Genetically Optimised Microorganism For Producing Molecules Of Interest
BOISART; Cedric ; et al.
U.S. patent application number 16/480569 was filed with the patent office on 2020-09-03 for genetically optimised microorganism for producing molecules of interest. The applicant listed for this patent is ENOBRAQ. Invention is credited to Cedric BOISART, Nicolas MORIN.
| Application Number | 20200277592 16/480569 |
| Document ID | / |
| Family ID | 1000004871394 |
| Filed Date | 2020-09-03 |




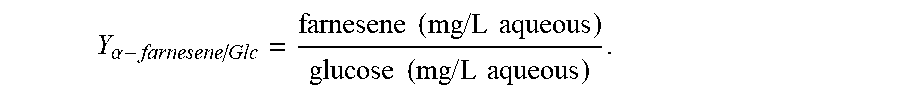


| United States Patent Application | 20200277592 |
| Kind Code | A1 |
| BOISART; Cedric ; et al. | September 3, 2020 |
GENETICALLY OPTIMISED MICROORGANISM FOR PRODUCING MOLECULES OF INTEREST
Abstract
The invention concerns a genetically modified microorganism expressing a functional type I or II RuBisCO enzyme and a functional phosphoribulokinase (PRK), and in which the glycolysis pathway is at least partially inhibited, said microorganism being genetically modified so as to produce an exogenous molecule and/or to overproduce an endogenous molecule. According to the invention, the oxidative branch of the pentose phosphate pathway may also be at least partially inhibited. The invention also concerns the use of such a genetically modified microorganism for the production or overproduction of a molecule of interest and processes for the synthesis or bioconversion of molecules of interest.
| Inventors: | BOISART; Cedric; (Belberaud, FR) ; MORIN; Nicolas; (Toulouse, FR) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 1000004871394 | ||||||||||
| Appl. No.: | 16/480569 | ||||||||||
| Filed: | January 26, 2018 | ||||||||||
| PCT Filed: | January 26, 2018 | ||||||||||
| PCT NO: | PCT/EP2018/052005 | ||||||||||
| 371 Date: | July 24, 2019 |
| Current U.S. Class: | 1/1 |
| Current CPC Class: | C12P 7/625 20130101; C12P 7/42 20130101; C12N 9/88 20130101; C12P 13/14 20130101; C12Y 207/01019 20130101; C12N 9/1205 20130101; C12Y 401/01039 20130101; C12P 13/001 20130101; C12P 7/62 20130101 |
| International Class: | C12N 9/88 20060101 C12N009/88; C12N 9/12 20060101 C12N009/12; C12P 7/62 20060101 C12P007/62; C12P 13/14 20060101 C12P013/14; C12P 13/00 20060101 C12P013/00; C12P 7/42 20060101 C12P007/42 |
Foreign Application Data
| Date | Code | Application Number |
|---|---|---|
| Jan 27, 2017 | FR | 1750694 |
Claims
1. A genetically modified microorganism for the production of an exogenous molecule of interest and/or to overproduce an endogenous molecule of interest, other than a RuBisCO or phosphoribulokinase enzyme, said microorganism expressing a functional RuBisCO enzyme and a functional phosphoribulokinase (PRK), and in which the glycolysis pathway is at least partially inhibited, upstream of the production of 1,3-biphospho-D-glycerate (1,3-BPG) or upstream of the production of 3-phosphoglycerate (3PG), and downstream of the production of glyceraldehyde-3-phosphate (G3P), wherein said microorganism is genetically modified so as to produce the exogenous molecule of interest and/or to overproduce the endogenous molecule of interest, other than a RuBisCO or phosphoribulokinase enzyme.
2. The genetically modified microorganism according to claim 1, wherein the oxidative branch of the pentose phosphate pathway is also at least partially inhibited.
3. The genetically modified microorganism according to claim 1, wherein said microorganism is genetically modified to express a recombinant RuBisCO enzyme and/or PRK.
4. The genetically modified microorganism according to claim 2, wherein said microorganism is genetically modified to inhibit the oxidative branch of the pentose phosphate pathway upstream of ribulose-5-phosphate production.
5. The genetically modified microorganism according to claim 1, wherein the exogenous molecule and/or the endogenous molecule is selected from amino acids, peptides, proteins, vitamins, sterols, flavonoids, terpenes, terpenoids, fatty acids, polyols and organic acids.
6. The genetically modified microorganism according to claim 1, wherein said microorganism is a eukaryotic cell or a prokaryotic cell.
7. The genetically modified microorganism according to claim 1, wherein the expression of the gene encoding glyceraldehyde 3-phosphate dehydrogenase is at least partially inhibited.
8. The genetically modified microorganism according to claim 1, wherein the expression of the gene encoding phosphoglycerate kinase is at least partially inhibited.
9. The genetically modified microorganism according to claim 7, wherein the expression of the gene encoding glucose-6-phosphate dehydrogenase or 6-phosphogluconolactonase or 6-phosphogluconate dehydrogenase is at least partially inhibited.
10. The genetically modified microorganism according to claim 1, wherein said microorganism is a yeast of the genus Saccharomyces cerevisiae genetically modified to express a functional type I or II RuBisCO and a functional phosphoribulokinase (PRK), and in which the expression of the TDH1, TDH2 and/or TDH3 gene is at least partially inhibited.
11. The genetically modified microorganism according to claim 1, wherein said microorganism is a Saccharomyces cerevisiae yeast genetically modified to express a functional type I or II RuBisCO and a functional phosphoribulokinase (PRK), and in which the expression of the PGK1 gene is at least partially inhibited.
12. The genetically modified microorganism according to claim 10, wherein the expression of the ZWF1 gene is at least partially inhibited.
13. The genetically modified microorganism according to claim 1, wherein said microorganism is a filamentous fungus of the genus Aspergillus genetically modified to express a functional type I or II RuBisCO and a functional phosphoribulokinase (PRK), and in which the expression of the pgk and gsdA genes is at least partially inhibited.
14. The genetically modified microorganism according to claim 1, wherein said microorganism is an E. coli bacterium genetically modified to express a functional type I or II RuBisCO and a functional phosphoribulokinase (PRK), and in which the expression of the gapA and/or pgk gene, and optionally the zwf gene, is at least partially inhibited.
15.-16. (canceled)
17. A biotechnological process for producing at least one molecule of interest, wherein it comprises a step of culturing a genetically modified microorganism as defined in claim 1, under conditions allowing the synthesis or bioconversion by said microorganism of said molecule of interest, and optionally a step of recovering and/or purifying said molecule of interest.
18. The biotechnological process according to claim 17, wherein the molecule of interest is selected from amino acids, peptides, proteins, vitamins, sterols, flavonoids, terpenes, terpenoids, fatty acids, polyols and organic acids.
19. The biotechnological process according to claim 17, wherein the molecule of interest is selected glutamate, citrate, itaconate or GABA.
20. The biotechnological process according to claim 17, wherein the microorganism is genetically modified to express at least one enzyme involved in the bioconversion or synthesis of said molecule of interest.
21. The biotechnological process according to claim 17, wherein the microorganism is genetically modified to at least partially inhibit an enzyme involved in the degradation of said molecule of interest.
22. A process for producing a molecule of interest comprising (i) inserting at least one sequence encoding an enzyme involved in the synthesis or bioconversion of said molecule of interest into a recombinant microorganism as defined in claim 1, (ii) culturing said microorganism under conditions allowing expression of said enzyme and optionally (iii) recovering and/or purifying said molecule of interest.
23. A process for producing a molecule of interest comprising (i) inhibiting the expression of at least one gene encoding an enzyme involved in the degradation of said molecule of interest in a recombinant microorganism as defined in claim 1, (ii) culturing said microorganism under conditions allowing expression of said enzyme and optionally (iii) recovering and/or purifying said molecule of interest.
24. The genetically modified microorganism of claim 1, wherein said microorganism is a eukaryotic cell selected from yeasts, fungi and microalgae.
25. The genetically modified microorganism of claim 1, wherein said microorganism is a bacterium.
Description
FIELD OF THE INVENTION
[0001] The invention concerns a genetically modified microorganism, capable of using carbon dioxide as an at least partial carbon source, for the production of molecules of interest. More specifically, the invention relates to a microorganism in which at least the glycolysis pathway is at least partially inhibited. The invention also relates to processes for the production of at least one molecule of interest using such a microorganism.
STATE OF THE ART
[0002] Over the past few years, a number of microbiological processes have been developed to enable the production of molecules of interest in large quantities.
[0003] For example, fermentation processes are used to produce molecules by a microorganism from a fermentable carbon source, such as glucose.
[0004] Bioconversion processes have also been developed to allow a microorganism to convert a co-substrate, not assimilable by said microorganism, into a molecule of interest. Here again, a carbon source is required, not for the actual production of the molecule of interest, but for the production of cofactors, and more particularly NADPH, that may be necessary for bioconversion. In general, the production yield of such microbiological processes is low, mainly due to the need for cofactors and the difficulty of balancing redox metabolic reactions. There is also the problem of the cost price of such molecules, since a source of carbon assimilable by the microorganism is still necessary. In other words, currently, in order to produce a molecule of interest with a microbiological process, it is necessary to provide a molecule (glucose, or other), certainly of lower industrial value, but which is sufficient to make the production of certain molecules not economically attractive.
[0005] At the same time, carbon dioxide (CO.sub.2), whose emissions into the atmosphere are constantly increasing, is used little, if at all, in current microbiological processes, while its consumption by microorganisms for the production of molecules of interest would not only reduce production costs, but also address certain ecological issues.
[0006] There is therefore still a need for microbiological processes to enable the production of molecules of interest in large quantities and with lower cost prices than with current processes.
SUMMARY OF THE INVENTION
[0007] The advantage of using non-photosynthetic microorganisms genetically modified to capture CO.sub.2 and use it as the main carbon source, in the same way as plants and photosynthetic microorganisms, has already been demonstrated. For example, microorganisms modified to express a functional RuBisCO (ribulose-1,5-bisphosphate carboxylase/oxygenase--EC 4.1.1.39) and a functional PRK (phosphoribulokinase--EC 2.7.1.19) to reproduce a partial Calvin cycle and convert ribulose-5-phosphate into two 3-phosphoglycerate molecules by capturing a carbon dioxide molecule have been developed.
[0008] By working on the solutions provided by the Calvin cycle to produce molecules of interest using CO.sub.2 as carbon source, the inventors discovered that it is possible to increase the production yield of molecules of interest by coupling part of the Calvin cycle (PRK/RuBisCO) to at least partial inhibition of glycolysis. The inventors have also discovered that it is possible to increase the consumption of exogenous CO.sub.2 during the production of molecules of interest, by also at least partially inhibiting the oxidative branch of the pentose phosphate pathway. The microorganisms thus developed make it possible to produce on a large scale and with an industrially attractive yield a large number of molecules of interest, such as amino acids, organic acids, terpenes, terpenoids, peptides, fatty acids, polyols, etc.
[0009] The invention thus relates to a genetically modified microorganism expressing a functional RuBisCO enzyme and a functional phosphoribulokinase (PRK), and in which the glycolysis pathway is at least partially inhibited, said microorganism being genetically modified so as to produce an exogenous molecule of interest and/or to overproduce an endogenous molecule of interest, other than a RuBisCO or phosphoribulokinase enzyme.
[0010] In one particular embodiment, the genetically modified microorganism has an oxidative branch of the pentose phosphate pathway that is also at least partially inhibited.
[0011] The invention also concerns the use of a genetically modified microorganism according to the invention, for the production or overproduction of a molecule of interest, preferentially selected from amino acids, peptides, proteins, vitamins, sterols, flavonoids, terpenes, terpenoids, fatty acids, polyols and organic acids.
[0012] The present invention also concerns a biotechnological process for producing or overproducing at least one molecule of interest, characterized in that it comprises a step of culturing a genetically modified microorganism according to the invention, under conditions allowing the synthesis or bioconversion, by said microorganism, of said molecule of interest, and optionally a step of recovering and/or purifying said molecule of interest.
[0013] It also concerns a process for producing a molecule of interest comprising (i) inserting at least one sequence encoding an enzyme involved in the synthesis or bioconversion of said molecule of interest into a recombinant microorganism according to the invention, (ii) culturing said microorganism under conditions allowing the expression of said enzyme and optionally (iii) recovering and/or purifying said molecule of interest.
DESCRIPTION OF THE FIGURES
[0014] FIG. 1: General diagram of glycolysis, the pentose phosphate pathway and the Entner-Doudoroff pathway;
[0015] FIG. 2: Schematic representation of inhibition of the glycolysis pathway, according to the invention;
[0016] FIG. 3: Schematic representation of inhibition of the glycolysis pathway, combined with inhibition of the oxidative branch of the pentose phosphate pathway, according to the invention.
DETAILED DESCRIPTION OF THE INVENTION
Definitions
[0017] The terms "recombinant microorganism", "modified microorganism" and "recombinant host cell" are used herein interchangeably and refer to microorganisms that have been genetically modified to express or overexpress endogenous nucleotide sequences, to express heterologous nucleotide sequences, or that have an altered expression of an endogenous gene. "Alteration" means that the expression of the gene, or level of an RNA molecule or equivalent RNA molecules encoding one or more polypeptides or polypeptide subunits, or the activity of one or more polypeptides or polypeptide subunits is regulated, so that the expression, the level or the activity is higher or lower than that observed in the absence of modification.
[0018] It is understood that the terms "recombinant microorganism", "modified microorganism" and "recombinant host cell" refer not only to the particular recombinant microorganism but to the progeny or the potential progeny of such a microorganism. As some modifications may occur in subsequent generations, due to mutation or environmental influences, these offspring may not be identical to the mother cell, but they are still understood within the scope of the term as used here.
[0019] In the context of the invention, an at least partially "inhibited" or "inactivated" metabolic pathway refers to an altered metabolic pathway that can no longer function properly in the microorganism considered, compared with the same wild-type microorganism (not genetically modified to inhibit said metabolic pathway). In particular, the metabolic pathway may be interrupted, leading to the accumulation of an intermediate metabolite. Such an interruption may be achieved, for example, by inhibiting the enzyme necessary for the degradation of an intermediate metabolite of the metabolic pathway considered and/or by inhibiting the expression of the gene encoding that enzyme. The metabolic pathway may also be attenuated, i.e. slowed down. Such attenuation may be achieved, for example, by partially inhibiting one or more enzymes involved in the metabolic pathway considered and/or partially inhibiting the expression of a gene encoding at least one of these enzymes and/or by exploiting the cofactors required for certain reactions. The expression "at least partially inhibited metabolic pathway" means that the level of the metabolic pathway considered is reduced by at least 20%, more preferentially at least 30%, 40%, 50%, or more, compared with the level in a wild-type microorganism. The reduction may be greater, and in particular be at least greater than 60%, 70%, 80%, 90%. According to the invention, inhibition may be total, in the sense that the metabolic pathway considered is no longer used at all by said microorganism. According to the invention, such inhibition may be temporary or permanent.
[0020] According to the invention, "inhibition of gene expression" means that the gene is no longer expressed in the microorganism considered or that its expression is reduced, compared with wild-type microorganisms (not genetically modified to inhibit gene expression), leading to the absence of production of the corresponding protein or to a significant decrease in its production, and in particular to a decrease of more than 20%, more preferentially 30%, 40%, 50%, 60%, 70%, 80%, 90%. In one embodiment, inhibition can be total, i.e. the protein encoded by said gene is no longer produced at all. Inhibition of gene expression can be achieved by deletion, mutation, insertion and/or substitution of one or more nucleotides in the gene considered. Preferentially, inhibition of gene expression is achieved by total deletion of the corresponding nucleotide sequence. According to the invention, any method of gene inhibition, known per se by the skilled person and applicable to a microorganism, may be used. For example, inhibition of gene expression can be achieved by homologous recombination (Datsenko et al., Proc Natl Acad Sci USA. 2000; 97:6640-5; Lodish et al., Molecular Cell Biology 4.sup.th ed. 2000. W. H. Freeman and Company. ISBN 0-7167-3136-3); random or directed mutagenesis to modify gene expression and/or encoded protein activity (Thomas et al., Cell. 1987; 51:503-12); modification of a promoter sequence of the gene to alter its expression (Kaufmann et al., Methods Mol Biol. 2011; 765:275-94. doi: 10.1007/978-1-61779-197-0_16); targeting induced local lesions in genomes (TILLING); conjugation, etc. Another particular approach is gene inactivation by insertion of a foreign sequence, for example by transposon mutagenesis using mobile genetic elements (transposons), of natural or artificial origin. According to another preferred embodiment, inhibition of gene expression is achieved by knock-out techniques. Inhibition of gene expression can also be achieved by extinguishing the gene using interfering, ribozyme or antisense RNA (Daneholt, 2006. Nobel Prize in Physiology or Medicine). In the context of the present invention, the term "interfering RNA" or "iRNA" refers to any iRNA molecule (for example single-stranded RNA or double-stranded RNA) that can block the expression of a target gene and/or facilitate the degradation of the corresponding mRNA. Gene inhibition can also be achieved by genome editing methods that allow direct genetic modification of a given genome, through the use of zinc finger nucleases (Kim et al., PNAS; 93: 1156-1160), transcription activator-like effector nucleases, or "TALEN" (Ousterout et al., Methods Mol Biol. 2016; 1338:27-42. doi: 10.1007/978-1-4939-2932-0_3), a system combining Cas9 nucleases with clustered regularly interspaced short palindromic repeats, or "CRISPR" (Mali et al., Nat Methods. 2013 October; 10(10):957-63. doi: 10.1038/nmeth.2649), or meganucleases (Daboussi et al., Nucleic Acids Res. 2012. 40:6367-79). Inhibition of gene expression can also be achieved by inactivating the protein encoded by said gene.
[0021] In the context of the invention, "NADPH-dependent" or "NADPH-consuming" biosynthesis or bioconversion means all biosynthesis or bioconversion pathways in which one or more enzymes require the concomitant supply of electrons obtained by the oxidation of an NADPH cofactor. "NADPH-dependent" biosynthesis or bioconversion pathways notably concern the synthesis of amino acids (e.g. arginine, lysine, methionine, threonine, proline, glutamate, homoserine, isoleucine, valine) .gamma.-aminobutyric acid, terpenoids and terpenes (e.g. farnesene), vitamins and precursors (e.g. pantoate, pantothenate, transneurosporene, phylloquinone, tocopherols), sterols (e.g. squalene, cholesterol, testosterone, progesterone, cortisone), flavonoids (e.g. frambinone, vestinone), organic acids (e.g. citric acid, succinic acid, oxalic acid, itaconic acid, coumaric acid, 3-hydroxypropionic acid), polyols (e.g. sorbitol, xylitol, glycerol), polyamines (e.g. spermidine), aromatic molecules from stereospecific hydroxylation, via an NADP-dependent cytochrome p450 (e.g. phenylpropanoids, terpenes, lipids, tannins, fragrances, hormones).
[0022] The term "exogenous" as used here in reference to various molecules (nucleotide sequences, peptides, enzymes, etc.) refers to molecules that are not normally or naturally found in and/or produced by the microorganism considered. Conversely, the term "endogenous" or "native" refers to various molecules (nucleotide sequences, peptides, enzymes, etc.), designating molecules that are normally or naturally found in and/or produced by the microorganism considered.
[0023] Microorganisms
[0024] The invention proposes genetically modified microorganisms for the production of a molecule of interest, endogenous or exogenous.
[0025] "Genetically modified" microorganism means that the genome of the microorganism has been modified to incorporate a nucleic sequence encoding an enzyme involved in the biosynthesis or bioconversion pathway of a molecule of interest, or encoding a biologically active fragment thereof. Said nucleic sequence may have been introduced into the genome of said microorganism or one of its ancestors, by any suitable molecular cloning method. In the context of the invention, the genome of the microorganism refers to all genetic material contained in the microorganism, including extrachromosomal genetic material contained, for example, in plasmids, episomes, synthetic chromosomes, etc. The introduced nucleic sequence may be a heterologous sequence, i.e. one that does not naturally exist in said microorganism, or a homologous sequence. Advantageously, a transcriptional unit with the nucleic sequence of interest is introduced into the genome of the microorganism, under the control of one or more promoters. Such a transcriptional unit also includes, advantageously, the usual sequences such as transcriptional terminators, and, if necessary, other transcription regulatory elements.
[0026] Promoters usable in the present invention include constitutive promoters, i.e. promoters that are active in most cellular states and environmental conditions, as well as inducible promoters that are activated or suppressed by exogenous physical or chemical stimuli, and therefore induce a variable state of expression depending on the presence or absence of these stimuli. For example, when the microorganism is a yeast, it is possible to use a constitutive promoter, such as that of a gene among TEF1, TDH3, PGI1, PGK, ADH1. Examples of inducible promoters that can be used in yeast are tetO-2, GAL10, GAL10-CYC1, PHO5.
[0027] In general, the genetically modified microorganism according to the invention has the following features: [0028] Expression of a functional RuBisCO (EC 4.1.1.39); [0029] Expression of a functional PRK (EC 2.7.1.19); [0030] At least partial inhibition of glycolysis; and [0031] Expression of at least one gene involved in the synthesis and/or bioconversion of a molecule of interest, and/or inhibition of at least one gene encoding activity competing with the synthesis and/or bioconversion of a molecule of interest.
[0032] According to the invention, any microorganism can be used. Preferentially the microorganism is a eukaryotic cell, preferentially selected from yeasts, fungi, microalgae or a prokaryotic cell, preferentially a bacterium or cyanobacterium.
[0033] In one embodiment, the genetically modified microorganism according to the invention is a yeast, preferentially selected from among the ascomycetes (Spermophthoraceae and Saccharomycetaceae), basidiomycetes (Leucosporidium, Rhodosporidium, Sporidiobolus, Filobasidium, and Filobasidiella) and deuteromycetes yeasts belonging to Fungi imperfecti (Sporobolomycetaceae, and Cryptococcaceae). Preferentially, the genetically modified yeast according to the invention belongs to the genus Pichia, Kluyveromyces, Saccharomyces, Schizosaccharomyces, Candida, Lipomyces, Rhodotorula, Rhodosporidium, Yarrowia, or Debaryomyces. More preferentially, the genetically modified yeast according to the invention is selected from Pichia pastoris, Kluyveromyces lactis, Kluyveromyces marxianus, Saccharomyces cerevisiae, Saccharomyces carlsbergensis, Saccharomyces diastaticus, Saccharomyces douglasii, Saccharomyces kluyveri, Saccharomyces norbensis, Saccharomyces oviformis, Schizosaccharomyces pombe, Candida albicans, Candida tropicalis, Rhodotorula glutinis, Rhodosporidium toruloides, Yarrowia lipolytica, Debaryomyces hansenii and Lipomyces starkeyi.
[0034] In another embodiment, the genetically modified microorganism according to the invention is a fungus, and more particularly a "filamentous" fungus. In the context of the invention, "filamentous fungi" refers to all filamentous forms of subdivision Eumycotina. For example, the genetically modified fungus according to the invention belongs to the genus Aspergillus, Trichoderma, Neurospora, Podospora, Endothia, Mucor, Cochliobolus or Pyricularia. Preferentially, the genetically modified fungus according to the invention is selected from Aspergillus nidulans, Aspergillus niger, Aspergillus awomari, Aspergillus oryzae, Aspergillus terreus, Neurospora crassa, Trichoderma reesei, and Trichoderma viride.
[0035] In another embodiment, the genetically modified microorganism according to the invention is a microalga. In the context of the invention, "microalga" refers to all eukaryotic microscopic algae, preferentially belonging to the classes or superclasses Chlorophyceae, Chrysophyceae, Prymnesiophyceae, Diatomae or Bacillariophyta, Euglenophyceae, Rhodophyceae, or Trebouxiophyceae. Preferentially, the genetically modified microalgae according to the invention are selected from Nannochloropsis sp. (e.g. Nannochloropsis oculata, Nannochloropsis gaditana, Nannochloropsis salina), Tetraselmis sp. (e.g. Tetraselmis suecica, Tetraselmis chuii), Chlorella sp. (e.g. Chlorella salina, Chlorella protothecoides, Chlorella ellipsoidea, Chlorella emersonii, Chlorella minutissima, Chlorella pyrenoidosa, Chlorella sorokiniana, Chlorella vulgaris), Chlamydomonas sp. (e.g. Chlamydomonas reinhardtii) Dunaliella sp. (e.g. Dunaliella tertiolecta, Dunaliella salina), Phaeodactulum tricornutum, Botrycoccus braunii, Chroomonas salina, Cyclotella cryptica, Cyclotella sp., Ettlia texensis, Euglena gracilis, Gymnodinium nelsoni, Haematococcus pluvialis, Isochrysis galbana, Monoraphidium minutum, Monoraphidium sp, Neochloris oleoabundans, Nitzschia laevis, Onoraphidium sp., Pavlova lutheri, Phaeodactylum tricornutum, Porphyridium cruentum, Scenedesmus sp. (e.g. Scenedesmus obliquuus, Scenedesmus quadricaulaula, Scenedesmus sp.), Stichococcus bacillaris, Spirulina platensis, Thalassiosira sp.
[0036] In one embodiment, the genetically modified microorganism according to the invention is a bacterium, preferentially selected from phyla Acidobacteria, Actinobacteria, Aquificae, Bacterioidetes, Chlamydia, Chlorobi, Chloroflexi, Chrysiogenetes, Cyanobacteria, Deferribacteres, Deinococcus-Thermus, Dictyoglomi, Fibrobacteres, Firmicutes, Fusobacteria, Gemmatimonadetes, Nitrospirae, Planctomycetes, Proteobacteria, Spirochaetes, Thermodesulfobacteria, Thermomicrobia, Thermotogae, or Verrucomicrobia. Preferably, the genetically modified bacterium according to the invention belongs to the genus Acaryochloris, Acetobacter, Actinobacillus, Agrobacterium, Alicyclobacillus, Anabaena, Anacystis, Anaerobiospirillum, Aquifex, Arthrobacter, Arthrospira, Azobacter, Bacillus, Brevibacterium, Burkholderia, Chlorobium, Chromatium, Chlorobaculum, Clostridium, Corynebacterium, Cupriavidus, Cyanothece, Enterobacter, Deinococcus, Erwinia, Escherichia, Geobacter, Gloeobacter, Gluconobacter, Hydrogenobacter, Klebsiella, Lactobacillus, Lactococcus, Mannheimia, Mesorhizobium, Methylobacterium, Microbacterium, Microcystis, Nitrobacter, Nitrosomonas, Nitrospina, Nitrospira, Nostoc, Phormidium, Prochlorococcus, Pseudomonas, Ralstonia, Rhizobium, Rhodobacter, Rhodococcus, Rhodopseudomonas, Rhodospirillum, Salmonella, Scenedesmun, Serratia, Shigella, Staphylococcus, Streptomyces, Synechoccus, Synechocystis, Thermosynechococcus, Trichodesmium, or Zymomonas. Also preferably, the genetically modified bacterium according to the invention is selected from the species Agrobacterium tumefaciens, Anaerobiospirillum succiniciproducens, Actinobacillus succinogenes, Aquifex aeolicus, Aquifex pyrophilus, Bacillus subtilis, Bacillus amyloliquefacines, Brevibacterium ammoniagenes, Brevibacterium immariophilum, Clostridium pasteurianum, Clostridium ljungdahlii, Clostridium acetobutylicum, Clostridium beigerinckii, Corynebacterium glutamicum, Cupriavidus necator, Cupriavidus metallidurans, Enterobacter sakazakii, Escherichia coli, Gluconobacter oxydans, Hydrogenobacter thermophilus, Klebsiella oxytoca, Lactococcus lactis, Lactobacillus plantarum, Mannheimia succiniciproducens, Mesorhizobium loti, Pseudomonas aeruginosa, Pseudomonas mevalonii, Pseudomonas pudica, Pseudomonas putida, Pseudomonas fluorescens, Rhizobium etli, Rhodobacter capsulatus, Rhodobacter sphaeroides, Rhodospirillum rubrum, Salmonella enterica, Salmonella enterica, Salmonella typhi, Salmonella typhimurium, Shigella dysenteriae, Shigella flexneri, Shigella sonnei, Staphylococcus aureus, Streptomyces coelicolor, Zymomonas mobilis, Acaryochloris marina, Anabaena variabilis, Arthrospira platensis, Arthrospira maxa, Chlorobium tepidum, Chlorobaculum sp., Cyanothece sp., Gloeobacter violaceus, Microcystis aeruginosa, Nostoc punctiforme, Prochlorococcus marinus, Synechococcus elongatus, Synechocystis sp., Thermosynechococcus elongatus, Trichodesmium erythraeum, and Rhodopseudomonas palustris.
[0037] Expression of a Functional RuBisCO and a Functional PRK
[0038] According to the invention, the microorganism can naturally express a functional RuBisCO and a functional PRK. This is the case, for example, for photosynthetic microorganisms such as microalgae and cyanobacteria.
[0039] There are several forms of RuBisCO in nature (Tabita et al., J Exp Bot. 2008; 59(7):1515-24. doi: 10.1093/jxb/erm361). Forms I, II and III catalyze the carboxylation and oxygenation reactions of ribulose-1,5-biphosphate. Form I is present in eukaryotes and bacteria. It consists of two types of subunits: large subunits (RbcL) and small subunits (RbcS). The functional enzyme complex is a hexadecamer consisting of eight L subunits and eight S subunits. The correct assembly of these subunits also requires the intervention of at least one specific chaperone: RbcX (Liu et al., Nature. 2010 Jan. 14; 463(7278):197-202. doi: 10.1038/nature08651). Form II is mainly found in proteobacteria, archaea (Archaea or archaebacteria) and dinoflagellate algae. Its structure is much simpler: it is a homodimer (formed by two identical RbcL subunits). Depending on the organism, the genes encoding a type I RuBisCO may be called rbcL/rbcS (for example Synechococcus elongatus), or cbxLC/cbxSC, cfxLC/cfxSC, cbbL/cbbS (for example Cupriavidus necator). Depending on the organism, the genes encoding a type II RuBisCO are generally called cbbM (for example Rhodospirillum rubrum). Form III is present in the archaea. It is generally found in the form of dimers of the RbcL subunit, or in pentamers of dimers. Depending on the organism, the genes encoding a type III RuBisCO may be called rbcL (for example Thermococcus kodakarensis), cbbL (for example Haloferax sp.).
[0040] Two classes of PRKs are known: class I enzymes found in proteobacteria are octamers, while class II enzymes found in cyanobacteria and plants are tetramers or dimers. Depending on the organism, the genes encoding a PRK may be called prk (for example Synechococcus elongatus), prkA (for example Chlamydomonas reinhardtii), prkB (for example Escherichia coli), prk1, prk2 (for example Leptolyngbya sp.), cbbP (for example Nitrobacter vulgaris) or cfxP (for example Cupriavidus necator).
