Signalling System
Pule; Martin ; et al.
U.S. patent application number 16/848717 was filed with the patent office on 2021-02-04 for signalling system. The applicant listed for this patent is UCL BUSINESS LTD. Invention is credited to Shaun Cordoba, Khai Kong, Martin Pule.
| Application Number | 20210032332 16/848717 |
| Document ID | / |
| Family ID | 1000005162160 |
| Filed Date | 2021-02-04 |
View All Diagrams
| United States Patent Application | 20210032332 |
| Kind Code | A1 |
| Pule; Martin ; et al. | February 4, 2021 |
SIGNALLING SYSTEM
Abstract
The present invention provides a chimeric antigen receptor (CAR) signalling system comprising; (i) a receptor component comprising an antigen binding domain, a transmembrane domain and a first binding domain; and (ii) an intracellular signalling component comprising a signalling domain and a second binding domain which specifically binds the first binding domain of the receptor component; wherein, binding of the first and second binding domains is disrupted by the presence of an agent, such that in the absence of the agent the receptor component and the signalling component heterodimerize and binding of the antigen binding domain to antigen results in signalling through the signalling domain, whereas in the presence of the agent the receptor component and the signalling component do not heterodimerize and binding of the antigen binding domain to antigen does not result in signalling through the signalling domain.
| Inventors: | Pule; Martin; (London, GB) ; Cordoba; Shaun; (London, GB) ; Kong; Khai; (London, GB) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 1000005162160 | ||||||||||
| Appl. No.: | 16/848717 | ||||||||||
| Filed: | April 14, 2020 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 15506383 | Feb 24, 2017 | 10654927 | ||
| PCT/GB2015/052494 | Aug 28, 2015 | |||
| 16848717 | ||||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | C07K 2319/80 20130101; C07K 2317/622 20130101; C07K 16/2803 20130101; C12N 2510/00 20130101; A61K 35/17 20130101; C07K 14/70514 20130101; A61K 31/65 20130101; C12N 5/0636 20130101; C07K 14/7051 20130101 |
| International Class: | C07K 16/28 20060101 C07K016/28; A61K 35/17 20060101 A61K035/17; C07K 14/725 20060101 C07K014/725; C12N 5/0783 20060101 C12N005/0783; A61K 31/65 20060101 A61K031/65; C07K 14/73 20060101 C07K014/73 |
Foreign Application Data
| Date | Code | Application Number |
|---|---|---|
| Aug 29, 2014 | GB | 1415347.2 |
Claims
1. A chimeric antigen receptor (CAR) system comprising; (i) a receptor component comprising an antigen binding domain, a transmembrane domain and a first binding domain; and (ii) an intracellular signalling component comprising a signalling domain and a second binding domain which specifically binds the first binding domain of the receptor component; wherein, binding of the first and second binding domains is disruptable by the presence of an agent, such that in the absence of the agent the receptor component and the intracellular signalling component heterodimerize and binding of the antigen binding domain to antigen results in signalling through the signalling domain, whereas in the presence of the agent the receptor component and the intracellular signalling component do not heterodimerize and binding of the antigen binding domain to antigen does not result in signalling through the signalling domain.
2-3. (canceled)
4. The CAR system according to claim 1 wherein the first binding domain comprises Tet Repressor Protein (TetR) or a variant thereof and the second binding domain comprises Transcription inducing peptide (TiP) or a variant thereof; or wherein the first binding domain comprises TiP or a variant thereof and the second binding domain comprises TetR or a variant thereof; and the agent is tetracycline, doxycycline or minocycline or an analogue thereof .
5-10. (canceled)
11. The CAR system according to claim 1, wherein the signalling domain of the intracellular signalling component comprises a single endodomain selected from CD3 zeta endodomain, CD28 endodomain, 41 BB endodomain and OX40 endodomain.
12. The CAR system according to claim 1, wherein the signalling domain of the intracellular signalling component comprises at least one of CD3 zeta endodomain, CD28 endodomain, 41 BB endodomain and OX40 endodomain.
13-18. (canceled)
19. A nucleic acid comprising a nucleotide sequence encoding a CAR system according to claim 1, wherein the receptor component and intracellular signalling component are co-expressed, joined by a self-cleaving peptide which is cleaved between the receptor component and the intracellular signalling component after translation.
20. A vector comprising a nucleic acid according to claim 19.
21. (canceled)
22. A T cell or NK cell which expresses a CAR system according to claim 1.
23. (canceled)
24. A pharmaceutical composition comprising a plurality of T cells or NK cells according to claim 22.
25. (canceled)
26. A method for treating a disease, which comprises the step of administering a pharmaceutical composition according to claim 24 to a subject.
27. A method for treating a disease, which comprises the following steps: (i) isolating a T cell or NK cell containing sample; (ii) transducing or transfecting the T or NK cells with a nucleic acid sequence according to claim 19 or a vector comprising the nucleic acid; and (iii) administering the T cells or NK cells from (ii) to a subject.
28. A method according to claim 26, which involves monitoring toxic activity in the subject and comprises the step of administering an agent for use in the CAR system to the subject to reduce adverse toxic effects.
29. A method according to claim 27, which involves monitoring the progression of disease and/or monitoring toxic activity in the subject and comprises the step of administering an agent for use in the CAR system to the subject to provide acceptable levels of disease progression and/or toxic activity.
30. The method according to claim 26, wherein the disease is cancer.
31-32. (canceled)
33. A method for making a T cell or NK cell, which comprises the step of introducing a nucleic acid according to claim 19 or a vector comprising the nucleic acid into a T cell or NK cell.
34. (canceled)
35. A method for inhibiting the CAR system according to claim 1 in a subject which comprises a T or NK cell which expresses the CAR signaling system, which method comprises the step of administering the agent to the subject.
Description
FIELD OF THE INVENTION
[0001] The present invention relates to a chimeric antigen receptor signalling system.
BACKGROUND TO THE INVENTION
[0002] Traditionally, antigen-specific T-cells have been generated by selective expansion of peripheral blood T-cells natively specific for the target antigen. However, it is difficult and quite often impossible to select and expand large numbers of T-cells specific for most cancer antigens. Gene-therapy with integrating vectors affords a solution to this problem as transgenic expression of Chimeric Antigen Receptor (CAR) allows generation of large numbers of T cells specific to any surface antigen by ex vivo viral vector transduction of a bulk population of peripheral blood T-cells.
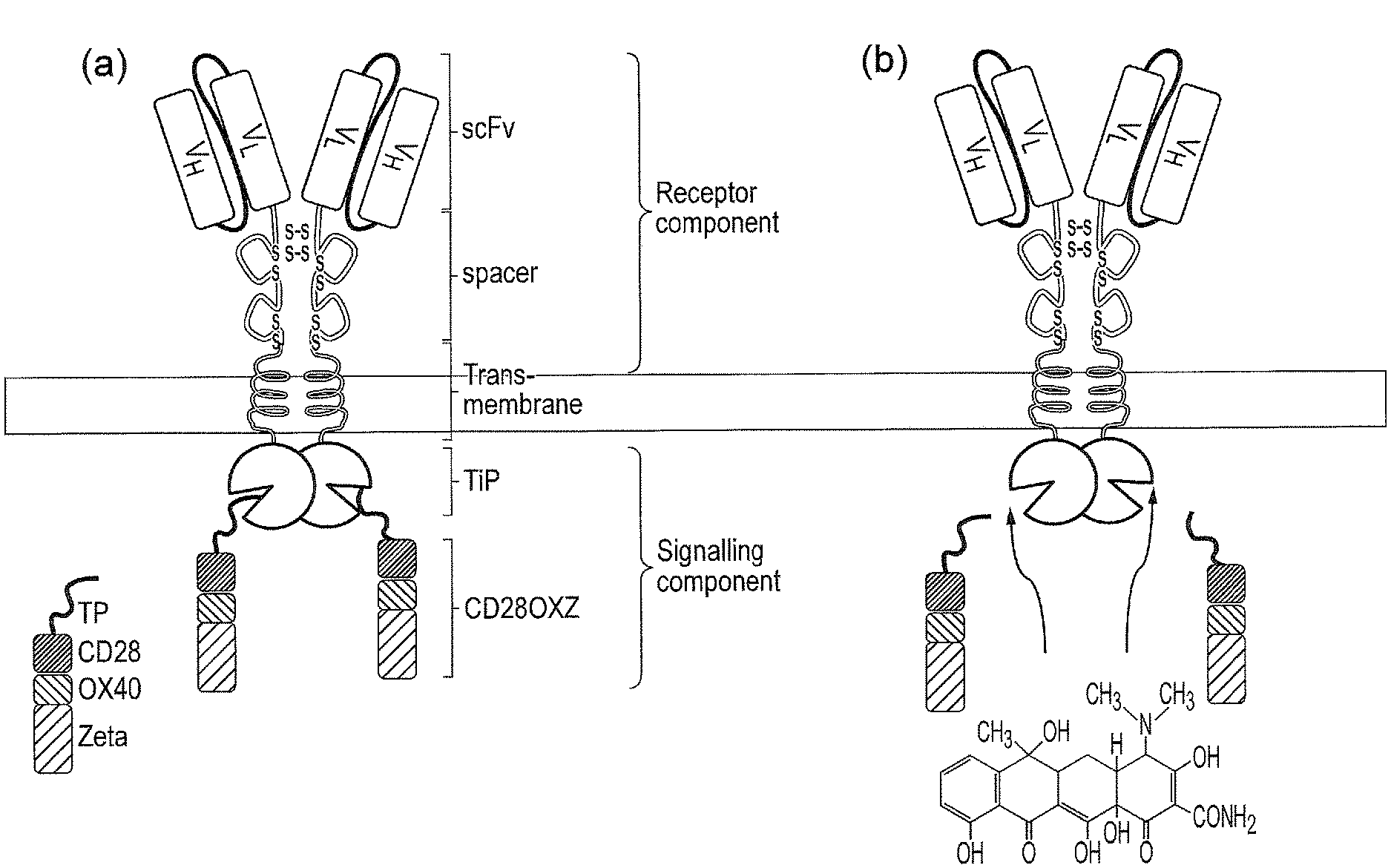
[0003] Chimeric antigen receptors are proteins which graft the specificity of a monoclonal antibody (mAb) to the effector function of a T-cell. Their usual form is that of a type I transmembrane domain protein with an antigen recognizing amino terminus, a spacer, a transmembrane domain all connected to a compound endodomain which transmits T-cell survival and activation signals (see FIG. 1A).
[0004] The most common forms of these molecules are fusions of single-chain variable fragments (scFv) derived from monoclonal antibodies which recognize a target antigen, fused via a spacer and a trans-membrane domain to a signalling endodomain. Such molecules result in activation of the T-cell in response to recognition by the scFv of its target. When T cells express such a CAR, they recognize and kill target cells that express the target antigen. Several CARs have been developed against tumour associated antigens, and adoptive transfer approaches using such CAR-expressing T cells are currently in clinical trial for the treatment of various cancers.
[0005] A number of toxicities have been reported from CAR studies, and additional theoretical toxicities exist. Such toxicities include immunological toxicity caused by sustained intense activation of the CAR T-cells resulting in a macrophage activation syndrome (MAS) and "On-target off-tumour" toxicity i.e. recognition of the target antigen on normal tissues. MAS is presumed to be caused by persistent antigen-driven activation and proliferation of T-cells which in turn release copious inflammatory cytokines leading to hyper-activation of macrophages and a feed-forward cycle of immune activation. A large spike in serum IL-6 is characteristic and the syndrome can result in a severe systemic illness requiring ICU admission.
[0006] On-target off-tumour toxicity has been reported with other CARs, for example a group of patients treated with a CAR against the renal cell carcinoma antigen CAIX developed unexpected and treatment limiting biliary toxicity. Two fatalities have been reported with CAR studies: one patient died of a respiratory distress syndrome which occurred immediately post-infusion of a large dose of 3rd generation anti-ERBB2 CAR T-cells; a further patient died in a different study after a possible cytokine storm following treatment of CLL with a second generation anti-CD19 CAR.
[0007] These toxicities are very difficult to predict even with detailed animal studies or non-human primate work. Crucially, unlike small molecules and biologics, CAR T-cells do not have a half-life and one cannot cease administration and wait for the agent to breakdown/become excreted. CAR T-cells are autonomous and can engraft and proliferate. Toxicity can therefore be progressive and fulminant.
[0008] Suicide genes are genetically expressed elements which can conditionally destroy cells which express them. Examples include Herpes-simplex virus thymidine kinase, which renders cells susceptible to Ganciclovir; inducible Caspase 9, which renders cells susceptible to a small molecular homodimerizer and CD20 and RQR8, which renders cells susceptible to Rituximab.
[0009] This technology adds a certain amount of safety to CAR T-cell therapy, however there are limitations. Firstly, it is a binary approach wherein all the CAR T-cells are destroyed upon addition of the suicide agent. In addition, medicinal therapeutics often have a therapeutic window. With a suicide gene the potency of the product cannot be tuned such that efficacy with tolerable toxicity can be achieved. Secondly, it is not clear whether a suicide gene would help with some of the immune-toxicities described above: for instance by the time a macrophage activation syndrome had been triggered, it may well no longer need the CAR T-cells to perpetuate and the suicide gene would no longer be helpful. The more acute cytokine release syndromes probably occur too quickly for the suicide gene to work.
[0010] There is thus a need for alternative methods for controlling CAR T-cells that are not associated with the disadvantages and problems mentioned above.
DESCRIPTION OF THE FIGURES
[0011] FIG. 1--a) Schematic diagram illustrating a classical CAR. (b) to (d): Different generations and permutations of CAR endodomains: (b) initial designs transmitted ITAM signals alone through Fc.epsilon.R1-.gamma. or CD3.zeta. endodomain, while later designs transmitted additional (c) one or (d) two co-stimulatory signals in the same compound endodomain.
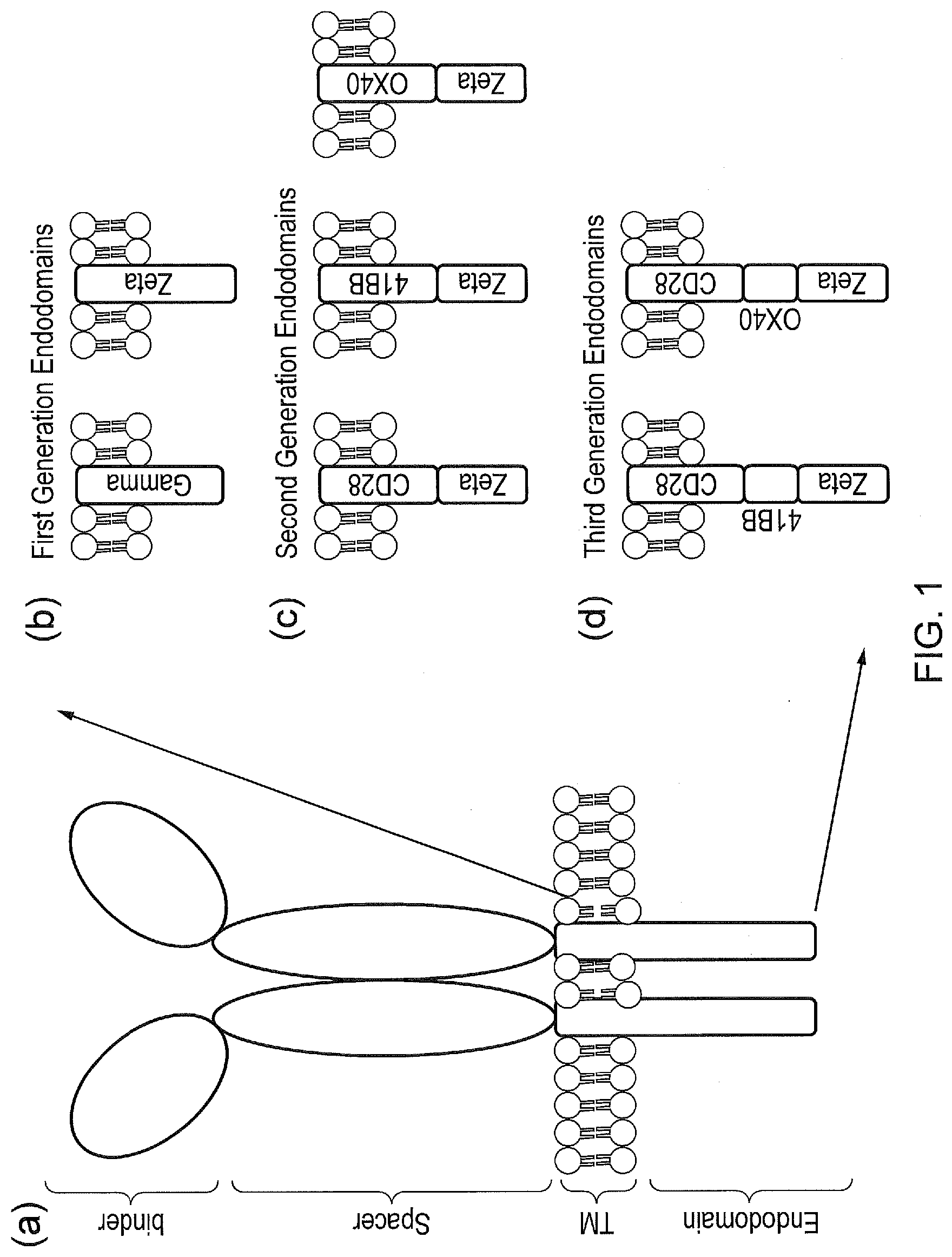
[0012] FIG. 2--Structures of TetR and TiP. (a) sequence of TiP attached at the amino-terminus of an arbitrary protein; (b) Crystallography derived structure of TiP interacting with TetR (from PDB 2NS8 and Luckner et al (J. Mol. Biol. 368, 780-790 (2007)). TiP can be seen engaged deep within the TetR homodimer associating with many of the residues tetracycline associates with.
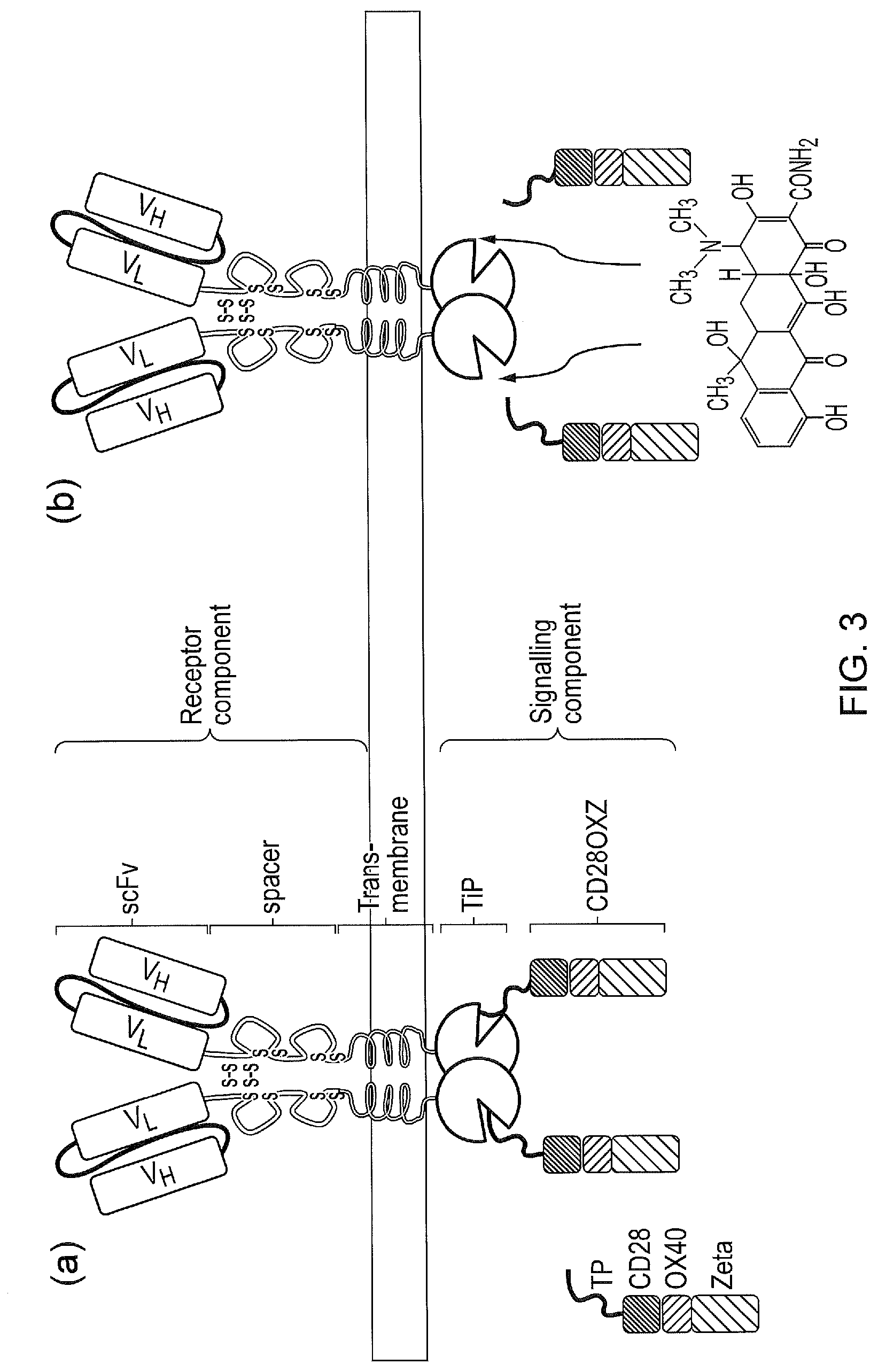
[0013] FIG. 3--(a) A membrane spanning receptor component comprises an extracellular antigen-binding domain, a transmembrane domain and an intracellular linker to TetR. A separate molecule, the signalling component, comprises an intracellular protein which is generated by fusion of TiP to one or several T-cell signalling domains. In the absence of tetracycline or tetracycline analogues, the receptor and the signalling components interact and in the presence of cognate antigen the system signals. (b) In the presence of tetracycline or tetracycline analogues, TiP is displaced from TetR and the receptor can not transmit signals even in the presence of cognate antigen.
