Aqueous Pharmaceutical Formulation Comprising Anti-pd-l1 Antibody Avelumab
RINALDI; Gianluca ; et al.
U.S. patent application number 17/116587 was filed with the patent office on 2021-04-08 for aqueous pharmaceutical formulation comprising anti-pd-l1 antibody avelumab. The applicant listed for this patent is MERCK PATENT GMBH, PFIZER INC.. Invention is credited to Alessandra DEL RIO, Silvia FRATAR-CANGELI, Gianluca RINALDI, Senta VOSS, Markus WEIGANDT.
| Application Number | 20210100903 17/116587 |
| Document ID | / |
| Family ID | 1000005290074 |
| Filed Date | 2021-04-08 |









View All Diagrams
| United States Patent Application | 20210100903 |
| Kind Code | A1 |
| RINALDI; Gianluca ; et al. | April 8, 2021 |
AQUEOUS PHARMACEUTICAL FORMULATION COMPRISING ANTI-PD-L1 ANTIBODY AVELUMAB
Abstract
The present invention relates to a novel anti-PD-L1 antibody formulation. In particular, the invention relates to an aqueous pharmaceutical formulation of the anti-PD-L1 antibody Avelumab.
| Inventors: | RINALDI; Gianluca; (Monterotondo, IT) ; DEL RIO; Alessandra; (Roma, IT) ; FRATAR-CANGELI; Silvia; (Ceprano, IT) ; VOSS; Senta; (Mainz, DE) ; WEIGANDT; Markus; (Mannheim, DE) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 1000005290074 | ||||||||||
| Appl. No.: | 17/116587 | ||||||||||
| Filed: | December 9, 2020 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 16060319 | Jun 7, 2018 | |||
| PCT/EP2016/002040 | Dec 5, 2016 | |||
| 17116587 | ||||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61K 2039/54 20130101; C07K 2317/21 20130101; C07K 2317/76 20130101; A61K 39/39591 20130101; C07K 16/2827 20130101 |
| International Class: | A61K 39/395 20060101 A61K039/395; C07K 16/28 20060101 C07K016/28 |
Foreign Application Data
| Date | Code | Application Number |
|---|---|---|
| Dec 7, 2015 | EP | 15198233.7 |
Claims
1-42. (canceled)
43. A method of treating cancer comprising administering an aqueous pharmaceutical antibody formulation to a patient, wherein the formulation comprises: (i) Avelumab in a concentration of 1 milligram/milliliter (mg/mL) to 30 mg/ml, as the antibody; (ii) acetate or histidine in a concentration of 5 millimolar (mM) to 15 mM as the buffering agent; (iii) D-mannitol or trehalose in a concentration of 240 mM to 320 mM, or a combination of arginine HCl in a concentration of 50 to 150 mM and glutamic acid in a concentration of 25 mM to 75 mM as a stabiliser; and (iv) Poloxamer 188 or Polysorbate 20 in a concentration of 0.25 mg/mL to 0.75 mg/mL, as surfactant, or no surfactant; wherein the formulation does not comprise methionine, and further wherein the formulation has a pH of 5.0 to 6.0.
44. The method of claim 43, wherein the formulation has a pH of 5.0 to 5.6.
45. The method of claim 43, wherein the formulation comprises Avelumab at a concentration of about 10 mg/mL to about 20 mg/mL.
46. The method of claim 43, wherein the formulation comprises acetate or histidine at a concentration of about 10 mM.
47. The method of claim 43, wherein the formulation comprises D-mannitol or trehalose at a concentration of about 280 mM, or for the combination of arginine HCl and glutamic acid, the formulation comprises arginine HCl at a concentration of about 150 mM and glutamic acid at a concentration of about 50 mM.
48. The method of claim 43, wherein the formulation comprises Poloxamer 188 or Polysorbate 20 at a concentration of about 0.5 mg/mL.
49. The method of claim 43, wherein the formulation has a pH of 5.2 (.+-.0.1) to 5.5 (.+-.0.1).
50. The method of claim 43, wherein the formulation comprises acetate in a concentration of about 10 mM, and not comprising any other buffering agent.
51. The method of claim 43, wherein the formulation comprises D-mannitol or trehalose in a concentration of about 280 mM, and not comprising any other stabiliser.
52. The method of claim 43, wherein the formulation comprises Polysorbate 20 or Poloxamer 188 in a concentration of about 0.5 mg/mL and does not comprise any other surfactant.
53. The method of claim 51, wherein the formulation comprises: (i) Avelumab in a concentration of 10 mg/mL; (ii) acetate in a concentration of 10 mM; (iii) D-mannitol or trehalose in a concentration of 280 mM; and (iv) Polysorbate 20 or Poloxamer 188 in a concentration of 0.5 mg/mL; wherein the formulation has a pH of 5.5 (.+-.0.1).
54. The method of claim 52, wherein the formulation consists of: (i) Avelumab in a concentration of 10 mg/mL; (ii) sodium acetate trihydrate in a concentration of 10 mM; (iii) D-mannitol or trehalose in a concentration of 280 mM; (iv) Polysorbate 20 or Poloxamer 188 in a concentration of 0.5 mg/mL; (v) HCl to adjust the pH; and (vi) water (for injection) as the solvent; wherein the formulation has a pH of 5.5 (.+-.0.1).
55. The method of claim 54, wherein the formulation consists of: (i) Avelumab in a concentration of 10 mg/mL; (ii) sodium acetate trihydrate in a concentration of 10 mM; (iii) trehalose dihydrate in a concentration of 280 mM; (iv) Polysorbate 20 in a concentration of 0.5 mg/mL; (v) HCl to adjust the pH; and (vi) water (for injection) as the diluent; wherein the formulation has a pH of 5.5 (.+-.0.1).
56. The method of claim 43, wherein the formulation comprises: (i) Avelumab in a concentration of about 20 mg/mL as the antibody; (ii) acetate in a concentration of about 10 mM as the buffering agent; (iii) D-mannitol or trehalose in a concentration of about 280 mM as a stabiliser; and (iv) Polysorbate 20 or Poloxamer 188 in a concentration of about 0.5 mg/mL as surfactant; wherein the formulation has a pH of 5.2 (.+-.0.1).
57. The method of claim 56, wherein the formulation comprises: (i) Avelumab in a concentration of 20 mg/mL; (ii) acetate in a concentration of 10 mM; (iii) D-mannitol or trehalose in a concentration of 280 mM; and (iv) Polysorbate 20 or Poloxamer 188 in a concentration of 0.5 mg/mL; wherein the formulation has a pH of 5.2 (.+-.0.1).
58. The method of claim 43, wherein the formulation does not comprise an antioxidant.
59. The method of claim 56, wherein the formulation consists of: (i) Avelumab in a concentration of 20 mg/mL; (ii) acetic acid in a concentration of 10 mM; (iii) D-mannitol or trehalose dihydrate in a concentration of 280 mM; (iv) Polysorbate 20 or Poloxamer 188 in a concentration of 0.5 mg/mL; (v) sodium acetate to adjust the pH; and (vi) water (for injection) as the diluent; wherein the formulation has a pH of 5.2 (.+-.0.1).
60. The method of claim 59, wherein the formulation consists of: (i) Avelumab in a concentration of 20 mg/mL; (ii) acetic acid in a concentration of 10 mM; (iii) D-mannitol in a concentration of 280 mM; (iv) Polysorbate 20 in a concentration of 0.5 mg/mL; (v) sodium acetate to adjust the pH; and (vi) water (for injection) as the diluent; wherein the formulation has a pH of 5.2 (.+-.0.1).
61. The method of claim 59, wherein the formulation consists of: (i) Avelumab in a concentration of 20 mg/mL; (ii) acetic acid in a concentration of 10 mM; (iii) trehalose dihydrate in a concentration of 280 mM; (iv) Polysorbate 20 in a concentration of 0.5 mg/mL; (v) sodium acetate to adjust the pH; and (vi) water (for injection) as the diluent; wherein the formulation has a pH of 5.2 (.+-.0.1).
62. The method of claim 59, wherein the formulation consists of: (i) Avelumab in a concentration of 20 mg/mL; (ii) acetic acid in a concentration of 10 mM; (iii) D-mannitol in a concentration of 280 mM; (iv) Poloxamer 188 in a concentration of 0.5 mg/mL; (v) sodium acetate to adjust the pH; and (vi) water (for injection) as the diluent; wherein the formulation has a pH of 5.2 (.+-.0.1).
63. The method of claim 59, wherein the formulation consists of: (i) Avelumab in a concentration of 20 mg/mL; (ii) acetic acid in a concentration of 10 mM; (iii) trehalose dihydrate in a concentration of 280 mM; (iv) Poloxamer 188 in a concentration of 0.5 mg/mL; (v) sodium acetate to adjust the pH; and (vi) water (for injection) as the diluent; wherein the formulation has a pH of 5.2 (.+-.0.1).
64. The method of claim 56, wherein the formulation consists of: (i) Avelumab in a concentration of 20 mg/mL; (ii) acetic acid in a concentration of 10 mM; (iii) D-mannitol in a concentration of 280 mM; (iv) Polysorbate 20 in a concentration of 0.5 mg/mL; (v) sodium hydroxide in a concentration of 7.5 mM; and (vi) water (for injection) as the diluent; wherein the formulation has a pH of 5.2 (.+-.0.1).
65. The method of claim 64, wherein the formulation is made by combining: (i) 20 mg/mL of Avelumab; (ii) 0.6 mg/mL of glacial acetic acid; (iii) 51 mg/mL of D-mannitol; (iv) 0.5 mg/mL of Polysorbate 20; (v) 0.3 mg/mL of sodium hydroxide; and (vi) water (for injection) as the diluent.
66. The method of claim 44, wherein the formulation consists of: (i) Avelumab in a concentration of 20 mg/mL; (ii) acetic acid in a concentration of 0.6 mg/mL; (iii) D-mannitol in a concentration of 51 mg/mL; (iv) Polysorbate 20 in a concentration of 0.5 mg/mL; (v) sodium hydroxide in a concentration of 0.3 mg/mL; and (vi) water (for injection) as the diluent; wherein the formulation has a pH of 5.0 to 5.6
67. The method of claim 43, wherein the Avelumab has the heavy chain sequence of either (SEQ ID NO:1) or (SEQ ID NO:2), the light chain sequence of (SEQ ID NO:3), and carries a glycosylation on Asn300 comprising FA2 and FA2G1 as the main glycan species, having a joint share of more than 70% of all glycan species.
68. The method of claim 67, wherein in the Avelumab glycosylation the FA2 has a share of 44% to 54% and the FA2G1 has a share of 25% to 41% of all glycan species.
69. The method of claim 68, wherein in the Avelumab glycosylation the FA2 has a share of 47% to 52% and the FA2G1 has a share of 29% to 37% of all glycan species.
70. The method of claim 67, wherein in the Avelumab glycosylation the FA2 has a share of about 49% and the FA2G1 has a share of about 30% to about 35% of all glycan species.
71. The method of claim 67, wherein the Avelumab glycosylation further comprises as minor glycan species A2 with a share of less than 5%, A2G1 with a share of less than 5%, A2G2 with a share of less than 5% and FA2G2 with a share of less than 7% of all glycan species.
72. The method of claim 71, wherein in the Avelumab glycosylation the A2 has a share of 3% to 5%, the A2G1 has a share of less than 4%, the A2G2 has a share of less than 3% and the FA2G2 has a share of 5% to 6% of all glycan species.
73. The method of claim 72, wherein in the Avelumab glycosylation the A2 has a share of about 3.5% to about 4.5%, the A2G1 has a share of about 0.5% to about 3.5%, the A2G2 has a share of less than 2.5% and the FA2G2 has a share of about 5.5% of all glycan species.
74. The method of claim 67, wherein the Avelumab has the heavy chain sequence of (SEQ ID NO:2).
75. The method of claim 43, wherein the formulation is administered by intravenous (IV) administration.
76. The method of claim 43, wherein the cancer is selected from non-small cell lung cancer, urothelial carcinoma, bladder cancer, mesothelioma, Merkel cell carcinoma, gastric or gastroesophageal junction cancer, ovarian cancer, breast cancer, thymoma, adenocarcinoma of the stomach, adrenocortical carcinoma, head and neck squamous cell carcinoma, renal cell carcinoma, melanoma, and/or classical Hodgkin's lymphoma.
77. A method of treating cancer comprising administering an aqueous pharmaceutical antibody formulation to a patient, wherein the formulation comprises: (i) Avelumab in a concentration of about 10 milligram/milliliter (mg/mL) as the antibody; (ii) acetate in a concentration of about 10 millimolar (mM) as the buffering agent; (iii) D-mannitol or trehalose in a concentration of about 280 mM as a stabiliser; and (iv) Polysorbate 20 or Poloxamer 188 in a concentration of about 0.5 mg/mL as surfactant; wherein the formulation does not comprise methionine, and further wherein the formulation has a pH of 5.5 (.+-.0.1).
78. The method of claim 77, wherein the formulation consists of: (i) Avelumab in a concentration of 10 mg/mL; (ii) sodium acetate trihydrate in a concentration of 10 mM; (iii) D-mannitol in a concentration of 280 mM; (iv) Polysorbate 20 in a concentration of 0.5 mg/mL; (v) HCl to adjust the pH; and (vi) water (for injection) as the diluent; wherein the formulation has a pH of 5.5 (.+-.0.1).
79. The method of claim 77, wherein the formulation is administered by intravenous (IV) administration.
80. The method of claim 77, wherein the cancer is selected from non-small cell lung cancer, urothelial carcinoma, bladder cancer, mesothelioma, Merkel cell carcinoma, gastric or gastroesophageal junction cancer, ovarian cancer, breast cancer, thymoma, adenocarcinoma of the stomach, adrenocortical carcinoma, head and neck squamous cell carcinoma, renal cell carcinoma, melanoma, and/or classical Hodgkin's lymphoma.
81. A method of treating cancer comprising administering an aqueous pharmaceutical antibody formulation to a patient, wherein the formulation consists of Avelumab in a concentration of 20 milligram/milliliter (mg/mL) as the active ingredient; and glacial acetic acid, D-mannitol, Polysorbate 20, sodium hydroxide and water for injection as the excipients; wherein the formulation has a pH of 5.0 to 5.6.
82. The method of claim 81, wherein the formulation has a pH of 5.2 (.+-.0.1).
83. The method of claim 81, wherein the formulation is administered by intravenous (IV) administration.
84. The method of claim 81, wherein the cancer is selected from non-small cell lung cancer, urothelial carcinoma, bladder cancer, mesothelioma, Merkel cell carcinoma, gastric or gastroesophageal junction cancer, ovarian cancer, breast cancer, thymoma, adenocarcinoma of the stomach, adrenocortical carcinoma, head and neck squamous cell carcinoma, renal cell carcinoma, melanoma, and/or classical Hodgkin's lymphoma.
Description
[0001] The present invention relates to a novel anti-PD-L1 antibody formulation. In particular, the invention relates to an aqueous pharmaceutical formulation of the anti-PD-L1 antibody Avelumab.
BACKGROUND OF THE INVENTION
[0002] The programmed death 1 (PD-1) receptor and PD-1 ligands 1 and 2 (PD-L1, PD-L2) play integral roles in immune regulation. Expressed on activated T cells, PD-1 is activated by PD-L1 and PD-L2 expressed by stromal cells, tumor cells, or both, initiating T-cell death and localized immune suppression (Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 1999;5:1365-69; Freeman G J, Long A J, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000;192:1027-34; Dong H, Strome S E, Salomao D R, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002; 8:793-800. [Erratum, Nat Med 2002;8:1039; Topalian S L, Drake C G, Pardoll D M. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol 2012;24:207-12), potentially providing an immune-tolerant environment for tumor development and growth. Conversely, inhibition of this interaction can enhance local T-cell responses and mediate antitumor activity in nonclinical animal models (Dong H, Strome S E, Salomao D R, et al. Nat Med 2002; 8:793-800. [Erratum, Nat Med 2002;8:1039; Iwai Y, Ishida M, Tanaka Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA 2002;99:12293-97). In the clinical setting, treatment with antibodies that block the PD-1-PD-L1 interaction have been reported to produce objective response rates of 7% to 38% in patients with advanced or metastatic solid tumors, with tolerable safety profiles (Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (Anti-PD-1) in melanoma. N Engl J Med 2013;369:134-44; Brahmer J R, Tykodi S S, Chow L Q, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366(26):2455-65; Topalian S L, Hodi F S, Brahmer J R, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366(26):2443-54; Herbst R S, Soria J-C, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563-67). Notably, responses appeared prolonged, with durations of 1 year or more for the majority of patients.
[0003] Avelumab (also known as MSB0010718C) is a fully human monoclonal antibody of the immunoglobulin (Ig) G1 isotype. Avelumab selectively binds to PD-L1 and competitively blocks its interaction with PD-1.
[0004] Compared with anti-PD-1 antibodies that target T-cells, Avelumab targets tumor cells, and therefore is expected to have fewer side effects, including a lower risk of autoimmune-related safety issues, as blockade of PD-L1 leaves the PD-L2-PD-1 pathway intact to promote peripheral self-tolerance (Latchman Y, Wood C R, Chernova T, et al. PD-L1 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2001;2(3):261-68).
