Methods For The Preparation Of Ribosides
Axt; Steven Donald ; et al.
U.S. patent application number 16/692966 was filed with the patent office on 2020-06-25 for methods for the preparation of ribosides. The applicant listed for this patent is Gilead Sciences, Inc.. Invention is credited to Steven Donald Axt, Pavel Robertovich Badalov, Katrien Brak, Silvio Campagna, Andrei Chtchemelinine, Edward Doerffler, Morin Mae Frick, Detian Gao, Lars V. Heumann, Brittanie Hoang, Willard Lew, Robert Ronald Milburn, Sean Timothy Neville, Bruce Ross, Erik Rueden, Robert William Scott, Dustin Siegel, Andrew C. Stevens, Clarissa Tadeus, Tiago Vieira, Andrew W. Waltman, Xianghong Wang, Mark Charles Whitcomb, Lydia Wolfe, Chia-Yun Yu.
| Application Number | 20200197422 16/692966 |
| Document ID | / |
| Family ID | 54557474 |
| Filed Date | 2020-06-25 |











View All Diagrams
| United States Patent Application | 20200197422 |
| Kind Code | A1 |
| Axt; Steven Donald ; et al. | June 25, 2020 |
METHODS FOR THE PREPARATION OF RIBOSIDES
Abstract
Provided are methods of preparing compounds and pharmaceutical compositions for treating Filoviridae virus infections The compounds, compositions, and methods provided are particularly useful for the treatment of Marburg virus, Ebola virus and Cueva virus infections.
| Inventors: | Axt; Steven Donald; (San Mateo, CA) ; Badalov; Pavel Robertovich; (Edmonton, CA) ; Brak; Katrien; (Belmont, CA) ; Campagna; Silvio; (Dover, DE) ; Chtchemelinine; Andrei; (San Mateo, CA) ; Doerffler; Edward; (Union City, CA) ; Frick; Morin Mae; (San Mateo, CA) ; Gao; Detian; (Edmonton, CA) ; Heumann; Lars V.; (Redwood City, CA) ; Hoang; Brittanie; (Berkeley, CA) ; Lew; Willard; (San Mateo, CA) ; Milburn; Robert Ronald; (San Mateo, CA) ; Neville; Sean Timothy; (San Mateo, CA) ; Ross; Bruce; (El Granada, CA) ; Rueden; Erik; (San Mateo, CA) ; Scott; Robert William; (San Mateo, CA) ; Siegel; Dustin; (Half Moon Bay, CA) ; Stevens; Andrew C.; (Edmonton, CA) ; Tadeus; Clarissa; (San Francisco, CA) ; Vieira; Tiago; (Edmonton, CA) ; Waltman; Andrew W.; (Kalamazoo, MI) ; Wang; Xianghong; (Dublin, CA) ; Whitcomb; Mark Charles; (Woodside, CA) ; Wolfe; Lydia; (San Mateo, CA) ; Yu; Chia-Yun; (Burlingame, CA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 54557474 | ||||||||||
| Appl. No.: | 16/692966 | ||||||||||
| Filed: | November 22, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 14926063 | Oct 29, 2015 | |||
| 16692966 | ||||
| 62105619 | Jan 20, 2015 | |||
| 62072331 | Oct 29, 2014 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61K 31/7056 20130101; A61K 31/665 20130101; C07H 15/18 20130101; C07D 519/00 20130101; A61K 31/683 20130101; A61P 31/12 20180101; A61K 45/06 20130101; C07F 9/2429 20130101; C07H 1/00 20130101; A61K 31/706 20130101; A61K 31/685 20130101; A61P 31/14 20180101; C07F 9/65616 20130101; A61K 31/53 20130101; A61K 31/675 20130101; C07D 487/04 20130101; C07H 11/00 20130101; A61K 31/6615 20130101; A61K 31/00 20130101; A61P 31/00 20180101; C07H 1/02 20130101; A61P 43/00 20180101; A61K 31/53 20130101; A61K 2300/00 20130101; A61K 31/675 20130101; A61K 2300/00 20130101; A61K 31/685 20130101; A61K 2300/00 20130101; A61K 31/706 20130101; A61K 2300/00 20130101; A61K 31/7056 20130101; A61K 2300/00 20130101 |
| International Class: | A61K 31/685 20060101 A61K031/685; A61K 31/706 20060101 A61K031/706; A61K 31/7056 20060101 A61K031/7056; A61K 31/53 20060101 A61K031/53; C07H 1/00 20060101 C07H001/00; C07H 1/02 20060101 C07H001/02; C07H 11/00 20060101 C07H011/00; C07H 15/18 20060101 C07H015/18; A61K 31/6615 20060101 A61K031/6615; A61K 31/665 20060101 A61K031/665; A61K 31/683 20060101 A61K031/683; C07D 487/04 20060101 C07D487/04; C07D 519/00 20060101 C07D519/00; C07F 9/24 20060101 C07F009/24; C07F 9/6561 20060101 C07F009/6561; A61K 31/675 20060101 A61K031/675; A61K 45/06 20060101 A61K045/06; A61K 31/00 20060101 A61K031/00 |
Claims
1.-42. (canceled)
43. A method of preparing a compound of Formula (VIII) having structure: ##STR00210## the method comprising: (a) (i) forming a first reaction mixture having BCl.sub.3, dichloromethane, and a compound of Formula (XI-a): ##STR00211## and (ii) combining K.sub.2CO.sub.3 with the first reaction mixture to prepare the compound of Formula (XI-b): ##STR00212## (b) forming a second reaction mixture having acetone, 2,2-dimethoxypropane, sulfuric acid, and the compound of Formula (XI-b) to prepare a compound of Formula (XI-c): ##STR00213## and (c) forming a third reaction mixture having: (i) MgCl.sub.2, (ii) diisopropylethylamine, (iii) the compound of Formula (XI-c), and (iv) a compound of Formula (X) having structure: ##STR00214## to prepare the compound of Formula (VIII).
44. The method of claim 43, further comprising preparing the compound of Formula (XI-a) by a method comprising: forming a reaction mixture with a cyanating agent, a Lewis acid, a Bronsted acid, a solvent, and a compound of Formula (V-a) or Formula (V-b) having structure: ##STR00215## to prepare the compound of Formula (XI-a).
45. The method of claim 44, wherein the cyanating agent is TMSCN, TBSCN, TESCN, HCN, KCN, NaCN, 4-toluenesulfonyl cyanide, CuCN, CuCN*LiCl, LiCN, Zn(CN).sub.2, K.sub.4 [Fe(CN).sub.6], tetrabutylammonium cyanide, tetramethylammonium cyanide, tetraethylammonium cyanide, tetrabutylammonium cyanide, tetraalkylammonium cyanide with alkyl independently being Me, Et, Pr, iPr, Bu, iBu, tertBu, Pent, Hex, tributyltin cyanide, trimethyltin cyanide, triethyltin cyanide, tripropyltin cyanide, trialkyltin cyanide with alkyl independently being Me, Et, Pr, iPr, Bu, iBu, tertBu, Pent, Hex, 2-hydroxy-2-methylpropanenitrile; or combinations thereof.
46. The method of claim 44, wherein the Lewis acid is TMSOTf, TMSOTf, TBSOTf, TESOTf, BF.sub.3, BF.sub.3-OEt.sub.2, BCl.sub.3, BF.sub.3-THF, MgCl.sub.2, MgI.sub.2, MgBr.sub.2, MgBr.sub.2-OEt.sub.2, ZnCl.sub.2, ZnBr.sub.2, ZnI.sub.2, LiCl, LiBr, LiI, AlCl.sub.3, AlBr.sub.3, AlI.sub.3, Me.sub.2Si(OTf).sub.2, Et.sub.2Si(OTf).sub.2, Pr.sub.2Si(OTf).sub.2, iPr.sub.2Si(OTf).sub.2, (tBu).sub.2Si(OTf).sub.2, (C.sub.6F.sub.5).sub.3B, MeSiCl.sub.3, Me.sub.2SiCl.sub.2, SiCl.sub.4, TMSCl, TMSI, TMSVr, TBSCl, TBSBr, TBSI, TESCl, TESBr, TESI, SmCl.sub.3, SmBr.sub.3, SmI.sub.2, SmI.sub.3, ScI.sub.3, ScBr.sub.3, ScI.sub.3, Sm(OTf).sub.3, Sc(OTf).sub.3, TiCl.sub.4, Ti(OiPr).sub.4, Ti(OiPr).sub.3Cl, Ti(OiPr).sub.2Cl.sub.2, Ti(OiPr)Cl.sub.3, Zn(BF.sub.4).sub.2, LiBF.sub.4, Mg(BF.sub.4).sub.2, ZrCl.sub.4, FeCl.sub.2, FeCl.sub.3, FeBr.sub.2, FeBr.sub.3, FeI.sub.2, FeI.sub.3, Cu(OTf), Cu(OTf).sub.2, 4-toluenesulfonylchloride, benzenesulfonylchloride, 4-toluenesulfonyl triflate, benzenesulfonyl triflate, methyl sulfonyl chloride, methylsulfonic anhydrate, InCl.sub.3, InBr.sub.3, InI.sub.3, In(OTf).sub.3, Mg(SO.sub.4).sub.2, NaSO.sub.4; or combinations thereof.
47. The method of claim 44, wherein the Bronsted acid is TFA, benzenesulfonic acid, HCl, 4-toluenesulfonic acid, triflic acid, trifluoroacetic acid, 4-nitrobenzoic acid, methylsoulfonic acid, sulfuric acid, phosphoric acid, HBr, acetic acid, formic acid, HI, trifluoromethylsulfonic acid, 4-fluorobenzoic acid, pivalic acid, HBF.sub.4, nitric acid, 4-chloro-benzoic acid, pentafluorophenol, HPF.sub.6, Camphorsulfonic acid; or combinations thereof.
48. The method of claim 44, wherein the solvent is DCM, THF, MeTHF, Et.sub.2O, MeCN, EtCN, toluene, benzene, chlorobenzene, nitrobenzene, fluorobenzene, methanol, ethanol, 2-propanol, propanol, butanol, MTBE, EtOAc, iPrOAc, Me.sub.2O, (TMS).sub.2O, acetone, 2-butanone, chloroform, 1,2-dichloroethane, diglyme, dioxane, acetic acid, formic acid, trifluoroacetic acid, methylisobutylketone, DMAc, DMF, NMP, DMSO; or combinations thereof.
49. The method of claim 44, further comprising preparing the compound of Formula (V-a) or Formula (V-b) by a method comprising: forming the reaction mixture with TMSCl, PhMgCl, iPrMgCl, an additive, a compound of Formula (VI-a) having structure: ##STR00216## and a compound of Formula (VII): ##STR00217## to prepare the compound of Formula (V-a) or Formula (V-b), wherein the additive is BF.sub.3-OEt.sub.2, Sm(OTf).sub.3, Sc(OTf).sub.3, FeCl.sub.3, LiCl, LiBr, TiCl(OiPr).sub.3, ScCl.sub.3, Bu.sub.4NBr+LaCl.sub.3-2LiCl, nLaCl.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, LaCl.sub.3+2LiCl, Sm(OTf).sub.3+LiCl, SmCl.sub.3, Bis[2-(N,N-dimethylamino)ethyl] ether, TMEDA, NdCl.sub.3, NdCl.sub.3+CsCl, nNdCl.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, NdCl.sub.3+2LiCl, NdCl.sub.3+LiBr, NdCl.sub.3+LiI, NdBr.sub.3, NdBr.sub.3+CsCl, nNdBr.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, NdBr.sub.3+2LiCl, NdBr.sub.3+LiBr, NdBr.sub.3+LiI, Nd(OTf).sub.3, CeCl.sub.3, CeCl.sub.3+CsCl, nCeCl.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, CeCl.sub.3+2LiCl, CeCl.sub.3+LiBr, CeCl.sub.3+LiI, CeBr.sub.3, Ce(OTf).sub.3, YCl.sub.3, YCl.sub.3+CsCl, nYCl.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, YCl.sub.3+2LiCl, YCl.sub.3+LiBr, YCl.sub.3+LiI, YBr.sub.3, YBr.sub.3+CsCl, nYBr.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, YBr.sub.3+2LiCl, YBr.sub.3+LiBr, YBr.sub.3+LiI, Y(OTf).sub.3, LaCl.sub.3, La(OTf).sub.3, MgCl.sub.2, TiCl.sub.4, SnCl.sub.4, AlCl.sub.3, Bu.sub.4NCl, Diethyleneglycol diethylether (DGDE), DGDE+Bu.sub.4NCl, DGDE+Bu.sub.4NBr, DGDE+Bu.sub.4NI, CaCl.sub.2, CaBr.sub.2, CaI.sub.2, Ca(OTf).sub.2, YCl.sub.3, YCl.sub.3-2LiCl, YCl.sub.3--LiCl or a combination thereof.
50. The method of claim 49, wherein the additive is LaCl.sub.3-2LiCl, YCl.sub.3, CeCl.sub.3, NdCl.sub.3, or LaCl.sub.3.
51. A method of preparing a compound of Formula V-a or V-b: ##STR00218## the method comprising: forming a reaction mixture comprising a deprotonating agent, a silylating agent, a coupling agent, an additive, a compound of Formula VI-a: ##STR00219## and a compound of Formula VII: ##STR00220## to prepare the compound of Formula V-a or V-b, wherein each R.sup.b is independently benzyl (Bn) or tert-butyldimethylsilyl (TBS); alternatively, two R.sup.b groups on adjacent carbons can be combined to form a --C(Me).sub.2-- group; and R.sup.10 is H or a silyl group; wherein the additive is BF.sub.3-OEt.sub.2, Sm(OTf).sub.3, Sc(OTf).sub.3, FeCl.sub.3, LiCl, LiBr, TiCl(OiPr).sub.3, ScCl.sub.3, Bu.sub.4NBr+LaCl.sub.3-2LiCl, nLaCl.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, LaCl.sub.3+2LiCl, Sm(OTf).sub.3+LiCl, SmCl.sub.3, Bis[2-(N,N-dimethylamino)ethyl] ether, TMEDA, NdCl.sub.3, NdCl.sub.3+CsCl, nNdCl.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, NdCl.sub.3+2LiCl, NdCl.sub.3+LiBr, NdCl.sub.3+LiI, NdBr.sub.3, NdBr.sub.3+CsCl, nNdBr.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, NdBr.sub.3+2LiCl, NdBr.sub.3+LiBr, NdBr.sub.3+LiI, Nd(OTf).sub.3, CeCl.sub.3, CeCl.sub.3+CsCl, nCeCl.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, CeCl.sub.3+2LiCl, CeCl.sub.3+LiBr, CeCl.sub.3+LiI, CeBr.sub.3, Ce(OTf).sub.3, YCl.sub.3, YCl.sub.3+CsCl, nYCl.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, YCl.sub.3+2LiCl, YCl.sub.3+LiBr, YCl.sub.3+LiI, YBr.sub.3, YBr.sub.3+CsCl, nYBr.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, YBr.sub.3+2LiCl, YBr.sub.3+LiBr, YBr.sub.3+LiI, Y(OTf).sub.3, LaCl.sub.3, La(OTf).sub.3, MgCl.sub.2, TiCl.sub.4, SnCl.sub.4, AlCl.sub.3, Bu.sub.4NCl, Diethyleneglycol diethylether (DGDE), DGDE+Bu.sub.4NCl, DGDE+Bu.sub.4NBr, DGDE+Bu.sub.4NI, CaCl.sub.2, CaBr.sub.2, CaI.sub.2, Ca(OTf).sub.2, YCl.sub.3, YCl.sub.3-2LiCl, YCl.sub.3--LiCl or a combination thereof.
52. The method of claim 51, wherein the deprotonating agent is sodium hydride (NaH), isopropylmagnesium chloride (iPrMgCl), tert-butylmagnesium chloride (tBuMgCl), phenylmagnesium chloride (PhMgCl), phenylmagnesium bromide (PhMgBr), butyllithium (BuLi), methyllithium (MeLi), methylmagnesium chloride (MeMgCl), methylmagnesium bromide (MeMgBr), tert-butyllithium (tBuLi), isopropyllithium (iPrLi), phenyllithium (PhLi), lithium hydride (LiH), potassium hydride (KH), ethyllithium (EtLi), ethylmagnesium bromide (EtMgBr), ethylmagnesium chloride (EtMgCl), propyllithium (PrLi), propylmagnesium bromide (PrMgBr), propylmagnesium chloride (PrMgCl), cyclohexanelithium (cyHexLi), cyclohexanemagnesium bromide (cyHexMgBr), cyclohexanemagnesium chloride (cyHexMgCl), or combinations thereof.
53. The method of claim 51, wherein the coupling agent is n-butyllithium (nBuLi), magnesium chloride (MgCl.sub.2), isopropylmagnesium chloride (iPrMgCl), isopropylmagnesium chloride-lithium chloride (iPrMgCl--LiCl), tert-butylmagnesium chloride (tBuMgCl), phenylmagnesium chloride (PhMgCl), methyllithium (MeLi), methylmagnesium chloride (MeMgCl), methylmagnesium bromide (MeMgBr), tert-butyllithium (tBuLi), isopropyllithium (iPrLi), phenyllithium (PhLi), lithium hydride (LiH), potassium hydride (KH), sodium hydride (NaH), ethyllithium (EtLi), ethylmagnesium bromide (EtMgBr), ethylmagnesium chloride (EtMgCl), propyllithium (PrLi), propylmagnesium bromide (PrMgBr), propylmagnesium chloride (PrMgCl), cyclohexanelithium (cyHexLi), cyclohexanemagnesium bromide (cyHexMgBr), cyclohexanemagnesium chloride (cyHexMgCl), or combinations thereof.
54. The method of claim 51, wherein the silylating agent is a tri-substituted silyl chloride, a tri-substituted silyl bromide, a tri-substituted silyl iodide, or a tri-substituted silyl fluoride.
55. The method of claim 51, wherein the deprotonating agent is PhMgCl; and the coupling agent is iPrMgCl or iPrMgCl--LiCl.
56. The method of claim 51, wherein the deprotonating agent is PhMgCl; the silylating agent is TMSCl; the coupling agent is iPrMgCl; and R.sup.b is benzyl.
57. The method of claim 51, wherein the compound of Formula (V-a) or Formula (V-b) has the structure: ##STR00221## the method comprising: forming the reaction mixture comprising TMSCl, PhMgCl, iPrMgCl, the additive, the compound of Formula (VI-a) having structure: ##STR00222## and the compound of Formula (VII): ##STR00223## to prepare the compound of Formula (V-a) or Formula (V-b).
58. The method of claim 51, wherein the compound of Formula (V-a) has the structure: ##STR00224## the method comprising: forming the reaction mixture comprising TMSCl, PhMgCl, iPrMgCl--LiCl, the additive, the compound of Formula (VI-a) having structure: ##STR00225## and the compound of Formula (VII): ##STR00226## to prepare the compound of Formula (V-a).
59. The method of claim 51, wherein the additive is LaCl.sub.3-2LiCl, LaCl.sub.3, CeCl.sub.3, NdCl.sub.3, or YCl.sub.3.
60. A compound of formula: ##STR00227## or a pharmaceutically acceptable salt thereof.
Description
CROSS-REFERENCES TO RELATED APPLICATIONS
[0001] This patent application is a Continuation of U.S. patent application Ser. No. 14/926,063, filed Oct. 29, 2015, which claims the benefit under 35 U.S.C. .sctn. 119(e) of U.S. Provisional Patent Application No. 62/072,331, filed Oct. 29, 2014, and U.S. Provisional Patent Application No. 62/105,619, filed Jan. 20, 2015. The foregoing patent applications are incorporated herein by reference in their entireties.
FIELD OF THE INVENTION
[0002] The invention relates generally to methods and compounds for treating Filoviridae virus infections, particularly methods and nucleosides for treating Ebola virus, Marburg virus and Cueva virus.
BACKGROUND OF THE INVENTION
[0003] Filoviruses (e.g., Ebola virus (EBOV) and Marburg virus (MARV)) are among the most lethal and destructive viruses. They cause severe, often fatal viral hemorrhagic fevers in humans and nonhuman primates (e.g., monkeys, gorillas, and chimpanzees). Filoviruses are of particular concern as possible biological weapons since they have the potential for aerosol dissemination and weaponization.
[0004] The incubation period for Filovirus infection ranges from 2 to 21 days. The onset of illness is abrupt and is characterized by high fever, headaches, joint and muscle aches, sore throat, fatigue, diarrhea, vomiting, and stomach pain. A rash, red eyes, hiccups and internal and external bleeding may be seen in some patients. Within one week of becoming infected with the virus, most patients experience chest pains and multiple organ failure, go into shock, and die. Some patients also experience blindness and extensive bleeding before dying.
[0005] Filoviridae are a family of RNA viruses. Two members of the Filoviridae family have been identified: EBOV and MARV. Two key pathogenic types of the Filoviridae family have been identified: Ebolavirus and MARV. There is one identified variant of MARV and five identified species of ebolavirus: Zaire (i.e. Ebola virus, EBOV), Sudan, Tai Forest, Bundibugyo, and Reston. The exact origin, locations, and natural habitat of Filoviridae are unknown. However, on the basis of available evidence and the nature of similar viruses, it is postulated that Filoviridae are zoonotic (i.e., animal-borne) and are normally maintained in an animal host that is native to the African continent.
[0006] For more than 30 years, ebolaviruses have been associated with periodic episodes of hemorrhagic fever in Central Africa that produce severe disease in infected patients. Mortality rates in outbreaks have ranged from 50% for the Sudan species of ebolavirus (SEBOV) to up to 90% for the Zaire species of ebolavirus (EBOV, ZEBOV) (Sanchez et al., Filoviridae: Marburg and Ebola Viruses, in Fields Virology (eds. Knipe, D. M. & Howley, P. M.) 1409-1448 (Lippincott Williams & Wilkins, Philadelphia)). An outbreak late in 2007 caused by an apparently new species of ebolavirus in Uganda resulted in a fatality rate of about 25% (Towner et al., PLoS Pathog., 4:e1000212 (2008)). ZEBOV has also decimated populations of wild apes in this same region of Africa (Walsh et al., Nature, 422:611-614 (2003)).
[0007] Prevention and treatment of Filovirus infections, including ebolaviruses (i.e. EBOV) presents many challenges. In fact, there are no vaccines or post exposure treatment modalities available for preventing or managing EBOV infections. Patients instead receive supportive therapy, i.e., electrolyte and fluid balancing, oxygen, blood pressure maintenance, and treatment for any secondary infections.
[0008] In view of the importance of novel therapeutics for treating Filoviridae infections, new efficient methods of producing ribosides, riboside phosphates and prodrugs are needed.
SUMMARY OF THE INVENTION
[0009] In some embodiments, the present invention provides a method of preparing a compound of Formula V:
##STR00001##
The method of making the compound of Formula V includes forming a reaction mixture having a coupling agent, a halo-silane, a compound of Formula VI:
##STR00002##
and a compound of Formula VII:
##STR00003##
under conditions suitable to prepare the compound of Formula V, wherein each PG is independently a hydroxy protecting group, alternatively, two PG groups on adjacent carbons can be combined to form a --C(R.sup.19).sub.2-- group, R.sup.10 is H or a silyl group, and R.sup.19 is H, C.sub.1-C.sub.8 alkyl, phenyl or substituted phenyl.
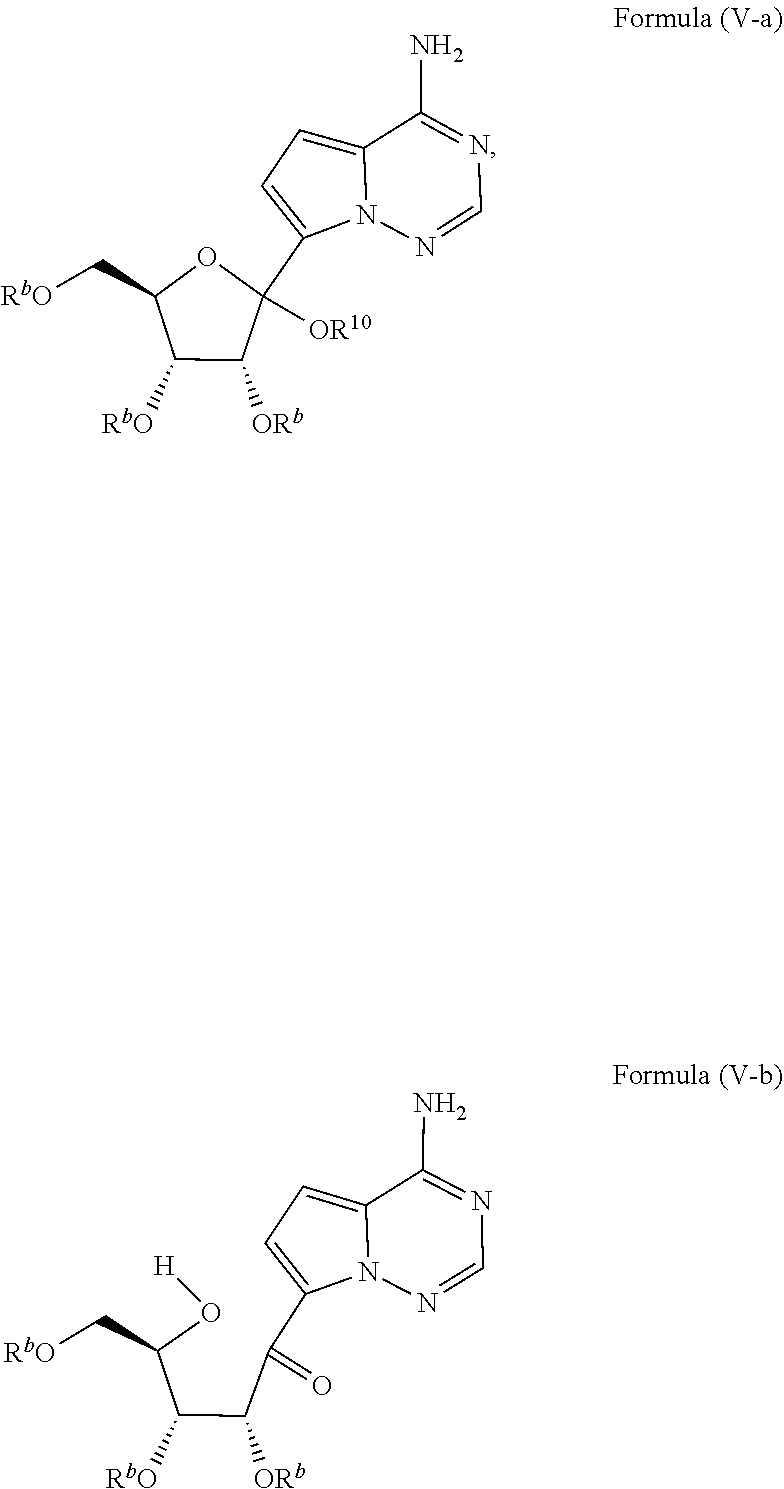
[0010] In some embodiments, the present invention provides a method of preparing a compound of Formula V-a or V-b:
##STR00004##
The method of making the compound of Formula V-a or Formula V-b comprises forming a reaction mixture having a deprotonating agent, a silylating agent, a coupling agent, an additive, a compound of Formula VI-a:
##STR00005##
and a compound of Formula VII:
##STR00006##
under conditions suitable to prepare the compound of Formula V-a or Formula V-b, wherein each R.sup.b is independently a hydroxy protecting group, alternatively, two R.sup.b groups on adjacent carbons can be combined to form a --C(R.sup.19).sub.2-- group, R.sup.10 is H or a silyl group, and R.sup.19 is H, C.sub.1-C.sub.8 alkyl, phenyl or substituted phenyl.
[0011] In some embodiments, the present invention provides a method of preparing a compound of Formula XI:
##STR00007##
wherein R.sup.c is H or a hydroxy protecting group, or two R.sup.c on adjacent carbons can be combined to form a --C(R.sup.19).sub.2-- group, and R.sup.19 is H or C.sub.1-C.sub.8 alkyl.
