Peptide Inhibitors Of Phosphoglycerate Mutase And Methods Of Use
Inglese; James ; et al.
U.S. patent application number 17/016168 was filed with the patent office on 2020-12-24 for peptide inhibitors of phosphoglycerate mutase and methods of use. This patent application is currently assigned to The United States of America, as represented by the Secretary, Dept. of Health and Human Services. The applicant listed for this patent is New England Biolabs, Inc., The United States of America, as represented by the Secretary, Dept. of Health and Human Services, The United States of America, as represented by the Secretary, Dept. of Health and Human Services, The University of Tokyo. Invention is credited to Clotilde Carlow, Patricia Dranchak, James Inglese, Zhiru Li, Ryan MacArthur, Hiroaki Suga, Hao Yu.
| Application Number | 20200399318 17/016168 |
| Document ID | / |
| Family ID | 1000005077178 |
| Filed Date | 2020-12-24 |








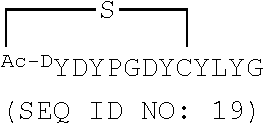



View All Diagrams
| United States Patent Application | 20200399318 |
| Kind Code | A1 |
| Inglese; James ; et al. | December 24, 2020 |
PEPTIDE INHIBITORS OF PHOSPHOGLYCERATE MUTASE AND METHODS OF USE
Abstract
Disclosed herein are isolated peptides inhibit activity of a cofactor-independent phosphoglycerate mutase. In some examples, the isolated peptide is 6-20 amino acids long and includes the amino acid sequence of any one of SEQ ID NOs: 1-22 or 54, an analog or derivative thereof, or a pharmaceutically acceptable salt or ester thereof. In some examples, the peptide is a cyclic peptide with an N-terminal ring of 6-15 amino acids (for example, 6-10 amino acids) and a C-terminal linear portion of 1-9 amino acids (for example, 3-8 amino acids. Also disclosed h are methods of treating or inhibiting an infection in a subject, including administering to the subject an effective amount of a composition including one of more of the disclosed peptides, or analogs or derivative thereof, or pharmaceutically acceptable salts or esters thereof.
| Inventors: | Inglese; James; (Bethesda, MD) ; Dranchak; Patricia; (Gaithersburg, MD) ; MacArthur; Ryan; (Odenton, MD) ; Suga; Hiroaki; (Tokyo, JP) ; Yu; Hao; (Tokyo, JP) ; Carlow; Clotilde; (South Hamilton, MA) ; Li; Zhiru; (Lexington, MA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Assignee: | The United States of America, as
represented by the Secretary, Dept. of Health and Human
Services Bethesda MD The University of Tokyo Tokyo MA New England Biolabs, Inc. Ipswich |
||||||||||
| Family ID: | 1000005077178 | ||||||||||
| Appl. No.: | 17/016168 | ||||||||||
| Filed: | September 9, 2020 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 16324424 | Feb 8, 2019 | 10808010 | ||
| PCT/US2017/046228 | Aug 10, 2017 | |||
| 17016168 | ||||
| 62373835 | Aug 11, 2016 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61P 33/10 20180101; A61K 45/06 20130101; C07K 7/56 20130101; Y02A 50/30 20180101; C07K 14/00 20130101; C07K 7/64 20130101; A61K 38/00 20130101; A61K 38/12 20130101; C07K 7/08 20130101; C07K 7/06 20130101 |
| International Class: | C07K 7/64 20060101 C07K007/64; C07K 7/08 20060101 C07K007/08; C07K 7/06 20060101 C07K007/06; C07K 7/56 20060101 C07K007/56; C07K 14/00 20060101 C07K014/00; A61P 33/10 20060101 A61P033/10; A61K 38/12 20060101 A61K038/12; A61K 45/06 20060101 A61K045/06 |
Claims
1. An isolated peptide comprising the amino acid sequence of any one of SEQ ID NOs: 7-9.
2. The isolated peptide of claim 1, wherein the N-terminal amino acid is D-N-chloroacetyl tyrosine.
3. The isolated peptide of claim 2, wherein the peptide comprises: TABLE-US-00013 (SEQ ID NO: 7) .sup.Ac-DYDYPGDYSYLYGTCG; (SEQ ID NO: 8) .sup.Ac-DYDYPGDYSYLY; or (SEQ ID NO: 9) .sup.Ac-DYLYGTCG.
4. A pharmaceutical composition comprising the isolated peptide of claim 1 and a pharmaceutically acceptable carrier.
5. A method of treating or inhibiting infection with an organism expressing at least one cofactor-independent phosphoglycerate mutase in a subject, comprising administering to the subject an effective amount of the isolated peptide of claim 1.
6. The method of claim 5, wherein the subject is infected with an organism selected from the group consisting of a nematode, a trypanosome, a helminth, a protozoan parasite, or a bacterium.
7. The method of claim 6, wherein the subject is infected with a nematode, and the nematode comprises one or more of Brugia malayi, Brugia timori, Wuchereria bancrofti, Onchocerca volvulus, Loa, Mansonella streptocerca, Mansonella perstans, Mansonella ozzardi, Dirofilaria immitis, Trichinella, Parafilaria bovicola, Onchocerca dermatan, Onchocerca ochengi, Onchocerca dukei, Stenofilaria assamensis, and Parafilaria multipapillosa.
8. The method of claim 6, wherein the subject is infected with a bacterium and the bacterium comprises one or more of Staphylococcus aureus, Bacillus anthracia, Streptococcus pneumoniae, and Escherichia coli.
9. The method of claim 5, wherein the peptide is administered to the subject orally, intravenously, or topically.
10. The method of claim 5, further comprising administering one or more of an antiparasitic or antibiotic agent to the subject.
11. An isolated cyclic peptide comprising the any one of the peptides in FIG. 21, SEQ ID NO: 21 or SEQ ID NO: 22.
12. The isolated cyclic peptide of claim 11, wherein the peptide comprises: ##STR00057##
13. A pharmaceutical composition comprising the isolated cyclic peptide of claim 11 and a pharmaceutically acceptable carrier.
14. A method of treating or inhibiting infection with an organism expressing at least one cofactor-independent phosphoglycerate mutase in a subject, comprising administering to the subject an effective amount of the isolated cyclic peptide of claim 11.
15. The method of claim 14, wherein the subject is infected with an organism selected from the group consisting of a nematode, a trypanosome, a helminth, a protozoan parasite, or a bacterium.
16. The method of claim 15, wherein the subject is infected with a nematode, and the nematode comprises one or more of Brugia malayi, Brugia timori, Wuchereria bancrofti, Onchocerca volvulus, Loa loa, Mansonella streptocerca, Mansonella perstans, Mansonella ozzardi, Dirofilaria immitis, Trichinella, Parafilaria bovicola, Onchocerca dermatan, Onchocerca ochengi, Onchocerca dukei, Stenofilaria assamensis, and Parafilaria multipapillosa.
17. The method of claim 15, wherein the subject is infected with a bacterium and the bacterium comprises one or more of Staphylococcus aureus, Bacillus anthracia, Streptococcus pneumoniae, and Escherichia coli.
18. The method of claim 14, wherein the cyclic peptide is administered to the subject orally, intravenously, or topically.
19. The method of claim 14, further comprising administering one or more of an antiparasitic or antibiotic agent to the subject.
Description
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application is a divisional of U.S. patent application Ser. No. 16/324,424, filed Feb. 8, 2019, which is the .sctn. 371 U.S. National Stage of International Application No. PCT/US2017/046228, filed Aug. 10, 2017, which was published in English under PCT Article 21(2), which in turn claims the benefit of U.S. Provisional Application No. 62/373,835, filed Aug. 11, 2016, which is herein incorporated by reference in its entirety.
FIELD
[0002] This disclosure relates to peptide inhibitors of phosphoglycerate mutase, particularly peptide inhibitors, and methods of their use for treating or inhibiting microbial infection or disease.
BACKGROUND
[0003] Nematode worms are the most abundant animal on earth and are found in widely different environments. They can be free-living or parasitic, infecting plants, animals, and humans. Parasitic nematode infection in humans can lead to a number of devastating diseases. Lymphatic filariasis and onchocerciasis are neglected tropical diseases caused by filarial nematode parasites that are transmitted to humans by insects. Collectively, they afflict 150 million people in over 80 countries and threaten the health of over 1.5 billion people. These infections are responsible for extreme infirmity, social stigma and severe economic consequences. The lymphatic dwelling parasites such as Wuchereria bancrofti and Brugia malayi are the cause of lymphedema, hydrocele, and in the most extreme cases, elephantiasis. Infection with Onchocerca volvulus can result in severe dermatitis and/or blindness. The mainstay of filarial disease control for several years has been a limited number of drugs, predominantly ivermectin together with albendazole (where onchocerciasis is endemic) or diethylcarbamazine citrate (where onchocerciasis is not present). These compounds mainly target the larval stages and require annual or semi-annual administration. Furthermore, there are reports of drug resistance emerging (Churcher et al., Proc. Natl. Acad. Sci. USA 106:16716-16721, 2009; Osei-Atweneboana et al., PLoS Negl. Trop. Dis. 5:e998, 2011). Therefore, new drugs with a novel mode of action are needed.
SUMMARY
[0004] Disclosed herein are compounds and compositions for inhibiting phosphoglycerate mutase (PGM) activity. In some examples, the compounds selectively inhibit cofactor-independent PGM (iPGM). In particular embodiments, the compounds include one or more cyclic peptides.
[0005] In some embodiments, the compounds or compositions include an isolated peptide (such as a linear or cyclic peptide) that selectively or specifically inhibits activity of an iPGM compared to a cofactor-dependent PGM (dPGM). In some examples, the isolated peptide is 6-20 amino acids long and includes the amino acid sequence of any one of SEQ ID NOs: 1-22 and 54-69, an analog or derivative thereof, or a pharmaceutically acceptable salt or ester thereof. In some examples, the peptide is a cyclic peptide with an N-terminal ring structure of 6-15 amino acids (for example, 6-10 amino acids) and a C-terminal linear portion of 1-9 amino acids (for example, 3-8 amino acids). In particular examples, the isolated peptide includes a peptide having the structure of any one of the peptides in Tables 2, 6, 8, or 9 or FIG. 12A, 12B, or 21, or an analog or derivative thereof, or a pharmaceutically acceptable salt or ester thereof.
[0006] Also disclosed herein are methods of treating or inhibiting an infection in a subject, including administering to the subject an effective amount of a composition including one of more of the disclosed peptides or analogs or derivatives thereof, or pharmaceutically acceptable salts or esters thereof. In some examples, the methods including treating infection with a nematode (for example, treating a filarial disease), infection with Leishmania, or infection with a bacterium (for example, treating infection with Staphylococcus aureus, Bacillus anthracia, or Streptococcus pneumoniae).
[0007] The foregoing and other features of the disclosure will become more apparent from the following detailed description, which proceeds with reference to the accompanying figures.
BRIEF DESCRIPTION OF THE DRAWINGS
[0008] FIGS. 1A-1C are a series of panels showing PGM coupled-enzyme assays. FIG. 1A is a graph showing a reaction time course (1 hour) as measured by a continuous NADH-dependent absorbance assay. The absorbance measured at 340 nm decreases as NADH is oxidized to NAD and is dependent on the pyruvic acid formed in the prior enzymatic reactions. FIG. 1B is a graph showing that the linear phase of assays occurs over the initial 15 minutes. FIG. 1C is a graph showing calibration of bioluminescent end-point HTS assays for seven PGM orthologs and pyruvate kinase (PK)-FLuc control. The light generated in the bioluminescence assay is based on the oxidation of luciferin by ATP and oxygen, and is dependent on the ATP generated in the prior enzymatic reactions. Enzyme concentrations (Bm iPGM, 5 nM; Ce iPGM, 5 nM; Di iPGM, 10 nM; Ec iPGM, 10 nM; Ov iPGM, 20 nM; Hs dPGM, 5 nM; Ec dPGM, 4 nM) were adjusted to give approximately equivalent RLU after a 5 min assay time. Open bars are `no PGM` controls for background measurements. Error bars are SD from 24 replicates.
[0009] FIGS. 2A-2C are a series of panels showing species-dependent PGM catalytic mechanisms and assay methods to detect 2- and 3-phosphoglycerate (2-PG, 3-PG). FIG. 2A is a schematic diagram showing isomerization catalyzed by PGMs illustrating the phosphohistidine enzyme/2,3-phosphoglycerate intermediate of human cofactor-dependent PGM (dPGM, top) and the phosphoserine enzyme intermediate of C. elegans cofactor independent PGM (iPGM, bottom). FIG. 2B is a schematic diagram showing coupling enzymes used in kinetic NADH absorbance (from generated pyruvate) and endpoint bioluminescence (from generated ATP) assays. Coupling enzymes and substrates for the absorbance assay are enolase, 2-PG, pyruvate kinase, phosphoenolpyruvic acid (PEP), ADP, lactate dehydrogenase, pyruvic acid, and NADH. Coupling enzymes and substrates for the bioluminescence assay are enolase, 2-PG, pyruvate kinase, phosphoenolpyruvic acid (PEP), ADP, luciferase, ATP, and luciferin (LH2). The products of the absorbance assay are lactate and NAD.sup.+. The products of the bioluminescence assay are oxyluciferin (L), CO.sub.2 and light (hv). FIG. 2C is a schematic of gradient elution moving boundary capillary electrophoresis (GEMBE) device used in the direct detection of 2-PG/3-PG (left) and 1.sup.st derivative of current detected for phosphoglycerate isomers (right).
[0010] FIGS. 3A and 3B are a series of panels showing separation of phosphoglycerates with GEMBE. FIG. 3A shows current (top) and 1.sup.st derivative (bottom) plots for the elution of 2- and 3-PG. FIG. 3B shows time courses for the conversion of 3-PG to 2-PG measured with GEMBE, for the C. elegans iPGM (top) and the B. malayi iPGM (bottom). Left axis, % conversion of (.circle-solid.) 2-PG, (.largecircle.) 3-PG; right axis, ratio (+) 2-PG to 3-PG.
[0011] FIGS. 4A-4C are a series of panels showing sequence alignments of DNA sequences from RaPID selections. FIG. 4A is an alignment of DNA sequences identified from 4th and 5th rounds of selection with .sup.DY library against B. malayi iPGM. Sequence 4-6, SEQ ID NO: 23; 4-9, SEQ ID NO: 24; 4-7, SEQ ID NO: 25; 4-13, SEQ ID NO: 26; 5-13, 5-15, 4-17, 4-18, and 5-10, SEQ ID NO: 27; 4-3, SEQ ID NO: 28; 4-8, SEQ ID NO: 29. FIG. 4B is an alignment of DNA sequences identified from 6th and 7th rounds of selection with .sup.DY library against C. elegans iPGM. Sequence 6-1, SEQ ID NO: 30; 6-4 and 6-5, SEQ ID NO: 31; 6-6 and 6-7, SEQ ID NO: 32; 6-9, SEQ ID NO: 30; 6-11, SEQ ID NO: 31, 6-12, SEQ ID NO: 30; 6-13 and 7-1, SEQ ID NO: 31; 7-2, SEQ ID NO: 33; 7-3, SEQ ID NO: 34; 7-4, SEQ ID NO: 35; 7-5, SEQ ID NO: 31; 7-6 and 7-7, SEQ ID NO: 30; 7-9 and 7-10, SEQ ID NO: 31; 7-12, SEQ ID NO: 30; 7-13, SEQ ID NO: 31; 7-14, SEQ ID NO: 36; 7-15, SEQ ID NO: 33; 6-14, 7-8, and 7-11, SEQ ID NO: 37. FIG. 4C is an alignment of DNA sequences identified from 5th and 6th rounds of selection with .sup.LY library against C. elegans iPGM. Sequences 5-1, 5-2, 5-3, 5-5, 5-8, 5-9, 5-10, 5-13, 5-14, SEQ ID NO: 38; 5-16, SEQ ID NO: 39; 5-19, SEQ ID NO: 38; 5-20, SEQ ID NO: 39; 5-21 and 5-22, SEQ ID NO: 38; 5-24, SEQ ID NO: 39; 5-28, 5-30, 6-1, 6-2, 6-3, 6-4, 6-5, and 6-6, SEQ ID NO: 38; 6-7, SEQ ID NO: 40; 6-8 and 6-9, SEQ ID NO: 38; 6-11, SEQ ID NO: 39; 6-12, SEQ ID NO: 41; 6-13, SEQ ID NO: 42; 6-14, SEQ ID NO: 40; 6-15, SEQ ID NO: 43; 6-16, SEQ ID NO: 39; 5-26, SEQ ID NO: 44. Mutations at second or third codon base are colored in purple or red, respectively.
[0012] FIG. 5 is a MSMS spectrum and fragment analysis of Ce-2 by MALDI-TOF/TOF. b-type, y-type and c-type fragment ions are indicated by number. The parent ion [M+H].sup.+ of Ce-2 is 1790.66. Ce-2 peptide sequence (SEQ ID NO: 2) is shown.
[0013] FIGS. 6A-6D are a series of panels showing crystals of apo C. elegans iPGM and in a complex with Ce-2d. FIG. 6A is a digital image of an initial sample of monoclinic P iPGM (iPGM-m) obtained from Wizard 3-4 D11 and FIG. 6B is a digital image showing crystals observed from the Hampton Additive HT screen D7. FIG. 6C is a digital image of crystals of an orthorhombic P iPGM (iPGM-o) obtained from Index HT F7. FIG. 6D is a digital image of crystals of a co-complex with Ce-2d.
[0014] FIGS. 7A-7C are a series of ribbon diagrams of asymmetric unit cell of C. elegans iPGM-m. FIG. 7A is a diagram of asymmetric unit of C. elegans iPGM-m showing subunit A (magenta) and B (cyan). The Mn.sup.2+ and Zn.sup.2+ ions are represented as blue spheres. FIG. 7B is a diagram showing a comparison of the NCS dimer subunits of C. elegans apo iPGM-m showing subunit A (magenta) and B (cyan) superimposed (top) and the same diagram rotated 90.degree. in the horizontal direction (bottom). FIG. 7C is a diagram showing superposition of C. elegans iPGM-m (magenta) and C. elegans iPGM-o (cyan).
[0015] FIGS. 8A-8C are a series of ribbon diagrams showing a comparison of C. elegans iPGM to Bacillus anthracis and Bacillus stearothermophilus iPGM. FIG. 8A shows a superposition of C. elegans apo iPGM-m showing subunit A (magenta) and iPGM from Bacillus anthracis (gray, PDB: 2IFY) superimposed. Mn.sup.2+ and Zn.sup.2+ ions are represented as spheres. FIG. 8B shows a superposition of C. elegans iPGM-m showing subunit A (magenta) and iPGM from Bacillus stearothermophilus (green, PDB: 1098) superimposed. FIG. 8C shows the same diagram as FIG. 8B, but rotated 90.degree. in the horizontal direction. The arrow indicates the conformational difference in the transferase domain.
[0016] FIGS. 9A-9C are a series of diagrams showing metal ion binding sites in iPGM. FIG. 9A shows phased anomalous difference map (mesh) at the metal binding sites of the C. elegans iPGM.circle-solid.Ce-2d complex contoured at 3.sigma.. FIG. 9B shows metal coordination in the C. elegans iPGM.circle-solid.Ce-2d complex. Contacts between the protein and Mn.sup.2+ (large dark sphere) ion, Zn.sup.2+ (large light sphere) ion, and water (small spheres) are shown. FIG. 9C shows the Mg.sup.2+ binding site for iPGM.circle-solid.Ce-2d. Mg.sup.2+ ion is depicted as a large sphere and water molecules as small spheres.
[0017] FIGS. 10A-10E are a series of panels showing pharmacologic-phylogenetic relationship of iPGM macrocyclic peptide inhibitors. FIG. 10A is a diagram of the structure of the Ce-2 macrocycle obtained from affinity selection and showing truncation to give Ce-2d and position of Cys14Ser substitution. The thioether bond and D-tyrosine are highlighted. FIGS. 10B-10D are a series of graphs showing IC.sub.50 concentration-response curves for characterization of Ce-2 (FIG. 10B), Ce-2d (FIG. 10C) and Ce-2S (FIG. 10D) on the iPGM orthologs and dPGM isozymes using the enzyme coupled bioluminescent assay. Plots are representatives from individual experiments (N.gtoreq.3), error bars are standard deviations values of technical replicates. Inset, titration of 0.5 nM, 5 nM, 50 nM, 0.5 .mu.M and 5 .mu.M C. elegans iPGM with Ce-2. FIG. 10E is a phylogenetic tree constructed for amino acid sequence alignments of seven species orthologs of PGM. Percentage bootstrap values based on 1,000 replicates are indicated at branch nodes.
[0018] FIGS. 11A-11C are a series of representative concentration response curves (CRCs) for iPGM inhibitory activity of Bm and Ce-cyclic peptides and GEMBE analysis of Ce-2 and Ce-2d. FIG. 11A is a series of CRCs for Bm-1 (top), Bm-4 (middle), and Ce-3 (bottom). FIG. 11B shows CRCs for Ce-2. Significant deviation from a hyperbolic response required a 5 parameter Hill equation to fit the iPGM data in this plot. Data from the iPGM orthologs and dPGM isozymes determined from the enzyme-coupled bioluminescent assay. The iPGM concentrations for C. elegans, B. malayi was 5 nM, for D. immitis, E. coli 10 nM, O. volvulus 20 nM, and E. coli, H. sapiens dPGM, 5 nM. Plots are representative from experimental replicates listed in Table 3, error bars are defined as described in Table 3. FIG. 11C is a graph showing GEMBE analysis of macrocyclic peptides Ce-2 and Ce-2d. Green: B. malayi iPGM, blue: C. elegans iPGM, amber: E. coli iPGM, black: H. sapiens dPGM, treated with inhibitor Ce-2 (squares) or Ce-2d (diamonds). pEC.sub.50 values from experimental replicates (N): Ce-2--(Bm iPGM) 8.37.+-.0.04 (2); (Ce iPGM) 8.36.+-.0.03 (1); (Ec iPGM) 8.49.+-.0.03 (3); (Hs dPGM) no apparent activity; Ce-2d--(Bm iPGM) 7.21.+-.0.19 (1); (Ce iPGM) 8.49.+-.0.02 (1); (Ec iPGM) 8.50.+-.0.02 (3); (Hs dPGM) no apparent activity. Error bars are standard deviation values of experimental replicates.
[0019] FIGS. 12A and 12B are a series of diagrams of macrocyclic peptide analogs. FIG. 12A shows a series of cyclic peptides and FIG. 12B shows a series of linear peptides. Hydroxyl groups and methyl groups are shaded to indicate substitutions.
[0020] FIGS. 13A and 13B are a series of graphs showing PGM panel activity for macrocyclic peptide analogs of Ce-2. FIG. 12A is a series of graphs showing activity of Ce-2S (a Cys14Ser substitution) and Ce-2a-g (C-terminal amino acid truncation analogs). FIG. 13B is a series of graphs showing activity of linear peptides Ce-L2, Ce-L2d, and Ce-2tail. B. malayi iPGM (.box-solid.), C. elegans iPGM (.tangle-solidup.), O. volvulus iPGM (), D. immitis iPGM (.diamond-solid.), E. coli iPGM (.circle-solid.), H. sapiens dPGM (.quadrature.), E. coli dPGM (.DELTA.) and PK-FLuc (.circle-solid.). Plots are representative from experimental replicates listed in Table 3, error bars are standard deviations values of technical two replicates.
[0021] FIG. 14 is a sequence alignment of iPGM orthologs from C. elegans (SEQ ID NO: 45), B. malayi (SEQ ID NO: 46), O. volvulus (SEQ ID NO: 47), D. immitis (SEQ ID NO: 48), E. coli (SEQ ID NO: 49), S. aureus (SEQ ID NO: 50), B. anthracia (SEQ ID NO: 51), L. mexicana (SEQ ID NO: 52), and T. brucei (SEQ ID NO: 53). Residues within 5 .ANG. of Ce-2d are colored orange. Residues identical between C. elegans and E. coli iPGM are colored yellow; grey indicates hinge regions, green and blue are amino acids that ligand metal ions.
[0022] FIGS. 15A-15C are a series of diagrams showing the structure of iPGM.circle-solid.Ce-2d complex asymmetric unit. FIG. 15A shows asymmetric unit of iPGM.circle-solid.Ce-2d showing subunit A (magenta) and B (cyan). The Mn.sup.2+ and Zn.sup.2+ ions are represented as blue and tan spheres, respectively, and the cyclic peptides bound to each subunit are drawn as gray spheres. FIG. 15B shows an electron density map (mesh, Fo-Fc omit) contoured at 3.sigma. for the peptide associated with subunit A (magenta) with numbering for the Ce-2d peptide. Residue Tyr11 is capped as an amide. FIG. 15C shows the .alpha.-helical structure of Ce-2d C-terminus. Backbone trace with side chains involved in .alpha.-helix formation shown.
[0023] FIGS. 16A-16E are a series of diagrams showing Ce-2d traps iPGM in an open conformation. FIG. 16A shows one subunit of the asymmetric unit showing the binding mode of the Ce-2d macrocycle to C. elegans iPGM. The Mn.sup.2+ and Zn.sup.2+ ions are represented as blue and tan spheres, respectively, and the bound macrocycle is drawn as CPK space-filling spheres in a cavity defined by iPGM residues within 5 .ANG. (transparent spheres). FIG. 16B shows superposition of C. elegans iPGM-o (cyan), C. elegans iPGM-m (tan) and C. elegans iPGM.circle-solid.Ce-2d (aquamarine). The Ce-2d peptide is represented as cylinders. FIG. 16C shows the macrocycle (cylinders) positioned within a cleft of iPGM represented as an electrostatic surface. FIG. 16D shows CPK space-filling representations of Ce-2d illustrating the `capping` orientation of the five tyrosine residues (1, 3, 7, 9 and 11) and FIG. 16E shows the edge-to-face interaction of Tyr 1 and Tyr 9. Additional residues are indicated.
[0024] FIGS. 17A-17E are a series of panels showing Ce-2d.circle-solid.iPGM interactions. FIG. 17A shows hydrogen bond interactions (black dashed lines) between C. elegans iPGM and Ce-2d. FIG. 17B shows direct interactions and water (spheres)-mediated contacts. FIG. 17C shows the distance between the C-terminal amide of Tyr11 and the Zn.sup.2+ and Mn.sup.2+ ion centers. FIG. 17D shows superimposed structure of C. elegans iPGM_Ce-2d with that of Staphylococcus aureus iPGM in 2-phosphoglyceric acid bound form (PDB: 4NWX). The following S. Aureus: C. elegans residue pairs were used for alignment: 123His147, 153-154Asp177-178, 191Arg216, 185Arg210, 257Arg284, and 260Arg287. Ce-2d and 2-PG are shown as CPK space filling models. The purple spheres are the Mn.sup.2+ ions of S. aureus iPGM and the blue and tan spheres are the Mn.sup.2+ and Zn.sup.2+ ion, respectively of C. elegans iPGM. FIG. 17E is an enlarged region from FIG. 17D, showing the relative locations of the 2-PG and Ce-2d as cylinder models with transparent van der Waals surfaces and alignment residue side chains clustering around 2-PG.
[0025] FIGS. 18A and 18B are diagrams showing the hydrophobic pocket occupied by Leu10 of Cd-2d. FIG. 18A shows C. elegans iPGM residues (light blue chain under transparent spheres) within 5 .ANG. of the Ce-2d macrocycle, shown as B-factor .alpha.-chain (gold) with Leu10 and thioether linkage in green. The iPGM Ala334 residue is shown as a CPK space fill and the Mn.sup.2+ and Zn.sup.2+ ions are represented as blue and tan spheres, respectively. FIG. 18B shows FIG. 18A without transparent spheres, for clearer view of iPGM side chains.
[0026] FIGS. 19A-19C are a series of panels showing the structural basis underlying the pharmacologic-phylogenetic Ce-2 macrocycle series-iPGM ortholog relationship. FIG. 19A is a graph showing relationship between Ce-2 macrocycle truncation series IC.sub.50s and iPGM orthologs. Analogs with no detectable inhibitory activity are indicated as inactive. Values are from Table 3, converted from pIC.sub.50 where IC.sub.50=10.sup.-pIC50. Error bars represent the SD values of the log normal distributed IC.sub.50s determined for the given peptide. FIG. 19B shows select amino acid sequence alignments of portions of iPGM orthologs (SEQ ID NOs: 45-49; full alignment shown in FIG. 14). iPGM residues within 5 .ANG. of Ce-2d are colored orange. Residues identical between C. elegans and E. coli iPGM are colored yellow; grey indicated hinge regions; green and blue are amino acids that ligand metal ions. FIG. 19C is a diagram showing the cavity formed from C. elegans iPGM residues (light blue chain under transparent spheres) within 5 .ANG. of the Ce-2d macrocycle shown as a worm .alpha.-chain (gold) representation scaled by B-factor with select side chains (Tyr3, Pro4, thioether linkage, and C-terminal Tyr11 amide) shown. The iPGM Ala334 residue is shown as a CPK space fill. Electrostatic surface of the Ce-2d binding cavity is also shown.