[0041] In the case where the microorganism used does not naturally express a functional RuBisCO and a functional PRK, said microorganism is genetically modified to express heterologous RuBisCO and PRK. Advantageously, in such a case, the microorganism is transformed so as to integrate into its genome one or more expression cassettes integrating the sequences encoding said proteins, and advantageously the appropriate transcription factors. Depending on the type of RuBisCO to be expressed, it may also be necessary to have one or more chaperone proteins expressed by the microorganism, in order to promote the proper assembly of the subunits forming the RuBisCO. This is particularly the case for type I RuBisCO, where the introduction and expression of genes encoding a specific chaperone (Rbcx) and generalist chaperones (GroES and GroEL, for example) are necessary to obtain a functional RuBisCO. Application WO2015/107496 describes in detail how to genetically modify a yeast to express a functional type I RuBisCO and PRK. It is also possible to refer to the method described in GUADALUPE-MEDINA et al. (Biotechnology for Biofuels, 6, 125, 2013).
[0042] In one embodiment, the microorganism is genetically modified to express a type I RuBisCO. In another embodiment, the microorganism is genetically modified to express a type II RuBisCO. In another embodiment, the microorganism is genetically modified to express a type III RuBisCO.
[0043] Tables 1 and 2 below list, as examples, sequences encoding RuBisCO and PRK that can be used to transform a microorganism to express a functional RuBisCO and a functional PRK.
TABLE-US-00001 TABLE 1 Examples of sequences encoding a RuBisCO Gene GenBank GI Organism rbcL BAD78320.1 56685098 Synechococcus elongatus rbcS BAD78319.1 56685097 Synechococcus elongatus cbbL2 CAJ96184.1 113529837 Cupriavidus necator cbbS P09658.2 6093937 Cupriavidus necator cbbM YP_427487.1 132036 Rhodospirillum rubrum cbbM Q21YM9.1 115502580 Rhodoferax ferrireducens cbbM Q479W5.1 115502578 Dechloromonas aromatica rbcL O93627.5 37087684 Thermococcus kodakarensis cbbL CQR50548.1 811260688 Haloferax sp. Arc-Hr
TABLE-US-00002 TABLE 2 Examples of sequences encoding a PRK Gene GenBank GI Organism prk BAD78757.1 56685535 Synechococcus elongatus cfXP P19923.3 125575 Cupriavidus necator PRK P09559.1 125579 Spinacia oleracea cbbP P37100.1 585367 Nitrobacter vulgaris
[0044] Inhibition of Glycolysis
[0045] According to the invention, the glycolysis pathway is at least partially inhibited, so that the microorganism is no longer able to use this metabolic pathway normally (FIG. 1--glycolysis). In other words, the microorganism no longer has the ability to assimilate glucose in a similar way to a wild-type microorganism, in which the glycolysis pathway has not been inhibited (independently of any other genetic modification).
[0046] In one particular embodiment, the microorganism is genetically modified to inhibit, totally or partially, glycolysis downstream of the production of glyceraldehyde-3-phosphate (G3P).
[0047] For example, glycolysis is inhibited upstream of the production of 1,3-biphospho-D-glycerate (1,3-BPG) or upstream of the production of 3-phosphoglycerate (3PG).
[0048] Depending on the microorganism, the reactions involved between glyceraldehyde-3-phosphate (G3P) and 3-phosphoglycerate (3PG) can be managed (i) by two enzymes acting concomitantly, glyceraldehyde-3-phosphate dehydrogenase (EC 1.2.1.12, abbreviated GAPDH or more rarely G3PDH) and phosphoglycerate kinase (E.C. 2.7.2.3, abbreviated PGK), or (ii) by a single non-phosphorylating glyceraldehyde 3-phosphate dehydrogenase enzyme (EC 1.2.1.9, abbreviated GAPN).
[0049] Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) catalyzes the reversible conversion of G3P to 1,3-biphospho-D-glycerate (1,3-BPG), using the pair NAD.sup.+/NADH as electron donor/acceptor in the direction of the reaction. Depending on the organism, the genes encoding GAPDH may be called gapA, gapB, gapC (e.g. Escherichia coli, Arabidopsis thaliana), GAPDH, GAPD, G3PD, GAPDHS (e.g. Homo sapiens), TDH1, TDH2, TDH3 (e.g. Saccharomyces cerevisiae), gap, gap2, gap3 (e.g. Mycobacterium sp., Nostoc sp.).
[0050] Phosphoglycerate kinase (PGK) catalyzes the reversible conversion of 1,3-BPG to 3PG, using the pair ATP/ADP as cofactor. Depending on the organism, the genes encoding PGK may be called PGK, PGK1, PGK1, PGK2, PGK3, pgkA, PGKB, PGKC, cbbK, cbbKC, cbbKP (e.g. Cupriavidus necator).
[0051] Non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase (GAPN) catalyzes the conversion of G3P to 3PG, without going through 1,3-BPG. This reaction is catalyzed in the presence of the cofactor pair NADP.sup.+/NADPH, which acts as an electron acceptor. Depending on the organism, the genes encoding GAPN may be called GAPN (e.g. Bacillus sp., Streptococcus sp.), GAPN1 (e.g. Chlamydomonas sp.).
[0052] In one particular example, the microorganism is genetically modified so that the expression of the gene encoding glyceraldehyde 3-phosphate dehydrogenase is at least partially inhibited. Preferentially, gene expression is completely inhibited.
[0053] Alternatively or additionally, the expression of the gene encoding phosphoglycerate kinase may also be at least partially inhibited. Preferentially, gene expression is completely inhibited.
[0054] Alternatively, the microorganism is genetically modified so that the expression of the gene encoding non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase is at least partially inhibited. Preferentially, gene expression is completely inhibited.
[0055] Tables 3, 4 and 5 below list, as examples, the sequences encoding a glyceraldehyde 3-phosphate dehydrogenase, a phosphoglycerate kinase and a non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase that can be inhibited depending on the target microorganism. The skilled person knows which gene corresponds to the enzyme of interest to be inhibited depending on the microorganism.
TABLE-US-00003 TABLE 3 Examples of sequences encoding a GAPDH Gene GenBank GI Organism gapA NP_416293.1 947679 Escherichia coli TDH1 NP_012483.3 398364523 Saccharomyces cerevisiae TDH2 NP_012542.1 6322468 Saccharomyces cerevisiae TDH3 NP_011708.3 398366083 Saccharomyces cerevisiae gap ECC36949.1 378544675 Mycobacterium tuberculosis gap2 P34917.2 92090599 Nostoc sp.
TABLE-US-00004 TABLE 4 Examples of sequences encoding a PGK Gene GenBank GI Organism pgk AKL94701.1 831186507 Clostridium aceticum PGK1 NP_009938.2 10383781 Saccharomyces cerevisiae pgk BAG04189.1 166089481 Microcystis aeruginosa PGKA AAG34561.2 22711882 Dictyostelium discoideum PGKB CAJ03534.1 68126221 Leishmania major cbbKC AAC43444.1 976365 Cupriavidus necator pgk CAK45271.1 4982539 Aspergillus niger pgk EAU38870.1 4354973 Aspergillus terreus
TABLE-US-00005 TABLE 5 Examples of sequences encoding a GAPN Gene GenBank GI Organism gapN CUB58597.1 924094571 Bacillus subtilis GAPN NP_358622.1 933338 Streptococcus pneumoniae GAPN1 EDP03116.1 542583 Chlamydomonas reinhardtii
[0056] In general, the production of 3-phosphoglycerate (3PG) is no longer possible through glycolysis, or at least significantly reduced, in the genetically modified microorganism according to the invention.
[0057] In a particular exemplary embodiment, the microorganism is a yeast of the genus Saccharomyces cerevisiae in which the expression of the TDH1 (Gene ID: 853395), TDH2 (Gene ID: 853465) and/or TDH3 gene (Gene ID: 853106) is at least partially inhibited.
[0058] In another particular exemplary embodiment, the microorganism is a yeast of the genus Saccharomyces cerevisiae in which the expression of the PGK1 gene (Gene ID: 5230) is at least partially inhibited.
[0059] In another exemplary embodiment, the microorganism is a yeast of the genus Saccharomyces cerevisiae in which the expression of the PGK1 gene (Gene ID: 5230), the expression of the TDH1 gene (Gene ID: 853395), TDH2 (Gene ID: 853465) and/or the expression of the TDH3 gene (Gene ID: 853106) are at least partially inhibited.
[0060] In a particular exemplary embodiment, the microorganism is an Escherichia coli bacterium in which the expression of the gapA gene (Gene ID: 947679) is at least partially inhibited.
[0061] In another particular exemplary embodiment, the microorganism is an Escherichia coli bacterium in which the expression of the pgk gene (Gene ID: 947414) is at least partially inhibited.
[0062] In another exemplary embodiment, the microorganism is an E. coli bacterium in which the expression of the pgk gene (Gene ID: 947414), and/or the expression of the gapA gene (Gene ID: 947679) are at least partially inhibited.
[0063] According to the invention, the genetically modified microorganism, which expresses a functional RuBisCO and a functional PRK, is on the other hand capable of producing 3PG by capturing CO.sub.2 from ribulose-5-phosphate produced by the pentose phosphate pathway (FIG. 2).
[0064] Since the enzymes necessary for the metabolism of 3PG to pyruvate are not inhibited in the microorganism, said microorganism can then metabolize 3PG to produce pyruvate and ATP.
[0065] Thus, the genetically modified microorganism is able to produce pyruvate and NADPH cofactors using CO.sub.2 as complementary carbon source.
[0066] In the context of the invention, "complementary" carbon source means that the microorganism uses CO.sub.2 as a partial carbon source, in addition to the carbon atoms provided by fermentable sugars (glucose, galactose, sucrose, fructose, etc.), which constitute the majority or main carbon source for pyruvate production.
[0067] Thus, the genetically modified microorganism according to the invention makes it possible to increase carbon yield, by fixing and using the CO.sub.2 normally lost during glucose metabolism via the pentose phosphate pathway, for the production of pyruvate (and subsequently molecules of interest).
[0068] Inhibition of the Oxidative Branch of the Pentose Phosphate Pathway
[0069] In one particular embodiment, the genetically modified microorganism according to the invention is also modified in such a way that the oxidative branch of the pentose phosphate pathway is also at least partially inhibited.
[0070] Preferentially, the microorganism is genetically modified to inhibit the oxidative branch of the pentose phosphate pathway upstream of ribulose-5-phosphate production (FIG. 1--pentose phosphate pathway).
[0071] The interruption of the oxidative branch of the pentose phosphate pathway upstream of ribulose-5-phosphate (Ru5P) production specifically targets one or more reactions in the Ru5P synthesis process from glucose-6-phosphate (G6P). This synthesis is generally catalyzed by the successive actions of three enzymes: (i) glucose-6-phosphate dehydrogenase (EC. 1.1.1.49, abbreviated G6PDH), (ii) 6-phosphogluconolactonase (E.C. 3.1.1.31, abbreviated PGL), and (iii) 6-phosphogluconate dehydrogenase (EC 1.1.1.44, abbreviated PGD).
[0072] Glucose-6-phosphate dehydrogenase (G6PDH) catalyzes the first reaction of the pentose phosphate pathway, i.e. the oxidation of glucose-6-phosphate to 6-phosphogluconolactone (6PGL), with concomitant reduction of one molecule of NADP to NADPH. Depending on the organism, the genes encoding G6PDH may be called G6PD (for example in Homo sapiens), G6pdx (for example in Musculus), gsdA (for example in Aspergillus nidulans), zwf (for example in Escherichia coli), or ZWF1 (for example in Saccharomyces cerevisiae).
[0073] 6-Phosphogluconolactonase (PGL) is a hydrolase that catalyzes the synthesis of 6-phosphogluconate (6PGA) from 6PGL. Depending on the organism, the genes encoding PGL may be called pgl (for example in Escherichia coli, Synechocystis sp.) pgls (for example in Rhodobacteraceae bacterium), or SOL (for example in Saccharomyces cerevisiae).
[0074] 6-Phosphogluconate dehydrogenase (PGD) is an oxidoreductase that catalyzes the synthesis of Ru5P from 6PGA, with concomitant reduction of an NADP molecule to NADPH and emission of a CO.sub.2 molecule. Depending on the organism, the genes encoding PGD may be called gnd (for example in Escherichia coli, Saccharomyces cerevisiae), PGD (for example in Homo sapiens), gntZ (for example in Bacillus subtilis), or 6-PGDH (for example in Lactobacillus paracollinoides).
[0075] In one particular example, the microorganism is genetically modified so that the expression of the gene encoding glucose-6-phosphate dehydrogenase is at least partially inhibited. Preferentially, gene expression is completely inhibited.
[0076] Alternatively or additionally, the microorganism is genetically modified so that the expression of the gene encoding 6-phosphogluconolactonase is at least partially inhibited. Preferentially, gene expression is completely inhibited.
[0077] Alternatively or additionally, the microorganism is genetically modified so that the expression of the gene encoding 6-phosphogluconate dehydrogenase is at least partially inhibited. Preferentially, gene expression is completely inhibited.
[0078] Tables 6, 7 and 8 below list, as examples, the sequences encoding a glucose-6-phosphate dehydrogenase, a 6-phosphogluconolactonase and a 6-phosphogluconate dehydrogenase that can be inhibited depending on the target microorganism. The skilled person knows which gene corresponds to the enzyme of interest to be inhibited depending on the microorganism.
TABLE-US-00006 TABLE 6 Examples of sequences encoding a G6PDH Gene GenBank GI Organism zwf BAA15660.1 946370 Escherichia coli ZWF1 NP_014158.1 6324088 Saccharomyces cerevisiae gsdA CAA54841.1 1523786 Aspergillus nidulans gsdA CAK37895.1 4979751 Aspergillus niger gsdA EAU38380.1 4316232 Aspergillus terreus
TABLE-US-00007 TABLE 7 Examples of sequences encoding a PGL Gene GenBank GI Organism pgl BAA35431.1 4062334 Escherichia coli pgl BAK51770.1 339275283 Synechocystis pgls KPQ07176.1 938272062 Rhodobacteraceae bacterium SOL3 KZV10901.1 1023943655 Saccharomyces cerevisiae
TABLE-US-00008 TABLE 8 Examples of sequences encoding a PGD Gene GenBank GI Organism gnd ALI40222.1 937519736 Escherichia coli GND1 EDN62420.1 151944127 Saccharomyces cerevisiae gntZ NP_391888.1 16081060 Bacillus subtilis 6-PGDH WP_054711110.1 938929230 Lactobacillus paracollinoides
[0079] In general, the production of ribulose-5-phosphate (Ru5P) is no longer possible through the pentose phosphate pathway, or at least significantly reduced, in the genetically modified microorganism according to the invention.
[0080] In a particular exemplary embodiment, the microorganism is a yeast of the genus Saccharomyces cerevisiae in which the expression of the ZWF1 gene is at least partially inhibited.
[0081] In one particular example, the yeast of the genus Saccharomyces cerevisiae is genetically modified so that the expression of the TDH1, TDH2, TDH3 and/or PGK1 genes, and the expression of the ZWF1 gene are at least partially inhibited.
[0082] In another particular exemplary embodiment, the microorganism is a bacterium of the genus Escherichia coli in which the expression of the zwf gene is at least partially inhibited.
[0083] In one particular example, the bacterium of the genus Escherichia coli is genetically modified so that the expression of the gapA and/or pgk genes, and the expression of the zwf gene are at least partially inhibited.
[0084] In another example, the microorganism is a filamentous fungus of the genus Aspergillus, such as Aspergillus niger or Aspergillus terreus, genetically modified so that the expression of the pgk and gsdA genes is partially inhibited.
[0085] According to the invention, the genetically modified microorganism, which expresses a functional RuBisCO and a functional PRK, and whose glycolysis pathway and oxidative branch of the pentose phosphate pathway are at least partially inhibited, is no longer capable of producing 3PG via the glycolysis pathway or Ru5P via the oxidative branch of the pentose phosphate pathway. On the other hand, it is capable of producing Ru5P by diverting the production of fructose-6-phosphate (F6P) and/or glyceraldehyde-3-phosphate (G3P), produced at the beginning of glycolysis (upstream of inhibition). This production is possible thanks to the enzymes transketolase (EC 2.2.1.1), transaldolase (EC 2.2.1.2), ribose-5-phosphate isomerase (EC 5.3.1.6), and ribulose-5-phosphate epimerase (EC 5.1.3.1) naturally present and active in the microorganisms (FIG. 3).
[0086] Since the enzymes necessary for the metabolism of 3PG to pyruvate are not inhibited in the microorganism according to the invention, said microorganism can then metabolize 3PG to produce pyruvate and ATP.
[0087] Thus, the genetically modified microorganism is able to produce pyruvate by using exogenous CO.sub.2 as complementary carbon source.
[0088] Thus, the genetically modified microorganism according to the invention makes it possible to increase the carbon yield, by fixing and using exogenous CO.sub.2, for the production of pyruvate (and subsequently molecules of interest). Here again, there is an increase in carbon yield.
[0089] Inhibition of the Entner-Doudoroff Pathway
[0090] In one particular embodiment, the genetically modified microorganism according to the invention has an Entner-Doudoroff pathway, and this is at least partially inhibited. This pathway, mainly found in bacteria (especially Gram-negative bacteria), is an alternative to glycolysis and the pentose pathway for the production of pyruvate from glucose. More precisely, this pathway connects to the pentose phosphate pathway at P-gluconate to feed glycolysis, particularly at pyruvate.
[0091] Preferentially, the microorganism is genetically modified to inhibit Entner-Doudoroff pathway reactions downstream of 6-phosphogluconate production. This inhibition eliminates a possible competing pathway, and ensures the availability of 6-phosphogluconate as a substrate for PRK/RuBisCO engineering.
[0092] The interruption of the Entner-Doudoroff pathway downstream of 6-phosphogluconate production specifically targets one or more reactions in the pyruvate synthesis process from 6-phosphogluconate. This synthesis is initiated by the successive actions of two enzymes: (i) 6-phosphogluconate dehydratase ("EDD"--EC. 4.2.1.12), and (ii) 2-dehydro-3-deoxy-phosphogluconate aldolase ("EDA"--E.C. 4.1.2.14).
[0093] 6-Phosphogluconate dehydratase catalyzes the dehydration of 6-phosphogluconate to 2-keto-3-deoxy-6-phosphogluconate. Depending on the organism, the genes encoding 6-phosphogluconate dehydratase may be called edd (GenBank NP_416365, for example, in Escherichia coli), or ilvD (for example, in Mycobacterium sp.).
[0094] 2-Dehydro-3-deoxy-phosphogluconate aldolase catalyzes the synthesis of a pyruvate molecule and a glyceraldehyde-3-phosphate molecule from the 2-keto-3-deoxy-6-phosphogluconate produced by 6-phosphogluconate dehydratase. Depending on the organism, the genes encoding 2-dehydro-3-deoxy-phosphogluconate aldolase may be called eda (GenBank NP_416364, for example, in Escherichia coli), or kdgA (for example in Thermoproteus tenax), or dgaF (for example in Salmonella typhimurium).
[0095] In one particular example, the microorganism is genetically modified so that the expression of the gene encoding 6-phosphogluconate dehydratase is at least partially inhibited. Preferentially, gene expression is completely inhibited.
[0096] Alternatively or additionally, the microorganism is genetically modified so that the expression of the gene encoding 2-dehydro-3-deoxy-phosphogluconate aldolase is at least partially inhibited. Preferentially, gene expression is completely inhibited.
[0097] Tables 9 and 10 below list, as examples, the sequences encoding a 6-phosphogluconate dehydratase and a 2-dehydro-3-deoxy-phosphogluconate aldolase that can be inhibited depending on the target microorganism. The skilled person knows which gene corresponds to the enzyme of interest to be inhibited depending on the microorganism.
TABLE-US-00009 TABLE 9 Examples of sequences encoding an EDD Gene GenBank GI Organism edd NP_416365.1 16129804 Escherichia coli ilvD CND70554.1 893638835 Mycobacterium tuberculosis edd AJQ65426.1 764046652 Salmonella enterica
TABLE-US-00010 TABLE 10 Examples of sequences encoding an EDA Gene GenBank GI Organism eda AKF72280.1 817591701 Escherichia coli kdgA Q704D1.1 74500902 Thermoproteus tenax eda O68283.2 81637643 Pseudomonas aeruginosa
[0098] In general, in this embodiment, pyruvate production is no longer possible via the Entner-Doudoroff pathway, or at least significantly reduced.
[0099] In a particular exemplary embodiment, the microorganism is a bacterium of the genus Escherichia coli in which the expression of the edd gene is at least partially inhibited.
[0100] In one particular example, the bacterium of the genus Escherichia coli is genetically modified so that the expression of the gapA, and edd genes are at least partially inhibited.
[0101] According to the invention, the genetically modified microorganism, which expresses a functional RuBisCO and a functional PRK, and whose glycolysis pathway and Entner-Doudoroff pathway are at least partially inhibited, is no longer capable of producing 3PG by glycolysis or pyruvate by the Entner-Doudoroff pathway. The carbon flow from glucose is therefore preferably directed towards PRK/RuBisCO engineering.
[0102] Production of Molecules of Interest
[0103] According to the invention, the genetically modified microorganism is transformed so as to produce an exogenous molecule of interest and/or to overproduce an endogenous molecule of interest.
[0104] In the context of the invention, molecule of interest preferentially refers to a small organic molecule with a molecular mass less than or equal to 0.8 kDa.
[0105] In general, genetic modifications made to the microorganism, as described above, improve the carbon yield of the synthesis and/or bioconversion pathways of molecules of interest.
[0106] In the context of the invention, "improved" yield refers to the quantity of the finished product. In general, in the context of the invention, the carbon yield corresponds to the ratio of quantity of finished product to quantity of fermentable sugar, particularly by weight. According to the invention, the carbon yield is increased in the genetically modified microorganisms according to the invention, compared with wild-type microorganisms, placed under identical culture conditions. Advantageously, the carbon yield is increased by 2%, 5%, 10%, 15%, 18%, 20%, or more. The genetically modified microorganism according to the invention may produce a larger quantity of molecules of interest (finished product) than heterologous molecules produced by a genetically modified microorganism simply to produce or overproduce that molecule. According to the invention, the genetically microorganism may also overproduce an endogenous molecule compared with the wild-type microorganism. The overproduction of an endogenous molecule is mainly understood in terms of quantities. Advantageously, the genetically modified microorganism produces at least 20%, 30%, 40%, 50%, or more by weight of the endogenous molecule than the wild-type microorganism. Advantageously, the microorganism according to the invention is genetically modified so as to produce or overproduce at least one molecule among amino acids, terpenoids, terpenes, vitamins and/or vitamin precursors, sterols, flavonoids, organic acids, polyols, polyamines, aromatic molecules obtained from stereospecific hydroxylation, via an NADP-dependent cytochrome p450, etc.
[0107] In one particular example, the microorganism is genetically modified to overproduce at least one amino acid, preferentially selected from arginine, lysine, methionine, threonine, proline, glutamate, homoserine, isoleucine, valine, and .gamma.-aminobutyric acid.
[0108] In one particular example, the microorganism is genetically modified to produce or overproduce molecules from the terpenoid pathway, such as farnesene, and from the terpene pathway.
[0109] In one particular example, the microorganism is genetically modified to produce or overproduce a vitamin or precursor, preferentially selected from pantoate, pantothenate, transneurosporene, phylloquinone and tocopherols.
[0110] In one particular example, the microorganism is genetically modified to produce or overproduce a sterol, preferentially selected from squalene, cholesterol, testosterone, progesterone and cortisone.
[0111] In one particular example, the microorganism is genetically modified to produce or overproduce a flavonoid, preferentially selected from frambinone and vestinone.
[0112] In one particular example, the microorganism is genetically modified to produce or overproduce an organic acid, preferentially selected from coumaric acid, 3-hydroxypropionic acid, citric acid, oxalic acid, succinic acid, and itaconic acid.
[0113] In one particular example, the microorganism is genetically modified to produce or overproduce a polyol, preferentially selected from sorbitol, xylitol and glycerol.
[0114] In one particular example, the microorganism is genetically modified to produce or overproduce a polyamine, preferentially spermidine.
[0115] In one particular example, the microorganism is genetically modified to produce or overproduce an aromatic molecule from a stereospecific hydroxylation, via an NADP-dependent cytochrome p450, preferentially selected from phenylpropanoids, terpenes, lipids, tannins, fragrances, hormones.
[0116] In the case where the molecule of interest is obtained by bioconversion, the genetically modified microorganism is advantageously cultured in a culture medium including the substrate to be converted. In general, the production or overproduction of a molecule of interest by a genetically modified microorganism according to the invention is obtained by culturing said microorganism in an appropriate culture medium known to the skilled person.
[0117] The term "appropriate culture medium" generally refers to a sterile culture medium providing essential or beneficial nutrients for the maintenance and/or growth of said microorganism, such as carbon sources; nitrogen sources such as ammonium sulfate; sources of phosphors, for example, potassium phosphate monobasic; trace elements, for example, salts of copper, iodide, iron, magnesium, zinc or molybdate; vitamins and other growth factors such as amino acids or other growth promoters. An antifoam agent can be added as needed. According to the invention, this appropriate culture medium may be chemically defined or complex. The culture medium may thus be identical or similar in composition to a synthetic medium, as defined by Verduyn et al. (Yeast. 1992. 8:501-17), adapted by Visser et al. (Biotechnology and bioengineering. 2002. 79:674-81), or commercially available such as yeast nitrogen base (YNB) medium (MP Biomedicals or Sigma-Aldrich).
[0118] In particular, the culture medium may include a simple carbon source, such as glucose, galactose, sucrose, molasses, or the by-products of these sugars, optionally supplemented with CO.sub.2 as carbon co-substrate. According to the present invention, the simple carbon source must allow the normal growth of the microorganism of interest. It is also possible, in some cases, to use a complex carbon source, such as lignocellulosic biomass, rice straw, or starch. The use of a complex carbon source usually requires pretreatment before use.
[0119] In one particular embodiment, the culture medium contains at least one carbon source among monosaccharides such as glucose, xylose or arabinose, disaccharides such as sucrose, organic acids such as acetate, butyrate, propionate or valerate to promote different kinds of polyhydroxyalkanoate (PHA), treated or untreated glycerol.
[0120] Depending on the molecules to be produced and/or overproduced, it is possible to exploit the supply of nutritional factors (N, O, P, S, K, Mg, Fe, Mn, Co, Cu, Ca, Sn; Koller et al., Microbiology Monographs, G.-Q. Chen, 14: 85-119, (2010)). This is particularly the case to promote the synthesis and intracellular accumulation of polyhydroalkanoate (PHA) including polyhydroxybutyrate (PHB).
[0121] According to the invention, any culture method allowing the production on an industrial scale of molecules of interest can be considered. Advantageously, the culture is done in bioreactors, especially in batch, fed-batch and/or continuous culture mode. Preferentially, the culture associated with the production of the molecule of interest is in fed-batch mode corresponding to a controlled supply of one or more substrates, for example by adding a concentrated glucose solution whose concentration can be between 200 g/L and 700 g/L. A controlled supply of vitamins during the process can also be beneficial to productivity (Alfenore et al., Appl Microbiol Biotechnol. 2002. 60:67-72). It is also possible to add an ammonium salt solution to limit the nitrogen supply.
[0122] Fermentation is generally carried out in bioreactors, with possible steps of solid and/or liquid precultures in Erlenmeyer flasks, with an appropriate culture medium containing at least a simple carbon source and/or an exogenous CO.sub.2 supply, necessary for the production of the molecule of interest.
[0123] In general, the culture conditions of the microorganisms according to the invention are easily adaptable by the skilled person, depending on the microorganism and/or the molecule to be produced/overproduced. For example, the culture temperature is between 20.degree. C. and 40.degree. C. for yeasts, preferably between 28.degree. C. and 35.degree. C., and more particularly around 30.degree. C., for S. cerevisiae. The culture temperature is between 25.degree. C. and 35.degree. C., preferably 30.degree. C., for Cupriavidus necator.
[0124] The invention therefore also relates to the use a genetically modified microorganism according to the invention, for the production or overproduction of a molecule of interest, preferentially selected from amino acids, peptides, proteins, vitamins, sterols, flavonoids, terpenes, terpenoids, fatty acids, polyols and organic acids.
[0125] The invention also relates to a biotechnological process for producing at least one molecule of interest, characterized in that it comprises a step of culturing a genetically modified microorganism according to the invention, under conditions allowing the synthesis or bioconversion, by said microorganism, of said molecule of interest, and optionally a step of recovering and/or purifying said molecule of interest.
[0126] In one particular embodiment, the microorganism is genetically modified to express at least one enzyme involved in the synthesis of said molecule of interest.
[0127] In another particular embodiment, the microorganism is genetically modified to express at least one enzyme involved in the bioconversion of said molecule of interest.
[0128] The invention also relates to a process for producing a molecule of interest comprising (i) inserting at least one sequence encoding an enzyme involved in the synthesis or bioconversion of said molecule of interest into a recombinant microorganism according to the invention, (ii) culturing said microorganism under conditions allowing the expression of said enzyme and optionally (iii) recovering and/or purifying said molecule of interest.
[0129] For example, it is possible to overproduce citrate by a fungus, particularly a filamentous fungus, such as Aspergillus niger, genetically modified to express a functional PRK and a functional type I or II RuBisCO, and in which the expression of the pgk (Gene ID: 4982539) and gsdA (Gene ID: 497979751) genes is at least partially inhibited.
[0130] It is also possible to overproduce itaconic acid by a fungus, particularly a filamentous fungus, such as Aspergillus terreus or Aspergillus niger, genetically modified to express a functional PRK and a functional type I or II RuBisCO, and in which the expression of the pgk (Gene ID: 4354973) and gsdA (Gene ID: 4316232) genes is at least partially inhibited.
[0131] Similarly, it is possible to produce farnesene by a yeast such as a yeast of the genus Saccharomyces cerevisiae genetically modified to express a functional PRK and a functional type I or II RuBisCO, a farnesene synthase and in which the expression of a PGK1 gene (Gene ID: 5230) is at least partially inhibited.
[0132] It is also possible to overproduce glutamate by a bacterium, such as a bacterium of the genus Escherichia coli, genetically modified to express a functional PRK and a functional type I or II RuBisCO, and in which the expression of the gapA gene (Gene ID: 947679) is at least partially inhibited. This overproduction can also occur in a strain where at least partial inhibition of the gapA gene is combined with at least partial inhibition of the zwf gene (Gene ID: 946370).
