Glycopeptide Derivative Compounds And Uses Thereof
HECKLER; Ryan ; et al.
U.S. patent application number 16/615232 was filed with the patent office on 2020-11-26 for glycopeptide derivative compounds and uses thereof. This patent application is currently assigned to Insmed Incorporated. The applicant listed for this patent is Insmed Incorporated. Invention is credited to Ryan HECKLER, Donna KONICEK, Vladimir MALININ, Walter PERKINS, Adam PLAUNT.
| Application Number | 20200368312 16/615232 |
| Document ID | / |
| Family ID | 1000005060764 |
| Filed Date | 2020-11-26 |











View All Diagrams
| United States Patent Application | 20200368312 |
| Kind Code | A1 |
| HECKLER; Ryan ; et al. | November 26, 2020 |
GLYCOPEPTIDE DERIVATIVE COMPOUNDS AND USES THEREOF
Abstract
Provided herein are methods and compositions for the treatment of Gram positive bacterial infections. The infection in some embodiments, is a pulmonary infection. The method for treating the bacterial infection, comprises in one embodiment, administering to a patient in need thereof, a composition comprising an effective amount of a compound a glycopeptide derivative of Formula (I) or (II), or a pharmaceutically acceptable salt of Formula (I) or (II).
| Inventors: | HECKLER; Ryan; (Bridgewater, NJ) ; KONICEK; Donna; (Bridgewater, NJ) ; PLAUNT; Adam; (Bridgewater, NJ) ; MALININ; Vladimir; (Bridgewater, NJ) ; PERKINS; Walter; (Bridgewater, NJ) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Assignee: | Insmed Incorporated Bridgewater NJ |
||||||||||
| Family ID: | 1000005060764 | ||||||||||
| Appl. No.: | 16/615232 | ||||||||||
| Filed: | May 22, 2018 | ||||||||||
| PCT Filed: | May 22, 2018 | ||||||||||
| PCT NO: | PCT/US2018/033953 | ||||||||||
| 371 Date: | November 20, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 62509378 | May 22, 2017 | |||
| 62518280 | Jun 12, 2017 | |||
| 62560413 | Sep 19, 2017 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61P 31/04 20180101; A61K 38/14 20130101; A61K 9/0073 20130101 |
| International Class: | A61K 38/14 20060101 A61K038/14; A61K 9/00 20060101 A61K009/00; A61P 31/04 20060101 A61P031/04 |
Claims
1. (canceled)
2. A method for treating a bacterial pulmonary infection in a patient in need thereof, comprising administering to the lungs of a patient via a nebulizer, metered dose inhaler or a dry powder inhaler, a composition comprising an effective amount of a compound of Formula (II), or a pharmaceutically acceptable salt thereof: ##STR00053## wherein: R.sup.1 is C.sub.1-C.sub.18 linear alkyl, C.sub.1-C.sub.18 branched alkyl, R.sup.5--Y--R.sup.6--(Z).sub.n, or ##STR00054## R.sup.2 is --OH or --NH--(CH.sub.2).sub.q--R.sup.7; R.sup.3 is H or ##STR00055## R.sup.4 is H or CH.sub.2NH--CH.sub.2--PO.sub.3H.sub.2; n is 1 or 2; q is 1, 2, 3, 4, or 5; X is O, S, NH or H.sub.2; each Z is independently selected from hydrogen, aryl, cycloalkyl, cycloalkenyl, heteroaryl and heterocyclic; R.sup.5 and R.sup.6 are independently selected from the group consisting of alkylene, alkenylene and alkynylene, wherein the alkylene, alkenylene and alkynylene groups are optionally substituted with from 1 to 3 substituents selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl R.sup.7 is --N(CH.sub.2).sub.2; --N(CH.sub.2).sub.3; or ##STR00056## Y is independently selected from the group consisting of oxygen, sulfur, --S--S--, --NR.sup.8--, --S(O)--, --SO.sub.2--, --OSO.sub.2--, --NR.sup.8SO.sub.2--, --SO.sub.2NR.sup.8--, --SO.sub.2O--, --P(O)(OR.sup.8)O--, --P(O)(OR.sup.8)NR--, --OP(O)(OR.sup.8)O--, --OP(O)(OR.sup.8)NR.sup.8--, --NR.sup.8C(O)NR.sup.8--, and --NR.sup.8SO.sub.2NR.sup.8--; and each R.sup.8 is independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, heteroaryl and heterocyclic.
3. The method of claim 2, wherein R.sup.3 is H.
4. The method of claim 2, wherein R.sup.3 is ##STR00057##
5. The method of claim 3, wherein R.sup.4 is H.
6. The method of claim 3, wherein R.sup.4 is CH.sub.2--NH--CH.sub.2--PO.sub.3H.sub.2.
7. The method of claim 1, wherein X is O.
8-12. (canceled)
13. The method of claim 1, wherein R.sup.1 is R.sup.5--Y--R.sup.6--(Z).sub.n and Y is --NH--.
14. (canceled)
15. The method of claim 7, wherein R.sup.1 is (CH.sub.2).sub.2--Y--R.sup.6--(Z).sub.n.
16-19. (canceled)
20. The method of claim 13, wherein R.sup.1 is R.sup.5--Y--(CH.sub.2).sub.10--(Z).
21. The method of claim 20, wherein (Z).sub.n is H and R.sup.4 is H.
22. The method of claim 1, wherein R.sup.2 is OH, R.sup.4 is H, Y is --NH--, and (Z).sub.n is H.
23-24. (canceled)
25. The method of claim 1, wherein R.sup.1 is n-decyl.
26-39. (canceled)
40. The method of claim 1, wherein R.sup.1 is (CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3.
41-43. (canceled)
44. The method of claim 40, wherein R.sup.2 is OH.
45-80. (canceled)
81. The method of claim 1, wherein the bacterial infection is a Gram positive bacterial pulmonary infection.
82-83. (canceled)
84. The method of claim 81, wherein the Gram-positive bacterial pulmonary infection is a Staphylococcus pulmonary infection.
85-86. (canceled)
87. The method of claim 84, wherein the Staphylococcus pulmonary infection is a Staphylococcus aureus (S. aureus) pulmonary infection.
88. The method of claim 87, wherein the S. aureus pulmonary infection is a methicillin-resistant S. aureus (MRSA) pulmonary infection.
89. The method of claim 87, wherein the S. aureus pulmonary infection is a methicillin-sensitive S. aureus (MSSA) pulmonary infection.
90-117. (canceled)
118. The method of claim 1, wherein the patient is a cystic fibrosis patient.
119-122. (canceled)
Description
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application claims priority from U.S. Provisional Application Ser. No. 62/509,378, filed May 22, 2017; U.S. Provisional Application Ser. No. 62/518,280, filed Jun. 12, 2017; and U.S. Provisional Application Ser. No. 62/560,413, filed Sep. 19, 2017, the disclosures of each of which is incorporated by reference herein in their entireties.
BACKGROUND OF THE INVENTION
[0002] The high frequency of multidrug resistant bacteria, and in particular, Gram-positive bacteria, both in the hospital setting and the community present a significant challenge for the management of infections (Krause et al. (2008). Antimicrobial Agents and Chemotherapy 52(7), pp. 2647-2652, incorporated by reference herein in its entirety for all purposes).
[0003] The treatment of invasive Staphylococcus aureus (S. aureus) infections has relied significantly on vancomycin. However, the treatment and management of such infections is a therapeutic challenge because certain S. aureus isolates, and in particular, methicillin-resistant S. aureus isolates, have been shown to be resistant to vancomycin (Shaw et al. (2005). Antimicrobial Agents and Chemotherapy 49(1), pp. 195-201; Mendes et al. (2015). Antimicrobial Agents and Chemotherapy 59(3), pp. 1811-1814, each of which is incorporated by reference herein in its entirety for all purposes).
[0004] Because of the resistance displayed by many Gram-positive organisms to antibiotics, and the general lack of susceptibility to existing antibiotics, there is a need for new therapeutic strategies to combat infections due to these bacteria. The present invention addresses this and other needs.
SUMMARY OF THE INVENTION
[0005] In one aspect of the invention, a method is provided for treating a bacterial infection in a patient in need thereof. In one aspect a method of the invention comprises administrating to the patient a composition comprising an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof:
##STR00001## [0006] wherein,
[0007] R.sup.1 is C.sub.1-C.sub.18 linear alkyl, C.sub.1-C.sub.18 branched alkyl, R.sup.5--Y--R.sup.6--(Z).sub.n, or
##STR00002##
[0008] R.sup.2 is --OH or --NH--(CH.sub.2).sub.q--R.sup.7;
[0009] R.sup.3 is H or
##STR00003##
[0010] R.sup.4 is H or CH.sub.2--NH--CH.sub.2--PO.sub.3H.sub.2;
[0011] n is 1 or 2;
[0012] each q is independently 1, 2, 3, 4, or 5;
[0013] X is O, S, NH or H.sub.2;
[0014] each Z is independently selected from hydrogen, aryl, cycloalkyl, cycloalkenyl, heteroaryl and heterocyclic;
[0015] R.sup.5 and R.sup.6 are independently selected from the group consisting of alkylene, alkenylene and alkynylene, wherein the alkylene, alkenylene and alkynylene groups are optionally substituted with from 1 to 3 substituents selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl
[0016] R.sup.7 is --N(CH.sub.2).sub.2; --N.sup.+(CH.sub.2).sub.3; or
##STR00004##
[0017] Y is independently selected from the group consisting of oxygen, sulfur, --S--S--, --NR.sup.8--, --S(O)--, --SO.sub.2--, --NR.sup.8C(O)--, --OSO.sub.2--, --OC(O)--, --NR.sup.8SO.sub.2--, --C(O)NR.sup.8--, --C(O)O--, --SO.sub.2NR.sup.8--, --SO.sub.2O--, --P(O)(OR.sup.8)O--, --P(O)(OR.sup.8)NR.sup.8--, --OP(O)(OR.sup.8)O--, --OP(O)(OR.sup.8)NR.sup.8--, --OC(O)O--, --NR.sup.8C(O)O--, --NR.sup.8C(O)NR.sup.8--, --OC(O)NR.sup.8-- and --NR.sup.8SO.sub.2NR.sup.8--; and
[0018] each R.sup.8 is independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, heteroaryl and heterocyclic.
[0019] In another aspect, a method of the invention comprises administrating to the patient a composition comprising an effective amount of a compound of Formula (II), a prodrug thereof, or a pharmaceutically acceptable salt thereof:
##STR00005## [0020] wherein,
[0021] R.sup.1 is C.sub.1-C.sub.18 linear alkyl, C.sub.1-C.sub.18 branched alkyl, R.sup.5--Y--R.sup.6--(Z).sub.n, or
##STR00006##
[0022] R.sup.2 is --OH or --NH--(CH.sub.2).sub.q--R.sup.7;
[0023] R.sup.3 is H or
##STR00007##
[0024] R.sup.4 is H or CH.sub.2--NH--CH.sub.2--PO.sub.3H.sub.2;
[0025] n is 1 or 2;
[0026] each q is independently 1, 2, 3, 4, or 5;
[0027] X is O, S, NH or H.sub.2;
[0028] each Z is independently selected from hydrogen, aryl, cycloalkyl, cycloalkenyl, heteroaryl and heterocyclic;
[0029] R.sup.5 and R.sup.6 are independently selected from the group consisting of alkylene, alkenylene and alkynylene, wherein the alkylene, alkenylene and alkynylene groups are optionally substituted with from 1 to 3 substituents selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl;
[0030] R.sup.7 is --N(CH.sub.2).sub.2; --N.sup.+(CH.sub.2).sub.3; or
##STR00008##
[0031] Y is independently selected from the group consisting of oxygen, sulfur, --S--S--, --NR.sup.8--, --S(O)--, --SO.sub.2--, --OSO.sub.2--, --NR.sup.8SO.sub.2--, --SO.sub.2NR.sup.8--, --SO.sub.2O--, --P(O)(OR.sup.8)O--, --P(O)(OR.sup.8)NR.sup.8--, --OP(O)(OR.sup.8)O--, --OP(O)(OR.sup.8)NR.sup.8--, --NR.sup.8C(O)NR.sup.8--, and --NR.sup.8SO.sub.2NR.sup.8--; and
[0032] each R.sup.8 is independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, heteroaryl and heterocyclic.
[0033] In one embodiment of the method for treating a bacterial infection, the composition comprises an effective amount of a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), wherein R.sup.1 is C.sub.6 to C.sub.16 linear alkyl. In a further embodiment, R.sup.1 is C.sub.6, C.sub.10 or C.sub.16 alkyl. In even a further embodiment, R is C.sub.10 alkyl. In a further embodiment, the bacterial infection is a pulmonary bacterial infection. In even a further embodiment, the administering comprises administering via inhalation.
[0034] In one embodiment, the method for treating a bacterial infection comprises administering to the patient in need thereof, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), where R.sup.1 is R.sup.5--Y--R.sup.6--(Z).sub.n. In a further embodiment, R.sup.5 is --(CH.sub.2).sub.2--, R.sup.6 is --(CH.sub.2).sub.10--, X is O; Y is NR.sup.8, Z is hydrogen and n is 1. In a further embodiment, R.sup.8 is hydrogen. As such, one embodiment of the invention includes a compound of Formula (I), Formula (II) or a pharmaceutically acceptable salt thereof, where R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3. In a further embodiment, the bacterial infection is a pulmonary bacterial infection. In even a further embodiment, the administering comprises administering via inhalation.
[0035] In one embodiment, the method for treating a bacterial infection comprises administering to the patient in need thereof, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), where R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3 and R.sup.3 and R.sup.4 are H. In a further embodiment, R.sup.2 is OH. In even a further embodiment, the administering comprises administering via the intravenous route. In a further embodiment, X is O.
[0036] In one embodiment, the method for treating a bacterial infection comprises administering to the patient in need thereof, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II) where R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3, R.sup.2 is --NH--(CH.sub.2).sub.q--R.sup.7, and R.sup.3 and R.sup.4 are H. In a further embodiment, the administering comprises administering via the intravenous or pulmonary route. In even a further embodiment, q is 2 or 3 and R.sup.7 is --N(CH.sub.2).sub.2. In a further embodiment, X is O.
[0037] In one embodiment of the methods provided herein, the composition administered to the patient comprises an effective amount of a compound of Formula (I) or Formula (II), where R.sup.1 is
##STR00009##
In a further embodiment, R.sup.2 is OH and R.sup.3 and R.sup.4 are H. In even a further embodiment, the halogen is Cl and q is 1 or 2. In a further embodiment, the administering comprises administering via the pulmonary or intravenous route. In a further embodiment, X is O and R.sup.1 is
##STR00010##
[0038] In one embodiment of the methods provided herein, the composition administered to the patient comprises an effective amount of a compound of Formula (I) or Formula (II), where R.sup.1 is
##STR00011##
R.sup.2 is OH and R.sup.3 is
##STR00012##
and R.sup.4 is H. In even a further embodiment, the halogen is Cl and q is 1 or 2. In a further embodiment, the administering comprises administering via the intravenous route. In a further embodiment, X is O and R.sup.1 is
##STR00013##
[0039] In yet another embodiment, the bacterial infection is a Gram-positive cocci infection and the composition administered to the patient in need thereof comprises an effective amount of a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), wherein R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3. In a further embodiment, the infection is a Gram-positive infection is a cocci infection, and in a further embodiment, is a vancomycin-resistant enterococci (VRE), methicillin-resistant Staphylococcus aureus (MRSA), methicillin-resistant Staphylococcus epidermidis (MRSE), vancomycin resistant Enterococcus faecium also resistant to teicoplanin (VRE Fm Van A), vancomycin resistant Enterococcus faecium sensitive to teicoplanin (VRE Fm Van B), vancomycin resistant Enterococcus faecalis also resistant to teicoplanin (VRE Fs Van A), vancomycin resistant Enterococcus faecalis sensitive to teicoplanin (VRE Fs Van B), or penicillin-resistant Streptococcus pneumoniae (PRSP).
[0040] In even another embodiment, the bacterial infection is a Gram-positive cocci infection and the composition administered to the patient in need thereof comprises an effective amount of a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), wherein R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3. In a further embodiment, the infection is erythromycin-resistant (erm.sup.R), vancomycin-intermediate S. aureus (VISA) heterogenous vancomycin-intermediate S. aureus (hVISA), S. epidermidis coagulase-negative staphylococci (CoNS), penicillin-intermediate S. pneumoniae (PISP), or penicillin-resistant S. pneumoniae (PRSP).
[0041] In even another embodiment, R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3 and the bacterial infection is Propionibacterium acnes (skin acne), Eggerthella lenta (bacteremia) or Peptostreptococcus anaerobius (gynecological infection). In a further embodiment, R.sup.2 is OH and R.sup.3 and R.sup.4 are H.
[0042] In one embodiment, the bacterial infection is a methicillin-resistant Staphylococcus aureus (MRSA) infection and the composition administered to the patient in need thereof comprises an effective amount of a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), wherein R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3. In a further embodiment, the administration is conducted via a nebulizer or a dry powder inhaler and the bacterial infection is a pulmonary infection. In another embodiment, administration is intravenous, R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3; R.sup.2 is OH and R.sup.3 and R.sup.4 are H. In a further embodiment, X is O.
BRIEF DESCRIPTION OF THE FIGURES
[0043] FIG. 1, top shows the reductive amination of vancomycin to arrive at a glycopeptide derivative. The reaction occurs at the primary amine of vancomycin. FIG. 1, bottom, shows a synthesis scheme for a chloroeremomycin derivative.
[0044] FIG. 2 shows synthesis schemes for making the glycopeptide derivative RV40.
[0045] FIG. 3 shows a synthesis scheme for making the glycopeptide derivative RV79.
[0046] FIG. 4 is a synthesis scheme for making alkyl vancomycin derivatives.
[0047] FIG. 5 shows one synthesis scheme for making decyl-vancomycin (Compound #5).
[0048] FIG. 6 is a bar graph showing the minimum inhibitory concentration (MIC) (.mu.g antibiotic/mL) for various antibiotics against 23 different S. aureus strains.
[0049] FIG. 7 is a scatter plot showing the minimum inhibitory concentration (MIC) (.mu.g antibiotic/mL) for various antibiotics against 23 different S. aureus strains. Data is plotted as geometric mean with a 95% confidence interval.
[0050] FIG. 8 is a bar graph showing the minimum inhibitory concentration (MIC) (.mu.g antibiotic/mL) for various antibiotics against 12 different MRSA strains.
[0051] FIG. 9 is a scatter plot showing the minimum inhibitory concentration (MIC) (.mu.g antibiotic/mL) for various antibiotics against 12 different MRSA strains. Data is plotted as geometric mean with a 95% confidence interval.
[0052] FIG. 10 is a graph showing the log reduction of in CFU/mL biofilm as a function of antibiotic concentration (.mu.g/mL).
[0053] FIG. 11 is a graph showing the log reduction of in CFU/mL biofilm as a function of antibiotic concentration (.mu.g/mL).
[0054] FIG. 12 is a graph showing bacterial burden in lung versus control in animal model of pulmonary MRSA infection. Dose based on body weight target. The geometric mean for control was 6.4 Log.sub.10 CFU/g lung versus 3.2 Log.sub.10 CFU/g lung for RV40 treatment. Error is 95% Cl of geometric mean. N=11 for control and n=10 for RV40 treatment. P<0.0001, Mann-Whitney U-Test.
[0055] FIG. 13 is a graph showing the difference in log reduction in CFU/g lung versus control treatment (nebulized inhaled saline) for various antibiotics. Dose based on body weight target. Data plotted as mean of log values and error is SEM. Vehicle and control for RV40 and ORI was bicine buffer, pH 9.2. Vehicle and control for Vancomycin treatments was saline. N=10 for RV40, n=11 for ORI, n=9 for VAN neb, and n=6 for VAN i.v.
[0056] FIG. 14 is a graph showing reduction in lung CFU for inhaled RV40 targeted delivered dosed at 10, 5, 2, and 1 mg/kg vs control. Drugs were administered via inhalation at 12 and 24 h after intranasal bacterial challenge with MRSA (USA300, ATCC BAA-1556) in neutropenic rats and CFUs were counted 36 h after challenge. Data plotted is average of Log CFU/g (n=10 for 10 mg/kg, n=9 for 5 and 2 mg/kg, and n=11 for 1 mg/kg groups). Error is SEM.
[0057] FIG. 15 is a graph showing the difference in log reduction in CFU/g lung versus control treatment (nebulized inhaled saline) for prophylactic dosing of RV40. Prophylactic dosing of inhaled RV40 reduces lung bacterial burden vs. control (inhaled saline) up to 5 days before infection. Single doses of RV40 (10 mg/kg delivered target) were administered by inhalation. Neutropenic rats were infected with MRSA (USA300, ATCC BAA-1556) on Day 0 and CFUs were counted 36 h after challenge. Data plotted as geometric mean of CFU/g. Error bars are 95% confidence interval (CI). Statistics based on one-way ANOVA (p=0.001) with post-hoc Bonferroni multiple comparison test. N=11 for treatment groups on Days -7, -5, -3, -1, n=10 for Day +0.5, and n=8 for control.
DETAILED DESCRIPTION OF THE INVENTION
[0058] The high frequency of multidrug resistant bacteria, and in particular, Gram-positive bacteria, both in the healthcare setting and the community present a significant challenge for the management of infections (Krause et al. (2008). Antimicrobial Agents and Chemotherapy 52(7), pp. 2647-2652, incorporated by reference herein in its entirety for all purposes). Moreover, methicillin resistant S. aureus (MRSA) infections in cystic fibrosis (CF) patients is a concern, and there is a lack of clinical data regarding approaches to eradicate such infections (Goss and Muhlebach (2011). Journal of Cystic Fibrosis 10, pp. 298-306, incorporated by reference herein in its entirety for all purposes).
[0059] The present invention addresses the need for new bacterial infection treatment methods, and in particular, bacterial infection treatment methods by delivering compounds of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II) to patients in need thereof, for example via the pulmonary or intravenous route.
[0060] In one aspect, the present invention relates to methods for treating bacterial infections, for example, Gram-positive bacterial infections and in some embodiments, Gram-positive bacterial pulmonary infections. The method, in one embodiment, comprises administering to a patient in need thereof, a composition comprising an effective amount of a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II). The composition can be administered by any route. In the case of a pulmonary infection, in one embodiment, the composition is administered via a nebulizer, dry powder inhaler or metered dose inhaler. In another embodiment, the composition is administered intravenously.
[0061] The compounds for use in the bacterial infection treatment methods, and the specific treatment methods, are discussed in detail below.
[0062] An "effective amount" of a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), is an amount that can provide the desired therapeutic response. The effective amount can refer to a single dose as part of multiple doses during an administration period, or as the total dosage of glycopeptide given during an administration period. A treatment regimen can include substantially the same dose for each glycopeptide administration, or can comprise at least one, at least two or at least three different dosages.
[0063] The term "alkyl" refers to a monoradical branched or unbranched saturated hydrocarbon chain having from 1 to 40 carbon atoms, e.g., from 1 to 10 carbon atoms, or from 1 to 6 carbon atoms. This term is exemplified by groups such as methyl, ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, n-hexyl, n-decyl, tetradecyl, and the like. Both linear and branched alkyl groups are encompassed by the term "alkyl".
[0064] The term "substituted alkyl" refers to an alkyl group as defined above, having from 1 to 8 substituents, e.g., from 1 to 5 substituents or from 1 to 3 substituents, selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-- heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl.
[0065] The term "alkylene" refers to a diradical of a branched or unbranched saturated hydrocarbon chain, for example, having from 1 to 40 carbon atoms, e.g., from 1 to 10 carbon atoms, or from 1 to 6 carbon atoms. This term is exemplified by groups such as methylene (CH.sub.2--), ethylene (--CH.sub.2CH.sub.2--), the propylene isomers (e.g., --CH.sub.2CH.sub.2CH.sub.2-- and --CH(CH.sub.3)CCH.sub.2--) and the like.