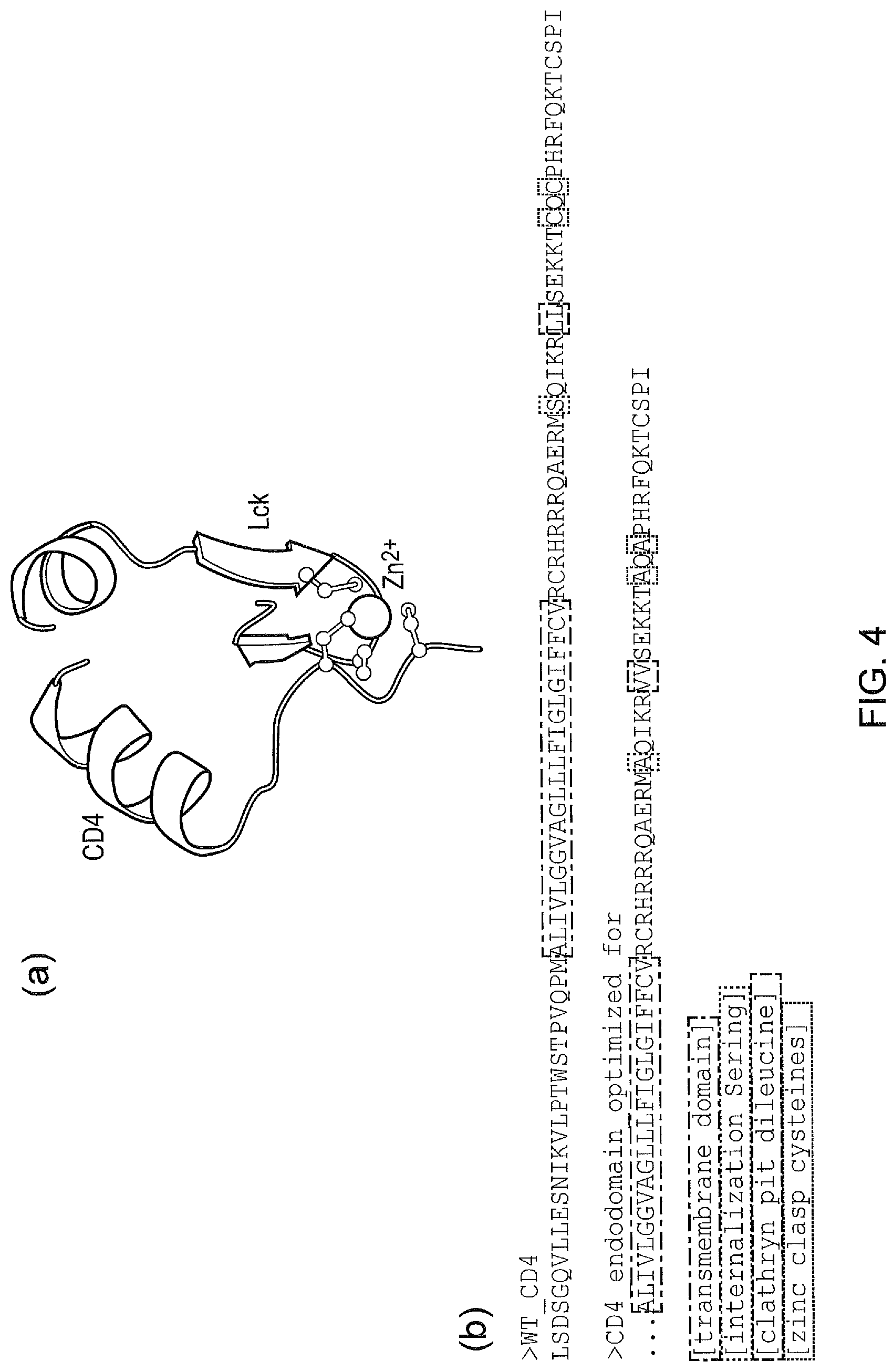
[0014] FIG. 4--Intracellular linker domain derived from CD4.
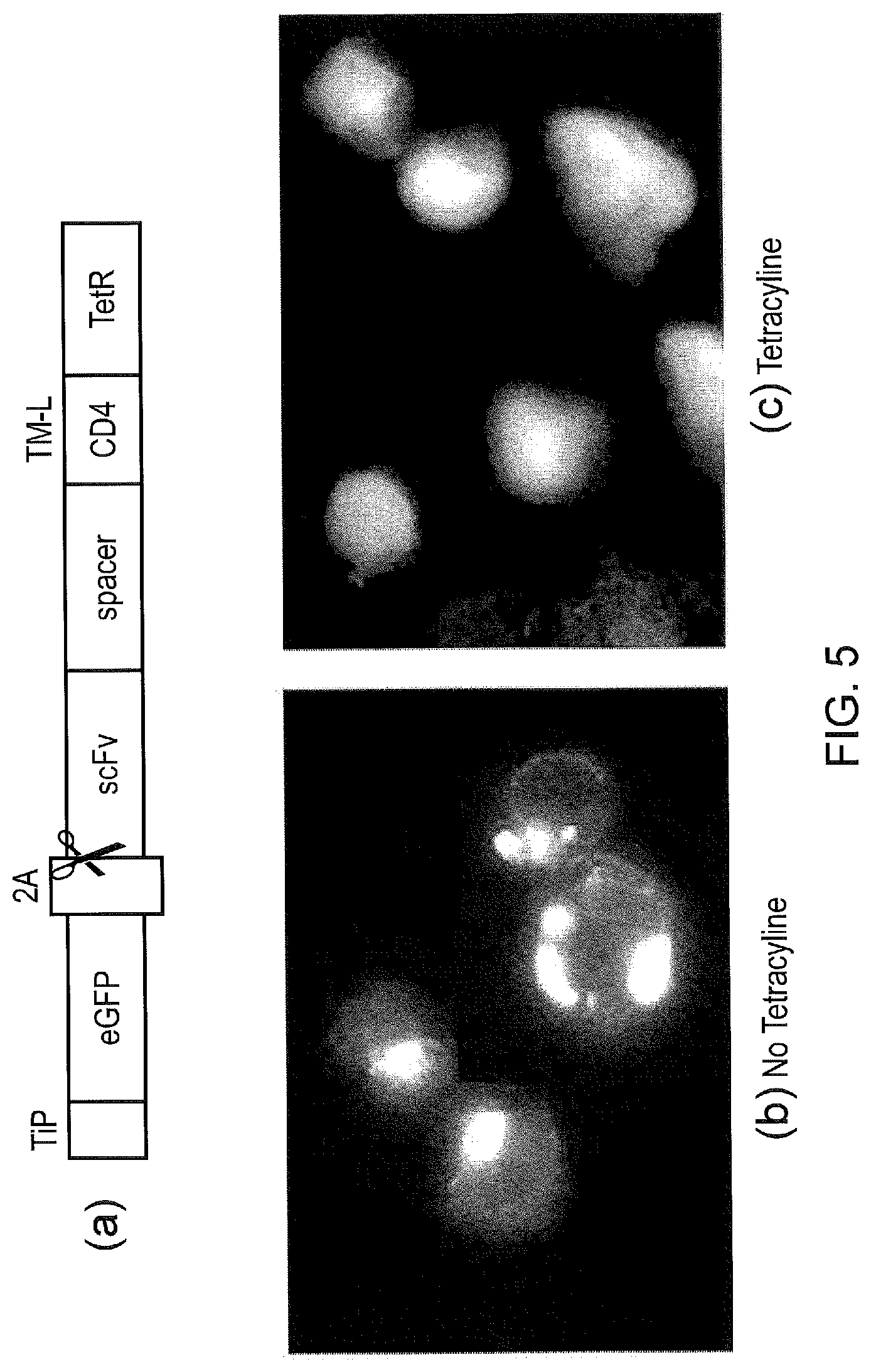
[0015] FIG. 5--Test construct with eGFP to demonstrate function of the system. (a) a bicistronic construct expressed as a single transcript which self-cleaves at the 2A site to yield: TiP fused to eGFP; and a CAR with TetR as its endodomain. (b) Fluorescent micrograph of SupT1 cells expressing this construct in the absence of tetracycline. The eGFP fluorescence can clearly be seen at the cell membrane; (c) Fluorescent micrograph of the same cells but now in the presence of tetracycline. Here, the eGFP is cytoplasmic showing that tetracycline has displaced TiP.
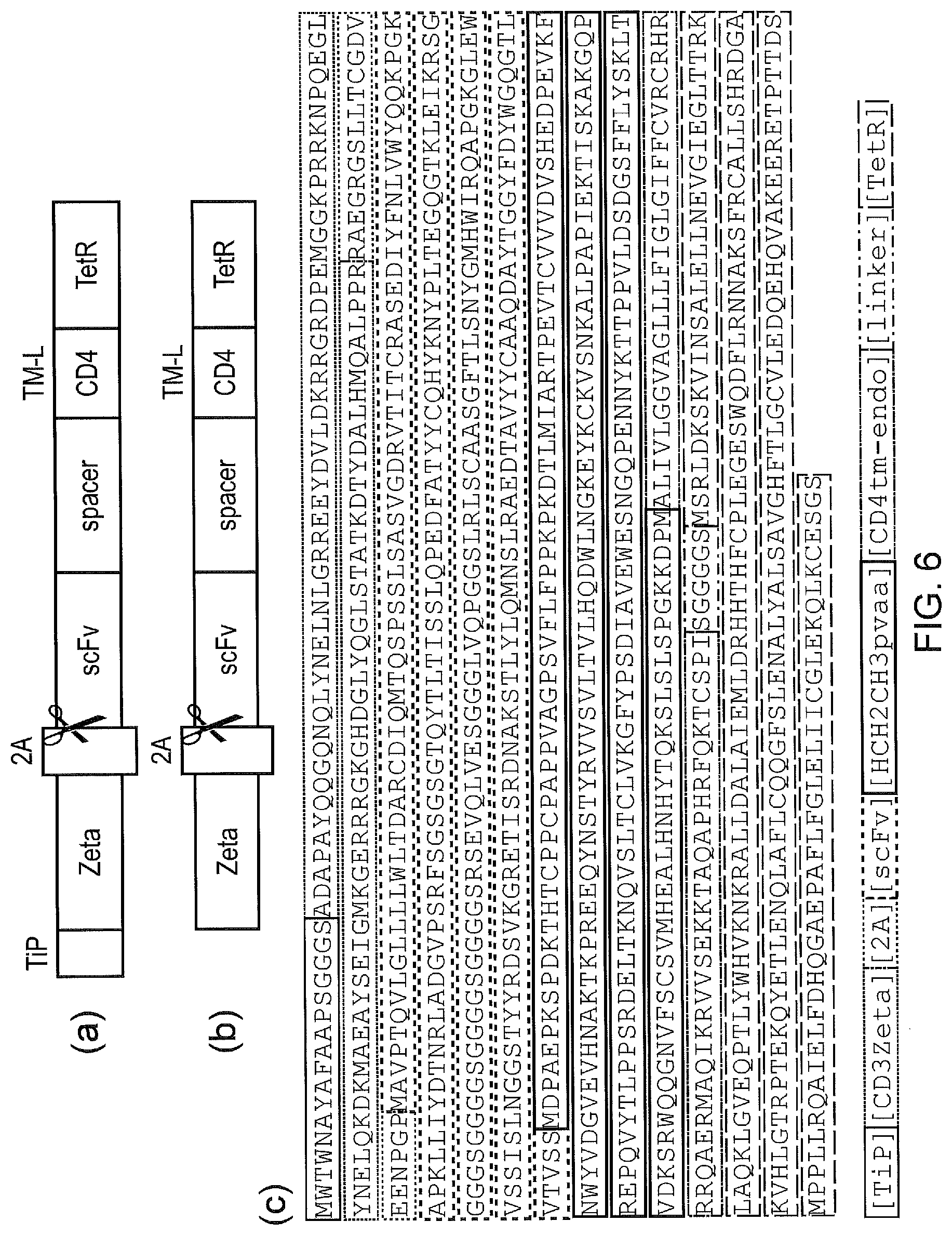
[0016] FIG. 6--Initial TetCAR construct and control (a) a bicistronic construct expressed as a single transcript which self-cleaves at the 2A site to yield: a signalling component which comprises TiP fused via a flexible linker to the endodomain of CD3-Zeta; and a receptor component which comprises a CD33 recognizing scFv, a spacer derived from the Fc domain of IgG1, a CD4 derived transmembrane and intracellular domain; and TetR. (b) a control was also constructed which was identical except TiP was absent from the signalling component. (c) annotated amino-acid sequence of the basic TetCAR is shown.
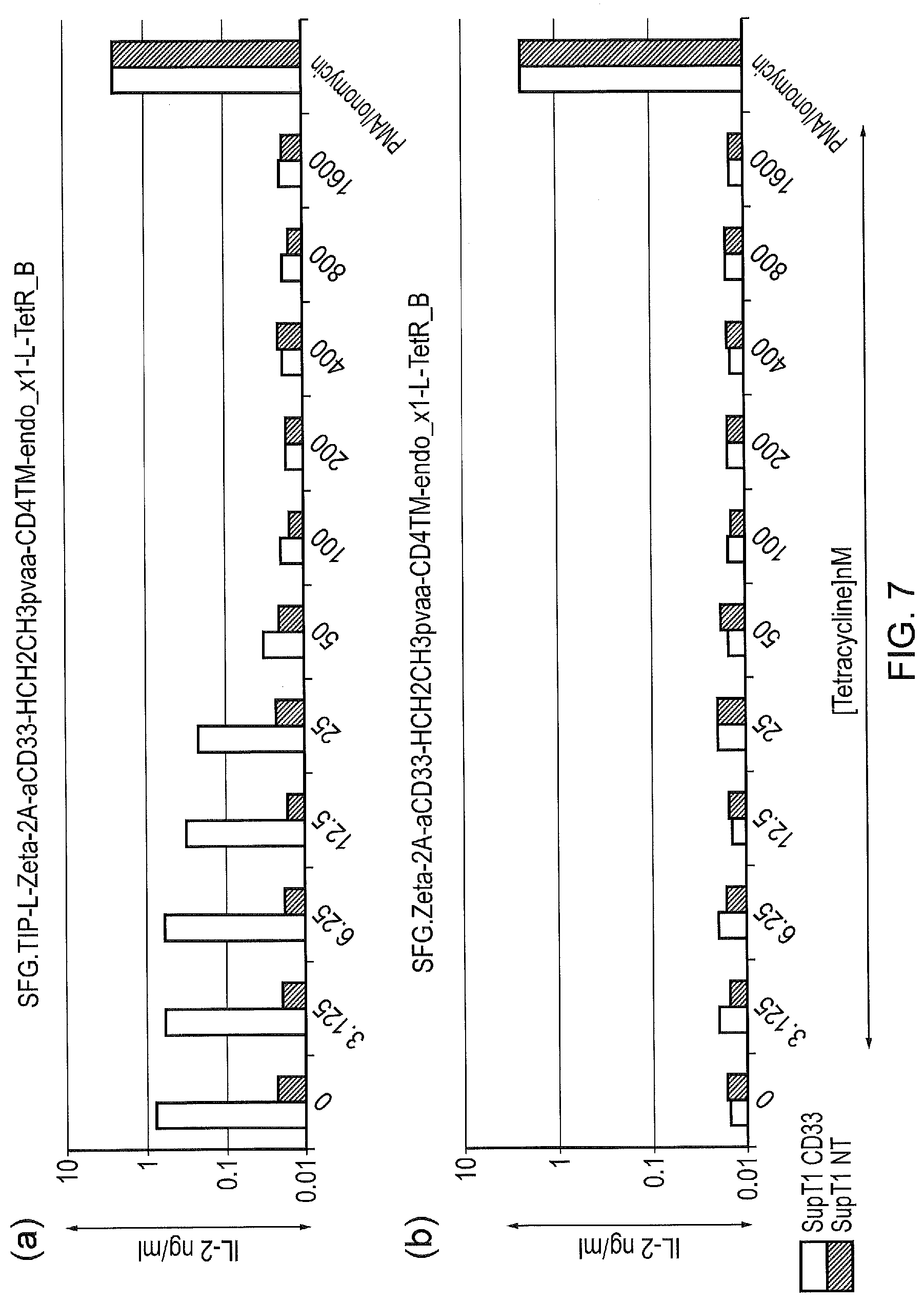
[0017] FIG. 7--Function of the initial TetR construct in comparison with control. (a) TetCAR was expressed in BW5 T-cells. These T-cells were challenged with wild-type SupT1 cells or SupT1 cells engineered to express CD33 in the absence of tetracycline or in the presence of increasing concentrations of tetracycline. T-cells challenged with wild-type SupT1 cells do not activate in either the presence or absence of Tetracyline; T-cells challenged with SupT1 cells expressing CD33 activate in the absence of Tetracycline, but activation is rapidly inhibited in the presence of tetracycline with activation fully inhibited in the presence of 100nM of Tetracycline. (b) Control TetCAR which lacks the TiP domain was transduced into BW5. Once again, these T-cells were challenged with wild-type SupT1 cells or SupT1 cells engineered to express CD33 in the absence or in the presence of increasing concentration of Tetracycline. A lack of TiP element in the signalling component resulted in no signalling in any conditions.