[0133] Similarly, it is also possible to overproduce .gamma.-aminobutyric acid by a bacterium, such as a bacterium of the genus Escherichia coli, genetically modified to express a functional PRK and a functional type I or II RuBisCO, as well as a glutamate decarboxylase gadB (Gene ID: 946058), and in which the expression of the gapA gene (Gene ID: 947679) is at least partially inhibited. This overproduction can also occur in a strain where at least partial inhibition of the gapA gene is combined with at least partial inhibition of the zwf gene (Gene ID: 946370).
[0134] Similarly, it is possible to overproduce succinic acid and oxalic acid by a bacterium, such as a bacterium of the genus Escherichia coli, genetically modified to express a functional PRK and a functional type I or II RuBisCO, as well as an enzymatic activity allowing the oxidation of glyoxylate to oxalate, preferentially a glyoxylate dehydrogenase FPGLOXDH1 (mRNA: BAH29964.1), a glyoxylate oxidase GLO (mRNA: AOW73106.1), or a lactate dehydrogenase LDHA (Gene ID: 3939), and in which the expression of the gapA (Gene ID: 947679) and zwf (Gene ID: 946370) genes is at least partially inhibited.
EXAMPLES
Example 1: Bioinformatics Analysis
[0135] a) Calculation of Theoretical Yields
[0136] i) Comparison of Carbon Fixation Yields from Glucose Between a Wild-Type Strain Using the Pentose Phosphate Pathway and Glycolysis and a Modified Strain According to the Invention
[0137] In order to evaluate the benefit of the modifications described according to the invention, theoretical yield calculations were carried out on the basis of the stoichiometry of the reactions involved.
[0138] Two scenarios were analyzed: the improvement provided by PRK-RuBisCO engineering (i) in a strain inhibited for glycolysis on the yield of a NADPH-dependent biosynthetic pathway (for example farnesene synthesis), and (ii) in a strain inhibited for glycolysis and for the oxidative branch of the pentose phosphate pathway on the yield of a biosynthetic pathway of interest (for example citrate synthesis).
[0139] In the context of the improvement of NADPH-dependent biosynthetic pathways, the theoretical balance of the formation of NADPH and glyceraldehyde-3-phosphate (G3-P) from glucose via the pentose phosphate pathway was calculated according to the following equation (1):
3Glucose+5ATP+6NADP.sup.++3H.sub.2O.fwdarw.5G3-P+5ADP+6NADPH+11H.sup.++3- CO.sub.2 (1)
[0140] Going down to pyruvate formation from G3P, we arrive at the following balance:
3Glucose+5ADP+6NADP.sup.++5NAD.sup.++5P.sub.i.fwdarw.5Pyruvate+5ATP+6NAD- PH+5NADH+11H.sup.++3CO.sub.2+2H.sub.2O (2)
[0141] If we normalize the balance for one mole of glucose, we obtain the following yield:
Glucose+1.67ADP+2NADP.sup.++1.67NAD.sup.++1.67P.sub.i.fwdarw.1.67Pyruvat- e+1.67ATP+2NADPH+1.67NADH+3.67H.sup.++CO.sub.2+0.67H.sub.2O (3)
[0142] Thus, by using the pentose phosphate pathway, 1.67 moles of pyruvate and 2 moles of NADPH are produced from one mole of glucose. However, one mole of carbon is lost by decarboxylation when ribulose-5-phosphate is formed by 6-phosphogluconate dehydrogenase (EC 1.1.1.44). In comparison, pyruvate formation by the glycolysis pathway gives the following yield:
Glucose+2ADP+2NAD.sup.++2P.sub.i.fwdarw.2Pyruvate+2ATP+2NADH+2H.sup.++2H- .sub.2O (4)
[0143] The maximum theoretical yield of pyruvate production by the pentose phosphate pathway is therefore 0.82 g.sub.pyruvate/g.sub.glucose (g of synthesized pyruvate, per g of glucose consumed), while it is 0.98 g.sub.pyruvate/g.sub.glucose by the glycolysis pathway.
[0144] By integrating PRK/RuBisCO engineering into a strain inhibited for glycolysis (for example .DELTA.PGK1 in S. cerevisiae yeast), the carbon fixation flux is redirected to the oxidative branch of the pentose phosphate pathway and then to PRK/RuBisCO engineering (see FIG. 2). This flux is related to the end of the glycolysis pathway, at the level of 3-phosphoglycerate (3PG) formation, with the following yield:
Glucose+2ATP+2NADP.sup.++2H.sub.2O.fwdarw.2 3PG+2ADP+2NADPH+6H.sup.+ (5)
[0145] Going down to pyruvate formation from 3PG, we arrive at the following balance:
Glucose+2NADP.sup.+.fwdarw.2Pyruvate+2NADPH+4H.sup.+ (6)
[0146] The integration of the modifications according to the invention into a microorganism makes it possible to recover the carbon molecule otherwise lost by decarboxylation in the pentose pathway. The maximum theoretical carbon fixation yield is therefore 0.98 g.sub.pyruvate/g.sub.glucose, which improves by 20.5% the yield obtained by the production of pyruvate by the pentose phosphate pathway, while producing NADPH.
[0147] In a second case (see FIG. 3), PRK/RuBisCO engineering is integrated into a strain that is both inhibited for glycolysis (for example .DELTA.PGK1 in the case of S. cerevisiae yeast) and for the oxidative branch of the pentose phosphate pathway (for example .DELTA.ZWF1 in the case of S. cerevisiae yeast). The theoretical balance of the formation of NADPH and 3-phosphoglycerate (3PG) from glucose then becomes
2.5Glucose+6ATP+3CO.sub.2+3H.sub.2O.fwdarw.6 3PG+6ADP+12H.sup.+ (7)
[0148] Going down to pyruvate formation from 3PG, we arrive at the following balance
2.5Glucose+3CO.sub.2.fwdarw.6Pyruvate++3H.sub.2O+6H.sup.+ (8)
[0149] If we normalize the balance for one mole of glucose, we obtain the following yield:
Glucose+1.2CO.sub.2.fwdarw.2.4Pyruvate+1.2H.sub.2O+2.4H.sup.+ (9)
[0150] The integration of the modifications according to the invention makes it possible to fix 1.2 additional carbon molecule per mole of glucose consumed. The corresponding maximum theoretical yield is 1.17 g.sub.pyruvate/g.sub.glucose, which is .about.20% improvement compared with the carbon fixation yield of glycolysis.
[0151] ii) Application to Citrate Production
[0152] In a second case, the calculation is applied to citrate production in S. cerevisiae yeast, in a wild-type strain and in a modified strain modified according to the invention incorporating PRK/RuBisCO engineering and deleted for the PGK1 gene so as to inhibit the glycolysis pathway, and for the ZWF1 gene to inhibit the oxidative branch of the pentose pathway.
[0153] The production of citrate from pyruvate is summarized by the following balance equation:
2Pyruvate+ATP+NAD.sup.++2H.sub.2O.fwdarw.Citrate+ADP+NADH+P.sub.i+3H.sup- .+ (11)
[0154] This synthesis does not require NADPH, but 2 moles of pyruvate. Optimally, a wild-type strain obtains these 2 moles of pyruvate by glycolysis, from one mole of glucose according to equation (4), with the following balance:
Glucose+ADP+3NAD.sup.++P.sub.i.fwdarw.Citrate+ATP+3NADH+5H.sup.+ (12)
[0155] The corresponding g.sub.citrate/g.sub.glucose yield is 1.07
[0156] In the context of a modified strain according to the invention, inhibited for the glycolysis pathway and the pentose phosphate pathway, the 2 pyruvates required are obtained with only 0.83 mole of glucose (see equation 9), with the following balance:
0.83Glucose+CO.sub.2+ATP+NAD.sup.++H.sub.2O.fwdarw.Citrate+ADP+NADH+P.su- b.i+5H.sup.+ (13)
[0157] The corresponding g.sub.citrate/g.sub.glucose yield is 1.28, a maximum theoretical increase of about 20% compared with the yield of the wild-type strain.
[0158] b) Simulation of Biosynthesis Yields by Flux Balance Analysis
[0159] In a bioinformatics approach, flux balance analyses (FBAs) were also performed to simulate the impact of the modifications described according to the invention on the yield of different biosynthetic pathways.
[0160] FBAs are based on mathematical models that simulate metabolic networks at the genome scale (Orth et al., Nat Biotechnol. 2010; 28: 245-248). Reconstructed networks contain the known metabolic reactions of a given organism and integrate the needs of the cell, in particular to ensure cell maintenance or growth. FBAs make it possible to calculate the flow of metabolites through these networks, making it possible to predict theoretical growth rates as well as metabolite production yields.
[0161] i) Procedure
[0162] FBA simulations were performed with the OptFlux software (Rocha et al., BMC Syst Biol. 2010 Apr. 19; 4:45. doi: 10.1186/1752-0509-4-45), and the Saccharomyces cerevisiae metabolic model iMM904 (Mo et al., BMC Syst Biol. 2009 Mar. 25; 3:37. doi: 10.1186/1752-0509-37). This model has been modified to include the improvements described according to the invention, including a heterologous CO.sub.2 fixation pathway with (i) the addition of a PRK-type reaction, (ii) the addition of a RuBisCO-type reaction.
[0163] In particular exemplary embodiments, the reactions necessary to simulate the production of molecules through heterologous pathways have also been added to the model.
[0164] In a particular exemplary embodiment, a farnesene synthase reaction (EC 4.2.3.46 or EC 4.2.3.47) has been added for the heterologous production of farnesene.
[0165] In a second particular exemplary embodiment, acetoacetyl-CoA reductase (EC 1.1.1.36) and poly-hydroxybutyrate synthase (EC 2.3.1.B2 or 2.3.1.B5) reactions were added to the model to simulate a heterologous production pathway of .beta.-hydroxybutyrate, the monomer of polyhydroxybutyrate.
[0166] In another particular exemplary embodiment, a glutamate decarboxylase reaction (EC 4.1.1.15) was added for the heterologous production of .gamma.-aminobutyric acid.
[0167] In another particular exemplary embodiment, an aconitate decarboxylase reaction (EC 4.1.1.6) was added for the heterologous production of itaconic acid.
[0168] In another particular exemplary embodiment, a lactate dehydrogenase reaction (EC 1.1.1.27) was added for the heterologous production of oxalate
[0169] The simulations were carried out by applying to the model a set of constraints reproducible by the skilled person, aimed at simulating the in vivo culture conditions of a strain of S. cerevisiae under the conditions described according to the invention (for example presence of unrestricted glucose in the medium, aerobic culture condition).
[0170] In particular exemplary embodiments, simulations are performed by virtually inactivating the reactions of the enzymes PGK1 (for example glutamate, .beta.-hydroxybutyric acid, farnesene) and ZWF1 (for example citrate production), in order to simulate the decreases in glycolysis activity and the pentose phosphate pathway, described according to the invention.
[0171] Simulations are carried out in parallel on an unmodified "wild-type strain" model in order to evaluate the impact of the improvements described according to the invention on the production yield of the biosynthetic pathways tested.
[0172] ii) Results
[0173] The theoretical yields obtained and the percentages of improvement provided by the invention are described in Table 11 below.
TABLE-US-00011 TABLE 11 Maximum theoretical production yields evaluated by FBA on a wild-type strain and a modified strain according to the modifications of the patent, for the production of different molecules. Percentage improvement in theoretical Maximum theoretical Maximum theoretical production mass production yields yields with a modified strain efficiency with a wild-type strain according to the invention g.sub.x/g.sub.GLUC Target Simulation Mol.sub.x/ CMol.sub.x/ Mol.sub.x/ CMol.sub.x/ provided by molecule conditions Mol.sub.GLUC CMol.sub.GLUC g.sub.x/g.sub.GLUC Mol.sub.GLUC CMol.sub.GLUC g.sub.x/g.sub.GLUC the invention Citrate .DELTA.PGK1, 1 1 1.07 1.2 1.2 1.28 +20% .DELTA.ZWF1, Itaconate .DELTA.PGK1, 1 0.83 0.72 1.2 1 0.87 +20% .DELTA.ZWF1, Glutamate .DELTA.PGK1 0.92 0.77 0.75 1.09 0.91 0.89 +18.7% .DELTA.PGK1, 1 0.83 0.82 1.2 1 0.98 +20% .DELTA.ZWF1, GABA .DELTA.PGK1, 1 0.67 0.57 1.2 0.8 0.69 +20% .DELTA.ZWF1, .beta.- .DELTA.PGK1 0.92 0.61 0.53 1.09 0.73 0.63 +18.2% Hydroxybutyric acid Farnesene .DELTA.PGK1 0.21 0.54 0.24 0.24 0.59 0.27 +12.5% Co-production Succinate .DELTA.PGK1, 1 0.67 0.66 1.2 0.8 0.79 +20% Oxalate .DELTA.ZWF1, 1 0.33 0.5 1.2 0.4 0.6 +20% Mol.sub.x//Mol.sub.GLUC: moles of molecule X produced, in relation to the moles of glucose consumed CMol.sub.x/CMol.sub.GLUC:: moles of carbon of molecule X produced, in relation to the moles of carbon of glucose consumed g.sub.x/g.sub.GLUC: g of molecule X produced, in relation to the g of glucose consumed.
Example 2: Improvement of Farnesene Production in S. cerevisiae
[0174] A Saccharomyces cerevisiae yeast strain, CEN.PK 1605 (Mat a HIS3 leu2-3.112 trp1-289 ura3-52 MAL.28c) derived from the commercial strain CEN.PK 113-7D (GenBank: JRIV00000000 is engineered to produce NADPH without CO.sub.2 loss and thus allow the improvement of alpha-farnesene production from glucose.
[0175] a) Inactivation of the Glycolysis Pathway
[0176] To that end, the glycolysis pathway was inactivated by deletion of the PGK1 gene. Once glycolysis is inhibited, the resulting yeast strain is no longer able to use glucose as a source of carbon and energy. It is therefore necessary to supply the biomass synthesis pathways with glycerol and the energy pathways with ethanol. The strains in which PGK1 is deleted are grown on YPGE (yeast extract peptone glycerol ethanol) medium.
[0177] The deletion of the PGK1 gene was obtained as follows:
[0178] The coding phase of the G418 resistance gene, derived from the KanMX cassette contained on plasmid pUG6 (P30114--Euroscarf), was amplified with the oligonucleotides CB101 (SEQ ID NO: 1) and CB102 (SEQ ID NO: 2):
TABLE-US-00012 SEQ ID NO: 1: CB101 (forward): 5'-ACAGATCATCAAGGAAGTAATTATCTACTTTTTACAACAAAT ATAAAACAATGGGTAAGGAAAAGACTCACGTTTC-3' SEQ ID NO: 2: CB102 (reverse): 5'-GGGAAAGAGAAAAGAAAAAAATTGATCTATCGATTTCAATTC AATTCAATTTAGAAAAACTCATCGAGCATCAAATGAAAC-3'
[0179] The underlined portion of the oligonucleotides is perfectly homologous to the Kan sequence and the rest of the sequence corresponds to the regions adjacent to the coding phase of the PGK1 gene on the Saccharomyces cerevisiae genome so as to generate a PCR amplicon containing at its ends homologous recombination sequences of the PGK1 gene locus.
[0180] For the transformation reaction according to the skilled man (Methods in Yeast Genetics, Cold Spring Harbor lab course manual, 1997; Gietz and Schiest, 1995, Methods in Molecular and Cellular Biology 5[5]:225-269), strain CEN.PK 1605 was grown in a volume of 50 mL of complex rich medium YPD (yeast extract peptone dextrose) at 30.degree. C. to an optical density at 600 nm of 0.8. The cells were centrifuged for 5 minutes at 2,500 rpm at room temperature.
[0181] The supernatant was removed and the cells were resuspended in 25 mL of sterile water and centrifuged again for 5 minutes at 2,500 rpm at room temperature. After removing the supernatant, the cells were resuspended in 400 .mu.L of 100 mM sterile lithium acetate.
[0182] At the same time, a transformation mix was prepared in a 2 mL tube as follows: 250 .mu.L of 50% PEG, 10 .mu.L of "carrier" DNA at 5 mg/mL, 36 .mu.L of 1 M lithium acetate, 5 or 10 .mu.L of purified PCR reaction (deletion cassette) and 350 .mu.L of water.
[0183] The resuspended cells (50 .mu.L) were added to the transformation mixture and incubated at 42.degree. C. for 40 minutes in a water bath.
[0184] After incubation, the tube was centrifuged for 1 minute at 5,000 rpm at room temperature and the supernatant was discarded. The cells were resuspended in 2 mL of YPGE (yeast extract peptone glycerol ethanol) medium, transferred to a 14 mL tube and incubated for 2 hours at 30.degree. C. at 200 rpm. The cells were then centrifuged for 1 minute at 5,000 rpm at room temperature. The supernatant was removed and the cells were resuspended in 1 mL of sterile water and centrifuged again for 1 minute and resuspended in 100 .mu.L of sterile water and spread over 180 .mu.g/mL YPGE+G418.
[0185] The colonies obtained were genotyped for the validation of the deletion of the PGK1 gene and referenced EQ-0134 (CEN.PK1605 .DELTA.pgk1::kan).
[0186] b) Introduction of PRK--RuBisCO--Alpha-Farnesene Synthase Enzymes
[0187] In order to reconstitute an alternative pathway to glycolysis and allow the .DELTA.pgk1 strain to grow on glucose, said strain has been modified to allow combinatorial expression of: [0188] a gene encoding a phosphoribulokinase PRK which is grafted onto the pentose phosphate pathway by consuming ribulose-5P to give ribulose-1.5bisP and [0189] a type I RuBisCO (with the structural genes RbcL and RbcS and the chaperones RbcX, GroES and GroEL). RuBisCO consumes ribulose-1.5bisP and one mole of CO.sub.2 to form 3-phosphoglycerate downstream of the PGK1 deletion in the glycolysis pathway.
[0190] This alternative pathway once again allows the strain to consume glucose as its main source of carbon and energy.
[0191] To produce alpha-farnesene, the yeast lacks the alpha-farnesene synthase gene (AFS1; SEQ ID NO: 71; GenBank accession number AY182241).
[0192] Also, the seven genes required for PRK-RuBisCO engineering (Table 12) were cloned on four plasmid vectors capable of autonomous replication, with compatible origins of replication and each carrying a different gene for complementation of auxotrophy or of antibiotic resistance, allowing the selection of strains containing the three or four plasmid constructs.
[0193] Two of these plasmids are single-copy, with an Ars/CEN origin of replication and the third is multicopy with a 2.mu. origin.
TABLE-US-00013 TABLE 12 Description of expression cassettes and plasmid composition Codon Auxo- optimi- Termi- trophic GenBank zation Promoter nator ori marker Plasmids RbcL BAD78320.1 Yes TDH3p ADH1t 2.mu. URA3 pFPP45 RbcS BAD78319.1 Yes TEF1p PGK1t 2.mu. URA3 pFPP45 RbcX BAD80711.1 Yes TEF1p PGK1t ARS-CEN6 LEU2 pFPP56 GroES U00096 No PGI1p CYC1t ARS-CEN6 LEU2 pFPP56 GroEL AP009048 No TDH3p ADH1t ARS-CEN6 LEU2 pFPP56 PRK BAD78757.1 Yes Tet-OFF CYC1t ARS416-CEN4 TRP1 pFPP20 alpha- AY182241 Yes TEF1p PGK1t 2.mu. NatMX pL4 farnesene synthase (AFS1) Empty Tet-OFF CYC1t ARS416-CEN4 TRP1 pCM185 Empty ARS-CEN6 LEU2 pFL36 Empty TEF1p PGK1t 2.mu. URA3 pV51TEF
[0194] Genes from Synechococcus elongatus such as rbcL, rbcS, rbcX and prk (as described in WO 2015107496 A1) and Malus domestica alpha-farnesene synthase (Tippmann et al., Biotechnol Bioeng. 2016 January; 113(1):72-81) have been optimized for the use of codons in Saccharomyces cerevisiae yeast.
[0195] According to the protocol previously described for yeast transformation, strain EQ-0134 was grown in a volume of 50 mL of complex rich medium YPGE (yeast extract peptone glycerol ethanol) at 30.degree. C. The cells are centrifuged for 5 minutes at 2,500 rpm at room temperature. The supernatant is removed and the cells are resuspended in 25 mL of sterile water and centrifuged again for 5 minutes at 2,500 rpm at room temperature. After removing the supernatant, the cells are resuspended in 400 .mu.L of 100 mM sterile lithium acetate. At the same time, the following transformation mix is prepared: 250 .mu.L of 50% PEG, 10 .mu.L of "carrier" DNA at 5 mg/mL, 36 .mu.L of 1 M lithium acetate, 10 .mu.L (3 .mu.g of one of the following combinations, pFPP45+pFPP56+pFPP20 or pL4+pFPP45+pFPP56+pFPP20) and 350 .mu.L of water.
[0196] The resuspended cells (50 .mu.L) were added to the transformation mixture and incubated at 42.degree. C. for 40 minutes in a water bath. After incubation, the tube was centrifuged for 1 minute at 5,000 rpm at room temperature and the supernatant was discarded. The cells were resuspended in 2 mL YNB (yeast nitrogen base including ammonium sulfate) with glycerol and ethanol, transferred to a 14 mL tube and incubated for 2 hours at 30.degree. C. under atmosphere enriched with 10% CO.sub.2. The final mix is spread on YNB agar medium including ammonium sulfate+CSM without LUW (leucine uracil, tryptophan)+nourseothricin if applicable, with glycerol and ethanol as carbon sources.
[0197] According to the previously described protocol, strain CEN.PK 1605 is transformed with the following plasmid combination: pL4+pFL36+pCM185+pV51TEF.
[0198] The clones obtained were genotyped for all engineering genes and then adapted on liquid medium YNB ammonium sulfate and glucose. [0199] EQ-0153 (CEN.PK1605 .DELTA.pgk1::kan) (pFPP45+pFPP56+pFPP20) [0200] EQ-0253 (CEN.PK1605 .DELTA.pgk1::kan) (pL4+pFPP56+pFPP20+pFPP45) [0201] EQ-0353 (CEN.PK1605) (pL4+pFL36+pCM185+pV51TEF)
[0202] c) Adaptation of Strains EQ-0153 and EQ-0253 to Growth in Liquid Medium with Glucose and CO.sub.2.
[0203] Batch-mode cultures in Erlenmeyer flasks are carried out with the appropriate culture medium and a 10% exogenous CO.sub.2 supply, in a shaking incubator (120 rpm, 30.degree. C.), with inoculation at 0.05 OD 600 nm measured using an EON spectrophotometer (BioTek Instruments). The strain of interest is grown on YNB+CSM-LUW medium with 10 g/L glycerol and 7.5 g/L ethanol, under conditions where PRK expression is not induced, and in the presence of nourseothricin if appropriate. Under these conditions, it is necessary to feed the strain before and after the deletion of the PGK1 gene.
[0204] After obtaining a sufficient quantity of biomass, cultures with a volume greater than or equal to 50 mL in Erlenmeyer flasks of at least 250 mL are inoculated in order to adapt the strain to the use of the PRK/RuBisCO engineering. This adaptation is carried out on YNB+CSM-LUW culture medium with 20 g/L glucose, in the presence of nourseothricin if necessary and an exogenous CO.sub.2 supply as described above.
[0205] After observation of a significant growth start, the strains are adapted to a minimum mineral medium free of the amino acids and nitrogenous bases included in the CSM-LUW, i.e. only YNB with 20 g/L glucose, nourseothricin if necessary and an exogenous CO.sub.2 supply as described above.
[0206] d) Production of Farnesene in Erlenmeyer Flasks
[0207] Saccharomyces cerevisiae strain EQ-0253, with a deletion in the glycolytic pathway at the PGK1 gene, is grown to produce farnesene while overproducing NADPH without CO.sub.2 loss, using a PRK and a RuBisCO.
[0208] This strain of interest is compared with a reference strain EQ-0353 producing farnesene following the introduction of a heterologous alpha-farnesene synthase, without deletion of PGK1 or addition of PRK and RuBisCO.
[0209] Strains EQ-0253 (CEN.PK1605 .DELTA.pgk1::kan) (pL4+pFPP56+pFPP20+pFPP45) and EQ-0353 (CEN.PK1605) (pL4+pFL36+pCM185+pV51TEF) were grown in a YNB medium with 20 g/L D-glucose, to which 100 .mu.g/L nourseothricin was added. A pre-culture containing 20 mL of culture medium was inoculated at 0.05 OD.sub.600 nm into a 250 mL baffled Erlenmeyer flask, shaken at 120 rpm for 24 h at 30.degree. C. in a Minitron incubator with an atmosphere regulated at 10% CO.sub.2. From the first pre-culture, 50 mL of medium was inoculated at 0.05 OD.sub.600 nm into a 250 mL Erlenmeyer and shaken at 120 rpm for 24 h at 30.degree. C., 10% CO.sub.2. The culture, also conducted in Erlenmeyer flasks (500 mL, baffled) from the second pre-culture, was inoculated at 0.05 OD.sub.600 nm into 100 mL of the same culture medium, to which 50 .mu.g/mL ampicillin, 10 .mu.L antifoam (Antifoam 204, Sigma, A6426) and 10% (v/v) dodecane were added (Tippman et al., Talanta (2016), 146: 100-106). The cultures were shaken at 120 rpm at 30.degree. C. in the presence of 10% CO.sub.2. Growth was monitored by measuring turbidity at 600 nm.
[0210] To extract farnesene, 500 .mu.L of organic phase was collected and centrifuged at 5,000 g for 5 min for complete separation of the two phases. The organic phase was stored at 4.degree. C. until GC-MS analysis. The detection and quantification of .alpha.-farnesene was performed by single quadrupole mass spectrometry. A Zebron ZB-FFAP column was used with hydrogen as the carrier gas at a fixed rate of 2.95 mL/min. The inlet temperature was 260.degree. C., 1 .mu.L of sample was injected in splitless mode. The initial oven temperature was 70.degree. C. (4 min) then it was gradually increased to 160.degree. C. (7.degree. C./min) then to 240.degree. C. (40.degree. C./min) where it was maintained for 1.05 min. For mass spectrometric detection, the transfer line and source temperatures were 250.degree. C. and 200.degree. C. respectively. The mass acquisition was made between t=10 min and t=20 min. An external calibration including seven points was performed using the farnesene isomer mix (Sigma, W383902) for the quantification of .alpha.-farnesene produced by the strains.
[0211] To quantify the glucose consumed by the strains, 500 .mu.L of culture medium was collected at the same farnesene extraction OD, centrifuged at 5,000 g, 5 min at 4.degree. C. The supernatant was filtered (Minicart RC4, Sartorius 0.45 .mu.m) and stored in a flask at -20.degree. C. The glucose contained in this sample was quantified by UltiMate 3000 HPLC-UV (Thermo Scientific) equipped with a pump, an 8.degree. C. refrigerated autosampler and a refractive index (RI) detector (Precision Instruments IOTA 2). A Rezex ROA-Organic Acid H.sup.+ column (8%) 150.times.7 8 mm, 8 .mu.m particle size (Phenomenex, 00H-0138-KO) was used with a Carbo-H pre-column 4.times.3.0 mm. The temperature of the column was 35.degree. C. and the flow rate was set at 0.5 mL/min. Isocratic elution was performed with an aqueous mobile phase at 5 mM H.sub.2SO.sub.4 and lasted 30 min A volume of 20 .mu.L was injected for each sample. The identification of compounds was based on the comparison of retention times with standards. The external calibration includes 10 points of variable glucose concentration (0-20 g/L).
[0212] The carbon yield Y.sub..alpha.-farnesene/Glc is calculated in grams of farnesene produced per gram of glucose consumed for both strains EQ-0253 and EQ-0353,
Y .alpha. - farnesene / Glc = farnesene ( mg / L aqueous ) glucose ( mg / L aqueous ) . ##EQU00001##
TABLE-US-00014 TABLE 13 Mass yield of .alpha.-farnesene to D-glucose Y.sub..alpha.-farnesene/glucose Yield Strains (.times.10.sup.-4) (g/g) improvement EQ-0253 12.5 +9.6% EQ-0353 11.4
[0213] The increase in the mass yield of .alpha.-farnesene to D-glucose observed was 9.6% for strain EQ-0253, compared with control strain EQ-0353.
Example 3: Improvement of Citrate Production in S. cerevisiae
[0214] a) Inactivation of the ZWF1 Gene and the IDH1 Gene in a Haploid Strain of Mating Type MAT a
[0215] Inactivation of the ZWF1 Gene
[0216] The coding phase of the hygromycin B resistance gene, derived from the hphMX cassette (loxP-pAgTEF1-hph-tAgTEF1-loxP) and contained on plasmid pUG75 (P30671)--Euroscarf), is amplified with the oligonucleotides Sdzwf1 and Rdzwf1 (Table 14). This makes it possible to generate a .DELTA.zwf1 PCR amplicon containing at its ends homologous recombination sequences of the glucose-6-phosphate dehydrogenase ZWF1 gene locus.
TABLE-US-00015 TABLE 14 Oligonucleotides Name Sequence Sdzwf1 AAGAGTAAATCCAATAGAATAGAAAACCACATAAGGCAAGA (SEQ ID TGGGTAAAAAGCCTGAACTCACCG NO: 3) Rdzwf1 ATTTCAGTGACTTAGCCGATAAATGAATGTGCTTGCATTTT (SEQ ID TTTATTCCTTTGCCCTCGGACG NO: 4) Sdpgk1 ACAGATCATCAAGGAAGTAATTATCTACTTTTTACAACAAA (SEQ ID TATAAAACAATGGGTAAGGAAAAGACTCACGTTTC NO: 5) Rdpgk1 GGGAAAGAGAAAAGAAAAAAATTGATCTATCGATTTCAATT (SEQ ID CAATTCAATTTAGAAAAACTCATCGAGCATCAAATGAAAC NO: 6) Sdidh1 TCTCCCTATCCTCATTCTTCTCCCTTTTCCTCCATAATTGT (SEQ ID AAGAGAAAAATGGGTACCACTCTTGACGACACGG NO: 7) Rdidh1 AATTTGAACACACTTAAGTTGCAGAACAAAAAAAAGGGGAA (SEQ ID TTGTTTTCATTAGGGGCAGGGCATGCTCATGTAGAGC NO: 8)
[0217] The underlined portion of the oligonucleotides corresponds to the portion perfectly homologous to the sequence of the selection gene, the rest of the sequence corresponding to the regions adjacent to the coding phase of the target gene to be deleted on the Saccharomyces cerevisiae genome.