[0066] The term "substituted alkylene" refers to an alkylene group, as defined above, having from 1 to 5 substituents, for example, from 1 to 3 substituents, selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl. Additionally, such substituted alkylene groups include those where 2 substituents on the alkylene group are fused to form one or more cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, heterocyclic or heteroaryl groups fused to the alkylene group. Such fused groups can contain from 1 to 3 fused ring structures. Additionally, the term substituted alkylene includes alkylene groups in which from 1 to 5 of the alkylene carbon atoms are replaced with oxygen, sulfur or NR-- where R is hydrogen or alkyl. Examples of substituted alkylenes are chloromethylene (--CH(Cl)--), aminoethylene (--CH(NH.sub.2)CH.sub.2--), 2-carboxypropylene isomers (--CH.sub.2CH(CO.sub.2H)CH.sub.2--), ethoxyethyl (--CH.sub.2CH.sub.2--O--CH.sub.2CH.sub.2--) and the like.
[0067] The term "alkaryl" refers to the groups -alkylene-aryl and substituted alkylene-aryl where alkylene, substituted alkylene and aryl are defined herein. Such alkaryl groups are exemplified by benzyl, phenethyl and the like.
[0068] The term "alkoxy" refers to the groups alkyl-O--, alkenyl-O--, cycloalkyl-O-cycloalkenyl-O--, and alkynyl-O--, where alkyl, alkenyl, cycloalkyl, cycloalkenyl, and alkynyl are as defined herein. Alkyl-O-- alkoxy groups include, e.g., methoxy, ethoxy, n-propoxy, iso-propoxy, n-butoxy, tert-butoxy, sec-butoxy, n-pentoxy, n-hexoxy, 1,2-dimethylbutoxy, and the like.
[0069] The term "substituted alkoxy" refers to the groups substituted alkyl-O--, substituted alkenyl-O--, substituted cycloalkyl-O--, substituted cycloalkenyl-O--, and substituted alkynyl-O-- where substituted alkyl, substituted alkenyl, substituted cycloalkyl, substituted cycloalkenyl and substituted alkynyl are as defined herein.
[0070] The term "alkylalkoxy" refers to the groups -alkylene-O-alkyl, alkylene-O-substituted alkyl, substituted alkylene-O-alkyl and substituted alkylene-O-substituted alkyl wherein alkyl, substituted alkyl, alkylene and substituted alkylene are as defined herein. Alkylalkoxy groups are also expressed as alkylene-O-alkyl and include, by way of example, methylenemethoxy (--CH.sub.2OCH.sub.3), ethylenemethoxy (--CH.sub.2CH.sub.2OCH.sub.3), n-propylene-iso-propoxy (--CH.sub.2CH.sub.2CH.sub.2OCH(CH.sub.3).sub.2), methylene-t-butoxy (--CH.sub.2--O--C(CH.sub.3).sub.3) and the like.
[0071] The term "alkenyl" refers to a monoradical of a branched or unbranched unsaturated hydrocarbon group having from 2 to 40 carbon atoms, e.g., 2 to 10 carbon atoms or 2 to 6 carbon atoms, and having at least 1 and in some embodiments, from 1-6 sites of vinyl unsaturation. Alkenyl groups include ethenyl (--CH.dbd.CH.sub.2), n-propenyl (--CH.sub.2CH.dbd.CH.sub.2), iso-propenyl (--C(CH.sub.3).dbd.CH.sub.2), and the like.
[0072] The term "substituted alkenyl" refers to an alkenyl group as defined above having from 1 to 5 substituents, and e.g., from 1 to 3 substituents, selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl.
[0073] The term "alkenylene" refers to a diradical of a branched or unbranched unsaturated hydrocarbon group having from 2 to 40 carbon atoms, for example from 2 to 10 carbon atoms or from 2 to 6 carbon atoms and having at least 1 and for example, from 1-6 sites of vinyl unsaturation. This term is exemplified by groups such as ethenylene (--CH.dbd.CH--), the propenylene isomers (e.g., --CH.sub.2CH.dbd.CH-- and --C(CH.sub.3).dbd.CH--) and the like.
[0074] The term "substituted alkenylene" refers to an alkenylene group as defined above having from 1 to 5 substituents, and for example, from 1 to 3 substituents, selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-- heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl. Additionally, such substituted alkenylene groups include those where 2 substituents on the alkenylene group are fused to form one or more cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, heterocyclic or heteroaryl groups fused to the alkenylene group.
[0075] The term "alkynyl" refers to a monoradical of an unsaturated hydrocarbon having from 2 to 40 carbon atoms, for example, from 2 to 20 carbon atoms, or from 2 to 6 carbon atoms and having at least 1 and in some embodiments from 1 to 6 sites of acetylene (triple bond) unsaturation. Representative alkynyl groups include ethynyl (--C.ident.CH), propargyl (--CH.sub.2C.ident.CH) and the like.
[0076] The term "substituted alkynyl" refers to an alkynyl group as defined above having from 1 to 5 substituents, for example, from 1 to 3 substituents, selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl.
[0077] The term "alkynylene" refers to a diradical of an unsaturated hydrocarbon having from 2 to 40 carbon atoms, for example from 2 to 10 carbon atoms or 2 to 6 carbon atoms and having at least 1 and in some embodiment, from 1-6 sites of acetylene (triple bond) unsaturation. Representative alkynylene groups include ethynylene (--C.ident.C--), propargylene (--CH.sub.2C.ident.C--).
[0078] The term "substituted alkynylene" refers to an alkynylene group as defined above having from 1 to 5 substituents, for example, from 1 to 3 substituents, selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-- heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl.
[0079] The term "acyl" refers to the groups HC(O)--, alkyl-C(O)--, substituted alkyl-C(O)--, cycloalkyl-C(O)--, substituted cycloalkyl-C(O)--, cycloalkenyl-C(O)--, substituted cycloalkenyl-C(O)--, aryl-C(O)--, heteroaryl-C(O)-- and heterocyclic-C(O)-- where alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, heteroaryl and heterocyclic are as defined herein.
[0080] The term "acylamino" or "aminocarbonyl" refers to the group --C(O)NRR where each R is independently hydrogen, alkyl, substituted alkyl, aryl, heteroaryl, heterocyclic or where both R groups are joined to form a heterocyclic group (e.g., morpholino) wherein alkyl, substituted alkyl, aryl, heteroaryl and heterocyclic are as defined herein.
[0081] The term "aminoacyl" refers to the group --NRC(O)R where each R is independently hydrogen, alkyl, substituted alkyl, aryl, heteroaryl, or heterocyclic wherein alkyl, substituted alkyl, aryl, heteroaryl and heterocyclic are as defined herein.
[0082] The term "aminoacyloxy" or "alkoxycarbonylamino" refers to the group --NRC(O)OR where each R is independently hydrogen, alkyl, substituted alkyl aryl, heteroaryl, or heterocyclic.
[0083] The term "acyloxy" refers to the groups alkyl-C(O)O--, substituted alkyl-C(O)O--, cycloalkyl-C(O)O--, substituted cycloalkyl-C(O)O--, aryl-C(O)O--, heteroaryl-C(O)O--, and heterocyclic-C(O)O-- wherein alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, aryl, heteroaryl, and heterocyclic are as defined herein.
[0084] The term "aryl" refers to an unsaturated aromatic carbocyclic group of from 6 to 20 carbon atoms having a single ring (e.g., phenyl) or multiple condensed (fused) rings (e.g., naphthyl or anthryl). Representative aryls include phenyl, naphthyl and the like. Unless otherwise constrained by the definition for the aryl substituent, such aryl groups can optionally be substituted with from 1 to 5 substituents, e.g., from 1 to 3 substituents, selected from the group consisting of acyloxy, hydroxy, thiol, acyl, alkyl, alkoxy, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, substituted alkyl, substituted alkoxy, substituted alkenyl, substituted alkynyl, substituted cycloalkyl, substituted cycloalkenyl, amino, substituted amino, aminoacyl, acylamino, alkaryl, aryl, aryloxy, azido, carboxyl, carboxylalkyl, cyano, halo, nitro, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, aminoacyloxy, oxyacylamino, sulfonamide, thioalkoxy, substituted thioalkoxy, thioaryloxy, thioheteroaryloxy, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl, --SO.sub.2-heteroaryl and trihalomethyl. In one embodiment, the aryl substituent is alkyl, alkoxy, halo, cyano, nitro, trihalomethyl, thioalkoxy or a combination thereof.
[0085] The term "aryloxy" refers to the group aryl-O-- wherein the aryl group is as defined above including optionally substituted aryl groups as also defined above.
[0086] The term "arylene" refers to the diradical derived from aryl (including substituted aryl) as defined above and is exemplified by 1,2-phenylene, 1,3-phenylene, 1,4-phenylene, 1,2-naphthylene and the like.
[0087] The term "amino" refers to the group --NH.sub.2.
[0088] The term "substituted amino" refers to the group --NRR where each R is independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, alkenyl, substituted alkenyl, cycloalkenyl, substituted cycloalkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl and heterocyclic provided that both R groups are not H.
[0089] "Amino acid" refers to any of the naturally occurring amino acids, synthetic amino acids, and derivatives thereof .alpha.-Amino acids comprise a carbon atom to which is bonded an amino group, a carboxy group, a hydrogen atom, and a distinctive group referred to as a "side chain". The side chains of naturally occurring amino acids are well known in the art and include, for example, hydrogen (e.g., glycine), alkyl (e.g., alanine, valine, leucine, isoleucine, proline), substituted alkyl (e.g., as in threonine, serine, methionine, cysteine, aspartic acid, asparagine, glutamic acid, glutamine, arginine, and lysine), alkaryl (e.g., phenylalanine and tryptophan), substituted arylalkyl (e.g., tyrosine), and heteroarylalkyl (e.g., histidine).
[0090] The term "carboxyalkyl" or "alkoxycarbonyl" refers to the groups "--C(O)O-alkyl", "--C(O)O-substituted alkyl", "--C(O)O-cycloalkyl", "--C(O)O-substituted cycloalkyl", "--C(O)O-- alkenyl", "--C(O)O-substituted alkenyl", "--C(O)O-alkynyl" and "--C(O)O-substituted alkynyl" where alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, alkenyl, substituted alkenyl, alkynyl and substituted alkynyl are as defined herein
[0091] The term "cycloalkyl" refers to cyclic alkyl groups of from 3 to 20 carbon atoms having a single cyclic ring or multiple condensed rings. Such cycloalkyl groups include, by way of example, single ring structures such as cyclopropyl, cyclobutyl, cyclopentyl, cyclooctyl, and the like, or multiple ring structures such as adamantanyl, and the like.
[0092] The term "substituted cycloalkyl" refers to cycloalkyl groups having from 1 to 5 substituents, and for example, from 1 to 3 substituents, selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl.
[0093] The term "cycloalkenyl" refers to cyclic alkenyl groups of from 4 to 20 carbon atoms having a single cyclic ring and at least one point of internal unsaturation. Examples of suitable cycloalkenyl groups include, e.g., cyclobut-2-enyl, cyclopent-3-enyl, cyclooct-3-enyl.
[0094] The term "substituted cycloalkenyl" refers to cycloalkenyl groups having from 1 to 5 substituents, and for example, from 1 to 3 substituents, selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl.
[0095] The term "halo" or "halogen" refers to fluoro, chloro, bromo and/or iodo.
[0096] "Haloalkyl" refers to alkyl as defined herein substituted by 1-4 halo groups as defined herein, which may be the same or different. Representative haloalkyl groups include, by way of example, trifluoromethyl, 3-fluorododecyl, 12,12,12-trifluorododecyl, 2-bromooctyl, 3-bromo-6-chloroheptyl, and the like.
[0097] The term "heteroaryl" refers to an aromatic group of from 1 to 15 carbon atoms and 1 to 4 heteroatoms selected from oxygen, nitrogen and sulfur within at least one ring moiety.
[0098] Unless otherwise constrained by the definition for the heteroaryl substituent, such heteroaryl groups can be optionally substituted with 1 to 5 substituents, for example from 1 to 3 substituents, selected from the group consisting of acyloxy, hydroxy, thiol, acyl, alkyl, alkoxy, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, substituted alkyl, substituted alkoxy, substituted alkenyl, substituted alkynyl, substituted cycloalkyl, substituted cycloalkenyl, amino, substituted amino, aminoacyl, acylamino, alkaryl, aryl, aryloxy, azido, carboxyl, carboxylalkyl, cyano, halo, nitro, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, aminoacyloxy, oxyacylamino, thioalkoxy, substituted thioalkoxy, thioaryloxy, thioheteroaryloxy, SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl and trihalomethyl. Representative aryl substituents include alkyl, alkoxy, halo, cyano, nitro, trihalomethyl, and thioalkoxy. Such heteroaryl groups can have a single ring (e.g., pyridyl or furyl) or multiple condensed rings (e.g., indolizinyl or benzothienyl). In one embodiment, the heteroaryl is pyridyl, pyrrolyl or furyl. "Heteroarylalkyl" refers to (heteroaryl)alkyl- where heteroaryl and alkyl are as defined herein. Representative examples include 2-pyridylmethyl and the like.
[0099] The term "heteroaryloxy" refers to the group heteroaryl-O--.
[0100] The term "heteroarylene" refers to the diradical group derived from heteroaryl (including substituted heteroaryl), as defined above, and is exemplified by the groups 2,6-pyridylene, 2,4-pyridiylene, 1,2-quinolinylene, 1,8-quinolinylene, 1,4-benzofuranylene, 2,5-pyridnylene, 2,5-indolenyl and the like.
[0101] The term "heterocycle" or "heterocyclic" refers to a monoradical saturated unsaturated group having a single ring or multiple condensed rings, from 1 to 40 carbon atoms and from 1 to 10 hetero atoms, for example from 1 to 4 heteroatoms, selected from nitrogen, sulfur, phosphorus, and/or oxygen within the ring.
[0102] Unless otherwise constrained by the definition for the heterocyclic substituent, such heterocyclic groups can be optionally substituted with 1 to 5, and for example, from 1 to 3 substituents, selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl. Such heterocyclic groups can have a single ring or multiple condensed rings. In one embodiment, the heterocyclic is morpholino or piperidinyl.
[0103] Examples of nitrogen heterocycles and heteroaryls include, but are not limited to, pyrrole, imidazole, pyrazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, phenanthroline, isothiazole, phenazine, isoxazole, phenoxazine, phenothiazine, imidazolidine, imidazoline, piperidine, piperazine, indoline, morpholino, piperidinyl, tetrahydrofuranyl, and the like as well as N-alkoxy-nitrogen containing heterocycles.
[0104] Another class of heterocyclics is known as "crown compounds" which refers to a specific class of heterocyclic compounds having one or more repeating units of the formula [(CH.sub.2--).sub.aA-] where a is equal to or greater than 2, and A at each separate occurrence can be 0, N, S or P. Examples of crown compounds include, by way of example only, [--(CH.sub.2).sub.3--NH-]3, [--((CH.sub.2).sub.2--O).sub.4--((CH.sub.2).sub.2--NH).sub.2] and the like. In one embodiment, the crown compound has from 4 to 10 heteroatoms and 8 to 40 carbon atoms.
[0105] The term "heterocyclooxy" refers to the group heterocyclic-O--.
[0106] The term "heterocyclene" refers to the diradical group formed from a heterocycle, as defined herein, and is exemplified by the groups 2,6-morpholino, 2,5-morpholino and the like.
[0107] The term "oxyacylamino" or "aminocarbonyloxy" refers to the group --OC(O)NRR where each R is independently hydrogen, alkyl, substituted alkyl, aryl, heteroaryl, or heterocyclic wherein alkyl, substituted alkyl, aryl, heteroaryl and heterocyclic are as defined herein.
[0108] The term "spiro-attached cycloalkyl group" refers to a cycloalkyl group attached to another ring via one carbon atom common to both rings.
[0109] The term "sulfonamide" refers to a group of the formula --SO.sub.2NRR, where each R is independently hydrogen, alkyl, substituted alkyl, aryl, heteroaryl, or heterocyclic wherein alkyl, substituted alkyl, aryl, heteroaryl and heterocyclic are as defined herein.
[0110] The term "thiol" refers to the group --SH.
[0111] The term "thioheteroaryloxy" refers to the group heteroaryl-S-- wherein the heteroaryl group is as defined above including optionally substituted aryl groups as also defined above.
[0112] As to any of the above groups which contain one or more substituents, it is understood that such groups do not contain any substitution or substitution patterns which are sterically impractical and/or synthetically non-feasible. In addition, the compounds of this invention include all stereochemical isomers arising from the substitution of these compounds.
[0113] "Glycopeptide" refers to heptapeptide antibiotics, characterized by a multi-ring peptide core optionally substituted with saccharide groups. Examples of glycopeptides included in this definition may be found in "Glycopeptides Classification, Occurrence, and Discovery", by Raymond C. Rao and Louise W. Crandall, ("Drugs and the Pharmaceutical Sciences" Volume 63, edited by Ramakrishnan Nagarajan, published by Marcal Dekker, Inc.), which is hereby incorporated by reference in its entirety. Representative glycopeptides include those identified as A477, A35512, A40926, A41030, A42867, A47934, A80407, A82846, A83850, A84575, AB-65, Actaplanin, Actinoidin, Ardacin, Avoparcin, Azureomycin, Balhimycin, Chloroorientiein, Chloropolysporin, Decaplanin, N-demethylvancomycin, Eremomycin, Galacardin, Helvecardin, Izupeptin, Kibdelin, LL-AM374, Mannopeptin, MM45289, MM47756, MM47761, MM49721, MM47766, MM55260, MM55266, MM55270, MM56597, MM56598, OA-7653, Orenticin, Parvodicin, Ristocetin, Ristomycin, Synmonicin, Teicoplanin, Telavancin, UK-68597, UK-69542, UK-72051, Vancomycin, and the like. The term "glycopeptide" as used herein is also intended to include the general class of peptides disclosed above on which the sugar moiety is absent, i.e., the aglycone series of glycopeptides. For example, removal of the disaccharide moiety appended to the phenol on vancomycin by mild hydrolysis gives vancomycin aglycone. Also within the scope of the invention are glycopeptides that have been further appended with additional saccharide residues, especially aminoglycosides, in a manner similar to vancosamine. In embodiments described herein, one or more of the aforementioned glycopeoptides can be used in combination with a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or (II).
[0114] "Pharmaceutically acceptable salt" includes both acid and base addition salts. A pharmaceutically acceptable addition salt refers to those salts which retain the biological effectiveness and properties of the free bases, which are not biologically or otherwise undesirable, and which are formed with inorganic acids such as, but are not limited to, hydrochloric acid (HCl), hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, and organic acids such as, but not limited to, acetic acid, 2,2-dichloroacetic acid, adipic acid, alginic acid, ascorbic acid, aspartic acid, benzenesulfonic acid, benzoic acid, 4-acetamidobenzoic acid, camphoric acid, camphor-10-sulfonic acid, capric acid, caproic acid, caprylic acid, carbonic acid, cinnamic acid, citric acid, cyclamic acid, dodecylsulfuric acid, ethane-1,2-disulfonic acid, ethanesulfonic acid, 2-hydroxyethanesulfonic acid, formic acid, fumaric acid, galactaric acid, gentisic acid, glucoheptonic acid, gluconic acid, glucuronic acid, glutamic acid, glutaric acid, 2-oxo-glutaric acid, glycerophosphoric acid, glycolic acid, hippuric acid, isobutyric acid, lactic acid (e.g., as lactate), lactobionic acid, lauric acid, maleic acid, malic acid, malonic acid, mandelic acid, methanesulfonic acid, mucic acid, naphthalene-1,5-disulfonic acid, naphthalene-2-sulfonic acid, 1-hydroxy-2-naphthoic acid, nicotinic acid, oleic acid, orotic acid, oxalic acid, palmitic acid, pamoic acid, propionic acid, pyroglutamic acid, pyruvic acid, salicylic acid, 4-aminosalicylic acid, sebacic acid, stearic acid, succinic acid, acetic acid (e.g., as acetate), tartaric acid, thiocyanic acid, p-toluenesulfonic acid, trifluoroacetic acid (TFA), undecylenic acid, and the like. In one embodiment, the pharmaceutically acceptable salt is HCl, TFA, lactate or acetate.
[0115] A pharmaceutically acceptable base addition salt retains the biological effectiveness and properties of the free acids, which are not biologically or otherwise undesirable. These salts are prepared from addition of an inorganic base or an organic base to the free acid. Salts derived from inorganic bases include, but are not limited to, the sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum salts and the like. Inorganic salts include the ammonium, sodium, potassium, calcium, and magnesium salts. Salts derived from organic bases include, but are not limited to, salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion exchange resins, such as ammonia, isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, diethanolamine, ethanolamine, deanol, 2-dimethylaminoethanol, 2-diethylaminoethanol, dicyclohexylamine, lysine, arginine, histidine, caffeine, procaine, hydrabamine, choline, betaine, benethamine, benzathine, ethylenediamine, glucosamine, methylglucamine, theobromine, triethanolamine, tromethamine, purines, piperazine, piperidine, N-ethylpiperidine, polyamine resins and the like. Organic bases that can be used to form a pharmaceutically acceptable salt include isopropylamine, diethylamine, ethanolamine, trimethylamine, dicyclohexylamine, choline and caffeine.
[0116] In one aspect of the invention, a method is provided for treating a bacterial infection in a patient in need thereof. The method comprises administrating to the patient a composition comprising an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof.
##STR00014## [0117] wherein,
[0118] R.sup.1 is C.sub.1-C.sub.18 linear alkyl, C.sub.1-C.sub.18 branched alkyl, R.sup.5--Y--R.sup.6--(Z).sub.n, or
##STR00015##
[0119] R.sup.2 is --OH or --NH--(CH.sub.2).sub.q--R.sup.7;
[0120] R.sup.3 is H or
##STR00016##
[0121] R.sup.4 is H or CH.sub.2--NH--CH.sub.2--PO.sub.3H.sub.2;
[0122] n is 1 or 2;
[0123] each q is independently 1, 2, 3, 4, or 5;
[0124] X is O, S, NH or H.sub.2;
[0125] each Z is independently selected from hydrogen, aryl, cycloalkyl, cycloalkenyl, heteroaryl and heterocyclic;
[0126] R.sup.5 and R.sup.6 are independently selected from the group consisting of alkylene, alkenylene and alkynylene, wherein the alkylene, alkenylene and alkynylene groups are optionally substituted with from 1 to 3 substituents selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl
[0127] R.sup.7 is --N(CH.sub.2).sub.2; --N.sup.+(CH.sub.2).sub.3; or
##STR00017##
Y is independently selected from the group consisting of oxygen, sulfur, --S--S--, --NR.sup.8--, --S(O)--, --SO.sub.2--, --NR.sup.8C(O)--, --OSO.sub.2--, --OC(O)--, --NR.sup.8SO.sub.2--, --C(O)NR.sup.8--, --C(O)O--, --SO.sub.2NR.sup.8--, --SO.sub.2O--, --P(O)(OR.sup.8)O--, --P(O)(OR.sup.8)NR.sup.8--, --OP(O)(OR.sup.8)O--, --OP(O)(OR.sup.8)NR.sup.8--, --OC(O)O--, --NR.sup.8C(O)O--, --NR.sup.8C(O)NR.sup.8--, --OC(O)NR.sup.8-- and --NR.sup.8SO.sub.2NR.sup.8--; and
[0128] each R.sup.8 is independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, heteroaryl and heterocyclic
[0129] Another aspect of the invention relates to a method of treating a patient for a bacterial infection. The method comprises administering a composition comprising an effective amount of a compound of Formula (II), or a pharmaceutically acceptable salt thereof, to the patient in need of treatment. Formula (II) is defined as follows:
##STR00018## [0130] wherein,
[0131] R.sup.1 is C.sub.1-C.sub.18 linear alkyl, C.sub.1-C.sub.18 branched alkyl, R.sup.5--Y--R.sup.6--(Z).sub.n, or
##STR00019##
[0132] R.sup.2 is --OH or --NH--(CH.sub.2).sub.q--R.sup.7;
[0133] R.sup.3 is H or
##STR00020##
[0134] R.sup.4 is H or CH.sub.2--NH--CH.sub.2--PO.sub.3H.sub.2;
[0135] n is 1 or 2;
[0136] each q is independently 1, 2, 3, 4, or 5;
[0137] X is O, S, NH or H.sub.2;
[0138] each Z is independently selected from hydrogen, aryl, cycloalkyl, cycloalkenyl, heteroaryl and heterocyclic;
[0139] R.sup.5 and R.sup.6 are independently selected from the group consisting of alkylene, alkenylene and alkynylene, wherein the alkylene, alkenylene and alkynylene groups are optionally substituted with from 1 to 3 substituents selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxy aminoacyl, azido, cyano, halogen, hydroxyl, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, --SO-alkyl, --SO-substituted alkyl, --SO-aryl, --SO-heteroaryl, --SO.sub.2-alkyl, --SO.sub.2-substituted alkyl, --SO.sub.2-aryl and --SO.sub.2-heteroaryl
[0140] R.sup.7 is --N(CH.sub.2).sub.2; --N.sup.+(CH.sub.2).sub.3; or
##STR00021##
[0141] Y is independently selected from the group consisting of oxygen, sulfur, --S--S--, --NR.sup.8--, --S(O)--, --SO.sub.2--, --OSO.sub.2--, --NR.sup.8SO.sub.2--, --SO.sub.2NR.sup.8--, --SO.sub.2O--, --P(O)(OR.sup.8)O--, --P(O)(OR.sup.8)NR.sup.8--, --OP(O)(OR.sup.8)O--, --OP(O)(OR.sup.8)NR.sup.8--, NR.sup.8C(O)NR.sup.1--, and --NR.sup.8SO.sub.2NR.sup.8--; and
[0142] each R.sup.8 is independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, heteroaryl and heterocyclic.