[0027] FIGS. 20A and 20B are a pair of diagrams showing modeling of C-terminal residues of Ce-2 onto Ce-2d. FIG. 20A shows the Ce-2d macrocycle as worm .alpha.-chain (gold) representation scaled by B-factor within a cavity of C. elegans iPGM residues (transparent spheres) formed from residues within 5 .ANG. of cyclic peptide. The C-terminal residues, -Gly12-Thr13-Cys14-Gly15 of Ce-2 were modeled onto the iPGM.circle-solid.Ce-2d complex and are shown as tan sticks extending from Ce-2d. Electrostatic surface of the binding cavity is also shown. FIG. 20B shows Ce-2 van der Waals radii using a CPK model. The Cys14 sulfhydryl is shown in yellow.
[0028] FIG. 21 is a schematic showing exemplary cyclic iPGM inhibitor peptides and analogs, based on the Ce-2d core structure.
[0029] FIGS. 22A and 22B are a series of plots showing Concentration response curves (FIG. 22A) and IC.sub.50 variation (FIG. 22B) for Ce-2 (right) and Ce-2d (left) tested across varying concentrations of 3-PG substrate. Fifteen minute post substrate addition as measured by a continuous NADH-dependent absorbance assay. The C. elegans iPGM concentration used was .about.5 and .about.15 nM for Ce-2 and Ce-2d respectively. Absorbance values were normalized to no enzyme control and plotted in GraphPad Prism using a 4-parameter logistic fit for the purpose of estimating IC.sub.50 values. Error bars represent standard deviation of four replicates. K.sub.M of 3-PG=200 .mu.M as calculated in this study.
[0030] FIGS. 23A-23C are a series of graphs showing Ce-2 and Ce-2d in vivo activity in C. elegans culture assay. L1 stage C. elegans were exposed to 5 .mu.M (FIG. 23A) or 10 .mu.M (FIG. 23B) Ce-2 or Ce-2d for 1, 2, and 7 days. L4 stage C. elegans were exposed to 50 .mu.M Ce-2, 50 .mu.M Ce-2d, or DMSO (FIG. 23C). Data represent mean.+-.s.d. of triplicate samples.
SEQUENCE LISTING
[0031] The nucleic acid and amino acid sequences provided herein and in the accompanying sequence listing are shown using standard letter abbreviations for nucleotide bases and amino acids, as defined in 37 C.F.R. 1.822. Only one strand of each nucleic acid sequence is shown, but the complementary strand is understood as included by any reference to the displayed strand.
[0032] The Sequence Listing is submitted as an ASCII text file in the form of the file named Sequence_Listing.txt, which was created on Sep. 9, 2020, and is .about.57 kilobytes, which is incorporated by reference herein.
[0033] SEQ ID NOs: 1-4 are amino acid sequences of cyclic peptides identified in RaPID screening with C. elegans iPGM.
[0034] SEQ ID NOs: 5 and 6 are amino acid sequences of modified cyclic peptides based on SEQ ID NO: 2.
[0035] SEQ ID NOs: 7-9 are amino acid sequences of linear peptides based on SEQ ID NO: 2.
[0036] SEQ ID NOs: 10-16 are amino acid sequences of cyclic peptides identified in RaPID screening with B. malayi iPGM.
[0037] SEQ ID NOs: 17-22 are amino acid sequences of modified cyclic peptides based on SEQ ID NO: 2.
[0038] SEQ ID NOs: 23-29 are nucleic acid sequences identified by RaPID selection against B. malayi iPGM.
[0039] SEQ ID NOs: 30-44 are nucleic acid sequences identified by RaPID selection against C. elegans iPGM.
[0040] SEQ ID NOs: 45-53 are amino acid sequences of iPGM orthologs.
[0041] SEQ ID NOs: 54-69 are the amino acid sequences of additional modified cyclic peptides based on SEQ ID NO: 2.
DETAILED DESCRIPTION
[0042] Enzymes essential for nematode survival but absent from humans represent potential targets for intervention. Essential nematode genes have been identified using comparative genomic studies of the free-living nematode Caenorhabditis elegans. As a result, several novel drug targets in filarial parasites have been proposed. Among the highest ranking is cofactor-independent phosphoglycerate mutase (iPGM) (EC 5.4.2.1). Silencing of ipgm in C. elegans and B. malayi leads to nematode death (Zhang et al., J. Biol. Chem. 279:37185-37190, 2004; Singh et al., Infect. Dis. Poverty 2:5, 2013), demonstrating the importance of this enzyme in nematode viability and, therefore, its potential as an anthelmintic drug target.
[0043] PGMs catalyze the interconversion of 2- and 3-phosphoglycerate (PG) in the glycolytic and gluconeogenic pathways. Although these pathways are highly conserved among different organisms, two distinct PGM isoenzymes are known to exist, namely iPGM and co-factor-dependent phosphoglycerate mutase (dPGM). The enzymes have no amino acid sequence similarity and differ in their mechanism of catalysis. iPGM is comprised of approximately 510 amino acids and catalyzes the intramolecular transfer of the phosphoryl group on the monophosphoglycerates through a phosphoserine intermediate and is the sole PGM in nematode (Jedrzejas et al., EMBO J. 19:1419-1431, 2000; Jedrzejas et al., J. Biol. Chem. 275:23146-23153, 2000). In contrast, dPGM is the form of enzyme in human that is composed of approximately 250 amino acids, and catalyzes the intermolecular transfer of the phosphoryl group between the monophosphoglycerates and the cofactor (2,3-diphosphoglycerate) through a phosphorylhistidine intermediate (Rigden et al., J. Mol. Biol. 315:1129-1143, 2002). While the two forms of PGM are distinct isozymes, the amino acid sequence of each isozyme family is conserved, when present, from bacteria to higher eukaryotes (Jedrzejas et al., Prog. Biophys. Mol. Biol. 73:263-287, 2000). The completely distinct structures and catalytic mechanism of iPGM and dPGM enzymes offer great promise for the discovery of inhibitors with high selectivity for the nematode enzymes. Furthermore, the high similarity in primary sequence and catalytic properties among the iPGMs indicates that a single inhibitor could be effective against a range of parasitic and microbial enzymes. However, iPGMs have been considered to be "undruggable," as high throughput screening (HTS) has to date identified only two low potency inhibitors of this enzyme (Crowther et al., PLoS Neglected Tropical Diseases 8:e2628, 2014).
[0044] Disclosed herein are a series of cyclic peptides and analogs that exhibit potent and isozyme-selective inhibition against iPGMs. The identification of the disclosed inhibitors of iPGM overturns the designation of this target as "undruggable" and provides novel anti-microbial therapeutics. The parental peptides, referred to in some instances herein as "ipglycermides," were identified from a library containing a trillion cyclic peptide members, each of which was displayed on a cognate mRNA template. These peptides have a unique lariat structure. In some examples the ring peptide consists of eight amino acid residues, one of which is D-tyrosine, closed by a thioether bond and the tail peptide consists of seven residues, one of which is L-cysteine (Cys) at the C-terminal region. Study of structure-activity relationships revealed that the ring peptide is involved in binding to iPGM orthologs, while the tail peptide region is involved in the nature of the inhibitory pharmacology and ortholog selectivity.
I. Abbreviations
[0045] dPGM cofactor-dependent phosphoglycerate mutase
[0046] GEMBE gradient elution moving boundary capillary electrophoresis
[0047] HTS high throughput screening
[0048] iPGM cofactor-independent phosphoglycerate mutase
[0049] PEP phosphoenolpyruvate
[0050] PG phosphoglycerate
[0051] PGM phosphoglycerate mutase
[0052] PK pyruvate kinase
[0053] RaPID Random non-standard Peptides Integrated Discovery
II. Terms
[0054] Unless otherwise noted, technical terms are used according to conventional usage. Definitions of common terms in molecular biology may be found in Benjamin Lewin, Genes VII, published by Oxford University Press, 2000 (ISBN 019879276X); Kendrew et al. (eds.), The Encyclopedia of Molecular Biology, published by Blackwell Publishers, 1994 (ISBN 0632021829); and Robert A. Meyers (ed.), Molecular Biology and Biotechnology: a Comprehensive Desk Reference, published by Wiley, John & Sons, Inc., 1995 (ISBN 0471186341); and other similar references.
[0055] Unless otherwise explained, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this disclosure belongs. The singular terms "a," "an," and "the" include plural referents unless context clearly indicates otherwise. Similarly, the word "or" is intended to include "and" unless the context clearly indicates otherwise. Hence "comprising A or B" means including A, or B, or A and B. It is further to be understood that all base sizes or amino acid sizes, and all molecular weight or molecular mass values, given for nucleic acids or polypeptides are approximate, and are provided for description. Although methods and materials similar or equivalent to those described herein can be used in the practice or testing of the present disclosure, suitable methods and materials are described below. All publications, patent applications, patents, and other references mentioned herein are incorporated by reference in their entirety. Sequences or structures associated with database accession numbers are herein incorporated by reference as present in the indicated database on Aug. 11, 2016, unless otherwise noted. In case of conflict, the present specification, including explanations of terms, will control. In addition, the materials, methods, and examples are illustrative only and not intended to be limiting.
[0056] In order to facilitate review of the various embodiments of the disclosure, the following explanations of specific terms are provided:
[0057] Analog or derivative: Compounds that differ from the disclosed peptides at one or more positions. An analog includes a molecule that differs in chemical structure from a parent compound, for example, differing by an increment in chemical structure (such as a difference in the length of an alkyl chain), a molecular fragment, a structure that differs by one or more functional groups, or a change in ionization. A derivative includes a biologically active molecule derived from the parent structure, for example, if one atom or group of atoms is replaced with another atom or group of atoms.
[0058] Cyclic peptide: A peptide with at least a portion forming a ring structure. In some examples, a cyclic peptide is fully cyclic, for example, having a head to tail cyclization. In other examples, a cyclic peptide includes both a ring structure and a linear peptide structure, such as formed by side chain to N-terminus, side chain to C-terminus, side chain to side chain, or backbone to backbone cyclization. In particular non-limiting examples, a cyclic peptide includes an N-terminal ring structure (e.g., formed by side chain to N-terminus cyclization) and a C-terminal linear portion (also referred to as a "tail").
[0059] Effective amount: A quantity of a specified agent sufficient to achieve a desired effect, such as an amount of an agent sufficient to inhibit or treat a disease without causing a substantial cytotoxic effect in a subject. For example, this may be the amount of an iPGM inhibitor useful for example, for reducing infection by, symptoms of, or transmission of microorganisms (such as parasites or bacteria).
[0060] Inhibiting or treating a disease: "Inhibiting" refers to reducing or delaying (or even preventing) the full development of a disease, disorder or condition, for example, in a subject who is at risk for a disease. "Treatment" refers to a therapeutic intervention that ameliorates a sign or symptom of a disease or pathological condition after it has begun to develop. As used herein, the term "ameliorating," with reference to a disease, pathological condition or symptom, refers to any observable beneficial effect of the treatment. The beneficial effect can be evidenced, for example, by a delayed onset of clinical symptoms of the disease in a susceptible subject, a reduction in severity of some or all clinical symptoms of the disease, a slower progression of the disease, a reduction in the number of relapses of the disease, an improvement in the overall health or well-being of the subject, or by other parameters known to one of ordinary skill in the art that are specific to the particular disease.
[0061] Isolated: An "isolated" biological component (such as a nucleic acid, protein, peptide, or pathogen) has been substantially separated or purified away from other biological components (such as cell debris, or other proteins or nucleic acids). Biological components that have been "isolated" include those components purified by standard purification methods. The term also embraces recombinant nucleic acids, peptides, or pathogens, as well as chemically synthesized nucleic acids or peptides. The term "isolated" (or "enriched" or "purified") does not require absolute purity, and can include molecules (such as peptides) that are at least 50% isolated, such as at least 75%, 80%, 90%, 95%, 98%, 99% or even 100% isolated.
[0062] Microorganism: An organism that can be seen only through a microscope. Microorganisms include bacteria, protozoa, algae, and fungi, as well as microscopic multicellular organisms, such as nematodes.
[0063] Nematode: A member of the phylum Nematoda, commonly referred to as roundworms. Nematodes include free-living species (such as the soil nematode C. elegans) and parasitic species. Species parasitic on humans include ascarids (e.g., Ascaris lumbricoides), filarias, hookworms (e.g., Ancylostoma duodenale or Necator americanus), pinworms, and whipworms. Exemplary species include Brugia malayi, Wuchereria bancrofti, and Brugia timori, which cause lymphedema, hydrocele, and in extreme cases, elephantiasis in humans. Onchocerca volvulus causes onchocerciasis ("river blindness") and severe dermatitis. Parasitic nematodes also infect companion animals and livestock, including dogs and cats (e.g., Dirofilaria immitis; heartworm), pigs (Trichinella spiralis), and sheep (e.g., Haemonchus contortus). There are also nematode species which are parasitic on insects and plants.
[0064] Pharmaceutically acceptable carrier: The pharmaceutically acceptable carriers (vehicles) useful in this disclosure are conventional. Remington: The Science and Practice of Pharmacy, The University of the Sciences in Philadelphia, Editor, Lippincott, Williams, & Wilkins, Philadelphia, Pa., 21' Edition (2005), describes compositions and formulations suitable for pharmaceutical delivery of one or more therapeutic compounds or molecules, such as one or more iPGM inhibitor peptides alone or in combination with additional pharmaceutical agents.
[0065] In general, the nature of the carrier will depend on the particular mode of administration being employed. For instance, parenteral formulations usually comprise injectable fluids that include pharmaceutically and physiologically acceptable fluids such as water, physiological saline, balanced salt solutions, aqueous dextrose, glycerol or the like as a vehicle. For solid compositions (e.g., powder, pill, tablet, or capsule forms), conventional non-toxic solid carriers can include, for example, pharmaceutical grades of mannitol, lactose, starch, or magnesium stearate. In addition to biologically-neutral carriers, pharmaceutical compositions to be administered can contain minor amounts of non-toxic auxiliary substances, such as wetting or emulsifying agents, preservatives, and pH buffering agents, and the like, for example sodium acetate or sorbitan monolaurate.
[0066] Phosphoglycerate mutase (PGM): An enzyme that catalyzes the reversible conversion of 3-phosphoglycerate to 2-phosphoglycerate. There are two classes of PGM. Cofactor-independent PGM (iPGM) (EC 5.4.2.12) and cofactor-dependent PGM (dPGM) (EC 5.4.2.11). iPGM is found in organisms including, but not limited to plants, algae, fungi, nematodes, and some bacteria (such as some Gram-positive bacteria). This enzyme catalyzes isomerization of 2-PG and 3-PG through a phosphoserine intermediate. dPGM is found in organisms including, but not limited to vertebrates, mollusks, crustaceans, insects, algae, fungi, and some bacteria (such as some Gram-negative bacteria). dPGM catalyzes isomerization of 2-PG and 3-PG through a phosphohistidine intermediate.
[0067] PGM nucleic acid and amino acid sequences are publicly available. Exemplary PGM amino acid sequences include GenBank Accession Nos. NP_871851 (Caenorhabditis elegans), AAQ97626 (Brugia malayi), AAV33247 (Onchocerca volvulus), AEA91534 (Dirofilaria immitis), NP_002620 (H. sapiens), and P37689 and P62707 (E. coli), all of which are incorporated herein by reference as present in GenBank on Aug. 11, 2016. One of ordinary skill in the art can identify additional PGM sequences and variants thereof.
[0068] Subject: Living multi-cellular vertebrate organisms, a category that includes, for example, mammals and birds. The term mammal includes both human and non-human mammals. Similarly, the term "subject" includes both human and veterinary or laboratory subjects, for example, humans, non-human primates, mice, rats, dogs, cats, sheep, pigs, horses, and cows.
III. Peptide Inhibitors of PGM
[0069] Disclosed herein are peptides that inhibit activity of PGM (for example, iPGM). In some examples, the peptides selectively or specifically inhibit activity of iPGM compared to dPGM. In some examples, an agent that selectively or specifically inhibits (for example, decreases activity of) iPGM is a compound that decreases activity of iPGM, but does not substantially decrease activity of dPGM. For example, a selective inhibitor of iPGM may decrease activity of iPGM, but not show any appreciable activity against dPGM. In other examples, a selective inhibitor of iPGM is an agent that decreases activity of iPGM by at least 2-fold more (for example, at least 5-fold more, at least 10-fold more, at least 20-fold more, 50-fold more, 100-fold more, 500-fold more, 1000-fold more, 2000-fold more, 5000-fold more, 10,000-fold more, or more) than it decreases activity of dPGM. The determination that a particular agent selectively or specifically inhibits iPGM may readily be made by using or adapting routine procedures for determining PGM activity. Exemplary methods of measuring iPGM and dPGM activity are described in Example 1, below.
[0070] In some embodiments, the disclosed peptides inhibit activity of iPGM with an IC.sub.50 of 100 .mu.M or less, such as 50 .mu.M or less, 10 .mu.M or less, 5 .mu.M or less, 1 .mu.M or less, 500 nM or less, 100 nM or less, 50 nM or less, 10 nM or less, 5 nM or less, 1 nM or less, or 100 pM or less. In other examples, the disclosed peptides inhibit activity of iPGM with an IC.sub.50 of about 1 pM to 100 .mu.M, for example, about 5 pM to 10 nM, about 10 pM to 50 nM, about 100 pM to 100 nM, about 1 nM to 100 nM, about 10 nM to 1 .mu.M, about 100 nM to 10 .mu.M, or about 1 .mu.M to 100 .mu.M.
[0071] In particular examples, the peptides are cyclic peptides; however, in some examples, linear peptides that inhibit iPGM activity are also disclosed herein. In some embodiments, the peptides are 6-20 amino acids long (such as 6-15, 8-20, or 10-20 amino acids long, for example, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 amino acids long). In some examples, the iPGM inhibitor peptide has a ring portion of 6-15 amino acids (such as 7-13, 8-12, 9-11, or 10-15 amino acids, for example, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 amino acids) and a linear portion of 1-9 amino acids (such as 1-5, 2-7, or 4-8 amino acids, for example, 1, 2, 3, 4, 5, 6, 7, 8, or 9 amino acids). In specific non-limiting examples, the iPGM inhibitor peptide has a cyclic portion of 8 amino acids and a linear portion of 3-7 amino acid. As discussed below, analogs or derivatives of the peptides (such as peptides with modified amino acid sequence and/or modifications at the N-terminus, C-terminus, peptide backbone, or side chains) are also disclosed herein. In some embodiments, the peptides or analogs or derivatives thereof include pharmaceutically acceptable salts or esters.
[0072] In some embodiments, the PGM inhibitors include or consist of the following cyclic peptides:
##STR00001## ##STR00002##
[0073] In other embodiments, the PGM inhibitors include or consist of the following linear peptide:
TABLE-US-00001 (SEQ ID NO: 7) YDYPGDYSYLYGTCG
[0074] In specific examples disclosed herein, the ring portion of the cyclic peptide is produced by forming a thioether linkage between a cysteine residue in the peptide and an N-terminal chloroacetyl group (for example, as shown in Tables 2, 6, 8, and 9 and FIGS. 12A and 21). However, additional linkers can be utilized to form the cyclic peptides. For example, in some examples, the ring portion of the peptide is formed using an aromatic or aliphatic linker. For example, the cyclic portion of the peptide can be formed using an aliphatic linker, such as a 1-6 carbon chain. In other examples, the linkage may include an aromatic group.
[0075] In addition, in specific examples disclosed herein, the cyclic peptides are formed by side chain to N-terminus cyclization (for example, as shown in Tables 2, 6, 8, and 9 and FIGS. 12A and 21). However, one of ordinary skill in the art will recognize that additional types of cyclization (such as head to tail, side chain to side chain, side chain to C-terminus, or backbone to backbone cyclization) can also be utilized to form cyclic iPGM inhibitor peptides.
[0076] Also disclosed are analogs or derivatives of iPGM inhibitors, such as those described herein. An analog includes a molecule that differs in chemical structure from a parent compound, for example, differing by an increment in chemical structure (such as a difference in the length of an alkyl chain), a molecular fragment, a structure that differs by one or more functional groups, or a change in ionization. A derivative includes a biologically active molecule derived from the parent structure, for example, if one atom or group of atoms is replaced with another atom or group of atoms. Thus, in some examples, analogs or derivatives include compounds that differ from the disclosed peptides by substitution, deletion, and/or addition of one or more amino acids. Analogs or derivatives also include compounds that differ from the disclosed peptides by one or more modifications, such as substitution, addition, and/or deletion of one or more atom or group of atoms at the N-terminus, C-terminus, peptide backbone, and/or amino acid side chain of the peptide. In addition, an analog or derivative may include a combination of one or more amino acids changes and one or more changes of an atom or group of atoms of the peptide.
[0077] In some examples, analogs of the disclosed peptides include modifications to allow cyclization of the peptide. Thus, in one non-limiting example, an analog includes an N-terminal acetyl group (for example, an N-terminal chloroacetyl), which permits peptide cyclization through a thioether bond to a cysteine residue in the peptide. In other examples, the peptides (such as any one of SEQ ID NOs: 1-22 or 54-69) include a C-terminal amide modification. In some examples, the peptides include all L-amino acids, while in other examples, analogs include one or more D-amino acids. In particular, non-limiting examples, analogs include D-tyrosine (e.g., D-N-chloroacetyl tyrosine) as the N-terminal amino acid, such as Ce2 or Ce-2d.
[0078] In still further examples, an analog of the disclosed peptides includes a peptide with one or more amino acid substitutions, deletion of one or more amino acids (including, but not limited to truncation mutants), and/or addition of one or more amino acids. In another example, a derivative of the disclosed peptides includes substitution of the sulfhydryl group of a cysteine residue (such as the cysteine residue in the linear portion of any one of the disclosed peptides) with a hydroxamic acid, diketo acid, or a metal ion chelator. In further examples the peptides include a modification to increase stability (for example, resistance to proteolysis), such as one or more N-methyl amides in the linear portion of the peptide (exemplified by those shown in Table 9). In yet further examples, a carbon chain (such as a 1-8 carbon chain) is added at the C-terminus of any of the cyclic peptides disclosed herein. In still further examples, an aminoalkyl thiol (such as a C1-C8 alkyl) is added to the C-terminus of any of the cyclic peptides disclosed herein. In some examples, the C-terminal carbon chain or aminoalkyl thiol includes a free thiol, an alkyl thioester (C1-C9) or an alkyl acid at the C-terminus. Exemplary analogs are shown in FIG. 21. These are illustrated with respect to the Ce-2d core structure, but corresponding modifications may be made to any of the peptides disclosed herein. Any combination of modifications or substitutions is contemplated herein.
[0079] In additional examples, an analog of the disclosed peptides includes replacing one or more (such as 1, 2, 3, 4, or 5) tyrosine residues with either 4-F-Phe or 4-MeO-Phe. In some examples, the tyrosine residue corresponds to Tyr1, Tyr3, Tyr9, or Tyr11 of a disclosed peptide (such as SEQ ID NO: 1). In an additional example, the tyrosine residue corresponds to Tyr7 of a disclosed peptide (such as SEQ ID NO: 2). The corresponding tyrosine residues in other peptides disclosed herein can be identified by one of ordinary skill in the art. Other modifications include increasing the basicity of the molecule by reducing negative charge, for example by replacing Asp2 with Asn or His and/or by including positive charges in the alkyl linker, such as tertiary amines.
[0080] The disclosed peptides and analogs or derivatives may be in the form of one or more pharmaceutically acceptable salts or esters. Pharmaceutically acceptable salts of the disclosed peptides include those formed from cations such as sodium, potassium, aluminum, calcium, lithium, magnesium, zinc, and from bases such as ammonia, ethylenediamine, N-methyl-glutamine, lysine, arginine, ornithine, choline, N,N'-dibenzylethylenediamine, chloroprocaine, diethanolamine, procaine, N-benzylphenethylamine, diethylamine, piperazine, tris(hydroxymethyl)aminomethane, and tetramethylammonium hydroxide. The salts may be prepared by standard procedures, for example by reacting the free acid with a suitable organic or inorganic base. Representative bases include ammonium hydroxide, sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide, magnesium hydroxide, ferrous hydroxide, zinc hydroxide, copper hydroxide, aluminum hydroxide, ferric hydroxide, isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, ethanolamine, 2-dimethylaminoethanol, 2-diethylaminoethanol, lysine, arginine, histidine, and the like. In one aspect, the reaction is conducted in water, alone or in combination with an inert, water-miscible organic solvent, at a temperature of from about 0.degree. C. to about 100.degree. C., such as at room temperature. The molar ratio of compounds to be used is chosen to provide the ratio desired for any particular salts. In some examples, the peptides can be treated with approximately one equivalent of pharmaceutically acceptable base to yield a neutral salt. Pharmaceutically acceptable salts are also inclusive of the free acid, base, and zwitterionic forms. Description of suitable pharmaceutically acceptable salts can be found in Handbook of Pharmaceutical Salts, Properties, Selection and Use, Wiley VCH (2002).
