Microarray Synthesis And Assembly Of Gene-length Polynucleotides
Oleinikov; Andrew V.
U.S. patent application number 17/019448 was filed with the patent office on 2021-03-04 for microarray synthesis and assembly of gene-length polynucleotides. This patent application is currently assigned to Gen9, Inc.. The applicant listed for this patent is Gen9, Inc.. Invention is credited to Andrew V. Oleinikov.
| Application Number | 20210062185 17/019448 |
| Document ID | / |
| Family ID | 1000005220480 |
| Filed Date | 2021-03-04 |











View All Diagrams
| United States Patent Application | 20210062185 |
| Kind Code | A1 |
| Oleinikov; Andrew V. | March 4, 2021 |
MICROARRAY SYNTHESIS AND ASSEMBLY OF GENE-LENGTH POLYNUCLEOTIDES
Abstract
There is disclosed a process for in vitro synthesis and assembly of long, gene-length polynucleotides based upon assembly of multiple shorter oligonucleotides synthesized in situ on a microarray platform. Specifically, there is disclosed a process for in situ synthesis of oligonucleotide fragments on a solid phase microarray platform and subsequent, "on device" assembly of larger polynucleotides composed of a plurality of shorter oligonucleotide fragments.
| Inventors: | Oleinikov; Andrew V.; (Boston, MA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Assignee: | Gen9, Inc. Boston MA |
||||||||||
| Family ID: | 1000005220480 | ||||||||||
| Appl. No.: | 17/019448 | ||||||||||
| Filed: | September 14, 2020 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 16793632 | Feb 18, 2020 | 10774325 | ||
| 17019448 | ||||
| 15440293 | Feb 23, 2017 | 10640764 | ||
| 16793632 | ||||
| 14701957 | May 1, 2015 | 10450560 | ||
| 15440293 | ||||
| 13617685 | Sep 14, 2012 | 9051666 | ||
| 14701957 | ||||
| 13250207 | Sep 30, 2011 | 9023601 | ||
| 13617685 | ||||
| 12488662 | Jun 22, 2009 | 8058004 | ||
| 13250207 | ||||
| 10243367 | Sep 12, 2002 | 7563600 | ||
| 12488662 | ||||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | B01J 2219/00378 20130101; B01J 2219/00585 20130101; B01J 2219/00454 20130101; B01J 2219/00626 20130101; B82Y 30/00 20130101; B01J 2219/00689 20130101; B01J 2219/00677 20130101; B01J 2219/00641 20130101; B01J 2219/00605 20130101; B01J 2219/00722 20130101; C12Q 1/6837 20130101; C40B 40/06 20130101; B01J 2219/005 20130101; B01J 19/0046 20130101; B01J 2219/00675 20130101; B01J 2219/00432 20130101; B01J 2219/00713 20130101; B01J 2219/00659 20130101; B01J 2219/00596 20130101; C40B 50/14 20130101; B01J 2219/00385 20130101; B01J 2219/00639 20130101; C40B 80/00 20130101; B01J 2219/00608 20130101; C12N 15/1068 20130101; B01J 2219/00527 20130101 |
| International Class: | C12N 15/10 20060101 C12N015/10; B82Y 30/00 20060101 B82Y030/00; C40B 80/00 20060101 C40B080/00; B01J 19/00 20060101 B01J019/00; C40B 50/14 20060101 C40B050/14; C12Q 1/6837 20060101 C12Q001/6837 |
Claims
1. (canceled)
2. A method for producing a polynucleotide comprising a target sequence, the method comprising: (a) providing a plurality of double-stranded oligonucleotides, each double-stranded oligonucleotide comprising: (i) an internal sequence identical to a portion of the target sequence, wherein the internal sequence, at one or both ends, comprises an overlapping sequence region that: is identical to an overlapping sequence region of another double-stranded oligonucleotide in the plurality of double-stranded oligonucleotides; and comprises a Type II restriction endonuclease cleavage site; and (ii) one or more flanking sequences, wherein each flanking sequence comprises a Type II restriction endonuclease recognition site corresponding to the Type II restriction endonuclease cleavage site of the internal sequence; (b) digesting the plurality of double-stranded oligonucleotides with a Type II restriction endonuclease that recognizes the Type II restriction endonuclease recognition site; and (c) ligating the digested oligonucleotides to produce the polynucleotide comprising the target sequence.
3. The method of claim 2, wherein each double-stranded oligonucleotide in (a) comprises a flanking sequence at each end.
4. The method of claim 2, wherein the plurality of double-stranded oligonucleotides are digested in (b) with a Type IIS restriction endonuclease.
5. The method of claim 2, wherein the Type IIS restriction endonuclease includes at least one of MylI, BspMI, BsaXI, BsrI, BmrI, BtsI, and Fok1.
6. The method of claim 2, wherein the method does not comprise PCR amplification prior to step (c).
7. The method of claim 2, further comprising: purifying the digested oligonucleotides prior to ligation in (c); amplifying the polynucleotide comprising the target sequence; or a combination thereof.
8. A method for producing a polynucleotide comprising a target sequence, the method comprising: (a) synthesizing on a surface a plurality of single-stranded oligonucleotides, each single-stranded oligonucleotide comprising: (i) an internal sequence identical to or complementary to a portion of the target sequence, wherein the internal sequence, at one or both ends, comprises an overlapping sequence region that is identical to or complementary to an overlapping sequence region of another single-stranded oligonucleotide in the plurality of single-stranded oligonucleotides; and (ii) flanking sequences, one at each end of the internal sequence, each flanking sequence comprising a primer binding site, and at least one flanking sequence further comprising a Type II restriction endonuclease recognition site; (b) amplifying the plurality of single-stranded oligonucleotides; (c) digesting the amplified oligonucleotides with a Type II restriction endonuclease that recognizes the Type II restriction endonuclease recognition site; and (d) ligating the digested oligonucleotides to produce the polynucleotide comprising the target sequence.
9. The method of claim 8, wherein each single-stranded oligonucleotide in the plurality of single-stranded oligonucleotides comprises flanking sequences, one at each end of the internal sequence, each flanking sequence comprising: (i) a primer binding site; and (ii) a Type II restriction endonuclease recognition site.
10. The method of claim 8, wherein the plurality of single-stranded oligonucleotides has been synthesized on a surface.
11. The method of claim 10, wherein the surface is a microarray or wherein the surface comprises beads.
12. The method of claim 8, wherein the amplified oligonucleotides are digested in (c) with a Type IIS restriction endonuclease.
13. The method of claim 12, wherein the Type IIS restriction endonuclease includes at least one of MylI, BspMI, BsaXI, BsrI, BmrI, BtsI, and Fok1.
14. The method of claim 8, further comprising: purifying the digested oligonucleotides prior to ligation in (d); amplifying the polynucleotide comprising the target sequence; or a combination thereof.
15. A method for producing a polynucleotide comprising a target sequence, the method comprising: (a) providing a plurality of oligonucleotides, each oligonucleotide comprising: (i) an internal sequence identical to or complementary to a portion of the target sequence, wherein the internal sequence, at one or both ends, comprises an overlapping sequence region that is identical to or complementary to an overlapping sequence region of another oligonucleotide in the plurality of oligonucleotides; and (ii) flanking sequences, one at each end of the internal sequence, each flanking sequence comprising a primer binding site, and at least one flanking sequence further comprising a Type II restriction endonuclease recognition site; (b) amplifying the plurality of oligonucleotides; (c) digesting the amplified oligonucleotides with a restriction endonuclease that recognizes the Type II restriction endonuclease recognition site; and (d) ligating the digested oligonucleotides to produce the polynucleotide comprising the target sequence.
16. The method of claim 15, wherein each oligonucleotide in the plurality of oligonucleotides comprises flanking sequences, one at each end of the internal sequence, each flanking sequence comprising: (i) a primer binding site; and (ii) a Type IIS restriction endonuclease recognition site.
17. The method of claim 15, wherein the plurality of oligonucleotides has been synthesized on a surface.
18. The method of claim 17, wherein the surface is a microarray or comprises beads.
19. The method of claim 15, further comprising: purifying the digested oligonucleotides prior to ligation in (d); amplifying the polynucleotide comprising the target sequence; or a combination thereof.
20. The method of claim 15, wherein the amplified oligonucleotides are digested in (c) with a Type IIS restriction endonuclease.
21. The method of claim 20, wherein the Type IIS restriction endonuclease includes at least one of MylI, BspMI, BsaXI, BsrI, BmrI, BtsI, and Fok1.
