Ret Inhibitor For Use In Treating Cancer Having A Ret Alteration
Evans Raab; Erica ; et al.
U.S. patent application number 17/044884 was filed with the patent office on 2021-04-08 for ret inhibitor for use in treating cancer having a ret alteration. The applicant listed for this patent is BLUEPRINT MEDICINES CORPORATION. Invention is credited to Erica Evans Raab, Beni B. Wolf.
| Application Number | 20210100795 17/044884 |
| Document ID | / |
| Family ID | 1000005307147 |
| Filed Date | 2021-04-08 |












View All Diagrams
| United States Patent Application | 20210100795 |
| Kind Code | A1 |
| Evans Raab; Erica ; et al. | April 8, 2021 |
RET INHIBITOR FOR USE IN TREATING CANCER HAVING A RET ALTERATION
Abstract
Disclosed herein is the treatment of a subject afflicted with a cancer having an activating RET alteration by administering an effective amount of a selective RET inhibitor, e.g., Compound 1 or pharmaceutically acceptable salts thereof, including, e.g., administering an amount of 300 mg to 400 mg of the selective RET inhibitor once daily.
| Inventors: | Evans Raab; Erica; (Cambridge, MA) ; Wolf; Beni B.; (Cambridge, MA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 1000005307147 | ||||||||||
| Appl. No.: | 17/044884 | ||||||||||
| Filed: | April 3, 2019 | ||||||||||
| PCT Filed: | April 3, 2019 | ||||||||||
| PCT NO: | PCT/US2019/025655 | ||||||||||
| 371 Date: | October 2, 2020 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 62741683 | Oct 5, 2018 | |||
| 62657605 | Apr 13, 2018 | |||
| 62656297 | Apr 11, 2018 | |||
| 62652284 | Apr 3, 2018 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61P 35/04 20180101; A61K 31/506 20130101; A61P 35/00 20180101 |
| International Class: | A61K 31/506 20060101 A61K031/506; A61P 35/00 20060101 A61P035/00; A61P 35/04 20060101 A61P035/04 |
Claims
1. A method of treating a subject afflicted with a cancer having an activating rearranged during transfection (RET) alteration, the method comprising administering to the subject a therapeutically effective amount of 300 to 400 mg of Compound 1 or a pharmaceutically acceptable salt thereof once daily.
2. The method of claim 1, wherein the amount administered is 300 mg.
3. The method of claim 1 or 2, wherein the amount administered is 400 mg.
4. The method of any one of claims 1-3, wherein the cancer is chosen from papillary thyroid carcinoma (PTC), medullary thyroid cancer (MTC), pheochromocytoma (PCC), pancreatic ductal adenocarcinoma, multiple endocrine neoplasia (MEN2A and MEN2B), metastatic breast cancer, testicular cancer, small cell lung cancer, non-small cell lung cancer (NSCLC), chronic myelomonocytic leukemia (CMML), colorectal cancer, ovarian cancer, inflammatory myofibroblastic tumor, and cancer of the salivary gland.
5. The method of any one of claims 1-3, wherein the cancer is chosen from esophageal cancer, skin cancer (non-melanoma), endometrial cancer, head and neck cancer, bladder cancer, prostate cancer, hematological cancer, leukemia, soft tissue sarcoma, renal cell carcinoma (RCC), non-Hodgkin lymphoma, hepatobiliary cancer, adrenocortical carcinoma, myelodysplasia (MDS), uterine sarcoma, germ cell tumor, cervical cancer, central nervous system cancer, bone cancer, ampullary carcinoma, gastrointestinal stromal tumor, small bowel cancer, mesothelioma, rectal cancer, paraganglioma, and intrahepatic bile duct cancer.
6. The method of any one of claims 1-3, wherein the cancer is chosen from adenocarcinoma, spitzoid neoplasm, lung adenocarcinoma, adenosquamous carcinoma, colon cancer, metastatic colon cancer, metastatic papillary thyroid cancer, diffuse sclerosing variant of papillary thyroid cancer, primary myelofibrosis with secondary acute myeloid leukemia, diffuse gastric cancer, thyroid gland carcinoma, and bronchioles lung cell carcinoma.
7. The method of any one of claims 1-3, wherein the cancer is chosen from hepatobiliary cancer, ampullary carcinoma, small bowel cancer, intrahepatic bile duct cancer, metastatic colon cancer, brain cancer associated with lung cancer, brain metastasis associated with lung cancer, and retropentoneal paraganglioma.
8. The method of any one of claims 1-3, wherein the cancer is chosen from medullary thyroid cancer (MTC) and non-small cell lung cancer (NSCLC).
9. The method of claim 8, wherein the cancer is chosen from sporadic MTC, metastatic RET-altered NSCLC, tyrosine kinase inhibitor (TKI)-refractory KIF5B-RET NSCLC, and KIF5B-RET NSCLC.
10. The method of any one of claims 1-3, wherein the cancer is chosen from a brain cancer associated with a lung cancer.
11. The method of claim 10, wherein the brain cancer is brain metastasis.
12. The method of any one of claims 1-11, wherein the activating RET alteration comprises a RET mutation or a RET gene rearrangement (fusion).
13. The method of any one of claims 1-11, wherein the activating RET alteration is a RET mutation.
14. The method of claim 12 or 13, wherein the RET mutation is a point mutation.
15. The method of any one of claims 12-14, wherein the RET mutation is a resistance mutation.
16. The method of any one of claims 12-15, wherein the RET alteration is a RET mutation chosen from Table 1.
17. The method of any one of claims 12-16, wherein the RET mutation is V804M, M918T, C634R, or C634W.
18. The method of any one of claims 1-4, 8, 9, and 12-16, wherein the cancer is RET-altered medullary thyroid cancer (MTC).
19. The method of claim 18, wherein the cancer is familial MTC.
20. The method of claim 18, wherein the cancer is sporadic MTC.
21. The method of any one of claims 1-3 and 12-19, wherein the cancer is MTC having a M918T mutation.
22. The method of any one of claims 1-3 and 12-19, wherein the cancer is MTC having a C634R mutation.
23. The method of any one of claims 1-3 and 12-19, wherein the cancer is MTC having a V804M mutation.
24. The method of any one of claims 1-3, 6, and 12-16, wherein the cancer is paraganglioma.
25. The method of claim 24, wherein the cancer is retropentoneal paraganglioma.
26. The method of any one of claims 1-3, 6, 12-16, 24, and 25, wherein the paraganglioma has a R77H mutation.
27. The method of any one of claims 1-11, wherein the activating RET alteration is a gene-rearrangement (fusion).
28. The method of claim 27, wherein the activating RET alteration is a fusion with a RET fusion partner chosen from Table 2.
29. The method of claim 27 or 28, wherein the fusion is KIF5B-RET, CCDC6-RET, KIAA1468-RET, or NCOA4-RET.
30. The method of any one of claims 1-4 and 27-29, wherein the cancer is RET-altered NSCLC.
31. The method of claim 30, wherein the cancer is NSCLC having a KIF5B-RET fusion.
32. The method of claim 30, wherein the cancer is NSCLC having a CCDC6-RET fusion.
33. The method of claim 30, wherein the cancer is NSCLC having a KIAA1468-RET fusion.
34. The method of claim 30, wherein the cancer is NSCLC having a RET fusion identified as FISH positive.
35. The method of claim 29 or 30, wherein the RET alteration is KIF5B-RET V804L (cabozantinib resistant).
36. The method of claim 29 or 30, wherein the RET alteration is CCDC6-RET V804M (ponatinib resistant).
37. The method of any one of claims 1-4 and 27-29, wherein the cancer is RET-altered PTC.
38. The method of claim 37, wherein the cancer is PTC having a CCDC6-RET fusion.
39. The method of claim 37, wherein the cancer is PTC having a NCOA4-RET fusion.
40. The method of any one of claims 1-3 and 27-29, wherein the cancer is RET-altered intrahepatic bile duct carcinoma.
41. The method of claim 40, wherein the cancer is intrahepatic bile duct carcinoma having a NCOA4-RET fusion.
42. The method of any one of claims 1-41, wherein the subject has not received prior treatment with a multikinase RET inhibitor.
43. The method of any one of claims 1-41 wherein the subject has received one or more prior treatments with a multikinase RET inhibitor.
44. The method of claim 43, wherein the multikinase RET inhibitor is chosen from lenvatinib, vandetanib, cabozantinib, and RXDX-105.
45. The method of any one of claims 1-41, wherein the subject has not received prior treatment with platinum.
46. The method of any one of claims 1-41, wherein the subject has received prior treatment with platinum.
47. The method of any one of claims 1-41, wherein the subject has received prior treatment with a selective RET inhibitor.
48. The method of any one of claims 1-47, wherein the subject has not received prior chemotherapy.
49. The method of any one of claims 1-47, wherein the subject has received prior chemotherapy.
50. The method of claim 49, wherein the prior chemotherapy is chosen from carboplatin, pemetrexed, abraxane, cisplatin, bevacizumab, and combinations thereof.
51. The method of any one of claims 1-42, wherein the subject has not received prior immunotherapy.
52. The method of any one of claims 1-42, wherein the subject has received prior immunotherapy.
53. The method of claim 52, wherein the prior immunotherapy is chosen from ipilimumab, pembrolizumab, nivolumab, MPDL3280A, MEDI4736, and combinations thereof.
54. A method of treating a subject afflicted with a brain cancer associated with a RET-altered lung cancer, the method comprising administering to the subject a therapeutically effective amount of Compound 1 or a pharmaceutically acceptable salt thereof.
55. The method of claim 54, wherein the brain cancer is brain metastasis.
56. A method of treating a subject afflicted with a cancer having an activating RET mutation, the comprising administering to the subject a physiologically effective amount of a RET inhibitor, wherein administration of the RET inhibitor is associated with a sustained down-regulation of at least one effect marker in the subject.
57. The method of claim 56, wherein the RET inhibitor is orally administered.
58. The method of claim 56 or 57, wherein the RET inhibitor is Compound 1 or a pharmaceutically acceptable salt thereof.
59. The method of any one of claims 56-58, wherein the effect marker is chosen from DUSP6 mRNA expression, SPRY4 mRNA expression, carcinoembryonic antigen level, and calcitonin level.