[0012] In some embodiments, the present invention provides a method of preparing a compound of Formula XI-a:
##STR00008##
wherein the method comprises forming a reaction mixture having a cyanating agent, a Lewis Acid, a Broenstedt acid, a solvent, and the compound of Formula V or V-b:
##STR00009##
under conditions suitable to prepare the compound of Formula XI, wherein R.sup.b is independently a hydroxy protecting group, alternatively, two R.sup.b groups on adjacent carbons can be combined to form a --C(R.sup.19).sub.2 group, R.sup.19 is H or a silyl group, and R.sup.19 is H, C.sub.1-C.sub.8 alkyl, phenyl or substituted phenyl.
[0013] In some embodiments, the present invention provides a method of preparing a compound of Formula XI-b:
##STR00010##
wherein the method comprises forming a reaction mixture having a Lewis Acid, a base, a solvent, a filtering agent, and the compound of Formula XI-a
##STR00011##
under conditions suitable to prepare the compound of Formula XI-b.
[0014] In some embodiments, the present invention provides a method of preparing a compound of Formula XI-c:
##STR00012##
wherein the method comprises forming a reaction mixture having a solvent, a reagent, and the compound of Formula XI-b
##STR00013##
under conditions suitable to prepare the compound of Formula XI-c.
[0015] In some embodiments, the present invention provides a method of preparing a compound of Formula VIII:
##STR00014##
wherein the method includes forming a reaction mixture including a coupling agent, a non-nucleophilic base, a compound of Formula IX:
##STR00015##
and a compound of Formula X:
##STR00016##
under conditions suitable to form the compound of Formula VIII, wherein each R.sup.a is H or PG, each PG group is a hydroxy protecting group, or both PG groups are combined to form --C(R.sup.19).sub.2--, R.sup.e1 and R.sup.e2 are each independently H, C.sub.1-C.sub.6 alkyl or benzyl, R.sup.f is H, C.sub.1-C.sub.8 alkyl, benzyl, C.sub.3-C.sub.6 cycloalkyl, or --CH.sub.2--C.sub.3-C.sub.6 cycloalkyl, R.sup.19 is H, C.sub.1-C.sub.8 alkyl, phenyl or substituted phenyl, and LG is a leaving group.
[0016] In some embodiments, the present invention provides a method of preparing a compound of Formula VIII:
##STR00017##
wherein the method comprises forming a reaction mixture including a coupling agent, a non-nucleophilic base, a compound of Formula IX-a:
##STR00018##
and a compound of Formula X:
##STR00019##
under conditions suitable to form the compound of Formula VIII, wherein R.sup.a is independently H or a hydroxy protecting group, or two R.sup.a on adjacent carbons can be combined to form a --C(R.sup.19).sub.2-- group, R.sup.35 is independently H or a hydroxy protecting group, or two R.sup.35 on adjacent carbons can be combined to form a --C(R.sup.19).sub.2-- group, R.sup.19 is H or C.sub.1-C.sub.8 alkyl, R.sup.e1 and R.sup.e2 are each independently H, C.sub.1-C.sub.6 alkyl or benzyl, R.sup.f is H, C.sub.1-C.sub.8 alkyl, benzyl, C.sub.3-C.sub.6 cycloalkyl, or --CH.sub.2--C.sub.3-C.sub.6 cycloalkyl, R.sup.19 is H, C.sub.1-C.sub.8 alkyl, phenyl or substituted phenyl, and LG is a leaving group.
[0017] In one embodiment, there is provided a method for the crystallization-induced dynamic resolution of (2S)-2-ethylbutyl 2-(((4-nitrophenoxy)(phenoxy)phosphoryl)amino)propanoate (Formula X-a):
##STR00020##
to provide (Formula X-b).
DETAILED DESCRIPTION OF THE INVENTION
I. Definitions
[0018] Unless stated otherwise, the following terms and phrases as used herein are intended to have the following meanings:
[0019] When trade names are used herein, applicants intend to independently include the trade name product and the active pharmaceutical ingredient(s) of the trade name product.
[0020] As used herein, "a compound of the invention" or "a compound of Formula V" means a compound of Formula V or a pharmaceutically acceptable salt or cocrystal, thereof. In some embodiments, "a compound of the invention" or "a compound of Formula V" means a compound of Formula V or a pharmaceutically acceptable salt, thereof. Similarly, with respect to isolatable intermediates, the phrase "a compound of Formula (number)" means a compound of that formula and pharmaceutically acceptable salts or cocrystals, thereof. In some embodiments, with respect to isolatable intermediates, the phrase "a compound of Formula (number)" means a compound of that formula and pharmaceutically acceptable salts, thereof.
[0021] "Alkyl" is hydrocarbon containing normal, secondary, tertiary or cyclic carbon atoms. For example, an alkyl group can have 1 to 20 carbon atoms (i.e, C.sub.1-C.sub.20 alkyl), 1 to 8 carbon atoms (i.e., C.sub.1-C.sub.8 alkyl), or 1 to 6 carbon atoms (i.e., C.sub.1-C.sub.6 alkyl). Examples of suitable alkyl groups include, but are not limited to, methyl (Me, --CH.sub.3), ethyl (Et, --CH.sub.2CH.sub.3), 1-propyl (n-Pr, n-propyl, --CH.sub.2CH.sub.2CH.sub.3), 2-propyl (i-Pr, i-propyl, --CH(CH.sub.3).sub.2), 1-butyl (n-Bu, n-butyl, --CH.sub.2CH.sub.2CH.sub.2CH.sub.3), 2-methyl-1-propyl (i-Bu, i-butyl, --CH.sub.2CH(CH.sub.3).sub.2), 2-butyl (s-Bu, s-butyl, --CH(CH.sub.3)CH.sub.2CH.sub.3), 2-methyl-2-propyl (t-Bu, --C(CH.sub.3).sub.3), 1-pentyl (n-pentyl, --CH.sub.2CH.sub.2CH.sub.2CH.sub.2CH.sub.3), 2-pentyl (--CH(CH.sub.3)CH.sub.2CH.sub.2CH.sub.3), 3-pentyl (--CH(CH.sub.2CH.sub.3).sub.2), 2-methyl-2-butyl (--C(CH.sub.3).sub.2CH.sub.2CH.sub.3), 3-methyl-2-butyl (--CH(CH.sub.3)CH(CH.sub.3).sub.2), 3-methyl-1-butyl (--CH.sub.2CH.sub.2CH(CH.sub.3).sub.2), 2-methyl-1-butyl (--CH.sub.2CH(CH.sub.3)CH.sub.2CH.sub.3), 1-hexyl (--CH.sub.2CH.sub.2CH.sub.2CH.sub.2CH.sub.2CH.sub.3), 2-hexyl (--CH(CH.sub.3)CH.sub.2CH.sub.2CH.sub.2CH.sub.3), 3-hexyl (--CH(CH.sub.2CH.sub.3)(CH.sub.2CH.sub.2CH.sub.3)), 2-methyl-2-pentyl (--C(CH.sub.3).sub.2CH.sub.2CH.sub.2CH.sub.3), 3-methyl-2-pentyl (--CH(CH.sub.3)CH(CH.sub.3)CH.sub.2CH.sub.3), 4-methyl-2-pentyl (--CH(CH.sub.3)CH.sub.2CH(CH.sub.3).sub.2), 3-methyl-3-pentyl (--C(CH.sub.3)(CH.sub.2CH.sub.3).sub.2), 2-methyl-3-pentyl (--CH(CH.sub.2CH.sub.3)CH(CH.sub.3).sub.2), 2,3-dimethyl-2-butyl (--C(CH.sub.3).sub.2CH(CH.sub.3).sub.2), 3,3-dimethyl-2-butyl (--CH(CH.sub.3)C(CH.sub.3).sub.3, and octyl (--(CH.sub.2).sub.7CH.sub.3).
[0022] "Alkoxy" means a group having the formula --O-alkyl, in which an alkyl group, as defined above, is attached to the parent molecule via an oxygen atom. The alkyl portion of an alkoxy group can have 1 to 20 carbon atoms (i.e., C.sub.1-C.sub.20 alkoxy), 1 to 12 carbon atoms (i.e., C.sub.1-C.sub.12 alkoxy), or 1 to 6 carbon atoms (i.e., C.sub.1-C.sub.6 alkoxy). Examples of suitable alkoxy groups include, but are not limited to, methoxy (--O--CH.sub.3 or --OMe), ethoxy (--OCH.sub.2CH.sub.3 or --OEt), t-butoxy (--O--C(CH.sub.3).sub.3 or --OtBu) and the like.
[0023] "Haloalkyl" is an alkyl group, as defined above, in which one or more hydrogen atoms of the alkyl group is replaced with a halogen atom. The alkyl portion of a haloalkyl group can have 1 to 20 carbon atoms (i.e., C.sub.1-C.sub.20 haloalkyl), 1 to 12 carbon atoms (i.e., C.sub.1-C.sub.12 haloalkyl), or 1 to 6 carbon atoms (i.e., C.sub.1-C.sub.6 alkyl). Examples of suitable haloalkyl groups include, but are not limited to, --CF.sub.3, --CHF.sub.2, --CFH.sub.2, --CH.sub.2CF.sub.3, and the like.
[0024] "Alkenyl" is a hydrocarbon containing normal, secondary, tertiary or cyclic carbon atoms with at least one site of unsaturation, i.e. a carbon-carbon, sp.sup.2 double bond. For example, an alkenyl group can have 2 to 20 carbon atoms (i.e., C.sub.2-C.sub.20 alkenyl), 2 to 8 carbon atoms (i.e., C.sub.2-C.sub.8 alkenyl), or 2 to 6 carbon atoms (i.e., C.sub.2-C.sub.6 alkenyl). Examples of suitable alkenyl groups include, but are not limited to, ethylene or vinyl (--CH.dbd.CH.sub.2), allyl (--CH.sub.2CH.dbd.CH.sub.2), cyclopentenyl (--C.sub.5H.sub.7), and 5-hexenyl (--CH.sub.2CH.sub.2CH.sub.2CH.sub.2CH.dbd.CH.sub.2).
[0025] "Alkynyl" is a hydrocarbon containing normal, secondary, tertiary or cyclic carbon atoms with at least one site of unsaturation, i.e. a carbon-carbon, sp triple bond. For example, an alkynyl group can have 2 to 20 carbon atoms (i.e., C.sub.2-C.sub.20 alkynyl), 2 to 8 carbon atoms (i.e., C.sub.2-C.sub.8 alkyne,), or 2 to 6 carbon atoms (i.e., C.sub.2-C.sub.6 alkynyl). Examples of suitable alkynyl groups include, but are not limited to, acetylenic (--C.ident.CH), propargyl (--CH.sub.2C.ident.CH), and the like.
[0026] "Alkylene" refers to a saturated, branched or straight chain or cyclic hydrocarbon radical having two monovalent radical centers derived by the removal of two hydrogen atoms from the same or two different carbon atoms of a parent alkane. For example, an alkylene group can have 1 to 20 carbon atoms, 1 to 10 carbon atoms, or 1 to 6 carbon atoms. Typical alkylene radicals include, but are not limited to, methylene (--CH.sub.2--), 1,1-ethyl (--CH(CH.sub.3)--), 1,2-ethyl (--CH.sub.2CH.sub.2--), 1,1-propyl (--CH(CH.sub.2CH.sub.3)--), 1,2-propyl (--CH.sub.2CH(CH.sub.3)--), 1,3-propyl (--CH.sub.2CH.sub.2CH.sub.2--), 1,4-butyl (--CH.sub.2CH.sub.2CH.sub.2CH.sub.2--), and the like.
[0027] "Alkenylene" refers to an unsaturated, branched or straight chain or cyclic hydrocarbon radical having two monovalent radical centers derived by the removal of two hydrogen atoms from the same or two different carbon atoms of a parent alkene. For example, and alkenylene group can have 1 to 20 carbon atoms, 1 to 10 carbon atoms, or 1 to 6 carbon atoms. Typical alkenylene radicals include, but are not limited to, 1,2-ethylene (--CH.dbd.CH--).
[0028] "Alkynylene" refers to an unsaturated, branched or straight chain or cyclic hydrocarbon radical having two monovalent radical centers derived by the removal of two hydrogen atoms from the same or two different carbon atoms of a parent alkyne. For example, an alkynylene group can have 1 to 20 carbon atoms, 1 to 10 carbon atoms, or 1 to 6 carbon atoms. Typical alkynylene radicals include, but are not limited to, acetylene (--C.ident.C--), propargyl (--CH.sub.2C.ident.C--), and 4-pentynyl (--CH.sub.2CH.sub.2CH.sub.2C.ident.C--).
[0029] "Amino" refers generally to a nitrogen radical which can be considered a derivative of ammonia, having the formula --N(X).sub.2, where each "X" is independently H, substituted or unsubstituted alkyl, substituted or unsubstituted carbocyclyl, substituted or unsubstituted heterocyclyl, etc. The hybridization of the nitrogen is approximately sp.sup.3. Nonlimiting types of amino include --NH.sub.2, --N(alkyl).sub.2, --NH(alkyl), --N(carbocyclyl).sub.2, --NH(carbocyclyl), --N(heterocyclyl).sub.2, --NH(heterocyclyl), --N(aryl).sub.2, --NH(aryl), --N(alkyl)(aryl), --N(alkyl)(heterocyclyl), --N(carbocyclyl)(heterocyclyl), --N(aryl)(heteroaryl), --N(alkyl)(heteroaryl), etc. The term "alkylamino" refers to an amino group substituted with at least one alkyl group. Nonlimiting examples of amino groups include --NH.sub.2, --NH(CH.sub.3), --N(CH.sub.3).sub.2, --NH(CH.sub.2CH.sub.3), --N(CH.sub.2CH.sub.3).sub.2, --NH(phenyl), --N(phenyl).sub.2, --NH(benzyl), --N(benzyl).sub.2, etc. Substituted alkylamino refers generally to alkylamino groups, as defined above, in which at least one substituted alkyl, as defined herein, is attached to the amino nitrogen atom. Non-limiting examples of substituted alkylamino includes --NH(alkylene-C(O)--OH), --NH(alkylene-C(O)--O-alkyl), --N(alkylene-C(O)--OH).sub.2, --N(alkylene-C(O)--O-alkyl).sub.2, etc.
[0030] "Aryl" means an aromatic hydrocarbon radical derived by the removal of one hydrogen atom from a single carbon atom of a parent aromatic ring system. For example, an aryl group can have 6 to 20 carbon atoms, 6 to 14 carbon atoms, or 6 to 10 carbon atoms. Typical aryl groups include, but are not limited to, radicals derived from benzene (e.g., phenyl), substituted benzene, naphthalene, anthracene, biphenyl, and the like. Further typical aryl groups include, but are not limited to, phenyl.
[0031] "Arylalkyl" refers to an acyclic alkyl radical in which one of the hydrogen atoms bonded to a carbon atom, typically a terminal or sp.sup.3 carbon atom, is replaced with an aryl radical. Typical arylalkyl groups include, but are not limited to, benzyl, 2-phenylethan-1-yl, naphthylmethyl, 2-naphthylethan-1-yl, naphthobenzyl, 2-naphthophenylethan-1-yl and the like. The arylalkyl group can comprise 7 to 20 carbon atoms, e.g., the alkyl moiety is 1 to 6 carbon atoms and the aryl moiety is 6 to 14 carbon atoms.
[0032] "Arylalkenyl" refers to an acyclic alkenyl radical in which one of the hydrogen atoms bonded to a carbon atom, typically a terminal or sp.sup.3 carbon atom, but also an sp.sup.2 carbon atom, is replaced with an aryl radical. The aryl portion of the arylalkenyl can include, for example, any of the aryl groups disclosed herein, and the alkenyl portion of the arylalkenyl can include, for example, any of the alkenyl groups disclosed herein. The arylalkenyl group can comprise 8 to 20 carbon atoms, e.g., the alkenyl moiety is 2 to 6 carbon atoms and the aryl moiety is 6 to 14 carbon atoms.
[0033] "Arylalkynyl" refers to an acyclic alkynyl radical in which one of the hydrogen atoms bonded to a carbon atom, typically a terminal or sp.sup.3 carbon atom, but also an sp carbon atom, is replaced with an aryl radical. The aryl portion of the arylalkynyl can include, for example, any of the aryl groups disclosed herein, and the alkynyl portion of the arylalkynyl can include, for example, any of the alkynyl groups disclosed herein. The arylalkynyl group can comprise 8 to 20 carbon atoms, e.g., the alkynyl moiety is 2 to 6 carbon atoms and the aryl moiety is 6 to 14 carbon atoms.
[0034] The term "substituted" in reference to alkyl, alkylene, aryl, arylalkyl, alkoxy, heterocyclyl, heteroaryl, carbocyclyl, etc., for example, "substituted alkyl", "substituted alkylene", "substituted aryl", "substituted arylalkyl", "substituted heterocyclyl", and "substituted carbocyclyl" means alkyl, alkylene, aryl, arylalkyl, heterocyclyl, carbocyclyl respectively, in which one or more hydrogen atoms are each independently replaced with a non-hydrogen substituent. The term "substituted phenyl" means phenyl, in which one or more hydrogen atoms are each independently replaced with a non-hydrogen substituent. Typical substituents include, but are not limited to, --X, --R.sup.b, --O.sup.-, .dbd.O, --OR.sup.b, --SR.sup.b, --S.sup.-, --NR.sup.b.sub.2, --N.sup.+R.sup.b.sub.3, =NR.sup.b, --CX.sub.3, --CN, --OCN, --SCN, --N.dbd.C.dbd.O, --NCS, --NO, --NO.sub.2, .dbd.N.sub.2, --N.sub.3, --NHC(.dbd.O)R.sup.b, --OC(.dbd.O)R.sup.b, --NHC(.dbd.O)NR.sup.b.sub.2, --S(.dbd.O).sub.2--, --S(.dbd.O).sub.2OH, --S(.dbd.O).sub.2R.sup.b, --OS(.dbd.O).sub.2OR.sup.b, --S(.dbd.O).sub.2NR.sup.b.sub.2, --S(.dbd.O)R.sup.b, --OP(.dbd.O)(OR.sup.b).sub.2, --P(.dbd.O)(OR.sup.b).sub.2, --P(.dbd.O)(O.sup.-).sub.2, --P(.dbd.O)(OH).sub.2, --P(O)(OR.sup.b)(O), --C(.dbd.O)R.sup.b, --C(.dbd.O)X, --C(S)R.sup.b, --C(O)OR.sup.b, --C(O)O.sup.-, --C(S)OR.sup.b, --C(O)SR.sup.b, --C(S)SR.sup.b, --C(O)NR.sup.b.sub.2, --C(S)NR.sup.b.sub.2, --C(.dbd.NR.sup.b)NR.sup.b.sub.2, where each X is independently a halogen: F, Cl, Br, or I; and each R.sup.b is independently H, alkyl, aryl, arylalkyl, a heterocycle, or a protecting group or prodrug moiety. Alkylene, alkenylene, and alkynylene groups may also be similarly substituted. Unless otherwise indicated, when the term "substituted" is used in conjunction with groups such as arylalkyl, which have two or more moieties capable of substitution, the substituents can be attached to the aryl moiety, the alkyl moiety, or both.
[0035] The term "prodrug" as used herein refers to any compound that when administered to a biological system generates the drug substance, i.e., active ingredient, as a result of spontaneous chemical reaction(s), enzyme catalyzed chemical reaction(s), photolysis, and/or metabolic chemical reaction(s). A prodrug is thus a covalently modified analog or latent form of a therapeutically active compound.
[0036] One skilled in the art will recognize that substituents and other moieties of the compounds of Formula I-IV should be selected in order to provide a compound which is sufficiently stable to provide a pharmaceutically useful compound which can be formulated into an acceptably stable pharmaceutical composition. Compounds of Formula I-IV which have such stability are contemplated as falling within the scope of the present invention.
[0037] "Heteroalkyl" refers to an alkyl group where one or more carbon atoms have been replaced with a heteroatom, such as, O, N, or S. For example, if the carbon atom of the alkyl group which is attached to the parent molecule is replaced with a heteroatom (e.g., O, N, or S) the resulting heteroalkyl groups are, respectively, an alkoxy group (e.g., --OCH.sub.3, etc.), an amine (e.g., --NHCH.sub.3, --N(CH.sub.3).sub.2, etc.), or a thioalkyl group (e.g., --SCH.sub.3). If a non-terminal carbon atom of the alkyl group which is not attached to the parent molecule is replaced with a heteroatom (e.g., O, N, or S) the resulting heteroalkyl groups are, respectively, an alkyl ether (e.g., --CH.sub.2CH.sub.2--O--CH.sub.3, etc.), an alkyl amine (e.g., --CH.sub.2NHCH.sub.3, --CH.sub.2N(CH.sub.3).sub.2, etc.), or a thioalkyl ether (e.g., --CH.sub.2--S--CH.sub.3). If a terminal carbon atom of the alkyl group is replaced with a heteroatom (e.g., O, N, or S), the resulting heteroalkyl groups are, respectively, a hydroxyalkyl group (e.g., --CH.sub.2CH.sub.2--OH), an aminoalkyl group (e.g., --CH.sub.2NH.sub.2), or an alkyl thiol group (e.g., --CH.sub.2CH.sub.2--SH). A heteroalkyl group can have, for example, 1 to 20 carbon atoms, 1 to 10 carbon atoms, or 1 to 6 carbon atoms. A C.sub.1-C.sub.6 heteroalkyl group means a heteroalkyl group having 1 to 6 carbon atoms.
[0038] "Heterocycle" or "heterocyclyl" as used herein includes by way of example and not limitation those heterocycles described in Paquette, Leo A.; Principles of Modern Heterocyclic Chemistry (W. A. Benjamin, New York, 1968), particularly Chapters 1, 3, 4, 6, 7, and 9; The Chemistry of Heterocyclic Compounds, A Series of Monographs" (John Wiley & Sons, New York, 1950 to present), in particular Volumes 13, 14, 16, 19, and 28; and J. Am. Chem. Soc. (1960) 82:5566. In one specific embodiment of the invention "heterocycle" includes a "carbocycle" as defined herein, wherein one or more (e.g. 1, 2, 3, or 4) carbon atoms have been replaced with a heteroatom (e.g. O, N, or S). The terms "heterocycle" or "heterocyclyl" includes saturated rings, partially unsaturated rings, and aromatic rings (i.e., heteroaromatic rings). Substituted heterocyclyls include, for example, heterocyclic rings substituted with any of the substituents disclosed herein including carbonyl groups. A non-limiting example of a carbonyl substituted heterocyclyl is:
##STR00021##
[0039] Examples of heterocycles include by way of example and not limitation pyridyl, dihydroypyridyl, tetrahydropyridyl (piperidyl), thiazolyl, tetrahydrothiophenyl, sulfur oxidized tetrahydrothiophenyl, pyrimidinyl, furanyl, thienyl, pyrrolyl, pyrazolyl, imidazolyl, tetrazolyl, benzofuranyl, thianaphthalenyl, indolyl, indolenyl, quinolinyl, isoquinolinyl, benzimidazolyl, piperidinyl, 4-piperidonyl, pyrrolidinyl, 2-pyrrolidonyl, pyrrolinyl, tetrahydrofuranyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl, decahydroquinolinyl, octahydroisoquinolinyl, azocinyl, triazinyl, 6H-1,2,5-thiadiazinyl, 2H,6H-1,5,2-dithiazinyl, thienyl, thianthrenyl, pyranyl, isobenzofuranyl, chromenyl, xanthenyl, phenoxathinyl, 2H-pyrrolyl, isothiazolyl, isoxazolyl, pyrazinyl, pyridazinyl, indolizinyl, isoindolyl, 3H-indolyl, 1H-indazoly, purinyl, 4H-quinolizinyl, phthalazinyl, naphthyridinyl, quinoxalinyl, quinazolinyl, cinnolinyl, pteridinyl, 4aH-carbazolyl, carbazolyl, .beta.-carbolinyl, phenanthridinyl, acridinyl, pyrimidinyl, phenanthrolinyl, phenazinyl, phenothiazinyl, furazanyl, phenoxazinyl, isochromanyl, chromanyl, imidazolidinyl, imidazolinyl, pyrazolidinyl, pyrazolinyl, piperazinyl, indolinyl, isoindolinyl, quinuclidinyl, morpholinyl, oxazolidinyl, benzotriazolyl, benzisoxazolyl, oxindolyl, benzoxazolinyl, isatinoyl, and bis-tetrahydrofuranyl:
##STR00022##
[0040] By way of example and not limitation, carbon bonded heterocycles are bonded at position 2, 3, 4, 5, or 6 of a pyridine, position 3, 4, 5, or 6 of a pyridazine, position 2, 4, 5, or 6 of a pyrimidine, position 2, 3, 5, or 6 of a pyrazine, position 2, 3, 4, or 5 of a furan, tetrahydrofuran, thiofuran, thiophene, pyrrole or tetrahydropyrrole, position 2, 4, or 5 of an oxazole, imidazole or thiazole, position 3, 4, or 5 of an isoxazole, pyrazole, or isothiazole, position 2 or 3 of an aziridine, position 2, 3, or 4 of an azetidine, position 2, 3, 4, 5, 6, 7, or 8 of a quinoline or position 1, 3, 4, 5, 6, 7, or 8 of an isoquinoline. Still more typically, carbon bonded heterocycles include 2-pyridyl, 3-pyridyl, 4-pyridyl, 5-pyridyl, 6-pyridyl, 3-pyridazinyl, 4-pyridazinyl, 5-pyridazinyl, 6-pyridazinyl, 2-pyrimidinyl, 4-pyrimidinyl, 5-pyrimidinyl, 6-pyrimidinyl, 2-pyrazinyl, 3-pyrazinyl, 5-pyrazinyl, 6-pyrazinyl, 2-thiazolyl, 4-thiazolyl, or 5-thiazolyl.
[0041] By way of example and not limitation, nitrogen bonded heterocycles are bonded at position 1 of an aziridine, azetidine, pyrrole, pyrrolidine, 2-pyrroline, 3-pyrroline, imidazole, imidazolidine, 2-imidazoline, 3-imidazoline, pyrazole, pyrazoline, 2-pyrazoline, 3-pyrazoline, piperidine, piperazine, indole, indoline, 1H-indazole, position 2 of a isoindole, or isoindoline, position 4 of a morpholine, and position 9 of a carbazole, or .beta.-carboline. Still more typically, nitrogen bonded heterocycles include 1-aziridyl, 1-azetedyl, 1-pyrrolyl, 1-imidazolyl, 1-pyrazolyl, and 1-piperidinyl.
[0042] "Heterocyclylalkyl" refers to an acyclic alkyl radical in which one of the hydrogen atoms bonded to a carbon atom, typically a terminal or sp.sup.3 carbon atom, is replaced with a heterocyclyl radical (i.e., a heterocyclyl-alkylene- moiety). Typical heterocyclyl alkyl groups include, but are not limited to heterocyclyl-CH.sub.2--, 2-(heterocyclyl)ethan-1-yl, and the like, wherein the "heterocyclyl" portion includes any of the heterocyclyl groups described above, including those described in Principles of Modern Heterocyclic Chemistry. One skilled in the art will also understand that the heterocyclyl group can be attached to the alkyl portion of the heterocyclyl alkyl by means of a carbon-carbon bond or a carbon-heteroatom bond, with the proviso that the resulting group is chemically stable. The heterocyclyl alkyl group comprises 3 to 20 carbon atoms, e.g., the alkyl portion of the arylalkyl group is 1 to 6 carbon atoms and the heterocyclyl moiety is 2 to 14 carbon atoms. Examples of heterocyclylalkyls include by way of example and not limitation 5-membered sulfur, oxygen, and/or nitrogen containing heterocycles such as thiazolylmethyl, 2-thiazolylethan-1-yl, imidazolylmethyl, oxazolylmethyl, thiadiazolylmethyl, etc., 6-membered sulfur, oxygen, and/or nitrogen containing heterocycles such as piperidinylmethyl, piperazinylmethyl, morpholinylmethyl, pyridinylmethyl, pyridizylmethyl, pyrimidylmethyl, pyrazinylmethyl, etc.
