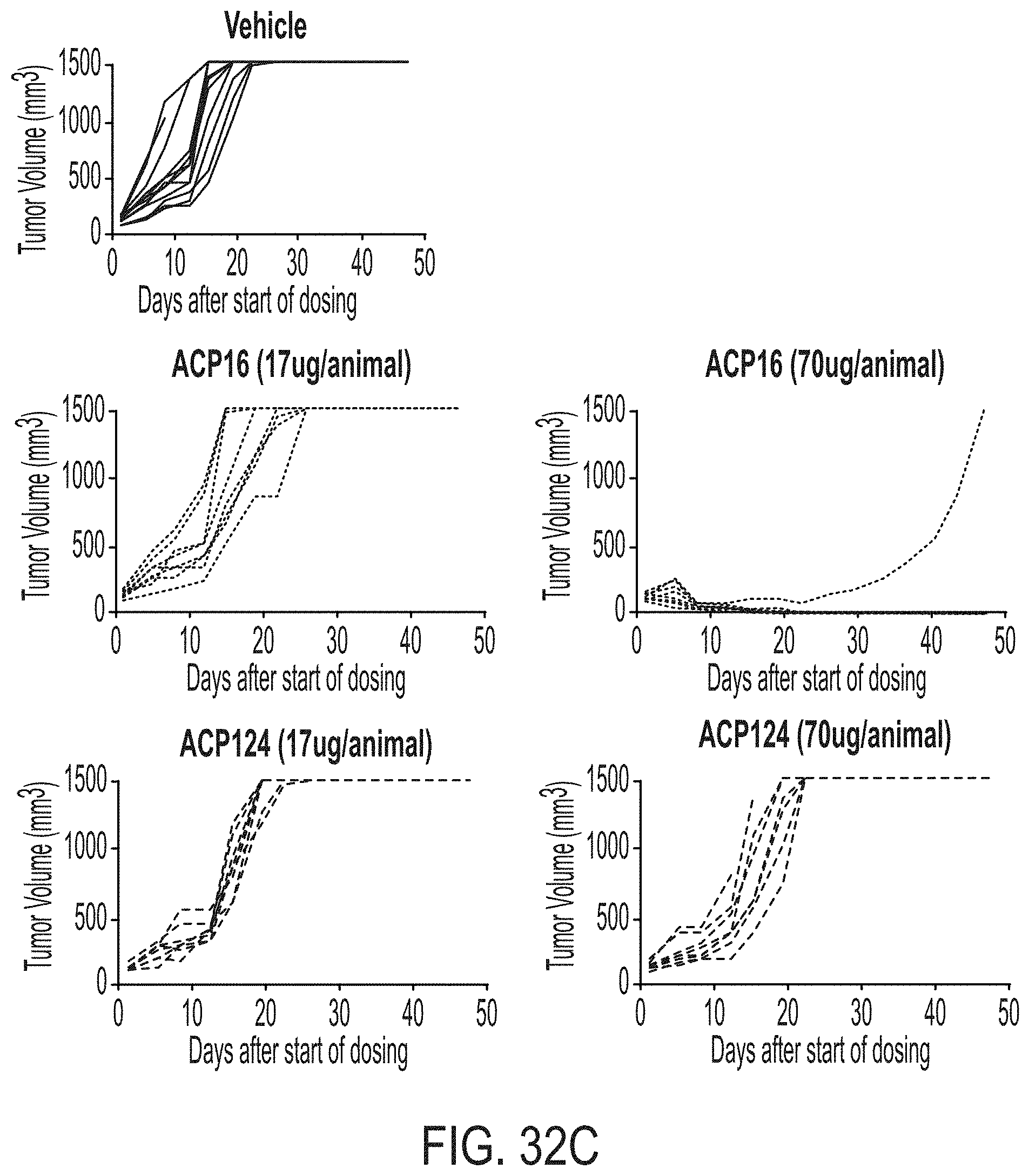
D00054

D00055

D00056

D00057

D00058

D00059

D00060

D00061

D00062

D00063