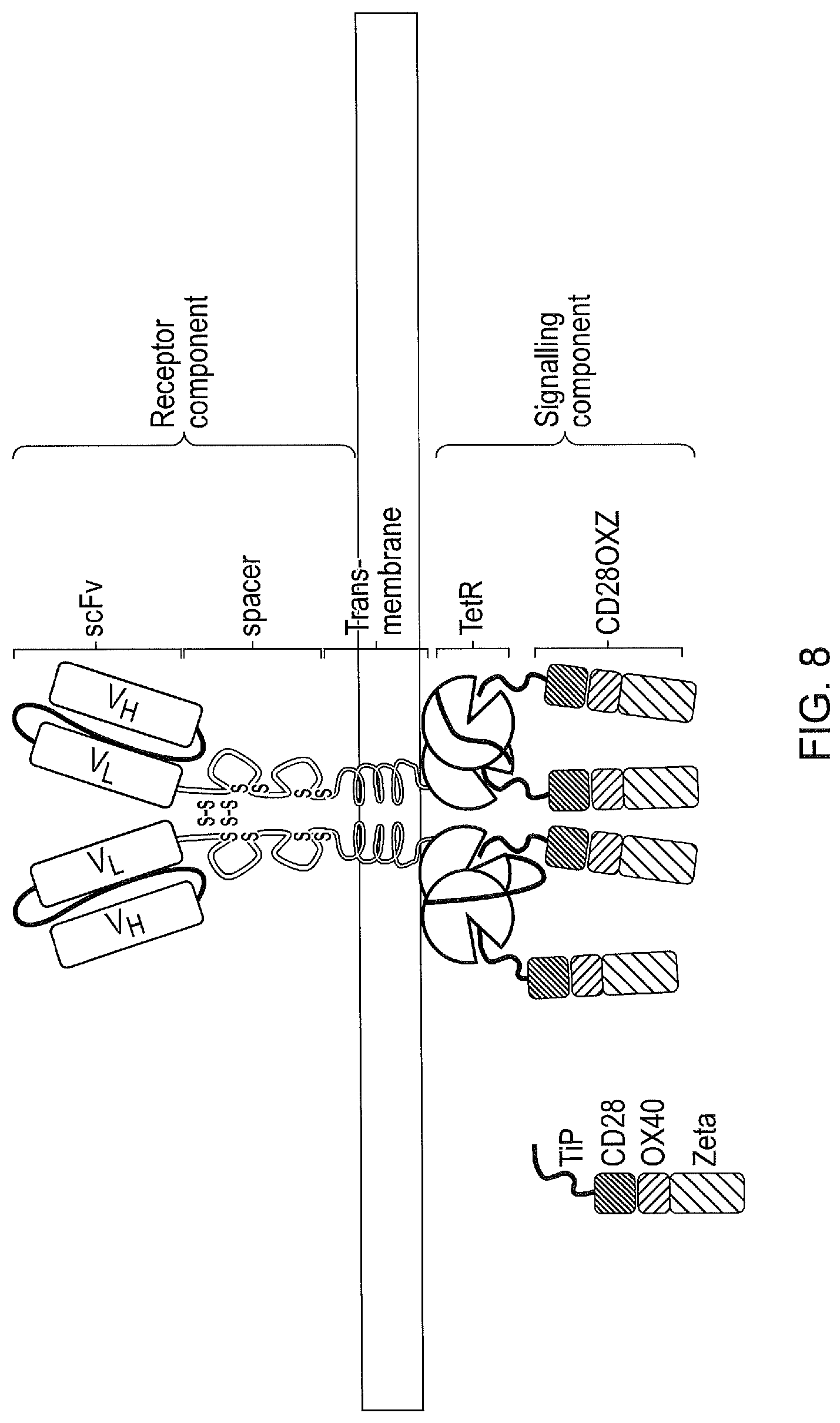
[0018] FIG. 8--Dual tetR domain tetCARs. tetR is expressed as a single-chain with two TetRs attached together. If tetR domains with differing affinity for tetracycline (and hence TiP) are used, the kinetics of Tetracycline mediated displacement of TiP can modulate the levels of signalling.
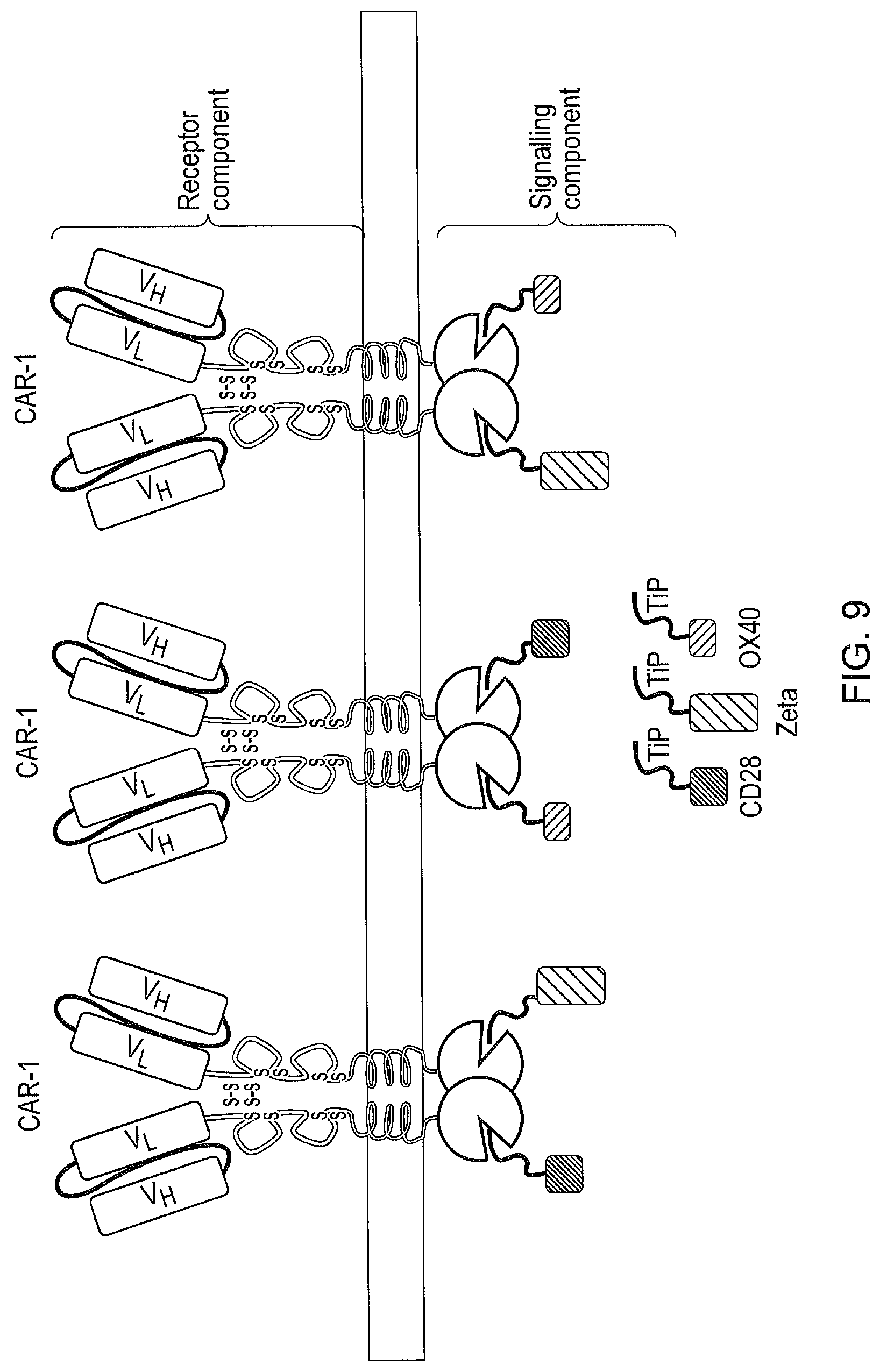
[0019] FIG. 9--A tetCAR signalling system utilising a plurality of signalling components containing single endodomains. A single CAR is expressed with many different signalling components all of which comprise TiP at their amino terminus but a different individual signalling domain, in contrast to a compound signalling domain. These randomly interact with the receptor component. Lack of steric interaction between the different signalling domains and their second messengers improves their function.
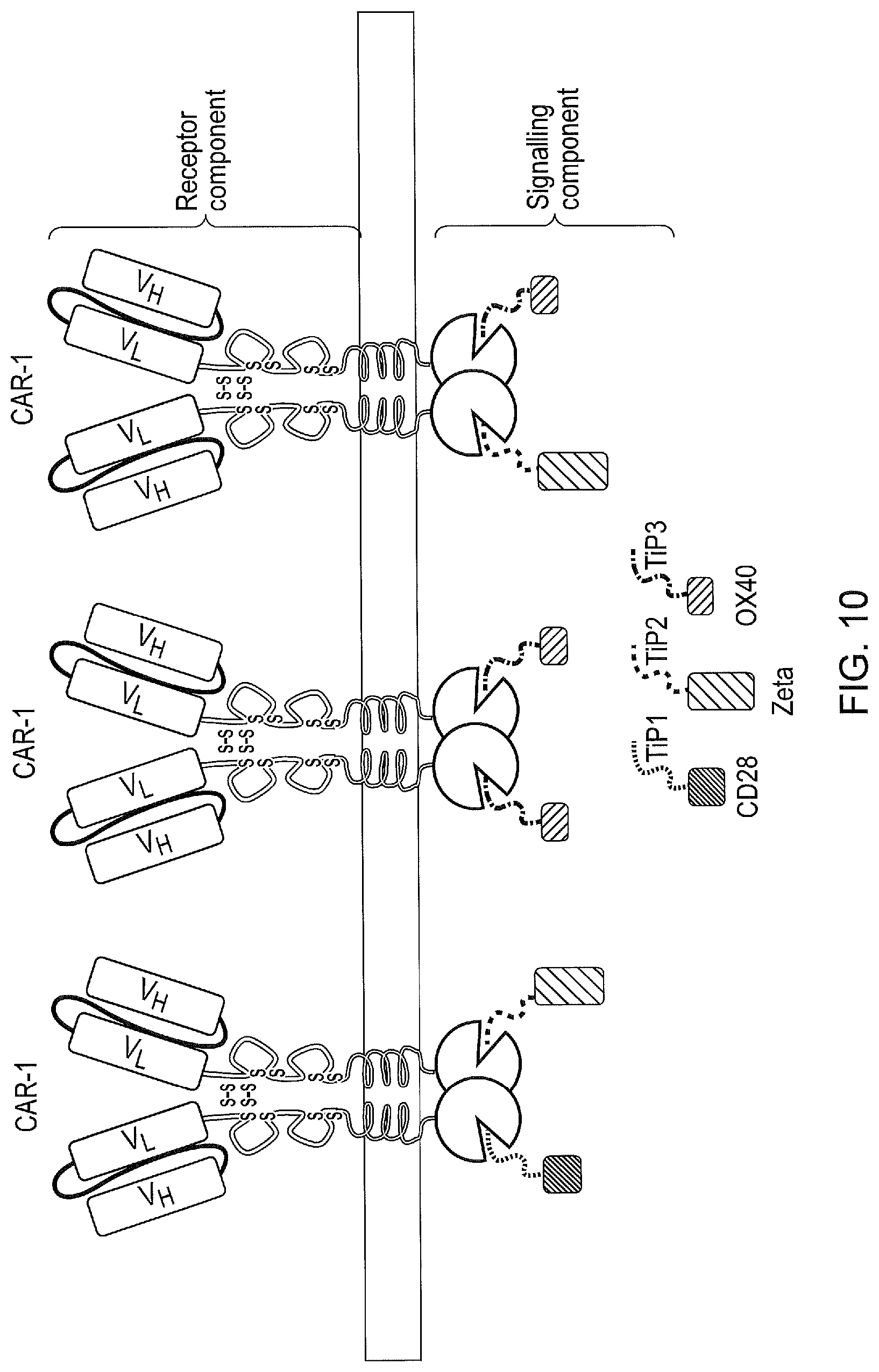
[0020] FIG. 10--A tetCAR signalling system utilising a plurality of signalling components containing single endodomains and different TiP domains. Each signalling component comprises of an individual signalling domain. Each signalling component also comprises of a TiP, however each TiP has different affinities to the TetR domain. Hence the stoichiometry of the interactions between the CAR and the signalling domains can be varied. In the example shown, the signalling system is constructed such that OX40>CD3Zeta>CD28.
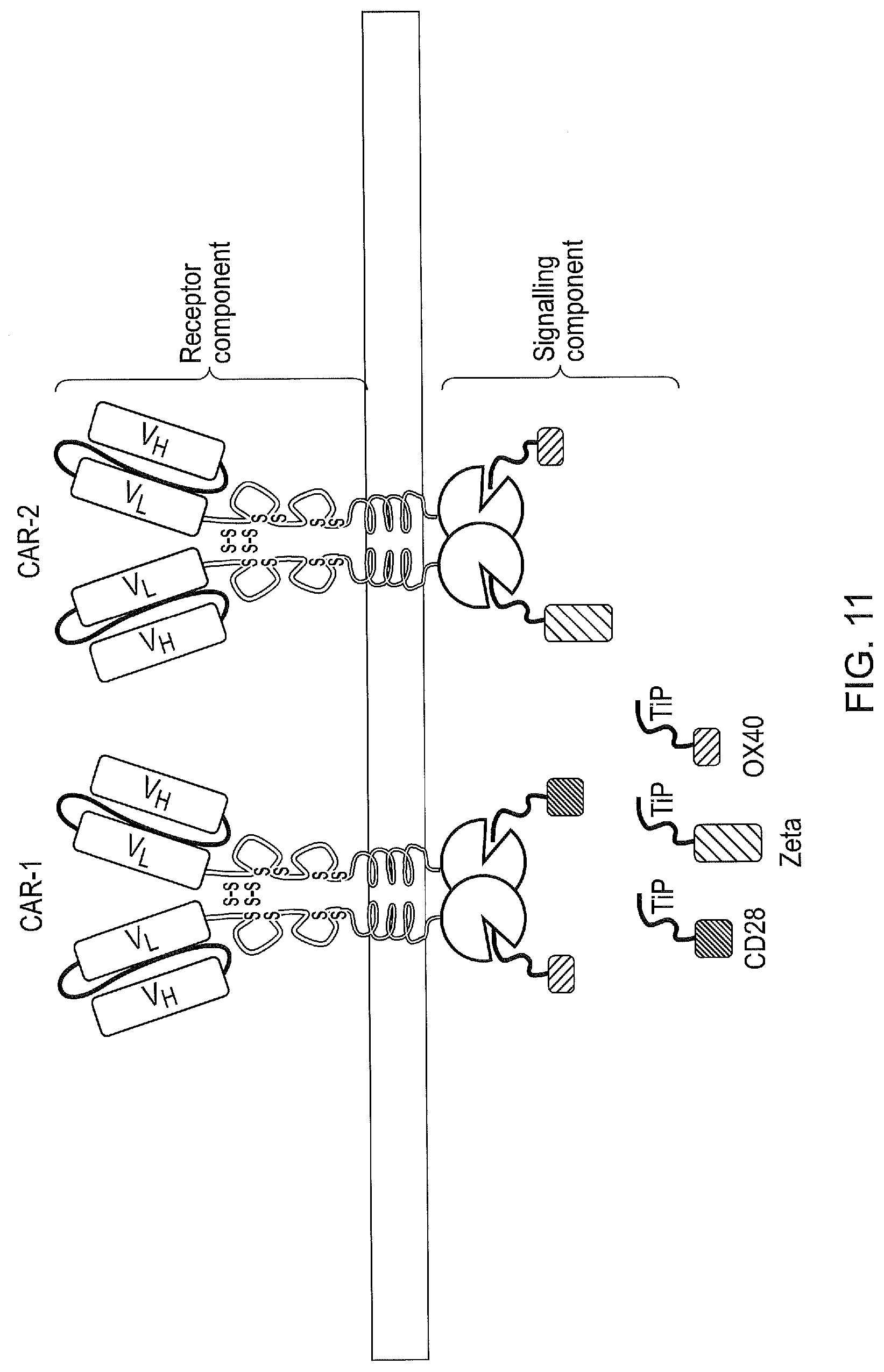
[0021] FIG. 11--A tetCAR signalling system utilising a plurality of receptor components and a plurality of signalling components, each signalling component containing a single endodomain.
[0022] FIG. 12--TetCAR signalling in primary cells (a) Different constructs tested: (i) Classic CAR; (ii) tetCAR; (iii) control tetCAR where TiP has been deleted. (b) non-transduced and SupT1.CD19 cells stained for CD19; (c) Non-transduced T-cells and T-cells transduced with the different CAR constructs stained with anti-Fc.
[0023] FIG. 13--Interferon-Gamma release from non-transduced T-cells, and T-cells transduced with the different CAR construct challenged ((i) Classical first generation CAR, (ii) tetCAR and (iii) control tetCAR), with SupT1 cells, SupT1.CD19 cells in different concentrations of Tetracyline.
[0024] FIG. 14--Killing of target cells. A chromium release assay was used to demonstrate killing of target cells (SupT1.CD19) in the absence of tetracycline. Key: (i)--regular CAR; (ii)--tetCAR; (iii)--control tetCAR (no TiP on endodomain).
SUMMARY OF ASPECTS OF THE INVENTION
[0025] The present inventors have found that it is possible to separate the antigen-recognition and signalling components of a CAR to produce a system in which signalling can be rapidly inhibited/terminated despite continued binding of antigen to an antigen-recognition component of the CAR system. This inhibition of signalling occurs in the presence of an agent, such as a small molecule, which inhibits the co-localisation and interaction which would otherwise occur between an extracellular antigen-binding component (referred to herein as the receptor component) and an intracellular signalling component of the CAR.
[0026] Thus in a first aspect the present invention provides a chimeric antigen receptor (CAR) system comprising; [0027] (i) a receptor component comprising an antigen binding domain, a transmembrane domain and a first binding domain; and [0028] (ii) an intracellular signalling component comprising a signalling domain and a second binding domain which specifically binds the first binding domain of the receptor component;
[0029] wherein, binding of the first and second binding domains is disrupted by the presence of an agent, such that in the absence of the agent the receptor component and the signalling component heterodimerize and binding of the antigen binding domain to antigen results in signalling through the signalling domain, whereas in the presence of the agent the receptor component and the signalling component do not heterodimerize and binding of the antigen binding domain to antigen does not result in signalling through the signalling domain.
[0030] The receptor component may comprise a linker between the transmembrane domain and the first binding domain.
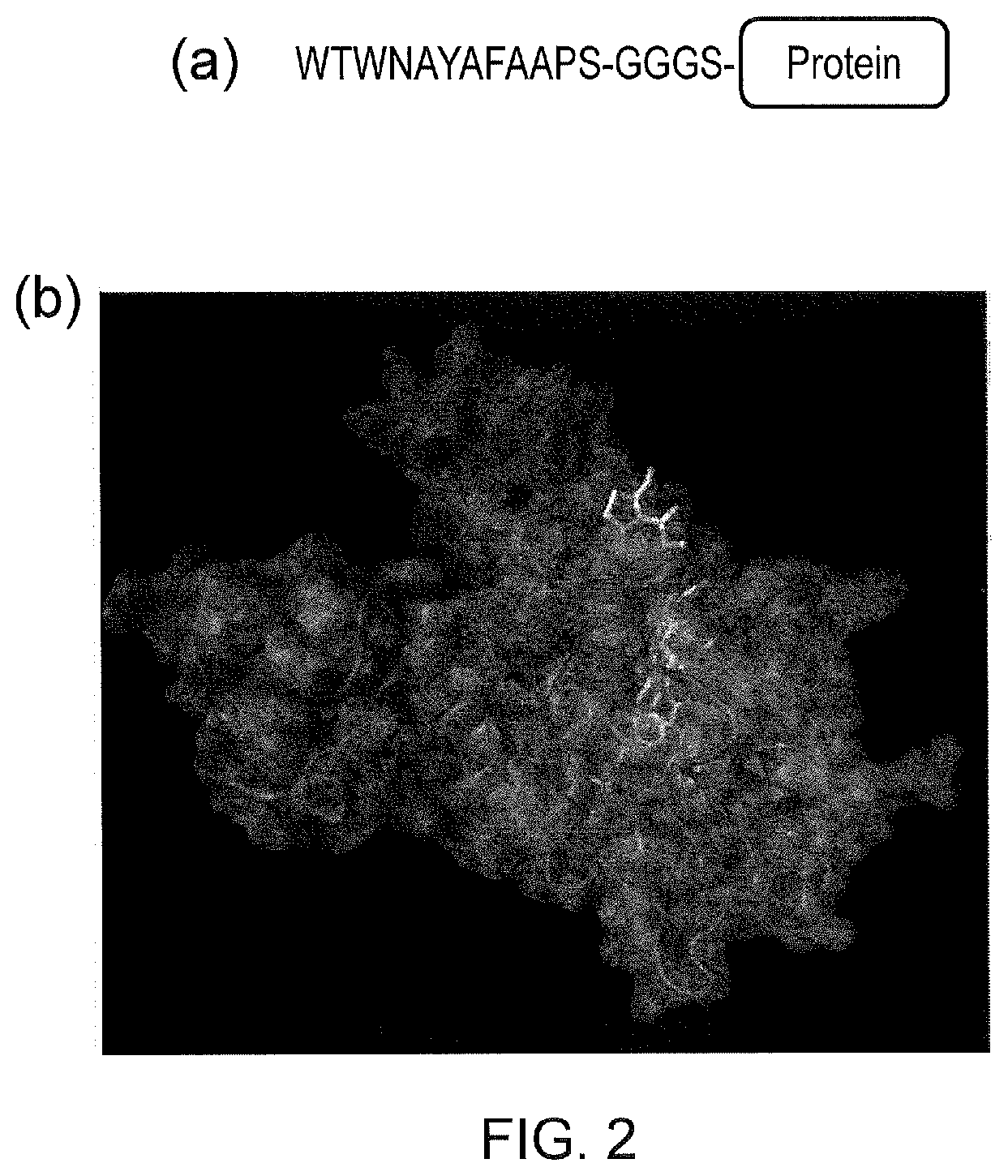
[0031] The linker may comprise or consist of the sequence shown as SEQ ID NO: 3.