[0043] "Heterocyclylalkenyl" refers to an acyclic alkenyl radical in which one of the hydrogen atoms bonded to a carbon atom, typically a terminal or sp.sup.3 carbon atom, but also a sp.sup.2 carbon atom, is replaced with a heterocyclyl radical (i.e., a heterocyclyl-alkenylene- moiety). The heterocyclyl portion of the heterocyclyl alkenyl group includes any of the heterocyclyl groups described herein, including those described in Principles of Modern Heterocyclic Chemistry, and the alkenyl portion of the heterocyclyl alkenyl group includes any of the alkenyl groups disclosed herein. One skilled in the art will also understand that the heterocyclyl group can be attached to the alkenyl portion of the heterocyclyl alkenyl by means of a carbon-carbon bond or a carbon-heteroatom bond, with the proviso that the resulting group is chemically stable. The heterocyclyl alkenyl group comprises 4 to 20 carbon atoms, e.g., the alkenyl portion of the heterocyclyl alkenyl group is 2 to 6 carbon atoms and the heterocyclyl moiety is 2 to 14 carbon atoms.
[0044] "Heterocyclylalkynyl" refers to an acyclic alkynyl radical in which one of the hydrogen atoms bonded to a carbon atom, typically a terminal or sp.sup.3 carbon atom, but also an sp carbon atom, is replaced with a heterocyclyl radical (i.e., a heterocyclyl-alkynylene- moiety). The heterocyclyl portion of the heterocyclyl alkynyl group includes any of the heterocyclyl groups described herein, including those described in Principles of Modern Heterocyclic Chemistry, and the alkynyl portion of the heterocyclyl alkynyl group includes any of the alkynyl groups disclosed herein. One skilled in the art will also understand that the heterocyclyl group can be attached to the alkynyl portion of the heterocyclyl alkynyl by means of a carbon-carbon bond or a carbon-heteroatom bond, with the proviso that the resulting group is chemically stable. The heterocyclyl alkynyl group comprises 4 to 20 carbon atoms, e.g., the alkynyl portion of the heterocyclyl alkynyl group is 2 to 6 carbon atoms and the heterocyclyl moiety is 2 to 14 carbon atoms.
[0045] "Heteroaryl" refers to an aromatic heterocyclyl having at least one heteroatom in the ring. Non-limiting examples of suitable heteroatoms which can be included in the aromatic ring include oxygen, sulfur, and nitrogen. Non-limiting examples of heteroaryl rings include all of those aromatic rings listed in the definition of "heterocyclyl", including pyridinyl, pyrrolyl, oxazolyl, indolyl, isoindolyl, purinyl, furanyl, thienyl, benzofuranyl, benzothiophenyl, carbazolyl, imidazolyl, thiazolyl, isoxazolyl, pyrazolyl, isothiazolyl, quinolyl, isoquinolyl, pyridazyl, pyrimidyl, pyrazyl, etc.
[0046] "Carbocycle" or "carbocyclyl" refers to a saturated (i.e., cycloalkyl), partially unsaturated (e.g., cycloakenyl, cycloalkadienyl, etc.) or aromatic ring having 3 to 7 carbon atoms as a monocycle, 7 to 12 carbon atoms as a bicycle, and up to about 20 carbon atoms as a polycycle. Monocyclic carbocycles have 3 to 7 ring atoms, still more typically 5 or 6 ring atoms. Bicyclic carbocycles have 7 to 12 ring atoms, e.g., arranged as a bicyclo [4,5], [5,5], [5,6] or [6,6] system, or 9 or 10 ring atoms arranged as a bicyclo [5,6] or [6,6] system, or spiro-fused rings. Non-limiting examples of monocyclic carbocycles include cyclopropyl, cyclobutyl, cyclopentyl, 1-cyclopent-1-enyl, 1-cyclopent-2-enyl, 1-cyclopent-3-enyl, cyclohexyl, 1-cyclohex-1-enyl, 1-cyclohex-2-enyl, 1-cyclohex-3-enyl, and phenyl. Non-limiting examples of bicyclo carbocycles includes naphthyl, tetrahydronapthalene, and decaline.
[0047] "Carbocyclylalkyl" refers to an acyclic akyl radical in which one of the hydrogen atoms bonded to a carbon atom is replaced with a carbocyclyl radical as described herein. Typical, but non-limiting, examples of carbocyclylalkyl groups include cyclopropylmethyl, cyclopropylethyl, cyclobutylmethyl, cyclopentylmethyl and cyclohexylmethyl.
[0048] "Arylheteroalkyl" refers to a heteroalkyl as defined herein, in which a hydrogen atom (which may be attached either to a carbon atom or a heteroatom) has been replaced with an aryl group as defined herein. The aryl groups may be bonded to a carbon atom of the heteroalkyl group, or to a heteroatom of the heteroalkyl group, provided that the resulting arylheteroalkyl group provides a chemically stable moiety. For example, an arylheteroalkyl group can have the general formulae -alkylene-O-aryl, -alkylene-O-alkylene-aryl, -alkylene-NH-aryl, -alkylene-NH-alkylene-aryl, -alkylene-S-aryl, -alkylene-S-alkylene-aryl, etc. In addition, any of the alkylene moieties in the general formulae above can be further substituted with any of the substituents defined or exemplified herein.
[0049] "Heteroarylalkyl" refers to an alkyl group, as defined herein, in which a hydrogen atom has been replaced with a heteroaryl group as defined herein. Non-limiting examples of heteroaryl alkyl include --CH.sub.2-pyridinyl, --CH.sub.2-pyrrolyl, --CH.sub.2-oxazolyl, --CH.sub.2-indolyl, --CH.sub.2-isoindolyl, --CH.sub.2-purinyl, --CH.sub.2-furanyl, --CH.sub.2-thienyl, --CH.sub.2-benzofuranyl, --CH.sub.2-benzothiophenyl, --CH.sub.2-carbazolyl, --CH.sub.2-imidazolyl, --CH.sub.2-thiazolyl, --CH.sub.2-isoxazolyl, --CH.sub.2-pyrazolyl, --CH.sub.2-isothiazolyl, --CH.sub.2-quinolyl, --CH.sub.2-isoquinolyl, --CH.sub.2-pyridazyl, --CH.sub.2-pyrimidyl, --CH.sub.2-pyrazyl, --CH(CH.sub.3)-pyridinyl, --CH(CH.sub.3)-pyrrolyl, --CH(CH.sub.3)-oxazolyl, --CH(CH.sub.3)-indolyl, --CH(CH.sub.3)-isoindolyl, --CH(CH.sub.3)-purinyl, --CH(CH.sub.3)-furanyl, --CH(CH.sub.3)-thienyl, --CH(CH.sub.3)-benzofuranyl, --CH(CH.sub.3)-benzothiophenyl, --CH(CH.sub.3)-carbazolyl, --CH(CH.sub.3)-imidazolyl, --CH(CH.sub.3)-thiazolyl, --CH(CH.sub.3)-isoxazolyl, --CH(CH.sub.3)-pyrazolyl, --CH(CH.sub.3)-isothiazolyl, --CH(CH.sub.3)-quinolyl, --CH(CH.sub.3)-isoquinolyl, --CH(CH.sub.3)-pyridazyl, --CH(CH.sub.3)-pyrimidyl, --CH(CH.sub.3)-pyrazyl, etc.
[0050] The term "optionally substituted" in reference to a particular moiety of the compound of Formula I-IV (e.g., an optionally substituted aryl group) refers to a moiety wherein all substituents are hydrogen or wherein one or more of the hydrogens of the moiety may be replaced by substituents such as those listed under the definition of "substituted".
[0051] The term "optionally replaced" in reference to a particular moiety of the compound of Formula I-IV (e.g., the carbon atoms of said (C.sub.1-C.sub.8)alkyl may be optionally replaced by --O--, --S--, or --NR.sup.a--) means that one or more of the methylene groups of the (C.sub.1-C.sub.8)alkyl may be replaced by 0, 1, 2, or more of the groups specified (e.g., --O--, --S--, or --NR.sup.a--).
[0052] The term "non-terminal carbon atom(s)" in reference to an alkyl, alkenyl, alkynyl, alkylene, alkenylene, or alkynylene moiety refers to the carbon atoms in the moiety that intervene between the first carbon atom of the moiety and the last carbon atom in the moiety. Therefore, by way of example and not limitation, in the alkyl moiety --CH.sub.2(C*)H.sub.2(C*)H.sub.2CH.sub.3 or alkylene moiety --CH.sub.2(C*)H.sub.2(C*)H.sub.2CH.sub.2-- the C* atoms would be considered to be the non-terminal carbon atoms.
[0053] Certain Q and Q.sup.1 alternatives are nitrogen oxides such as .sup.+N(O)(R) or .sup.+N(O)(OR). These nitrogen oxides, as shown here attached to a carbon atom, can also be represented by charge separated groups such as
##STR00023##
respectively, and are intended to be equivalent to the aforementioned representations for the purposes of describing this invention.
[0054] "Linker" or "link" means a chemical moiety comprising a covalent bond or a chain of atoms. Linkers include repeating units of alkyloxy (e.g. polyethyleneoxy, PEG, polymethyleneoxy) and alkylamino (e.g. polyethyleneamino, Jeffamine.TM.); and diacid ester and amides including succinate, succinamide, diglycolate, malonate, and caproamide.
[0055] The terms such as "oxygen-linked", "nitrogen-linked", "carbon-linked", "sulfur-linked", or "phosphorous-linked" mean that if a bond between two moieties can be formed by using more than one type of atom in a moiety, then the bond formed between the moieties is through the atom specified. For example, a nitrogen-linked amino acid would be bonded through a nitrogen atom of the amino acid rather than through an oxygen or carbon atom of the amino acid.
[0056] In some embodiments of the compounds of Formula I-IV, one or more of Z.sup.1 or Z.sup.2 are independently a radical of a nitrogen-linked naturally occurring .alpha.-amino acid ester. Examples of naturally occurring amino acids include isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine, alanine, asparagine, aspartic acid, cysteine, glutamic acid, glutamine, glycine, proline, selenocysteine, serine, tyrosine, arginine, histidine, ornithine and taurine. The esters of these amino acids comprise any of those described for the substituent R, particularly those in which R is optionally substituted (C.sub.1-C.sub.8)alkyl.
[0057] The term "purine" or "pyrimidine" base comprises, but is not limited to, adenine, N.sup.6-alkylpurines, N.sup.6-acylpurines (wherein acyl is C(O)(alkyl, aryl, alkylaryl, or arylalkyl), N.sup.6-benzylpurine, N.sup.6-halopurine, N.sup.6-vinylpurine, N.sup.6-acetylenic purine, N.sup.6-acyl purine, N.sup.6-hydroxyalkyl purine, N.sup.6-allylaminopurine, N.sup.6-thioallyl purine, N.sup.2-alkylpurines, N.sup.2-alkyl-6-thiopurines, thymine, cytosine, 5-fluorocytosine, 5-methylcytosine, 6-azapyrimidine, including 6-azacytosine, 2- and/or 4-mercaptopyrmidine, uracil, 5-halouracil, including 5-fluorouracil, C.sup.5-alkylpyrimidines, C.sup.5-benzylpyrimidines, C.sup.5-halopyrimidines, C.sup.5-vinylpyrimidine, C.sup.5-acetylenic pyrimidine, C.sup.5-acyl pyrimidine, C.sup.5-hydroxyalkyl purine, C.sup.5-amidopyrimidine, C.sup.5-cyanopyrimidine, C.sup.5-5-iodopyrimidine, C.sup.6-iodo-pyrimidine, C.sup.5--Br-vinyl pyrimidine, C.sup.6--Br-- vinyl pyriniidine, C.sup.5-nitropyrimidine, C.sup.5-amino-pyrimidine, N.sup.2-alkylpurines, N.sup.2-alkyl-6-thiopurines, 5-azacytidinyl, 5-azauracilyl, triazolopyridinyl, imidazolopyridinyl, pyrrolopyrimidinyl, and pyrazolopyrimidinyl. Purine bases include, but are not limited to, guanine, adenine, hypoxanthine, 2,6-diaminopurine, and 6-chloropurine. The purine and pyrimidine bases are linked to the ribose sugar, or analog thereof, through a nitrogen atom of the base. Functional oxygen and nitrogen groups on the base can be protected as necessary or desired. Suitable protecting groups are well known to those skilled in the art, and include trimethylsilyl, dimethylhexylsilyl, t-butyldimethylsilyl, and t-butyldiphenylsilyl, trityl, alkyl groups, and acyl groups such as acetyl and propionyl, methanesulfonyl, and p-toluenesulfonyl.
[0058] Unless otherwise specified, the carbon atoms of the compounds of Formula I-IV are intended to have a valence of four. In some chemical structure representations where carbon atoms do not have a sufficient number of variables attached to produce a valence of four, the remaining carbon substituents needed to provide a valence of four should be assumed to be hydrogen. For example,
##STR00024##
has the same meaning as
##STR00025##
[0059] "Protecting group" refers to a moiety of a compound that masks or alters the properties of a functional group or the properties of the compound as a whole. The chemical substructure of a protecting group varies widely. One function of a protecting group is to serve as an intermediate in the synthesis of the parental drug substance. Chemical protecting groups and strategies for protection/deprotection are well known in the art. See: "Protective Groups in Organic Chemistry", Theodora W. Greene (John Wiley & Sons, Inc., New York, 1991. See also Protective Groups in Organic Chemistry, Peter G. M. Wuts and Theodora W. Greene, 4th Ed., 2006. Protecting groups are often utilized to mask the reactivity of certain functional groups, to assist in the efficiency of desired chemical reactions, e.g. making and breaking chemical bonds in an ordered and planned fashion. Protection of functional groups of a compound alters other physical properties besides the reactivity of the protected functional group, such as the polarity, lipophilicity (hydrophobicity), and other properties which can be measured by common analytical tools. Chemically protected intermediates may themselves be biologically active or inactive. "Hydroxy protecting groups" refers to those protecting groups useful for protecting hydroxy groups (--OH).
[0060] Protected compounds may also exhibit altered, and in some cases, optimized properties in vitro and in vivo, such as passage through cellular membranes and resistance to enzymatic degradation or sequestration. In this role, protected compounds with intended therapeutic effects may be referred to as prodrugs. Another function of a protecting group is to convert the parental drug into a prodrug, whereby the parental drug is released upon conversion of the prodrug in vivo. Because active prodrugs may be absorbed more effectively than the parental drug, prodrugs may possess greater potency in vivo than the parental drug. Protecting groups are removed either in vitro, in the instance of chemical intermediates, or in vivo, in the case of prodrugs. With chemical intermediates, it is not particularly important that the resulting products after deprotection, e.g. alcohols, be physiologically acceptable, although in general it is more desirable if the products are pharmacologically innocuous.
[0061] The term "chiral" refers to molecules which have the property of non-superimposability of the mirror image partner, while the term "achiral" refers to molecules which are superimposable on their mirror image partner.
[0062] The term "stereoisomers" refers to compounds which have identical chemical constitution, but differ with regard to the arrangement of the atoms or groups in space.
[0063] "Diastereomer" refers to a stereoisomer with two or more centers of chirality and whose molecules are not mirror images of one another. Diastereomers have different physical properties, e.g. melting points, boiling points, spectral properties, reactivities and biological properties. For example, the compounds of Formula I-IV may have a chiral phosphorus atom when R.sup.7 is
##STR00026##
and Z.sup.1 and Z.sup.2 are different. When at least one of either Z.sup.1 or Z.sup.2 also has a chiral center, for example with Z.sup.1 or Z.sup.2 is a nitrogen-linked, chiral, naturally occurring .alpha.-amino acid ester, then the compound of Formula I-IV will exists as diastereomers because there are two centers of chirality in the molecule. All such diastereomers and their uses described herein are encompassed by the instant invention. Mixtures of diastereomers may be separate under high resolution analytical procedures such as electrophoresis, crystallization and/or chromatography. Diastereomers may have different physical attributes such as, but not limited to, solubility, chemical stabilities and crystallinity and may also have different biological properties such as, but not limited to, enzymatic stability, absorption and metabolic stability.
[0064] "Enantiomers" refer to two stereoisomers of a compound which are non-superimposable mirror images of one another.
[0065] The modifier "about" used in connection with a quantity is inclusive of the stated value and has the meaning dictated by the context (e.g., includes the degree of error associated with measurement of the particular quantity).
[0066] The term "treating", as used herein, unless otherwise indicated, means reversing, alleviating, inhibiting the progress of, or preventing the disorder or condition to which such term applies, or one or more symptoms of such disorder or condition. The term "treatment", as used herein, refers to the act of treating, as "treating" is defined immediately above.
[0067] The term "therapeutically effective amount", as used herein, is the amount of compound of Formula I-IV present in a composition described herein that is needed to provide a desired level of drug in the secretions and tissues of the airways and lungs, or alternatively, in the bloodstream of a subject to be treated to give an anticipated physiological response or desired biological effect when such a composition is administered by the chosen route of administration. The precise amount will depend upon numerous factors, for example the particular compound of Formula I-IV, the specific activity of the composition, the delivery device employed, the physical characteristics of the composition, its intended use, as well as patient considerations such as severity of the disease state, patient cooperation, etc., and can readily be determined by one skilled in the art based upon the information provided herein.
[0068] The term "normal saline" means a water solution containing 0.9% (w/v) NaCl.
[0069] The term "hypertonic saline" means a water solution containing greater than 0.9% (w/v) NaCl. For example, 3% hypertonic saline would contain 3% (w/v) NaCl.
[0070] "Forming a reaction mixture" refers to the process of bringing into contact at least two distinct species such that they mix together and can react. It should be appreciated, however, the resulting reaction product can be produced directly from a reaction between the added reagents or from an intermediate from one or more of the added reagents which can be produced in the reaction mixture.
[0071] "Coupling agent" refers to an agent capable of coupling two disparate compounds. Coupling agents can be catalytic or stoichiometric. For example, the coupling agents can be a lithium based coupling agent or a magnesium based coupling agent such as a Grignard reagent. Exemplary coupling agents include, but are not limited to, n-BuLi, MgCl.sub.2, iPrMgCl, tBuMgCl, PhMgCl or combinations thereof.
[0072] "Silane" refers to a silicon containing group having the formula SiR.sub.4, where each R group can be alkyl, alkenyl, cycloalkyl, phenyl, or other silicon containing groups. When the silane is linked to another compound, the silane is referred to as a "silyl" and has the formula --SiR.sub.3.
[0073] "Halo-silane" refers to a silane having at least one halogen group linked to the silicon atom. Representative halo-silanes have the formula Halo-SiR.sub.3, where each R group can be alkyl, alkenyl, cycloalkyl, phenyl, or other silicon containing groups. Specific halo-silanes include Cl--Si(CH.sub.3).sub.3, and Cl--Si(CH.sub.3).sub.2CH.sub.2CH.sub.2Si(CH.sub.3).sub.2--Cl.
[0074] "Non-nucleophilic base" refers to an electron donor, a Lewis base, such as nitrogen bases including triethylamine, diisopropylethyl amine, N,N-diethylaniline, pyridine, 2,6-lutidine, 2,4,6-collidine, 4-dimethylaminopyridine, and quinuclidine.
[0075] "Leaving group" refers to groups that maintain the bonding electron pair during heterolytic bond cleavage. For example, a leaving group is readily displaced during a nucleophilic displacement reaction. Suitable leaving groups include, but are not limited to, chloride, bromide, mesylate, tosylate, triflate, 4-nitrobenzenesulfonate, 4-chlorobenzenesulfonate, 4-nitrophenoxy, pentafluorophenoxy, etc. One of skill in the art will recognize other leaving groups useful in the present invention.
[0076] "Deprotection agent" refers to any agent capable of removing a protecting group. The deprotection agent will depend on the type of protecting group used. Representative deprotection agents are known in the art and can be found in Protective Groups in Organic Chemistry, Peter G. M. Wuts and Theodora W. Greene, 4th Ed., 2006.
II. Preparation of Compounds
[0077] The compounds of the present invention can be prepared by a variety of means. For example, protected nucleosides of Formula V can be prepared by reaction of a protected lactone with an iodo-substituted base under suitable coupling conditions. The nucleosides can then be modified with a prodrug moiety by reaction of a partially protected nucleoside with a suitable prodrug moiety, following be removal of the protecting groups, to afford the compounds of the present invention.
[0078] A. Preparation of Nucleosides Via Iodo-Base
[0079] In one embodiment, the present invention provides a method of preparing a compound of Formula V:
##STR00027##
The method of making the compound of Formula V includes forming a reaction mixture having a coupling agent, a halo-silane, a compound of Formula VI:
##STR00028##
and a compound of Formula VII:
##STR00029##
under conditions suitable to prepare the compound of Formula V, wherein each PG is independently a hydroxy protecting group, alternatively, two PG groups on adjacent carbons can be combined to form a --C(R.sup.19).sub.2-- group, R.sup.10 is H or a silyl group, and R.sup.19 is H, C.sub.1-C.sub.8 alkyl, phenyl or substituted phenyl.
[0080] Any suitable coupling agent can be used in the method of making the compound of Formula V. The coupling agent can be a lithium coupling agent, a sodium coupling agent, a magnesium coupling agent, or others. For example, the coupling agent can be a deprotonating agent such as n-butyl lithium (n-BuLi), sodium hydride (NaH), lithium aluminum hydride (LAH or LiAlH.sub.4), and others. The coupling agent can also be a magnesium based coupling agent such as, but not limited to, MgCl.sub.2, iPrMgCl, tBuMgCl, PhMgCl, or combinations thereof. In some embodiments, the coupling agent can be a lithium coupling agent or a magnesium coupling agent. In some embodiments, the coupling agent can be n-BuLi, MgCl.sub.2, iPrMgCl, tBuMgCl, PhMgCl, or combinations thereof. In some embodiments, the coupling agent can be n-BuLi. In some embodiments, the coupling agent can be PhMgCl and iPrMgCl.
[0081] The coupling agent can be present in any suitable amount. For example, the coupling agent can be present in an amount of at least 1.0 eq. (mol/mol) to the compound of Formula V, such as about 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The coupling agent can also be present in an amount of from about 1.0 to about 10.0 eq. (mol/mol) to the compound of Formula V, such as of from about 1.0 to about 5.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the coupling agent can be present in an amount of from about 1.0 to about 5.0 eq. (mol/mol) to the compound of Formula V. In some embodiments, the coupling agent can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula V.
[0082] Any suitable halo-silane can be used in the method of making the compound of Formula V. For example, the halo-silane can be a fluoro-silane, a chloro-silane, a bromo-silane or an iodo-silane. The silane portion can have any suitable substituents, such as alkyl, alkenyl, alkynyl, cycloalkyl, or phenyl. Exemplary halo-silanes include, but are not limited to, Cl--Si(CH.sub.3).sub.3, or Cl--Si(CH.sub.3).sub.2CH.sub.2CH.sub.2Si(CH.sub.3).sub.2--Cl. In some embodiments, the halo-silane can be a chloro-silane. In some embodiments, the halo-silane can be Cl--Si(CH.sub.3).sub.3, or Cl--Si(CH.sub.3).sub.2CH.sub.2CH.sub.2Si(CH.sub.3).sub.2--Cl. In some embodiments, the halo-silane can be TMSCl.
[0083] The silyl group of R.sup.10 can be any suitable group, but can depend on the choice of the halo-silane. For example, when the halo-silane is TMSCl, the silyl group can be trimethylsilyl.
[0084] The halo-silane can be present in any suitable amount. For example, the halo-silane can be present in an amount of at least 1.0 eq. (mol/mol) to the compound of Formula V, such as about 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The halo-silane can also be present in an amount of from about 1.0 to about 10.0 eq. (mol/mol) to the compound of Formula V, such as of from about 1.0 to about 5.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the halo-silane can be present in an amount of from about 1.0 to about 5.0 eq. (mol/mol) to the compound of Formula V. In some embodiments, the halo-silane can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula V.
[0085] The hydroxy protecting group can be any protecting group suitable for a hydroxy functional group. Representative hydroxy protecting groups include, but are not limited to, silanes such as trimethyl silane (TMS), t-butyl dimethyl silane (TBDMS), or t-butyl diphenyl silane (TBDPS), ethers such as methyl-methoxy (MOM), tetrahydropyran (THP), t-butyl, allyl, or benzyl, and esters such as acetyl, pivaloyl, or benzoyl. In some embodiments, the hydroxy protecting group can be trimethyl silane (TMS), t-butyl dimethyl silane (TBDMS), t-butyl diphenyl silane (TBDPS), methyl-methoxy (MOM), tetrahydropyran (THP), t-butyl, allyl, benzyl, acetyl, pivaloyl, or benzoyl. In some embodiments, the hydroxy protecting group can be benzyl.
[0086] Hydroxy groups on adjacent carbons, referred to as 1,2-hydroxy groups, can form a cyclic protecting group called an acetonide by reaction with a ketone of di-ether. Exemplary acetonides include, but are not limited to acetonide and benzylidene acetal. In some embodiments, the hydroxy protecting groups of hydroxy groups on adjacent carbons can be combined to form acetonide.
[0087] When the R.sup.19 group is C.sub.1-C.sub.8 alkyl, R.sup.19 can be methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, sec-buty, t-butyl, pentyl, iso-pentyl, neo-pentyl, hexyl, isohexyl, neohexyl, septyl or octyl. In some embodiments, the R.sup.19 group can be methyl.
[0088] Any suitable solvent can be used in the method of the present invention. Representative solvents include, but are not limited to, pentane, pentanes, hexane, hexanes, heptane, heptanes, petroleum ether, cyclopentanes, cyclohexanes, benzene, toluene, xylene, trifluoromethylbenzene, halobenzenes such as chlorobenzene, fluorobenzene, dichlorobenzene and difluorobenzene, methylene chloride, chloroform, acetone, ethyl acetate, diethyl ether, tetrahydrofuran, or combinations thereof. In some embodiments, the solvent can be tetrahydrofuran.
[0089] The reaction mixture of the method can be at any suitable temperature. For example, the temperature of the reaction mixture can be of from about -78.degree. C. to about 100.degree. C., or of from about -50.degree. C. to about 100.degree. C., or of from about -25.degree. C. to about 50.degree. C., or of from about -10.degree. C. to about 25.degree. C., or of from about 0.degree. C. to about 20.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about 0.degree. C. to about 20.degree. C.
[0090] The reaction mixture of the method can be at any suitable pressure. For example, the reaction mixture can be at atmospheric pressure. The reaction mixture can be also be exposed to any suitable environment, such as atmospheric gasses, or inert gasses such as nitrogen or argon.
[0091] The method of the present invention can provide the compound of Formula V in any suitable yield. For example, the compound of Formula V can be prepared in a yield of at least about 50%, 55, 60, 65, 70, 75, 80, 85, 90 or at least about 95%.
[0092] The method of the present invention can provide the compound of Formula V in any suitable purity. For example, the compound of Formula V can be prepared in a purity of at least about 90, 95, 96, 97, 98 or at least about 99%. In some embodiments, the compound of Formula V can be prepared in at least 95% purity. In some embodiments, the compound of Formula V can be prepared in at least 98% purity. In some embodiments, the compound of Formula V can be prepared in at least 99% purity.
[0093] In some embodiments, the method including preparing the compound of Formula V:
##STR00030##
wherein the method includes forming the reaction mixture having TMSCl, PhMgCl, iPrMgCl, the compound of Formula VI:
##STR00031##
and the compound of Formula VII:
##STR00032##
under conditions suitable to prepare the compound of Formula V.