[0081] Pharmaceutically acceptable esters include, but are not limited to, methyl, ethyl, propyl, butyl, pentyl, hexyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclopentenyl, cyclopentadienyl, cyclohexenyl, cyclohexadienyl, phenyl, pyridinyl, benzyl, and the like. Pharmaceutically acceptable esters can be prepared by, for example, by treating the compound with an appropriate amount of carboxylic acid, ester, acid chloride, acid anhydride, or mixed anhydride agent that will provide the corresponding pharmaceutically acceptable ester. Typical agents that can be used to prepare pharmaceutically acceptable esters include, for example, acetic acid, acetic anhydride, acetyl chloride, benzylhalide, benzaldehyde, benzoylchloride, methyl ethylanhydride, methyl phenylanhydride, methyl iodide, and the like. In one non-limiting example, a pharmaceutically acceptable ester of the disclosed peptides is a cysteine ester (such as a cysteine alkyl ester, cysteine ethyl ester, or N-acetyl cysteine ester) or the esters shown in FIG. 21.
IV. Methods of Treating or Inhibiting Disorders
[0082] Disclosed herein are methods of treating or inhibiting disorders (such as infection) using the inhibitors of PGM described herein. In some embodiments, the methods include treating or inhibiting infection with and/or disease caused by organisms that express a PGM, such as iPGM. Organisms that express iPGM include, but are not limited to nematodes, fungi, bacteria (including Gram-positive and Gram-negative bacteria), trypanosomes, protozoan parasites, helminths, plants, and algae. In some non-limiting examples, the methods include treating or inhibiting infection with nematodes (including but not limited to Brugia malayi, Brugia timori, Wuchereria bancrofti, Onchocerca volvulus, Ascaris lumbricoides, Ancylostoma duodenale, Necator americanus, Loa loa, Mansonella streptocerca, Mansonella perstans, Mansonella ozzardi, Dirofilaria immitis, Trichinella, Parafilaria bovicola, Onchocerca dermatan, Onchocerca ochengi, Onchocerca dukei, Stenofilaria assamensis, and Parafilaria multipapillosa). In other non-limiting examples, the methods include treating or inhibiting infection with Gram-positive bacteria expressing an iPGM (including, but not limited to Staphylococcus aureus, Streptococcus pneumoniae, and Bacillus anthracis). In other examples, the methods include treating or inhibiting an infection and/or disease caused by an organism expressing an iPGM, for example, trypanosomes (such as Trypanosoma brucei or Trypanosoma cruzi) or protozoan parasites (such as Leishmania mexicana, L. major, L. tropica, L. aethiopica, L. braziliensis, L. donovani, Plasmodium falciparum, P. vivax, P. ovale, P. malariae, or P. knowlesi), Babesia, or Giardia. In another example, the methods include treating or inhibiting infection with Gram-negative bacteria that express iPGM, including Escherichia coli. In some examples, the infection and/or disease is caused by an organism expressing only iPGM, while in other examples, the infection and/or disease is caused by an organism expressing both iPGM and dPGM.
[0083] In particular examples, the methods include administering to a subject an effective amount of a composition that includes one or more iPGM inhibitor peptides disclosed herein. In some examples, subject is infected with one or more of a nematode, helminth, trypanosome, protozoan parasite, or bacteria. In particular examples, the subject has a disease caused by a nematode, including but not limited to lymphatic filariasis (for example, lymphedema, hydrocele, and/or elephantiasis), subcutaneous filariasis (such as dermatitis and/or blindness), or serous cavity filariasis. In other examples, the subject has heart filariasis (heartworm, for example, in dogs or cats), verminous hemorrhagic dermatitis (cattle), intradermal onchocercosis (cattle), "summer bleeding" (horses, cause by Parafilaria multipapillosa), or trichnosis. In other examples, the subject has a disease caused by a protozoan parasite, including but not limited to sleeping sickness (or Nagana, in cattle), Chagas' disease, leishmaniasis, giardiasis, babesiosis, or malaria. In still further examples, the subject has a bacterial infection, including but not limited to infection with Staphylococcus aureus, Bacillus anthracis, or Streptococcus pneumoniae.
[0084] In some embodiments, the subject is administered an effective amount of a composition including one or more cyclic peptide inhibitors of iPGM, for example one or more cyclic peptides having the amino acid sequence of any one of SEQ ID NOs: 1-6, 10-22, and 54-69 (for example, one or more of the cyclic peptides shown in Tables 2, 6, 8, or 9, or FIG. 12A-12B or 21) or an analog or derivative thereof, or a pharmaceutically acceptable salt or ester thereof. In other examples, the subject is administered an effective amount of a composition including one or more linear peptide inhibitors of iPGM, for example, one or more peptides having the amino acid sequence of SEQ ID NOs: 7-9 or an analog or derivative thereof, or a pharmaceutically acceptable salt or ester thereof. In other examples, the subject is administered an effective amount of a composition including one or more cyclic peptide iPGM inhibitors and one or more linear peptide iPGM inhibitors, such as those disclosed herein. In particular non-limiting examples, the subject is administered an effective amount of a composition including peptide Ce-2 (SEQ ID NO: 2 with a cyclization between the N-terminus and Cys8) or an analog or derivative thereof, or a pharmaceutically acceptable salt or ester thereof.
[0085] The disclosed peptides can be administered by any means known to one of skill in the art, such as by intramuscular, subcutaneous, intraperitoneal, or intravenous injection, but even oral, nasal, or anal administration is contemplated. The disclosed peptides can also be administered topically, transdermally, or by local injection. In some embodiments, administration is orally, by intravenous injection, or topically. To extend the time during which the peptide is available to inhibit or treat an infection, the peptide can be provided as an implant, an oily injection, or as a particulate system. The particulate system can be a microparticle, a microcapsule, a microsphere, a nanoparticle, a nanocapsule, or similar particle. One of ordinary skill in the art is aware of methods of administering peptides to a subject. See, e.g., Banga, "Parenteral Controlled Delivery of Therapeutic Peptides and Proteins," in Therapeutic Peptides and Proteins, Technomic Publishing Co., Inc., Lancaster, Pa., 1995.
[0086] In some examples, the provided peptides are combined with a pharmaceutically acceptable carrier or vehicle for administration to human or animal subjects. In some embodiments, more than one disclosed peptide (for example, 1, 2, 3, 4, 5, or more peptides) can be combined to form a single preparation. Examples of suitable pharmaceutically acceptable carriers, vehicles, or excipients include sterile aqueous or non-aqueous solutions, suspensions, and/or emulsions. Examples of non-aqueous solvents include propylene glycol, polyethylene glycol, vegetable oils such as olive oil, and injectable organic esters such as ethyl oleate. Aqueous carriers include water, alcoholic/aqueous solutions, emulsions or suspensions, including saline and buffered media. Parenteral vehicles include sodium chloride solution, Ringer's dextrose, dextrose and sodium chloride, lactated Ringer's, or fixed oils. Intravenous vehicles include fluid and nutrient replenishers, electrolyte replenishers (such as those based on Ringer's dextrose), and the like. Preservatives and other additives may also be present such as, for example, antimicrobials, anti-oxidants, chelating agents, and inert gases and the like.
[0087] The peptides and pharmaceutical compositions provided herein may be administered through different routes, such as oral, including buccal and sublingual, rectal, parenteral, aerosol, nasal, intravenous, intramuscular, subcutaneous, intradermal, and topical. They may be administered in different forms, including but not limited to solutions, emulsions and suspensions, microspheres, particles, microparticles, nanoparticles, and liposomes.
[0088] In another embodiment, it may be desirable to administer the peptides or pharmaceutical compositions locally to the area in need of treatment. This may be achieved by, for example, and not by way of limitation, local or regional infusion or perfusion, topical application, injection, catheter, suppository, or implant (e.g., implants formed from porous, non-porous, or gelatinous materials, including membranes, such as sialastic membranes or fibers), and the like.
[0089] In a specific embodiment, one or more of the disclosed peptides may be associated either by coating or impregnating an implant. In an example, the implant can be partially or completely coated with the peptide. The peptide may be attached to the implant by any chemical or mechanical bond or force, including linking agents. Alternatively, the coating may be directly linked (tethered) to the surface, such as through silane groups. In other examples, the implant may be impregnated with at least one peptide by methods known to those of skill in the art so that multiple surfaces (such as the outer and inner surfaces) of the implant include the peptide. In an additional embodiment, the implant may be coated or impregnated with materials in addition to the disclosed peptides to further enhance their bio-utility. Examples of suitable coatings are medicated coatings, drug-eluting coatings, hydrophilic coatings, or smoothing coatings.
[0090] In one embodiment, administration can be by direct injection at the site of a tissue that is to be treated. In another embodiment, the pharmaceutical compositions are delivered in a vesicle, in particular liposomes (see, e.g., Langer, Science 249:1527-1533, 1990; Treat et al., in Liposomes in the Therapy of Infectious Disease and Cancer, Lopez-Berestein and Fidler (eds.), Liss, N.Y., pp. 353-365, 1989).
[0091] In yet another embodiment, the pharmaceutical compositions can be delivered in a controlled release system. In one embodiment, a pump can be used (see, e.g., Langer Science 249:1527-1533, 1990; Sefton Crit. Rev. Biomed. Eng. 14:201-240, 1987; Buchwald et al., Surgery 88:507-516, 1980; Saudek et al., N Engl. J. Med. 321:574-579, 1989). In another embodiment, polymeric materials can be used (see, e.g., Ranger et al., Macromol. Sci. Rev. Macromol. Chem. 23:61-64, 1983; Levy et al., Science 228:190-192, 1985; During et al., Ann. Neurol. 25:351-356, 1989; and Howard et al., J. Neurosurg. 71:105-112, 1989). Other controlled release systems, such as those discussed in the review by Langer (Science 249:1527-1533, 1990), can also be used.
[0092] Pharmaceutical compositions for oral use can also be formulated, for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsion hard or soft capsules, or syrups or elixirs. Such compositions can be prepared according to standard methods known to the art for the manufacture of pharmaceutical compositions and may contain one or more agents selected from the group of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations. Tablets contain the active ingredient in admixture with suitable non-toxic pharmaceutically acceptable excipients including, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, such as corn starch, or alginic acid; binding agents, such as starch, gelatin or acacia, and lubricating agents, such as magnesium stearate, stearic acid or talc. The tablets can be uncoated, or they may be coated by known techniques in order to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period. For example, a time delay material such as glyceryl monostearate or glyceryl distearate may be employed. Pharmaceutical compositions for oral use can also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium such as peanut oil, liquid paraffin or olive oil.
[0093] The peptides can be conveniently presented in unit dosage form and prepared using conventional pharmaceutical techniques. Such techniques include the step of bringing into association the active ingredient and the pharmaceutical carrier(s) or excipient(s). In general, the formulations are prepared by uniformly and intimately bringing into association the active ingredient with liquid carriers. Formulations suitable for parenteral administration include aqueous and non-aqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostatic agents, and/or solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents. The formulations may be presented in unit-dose or multi-dose containers, for example, sealed ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of a sterile liquid carrier, for example, water for injections, immediately prior to use. Injection solutions and suspensions may be prepared from sterile powders, granules and tablets commonly used by one of ordinary skill in the art.
[0094] In certain embodiments, unit dosage formulations are those containing a dose or unit, or an appropriate fraction thereof, of the administered ingredient. It should be understood that in addition to the ingredients particularly mentioned above, formulations encompassed herein may include other agents commonly used by one of ordinary skill in the art.
[0095] The amount of the peptide(s) that will be effective depends on the nature of the disorder or condition to be treated, as well as the stage of the disorder or condition. Effective amounts can be determined by standard clinical techniques. The precise dose of the peptide(s) to be employed in the formulation will also depend on the route of administration, and should be decided according to the judgment of the health care practitioner and each subject's circumstances. An example of such a dosage range is 1 .mu.g/kg to 200 mg/kg body weight (for example, about 5 .mu.g/kg to 1 mg/kg, about 10 .mu.g/kg to 5 mg/kg, about 100 .mu.g/kg to 20 mg/kg, about 0.2 to 100 mg/kg, about 0.5 to 50 mg/kg, about 1 to 25 mg/kg, about 5 to 75 mg/kg, about 50 to 150 mg/kg, or about 100 to 200 mg/kg) in single or divided doses. Another example of a dosage range is 1 .mu.g/kg to 100 mg/kg body weight (for example, about 1 to 100 .mu.g/kg, 10 .mu.g/kg to 1 mg/kg, 100 .mu.g/kg to 5 mg/kg, 1 to 10 mg/kg, about 5 to 25 mg/kg, about 20 to 50 mg/kg, about 40 to 80 mg/kg, or about 60 to 100 mg/kg) in single or divided doses. For example, a suitable dose is about 0.1 mg/kg, about 0.2 mg/kg, about 0.5 mg/kg, about 0.75 mg/kg, about 1 mg/kg, about 2 mg/kg, about 5 mg/kg, about 7.5 mg/kg, about 10 mg/kg, about 15 mg/kg, about 25 mg/kg, about 50 mg/kg, about 75 mg/kg, or about 100 mg/kg. Unit dosage forms are also possible, for example 0.01 mg, 0.05 mg, 0.1 mg, 0.25 mg, 0.5 mg, 1 mg, 2 mg, 5 mg, 10 mg, 15 mg, 20 mg, 25 mg, 50 mg, 100 mg, 150 mg, 200 mg, 500 mg, or up to 1000 mg per dose. However, other higher or lower dosages also could be used, as can be determined by in vitro and/or in vivo testing.
[0096] The specific dose level and frequency of dosage for any particular subject may be varied and will depend upon a variety of factors, including the activity of the specific compound, the metabolic stability and length of action of that compound, the particular disease or disorder to be treated, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, and/or any drug combinations administered. Treatment can involve daily or multi-daily, weekly, bi-monthly, or monthly doses of compound(s) over a period of a few days or weeks to months, or even years.
[0097] The pharmaceutical compositions of the present disclosure can be administered at about the same dose throughout a treatment period, in an escalating dose regimen, or in a loading-dose regime (e.g., in which the loading dose is about two to five times the maintenance dose). In some embodiments, the dose is varied during the course of a treatment based on the condition of the subject being treated, the severity of the disease or condition, the apparent response to the therapy, and/or other factors as judged by one of ordinary skill in the art.
[0098] The disclosed peptides can be used alone or in combination therapy with other compositions or drugs used to treat the described conditions. Such combination therapies include, but are not limited to simultaneous or sequential administration of the drugs involved. For example, in the treatment of nematodes, the peptide formulations can be administered in combination with any one or more of the anti-filarial therapies currently in use, for example, diethylcarbamazine, albendazole, levamisole, doxycycline, and/or ivermectin. Similarly, in the treatment of bacterial infection, the peptide formulations can be administered in combination with one or more antibiotics. One of ordinary skill in the art can identify appropriate combination therapies based on the infection or disease being treated.
EXAMPLES
[0099] The following examples are provided to illustrate certain particular features and/or embodiments. These examples should not be construed to limit the disclosure to the particular features or embodiments described.
Example 1
Materials and Methods
[0100] Preparation of PGM enzymes: All PGM enzymes were cloned into pET21a(+) and expressed in the Escherichia coli strain C2566/T7 Express (fhuA2 lacZ::T7 gene1 [lon] ompT gal sulA11 R(mcr-73::miniTn10--TetS)2[dcm] R(zgb-210::Tn10--TetS) endA1 .DELTA.(mcrC-mrr)114::IS10) (New England Biolabs) and expressed and purified as previously described (Raverdy et al., Mol. Biochem. Parasitol. 156:210-216, 2007; Zhang et al., J. Biol. Chem. 279:37185-37190, 2004). Briefly, optimum conditions for production of soluble recombinant iPGM involved growth of cultures at 37.degree. C. to 0.6.sub.OD600, induction with 0.1 mM IPTG overnight at 16.degree. C. The His-tagged proteins were purified on a 5 ml HiTrap.TM. chelating HP column (GE Healthcare; Pittsburgh, Pa.) using an AKTA FPLC following manufacturer's instructions. After application of the sample, the column was washed with five column volumes of buffer A (20 mM NaPO.sub.4, 300 mM NaCl, 10 mM imidazole, pH 7.4) followed by 10 column volumes of 92% buffer A:8% buffer B (20 mM NaPO.sub.4, 300 mM NaCl, 400 mM imidazole, pH 7.4). Protein was then eluted using a linear gradient (8-100%) of buffer B equivalent to 40-400 mM imidazole.
[0101] Fractions containing iPGM-His6.times. were pooled, dialyzed against dialysis buffer (40 mM Tris-HCl, 200 mM NaCl and 50% glycerol, pH 7.5) and stored at -20.degree. C. prior to use. Purity of the protein was estimated by 4-20% SDS-PAGE and the protein concentration was determined using the Bradford assay. Protein concentrations were 10 mg/ml or higher.
[0102] The sequences encoding the PGMs used in this study have the following NCBI (GenBank) Accession numbers: C. elegans iPGM, long form, NP_871851.1; C. elegans iPGM, short form, NP_491896.1; B. malayi iPGM AAQ97626.1; O. volvulus iPGM, AAV33247.1; Dirofilaria immitis iPGM, AEA91534.1; Homo sapiens dPGM, NP_002620.1; E. coli iPGM, P37689.1; and E. coli dPGM, P62707.2, all of which are incorporated by reference herein as present in GenBank on Aug. 11, 2016.
[0103] iPGM and dPGM Assays: Phosphoglycerate mutase activity was measured either as a continuous or endpoint output assay. The continuous assay was based on lactate dehydrogenase oxidation of NADH as monitored at an absorbance of 340 nm using pyruvate supplied through a series of coupling enzymes as previously described for C. elegans and B. malayi iPGMs (Zhang et al., J. Biol. Chem. 279:37185-37190, 2004; White et al., Eur. J. Biochem. 207:709-714, 1992), adapted here in 1536-well microtiter plate format for the PGM orthologs and isozymes used in this study. Initial rate conditions determined from the continuous assay were used to calibrate an end-point bioluminescent assay format for a PGM profiling panel to evaluate the inhibitors.
[0104] 1536-well format kinetic assay: Briefly, the forward glycolytic PGM catalyzed conversion of 3-phosphoglycerate (3-PG) to 2-phosphoglycerate (2-PG) was measured indirectly by monitoring the consumption of NADH through a coupled enzyme reaction. Four .mu.L of the respective PGM enzyme was dispensed into black clear-bottom 1536 well plates (Cat#789092-F, Greiner Bio-One North America) in a pH 8.0 assay buffer with the BioRaptr.TM. FRD microfluidic workstation (Beckman Coulter, Brea, Calif.), for a final concentration of 30 mM Tris-HCl, 5 mM MgSO.sub.4, 20 mM KCl and 0.08% BSA. Two .mu.l of 3-PG substrate was added to each enzyme solution in a coupled enzyme assay buffer using the BioRaptr.TM. FRD, for a final assay concentration of 3 mM ADP, 500 .mu.M NADH, 0.3 units enolase, 0.3 units pyruvate kinase, and 0.3 units lactate dehydrogenase. To confirm the 3-PG apparent K.sub.m for several PGMs, 4 .mu.L of B. malayi iPGM, C. elegans iPGM, or H. sapiens dPGM enzymes were dispensed as above at a final concentration of 1 nM in pH 8.0 assay buffer. Two .mu.l of an 11-point titration series of 3-PG substrate ranging from 0.083-2.0 mM was added to each enzyme solution in the coupled enzyme assay buffer. A 60 min time course was read for each enzyme-substrate solution at an absorbance of 340 nm on an Infinite.RTM. M1000 PRO microplate reader (Tecan Group Ltd), as shown in FIG. 1A. NADH consumption was plotted as absorbance (340 nm) vs. time (seconds) in GraphPad Prism software (GraphPad Software, Inc.) for each of the enzyme-substrate titrations and the slope of the linear phase for each substrate concentration was calculated for the respective enzymes (FIG. 1B). The initial rate (vi) for each enzyme-substrate reaction was determined and plotted against molar substrate concentration to generate Michaelis-Menton curves, while the reciprocal of the rate and molar substrate concentrations were re-plotted as Lineweaver-Burk graphs in GraphPad Prism and apparent K.sub.m values were estimated for each respective enzyme.
[0105] 1536-well format luminescence assay: ATP generated from the pyruvate kinase (PK) catalyzed conversion of phosphoenolpyruvate (PEP) to pyruvate was utilized to configure a luminescence output for the PGM enzyme panel. Various concentrations of B. malayi iPGM, C. elegans iPGM, and H. sapiens dPGM enzymes ranging from 1-5 nM final concentrations were dispensed in a total volume of 4 .mu.l of the above assay buffer into respective wells of 1536-well white/solid bottom plates (Cat #789173-F, Greiner Bio-One North America) using the BioRaptr.TM. FRD workstation. Two .mu.l of 3-PG substrate solution prepared at the estimated K.sub.m concentration was added to each enzyme solution as described above, for a final assay concentration of 0.4 mM 3-PG, 3 mM ADP, 0.3 units enolase, and 0.3 units PK. Enzyme-substrate solutions were incubated at room temperature for 5 min, 4 .mu.l Kinase-Glo.RTM. Plus reagent (Promega Corporation, Madison, Wis.) was added to each reaction with the BioRaptr.TM. FRD workstation, plates were incubated at room temperature for 10 min protected from light, and a luciferase-based ATP detection read-out was measured by a ViewLux.RTM. plate reader (PerkinElmer, Waltham, Mass.). To expand the PGM enzyme selectivity panel to additional PGMs, luminescence measurements for O. volvulus iPGM, D. immitis iPGM, E. coli iPGM, and E. coli dPGM enzymes were measured as described above for each enzyme titrated from 1-20 nM in the presence of 0.4 mmol/L substrate. The enzyme concentration for each PGM that generated relatively equivalent luminescence RLU across the panel was selected for macrocyclic peptide and peptide analog profiling.
[0106] The pyruvate kinase (PK) coupling enzyme was included as an additional specificity control in the enzyme panel. PK concentrations of 0.3 units (.about.930 nM) or 0.15 units (.about.460 nM) were dispensed in a total volume of 4 .mu.l of the above assay buffer into respective wells of 1536-well white/solid bottom plates as previously described. Two .mu.1 of PEP substrate solution prepared at an equivalent concentration to 3-PG substrate were added to the PK enzyme solution as described above, for a final assay concentration of 0.4 mM PEP and 3 mM ADP. The protocol for this assay profile is given in Table 1.
[0107] SPPS cyclic peptides were tested under initial assay conditions described in Table 1 designed to give robust and uniform signal to background across the 7 enzyme PGM panel. Concentration response curves (CRCs) were fit using a 4 or 5 parameter Hill equation (below). Model selection was determined by an extra-sum-of-squares F test for each ortholog condition. The majority of cyclic peptides displayed hyperbolic responses; those with steep Hill slopes or requiring a 5 parameter fit were revaluated at assay conditions employing lower iPGM concentrations.
[0108] 5 Parameter Hill Equation:
Y = ( S max - S 0 ) [ 1 + 10 ( n ( Log Xb ) - X ) ] S ##EQU00001##
Where
[0109] Log Xb = Log EC 50 + 1 n Log ( 2 ( 1 S ) - 1 ) ##EQU00002##
Where S0 is the signal at zero concentration, Smax is the signal at infinite concentration, n is the Hill slope, LogEC.sub.50 is the log of the concentration at half-maximal signal, X is the log of the concentration, and S is the asymmetry parameter.
[0110] C. elegans iPGM titration: The 1536-well format luminescence assay (above) was adapted to evaluate enzyme concentration on IC50 of Ce-2. C. elegans iPGM 10 nM to 50 pM concentration range was tested across a 16-pt 1:3 dilution of Ce-2 from 3.83 .mu.M to 0.27 pM final concentration, while 1 .mu.M to 50 nM enzyme concentration range was tested across a 16-pt 1:2 dilution of Ce-2 from 3.83 .mu.M to 117 pM final concentration. C. elegans iPGM concentrations of 1 nM-50 pM were incubated with 0.4 mM 3PG substrate solution for 15 min at room temperature and read on the ViewLux plate reader with standard assay settings (1 sec exp, medium gain, slow speed, 2.times. binning) 100 nM-5 nM enzyme concentrations were incubated with 0.4 m 3PG substrate solution for 5 min at room temperature and read on the ViewLux with standard assay settings. Enzyme concentrations of 500 nM and 1 .mu.M were incubated with substrate for 5 min as above, but ViewLux plate reader settings were reduced to eliminate overexposure (1 sec exp, medium gain, medium speed, 2.times. binning) For the assay, 23 nL of the peptide titration series from a 16-point compound dispense plate were simultaneously transferred to 1536-well assay plates (Cat#789092-F, Greiner Bio-One North America) using a 1536-pin tool (Wako) for a final concentration range of 3.8 .mu.M-0.27 pM.