D00064

D00065

D00066

D00067

D00068

D00069

D00070

D00071
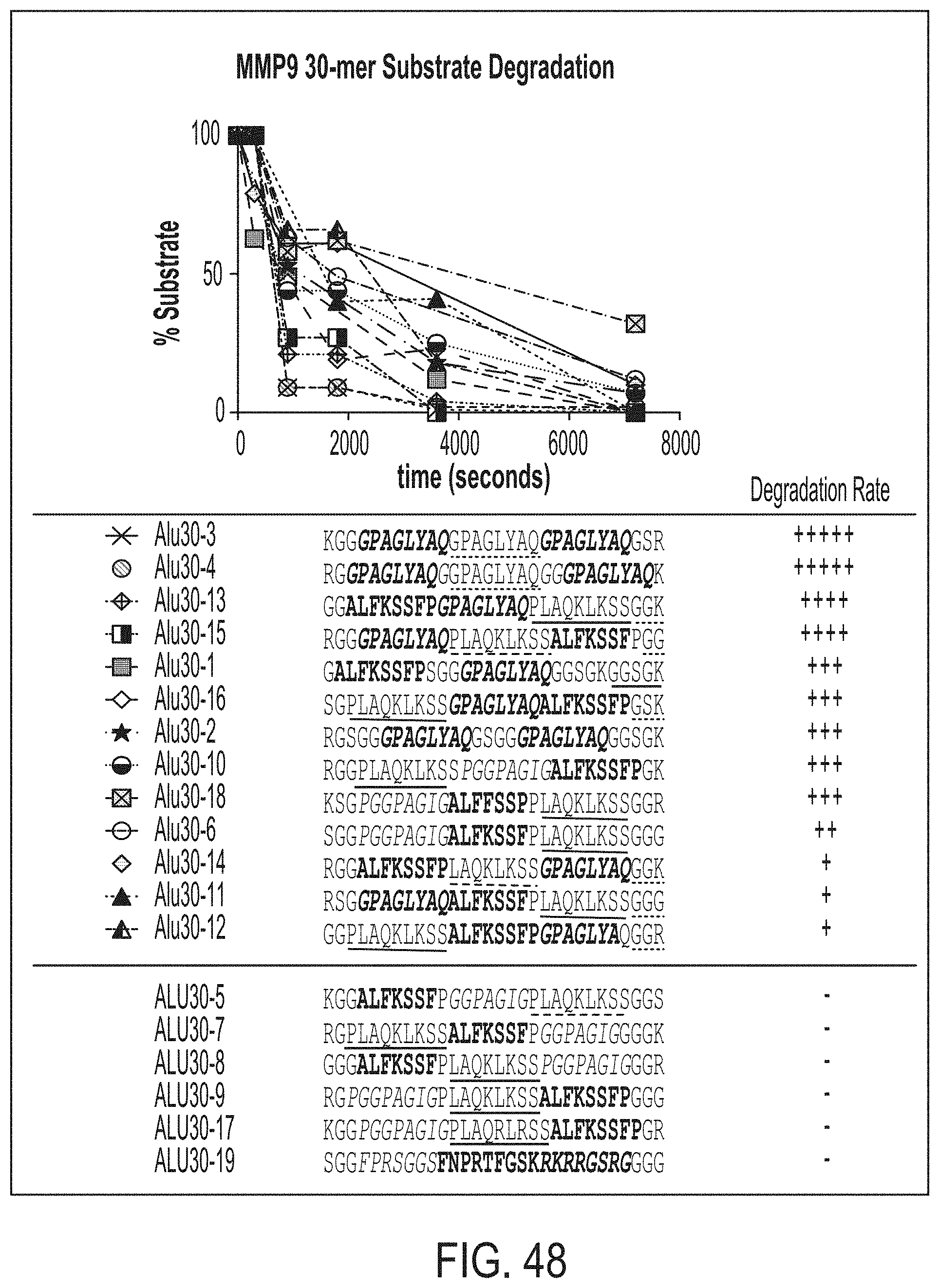
D00072

D00073