[0032] The first binding domain may comprise Tet Repressor Protein (TetR) or a variant thereof and the second binding domain may comprise TetR inducing Peptide (TiP, as described by Klotzsche et al; The Journal of biological chemistry; 2005; 280(26); 24591-9) (TiP); or vice versa. In this case the agent may be tetracycline, doxycycline or minocycline or an analogue thereof.
[0033] The receptor component may comprise two first binding domains which are TetR domains. The two TetR domains may be separated by a linker. Each TetR domain may have a different affinity for the agent.
[0034] The CAR system of the first aspect of the invention may comprise multiple receptor components, each recognizing a different antigen.
[0035] The first binding domains of the multiple receptor components may differ in binding to the second binding domain of the signalling component such that each antigen propagates different signalling strengths.
[0036] The first binding domains of the multiple receptor components may differ in binding to the agent such that each antigen propagates different signalling strengths in the presence of the agent.
[0037] The signalling domain of the signalling component may comprise a single endodomain selected from CD3 zeta endodomain, CD28 endodomain, 41BB endodomain and OX40 endodomain.
[0038] The signalling domain of the signalling component may comprise at least one of CD3 zeta endodomain, CD28 endodomain, 41BB endodomain and OX40 endodomain.
[0039] The CAR system of the first aspect of the invention may comprise a plurality of signalling components, each comprising a signalling domain and a second binding domain, wherein the second binding domains each recognise the same first binding domain of the receptor component but the signalling domains comprise different endodomains.
[0040] The plurality of signalling components may comprise a plurality of second binding domains, each of which independently recognises the first binding domain of the receptor component with different affinities.
[0041] In a second aspect the present invention provides a receptor component suitable for use in the CAR system of the first aspect of the invention which comprises an antigen-binding domain, a transmembrane domain and a first binding domain.
[0042] In a third aspect the present invention provides a signalling component suitable for use in the CAR system of the first aspect of the invention which comprises a signalling domain and a second binding domain.
[0043] In a fourth aspect the present invention provides a nucleic acid sequence encoding the receptor component according to the second aspect of the invention.
[0044] In a fifth aspect the present invention provides a nucleic acid sequence encoding the signalling component according to the third aspect of the invention.
[0045] In a sixth aspect the present invention provides a nucleic acid sequence encoding a CAR system of the first aspect of the invention, wherein the receptor component and signalling component are co-expressed by means of a self-cleaving peptide which is cleaved between the receptor component and the signalling component after translation.
[0046] In a seventh aspect the present invention provides a vector comprising a nucleic acid sequence according to the fourth to sixth aspects of the invention.
[0047] In an eighth aspect the present invention provides a retroviral vector or a lentiviral vector or a transposon comprising a vector according to the seventh aspect of the invention.
[0048] In a ninth aspect the present invention provides a T cell or NK cell which expresses a receptor component according to the second aspect of the invention and a signalling component according to the third aspect of the invention.
[0049] The T cell or NK cell may comprise a nucleic acid according to the fourth to sixth aspects of the invention or a vector according to the seventh or eighth aspect of the invention.
[0050] In a tenth aspect the present invention provides a pharmaceutical composition comprising a plurality of T cells or NK cells according to the ninth aspect of the invention.
[0051] In an eleventh aspect the present invention provides a pharmaceutical composition according to the tenth aspect of the invention for use in treating and/or preventing a disease.
[0052] In a twelfth aspect the present invention relates to a method for treating and/or preventing a disease, which comprises the step of administering a pharmaceutical composition according to the tenth aspect of the invention to a subject.
[0053] The method according to the twelfth aspect of the invention may comprise the following steps: [0054] (i) isolation of a T cell or NK containing sample; [0055] (ii) transduction or transfection of the T or NK cells with a nucleic acid sequence according to any of the fourth to sixth aspects of the invention or a vector according to the seventh or eighth aspect of the invention; and [0056] (iii) administering the T cells or NK cells from (ii) to a subject.
[0057] The method may involve administration of T cells/NK cells to a subject, which Tcells/NK cells have been previously isolated from the subject and transduced/transfected with a nucleic acid sequence according to any of the fourth to sixth aspects of the invention or a vector according to the seventh or eighth aspect of the invention.
[0058] The method according to the twelfth aspect of the invention may involve monitoring toxic activity in the subject and comprise the step of administering an agent for use in the CAR system of the first aspect of the invention to the subject to reduce adverse toxic effects.
[0059] The method may involve monitoring the progression of disease and/or monitoring toxic activity in the subject and comprise the step of administering an agent for use in the CAR system of the first aspect of the invention to the subject to provide acceptable levels of disease progression and/or toxic activity.
[0060] In the use of a pharmaceutical composition according to the tenth aspect of the invention or a method according to the twelfth aspect of the invention, the disease may be cancer.
[0061] In a thirteenth aspect the present invention relates to the use of a pharmaceutical composition according to the tenth aspect of the invention in the manufacture of a medicament for the treatment and/or prevention of a disease.
[0062] In a fourteenth aspect the present invention provides a kit which comprises a nucleic acid according to the fourth to sixth aspects of the invention or a vector according to the seventh of eighth aspect of the invention.
[0063] In a fifteenth aspect the present invention relates to a method for making a T or NK cell according to the ninth aspect of the invention, which comprises the step of introducing a nucleic acid sequence according to fourth to sixth aspect of the invention or the vector according to the seventh or eighth aspect of the invention into a T or NK cell.
[0064] The T or NK cell may be from a sample isolated from a subject.
[0065] In a sixteenth aspect the present invention relates to a method for inhibiting the CAR system according to the first aspect of the invention in a subject which comprises a T or NK cell according to the ninth aspect of the invention which method comprises the step of administering the agent to the subject.
[0066] The present invention therefore provides a CAR system in which signalling can be inhibited in the presence of an agent, for example a small molecule, which prevents co-localisation of the receptor component and signalling component. This allows CAR signalling and thus the potency of CAR cells to be reversibly terminated in a controllable manner in order to avoid potential toxic effects associated with unabated CAR signalling. Further the present system also allows the potency of CAR cells to be controlled pharmacologically and tuned to an acceptable balance between achieving the desired therapeutic effect and avoiding unwanted toxicities.
DETAILED DESCRIPTION
Chimeric Antigen Receptors (CARs)
[0067] Classical CARs, which are shown schematically in FIG. 1, are chimeric type I trans-membrane proteins which connect an extracellular antigen-recognizing domain (binder) to an intracellular signalling domain (endodomain). The binder is typically a single-chain variable fragment (scFv) derived from a monoclonal antibody (mAb), but it can be based on other formats which comprise an antibody-like antigen binding site. A spacer domain may be necessary to isolate the binder from the membrane and to allow it a suitable orientation. A common spacer domain used is the Fc of IgG1. More compact spacers can suffice e.g. the stalk from CD8a and even just the IgG1 hinge alone, depending on the antigen. A trans-membrane domain anchors the protein in the cell membrane and connects the spacer to the endodomain.
[0068] Early CAR designs had endodomains derived from the intracellular parts of either the y chain of the Fc.epsilon.R1 or CD3.zeta.. Consequently, these first generation receptors transmitted immunological signal 1, which was sufficient to trigger T-cell killing of cognate target cells but failed to fully activate the T-cell to proliferate and survive. To overcome this limitation, compound endodomains have been constructed: fusion of the intracellular part of a T-cell co-stimulatory molecule to that of CD3 results in second generation receptors which can transmit an activating and co-stimulatory signal simultaneously after antigen recognition.
[0069] The co-stimulatory domain most commonly used is that of CD28. This supplies the most potent co-stimulatory signal - namely immunological signal 2, which triggers T-cell proliferation. Some receptors have also been described which include TNF receptor family endodomains, such as the closely related OX40 and 41BB which transmit survival signals. Even more potent third generation CARs have now been described which have endodomains capable of transmitting activation, proliferation and survival signals.
[0070] CAR-encoding nucleic acids may be transferred to T cells using, for example, retroviral vectors. In this way, a large number of antigen-specific T cells can be generated for adoptive cell transfer. When the CAR binds the target-antigen, this results in the transmission of an activating signal to the T-cell it is expressed on. Thus the CAR directs the specificity and cytotoxicity of the T cell towards cells expressing the targeted antigen.
[0071] In a first aspect, the present invention relates to a CAR system in which the antigen-recognizing/antigen binding domain and transmembrane domain are provided on a first molecule (termed herein `receptor component`), which localizes to the cell membrane. The intracellular signalling domain is provided on a second, intracellular molecule (termed herein `signalling component`).
[0072] Importantly, the receptor component comprises a first binding domain and the signalling component comprises a second binding domain which specifically binds to the first binding domain of the receptor component. Thus binding of the first binding domain to the second binding domain causes heterodimerization and co-localization of the receptor component and the signalling component. When antigen binds to the antigen binding domain of the receptor component there is signalling through the signalling component.
[0073] The first or second binding domain is also capable of binding a further agent in addition to the reciprocal binding domain. The further agent may be, for example, a small molecule. The binding between the agent and the first or second binding domain is of a higher affinity than the binding between the first binding domain and the second binding domain. Thus, when the agent is present it preferentially binds to the first or second binding domain and inhibits/disrupts the heterodimerization between the receptor component and the signalling component. When antigen binds to the antigen binding domain of the receptor component in the presence of the further agent there is no signalling through the signalling component.
[0074] Specifically, in the presence of the agent, the receptor component and signalling component are located in a stochastically dispersed manner and binding of antigen by the antigen-binding domain of the receptor component does not result in signalling through the signaling component.
[0075] Herein `co-localization` or `heterodimerization` of the receptor and signalling components is analogous to ligation/recruitment of the signalling component to the receptor component via binding of the first binding domain of the receptor component and the second binding domain of the signalling component.
[0076] Antigen binding by the receptor component in the presence of the agent may be termed as resulting in `non-productive` signalling through the signalling component. Such signalling does not result in cell activation, for example T cell activation. Antigen binding by the receptor component in the absence of the agent may be termed as resulting in `productive` signalling through the signalling component. This signalling results in T-cell activation, triggering for example target cell killing and T cell activation.
[0077] Antigen binding by the receptor component in the absence of the agent may result in signalling through the signalling component which is 2, 5, 10, 50, 100, 1,000 or 10,000-fold higher than the signalling which occurs when antigen is bound by the receptor component in the presence of the agent.
[0078] Signalling through the signalling component may be determined by a variety of methods known in the art. Such methods include assaying signal transduction, for example assaying levels of specific protein tyrosine kinases (PTKs), breakdown of phosphatidylinositol 4,5-biphosphate (PIP.sub.2), activation of protein kinase C (PKC) and elevation of intracellular calcium ion concentration. Functional readouts, such as clonal expansion of T cells, upregulation of activation markers on the cell surface, differentiation into effector cells and induction of cytotoxicity or cytokine secretion may also be utilised. As an illustration, in the present examples the inventors determined levels of interleukin-2 (IL-2) produced by T-cells expressing a receptor component and signalling component of the CAR system according to the present invention upon binding of antigen to the receptor component in the presence of varying concentrations of an agent.
First Binding Domain, Second Binding Domain and Agent
[0079] The first binding domain, second binding domain and agent of the present CAR system may be any combination of molecules/peptides/domains which enable the selective co-localization and dimerization of the receptor component and signalling component in the absence of the agent.
[0080] As such, the first binding domain and second binding domain are capable of specifically binding.
[0081] The signalling system of the present invention is not limited by the arrangement of a specific dimerization system. The receptor component may comprise either the first binding domain or the second binding domain of a given dimerization system so long as the signalling component comprises the corresponding, complementary binding domain which enables the receptor component and signalling component to co-localize in the absence of the agent.
[0082] The first binding domain and second binding domain may be a peptide domain and a peptide binding domain; or vice versa. The peptide domain and peptide binding domain may be any combination of peptides/domains which are capable of specific binding.
[0083] The agent is a molecule, for example a small molecule, which is capable of specifically binding to the first binding domain or the second binding domain at a higher affinity than the binding between the first binding domain and the second binding domain.
[0084] For example, the binding system may be based on a peptide:peptide binding domain system. The first or second binding domain may comprise the peptide binding domain and the other binding domain may comprise a peptide mimic which binds the peptide binding domain with lower affinity than the peptide. The use of peptide as agent disrupts the binding of the peptide mimic to the peptide binding domain through competitive binding. The peptide mimic may have a similar amino acid sequence to the "wild-type" peptide, but with one of more amino acid changes to reduce binding affinity for the peptide binding domain.
[0085] For example, the agent may bind the first binding domain or the second binding domain with at least 10, 20, 50, 100, 1000 or 10000-fold greater affinity than the affinity between the first binding domain and the second binding domain.
[0086] The agent may be any pharmaceutically acceptable molecule which preferentially binds the first binding domain or the second binding domain with a higher affinity than the affinity between the first binding domain and the second binding domain.
[0087] The agent is capable of being delivered to the cytoplasm of a target cell and being available for intracellular binding.
[0088] The agent may be capable of crossing the blood-brain barrier.
[0089] Small molecule systems for controlling the co-localization of peptides are known in the art, for example the Tet repressor (TetR), TetR interacting protein (TiP), tetracycline system (Klotzsche et al.; J. Biol. Chem. 280, 24591-24599 (2005); Luckner et al.; J. Mol. Biol. 368, 780-790 (2007)).
The Tet Repressor (TetR) System
[0090] The Tet operon is a well-known biological operon which has been adapted for use in mammalian cells. The TetR binds tetracycline as a homodimer and undergoes a conformational change which then modulates the DNA binding of the TetR molecules. Klotzsche et al. (as above), described a phage-display derived peptide which activates the TetR. This protein (TetR interacting protein/TiP) has a binding site in TetR which overlaps, but is not identical to, the tetracycline binding site (Luckner et al.; as above). Thus TiP and tetracycline compete for binding of TetR.
[0091] In the present CAR system the first binding domain of the receptor component may be TetR or TiP, providing that the second binding domain of the signalling component is the corresponding, complementary binding partner. For example if the first binding domain of the receptor component is TetR, the second binding domain of the signalling component is TiP. If the first binding domain of the receptor component is TiP, the second binding domain of the signalling component is TetR.
[0092] For example, the first binding domain or second binding domain may comprise the sequence shown as SEQ ID NO: 1 or SEQ ID NO: 2:
TABLE-US-00001 -TetR SEQ ID NO: 1 MSRLDKSKVINSALELLNEVGIEGLTTRKLAQKLGVEQPTLYWHVKNKRA LLDALAIEMLDRHHTHFCPLEGESWQDFLRNNAKSFRCALLSHRDGAKVH LGTRPTEKQYETLENQLAFLCQQGFSLENALYALSAVGH -TiP SEQ ID NO: 2 MWTWNAYAFAAPSGGGS
[0093] TetR must homodimerize in order to function. Thus when the first binding domain on the receptor component is TetR, the receptor component may comprise a linker between the transmembrane domain and the first binding domain (TetR). The linker enables TetR to homodimerize with a TetR from a neighbouring receptor component and orient in the correct direction.
[0094] The linker may be the sequence shown as SEQ ID NO: 3.
TABLE-US-00002 -modified CD4 endodomain SEQ ID NO: 3 ALIVLGGVAGLLLFIGLGIFFCVRCRHRRRQAERMAQIKRVVSEKKTAQA PHRFQKTCSPI
[0095] The linker may alternatively comprise an alternative linker sequence which has similar length and/or domain spacing properties as the sequence shown as SEQ ID NO: 3.
[0096] The linker may have at least 80%, 85%, 90%, 95%, 98% or 99% sequence identity to SEQ ID NO: 3 providing it provides the function of enabling TetR to homodimerize with a TetR from a neighbouring receptor component and orient in the correct direction.
[0097] One potential disadvantage of the TetR/TiP system is TetR is xenogenic and immunogenic.
[0098] The TetR sequence may therefore be a variant which is less immunogenic but retains the ability to specifically bind TiP.
[0099] Where the first and second binding domains are TetR or TiP or a variant thereof, the agent may be tetracycline, doxycycline, minocycline or an analogue thereof.
[0100] An analogue refers to a variant of tetracycline, doxycycline or minocycline which retains the ability to specifically bind to TetR.
[0101] Other combinations of binding domains and agents which may be used in the present CAR system are known in the art. For example, the CAR system may use a streptavidin/biotin-based binding system.
Streptavidin-Binding Epitope
[0102] The first or second binding domain may comprise one or more streptavidin-binding epitope(s). The other binding domain may comprise a biotin mimic.
[0103] Streptavidin is a 52.8 kDa protein from the bacterium Streptomyces avidinii. Streptavidin homo-tetramers have a very high affinity for biotin (vitamin B7 or vitamin H), with a dissociation constant (Kd) .about.10.sup.15 M. The biotin mimic has a lower affinity for streptavidin than wild-type biotin, so that biotin itself can be used as the agent to disrupt or prevent heterodimerisation between the streptavidin domain and the biotin mimic domain. The biotin mimic may bind streptavidin with for example with a Kd of 1 nM to 100 uM.
[0104] The `biotin mimic` domain may, for example, comprise a short peptide sequence (for example 6 to 20, 6 to 18, 8 to 18 or 8 to 15 amino acids) which specifically binds to streptavidin.
[0105] The biotin mimic may comprise a sequence as shown in Table 1.
TABLE-US-00003 TABLE 1 Biotin mimicking peptides. name Sequence affinity Long nanotag DVEAWLDERVPLVET (SEQ ID NO: 4) 3.6 nM Short nanotag DVEAWLGAR (SEQ ID NO: 5) 17 nM Streptag WRHPQFGG (SEQ ID NO: 6) 72 uM streptagII WSHPQFEK (SEQ ID NO: 7) SBP-tag MDEKTTGWRGGHVVEGLAGELEQLRARLEHHPQGQREP 2.5 nM (SEQ ID NO: 8) ccstreptag CHPQGPPC (SEQ ID NO: 9) 230 nM flankedccstreptag AECHPQGPPCIEGRK (SEQ ID NO: 10)
[0106] The biotin mimic may be selected from the following group: Streptagll, Flankedccstreptag and ccstreptag.
[0107] The streptavidin domain may comprise streptavidin having the sequence shown as SEQ ID No. 11 or a fragment or variant thereof which retains the ability to bind biotin.
[0108] Full length Streptavidin has 159 amino acids. The N and C termini of the 159 residue full-length protein are processed to give a shorter `core` streptavidin, usually composed of residues 13-139; removal of the N and C termini is necessary for the high biotin-binding affinity.
[0109] The sequence of "core" streptavidin (residues 13-139) is shown as SEQ ID No. 11
TABLE-US-00004 SEQ ID No. 11 EAGITGTWYNQLGSTFIVTAGADGALTGTYESAVGNAESRYVLTGRYDSA PATDGSGTALGWTVAWKNNYRNAHSATTWSGQYVGGAEARINTQWLLTSG TTEANAWKSTLVGHDTFTKVKPSAAS
[0110] Streptavidin exists in nature as a homo-tetramer. The secondary structure of a streptavidin monomer is composed of eight antiparallel .beta.-strands, which fold to give an antiparallel beta barrel tertiary structure. A biotin binding-site is located at one end of each .beta.-barrel. Four identical streptavidin monomers (i.e. four identical .beta.-barrels) associate to give streptavidin's tetrameric quaternary structure. The biotin binding-site in each barrel consists of residues from the interior of the barrel, together with a conserved Trp120 from neighbouring subunit. In this way, each subunit contributes to the binding site on the neighbouring subunit, and so the tetramer can also be considered a dimer of functional dimers.
[0111] The streptavidin domain of the CAR system of the present invention may consist essentially of a streptavidin monomer, dimer or tetramer.
[0112] The sequence of the streptavidin monomer, dimer or tetramer may comprise all or part of the sequence shown as SEQ ID No. 11, or a variant thereof which retains the capacity to bind biotin.