60. The method of any one of claims 56-58, wherein the effect marker is KIF5B ctDNA level or TP53 ctDNA level.
61. The method of any one of claims 56-59, wherein the amount administered to the subject produces a greater than 95% down-regulation of at least one effect marker.
62. The method of any one of claims 56-59, wherein the amount administered to the subject produces a greater than 94%, greater than 93%, greater than 92%, greater than 91%, greater than 90%, greater than 89%, greater than 88%, greater than 87%, greater than 86% greater than 85%, greater than 80%, greater than 75%, greater than 70%, greater than 65%, greater than 60%, greater than 55%, or greater than 50% down-regulation in at least one effect marker.
63. The method of claim 61, wherein the amount administered to the subject produces a greater than 89%, greater than 88%, greater than 87%, greater than 86%, greater than 85%, greater than 80%, greater than 75%, or greater than 70% down-regulation in at least one effect marker.
64. The method of any one of claims 56-59, wherein at least two effect markers are down-regulated.
Description
[0001] This application claims priority to U.S. Provisional Application No. 62/652,284, filed Apr. 3, 2018, U.S. Provisional Application No. 62/656,297, filed Apr. 11, 2018, U.S. Provisional Application No. 62/657,605, filed Apr. 13, 2018, and U.S. Provisional Application No. 62/741,683, filed Oct. 5, 2018, the contents of which are incorporated by reference herein in its entirety.
[0002] This disclosure relates to methods for treating a subject afflicted with a cancer having an activating RET alteration by administering an effective amount of a selective RET inhibitor, i.e., a compound which is specifically designed to selectively target one or more RET or RET-altered kinases. As used herein, the term "afflicted with a cancer" means having a cancer. Said another way, a subject afflicted with a cancer has a cancer. More specifically, the methods described herein relate to treating a subject having a cancer characterized by an activating RET alteration. In some embodiments, the selective RET inhibitor is Compound 1 or pharmaceutically acceptable salts thereof. In some embodiments, the selective RET inhibitor is administered once daily. In some embodiments, the effective amount is 60 mg to 400 mg, 100 mg to 400 mg, 300 mg, or 400 mg. In some embodiments, the effective amount is 60 mg to 400 mg, 100 mg to 400 mg, 300 mg, or 400 mg administered once daily. In some embodiments, the cancer is a RET-altered solid tumor, a RET-altered non-small cell lung cancer, or a RET-altered thyroid cancer. In some embodiments, the cancer is a brain cancer, wherein the brain cancer is associated with non-small cell lung cancer. This disclosure also relates to methods of treating RET-altered cancers by administering a physiological effective dose of a selective RET inhibitor that produces a sustained down-regulation of at least one effect marker.
[0003] The receptor tyrosine kinase (RTK) RET, along with glial cell line-derived neurotrophic factors (GDNF) and GDNF family receptors-.alpha. (GFR.alpha.), is required for the development, maturation, and maintenance of several neural, neuroendocrine, and genitourinary tissue types. However, increasing evidence implicates aberrant activation of RET as a critical driver of tumor growth and proliferation across a broad number of solid tumors (Mulligan L M., Nat. Rev. Cancer. 14:173-186 (2014)). Oncogenic RET activation occurs via gain of function mutation or RET gene rearrangement resulting in the production of a RET fusion protein with constitutively active RET signaling that promotes ligand-independent tumor growth. Oncogenic RET activation was initially described in hereditary and sporadic thyroid cancers and subsequently in non-small cell lung cancer (NSCLC).
[0004] Oncogenic RET rearrangements have been identified in 1-2% of NSCLC (Lipson, D. et al., Nat. Med. 18:382-384 (2012); Takeuchi, K. et al., Nat. Med. 18:378-381 (2012); Stransky, N. et al., Nat. Commun. 5:4846 (2014)). This generates a constitutively active kinase that promotes tumorigenesis. As with anaplastic lymphoma kinase (ALK) and c-ros oncogene (ROS) 1-rearranged NSCLC, RET-rearranged NSCLC typically has adenocarcinoma histology (though occasionally squamous) and occurs in young, non-smoking patients. Because diagnostic testing for RET is not standard of care, RET-rearranged patients with advanced NSCLC are treated per NCCN guidelines for epidermal growth factor receptor (EGFR-) and ALK-negative adenocarcinoma. This usually includes chemotherapy with a platinum doublet or more recently with a checkpoint inhibitor however, clinical response and overall survival specifically in RET-rearranged NSCLC with these agents is not well understood. Subsequent therapy beyond chemotherapy and checkpoint inhibitors for refractory patients per NCCN guidelines is best supportive care or clinical trial.
[0005] Initial case reports and single-arm studies with the multikinase RET inhibitors (MKIs) cabozantinib, vandetanib, sorafenib, and alectinib in patients with known RET-rearranged NSCLC have demonstrated clinical activity, suggesting that RET may be a valid target in NSCLC. Although encouraging response rates (.about.12%-60%) (Horiike A et al., Lung Cancer 93:43-6 (March 2016); Lin J J et al., J Thorac Oncol. 11(11):2027-32 (November 2016); Gautshi O et al., J Clin Oncol. 34 (suppl; abstr 9014) (2016)) have been observed in these early studies, duration of response is typically less than a year. MKI treatment was associated with significant toxicity, requiring dose interruption and/or dose modification, which likely limit exposures required to effectively inhibit RET.
[0006] Oncogenic RET activation is also associated with thyroid cancer. Thyroid cancer consists primarily of differentiated thyroid cancer (DTC; .about.90% of cases), medullary thyroid cancer (MTC; .about.5% of cases), and anaplastic thyroid cancer (<5% of cases). DTC arises sporadically from thyroid follicular cells and consists of papillary thyroid cancer (PTC) (.about.80% of all thyroid cancer cases) and follicular thyroid cancer. In contrast, MTC arises from parafollicular C cells and occurs in both hereditary and sporadic forms. Oncogenic RET activation has been implicated as a driver in both MTC and PTC.
[0007] Recurrent gene rearrangements involving RET and a dimerization domain-encoding gene have been identified in approximately 5%-20% of sporadic papillary tumors in adults. Kinase-activating RET mutations occur in nearly all cases of hereditary MTC (87%-97%) (Machens A et al., N Engl J Med 349:1517-25 (2003); Mulligan L M et al., Nature 363(6428):458-60 (1993 Jun. 3); Mulligan L M et al., J Int Med. 238(4):343-346 (1995)) and approximately 43%-65% of sporadic MTC (Elisei R. et al., J Clin Endocrinol Metab. 93:682-687 (2008); Moura M M et al., British Journal of Cancer 100:1777-1783 (2009)). These RET mutations occur in the extracellular domain (primarily at the C634 position) which promote ligand-independent dimerization and activation of RET, and kinase domains mutations (primarily M918T, A883F or V804L/M) which promote RET auto-activation and consequent oncogenic signaling (Romei C et al., Nat Rev Endocrinol. 12(4):192-202 (2016 April)).
[0008] Both PTC and MTC are treated with surgery when localized (Fagin J A & Wells S A Jr., N Engl J Med. 375(11):1054-67 (2016 Sep. 15)). Ablative therapy with radioactive iodine (RAI) is effective in PTC patients with recurrence; however, patients eventually become refractory to RAI. As MTC arises from follicular C-cells, RAI is not effective. Once advanced, RAI-refractory PTC and MTC are poorly responsive to chemotherapy and systemic treatment with a small molecule MKI is the standard of care for both. Sorafenib and lenvatinib are approved MKIs for progressive and/or symptomatic RAI-refractory PTC. Cabozantinib and vandetanib are approved MKIs for advanced MTC and are used regardless of RET mutational status. MKIs used to treat thyroid cancer have broad activity against many kinases (e.g., RAF, MET, EGFR, VEGFR1-3, PDGFR, RET and others), and are associated with significant dermatologic, cardiovascular, and gastrointestinal side effects. Therefore, National Clinical Practice Guidelines in Oncology from the National Comprehensive Cancer Network (available at https://www.nccn.org/professionals/physician_gls/f_guidelines.asp) recommends careful monitoring and dose interruption and/or dose modification for drug-related side effects with these agents. For patients with disease progression on MKI therapy or MKI intolerance, there are no effective therapies and NCCN guidelines recommend clinical trial participation.
[0009] Given the strong genetic and preclinical evidence that activated RET is an oncogenic disease driver, the lack of selective RET inhibitors available, and the poor prognosis of many patients with RET-altered tumors, a need remains for identifying dosing amounts and schedules with the appropriate safety, exposures, and tolerability for selective RET inhibitors for the treatment of RET-altered cancers.
[0010] Small molecule compounds that selectively inhibit RET are a desirable means for treating cancers having an activating RET alteration. One small molecule is (S,4R)-N-((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methox- y-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexan- ecarboxamide (Compound 1). Compound 1 has the chemical structure:
##STR00001##
[0011] In March 2017, Compound 1 (also known as BLU-667) entered Phase I clinical trials in the United States for the treatment of patients with thyroid cancer, non-small cell lung cancer, and other advanced solid tumors (NCT03037385). WO 2017/079140, incorporated herein by reference, describes the synthesis of Compound 1 (Example Compound 130) and also discloses the therapeutic activity of this molecule to inhibit, regulate, and/or modulate RET kinase (Assays, Example 10 on pp. 72-74).
BRIEF DESCRIPTION OF THE FIGURES
[0012] FIGS. 1A, 1B, and 1C are a series of bar graphs which show the impact of Compound 1 on expression of DUSP6 and SPRY4 in LC2/ad (FIG. 1A), MZ-CRC-1 (FIG. 1B), and TT (FIG. 1C) cells.
[0013] FIG. 2 is a bar graph which shows the sustained decrease in expression of the MAPK target genes DUSP6 and SPRY4 in a KIF5B-RET NSCLC PDX model.
[0014] FIG. 3 is a graph which shows in vivo anti-tumor activity of Compound 1 in a cabozantinib-resistant tumor model generated from an engineered KIF5B-RET V804L cell line.