[0218] The previously described strain CEN.PK 1605 (Mat a HISS leu2-3.112 trp1-289 ura3-52 MAL.28c) derived from the commercial strain CEN.PK 113-7D (GenBank: JRIV00000000) is transformed with the .DELTA.zwf1 PCR fragment described above.
[0219] For the transformation reaction, strain CEN.PK 1605 is grown in a volume of 50 mL of complex rich medium YPD (yeast extract peptone dextrose, here 20 g/L glucose) at 30.degree. C. to an optical density at 600 nm of 0.8. The cells are centrifuged for 5 minutes at 2,500 rpm at room temperature. The supernatant is removed and the cells are resuspended in 25 mL of sterile water and centrifuged again for 5 minutes at 2,500 rpm at room temperature. After removing the supernatant, the cells are resuspended in 400 .mu.L of 100 mM sterile lithium acetate.
[0220] At the same time, a transformation mix is prepared in a 2 mL tube as follows: 250 .mu.L of 50% PEG, 10 .mu.L of "carrier" DNA at 5 mg/mL, 36 .mu.L of 1 M lithium acetate, 10 .mu.L of purified PCR reaction (deletion cassette) and 350 .mu.L of water.
[0221] The resuspended cells (50 .mu.L) are added to the transformation mixture and incubated at 42.degree. C. for 40 minutes in a water bath. After incubation, the tube is centrifuged for 1 minute at 5,000 rpm at room temperature and the supernatant is discarded. The cells are resuspended in 2 mL of YPD (yeast extract peptone dextrose) medium, transferred to a 14 mL tube and incubated for 2 hours at 30.degree. C. at 200 rpm. The cells are then centrifuged for 1 minute at 5,000 rpm at room temperature. The supernatant is removed and the cells are resuspended in 1 mL of sterile water and centrifuged again for 1 minute and resuspended in 100 .mu.L of sterile water and spread on YPD+HygromycinB (200 .mu.g/mL).
[0222] The colonies obtained were genotyped for the validation of the deletion of the ZWF1 gene and referenced EQSC-002 (CEN.PK 1605 .DELTA.zwf1::hph).
[0223] Inactivation of the IDH1 Gene
[0224] Inactivation of this gene allows citrate to accumulate (Rodriguez et al., Microb Cell Fact. 2016 Mar. 3; 15:48).
[0225] The coding phase of the nourseothricin resistance gene, derived from the natMX cassette (loxP-pAgTEF1-nat-tAgTEF1-loxP) contained on the plasmid (pUG74 (P30670)--Euroscarf) is amplified with the oligonucleotides Sdidh1 and Rdidh1 (Table 13). This makes it possible to generate a .DELTA.idh1 PCR amplicon containing at its ends homologous recombination sequences of the isocitrate dehydrogenase IDH1 subunit gene locus.
[0226] The strains previously described, EQSC-002 (CEN.PK 1605 .DELTA.zwf1::hph) and CEN.PK 1605 (Mat a HISS leu2-3.112 trp1-289 ura3-52 MAL.28c) derived from the commercial strain CEN.PK 113-7D (GenBank: JRIV00000000) are transformed with the PCR fragment for inactivation of the IDH1 gene.
[0227] For the transformation reaction, strains EQSC-002 and CEN.PK1605 are grown in a volume of 50 mL of complex rich medium YPD (yeast extract peptone dextrose, here 20 g/L glucose) at 30.degree. C. to an optical density at 600 nm of 0.8. The cells are centrifuged for 5 minutes at 2,500 rpm at room temperature. The supernatant is removed and the cells are resuspended in 25 mL of sterile water and centrifuged again for 5 minutes at 2,500 rpm at room temperature. After removing the supernatant, the cells are resuspended in 400 .mu.L of 100 mM sterile lithium acetate.
[0228] At the same time, a transformation mix is prepared in a 2 mL tube as follows: 250 .mu.L of 50% PEG, 10 .mu.L of "carrier" DNA at 5 mg/mL, 36 .mu.L of 1 M lithium acetate, 10 .mu.L of purified PCR reaction (deletion cassette) and 350 .mu.L of water.
[0229] The resuspended cells (50 .mu.L) are added to the transformation mixture and incubated at 42.degree. C. for 40 minutes in a water bath. After incubation, the tube is centrifuged for 1 minute at 5,000 rpm at room temperature and the supernatant is discarded. The cells are resuspended in 2 mL of YPD (yeast extract peptone dextrose), transferred to a 14 mL tube and incubated for 2 hours at 30.degree. C. at 200 rpm. The cells are then centrifuged for 1 minute at 5,000 rpm at room temperature. The supernatant is removed and the cells are resuspended in 1 mL of sterile water and centrifuged again for 1 minute and resuspended in 100 .mu.L of sterile water and spread on YPD+HygromycinB 200 .mu.g/mL, 50 nourseothricin.
[0230] The colonies obtained were genotyped for the validation of the deletion of the IDH1 gene and are called EQSC-003 (CEN.PK 1605 .DELTA.zwf1::hph, .DELTA.idh1::nat) and EQSC-005 (CEN.PK 1605 .DELTA.idh1::nat)
[0231] b) Inactivation of the PGK1 Gene in a Haploid Strain of Mating Type MAT Alpha
[0232] The coding phase of the G418 resistance gene from the KanMX cassette (loxP-pAgTEF1-kanMX-tAgTEF1-loxP) contained on plasmid pUG6 (P30114)--Euroscarf is amplified with the oligonucleotides Sdpgk1 and Rdpgk1 (Table 13) to generate a .DELTA.pgk1 PCR amplicon containing at its ends homologous recombination sequences of the 3-phosphoglycerate kinase PGK1 gene locus.
[0233] Strain CEN.PK 1606 (Mat alpha HIS3 leu2-3.112 trp1-289 ura3-52 MAL.28c) derived from the commercial strain CEN.PK 113-7D (GenBank: JRIV00000000) is transformed with the PCR fragment for inactivation of the PGK1 gene.
[0234] For the transformation reaction, strain CEN.PK 1606 is grown in a volume of 50 mL of complex rich medium YPD (yeast extract peptone dextrose, here 20 g/L glucose) at 30.degree. C. to an optical density at 600 nm of 0.8. The cells are centrifuged for 5 minutes at 2,500 rpm at room temperature. The supernatant is removed and the cells are resuspended in 25 mL of sterile water and centrifuged again for 5 minutes at 2,500 rpm at room temperature. After removing the supernatant, the cells are resuspended in 400 .mu.L of 100 mM sterile lithium acetate.
[0235] At the same time, a transformation mix is prepared in a 2 mL tube as follows: 250 .mu.L of 50% PEG, 10 .mu.L of "carrier" DNA at 5 mg/mL, 36 .mu.L of 1 M lithium acetate, 10 .mu.L of purified PCR reaction (deletion cassette) and 350 .mu.L of water.
[0236] The resuspended cells (50 .mu.L) are added to the transformation mixture and incubated at 42.degree. C. for 40 minutes in a water bath. After incubation, the tube is centrifuged for 1 minute at 5,000 rpm at room temperature and the supernatant is discarded. The cells are resuspended in 2 mL of YPGE (yeast extract peptone 20 g/L glycerol, 30 g/L ethanol), transferred to a 14 mL tube and incubated for 2 hours at 30.degree. C. at 200 rpm. The cells are then centrifuged for 1 minute at 5,000 rpm at room temperature. The supernatant is removed and the cells are resuspended in 1 mL of sterile water and centrifuged again for 1 minute and resuspended in 100 .mu.L of sterile water and spread over YPGE+150 .mu.g/mL G418.
[0237] The colonies obtained were genotyped for the validation of the deletion of the PGK1 gene and referenced EQSC-008 (CEN.PK 1605, .DELTA.pgk1::kan).
[0238] c) Construction of a Strain in which IDH1, ZWF1 and PGK1 have been Inactivated by Crossing
[0239] The haploid strains of opposite mating types EQSC-003 (CEN.PK 1605 .DELTA.zwf1::hph, .DELTA.idh1::nat) and EQSC-008 (CEN.PK 1606 .DELTA.pgk1::kan) are grown overnight on agar medium:YPD (yeast extract peptone dextrose) for strain EQSC-008 and YPGE (yeast extract peptone glycerol ethanol) for strain EQSC003, at 30.degree. C. Then the two strains are crossed by direct contact on YPGE (yeast extract peptone glycerol ethanol) agar medium+150 .mu.g/mL G418+200 .mu.g/mL hygromycin B. The G418 and hygromycin B double selection eliminates the two parental strains, only the MAT a/MAT alpha, ZWF1/.DELTA.zwf1::hph, IDH1/.DELTA.idh1::nat, PGK1/.DELTA.pgk1::kan diploid strains grow on this medium. An isolated diploid clone from this crossing is collected. The presence of the three cassettes .DELTA.zwf1::hph, .DELTA.idh1::nat, .DELTA.pgk1::kan is validated by growth tests on YPGE (yeast extract peptone glycerol ethanol) agar medium supplemented with 150 .mu.g/mL G418 or 200 .mu.g/mL hygromycin B or 50 .mu.g/mL nourseothricin. The strain obtained is referenced EQSC-009 (CEN.PK 1607, MAT a/MAT alpha, ZWF1/.DELTA.zwf1::hph, IDH1/.DELTA.idh1::nat, PGK1/.DELTA.pgk1::kan).
[0240] The previously described strain EQSC-009 (CEN.PK 1607, MAT a/MAT alpha, ZWF1/.DELTA.zwf1::hph, IDH1/.DELTA.idh1::nat, PGK1/.DELTA.pgk1::kan) is grown on YPGE (yeast extract peptone glycerol ethanol) agar medium overnight at 30.degree. C. The cells are then placed in liquid culture in a deficient medium (Sporulation Medium, 1% potassium acetate+leucine+uracil+tryptophan) to induce meiosis of the diploid cells and thus lead to the formation of tetrads containing four haploid spores. The tetrads are spread on YNB.GE medium (yeast nitrogen base, glycerol, ethanol)+leucine+uracil+tryptophan+1 g/L glutamic acid+20 mg/L methionine+40 mg/L cysteine and immediately dissected (using a microdissector) to isolate the spores on the same medium. The spores are germinated for several days at 30.degree. C. The genetic content of the haploid cells thus obtained is tested by growth on selective media: YPGE (yeast extract peptone glycerol ethanol) supplemented with 150 .mu.g/mL G418 or 200 .mu.g/mL hygromycin B or 50 .mu.g/mL nourseothricin and their mating type is tested by crossing with two tectrix strains of mating type MAT a or MAT alpha. The colonies obtained are genotyped for the validation of the deletion of the PGK1, IDH1, ZWF1 genes and the absence of transcripts corresponding to these genes is validated by real-time PCR after reverse transcription of ribonucleic acids. One of the strains obtained is referenced EQSC-004 (CEN.PK 1606 MAT alpha .DELTA.zwf1::hph, .DELTA.idh1::nat, .DELTA.pgk1::kan)
[0241] d) Introduction of PRK-RuBisCO Enzymes
[0242] The six genes required for PRK-RuBisCO engineering (Table 15 below) are cloned on three plasmid vectors capable of autonomous replication, with compatible origins of replication and each carrying a different auxotrophic complementation gene, allowing the selection of strains containing the three plasmid constructs (see WO 2015107496). Two of these plasmids are single-copy with an ARS/CEN origin of replication and the third is multicopy with a 2.mu. origin.
TABLE-US-00016 TABLE 15 Description of expression cassettes and plasmid composition Codon Auxo- optimi- Termi- trophic GenBank zation Promoter nator ori marker Plasmids RbcL BAD78320.1 Yes TDH3p ADH1t 2.mu. URA3 pFPP45 RbcS BAD78319.1 Yes TEF1p PGK1t 2.mu. URA3 pFPP45 RbcX BAD80711.1 Yes TEF1p PGK1t ARS-CEN6 LEU2 pFPP56 GroES U00096 No PGI1p CYC1t ARS-CEN6 LEU2 pFPP56 GroEL AP009048 No TDH3p ADH1t ARS-CEN6 LEU2 pFPP56 PRK BAD78757.1 Yes Tet-OFF CYC1t ARS416-CEN4 TRP1 pFPP20 Empty Tet-OFF ARS416-CEN4 TRP1 pCM185 Empty TEF1p PGK1t 2.mu. URA3 pV51TEF Empty ARS-CEN6 LEU2 pFL36
[0243] According to the transformation protocol previously described, strain EQSC-004 (CEN.PK 1606 .DELTA.zwf1::hph, .DELTA.idh1::nat, .DELTA.pgk1::kan) was grown in a volume of 50 mL of complex rich medium YPGE (yeast extract peptone glycerol ethanol) at 30.degree. C. to an optical density at 600 nm of 0.8. The cells are centrifuged for 5 minutes at 2,500 rpm at room temperature. The supernatant is removed and the cells are resuspended in 25 mL of sterile water and centrifuged again for 5 minutes at 2,500 rpm at room temperature. After removing the supernatant, the cells are resuspended in 400 .mu.L of 100 mM sterile lithium acetate.
[0244] At the same time, a transformation mix is prepared in a 2 mL tube as follows: 250 .mu.L of 50% PEG, 10 .mu.L of "carrier" DNA at 5 mg/mL, 36 .mu.L of 1 M lithium acetate, 10 .mu.L (3 .mu.g) of a combination of pFPP45+pFPP56+pFPP20 and 350 .mu.L of water.
[0245] The resuspended cells (50 .mu.L) are added to the transformation mixture and incubated at 42.degree. C. for 40 minutes in a water bath. After incubation, the tube is centrifuged for 1 minute at 5,000 rpm at room temperature and the supernatant is discarded. The cells are resuspended in 2 mL of YPGE (yeast extract peptone glycerol ethanol)+2 mg/L doxycycline, transferred into a 14 mL tube and incubated for 2 hours at 30.degree. C. at 200 rpm. The cells are then centrifuged for 1 minute at 5,000 rpm at room temperature. The supernatant is removed and the cells are resuspended in 1 mL of sterile water and centrifuged again for 1 minute and resuspended in 100 .mu.L of sterile water and spread over YNB.GE (yeast nitrogen base, glycerol, ethanol)+1 g/L glutamic acid+20 mg/L methionine+40 mg/L cysteine+2 mg/L doxycycline. The strain obtained is referenced: EQSC-006 (CEN.PK 1606 .DELTA.zwf1::hph, .DELTA.idh1::nat, .DELTA.pgk1::kan) (pFPP45+pFPP56+pFPP20).
[0246] According to the transformation protocol previously described, strain EQSC-005 (CEN.PK 1605 .DELTA.idh1::nat) was grown in a volume of 50 mL of complex rich medium YPGE (yeast extract peptone glycerol ethanol) at 30.degree. C. to an optical density at 600 nm of 0.8. The cells are centrifuged for 5 minutes at 2,500 rpm at room temperature. The supernatant is removed and the cells are resuspended in 25 mL of sterile water and centrifuged again for 5 minutes at 2,500 rpm at room temperature. After removing the supernatant, the cells are resuspended in 400 .mu.L of 100 mM sterile lithium acetate.
[0247] At the same time, a transformation mix is prepared in a 2 mL tube as follows: 250 .mu.L of 50% PEG, 10 .mu.L of "carrier" DNA at 5 mg/mL, 36 .mu.L of 1 M lithium acetate, 10 .mu.L (3 .mu.g) of a combination of pV51TEF+pFL36+pCM185 and 350 .mu.L of water.
[0248] The resuspended cells (50 .mu.L) are added to the transformation mixture and incubated at 42.degree. C. for 40 minutes in a water bath. After incubation, the tube is centrifuged for 1 minute at 5,000 rpm at room temperature and the supernatant is discarded. The cells are resuspended in 2 mL of YPD (yeast extract peptone dextrose), transferred to a 14 mL tube and incubated for 2 hours at 30.degree. C. at 200 rpm. The cells are then centrifuged for 1 minute at 5,000 rpm at room temperature. The supernatant is removed and the cells are resuspended in 1 mL of sterile water and centrifuged again for 1 minute and resuspended in 100 .mu.L of sterile water and spread on YNBD (yeast nitrogen base dextrose)+2 mg/L doxycycline. The strain obtained is referenced: EQSC-007 (CEN.PK 1605 .DELTA.idh1::nat) (pV51TEF+pFL36+pCM185).
[0249] d) Adaptation and Evolution Phase of Strains EQSC-006 and EQSC-007
[0250] Adaptation of strains EQSC-006 and EQSC-007 to growth on YNB (yeast nitrogen base) liquid medium with glucose and CO.sub.2.
[0251] Batch-mode cultures in Erlenmeyer flasks are carried out with the appropriate culture medium and a 10% exogenous CO.sub.2 supply, in a shaking incubator (120 rpm, 30.degree. C.), with inoculation at 0.05 OD 600 nm measured using an EON spectrophotometer (BioTek Instruments). The strain of interest is grown on YNB+CSM-LUW medium with 10 g/L glycerol and 7.5 g/L ethanol, +50 mg/L glutamate under conditions where PRK expression is not induced.
[0252] After obtaining a sufficient quantity of biomass, cultures with a volume greater than or equal to 50 mL in Erlenmeyer flasks of at least 250 mL are inoculated in order to adapt the strain to the use of the PRK/RuBisCO engineering. This adaptation is carried out on YNB+CSM-LUW culture medium with 20 g/L glucose, 50 mg/L glutamate and an exogenous CO.sub.2 supply as described above.
[0253] After observation of a significant growth start, the strains are adapted to a minimum mineral medium free of all amino acids except those indicated below, and nitrogenous bases included in the CSM-LUW, i.e. only YNB with, in final concentrations, 20 g/L glucose, 1 g/L glutamate, 40 mg/L L-cysteine and 20 mg/L L-methionine and an exogenous CO.sub.2 supply as described above.
[0254] e) Production of Citrate in Erlenmeyer Flasks
[0255] Saccharomyces cerevisiae strain EQSC-006, with a deletion in the glycolytic pathway at the PGK1 gene, in the oxidative part of the pentose phosphate pathway and in the Krebs cycle, is grown to produce citrate without CO.sub.2 loss, using PRK and RuBisCO. This strain of interest is compared with a reference strain EQSC-007 producing citrate following inactivation of the IDH1 gene, without deletion of PGK1 or ZWF1 or addition of PRK and RuBisCO.
[0256] Strains EQSC-006 (CEN.PK 1605 .DELTA.zwf1::hph, .DELTA.idh1::nat, .DELTA.pgk1::kan, pFPP45+pFPP56+pFPP20) and EQSC-007 (CEN.PK 1605 .DELTA.idh1::nat, pV51TEF+pFL36+pCM185) were cultured in yeast nitrogen base (YNB) medium supplemented with 20 g/L D-glucose (YNB D20).
[0257] In order to establish the citrate to glucose mass yields, a pre-culture containing 20 mL of culture medium was inoculated at 0.05 OD.sub.600 nm into a 250 mL baffled Erlenmeyer flask, shaken at 120 rpm at 30.degree. C. From the first pre-culture, 50 mL of medium was inoculated at 0.05 OD.sub.600 nm into a 250 mL Erlenmeyer flask and shaken at 120 rpm, at 30.degree. C. The culture was carried out in Erlenmeyer flasks (500 mL, baffled) from the second pre-culture, inoculated at 0.05 OD.sub.600 nm into 100 mL of the same medium, at 30.degree. C., 120 rpm. Growth was monitored by measuring turbidity at 600 nm.
[0258] For citrate quantification, 500 .mu.L of culture medium was collected, centrifuged at 5,000 g, 5 min, 4.degree. C. The supernatant was filtered (Minicart RC4, Sartorius 0.45 .mu.m) and stored in a flask at -20.degree. C. before HPLC analysis (Thermo Scientific UltiMate 3000 HPLC) coupled to a single quadrupole mass spectrometer. Each sample (20 .mu.L) was injected into an Aminex HPX-87H H.sup.+ column, 300 mm.times.7.8 mm (Bio-Rad, 125-0140). An isocratic elution at a flow rate of 0.5 mL/min was carried out with an aqueous solution of 0.037% formic acid (v/v) whose pH was adjusted to 4.5 with ammonium hydroxide. The column oven temperature was 65.degree. C. The mass spectrometry analytical conditions were: negative electrospray mode, source temperature 450.degree. C., needle voltage 3 kV, cone voltage 50 V. A seven-point external calibration was performed using a commercial sodium citrate solution.
[0259] To quantify the glucose consumed by the strains, 500 .mu.L of the culture medium was collected, at the same culture OD.sub.600 nm as for citrate quantification, centrifuged at 5,000 g, 5 min at 4.degree. C. The supernatant was filtered (Minicart RC4, Sartorius 0.45 .mu.m) and stored in a flask at -20.degree. C. The glucose contained in this sample was quantified by HPLC-RI UltiMate 3000 (Thermo Scientific) equipped with a pump, an 8.degree. C. refrigerated autosampler and a refractive index (RI) detector (Precision Instruments IOTA 2). A Rezex ROA-Organic Acid H.sup.+ column (8%) 150.times.7.8 mm, 8 .mu.m particle size (Phenomenex, 00H-0138-KO) was used with a Carbo-H 4.times.3.0 mm pre-column. The column oven temperature was 35.degree. C. and the flow rate was set at 0.5 mL/min A 30 min isocratic elution was performed with an aqueous mobile phase at 5 mM H.sub.2SO.sub.4. A volume of 20 .mu.L was injected for each sample. The identification of the compounds was based on the comparison of retention times with standards. The external calibration included 10 points of variable glucose concentration (0 to 20 g/L).
[0260] The Y.sub.citrate/Glc mass yield was calculated in grams of citrate produced per gram of glucose consumed for both strains EQSC-006 and EQSC-007,
Y c i t r a t e / G l c = citrate ( mg / L aqueous ) glucose ( mg / Laqueous ) . ##EQU00002##
TABLE-US-00017 TABLE 16 Mass yield, citrate to D-glucose Y.sub.citrate/glucose Yield Strains (.times.10.sup.-3) (g/g) improvement EQSC-006 2.1 +19.5% EQSC-007 1.8
[0261] A 19.5% increase in the citrate to D-glucose mass yield was observed for strain EQSC-006 compared with control strain EQSC-007.
Example 4: Improvement of Glutamate Production in E. coli
[0262] Deletion of the alpha-ketoglutarate dehydrogenase gene increases glutamate production (Usuda et al. J Biotechnol. 2010 May 3; 147(1):17-30. doi: 10.1016/j.jbiotec.2010.02.018).
[0263] In these examples, Escherichia coli strain K12 MG1655 with a deleted sucA gene was used. This strain is derived from a gene deletion bank (Baba et al. Mol Syst Biol. 2006; 2:2006.0008) in Escherichia coli and supplied by the coli Genetic Stock Center under the name JW0715-2 and with reference 8786. (JW0715-2: MG1655 .DELTA.sucA::Kan)
[0264] 4A] Improvement of Glutamate Production by Inactivation of Glycolysis
[0265] a) Removal of the Selection Cassette by Specific Recombination of FTR Regions by Flp Recombinase
[0266] In order to be able to reuse the same deletion strategy as that used to construct strain JW0715-2 above (Rodriguez et al., 2016), the selection cassette was deleted using a recombinase.
[0267] Plasmid p707-Flpe (provided in the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit by Gene Bridges) is transformed by electroporation according to the kit protocol. The cells are selected on LB agar supplemented with 0.2% glucose, 0.0003% tetracycline and added with 0.3% L-arabinose. A counter-selection of the clones obtained is carried out by verifying that they are no longer able to grow on the same medium supplemented with 0.0015% kanamycin.
[0268] The strain obtained is called EQ.EC002: MG1655 .DELTA.sucA
[0269] b) Deletion of the Edd-Eda Operon Encoding the Entner-Doudoroff Metabolic Pathway
[0270] The deletion of the edd-eda operon is performed by homologous recombination and the use of the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit (Gene Bridges) according to the supplier's protocol. [0271] 1. Oligonucleotides designed to amplify an FRT-PKG-gb2-neo-FRT resistance gene expression cassette and having a 5' sequence homologous over 50 nucleotides to the adjacent regions of the deletion locus, i.e. at positions 1932065-1932115 and 1934604-1934654 on the chromosome thus generating recombination arms of the cassette on the bacterial genome on either side of the entire operon. [0272] 2. The Escherichia coli K-12 strain EQ.EC002 is transformed by electroporation with plasmid pRedET according to the kit protocol. The colonies obtained are selected on rich complex medium LB agar with 0.2% glucose, 0.0003% tetracycline. [0273] 3. Transformation of the amplicon obtained in the first step in the presence of RedET recombinase, induced by 0.3% arabinose in liquid LB for 1 h. To that end, a second electroporation of the cells expressing RedET by the deletion cassette is performed and the colonies are selected on LB agar supplemented with 0.2% glucose, 0.0003% tetracycline and added with 0.3% L-arabinose and 0.0015% kanamycin. [0274] 4. Plasmid p707-Flpe (provided in the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit by Gene Bridges) is transformed by electroporation according to the kit protocol. The cells are selected on LB agar supplemented with 0.2% glucose, 0.0003% tetracycline and added with 0.3% L-arabinose. A counter-selection of the clones obtained is carried out by verifying that they are no longer able to grow on the same medium supplemented with 0.0015% kanamycin. [0275] 5. The strain obtained is called EQ.EC003: MG1655 .DELTA.sucA .DELTA.edd-eda
[0276] c) Deletion of the gapA Gene
[0277] The deletion of the gapA gene is performed by homologous recombination and the use of the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit (Gene Bridges) according to the supplier's protocol. [0278] 1. Oligonucleotides designed to amplify an FRT-PKG-gb2-neo-FRT resistance gene expression cassette and having a 5' sequence homologous over 50 nucleotides to the adjacent regions of the deletion locus, i.e. the coding phase of the gene (gapA) (GenBank: X02662.1) thus generating recombination arms of the cassette on the bacterial genome. [0279] 2. The Escherichia coli K-12 strain EQ.EC003 is transformed by electroporation with plasmid pRedET according to the kit protocol. The colonies obtained are selected on rich complex medium LB agar with 0.2% glucose, 0.0003% tetracycline. [0280] 3. Transformation of the amplicon obtained in the first step in the presence of RedET recombinase which will be induced by 0.3% arabinose in liquid LB for 1 h. To that end, a second electroporation of the cells expressing RedET by the deletion cassette is performed and the colonies are selected on LB agar supplemented with 0.2% glycerol and 0.3% pyruvate, 0.0003% tetracycline and added with 0.3% L-arabinose and 0.0015% kanamycin.
[0281] Deletions are verified by genotyping and sequencing and the name of the strains obtained is [0282] EQ.EC002: MG1655 .DELTA.sucA [0283] EQ.EC003: MG1655 .DELTA.sucA .DELTA.edd-eda [0284] EQ.EC004: MG1655 .DELTA.sucA .DELTA.edd-eda .DELTA.gapA::kan
[0285] d) Insertion of the Engineering Required for CO.sub.2 Fixation
[0286] For the recombinant expression of the different components of a type I RuBisCO in E. coli, the genes described in Table 17 below are cloned as a synthetic operon containing the genes described in Table 18 below.
TABLE-US-00018 TABLE 17 Genes encoding a PRK and type I RuBisCO system Genes GenBank Organism rbcL BAD78320.1 Synechococcus elongatus rbcS BAD78319.1 Synechococcus elongatus rbcX BAD80711.1 Synechococcus elongatus Prk BAD78757.1 Synechococcus elongatus
TABLE-US-00019 TABLE 18 Composition of the expression cassettes Structure of the synthetic operon in vector pZA11 Plasmid Gene A RBS1 Gene B RBS2 Gene C RBS3 Gene D RBS4 Gene E pZA11 pEQEC005 rbcS D rbcL B rbcX F pEQEC006 rbcS D rbcL B rbcX F prk pEQEC008 prk
[0287] To control the expression level of these genes, ribosome binding sequences (RBS) presented in Table 19 below, with variable translation efficiencies (Levin-Karp et al., ACS Synth Biol. 2013 Jun. 21; 2(6):327-36. doi: 10.1021/sb400002n; Zelcbuch et al., Nucleic Acids Res. 2013 May; 41(9):e98) are inserted between the coding phase for each gene. The succession of each coding phase interspersed by an RBS sequence is constructed by successive insertions into a pZA11 vector (Expressys) that contains a PLtetO-1 promoter, a p15A origin of replication and an ampicillin resistance gene.
TABLE-US-00020 TABLE 19 RBS intercistronic sequences Name RBS sequences A (SEQ ID NO: 9) AGGAGGTTTGGA B (SEQ ID NO: 10) AACAAAATGAGGAGGTACTGAG C (SEQ ID NO: 11) AAGTTAAGAGGCAAGA D (SEQ ID NO: 12) TTCGCAGGGGGAAG E (SEQ ID NO: 13) TAAGCAGGACCGGCGGCG F (SEQ ID NO: 14) CACCATACACTG
[0288] Several strains are produced by electroporating the different vectors presented according to the above plan
[0289] EQ.EC 005.fwdarw.(EQ.EC 003+pZA11): MG1655 .DELTA.sucA .DELTA.edd-eda
[0290] EQ.EC 006.fwdarw.(EQ.EC 004+pEQEC005): MG1655 .DELTA.sucA .DELTA.edd-eda .DELTA.gapA::kan (RuBisCO)
[0291] EQ.EC 007.fwdarw.(EQ.EC 004+pEQEC006): MG1655 .DELTA.sucA .DELTA.edd-eda .DELTA.gapA::kan (RuBisCO+PRK)
[0292] EQ.EC 009.fwdarw.(EQ.EC 004+pEQEC008): MG1655 .DELTA.sucA .DELTA.edd-eda .DELTA.gapA::kan (PRK)
[0293] Clones are selected on LB medium supplemented with 2 g/L glycerol and 5 g/L pyruvate and with 100 mg/L ampicillin. After obtaining a sufficient quantity of biomass, cultures with a volume greater than or equal to 50 mL in a minimum 250 mL Erlenmeyer flask are inoculated in order to adapt the strain to the use of the PRK/RuBisCO engineering. This adaptation is carried out on LB culture medium with 2 g/L glucose, and an exogenous CO.sub.2 supply at 37.degree. C. as described above.
[0294] e) Glutamate Production
[0295] For glutamate production, cells from 500 mL of LB culture are inoculated into 20 mL of MS medium (40 g/L glucose, 1 g/L MgSO.sub.4.7H.sub.2O, 20 g/L (NH.sub.4).sub.2SO.sub.4, 1 g/L KH.sub.2PO.sub.4, 10 mg/L FeSO.sub.4.7H.sub.2O, 10 mg/L MnSO.sub.4.7H.sub.2O, 2 g/L yeast extract, 30 g/L CaCO.sub.3, 100 mg/L ampicillin at a pressure of 0.1 atmosphere CO.sub.2.