TABLE-US-00002 TABLE 1 PGM ortholog and isozyme 1536-well plate assay protocol Step Parameter Value Target Description 1a Reagent 4 .mu.L B. malayi No Enzyme control and iPGM Enzyme solutions (5 nM 1b Reagent 4 .mu.L C. elegans B. malayi iPGM, 5 iPGM nM C. elegans iPGM, 1c Reagent 4 .mu.L H. sapiens 5 nM H. sapiens dPGM, iPGM 20 nM O. volvulus 1d Reagent 4 .mu.L O. volvulus iPGM, 10 nM D. immitis iPGM iPGM, 10 nM E. coli 1e Reagent 4 .mu.L D. immitis iPGM, and 4 nM E. coli iPGM dPGM final 1f Reagent 4 .mu.L E. coli iPGM concentrations); 1g Reagent 4 .mu.L E. coli dPGM white/solid bottom base plate (Greiner) 1h Reagent 4 .mu.L PK-Fluc ctrl 0.15 U PK final concentrations; white/solid bottom high base plate (Greiner) 2 Cyclic 23 nL Cyclic Peptides peptides profiled across iPGM orthologs and dPGM isozymes (5 mM-84.7 nM; 19.1 .mu.M-324.6 pM; 11-pt 1:3 titration series) or vehicle (DMSO) control; Peptides transfer by Pintool; Control = No Enzyme 3 Incubation 20-30 Peptide interaction min with enzyme 4 Reagent 2 .mu.L 3-phoshoglycerate (3PG) and PEP; PEP only for PK control 5 Incubation 10 min Ambient temperature; dark 6 Reagent 3 .mu.L Kinase-Glo Plus reagent 7 Incubation 20-30 Ambient temperature; dark min 8 Measurement ViewLux Luminescence mode, 1 sec exposure; gain = med; speed = slow; binning = 2X Step Notes 1 Assay buffer: 30 mM Tris-HCl, pH 8.0, 5 mM MgSO.sub.4, 20 mM KCl, 0.12% BSA + 6-30 nM enzyme or 0.0375 units/.mu.L pyruvate kinase 5X Assay Buffer: 150 mM Tris-HCl, pH 8, 25 mM MgSO.sub.4, 100 mM KCl, 0.6% BSA 4 Substrate buffer for PGM enzymes: 30 mM Tris-HCl, pH 8.0, 5 mM MgSO.sub.4, 20 mM KCl, 9 mM ADP, and 0.15 units/.mu.L each of enolase and pyruvate kinase + 1.2 mM 3PG Substrate buffer for PK enzyme: 30 mM Tris-HCl, pH 8.0, 5 mM MgSO.sub.4, 20 mM KCl + 1.2 mM PEP 5X Assay buffer: 150 mM Tris-HCl, pH 8, 25 mM MgSO.sub.4, 100 mM KCl Final PGM assay buffer concentrations: 30 mM Tris-HCl, pH 8.0, 5 mM MgSO.sub.4, 20 mM KCl, 3 mM ADP, and 0.3 units each of enolase and pyruvate kinase + 4- 20 nM PGM enzyme and 0.4 mM 3PG (or 0.15 units PK + 0.4 mM PEP)
[0111] Gradient elution moving boundary electrophoresis (GEMBE): GEMBE was used for the direct monitoring of the activity of the enzyme iPGMs via label-free measurement of the substrate and product, 2-PG and 3-PG. A custom built apparatus was used to perform gradient elution moving boundary electrophoresis (Strychalski et al., Anal. Chem. 83:6316-6322, 2011). The separation channel consisted of a 5 cm length of capillary (360 .mu.m OD, 15 .mu.m ID) joining a custom machined 200 .mu.L sample reservoir and 2000 .mu.L buffer reservoir. Pressure in the headspace of the buffer reservoir was controlled using a Series 600 automated pressure calibrator (Mensor, San Marcos, Tex.). Platinum electrodes were inserted into the reservoirs to apply voltage across the separation channel. The capillary passed through a TraceDec.RTM. capacitively-coupled contactless conductivity detector (Innovative Sensor Technologies, Strasshof, Austria) with the detection spot located approximately 2 cm from the sample reservoir. The buffer reservoir was filled with electrophoresis buffer (30 mM Tris-HCl pH 8.0, 20 mM MgCl.sub.2), and enzyme reactions were mixed and run directly in the sample reservoir. The magnesium concentration in the electrophoresis buffer was chosen to optimize the resolution between the 2-PG and 3-PG signals (Schaeper et al., J. Capillary Electrophor. 3:215-221, 1996) in the GEMBE electopherogram (FIG. 2C and FIGS. 3A-3B).
[0112] The equilibrium ratio of 2-PG to 3-PG predicted from the standard free energy is approximately 1:7 at room temperature (Clarke et al., Biochem. J. 139:491-497, 1974). Consequently, with the GEMBE assay, the typical change in signal is approximately 11.times. larger for the reaction starting with 2-PG and converting to 3-PG than for the reaction starting with 3-PG and converting to 2-PG. For the cofactor independent enzymes (B. malayi iPGM, C. elegans iPGM, and E. coli iPGM), the mutase reaction was found to be reversible in the GEMBE assay. Therefore, reactions with those enzymes were run starting with pure 2-PG and monitoring the conversion of 2-PG to 3-PG to maximize signal. For the cofactor dependent enzyme, H. sapiens dPGM, the mutase reaction was found to be irreversible in the GEMBE assay, with much faster reaction rates found for the conversion of 3-PG to 2-PG. Reactions with that enzyme were therefore started with pure 3-PG and the conversion of 3-PG to 2-PG was monitored.
[0113] The analytical separation of the product and substrate was carried out as follows. The buffer reservoir pressure was maintained at 30 kPa between separations and during sample loading. Once a sample was loaded and the GEMBE separation initiated (as described below), the pressure was reduced to 20 kPa for 30 s with the high voltage off. The high voltage (+2 kV) was then turned on, and the pressure was further reduced to a starting pressure of between 750 Pa and 2500 Pa and held constant for approximately 14 seconds. These results were obtained over several months using different capillaries with nominally identical properties. Because of slight differences in the electro-osmotic properties and inner diameter of the capillaries, the optimal starting pressure varied between sets of analyses. The pressure was then reduced at a rate of 12.5 Pa/s until both 2-PG and 3-PG had been detected (216 s to 240 s). The pressure was then increase to 20 kPa and held constant for 10 s. The high voltage was turned off, and the pressure was increased to 30 kPa for at least 30 s before the start of the next GEMBE separation. The GEMBE separation was repeated 5 or 6 times for each sample, to monitor the conversion of substrate to product over a period of approximately 25 minutes.
[0114] Stock solutions of 2-PG and 3-PG were prepared at concentrations of 4 mM in electrophoresis buffer (30 mM Tris-HCl pH 8.0, 20 mM MgCl.sub.2). Enzyme dilution buffer was prepared with 30 mM Tris-HCl pH 8.0, 20 mM MgCl.sub.2, 6.4 mg/mL BSA Inhibitor solutions were prepared in DMSO by 2-fold serial dilution to cover a range of at least 100-fold in concentration. PGM enzyme stock solutions were in 50% glycerol and were stored at -20.degree. C. Final enzyme concentrations used were chosen to give similar reaction rates for the GEMBE assays. Before each enzyme reaction mixture was loaded, the sample reservoir was rinsed with electrophoresis buffer.
[0115] For the GEMBE measurements with Ce-2 and Ce-2d, the enzyme reactions were mixed and initiated according to the following: working solutions of C. elegans, B. malayi, E. coli iPGM, and H. sapiens dPGM were prepared by volumetric dilution from 93, 136, 62 and 125 .mu.M stocks with enzyme dilution buffer to enzyme concentration of 47, 34, 250 and 21 nM respectively. To initiate a reaction, 159 .mu.L of electrophoresis buffer was added to the sample reservoir, followed by 1 .mu.L of inhibitor solution in DMSO (or pure DMSO for no-inhibitor controls) and 20 .mu.L of the enzyme working solution. Mixing was achieved with vigorous pipetting. Five minutes after addition of the enzyme, 20 .mu.L of substrate (2-PG or 3-PG) stock solution was added, and the sample was again mixed with pipetting. Forty-five seconds after addition of the substrate, the first analytical separation was started. The final concentrations of all components in the reaction were: 30 mM Tris HCl, 20 mM MgCl.sub.2, 0.64 mg/mL BSA, 400 .mu.M substrate, 0.5% v/v DMSO, inhibitor ranging from 195 pM to 2.5 .mu.M, and either 3.4 nM B. malayi iPGM, 4.6 nM C. elegans iPGM, 25 nM E. coli iPGM, or 2.1 nM H. sapiens dPGM.
[0116] Analysis of the GEMBE data and calculation of reaction rates was similar to that previously reported (Ross et al., Anal. Chem. 80:9467-9474, 2008). Briefly, the detector signal vs. time data for each electropherogram (see FIG. 3A, for example) was fit to a functional form consisting of the sum of three complementary error functions and a quadratic baseline:
signal ( t ) = A 0 + A 1 t + A 2 t 2 + C 1 2 erfc ( t - t 1 2 .sigma. 1 ) + C 2 2 erfc ( t - t 2 2 .sigma. 2 ) + C 3 2 erfc ( t - t 3 2 .sigma. 3 ) ##EQU00003##
The three error functions correspond to 2-PG, 3-PG, and an unknown species present in the enzyme stock solutions. Calibration measurements indicated that the resulting best fit values for C.sub.2 and C3 were proportional to the concentration of the analytes, 2-PG and 3-PG, respectively. The percent conversion was then calculated from:
percent conversion = C 3 C 2 + C 2 100 ##EQU00004##
The reaction rate was determined by the slope of a linear fit to the percent conversion vs. reaction time data for the first four GEMBE separations with each sample. The reaction rate was normalized by the rate of reaction from a no inhibitor control. Data were modeled to a four parameter Hill equation for determination of IC.sub.50s using Prism GraphPad.
[0117] Macrocyclic library design: Two thioether-macrocyclic peptide libraries were constructed with either N-(2-chloroacetyl)-L-tyrosine (ClAc.sup.LY) or N-(2-chloroacetyl)-D-tyrosine (ClAc.sup.DY) as an initiator by using the Flexible in vitro Translation (FIT) system (Goto et al., Nat. Protoc. 6:779-790, 2011). The corresponding mRNA library is designed to have an AUG (ClAc.sup.L/DY) initiator codon followed by 4-12 NNK random codons (N=G, C, A or U; K=G or U), which code random proteinogenic amino acid residues, followed by a fixed UGC codon that assigns Cys. The theoretical diversity of the macrocycles based on the quantitative assessment of efficiencies of the individual transformation steps (see below) is at least 10.sup.12. After in vitro translation, a thioether bond formed spontaneously between the N-terminal ClAc group of the initiator .sup.L/DTyr residue and the sulfhydryl group of a downstream Cys residue.
[0118] Affinity selection and enrichment: Affinity selections were independently performed with .sup.DY library against B. malayi iPGM (His10-tagged) and .sup.D/LY libraries against C. elegans iPGM (His10-tagged) by employing the Random non-standard Peptides Integrated Discovery (RaPID) system. The mRNA library, ClAc-L-Tyr-tRNA.sup.fMet.sub.CAU and ClAc-.sub.D-Tyr-tRNA.sup.fMet.sub.CAU were prepared as reported (Hayashi et al., ACS Chem. Biol. 7:607-613, 2012; Hipolito et al., Molecules 18:10514-10530, 2013). One .mu.M mRNAs library were ligated with 1.5 .mu.M puromycin linker using a T4 RNA ligase at 25.degree. C. for 30 min and purified. Then, 1.4 .mu.M mRNA-puromycin conjugate and 50 .mu.M ClAc-.sub.L-Tyr-tRNA.sup.fMet.sub.CAU or ClAc-.sub.D-Tyr-tRNA.sup.fMet.sub.CAU were used in a methionine-deficient FIT system to generate respective peptide libraries. The in vitro translation reaction were performed at 37.degree. C. for 30 min with an extra incubation at 25.degree. C. After an addition of EDTA solution (200 mM, 15 .mu.L), the reaction solution was incubated at 37.degree. C. for 30 min to facilitate macrocyclization and subject to pre-washed Sephadex G-25 columns to remove salts. The desalted solution of peptide-mRNA was applied to Dynabeads.RTM. His-tag Isolation & Pulldown magnetic beads (ThermoFisher Scientific) to remove undesired beads binders. This process is called pre-clearance and was repeated trice. After the pre-clearance, the peptide-mRNA solution was incubated with B. malayi iPGM- or C. elegans iPGM-immobilized Dynabeads for 30 min at 4.degree. C. to obtain iPGM-binders. This process is referred to as positive selection. The selected fused peptide-mRNAs on the beads were reverse transcribed by M-MLV reverse transcriptase (Promega, Madison, Wis.) for 1 h at 42.degree. C. The fused peptide-cDNAs were isolated from the beads by using in 1.times.PCR reaction buffer and heated 5 min at 95.degree. C. The amount of eluted cDNAs were measured by quantitative PCR. The remaining cDNAs were amplified by PCR, purified and transcribed into mRNAs as a library for the next round of selection. The library preparation, preclearance and positive selection were one round of the enrichment processes. Significant cDNAs enrichments were observed at the sixth round and seventh round for B. malayi iPGM and C. elegans iPGM, respectively. The recovered cDNAs were ligated into the pGEM-T-Easy vector (Promega), using TA-cloning. The vectors were cloned into DH5.alpha. competent cells; individual clones were picked and sequenced (FIGS. 4A-4C).
[0119] Chemical synthesis of macrocycles and analogs: Macrocycles were chemically synthesized using a Syro Wave.TM. automated peptide synthesizer (Biotage, Charlotte, N.C.) by Fmoc solid-phase peptide synthesis as previously described (Morimoto et al., Angew Chem. Int. Ed. Engl. 51:3423-3427, 2012; Yamagata et al., Structure 22:345-352, 2012). Briefly, the chloroacetyl group or acetyl group was coupled onto the N-terminal amide group for the formation of cyclic or linear peptide analogs respectively after the automated synthesis. Peptides were cleaved by a solution of 92.5% trifluoroacetic acid (TFA), 2.5% water, 2.5% triisopropylsilane, and 2.5% ethanedithiol and precipitated by diethyl ether. To conduct the cyclization reaction, peptide pellet was dissolved in 10 mL DMSO/0.1% TFA in water (1:1), adjusted the pH>8 by addition of triethylamine and incubated for 1 h at 25.degree. C. This cyclization reaction was quenched by addition of TFA to acidify the peptide suspensions. Then peptides were purified by reverse-phase HPLC (RP-HPLC) and molecular masses were verified by MALDI-TOF mass spectrometry, using a microflex or ultraflex instrument (Bruker Daltonics, Billerica, Mass.) (FIG. 5 and Table 2).
[0120] All peptides were chemically synthesized on a 25 .mu.mole scale using a Syro Wave automated peptide synthesizer (Biotage) by Fmoc solid phase peptide chemical synthesis (SPPS). Firstly, NovaPEG Rink Amide resins were incubated with N,N-dimethylformamide (DMF) with rotation at ambient temperature for 30 min and washed 5 times with DMF. Coupling of each Fmoc-protected amino acid was performed on the engorged resin with a solution of 300 .mu.L 0.5 M Fmoc-protected amino acid, 300 .mu.L 0.5 M 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and 1-hydroxybenzotriazole (HOBt), and 150 .mu.L 0.5 M N,N-diisopropylethylamine (DIPEA) in DMF and reacted for 1 hour at ambient temperature. After washing the resins with 1 mL DMF five times, Fmoc-deprotection was performed by incubating the resin with 600 .mu.L 40% piperidine in DMF (vol/vol) and reacted for 30 min at ambient temperature. Each peptide was synthesized using the appropriately protected amino acid monomers corresponding to sequences in Tables 6 and 8 by repeating the Fmoc-protected amino acid coupling and Fmoc-deprotection steps accordingly. The N-terminal .alpha.-amino group of the synthesized peptides on the resin was chloroacetylated by incubating with a solution of 500 .mu.L 0.5 M chloroacetyl N-hydroxysuccinimide (NHS) ester in N-methylpyrrolidone (NMP) with rotation for 60 min at ambient temperature. For the synthesis of Ce-L2 and Ce-L2d, the N-terminal .alpha.-amino group was acetylated by incubating with a solution of 500 .mu.L 0.5 M acetic anhydride and 0.25 M DIPEA in NMP with rotation for 60 min at ambient temperature. After washing the resin with 5.times.1 mL DMF, peptides were fully deprotected and cleaved from resin by incubating with a solution of 2 mL trifluoroacetic acid (TFA), water, triisopropylsilane (TIS) and ethanedithiol (EDT) (92.5:2.5:2.5:2.5) with rotation for 3 hours at ambient temperature and precipitated with diethyl ether. The peptide pellet was dissolved in 10 mL DMSO/0.1% TFA in water (1:1), and the pH adjusted to >8 by addition of triethylamine (TEA), and incubated at ambient temperature for 1 h to enhance the cyclization via a thioether bond formation between N-terminal chloroacetamide group and cysteine sulfhydryl group. Peptide mass and cyclization was confirmed by MALDI-TOF MS analysis. The cyclization reaction was quenched by addition of TFA to acidify the peptide suspensions. Peptides were then purified by reverse-phase HPLC (Table 4), molecular masses were verified by MALDI-TOF MS analysis (Table 4), using a microflex or autoflex instrument (Bruker Daltonics). Ring junction confirmed by MSMS spectrum and fragment analysis (FIG. 5).
TABLE-US-00003 TABLE 2 LC-MS of representative peptide analogs chemically synthesized Peptide Purity Retention MALDI-TOF ID Sequence* (%) time (min) analysis (Calc./Obs.) Ce-1 ##STR00003## >90 20.3 1764.65/1764.09 Ce-2 ##STR00004## >90 21.9 1790.66/1790.10 Ce-3 ##STR00005## >85 25.3 1655.86/1657.20 Ce-4 ##STR00006## >90 23.9 1643.82/1645.16 Ce-2a ##STR00007## >90 20.8 1734.62/1733.22 Ce-2b ##STR00008## >95 21.4 1631.61/1630.15 Ce-2c ##STR00009## >95 21.5 1530.56/1529.05 Ce-2d ##STR00010## >95 21.7 1472.56/1472.70 Ce-2e ##STR00011## >90 20.4 1310.48/1308.75 Ce-2f ##STR00012## >90 19.0 1197.40/1195.72 Ce-2g ##STR00013## >90 20.9 1034.33/1032.62 Ce-2S ##STR00014## >95 20.9 1774.68/1775.06 Ce-L2 .sup.Ac-DYDYPGDYSYLYGTCG >95 21.5 1776.70/1776.47 (SEQ ID NO: 7) Ce-L2d .sup.Ac-DYDYPGDYSYLY >95 21.4 1458.60/1458.70 (SEQ ID NO: 8) Ce-2tail .sup.Ac-DYLYGTCG >95 18.8 816.35/815.94 (SEQ ID NO: 9) Bm-1 ##STR00015## >95 21.9 1692.71/1693.56 Bm-2 ##STR00016## >90 21.8 1754.87/1755.95 Bm-3 ##STR00017## >90 24.4 1601.68/1602.53 Bm-4 ##STR00018## >90 22.1 1338.50/1339.27 Bm-5 ##STR00019## >90 24.2 1297.47/1298.65 Bm-6 ##STR00020## >90 21.9 1295.46/1296.39 Bm-7 ##STR00021## >95 21.6 1308.54/1309.70 *Each peptide had a C-terminal amide
[0121] Macrocyclic peptide characterization across PGM orthologs: Macrocyclic peptide solutions were prepared in DMSO at a concentration of 5 mM and titrated as an 11-point 1:3, or 16-point 1:2 series. For the 11-point titration series, compound dispense plates were prepared by NCATS compound management in 1536-well polypropylene deep well, v-bottom plates (Greiner Bio-One, #782270) in a single interweaved row-wise pattern per macrocyclic peptide resulting in a concentration range of 5 mM to 84.7 nM. For the 16-point titration series, compound dispense plate were prepared by hand down a single column per peptide of 384-well polypropylene deep well, v-bottom plates (Greiner Bio-One, #781270), and transferred to 1536-well polypropylene deep well, v-bottom plates with a multichannel pipette in duplicate for a concentration range of 5 mM to 152.6 nM. Each macrocyclic peptide was characterized across five iPGM orthologs, two dPGM isozymes, and the PK-FLuc control in the Kinase-Glo Plus coupled-enzyme assay described above. For the assay, 23 nL of the peptide titration series from either the 11-point or 16-point compound dispense plate were simultaneously transferred to 1536-well assay plates (Cat#789092-F, Greiner Bio-One North America) using a 1536-pin tool (Wako) for a final concentration range of 19.2 .mu.M-0.33 nM or 19.2 .mu.M-0.58 nM, respectively.
[0122] Curve fitting: All concentration response curves reporter were generated using GraphPad Prism 5 employing the sigmoidal dose-response (variable slope) curve fitting function:
Y = Bottom + ( Top - Bottom ) 1 + 10 ( Log EC 50 - X ) + Hill Slope ##EQU00005##
[0123] Error analysis: The concentration of cyclic peptide or cyclic peptide analogs resulting in an inhibition of 50% of the indicated PGM activity tested are reported as pIC.sub.50 values in Table 3. The number of independent experiments each peptide was tested in is indicated in the last column of Table 3 under `N.` Inactive peptides were tested once. All experiments with reported standard deviation (SD) for error bars in FIG. 10B were conducted with two technical replicates and are representative plots from N>3 independent experiments. The data used to construct FIG. 19A were from Table 3 converted from pIC.sub.50 where IC.sub.50=10.sup.-pIC50 Error bars represent the SD values of the log normal distributed IC.sub.50s determined for the given peptide, such that IC.sub.50low=10.sup.-(pIC50+SD) and IC.sub.50high=10 .sup.-(pIC50-SD).
TABLE-US-00004 TABLE 3 Activity (pIC.sub.50 and Max response) of macrocyclic peptides on PGM panel iPGM Ortholog B. malayi iPGM C. elegans iPGM O. volvulus iPGM D. immitis iPGM E. coli iPGM Compound Max Max Max Max Max ID Inhibition PIC50, M N Inhibition PIC50, M N Inhibition PIC50, M N Inhibition PIC50, M N Inhibition PIC50, M N Bm-1 -82.8 .+-. 0.9 5.89 .+-. 0.27 2 -60.5 .+-. 2.8 5.30 .+-. 0.08 2 -82.1 .+-. 5.4 5.88 .+-. 0.53 2 -85.9 .+-. 0.2 5.99 .+-. 0.41 2 -60.6 .+-. 9.0 5.31 .+-. 0.06 2 Bm-2 -0.1 .+-. 2.3 NA 2 -7.9 .+-. 6.2 NA 2 -15.2 .+-. 26.6 NA 2 -10.5 .+-. 20.0 NA 2 -12.3 .+-. 10.6 NA 2 Bm-3 -86.4 .+-. 0.6 5.56 .+-. 0.29 2 -66.1 .+-. 7.5 5.05 .+-. 0.14 2 -89.2 .+-. 2.0 5.62 .+-. 0.45 2 -89.4 .+-. 2.8 5.73 .+-. 0.47 2 -60.0 .+-. 3.5 5.03 .+-. 0.04 2 Bm-4 -86.3 .+-. 1.4 6.24 .+-. 0.41 2 -75.5 .+-. 4.3 5.61 .+-. 0.29 2 -85.7 .+-. 6.5 6.10 .+-. 0.63 2 -88.1 .+-. 0.8 6.21 .+-. 0.44 2 -76.3 .+-. 8.3 5.56 .+-. 0.16 2 Bm-5 -84.7 .+-. 1.2 6.16 .+-. 0.36 2 -73.9 .+-. 4.8 5.60 .+-. 0.29 2 -83.1 .+-. 7.3 5.99 .+-. 0.69 2 -87.1 .+-. 3.2 6.10 .+-. 0.56 2 -74.3 .+-. 9.5 5.54 .+-. 0.20 2 Bm-6 -86.8 .+-. 0.6 6.16 .+-. 0.30 2 -76.4 .+-. 1.88 5.58 + 0.25 2 -86.0 .+-. 3.2 6.08 .+-. 0.53 2 -90.2 .+-. 1.2 6.26 .+-. 0.43 2 -77.4 .+-. 4.3 5.55 .+-. 0.07 2 Bm-7 -74.0 .+-. 10.1 5.28 .+-. 0.39 2 -78.0 .+-. 2.0 5.13 .+-. 0.18 2 -78.0 .+-. 19.9 5.37 .+-. 0.52 2 -83.1 .+-. 9.7 5.42 .+-. 0.46 2 -69.7 .+-. 12.3 5.12 .+-. 0.09 2 Bm-4a -88.0 .+-. 0.4 6.31 .+-. 0.29 2 -79.0 .+-. 0.1 5.61 .+-. 0.23 2 -87.8 .+-. 4.1 6.14 .+-. 0.62 2 -90.5 .+-. 1.5 6.28 .+-. 0.40 2 -78.3 .+-. 3.4 5.58 .+-. 0.19 2 Bm-4b 3.1 .+-. 2.5 NA 2 2.1 .+-. 5.6 NA 2 -4.1 .+-. 19.1 NA 2 3.9 .+-. 10.9 NA 2 13.6 .+-. 2.3 NA 2 Bm-4c 4.2 .+-. 7.0 NA 2 2.1 .+-. 10.6 NA 2 8.7 .+-. 11.6 NA 2 6.5 .+-. 10.0 NA 2 6.5 .+-. 8.2 NA 2 Bm-4d 11.2 NA 1 22.7 NA 1 16.9 NA 1 0.7 NA 1 -5.7 NA 1 Bm-4e -5.9 .+-. 4.1 NA 2 -33.6 .+-. 33.8 2.50 .+-. 3.53 1 -14.9 .+-. 30.9 NA 2 -21.4 .+-. 37.3 4.67 2 -20.9 .+-. 4.5 5.2 2 Ce-1 -94 .+-. 3.4 7.92 .+-. 0.38 2 -95.8 .+-. 3 5 8.40 .+-. 0.07 1 -95.4 .+-. 2.6 7.61 .+-. 0.65 2 -95.0 .+-. 6.0 8.04 .+-. 0.41 2 -96.5 .+-. 4.2 8.38 .+-. 0.10 2 Ce-2 -98.8 .+-. 3.9 8.27 .+-. 0.47 9 -100.2 .+-. 6.6 8.65 .+-. 0.55 9 -98.2 .+-. 3.4 7.72 .+-. 0.69 9 -101.1 .+-. 5.5 8.21 .+-. 0.60 9 -101.7 .+-. 5.9 8.78 .+-. 0.43 9 Ce-2* -101.7 .+-. 1.0 9.02 .+-. 0.04 4 -100.9 .+-. 2.4 9.60 .+-. 0.15 4 -101.5 .+-. 3.4 9.18 .+-. 0.08 4 -98.9 .+-. 5.8 9.36 .+-. 0.11 4 -105.7 .+-. 15.1 9.26 .+-. 0.04 4 Ce-3 -13.3 .+-. 2.0 NA 2 -92.3 .+-. 0.4 6.33 .+-. 0.18 2 -22.9 .+-. 30.8 5.26 .+-. 0.18* 2 -30.1 .+-. 32.2 5.18 .+-. 0.03 2 -91.4 .+-. 1.3 6.27 .+-. 0.10 2 Ce-4 -2.9 .+-. 3.3 NA 2 -57.0 .+-. 17.1 5.22 .+-. 0.17 2 -23.0 .+-. 35.7 5.37 .+-. 0.14 2 -22.1 .+-. 35.5 5.28 .+-. 0.06 2 -44.3 .+-. 1.9 5.28 .+-. 0.02 2 Ce-2a -99.2 .+-. 5.3 8.07 .+-. 0.10 3 100.7 .+-. 6.9 8.39 .+-. 0.11 3 -97.7 .+-. 4.4 7.31 .+-. 0.09 3 -100.1 .+-. 5.6 7.82 .+-. 0.29 3 -99.2 .+-. 8.5 8.51 .+-. 0.14 3 Ce-2a* -100.3 .+-. 0.8 9.49 .+-. 0.08 4 -99.6 .+-. 3.9 9.76 .+-. 0.05 4 -97.5 .+-. 2.7 9.68 .+-. 0.09 4 -92.5 .+-. 4.0 9.68 .+-. 0.09 4 -84.1 .+-. 9.5 9.58 .+-. 0.02 4 Ce-2b -91.8 .+-. 5.4 6.16 .+-. 0.14 2 -97.9 .+-. 8.0 7.80 .+-. 0.20 2 -80.1 .+-. 2.1 5.61 .+-. 0.02 2 -90.8 .+-. 4.9 5.96 .+-. 0.10 2 -95.3 .+-. 9.0 7.98 .+-. 0.13 2 Ce-2c -88.2 5.93 .+-. 0.06 1 -92.4 7.64 .+-. 0.03 1 -78.4 5.41 .+-. 0.05 1 -85.3 5.70 .+-. 0.05 1 -89.1 7.98 .+-. 0.12 1 Ce-2d -98.9 .+-. 4.6 7.17 .+-. 0.42 6 -102.6 .+-. 6.6 8.56 .+-. 0.39 6 -97.0 .+-. 5.4 6.72 .+-. 0.60 6 -101.7 .+-. 6.0 7.04 .+-. 0.44 6 -102.0 .+-. 6.9 8.71 .+-. 0.31 6 Ce-2d* -100.2 .+-. 1.4 7.29 .+-. 0.04 4 -100.4 .+-. 4.0 9.03 .+-. 0.06 4 -99.1 .+-. 3.8 7.13 .+-. 0.05 4 -94.0 .+-. 8.2 6.20 .+-. 0.13 4 -99.4 .+-. 13.3 7.98 .+-. 0.27 4 Ce-2e -82.7 .+-. 6.1 5.53 .+-. 0.23 2 -97.2 .+-. 8.2 7.16 .+-. 0.26 2 -55.5 .+-. 1.4 4.72 .+-. 0.26 2 7.6 .+-. 7.4 5.32 .+-. 0.05 2 -94.5 .+-. 9.0 7.27 .+-. 0.16 2 Ce-2f -14.8 NA 1 -79.6 4.29 .+-. 0.13 1 4.3 NA 1 -3.9 NA 1 -79.6 4.59 .+-. 0.10 1 Ce-2g 6.9 NA 1 -18.7 NA 1 7.4 NA 1 7.4 NA 1 -27.0 NA 1 Ce-L2 -48.2 .+-. 18.2 4.89 .+-. 0.21 1 -95.4 .+-. 8.7 5.94 .+-. 0.36 3 -40.2 .+-. 27.6 5.18 3 -44 .+-. 22.6 5.18 .+-. 0.17 3 -95.1 .+-. 5.2 5.88 .+-. 0.39 3 Ce-L2d -7.3 .+-. 2.0 NA 3 -20.9 .+-. 9.2 NA 3 -15.7 .+-. 8.3 NA 3 -5.2 .+-. 4.9 NA 3 -21.1 .+-. 2.4 NA 3 Ce-2tail -10.2 .+-. 3.3 NA 3 -8.1 .+-. 13.9 NA 3 -11.6 .+-. 5.3 NA 3 -1.9 .+-. 10.3 NA 3 -21.1 .+-. 2.7 NA 3 Ce-2S -97.7 .+-. 0.6 6.28 .+-. 0.17 3 -105 .+-. 4.7 8.08 .+-. 0.04 3 -97.0 .+-. 1.6 5.99 .+-. 0.28 3 -98.5 .+-. 0.6 6.01 .+-. 0.28 3 -104.6 .+-. 2.5 8.00 .+-. 0.10 3 Ce-2S* -77.1 .+-. 7.5 6.39 .+-. 0.04 4 -98.6 .+-. 4.9 7.92 .+-. 0.08 4 -69.8 .+-. 9.3 6.35 .+-. 0.08 4 -63.7 .+-. 11.4 6.24 .+-. 0.23 4 -68.1 .+-. 8.9 7.98 .+-. 0.27 4 PGM Isozyme Control H. sapiens dPGM E. coli dPGM PK-Fluc Compound Max Max Max ID Inhibition pIC50, M N Inhibition pIC50, M N Inhibition pIC50, M N Bm-1 3.0 .+-. 0.2 NA 2 7.8 .+-. 4.3 NA 2 10.4 .+-. 9.0 NA 2 Bm-2 -0.1 .+-. 1.3 NA 2 -0.1 .+-. 1.3 NA 2 -0.1 .+-. 2.3 NA 2 Bm-3 5.1 .+-. 0.9 NA 2 8.4 .+-. 1.0 NA 2 7.9 .+-. 7.9 NA 2 Bm-4 6.1 .+-. 2.2 NA 2 7.3 .+-. 3.1 NA 2 9.6 .+-. 6.4 NA 2 Bm-5 5.5 .+-. 0.8 NA 2 3.8 .+-. 4.4 NA 2 7.3 .+-. 9.7 NA 2 Bm-6 1.4 .+-. 3 8 NA 2 7.6 .+-. 1.1 NA 2 5.7 .+-. 2.8 NA 2 Bm-7 7.1 .+-. 2.6 NA 2 9.3 .+-. 2.2 NA 2 2.7 .+-. 3.2 NA 2 Bm-4a 5.8 .+-. 0.4 NA 2 8.8 .+-. 0.7 NA 2 10.7 .+-. 2.2 NA 2 Bm-4b 6.1 .+-. 3.1 NA 2 9.8 .+-. 0.4 NA 2 9.9 .+-. 0.4 NA 2 Bm-4c 8.2 .+-. 0.2 NA 2 8.1 .+-. 4.6 NA 2 6.1 .+-. 10.0 NA 2 Bm-4d -1.1 NA 1.0 -1.8 NA 1 4.4 NA 1 Bm-4e 3.2 .+-. 2.9 NA 2 8.1 .+-. 3.0 NA 2 11.5 .+-. 5.2 NA 2 Ce-1 4.0 .+-. 1.1 NA 2 5.0 .+-. 10.4 NA 2 7.6 .+-. 17.6 NA 2 Ce-2 -0.3 .+-. 6.3 NA 9 -5.9 .+-. 6.9 NA 9 1.4 .+-. 3.6 NA 9 Ce-2* 1.9 .+-. 2.7 NA 4 2.7 .+-. 0.7 NA 4 NT NT 0 Ce-3 1.6 .+-. 0.7 NA 2 9.1 .+-. 0.8 NA 2 9.5 .+-. 5.5 NA 2 Ce-4 5.6 .+-. 3 5 NA 2 8.5 .+-. 3.3 NA 2 4.7 .+-. 3.7 NA 2 Ce-2a -1.6 .+-. 4.4 NA 3 -0.9 .+-. 8.3 NA 3 4.8 .+-. 7.5 NA 3 Ce-2a* 20.8 .+-. 1.7 NA 4 9.0 .+-. 2.0 NA 4 NT NT 0 Ce-2b 3.0 .+-. 3.9 NA 2 3.4 .+-. 8.1 NA 2 7.2 .+-. 12.8 NA 2 Ce-2c 9.6 NA 1 9.7 NA 1 13.4 NA 1 Ce-2d -1.2 .+-. 5.6 NA 6 -6.3 .+-. 9.9 NA 6 0.0 .+-. 7.1 NA 6 Ce-2d* 7.9 .+-. 5.2 NA 4 2.3 .+-. 1.0 NA 4 NT NT 0 Ce-2e 5.3 .+-. 3.5 NA 2 3.8 .+-. 5.9 NA 2 8.9 .+-. 11.2 NA 2 Ce-2f 7.5 NA 1 10.0 NA 1 18.1 NA 1 Ce-2g 7.4 NA 1 10.2 NA 1 15.8 NA 1 Ce-L2 -1.4 .+-. 6.9 NA 3 -14.1 .+-. 1.3 NA 3 -3.0 .+-. 1.2 NA 3 Ce-L2d -1.1 .+-. 8.5 NA 3 -13.4 .+-. 3.2 NA 3 -2.5 .+-. 2.3 NA 3 Ce-2tail -0.4 .+-. 7.4 NA 3 -13.9 .+-. 3.8 NA 3 -2.9 .+-. 3.4 NA 3 Ce-2S 1.6 .+-. 7.0 NA 3 -12.1 .+-. 5.1 NA 3 -1.4 .+-. 4.4 NA 3 Ce-2S* 30.5 .+-. 5.2 NA 4 12.4 .+-. 1.6 NA 4 NT NT 0 Error was determined as follows: values for samples with N .gtoreq. 2 experiments (.gtoreq.4 replicates) represent s.d. For samples tested twice (N = 1) the standard error is determined from the nonlinear fit of the standard Hill equation to the aggregated data from the technical replicates (n = 2). NA = no appreciable inhibition, NT = not tested. *Represent values obtained for iPGM concentrations of 500 pM to better estimate potency of the higher affinity ligands, Ce-2 and Ce-2a; lower affinity ligands, Ce-2d and Ce-S re-tested as well for comparison.