[0015] FIG. 4A is a graph which shows tumor size and levels of calcitonin and CEA (carcinoembryonic antigen) decrease over the course of treatment with Compound 1. The RET-mutant MTC patient (RET L629P, D631_R635DELINSG, V637R MTC) was treated with 60 mg once daily and then received successive dose escalation up to 300 mg once daily. FIG. 4B is a CT scan of the same RET-mutant MTC patient of FIG. 4A at baseline (top) and after 8 weeks of Compound 1 treatment (bottom) demonstrating rapid reduction in tumor growth.
[0016] FIG. 4C is a graph which shows tumor size and the levels of calcitonin and CEA decrease in a patient with RET M918T-mutant MTC over the course of treatment with Compound 1 with 300 mg once daily. FIG. 4D is a CT scan of the RET M918T-mutant patient of FIG. 4C's tumor at baseline (top) and after 24 weeks of Compound 1 treatment (bottom). FIG. 4E is a graph which shows ctDNA analysis of RET M918T levels in plasma from an MTC patient during treatment. Pre- and post-treatment tumor biopsy revealed a 93% decrease in DUSP6 and 86% decrease in SPRY4 mRNA expression after 28 days of treatment with Compound 1.
[0017] FIG. 5A is a graph which shows lung tumor and KIF5B-RET and TP53 ctDNA reduction over the course of treatment with 200 mg once daily Compound 1; FIG. 5B is a CT scan which illustrates tumor at baseline (top) and after 32 weeks of Compound 1 treatment (bottom).
[0018] FIG. 6A is a graph which shows the mean plasma concentration (ng/mL) vs. time (h); FIG. 6B is a bar graph which shows the percent change from baseline in mean gene expression levels of DUSP6 and SPRY4.
[0019] FIG. 7A is a bar graph which shows dose-dependent reduction in CEA in patients measured on cycle 2, day 1. FIG. 7B is a bar graph which shows dose-dependent reduction in calcitonin in patients measured on cycle 2 day 1.
[0020] FIG. 8 is a waterfall plot which shows maximum tumor reduction-sum of diameter change from baseline percent--from patients in the phase I clinical study. Data cut-off: Apr. 6, 2018.
[0021] FIG. 9A is a brain CT scan at baseline prior to treatment with Compound 1. FIG. 9B is a brain CT scan after 8 weeks of treatment with Compound 1 treatment.
[0022] FIG. 10 is a chart which shows patient response rate in RET-altered NSCLC. Data cut-off: Apr. 6, 2018.
[0023] FIG. 11A is a CT scan at baseline prior to treatment with Compound 1. FIG. 11B is a CT scan after 8 weeks of treatment with Compound 1. FIG. 11C is a CT scan at baseline prior to treatment with Compound 1. FIG. 11D is a CT scan after 8 weeks of treatment with Compound 1.
[0024] FIG. 12 is a graph which shows that the response rate in medullary thyroid cancer patients increases with dose and duration of therapy. Specifically, the graph shows the response rate for dosing Compound 1 at 60 to 200 mg once daily and 300/400 mg once daily over a period of 8 to 24+ weeks.
[0025] FIG. 13 is a CT scan at baseline (BSL) and after 5 months of treatment with Compound 1 at 400 mg once daily.
ABBREVIATIONS AND DEFINITIONS
[0026] The following abbreviations and terms have the indicated means throughout:
[0027] "Compound 1" is (1S,4R)-N-((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridine-3-yl)ethyl)-1-meth- oxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohex- anecarboxamide:
##STR00002##
[0028] As used herein, "DOR" means duration of response.
[0029] As used herein, "PD" means progressive disease.
[0030] As used herein, "SD" means stable disease.
[0031] As used herein, "CR" means complete response.
[0032] As used herein, "ORR" means overall all response rate.
[0033] As used herein, "CBR" means clinical benefit rate.
[0034] As used herein, "PFS" means progression free survival.
[0035] As used herein, a "fusion" is a protein that results from a chromosomal translocation in which two genes are joined with an in-frame coding sequence and results in a chimeric protein. In some embodiments, a fusion is a chromosomal translocation where the kinase domain of one protein fuses to a dimerization domain of another gene.
[0036] As used herein, a "RET-altered cancer" is a cancer having an activating rearranged during transfection (RET) alteration, which drives tumorigenesis. Non-limiting examples of activating RET alterations include mutations, fusions, and copy number variations.
[0037] As used herein, a "RET fusion" is a gene rearrangement. RET rearrangements create a fusion protein juxtaposing the RET kinase domain and a dimerization domain of another protein, creating a constitutively activated dimer, which drives tumorigenesis.
[0038] As used herein, a "RET fusion protein" is the result of a gene rearrangement.
[0039] As used herein, a "RET activating mutation" means a mutation in RET kinase which promotes ligand-independent, constitutive RET kinase activation, which drives tumorigenesis. For example, RET mutations can occur in the extracellular cysteine residues (e.g., C620R or C634R/W), which trigger aberrant receptor dimerization, or RET mutations can occur in the intracellular kinase domain.
[0040] As used herein, a "RET inhibitor" is a compound which inhibits the activity of RET kinase. RET kinase is wild-type RET kinase and/or one or more RET-altered kinases (e.g., RET fusion, RET mutation, or RET copy number variation).
[0041] Examples of RET inhibitors include, but are not limited to, Compound 1, LOXO-292 (selpercatinib), cabozantinib, vandetanib, alectinib, sorafenib, levatinib, ponatinib, dovitinib, sunitinib, foretinib, sitravatinib, DS-5010 (BOS172738), and RXDX-105.
[0042] In some embodiments, a RET inhibitor may also inhibit other kinases. As used herein, a "multi-kinase RET inhibitor" is a compound which inhibits wild type RET kinase and inhibits at least one other kinase equally or more potently than wild type RET kinase. Examples of multikinase RET inhibitors include: cabozantinib; vandetanib; alectinib; sorafenib; levatinib, ponatinib; dovitinib; sunitinib; foretinib; sitravatinib; DS-5010; and RXDX-105.
[0043] As used herein, the term "selective RET inhibitor" means a compound which selectively inhibits RET kinase. RET kinase can include RET wild type kinase and/or one or more RET-altered kinases (e.g., RET fusion, RET mutation, or RET copy number variation). A selective RET inhibitor's inhibitory activity against RET kinase is more potent in terms of IC.sub.50 value (i.e., the IC.sub.50 value is subnanomolar) when compared with its inhibitory activity against many other kinases (e.g., KDR, VEGFR-2, ABL, EGFR, FGFR2, HER2, IGFIR, JAKI, KIT, MET, AKTI, MEKI). Potency can be measured using known biochemical assays. Examples of selective RET inhibitors include Compound 1 and selpercatinib.
[0044] As used herein, the term "subject" or "patient" refers to organisms to be treated by the methods of the present disclosure. Such organisms include, but are not limited to, mammals (e.g., murines, simians, equines, bovines, porcines, canines, felines, and the like), and in some embodiments, humans.
[0045] Many cancers have been linked to aberrant RET expression (Kato et al., Clin. Cancer Res. 23(8):1988-97 (2017)). Non-limiting examples of "cancer" as used herein include lung cancer, head and neck cancer, gastrointestinal cancer, breast cancer, skin cancer, genitourinary tract cancer, gynecological cancer, hematological cancer, central nervous system (CNS) cancer, peripheral nervous system cancer, endometrial cancer, colorectal cancer, bone cancer, sarcoma, spitzoid neoplasm, adenosquamous carcinoma, pheochromocytoma (PCC), hepatocellular carcinoma, multiple endocrine neoplasia (MEN2A and MEN2B), and inflammatory myofibroblastic tumor. For other examples, see Nature Reviews Cancer 14:173-86(2014).
[0046] Additional non-limiting examples of cancer include hemangiopericytoma, differentiated thyroid carcinoma, anaplastic thyroid carcinoma, lung carcinosarcoma, ureter urothelial carcinoma, uterine carcinosarcoma, basal cell carcinoma, Merkel cell carcinoma, atypical lung carcinoma, fallopian tube adenocarcinoma, ovarian epithelial carcinoma, salivary gland adenocarcinoma, meningioma, duodenal adenocarcinoma, cervical adenocarcinoma, adrenal carcinoma, gastroesophageal junction carcinoma, cutaneous squamous cell carcinoma, pancreatic ductal adenocarcinoma, prostate adenocarcinoma, esophageal adenocarcinoma, endometrial adenocarcinoma, ovarian serous carcinoma, carcinoma unknown primary, bladder urothelial (transition cell) carcinoma, lung squamous cell carcinoma, colorectal adenocarcinoma, head and neck squamous cell carcinoma, and gastric adenocarcinoma.
[0047] In some embodiments, the cancer is liver cholangiocarcinoma. In some embodiments, the cancer is duodenum adenocarcinoma. In some embodiments, the cancer is uterus endometrial adenocarcinoma endometrioid.
[0048] In some embodiments, MEN2A is associated with pheochromocytoma and parathyroidhyperplasia.
[0049] In some embodiments, MEN2B is associated with mucosal neuromas, pheochromocytomas, intestinal ganglioneuromas and marfanoid habitus.
[0050] In some embodiments, the lung cancer is chosen from small cell lung cancer (SCLC), lung adenocarcinoma, non-small cell lung cancer (NSCLC), bronchioles lung cell carcinoma, and mesothelioma. In some embodiments, the lung cancer is SCLC. In some embodiments, the lung cancer is NSCLC.
[0051] In some embodiments, the head and neck cancer is chosen from thyroid cancer and cancer of the salivary gland. In some embodiments, the thyroid cancer is chosen from papillary thyroid carcinoma (PTC), metastatic papillary thyroid cancer, medullary thyroid cancer (MTC), diffuse sclerosing variant of papillary thyroid cancer, and thyroid gland carcinoma. In some embodiments, the cancer is familial medullary thyroid cancer. In some embodiments, the thyroid cancer is PTC. In some embodiments, the thyroid cancer is MTC.