[0005] Avelumab is currently being tested in the clinic in a number of cancer types including non-small cell lung cancer, urothelial carcinoma, mesothelioma, Merkel cell carcinoma, gastric or gastroesophageal junction cancer, ovarian cancer, and breast cancer.
[0006] The amino acid sequences of Avelumab and sequence variants and antigen binding fragments thereof, are disclosed in WO2013079174, where the antibody having the amino acid sequence of Avelumab is referred to as A09-246-2. Also disclosed are methods of manufacturing and certain medical uses.
[0007] Further medical uses of Avelumab are described in WO2016137985, PCT/IB2016/052748, PCT/US2016/037498, PCT/US2016/053939, U.S. patent application Ser. No. 62/341,921.
[0008] WO2013079174 also describes in section 2.4 a human aqueous formulation of an antibody having the amino acid sequence of Avelumab. This formulation comprises the antibody in a concentration of 10 mg/ml, methionine as an antioxidant and has a pH of 5.5.
[0009] A formulation study for an aglycosylated anti-PD-L1 antibody of the IgG1 type is described in WO2015048520, where a formulation with a pH of 5.8 was selected for clinical studies.
DESCRIPTION OF THE INVENTION
[0010] As Avelumab is generally delivered to a patient via intravenous infusion, and is thus provided in an aqueous form, the present invention relates to further aqueous formulations that are suitable to stabilize Avelumab with its post-translational modifications, and at higher concentrations as disclosed in WO2013079174.
[0011] FIG. 1a (SEQ ID NO:1) shows the full length heavy chain sequence of Avelumab, as expressed by the CHO cells used as the host organism.
[0012] It is frequently observed, however, that in the course of antibody production the C-terminal lysine (K) of the heavy chain is cleaved off. Located in the Fc part, this modification has no influence on the antibody-antigen binding. Therefore, in some embodiments the C-terminal lysine (K) of the heavy chain sequence of Avelumab is absent. The heavy chain sequence of Avelumab without the C-terminal lysine is shown in FIG. 1b (SEQ ID NO:2).
[0013] FIG. 2 (SEQ ID NO:3) shows the full length light chain sequence of Avelumab.
[0014] A post-translational modification of high relevance is glycosylation.
[0015] Most of the soluble and membrane-bound proteins that are made in the endoplasmatic reticulum of eukaryotic cells undergo glycosylation, where enzymes called glycosyltransferases attach one or more sugar units to specific glycosylation sites of the proteins. Most frequently, the points of attachment are NH.sub.2 or OH groups, leading to N-linked or O-linked glycosylation.
[0016] This applies also to proteins, such as antibodies, which are recombinantly produced in eukaryotic host cells. Recombinant IgG antibodies contain a conserved N-linked glycosylation site at a certain asparagine residue of the Fc region in the CH2 domain. There are many known physical functions of N-linked glycosylation in an antibody such as affecting its solubility and stability, protease resistance, binding to Fc receptors, cellular transport and circulatory half-life in vivo (Hamm M. et al., Pharmaceuticals 2013, 6, 393-406). IgG antibody N-glycan structures are predominantly biantennary complex-type structures, comprising b-D-N-acetylglucosamine (GlcNac), mannose (Man) and frequently galactose (Gal) and fucose (Fuc) units.
[0017] In Avelumab the single glycosylation site is Asn300, located in the CH2 domain of both heavy chains. Details of the glycosylation are described in Example 1.
[0018] Since glycosylation affects the solubility and stability of an antibody, it is prudent to take this parameter into account when a stable, pharmaceutically suitable formulation of the antibody is to be developed.
[0019] Surprisingly, it has been found by the inventors of the present patent application that it is possible to stabilize Avelumab, fully characterized by its amino acid sequence and its post-translational modifications, in a number of aqueous formulations without the presence of an antioxidant, at pH values as low as 5.2.
FIGURES
[0020] FIG. 1a: Heavy chain sequence of Avelumab (SEQ ID NO:1)
[0021] FIG. 1b: Heavy chain sequence of Avelumab, lacking the C-terminal K (SEQ ID NO:2)
[0022] FIG. 2: Light chain sequence of Avelumab (SEQ ID NO:3)
[0023] FIG. 3: Secondary structure of Avelumab
[0024] FIG. 4: 2AB HILIC-UPLC Chromatogram of Avelumab Glycans
[0025] FIG. 5: Numbering of the peaks of FIG. 4
[0026] FIG. 6: Total aggregates by SE-HPLC of DoE2 formulations (40.degree. C.)
[0027] FIG. 7: Total aggregates by SE-HPLC of DoE2 formulations (25.degree. C.)
[0028] FIG. 8: Fragments by Bioanalyzer of DoE2 formulations (40.degree. C.)
[0029] FIG. 9: Fragments by Bioanalyzer of DoE2 formulations (25.degree. C.)
[0030] FIG. 10: Acidic cluster and main peak abundance of DoE2 (25.degree. C.)
[0031] FIG. 11: Long Term Stability LMWs (%) at 2-8.degree. C.
[0032] FIG. 12: Long Term Stability Sub-visible particles .gtoreq.10 .mu.m at 2-8.degree. C.
[0033] FIG. 13: Long Term Stability Sub-visible particles .gtoreq.25 .mu.m at 2-8.degree. C.
[0034] FIG. 14: Long Term Stability Acidic cluster (%) at 2-8.degree. C.
[0035] FIG. 15: Long Term Stability Main peak (%) at 2-8.degree. C.
[0036] FIG. 16: Long Term Stability Basic cluster (%) at 2-8.degree. C.
[0037] FIG. 17: Long Term Stability LMWs (%) at 25.degree. C.
[0038] FIG. 18: Long Term Stability Sub-visible particles .gtoreq.10 .mu.m at 25.degree. C.
[0039] FIG. 19: Long Term Stability Sub-visible particles .gtoreq.25 .mu.m at 25 .degree. C.
[0040] FIG. 20: Long Term Stability Acidic cluster (%) at 25.degree. C.
[0041] FIG. 21: Long Term Stability Main peak (%) at 25.degree. C.
[0042] FIG. 22: Long Term Stability Basic cluster (%) at 25.degree. C.
[0043] FIG. 23: Long Term Stability LMWs (%) at 40.degree. C.
[0044] FIG. 24: Long Term Stability Sub-visible particles .gtoreq.10 .mu.m at 40 .degree. C.
[0045] FIG. 25: Long Term Stability Sub-visible particles .gtoreq.25 .mu.m at 40.degree. C.
[0046] FIG. 26: Long Term Stability Acidic cluster (%) at 40.degree. C.
[0047] FIG. 27: Long Term Stability Main peak (%) at 40.degree. C.
[0048] FIG. 28: Long Term Stability Basic cluster (%) at 40.degree. C.
DEFINITIONS
[0049] Unless otherwise stated, the following terms used in the specification and claims have the following meanings set out below.
[0050] References herein to "Avelumab" include the anti-PD-L1 antibody of the IgG1 type as defined in WO2013079174 by its amino acid sequence, and as defined in the present patent application by its amino acid sequence and by its post-translational modifications. References herein to "Avelumab" may include biosimilars which, for instance, may share at least 75%, suitably at least 80%, suitably at least 85%, suitably at least 90%, suitably at least 95%, suitably at least 96%, suitably at least 97%, suitably at least 98% or most suitably at least 99% amino acid sequence identity with the amino acid sequences disclosed in WO2013079174. Alternatively or additionally, references herein to "Avelumab" may include biosimilars which differ in the post-translational modifications, especially in the glycosylation pattern, herein disclosed.
[0051] The term "biosimilar" (also known as follow-on biologics) is well known in the art, and the skilled person would readily appreciate when a drug substance would be considered a biosimilar of Avelumab. The term "biosimilar" is generally used to. describe subsequent versions (generally from a different source) of "innovator biopharmaceutical products" ("biologics" whose drug substance is made by a living organism or derived from a living organism or through recombinant DNA or controlled gene expression methodologies) that have been previously officially granted marketing authorisation. Since biologics have a high degree of molecular complexity, and are generally sensitive to changes in manufacturing processes (e.g. if different cell lines are used in their production), and since subsequent follow-on manufacturers generally do not have access to the originator's molecular clone, cell bank, know-how regarding the fermentation and purification process, nor to the active drug substance itself (only the innovator's commercialized drug product), any "biosimilar" is unlikely to be exactly the same as the innovator drug product. Herein, the term "buffer" or "buffer solution" refers to a generally aqueous solution comprising a mixture of an acid (usually a weak acid, e.g. acetic acid, citric acid, imidazolium form of histidine) and its conjugate base (e.g. an acetate or citrate salt, for example, sodium acetate, sodium citrate, or histidine) or alternatively a mixture of a base (usually a weak base, e.g. histidine) and its conjugate acid (e.g. protonated histidine salt). The pH of a "buffer solution" will change very only slightly upon addition of a small quantity of strong acid or base due to the "buffering effect" imparted by the "buffering agent".
[0052] Herein, a "buffer system" comprises one or more buffering agent(s) and/or an acid/base conjugate(s) thereof, and more suitably comprises one or more buffering agent(s) and an acid/base conjugate(s) thereof, and most suitably comprises one buffering agent only and an acid/base conjugate thereof. Unless stated otherwise, any concentrations stipulated herein in relation to a "buffer system" (i.e. a buffer concentration) suitably refers to the combined concentration of the buffering agent(s) and/or acid/base conjugate(s) thereof. In other words, concentrations stipulated herein in relation to a "buffer system" suitably refer to the combined concentration of all the relevant buffering species (i.e. the species in dynamic equilibrium with one another, e.g. citrate/citric acid). As such, a given concentration of a histidine buffer system generally relates to the combined concentration of histidine and the imidazolium form of histidine. However, in the case of histidine, such concentrations are usually straightforward to calculate by reference to the input quantities of histidine or a salt thereof. The overall pH of the composition comprising the relevant buffer system is generally a reflection of the equilibrium concentration of each of the relevant buffering species (i.e. the balance of buffering agent(s) to acid/base conjugate(s) thereof).
[0053] Herein, the term "buffering agent" refers to an acid or base component (usually a weak acid or weak base) of a buffer or buffer solution. A buffering agent helps maintain the pH of a given solution at or near to a pre-determined value, and the buffering agents are generally chosen to complement the pre-determined value. A buffering agent is suitably a single compound which gives rise to a desired buffering effect, especially when said buffering agent is mixed with (and suitably capable of proton exchange with) an appropriate amount (depending on the pre-determined pH desired) of its corresponding "acid/base conjugate", or if the required amount of its corresponding "acid/base conjugate" is formed in situ--this may be achieved by adding strong acid or base until the required pH is reached. For example in the sodium acetate buffer system, it is possible to start out with a solution of sodium acetate (basic) which is then acidified with, e.g., hydrochloric acid, or to a solution of acetic acid (acidic), sodium hydroxide or sodium acetate is added until the desired pH is reached.
[0054] Generally, a "stabiliser" refers to a component which facilitates maintenance of the structural integrity of the biopharmaceutical drug, particularly during freezing and/or lyophilization and/or storage (especially when exposed to stress). This stabilising effect may arise for a variety of reasons, though typically such stabilisers may act as osmolytes which mitigate against protein denaturation. As used herein, stabilisers are amino acids (i.e. free amino acids not part of a peptide or protein--e.g. glycine, arginine, histidine, aspartic acid, lysine) and sugar stabilisers, such as a sugar polyol (e.g. mannitol, sorbitol), and/or a disaccharide (e.g. trehalose, sucrose, maltose, lactose).
[0055] Agents used as buffering agents, antioxidants or surfactants according to the invention, are excluded from the meaning of the term "stabilisers" as used herein, even if they may exhibit, i.a. stabilising activity.
[0056] Herein, the term "surfactant" refers to a surface-active agent, preferably a nonionic surfactant. Examples of surfactants used herein include polysorbate (for example, polysorbate 20 (polyoxyethylene (20) sorbitan monolaurate) also known under the tradename Tween 20); poloxamer (e.g. poloxamer 188, a non-ionic triblock copolymers composed of a central hydrophobic chain of polyoxypropylene (poly(propylene oxide)) flanked by two hydrophilic chains of polyoxyethylene (poly(ethylene oxide)), also known under the tradename Lutrol F 68).
[0057] Herein, the term "stable" generally refers to the physical stability and/or chemical stability and/or biological stability of a component, typically an active or composition thereof, during preservation/storage.
[0058] Agents used as buffering agents, antioxidants or stabilisers according to the invention, are excluded from the meaning of the term "surfactants" as used herein, even if they may exhibit, i.a. surfactant activity.
[0059] Herein, the term "antioxidant" refers to an agent capable of preventing or decreasing oxidation of the biopharmaceutical drug to be stabilized in the formulation.
[0060] Antioxidants include radical scavengers (e.g. ascorbic acid, BHT, sodium sulfite, p-amino benzoic acid, glutathione or propyl gallate), chelating agents (e.g. EDTA or citric acid) or chain terminators (e.g. methionine or N-acetyl cysteine).
[0061] Agents used as buffering agents, stabilisers or surfactants according to the invention, are excluded from the meaning of the term "antioxidants" as used herein, even if they may exhibit, i.a. antioxidative activity.
[0062] A "diluent" is an agent that constitutes the balance of ingredients in any liquid pharmaceutical composition, for instance so that the weight percentages total 100%. Herein, the liquid pharmaceutical composition is an aqueous pharmaceutical composition, so that a "diluent" as used herein is water, preferably water for injection (WFI).
[0063] Herein, the term "particle size" or "pore size" refers respectively to the length of the longest dimension of a given particle or pore. Both sizes may be measured using a laser particle size analyser and/or electron microscopes (e.g. tunneling electron microscope, TEM, or scanning electron microscope, SEM). The particle count (for any given size) can be obtained using the protocols and equipment outlined in the Examples, which relates to the particle count of sub-visible particles.
[0064] Herein, the term "about" refers to the usual error range for the respective value readily known to the skilled person in this technical field. Reference to "about" a value or parameter herein includes (and describes) embodiments that are directed to that value or parameter per se. In case of doubt, or should there be no art recognized common understanding regarding the error range for a certain value or parameter, "about" means .+-.5% of this value or parameter.
[0065] Herein, the term "percent share" in connection with glycan species refers directly to the number of different species. For example the term "said FA2G1 has a share of 25%-41% of all glycan species" means that in 50 antibody molecules analysed, having 100 heavy chains, 25-41 of the heavy chains will exhibit the FA2G1 glycosylation pattern.
[0066] It is to be appreciated that references to "treating" or "treatment" include prophylaxis as well as the alleviation of established symptoms of a condition. "Treating" or "treatment" of a state, disorder or condition therefore includes: (1) preventing or delaying the appearance of clinical symptoms of the state, disorder or condition developing in a human that may be afflicted with or predisposed to the state, disorder or condition but does not yet experience or display clinical or subclinical symptoms of the state, disorder or condition, (2) inhibiting the state, disorder or condition, i.e., arresting, reducing or delaying the development of the disease or a relapse thereof (in case of maintenance treatment) or at least one clinical or subclinical symptom thereof, or (3) relieving or attenuating the disease, i.e., causing regression of the state, disorder or condition or at least one of its clinical or subclinical symptoms.
[0067] Aqueous anti-PD-L1 Antibody Formulation
[0068] In a first aspect, the invention provides a novel aqueous pharmaceutical antibody formulation, comprising:
[0069] (i) Avelumab in a concentration of 1 mg/mL to 30 mg/mL as the antibody;