[0143] Compounds of Formula (I) and Formula (II) are synthesized, in one embodiment, by the methods provided in U.S. Pat. Nos. 6,455,669 and/or 7,160,984, the disclosure of each of which is incorporated by reference herein in their entireties. Further synthesis methods are provided in the Example section, herein. Other preparation steps and methods that can be employed are disclosed in U.S. Pat. No. 6,392,012; U.S. Patent Application Publication No. 2017/0152291; U.S. Patent Application Publication No. 2016/0272682, each of which is hereby incorporated by reference in their entirety for all purposes. Methods described in International Publication No. WO 2018/08197, the disclosure of which is incorporated by reference in its entirety, can also be employed. Synthesis schemes are also provided at the Example section, herein.
[0144] In one embodiment, compounds of Formula (I) and Formula (II), e.g., where R.sup.1 is
##STR00022##
and R.sup.2 is OH, are synthesized according to the methods provided in U.S. Patent Application Publication No. 2017/0152291, the disclosure of which is incorporated by reference in its entirety.
[0145] In embodiments, where R.sup.2 is --NH--(CH.sub.2).sub.q--R.sup.7, the amide coupling can be carried out as described in Yarlagadda et al. (2014). J. Med. Chem. 57, pp. 4558-4568, the disclosure of which is incorporated by reference herein in its entirety for all purposes. For example, a solution of vancomycin or other glycopeptide derivative (e.g., a compound of Formula (I) or Formula (II), where R.sup.1 is
##STR00023##
and X is O) can be treated with a solution of --NH--(CH.sub.2).sub.q--R.sup.7 (e.g., a solution of --NH--(CH.sub.2).sub.3--N(CH.sub.2).sub.2, --NH--(CH.sub.2).sub.3--N.sup.+(CH.sub.2).sub.3, or
##STR00024##
N-methylmorpholine and HBTU at 25.degree. C. The reaction mixture can be stirred at 25.degree. C. for 5 min and quenched with the addition of 50% MeOH in H.sub.2O at 25.degree. C. The mixture can be purified by semi-preparative reverse-phase HPLC to afford the compound as a white film.
[0146] In one embodiment, the method for treating a bacterial infection comprises administering to the patient in need thereof, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), where R.sup.1 does not include a physiologically cleavable functional group. Stated another way, the R.sup.1 group, in one embodiment, is not subject to hydrolysis or enzymatic cleavage in vivo.
[0147] In another embodiment, the method for treating a bacterial infection comprises administering to the patient in need thereof, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), where R.sup.1 does not include an amide or ester moiety.
[0148] In one embodiment, the method for treating a bacterial infection comprises administering to the patient in need thereof, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), where R.sup.1 is R.sup.5--Y--R.sup.6--(Z).sub.n. In a further embodiment, R.sup.5 is --(CH.sub.2).sub.2--, R.sup.6 is --(CH.sub.2).sub.10--, X is O, Y is NR, Z is hydrogen and n is 1. In a further embodiment, R.sup.8 is hydrogen. As such, one embodiment of the method provided herein includes delivering to a patient a composition comprising an effective amount of a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), where R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3. In a further embodiment, X is O, R.sup.2 is OH and R.sup.3 and R.sup.4 are H. In even a further embodiment, administration is via the intravenous or pulmonary route.
[0149] In one embodiment of the method, a patient is administered a composition comprising an effective amount of a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), where R.sup.1 is --CH.sub.2--NH--(CH.sub.2).sub.10--CH.sub.3. In a further embodiment, X is O, R.sup.2 is OH and R.sup.3 and R.sup.4 are H. In even a further embodiment, administration is via the intravenous or pulmonary route.
[0150] In one embodiment of the method, a patient is administered a composition comprising an effective amount of a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), or a pharmaceutically acceptable salt thereof, where R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.10--CH.sub.3. In a further embodiment, X is O, R.sup.2 is OH and R.sup.3 and R.sup.4 are H. In even a further embodiment, administration is via the intravenous or pulmonary route.
[0151] In one embodiment of the method, a patient is administered a composition comprising an effective amount of a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), where R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.11--CH.sub.3. In a further embodiment, X is O, R.sup.2 is OH and R.sup.3 and R.sup.4 are H. In even a further embodiment, administration is via the intravenous or pulmonary route.
[0152] In another embodiment, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), is administered to the patient in need thereof, where R.sup.1 is
##STR00025##
X is O or H.sub.2; and R.sup.2 is --NH--(CH.sub.2).sub.q--R.sup.7. In a further embodiment, R.sup.2 is --NH--(CH.sub.2).sub.3--R.sup.7. In a further embodiment, R.sup.1 is
##STR00026##
and R.sup.7 is --N.sup.+(CH.sub.2).sub.3 or --N(CH.sub.2).sub.2.
[0153] In yet another embodiment, R is C.sub.10-C.sub.16 alkyl. In even a further embodiment, R.sup.1 is C.sub.10 alkyl.
[0154] In yet another embodiment, a composition comprising an effective amount of a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), is delivered to the patient, where R.sup.2 is OH, R.sup.3 and R.sup.4 are H and X is O. In a further embodiment, R.sup.1 is
##STR00027##
or R.sup.5--Y--R.sup.6--(Z).sub.n. In even a further embodiment, R.sup.1 is R.sup.5--Y--R.sup.6--(Z).sub.1, R.sup.5 is methylene, ethylene or propylene; R.sup.6 is --(CH.sub.2).sub.9--, --(CH.sub.2).sub.10--, --(CH.sub.2).sub.11--, or --(CH.sub.2).sub.12--, Z is H and n is 1.
[0155] In yet another embodiment, of the bacterial infection treatment methods, an effective amount of a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II) is provided, wherein one or more hydrogen atoms is replaced with a deuterium atom.
[0156] In one embodiment, the method for treating the bacterial infection comprises administering to the patient in need thereof, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), where R.sup.1 is R.sup.5--Y--R.sup.6--(Z).sub.n. In a further embodiment, R.sup.5 is --(CH.sub.2).sub.2--, R.sup.6 is --(CH.sub.2).sub.10--, Y is NR.sup.8, Z is hydrogen and n is 1. In a further embodiment, R.sup.8 is hydrogen.
[0157] In one embodiment of the methods provided herein, R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3.
[0158] Exemplary embodiments of the compound of Formula (I) or Formula (II), for use in methods of treating bacterial infections, are provided in Table 1, below. It should be noted that the compound can also be provided as a pharmaceutically acceptable salt. The compounds in Table 1 are identified by their respective R.sup.1, R.sup.2 and X groups. Compounds of Table 1, in one embodiment, are defined as having R.sup.3 and R.sup.4 as both H. In another embodiment, a compound of Table 1 is administered, where R.sup.3 is
##STR00028##
and R.sup.4 is H. In yet another embodiment, a compound of Table 1 is administered, where R.sup.3 is H and R.sup.4 is CH.sub.2--NH--CH.sub.2--PO.sub.3H.sub.2. In even another embodiment, a compound of Table 1 is administered, where R.sup.3 is
##STR00029##
and R.sup.4 is CH.sub.2--NH--CH.sub.2--PO.sub.3H.sub.2.
TABLE-US-00001 TABLE 1 Exemplary compounds of Formula (I) or Formula (II) for use with the invention. Com- pound # R.sup.2 X R.sup.1 1. OH O --(CH.sub.2).sub.5--CH.sub.3 (n-hexyl) 2. OH O --(CH.sub.2).sub.6--CH.sub.3 (n-heptyl) 3. OH O --(CH.sub.2).sub.7--CH.sub.3 (n-octyl) 4. OH O --(CH.sub.2).sub.8--CH.sub.3 (n-nonyl) 5. OH O --(CH.sub.2).sub.9--CH.sub.3 (n-decyl) 6. OH O --(CH.sub.2).sub.10--CH.sub.3 (n-undecyl) 7. OH O --(CH.sub.2).sub.11--CH.sub.3 (n-dodecyl) 8. OH O --(CH.sub.2).sub.12--CH.sub.3 (n-tridecyl) 9. OH O --(CH.sub.2).sub.13--CH.sub.3 (n-butadecyl) 10. OH O --(CH.sub.2).sub.14--CH.sub.3 (n-pentadecyl) 11. OH O --(CH.sub.2).sub.15--CH.sub.3 (n-hexadecyl) 12. OH O --(CH.sub.2).sub.16--CH.sub.3 (n-heptadecyl) 13. OH O --(CH.sub.2).sub.17--CH.sub.3 (n-octadecyl) 14. OH O --CH.sub.2--NH--(CH.sub.2).sub.5--CH.sub.3 15. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.5--CH.sub.3 16. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.6--CH.sub.3 17. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.7--CH.sub.3 18. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.8--CH.sub.3 19. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.9--CH.sub.3 20. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.10--CH.sub.3 21. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.11--CH.sub.3 22. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.12--CH.sub.3 23. OH O --(CH.sub.2).sub.2--OSO.sub.2--(CH.sub.2).sub.5--CH.sub.3 24. OH O --(CH.sub.2).sub.2--OSO.sub.2--(CH.sub.2).sub.6--CH.sub.3 25. OH O --(CH.sub.2).sub.2--OSO.sub.2--(CH.sub.2).sub.7--CH.sub.3 26. OH O --(CH.sub.2).sub.2--OSO.sub.2--(CH.sub.2).sub.8--CH.sub.3 27. OH O --(CH.sub.2).sub.2--OSO.sub.2--(CH.sub.2).sub.9--CH.sub.3 28. OH O --(CH.sub.2).sub.2--OSO.sub.2--(CH.sub.2).sub.10--CH.sub.3 29. OH O --(CH.sub.2).sub.2--OSO.sub.2--(CH.sub.2).sub.11--CH.sub.3 30. OH O --(CH.sub.2).sub.2--OSO.sub.2--(CH.sub.2).sub.12--CH.sub.3 31. OH O --(CH.sub.2).sub.2--OSO.sub.2--(CH.sub.2).sub.13--CH.sub.3 32. OH O --(CH.sub.2).sub.2--OSO.sub.2--(CH.sub.2).sub.14--CH.sub.3 33. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.2--CH.sub.3 34. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.3--CH.sub.3 35. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.4--CH.sub.3 36. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.5--CH.sub.3 37. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.6--CH.sub.3 38. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.7--CH.sub.3 39. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.8--CH.sub.3 40. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3 41. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.10--CH.sub.3 42. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.11--CH.sub.3 43. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.12--CH.sub.3 44. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.13--CH.sub.3 45. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.14--CH.sub.3 46. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.5--CH.sub.3 47. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.6--CH.sub.3 48. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.7--CH.sub.3 49. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.8--CH.sub.3 50. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.9--CH.sub.3 51. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.10--CH.sub.3 52. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.11--CH.sub.3 53. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.12--CH.sub.3 54. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.13--CH.sub.3 55. OH O --(CH.sub.2).sub.2--C(O)O--(CH.sub.2).sub.5--CH.sub.3 56. OH O --(CH.sub.2).sub.2--C(O)O--(CH.sub.2).sub.6--CH.sub.3 57. OH O --(CH.sub.2).sub.2--C(O)O--(CH.sub.2).sub.7--CH.sub.3 58. OH O --(CH.sub.2).sub.2--C(O)O--(CH.sub.2).sub.8--CH.sub.3 59. OH O --(CH.sub.2).sub.2--C(O)O--(CH.sub.2).sub.9--CH.sub.3 60. OH O --(CH.sub.2).sub.2--C(O)O--(CH.sub.2).sub.10--CH.sub.3 61. OH O --(CH.sub.2).sub.2--C(O)O--(CH.sub.2).sub.11--CH.sub.3 62. OH O --(CH.sub.2).sub.2--C(O)O--(CH.sub.2).sub.12--CH.sub.3 63. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.5--CH.sub.3 64. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.6--CH.sub.3 65. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.7--CH.sub.3 66. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.8--CH.sub.3 67. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.9--CH.sub.3 68. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.10--CH.sub.3 69. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.11--CH.sub.3 70. OH O --(CH.sub.2).sub.2--NHSO.sub.2--(CH.sub.2).sub.12--CH.sub.3 71. OH O --(CH.sub.2).sub.2--NHC(O)--(CH.sub.2).sub.5--CH.sub.3 72. OH O --(CH.sub.2).sub.2--NHC(O)--(CH.sub.2).sub.6--CH.sub.3 73. OH O --(CH.sub.2).sub.2--NHC(O)--(CH.sub.2).sub.7--CH.sub.3 74. OH O --(CH.sub.2).sub.2--NHC(O)--(CH.sub.2).sub.8--CH.sub.3 75. OH O --(CH.sub.2).sub.2--NHC(O)--(CH.sub.2).sub.9--CH.sub.3 76. OH O --(CH.sub.2).sub.2--NHC(O)--(CH.sub.2).sub.10--CH.sub.3 77. OH O --(CH.sub.2).sub.2--NHC(O)--(CH.sub.2).sub.11--CH.sub.3 78. OH O --(CH.sub.2).sub.2--NHC(O)--(CH.sub.2).sub.12--CH.sub.3 79. OH O ##STR00030## 80. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.6--CH.sub.3 81. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.7--CH.sub.3 82. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.8--CH.sub.3 83. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.9--CH.sub.3 84. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.10--CH.sub.3 85. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.11--CH.sub.3 86. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.12--CH.sub.3 87. OH O --(CH.sub.2).sub.2--C(O)NH--(CH.sub.2).sub.5--CH.sub.3 88. OH O --(CH.sub.2).sub.2--C(O)NH--(CH.sub.2).sub.6--CH.sub.3 89. OH O --(CH.sub.2).sub.2--C(O)NH--(CH.sub.2).sub.7--CH.sub.3 90. OH O --(CH.sub.2).sub.2--C(O)NH--(CH.sub.2).sub.8--CH.sub.3 91. OH O --(CH.sub.2).sub.2--C(O)NH--(CH.sub.2).sub.9--CH.sub.3 92. OH O --(CH.sub.2).sub.2--C(O)NH--(CH.sub.2).sub.10--CH.sub.3 93. OH O --(CH.sub.2).sub.2--C(O)NH--(CH.sub.2).sub.11--CH.sub.3 94. OH O --(CH.sub.2).sub.2--C(O)NH--(CH.sub.2).sub.12--CH.sub.3 95. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.5--CH.sub.3 96. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.6--CH.sub.3 97. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.7--CH.sub.3 98. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.8--CH.sub.3 99. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.9--CH.sub.3 100. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.10--CH.sub.3 101. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.11--CH.sub.3 102. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.12--CH.sub.3 103. OH O --(CH.sub.2).sub.3--NH--(CH.sub.2).sub.5--CH.sub.3 104. OH O --(CH.sub.2).sub.3--NH--(CH.sub.2).sub.6--CH.sub.3 105. OH O --(CH.sub.2).sub.3--NH--(CH.sub.2).sub.7--CH.sub.3 106. OH O --(CH.sub.2).sub.3--NH--(CH.sub.2).sub.8--CH.sub.3 107. OH O --(CH.sub.2).sub.3--NH--(CH.sub.2).sub.9--CH.sub.3 108. OH O --(CH.sub.2).sub.3--NH--(CH.sub.2).sub.10--CH.sub.3 109. OH O --(CH.sub.2).sub.3--NH--(CH.sub.2).sub.11--CH.sub.3 110. OH O --(CH.sub.2).sub.3--NH--(CH.sub.2).sub.12--CH.sub.3 111. OH O --(CH.sub.2).sub.4--NH--(CH.sub.2).sub.5--CH.sub.3 112. OH O --(CH.sub.2).sub.4--NH--(CH.sub.2).sub.6--CH.sub.3 113. OH O --(CH.sub.2).sub.4--NH--(CH.sub.2).sub.7--CH.sub.3 114. OH O --(CH.sub.2).sub.4--NH--(CH.sub.2).sub.8--CH.sub.3 115. OH O --(CH.sub.2).sub.4--NH--(CH.sub.2).sub.9--CH.sub.3 116. OH O --(CH.sub.2).sub.4--NH--(CH.sub.2).sub.10--CH.sub.3 117. OH O --(CH.sub.2).sub.4--NH--(CH.sub.2).sub.11--CH.sub.3 118. OH O --(CH.sub.2).sub.4--NH--(CH.sub.2).sub.12--CH.sub.3 119. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.5--CH.sub.3 120. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.6--CH.sub.3 121. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.7--CH.sub.3 122. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.8--CH.sub.3 123. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.9--CH.sub.3 124. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.10--CH.sub.3 125. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.11--CH.sub.3 126. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.12--CH.sub.3 127. OH O --(CH.sub.2).sub.2--N[(CH.sub.2).sub.9CH.sub.3].sub.2 128. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.5--CH(CH.sub.3).sub.2 129. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.6--CH(CH.sub.3).sub.2 130. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.7--CH(CH.sub.3).sub.2 131. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.8--CH(CH.sub.3).sub.2 132. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.9--CH(CH.sub.3).sub.2 133. OH O --(CH.sub.2).sub.5--NH--(CH.sub.2).sub.10--CH(CH.sub.3).sub.2 134. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--Ph 135. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.2--Ph 136. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.3--Ph 137. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.4--Ph 138. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.5--Ph 139. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.6--Ph 140. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.7--Ph 141. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.8--Ph 142. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4- [(CH.sub.3).sub.2CHCH.sub.2--]Ph 143. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-(Ph--S--)Ph 144. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-(4-CF.sub.3--Ph)Ph 145. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-{4- [CH.sub.3(CH.sub.2).sub.4O--]--Ph}--Ph 146. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-Cl--Ph 147. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.2--4-Cl--Ph 148. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.3--4-Cl--Ph 149. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.4--4-Cl--Ph 150. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.5--4-Cl--Ph 151. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.6--4-Cl--Ph 152. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.7--4-Cl--Ph 153. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.8--4-Cl--Ph 154. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-Ph--Ph 155. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-(4-Cl--Ph)--Ph 156. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3(CH.sub.2).sub.2O--- ]Ph 157. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3(CH.sub.2).sub.3O--- ]Ph 158. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3(CH.sub.2).sub.4O--- ]Ph 159. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3(CH.sub.2).sub.5O--- ]Ph 160. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3(CH.sub.2).sub.6O--- ]Ph 161. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3(CH.sub.2).sub.7O--- ]Ph 162. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3(CH.sub.2).sub.8O--- ]Ph 163. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3(CH.sub.2).sub.2--]- Ph 164. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3(CH.sub.2).sub.3--]- Ph 165. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3(CH.sub.2).sub.4--]- Ph 166. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3(CH.sub.2).sub.5--]- Ph 167. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3(CH.sub.2).sub.6--]- Ph 168. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--3-[Ph--S--]Ph 169. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[Ph--O--]Ph 170. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3CH.sub.2Ph--O--]Ph 171. OH O --(CH.sub.2).sub.2--NH--CH.sub.2-- 4-[CH.sub.3(CH.sub.2).sub.2Ph--O--]Ph 172. OH O --(CH.sub.2).sub.2--NH--CH.sub.2-- 4-[CH.sub.3(CH.sub.2).sub.3Ph--O--]Ph 173. OH O --(CH.sub.2).sub.2--NH--CH.sub.2-- 4-[CH.sub.3(CH.sub.2).sub.4Ph--O--]Ph 174. OH O --(CH.sub.2).sub.2--NH--CH.sub.2-- 4-[CH.sub.3(CH.sub.2).sub.5Ph--O--]Ph 175. OH O --(CH.sub.2).sub.2--NH--CH.sub.2-- 4-[CH.sub.3(CH.sub.2).sub.6Ph--O--]Ph 176. OH O --(CH.sub.2).sub.2--NH--CH.sub.2-- 4-[CH.sub.3(CH.sub.2).sub.7Ph--O--]Ph 177. OH O --(CH.sub.2).sub.2--NH--CH.sub.2-- 4-[CH.sub.3(CH.sub.2).sub.8Ph--O--]Ph 178. OH O --(CH.sub.2).sub.2--NH--CH.sub.2-- 4-[CH.sub.3(CH.sub.2).sub.9Ph--O--]Ph 179. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--3-[Ph--S--]Ph 180. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[Ph--S--]Ph 181. OH O --(CH.sub.2).sub.2--NH--CH.sub.2-cyclopropyl 182. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.2-cyclopropyl 183. OH O --(CH.sub.2).sub.2--NH--CH.sub.2-cyclopentyl 184. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.2-cyclopentyl 185. OH O --(CH.sub.2).sub.2--NH--CH.sub.2-cyclohexyl 186. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.2-cyclohexyl 187. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.8--CH.dbd.CH.sub.2 188. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.8--CH(OH)--CH.sub.3 189. OH O --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.3CH.dbd.CH(CH.sub.2).sub.- 4CH.sub.3 (trans) 190. OH O --(CH.sub.2).sub.2--NH--CH.sub.2CH.dbd.C(CH.sub.3)(CH.sub.2).sub- .2-- CH.dbd.C(CH.sub.3).sub.2 (trans, trans) 191. OH O --(CH.sub.2).sub.2--NHC(O)--(CH.sub.2).sub.6--CH(CH.sub.3)CH.sub- .3 192. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.8Ph 193. OH O --(CH.sub.2).sub.2NH--CH.sub.2--4-[(CH.sub.3).sub.3CO]--Ph 194. OH O --(CH.sub.2).sub.2--S--(CHO).sub.3CH.ident.CH(CH.sub.2).sub.4CH.- sub.3 (trans) 195. OH O --(CH.sub.2).sub.2NH--CH.sub.2--3,4-di-(CH.sub.3CH.sub.2O)--Ph 196. OH O --(CH.sub.2).sub.2--S--CH.sub.2CH.sub.2(CF.sub.2).sub.5CF.sub.3 197. OH O --(CH.sub.2).sub.2NH--CH.sub.2--4-[(CH.sub.3).sub.2CH]--Ph 198. OH O --(CH.sub.2).sub.2--S--CH.sub.2--4-[(CH.sub.3).sub.2CHCH.sub.2--- ]Ph 199. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[CH.sub.3(CH.sub.2).sub.3C.i- dent.C]--Ph 200. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.11CH.sub.3 201. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[(CH.sub.3).sub.2CHO]--Ph 202. OH O --(CH.sub.2).sub.2--S--(CH.sub.2)8CH.sub.3 203. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-(Ph--.ident.C)--Ph 204. OH O --(CH.sub.2).sub.2--S--CH.sub.23,4-di(PhCH.sub.2O--)Ph 205. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[(CH.sub.3).sub.3C]--Ph 206. OH O --(CH.sub.2).sub.3--S--(CH.sub.2).sub.8Ph 207. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--5-(PhC.ident.C)- thiophen-2-yl 208. OH O --(CH.sub.2).sub.3--S--(CH.sub.2).sub.8CH.sub.3
209. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-(PhCH.ident.CH--)Ph (trans) 210. OH O --(CH.sub.2).sub.3--S--(CH.sub.2).sub.9CH.sub.3 211. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--(CH.ident.CH).sub.4--CH.sub.3 (trans, trans, trans, trans) 212. OH O --(CH.sub.2).sub.3--S--(CH.sub.2).sub.6Ph 213. OH O --(CH.sub.2).sub.2--N(C(O)Ph)--(CH.sub.2).sub.9CH.sub.3 214. OH O --(CH.sub.2).sub.4--S--(CH.sub.2).sub.7CH.sub.3 215. OH O --(CH.sub.2).sub.2--NH--CH.sub.2--4-[4-(CH.sub.3).sub.3C- thiozol-2-yl]--Ph 216. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.6Ph 217. OH O --(CH.sub.2).sub.2--N[(CH.sub.2).sub.9CH.sub.3]-- C(O)CHrS-4-pyridyl 218. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.10Ph 219. OH O --(CH.sub.2).sub.2--N[(CH.sub.2).sub.9CH.sub.3]-- C(O)-2-[PhCH(CH.sub.3)NHC(O)--]Ph (R isomer) 220. OH O --(CH.sub.2).sub.3--S--CH.sub.2-- 4-[(CH.sub.3).sub.2CHCH.sub.2--]Ph 221. OH O --(CH.sub.2).sub.2--N[(CH.sub.2).sub.9CH.sub.3]-- C(O)-(1-PhCH.sub.2OC(O)--2- oxoimidazolidin-5-yl) (S isomer) 222. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.3CH.ident.CH(CH.sub.2).sub- .4CH.sub.3 (trans) 223. OH O --(CH.sub.2).sub.2--N[(CH.sub.2).sub.9CH.sub.3]-- C(O)-1-HO-cyclopropyl 224. OH O --(CH.sub.2).sub.2--S--CH.sub.2-4-[3,4- di-Cl--PhCH.sub.2O--]Ph 225. OH O --(CH.sub.2).sub.2--N(C(O)CH.sub.2-- naphth-2-yl)--(CH.sub.2).sub.9CH 226. OH O --(CH.sub.2).sub.3--S--CH.sub.2-4-[3,4- di-Cl--PhCH.sub.2O--]Ph 227. OH O --(CH.sub.2).sub.2--N[C(O)(CH.sub.2).sub.9CH.sub.2OH]--(CH.sub.2- ).sub.9CH.sub.3 228. OH O --(CH.sub.2).sub.2--SO--4-(4-Cl--Ph)--Ph 229. OH O --CH.sub.2CH.sub.2--N[C(O)CH.sub.2(OCH.sub.2CH.sub.2).sub.2 OCH.sub.3]--(CH.sub.2).sub.9CH.sub.3 230. OH O --(CH.sub.2).sub.3--SO--4-(4-Cl--Ph)--Ph 231. OH O --(CH.sub.2).sub.2--N[C(O)CH.sub.2CH(Ph).sub.2]-- (CH.sub.2).sub.9CH.sub.3 232. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.10CH.sub.3 233. OH O --(CH.sub.2).sub.2--N(C(O)CH.sub.2--3-HO--Ph)-- (CH.sub.2).sub.9CH.sub.3 234. OH O --(CH.sub.2).sub.3--S--(CH.sub.2).sub.10CH.sub.3 235. OH O --(CH.sub.2).sub.2--N(C(O)CH.sub.2--NHC(O)--3- CH.sub.3--Ph)--(CH.sub.2).sub.9CH.sub.3 236. OH O --(CH.sub.2).sub.3--S--CH.sub.2-4-(CH.sub.3(CH.sub.2).sub.4O--]P- h 237. OH O --(CH.sub.2).sub.2--N(C(O)CH.sub.2CH.sub.2--O--Ph)-- (CH.sub.2).sub.9CH.sub.3 238. OH O --(CH.sub.2).sub.3--S--CH.sub.2CH.ident.CH--CH.ident. CH(CH.sub.2).sub.4CH.sub.3(trans, trans) 239. OH O --(CH.sub.2).sub.2--N(C(O)CH.sub.2CH.sub.2-- 3-pyridyl)--(CH.sub.2).sub.9CH.sub.3 240. OH O --(CH.sub.2).sub.2--S--CHr4-[4-Cl--PhCH.sub.2O--]Ph 241. OH O --(CH.sub.2).sub.2--N(C(O)(CH.sub.2).sub.3-- 4-CH.sub.3O--Ph)--(CH.sub.2).sub.9CH.sub.3 242. OH O --(CH.sub.2).sub.3--S--CH.sub.24-[4-Cl--PhCH.sub.2O--]Ph 243. OH O --(CH.sub.2).sub.2--N(C(O)-indol-2- yl)-(CH.sub.2).sub.9CH.sub.3 244. OH O --(CH.sub.2).sub.3--S--CH.sub.2-4-(4-CF.sub.3--Ph--)Ph 245. OH O --(CH.sub.2).sub.2--N{C(O)-1- [CH.sub.3COC(O)--]-pyrrolidin-2-yl}-(CH.sub.2).sub.9CH.sub.3 246. OH O --(CH.sub.2).sub.3--S--CH.sub.2-4- (4-F--PhSO.sub.2NH--)Ph 247. OH O --(CH.sub.2).sub.2--N(C(O)CH.sub.2--NHC(O)--CH.dbd.CH- furan-2-yl)-(CH.sub.2).sub.9CH.sub.3 (trans) 248. OH O --(CH.sub.2).sub.3--S--(CH.sub.2).sub.8CH.sub.3 249. OH O --(CH.sub.2).sub.2--N[C(O)-1-CH.sub.3CHr 7-CH.sub.3-4-oxo- 1,4-dihydro[1,8]naphthyridin-3-yl]-(CH.sub.2).sub.9CH.sub.3 250. OH O --(CH.sub.2).sub.3--S(O)--(CH.sub.2).sub.6Ph 251. OH O --(CH.sub.2).sub.2--N(C(O)-1,3-benzodioxol-5-yl)- (CH.sub.2).sub.9CH.sub.3 252. OH O --(CH.sub.2).sub.2--S(O)--(CH.sub.2).sub.8Ph 253. OH O --(CH.sub.2).sub.2--N(C(O)CH.sub.2-4-oxo-2- thiooxothiazolidin-.sub.3-yl)-(CH.sub.2).sub.9CH.sub.3 254. OH O --(CH.sub.2).sub.2--S--(CH.sub.2).sub.3-4-Cl--Ph 255. OH O --(CH.sub.2).sub.2--N(C(O)-3,4,5-tri-HO-cyclohex-1-en- 1-yl)-(CH.sub.2).sub.9CH.sub.3 (R,S,R isomer) 256. OH O --(CH.sub.2).sub.2--S--(CH.sub.2)c4-Cl--Ph 257. OH O --(CH.sub.2).sub.2--N(C(O)CH.sub.2CH.sub.2C(O)NH.sub.2)-- (CH.sub.2).sub.9CH.sub.3 258. OH O --(CH.sub.2).sub.2--SO.sub.2--(CH.sub.2).sub.9CH.sub.3 259. OH O --(CH.sub.2).sub.2--N(C(O)(CH.sub.2)5--CH.sub.3-2,4-dioxo- 3,4-dihydropyrimidin-1-yl)-(CH.sub.2).sub.9CH.sub.3 260. OH O --(CH.sub.2).sub.2--NHC(O)--CH.sub.2CH(CH.sub.2CH.sub.2Ph)-- {3-[4-(9H-flouren-9-ylCH.sub.2OC(O)NH(CH.sub.2).sub.4--]- 1,4-dioxohexahydro-1,2-.alpha.-pyrazin-2-yl} (S,S,S isomer) 261. OH O --(CH.sub.2).sub.2--N(C(O)CH.ident.CH--imidazol-4-yl)- (CH.sub.2).sub.9CH.sub.3 (trans) 262. OH O --(CH.sub.2).sub.2--NHSO.sub.2--4-(2-Cl--Ph)--Ph 263. OH O --(CH.sub.2).sub.2--N[C(O)CH(CH.sub.2CH.sub.2C(O)NH.sub.2)-- NHC(O)O--CH.sub.2Ph]--(CH.sub.2).sub.9CH.sub.3(S isomer) 264. OH O --(CH.sub.2).sub.2--NHSO.sub.2--4-[4-(CH.sub.3).sub.3C--Ph]--Ph 265. OH O --(CH.sub.2).sub.2--N[C(O)CH(CH.sub.2OH)-- NHC(O)O--CH.sub.2Ph]--(CH.sub.2).sub.9CH.sub.3(S isomer) 266. OH O --(CH.sub.2).sub.2--NHSO.sub.2--4-[4-(Ph)--Ph--]Ph 267. OH O --(CH.sub.2).sub.2--N[C(O)CH[CH(OH)CH.sub.3] NH--C(O)O--CH.sub.2Ph]--(CH.sub.2).sub.9CH.sub.3(S isomer) 268. OH O --(CH.sub.2).sub.2--NH--4-(4-CF.sub.3--Ph)--Ph 269. OH O --(CH.sub.2).sub.2--N(C(O)CH.sub.2NHSO.sub.2-4- CH.sub.3--Ph)--(CH.sub.2).sub.9CH.sub.3(S isomer) 270. OH O --(CH.sub.2).sub.2--N(C(O)CH(NH.sub.2)CHr.sub.3-- HO--Ph)--(CH.sub.2).sub.9CH.sub.3 271. OH O --(CH.sub.2).sub.2--N(C(O)(CH.sub.2).sub.3--NH.sub.2)--(CH.sub.2- ).sub.9CH.sub.3 272. OH O --(CH.sub.2).sub.2--N(C(O)CH(NH.sub.2)CH.sub.3)-- (CH.sub.2).sub.9CH.sub.3(R isomer) 273. OH O --(CH.sub.2).sub.2--N(C(O)--pyrrolidin-2-yl)- (CH.sub.2).sub.9CH.sub.3 274. OH O --(CH.sub.2).sub.2--N[C(O)CH(CH.sub.2OH)NHC(O)-- CH.sub.3]--(CH.sub.2).sub.9CH.sub.3(S isomer) 275. OH O --(CH.sub.2).sub.2--N(C(O)--pyrrolidin-2-yl)- (CH.sub.2).sub.9CH.sub.3(R isomer) 276. OH O --(CH.sub.2).sub.2--N[C(O)CH(NHC(O)CH.sub.3-- (CH.sub.2).sub.3--NHC(NH)NH.sub.2]--(CH.sub.2).sub.9CH.sub.3(S isomer) 277. OH O --(CH.sub.2).sub.2--N(C(O)CH(NH.sub.2)(CH.sub.2)4- NH.sub.2)--(CH.sub.2).sub.9CH.sub.3(S isomer) 278. OH O --(CH.sub.2).sub.2--N(C(O)CH.sub.2NHC(O)CH.sub.3)-- (CH.sub.2).sub.9CH.sub.3 279. OH O --(CH.sub.2).sub.2--N(C(O)-5-oxopyrrolidin-2-yl)- (CH.sub.2).sub.9CH.sub.3(R isomer) 280. OH O --(CH.sub.2).sub.2--N(C(O)CH(CH.sub.3)OC(O)CH-- (NH.sub.2)(CH.sub.3)--(CH.sub.2).sub.9CH.sub.3(R,R isomer) 281. --N(CH.sub.2).sub.2 O ##STR00031## 282. --N(CH.sub.2).sub.2 NH ##STR00032## 283. --N(CH.sub.2).sub.2 S ##STR00033## 284. --N(CH.sub.2).sub.2 H.sub.2 ##STR00034## 285. ##STR00035## O ##STR00036## 286. ##STR00037## NH ##STR00038## 287. ##STR00039## S ##STR00040## 288. ##STR00041## H.sub.2 ##STR00042## 289. --N.sup.+(CH.sub.2).sub.3 O ##STR00043## 290. --N.sup.+(CH.sub.2).sub.3 NH ##STR00044## 291. --N.sup.+(CH.sub.2).sub.3 S ##STR00045## 292. --N.sup.+(CH.sub.2).sub.3 H.sub.2 ##STR00046## 293. OH O ##STR00047## 294. OH O --(CH.sub.2).sub.2--OC(O)--(CH.sub.2).sub.5--CH.sub.3 Ph- phenyl
[0159] In one embodiment, the compound is a prodrug of Formula (I) or Formula (II), for example an alkyl ester prodrug. The alkyl group in one embodiment is a straight chain C.sub.1-C.sub.20 alkyl or a branched C.sub.1-C.sub.20 alkyl. The alkyl ester attachment can be at any oxygen in the molecule, determined by the user of the method.
[0160] In one embodiment of the invention, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), is delivered to a patient in need thereof, wherein, X is O, R.sup.1 is a C.sub.1-C.sub.18 linear alkyl, R.sup.2 is OH, and R.sup.3 and R.sup.4 are H.
[0161] R.sup.1 in a further embodiment is a C.sub.7-C.sub.17 linear alkyl; C.sub.7-C.sub.10 or C.sub.6-C.sub.10 linear alkyl.
[0162] In yet another embodiment, a compound of Formula (I), Formula (II) a prodrug thereof, or a pharmaceutically acceptable salt thereof, is delivered to a patient in need thereof, wherein, X is O, R.sup.1 is R.sup.5--Y--R.sup.6--(Z).sub.n, R.sup.2 is OH, and R.sup.3 and R.sup.4 are H.
[0163] In a further embodiment, R.sup.5 is --(CH.sub.2).sub.2--, R.sup.6 is --(CH.sub.2).sub.10--, Y is NR, Z is hydrogen and n is 1. In a further embodiment, R.sup.8 is hydrogen and X is O. In even a further embodiment, the administering is intravenous or via the pulmonary route.
[0164] In one embodiment of the invention, a compound of Formula (I), Formula (II) or a pharmaceutically acceptable salt thereof, is delivered to a patient in need of bacterial infection treatment, where R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3, X is O, R.sup.2 is --NH--(CH.sub.2).sub.q--R.sup.7, and R.sup.3 and R.sup.4 are H. In a further embodiment, q is 2 or 3 and R.sup.1 is --N(CH.sub.2).sub.2.
[0165] In one embodiment of the invention, a compound of Formula (I), Formula (II) or a pharmaceutically acceptable salt thereof, is delivered to a patient in need of bacterial infection treatment, where R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3, X is O, R.sup.2 is OH, R.sup.3 is
##STR00048##
and R.sup.4 is H.
[0166] In one embodiment, a compound of Formula (I), Formula (II) or a pharmaceutically acceptable salt thereof, is delivered to a patient in need of bacterial infection treatment, where R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3, X is O, R.sup.2 is OH, and R.sup.3 is H and R.sup.4 is CH.sub.2--NH--CH.sub.2--PO.sub.3H.sub.2.
[0167] In yet another embodiment, a compound of Formula (I) or Formula (II) is provided, wherein one or more hydrogen atoms is replaced with a deuterium atom. In a further embodiment, R.sup.2--Y--R.sup.3--(Z) is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3.
[0168] In one embodiment of the treatment methods provided herein, the compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), is defined as follows: R.sup.1 is (CH.sub.2).sub.n1--Y--(CH.sub.2).sub.n2--CH.sub.3, R.sup.2 is OH, R.sup.3 and R.sup.4 are H, n2 is an integer selected from 1 to 6 and n3 is an integer from 1 to 15. In a further embodiment, X is O. In even a further embodiment, the administering is intravenous or via the pulmonary route.
[0169] In one embodiment, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), is delivered to a patient in need of bacterial infection treatment, where R.sup.1 is (CH.sub.2)--Y--(CH.sub.2).sub.n2--CH.sub.3, R.sup.2 is OH, R.sup.3 and R.sup.4 are H and X is O. In a further embodiment, Y is oxygen, sulfur, --S--S--, --NH--, --S(O)-- or --SO.sub.2--. In a further embodiment, Y is --NH--.
[0170] In one embodiment, a compound of Formula (I), or a pharmaceutically acceptable salt thereof, is delivered to a patient in need of bacterial infection treatment, where R.sup.1 is (CH.sub.2).sub.2--Y--(CH.sub.2).sub.n2--CH.sub.3, R.sup.2 is OH, R.sup.3 and R.sup.4 are H and X is O. In a further embodiment, Y is oxygen, sulfur, --S--S--, --NH--, --S(O)-- or --SO.sub.2--. In a further embodiment, Y is --NH--.
[0171] In one embodiment, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), is delivered to a patient in need of bacterial infection treatment, where R.sup.1 is (CH.sub.2).sub.3--Y--(CH.sub.2).sub.2--CH.sub.3, R.sup.2 is OH, R.sup.3 and R.sup.4 are H and X is O. In a further embodiment, Y is oxygen, sulfur, --S--S--, --NH--, --S(O)-- or --SO.sub.2--. In a further embodiment, Y is --NH--.
[0172] In one embodiment, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), is delivered to a patient in need of bacterial infection treatment, where R.sup.1 is (CH.sub.2).sub.1-3--Y--(CH.sub.2).sub.8--CH.sub.3, R.sup.2 is OH, R.sup.3 and R.sup.4 are H and X is O. In a further embodiment, Y is oxygen, sulfur, --S--S--, --NH--, --S(O)-- or --SO.sub.2--. In a further embodiment, Y is --NH--.
[0173] In one embodiment, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), is delivered to a patient in need of bacterial infection treatment, where R.sup.1 is (CH.sub.2).sub.1-3--Y--(CH.sub.2).sub.9--CH.sub.3, R.sup.2 is OH, R.sup.3 and R.sup.4 are H and X is O. In a further embodiment, Y is oxygen, sulfur, --S--S--, --NH--, --S(O)-- or --SO.sub.2--. In a further embodiment, Y is --NH--.
[0174] In one embodiment, a compound of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II), is delivered to a patient in need of bacterial infection treatment, where R.sup.1 is (CH.sub.2).sub.2--Y--(CH.sub.2).sub.1--CH.sub.3, R.sup.2 is OH, R.sup.3 and R.sup.4 are H and X is O. In a further embodiment, Y is oxygen, sulfur, --S--S--, --NH--, --S(O)-- or --SO.sub.2--. In a further embodiment, Y is --NH--.
[0175] Compositions provided herein can be in the form of a solution, suspension or dry powder. Compositions can be administered by techniques known in the art, including, but not limited to intramuscular, intravenous, intratracheal, intranasal, intraocular, intraperitoneal, subcutaneous, and transdermal routes. In addition, as discussed throughout, the compositions can also be administered via the pulmonary route, e.g., via inhalation with a nebulizer or a dry powder inhaler.
[0176] In one embodiment, the composition provided herein comprises a plurality of nanoparticles of the antibiotic of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II) in association with a polymer. The mean diameter of the plurality of nanoparticles, in one embodiment, is from about 50 nm to about 900 nm, for example from about 10 nm to about 800 nm, from about 100 nm to about 700 nm, from about 100 nm to about 600 nm or from about 100 nm to about 500 nm.
[0177] In one embodiment, the plurality of nanoparticles comprise a biodegradable polymer and the antibiotic of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II). In a further embodiment, the biodegradable polymer is poly(D,L-lactide), poly(lactic acid) (PLA), poly(D,L-glycolide) (PLG), poly(lactide-co-glycolide) (PLGA), poly-(cyanoacrylate) (PCA), or a combination thereof.
[0178] In even a further embodiment, the biodegradable polymer is poly(lactic-co-glycolitic acid) (PLGA).
[0179] Nanoparticle compositions can be prepared according to methods known to those of ordinary skill in the art. For example, coacervation, solvent evaporation, emulsification, in situ polymerization, or a combination thereof can be employed (see, e.g., Soppimath et al. (2001). Journal of Controlled Release 70, pp. 1-20, incorporated by reference herein in its entirety for all purposes).
[0180] The amount of polymer in the composition can be adjusted, for example, to adjust the release profile of the compound of Formula from the composition.
[0181] In one embodiment, a dry powder composition disclosed in one of U.S. Pat. Nos. 5,874,064, 5,855,913 and/or U.S. Patent Application Publication No. 2008/0160092 is used to formulate one of the glycopeptides of Formula (I), Formula (II), or a pharmaceutically acceptable salt of Formula (I) or Formula (II). The disclosures of U.S. Pat. Nos. 5,874,064, 5,855,913 and U.S. Patent Application Publication No. 2008/0160092 are each incorporated by reference herein in their entireties for all purposes.
[0182] In one embodiment, the composition delivered via the methods provided herein are spray dried, hollow and porous particulate compositions. For example, the hollow and porous particulate compositions as disclosed in WO 1999/16419, hereby incorporated in its entirety by reference for all purposes, can be employed. Such particulate compositions comprise particles having a relatively thin porous wall defining a large internal void, although, other void containing or perforated structures are contemplated as well.
[0183] Compositions delivered via the methods provided herein, in one embodiment, yield powders with bulk densities less than 0.5 g/cm.sup.3 or 0.3 g/cm.sup.3, for example, less 0.1 g/cm3, or less than 0.05 g/cm.sup.3. By providing particles with very low bulk density, the minimum powder mass that can be filled into a unit dose container is reduced, which eliminates the need for carrier particles. Moreover, the elimination of carrier particles, without wishing to be bound by theory, can minimize throat deposition and any "gag" effect, since the large lactose particles can impact the throat and upper airways due to their size.
[0184] In some embodiments, the particulate compositions delivered via the methods provided herein comprise a structural matrix that exhibits, defines or comprises voids, pores, defects, hollows, spaces, interstitial spaces, apertures, perforations or holes. The particulate compositions in one embodiment, are provided in a "dry" state. That is, the particulate composition possesses a moisture content that allows the powder to remain chemically and physically stable during storage at ambient temperature and easily dispersible. As such, the moisture content of the microparticles is typically less than 6% by weight, and for example, less 3% by weight. In some embodiments, the moisture content is as low as 1% by weight. The moisture content is, at least in part, dictated by the formulation and is controlled by the process conditions employed, e.g., inlet temperature, feed concentration, pump rate, and blowing agent type, concentration and post drying.
[0185] Reduction in bound water can lead to improvements in the dispersibility and flowability of phospholipid based powders, leading to the potential for highly efficient delivery of powdered lung surfactants or particulate composition comprising active agent dispersed in the phospholipid.
[0186] The composition administered via the methods provided herein, in one embodiment, is a particulate composition comprising a compound of Formula (I) or Formula (II), a phospholipid and a polyvalent cation. In particular, the compositions of the present invention can employ polyvalent cations in phospholipid-containing, dispersible particulate compositions for pulmonary administration to the respiratory tract for local or systemic therapy via aerosolization.
