Pd-l1 Antagonist Combination Treatments
ANDREWS; Glen Ian ; et al.
U.S. patent application number 16/952620 was filed with the patent office on 2021-03-11 for pd-l1 antagonist combination treatments. The applicant listed for this patent is MERCK PATENT GMBH, PFIZER INC.. Invention is credited to Glen Ian ANDREWS, Shihao CHEN, Alessandra DI PIETRO, David FONTANA, Zelanna GOLDBERG, Chia-Yang LIN, Hua LONG, Marcella MARTIGNONI, Dimitry Serge Antoine NUYTEN, Aron David THALL, Adrian WOOLFSON.
| Application Number | 20210069326 16/952620 |
| Document ID | / |
| Family ID | 1000005226055 |
| Filed Date | 2021-03-11 |



| United States Patent Application | 20210069326 |
| Kind Code | A1 |
| ANDREWS; Glen Ian ; et al. | March 11, 2021 |
PD-L1 ANTAGONIST COMBINATION TREATMENTS
Abstract
The present disclosure describes combination therapies comprising an antagonist of Programmed Death Ligand 1 receptor (PD-L1) and another therapeutic agent, and the use of the combination therapies for the treatment of cancer.
| Inventors: | ANDREWS; Glen Ian; (San Diego, CA) ; CHEN; Shihao; (Foster City, CA) ; DI PIETRO; Alessandra; (Opera, IT) ; FONTANA; David; (Clyde Hill, WA) ; GOLDBERG; Zelanna; (San Diego, CA) ; LIN; Chia-Yang; (Palo Alto, CA) ; LONG; Hua; (San Carlos, CA) ; MARTIGNONI; Marcella; (Milan, IT) ; NUYTEN; Dimitry Serge Antoine; (San Francisco, CA) ; THALL; Aron David; (San Diego, CA) ; WOOLFSON; Adrian; (New York, NY) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 1000005226055 | ||||||||||
| Appl. No.: | 16/952620 | ||||||||||
| Filed: | November 19, 2020 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 15736615 | Dec 14, 2017 | 10869924 | ||
| PCT/US2016/037498 | Jun 15, 2016 | |||
| 16952620 | ||||
| 62180543 | Jun 16, 2015 | |||
| 62219995 | Sep 17, 2015 | |||
| 62286501 | Jan 25, 2016 | |||
| 62337489 | May 17, 2016 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61K 39/39558 20130101; C07K 16/2827 20130101; A61K 2039/505 20130101; C07K 2317/76 20130101; A61K 45/06 20130101; C07K 16/2887 20130101; A61K 33/24 20130101; A61P 35/00 20180101; A61K 39/3955 20130101; A61K 31/4184 20130101; A61K 31/4439 20130101; C07K 16/2878 20130101; C07K 2317/21 20130101; A61K 31/706 20130101; A61K 2039/507 20130101; C07K 16/243 20130101 |
| International Class: | A61K 39/395 20060101 A61K039/395; A61K 31/4439 20060101 A61K031/4439; C07K 16/24 20060101 C07K016/24; C07K 16/28 20060101 C07K016/28; A61K 45/06 20060101 A61K045/06; A61P 35/00 20060101 A61P035/00; A61K 31/4184 20060101 A61K031/4184; A61K 31/706 20060101 A61K031/706; A61K 33/24 20060101 A61K033/24 |
Claims
1-25. (canceled)
26. A method for treating a cancer in a subject comprising administering to the subject a combination therapy which comprises an antagonist of a Programmed Death Ligand 1 protein (PD-L1) and a second agent, wherein the second agent is an anti-4-1BB antibody, an anti-M-CSF antibody, or an anti-OX40 antibody.
27. The method of claim 26, wherein the PD-L1 antagonist is an anti-PD-L1 monoclonal antibody comprising three CDRs from a heavy chain variable region comprising the amino acid sequence shown in SEQ ID NO: 8 and three CDRs from a light chain variable region comprising the amino acid sequence shown in SEQ ID NO: 9; wherein the anti-4-1BB antibody comprises three CDRs from a heavy chain variable region comprising the amino acid sequence shown in SEQ ID NO: 18 and three CDRs from a light chain variable region comprising the amino acid sequence shown in SEQ ID NO: 19; wherein the anti-M-CSF antibody comprises three CDRs from a heavy chain variable region comprising the amino acid sequence shown in SEQ ID NO: 30 and three CDRs from a light chain variable region comprising the amino acid sequence shown in SEQ ID NO: 31; and wherein the anti-OX40 comprises three CDRs from a heavy chain variable region comprising the amino acid sequence shown in SEQ ID NO: 38 and three CDRs from a light chain variable region comprising the amino acid sequence shown in SEQ ID NO: 39.
28. The method of claim 26 or 27, wherein the second agent is an anti-4-1BB antibody.
29. The method of claim 28, wherein the PD-Ll antagonist is administered as a 1-hour intravenous infusion every 2 weeks at a dose of 10 mg/kg.
30. The method of claim 29, wherein the anti-4-1BB antibody is administered at 100 mg as a 1-hour IV infusion once every 4 weeks on Day 1 of each cycle.
31. The method of claim 30, wherein when the anti-4-1BB antibody and the PD-L1 antagonist are both administered on the same day, the anti-4-1BB antibody is administered first, followed by the avelumab infusion no more than 30 minutes after the end of the anti-4-1BB antibody infusion.
32. (canceled)
33. The method of claim 28, wherein the combination therapy further comprises a third agent, wherein the third agent is an anti-M-CSF antibody or an anti-OX40 antibody.
34. The method of claim 33, wherein the anti-M-CSF antibody comprises a heavy chain variable region and a light chain variable region comprising the amino acid sequences shown in SEQ ID NO: 30 and SEQ ID NO: 31, respectively.
35. The method of claim 33, wherein the anti-OX40 antibody comprises a heavy chain variable region and a light chain variable region comprising the amino acid sequences shown in SEQ ID NO: 38 and SEQ ID NO: 39, respectively.
36. The method of claim 26, wherein the subject is a human.
37. The method of claim 26, wherein the cancer is a solid tumor.
38. The method of claim 26, wherein the PD-L1 antagonist is avelumab.
39. The method of claim 38, wherein the PD-L1 antagonist is administered as an initial dose of at least about 5 mg/kg, or about 10 mg/kg.
40. The method of claim 39, wherein the PD-L1 antagonist is administered about once a week, or about once every two, three, four, or five weeks; and the second agent is administered about once a week, or about once every two, three, four, or five weeks.
41. The method of claim 40, wherein the PD-L1 antagonist is administered about once every two weeks; and the second agent is administered about once every two weeks.
42. The method of claim 26, further comprising administering a chemotherapy, radiotherapy, or chemoradiotherapy to the subject.
43. The method of claim 42, wherein the chemoradiotherapy comprises cisplatin and intensely modulated radiation therapy (IMIRT).
44. The method of claim 26, wherein the cancer is diffuse large B-cell lymphoma (DLBCL) or Squamous Cell Carcinoma of the Head and Neck (SCCHN).
45-120. (canceled)
121. The method of claim 26, wherein the cancer is selected from the group consisting of bladder cancer, breast cancer, colon cancer, clear cell kidney cancer, head/neck squamous cell carcinoma, lung squamous cell carcinoma, malignant melanoma, non-small-cell lung cancer (NSCLC), ovarian cancer, pancreatic cancer, prostate cancer, renal cell carcinoma, small-cell lung cancer (SCLC), triple negative breast cancer, acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), chronic myeloid leukemia (CML), diffuse large B-cell lymphoma (DLBCL), follicular lymphoma, Hodgkin's lymphoma (HL), mantle cell lymphoma (MCL), multiple myeloma (MM), myeloid cell leukemia-1 protein (Mcl-1), myelodysplastic syndrome (MDS), non-Hodgkin's lymphoma (NHL), small lymphocytic lymphoma (SLL), Merkel cell carcinoma, Squamous Cell Carcinoma of the Head and Neck (SCCHN), and adrenocortical carcinoma (ACC).
122-123. (canceled)
124. The method of claim 121, wherein the cancer is advanced NSCLC, RCC, or urothelial cancer (UC), and wherein the cancer has progressed on one or more prior therapies.
Description
FIELD
[0001] The present invention relates to combination therapies useful for the treatment of cancer. In particular, the invention relates to a combination therapy which comprises an antagonist of a Programmed Death-Ligand 1 protein (PD-L1) and one or more additional therapeutic agent(s).
BACKGROUND
[0002] Renal cell carcinoma (RCC) is the most common kidney cancer and constitutes about 3% of all malignant tumors in adults. Until 2005, interferon-alpha (IFN-.alpha.) and high-dose interleukin (IL)-2 therapies were the standards of care for patients with advanced RCC (aRCC), albeit with modest efficacy. Since then, development and approval of multiple vascular endothelial growth factor (VEGF) pathway and mammalian target of rapamycin (mTOR) inhibitors have significantly improved the outcomes of aRCC patients. These agents include the VEGF receptor (VEGFR) tyrosine kinase inhibitors (TKIs) sunitinib, pazopanib, axitinib and sorafenib, the mTOR inhibitors temsirolimus and everolimus, and the anti-VEGF monoclonal antibody bevacizumab. However, despite the substantial improvement of patient outcomes with these agents, durable and complete responses in aRCC patients are uncommon; the majority of patients will eventually develop resistance, exhibit disease progression while on therapy, and succumb to death due to metastatic disease.
[0003] The programmed death 1 (PD-1) receptor and PD-1 ligands 1 and 2 (PD-L1 and PD-L2, respectively) play integral roles in immune regulation. Expressed on activated T cells, PD-1 is activated by PD-L1 (also known as B7-H1) and PD-L2 expressed by stromal cells, tumor cells, or both, initiating T-cell death and localized immune suppression (Dong et al., Nat Med 1999; 5:1365-69; Freeman et al. J Exp Med 2000; 192:1027-34), potentially providing an immune-toleraant environment for tumor development and growth. Conversely, inhibition of this interaction can enhance local T-cell responses and mediate antitumor activity in nonclinical animal models (Iwai Y, et al. Proc Natl Acad Sci USA 2002; 99:12293-97). Avelumab is a fully human mAb of the IgG1 isotype that specifically targets and blocks PD-L1. Avelumab is the International Nonproprietary Name (INN) for the anti-PD-L1 monoclonal antibody MSB0010718C.
[0004] Axitinib is a VEGF receptor (VEGFR) TKI. The antitumor activity of single-agent axitinib 5 mg twice daily (BID) in previously untreated patients with clear cell aRCC was assessed against sorafenib in a randomized, open-label, Phase 3 trial. Although the study did not demonstrate a statistically significant difference in progression-free survival (PFS) between patients treated with axitinib or sorafenib, axitinib was associated with a longer median PFS (mPFS) time (mPFS of 10.1 months (95% CI 7.2,12.1) with axitinib vs. 6.5 months (95% CI 4.7, 8.3) with sorafenib, stratified hazard ratio 0.77 (95% CI 0.56, 1.05).
[0005] 4-1BB (CD137 and TNFRSF9), which was first identified as an inducible costimulatory receptor expressed on activated T cells, is a membrane spanning glycoprotein of the Tumor Necrosis Factor (TNF) receptor superfamily. Current understanding of 4-1 BB indicates that expression is generally activation dependent and encompasses a broad subset of immune cells including activated NK and NKT cells; regulatory T cells; dendritic cells (DC) including follicular DC; stimulated mast cells, differentiating myeloid cells, monocytes, neutrophils, eosinophils, and activated B cells. 4-1BB expression has also been demonstrated on tumor vasculature (19-20) and atherosclerotic endothelium. The ligand that stimulates 4-1 BB (4-1 BBL) is expressed on activated antigen presenting cells (APCs), myeloid progenitor cells and hematopoietic stem cells. 4-1BB agonist mAbs increase costimulatory molecule expression and markedly enhance cytolytic T lymphocyte responses, resulting in anti-tumor efficacy in various models. 4-1 BB agonist mAbs have demonstrated efficacy in prophylactic and therapeutic settings and both monotherapy and combination therapy tumor models and have established durable anti-tumor protective T cell memory responses
[0006] Macrophage colony stimulating factor (M-CSF) is a member of the family of proteins referred to as colony stimulating factors (CSFs). M-CSF, also known as CSF-1, is a secreted or a cell surface glycoprotein comprised of two subunits that are joined by a disulfide bond with a total molecular mass varying from 40 to 90 kD ((Stanley E. R., et al., Mol. Reprod. Dev., 46:4-10 (1997)). Similar to other CSFs, M-CSF is produced by macrophages, monocytes, and human joint tissue cells, such as chondrocytes and synovial fibroblasts, in response to proteins such as interleukin-1 or tumor necrosis factor-alpha. M-CSF stimulates the formation of macrophage colonies from pluripotent hematopoietic progenitor stem cells (Stanley E. R., et al., Mol. Reprod. Dev., 46:4-10 (1997)). M-CSF typically bind to its receptor, c-fms, in order to exert a biological effect. c-fms contains five extracellular Ig domains, one transmembrane domain, and an intracellular domain with two kinase domains. Upon M-CSF binding to c-fms, the receptor homo-dimerizes and initiates a cascade of signal transduction pathways including the JAK/STAT, PI3K, and ERK pathways.
[0007] The OX40 receptor (OX40, also known as CD134, TNFRSF4, ACT-4, ACT35, and TXGP1L) is a member of the TNF receptor superfamily. OX40 is found to be expressed on activated CD4+T-cells. High numbers of OX40+ T cells have been demonstrated within tumors (tumor infiltrating lymphocytes) and in the draining lymph nodes of cancer patients (Weinberg, A. et al., J. Immunol. 164: 2160-69, 2000; Petty, J. et al., Am. J. Surg. 183: 512-518, 2002). It was shown in tumor models in mice that engagement of OX40 in vivo during tumor priming significantly delayed and prevented the appearance of tumors as compared to control treated mice (Weinberg et al., 2000). Therefore, it has been contemplated to enhance the immune response of a mammal to an antigen by engaging OX40 through the use of an OX40 binding agent (WO 99/42585; Weinberg et al., 2000).
[0008] The rituximab antibody is a genetically engineered chimeric murine/human monoclonal antibody directed against the CD20 antigen. Rituximab is the antibody called "C2B8" in U.S. Pat. No. 5,736,137 issued Apr. 7, 1998 (Anderson et al.). rituximab is indicated for the treatment of patients with relapsed or refractory low-grade or follicular, CD20 positive, B cell non-Hodgkin's lymphoma. In vitro mechanism of action studies have demonstrated that rituximab binds human complement and lyses lymphoid B cell lines through complement-dependent cytotoxicity (CDC) (Reff et al. Blood 83(2):435-445 (1994)). Additionally, it has significant activity in assays for antibody-dependent cellular cytotoxicity (ADCC).
[0009] There is a need for improved therapies for the treatment of cancers. Furthermore, there is a need for therapies having greater efficacy than existing therapies. Preferred combination therapies of the present invention show greater efficacy than treatment with either therapeutic agent alone.
SUMMARY
[0010] This invention relates to therapeutic regimens for treatment of cancer.
[0011] Provided herein are methods for treating a cancer in a subject. Also provided are methods of inhibiting tumor growth or progression in a subject who has malignant cells.
[0012] Also provided are methods of inhibiting metastasis of malignant cells in a subject. Also provided are methods of inducing tumor regression in a subject who has malignant cells.
[0013] In some embodiments, the method comprises administering to the subject a combination therapy which comprises a PD-L1 antagonist and a VEGFR inhibitor. In some embodiments, the invention provides a medicament comprising a PD-L1 antagonist for use in combination with a VEGFR inhibitor for treating a cancer. In some embodiments, the invention provides a medicament comprising a VEGFR inhibitor for use in combination with a PD-L1 antagonist for treating a cancer. Other embodiments provide use of a PD-L1 antagonist in the manufacture of medicament for treating a cancer in a subject when administered in combination with a VEGFR inhibitor and use of a VEGFR inhibitor in the manufacture of a medicament for treating a cancer in a subject when administered in combination with a PD-L1 antagonist. In some embodiments, the invention provides use of a PD-L1 antagonist and a VEGFR inhibitor in the manufacture of medicaments for treating a cancer in a subject. In some embodiments, the medicaments comprise a kit, and the kit also comprises a package insert comprising instructions for using the PD-L1 antagonist in combination with a VEGFR inhibitor to treat a cancer in a subject. In all of the above embodiments of the treatment method, medicaments and uses herein, the VEGFR inhibitor is N-methyl-2-[3-((E)-2-pyridin-2-yl-vinyl)-1H-indazol-6-ylsulfanyl]-benz- amide or a pharmaceutically acceptable salt thereof.
[0014] Also provided are kits comprising a first container, a second container and a package insert, wherein the first container comprises at least one dose of a medicament comprising an anti-PD-L1 antagonist, the second container comprises at least one dose of a medicament comprising a VEGFR inhibitor, and the package insert comprises instructions for treating a subject for cancer using the medicaments.
[0015] In some embodiments of the above methods, medicaments, uses or kits, the VEFR inhibitor can be axitinib and can be formulated as a 1 mg tablet, 3 mg tablet, or a 5 mg tablet.
[0016] In some embodiments, the method comprises administering to the subject a combination therapy which comprises a PD-L1 antagonist and an anti-4-1BB antibody.
[0017] In some embodiments, the method comprises administering to the subject a combination therapy which comprises a PD-L1 antagonist and an anti-M-CSF antibody. In some embodiments, the method comprises administering to the subject a combination therapy which comprises a PD-L1 antagonist and an anti-OX40 antibody. In some embodiments, the method comprises administering to the subject a combination therapy which comprises a PD-L1 antagonist, an anti-4-1 BB antibody, and an anti-M-CSF antibody. In some embodiments, the method comprises administering to the subject a combination therapy which comprises a PD-L1 antagonist, an anti-4-1 BB antibody, and an anti-OX40 antibody. In some embodiments, the method comprises administering to the subject a combination therapy which comprises a PD-L1 antagonist and a CD20 antagonist. In some embodiments, the method comprises administering to the subject a combination therapy which comprises a PD-L1 antagonist, a CD20 antagonist, and an anti-4-1 BB antibody. In some embodiments, the PD-L1 antagonist is avelumab and the CD20 antagonist is rituximab. In some embodiments, the anti-4-1BB antibody is PF-05082566. In some embodiments, the method comprises administering rituximab at a dose of IV on Day 1 of a 28 day cycle, PF-05082566 at a fixed dose of 100 mg as a 1 hour IV infusion on Day 2 of each cycle, and avelumab as a 1 hour IV infusion on Day 2 and Day 16 of each cycle at a dose of 10 mg/kg. In some embodiments, the method comprises administering rituximab at a dose of IV on Day 1 of a 28 day cycle, PF-05082566 at a fixed dose of 100 mg as a 1 hour IV infusion on Day 1 of each cycle, and avelumab as a 1 hour IV infusion on Day 2 and Day 16 of each cycle at a dose of 10 mg/kg. In some embodiments, the method comprises administering rituximab at a dose of IV on Day 1 of a 28 day cycle, PF-05082566 at a fixed dose of 100 mg as a 1 hour IV infusion on Day 1 of each cycle, and avelumab as a 1 hour IV infusion on Day 1 and Day 15 of each cycle at a dose of 10 mg/kg. In some embodiments, the method comprises administering rituximab at a dose of IV on Day 1 of a 28 day cycle, PF-05082566 at a fixed dose of 100 mg as a 1 hour IV infusion on Day 2 of each cycle, and avelumab as a 1 hour IV infusion on Day 1 and Day 15 of each cycle at a dose of 10 mg/kg. In some embodiments, avelumab is administered at least 3 hours after PF-05082566 when avelumab and PF-05082566 are administered on the same day. In some embodiments, avelumab is administered about 60 minutes after PF-05082566 when avelumab and PF-05082566 are administered on the same day. In some embodiments, avelumab is administered about 30 minutes after PF-05082566 when avelumab and PF-05082566 are administered on the same day. In some embodiments, the cancer is R/R DLBCL.
[0018] In some embodiments, the method comprises administering to the subject a combination therapy which comprises a PD-L1 antagonist, a CD20 antagonist, and bendamustine. In some embodiments, the method comprises administering to the subject a combination therapy which comprises a PD-L1 antagonist, a CD20 antagonist, and bendamustine. In some embodiments, the PD-L1 antagonist is avelumab and the CD20 antagonist is rituximab. In some embodiments, the method comprises administering rituximab at a dose of 375 mg/m.sup.2 IV on Day 1 of a 28 day cycle, bendamustine at a dose of 90 mg/m.sup.2 IV on Day 2 and Day 3 of each 28 day cycle, and avelumab as a 1 hour IV infusion on Day 2 and Day 16 of each cycle at a dose of 10 mg/kg. In some embodiments, the method comprises administering rituximab at a dose of IV on Day 1 of a 28 day cycle, bendamustine at a dose of 90 mg/m.sup.2 IV on Day 1 and Day 2 of each 28 day cycle, and avelumab as a 1 hour IV infusion on Day 2 and Day 16 of each cycle at a dose of 10 mg/kg. In some embodiments, the method comprises administering rituximab at a dose of IV on Day 1 of a 28 day cycle, bendamustine at a dose of 90 mg/m.sup.2 IV on Day 2 and Day 3 of each 28 day cycle, and avelumab as a 1 hour IV infusion on Day 1 and Day 15 of each cycle at a dose of 10 mg/kg. In some embodiments, the method comprises administering rituximab at a dose of IV on Day 1 of a 28 day cycle, bendamustine at a dose of 90 mg/m.sup.2 IV on Day 1 and Day 2 of each 28 day cycle, and avelumab as a 1 hour IV infusion on Day 1 and Day 15 of each cycle at a dose of 10 mg/kg. In some embodiments, avelumab is administered at least 3 hours after bendamustine when avelumab and bendamustine are administered on the same day. In some embodiments, the cancer is R/R DLBCL.
[0019] In some embodiments, the method comprises administering to the subject a combination therapy which comprises a PD-L1 antagonist, azacitidine, and an anti-4-1BB antibody. In some embodiments, the method comprises administering to the subject a combination therapy which comprises avelumab, azacitidine, and PF-05082566. In some embodiments, the method comprises administering azacitidine at a daily dose of 75 mg/m.sup.2 subcutaneously (SC) each day from Day 1 to Day 7 of a 28 day cycle, PF-05082566 at a fixed dose of 100 mg as a 1 hour IV infusion on Day 2 of each cycle, and avelumab as a 1 hour IV infusion on Day 2 and Day 16 of each cycle at a dose of 10 mg/kg. In some embodiments, the method comprises administering azacitidine at a daily dose of 75 mg/m.sup.2 SC each day from Day 1 to Day 7 of a 28 day cycle, PF-05082566 at a fixed dose of 100 mg as a 1 hour IV infusion on Day 1 of each cycle, and avelumab as a 1 hour IV infusion on Day 2 and Day 16 of each cycle at a dose of 10 mg/kg. In some embodiments, the method comprises administering azacitidine at a daily dose of 75 mg/m.sup.2 SC each day from Day 1 to Day 7 of a 28 day cycle, PF-05082566 at a fixed dose of 100 mg as a 1 hour IV infusion on Day 1 of each cycle, and avelumab as a 1 hour IV infusion on Day 1 and Day 15 of each cycle at a dose of 10 mg/kg. In some embodiments, the method comprises administering azacitidine at a daily dose of 75 mg/m.sup.2 SC each day from Day 1 to Day 7 of a 28 day cycle, PF-05082566 at a fixed dose of 100 mg as a 1 hour IV infusion on Day 2 of each cycle, and avelumab as a 1 hour IV infusion on Day 1 and Day 15 of each cycle at a dose of 10 mg/kg. In some embodiments, on the days when avelumab is administered on the same day as azacitidine, avelumab is administered at least 3 hours after administration of azacitidine. In some embodiments, avelumab is administered at least 3 hours after PF-05082566 when avelumab and PF-05082566 are administered on the same day. In some embodiments, avelumab is administered about 60 minutes after PF-05082566 when avelumab and PF-05082566 are administered on the same day. In some embodiments, avelumab is administered about 30 minutes after PF-05082566 when avelumab and PF-05082566 are administered on the same day. In some embodiments, the cancer is R/R DLBCL.
[0020] In some embodiments, the method comprises administering to the subject a combination therapy which comprises avelumab and PF-05082566. In some embodiments, the cancer is advanced NSCLC, RCC, or urothelial cancer which was resistant (responded and then progressed) or refractory (never responded) to prior therapy(ies), including for example a single-agent immune checkpoint inhibitor (e.g., anti-PD-1 antibody, anti-PD-L1 antibody, or anti-CTLA-4 antibody treatment). In some embodiments, avelumab is administered as a 1 hour IV infusion every 2 weeks at a dose of 10 mg/kg, PF-05082566 is administered at fixed dose of 10 mg as a 1 hour IV infusion once every four weeks on Day 1 of each cycle, and on days when both avelumab and PF-05082566 are administered, PF-05082566 is administered first, followed by avelumab infusion within 30 mintues after the end of the PF-05082566 infusion.
[0021] In some embodiments, the method comprises administering to the subject a combination therapy which comprises avelumab and chemoradiotherapy. In some embodiments, the chemoradiotherapy comprises cisplatin and definitive radiation therapy. In some embodiments, subject has locally-advanced squamous cell carcinoma of the head and neck (SCCHN). In some embodiments, the SCCHN is localized to the oral cavity, oropharynx, larynx, or hypopharynx. In some embodiments, the method comprises a lead-in phase and a chemoradiotherapy (CRT) phase, wherein the lead-in phase begins seven days prior to initiation of the CRT phase. In some embodiments, avelumab is administered at a dose of 10 mg/kg on Day 1 of the lead-in phase' and on Day 8, Day 29, and Day 39 of the CRT phase; cisplatin is administered at a dose of 100 mg/m.sup.2 on Day 1, Day 22, and Day 23 of the CRT phase; and radiation therapy is 70 Gy/33-35 fractions/day, 5 fractions/week intensity modulated radiation therapy (IMRT). In some embodiments, the method comprises a maintenance phase which begins two weeks after completion of the CRT phase. In some embodiments the maintenance phase comprises administration of avelumab at a dose of 10 mg/kg every two weeks (Q2W) after completion of the CRT phase.
[0022] In all of the above treatment methods, medicaments and uses, the PD-L1 antagonist inhibits the binding of PD-L1 to PD-1. In some embodiments of the above treatment methods, medicaments and uses, the PD-L1 antagonist is a monoclonal antibody, or an antigen binding fragment thereof, which specifically binds to PD-L1 or to PD-L1 and blocks the binding of PD-L1 to PD-1. In some embodiments, the PD-L1 antagonist is an anti-PD-L1 antibody which comprises three complementarity determining regions (CDRs) from a heavy chain variable region comprising the amino acid sequence shown in SEQ ID NO: 8 and three CDRs from a light chain variable region comprising the amino acid sequences shown in SEQ ID NO: 9. In some embodiments, the PD-L1 antagonist is an anti-PD-L1 antibody which comprises heavy and light chain variable regions comprising the amino acid sequences shown in SEQ ID NO: 8 and SEQ ID NO: 9, respectively.