[0052] In some embodiments, the gastrointestinal cancer is chosen from esophageal cancer, esophagogastric cancer, gastrointestinal stromal tumor (e.g., imatinib-resistant gastrointestinal stromal tumor), small bowel cancer, diffuse gastric cancer, and ampullary carcinoma.
[0053] In some embodiments, the breast cancer is metastatic breast cancer. In some embodiments, skin cancer is melanoma or non-melanoma.
[0054] In some embodiments, the genitourinary tract cancer is chosen from colon cancer, metastatic colon cancer, bladder cancer, renal cell carcinoma (RCC), prostate cancer, hepatobiliary cancer, intrahepatic bile duct cancer, adrenocortical carcinoma, pancreatic cancer, and pancreatic ductal adenocarcinoma.
[0055] In some embodiments, the gynecological cancer is chosen from uterine sarcoma, germ cell tumor, cervical cancer, rectal cancer, testicular cancer, and ovarian cancer. In some embodiments, the hematological cancer is chosen from leukemia, primary myelofibrosis with secondary acute myeloid leukemia, myelodysplasia (MDS), non-Hodgkin lymphoma, chronic myeloid leukemia, Philadelphia chromosome-positive acute lymphoblastic leukemia, and chronic myelomonocytic leukemia (CMML).
[0056] In some embodiments, the peripheral nervous system cancer is paraganglioma. In some embodiments, the endometrial cancer isendometrial adenocarcinoma. In some embodiments, the sarcoma is a soft tissue sarcoma.
[0057] In some embodiments, the central nervous system (CNS) cancer is chosen from brain cancer associated with lung cancer and glioma.
[0058] Lung cancer is known to spread to the brain in about 40 percent of cases in which a metastasis has occurred. With lung cancer, this is considered stage 4 of the disease, and the average survival time with brain metastases is usually less than a year. Lung cancers with metastases to the brain have a relatively poor prognosis, e.g., chemotherapy drugs. Brain metastases are difficult to treat for many reasons. Often, by the time the patient first exhibits symptoms, they already have multiple lesions. Brain metastases tend to be very aggressive. The brain has many defenses to reduce the penetration of harmful substances. Specifically, the blood-brain-barrier prevents many medications, e.g., compounds from entering the brain. Treatment options may damage surrounding normal tissue and have a significant impact on the quality of life. In particular, there is a need to provide compounds that can be administered at a safe dose, with good tolerability, and which penetrate the brain for treatment of brain metastases.
[0059] In some embodiments, the cancer is brain metastasis associated with lung cancer.
[0060] In some embodiments, the cancer is a "RET-altered cancer," which, as used herein, means the cancer has an activating RET alteration. In some embodiments, the RET-altered cancer has a RET mutation or a RET gene rearrangement. In some embodiments, the RET-altered cancer is a RET-altered solid tumor.
[0061] As used herein, the term "effective amount" refers to the amount of a selective RET inhibitor (e.g., Compound 1 or a pharmaceutically acceptable salt thereof) sufficient to effect beneficial or desired results. Beneficial or desired results may be a therapeutic benefit or result or a physiological benefit or result. An effective amount can be administered in one or more administrations, applications, or dosages and is not intended to be limited to a specific formulation or administration route.
[0062] As used herein, the term "therapeutically effective amount" refers to the amount of a selective inhibitor (e.g., Compound 1 or a pharmaceutically acceptable salt thereof) sufficient to effect beneficial or desired therapeutic results in a subject. A therapeutically effective amount can be administered to a subject in need thereof in one or more administrations, applications, or dosages and is not intended to be limited to a specific formulation or administration route. In some embodiments, a therapeutically effective amount provides the desired safety, exposure, and tolerability. Selecting the therapeutically effective amount, i.e., the right dose for administering a compound, is a required step in the development of a pharmaceutical drug for clinical use. Without adequate information on dosage, it is not possible for doctors to prescribe a particular drug to patients. Therefore, determining the correct drug dosage is a key question that can only be answered in clinical studies. If the dose and frequency of administration that allows safe and predictable administration cannot be identified, then the compound cannot be a medically useful or commercially viable pharmaceutical product.
[0063] As used herein, the term "physiologically effective amount" refers to the amount of a selective inhibitor (e.g., Compound 1 or a pharmaceutically acceptable salt thereof) sufficient to effect beneficial or desired physiological result in a subject. A physiological result may be a sustained down-regulation of at least one effect marker in the subject.
[0064] As used herein, the term "treating" includes any effect, e.g., lessening, reducing, modulating, ameliorating, or eliminating, that results in the improvement of the condition, disease, disorder, and the like, or ameliorating a symptom thereof.
[0065] As used herein, an "effect marker" means DUSP6 mRNA expression, SPRY4 mRNA expression, CEA, calcitonin, KIF5B ctDNA or TP53 ctDNA.
[0066] Some example embodiments of the disclosure include the following:
1. A method of treating a subject afflicted with a cancer having an activating rearranged during transfection (RET) alteration, the method comprising administering to the subject a therapeutically effective amount of 300 to 400 mg of Compound 1 or a pharmaceutically acceptable salt thereof once daily. 2. The method of embodiment 1, wherein the amount administered is 300 mg. 3. The method of embodiment 1 or 2, wherein the amount administered is 400 mg. 4. The method of any one of embodiments 1-3, wherein the cancer is chosen from papillary thyroid carcinoma (PTC), medullary thyroid cancer (MTC), pheochromocytoma (PCC), pancreatic ductal adenocarcinoma, multiple endocrine neoplasia (MEN2A and MEN2B), metastatic breast cancer, testicular cancer, small cell lung cancer, non-small cell lung cancer (NSCLC), chronic myelomonocytic leukemia (CMML), colorectal cancer, ovarian cancer, inflammatory myofibroblastic tumor, and cancer of the salivary gland. 5. The method of any one of embodiments 1-3, wherein the cancer is chosen from esophageal cancer, skin cancer (non-melanoma), endometrial cancer, head and neck cancer, bladder cancer, prostate cancer, hematological cancer, leukemia, soft tissue sarcoma, renal cell carcinoma (RCC), non-Hodgkin lymphoma, hepatobiliary cancer, adrenocortical carcinoma, myelodysplasia (MDS), uterine sarcoma, germ cell tumor, cervical cancer, central nervous system cancer, bone cancer, ampullary carcinoma, gastrointestinal stromal tumor, small bowel cancer, mesothelioma, rectal cancer, paraganglioma, and intrahepatic bile duct cancer. 6. The method of any one of embodiments 1-3, wherein the cancer is chosen from adenocarcinoma, spitzoid neoplasm, lung adenocarcinoma, adenosquamous carcinoma, colon cancer, metastatic colon cancer, metastatic papillary thyroid cancer, diffuse sclerosing variant of papillary thyroid cancer, primary myelofibrosis with secondary acute myeloid leukemia, diffuse gastric cancer, thyroid gland carcinoma, and bronchioles lung cell carcinoma. 7. The method of any one of embodiments 1-3, wherein the cancer is chosen from hepatobiliary cancer, ampullary carcinoma, small bowel cancer, intrahepatic bile duct cancer, metastatic colon cancer, brain cancer associated with lung cancer, brain metastasis associated with lung cancer, and retropentoneal paraganglioma. 8. The method of any one of embodiments 1-3, wherein the cancer is chosen from medullary thyroid cancer (MTC) and non-small cell lung cancer (NSCLC). 9. The method of embodiment 8, wherein the cancer is chosen from sporadic MTC, metastatic RET-altered NSCLC, tyrosine kinase inhibitor (TKI)-refractory KIF5B-RET NSCLC, and KIF5B-RET NSCLC. 10. The method of any one of embodiments 1-3, wherein the cancer is chosen from a brain cancer associated with a lung cancer. 11. The method of embodiment 10, wherein the brain cancer is brain metastasis. 12. The method of any one of embodiments 1-11, wherein the activating RET alteration comprises a RET mutation or a RET gene rearrangement (fusion). 13. The method of any one of embodiments 1-11, wherein the activating RET alteration is a RET mutation. 14. The method of embodiment 12 or 13, wherein the RET mutation is a point mutation. 15. The method of any one of embodiments 12-14, wherein the RET mutation is a resistance mutation. 16. The method of any one of embodiments 12-15, wherein the RET alteration is a RET mutation chosen from Table 1. 17. The method of any one of embodiments 12-16, wherein the RET mutation is V804M, M918T, C634R, or C634W. 18. The method of any one of embodiments 1-4, 8, 9, and 12-16, wherein the cancer is RET-altered medullary thyroid cancer (MTC). 19. The method of embodiment 18, wherein the cancer is familial MTC. 20. The method of embodiment 18, wherein the cancer is sporadic MTC. 21. The method of any one of embodiments 1-3 and 12-19, wherein the cancer is MTC having a M918T mutation. 22. The method of any one of embodiments 1-3 and 12-19, wherein the cancer is MTC having a C634R mutation. 23. The method of any one of embodiments 1-3 and 12-19, wherein the cancer is MTC having a V804M mutation. 24. The method of any one of embodiments 1-3, 6, and 12-16, wherein the cancer is paraganglioma. 25. The method of embodiment 24, wherein the cancer is retropentoneal paraganglioma. 26. The method of any one of embodiments 1-3, 6, 12-16, 24, and 25, wherein the paraganglioma has a R77H mutation. 27. The method of any one of embodiments 1-11, wherein the activating RET alteration is a gene-rearrangement (fusion). 28. The method of embodiment 27, wherein the activating RET alteration is a fusion with a RET fusion partner chosen from Table 2. 29. The method of embodiment 27 or 28, wherein the fusion is KIF5B-RET, CCDC6-RET, KIAA1468-RET, or NCOA4-RET. 30. The method of any one of embodiments 1-4 and 27-29, wherein the cancer is RET-altered NSCLC. 31. The method of embodiment 30, wherein the cancer is NSCLC having a KIF5B-RET fusion. 32. The method of embodiment 30, wherein the cancer is NSCLC having a CCDC6-RET fusion. 33. The method of embodiment 30, wherein the cancer is NSCLC having a KIAA1468-RET fusion. 34. The method of embodiment 30, wherein the cancer is NSCLC having a RET fusion identified as FISH positive. 35. The method of embodiment 29 or 30, wherein the RET alteration is KIF5B-RET V804L (cabozantinib resistant). 