[0187] Without wishing to be bound by theory, it is thought that the use of a polyvalent cation in the form of a hygroscopic salt such as calcium chloride stabilizes a dry powder prone to moisture induced stabilization. Without wishing to be bound by theory, it is thought that such cations intercalate the phospholipid membrane, thereby interacting directly with the negatively charged portion of the zwitterionic headgroup. The result of this interaction is increased dehydration of the headgroup area and condensation of the acyl-chain packing, all of which leads to increased thermodynamic stability of the phospholipids. Other benefits of such dry powder compositions are provided in U.S. Pat. No. 7,442,388, the disclosure of which is incorporated herein in its entirety for all purposes.
[0188] The polyvalent cation for use in the present invention in one embodiment, is a divalent cation. In a further embodiment, the divalent cation is calcium, magnesium, zinc or iron. The polyvalent cation is present in one embodiment, to increase the Tm of the phospholipid such that the particulate composition exhibits a Tm which is greater than its storage temperature Ts by at least 20.degree. C. The molar ratio of polyvalent cation to phospholipid in one embodiment, is 0.05, e.g., from about 0.05 to about 2.0, or from about 0.25 to about 1.0. In one embodiment, the molar ratio of polyvalent cation to phospholipid is about 0.50. In one embodiment, the polyvalent cation is calcium and is provided as calcium chloride.
[0189] According to one embodiment, the phospholipid is a saturated phospholipid. In a further embodiment, the saturated phospholipid is a saturated phosphatidylcholine. Acyl chain lengths that can be employed range from about C.sub.16 to C.sub.22. For example, in one embodiment an acyl chain length of 16:0 or 18:0 (i.e., palmitoyl and stearoyl) is employed. In one phospholipid embodiment, a natural or synthetic lung surfactant is provided as the phospholipid component. In this embodiment, the phospholipid can make up to 90 to 99.9% w/w of the lung surfactant. Suitable phospholipids according to this aspect of the invention include natural or synthetic lung surfactants such as those commercially available under the trademarks ExoSurf, InfaSurf.RTM. (Ony, Inc.), Survanta, CuroSurf, and ALEC.
[0190] The Tm of the phospholipid-glycopeptide particles, in one embodiment, is manipulated by varying the amount of polyvalent cations in the composition.
[0191] Phospholipids from both natural and synthetic sources are compatible with the compositions administered by the methods provided herein, and may be used in varying concentrations to form the structural matrix. Generally compatible phospholipids comprise those that have a gel to liquid crystal phase transition greater than about 40.degree. C. The incorporated phospholipids in one embodiment, are relatively long chain (i.e., C.sub.16-C.sub.22) saturated lipids and in a further embodiment, comprise saturated phospholipids. In even a further embodiment, the saturated phospholipid is a saturated phosphatidylcholine. In even a further embodiment, the saturated phosphatidylcholine has an acyl chain lengths of 16:0 or 18:0 (palmitoyl or stearoyl). Exemplary phospholipids useful in the disclosed stabilized preparations comprise, phosphoglycerides such as dipalmitoylphosphatidylcholine (DPPC), disteroylphosphatidylcholine (DSPC), diarachidoylphosphatidylcholine dibehenoylphosphatidylcholine, diphosphatidyl glycerol, short-chain phosphatidylcholines, long-chain saturated phosphatidylethanolamines, long-chain saturated phosphatidylserines, long-chain saturated phosphatidylglycerols, long-chain saturated phosphatidylinositols.
[0192] In addition to the phospholipid, a co-surfactant or combinations of surfactants, including the use of one or more in the liquid phase and one or more associated with the particulate compositions can be used in the compositions delivered via the methods provided herein. By "associated with or comprise" it is meant that the particulate compositions may incorporate, adsorb, absorb, be coated with or be formed by the surfactant. Surfactants include fluorinated and nonfluorinated compounds and can include saturated and unsaturated lipids, nonionic detergents, nonionic block copolymers, ionic surfactants and combinations thereof. In one embodiment comprising stabilized dispersions, nonfluorinated surfactants are relatively insoluble in the suspension medium.
[0193] Compatible nonionic detergents suitable as co-surfactants in the compositions provided herein include sorbitan esters including sorbitan trioleate (Span.TM. 85), sorbitan sesquioleate, sorbitan monooleate, sorbitan monolaurate, polyoxyethylene (20) (Brij.RTM. S20), sorbitan monolaurate, and polyoxyethylene (20) sorbitan monooleate, oleyl polyoxyethylene (2) ether, stearyl polyoxyethylene (2) ether, lauryl polyoxyethylene (4) ether, glycerol esters, and sucrose esters. Block copolymers include diblock and triblock copolymers of polyoxyethylene and polyoxypropylene, including poloxamer 188 (Pluronic.RTM. F-68), poloxamer 407 (Pluronic.RTM. F-127), and poloxamer 338. Ionic surfactants such as sodium sulfosuccinate, and fatty acid soaps may also be utilized.
[0194] The phospholipid-glycopeptide particulate compositions can include additional lipids such as a glycolipid, ganglioside GM1, sphingomyelin, phosphatidic acid, cardiolipin; a lipid bearing a polymer chain such as polyethylene glycol, chitin, hyaluronic acid, or polyvinylpyrrolidone; a lipid bearing sulfonated mono-, di-, and polysaccharides; a fatty acid such as palmitic acid, stearic acid, and/or oleic acid; cholesterol, cholesterol esters, and cholesterol hemisuccinate.
[0195] In addition to the phospholipid and polyvalent cation, the particulate composition delivered via the methods provided herein can also include a biocompatible, and in some embodiments, biodegradable polymer, copolymer, or blend or other combination thereof. The polymer in one embodiment is a polylactide, polylactide-glycolide, cyclodextrin, polyacrylate, methylcellulose, carboxymethylcellulose, polyvinyl alcohol, polyanhydride, polylactam, polyvinyl pyrrolidone, polysaccharide (e.g., dextran, starch, chitin, chitosan), hyaluronic acid, protein (e.g., albumin, collagen, gelatin, etc.).
[0196] Besides the aforementioned polymer materials and surfactants, other excipients can be added to a particulate composition, for example, to improve particle rigidity, production yield, emitted dose and deposition, shelf-life and/or patient acceptance. Such optional excipients include, but are not limited to: coloring agents, taste masking agents, buffers, hygroscopic agents, antioxidants, and chemical stabilizers. Other excipients may include, but are not limited to, carbohydrates including monosaccharides, disaccharides and polysaccharides. For example, monosaccharides such as dextrose (anhydrous and monohydrate), galactose, mannitol, D-mannose, sorbitol, sorbose and the like; disaccharides such as lactose, maltose, sucrose, trehalose, and the like; trisaccharides such as raffinose and the like; and other carbohydrates such as starches (hydroxyethylstarch), cyclodextrins and maltodextrins. Mixtures of carbohydrates and amino acids are further held to be within the scope of the present invention. The inclusion of both inorganic (e.g., sodium chloride), organic acids and their salts (e.g., carboxylic acids and their salts such as sodium citrate, sodium ascorbate, magnesium gluconate, sodium gluconate, tromethamine hydrochloride, etc.) and buffers can also be undertaken. Salts and/or organic solids such as ammonium carbonate, ammonium acetate, ammonium chloride or camphor can also be employed.
[0197] According to one embodiment, the particulate compositions may be used in the form of dry powders or in the form of stabilized dispersions comprising a non-aqueous phase. The dispersions or powders of the present invention may be used in conjunction with metered dose inhalers (MDIs), dry powder inhalers (DPIs), atomizers, or nebulizers to provide for pulmonary delivery.
[0198] While several procedures are generally compatible with making certain dry powder compositions described herein, spray drying is a particularly useful method. As is well known, spray drying is a one-step process that converts a liquid feed to a dried particulate form. With respect to pharmaceutical applications, it will be appreciated that spray drying has been used to provide powdered material for various administrative routes including inhalation. See, for example, M. Sacchetti and M. M. Van Oort in: Inhalation Aerosols: Physical and Biological Basis for Therapy, A. J. Hickey, ed. Marcel Dekkar, New York, 1996, which is incorporated herein by reference in its entirety for all purposes. In general, spray drying consists of bringing together a highly dispersed liquid, and a sufficient volume of hot air to produce evaporation and drying of the liquid droplets. The preparation to be spray dried or feed (or feed stock) can be any solution, suspension, slurry, colloidal dispersion, or paste that may be atomized using the selected spray drying apparatus. In one embodiment, the feed stock comprises a colloidal system such as an emulsion, reverse emulsion, microemulsion, multiple emulsion, particulate dispersion, or slurry. Typically, the feed is sprayed into a current of warm filtered air that evaporates the solvent and conveys the dried product to a collector. The spent air is then exhausted with the solvent.
[0199] It will further be appreciated that spray dryers, and specifically their atomizers, may be modified or customized for specialized applications, e.g., the simultaneous spraying of two solutions using a double nozzle technique. More specifically, a water-in-oil emulsion can be atomized from one nozzle and a solution containing an anti-adherent such as mannitol can be co-atomized from a second nozzle. In one embodiment, it may be desirable to push the feed solution though a custom designed nozzle using a high pressure liquid chromatography (HPLC) pump. Examples of spray drying methods and systems suitable for making the dry powders of the present invention are disclosed in U.S. Pat. Nos. 6,077,543, 6,051,256, 6,001,336, 5,985,248, and 5,976,574, each of which is incorporated in their entirety by reference for all purposes.
[0200] While the resulting spray-dried powdered particles typically are approximately spherical in shape, nearly uniform in size and frequently are hollow, there may be some degree of irregularity in shape depending upon the incorporated glycopeptide of Formula (I) or Formula (II) and the spray drying conditions. In one embodiment, an inflating agent (or blowing agent) is used in the spray-dried powder production, e.g., as disclosed in WO 99/16419, incorporated by reference herein in its entirety for all purposes. Additionally, an emulsion can be included with the inflating agent as the disperse or continuous phase. The inflating agent can be dispersed with a surfactant solution, using, for instance, a commercially available microfluidizer at a pressure of about 5000 to 15,000 PSI. This process forms an emulsion, and in some embodiments, an emulsion stabilized by an incorporated surfactant, and can comprise submicron droplets of water immiscible blowing agent dispersed in an aqueous continuous phase. The blowing agent in one embodiment, is a fluorinated compound (e.g., perfluorohexane, perfluorooctyl bromide, perfluorooctyl ethane, perfluorodecalin, perfluorobutyl ethane) which vaporizes during the spray-drying process, leaving behind generally hollow, porous aerodynamically light microspheres. Other suitable liquid blowing agents include nonfluorinated oils, chloroform, Freons, ethyl acetate, alcohols and hydrocarbons. Nitrogen and carbon dioxide gases are also contemplated as a suitable blowing agent. Perfluorooctyl ethane is the blowing agent, in one embodiment.
[0201] Whatever components are selected, the first step in particulate production in one embodiment, comprises feed stock preparation. The selected glycopeptide is dissolved in a solvent, for example water, dimethylformamide (DMF), dimethyl sulfoxide (DMSO), acetonitrile, ethanol, methanol, or combinations thereof, to produce a concentrated solution. The polyvalent cation may be added to the glycopeptide solution or may be added to the phospholipid emulsion as discussed below. The glycopeptide may also be dispersed directly in the emulsion, particularly in the case of water insoluble agents. Alternatively, the glycopeptide is incorporated in the form of a solid particulate dispersion. The concentration of the glycopeptide used is dependent on the amount of glycopeptide required in the final powder and the performance of the delivery device employed (e.g., the fine particle dose for a MDI or DPI). As needed, cosurfactants such as poloxamer 188 or span 80 may be dispersed into this annex solution. Additionally, excipients such as sugars and starches can also be added.
[0202] In one embodiment, a polyvalent cation-containing oil-in-water emulsion is then formed in a separate vessel. The oil employed in one embodiment, is a fluorocarbon (e.g., perfluorooctyl bromide, perfluorooctyl ethane, perfluorodecalin) which is emulsified with a phospholipid. For example, polyvalent cation and phospholipid may be homogenized in hot distilled water (e.g., 60.degree. C.) using a suitable high shear mechanical mixer (e.g., Ultra-Turrax model T-25 mixer) at 8000 rpm for 2 to 5 minutes. In one embodiment, 5 to 25 g of fluorocarbon is added dropwise to the dispersed surfactant solution while mixing. The resulting polyvalent cation-containing perfluorocarbon in water emulsion is then processed using a high pressure homogenizer to reduce the particle size. In one embodiment, the emulsion is processed at 12,000 to 18,000 PSI, 5 discrete passes and kept at 50 to 80.degree. C.
[0203] The glycopeptide solution (or suspension) and perfluorocarbon emulsion are then combined and fed into the spray dryer. In one embodiment, the two preparations are miscible. While the glycopeptide is solubilized separately for the purposes of the instant discussion it will be appreciated that, in other embodiments, the glycopeptide may be solubilized (or dispersed) directly in the emulsion. In such cases, the glycopeptide emulsion is simply spray dried without combining a separate glycopeptide preparation.
[0204] Operating conditions such as inlet and outlet temperature, feed rate, atomization pressure, flow rate of the drying air, and nozzle configuration can be adjusted in accordance with the manufacturer's guidelines in order to produce the desired particle size, and production yield of the resulting dry particles. The selection of appropriate apparatus and processing conditions are well within the purview of a skilled artisan. In one embodiment, the particulate composition comprises hollow, porous spray dried micro- or nano-particles.
[0205] Along with spray drying, particulate compositions useful in the present invention may be formed by lyophilization. Those skilled in the art will appreciate that lyophilization is a freeze-drying process in which water is sublimed from the composition after it is frozen. Methods for providing lyophilized particulates are known to those of skill in the art. The lyophilized cake containing a fine foam-like structure can be micronized using techniques known in the art.
[0206] Besides the aforementioned techniques, the glycopeptide particulate compositions or glycopeptide particles provided herein may be formed using a method where a feed solution (either emulsion or aqueous) containing wall forming agents is rapidly added to a reservoir of heated oil (e.g., perflubron or other high boiling FCs) under reduced pressure. The water and volatile solvents of the feed solution rapidly boils and are evaporated. In one embodiment, the wall forming agents are insoluble in the heated oil. The resulting particles can then separated from the heated oil using a filtering technique and then dried under vacuum.
[0207] In another embodiment, the particulate compositions of the present invention may also be formed using a double emulsion method. In the double emulsion method, the medicament is first dispersed in a polymer dissolved in an organic solvent (e.g., methylene chloride, ethyl acetate) by sonication or homogenization. This primary emulsion is then stabilized by forming a multiple emulsion in a continuous aqueous phase containing an emulsifier such as polyvinylalcohol. Evaporation or extraction using conventional techniques and apparatus then removes the organic solvent. The resulting particles are washed, filtered and dried prior to combining them with an appropriate suspension medium.
[0208] In order to maximize dispersibility, dispersion stability and optimize distribution upon administration, the mean geometric particle size of the particulate compositions in one embodiment, is from about 0.5-50 .mu.m, for example from about 0.5 .mu.m to about 10 .mu.m or from about 0.5 to about 5 .mu.m. In one embodiment, the mean geometric particle size (or diameter) of the particulate compositions is less than 20 .mu.m or less than 10 .mu.m. In a further embodiment, the mean geometric diameter is .ltoreq.about 7 .mu.m or .ltoreq.5 .mu.m. In even a further embodiment, the mass geometric diameter is .ltoreq.about 2.5 .mu.m. In one embodiment, the particulate composition comprises a powder of dry, hollow, porous spherical shells of from about 0.1 to about 10 .mu.m, e.g., from about 0.5 to about 5 .mu.m in diameter, with shell thicknesses of approximately 0.1 .mu.m to about 0.5 .mu.m.
[0209] In addition to the glycopeptides of Formula (I), Formula (II) or a pharmaceutically acceptable salt thereof, one or more additional antiinfectives can be included in the composition administered to the patient in need thereof, either in the same composition, or a different composition. Additional antiinfectives include an additional glycopeptide, for example, one of the glycopeptides described herein. Other additional antiinfectives include but are not limited to aminoglycosides (e.g., dibekacin, K-4619, sisomicin, amikacin, dactimicin, isepamicin, rhodestreptomycin, apramycin, etimicin, KA-5685, sorbistin, arbekacin, framycetin, kanamycin, spectinomycin, astromicin, gentamicin, neomycin, sporaricin, bekanamycin, H107, netilmicin, streptomycin, boholmycin, hygromycin, paromomycin, tobramycin, brulamycin, hygromycin B, plazomicin, verdamicin, capreomycin, inosamycin, ribostamycin, vertilmicin), tetracyclines (e.g., chlortetracycline, oxytetracycline, methacycline, doxycycline, minocycline), sulfonamides (e.g., sulfanilamide, sulfadiazine, sulfamethaoxazole, sulfisoxazole, sulfacetamide), paraaminobenzoic acid, diaminopyrimidines (e.g., trimethoprim), quinolones (e.g., nalidixic acid, cinoxacin, ciprofloxacin and norfloxacin), penicillins (e.g., penicillin G, penicillin V, ampicillin, amoxicillin, bacampicillin, carbenicillin, carbenicillin indanyl, ticarcillin, azlocillin, mezlocillin, piperacillin), penicillinase resistant penicillin (e.g., methicillin, oxacillin, cloxacillin, dicloxacillin, nafcillin), first generation cephalosporins (e.g., cefadroxil, cephalexin, cephradine, cephalothin, cephapirin, cefazolin), second generation cephalosporins (e.g., cefaclor, cefamandole, cefonicid, cefoxitin, cefotetan, cefuroxime, cefuroxime axetil; cefmetazole, cefprozil, loracarbef, ceforanide), third generation cephalosporins (e.g., cefepime, cefoperazone, cefotaxime, ceftizoxime, ceftriaxone, ceftazidime, cefixime, cefpodoxime, ceftibuten), other .beta.-lactams (e.g., imipenem, meropenem, aztreonam, clavulanic acid, sulbactam, tazobactam, and the like), betalactamase inhibitors (e.g., clavulanic acid), chlorampheriicol, macrolides (e.g., erythromycin, azithromycin, clarithromycin), lincomycin, clindamycin, spectinomycin, polymyxin B, polymixins (e.g., polymyxin A, B, C, D, E1 (colistin A), or E2, colistin B or C) colistin, vancomycin, telavancin, bacitracin, isoniazid, rifampin, ethambutol, ethionamide, aminosalicylic acid, cycloserine, capreomycin, sulfones (e.g., dapsone, sulfoxone sodium, and the like), clofazimine, thalidomide.
[0210] In one embodiment, the compound of Formula (I) or (II), or pharmaceutically acceptable salt of Formula (I) or (II), is administered in combination with an aminoglycoside. In a further embodiment, the compound is a compound of Formula (I) or Formula (I) wherein R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3. The aminoglycoside, in a further embodiment, is dibekacin, K-4619, sisomicin, amikacin, dactimicin, isepamicin, rhodestreptomycin, apramycin, etimicin, KA-5685, sorbistin, arbekacin, framycetin, kanamycin, spectinomycin, astromicin, gentamicin, neomycin, sporaricin, bekanamycin, H107, netilmicin, streptomycin, boholmycin, hygromycin, paromomycin, tobramycin, brulamycin, hygromycin B, plazomicin, verdamicin, capreomycin, inosamycin, ribostamycin or vertilmicin. In a further embodiment, the aminoglycoside is amikacin or gentamicin. In a further embodiment, the aminoglycoside is gentamicin.
[0211] Methods for treating bacterial infections, e.g., those caused by Gram-positive microorganisms, are provided. Without wishing to be bound by a particular theory, it is believed that the R.sup.1 groups conjugated to the glycopeptides provided herein facilitate cellular uptake of the glycopeptide at the site of infection, for example, macrophage uptake.
[0212] In one embodiment, the infection is a Gram-positive cocci infection, for example, a Staphylococcus, Enterococcus or Streptococcus infection. Streptococcus pneumoniae is treated, in one embodiment, in a patient that has been diagnosed with community-acquired pneumonia or purulent meningitis. An Enterococcus infection is treated, in one embodiment, in a patient that has been diagnosed with a urinary-catheter related infection. A Staphylococcus infection, e.g., S. aureus is treated in one embodiment, in a patient that has been diagnosed with mechanical ventilation-associated pneumonia.
[0213] Over the past few decades, there has been a decrease in the susceptibility of Gram-positive cocci to antibacterials for the treatment of infection. See, e.g., Alvarez-Lerma et al. (2006) Drugs 66, pp. 751-768, incorporated by reference herein in its entirety for all purposes. As such, in one aspect, the present invention addresses this need by providing a composition comprising an effective amount of a compound of Formula (I), Formula (II) or a pharmaceutically acceptable salt thereof, in a method for treating a patient in need thereof for a Gram-positive cocci infection that is resistant to a different antibacterial. For example, in one embodiment, the Gram-positive cocci infection is a penicillin resistant or a vancomycin resistant bacterial infection. In a further embodiment, the resistant bacterial infection is a methicillin-resistant Staphylococcus infection, e.g., methicillin-resistant S. aureus or a methicillin-resistant Staphylococcus epidermidis infection. In another embodiment, the resistant bacterial infection is an oxacillin-resistant Staphylococcus (e.g., S. aureus) infection, a vancomycin-resistant Enterococcus infection or a penicillin-resistant Streptococcus (e.g., S. pneumoniae) infection. In yet another embodiment, the Gram-positive cocci infection is a vancomycin-resistant enterococci (VRE), methicillin-resistant Staphylococcus aureus (MRSA), methicillin-resistant Staphylococcus epidermidis (MRSE), vancomycin resistant Enterococcus faecium also resistant to teicoplanin (VRE Fm Van A), vancomycin resistant Enterococcus faecium sensitive to teicoplanin (VRE Fm Van B), vancomycin resistant Enterococcus faecalis also resistant to teicoplanin (VRE Fs Van A), vancomycin resistant Enterococcus faecalis sensitive to teicoplanin (VRE Fs Van B), or penicillin-resistant Streptococcus pneumoniae (PSRP).
[0214] According to one embodiment, a method is provided to treat an infection due to a Gram-positive bacterium, including, but not limited to, genera Staphylococcus, Streptococcus, Enlerococcus, Bacillus, Corynebaclerium, Nocardia, Clostridium, and Listeria. In one embodiment, the infection is due to a Gram-positive Cocci bacterium. In a further embodiment, the infection is a pulmonary infection. In another embodiment, the infection is a Clostridium difficile infection.
[0215] In even another embodiment, the bacterial infection is Propionibacterium acnes (skin acne), Eggerthella lenta (bacteremia) or Peptostreptococcus anaerobius (gynecological infection). In a further embodiment, the composition administered to the patient in need thereof comprises a compound of Formula (I) or Formula (II) wherein R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3 and X is O.
[0216] Staphylococcus is Gram positive non-motile bacteria that colonizes skin and mucus membranes. Staphylococci are spherical and occur in microscopic clusters resembling grapes. The natural habitat of Staphylococcus is nose; it can be isolated in 50% of normal individuals. 20% of people are skin carriers and 10% of people harbor Staphylococcus in their intestines. Examples of Staphylococci infections treatable with the methods and compositions provided herein, include S. aureus, S. epidermidis, S. auricularis, S. carnosus, S. haemolyticus, S. hyicus, S. intermedius, S. lugdunensis, S. saprophytics, S. sciuri, S. simulans, and S. warneri.
[0217] While there have been about 20 species of Staphylococcus reported, only Staphylococcus aureus and Staphylococcus epidermis are known to be significant in their interactions with humans.
[0218] In one embodiment, the Staphylococcus species is resistant to a penicillin such as methicillin. In a further embodiment, the Staphylococcus species is methicillin-resistant Staphylococcus aureus (MRSA) or methicillin-resistant Staphylococcus epidermidis (MRSE). The Staphylococcus infection, in another embodiment, is a methicillin-sensitive S. aureus (MSSA) infection, a vancomycin-intermediate S. aureus (VISA) infection, or a vancomycin-resistant S. aureus (VRSA) infection.