[0070] (ii) acetate or histidine in a concentration of 5 mM to 15 mM as the buffering agent;
[0071] (iii) D-mannitol or trehalose in a concentration of 240 mM to 320 mM, or a combination of arginine HCl in a concentration of 50 to 150 mM and glutamic acid in a concentration of 25 mM to 75 mM as a stabiliser;
[0072] (iv) Poloxamer 188 or Polysorbate 20 in a concentration of 0.25 mg/mL to 0.75 mg/mL, as surfactant, or no surfactant;
[0073] wherein the formulation does not comprise methionine, and
[0074] further wherein the formulation has a pH of 5.0 to 6.0, preferably, 5.0 to 5.6.
[0075] In a preferred embodiment the formulation does not comprise any antioxidant.
[0076] In an embodiment the concentration of Avelumab in the said formulation is about 10 mg/mL to about 20 mg/mL.
[0077] In another embodiment the concentration of acetate or histidine in the said formulation is about 10 mM.
[0078] In yet another embodiment the concentration of D-mannitol or trehalose in the said formulation is about 280 mM, or for the combination of arginine HCl and glutamic acid, the concentration of arginine HCl is about 150 mM and the concentration of glutamic acid is about 50 mM.
[0079] In yet another embodiment the concentration of Poloxamer 188 or Polysorbate 20 in the said formulation is about 0.5 mg/mL.
[0080] In yet another embodiment the pH of the said formulation is 5.2 (.+-.0.1) to 5.5 (.+-.0.1).
[0081] In a preferred embodiment the said formulation comprises acetate in a concentration of about 10 mM, and does not comprise any other buffering agent.
[0082] In another preferred embodiment the said formulation comprises D-mannitol or trehalose in a concentration of about 280 mM, and does not comprise any other stabiliser.
[0083] In yet another preferred embodiment the said formulation comprises Polysorbate 20 or Poloxamer 188 in a concentration of about 0.5 mg/mL, and does not comprise any other surfactant.
[0084] In an embodiment the said formulation comprises:
[0085] (i) Avelumab in a concentration of about 10 mg/mL as the antibody;
[0086] (ii) acetate in a concentration of about 10 mM as the buffering agent;
[0087] (iii) D-mannitol or trehalose in a concentration of about 280 mM as a stabiliser;
[0088] (iv) Polysorbate 20 or Poloxamer 188 in a concentration of about 0.5 mg/mL as surfactant;
[0089] and does not comprise methionine, and has a pH of about 5.5.
[0090] In a preferred embodiment the said formulation comprises:
[0091] (i) Avelumab in a concentration of 10 mg/mL;
[0092] (ii) acetate in a concentration of 10 mM;
[0093] (iii) D-mannitol or trehalose in a concentration of 280 mM;
[0094] (iv) Polysorbate 20 or Poloxamer 188 in a concentration of 0.5 mg/mL;
[0095] and has a pH of 5.5 (.+-.0.1).
[0096] In a preferred embodiment the said formulation consists of:
[0097] (i) Avelumab in a concentration of 10 mg/mL;
[0098] (ii) sodium acetate trihydrate in a concentration of 10 mM;
[0099] (iii) D-mannitol or trehalose in a concentration of 280 mM;
[0100] (iv) Polysorbate 20 or Poloxamer 188 in a concentration of 0.5 mg/mL;
[0101] (v) HCl to adjust the pH;
[0102] (vi) water (for injection) as the solvent;
[0103] and has a pH of 5.5 (.+-.0.1).
[0104] In a preferred embodiment the said formulation consists of:
[0105] (i) Avelumab in a concentration of 10 mg/mL;
[0106] (ii) sodium acetate trihydrate in a concentration of 10 mM;
[0107] (iii) trehalose dihydrate in a concentration of 280 mM;
[0108] (iv) Polysorbate 20 in a concentration of 0.5 mg/mL;
[0109] (v) HCl to adjust the pH;
[0110] (vi) water (for injection) as the diluent;
[0111] and has a pH of 5.5 (.+-.0.1).
[0112] In a more preferred embodiment the said formulation consists of:
[0113] (i) Avelumab in a concentration of 10 mg/mL;
[0114] (ii) sodium acetate trihydrate in a concentration of 10 mM;
[0115] (iii) D-mannitol in a concentration of 280 mM;
[0116] (iv) Polysorbate 20 in a concentration of 0.5 mg/mL;
[0117] (v) HCl to adjust the pH;
[0118] (vi) water (for injection) as the diluent;
[0119] and has a pH of 5.5 (.+-.0.1).
[0120] In another embodiment the said formulation comprises:
[0121] (i) Avelumab in a concentration of about 20 mg/mL as the antibody;
[0122] (ii) acetate in a concentration of about 10 mM as the buffering agent;
[0123] (iii) D-mannitol or trehalose in a concentration of about 280 mM as a stabiliser;
[0124] (iv) Polysorbate 20 or Poloxamer 188 in a concentration of about 0.5 mg/mL as surfactant;
[0125] and does not comprise methionine, and has a pH of 5.2 (.+-.0.1).
[0126] In a preferred embodiment the said formulation comprises:
[0127] (i) Avelumab in a concentration of 20 mg/mL;
[0128] (ii) acetate in a concentration of 10 mM;
[0129] (iii) D-mannitol or trehalose in a concentration of 280 mM;
[0130] (iv) Polysorbate 20 or Poloxamer 188 in a concentration of 0.5 mg/mL;
[0131] and has a pH of 5.5 (.+-.0.1).
[0132] In a preferred embodiment the said formulation comprises:
[0133] (i) Avelumab in a concentration of 20 mg/mL;
[0134] (ii) acetic acid in a concentration of 10 mM;
[0135] (iii) D-mannitol or trehalose dihydrate in a concentration of 280 mM;
[0136] (iv) Polysorbate 20 or Poloxamer 188 in a concentration of 0.5 mg/mL;
[0137] (v) sodium acetate to adjust the pH;
[0138] (vi) water (for injection) as the diluent;
[0139] and has a pH of 5.2 (.+-.0.1).
[0140] In a more preferred embodiment the said formulation consists of:
[0141] (i) Avelumab in a concentration of 20 mg/mL;
[0142] (ii) acetic acid in a concentration of 10 mM;
[0143] (iii) D-mannitol in a concentration of 280 mM;
[0144] (iv) Polysorbate 20 in a concentration of 0.5 mg/mL;
[0145] (v) sodium acetate to adjust the pH;
[0146] (vi) water (for injection) as the diluent;
[0147] and has a pH of 5.2 (.+-.0.1).
[0148] In a more preferred embodiment the said formulation consists of:
[0149] (i) Avelumab in a concentration of 20 mg/mL;
[0150] (ii) acetic acid in a concentration of 10 mM;
[0151] (iii) trehalose dihydrate in a concentration of 280 mM;
[0152] (iv) Polysorbate 20 in a concentration of 0.5 mg/mL;
[0153] (v) sodium acetate to adjust the pH;
[0154] (vi) water (for injection) as the diluent;
[0155] and has a pH of 5.2 (.+-.0.1).
[0156] In a more preferred embodiment the said formulation consists of:
[0157] (i) Avelumab in a concentration of 20 mg/mL;
[0158] (ii) acetic acid in a concentration of 10 mM;
[0159] (iii) D-mannitol in a concentration of 280 mM;
[0160] (iv) Poloxamer 188 in a concentration of 0.5 mg/mL;
[0161] (v) sodium acetate to adjust the pH;
[0162] (vi) water (for injection) as the diluent;
[0163] and has a pH of 5.2 (.+-.0.1).
[0164] In a more preferred embodiment the said formulation consists of:
[0165] (i) Avelumab in a concentration of 20 mg/mL;
[0166] (ii) acetic acid in a concentration of 10 mM;
[0167] (iii) trehalose dihydrate in a concentration of 280 mM;
[0168] (iv) Poloxamer 188 in a concentration of 0.5 mg/mL;
[0169] (v) sodium acetate to adjust the pH;
[0170] (vi) water (for injection) as the diluent;
[0171] and has a pH of 5.2 (.+-.0.1).
[0172] In a preferred embodiment, the said formulation consists of:
[0173] (i) Avelumab in a concentration of 20 mg/mL;
[0174] (ii) acetic acid in a concentration of 10 mM (0.6 mg/mL);
[0175] (iii) D-mannitol in a concentration of 280 mM (51 mg/mL);
[0176] (iv) Polysorbate 20 in a concentration of 0.5 mg/mL;
[0177] (v) sodium hydroxide in a concentration of 7.5 mM (0.3 mg/mL);
[0178] (vi) water (for injection) as the diluent;
[0179] and has a pH of 5.0 to 5.6, preferably 5.2 (.+-.0.1).
[0180] In a preferred embodiment, the latter formulation is made by combining:
[0181] (i) 20 mg/mL of Avelumab;
[0182] (ii) 0.6 mg/mL of glacial acetic acid;
[0183] (iii) 51 mg/mL of D-mannitol;
[0184] (iv) 0.5 mg/mL of Polysorbate 20;
[0185] (v) 0.3 mg/mL of sodium hydroxide;
[0186] (vi) water (for injection) as the diluent;
[0187] to obtain the desired volume of the formulation.
[0188] In a further embodiment, the invention concerns an aqueous pharmaceutical antibody formulation, whose pH is adjusted with sodium hydroxide. Therefore, the formulation consists of Avelumab in a concentration of 20 mg/mL as the active ingredient; and glacial acetic acid, D-mannitol, Polysorbate 20, sodium hydroxide and water for injection as the excipients; wherein the formulation has a pH of 5.0 to 5.6, preferably 5.2 (.+-.0.1).
[0189] In a preferred embodiment, the formulation has a osmolality between 270 and 330 mOsm/kg.
[0190] In an embodiment said Avelumab in the formulations as described above has the heavy chain sequence of either FIG. 1a (SEQ ID NO:1) or FIG. 1b (SEQ ID NO:2), the light chain sequence of FIG. 2 (SEQ ID NO:3), and carries a glycosylation on Asn300 comprising FA2 and FA2G1 as the main glycan species, having a joint share of >70% of all glycan species.
[0191] In a preferred embodiment, in the Avelumab glycosylation the said FA2 has a share of 44%-54% and said FA2G1 has a share of 25%-41% of all glycan species.
[0192] In a preferred embodiment, in the Avelumab glycosylation the said FA2 has a share of 47%-52% and said FA2G1 has a share of 29%-37% of all glycan species.
[0193] In a preferred embodiment, in the Avelumab glycosylation the said FA2 has a share of about 49% and said FA2G1 has a share of about 30%-about 35% of all glycan species.
[0194] In a preferred embodiment the Avelumab glycosylation further comprises as minor glycan species A2 with a share of <5%, A2G1 with a share of <5%, A2G2 with a share of <5% and FA2G2 with a share of <7% of all glycan species.
[0195] In a preferred embodiment, in the Avelumab glycosylation said A2 has a share of 3%-5%, said A2G1 has a share of <4%, said A2G2 has a share of <3% and said FA2G2 has a share of 5%-6% of all glycan species.
[0196] In a preferred embodiment, in the Avelumab glycosylation said A2 has a share of about 3.5%-about 4.5%, said A2G1 has a share of about 0.5%-about 3.5%, said A2G2 has a share of <2.5% and said FA2G2 has a share of about 5.5% of all glycan species.
[0197] In an embodiment the said Avelumab in the formulation as described above has the heavy chain sequence of FIG. 1b (SEQ ID NO:2).
[0198] In an embodiment the Avelumab formulation as described above is for intravenous (IV) administration.
[0199] Drug-delivery Device
[0200] In a second aspect the present invention provides a drug delivery device comprising a liquid pharmaceutical composition as defined herein. Suitably the drug delivery device comprises a chamber within which the pharmaceutical composition resides. Suitably the drug delivery device is sterile.
[0201] The drug delivery device may a vial, ampoule, syringe, injection pen (e.g. essentially incorporating a syringe), or i.v. (intravenous) bag.
[0202] The aqueous pharmaceutical formulations are parenterally administered, preferably via sub-cutaneous injection, intramuscular injection, i.v. injection or i.v. infusion. The most preferred way of administration is i.v. infusion.
[0203] In a preferred embodiment, the drug delivery device is a vial containing the formulation as described above.
[0204] In a more preferred embodiment the said vial contains 200 mg avelumab in 10 mL of solution for a concentration of 20 mg/mL.
[0205] In an even more preferred embodiment the vial is a glass vial.
[0206] Medical Treatment
[0207] In a third aspect, the invention provides a method of treating cancer comprising administering the formulation as described above to a patient.
[0208] In an embodiment the cancer to be treated is selected from non-small cell lung cancer, urothelial carcinoma, bladder cancer, mesothelioma, Merkel cell carcinoma, gastric or gastroesophageal junction cancer, ovarian cancer, breast cancer, thymoma, adenocarcinoma of the stomach, adrenocortical carcinoma, head and neck squamous cell carcinoma, renal cell carcinoma, melanoma, and/or classical Hodgkin's lymphoma.
ABBREVIATIONS
[0209] ANOVA Analysis of variance
[0210] CD Circular Dichroism
[0211] CE Capillary Electrophoresis
[0212] DoE Design of Experiments
[0213] DP Drug Product
[0214] DS Drug Substance
[0215] DSF Differential Scanning Fluorimetry
[0216] DTT Dithiothreitol
[0217] ESI Electrospray Ionization
[0218] HILIC Hydrophilic Interaction Liquid Chromatography
[0219] HMWs Higher Molecular Weights
[0220] HPLC High Performance Liquid Chromatography
[0221] iCE Capillary Isoelectric Focusing
[0222] LC Liquid Chromatography
[0223] LMWs Lower Molecular Weights
[0224] MALDI Matrix-Assisted Laser Desorption Ionization
[0225] MS Mass Spectrometry
[0226] NTU Nephelometry Turbidity Units
[0227] OD Optical density
[0228] PBS Poly Buffer Saline
[0229] PES Polyethersulfone
[0230] PVDF Polyvinylidene Fluoride
[0231] SDS-PAGE Sodium Dodecyl Sulphate-PolyAcrylamide Gel Electrophoresis
[0232] SE Size Exclusion
[0233] TOF Time of Flight
[0234] UPLC Ultra Performance Liquid Chromatography
[0235] RH Residual Humidity
[0236] UV Ultraviolet
EXAMPLES
[0237] Methods Used to Determine Stability
[0238] In order to assess the stability of the antibody formulations tested, and select the best candidates, thermal stress, mechanical stress, light exposure, osmolality, turbidity, protein content, total aggregates, fragmentation, pH, isoforms, circular dichroism, sub-visible particles and biological activity were determined as stability parameters according to the following protocols:
[0239] Thermal Stress:
[0240] At 40.degree. C.: the samples in the original vial container were incubated in a thermostatic cabinet at a temperature of 40.degree. C..+-.2.degree. C. (RH 75%.+-.5%) and withdrawn at pre-determined time points.
[0241] At 25.degree. C.: the samples in the original vial container were incubated in a thermostatic cabinet at a temperature of 25.degree. C..+-.2.degree. C. (RH 60%.+-.5%) and withdrawn at pre-determined time points.
[0242] Mechanical Stress:
[0243] The samples in the original vial container were placed on an orbital shaker maintained at 300 rpm for up to 24 hours (room temperature).
[0244] Light Exposure:
[0245] The samples in the original vial container were exposed to a light source for 7 hours adjusting the irradiance level in the Suntest machine to 765 W/m.sup.2 (radiation wavelength between 320 nm and 800 nm).
[0246] Osmolality:
[0247] Normal human plasma has an osmolality of about 280 mOSm/kg (Medical. Physiology--Principles for Clinical Medicine. Edited by Rodney A. Rhoades PhD, David R. Bell PhD). In general, solutions with osmolality close to 300 mOsm/kg are to be targeted when developing parenteral formulations. Acceptable ranges (as per product specifications) are 250-400 mOsm/kg.
[0248] Here, osmolality was determined by a cryoscopic method determining the freezing point depression of water solutions after addition of solutes. Amount of solutes, and hence the observed osmolality value is proportional to the observed freezing point depression of the compounded solution.
[0249] Turbidity:
[0250] The turbidity of the solutions were determined with a nephelometer with the capability to measure scattered or attenuated light (Hach Lange Model 2100AN). About 3 mL of solution in reduced volume cuvettes were illuminated by an 870.+-.30 nm light emitting diode (LED) assembly. A detector monitors the scattered light and provided the turbidity (NTU) of the solution by comparison with a series of standards of known turbidity.
[0251] Protein Content:
[0252] Protein content was determined via the optical density of solutions (diluted to .about.0.5 mg/mL protein concentration with relevant buffer) at 280 nm and 320 nm in 1 cm path length quartz cuvettes. Assuming a molar extinction coefficient of 1.46 cm.sup.2/mg, protein concentration was obtained by applying the formula: (A280-A320)/(1.46 cm.sup.2/mg.times.1 cm).
[0253] Total Aggregates:
[0254] The amount of aggregates was determined by the SE-HPLC method. A sample volume of 20 .mu.L (sample diluted to about 0.5 mL with PBS) was injected in a TSK gel Super SW3000 4.6 mm.times.30 cm (cod. 18675) kept at a temperature of 22.+-.5.degree. C. at a flow rate of 0.35 mL/min (mobile phase was 50 mM sodium phosphate+0.4 sodium perchlorate at pH 6.3.+-.0.1). UV detection at 214 nm.
[0255] Fragmentation:
[0256] Low molecular species (or fragments) were determined by Bioanalyzer. Samples are analyzed at a concentration ranging between 1.25-3.75 mg/mL (dilutions made with purified water). 34 of each diluted sample were merged with 2 .mu.L of the corresponding sample buffer (with the addition of DTT when tests were conducted under reducing conditions) and 1 .mu.L of a 60 mM maleimide solution. The samples were heated for 5 minutes at 70.degree. C., then 84 .mu.L of purified water were added and the solutions vortexed and spun down. 6 .mu.L were loaded onto the chip (0.25-0.75 .mu.g of protein). The chip was placed into the Agilent 2100 Bioanalyzer and the analysis started within the following five minutes.