36. The method of embodiment 29 or 30, wherein the RET alteration is CCDC6-RET V804M (ponatinib resistant). 37. The method of any one of embodiments 1-4 and 27-29, wherein the cancer is RET-altered PTC. 38. The method of embodiment 37, wherein the cancer is PTC having a CCDC6-RET fusion. 39. The method of embodiment 37, wherein the cancer is PTC having a NCOA4-RET fusion. 40. The method of any one of embodiments 1-3 and 27-29, wherein the cancer is RET-altered intrahepatic bile duct carcinoma. 41. The method of embodiment 40, wherein the cancer is intrahepatic bile duct carcinoma having a NCOA4-RET fusion. 42. The method of any one of embodiments 1-41, wherein the subject has not received prior treatment with a multikinase RET inhibitor. 43. The method of any one of embodiments 1-41 wherein the subject has received one or more prior treatments with a multikinase RET inhibitor. 44. The method of embodiment 43, wherein the multikinase RET inhibitor is chosen from lenvatinib, vandetanib, cabozantinib, and RXDX-105. 45. The method of any one of embodiments 1-41, wherein the subject has not received prior treatment with platinum. 46. The method of any one of embodiments 1-41, wherein the subject has received prior treatment with platinum. 47. The method of any one of embodiments 1-41, wherein the subject has received prior treatment with a selective RET inhibitor. 48. The method of any one of embodiments 1-47, wherein the subject has not received prior chemotherapy. 49. The method of any one of embodiments 1-47, wherein the subject has received prior chemotherapy. 50. The method of embodiment 49, wherein the prior chemotherapy is chosen from carboplatin, pemetrexed, abraxane, cisplatin, bevacizumab, and combinations thereof. 51. The method of any one of embodiments 1-42, wherein the subject has not received prior immunotherapy. 52. The method of any one of embodiments 1-42, wherein the subject has received prior immunotherapy. 53. The method of embodiment 52, wherein the prior immunotherapy is chosen from ipilimumab, pembrolizumab, nivolumab, MPDL3280A, MEDI4736, and combinations thereof. 54. A method of treating a subject afflicted with a brain cancer associated with a RET-altered lung cancer, the method comprising administering to the subject a therapeutically effective amount of Compound 1 or a pharmaceutically acceptable salt thereof. 55. The method of embodiment 54, wherein the brain cancer is brain metastasis. 56. A method of treating a subject afflicted with a cancer having an activating RET mutation, the comprising administering to the subject a physiologically effective amount of a RET inhibitor, wherein administration of the RET inhibitor is associated with a sustained down-regulation of at least one effect marker in the subject. 57. The method of embodiment 56, wherein the RET inhibitor is orally administered. 58. The method of embodiment 56 or 57, wherein the RET inhibitor is Compound 1 or a pharmaceutically acceptable salt thereof. 59. The method of any one of embodiments 56-58, wherein the effect marker is chosen from DUSP6 mRNA expression, SPRY4 mRNA expression, carcinoembryonic antigen level, and calcitonin level. 60. The method of any one of embodiments 56-58, wherein the effect marker is KIF5B ctDNA level or TP53 ctDNA level. 61. The method of any one of embodiments 56-59, wherein the amount administered to the subject produces a greater than 95% down-regulation of at least one effect marker. 62. The method of any one of embodiments 56-59, wherein the amount administered to the subject produces a greater than 94%, greater than 93%, greater than 92%, greater than 91%, greater than 90%, greater than 89%, greater than 88%, greater than 87%, greater than 86% greater than 85%, greater than 80%, greater than 75%, greater than 70%, greater than 65%, greater than 60%, greater than 55%, or greater than 50% down-regulation in at least one effect marker. 63. The method of embodiment 61, wherein the amount administered to the subject produces a greater than 89%, greater than 88%, greater than 87%, greater than 86%, greater than 85%, greater than 80%, greater than 75%, or greater than 70% down-regulation in at least one effect marker. 64. The method of any one of embodiments 56-59, wherein at least two effect markers are down-regulated.
TABLE-US-00001 TABLE 1 RET Point Mutations. Example RET Point Mutation Example RET Point Mutation Amino acid position 2 Amino acid position 665 (e.g., H665Q) Amino acid position 3 Amino acid position 666 (e.g., K666E, K666M, or K666N) Amino acid position 4 Amino acid position 686 (e.g., S686N) Amino acid position 5 Amino acid position 691 (e.g., G691S) Amino acid position 6 Amino acid position 694 (e.g., R694Q) Amino acid position 7 Amino acid position 700 (e.g., M700L) Amino acid position 8 Amino acid position 706 (e.g., V706M or V706A) Amino acid position 11 Amino acid position 713 splice variant (e.g., E713K) Amino acid position 12 Amino acid position 736 (e.g., G736R) Amino acid position 13 Amino acid position 748 (e.g., G748C) Amino acid position 20 Amino acid position 750 (e.g., A750P) Amino acid position 32 (e.g., S32L) Amino acid position 765 (e.g., S765P) Amino acid position 34 (e.g., D34S) Amino acid position 766 (e.g., P766S or P766M6) Amino acid position 40 (e.g., L40P) Amino acid position 768 (e.g., E768Q or E768D) Amino acid position 64 (e.g., P64L) Amino acid position 769 (e.g., L769L) Amino acid position 67 (e.g., R67H) Amino acid position 770 (e.g., R770Q) Amino acid position 114 (e.g., R114H) Amino acid position 771 (e.g., D771N) Amino acid position 136 (e.g., glutamic Amino acid position 777 (e.g., N777S) acid to stop codon) Amino acid position 145 (e.g., V145G) Amino acid position 778 (e.g., V778I) Amino acid position 180 (e.g., arginine Amino acid position 781 (e.g., Q781R) to stop codon) Amino acid position 200 Amino acid position 790 (e.g., L790F) Amino acid position 292 (e.g., V292M) Amino acid position 791 (e.g., Y791F or Y791N) Amino acid position 294 Amino acid position 802 Amino acid position 321 (e.g., G321R) Amino acid position 804 (e.g., V804L, V804M, V804M, or V804E) Amino acid position 330 (e.g., R330Q) Amino acid position 805 (e.g., E805K) Amino acid position 338 (e.g., T338I) Amino acid position 806 (e.g., E806C, Y806E, Y806F, Y806S, Y806G, Y806H, Y806N, or Y806C) Amino acid position 360 (e.g., R360W) Amino acid position 818 (e.g., E818K) Amino acid position 373 (e.g., alanine Amino acid position 819 (e.g., S819I) to frameshift) Amino acid position 388 (e.g., V388A) Amino acid position 393 (e.g., F393L) Amino acid position 823 (e.g., G823E) Amino acid position 432 Amino acid position 826 (e.g., Y826M) .DELTA. Amino acid residues 505-506 (6- Amino acid position 833 (e.g., R833C) Base Pair In-Frame Germline Deletion in Exon 7) Amino acid position 510 (e.g., A510V) Amino acid position 841 (e.g., P841L or P841P) Amino acid position 511 (e.g., E511K) Amino acid position 843 (e.g., E843D) Amino acid position 513 (e.g., A513D) Amino acid position 844 (e.g., R844W, R844Q, or R844L) Amino acid position 515 (e.g., C515S, Amino acid position 848 (e.g., M848T) C515W) Amino acid position 525 (e.g., R525W) Amino acid position 852 (e.g., 1852M) Amino acid position 531 (e.g., C531R, Amino acid position 866 (e.g., A866W) or 9 base pair duplication) Amino acid position 532 (e.g., Amino acid position 873 (e.g., R873W) duplication) Amino acid position 533 (e.g G533C or Amino acid position 876 (e.g., A876V) G533S) Amino acid position 550 (e.g., G550E) Amino acid position 881 (e.g., L881V) Amino acid position 591 (e.g., V591I) Amino acid position 882 Amino acid position 593 (e.g., G593E) Amino acid position 883 (e.g., A883F, A883S, A883T, or A883T*) Amino acid position 600 (e.g., R600Q) Amino acid position 884 (e.g., E884K) Amino acid position 602 (e.g., I602V) Amino acid position 886 (e.g., R886W) Amino acid position 603 (e.g., K603Q Amino acid position 891 (e.g., S891A) or K603E2) Amino acid position 606 (e.g., Y606C) Amino acid position 897 (e.g., R897Q) Amino acid position 609 (e.g., C609Y, Amino acid position 898 (e.g., D898V) C609S, C609G, C609R, C609F, or C609W) Amino acid position 611 (e.g., C611R, Amino acid position 901 (e.g., E901K) C611S, C611G, C611Y, C611F, or C611W) Amino acid position 618 (e.g., C618S, Amino acid position 904 (e.g., S904F or C618Y, C618R, C618Y, C618G, C618F, S904C2) C618W) Amino acid position 619 (e.g., F619F) Amino acid position 907 (e.g., K907E or K907M) Amino acid position 620 (e.g., C620S, Amino acid position 908 (e.g., R908K) C690W, C670R, C620G, C620L, C620Y, C620F) Amino acid position 623 (e.g., E623K) Amino acid position 911 (e.g., G911D) Amino acid position 624 (e.g., D624N) Amino acid position 912 (e.g., R912P, R912Q) Amino acid position 629 (e.g., L629P) Amino acid position 630 (e.g., C630A, Amino acid position 918 (e.g., M918T, M918V, C630R, C6305, C630Y, or C630F) or M918L6) Amino acid position 631 (e.g., D631N, Amino acid position 919 (e.g., A919V) D631Y, D631A, D631G, D631V, or D631E, D631_R635DELINSG) Amino acid position 632 (e.g., E632K Amino acid position 921 (e.g., E921K) or E632G5) .DELTA. Amino acid residues 632-633 (6- Amino acid position 922 (e.g., S922P or S922Y) Base Pair In-Frame Germline Deletion in Exon 11) Amino acid position 633 (e.g., 9 base Amino acid position 930 (e.g., T930M) pair duplication) Amino acid position 634 (e.g., C634W, Amino acid position 961 (e.g., F961L) C634Y, C634S, C634R, C634F, C634G, C634L, C634A, or C634T, or an insertion ELCR2, or a 12 base pair duplication) Amino acid position 635 (e.g., R6356) Amino acid position 972 (e.g., R972G) Amino acid position 636 (e.g., T636P Amino acid position 982 (e.g., R982C) or T636M4) Amino acid position 637 (e.g., V637R) Amino acid position 640 (e.g., A640G) Amino acid position 1009 (e.g., M1009V) Amino acid position 641 (e.g., A641S Amino acid position 1017 (e.g., Dl017N) or A641T8) Amino acid position 648 (e.g., V6481) Amino acid position 1041 (e.g., V1041G) Amino acid position 649 (e.g., S649L) Amino acid position 1064 (e.g., M1064T) Amino acid position 664 (e.g., A664D) RET + 3 Amino acid position 629 (e.g., L629P) Amino acid position 637 (e.g., V637R)
[0067] Some of the RET point mutations in Table 1 are discussed in: U.S. Patent Application Publication No. 2014/0272951; Krampitz et al., Cancer 120:1920-31 (2014); Latteyer et al., J Clin. Endocrinol. Metab. 101(3): 1016-22 (2016); Silva et al. Endocrine 49.2:366-72 (2015); Jovanovic et al., Prilozi 36(1):93-107 (2015); Qi et al., Oncotarget 6(32):33993-4003 (2015); Kim et al. ACTA ENDOCRINOLOGICA-BUCHAREST 11.2, 189-194, (2015); Cecchirini et al. Oncogene, 14:2609-12 (1997); Karrasch et al., Eur. Thyroid J 5(1):73-77 (2016); Scollo et al., Endocr. J63:87-91 (2016); and Wells et al., Thyroid 25:567-610(2015).