[0094] In some embodiments, the present invention provides the compound:
##STR00033##
[0095] In some embodiments, the present invention provides a method of preparing a compound of Formula V-a or V-b:
##STR00034##
The method of making the compound of Formula V-a or Formula V-b comprises forming a reaction mixture having a deprotonating agent, a silylating agent, a coupling agent, an additive, a compound of Formula VI-a:
##STR00035##
and a compound of Formula VII:
##STR00036##
under conditions suitable to prepare the compound of Formula V-a or Formula V-b, wherein each R.sup.b is independently a hydroxy protecting group, alternatively, two R.sup.b groups on adjacent carbons can be combined to form a --C(R.sup.19).sub.2-- group, R.sup.10 is H or a silyl group, and R.sup.19 is H, C.sub.1-C.sub.8 alkyl, phenyl or substituted phenyl.
[0096] Any suitable deprotonating agent can be used in the method of making the compound of Formula V-a or Formula V-b. The deprotonating agent can be a sodium deprotonating agent, a magnesium based deprotonating agent, lithium based deprotonating agent, potassium based deprotonating agent, or others. For example, the deprotonating agent can be sodium hydride (NaH), isopropylmagnesium chloride (iPrMgCl), tert-butylmagnesium chloride (tBuMgCl), phenylmagnesium chloride (PhMgCl), phenylmagnesium bromide (PhMgBr), butyllithium (BuLi), methyllithium (MeLi), methylmagnesium chloride (MeMgCl), methylmagnesium bromide (MeMgBr), tert-butyllithium (tBuLi), isopropyllithium (iPrLi), phenyllithium (PhLi), lithium hydride (LiH), potassium hydride (KH), ethyllithium (EtLi), ethylmagnesium bromide (EtMgBr), ethylmagnesium chloride (EtMgCl), propyllithium (PrLi), propylmagnesium bromide (PrMgBr), propylmagnesium chloride (PrMgCl), cyclohexanelithium (cyHexLi), cyclohexanemagnesium bromide (cyHexMgBr), cyclohexanemagnesium chloride (cyHexMgCl), or combinations thereof. In some embodiments, the deprotonating agent can be PhMgCl.
[0097] The deprotonating agent can be present in any suitable amount. For example, the deprotonating agent can be present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula VII, such as about 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The deprotonating agent can also be present in an amount of from about 0.1 to about 10.0 eq. (mol/mol) to the compound of Formula VII, such as of from about 0.1 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the deprotonating agent can be present in an amount from about 0.1 to 1.0 eq. (mol/mol) to the compound of Formula VII. In some embodiments, the deprotonating agent can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula VII.
[0098] Any suitable silylating agent can be used in the method of making the compound of Formula V-a or Formula V-b. For example, the silylating agent can be a fluoro-silane, a chloro-silane, a bromo-silane or an iodo-silane. For example, the silylating agent can be a tri-substituted silyl chloride, a tri-substituted silyl bromide, a tri-substituted silyl iodide, or a tri-substituted silyl fluoride. The silyl portion can have any suitable substituents, such as alkyl, alkenyl, alkynyl, cycloalkyl, or phenyl. Exemplary silylating agents include, but are not limited to, Cl--Si(CH.sub.3).sub.3, Cl--Si(CH.sub.3).sub.2CH.sub.2CH.sub.2Si(CH.sub.3).sub.2--Cl, or tert-butyldiphenylsilyl (TBDPS). In some embodiments, the silylating agent can be a chloro-silane. In some embodiments, the silylating agent can be Cl--Si(CH.sub.3).sub.3, or Cl--Si(CH.sub.3).sub.2CH.sub.2CH.sub.2Si(CH.sub.3).sub.2--Cl. In some embodiments, the silylating agent can be TMSCl.
[0099] The silyl group of R.sup.10 can be any suitable group, but can depend on the choice of the silylating agent. For example, when the silylating agent is TMSCl, the silyl group can be trimethylsilyl.
[0100] The silylating agent can be present in any suitable amount. For example, the silylating agent can be present in an amount of at least 0.0 eq. (mol/mol) to the compound of Formula VII, such as about 0.0, 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The silylating agent can also be present in an amount of from about 0.0 to about 10.0 eq. (mol/mol) to the compound of Formula VII, such as of from about 0.0 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the silylating agent can be present in an amount from about 0.0 to 1.0 eq. (mol/mol) to the compound of Formula VII. In some embodiments, the silylating agent can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula VII.
[0101] Any suitable coupling agent can be used in the method of making the compound of Formula V-a or Formula V-b. The coupling agent can be a lithium coupling agent, a magnesium based deprotonating agent, or others. For example, the coupling agent can be n-butyllithium (nBuLi), magnesium chloride (MgCl.sub.2), isopropylmagnesium chloride (iPrMgCl), isopropylmagnesium chloride-lithium chloride (iPrMgCl--LiCl), tert-butylmagnesium chloride (tBuMgCl), phenylmagnesium chloride (PhMgCl), methyllithium (MeLi), methylmagnesium chloride (MeMgCl), methylmagnesium bromide (MeMgBr), tert-butyllithium (tBuLi), isopropyllithium (iPrLi), phenyllithium (PhLi), lithium hydride (LiH), potassium hydride (KH), sodium hydride (NaH), ethyllithium (EtLi), ethylmagnesium bromide (EtMgBr), ethylmagnesium chloride (EtMgCl), propyllithium (PrLi), propylmagnesium bromide (PrMgBr), propylmagnesium chloride (PrMgCl), cyclohexanelithium (cyHexLi), cyclohexanemagnesium bromide (cyHexMgBr), cyclohexanemagnesium chloride (cyHexMgCl), or combinations thereof. In some embodiments, the coupling agent can be iPrMgCl.
[0102] The coupling agent can be present in any suitable amount. For example, the coupling agent can be present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula VII, such as about 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The coupling agent can also be present in an amount of from about 0.1 to about 10.0 eq. (mol/mol) to the compound of Formula VII, such as of from about 0.1 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the coupling agent can be present in an amount from about 0.1 to 1.0 eq. (mol/mol) to the compound of Formula VII. In some embodiments, the coupling agent can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula VII.
[0103] Any suitable additive can be used in the method of making the compound of Formula V-a of Formula V-b. In some embodiments, the additive is a Lewis Acid. In some embodiments, the additive can be BF.sub.3-OEt.sub.2, SmOTf).sub.3, Sc(OTf).sub.3, FeCl.sub.3, LiCl, LiBr, TiCl(OiPr).sub.3, ScCl.sub.3, Bu.sub.4NBr+LaCl.sub.3-2LiCl, nLaCl.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, LaCl.sub.3+2LiCl, Sm(OTf).sub.3+LiCl, SmCl.sub.3, Bis[2-(N,N-dimethylamino)ethyl]ether, TMEDA, NdCl.sub.3, NdCl.sub.3+CsCl, nNdCl.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, NdCl.sub.3+2LiCl, NdCl.sub.3+LiBr, NdCl.sub.3+LiI, NdBr.sub.3, NdBr.sub.3+CsCl, nNdBr.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, NdBr.sub.3+2LiCl, NdBr.sub.3+LiBr, NdBr.sub.3+LiI, Nd(OTf).sub.3, CeCl.sub.3, CeCl.sub.3+CsCl, nCeCl.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, CeCl.sub.3+2LiCl, CeCl.sub.3+LiBr, CeCl.sub.3+LiI, CeBr.sub.3, Ce(OTf).sub.3, YCl.sub.3, YCl.sub.3+CsCl, nYCl.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, YCl.sub.3+2LiCl, YCl.sub.3+LiBr, YCl.sub.3+LiI, YBr.sub.3, YBr.sub.3+CsCl, nYBr.sub.3+mLiCl, wherein m is 0.5 to 50, n is 1 to 100, YBr.sub.3+2LiCl, YBr.sub.3+LiBr, YBr.sub.3+LiI, Y(OTf).sub.3, LaCl.sub.3, La(OTf).sub.3, MgCl.sub.2, TiCl.sub.4, SnCl.sub.4, AlCl.sub.3, Bu.sub.4NCl, Diethyleneglycol diethylether (DGDE), DGDE+Bu.sub.4NCl, DGDE+Bu.sub.4NBr, DGDE+Bu.sub.4NI, CaCl.sub.2, CaBr.sub.2, CaI.sub.2, Ca(OTf).sub.2, YCl.sub.3, YCl.sub.3-2LiCl, YCl.sub.3--LiCl or a combination thereof. In some embodiments, the additive can be LiCl, Ca(OTf).sub.2, CaCl.sub.2 and MgCl.sub.2, CeCl.sub.3, LaCl.sub.3, or a combination thereof. In some embodiments, the additive can be YCl.sub.3, CeCl.sub.3, NdCl.sub.3, LaCl.sub.3, or a combination thereof.
[0104] The additive can be present in any suitable amount. For example, the additive can be present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula VII, such as about 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The additive can also be present in an amount of from about 0.1 to about 10.0 eq. (mol/mol) to the compound of Formula VII, such as of from about 0.1 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the additive can be present in an amount from about 0.1 to 1.0 eq. (mol/mol) to the compound of Formula VII. In some embodiments, the additive can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula VII.
[0105] In some embodiments, the additive is LaCl.sub.3-2LiCl and is present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula VII, such as about 0, 0.1, 0.3, 0.5, 1.0, 2, or about 2.0 eq. (mol/mol). In some embodiments, the additive is LaCl.sub.3-2LiCl and is present in an amount of from about 0 to about 2.0 eq. (mol/mol) to the compound of Formula VII, such as of from about 0 to about 0.3 eq. (mol/mol), or of from about 0 to about 0.5 eq. (mol/mol). In some embodiments, the additive is LaCl.sub.3-2LiCl and is present in an amount from about 0 to 0.5 eq. (mol/mol) to the compound of Formula VII. In some embodiments, the the additive is LaCl.sub.3-2LiCl and is present in an amount of about 0.5 eq. (mol/mol) to the compound of Formula VII.
[0106] In some embodiments, the additive is CeCl.sub.3 and is present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula VII, such as about 0, 0.1, 0.3, 0.5, 1.0, 2, or about 2.0 eq. (mol/mol). In some embodiments, the additive is CeCl.sub.3 and is present in an amount of from about 0 to about 2.0 eq. (mol/mol) to the compound of Formula VII, such as of from about 0 to about 0.3 eq. (mol/mol), or of from about 0 to about 0.5 eq. (mol/mol). In some embodiments, the additive is CeCl.sub.3 and is present in an amount from about 0 to 0.5 eq. (mol/mol) to the compound of Formula VII. In some embodiments, the the additive is CeCl.sub.3 and is present in an amount of about 0.5 eq. (mol/mol) to the compound of Formula VII.
[0107] In some embodiments, the additive is NdCl.sub.3 and is present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula VII, such as about 0, 0.1, 0.3, 0.5, 1.0, 2, or about 2.0 eq. (mol/mol). In some embodiments, the additive is NdCl.sub.3 and is present in an amount of from about 0 to about 2.0 eq. (mol/mol) to the compound of Formula VII, such as of from about 0 to about 0.3 eq. (mol/mol), or of from about 0 to about 0.5 eq. (mol/mol). In some embodiments, the additive is NdCl.sub.3 and is present in an amount from about 0 to 0.5 eq. (mol/mol) to the compound of Formula VII. In some embodiments, the the additive is NdCl.sub.3 and is present in an amount of about 0.5 eq. (mol/mol) to the compound of Formula VII.
[0108] In some embodiments, the additive is YCl.sub.3 and is present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula VII, such as about 0, 0.1, 0.3, 0.5, 1.0, 2, or about 2.0 eq. (mol/mol). In some embodiments, the additive is YCl.sub.3 and is present in an amount of from about 0 to about 2.0 eq. (mol/mol) to the compound of Formula VII, such as of from about 0 to about 0.3 eq. (mol/mol), or of from about 0 to about 0.5 eq. (mol/mol). In some embodiments, the additive is YCl.sub.3 and is present in an amount from about 0 to 0.5 eq. (mol/mol) to the compound of Formula VII. In some embodiments, the the additive is YCl.sub.3 and is present in an amount of about 0.5 eq. (mol/mol) to the compound of Formula VII.
[0109] When the R.sup.19 group is C.sub.1-C.sub.8 alkyl, R.sup.19 can be methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, sec-buty, t-butyl, pentyl, iso-pentyl, neo-pentyl, hexyl, isohexyl, neohexyl, septyl or octyl. In some embodiments, the R.sup.19 group can be methyl.
[0110] When the R.sup.b group is a hydroxy protecting group, R.sup.b can be any example protecting group described in Protective Groups in Organic Chemistry, Peter G. M. Wuts and Theodora W. Greene, 4th Ed., 2006. In some embodiments, the R.sup.b group can be benzyl. In some embodiments, the R.sup.b group can be TBS.
[0111] The hydroxy protecting group can be any protecting group suitable for a hydroxy functional group. Representative hydroxy protecting groups include, but are not limited to, silanes, ethers, esters, or others. Representative hydroxy protecting groups include, but are not limited to trimethyl silane (TMS), t-butyl dimethyl silane (TBDMS), t-butyl diphenyl silane (TBDPS), methyl-methoxy (MOM), tetrahydropyran (THP), t-butyl, allyl, benzyl, acetyl, pivaloyl, or benzoyl. In some embodiments, the hydroxy protecting group can be trimethyl silane (TMS), t-butyl dimethyl silane (TBDMS), t-butyl diphenyl silane (TBDPS), methyl-methoxy (MOM), tetrahydropyran (THP), t-butyl, allyl, benzyl, acetyl, pivaloyl, or benzoyl. In some embodiments, the hydroxy protecting group can be benzyl. In some embodiments, the hydroxy protecting group can be TBS.
[0112] Hydroxy groups on adjacent carbons, referred to as 1,2-hydroxy groups, can form a cyclic protecting group called an acetal or a ketal by reaction with an aldehyde, an acetale, aketoneor a ketal. Exemplary acetals and ketals include, but are not limited to a, benzylidene acetal and an acetonide. In some embodiments, the hydroxy protecting groups of hydroxy groups on adjacent carbons can be combined to form acetonide.
[0113] Any suitable solvent can be used in the method of the present invention. Representative solvents include, but are not limited to, pentane, pentanes, hexane, hexanes, heptane, heptanes, petroleum ether, cyclopentanes, cyclohexanes, benzene, toluene, xylene, dichloromethane, trifluoromethylbenzene, halobenzenes such as chlorobenzene, fluorobenzene, dichlorobenzene and difluorobenzene, methylene chloride, chloroform, acetone, ethyl acetate, diethyl ether, tetrahydrofuran (THF), 2-methyltetrahydrofuran, dibutyl ether, diisopropyl ether, methyl tert-butyl ether, dimethoxyethane, dioxanes (1.4 dioxane), N-methyl pyrrolidinone (NMP), diisopropyl ether, or combinations thereof. In certain embodiments, the solvent can be THF, MeTHF, toluene, THF+dioxane, THF+pyridine, or THF+DCM, or combinations thereof. In some embodiments, the solvent can be THF.
[0114] The reaction mixture of the method can be at any suitable temperature. For example, the temperature of the reaction mixture can be from about -78.degree. C. to about 100.degree. C., or from about -50.degree. C. to about 100.degree. C., or from about -25.degree. C. to about 50.degree. C., or from about -10.degree. C. to about 25.degree. C., or from about 0.degree. C. to about 20.degree. C. In some embodiments, the temperature of the reaction mixture can be from about 0.degree. C. to about 20.degree. C. In some embodiments, the temperature of the reaction mixture can be from about -30.degree. C. to about -10.degree. C.
[0115] The reaction mixture of the method can be at any suitable pressure. For example, the reaction mixture can be at atmospheric pressure. The reaction mixture can be also be exposed to any suitable environment, such as atmospheric gasses, or inert gasses such as nitrogen or argon.
[0116] The method of the present invention can provide the compound of Formula V-a or Formula V-b in any suitable yield. For example, the compound of Formula V-a or Formula V-b can be prepared in a yield of at least about 50%, 55, 60, 65, 70, 75, 80, 85, 90 or at least about 95%.
[0117] The method of the present invention can provide the compound of Formula V-a or Formula V-b in any suitable purity. For example, the compound of Formula V-a or Formula V-b can be prepared in a purity of at least about 90, 95, 96, 97, 98 or at least about 99%. In some embodiments, the compound of Formula V-a or Formula V-b can be prepared in at least 95% purity. In some embodiments, the compound of Formula V-a or Formula V-b can be prepared in at least 98% purity. In some embodiments, the compound of Formula V-a or Formula V-b can be prepared in at least 99% purity.
[0118] In some embodiments, the method comprises preparing the compound of Formula V-a or Formula V-b:
##STR00037##
wherein the method comprises forming the reaction mixture having TMSCl, PhMgCl, iPrMgCl, LaCl.sub.3-2LiCl the compound of Formula VI:
##STR00038##
and the compound of Formula VII:
##STR00039##
under conditions suitable to prepare the compound of Formula V-a or Formula V-b.
[0119] In some embodiments, the method comprises preparing the compound of Formula V-a or Formula V-b:
##STR00040##
wherein the method comprises forming the reaction mixture having TMSCl, PhMgCl, iPrMgCl, CeCl.sub.3 the compound of Formula VI:
##STR00041##
and the compound of Formula VII:
##STR00042##
under conditions suitable to prepare the compound of Formula V-a or Formula V-b.
[0120] In some embodiments, the method comprises preparing the compound of Formula V-a or Formula V-b:
##STR00043##
wherein the method comprises forming the reaction mixture having TMSCl, PhMgCl, iPrMgCl, NdCl.sub.3 the compound of Formula VI:
##STR00044##
and the compound of Formula VII:
##STR00045##
under conditions suitable to prepare the compound of Formula V-a or Formula V-b.
[0121] In some embodiments, the method comprises preparing the compound of Formula V-a or Formula V-b:
##STR00046##
wherein the method comprises forming the reaction mixture having TMSCl, PhMgCl, iPrMgCl, YCl.sub.3 the compound of Formula VI:
##STR00047##
and the compound of Formula VII:
##STR00048##
under conditions suitable to prepare the compound of Formula V-a or Formula V-b.
[0122] In some embodiments, the method comprises preparing the compound of Formula V-a:
##STR00049##
wherein the method comprises forming the reaction mixture having TMSCl, PhMgCl, iPrMgCl--LiCl, LaCl.sub.3-2LiCl the compound of Formula VI:
##STR00050##
and the compound of Formula VII:
##STR00051##
under conditions suitable to prepare the compound of Formula V-a.
[0123] B. Preparation of Cyano Nucleosides
[0124] In some embodiments, the present invention provides a method of preparing a compound of Formula XI:
##STR00052##
wherein R.sup.c is H or a hydroxy protecting group, or two R.sup.c on adjacent carbons can be combined to form a --C(R.sup.19).sub.2-- group, and R.sup.19 is H or C.sub.1-C.sub.8 alkyl.
[0125] When the R.sup.19 group is C.sub.1-C.sub.8 alkyl, R.sup.19 can be methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, sec-buty, t-butyl, pentyl, iso-pentyl, neo-pentyl, hexyl, isohexyl, neohexyl, septyl or octyl. In some embodiments, the R.sup.19 group can be methyl.
[0126] When the R.sup.c group is a hydroxy protecting group, the hydroxy protecting group can be any protecting group suitable for a hydroxy functional group. Representative hydroxy protecting groups include, but are not limited to, silanes, ethers, esters, or others. Representative hydroxy protecting groups include, but are not limited to trimethyl silane (TMS), t-butyl dimethyl silane (TBDMS), t-butyl diphenyl silane (TBDPS), methyl-methoxy (MOM), tetrahydropyran (THP), t-butyl, allyl, benzyl, acetyl, pivaloyl, or benzoyl. In some embodiments, the hydroxy protecting group can be trimethyl silane (TMS), t-butyl dimethyl silane (TBDMS), t-butyl diphenyl silane (TBDPS), methyl-methoxy (MOM), tetrahydropyran (THP), t-butyl, allyl, benzyl, acetyl, pivaloyl, or benzoyl. In some embodiments, the hydroxy protecting group can be benzyl. In some embodiments, the hydroxy protecting group can be TBS
[0127] Hydroxy groups on adjacent carbons, referred to as 1,2-hydroxy groups, can form a cyclic protecting group called an acetal or a ketal by reaction with an aldehyde, an acetale, aketoneor a ketal. Exemplary acetals and ketals include, but are not limited to a, benzylidene acetal and an acetonide. In some embodiments, the hydroxy protecting groups of hydroxy groups on adjacent carbons can be combined to form acetonide.
[0128] In some embodiments, the present invention provides a method of preparing a compound of Formula XI-a:
##STR00053##
wherein the method comprises forming a reaction mixture having a cyanating agent, a Lewis Acid, a Broenstedt acid, a solvent, and the compound of Formula V-a or V-b:
##STR00054##
under conditions suitable to prepare the compound of Formula XI-a, wherein R.sup.b is independently a hydroxy protecting group, alternatively, two R.sup.b groups on adjacent carbons can be combined to form a --C(R.sup.19).sub.2 group, R.sup.19 is H or a silyl group, and R.sup.19 is H, C.sub.1-C.sub.8 alkyl, phenyl or substituted phenyl.
[0129] Any suitable cyanating agent can be used in the method of making the compound of Formula XI-a. For example, the cyanating agent can be TMSCN, TBSCN, TESCN, HCN, KCN, NaCN, 4-toluenesulfonyl cyanide, CuCN, CuCn*LiCl, LiCN, Zn(CN).sub.2, K.sub.4[Fe(CN).sub.6], tetrabutylammonium cyanide, tetrmethylammonium cyanide, tetraethylammonium cyanide, tetrabutylammonium cyanide, (including tetraalkylammonium cyanide with alkyl independently being Me, Et, Pr, iPr, Bu, iBu, tertBu, Pent, Hex), tributyltn cyanide, trimethyltin cyanide, triethyltin cyanide, tripropyltin cyanide, (including trialkyltin cyanide cyanide with alkyl independently being Me, Et, Pr, iPr, Bu, iBu, tertBu, Pent, Hex), 2-hydroxy-2-methylpropanenitrile; or combinations thereof. In some embodiments, the cyanating agent can be TMSCN.
[0130] The cyanating agent can be present in any suitable amount. For example, the cyanating agent can be present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula V-a or Formula V-b, such as about 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The cyanating agent can also be present in an amount of from about 0.1 to about 10.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b, such as of from about 0.1 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the cyanating agent can be present in an amount from about 0.1 to 1.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b. In some embodiments, the cyanating agent can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b.
[0131] Any suitable Lewis Acid can be used in the method of making the compound of Formula XI-a. For example, the Lewis Acid can be TMSOTf, TMSOTf, TBSOTf, TESOTf, BF.sub.3, BF.sub.3-OEt.sub.2, BCl.sub.3, BF.sub.3-THF, MgCl.sub.2, MgI.sub.2, MgBr.sub.2, MgBr.sub.2-OEt.sub.2, ZnCl.sub.2, ZnBr.sub.2, ZnI.sub.2, LiCl, LiBr, LiI, AlCl.sub.3, AlBr.sub.3, AlI.sub.3, Me.sub.2Si(OTf).sub.2, Et.sub.2Si(OTf).sub.2, Pr.sub.2Si(OTf).sub.2, iPr.sub.2Si(OTf).sub.2, (tBu).sub.2Si(OTf).sub.2, (C.sub.6F.sub.5).sub.3B, MeSiCl.sub.3, Me.sub.2SiCl.sub.2, SiCl.sub.4, TMSCl, TMSI, TMSVr, TBSCl, TBSBr, TBSI, TESCl, TESBr, TESI, SmCl.sub.3, SmBr.sub.3, SmI.sub.2, SmI.sub.3, ScI.sub.3, ScBr.sub.3, ScI.sub.3, Sm(OTf).sub.3, Sc(OTf).sub.3, TiCl.sub.4, Ti(OiPr).sub.4, Ti(OiPr).sub.3Cl, Ti(OiPr).sub.2Cl.sub.2, Ti(OiPr)Cl.sub.3, Zn(BF.sub.4).sub.2, LiBF.sub.4, Mg(BF.sub.4).sub.2, ZrCl.sub.4, FeCl.sub.2, FeCl.sub.3, FeBr.sub.2, FeBr.sub.3, FeI.sub.2, FeI.sub.3, Cu(OTf), Cu(OTf).sub.2, 4-toluenesulfonylchoride, benzenesulfonylchlopride, 4-toluenesulfonyl triflate, benzenesulfonyl triflate, methylsulfonyl chloride, methylsulfonic anhydrate, InCl.sub.3, InBr.sub.3, InI.sub.3, In(OTf).sub.3, Mg(SO.sub.4).sub.2, NaSO.sub.4; or combinations thereof. In some embodiments, the Lewis Acid can be TMSOTf. In some embodiments, the following may be used in the method of making the compound of Formula XI-a instead of a Lewis Acid: dicyclohexylcarbodiimide, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide, benzenesulfonic acid, HCl, 4-toluenesulfonic acid, triflic acid, trifluoroacetic acid, 4-nitrobenzolic acid, methylsoulfonic acid, sulfuric acid, phosphoric acid, HBr, acetic acid, formic acid, HI; or combinations thereof.
[0132] The Lewis Acid can be present in any suitable amount. For example, the Lewis Acid can be present in an amount of at least 0.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b, such as about 0.0, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The Lewis Acid can also be present in an amount of from about 0.0 to about 10.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b, such as of from about 0.0 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the Lewis Acid can be present in an amount from about 0.0 to 1.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b. In some embodiments, the Lewis Acid can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b.
[0133] Any suitable Broenstedt acid can be used in the method of making the compound of Formula XI-a. For example, the Broenstedt acid can be TFA, benzenesulfonic acid, HCl, 4-toluenesulfonic acid, triflic acid, trifluoroacetic acid, 4-nitrobenzoic acid, methylsoulfonic acid, sulfuric acid, phosphoric acid, HBr, acetic acid, formic acid, HI, trifluoromethylsulfonic acid, 4-fluorobenzoic acid, pivalic acid, HBF.sub.4, nitric acid, 4-chloro-benzoic acid, pentafluorophenol, HPF.sub.6, Camphorsulfonic acid; or combinations thereof. In some embodiments, the Broenstedt acid can be TFA.
[0134] The Broenstedt acid can be present in any suitable amount. For example, the Broenstedt acid can be present in an amount of at least about 0.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b, such as about 0.0, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The Broenstedt acid can also be present in an amount of from about 0.0 to about 10.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b, such as of from about 0.0 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the Broenstedt acid can be present in an amount from about 0.0 to about 1.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b. In some embodiments, the Broenstedt acid can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b.
[0135] Any suitable solvent can be used in the method of making the compound of Formula XI-a. For example, the solvent can be DCM, THF, MeTHF, Et.sub.2O, MeCN, EtCN, toluene, benzene, chlorobenzene, nitrobenzene, flurorbenzene, methanol, ethanol, 2-propanol, propanol, butanol, MTBE, EtOAc, iPrOAc, Me2O, (TMS)2O, acetone, 2-butanone, chloroform, 1,2-dichloroethane, diglyme, dioxane, acetic acid, formic acid, trifluoroacetic acid, methylisobutylketone, DMAc, DMF, NMP, DMSO; or combinations thereof. In some embodiments, the solvent can be DCM.
[0136] The solvent can be present in any suitable amount. For example, the solvent can be present in an amount of at least 0.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b, such as about 0.0, 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The solvent can also be present in an amount of from about 0.0 to about 10.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b, such as of from about 0.0 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the solvent can be present in an amount from about 0.1 to about 1.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b. In some embodiments, the solvent can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula V-a or Formula V-b.
[0137] The reaction mixture of the method can be at any suitable temperature. For example, the temperature of the reaction mixture can be of from about -150.degree. C. to about 0.degree. C., or of from about -120.degree. C. to about 0.degree. C., or of from about -100.degree. C. to about 0.degree. C., or of from about -100.degree. C. to about -50.degree. C., or of from about -100.degree. C. to about -70.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about -120.degree. C. to about -70.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about -120.degree. C. to about -100.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about -80.degree. C. to about -30.degree. C.