Description
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application is a continuation of U.S. application Ser. No. 16/793,632 filed Feb. 18, 2020, which is a continuation of U.S. patent application Ser. No. 15/440,293 filed Feb. 23, 2017, now U.S. Pat. No. 10,640,764, which is a continuation of U.S. patent application Ser. No. 14/701,957 filed May 1, 2015, now U.S. Pat. No. 10,450,560, which is a continuation of U.S. patent application Ser. No. 13/617,685 filed Sep. 14, 2012, now U.S. Pat. No. 9,051,666, which is a continuation of U.S. patent application Ser. No. 13/250,207 filed Sep. 30, 2011, now U.S. Pat. No. 9,023,601, which is a continuation of U.S. patent application Ser. No. 12/488,662 filed Jun. 22, 2009, now U.S. Pat. No. 8,058,004, which is a continuation of U.S. patent application Ser. No. 10/243,367 filed Sep. 12, 2002, now U.S. Pat. No. 7,563,600, the content of all of which applications is incorporated herein by reference in their entirety.
TECHNICAL FIELD OF THE INVENTION
[0002] The present invention provides a process for in vitro synthesis and assembly of long, gene-length polynucleotides based upon assembly of multiple shorter oligonucleotides synthesized in situ on a microarray platform. Specifically, the present invention provides a process for in situ synthesis of oligonucleotide sequence fragments on a solid phase microarray platform and subsequent, "on chip" assembly of larger polynucleotides composed of a plurality of smaller oligonucleotide sequence fragments.
BACKGROUND OF THE INVENTION
[0003] In the world of microarrays, biological molecules (e.g., oligonucleotides, polypeptides and the like) are placed onto surfaces at defined locations for potential binding with target samples of nucleotides or receptors. Microarrays are miniaturized arrays of biomolecules available or being developed on a variety of platforms. Much of the initial focus for these microarrays have been in genomics with an emphasis of single nucleotide polymorphisms (SNPs) and genomic DNA detection/validation, functional genomics and proteomics (Wilgenbus and Lichter, J Mal. Med. 77:761, 1999; Ashfari et al., Cancer Res. 59:4759, 1999; Kurian et al., J Pathol. 187:267, 1999; Hacia, Nature Genetics 21 suppl.:42, 1999; Hacia et al., Mol. Psychiatry 3:483, 1998; and Johnson, Curr. Biol. 26:R171, 1998).
[0004] There are, in general, three categories of microarrays (also called "biochips" and "DNA Arrays" and "Gene Chips" but this descriptive name has been attempted to be a trademark) having oligonucleotide content. Most often, the oligonucleotide microarrays have a solid surface, usually silicon-based and most often a glass microscopic slide. Oligonucleotide microarrays are often made by different techniques, including (1) "spotting" by depositing single nucleotides for in situ synthesis or completed oligonucleotides by physical means (ink jet printing and the like), (2) photolithographic techniques for in situ oligonucleotide synthesis (see, for example, Fodor U.S. Patent '934 and the additional patents that claim priority from this priority document, (3) electrochemical in situ synthesis based upon pH based removal of blocking chemical functional groups (see, for example, Montgomery U.S. Pat. No. 6,092,302 the disclosure of which is incorporated by reference herein and Southern U.S. Pat. No. 5,667,667), and (4) electric field attraction/repulsion of fully-formed oligonucleotides (see, for example, Hollis et al., U.S. Pat. No. 5,653,939 and its duplicate Heller U.S. Pat. No. 5,929,208). Only the first three basic techniques can form oligonucleotides in situ e.g., building each oligonucleotide, nucleotide-by-nucleotide, on the microarray surface without placing or attracting fully formed oligonucleotides.
[0005] With regard to placing fully formed oligonucleotides at specific locations, various micro-spotting techniques using computer-controlled plotters or even ink-jet printers have been developed to spot oligonucleotides at defined locations. One technique loads glass fibers having multiple capillaries drilled through them with different oligonucleotides loaded into each capillary tube. Microarray chips, often simply glass microscope slides, are then stamped out much like a rubber stamp on each sheet of paper of glass slide. It is also possible to use "spotting" techniques to build oligonucleotides in situ. Essentially, this involves "spotting" relevant single nucleotides at the exact location or region on a slide (preferably a glass slide) where a particular sequence of oligonucleotide is to be built. Therefore, irrespective of whether or not fully formed oligonucleotides or single nucleotides are added for in situ synthesis, spotting techniques involve the precise placement of materials at specific sites or regions using automated techniques.
[0006] Another technique involves a photolithography process involving photomasks to build oligonucleotides in situ, base-by-base, by providing a series of precise photomasks coordinated with single nucleotide bases having light-cleavable blocking groups. This technique is described in Fodor et al., U.S. Pat. No. 5,445,934 and its various progeny patents. Essentially, this technique provides for "solid-phase chemistry, photolabile protecting groups, and photolithography . . . to achieve light-directed spatially-addressable parallel chemical synthesis."
[0007] The electrochemistry platform (Montgomery U.S. Pat. No. 6,092,302, the disclosure of which is incorporated by reference herein) provides a microarray based upon a semiconductor chip platform having a plurality of microelectrodes. This chip design uses Complimentary Metal Oxide Semiconductor (CMOS) technology to create high-density arrays of microelectrodes with parallel addressing for selecting and controlling individual microelectrodes within the array. The electrodes turned on with current flow generate electrochemical reagents (particularly acidic protons) to alter the pH in a small "virtual flask" region or volume adjacent to the electrode. The microarray is coated with a porous matrix for a reaction layer material. Thickness and porosity of the material is carefully controlled and biomolecules are synthesized within volumes of the porous matrix whose pH has been altered through controlled diffusion of protons generated electrochemically and whose diffusion is limited by diffusion coefficients and the buffering capacities of solutions. However, in order to function properly, the microarray biochips using electrochemistry means for in situ synthesis has to alternate anodes and cathodes in the array in order to generated needed protons (acids) at the anodes so that the protons and other acidic electrochemically generated acidic reagents will cause an acid pH shift and remove a blocking group from a growing oligomer.
Gene Assembly
[0008] The preparation of arbitrary polynucleotide sequences is useful in a "postgenomic" era because it provides any desirable gene oligonucleotide or its fragment, or even whole genome material of plasmids, phages and viruses. Such polynucleotides are long, such as in excess of 1000 bases in length. In vitro synthesis of oligonucleotides (given even the best yield conditions of phosphoramidite chemistry) would not be feasible because each base addition reaction is less than 100% yield. Therefore, researchers desiring to obtain long polynucleotides of gene length or longer had to turn to nature or gene isolation techniques to obtain polynucleotides of such length. For the purposes of this patent application, the term "polynucleotide" shall be used to refer to nucleic acids (either single stranded or double stranded) that are sufficiently long so as to be practically not feasible to make in vitro through single base addition. In view of the exponential drop-off in yields from nucleic acid synthesis chemistries, such as phosphoramidite chemistry, such polynucleotides generally have greater than 100 bases and often greater than 200 bases in length. It should be noted that many commercially useful gene cDNA's often have lengths in excess of 1000 bases.
[0009] Moreover, the term "oligonucleotides" or shorter term "oligos" shall be used to refer to shorter length single stranded or double stranded nucleic acids capable of in vitro synthesis and generally shorter than 150 bases in length. While it is theoretically possible to synthesize polynucleotides through single base addition, the yield losses make it a practical impossibility beyond 150 bases and certainly longer than 250 bases.
[0010] However, knowledge of the precise structure of the genetic material is often not sufficient to obtain this material from natural sources. Mature cDNA, which is a copy of an mRNA molecule, can be obtained if the starting material contains the desired mRNA. However, it is not always known if the particular mRNA is present in a sample or the amount of the mRNA might be too low to obtain the corresponding cDNA without significant difficulties. Also, different levels of homology or splice variants may interfere with obtaining one particular species of mRNA. On the other hand many genomic materials might be not appropriate to prepare mature gene (cDNA) due to exon-intron structure of genes in many different genomes.
[0011] In addition, there is a need in the art for polynucleotides not existing in nature to improve genomic research performance. In general, the ability to obtain a polynucleotide of any desired sequence just knowing the primary structure, for a reasonable price, in a short period of time, will significantly move forward several fields of biomedical research and clinical practice.