[0113] A variant streptavidin sequence may have at least 70, 80, 90, 95 or 99% identity to SEQ ID No. 11 or a functional portion thereof. Variant streptavidin may comprise one or more of the following amino acids, which are involved in biotin binding: residues Asn23, Tyr43, Ser27, Ser45, Asn49, Ser88, Thr90 and Asp128. Variany streptavidin may, for example, comprise all 8 of these residues. Where variant streptavidin is present in the binding domain as a dimer or teramer, it may also comprise Trp120 which is involved in biotin binding by the neighbouring subunit.
[0114] Small molecules agents which disrupt protein-protein interactions have long been developed for pharmaceutical purpose (reviewed by Vassilev et al; Small-Molecule Inhibitors of Protein-Protein Interactions ISBN: 978-3-642-17082-9). A CAR system as described may use such a small molecule. The proteins or peptides whose interaction is disrupted (or relevant fragments of these proteins) can be used as the first and/or second binding domains and the small molecule may be used as the agent which inhibits CAR activation. Such a system may be varied by altering the small molecule and proteins such the system functions as described but the small molecule is devoid of unwanted pharmacological activity (e.g. in a manner similar to that described by Rivera et al (Nature Med; 1996; 2; 1028-1032).
[0115] A list of proteins/peptides whose interaction is disruptable using an agent such as a small molecule is given in Table 2. These disputable protein-protein interactions (PPI) may be used in the CAR system of the present invention. Further information on these PPIs is available from White et al 2008 (Expert Rev. Mol. Med. 10: e8).
TABLE-US-00005 TABLE 2 Interacting Protein 1 Interacting Protein 2 Inhibitor of PPI p53 MDM2 Nutlin Anti-apoptotic Bcl2 Apoptotic Bcl2 GX015 and member member ABT-737 Caspase-3, -7 or -9 X-linked inhibitor of DIABLO and apoptosis protein (XIAP) DIABLO mimetics RAS RAF Furano-indene derivative FR2-7 PD2 domain of DVL FJ9 T-cell factor (TCF) Cyclic AMP response ICG-001 element binding protein (CBP)
[0116] Second binding domains which competitively bind to the same first binding domain as the agents described above, and thus may be used to co-localise the receptor component and signalling component of the present signalling system in the absence of the agent, may be identified using techniques and methods which are well known in the art. For example such second binding domains may be identified by display of a single domain VHH library.
[0117] The first binding domain and/or second binding domain of the present signalling system may comprise a variant(s) which is able to specifically bind to the reciprocal binding domain and thus facilitate co-localisation of the receptor component and signalling component.
[0118] Variant sequences may have at least 80%, 85%, 90%, 95%, 98% or 99% sequence identity to the wild-type sequence, provided that the sequences provide an effective dimerization system. That is, provided that the sequences facilitate sufficient co-localisation of the receptor and signalling components, in the absence of the agent, for productive signalling to occur upon binding of the antigen-binding domain to antigen.
[0119] The present invention also relates to a method for inhibiting the CAR system of the first aspect of the invention, which method comprises the step of administering the agent. As described above, administration of the agent results in a disruption of the co-localization between the receptor component and the signalling component, such that signalling through the signalling component is inhibited even upon binding of antigen to the antigen binding domain.
[0120] The first and second binding domains may facilitate signalling through the CAR system which is proportional to the concentration of the agent which is present. Thus, whilst the agent binds the first binding domain or the second binding domain with a higher affinity than binding affinity between the first and second binding domains, co-localization of the receptor and signalling components may not be completely ablated in the presence of low concentrations of the agent. For example, low concentrations of the agent may decrease the total level of signalling in response to antigen without completely inhibiting it. The specific concentrations of agent will differ depending on the level of signalling required and the specific binding domains and agent. Levels of signalling and the correlation with concentration of agent can be determined using methods known in the art, as described above.
Receptor Component
[0121] The present invention provides a receptor component comprising an antigen-binding domain, an optional spacer domain, a transmembrane domain and a first biding domain. When expressed in a cell, the receptor component localises to the cell membrane. Here, the antigen-binding domain of the molecule is orientated on the extracellular side of the membrane and the first binding domain is localised to the intracellular side of the membrane.
[0122] The receptor component therefore provides the antigen-binding function of the CAR system of the present invention.
Antigen Binding Domain
[0123] The antigen-binding domain is the portion of a classical CAR which recognizes antigen. In the signalling system of the present invention the antigen-binding is located within the receptor component.
[0124] Numerous antigen-binding domains are known in the art, including those based on the antigen binding site of an antibody, antibody mimetics, and T-cell receptors. For example, the antigen-binding domain may comprise: a single-chain variable fragment (scFv) derived from a monoclonal antibody; a natural ligand of the target antigen; a peptide with sufficient affinity for the target; a single domain binder such as a camelid; an artificial binder single as a Darpin; or a single-chain derived from a T-cell receptor.
[0125] Various tumour associated antigens (TAA) are known, as shown in the following Table 1. The antigen-binding domain used in the present invention may be a domain which is capable of binding a TAA as indicated therein.
TABLE-US-00006 TABLE 1 Cancer type TAA Diffuse Large B-cell Lymphoma CD19, CD20 Breast cancer ErbB2, MUC1 AML CD13, CD33 Neuroblastoma GD2, NCAM, ALK, GD2 B-CLL CD19, CD52, CD160 Colorectal cancer Folate binding protein, CA-125 Chronic Lymphocytic Leukaemia CD5, CD19 Glioma EGFR, Vimentin Multiple myeloma BCMA, CD138 Renal Cell Carcinoma Carbonic anhydrase IX, G250 Prostate cancer PSMA Bowel cancer A33
Transmemebrane Domain
[0126] The transmembrane domain is the sequence of a classical CAR that spans the membrane. In the signalling system of the present invention the transmembrane domain is located in the receptor component. It may comprise a hydrophobic alpha helix. The transmembrane domain may be derived from CD28, which gives good receptor stability.
Signal Peptide
[0127] The receptor component of the CAR system of the present invention may comprise a signal peptide so that when the receptor component is expressed in a cell, such as a T-cell, the nascent protein is directed to the endoplasmic reticulum and subsequently to the cell surface, where it is expressed.
[0128] The core of the signal peptide may contain a long stretch of hydrophobic amino acids that has a tendency to form a single alpha-helix. The signal peptide may begin with a short positively charged stretch of amino acids, which helps to enforce proper topology of the polypeptide during translocation. At the end of the signal peptide there is typically a stretch of amino acids that is recognized and cleaved by signal peptidase. Signal peptidase may cleave either during or after completion of translocation to generate a free signal peptide and a mature protein. The free signal peptides are then digested by specific proteases.
[0129] The signal peptide may be at the amino terminus of the molecule.
[0130] The signal peptide may comprise the sequence shown as SEQ ID NO: 12, 13 or 14 or a variant thereof having 5, 4, 3, 2 or 1 amino acid mutations (insertions, substitutions or additions) provided that the signal peptide still functions to cause cell surface expression of the CAR.
TABLE-US-00007 SEQ ID NO: 12: MGTSLLCWMALCLLGADHADG
[0131] The signal peptide of SEQ ID NO: 12 is compact and highly efficient. It is predicted to give about 95% cleavage after the terminal glycine, giving efficient removal by signal peptidase.
TABLE-US-00008 SEQ ID NO: 13: MSLPVTALLLPLALLLHAARP
[0132] The signal peptide of SEQ ID NO: 13 is derived from IgG1.
TABLE-US-00009 SEQ ID NO: 14: MAVPTQVLGLLLLWLTDARC
[0133] The signal peptide of SEQ ID NO: 14 is derived from CD8.
Spacer Domain
[0134] The CAR system described herein may comprise a spacer sequence to connect the antigen-binding domain with the transmembrane domain in the receptor component. A flexible spacer allows the antigen-binding domain to orient in different directions to facilitate binding.
[0135] The spacer sequence may, for example, comprise an IgG1 Fc region, an IgG1 hinge or a human CD8 stalk or the mouse CD8 stalk. The spacer may alternatively comprise an alternative linker sequence which has similar length and/or domain spacing properties as an IgG1 Fc region, an IgG1 hinge or a CD8 stalk. A human IgG1 spacer may be altered to remove Fc binding motifs.
[0136] Examples of amino acid sequences for these spacers are given below:
TABLE-US-00010 (hinge-CH2CH3 of human IgG1) SEQ ID NO: 15 AEPKSPDKTHTCPPCPAPPVAGPSVFLFPPKPKDTLMIARTPEVTCVVV DVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDW LNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQ VSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLT VDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGKKD SEQ ID NO: 16 (human CD8 stalk): TTTPAPRPPTPAPTIASQPLSLRPEACRPAAGGAVHTRGLDFACDI SEQ ID NO: 17 (human IgG1 hinge): AEPKSPDKTHTCPPCPKDPK (CD2 ectodomain) SEQ ID NO: 18 KEITNALETVVGALGQDINLDIPSFQMSDDIDDIKWEKTSDKKKIAQFR KEKETFKEKDTYKLFKNGTLKIKHLKTDDQDIYKVSIYDTKGKNVLEKI FDLKIQERVSKPKISVVTCINTTLTCEVIVINGTDPELNLYQDGKHLKL SQRVITHKVVTTSLSAKFKCTAGNKVSKESSVEPVSCPEKGLD (CD34 ectodomain) SEQ ID NO: 19 SLDNNGTATPELPTQGTFSNVSTNVSYQETTTPSTLGSTSLHPVSQHGN EATTNITETTVKFTSTSVITSVYGNTNSSVQSQTSVISTVFTTPANVST PETTLKPSLSPGNVSDLSTTSTSLATSPTKPYTSSSPILSDIKAEIKCS GIREVKLTQGICLEQNKTSSCAEFKKDRGEGLARVLCGEEQADADAGAQ VCSLLLAQSEVRPQCLLLVLANRTEISSKLQLMKKHQSDLKKLGILDFT EQDVASHQSYSQKT
Receptor Component Comprising a Plurality of First Binding Domains
[0137] The receptor component may comprise a plurality of first binding domains and thus be capable of recruiting more than one signalling component.
[0138] The plurality of first binding domains may be present in a single intracellular domain of the receptor component.
[0139] The receptor component may comprise an appropriate number of transmembrane domains such that each first binding domain is orientated on the intracellular side of the cell membrane. For example the receptor component may comprise 3, 5, 7, 9, 11, or more transmembrane domains. In this way, a single receptor component may recruit multiple signalling components amplifying signalling in response to antigen.
[0140] The first binding domains may each be variants which have a different affinity for the second binding domain of the signalling component.
Multiple Receptor Components
[0141] In another embodiment of the invention, the CAR system may comprise two or more receptor components each recognizing different antigens but comprising of the same intracellular first binding domain. Such a CAR system would be capable of recognizing multiple antigens (FIG. 11). This might be useful for instance in avoiding tumour escape. In a further related aspect of the invention, the first binding domains of the receptor components differ in residues which dictate their affinity for the second binding domain of the signalling component. In this way, a CAR system can be tuned such that signalling in response to one antigen is greater or lesser than the response to another (FIG. 11). This might be useful for instance when targeting two tumour antigens simultaneously but one is expressed at a higher density than the other. Response to this antigen could be tuned down to avoid toxicity caused by over-stimulation.
[0142] Methods suitable for altering the amino acid residues of the first or second binding domain such that the binding affinity between the two domains is altered are known in the art and include substitution, addition and removal of amino acids using both targeted and random mutagenesis. Methods for determining the binding affinity between a first binding domain and a second binding domain are also well known in the art and include bioinformatics prediction of protein-protein interactions, affinity electrophoresis, surface plasma resonance, bio-layer interferometry, dual polarisation interferometry, static light scattering and dynamic light scattering.
Signalling Component
[0143] The present invention also provides a signalling component comprising a signalling domain and a second binding domain. The signalling component is a soluble molecule and thus localises to the cytoplasm when it is expressed in a cell, for example a T cell.
[0144] No signalling occurs through the signalling domain of the signalling component unless it is co-localised with the receptor component provided by the present invention. Such co-localisation occurs only in the absence of the agent, as described above.
Intracellular Signalling Domain
[0145] The intracellular signalling domain is the signal-transmission portion of a classical CAR. In the signalling system of the present invention the intracellular signalling domain (signalling domain) is located in the signalling component. In the absence of the agent, the membrane-bound, receptor component and the intracellular signalling component are brought into proximity. After antigen recognition, receptors cluster, native CD45 and CD148 are excluded from the synapse and a signal is transmitted to the cell.
[0146] As such the signalling domain of the signalling component is analogous to the endodomain of a classical CAR molecule.
[0147] The most commonly used signalling domain component is that of CD3-zeta endodomain, which contains 3 ITAMs. This transmits an activation signal to the T cell after antigen is bound. CD3-zeta may not provide a fully competent activation signal and additional co-stimulatory signalling may be needed. For example, chimeric CD28 and OX40 can be used with CD3-Zeta to transmit a proliferative/survival signal, or all three can be used together (illustrated in FIG. 1B).
[0148] The signalling component described herein comprises a signalling domain, it may comprise the CD3-Zeta endodomain alone, the CD3-Zeta endodomain with that of either CD28 or
[0149] OX40 or the CD28 endodomain and OX40 and CD3-Zeta endodomain (FIG. 3A).
[0150] The signalling component of a CAR system according to the present invention may comprise the sequence shown as SEQ ID NO: 20, 21 or 22 or a variant thereof having at least 80% sequence identity.
TABLE-US-00011 -CD3 Z endodomain SEQ ID NO: 20 RVKFSRSADAPAYQQGQNQLYNELNLGRREEYDVLDKRRGRDPEMGGKPR RKNPQEGLYNELQKDKMAEAYSEIGMKGERRRGKGHDGLYQGLSTATKDT YDALHMQALPPR -CD28 and CD3 Zeta endodomains SEQ ID NO: 21 SKRSRLLHSDYMNMTPRRPGPTRKHYQPYAPPRDFAAYRSRVKFSRSADA PAYQQGQNQLYNELNLGRREEYDVLDKRRGRDPEMGGKPRRKNPQEGLYN ELQKDKMAEAYSEIGMKGERRRGKGHDGLYQGLSTATKDTYDALHMQALP PR -CD28, OX40 and CD3 Zeta endodomains SEQ ID NO: 22 SKRSRLLHSDYMNMTPRRPGPTRKHYQPYAPPRDFAAYRSRDQRLPPDAH KPPGGGSFRTPIQEEQADAHSTLAKIRVKFSRSADAPAYQQGQNQLYNEL NLGRREEYDVLDKRRGRDPEMGGKPRRKNPQEGLYNELQKDKMAEAYSEI GMKGERRRGKGHDGLYQGLSTATKDTYDALHMQALPPR
[0151] A variant sequence may have at least 80%, 85%, 90%, 95%, 98% or 99% sequence identity to SEQ ID NO: 20, 21 or 22, provided that the sequence provides an effective intracellular signalling domain.
Multiple Signalling Components
[0152] The signalling system according to the first aspect of the present invention may comprise a plurality of signalling components, each comprising a signalling domain and a second binding domain, wherein each second binding domain is bound by the same first binding domain of the receptor component but the signalling domains comprise different endodomains (FIG. 9). In this way, multiple different endodomains can be activated simultaneously. This is advantageous over a compound signalling domain since each signalling domain remains unencumbered from other signalling domains.
[0153] If each signalling component comprises a second binding domain which differs in residues which alter their affinity to the first binding domain of the receptor component, the signalling components comprising different signalling domains ligate to the first binding domain with differing kinetics (FIG. 10). This allows greater control over the signalling in response to antigen-binding by the receptor component as different signalling components are recruited to the receptor component in varying kinetics/dynamics. This is advantageous since rather than a fixed equal ratio of signal transmitted by a compound endodomain, an optimal T-cell activation signal may require different proportions of different immunological signals.
Nucleic Acid
[0154] The present invention further provides a nucleic acid encoding the receptor component of the second aspect and a nucleic acid encoding a signalling component of the third aspect.
[0155] As used herein, the terms "polynucleotide", "nucleotide", and "nucleic acid" are intended to be synonymous with each other.
[0156] It will be understood by a skilled person that numerous different polynucleotides and nucleic acids can encode the same polypeptide as a result of the degeneracy of the genetic code. In addition, it is to be understood that skilled persons may, using routine techniques, make nucleotide substitutions that do not affect the polypeptide sequence encoded by the polynucleotides described here to reflect the codon usage of any particular host organism in which the polypeptides are to be expressed.
[0157] Nucleic acids according to the invention may comprise DNA or RNA. They may be single-stranded or double-stranded. They may also be polynucleotides which include within them synthetic or modified nucleotides. A number of different types of modification to oligonucleotides are known in the art. These include methylphosphonate and phosphorothioate backbones, addition of acridine or polylysine chains at the 3' and/or 5' ends of the molecule. For the purposes of the use as described herein, it is to be understood that the polynucleotides may be modified by any method available in the art. Such modifications may be carried out in order to enhance the in vivo activity or life span of polynucleotides of interest.
[0158] The terms "variant", "homologue" or "derivative" in relation to a nucleotide sequence include any substitution of, variation of, modification of, replacement of, deletion of or addition of one (or more) nucleic acid from or to the sequence.
[0159] The nucleic acid of the invention may be a nucleic acid which encodes both the receptor component and the signalling component.
[0160] The nucleic acid may produce a polypeptide which comprises the receptor component and the signalling component joined by a cleavage site. The cleavage site may be self-cleaving, such that when the polypeptide is produced, it is immediately cleaved into the receptor component and the signalling component without the need for any external cleavage activity.
[0161] Various self-cleaving sites are known, including the Foot-and-Mouth disease virus (FMDV) 2a self-cleaving peptide, which has the sequence shown:
TABLE-US-00012 SEQ ID NO: 23 RAEGRGSLLTCGDVEENPGP. or SEQ ID NO: 24 QCTNYALLKLAGDVESNPGP
[0162] The nucleic acid may produce a polypeptide which comprises the sequence shown as SEQ ID NO: 25.
TABLE-US-00013 SEQ ID NO: 25 MWTWNAYAFAAPSGGGSADAPAYQQGQNQLYNELNLGRREEYDVLDKRRGR DPEMGGKPRRKNPQEGLYNELQKDKMAEAYSEIGMKGERRRGKGHDGLYQG LSTATKDTYDALHMQALPPRRAEGRGSLLTCGDVEENPGPMAVPTQVLGLL LLWLTDARCDIQMTQSPSSLSASVGDRVTITCRASEDIYFNLVWYQQKPGK APKLLIYDTNRLADGVPSRFSGSGSGTQYTLTISSLQPEDFATYYCQHYKN YPLTFGQGTKLEIKRSGGGGSGGGGSGGGGSGGGGSRSEVQLVESGGGLVQ PGGSLRLSGAASGFTLSNYGMHWIRQAPGKGLEWVSSISLNGGSTYYRDSV KGRFTISRDNAKSTLYLQMNSLRAEDTAVYYCAAQDAYTGGYFDYWGQGTL VTVSSMDPAEPKSPDKTHTCPPCPAPPVAGPSVFLFPPKPKDTLMIARTPE VTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVL HQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTK NQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLT VDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGKKDPM..SGGGGSMSR LDKSKVINSALELLNEVGIEGLTTRKLAQKLGVEQPTLYWHVKNKRALLDA LAIEMLDRHHTHFCPLEGESWQDFLRNNAKSFRCALLSHRDGAKVHLGTRP TEKQYETLENQLAFLCQQGFSLENALYALSAVGHFTLGCVLEDQEHQVAKE ERETPTTDSMPPLLRQAIELFDHQGAEPAFLFGLELIICGLEKQLKCESGS
[0163] Wherein indicates the position where an antigen binding domain sequence may be included. Any antigen binding domain may be included, for example an scFV, as described herein.
[0164] The co-expressing sequence may be an internal ribosome entry sequence (IRES). The co-expressing sequence may be an internal promoter.
[0165] The present invention also provides a kit comprising a nucleic acid encoding the receptor component of the second aspect and/or a nucleic acid encoding a signalling component of the third aspect.
Vector
[0166] The present invention also provides a vector, or kit of vectors which comprises one or more nucleic acid sequence(s) encoding a receptor component of the second aspect and/or signalling component of the third aspect of the invention. Such a vector may be used to introduce the nucleic acid sequence(s) into a host cell so that it expresses the receptor component and signalling component of the CAR system according to the first aspect of the invention.