[0219] S. aureus colonizes mainly the nasal passages, but it may be found regularly in most anatomical locales, including skin oral cavity, and gastrointestinal tract. In one embodiment, a S. aureus infection is treated with one of the methods and/or compositions provided herein. In a further embodiment, the S. aureus infection is a methicillin-resistant Staphylococcus aureus (MRSA) infection. In another embodiment, the S. aureus infection is a S. aureus (VISA) infection, or a vancomycin-resistant S. aureus (VRSA) infection.
[0220] The S. aureus infection can be a healthcare associated, i.e., acquired in a hospital or other healthcare setting, or community-acquired.
[0221] In one embodiment, the Staphylococcal infection treated with one of the methods and/or compositions provided herein, causes endocarditis or septicemia (sepsis). As such, the patient in need of treatment with one of the methods and/or compositions provided herein, in one embodiment, is an endocarditis patient. In another embodiment, the patient is a septicemia (sepsis) patient.
[0222] In one embodiment, the bacterial infection is erythromycin-resistant (erm.sup.R), vancomycin-intermediate S. aureus (VISA) heterogenous vancomycin-intermediate S. aureus (hVISA), S. epidermidis coagulase-negative staphylococci (CoNS), penicillin-intermediate S. pneumoniae (PISP), or penicillin-resistant S. pneumoniae (PRSP). In even a further embodiment, the administering comprises administering via inhalation. In even a further embodiment, the compound of Formula (I) or Formula (II) is a compound wherein R.sup.1 is --(CH.sub.2).sub.2--NH--(CH.sub.2).sub.9--CH.sub.3 or
##STR00049##
[0223] Streptococci are Gram-positive, non-motile cocci that divide in one plane, producing chains of cells. The primary pathogens include S. pyrogenes and S. pneumoniae but other species can be opportunistic. S. pyrogenes is the leading cause of bacterial pharyngitis and tonsillitis. It can also produce sinusitis, otitis, arthritis, and bone infections. Some strains prefer skin, producing either superficial (impetigo) or deep (cellulitis) infections.
[0224] S. pneumoniae is the major cause of bacterial pneumonia in adults, and in one embodiment, an infection due to S. pneumoniae is treated via one of the methods and/or compositions provided herein. Its virulence is dictated by its capsule. Toxins produced by streptococci include: streptolysins (S & O), NADase, hyaluronidase, streptokinase, DNAses, erythrogenic toxin (which causes scarlet fever rash by producing damage to blood vessels; requires that bacterial cells are lysogenized by phage that encodes toxin). Examples of Streptococcus infections treatable with the compositions and methods provided herein include, S. agalactiae, S. anginosus, S. bovis, S. canis, S. constellatus, S. dysgalactiae, S. equi, S. equinus, S. Mae, S. intermedins, S. mitis, S. mutans, S. oralis, S. parasanguinis, S. peroris, S. pneumoniae, S. pyogenes, S. ratti, S. salivarius, S. salivarius ssp. thermophilics, S. sanguinis, S. sobrinus, S. suis, S. uteris, S. vestibularis, S. viridans, and S. zooepidemicus.
[0225] The genus Enterococci consists of Gram-positive, facultatively anaerobic organisms that are ovoid in shape and appear on smear in short chains, in pairs, or as single cells. Enterococci are human pathogens that are increasingly resistant to antimicrobial agents. Examples of Enterococci treatable with the methods and compositions provided herein are E. avium, E. durans, E. faecalis, E. faecium, E. gallinarum, and E. solitarius.
[0226] In one embodiment of the methods provided herein, a patient in need thereof is treated for an Enterococcus faecalis (E. faecalis) infection. In a further embodiment, the infection is a pulmonary infection. In another embodiment, a patient in need thereof is treated for an Enterococcus faecium (E. faecium) infection. In a further embodiment, the infection is a pulmonary infection. In one embodiment, a patient in need thereof is treated for an Enterococcus infection that is resistant or sensitive to vancomycin or resistant or sensitive to penicillin. In a further embodiment, the infection is a E. faecalis or E. faecium infection.
[0227] Bacteria of the genus Bacillus are aerobic, endospore-forming, Gram-positive rods, and infections due to such bacteria are treatable via the methods and compositions provided herein. Bacillus species can be found in soil, air, and water where they are involved in a range of chemical transformations. In one embodiment, a method is provided herein to treat a Bacillus anthracis (B. anthracis) infection with a glycopeptide composition. Bacillus anthracis, the infection that causes Anthrax, is acquired via direct contact with infected herbivores or indirectly via their products. The clinical forms include cutaneous anthrax, from handling infected material, intestinal anthrax, from eating infected meat, and pulmonary anthrax from inhaling spore-laden dust. The route of administration of the glycopeptide will vary depending on how the patient acquires the B. anthracis infection. For example, in the case of pulmonary anthrax, the patient, in one embodiment, is treated via a dry powder inhaler (DPI), nebulizer or metered dose inhaler (MDI).
[0228] Several other Bacillus species, in particular, B. cereus, B. subtilis and B. licheniformis, are associated periodically with bacteremia/septicemia, endocarditis, meningitis, and infections of wounds, the ears, eyes, respiratory tract, urinary tract, and gastrointestinal tract, and are therefore treatable with the methods and compositions provided herein. Examples of pathogenic Bacillus species whose infection is treatable with the methods and compositions provided herein, include, but are not limited to, B. anthracis, B. cereus and B. coagulans.
[0229] Corynebacteria are small, generally non-motile, Gram-positive, non sporalating, pleomorphic bacilli and infections due to these bacteria are treatable via the methods provided herein. Corynebacterium diphtheria is the etiological agent of diphtheria, an upper respiratory disease mainly affecting children, and is treatable via the methods provided herein. Examples of other Corynebacteria species treatable with the methods and compositions provided herein include Corynebacterium diphtheria, Corynebacterium pseudotuberculosis, Corynebacterium tenuis, Corynebacterium striatum, and Corynebacterium minutissimum.
[0230] The bacteria of the genus Nocardia are Gram-positive, partially acid-fast rods, which grow slowly in branching chains resembling fungal hyphae. Three species cause nearly all human infections: N. asteroides, N. brasiliensis, and N. caviae, and patients with such infections can be treated with the compositions and methods provided herein. Infection is by inhalation of airborne bacilli from an environmental source (soil or organic material). Other Nocardial species treatable with the methods provided herein include N. aerocolonigenes, N. africana, N. argentinensis, N. asteroides, N. blackwellu, N. brasiliensis, N. brevicalena, N. cornea, N. caviae, N. cerradoensis, N. corallina, N. cyriacigeorgica, N. dassonvillei, N. elegans, N. farcinica, N. nigiitansis, N. nova, N. opaca, N. otitidis-cavarium, N. paucivorans, N. pseudobrasiliensis, N. rubra, N. transvelencesis, N. unif ormis, N. vaccinii, and N. veterana.
[0231] Clostridia are spore-forming, Gram-positive anaerobes, and infections due to such bacteria are treatable via the methods and compositions provided herein. In one embodiment, one of the methods provided herein are used to treat a Clostridium tetani (C. tetani) infection, the etiological agent of tetanus. In another embodiment, one of the methods provided herein is used to treat a Clostridium botidinum (C. botidinum) infection, the etiological agent of botulism. In yet another embodiment, one of the methods provided herein is used to treat a C. perfringens infection, one of the etiological agents of gas gangrene. Other Clostridium species treatable with the methods of the present invention, include, C. difficile, C. perfringens, and/or C. sordelii. In one embodiment, the infection to be treated is a C. difficile infection.
[0232] Listeria are non spore-forming, nonbranching Gram-positive rods that occur individually or form short chains. Listeria monocytogenes (L. monocytogenes) is the causative agent of listeriosis, and in one embodiment, a patient infected with L. monocytogenes is treated with one of the methods and compositions provided herein. Examples of Listeria species treatable with the methods and compositions provided herein, include L. grayi, L. innocua, L. ivanovii, L. monocytogenes, L. seeligeri, L. murrayi, and L. welshimeri.
[0233] The bacterial infection in one embodiment, is a respiratory tract infection. In a further embodiment, the infection is a resistant bacterial infection, for example, one of the infections provided above. The patient treatable by the methods provided herein, in one embodiment, has been diagnosed with a community-acquired respiratory tract infection, e.g., pneumonia. In one embodiment, the bacterial infection treated in the pneumonia patient is a S. pneumoniae infection. In another embodiment, the bacterial infection treated in the pneumonia patient is Mycoplasma pneumonia or a Legionella species. In another embodiment, the bacterial infection in the pneumonia patient is penicillin resistant, e.g., penicillin-resistant S. pneumoniae.
[0234] The bacterial infection, in one embodiment, is a hospital acquired infection (HAI), or acquired in another health care facility, e.g., a nursing home, rehabilitation facility, outpatient clinic, etc. Such infections are also referred to as nosocomial infections. In a further embodiment, the infection is a respiratory tract infection or a skin infection. In one embodiment, the HAI is pneumonia. In a further embodiment, the pneumonia is due to S. aureus, e.g., MRSA.
[0235] The inhalation delivery device employed in embodiments of the methods provided herein can be a nebulizer, dry powder inhaler (DPI), or a metered dose inhaler (MDI), or any other suitable inhalation delivery device known to one of ordinary skill in the art. The device can contain and be used to deliver a single dose of the composition or the device can contain and be used to deliver multi-doses of the composition of the present invention.
[0236] According to one embodiment, a dry powder particulate composition is delivered to a patient in need thereof via a metered dose inhaler (MDI), dry powder inhaler (DPI), atomizer, nebulizer or liquid dose instillation (LDI) technique to provide for glycopeptide delivery. With respect to inhalation therapies, those skilled in the art will appreciate that where a hollow and porous microparticle composition is employed, the composition is particularly amenable for delivery via a DPI. Conventional DPIs comprise powdered formulations and devices where a predetermined dose of medicament, either alone or in a blend with lactose carrier particles, is delivered as an aerosol of dry powder for inhalation.
[0237] The medicament is formulated in a way such that it readily disperses into discrete particles with an MMD between 0.5 to 20 .mu.m, for example from 0.5-5 .mu.m, and are further characterized by an aerosol particle size distribution less than about 10 .mu.m mass median aerodynamic diameter (MMAD), and in some embodiments, less than 5.0 .mu.m. The MMAD of the powders will characteristically range from about 0.5-10 .mu.m, from about 0.5-5.0 .mu.m, or from about 0.5-4.0 .mu.m.
[0238] The powder is actuated either by inspiration or by some external delivery force, such as pressurized air. Examples of DPIs suitable for administration of the particulate compositions of the present invention are disclosed in U.S. Pat. Nos. 5,740,794, 5,785,049, 5,673,686, and 4,995,385 and PCT application Nos. 00/72904, 00/21594, and 01/00263, the disclosure of each of which is incorporated by reference in their entireties for all purposes. DPI formulations are typically packaged in single dose units such as those disclosed in the aforementioned patents or they employ reservoir systems capable of metering multiple doses with manual transfer of the dose to the device.
[0239] The compositions disclosed herein may also be administered to the nasal or pulmonary air passages of a patient via aerosolization, such as with a metered dose inhaler (MDI). Breath activated MDIs are also compatible with the methods provided herein.
[0240] Along with the aforementioned embodiments, the compositions disclosed herein may be delivered to a patient in need thereof via a nebulizer, e.g., a nebulizer disclosed in PCT WO 99/16420, the disclosure of which is hereby incorporated in its entirety by reference, in order to provide an aerosolized medicament that may be administered to the pulmonary air passages of the patient. A nebulizer type inhalation delivery device can contain the compositions of the present invention as a solution, usually aqueous, or a suspension. For example, the prostacyclin compound or composition can be suspended in saline and loaded into the inhalation delivery device. In generating the nebulized spray of the compositions for inhalation, the nebulizer delivery device may be driven ultrasonically, by compressed air, by other gases, electronically or mechanically (e.g., vibrating mesh or aperture plate). Vibrating mesh nebulizers generate fine particle, low velocity aerosol, and nebulize therapeutic solutions and suspensions at a faster rate than conventional jet or ultrasonic nebulizers. Accordingly, the duration of treatment can be shortened with a vibrating mesh nebulizer, as compared to a jet or ultrasonic nebulizer. Vibrating mesh nebulizers amenable for use with the methods described herein include the Philips Respironics I-Neb.RTM., the Omron MicroAir, the Nektar Aeroneb.RTM., and the Pari eFlow.RTM..
[0241] The nebulizer may be portable and hand held in design, and may be equipped with a self contained electrical unit. The nebulizer device may comprise a nozzle that has two coincident outlet channels of defined aperture size through which the liquid formulation can be accelerated. This results in impaction of the two streams and atomization of the formulation. The nebulizer may use a mechanical actuator to force the liquid formulation through a multiorifice nozzle of defined aperture size(s) to produce an aerosol of the formulation for inhalation. In the design of single dose nebulizers, blister packs containing single doses of the formulation may be employed.
[0242] In the present invention, the nebulizer may be employed to ensure the sizing of particles is optimal for positioning of the particle within, for example, the pulmonary membrane.
[0243] Upon nebulization, the nebulized composition (also referred to as "aerosolized composition") is in the form of aerosolized particles. The aerosolized composition can be characterized by the particle size of the aerosol, for example, by measuring the "mass median aerodynamic diameter" or "fine particle fraction" associated with the aerosolized composition. "Mass median aerodynamic diameter" or "MMAD" is normalized regarding the aerodynamic separation of aqua aerosol droplets and is determined by impactor measurements, e.g., the Andersen Cascade Impactor (ACI) or the Next Generation Impactor (NGI). The gas flow rate, in one embodiment, is 8 Liter per minute for the ACI and 15 liters per minute for the NGI.
[0244] "Geometric standard deviation" or "GSD" is a measure of the spread of an aerodynamic particle size distribution. Low GSDs characterize a narrow droplet size distribution (homogeneously sized droplets), which is advantageous for targeting aerosol to the respiratory system. The average droplet size of the nebulized composition provided herein, in one embodiment is less than 5 .mu.m or about 1 .mu.m to about 5 .mu.m, and has a GSD in a range of 1.0 to 2.2, or about 1.0 to about 2.2, or 1.5 to 2.2, or about 1.5 to about 2.2.
[0245] "Fine particle fraction" or "FPF," as used herein, refers to the fraction of the aerosol having a particle size less than 5 .mu.m in diameter, as measured by cascade impaction. FPF is usually expressed as a percentage.
[0246] In one embodiment, the mass median aerodynamic diameter (MMAD) of the nebulized composition is about 1 .mu.m to about 5 .mu.m, or about 1 .mu.m to about 4 .mu.m, or about 1 .mu.m to about 3 .mu.m or about 1 .mu.m to about 2 .mu.m, as measured by the Anderson Cascade Impactor (ACI) or Next Generation Impactor (NGI). In another embodiment, the MMAD of the nebulized composition is about 5 .mu.m or less, about 4 .mu.m or less, about 3 .mu.m or less, about 2 .mu.m or less, or about 1 .mu.m or less, as measured by cascade impaction, for example, by the ACI or NGI.
[0247] In one embodiment, the MMAD of the aerosol of the pharmaceutical composition is less than about 4.9 .mu.m, less than about 4.5 .mu.m, less than about 4.3 .mu.m, less than about 4.2 .mu.m, less than about 4.1 .mu.m, less than about 4.0 .mu.m or less than about 3.5 .mu.m, as measured by cascade impaction.
[0248] In one embodiment, the MMAD of the aerosol of the pharmaceutical composition is about 1.0 .mu.m to about 5.0 .mu.m, about 2.0 .mu.m to about 4.5 .mu.m, about 2.5 .mu.m to about 4.0 .mu.m, about 3.0 .mu.m to about 4.0 .mu.m or about 3.5 .mu.m to about 4.5 .mu.m, as measured by cascade impaction (e.g., by the ACI or NGI).
[0249] In one embodiment, the FPF of the aerosolized composition is greater than or equal to about 50%, as measured by the ACI or NGI, greater than or equal to about 60%, as measured by the ACI or NGI or greater than or equal to about 70%, as measured by the ACI or NGI. In another embodiment, the FPF of the aerosolized composition is about 50% to about 80%, or about 50% to about 70% or about 50% to about 60%, as measured by the NGI or ACI.
[0250] In one embodiment, a metered dose inhalator (MDI) is employed as the inhalation delivery device for the compositions of the present invention. In a further embodiment, the prostacyclin compound is suspended in a propellant (e.g., hydrofluorocarbon) prior to loading into the MDI. The basic structure of the MDI comprises a metering valve, an actuator and a container. A propellant is used to discharge the formulation from the device. The composition may consist of particles of a defined size suspended in the pressurized propellant(s) liquid, or the composition can be in a solution or suspension of pressurized liquid propellant(s). The propellants used are primarily atmospheric friendly hydroflourocarbons (HFCs) such as 134a and 227. The device of the inhalation system may deliver a single dose via, e.g., a blister pack, or it may be multi dose in design. The pressurized metered dose inhalator of the inhalation system can be breath actuated to deliver an accurate dose of the lipid-containing formulation. To insure accuracy of dosing, the delivery of the formulation may be programmed via a microprocessor to occur at a certain point in the inhalation cycle. The MDI may be portable and hand held.
[0251] In one embodiment, a dry powder inhaler (DPI) is employed as the inhalation delivery device for the compositions of the present invention.
[0252] In one embodiment, the DPI generates particles having an MMAD of from about 1 .mu.m to about 10 .mu.m, or about 1 .mu.m to about 9 .mu.m, or about 1 .mu.m to about 8 .mu.m, or about 1 .mu.m to about 7 .mu.m, or about 1 .mu.m to about 6 .mu.m, or about 1 .mu.m to about 5 .mu.m, or about 1 .mu.m to about 4 .mu.m, or about 1 .mu.m to about 3 .mu.m, or about 1 .mu.m to about 2 .mu.m in diameter, as measured by the NGI or AC. In another embodiment, the DPI generates particles having an MMAD of from about 1 .mu.m to about 10 .mu.m, or about 2 .mu.m to about 10 .mu.m, or about 3 .mu.m to about 10 .mu.m, or about 4 .mu.m to about 10 .mu.m, or about 5 .mu.m to about 10 .mu.m, or about 6 .mu.m to about 10 .mu.m, or about 7 .mu.m to about 10 .mu.m, or about 8 .mu.m to about 10 .mu.m, or about 9 .mu.m to about 10 .mu.m, as measured by the NGI or ACI.
[0253] In one embodiment, the MMAD of the particles generated by the DPI is about 1 .mu.m or less, about 9 .mu.m or less, about 8 .mu.m or less, about 7 .mu.m or less, 6 .mu.m or less, 5 .mu.m or less, about 4 .mu.m or less, about 3 .mu.m or less, about 2 .mu.m or less, or about 1 .mu.m or less, as measured by the NGI or ACI.
[0254] In one embodiment, each administration comprises 1 to 5 doses (puffs) from a DPI, for example 1 dose (1 puff), 2 dose (2 puffs), 3 doses (3 puffs), 4 doses (4 puffs) or 5 doses (5 puffs). The DPI, in one embodiment, is small and transportable by the patient.
[0255] In one embodiment, the MMAD of the particles generated by the DPI is less than about 9.9 .mu.m, less than about 9.5 .mu.m, less than about 9.3 .mu.m, less than about 9.2 .mu.m, less than about 9.1 .mu.m, less than about 9.0 .mu.m, less than about 8.5 .mu.m, less than about 8.3 .mu.m, less than about 8.2 .mu.m, less than about 8.1 .mu.m, less than about 8.0 .mu.m, less than about 7.5 .mu.m, less than about 7.3 .mu.m, less than about 7.2 .mu.m, less than about 7.1 .mu.m, less than about 7.0 .mu.m, less than about 6.5 .mu.m, less than about 6.3 .mu.m, less than about 6.2 .mu.m, less than about 6.1 .mu.m, less than about 6.0 .mu.m, less than about 5.5 .mu.m, less than about 5.3 .mu.m, less than about 5.2 .mu.m, less than about 5.1 .mu.m, less than about 5.0 .mu.m, less than about 4.5 .mu.m, less than about 4.3 .mu.m, less than about 4.2 .mu.m, less than about 4.1 .mu.m, less than about 4.0 .mu.m or less than about 3.5 .mu.m, as measured by the NGI or ACI.
[0256] In one embodiment, the MMAD of the particles generated by the DPI is about 1.0 .mu.m to about 10.0 .mu.m, about 2.0 .mu.m to about 9.5 .mu.m, about 2.5 .mu.m to about 9.0 .mu.m, about 3.0 .mu.m to about 9.0 .mu.m, about 3.5 .mu.m to about 8.5 .mu.m or about 4.0 .mu.m to about 8.0 .mu.m.
[0257] In one embodiment, the FPF of the prostacyclin particulate composition generated by the DPI is greater than or equal to about 40%, as measured by the ACI or NGI, greater than or equal to about 50%, as measured by the ACI or NGI, greater than or equal to about 60%, as measured by the ACI or NGI, or greater than or equal to about 70%, as measured by the ACI or NGI. In another embodiment, the FPF of the aerosolized composition is about 40% to about 70%, or about 50% to about 70% or about 40% to about 60%, as measured by the NGI or ACI.
EXAMPLES
[0258] The present invention is further illustrated by reference to the following Examples. However, it should be noted that these Examples, like the embodiments described above, are illustrative and are not to be construed as restricting the scope of the invention in any way.
Example 1--Synthesis of Glycopeptide Derivative Via Reductive Amination
[0259] Glycopeptide derivatives were prepared as follows. The synthesis scheme is also provided at FIG. 1.
[0260] To a reactor vessel equipped with temperature control and agitation was added anhydrous DMF and DIPEA. The resulting solution was heated to 65.degree. C. with agitation and Vancomycin HCl or telavancin HCl was added slowly in portions. Heating was continued until all of vancomycin HCl or telavancin HCl had dissolved (5-10 min).
[0261] The beige colored solution was allowed to cool after which a solution of the desired aldehyde dissolved in DMF was added over 5-10 min. The resulting solution was allowed to stir overnight, typically producing a clear red-yellow solution. MeOH and TFA were introduced and stirring was further continued for at least 2 h. At the end of the stirring period, the imine forming reaction mixture was analyzed by HPLC which was characteristically typical. Borane tert-butylamine complex was added in portions and the reaction mixture was stirred at ambient temperature for an additional 2 h after which an in-process HPLC analysis of the reaction mixture indicated a near quantitative reduction of the intermediate imine group. After the reaction was over, the reaction mixture was purified using reverse phase C18 column chromatography (Phenomenex Luna 10 uM PREP C18(2) 250.times.21.2 mm column) using gradients of water and acetonitrile, each containing 0.1% (v/v) of TFA. Fractions were evaluated using HPLC and then pertinent fractions containing the target product were pooled together for the isolation of the product via lyophilization. Typical products were isolated as fluffy white solids. The procedure is shown below in Scheme 1 with vancomycin HCl as a representative starting compound.
Example 2--Synthesis of Vancomycin Derivative RV40 (Compound 40)
[0262] General Synthesis:
[0263] To a temperature controlled reactor vessel equipped with an overhead stirrer was added a suitable reaction solvent (DMF or DMA) and an organic base (typically DIPEA). The temperature was increased to approximately 60.degree. C. and vancomycin HCl was added. The warm reaction mixture was agitated at elevated temperature for approximately 20 minutes at which point all vancomycin HCl had dissolved and the reaction mixture was returned to room temperature. To the reaction mixture was then added 9H-fluoren-9-ylmethyl N-decyl-N-(2-oxoethyl)carbamate (N-Fmoc-N-decylaminoacetaldehyde) dissolved in a suitable reaction solvent (DMF or DMA). The reaction mixture was agitated with an overhead stirrer overnight at which point a suitable reducing agent, acid catalyst (e.g., TFA), and a protic solvent (e.g., MeOH) were added. The reaction mixture was agitated by an overhead stirrer at room temperature for approximately two hours at which point solvent volume was reduced by half via rotary evaporation. To the concentrated reaction mixture was then added an organic base to remove the FMOC protecting group and yield crude product (Compound 40, also referred to as "RV40", see also Table 1). Solvent was then evaporated by rotary evaporation and the crude material was dry-packed using C18 silica and purified via reverse phase C18 flash chromatography to isolate product with >97% purity. Solvent was removed from the purified material using a combination of techniques including rotary evaporation, lyophilization, and spray drying to yield product (Compound 40 or RV40) as a white powder, typically in 40-75% overall yield. Suitable solvents include N,N-Dimethylacetamide, N,N-Dimethylformamide, N,N-Dimethylacetamide or a combination thereof. Suitable organic bases include N,N-diisopropylethylamine or trimethylamine. Suitable reducing agents include NaBH.sub.4, NaBH.sub.3CN, Borane-pyridine complex, or Borane-.sup.tertbutylamine complex. Suitable organic bases for FMOC deprotection include piperidine, methylamine, and .sup.tertbutylamine.