[0012] Assembly of long arbitrary polynucleotides from oligonucleotides synthesized by organic synthesis and individually purified has other problems. The assembly can be performed using PCR or ligation methods. The synthesis and purification of many different oligonucleotides by conventional methods (even using multi-channel synthesizers) are laborious and expensive procedures. The current price of assembled polynucleotide on the market is about $12-25 per base pair, which can be considerable for assembling larger polvnucleotides. Very often the amount of conventionally synthesized oligonucleotides would be excessive. This also contributes to the cost of the final product.
[0013] Therefore, there is a need in the art to provide cost-effective polynucleotides by procedures that are not as cumbersome and labor-intensive as present methods to be able to provide polynucleotides at costs below $1 per base or 1-20 times less than current methods. The present invention was made to address this need.
SUMMARY OF THE INVENTION
[0014] The present invention provides a process for the assembly of oligonucleotides synthesized on microarrays into a polynucleotide sequence. The desired target polynucleotide sequence is dissected into pieces of overlapping oligonucleotides. In the first embodiment these oligonucleotides are synthesized in situ, in parallel on a microarray chip in a non-cleavable form. A primer extension process assembles the target polynucleotides. The primer extension process uses starting primers that are specific for the appropriate sequences. The last step is PCR amplification of the final polynucleotide product. Preferably, the polynucleotide product is a cDNA suitable for transcription purposes and further comprising a promoter sequence for transcription.
[0015] The present invention provides a process for assembling a polynucleotide from a plurality of oligonucleotides comprising: [0016] (a) synthesizing or spotting a plurality of oligonucleotide sequences on a microarray device or bead device having a solid or porous surface, wherein a first oligonucleotide is oligo 1 and a second oligonucleotide is oligo 2 and so on, wherein the plurality of oligonucleotide sequences are attached to the solid or porous surface, and wherein the first oligonucleotide sequence has an overlapping sequence region of from about 10 to about 50 bases that is the same or substantially the same as a region of a second oligonucleotide sequence, and wherein the second oligonucleotide sequence has an overlapping region with a third oligonucleotide sequence and so on; [0017] (b) forming complementary oligo 1 by extending primer 1, wherein primer 1 is complementary to oligo 1; [0018] (c) disassociating complementary oligo 1 from oligo 1 and annealing complementary oligo 1 to both oligo 1 and to the overlapping region of oligo 2, wherein the annealing of complementary oligo 1 to oligo 2 serves as a primer for extension for forming complementary oligo 1+2; [0019] (d) repeating the primer extension cycles of step (c) until a full-length polynucleotide is produced; and [0020] (e) amplifying the assembled complementary full length polynucleotide to produce a full length polynucleotide in desired quantities.
[0021] Preferably, the solid or porous surface is in the form of a microarray device. Most preferably, the microarray device is a semiconductor device having a plurality of electrodes for synthesizing oligonucleotides in situ using electrochemical means to couple and decouple nucleotide bases. Preferably, the primer extension reaction is conducted through a sequential process of melting, annealing and then extension. Most preferably, the primer extension reaction is conducted in a PCR amplification device using the microarray having the plurality of oligonucleotides bound thereto.
[0022] The present invention further provides a process for assembling a polynucleotide from a plurality of oligonucleotides comprising: [0023] (a) synthesizing in situ or spotting a plurality of oligonucleotide sequences on a microarray device or bead device each having a solid or porous surface, wherein the plurality of oligonucleotide sequences are attached to the solid or porous surface, and wherein each oligonucleotide sequence has an overlapping region corresponding to a next oligonucleotide sequence within the sequence and further comprises two flanking sequences, one at the 3' end and the other at the 5' end of each oligonucleotide, wherein each flanking sequence is from about 7 to about 50 bases and comprising a primer region and a sequence segment having a restriction enzyme cleavable site; [0024] (b) amplifying each oligonucleotide using the primer regions of the flanking sequence to form double stranded (ds) oligonucleotides; [0025] (c) cleaving the oligonucleotide sequences at the restriction enzyme cleavable site; and [0026] (d) assembling the cleaved oligonucleotide sequences through the overlapping regions to form a full length polynucleotide.
[0027] Preferably, the flanking sequence is from about 10 to about 20 bases in length. Preferably, the restriction enzyme cleavable site is a class ii endonuclease restriction site sequence capable of being cleaved by its corresponding class II restriction endonuclease enzyme. Most preferably, the restriction endonuclease class II site corresponds to restriction sites for a restriction endonuclease class II enzyme selected from the group consisting of Mly I, BspM I, Bae I, BsaX I, Bsr I, Bmr I, Btr I, Bts I, Fok I, and combinations thereof. Preferably, the flanking sequence further comprises a binding moiety used to purify cleaved oligonucleotides from flanking sequences. Preferably, the process further comprises the step of labeling the flanking sequence during the amplification step (b) using primer sequences labeled with binding moieties. Most preferably, a binding moiety is a small molecule able to be captured, such as biotin captured by avidin or streptavidin, or fluorescein able to be captured by an anti-fluorescein antibody.
[0028] The present invention further provides a process for assembling a polynucleotide from a plurality of oligonucleotides comprising: [0029] (a) synthesizing in situ or spotting a plurality of oligonucleotide sequences on a microarray device or bead device each having a solid or porous surface, wherein the plurality of oligonucleotide sequences are attached to the solid or porous surface, and wherein each oligonucleotide sequence has an overlapping region corresponding to a next oligonucleotide sequence within the sequence, and further comprises a sequence segment having a cleavable linker moiety; [0030] (b) cleaving the oligonucleotide sequences at the cleavable linker site to cleave each oligonucleotide complex from the microarray or bead solid surface to form a soluble mixture of oligonucleotides, each having an overlapping sequence; and [0031] (c) assembling the oligonucleotide sequences through the overlapping regions to form a full length polynucleotide.
[0032] Preferably, the cleavable linker is a chemical composition having a succinate moiety bound to a nucleotide moiety such that cleavage produces a 3'hydroxy nucleotide. Most preferably, the cleavable linker is selected from the group consisting of 5'-dimethoxytrityl-thymidine-3'succinate, 4-N-benzoyl-5'-dimethoxytrityl-deoxycytidine-3'-succinate, 1-N-benzoyl-5'-dimethoxytrityl-deoxyadenosine-3'-succinate, 2-N-isobutyryl-5'-dimethoxytrityl-deoxyguanosine-3'-succinate, and combinations thereof.
[0033] The present invention further provides a process for assembling a polynucleotide from a plurality of oligonucleotides comprising: [0034] (a) synthesizing in situ or spotting a plurality of oligonucleotide sequences on a microarray device or bead device each having a solid or porous surface, wherein the plurality of oligonucleotide sequences are attached to the solid or porous surface, and wherein each oligonucleotide sequence has a flanking region at an end attached to the solid or porous surface, and a specific region designed by dissecting the polynucleotide sequence into a plurality of overlapping oligonucleotides, wherein a first overlapping sequence on a first oligonucleotide corresponds to a second overlapping sequence of a second oligonucleotide, and wherein the flanking sequence comprises a sequence segment having a restriction endonuclease (RE) recognition sequence capable of being cleaved by a corresponding RE enzyme; [0035] (b) hybridizing an oligonucleotide sequence complementary to the flanking region to form a double stranded sequence capable of interacting with the corresponding RE enzyme; [0036] (c) digesting the plurality of oligonucleotides to cleave them from the microarray device or beads into a solution; and [0037] (d) assembling the oligonucleotide mixture through the overlapping regions to form a full length polynucleotide.
[0038] Preferably, the flanking sequence is from about 10 to about 20 bases in length. Preferably, the restriction enzyme cleavable site is a class II endonuclease restriction site sequence capable of being cleaved by its corresponding class II restriction endonuclease enzyme. Most preferably, the restriction endonuclease class II site corresponds to restriction sites for a restriction endonuclease class II enzyme selected from the group consisting of Mly I, BspM I, Bae I, BsaX I, Bsr I, Bmr I, Btr I, Bts I, Fok I, and combinations thereof. Preferably, the process further comprises a final step of amplifying the polynucleotide sequence using primers located at both ends of the polynucleotide.
[0039] The present invention further provides a process for creating a mixture of oligonucleotide sequences in solution comprising: [0040] (a) synthesizing in situ or spotting a plurality of oligonucleotide sequences on a microarray device or bead device each having a solid or porous surface, wherein the plurality of oligonucleotide sequences are attached to the solid or porous surface, and wherein each oligonucleotide sequence further comprises two flanking sequences, one at the 3' end and the other at the 5' end of each oligonucleotide, wherein each flanking sequence is from about 7 to about 50 bases and comprising a primer region and a sequence segment having a restriction enzyme cleavable site; [0041] (b) amplifying each oligonucleotide using the primer regions of the flanking sequence to form a double stranded (ds) oligonucleotides; and [0042] (c) cleaving the double stranded oligonucleotide sequences at the restriction enzyme cleavable site.