[0257] Isoforms:
[0258] Isoforms distribution was determined by iCE. An Fc coated capillary cartridge (100 mm internal diameter and 50 mm length) was used. The separation is conducted using a 100 mM NaOH solution in 0.1% methylcellulose as a cathodic solution and a 80 mM o-phosphoric acid in 0.1% methylcellulose as an anodic solution. The samples were prepared starting from 80 .mu.L of master mix solution (obtained mixing 700 .mu.L of 0.1% methylcellulose, 10 .mu.L of Pharmalyte 5-8, 70 .mu.L of Pharmalyte 8-10.5, 10 .mu.L of a 7.65 pl marker and 104 of a 9.77 pl marker), to which the suitable volume of washed Avelumab sample (corresponding to 200 .mu.g of protein after washing to remove formulation components) was added. An amount of purified water corresponding to (120 .mu.L--volume of washed Avelumab sample added at the previous step) is added. The separation is conducted at a detection wavelength of 280 nm setting pre-focusing and focusing times of 1 and 15 minutes respectively and pre-focusing and focusing voltages of 1500 V and 3000 V respectively. Samples were injected at a pressure of 1000 mBar.
[0259] pH: was determined by conventional potentiometry.
[0260] Circular Dichroism (CD):
[0261] Investigations on tertiary structure of Avelumab were carried out using a CD spectropolarimeter by Jasco (mod. J810) in the near UV range (320-250 nm). Samples were diluted to 1.5 mg/mL protein concentration with purified water and, once filled in 1 cm-path length quartz cuvettes, analyzed at room temperature, at a scanning speed of 20 nm/min, with a data pitch of 0.5 nm, integration time of 8 s and standard sensitivity.
[0262] Sub-visible Particles:
[0263] Sub-visible particles were counted through the technique of light obscuration method using a Pamas SVSS-C particle counter. Samples were diluted 5-fold with purified water to obtain a final volume of at least 25 mL to be tested.
[0264] Biological Activity:
[0265] For the long term stability studies described in Example 5 biological activity was measured as an additional stability parameter.
[0266] The method used is based on the ability of Avelumab, absorbed on an ELISA plate, to bind in a dose-dependent manner its antigen PD-L1 present on the cell line HEK-293 (hPDL1, permanently transfected with PD-L1). Dosages used were 400, 200, 100, 50, 25, 12.5, 6.25 and 3.12 ng/mL. From the data obtained EC.sub.50 values were calculated. The biological activity (potency) of the samples is expressed as the percentage of bioactivity of the sample against the standard and is calculated as follows: Potency (sample) [%]=(EC.sub.50 (sample)/EC.sub.50 (standard))*100.7.
[0267] Methods of Manufacturing
[0268] The present invention also provides a method of manufacturing an aqueous pharmaceutical formulation as defined herein. The method suitably comprises mixing together, in any particular order deemed appropriate, any relevant components required to form the aqueous pharmaceutical formulation. The skilled person may refer to the examples or techniques well known in the art for forming aqueous pharmaceutical formulations (especially those for injection via syringe, or i.v. infusion).
[0269] The method may involve first preparing a pre-mixture (or pre-solution) of some or all components (optionally with some or all of the diluent) excluding Avelumab, and Avelumab may then itself (optionally with or pre-dissolved in some of the diluent) be mixed with the pre-mixture (or pre-solution) to afford the aqueous pharmaceutical formulation, or a composition to which final components are then added to furnish the final aqueous pharmaceutical formulation. Preferably, the method involves forming a buffer system, suitably a buffer system comprising a buffering agent as defined herein. The buffer system is suitably formed in a pre-mixture prior to the addition of Avelumab. The buffer system may be formed through simply mixing the buffering agent (supplied ready-made) with its acid/base conjugate (suitably in appropriate relative quantities to provide the desired pH--this can be determined by the skilled person either theoretically or experimentally). In the case of an acetate buffer system, this means e.g. mixing sodium acetate with HCl, or mixing acetic acid with NaOH or acetate. The pH of either the pre-mixture of final aqueous pharmaceutical formulation may be judiciously adjusted by adding the required quantity of base or acid, or a quantity of buffering agent or acid/base conjugate.
[0270] In certain embodiments, the buffering agent and/or buffer system is pre-formed as a separate mixture, and the buffer system is transferred to a precursor of the aqueous pharmaceutical formulation (comprising some or all components save for the buffering agent and/or buffer system, suitably comprising Avelumab and potentially only Avelumab) via buffer exchange (e.g. using diafiltration until the relevant concentrations or osmolality is reached). Additional excipients may be added thereafter if necessary in order to produce the final liquid pharmaceutical composition. The pH may be adjusted once or before all the components are present.
[0271] Any, some, or all components may be pre-dissolved or pre-mixed with a diluent prior to mixing with other components.
[0272] The final aqueous pharmaceutical formulation may be filtered, suitably to remove particulate matter. Suitably filtration is through filters sized at or below 1 .mu.m, suitably at 0.22 .mu.m. Suitably, filtration is through either PES filters or PVDF filters, suitably with 0.22 .mu.m PES filters.
[0273] The person of skill in the art is well aware how an aqueous pharmaceutical formulation can be used to prepare an IV solution, so that the antibody drug substance can be administered intravenously.
[0274] The preparation of the IV solution typically consists of a certain amount of solution being withdrawn from saline bags (e.g. 0.9% or 0.45% saline) with a plastic syringe (PP) and a needle and replaced with aqueous pharmaceutical formulation. The amount of solution replaced will depend on the body weight of the patients.
Example 1
Structure of Avelumab
[0275] 1.1 Primary Structure
[0276] Avelumab is an IgG with two heavy and two light chain molecules. The amino acid sequences of the two chains are shown in FIGS. 1a (SEQ ID NO:1)/1b (SEQ ID NO:2) and 2 (SEQ ID NO:3), respectively.
[0277] 1.2 Secondary Structure
[0278] LC-MS and MS/MS methods were used to confirm the intact chains of the molecule and the presence of post-translational modifications to the proteins. The secondary structure of the Avelumab molecule subunits are shown in FIG. 3.
[0279] As confirmed by UPLC-Q-TOF mass spectrometry of peptides obtained by trypsin digestion, the disulfide bonds Cys21-Cys96,Cys21-Cys90, Cys147-Cys203, Cys138-Cys197, Cys215-Cys223, Cys229-Cys229, Cys232-Cys232, Cys264-Cys324 and Cys370-Cys428 are forming the nine typical IgG bonding pattern.
[0280] 1.3 Glycosylation
[0281] The molecule contains one N-glycosylation site on Asn300 of the heavy chain. As determined by peptide mapping, the main structure identified by MALDI-TOF was a complex, biantennary type core fucosylated oligosaccaride with zero (G0F), one (G1F), or two galactose (G2F) residues. The main species are G0F and G1F.
[0282] Avelumab glycans fluorescence labeled by 2-aminobenzamide have been analysed by HILIC-UPLC-ESI-Q-TOF. FIG. 4 shows the UPLC profile of the glycan species found.
TABLE-US-00001 TABLE 1 Peak identification of 2AB HILIC-UPLC chromatogram RT Measured Expected Oxford Peak (min) MW MW Identification nomenclature Identification by 1a 5.99 1380.52 (M + H) 1380.54 (M + H) ##STR00001## FA1 Manually identified by MS 2 6.01 1437.54 1437.56 ##STR00002## A2 Manually identified by MS 3 7.02 1583.74 (M + H) 1583.62 (M + H) ##STR00003## FA2 MS in source fragmentation by GlycoworkBench 4 7.77 1355.57 (M + H) 1355.51 (M + H) ##STR00004## M5 Manually identified by MS 5 8.16 1599.77 (M + H) 1599.62 (M + H) ##STR00005## A2G1 Manually identified by MS 6 9.82 1744.79 1744.67 ##STR00006## FA2G1 MS in source fragmentation by GlycoworkBench 1462.90 1462.54 ##STR00007## FA2 freeEnd GlycoworkBench identified by MS 7 10.07 1744.80 1744.67 ##STR00008## FA2G1 MS in source fragmentation by GlycoworkBench 1462.91 1462.54 ##STR00009## FA2 freeEnd GlycoworkBench identified by MS 8 10.44 1462.90 1462.54 ##STR00010## FA2 freeEnd GlycoworkBench identified by MS 1744.79 1744.67 ##STR00011## FA2G1 Manually identified by MS 9 12.15 1177.50 (M + H) 1177.46 (M + H) ##STR00012## FM3 GlycoworkBench identified by MS 10 16.66 No No ionization ionization 11 13.42 1906.33 1906.72 ##STR00013## FA2G2 MS in source fragmentation by GlycoworkBench 1624.71 1624.59 ##STR00014## FA2G1 freeEnd GlycoworkBench identified by MS 12 13.71 954.40 (M + 2H)/2 954.36 (M + 2H)/2 ##STR00015## FA2G2 Manually identified by MS 1626.69 1626.61 ##STR00016## FA2G1 redEnd GlycoworkBench identified by MS 13 17.46 1099.97 (M + 2H)/2 1099.91 (M + 2H)/2 ##STR00017## FA2G2S MS in source fragmentation by GlycoworkBench 14 18.54 1079.91 (M + 2H)/2 1079.86 (M + 2H)/2 ##STR00018## FA2G2S freeEnd + S (probable- small traces) Manually identified by MS 15 21.04 2489.05 2488.91 ##STR00019## FA2G2S2 Manually identified by MS
[0283] The geometric shapes representing the glycan building blocks correspond to the following molecular entities:
[0284] Man .DELTA. Fuc .largecircle. Gal .quadrature. GalNAc GlcNAc .diamond. NANA NGNA
[0285] Man: mannose, Fuc: fucose, Gal: galactose, GalNAc: N-Acetylgalactosamine, NANA: sialic acid, NGNA: N-glycolylneuraminic acid
[0286] The glycan nomenclature used follows the Oxford Notation as proposed by Harvey et al. (Proteomics 2009, 9, 3796-3801). In species containing fucose (FA2, FA2G1, FA2G2), the Fuc-GlcNAc connectivity is a1-6. In species having a terminal GlcNAc, the GlcNAc-Man connectivity is 81-2. In species containing galactose, the Gal-GlcNAc connectivity is .beta.1-4.
[0287] The reported chromatographic profile has been integrated and yielded the Glycan Species Distribution of Avelumab as shown in Table 2a.
TABLE-US-00002 TABLE 2a A2 FA2 A2G1 FA2G1 A2G2 FA2G2 M5** 3.6 48.7 3.4 35.6 2.3 5.4 1.0 **Probably Mannose 5, coelution with biantennary mono-galactosylated species
[0288] The glycan mapping analysis confirmed the identification carried out by peptide mapping (that allowed to identify the two main glycan species), in addition secondary and minor species were also characterized by this method, specific for glycan analysis.
[0289] In another measurement the following Glycan Species Distribution was observed.
TABLE-US-00003 TABLE 2b A2 FA2 A2G1 FA2G1 A2G2 FA2G2 4.0 50.2 1.0 30.0 0.1 5.6
Example 2
DoE1 Screening
[0290] A first Design of Experiment screening DoE1 at 10 mg/mL Avelumab assessed the impact of several factors such as varying buffer type/pH, excipients, surfactant type and relevant concentration. The study led to the selection of the optimal conditions which can maximize protein stability.
[0291] In DoE1 the following factors were taken into account for investigation: [0292] Buffer type and pH: acetate, citrate and histidine buffers to be evaluated in the pH range 5.0-6.0. [0293] Excipients: 3 different excipients were considered in order to give indications as to whether sugars/polyols or amino acids are to be preferred for compounding in the formula, [0294] Surfactant type and concentration: two alternative surfactants (Tween 20 and Poloxamer 188) to be evaluated at varying concentrations (0-1 mg/mL).
[0295] The study was conducted in DINER vials (Schott) at a protein concentration of 10 mg/mL with filling volumes of 8 mL (80 mg/vial).
[0296] Table 3 illustrates the selection of DoE1 formulas investigated.
[0297] DoE1 allowed a selection of suitable buffer/pH, excipient type and surfactant type to be made, that were used for the subsequent DoE2 study described in Example 3.
TABLE-US-00004 TABLE 3 DoE1 screening formulations Surfactant Avelumab Buffer concentration ID (mg/mL) pH (10 mM) Excipient Surfactant (mg/mL) DoE1-1 10 5.00 Acetate Mannitol (51 mg/mL.sup.1) Poloxamer 188 0.5 DoE1-2 10 5.00 Acetate Trehalose dihydrate (106 Tween 20 0.5 mg/mL.sup.1) DoE1-3 10 5.00 Citrate Mannitol (51 mg/mL.sup.1) Poloxamer 188 0.2 DoE1-4 10 5.25 Acetate Trehalose dihydrate (106 Tween 20 0.2 mg/mL.sup.1) DoE1-5 10 5.25 Acetate Arginine HCl (21.1 mg/mL.sup.2) + Poloxamer 188 0.2 Glutamic acid (7.4 mg/mL.sup.3) DoE1-6 10 5.25 Citrate Arginine HCl (21.1 mg/mL.sup.2) + -- -- Glutamic acid (7.4 mg/mL.sup.3) DoE1-7 10 5.25 Citrate Mannitol (51 mg/mL.sup.1) Tween 20 0.2 DoE1-8 10 5.50 Acetate Mannitol (51 mg/mL.sup.1) Tween 20 0.5 DoE1-9 10 5.50 Acetate Trehalose dihydrate (106 -- -- mg/mL.sup.1) DoE1-10 10 5.50 Citrate Trehalose dihydrate (106 Poloxamer 188 1 mg/mL.sup.1) DoE1-11 10 5.50 Citrate Arginine HCl (21.1 mg/mL.sup.2) + Tween 20 0.2 Glutamic acid (7.4 mg/mL.sup.3) DoE1-12 10 5.75 Citrate Trehalose dihydrate (106 Tween 20 1 mg/mL) DoE1-13 10 5.75 Citrate Mannitol (51 mg/mL) -- -- DoE1-14 10 5.75 Histidine Arginine HCl (21.1 mg/mL) + Poloxamer 188 0.5 Glutamic acid (7.4 mg/mL) DoE1-15 10 5.75 Histidine Mannitol (51 mg/mL) Tween 20 1 DoE1-16 10 6.00 Citrate Arginine HCl (21.1 mg/mL) + Tween 20 1 Glutamic acid (7.4 mg/mL) DoE1-17 10 6.00 Citrate Trehalose dihydrate (106 Poloxamer 188 0.2 mg/mL) DoE1-18 10 6.00 Histidine Arginine HCl (21.1 mg/mL) + Poloxamer 188 1 Glutamic acid (7.4 mg/mL) DoE1-19 10 6.00 Histidine Trehalose dihydrate (106 Poloxamer 188 0.5 mg/mL) Reference .sup.4 10 5.50 Acetate Mannitol (51 mg/mL)/L- Tween 20 0.5 Methionine (0.21 mg/mL) .sup.1Corresponds to 280 mM .sup.2Corresponds to 150 mM .sup.3Corresponds to 50 mM .sup.4 Formulation disclosed in WO2013079174
[0298] 2.1 Manufacturing
[0299] The pre-formulated drug substance (DS) (10 (.+-.1) mg/mL Avelumab, 1.36 mg/mL Sodium acetate trihydrate, 51 mg/mL D-Mannitol, 0.21 mg/mL L-Methionine, hydrochloric acid q.b. to pH 5.5) was buffer exchanged by tangential flow filtration (using Pellicon XL Biomax Cassettes with a 10 kDa cut-off) in the three buffers: 10 mM sodium acetate pH 5.0, 10 mM sodium citrate pH 5.0 and 10 mM histidine pH 5.75 until a three-fold volume exchange was achieved. At each step the DS solution was diluted 5-fold with relevant buffer. Final target protein concentration in the exchanged DS material was >10 mg/mL. The required excipients were then added to the relevant buffer-exchanged DS material, pH and final solution weight adjusted to the target so as to obtain the DP compositions listed in Table 3.
[0300] The sequence of addition of ingredients to the exchanged DS solutions was as follows:
[0301] Add D-Mannitol or Trehalose dihydrate or Arginine HCl+Glutamic acid to the exchanged DS solution, stir until complete dissolution, add L-Methionine and stir until complete dissolution (only for Reference), add Poloxamer 188 or Polysorbate 20 (50 mg/mL stock solution), stir until complete dissolution, check pH and adjust to target with sodium hydroxide.
[0302] Drug product (DP) solutions were filled (8 mL) in DINER vials (Schott).
[0303] Visual inspection during the DS diafiltration process revealed that sodium citrate buffer caused generally higher opalescence, whilst remarkably clearer solutions were obtained when exchanges were made in sodium acetate and in histidine buffers.
[0304] In Table 4, the results of the experiments carried out to determine protein recovery, osmolality (Osmomat 030/D, Gonotec) and turbidity of the three DS materials upon buffer exchange are shown. Satisfactory protein recoveries (>89%) and final osmolality values (<61 mOsm/kg) were obtained. Turbidity analyses confirmed the higher opalescence of the DS exchanged in sodium citrate.
TABLE-US-00005 TABLE 4 Results of recovery (by OD), osmolality and turbidity experiments conducted on DS materials after buffer exchange. Recovery Osmolality Turbidity Buffer (%) (mOsm/kg) (NTU) Acetate 96 29 3 Citrate 89 38 30 Histidine 93 61 6
[0305] 2.2. Osmolality
[0306] The osmolality values of the DP formulations relevant to the DoE1 screening were comprised in the range 299-396 mOsm/kg, with most formulations having osmolalities below around 360 mOsm/kg.
[0307] The measurements were carried out at time 0, upon manufacturing completion.
[0308] The values obtained were in line with target (acceptable range 250-400 mOsm/Kg). Solutions containing Trehalose dihydrate showing higher values (close to 400 mOsm/kg) due to effect of this ingredient on freezing point and subsequent (apparent) increase in osmolality.
[0309] 2.3 Thermal Stress