[0264] Salt Forms:
[0265] Control over the salt form and associated counter-ions for alkyl-vancomycin derivatives was managed by altering the acid species used during flash chromatography. Lactate, Acetate, HCl, and TFA salts have been prepared. To isolate free base derivatives of alkyl vancomycin derivatives the pH of purified material was adjusted between 7-8 to induce precipitation; purified free base material was then collected by filtration, rotary evaporation, lyophilization, or spray drying.
[0266] One synthetic scheme for arriving at compound 40 (RV40) is provided at FIG. 2 (top). Here, a jacketed 1 L reactor vessel was equipped with an overhead stirrer and connected to a recirculating water bath calibrated to 65.degree. C. To the warm reaction vessel was added N,N-Dimethylformamide (75 mL) and DIPEA (640 .mu.L, 3.7 mmol, 2.0 equivalents). Solvent was allowed to stir for 20 minutes and warmed to 65.degree. C., at which point vancomycin HCl (2.70 g, 1.8 mmol, 1.00 equivalents) was added to the reaction mixture. Once all vancomycin HCl had dissolved the temperature was reduced to 25.degree. C. and 9H-fluoren-9-ylmethyl N-decyl-N-(2-oxoethyl)carbamate (890 mg, 2.1 mmol, 1.15 equivalents) dissolved in N,N-Dimethylformamide (20 mL) was added. The reaction mixture was allowed to stir at 25.degree. C. for 18 hrs. To the reaction mixture was then added NaBH.sub.3CN (330 mg, 5.3 mmol, 2.89 equivalents), MeOH (75 mL), and TFA (3.0 mL, 5.5 mmol, 3.00 equivalents). The reaction mixture was allowed to stir for 3 hr at RT at which point solvent volume was reduced by half via rotary evaporation. To the concentrated reaction mixture was then added piperidine (360 .mu.L, 3.7 mmol, 2.00 equivalents) with stirring. Reaction progress was monitored by HPLC. Once HPLC analysis indicated complete deprotection, solvent was removed from the reaction mixture under reduced pressure to yield crude product (Compound 40) as an off-white solid. The crude material was dry-packed using C18 silica and purified via reverse phase C18 flash chromatography to isolate product with >97% purity.
Example 3--Synthesis of Vancomycin Derivative RV40 (Compound 40)
[0267] General Synthesis:
[0268] To a temperature controlled reactor vessel equipped with an overhead stirrer was added a suitable reaction solvent (DMF or DMA) and an organic base (typically DIPEA). The temperature was increased to approximately 60.degree. C. and vancomycin HCl was added. The warm reaction mixture was agitated at elevated temperature for approximately 20 minutes at which point all vancomycin HCl had dissolved and the reaction mixture was returned to room temperature. To the reaction mixture was then added 9H-fluoren-9-ylmethyl N-decyl-N-(2-oxoethyl)carbamate (N-Fmoc-N-decylaminoacetaldehyde) dissolved in a suitable reaction solvent (DMF or DMA). The reaction mixture was agitated with an overhead stirrer overnight. To the reaction mixture was added a protic solvent (e.g., MeOH) and an acid catalyst (e.g., TFA) and the reaction mixture was allowed to stir for 15 minutes prior to addition of a suitable reducing agent (e.g., borane tertbutylamine complex).
[0269] The reaction mixture was agitated by an overhead stirrer at room temperature for approximately two hours at which point an organic base (e.g., tertbutylamine) was added to remove the FMOC protecting group. The temperature was increased to 55.degree. C. and the mixture was allowed to stir for 2 h. Solvent was then evaporated by rotary evaporation and the crude material was dry-packed using C18 silica and purified via reverse phase C18 flash chromatography to isolate product with >97% purity. Solvent was removed from the purified material using a combination of techniques including rotary evaporation, lyophilization, and spray drying to yield product (RV40) as a white powder, typically in 75% overall yield. Suitable solvents include N,N-Dimethylacetamide, N,N-Dimethylformamide, N,N-Dimethylacetamide or a combination thereof. Suitable organic bases include N,N-diisopropylethylamine or trimethylamine. Suitable reducing agents include NaBH.sub.4, NaBH.sub.3CN, Borane-pyridine complex, or Borane-.sup.tertbutylamine complex. Suitable organic bases for FMOC deprotection include piperidine, methylamine, and .sup.tertbutylamine.
[0270] Salt Forms:
[0271] Control over the salt form and associated counter-ions for alkyl-vancomycin derivatives was managed by altering the acid species used during flash chromatography. Lactate, Acetate, HCl, and TFA salts have been prepared. To isolate free base derivatives of the vancomycin derivative, the pH of purified material was adjusted between 7-8 to induce precipitation; purified free base material was then collected by filtration, rotary evaporation, lyophilization, or spray drying.
[0272] One synthetic scheme for arriving at compound 40 (RV40) is provided at FIG. 2, bottom, and is described in further detail below. To a 400 mL reactor vessel equipped with an overhead stirrer, a thermometer, and a pH meter was added DMF (50 mL) and DIPEA (1.17 mL, 6.73 mmol, 2.00 equivalents). The reaction mixture was heated to 55.degree. C. at which point vancomycin HCl (5.0 g, 3.37 mmol, 1.0 equivalents) were added. The mixture was stirred at 55.degree. C. for about 15 min., or until all of the vancomycin dissolved, at which point the temperature was reduced to 25.degree. C. To the reaction mixture was added a solution of N-Fmoc-decylaminoacetaldehyde (1.63 g, 3.87 mmol, 1.15 equivalents) dissolved in DMF (16.32 mL). The reaction mixture was allowed to stir at 25.degree. C. for 18 h. To the reaction mixture was added MeOH (14.0 mL) and TFA (1.03 mL, 13.46 mmol, 4.00 equivalent) and the mixture was allowed to stir at 25.degree. C. for 15 min., at which point Borane tert-butylamine complex (294 mg, 3.37 mmol, 1.0 equivalents) were added. The reaction mixture was allowed to stir at 25.degree. C. for 2 h, at which point tert-butylamine (4.24 mL, 40.38 mmol, 12.0 equivalents) was added, and the temperature was increased to 55.degree. C. The reaction mixture was allowed to stir at 55.degree. C. for 2 h. C18 functionalized silica gel was then added to the reaction mixture and solvent was removed under reduced pressure. The dry-packed material was purified using reverse phase C18 flash chromatography (Biotage.RTM. SNAP-KP-C18-HS column).
Example 4--Preparation of Monolactate Salt of RV40
[0273] A 3 L three-necked flask was equipped with a mechanical stirrer, a nitrogen inlet, a condenser and an addition funnel. Anhydrous DMF (900 mL) and DIPEA (21.06 mL, 0.12 mol) were charged. The resulting solution was heated to 55-60.degree. C. and vancomycin HCl (90.0 g, 0.06 mol) was added in portions. Heating was continued until all of vancomycin HCl had dissolved (15-30 min). The beige colored solution was allowed to cool to ambient temperature after which a solution of N--FMOC--N-decylaminoacetaldehyde (29.34 g, 0.069 mol) and DMF (293.4 mL) was added via the addition funnel over 5-10 min. The resulting solution was allowed to stir overnight to give a clear red-yellow solution. An in-process HPLC analysis of the reaction mixture at the end of the stirring period was typical. MeOH (252 mL) and TFA (18.54 mL, 0.24 mol) were introduced and stirring was further continued for at least 2 h. At the end of the stirring period, the imine forming reaction mixture was analyzed by HPLC which was characteristically typical. Borane tert-butylamine complex (5.28 g, 0.61 mol) was added in portions and the reaction mixture was stirred at ambient temperature for an additional 2 h after which an in-process HPLC analysis of the reaction mixture indicated a near quantitative reduction of the intermediate imine group with less than 3% of the unreacted vancomycin remaining. Tert-Butylamine (76.32 mL, 0.73 mol) was added via the addition funnel and the resulting reaction was heated to 55.degree. C. The stirring was continued at 55.degree. C. and progress of the FMOC group deprotection reaction was monitored by HPLC.
[0274] After the reaction was over (about 2 h), heating was removed and C18 silica gel (C-18 (Carbon 17%) 60A, 40-63 .mu.m, 270 g) was added and the mixture was concentrated on a rotavap at 52.degree. C./15 torr until free-flowing solids of C-18 silica adsorbed crude RV40 were obtained (3-7 h). The C-18 silica adsorbed crude RV40 (compound 40) was divided into three equal parts and each part-lot was purified by means of Biotage chromatography on a Biotage SNAP ULTRA C18 1850 g Cartridge (Biotage HP-Sphere C18 25 .mu.m) using gradients of water and acetonitrile, each containing 0.1% (v/v) of an 85% L-(+)-Lactic acid solution in water, and collecting 240 mL fractions. Each part lot required 50 liters of eluents. After each Biotage run, the C-18 column was conditioned for the next run by running through 60 liters of methanol. Fractions were evaluated using HPLC and then pertinent fractions containing RV40 were pooled together for the isolation of the product via lyophilization.
[0275] Lyophilization provided RV40 lactate salt as a white solid. The lyophilized RV40 lactate at this point typically contained excess lactic acid and also contained lactic acid related impurities arising from its self-condensation reactions. The isolated RV40 lactate from this run was combined with two other batches of similarly isolated lyophilized RV40 lactate to form a composite batch of RV40 lactate totaling 105 g (lot 637-140A). The excess lactic acid and its related impurities present in the above composite batch of RV40 lactate were removed via trituration with THF and then the final triturated material (RV40 mono lactate salts) was subjected to re-lyophilization to remove the trapped residual THF; both steps are described below.
[0276] A 5 L three-necked flask was equipped with a mechanical stirrer, a nitrogen inlet, and a condenser. RV40 lactate salts (105 g) and inhibitor-free anhydrous THF (1 L) were charged. The resulting mixture was stirred under nitrogen. After stirring overnight, the resulting mixture was filtered using a medium frit Buchner filter funnel. The filtered cake was washed with THF (250 mL). The filtered cake was dried on the filter funnel by pulling vacuum under nitrogen. After drying for 5 h the cake was analyzed by 1H NMR for the residual levels of lactic acid which were measured as 3.5 equivalents. The process of trituration with THF was repeated two more times after which the isolated product was determined to contain estimated 1 equivalent of lactic acid/lactate and THF. The isolated material was re-lyophilized to remove the residual THF as follows:
[0277] The above THF-triturated material was first dissolved in aqueous acetonitrile (3:1 water:acetonitrile) at a concentration of 8.1 mL per gram and then lyophilized in batches using multiple flasks. Typically, about 10-12 grams (maximum) of the material was charged into each 2 L flask followed by aqueous acetonitrile (125 mL) to prepare a solution which was lyophilized. At the end of the lyophilization and drying, product was analyzed by NMR for THF levels to determine whether lyophilization was needed to be repeated. In the current case, contents of each flask were lyophilized once more (after re-dissolving in 125 mL of aqueous acetonitrile) when no remaining THF could be detected by NMR. The final lyophilized product at this point contained an average of 0.8 wt. % acetonitrile as estimated by NMR. The contents of each flask were pulverized into smaller particles using spatula and then placed on high vacuum pumps to remove acetonitrile. No further reduction in acetonitrile levels was observed after 56-60 h on the vacuum pumps. Contents of each flasks were combined to provide a total of 74.3 g (35.5% yield based on the total conversion of 180 g of vancomycin HCl) of a composite batch of RV40 mono lactate salts as white solid which was found to be >99 area % pure by HPLC and contained one equivalent of lactate as determined by 1HNMR (DMSO-d6) analysis. The water content in the product was found at 5.6 wt. % as determined by K--F analysis.
Example 5--Synthesis of Vancomycin Derivative RV79
[0278] The synthesis scheme for arriving at the glycopeptide derivative RV79 is described below, and also provided at FIG. 3. To a 40 mL vial equipped a stir bar was added anhydrous DMF (20 mL) and DIPEA (0.20 mL). The resulting solution was heated to 65.degree. C. on an incubated shaker and vancomycin HCl (700 mg, 0.462 mmol) was added slowly in portions. Heating was continued until all of vancomycin HCl had dissolved (5-10 min). The beige colored solution was allowed to cool to room temperature at which point 4'-Chloro-biphenyl-4-carbaldehyde (0.1 g, 0.462 mmol) was added to the reaction mixture. The reaction mixture was allowed to stir overnight. MeOH (1.5 mL) and TFA (0.14 mL, 1.8 mmol) were introduced and stirring was further continued for at least 2 h. Borane tert-butylamine complex (40 mg, 0.46 mmol) was added in portions and the reaction mixture was stirred at ambient temperature for an additional 2 h. After the reaction completed, the reaction mixture is purified using reverse phase C18 column chromatography (Phenomenex Luna 10 uM PREP C18(2) 250.times.21.2 mm column) using gradients of water and acetonitrile, each containing 0.1% (v/v) of TFA. Fractions were evaluated using HPLC and then pertinent fractions containing RV79 were pooled together for isolation of the product via lyophilization. The target compound, RV79 (81.2 mg, 0.05 mmol, 10% overall yield), was obtained as a white solid in >97% purity (by HPLC). The reaction scheme is shown at FIG. 3.
Example 6--Synthesis of Alkyl-Vancomycin Derivatives
[0279] Alkyl vancomycin derivatives were prepared according to the procedure disclosed in Nagarajan et al., with slight modifications (Nagarajan et al. (1989). The Journal of Antibiotics 42(1), pp. 63-72, incorporated by reference herein in its entirety for all purposes).
[0280] The general synthesis for alkyl vancomycin derivatives is shown in FIG. 4. Briefly, to a temperature controlled reactor vessel was added vancomycin HCl, a suitable reaction solvent, an organic base, and the appropriate aldehyde. The reaction mixture was agitated with an overhead stirrer at elevated temperature and reaction progress was monitored via HPLC looking at consumption of vancomycin and imine formation. To the reactor vessel was then added a suitable reducing agent, acid catalyst (TFA), and a protic solvent (MeOH). The reaction mixture was agitated by an overhead stirrer for approximately 2 h. The reaction mixture was then either poured into water to induce precipitation of the alkyl vancomycin derivative, or solvent was removed under reduced pressure.
[0281] The crude material was dissolved in a suitable mobile phase and purified via preparative chromatography. Solvent was removed from the purified material using a combination of techniques including rotary evaporation, lyophilization, and spray drying to yield the vancomycin alkyl derivative as a white powder, typically in 40-60% overall yield. Suitable solvents include either N,N-Dimethylformamide or N,N-Dimethylacetamide. Suitable organic bases include N,N-diisopropylethylamine or trimethylamine. Suitable reducing agents include NaBH.sub.4, NaBH.sub.3CN, Borane-pyridine complex, or Borane-.sup.tertbutylamine complex.
[0282] Synthesis of N-Decyl Vancomycin (Compound 5):
[0283] The synthetic route to Compound 5, decyl vancomycin, is provided at FIG. 5. A jacketed 1 L reactor vessel was equipped with an overhead stirrer and connected to a recirculating water bath calibrated to 65.degree. C. To the warm reaction vessel was added N,N-Dimethylacetamide (160 mL) and DIPEA (6.8 mL, 39.0 mmol, 2.92 equivalents), the solvents were allowed to stir for approximately 20 minutes. Once the solvent temperature had reached 65.degree. C., vancomycin HCl (19.8 g, 13.38 mmol, 1.00 equivalents) was added to the reactor vessel. To the reactor vessel was added 1-Decanal (2.54 mL, 13.50 mmol, 1.01 equivalents) and the reaction mixture was allowed to stir for 2 hours at 65 C.degree.. To the reaction mixture was then added NaBH.sub.3CN (2.31 g, 36.77 mmol, 2.75 equivalents), MeOH (100 mL), and TFA (3.1 mL, 40.48 mmol, 3.03 equivalents). The reaction mixture was allowed to stir for 2 hours while cooling to room temperature. The reaction mixture was then poured into acetonitrile (1 L) to induce precipitation. The decant was removed and the remaining off-white slurry was centrifuged and decanted to remove excess solvent and produce a slurry containing N-decyl vancomycin and unreacted vancomycin. Crude N-decyl vancomycin as dissolved in 30:70 acetonitrile:H.sub.2O with 0.05% HOAc and purified using reverse phase C18 preparative HPLC. Pure fractions were subjected to rotary evaporation to remove organics and the flash-frozen and lyophilized to isolate purified N-decyl vancomycin as a fluffy white powder.
Example 7--Synthesis of Chloroeremomycin Derivative RV79
[0284] To a 20 mL scintillation vial equipped with a stir bar was added chloroeremomycin and a solution of copper (II) acetate in MeOH. The reaction mixture was stirred at room temperature until the chloroeremomycin had dissolved. To the reaction mixture was then added the appropriate aldehyde and sodium cyanoborohydride as a 1M solution in THF. The reaction mixture was transferred to an incubated shaker set to 45.degree. C. and reaction progress was monitored by HPLC. In some instances, it was necessary to add an additional aliquot of aldehyde reagent. The reaction mixture was allowed to shake overnight at 45.degree. C. The reaction mixture was cooled to RT and sodium borohydride was added to convert residual aldehyde reagent to the corresponding alcohol. The pH was adjusted to between 7-8 using either acetic acid or 0.1M NaOH and volatile solvents were removed by blowing N.sub.2(g) with gentle heat. To the reaction mixture was added acetonitrile to precipitate the crude product as an off-white solid. The reaction mixture was centrifuged and the liquid was decanted. The solid was dissolved in 10% MeCN/H.sub.2O containing 0.1% phosphoric acid to decomplex the copper at which point the solution briefly turned purple and then took on a yellow tinge. Preparatory HPLC was used to purify final product and LCMS was used to confirm compound identity and purity.
[0285] A diagram of the reaction is provided at FIG. 1, bottom.
Example 8--C-terminus Modification of Glyconentide Derivative
[0286] To a round bottom flask equipped with a stir bar was added a LPGC derivative, a 1:1 solution of DMF:DMSO, and DIPEA. To the reaction mixture was then added HBTU and the appropriate amine (e.g., 3-(dimethylamino)-1-propylamine). Reaction progress was monitored by HPLC. Once complete, the reaction was quenched upon addition of 1:1 H.sub.2O:MeOH. The crude material was then purified using reverse phase C18 preparatory HPLC. Purified fractions were lyophilized to yield the target products, typically as a white fluffy powder in modest yield and high purity.
Example 9--Resorcinol-Like Modification of Glycopeptide Derivative
[0287] To a round bottom flask equipped with a stir bar was added (Aminomethyl)phosphoric acid, water, and DIPEA. The reaction mixture was allowed to stir for 15 minutes at room temperature. To the reaction mixture was then added acetonitrile and formaldehyde, 37% solution in H.sub.2O. The reaction mixture was allowed to stir for an additional 15 min. at which point a glycopeptide derivative and additional DIPEA were added. Reaction progress was closely monitored using HPLC. Once complete the reaction mixture was purified using reverse phase C18 preparative HPLC. Purified fractions were lyophilized to yield the target product as a white fluffy powder.
Example 10--Minimum Inhibitory Concentration (MIC) of Glycopeptide Derivatives
[0288] MIC Testing: Glycopeptide compounds were dissolved in 100% DMSO. In vitro activities were determined using CLSI-guided broth susceptibility testing to measure drug minimum inhibitory concentrations (MICs) of the compounds against the quality control strain ATCC 29213 (MSSA) and the MRSA isolate ATCC BAA-1556. The minimal inhibitory concentrations MICs are summarized in Table 2. Glycopeptides are defined as compounds of Formula (I), and their respective R.sup.1, R.sup.2, R.sup.3 and R.sup.4 groups. X is --O-- for each compound in Table 2.
TABLE-US-00002 TABLE 2 MIC Values .mu.g/mL MRSA MRSA Glycopeptide Structure (Formula (I)) CMPD 1556 29213 R.sup.1 R.sup.2 R.sup.3 R.sup.4 RV41 0.063 0.031 (CH.sub.2).sub.9CH.sub.3 H H H RV57 0.250 0.125 (CH.sub.2).sub.7CH.sub.3 H H H RV58 1.000 1.000 (CH.sub.2).sub.5CH.sub.3 H H H RV79 0.016 0.016 ##STR00050## H H H RV84 1.000 1.000 (CH2).sub.2NH(CH.sub.2).sub.9CH.sub.3 H H ##STR00051## RV85 0.125 0.125 (CH.sub.2).sub.9CH.sub.3 H H ##STR00052## RV45 0.063 0.125 (CH.sub.2).sub.2NH(CH.sub.2).sub.9CH.sub.3 H NH(CH.sub.2).sub.3N(CH.sub.3).sub.2 H Tela- 0.063 0.063 (CH.sub.2).sub.2NH(CH.sub.2).sub.9CH.sub.3 CH.sub.2--NH--CH.sub.2--PO.sub.3H.sub.2 H H vancin
Example 11--In Vitro Activity of Glycopeptide Agents Against Gram Positive Bacteria
[0289] The susceptibility of a variety of Staphylococcus aureus, including reference methicillin-resistant (MRSA) and vancomycin-intermediate (VISA) isolates to various antibiotic compounds was assessed.
[0290] Broth microdilution MIC testing was conducted in accordance with guidelines from the Clinical and Laboratory Standards Institute (CLSI; 1, 2) and included the comparators telavancin (TLV), vancomycin (VAN), tigecycline (TGC), and linezolid (LNZ). In addition, the susceptibility of other Gram-positive bacteria (Enterococci, Streptococci, and Clostridium difficile) to the test agents and comparators was also determined.
Materials and Methods
[0291] Test compounds. The 6 test agents and the comparators are detailed in Table 3 below.
TABLE-US-00003 TABLE 3 Test compounds Stock Solution Compound Solvent/Diluent .mu.g/mL Compound 5 DMSO 6400 Compound 40 (RV40) DMSO 3200-6400 Telavancin (TLV) DMSO 800 Tigecycline (TGC) H.sub.2O 800-3200 Vancomycin (VAN) H.sub.2O 6400 Linezolid (LNZ) H.sub.2O 1600 Oritavancin (ORI) 0.002% P80 800 DMSO: Dimethyl sulfoxide (Sigma, St. Louis, MO; Cat. No. 472301) P80: Polysorbate-80 (Spectrum, New Brunswick, NJ; Cat. No. P0138)
[0292] Isolates.
[0293] The test organisms were originally received from clinical sources, the American Type Culture Collection (ATCC, Manassas, Va.), and the Network on Antimicrobial Resistance in S. aureus (NARSA; BEI Resources, Manassas, Va.). Upon receipt, the organisms were sub-cultured onto an appropriate agar medium. Following incubation, colonies were harvested from these plates and cell suspensions prepared and frozen at -80.degree. C. with a cryoprotectant. On the day prior to the assay, frozen stocks of isolates were streaked onto Trypticase Soy Agar with 5% sheep blood (Remel, Lenexa, Kans.; Lot No. 964323) and incubated overnight at 35.degree. C. in ambient atmosphere, with the exception of Streptococci which were incubated overnight at 35.degree. C. in 5% CO.sub.2, and C. difficile which was streaked onto Brucella Agar (Becton Dickinson, Sparks, Md.; Lot No. 6168880) and incubated anaerobically at 35.degree. C. for 48 h.