[0043] Preferably, the flanking sequence is from about 10 to about 20 bases in length. Preferably, the restriction enzyme cleavable site is a class II endonuclease restriction site sequence capable of being cleaved by its corresponding class II restriction endonuclease enzyme. Most preferably, the restriction endonuclease class II site corresponds to restriction sites for a restriction endonuclease class II enzyme selected from the group consisting of Mly I, BspM I, Bae I, BsaX I, Bsr I, Bmr I, Btr I, Bts I, Fok I, and combinations thereof. Preferably, the flanking sequence further comprises a binding moiety used to purify cleaved oligonucleotides from flanking sequences. Preferably, the process further comprises the step of labeling the flanking sequence during the amplification step (h) using primer sequences labeled with binding moieties. Most preferably, a binding moiety is a small molecule able to be captured, such as biotin captured by avidin or streptavidin, or fluorescein able to be captured by an anti-fluorescein antibody.
[0044] The present invention further provides a process for creating a mixture of oligonucleotide sequences in solution comprising: [0045] (a) synthesizing in situ or spotting a plurality of oligonucleotide sequences on a microarray device or bead device each having a solid or porous surface, wherein the plurality of oligonucleotide sequences are attached to the solid or porous surface, and wherein each oligonucleotide sequence has a sequence segment having a cleavable linker moiety; [0046] (b) cleaving the oligonucleotide sequences at the cleavable linker site to cleave each oligonucleotide sequence from the microarray or bead solid surface to form a soluble mixture of oligonucleotides.
[0047] Preferably, the cleavable linker is a chemical composition having a succinate moiety bound to a nucleotide moiety such that cleavage produces a 3'hydroxy nucleotide. Most preferably, the cleavable linker is selected from the group consisting of 5'-dimethoxytrityl-thymidine-3'succinate, 4-N-benzoyl-5'-dimethoxytrityl-deoxycytidine-3'-succinate, 1-N-benzoyl-5'-dimethoxytrityl-deoxyadenosine-3'-succinate, 2-N-isobutyryl-5'-dimethoxytrityl-deoxyguanosine-3'-succinate, and combinations thereof.
[0048] The present invention further provides a process for creating a mixture of oligonucleotide sequences in solution comprising: [0049] (a) synthesizing in situ or spotting a plurality of oligonucleotide sequences on a microarray device or bead device each having a solid or porous surface, wherein the plurality of oligonucleotide sequences are attached to the solid or porous surface, and wherein each oligonucleotide sequence has a flanking region at an end attached to the solid or porous surface, and a specific region, wherein the flanking sequence comprises a sequence segment having a restriction endonuclease (RE) recognition sequence capable of being cleaved by a corresponding RE enzyme; [0050] (b) hybridizing an oligonucleotide sequence complementary to the flanking region to form a double stranded sequence capable of interacting with the corresponding RE enzyme; [0051] (c) digesting the plurality of oligonucleotides to cleave them from the microarray device or beads into a solution.
[0052] Preferably, the flanking sequence is from about 10 to about 20 bases in length. Preferably, the restriction enzyme cleavable site is a class II endonuclease restriction site sequence capable of being cleaved by its corresponding class II restriction endonuclease enzyme. Most preferably, the restriction endonuclease class II site corresponds to restriction sites for a restriction endonuclease class II enzyme selected from the group consisting of Mly I, BspM I, Bae I, BsaX I, Bsr I, Bmr I, Btr I, Bts I, Fok I, and combinations thereof.
BRIEF DESCRIPTION OF THE DRAWINGS
[0053] FIGS. 1A-1C show schematics of gene assembly on a microarray device surface or porous matrix. In FIG. 1A, the target gene sequence is dissected into number of overlapping oligonucleotides. The 3' and 5' are the ends of the shown strand. FIG. 1A also shows, relative to the target sequence, primer Pr1; extension product of primer Pr1, which is complementary to oligonucleotide 1; and extension product of complementary oligonucleotide 1, which is complementary to oligonucleotides 1+2. FIG. 1B illustrates one embodiment of the initial steps of an assembly process. In step 1 of assembly, Primer Pr1 is annealed to oligonucleotide 1 and extended by appropriate polymerase enzyme into product complementary to oligonucleotide 1. The second step is melting, re-annealing and extension (i.e., amplification) to lead to production of larger amount of Pr1 extension product (complementary oligonucleotide 1), re-association of the complementary oligonucleotide 1 with oligonucleotide 1, and to annealing of the complementary oligonucleotide 1 with oligonucleotide 2 followed by its extension into product complementary to oligonucleotides 1+2. FIG. 1C shows a continuation of the assembly process from FIG. 1B. Specifically, step 3 of the process (i.e., melting, re-annealing and extension) leads to the same products as step 2 plus a product complementary to oligonucleotides 1+2+3. Cycles (steps) are repeated until a full-length complementary polynucleotide is formed. The final step is preparation of the final target polynucleotide molecule in desirable amounts by amplification (i.e., PCR) using two primers complementary to the ends of this molecule (PrX and PrY).
[0054] FIG. 2 shows a second embodiment of the inventive gene assembly process using oligonucleotides synthesized in situ onto a microarray device, each having a flanking sequence region containing a restriction enzyme cleavage site, followed by a PCR amplification step and followed by a REIT restriction enzyme cleavage step.
[0055] FIGS. 3A-3C show schematics for gene assembly using oligos synthesized and then cleaved from a microarray device. Specifically, in FIG. 3A, oligonucleotide sequences are connected to the microarray device through a cleavable linker (CL) moiety. An example of a cleavable linker moiety is provided in FIG. 3A. The cleavable linkers are molecules that can withstand the oligonucleotide synthesis process (i.e., phosphoramidite chemistry) and then can be cleaved to release oligonucleotide fragments. Chemical cleavage at cleavable linker CL recreates usual 3' end of specific oligos 1 through N. These oligonucleotides are released into a mixture. The mixture of oligonucleotides is subsequently assembled into full-length polynucleotide molecules. FIG. 3B, oligonucleotide sequences are connected to the microarray device through additional flanking sequence containing a restriction enzyme (RE) sequence site. Another oligonucleotide sequence, complementary to the flanking sequence region, is hybridized to the oligonucleotides on the microarray device. This recreates a "ds" or double-stranded oligonucleotide structure, each having a RE sequence recognition region in the flanking sequence region. Digestion of this ds oligonucleotides with the corresponding RE enzymes at the RE recognition sites in the flanking sequence regions releases the specific oligonucleotides 1 through N. When assembled, oligonucleotide sequences 1 through N form a full-length polynucleotide molecule. FIG. 3C: Cleavable linker for oligonucleotide synthesis.