[0310] 2.3.1 Protein Content
[0311] As determined by OD measurements, the time 0 content values were in line with theoretical values (10 mg/mL). No significant changes were observed after 1 month at 40.degree. C.
[0312] 2.3.2 Total Aggregates
[0313] Total aggregates DoE1 formulations were determined by SE-HPLC at time 0 and after 2 and 4 weeks of storage at 40.degree. C.
[0314] No statistically significant variations in terms of aggregates upon thermal stress at 40.degree. C. could be highlighted, thus indicating that the different matrices tested led to invariant/negligible changes in the aggregation pattern.
[0315] 2.3.3 Fragmentation
[0316] Fragmentation by Bioanalyzer (2100 Bioanalyzer, Agilent) in DoE1 formulations was determined at time 0 and after 2 and 4 weeks of storage at 40.degree. C.
[0317] The data indicated that: [0318] pH is a critical factor to protein fragmentation at 40.degree. C. At pH>5.75, fragmentation tends to significantly increase (most typically in formulations from DoE1-13 to DoE1-19, in citrate and histidine buffers). [0319] The formulations presenting the lowest variations in fragmentation are those in a pH range of 5.0-5.75 preferably in presence of either D-Mannitol or Trehalose dihydrate (DoE1-2-8-9-10-12). [0320] Formulation DoE1-7 (citrate buffer at pH 5.25, in presence of D-Mannitol and Tween 20) presented abnormal profiles with consistent peak doubling (some issues might be related to usage of citrate as a buffering agent in terms of fragmentation, in addition to those already highlighted during manufacturing with the increase in turbidity/opalescence).
[0321] 2.3.4 Turbidity
[0322] Turbidity by nephelometry in DoE1 formulations was determined at time 0 and after 2 and 4 weeks of storage at 40.degree. C.
[0323] Opalescence/strong opalescence consistently observed in all DP formulations containing citrate as a pH buffering agent.
[0324] All formulations in sodium acetate and histidine were found to be clear/slightly opalescent with no significant changes observed over 1 month of storage at 40.degree. C.
[0325] 2.3.5 pH
[0326] No pH changes were observed.
[0327] 2.4 Mechanical Stress
[0328] The DoE1 formulations were subjected to 24-hour orbital shaking in vials at 300 rpm (room temperature). Upon stress termination the samples were tested for aggregates and opalescence.
[0329] 2.4.1 Total Aggregates
[0330] Total aggregates were determined by SE-HPLC after mechanical stress and compared to time 0 results. Negligible changes were observed.
[0331] 2.4.2 Turbidity
[0332] Turbidity of DoE1 formulations was determined by nephelometry (2100AN IS, Hach Lange) after mechanical stress and compared to time 0 results. The data were evaluated by ANOVA and a moderately significant impact deriving from surfactant presence (0.01<p-value<0.05) was observed. Either Tween 20 or Poloxamer 188 can help minimize turbidity changes after mechanical stress.
[0333] 2.5 Light Exposure
[0334] The DoE1 formulations were subjected to 7-hour irradiation at 765 W/m.sup.2 (Suntest CPS, Atlas). Upon light stress termination the samples were tested for aggregates, opalescence, pH and isoforms profile.
[0335] 2.5.1 Total Aggregates
[0336] Using SE-HPLC (Alliance, Waters) slight variations were observed, most frequently when sodium citrate buffer is used (p-value<0.01).
[0337] Sodium acetate and histidine are the buffers to be preferred in order to minimize aggregation changes.
[0338] 2.5.2 Turbidity
[0339] As determined by nephelometry the most evident turbidity increases were typically found in citrate buffer at pH values>5.75 (DoE1-13 and DoE1-16 and DoE1-17).
[0340] 2.5.3 pH
[0341] No changes were observed.
[0342] 2.6 DoE1: Outcome
[0343] The data obtained in the frame of the thermal, mechanical and light stress were evaluated in order to determine conditions that provide maximal protein resistance against stresses.
[0344] The results of the analysis are reported in Table 5.
TABLE-US-00006 TABLE 5 Components of highly stabilized Avelumab formulations at 10 mg/mL protein concentration ID# Buffer pH Excipient Surfactant Extrapolated 10 mM Acetate 5.20 Trehalose Tween 20 dihydrate (0.5 mg/mL) (280 mM) DoE1-4 10 mM Acetate 5.25 Trehalose Tween 20 dihydrate (0.2 mg/mL) (280 mM)
[0345] The extrapolated formulation is highlighted in green (ID#=Extrapolated), whilst the most similar formula in the set of those tested is the DoE1-4, also reported.
[0346] These data demonstrate that acetate buffer pH 5.0-5.5 provides improved protein stability, and that surfactant presence, such as either Tween 20 or Poloxamer 188, at concentrations higher than 0.2 mg/mL, is also important for improved protein stability in the formulation.
Example 3
[0347] A second DoE screening "DoE2" aimed at fine-tuning the formulations selected upon DoE1 completion and concurrently increasing protein concentration to 20 mg/mL.
[0348] With this second formulation screening, six formulations at 20 mg/mL protein concentration varying in excipients (D-Mannitol, Trehalose dihydrate) and surfactant (no surfactant, Poloxamer 188 or Polysorbate 20 at 0.5 mg/mL) in presence of 10 mM sodium acetate buffer pH 5.2 were tested after thermal stress (1 month at 40.degree. C., 8 weeks at 25.degree. C. and 2-8.degree. C.) and mechanical shaking (24 hours at 300 rpm, room temperature). The relevant compositions are listed in Table 6.
TABLE-US-00007 TABLE 6 DoE2 screening formulations (protein concentration = 20 mg/mL) Avelumab ID (mg/mL) Buffer Excipient Surfactant DoE2-1 20 10 mM Mannitol -- acetate (51 mg/mL.sup.1) pH 5.2 DoE2-2 20 10 mM Trehalose dihydrate -- acetate (106 mg/mL.sup.1) pH 5.2 DoE2-3 20 10 mM Mannitol Tween 20 acetate (51 mg/mL.sup.1) (0.5 mg/mL) pH 5.2 DoE2-4 20 10 mM Trehalose dihydrate Tween 20 acetate (106 mg/mL.sup.1) (0.5 mg/mL) pH 5.2 DoE2-5 20 10 mM Mannitol Poloxamer 188 acetate (51 mg/mL.sup.1) (0.5 mg/mL) pH 5.2 DoE2-6 20 10 mM Trehalose dihydrate Poloxamer 188 acetate (106 mg/mL.sup.1) (0.5 mg/mL) pH 5.2 DoE1-8 20 10 mM Mannitol Tween 20 acetate (51 mg/mL.sup.1) (0.5 mg/mL) pH 5.5 Reference 20 10 mM Mannitol Tween 20 acetate (51 mg/mL)/L- (0.5 mg/mL) pH 5.5 Methionine (0.21 mg/mL)
[0349] The DoE2 study was conducted to comparatively evaluate the effect of D-Mannitol vs. Trehalose dihydrate, and the impact of surfactant (either Tween 20 or Poloxamer 188, or no surfactant) in sodium acetate buffer at pH 5.2, at the increased protein concentration of 20 mg/mL. Two pH 5.5 reference samples have been included in the design: "Reference" with L-Methionine, and a reference formulation without L-Methionine, corresponding to DoE1-8.
[0350] 3.1 Manufacturing
[0351] The pre-formulated drug substance (DS) (27.1 mg/mL Avelumab in 10 mM sodium acetate pH 5.5) was used. The required excipients were then added to the DS material.
[0352] The sequence of addition of ingredients to the DS solution was as follows:
[0353] Add D-Mannitol or Trehalose dihydrate, stir until complete dissolution, add Poloxamer 188 or Polysorbate 20 (20 mg/mL stock solution), stir until complete dissolution, add L-Methionine and stir until complete dissolution (only for Reference), stir until complete dissolution, check pH and adjust to target with sodium hydroxide or diluted acetic acid.
[0354] The solutions were weight adjusted to the target with relevant buffer so as to obtain the DP compositions listed in Table 7.
[0355] DP solutions were filled (8 mL) in DIN6R vials.
[0356] 3.2 Thermal Stress
[0357] 3.2.1 Protein Content
[0358] No protein content (OD, Lambda 35, Perkin Elmer) changes observed over 4 weeks at 40.degree. C. (Table 7) and 8 weeks at 25.degree. C. (Table 8).
TABLE-US-00008 TABLE 7 Protein content (mg/mL) by OD of DoE2 formulations (thermal stress at 40.degree. C.) Protein conc Excipient Time 2 weeks 4 weeks # ID (mg/mL) Buffer (280 mM) Surfactant 0 (40.degree. C.) (40.degree. C.) 1 DoE2 - 1 20 10 mM acetate D-Mannitol No 22.3 20.0 20.9 pH 5.2 2 DoE2 - 2 20 10 mM acetate Trehalose No 22.0 20.6 21.6 pH 5.2 dihydrate 3 DoE2 - 3 20 10 mM acetate D-Mannitol Tween 20 (0.5 21.9 20.5 21.6 pH 5.2 mg/mL) 4 DoE2 - 4 20 10 mM acetate Trehalose Tween 20 (0.5 22.1 20.5 22.3 pH 5.2 dihydrate mg/mL) 5 DoE2 - 5 20 10 mM acetate D-Mannitol Lutrol F-68 21.7 20.7 22.8 pH 5.2 (0.5 mg/mL) 6 DoE2 - 6 20 10 mM acetate Trehalose Lutrol F-68 22.7 21.3 22.5 pH 5.2 dihydrate (0.5 mg/mL) 7 DoE1 - 8 20 10 mM acetate D-Mannitol Tween 20 (0.5 21.5 20.5 23.5 pH 5.5 mg/mL) 8 Reference 20 10 mM acetate D-Mannitol + Tween 20 (0.5 21.5 20.4 23.3 pH 5.5 L-Methionine mg/mL)
TABLE-US-00009 TABLE 8 Protein content (mg/mL) by OD of DoE2 formulations (thermal stress at 25.degree. C.) Protein conc Excipient Time 8 weeks # ID (mg/mL) Buffer (280 mM) Surfactant 0 (25.degree. C.) 1 DoE2 - 1 20 10 mM acetate D-Mannitol No 22.3 20.6 pH 5.2 2 DoE2 - 2 20 10 mM acetate Trehalose No 22.0 21.0 pH 5.2 dihydrate 3 DoE2 - 3 20 10 mM acetate D-Mannitol Tween 20 (0.5 21.9 21.3 pH 5.2 mg/mL) 4 DoE2 - 4 20 10 mM acetate Trehalose Tween 20 (0.5 22.1 21.5 pH 5.2 dihydrate mg/mL) 5 DoE2 - 5 20 10 mM acetate D-Mannitol Lutrol F-68 21.7 20.5 pH 5.2 (0.5 mg/mL) 6 DoE2 - 6 20 10 mM acetate Trehalose Lutrol F-68 22.7 21.0 pH 5.2 dihydrate (0.5 mg/mL) 7 DoE1 - 8 20 10 mM acetate D-Mannitol Tween 20 (0.5 21.5 21.1 pH 5.5 mg/mL) 8 Reference 20 10 mM acetate D-Mannitol + Tween 20 (0.5 21.5 21.2 pH 5.5 L-Methionine mg/mL)
[0359] 3.2.2 Total Aggregates
[0360] Total aggregates determined by SE-HPLC over stability at 40.degree. C. and 25.degree. C. are represented in FIGS. 6 and 7 respectively. Only minor, non-significant changes in aggregation were observed.
[0361] 3.2.3 Fragmentation by Bioanalyzer
[0362] Fragments were evaluated over 1 month at 40.degree. C. and after 2 months at 25.degree. C. The relevant results are shown in FIG. 8 and FIG. 9 respectively.
[0363] At 40.degree. C., aside from formulation DoE2-1, which presented an amount of fragments higher than 7% after 1 month, the other formulas were observed to have similar behavior (4-6% in fragments after 1 month) with slightly better performances of formulations DoE2-4, DoE2-5 and DoE2-6 (4.0-4.5% in fragments after 1 month at 40.degree. C.).
[0364] At 25.degree. C., similar fragmentation percentages were found after 2 months (4.6-6.1%) 3.2.4 Isoforms profile
[0365] The isoforms profile by iCE280 (Fast IEF Analyzer, Convergent Bioscience) in DoE2 formulations was determined at time 0 and after 4 weeks of storage at 40.degree. C. Upon storage at 40.degree. C. typically increases in the acidic cluster can be determined, while a concurrent decrease in the basic isoforms is observed.
[0366] The isoforms profiles were evaluated over 1 month at 40.degree. C. (Table 9) and after 8 weeks at 25.degree. C. (FIG. 10).
[0367] Comparable variations were observed in all samples at both stressing conditions.
TABLE-US-00010 TABLE 9 iCE280 results for DoE2 formulations after 4 weeks at 40.degree. C. Time 0 4 weeks at 40.degree. C. Acidic Main Basic Acidic Main Basic forms peak forms forms peak forms (%) (%) (%) (%) (%) (%) DoE2 - 1 32.3 36.0 31.7 40.3 31.9 27.8 DoE2 - 2 32.0 37.7 30.4 38.0 33.6 28.5 DoE2 - 3 32.2 36.7 31.1 39.9 32.7 27.5 DoE2 - 4 32.7 36.7 30.6 39.7 33.0 27.3 DoE2 - 5 32.5 37.4 30.2 38.1 33.4 28.5 DoE2 - 6 32.4 37.0 30.7 38.3 33.7 28.0 DoE1 - 8 33.2 36.9 30.0 38.8 33.5 27.7 Reference 32.2 36.2 31.7 37.7 33.4 28.9
[0368] 3.2.6 Turbidity
[0369] No variations observed after 1 month at 40.degree. C. (Table 8) and 2 months at 25.degree. C. (Table 9).
TABLE-US-00011 TABLE 10 Turbidity of DoE2 formulations after 1 month at 40.degree. C. Protein conc Excipient Time 2 weeks 4 weeks # ID (mg/mL) Buffer (280 mM) Surfactant 0 (40.degree. C.) (40.degree. C.) 1 DoE2 - 1 20 10 mM acetate D-Mannitol No 2 2 2 pH 5.2 2 DoE2 - 2 20 10 mM acetate Trehalose No 2 2 2 pH 5.2 dihydrate 3 DoE2 - 3 20 10 mM acetate D-Mannitol Tween 20 (0.5 2 2 2 pH 5.2 mg/mL) 4 DoE2 - 4 20 10 mM acetate Trehalose Tween 20 (0.5 2 2 2 pH 5.2 dihydrate mg/mL) 5 DoE2 - 5 20 10 mM acetate D-Mannitol Lutrol F-68 2 2 2 pH 5.2 (0.5 mg/mL) 6 DoE2 - 6 20 10 mM acetate Trehalose Lutrol F-68 2 2 2 pH 5.2 dihydrate (0.5 mg/mL) 7 DoE1 - 8 20 10 mM acetate D-Mannitol Tween 20 (0.5 2 2 2 pH 5.5 mg/mL) 8 Reference 20 10 mM acetate D-Mannitol + Tween 20 (0.5 3 3 2 pH 5.5 L-Methionine mg/mL)
TABLE-US-00012 TABLE 11 Turbidity of DoE2 formulations after 2 months at 25.degree. C. Protein conc Excipient Time 8 weeks # ID (mg/mL) Buffer (280 mM) Surfactant 0 (25.degree. C.) 1 DoE2 - 1 20 10 mM acetate D-Mannitol No 2 2 pH 5.2 2 DoE2 - 2 20 10 mM acetate Trehalose No 2 2 pH 5.2 dihydrate 3 DoE2 - 3 20 10 mM acetate D-Mannitol Tween 20 (0.5 2 2 pH 5.2 mg/mL) 4 DoE2 - 4 20 10 mM acetate Trehalose Tween 20 (0.5 2 2 pH 5.2 dihydrate mg/mL) 5 DoE2 - 5 20 10 mM acetate D-Mannitol Lutrol F-68 2 2 pH 5.2 (0.5 mg/mL) 6 DoE2 - 6 20 10 mM acetate Trehalose Lutrol F-68 2 2 pH 5.2 dihydrate (0.5 mg/mL) 7 DoE1 - 8 20 10 mM acetate D-Mannitol Tween 20 (0.5 2 3 pH 5.5 mg/mL) 8 Reference 20 10 mM acetate D-Mannitol + Tween 20 (0.5 3 3 pH 5.5 L-Methionine mg/mL)
[0370] 3.2.7 pH
[0371] No variations observed after 1 month at 40.degree. C. (Table 12) and 2 months at 25.degree. C. (Table 13).
TABLE-US-00013 TABLE 12 pH of DoE2 formulations after 1 month at 40.degree. C. Protein conc Excipient Time 2 weeks 4 weeks # ID (mg/mL) Buffer (280 mM) Surfactant 0 (40.degree. C.) (40.degree. C.) 1 DoE2 - 1 20 10 mM acetate D-Mannitol No 5.2 5.2 5.2 pH 5.2 2 DoE2 - 2 20 10 mM acetate Trehalose No 5.2 5.2 5.2 pH 5.2 dihydrate 3 DoE2 - 3 20 10 mM acetate D-Mannitol Tween 20 (0.5 5.2 5.2 5.2 pH 5.2 mg/mL) 4 DoE2 - 4 20 10 mM acetate Trehalose Tween 20 (0.5 5.2 5.2 5.2 pH 5.2 dihydrate mg/mL) 5 DoE2 - 5 20 10 mM acetate D-Mannitol Lutrol F-68 5.2 5.2 5.2 pH 5.2 (0.5 mg/mL) 6 DoE2 - 6 20 10 mM acetate Trehalose Lutrol F-68 5.2 5.2 5.2 pH 5.2 dihydrate (0.5 mg/mL) 7 DoE1 - 8 20 10 mM acetate D-Mannitol Tween 20 (0.5 5.5 5.5 5.5 pH 5.5 mg/mL) 8 Reference 20 10 mM acetate D-Mannitol + Tween 20 (0.5 5.5 5.5 5.5 pH 5.5 L-Methionine mg/mL)
TABLE-US-00014 TABLE 13 pH of DoE2 formulations after 2 months at 25.degree. C. Protein conc Excipient Time 8 weeks # ID (mg/mL) Buffer (280 mM) Surfactant 0 (25.degree. C.) 1 DoE2 - 1 20 10 mM acetate D-Mannitol No 5.2 5.2 pH 5.2 2 DoE2 - 2 20 10 mM acetate Trehalose No 5.2 5.2 pH 5.2 dihydrate 3 DoE2 - 3 20 10 mM acetate D-Mannitol Tween 20 (0.5 5.2 5.3 pH 5.2 mg/mL) 4 DoE2 - 4 20 10 mM acetate Trehalose Tween 20 (0.5 5.2 5.2 pH 5.2 dihydrate mg/mL) 5 DoE2 - 5 20 10 mM acetate D-Mannitol Lutrol F-68 5.2 5.2 pH 5.2 (0.5 mg/mL) 6 DoE2 - 6 20 10 mM acetate Trehalose Lutrol F-68 5.2 5.2 pH 5.2 dihydrate (0.5 mg/mL) 7 DoE1 - 8 20 10 mM acetate D-Mannitol Tween 20 (0.5 5.5 5.5 pH 5.5 mg/mL) 8 Reference 20 10 mM acetate D-Mannitol + Tween 20 (0.5 5.5 5.6 pH 5.5 L-Methionine mg/mL)
[0372] 3.2.8 Circular Dichroism
[0373] CD spectra (J-810 Spectropolarimeter, Jasco) of DoE2 formulations were collected at time 0 and after 4 weeks at 40.degree. C. and 8 weeks at 25.degree. C. in the near UV range.
[0374] Protein in all formulation generally retains its tertiary structure after 4 weeks at 40.degree. C. and 8 weeks at 25.degree. C.
[0375] 3.2.9 Sub-visible Particles
[0376] The sub-visible particles of the DoE2 formulations after 8 weeks of storage at 2-8.degree. C. were determined. The results are shown in Table 14. The values were found within European Pharmacopoeia limits (for solutions supplied in containers with a nominal content of less than 100 mL).
TABLE-US-00015 TABLE 14 Sub-visible particles of DoE2 formulations after 8 weeks at 2-8.degree. C. Sub-visible Sub-visible particles >10 particles >25 Protein conc Excipient .mu.m (per .mu.m (per # ID (mg/mL) Buffer (280 mM) Surfactant container) container) 1 DoE2 - 1 20 10 mM acetate D-Mannitol No 754 33 pH 5.2 2 DoE2 - 2 20 10 mM acetate Trehalose No 716 14 pH 5.2 dihydrate 3 DoE2 - 3 20 10 mM acetate D-Mannitol Tween 20 (0.5 597 24 pH 5.2 mg/mL) 4 DoE2 - 4 20 10 mM acetate Trehalose Tween 20 (0.5 1839 100 pH 5.2 dihydrate mg/mL) 5 DoE2 - 5 20 10 mM acetate D-Mannitol Lutrol F-68 431 38 pH 5.2 (0.5 mg/mL) 6 DoE2 - 6 20 10 mM acetate Trehalose Lutrol F-68 521 28 pH 5.2 dihydrate (0.5 mg/mL) 7 DoE1 - 8 20 10 mM acetate D-Mannitol Tween 20 (0.5 915 14 pH 5.5 mg/mL) 8 Reference 20 10 mM acetate D-Mannitol + Tween 20 (0.5 1873 52 pH 5.5 L-Methionine mg/mL)
[0377] 3.3 Mechanical Stress
[0378] 3.3.1 Fragmentation by Bioanalyzer
[0379] After 24 hours at 300 rpm, slight variations in fragments (Table 15) were observed in all samples (up to 5.0-6.5%) with no specific relation to the specific compositions tested.