[0023] In some embodiments, the invention provides a medicament comprising a PD-L1 antagonist for use in combination with an anti-4-1BB antibody for treating a cancer.
[0024] In some embodiments, the invention provides a medicament comprising an anti-4-1BB antibody for use in combination with a PD-L1 antagonist for treating a cancer.
[0025] Other embodiments provide use of a PD-L1 antagonist in the manufacture of medicament for treating a cancer in a subject when administered in combination with an anti-4-1BB antibody and use of an anti-4-1BB antibody in the manufacture of a medicament for treating a cancer in a subject when administered in combination with a PD-L1 antagonist.
[0026] In some embodiments, the invention provides use of a PD-L1 antagonist and an anti-4-1BB antibody in the manufacture of medicaments for treating a cancer in a subject. In some embodiments, the medicaments comprise a kit, and the kit also comprises a package insert comprising instructions for using the PD-L1 antagonist in combination with an anti-4-1 BB antibody to treat a cancer in a subject.
[0027] In some embodiments, the invention provides a medicament comprising a PD-L1 antagonist for use in combination with an anti-M-CSF antibody for treating a cancer.
[0028] In some embodiments, the invention provides a medicament comprising an anti-M-CSF antibody for use in combination with a PD-L1 antagonist for treating a cancer.
[0029] Other embodiments provide use of a PD-L1 antagonist in the manufacture of medicament for treating a cancer in a subject when administered in combination with an anti-M-CSF antibody and use of an anti-M-CSF antibody in the manufacture of a medicament for treating a cancer in a subject when administered in combination with a PD-L1 antagonist.
[0030] In some embodiments, the invention provides use of a PD-L1 antagonist and an anti-M-CSF antibody in the manufacture of medicaments for treating a cancer in a subject. In some embodiments, the medicaments comprise a kit, and the kit also comprises a package insert comprising instructions for using the PD-L1 antagonist in combination with an anti-M-CSF antibody to treat a cancer in a subject.
[0031] In some embodiments, the invention provides a medicament comprising a PD-L1 antagonist for use in combination with an anti-OX40 antibody for treating a cancer.
[0032] In some embodiments, the invention provides a medicament comprising an anti-OX40 antibody for use in combination with a PD-L1 antagonist for treating a cancer.
[0033] Other embodiments provide use of a PD-L1 antagonist in the manufacture of medicament for treating a cancer in a subject when administered in combination with an anti-OX40 antibody and use of an anti-OX40 antibody in the manufacture of a medicament for treating a cancer in a subject when administered in combination with a PD-L1 antagonist.
[0034] In some embodiments, the invention provides use of a PD-L1 antagonist and an anti-OX40 antibody in the manufacture of medicaments for treating a cancer in a subject. In some embodiments, the medicaments comprise a kit, and the kit also comprises a package insert comprising instructions for using the PD-L1 antagonist in combination with an anti-OX40 antibody to treat a cancer in a subject.
[0035] In some embodiments, the invention provides a medicament comprising a PD-L1 antagonist for use in combination with an anti-M-CSF antibody for treating a cancer.
[0036] In some embodiments, the invention provides a medicament comprising an anti-M-CSF antibody for use in combination with a PD-L1 antagonist for treating a cancer.
[0037] Other embodiments provide use of a PD-L1 antagonist in the manufacture of medicament for treating a cancer in a subject when administered in combination with an anti-M-CSF antibody and use of an anti-M-CSF antibody in the manufacture of a medicament for treating a cancer in a subject when administered in combination with a PD-L1 antagonist.
[0038] In some embodiments, the invention provides use of a PD-L1 antagonist and an anti-M-CSF antibody in the manufacture of medicaments for treating a cancer in a subject. In some embodiments, the medicaments comprise a kit, and the kit also comprises a package insert comprising instructions for using the PD-L1 antagonist in combination with an anti-M-CSF antibody to treat a cancer in a subject.
[0039] In some embodiments, the invention provides a medicament comprising a PD-L1 antagonist for use in combination with an anti-OX40 antibody for treating a cancer.
[0040] In some embodiments, the invention provides a medicament comprising an anti-OX40 antibody for use in combination with a PD-L1 antagonist for treating a cancer.
[0041] Other embodiments provide use of a PD-L1 antagonist in the manufacture of medicament for treating a cancer in a subject when administered in combination with an anti-OX40 antibody and use of an anti-OX40 antibody in the manufacture of a medicament for treating a cancer in a subject when administered in combination with a PD-L1 antagonist.
[0042] In some embodiments, the invention provides use of a PD-L1 antagonist and an anti-OX40 antibody in the manufacture of medicaments for treating a cancer in a subject. In some embodiments, the medicaments comprise a kit, and the kit also comprises a package insert comprising instructions for using the PD-L1 antagonist in combination with an anti-OX40 antibody to treat a cancer in a subject.
[0043] In some embodiments, the invention provides a medicament comprising a PD-L1 antagonist for use in combination with an anti-4-1BB antibody and an anti-M-CSF antibody for treating a cancer.
[0044] In some embodiments, the invention provides a medicament comprising an anti-4-1BB antibody and an anti-M-CSF antibody for use in combination with a PD-L1 antagonist for treating a cancer.
[0045] Other embodiments provide use of a PD-L1 antagonist in the manufacture of medicament for treating a cancer in a subject when administered in combination with an anti-4-1BB antibody and an anti-M-CSF antibody and use of an anti-4-1BB antibody and an anti-M-CSF antibody in the manufacture of a medicament for treating a cancer in a subject when administered in combination with a PD-L1 antagonist.
[0046] In some embodiments, the invention provides use of a PD-L1 antagonist and an anti-4-1BB antibody and an anti-M-CSF antibody in the manufacture of medicaments for treating a cancer in a subject. In some embodiments, the medicaments comprise a kit, and the kit also comprises a package insert comprising instructions for using the PD-L1 antagonist in combination with an anti-4-1BB antibody and an anti-M-CSF antibody to treat a cancer in a subject.
[0047] In some embodiments, the invention provides a medicament comprising a PD-L1 antagonist for use in combination with an anti-4-1 BB antibody and an anti-OX40 antibody for treating a cancer.
[0048] In some embodiments, the invention provides a medicament comprising an anti-4-1BB antibody and an anti-OX40 antibody for use in combination with a PD-L1 antagonist for treating a cancer.
[0049] Other embodiments provide use of a PD-L1 antagonist in the manufacture of medicament for treating a cancer in a subject when administered in combination with an anti-4-1 BB antibody and an anti-OX40 antibody and use of an anti-4-1 BB antibody and an anti-OX40 antibody in the manufacture of a medicament for treating a cancer in a subject when administered in combination with a PD-L1 antagonist.
[0050] In some embodiments, the invention provides use of a PD-L1 antagonist and an anti-4-1 BB antibody and an anti-OX40 antibody in the manufacture of medicaments for treating a cancer in a subject. In some embodiments, the medicaments comprise a kit, and the kit also comprises a package insert comprising instructions for using the PD-L1 antagonist in combination with an anti-4-1BB antibody and an anti-OX40 antibody to treat a cancer in a subject.
[0051] In all of the above treatment methods, medicaments and uses, the PD-L1 antagonist inhibits the binding of PD-L1 to PD-1. In some embodiments of the above treatment methods, medicaments and uses, the PD-L1 antagonist is a monoclonal antibody, or an antigen binding fragment thereof, which specifically binds to PD-L1 or to PD-L1 and blocks the binding of PD-L1 to PD-1. In some embodiments, the PD-L1 antagonist is an anti-PD-L1 antibody which comprises three CDRs from a heavy chain variable region comprising the amino acid sequence shown in SEQ ID NO: 8 and three CDRs from a light chain variable region comprising the amino acid sequence shown in SEQ ID NO: 9. In some embodiments, the PD-L1 antagonist is an anti-PD-L1 antibody which comprises heavy and light chain variable regions comprising the amino acid sequences shown in SEQ ID NO: 8 and SEQ ID NO: 9, respectively. In some embodiments, the anti-PD-L1 antibody is Avelumab.
[0052] In some embodiments, the anti-4-1BB antibody can comprise a heavy chain variable region comprising three CDRs from the heavy chain variable region having the amino acid sequence shown in SEQ ID NO: 18, and a light chain variable region comprising three CDRs from the light chain variable region having the amino acid sequence shown in SEQ ID NO: 19. In some embodiments, the anti-4-1BB antibody can comprise heavy and light chain variable regions comprising the amino acid sequences shown in SEQ ID NO: 18 and SEQ ID NO: 19, respectively. In some embodiments, the anti-4-1 BB antibody is PF-05082566.
[0053] In some embodiments, the anti-M-CSF antibody can comprise a heavy chain variable region comprising three CDRs from the heavy chain variable region having the amino acid sequence shown in SEQ ID NO: 30, and a light chain variable region comprising three CDRs from the light chain variable region having the amino acid sequence shown in SEQ ID NO: 31. In some embodiments, the anti-M-CSF antibody can comprise heavy and light chain variable regions comprising the amino acid sequences shown in SEQ ID NO: 30 and SEQ ID NO: 31, respectively. In some embodiments, the anti-M-CSF antibody is PD-0360324.
[0054] In some embodiments, the anti-OX40 antibody can comprise a heavy chain variable region comprising three CDRs from the heavy chain variable region having the amino acid sequence shown in SEQ ID NO: 38, and a light chain variable region comprising three CDRs from the light chain variable region having the amino acid sequence shown in SEQ ID NO: 39. In some embodiments, the anti-OX40 antibody can comprise a heavy chain variable region comprising the amino acid sequence shown in SEQ ID NO: 38, and a light chain variable region comprising the amino acid sequence shown in SEQ ID NO: 39. In some embodiments, the anti-OX40 antibody is PF-04518600.
[0055] In some embodiments of the above treatment methods, medicaments and uses of the invention, the individual is a human and the cancer is a solid tumor. In some embodiments, the solid tumor is renal cell carcinoma (RCC), bladder cancer, breast cancer, clear cell kidney cancer, head/neck squamous cell carcinoma (SCCHN), lung squamous cell carcinoma, malignant melanoma, non-small-cell lung cancer (NSCLC), ovarian cancer, pancreatic cancer, prostate cancer, small-cell lung cancer (SCLC) or triple negative breast cancer.
[0056] In other embodiments of the above treatment methods, medicaments and uses of the invention, the individual is a human and the cancer is a Heme malignancy and in some embodiments, the Heme malignancy is acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), chronic myeloid leukemia (CML), diffuse large B-cell lymphoma (DLBCL), EBV-positive DLBCL, primary mediastinal large B-cell lymphoma, T-cell/histiocyte-rich large B-cell lymphoma, follicular lymphoma, Hodgkin's lymphoma (HL), mantle cell lymphoma (MCL), multiple myeloma (MM), myeloid cell leukemia-1 protein (Mcl-1), myelodysplastic syndrome (MDS), non-Hodgkin's lymphoma (NHL), or small lymphocytic lymphoma (SLL).
[0057] Also, in some embodiments of any of the above treatment methods, medicaments and uses, the cancer tests positive for the expression of one or both of PD-L1 and PD-L2. In still other embodiments, the cancer has elevated PD-L1 expression.
[0058] In some embodiments of the above treatment methods, medicaments and uses, the subject is a human and the cancer is RCC that tests positive for human PD-L1.
[0059] In some embodiments of the above treatment methods, medicaments and uses, the cancer is advanced RCC with clear cell subtype and is present in a human who has not been previously treated for RCC.
[0060] In some embodiments of the above treatment methods, medicaments and uses, the cancer is relapsed or refractory (R/R) cancer. In some embodiments, the R/R cancer is R/R DLBCL.
[0061] In some embodiments of the above treatment methods, medicaments and uses, the cancer is locally advanced cancer. In some embodiments, the locally advanced cancer is locally advanced SCCHN. In some embodiments, the SCCHN is localized to the oral cavity, oropharynx, larynx, or hypopharynx.
BRIEF DESCRIPTION OF THE FIGURES/DRAWINGS
[0062] FIG. 1 depicts a graph summarizing infiltration of T cells in response to treatment.
[0063] FIG. 2 depicts a graph summarizing ratio of CD8+ T cells/Treg in response to treatment.
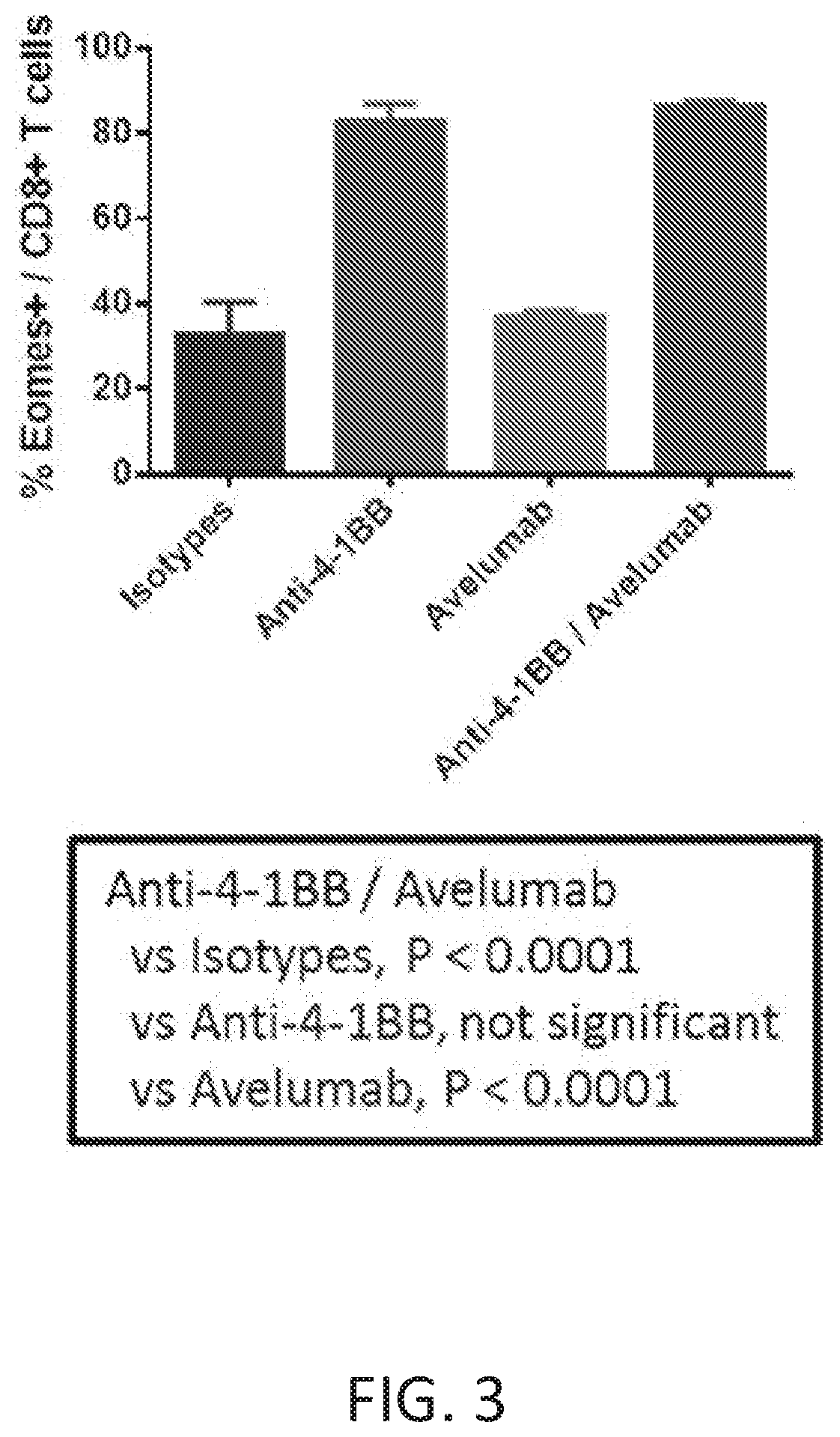
[0064] FIG. 3 depicts a graph summarizing Eomes induction in response to treatment.
DETAILED DESCRIPTION
I. DEFINITIONS
[0065] So that the invention may be more readily understood, certain technical and scientific terms are specifically defined below. Unless specifically defined elsewhere in this document, all other technical and scientific terms used herein have the meaning commonly understood by one of ordinary skill in the art to which this invention belongs.
[0066] "About" when used to modify a numerically defined parameter (e.g., the dose of a PD-L1 antagonist or VEGFR inhibitor, or the length of treatment time with a combination therapy described herein) means that the parameter may vary by as much as 10% below or above the stated numerical value for that parameter. For example, a dose of about 5 mg/kg may vary between 4.5 mg/kg and 5.5 mg/kg.
[0067] As used herein, including the appended claims, the singular forms of words such as "a," "an," and "the," include their corresponding plural references unless the context clearly dictates otherwise.
[0068] "Administration" and "treatment," as it applies to an animal, human, experimental subject, cell, tissue, organ, or biological fluid, refers to contact of an exogenous pharmaceutical, therapeutic, diagnostic agent, or composition to the animal, human, subject, cell, tissue, organ, or biological fluid. Treatment of a cell encompasses contact of a reagent to the cell, as well as contact of a reagent to a fluid, where the fluid is in contact with the cell. "Administration" and "treatment" also means in vitro and ex vivo treatments, e.g., of a cell, by a reagent, diagnostic, binding compound, or by another cell. The term "subject" includes any organism, preferably an animal, more preferably a mammal (e.g., rat, mouse, dog, cat, rabbit) and most preferably a human.
[0069] An "antibody" is an immunoglobulin molecule capable of specific binding to a target, such as a carbohydrate, polynucleotide, lipid, polypeptide, etc., through at least one antigen recognition site, located in the variable region of the immunoglobulin molecule. As used herein, the term encompasses not only intact polyclonal or monoclonal antibodies, but also fragments thereof (such as Fab, Fab', F(ab')2, Fv), single chain (scFv) and domain antibodies (including, for example, shark and camelid antibodies), and fusion proteins comprising an antibody, and any other modified configuration of the immunoglobulin molecule that comprises an antigen recognition site. An antibody includes an antibody of any class, such as IgG, IgA, or IgM (or sub-class thereof), and the antibody need not be of any particular class. Depending on the antibody amino acid sequence of the constant region of its heavy chains, immunoglobulins can be assigned to different classes. There are five major classes of immunoglobulins: IgA, IgD, IgE, IgG, and IgM, and several of these may be further divided into subclasses (isotypes), e.g., IgG1, IgG2, IgG3, IgG4, IgA1 and IgA2. The heavy-chain constant regions that correspond to the different classes of immunoglobulins are called alpha, delta, epsilon, gamma, and mu, respectively. The subunit structures and three-dimensional configurations of different classes of immunoglobulins are well known.
[0070] The term "antigen binding fragment" or "antigen binding portion" of an antibody, as used herein, refers to one or more fragments of an intact antibody that retain the ability to specifically bind to a given antigen (e.g., PD-L1). Antigen binding functions of an antibody can be performed by fragments of an intact antibody. Examples of binding fragments encompassed within the term "antigen binding fragment" of an antibody include Fab; Fab'; F(ab')2; an Fd fragment consisting of the VH and CH1 domains; an Fv fragment consisting of the VL and VH domains of a single arm of an antibody; a single domain antibody (dAb) fragment (Ward et al., Nature 341:544-546, 1989), and an isolated complementarity determining region (CDR).
[0071] An antibody, an antibody conjugate, or a polypeptide that "preferentially binds" or "specifically binds" (used interchangeably herein) to a target (e.g., PD-L1 protein) is a term well understood in the art, and methods to determine such specific or preferential binding are also well known in the art. A molecule is said to exhibit "specific binding" or "preferential binding" if it reacts or associates more frequently, more rapidly, with greater duration and/or with greater affinity with a particular cell or substance than it does with alternative cells or substances. An antibody "specifically binds" or "preferentially binds" to a target if it binds with greater affinity, avidity, more readily, and/or with greater duration than it binds to other substances. For example, an antibody that specifically or preferentially binds to a PD-L1 epitope is an antibody that binds this epitope with greater affinity, avidity, more readily, and/or with greater duration than it binds to other PD-L1 epitopes or non-PD-L1 epitopes. It is also understood that by reading this definition, for example, an antibody (or moiety or epitope) that specifically or preferentially binds to a first target may or may not specifically or preferentially bind to a second target. As such, "specific binding" or "preferential binding" does not necessarily require (although it can include) exclusive binding. Generally, but not necessarily, reference to binding means preferential binding.
[0072] A "variable region" of an antibody refers to the variable region of the antibody light chain or the variable region of the antibody heavy chain, either alone or in combination. As known in the art, the variable regions of the heavy and light chain each consist of four framework regions (FR) connected by three complementarity determining regions (CDRs) also known as hypervariable regions. The CDRs in each chain are held together in close proximity by the FRs and, with the CDRs from the other chain, contribute to the formation of the antigen binding site of antibodies. There are at least two techniques for determining CDRs: (1) an approach based on cross-species sequence variability (i.e., Kabat et al. Sequences of Proteins of Immunological Interest, (5th ed., 1991, National Institutes of Health, Bethesda Md.)); and (2) an approach based on crystallographic studies of antigen-antibody complexes (Al-lazikani et al., 1997, J. Molec. Biol. 273:927-948). As used herein, a CDR may refer to CDRs defined by either approach or by a combination of both approaches.
[0073] A "CDR" of a variable domain are amino acid residues within the variable region that are identified in accordance with the definitions of the Kabat, Chothia, the accumulation of both Kabat and Chothia, AbM, contact, and/or conformational definitions or any method of CDR determination well known in the art. Antibody CDRs may be identified as the hypervariable regions originally defined by Kabat et al. See, e.g., Kabat et al., 1992, Sequences of Proteins of Immunological Interest, 5th ed., Public Health Service, NIH, Washington D.C. The positions of the CDRs may also be identified as the structural loop structures originally described by Chothia and others. See, e.g., Chothia et al., Nature 342:877-883, 1989. Other approaches to CDR identification include the "AbM definition," which is a compromise between Kabat and Chothia and is derived using Oxford Molecular's AbM antibody modeling software (now Accelrys.RTM.), or the "contact definition" of CDRs based on observed antigen contacts, set forth in MacCallum et al., J. Mol. Biol., 262:732-745, 1996. In another approach, referred to herein as the "conformational definition" of CDRs, the positions of the CDRs may be identified as the residues that make enthalpic contributions to antigen binding. See, e.g., Makabe et al., Journal of Biological Chemistry, 283:1156-1166, 2008. Still other CDR boundary definitions may not strictly follow one of the above approaches, but will nonetheless overlap with at least a portion of the Kabat CDRs, although they may be shortened or lengthened in light of prediction or experimental findings that particular residues or groups of residues or even entire CDRs do not significantly impact antigen binding. As used herein, a CDR may refer to CDRs defined by any approach known in the art, including combinations of approaches. The methods used herein may utilize CDRs defined according to any of these approaches. For any given embodiment containing more than one CDR, the CDRs may be defined in accordance with any of Kabat, Chothia, extended, AbM, contact, and/or conformational definitions.
[0074] "Isolated antibody" and "isolated antibody fragment" refers to the purification status and in such context means the named molecule is substantially free of other biological molecules such as nucleic acids, proteins, lipids, carbohydrates, or other material such as cellular debris and growth media. Generally, the term "isolated" is not intended to refer to a complete absence of such material or to an absence of water, buffers, or salts, unless they are present in amounts that substantially interfere with experimental or therapeutic use of the binding compound as described herein.
[0075] "Monoclonal antibody" or "mAb" or "Mab", as used herein, refers to a population of substantially homogeneous antibodies, i.e., the antibody molecules comprising the population are identical in amino acid sequence except for possible naturally occurring mutations that may be present in minor amounts. In contrast, conventional (polyclonal) antibody preparations typically include a multitude of different antibodies having different amino acid sequences in their variable domains, particularly their CDRs, which are often specific for different epitopes. The modifier "monoclonal" indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies, and is not to be construed as requiring production of the antibody by any particular method. For example, the monoclonal antibodies to be used in accordance with the present invention may be made by the hybridoma method first described by Kohler et al. (1975) Nature 256: 495, or may be made by recombinant DNA methods (see, e.g., U.S. Pat. No. 4,816,567). The "monoclonal antibodies" may also be isolated from phage antibody libraries using the techniques described in Clackson et al. (1991) Nature 352: 624-628 and Marks et al. (1991) J. Mol. Biol. 222: 581-597, for example. See also Presta (2005) J. Allergy Clin. Immunol. 116:731.
[0076] "Chimeric antibody" refers to an antibody in which a portion of the heavy and/or light chain is identical with or homologous to corresponding sequences in an antibody derived from a particular species (e.g., human) or belonging to a particular antibody class or subclass, while the remainder of the chain(s) is identical with or homologous to corresponding sequences in an antibody derived from another species (e.g., mouse) or belonging to another antibody class or subclass, as well as fragments of such antibodies, so long as they exhibit the desired biological activity.
[0077] "Human antibody" refers to an antibody that comprises human immunoglobulin protein sequences only. A human antibody may contain murine carbohydrate chains if produced in a mouse, in a mouse cell, or in a hybridoma derived from a mouse cell. Similarly, "mouse antibody" or "rat antibody" refer to an antibody that comprises only mouse or rat immunoglobulin sequences, respectively.
[0078] "Humanized antibody" refers to forms of antibodies that contain sequences from non-human (e.g., murine) antibodies as well as human antibodies. Such antibodies contain minimal sequence derived from non-human immunoglobulin. In general, the humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non-human immunoglobulin and all or substantially all of the FR regions are those of a human immunoglobulin sequence. The humanized antibody optionally also will comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin. The prefix "hum", "hu" or "h" is added to antibody clone designations when necessary to distinguish humanized antibodies from parental rodent antibodies. The humanized forms of rodent antibodies will generally comprise the same CDR sequences of the parental rodent antibodies, although certain amino acid substitutions may be included to increase affinity, increase stability of the humanized antibody, or for other reasons.
[0079] The terms "cancer", "cancerous", or "malignant" refer to or describe the physiological condition in mammals that is typically characterized by unregulated cell growth. Examples of cancer include but are not limited to, carcinoma, lymphoma, leukemia, blastoma, and sarcoma. More particular examples of such cancers include squamous cell carcinoma, myeloma, small-cell lung cancer, non-small cell lung cancer, glioma, hodgkin's lymphoma, non-hodgkin's lymphoma, acute myeloid leukemia (AML), multiple myeloma, gastrointestinal (tract) cancer, renal cancer, ovarian cancer, liver cancer, lymphoblastic leukemia, lymphocytic leukemia, colorectal cancer, endometrial cancer, kidney cancer, prostate cancer, thyroid cancer, melanoma, chondrosarcoma, neuroblastoma, pancreatic cancer, glioblastoma multiforme, cervical cancer, brain cancer, stomach cancer, bladder cancer, hepatoma, breast cancer, colon carcinoma, and head and neck cancer. Another particular example of cancer includes renal cell carcinoma.
[0080] "Biotherapeutic agent" means a biological molecule, such as an antibody or fusion protein, that blocks ligand/receptor signaling in any biological pathway that supports tumor maintenance and/or growth or suppresses the anti-tumor immune response.