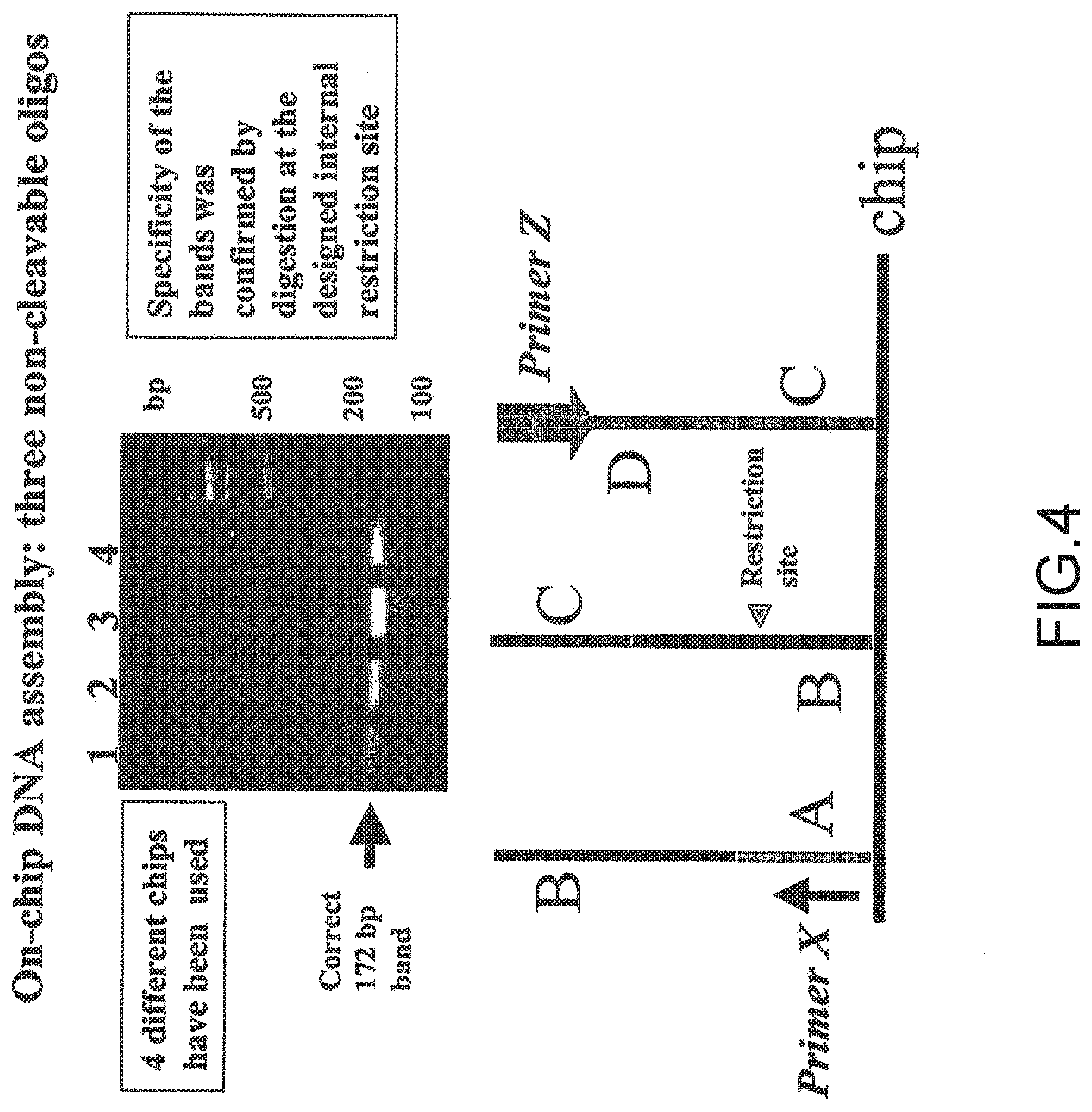
[0056] FIG. 4 shows the assembly of a polynucleotide from three oligonucleotide fragments wherein each oligonucleotide fragment was synthesized in situ on a microarray device. The fully assembled polynucleotide was 172 mers in length, a length not practically achievable by in situ synthesis. The first embodiment inventive process was used in this example.
[0057] FIG. 5 shows the oligonucleotide sequences (SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3) used to assemble the 172-mer polynucleotide (SEQ ID NO: 4) of FIG. 4. The sequences of primers X and Z are underlined. The Hpa II restriction site is indicated by italic underlined letters.
[0058] FIG. 6 shows a scheme for preparing the sequences of flanking regions (SEQ ID NO: 52 and SEQ ID NO: 53) and primers used for preparation of specific oligonucleotide for assembly using the REII enzyme MlyI. Primer 1 is complementary to the oligonucleotide strand on a microarray device and contains a Biotin-TEG (triethylene glycol) moiety. Primer 2 is the same strand as the oligonucleotide strand on microarray device and contains Biotin-TEG moiety. Any sequence between the primers can be used and is just designated by a string of N's.
[0059] FIG. 7 shows the results of PCR and Mly I digestion of an oligonucleotide sequence as described in FIG. 6. The clean bands show the ability to obtain pure oligonucleotides using the second embodiment of the inventive process to cleave off oligonucleotide sequences using appropriate restriction enzymes.
[0060] FIG. 8 shows the sequences from nine oligonucleotides fragments (consecutively numbered 1-9 and having SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, SEQ ID NO: 8, SEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, and SEQ ID NO: 13, respectively) used to assemble a 290 bp polynucleotide. The flanking regions are shown in bold and underlined. The process used for polynucleotide assembly was the second embodiment. The overlapping regions further contained a cleavable site as the Mly I recognition site for the Mly I class II restriction endonuclease.
[0061] FIG. 9 shows a schematic in the top panel for assembling a polynucleotide from nine oligonucleotides. Nine oligonucleotide sequences, shown in FIG. 8, were amplified by PCR using primers 1 and 2 (as described in FIG. 6) into ds DNA fragments containing the same flanking regions and specific overlapping sequences, digested with Mly I enzyme to remove flanking sequences, and used for assembly of 290 by DNA fragment. The columns in the gel shown are M--markers, 1--negative control, assembly without primers FP1 and FP2, 2--negative control, assembly without specific oligos, 3--assembly of 290 bp fragment from specific oligos plus amplification with FP1 and FP2 primers. The band in column 3 shows a high efficiency of the inventive polynucleotide assembly process.
[0062] FIG. 10 shows a sequence of an assembled polynucleotide in Example 4, broken down into its component oligonucleotides (Fragment 1, SEQ ID NO: 14, Fragment 2, SEQ ID NO: 15, Fragment 3, SEQ ID NO: 16, Fragment 4, SEQ ID NO: 17, Fragment 5, SEQ ID NO: 18, Fragment 6, SEQ ID NO: 19, Fragment 7, SEQ ID NO: 20, Fragment 8, SEQ ID NO: 21, Fragment 9, SEQ ID NO: 22, Fragment 1F, SEQ ID NO: 23, Fragment 2F, SEQ ID NO: 24, Fragment 3F, SEQ NO: 25, Fragment 4F, SEQ ID NO: 26, Fragment 5F, SEQ ID NO: 27, Fragment 6F, SEQ ID NO: 28, Fragment 7F, SEQ ID NO: 29, Fragment 8F, SEQ ID NO: 30, Fragment 9F, SEQ ID NO: 31, Fragment 10F, SEQ ID NO: 32, Fragment 11F, SEQ ID NO: 33, Fragment 12F, SEQ ID NO: 34, Fragment 13F, SEQ ID NO: 35, Fragment 14F, SEQ ID NO: 36, Fragment 15F, SEQ ID NO: 37, Fragment 16F, SEQ ID NO: 38, Fragment 17F, SEQ ID NO: 39, Fragment 18F, SEQ ID NO: 40, Fragment 19F, SEQ ID NO: 41, Fragment 20F, SEQ ID NO: 42, Fragment 21F, SEQ ID NO: 43, Fragment 22F, SEQ ID NO: 44, Fragment 23F, SEQ ID NO: 45, Fragment 24F, SEQ ID NO: 46, Fragment 25F, SEQ ID NO: 47, Fragment 26F, SEQ ID NO: 48, Fragment 27F, SEQ ID NO: 49, Fragment 28F, SEQ ID NO: 50, Fragment 29F, SEQ ID NO: 51).
DETAILED DESCRIPTION OF THE INVENTION
[0063] The present invention describes the preparation of a polynucleotide sequence (also called "gene") using assembly of overlapping shorter oligonucleotides synthesized or spotted on microarray devices or on solid surface bead devices. The shorter oligonucleotides include sequence regions having overlapping regions to assist in assembly into the sequence of the desired polynucleotide. Overlapping regions refer to sequence regions at either a 3' end or a 5' end of a first oligonucleotide sequence that is the same as part of the second oligonucleotide and has the same direction (relative to 3' to 5' or 5' to 3' direction), and will hybridize to the 5' end or 3' end of a second oligonucleotide sequence or its complementary sequence (second embodiment), and a second oligonucleotide sequence to a third oligonucleotide sequence, and so on. In order to design or develop a microarray device or bead device to be used for polynucleotide assembly, the polynucleotide sequence is divided (or dissected) into a number of overlapping oligonucleotides segments, each with lengths preferably from 20 to 1000 bases, and most preferably from 20 to 200 bases (FIG. 1A). The overlap between oligonucleotide segments is 5 or more bases, preferably 15-25 bases to that proper hybridization of first to second, second to third, third to fourth and so on occurs. These oligonucleotides (or oligos) are preferably synthesized on a microarray device using any available method (i.e., electrochemical in situ synthesis, photolithography in situ synthesis, ink-jet printing, spotting, etc.). The direction of synthesis relative to the microarray device surface or porous matrix covering a microarray device can be from 3' to 5' or from 5' to 3'. Preferably, in situ synthesis is done in the 3' to 5' direction.
[0064] In the first embodiment the inventive gene/polynucleotide assembly process uses oligonucleotides immobilized on a microarray device. The microarray device itself or a porous reaction layer with immobilized oligonucleotides can be used for the inventive gene/polynucleotide assembly process.