TABLE-US-00016 TABLE 15 Fragments (%) by Bioanalyzer of DoE2 formulations after 24-hour shaking (300 rpm; room temperature) Protein conc Excipient Time 24 H # ID (mg/mL) Buffer (280 mM) Surfactant 0 300 RPM 1 DoE2 - 1 20 10 mM acetate D-Mannitol No 4.9 5.5 pH 5.2 2 DoE2 - 2 20 10 mM acetate Trehalose No 4.6 5.0 pH 5.2 dihydrate 3 DoE2 - 3 20 10 mM acetate D-Mannitol Tween 20 (0.5 4.7 5.7 pH 5.2 mg/mL) 4 DoE2 - 4 20 10 mM acetate Trehalose Tween 20 (0.5 4.6 6.5 pH 5.2 dihydrate mg/mL) 5 DoE2 - 5 20 10 mM acetate D-Mannitol Lutrol F-68 5.1 6.2 pH 5.2 (0.5 mg/mL) 6 DoE2 - 6 20 10 mM acetate Trehalose Lutrol F-68 5.2 5.3 pH 5.2 dihydrate (0.5 mg/mL) 7 DoE1 - 8 20 10 mM acetate D-Mannitol Tween 20 (0.5 3.5 5.4 pH 5.5 mg/mL) 8 Reference 20 10 mM acetate D-Mannitol + Tween 20 (0.5 3.4 5.4 pH 5.5 L-Methionine mg/mL)
[0380] 3.3.2 Aggregates
[0381] No changes were observed after mechanical shaking (Table 16).
TABLE-US-00017 TABLE 16 Aggregates (%) by SE-HPLC of DoE2 formulations after 24-hour shaking (300 rpm; room temperature) Protein conc Excipient Time 24 H # ID (mg/mL) Buffer (280 mM) Surfactant 0 300 RPM 1 DoE2 - 1 20 10 mM acetate D-Mannitol No 1.5 1.5 pH 5.2 2 DoE2 - 2 20 10 mM acetate Trehalose No 1.5 1.5 pH 5.2 dihydrate 3 DoE2 - 3 20 10 mM acetate D-Mannitol Tween 20 (0.5 1.6 1.5 pH 5.2 mg/mL) 4 DoE2 - 4 20 10 mM acetate Trehalose Tween 20 (0.5 1.6 1.5 pH 5.2 dihydrate mg/mL) 5 DoE2 - 5 20 10 mM acetate D-Mannitol Lutrol F-68 1.6 1.5 pH 5.2 (0.5 mg/mL) 6 DoE2 - 6 20 10 mM acetate Trehalose Lutrol F-68 1.6 1.6 pH 5.2 dihydrate (0.5 mg/mL) 7 DoE1 - 8 20 10 mM acetate D-Mannitol Tween 20 (0.5 1.6 1.6 pH 5.5 mg/mL) 8 Reference 20 10 mM acetate D-Mannitol + Tween 20 (0.5 1.6 1.6 pH 5.5 L-Methionine mg/mL)
[0382] 3.3.3 pH
[0383] No changes were observed after mechanical shaking (Table 17).
TABLE-US-00018 TABLE 17 pH of DoE2 formulations after 24-hour shaking (300 rpm; room temperature) Protein conc Excipient Time 24 H # ID (mg/mL) Buffer (280 mM) Surfactant 0 300 RPM 1 DoE2 - 1 20 10 mM acetate D-Mannitol No 5.2 5.2 pH 5.2 2 DoE2 - 2 20 10 mM acetate Trehalose No 5.2 5.2 pH 5.2 dihydrate 3 DoE2 - 3 20 10 mM acetate D-Mannitol Tween 20 (0.5 5.2 5.2 pH 5.2 mg/mL) 4 DoE2 - 4 20 10 mM acetate Trehalose Tween 20 (0.5 5.2 5.2 pH 5.2 dihydrate mg/mL) 5 DoE2 - 5 20 10 mM acetate D-Mannitol Lutrol F-68 5.2 5.2 pH 5.2 (0.5 mg/mL) 6 DoE2 - 6 20 10 mM acetate Trehalose Lutrol F-68 5.2 5.2 pH 5.2 dihydrate (0.5 mg/mL) 7 DoE1 - 8 20 10 mM acetate D-Mannitol Tween 20 (0.5 5.5 5.5 pH 5.5 mg/mL) 8 Referenee 20 10 mM acetate D-Mannitol + Tween 20 (0.5 5.5 5.5 pH 5.5 L-Methionine mg/mL)
[0384] 3.4 Turbidity
[0385] No changes were observed after mechanical shaking (Table 18).
TABLE-US-00019 TABLE 18 Turbidity (NTU) of DoE2 formulations after 24-hour shaking (300 rpm; room temperature) Protein conc Excipient Time 24 h # ID (mg/mL) Buffer (280 mM) Surfactant 0 300 rpm 1 DoE2 - 1 20 10 mM acetate D-Mannitol No 2 2 pH 5.2 2 DoE2 - 2 20 10 mM acetate Trehalose No 2 2 pH 5.2 dihydrate 3 DoE2 - 3 20 10 mM acetate D-Mannitol Tween 20 (0.5 2 2 pH 5.2 mg/mL) 4 DoE2 - 4 20 10 mM acetate Trehalose Tween 20 (0.5 2 2 pH 5.2 dihydrate mg/mL) 5 DoE2 - 5 20 10 mM acetate D-Mannitol Lutrol F-68 2 2 pH 5.2 (0.5 mg/mL) 6 DoE2 - 6 20 10 mM acetate Trehalose Lutrol F-68 2 2 pH 5.2 dihydrate (0.5 mg/mL) 7 DoE1 - 8 20 10 mM acetate D-Mannitol Tween 20 (0.5 2 2 pH 5.5 mg/mL) 8 Reference 20 10 mM acetate D-Mannitol + Tween 20 (0.5 3 2 pH 5.5 L-Methionine mg/mL)
[0386] 3.5 DoE2: Outcome
[0387] These results demonstrate that pH 5.2 (extrapolated from DoE1) does not impact fragmentation and therefore is suitable for use in a stable formulation. Optimal pH for preserving protein stability was demonstrated to be in the range 5.0-5.5 (DoE1). In contrast, pH values of 5.6-5.7 could result in higher fragmentation.
[0388] Mannitol and Trehalose dihydrate resulted in similar behavior.
[0389] No superiority of Poloxamer 188 over Tween 20 was found.
[0390] These results also demonstrate that higher protein concentration (20 mg/mL) in the DoE2 formulation is feasible with no observed or anticipated stability issues.
[0391] DoE2: Formulation 3 (the formula most preferred and finally selected for further use at 20 mg/mL) was compared in terms of isoforms profiles to the reference formula at time 0, after 4 weeks at 40.degree. C. and 8 weeks at 25.degree. C. in order to evaluate whether different behavior between the two formulas are present over stability time at different conditions. The results are presented in Table 19.
TABLE-US-00020 TABLE 19 Isoforms profiles by iCE280 of DoE2 Formulation 3 and Reference formulation at time 0, after 4 weeks (40.degree. C.) and 8 weeks (25.degree. C.) Time 4 weeks 8 weeks ID# Buffer pH Excipient Surfactant Cluster 0 (40.degree. C.) (25.degree. C.) DoE2 - 3 10 mM 5.20 D-Mannitol Tween 20 1 1.6 3.1 2.9 Acetate (0.5 mg/mL) 2 4.5 5.3 7.1 3 9.9 10.8 9.1 4 16.2 20.7 18.0 5 36.7 32.7 34.9 6 22.2 19.7 20.3 7 8.9 7.8 7.7 Reference 10 mM 5.50 Mannitol + Tween 20 1 1.8 2.3 3.4 Acetate L-Methionine (0.5 mg/mL) 2 4.3 5.0 8.1 3 9.8 10.6 10.0 4 16.3 19.8 16.9 5 36.2 33.4 34.7 6 22.6 21.1 19.6 7 9.1 7.8 7.4
[0392] Also the additional timepoint (8 weeks) at 25.degree. C. highlighted no major issues deriving from the reduced pH with respect to the reference formulation.
Example 4
Effect of Antioxidant (L-Methionine)
[0393] As methionine was used in the formulation disclosed in WO2013079174, the present Avelumab formulation development aimed to also clarify the impact of this compound as an antioxidant.
[0394] The 10 mg/mL samples (from the DoE1 set) were 2-fold diluted with 200 .mu.L of 6% H.sub.2O.sub.2, obtaining a final protein concentration of about 5 mg/mL and 3% H.sub.2O.sub.2, and then incubated 3 h at 5.degree. C. At the end of the incubation the sample was washed versus water by ultracentrifugation using an Amicon Ultra (Millipore) 4 mL 10 kDa (4 washes 1 mL each step). The final protein concentration after Amicon treatment was about 10 mg/mL.
[0395] DoE1: Formulation 8 is identical to Reference formula of DoE2, except for the presence of L-Methionine: the forced oxidation with H.sub.2O.sub.2 (3 hours at 2-8.degree. C.) of the two formulas and following testing by iCE280 (oxidation generally leads to increase in more acidic species in electropherograms) and Bioanalyzer aimed to determine whether any differences arise in the two formulations due to the presence of the antioxidant agent. The results are presented in Tables 20 and 21.
TABLE-US-00021 TABLE 20 Isoforms profiles by iCE280 of DoE1 Formulation 8 and Reference formulation after forced oxidation treatment. Upper table: samples stored at 2-8.degree. C. Bottom table: sample stored 4 weeks at 40.degree. C. + 6 weeks at 2-8.degree. C. Oxidised with 3% H.sub.2O.sub.2 Anti PD-L1 (after 10 week ID# Buffer pH (mg/mL) Excipient Surfactant Cluster storage at 2-8.degree. C.) DoE1 - 8 10 mM 5.50 10 Mannitol Tween 20 1 2.2 Acetate (280 mM) (0.5 mg/mL) 2 4.5 3 9.8 4 23.2 5 39.0 5 16.6 7 4.8 Reference 10 mM 5.50 10 Mannitol Tween 20 1 2.2 Acetate (280 mM) + (0.5 mg/mL) 2 4.5 L-Methionine 3 10.1 (1.4 mM) 4 23.7 5 37.9 6 16.8 7 4.8 Oxidised with 3% H.sub.2O.sub.2 (after 4 week Anti PD-L1 storage at 40.degree. C. + ID# Buffer pH (mg/mL) Excipient Surfactant Cluster 6 weeks at 2-8.degree. C.) DoE1 - 8 10 mM 5.50 10 Mannitol Tween 20 1 2.5 Acetate (280 mM) (0.5 mg/mL) 2 5.9 3 11.4 4 27.2 5 34.5 6 14.6 7 4.0 Reference 10 mM 5.50 10 Mannitol Tween 20 1 2.7 Acetate (280 mM) + (0.5 mg/mL) 2 5.9 L-Methionine 3 11.1 (1.4 mM) 4 26.7 5 35.8 6 14.4 7 3.4
[0396] Comparable acidic clusters abundances were observed for the two formulations (with or w/o methionine).
[0397] Fragments by Bioanalyzer were also tested for these samples (Table 21):
[0398] comparable levels of fragmentation were observed for the two formulations (with or w/o methionine).
TABLE-US-00022 TABLE 21 Fragments by Bioanalyzer of DoE1 Formulation 8 and Reference formulation after forced oxidation treatment. Upper table: samples stored at 2-8.degree. C. Bottom table: sample stored 4 weeks at 40.degree. C. + 6 weeks at 2-8.degree. C. Oxidized with 3% H.sub.2O.sub.2 Anti PD-L1 (after 10 week ID# Buffer pH (mg/mL) Excipient Surfactant storage at 2-8.degree. C.) DoE1 - 8 10 mM 5.50 10 Mannitol Tween 20 2.4 Acetate (280 mM) (0.5 mg/mL) Reference 10 mM 5.50 10 Mannitol Tween 20 2.5 Acetate (280 mM) + (0.5 mg/mL) L-Methionine (1.4 mM) Oxidised with 3% H.sub.2O.sub.2 (after 4 week AntiPD-L1 storage at 40.degree. C. + ID# Buffer pH (mg/mL) Excipient Surfactant 6 weeks at 2-8.degree. C.) DoE1 - 8 10 mM 5.50 10 Mannitol Tween 20 2.5 Acetate (280 mM) (0.5 mg/mL) Reference 10 mM 5.50 10 Mannitol Tween 20 3.0 Acetate (280 mM) + (0.5 mg/mL) L-Methionine (1.4 mM)
[0399] These results suggest that an antioxidant is not needed to stabilize Avelumab and can, therefore, be omitted from the formulation.
Example 5
Long Term Stability Studies 5.1 Drug Product Compositions and Strengths
[0400] The Avelumab formulations 1, 2, 3, 4 and 5 listed in Table 22 were manufactured and used for a long term stability study. The manufacturing process included a compounding followed by a sterilizing double-filtration step through a 0.22 .mu.m membrane (PES and PVDF filters were tested) before the final filling in vials. Formulation 5 corresponds to the Reference used also in the DoE-1 and -2 studies as described in Example 2 and 3.
TABLE-US-00023 TABLE 22 DP compositions DP Compositions Formu- Formu- Formu- Formu- Refer- lation 1 lation 2 lation 3 lation 4 ence (DP 01- (DP 02- (DP 03- (DP 04- (DP 05- Ingredient(s) 190214) 190214) 180214) 180214) 190214) Avelumab 20 20 10 10 10 mg/mL mg/mL mg/mL mg/mL mg/mL Sodium 10 mM 10 mM 10 mM 10 mM 10 mM acetate buffer Mannitol 51 0 51 0 51 mg/mL mg/mL mg/mL mg/mL mg/mL Trehalose 0 106 0 106 0 Dihydrate mg/mL mg/mL mg/mL mg/mL mg/mL Polysorbate 20 0.5 0.5 0.5 0.5 0.5 mg/mL mg/mL mg/mL mg/mL mg/mL L-Methionine 0 0 0 0 1.4 mM sodium q.s to pH q.s to pH q.s to pH q.s to pH q.s to pH hydroxide or 5.2 .+-. 0.1 5.2 .+-. 0.1 5.2 .+-. 0.1 5.2 .+-. 0.1 5.5 .+-. 0.1 hydrochloric acid Filling volume 10 mL 10 mL 20 mL 20 mL 8 mL (in Type I glass vials)
[0401] Upon manufacturing (time 0), the osmolality was determined and found in line with expected value (range: 320-350 mOsm/kg).
[0402] 5.2 Stability Study Plan and Duration
[0403] Concerning the stability of the formulations, the study schedule, the storage conditions and the tests to be applied are summarized in Table 23. For each time point the table indicates the storage condition to be tested.
[0404] The storage of the samples has been carried out with the vials in the upright position. The study is to be conducted over 1 month at 40.degree. C., 6 months at accelerated conditions (at 25.degree. C.) and 12 months at long term conditions (2-8.degree. C.).
TABLE-US-00024 TABLE 23 Stability Plan T = 0.5 M 1 M 2 M 3 M 6 M 9 M 12 M Test 0 (2 wk) (4 wk) (8 wk) (13 wk) (26 wk) (39 wk) (52 wk) Colour X 40.degree. C. 25.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 40.degree. C. 25.degree. C. 25.degree. C. 25.degree. C. Turbidity X 40.degree. C. 25.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 40.degree. C. 25.degree. C. 25.degree. C. 25.degree. C. pH X 40.degree. C. 25.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 40.degree. C. 25.degree. C. 25.degree. C. 25.degree. C. Content A280- X 40.degree. C. 25.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. A320 40.degree. C. 25.degree. C. 25.degree. C. 25.degree. C. SE-HPLC X 40.degree. C. 25.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 40.degree. C. 25.degree. C. 25.degree. C. 25.degree. C. SDS-page red X 40.degree. C. 25.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 40.degree. C. 25.degree. C. 25.degree. C. 25.degree. C. SDS-page non- X 40.degree. C. 25.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. red 40.degree. C. 25.degree. C. 25.degree. C. 25.degree. C. iCE-280 X 40.degree. C. 25.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 40.degree. C. 25.degree. C. 25.degree. C. 25.degree. C. Osmolarity X 40.degree. C. N/A N/A N/A N/A N/A N/A Subvisible X 40.degree. C. 25.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. particles 40.degree. C. 25.degree. C. 25.degree. C. 25.degree. C. Potency X 40.degree. C. 25.degree. C. N/A 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 2-8.degree. C. 40.degree. C. 25.degree. C. 25.degree. C.
[0405] Data were collected at 40.degree. C. (up to 1 month), 25.degree. C. (up to 6 months) and 2-8.degree. C. (up to 12 months).
[0406] 5.3 Stability at 2-8.degree. C.
[0407] 5.3.1 Degree of Coloration by Visual Inspection
[0408] No changes observed over stability. All solutions remain clearer than clearest standard solution (<Y7). Values within specifications.
[0409] 5.3.2 Degree of Opalescence by Nephelometry
[0410] All solutions show turbidity comprised in the range of clear solutions (1-3 NTU). Values within specifications.
[0411] 5.3.3 pH
[0412] No changes observed over stability. All solutions show pH values in line with target (5.2.+-.0.1 for formulations 1 to 4 and 5.5.+-.0.1 for Reference DP). Values within specifications.
[0413] 5.3.4 Protein Content by OD
[0414] Concentration of formulations 1 and 2 (target concentration=20 mg/mL) was found in the range 18.7-19.8 mg/mL (within .+-.10% limits with respect to target) during the study, with no significant changes over time.
[0415] Concentration of formulations 3 and 4 and Reference DP (target concentration=10 mg/mL) was found in the range 9.3-10.2 mg/mL during the study; no significant changes found.
[0416] Protein content remains therefore unaltered over 12 month stability at 2-8 .degree. C. (Values within specifications).
[0417] 5.3.5 Dimers and HMWs by SE-HPLC
[0418] No changes in aggregation over 12 months at 2-8 .degree. C. with respect to time 0. Values within specifications.
[0419] 5.3.6 Fragments (LMWs) by SDS-PAGE N-Red
[0420] As shown in FIG. 11 the samples showed a time 0 value of LMWs by SDS-PAGE N-RED in the range 11.9-16.2%, followed by a +5-7% increase at the next point (8 weeks) and by minor changes over the rest of stability, up to six months.
[0421] 5.3.7 Sub-visible Particles
[0422] As for sub-visible particles per container, the counts were below the limits set by United States, European and Japanese Pharmacopoeia for solutions for infusion or injection with nominal content of less than 100 mL (6000 per container equal to or greater than 10 .mu.m and 600 per container equal to or greater than 25 .mu.m). The relevant bar charts for the two particle size ranges are shown in FIG. 12 and FIG. 13 respectively for sub-visible particles 10 .mu.m and sub-visible particles 25 .mu.m.
[0423] No changes in sub-visible particles upon storage were highlighted.
[0424] 5.3.8 Biological Activity
[0425] Bioactivity values were typically in the range 89-110% for all time points tested in the course of the stability study. No decrease observed upon storage.
[0426] 5.3.9 Isoforms Pattern
[0427] The results from iCE280 experiments are reported in FIG. 14 (acidic cluster, sum of peaks 1-2-3-4), FIG. 15 (main peak) and FIG. 16 (basic cluster, sum of peaks 6-7). Isoforms profile is retained throughout the 12 month stability period. At refrigerated conditions, no impact of pH on antibody's isoforms is observed.
[0428] 5.3.10 2-8.degree. C. Stability Outcome
[0429] None of the physico-chemical properties of the five formulations tested was found to undergo significant changes over the 12 month stability at 2-8.degree. C. This is surprising especially for the isoforms patterns, as in formulations 1 to 4 no methionine is present.
[0430] 5.4 Stability at 25.degree. C.
[0431] 5.4.1 Degree of Coloration by Visual Inspection
[0432] No changes observed over stability. All solutions remain clearer than clearest standard solution (<Y7). Values within specifications.
[0433] 5.4.2 Degree of Opalescence by Nephelometry
[0434] All solutions show turbidity comprised between 1-3 NTU (clear solutions range). Values within specifications.
[0435] 5.4.3 pH
[0436] No changes observed over stability. All solutions show pH values in line with target (5.2.+-.0.1 for formulations 1-2-3-4 and 5.5.+-.0.1 for Reference DP). Values within specifications.
[0437] 5.4.4 Protein Content by OD
[0438] Concentration of formulations 1 and 2 (target concentration=20 mg/mL) was found in the range 18.5-20.0 mg/mL (within .+-.10% limits with respect to target) during the study, with no significant changes over time.
[0439] Concentration of formulations 3 and 4 and Reference DP (target concentration=10 mg/mL) was found in the range 9.5-10.0 mg/mL during the study; no significant changes found.
[0440] Protein content remains therefore unaltered over six-month stability at 25.degree. C. Values within specifications.