[0081] "Chemotherapeutic agent" is a chemical compound useful in the treatment of cancer. Classes of chemotherapeutic agents include, but are not limited to: alkylating agents, antimetabolites, kinase inhibitors, spindle poison plant alkaloids, cytotoxic/antitumor antibiotics, topisomerase inhibitors, photosensitizers, anti-estrogens and selective estrogen receptor modulators (SERMs), anti-progesterones, estrogen receptor down-regulators (ERDs), estrogen receptor antagonists, leutinizing hormone-releasing hormone agonists, anti-androgens, aromatase inhibitors, EGFR inhibitors, VEGF inhibitors, and anti-sense oligonucleotides that inhibit expression of genes implicated in abnormal cell proliferation or tumor growth. Chemotherapeutic agents useful in the treatment methods of the present invention include cytostatic and/or cytotoxic agents.
[0082] "Conservatively modified variants" or "conservative substitution" refers to substitutions of amino acids in a protein with other amino acids having similar characteristics (e.g. charge, side-chain size, hydrophobicity/hydrophilicity, backbone conformation and rigidity, etc.), such that the changes can frequently be made without altering the biological activity or other desired property of the protein, such as antigen affinity and/or specificity. Those of skill in this art recognize that, in general, single amino acid substitutions in non-essential regions of a polypeptide do not substantially alter biological activity (see, e.g., Watson et al. (1987) Molecular Biology of the Gene, The Benjamin/Cummings Pub. Co., p. 224 (4th Ed.)). In addition, substitutions of structurally or functionally similar amino acids are less likely to disrupt biological activity.
[0083] Exemplary conservative substitutions are set forth in Table 1 below.
TABLE-US-00001 TABLE 1 Exemplary Conservative Amino Acid Substitutions Original residue Conservative substitution Ala (A) Gly; Ser Arg (R) Lys; His Asn (N) Gln; His Asp (D) Glu; Asn Cys (C) Ser; Ala Gln (Q) Asn Glu (E) Asp; Gln Gly (G) Ala His (H) Asn; Gln Ile (I) Leu; Val Leu (L) Ile; Val Lys (K) Arg; His Met (M) Leu; Ile; Tyr Phe (F) Tyr; Met; Leu Pro (P) Ala Ser (S) Thr Thr (T) Ser Trp (W) Tyr; Phe Tyr (Y) Trp; Phe Val (V) Ile; Leu
[0084] "Consists essentially of," and variations such as "consist essentially of" or "consisting essentially of," as used throughout the specification and claims, indicate the inclusion of any recited elements or group of elements, and the optional inclusion of other elements, of similar or different nature than the recited elements, that do not materially change the basic or novel properties of the specified dosage regimen, method, or composition. As a non-limiting example, a PD-L1 antagonist that consists essentially of a recited amino acid sequence may also include one or more amino acids, including substitutions of one or more amino acid residues, which do not materially affect the properties of the binding compound.
[0085] "Diagnostic anti-PD-L1 monoclonal antibody" means a mAb which specifically binds to PD-L1 that is expressed on the surface of certain mammalian cells. A mature PD-L1 lacks the presecretory leader sequence, also referred to as leader peptide The terms "PD-L1" and "mature PD-L1" are used interchangeably herein, and shall be understood to mean the same molecule unless otherwise indicated or readily apparent from the context.
[0086] As used herein, an anti-human PD-L1 mAb or a diagnostic anti-hPD-L1 mAb refers to a monoclonal antibody that specifically binds to mature human PD-L1. A mature human PD-L1 molecule consists of amino acids 19-290 of the following sequence (SEQ ID NO: 1): MRIFAVFIFMTYWHLLNAFTVTVPKDLYVVEYGSNMTIEC KFPVEKQLDLAALIVYWEMEDKN I IQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAA LQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYNKINQRILVVDPVTSEHELTCQA EGYPKAEVIWTSSDHQVLSG KTTTTNSKREEKLFNVTSTLRI NTTTN E I FYCTFRRLDP EENHTAELVIPELPLAHPPNERTHLVILGAILLCLGVALTFIFRLRKGRMMDVKKCGIQD TNSKKQSDTHLEET (SEQ ID NO: 1).
[0087] "Homology" refers to sequence similarity between two polypeptide sequences when they are optimally aligned. When a position in both of the two compared sequences is occupied by the same amino acid monomer subunit, e.g., if a position in a light chain CDR of two different Abs is occupied by alanine, then the two Abs are homologous at that position. The percent of homology is the number of homologous positions shared by the two sequences divided by the total number of positions compared.times.100. For example, if 8 of 10 of the positions in two sequences are matched or homologous when the sequences are optimally aligned then the two sequences are 80% homologous. Generally, the comparison is made when two sequences are aligned to give maximum percent homology. For example, the comparison can be performed by a BLAST algorithm wherein the parameters of the algorithm are selected to give the largest match between the respective sequences over the entire length of the respective reference sequences.
[0088] The following references relate to BLAST algorithms often used for sequence analysis: BLAST ALGORITHMS: Altschul, S. F., et al., (1990) J. Mol. Biol. 215:403-410; Gish, W., et al., (1993) Nature Genet. 3:266-272; Madden, T. L., et al., (1996) Meth. Enzymol. 266:131-141; Altschul, S. F., et al., (1997) Nucleic Acids Res. 25:3389-3402; Zhang, J., et al., (1997) Genome Res. 7:649-656; Wootton, J. C., et al., (1993) Comput. Chem. 17:149-163; Hancock, J. M. et al., (1994) Comput. Appl. Biosci. 10:67-70; ALIGNMENT SCORING SYSTEMS: Dayhoff, M. O., et al., "A model of evolutionary change in proteins." in Atlas of Protein Sequence and Structure, (1978) vol. 5, suppl. 3. M. O. Dayhoff (ed.), pp. 345-352, Natl. Biomed. Res. Found., Washington, D.C.; Schwartz, R. M., et al., "Matrices for detecting distant relationships." in Atlas of Protein Sequence and Structure, (1978) vol. 5, suppl. 3.'' M. O. Dayhoff (ed.), pp. 353-358, Natl. Biomed. Res. Found., Washington, D.C.; Altschul, S. F., (1991) J. Mol. Biol. 219:555-565; States, D. J., et al., (1991) Methods 3:66-70; Henikoff, S., et al., (1992) Proc. Natl. Acad. Sci. USA 89:10915-10919; Altschul, S. F., et al., (1993) J. Mol. Evol. 36:290-300; ALIGNMENT STATISTICS: Karlin, S., et al., (1990) Proc. Natl. Acad. Sci. USA 87:2264-2268; Karlin, S., et al., (1993) Proc. Natl. Acad. Sci. USA 90:5873-5877; Dembo, A., et al., (1994) Ann. Prob. 22:2022-2039; and Altschul, S. F. "Evaluating the statistical significance of multiple distinct local alignments." in Theoretical and Computational Methods in Genome Research (S. Suhai, ed.), (1997) pp. 1-14, Plenum, New York.
[0089] "Patient" or "subject" refers to any single subject for which therapy is desired or that is participating in a clinical trial, epidemiological study or used as a control, including humans and mammalian veterinary patients such as cattle, horses, dogs, and cats.
[0090] "PD-L1 antagonist" means any chemical compound or biological molecule that blocks binding of PD-L1 expressed on a cancer cell to PD-1. In any of the treatment method, medicaments and uses of the present invention in which a human subject is being treated, the PD-L1 antagonist blocks binding of human PD-L1 to human PD-1.
[0091] PD-L1 antagonists useful in the any of the treatment methods, medicaments, and uses of the present invention include a monoclonal antibody (mAb) which specifically binds to PD-L1, and preferably specifically binds to human PD-L1. The mAb may be a human antibody, a humanized antibody or a chimeric antibody, and may include a human constant region. In some embodiments the human constant region is selected from the group consisting of IgG1, IgG2, IgG3 and IgG4 constant regions, and in preferred embodiments, the human constant region is an IgG1 or IgG4 constant region. In some embodiments, the antigen binding fragment is selected from the group consisting of Fab, Fab'-SH, F(ab')2, scFv and Fv fragments.
[0092] Examples of mAbs that bind to human PD-L1, and useful in the treatment method, medicaments and uses of the present invention, are described in WO2013079174, WO2015061668, WO2010089411, WO/2007/005874, WO/2010/036959, WO/2014/100079, WO2013/019906, WO/2010/077634, and U.S. Pat. Nos. 8,552,154, 8,779,108, and 8,383,796. Specific anti-human PD-L1 mAbs useful as the PD-L1 antagonist in the treatment method, medicaments and uses of the present invention include, for example without limitation: avelumab (MSB0010718C), nivolumab (BMS-936558), MPDL3280A (an IgG1-engineered, anti-PD-L1 antibody), BMS-936559 (a fully human, anti-PD-L1, IgG4 monoclonal antibody), MED14736 (an engineered IgG1 kappa monoclonal antibody with triple mutations in the Fc domain to remove antibody-dependent, cell-mediated cytotoxic activity), and an antibody which comprises the heavy chain and light chain variable regions of SEQ ID NO:24 and SEQ ID NO:21, respectively, of WO2013/019906.
[0093] Other PD-L1 antagonists useful in the any of the treatment method, medicaments and uses of the present invention include an immunoadhesin that specifically binds to PD-L1, and preferably specifically binds to human PD-L1, e.g., a fusion protein containing the PD-L1 binding portion of PD-1 fused to a constant region such as an Fc region of an immunoglobulin molecule.
[0094] Table 2 below provides exemplary anti-PD-L1 antibody sequences for use in the treatment method, medicaments and uses of the present invention.
TABLE-US-00002 TABLE 2 EXEMPLARY ANTI-HUMAN PD-L1 MONOCLONAL ANTIBODY SEQUENCES Heavy chain CDR1 SYIMM (SEQ ID NO: 2) (CDRH1) Heavy chain CDR2 SIYPSGGITFY (SEQ ID NO: 3) (CDRH2) Heavy chain CDR3 IKLGTVTTVDY (SEQ ID NO: 4) (CD RH3) Light chain CDR1 TGTSSDVGGYNYVS (SEQ ID NO: 5) (CDRL1) Light chain CDR2 DVSNRPS (SEQ ID NO: 6) (CDRL2) Light chain CDR3 SSYTSSSTRV (SEQ ID NO: 7) (CDRL3) Heavy chain EVQLLESGGGLVQPGGSLRLSCAASGFTFSSYIMMWVRQAPGKGL variable region (VR) EWVSSIYPSGGITFYADKGRFTISRDNSKNTLYLQMNSLRAEDTAVY YCARIKLGTVTTVDYWGQGTLVTVSS (SEQ ID NO: 8) Light chain VR QSALTQPASVSGSPGQSITISCTGTSSDVGGYNYVSWYQQHPGKA PKLMIYDVSNRPSGVSNRFSGSKSGNTASLTISGLQAEDEADYYCS SYTSSSTRVFGTGTKVTVL (SEQ ID NO: 9) Heavy chain EVQLLESGGGLVQPGGSLRLSCAASGFTFSSYIMMWVRQAPGKGL EWVSSIYPSGGITFYADTVKGRFTISRDNSKNTLYLQMNSLRAEDTA VYYCARIKLGTVTTVDYWGQGTLVTVSSASTKGPSVFPLAPSSKSTS GGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSL SSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPP CPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKF NWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYK CKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTC LVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVD KSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK (SEQ ID NO: 10) Light chain QSALTQPASVSGSPGQSITISCTGTSSDVGGYNYVSWYQQHPGKA PKLMIYDVSNRPSGVSNRFSGSKSGNTASLTISGLQAEDEADYYCS SYTSSSTRVFGTGTKVTVLGQPKANPTVTLFPPSSEELQANKATLVC LISDFYPGAVTVAWKADGSPVKAGVETTKPSKQSNNKYAASSYLSL TPEQWKSHRSYSCQVTHEGSTVEKTVAPTECS (SEQ ID NO: 11)
[0095] "PD-L1" expression as used herein means any detectable level of expression of PD-L1 protein on the cell surface or of PD-L1 mRNA within a cell or tissue. PD-L1 protein expression may be detected with a diagnostic PD-L1 antibody in an IHC assay of a tumor tissue section or by flow cytometry. Alternatively, PD-L1 protein expression by tumor cells may be detected by PET imaging, using a binding agent (e.g., antibody fragment, affibody and the like) that specifically binds to PD-L1. Techniques for detecting and measuring PD-L1 mRNA expression include RT-PCR and real-time quantitative RT-PCR.
[0096] Several approaches have been described for quantifying PD-L1 protein expression in IHC assays of tumor tissue sections. See, e.g., Thompson, R. H., et al., PNAS 101 (49); 17174-17179 (2004); Thompson, R. H. et al., Cancer Res. 66:3381-3385 (2006); Gadiot, J., et al., Cancer 117:2192-2201 (2011); Taube, J. M. et al., Sci Trans! Med 4, 127ra37 (2012); and Toplian, S. L. et al., New Eng. J Med. 366 (26): 2443-2454 (2012).
[0097] One approach employs a simple binary end-point of positive or negative for PD-L1 expression, with a positive result defined in terms of the percentage of tumor cells that exhibit histologic evidence of cell-surface membrane staining. A tumor tissue section is counted as positive for PD-L1 expression is at least 1%, and preferably 5% of total tumor cells.
[0098] In another approach, PD-L1 expression in the tumor tissue section is quantified in the tumor cells as well as in infiltrating immune cells, which predominantly comprise lymphocytes. The percentage of tumor cells and infiltrating immune cells that exhibit membrane staining are separately quantified as <5%, 5 to 9%, and then in 10% increments up to 100%. For tumor cells, PD-L1 expression is counted as negative if the score is <5% score and positive if the score is .gtoreq.5%. PD-L1 expression in the immune infiltrate is reported as a semi-quantitative measurement called the adjusted inflammation score (AIS), which is determined by multiplying the percent of membrane staining cells by the intensity of the infiltrate, which is graded as none (0), mild (score of 1, rare lymphocytes), moderate (score of 2, focal infiltration of tumor by lymphohistiocytic aggregates), or severe (score of 3, diffuse infiltration). A tumor tissue section is counted as positive for PD-L1 expression by immune infiltrates if the AIS is ? 5.
[0099] The level of PD-L1 mRNA expression may be compared to the mRNA expression levels of one or more reference genes that are frequently used in quantitative RT-PCR, such as ubiquitin C.
[0100] In some embodiments, a level of PD-L1 expression (protein and/or mRNA) by malignant cells and/or by infiltrating immune cells within a tumor is determined to be "overexpressed" or "elevated" based on comparison with the level of PD-L1 expression (protein and/ or mRNA) by an appropriate control. For example, a control PD-L1 protein or mRNA expression level may be the level quantified in nonmalignant cells of the same type or in a section from a matched normal tissue.
[0101] "RECIST 1.1 Response Criteria" as used herein means the definitions set forth in Eisenhauer et al., E.A. et al., Eur. J Cancer 45:228-247 (2009) for target lesions or nontarget lesions, as appropriate based on the context in which response is being measured.
[0102] "Sustained response" means a sustained therapeutic effect after cessation of treatment with a therapeutic agent, or a combination therapy described herein. In some embodiments, the sustained response has a duration that is at least the same as the treatment duration, or at least 1.5, 2.0, 2.5 or 3 times longer than the treatment duration.
[0103] "Tissue Section" refers to a single part or piece of a tissue sample, e.g., a thin slice of tissue cut from a sample of a normal tissue or of a tumor.
[0104] "Treat" or "treating" a cancer as used herein means to administer a combination therapy of a PD-L1 antagonist and another therapeutic agent to a subject having a cancer, or diagnosed with a cancer, to achieve at least one positive therapeutic effect, such as for example, reduced number of cancer cells, reduced tumor size, reduced rate of cancer cell infiltration into peripheral organs, or reduced rate of tumor metastasis or tumor growth. Positive therapeutic effects in cancer can be measured in a number of ways (See, W. A. Weber, J. Nucl. Med. 50:1S-10S (2009)). For example, with respect to tumor growth inhibition, according to National Cancer Institute (NCI) standards, a T/C less than or equal to 42% is the minimum level of anti-tumor activity. A T/C <10% is considered a high anti-tumor activity level, with T/C (%)=Median tumor volume of the treated/Median tumor volume of the control.times.100. In some embodiments, the treatment achieved by a combination of the invention is any of partial response (PR), complete response (CR), overall response (OR), progression free survival (PFS), disease free survival (DFS) and overall survival (OS). PFS, also referred to as "Time to Tumor Progression" indicates the length of time during and after treatment that the cancer does not grow, and includes the amount of time patients have experienced a CR or PR, as well as the amount of time patients have experienced stable disease (SD). DFS refers to the length of time during and after treatment that the patient remains free of disease. OS refers to a prolongation in life expectancy as compared to naive or untreated subjects or patients. In some embodiments, response to a combination of the invention is any of PR, CR, PFS, DFS, OR, or OS that is assessed using Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 response criteria. The treatment regimen for a combination of the invention that is effective to treat a cancer patient may vary according to factors such as the disease state, age, and weight of the patient, and the ability of the therapy to elicit an anti-cancer response in the subject. While an embodiment of any of the aspects of the invention may not be effective in achieving a positive therapeutic effect in every subject, it should do so in a statistically significant number of subjects as determined by any statistical test known in the art such as the Student's t-test, the chi2-test, the U-test according to Mann and Whitney, the Kruskal-Wallis test (H-test), Jonckheere-Terpstra-test and the Wilcoxon-test.
[0105] The terms "treatment regimen", "dosing protocol" and dosing regimen are used interchangeably to refer to the dose and timing of administration of each therapeutic agent in a combination of the invention.
[0106] As used herein, "treatment" is an approach for obtaining beneficial or desired clinical results. For purposes of this invention, beneficial or desired clinical results include, but are not limited to, one or more of the following: reducing the proliferation of (or destroying) neoplastic or cancerous cells, inhibiting metastasis of neoplastic cells, shrinking or decreasing the size of tumor, remission of a PD-L1 associated disease (e.g., cancer), decreasing symptoms resulting from a PD-L1 associated disease (e.g., cancer), increasing the quality of life of those suffering from a PD-L1 associated disease (e.g., cancer), decreasing the dose of other medications required to treat a PD-L1 associated disease (e.g., cancer), delaying the progression of a PD-L1 associated disease (e.g., cancer), curing a PD-L1 associated disease (e.g., cancer), and/or prolong survival of patients having a PD-L1 associated disease (e.g., cancer).
[0107] "Ameliorating" means a lessening or improvement of one or more symptoms as compared to not administering a PD-L1 antibody. "Ameliorating" also includes shortening or reduction in duration of a symptom.
[0108] As used herein, an "effective dosage" or "effective amount" of drug, compound, or pharmaceutical composition is an amount sufficient to effect any one or more beneficial or desired results. For prophylactic use, beneficial or desired results include eliminating or reducing the risk, lessening the severity, or delaying the outset of the disease, including biochemical, histological and/or behavioral symptoms of the disease, its complications and intermediate pathological phenotypes presenting during development of the disease. For therapeutic use, beneficial or desired results include clinical results such as reducing incidence or amelioration of one or more symptoms of various PD-L1 associated diseases or conditions (such as for example advanced RCC), decreasing the dose of other medications required to treat the disease, enhancing the effect of another medication, and/or delaying the progression of the PD-L1 associated disease of patients. An effective dosage can be administered in one or more administrations. For purposes of this invention, an effective dosage of drug, compound, or pharmaceutical composition is an amount sufficient to accomplish prophylactic or therapeutic treatment either directly or indirectly. As is understood in the clinical context, an effective dosage of a drug, compound, or pharmaceutical composition may or may not be achieved in conjunction with another drug, compound, or pharmaceutical composition. Thus, an "effective dosage" may be considered in the context of administering one or more therapeutic agents, and a single agent may be considered to be given in an effective amount if, in conjunction with one or more other agents, a desirable result may be or is achieved.
[0109] "Tumor" as it applies to a subject diagnosed with, or suspected of having, a cancer refers to a malignant or potentially malignant neoplasm or tissue mass of any size, and includes primary tumors and secondary neoplasms. A solid tumor is an abnormal growth or mass of tissue that usually does not contain cysts or liquid areas. Different types of solid tumors are named for the type of cells that form them. Examples of solid tumors are sarcomas, carcinomas, and lymphomas. Leukemias (cancers of the blood) generally do not form solid tumors (National Cancer Institute, Dictionary of Cancer Terms).
[0110] "Tumor burden" also referred to as "tumor load", refers to the total amount of tumor material distributed throughout the body. Tumor burden refers to the total number of cancer cells or the total size of tumor(s), throughout the body, including lymph nodes and bone narrow. Tumor burden can be determined by a variety of methods known in the art, such as, e.g. by measuring the dimensions of tumor(s) upon removal from the subject, e.g., using calipers, or while in the body using imaging techniques, e.g., ultrasound, bone scan, computed tomography (CT) or magnetic resonance imaging (MRI) scans.
[0111] The term "tumor size" refers to the total size of the tumor which can be measured as the length and width of a tumor. Tumor size may be determined by a variety of methods known in the art, such as, e.g. by measuring the dimensions of tumor(s) upon removal from the subject, e.g., using calipers, or while in the body using imaging techniques, e.g., bone scan, ultrasound, CT or MRI scans.
[0112] "Variable regions" or "V region" as used herein means the segment of IgG chains which is variable in sequence between different antibodies. It extends to Kabat residue 109 in the light chain and 113 in the heavy chain.
[0113] "VEGFR inhibitor" means a small molecule inhibitor of vascular endothelial growth factor (VEGF) receptor or a monoclonal antibody against vascular endothelial growth factor (VEGF). In an embodiment, a "VEGFR inhibitor" means a small molecule inhibitor of vascular endothelial growth factor (VEGF) receptor. Specific VEGFR inhibitors useful as the VEGFR inhibitor in the treatment method, medicaments and uses of the present invention, include axitinib, sunitinib, sorafenib, tivozanib, and bevacizumab. In an embodiment, specific VEGFR inhibitors useful as the VEGFR inhibitor in the treatment method, medicaments and uses of the present invention, include axitinib, sunitinib, sorafenib, and tivozanib.
[0114] In an embodiment of the treatment method, medicaments and uses of the present invention, the VEGFR inhibitor is the compound, N-methyl-2-[3-((E)-2-pyridin-2-yl-vinyl)-1 H-indazol-6-ylsulfanyl]-benzamide or 6-[2-(methylcarbamoyl)phenylsulfanyl]-3-E-[2-(pyridin-2-ypethenyl]indazol- e, of the following structure:
##STR00001##
which is known as axitinib or AG-013736.
[0115] Axitinib is a potent and selective inhibitor of vascular endothelial growth factor (VEGF) receptors 1, 2 and 3. These receptors are implicated in pathologic angiogenesis, tumor growth, and metastatic progression of cancer. Axitinib has been shown to potently inhibit VEGF-mediated endothelial cell proliferation and survival (Hu-Lowe, D. D., et al., Clin Cancer Res 14: 7272-7283 (2008); Solowiej, S., et al., Biochemistry 48: 7019-31 (2009)). Clinical trials are currently on-going or have been conducted to study the use of axitinib for the treatment of various cancers, including liver cancer, melanoma, mesothelioma, non-small cell lung cancer, prostate cancer, renal cell carcinoma, soft tissue sarcomas and solid tumors. Inlyta.RTM. (axitinib) has been approved in the United States, Europe, Japan and other jurisdictions for the treatment of renal cell carcinoma.
[0116] Axitinib, as well as pharmaceutically acceptable salts thereof, is described in U.S. Pat. No. 6,534,524. Methods of making axitinib are described in U.S. Pat. Nos. 6,884,890 and 7,232,910, in U.S. Publication Nos. 2006-0091067 and 2007-0203196 and in International Publication No. WO 2006/048745. Dosage forms of axitinib are described in U.S. Publication No. 2004-0224988. Polymorphic forms and pharmaceutical compositions of axitinib are also described in U.S. Publication Nos. 2006-0094763, 2008-0274192 and 2010-0179329 and International Publication No. WO 2013/046133. The patents and patent applications listed above are incorporated herein by reference.
[0117] Axitinib is understood to include reference to salts thereof, unless otherwise indicated. Axitinib is basic in nature and capable of forming a wide variety of salts with various inorganic and organic acids. The term "salt(s)", as employed herein, denotes acidic salts formed with inorganic and/or organic acids. Pharmaceutically acceptable salts of axitinib may be formed, for example, by reacting axitinib with an amount of acid, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization.
[0118] Exemplary acid addition salts of the compound of Formula I include acetates, ascorbates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, fumarates, hydrochlorides, hydrobromides, hydroiodides, lactates, maleates, methanesulfonates, naphthalenesulfonates, nitrates, oxalates, phosphates, propionates, salicylates, succinates, sulfates, tartarates, thiocyanates, toluenesulfonates (also known as tosylates,) and the like. Additionally, acids which are generally considered suitable for the formation of pharmaceutically useful salts from basic pharmaceutical compounds are discussed, for example, by S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66(1) 1-19; P. Gould, International J. of Pharmaceutics (1986) 33 201-217; Anderson et al, The Practice of
[0119] Medicinal Chemistry (1996), Academic Press, New York; and in The Orange Book (Food & Drug Administration, Washington, D.C. on their website). These disclosures are incorporated herein by reference thereto.
[0120] All such acid salts are intended to be pharmaceutically acceptable salts within the scope of axitinib, as used in the present invention and all acid salts are considered equivalent to the free forms of the corresponding compound for purposes of the invention.
[0121] Prodrugs of axitinib are also contemplated for use in the methods, medicaments and uses of the present invention. The term "prodrug", as employed herein, denotes a compound that is a drug precursor which, upon administration to a subject, undergoes chemical conversion by metabolic or chemical processes to yield axitinib or a salt thereof. A discussion of prodrugs is provided in T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems (1987) 14 of the A.C.S. Symposium Series, and in
[0122] Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press, both of which are incorporated herein by reference thereto.
[0123] The term "4-1 BB antibody" as used herein means an antibody, as defined herein, capable of binding to human 4-1BB receptor.
[0124] The terms "4-1 BB" and "4-1 BB receptor" are used interchangeably in the present application, and refer to any form of 4-1 BB receptor, as well as variants, isoforms, and species homologs thereof that retain at least a part of the activity of4-1 BB receptor. Accordingly, a binding molecule, as defined and disclosed herein, may also bind 4-1 BB from species other than human. In other cases, a binding molecule may be completely specific for the human 4-1 BB and may not exhibit species or other types of cross-reactivity. Unless indicated differently, such as by specific reference to human4-166,4-1BB includes all mammalian species of native sequence4-1BB, e.g., human, canine, feline, equine and bovine. One exemplary human 4-1BB is a 255 amino acid protein (Accession No. NM_001561; NP_001552).
[0125] 4-1BB comprises a signal sequence (amino acid residues 1-17), followed by an extracellular domain (169 amino acids), a transmembrane region (27 amino acids), and an intracellular domain (42 amino acids) (Cheuk ATC et al. 2004 Cancer Gene Therapy 11: 215-226). The receptor is expressed on the cell surface in monomer and dimer forms and likely trimerizes with 4-1 BB ligand to signal.