[0296] Residual glutamate and glucose are measured with a bioanalyzer (Sakura Seiki). The carbon yield Y.sub.p/s is calculated in grams of glutamate produced per gram of glucose consumed.
[0297] This yield increases significantly by 10% for strains EQ.EC 007 (RuBisCO+PRK) compared with the control strains EQ.EC 005 (empty), EQ.EC 006 (RuBisCO only). The control strain EQ.EC 009 (PRK alone) is not viable.
[0298] 4B] Improvement of Production by Inactivation of Glycolysis and of the Pentose Phosphate Oxidative Pathway
[0299] a) Removal of the Selection Cassette by Specific Recombination of FTR Regions by Flp Recombinase
[0300] This step is performed in the same way as example 4A] above.
[0301] The strain obtained is called EQ.EC002: MG1655 .DELTA.sucA
[0302] b) Deletion of the zwf Gene
[0303] The deletion of the zwf gene (GeneID: 946370) is performed by homologous recombination and the use of the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit (Gene Bridges) according to the supplier's protocol, as detailed in Example 4A].
[0304] The strain obtained is called EQ.EC010: MG1655 .DELTA.sucA .DELTA.zwf
[0305] c) Deletion of the gapA Gene
[0306] The deletion of the gapA gene in the Escherichia coli K-12 strain EQ.EC010 is performed by homologous recombination and the use of the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit (Gene Bridges) according to the supplier's protocol, as detailed in Example 4A].
[0307] Deletions are verified by genotyping and sequencing and the name of the strains obtained is: [0308] EQ.EC002: MG1655 .DELTA.sucA [0309] EQ.EC010: MG1655 .DELTA.sucA .DELTA.zwf [0310] EQ.EC011: MG1655 .DELTA.sucA .DELTA.zwf .DELTA.gapA
[0311] d) Insertion of the Engineering Required for CO.sub.2 Fixation
[0312] For the recombinant expression of the different components of the functional PRK/RuBisCO system in E. coli, the genes described in Table 20 and encoding a type I RuBisCO, a phosphoribulokinase, a chaperone and a carbonic anhydrase are cloned as a synthetic operon containing the genes described above (Table 21).
TABLE-US-00021 TABLE 20 Genes encoding a type I RuBisCO, a phosphoribulokinase and a carbonic anhydrase Genes GenBank Organism rbcL BAD78320.1 Synechococcus elongatus rbcS BAD78319.1 Synechococcus elongatus rbcX BAD80711.1 Synechococcus elongatus Prk BAD78757.1 Synechococcus elongatus icfA WP_011378036.1 Synechococcus elongatus
TABLE-US-00022 TABLE 21 Plasmid names and expression cassette composition Structure of the synthetic operon in vector pZA11 Plasmid Gene A RBS1 Gene B RBS2 Gene C RBS3 Gene D RBS4 Gene E pZA11 pEQEC006 rbcS D rbcL B rbcX F prk pEQEC007 rbcS D rbcL B rbcX F prk A icfA
[0313] To control the expression level of these genes, ribosome binding sequences (RBS) presented in Table 17 (see Example 4A]), with variable translation efficiencies (Levin-Karp et al., ACS Synth Biol. 2013 Jun. 21; 2(6):327-36. doi: 10.1021/sb400002n; Zelcbuch et al., Nucleic Acids Res. 2013 May; 41(9):e98) are inserted between the coding phase of each gene. The succession of each coding phase interspersed by an RBS sequence is constructed by successive insertions into a pZA11 vector (Expressys) that contains a PLtetO-1 promoter, a p15A origin of replication and an ampicillin resistance gene. The addition of a carbonic anhydrase (icfA) also allows an inter-conversion of bicarbonate ions into available CO.sub.2 molecules and improves the efficiency of RuBisCO.
[0314] Several strains are produced by electroporating the different vectors presented according to the plan below
[0315] EQ.EC 012.fwdarw.(EQ.EC 002+pZA11): MG1655 .DELTA.sucA
[0316] EQ.EC 014.fwdarw.(EQ.EC 011+pEQEC006): MG1655 .DELTA.sucA .DELTA.zwf .DELTA.gapA (RuBisCO+PRK)
[0317] EQ.EC 015.fwdarw.(EQ.EC 011+pEQEC007): MG1655 .DELTA.sucA .DELTA.zwf .DELTA.gapA (RuBisCO+PRK+carbonic anhydrase)
[0318] After transformation, clones are selected on LB glycerol, pyruvate medium supplemented with 100 mg/L ampicillin. An adaptation and evolution phase of the strains with PRK and RuBisCO engineering is performed as described in Example 4A].
[0319] e) Glutamate Production
[0320] For glutamate production, cells from 500 mL of LB culture are inoculated into 20 mL of MS medium (40 g/L glucose, 1 g/L MgSO.sub.4.7H.sub.2O, 20 g/L (NH4).sub.2SO.sub.4, 1 g/L KH.sub.2PO.sub.4, 10 mg/L FeSO.sub.4.7H.sub.2O, 10 mg/L MnSO.sub.4.7H.sub.2O, 2 g/L yeast extract, 30 g/L CaCO.sub.3, 100 mg/L ampicillin at a pressure of 0.1 atmosphere CO.sub.2.
[0321] Residual glutamate and glucose are measured with a bioanalyzer (YSI Inc.). The carbon yield Y.sub.p/s is calculated in grams of glutamate produced per gram of glucose consumed.
[0322] This yield increases significantly by 15% for strains EQ.EC 014 (RuBisCO+PRK) and EQ.EC 015 (RuBisCO+PRK+carbonic anhydrase) compared with the control strains EQ.EC 012 (empty).
[0323] 4C] Improvement of Production by Inactivation of Glycolysis and Oxidative Pentose Phosphate Pathway, and Overexpression of Pyruvate Decarboxylase and Glutamate Dehydrogenase.
[0324] a) Removal of the Selection Cassette by Specific Recombination of FTR Regions by Flp Recombinase
[0325] This step is performed in the same way as example 4A] above.
[0326] The strain obtained is called EQ.EC002: MG1655 .DELTA.sucA
[0327] b) Deletion of the zwf Gene
[0328] The deletion of the zwf gene (GeneID: 946370) is performed by homologous recombination and the use of the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit (Gene Bridges) according to the supplier's protocol, as detailed in Example 4A]. The strain obtained is called EQ.EC010: MG1655 .DELTA.sucA .DELTA.zwf
[0329] c) Deletion of the gapA Gene
[0330] The deletion of the gapA gene in the Escherichia coli K-12 strain EQ.EC010 is performed by homologous recombination and the use of the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit (Gene Bridges) according to the supplier's protocol, as detailed in Example 4A]. Deletions are verified by genotyping and sequencing and the name of the strains obtained is: [0331] EQ.EC002: MG1655 .DELTA.sucA [0332] EQ.EC010: MG1655 .DELTA.sucA .DELTA.zwf [0333] EQ.EC011: MG1655 .DELTA.sucA .DELTA.zwf .DELTA.gapA
[0334] d) Insertion of the Engineering Necessary for CO.sub.2 Fixation
[0335] For the recombinant expression of the different components of the functional PRK/RuBisCO system in E. coli, the genes described in Table 22 and encoding a type II RuBisCO, a phosphoribulokinase and a carbonic anhydrase are cloned as a synthetic operon containing the genes described above (Table 23).
TABLE-US-00023 TABLE 22 Genes encoding a type II RuBisCO, a phosphoribulokinase, a carbonic anhydrase, a glutamate dehydrogenase and a pyruvate carboxylase Genes GenBank Organism cbbM YP_427487.1 Rhodospirillum rubrum Prk BAD78757.1 Synechococcus elongatus CA YP_427143.1 Rhodospirillum rubrum gdhA NP_416275.1 Escherichia coli K-12 pycA NP_389369.1 Bacillus subtilis
TABLE-US-00024 TABLE 23 Plasmid names and expression cassette composition Structure of the synthetic operon in vector pZA11 Plasmid Gene A RBS1 Gene B RBS2 Gene C RBS3 Gene D RBS4 Gene E pZA11 pEQEC009 cbbM B gdhA C pycA E prk pEQEC010 cbbM B gdhA C pycA E prk D CA pEQEC011 B gdhA C pycA
[0336] To control the expression level of these genes, ribosome binding sequences (RBS) presented in Table 17 (see Example 4A]), with variable translation efficiencies, are inserted between the coding phase of each gene. The succession of each coding phase interspersed by an RBS sequence is constructed by successive insertions into a pZA11 vector (Expressys) that contains a PLtetO-1 promoter, a p15A origin of replication and an ampicillin resistance gene. The addition of a glutamate dehydrogenase (gdhA) and a pyruvate carboxylase (pycA) allows a better production of glutamic acid. The addition of a carbonic anhydrase (CA) also allows an interconversion of bicarbonate ions into available CO.sub.2 molecules and improves the efficiency of RuBisCO.
[0337] Several strains are produced by electroporating the different vectors presented according to the plan below:
[0338] EQ.EC 016.fwdarw.(EQ.EC 002+pEQEC011): MG1655 .DELTA.sucA (glutamate dehydrogenase+pyruvate carboxylase)
[0339] EQ.EC 017.fwdarw.(EQ.EC 011+pEQEC009): MG1655 .DELTA.sucA .DELTA.zwf .DELTA.gapA (RuBisCO+PRK+glutamate dehydrogenase+pyruvate carboxylase)
[0340] EQ.EC 018.fwdarw.(EQ.EC 011+pEQEC010): MG1655 .DELTA.sucA .DELTA.zwf .DELTA.gapA (RuBisCO+PRK+carbonic anhydrase+glutamate dehydrogenase+pyruvate carboxylase+carbonic anhydrase)
[0341] After transformation, clones are selected on LB glycerol, pyruvate medium supplemented with 100 mg/L ampicillin. An adaptation and evolution phase of the strains with PRK and RuBisCO engineering is performed as described in Example 4A].
[0342] e) Glutamate Production
[0343] For glutamate production, cells from 500 mL of LB culture are inoculated into 20 mL of MS medium (40 g/L glucose, 1 g/L MgSO.sub.4.7H.sub.2O, 20 g/L (NH.sub.4).sub.2SO.sub.4, 1 g/L KH.sub.2PO.sub.4, 10 mg/L FeSO.sub.4.7H.sub.2O, 10 mg/L MnSO.sub.4.7H.sub.2O, 2 g/L yeast extract, 30 g/L CaCO.sub.3, 100 mg/L ampicillin at a pressure of 0.1 atmosphere CO.sub.2.
[0344] Residual glutamate and glucose are measured with a bioanalyzer (YSI Inc.). The carbon yield Y.sub.p/s is calculated in grams of glutamate produced per gram of glucose consumed.
[0345] This yield increases significantly by 15% for strains EQ.EC 017 and EQ.EC 018 compared with the control strain EQ.EC 016.
Example 5: Improvement of Polyhydroxybutyrate Production in C. necator
[0346] The increase in reducing power obtained through the genetic modifications proposed according to the invention may also have a considerable gain over existing metabolic pathways.
[0347] This is the case for the bacterial strain Cupriavidus necator ATCC 17699 which naturally produces polyhydroxybutyrate (PHB). This bacterium is capable of developing under both autotrophic and heterotrophic conditions. The deletion of the gapA gene (glyceraldehyde-3-phosphate dehydrogenase NC_008313.1) diverts the metabolic flux to the pentose phosphate pathway and increases the pool of NADPH reduced nucleotides thus increasing the PHB production yield.
[0348] This C. necator H16 strain has a megaplasmid pHG1 and two chromosomes. The deletion of the gapA gene is performed by generating a vector containing the Bacillus subtilis suicide gene sacB for Gram-negative bacteria (Quandt et al., Gene. 1993 May 15; 127(1):15-21; Lindenkamp et al., Appl Environ Microbiol. 2010 August; 76(16):5373-82 and Appl Environ Microbiol. 2012 August; 78(15):5375-83).
[0349] a) Inactivation of the Entner-Doudoroff Metabolic Pathway
[0350] Two PCR amplicons corresponding to adjacent regions of the edd and eda genes (upstream of edd and downstream of eda) are cloned by restriction according to the procedure described in Srinivasan et al. (Appl Environ Microbiol. 2002 December; 68(12):5925-32), in plasmid pJQ200mp18Cm.
[0351] The modified plasmid pJQ200mp18Cm::.DELTA.edd-eda is then transformed into an E. coli strain S17-1 by the calcium chloride transformation method. The transfer of genetic material into C. necator is done by conjugation by depositing on agar a spot of C. necator culture on a dish containing a cell monolayer of S17-1 bacteria. Selection is made on nutrient broth (NT) medium at 30.degree. C. in the presence of 10% sucrose for purposes of selection (Hogrefe et al., J Bacteriol. 1984 April; 158(1):43-8) and validated on a mineral medium containing 50 .mu.g/mL chloramphenicol.
[0352] The deletions are validated by genotyping and sequencing. The resulting strain EQCN_002 therefore has deletions of the genes of the Entner-Doudoroff metabolic pathway edd-eda. EQCN_002: H16 .DELTA.edd-eda.
[0353] b) Inactivation of the Glycolysis Pathway
[0354] Two PCR amplicons corresponding to adjacent regions of the gapA gene are cloned by restriction according to the procedure described in Lindenkamp et al. 2012, in plasmid pjQ200mp18Tc.
[0355] The modified plasmid pjQ200mp18Tc::.DELTA.gapA is then transformed into an E. coli strain S17-1 by the calcium chloride transformation method. The transfer of genetic material is done by conjugation by depositing on agar a spot of C. necator culture on a plate containing a cell monolayer of S17-1 bacteria. Selection is made on nutrient broth (NT) medium at 30.degree. in the presence of 10% sucrose for purposes of selection (Hogrefe et al., J Bacteriol. 1984 April; 158(1):43-8) and validated on a mineral medium containing 25 .mu.g/mL tetracycline.
[0356] The deletions are validated by genotyping and sequencing. The strain obtained, EQCN_003, therefore has a deletion of the gapA gene. EQCN_003: H16 .DELTA.edd-eda .DELTA.gapA.
[0357] Strain EQCN_003, with a deletion in the glycolytic pathway at the gapA gene and in the Entner-Doudoroff pathway at the edd-eda genes, is grown to improve PHB production yield by fixing exogenous CO.sub.2 via the use of the PRK and RuBisCO enzymes.
[0358] b) Production of PHB in a Bioreactor
[0359] The inoculum from a frozen stock is spread on solid medium at a rate of 50 to 100 .mu.L from a cryotube incubated at 30.degree. C. for 48 to 96 h in the presence of fructose. The expression of genes encoding RuBisCO and PRK are maintained in C. necator under heterotrophic aerobic conditions (Rie Shimizu et al., Sci Rep. 2015; 5: 11617. Published online 2015 Jul. 1).
[0360] Batch cultures in Erlenmeyer flasks (10 mL in 50 mL, then 50 mL in 250 mL) are carried out with the appropriate culture medium, in 20 g/L fructose and a 10% exogenous CO.sub.2 supply in a shaking incubator (100-200 rpm, 30.degree. C.), with a minimum inoculation of 0.01.
[0361] The strain of interest EQCN_003 improving PHB production yield is compared with a reference strain H16 naturally accumulating PHB under heterotrophic conditions in the presence of a nutritional limitation.
[0362] The productivity of the strains is compared in bioreactors. Cultures carried out in bioreactors are seeded from solid and/or liquid amplification chains in Erlenmeyer flasks under the conditions described above. The bioreactors, of type My-control (Applikon Biotechnology, Delft, Netherlands) 750 mL or Biostat B (Sartorius Stedim, Gottingen, Germany) 2.5 L, are seeded at a density equivalent to 0.01 OD.sub.620 nm.
[0363] The accumulation of PHB is decoupled from growth. The culture is regulated at 30.degree. C., aeration is between 0.1 VVM (gas volume/liquid volume/min) and 1 VVM in order to maintain a minimum dissolved oxygen concentration above 20% (30.degree. C., 1 bar), shaking is adapted according to the scale of the bioreactor used. The inlet gas flow consists of air optionally supplemented with CO.sub.2. CO.sub.2 supplementation is between 1% and 10%. The pH is adjusted to 7 with a 14% or 7% ammonia solution. The fed-batch culture method allows a supply of non-limiting carbon substrate combined with a limitation of phosphorus or nitrogen, while maintaining a constant carbon/phosphorus or carbon/nitrogen ratio. PHB extraction and quantification are performed according to the method of Brandl et al. (Appl Environ Microbiol. 2013 July; 79(14):4433-9). The protocol consists in adding 1 mL of chloroform to 10 mg of lyophilized cells, followed by 850 .mu.L of methanol and 150 .mu.L of sulfuric acid. The mixture is heated for 2.5 h at 100.degree. C., cooled and 500 .mu.L of water is added. The two phases are separated by centrifugation and the organic phase is dried by adding sodium sulfate The samples are filtered and analyzed as described by Muller et al. (Appl Environ Microbiol. 2013 July; 79(14):4433-9).
[0364] A comparison of wild-type C. necator H16 cultures and strain EQCN_003: H16 .DELTA.edd-eda .DELTA.gapA shows a 5% increase in carbon yield, corresponding here to the ratio grams of PHB per gram of fructose consumed.
Example 6: Improvement of GABA Production in E. coli
[0365] An Escherichia coli K-12 strain, genetically modified to increase the yield of its glutamate production according to example 4B], can also be modified to allow the constitutive expression of a glutamate decarboxylase gadB (Gene ID: 946058) and thus increase the production yield of .gamma.-aminobutyric acid.
[0366] The deletion of the alpha-ketoglutarate dehydrogenase gene also increases glutamate production (Usuda et al. J Biotechnol. 2010 May 3; 147(1):17-30. doi: 10.1016/j.jbiotec.2010.02.018).
[0367] In this example, the following strains are used, obtained from example 4B]: [0368] EQ.EC002: MG1655_.DELTA.sucA [0369] EQ.EC010: MG1655_.DELTA.sucA .DELTA.zwf [0370] EQ.EC011: MG1655 .DELTA.sucA .DELTA.zwf .DELTA.gapA
[0371] a) Constitutive Overexpression of the gadB Gene
[0372] Overexpression of the gadB gene is subcloned into a bacterial expression vector pZE21MCS (EXPRESSYS). This vector has a ColE1 origin of replication and a kanamycin antibiotic resistance gene.
[0373] Rapidly, the coding phase of the gadB gene (Gene ID: 946058) is amplified from the genome of strain MG1655 .DELTA.sucA with primers homologous to the Escherichia coli K-12 genome covering positions 1570595 to 1570645 and 1572095 to 1572045. Each of these primers is coupled to floating sequences homologous over 18 nucleotides at the ends of the fragment obtained by amplifying vector pZE21MCS excluding the multiple cloning site. The two amplicons are combined according to the protocol of the In-Fusion.RTM. HD Cloning Kit User Manual--Clontech to form plasmid pEQEC030 allowing the constitutive overexpression of the gadB gene.
[0374] b) Insertion of the Engineering Required for CO.sub.2 Fixation
[0375] For the recombinant expression of the different components of a functional type I RuBisCO in E. coli, the genes described in Table 17 (Example 4A]), are cloned as a synthetic operon following the construction structure described in Table 22.
[0376] Assembly of the Different Vectors
[0377] The coding sequences (CDS) of the genes described in Table 24 are amplified and assembled into blocks according to the protocol provided with the NEBuilder.RTM. HiFi DNA Assembly Master Mix Kit (E2321) so as to obtain three integration blocks described in Table 24. Each block is then amplified according to the protocol of the In-Fusion.RTM. HD Cloning Kit User Manual--Clontech to form the plasmids described below in Table 24.
TABLE-US-00025 TABLE 24 Composition of expression cassettes Structure of the synthetic operon in vector pZA11 Block I Block II Block III Plasmid CDS A RBS1 CDS B RBS2 CDS C RBS3 CDS D pZA11 pEQEC006 rbcS D rbcL B rbcX F prk
[0378] To control the expression level of these genes, ribosome binding sequences (RBS) presented in Table 19 (Example 4B]), with variable translation efficiencies (Levin-Karp et al., ACS Synth Biol. 2013 Jun. 21; 2(6):327-36. doi: 10.1021/sb400002n; Zelcbuch et al., Nucleic Acids Res. 2013 May; 41(9):e98) are inserted between the coding phase for each gene. The succession of each coding phase interspersed by an RBS sequence is constructed by successive insertions into a pZA11 vector (Expressys) that contains a PLtetO-1 promoter, a p15A origin of replication and an ampicillin resistance gene. The addition of a glutamate decarboxylase (gadB) also allows a conversion of glutamate to gamma-aminobutyrate (GABA).
[0379] Several strains are produced by electroporating the different vectors presented according to the plan below [0380] EQ.EC 013.fwdarw.(EQ.EC 002+pZA11+pEQ030): MG1655 .DELTA.sucA+(gadB) [0381] EQ.EC 020.fwdarw.(EQ.EC 011+pEQ030+pEQEC006): MG1655 .DELTA.sucA .DELTA.zwf .DELTA.gapA+(gadB)+(RuBisCO+PRK)
[0382] After transformation, clones are selected on LB glycerol, pyruvate medium supplemented with 100 mg/L ampicillin and 30 mg/L kanamycin. An adaptation and evolution phase of the strains with PRK and RuBisCO engineering is performed as described in Example 4A].
[0383] c) GABA Production
[0384] For the production of GABA, cells from 500 mL of LB culture are inoculated into 20 mL of MS medium (40 g/L glucose, 1 g/L MgSO.sub.4.7H.sub.2O, 20 g/L (NH.sub.4).sub.2SO.sub.4, 1 g/L KH.sub.2PO.sub.4, 10 mg/L FeSO.sub.4.7H.sub.2O, 10 mg/L MnSO.sub.4.7H.sub.2O, 2 g/L yeast extract, 30 g/L CaCO.sub.3, 100 mg/L ampicillin and 30 mg/L kanamycin at a pressure of 0.1 atmosphere CO.sub.2, at 30.degree. C. at pH 3.5.
[0385] The GABA concentration is measured by high-performance liquid chromatography (HPLC), using an OptimaPak C18 column (4.6.times.150 mm, RS Tech Corporation, Daejeon, Korea). The samples are centrifuged at 12,000 rpm for 5 minutes, 100 .mu.L of the supernatant transferred into a new Eppendorf tube. The following reagents are added to these tubes: 200 .mu.L of 1 M sodium bicarbonate buffer (pH 9.8), 100 .mu.L of 80 g/L dansyl chloride in acetonitrile and 600 .mu.L of double-distilled water. The mixture is incubated at 80.degree. C. for 40 minutes. The reaction is stopped by adding 100 .mu.L of 2% acetic acid. The mixture is centrifuged at 12,000 rpm for 5 minutes. The supernatant is then filtered through a 0.2 .mu.m Millipore filter and analyzed by HPLC on an Agilent system using a UV detector. Derivatized samples are separated using a binary non-linear gradient using eluent A [tetrahydrofuran/methanol/sodium acetate 50 mM at pH 6.2 (5: 75: 420, by volume)] and eluent B (methanol). Residual glucose is measured with a bioanalyzer (YSI Inc.).
[0386] The carbon yield Y.sub.p/s is calculated in grams of GABA produced per gram of glucose consumed.
[0387] This yield increases significantly by 15% for strain EQ.EC 020 .DELTA.sucA .DELTA.zwf .DELTA.gapA (RuBisCO+PRK)+(GadB) compared with the control strains EQ.EC 013 .DELTA.sucA (GadB).
Example 7: Improvement of Succinate and Oxalate Production in E. coli
[0388] An Escherichia coli K-12 strain, genetically modified to allow constitutive expression of a glyoxylate dehydrogenase FPGLOXDH1 (Gene ID: 946058) from Fomitopsis palustris, to reduce expression of the icd gene (Gene ID: 945702), and to inactivate the aceB (GeneID 948512) and sdhA (Gene ID: 945402) genes, would increase succinate and oxalic acid production yield.
[0389] The reduction in isocitrate dehydrogenase (icd) expression allows the metabolic flux to be redirected to the glyoxylic shunt. Inactivation of malate synthase (aceB) and succinate dehydrogenase (sdhA) prevents the glyoxylate and succinate, respectively, produced from being re-consumed. Deletion of the succinate dehydrogenase gene increases succinate production under aerobic conditions (Yang et al., Microbiol res. 2014 May-June; 169(5-6):432-40). Deletion of the malate synthase gene allows the accumulation of glyoxylate which will be converted to oxalate by the constitutive expression of glyoxylate dehydrogenase.
[0390] In this example, an Escherichia coli K-12 strain MG1655 in which the sdhA gene has been deleted is used. This strain is derived from a gene deletion bank (Baba et al. Mol Syst Biol. 2006; 2:2006.0008) in Escherichia coli K-12 and supplied by the coli Genetic Stock Center under the name JW0715-2 and with reference 8302. (JW0713-1: MG1655 .DELTA.sdhA::Kan).
[0391] a) Removal of the Selection Cassette by Specific Recombination of FTR Regions by Flp Recombinase
[0392] In order to be able to reuse the same deletion strategy as that used to construct strain JW0715-2 above (Rodriguez et al., 2016), the selection cassette is deleted using a recombinase.
[0393] Plasmid p707-Flpe (provided in the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit by Gene Bridges) is transformed by electroporation according to the kit protocol. The cells are selected on LB agar supplemented with 0.2% glucose, 0.0003% tetracycline and added with 0.3% L-arabinose. A counter-selection of the clones obtained is carried out by verifying that they are no longer able to grow on the same medium supplemented with 0.0015% kanamycin.
[0394] The strain obtained is called EQ.EC040: MG1655 .DELTA.sdhA
[0395] b) Deletion of the aceB Gene
[0396] The deletion of the aceB gene (GeneID 948512) is performed by homologous recombination and the use of the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit (Gene Bridges) according to the supplier's protocol.
[0397] Oligonucleotides designed to amplify an FRT-PKG-gb2-neo-FRT resistance gene expression cassette and having a 5' sequence homologous over 50 nucleotides to the adjacent regions of the deletion locus, i.e. at positions 4215428 to 4215478.and 4217129.to 4217079 on the chromosome thus generating recombination arms of the cassette on the bacterial genome on either side of the aceB gene coding sequence.
[0398] The Escherichia coli K-12 strain EQ.EC040 is transformed by electroporation with plasmid pRedET according to the kit protocol. The colonies obtained are selected on rich complex medium LB agar with 0.2% glucose, 0.0003% tetracycline.
[0399] Transformation of the amplicon obtained in the first step in the presence of RedET recombinase, induced by 0.3% arabinose in liquid LB for 1 h. To that end, a second transformation of the deletion cassette is performed by electroporation in cells expressing RedET and the colonies are selected on LB agar supplemented with 0.2% glucose, 0.0003% tetracycline and added with 0.3% L-arabinose and 0.0015% kanamycin.
[0400] Plasmid p707-Flpe (provided in the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit by Gene Bridges) is transformed by electroporation according to the kit protocol. The cells are selected on LB agar supplemented with 0.2% glucose, 0.0003% tetracycline and added with 0.3% L-arabinose. A counter-selection of the clones obtained is carried out by verifying that they are no longer able to grow on the same medium supplemented with 0.0015% kanamycin.
[0401] The strain obtained is called EQ.EC041: MG1655 .DELTA.sdhA .DELTA.aceB
[0402] c) Change in the icd Gene Promoter
[0403] i. Strategy
[0404] The replacement of the native promoter of the icd gene (Gene ID: 945702) by a weaker promoter is performed by homologous recombination and the use of the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit (Gene Bridges) according to the supplier's protocol.
[0405] ii. Introduction of the Weak Promoter P oxb1
[0406] The icd gene promoter is replaced by a cassette coupling the promoter P.sub.oxb1, characterized as weak, and an antibiotic resistance gene cassette to allow the selection of the insertion of the P.sub.oxb1 cassette with an antibiotic resistance gene.
[0407] Oligonucleotides designed to amplify an FRT-PKG-gb2-neo-FRT resistance gene expression cassette and having a 5' sequence homologous over 50 nucleotides to the left adjacent region of the P.sub.icd promoter locus (Genomic target LA) for the sense oligo, i.e. at positions 1194911 to 1194961 on the genome, and the Spacer R sequence (Table 23) for the reverse oligo allow amplification of a fragment allowing assembly with the P.sub.oxb1 fragment.
[0408] Oligonucleotides designed to amplify the P.sub.oxb1 promoter from plasmid PSF-OXB1 (Sigma # OGS553) and having a 5' sequence homologous over 50 nucleotides to the right adjacent region of the P.sub.icd promoter locus (Genomic target RA) for the reverse oligo, i.e. at positions 1195173 to 1195123 on the genome, and the Spacer S sequence (Table 25) for the oligo produce amplification of the P.sub.oxb1 fragment.
[0409] The amplification of a fusion fragment using the NEBuilder.RTM. HiFi DNA Assembly Master Mix Kit (E2321) allows the replacement promoter to be combined with an antibiotic selection cassette.
TABLE-US-00026 TABLE 25 Primer sequences for amplifying the OXB1 gene promoter Sequences of homology Name with vector PSF-OXB1 POXB1-S TCGTTGCGTTACACACAC (SEQ ID NO: 15) POXB1-R TGTGTCGAGTGGATGGTAG (SEQ ID NO: 16) Spacer S GCATGAATTCG (SEQ ID NO: 17) Spacer R CGAATTCATGC (SEQ ID NO: 18)
[0410] The Escherichia coli K-12 strain EQ.EC041 is transformed by electroporation with plasmid pRedET according to the kit protocol. The colonies obtained are selected on rich complex medium LB agar with 0.2% glucose, 0.0003% tetracycline.
[0411] Transformation of the amplicon obtained in the first step in the presence of RedET recombinase, induced by 0.3% arabinose in liquid LB for 1 hour. To that end, a second transformation of the deletion cassette is performed by electroporation in cells expressing RedET and the colonies are selected on LB agar supplemented with 0.2% glucose, 0.0003% tetracycline and added with 0.3% L-arabinose and 0.0015% kanamycin.
[0412] Plasmid p707-Flpe (provided in the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit by Gene Bridges) is transformed by electroporation according to the kit protocol. The cells are selected on LB agar supplemented with 0.2% glucose, 0.0003% tetracycline and added with 0.3% L-arabinose. A counter-selection of the clones obtained is carried out by verifying that they are no longer able to grow on the same medium supplemented with 0.0015% kanamycin.