[0441] 5.4.5 Dimers and HMWs by SE-HPLC
[0442] No changes in aggregation over six months at 25.degree. C. with respect to time 0.
[0443] Aggregation lower than specification limit (not more than 5%) was found throughout the study.
[0444] 5.4.6 Fragments (LMWs) by SDS-PAGE N-Red
[0445] The samples showed a time 0 value of LMWs by SDS-PAGE N-RED in the range 11.9-16.2%, followed by step-wise increase at the next point (4 weeks) followed by minor changes over the rest of stability, up to six months (FIG. 17).
[0446] 5.4.7 Sub-visible Particles
[0447] As for sub-visible particles per container, the counts were below the limits set by United States, European and Japanese Pharmacopoeia for solutions for infusion or injection with nominal content of less than 100 mL (6000 per container equal to or greater than 10 .mu.m and 600 per container equal to or greater than 25 .mu.m). The relevant bar-charts are shown in FIG. 18 and FIG. 19.
[0448] No changes in sub-visible particles upon stability at 25.degree. C. were highlighted.
[0449] 5.4.8 Biological Activity
[0450] Bioactivity values were typically in the range 90-110% for all time points tested in the course of the stability study. No decreases observed upon stability at 25.degree. C.
[0451] 5.4.9 Isoforms Pattern
[0452] The results from iCE280 experiments are reported in FIG. 20 (acidic cluster, sum of peaks 1-2-3-4), FIG. 21 (main peak) and FIG. 22 (basic cluster, sum of peaks 6-7).
[0453] Acidic cluster tends to increase over storage at 25.degree. C. All samples show acidic cluster increase of about +10% after six months at 25.degree. C. and concurrent decrease in main peak (-5% after 6 months) and basic cluster (-5% after 6 months).
[0454] 5.4.10 25.degree. C. Stability Outcome
[0455] Over 6-month stability at 25.degree. C., the five formulations tested showed no changes in terms of protein content, appearance, clarity, pH, aggregates, sub-visible particles and bioactivity with respect to time 0.
[0456] Fragments were found to increase by +5 percentage points according to SDS-PAGE N-RED after six-months at 25.degree. C., while no statistically significant changes were highlighted by Bioanalyzer.
[0457] Similar behavior in isoforms profile by iCE280: acidic cluster of all formulations tend to increase by +10% over the six month-study, with concurrent decreases in main peak and basic cluster.
[0458] 5.5 Stability at 40.degree. C.
[0459] 5.5.1 Degree of Coloration by Visual Inspection
[0460] No changes observed over stability. All solutions remain clearer than clearest standard solution (<Y7).
[0461] 5.5.2 Degree of Opalescence by Nephelometry
[0462] No changes observed over stability. All solutions show turbidity comprised of 2 NTU (clear solutions range). Values within specifications.
[0463] 5.5.3 pH
[0464] No changes observed over stability. All solutions show pH values in line with target (5.2.+-.0.1 for formulations 1-2-3-4 and 5.5.+-.0.1 for Reference DP). Values within specifications.
[0465] 5.5.4 Protein Content by OD
[0466] Concentration of formulations 1 and 2 (target concentration=20 mg/mL) was found in the range 18.0-19.0 mg/mL (within .+-.10% limits with respect to target) during the study, with no tendency towards loss in protein over time.
[0467] Concentration of formulations 3 and 4 and Reference DP (target concentration=10 mg/mL) was found in the range 9.5-10.0 mg/mL during the study, with no tendency towards loss in protein over time. Values within specifications.
[0468] Heat stress is, in conclusion, not detrimental to protein content at the conditions tested (up to 1 month at 40.degree. C.).
[0469] 5.5.5 Dimers and HMWs by SE-HPLC
[0470] No major changes in aggregation were highlighted after 1 month. All values below 1% total aggregates after 1 month (lower than specification limits, that is not more than 5%). 5.5.6 Fragments (LMWs) by SDS-PAGE N-Red, Bioanalyzer
[0471] Given the variability of the SDS-PAGE N-RED method (for instance, time 0 values of 11.9 and 14.5 were determined for DP 01-190214 and DP 02-190214 respectively) it can be concluded that no major changes occur during the study at 40 .degree. C. (FIG. 23).
[0472] 5.5.7 Sub-visible Particles
[0473] As for sub-visible particles per container, the counts were abundantly below the limits set by United States, European and JP Pharmacopoeia for solutions for infusion or injection with nominal content of less than 100 mL (6000 per container equal to or greater than 10 .mu.m and 600 per container equal to or greater than 25 .mu.m). Relevant bar charts shown in FIG. 24 and FIG. 25.
[0474] No changes in sub-visible particles upon thermal stress were highlighted.
[0475] 5.5.8 Biological Activity
[0476] Bioactivity values were typically in the range 99-120% for all time points tested in the course of the stability study. No decrease observed upon thermal stress in the samples.
[0477] 5.5.9 Isoforms Pattern
[0478] The results from iCE280 experiments are reported in FIG. 26 (acidic cluster, sum of peaks 1-2-3-4), FIG. 27 (main peak) and FIG. 28 (basic cluster, sum of peaks 6-7).
[0479] Acidic cluster tends to increase over storage at 40.degree. C.
[0480] Main peak variations (Fehler! Verweisquelle konnte nicht gefunden werden.) confirmed a slightly better stability of new formulas at 10 mg/mL and identical behavior of the remaining compositions.
[0481] Results obtained with basic cluster determination confirmed the above described results.
[0482] Up to two weeks, similar behavior was observed in the five compositions. At higher stability times, slight differentiation arises between the 20 mg/mL and the 10 mg/mL Avelumab DP (slightly better resistance in formulas at 10 mg/mL).
[0483] 5.5.10 40.degree. C. Stability Outcome
[0484] At 40.degree. C. (1 month), the five formulations tested showed no changes in terms of protein content, appearance, clarity, pH, aggregates, sub-visible particles and bioactivity with respect to time 0.
[0485] Small differences between 10 mg/mL and 20 mg/mL DP formulations highlighted by iCE280 (acidic cluster tends to undergo some increase upon storage, slightly more evident in 20 mg/mL than in 10 mg/mL DP formulations).
[0486] 5.6 Conclusions
[0487] 5.6.1 Stability at 2-8.degree. C. (12 Months)
[0488] All formulations were found to be stable: no significant changes observed in terms of appearance, turbidity (by nephelometry), sub-visible particles, pH, protein content (by OD), aggregation (by SE-HPLC), fragments (by SDS-PAGE N-RED and Bioanalyzer), isoforms profile (by iCE280) and biological activity (by bioassay) with respect to time 0.
[0489] 5.6.2 Stability at 25.degree. C. (6 Months)
[0490] No changes in terms of protein content, appearance, clarity, pH, aggregates, sub-visible particles and bioactivity with respect to time 0.
[0491] Fragments were found to increase by +5% according to SDS-PAGE N-RED after six-months at 25.degree. C., while no statistically significant changes were highlighted by Bioanalyzer (a method used as an additional tool to add robustness to conclusions on fragmentation occurrence).
[0492] A similar behavior was observed in isoforms profile by iCE280: acidic cluster of all formulations tend to increase by +10% over the six month-study, with concurrent decreases in main peak and basic cluster.
[0493] 5.6.3 Stability at 40.degree. C. (1 Month)
[0494] No changes in terms of protein content, appearance, clarity, pH, aggregates, sub-visible particles and bioactivity with respect to time 0,
[0495] Small differences between 10 mg/mL and 20 mg/mL DP formulations highlighted by iCE280 (acidic cluster tends to undergo some increase upon storage, slightly more evident in 20 mg/mL than in 10 mg/mL DP formulations) 5.7 Stability over 24 Months 5.7.1 Manufacturing of DP Compositions
[0496] The following DP compositions were manufactured and their stability studied over a period of 24 months:
TABLE-US-00025 TABLE 24 DP compositions DP Compositions Ingredient(s) DP 01-160414 DP 02-160414 avelumab 20 mg/mL 20 mg/mL Acetate Acid Glacial (100%) 0.60 mg/mL * 0.60 mg/mL * Mannitol 51 mg/mL 51 mg/mL Polysorbate 20 0.5 mg/mL 0.5 mg/mL sodium hydroxide 0.30 mg/mL ** 0.30 mg/mL ** Filling volume 10 mL 30 mL Strength 200 mg/vial 600 mg/vial * Corresponding to 10 mM Sodium Acetate ** Final pH: 5.2
[0497] Both formulations correspond to formulation DP 01-190214 as shown in Table 22. The only difference is that a fixed amount of 0.3 mg/mL (7.5 mM) of sodium hydroxide was used to yield a pH of 5.2 when combined with 0.6 mg/mL glacial acetic acid. The sole difference between formulations DP 01-160414 and DP 02-160414 is that the latter formulation has a volume of 30 mL per vial, while the former has 10 mL per vial.
[0498] Both formulations were double-filtered through a 0.22 .mu.m PVDF membrane, followed by the manual filling in vials. Protein content was tested before and after filtration; the relevant results indicate that no loss of protein occurs upon double aseptic filtration.
[0499] Stability data up to 24 months (at +5.+-.3.degree. C.) and up to 6 months at +25.degree. C..+-.2.degree. C. (RH 60%.+-.5%) have been collected on the two formulations in the respective final containers (vials).
[0500] 5.7.2 Stability up to 24 Months (at +5.+-.3.degree. C.)
[0501] At +5.+-.3.degree. C., up to 24 months, no changes in protein content (by OD), HMWs (by SE-HPLC), turbidity (by nephelometry), particles formation (by light obscuration), degree of coloration (by visual inspection), and biopotency were observed. Slight increase in acidic isoforms (+5% observed for all compositions after 2 years).
[0502] No statistically significant changes were observed in terms of fragmentation by SDS-PAGE N-RED, Bioanalyzer and CE-SDS N-RED.
[0503] 5.7.3 Stability up to 6 Months at +25.degree. C..+-.2.degree. C. (RH 60%.+-.5%)
[0504] At +25.degree. C..+-.2.degree. C. (RH 60%.+-.5%), up to 6 months, no changes in protein content (by OD), HMWs (by SE-HPLC), turbidity (by nephelometry), particles formation (by light obscuration), isoforms profile (by iCE280), degree of coloration (by visual inspection), electrophoretic purity (by SDS/-PAGE RED) and biopotency were observed. Similarly to stability at 5 .degree. C., no statistically significant increase in fragmentation was observed at +25.degree. C..+-.2.degree. C. (RH 60%.+-.5%) (results confirmed by Bioanalyzer).
[0505] 5.7.4 Holding Time
[0506] Holding time before filtration (in bags, up to 24 hours at room temperatures), holding time after filtration (in bags, up to 72 hours at room temperature) and shaking (up to 24 hours at 200 rpm at room temperature) showed no significant changes in protein content, particles formation, aggregates and turbidity, thus indicating no major issues that may arise during standard times of operations typically considered during manufacturing process.
[0507] 5.7.5 Freeze/Thaw Study
[0508] A freeze/thaw study evidenced that the tested formulations can safely be frozen at -80.degree. C. and then allowed to warm up to +5.+-.3.degree. C., or +25.degree. C., with no major changes occurring to the protein.
Sequence CWU 1
1
31450PRTArtificial SequenceHeavy chain sequence of Avelumab 1Glu
Val Gln Leu Leu Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly1 5 10
15Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Ser Ser Tyr
20 25 30Ile Met Met Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp
Val 35 40 45Ser Ser Ile Tyr Pro Ser Gly Gly Ile Thr Phe Tyr Ala Asp
Thr Val 50 55 60Lys Gly Arg Phe Thr Ile Ser Arg Asp Asn Ser Lys Asn
Thr Leu Tyr65 70 75 80Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr
Ala Val Tyr Tyr Cys 85 90 95Ala Arg Ile Lys Leu Gly Thr Val Thr Thr
Val Asp Tyr Trp Gly Gln 100 105 110Gly Thr Leu Val Thr Val Ser Ser
Ala Ser Thr Lys Gly Pro Ser Val 115 120 125Phe Pro Leu Ala Pro Ser
Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala 130 135 140Leu Gly Cys Leu
Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser145 150 155 160Trp
Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val 165 170
175Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro
180 185 190Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn
His Lys 195 200 205Pro Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro
Lys Ser Cys Asp 210 215 220Lys Thr His Thr Cys Pro Pro Cys Pro Ala
Pro Glu Leu Leu Gly Gly225 230 235 240Pro Ser Val Phe Leu Phe Pro
Pro Lys Pro Lys Asp Thr Leu Met Ile 245 250 255Ser Arg Thr Pro Glu
Val Thr Cys Val Val Val Asp Val Ser His Glu 260 265 270Asp Pro Glu
Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His 275 280 285Asn
Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg 290 295
300Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly
Lys305 310 315 320Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro
Ala Pro Ile Glu 325 330 335Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro
Arg Glu Pro Gln Val Tyr 340 345 350Thr Leu Pro Pro Ser Arg Asp Glu
Leu Thr Lys Asn Gln Val Ser Leu 355 360 365Thr Cys Leu Val Lys Gly
Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp 370 375 380Glu Ser Asn Gly
Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val385 390 395 400Leu
Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp 405 410
415Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His
420 425 430Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu
Ser Pro 435 440 445Gly Lys 4502449PRTArtificial SequenceHeavy chain
sequence of Avelumab, lacking the C-terminal K 2Glu Val Gln Leu Leu
Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly1 5 10 15Ser Leu Arg Leu
Ser Cys Ala Ala Ser Gly Phe Thr Phe Ser Ser Tyr 20 25 30Ile Met Met
Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val 35 40 45Ser Ser
Ile Tyr Pro Ser Gly Gly Ile Thr Phe Tyr Ala Asp Thr Val 50 55 60Lys
Gly Arg Phe Thr Ile Ser Arg Asp Asn Ser Lys Asn Thr Leu Tyr65 70 75
80Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95Ala Arg Ile Lys Leu Gly Thr Val Thr Thr Val Asp Tyr Trp Gly
Gln 100 105 110Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly
Pro Ser Val 115 120 125Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser
Gly Gly Thr Ala Ala 130 135 140Leu Gly Cys Leu Val Lys Asp Tyr Phe
Pro Glu Pro Val Thr Val Ser145 150 155 160Trp Asn Ser Gly Ala Leu
Thr Ser Gly Val His Thr Phe Pro Ala Val 165 170 175Leu Gln Ser Ser
Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro 180 185 190Ser Ser
Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys 195 200
205Pro Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp
210 215 220Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu
Gly Gly225 230 235 240Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys
Asp Thr Leu Met Ile 245 250 255Ser Arg Thr Pro Glu Val Thr Cys Val
Val Val Asp Val Ser His Glu 260 265 270Asp Pro Glu Val Lys Phe Asn
Trp Tyr Val Asp Gly Val Glu Val His 275 280 285Asn Ala Lys Thr Lys
Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg 290 295 300Val Val Ser
Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys305 310 315
320Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu
325 330 335Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln
Val Tyr 340 345 350Thr Leu Pro Pro Ser Arg Asp Glu Leu Thr Lys Asn
Gln Val Ser Leu 355 360 365Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser
Asp Ile Ala Val Glu Trp 370 375 380Glu Ser Asn Gly Gln Pro Glu Asn
Asn Tyr Lys Thr Thr Pro Pro Val385 390 395 400Leu Asp Ser Asp Gly
Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp 405 410 415Lys Ser Arg
Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His 420 425 430Glu
Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro 435 440
445Gly3216PRTArtificial SequenceLight chain sequence of Avelumab
3Gln Ser Ala Leu Thr Gln Pro Ala Ser Val Ser Gly Ser Pro Gly Gln1 5
10 15Ser Ile Thr Ile Ser Cys Thr Gly Thr Ser Ser Asp Val Gly Gly
Tyr 20 25 30Asn Tyr Val Ser Trp Tyr Gln Gln His Pro Gly Lys Ala Pro
Lys Leu 35 40 45Met Ile Tyr Asp Val Ser Asn Arg Pro Ser Gly Val Ser
Asn Arg Phe 50 55 60Ser Gly Ser Lys Ser Gly Asn Thr Ala Ser Leu Thr
Ile Ser Gly Leu65 70 75 80Gln Ala Glu Asp Glu Ala Asp Tyr Tyr Cys
Ser Ser Tyr Thr Ser Ser 85 90 95Ser Thr Arg Val Phe Gly Thr Gly Thr
Lys Val Thr Val Leu Gly Gln 100 105 110Pro Lys Ala Asn Pro Thr Val
Thr Leu Phe Pro Pro Ser Ser Glu Glu 115 120 125Leu Gln Ala Asn Lys
Ala Thr Leu Val Cys Leu Ile Ser Asp Phe Tyr 130 135 140Pro Gly Ala
Val Thr Val Ala Trp Lys Ala Asp Gly Ser Pro Val Lys145 150 155
160Ala Gly Val Glu Thr Thr Lys Pro Ser Lys Gln Ser Asn Asn Lys Tyr
165 170 175Ala Ala Ser Ser Tyr Leu Ser Leu Thr Pro Glu Gln Trp Lys
Ser His 180 185 190Arg Ser Tyr Ser Cys Gln Val Thr His Glu Gly Ser
Thr Val Glu Lys 195 200 205Thr Val Ala Pro Thr Glu Cys Ser 210
215