[0126] "4-1 BB agonist" as used herein means, any chemical compound or biological molecule, as defined herein, which upon binding to 4-1 BB, (1) stimulates or activates 4-1 BB, (2) enhances, increases, promotes, induces, or prolongs an activity, function, or presence of 4-1 BB, or (3) enhances, increases, promotes, or induces the expression of 4-1BB. 4-1BB agonists useful in the any of the treatment method, medicaments and uses of the present invention include a monoclonal antibody (mAb), or antigen binding fragment thereof, which specifically binds to 4-1 BB. Alternative names or synonyms for 4-1BB include CD137 and TNFRSF9. In any of the treatment method, medicaments and uses of the present invention in which a human individual is being treated, the 4-1BB agonists increase a 4-1BB-mediated response. In some embodiments of the treatment method, medicaments and uses of the present invention, 4-1 BB agonists markedly enhance cytotoxic T-cell responses, resulting in anti-tumor activity in several models.
[0127] Human 4-1 BB comprises a signal sequence (amino acid residues 1-17), followed by an extracellular domain (169 amino acids), a transmembrane region (27 amino acids), and an intracellular domain (42 amino acids) (Cheuk A T C et al. 2004 Cancer Gene Therapy 11: 215-226). The receptor is expressed on the cell surface in monomer and dimer forms and likely trimerizes with 4-1 BB ligand to signal.
[0128] Examples of mAbs that bind to human 4-1BB, and useful in the treatment method, medicaments and uses of the present invention, are described in U.S. Pat. No. 8,337,850 and US20130078240. In some embodiments an anti-4-1BB antibody useful in the treatment, method, medicaments and uses disclosed herein is a fully humanized IgG2 agonist monoclonal antibody comprising a heavy chain variable region and a light chain variable region comprising the amino acid sequences shown in SEQ ID NO: 18 and SEQ ID NO: 19, respectively.
[0129] Table 3A below provides exemplary anti-4-1BB antibody sequences for use in the treatment method, medicaments and uses of the present invention.
TABLE-US-00003 TABLE 3A EXEMPLARY ANTI-HUMAN 4-1BB MONOCLONAL ANTIBODY SEQUENCES CDRH1 STYWIS (SEQ ID NO: 12) CDRH2 KIYPGDSYTNYSPSFQG (SEQ ID NO: 13) CDRH3 RGYGIFDY (SEQ ID NO: 14) CDRL1 SGDNIGDQYAH (SEQ ID NO: 15) CDRL2 QDKNRPS (SEQ ID NO: 16) CDRL3 ATYTGFGSLAV (SEQ ID NO: 17) Heavy chain VR EVQLVQSGAEVKKPGESLRISCKGSGYSFSTYWISWVRQMPGKGL EWMGKIYPGDSYTNYSPSFQGQVTISADKSISTAYLQWSSLKASDT AMYYCARGYGIFDYWGQGTLVTVSS (SEQ ID NO: 18) Light chain VR SYELTQPPSVSVSPGQTASITCSGDNIGDQYAHWYQQKPGQSPVL VIYQDKNRPSGIPERFSGSNSGNTATLTISGTQAMDEADYYCATYT GFGSLAVFGGGTKLTVL (SEQ ID NO: 19) Heavy chain EVQLVQSGAEVKKPGESLRISCKGSGYSFSTYWISWVRQMPGKGL EWMGKIYPGDSYTNYSPSFQGQVTISADKSISTAYLQWSSLKASDT AMYYCARGYGIFDYWGQGTLVTVSSastkgpsvfplapcsrstses taalgclvkdyfpepvtvswnsgaltsgvhtfpavlqssglyslss vvtvpssnfgtqtytcnvdhkpsntkvdktverkccvecppcpapp vagpsvflfppkpkdtlmisrtpevtcvvvdvshedpevqfnwyvd gvevhnaktkpreeqfnstfrvvsvltvvhqdwlngkeykckvsnk glpapiektisktkgqprepqvytlppsreemtknqvsltclvkgf ypsdiavewesngqpennykttppmldsdgsfflyskltvdksrwq qgnvfscsvmhealhnhytqkslslspgk (SEQ ID NO: 20) Light chain SYELTQPPSVSVSPGQTASITCSGDNIGDQYAHWYQQKPGQSPVL VIYQDKNRPSGIPERFSGSNSGNTATLTISGTQAMDEADYYCATYT GFGSLAVFGGGTKLTVLgqpkaapsvtlfppsseelqankatlycl isdfypgavtvawkadsspvkagvetttpskqsnnkyaassylslt peqwkshrsyscqvthegstvektvaptecs (SEQ ID NO: 21)
[0130] The term "M-CSF antibody" as used herein means an antibody, as defined herein, capable of binding to human M-CSF receptor.
[0131] The terms "M-CSF" and "M-CSF receptor" are used interchangeably in the present application, and refer to any form of M-CSF receptor, as well as variants, isoforms, and species homologs thereof that retain at least a part of the activity of M-CSF receptor. Accordingly, a binding molecule, as defined and disclosed herein, may also bind M-CSF from species other than human. In other cases, a binding molecule may be completely specific for the human M-CSF and may not exhibit species or other types of cross-reactivity. Unless indicated differently, such as by specific reference to human M-CSF, M-CSF includes all mammalian species of native sequence M-CSF, e.g., human, canine, feline, equine and bovine. One exemplary human M-CSF is a 554 amino acid protein (UniProt Accession No. P09603).
[0132] "M-CSF antagonist antibody" as used herein means, any antibody, as defined herein, which upon binding to M-CSF, inhibits the binding of a M-CSF to c-fms receptor and blocks or prevents activation of c-fms. M-CSF antagonists useful in the any of the treatment method, medicaments and uses of the present invention include a monoclonal antibody (mAb) which specifically binds to M-CSF.
[0133] Examples of mAbs that bind to human M-CSF, and useful in the treatment method, medicaments and uses of the present invention, are described in, for example, U.S. Pat. No. 7,326,414, PCT Patent Application Publication No. WO2014167088, and U.S. Patent Application Publication No. 20140242071. In some embodiments an anti-M-CSF antibody useful in the treatment, method, medicaments and uses disclosed herein is a fully human IgG2 antagonist monoclonal antibody comprising a heavy chain variable region and a light chain variable region comprising the amino acid sequences shown in SEQ ID NO: 30 and SEQ ID NO: 31, respectively.
[0134] Table 3B below provides exemplary anti-M-CSF antibody sequences for use in the treatment method, medicaments and uses of the present invention.
TABLE-US-00004 TABLE 3B EXEMPLARY ANTI-HUMAN M-CSF MONOCLONAL ANTIBODY SEQUENCES CDRH1 SFSMT (SEQ ID NO: 24) CDRH2 YISSRSSTISYADSVKG (SEQ ID NO: 25) CDRH3 DPLLAGATFFDY (SEQ ID NO: 26) CDRL1 RASQSVSSSYLA (SEQ ID NO: 27) CDRL2 GASSRAT (SEQ ID NO: 28) CDRL3 QQYGSSPLT (SEQ ID NO: 29) Heavy chain VR MELGLCWVFLVAILEGVQCEVQLVESGGGLVQPGGSLRLSCAASG FTFSSFSMTWVRQAPGKGLEWVSYISSRSSTISYADSVKGRFTISR DNAKNSLYLQMNSLRDEDTAVYYCARDPLLAGATFFDYWGQGTLV TVSSA (SEQ ID NO: 30) Light chain VR METPAQLLFLLLLWLPDTTGEFVLTQSPGTLSLSPGERATLSCRAS QSVSSSYLAWYQQKPGQAPRLLIYGASSRATGIPDRFSGSGSGTD FTLTISRLEPEDFAVYYCQQYGSSPLTFGGGTKVEIK (SEQ ID NO: 31) Heavy chain MELGLCWVFLVAILEGVQCEVQLVESGGGLVQPGGSLRLSCAASG FTFSSFSMTWVRQAPGKGLEWVSYISSRSSTISYADSVKGRFTISR DNAKNSLYLQMNSLRDEDTAVYYCARDPLLAGATFFDYWGQGTLV TVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWN SGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHK PSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISR TPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTF RVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREP QVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNY KTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHY TQKSLSLSPGK (SEQ ID NO: 22) Light chain METPAQLLFLLLLWLPDTTGEFVLTQSPGTLSLSPGERATLSCRAS QSVSSSYLAWYQQKPGQAPRLLIYGASSRATGIPDRFSGSGSGTD FTLTISRLEPEDFAVYYCQQYGSSPLTFGGGTKVEIKRTVAAPSVFIF PPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESV TEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSF NRGEC (SEQ ID NO: 23)
[0135] The term "OX40 antibody" as used herein means an antibody, as defined herein, capable of binding to human OX40 receptor.
[0136] The terms "OX40" and "OX40 receptor" are used interchangeably in the present application, and refer to any form of OX40 receptor, as well as variants, isoforms, and species homologs thereof that retain at least a part of the activity of OX40 receptor. Accordingly, a binding molecule, as defined and disclosed herein, may also bind OX40 from species other than human. In other cases, a binding molecule may be completely specific for the human OX40 and may not exhibit species or other types of cross-reactivity. Unless indicated differently, such as by specific reference to human OX40, 0X40 includes all mammalian species of native sequence OX40, e.g., human, canine, feline, equine and bovine. One exemplary human OX40 is a 277 amino acid protein (UniProt Accession No. P43489).
[0137] "OX40 agonist antibody" as used herein means, any antibody, as defined herein, which upon binding to OX40, (1) stimulates or activates OX40, (2) enhances, increases, promotes, induces, or prolongs an activity, function, or presence of OX40, or (3) enhances, increases, promotes, or induces the expression of OX40. 0X40 agonists useful in the any of the treatment method, medicaments and uses of the present invention include a monoclonal antibody (mAb) which specifically binds to OX40.
[0138] Examples of mAbs that bind to human OX40, and useful in the treatment method, medicaments and uses of the present invention, are described in, for example, U.S. Pat. No. 7,960,515, PCT Patent Application Publication Nos. WO2013028231 and WO2013/119202, and U.S. Patent Application Publication No. 20150190506. In some embodiments an anti-OX40 antibody useful in the treatment, method, medicaments and uses disclosed herein is a fully human agonist monoclonal antibody comprising a heavy chain variable region and a light chain variable region comprising the amino acid sequences shown in SEQ ID NO: 38 and SEQ ID NO: 39, respectively. In some embodiments, the anti-OX40 antibody is a fully human IgG2 or IgG1 antibody.
[0139] Table 3C below provides exemplary anti-OX40 antibody sequences for use in the treatment method, medicaments and uses of the present invention.
TABLE-US-00005 TABLE 3C EXEMPLARY ANTI-HUMAN OX40 MONOCLONAL ANTIBODY SEQUENCES CDRH1 SYSMN (SEQ ID NO: 32) CDRH2 YISSSSSTIDYADSVKG (SEQ ID NO: 33) CDRH3 ESGWYLFDY (SEQ ID NO: 34) CDRL1 RASQGISSWLA (SEQ ID NO: 35) CDRL2 AASSLQS (SEQ ID NO: 36) CDRL3 QQYNSYPPT (SEQ ID NO: 37) Heavy chain VR EVQLVESGGGLVQPGGSLRLSCAASGFTFSSYSMNWVRQAPGKG LEWVSYISSSSSTIDYADSVKGRFTISRDNAKNSLYLQMNSLRDEDT AVYYCARESGWYLFDYWGQGTLVTVSS (SEQ ID NO: 38) Light chain VR DIQMTQSPSSLSASVGDRVTITCRASQGISSWLAWYQQKPEKAPKS LIYAASSLQSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYNS YPPTFGGGTKVEIK (SEQ ID NO: 39) Heavy chain EVQLVESGGGLVQPGGSLRLSCAASGFTFSSYSMNWVRQAPGKG LEWVSYISSSSSTIDYADSVKGRFTISRDNAKNSLYLQMNSLRDEDT AVYYCARESGWYLFDYWGQGTLVTVSSastkgpsvfplapcsrstse staalgclvkdyfpepvtvswnsgaltsgvhtfpavlqssglyslss vvtvpssnfgtqtytcnvdhkpsntkvdktverkccvecppcpappv agpsvflfppkpkdtlmisrtpevtcvvvdvshedpevqfnwyvdgv evhnaktkpreeqfnstfrvvsvltvvhqdwlngkeykckvsnkglp apiektisktkgqprepqvytlppsreemtknqvsltclvkgfypsd iavewesngqpennykttppmldsdgsfflyskltvdksrwqqgnvf scsvmhealhnhytqkslslspgk (SEQ ID NO: 40) Light chain DIQMTQSPSSLSASVGDRVTITCRASQGISSWLAWYQQKPEKAPKS LIYAASSLQSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYNS YPPTFGGGTKVEIKrtvaapsvfifppsdeqlksgtasvvcllnnfy preakvqwkvdnalqsgnsqesvteqdskdstyslsstltlskadye khkvyacevthqglsspvtksfnrgec (SEQ ID NO: 41)
[0140] The "CD20" antigen is a "35 kDa, non-glycosylated phosphoprotein found on the surface of greater than 90% of B cells from peripheral blood or lymphoid organs. CD20 is expressed during early pre-B cell development and remains until plasma cell differentiation. CD20 is present on both normal B cells as well as malignant B cells. Other names for CD20 in the literature include "B-lymphocyte-restricted antigen" and "Bp35". The CD20 antigen is described in Clark et al. PNAS (USA) 82:1766 (1985), for example.
[0141] Unless otherwise defined, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. In case of conflict, the present specification, including definitions, will control. Throughout this specification and claims, the word "comprise," or variations such as "comprises" or "comprising" will be understood to imply the inclusion of a stated integer or group of integers but not the exclusion of any other integer or group of integers. Unless otherwise required by context, singular terms shall include pluralities and plural terms shall include the singular.
[0142] Exemplary methods and materials are described herein, although methods and materials similar or equivalent to those described herein can also be used in the practice or testing of the invention. The materials, methods, and examples are illustrative only and not intended to be limiting.
II. METHODS, USES AND MEDICAMENTS
[0143] In one aspect of the invention, the invention provides a method for treating a cancer in a subject comprising administering to the subject a combination therapy which comprises a PD-L1 antagonist and a VEGR inhibitor.
[0144] In another aspect of the invention, the invention provides a method for treating a cancer in a subject comprising administering to the subject a combination therapy which comprises a PD-L1 antagonist and an anti-4-1 BB antibody.
[0145] In another aspect of the invention, the invention provides a method for treating a cancer in a subject comprising administering to the subject a combination therapy which comprises a PD-L1 antagonist and an anti-M-CSF antibody.
[0146] In another aspect of the invention, the invention provides a method for treating a cancer in a subject comprising administering to the subject a combination therapy which comprises a PD-L1 antagonist and an anti-OX40 antibody.
[0147] In another aspect of the invention, the invention provides a method for treating a cancer in a subject comprising administering to the subject a combination therapy which comprises a PD-L1 antagonist, an anti-4-1 BB antibody, and an anti-M-CSF antibody.
[0148] In another aspect of the invention, the invention provides a method for treating a cancer in a subject comprising administering to the subject a combination therapy which comprises a PD-L1 antagonist, an anti-4-1 BB antibody, and an anti-OX40 antibody.
[0149] In another aspect of the invention, the invention provides a method for treating a cancer in a subject comprising administering to the subject a combination therapy which comprises a PD-L1 antagonist, an anti-4-1 BB antibody, and a CD20 antagonist. In some embodiments, the PD-L1 antagonist is avelumab, the anti-4-1 BB antibody is PF-05082566, and the CD20 antagonist is rituximab. In some embodiments, the method comprises a 28-day cycle wherein rituximab is administered on Day 1 of each 28-day cycle at a dose of 375 mg/m.sup.2, PF-05082566 is administered on Day 1 or Day 2 at a fixed dose of 100 mg, and avelumab is administered at a dose of 10 mg/kg on Day 2 and Day 15 or 16 of each 28-day cycle. In some embodiments on Day 2, avelumab is administered at least 3 hours after administration of PF-05082566. In some embodiments on Day 2, avelumab is administered about 30 minutes after administration of PF-05082566. In some embodiments on Day 2, avelumab is administered about 60 minutes after administration of PF-05082566.
[0150] In another aspect of the invention, the invention provides a method for treating a cancer in a subject comprising administering to the subject a combination therapy which comprises a PD-L1 antagonist, an anti-4-1BB antibody, and azacitidine. In some embodiments, the PD-L1 antagonist is avelumab, and the anti-4-1BB antibody is PF-05082566. In some embodiments, the method comprises a 28-day cycle wherein azacitidine is administered subcutaneously at a daily dose of 75 mg/m.sup.2 on Day 1 to Day 7 consecutively of each 28-day cycle, PF-05082566 is administered intravenously at a fixed dose of 100 mg on Day 1 or Day 2, and avelumab is administered at a dose of 10 mg/kg on Day 2 and either Day 15 or Day 16 of each 28-day cycle. In some embodiments on Day 2, avelumab is administered at least 3 hours after administration of PF-05082566. In some embodiments on Day 2, avelumab is administered about 30 minutes after administration of PF-05082566. In some embodiments on Day 2, avelumab is administered about 60 minutes after administration of PF-05082566. In some embodiments on Day 2, avelumab is administered at least 3 hours after administration of PF-05082566. In some embodiments on Day 2, avelumab is administered about 30 minutes after administration of PF-05082566. In some embodiments on Day 2, avelumab is administered about 60 minutes after administration of PF-05082566. In some embodiments, azacitidine is administered at least 3 hours prior to PF-05082566 when dosed on the same day.
[0151] In another aspect of the invention, the invention provides a method for treating a cancer in a subject comprising administering to the subject a combination therapy which comprises a PD-L1 antagonist, bendamustine, and a CD20 antagonist. In some embodiments, the PD-L1 antagonist is avelumab, and the CD20 antagonist is rituximab. In some embodiments, the method comprises a 28-day cycle wherein rituximab is administered on Day 1 of each 28-day cycle at a dose of 375 mg/m.sup.2, bendamustine is administered intravenously at a dose of 90 mg/m.sup.2 on Day 2 and Day 3, and avelumab is administered at a dose of 10 mg/kg on Day 2 and Day 15 or 16 of each 28-day cycle. In some embodiments, the method comprises a 28-day cycle wherein rituximab is administered on Day 1 of each 28-day cycle at a dose of 375 mg/m.sup.2, bendamustine is administered intravenously at a dose of 90 mg/m.sup.2 on Day 1 and Day 2, and avelumab is administered at a dose of 10 mg/kg on Day 2 and Day 15 or 16 of each 28-day cycle. In some embodiments on Day 2, avelumab is administered at least 3 hours after administration of bendamustine. In some embodiments on Day 2, avelumab is administered about 30 minutes after administration of bendamustine. In some embodiments on Day 2, avelumab is administered about 60 minutes after administration of bendamustine.
[0152] In another aspect of the invention, the invention provides a method for treating a cancer in a subject comprising administering to the subject a combination therapy which comprises a PD-L1 antagonist and chemoradiotherapy.
[0153] The combination therapy may also comprise one or more additional therapeutic agents. The additional therapeutic agent may be, e.g., a chemotherapeutic other than a VEGR inhibitor, a biotherapeutic agent (including but not limited to antibodies to VEGF, EGFR, Her2/neu, other growth factor receptors, CD40, CD-40L, CTLA-4, and ICOS), an immunogenic agent (for example, attenuated cancerous cells, tumor antigens, antigen presenting cells such as dendritic cells pulsed with tumor derived antigen or nucleic acids, immune stimulating cytokines (for example, IL-2, IFNa2, GM-CSF), a chimeric antigen receptior (CAR)-T cell, and cells transfected with genes encoding immune stimulating cytokines such as but not limited to GM-CSF).
[0154] Examples of chemotherapeutic agents include alkylating agents such as thiotepa and cyclosphosphamide; alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa; ethylenimines and methylamelamines including altretamine, triethylenemelamine, trietylenephosphoramide, triethylenethiophosphoramide and trimethylolomelamine;
[0155] acetogenins (especially bullatacin and bullatacinone); a camptothecin (including the synthetic analogue topotecan); bryostatin; callystatin; CC-1065 (including its adozelesin, carzelesin and bizelesin synthetic analogues); cryptophycins (particularly cryptophycin 1 and cryptophycin 8); dolastatin; duocarmycin (including the synthetic analogues, KW-2189 and CBI-TMI); eleutherobin; pancratistatin; a sarcodictyin; spongistatin; nitrogen mustards such as chlorambucil, chlornaphazine, cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosureas such as carmustine, chlorozotocin, fotemustine, lomustine, nimustine, ranimustine; antibiotics such as the enediyne antibiotics (e.g. calicheamicin, especially calicheamicin gamma1 I and calicheamicin phil1, see, e.g., Agnew, Chem. Intl. Ed. Engl., 33:183-186 (1994); dynemicin, including dynemicin A; bisphosphonates, such as clodronate; an esperamicin; as well as neocarzinostatin chromophore and related chromoprotein enediyne antibiotic chromomophores), aclacinomysins, actinomycin, authramycin, azaserine, bleomycins, cactinomycin, carabicin, caminomycin, carzinophilin, chromomycins, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L-norleucine, doxorubicin (including morpholino-doxorubicin, cyanomorpholino-doxorubicin, 2-pyrrolino-doxorubicin and deoxydoxorubicin), epirubicin, esorubicin, idarubicin, marcellomycin, mitomycins such as mitomycin C, mycophenolic acid, nogalamycin, olivomycins, peplomycin, potfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin; anti-metabolites such as methotrexate and 5-fluorouracil (5-FU); folic acid analogues such as denopterin, methotrexate, pteropterin, trimetrexate; purine analogs such as fludarabine, 6-mercaptopurine, thiamiprine, thioguanine; pyrimidine analogs such as ancitabine, 6-azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine; androgens such as calusterone, dromostanolone propionate, epitiostanol, mepitiostane, testolactone; anti-adrenals such as aminoglutethimide, mitotane, trilostane; folic acid replenisher such as frolinic acid; aceglatone; aldophosphamide glycoside; aminolevulinic acid; eniluracil; amsacrine; bestrabucil; bisantrene; edatraxate; defofamine; demecolcine; diaziquone; elformithine; elliptinium acetate; an epothilone; etoglucid; gallium nitrate; hydroxyurea; lentinan; lonidamine; maytansinoids such as maytansine and ansamitocins; mitoguazone; mitoxantrone; mopidamol; nitracrine; pentostatin; phenamet; pirarubicin; losoxantrone; podophyllinic acid; 2-ethylhydrazide; procarbazine; razoxane; rhizoxin; sizofuran; spirogermanium; tenuazonic acid; triaziquone; 2, 2',2''-trichlorotriethylamine; trichothecenes (especially T-2 toxin, verracurin A, roridin A and anguidine); urethan; vindesine; dacarbazine; mannomustine; mitobronitol; mitolactol; pipobroman; gacytosine; arabinoside ("Ara-C"); cyclophosphamide; thiotepa; taxoids, e.g. paclitaxel and doxetaxel; chlorambucil; gemcitabine; 6-thioguanine; mercaptopurine; methotrexate; platinum analogs such as carboplatin; vinblastine; platinum; etoposide (VP-16); ifosfamide; mitoxantrone; vincristine; vinorelbine; novantrone; teniposide; edatrexate; daunomycin; aminopterin; xeloda; ibandronate; CPT-11; topoisomerase inhibitor RFS 2000; difluoromethylornithine (DMFO); retinoids such as retinoic acid; capecitabine; and pharmaceutically acceptable salts, acids or derivatives of any of the above. Also included are anti-hormonal agents that act to regulate or inhibit hormone action on tumors such as anti-estrogens and selective estrogen receptor modulators (SERMs), including, for example, tamoxifen, raloxifene, droloxifene, 4-hydroxytamoxifen, trioxifene, keoxifene, LY117018, onapristone, and toremifene (Fareston); aromatase inhibitors that inhibit the enzyme aromatase, which regulates estrogen production in the adrenal glands, such as, for example, 4(5)-imidazoles, aminoglutethimide, megestrol acetate, exemestane, formestane, fadrozole, vorozole, letrozole, and anastrozole; and anti-androgens such as flutamide, nilutamide, bicalutamide, leuprolide, and goserelin; and pharmaceutically acceptable salts, acids or derivatives of any of the above.
[0156] Each therapeutic agent in a combination therapy of the invention may be administered either alone or in a medicament (also referred to herein as a pharmaceutical composition) which comprises the therapeutic agent and one or more pharmaceutically acceptable carriers, excipients and diluents, according to standard pharmaceutical practice.
[0157] Each therapeutic agent in a combination therapy of the invention may be administered simultaneously (i.e., in the same medicament), concurrently (i.e., in separate medicaments administered one right after the other in any order) or sequentially in any order. Sequential administration is particularly useful when the therapeutic agents in the combination therapy are in different dosage forms (one agent is a tablet or capsule and another agent is a sterile liquid) and/or are administered on different dosing schedules, e.g., a chemotherapeutic that is administered at least daily and a biotherapeutic that is administered less frequently, such as once weekly, once every two weeks, or once every three weeks.
[0158] In some embodiments, the VEGFR inhibitor or anti-4-1BB antibody is administered before administration of the PD-L1 antagonist, while in other embodiments, the VEGFR inhibitor or anti-4-1BB antibody is administered after administration of the PD-L1 antagonist.
[0159] In some embodiments, at least one of the therapeutic agents in the combination therapy is administered using the same dosage regimen (dose, frequency and duration of treatment) that is typically employed when the agent is used as monotherapy for treating the same cancer. In other embodiments, the patient receives a lower total amount of at least one of the therapeutic agents in the combination therapy than when the agent is used as monotherapy, e.g., smaller doses, less frequent doses, and/or shorter treatment duration.
[0160] Each small molecule therapeutic agent in a combination therapy of the invention can be administered orally or parenterally, including the intravenous, intramuscular, intraperitoneal, subcutaneous, rectal, topical, and transdermal routes of administration.
[0161] A combination therapy of the invention may be used prior to or following surgery to remove a tumor and may be used prior to, during or after radiation therapy.
[0162] In some embodiments, a combination therapy of the invention is administered to a patient who has not been previously treated with a biotherapeutic or chemotherapeutic agent, i.e., is treatment-naive. In other embodiments, the combination therapy is administered to a patient who failed to achieve a sustained response after prior therapy with a biotherapeutic or chemotherapeutic agent, i.e., is treatment-experienced.
[0163] A combination therapy of the invention is typically used to treat a tumor that is large enough to be found by palpation or by imaging techniques well known in the art, such as MRI, ultrasound, or CAT scan. In some embodiments, a combination therapy of the invention is used to treat an advanced stage tumor having dimensions of at least about 200 mm.sup.3, 300 mm.sup.3, 400 mm.sup.3, 500 mm.sup.3, 750 mm.sup.3, or up to 1000 mm.sup.3.
[0164] In some embodiments, a combination therapy of the invention is administered to a human patient who has a cancer that tests positive for PD-L1 expression. In some embodiments, PD-L1 expression can be detected using a diagnostic anti-human PD-L1 antibody, or antigen binding fragment thereof, in an IHC assay on an FFPE or frozen tissue section of a tumor sample removed from the patient. Typically, the patient's physician would order a diagnostic test to determine PD-L1 expression in a tumor tissue sample removed from the patient prior to initiation of treatment with the PD-L1 antagonist and VEGFR inhibitor, but it is envisioned that the physician could order the first or subsequent diagnostic tests at any time after initiation of treatment, such as for example after completion of a treatment cycle.