[0138] The reaction mixture of the method can be at any suitable pressure. For example, the reaction mixture can be at atmospheric pressure. The reaction mixture can be also be exposed to any suitable environment, such as atmospheric gasses, or inert gasses such as nitrogen or argon.
[0139] The method of the present invention can provide the compound of Formula XI-a in any suitable yield. For example, the compound of Formula XI-a can be prepared in a yield of at least about 50%, 55, 60, 65, 70, 75, 80, 85, 90 or at least about 95%.
[0140] The method of the present invention can provide the compound of Formula XI-a in any suitable purity. For example, the compound of Formula XI-a can be prepared in a purity of at least about 90, 95, 96, 97, 98 or at least about 99%. In some embodiments, the compound of Formula XI-a can be prepared in at least about 95% purity. In some embodiments, the compound of Formula XI-a can be prepared in at least about 98% purity. In some embodiments, the compound of Formula XI-a can be prepared in at least about 99% purity.
[0141] In some embodiments, the method of the present invention can be performed as a batch mode process. In some embodiments, the method of the present invention can be performed as a flow process.
[0142] In some embodiments, the method comprises preparing the compound of Formula XI-a:
##STR00055##
wherein the method comprises forming the reaction mixture having TFA, TMSCN, TMSOTf and the compound of Formula Va or Formula V-b:
##STR00056##
under conditions suitable to prepare the compound of Formula XI-a. In certain embodiments, the method of preparing Formula XI-a is performed between about -120.degree. C. and about 20.degree. C. In another embodiment, the method of preparing Formula XI-a is performed between about -120.degree. C. and about 0.degree. C. In another embodiment, the method of preparing Formula XI-a is performed between about -40.degree. C. and about -20.degree. C.
[0143] In some embodiments, the present invention provides a method of preparing a compound of Formula XI-a.sup.2:
##STR00057##
wherein the method comprises forming a reaction mixture having a cyanating agent, a Lewis Acid, a Broenstedt acid, solvent, and the compound of Formula V-a:
##STR00058##
under conditions suitable to prepare the compound of Formula XI-a.sup.2, wherein R.sup.b is independently a hydroxy protecting group, alternatively, two R.sup.b groups on adjacent carbons can be combined to form a --C(R.sup.19).sub.2 group, R.sup.10 is H or a silyl group, and R.sup.19 is H, C.sub.1-C.sub.8 alkyl, phenyl or substituted phenyl.
[0144] Any suitable cyanating agent can be used in the method of making the compound of Formula XI-a.sup.2. For example, the cyanating agent can be TMSCN, TBSCN, TESCN, HCN, KCN, NaCN, 4-toluenesulfonyl cyanide, CuCN, CuCn*LiCl, LiCN, Zn(CN).sub.2, K.sub.4[Fe(CN).sub.6], tetrabutylammonium cyanide, tetrmethylammonium cyanide, tetraethylammonium cyanide, tetrabutylammonium cyanide, (including tetraalkylammonium cyanide with alkyl independently being Me, Et, Pr, iPr, Bu, iBu, tertBu, Pent, Hex), tributyltn cyanide, trimethyltin cyanide, triethyltin cyanide, tripropyltin cyanide, (including trialkyltin cyanide cyanide with alkyl independently being Me, Et, Pr, iPr, Bu, iBu, tertBu, Pent, Hex), 2-hydroxy-2-methylpropanenitrile; or combinations thereof. In some embodiments, the cyanating agent can be TMSCN.
[0145] The cyanating agent can be present in any suitable amount. For example, the cyanating agent can be present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula V-a, such as about 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The cyanating agent can also be present in an amount of from about 0.1 to about 10.0 eq. (mol/mol) to the compound of Formula V-a, such as of from about 0.1 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the cyanating agent can be present in an amount from about 0.1 to 1.0 eq. (mol/mol) to the compound of Formula V-a. In some embodiments, the cyanating agent can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula V-a.
[0146] Any suitable Lewis Acid can be used in the method of making the compound of Formula XI-a.sup.2. For example, the Lewis Acid can be TMSOTf, TMSOTf, TBSOTf, TESOTf, BF.sub.3, BF.sub.3-OEt.sub.2, BCl.sub.3, BF.sub.3-THF, MgCl.sub.2, MgI.sub.2, MgBr.sub.2, MgBr.sub.2-OEt.sub.2, ZnCl.sub.2, ZnBr.sub.2, ZnI.sub.2, LiCl, LiBr, LiI, AlCl.sub.3, AlBr.sub.3, AlI.sub.3, Me.sub.2Si(OTf).sub.2, Et.sub.2Si(OTf).sub.2, Pr.sub.2Si(OTf).sub.2, iPr.sub.2Si(OTf).sub.2, (tBu).sub.2Si(OTf).sub.2, (C.sub.6F.sub.5).sub.3B, MeSiCl.sub.3, Me.sub.2SiCl.sub.2, SiCl.sub.4, TMSCl, TMSI, TMSVr, TBSCl, TBSBr, TBSI, TESCl, TESBr, TESI, SmCl.sub.3, SmBr.sub.3, SmI.sub.2, SmI.sub.3, ScI.sub.3, ScBr.sub.3, ScI.sub.3, Sm(OTf).sub.3, Sc(OTf).sub.3, TiCl.sub.4, Ti(OiPr).sub.4, Ti(OiPr).sub.3Cl, Ti(OiPr).sub.2Cl.sub.2, Ti(OiPr)Cl.sub.3, Zn(BF.sub.4).sub.2, LiBF.sub.4, Mg(BF.sub.4).sub.2, ZrCl.sub.4, FeCl.sub.2, FeCl.sub.3, FeBr.sub.2, FeBr.sub.3, FeI.sub.2, FeI.sub.3, Cu(OTf), Cu(OTf).sub.2, 4-toluenesulfonylchoride, benzenesulfonylchlopride, 4-toluenesulfonyl triflate, benzenesulfonyl triflate, methylsulfonyl chloride, methylsulfonic anhydrate, InCl.sub.3, InBr.sub.3, InI.sub.3, In(OTf).sub.3, Mg(SO.sub.4).sub.2, NaSO.sub.4; or combinations thereof. In some embodiments, the Lewis Acid can be TMSOTf. In some embodiments, the following may be used in the method of making the compound of Formula XI-a.sup.2 instead of a Lewis Acid: dicyclohexylcarbodiimide, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide, benzenesulfonic acid, HCl, 4-toluenesulfonic acid, triflic acid, trifluoroacetic acid, 4-nitrobenzolic acid, methylsoulfonic acid, sulfuric acid, phosphoric acid, HBr, acetic acid, formic acid, HI; or combinations thereof.
[0147] The Lewis Acid can be present in any suitable amount. For example, the Lewis Acid can be present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula V-a, such as about 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The Lewis Acid can also be present in an amount of from about 0.1 to about 10.0 eq. (mol/mol) to the compound of Formula V-a, such as of from about 0.1 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the Lewis Acid can be present in an amount from about 0.1 to 1.0 eq. (mol/mol) to the compound of Formula V-a. In some embodiments, the Lewis Acid can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula V-a.
[0148] Any suitable Broenstedt acid can be used in the method of making the compound of Formula XI-a.sup.2. For example, the Broenstedt acid can be TFA, benzenesulfonic acid, HCl, 4-toluenesulfonic acid, triflic acid, trifluoroacetic acid, 4-nitrobenzoic acid, methylsoulfonic acid, sulfuric acid, phosphoric acid, HBr, acetic acid, formic acid, HI, trifluoromethylsulfonic acid, 4-fluorobenzoic acid, pivalic acid, HBF.sub.4, nitric acid, 4-chloro-benzoic acid, pentafluorophenol, HPF.sub.6, Camphorsulfonic acid; or combinations thereof. In some embodiments, the Broenstedt acid can be TFA.
[0149] The Broenstedt acid can be present in any suitable amount. For example, the Broenstedt acid can be present in an amount of at least about 0.1 eq. (mol/mol) to the compound of Formula V-a, such as about 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The Broenstedt acid can also be present in an amount of from about 0.1 to about 10.0 eq. (mol/mol) to the compound of Formula V-a, such as of from about 0.1 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the Broenstedt acid can be present in an amount from about 0.1 to about 1.0 eq. (mol/mol) to the compound of Formula V-a. In some embodiments, the Broenstedt acid can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula V-a.
[0150] Any suitable solvent can be used in the method of making the compound of Formula XI or XI-a.sup.2. For example, the solvent can be DCM, THF, MeTHF, Et.sub.2O, MeCN, EtCN, toluene, benzene, chlorobenzene, nitrobenzene, flurorbenzene, methanol, ethanol, 2-propanol, propanol, butanol, MTBE, EtOAc, iPrOAc, Me2O, (TMS)2O, acetone, 2-butanone, chloroform, 1,2-dichloroethane, diglyme, dioxane, acetic acid, formic acid, trifluoroacetic acid, methylisobutylketone, DMAc, DMF, NMP, DMSO; or combinations thereof. In some embodiments, the solvent can be DCM.
[0151] The solvent can be present in any suitable amount. For example, the solvent can be present in an amount of at least 0.0 eq. (mol/mol) to the compound of Formula V-a, such as about 0.0, 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The solvent can also be present in an amount of from about 0.0 to about 10.0 eq. (mol/mol) to the compound of Formula V-a, such as of from about 0.0 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the solvent can be present in an amount from about 0.1 to about 1.0 eq. (mol/mol) to the compound of Formula V-a. In some embodiments, the solvent can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula V-a.
[0152] The reaction mixture of the method can be at any suitable temperature. For example, the temperature of the reaction mixture can be of from about -150.degree. C. to about 0.degree. C., or of from about -120.degree. C. to about 0.degree. C., or of from about -100.degree. C. to about 0.degree. C., or of from about -100.degree. C. to about -50.degree. C., or of from about -100.degree. C. to about -70.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about -120.degree. C. to about -70.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about -120.degree. C. to about -100.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about -80.degree. C. to about -30.degree. C.
[0153] The reaction mixture of the method can be at any suitable pressure. For example, the reaction mixture can be at atmospheric pressure. The reaction mixture can be also be exposed to any suitable environment, such as atmospheric gasses, or inert gasses such as nitrogen or argon.
[0154] The method of the present invention can provide the compound of Formula XI-a.sup.2 in any suitable yield. For example, the compound of Formula XI-a.sup.2 can be prepared in a yield of at least about 50%, 55, 60, 65, 70, 75, 80, 85, 90 or at least about 95%.
[0155] The method of the present invention can provide the compound of Formula XI-a.sup.2 in any suitable purity. For example, the compound of Formula XI-a.sup.2 can be prepared in a purity of at least about 90, 95, 96, 97, 98 or at least about 99%. In some embodiments, the compound of Formula XI-a.sup.2 can be prepared in at least about 95% purity. In some embodiments, the compound of Formula XI-a.sup.2 can be prepared in at least about 98% purity. In some embodiments, the compound of Formula XI-a.sup.2 can be prepared in at least about 99% purity.
[0156] In some embodiments, the method of the present invention can be performed as a batch mode process. In some embodiments, the method of the present invention can be performed as a flow process.
[0157] In some embodiments, the method comprises preparing the compound of Formula XI-a.sup.2:
##STR00059##
wherein the method comprises forming the reaction mixture having TFA, TMSCN, TMSOTf and the compound of Formula Va:
##STR00060##
under conditions suitable to prepare the compound of Formula XI-a.sup.2. In certain embodiments, the method of preparing Formula XI-a.sup.2 is performed between about -120.degree. C. and about 20.degree. C. In another embodiment, the method of preparing Formula XI-a.sup.2 is performed between about -120.degree. C. and about 0.degree. C. In another embodiment, the method of preparing Formula XI-a.sup.2 is performed between about -40.degree. C. and about -20.degree. C.
[0158] In some embodiments, the present invention provides a method of preparing a compound of Formula XI-b:
##STR00061##
wherein the method comprises forming a reaction mixture having a Lewis Acid, a base, a solvent, a filtering agent, and the compound of Formula XI-a
##STR00062##
under conditions suitable to prepare the compound of Formula XI-b.
[0159] Any suitable Lewis Acid can be used in the method of making the compound of Formula XI-b. For example, the Lewis Acid can be TMSOTf, TMSOTf, TBSOTf, TESOTf, BF.sub.3, BF.sub.3-OEt.sub.2, BCl.sub.3, BF.sub.3-THF, MgCl.sub.2, MgI.sub.2, MgBr.sub.2, MgBr.sub.2-OEt.sub.2, ZnCl.sub.2, ZnBr.sub.2, ZnI.sub.2, LiCl, LiBr, LiI, AlCl.sub.3, AlBr.sub.3, AlI.sub.3, Me.sub.2Si(OTf).sub.2, Et.sub.2Si(OTf).sub.2, Pr.sub.2Si(OTf).sub.2, iPr.sub.2Si(OTf).sub.2, (tBu).sub.2Si(OTf).sub.2, (C.sub.6F.sub.5).sub.3B, MeSiCl.sub.3, Me.sub.2SiCl.sub.2, SiCl.sub.4, TMSCl, TMSI, TMSVr, TBSCl, TBSBr, TBSI, TESCl, TESBr, TESI, SmCl.sub.3, SmBr.sub.3, SmI.sub.2, SmI.sub.3, ScI.sub.3, ScBr.sub.3, ScI.sub.3, Sm(OTf).sub.3, Sc(OTf).sub.3, TiCl.sub.4, Ti(OiPr).sub.4, Ti(OiPr).sub.3Cl, Ti(OiPr).sub.2Cl.sub.2, Ti(OiPr)Cl.sub.3, Zn(BF.sub.4).sub.2, LiBF.sub.4, Mg(BF.sub.4).sub.2, ZrCl.sub.4, FeCl.sub.2, FeCl.sub.3, FeBr.sub.2, FeBr.sub.3, FeI.sub.2, FeI.sub.3, Cu(OTf), Cu(OTf).sub.2, 4-toluenesulfonylchoride, benzenesulfonylchlopride, 4-toluenesulfonyl triflate, benzenesulfonyl triflate, methylsulfonyl chloride, methylsulfonic anhydrate, InCl.sub.3, InBr.sub.3, InI.sub.3, In(OTf).sub.3, Mg(SO.sub.4).sub.2, NaSO.sub.4; or combinations thereof. In some embodiments, the Lewis Acid can be BCL.sub.3. In some embodiments, the following may be used in the method of making the compound of Formula XI-b instead of a Lewis Acid: dicyclohexylcarbodiimide, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide, benzenesulfonic acid, HCl, 4-toluenesulfonic acid, triflic acid, trifluoroacetic acid, 4-nitrobenzolic acid, methylsoulfonic acid, sulfuric acid, phosphoric acid, HBr, acetic acid, formic acid, HI; or combinations thereof.
[0160] The Lewis Acid can be present in any suitable amount. For example, the Lewis Acid can be present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula XI-a, such as about 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The Lewis Acid can also be present in an amount of from about 0.1 to about 10.0 eq. (mol/mol) to the compound of Formula XI-a, such as of from about 0.1 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the Lewis Acid can be present in an amount from about 0.1 to 1.0 eq. (mol/mol) to the compound of Formula XI-a. In some embodiments, the Lewis Acid can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula XI-a.
[0161] Any suitable base can be used in the method of making the compound of Formula XI-b. For example, the base can be (C.sub.1-8Alkyl).sub.3N. In some embodiments, the base can be Et.sub.3N.
[0162] The base can be present in any suitable amount. For example, the base can be present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula XI-a, such as about 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The base can also be present in an amount of from about 0.1 to about 10.0 eq. (mol/mol) to the compound of Formula XI-a, such as of from about 0.1 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the base can be present in an amount from about 0.1 to 1.0 eq. (mol/mol) to the compound of Formula XI-a. In some embodiments, the base can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula XI-a.
[0163] Any suitable solvent can be used in the method of making the compound of Formula XI-b. For example, the solvent can be MeOH, DCM, THF, MeTHF, Et.sub.2O, MeCN, EtCN, toluene, benzene, chlorobenzene, nitrobenzene, flurorbenzene, methanol, ethanol, 2-propanol, propanol, butanol, MTBE, EtOAc, iPrOAc, Me2O, (TMS)2O, acetone, 2-butanone, chloroform, 1,2-dichloroethane, diglyme, dioxane, acetic acid, formic acid, trifluoroacetic acid, methylisobutylketone, DMAc, DMF, NMP, DMSO; or combinations thereof. In some embodiments, the solvent can be MeOH.
[0164] The solvent can be present in any suitable amount. For example, the solvent can be present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula XI-a, such as about 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The solvent can also be present in an amount of from about 0.1 to about 10.0 eq. (mol/mol) to the compound of Formula XI-a, such as of from about 0.1 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the solvent can be present in an amount from about 0.1 to about 1.0 eq. (mol/mol) to the compound of Formula XI-a. In some embodiments, the solvent can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula XI-a.
[0165] Any suitable filtering agent can be used in the method of making the compound of Formula XI-b. For example, the filtering agent can be silica gel, Celite.RTM. or combinations thereof. In some embodiments, the filtering agent can be Celite.RTM..
[0166] The filtering agent can be present in any suitable amount. For example, the filtering agent can be present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula XI-a, such as about 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The filtering agent can also be present in an amount of from about 0.1 to about 10.0 eq. (mol/mol) to the compound of Formula XI-a, such as of from about 0.1 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the filtering agent can be present in an amount from about 0.1 to about 1.0 eq. (mol/mol) to the compound of Formula XI-a. In some embodiments, the filtering agent can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula XI-a.
[0167] The reaction mixture of the method can be at any suitable temperature. For example, the temperature of the reaction mixture can be of from about -50.degree. C. to about 0.degree. C., or of from about -40.degree. C. to about 0.degree. C., or of from about -30.degree. C. to about 0.degree. C., or of from about -20.degree. C. to about 0.degree. C., or of from about -20.degree. C. to about -10.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about -30.degree. C. to about 0.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about -20.degree. C. to about -10.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about -25.degree. C. to about -15.degree. C.
[0168] The reaction mixture of the method can be at any suitable pressure. For example, the reaction mixture can be at atmospheric pressure. The reaction mixture can be also be exposed to any suitable environment, such as atmospheric gasses, or inert gasses such as nitrogen or argon.
[0169] The method of the present invention can provide the compound of Formula XI-b in any suitable yield. For example, the compound of Formula XI-b can be prepared in a yield of at least about 50%, 55, 60, 65, 70, 75, 80, 85, 90 or at least about 95%.
[0170] The method of the present invention can provide the compound of Formula XI-b in any suitable purity. For example, the compound of Formula XI-b can be prepared in a purity of at least about 90, 95, 96, 97, 98 or at least about 99%. In some embodiments, the compound of Formula XI-b can be prepared in at least about 95% purity. In some embodiments, the compound of Formula XI-b can be prepared in at least about 98% purity. In some embodiments, the compound of Formula XI-b can be prepared in at least about 99% purity.
[0171] In some embodiments, the present invention provides a method of preparing a compound of Formula XI-b:
##STR00063##
wherein the method comprises forming a reaction mixture having BCL.sub.3, Et.sub.2N, MeOH, Celite.RTM., and the compound of Formula XI-a
##STR00064##
under conditions suitable to prepare the compound of Formula XI-b. In certain embodiments, the method of preparing Formula XI-b is performed between about -30.degree. C. and about 0.degree. C. In another embodiment, the method of preparing Formula XI is performed between about -20.degree. C. and about 0.degree. C.
[0172] In some embodiments, the present invention provides a method of preparing a compound of Formula XI-c:
##STR00065##
wherein the method comprises forming a reaction mixture having a solvent, a reagent, an acid, and the compound of Formula XI-b
##STR00066##
under conditions suitable to prepare the compound of Formula XI-c.
[0173] Any suitable solvent can be used in the method of making the compound of Formula XI-c. For example, the solvent can be acetone, MeOH, DCM, THF, MeTHF, Et.sub.2O, MeCN, EtCN, toluene, benzene, chlorobenzene, nitrobenzene, flurorbenzene, methanol, ethanol, 2-propanol, propanol, butanol, MTBE, EtOAc, iPrOAc, Me2O, (TMS)2O, acetone, 2-butanone, chloroform, 1,2-dichloroethane, diglyme, dioxane, acetic acid, formic acid, trifluoroacetic acid, methylisobutylketone, DMAc, DMF, NMP, DMSO; or combinations thereof. In some embodiments, the solvent can be acetone.
[0174] The solvent can be present in any suitable amount. For example, the solvent can be present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula XI-b, such as about 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The solvent can also be present in an amount of from about 0.1 to about 10.0 eq. (mol/mol) to the compound of Formula XI-b, such as of from about 0.1 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the solvent can be present in an amount from about 0.1 to 1.0 eq. (mol/mol) to the compound of Formula XI-b. In some embodiments, the solvent can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula XI-b.
[0175] Any suitable reagent can be used in the method of making the compound of Formula XI-c. For example, the reagent can be 2,2-dimethoxypropane, acetone, 2-methoxypropene, 2,2-diethylpropane, 2-ethoxypropene, 2,2-dimethyl-1,3-dioxolane, 2,2-dimethyl-1,3-dioxane; or combinations thereof. In some embodiments, the reagent can be 2,2-dimethoxypropane.
[0176] The reagent can be present in any suitable amount. For example, the reagent can be present in an amount of at least 0.1 eq. (mol/mol) to the compound of Formula XI-b, such as about 0.1, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The reagent can also be present in an amount of from about 0.1 to about 10.0 eq. (mol/mol) to the compound of Formula XI-b, such as of from about 0.1 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the reagent can be present in an amount from about 0.1 to 1.0 eq. (mol/mol) to the compound of Formula XI-b. In some embodiments, the reagent can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula XI-b.
[0177] Any suitable acid can be used in the method of making the compound of Formula XI-c. For example, the acid can be TMSOTf, TMSOTf, TBSOTf, TESOTf, BF.sub.3, BF.sub.3-OEt.sub.2, BCl.sub.3, BF.sub.3-THF, MgCl.sub.2, MgI.sub.2, MgBr.sub.2, MgBr.sub.2-OEt.sub.2, ZnCl.sub.2, ZnBr.sub.2, ZnI.sub.2, LiCl, LiBr, LiI, AlCl.sub.3, AlBr.sub.3, AlI.sub.3, Me.sub.2Si(OTf).sub.2, Et.sub.2Si(OTf).sub.2, Pr.sub.2Si(OTf).sub.2, iPr.sub.2Si(OTf).sub.2, (tBu).sub.2Si(OTf).sub.2, (C.sub.6F.sub.5).sub.3B, MeSiCl.sub.3, Me.sub.2SiCl.sub.2, SiCl.sub.4, TMSCl, TMSI, TMSVr, TBSCl, TBSBr, TBSI, TESCl, TESBr, TESI, SmCl.sub.3, SmBr.sub.3, SmI.sub.2, SmI.sub.3, ScI.sub.3, ScBr.sub.3, ScI.sub.3, Sm(OTf).sub.3, Sc(OTf).sub.3, TiCl.sub.4, Ti(OiPr).sub.4, Ti(OiPr).sub.3Cl, Ti(OiPr).sub.2Cl.sub.2, Ti(OiPr)Cl.sub.3, Zn(BF.sub.4).sub.2, LiBF.sub.4, Mg(BF.sub.4).sub.2, ZrCl.sub.4, FeCl.sub.2, FeCl.sub.3, FeBr.sub.2, FeBr.sub.3, FeI.sub.2, FeI.sub.3, Cu(OTf), Cu(OTf).sub.2, 4-toluenesulfonylchoride, benzenesulfonylchlopride, 4-toluenesulfonyl triflate, benzenesulfonyl triflate, methylsulfonyl chloride, methylsulfonic anhydrate, InCl.sub.3, InBr.sub.3, InI.sub.3, In(OTf).sub.3, Mg(SO.sub.4).sub.2, NaSO.sub.4, dicyclohexylcarbodiimide, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide, benzenesulfonic acid, HCl, 4-toluenesulfonic acid, triflic acid, trifluoroacetic acid, 4-nitrobenzolic acid, methylsoulfonic acid, sulfuric acid, phosphoric acid, HBr, acetic acid, formic acid, HI, TFA, benzenesulfonic acid, HCl, 4-toluenesulfonic acid, triflic acid, trifluoroacetic acid, 4-nitrobenzoic acid, methylsoulfonic acid, sulfuric acid, phosphoric acid, HBr, acetic acid, formic acid, HI, trifluoromethylsulfonic acid, 4-fluorobenzoic acid, pivalic acid, HBF.sub.4, nitric acid, 4-chloro-benzoic acid, pentafluorophenol, HPF.sub.6, Camphorsulfonic acid; or combinations thereof. In some embodiments, the acid can be sulfuric acid.
[0178] The acid can be present in any suitable amount. For example, the acid can be present in an amount of at least 0.0 eq. (mol/mol) to the compound of Formula XI-b, such as about 0.0, 0.5, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, or about 10.0 eq. (mol/mol). The acid can also be present in an amount of from about 0.0 to about 10.0 eq. (mol/mol) to the compound of Formula XI-b, such as of from about 0.0 to about 3.0 eq. (mol/mol), or of from about 1.0 to about 2.0 eq. (mol/mol). In some embodiments, the acid can be present in an amount from about 0.0 to 1.0 eq. (mol/mol) to the compound of Formula XI-b. In some embodiments, the acid can be present in an amount of from about 1.0 to about 2.0 eq. (mol/mol) to the compound of Formula XI-b.
[0179] The reaction mixture of the method can be at any suitable temperature. For example, the temperature of the reaction mixture can be of from about -50.degree. C. to about 50.degree. C., or of from about 0.degree. C. to about 50.degree. C., or of from about 0.degree. C. to about 40.degree. C., or of from about 0.degree. C. to about 30.degree. C., or of from about 0.degree. C. to about 25.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about 0.degree. C. to about 23.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about 0.degree. C. to about 25.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about 0.degree. C. to about 30.degree. C.
[0180] The reaction mixture of the method can be at any suitable pressure. For example, the reaction mixture can be at atmospheric pressure. The reaction mixture can be also be exposed to any suitable environment, such as atmospheric gasses, or inert gasses such as nitrogen or argon.
[0181] The method of the present invention can provide the compound of Formula XI-c in any suitable yield. For example, the compound of Formula XI-c can be prepared in a yield of at least about 50%, 55, 60, 65, 70, 75, 80, 85, 90 or at least about 95%.
[0182] The method of the present invention can provide the compound of Formula XI-c in any suitable purity. For example, the compound of Formula XI-c can be prepared in a purity of at least about 90, 95, 96, 97, 98 or at least about 99%. In some embodiments, the compound of Formula XI-b can be prepared in at least about 95% purity. In some embodiments, the compound of Formula XI-b can be prepared in at least about 98% purity. In some embodiments, the compound of Formula XI-b can be prepared in at least about 99% purity.
[0183] In some embodiments, the method comprises preparing the compound of Formula XI-c:
##STR00067##
wherein the method comprises forming a reaction mixture having acetone, 2,2-dimethoxypropane, sulfuric acid, and the compound of Formula XI-b
##STR00068##
under conditions suitable to prepare the compound of Formula XI-c. In certain embodiments, the method of preparing Formula XI-c is performed between about 0.degree. C. and about 30.degree. C. In another embodiment, the method of preparing Formula XI is performed between about 10.degree. C. and about 30 C.
[0184] C. Addition of Prodrug Moiety
[0185] The present invention also provides a method of coupling a prodrug moiety to a nucleoside to provide a compound of the present invention. In some embodiments, the present invention provides a method of preparing a compound of Formula VIII:
##STR00069##
wherein the method includes forming a reaction mixture including a coupling agent, a non-nucleophilic base, a compound of Formula IX:
##STR00070##
and a compound of Formula X:
##STR00071##
under conditions suitable to form the compound of Formula VIII, wherein each R.sup.a is H or PG, each PG group is a hydroxy protecting group, or both PG groups are combined to form --C(R.sup.19).sub.2--, R.sup.e1 and R.sup.e2 are each independently H, C.sub.1-C.sub.6 alkyl or benzyl, R.sup.f is H, C.sub.1-C.sub.8 alkyl, benzyl, C.sub.3-C.sub.6 cycloalkyl, or --CH.sub.2--C.sub.3-C.sub.6 cycloalkyl, R.sup.19 is H, C.sub.1-C.sub.8 alkyl, phenyl or substituted phenyl, and LG is a leaving group.