[0068] R525W and A513D may act in combination with S891A to enhance oncogenic activity.
TABLE-US-00002 TABLE 2 RET Fusions. RET fusion partner Exemplary cancers in which the fusion is found BCR Chronic Myelomonocytic Leukemia (CMML) CLIP 1 Adenocarcinoma KIFSB NSCLC, Ovarian Cancer, Spitzoid Neoplasm; Lung Adenocarcinoma, Adenosquamous Carcinomas CCDC6 NSCLC, Colon Cancer, Papillary Thyroid Cancer; Adeno- carcinoma; Lung Adenocarcinoma; Metastatic Colorectal Cancer; Adenosquamous Carcinoma, Metastatic papillary thyroid cancer PTClex9 Metastatic papillary thyroid cancer NCOA4 Papillary Thyroid Cancer, NSCLC, Colon Cancer, Salivary Gland Cancer, Metastatic Colorectal Cancer; Lung Adenocarcinoma, Adenosquamous Carcinomas; Diffuse Sclerosing Variant of Papillary Thyroid Cancer TRIM33 NSCLC, Papillary Thyroid Cancer ERC1 Papillary Thyroid Cancer, Breast Cancer FGFRIOP CMML, Primary Myelofibrosis with secondary Acute Myeloid Leukemia MBD1 Papillary Thyroid Cancer RAB61P2 Papillary Thyroid Cancer PRKAR1A Papillary Thyroid Cancer TRIM24 Papillary Thyroid Cancer KTN1 Papillary Thyroid Cancer GOLGA5 Papillary Thyroid Cancer, Spitzoid Neoplasms HOOK3 Papillary Thyroid Cancer KIAA1468 Papillary Thyroid Cancer, Lung Adenocarcinoma TRIM27 Papillary Thyroid Cancer AKAP13 Papillary Thyroid Cancer FKBP15 Papillary Thyroid Cancer SPECC1L Papillary Thyroid Cancer, Thyroid Gland Carcinoma TBL1XR1 Papillary Thyroid Cancer, Thyroid Gland Carcinoma CEP55 Diffuse Gastric Cancer CUX1 Lung Adenocarcinoma ACBD5 Papillary Thyroid Carcinoma MYH13 Medullary Thyroid Carcinoma PIBF1 Bronchiolus Lung Cell Carcinoma KIAA1217 Papillary Thyroid Cancer, Lung Adenocarcinoma, NSCLC MPRIP NSCLC
[0069] Some of the RET fusions in Table 2 are discussed in: Grubbs et al., J Clin Endocrinol Metab, 100:788-93 (2015); Halkova et al., Human Pathology 46:1962-69 (2015); U.S. Pat. Nos. 9,297,011; 9,216,172; Le Rolle et al., Oncotarget 6(30):28929-37 (2015); Antonescu et al., Am J Surg Pathol 39(7):957-67 (2015); U.S. Patent Application Publication No. 2015/0177246; U.S. Patent Application Publication No. 2015/0057335; Japanese Patent Application Publication No. 2015/109806A; Chinese Patent Application Publication No. 105255927A; Fang, et al., Journal of Thoracic Oncology 11.2 (2016): S21-S22; European Patent Application Publication No. EP3037547A1; Lee et al., Oncotarget DOI: 10.18632/oncotarget.9137, e-published ahead of printing, 2016; Saito et al., Cancer Science 107:713-20 (2016); Pirker et al., Transl Lung Cancer Res, 4(6):797-800 (2015); and Joung et al., Histopathology 69(1):45-53 (2016).
[0070] A person of ordinary skill in the art may determine if a subject possesses a RET-altered cell, cancer, gene, or gene product, e.g., having a mutation, e.g., a fusion, deletion, insertion, translocation, frameshift, duplication, point mutation, and/or rearrangement, e.g., using a method selected from hybridization-based methods, amplification-based methods, microarray analysis, flow cytometry analysis, DNA sequencing, next-generation sequencing (NGS), primer extension, PCR, in situ hybridization, fluorescent in situ hybridization, dot blot, and Southern blot.
[0071] To detect a fusion, primary tumor samples may be collected from a subject. The samples are processed, the nucleic acids are isolated using techniques known in the art, then the nucleic acids are sequenced using methods known in the art. Sequences are then mapped to individual exons, and measures of transcriptional expression (such as RPKM, or reads per kilobase per million reads mapped), are quantified. Raw sequences and exon array data are available from sources such as TCGA, ICGC, and the NCBI Gene Expression Omnibus (GEO). For a given sample, individual exon coordinates are annotated with gene identifier information, and exons belonging to kinase domains are flagged. The exon levels are then z-score normalized across all tumors samples.
[0072] Next, genes in which 5' exons are expressed at significantly different levels than 3' exons are identified. A sliding frame is used to identify the breakpoint within an individual sample. Specifically, at each iteration, an incremental breakpoint divides the gene into 5' and 3' regions, and a t-statistic is used to measure the difference in expression (if any) between the two regions. The breakpoint with the maximal t-statistic is chosen as the likely fusion breakpoint. As used herein, "breakpoint" is the boundary at which two different genes are fused. It is sometimes referred to as a "fusion point." The location where the difference in exon expression is maximal between 5' and 3' is the inferred breakpoint of the fusion. Thousands of tumor samples can be rapidly profiled in this manner, generating a list of fusion candidates (ranked by t-statistic). High-ranking candidates can then be validated, and fusion partners identified by examining the raw RNA-seq data sets, and identifying chimeric pairs and/or split reads which support the fusion. Candidate fusions can then be experimentally confirmed as described below.
[0073] Alternatively, the methods described in Wang L et al., Genes Chromosomes Cancer 51(2):127-39 (2012). doi: 10.1002/gcc.20937, Epub 2011 Oct. 27; and Suehara Y et al., Clin Cancer Res. 18(24):6599-608 (2012). doi: 10.1158/1078-0432.CCR-12-0838, Epub 2012 Oct. 10 can also be used.
[0074] It has been proposed that the inclusion of a pharmacodynamic assessment of molecularly targeted therapies in clinical trials can streamline the drug development process (Tan D S et al., Cancer J 15(5):406-20 (2009); Sarker D & Workman P. Adv Cancer Res 96:213-68 (2007)). Pharmacodynamic biomarkers have been successfully utilized for the clinical development of kinase inhibitors, including imatinib and gefitinib (Sarker D & Workman P. Adv Cancer Res 96:213-68 (2007); Baselga J et al., J Clin Oncol 23(23):5323-33 (2005); Druker B J et al., N Engl J Med 344(14):1031-7 (2001)). As described herein, Compound 1 dose-dependently inhibited RET and SHC activation, which mirrored the inhibition of DUSP6 and SPRY4 transcription across RET-driven preclinical models, indicating that these transcripts can serve as biomarkers for RET inhibitory activity. The translational capability of these markers was established in this study in which MTC tumor shrinkage induced by Compound 1 treatment was associated with efficient inhibition of DUSP6 and SPRY4 expression within the tumor tissue. To Applicant's knowledge, this represents the first confirmation of RET target engagement by a small molecule inhibitor, multi-targeted or selective, within the clinical setting. These effect markers may be used to more precisely define the optimal dose and schedule required for effective RET inhibition.
[0075] While it is possible for Compound 1 to be administered alone, in some embodiments, Compound 1 can be administered as a pharmaceutical formulation, wherein Compound 1 is combined with one or more pharmaceutically acceptable excipients or carriers. Compound 1 may be formulated for administration in any convenient way for use in human or veterinary medicine. In certain embodiments, the compound included in the pharmaceutical preparation may be active itself, or may be a prodrug, e.g., capable of being converted to an active compound in a physiological setting.
[0076] The phrase "pharmaceutically acceptable" is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
[0077] Examples of pharmaceutically acceptable carriers include: (1) sugars, such as lactose, glucose, and sucrose; (2) starches, such as corn starch and potato starch; (3) cellulose and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose, and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients, such as cocoa butter and suppository waxes; (9) oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil, and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol, and polyethylene glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar, (14) buffering agents, such as magnesium hydroxide and aluminum hydroxide; (15) alginic acid; (16) pyrogen-free water; (17) isotonic saline; (18) Ringer's solution; (19) ethyl alcohol; (20) phosphate buffer solutions; (21) cyclodextrins such as Captisol.RTM.; and (22) other non-toxic compatible substances employed in pharmaceutical formulations.