[0167] The vector may, for example, be a plasmid or a viral vector, such as a retroviral vector or a lentiviral vector, or a transposon based vector or synthetic mRNA.
[0168] The vector may be capable of transfecting or transducing a T cell or a NK cell.
Cytolytic Immune Cell
[0169] The present invention also relates to an immune cell comprising the CAR system according to the first aspect of the invention.
[0170] The cytolytic immune cell may comprise a nucleic acid or a vector of the present invention.
[0171] The cytolytic immune cell may comprise a receptor component and a signalling component of the present invention.
[0172] The cytolytic immune cell may comprise at least one signalling component of the present invention. For example the cytolytic immune cell may comprise one, two, three, four, five, up to a plurality of signalling components of the present invention.
[0173] The cytolytic immune cell may comprise at least one receptor component of the present invention. For example the cytolytic immune cell may comprise one, two, three, four, five, up to a plurality of receptor components of the present invention.
[0174] Cytolytic immune cells can be T cells or T lymphocytes which are a type of lymphocyte that play a central role in cell-mediated immunity. They can be distinguished from other lymphocytes, such as B cells and natural killer cells (NK cells), by the presence of a T-cell receptor (TCR) on the cell surface. There are various types of T cell, as summarised below.
[0175] Helper T helper cells (TH cells) assist other white blood cells in immunologic processes, including maturation of B cells into plasma cells and memory B cells, and activation of cytotoxic T cells and macrophages. TH cells express CD4 on their surface. TH cells become activated when they are presented with peptide antigens by MHC class II molecules on the surface of antigen presenting cells (APCs). These cells can differentiate into one of several subtypes, including TH1, TH2, TH3, TH17, Th9, or TFH, which secrete different cytokines to facilitate different types of immune responses.
[0176] Cytolytic T cells (TC cells, or CTLs) destroy virally infected cells and tumor cells, and are also implicated in transplant rejection. CTLs express the CD8 at their surface. These cells recognize their targets by binding to antigen associated with MHC class I, which is present on the surface of all nucleated cells. Through IL-10, adenosine and other molecules secreted by regulatory T cells, the CD8+ cells can be inactivated to an anergic state, which prevent autoimmune diseases such as experimental autoimmune encephalomyelitis.
[0177] Memory T cells are a subset of antigen-specific T cells that persist long-term after an infection has resolved. They quickly expand to large numbers of effector T cells upon re-exposure to their cognate antigen, thus providing the immune system with "memory" against past infections. Memory T cells comprise three subtypes: central memory T cells (TCM cells) and two types of effector memory T cells (TEM cells and TEMRA cells). Memory cells may be either CD4+ or CD8+. Memory T cells typically express the cell surface protein CD45RO.
[0178] Regulatory T cells (Treg cells), formerly known as suppressor T cells, are crucial for the maintenance of immunological tolerance. Their major role is to shut down T cell-mediated immunity toward the end of an immune reaction and to suppress auto-reactive T cells that escaped the process of negative selection in the thymus.
[0179] Two major classes of CD4+ Treg cells have been described--naturally occurring Treg cells and adaptive Treg cells.
[0180] Naturally occurring Treg cells (also known as CD4+ CD25+ FoxP3+ Treg cells) arise in the thymus and have been linked to interactions between developing T cells with both myeloid (CD11c+) and plasmacytoid (CD123+) dendritic cells that have been activated with TSLP. Naturally occurring Treg cells can be distinguished from other T cells by the presence of an intracellular molecule called FoxP3. Mutations of the FOXP3 gene can prevent regulatory T cell development, causing the fatal autoimmune disease IPEX.
[0181] Adaptive Treg cells (also known as Tr1 cells or Th3 cells) may originate during a normal immune response.
[0182] Natural Killer Cells (or NK cells) are a type of cytolytic cell which form part of the innate immune system. NK cells provide rapid responses to innate signals from virally infected cells in an MHC independent manner
[0183] NK cells (belonging to the group of innate lymphoid cells) are defined as large granular lymphocytes (LGL) and constitute the third kind of cells differentiated from the common lymphoid progenitor generating B and T lymphocytes. NK cells are known to differentiate and mature in the bone marrow, lymph node, spleen, tonsils and thymus where they then enter into the circulation.
[0184] The CAR cells of the invention may be any of the cell types mentioned above.
[0185] T or NK cells expressing the molecules of the CAR system according to the first aspect of the invention may either be created ex vivo either from a patient's own peripheral blood (1st party), or in the setting of a haematopoietic stem cell transplant from donor peripheral blood (2nd party), or peripheral blood from an unconnected donor (3rd party).
[0186] Alternatively, T or NK cells expressing the molecules of the CAR system according to the first aspect of the invention may be derived from ex vivo differentiation of inducible progenitor cells or embryonic progenitor cells to T cells. Alternatively, an immortalized T-cell line which retains its lytic function and could act as a therapeutic may be used.
[0187] In all these embodiments, CAR cells are generated by introducing DNA or RNA coding for the receptor component and signalling component by one of many means including transduction with a viral vector, transfection with DNA or RNA.
[0188] The CAR cell of the invention may be an ex vivo T or NK cell from a subject. The T or NK cell may be from a peripheral blood mononuclear cell (PBMC) sample. T or NK cells may be activated and/or expanded prior to being transduced with nucleic acid encoding the molecules providing the CAR system according to the first aspect of the invention, for example by treatment with an anti-CD3 monoclonal antibody.
[0189] The T or NK cell of the invention may be made by: [0190] (i) isolation of a T or NK cell-containing sample from a subject or other sources listed above; and [0191] (ii) transduction or transfection of the T or NK cells with one or more a nucleic acid sequence(s) encoding the receptor component and/or signalling component of the CAR system according to the second and third aspects of the invention.
[0192] The T or NK cells may then by purified, for example, selected on the basis of expression of the antigen-binding domain of the antigen-binding polypeptide.
[0193] The present invention also provides a kit which comprises a T or NK cell comprising the CAR system according to the first aspect of the invention.
Pharmaceutical Composition
[0194] The present invention also relates to a pharmaceutical composition containing a plurality of cytolytic immune cells expressing the components of the CAR system of the first aspect of the invention. The pharmaceutical composition may additionally comprise a pharmaceutically acceptable carrier, diluent or excipient. The pharmaceutical composition may optionally comprise one or more further pharmaceutically active polypeptides and/or compounds. Such a formulation may, for example, be in a form suitable for intravenous infusion.
Method of Treatment
[0195] The present invention provides a method for treating and/or preventing a disease which comprises the step of administering the cytolytic immune cells of the present invention (for example in a pharmaceutical composition as described above) to a subject.
[0196] A method for treating a disease relates to the therapeutic use of the cytolytic immune cells of the present invention. Herein the cells may be administered to a subject having an existing disease or condition in order to lessen, reduce or improve at least one symptom associated with the disease and/or to slow down, reduce or block the progression of the disease.
[0197] The method for preventing a disease relates to the prophylactic use of the cytolytic immune cells of the present invention. Herein such cells may be administered to a subject who has not yet contracted the disease and/or who is not showing any symptoms of the disease to prevent or impair the cause of the disease or to reduce or prevent development of at least one symptom associated with the disease. The subject may have a predisposition for, or be thought to be at risk of developing, the disease.
[0198] The method may involve the steps of: [0199] (i) isolating a T or NK cell-containing sample; [0200] (ii) transducing or transfecting such cells with a nucleic acid sequence or vector provided by the present invention; [0201] (iii) administering the cells from (ii) to a subject.
[0202] The T or NK cell-containing sample may be isolated from a subject or from other sources, for example as described above. The T or NK cells may be isolated from a subject's own peripheral blood (1st party), or in the setting of a haematopoietic stem cell transplant from donor peripheral blood (2nd party), or peripheral blood from an unconnected donor (3rd party).
[0203] The methods provided by the present invention for treating a disease may involve monitoring the progression of the disease and any toxic activity and administering an agent suitable for use in the CAR system according to the first aspect of the invention to inhibit CAR signalling and thereby reduce or lessen any adverse toxic effects.
[0204] The methods provided by the present invention for treating a disease may involve monitoring the progression of the disease and monitoring any toxic activity and adjusting the dose of the agent administered to the subject to provide acceptable levels of disease progression and toxic activity.
[0205] Monitoring the progression of the disease means to assess the symptoms associated with the disease over time to determine if they are reducing/improving or increasing/worsening.
[0206] Toxic activities relate to adverse effects caused by the CAR cells of the invention following their administration to a subject. Toxic activities may include, for example, immunological toxicity, biliary toxicity and respiratory distress syndrome.
[0207] The level of signalling through the signalling system of the first aspect of the invention, and therefore the level of activation of CAR cells expressing the signalling system, may be adjusted by altering the amount of agent present, or the amount of time the agent is present. In the present method the level of CAR cell activation may be augmented by decreasing the dose of agent administered to the subject or decreasing the frequency of its administration. Conversely, the level of CAR cell activation may be reduced by increasing the dose of the agent, or the frequency of administration to the subject.
[0208] Higher levels of CAR cell activation are likely to be associated with reduced disease progression but increased toxic activities, whilst lower levels of CAR cell activation are likely to be associated with increased disease progression but reduced toxic activities.
[0209] The present invention also provides a method for treating and/or preventing a disease in a subject which subject comprises cells of the invention, which method comprises the step of administering an agent suitable for use in the CAR system according to the first aspect to the subject. As such, this method involves administering a suitable agent to a subject which already comprises CAR cells of the present invention.
[0210] As such the dose of agent administered to a subject, or the frequency of administration, may be altered in order to provide an acceptable level of both disease progression and toxic activity. The specific level of disease progression and toxic activities determined to be `acceptable` will vary according to the specific circumstances and should be assessed on such a basis. The present invention provides a method for altering the activation level of the
[0211] CAR cells in order to achieve this appropriate level.
[0212] The agent may be administered in the form of a pharmaceutical composition. The pharmaceutical composition may additionally comprise a pharmaceutically acceptable carrier, diluent or excipient. The pharmaceutical composition may optionally comprise one or more further pharmaceutically active polypeptides and/or compounds. Such a formulation may, for example, be in a form suitable for intravenous infusion.
[0213] The present invention provides a CAR cell of the present invention for use in treating and/or preventing a disease.
[0214] The invention also relates to the use of a CAR cell of the present invention in the manufacture of a medicament for the treatment and/or prevention of a disease.
[0215] The present invention also provides an agent suitable for inhibiting a CAR system according to the first aspect of the invention for use in treating and/or preventing a disease.
[0216] The present invention also provides an agent for use in inhibiting a CAR system according to the first aspect of the invention in a CAR cell.
[0217] The invention also provides the use of an agent suitable for inhibiting a CAR system according to the first aspect of the invention in the manufacture of a medicament for the treatment and/or prevention of a disease.
[0218] The disease to be treated and/or prevented by the methods of the present invention may be an infection, such as a viral infection.
[0219] The methods of the invention may also be for the control of pathogenic immune responses, for example in autoimmune diseases, allergies and graft-vs-host rejection.
[0220] The methods may be for the treatment of a cancerous disease, such as bladder cancer, breast cancer, colon cancer, endometrial cancer, kidney cancer (renal cell), leukaemia, lung cancer, melanoma, non-Hodgkin lymphoma, pancreatic cancer, prostate cancer and thyroid cancer.
[0221] The CAR cells of the present invention may be capable of killing target cells, such as cancer cells. The target cell may be recognisable by expression of a TAA, for example the expression of a TAA provided above in Table 1.
[0222] The CAR cells and pharmaceutical compositions of present invention may be for use in the treatment and/or prevention of the diseases described above.
[0223] The CAR cells and pharmaceutical compositions of present invention may be for use in any of the methods described above.
[0224] The invention will now be further described by way of Examples, which are meant to serve to assist one of ordinary skill in the art in carrying out the invention and are not intended in any way to limit the scope of the invention.
EXAMPLES
Example 1--Functionality of the TetCAR Signalling System
[0225] A bicistronic construct was expressed as a single transcript which self-cleaves at the 2A site to yield TiP fused to eGFP and a CAR with TetR as its endodomain (FIG. 5a).
[0226] Fluorescent microscopy of SupT1 cells expressing this construct in the absence of tetracycline demonstrated that eGFP fluorescence can clearly be seen at the cell membrane (FIG. 5b); whilst in the presence of tetracycline the eGFP was cytoplasmic (FIG. 5c). These data demonstrate that tetracycline has displaced TiP from the TetR CAR.
Example 2--Signalling Through the TetCAR System
[0227] A bicistronic construct was expressed in BW5 T cells as a single transcript which self-cleaves at the 2A site to yield a signalling component which comprises TiP fused via a flexible linker to the endodomain of CD3-Zeta; and a receptor component which comprises a CD33 recognizing scFv, a spacer derived from the Fc domain of IgG1, a CD4 derived transmembrane and intracellular domain; and TetR (FIG. 6a). A control was also expressed which was identical except that TiP was absent from the signalling component (FIG. 6b).
[0228] The BW5 T-cells were challenged with wild-type SupT1 cells or SupT1 cells engineered to express CD33 in the absence of tetracycline or in the presence of increasing concentrations of tetracycline. T-cells challenged with wild-type SupT1 cells did not activate in either the presence or absence of Tetracyline; T-cells challenged with SupT1 cells expressing CD33 were activated in the absence of Tetracycline, but activation is rapidly inhibited in the presence of tetracycline with activation fully inhibited in the presence of 100 nM of tetracycline (FIG. 7a).
[0229] Control TetCAR which lacks the TiP domain was also transduced into BW5. Once again, these T-cells were challenged with wild-type SupT1 cells or SupT1 cells engineered to express CD33 in the absence or in the presence of increasing concentration of Tetracycline. A lack of TiP element in signalling component resulted in no signalling in any conditions (FIG. 7b).
Example 3--Signalling of the TetCAR System in Primary T Cells
[0230] SupT1 cells (which are CD19 negative), were engineered to be CD19 positive giving target negative and positive cell lines which were as similar as possible. Primary human T-cells from 3 donors were transduced with three CAR constructs: (i) "Classical" 1st generation anti-CD19 CAR; (ii) 1st generation anti-CD19 tetCAR; (iii) Control anti-CD19 tetCAR where TiP is missing from endodomain. Non-transduced T-cells and T-cells transduced with the different CAR constructs were challenged 1:1 with either SupT1 cells or SupT1.CD19 cells in the presence of different concentrations of Tetracycline. Supernatant was sampled 48 hours after challenge. Supernatant from background (T-cells alone), and maximum (T-cells stimulated with PMA/Ionomycin) was also samples. Interferon-gamma was measured in supernatants by ELISA (FIG. 13). "Classical" CAR T-cells were activated by SupT1.CD19 irrespective of tetracycline. TetCAR T-cell were activated by SupT1.CD19 cells but activation was inhibited by Tetracycline. The control TetCAR and NT T-cells did not respond to SupT1.CD19 cells.
Example 4--Killing of Target Cells
[0231] Following on from the interferon-gamma release study described in Example 3, killing of target cells was demonstrated using a chromium release assay. SupT1 and SupT1.CD19 cells were loaded with .sup.51Cr and incubated with control and Tet-CAR T-cells for 4 hours in the presence or absence of tetracycline. Lysis of target cells was determined by counting .sup.51Cr in the supernatant. The results are shown in FIG. 14. It was shown that Tet-CAR T-cells lysed SupT1.CD19 target cells only in the absence of Tetracycline.
[0232] All publications mentioned in the above specification are herein incorporated by reference. Various modifications and variations of the described methods and system of the invention will be apparent to those skilled in the art without departing from the scope and spirit of the invention. Although the invention has been described in connection with specific preferred embodiments, it should be understood that the invention as claimed should not be unduly limited to such specific embodiments. Indeed, various modifications of the described modes for carrying out the invention which are obvious to those skilled in molecular biology, cellular immunology or related fields are intended to be within the scope of the following claims.