[0294] The following S. aureus isolates and associated phenotypes were evaluated against the aforementioned antibiotics (Table 4).
TABLE-US-00004 TABLE 4 MIC (.mu.g/mL) Organism Isolate No. Phenotype LNZ* VAN* TCG* ORI #5 TLV #40* S. aureus MMX MSSA 1.5 1 0.12 0.025 0.06 0.12 0.015 2490 S. aureus MMX MSSA 3 0.5 0.12 0.06 0.06 0.06 0.008 7907 S. aureus MMX MSSA 2 1 0.12 0.06 0.12 0.12 0.015 7908 S. aureus MMX MRSA 3 1 0.09 0.06 0.06 0.06 0.008 2010 S. aureus MMX MRSA 2 1 0.12 0.03 0.12 0.06 0.008 2011ATCC BAA-1756 S. aureus MMX MRSA 3 0.5 0.185 0.12 0.06 0.06 0.008 3982 S. aureus MMX MRSA 1.5 1 0.12 0.06 0.06 0.06 0.008 4675ATCC BAA-1556 S. aureus MMX MRSA 1 1 0.25 0.12 0.06 0.06 0.015 5717 ATCC 33591 S. aureus MMX MRSA 2.5 0.5 0.185 0.03 0.06 0.06 0.008 5985 S. aureus MMX MRSA 1.5 0.5 0.12 0.12 0.06 0.06 0.008 5999 S. aureus MMX MRSA 1.5 0.5 0.12 0.12 0.06 0.06 0.008 7899 S. aureus MMX MRSA 2 0.5 0.185 0.12 0.12 0.06 0.008 7900 S. aureus MMX MRSA 2 0.5 0.12 0.5 0.06 0.03 0.008 7901 S. aureus MMX MRSA 2 1 0.12 0.5 0.25 0.06 0.015 7902 S. aureus MMX MRSA 1.5 1 0.12 0.06 0.06 0.06 0.008 7903 S. aureus MMX hVISA 1.5 4 0.25 0.5 0.5 0.25 0.03 4665 S. aureus MMX Mu3; hVISA 1.5 1 0.75 0.12 0.06 0.12 0.015 5989 S. aureus MMX Mu50; VISA 1.25 8 0.5 1 0.5 0.5 0.06 1723 S. aureus MMX VISA; 1.5 4 0.045 1 0.25 0.5 0.06 2124 DaptoNS S. aureus MMX VISA 1.5 4 0.12 0.25 0.5 0.12 0.03 4658 S. aureus MMX VISA 2.5 4 0.09 1 0.12 0.12 0.03 4660 S. aureus MMX 100 MSSA 4 1 0.25 0.03 0.06 0.06 0.008 ATCC 29213 *LNZ and TGC values are average MICs from n = 2 experiments; all others are n = 1 experiment. **In Figure 4, strains listed here are arranged left to right in the bar plots.
[0295] S. aureus ATCC 29213 was included during the testing of S. aureus for purposes of quality control (Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing: Twenty-Seventh Informational Supplement. CLSI document M100-S27. CLSI, 950 West Valley Road, Suite 2500, Wayne, Pa. 19087 USA, 2017; CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard-Tenth Edition. CLSI document M07-A10. CLSI, 950 West Valley Road, Suite 2500, Wayne, Pa. 19087-1898 USA, 2015).
[0296] A summary of the subset of MRSA strains is also provided in Table 5.
TABLE-US-00005 TABLE 5 MIC (.mu.g/mL) #40 Organism Isolate No. Phenotype LNZ* VAN* TCG* ORI #5 TLV (RV40)* S. aureus MMX MRSA 3 1 0.09 0.06 0.06 0.06 0.008 2010 S. aureus MMX MRSA 2 1 0.12 0.03 0.12 0.06 0.008 2011ATCC BAA-1756 S. aureus MMX MRSA 3 0.5 0.185 0.12 0.06 0.06 0.008 3982 S. aureus MMX MRSA 1.5 1 0.12 0.06 0.06 0.06 0.008 4675ATCC BAA-1556 S. aureus MMX MRSA 1 1 0.25 0.12 0.06 0.06 0.015 5717 ATCC 33591 S. aureus MMX MRSA 2.5 0.5 0.185 0.03 0.06 0.06 0.008 5985 S. aureus MMX MRSA 1.5 0.5 0.12 0.12 0.06 0.06 0.008 5999 S. aureus MMX MRSA 1.5 0.5 0.12 0.12 0.06 0.06 0.008 7899 S. aureus MMX MRSA 2 0.5 0.185 0.12 0.12 0.06 0.008 7900 S. aureus MMX MRSA 2 0.5 0.12 0.5 0.06 0.03 0.008 7901 S. aureus MMX MRSA 2 1 0.12 0.5 0.25 0.06 0.015 7902 S. aureus MMX MRSA 1.5 1 0.12 0.06 0.06 0.06 0.008 7903 *LNZ and TGC values are average MICs from n = 2 experiments; all others are n = 1 experiment. **In Figure 6, strains listed here are arranged left to right in the bar plots.
[0297] With respect to the MRSA strains (Table 5), Compound 40 (RV40) was found to be more active than the respective comparator drug by the factors provided in Table 6.
TABLE-US-00006 TABLE 6 RV40 activity Comparator as compared Drug to Comparator LNZ 214 .times. VAN 86 .times. TGC 16 .times. ORI 17 .times. Compound 5 9 .times. TLV 6 .times.
[0298] A summary of the subset of Gram Positive strains other than those of the S. aureus species is provided below in Table 7. Blank entries indicate that the respective antibiotic was not tested against the respective organism.
TABLE-US-00007 TABLE 7 MIC Organism Isolate No. Type LNZ VAN TGC ORI #5 TLV #40 S. epidermidis MMX 762 MRSE 2 2 0.06 0.25 0.12 0.12 0.008 (CoNS) S. epidermidis MMX 5145 MRSE 0.5 1 0.12 0.12 0.06 0.03 0.004 (CoNS) S. lugdenensis MMX 8724 -- 0.5 0.5 0.06 .ltoreq.0.008 0.03 0.06 0.004 (CoNS) S. haemolyticus MMX 529 -- 0.5 1 0.06 0.12 0.06 0.06 0.015 (CoNS) ATCC 29970 S. hominis MMX 667 -- 1 0.5 0.12 0.06 0.06 0.06 0.015 (CoNS) ATCC 27844 E. faecalis MMX 101 VSE 2 2 0.06 .ltoreq.0.008 0.06 0.12 0.015 ATCC 29212 E. faecalis MMX 4176 -- 4 1 0.12 0.03 -- 0.25 0.03 E. faecalis MMX 1086 VanA VRE 2 >64 0.12 1 0.06 1 0.5 E. faecium MMX 4204 -- 2 0.5 0.06 0.015 -- 0.06 0.004 E. faecium MMX 851 VanA VRE 2 >64 0.06 0.12 2 2 1 E. faecium MMX 173 VanB VRE 4 >64 0.06 .ltoreq.0.008 0.12 0.06 0.008 S. pneumoniae MMX 1195 PISP 1 0.25 0.06 .ltoreq.0.008 0.015 0.03 0.004 ATCC 49619 S. pneumoniae MMX 747 -- 1 0.12 0.03 .ltoreq.0.008 -- 0.015 0.004 S. pneumoniae MMX 432 PRSP 1 0.25 0.03 .ltoreq.0.008 -- 0.03 0.008 S. pyogenes MMX 404 -- 2 0.25 0.015 2 -- 0.06 0.06 ATCC 19615 S. pyogenes MMX 946 ermR 1 0.25 0.015 0.25 0.015 0.06 0.008 S. pyogenes MMX 8778 -- 1 0.25 0.03 1 -- 0.06 0.015 C. difficile MMX 4381 toxinAB- 1 0.25 0.18 1 0.06 0.12 0.06 ATCC 700057 negative C. difficile MMX 4994 ribotype 027 -- 0.25 0.03 0.5 0.06 0.12 0.06 ATCC BAA-1805 C. difficile MMX 5668 NAP1/027 -- 1 0.03 2 0.5 0.12 0.12 ATCC BAA-1870 S. agalactiae MMX 427 -- -- 0.25 -- 1 0.03 0.06 0.015 ATCC 13813 S. agalactiae MMX 4088 -- 1 0.25 0.03 0.12 0.015 0.06 0.008 S. agalactiae MMX 4115 erm.sup.R 1 0.25 0.03 0.25 0.015 0.06 0.008 S. dysgalactiae MMX 5121 -- 1 0.25 0.06 0.05 0.015 0.12 0.008 S. dysgalactiae MMX 5123 -- 1 0.5 0.015 2 0.03 0.25 0.12 S. dysgalactiae MMX 5124 -- 1 0.25 0.12 1 0.015 0.03 0.015 S. anginosus MMX 1201 -- 0.5 0.5 0.008 0.5 0.03 0.06 0.015 (AGS) ATCC 33397 S. constellatus MMX 5677 -- 0.5 0.25 0.03 .ltoreq.0.008 0.03 0.03 0.008 (AGS) S. mitis (MGS) MMX 1205 -- -- 0.5 -- 0.03 0.03 0.06 0.008 ATCC 49456 S. mitis (MGS) MMX 5798 -- 0.5 0.25 0.03 0.25 0.015 -0.03 0.03 S. oralis (MGS) MMX 5821 -- 1 0.25 0.06 .ltoreq.0.008 0.03 0.06 0.015 C. perfringens MMX 8351 -- -- 0.5 0.12 0.015 0.06 0.015 0.015 ATCC 13124 P. micros MMX 3546 -- -- 0.5 0.015 0.03 0.12 0.06 0.015 P. anaerobius MMX 1208 -- -- 0.25 0.03 0.03 0.03 0.03 0.008 ATCC 27337 P. acnes MMX 7942 -- -- 0.25 0.03 .ltoreq.0.008 0.06 0.03 0.008 ATCC 6919 P. acnes MMX 7946 -- -- 0.25 0.03 .ltoreq.0.008 0.06 0.015 0.008 ATCC 11827 E. lenta MMX 1287 -- -- -- 0.25 -- 0.12 -- -- ATCC 43055
[0299] Test Media.
[0300] The medium employed for the MIC assay was cation-adjusted Mueller-Hinton Broth (MHBII; BD; Lot No. 6117994), excluding C. difficile which were tested in supplemented Brucella Broth (SBB). For Streptococcus isolates, the MHBII was supplemented with 3% Laked Horse Blood (Cleveland Scientific; Bath, Ohio; Lot No. 333835). For testing C. difficile, Brucella Broth (BD; Lot No. 6155858) was supplemented with vitamin K (Sigma, St. Louis, Mo.; Lot No. 108K1088), hemin (Sigma; Lot No. SLB14685V), and 5% Laked Horse Blood. Test media was prepared fresh on each day of testing and was supplemented with 0.002% polysorbate-80 (v/v) for the testing of telavancin per CLSI (Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing: Twenty-Seventh Informational Supplement. CLSI document M100-S27. CLSI, 950 West Valley Road, Suite 2500, Wayne, Pa. 19087 USA, 2017; CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard--Tenth Edition. CLSI document M07-A10. CLSI, 950 West Valley Road, Suite 2500, Wayne, Pa. 19087-1898 USA, 2015) and for the testing of oritavancin, Compound 40 and Compound 5.
[0301] Test Procedure.
[0302] MIC values were determined using a broth microdilution procedure described by CLSI (Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing: Twenty-Seventh Informational Supplement. CLSI document M100-S27. CLSI, 950 West Valley Road, Suite 2500, Wayne, Pa. 19087 USA, 2017). Automated liquid handlers (Multidrop 384, Labsystems, Helsinki, Finland; Biomek 2000 and Biomek FX, Beckman Coulter, Fullerton Calif.) were used to conduct serial dilutions and liquid transfers.
[0303] To prepare the drug mother plates, which would provide the serial drug dilutions for the replicate daughter plates, the wells of columns 2-12 of standard 96-well microdilution plates (Costar 3795) were filled with 150 .mu.L of the designated diluent for each row of drug. The test articles and comparator compounds (300 .mu.L at 100.times. the highest concentration to be tested) were dispensed into the appropriate wells in column 1. The Biomek 2000 was then used to make 2-fold serial dilutions in the mother plates from column 1 through column 11. The wells of Column 12 contained no drug and served as the organism growth control wells for the assay.
[0304] The daughter plates were loaded with 190 .mu.L per well of the appropriate test medium containing 0.002% polysorbate-80 (v/v) for telavancin, oritavancin, Compound 40, and Compound 5, using the Multidrop 384. The daughter plates were prepared on the Biomek FX instrument which transferred 2 .mu.L of drug solution from each well of a mother plate to the corresponding well of each daughter plate in a single step. The daughter plates for C. difficile were placed in the anaerobe chamber and allowed to reduce for one hour prior to inoculation.
[0305] A standardized inoculum of each organism was prepared per CLSI methods (CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically;
[0306] Approved Standard-Tenth Edition. CLSI document M07-A10. CLSI, 950 West Valley Road, Suite 2500, Wayne, Pa. 19087-1898 USA, 2015). Bacterial suspensions were prepared in MHBII (or in the case of C. difficile SBB without blood) to equal the turbidity of a 0.5 McFarland standard. The 0.5 McFarland suspensions were further diluted 1:20 (or in the case of C. difficile, 1:10) in the appropriate test medium. The inoculum for each organism was dispensed into sterile reservoirs (Beckman Coulter 372788), and the Biomek 2000 was used to deliver 10 .mu.L of standardized inoculum into each well resulting in a final test concentration of approximately 5.times.10.sup.5 CFU/mL. Daughter plates were placed on the Biomek 2000 work surface reversed so that inoculation took place from low to high drug concentration. For C. difficile, inoculum preparation and the inoculation of the daughter plates was carried out by hand in the anaerobe chamber.
[0307] Plates were stacked 3 high, covered with a lid on the top plate, placed in plastic bags, and incubated at 35.degree. C. in ambient atmosphere for approximately 18-20 hr for telavancin, Compound 40, Compound 5, oritavancin, linezolid, and tigecycline, or 24 hr for vancomycin with the exception of C. difficile plates which were incubated at 35.degree. C. anaerobically for 48 h. Following incubation, the microplates were removed from the incubator and viewed from the bottom using a plate viewer. For each of the test compounds, an un-inoculated solubility control plate for each test medium was observed for evidence of drug precipitation. The MIC was read and recorded as the lowest concentration of drug that inhibited visible growth. For linezolid, pinpoint trailing was ignored when reading the MIC per CLSI (CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard-Tenth Edition. CLSI document M07-A10. CLSI, 950 West Valley Road, Suite 2500, Wayne, Pa. 19087-1898 USA, 2015).
Results
[0308] No precipitation was observed for the comparators during the assay with the un-inoculated solubility controls. Some precipitation was noted for Compound 40 and Compound 5 at the top concentration tested (64 .mu.g/mL) in MHBII with blood and supplemented Brucella broth, and for Compound 40 (RV40) in HTM. However, this precipitation did not interfere with the reading of MICs. The MICs of comparators against ATCC quality control organisms were within the established CLSI QC ranges (CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Twenty-seventh Informational Supplement. CLSI document M100-S27. CLSI, 940 West Valley Road, Suite 1400, Wayne, Pa. 19087-1898 USA, 2017.), thus validating the assay.
[0309] The observed MIC values for the S. aureus strains against the various antibiotics is provided at Tables 3-5. These data are also provided at FIGS. 6 and 7. Data for the 12 MRSA strains are provided at Tables 4-5, and FIGS. 8 and 9.
[0310] The observed MIC values for the Gram positive strains other than S. aureus against the various antibiotics is provided at Table 6. Compound 40 was the most potent compound across the evaluated Streptococci, Enterococci, and C. difficile test isolates. The activity observed with Compound 40 was 5-fold greater than that of the telavancin, a trend consistent with the data observed with S. aureus where Compound 40 was 3-fold great than that of telavancin (Table 1). Both telavancin and Compound 40 had potent activity against vancomycin-resistant enterococci (VRE) though MIC values for VRE were elevated relative to vancomycin-susceptible enterococci (VSE).
Example 12--MRSA 1556 Biofilm Eradication
[0311] Vancomycin (Vanc), telavancin (TLV), oritivancin (ORI) and RV40 (compound of Formula (I) or (II) where R.sup.1 is (CH.sub.2)--NH--(CH.sub.2).sub.9--CH.sub.3, R.sup.2 is OH, R.sup.3 and R.sup.4 are H and X is O) were tested for their ability to eradicate MRSA 1556 biofilm.
[0312] For biofilm development, empty 96-well plates or cystic fibrosis bronchial epithelial (CFBE) cells that were seeded in a 24-well plate were inoculated with MRSA 1556 overnight culture for 6 h followed by 16 h antibiotic treatment. After 16 h incubation, planktonic bacteria were removed and biofilm was disrupted by scrapping method and collected for CFU count. The results are provided in FIG. 10 (plastic biofilm) and FIG. 11 (cell biofilm). RV40 killed MRSA 1556 biofilm that formed on plastic significantly at 0.3-10 .mu.g/ml compared to telavancin and vancomycin (FIG. 10). RV40 was more potent to kill MRSA 1556 biofilm developed in a co-culture with CFBE cells compared to vancomycin, telavancin and oritavancin with >3 log CFU/ml reduction at 20 .mu.g/ml (FIG. 11).
Example 13--In Vivo Activity of Glycopeptide Agents Against MRSA Organisms
[0313] Male Sprague Dawley rats (179-200 g) were rendered neutropenic through a series of cyclophosphamide injections (IP) at 150 mg/kg (Day -4) and 100 mg/kg (Day -1). They were then challenged with Methicillin-Resistant Staphylococcus aureus (MRSA) (ATCC-BAA-1556; TPPS 1062) at 8 log 10 via intranasal (IN) instillation on study Day 0.
[0314] Rats were treated with vehicle control (bicine; pH 9.2) or RV40 (compound 40) (in bicine; pH 9.2) via nebulization using CH Technologies 12 Port Module Oral-Nasal Aerosol and Respiratory Exposure Systems (ONARES) connected to an Aeroneb Pro nebulizer at 12 h and 24 h post-challenge. At 36 hours, post-challenge lungs were collected for CFU enumeration. The results are shown in FIG. 12.
[0315] For the data reported in FIG. 13, At 36 h, post-challenge lungs were collected for CFU enumeration. Drugs that were nebulized were done using the same procedures described for the data in FIG. 12. Results are listed as the .DELTA. Log reduction in lung CFU versus control (FIG. 13).
[0316] The same animal model from FIG. 12 was used to acquire the data in FIG. 14 regarding the dose response of inhaled RV40 in reducing lung MRSA CFUs. Again, animals were treated with the drug at 12 h and 24 h post-challenge. At 36 hours, post-challenge lungs were collected for CFU enumeration. Here, animals were dosed with RV40 at body-weight targets of 1, 2, 5, and 10 mg/kg using the same nebulization procedures described for the data in FIG. 12. Results are listed as the .DELTA. Log reduction in lung CFU versus control (FIG. 14). Data is plotted as mean and error is SEM.
[0317] The same animal model from FIG. 12 was used to acquire the data in FIG. 15 regarding prophylactic dosing of inhaled RV40 to reduce lung MRSA CFUs. Here, animals were administered single doses of nebulized inhaled RV40 (10 mg/kg body weight target) at 7, 5, 3, and 1 days before bacterial challenge and at 0.5 days after bacterial challenge. At 36 hours post-challenge lungs were collected for CFU enumeration. Dosing was done using the same nebulization procedures described for the data in FIG. 12. The results are provided at FIG. 15. Data is plotted as geometric mean with 95% CI. Statistics based on one-way ANOVA (p=0.001) with post-hoc Bonferroni multiple comparison test. N=11 for treatment groups on Days -7, -5, -3, -1, n=10 for Day +0.5, and n=8 for control.
[0318] All, documents, patents, patent applications, publications, product descriptions, and protocols which are cited throughout this application are incorporated herein by reference in their entireties for all purposes.
[0319] The embodiments illustrated and discussed in this specification are intended only to teach those skilled in the art the best way known to the inventors to make and use the invention. Modifications and variation of the above-described embodiments of the invention are possible without departing from the invention, as appreciated by those skilled in the art in light of the above teachings. It is therefore understood that, within the scope of the claims and their equivalents, the invention may be practiced otherwise than as specifically described.
* * * * *








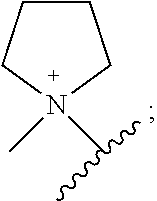



































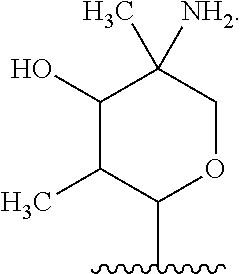
D00001

D00002

D00003
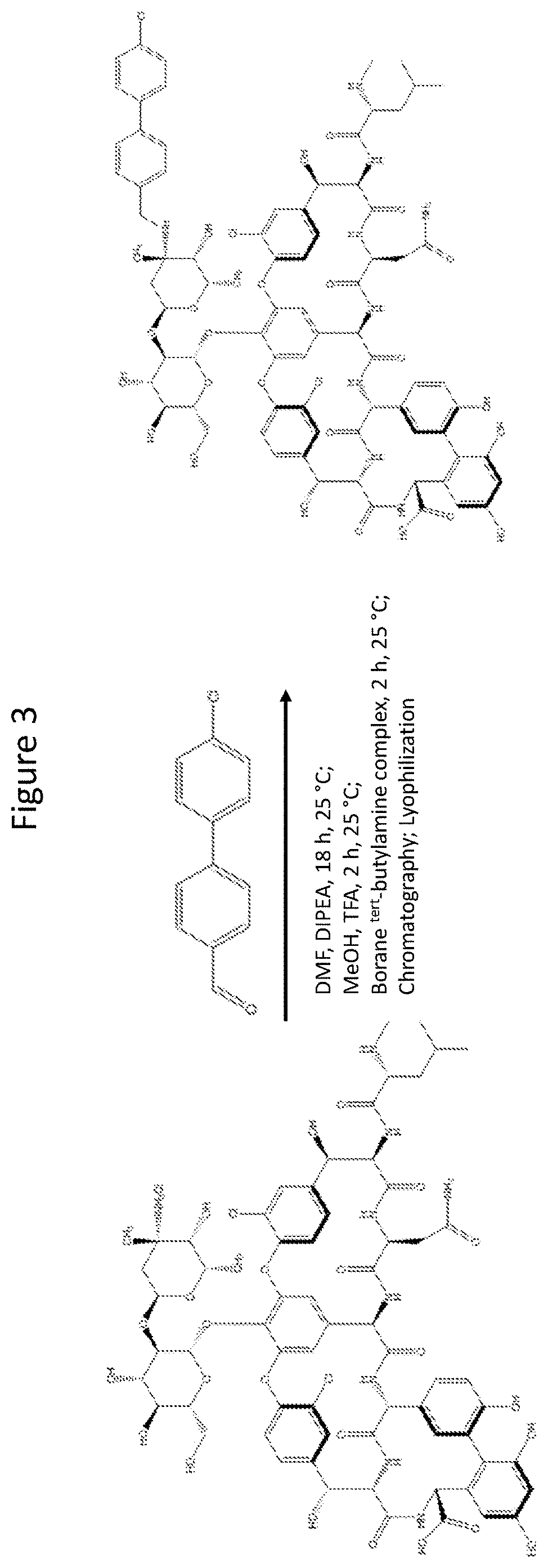
D00004

D00005

D00006

D00007

D00008

D00009

D00010

XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.