[0124] Exclusion criteria: A data point would be eliminated from the curve fit if the value was determined to be an outlier based on the criteria described in Southall et al. (Handbook of Drug Screening, pp. 442-464, 2009) Potential reasons for a data point to be eliminated from a curve fit would include, for example, known failure of compound transfer or under dispensing of assay reagent to the test well of the 1536-well assay plate. No data points needed to be excluded in the concentration response curves presented in this study.
[0125] Size exclusion chromatography: Samples were analyzed and fractionated on a Superdex.RTM. 75 16/600 column using an AKTA.RTM. Pure chromatography system (GE Healthcare Bio-Sciences, Pittsburgh, Pa.). Samples, 500 .mu.L, were eluted at 1 mL/min in buffer containing 30 mM Tris, 150 mM NaCl and 2 mM MgSO.sub.4 at 4.degree. C. Elution profile absorbance was recorded with in-line detection at 280 and 500 nm, and 2 mL fractions were collected in 96 deep-well plates.
[0126] Polyacrylamide gel electrophoresis: Protein (10 .mu.L/lane) was electrophoresed on Criterion.TM. TGX.TM. 12% precast polyacrylamide mini slab gels (Bio-Rad, Hercules, Calif.) in 1.times. Tris running buffer, ambient temperature at 200V for 45 min Samples, 100 .mu.L aliquots from size exclusion column fractions plus 33 .mu.L 4.times. Laemmeli sample buffer (Bio-Rad 161-0747).+-.2% .beta.ME, were heated for 10 min at 95.degree. C. Gels were fixed for 30 mins in 50% methanol 10% acetic acid then stained with Coomassie Brilliant Blue G-250 colloidal protein stain (Sigma-Aldrich B8522) overnight and imaged on a Bio-Rad ChemiDoc.TM. imaging system or HP flatbed scanner. Molecular weight ladder was Bio-Rad catalog number 161-0376.
[0127] Phylogenetic Tree Construction: The protein sequences for the seven PGM orthologs were aligned using Clustal Omega multiple sequence alignment analysis (available on the World Wide Web at ebi.ac.uk/Tools/msa/clustalo/). The accession numbers for the sequence alignment are NP_871851.1 (Caenorhabditis elegans), AAQ97626.1 (Brugia malayi), AAV33247.1 (Onchocerca volvulus), AEA91534.1 (Dirofilaria immitis), NP_002620.1 (Homo sapiens), P37689.1 (Escherichia coli 2, 3-bisphosphoglycerate-independent phosphoglycerate mutase), and P62707.2 (Escherichia coli 2, 3-bisphosphoglycerate-dependent phosphoglycerate mutase), all of which are incorporated herein by reference as present in GenBank on Aug. 11, 2016. The Pearson/FASTA alignment was uploaded to RAxML BlackBox (available on the World Wide Web at genome.jp/tools/raxml) for tree construction. Gamma model of rate of heterogeneity and the BLOSUM62 protein substitution matrix with a maximum likelihood search were applied for tree building. No outgroup was selected for tree rooting. A rapid algorithm bootstrapping analysis was performed with 1,000 replicates.
[0128] Crystallization and Data Collection: Purified full length apo iPGM from Caenorhabditis elegans spanning and harboring a C-terminal hexahistidine tag was concentrated to 11.6 mg/mL in 200 mM NaCl, 20 mM Tris pH 7.5, 2 mM TCEP. Another sample of iPGM from Caenorhabditis elegans spanning residues M19 to 1539, for preparation of the peptide complex, was concentrated to 10.8 mg/mL in 150 mM NaCl, 30 mM Tris pH 8.0 for crystallization screening. To prepare the Ce-2d cyclic peptide complex a 50 mM stock solution of peptide Ce-2d was prepared in DMSO, mixed in a 1:1.5 (protein:cyclic peptide) molar ratio and incubated on ice for 30 minutes prior to screening. All crystallization experiments were conducted with Compact 300 (Rigaku Reagents) sitting drop vapor diffusion plates at 20.degree. C. using equal volumes of protein and crystallization solution equilibrated against 75 .mu.L of the latter.
[0129] Apo iPGM: Native C. elegans iPGM yielded crystals that formed plate clusters which represented two crystal forms. Monoclinic P crystals (C. elegans iPGM-m) were obtained in approximately 4 weeks (FIG. 6A) from the Wizard 3-4 screen (Rigaku Reagents) condition D11 (30% (w/v) PEG 5000 MME, 100 mM MES pH 6.5, 200 mM ammonium sulfate). The sample was subjected to refinement screening using the Additive Screen HT (Hampton Research). After approximately two weeks, single plate shape crystals (FIG. 6B) were observed from a condition consisting of 30% (w/v) PEG 5000 MME, 100 mM MES pH 6.5, 200 mM ammonium sulfate, 100 mM guanidine-HCl. A second crystal form belonging to an orthorhombic P lattice (C. elegans iPGM-o) was observed after 6 months (FIG. 6C) from the Index HT (Hampton Research) condition F7 (25% (w/v) PEG 3350, 100 mM Bis-Tris pH 6.5, 200 mM ammonium sulfate). Samples were transferred to a fresh drop composed of 75% crystallization solution and 25% PEG 400 and stored in liquid nitrogen.
[0130] iPGM.circle-solid.Ce-2d complex: C. elegans Met19 iPGM prepared in a 1:1.5 ratio with Ce-2d yielded crystals displaying a needle morphology after 7 days (FIG. 6D) from the Crystal Screen HT (Hampton Research) condition A6 (30% (w/v) PEG 4000, 100 mM Tris pH 8.5, 200 mM MgCl.sub.2. Samples were transferred to a fresh drop composed of 80% crystallization solution and 20% glycerol and stored in liquid nitrogen.
[0131] Structure Solution and Refinement: X-ray diffraction data were collected at the Advanced Photon Source beamline 17-ID using a Dectris Pilatus 6M pixel array detector. Intensities were integrated using XDS (Kabsch J. Appl. Crystallogr. 21:67-71, 1988; Kabsch, Acta Crystallogr. D 66:125-132, 2010) via Autoproc (Vonrhein et al., Acta Crystallogr. D 67:293-302, 2011) and the Laue class analysis and data scaling were performed with Aimless19 which suggested that the highest probability Laue class was 2/m for iPGM-m and mmm for iPGM-o. The Matthew's coefficient (Matthews et al., J. Mol. Biol. 33:491, 1968) indicated that there were two molecules in the asymmetric unit (V.sub.m=2.7 .ANG..sup.3/Da, % solvent=54%) and (V.sub.m=2.5 .ANG..sup.3/Da, % solvent=50%) for C. elegans iPGM-m and mmm iPGM-o respectively. Structure solution for iPGM-m was conducted by molecular replacement with Balbes (Long et al., Acta Crystallogr. D 64:125-132, 2008) which generated a search model using a previously determined iPGM structure (PDB: 1098; Rigden et al., J. Mol. Biol. 328:909-920, 2003). Searches were conducted in space groups P2 and P2.sub.1 and the top solution was obtained in the latter space group which was used from this point forward. Initial refinement of the model with Refmac (Murshudov et al., Acta Crystallogr. D 53:240-255, 1997) converged at R/R.sub.free of 34%/37%. For iPGM-o, molecular replacement was conducted using Phaser (Mccoy et al., J. Appl. Crystallogr. 40:658-674, 2007) in all possible space groups with 222 point symmetry using PDB 2IFY (Nukui et al., Biophys. J. 92:977-988, 2007) as the search model. The top solution was obtained in the space group P2.sub.12.sub.12.sub.1. The models were improved by automated model building with Phenix (Adams et al., Acta Crystallogr. D 66:213-221, 2010).
[0132] Structure solution for iPGM.circle-solid.Ce-2d was conducted by molecular replacement using a single subunit of iPGM-o as the search model. Searches were conducted in space groups P2 and P2.sub.1 (V.sub.m=2.3 .ANG..sup.3/Da, % solvent=47%) for two molecules in the asymmetric unit and the top solution was obtained in P2 which was used from this point forward. Initial refinement of the model was carried out with Refmac (Murshudov et al., Acta Crystallogr. D 53:240-255, 1997) and was improved by automated model building with Apr/warp (Langer et al., Nat. Protoc. 3:1171-1179, 2008). Subsequent refinement and manual model building were carried out with Phenix and Coot (Emsley et al., Acta Crystallogr. D 66:486-501, 2010), respectively. Disordered side chains were truncated to the point for which electron density could be observed. Structure validation was conducted with Molprobity (Chen et al., Acta Crystallogr. D 66:12-21, 2010) and figures were prepared using the CCP4MG package (Potterton et al., Acta Crystallogr. D 60:2288-2294, 2004). Superposition of iPGM structures was conducted using GESAMT (Krissinel et al., J. Mol. Biochem. 1:76-85, 2012) via the CCP4 (Winn et al., Acta Crystallogr. D 67:235-242, 2011) interface. Relevant crystallographic data are provided in Table 4.
TABLE-US-00005 TABLE 4 X-ray data collection and refinement statistics C. elegans C. elegans C. elegans Data Collection iPGM-m iPGM-o iPGM.quadrature.Ce-2d Unit-cell parameters a = 67.85, a = 70.32, a = 73.78, (.ANG.,.degree.) b = 94.38, b = 98.85, b = 75.83, c = 102.2, c = 173.10 c = 101.42, .beta. = 96.6 .beta. = 95.7 Space group P2.sub.1 P2.sub.12.sub.12.sub.1 P2 Resolution (.ANG.).sup.a 46.48-2.95 98.85-2.45 48.17-1.95 (3.13-2.95) (2.54-2.45) (1.99-1.95) Wavelength (.ANG.) 1.0000 1.0000 1.0000 Temperature (K) 100 100 100 Observed reflections 90,718 217,179 275,647 Unique reflections 27,020 43,707 80,429 <I/.sigma.(I)>.sup.a 9.0 (2.3) 7.6 (1.9) 7.8 (2.2) Completeness (%).sup.a 99.7 (99.9) 97.5 (99.4) 99.0 (98.1) Multiplicity.sup.a 3.4 (3.5) 5.0 (4.7) 3.4 (3.4) R.sub.merge(%).sup.a,b 14.1 (62.5) 20.5 (84.0) 14.6 (60.3) R.sub.meas(%).sup.a,d 16.8 (74.0) 22.9 (92.6) 17.4 (71.8) R.sub.pim(%).sup.a,d 9.0 (39.4) 9.9 (41.7) 9.2 (38.5) CC.sub.1/2.sup.a,e 0.985 (0.779) 0.976 (0.601) 0.988 (0.811) Refinement Resolution (.ANG.).sup.a 42.98-2.95 40.44-2.45 35.49-1.95 Reflections 25,639/1,353 41,536/2,137 76,298/4,113 (working/test).sup.a R .sub.factor/R .sub.free (%).sup.a,c 18.4/23.7 18.5/26.0 15.3/20.3 No. of atoms 7,837/0/ 7,886/0/2/ 8,001/210/2/ (Protein/Peptide/Mn.sup.2+/ 2/2/0 2/234 2/735 Zn.sup.2+/water) Model Quality R.m.s deviations Bond lengths (.ANG.) 0.008 0.009 0.018 Bond angles (.degree.) 1.013 0.936 1.029 Average B-factor (.ANG..sup.2) All Atoms 48.6 25.2 19.3 Protein 48.7 25.2 18.8 Peptide -- -- 18.2 Mn.sup.2+/Zn.sup.2+ 49.2/48.3 38.4/27.0 17.5/17.6 Water -- 20.9 26 Coordinate 0.35 0.35 0.2 error(maximum likelihood) (.ANG.) Ramachandran Plot Most favored (%) 97.6 96.6 98 Additionally allowed (%) 1.8 3.1 1.7 .sup.aValues in parenthesis are for the highest resolution shell. .sup.bR.sub.merge = .SIGMA..sub.hkl.SIGMA..sub.i |l(hkl) - <l(hkl)>|/.SIGMA..sub.hkl.SIGMA..sub.ii(hkl), where I(hkl) is the intensity measured for the ith reflection and <I(hkl)> is the average intensity of all reflections with indices hkl. .sup.cR.sub.factor = .SIGMA..sub.hkl||F.sub.obs (hkl) | - |F.sub.calc (hkl) ||/.SIGMA..sub.hkl|F.sub.obs (hkl)|; Rfree is calculated in an identical manner using 5% of randomly selected reflections that were not included in the refinement. .sup.dR.sub.meas = redundancy-independent (multiplicity-weighted) R.sub.merge R.sub.pim = precision-indicating (multiplicity-weighted) R.sub.merge. .sup.eCC.sub.1/2 is the correlation coefficient of the mean intensities between two random half-sets of data.
[0133] Crystallographic Analysis: The final model of iPGM-m consisted of two subunits with two Mn.sup.2+ and Zn.sup.2+ ions modeled within domain A of each subunit (FIG. 7A) and the first 20 residues of the N-terminus and last 13 residues of the C-terminus were disordered and could not be modeled. The two subunits are nearly identical with an RMSD deviation of 0.58 .ANG. between Ca atoms for 517 residues aligned using GESAMT (Krissinel et al., J. Mol. Biochem. 1:76-85, 2012) (FIG. 7B). Crystals of the orthorhombic form (C. elegans iPGM-o) were obtained after approximately 6 months and diffracted to higher resolution than iPGM-m. Similarly, the N and C-terminal residues were disordered in the iPGM-o as well. The subunits of iPGM-o were structurally very similar with an RMSD deviation of 0.77 .ANG. between Ca atoms for 520 residues aligned. In addition, the structures of iPGM-o and iPGM-m were quite similar with an RMSD deviation of 1.85 .ANG. between Ca atoms for 495 residues aligned. However, the phosphatase domains are much more closely aligned whereas the transferase domains exhibit a slight shift (FIG. 8C) which may be due to the conformational flexibility of this domain. The structure of iPGM-o also contains two metal ions in the substrate binding site which bind in a similar manner to iPGM-m (see below).
[0134] Comparison of C. elegans iPGM-m to other apo iPGM structures: Superposition of C. elegans iPGM-m with iPGM from Bacillus anthracia (PDB: 2IFY) yielded an RMSD of 2.76 .ANG. between Ca atoms (476 residues aligned). Although the deviation from superposition was somewhat large between the two structures, the overall fold was very similar between the two structures (FIGS. 8A-8C). The structure of C. elegans iPGM-m was also compared with that of a substrate bound iPGM from Bacillus stearothermophilus (PDB: 1098). Superposition yielded an RMSD of 1.07 .ANG. between Ca atoms. However, only 282 residues could be aligned for residues in the transferase domain since substrate binding produced a large conformational change in the phosphatase domain (FIG. 8B).
[0135] Metal ion sites: Large peaks of positive electron density (Fo-Fc) were observed in the metal binding sites of the phosphatase domain following refinement FIG. 9A. This region is occupied by Asp 426 and His 430 (site 1) and Asp 37, Ser 86, Asp 467 and His 468 (site 2). When these sites were refined as Mg.sup.2+ ions, residual positive electron density was observed suggesting that a larger metal ion was present. Modelling Mn.sup.2+ ions at each site resulted in a B-factor of approximately 17 .ANG..sup.2 at site 1, but site 2 contained some positive electron density and a B-factor of approximately 7 .ANG..sup.2. Additionally, the coordinating distances between the surrounding residues and the ion at site 2 were approximately 1.9 .ANG. to 2.0 .ANG. which are shorter than expected for a Mn.sup.2+ ion (2.1 .ANG. to 2.2 .ANG.). Phased anomalous difference maps were calculated, using data collected at a wavelength of 1.0000 .ANG., which yielded peak heights of: site 1 (5.7.sigma.) and site 2 (11.7.sigma.) as shown in FIG. 9A. Zn.sup.2+ ions may occupy site 2 based on the coordinating distances and anomalous signal, and homology of the iPGM phosphatase domain to that of E. coli alkaline phosphatase (Jedrzejas Prog. Biophys. Mol. Biol. 73:263-287, 2000). The theoretical anomalous signal at a wavelength of 1.0000 .ANG., is 2.6 e.sup.- and 1.4 e.sup.- for Zn.sup.2+ and Mn.sup.2+ ions, respectively. Additionally, data were collected at a wavelength of 1.9016 .ANG. which is on the low energy side of the Mn.sup.2+ absorption edge. The theoretical anomalous signal at this wavelength is 1.1 e.sup.- and 0.49 e.sup.- for Zn.sup.2+ and Mn.sup.2+ ions respectively. A phased anomalous difference map was calculated using the 1.9016 .ANG. data and produced no peaks at site 1 whereas peaks of approximately 5.sigma. were observed at site 2. Subsequent refinement of Mn.sup.2+ and Zn.sup.2+ ions at site1 and site2, respectively yielded no residual positive electron density which further supported the assignments for these sites. The coordination of these metals by C. elegans iPGM is shown in FIG. 9B and the distances are listed in Table 5. It should be noted that an Mg.sup.2+ binding site, from the crystallization solution, was also observed as shown in FIG. 9C.
TABLE-US-00006 TABLE 5 H-bonding and metal coordination distances in the C. elegans iPGM Ce-2d structure Ce-2d Residue iPGM Distance Min Closest or metalion Residue Residue Pair Atom Filter .ltoreq. 5.ANG. .sub.D-TYR 1 ASN336 3.802 O .sub.D-TYR1/O ASN336 .sub.D-TYR 1 PHE365 3.1853 O .sub.D-TYR1/CZ PHE365 .sub.D-TYR 1 GLY369 3.2692 O .sub.D-TYR1/O GLY369 .sub.D-TYR 1 GLY370 2.4339 O .sub.D-TYR1/O GLY370 .sub.D-TYR 1 PHE366 2.7405 CE1 .sub.D-TYR1/CE1 PHE366 .sub.D-TYR 1 GLU 87 2.4137 OH .sub.D-TYR1/CB GLU87 .sub.D-TYR 1 LEU 91 3.7614 OH .sub.D-TYR1/CA LEU91 ASP 2 ARG289 2.6574 OD2 ASP2/NH2 ALA334 TYR 3 ALA 334 2.4222 CD1 TYR3/N ALA334 TYR 3 ARG289 4.541 CD2 TYR3/NE ARG289 TYR 3 PRO333 2.4141 CD2 TYR3/CB PRO333 TYR 3 GLN320 3.3003 CE2 TYR3/O GLN320 TYR 3 ASP 102 2.5979 OH TYR3/OD2 ASP102 TYR 3 TYR283 3.0582 OH TYR3/O TYR283 TYR 3 ARG284 3.2451 OH TYR3/C ARG284 TYR 3 ALA 285 2.6402 OH TYR3/N ALA285 TYR 3 ASP 286 3.1893 OH TYR3/N ASP286 TYR 3 THR319 3.1545 OH TYR3/CB THR319 PRO 4 ALA 334 2.3729 O PRO4/CB ALA334 PRO 4 ASN336 2.6715 CG PRO4/CB ASN336 GLY 5 ILE 99 2.4236 N GLYS/CG2 ILE99 GLY 5 TYR100 2.5803 O GLYS/O TYR 100 GLY 5 GLN101 3.2351 O GLY5/CA GLN101 GLY 5 ASP102 2.8573 O GLY5/N ASP102 TYR 7 GLN101 2.7272 CA TYR7/NE2 GLN101 TYR 7 ILE 99 3.5024 O TYR7/C ILE99 TYR 7 ARG284 2.4231 CD1 TYR7/NH2 ARG284 TYR 7 ASP286 2.5816 CE2 TYR7/OD2 ASP286 TYR 9 VAL88 2.1049 O TYR9/CG2 VAL88 TYR 9 ILE 99 2.9585 CG TYR9/CB ILE99 TYR 9 LEU91 2.0412 CE1 TYR9/CD2 LEU91 TYR 9 ASN336 2.9207 OH TYR9/ND2 ASN336 TYR 9 GLY369 3.6132 OH TYR9/C GLY369 TYR 9 GLY370 3.3093 OH TYR9/CA GLY370 LEU 10 ILE 99 3.3204 N LEU10/O ILE99 LEU 10 LEU 79 2.2841 CA LEU10/CD2 LEU79 LEU 10 LEU 82 3.2601 O LEU10/CD2 LEU82 LEU 10 TYR100 3.7162 CD1 LEU10/CD1 TYR100 LEU 10 GLN101 2.5471 CD1 LEU10/OE1 GLN101 LEU 10 ILE103 4.0535 CD1 LEU10/CD1 ILE103 LEU 10 PRO 79 2.948 CD2 LEU10/CD PRO79 TYR 11 VAL88 2.6738 C TYR11/CB VAL88 TYR 11 ASN 85 1.8956 O TYR11/CG ASN85 TYR 11 HIS485 4.5843 O TYR11/CE1 HIS485 TYR 11 GLU 87 3.02 OXT TYR11/OE1 GLU87 Mn2+ Asp426 2.14 Asp/426/OD2 Mn2+ His430 2.21 His430/NE2 Mn2+ His485 2.13 His485/NE2 Mn2+ H20 2.09, 4.45, 23.9 H20 Zn2+ Asp37 1.95 OD1 Asp37 Zn2+ Ser86 1.99 OG Ser86 Zn2+ His468 2 NE2 His468 Zn2+ Asp467 1.91 OD2 Asp467
[0136] Database deposition: Coordinates and structure factors have been deposited to the Worldwide Protein Databank with the following accession codes: C. elegans apo iPGM-m (PDB 5KGM), C. elegans apo iPGM-o (PDB 5KGL) and the complex C. elegans Met19 iPGM.circle-solid.Ce-2d (PDB 5KGN), all of which are incorporated herein by reference.
Example 2
Identification of High Affinity iPGM Ligands
[0137] A recently reported attempt to obtain small molecule inhibitors against iPGM from a combined library of 380,000 compounds by Genzyme Corporation and the National Center for Drug Screening in Shanghai resulted in only two low potency compounds, apparent metal ion chelators (Crowther et al., PLoS Neglected Trop. Dis. 8:e2628, 2014). Given the apparent refractory nature of iPGM toward small molecule inhibition outside of metal ion ligands, and the difficulty of identifying chemotypes of the alkaline phosphatase superfamily enzyme class from HTS with sub-micromolar optimization potential (Narisawa et al., J. Bone Miner. Res. 22:1700-1710, 2007), we approached the problem through the complementary method of affinity selection using an in-vitro display system, referred to as RaPID (Random non-standard Peptides Integrated Discovery).
[0138] The RaPID system enabled us to exploit the diversity of macrocyclic peptide populations numbering in a trillion unique members and enrich for and amplify low abundance, high affinity ligands (Bashiruddin et al., Curr. Opin. Chem. Biol. 24:131-138, 2015). For this particular selection campaign, we utilized a thioether-cyclic peptide library initiated with either L- or D-tyrosine (L/D-Tyr) and performed selection of high affinity ligands against two protein targets, B. malayi iPGM and C. elegans iPGM, which were individually immobilized on magnetic beads via the His6 tag at the C-terminus of these recombinant enzymes. The sequence alignments (FIGS. 4A-4C) from 69 RaPID-derived clones resulted in 11 independent sequence families, corresponding to macrocyclic peptides of a lariat structure with ring sizes ranging between 7-13 amino acids and C-terminal tails of 1-7 amino acids (Table 6).
[0139] It should be noted that the cyclic peptides as isolated by RaPID are tethered at their carboxyl terminus via puromycin to the encoding mRNA. Any effect of the tethered nucleic acid during cyclic peptide binding to their target, either to facilitate binding or block possible productive target-cyclic peptide interactions is an inherent property of mRNA-display technology. Significant binding contributions made via the nucleic acid will not be present in samples made by the solid-phase peptide synthesis step.
TABLE-US-00007 TABLE 6 Inhibitory activity of RaPID selected macrocyclic peptides Ring pIC.sub.50 size/ C. B. O. D. E. H. Protein tail elegans malayi volvulus immitis coli sapiens E. coli ID* Sequence length iPGM iPGM iPGM iPGM iPGM dPGM dPGM Bm-1 ##STR00022## 12/2 5.30 5.89 5.88 5.99 5.31 NA NA Bm-2 ##STR00023## 13/1 NA NA NA NA NA NA NA Bm-3 ##STR00024## 12/2 5.05 5.56 5.62 5.73 5.03 NA NA Bm-4 ##STR00025## 7/4 5.61 6.24 6.10 6.21 5.56 NA NA Bm-5 ##STR00026## 7/4 5.60 6.16 5.99 6.10 5.54 NA NA Bm-6 ##STR00027## 7/4 5.58 6.16 6.08 6.26 5.55 NA NA Bm-7 ##STR00028## 7/4 5.13 5.28 5.37 5.42 5.12 NA NA Ce-1 ##STR00029## 8/7 8.40 7.92 7.62 8.04 8.38 NA NA Ce-2 ##STR00030## 8/7 8.65 8.27 7.72 8.21 8.78 NA NA Ce-3 ##STR00031## 13/1 6.33 NA NA NA 6.27 NA NA Ce-4 ##STR00032## 13/1 5.22 NA NA NA 5.28 NA NA Each peptide had a C-terminal amide. Peptide sequences identified from the pool in round 6 and 7 for B. malayli and C. elegans iPGM, respectively. pIC.sub.50 = -log IC.sub.50. NA = No appreciable inhibitory activity at highest concentration tested. *Ce-1 is ipglycermide A and Ce-2 is ipglycermide B.
Example 3
Functional Evaluation of Cyclic Peptide PGM Inhibitors
[0140] To efficiently profile the activity of the cyclic peptides derived from in vitro selection, several phosphoglycerate mutase enzymes from a range of species were evaluated, including the parasite target, B. malayi iPGM and filarial orthologs (Onchocerca volvulus, Dirofilaria immitis), the corresponding model organism C. elegans iPGM ortholog, and the H. sapiens anti-target dPGM isozyme. Both iPGM and dPGM enzymes from E. coli were also included. Low volume, 1536-well plate format kinetic and endpoint assays for the PGM-catalyzed conversion of 3-PG to 2-PG were developed (FIG. 2A). Each assay utilized a coupled enzyme approach where the product 2-PG is driven through phosphoenol pyruvate (PEP) to pyruvate and ATP via enolase and pyruvate kinase, respectively (Feraudi et al., J. Clin. Chem. Clin. Biochem. 21:193-197, 1983). A kinetic absorbance output was achieved using lactate dehydrogenase-mediated changes in NADH concentration through pyruvate conversion to lactate (FIG. 2B) (Fuad et al., Metallomics 3:1310-1317, 2011; White et al., Eur. J. Biochem. 207:709-714, 1992). For a bioluminescent endpoint assay the ATP produced is used in light production by Firefly luciferase and luciferin (FIG. 2B, Table 1). The continuous NADH-dependent absorbance assay was used to determine the relative activity and 3-PG K.sub.M for each of the seven PGM orthologs and isozymes being used in this study. Each of these enzymes and conditions was used to calibrate the bioluminescent end-point assay that gave a 3.5-5.4-fold ratio of signal-to-background following a five minute incubation (FIG. 1C and Table 7).
TABLE-US-00008 TABLE 7 Assay summary statistics Output Control PGM Signal (RLU) S:B CV Z' Factor condition B. malayi 22000 .+-. 6500 4.4 .+-. 1.8 3.4 .+-. 1.0 0.81 .+-. 0.06 No Enzyme iPGM C. elegans 19000 .+-. 5400 3.6 .+-. 1.5 4.0 .+-. 1.3 0.76 .+-. 0.11 No Enzyme iPGM O. volvulus 25000 .+-. 6300 4.7 .+-. 2.2 4.7 .+-. 2.1 0.79 .+-. 0.10 No Enzyme iPGM D. immitis 20000 .+-. 7000 3.8 .+-. 2.2 4.2 .+-. 2.4 0.74 .+-. 0.15 No Enzyme iPGM E. coli 1700 .+-. 4100 3.1 .+-. 1.2 4.0 .+-. 2.1 0.72 .+-. 0.16 No Enzyme iPGM H. sapiens 19000 .+-. 3200 3.7 .+-. 1.4 4.0 .+-. 0.9 0.75 .+-. 0.10 No Enzyme dPGM E. coli 23000 .+-. 3900 3.8 .+-. 1.5 3.7 .+-. 1.3 0.77 .+-. 0.11 No Enzyme dPGM PK-FLuc 29000 .+-. 5300 5.2 .+-. 2.0 3.3 .+-. 1.0 0.77 .+-. 0.18 No Enzyme
[0141] For the direct evaluation of compounds and cyclic peptides on the target enzyme, gradient elution moving boundary electrophoresis (GEMBE) (Shackman et al., Anal. Chem. 79:565-571, 2007; Strychalski et al., Anal. Chem. 81:10201-10207, 2009) was used to enact an electrophoretic separation of 3-PG from 2-PG (FIG. 2C, FIG. 3A). The method provides a direct, label-free measurement of the substrate and product, 3-PG and 2-PG (FIG. 3B top). Baseline separation of the isomers was clearly apparent in the first derivative plot (FIG. 3B, bottom) for equimolar amounts of 3-PG and 2-PG. Using the established separation conditions, a time course for conversion of 2-PG to 3-PG was demonstrated with the C. elegans and B. malayi iPGMs. GEMBE, while low throughput, provides an ideal orthogonal validation of the activity of the iPGM inhibitors described in this study.
Example 4
Identification of a Potent and Selective Class of iPGM Inhibitors
[0142] The parasitic target B. malayi iPGM was initially panned against the macrocyclic peptide library to obtain seven macrocycles represented by tetradeca-through undecapeptides, Bm-1-Bm-7 (Table 6). From the nucleic acid sequences (FIGS. 4A-4C) encoding these cyclic peptides sufficient quantities for evaluation as inhibitors of the enzymatic activity of a panel of seven PGM orthologs and isozymes were synthesized (Table 6). Concentration response curves obtained for Bm-1-7 across the PGM panel primarily revealed a selective, modestly potent macrocyclic series. All except Bm-2 (for which RaPID selection may have been influenced by the tethered nucleic acid not present in the resynthesized cyclic peptide) inhibited the iPGM orthologs in the high nanomolar to low micromolar range (Table 6), and further showed complete selectivity for iPGM vs. dPGM enzymes. These cyclic peptides comprised two groups, one having large rings of 12 or 13 amino acids with short 1-2 amino acid tails (Bm-1-Bm-3) and the second having 7-membered rings with C-terminal extensions of between 3-4 amino acids (Bm-4-Bm-7), and all seven peptides contained a cysteine at the penultimate position. Because Bm-4 represented the shorter, potent undecapeptide class of sequences among the cyclic peptides it was selected as a template for additional study through C-terminal truncation analogs. Additionally, to probe the importance of the free sulfhydryl group of the tail cysteine, Cys10 was replaced with Ser. Interestingly, the Cys10Ser replacement and elimination of all but the terminal Gly11 resulted in inactive macrocycles (Table 3).