[0186] Any suitable coupling agent can be used in the method of making the compound of Formula VIII, as described above for the method of making the compound of Formula V. In some embodiments, the coupling agent can be a magnesium coupling agent. In some embodiments, the coupling agent can be MgCl.sub.2, iPrMgCl, tBuMgCl, PhMgCl, or combinations thereof. In some embodiments, the coupling agent can be MgCl.sub.2.
[0187] Any suitable non-nucleophilic base can be used in the method of making the compound of Formula VIII. Representative non-nucleophilic bases include, but are not limited to, triethylamine, diisopropylethyl amine, N,N-diethylaniline, pyridine, 2,6-lutidine, 2,4,6-collidine, 4-dimethylaminopyridine, and quinuclidine. In some embodiments, the non-nucleophilic base can be di-isopropyl ethyl amine (DIPEA).
[0188] The protecting groups PG can be any suitable hydroxy protecting groups, as described above for the method of making the compound of Formula V. Exemplary protecting groups PG can be benzyl, or the PG groups can be combined to form an acetonide. Exemplary acetonides include, but are not limited to acetonide and benzylidene acetal. In some embodiments, the hydroxy protecting groups of hydroxy groups on adjacent carbons can be combined to form acetonide. In some embodiments, the PG groups are combined to form --C(R.sup.19).sub.2--. In some embodiments, each R.sup.a is the protecting group PG where the PG groups are combined to form --C(Me).sub.2-.
[0189] When the R.sup.e group is C.sub.1-C.sub.8 alkyl, each R.sup.e can be methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, sec-buty, t-butyl, pentyl, iso-pentyl, neo-pentyl, hexyl, isohexyl, neohexyl, septyl or octyl. In some embodiments, each R.sup.e group can be methyl.
[0190] When the R.sup.f group is C.sub.1-C.sub.8 alkyl, R.sup.f can be methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, sec-buty, t-butyl, pentyl, iso-pentyl, neo-pentyl, hexyl, isohexyl, neohexyl, septyl or octyl. In some embodiments, the R.sup.f group can be methyl, ethyl, isopropyl, t-butyl, or iso-hexyl. When the R.sup.f group is C.sub.3-C.sub.6 cycloalkyl, R.sup.f can be cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl. In some embodiments, R.sup.f can be cyclobutyl, cyclopentyl or cyclohexyl.
[0191] When the R.sup.19 group is C.sub.1-C.sub.8 alkyl, R.sup.19 can be methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, sec-buty, t-butyl, pentyl, iso-pentyl, neo-pentyl, hexyl, isohexyl, neohexyl, septyl or octyl. In some embodiments, the R.sup.19 group can be methyl.
[0192] The leaving group can be any suitable leaving group. Suitable leaving groups LG include, but are not limited to, chloride, bromide, mesylate, tosylate, triflate, 4-nitrobenzenesulfonate, 4-chlorobenzenesulfonate, 4-nitrophenoxy, pentafluorophenoxy, etc. In some embodiments, the leaving group LG can be 4-nitrophenoxy or pentafluorophenoxy. In some embodiments, the leaving group LG can be 4-nitrophenoxy.
[0193] In some embodiments, each R.sup.a is PG where the PG groups are combined to form --C(R.sup.19).sub.2--, R.sup.f is C.sub.1-C.sub.8 alkyl, R.sup.19 is C.sub.1-C.sub.8 alkyl, and the leaving group LG is 4-nitrophenoxy or pentafluorophenoxy.
[0194] In some embodiments, the coupling agent is MgCl.sub.2, and the non-nucleophilic base is di-isopropyl ethyl amine.
[0195] In some embodiments, the compound of Formula VIII can be
##STR00072##
In some embodiments, the compound of Formula VIII can be
##STR00073##
In some embodiments, the compound of Formula VIII can be
##STR00074##
[0196] In some embodiments, the method of making the compound Formula VIII includes forming the reaction mixture including MgCl.sub.2, DIPEA, the compound of Formula IX:
##STR00075##
and the compound of Formula X:
##STR00076##
under conditions suitable to form the compound of Formula VIII:
##STR00077##
[0197] When the R.sup.a groups of the compound of Formula VIII are the hydroxy protecting groups PG, the method can include the additional step of removing the protecting groups to form the compound of Formula VIII where each R.sup.a is H. In some embodiments, the method of preparing the compound of Formula VIII includes forming a second reaction mixture including a deprotection agent and the compound Formula VIII wherein each R.sup.a group is the protecting group PG, under suitable conditions to form the compound of Formula VIII where each R.sup.a is H. The deprotection agent can be any suitable agent to remove the protecting groups PG such as hydrogen and a hydrogenation catalyst, or acid. For example, if the protecting group PG is benzyl, the deprotection agent can be hydrogen and platinum on carbon. Alternatively, when the protecting group PG is an acetonide, the deprotection agent can be an acid. Representative acids include, but are not limited to, acetic acid, glacial acetic acid, trifluoroacetic acid (TFA), hydrochloric acid, concentrated hydrochloric acid, and others. In some embodiments, the method of preparing the compound of Formula VIII includes forming a second reaction mixture including an acid and the compound Formula VIII wherein the R.sup.a groups are combined to form --C(R.sup.19).sub.2--, under suitable conditions to form the compound of Formula VIII where each R.sup.a is H. In some embodiments, the acid can be hydrochloric acid.
[0198] Any suitable solvent can be used in the method of the present invention. Representative solvents include, but are not limited to, pentane, pentanes, hexane, hexanes, heptane, heptanes, petroleum ether, cyclopentanes, cyclohexanes, benzene, toluene, xylene, trifluoromethylbenzene, halobenzenes such as chlorobenzene, fluorobenzene, dichlorobenzene and difluorobenzene, methylene chloride, chloroform, acetone, ethyl acetate, diethyl ether, tetrahydrofuran, acetonitrile, or combinations thereof. In some embodiments, the solvent can be acetonitrile.
[0199] The reaction mixture of the method can be at any suitable temperature. For example, the temperature of the reaction mixture can be of from about -78.degree. C. to about 100.degree. C., or of from about -50.degree. C. to about 100.degree. C., or of from about -25.degree. C. to about 50.degree. C., or of from about -10.degree. C. to about 25.degree. C., or of from about 0.degree. C. to about 20.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about 0.degree. C. to about 20.degree. C.
[0200] The reaction mixture of the method can be at any suitable pressure. For example, the reaction mixture can be at atmospheric pressure. The reaction mixture can be also be exposed to any suitable environment, such as atmospheric gasses, or inert gasses such as nitrogen or argon.
[0201] The method of the present invention can provide the compound of Formula VIII in any suitable yield. For example, the compound of Formula VIII can be prepared in a yield of at least about 50%, 55, 60, 65, 70, 75, 80, 85, 90 or at least about 95%.
[0202] The method of the present invention can provide the compound of Formula VIII in any suitable purity. For example, the compound of Formula VIII can be prepared in a purity of at least about 90, 95, 96, 97, 98 or at least about 99%. In some embodiments, the compound of Formula VIII can be prepared in at least 95% purity. In some embodiments, the compound of Formula VIII can be prepared in at least 98% purity. In some embodiments, the compound of Formula VIII can be prepared in at least 99% purity.
[0203] In some embodiments, the present invention provides the compound
##STR00078##
[0204] In some embodiments, the present invention provides a method of preparing a compound of Formula VIII:
##STR00079##
wherein the method comprises forming a reaction mixture including a coupling agent, a non-nucleophilic base, a compound of Formula IX-a:
##STR00080##
and a compound of Formula X:
##STR00081##
under conditions suitable to form the compound of Formula VIII, wherein R.sup.a is independently H or a hydroxy protecting group, or two R.sup.a on adjacent carbons can be combined to form a --C(R.sup.19).sub.2-- group, R.sup.35 is independently H or a hydroxy protecting group, or two R.sup.35 on adjacent carbons can be combined to form a --C(R.sup.19).sub.2-- group, R.sup.19 is H or C.sub.1-C.sub.8 alkyl, R.sup.e1 and R.sup.e2 are each independently H, C.sub.1-C.sub.6 alkyl or benzyl, R.sup.f is H, C.sub.1-C.sub.8 alkyl, benzyl, C.sub.3-C.sub.6 cycloalkyl, or --CH.sub.2--C.sub.3-C.sub.6 cycloalkyl, R.sup.19 is H, C.sub.1-C.sub.8 alkyl, phenyl or substituted phenyl, and LG is a leaving group.
[0205] Any suitable coupling agent can be used in the method of making the compound of Formula VIII, as described above for the method of making the compound of Formula V. In some embodiments, the coupling agent can be a magnesium coupling agent. In some embodiments, the coupling agent can be MgCl.sub.2, iPrMgCl, tBuMgCl, PhMgCl, or combinations thereof. In some embodiments, the coupling agent can be MgCl.sub.2.
[0206] Any suitable non-nucleophilic base can be used in the method of making the compound of Formula VIII. Representative non-nucleophilic bases include, but are not limited to, triethylamine, diisopropylethyl amine, N,N-diethylaniline, pyridine, 2,6-lutidine, 2,4,6-collidine, 4-dimethylaminopyridine, and quinuclidine. In some embodiments, the non-nucleophilic base can be di-isopropyl ethyl amine (DIPEA).
[0207] The hydroxy protecting groups, as described above for the method of making the compound of Formula V. Exemplary hydroxy protecting group can be benzyl, SiR.sub.3, wherein each R group can be hydrogen, alkyl, alkenyl, cycloalkyl, phenyl, or other silicon containing groups, or the PG groups can be combined to form an acetonide. Exemplary silanes include, but are not limited to tert-butyldimethylsilyl (TBS). Exemplary acetonides include, but are not limited to acetonide and benzylidene acetal. In some embodiments, the hydroxy protecting groups of hydroxy groups on adjacent carbons can be combined to form acetonide. In some embodiments, the PG groups are combined to form --C(R.sup.19).sub.2--. In some embodiments, each R.sup.a is the protecting group PG where the PG groups are combined to form --C(Me).sub.2-. In other embodiments, PG is a SiR.sub.3. In other embodiments, PG is tert-butyldimethylsilyl (TBS).
[0208] When the R.sup.e group is C.sub.1-C.sub.8 alkyl, each R.sup.e can be methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, sec-buty, t-butyl, pentyl, iso-pentyl, neo-pentyl, hexyl, isohexyl, neohexyl, septyl or octyl. In some embodiments, each R.sup.e group can be methyl.
[0209] When the R.sup.f group is C.sub.1-C.sub.8 alkyl, R.sup.f can be methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, sec-buty, t-butyl, pentyl, iso-pentyl, neo-pentyl, hexyl, isohexyl, neohexyl, septyl or octyl. In some embodiments, the R.sup.f group can be methyl, ethyl, isopropyl, t-butyl, or iso-hexyl. When the R.sup.f group is C.sub.3-C.sub.6 cycloalkyl, R.sup.f can be cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl. In some embodiments, R.sup.f can be cyclobutyl, cyclopentyl or cyclohexyl.
[0210] When the R.sup.19 group is C.sub.1-C.sub.8 alkyl, R.sup.19 can be methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, sec-buty, t-butyl, pentyl, iso-pentyl, neo-pentyl, hexyl, isohexyl, neohexyl, septyl or octyl. In some embodiments, the R.sup.19 group can be methyl.
[0211] When the R.sup.35 group is a hydroxy protecting group, R.sup.35 can be any example protecting group described in Protective Groups in Organic Chemistry, Peter G. M. Wuts and Theodora W. Greene, 4th Ed., 2006. In some embodiments, the R.sup.35 group can be benzyl. In some embodiments, the R.sup.35 group can be TBS.
[0212] The leaving group can be any suitable leaving group. Suitable leaving groups LG include, but are not limited to, chloride, bromide, mesylate, tosylate, triflate, 4-nitrobenzenesulfonate, 4-chlorobenzenesulfonate, 4-nitrophenoxy, pentafluorophenoxy, etc. In some embodiments, the leaving group LG can be 4-nitrophenoxy or pentafluorophenoxy. In some embodiments, the leaving group LG can be 4-nitrophenoxy.
[0213] In some embodiments, each R.sup.a is PG where the PG groups are combined to form --C(R.sup.19).sub.2--, R.sup.f is C.sub.1-C.sub.8 alkyl, R.sup.19 is C.sub.1-C.sub.8 alkyl, and the leaving group LG is 4-nitrophenoxy or pentafluorophenoxy.
[0214] In some embodiments, the coupling agent is MgCl.sub.2, and the non-nucleophilic base is di-isopropyl ethyl amine.
[0215] In some embodiments, the compound of Formula VIII can be
##STR00082##
In some embodiments, the compound of Formula VIII can be
##STR00083##
In some embodiments, the compound of Formula VIII can be
##STR00084##
[0216] In some embodiments, the method of making the compound Formula VIII comprises forming the reaction mixture including MgCl.sub.2, DIPEA, the compound of Formula IX:
##STR00085##
and the compound of Formula X:
##STR00086##
under conditions suitable to form the compound of Formula VIII:
##STR00087##
[0217] When the R.sup.a groups of the compound of Formula VIII are the hydroxy protecting groups PG, the method can include the additional step of removing the protecting groups to form the compound of Formula VIII where each R.sup.a is H. In some embodiments, the method of preparing the compound of Formula VIII comprises forming a second reaction mixture including a deprotection agent and the compound Formula VIII wherein each R.sup.a group is the protecting group PG, under suitable conditions to form the compound of Formula VIII where each R.sup.a is H. The deprotection agent can be any suitable agent to remove the protecting groups PG such as hydrogen and a hydrogenation catalyst, or acid. For example, if the protecting group PG is benzyl, the deprotection agent can be hydrogen and platinum on carbon. Alternatively, when the protecting group PG is an acetonide, the deprotection agent can be an acid. Representative acids include, but are not limited to, acetic acid, glacial acetic acid, trifluoroacetic acid (TFA), hydrochloric acid, concentrated hydrochloric acid, formic acids, toluenesulfonic acid, sulfuric acid, and others. Additional representative acids include, but are not limited to those found in Greene, T. W.; Wuts, P. G. M. Protective Groups In Organic Synthesis, 4th Ed., John Wiley & Sons: New York, 2006. In some embodiments, the method of preparing the compound of Formula VIII comprises forming a second reaction mixture including an acid and the compound Formula VIII wherein the R.sup.a groups are combined to form --C(R.sup.19).sub.2--, under suitable conditions to form the compound of Formula VIII where each R.sup.a is H. In some embodiments, the acid can be hydrochloric acid. Alternatively, when the protecting group PG is SiR.sub.3, the deprotection agent can be TBAF, pyridine HF, HCl, TsOH, camphor sulfonic acid, AcCl in MeOH, BF.sup.3 OEt.sup.2, TFA, AGOG, Formic Acid, HBr, F, HF, Et.sub.3N--HF, KF--H.sub.2O, KHF.sub.2, NaF, LiF, LiCl, LiBr, LiI, and others.
[0218] Any suitable solvent can be used in the method of the present invention.
[0219] Representative solvents include, but are not limited to, pentane, pentanes, hexane, hexanes, heptane, heptanes, petroleum ether, cyclopentanes, cyclohexanes, benzene, toluene, xylene, trifluoromethylbenzene, halobenzenes such as chlorobenzene, fluorobenzene, dichlorobenzene and difluorobenzene, methylene chloride, chloroform, acetone, ethyl acetate, diethyl ether, tetrahydrofuran, acetonitrile, or combinations thereof. In some embodiments, the solvent can be acetonitrile. In some embodiments, the solvent can be MeCN. In some embodiments, the solvent can be tetrahydrofuran.
[0220] The reaction mixture of the method can be at any suitable temperature. For example, the temperature of the reaction mixture can be of from about -78.degree. C. to about 100.degree. C., or of from about -50.degree. C. to about 100.degree. C., or of from about -25.degree. C. to about 50.degree. C., or of from about -10.degree. C. to about 25.degree. C., or of from about 0.degree. C. to about 20.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about 0.degree. C. to about 20.degree. C.
[0221] The reaction mixture of the method can be at any suitable pressure. For example, the reaction mixture can be at atmospheric pressure. The reaction mixture can also be exposed to any suitable environment, such as atmospheric gasses, or inert gasses such as nitrogen or argon.
[0222] The method of the present invention can provide the compound of Formula VIII in any suitable yield. For example, the compound of Formula VIII can be prepared in a yield of at least about 50%, 55, 60, 65, 70, 75, 80, 85, 90 or at least about 95%.
[0223] The method of the present invention can provide the compound of Formula VIII in any suitable purity. For example, the compound of Formula VIII can be prepared in a purity of at least about 90, 95, 96, 97, 98 or at least about 99%. In some embodiments, the compound of Formula VIII can be prepared in at least about 95% purity. In some embodiments, the compound of Formula VIII can be prepared in at least about 98% purity. In some embodiments, the compound of Formula VIII can be prepared in at least about 99% purity.
In some embodiments, the compound of Formula VIII can be
##STR00088##
[0224] In some embodiments, the method of making the compound Formula VIII comprises forming the reaction mixture including MgCl.sub.2, DIPEA, the compound of Formula IX-a.sup.2:
##STR00089##
and the compound of Formula X:
##STR00090##
under conditions suitable to form the compound of Formula VIII:
##STR00091##
[0225] The method can include the additional step of removing the protecting groups to form the compound of Formula VIII where each TBS is H.
[0226] The reaction mixture of the method can be at any suitable temperature. For example, the temperature of the reaction mixture can be of from about -78.degree. C. to about 100.degree. C., or of from about -50.degree. C. to about 100.degree. C., or of from about -25.degree. C. to about 50.degree. C., or of from about -10.degree. C. to about 25.degree. C., or of from about 0.degree. C. to about 20.degree. C. In some embodiments, the temperature of the reaction mixture can be of from about 0.degree. C. to about 20.degree. C.
[0227] The reaction mixture of the method can be at any suitable pressure. For example, the reaction mixture can be at atmospheric pressure. The reaction mixture can be also be exposed to any suitable environment, such as atmospheric gasses, or inert gasses such as nitrogen or argon.
[0228] The method of the present invention can provide the compound of Formula VIII in any suitable yield. For example, the compound of Formula VIII can be prepared in a yield of at least about 50%, 55, 60, 65, 70, 75, 80, 85, 90 or at least about 95%.
[0229] The method of the present invention can provide the compound of Formula VIII in any suitable purity. For example, the compound of Formula VIII can be prepared in a purity of at least about 90, 95, 96, 97, 98 or at least about 99%. In some embodiments, the compound of Formula VIII can be prepared in at least about 95% purity. In some embodiments, the compound of Formula VIII can be prepared in at least about 98% purity. In some embodiments, the compound of Formula VIII can be prepared in at least about 99% purity.
[0230] D. Preparation of Formula X-b by Crystallization-Induced Dynamic Resolution
[0231] In one embodiment, there is provided a method for the crystallization-induced dynamic resolution of (2S)-2-ethylbutyl 2-(((4-nitrophenoxy)(phenoxy)phosphoryl)amino)propanoate (Formula X-a):
##STR00092##
to provide (S)-2-ethylbutyl 2-(((S)-(4-nitrophenoxy)(phenoxy)phosphoryl)amino)propanoate (Formula X-b). The method comprises subjecting a solution comprising: a) a suitable solvent; b) a suitable base; c) (2S)-2-ethylbutyl 2-(((4-nitrophenoxy)(phenoxy)phosphoryl)amino) propanoate; and, optionally, d) one or more seed crystals of (S)-2-ethylbutyl 2-(((S)-(4-nitrophenoxy)(phenoxy)phosphoryl)amino)propanoate, to conditions that provide for the epimerization of the phosphorus center, under conditions that also provide selective crystallization of (S)-2-ethylbutyl 2-(((S)-(4-nitrophenoxy)(phenoxy)phosphoryl)amino) propanoate.
[0232] The crystallization can be carried out in any suitable solvent. For example, it can be carried out in an aprotic organic solvent, or in a mixture thereof. For example, the aprotic organic solvent may comprise ethyl acetate, methyl acetate, propyl acetate, isopropyl acetate, diethyl ether, diisopropyl ether, tetrahydrofuran, dichloromethane, acetone, methyl ethyl ketone, methyl tert-butylether, toluene, or acetonitrile, or a mixture thereof. In one embodiment, the solvent comprises acetonitrile.
[0233] The resolution can be carried out in the presence of any suitable base. For example, the resolution can be carried out in the presence of a base selected from 1,5-diazobicyclo[4.3.0]non-5-ene (DBN), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD), triethylamine (Et.sub.3N), Hunig's Base (iPr.sub.2NEt), tetramethylguanidine, a Verkade base (e.g., 2,8,9-triisopropyl-2,5,8,9-tetraaza-1-phosphabicyclo[3.3.3]undecane, and 2,8,9-triisobutyl-2,5,8,9-tetraaza-1-phosphabicyclo [3.3.3]undecane), a metal carbonate (e.g., M.sub.xCO.sub.3), a metal phenoxide (M.sup.+-OPh), and PhOTMS in combination with a fluoride ion source (e.g., R.sub.4N.sup.+-F, TASF (tris(dimethylamino) sulfonium difluorotrimethylsilicate), or TBAT (tetrabutylammonium triphenyldifluorosilicate), and mixtures thereof, wherein each M is a suitable metal such as an alkali metal or an alkaline earth metal, and each R is, for example, a (C.sub.1-C.sub.6) alkyl. In one specific embodiment, the base is DBU.
[0234] The resolution can also be carried out at any suitable temperature, for example, a temperature in the range of from about 0.degree. C. to about 50.degree. C. In one specific embodiment, the resolution is carried out at a temperature of about 0.degree. C.
[0235] In one specific embodiment, the resolution is carried out in the presence of phenol.
[0236] The percentage of (S)-2-ethylbutyl 2-(((S)-(4-nitrophenoxy)(phenoxy)phosphoryl) amino) propanoate in the starting diastereomeric mixture can be anywhere in the range from about 0% to about 99%. In one embodiment of the invention, the percentage of (S)-2-ethylbutyl 2-(((S)-(4-nitrophenoxy)(phenoxy)phosphoryl)amino)propanoate in the starting diastereomeric mixture is in the range from about 0% to about 20%. In one embodiment, the percentage of Compound (S)-2-ethylbutyl 2-(((S)-(4-nitrophenoxy)(phenoxy) phosphoryl)amino)propanoate in the starting diastereomeric mixture is in the range from about 20% to about 99%. In one embodiment, the percentage of Compound (S)-2-ethylbutyl 2-(((S)-(4-nitrophenoxy)(phenoxy)phosphoryl)amino)propanoate in the starting diastereomeric mixture is in the range from about 50% to about 99%. In one embodiment, the final Compound (S)-2-ethylbutyl 2-(((S)-(4-nitrophenoxy)(phenoxy)phosphoryl)amino)propanoate is at least about 90%, about 95%, about 97%, or about 99% diastereomerically pure. In one embodiment, the final Compound (S)-2-ethylbutyl 2-(((S)-(4-nitrophenoxy)(phenoxy)phosphoryl)amino) propanoate contains less than 1% of any diastereomeric impurities. In one embodiment, the final Compound (S)-2-ethylbutyl 2-(((S)-(4-nitrophenoxy)(phenoxy)phosphoryl)amino) propanoate is free of any detectable diastereomeric impurities.
Examples
[0237] Certain abbreviations and acronyms are used in describing the experimental details. Although most of these would be understood by one skilled in the art, Table 1 contains a list of many of these abbreviations and acronyms.
TABLE-US-00001 TABLE 1 List of abbreviations and acronyms. Abbreviation Meaning Ac.sub.2O acetic anhydride AIBN 2,2'-azobis (2-methylpropionitrile) Bn benzyl BnBr benzylbromide BSA bis (trimethylsilyl) acetamide BzCl benzoyl chloride CDI carbonyl diimidazole DABCO 1,4-diazabicyclo[2.2.2]octane DBN 1,5-diazabicyclo[4.3.0]non-5-ene DDQ 2,3-dichloro-5,6-dicyano-1,4-benzoquinone DBU 1,5-diazabicyclo[5.4.0]undec-5-ene DCA dichloroacetamide DCC dicyclohexylcarbodiimide DCM dichloromethane DMAP 4-dimethylaminopyridine DME 1,2-dimethoxyethane DMTCl dimethoxytrityl chloride DMSO dimethylsulfoxide DMTr 4, 4'-dimethoxytrityl DMF dimethylformamide EtOAc ethyl acetate ESI electrospray ionization EtOAc ethyl acetate HMDS hexamethyldisilazane HPLC High pressure liquid chromatography LDA lithium diisopropylamide LRMS low resolution mass spectrum MCPBA meta-chloroperbenzoic acid MeCN acetonitrile MeOH methanol MMTC mono methoxytrityl chloride m/z or m/e mass to charge ratio MH.sup.+ mass plus 1 MH.sup.- mass minus 1 MsOH methanesulfonic acid MS or ms mass spectrum MTBE tert-butylmethyl ether NBS N-bromosuccinimide Ph phenyl rt or r.t. room temperature TBAF tetrabutylammonium fluoride THF tetrahydrofuran TMSCl chlorotrimethylsilane TMSBr bromotrimethylsilane TMSI iodotrimethylsilane TMSOTf (trimethylsily) trifluoromethylsulfonate TEA triethylamine TBA tributylamine TBAP tributylammonium pyrophosphate TBSCl t-butyldimethylsilyl chloride TEAB triethylammonium bicarbonate TFA trifluoroacetic acid TLC or tlc thin layer chromatography Tr triphenylmethyl Tol 4-methylbenzoyl Turbo Grignard 1:1 mixture of isopropylmagnesium chloride and lithium chloride .delta. parts per million down field from tetramethylsilane
[0238] E. Preparation of Compounds
Example 1. (2S)-ethyl 2-(chloro(phenoxy)phosphorylamino)propanoate (Chloridate A)
##STR00093##
[0240] Ethyl alanine ester hydrochloride salt (1.69 g, 11 mmol) was dissolved in anhydrous CH.sub.2Cl.sub.2 (10 mL) and the mixture stirred with cooling to 0.degree. C. under N.sub.2(g). Phenyl dichlorophosphate (1.49 mL, 10 mmol) was added followed by dropwise addition of Et.sub.3N over about 10 min. The reaction mixture was then slowly warmed to RT and stirred for about 12 h. Anhydrous Et.sub.2O (50 mL) was added and the mixture stirred for about 30 min. The solid that formed was removed by filtration, and the filtrate concentrated under reduced pressure. The residue was subjected to silica gel chromatography eluting with 0-50% EtOAc in hexanes to provide intermediate A. .sup.1H NMR (300 MHz, CDCl.sub.3) .delta. 7.39-7.27 (m, 5H), 4.27 (m, 3H), 1.52 (m, 3H), 1.32 (m, 3H). .sup.31P NMR (121.4 MHz, CDCl.sub.3) .delta. 8.2, 7.8.
Example 2. (2S)-2-ethylbutyl 2-(chloro(phenoxy)phosphorylamino)propanoate (Chloridate B)
##STR00094##
[0242] The 2-ethylbutyl alanine chlorophosphoramidate ester B was prepared using the same procedure as chloridate A except substituting 2-ethylbutyl alanine ester for ethyl alanine ester. The material is used crude in the next reaction. Treatment with methanol or ethanol forms the displaced product with the requisite LCMS signal.
Example 3. (2S)-isopropyl 2-(chloro(phenoxy)phosphorylamino)propanoate (Chloridate
##STR00095##
[0244] The isopropyl alanine chlorophosphoramidate ester C was prepared using the same procedure as chloridate A except substituting isopropyl alanine ester for the ethyl alanine ester. The material is used crude in the next reaction. Treatment with methanol or ethanol forms the displaced product with the requisite LCMS signal.