[0413] The strain obtained is called EQ.EC042: MG1655 .DELTA.sdhA .DELTA.aceB P.sub.icd::P.sub.oxb1
[0414] d) Deletion of the zwf Gene
[0415] The deletion of the zwf gene (GeneID: 946370) is performed by homologous recombination and the use of the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit (Gene Bridges) according to the supplier's protocol.
[0416] Oligonucleotides designed to amplify an FRT-PKG-gb2-neo-FRT resistance gene expression cassette and having a 5' sequence homologous over 50 nucleotides to the adjacent regions of the deletion locus, i.e. at positions 1934789 to 1934839 and 1936364 to 1936314 on the chromosome thus generating recombination arms of the cassette on the bacterial genome on either side of the entire operon.
[0417] The Escherichia coli K-12 strain EQ.EC042 is transformed by electroporation with plasmid pRedET according to the kit protocol. The colonies obtained are selected on rich complex medium LB agar with 0.2% glucose, 0.0003% tetracycline.
[0418] Transformation of the amplicon obtained in the first step in the presence of RedET recombinase, induced by 0.3% arabinose in liquid LB for 1 h. To that end, a second transformation of the deletion cassette is performed by electroporation in cells expressing RedET and the colonies are selected on LB agar supplemented with 0.2% glucose, 0.0003% tetracycline and added with 0.3% L-arabinose and 0.0015% kanamycin.
[0419] Plasmid p707-Flpe (provided in the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit by Gene Bridges) is transformed by electroporation according to the kit protocol. The cells are selected on LB agar supplemented with 0.2% glucose, 0.0003% tetracycline and added with 0.3% L-arabinose. A counter-selection of the clones obtained is carried out by verifying that they are no longer able to grow on the same medium supplemented with 0.0015% kanamycin.
[0420] The strain obtained is called EQ.EC043: MG1655 .DELTA.sdhA .DELTA.aceB P.sub.icd::P.sub.oxb1 .DELTA.zwf
[0421] e) Deletion of the gapA Gene
[0422] The deletion of the gapA gene is performed by homologous recombination and the use of the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit (Gene Bridges) according to the supplier's protocol.
[0423] Oligonucleotides designed to amplify an FRT-PKG-gb2-neo-FRT resistance gene expression cassette and having a 5' sequence homologous over 50 nucleotides to the adjacent regions of the deletion locus, i.e. the coding phase of the gene (gapA) (GenBank: X02662.1) thus generating recombination arms of the cassette on the bacterial genome.
[0424] The Escherichia coli K-12 strain EQ.EC043 is transformed by electroporation with plasmid pRedET according to the kit protocol. The colonies obtained are selected on rich complex medium LB agar with 0.2% glucose, 0.0003% tetracycline.
[0425] Transformation of the amplicon obtained in the first step in the presence of RedET recombinase is induced by 0.3% arabinose in liquid LB for 1 h. To that end, a second electroporation of the cells expressing RedET by the deletion cassette is performed and the colonies are selected on LB agar supplemented with 0.2% glycerol and 0.3% pyruvate, 0.0003% tetracycline and added with 0.3% L-arabinose and 0.0015% kanamycin.
[0426] Plasmid p707-Flpe (provided in the Quick & Easy E. coli Gene Deletion Red.RTM./ET.RTM. Recombination Kit by Gene Bridges) is transformed by electroporation according to the kit protocol. The cells are selected on LB agar supplemented with 0.2% glucose, 0.0003% tetracycline and added with 0.3% L-arabinose. A counter-selection of the clones obtained is carried out by verifying that they are no longer able to grow on the same medium supplemented with 0.0015% kanamycin.
[0427] The strain obtained is called EQ.EC044: MG1655 .DELTA.sdhA .DELTA.aceB P.sub.icd::P.sub.oxb1 .DELTA.zwf .DELTA.gapA
[0428] f) Constitutive Overexpression of the FPGLOXDH1 and aceA Genes
[0429] The coding sequences (CDS) of the FPGLOXDH1 (Gene ID: 946058) and aceA (Gene ID: 948517) genes subcloned into a bacterial expression vector pZE21MCS (EXPRESSYS) as synthetic operons according to the structure described in Table 24. This vector has a ColE1 origin of replication and a kanamycin antibiotic resistance gene.
[0430] Each of these primers is coupled to floating sequences homologous over 18 nucleotides at the ends of the fragment obtained by amplifying vector pZE21MCS excluding the multiple cloning site. The two amplicons are combined according to the protocol of the In-Fusion.RTM. HD Cloning Kit User Manual--Clontech to form plasmid pEQEC035 allowing the constitutive overexpression of the FPGLOXDH1 and aceA genes.
[0431] g) Insertion of the Engineering Required for CO.sub.2 Fixation
[0432] For the recombinant expression of the different components of a functional type I RuBisCO in E. coli, the genes described in Table 17 (Example 4A]), are cloned in the form of a synthetic operon.
[0433] The coding sequences (CDS) of the genes described in the Table 2 are amplified and assembled into blocks according to the protocol provided with the NEBuilder.RTM. HiFi DNA Assembly Master Mix Kit (E2321) to obtain three integration blocks described in Table 26. Each block is then amplified according to the protocol of the In-Fusion.RTM. HD Cloning Kit User Manual--Clontech to form the plasmids described below in Table 24.
TABLE-US-00027 TABLE 26 Composition of expression cassettes Structure of the synthetic operon Block I Block II Block III Plasmid Vector type CDS A RBS1 CDS B RBS2 CDS C RBS3 CDS D pZA11 pZA11 pEQEC006 pZA11 rbcS D rbcL B rbcX F prk pZE21MCS pZE21MCS pEQEC035 pZE21MCS FPGLOXDH1 D aceA
[0434] To control the expression level of these genes, ribosome binding sequences (RBS) presented in Table 19 (Example 4B]), with variable translation efficiencies (Levin-Karp et al., ACS Synth Biol. 2013 Jun. 21; 2(6):327-36. doi: 10.1021/sb400002n; Zelcbuch et al., Nucleic Acids Res. 2013 May; 41(9):e98) are inserted between the coding phase for each gene. The succession of each coding phase interspersed by an RBS sequence is constructed by successive insertions into a pZA11 vector (Expressys) that contains a PLtetO-1 promoter, a p15A origin of replication and an ampicillin resistance gene.
[0435] Several strains are produced by electroporating the different vectors presented according to the plan below
[0436] EQ.EC045.fwdarw.(EQ.EC042+pZA11+pZE21MCS): MG1655 .DELTA.sdhA .DELTA.aceB P.sub.icd::P.sub.oxb1
[0437] EQ.EC046.fwdarw.(EQ.EC045+pEQEC006+pEQEC035): MG1655 .DELTA.sdhA .DELTA.aceB P.sub.icd::P.sub.oxb1 .DELTA.zwf .DELTA.gapA+(FPGLOXDH1+aceA)+(RuBisCO+PRK)
[0438] After transformation, clones are selected on LB glycerol, pyruvate medium supplemented with 100 mg/L ampicillin and 30 mg/L kanamycin. An adaptation and evolution phase of the strains with PRK and RuBisCO engineering is performed as described in Example 4A].
[0439] h) Production of Succinate and Oxalate
[0440] For the production of succinate and oxalate, cells from 500 mL of LB culture are inoculated into 20 mL of MS medium (40 g/L glucose, 1 g/L MgSO.sub.4.7H.sub.2O, 20 g/L (NH.sub.4).sub.2SO.sub.4, 1 g/L KH.sub.2PO.sub.4, 10 mg/L FeSO.sub.4.7H.sub.2O, 10 mg/L MnSO.sub.4.7H.sub.2O, 2 g/L yeast extract, 30 g/L CaCO.sub.3, 100 mg/L ampicillin and 30 mg/L kanamycin at a pressure of 0.1 atmosphere CO.sub.2, at 30.degree. C. at pH 3.5.
[0441] The succinate concentration is measured by high-performance liquid chromatography (HPLC), culture samples are centrifuged at 12,000 g for 5 min.
[0442] i. Succinate Determination
[0443] The culture supernatant is filtered through a 0.2 .mu.m Millipore filter and analyzed on an Agilent HPLC system (series 1100) equipped with a cation-exchange column. (Aminex HPX87-H, Bio-Rad, Hercules, Calif., USA), a UV absorbance detector (Agilent Technologies, G1315D) and a refractive index (RI) detector (Agilent Technologies, HP1047A). The samples are separated on a 5 mM H.sub.2S0.sub.4 mobile phase at a flow rate of 0.4 mL/min. The column oven temperature is 65.degree. C.
[0444] Residual glucose is measured with a bioanalyzer (Ysi Inc.) or by HPLC-refractometry with an Aminex HPX87-H column.
[0445] The carbon yield Y.sub.p/s is calculated in grams of succinate produced per gram of glucose consumed.
[0446] This yield increases significantly by 6% for the engineering strain EQ.EC046 compared with the control strain EQ.EC045 (empty).
[0447] ii) Oxalate Determination
[0448] The pellets are washed twice with 10 mM potassium phosphate buffer (pH 7.5) containing 2 mM EDTA and stored at -20.degree. C. Samples (1 mL) are transferred into a tube pre-cooled with 0.75 g of glass beads (425-600 .mu.m) and introduced into a Fast Prep homogenizer (Thermo Scientific, Erembodegem, Netherlands) and subjected to 4 bursts of 20 s at speed control 6. The lysates are centrifuged for 20 min at 4.degree. C. and 36,000 g. Total protein determinations are performed according to the Lowry method (Lowry et al., 1951). Oxaloacetate acetyl hydrolase (EC 3.7.1.1.1) activity is measured using a modification of the direct optical determination of oxaloacetate (OAA) at 255 nm as described in (Lenz et al., 1976). The disappearance of the OAA enol tautomer is checked at 255 nm at 25.degree. C. in a Hitachi Model 100-60 spectrophotometer (Hitachi, Tokyo, Japan), using quartz cuvettes. The 1 mL reaction mixture contains 100 mM imidazole-HCl (pH 7.5), 0.9 mM MnCl.sub.2.2H.sub.2O, 1 mM OAA, 20 .mu.L cell extract (controls with different volumes of cell extracts confirm the linear relationship between enzyme activity and the amount of cell extract). The reaction is started by adding the cell extract.
[0449] The carbon yield Y.sub.p/s is calculated in grams of doxalate produced per gram of glucose consumed.
[0450] This efficiency increases significantly by 3% for the engineering strain EQ.EC046 compared with the control strain EQ.EC045 (empty).
Example 8: Improvement of Citrate Production in Aspergillus niger
[0451] a) Strategy
[0452] The inactivation of the pgkA gene (Locus tag An08g02260), leading to the non-functionality of the glycolysis pathway, and that of the gsdA gene (Locus tag An02g12140), inhibiting the oxidative part of the pentose phosphate pathway, are used to integrate the six genes for the functional expression of the PRK and RuBisCO enzymes, namely RbcS, RbcL, RbcX, GroES, GroEL and PRK for CO.sub.2 fixation.
[0453] b) DNA Constructs
[0454] i) Guide RNA Sequences for Targeting the Gene to be Inactivated
[0455] In each of these two genes, a sequence of 20 nucleotides punctuated by an NGG motif (CRISPR target sequence underlined) was determined (Table 27). In both cases, this sequence is specific to the targeted gene but also unique in the Aspergillus niger genome. These sequences are used to express a guide RNA (gRNA) which, by forming a heteroduplex with the homologous region of the Aspergillus niger genome, directs the action of the CAS9 endonuclease to induce a double-stranded break specifically on the chosen locus.
TABLE-US-00028 TABLE 27 gRNA target sequence Reference Locus CRISPR Locus Gene genome tag sequences 1 pgkA A. niger An08g CAACAAGGCCACTGG CBS 02260) TGGCCAGG 513-88 (SEQ ID NO: 19) 2 gsdA A. niger An02g CATTTCCGGTCAATA CBS 12140) TGACAAGG 513-88 (SEQ ID NO: 20)
[0456] Plasmid pFC332 (Addgene #87845) described in Sarkari et al. (Bioresour Technol. 2017 December; 245(Pt B):1327-1333) contains a gRNA expression cassette, a cassette allowing the functional expression of the Cas9 endonuclease and an Hph cassette allowing the selection of this plasmid. The plasmid also contains the fragment AMA1_2.8 which allows transient propagation of the plasmid. Finally, an origin of replication for E. coli is also present.
[0457] In order to target another gene, the gRNA cassette between FS A and FS B can be easily exchanged. This plasmid is modified by amplifying the different parts of this plasmid, in order to eliminate the antibiotic selection cassette and modify the 20 nucleotides allowing the specificity of gRNA in favor of the sequences described in Table 27 to form plasmids pEQ0610 to target pgkA and pEQ0611 to target gsdA.
[0458] Donor Plasmid
[0459] Regions of Homologies with the Genome
[0460] The donor plasmid consists of an In-Fusion.RTM. HD Cloning Kit User Manual--Clontech assembly between plasmid pUC19 (GenBank: M77789.2) and the genomic targeting sequences (LA and RA) of approximately 1500 bp each, homologous to the locus chosen for integration. The LA and RA sequences are adjacent at 5' and 3' respectively to the locus sequence targeted by the guide RNA. The genomic DNA/guide RNA heterodimer is recognized by the Cas9 endonuclease for double-stranded cleavage (locus 1: pgkA; locus 2: gsdA) (Table 28). The RA and LA fragments are amplified with primers for the pgkA gene and the gsdA gene (Table 29). The amplicon sequences are given in the sequence listing (SEQ ID NO: 55 to SEQ ID NO: 58). An extension of 18 nucleotides on all forward primers of the three fragments is added according to the protocol of the In-Fusion.RTM. HD Cloning Kit User Manual--Clontech, to allow a functional assembly of the plasmids (pEQ0600 or pEQ0601) and the introduction of two restriction sites for type II restriction endonucleases (restriction enzymes I-CeuI and I-Sce)I which have large asymmetric recognition sites (12 to 40 base pairs). These are recognition sequences of 18 base pairs, so rare. The fact that the cleavage is asymmetric at the reconnaissance site allows the release of a fragment lacking sequences from bacterial vector pUC19. These two enzymes allow the integration block to be extracted by restriction after amplification by cloning in E. coli.
TABLE-US-00029 TABLE 28 Amplification of regions of homologies for the pgkA gene Primer Amplicon Alias position Primer sequence 5' pgkA_ LA1 Forward GGATCGCAGATACGG A. niger TCGC (SEQ ID NO: 21) Reverse CCTCGGTGAAGACAA CGCTG (SEQ ID NO: 22) 3' pgkA_ RA1 Forward CTCCTTGAGAACCTG A. niger CGTTTCC (SEQ ID NO: 23) Reverse CTGAAGTACGTTTTC CCAAGCC (SEQ ID NO: 24)
TABLE-US-00030 TABLE 29 Amplification of regions of homologies for the gsdA gene Primer Amplicon Alias position Primer sequence 5' gsdA_ LA2 Forward CGTTATCACAAAGAA A. niger GCCAGGTCC (SEQ ID NO: 25) Reverse GCTGCTCTTCGATTT CCTTGGT (SEQ ID NO: 26) 3' gsdA_ RA2 Forward TCATCAACCTCAACA A. niger AGCACCTC (SEQ ID NO: 27) Reverse GTGAAGACAGCGGCG GTCC (SEQ ID NO: 28)
[0461] Engineering Expression Cassettes
[0462] The promoters and terminators are identified on the basis of GenBank data. The selected promoters are determined from the +1 transcription point and go up 1.4 kb upstream in order to cover both the "core" sequences (TATA box) and the trans-activating sequences allowing the optimal functionality of the promoter concerned.
[0463] For the terminators, the cut-off is made 500 bp after the stop codon of the gene.
[0464] The structure of each integration block of four expression cassettes is defined as follows: the first level consists of simple elements, namely promoters, coding sequences (CDS) and terminators. The promoter (Table 30) and terminator (Table 31) elements, whose sequences are provided in the sequence listing (SEQ ID NO: 59 to 62), are amplified and assembled with the engineering CDS according to Table 32. The CDS, whose sequences are provided in the sequence listing (SEQ ID NO: 63 to 66), are amplified according to the protocol provided with the NEBuilder.RTM. HiFi DNA Assembly Master Mix Kit (E2321) to obtain the functional expression cassettes compiled in the table. Each integration block of four genes is organized to include four different pairs (promoter/terminator) in order to limit trans interference. Each integration block of six genes is organized to include six different pairs (promoter/terminator) in order to limit transcriptional interference
[0465] Donor Fragment for Insertion into the Target Locus of the Genome
[0466] The different multiple expression cassettes (RbcS, RbcL and RbcX) or (GroES, GroEL and PRK) are amplified and assembled around an antibiotic selection cassette (Table), according to the protocol of the In-Fusion.RTM. HD Cloning Kit User Manual--Clontech, to form donor plasmids (pEQ0602 or pEQ0603).
TABLE-US-00031 TABLE 30 Native location of Aspergillus niger promoters used in genomic combinatorics to insert the six genes of the CO.sub.2 fixation engineering into the Aspergillus niger genome. Pro- Gene Reverse Forward moters Organism ID primer primer PmbfA A. niger An02g TTTGAAGATGGA GCCATGAAATC CBS 12390 TGAGAAGTCGG CAATCATTTCC 513-88 (SEQ ID (SEQ ID NO: 33) NO: 29) PcoxA A. niger An07g TGTCCTGGTGGG GACGGCATTTG CBS 07390 TGGGTTG AGCAACATC 513-88 (SEQ ID (SEQ ID NO: 34) NO: 30) PsrpB A. niger An16g CTCGAACGAGAA TTGGCAGGGTC CBS 08910 TGGGAACC ACGTAGCC 513-88 (SEQ ID (SEQ ID NO: 35) NO: 31) PtvdA A. niger An04g GGCGGAATGAGA TTAGTCCATTC CBS 01530 TGCGACAG AGCAAGCTGCC 513-88 (SEQ ID (SEQ ID NO: 36) NO: 32)
TABLE-US-00032 TABLE 31 Native location of Aspergillus Niger terminators used in genomic combinatorics to insert the six genes of the CO.sub.2 fixation engineering into the Aspergillus niger genome. Ter- Gene Forward Reverse minators Organism ID primer primer TtrpC A. AN0648 TGATTTAATA GGGTAAACG nidulans GCTCCATGTC ACTCATAGG FGSC A4 AACAAG AGA (SEQ ID (SEQ ID NO: 37) NO: 41) TniaD A. AN1006 ACGGGTTCGC GGGATATTT nidulans ATAGGTTTGG GACACGATT FGSC A4 (SEQ ID CTGAGG NO: 38) (SEQ ID NO: 42) TgiaA A. niger An03g CGACCGCGAC CCGGAGATC CBS 513- 06550 GGTGACTGAC CTGATCATC 88 (SEQ ID CG NO: 39) (SEQ ID NO: 43) TgpdA A. niger An16g GAATCAGGAC CGTGGTCTA CBS 513- 01830 GGCAAACTGA GCTGCCCTC 88 AT C (SEQ ID (SEQ ID NO: 40) NO: 44)
TABLE-US-00033 TABLE 32 Assembly of expression cassettes Codon Expression optimi- Termi- cassette Gene GenBank zation Promoter nator CAS 1 RbcL BAD78320.1 Yes PmbfA.sub.p trpct CAS 2 RbcS BAD78319.1 Yes PcoxA.sub.p TniaD CAS 3 RbcX BAD80711.1 Yes PsrpB.sub.p glaAt CAS 4 Hph pUG75(P30671) No picdA.sub.p TgpdA CAS 5 GroES U00096 No PmbfA.sub.p trpct CAS 6 GroEL AP009048 No PcoxA.sub.p TniaD CAS 7 PRK BAD78757.1 Yes PsrpB.sub.p glaAt CAS 8 Ble pUG66(P30116) No picdA.sub.p TgpdA
TABLE-US-00034 TABLE 33 Plasmid assembly Genomic Genomic Selection Plasmid sequence Promoter Gene Terminator sequence ori marker pEQ0600 LA1 RA1 coli Ampicillin pEQ0601 LA2 RA2 coli Ampicillin pEQ0602 LA2 PmbfA.sub.p RbcL trpct RA2 coli Ampicillin and PcoxA.sub.p RbcS TniaD coli hydromycin B PsrpB.sub.p RbcX glaAt coli picdA.sub.p Hph TgpdA coli pEQ0603 LA1 PmbfA.sub.p GroES Trpct RA1 coli Ampicillin and PcoxA.sub.p GroEL TniaD coli bleomycin PsrpBp PRK glaAt coli picdAp Blue TgpdA coli
[0467] c) Transformation of Aspergillus niger
[0468] The transformation of DNA in Aspergillus niger is constrained by the presence of the fungal cell wall, and is extremely ineffective compared with yeast or Escherichia coli. Nevertheless, the transformation of protoplasts prepared from fungal hyphae or conidiospores to germination by treatment with cell wall degrading enzymes such as the cocktail consisting of Lysing Enzyme.RTM. from Trichoderma harzianum, chitinase from Streptomyces griseus and .beta.-glucuronidase from Helix pomatia (de Bekker et al., J Microbiol Methods. 2009 March; 76(3):305-6) allows transformants to be produced.
[0469] The A. niger strain CBS 513-88 is grown at 30.degree. C. in a 1 L Erlenmeyer flask with 250 mL of transformation medium (Kusters-van Someren et al., Curr Genet. 1991 September; 20(4):293-9). After growth for 16 h at 250 rpm, the mycelium is collected by filtration on Miracloth (Calbiochem) and washed with deionized water. Protoplasts are prepared in the presence of 5 g/L lysis enzymes from Trichoderma harzianum (Sigma Saint Louis, Mo., USA), 0.075 Uml-1 chitinase from Streptomyces griseus (Sigma) and 460 Uml-1 glucuronidase from Helix pomatia (Sigma) in KMC (0.7 M KCl, 50 mM CaCl.sub.2, 20 mM Mes/NaOH, pH 5.8) for 2 hours at 37.degree. C. and 120 rpm. Protoplasting is monitored every 30 minutes with a microscope. The protoplasts are filtered through a Miracloth filter and collected by centrifugation at 2000.times.g and 4.degree. C. for 10 minutes. The protoplasts are washed with cold STC (1.2 M sorbitol, 10 mM Tris/HCl, 50 mM CaCl.sub.2, pH 7.5) and then resuspended in 100 pi of STC and used directly for the transformation.
[0470] In order to integrate a metabolic pathway into the A. niger genome, co-transformation of a plasmid and a linear fragment is required. Plasmid pEQ0610 is co-transformed with a donor fragment to integrate part of the engineering into the genome while inactivating the pgkA gene. Similarly, plasmid pEQ0611 is co-transformed with a donor fragment to integrate the other part of the engineering into the genome while inactivating the gsdA gene. These sequences serve both as matrices for homologous recombination and as selection markers: during integration with functional expression of the antibiotic resistance genes Hph or Ble. The strains are directly selected on minimal medium plates with an addition of hygromycin B or bleomycin allowing direct selection on the integration event. Due to the presence of the origin of replication AMA1_2.8, plasmid pCAS_pyrG2 is easily lost causing only transient expression of the Cas9 protein, thus reducing the risk of non-targeted adverse effects.
[0471] Linear cassettes (10 .mu.g) and plasmid (5 .mu.g) are mixed with 100 .mu.L of STC solution containing at least 10.sup.7 protoplasts and 330 .mu.L of freshly prepared polyethylene glycol (PEG) solution (25% PEG 6000, 50 mM CaCl.sub.2, 10 mM Tris/HCl, pH 7.5) and kept on ice for 20 minutes. After mixing with an additional 2 mL PEG solution and incubating at room temperature for 10 minutes, the protoplast mixture is diluted with 4 mL of STC.
[0472] The selection of transformants is carried out on MM plates with 150 .mu.g/mL hygromycin B added or MM plates with 50 .mu.g/mL bleomycin added. All transformants are purified by isolating single colonies from the selection medium at least twice. The insertion of the fragments is verified by sequencing the target locus with the appropriate control primers. Genomic DNA from fungal cells is isolated with a modified protocol, using the Wizard.RTM. Genomic DNA Purification Kit (Promega, Wisconsin, USA). The mycelium is cultured overnight in CM (30.degree. C., 150 rpm) in 290 pi of 50 mM EDTA solution and 10 pi of lyticase (10 mg/mL) to remove the cell wall. After 90 minutes of incubation at 37.degree. C., the suspension is centrifuged and the supernatant is discarded. The mycelium pellet is resuspended in 300 .mu.L of nuclei lysis solution and 100 .mu.L of protein precipitation solution. The samples are incubated on ice for 5 minutes and centrifuged. The DNA is precipitated with isopropanol and washed with 70% ethanol. The DNA pellet is rehydrated with a DNA rehydration solution containing RNase (100 .mu.g/mL). The successful transformation and integration of the expression cassettes was verified by PCR.
TABLE-US-00035 TABLE 34 Strains used for the yield study Strains Genome Genetic modification EQ1500 A. niger CBS 513-88 EQ1501 A. niger gsdA :: PmbfA.sub.p-RbcL-trpc; PcoxA.sub.p-RbcS-TniaD; CBS picdA.sub.p-Hph-TgpdA; PsrpB.sub.p-RbcX-glaAt 513-88 EQ1502 A. niger gsdA :: PmbfA.sub.p-RbcL-trpc; PcoxA.sub.p-RbcS-TniaD; CBS picdA.sub.p-Hph-TgpdA; PsrpB.sub.p-RbcX-glaAt 513-88 pgkA :: PmbfA.sub.p-GrES-trpc; PcoxA.sub.p-GroEL-TniaD; picdA.sub.p-Ble-TgpdA; PsrpB.sub.p-PRK-glaAt
[0473] Conidia (10.sup.8/L) from strains EQ1500 and EQ1502 are inoculated and cultured at 30.degree. C. on a rotary shaker (180 rpm) in shaker flasks containing Vogel medium without MnSO.sub.4 with a total glucose content of 15% and a total nitrogen content of 0.2% and 10% CO.sub.2. The determination of glucose and organic acids was performed as described above (Blumhoff et al., 2013; Steiger et al., 2016) on an HPLC (Shimadzu, Kyoto; Japan) equipped with an Aminex HPX-87 H column (300.times.7.8 mm, Bio-Rad, Hercules, Calif.). A refractive index detector (RID-10 A, Shimadzu) is used for the detection of glucose and citric acid, while a PDA detector (SPD-M20A, Shimadzu) at 300 nm is used to detect cis-aconitic and trans-aconitic acid. The column is used at 60.degree. C. at a flow rate of 0.6 mL/min and with a 0.004 M H.sub.2SO.sub.4 aqueous solution as mobile phase. The culture was carried out in three biological replicates.
[0474] d) Analytical Method
[0475] For the quantification of extracellular metabolites, a culture sample is centrifuged at 14,000.times.g for 5 min. The supernatant is filtered through a filter with a 0.45 pm pore size. The filtrate is maintained at -20.degree. C. until analysis. The concentration of citrate and of oxalate is detected and quantified with ultraviolet light at 210 nm using an Amethyst C18-H column (250.times.4.6 mm, Sepax Technologies, Newark, Del., USA). Elution is carried out at 30.degree. C. with 0.03% H.sub.3P0.sub.4 at a flow rate of 0.8 mL/min. Reducing sugar is detected with the 3,5-dinitrosalicylic acid method. Biomass determination: 5 mL of sample is filtered through Miracloth (Calbiochem, San Diego, Calif., USA) to collect hyphae and washed with distilled water. The hyphae are heated to 105.degree. C. in a "Miracloth". For the calculation of the dry cell weight (DCW), the weight of Miracloth is measured beforehand and subtracted from the total weight to give the net weight, then the net weight per unit volume is calculated as DCW.
[0476] After complete analysis, the comparison of citric acid production yield as a function of glucose consumption is 18% higher in the engineered strain EQ1502 than in the wild-type strain EQ1500.
Example 9: Improvement of Itaconate Production in Aspergillus terreus
[0477] a) Strategy
[0478] Inactivation of the pgkA gene (Locus tag (ATEG_00224), leading to the non-functionality of the glycolysis pathway, and that of the gsdA gene (Locus tag ATEG_01623), inhibiting the oxidative part of the phosphate pentose pathway, are used to integrate the six genes allowing the functional expression of the PRK and RuBisCO enzymes, namely rbcS, rbcL, rbcX, groES, groEL and prk allowing CO.sub.2 fixation.
[0479] b) DNA Constructs
[0480] i) RNA Guide Sequences to Target the Gene to be Inactivated
[0481] In each of these two genes, a sequence of 20 nucleotides punctuated by an NGG motif (CRISPR target sequence underlined) was determined (Table 35). In both cases, this sequence is specific to the targeted gene but also unique in the Aspergillus terreus genome. These sequences are used to express a guide RNA (gRNA) which, by forming a heteroduplex with the homologous region of the Aspergillus terreus genome, directs the action of the CAS9 endonuclesae to induce a double-stranded break specifically on the selected locus. For pgkA, the sequence identified in the second intron, the first 20 nucleotides have a unique pattern in the genome, even allowing two mismatches. For gsdA, the sequence identified in the fourth intron, the first 20 nucleotides have a unique pattern in the genome, even allowing two mismatches.
TABLE-US-00036 TABLE 35 Guide RNA target sequence Reference Locus Locus Gene genome tag CRISPR sequences 3 pgkA A. (ATEG_ CTGCGTCGGCAA terreus 00224) GGAAGTTGAGG NIH262 (SEQ ID NO: 45) 4 gsdA A. (ATEG_ CATCAGCGGCCA terreus 01623) ATATGACAAGG NIH2624 (SEQ ID NO: 46)
[0482] Plasmid pFC332 (Addgene #87845) described in Sakari et al. (Bioresour technol. 2017; 245(Pt B):1327-1333) contains a gRNA expression cassette, a cassette for the functional expression of the Cas9 endonuclease and an Hph cassette for the selection of this plasmid. The plasmid also contains the fragment AMA1_2.8 which allows transient propagation of the plasmid. Finally, an origin of replication for E. coli is also present.
[0483] In order to target another gene, the gRNA cassette between FS A and FS B can be easily exchanged. Thus, this plasmid is modified by amplifying the different parts of this plasmid in order to eliminate the antibiotic selection cassette and to modify the 20 nucleotides allowing the specificity of gRNA in favor of the sequences described in Table 35 to form plasmids pEQ0615 to target pgkA and pEQ0616 to target gsdA in the Aspergillus terreus genome.