[0065] With regard to FIG. 1B, the process comprises several repeated steps of melting, annealing and extension (FIG. 1B), which can be performed in any thermal cycler instrument. The cycling program is similar to the programs used for PCR. At the first step of gene/polynucleotide assembly, primer Pr1 is added and anneals to oligonucleotide 1 on the microarray device and then extends by appropriate polymerase enzyme into product complementary to oligonucleotide 1 (called complementary oligonucleotide 1). At the second step of the process the product complementary to oligonucleotide 1 is melted from oligonucleotide 1, primer Pr1 is annealed again to the oligonucleotide 1 as well as product complementary to oligonucleotide 1 is partially re-anneals to oligonucleotide 1 and partially anneals to oligonucleotide 2 due to an overlapping sequence region between oligonucleotide 1 and oligonucleotide 2. Extension of Pr1 leads to production of an additional amount of Pr1 extension product (complementary oligonucleotide 1). The annealing of the complementary oligonucleotide 1 to oligonucleotide 2 followed by its extension leads to product complementary to oligonucleotides 1+2 (called complementary oligonucleotides 1+2). Similarly, at step 3 of the process melting, re-annealing and extension lead to the same products as at step 2 plus a product complementary to oligonucleotides 1+2+3. These cycles of melting, annealing and extension are repeated until full-length polynucleotide is formed. The number of cycles should be equal or more than the number of oligos on microarray device. After formation, the final target polynucleotide molecule is amplified by a PCR process with two primers complementary to the ends of this molecule to the desirable amounts.
[0066] In a second embodiment, a plurality of oligonucleotides that together comprise (with overlapping regions) the target polynucleotide sequence are synthesized on a microarray device (or can be synthesized on beads as a solid substrate), wherein each oligonucleotide sequence further comprises flanking short sequence regions, wherein each flanking sequence region comprises one or a plurality of sequence sites for restriction endonuclease, preferably endonuclease class II (ERII) enzymes. Each oligonucleotide is amplified by PCR using appropriate oligonucleotide primers to the flanking sequence regions to form a preparation of a plurality of oligonucleotides. The preparation of oligonucleotides is treated then with appropriate REII enzyme(s) (specific to the restriction sequences in the flanking sequence regions) to produce flanking fragments and overlapping oligonucleotides that, together comprise the desired polynucleotide sequence. Flanking fragments and PCR primers are removed from the mixture, if desired, by different methods based on size or specific labeling of the PCR primers. The oligonucleotides resembling the desired target polynucleotide then assembled into the final target polynucleotide molecule using repetition of the primer extension method and PCR amplification of the final molecule.
[0067] Specifically, in the second embodiment, the assembly process initially uses oligonucleotides immobilized on a microarray device or beads, via immobilization techniques, such as spotting or ink-jet printing or by direct in situ synthesis of the microarray device using various techniques, such as photolithography or electrochemical synthesis. The overlapping oligonucleotide sequences are designed having an overlapping region and one or two flanking sequence regions comprising a restriction class II recognition site (FIG. 2A). The assembled oligonucleotides together comprise the target polynucleotide sequence.
[0068] The length of flanking sequences is at least the length of REII recognition site. The flanking sequences are designed to have minimal homology to the specific oligonucleotide sequences regions on the microarray device. The flanking sequences can be the same for each oligonucleotide fragment, or be two or more different sequences. For example, a pair of appropriate primers, called Pr1 and Pr2, was designed to amplify each oligonucleotide on a microarray device (FIG. 2) by PCR. Each primer may contain a binding moiety, such as biotin, that does not affect their ability to serve as primers. After PCR amplification the amplified ds copy of each oligonucleotide was present in the reaction mixture. This reaction mixture was treated with the appropriate REII enzyme or enzymes specific for the restriction sites in the flanking sequence regions. The digestion sites for REII were designed, after cleavage, to produce the desired specific oligonucleotide sequence fragments that, when assembled will form the target polynucleotide sequence. As a result of digestion a mixture of specific double stranded (ds) overlapping oligonucleotide sequence fragments resembling the structure of desired target polynucleotide, and ds flanking sequences were formed. If desired, these flanking sequences and residual primers are removed from the mixture using specific absorption through specific moieties introduced in the primers (such as, for example, by absorption on avidin beads for biotin-labeled, primers), or based on the size difference of the specific oligos and flanking sequences and primers. The mixture of specific oligonucleotide sequences resembling target gene sequence is used to assemble the final target polynucleotide molecule using repeated cycles of melting, self-annealing and polymerase extension followed by PCR amplification of the final target polynucleotide molecule with appropriate PCR primers designed to amplify. This final PCR amplification step is routinely done in the art and described in, for example, Mullis et al., Cold Spring Harb. Symp. Quant. Biol. 51 Pt 1:263-73, 1986; and Saiki et al,, Science 239:487-91, 1988. PCR amplification steps generally follow manufacturer's instructions. Briefly, A process for amplifying any target nucleic acid sequence contained in a nucleic acid or mixture thereof comprises treating separate complementary strands of the nucleic acid with a molar excess of two oligonucleotide primers and extending the primers with a thermostable enzyme to form complementary primer extension products which act as templates for synthesizing the desired nucleic acid sequence. The amplified sequence can be readily detected. The steps of the reaction can be repeated as often as desired and involve temperature cycling to effect hybridization, promotion of activity of the enzyme, and denaturation of the hybrids formed.
[0069] In another embodiment for the assembly step, oligonucleotide sequences that together comprise the target polynucleotide molecule are assembled using a ligase chain reaction as described in Au et al., Biochem. Biophys. Res. Commun. 248:200-3, 1998. Briefly, short oligonucleotides are joined through ligase chain reaction (LCR) in high stringency conditions to make "unit fragments" (Fifty microliters of reaction mixture contained 2.2 mM of each oligo, 8 units Pfu DNA ligase (Stratagene La Jolla, Calif.) and reaction buffer provided with the enzyme. LCR was conducted as follows: 95.degree. C. 1 min; 55.degree. C. 1.5 min, 70.degree. C. 1.5 min, 95.degree. C. 30 sec for 15 cycles; 55.degree. C. 2 min; 70.degree. C. 2 min, which are then fused to form a full-length gene sequence by polymerase chain reaction.
[0070] In another embodiment the ds oligonucleotide sequences are assembled after preparation by chain ligation cloning as described in Pachuk et al., Gene 243:19-25, 2000; and U.S. Pat. No. 6,143,527 (the disclosure of which is incorporated by reference herein). Briefly, chain reaction cloning allows ligation of double-stranded DNA molecules by DNA ligases and bridging oligonucleotides. Double-stranded nucleic acid molecules are denatured into single-stranded molecules. The ends of the molecules are brought together by hybridization to a template. The template ensures that the two single-stranded nucleic acid molecules are aligned correctly. DNA ligase joins the two nucleic acid molecules into a single, larger, composite nucleic acid molecule. The nucleic acid molecules are subsequently denatured so that the composite molecule formed by the ligated nucleic acid molecules and the template cease to hybridize to each. Each composite molecule then serves as a template for orienting unligated, single-stranded nucleic acid molecules. After several cycles, composite nucleic acid molecules are generated from smaller nucleic acid molecules. A number of applications are disclosed for chain reaction cloning including site-specific ligation of DNA fragments generated by restriction enzyme digestion, DNAse digestion, chemical cleavage, enzymatic or chemical synthesis, and PCR amplification.
[0071] With regard to the second embodiment of the inventive process (illustrated in FIG. 2), a target polynucleotide gene sequence (either strand) is divided into number of overlapping oligonucleotide sequences by hand or with a software program, as shown in FIGS. 1A-1C. These oligonucleotide sequences, plus flanking sequences A and B (having one or a plurality of restriction enzyme sites in the flanking region sequence), are synthesized (in situ) on microarray device, or on a bead solid surface using standard in situ synthesis techniques, or spotted (pre-synthesized) onto a microarray device using standard oligonucleotide synthesis procedures with standard spotting (e.g., computer-aided or ink jet printing) techniques. The oligonucleotide sequences are amplified, preferably using a PCR process with a pair of primers (Pr1 and Pr2). The primers are optionally labeled with specific binding moieties, such as biotin. The resulting amplified mixture of different amplified oligonucleotide sequences are double stranded (ds). The mixture of ds oligonucleotide sequences are treated with an appropriate restriction enzyme, such as an REII restriction enzyme (e.g.,Mly I enzyme), to produce mixture of different double stranded (ds) overlapping oligonucleotide sequences that can be assembled into the structure of the desired polynucleotide (gene) and ds flanking sequences. Optionally, the flanking sequences and residual primers are removed from the ds oligonucleotide sequence mixture, preferably by a process of specific absorption using specific binding moieties introduced in the primers (e.g., biotin), or by a process of size fractionation based on the size differences of the specific oligonucleotide sequences and flanking sequences. The mixture of specific oligonucleotide sequences is assembled, for example, by a process of repeated cycles of melting, self-annealing and polymerase extension followed by PCR amplification of the final molecule with appropriate PCR primers designed to amplify this complete molecule (e.g., as described in Mullis et al., Cold Spring Harb. Symp. Quant. Biol. 51 Pt 1:263-73, 1986; and Saiki et al., Science 239:487-91, 1988).