Example 4. (2R, 3R, 4S, 5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-5-(hydroxy- methyl)tetrahydrofuran-2-carbonitrile (Compound 1)
##STR00096##
[0246] The preparation of (2R, 3R, 4S, 5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-5-(hydroxy- methyl)tetrahydrofuran-2-carbonitrile is described below.
##STR00097##
[0247] The commercially available lactol (10 g, 23.8 mmol) was dissolved in anhydrous DMSO (30 mL) under N.sub.2(g). Ac.sub.2O (20 mL) was added and the resultant reaction mixture stirred at RT for about 48 h. The reaction mixture was poured onto ice H.sub.2O (500 mL) and the mixture stirred for 20 min. The mixture was extracted with EtOAc (3.times.200 mL) and the combined organic extracts were then washed with H.sub.2O (3.times.200 mL). The organic extract was dried over anhydrous MgSO.sub.4, filtered and concentrated under reduced pressure. The residue was dissolved in CH.sub.2Cl.sub.2 and subjected to silica gel chromatography eluting with 25% EtOAc in hexanes to provide the lactone. .sup.1H NMR (400 MHz, DMSO) .delta. 7.30-7.34 (m, 13H), 7.19-7.21 (m, 2H), 4.55-4.72 (m, 6H), 4.47 (s, 2H), 4.28 (d, J=3.9 Hz, 1H), 3.66 (m, 2H). LCMS m/z 436.1 [M+H.sub.2O], 435.2 [M+OH]- Tr=2.82 min. HPLC Tr=4.59 [2-98% ACN in H2) over 5 min at 2 mL/min flow.
##STR00098##
[0248] The bromopyrazole (prepared according to WO2009/132135) (0.5 g, 2.4 mmol) was suspended in anhydrous THF (10 mL) under N.sub.2(g). The suspension was stirred and TMSCl (0.67 mL, 5.28 mmol) was added. The mixture was stirred for 20 min. at RT and then cooled to about -78.degree. C. after which time a solution of n-BuLi (6 mL, 1.6 N in hexanes, 9.6 mmol) was added slowly. The reaction mixture was stirred for 10 min. at about -78.degree. C. and then the lactone (1 g, 2.4 mmol) was added via syringe. When the reaction was complete as measured by LCMS, AcOH was added to quench the reaction. The mixture was concentrated under reduced pressure and the residue dissolved in a mixture of CH.sub.2Cl.sub.2 and H.sub.2O (100 mL, 1:1). The organic layer was separated and washed with H.sub.2O (50 mL). The organic layer was then dried over anhydrous MgSO.sub.4, filtered and concentrated under reduced pressure. The residue was subjected to silica gel chromatography eluting with 0-50% EtOAc in hexanes to provide the product as a 1:1 mixture of anomers. LCMS m/z 553 [M+H].
##STR00099##
[0249] The hydroxy nucleoside (1.1 g, 2.0 mmol) was dissolved in anhydrous CH.sub.2Cl.sub.2 (40 mL) and the solution cooled with stirring to about -78.degree. C. under N.sub.2(g). TMSCN (0.931 mL, 7 mmol) was added and the mixture stirred for a further 10 min. TMSOTf (1.63 mL, 9.0 mmol) was slowly added to the reaction and the mixture stirred for 1 h. The reaction mixture was then diluted with CH.sub.2Cl.sub.2 (120 mL) and aqueous NaHCO.sub.3 (120 mL) was added to quench the reaction. The reaction mixture was stirred for a further 10 min and the organic layer separated. The aqueous layer was extracted with CH.sub.2Cl.sub.2 (150 mL) and the combined organic extracts dried over anhydrous MgSO.sub.4, filtered and concentrated under reduced pressure. The residue was dissolved in a minimal amount of CH.sub.2Cl.sub.2 and subjected to silica gel chromatography eluting with a gradient of 0-75% EtOAc and hexanes to provide the tribenzyl cyano nucleoside as a mixture of anomers. .sup.1H NMR (300 MHz, CD.sub.3CN) .delta. 7.94 (s, 0.5H), 7.88 (s, 0.5H), 7.29-7.43 (m, 13H), 7.11-7.19 (m, 1H), 6.82-6.88 (m, 1H), 6.70-6.76 (m, 1H), 6.41 (bs, 2H), 5.10 (d, J=3.9 Hz, 0.5H), 4.96 (d, J=5.1 Hz, 0.5H), 4.31-4.85 (m, 7H), 4.09-4.18 (m, 2H), 3.61-3.90 (m, 2H). LCMS m/z 562 [M+H].
##STR00100##
[0250] The tribenzyl cyano nucleoside (70 mg, 0.124 mmol) was dissolved in anhydrous CH.sub.2Cl.sub.2 (2 mL) and cooled to about -20.degree. C. under N.sub.2(g). A solution of BCl.sub.3 (1N in CH.sub.2Cl.sub.2, 0.506 mL, 0.506 mmol) was added and the reaction mixture stirred for 1 h. at -78.degree. C. When the reaction was complete by LC/MS, MeOH was added to quench the reaction. The reaction mixture was allowed to warm to RT and the solvent removed under reduced pressure. The residue was subjected to C18 reverse phase HPLC, eluting for 5 min with H.sub.2O (0.1% TFA), followed by a gradient of 0-70% MeCN in H.sub.2O (0.1% TFA) over 35 min, to elute the .alpha.-anomer, and .beta.-anomer 1. (.alpha.-anomer) .sup.1H NMR (300 MHz, D.sub.2O) .delta. 7.96 (s, 1H), 7.20 (d, J=4.8 Hz, 1H), 6.91 (d, J=4.8 Hz, 1H), 4.97 (d, J=4.4 Hz, 1H), 4.56-4.62 (m, 1H), 4.08-4.14 (m, 1H), 3.90 (dd, J=12.9, 2.4 Hz, 1H), 3.70 (dd, J=13.2, 4.5 Hz, 1H). (.beta.-anomer) .sup.1H NMR (400 MHz, DMSO) .delta. 7.91 (s, 1H), 7.80-8.00 (br s, 2H), 6.85-6.89 (m, 2H), 6.07 (d, J=6.0 Hz, 1H), 5.17 (br s, 1H), 4.90 (br s, 1H), 4.63 (t, J=3.9 Hz, 1H), 4.02-4.06 (m, 1H), 3.94 (br s, 1H), 3.48-3.64 (m, 2H). LCMS m/z 292.2 [M+H], 290.0 [M-H]. Tr=0.35 min. 13C NMR (400 MHZ, DMSO), 156.0, 148.3, 124.3, 117.8, 117.0, 111.2, 101.3, 85.8, 79.0, 74.7, 70.5, 61.4. HPLC Tr=1.32 min
Example 4-a. (2R, 3R, 4S, 5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-5-(hydroxy- methyl)tetrahydrofuran-2-carbonitrile (Compound 1)
##STR00101##
[0252] The preparation of (2R, 3R, 4S, 5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-5-(hydroxy- methyl)tetrahydrofuran-2-carbonitrile is described below.
Preparation of (3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyloxy- )-5-((benzyloxy)methyl)tetrahydrofuran-2-ol Using LaCl.sub.3-2LiCl
##STR00102##
[0254] A solution of 7-iodopyrrolo[2,1-f][1,2,4]triazin-4-amine (7.5 g, 28.8 mmol, 1.0 equiv) was prepared in THF (67 mL). The solution was cooled to about 0.degree. C., and TMSCl (3.3 mL, 30.3 mmol, 1.05 equiv) was added. The reaction mixture was stirred for about 30 min, and then PhMgCl (2 M in THF; 28 mL, 56.8 mmol, 1.97 equiv) was added while maintaining an internal temperature below 5.degree. C. The reaction mixture was agitated at about 0.degree. C. for about 35 min, and then cooled to about -15.degree. C. iPrMgCl (2 M in THF, 14 mL, 30.2 mmol, 1.05 equiv) was then added while maintaining an internal temperature below about -10.degree. C. After approximately 15 minutes at about -15.degree. C., LaCl.sub.3-2LiCl (0.6 M in THF, 50 mL, 14.4 mmol, 0.5 equiv) was added while maintaining an internal temperature below about -15.degree. C. The reaction mixture was agitated for about 25 min at about -20.degree. C.
[0255] In a separate flask, a solution of (3R,4R,5R)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl)dihydrofuran-2(3H)-one (10.0 g, 23.9 mmol, 0.83 equiv) was prepared in THF (45 mL). The solution was cooled to about -20.degree. C., and then transferred to the Grignard solution while maintaining an internal temperature below about -15.degree. C. The resulting reaction mixture was agitated at about -20.degree. C. for about 30 min.
[0256] The reaction was quenched with 2 M HCl (53 mL), and the mixture warmed to about 15.degree. C. iPrOAc (38 mL) was added, and the organic and aqueous phases were separated. The bottom aqueous layer was discharged, and the upper organic layer was washed sequentially with 2.5 wt % NaHCO.sub.3 (53 mL), 2.5 wt % NaHCO.sub.3 (53 mL), and 10 wt % NaCl (53 mL).
[0257] The organic phase was concentrated to about 45 mL, and then diluted with iPrOAc (75 mL). The solution was concentrated again to about 45 mL, and then diluted with iPrOAc (23 mL). The solution was concentrated to about 45 mL, and then filtered over a pad of Celite. The filtered solution was concentrated to about 26 mL, and then diluted with MTBE (75 mL). After 2 h, heptane (23 mL) was slowly added and the slurry was stirred at about 25.degree. C. for about 2 h, and was then cooled to about -5.degree. C. over about 8 h. The solids were isolated by filtration, and the filter cake was washed with MTBE/heptane (4:1, 23 mL). The solids were dried in a vacuum oven at no more than about 35.degree. C. to afford (3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyloxy- )-5-((benzyloxy)methyl)tetrahydrofuran-2-ol.
Preparation of (3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyloxy- )-5-((benzyloxy)methyl)tetrahydrofuran-2-ol Using CeCl.sub.3
##STR00103##
[0259] The iodopyrazole (5.02 g, 19.3 mmol) was dissolved in THF (45 g) and the solution was cooled to about 0.degree. C. with stirring. TMSCl (2.04 g, 18.7 mmol) was added, and after about 1 h phenyl magnesium chloride (2.0 M in THF, 19.9 g, 38.2 mmol) was added. The reaction mixture was cooled to about -20.degree. C. and iso-propyl magnesium chloride (2.0 M in THF, 9.99 g, 20.5 mmol) was added slowly. After about 30 min, the reaction mixture was transferred to a mixture of anhydrous cerium chloride (4.75 g, 19.3 mmol) in THF (22 g) at about -20.degree. C. After about 1.5 h a solution of lactone (6.73 g, 16.1 mmol) in THF (22 g) was added slowly, and the resulting reaction mixture was stirred for about 1 h. 2 M HCl (41 g) was added, the mixture was warmed to about 15.degree. C., and iso-propyl acetate (35 g) was added. The layers were separated and the organic layer was washed with 2.5% NaHCO.sub.3 (2.times.40 g), 10% NaCl (1.times.35 g) and concentrated to about 30 mL volume. iso-Propyl acetate (44 g) was charged and the solution was concentrated to about 30 mL volume. iso-Propyl acetate (43 g) was charged and the solution was concentrated to about 30 mL volume. The solution was filtered and the filtrate was concentrated to about 18 mL volume. tert-Butylmethyl ether (37 g) was added followed by product seed crystals (10.7 mg). After about 14 h n-heptane (10.5 g) was added and the mixture was cooled to about -5.degree. C. and filtered. The solids were washed with tert-butylmethyl ether (9 g) at about -5.degree. C. and dried under vacuum at about 34.degree. C. for about 15 h to provide the product.
Preparation of (3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyloxy- )-5-((benzyloxy)methyl)tetrahydrofuran-2-ol Using CeCl.sub.3 and iPrMgCl--LiCl
##STR00104##
[0261] The iodopyrazole (5.03 g, 19.3 mmol) was dissolved in THF (45 g) and the solution was cooled to about 0.degree. C. with stirring under N.sub.2(g). TMSCl (2.06 g, 19.0 mmol) was added, and after about 1 h phenyl magnesium chloride (2.0 M in THF, 20.23 g, 38.8 mmol) was added. The reaction mixture was cooled to about -20.degree. C. and iso-propyl magnesium chloride-lithium chloride complex (2.0 M in THF, 15.37 g, 21.0 mmol) was added slowly. After about 1 h, the reaction mixture was transferred to a mixture of cerium chloride (4.77 g, 19.4 mmol) in THF (22 g) at about -20.degree. C. After about 1 h a solution of lactone (6.75 g, 16.1 mmol) in THF (23 g) was added slowly, and the resulting reaction mixture was stirred for about 1.5 h. 2 M HCl (40 g) was added, the mixture was warmed to about 15.degree. C. and iso-propyl acetate (35 g) was added. The layers were separated and the organic layer was washed with 2.5% NaHCO.sub.3 (2.times.40 g), 10% NaCl (1.times.36 g) and concentrated to about 30 mL volume. iso-Propyl acetate (44 g) was added and the solution was concentrated to about 30 mL volume. The solution was filtered and the filtrate was concentrated to about 18 mL volume. tert-Butylmethyl ether (37 g) was added followed by product seed crystals (10.5 mg). After about 14 h n-heptane (11 g) was added and the mixture was cooled to about -5.degree. C. and filtered. The solids were washed with tert-butylmethyl ether (9 g) at about -5.degree. C. and dried under vacuum at about 34.degree. C. for about 15 h to provide the product.
Preparation of (3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyloxy- )-5-((benzyloxy)methyl)tetrahydrofuran-2-ol Using YCl.sub.3
##STR00105##
[0263] The iodopyrazole (4.99 g, 19.2 mmol) was dissolved in THF (44 g) and the solution was cooled to about 0.degree. C. with stirring. TMSCl (2.45 mL, 19.4 mmol) was added, and after about 30 min phenyl magnesium chloride (2.0 M in THF, 20.29 g, 39.0 mmol) was added. The reaction mixture was cooled to about -20.degree. C. and iso-propyl magnesium chloride (2.0 M in THF, 9.85 g, 20.1 mmol) was added slowly. After about 30 min, the reaction mixture was transferred into a mixture of anhydrous yttrium chloride (3.76 g, 19.3 mmol) and lactone (6.68 g, 16.0 mml) in THF (24 g) at about -20.degree. C. After about 2.5 h 2 M HCl (30 g) was added, the mixture was warmed to about 15.degree. C., and iso-propyl acetate (22 g) was added. The layers were separated and the organic layer was washed with 2.5% NaHCO.sub.3 (2.times.40 g), 10% NaCl (1.times.35 g) and concentrated to about 30 mL volume. iso-Propyl acetate (44 g) was charged and the solution was concentrated to about 30 mL volume. iso-Propyl acetate (45 g) was charged and the solution was concentrated to about 30 mL volume. The solution was filtered and the filtrate was concentrated to about 18 mL volume. tert-Butylmethyl ether (37 g) was added followed by product seed crystals (11.5 mg). After about 1 h n-heptane (15 mL) was added and the mixture was cooled to about -5.degree. C. and agitated for about 17 h. The slurry was filtered and the solids were washed with a tert-butylmethyl ether (8 g)/n-heptane (2 g) mixture precooled to about -5.degree. C. The resulting solids were dried under vacuum at about 34.degree. C. for about 22 h to afford the product.
Preparation of (3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyloxy- )-5-((benzyloxy)methyl)tetrahydrofuran-2-ol Using NdCl.sub.3
##STR00106##
[0265] The iodopyrazole (5.02 g, 19.3 mmol) was dissolved in THF (38 g) and the solution was cooled to about 0.degree. C. with stirring under N.sub.2(g). TMSCl (2.45 mL, 19.4 mmol) was added, and after about 1 h phenylmagnesium chloride (2.0 M in THF, 19.75 g, 38.0 mmol) was added. The reaction mixture was cooled to about -20.degree. C. and iso-propylmagnesium chloride (2.0 M in THF, 9.40 g, 19.2 mmol) was added slowly. After about 1.5 h, the reaction mixture was transferred into a mixture of anhydrous neodymium (III) chloride (4.03 g, 16.1 mmol) and lactone (6.70 g, 16.0 mml) in THF (22 g) at about -20.degree. C. After about 1.5 h the reaction mixture was warmed to -10.degree. C. and, after an additional 2 h, 2 M HCl (36 g) was added. The mixture was warmed to about 15.degree. C. and iso-propyl acetate (23 g) was added. The layers were separated and the organic layer was washed with 2.5% NaHCO.sub.3 (2.times.44 g), 10% NaCl (1.times.41 g) and concentrated to about 30 mL volume. iso-Propyl acetate (44 g) was charged and the solution was concentrated to about 30 mL volume. iso-Propyl acetate (45 g) was charged and the solution was concentrated to about 30 mL volume. The solution was filtered and the filtrate was concentrated to about 18 mL volume. tert-Butylmethyl ether (37 g) was added followed by product seed crystals (11.9 mg). After about 1 h n-heptane (15 mL) was added and the mixture was cooled to about -5.degree. C. and agitated for about 15 h. The slurry was filtered and the solids were washed with a tert-butylmethyl ether (8 g)/n-heptane (11 g) mixture precooled to about -5.degree. C. The resulting solids were dried under vacuum at about 34.degree. C. for about 25 h to afford the product.
Preparation of (2R,3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyl- oxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-carbonitrile
##STR00107##
[0267] To a pre-cooled (-40.degree. C.) solution of (3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyloxy- )-5-((benzyloxy)methyl)tetrahydrofuran-2-ol (10.0 grams, 18.1 mmols, 1.0 equiv.) in DCM (100 mL) was charged trifluoroacetic acid (6.19 grams, 54.3 mmols, 3.0 equiv.), followed by a pre-cooled (-30.degree. C.) solution of TMSOTf (24.1 grams, 108.6 mmols, 6.0 equiv.) and TMSCN (10.8 grams, 108.6 mmols, 6.0 equiv.) in DCM (50 mL) while maintaining the internal temperature below about -25.degree. C. The reaction mixture was agitated at below about -30.degree. C. for no less than 10 minutes and quenched into a pre-cooled (about -10.degree. C.) solution of 20 wt. % KOH aq. (120 mL). The bi-phasic mixture was warmed to ambient temperature. The organic layer was separated and washed with 10 wt. % NaCl aq. (3.times.50 mL). The organic phase was filtered, concentrated under vacuum to about 50 mL, re-diluted with toluene (200 mL) and concentrated under vacuum to 140 mL at about 50.degree. C. The solution was seeded with (2R,3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyl- oxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-carbonitrile at about 55.degree. C. Agitated at about 55.degree. C. for about an hour and cooled to about 0.degree. C. over about 6 hours. The solids were isolated by filtration and the filter cake was washed with toluene (30 mL). The solids were dried under vacuum at about 50.degree. C.
Preparation of (2R,3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyl- oxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-carbonitrile Via Flow Chemistry
##STR00108##
[0269] Solutions of (3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyloxy- )-5-((benzyloxy)methyl)tetrahydrofuran-2-ol (23.0 g in 460.07 g of DCM), TMSOTf (55.81 g in 138.07 g of DCM) and TMSCN (25.03 g in 138.10 g of DCM) were sequentially pumped, into a tube reactor at about -40.degree. C. The reaction mixture was collected in a flask, kept in ice bath, containing 20% KOH aqueous solution (46.91 g KOH and 210 g of water). The layers were separated and the organic phase was sequentially washed with 10% KOH aqueous solution (10 g KOH and 90 mL of water) and with 10% brine (2.times.100 g). The organic phase was concentrated under vacuum to about 4 volumes, isopropyl alcohol was charged (162.89 g) and the mixture was concentrated under vacuum to about 10 volumes. The contents were warmed to about 60.degree. C., then adjusted to about 0.degree. C. over about 6.5 h and agitated at about 0.degree. C. for about 15.5 h. The resulting slurry was filtered, the solids were rinsed with isopropyl alcohol (61.79 g) and then dried at about 50.degree. C. under reduced pressure overnight to afford the product.
Preparation of (2R, 3R, 4S, 5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-5-(hydroxy- methyl)tetrahydrofuran-2-carbonitrile
##STR00109##
[0271] The tribenzyl cyano nucleoside (48.8 g, 86.9 mmol, 1.0 equiv.) was dissolved in anhydrous CH.sub.2Cl.sub.2 (244 mL) and cooled to about -20.degree. C. A solution of BCl.sub.3 (1M in CH.sub.2Cl.sub.2, 295 mL, 295 mmol, 3.4 equiv.) was added dropwise, maintaining the internal temperature below about -15.degree. C. Following addition, the reaction mixture was stirred for 1 h at about -20.degree. C. MeOH (340 ml) was added dropwise, maintaining the internal temperature below -15.degree. C. The resulting solution was distilled to about 250 ml, then refilled with about 250 ml MeOH. The resulting solution was again distilled to about 250 ml, then refilled with about 250 ml MeOH, and finally distilled to about 125 ml. Water (125 ml) was added, followed by K.sub.2CO.sub.3 solution (20 wt % in water, 125 ml). The pH was checked, and found to be .about.3. K.sub.2CO.sub.3 solution was added (20 wt % in water, 50 ml), and the pH was found to be .about.8. The resulting slurry was stirred overnight, then filtered and washed with water (50 ml) and MeOH (50 ml). The wet product cake was dried overnight at about 40.degree. C. overnight. .sup.1H NMR (300 MHz, D.sub.2O) .delta. 7.96 (s, 1H), 7.20 (d, J=4.8 Hz, 1H), 6.91 (d, J=4.8 Hz, 1H), 4.97 (d, J=4.4 Hz, 1H), 4.56-4.62 (m, 1H), 4.08-4.14 (m, 1H), 3.90 (dd, J=12.9, 2.4 Hz, 1H), 3.70 (dd, J=13.2, 4.5 Hz, 1H).
Example 5. (2R,3R,4R,5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-3-fl- uoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile (Compound 2)
##STR00110##
[0273] The preparation of (2R,3R,4R,5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-3-fluoro-4-hyd- roxy-5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile is described below.
##STR00111##
2-Deoxy-2-fluoro-4,5-O,O-dibenzyl-D-arabinose
[0274] 1'-Methoxy-2-deoxy-2-fluoro-4,5-O,O-dibenzyl-D-arabinose (1.0 g, 2.88 mmol) in TFA (13.5 mL) was treated with H.sub.2O (1.5 mL) and the resultant mixture stirred for 5 h. The mixture was then diluted with EtOAc (100 mL) and treated with saturated NaHCO.sub.3 (50 mL). The organic layer was separated and washed with NaCl (50 mL), dried over anhydrous MgSO.sub.4, filtered and concentrated under reduced pressure. The residue was subjected to silica gel chromatography (80 g SiO.sub.2 Combiflash HP Gold Column) eluting with 0-100% EtOAc in hexanes to afford 2-deoxy-2-fluoro-4,5-O,O-dibenzyl-D-arabinose as a white solid: R.sub.f=0.52 (25% EtOAc in hexanes). .sup.1H NMR (300 MHz, CDCl.sub.3) .delta. 7.30 (m, 10H), 5.35 (m, 1H), 4.68-4.29 (m, 7H), 3.70 (d, J=10.5 Hz, 1H), 3.50 (d, J=10.5 Hz, 2H). .sup.19F NMR (282.2 MHz, CDCl.sub.3) .delta. -207 (m), -211 (m). LCMS m/z 350 [M+H.sub.2O].
##STR00112##
(3R, 4R, 5R)-4-(benzyloxy)-5-(benzyloxymethyl)-3-fluorodihydrofuran-2(3H)- -one
[0275] 2-Deoxy-2-fluoro-4,5-O,O-dibenzyl-D-arabinose (4.3 g, 12.8 mmol) was dissolved in CH.sub.2Cl.sub.2 (85 mL) was treated with 4 .ANG. MS (10 g) and pyridinium dichromate (14.4 g, 38.3 mmol). The resultant mixture was stirred for 24 h and then filtered through a pad of Celite.RTM.. The eluant was concentrated under reduced pressure and the residue subjected to silica gel chromatography (120 g SiO.sub.2 HP Gold Combiflash Column) eluting with 0-100% EtOAc in hexanes to afford (3R, 4R, 5R)-4-(benzyloxy)-5-(benzyloxymethyl)-3-fluorodihydrofuran-2(3H)-one as a clear oil (3.5 g, 83%): R.sub.f=0.25 (25% EtOAc in hexanes). .sup.1H NMR (300 MHz, CDCl.sub.3) .delta. 7.37 (m, 10H), 5.45 (dd, J=49, 5.7, Hz, 1H), 4.85 (d, J=11.7 Hz, 1H), 4.52 (m, 4H), 4.29 (d, J=5.4 Hz, 1H), 2.08 (dd, J=15.3, 10.2 Hz, 2H). .sup.19F NMR (282.2 MHz, CDCl.sub.3) .delta. -216. LCMS m/z 348 [M+H.sub.2O]. HPLC (6-98% MeCN--H.sub.2O gradient, 0.05% TFA modifier) t.sub.R=5.29 min. Phenomenex Synergi 4 m Hydro-RP 80 A, 50.times.4.60 mm, 4 micron; 2 mL/min flow rate
##STR00113##
(3R, 4R, 5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-4-(benzyloxy)-5- -(benzyloxymethyl)-3-fluorotetrahydrofuran-2-ol
[0276] 7-Bromopyrrolo[1,2-f][1,2,4]-triazin-4-amine (68 mg, 0.319 mmol) in THF (1.4 mL) was treated with TMSCl (89 .mu.L, 0.703 mmol) and the mixture stirred for 2 h. The mixture was then cooled to about -78.degree. C. and treated with nBuLi (1.0 M in hexanes, 1.09 mL, 1.09 mmol). The solution was stirred for about 30 min and then treated with (3R, 4R, 5R)-4-(benzyloxy)-5-(benzyloxymethyl)-3-fluorodihydrofuran-2(3H)-one (106 mg, 0.319 mmol) dropwise in THF (1.4 mL). The resultant mixture was stirred for 30 min and then AcOH (83 .mu.L, 1.44 mmol) in THF (1.0 mL) was added to quench the reaction. The mixture was warmed to RT and then concentrated under reduced pressure. The residue was diluted with EtOAc (100 mL) and washed with saturated NaCl solution (50 mL). The organic layer was dried over anhydrous MgSO.sub.4, filtered and concentrated under reduced pressure. The residue was subjected to silica gel chromatography (40 g SiO.sub.2 HP Gold Combiflash Column) eluting with 0-100% EtOAc in hexanes followed by a 0-100% gradient of (20% MeOH in EtOAc) in EtOAc to afford (3R, 4R, 5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-4-(benzyloxy)-5-(benzylo- xymethyl)-3-fluorotetrahydrofuran-2-ol as a white solid (68 mg, 44%, 60/40 mixture of .alpha./.beta. isomers). R.sub.f=0.32 (EtOAc). .sup.1H NMR (300 MHz, CDCl.sub.3) .delta. 8.05 (s, 1H), 7.86 (s, 1H), 7.81 (s, 1H), 7.64 (s, 1H), 7.26 (m, 10H), 6.95 (m, 1H), 6.71 (m, 1H), 6.08 (m, 1H), 5.34 (m, 1H), 4.65 (m, 6H), 4.71 (m, 2H). .sup.19F NMR (282.2 MHz, CDCl.sub.3) .delta. -211 (m). LCMS m/z 465 [M+H]. HPLC (6-98% MeCN--H.sub.2O gradient, 0.05% TFA modifier) t.sub.R=4.37 min. (.alpha.-isomer), 4.54 min. (.beta.-isomer).