Sequence CWU 1
1
291139PRTArtificial SequenceTetR binding domain 1Met Ser Arg Leu
Asp Lys Ser Lys Val Ile Asn Ser Ala Leu Glu Leu1 5 10 15Leu Asn Glu
Val Gly Ile Glu Gly Leu Thr Thr Arg Lys Leu Ala Gln 20 25 30Lys Leu
Gly Val Glu Gln Pro Thr Leu Tyr Trp His Val Lys Asn Lys 35 40 45Arg
Ala Leu Leu Asp Ala Leu Ala Ile Glu Met Leu Asp Arg His His 50 55
60Thr His Phe Cys Pro Leu Glu Gly Glu Ser Trp Gln Asp Phe Leu Arg65
70 75 80Asn Asn Ala Lys Ser Phe Arg Cys Ala Leu Leu Ser His Arg Asp
Gly 85 90 95Ala Lys Val His Leu Gly Thr Arg Pro Thr Glu Lys Gln Tyr
Glu Thr 100 105 110Leu Glu Asn Gln Leu Ala Phe Leu Cys Gln Gln Gly
Phe Ser Leu Glu 115 120 125Asn Ala Leu Tyr Ala Leu Ser Ala Val Gly
His 130 135217PRTArtificial SequenceTiP binding domain 2Met Trp Thr
Trp Asn Ala Tyr Ala Phe Ala Ala Pro Ser Gly Gly Gly1 5 10
15Ser361PRTArtificial SequenceModified CD4 endodomain linker 3Ala
Leu Ile Val Leu Gly Gly Val Ala Gly Leu Leu Leu Phe Ile Gly1 5 10
15Leu Gly Ile Phe Phe Cys Val Arg Cys Arg His Arg Arg Arg Gln Ala
20 25 30Glu Arg Met Ala Gln Ile Lys Arg Val Val Ser Glu Lys Lys Thr
Ala 35 40 45Gln Ala Pro His Arg Phe Gln Lys Thr Cys Ser Pro Ile 50
55 60415PRTArtificial SequenceBiotin mimicking peptide, long
nanotag 4Asp Val Glu Ala Trp Leu Asp Glu Arg Val Pro Leu Val Glu
Thr1 5 10 1559PRTArtificial SequenceBiotin mimicking peptide, short
nanotag 5Asp Val Glu Ala Trp Leu Gly Ala Arg1 568PRTArtificial
SequenceBiotin mimicking peptide, streptag 6Trp Arg His Pro Gln Phe
Gly Gly1 578PRTArtificial SequenceBiotin mimicking peptide,
streptagII 7Trp Ser His Pro Gln Phe Glu Lys1 5838PRTArtificial
SequenceBiotin mimicking peptide, SBP-tag 8Met Asp Glu Lys Thr Thr
Gly Trp Arg Gly Gly His Val Val Glu Gly1 5 10 15Leu Ala Gly Glu Leu
Glu Gln Leu Arg Ala Arg Leu Glu His His Pro 20 25 30Gln Gly Gln Arg
Glu Pro 3598PRTArtificial SequenceBiotin mimicking peptide,
ccstreptag 9Cys His Pro Gln Gly Pro Pro Cys1 51015PRTArtificial
SequenceBiotin mimicking peptide, flankedccstreptag 10Ala Glu Cys
His Pro Gln Gly Pro Pro Cys Ile Glu Gly Arg Lys1 5 10
1511126PRTArtificial SequenceCore streptavidin sequence 11Glu Ala
Gly Ile Thr Gly Thr Trp Tyr Asn Gln Leu Gly Ser Thr Phe1 5 10 15Ile
Val Thr Ala Gly Ala Asp Gly Ala Leu Thr Gly Thr Tyr Glu Ser 20 25
30Ala Val Gly Asn Ala Glu Ser Arg Tyr Val Leu Thr Gly Arg Tyr Asp
35 40 45Ser Ala Pro Ala Thr Asp Gly Ser Gly Thr Ala Leu Gly Trp Thr
Val 50 55 60Ala Trp Lys Asn Asn Tyr Arg Asn Ala His Ser Ala Thr Thr
Trp Ser65 70 75 80Gly Gln Tyr Val Gly Gly Ala Glu Ala Arg Ile Asn
Thr Gln Trp Leu 85 90 95Leu Thr Ser Gly Thr Thr Glu Ala Asn Ala Trp
Lys Ser Thr Leu Val 100 105 110Gly His Asp Thr Phe Thr Lys Val Lys
Pro Ser Ala Ala Ser 115 120 1251221PRTArtificial Sequencesignal
peptide 12Met Gly Thr Ser Leu Leu Cys Trp Met Ala Leu Cys Leu Leu
Gly Ala1 5 10 15Asp His Ala Asp Gly 201321PRTArtificial
Sequencesignal peptide 13Met Ser Leu Pro Val Thr Ala Leu Leu Leu
Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg Pro
201420PRTArtificial Sequencesignal peptide 14Met Ala Val Pro Thr
Gln Val Leu Gly Leu Leu Leu Leu Trp Leu Thr1 5 10 15Asp Ala Arg Cys
2015234PRTArtificial Sequencespacer sequence, hinge-CH2CH3 of human
IgG1 15Ala Glu Pro Lys Ser Pro Asp Lys Thr His Thr Cys Pro Pro Cys
Pro1 5 10 15Ala Pro Pro Val Ala Gly Pro Ser Val Phe Leu Phe Pro Pro
Lys Pro 20 25 30Lys Asp Thr Leu Met Ile Ala Arg Thr Pro Glu Val Thr
Cys Val Val 35 40 45Val Asp Val Ser His Glu Asp Pro Glu Val Lys Phe
Asn Trp Tyr Val 50 55 60Asp Gly Val Glu Val His Asn Ala Lys Thr Lys
Pro Arg Glu Glu Gln65 70 75 80Tyr Asn Ser Thr Tyr Arg Val Val Ser
Val Leu Thr Val Leu His Gln 85 90 95Asp Trp Leu Asn Gly Lys Glu Tyr
Lys Cys Lys Val Ser Asn Lys Ala 100 105 110Leu Pro Ala Pro Ile Glu
Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro 115 120 125Arg Glu Pro Gln
Val Tyr Thr Leu Pro Pro Ser Arg Asp Glu Leu Thr 130 135 140Lys Asn
Gln Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser145 150 155
160Asp Ile Ala Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr
165 170 175Lys Thr Thr Pro Pro Val Leu Asp Ser Asp Gly Ser Phe Phe
Leu Tyr 180 185 190Ser Lys Leu Thr Val Asp Lys Ser Arg Trp Gln Gln
Gly Asn Val Phe 195 200 205Ser Cys Ser Val Met His Glu Ala Leu His
Asn His Tyr Thr Gln Lys 210 215 220Ser Leu Ser Leu Ser Pro Gly Lys
Lys Asp225 2301646PRTArtificial Sequencespacer sequence, human CD8
stalk 16Thr Thr Thr Pro Ala Pro Arg Pro Pro Thr Pro Ala Pro Thr Ile
Ala1 5 10 15Ser Gln Pro Leu Ser Leu Arg Pro Glu Ala Cys Arg Pro Ala
Ala Gly 20 25 30Gly Ala Val His Thr Arg Gly Leu Asp Phe Ala Cys Asp
Ile 35 40 451720PRTArtificial Sequencespacer sequence, human IgG1
hinge 17Ala Glu Pro Lys Ser Pro Asp Lys Thr His Thr Cys Pro Pro Cys
Pro1 5 10 15Lys Asp Pro Lys 2018185PRTArtificial Sequencespacer
sequence, CD2 ectodomain 18Lys Glu Ile Thr Asn Ala Leu Glu Thr Trp
Gly Ala Leu Gly Gln Asp1 5 10 15Ile Asn Leu Asp Ile Pro Ser Phe Gln
Met Ser Asp Asp Ile Asp Asp 20 25 30Ile Lys Trp Glu Lys Thr Ser Asp
Lys Lys Lys Ile Ala Gln Phe Arg 35 40 45Lys Glu Lys Glu Thr Phe Lys
Glu Lys Asp Thr Tyr Lys Leu Phe Lys 50 55 60Asn Gly Thr Leu Lys Ile
Lys His Leu Lys Thr Asp Asp Gln Asp Ile65 70 75 80Tyr Lys Val Ser
Ile Tyr Asp Thr Lys Gly Lys Asn Val Leu Glu Lys 85 90 95Ile Phe Asp
Leu Lys Ile Gln Glu Arg Val Ser Lys Pro Lys Ile Ser 100 105 110Trp
Thr Cys Ile Asn Thr Thr Leu Thr Cys Glu Val Met Asn Gly Thr 115 120
125Asp Pro Glu Leu Asn Leu Tyr Gln Asp Gly Lys His Leu Lys Leu Ser
130 135 140Gln Arg Val Ile Thr His Lys Trp Thr Thr Ser Leu Ser Ala
Lys Phe145 150 155 160Lys Cys Thr Ala Gly Asn Lys Val Ser Lys Glu
Ser Ser Val Glu Pro 165 170 175Val Ser Cys Pro Glu Lys Gly Leu Asp
180 18519259PRTArtificial Sequencespacer sequence, CD34 ectodomain
19Ser Leu Asp Asn Asn Gly Thr Ala Thr Pro Glu Leu Pro Thr Gln Gly1
5 10 15Thr Phe Ser Asn Val Ser Thr Asn Val Ser Tyr Gln Glu Thr Thr
Thr 20 25 30Pro Ser Thr Leu Gly Ser Thr Ser Leu His Pro Val Ser Gln
His Gly 35 40 45Asn Glu Ala Thr Thr Asn Ile Thr Glu Thr Thr Val Lys
Phe Thr Ser 50 55 60Thr Ser Val Ile Thr Ser Val Tyr Gly Asn Thr Asn
Ser Ser Val Gln65 70 75 80Ser Gln Thr Ser Val Ile Ser Thr Val Phe
Thr Thr Pro Ala Asn Val 85 90 95Ser Thr Pro Glu Thr Thr Leu Lys Pro
Ser Leu Ser Pro Gly Asn Val 100 105 110Ser Asp Leu Ser Thr Thr Ser
Thr Ser Leu Ala Thr Ser Pro Thr Lys 115 120 125Pro Tyr Thr Ser Ser
Ser Pro Ile Leu Ser Asp Ile Lys Ala Glu Ile 130 135 140Lys Cys Ser
Gly Ile Arg Glu Val Lys Leu Thr Gln Gly Ile Cys Leu145 150 155
160Glu Gln Asn Lys Thr Ser Ser Cys Ala Glu Phe Lys Lys Asp Arg Gly
165 170 175Glu Gly Leu Ala Arg Val Leu Cys Gly Glu Glu Gln Ala Asp
Ala Asp 180 185 190Ala Gly Ala Gln Val Cys Ser Leu Leu Leu Ala Gln
Ser Glu Val Arg 195 200 205Pro Gln Cys Leu Leu Leu Val Leu Ala Asn
Arg Thr Glu Ile Ser Ser 210 215 220Lys Leu Gln Leu Met Lys Lys His
Gln Ser Asp Leu Lys Lys Leu Gly225 230 235 240Ile Leu Asp Phe Thr
Glu Gln Asp Val Ala Ser His Gln Ser Tyr Ser 245 250 255Gln Lys
Thr20112PRTArtificial Sequencesignalling component, CD3 Z
endodomain 20Arg Val Lys Phe Ser Arg Ser Ala Asp Ala Pro Ala Tyr
Gln Gln Gly1 5 10 15Gln Asn Gln Leu Tyr Asn Glu Leu Asn Leu Gly Arg
Arg Glu Glu Tyr 20 25 30Asp Val Leu Asp Lys Arg Arg Gly Arg Asp Pro
Glu Met Gly Gly Lys 35 40 45Pro Arg Arg Lys Asn Pro Gln Glu Gly Leu
Tyr Asn Glu Leu Gln Lys 50 55 60Asp Lys Met Ala Glu Ala Tyr Ser Glu
Ile Gly Met Lys Gly Glu Arg65 70 75 80Arg Arg Gly Lys Gly His Asp
Gly Leu Tyr Gln Gly Leu Ser Thr Ala 85 90 95Thr Lys Asp Thr Tyr Asp
Ala Leu His Met Gln Ala Leu Pro Pro Arg 100 105
11021152PRTArtificial Sequencesignalling component, CD28 and CD3
Zeta endodomains 21Ser Lys Arg Ser Arg Leu Leu His Ser Asp Tyr Met
Asn Met Thr Pro1 5 10 15Arg Arg Pro Gly Pro Thr Arg Lys His Tyr Gln
Pro Tyr Ala Pro Pro 20 25 30Arg Asp Phe Ala Ala Tyr Arg Ser Arg Val
Lys Phe Ser Arg Ser Ala 35 40 45Asp Ala Pro Ala Tyr Gln Gln Gly Gln
Asn Gln Leu Tyr Asn Glu Leu 50 55 60Asn Leu Gly Arg Arg Glu Glu Tyr
Asp Val Leu Asp Lys Arg Arg Gly65 70 75 80Arg Asp Pro Glu Met Gly
Gly Lys Pro Arg Arg Lys Asn Pro Gln Glu 85 90 95Gly Leu Tyr Asn Glu
Leu Gln Lys Asp Lys Met Ala Glu Ala Tyr Ser 100 105 110Glu Ile Gly
Met Lys Gly Glu Arg Arg Arg Gly Lys Gly His Asp Gly 115 120 125Leu
Tyr Gln Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr Asp Ala Leu 130 135
140His Met Gln Ala Leu Pro Pro Arg145 15022188PRTArtificial
Sequencesignalling component, CD28, OX40 and CD3 Zeta endodomains
22Ser Lys Arg Ser Arg Leu Leu His Ser Asp Tyr Met Asn Met Thr Pro1
5 10 15Arg Arg Pro Gly Pro Thr Arg Lys His Tyr Gln Pro Tyr Ala Pro
Pro 20 25 30Arg Asp Phe Ala Ala Tyr Arg Ser Arg Asp Gln Arg Leu Pro
Pro Asp 35 40 45Ala His Lys Pro Pro Gly Gly Gly Ser Phe Arg Thr Pro
Ile Gln Glu 50 55 60Glu Gln Ala Asp Ala His Ser Thr Leu Ala Lys Ile
Arg Val Lys Phe65 70 75 80Ser Arg Ser Ala Asp Ala Pro Ala Tyr Gln
Gln Gly Gln Asn Gln Leu 85 90 95Tyr Asn Glu Leu Asn Leu Gly Arg Arg
Glu Glu Tyr Asp Val Leu Asp 100 105 110Lys Arg Arg Gly Arg Asp Pro
Glu Met Gly Gly Lys Pro Arg Arg Lys 115 120 125Asn Pro Gln Glu Gly
Leu Tyr Asn Glu Leu Gln Lys Asp Lys Met Ala 130 135 140Glu Ala Tyr
Ser Glu Ile Gly Met Lys Gly Glu Arg Arg Arg Gly Lys145 150 155
160Gly His Asp Gly Leu Tyr Gln Gly Leu Ser Thr Ala Thr Lys Asp Thr
165 170 175Tyr Asp Ala Leu His Met Gln Ala Leu Pro Pro Arg 180
1852320PRTArtificial Sequenceself-cleaving site, 2a self-cleaving
peptide 23Arg Ala Glu Gly Arg Gly Ser Leu Leu Thr Cys Gly Asp Val
Glu Glu1 5 10 15Asn Pro Gly Pro 202420PRTArtificial
Sequenceself-cleaving site, 2a self-cleaving peptide 24Gln Cys Thr
Asn Tyr Ala Leu Leu Lys Leu Ala Gly Asp Val Glu Ser1 5 10 15Asn Pro
Gly Pro 2025652PRTArtificial Sequencechimeric antigen receptor