D00000

D00001

D00002

D00003

D00004

D00005

D00006
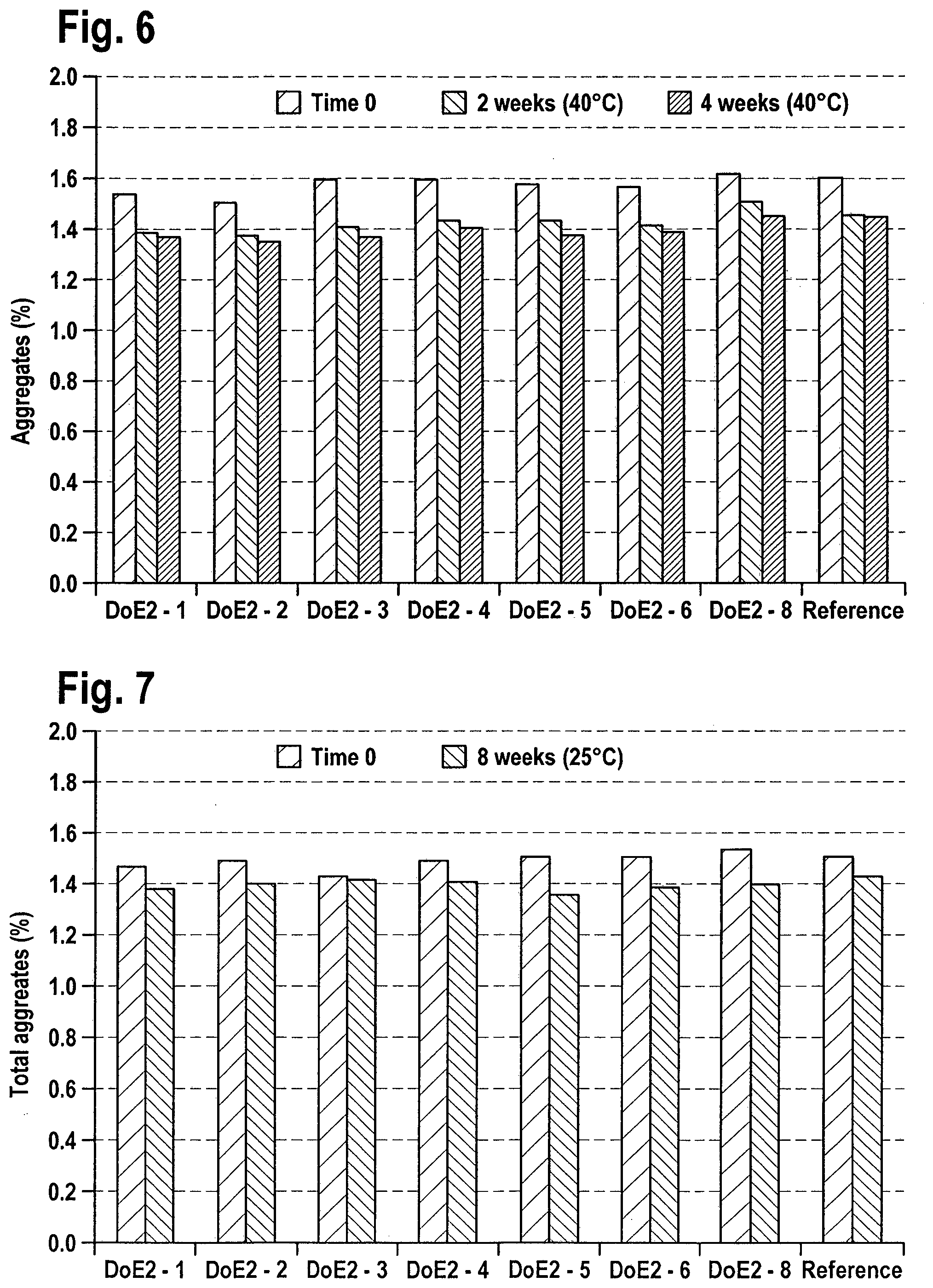
D00007

D00008

D00009

D00010

D00011
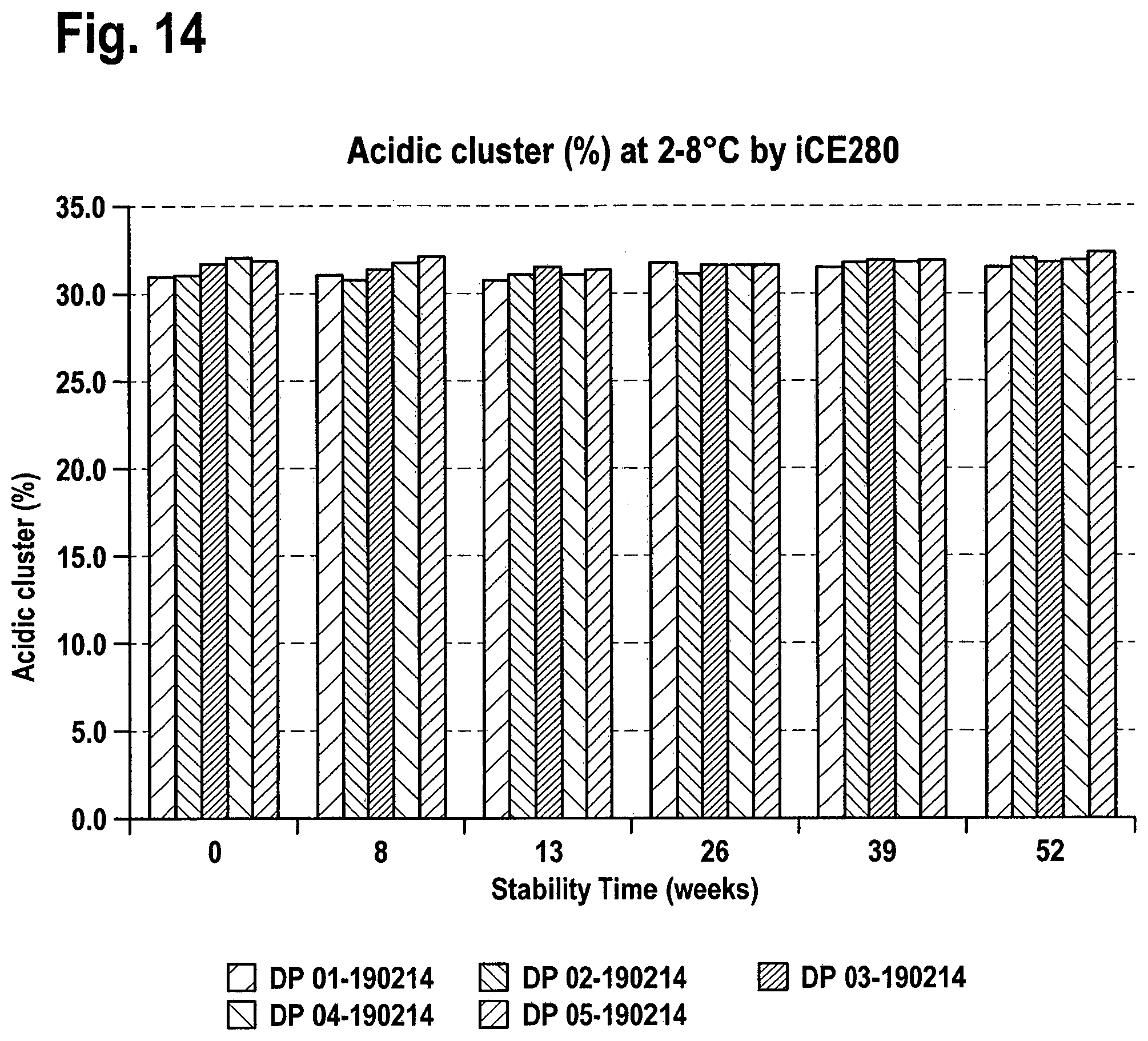
D00012

D00013

D00014

D00015
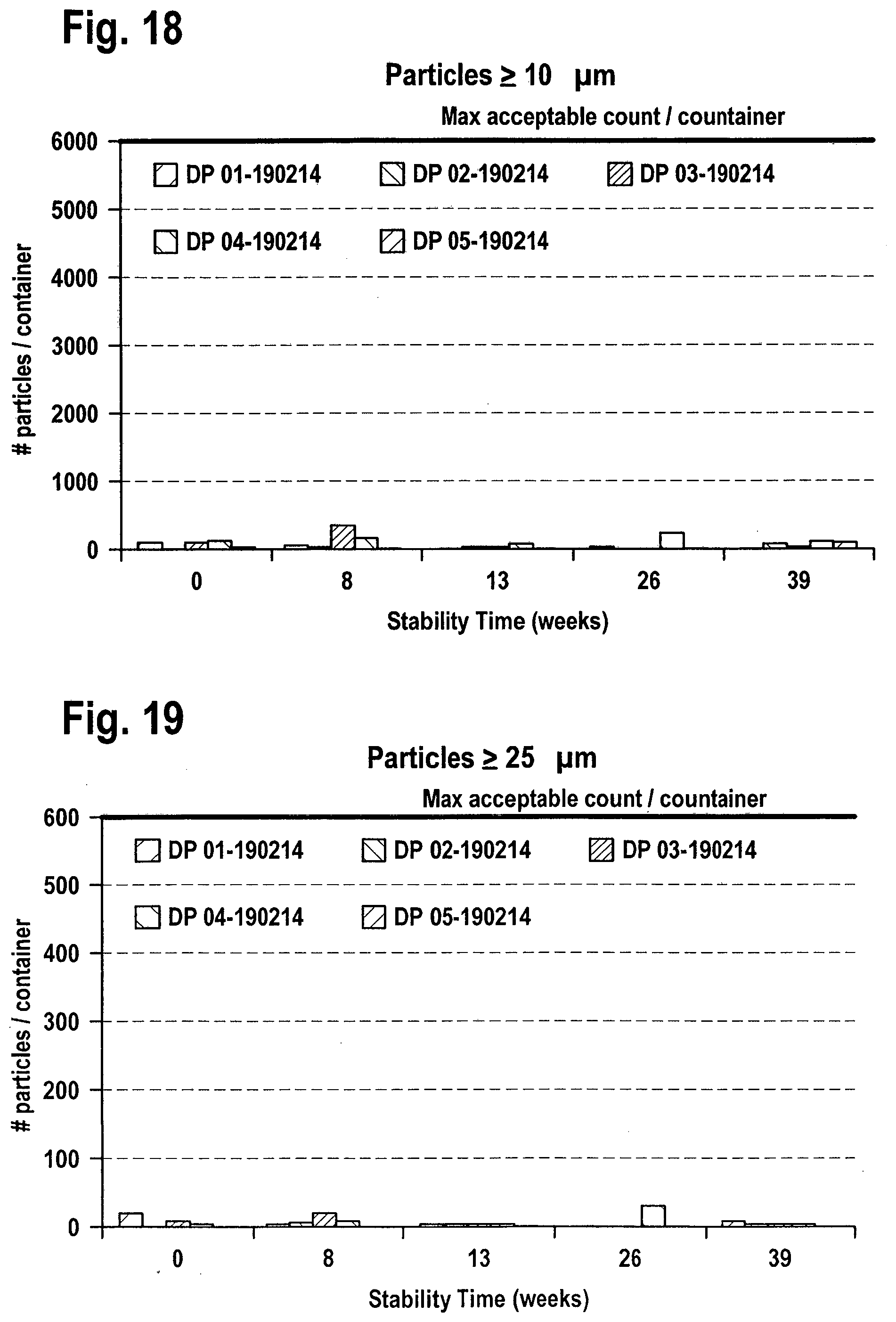
D00016

D00017

D00018

D00019

D00020

D00021

P00001

P00002

P00003

S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.