[0143] The Bm-series cyclic peptides were relatively low potency. Additionally B. malayi has proven refractory to crystallographic structure determination. However, successful crystals were obtained for C. elegans iPGM and motivated RaPID targeting of this ortholog towards the deduction of design rules guiding macrocycle-iPGM interactions. A second affinity selection experiment using the C. elegans model organism iPGM yielded four macrocyclic peptides, either pentadecapeptides, ipglycermides A (Ce-1) and B (Ce-2, FIG. 10A) or tetradecapeptides (Ce-3, Ce-4). As in the corresponding B. malayi-derived series ring systems the macrocycles are completely inactive toward the dPGM isozymes. Two of these, Ce-1 and Ce-2, ipglycermide A and B, respectively, were exceedingly potent inhibitors of the iPGM orthologues nominally exhibiting low nM activity (2-20 nM) under the initial conditions (that is, the [iPGM] for C. elegans, B. malayi was 5 nM, for D. immitis, E. coli 10 nM and O. volvulus 20 nM) used in the endpoint profiling assays, and were recapitulated in the GEMBE-based assay for the selected PGMs shown in FIGS. 11A-11B. The steep concentration response curves observed at enzyme concentrations 5 nM and higher for Ce-2 (FIGS. 10B-10D, 11A, 11B) suggest stoichiometric titration of the enzymes, which would occur under conditions where [E]>K.sub.d of the ligand, and therefore lead to an underestimation of potency. This was supported by the hyperbolic response of the concentration response curves and leftward shift in ipglycermide B IC.sub.50 as the iPGM concentration was taken below 5 nM (FIG. 10B). Using a quadratic model to account for stoichiometric binding at high iPGM concentration the family of concentration response curves in FIG. 10B was used to estimate an effective K.sub.d of 73.+-.15 pM for ipglycermide B.
Example 5
Ipglycermide B (Ce-2) Analogs Define a Pharmacologic-Phylogenetic Relationship to iPGM Orthologs
[0144] Ce-2 was chosen as a template for a structure activity relationship study involving a C-terminal truncation/substitution series and used to define the minimal sequence needed for activity. This cyclic peptide was also used to explore linear analogs to determine the effect on affinity from conformational constraining the sequence (see Table 8 and FIG. 12A-12B). While removing the majority of the linear sequence of Ce-2 resulted in loss or greatly diminished iPGM inhibitory activity (Ce-2e to Ce-2g), truncated analogs Ce-2a to Ce-2d resulted in a broadened range of activity among the iPGM orthologs. Of particular note is the retention of nanomolar activity of analog Ce-2d for C. elegans and E. coli iPGM, while the activity for B. malayi, O. volvulus, and D. immitis approach IC.sub.50s in the micromolar range (FIG. 10B and FIGS. 13A-13B). This separation of ortholog activity closely parallels the phylogenetic differences between the amino acid sequences of these enzymes (FIG. 10C).
TABLE-US-00009 TABLE 8 Inhibitory properties of ipglycermide B (Ce-2) analogs pIC.sub.50 C. B. O. D. E. H. Protein elegans malayi volvulus immitis coli sapiens E. coli ID* Sequence iPGM iPGM iPGM iPGM iPGM dPGM dPGM Ce-2 ##STR00033## 9.60 9.02 9.18 9.36 9.26 NA NA Ce-2S ##STR00034## 8.08 6.28 5.99 6.01 8.00 NA NA Ce-2a ##STR00035## 9.76 9.49 9.68 9.91 9.58 NA NA Ce-2b ##STR00036## 7.80 6.16 5.61 5.96 7.98 NA NA Ce-2c ##STR00037## 7.64 5.93 5.41 5.70 7.98 NA NA Ce-2d ##STR00038## 9.03 7.29 7.13 6.20 7.98 NA NA Ce-2e ##STR00039## 7.16 5.53 4.72 5.32 7.27 NA NA Ce-2f ##STR00040## 4.29 NA NA NA 4.59 NA NA Ce-2g ##STR00041## NA NA NA NA NA NA NA Ce-L2 .sup.Ac-DYDYPGDYSYLYGTCG 5.94 4.89* 5.18* 5.18* 5.88 NA NA (SEQ ID NO: 7) Ce-L2d .sup.Ac-DYDYPGDYSYLY NA NA NA NA NA NA NA (SEQ ID NO: 8) Ce-2tail .sup.Ac-DYLYGTCG NA NA NA NA NA NA NA (SEQ ID NO: 9) NA = no appreciable activity. Each peptide had a C-terminal amide *Estimated from incomplete concentration response curves (FIG. 13B)
[0145] Other than Ce-2, only Ce-2a, resulting from Gly14 truncation, retained subnanomolar potency against the iPGM orthologs (Table 8) pointing to Cys14 as a key determinant in high-affinity binding. Isosteric replacement of Cys14 with Ser in Ce-2 to generate Ce-2S caused an approximately 100-fold decrease in inhibitory activity (IC.sub.50.about.10 nM) for the C. elegans and E. coli iPGMs, and comparable to Ce-2d a separation in potency of 100-fold between the iPGMs of C. elegans and E. coli versus B. malayi, O. volvulus, and D. immitis (FIG. 10B). These results indicate that the high-affinity binding of Ce-2 is dependent on its Cys14 thiol, possibly involving a sulfur-transition metal ion interaction at the catalytic center of iPGM. Finally, while Cys14 contributes to important binding interactions, a peptide devoid of the macrocyclic core comprised solely of Ce-2 residues 9-14 alone (Ce-2tail) was inactive on all PGMs (Table 8; FIG. 13B). The large entropic contribution of macrocyclization of the peptide was demonstrated by comparing the IC.sub.50s between Ce-2 and a linear form of Ce-2 made by a Cys8Ser substitution (Ce-L2) for iPGMs on which there was measurable activity of Ce-L2. The most reliable data from C. elegans, O. volvulus, and E. coli iPGM (see FIG. 13B and Table 8) allowed a calculation of between a 2000- and 10,000-fold enhancement in affinity attributable to reduction of random states by macrocycle formation, though a potential steric clash between hydrogens replacing the thioether bond may contribute as well to this greatly decreased inhibitory activity. A related peptide derived from a linear form of Ce-2d, Ce-L2d, was completely inactive on all iPGMs, further supporting the importance of two functional lariat macrocycle sub-domains, the ring system and C-terminus, both necessary to engender high affinity binding to the Ce-2 macrocycle.
Example 6
Structure of Nematode iPGM
[0146] The iPGMs are monomeric bi-domain enzymes where a phosphatase domain, structurally related to the alkaline phosphatase family of binuclear metalloenzymes, is connected by two hinge peptides to a phosphotransferase domain (Jedrzej as et al., EMBO J. 19:1419-1431, 2000). X-ray crystal structures have been obtained for the enzymes derived from two trypanosomatids and several bacterial species (Jedrzejas et al., EMBO J. 19:1419-1431, 2000; Mercaldi et al., FEBS J. 279:2012-2021, 2012; Nowicki et al., J. Mol. Biol. 394:535-543, 2009; Nukui et al., Biophys. J. 92:977-988, 2007).
[0147] To develop a structural model delineating the molecular interactions mediating the pharmacologic-phylogenetic relationship between the macrocycles and iPGM orthologs we attempted to co-crystallize Ce-2 with B. malayi and C. elegans iPGM, but failed to obtain crystals. Although soaking of pre-formed iPGM crystals with Ce-2 caused the crystals to shatter, two apo crystal forms were obtained, monoclinic P (iPGM-m) and orthorhombic P (iPGM-o) lattices (FIG. 6A-6C), of native C. elegans iPGM providing the first structure of a nematode iPGM (Table 4 and FIG. 7A-7C). As anticipated from primary amino acid sequence homology among iPGM orthologs, C. elegans iPGM is quite similar to other iPGMs of bacterial origin (FIG. 14). Superposition of the monoclinic C. elegans apo iPGM (PDB: 5KGM) structure with Bacillus anthracia apo iPGM (PDB: 2IFY) yielded an RMSD of 2.76 .ANG. between Ca atoms (476 residues aligned). Though the deviation from superposition is fairly large between the two structures, the overall fold is very similar (FIGS. 8A-8C). The structure of monoclinic C. elegans iPGM was also compared with that of a substrate bound iPGM from Bacillus stearothermophilus (PDB: 1098). Superposition yielded an RMSD of 1.07 .ANG. between Ca atoms. However, only 282 residues could be aligned for residues in the transferase domain since substrate binding produces a large conformational change in the phosphatase domain (FIGS. 8A-8C). Similar to other iPGMs, Mn.sup.2+ occupies one of the two phosphatase domain metal ion binding sites while, as in alkaline phosphatase, a Zn.sup.2+ ion was found in the second binding site of C. elegans iPGM. The identity of these transition metal ions was verified from phased anomalous difference maps calculated from data collected at wavelengths of 1.0000 .ANG. and 1.9016 .ANG.. These metal ions contact the histidine and aspartate triads as shown in FIGS. 9A-9C at coordination distances listed in Table 5, and the catalytic Ser86 nucleophile coordinates to the Zn.sup.2+ ion.
Example 7
Co-Crystal Structure of iPGM.circle-solid.Macrocyclic Complex Elucidates Locked-Open Inhibitory Mechanism
[0148] From the apo iPGM structure it was observed that the N-terminal 18 amino acids, unique to C. elegans iPGM, were disordered. In a subsequent crystallization effort, using an 18 amino acid N-terminal truncated form of C. elegans iPGM, a pre-formed Ce-2 complex purified by sizing chromatography was prepared, as well as a mixture with Ce-2d. The latter resulted in needle like crystals diffracting to 1.95 .ANG. (FIG. 6D and Table 4). The final model of the iPGM.circle-solid.Ce-2d complex (PDB: 5KGN) contained two molecules in the asymmetric unit (FIG. 15A), articulating an inter-domain binding mode with the macrocycle cradled in a pocket shaped from the hinge peptides and adjacent phosphatase and transferase domain surfaces (FIG. 16A). The two subunits of the asymmetric unit are nearly identical with an RMSD deviation of 0.20 .ANG. between Ca atoms for 520 residues aligned. Therefore all subsequent analyses were carried out using subunit A of the model. The structure of iPGM.circle-solid.Ce-2d was compared with the aforementioned apo iPGM-m and iPGM-o (PDB: 5KGL) structures. Superposition yielded an RMSD deviation of 1.98 .ANG. (503 residues) and 2.05 .ANG. (502 residues), respectively. Although the RMSD deviations are somewhat large, the overall structures are remarkably similar (FIG. 16B) with slight displacement of secondary structure elements due to the high flexibility of iPGM. The cavity that accommodates the binding of the Ce-2d cyclic peptide is very similar amongst all of the structures with no dramatic conformational changes observed to accommodate binding of the peptide. Rather, it appears that the peptide adopts an optimal fit within this cavity as might be expected from the affinity selection approach used here to discover the parent macrocycle. This region of iPGM forms a somewhat negative asymmetrically charged pocket that accommodates the polar residues of Ce-2d as shown in FIG. 16C. Ce-2d has a total surface area of 432.1 .ANG..sup.2 and contacts iPGM with a total area of 127.1 .ANG..sup.2 as calculated using Areaimol (Lee et al., J. Mol. Biol. 55:379-400, 1971), which provides information regarding total area, contact area, and the solvent exposed area of a surface. A relatively small region of the total Ce-2d peptide surface makes direct contact to iPGM (127.1 .ANG..sup.2) with the remaining area (305 .ANG..sup.2) exposed to solvent as it is positioned within an open pocket between the phosphatase and transferase domains. On the basis of molecular weight (1501.6) and its 27 ring atoms, Ce-2d can be categorized as a large macrocycle. With 29% of its surface buried, Ce-2d solvent exposes slightly more surface area than comparably sized macrocycles. The electron density for the cyclic peptide was prominent for all of the residues except for the terminal tyrosine side chain which was somewhat disordered, while the C-terminal 4 residues form a short .alpha.-helix (FIGS. 15B and 15C). From the CPK representation of Ce-2d (FIGS. 16C and 16D) the orientation of the tyrosine side chains 3, 7 and 11 can be seen enfolding the cyclic peptide (FIG. 16D) while tyrosine side chains 1 and 9 engage in an edge-to-face interaction (FIG. 16E). In Ce-1, His7 replaces Tyr7 maintaining similar activity (Table 6). The three extra-cyclic C-terminal residues, Tyr9, Leu10 and Tyr11 are wrapped close to the core macrocycle, with the carboxamide of Tyr11 visible on the exterior surface of this compact structure and pointing toward the metal ion active site (FIG. 16E and FIGS. 17A-17C). Notably, the Ce-2d C-terminal amide of the Tyr11 residue is 6.5 .ANG. and 8.4 .ANG. from the Mn.sup.2+ and Zn.sup.2+ ions, respectively (FIG. 17C). Thus, it is feasible that the longer C-terminus of Ce-2 (and Ce-1) could potentially extend from this cavity, positioning Cys14 within coordination distance to either metal ion.
[0149] The Ce-2d macrocycle forms direct hydrogen bonds with C. elegans iPGM as well as water mediated contacts as depicted in FIGS. 17A and 17B and detailed in Table 5. Two of these key H-bonds are made with C-terminal tail residues, Tyr9 and the carboxamide of Tyr11. Others include, iPGM Arg289 which forms two H-bonds with Ce-2d ring system residues, one directly with Asp2 and one water-mediated through the Tyr3 hydroxyl (FIG. 17B), while a bifurcated Gly5 carbonyl H-bond occurs with iPGM via Gln101 and Asp102 (FIG. 17B). Hydrophobic interactions were also observed, for example Leu10 of the macrocycle sits in a small pocket formed by iPGM Ile103, Leu78 and Leu82 (FIG. 18A-18B), while Ile99 of the enzyme is within 3.5 .ANG. or less of cyclic peptide residues Tyr9, Leu10 and Tyr7.
[0150] To explore the orientation of Ce-2d binding relative to phosphoglycerate, we superimposed the S. aureus iPGM 2-PG-bound and C. elegans iPGM Ce-2d crystal structures using the His, Asp, Arg phosphoglycerate binding residues as alignment points in both structures (FIGS. 17D and 17E). The model places the macrocycle at a site non-overlapping with phosphoglycerate supporting an allosteric binding mode for the ipglycermides. This result is consistent with the independence of Ce-2d and Ce-2 IC50 on 3-PG substrate concentration (FIG. 22A-22B).
[0151] To gain insight into the mechanism underlying the iPGM ortholog selectivity of the Ce-2 series observed in FIG. 19A a binding cavity from residues within 5 .ANG. of the Ce-2d macrocycle (shaded orange in the protein sequence alignment shown in FIG. 19B) was defined. In the cavity defined by these amino acids Ce-2d was projected as a worm representation .alpha.-chain scaled by B-factor (gold) with several side chains, Tyr3, Pro4, Tyr11 amide and the thioether linkage shown in green (FIG. 19C), from which several salient observations were made. As previously discussed, truncation of Ce-2 beyond Cys14 resulted in a .about.10-fold potency decrease (Ce-2b, c) until Tyr11 becomes the C-terminal residue, at which point potency for C. elegans and E. coli iPGM was recovered (Ce-2d), but only marginally so for the B. malayi, O. volvulus, and D. immitis orthologs. The improvement of inhibitory potency was likely the result of a new H-bond made possible by the C-terminal Tyr11 amide of Ce-2d with the highly conserved Glu87 of the phosphatase domain (FIG. 19C). Subsequent removal of Tyr11 resulted in nearly a 100-fold potency decrease, probably a consequence of the Tyr11 amide--Glu87 H-bond forfeiture. Continued truncation led to virtual inactivation of the macrocycle (Ce-2f, g). A possible explanation for the dramatic separation of Ce-2d inhibitory potency between C. elegans and E. coli vs. B. malayi, O. volvulus, and D. immitis iPGM orthologs may be in part mediated by Ala334 within hinge 2 of C. elegans and E. coli iPGM, but replaced by a glutamate in the B. malayi, O. volvulus, and D. immitis iPGM orthologs. Ala334 is <2.5 .ANG. from Ce-2d Pro4 and Tyr3, thus the larger volume occupied by a glutamic acid residue may create a steric clash only partially compensated for by the Tyr11 amide H-bond. Sequence differences outside the binding cavity between C. elegans and E. coli vs. the B. malayi, O. volvulus, and D. immitis iPGMs highlighted in yellow in FIG. 14 could also contribute to ortholog selectivity.
[0152] From the progressive C-terminal truncation of ipglycermide B (Ce-2) it became apparent that Cys14 engendered the pan-ortholog potency and steep Hill coefficient (>1) to the macrocycle. This observation is consistent with a cysteinyl thiolate functioning as a potential catalytic-site metal ion ligand, particularly plausible with a borderline hard/soft Lewis acid Zn.sup.2+ at the iPGM active site suggested by the crystallographic findings herein, and its occurrence in related metalloenzymes, e.g., E. coli AP (Christianson et al., Ann. Rev. Biochem. 68:33-57, 1999). Loss of the sulfhydryl side chain as a consequence of truncation or Cys14Ser substitution results in a more graded Hill slope (<1) of the concentration response curves and potency discrimination among the iPGM orthologs (FIG. 13A). A clear correspondence can be seen between potency and the phylogenetic relationship of the iPGMs (determined from FIG. 10C) as follows: IC.sub.50.sup.Ce=IC.sub.50.sup.Ec>>IC.sub.50.sup.Bm>IC.sub.50.su- p.Di.apprxeq.IC.sub.50.sup.Ov. Taken together these results suggest a positive cooperative binding mechanism whereby the cyclic sequence and majority of the C-terminal extension bind to iPGM positioning the sulfhydryl of Cys14 within coordinating distance to the metal ion site. This mechanism would be consistent with a binding mode resulting in the step-like or ultrasensitive concentration response profile reflected in the steep Hill slope (Shoichet J. Med. Chem. 49:7274-7277, 2006; Zhang et al., Open Biol. 3:130031, 2013). Modeling of the C-terminal four amino acid residues of Ce-2 onto the iPGM.circle-solid.Ce-2d crystal structure positions the thiolate of Cys14 within coordination distance of the Mn.sup.2+ ion as illustrated in FIGS. 20A-20B. Interaction with the Zn.sup.2+ ion would likely require a conformational adjustment in the enzyme possibly explaining the instability of the C. elegans apo iPGM crystals upon soaking with Ce-2.
Example 8
iPGM Inhibition by Methylated Peptide Analogs
[0153] A series of analogs of Ce-2 (SEQ ID NO: 2) including one or more methylated amide bonds on the linear portion of the peptide were synthesized. The peptides are shown in Table 9. The activity of the analogs was determined as described in Example 1 and results are shown in Table 10.
TABLE-US-00010 TABLE 9 Ce-2 N-methyl amide analogs Protein ID Sequence SEQ ID NO: Ce-2h ##STR00042## 55 Ce-2i ##STR00043## 56 Ce-2j ##STR00044## 57 Ce-2k ##STR00045## 58 Ce-2l ##STR00046## 59 Ce-2m ##STR00047## 60 Ce-2n ##STR00048## 61 Ce-2o ##STR00049## 62 Ce-2p ##STR00050## 63 Ce-2q ##STR00051## 64 Ce-2r ##STR00052## 65 Ce-2d1 ##STR00053## 66 Ce-2b1 ##STR00054## 67 Ce-2b2 ##STR00055## 68 Ce-2b3 ##STR00056## 69 Each peptide had a C-terminal amide
TABLE-US-00011 TABLE 10 Inhibitory properties of Ce-2 N-methyl amide analogs iPGM Ortholog PGM Isozyme Control Bm iPGM Ce iPGM Ov iPGM Di iPGM Ec iPGM Hs dPGM Ec dPGM PK-Fluc Max Max Max Max Max Max Max Max ID Inhib pIC.sub.50 Inhib pIC.sub.50 Inhib pIC.sub.50 Inhib pIC.sub.50 Inhib pIC.sub.50 Inhib pIC.sub.50 Inhib pIC.sub.50 Inhib pIC.sub.50 N Ce-2h -0.7 NA -28.8 5.05 4.2 NA 4.7 NA -37.7 5.05 2.0 NA 1.2 NA 1.5 NA 1 Ce-2i -19.8 5.25 -58.9 5 1.8 NA -14.0 5.25 -82.7 NA 7.3 NA 4.6 NA 2.4 NA 1 Ce-2j -93.2 7 -94.2 8 -92.2 6.35 -94.2 6.8 -93.7 8.2 5.9 NA 2.8 NA 1.9 NA 1 Ce-2k -86.1 5.4 -93.3 6.2 -77.8 5.05 -85.6 5.4 -92.0 6.4 3.4 NA 8.5 NA 6.9 NA 1 Ce-2l -93.9 7.95 -94.4 8.5 -93.1 7.15 -93.7 7.75 -92.0 8.65 5.0 NA 10.3 NA 5.5 NA 1 Ce-2m -94.5 7.8 -94.9 8.2 -94.2 6.95 -96.0 7.6 -95.7 8.45 6.7 NA 3.5 NA 0.9 NA 1 Ce-2n -93.4 7.91 -92.9 8.46 -92.4 7.11 -93.4 7.63 -91.4 8.67 6.3 NA 2.4 NA -0.4 NA 3 Ce-2o -97.7 7.95 -98.7 8.39 -96.3 7.22 -98.3 7.72 -96.8 8.55 2.3 NA -1.8 NA 1.6 NA 2 Ce-2p -93.7 7.35 -92.8 7.88 -92.3 6.6 -93.5 7.01 -90.9 8.12 6.8 NA 5.1 NA 3.2 NA 1 Ce-2q -93.1 7.43 -92.7 8.19 -92.0 6.67 -93.1 7.08 -90.0 8.51 6.0 NA 4.5 NA 8.1 NA 1 Ce-2r -97.7 7.84 -99.1 8.51 -96 7.08 -97.8 7.57 -98.5 8.61 -2.2 NA -2.7 NA 5.8 NA 2 Ce-2d1 -29.6 NA -91.8 5.95 -2.1 NA -20.2 NA -91.0 6.02 4.5 NA -0.6 NA 1.7 NA 1 Ce-2b1 -72.0 5.37 -100.3 7.06 -24.3 NA -61.1 4.49 -98.7 7.07 -0.4 NA 0.1 NA 2.2 NA 1 Ce-2b2 -43.2 NA -97.0 6.35 2.2 NA -26.0 NA -96.6 6.38 4.4 NA 0.5 NA 2.9 NA 1 Ce-2b3 -167.4 NA -88.3 5.9 9.5 NA -8.9 NA -88.4 5.95 11.5 NA 7.9 NA 5.7 NA 1
Example 9
Comparison of N-Terminal L- and D-Amino Acids
[0154] Peptides with an N-terminal L- or D-amino acid were synthesized and tested for inhibition of PGM, as described in Example 1. Ce-2 and Ce-2d have N-terminal D-N-chloroacetyl tyrosine, as discussed above. The same peptides, with all L-amino acids (having an N-terminal L-N-chloroacetyl tyrosine (L-Tyr1-Ce2 and L-Tyr1-Ce-2d)) were also tested. As shown in Table 11, the peptides with all L-amino acids retained the majority of the activity of Ce-2 and Ce-2d. No appreciable activity was observed against Homo sapiens or E. coli dPGM.
TABLE-US-00012 TABLE 11 Inhibitory properties of Ce-2 and Ce-2d analogs C. elegans iPGM (5nM) E. coli iPGM B. malayi iPGM D. immitis iPGM O. volvulus iPGM pIC.sub.50 M nM pIC.sub.50 M nM pIC.sub.50 M nM pIC.sub.50 M nM pIC.sub.50 M nM Ce-2 8.63 2.34E-09 2.34 9.64 2.32E-10 0.23 7.78 1.68E-08 16.76 8.22 6.03E-09 6.03 7.63 2.35E-08 23.49 Ce-2d 8.41 3.88E-09 3.88 9.12 7.67E-10 0.77 6.83 1.48E-07 148.4 6.82 1.52E-07 151.8 6.42 3.83E-07 383.4 .sup.LTyr.sub.1- 8.55 2.83E-09 2.83 9.52 3.02E-10 0.3 7.75 1.79E-08 17.86 8.09 8.14E-09 8.14 7.46 3.43E-08 34.34 Ce-2 .sup.LTyr.sub.1- 7.86 1.40E-08 14 8.24 5.76E-09 5.76 6.02 9.49E-07 949 6.01 9.89E-07 989 5.73 1.85E-06 1,853 Ce-2d iPGMs: Ce, 5 nM C. elegans; Ec, 10 nM E. coli; Bm, 5 nM B. malayi; Di, 10 nM D. immitis; Ov, 15 nM O. volvulus; dPGMs
Example 10
Assessing Effect of Peptides on C. elegans
[0155] C. elegans strain N2 (wild type) was used for compound testing. Worms were handled using standard methods. They were cultivated on nematode growth medium (NGM) plates in a 20.degree. C. incubator and fed on living Escherichia coli strain OP50. The E. coli OP50 cells were precultured overnight at 37.degree. C. before spreading on the surface of NGM plates. Synchronous liquid culture of L1 C. elegans obtained through egg bleaching and individual L4 picked from NGM plates under the microscope were used for testing. Either 20 L1s or 1 L4 were placed in individual wells of 96-well plate in 100 .mu.L of S medium supplied with dead E. coli HB101 as food source. Compounds were added into the well at different concentrations and incubated with worms at 20.degree. C. for up to 7 days. The effect of compounds on worm health was measured by food consumption, development and reproduction. Food consumption was measured by a decline in OD600 nm readings monitored using a SpectraMax M5 microplate reader. Worm development and F1 progeny production were monitored visually under a microscope.
[0156] FIGS. 23A and 23B show the effect of exposure of C. elegans to 5 .mu.M or 10 .mu.M Ce-2 or Ce-2d for 1-7 days. Microscopic examination showed slightly decreased numbers of F1 progeny in the presence of the peptide. Exposure to 50 .mu.M peptide showed little effect on the adult stage (FIG. 23C). Though little in vivo efficacy was observed with these two peptides, other peptides, modes of administration, or alternative culturing conditions (e.g. axenic growth conditions) more closely mimicking that of parasitic species may have greater effects.
Example 11
Additional Assessment of Peptide Effect on C. elegans
[0157] C. elegans are handled and cultivated as described in Example 10. Compounds are microinjected in C. elegans using standard techniques. Worm development and F1 progeny production are monitored as described in Example 9. Active compounds are identified by impacts on development, worm viability (e.g., decreased viability), and progeny production.
[0158] In view of the many possible embodiments to which the principles of the disclosure may be applied, it should be recognized that the illustrated embodiments are only examples and should not be taken as limiting the scope of the invention. Rather, the scope of the invention is defined by the following claims. We therefore claim as our invention all that comes within the scope and spirit of these claims.
Sequence CWU 1
1
69115PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(8)cyclic linkage 1Tyr Asp Tyr Pro Gly Asp
His Cys Tyr Leu Tyr Gly Thr Cys Gly1 5 10 15215PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(8)cyclic linkage 2Tyr
Asp Tyr Pro Gly Asp Tyr Cys Tyr Leu Tyr Gly Thr Cys Gly1 5 10
15314PRTArtificial SequenceSynthetic peptidemisc(1)..(13)cyclic
linkage 3Tyr Ile Thr Leu Ala Asn Pro Phe Arg Ile Leu His Cys Gly1 5
10414PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(13)cyclic linkage 4Tyr Thr Thr Leu Ala Asn
Pro Phe Arg Ile Leu His Cys Gly1 5 10511PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(8)cyclic linkage 5Tyr
Asp Tyr Pro Gly Asp Tyr Cys Tyr Leu Tyr1 5 10615PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(8)cyclic linkage 6Tyr
Asp Tyr Pro Gly Asp Tyr Cys Tyr Leu Tyr Gly Thr Ser Gly1 5 10
15715PRTArtificial SequenceSynthetic peptide 7Tyr Asp Tyr Pro Gly
Asp Tyr Ser Tyr Leu Tyr Gly Thr Cys Gly1 5 10 15811PRTArtificial
SequenceSynthetic
peptideMISC_FEATURE(1)..(1)D-TyrosineMISC_FEATURE(1)..(1)N-chloroacetyl
modification 8Tyr Asp Tyr Pro Gly Asp Tyr Ser Tyr Leu Tyr1 5
1097PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(1)D-TyrosineMISC_FEATURE(1)..(1)N-chloroacetyl
modification 9Tyr Leu Tyr Gly Thr Cys Gly1 51014PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(12)cyclic linkage 10Tyr
Ser Trp Pro Asn Ala Pro Glu Ile Trp Lys Cys Cys Gly1 5
101114PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(13)cyclic linkage 11Tyr Asp Leu Arg Thr
Pro Trp Leu Lys Arg His Ala Cys Gly1 5 101213PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(12)cyclic linkage 12Tyr
Gln Asn Arg Ser Ile Trp Leu Tyr Gly Cys Cys Gly1 5
101311PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(7)cyclic linkage 13Tyr Leu Glu Trp Pro Asn
Cys Asn Thr Cys Gly1 5 101411PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(7)cyclic linkage 14Tyr Leu Asp Trp Pro Asn
Cys Ser Thr Cys Gly1 5 101511PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(7)cyclic linkage 15Tyr Pro Glu Trp Pro Asn
Cys Ser Thr Cys Gly1 5 101611PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(7)cyclic linkage 16Tyr Ala Val Trp Pro Asn
Cys Arg Thr Cys Gly1 5 101714PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(8)cyclic linkage 17Tyr Asp Tyr Pro Gly Asp
Tyr Cys Tyr Leu Tyr Gly Thr Cys1 5 101813PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(8)cyclic linkage 18Tyr
Asp Tyr Pro Gly Asp Tyr Cys Tyr Leu Tyr Gly Thr1 5
101912PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(8)cyclic linkage 19Tyr Asp Tyr Pro Gly Asp
Tyr Cys Tyr Leu Tyr Gly1 5 102010PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(8)cyclic linkage 20Tyr Asp Tyr Pro Gly Asp
Tyr Cys Tyr Leu1 5 10219PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(1)D-TyrosineMISC_FEATURE(1)..(1)N-chloroacetyl
modificationMISC_FEATURE(1)..(8)cyclic linkage 21Tyr Asp Tyr Pro
Gly Asp Tyr Cys Tyr1 5228PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(1)D-TyrosineMISC_FEATURE(1)..(1)N-chloroacetyl
modificationMISC_FEATURE(1)..(8)cyclic linkage 22Tyr Asp Tyr Pro
Gly Asp Tyr Cys1 52342DNAArtificial SequenceSynthetic nucleic acid
23atgagttggc ctaatgctcc ggagatttgg aagtgttgcg gc
422442DNAArtificial SequenceSynthetic nucleic acid 24atggatctta
ggacgccttg gttgaagcgg catgcttgcg gc 422539DNAArtificial
SequenceSynthetic nucleic acid 25atgcagaata ggtcgatttg gctgtatggt