(CAR) sequence 25Met Trp Thr Trp Asn Ala Tyr Ala Phe Ala Ala Pro
Ser Gly Gly Gly1 5 10 15Ser Ala Asp Ala Pro Ala Tyr Gln Gln Gly Gln
Asn Gln Leu Tyr Asn 20 25 30Glu Leu Asn Leu Gly Arg Arg Glu Glu Tyr
Asp Val Leu Asp Lys Arg 35 40 45Arg Gly Arg Asp Pro Glu Met Gly Gly
Lys Pro Arg Arg Lys Asn Pro 50 55 60Gln Glu Gly Leu Tyr Asn Glu Leu
Gln Lys Asp Lys Met Ala Glu Ala65 70 75 80Tyr Ser Glu Ile Gly Met
Lys Gly Glu Arg Arg Arg Gly Lys Gly His 85 90 95Asp Gly Leu Tyr Gln
Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr Asp 100 105 110Ala Leu His
Met Gln Ala Leu Pro Pro Arg Arg Ala Glu Gly Arg Gly 115 120 125Ser
Leu Leu Thr Cys Gly Asp Val Glu Glu Asn Pro Gly Pro Met Ala 130 135
140Val Pro Thr Gln Val Leu Gly Leu Leu Leu Leu Trp Leu Thr Asp
Ala145 150 155 160Arg Cys Asp Ile Gln Met Thr Gln Ser Pro Ser Ser
Leu Ser Ala Ser 165 170 175Val Gly Asp Arg Val Thr Ile Thr Cys Arg
Ala Ser Glu Asp Ile Tyr 180 185 190Phe Asn Leu Val Trp Tyr Gln Gln
Lys Pro Gly Lys Ala Pro Lys Leu 195 200 205Leu Ile Tyr Asp Thr Asn
Arg Leu Ala Asp Gly Val Pro Ser Arg Phe 210 215 220Ser Gly Ser Gly
Ser Gly Thr Gln Tyr Thr Leu Thr Ile Ser Ser Leu225 230 235 240Gln
Pro Glu Asp Phe Ala Thr Tyr Tyr Cys Gln His Tyr Lys Asn Tyr 245 250
255Pro Leu Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys Arg Ser Gly
260 265 270Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser
Gly Gly 275 280 285Gly Gly Ser Arg Ser Glu Val Gln Leu Val Glu Ser
Gly Gly Gly Leu 290 295 300Val Gln Pro Gly Gly Ser Leu Arg Leu Ser
Cys Ala Ala Ser Gly Phe305 310 315 320Thr Leu Ser Asn Tyr Gly Met
His Trp Ile Arg Gln Ala Pro Gly Lys 325 330 335Gly Leu Glu Trp Val
Ser Ser Ile Ser Leu Asn Gly Gly Ser Thr Tyr 340 345 350Tyr Arg Asp
Ser Val Lys Gly Arg Phe Thr Ile Ser Arg Asp Asn Ala 355 360 365Lys
Ser Thr Leu Tyr Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr 370 375
380Ala Val Tyr Tyr Cys Ala Ala Gln Asp Ala Tyr Thr Gly Gly Tyr
Phe385 390 395 400Asp Tyr Trp Gly Gln Gly Thr Leu Val Thr Val Ser
Ser Met Asp Pro 405 410 415Ala Glu Pro Lys Ser Pro Asp Lys Thr His
Thr Cys Pro Pro Cys Pro 420 425 430Ala Pro Pro Val Ala Gly Pro Ser
Val Phe Leu Phe Pro Pro Lys Pro 435 440 445Lys Asp Thr Leu Met Ile
Ala Arg Thr Pro Glu Val Thr Cys Val Val 450 455 460Val Asp Val Ser
His Glu Asp Pro Glu Val Lys Phe Asn Trp Tyr Val465 470 475 480Asp
Gly Val Glu Val His Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln 485 490
495Tyr Asn Ser Thr Tyr Arg Val Val Ser Val Leu Thr Val Leu His Gln
500 505 510Asp Trp Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val Ser
Asn
Lys Ala 515 520 525Leu Pro Ala Pro Ile Glu Lys Thr Ile Ser Lys Ala
Lys Gly Gln Pro 530 535 540Arg Glu Pro Gln Val Tyr Thr Leu Pro Pro
Ser Arg Asp Glu Leu Thr545 550 555 560Lys Asn Gln Val Ser Leu Thr
Cys Leu Val Lys Gly Phe Tyr Pro Ser 565 570 575Asp Ile Ala Val Glu
Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr 580 585 590Lys Thr Thr
Pro Pro Val Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr 595 600 605Ser
Lys Leu Thr Val Asp Lys Ser Arg Trp Gln Gln Gly Asn Val Phe 610 615
620Ser Cys Ser Val Met His Glu Ala Leu His Asn His Tyr Thr Gln
Lys625 630 635 640Ser Leu Ser Leu Ser Pro Gly Lys Lys Asp Pro Met
645 65026213PRTArtificial Sequencechimeric antigen receptor (CAR)
sequence 26Ser Gly Gly Gly Gly Ser Met Ser Arg Leu Asp Lys Ser Lys
Val Ile1 5 10 15Asn Ser Ala Leu Glu Leu Leu Asn Glu Val Gly Ile Glu
Gly Leu Thr 20 25 30Thr Arg Lys Leu Ala Gln Lys Leu Gly Val Glu Gln
Pro Thr Leu Tyr 35 40 45Trp His Val Lys Asn Lys Arg Ala Leu Leu Asp
Ala Leu Ala Ile Glu 50 55 60Met Leu Asp Arg His His Thr His Phe Cys
Pro Leu Glu Gly Glu Ser65 70 75 80Trp Gln Asp Phe Leu Arg Asn Asn
Ala Lys Ser Phe Arg Cys Ala Leu 85 90 95Leu Ser His Arg Asp Gly Ala
Lys Val His Leu Gly Thr Arg Pro Thr 100 105 110Glu Lys Gln Tyr Glu
Thr Leu Glu Asn Gln Leu Ala Phe Leu Cys Gln 115 120 125Gln Gly Phe
Ser Leu Glu Asn Ala Leu Tyr Ala Leu Ser Ala Val Gly 130 135 140His
Phe Thr Leu Gly Cys Val Leu Glu Asp Gln Glu His Gln Val Ala145 150
155 160Lys Glu Glu Arg Glu Thr Pro Thr Thr Asp Ser Met Pro Pro Leu
Leu 165 170 175Arg Gln Ala Ile Glu Leu Phe Asp His Gln Gly Ala Glu
Pro Ala Phe 180 185 190Leu Phe Gly Leu Glu Leu Ile Ile Cys Gly Leu
Glu Lys Gln Leu Lys 195 200 205Cys Glu Ser Gly Ser
2102716PRTArtificial Sequencesequence of TiP 27Trp Thr Trp Asn Ala
Tyr Ala Phe Ala Ala Pro Ser Gly Gly Gly Ser1 5 10 152887PRTHomo
sapiens 28Leu Ser Asp Ser Gly Gln Val Leu Leu Glu Ser Asn Ile Lys
Val Leu1 5 10 15Pro Thr Trp Ser Thr Pro Val Gln Pro Met Ala Leu Ile
Val Leu Gly 20 25 30Gly Val Ala Gly Leu Leu Leu Phe Ile Gly Leu Gly
Ile Phe Phe Cys 35 40 45Val Arg Cys Arg His Arg Arg Arg Gln Ala Glu
Arg Met Ser Gln Ile 50 55 60Lys Arg Leu Leu Ser Glu Lys Lys Thr Cys
Gln Cys Pro His Arg Phe65 70 75 80Gln Lys Thr Cys Ser Pro Ile
8529926PRTArtificial Sequencebasic TetCAR sequence 29Met Trp Thr
Trp Asn Ala Tyr Ala Phe Ala Ala Pro Ser Gly Gly Gly1 5 10 15Ser Ala
Asp Ala Pro Ala Tyr Gln Gln Gly Gln Asn Gln Leu Tyr Asn 20 25 30Glu
Leu Asn Leu Gly Arg Arg Glu Glu Tyr Asp Val Leu Asp Lys Arg 35 40
45Arg Gly Arg Asp Pro Glu Met Gly Gly Lys Pro Arg Arg Lys Asn Pro
50 55 60Gln Glu Gly Leu Tyr Asn Glu Leu Gln Lys Asp Lys Met Ala Glu
Ala65 70 75 80Tyr Ser Glu Ile Gly Met Lys Gly Glu Arg Arg Arg Gly
Lys Gly His 85 90 95Asp Gly Leu Tyr Gln Gly Leu Ser Thr Ala Thr Lys
Asp Thr Tyr Asp 100 105 110Ala Leu His Met Gln Ala Leu Pro Pro Arg
Arg Ala Glu Gly Arg Gly 115 120 125Ser Leu Leu Thr Cys Gly Asp Val
Glu Glu Asn Pro Gly Pro Met Ala 130 135 140Val Pro Thr Gln Val Leu
Gly Leu Leu Leu Leu Trp Leu Thr Asp Ala145 150 155 160Arg Cys Asp
Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser 165 170 175Val
Gly Asp Arg Val Thr Ile Thr Cys Arg Ala Ser Glu Asp Ile Tyr 180 185
190Phe Asn Leu Val Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu
195 200 205Leu Ile Tyr Asp Thr Asn Arg Leu Ala Asp Gly Val Pro Ser
Arg Phe 210 215 220Ser Gly Ser Gly Ser Gly Thr Gln Tyr Thr Leu Thr
Ile Ser Ser Leu225 230 235 240Gln Pro Glu Asp Phe Ala Thr Tyr Tyr
Cys Gln His Tyr Lys Asn Tyr 245 250 255Pro Leu Thr Phe Gly Gln Gly
Thr Lys Leu Glu Ile Lys Arg Ser Gly 260 265 270Gly Gly Gly Ser Gly
Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly 275 280 285Gly Gly Ser
Arg Ser Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu 290 295 300Val
Gln Pro Gly Gly Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe305 310
315 320Thr Leu Ser Asn Tyr Gly Met His Trp Ile Arg Gln Ala Pro Gly
Lys 325 330 335Gly Leu Glu Trp Val Ser Ser Ile Ser Leu Asn Gly Gly
Ser Thr Tyr 340 345 350Tyr Arg Asp Ser Val Lys Gly Arg Phe Thr Ile
Ser Arg Asp Asn Ala 355 360 365Lys Ser Thr Leu Tyr Leu Gln Met Asn
Ser Leu Arg Ala Glu Asp Thr 370 375 380Ala Val Tyr Tyr Cys Ala Ala
Gln Asp Ala Tyr Thr Gly Gly Tyr Phe385 390 395 400Asp Tyr Trp Gly
Gln Gly Thr Leu Val Thr Val Ser Ser Met Asp Pro 405 410 415Ala Glu
Pro Lys Ser Pro Asp Lys Thr His Thr Cys Pro Pro Cys Pro 420 425
430Ala Pro Pro Val Ala Gly Pro Ser Val Phe Leu Phe Pro Pro Lys Pro
435 440 445Lys Asp Thr Leu Met Ile Ala Arg Thr Pro Glu Val Thr Cys
Val Val 450 455 460Val Asp Val Ser His Glu Asp Pro Glu Val Lys Phe
Asn Trp Tyr Val465 470 475 480Asp Gly Val Glu Val His Asn Ala Lys
Thr Lys Pro Arg Glu Glu Gln 485 490 495Tyr Asn Ser Thr Tyr Arg Val
Val Ser Val Leu Thr Val Leu His Gln 500 505 510Asp Trp Leu Asn Gly
Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala 515 520 525Leu Pro Ala
Pro Ile Glu Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro 530 535 540Arg
Glu Pro Gln Val Tyr Thr Leu Pro Pro Ser Arg Asp Glu Leu Thr545 550
555 560Lys Asn Gln Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro
Ser 565 570 575Asp Ile Ala Val Glu Trp Glu Ser Asn Gly Gln Pro Glu
Asn Asn Tyr 580 585 590Lys Thr Thr Pro Pro Val Leu Asp Ser Asp Gly
Ser Phe Phe Leu Tyr 595 600 605Ser Lys Leu Thr Val Asp Lys Ser Arg
Trp Gln Gln Gly Asn Val Phe 610 615 620Ser Cys Ser Val Met His Glu
Ala Leu His Asn His Tyr Thr Gln Lys625 630 635 640Ser Leu Ser Leu
Ser Pro Gly Lys Lys Asp Pro Met Ala Leu Ile Val 645 650 655Leu Gly
Gly Val Ala Gly Leu Leu Leu Phe Ile Gly Leu Gly Ile Phe 660 665
670Phe Cys Val Arg Cys Arg His Arg Arg Arg Gln Ala Glu Arg Met Ala
675 680 685Gln Ile Lys Arg Val Val Ser Glu Lys Lys Thr Ala Gln Ala
Pro His 690 695 700Arg Phe Gln Lys Thr Cys Ser Pro Ile Ser Gly Gly
Gly Gly Ser Met705 710 715 720Ser Arg Leu Asp Lys Ser Lys Val Ile
Asn Ser Ala Leu Glu Leu Leu 725 730 735Asn Glu Val Gly Ile Glu Gly
Leu Thr Thr Arg Lys Leu Ala Gln Lys 740 745 750Leu Gly Val Glu Gln
Pro Thr Leu Tyr Trp His Val Lys Asn Lys Arg 755 760 765Ala Leu Leu
Asp Ala Leu Ala Ile Glu Met Leu Asp Arg His His Thr 770 775 780His
Phe Cys Pro Leu Glu Gly Glu Ser Trp Gln Asp Phe Leu Arg Asn785 790
795 800Asn Ala Lys Ser Phe Arg Cys Ala Leu Leu Ser His Arg Asp Gly
Ala 805 810 815Lys Val His Leu Gly Thr Arg Pro Thr Glu Lys Gln Tyr
Glu Thr Leu 820 825 830Glu Asn Gln Leu Ala Phe Leu Cys Gln Gln Gly
Phe Ser Leu Glu Asn 835 840 845Ala Leu Tyr Ala Leu Ser Ala Val Gly
His Phe Thr Leu Gly Cys Val 850 855 860Leu Glu Asp Gln Glu His Gln
Val Ala Lys Glu Glu Arg Glu Thr Pro865 870 875 880Thr Thr Asp Ser
Met Pro Pro Leu Leu Arg Gln Ala Ile Glu Leu Phe 885 890 895Asp His
Gln Gly Ala Glu Pro Ala Phe Leu Phe Gly Leu Glu Leu Ile 900 905
910Ile Cys Gly Leu Glu Lys Gln Leu Lys Cys Glu Ser Gly Ser 915 920
925
D00000
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
D00012
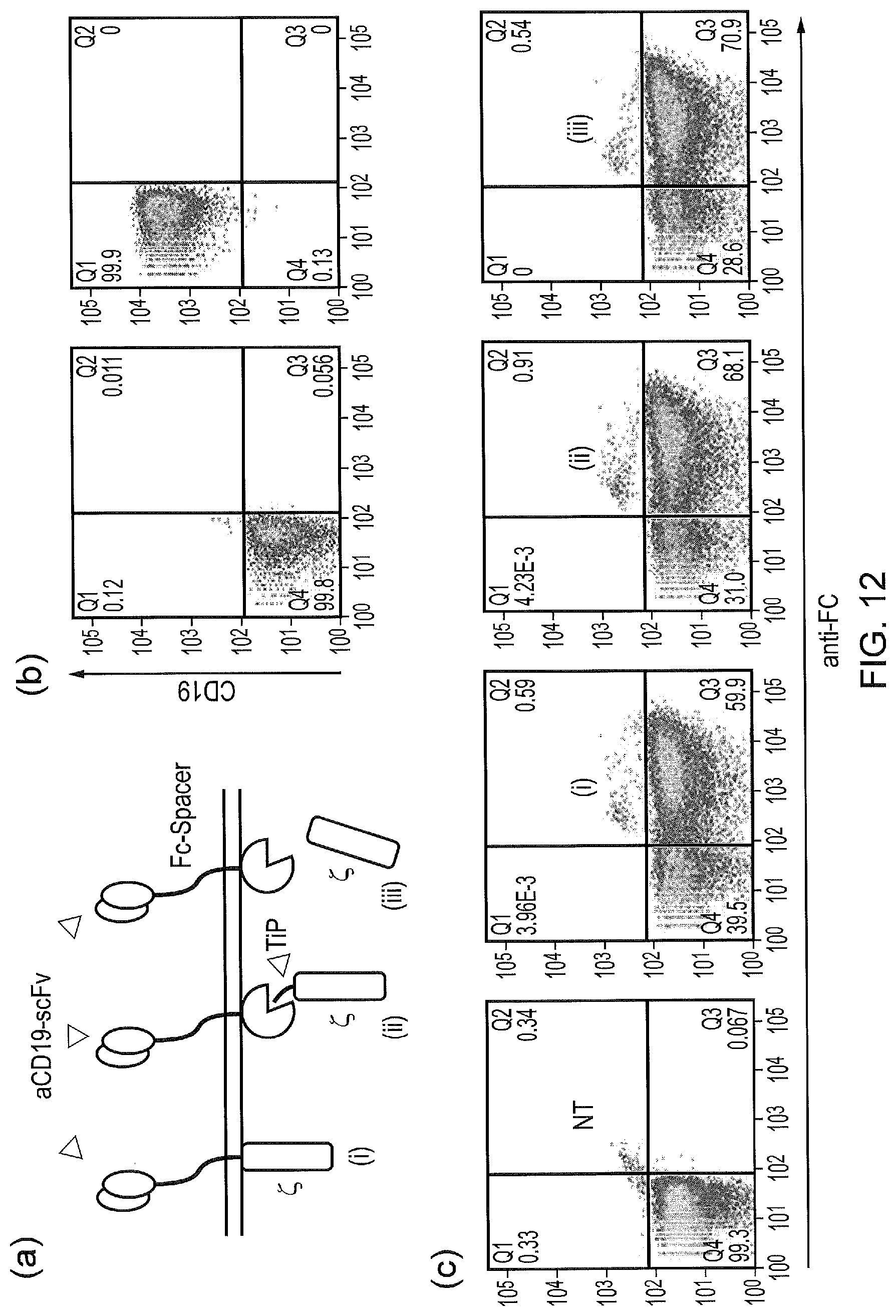
D00013
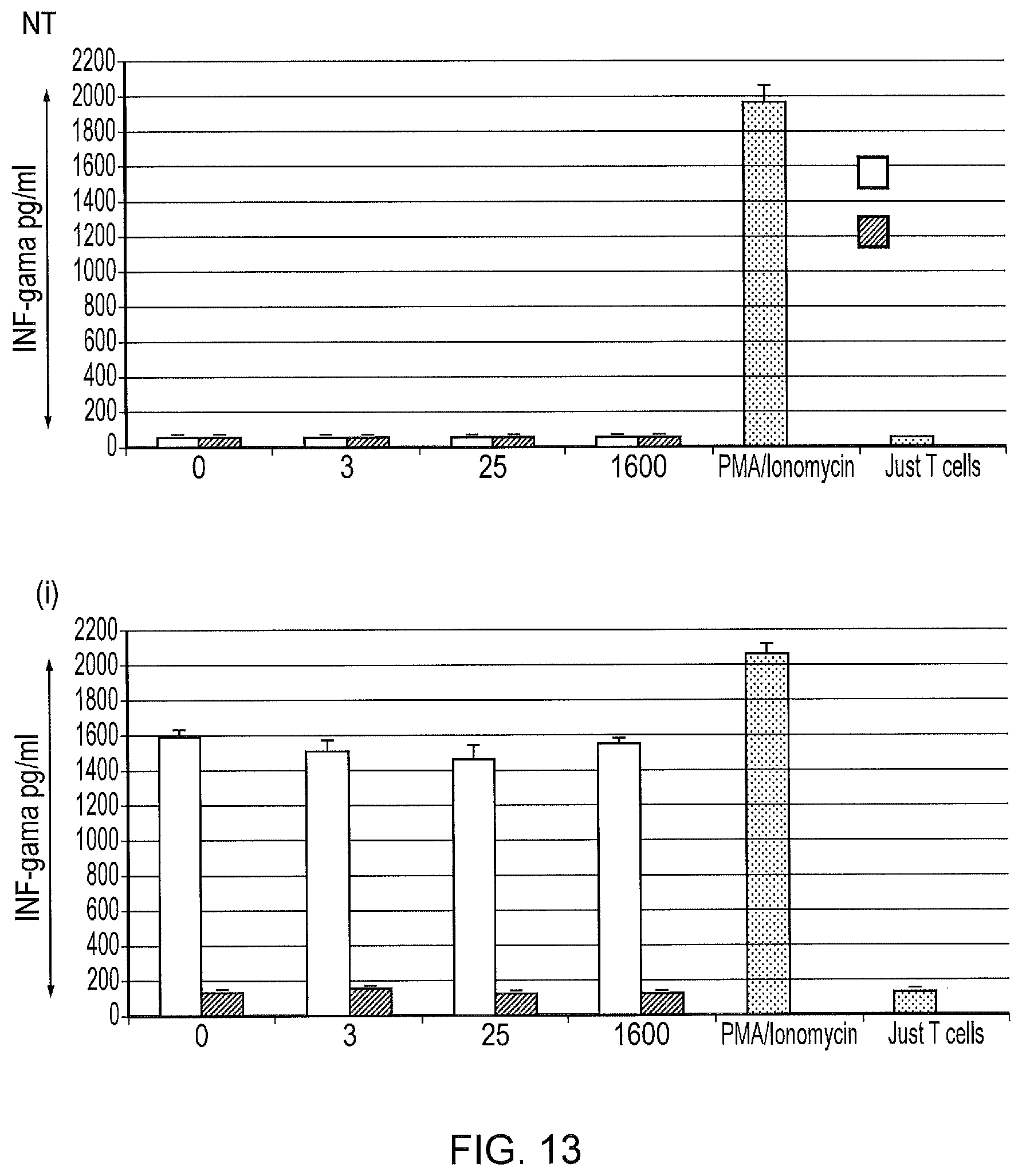
D00014
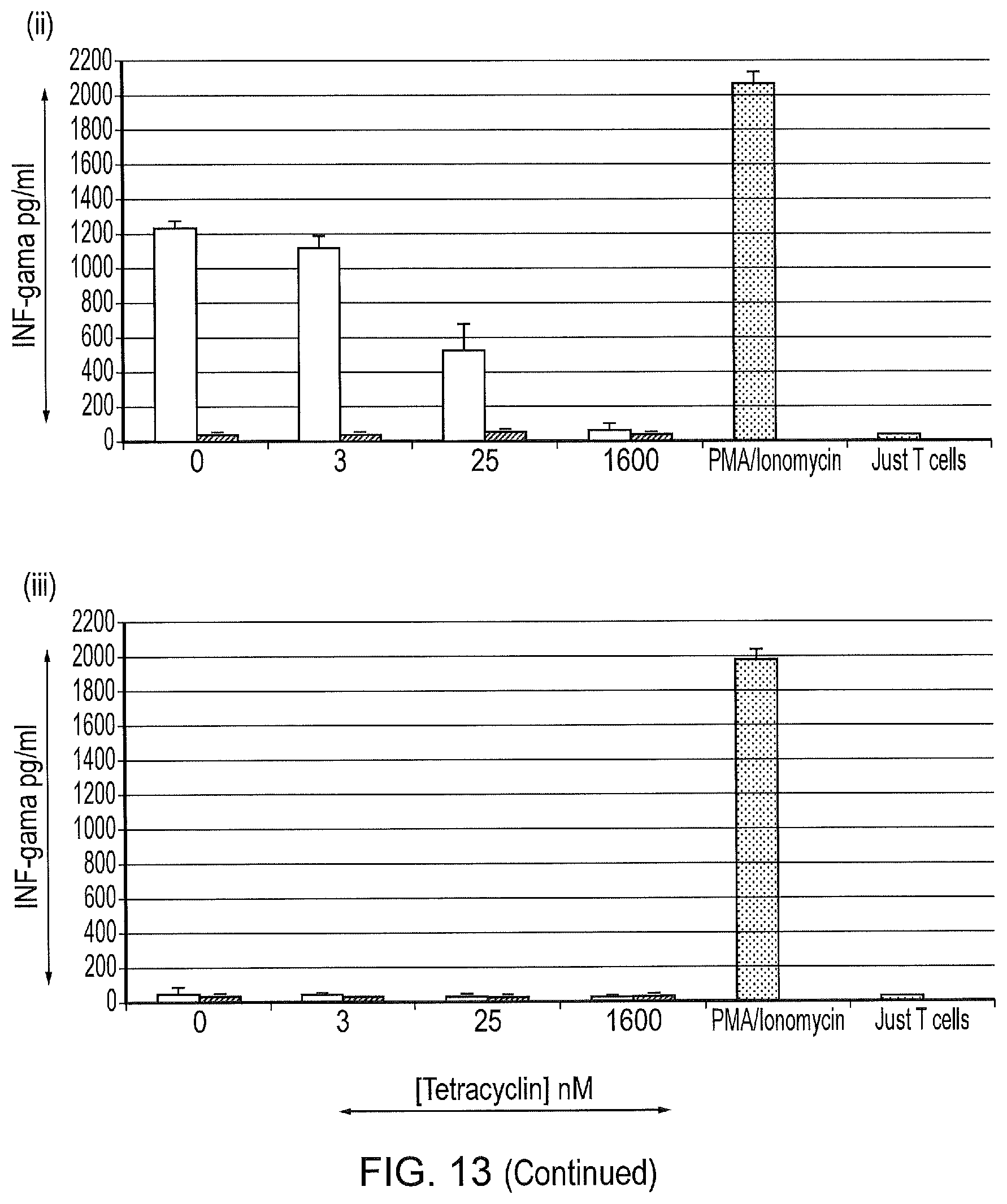
D00015
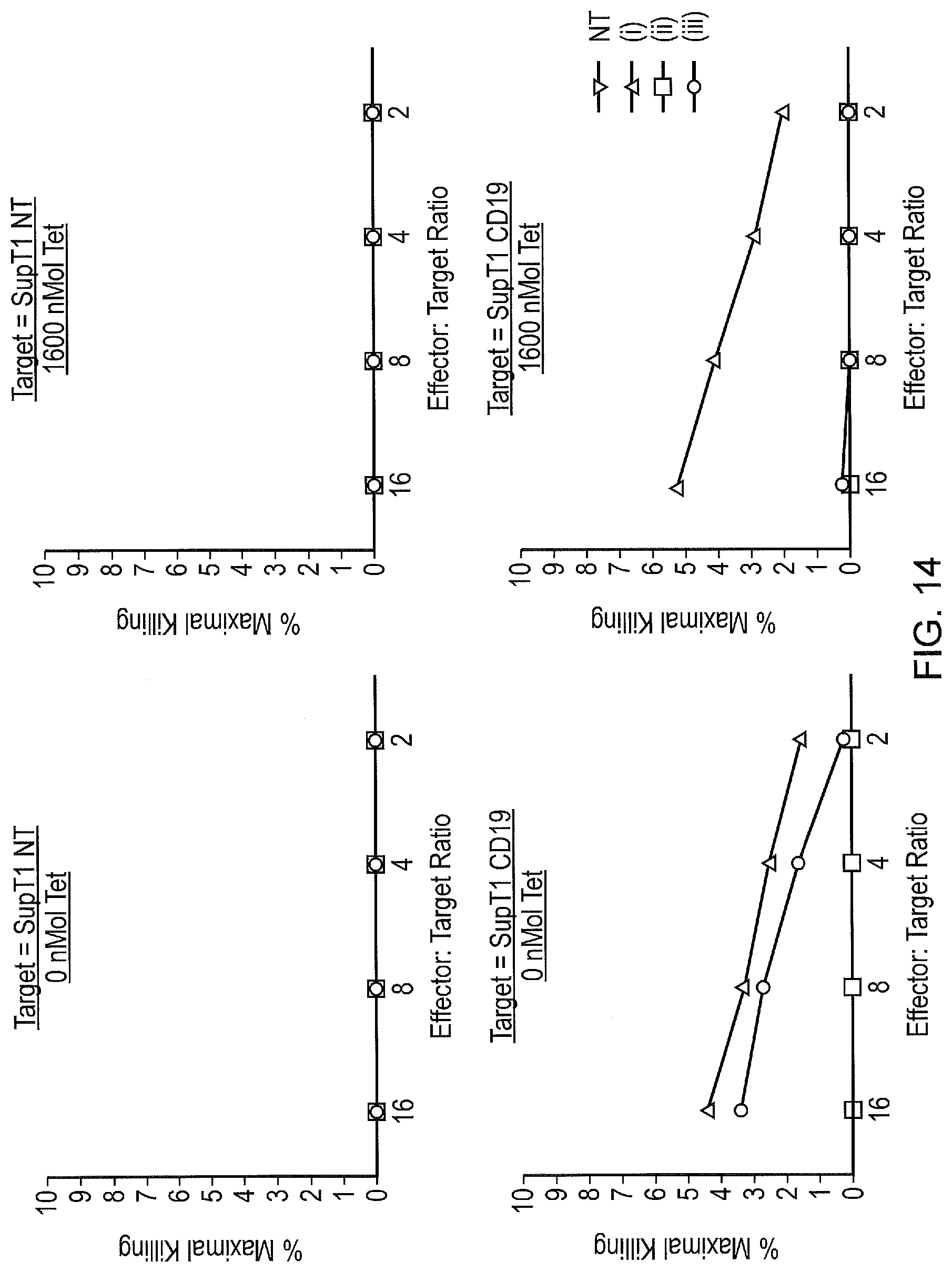
S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.