[0165] Selecting a dosage regimen (also referred to herein as an administration regimen) for a combination therapy of the invention depends on several factors, including the serum or tissue turnover rate of the entity, the level of symptoms, the immunogenicity of the entity, and the accessibility of the target cells, tissue or organ in the subject being treated. Preferably, a dosage regimen maximizes the amount of each therapeutic agent delivered to the patient consistent with an acceptable level of side effects. Accordingly, the dose amount and dosing frequency of each biotherapeutic and chemotherapeutic agent in the combination depends in part on the particular therapeutic agent, the severity of the cancer being treated, and patient characteristics. Guidance in selecting appropriate doses of antibodies, cytokines, and small molecules are available. See, e.g., Wawrzynczak (1996) Antibody Therapy, Bios Scientific Pub. Ltd, Oxfordshire, UK; Kresina (ed.) (1991) Monoclonal Antibodies, Cytokines and Arthritis, Marcel Dekker, New York, N.Y.; Bach (ed.) (1993) Monoclonal Antibodies and Peptide Therapy in Autoimmune Diseases, Marcel Dekker, New York, N.Y.; Baert et al. (2003) New Engl. J. Med. 348:601-608; Milgrom et al. (1999) New Engl. J. Med. 341:1966-1973; Slamon et al. (2001) New Engl. J. Med. 344:783-792; Beniaminovitz et al. (2000) New Engl. J. Med. 342:613-619; Ghosh et al. (2003) New Engl. J. Med.
[0166] 348:24-32; Lipsky et al. (2000) New Engl. J. Med. 343:1594-1602; Physicians' Desk Reference 2003 (Physicians' Desk Reference, 57th Ed); Medical Economics Company; ISBN: 1563634457; 57th edition (November 2002). Determination of the appropriate dosage regimen may be made by the clinician, e.g., using parameters or factors known or suspected in the art to affect treatment or predicted to affect treatment, and will depend, for example, the patient's clinical history (e.g., previous therapy), the type and stage of the cancer to be treated and biomarkers of response to one or more of the therapeutic agents in the combination therapy.
[0167] Biotherapeutic agents in a combination therapy of the invention may be administered by continuous infusion, or by doses at intervals of, e.g., daily, every other day, three times per week, or one time each week, two weeks, three weeks, monthly, bimonthly, etc. A total weekly dose is generally at least 0.05 .mu.g/kg, 0.2 .mu.g/kg, 0.5 .mu.g/kg, 1 .mu.g/kg, 10 .mu.g/kg, 100 .mu.g/kg, 0.2 mg/kg, 1.0 mg/kg, 2.0 mg/kg, 10 mg/kg, 25 mg/kg, 50 mg/kg body weight or more. See, e.g., Yang et al. (2003) New Engl. J. Med. 349:427-434; Herold et al. (2002) New Engl. J. Med. 346:1692-1698; Liu et al. (1999) J. Neurol.
[0168] Neurosurg. Psych. 67:451-456; Portielji et al. (20003) Cancer Immunol. Immunother. 52:133-144.
[0169] In some embodiments that employ an anti-human PD-L1 mAb as the PD-L1 antagonist in the combination therapy, the dosing regimen will comprise administering the anti-human PD-L1 mAb at a dose of about 1, 2, 3, 5 or 10 mg/kg at intervals of about 14 days (.+-.2 days) or about 21 days (.+-.2 days) or about 30 days (.+-.2 days) throughout the course of treatment.
[0170] In other embodiments that employ an anti-human PD-L1 mAb as the PD-L1 antagonist in the combination therapy, the dosing regimen will comprise administering the anti-human PD-L1 mAb at a dose of from about 0.005 mg/kg to about 10 mg/kg, with intra-patient dose escalation. In other escalating dose embodiments, the interval between doses will be progressively shortened, e.g., about 30 days (.+-.2 days) between the first and second dose, about 14 days (.+-.2 days) between the second and third doses. In certain embodiments, the dosing interval will be about 14 days (.+-.2 days), for doses subsequent to the second dose.
[0171] In certain embodiments, a subject will be administered an intravenous (IV) infusion of a medicament comprising any of the PD-L1 antagonists described herein.
[0172] In some embodiments, the PD-L1 antagonist in the combination therapy is avelumab, which is administered intravenously at a dose selected from the group consisting of: about 1 mg/kg Q2W (Q2W=one dose every two weeks), about 2 mg/kg Q2W, about 3 mg/kg Q2W, about 5 mg/kg Q2W, about 10 mg Q2W, about 1 mg/kg Q3W (Q3W=one dose every three weeks), about 2 mg/kg Q3W, about 3 mg/kg Q3W, about 5 mg/kg Q3W, and about 10 mg Q3W.
[0173] In some embodiments of the invention, the PD-L1 antagonist in the combination therapy is avelumab, which is administered in a liquid medicament at a dose selected from the group consisting of about 1 mg/kg Q2W, about 2 mg/kg Q2W, about 3 mg/kg Q2W, about 5 mg/kg Q2W, about 10 mg Q2W, about 1 mg/kg Q3W, about 2 mg/kg Q3W, about 3 mg/kg 03W, about 5 mg/kg Q3W, and about 10 mg Q3W.
[0174] In some embodiments, a treatment cycle begins with the first day of combination treatment and last for 2 weeks. In such embodiments, the combination therapy is preferably administered for at least 12 weeks (6 cycles of treatment), more preferably at least 24 weeks, and even more preferably at least 2 weeks after the patient achieves a CR.
[0175] In some embodiments, the 4-1BB agonist in the combination therapy comprises an anti-4-1 BB monoclonal antibody comprising heavy chain variable region and a light chain variable region comprising the amino acid sequences shown in SEQ ID NO: 18 and SEQ ID NO: 19, respectively, and is administered in a liquid medicament at a dose selected from the group consisting of 1 mg/kg Q2W, 2 mg/kg Q2W, 3 mg/kg Q2W, 5 mg/kg Q2W, 10 mg Q2W, 1 mg/kg Q3W, 2 mg/kg Q3W, 3 mg/kg Q3W, 5 mg/kg Q3W, and 10 mg Q3W. In some embodiments, the anti-4-1BB monoclonal antibody is administered as a liquid medicament, and the selected dose of the medicament is administered by IV infusion over a time period of about 60 minutes.
[0176] In some embodiments, the anti-4-1 BB monoclonal antibody is administered at a starting dose of about 0.6 mg/kg Q4W and avelumab is administered at a starting dose of 10 mg/kg Q2W, and if the starting dose combination is not tolerated by the patient, then the dose of avelumab is reduced to 5 mg/kg Q2W and/or the dose of the anti-4-1 BB monoclonal antibody is reduced to 0.3 mg/kg Q4W.
[0177] In some embodiments, the patient is selected for treatment with the combination therapy of the invention is the patient has been diagnosed with advanced RCC with predominantly clear cell subtype, and the primary tumor has been resected. In some embodiments, the patient has not received prior systemic therapy for advanced RCC.
[0178] The present invention also provides a medicament which comprises a PD-L1 antagonist as described above and a pharmaceutically acceptable excipient. When the PD-L1 antagonist is a biotherapeutic agent, e.g., a mAb, the antagonist may be produced in CHO cells using conventional cell culture and recovery/purification technologies.
[0179] In some embodiments, a medicament comprising an anti-PD-L1 antibody as the PD-L1 antagonist may be provided as a liquid formulation or prepared by reconstituting a lyophilized powder with sterile water for injection prior to use.
[0180] The present invention also provides a medicament which comprises axitinib and a pharmaceutically acceptable excipient.
[0181] The anti-PD-L1 and VEGFR inhibitor medicaments described herein may be provided as a kit which comprises a first container and a second container and a package insert. The first container contains at least one dose of a medicament comprising an anti-PD-L1 antagonist, the second container contains at least one dose of a medicament comprising a VEGFR inhibitor, and the package insert, or label, which comprises instructions for treating a patient for cancer using the medicaments. The first and second containers may be comprised of the same or different shape (e.g., vials, syringes and bottles) and/or material (e.g., plastic or glass). The kit may further comprise other materials that may be useful in administering the medicaments, such as diluents, filters, IV bags and lines, needles and syringes. In some embodiments of the kit, the anti-PD-L1 antagonist is an anti-PD-L1 antibody and the instructions state that the medicaments are intended for use in treating a patient having a cancer that tests positive for PD-L1 expression by an IHC assay.
[0182] The anti-PD-L1 and anti-4-1BB antibody medicaments described herein may be provided as a kit which comprises a first container and a second container and a package insert. The first container contains at least one dose of a medicament comprising an anti-PD-L1 antagonist, the second container contains at least one dose of a medicament comprising an anti-4-1 BB antibody, and the package insert, or label, which comprises instructions for treating a patient for cancer using the medicaments. The first and second containers may be comprised of the same or different shape (e.g., vials, syringes and bottles) and/or material (e.g., plastic or glass). The kit may further comprise other materials that may be useful in administering the medicaments, such as diluents, filters, IV bags and lines, needles and syringes. In some embodiments of the kit, the anti-PD-L1 antagonist is an anti-PD-L1 antibody and the instructions state that the medicaments are intended for use in treating a patient having a cancer that tests positive for PD-L1 expression by an IHC assay.
[0183] The anti-PD-L1 antibody and CD20 antagonist medicaments described herein may be provided as a kit which comprises a first container and a second container and a package insert. The first container contains at least one dose of a medicament comprising an anti-PD-L1 antagonist, the second container contains at least one dose of a medicament comprising a CD20 antagonist, and the package insert, or label, which comprises instructions for treating a patient for cancer using the medicaments. The first and second containers may be comprised of the same or different shape (e.g., vials, syringes and bottles) and/or material (e.g., plastic or glass). The kit may further comprise other materials that may be useful in administering the medicaments, such as diluents, filters, IV bags and lines, needles and syringes. In some embodiments of the kit, the anti-PD-L1 antagonist is an anti-PD-L1 antibody and the instructions state that the medicaments are intended for use in treating a patient having a cancer that tests positive for PD-L1 expression by an IHC assay.
[0184] These and other aspects of the invention, including the exemplary specific embodiments listed below, will be apparent from the teachings contained herein.
III. GENERAL METHODS
[0185] Standard methods in molecular biology are described Sambrook, Fritsch and Maniatis (1982 & 1989 2nd Edition, 2001 3rd Edition) Molecular Cloning, A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.; Sambrook and Russell (2001) Molecular Cloning, 3rd ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.; Wu (1993) Recombinant DNA, Vol. 217, Academic Press, San Diego, Calif.). Standard methods also appear in Ausbel, et al. (2001) Current Protocols in Molecular Biology, Vols.1-4, John Wiley and Sons, Inc. New York, N.Y., which describes cloning in bacterial cells and DNA mutagenesis (Vol. 1), cloning in mammalian cells and yeast (Vol. 2), glycoconjugates and protein expression (Vol. 3), and bioinformatics (Vol. 4).
[0186] Methods for protein purification including immunoprecipitation, chromatography, electrophoresis, centrifugation, and crystallization are described (Coligan, et al. (2000) Current Protocols in Protein Science, Vol. 1, John Wiley and Sons, Inc., New York). Chemical analysis, chemical modification, post-translational modification, production of fusion proteins, glycosylation of proteins are described (see, e.g., Coligan, et al. (2000) Current Protocols in Protein Science, Vol. 2, John Wiley and Sons, Inc., New York; Ausubel, et al. (2001) Current Protocols in Molecular Biology, Vol. 3, John Wiley and Sons, Inc., NY, N.Y., pp. 16.0.5-16.22.17; Sigma-Aldrich, Co. (2001) Products for Life Science Research, St. Louis, Mo.; pp. 45-89; Amersham Pharmacia Biotech (2001) BioDirectory, Piscataway, N.J., pp. 384-391). Production, purification, and fragmentation of polyclonal and monoclonal antibodies are described (Coligan, et al. (2001) Current Protcols in Immunology, Vol. 1, John Wiley and Sons, Inc., New York; Harlow and Lane (1999) Using Antibodies, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.; Harlow and Lane, supra). Standard techniques for characterizing ligand/receptor interactions are available (see, e.g., Coligan, et al. (2001) Current Protocols in Immunology, Vol. 4, John Wiley, Inc., New York).
[0187] Monoclonal, polyclonal, and humanized antibodies can be prepared (see, e.g., Sheperd and Dean (eds.) (2000) Monoclonal Antibodies, Oxford Univ. Press, New York, N.Y.; Kontermann and Dubel (eds.) (2001) Antibody Engineering, Springer-Verlag, New York; Harlow and Lane (1988) Antibodies A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., pp. 139-243; Carpenter, et al. (2000) J. Immunol. 165:6205; He, et al. (1998) J. Immunol. 160:1029; Tang et al. (1999) J. Biol. Chem. 274:27371-27378; Baca et al. (1997) J. Biol. Chem. 272:10678-10684; Chothia et al. (1989) Nature 342:877-883; Foote and Winter (1992) J. Mol. Biol. 224:487-499; U.S. Pat. No. 6,329,511).
[0188] An alternative to humanization is to use human antibody libraries displayed on phage or human antibody libraries in transgenic mice (Vaughan et al. (1996) Nature Biotechnol. 14:309-314; Barbas (1995) Nature Medicine 1:837-839; Mendez et al. (1997) Nature Genetics 15:146-156; Hoogenboom and Chames (2000) Immunol. Today 21:371-377; Barbas et al. (2001) Phage Display: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York; Kay et al. (1996) Phage Display of Peptides and Proteins: A Laboratory Manual, Academic Press, San Diego, Calif.; de Bruin et al. (1999) Nature Biotechnol. 17:397-399).
[0189] Purification of antigen is not necessary for the generation of antibodies. Animals can be immunized with cells bearing the antigen of interest. Splenocytes can then be isolated from the immunized animals, and the splenocytes can fused with a myeloma cell line to produce a hybridoma (see, e.g., Meyaard et al. (1997) Immunity 7:283-290; Wright et al. (2000) Immunity 13:233-242; Preston et al., supra; Kaithamana et al. (1999) J. Immunol. 163:5157-5164).
[0190] Antibodies can be conjugated, e.g., to small drug molecules, enzymes, liposomes, polyethylene glycol (PEG). Antibodies are useful for therapeutic, diagnostic, kit or other purposes, and include antibodies coupled, e.g., to dyes, radioisotopes, enzymes, or metals, e.g., colloidal gold (see, e.g., Le Doussal et al. (1991) J. Immunol. 146:169-175; Gibellini et al. (1998) J. Immunol. 160:3891-3898; Hsing and Bishop (1999) J. Immunol. 162:2804-2811; Everts et al. (2002) J. Immunol. 168:883-889).
[0191] Methods for flow cytometry, including fluorescence activated cell sorting (FACS), are available (see, e.g., Owens, et al. (1994) Flow Cytometry Principles for Clinical Laboratory Practice, John Wiley and Sons, Hoboken, N.J.; Givan (2001) Flow Cytometry, 2nd ed.; Wiley-Liss, Hoboken, N.J.; Shapiro (2003) Practical Flow Cytometry, John Wiley and Sons, Hoboken, N.J.). Fluorescent reagents suitable for modifying nucleic acids, including nucleic acid primers and probes, polypeptides, and antibodies, for use, e.g., as diagnostic reagents, are available (Molecular Probesy (2003) Catalogue, Molecular Probes, Inc., Eugene, Oreg.; Sigma-Aldrich (2003) Catalogue, St. Louis, Mo.).
[0192] Standard methods of histology of the immune system are described (see, e.g., Muller-Harmelink (ed.) (1986) Human Thymus: Histopathology and Pathology, Springer Verlag, New York, N.Y.; Hiatt, et al. (2000) Color Atlas of Histology, Lippincott, Williams, and Wilkins, Phila, Pa.; Louis, et al. (2002) Basic Histology: Text and Atlas, McGraw-Hill, New York, N.Y.).
[0193] Software packages and databases for determining, e.g., antigenic fragments, leader sequences, protein folding, functional domains, glycosylation sites, and sequence alignments, are available (see, e.g., GenBank, Vector NTI.RTM. Suite (Informax, Inc, Bethesda, Md.); GCG Wisconsin Package (Accelrys, Inc., San Diego, Calif.); DeCypher.RTM. (TimeLogic Corp., Crystal Bay, Nev.); Menne, et al. (2000) Bioinformatics 16: 741-742; Menne, et al. (2000) Bioinformatics Applications Note 16:741-742; Wren, et al. (2002) Comput. Methods Programs Biomed. 68:177-181; von Heijne (1983) Eur. J. Biochem. 133:17-21; von Heijne (1986) Nucleic Acids Res. 14:4683-4690).
IV. EXAMPLES
Example 1
Combination Treatment with Avelumab and Axitinib
[0194] This example illustrates a clinical trial study to evaluate safety, efficacy, pharmacokinetics, and pharmacodynamics of avelumab (MSB0010718C) in combination with axitinib (AG-013736) in patients with previously untreated advanced renal cell carcinoma (aRCC).
[0195] This study is an open-label, multi-center, multiple-dose trial designed to estimate the maximum tolerated dose (MTD) and select the recommended phase 2 dose (RP2D) of avelumab (MSB0010718C) in combination with axitinib (AG-013736). Once the MTD of avelumab administered in combination with axitinib is estimated (dose finding portion), the dose expansion phase will be opened to further characterize the combination in term of safety profile, anti-tumor activity, pharmacokinetics, pharmacodynamics and biomarker modulation. Protocol design is set forth in Table 4.
[0196] The Dose Finding Phase will estimate the MTD and RP2D in patients with aRCC with clear cell histology who did not receive prior systemic therapy for advanced disease, using the modified toxicity probability interval (mTPI) method.35 Dose finding will follow an "Up-and-Down" design, with up to 4 potential dose levels (DL) to be tested, shown in Table 4.
[0197] The Dose Finding Phase will lead to the identification of an Expansion Test Dose for avelumab in combination with axitinib in patients with aRCC who did not receive prior systemic therapy for their advanced disease. The Expansion Test Dose will either be the MTD (i.e., the highest dose of avelumab and axitinib associated with the occurrence of DLTs in <33% of patients) or the RP2D, i.e., the highest tested dose that is declared safe and tolerable by the investigators and sponsor. Once the Expansion Test Dose is identified, the Dose Expansion Phase will be opened, and avelumab in combination with axitinib will be evaluated in up to approximately 20-40 patients with previously untreated aRCC.
TABLE-US-00006 TABLE 4 Arms Assigned Interventions Dose finding Group 1: avelumab 10 mg/kg IV Q2W; axitinib 5 mg oral BID phase Group 2: avelumab 5 mg/kg IV Q2W; axitinib 5 mg oral BID Group 3: avelumab 10 mg/kg IV Q2W; axitinib 3 mg oral BID Group 4: avelumab 5 mg/kg IV Q2W; axitinib 3 mg oral BID Dose expansion Group 1: avelumab 10 mg/kg IV Q2W; axitinib 5 mg oral BID phase Group 2: avelumab 5 mg/kg IV Q2W; axitinib 5 mg oral BID Group 3: avelumab 10 mg/kg IV Q2W; axitinib 3 mg oral BID Group 4: avelumab 5 mg/kg IV Q2W; axitinib 3 mg oral BID
[0198] Inclusion Criteria: Histologically or cytologically confirmed advanced RCC with clear cell component. Primary tumor resected. Mandatory archival formalin fixed, paraffin embedded (FFPE) tumor tissue block from primary tumor resection specimen (all patients). For Extension Cohort only, mandatory de novo tumor biopsy from a locally recurrent or metastatic lesion unless obtained from a procedure performed within 6 months of study entry and if the patient has received no intervening systemic anti-cancer treatment. At least one measureable lesion as defined by RECIST version 1.1. Age.gtoreq.18 years. Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1. Adequate bone marrow function, renal and liver functions.
[0199] The number of patients to be enrolled in the Dose Finding Phase will depend on the observed safety profile, and the number of tested dose levels. Up to approximately 55 patients (including Dose Finding Phase and Dose Expansion Phase) are projected to be enrolled in the study.
[0200] Study Treatment: Axitinib will be given orally (PO) twice daily (BID), with or without food, on a continuous dosing schedule. Avelumab will be given as a 1-hour intravenous infusion (IV) every two weeks (Q2W). In all patients, treatment with study drugs may continue until confirmed disease progression, patient refusal, patient lost to follow up, unacceptable toxicity, or the study is terminated by the sponsor, whichever comes first.
[0201] In order to mitigate avelumab infusion-related reactions, a premedication regimen of 25 to 50 mg IV or oral equivalent diphenhydramine and 650 mg IV or oral equivalent acetaminophen/paracetamol (as per local practice) may be administered approximately 30 to 60 minutes prior to each dose of avelumab. This may be modified based on local treatment standards and guidelines, as appropriate.
[0202] Tumor Assessment: Anti-tumor activity will be assessed by radiological tumor assessments at 6-week intervals, using RECIST version 1.1. Complete and partial responses will be be confirmed on repeated imaging at least at 4 weeks after initial documentation. After 1 year from enrollment in the study, tumor assessments should be conducted less frequently, i.e., at 12-week intervals. In addition, radiological tumor assessments will also be conducted whenever disease progression is suspected (e.g., symptomatic deterioration), and at the time of End of Treatment/Withdrawal (if not done in the previous 6 weeks). If radiologic imaging shows progressive disease (PD), tumor assessment should be repeated at least .gtoreq.4 weeks later in order to confirm PD.
[0203] Brain Computerized Tomography (CT) or Magnetic Resonance Imaging (MRI) scans are required at baseline and when there is a suspected brain metastasis. Bone scan (bone scintigraphy) or 18fluorodeoxyglucose-positron emission tomography/CT(18FDG-PET/CT) are required at baseline, then every 16 weeks only if bone metastases are present at baseline. Otherwise, bone imaging is required only if new bone metastases are suspected. Bone imaging is also required at the time of confirmation of CR for patients who have bone metastases.
[0204] Pharmacokinetic/Immunogenicity Assessments: PK/immunogenicity sampling will be collected. To understand the PK effects of avelumab on axitinib, a 7-day lead-in period with single-agent axitinib will be included prior to Cycle 1 in all patients in the Dose Finding Phase and in at least 8 patients in the Dose Expansion Phase of the study. Since avelumab has a long half-life (3-5 days), it would not be feasible to run a lead-in to study the PK of avelumab alone. Therefore, the effect of axitinib on avelumab will be evaluated by comparing avelumab trough concentrations at steady state in the presence of axitinib with those reported for avelumab alone in prior studies.
[0205] Biomarker Assessments: A key objective of the biomarker analyses that will be performed in this study is to investigate biomarkers that are potentially predictive of treatment benefit with the combination of avelumab and axitinib. In addition, biomarker studies of tumor and blood biospecimens will be carried out to help further understand the mechanism of action of the avelumab in combination with axitinib, as well as potential mechanisms of resistance.
[0206] Tumor biospecimens from archived tissue samples and metastatic lesions will be used to analyze candidate DNA, RNA, or protein markers, or a relevant signature of markers, for their ability to identify those patients who are most likely to benefit from treatment with the study drugs. Markers that may be analyzed include, but not be limited to, PD-L1 expression tumor-infiltrating CD8+ T lymphocytes, and T-cell receptor gene sequence quantitation. Optional tumor biopsies obtained upon disease progression will be used to investigate acquired mechanisms of resistance. Only core needle or excisional biopsies, or resection specimen are suitable.
[0207] Peripheral Blood: Specimens will be retained as whole blood, serum, and plasma in a biobank for exploratory biomarker assessments, unless prohibited by local regulation or by decision of the Institutional Review Board or Ethics Committee. Samples may be used to identify or characterize cells, DNA, RNA, or protein markers known or suspected to be of relevance to the mechanisms of action, or the development of resistance to avelumab used in combination with axitinib. These include biomarkers that may aid in the identification of those patients who might preferentially benefit from treatment with avelumab in combination with axitinib, including but not limited to biomarkers related to anti-tumor immune response or target modulation, such as soluble VEGF-A, IL-8, IFN.gamma. and/or tissue FoxP3, PD-1, PD-L2. Biospecimens should be obtained pre-dose and at the same time as PK samples whenever possible.
Example 2
Combination Treatment with Axitinib and Avelumab Versus Sunitinib
[0208] This example illustrates a clinical trial study to evaluate safety and efficacy of avelumab (MSB0010718C) in combination with axitinib (AG-013736) and to demonstrate the superiority of this combination versus standard-of-care sunitinib monotherapy in the first-line treatment of patients with advanced RCC (aRCC). Sunitinib malate (SUTENT.RTM.) is an oral multitargeted TKI of stem cell receptor factor (KIT), platelet derived growth factor-receptors (PDGFRs), VEGFRs, glial cell-line neurotrophic factor receptor (RET), and FMS-like tyrosine kinase 3 (FLT3), and colony stimulating factor receptor Type 1 (CSR-1 R) approved multinationally for the treatment of aRCC, imatinib-resistant or intolerant gastrointestinal stromal tumor (GIST), and unresectable, well-differentiated metastatic pancreatic neuroendocrine tumors (NET).
[0209] The study is a Phase 3, randomized, multination, multicenter, open-label, parallel 2-arm study in which approximately 465 patients are planned to be randomized to receive avelumab in combination with axitinib or sunitinib monotherapy: Arm A: avelumab in combination with axitinib; Arm B: sunitinib. Patients will be stratified according to ECOG performance status (0 versus 1) and LDH (>1.5 ULN vs. .ltoreq.1.5 ULN). In arm A (avelumab in combination with axitinib), avelumab will be given as a 1 hour intravenous infusion (IV) every 2 weeks in a 6-week cycle. Axitinib will be given orally (PO) twice daily (BID), with or without food, on a continuous dosing schedule.
[0210] Treatment with study drugs may continue until confirmed disease progression, patient refusal, patient lost to follow up, unacceptable toxicity, or the study is terminated by the sponsor, whichever comes first. Axitinib treatment may be adjusted by dosing interruption with or without dose reduction. Intrapatient axitinib dose escalation may occur if the intrapatient escalation criteria are met.
[0211] Study Treatment: Axitinib will be given orally twice daily PO on a continuous daily dosing schedule. Avelumab will be given as a 1 hour intravenous infusion every 2 weeks in a 6-week cycle. Sunitinib will be given orally 50 mg taken once daily, on a schedule 4 weeks on treatment followed by 2 weeks off (Schedule 4/2). Patients who develop disease progression on study treatment but are otherwise continuing to derive clinical benefit from study treatment will be eligible to continue with avelumab combined with axitinib, or single-agent avelumab, or single-agent axitinib, or single-agent sunitinib provided that the treating physician has determined that the benefit/risk for doing so is favorable.