tgttgcggc 392633DNAArtificial SequenceSynthetic nucleic acid
26atgcttgagt ggccgaattg taatacttgc ggc 332733DNAArtificial
SequenceSynthetic nucleic acid 27atgcttgatt ggcctaattg tagtacttgc
ggc 332833DNAArtificial SequenceSynthetic nucleic acid 28atgcctgagt
ggccgaattg tagtacttgc ggc 332933DNAArtificial SequenceSynthetic
nucleic acid 29atggctgttt ggcctaattg taggacgtgc ggc
333045DNAArtificial SequenceSynthetic nucleic acid 30atggattatc
cgggtgatca ttgttatctt tatgggacct gcggc 453145DNAArtificial
SequenceSynthetic nucleic acid 31atggattatc cgggtgatca ttgttatctt
tatgggactt gcggc 453245DNAArtificial SequenceSynthetic nucleic acid
32atggattatc ctggtgatca ttgttatctt tatgggactt gcggc
453345DNAArtificial SequenceSynthetic nucleic acid 33atggattatc
caggtgatca ttgttatctt tatgggacct gcggc 453445DNAArtificial
SequenceSynthetic nucleic acid 34atggattatc cgggagatca ttgttatctt
tatgggactt gcggc 453545DNAArtificial SequenceSynthetic nucleic acid
35atggattatc cgggagatca ttgttatctt tatgggacct gcggc
453645DNAArtificial SequenceSynthetic nucleic acid 36atggattatc
caggtgatca ttgttatctt tatgggacat gcggc 453745DNAArtificial
SequenceSynthetic nucleic acid 37atggattatc cgggtgatta ttgttatctt
tatgggactt gcggc 453842DNAArtificial SequenceSynthetic nucleic acid
38atgattacgc ttgcgaatcc ttttcgtatt ttgcattgcg gc
423942DNAArtificial SequenceSynthetic nucleic acid 39atgattacgc
ttgcgaatcc ttttcgtatt ttgcactgcg gc 424042DNAArtificial
SequenceSynthetic nucleic acid 40atgattacgc ttgcgaatcc ttttcgcatt
ttgcactgcg gc 424142DNAArtificial SequenceSynthetic nucleic acid
41atgattacgc ttgcgaatcc gttccgtatt ttgcattgcg gc
424242DNAArtificial SequenceSynthetic nucleic acid 42atgattacgc
ttgcgaatcc ttttcgcatt ttgcattgcg gc 424342DNAArtificial
SequenceSynthetic nucleic acid 43atgattacgc ttgcgaatcc ttttcgtatt
ttacattgcg gc 424442DNAArtificial SequenceSynthetic nucleic acid
44atgactacgc ttgcgaatcc ttttcgtatc ttgcattgcg gc
4245539PRTCaenorhabditis elegans 45Met Phe Val Ala Leu Gly Ala Gln
Ile Tyr Arg Gln Tyr Phe Gly Arg1 5 10 15Arg Gly Met Ala Met Ala Asn
Asn Ser Ser Val Ala Asn Lys Val Cys 20 25 30Leu Ile Val Ile Asp Gly
Trp Gly Val Ser Glu Asp Pro Tyr Gly Asn 35 40 45Ala Ile Leu Asn Ala
Gln Thr Pro Val Met Asp Lys Leu Cys Ser Gly 50 55 60Asn Trp Ala Gln
Ile Glu Ala His Gly Leu His Val Gly Leu Pro Glu65 70 75 80Gly Leu
Met Gly Asn Ser Glu Val Gly His Leu Asn Ile Gly Ala Gly 85 90 95Arg
Val Ile Tyr Gln Asp Ile Val Arg Ile Asn Leu Ala Val Lys Asn 100 105
110Asn Lys Phe Val Thr Asn Glu Ser Leu Val Asp Ala Cys Asp Arg Ala
115 120 125Lys Asn Gly Asn Gly Arg Leu His Leu Ala Gly Leu Val Ser
Asp Gly 130 135 140Gly Val His Ser His Ile Asp His Met Phe Ala Leu
Val Lys Ala Ile145 150 155 160Lys Glu Leu Gly Val Pro Glu Leu Tyr
Leu His Phe Tyr Gly Asp Gly 165 170 175Arg Asp Thr Ser Pro Asn Ser
Gly Val Gly Phe Leu Glu Gln Thr Leu 180 185 190Glu Phe Leu Glu Lys
Thr Thr Gly Tyr Gly Lys Leu Ala Thr Val Val 195 200 205Gly Arg Tyr
Tyr Ala Met Asp Arg Asp Asn Arg Trp Glu Arg Ile Asn 210 215 220Val
Ala Tyr Glu Ala Met Ile Gly Gly Val Gly Glu Thr Ser Asp Glu225 230
235 240Ala Gly Val Val Glu Val Val Arg Lys Arg Tyr Ala Ala Asp Glu
Thr 245 250 255Asp Glu Phe Leu Lys Pro Ile Ile Leu Gln Gly Glu Lys
Gly Arg Val 260 265 270Gln Asn Asp Asp Thr Ile Ile Phe Phe Asp Tyr
Arg Ala Asp Arg Met 275 280 285Arg Glu Ile Ser Ala Ala Met Gly Met
Asp Arg Tyr Lys Asp Cys Asn 290 295 300Ser Lys Leu Ala His Pro Ser
Asn Leu Gln Val Tyr Gly Met Thr Gln305 310 315 320Tyr Lys Ala Glu
Phe Pro Phe Lys Ser Leu Phe Pro Pro Ala Ser Asn 325 330 335Lys Asn
Val Leu Ala Glu Trp Leu Ala Glu Gln Lys Val Ser Gln Phe 340 345
350His Cys Ala Glu Thr Glu Lys Tyr Ala His Val Thr Phe Phe Phe Asn
355 360 365Gly Gly Leu Glu Lys Gln Phe Glu Gly Glu Glu Arg Cys Leu
Val Pro 370 375 380Ser Pro Lys Val Ala Thr Tyr Asp Leu Gln Pro Glu
Met Ser Ala Ala385 390 395 400Gly Val Ala Asp Lys Met Ile Glu Gln
Leu Glu Ala Gly Thr His Pro 405 410 415Phe Ile Met Cys Asn Phe Ala
Pro Pro Asp Met Val Gly His Thr Gly 420 425 430Val Tyr Glu Ala Ala
Val Lys Ala Cys Glu Ala Thr Asp Ile Ala Ile 435 440 445Gly Arg Ile
Tyr Glu Ala Thr Gln Lys His Gly Tyr Ser Leu Met Val 450 455 460Thr
Ala Asp His Gly Asn Ala Glu Lys Met Lys Ala Pro Asp Gly Gly465 470
475 480Lys His Thr Ala His Thr Cys Tyr Arg Val Pro Leu Thr Leu Ser
His 485 490 495Pro Gly Phe Lys Phe Val Asp Pro Ala Asp Arg His Pro
Ala Leu Cys 500 505 510Asp Val Ala Pro Thr Val Leu Ala Ile Met Gly
Leu Pro Gln Pro Ala 515 520 525Glu Met Thr Gly Val Ser Ile Val Gln
Lys Ile 530 53546515PRTBrugia malayi 46Met Ala Glu Ala Lys Asn Arg
Val Cys Leu Val Val Ile Asp Gly Trp1 5 10 15Gly Ile Ser Asn Glu Thr
Lys Gly Asn Ala Ile Leu Asn Ala Lys Thr 20 25 30Pro Val Met Asp Glu
Leu Cys Val Met Asn Ser His Pro Ile Gln Ala 35 40 45His Gly Leu His
Val Gly Leu Pro Glu Gly Leu Met Gly Asn Ser Glu 50 55 60Val Gly His
Leu Asn Ile Gly Ala Gly Arg Val Val Tyr Gln Asp Ile65 70 75 80Val
Arg Ile Asn Leu Ala Val Lys Asn Lys Thr Leu Val Glu Asn Lys 85 90
95His Leu Lys Glu Ala Ala Glu Arg Ala Ile Lys Gly Asn Gly Arg Met
100 105 110His Leu Cys Gly Leu Val Ser Asp Gly Gly Val His Ser His
Ile Asp 115 120 125His Leu Phe Ala Leu Ile Thr Ala Leu Lys Gln Leu
Lys Val Pro Lys 130 135 140Leu Tyr Ile Gln Phe Phe Gly Asp Gly Arg
Asp Thr Ser Pro Thr Ser145 150 155 160Gly Val Gly Phe Leu Gln Gln
Leu Ile Asp Phe Val Asn Lys Glu Gln 165 170 175Tyr Gly Glu Ile Ser
Thr Ile Val Gly Arg Tyr Tyr Ala Met Asp Arg 180 185 190Asp Lys Arg
Trp Glu Arg Ile Arg Val Cys Tyr Asp Ala Leu Ile Gly 195 200 205Gly
Val Gly Glu Lys Thr Thr Ile Asp Lys Ala Ile Asp Val Ile Lys 210 215
220Gly Arg Tyr Ala Lys Asp Glu Thr Asp Glu Phe Leu Lys Pro Ile
Ile225 230 235 240Leu Ser Asp Glu Gly Arg Thr Lys Asp Gly Asp Thr
Leu Ile Phe Phe 245 250 255Asp Tyr Arg Ala Asp Arg Met Arg Glu Ile
Thr Glu Cys Met Gly Met 260 265 270Glu Arg Tyr Lys Asp Leu Asn Ser
Asn Ile Lys His Pro Lys Asn Met 275 280 285Gln Val Ile Gly Met Thr
Gln Tyr Lys Ala Glu Phe Thr Phe Pro Ala 290 295 300Leu Phe Pro Pro
Glu Ser His Lys Asn Val Leu Ala Glu Trp Leu Ser305 310 315 320Val
Asn Gly Leu Thr Gln Phe His Cys Ala Glu Thr Glu Lys Tyr Ala 325 330
335His Val Thr Phe Phe Phe Asn Gly Gly Val Glu Lys Gln Phe Ala Asn
340 345 350Glu Glu Arg Cys Leu Val Val Ser Pro Lys Val Ala Thr Tyr
Asp Leu 355 360 365Glu Pro Pro Met Ser Ser Ala Ala Val Ala Asp Lys
Val Ile Glu Gln 370 375 380Leu His Met Lys Lys His Pro Phe Val Met
Cys Asn Phe Ala Pro Pro385 390 395 400Asp Met Val Gly His Thr Gly
Val Tyr Glu Ala Ala Val Lys Ala Val 405 410 415Glu Ala Thr Asp Ile
Ala Ile Gly Arg Ile Tyr Glu Ala Cys Lys Lys 420 425 430Asn Asp Tyr
Ile Leu Met Val Thr Ala Asp His Gly Asn Ala Glu Lys 435 440 445Met
Met Ala Pro Asp Gly Ser Lys His Thr Ala His Thr Cys Asn Leu 450 455
460Val Pro Phe Thr Cys Ser Ser Met Lys Tyr Lys Phe Met Asp Lys
Leu465 470 475 480Pro Asp Arg Glu Met Ala Leu Cys Asp Val Ala Pro
Thr Val Leu Lys 485 490 495Val Met Gly Val Pro Leu Pro Ser Glu Met
Thr Gly Gln Pro Leu Val 500 505 510Asn Glu Ala
51547515PRTOnchocerca volvulus 47Met Ser Glu Val Lys Asn Arg Val
Cys Leu Val Val Ile Asp Gly Trp1 5 10 15Gly Ile Ser Asn Glu Ser Lys
Gly Asn Ala Ile Leu Asn Ala Lys Thr 20 25 30Pro Val Met Asp Glu Leu
Cys Ala Leu Asn Ser His Pro Ile Glu Ala 35 40 45His Gly Leu His Val
Gly Leu Pro Glu Gly Leu Met Gly Asn Ser Glu 50 55 60Val Gly His Leu
Asn Ile Gly Ala Gly Arg Val Val Tyr Gln Asp Ile65 70 75 80Val Arg
Ile Asn Leu Ala Val Lys Asn Lys Thr Leu Val Glu Asn Lys 85 90 95His
Leu Lys Glu Ala Ala Glu Arg Ala Ile Lys Gly Asn Gly Arg Ile 100 105
110His Leu Cys Gly Leu Val Ser Asp Gly Gly Val His Ser His Ile Asp
115 120 125His Leu Phe Ala Leu Ile Thr Ala Leu Lys Gln Leu Lys Val
Pro Gln 130 135 140Leu Tyr Ile His Phe Phe Gly Asp Gly Arg Asp Thr
Ser Pro Thr Ser145 150 155 160Gly Val Gly Phe Leu Gln Gln Leu Ile
Asp Phe Val Asn Lys Glu Gln 165 170 175Tyr Gly Glu Ile Ala Thr Ile
Val Gly Arg Tyr Tyr Ala Met Asp Arg 180 185 190Asp Lys Arg Trp Glu
Arg Ile Arg Val Cys Tyr Asp Ala Leu Ile Ala 195 200 205Gly Val Gly
Glu Lys Thr Thr Ile Asp Lys Ala Ile Asp Val Ile Lys 210 215 220Gly
Arg Tyr Ala Lys Asp Glu Thr Asp Glu Phe Leu Lys Pro Ile Ile225 230
235 240Leu Ser Asp Lys Gly Arg Thr Lys Asp Gly Asp Thr Leu Ile Phe
Phe 245 250 255Asp Tyr Arg Ala Asp Arg Met Arg Glu Ile Thr Glu Cys
Met Gly Met 260 265 270Glu Arg Tyr Lys Asp Leu Lys Ser Asp Ile Lys
His Pro Lys Asp Met 275 280 285Gln Val Ile Gly Met Thr Gln Tyr Lys
Ala Glu Phe Thr Phe Pro Ala 290 295 300Leu Phe Pro Pro Glu Ser His
Lys Asn Val Leu Ala Glu Trp Leu Ser305 310 315 320Val Lys Gly Leu
Thr Gln Phe His Cys Ala Glu Thr Glu Lys Tyr Ala 325 330 335His Val
Thr Phe Phe Phe Asn Gly Gly Val Glu Lys Gln Phe Glu Asn 340 345
350Glu Glu Arg Cys Leu Val Pro Ser Pro Lys Val Ala Thr Tyr Asp Leu
355 360 365Glu Pro Ala Met Ser Ser Ala Gly Val Ala Asp Lys Met Ile
Glu Gln 370 375 380Leu Asn Arg Lys Ala His Ala Phe Ile Met Cys Asn
Phe Ala Pro Pro385 390 395 400Asp Met Val Gly His Thr Gly Val Tyr
Glu Ala Ala Val Lys Ala Val 405 410
415Glu Ala Thr Asp Ile Ala Ile Gly Arg Ile Tyr Glu Ala Cys Lys Lys
420 425 430Asn Asp Tyr Val Leu Met Val Thr Ala Asp His Gly Asn Ala
Glu Lys 435 440 445Met Ile Ala Pro Asp Gly Gly Lys His Thr Ala His
Thr Cys Asn Leu 450 455 460Val Pro Phe Thr Cys Ser Ser Leu Lys Phe
Lys Phe Met Asp Lys Leu465 470 475 480Pro Asp Arg Glu Met Ala Leu
Cys Asp Val Ala Pro Thr Val Leu Lys 485 490 495Val Leu Gly Leu Pro
Leu Pro Ser Glu Met Thr Gly Lys Pro Val Val 500 505 510Ile Glu Val
51548515PRTDirofilaria immitis 48Met Ala Glu Ala Lys Asn Arg Val
Cys Leu Val Val Ile Asp Gly Trp1 5 10 15Gly Ile Ser Asn Glu Ser Lys
Gly Asn Ala Ile Leu Asn Ala Lys Thr 20 25 30Pro Ile Met Asp Glu Leu
Cys Ala Leu Asn Ser His Pro Ile Glu Ala 35 40 45His Gly Leu His Val
Gly Leu Pro Glu Gly Leu Met Gly Asn Ser Glu 50 55 60Val Gly His Leu
Asn Ile Gly Ala Gly Arg Val Val Tyr Gln Asp Ile65 70 75 80Val Arg
Ile Asn Leu Ala Val Lys Asn Lys Thr Leu Val Glu Asn Lys 85 90 95His
Leu Lys Glu Ala Ala Glu Arg Ala Ile Lys Gly Asn Gly Arg Ile 100 105
110His Leu Cys Gly Leu Val Ser Asp Gly Gly Val His Ser His Ile Asp
115 120 125His Leu Phe Ala Leu Val Thr Ala Leu Lys Gln Leu Lys Val
Pro Gln 130 135 140Leu Phe Ile His Phe Phe Gly Asp Gly Arg Asp Thr
Ser Pro Thr Ser145 150 155 160Gly Val Gly Phe Leu Glu Gln Leu Ile
Asp Phe Val Asn Lys Glu Gln 165 170 175Tyr Gly Val Ile Ala Thr Ile
Val Gly Arg Tyr Tyr Ala Met Asp Arg 180 185 190Asp Lys Arg Trp Glu
Arg Ile Arg Val Cys Tyr Asp Ala Leu Ile Ala 195 200 205Gly Val Gly
Glu Lys Ala Thr Ile Asp Lys Ala Val Asp Val Ile Lys 210 215 220Ser
Arg Tyr Ala Lys Asp Glu Thr Asp Glu Phe Leu Lys Pro Ile Ile225 230
235 240Leu Ser Asp Glu Gly Arg Thr Lys Asp Gly Asp Thr Leu Ile Phe
Phe 245 250 255Asp Tyr Arg Ala Asp Arg Met Arg Glu Ile Thr Glu Cys
Met Gly Met 260 265 270Glu Arg Tyr Lys Asp Leu Lys Ser Asp Ile Lys
His Pro Lys Asn Met 275 280 285Gln Val Ile Gly Met Thr Gln Tyr Lys
Ala Glu Phe Thr Phe Pro Ala 290 295 300Leu Phe Pro Pro Glu Ser His
Lys Asn Val Leu Ala Glu Trp Leu Ser305 310 315 320Val Asn Gly Val
Thr Gln Phe His Cys Ala Glu Thr Glu Lys Tyr Ala 325 330 335His Val
Thr Phe Phe Phe Asn Gly Gly Val Glu Lys Gln Phe Glu Asn 340 345
350Glu Glu Arg Cys Leu Val Ala Ser Pro Lys Val Ala Thr Tyr Asp Leu
355 360 365Asp Pro Pro Met Ser Ser Ala Gly Val Ala Asp Lys Met Ile
Glu Gln 370 375 380Leu Asp Arg Lys Ala His Ala Phe Val Met Cys Asn
Phe Ala Pro Pro385 390 395 400Asp Met Val Gly His Thr Gly Val Tyr
Glu Ala Ala Val Lys Ala Val 405 410 415Glu Ala Thr Asp Ile Ala Ile
Gly Arg Ile Tyr Glu Ala Cys Lys Lys 420 425 430Asn Asp Tyr Ile Leu
Met Val Thr Ala Asp His Gly Asn Ala Glu Lys 435 440 445Met Met Ala
Pro Asp Gly Ser Lys His Thr Ala His Thr Cys Asn Leu 450 455 460Val
Pro Phe Thr Cys Ser Ser Met Lys Phe Lys Phe Met Asp Lys Leu465 470
475 480Pro Asp Arg Glu Met Ala Leu Cys Asp Val Ala Pro Thr Val Leu
Lys 485 490 495Val Met Gly Leu Pro Leu Pro Pro Glu Met Thr Gly Lys
Pro Val Val 500 505 510Ile Glu Val 51549514PRTEscherichia coli
49Met Leu Val Ser Lys Lys Pro Met Val Leu Val Ile Leu Asp Gly Tyr1
5 10 15Gly Tyr Arg Glu Glu Gln Gln Asp Asn Ala Ile Phe Ser Ala Lys
Thr 20 25 30Pro Val Met Asp Ala Leu Trp Ala Asn Arg Pro His Thr Leu
Ile Asp 35 40 45Ala Ser Gly Leu Glu Val Gly Leu Pro Asp Arg Gln Met
Gly Asn Ser 50 55 60Glu Val Gly His Val Asn Leu Gly Ala Gly Arg Ile
Val Tyr Gln Asp65 70 75 80Leu Thr Arg Leu Asp Val Glu Ile Lys Asp
Arg Ala Phe Phe Ala Asn 85 90 95Pro Val Leu Thr Gly Ala Val Asp Lys
Ala Lys Asn Ala Gly Lys Ala 100 105 110Val His Ile Met Gly Leu Leu
Ser Ala Gly Gly Val His Ser His Glu 115 120 125Asp His Ile Met Ala
Met Val Glu Leu Ala Ala Glu Arg Gly Ala Glu 130 135 140Lys Ile Tyr
Leu His Ala Phe Leu Asp Gly Arg Asp Thr Pro Pro Arg145 150 155
160Ser Ala Glu Ser Ser Leu Lys Lys Phe Glu Glu Lys Phe Ala Ala Leu
165 170 175Gly Lys Gly Arg Val Ala Ser Ile Ile Gly Arg Tyr Tyr Ala
Met Asp 180 185 190Arg Asp Asn Arg Trp Asp Arg Val Glu Lys Ala Tyr
Asp Leu Leu Thr 195 200 205Leu Ala Gln Gly Glu Phe Gln Ala Asp Thr
Ala Val Ala Gly Leu Gln 210 215 220Ala Ala Tyr Ala Arg Asp Glu Asn
Asp Glu Phe Val Lys Ala Thr Val225 230 235 240Ile Arg Ala Glu Gly
Gln Pro Asp Ala Ala Met Glu Asp Gly Asp Ala 245 250 255Leu Ile Phe
Met Asn Phe Arg Ala Asp Arg Ala Arg Glu Ile Thr Arg 260 265 270Ala
Phe Val Asn Ala Asp Phe Asp Gly Phe Ala Arg Lys Lys Val Val 275 280
285Asn Val Asp Phe Val Met Leu Thr Glu Tyr Ala Ala Asp Ile Lys Thr
290 295 300Ala Val Ala Tyr Pro Pro Ala Ser Leu Val Asn Thr Phe Gly
Glu Trp305 310 315 320Met Ala Lys Asn Asp Lys Thr Gln Leu Arg Ile
Ser Glu Thr Glu Lys 325 330 335Tyr Ala His Val Thr Phe Phe Phe Asn
Gly Gly Val Glu Glu Ser Phe 340 345 350Lys Gly Glu Asp Arg Ile Leu
Ile Asn Ser Pro Lys Val Ala Thr Tyr 355 360 365Asp Leu Gln Pro Glu
Met Ser Ser Ala Glu Leu Thr Glu Lys Leu Val 370 375 380Ala Ala Ile
Lys Ser Gly Lys Tyr Asp Thr Ile Ile Cys Asn Tyr Pro385 390 395
400Asn Gly Asp Met Val Gly His Thr Gly Val Met Glu Ala Ala Val Lys
405 410 415Ala Val Glu Ala Leu Asp His Cys Val Glu Glu Val Ala Lys
Ala Val 420 425 430Glu Ser Val Gly Gly Gln Leu Leu Ile Thr Ala Asp
His Gly Asn Ala 435 440 445Glu Gln Met Arg Asp Pro Ala Thr Gly Gln
Ala His Thr Ala His Thr 450 455 460Asn Leu Pro Val Pro Leu Ile Tyr
Val Gly Asp Lys Asn Val Lys Ala465 470 475 480Val Glu Gly Gly Lys
Leu Ser Asp Ile Ala Pro Thr Met Leu Ser Leu 485 490 495Met Gly Met
Glu Ile Pro Gln Glu Met Thr Gly Lys Pro Leu Phe Ile 500 505 510Val
Glu50505PRTStaphylococcus aureus 50Met Ala Lys Lys Pro Thr Ala Leu
Ile Ile Leu Asp Gly Phe Ala Asn1 5 10 15Arg Glu Ser Glu His Gly Asn
Ala Val Lys Leu Ala Asn Lys Pro Asn 20 25 30Phe Asp Arg Tyr Tyr Asn
Lys Tyr Pro Thr Thr Gln Ile Glu Ala Ser 35 40 45Gly Leu Asp Val Gly
Leu Pro Glu Gly Gln Met Gly Asn Ser Glu Val 50 55 60Gly His Met Asn
Ile Gly Ala Gly Arg Ile Val Tyr Gln Ser Leu Thr65 70 75 80Arg Ile
Asn Lys Ser Ile Glu Asp Gly Asp Phe Phe Glu Asn Asp Val 85 90 95Leu
Asn Asn Ala Ile Ala His Val Asn Ser His Asp Ser Ala Leu His 100 105
110Ile Phe Gly Leu Leu Ser Asp Gly Gly Val His Ser His Tyr Lys His
115 120 125Leu Phe Ala Leu Leu Glu Leu Ala Lys Lys Gln Gly Val Glu
Lys Val 130 135 140Tyr Val His Ala Phe Leu Asp Gly Arg Asp Val Asp
Gln Lys Ser Ala145 150 155 160Leu Lys Tyr Ile Glu Glu Thr Glu Ala
Lys Phe Asn Glu Leu Gly Ile 165 170 175Gly Gln Phe Ala Ser Val Ser
Gly Arg Tyr Tyr Ala Met Asp Arg Asp 180 185 190Lys Arg Trp Glu Arg
Glu Glu Lys Ala Tyr Asn Ala Ile Arg Asn Phe 195 200 205Asp Ala Pro
Thr Tyr Ala Thr Ala Lys Glu Gly Val Glu Ala Ser Tyr 210 215 220Asn
Glu Gly Leu Thr Asp Glu Phe Val Val Pro Phe Ile Val Glu Asn225 230
235 240Gln Asn Asp Gly Val Asn Asp Gly Asp Ala Val Ile Phe Tyr Asn
Phe 245 250 255Arg Pro Asp Arg Ala Ala Gln Leu Ser Glu Ile Phe Ala
Asn Arg Ala 260 265 270Phe Glu Gly Phe Lys Val Glu Gln Val Lys Asp
Leu Phe Tyr Ala Thr 275 280 285Phe Thr Lys Tyr Asn Asp Asn Ile Asp
Ala Ala Ile Val Phe Glu Lys 290 295 300Val Asp Leu Asn Asn Thr Ile
Gly Glu Ile Ala Gln Asn Asn Asn Leu305 310 315 320Thr Gln Leu Arg
Ile Ala Glu Thr Glu Lys Tyr Pro His Val Thr Tyr 325 330 335Phe Met
Ser Gly Gly Arg Asn Glu Glu Phe Lys Gly Glu Arg Arg Arg 340 345
350Leu Ile Asp Ser Pro Lys Val Ala Thr Tyr Asp Leu Lys Pro Glu Met
355 360 365Ser Ala Tyr Glu Val Lys Asp Ala Leu Leu Glu Glu Leu Asn
Lys Gly 370 375 380Asp Leu Asp Leu Ile Ile Leu Asn Phe Ala Asn Pro
Asp Met Val Gly385 390 395 400His Ser Gly Met Leu Glu Pro Thr Ile
Lys Ala Ile Glu Ala Val Asp 405 410 415Glu Cys Leu Gly Glu Val Val
Asp Lys Ile Leu Asp Met Asp Gly Tyr 420 425 430Ala Ile Ile Thr Ala
Asp His Gly Asn Ser Asp Gln Val Leu Thr Asp 435 440 445Asp Asp Gln
Pro Met Thr Thr His Thr Thr Asn Pro Val Pro Val Ile 450 455 460Val
Thr Lys Glu Gly Val Thr Leu Arg Glu Thr Gly Arg Leu Gly Asp465 470
475 480Leu Ala Pro Thr Leu Leu Asp Leu Leu Asn Val Glu Gln Pro Glu
Asp 485 490 495Met Thr Gly Glu Ser Leu Ile Lys His 500
50551509PRTBacillus anthracis 51Met Arg Lys Pro Thr Ala Leu Ile Ile
Leu Asp Gly Phe Gly Leu Arg1 5 10 15Glu Glu Thr Tyr Gly Asn Ala Val
Ala Gln Ala Lys Lys Pro Asn Phe 20 25 30Asp Gly Tyr Trp Asn Lys Phe
Pro His Thr Thr Leu Thr Ala Cys Gly 35 40 45Glu Ala Val Gly Leu Pro
Glu Gly Gln Met Gly Asn Ser Glu Val Gly 50 55 60His Leu Asn Ile Gly
Ala Gly Arg Ile Val Tyr Gln Ser Leu Thr Arg65 70 75 80Val Asn Val
Ala Ile Arg Glu Gly Glu Phe Asp Lys Asn Glu Thr Phe 85 90 95Gln Ser
Ala Ile Lys Ser Val Lys Glu Lys Gly Thr Ala Leu His Leu 100 105
110Phe Gly Leu Leu Ser Asp Gly Gly Val His Ser His Met Asn His Met
115 120 125Phe Ala Leu Leu Arg Leu Ala Ala Lys Glu Gly Val Glu Lys
Val Tyr 130 135 140Ile His Ala Phe Leu Asp Gly Arg Asp Val Gly Pro
Lys Thr Ala Gln145 150 155 160Ser Tyr Ile Asp Ala Thr Asn Glu Val
Ile Lys Glu Thr Gly Val Gly 165 170 175Gln Phe Ala Thr Ile Ser Gly
Arg Tyr Tyr Ser Met Asp Arg Asp Lys 180 185 190Arg Trp Asp Arg Val
Glu Lys Cys Tyr Arg Ala Met Val Asn Gly Glu 195 200 205Gly Pro Thr
Tyr Lys Ser Ala Glu Glu Cys Val Glu Asp Ser Tyr Ala 210 215 220Asn
Gly Ile Tyr Asp Glu Phe Val Leu Pro Ser Val Ile Val Asn Glu225 230
235 240Asp Asn Thr Pro Val Ala Thr Ile Asn Asp Asp Asp Ala Val Ile
Phe 245 250 255Tyr Asn Phe Arg Pro Asp Arg Ala Ile Gln Ile Ala Arg
Val Phe Thr 260 265 270Asn Gly Asp Phe Arg Glu Phe Asp Arg Gly Glu
Lys Val Pro His Ile 275 280 285Pro Glu Phe Val Cys Met Thr His Phe
Ser Glu Thr Val Asp Gly Tyr 290 295 300Val Ala Phe Lys Pro Met Asn
Leu Asp Asn Thr Leu Gly Glu Val Val305 310 315 320Ala Gln Ala Gly
Leu Lys Gln Leu Arg Ile Ala Glu Thr Glu Lys Tyr 325 330 335Pro His
Val Thr Phe Phe Phe Ser Gly Gly Arg Glu Ala Glu Phe Pro 340 345
350Gly Glu Glu Arg Ile Leu Ile Asn Ser Pro Lys Val Ala Thr Tyr Asp
355 360 365Leu Lys Pro Glu Met Ser Ile Tyr Glu Val Thr Asp Ala Leu
Val Asn 370 375 380Glu Ile Glu Asn Asp Lys His Asp Val Ile Ile Leu
Asn Phe Ala Asn385 390 395 400Cys Asp Met Val Gly His Ser Gly Met
Met Glu Pro Thr Ile Lys Ala 405 410 415Val Glu Ala Thr Asp Glu Cys
Leu Gly Lys Val Val Glu Ala Ile Leu 420 425 430Ala Lys Asp Gly Val
Ala Leu Ile Thr Ala Asp His Gly Asn Ala Asp 435 440 445Glu Glu Leu
Thr Ser Glu Gly Glu Pro Met Thr Ala His Thr Thr Asn 450 455 460Pro
Val Pro Phe Ile Val Thr Lys Asn Asp Val Glu Leu Arg Glu Asp465 470
475 480Gly Ile Leu Gly Asp Ile Ala Pro Thr Met Leu Thr Leu Leu Gly
Val 485 490 495Glu Gln Pro Lys Glu Met Thr Gly Lys Thr Ile Ile Lys
500 50552553PRTLeishmania mexicana 52Met Ser Ala Leu Leu Leu Lys
Pro His Lys Asp Leu Pro Arg Arg Thr1 5 10 15Val Leu Ile Val Val Met
Asp Gly Leu Gly Ile Gly Pro Glu Asp Asp 20 25 30Tyr Asp Ala Val His
Met Ala Ser Thr Pro Phe Met Asp Ala His Arg 35 40 45Arg Asp Asn Arg
His Phe Arg Cys Val Arg Ala His Gly Thr Ala Val 50 55 60Gly Leu Pro
Thr Asp Ala Asp Met Gly Asn Ser Glu Val Gly His Asn65 70 75 80Ala
Leu Gly Ala Gly Arg Val Ala Leu Gln Gly Ala Ser Leu Val Asp 85 90
95Asp Ala Ile Lys Ser Gly Glu Ile Tyr Thr Gly Glu Gly Tyr Arg Tyr
100 105 110Leu His Gly Ala Phe Ser Lys Glu Gly Ser Thr Leu His Leu
Ile Gly 115 120 125Leu Leu Ser Asp Gly Gly Val His Ser Arg Asp Asn
Gln Ile Tyr Ser 130 135 140Ile Ile Glu His Ala Val Lys Asp Gly Ala
Lys Arg Ile Arg Val His145 150 155 160Ala Leu Tyr Asp Gly Arg Asp
Val Pro Asp Gly Ser Ser Phe Arg Phe 165 170 175Thr Asp Glu Leu Glu
Ala Val Leu Ala Lys Val Arg Gln Asn Gly Cys 180 185 190Asp Ala Ala
Ile Ala Ser Gly Gly Gly Arg Met Phe Val Thr Met Asp 195 200 205Arg
Tyr Asp Ala Asp Trp Ser Ile Val Glu Arg Gly Trp Arg Ala Gln 210 215
220Val Leu Gly Asp Ala Arg His Phe His Ser Ala Lys Glu Ala Ile
Thr225 230 235 240Thr Phe Arg Glu Glu Asp Pro Lys Val Thr Asp Gln
Tyr Tyr Pro Pro 245 250 255Phe Ile Val Val Asp Glu Gln Asp Lys Pro
Leu Gly Thr Ile Glu Asp 260 265 270Gly Asp Ala Val Leu Cys Val Asn
Phe Arg Gly Asp Arg Val Ile Glu 275 280 285Met Thr Arg Ala Phe Glu
Asp Glu Asp Phe Asn Lys Phe Asp Arg Val 290 295 300Arg Val Pro Lys
Val Arg Tyr Ala Gly Met Met Arg Tyr Asp Gly
Asp305 310 315 320Leu Gly Ile Pro Asn Asn Phe Leu Val Pro Pro Pro
Lys Leu Thr Arg 325 330 335Val Ser Glu Glu Tyr Leu Cys Gly Ser Gly
Leu Asn Ile Phe Ala Cys 340 345 350Ser Glu Thr Gln Lys Phe Gly His
Val Thr Tyr Phe Trp Asn Gly Asn 355 360 365Arg Ser Gly Lys Ile Asp
Glu Lys His Glu Thr Phe Lys Glu Val Pro 370 375 380Ser Asp Arg Val
Gln Phe Asn Glu Lys Pro Arg Met Gln Ser Ala Ala385 390 395 400Ile
Thr Glu Ala Ala Ile Glu Ala Leu Lys Ser Gly Met Tyr Asn Val 405 410
415Val Arg Ile Asn Phe Pro Asn Gly Asp Met Val Gly His Thr Gly Asp
420 425 430Leu Lys Ala Thr Ile Thr Gly Val Glu Ala Val Asp Glu Ser
Leu Ala 435 440 445Lys Leu Lys Asp Ala Val Asp Ser Val Asn Gly Val
Tyr Ile Val Thr 450 455 460Ala Asp His Gly Asn Ser Asp Asp Met Ala
Gln Arg Asp Lys Lys Gly465 470 475 480Lys Pro Met Lys Asp Gly Asn
Gly Asn Val Leu Pro Leu Thr Ser His 485 490 495Thr Leu Ser Pro Val
Pro Val Phe Ile Gly Gly Ala Gly Leu Asp Pro 500 505 510Arg Val Ala
Met Arg Thr Asp Leu Pro Ala Ala Gly Leu Ala Asn Val 515 520 525Thr
Ala Thr Phe Ile Asn Leu Leu Gly Phe Glu Ala Pro Glu Asp Tyr 530 535
540Glu Pro Ser Leu Ile Tyr Val Glu Lys545 55053551PRTTrypanosoma
brucei 53Met Ala Leu Thr Leu Ala Ala His Lys Thr Leu Pro Arg Arg
Lys Leu1 5 10 15Val Leu Val Val Leu Asp Gly Val Gly Ile Gly Pro Arg
Asp Glu Tyr 20 25 30Asp Ala Val His Val Ala Lys Thr Pro Leu Met Asp
Ala Leu Phe Asn 35 40 45Asp Pro Lys His Phe Arg Ser Ile Cys Ala His
Gly Thr Ala Val Gly 50 55 60Leu Pro Thr Asp Ala Asp Met Gly Asn Ser
Glu Val Gly His Asn Ala65 70 75 80Leu Gly Ala Gly Arg Val Val Leu
Gln Gly Ala Ser Leu Val Asp Asp 85 90 95Ala Leu Glu Ser Gly Glu Ile
Phe Thr Ser Glu Gly Tyr Arg Tyr Leu 100 105 110His Gly Ala Phe Ser
Gln Pro Gly Arg Thr Leu His Leu Ile Gly Leu 115 120 125Leu Ser Asp
Gly Gly Val His Ser Arg Asp Asn Gln Val Tyr Gln Ile 130 135 140Leu
Lys His Ala Gly Ala Asn Gly Ala Lys Arg Ile Arg Val His Ala145 150
155 160Leu Tyr Asp Gly Arg Asp Val Pro Asp Lys Thr Ser Phe Lys Phe
Thr 165 170 175Asp Glu Leu Glu Glu Val Leu Ala Lys Leu Arg Glu Gly
Gly Cys Asp 180 185 190Ala Arg Ile Ala Ser Gly Gly Gly Arg Met Phe
Val Thr Met Asp Arg 195 200 205Tyr Glu Ala Asp Trp Ser Ile Val Glu
Arg Gly Trp Arg Ala Gln Val 210 215 220Leu Gly Glu Gly Arg Ala Phe
Lys Ser Ala Arg Glu Ala Leu Thr Lys225 230 235 240Phe Arg Glu Glu
Asp Ala Asn Ile Ser Asp Gln Tyr Tyr Pro Pro Phe 245 250 255Val Ile
Ala Gly Asp Asp Gly Arg Pro Ile Gly Thr Ile Glu Asp Gly 260 265
270Asp Ala Val Leu Cys Phe Asn Phe Arg Gly Asp Arg Val Ile Glu Met
275 280 285Ser Arg Ala Phe Glu Glu Glu Glu Phe Asp Lys Phe Asn Arg
Val Arg 290 295 300Leu Pro Lys Val Arg Tyr Ala Gly Met Met Arg Tyr
Asp Gly Asp Leu305 310 315 320Gly Ile Pro Asn Asn Phe Leu Val Pro
Pro Pro Lys Leu Thr Arg Thr 325 330 335Ser Glu Glu Tyr Leu Ile Gly
Ser Gly Cys Asn Ile Phe Ala Leu Ser 340 345 350Glu Thr Gln Lys Phe
Gly His Val Thr Tyr Phe Trp Asn Gly Asn Arg 355 360 365Ser Gly Lys
Leu Ser Glu Glu Arg Glu Thr Phe Cys Glu Ile Pro Ser 370 375 380Asp
Arg Val Gln Phe Asn Gln Lys Pro Leu Met Lys Ser Lys Glu Ile385 390
395 400Thr Asp Ala Ala Val Asp Ala Ile Lys Ser Gly Lys Tyr Asp Met
Ile 405 410 415Arg Ile Asn Tyr Pro Asn Gly Asp Met Val Gly His Thr
Gly Asp Leu 420 425 430Lys Ala Thr Ile Thr Ser Leu Glu Ala Val Asp
Gln Ser Leu Gln Arg 435 440 445Leu Lys Glu Ala Val Asp Ser Val Asn
Gly Val Phe Leu Ile Thr Ala 450 455 460Asp His Gly Asn Ser Asp Asp
Met Val Gln Arg Asp Lys Lys Gly Lys465 470 475 480Pro Val Arg Asp
Ala Glu Gly Asn Leu Met Pro Leu Thr Ser His Thr 485 490 495Leu Ala
Pro Val Pro Val Phe Ile Gly Gly Ala Gly Leu Asp Pro Arg 500 505
510Val Gln Met Arg Thr Asp Leu Pro Arg Ala Gly Leu Ala Asn Val Thr
515 520 525Ala Thr Phe Ile Asn Leu Met Gly Phe Glu Ala Pro Ser Asp
Tyr Glu 530 535 540Pro Ser Leu Ile Glu Val Ala545
5505411PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(8)cyclic linkage 54Tyr Asp Tyr Pro Gly Asp
His Cys Tyr Leu Tyr1 5 105515PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(8)cyclic linkageMOD_RES(9)..(9)METHYLATION
55Tyr Asp Tyr Pro Gly Asp Tyr Cys Tyr Leu Tyr Gly Thr Cys Gly1 5 10
155615PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(8)cyclic
linkageMOD_RES(10)..(10)METHYLATION 56Tyr Asp Tyr Pro Gly Asp Tyr
Cys Tyr Leu Tyr Gly Thr Cys Gly1 5 10 155715PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(8)cyclic
linkageMOD_RES(11)..(11)METHYLATION 57Tyr Asp Tyr Pro Gly Asp Tyr
Cys Tyr Leu Tyr Gly Thr Cys Gly1 5 10 155815PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(8)cyclic
linkageMOD_RES(12)..(12)METHYLATION 58Tyr Asp Tyr Pro Gly Asp Tyr
Cys Tyr Leu Tyr Gly Thr Cys Gly1 5 10 155915PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(8)cyclic
linkageMOD_RES(13)..(13)METHYLATION 59Tyr Asp Tyr Pro Gly Asp Tyr
Cys Tyr Leu Tyr Gly Thr Cys Gly1 5 10 156015PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(8)cyclic
linkageMOD_RES(14)..(14)METHYLATION 60Tyr Asp Tyr Pro Gly Asp Tyr
Cys Tyr Leu Tyr Gly Thr Cys Gly1 5 10 156115PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(8)cyclic
linkageMOD_RES(15)..(15)METHYLATION 61Tyr Asp Tyr Pro Gly Asp Tyr
Cys Tyr Leu Tyr Gly Thr Cys Gly1 5 10 156215PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(8)cylcic
linkageMOD_RES(13)..(15)METHYLATION 62Tyr Asp Tyr Pro Gly Asp Tyr
Cys Tyr Leu Tyr Gly Thr Cys Gly1 5 10 156315PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(8)cyclic
linkageMOD_RES(14)..(15)METHYLATION 63Tyr Asp Tyr Pro Gly Asp Tyr
Cys Tyr Leu Tyr Gly Thr Cys Gly1 5 10 156419PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(8)cyclic
linkageMOD_RES(13)..(13)METHYLATIONMOD_RES(15)..(15)METHYLATION
64Tyr Asp Tyr Pro Gly Asp Tyr Cys Tyr Leu Tyr Gly Met Glu Thr Cys1
5 10 15Met Glu Gly6515PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(8)cyclic
linkageMOD_RES(13)..(14)METHYLATION 65Tyr Asp Tyr Pro Gly Asp Tyr
Cys Tyr Leu Tyr Gly Thr Cys Gly1 5 10 156611PRTArtificial
SequenceSynthetic peptideMISC_FEATURE(1)..(8)cyclic
linkageMOD_RES(11)..(11)METHYLATION 66Tyr Asp Tyr Pro Gly Asp Tyr
Cys Tyr Leu Tyr1 5 106713PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(8)cyclic
linkageMOD_RES(13)..(13)METHYLATION 67Tyr Asp Tyr Pro Gly Asp Tyr
Cys Tyr Leu Tyr Gly Thr1 5 106813PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(8)cyclic
linkageMOD_RES(11)..(11)METHYLATION 68Tyr Asp Tyr Pro Gly Asp Tyr
Cys Tyr Leu Tyr Gly Thr1 5 106913PRTArtificial SequenceSynthetic
peptideMISC_FEATURE(1)..(8)cyclic
linkageMOD_RES(11)..(11)METHYLATIONMOD_RES(13)..(13)METHYLATION
69Tyr Asp Tyr Pro Gly Asp Tyr Cys Tyr Leu Tyr Gly Thr1 5 10