D00074

D00075

D00076

D00077

D00078

D00079

D00080

D00081
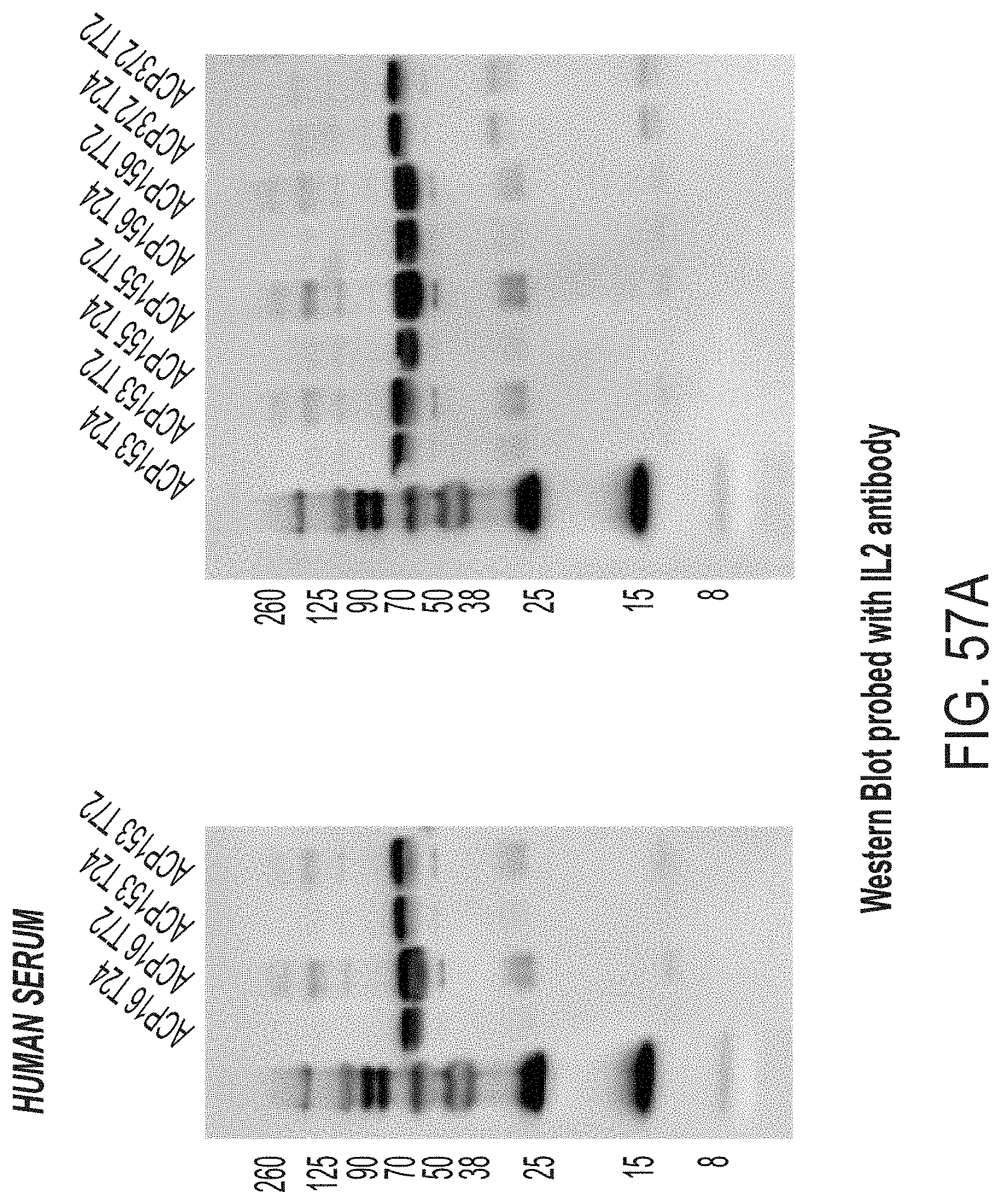
D00082