[0212] Tumor Assessments: Anti-tumor activity will be assessed by radiological tumor assessments and will be based on RECIST guidelines version 1.1 for primary and secondary endpoints and on immune-related RECIST (irRECIST) guidelines for exploratory endpoints. Tumor assessments will be performed every 6 weeks (Q6W) up to 1 year from first dose therapy; thereafter, tumor assessments will be performed every 2 cycles. In addition, radiological tumor assessments will also be conducted whenever disease progression is suspected (e.g., symptomatic deterioration), at the time of the End of Treatment/Withdrawal visit (if not done in the previous 6 weeks), and during the Short term Follow-up period (at the 90-day visit only); subsequent tumor assessments during the Long term Follow-up period can be collected in absence of withdrawal of consent, regardless of initiation of subsequent anti-cancer therapies.
[0213] Tumor assessments will include all known or suspected disease sites. Imaging may include chest, abdomen, and pelvis CT or MRI scans; brain CT or MRI scans (required at baseline and when suspected brain metastasis) and bone scans or 18FDG PET (required at baseline then every 16 weeks only if bone metastases are present at baseline). Otherwise, bone imaging is required only if new bone metastasis are suspected and at the time of confirmation of complete response for patients who have bone metastases. The CT scans should be performed with contrast agents unless contraindicated for medical reasons. The same imaging technique used to characterize each identified and reported lesion at baseline will be employed in the following tumor assessments. Antitumor activity will be assessed through radiological tumor assessments conducted at baseline, at 6 weeks after the first dose of therapy, then every 6 weeks up to 1 year from the first dose of therapy and every 12 weeks thereafter, (if not done in the previous 6 weeks), and during the Short term Follow-up period (at the 90-day visit only); subsequent tumor assessments during the Long term Follow-up period can be collected in absence of withdrawal of consent, regardless of initiation of subsequent anti-cancer therapies. Further imaging assessments may be performed at any time if clinically indicated (e.g., suspected PD, symptomatic deterioration, etc.). Assessment of response will be made using RECIST version 1.1 and as per immune-related response criteria (irRC) (Nishino 2013). All radiographic images will be collected and may be objectively verified by a BICR independent third-party core imaging laboratory.
[0214] Primary Endpoint: Progression-Free Survival (PFS) as assessed by Blinded
[0215] Independent Central Review (BICR) per RECIST v1.1. Secondary Endpoints: Overall Survival (OS); objective tumor response rate (OR), as assessed by BICR per RECIST version 1.1.; disease Control (DC), as assessed by BICR per RECIST version 1.1.; time to event: time to response (TTR), Duration of Response (DR); adverse Events (AEs) as characterized by type, frequency, severity (as graded by National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE v.4.03), timing, seriousness, and relationship to study therapy; Laboratory abnormalities as characterized by type, frequency, severity (as graded by NCI CTCAE v.4.03), and timing; PK parameters including trough concentrations (Ctrough) of avelumab and trough concentrations (Ctrough) and maximum concentrations (Cmax) of axitinib; tumor tissue biomarker status (i.e., positive or negative; based on for example, PD-L1 expression and/or quantitation of tumor infiltrating CD8+T lymphocytes as assessed by immunohistochemistry); measures of clinical outcome (PFS, OS, OR, DCR, DR and TTR) in biomarker-positive and biomarker-negative sub-groups; anti-drug antibodies (ADAs; neutralizing antibodies) of avelumab when in combination with axitinib; patient-Reported Outcomes (PRO): FACT-Kidney Symptom Index (FKSI-19), EuroQol 5 Dimension (EQ 5D).
Example 3
Combination Treatment with Anti-4-1 BB Antibody and Avelumab
[0216] This example illustrates the therapeutic activity of anti-4-1 BB antibody and avelumab combination therapy in murine B16F10 melanoma and MC38 colon carcinoma models.
[0217] Six (6)- to 8-week old female C57BL/6 mice were purchased from the Jackson
[0218] Laboratories. All animals were housed in a pathogen free vivarium facility at Rinat and experiments were conducted according to the protocols in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines.
[0219] The B16F10 melanoma cell line was purchased from American Type Culture Collection (ATCC). The MC38 colon carcinoma cell line was kindly provided by Dr. Antoni Ribas at University of California, Los Angeles, CA. Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine at 37.degree. C. in 5% carbon dioxide (CO.sub.2), and IMPACT-tested for pathogens at Research Animal Diagnostic Laboratory (RADIL) (Columbia, Mo.). Pathogen-free cells growing in an exponential growth phase were harvested and used for tumor inoculation.
[0220] Antibodies used for cell surface or intracellular staining were purchased from BD Biosciences or eBioscience. They were rat anti-mouse CD4-PerCP-Cy5.5 (clone RM4-5, BD Biosciences), rat anti-mouse CD8a-APC-H7 (clone 53-6.7, BD Biosciences), rat anti-mouse CD25-PE-Cy7 (clone PC61, BD Biosciences), rat anti-mouse CD45-BV510 (clone 30-F11, BD Biosciences), rat anti-mouse CD90.2-FITC (clone 53-2.1, BD Biosciences), rat anti-mouse Eomes-PE (clone: Dan11mag, eBioscience), rat anti-mouse FoxP3-eFluor450 (clone FJK-16s, eBioscience), and rat anti-mouse NKp46-BV421 or -AF647 (clone 29A1.4, BD Biosciences). Live cells were separated from dead cells using LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen).
[0221] Therapeutic mouse anti-mouse 4-1BB mAb (mouse immunoglobulin G1 [mIgG1]), derived from the parental clone MAB9371 (R&D Systems), was prepared in-house. Avelumab was provided by Merck Serono. Isotype control mIgG1 (clone: MOPC-21) was purchased from BioXcell. Human IgG1 was prepared in-house. Anti-4-1BB and avelumab were diluted to concentrations of 0.1 mg/mL and 1 mg/mL, respectively, in phosphate buffered saline (PBS) (Life Technologies), and dosed at 0.2 mL per mouse intraperitoneally (ip) for 3 doses 3 to 4 days apart.
[0222] C57BL/6 mice were inoculated subcutaneously at the right flank with 0.2.times.10.sup.6 B16F10 or 0.5.times.10.sup.6 MC38 cells in 0.1 mL of serum-free DMEM. When tumors reached target size, mice were randomized into treatment groups. Treatment was started on the same day as randomization. Tumor size was measured twice weekly in 2 dimensions using a caliper, and the volume was expressed in cubic millimeters using the formula: V=0.5 L.times.W.sup.2 where L is the longest diameter of the tumor and W is the diameter perpendicular to L. Body weight was recorded weekly.
[0223] Tumors were disseminated into single cell suspension using gentle MACS and Miltenyi Mouse Dissociation Kit (Miltenyi Biotec) according to manufacturer's protocol with modification. Ammonium-Chloride-Potassium (ACK) Lysing Buffer (Life
[0224] Technologies) was used to remove red blood cells. Cells were washed twice with FACS staining buffer (PBS supplemented with 2% FBS and 0.9% sodium azide [NaN3]), and finally resuspended in FACS staining buffer.
[0225] An aliquot of cells was pre-incubated with 10 .mu.g/mL of mouse BD Fc Block (BD Biosciences) for 10 minutes before phenotyping mAbs were added to specifically stain immune cells. Cell surface antigens were labeled by incubating cells at 4.degree. C. for 30 minutes. After removing unbound mAbs, cells were washed twice with FACS staining buffer, fixed in fixative buffer (PBS+2% FBS+1% paraformaldehyde), and stored at 4.degree. C. in the dark until analyzed by flow cytometry. Intracellular staining was carried out using Foxp3/Transcription Factor Staining Buffer set (eBioscience) according to the manufacturer's protocol. Flow cytometry data were acquired using LSR Fortessa (BD Biosciences) and analyzed using FlowJo (TreeStar Inc.). Results were expressed as mean.+-.SEM. Statistical analyses were performed using GraphPad Prism 6.0. One-way or 2-way ANOVA was applied to compare the statistical differences among multiple groups relative to the isotype control. P<0.05 was considered as significant difference.
[0226] Two murine models were used to evaluate the therapeutic efficacy of anti-4-1 BB in combination with avelumab. In the B16F10 melanoma model, the average starting tumor size was 67 to 78 mm.sup.3 (range 44 to 114 mm.sup.3; n=7 animals per group) (Table 5). By Day 26 post tumor inoculation, the tumors for isotype, anti-4-1BB alone, and avelumab alone groups reached an average of 1206.+-.397 mm.sup.3, 1979.+-.425 mm.sup.3, and 2112.+-.429 mm.sup.3, respectively (Table 5). By contrast, dramatic tumor suppression (average of 341.+-.146 mm.sup.3) was observed when animals were administered with anti-4-1BB and avelumab concurrently (n <0.0001 vs sinale aaent alone arouasl (Table 5).
TABLE-US-00007 TABLE 5 Tumor Measurements (Mean .+-. SEM) of Subcutaneous B16F10 Melanoma over Time Isotypes Anti-4-1BB Avelumab Anti-4-1BB/Avelumab Days Mean SEM N Mean SEM N Mean SEM N Mean SEM N 13 67 4 7 69 6 7 78 8 7 70 10 7 17 251 109 7 364 87 7 327 78 7 219 57 7 20 475 222 7 725 266 7 654 174 7 272 94 7 24 909 368 7 1511 417 7 1304 274 7 243 106 6 26 1206 397 7 1979 425 7 2112 429 7 341 146 6 Tumor volume is expressed in mm.sup.3. N = Number of animals within each group; SEM = Standard error of the mean.
[0227] In the MC38 colon carcinoma model, the average starting tumor size was approximately 60 mm.sup.3 (range 41-92 mm.sup.3; n=10 animals per group) (Table 6). At the end of study (Day 23 post tumor implantation), the average tumor volumes of isotype, anti-4-1 BB alone, avelumab alone, and anti-4-1 BB antibody/avelumab combination groups were 1177.+-.252 mm.sup.3, 1093.+-.183 mm.sup.3, 901.+-.206 mm.sup.3, and 530.+-.190 mm.sup.3, respectively (Table 6). The reduction in tumor size by the combination treatment was significant, compared to the isotype control (p<0.001) and 4-1BB alone groups (p<0.01), but not to the avelumab group (p>0.05) (Table 6).
TABLE-US-00008 TABLE 6 Tumor Measurements (Mean .+-. SEM) of Subcutaneous MC38 Colon Carcinoma over Time Isotypes Anti-4-1BB Avelumab Anti-4-1BB/Avelumab Days Mean SEM N Mean SEM N Mean SEM N Mean SEM N 7 60 5 10 62 3 10 63 5 10 64 5 10 10 130 21 10 122 15 10 127 19 10 117 13 10 14 357 72 10 250 30 10 254 42 10 146 42 10 16 501 108 10 355 56 10 384 86 10 176 64 10 18 680 148 10 508 76 10 523 114 10 246 93 10 21 987 236 9 785 143 10 714 158 9 416 149 10 23 1177 252 9 1093 183 10 901 206 9 530 190 10 Tumor volume is expressed in mm.sup.3. N = Number of animals within each group; SEM = Standard error of the mean.
[0228] Tumor-infiltrating lymphocytes (TILs) were isolated from MC38 tumors after treatment and analyzed for markers associated with anti-tumor immune response. The combination treatment facilitated the infiltration of T cells into tumors with an average of 53% of total CD45+ cells, while T-cell frequency (of CD45+ cells) was 25%, 31%, and 36% in the isotype, anti-4 1 BB antibody treatment alone, and avelumab alone groups, respectively (FIG. 1). The ratio of CD8+ T cells/regulatory T cell (Treg) in the isotype and avelumab groups was 1.2 and 2.5, respectively. This ratio increased to 10 and 21 in anti-4-1 BB antibody treatment alone and in combination with avelumab, respectively (FIG. 2). Furthermore, the induction of Eomes, a marker associated with T-cell effector/memory differentiation, was observed in the anti-4-1 BB antibody treatment alone and anti-4-1 BB and avelumab combination groups (FIG. 3).
[0229] These results demonstrate that treatment with anti-4-1 BB antibody in combination with avelumab has a synergistic anti-tumor effect accompanied by the enrichment of T cells in tumor, increased CD8+ T cell/regulatory T cell (Treg) ratio, and induction of eomesodermin (Eomes) expression. Furthermore, the combination therapy elicited an anti-tumor immune response in the tumor microenvironment.
Example 4
Combination Treatment of Advanced Malignancies with Avelumab and PF-05082566
[0230] This example illustrates a clinical trial study to evaluate safety, efficacy, pharmacokinetics, and pharmacodynamics of avelumab (MSB0010718C) in combination with PF-05082566, an anti-4-1BB agonist IgG2 antibody, in patients with with locally advanced or metastatic solid tumors (e.g., non-small cell lung cancer (NSCLC), melanoma, and squamous cell carcinoma (SCCHN)). Protocol design is set forth in Table 7.
TABLE-US-00009 TABLE 7 Arms Assigned Interventions Cohort A1: NSCLC patients Avelumab 10 mg/kg IV Q2W; PF- treated with 05082566 500 mg IV every 4 weeks. 10 mg/kg avelumab + Treatment with the combination of 500 mg PF-05082566 avelumab with PF-05082566 will continue until disease progression. Cohort A2: NSCLC patients Avelumab 10 mg/kg IV Q2W; PF- treated with 05082566 100 mg IV every 4 weeks. 10 mg/kg avelumab + Treatment with the combination of 100 mg PF-05082566 avelumab with PF-05082566 will continue until disease progression. Cohort A3: NSCLC patients Avelumab 10 mg/kg IV Q2W; PF- treated with 05082566 20 mg IV every 4 weeks. 10 mg/kg avelumab + Treatment with the combination of 20 mg PF-05082566 avelumab with PF-05082566 will continue until disease progression. Cohort A4: Melanoma patients Avelumab 10 mg/kg IV Q2W; PF- treated with 05082566 100 mg IV every 4 weeks. 10 mg/kg avelumab + Treatment with the combination of 100 mg PF-05082566 avelumab with PF-05082566 will continue until disease progression. Cohort A5: SCCHN patients Avelumab 10 mg/kg IV Q2W; PF- treated with 05082566 100 mg IV every 4 weeks. 10 mg/kg avelumab + Treatment with the combination of 100 mg PF-05082566 avelumab with PF-05082566 will continue until disease progression.
Example 5
Combination Treatment of Cancer with Avelumab, Anti-4-1 BB Antibody, and Anti-M-CSF Antibody
[0231] This example illustrates the therapeutic activity of anti-4-1 BB antibody, anti-M-CSF antibody, and the anti-PD-L1 antibody Avelumab triple combination therapy in murine MC38 colon carcinoma models.
[0232] Six (6)- to 8-week old female C57BL/6 mice were purchased from the Jackson Laboratories. All animals were housed in a pathogen free vivarium facility at Rinat and experiments were conducted according to the protocols in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines.
[0233] The MC38 colon carcinoma cell line was kindly provided by Dr. Antoni Ribas at University of California, Los Angeles, Calif. Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L glutamine at 37.degree. C. in 5% carbon dioxide (CO2), and IMPACT-tested for pathogens at Research Animal Diagnostic Laboratory (RADIL) (Columbia, Mo.). Pathogen-free cells growing in an exponential growth phase were harvested and used for tumor inoculation.
[0234] Therapeutic mouse anti-mouse 4-1BB mAb (mouse immunoglobulin G1 [mIgG1]), derived from the parental clone MAB9371 (R&D Systems), was prepared in-house. Avelumab was provided by Merck Serono. Rat anti-mouse M-CSF (clone 5A1), rat IgG1 (clone HRPN) and mIgG1 (clone: MOPC-21) isotype controls were purchased from BioXcell. Human IgG1 isotype was prepared in-house. Anti-4-1 BB, avelumab and anti-M-CSF mAbs were diluted to concentrations of 0.1 mg/mL and 1 mg/mL, and 1.5 mg/mL, respectively, in phosphate buffered saline (PBS) (Life Technologies), and dosed at 0.2 mL per mouse intraperitoneally (ip) for 3 doses 3 to 4 days apart.
[0235] C57BL/6 mice were inoculated subcutaneously at the right flank with 0.5.times.106 MC38 cells in 0.1 mL of DMEM. When tumors reached an average of .about.60 mm.sup.3 (range 41-93 mm.sup.3), mice were randomized into groups of 10 animals per group, and treatment was started at the same day. Tumor size was measured in two dimensions using a caliper, and the volume was expressed in mm.sup.3 using the formula: V=0.5 L.times.W2 where L and W are the long and short diameters of the tumor, respectively. Body weight was recorded weekly.
[0236] Results were expressed as mean.+-.SEM (Table 8). Statistical analyses were performed using GraphPad Prism 6.0. One-way or two-way ANOVA was applied to compare the statistical differences among multiple groups relative to isotype controls. P<0.05 was considered as significant difference.
TABLE-US-00010 TABLE 8 Days Post-Tumor Mean Tumor Size Inoculation (mm.sup.3) SEM N Group 1. Isotype control 7 60 5 10 10 130 21 10 14 357 72 10 16 501 108 10 18 680 148 10 21 987 236 9 23 1177 252 9 Group 2. Anti-4-1BB antibody (1 mg/kg) 7 62 3 10 10 122 15 10 14 250 30 10 16 355 56 10 18 508 76 10 21 785 143 10 23 1093 183 10 Group 3. Anti-M-CSF antibody (15 mg/kg) 7 58 4 10 10 138 27 10 14 196 32 10 16 268 43 10 18 350 56 10 21 432 84 9 23 572 123 9 Group 4. Anti-PD-L1 antibody (Avelumab, 10 mg/kg) 7 63 5 10 10 127 19 10 14 254 42 10 16 384 86 10 18 523 114 10 21 714 158 9 23 901 206 9 Group 5. Anti-4-1BB antibody (1 mg/kg) + Anti-PD-L1 antibody (Avelumab, 10 mg/kg) 7 64 5 10 10 117 13 10 14 146 42 10 16 176 64 10 18 246 93 10 21 416 149 10 23 530 190 10 Group 6. Anti-M-CSF antibody (15 mg/kg) + Anti-PD-L1 antibody (Avelumab, 10 mg/kg) 7 62 4 10 10 106 10 10 14 182 29 10 16 211 32 9 18 297 65 9 21 436 112 9 23 499 145 9 Group 7. Anti-4-1BB antibody (1 mg/kg) + Anti-M-CSFantibody (15 mg/kg) + Anti-PD-L1 antibody (Avelumab, 10 mg/kg) 7 61 4 10 10 120 16 10 14 139 15 10 16 145 20 10 18 166 20 10 21 214 28 10 23 277 39 10
[0237] Treatment with the triple combination anti-4-1BB antibody, Avelumab, and anti-M-CSF antibody delayed MC38 tumor growth compared to isotype control (Table 8). The triple antibody combination (Table 8, Group 7) was more efficacious that either double combination of avelumab and anti-4-1BB antibody (Table 8, Group 5) or avelumab and anti-CSF-1 antibody (Table 8, Group 6). For example, at day 23 post-tumor inoculation, tumors in animals treated with the triple combination of avelumab, anti-4-1BB antibody, and anti-CSF-1 antibody had a mean size of 277 mm.sup.3. In comparison, tumors in animals treated with either the double combination of avelumab and anti-4-1 BB antibody or avelumab and anti-CSF-1 antibody had a mean size of 530 mm.sup.3 and 499 mm.sup.3, respectively, at day 23. Tumors in animals given isotype control had a mean size of 1177 mm.sup.3 at day 23. Tumors in animals given anti-4-1 BB antibody had a mean size of 1093 mm.sup.3 at day 23. Tumors in animals given anti-CSF-1 antibody had a mean size of 572 mm.sup.3 at day 23. Tumors in animals given anti-PD-L1 antibody (Avelumab) had a mean size of 901 mm.sup.3 at day 23. These results demonstrate that treatment with the triple combination of anti-4-1 BB antibody, Avelumab, and anti-M-CSF-antibody is more efficacious in treating cancer than single antibody or double antibody combination treatment.
Example 6
Combination Treatment of Colon Carcinoma with Avelumab, Anti-4-1 BB Antibody, and Anti-OX40 Antibody
[0238] This example illustrates the therapeutic activity of the anti-PD-L1 antibody Avelumab, anti-4-1 BB antibody, and anti-OX40 antibody triple combination therapy in murine cancer models.
[0239] Two murine models were used to evaluate the therapeutic efficacy of combinatorial treatment of anti-OX40 antibody, anti-4-1 BB and Avelumab. Six (6)- to 8-week old female C57BL/6 mice or Balb/C mice were purchased from the Jackson Laboratories. All animals were housed in a pathogen free vivarium facility at Rinat and experiments were conducted according to the protocols in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines.
[0240] The B16F10 melanoma cell line was purchased from American Type Culture Collection (ATCC). The MC38 colon carcinoma cell line was kindly provided by Dr. Antoni Ribas at University of California, Los Angeles, Calif. Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine at 37.degree. C. in 5% carbon dioxide (CO.sub.2). Cells growing in an exponential growth phase were harvested and used for tumor inoculation.
[0241] Therapeutic mouse anti-OX40 antibodies with either the mIgG1 or the mIgG2a isotype (anti-OX40 mIgG1 and anti-OX40 mIgG2a, respectively) were derived from parental clone OX86 in house. Therapeutic mouse anti-mouse 4-1 BB antibody (mouse immunoglobulin G1 [mIgG1]), derived from the parental clone MAB9371 (R&D Systems), was prepared in-house. Avelumab was provided by Merck Serono. Isotype control mIgG1 (clone: MOPC-21) and mIgG2a (C1.18.4) was purchased from BioXcell. Human IgG1 was prepared in-house. Anti-OX40 antibody, anti-4-1BB antibody, and avelumab were dosed at 3 mg/kg, 1 mg/kg and 20 mg/kg in the B16F10 model and 1 mg/kg, 1 mg/kg and 10 mg/kg in the MC38 model, respectively, in phosphate buffered saline (PBS) (Life Technologies), and dosed at 0.2 mL per mouse intraperitoneally (ip) for 3 doses 3 to 4 days apart.
[0242] C57BL/6 mice were inoculated subcutaneously at the right flank with 0.3.times.10.sup.6 B16F10 cells in 0.1 mL of PBS. Balb/C mice were inoculated subcutaneously at the right flank with 0.5.times.10.sup.6 MC38 cells in 0.1 mL of PBS. When tumors reached target size, mice were randomized into treatment groups. Treatment was started on the same day as randomization. Tumor size was measured twice weekly in 2 dimensions using a caliper, and the volume was calculated in cubic millimeters using the formula: V=0.5 L.times.W.sup.2 where L is the longest diameter of the tumor and W is the diameter perpendicular to L. Body weight was recorded weekly.
[0243] Results are summarized in Table 9 (B16F10 melanoma) and Table 10 (MC38 colon carcinoma) below (mean tumor size.+-.SEM). Statistical analyses were performed using GraphPad Prism 6.0. 2-way ANOVA was applied to compare the statistical differences among multiple groups relative to the isotype control or other treatment groups. P<0.05 was considered as significant difference. Tumor measurements are in mm.sup.3.
TABLE-US-00011 TABLE 9 Tumor Measurements of Subcutaneous B16F10 Melanoma over Time Days Post-Tumor Mean Tumor Size Inoculation (mm.sup.3) SEM N Group 1. Isotype control 12 74 11 8 15 214 46 8 18 392 67 8 22 1015 204 8 25 1897 310 8 29 2233 249 8 32 2311 228 8 Group 2. Anti-4-1BB antibody 12 73 9 8 15 282 67 8 18 413 98 8 22 742 155 8 25 1392 278 8 29 2620 518 8 32 2759 493 8 Group 3. Anti-OX40 mIgG2a antibody 12 71 7 9 15 198 51 9 18 370 105 9 22 783 293 9 25 1147 283 9 29 2046 433 9 32 2576 360 9 Group 4. Avelumab 12 77 15 5 15 236 71 5 18 396 137 5 22 750 134 5 25 1291 210 5 29 2159 326 5 32 2352 264 5 Group 5. Anti-4-1BB antibody + Anti-OX40 mIgG2a antibody 12 78 14 9 15 155 23 9 18 313 50 9 22 595 87 9 25 861 65 9 29 1453 137 9 32 2003 245 9 Group 6. Anti-OX40 mIgG1 antibody + Avelumab 12 76 15 8 15 228 77 8 18 336 80 8 22 648 149 8 25 1009 248 8 29 1381 228 8 32 1908 261 8 Group 7. Avelumab + Anti-OX40 mIgG2a antibody 12 75 11 8 15 184 37 8 18 297 61 8 22 505 111 8 25 833 191 8 29 1731 392 8 32 2056 371 8 Group 8. Avelumab + Anti-4-1BB antibody 12 73 10 8 15 229 52 8 18 274 52 8 22 537 117 8 25 803 192 8 29 1435 305 8 32 1572 307 8 Group 9. Avelumab + Anti-4-1BB antibody + Anti-OX40 mIgG1 antibody 12 72 9 9 15 176 32 9 18 228 60 9 22 373 114 9 25 585 192 9 29 788 267 9 32 979 329 9 Group 10. Avelumab + Anti-4-1BB antibody + Anti-OX40 mIgG2a antibody 12 74 10 9 15 104 17 9 18 120 17 9 22 155 49 9 25 208 54 9 29 365 93 9 32 442 114 9
TABLE-US-00012 TABLE 10 Tumor Measurements of Subcutaneous MC38 Colon Carcinoma over Time Days Post-Tumor Mean Tumor Size Inoculation (mm.sup.3) SEM N Group 1. Isotype control 10 85 7 9 13 162 23 9 16 305 41 9 21 696 66 9 24 1064 112 9 28 1830 214 9 Group 2. Anti-OX40 mIgG1 antibody 10 85 6 9 13 160 15 9 16 280 28 9 21 751 79 9 24 1238 139 9 28 2223 270 9 Group 3. Anti-OX40 mIgG2a antibody 10 85 7 9 13 154 11 9 16 247 18 9 21 455 64 9 24 648 102 9 28 1053 181 9 Group 4. Anti-4-1BB antibody 10 84 7 8 13 161 11 8 16 264 19 8 21 585 37 8 24 909 65 8 28 1494 129 8 Group 5. Anti-OX40 mIgG1 antibody + Anti-4-1BB antibody 10 85 7 9 13 171 11 9 16 246 20 9 21 492 27 9 24 737 62 9 28 1241 217 9 Group 6. Anti-OX40 mIgG2a antibody + Anti-4-1BB antibody 10 85 7 8 13 175 15 8 16 248 29 8 21 387 74 8 24 567 119 8 28 854 163 8 Group 7. Anti-4-1BB antibody + Avelumab 10 85 6 9 13 152 8 9 16 195 27 9 21 349 89 9 24 573 157 9 28 1026 255 9 Group 8. Anti-OX40 mIgG1 antibody + Anti-4-1BB antibody + Avelumab 10 85 6 9 13 167 12 9 16 170 32 9 21 228 65 9 24 304 86 9 28 448 108 9 Group 9. Anti-OX40 mIgG2a antibody + Anti-4-1BB antibody + Avelumab 10 85 6 9 13 153 17 9 16 127 23 9 21 116 37 9 24 165 67 9 28 260 107 9
[0244] Two murine models were used to evaluate the therapeutic efficacy of triple combinatorial treatment of anti-OX40 antibody, anti-4-1 BB antibody, and Avelumab. In the B16F10 melanoma model, the average tumor size when treatment was started was 71-78 mm.sup.3 (Table 9). By day 32 post tumor innoculation, the tumors in animals treated with isotype control, anti-4-1BB antibody alone, avelumab alone, anti-OX40 mIgG2a antibody alone and anti-OX40 mIgG1 antibody plus Avelumab groups were either very close to or over 2000 mm.sup.3; they were 2311.+-.228 mm.sup.3, 2759.+-.493 mm.sup.3, 2352.+-.264 mm.sup.3, 2576.+-.360 mm.sup.3 and 1908.+-.261 mm.sup.3, respectively. Treatment of animals with anti-4-1 BB antibody plus anti-OX40 mIgG2a antibody, anti-OX40 mIgG2a antibody plus Avelumab, or anti-4-1 BB antibody plus Avelumab had better treatment efficacy by day 25 as comparing to isotype control treated animals; however the difference in tumor size became insignificant on day 32. By contrast, dramatic tumor suppression was observed when animals were administered Avelumab, anti-4-1 BB antibody, and anti-OX40 mIgG1 antibody concurrently (Table 9, Group 9), or Avelumab, anti-4-1BB antibody, and anti-OX40 mIgG2a antibody concurrently (Table 9, Group 10). Tumors were 979 .+-.329 mm.sup.3 (Table 9, Group 9; p<0.001 vs isotype control and single agent alone groups) and 442.+-.114 mm.sup.3 (Table 9, Group 10; p<0.00001 vs isotype control and single agent alone groups), respectively. In the case of triple combination with anti-4-1BB antibody, anti-OX40 mIgG2a antibody, and Avelumab combination, it is also significantly better than the double combination groups (p<0.01) (Table 9).