[0072] In yet another embodiment of the inventive process (illustrated in FIGS. 3A-3B), the oligonucleotide sequences comprising the target polynucleotide sequence are synthesized on a microarray device or bead solid support, each oligonucleotide having a cleavable linker moiety synthesized within the sequence, such that after synthesis, oligonucleotides can be cleaved from the microarray device into a solution. Examples of appropriate cleavable linker moieties are shown in FIG. 3A. In addition to this method of cleavage, a sequence containing RE enzyme site can be synthesized at the ends of oligonucleotides attached to the microarray device. These oligonucleotides on the microarray device then hybridize with an oligonucleotide complementary to this additional flanking sequence and treated with an RE enzyme specific for the RE enzyme site. This process releases oligonucleotide fragments resembling the structure of the target polynucleotide. This set of oligonucleotides then can be assembled into the final polynucleotide molecule using any one of the methods or combination of the methods of ligation, primer extension and PCR.
[0073] In a third embodiment of the inventive process, a plurality of oligonucleotides that can be assembled into a full length polynucleotide are synthesized on a microarray device (or beads having a solid surface) having specific cleavable linker moieties (FIG. 3A) or capable of being cleaved from the solid support of the microarray device or beads by a chemical treatment. The net effect is to recreate the functional 3' ends and 5' ends of each specific oligonucleotide sequence. After treatment to cleave them, the oligonucleotides (each having overlapping regions) are released into a mixture and used for full-length polynucleotide gene assembly using any of the gene assembly processes described herein.
[0074] Specifically, in the third embodiment and as illustrated in FIGS. 3A-3B, a target polynucleotide sequence is dissected into number of overlapping oligonucleotide sequences by a software program or on paper, but not necessarily physically in a laboratory. These oligonucleotide sequences are physically synthesized on a microarray device. In alternative A, the oligonucleotide sequences are connected to the microarray device through cleavable linker moiety. Chemical cleavage under basic conditions (e.g., through addition of ammonia), at cleavable linker CL recreates the usual 3' end of the specific oligonucleotide sequences 1 through N. Oligonucleotide sequences 1 through N are released into a mixture. The mixture of oligonucleotide sequences is used for polynucleotide assembly.
[0075] In alternative B, oligonucleotide sequences are connected to a microarray device through additional flanking sequence regions containing a restriction enzyme (RE) sequence site. A second oligonucleotide fragment, complementary to the flanking sequence, is hybridized to the oligonucleotides on the microarray device. This recreates a ds structure at the flanking sequence region, including the RE recognition site. Digestion of this ds DNA structure with RE enzyme specific to the RE recognition site in the flanking sequence region will release specific oligonucleotides 1 through N into a mixture solution. The oligonucleotides l through N are able to assemble into a polynucleotide molecule in solution.
[0076] In another example of alternative B, oligonucleotides that together assemble into the polynucleotide are synthesized on a microarray device, each having a flanking sequence on the microarray side. The flanking sequence further comprises a restriction endonuclease (RE) recognition site (see FIG. 3B). Oligonucleotides complementary to the flanking sequence region are added and hybridized to the oligonucleotides on microarray device. After hybridization a RE (restriction enzyme specific to the RE sequence in the flanking region) is added to the microarray device. Specific oligonucleotide sequences are released from the microarray device as a result of RE digestion into a mixture. The mixture of specific oligonucleotide sequences ARE assembled into the full-length polynucleotide sequence.
Example 1
[0077] This example illustrates assembly of 172-mer polynucleotide sequence from non-cleavable oligonucleotide sequences synthesized on a microarray device according to the first embodiment inventive process (FIGS. 4 and 5). Three oligonucleotides (sequences shown in FIG. 5) were synthesized in situ on a microarray device according to an electrochemical process (see U.S. Pat. No. 6,093,302, the disclosure of which is incorporated by reference herein). The oligonucleotide sequences synthesized were amplified by a PCR reaction with primers X (complementary to the strand of oligo #1) and Z (same strand as Oligo #3) (FIG. 5). After 45 cycles of PCR using a PCR kit with AmplyGold.RTM. enzyme (Applied Biosystems) a correct DNA fragment of 172 by was synthesized (FIG. 4). Its subsequent digestion confirmed the specificity of this enzyme with Hpa II producing two fragments of 106 bp and 68 bp.
Example 2
[0078] This example illustrates the second embodiment of the inventive process for preparing oligonucleotides for assembly into full-length polynucleotides by PCR and REII (restriction enzyme) digestion. A single oligonucleotide sequence was synthesized on a microarray device according to the procedure in Example 1 (see FIGS. 2 and 6). The oligonucleotide sequence further comprised 2 flanking sequences, each having a recognition site for a Mly I restriction enzyme. This microarray device was subject to a PCR (25 cycles) reaction with two primers (shown in FIG. 7) to produce an amplified PCR fragment mixture. The amplified PM fragment obtained was digested by Mly I restriction enzyme and purified by a PCR purification kit (Qiagen) to produce specific oligonucleotides ready for assembly (FIG. 7). Similarly, this specific oligonucleotide was purified from the flanking sequences by absorption of the digestion mixture by Streptavidin-agarose (Sigma).
Example 3
[0079] This example illustrates the assembly of a 290 bp polynucleotide sequence from 9 oligonucleotide sequences, each having flanking sequences containing a MlyI restriction site. Each of the nine different oligonucleotide sequences was synthesized on a microarray device through an in situ electrochemistry process as described in example 1 herein.
[0080] The microarray device containing the nine specific oligonucleotide sequences (with flanking sequences as shown in FIG. 8) was used for PCR amplification of each oligonucleotide sequence using two primers, Primer 1 and 2, described in FIG. 6 to form a mixture of ds oligonucleotide sequences. The primers were complementary to the flanking sequences. The mixture of the amplified ds oligonucleotide sequences was digested by Mly I enzyme. Specific ds oligonucleotide sequences were purified and then assembled into the final 290 bp polynucleotide sequence in two steps as described in FIG. 2 and shown schematically in FIG. 9. At the first step of assembly 20 cycles of melting-annealing-extension were used. The final product was amplified using two primers FP1 and FP2 (FIG. 9) in 25 cycles of PCR into a 290 bp polynucleotide DNA.
Example 4
[0081] This example illustrates the creation of a cDNA polynucleotide sequence capable of coding on expression for fusion protein MIP-GFP-FLAG (Macrophage Inflammation Protein-Green Fluorescence Protein-FLAG peptide) using thirty-eight overlapping oligonucleotide sequences (FIG. 10). The 38 oligonucleotides were synthesized on a microarray device using an electrochemical in situ synthesis approach, as described in example 1. Each oligonucleotide sequence contained a cleavable linker moiety (see FIG. 3A) at their 3' end. After simultaneous deprotection and cleavage of these oligonucleotide sequences by concentrated ammonia, the mixture of oligonucleotide sequences was purified by gel-filtration through the spin column. The purified oligonucleotide sequences were assembled into a polynucleotide by a process shown schematically in FIGS. 3A-3B. The resulting DNA polynucleotide was 965 bp and contained both a T7 RNA-polymerase promoter and a coding sequence for MIP-GFP-FLAG fusion protein. The polynucleotide assembled in this example was used in a standard transcription/translation reaction and produced the appropriate MIP-GFP-FLAG fusion protein. The translated protein was purified from the reaction mixture using anti-FLAG resin (Sigma). The functional protein possessed green fluorescence signal in appropriate blue light. Accordingly, this experiment demonstrated that the inventive gene assembly process provided the correct DNA sequence coding for the functional protein.