[0484] ii) Donor Plasmid
[0485] Regions of Homology with the Genome
[0486] The donor plasmid consists of an In-Fusion.RTM. HD Cloning Kit User Manual--Clontech assembly between plasmid pUC19 (GenBank: M77789.2) and genomic targeting sequences (LA and RA) of approximately 1500 bp each homologous to the locus chosen for integration. The LA and RA sequences are adjacent at 5' and 3' respectively to the locus sequence targeted by the guide RNA. The genomic DNA/guide RNA heterodimer is recognized by the Cas9 endonuclease for double-stranded cleavage (locus 1: pgkA; locus 2: gsdA) (Table 35). The RA and LA fragments are amplified with the primers described in Table 36, for the pgkA gene, and Table 37, for the gsdA gene. The amplicon sequences are in the sequence listing (SEQ ID NO: 67 to 70).
[0487] An extension of 18 nucleotides on all forward primers of the three fragments is added according to the protocol of the In-Fusion.RTM. HD Cloning Kit User Manual--Clontech to allow a functional assembly of the plasmids (pEQ0604 or pEQ0605) (33) and the introduction of two restriction sites for type II restriction endonucleases (restriction enzymes I-CeuI and I-SceI) which have large asymmetric recognition sites (12 to 40 base pairs). These are recognition sequences of 18 base pairs, therefore rare and not present in the described assembly. The fact that the cleavage is asymmetric at the reconnaissance site allows a fragment devoid of sequences to be released from the bacterial vector pUC19. These two enzymes allow the integration block to be extracted by restriction after amplification by cloning in E. coli.
TABLE-US-00037 TABLE 36 Amplification of regions of homologies for the pgkA gene Primer Amplicon position Primer sequence 5' pgkA_ Forward CTTGGGGAATTGG Aterreus GACACG (SEQ ID NO: 47) Reverse TCTTGCCGATGAG CTTCTCC (SEQ ID NO: 48) 3' pgkA_ Forward CAGATCATCCTCC Aterreus TGGAGAACC (SEQ ID NO: 49) Reverse ACGGCACGAATGT TCACCTG (SEQ ID NO: 50)
TABLE-US-00038 TABLE 37 Amplification of regions of homologies for the gsdA gene Primer Amplicon position Primer sequence 5' gsdA_ Forward ATTGGAAGCTGGCTCT Attereus ATCTCACC (SEQ ID NO: 51) Reverse GCTGTTCTTCGATTTC CTTGGTG (SEQ ID NO: 52) 3' gsdA_ Forward TCAACCTCACCAAGCA Aterreus CCTCG (SEQ ID NO: 53) Reverse CAAACAGCCCGTCGCA ACTG (SEQ ID NO: 54)
[0488] Engineering Expression Cassettes
[0489] Promoters and terminators are identified on the basis of GenBank data. The selected promoters are determined from the +1 transcription point and go up 1.4 kb upstream in order to cover both the "core" sequences (TATA box) and the trans-activating sequences allowing the optimal functionality of the promoter concerned.
[0490] For the terminators, the cut-off is made 500 bp after the stop codon of the gene.
[0491] The structure of each integration block of four expression cassettes is defined as follows: the first level consists of simple elements, namely promoters, coding sequences (CDS) and terminators. The promoter (Table 30) and terminator (Table 31) elements are amplified and assembled with the engineering CDS according to Table 32. The CDS are amplified according to the protocol provided with the NEBuilder.RTM. HiFi DNA Assembly Master Mix Kit (E2321) in order to obtain the functional expression cassettes compiled in the table. Each integration block of four genes is organized to include four different terminator promoter pairs in order to limit trans interference Each integration block of six genes is organized to include six different terminator promoter pairs in order to limit transcriptional interference.
[0492] Donor Fragment for Insertion into the Target Locus of the Genome
[0493] The different multiple expression cassettes (RbcS, RbcL and RbcX or GroES, GroEL and PRK are amplified and assembled around an antibiotic selection cassette (Table 38), according to the protocol of the In-Fusion.RTM. HD Cloning Kit User Manual--Clontech, to form donor plasmids (pEQ0606 or pEQ0607).
TABLE-US-00039 TABLE 38 Plasmid assembly Genomic Genomic Selection Plasmids sequence Promoter Gene Terminator sequence ori marker pEQ0604 LA4 RA4 coli Ampicillin pEQ0605 LA3 RA3 coli Ampicillin pEQ0606 LA4 PmbfA.sub.p rbcL trpct RA4 coli Ampicillin and PcoxA.sub.p rbcS TniaD coli hydromycin B PsrpB.sub.p rbcX glaAt coli picdA.sub.p Hph TgpdA coli pEQ0607 LA3 PmbfA.sub.p groES trpct RA3 coli Ampicillin and PcoxA.sub.p groEL TniaD coli bleomycin PsrpB.sub.p prk glaAt coli picdA.sub.p Ble TgpdA coli
[0494] c) Transformation of Aspergillus terreus
[0495] The transformation of Aspergillus terreus DNA is carried out in accordance with the strategy applied for Aspergillus niger (Example 8) using A. terreus strain NIH262.
TABLE-US-00040 TABLE 39 Strains used for the yield study Strains Genome Genetic modification EQ1600 A. terreus NIH262 EQ1601 A. terreus gsdA :: PmbfA.sub.p-RbcL-trpc; PcoxA.sub.p-RbcS-TniaD; NIH262 picdA.sub.p-Hph-TgpdA; PsrpB.sub.p-RbcX-glaAt EQ1602 A. terreus gsdA :: PmbfA.sub.p-RbcL-trpc; PcoxA.sub.p-RbcS-TniaD; NIH262 picdA.sub.p-Hph-TgpdA; PsrpB.sub.p-RbcX-glaAt pgkA :: PmbfA.sub.p-GrES-trpc; PcoxA.sub.p-GroEL-TniaD; picdA.sub.p-Ble-TgpdA; PsrpB.sub.p-PRK-glaAt
[0496] Culture of A. terreus strains EQ1600 and EQ1602 on 3% glucose.
[0497] The optimized media composition described by Hevekerl et al. (Appl Microbiol Biotechnol. 2014; 98:6983-6989) is used. It contains 0.8 g KH.sub.2P0.sub.4, 3 g NH.sub.4N0.sub.3, 1 g MgSO.sub.4.7H20, 5 g CaCl.sub.2.2 H.sub.20, 1.67 mg FeCl.sub.3.6H.sub.2O, 8 mg ZnSO.sub.4.7H.sub.2O and 15 mg CuSO.sub.4.7H2O per liter. To mimic the typical sugar concentration obtained from wheat straw hydrolysate (150 g/L) pretreated with dilute acid (0.75% v/v, 160.degree. C., 10 min) and enzymatically saccharified (pH 5.0, 45.degree. C., 72 h), an adequate amount of glucose up to 30 g/L is used. Sugars and all other components are added from sterile stock solutions. The pH of the medium without CaCl.sub.2 is adjusted to 3.1 with 0.5 M H.sub.2SO.sub.4 before inoculating the spore preparation for strains EQ1600 and EQ1602. The culture is carried out under shaking with 25 mL of medium in 125 mL Erlenmeyer flasks at 33.degree. C. in a rotary shaker at 200 rpm for 7-10 days in an environment of 10% CO.sub.2. The pH is not checked during fermentation. Shaking of the flasks is maintained during sampling for time studies to ensure a continuous supply of oxygen. All experiments are carried out in triplicate. All media components are obtained from Sigma Chemical, St. Louis, Mo. For these experiments, each sugar was dissolved in deionized water and passed through a column (440.times.45 mm) of Dowex 50-X8 (100/200 mesh) cation-exchange resin (Bio-Rad Laboratories, Hercules, Calif.) to remove manganese, if necessary.
[0498] d) Analytical Procedures
[0499] The concentration of the cell mass is determined from the dry cell weight. The cell mass present in the fermentation broth is collected by centrifugation at 10,000 g for 10 minutes and carefully rinsed three times with deionized water. The rinsed cell mass was completely dried at 80.degree. C. until a constant weight was obtained. The fermentation broth after centrifugation (10,000 g, 10 min) is stored at -20.degree. C. before analysis of glucose, itaconic acid and by-products (succinic acid, .alpha.-ketoglutaric acid, malic acid, cis-aconitic acid, and trans-aconitic acid) using high-performance liquid chromatography (HPLC). A Shimadzu Prominence HPLC system (Shimadzu America, Inc., Columbia, Md.) is used. Two columns (Aminex HPX-87P column, 300.times.7.8 mm with ash removal cartridge and Carbo-P protection cartridge, and one Aminex HPX 87H column, 300.times.7.8 mm with Microguard Cation H cartridge (Bio-Rad)) are used for the analysis of sugars and organic acids, respectively. The Aminex HPX 87P column is maintained at 85.degree. C. and glucose is eluted with Milli-Q acidified deionized water (Millipore, Bedford, Mass.) at a flow rate of 0.6 mL/min.
[0500] The Aminex HPX 87H column is maintained at 65.degree. C. and sugars and organic acids are eluted with 5 mM H.sub.2SO.sub.4 prepared using Milli-Q deionized filtered water at a rate of 0.5 mL/min. Detection is carried out using a refractive index detector for sugars and a 210 nm UV detector for organic acids. Propionic acid (1%, weight/volume) is used as internal standard to estimate the liquid lost during aerobic fermentation for 7-10 days at 33.degree. C. under 10% CO.sub.2. All HPLC standards, including organic acids, are purchased from Sigma. The manganese concentration (ppb level) is determined using an Optima 7000DV (Perkin-Elmer, Waltham, Mass.) inductively coupled plasma optical emission spectrometer (ICP-OES) by the procedure described by Bakota et al. (Eur J Lipid Sci Technol. 2015; 117:1452-1462.
[0501] Based on the results of the production of itaconic acid from glucose, a mass yield increment of itaconic acid from glucose of 15% is observed for the engineered strain EQ1602 compared with the reference strain EQ1600.
Sequence CWU 1
1
71176DNAartificial sequenceoligonucleotide CB101 (forward)
1acagatcatc aaggaagtaa ttatctactt tttacaacaa atataaaaca atgggtaagg
60aaaagactca cgtttc 76281DNAartificial sequenceoligonucleotide
CB102 (reverse) 2gggaaagaga aaagaaaaaa attgatctat cgatttcaat
tcaattcaat ttagaaaaac 60tcatcgagca tcaaatgaaa c 81365DNAartificial
sequencesynthetic oligonucleotide 3aagagtaaat ccaatagaat agaaaaccac
ataaggcaag atgggtaaaa agcctgaact 60caccg 65463DNAartificial
sequencesynthetic oligonucleotide 4atttcagtga cttagccgat aaatgaatgt
gcttgcattt ttttattcct ttgccctcgg 60acg 63576DNAartificial
sequencesynthetic oligonucleotide 5acagatcatc aaggaagtaa ttatctactt
tttacaacaa atataaaaca atgggtaagg 60aaaagactca cgtttc
76681DNAartificial sequencesynthetic oligonucleotide 6gggaaagaga
aaagaaaaaa attgatctat cgatttcaat tcaattcaat ttagaaaaac 60tcatcgagca
tcaaatgaaa c 81775DNAartificial sequencesynthetic oligonucleotide
7tctccctatc ctcattcttc tcccttttcc tccataattg taagagaaaa atgggtacca
60ctcttgacga cacgg 75878DNAartificial sequencesynthetic
Oligonucleotide 8aatttgaaca cacttaagtt gcagaacaaa aaaaagggga
attgttttca ttaggggcag 60ggcatgctca tgtagagc 78912DNAartificial
sequenceribosome binding sequence 9aggaggtttg ga
121022DNAartificial sequenceribosome binding sequence 10aacaaaatga
ggaggtactg ag 221116DNAartificial sequenceribosome binding sequence
11aagttaagag gcaaga 161214DNAartificial sequenceribosome binding
sequence 12ttcgcagggg gaag 141318DNAartificial sequenceribosome
binding sequence 13taagcaggac cggcggcg 181412DNAartificial
sequenceribosome binding sequence 14caccatacac tg
121518DNAartificial sequenceoligonucleotide POXB1-S 15tcgttgcgtt
acacacac 181619DNAartificial sequenceoligonucleotide POXB1-R
16tgtgtcgagt ggatggtag 191711DNAartificial sequenceoligonucleotide
Spacer S 17gcatgaattc g 111811DNAartificial sequenceoligonucleotide
Spacer R 18cgaattcatg c 111923DNAartificial sequencegRNA target
sequence for CRISPR pgkA 19caacaaggcc actggtggcc agg
232023DNAartificial sequencegRNA target sequence for CRISPR gsdA
20catttccggt caatatgaca agg 232119DNAartificial sequenceprimer LA1
forward 21ggatcgcaga tacggtcgc 192220DNAartificial sequenceprimer
LA1 reverse 22cctcggtgaa gacaacgctg 202322DNAartificial
sequenceprimer RA1 forward 23ctccttgaga acctgcgttt cc
222422DNAartificial sequenceprimer RA1 reverse 24ctgaagtacg
ttttcccaag cc 222524DNAartificial sequenceprimer LA2 forward
25cgttatcaca aagaagccag gtcc 242622DNAartificial sequenceprimer LA2
reverse 26gctgctcttc gatttccttg gt 222723DNAartificial
sequenceprimer RA2 forward 27tcatcaacct caacaagcac ctc
232819DNAartificial sequenceprimer RA2 reverse 28gtgaagacag
cggcggtcc 192922DNAartificial sequenceprimer 29gccatgaaat
ccaatcattt cc 223020DNAartificial sequenceprimer 30gacggcattt
gagcaacatc 203122DNAartificial sequenceprimer 31ttagtccatt
cagcaagctg cc 223222DNAartificial sequenceprimer 32ttagtccatt
cagcaagctg cc 223323DNAartificial sequenceprimer 33tttgaagatg
gatgagaagt cgg 233419DNAartificial sequenceprimer 34tgtcctggtg
ggtgggttg 193520DNAartificial sequenceprimer 35ctcgaacgag
aatgggaacc 203620DNAartificial sequenceprimer 36ggcggaatga
gatgcgacag 203726DNAartificial sequenceprimer 37tgatttaata
gctccatgtc aacaag 263820DNAartificial sequenceprimer 38acgggttcgc
ataggtttgg 203920DNAartificial sequenceprimer 39cgaccgcgac
ggtgactgac 204022DNAartificial sequenceprimer 40gaatcaggac
ggcaaactga at 224125DNAartificial sequenceprimer 41gggtaaacga
ctcataggag agttg 254224DNAartificial sequenceprimer 42gggatatttg
acacgattct gagg 244320DNAartificial sequenceprimer 43ccggagatcc
tgatcatccg 204419DNAartificial sequenceprimer 44cgtggtctag
ctgccctcc 194523DNAartificial sequencesequence for CRISPR pgkA
45ctgcgtcggc aaggaagttg agg 234623DNAartificial sequencesequence
for CRISPR gsdA 46catcagcggc caatatgaca agg 234719DNAartificial
sequenceprimer 47cttggggaat tgggacacg 194820DNAartificial
sequenceprimer 48tcttgccgat gagcttctcc 204922DNAartificial
sequenceprimer 49cagatcatcc tcctggagaa cc 225020DNAartificial
sequenceprimer 50acggcacgaa tgttcacctg 205124DNAartificial
sequenceprimer 51attggaagct ggctctatct cacc 245223DNAartificial
sequenceprimer 52gctgttcttc gatttccttg gtg 235321DNAartificial
sequenceprimer 53tcaacctcac caagcacctc g 215420DNAartificial
sequenceprimer 54caaacagccc gtcgcaactg 20551881DNAartificial
sequenceamplicon 5'pgkA A. niger 55ggatcgcaga tacggtcgca cagttaatca
cggctttcaa ggctcgtgga ggacgcaggc 60cctctgacag taacgatctt atttcaattg
agatccatga cctcgataaa gagaagccgc 120ctgctcttgg agagctggga
catgctagac atatgccgcc gcccttcgac ttcgagcgcc 180tgactcgctt
tatgtcgtac ggtttcttca tggccccggt tcagttccat tggttcggtt
240tcttgtccag ggcgttcccc cttaccaaga ggaacccgtc gattcccgcg
ctgaagagag 300tgtgcgtgga tcagctgatg ttcgctcctt ttggtatgaa
ctgtctccga aggaagaaga 360aagctctttt ctcttagcta atattattac
caggtctggc gtgcttcttc tccttcatga 420cggttgcaga gggaggagga
aggagagcct tgacgcgcaa gttccaggac gtctatctgc 480caaccctcaa
ggccaacttt gtcctttggc ccgctgttca gattctgaac ttccgtgtcg
540tacctatcca gttccagatt gtgagtacta ttgtgataaa actcttgtgc
actgttatac 600tgatatttct tttttcttcc agcccttcgt gtcttccgtc
ggaatcgcct ggactgcgta 660tctgtcgctg accaactctt cggaggagga
gtaatggtag tagcgggctc atattttggc 720tcccacaagg ttcccaatcg
tttcttctgt caattgtctg tcattcttcc ttcccctgcg 780tcgtttcgtg
cttactgggc gctgtaaatg aacttcgggg gttctgttat tcttcctctc
840atgggtggtg tataggtctt cgtccagcag tgtgggtaca ccggagtcaa
ttgcattgat 900cgataaacca tgtcgacaaa atgaattata caccattgtt
tttcgggttt gaaccaggca 960cgaatgatgg aatgtctcct gtaggcgggg
ttgtccccac gagcgagccg gcatccatag 1020acgagaaata tcggaacgat
tgcttgatag atccaccccc ggacgcagca gaagctccca 1080tcgcagcaat
gatccgaggc taaggggctc cgaacggggc ataacaggca taatgttcac
1140ccctgaggcc ccggaccctt ccgtcatcga ttcgcgggcg ctgacatcag
tcccgcatca 1200gcccgaggcg cccgcgattt caacttctct caccggctgc
cccaccacca gcccatacca 1260cttttacccc gccgccgaac cgaatgaagt
cgtagcttcc agtgaccaga ctcgctcacc 1320gcggacctat taagtaggca
tttttcccaa tctcctcatc ccccgtggtg ctactaatac 1380tacgtctctc
cccctcccaa tttctccttt ctctcttttt ttgtcacccc acttccctat
1440cttcccctca catcctctaa ccccgtccga ctgtcgtaga accgtcgttc
atcatgtctc 1500ttaccaacaa gctcgctatc accgatgtcg acctcaagga
caagcgtgtc ctgatccggg 1560tatgttgttg ccccggaaac ccccaccttt
tcatcagtca ccgtcgtggg gtttaagatg 1620agccttcaat gctaactagt
ctgctcactg gacaggtcga cttcaacgtc cccctcaatg 1680acaagaagga
gatcaccaac aaccagcgta tcgtcggtgc tctgcccacc atcaagtacg
1740ctatcgagaa tggtgccaag gccgttgtcc tcatgtccca cctgggccgc
cccgacggca 1800agaagaacga caagtacagc ctgaagcccg ttgttgccga
gctggagaag ctgcttggcc 1860gcagcgttgt cttcaccgag g
1881561883DNAartificial sequenceamplicon 3'pgkA A. niger
56ctccttgaga acctgcgttt ccacgctgag gaggagggca gctccaagga tgccgagggc
60aagaaggtca aggccgacaa ggagaaggtc gctgagttcc gcaagggtct gactgctctt
120ggtgatgtct acatcagtaa gtcatccttt tctcacacct tccatcttgg
gaggccacta 180gtttatgagc tgtgtactaa tattccacac cacagacgac
gctttcggca ctgcccaccg 240tgctcactct tccatggtcg gtgttgacct
tcctcagaag gcttccggtt tcctcgtgaa 300gaaggagctc gagtacttcg
ccaaggctct cgagagcccc cagcgcccct tccttgccat 360cctgggtggt
gccaaggtct ctgacaagat ccagctgatc gacaacctgc tgcccaaggt
420caacagcctg atcatcaccg gtgccatggc tttcactttc aagaagaccc
tcgagggtgt 480caagatcgga aacagtctct tcgacgaggc tggcagcaag
atcgttggcg aggtcgtcga 540gaaggccaag aagcacgacg tcaagatcgt
tctgcccgtc gactacgtca ctgccgacaa 600gttcgctgct gatgccaaga
ccggcactgc taccgacgct gagggtattc ccgacggcta 660catgggtctg
gatgttggcg agaagagtgt tgagctctac aagcagacca ttgctgaggc
720caagaccatc ctctggaacg gtccccccgg tgtcttcgag ttggagccct
tcgccaacgg 780caccaagaag accctcgatg ccgtcgtctc cgctgctcag
tccggctcca tcgtcatcat 840cggaggtggt gacactgcca ccgttgccgc
caagtacggt gtcgaagaca agcttagcca 900cgtctccacc ggtggtggtg
cctctctgga gctcctggag ggcaaggagc tgcccggtgt 960tgctgccctg
tcgagtaagt aaatttgaac aaacatatta ccactctgga tatgcggaga
1020tgctgcaaga caaaatccgt ccatgtttct ttggaggagg atgagctgtg
gttgagcttc 1080catgctcggg actagtggac aggcggcttg tgtagtacct
gtcagccttc ccggcgcccc 1140ttcaagacag ggaacaattt tacgaatgta
atgtacaaag aatgataatt aatctcaaca 1200aaatacgcgt ttaccttcat
tatcaagatg atgcaatctc atgactagtg acgggtccga 1260agctgtgagt
ctagttttat caaatgcgag ctacgaagag ggggccacaa gttgatgggc
1320ggtggacttg atcctttcag taatgggtgt gtaaataatc gtagaaagta
cataatgtgc 1380tcatgcagca catacaccgc ttccagcgaa gtgctagcct
gtaccagatc tcccaccata 1440gacgacttat cctctgcaga cgaatcctcc
acaaccacgc agctgggcta gccaccggtc 1500cggcaccgcc aaagcagacg
aatctcttac tcaaacaagt ctatctagct ccaacattgt 1560tcatcgtctc
gctaccgagc cgcgtgaggg taaagctgca cgtgtaatca agtgattaat
1620gcttcgtgga atttgcgtgt gttcatctca attaaaccga taacccgtca
tctaacataa 1680tctattgatt agacgccccg accttcttca gctcctctct
gtagtttgaa gcaaacatgt 1740tagtccaatt tgagctttag atgaggtgta
gatcaatggt aacatattgc gctcatagca 1800ttgaccgatg aggtttggtt
tggttcggtt tggttcggtt tggtttggtt cggtttggct 1860tggcttggga
aaacgtactt cag 1883571814DNAartificial sequenceamplicon 5'gsdA A.