[0245] In the MC38 colon carcinoma model, the average tumor size when treatment was started was 84-85 mm.sup.3. By day 28 post tumor implantation, tumors in animals treated with anti-OX40 mIgG2a antibody (Table 10, Group 3), anti-OX40 mIgG1 antibody plus anti-4-1BB antibody (Table 10, Group 5), anti-OX40 mIgG2a plus anti-4-1BB antibody (Table 10, Group 6), or anti-4-1BB antibody plus Avelumab (Table 10, Group 7) had tumors sizes of 1053 .+-.181 mm.sup.3, 1241 .+-.217 mm.sup.3, 854.+-.163 mm.sup.3 and 1026.+-.255 mm.sup.3, respectively, which is significantly lower than that of the isotype control treated group (1830.+-.214 mm.sup.3) (p<0.001) (Table 10, Group 1). Treatment with anti-OX40 mIgG1 antibody alone (Table 10, Group 2) or anti-4-1BB antibody alone (Table 10, Group 4) did not inhibit tumor growth. By contrast, treatment with the triple combination of anti-4-1BB antibody and Avelumab with either anti-OX40 mIgG1 antibody (Table 10, Group 8) or anti-OX40 mIgG2a antibody (Table 10, Group 9) antibody significantly inhibited tumor growth with the tumor size averaging 448.+-.108 mm.sup.3 and 260.+-.107 mm.sup.3, respectively. In both cases this is not only significant comparing to the isotype control group (p<0.0001), both triple combinations were also significantly better than any of the double combinations (p<0.001) (Table 10).
[0246] These results demonstrate that treatment with the triple combination of anti-4-1 BB antibody, Avelumab, and anti-OX40 antibody is more efficacious in treating cancer than single antibody or double antibody combination treatment.
Example 7
Combination Treatment of Relapsed or Refractory (R/R) Diffuse Large B-Cell Lymphoma (DLBCL) with Avelumab in Combination with Anti-4-1BB Antibody, Azacitidine, Anti-CD20 Antagonist Antibody, and/or Conventional Chemotherapy (Bendamustine).
[0247] In this study example, three treatment regiments are illustrated: [0248] Avelumab in combination with rituximab and PF-05082566 for the treatment of patients with relapsed or refractory DLBCL [0249] Avelumab in combination with azacitidine and PF-05082566 for the treatment of patients with relapsed or refractory DLBCL [0250] Avelumab in combination with rituximab and bendamustine is indicated for the treatment of patients with relapsed or refractory DLBCL
[0251] The target population for the study is patients with R/R DLBCL defined as follows: (i) patients with R/R DLBCL following failure of at least 2 lines (and a maximum of 4 lines) of prior rituximab/multi-agent chemotherapy and/or (ii) failure of ASCT, or (iii) who are not candidates for ASCT (refusal or no available donor), or (iv) who are not candidates for intensive second-line chemotherapy.
[0252] The current NCCN Guidelines (version 1.2016) for DLBCL recommend treatment with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in patients with newly diagnosed disease in all stages of disease, or mini-CHOP in patients >80 years with comorbidities. Approximately 60% of patients with DLBCL are expected to be cured following treatment with R-CHOP. Thirty to 50% of those with advanced disease will, however, have disease that is either primary refractory (-15%) or resistant (.about.25%) to R-CHOP (NCCN Guidelines, 2016; Sehn & Gascoyne, 2015; Vacirca et al, 2014).
[0253] High-dose chemotherapy followed by ASCT provides the best chance of a cure in patients with R/R DLBCL in the second-line setting; however, due to advanced age and/or comorbidities, only approximately 50% of patients for whom first-line R-CHOP failures are fit for high-dose chemotherapy, and of these, only about .about.50% have chemosensitive disease in the second-line setting and are suitable for ASCT (Sehn & Gascoyne, 2015). Even if eligible for high-dose chemotherapy, patients may refuse ASCT, lack a good donor, or be ineligible due to a variety of comorbidities. Even in patients treatmed with high-dose chemotherapy followed by ASCT, only a minority (<10%) are cured.
[0254] The following rituximab-containing chemotherapy regimens are currently recommended by the NCCN Guidelines (version 1.2016) for second-line salvage therapy and beyond in patients who are not eligible for high-dose chemotherapy and ASCT: bendamustine.+-.rituximab, brentuximab, cyclophosphamide/etoposide/procarbazine/prednisone (CEPP), cyclophosphamide/etoposide/ vincristine/prednisone (CEOP), dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide and doxorubicin (DA-EPOCH).+-.rituximab, gemcitabine, dexamethasone and cisplatin (GDP).+-.rituximab, gemcitabine/oxaliplatin.+-.rituximab, lenalidomide.+-.rituximab, and rituximab (NCCN Guidelines, 2016).
[0255] The outcome of patients for whom treatment with R-CHOP failures, and who are not eligible for high-dose chemotherapy or ASCT is dismal, with a median PFS of 3.6 months (Vacirca et al, 2014). The treatment options for these patients remain very limited, and there is consequently a high unmet medical need in patients with R/R DLBCL for the development of more effective salvage strategies that can prolong PFS and overall survival (OS).
[0256] The proposed Study is a multicenter, international, parallel design, randomized, open-label, 2-component (Phase 1b followed by Phase 3) study of avelumab in various combinations for the treatment of R/R DLBCL. Agents that will be tested include:
[0257] (i) PF-05082566, a novel fully human IgG2 monoclonal antibody agonist of 4-1BB
[0258] (ii) Azacitidine, a DNA methyltransferase inhibitor (DNMTi) and epigenetic agent which has been shown to have potential immune priming activity through various mechanisms including the induction of PD-1 on tumor infiltrating lymphocytes (TILs) and PD-L1 on tumor cells as well as the induction of tumor neo-antigen expression,
[0259] (iii) Rituximab, a CD20 antagonist antibody, and
[0260] (iv) Bendamustine, an alkylatingchemotherapy agent which is one of the National Comprehensive Cancer Network (NCCN) recommended agents for the salvage therapy of patients with DLBCL who are ineligible for high dose chemotherapy and autologous stem cell transplant (ASCT).
[0261] The treatment regimens proposed in the study include avelumab combined with:
[0262] (i) Rituximab and PF-05082566
[0263] (ii) Azacitidine and PF-05082566, and
[0264] (iii) Rituximab and bendamustine
[0265] In Phase 3, patients will be randomized in a 1:1 ratio to the treatment regimen selected in Phase 1b versus the Investigator's Choice standard of care (SOC) treatment to determine whether the selected treatment regimen is superior to the Investigator's Choice SOC treatment in prolonging progression-free survival (PFS).
[0266] The target study population of this Phase 1b/Phase 3 registrational study will comprise patients with R/R DLBCL who have completed at least 2 (but not more than 4) lines of prior rituximab/multi-agent chemotherapy, or in whom ASCT has been a failure , or who are not candidates for ASCT, or who are not eligible for intensive chemotherapy. The study will assess the safety, efficacy, pharmacokinetics (PK), immunogenicity, and patient reported outcomes.
[0267] The primary objective of the Phase 1b component is to make a preliminary assessment of safety for each combination regimen. Each arm without a significant safety signal among the first 6 patients will then be expanded to a total of 28 patients per arm in order to select a treatment regimen to be advanced to the Phase 3 component of the study. This decision will be based upon the investigator observed objective response rate (ORR) and safety profile of each combination regimen. The combination regimens to be assessed in the Phase 1b component of the study in 28-day cycles include:
[0268] Arm A: Avelumab/Rituximab/PF-05082566 (4-1 BB)
[0269] (i) Rituximab 375 mg/m.sup.2 (IV) in the morning on Day 1 of each 28-day cycle. Rituximab is administered for a maximum of 8 cycles.
[0270] Rituximab will be administered at least 3 hours prior to PF-05082566 when dosed on the same day.
[0271] (ii) PF-05082566 100 mg fixed dose (IV) in the morning on Day 2 of Cycles 1 and 2 of each 28-day cycle. If PF-05082566 is well tolerated in Cycles 1 and 2, administration of PF-05082566 may be on Day 1 in Cycle 3 (and all subsequent cycles).
[0272] PF-05082566 will be administered at least 3 hours prior to avelumab in Cycle 1. If PF-05082566 is well tolerated in Cycle 1, in Cycle 2 and all subsequent cycles the window of dose administration between PF-05082566 and avelumab may be decreased from at least 3 hours apart to 30-60 minutes apart.
[0273] (iii) Avelumab 10 mg/kg (IV) every 2-weeks Day 2 and Day 16 of each 28-day cycle in Cycle 1 and Cycle 2. If avelumab is well tolerated in Cycle 1 and 2, administration of avelumab may be on Day 1 and Day 15 in Cycle 3 (and all subsequent cycles).
[0274] Avelumab will be administered at least 3 hours after PF-05082566 in Cycle 1 and Cycle 2. If avelumab is well tolerated in Cycle 1 Day 2, in Cycle 2 Day 2 and subsequent cycles the window of dose administration between avelumab and PF-05082566 may be decreased from at least 3 hours apart to 30-60 minutes apart.
[0275] Arm B: Avelumab/Azacitidine/PF-05082566 (4-1 BB)
[0276] (i) Azacitidine 75 mg/m.sup.2 (SC) in the morning on Day 1-Day 7 consecutively of each 28-day cycle. Azacitidine is administered for a maximum of 6 cycles.
[0277] Azacitidine will be administered at least 3 hours prior to PF-05082566 when dosed on the same day.
[0278] (ii) PF-05082566 100 mg fixed dose (IV) in the morning on Day 2 for Cycle 1 and Cycle 2, of each 28-day cycle. If PF-05082566 is well tolerated in Cycle 1 and 2, PF-05082566 may be administered on Day 1 commencing with Cycle 3 (and subsequent cycles).
[0279] PF-05082566 should be administered at least 3 hours prior to avelumab administration. If PF-05082566 is well tolerated in Cycle 1, in Cycle 2 and all subsequent cycles the window of dose administration between PF-05082566 and avelumab may be decreased from at least 3 hours apart to 30-60 minutes apart.
[0280] (iii) Avelumab 10 mg/kg every 2-weeks (IV) Day 2 and Day 16 of each 28-day cycle in Cycle 1 and Cycle 2. If avelumab is well tolerated in Cycle 1 and 2, avelumab may be administered on Day 1 and Day 15 in Cycle 3 (and all subsequent cycles).
[0281] Avelumab administration should be at least 3 hours after PF-05082566 in Cycle 1 and Cycle 2. If avelumab is well tolerated in Cycle 1 Day 2, in Cycle 2 Day 2 and subsequent cycles the window of dose administration between avelumab and PF-05082566 may be decreased from at least 3 hours apart to 30-60 minutes apart.
[0282] Arm C: Avelumab/Bendamustine/Rituximab
[0283] (i) Rituximab 375 mg/m.sup.2 (IV) in the morning on Day 1 of each 28-day cycle. Rituximab is administered for a maximum of 8 cycles.
[0284] (ii) Bendamustine 90 mg/m.sup.2 (IV) on Day 2 and Day 3 of each 28-day cycle in Cycle 1 and Cycle 2. If bendamustine is well tolerated in Cycle 1 and 2, bendamustine may be administered on Day 1 and Day 2 in Cycle 3 (and all subsequent cycles). Bendamustine is administered for a maximum of 6 cycles.
[0285] (iii) Avelumab 10 mg/kg every 2-weeks (IV) Day 2 and Day 16 of each 28-day cycle in Cycle 1 and Cycle 2. If avelumab is well tolerated in Cycle 1 and 2, avelumab may be administered on Day 1 and Day 15 in Cycle 3 (and all subsequent cycles). Avelumab administration should be at least 3 hours after bendamustine.
[0286] In Phase 3 (N=220), the primary objective is to demonstrate superiority in PFS (as assessed by Blinded Independent Central Review [BICR]) of the combination regimen identified in Phase 1b, over the control treatment, namely Investigator's Choice SOC chemotherapy (comprising rituximab/bendamustine or rituximab/ gemcitabine/oxaliplatin).
[0287] The following treatment regimens will be assessed in the Phase 3 component of the study, with all treatments being administered in 28-day cycles:
[0288] Arm D (N=110): Regimen Selected from Phase 1b
[0289] Arm D will be one of the treatment regimens assessed in Phase 1b, ie, Arm A, B, or C, selected based on safety and efficacy assessments.
[0290] Cohort E (N=110): Investigator's Choice Option Between the Following Standard of Care Regimens:
[0291] (i) Rituximab/bendamustine [0292] Rituximab 375 mg/m.sup.2 IV Day 1 [0293] Bendamustine 120 mg/m.sup.2 IV Day 1 and Day 2
[0294] (ii) Rituximab/gemcitabine/oxaliplatin [0295] Rituximab 375 mg/m.sup.2 IV Day 1 [0296] Gemcitabine 1000 mg/m.sup.2 IV on Day 2 and Day 17 [0297] Oxaliplatin 100 mg/m.sup.2 IV on Day 2 and Day 17
Example 8
Combination Treatment of Patients with Advanced Malignancies whose Disease has Progressed on an Immune Checkpoint Inhibitor with Avelumab in Combination with Anti-4-1 BB Antibody.
[0298] This example illustrates a Phase 2 study to assess safety and efficacy of avelumab (MSB0010718C) in combination with anti-4-1 BB agonist antibody PF-05082566 in patients with advanced NSCLC, RCC, or urothelial cancer (UC) whose disease has progressed on prior therapy(ies), including a single-agent immune checkpoint inhibitor.
[0299] The objective of this study is to evaluate the Objective Response Rate (ORR) based on RECIST 1.1 of avelumab plus PF-05082566. Patients must have advanced NSCLC, RCC, or urothelial cancer which was resistant (responded and then progressed) or refractory (never responded) to prior therapy(ies), including a single-agent immune checkpoint inhibitor (e.g.,anti-PD-1/anti-PD-L1 or anti-CTLA-4).
[0300] Avelumab will be given as a 1-hour intravenous infusion every 2 weeks at a dose of 10 mg/kg in all three cohorts. PF-05082566 will be administered at 100 mg as a 1-hour IV infusion once every 4 weeks on Day 1 of each cycle.
[0301] On days when both drugs are administered, PF 05082566 will be administered first, followed by the avelumab infusion no more than 30 minutes after the end of the PF-05082566 infusion.
[0302] Dosing will continue until disease progression is confirmed by the investigator, patient refusal, unacceptable toxicity, patient is lost to follow-up, or until the study is terminated by the Sponsor, whichever occurs first.
[0303] The combination of avelumab plus anti-4-1BB antibody PF-05082566 and anti-OX40 antibody PF-04518600 has been evaluated for cytokine release using the standard human PBMC in vitro test. The cytokine release assay was completed for PF-05082566 alone and in combination with avelumab and PF-04518600. Results for the PF-05082566 antibody alone did not show a significant increase in cytokine release. In addition, there was no additive effect on cytokine release when the three monoclonal antibodies were combined.
[0304] ORR estimation will be the primary objective in any potential evaluation of avelumab in combination with immunotherapy other that PF-05082566. In each case, the ORR will be evaluated with the totality of the data for potential cohort expansion or testing of multiple tumor types and/or other combination immunotherapeutic agents.
Example 9
Randomized, Phase 3 Study of Avelumab (MSB0010718C) in Combination with Standard of Care Chemoradiotherapy (Cisplatin and Definitive Radiation Therapy) Versus Standard-of-Care Chemoradiotherapy in the Front-Line Treatment of Patients with Locally Advanced Squamous Cell Carcinoma of the Head and Neck
[0305] This example illustrates a Phase 3, multicenter, multinational, randomized, placebo controlled study of avelumab (MSB0010718C) in combination with standard of care (SOC) chemoradiotherapy (cisplatin and definitive radiation therapy) versus SOC chemotherapy for front-line treatment of patients with locally-advanced squamous cell carcinoma of the head and neck.
[0306] Approximately 640 patients who have received no prior therapy for their SCCHN (oral cavity, oropharynx, larynx, or hypopharynx) HPV-: Stage III, IVa, or IVb or HPV+: T4 or N3 who are eligible for definitive chemoradiotherapy with cisplatin will be randomized 1:1 to treatment with avelumab+SOC chemoradiotherapy vs. placebo+chemoradiotherapy followed by maintenance avelumab or placebo for up to 1 year. Patients will be stratified based on: [0307] Tumor (T) stage (<T4 vs T4); [0308] Nodal (N) stage (N0 vs N1/N2a/N2b vs N2c/N3)
[0309] Tumor assessment will occur every 12 weeks following the completion of definitive chemoradiotherapy for 2 years, and then every 16 weeks thereafter.
[0310] A blinded independent review committee (BICR) will review tumor assessments in addition to investigator reviews.
[0311] When the study treatment is discontinued for reasons other than progressive disease (PD), patient withdrawal of consent, or death, patients will be followed and have tumor assessments performed every 12 weeks until: 1) PD, 2) death, 3) patient withdrawal of consent from study, or 4) 2 years from completion of chemoradiotherapy have passed after which tumor assessments can be every 16 weeks, whichever occurs first. [0312] Arm A: Avelumab (MSB0010718C) +SOC Chemoradiotherapy (CRT). In this study, the lead-in phase is to start seven days prior to initiation of the CRT phase. The maintenance phase will start after completion of the CRT phase (i.e., two weeks following completion of CRT). [0313] Cisplatin 100 mg/m.sup.2 Days 1, 22, 43. Administered in 500 ml normal saline over a 60-120 minute infusion with an additional 1 to 1.5 L of fluid given post-hydration. [0314] Radiation therapy (RT) 70 Gy/33-35 fractions/day, 5 fractions/week intensity modulated radiation therapy (IMRT) [0315] Avelumab: 10 mg/kg administered on Day 1 of the lead-in phase' and Days 8, 29, 39 of the CRT phase, and every 2 weeks (Q2W) thereafter for up to 12 months. [0316] Arm B: SOC Chemoradiotherapy. [0317] Cisplatin 100 mg/m.sup.2 Days 1, 22, 43 [0318] RT 70 Gy/33-35 fractions/day, 5 fractions/week IMRT [0319] Placebo: Day 1 of the lead-in phase, Days 8, 29, 39, and Q2W thereafter for up to 12 months.
[0320] Avelumab and placebo will be administered as IV infusion.
[0321] Patients will receive study treatment until: 1) 12 months after start of maintenance therapy (study intervention completion), 2) PD 3) death, 4) patient withdrawal of consent, 5) patient is lost to follow-up, 6) unacceptable toxicity occurs, or 7) the study is terminated by the Sponsor, whichever occurs first.
[0322] The dose of cisplatin may be modified on Days 22 and/or 43 for toxicity as follows: starting dose level is 100 100 mg/m.sup.2, dose level -1 is 75 mg/m.sup.2, and dose level -2 is 50 mg/m.sup.2.
[0323] Peripheral blood and additional tumor tissue biomarkers consisting of the levels of cells, deoxyribonucleic acid (DNA), ribonucleic acid (RNA), or proteins that may be related to anti-tumor immune response and/or response to or disease progression on avelumab, such as genes related to IFN-.gamma. or transforming growth factor (TGF)-.beta..
Example 10
Phase 1b Dose-Finding Study of Avelumab (MS1300107180; Anti-PD-L1)+Axitinib in Treatment-Naive Patients with Advanced Renal Cell Carcinoma
[0324] This example illustrates results from the study described in Example 1 above. Eligible patients have histologically confirmed aRCC with a clear-cell component, primary tumour resection, .gtoreq.1 measurable lesion, archival/fresh tumour biopsy, ECOG PS .ltoreq.1, no preexisting uncontrolled hypertension, and no prior systemic therapy for aRCC. To determine dose modifications for future cohorts, dose escalation/de-escalation rules that follow the modified toxicity probability interval method were used. Adverse events (AEs) were graded by NCI CTCAE v4. Objective response rates (ORR; RECIST v1.1) were evaluated.
[0325] The starting dose of avelumab 10 mg/kg (1h IV infusion) Q2W+axitinib 5 mg PO BID met MTD criteria. By 5 Apr. 2016, 6 pts (median age 59.5 [range, 45-73]) have been treated with avelumab for a median of 17.0 wks (range, 11.9-21.7) and with axitinib for 16.3 wks (range, 12.7-22.7). One DLT of grade 3 proteinuria occurred. The most common treatment-related (TR) AEs of any grade were dysphonia (n=4), hypertension (n=4), fatigue (n=3), and headache (n=3). Grade 3-4 TRAEs were hypertension (n=2), hand-foot syndrome (n=1), elevated lipase (n=1), and proteinuria (n=1). Confirmed ORR is 83.3% (95% CI: 35.9, 99.6) based on 5 PRs and stable disease in 1 pt.
[0326] The MTD/RP2D for this expansion phase and further studies in aRCC has been confirmed as avelumab 10 mg/kg IV Q2W+axitinib 5 mg PO BID continuously. The regimen has shown preliminary antitumour activity in treatment-naive pts with aRCC. Enrollment is ongoing in the expansion cohort. These results demonstrate the efficacy and safety of avelumab+axitinib vs current monotherapies for aRCC.
[0327] Although the disclosed teachings have been described with reference to various applications, methods, kits, and compositions, it will be appreciated that various changes and modifications can be made without departing from the teachings herein and the claimed invention below. The foregoing examples are provided to better illustrate the disclosed teachings and are not intended to limit the scope of the teachings presented herein. While the present teachings have been described in terms of these exemplary embodiments, the skilled artisan will readily understand that numerous variations and modifications of these exemplary embodiments are possible without undue experimentation. All such variations and modifications are within the scope of the current teachings.
[0328] All references cited herein, including patents, patent applications, papers, text books, and the like, and the references cited therein, to the extent that they are not already, are hereby incorporated by reference in their entirety. In the event that one or more of the incorporated literature and similar materials differs from or contradicts this application, including but not limited to defined terms, term usage, described techniques, or the like, this application controls.
[0329] The foregoing description and Examples detail certain specific embodiments of the invention and describes the best mode contemplated by the inventors. It will be appreciated, however, that no matter how detailed the foregoing may appear in text, the invention may be practiced in many ways and the invention should be construed in accordance with the appended claims and any equivalents thereof.