[0078] Examples of pharmaceutically acceptable antioxidants include: (1) water soluble antioxidants, such as ascorbic acid, cysteine hydrochloride, sodium bisulfate, sodium metabisulfite, sodium sulfite, and the like; (2) oil-soluble antioxidants, such as ascorbyl palmitate, butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), lecithin, propyl gallate, alpha-tocopherol, and the like; and (3) metal chelating agents, such as citric acid, ethylenediamine tetraacetic acid (EDTA), sorbitol, tartaric acid, phosphoric acid, and the like.
[0079] Solid dosage forms (e.g., capsules, tablets, pills, dragees, powders, granules, and the like) can include one or more pharmaceutically acceptable carriers, such as sodium citrate or dicalcium phosphate, and/or any of the following: (1) fillers or extenders, such as starches, lactose, sucrose, glucose, mannitol, and/or silicic acid; (2) binders, such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinyl pyrrolidone, sucrose, and/or acacia; (3) humectants, such as glycerol; (4) disintegrating agents, such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate; (5) solution retarding agents, such as paraffin; (6) absorption accelerators, such as quaternary ammonium compounds; (7) wetting agents, such as, for example, cetyl alcohol and glycerol monostearate; (8) absorbents, such as kaolin and bentonite clay; (9) lubricants, such a talc, calcium stearate, magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate, and mixtures thereof; and (10) coloring agents.
[0080] Liquid dosage forms can include pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups, and elixirs. In addition to the active ingredient, the liquid dosage forms may contain inert diluents commonly used in the art, such as, for example, water or other solvents, solubilizing agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofuryl alcohol, polyethylene glycols, and fatty acid esters of sorbitan, and mixtures thereof.
[0081] Suspensions, in addition to the active compounds, may contain suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
[0082] Ointments, pastes, creams, and gels may contain, in addition to an active compound, excipients, such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc, and zinc oxide, or mixtures thereof.
[0083] Powders and sprays can contain, in addition to an active compound, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates, and polyamide powder, or mixtures of these substances. Sprays can additionally contain customary propellants, such as chlorofluorohydrocarbons and volatile unsubstituted hydrocarbons, such as butane and propane.
[0084] Dosage forms for the topical or transdermal administration of Compound 1 include powders, sprays, ointments, pastes, creams, lotions, gels, solutions, patches, and inhalants. The active compound may be mixed under sterile conditions with a pharmaceutically acceptable carrier, and with any preservatives, buffers, or propellants that may be required.
[0085] When Compound 1 is administered as a pharmaceutical, to humans and animals, it can be given per se or as a pharmaceutical composition containing, for example, 0.1 to 99.5% (such as 0.5 to 90%) of active ingredient in combination with a pharmaceutically acceptable carrier.
[0086] The formulations can be administered topically, orally, transdermally, rectally, vaginally, parentally, intranasally, intrapulmonary, intraocularly, intravenously, intramuscularly, intraarterially, intrathecally, intracapsularly, intradermally, intraperitoneally, subcutaneously, subcuticularly, or by inhalation.
[0087] The present disclosure is further illustrated by the following examples which should not be construed as further limiting. The contents of all references cited throughout this application are expressly incorporated herein by reference.
EXAMPLES
Example 1: DUSP6 and SPRY4 Expression Analysis
[0088] Cells were treated with the indicated compounds for 7 hours before lysis with Buffer RLT (QIAGEN, Hilden, Germany) containing 1% .beta.-mercaptoethanol. Total RNA was isolated using the Rneasy Plus Mini kit (QIAGEN, Hilden, Germany) according to the manufacturer's instructions. First-strand cDNA was synthesized using the SuperScript VILO Master Mix (Thermo Fisher Scientific, Waltham, Mass.) according to the manufacturer's instructions. Real-time qPCR was run on ViiA 7 Real Time PCR System (Thermo Fisher Scientific). For qRT-PCR, the expression of the reference gene glucuronidase beta (GUSB) was used to normalize expression of the target genes DUSP6, SPRY4, and glycogen synthase kinase 3 beta (GSK3B). Replicate qRT-PCR reactions were analyzed for each sample, and QuantStudio Real-Time PCR software (Life Technologies, Carlsbad, Calif.) normalized the average expression of DUSP6, SPRY4, or GSK3B to the average expression of the reference gene GUSB in each sample. FIGS. 1A-1C show relative transcript expression of RET pathway targets DUSP6 and SPRY4 and AKT-pathway target GSK3B 7 hours after treatment of L2C/ad cells (FIG. 1A), MZ-CRC-1 cells (FIG. 1B), or TT MTC cells (FIG. 1C) with Compound 1 or cabozantinib. FIG. 2 shows relative transcript expression of DUSP6, SPRY4 and GSK3B from KIF5B-RET NSCLC PDX. Tumors collected at the indicated times (hours) after administration of last dose. Data are the mean+SD. *P<0.05, **P<0.01, ***P<0.001, 2-sided Student's t-test. SD, standard deviation.
Example 2: Generation of KIF5B-RET Ba/F3 Cells and ENU Mutagenesis Assays
[0089] The DNA encoding the amino acid sequence of human KIF5B-RET variant 1 was placed in a lentivirus vector under a doxycycline-inducible promoter to maximize expression with a carboxyl-terminal FLAG epitope to facilitate immunodetection of the fusion by anti-FLAG antibodies. Lentiviral-mediated gene transduction was used to express KIF5B-RET in Ba/F3 cells, KIF5B-RET dependent cells were selected by IL-3 withdrawal and confirmed to express the KIF5B-RET fusion protein by immunoblot analysis. To generate Ba/F3 cells carrying V804 substitutions, WT KIF5B-RET Ba/F3 cells were mutagenized overnight with ENU and plated in 96-well plates for a period of 2 weeks in the presence of 6 concentrations of MKIs (ponatinib, regorafenib, cabozantinib, or vandetanib). The concentrations chosen ranged from 2.times.-64.times. the proliferation IC.sub.50 for each compound: 125 nM to 4 .mu.mol/L cabozantinib, 20 to 640 nM ponatinib, and 250 nM to 8 .mu.mol/L vandetanib. Genomic DNA was isolated from resistant clones, and Sanger sequencing was used to identify those that harbored substitutions. FIG. 3 shows antitumor activity of Compound 1 compared with cabozantinib in KIF5B-RET V804L Ba/F3 allografts.
Example 3: Phase I Study
[0090] A phase I, first-in-human study (NCT03037385) to define the maximum tolerated dose, safety profile, pharmacokinetics, and preliminary anti-tumor activity of Compound 1 in advanced, RET-altered NSCLC, MTC and other solid tumors was initiated. Prior to study entry, written informed consent was obtained from all patients for treatment with Compound 1 and collection of blood and tumor samples for exploratory biomarker analyses to characterize potential predictive biomarkers of safety and efficacy. Adult patients (18 years of age) must have had advanced, unresectable solid tumors, with an Eastern Cooperative Oncology Group performance status of 0 to 2, and adequate bone marrow, hepatic, renal, and cardiac function. Compound 1 was administered orally, once daily, on a 4-week cycle using a Bayesian Optimal Interval Design. At dose levels .gtoreq.120 mg, documented RET-alteration was additionally required for study entry. Adverse events were graded per Common Terminology Criteria for Adverse Events (CTCAE). Radiographic response by computed tomography was evaluated RECIST version 1.1 (European Journal of Cancer 45: 228-247 (2009)). Levels of ctDNA in plasma were assessed using the PlasmaSELECT.TM.-R64 NGS panel (Personal Genome Diagnostics, Baltimore, Md.). Serum calcitonin levels in MTC patients were measured by ELISA (Medpace, Cincinnati, Ohio). Tumor DUSP6/SPRY4 levels were analyzed by qRT-PCR (Molecular MD, Portland, Oreg.).
Case Studies
[0091] Patient 1 was a 27-year-old patient with sporadic MTC harboring multiple RET mutations (L629P, D631_R635DELINSG, and V637R). The patient was tyrosine kinase inhibitor naive prior to the start of Compound 1 treatment with highly invasive disease that required emergent tracheostomy and extensive surgery, including total thyroidectomy, central neck dissection, bilateral levels 1 through 4 neck dissection, total thymectomy, and median sternotomy. The postoperative course was complicated by chylothorax. Multidisciplinary medical consensus was against radiotherapy to the neck, and restaging scans showed left paratracheal disease with tracheal and esophageal invasion as well as metastatic disease to the lungs and liver. The two FDA approved multi-kinase drugs for MTC (vandetanib and cabozantinib) were not considered appropriate for this patient given the associated risk of VEGFR-related toxicities that can include impaired wound healing, and increase the risk of fistula formation and hemorrhage (CAPRELSA (vandetanib) [package insert]. Cambridge, Mass.: Sanofi Genzyme; 2016; COMETRIQ (cabozantinib) [package insert]. South San Francisco, Calif.: Exelixix, Inc.; 2018). Therefore, the patient was enrolled on the Compound 1 clinical trial and began treatment at the second dose level (60 mg, QD). Remarkably, after 28 days of Compound 1 therapy, there was a >90% reduction in the serum tumor marker calcitonin (FIG. 4A). After 8 weeks, target lesions were reduced by 19%. After successive dose escalations of Compound 1 to 200 mg QD, the patient achieved partial response with >30% tumor reduction per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 (FIG. 4B). This patient subsequently escalated to 300 mg QD Compound 1 and achieved a confirmed partial response (47% maximal reduction) at 10 months. Overall, carcinoembryonic antigen (CEA) levels decreased by 57% over this period. Improved health status with Compound 1 treatment allowed for removal of the patient's tracheostomy tube and a return to baseline body weight after several kilograms of weight loss prior to treatment. Compound 1 has been well tolerated throughout II months of continuous treatment with the only drug-related adverse event being transient grade 1 decrease in white blood cells, which resolved without drug interruption or dose modification. As of Apr. 13, 2018, the patient remains on therapy.