D00083

D00084

D00085

D00086

D00087
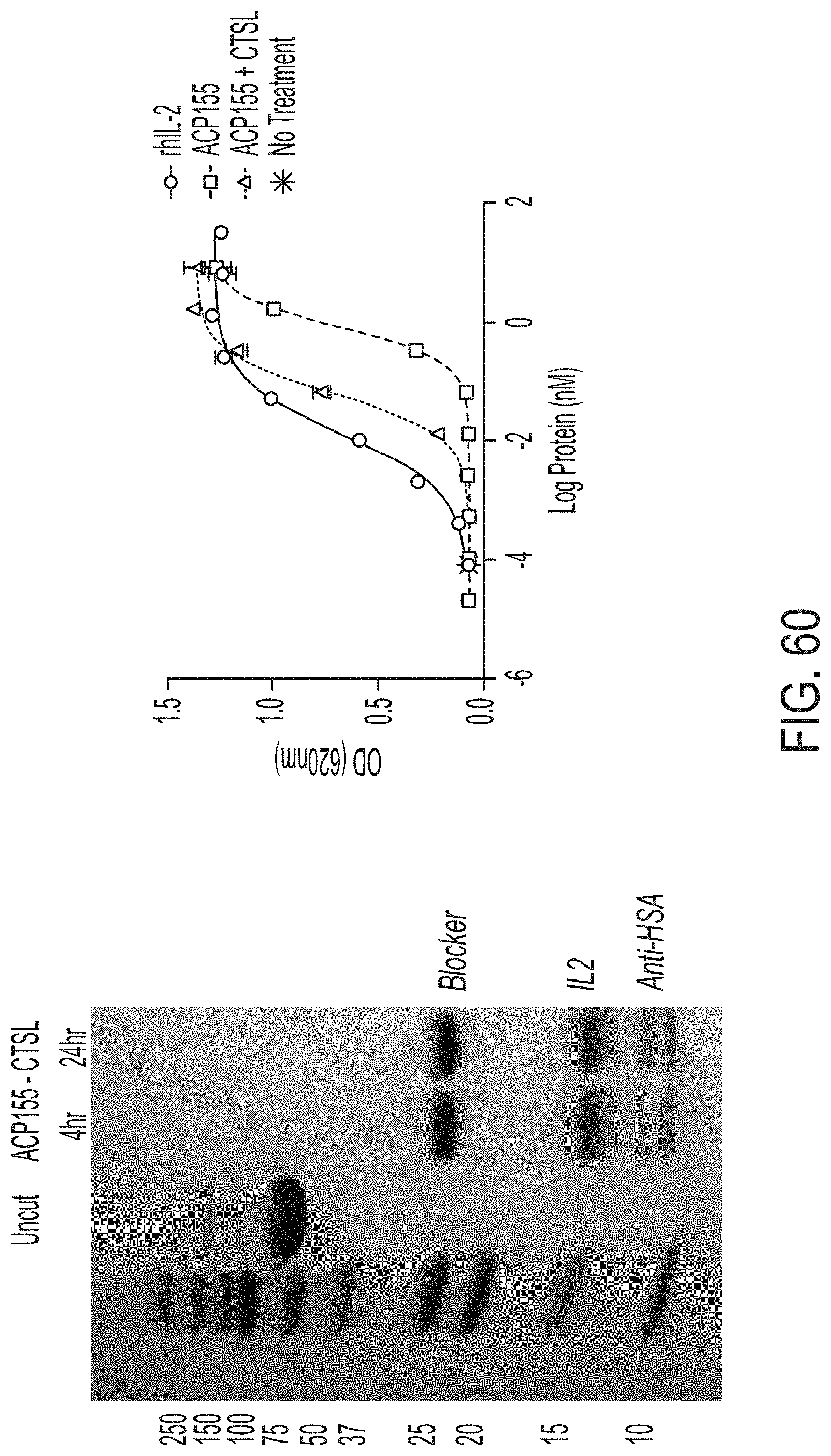
D00088
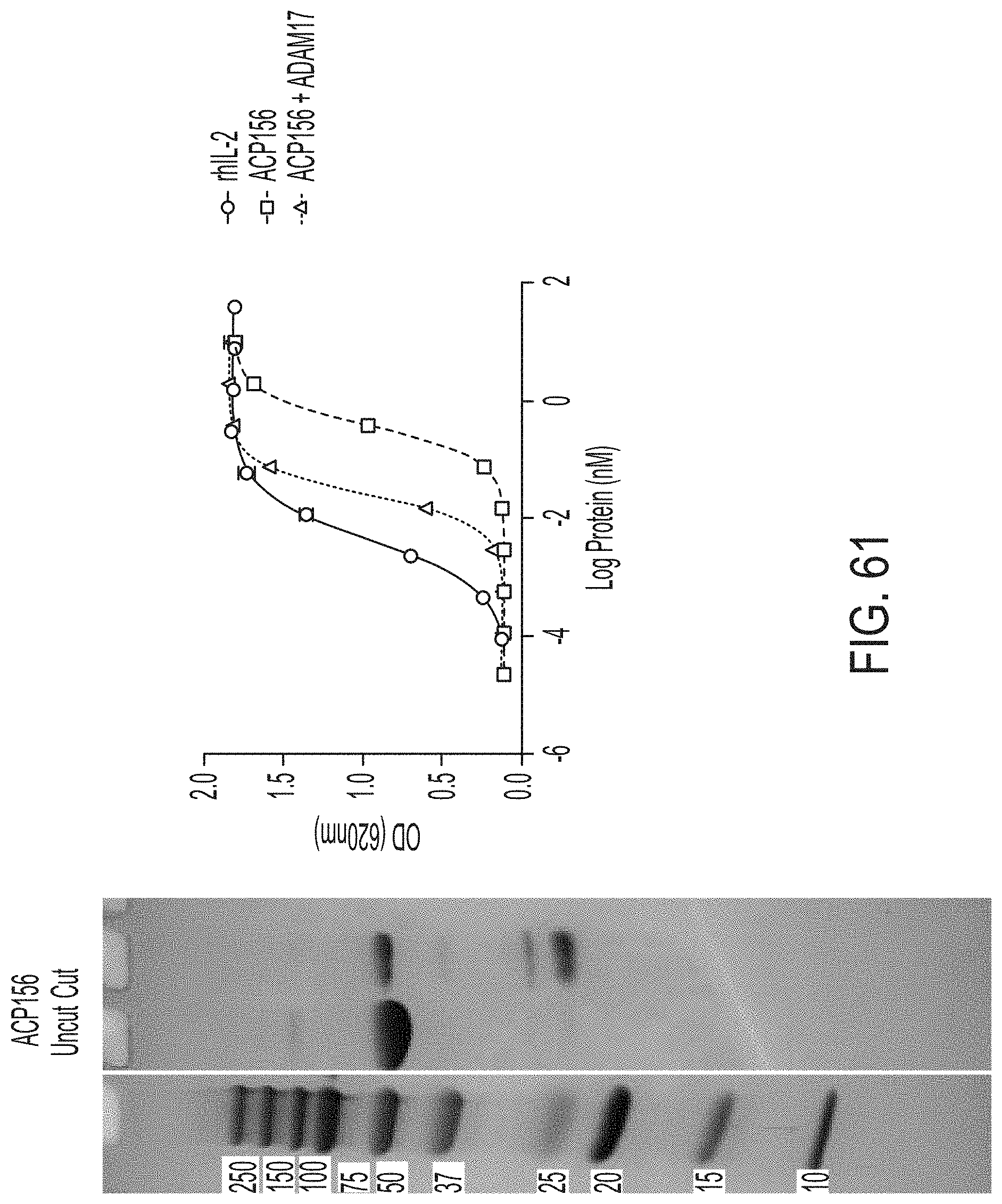
D00089

D00090

D00091

D00092

D00093

D00094



P00899

XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.