Sequence CWU 1
1
53182DNAArtificial SequenceSynthetic construct 1taattatgct
gagtgatatc cctttctacc tgtgcggctg gcggacgacg aagtcgaatg 60tggagggccg
tctaaggtgt ct 82284DNAArtificial SequenceSynthetic construct
2ggacgacgaa gtcgaatgtg gagggccgtc taaggtgtct taaagtatcg actgatgaaa
60ctctgctcgt cggtcacgag gttc 84386DNAArtificial SequenceSynthetic
construct 3gtatcgactg atgaaactct gctcgtcggt cacgaggttc cctcgaccac
cgcatgatgt 60ttctgctact gctgttcacg attatc 864142DNAArtificial
SequenceSynthetic construct 4taattatgct gagtgatatc cctttctacc
tgtgcggctg aagtcgaatg tggagggccg 60tctaaggtgt cttaaagtat aactctgctc
gtcggtcacg aggttccctc gaccaccgca 120gctactgctg ttcacgatta tc
142589DNAArtificial SequenceSynthetic construct 5ccatcacgct
gagtcttacg tacgtaatac gactcactat agggaaagtc gccaccatgg 60acacgccgac
gagacgactc ctaatcgaa 89685DNAArtificial SequenceSynthetic construct
6ccatcacgct gagtcttacg cgcctgctgc ttcagctaca cctcccggca gattccacag
60aatttcgaga cgactcctaa tcgaa 85787DNAArtificial SequenceSynthetic
construct 7ccatcacgct gagtcttacg atagctgact actttgagac gagcagccag
tgctccaagc 60ccggtgtcga gacgactcct aatcgaa 87881DNAArtificial
SequenceSynthetic construct 8ccatcacgct gagtcttacg atcttcctaa
ccaagcgaag ccggcaggtc tgtgctgacc 60ccgagacgac tcctaatcga a
819130DNAArtificial SequenceSynthetic construct 9ccatcacgct
gagtcttacg caggcactca gctctacggg gccgtcgccg atgggggtgt 60tctgctggta
gtggtcggcg agctgcatat ttctggaccc actcctcact gagacgactc
120ctaatcgaac 1301083DNAArtificial SequenceSynthetic construct
10ccatcacgct gagtcttacg atatttctgg acccactcct cactggggtc agcacagacc
60tgccgagacg actcctaatc gaa 831184DNAArtificial SequenceSynthetic
construct 11ccatcacgct gagtcttacg ggcttcgctt ggttaggaag atgacaccgg
gcttggagca 60ctggcgagac gactcctaat cgaa 841284DNAArtificial
SequenceSynthetic construct 12ccatcacgct gagtcttacg tgctcgtctc
aaagtagtca gctatgaaat tctgtggaat 60ctgccgagac gactcctaat cgaa
841388DNAArtificial SequenceSynthetic construct 13ccatcacgct
gagtcttacg gggaggtgta gctgaagcag caggcggtcg gcgtgtccat 60ggtggcgacg
agacgactcc taatcgaa 881450DNAArtificial SequenceSynthetic construct
14tacgtaatac gactcactat agggaaagtc gccaccatgg acacgccgac
501546DNAArtificial SequenceSynthetic construct 15cgcctgctgc
ttcagctaca cctcccggca gattccacag aatttc 461648DNAArtificial
SequenceSynthetic construct 16atagctgact actttgagac gagcagccag
tgctccaagc ccggtgtc 481742DNAArtificial SequenceSynthetic construct
17atcttcctaa ccaagcgaag ccggcaggtc tgtgctgacc cc
421847DNAArtificial SequenceSynthetic construct 18agtgaggagt
gggtccagaa atatgtcagc gacctagagc tgagtgc 471944DNAArtificial
SequenceSynthetic construct 19atatttctgg acccactcct cactggggtc
agcacagacc tgcc 442045DNAArtificial SequenceSynthetic construct
20ggcttcgctt ggttaggaag atgacaccgg gcttggagca ctggc
452145DNAArtificial SequenceSynthetic construct 21tgctcgtctc
aaagtagtca gctatgaaat tctgtggaat ctgcc 452249DNAArtificial
SequenceSynthetic construct 22gggaggtgta gctgaagcag caggcggtcg
gcgtgtccat ggtggcgac 492354DNAArtificial SequenceSynthetic
construct 23ggtgaacagc tcctcgccct tgctcaccat ggcactcagc tctaggtcgc
tgac 542452DNAArtificial SequenceSynthetic construct 24catggtgagc
aagggcgagg agctgttcac cggggtggtg cccatcctgg tc 522550DNAArtificial
SequenceSynthetic construct 25ttgtggccgt ttacgtcgcc gtccagctcg
accaggatgg gcaccacccc 502651DNAArtificial SequenceSynthetic
construct 26gagctggacg gcgacgtaaa cggccacaag ttcagcgtgt ccggcgaggg
c 512745DNAArtificial SequenceSynthetic construct 27ttgccgtagg
tggcatcgcc ctcgccctcg ccggacacgc tgaac 452848DNAArtificial
SequenceSynthetic construct 28gagggcgatg ccacctacgg caagctgacc
ctgaagttca tctgcacc 482948DNAArtificial SequenceSynthetic construct
29cagggcacgg gcagcttgcc ggtggtgcag atgaacttca gggtcagc
483054DNAArtificial SequenceSynthetic construct 30accggcaagc
tgcccgtgcc ctggcccacc ctcgtgacca ccctgaccta cggc
543155DNAArtificial SequenceSynthetic construct 31ggggtagcgg
ctgaagcact gcacgccgta ggtcagggtg gtcacgaggg tgggc
553248DNAArtificial SequenceSynthetic construct 32gtgcagtgct
tcagccgcta ccccgaccac atgaagcagc acgacttc 483351DNAArtificial
SequenceSynthetic construct 33gtagccttcg ggcatggcgg acttgaagaa
gtcgtgctgc ttcatgtggt c 513451DNAArtificial SequenceSynthetic
construct 34ttcaagtccg ccatgcccga aggctacgtc caggagcgca ccatcttctt
c 513549DNAArtificial SequenceSynthetic construct 35gggtcttgta
gttgccgtcg tccttgaaga agatggtgcg ctcctggac 493648DNAArtificial
SequenceSynthetic construct 36aaggacgacg gcaactacaa gacccgcgcc
gaggtgaagt tcgagggc 483749DNAArtificial SequenceSynthetic construct
37agctcgatgc ggttcaccag ggtgtcgccc tcgaacttca cctcggcgc
493851DNAArtificial SequenceSynthetic construct 38gacaccctgg
tgaaccgcat cgagctgaag ggcatcgact tcaaggagga c 513951DNAArtificial
SequenceSynthetic construct 39tccagcttgt gccccaggat gttgccgtcc
tccttgaagt cgatgccctt c 514051DNAArtificial SequenceSynthetic
construct 40ggcaacatcc tggggcacaa gctggagtac aactacaaca gccacaacgt
c 514152DNAArtificial SequenceSynthetic construct 41gttcttctgc
ttgtcggcca tgatatagac gttgtggctg ttgtagttgt ac 524251DNAArtificial
SequenceSynthetic construct 42tatatcatgg ccgacaagca gaagaacggc
atcaaggtga acttcaagat c 514350DNAArtificial SequenceSynthetic
construct 43acgctgccgt cctcgatgtt gtggcggatc ttgaagttca ccttgatgcc
504449DNAArtificial SequenceSynthetic construct 44cgccacaaca
tcgaggacgg cagcgtgcag ctcgccgacc actaccagc 494551DNAArtificial
SequenceSynthetic construct 45acggggccgt cgccgatggg ggtgttctgc
tggtagtggt cggcgagctg c 514651DNAArtificial SequenceSynthetic
construct 46agaacacccc catcggcgac ggccccgtgc tgctgcccga caaccactac
c 514749DNAArtificial SequenceSynthetic construct 47tttgctcagg
gcggactggg tgctcaggta gtggttgtcg ggcagcagc 494850DNAArtificial
SequenceSynthetic construct 48tgagcaccca gtccgccctg agcaaagacc
ccaacgagaa gcgcgatcac 504951DNAArtificial SequenceSynthetic
construct 49ggcggtcacg aactccagca ggaccatgtg atcgcgcttc tcgttggggt
c 515051DNAArtificial SequenceSynthetic construct 50atggtcctgc
tggagttcgt gaccgccgcc gggatcactc tcggcatgga c 515149DNAArtificial
SequenceSynthetic construct 51ggcggccgct ttacttgtac agctcgtcca
tgccgagagt gatcccggc 495220DNAArtificial SequenceSynthetic
construct 52gagacgactc ctaatcgaac 205320DNAArtificial
SequenceSynthetic construct 53ccatcacgct gagtcttacg 20
D00000
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
D00012

D00013

D00014

S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.