niger 57cgttatcaca aagaagccag gtcctgagct gacctacctg catgaatggg
ctgggggctg 60ctccacaatt gccagtaaac tcgaggtgcg cgtgccccgc gcatataacc
aatgtagtac 120agcgtcagac cgcaagaaac tcggcacctc tgcatttgcg
gctttccttt gttttaattc 180cagaggataa aactgtcaga tcagggtcgg
agggttaata ttactgggat aagttgacgc 240ggggggttcc gcatgcaaca
gtaaggtgta ccttaagatg ggcatcatcc gttccatgtg 300tggttctcag
tggagctggg aggagattta cagcggacct ggctcggata aatcagtccg
360tctagaaaag aaagggctgt tagttcaaac gatcatgctt ctgaaagaca
gatagagtaa 420gtcgagtgga ggatgcttca tcgtaagcac tgatttagag
atatctagat tgtctcaagt 480ggtagatagt agagtaagac tcatcgcgtt
acctagcgat gatataagag atgggttgcc 540acaacgggct ccgaaagaga
gaaggagtac tacagtatga gtgggaacgg aggacctgac 600aatttagggg
atgaatgcta gggatgaaaa aggaagcatt tcccggagta atcataccag
660ggaaatactg gataagttga ggtaaactag caggcagtgt gtcttgagtg
atgtaaaata 720accccgaata atagaattgg ataacaacta ctactcactc
ctcacggggt cccgcggcag 780caatcgacgt agtggaagaa cccaagccgg
gcttcccagt aacaaagtag taacaaagct 840gccccacccg ggctcactca
cttttgccca ccctgcagcc agcagctcct ctcctcgacg 900agaggccctc
cggtcttaaa aagtacttgc tccgccggaa ctgttgggat ttttccaaca
960aacctctctg tccttgctgt ctccctgtat cctctttatt tcctcctctt
ccctcctcca 1020ccgaatctct cacctttcct tcccatctcg tggttgttca
cacatcagta aaacatggcc 1080agcacaatag cacgcactga ggaacgccag
aatgctgggt gagttttgcg gtctccctct 1140ctgacatcac acccctcctc
ccactcccgt ccctcctgcc cgccgcccag acgtgaggat 1200tcaccaccca
cgactctcca taacaagccc cgtcgcccaa acattcactg gcaggcttcc
1260cgctttccat tattcttcaa ttcgtcacca ggattactcc ttcgggctta
acgaaaggac 1320tatccttctg actcaccacc aaccctcact gcccctcctg
catgctgtag cggactgcgg 1380gcaccgacct gcatcatcga tctacacccg
atccctgtga cattatattc gtcaagctat 1440agcctagcta acatggatgt
tttacgtagc accatggagc tcaaagatga cactgtcatc 1500atagtactgg
gtgcctccgg agatcttgca aagaagaaga ccgtcagtga cgaccccctg
1560attcatgttg acctgacaga aagctaacct tttacagttc ccggcccttt
tcggccttgt 1620atgtcctctc ccagatccaa ttgcagtttg actcaccagt
atggttgctg atttgcgctt 1680ccagtatcgc aacaagttcc tccccaaggg
aatcaagatc gtcggatatg cccggacaaa 1740catggaccat gaggagtacc
tgaggcgtgt gcgctcatac atcaagaccc ctaccaagga 1800aatcgaagag cagc
1814581837DNAartificial sequenceamplicon 3'gsdA A. niger
58tcatcaacct caacaagcac ctcgaggaga ttgagaaggg ccagaaggag cagaacagaa
60tctactacat ggccctcccc cccagcgttt tcaccaccgt ttccgaccaa cttaagcgca
120actgctaccc caagaacggc gttgcccgta tcatcgtgag tcaatcctgg
gctggtatca 180ccctgccatt ggtcattatt cttactcgct tgttttccta
tttcacaggt agagaagcct 240ttcggcaagg accttcagag ctcgcgcgat
ctccaaaaag ccctggagcc taactggaag 300gaagaggaga tcttccgtat
cgaccactac ctgggtaagg agatggtcaa gaacatcctt 360atcatgcgct
tcggaaacga attcttcaac gccacctgga accgtcacca catcgataac
420gttcaggtac gaccttgcgc tatccaattg gcctattgat ttacttgcta
aattgtcgct 480tctatcatta gatcacattc aaggagccct tcggcactga
gggacgtggt ggttacttcg 540atgaattcgg catcatccgt gatgtcatgc
agaaccgtac gttcaaagtc acgctcgaca 600tctccgacat gatgctgata
aaaatctctc ctagaccttc tccaggtgtt gacgctgctc 660gctatggagc
gccccatttc cttctccgcc gaggacatcc gtgacgagaa ggtacagtgt
720gcgcttgact attggttgtg ctgggttact gacacttaac caggttcgtg
tcctccgtgc 780gatggacgcc attgagccca agaacgtcat tattggccag
tacggaaagt ctctggatgg 840cagcaagccc gcctacaagg aggacgagac
cgttccccag gattcccgct gccccacctt 900ctgcgctatg gtcgcctaca
tcaagaacga gaggtgggac ggtgttcctt tcatcatgaa 960ggctggcaag
ggtatgtacc tctttccaag cgatcatagc accgattggt atactaataa
1020ttcgcagcct tgaacgagca gaagaccgag atccgtatcc agttccgtga
cgttacctcc 1080ggaattttca aggacatccc tcgcaacgag ctcgttatcc
gcgtccagcc caacgagtcc 1140gtgtacatca agatgaactc caagctgcct
ggcctgtcca tgcagacggt tgtgactgag 1200ctcgacctca cctaccgccg
ccgcttctcc gacctcaaga tccccgaagc ctacgagtct 1260ctgatcctgg
atgctctgaa gggcgaccac tccaacttcg tccgtgacga tgagctggat
1320gccagctgga ggatcttcac ccctctcctg cactacctgg atgacaacaa
ggagatcatc 1380cccatggaat acccctacgg tacgtgcact tcttgcaatt
tgtctaaatc gcttacatac 1440tgaccaacgc gcaggctccc gcggacccgc
cgtccttgat gacttcaccg cgtccttcgg 1500ctacaagttc agcgatgctg
ctggctacca gtggcccttg acttccaccc ccaaccgtct 1560gtaaataagg
gcggtcggca ggttatgacg gatgaggatg aaaaaaaaat tattgccaaa
1620aaaggctaaa aaaaagatgt taatgcgatt gattttcggt cgagaatcat
ggtatgacgg 1680ggcatctggg atgatatgac agaaatgaag cactcgggac
tatttatcgg tcggctgggc 1740aataactgga gttatctatt cgcaacccct
ttttagaaac gaatgcagag aacgtaacga 1800accacccccg gtcggttggg
accgccgctg tcttcac 1837591494DNAartificial sequencepromoter PmbfA
59gccatgaaat ccaatcattt ccttctggcc gccctcgggc aagagatagt gccgcagagg
60tctctcacag catctacatc tgcgaccgca acagccacca agcgaggcgc acatgagctt
120gtcctcctcc catgccaaag tttggccctc ttcgtttctg tgatgctgaa
ggaagtcaaa 180ctcgtcgatg ataggaccag atggtttgtc aagggtcaac
gctttccatg ccttctggca 240ccggtagtaa tgctcttctg caagggagac
ttgacgtttc ggatccgcgg gccccgggac 300atgctggaag ggattttctg
gctcaatacc acgtctgtat ttgacccttt ccagacagtt 360aatccgctgc
aggagggcga actgtagctc ctcgttctcc ttgtagcgct tgatccagtc
420tttttggatg ttgcacttgc ttggcctatg cttctcatat aatcttgccc
tgtcatagag 480acgacgtctg agattgtagc gttcgtcttt gatcacccgg
agccagatag gcctgagtat 540atctgacatt agatcaaagg gtctgtggat
agtctccttc agcatcagcg acgcatgtga 600ctcgcaagtc ggagagagct
tgtgggtggt catctttgat ggcgtcctct gctttccctt 660gattttcgtt
gattgttttt cgaaagttaa gtctggcagt caagagaatc cttctgccag
720acattatatt tacgtatact gacgtagtag aaacagcgtc aggatgagga
catggtgtgt 780gctggaccac ggaatcatag ttcatcagta tattgggttg
gacaaataac gctgagcatg 840tatatgtctt tacacactat aaaagccagc
gaacgccaat aaaatagggc atattgatgt 900gaaaatatga caccagttaa
aagcagtgta ttgattttat ctctcttcac ctcggaccta 960tactaccgta
tacaagactc aacttacttc cagatatagt aatatacacc ctatggacga
1020accagcacaa taattacagc caaacaacac cacccaaatg gcatattcct
aatcagcact 1080aagcacaaat accactgtca tcacagcata atcaataaga
atcccagaca accgactcac 1140tctgactcac cttacacaaa cccccaagca
aagcgcagcc cagaacctca gccaacaatc 1200gggcaacgta cggggaaaga
ttggccgatc catgatgtca gcagccctaa cccaaagcgg 1260actagcgcat
accgcccctc
tgactccgcc atcccagggc tcgagaagct tccgtggcgt 1320cgatataaat
tcagcgggcc ttgaacatcc ctccttacga cacacctcac gcgatcgatt
1380ttgacactca cgcaccgcca ccctcacatc ctccacccac accacacccc
ttaatcaacc 1440caccatcacc gctagaacgt ctatctcatc accgacttct
catccatctt caaa 1494601498DNAartificial sequencepromoter PcoxA
60gacggcattt gagcaacatc atgcgaacgc gaccgtgtta tcagcagcca ctcagcaacc
60gtcagttcat gatatgtcgc agagcgttgt cctcggcccg ccccagatgc tcccacgaca
120ccttccagtg cagtatccac ctgccagctt cgaaatcccc cctatccagc
gcgagaaaaa 180acgccatcac tcggagaccg aagaggatgg gaaccccgac
catggcctgc ggccgcagct 240atcagtcctc tcagctgacg cccctcacga
accctcgcac tctcccgaaa tgctcttcgg 300tgtccatggc gcatcatctc
agcagcatcc catgtcaaac catggctttg ggcccacgga 360ctctgtggcc
ctgccgcaac atcaccatca ccatcgactc ccgccccatg cagcgctgcg
420cgccccaggg cgtcatggag tgaatgtgga atctcctcct ttgccctcgg
gcccgccgag 480tgtggtgggc cagcctggaa tgcctgatcc agcccccagg
cctcgaggac caaaactgaa 540gtttactccc gaagaggacg ctctactggt
gtagttgaag gaaaacaaga acttgacgtg 600gaagcaaatt gcagacttct
tcccgggccg aacgagcggt accttgcaag tccgatactg 660caccaagctg
aaggctaagg atgtagcttg gagtgacgaa atggttcgat ttgctcctga
720tgtatttcta cgcctgtctc acacatgcta atgaagggaa taggtacaaa
ggctgcagcg 780ggcaatgcac gagtacgaga acgatcggtg gcgcatcatt
gcagggaagg ttggaaatgg 840cttcacccca gctgcttgcc gcgagaaagc
catgcagctc catgagtaaa agcgttgggg 900aattttcata tttatatcta
ctgtcgccag attcggccct gcttggaccc tctgatctcc 960ttactctcca
tattggttca aatgtcgggt ctccgatagg gctggtggtg caggcttgtt
1020gtaggcacgg gaggatgatc agcataactc tgagtcacta tagggacggg
ttgatgtaag 1080gtattaagtg atgtatgata attcatttta gcccggggga
acatatggcg ccggcatttg 1140ttcgttcgca atgaaccgac actagcgtcc
gctctcgcag tttagcaccg gctgatcccg 1200ggctgaacgc ggccattgct
cggccggggc atgtgttcct tatctacggc agaccgcaga 1260agaccactgg
agcagattat agaccctaag ccctaagccg gacacccaat cgagtaggtc
1320tgcggaccag gtcactgcgg gcagccggag aagctccgca accaatcaat
ccccggcgct 1380gactaagggc aggcgaccac gggccgaagc ggcttcaaac
tcacctcaac ctccaaactc 1440cctcatctcc aaacgtcctt gccttgtctg
ccgtcattgc aacccaccca ccaggaca 1498611490DNAartificial
sequencepromoter PsrpB 61ttggcagggt cacgtagccg taattatttt
cggggaaggt tggaatgcaa tggaaggaga 60tttccgtagc tagggctttg atcgatgcgg
ggagcactgc cggtaggagg tctggggtga 120atggggtgat atgcaggcgc
ttcgtatcgg acggtgtggt cgtcatttgc ccaatagata 180gttagataga
tacctgagta cggtagcagt gcaggtgacg gctaagaagt cggagggaaa
240aaggtgcagt cacaagcgca ttcagcctaa caagtgtctt tgatactcgg
tgagaaacaa 300acttgagtag aataagacag aaagttcttg tgaatggtca
caatgggctt ccaacgaagc 360atcaagcaga ccctgttgca atagatattc
caagaccgaa aaattaatga taggatcagt 420tattggccga gggattttcc
gggccgccaa gaccgggtta tggagatgtg gcgcaggcat 480gccatcctca
gccacaggtt tctgtgacat cccaaaagca ttgatcgaag ttggtataag
540tttcattcta tctaccatgg tgacaaggaa gtacgggtgt agaaaagaaa
aatctggtag 600gaatagctca gcaacaaatg gcggaatgat tgatgtaaga
ctcgatgtat ccactggaac 660gagatgcaag ttgcaacagc aataaatgga
tttcagcctc cattacaatg taacagtcgg 720gccgatactc agccggagca
ggatttggcg ggtgaatagt ggatccggag agaaacgacc 780aggtaatctt
tcgtacggga ccagacccga cccggcctgc tttttagtta ccagctgtta
840cttgtgtaat ccccgtaaaa cgatcagtaa ctgccattga tcttcctgct
cttttccctt 900attccctttt ccccctttga aacttatttt cttcttcctc
ttcatcgctt aactacttaa 960gtactaggat tctcactcgc cactcttccc
caatatctaa aagtagtctt gctacgaaga 1020tcccttcccc ctacattact
cctcctcctt caacacaccc acccccccct gatccggccc 1080cataccagtc
ttcccgcggc taactaaagc ccgcacgtct gatctcatcg ccgcttccag
1140cttcgacctc agtcgctcac atggccactc ggattcctta gcatcatctc
tttttttccc 1200atcccctccc cgccctacca actgagggtc ctctgaagtg
tgctccacat ttccttccct 1260tcacttattt tggatcctca ttttctttct
tcctctgttt cggggcgttc ttcaacatcg 1320ctacttagtc acttctctcc
tctcattacc ggacgggaac ttcgctccct tctccgcttt 1380cttatccgga
ccgcctcttg ccaatctcac catcgatcct aacccgtcat aatccagtca
1440ctcaacccta ctattgtcga catacacgtc ggttcccatt ctcgttcgag
1490621500DNAartificial sequencePpromoter PtvdA 62ttagtccatt
cagcaagctg ccgttgggat ccttggcttc agccgtcaag ggctggcctc 60gcttgagcgg
ctctttgctg aggaacacgg agaaaaagag gttagctgtc tgccagcaga
120gaaccaggag gcctcctgaa atcaaggttc gcacgataac tccgaggatt
ggcgggataa 180tgccgggagg ggatgcagcg gatctcggga aatcccagaa
tagctttgca aagtacagtg 240tagtgctcca tgccgggcgc cggaggacga
aggcgtatgc gaatgggcaa agaatggaaa 300ccaagatact ccgtttcatc
ccatctttga tgagctgcgg caatgctgac tggaggcgtt 360tagagatcga
ttgctcctgc cggctatctt catcagcgga gccagcgacc ctcctggcaa
420cgggaagagg gacgcgatcg tagtcgtagt aaagatgaac cactgactgc
gcgactgcca 480ggagaaagtg gtaggtataa aggtagattg ctctttcgtt
taagcttgct cgttcgtgag 540gtctagcata actagttagg cgagttcaag
agaaccgctt gagctcgtcg tcgcaggaaa 600catacctagc tcggttcacc
aattccaggt gtgcgctact tgaagaagac cacctgtaga 660tttcagtgaa
ccaccaggcg gagaacaagt accagccaaa agtctgtata acgtcgaggg
720gaacgaggta tttgaaggtg ctaagcgggg aggaggttgt cctggcaccg
atgtgcatct 780gaccaacacg gaggacgaag acaacgagac tgcagaggaa
gagaagaact gtgcgaactc 840cacatcctcc aatggggaac caagcccaca
agactatcga gatatcacag ctggttagcg 900atcacagctt gtcaaacgcc
gctcggtgtt ggtgtaaact tacaagatga cttatcgcca 960atagcaaatg
ccactacata gcagaccaga agcgccagtg ccgacgcatg aacgaatcgt
1020ctgtgcagcg cagaggtcaa gatgcgccga tacgggcgag gctgtgctgc
agccatggtg 1080caaggtggaa gagggactgt gagatctcta gggagagaag
agacaaagca atttcactgc 1140atgatcagat gacaggagga attgaggtat
ccggtcagcc tgtcagaacg gaaagctccg 1200aaggggggca agtggagctt
caaggaggag tgtcggcgtc ggatgcgaca ataaagtgaa 1260gcttttcggg
caaagcatca aaacacgcac cagccacaaa cggcatgtca tgtgacaccc
1320caccagccat cgatggcgtc aggtgagtgg cagcctgcga atcatggcat
ctgtgcggcc 1380aggcaaccgt cgacacctga cagcgataac gtactactac
tttagcaggg agagagccgg 1440ccggttgcat cggccgaatg atcccacctt
catccatcca ctgtcgcatc tcattccgcc 1500631064DNAartificial
sequenceterminator TglaA 63cgaccgcgac ggtgactgac acctggcggt
agacaatcaa tccatttcgc tatagttaaa 60ggatggggat gagggcaatt ggttatatga
tcatgtatgt agtgggtgtg cataatagta 120gtgaaatgga agccaagtca
tgtgattgta atcgaccgac ggaattgagg atatccggaa 180atacagacac
cgtgaaagcc atggtctttc cttcgtgtag aagaccagac agacagtccc
240tgatttaccc ttgcacaaag cactagaaaa ttagcattcc atccttctct
gcttgctctg 300ctgatatcac tgtcattcaa tgcatagcca tgagctcatc
ttagatccaa gcacgtaatt 360ccatagccga ggtccacagt ggagcagcaa
cattccccat cattgctttc cccaggggcc 420tcccaacgac taaatcaaga
gtatatctct accgtccaat agatcgtctt cgcttcaaaa 480tctttgacaa
ttccaagagg gtccccatcc atcaaaccca gttcaataat agccgagatg
540catggtggag tcaattaggc agtattgctg gaatgtcggg gccagttggc
ccggtggtca 600ttggccgcct gtgatgccat ctgccactaa atccgatcat
tgatccaccg cccacgaggc 660gcgtctttgc tttttgcgcg gcgtccaggt
tcaactctct ctgcagctcc agtccaacgc 720tgactgacta gtttacctac
tggtctgatc ggctccatca gagctatggc gttatcccgt 780gccgttgctg
cgcaatcgct atcttgatcg caaccttgaa ctcactcttg ttttaatagt
840gatcttggtg acggagtgtc ggtgagtgac aaccaacatc gtgcaaggga
gattgatacg 900gaattgtcgc tcccatcatg atgttcttgc cggctttgtt
ggccctattc gtgggatgcg 960atgccctcgc tgtgcagcag caggtactgc
tggatgagga gccatcggtc tctgcacgca 1020aacccaactt cctcttcatt
ctcacggatg atcaggatct ccgg 106464549DNAartificial
sequenceterminator TniaD 64acgggttcgc ataggtttgg ggttgtatct
tggcgttggg acggactggg tatggtgttt 60cttttggata tatgacatga tatgtacacg
gccgtgaatc tttaacttta tatcattata 120gaaatgcact tgcacatttc
aacacgctgc gagcagaatc tcgaagattg ttccgcaagt 180attagatcat
gagagcattt tcatttcctt tcaggcagtg ggagtaggcc atcctgaaaa
240caaggcggcc actgtagact agtaatacct cttcatatcc aaccttacca
gaagatgatc 300aaacacattc gcagatccac ctctcgccgc gaaagccatc
ccggcccttc cggtacccaa 360tgcctttgat gacgatgcca ccgcccccct
ggcaccatct aggctacgca aatacccaac 420aaaagcggcc atttgcttcc
taaacactgc atacaaccgc ttgaccatgg cttcactctc 480gtgcctcacg
ccatgtgtcc catccttgcg cgacagaggc gcaaacctca gaatcgtgtc 540aaatatccc
54965580DNAartificial sequenceterminator TgpdA 65gaatcaggac
ggcaaactga attcagaagt gtgctgtgag tgagactgat tgccgagcgc 60agacgactct
cgtggaaccc ggcttgtgga gaagcttgag aaggtcttaa ctcctagcgt
120aaaagctcat gatgacgtac aatttaatga aatgatacaa tgttcatatt
tcccgttcaa 180atttccggcc ttggtcagtg cgtaagatgt ccacgattga
atactaactc agtatgggtt 240tggtagcatt ggcaatgtag ttataagcat
gcaccggttg aagacgtcgg ccccagatgc 300aatgctgcgg tggtgactaa
gctctgcagt gaatggaatg cgtttctttg atcgacttcg 360gcgtgccgcg
ggattttctc ggcgcttcta ctggtgcaga aaggacgata ccactggctt
420tcggtccatg ccacatccca gtctcccggg aaattcattg catactttaa
gaaacaaact 480gatctccata atttccgtct ttagagttca cttggtactt
ttgggtggat cgaggggtgt 540ccgcggccat ccaagtcacg tggagggcag
ctagaccacg 58066528DNAartificial sequenceterminator TtrpC
66tgatttaata gctccatgtc aacaagaata aaacgcgttt cgggtttacc tcttccagat
60acagctcatc tgcaatgcat taatgcattg gacctcgcaa ccctagtacg cccttcaggc
120tccggcgaag cagaagaata gcttagcaga gtctattttc attttcggga
gacgagatca 180agcagatcaa cggtcgtcaa gagacctacg agactgagga
atccgctctt ggctccacgc 240gactatatat ttgtctctaa ttgtactttg
acatgctcct cttctttact ctgatagctt 300gactatgaaa attccgtcac
cagcccctgg gttcgcaaag ataattgcac tgtttcttcc 360ttgaactctc
aagcctacag gacacacatt catcgtaggt ataaacctcg aaaatcattc
420ctactaagat gggtatacaa tagtaaccat ggttgcctag tgaatgctcc
gtaacaccca 480atacgccggc cgaaactttt ttacaactct cctatgagtc gtttaccc
528671713DNAartificial sequenceamplicon 5'pgkA A. terreus
67cttggggaat tgggacacgc caggaaccta ccgcccccgt ttgattttga gcggttgacc
60cgtttcatgt cgtacggttt cttcatggcc ccggtgcagt tccagtggtt cgggttccta
120tcgaggacct tccctctcac caagaagaac ccgaccatcc cggctctgaa
gcgggtggcg 180gttgatcagc ttatgttcgc tccgttcggt atggacctga
tgttgtgcca cgagaaaaag 240gatatcccgc taatagaaat gaatataggt
ctggtctgtt tcttcacctt catgacaatt 300gctgagggtg gcggacggag
agccttgact cgcaagttcc aggatgtgta cctgccgaca 360ctcaaggcga
actttgtgct ctggcctgcg gtgcaaatcc tgaacttccg ggtggttccc
420atccaattcc agattgtgag tttgctcttc tcccgtgacc cgacgcgttt
tcattagtta 480ttgtatactg attgcttttt ctttcacagc catttgtgtc
gtcggtgggt atcgcatgga 540ccgcatacct gtctctgacc aactcttccg
aggaggagta aggtaaggta cgggttttct 600cgttgcattg gatggttatc
acttgatacc tatgtttacc tggtcggccc ttggctttgg 660aaatcttctt
tgtggattct ttcttctttc cttgtgttgc gtggggtatg tttacggcgt
720tttcaatgct gttttggggg gttctgtttc tcattggtgg tgtccggtct
gtcacagggt 780actctcaaag gtttcttcgt taggtattca aaaggcgcac
aaatgctgga tttagatcat 840caacccaagc ggaagagact attcaaagga
gtctccattc gcagggatat ggaactgcca 900ttctatgacg ctttcgggag
aaatttccag gattttcgta ggtcgagaca cccacagagg 960cacatgtagg
gaccgcgggg gacgatgatc ctatgaatgc ccgtacgacg agaattccga
1020agcggctgtt tccaattccc gcgagagatc cgaggctatc gagaacgcat
agggacaatg 1080ttcacccctg agagcccgga cccttccgtc atcgatcgcg
gtggactgac atcagtcccg 1140catcagcccg atgcgtcagc gaatctctca
ccggacgccc caccaaacac tgctgctacc 1200ccgccgctgg gccgaagtta
tcatatgggc tcaccgtgaa cggattgagc atttttccct 1260ccttcttcca
attcacctca ccatctctcc tgcgaggtac tgctccctat ttctatccct
1320ctagacccct gtggtatcgc tgacccgacg ccagctcctc cctctccaac
ttcttcacca 1380tgtctcttag caacaagctt gccatcactg acgtcgacct
caaggacaag cgtgtcttga 1440tccgggtatg tcacctccaa tggctgtggt
atgatcggtc aatgctaacc acgctcacct 1500cccaggtcga cttcaacgtc
cctctcgatg acaacaagaa cgtcaccaac ccccagcgta 1560tcgttggtgc
tctccccacc atcaagtacg ctgtcgagaa tggcgccaag gccgttgtcc
1620tcatgtccca cctcggtcgt cctgacggca agaagaaccc caagtacagc
ctgaagcccg 1680tcgtgcccgt cctggagaag ctcatcggca aga
1713681820DNAartificial sequenceamplicon 3'pgkA A. terreus
68cagatcatcc tcctggagaa cctgcgtttc cacgccgagg aggagggcag ctccaaggac
60gccgagggca agaaggtcaa ggccgacaag gccgccgtcg aggagttccg caagggactg
120accgctcttg gtgacatcta catcagtaag tctatttccc ctttggccca
tggaccgtaa 180ttgggtttgt ccatactaac gataccatcc agacgatgcc
tttggaaccg cccaccgtgc 240ccacagctcc atggtcggcg tcgacctccc
tcagaaggct tccggcttcc tggtgaagaa 300ggagcttgac tacttcgcca
aggccctcga gaacccctct cgtcccttcc tggccatcct 360cggtggcgct
aaggtctccg acaagatcca gctgattgat aacctcctcc ccaaggtcaa
420cagtctgatc atcaccggag ccatggcctt taccttcaag aagaccctgg
agaacgtcaa 480gatcggcaag agtcttttcg acgaggccgg cagcaagatc
gtccccgaga tcgtcgagaa 540ggctaagaag aacaacgtga agatcgtcct
tcctgtcgac tacgtcactg ccgacaattt 600cgctgccgac gccaagaccg
gctacgctac cgatgccgac ggcatccctg acggcttcat 660gggtctggat
gtcggcgaga agagtgtcga gctgtacaag cagactatcg ccgaggccaa
720gacgatcctc tggaacggcc cctgcggtgt cttcgagatg gagcccttcg
ccaacggcac 780caagaagacc ctcgacaacg tcgttgccgc cgctcagtcc
ggctccattg tcatcatcgg 840tggtggtgac actgctaccg tggccgccaa
gtacaacgcc gaggacaagc tcagccacgt 900ctccactggc ggtggtgcct
ctctggagct tctggagggc aaggagctgc ctggtgtcac 960tgctctgtca
agtaagtaaa cttttacgac catgtaatgt gttggaggga cgagttgcag
1020aatgagatcg tgcttgtcca tgtgtattgg gcgagccatg gctgcatcag
gccgtgtttt 1080tttagcgtcc ctctccatcc cgtcgaggga tggccttttc
tatgatgtaa tgtacaggca 1140ttaaaattaa tcaaaataag cgaaatgatt
ttgtttctcg attcaagaag ggttctgatg 1200aagtaatggc tactagtgac
gggatctaaa ctgtgagatc agacgcttgt cgttcccatt 1260gaaatggata
ttcttattgc gtagagcttc tcttatcttt tctatcaaac aattgttttc
1320ataggatgtc cgggagccat acaccgccag cgtccgtcac gatcatatat
gcctggtaga 1380tctgggccaa ggaccccggt cgcttctcca gtgggctggg
cgccgttccc cttgacgaac 1440gaacggccat gacgatccca ggtgaacgca
aggccgggcg aactaccacc atagaacgga 1500catgatgtcc taataaagaa
gaatgtagca tcttgatgat atagcttgtg ttccatcatg 1560gaaaaagagg
gcaaaaatag ataattctta tcgttcgttc attcgcgttc gggatattca
1620gagtaccaca tgtccatccg tccattagtg agatgcctcg accttttcca
gttcctctct 1680gtagcacgaa gagaaatacg ttagtgttgc gtaggcgtca
gaggaagaag gggagcaacc 1740gcactttagc tgtttcgaaa agtcctccgc
cgcatcgtcg tcgtcccagc tctcctccca 1800caggtgaaca ttcgtgccgt
1820691817DNAartificial sequenceamplicon 5'gsdA A. terreus
69attggaagct ggctctatct cacccctcct gacgaagacc tcagcagtgt acggtacact
60ggagttaagc tgtccgtctc cgcggacacc tgacgagctt agatagaggg tgggttggat
120tatggaccac accccggccg cctttatatt tctttttctc cttttttgat
gtcctggcct 180tcgcctatca gaggagaatc ccactagatc ctaaactgac
tgggcgtaag attactagta 240ccatgtagca tttggcgcgg ggctaggagg
tcagcatatc caagatgtgc ctctagagat 300gcgtctccgg tccatgtacg
gttcccggtg gagctcactg ggttgaaatt tccagcggag 360taggctcgga
taaaactagt gttaagaagc agttgagcag aagggttcgg atgattgggg
420acggtagtaa tacaaggtac aagcaccggc atcgggaagg atttccttcg
tgtcctctat 480atccgagtgg gagggagtat gaacgggctg actgagtacc
taggcaggcg tgagagggat 540gatataatac cagtagctgt cgccacggcg
gtggattcta gggagtaact actacgtaga 600tgatgaaaag gaaaaagaac
tgataaaaga tgacagatta tctatctccg gaaaaagggc 660gggcacccga
ggctatagtt atactacgaa gcataaataa aataataaaa acacctaatt
720aaatccaaca cgaagatatc ctaggttccc aaagcccagt tgtatgtaat
ttctctccag 780ttaggttttt tcttacccag gccaccggac ggggctcgcg
gcaacaatcg acgccccacc 840gtgtactgta atcgagagtc accctgcagc
aactgcagct cctcgactgc tgctacgact 900aagcagagtc cctctcacgg
cgttaaaaag tacttgctcc gccggaactg ttggggattt 960ttccaacaaa
cctcttctct ttcctcctct actccctcgt ctctccttca ttccaagctt
1020catttccctt ccttaacagt ccgttgttca tttttggtgc ctgcacaaac
tcccgtccgt 1080caccatgacc agcacaatag cccgcactga ggaacgccag
aacgctgggt gagtcttgac 1140tgccgccgcc gcctccctct ctgacatcac
acccctcctc cgtccccctc tacgtctttc 1200ttttcttccc acccgtctgc
cacctgctcg atcgccttcc tcaggacagt caatatcagt 1260caattcgacc
ccagattttc cacttccctt cgttgaatgc cccgagctct tccaatttgc
1320tttttagaaa ttgtcgagca gctctagttt tcaattcccc cttcttccga
tcgcgctggc 1380aaatccctgt gatcgacatc atacagcttc ccctgctcag
ccagtaacct tcgattcgct 1440actaacatgg atgtctcgcg tagcaccatt
gaactgaaag atgacacggt catcgtggtc 1500ctcggtgcct ccggagatct
cgcaaagaag aagacggtag gtgttctacc ctaaaaaggc 1560ttggccggtg
aagccgatcg gagactgact aggttccata gttccctgca ctgttcggtc
1620ttgtaagtat acgatgacgg tccgagtaaa gtatcgtcgc gtcggttggc
attgaccatg 1680catttagtac cgtaacaaat tcctccccaa ggggatcaag
atcgtcggat atgctcggac 1740gaacatggac cacgaagaat atctgagacg
ggtgcgctcg tacatcaaga cccccaccaa 1800ggaaatcgaa gaacagc
1817701825DNAartificial sequencesynthetic oligonucleotide
70tcaacctcac caagcacctc gaggacgtcg agaagggcca taaggaacag aacagagtct
60tctacatggc gctgcctcct agcgtcttca ttaccgtgtc ggatcaattg aagagaaact
120gctaccccaa gaacggcatt gcccgtatta ttgtgagtcg ccaacacaaa
agctttgttt 180ctaccgaggc atatgcttac gcgtgttttc tttgtgtcta
caggtcgaga agcccttcgg 240caaggatctc cagagttctc gtgacctcca
gaaggccctt gagcccaact ggaaggagga 300ggagatcttc cgtattgacc
actacctggg taaggagatg gtcaagaaca ttctcatcat 360gcgcttcgga
aacgagttct tcaacgctac ctggaaccgt caccacattg ataacgtgca
420ggtatgagtt gaatgttttc aggacgaccg gtgcagatgc taagctcttg
tcgcagatca 480cattcaagga gccgttcggc acggagggcc gtggaggtta
ctttgatgag ttcggcatca 540ttcgtgatgt catgcagaac cgtatgtcca
acaaaaaaat gaccttgacg gctaatgatg 600gcaagtgcta atgtccttat
ctagaccttc tgcaggtgtt gacgcttctt gccatggagc 660gtcccatctc
tttctctgct gaagacattc gtgatgagaa ggtatttttg ccgatccctt
720atcccaccgg cggcaactaa cgtaagtgac aggtgcgtgt tctgcgcgcg
atggacccca 780ttgagccaaa gaacgtcatc atcggccagt acggaaagtc
gctcgacggt agcaagcccg 840cctacaagga ggatgatact gtccctcaag
actcgcgctg cccgactttc tgcgccatgg 900ttgcgtacat caagaacgag
agatgggatg gcgttccctt catcatgaag gccggtaaag 960gtatgtatcc
tgttctctat ggcagcacgc cctggacttg gtgtactcac atcttcgaca
1020gccctgaacg agcagaagac cgagatccgt atccagttcc gtgacgtcac
ttccggcatc 1080ttcaaggaca tcccccgcaa cgaacttgtt attcgcgtcc
agcccaacga gtcggtgtac 1140attaagatga actccaagct gccgggtctc
tccatgcaga cggtcgttac cgagcttgac 1200cttacctacc gccgccgttt
ctccgacctc aagatcccgg aggcttacga gtctctgatt 1260ctggatgctc
tgaagggtga ccactcgaac tttgtccgtg acgatgaact tgactcgagc
1320tggaagatct tcacccctct gctgcactac ctggacgaca acaaggagat
catccccatg 1380gaatacccct acggtaagcg gcgaatgcgc catctgcgaa
acaaaatatt tcttttgagt 1440tccagtactg acaatgttat caggctctcg
cggacctgct gtgctcgacg acttcaccgc 1500atcgttcggg tacaagttca
gtgatgctgc tggctaccag tggcccctga cttcggcccc 1560caacagactg
taaatgaaaa atattatgat cgtttaagca atgaaaaggg aaagaaggaa
1620aatgggttaa tgtgagcact accgggtttc atagcatgat cggatggtcc
tcgaggttgg 1680gatggcacga tatttggaga cgggttttcc agatcggcca
tggtggacca gacgctcccc 1740tgggtcccta gaaacgaatg aagtgactgt
tacgaatcaa acacgatgat attgattccc 1800tcttgcagtt gcgacgggct gtttg
1825711731DNAartificial sequencesynthetic oligonucleotide
71atggaattca gagtccactt acaagccgat aacgaacaaa agatattcca aaaccaaatg
60aaaccagaac cagaagcctc ctacttaatt aatcaaagaa gatcagctaa ctacaagcca
120aacatctgga agaacgattt cttggatcaa tcattgatct ctaagtacga
tggtgacgaa 180tacagaaagt tatcagaaaa gttgatcgaa gaagttaaga
tctatatttc tgctgaaact 240atggatttgg ttgcaaagtt ggaattgatc
gattctgtta gaaagttggg tttagctaat 300ttgttcgaaa aggaaattaa
agaagcattg gattcaatcg ctgcaatcga atctgataat 360ttgggtacta
gagatgattt gtacggtaca gctttgcatt tcaagatttt gagacaacat
420ggttacaaag tttcacaaga tatcttcggt agattcatgg atgaaaaggg
tactttggaa 480aaccatcatt tcgctcattt gaagggcatg ttggaattgt
tcgaagcatc taatttgggt 540ttcgaaggtg aagatatctt ggatgaagct
aaagcatcat tgacattagc tttgagagat 600tctggtcata tctgttaccc
agattcaaat ttgtctagag atgttgttca ttcattagaa 660ttgccatctc
atagaagagt tcaatggttc gatgttaagt ggcaaattaa tgcttacgaa
720aaggatatct gtagagttaa cgctactttg ttggaattgg caaagttgaa
cttcaatgtt 780gttcaagcac aattgcaaaa gaatttgaga gaagcttcta
gatggtgggc aaatttgggt 840atcgctgata atttgaagtt cgctagagat
agattggttg aatgttttgc ttgtgcagtt 900ggtgttgcat tcgaaccaga
acattcttct tttagaatct gtttgacaaa agttattaat 960ttggttttga
ttattgatga tgtttatgat atctatggtt cagaagaaga attgaagcat
1020ttcactaatg ctgttgatag atgggattct agagaaacag aacaattgcc
agaatgtatg 1080aagatgtgtt tccaagtttt gtacaacact acatgtgaaa
tcgcaagaga aatcgaagaa 1140gaaaatggtt ggaaccaagt tttgccacaa
ttgactaagg tttgggcaga tttttgtaaa 1200gctttgttgg ttgaagcaga
atggtacaat aagtcacata tcccaacatt ggaagaatac 1260ttgagaaacg
gttgtatctc ttcatctgtt tctgttttgt tggttcattc tttcttttct
1320atcactcatg aaggtacaaa ggaaatggct gatttcttgc ataagaacga
agatttgttg 1380tacaacatct cattaattgt tagattgaat aatgatttgg
gtacttcagc tgcagaacaa 1440gaaagaggtg actctccatc atctatcgtt
tgttacatga gagaagttaa tgcttctgaa 1500gaaacagcaa gaaagaatat
caagggtatg atcgataacg cttggaagaa agttaacggt 1560aaatgtttca
ctacaaacca agttccattt ttatcatctt ttatgaataa tgcaacaaat
1620atggctagag ttgcacattc tttgtacaaa gatggtgacg gttttggtga
ccaagaaaaa 1680ggtccaagaa cccacatatt atcattatta ttccaaccat
tagtaaactg a 1731
D00000
D00001
D00002
D00003
P00999
S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.