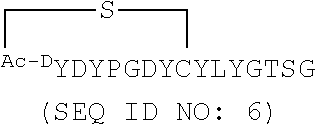






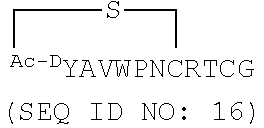




























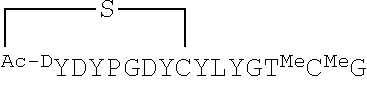







D00001

D00002

D00003

D00004

D00005

D00006

D00007

D00008

D00009

D00010
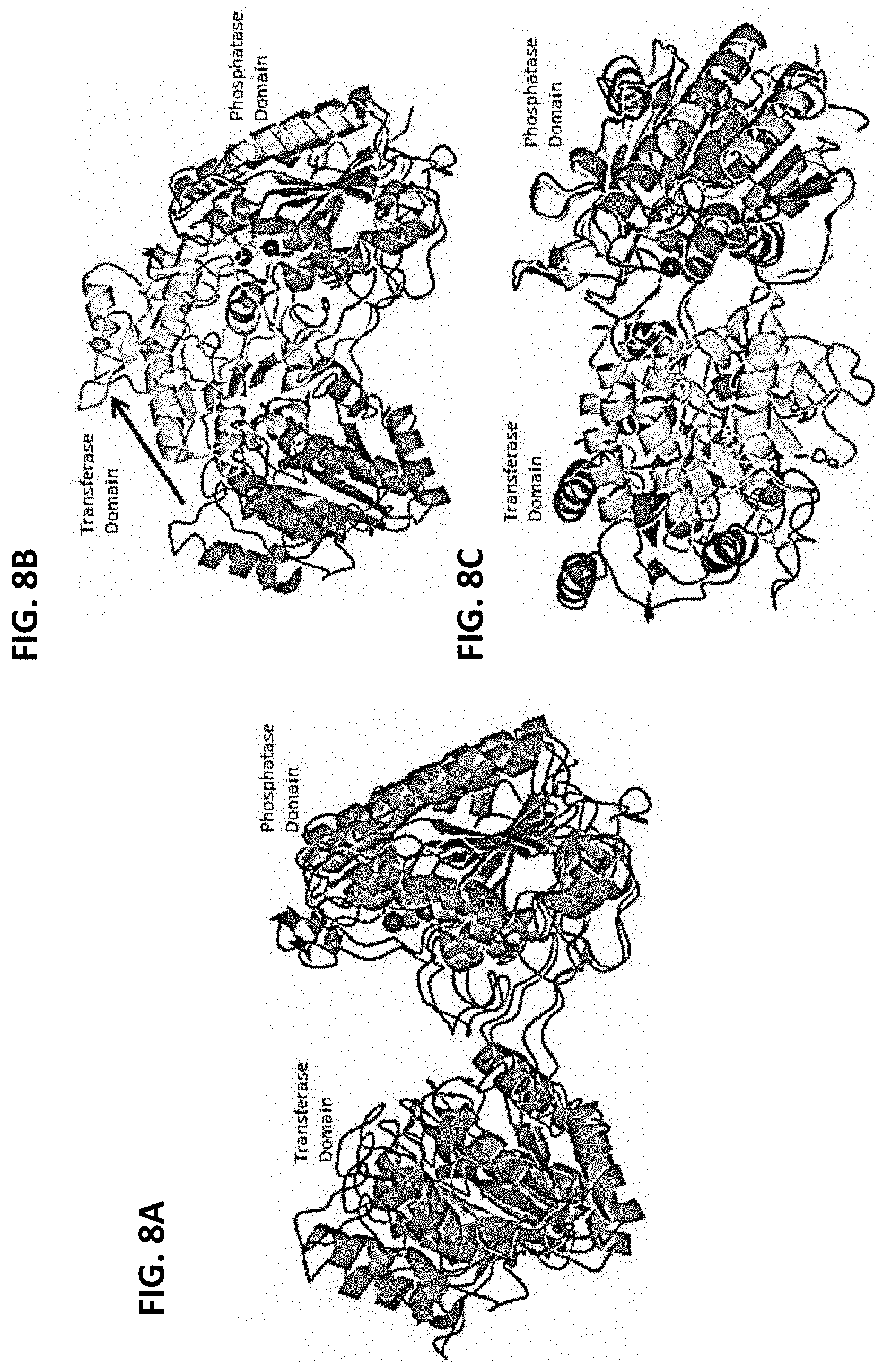
D00011

D00012

D00013

D00014

D00015

D00016

D00017

D00018

D00019

D00020

D00021

D00022

D00023

D00024

D00025

D00026

D00027

D00028

D00029

D00030

D00031

D00032

D00033
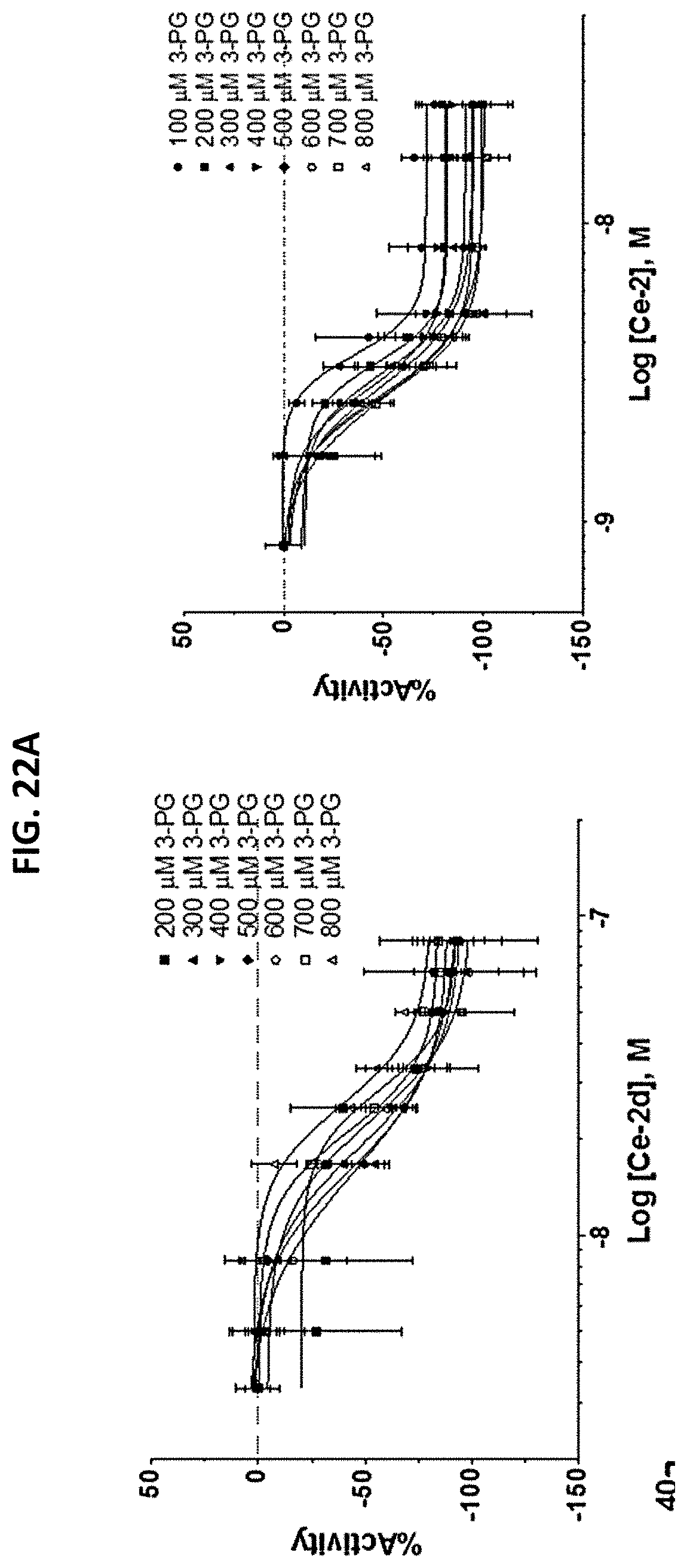
D00034

D00035

D00036




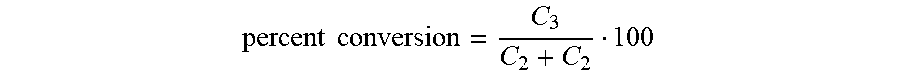

S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.