Sequence CWU 1
1
411290PRThomo sapiens 1Met Arg Ile Phe Ala Val Phe Ile Phe Met Thr
Tyr Trp His Leu Leu1 5 10 15Asn Ala Phe Thr Val Thr Val Pro Lys Asp
Leu Tyr Val Val Glu Tyr 20 25 30Gly Ser Asn Met Thr Ile Glu Cys Lys
Phe Pro Val Glu Lys Gln Leu 35 40 45Asp Leu Ala Ala Leu Ile Val Tyr
Trp Glu Met Glu Asp Lys Asn Ile 50 55 60Ile Gln Phe Val His Gly Glu
Glu Asp Leu Lys Val Gln His Ser Ser65 70 75 80Tyr Arg Gln Arg Ala
Arg Leu Leu Lys Asp Gln Leu Ser Leu Gly Asn 85 90 95Ala Ala Leu Gln
Ile Thr Asp Val Lys Leu Gln Asp Ala Gly Val Tyr 100 105 110Arg Cys
Met Ile Ser Tyr Gly Gly Ala Asp Tyr Lys Arg Ile Thr Val 115 120
125Lys Val Asn Ala Pro Tyr Asn Lys Ile Asn Gln Arg Ile Leu Val Val
130 135 140Asp Pro Val Thr Ser Glu His Glu Leu Thr Cys Gln Ala Glu
Gly Tyr145 150 155 160Pro Lys Ala Glu Val Ile Trp Thr Ser Ser Asp
His Gln Val Leu Ser 165 170 175Gly Lys Thr Thr Thr Thr Asn Ser Lys
Arg Glu Glu Lys Leu Phe Asn 180 185 190Val Thr Ser Thr Leu Arg Ile
Asn Thr Thr Thr Asn Glu Ile Phe Tyr 195 200 205Cys Thr Phe Arg Arg
Leu Asp Pro Glu Glu Asn His Thr Ala Glu Leu 210 215 220Val Ile Pro
Glu Leu Pro Leu Ala His Pro Pro Asn Glu Arg Thr His225 230 235
240Leu Val Ile Leu Gly Ala Ile Leu Leu Cys Leu Gly Val Ala Leu Thr
245 250 255Phe Ile Phe Arg Leu Arg Lys Gly Arg Met Met Asp Val Lys
Lys Cys 260 265 270Gly Ile Gln Asp Thr Asn Ser Lys Lys Gln Ser Asp
Thr His Leu Glu 275 280 285Glu Thr 29025PRThomo sapiens 2Ser Tyr
Ile Met Met1 5311PRThomo sapiens 3Ser Ile Tyr Pro Ser Gly Gly Ile
Thr Phe Tyr1 5 10411PRThomo sapiens 4Ile Lys Leu Gly Thr Val Thr
Thr Val Asp Tyr1 5 10514PRThomo sapiens 5Thr Gly Thr Ser Ser Asp
Val Gly Gly Tyr Asn Tyr Val Ser1 5 1067PRThomo sapiens 6Asp Val Ser
Asn Arg Pro Ser1 5710PRThomo sapiens 7Ser Ser Tyr Thr Ser Ser Ser
Thr Arg Val1 5 108118PRThomo sapiens 8Glu Val Gln Leu Leu Glu Ser
Gly Gly Gly Leu Val Gln Pro Gly Gly1 5 10 15Ser Leu Arg Leu Ser Cys
Ala Ala Ser Gly Phe Thr Phe Ser Ser Tyr 20 25 30Ile Met Met Trp Val
Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val 35 40 45Ser Ser Ile Tyr
Pro Ser Gly Gly Ile Thr Phe Tyr Ala Asp Lys Gly 50 55 60Arg Phe Thr
Ile Ser Arg Asp Asn Ser Lys Asn Thr Leu Tyr Leu Gln65 70 75 80Met
Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys Ala Arg 85 90
95Ile Lys Leu Gly Thr Val Thr Thr Val Asp Tyr Trp Gly Gln Gly Thr
100 105 110Leu Val Thr Val Ser Ser 1159110PRTHomo sapiens 9Gln Ser
Ala Leu Thr Gln Pro Ala Ser Val Ser Gly Ser Pro Gly Gln1 5 10 15Ser
Ile Thr Ile Ser Cys Thr Gly Thr Ser Ser Asp Val Gly Gly Tyr 20 25
30Asn Tyr Val Ser Trp Tyr Gln Gln His Pro Gly Lys Ala Pro Lys Leu
35 40 45Met Ile Tyr Asp Val Ser Asn Arg Pro Ser Gly Val Ser Asn Arg
Phe 50 55 60Ser Gly Ser Lys Ser Gly Asn Thr Ala Ser Leu Thr Ile Ser
Gly Leu65 70 75 80Gln Ala Glu Asp Glu Ala Asp Tyr Tyr Cys Ser Ser
Tyr Thr Ser Ser 85 90 95Ser Thr Arg Val Phe Gly Thr Gly Thr Lys Val
Thr Val Leu 100 105 11010450PRTHomo sapiens 10Glu Val Gln Leu Leu
Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly1 5 10 15Ser Leu Arg Leu
Ser Cys Ala Ala Ser Gly Phe Thr Phe Ser Ser Tyr 20 25 30Ile Met Met
Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val 35 40 45Ser Ser
Ile Tyr Pro Ser Gly Gly Ile Thr Phe Tyr Ala Asp Thr Val 50 55 60Lys
Gly Arg Phe Thr Ile Ser Arg Asp Asn Ser Lys Asn Thr Leu Tyr65 70 75
80Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95Ala Arg Ile Lys Leu Gly Thr Val Thr Thr Val Asp Tyr Trp Gly
Gln 100 105 110Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly
Pro Ser Val 115 120 125Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser
Gly Gly Thr Ala Ala 130 135 140Leu Gly Cys Leu Val Lys Asp Tyr Phe
Pro Glu Pro Val Thr Val Ser145 150 155 160Trp Asn Ser Gly Ala Leu
Thr Ser Gly Val His Thr Phe Pro Ala Val 165 170 175Leu Gln Ser Ser
Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro 180 185 190Ser Ser
Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys 195 200
205Pro Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp
210 215 220Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu
Gly Gly225 230 235 240Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys
Asp Thr Leu Met Ile 245 250 255Ser Arg Thr Pro Glu Val Thr Cys Val
Val Val Asp Val Ser His Glu 260 265 270Asp Pro Glu Val Lys Phe Asn
Trp Tyr Val Asp Gly Val Glu Val His 275 280 285Asn Ala Lys Thr Lys
Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg 290 295 300Val Val Ser
Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys305 310 315
320Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu
325 330 335Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln
Val Tyr 340 345 350Thr Leu Pro Pro Ser Arg Asp Glu Leu Thr Lys Asn
Gln Val Ser Leu 355 360 365Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser
Asp Ile Ala Val Glu Trp 370 375 380Glu Ser Asn Gly Gln Pro Glu Asn
Asn Tyr Lys Thr Thr Pro Pro Val385 390 395 400Leu Asp Ser Asp Gly
Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp 405 410 415Lys Ser Arg
Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His 420 425 430Glu
Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro 435 440
445Gly Lys 45011216PRTHomo sapiens 11Gln Ser Ala Leu Thr Gln Pro
Ala Ser Val Ser Gly Ser Pro Gly Gln1 5 10 15Ser Ile Thr Ile Ser Cys
Thr Gly Thr Ser Ser Asp Val Gly Gly Tyr 20 25 30Asn Tyr Val Ser Trp
Tyr Gln Gln His Pro Gly Lys Ala Pro Lys Leu 35 40 45Met Ile Tyr Asp
Val Ser Asn Arg Pro Ser Gly Val Ser Asn Arg Phe 50 55 60Ser Gly Ser
Lys Ser Gly Asn Thr Ala Ser Leu Thr Ile Ser Gly Leu65 70 75 80Gln
Ala Glu Asp Glu Ala Asp Tyr Tyr Cys Ser Ser Tyr Thr Ser Ser 85 90
95Ser Thr Arg Val Phe Gly Thr Gly Thr Lys Val Thr Val Leu Gly Gln
100 105 110Pro Lys Ala Asn Pro Thr Val Thr Leu Phe Pro Pro Ser Ser
Glu Glu 115 120 125Leu Gln Ala Asn Lys Ala Thr Leu Val Cys Leu Ile
Ser Asp Phe Tyr 130 135 140Pro Gly Ala Val Thr Val Ala Trp Lys Ala
Asp Gly Ser Pro Val Lys145 150 155 160Ala Gly Val Glu Thr Thr Lys
Pro Ser Lys Gln Ser Asn Asn Lys Tyr 165 170 175Ala Ala Ser Ser Tyr
Leu Ser Leu Thr Pro Glu Gln Trp Lys Ser His 180 185 190Arg Ser Tyr
Ser Cys Gln Val Thr His Glu Gly Ser Thr Val Glu Lys 195 200 205Thr
Val Ala Pro Thr Glu Cys Ser 210 215126PRTArtificial
SequenceHumanized antibody sequence 12Ser Thr Tyr Trp Ile Ser1
51317PRTArtificial SequenceHumanized antibody sequence 13Lys Ile
Tyr Pro Gly Asp Ser Tyr Thr Asn Tyr Ser Pro Ser Phe Gln1 5 10
15Gly148PRTArtificial SequenceHumanized antibody sequence 14Arg Gly
Tyr Gly Ile Phe Asp Tyr1 51511PRTArtificial SequenceHumanized
antibody sequence 15Ser Gly Asp Asn Ile Gly Asp Gln Tyr Ala His1 5
10167PRTArtificial SequenceHumanized antibody sequence 16Gln Asp
Lys Asn Arg Pro Ser1 51711PRTArtificial SequenceHumanized antibody
sequence 17Ala Thr Tyr Thr Gly Phe Gly Ser Leu Ala Val1 5
1018116PRTArtificial SequenceHumanized antibody sequence 18Glu Val
Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Glu1 5 10 15Ser
Leu Arg Ile Ser Cys Lys Gly Ser Gly Tyr Ser Phe Ser Thr Tyr 20 25
30Trp Ile Ser Trp Val Arg Gln Met Pro Gly Lys Gly Leu Glu Trp Met
35 40 45Gly Lys Ile Tyr Pro Gly Asp Ser Tyr Thr Asn Tyr Ser Pro Ser
Phe 50 55 60Gln Gly Gln Val Thr Ile Ser Ala Asp Lys Ser Ile Ser Thr
Ala Tyr65 70 75 80Leu Gln Trp Ser Ser Leu Lys Ala Ser Asp Thr Ala
Met Tyr Tyr Cys 85 90 95Ala Arg Gly Tyr Gly Ile Phe Asp Tyr Trp Gly
Gln Gly Thr Leu Val 100 105 110Thr Val Ser Ser
11519108PRTArtificial SequenceHumanized antibody sequence 19Ser Tyr
Glu Leu Thr Gln Pro Pro Ser Val Ser Val Ser Pro Gly Gln1 5 10 15Thr
Ala Ser Ile Thr Cys Ser Gly Asp Asn Ile Gly Asp Gln Tyr Ala 20 25
30His Trp Tyr Gln Gln Lys Pro Gly Gln Ser Pro Val Leu Val Ile Tyr
35 40 45Gln Asp Lys Asn Arg Pro Ser Gly Ile Pro Glu Arg Phe Ser Gly
Ser 50 55 60Asn Ser Gly Asn Thr Ala Thr Leu Thr Ile Ser Gly Thr Gln
Ala Met65 70 75 80Asp Glu Ala Asp Tyr Tyr Cys Ala Thr Tyr Thr Gly
Phe Gly Ser Leu 85 90 95Ala Val Phe Gly Gly Gly Thr Lys Leu Thr Val
Leu 100 10520442PRTArtificial SequenceHumanized antibody sequence
20Glu Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Glu1
5 10 15Ser Leu Arg Ile Ser Cys Lys Gly Ser Gly Tyr Ser Phe Ser Thr
Tyr 20 25 30Trp Ile Ser Trp Val Arg Gln Met Pro Gly Lys Gly Leu Glu
Trp Met 35 40 45Gly Lys Ile Tyr Pro Gly Asp Ser Tyr Thr Asn Tyr Ser
Pro Ser Phe 50 55 60Gln Gly Gln Val Thr Ile Ser Ala Asp Lys Ser Ile
Ser Thr Ala Tyr65 70 75 80Leu Gln Trp Ser Ser Leu Lys Ala Ser Asp
Thr Ala Met Tyr Tyr Cys 85 90 95Ala Arg Gly Tyr Gly Ile Phe Asp Tyr
Trp Gly Gln Gly Thr Leu Val 100 105 110Thr Val Ser Ser Ala Ser Thr
Lys Gly Pro Ser Val Phe Pro Leu Ala 115 120 125Pro Cys Ser Arg Ser
Thr Ser Glu Ser Thr Ala Ala Leu Gly Cys Leu 130 135 140Val Lys Asp
Tyr Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly145 150 155
160Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser
165 170 175Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser Ser
Asn Phe 180 185 190Gly Thr Gln Thr Tyr Thr Cys Asn Val Asp His Lys
Pro Ser Asn Thr 195 200 205Lys Val Asp Lys Thr Val Glu Arg Lys Cys
Cys Val Glu Cys Pro Pro 210 215 220Cys Pro Ala Pro Pro Val Ala Gly
Pro Ser Val Phe Leu Phe Pro Pro225 230 235 240Lys Pro Lys Asp Thr
Leu Met Ile Ser Arg Thr Pro Glu Val Thr Cys 245 250 255Val Val Val
Asp Val Ser His Glu Asp Pro Glu Val Gln Phe Asn Trp 260 265 270Tyr
Val Asp Gly Val Glu Val His Asn Ala Lys Thr Lys Pro Arg Glu 275 280
285Glu Gln Phe Asn Ser Thr Phe Arg Val Val Ser Val Leu Thr Val Val
290 295 300His Gln Asp Trp Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val
Ser Asn305 310 315 320Lys Gly Leu Pro Ala Pro Ile Glu Lys Thr Ile
Ser Lys Thr Lys Gly 325 330 335Gln Pro Arg Glu Pro Gln Val Tyr Thr
Leu Pro Pro Ser Arg Glu Glu 340 345 350Met Thr Lys Asn Gln Val Ser
Leu Thr Cys Leu Val Lys Gly Phe Tyr 355 360 365Pro Ser Asp Ile Ala
Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn 370 375 380Asn Tyr Lys
Thr Thr Pro Pro Met Leu Asp Ser Asp Gly Ser Phe Phe385 390 395
400Leu Tyr Ser Lys Leu Thr Val Asp Lys Ser Arg Trp Gln Gln Gly Asn
405 410 415Val Phe Ser Cys Ser Val Met His Glu Ala Leu His Asn His
Tyr Thr 420 425 430Gln Lys Ser Leu Ser Leu Ser Pro Gly Lys 435
44021214PRTArtificial SequenceHumanized antibody sequence 21Ser Tyr
Glu Leu Thr Gln Pro Pro Ser Val Ser Val Ser Pro Gly Gln1 5 10 15Thr
Ala Ser Ile Thr Cys Ser Gly Asp Asn Ile Gly Asp Gln Tyr Ala 20 25
30His Trp Tyr Gln Gln Lys Pro Gly Gln Ser Pro Val Leu Val Ile Tyr
35 40 45Gln Asp Lys Asn Arg Pro Ser Gly Ile Pro Glu Arg Phe Ser Gly
Ser 50 55 60Asn Ser Gly Asn Thr Ala Thr Leu Thr Ile Ser Gly Thr Gln
Ala Met65 70 75 80Asp Glu Ala Asp Tyr Tyr Cys Ala Thr Tyr Thr Gly
Phe Gly Ser Leu 85 90 95Ala Val Phe Gly Gly Gly Thr Lys Leu Thr Val
Leu Gly Gln Pro Lys 100 105 110Ala Ala Pro Ser Val Thr Leu Phe Pro
Pro Ser Ser Glu Glu Leu Gln 115 120 125Ala Asn Lys Ala Thr Leu Val
Cys Leu Ile Ser Asp Phe Tyr Pro Gly 130 135 140Ala Val Thr Val Ala
Trp Lys Ala Asp Ser Ser Pro Val Lys Ala Gly145 150 155 160Val Glu
Thr Thr Thr Pro Ser Lys Gln Ser Asn Asn Lys Tyr Ala Ala 165 170
175Ser Ser Tyr Leu Ser Leu Thr Pro Glu Gln Trp Lys Ser His Arg Ser
180 185 190Tyr Ser Cys Gln Val Thr His Glu Gly Ser Thr Val Glu Lys
Thr Val 195 200 205Ala Pro Thr Glu Cys Ser 21022466PRTHomo sapiens
22Met Glu Leu Gly Leu Cys Trp Val Phe Leu Val Ala Ile Leu Glu Gly1
5 10 15Val Gln Cys Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val
Gln 20 25 30Pro Gly Gly Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe
Thr Phe 35 40 45Ser Ser Phe Ser Met Thr Trp Val Arg Gln Ala Pro Gly
Lys Gly Leu 50 55 60Glu Trp Val Ser Tyr Ile Ser Ser Arg Ser Ser Thr
Ile Ser Tyr Ala65 70 75 80Asp Ser Val Lys Gly Arg Phe Thr Ile Ser
Arg Asp Asn Ala Lys Asn 85 90 95Ser Leu Tyr Leu Gln Met Asn Ser Leu
Arg Asp Glu Asp Thr Ala Val 100 105 110Tyr Tyr Cys Ala Arg Asp Pro
Leu Leu Ala Gly Ala Thr Phe Phe Asp 115 120 125Tyr Trp Gly Gln Gly
Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys 130 135 140Gly Pro Ser
Val Phe Pro Leu Ala Pro Cys Ser Arg Ser Thr Ser Glu145 150 155
160Ser Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro
165 170 175Val Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val
His Thr 180 185 190Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr
Ser Leu Ser Ser Val 195 200 205Val Thr Val Pro Ser Ser Asn Phe Gly
Thr Gln Thr Tyr Thr Cys Asn 210 215 220Val Asp His Lys Pro Ser Asn
Thr Lys Val Asp Lys Thr Val Glu Arg225 230 235 240Lys Cys Cys Val
Glu Cys Pro Pro Cys Pro Ala Pro Pro Val Ala Gly 245 250 255Pro Ser
Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile 260 265
270Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu
275 280 285Asp Pro Glu Val Gln Phe Asn Trp Tyr Val Asp Gly Val Glu
Val His 290 295 300Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn
Ser Thr Phe Arg305 310 315 320Val Val Ser Val Leu Thr Val Val His
Gln Asp Trp Leu Asn Gly Lys 325 330 335Glu Tyr Lys Cys Lys Val Ser
Asn Lys Gly Leu Pro Ala Pro Ile Glu 340 345 350Lys Thr Ile Ser Lys
Thr Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr 355 360 365Thr Leu Pro
Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu 370 375 380Thr
Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp385 390
395 400Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro
Met 405 410 415Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu
Thr Val Asp 420 425 430Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser
Cys Ser Val Met His 435 440 445Glu Ala Leu His Asn His Tyr Thr Gln
Lys Ser Leu Ser Leu Ser Pro 450 455 460Gly Lys46523235PRTHomo
sapiens 23Met Glu Thr Pro Ala Gln Leu Leu Phe Leu Leu Leu Leu Trp
Leu Pro1 5 10 15Asp Thr Thr Gly Glu Phe Val Leu Thr Gln Ser Pro Gly
Thr Leu Ser 20 25 30Leu Ser Pro Gly Glu Arg Ala Thr Leu Ser Cys Arg
Ala Ser Gln Ser 35 40 45Val Ser Ser Ser Tyr Leu Ala Trp Tyr Gln Gln
Lys Pro Gly Gln Ala 50 55 60Pro Arg Leu Leu Ile Tyr Gly Ala Ser Ser
Arg Ala Thr Gly Ile Pro65 70 75 80Asp Arg Phe Ser Gly Ser Gly Ser
Gly Thr Asp Phe Thr Leu Thr Ile 85 90 95Ser Arg Leu Glu Pro Glu Asp
Phe Ala Val Tyr Tyr Cys Gln Gln Tyr 100 105 110Gly Ser Ser Pro Leu
Thr Phe Gly Gly Gly Thr Lys Val Glu Ile Lys 115 120 125Arg Thr Val
Ala Ala Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu 130 135 140Gln
Leu Lys Ser Gly Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe145 150
155 160Tyr Pro Arg Glu Ala Lys Val Gln Trp Lys Val Asp Asn Ala Leu
Gln 165 170 175Ser Gly Asn Ser Gln Glu Ser Val Thr Glu Gln Asp Ser
Lys Asp Ser 180 185 190Thr Tyr Ser Leu Ser Ser Thr Leu Thr Leu Ser
Lys Ala Asp Tyr Glu 195 200 205Lys His Lys Val Tyr Ala Cys Glu Val
Thr His Gln Gly Leu Ser Ser 210 215 220Pro Val Thr Lys Ser Phe Asn
Arg Gly Glu Cys225 230 235245PRTHomo sapiens 24Ser Phe Ser Met Thr1
52517PRTHomo sapiens 25Tyr Ile Ser Ser Arg Ser Ser Thr Ile Ser Tyr
Ala Asp Ser Val Lys1 5 10 15Gly2612PRTHomo sapiens 26Asp Pro Leu
Leu Ala Gly Ala Thr Phe Phe Asp Tyr1 5 102712PRTHomo sapiens 27Arg
Ala Ser Gln Ser Val Ser Ser Ser Tyr Leu Ala1 5 10287PRTArtificial
SequenceMCSF antibody CDRL2 sequence 28Gly Ala Ser Ser Arg Ala Thr1
5299PRTHomo sapiens 29Gln Gln Tyr Gly Ser Ser Pro Leu Thr1
530141PRTArtificial SequenceMCSF antibody heavy chain variable
region sequence 30Met Glu Leu Gly Leu Cys Trp Val Phe Leu Val Ala
Ile Leu Glu Gly1 5 10 15Val Gln Cys Glu Val Gln Leu Val Glu Ser Gly
Gly Gly Leu Val Gln 20 25 30Pro Gly Gly Ser Leu Arg Leu Ser Cys Ala
Ala Ser Gly Phe Thr Phe 35 40 45Ser Ser Phe Ser Met Thr Trp Val Arg
Gln Ala Pro Gly Lys Gly Leu 50 55 60Glu Trp Val Ser Tyr Ile Ser Ser
Arg Ser Ser Thr Ile Ser Tyr Ala65 70 75 80Asp Ser Val Lys Gly Arg
Phe Thr Ile Ser Arg Asp Asn Ala Lys Asn 85 90 95Ser Leu Tyr Leu Gln
Met Asn Ser Leu Arg Asp Glu Asp Thr Ala Val 100 105 110Tyr Tyr Cys
Ala Arg Asp Pro Leu Leu Ala Gly Ala Thr Phe Phe Asp 115 120 125Tyr
Trp Gly Gln Gly Thr Leu Val Thr Val Ser Ser Ala 130 135
14031128PRTHomo sapiens 31Met Glu Thr Pro Ala Gln Leu Leu Phe Leu
Leu Leu Leu Trp Leu Pro1 5 10 15Asp Thr Thr Gly Glu Phe Val Leu Thr
Gln Ser Pro Gly Thr Leu Ser 20 25 30Leu Ser Pro Gly Glu Arg Ala Thr
Leu Ser Cys Arg Ala Ser Gln Ser 35 40 45Val Ser Ser Ser Tyr Leu Ala
Trp Tyr Gln Gln Lys Pro Gly Gln Ala 50 55 60Pro Arg Leu Leu Ile Tyr
Gly Ala Ser Ser Arg Ala Thr Gly Ile Pro65 70 75 80Asp Arg Phe Ser
Gly Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile 85 90 95Ser Arg Leu
Glu Pro Glu Asp Phe Ala Val Tyr Tyr Cys Gln Gln Tyr 100 105 110Gly
Ser Ser Pro Leu Thr Phe Gly Gly Gly Thr Lys Val Glu Ile Lys 115 120
125325PRTHomo sapiens 32Ser Tyr Ser Met Asn1 53317PRTHomo sapiens
33Tyr Ile Ser Ser Ser Ser Ser Thr Ile Asp Tyr Ala Asp Ser Val Lys1
5 10 15Gly349PRTHomo sapiens 34Glu Ser Gly Trp Tyr Leu Phe Asp Tyr1
53511PRTHomo sapiens 35Arg Ala Ser Gln Gly Ile Ser Ser Trp Leu Ala1
5 10367PRTHomo sapiens 36Ala Ala Ser Ser Leu Gln Ser1 5379PRTHomo
sapiens 37Gln Gln Tyr Asn Ser Tyr Pro Pro Thr1 538118PRTHomo
sapiens 38Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro
Gly Gly1 5 10 15Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe
Ser Ser Tyr 20 25 30Ser Met Asn Trp Val Arg Gln Ala Pro Gly Lys Gly
Leu Glu Trp Val 35 40 45Ser Tyr Ile Ser Ser Ser Ser Ser Thr Ile Asp
Tyr Ala Asp Ser Val 50 55 60Lys Gly Arg Phe Thr Ile Ser Arg Asp Asn
Ala Lys Asn Ser Leu Tyr65 70 75 80Leu Gln Met Asn Ser Leu Arg Asp
Glu Asp Thr Ala Val Tyr Tyr Cys 85 90 95Ala Arg Glu Ser Gly Trp Tyr
Leu Phe Asp Tyr Trp Gly Gln Gly Thr 100 105 110Leu Val Thr Val Ser
Ser 11539107PRTHomo sapiens 39Asp Ile Gln Met Thr Gln Ser Pro Ser
Ser Leu Ser Ala Ser Val Gly1 5 10 15Asp Arg Val Thr Ile Thr Cys Arg
Ala Ser Gln Gly Ile Ser Ser Trp 20 25 30Leu Ala Trp Tyr Gln Gln Lys
Pro Glu Lys Ala Pro Lys Ser Leu Ile 35 40 45Tyr Ala Ala Ser Ser Leu
Gln Ser Gly Val Pro Ser Arg Phe Ser Gly 50 55 60Ser Gly Ser Gly Thr
Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro65 70 75 80Glu Asp Phe
Ala Thr Tyr Tyr Cys Gln Gln Tyr Asn Ser Tyr Pro Pro 85 90 95Thr Phe
Gly Gly Gly Thr Lys Val Glu Ile Lys 100 10540444PRTHomo sapiens
40Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly1
5 10 15Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Ser Ser
Tyr 20 25 30Ser Met Asn Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu
Trp Val 35 40 45Ser Tyr Ile Ser Ser Ser Ser Ser Thr Ile Asp Tyr Ala
Asp Ser Val 50 55 60Lys Gly Arg Phe Thr Ile Ser Arg Asp Asn Ala Lys
Asn Ser Leu Tyr65 70 75 80Leu Gln Met Asn Ser Leu Arg Asp Glu Asp
Thr Ala Val Tyr Tyr Cys 85 90 95Ala Arg Glu Ser Gly Trp Tyr Leu Phe
Asp Tyr Trp Gly Gln Gly Thr 100 105 110Leu Val Thr Val Ser Ser Ala
Ser Thr Lys Gly Pro Ser Val Phe Pro 115 120 125Leu Ala Pro Cys Ser
Arg Ser Thr Ser Glu Ser Thr Ala Ala Leu Gly 130 135 140Cys Leu Val
Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp Asn145 150 155
160Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu Gln
165 170 175Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro
Ser Ser 180 185 190Asn Phe Gly Thr Gln Thr Tyr Thr Cys Asn Val Asp
His Lys Pro Ser 195 200 205Asn Thr Lys Val Asp Lys Thr Val Glu Arg
Lys Cys Cys Val Glu Cys 210 215 220Pro Pro Cys Pro Ala Pro Pro Val
Ala Gly Pro Ser Val Phe Leu Phe225 230 235 240Pro Pro Lys Pro Lys
Asp Thr Leu Met Ile Ser Arg Thr Pro Glu Val 245 250 255Thr Cys Val
Val Val Asp Val Ser His Glu Asp Pro Glu Val Gln Phe 260 265 270Asn
Trp Tyr Val Asp Gly Val Glu Val His Asn Ala Lys Thr Lys Pro 275 280
285Arg Glu Glu Gln Phe Asn Ser Thr Phe Arg Val Val Ser Val Leu Thr
290 295 300Val Val His Gln Asp Trp Leu Asn Gly Lys Glu Tyr Lys Cys
Lys Val305 310 315 320Ser Asn Lys Gly Leu Pro Ala Pro Ile Glu Lys
Thr Ile Ser Lys Thr 325 330 335Lys Gly Gln Pro Arg Glu Pro Gln Val
Tyr Thr Leu Pro Pro Ser Arg 340 345 350Glu Glu Met Thr Lys Asn Gln
Val Ser Leu Thr Cys Leu Val Lys Gly 355 360 365Phe Tyr Pro Ser Asp
Ile Ala Val Glu Trp Glu Ser Asn Gly Gln Pro 370 375 380Glu Asn Asn
Tyr Lys Thr Thr Pro Pro Met Leu Asp Ser Asp Gly Ser385 390 395
400Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys Ser Arg Trp Gln Gln
405 410 415Gly Asn Val Phe Ser Cys Ser Val Met His Glu Ala Leu His
Asn His 420 425 430Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro Gly Lys
435 44041214PRTHomo sapiens 41Asp Ile Gln Met Thr Gln Ser Pro Ser
Ser Leu Ser Ala Ser Val Gly1 5 10 15Asp Arg Val Thr Ile Thr Cys Arg
Ala Ser Gln Gly Ile Ser Ser Trp 20 25 30Leu Ala Trp Tyr Gln Gln Lys
Pro Glu Lys Ala Pro Lys Ser Leu Ile 35 40 45Tyr Ala Ala Ser Ser Leu
Gln Ser Gly Val Pro Ser Arg Phe Ser Gly 50 55 60Ser Gly Ser Gly Thr
Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro65 70 75 80Glu Asp Phe
Ala Thr Tyr Tyr Cys Gln Gln Tyr Asn Ser Tyr Pro Pro 85 90 95Thr Phe
Gly Gly Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala 100 105
110Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg
Glu Ala 130 135 140Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser
Gly Asn Ser Gln145 150 155 160Glu Ser Val Thr Glu Gln Asp Ser Lys
Asp Ser Thr Tyr Ser Leu Ser 165 170 175Ser Thr Leu Thr Leu Ser Lys
Ala Asp Tyr Glu Lys His Lys Val Tyr 180 185 190Ala Cys Glu Val Thr
His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser 195 200 205Phe Asn Arg
Gly Glu Cys 210
D00001
D00002
D00003
S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.