[0092] Patient 2 was a 56-year-old with sporadic RET M918T-mutant MTC, who had responded and then progressed on vandetanib, initiated therapy with Compound 1, 300 mg QD. Early signals of clinical activity emerged within the first few weeks of Compound 1 treatment: serum calcitonin decreased >90% and CEA decreased by 75% after 28 days (FIG. 4C). RET M918T circulating tumor DNA (ctDNA) decreased by 47% after 28 days and was not detectable after 56 days. Paired tumor biopsies collected pretreatment and 28 days post-treatment demonstrated a 93% reduction in DUSP6 and an 86% reduction in SPRY4 mRNA expression, confirming RET-pathway inhibition within the tumor (FIG. 4E). Importantly, these indications of activity were confirmed by radiographic response (-35%) per RECIST 1.1 after 8 weeks (FIG. 4D). The patient tolerated Compound 1 treatment well without dose interruption; drug-related adverse events were grade 1 nausea and hyperphosphatemia. The patient continues on therapy at 8 months with a confirmed partial response (maximum 47% reduction) as of Apr. 13, 2018.
[0093] Patient 3 was a 37-year-old patient with metastatic RET-altered NSCLC, who had progressed on cisplatin, pemetrexed, and bevacizumab, had tumor tissue test positive for a RET fusion via FISH analysis. The patient initiated treatment with 200 mg QD Compound 1, and ctDNA analysis at baseline revealed a canonical KIF5B-RET fusion and co-occurring TP53 mutation. Tumor reduction (-25%) was noted at first radiographic assessment after 8 weeks of treatment and correlated with a concomitant decline in KIF5B-RET and TP53 ctDNA levels (FIG. 5A). The patient achieved a partial response on the second radiographic assessment after 16 weeks (FIG. 5B) and continues on treatment through 10 months with a confirmed partial response as of Apr. 13, 2018. As observed with the MTC patients described above, Compound 1 has been well tolerated, with all drug-related adverse events being grade 1 and including constipation (resolved), dry skin, rash, and leukopenia.
[0094] Patient 4 was a 69-year-old patient with NSCLC, who had prior lung resection nephrectomy, and pleural drainage. The patient initiated treatment with 400 mg QD Compound 1. Tumor reduction was noted against KIF5B-RET NSCLC brain metastases (FIG. 9). Specifically, evidence of intracranial anti-tumor activity was observed in the patient. At baseline, the patient had an approximately 6 mm metastatic lesion in the brain, which appeared to resolve after 8 weeks on treatment. At the time of the 8-week assessment, the patient was determined to have stable disease.
[0095] Patient 5 was a 74-year-old former smoker with locally advanced KIF5B-RET NSCLC. The patient's CT scans are shown in FIGS. 11A-11D. The patient had received concurrent chemoradiation with cisplatin and pemetrexed, was then treated with carboplatin and nab-paclitaxel and eventually progressed. Next generation sequencing of the tumor tissue, along with FISH, revealed a KIF5B-RET fusion, and the patient was enrolled on a clinical trial testing a combination regimen of vandetanib and everolimus (NCT01582191). The patient achieved a partial response, but restaging scans performed after 11 cycles showed progressive disease, which was associated with clinical symptoms of increasing dyspnea and worsening performance status. The patient was then enrolled on the phase 1 trial of Compound 1. After 16 weeks of treatment with Compound 1 (300 mg QD), the patient had a partial response with 34% reduction of tumor volume (FIGS. 11C and 11D) and improvement of dyspnea and performance status. Compound 1 has been well tolerated throughout treatment, and the patient has not experienced drug-related adverse events as of Apr. 13, 2018.
[0096] Patient 6 was a 23-year old woman with PTC, sclerosing variant (CCDC6-RET fusion), who presented 6 years ago with symptomatic diffuse lung metastases requiring supplemental oxygen, since diagnosis. She had progressed on sorafenib and lenvatinib. She initiated treatment with Compound 1 at 400 mg once daily. FIG. 13 shows tumor reduction after 5 months of treatment with Compound 1. Within 5 months, she was weaned to room air.
Measuring ctDNA Levels
[0097] Levels of one example effect marker, ctDNA in plasma (e.g., KIF5B or TP53 ctDNA), may be assessed using the PlasmaSELECT.TM.-R64 NGS panel (Personal Genome Diagnostics, Baltimore, Md.). PlasmaSELECT.TM. 64 analyzes circulating tumor DNA for genetic alterations in cancer. Specifically, PlasmaSELECT.TM. 64 evaluates a targeted panel of 64 well-characterized cancer genes. Cell-free DNA is extracted from plasma and prepared using proprietary methods that accommodate low abundance sample DNA. Samples are then processed using a proprietary capture process and high coverage next-generation sequencing.
Steady State Plasma Concentration, RET IC.sub.90 and Brain IC.sub.90 (Predicted)
[0098] Blood samples were collected at pre-determined time points from patients dosed with 30 to 600 mg Compound 1 orally once daily. Plasma samples were analyzed for Compound 1 using a validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) method. The plasma Compound 1 concentration-time data were graphed using Phoenix WinNonlin.COPYRGT. (Version 6.4, Certara L. P.) or Graphpad Prism (Version 7.02). FIG. 6A shows the plasma concentration-time profile of Compound 1 at steady state. The RET IC.sub.90 and brain IC.sub.90 (predicted) are based on projections and extrapolations based on PK and PD data in animals.
[0099] A twice a day (BID) dosing schedule was also explored as part of the phase I clinical trial. The BID dosing schedule started at a 300 mg total daily dose (200 mg in the morning, 100 mg in the evening). A total of 9 patients were enrolled into the BID dose escalation: 4 patients at 300 mg total daily dose (200 mg in the morning, 100 mg in the evening) and 5 patients at 200 mg total daily dose (100 mg BID). Of the first 4 patients enrolled at the 300 mg total daily dose, 2 patients experienced dose limiting toxicities (DLTs) of Grade 3 hypertension and the dose was subsequently de-escalated to 100 mg BID. Two of 5 patients at 100 mg BID experienced DLTs, including 1 patient with Grade 3 hypertension and 1 patient with Grade 3 tumor lysis syndrome. Based on overall safety, exposure, and tolerability, QD was the superior dosing schedule and chosen for the dose expansion.
[0100] All publications and patents mentioned herein are hereby incorporated by reference in their entirety.
* * * * *
References
D00000
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
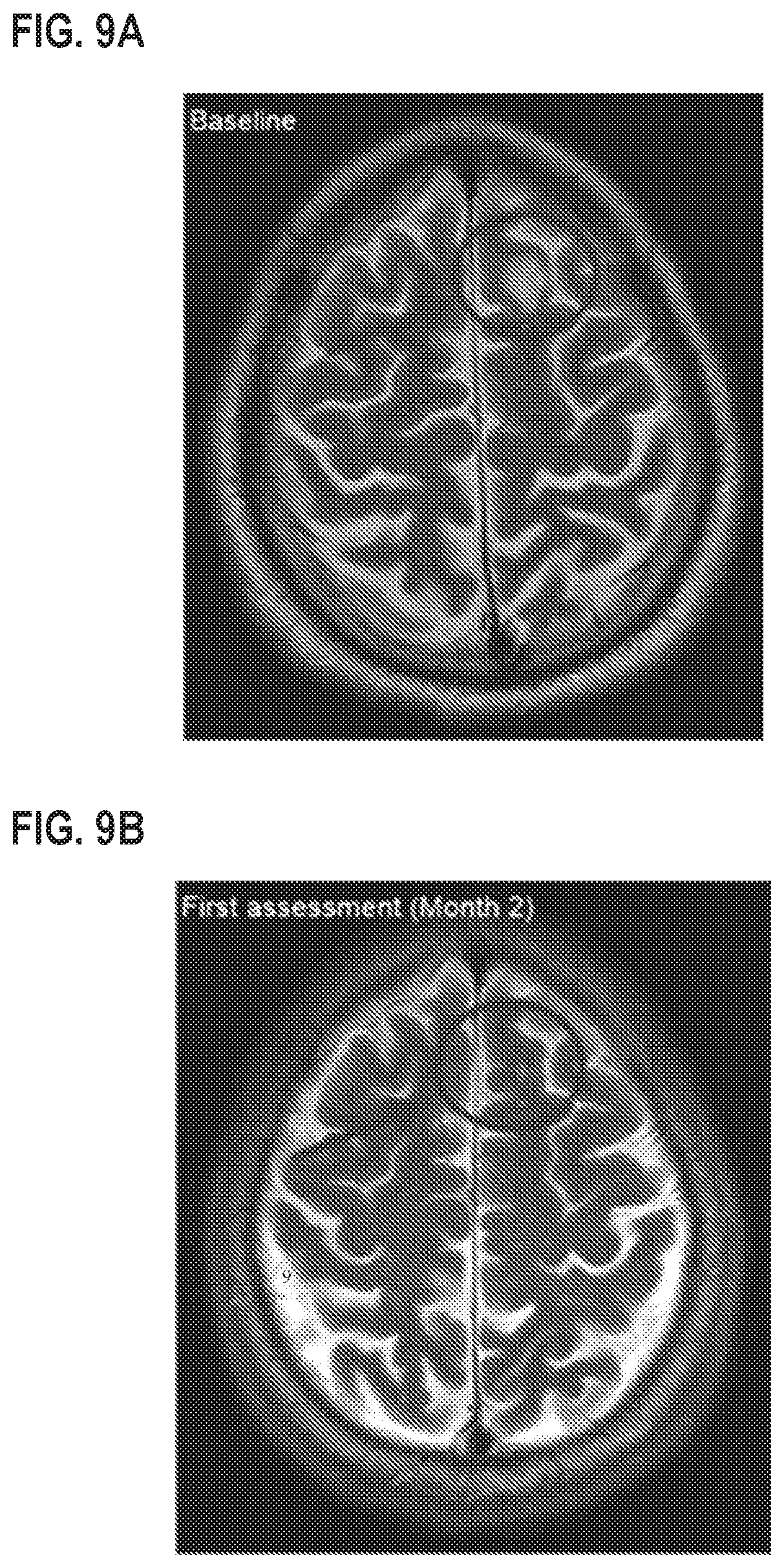
D00011

D00012

D00013

D00014

D00015

XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.