##STR00114##
(3R, 4R, 5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-4-(benzyloxy)-5- -(benzyloxymethyl)-3-fluorotetrahydrofuran-2-carbonitrile
[0277] (3R, 4R, 5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-4-(benzyloxy)-5-(benzylo- xymethyl)-3-fluorotetrahydrofuran-2-ol (195 mg, 0.42 mmol) was dissolved in MeCN (1.4 mL) was treated with TMSCN (336 .mu.L, 2.52 mmol) and In(OTf).sub.3 (708 mg, 1.26 mmol). The solution was stirred at about 70.degree. C. for 18 h and then cooled to about 0.degree. C. The mixture was treated with saturated NaHCO.sub.3 solution (20 drops) then warmed to RT and diluted with EtOAc (100 mL) and H.sub.2O (50 mL). The organic layer was separated and washed with saturated NaCl solution (50 mL), dried over MgSO.sub.4, filtered and concentrated under reduced pressure. The residue was subjected to silica gel chromatography (40 g SiO.sub.2 HP Gold Combiflash Column) eluting with 0-100% EtOAc in hexanes to afford (3R, 4R, 5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-4-(benzyloxy)-5- -(benzyloxymethyl)-3-fluorotetrahydrofuran-2-carbonitrile as a white solid (60/40 mixture of .alpha./.beta. isomers). Data for both isomers: R.sub.f=0.53 (EtOAc). .sup.1H NMR (300 MHz, CDCl.sub.3) .delta. 8.01 (s, 1H), 7.94 (s, 1H), 7.30 (m, 10H), 7.00 (d, J=4.5 Hz, 1H), 6.93 (d, J=4.8 Hz, 1H), 6.87 (d, J=5.4 Hz, 1H), 6.70 (d, J=4.8 Hz, 1H), 5.85 (dd, J=52, 3.3 Hz, 1H), 5.55 (dd, J=53, 4.5 Hz, 1H), 4.71 (m, 7H), 3.87 (m, 2H), 3.72 (m, 2H). .sup.19F NMR (282.2 MHz, CDCl.sub.3) .delta. -196 (m), -203 (m). LCMS m/z 474 [M+H]. HPLC (6-98% MeCN--H.sub.2O gradient, 0.05% TFA modifier) t.sub.R=4.98 min.
##STR00115##
(2R, 3R, 4R, 5R)-2-(4-aminopyrrolo[1,2,4][1,2,4]triazin-7-yl)-3-fluoro-4-hydroxy-5-(hy- droxymethyl)tetrahydrofuran-2-carbonitrile (2)
[0278] (3R, 4R, 5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-4-(benzyloxy)-5-(benzylo- xymethyl)-3-fluorotetrahydrofuran-2-carbonitrile (110 mg, 0.23 mmol) was dissolved in CH.sub.2Cl.sub.2 (1.5 mL) and cooled to about 0.degree. C. The reaction mixture was treated with BCl.sub.3 (1.0 M in CH.sub.2Cl.sub.2, 766 .mu.L, 0.77 mmol) and stirred for 2 h. The mixture was then cooled to about -78.degree. C. and treated with Et.sub.3N (340 .mu.L, 2.44 mmol) followed by MeOH (2 mL) before allowing to warm to RT. The reaction was concentrated under reduced pressure and then co-evaporated with MeOH (3.times.5 mL). The residue was then suspended in H.sub.2O (5 mL) and treated with NaHCO.sub.3 (1 g). The solution was stirred for 10 min and then concentrated under reduced pressure. The residue was filtered and washed with MeOH (3.times.10 mL) on a fritted glass funnel (coarse) and the eluant concentrated under reduced pressure. The residue was subjected to reverse phase HPLC (6-98% MeCN in H.sub.2O gradient with 0.05% TFA modifier) to afford (2R, 3R, 4R, 5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-3-fluoro-4-hydroxy-5-(hy- droxymethyl)tetrahydrofuran-2-carbonitrile 2 as a white solid and the .alpha.-isomer. Data for the .beta.-isomer: R.sub.f=0.13 (10% MeOH in EtOAc). .sup.1H NMR (300 MHz, CD.sub.3OD) .delta. 8.09 (s, 1H), 7.28 (d, J=5.1 Hz, 1H), 7.17 (d, J=5.1 Hz, 1H), 5.42 (dd, J=53, 3.3 Hz, 1H), 4.20 (m, 2H), 3.99 (d, J=3.6 Hz, 1H), 3.77 (d, J=3.6 Hz, 1H). .sup.19F NMR (282.2 MHz, CDCl.sub.3) .delta. -197 (m). LCMS m/z 294 [M+H]. HPLC (2-98% MeCN--H.sub.2O gradient, 0.05% TFA modifier) t.sub.R=1.49 min.
Example 6. (2R, 3R, 4R, 5S)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-4-fluoro-2-(hydroxymethy- l)-5-methyltetrahydrofuran-3-ol (Compound 3)
##STR00116##
[0280] The preparation of (2R, 3R, 4R, 5S)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-4-fluoro-2-(hydroxymethy- l)-5-methyltetrahydrofuran-3-ol is described below.
##STR00117##
[0281] The starting nucleoside (prepared as described in the synthesis of compound 2) (0.355 g, 0.765 mmol) was dissolved in anhydrous THF (35 mL) and cooled to about 0.degree. C. with stirring under N.sub.2(g). A solution of methyl magnesium chloride (2 mL, 6 mmol) (3N in THF) was added and the resultant mixture stirred overnight. Acetic acid (7 mmol) was added to quench the reaction and then the solvents were removed by rotory under reduced pressure. The residue was re-dissolved in CH.sub.2Cl.sub.2 and the solution subjected to a plug of silica gel to isolate the product (0.355 g) as a crude mixture. LC/MS (m/z: 480, M.sup.+1). The crude material was dissolved in anhydrous CH.sub.2Cl.sub.2 (20 mL) and placed under N.sub.2(g). The solution was stirred and treated with methanesulfonic acid (0.2 mL, 2.74 mmol). The reaction mixture was stirred for about 12 h at RT and then quenched by the addition of Et.sub.3N (3.5 mmol). The mixture was concentrated under reduced pressure and the residue subjected to silica gel chromatography to provide the methyl substituted nucleoside as a 4:1 mixture of beta- and alpha-anomers respectively. .sup.1H NMR (300 MHz, CD.sub.3CN) major anomer .delta. 7.87 (s, 1H), 7.27-7.40 (m, 10H), 6.77 (d, J=4.5 HZ, 1H), 6.70 (d, J=4.5 Hz, 1H), 6.23 (br s, 2H), 5.53 (dd, J=55, 3.3 Hz, 1H), 4.42-4.75 (m, 4H), 4.19-4.26 (m, 1H), 3.65-4.00 (m, 3H), 1.74 (d, J=3.9 Hz, 3H). .sup.19F NMR (282.2 MHz, CD.sub.3CN) major anomer .delta. -207 (m, 1F). LCMS m/z 463 [M+H].
##STR00118##
[0282] The benzylated nucleoside material (0.134 g, 0.290 mmol), Degussa catalyst (0.268 g) and AcOH (30 mL) were mixed together. The reaction atmosphere was charged with H.sub.2 (g) and the reaction stirred for about 2 h. The catalyst was removed by filtration and the mixture concentrated under reduced pressure. The residue was dissolved in a minimal amount of H.sub.2O and subjected to reverse phase HPLC (C.sup.18 hydro RP column) to isolate the .beta.-anomer 3. .sup.1H NMR (300 MHz, D.sub.2O) .delta. 7.87 (s, 1H), 7.22 (d, J=4.8 Hz, 1H), 6.87 (d, J=4.8 Hz, 1H), 5.35 (dd, J=54, 3.6 Hz, 1H), 3.97-4.10 (m, 2H), 3.81 (dd, J=12.6, 2.1 Hz, 1H), 3.64 (dd, J=12.6, 4.8 Hz, 1H), 1.65 (d, J=4.2 Hz, 3H). .sup.19F NMR (282.2 MHz, CD.sub.3CN) 8-207 (m, 1F).
[0283] A small amount of alpha anomer was characterized as follows. .sup.1H NMR (300 MHz, D.sub.2O) .delta. 7.86 (s, 1H), 7.26 (d, J=4.8 Hz, 1H), 6.85 (d, J=4.8 Hz, 1H), 5.31 (dd, J=54, 3.9 Hz, 1H), 4.39 (ddd, J=26.1, 9.9, 3.6 Hz, 2H), 4.00-4.05 (m, 1H), 3.90 (dd, J=12.3, 2.1 Hz, 1H), 3.66 (dd, J=12.6, 4.8, 1H), 1.56 (s, 3H). .sup.19F NMR (282.2 MHz, CD.sub.3CN) 8-198 (dd, J=54, 26 Hz, 1F).
Example 7. (2S)-isopropyl 2-(((((2R,3R,4R,5S)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-4-fluoro- -3-hydroxy-5-methyltetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)- propanoate (Compound 4)
##STR00119##
[0285] The nucleoside 3 (0.011 g, 0.04 mmol) was dissolved in trimethylphosphate (2 mL) and cooled to 0.degree. C. The mixture was stirred under an atmosphere of N.sub.2(g) and 1-Methylimidazole (0.320 mL, 5 mmol) followed by the alaninylmonoisopropyl, monophenol phosphorchloridate C (0.240 mL, 4.4 mmol) was added. The reaction mixture was stirred for 2 h. at 0.degree. C. and then allowed to warm slowly to RT. while monitoring by LC/MS. When complete by LCMS, the reaction mixture was treated with H.sub.2O (5 mL) and then concentrated under reduced pressure. The residue was dissolved in CH.sub.2Cl.sub.2 and subjected to silica gel chromatography eluting with 0-100% EtOAc in hexanes. The product fractions were collected and concentrated. The residue was subjected to prep HPLC to yield the alanine isopropyl monoamidate prodrug 4 as a mixture of isomers. .sup.1H NMR (300 MHz, CD.sub.3CN) .delta. 7.87 (s, 1H), 7.17-7.44 (m, 5H), 6.71-6.83 (m, 2H), 6.14 (br, s, 2H), 5.38 (dd, J=56, 3.3 Hz, 1H), 4.92-5.01 (m, 1H), 3.86-4.46 (m, 6H), 3.58 (m, 1H), 1.73 (m, 3H), 1.18-1.34 (m, 9H). LCMS m/z 552 [M+H].
Example 8. (2S)-ethyl 2-(((((2R,3R,4R,5S)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-4-fluoro- -3-hydroxy-5-methyltetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)- propanoate (Compound 5)
##STR00120##
[0287] The nucleoside 3 (0.026 g, 0.092 mmol) was dissolved in trimethylphosphate (2 mL) and cooled to 0.degree. C. The mixture was stirred under N.sub.2(g) and 1-methylimidazole (0.062 mL, 0.763 mmol) followed by the chloridate A (0.160 g, 0.552 mmol) were added. The reaction mixture was stirred for 2 h. at 0.degree. C. and then allowed to warm slowly to RT. H.sub.2O (5 mL) was added to quench the reaction and then the mixture concentrated under reduced pressure. The residue was dissolved in CH.sub.2Cl.sub.2 and subjected to silica gel chromatography eluting with 0-100% EtOAc in hexanes. The product fractions were collected and concentrated. Crude product was eluted using 0 to 100 percent EtOAc in hexanes. The crude product was collected and concentrated under reduced pressure. The residue was subjected to prep HPLC to yield compound 5. LCMS m/z 538 [M+H].
Example 9. ((2R, 3R, 4R, 5S)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-4-fluoro-3-hydroxy-5-met- hyltetrahydrofuran-2-yl)methyl Tetrahydrogen Triphosphate (Compound 6)
##STR00121##
[0289] The nucleoside 3 (0.022 g, 0.056 mmol) was dissolved in trimethylphosphate (1 mL) and stirred under N.sub.2(g). Phosphorous oxychloride (0.067 mL, 0.73 mmol) was added and the mixture stirred for about 2 h. Monitoring by analytical ion-exchange column determined the time at which >80 percent of monophosphate was formed. A solution of tributylamine (0.44 mL, 1.85 mmol) and triethylammonium pyrophosphate (0.327 g, 0.72 mmol) dissolved in anhydrous DMF (1 mL) was added. The reaction mixture was stirred for 20 min and then quenched by the addition of 1N triethylammonium bicarbonate solution in H.sub.2O (5 mL). The mixture was concentrated under reduced pressure and the residue re-dissolved in H.sub.2O. The solution was subjected to ion exchange chromatography to yield the title product compound 6. LCMS m/z 521 [M-H]. Tr=0.41. HPLC ion exchange TR=9.40 min
Example 10. (2R,3R,5S)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-3-hydroxy-5-(hydr- oxymethyl)-tetrahydrofuran-2-carbonitrile (Compound 7)
##STR00122##
[0291] The preparation of (2R,3R,5S)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-3-hydroxy-5-(hydr- oxymethyl)-tetrahydrofuran-2-carbonitrile is described below.
##STR00123##
[0292] ((3.alpha.R,5S,6.alpha.R)-2,2-dimethyl-tetrahydrofuro[2,3-d][1,3]di- oxol-5-yl)methanol
[0293] The acetate material (1.2 g, 5.5 mmol) (J. Org. Chem. 1985, 50, 3457, De Bernardo et al) was dissolved in a 1:1 mixture MeOH and THF (10 mL). A 1N solution of NaOH(aq) (10 mL) was added until the pH was 13. The reaction mixture was stirred for about 2 h and then neutralized to pH 8-9 by the addition of AcOH. The mixture was extracted with EtOAc (10.times.30 mL) and the combined organic extracts dried over anhydrous Na.sub.2SO.sub.4, filtered and concentrated under reduced pressure. The residue was subjected to silica gel chromatography eluting with 0-70% EtOAc in hexanes to give the desired product (866 mg, 90%). .sup.1H NMR (300 MHz, CDCl.sub.3) .delta. 5.84 (d, J=3.6 Hz, 1H), 4.78 (t, J=4.5 Hz, 1H), 4.38 (m, 1H), 3.93-3.54 (m, 2H), 2.04-1.84 (m, 2H), 1.52 (s, 3H), 1.33 (s, 3H).
##STR00124##
(3.alpha.R,5S,6.alpha.R)-5-(benzyloxymethyl)-2,2-dimethyl-tetrahydrofuro[- 2,3-d][1,3]dioxole
[0294] Sodium hydride (188 mg, 7.46 mmol) was dissolved in anhydrous THF (5 mL) and stirred under N.sub.2(g) at RT. The alcohol (866 mg, 4.97 mmol) was dissolved in anhydrous THF (3 mL) and then added in portions over 5 min. to the sodium hydride mixture. The resultant mixture was stirred for about 20 min. and then benzyl bromide (892 .mu.L, 7.46 mmol) was added. The reaction was stirred for about 2 h and then poured onto a mixture of ice cold aqueous NaHCO.sub.3 and EtOAc (30 mL). The organic layer was separated and then the aqueous layer re-extracted with EtOAc (30 mL). The combined organic extracts were dried over anhydrous Na.sub.2SO.sub.4, filtered and concentrated under reduced pressure. The residue was subjected to silica gel chromatography eluting with 0-40% EtOAc in hexanes to give the benzyl ether product. .sup.1H NMR (300 MHz, CDCl.sub.3) .delta. 7.35-7.27 (m, 5H), 5.86 (d, J=3.6 Hz, 1H), 4.74 (t, J=4.2 Hz, 1H), 4.60 (s, 2H), 4.42 (m, 1H), 3.69-3.53 (m, 2H), 2.10-2.04 (m, 1H), 1.83-1.77 (m, 1H), 1.52 (s, 3H), 1.33 (s, 3H).
##STR00125##
(3R,5S)-5-(benzyloxymethyl)-tetrahydrofuran-2,3-diol
[0295] The benzyl ether (910 mg, 3.44 mmol) was dissolved in a 1:1 AcOH and H.sub.2O (20 mL) mixture and stirred at about 60.degree. C. for about 7 h. The mixture was concentrated under reduced pressure and the residue subjected to silica gel chromatography eluting with 0-70% EtOAc in hexanes to give the diol product (705 mg, 91%). .sup.1H NMR (300 MHz, CDCl.sub.3) .delta. 7.36-7.27 (m, 5H), 5.40 (d, J=3.9 Hz, 0.5H), 5.17 (s, 0.5H), 4.67-4.56 (m, 3H), 4.33 (m, 0.5H), 4.24 (d, J=4.8 Hz, 0.5H), 3.71-3.67 (m, 1H), 3.56-3.42 (m, 2H), 2.31-2.22 (m, 1H), 2.08-1.89 (m, 2H).
##STR00126##
(3R,5S)-5-(benzyloxymethyl)-3-hydroxy-dihydrofuran-2(3H)-one
[0296] The diol (705 mg, 3.14 mmol) was dissolved in benzene (30 mL) and treated with a silver carbonate celite mixture (3.46 g, 6.28 mmol). The resultant mixture was stirred at about 80.degree. C. under N.sub.2(g) for about 2 h. The mixture was then cooled to RT, filtered and concentrated under reduced pressure. The residue was subjected to silica gel chromatography eluting with 0-70% EtOAc in hexanes to give the lactone product. .sup.1H NMR (300 MHz, CDCl.sub.3) .delta. 7.39-7.27 (m, 5H), 4.75-4.68 (m, 1H), 4.60-4.49 (m, 2H), 3.74-3.54 (m, 2H), 2.61-2.35 (m, 2H), 2.38-2.28 (m, 1H).
##STR00127##
(3R, 5S)-3-(benzyloxy)-5-(benzyloxymethyl)-dihydrofuran-2(3H)-one
[0297] The lactone (600 mg, 2.7 mmol) was dissolved in EtOAc (30 mL) and treated with silver oxide (626 mg, 2.7 mmol) followed by benzyl bromide (387 .mu.L, 3.24 mmol). The reaction mixture was then stirred at about 50.degree. C. under N.sub.2(g) for about 8 h. Additional silver oxide (300 mg) was then added and the resultant mixture stirred at about 50.degree. C. for about 16 h. Additional benzyl bromide (50 uL) and silver oxide (150 mg) were added and the mixture stirred for an additional about 8 h. The reaction mixture was allowed to cool, filtered and then concentrated under reduced pressure. The residue was subjected to silica gel chromatography eluting with 0-20% EtOAc in hexanes to give the title product. .sup.1H NMR (300 MHz, CDCl.sub.3) .delta. 7.39-7.27 (m, 10H), 4.99 (d, J=11.4 Hz, 1H), 4.72 (m, 2H), 4.56 (m, 2H), 4.39 (t, J=8.1 Hz, 1H), 3.72-3.51 (m, 2H), 2.42-2.25 (m, 2H).
##STR00128##
(3R,5S)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-3-(benzyloxy)-5-(ben- zyloxymethyl)-tetrahydrofuran-2-ol
[0298] The 7-bromopyrrolo[1,2-f][1,2,4]triazin-4-amine (607 mg, 2.85 mmol) was dissolved in anhydrous THF (10 mL) and stirred under Ar(g) at RT. TMSCl (1.1 mL, 8.55 mmol) was added dropwise and the mixture stirred for about 2 h. The reaction was concentrated under reduced pressure and then dried under high vacuum. The residue was suspended in THF (20 mL) and stirred under Ar(g) at about -78.degree. C. A 2.5M n-BuLi solution in hexane (2.28 mL, 5.7 mmol) was added dropwise over about 10 min. and the resultant mixture stirred for about 60 min. The lactone (742 mg, 2.37 mmol) dissolved in anhydrous THF (7 mL) was added to the above mixture over about 20 min. The reaction mixture was stirred for about 2 h. and then quenched with AcOH until pH was 5-6. The mixture was allowed to warm to RT and then diluted with EtOAc. The solution was washed with saturated NaHCO.sub.3 solution, saturated NaCl, dried over anhydrous Na.sub.2SO.sub.4 and concentrated under reduced pressure. The residue was subjected to silica gel chromatography eluting with 0-80% EtOAc in hexanes to give the title product. LCMS m/z 447.2 [M+H], 445.1 [M-H].
##STR00129##
(3R,5S)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-3-(benzyloxy)-5-(ben- zyloxymethyl)-tetrahydrofuran-2-carbonitrile
[0299] The alcohol (250 mg, 0.56 mmol) was dissolved in anhydrous CH.sub.2Cl.sub.2 (10 mL) and stirred under Ar(g) at about -15.degree. C. TMSCN (448 .mu.L, 3.36 mmol) was added dropwise and the mixture stirred for about 10 min. TMSOTf (466 .mu.L, 2.58 mmol) was added dropwise over 10 min and the resultant mixture stirred for about 90 min. at about -15.degree. C. Additional TMSCN (224 .mu.L, 3 eq.) and TMSOTf (202 .mu.L, 2 eq.) was added and stirring continued for about 5 h. Saturated aqueous NaHCO.sub.3 solution was added to quench the reaction and the mixture stirred for about 10 min. The organic layer was separated and washed with saturated aqueous NaHCO.sub.3 solution, saturated NaCl solution, dried over anhydrous Na.sub.2SO.sub.4, filtered and concentrated under reduced pressure. The residue was subjected to silica gel chromatography eluting with 0-70% EtOAc in hexanes to give the title product. LCMS m/z 456.3 [M+H], 454.1 [M-H].
##STR00130##
(2R,3R,5S)2-(4-aminopyrrolo[1,2,4][1,2,4]triazin-7-yl)-3-hydroxy-5-(hydro- xymethyl)-tetrahydrofuran-2-carbonitrile (7)
[0300] The benzyl ether (150 mg, 0.329 mmol) was dissolved in anhydrous CH.sub.2Cl.sub.2 (2 mL) and the mixture stirred under Ar(g) at about -20.degree. C. A 1M BCl.sub.3 solution in CH.sub.2Cl.sub.2 (724 .mu.L, 0.724 mmol) was added dropwise and the resultant mixture stirred for about 2 h. Additional 1M BCl.sub.3 in CH.sub.2Cl.sub.2 (724 .mu.L, 0.724 mmol) was added and stirring continued for 2 h. The mixture was then cooled to about -78.degree. C. and slowly treated with a 2:1 mixture of Et.sub.3N and MeOH (3 mL). The mixture was stirred for about 10 min and then treated with MeOH (10 mL). The reaction was allowed to warm to RT and then concentrated under reduced pressure. The residue was dissolved in MeOH and concentrated under reduced pressure. The residue was dissolved in MeOH again and treated with solid NaHCO.sub.3. The mixture was stirred for about 5 min and then the solid removed by filtration. The solution was concentrated under reduced pressure and subjected to preparative HPLC to provide the desired product 7. .sup.1H NMR (300 MHz, D.sub.2O) .delta. 7.71 (s, 1H), 6.75 (d, J=4.5 Hz, 1H), 6.65 (d, J=4.8 Hz, 1H), 4.91 (t, J=6.3 Hz, 1H), 4.57 (m, 1H), 3.67-3.47 (m, 2H), 2.18 (m, 2H). LCMS m/z 276.1 [M+H], 274.0 [M-H].
Example 11. (2S)-isopropyl 2-((((2R,3S,4R,5R)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-5-cyano-3- ,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)-phosphorylamino)propano- ate (Compound 8)
##STR00131##
[0302] The nucleoside 1 (45 mg, 0.15 mmol) was dissolved in anhydrous trimethyl phosphate (0.5 mL) and the solution stirred under N.sub.2(g) at about 0.degree. C. Methyl imidazole (36 .mu.L, 0.45 mmol) was added to the solution. Chlorophosphoramidate C (69 mg, 0.225 mmol) was dissolved in anhydrous THF (0.25 mL) and added dropwise to the nucleoside mixture. When the reaction was complete by LCMS, the reaction mixture was diluted with EtOAc and washed with saturated aqueous NaHCO.sub.3 solution, saturated NaCl, dried over anhydrous Na.sub.2SO.sub.4, filtered and concentrated under reduced pressure. The residue was subjected to silica gel chromatography eluting with 0-5% MeOH in CH.sub.2Cl.sub.2 followed by preparative HPLC to give the product. .sup.1H NMR (300 MHz, CD.sub.3OD) .delta. 7.95 (m, 1H), 7.31-6.97 (m, 7H), 4.94 (m, 1H), 4.78 (m, 1H), 4.43 (m, 3H), 4.20 (m, 1H), 3.80 (d, 1H), 1.30-1.18 (m, 9H). .sup.31P NMR (121.4 MHz, CD.sub.3OD) .delta. 3.8. LCMS m/z 561.0 [M+H], 559.0 [M-H].
Example 12. (2S)-2-ethylbutyl 2-((((2R,3S,4R,5R)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-5-cyano-3- ,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphorylamino)propanoa- te (Compound 9)
[0303] Compound 9 can be prepared by several methods described below.
Procedure 1
##STR00132##
[0305] Prepared from Compound 1 and chloridate B according to the same method as for the preparation of compound 8. .sup.1H NMR (300 MHz, CD.sub.3OD) .delta. 7.87 (m, 1H), 7.31-7.16 (m, 5H), 6.92-6.89 (m, 2H), 4.78 (m, 1H), 4.50-3.80 (m, 7H), 1.45-1.24 (m, 8H), 0.95-0.84 (m, 6H). .sup.31P NMR (121.4 MHz, CD.sub.3OD) .delta. 3.7. LCMS m/z 603.1 [M+H], 601.0 [M-H].
Procedure 2
##STR00133##
[0306] (2S)-2-ethylbutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino) Propanoate
[0307] (2S)-2-ethylbutyl 2-(((4-nitrophenoxy)(phenoxy)phosphoryl)amino)propanoate (1.08 g, 2.4 mmol) was dissolved in anhydrous DMF (9 mL) and stirred under a nitrogen atmosphere at RT. (2R,3R,4S,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-- 5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile (350 mg, 1.2 mmol) was added to the reaction mixture in one portion. A solution of t-butylmagnesium chloride in THF (1M, 1.8 mL, 1.8 mmol) was then added to the reaction dropwise over about 10 minutes. The reaction was stirred for about 2 h, at which point the reaction mixture was diluted with ethyl acetate (50 mL) and washed with saturated aqueous sodium bicarbonate solution (3.times.15 mL) followed by saturated aqueous sodium chloride solution (15 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting oil was purified with silica gel column chromatography (0-10% MeOH in DCM) to afford (2S)-2-ethylbutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino) propanoate (311 mg, 43%, 1:0.4 diastereomeric mixture at phosphorus) as a white solid. .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 7.85 (m, 1H), 7.34-7.23 (m, 2H), 7.21-7.09 (m, 3H), 6.94-6.84 (m, 2H), 4.78 (d, J=5.4 Hz, 1H), 4.46-4.33 (m, 2H), 4.33-4.24 (m, 1H), 4.18 (m, 1H), 4.05-3.80 (m, 3H), 1.52-1.39 (m, 1H), 1.38-1.20 (m, 7H), 0.85 (m, 6H). .sup.31P NMR (162 MHz, CD.sub.3OD) .delta. 3.71, 3.65. LCMS m/z 603.1 [M+H], 600.9 [M-H]. HPLC (2-98% MeCN--H.sub.2O gradient with 0.1% TFA modifier over 8.5 min, 1.5 mL/min, Column: Phenomenex Kinetex C18, 2.6 um 100 .ANG., 4.6.times.100 mm) t.sub.R=5.544 min, 5.601 min
Separation of the (S) and (R) Diastereomers
[0308] (2S)-2-ethylbutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino) propanoate was dissolved in acetonitrile. The resulting solution was loaded onto Lux Cellulose-2 chiral column, equilibrated in acetonitrile, and eluted with isocratic acetonitrile/methanol (95:5 vol/vol). The first eluting diastereomer had a retention time of 17.4 min, and the second eluting diastereomer had a retention time of 25.0 min.
[0309] First Eluting Diastereomer is (S)-2-ethylbutyl 2-(((R)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cy- ano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)pr- opanoate:
##STR00134##
[0310] .sup.1HNMR (400 MHz, CD.sub.3OD) .delta. 8.05 (s, 1H), 7.36 (d, J=4.8 Hz, 1H), 7.29 (br t, J=7.8 Hz, 2H), 7.19-7.13 (m, 3H), 7.11 (d, J=4.8 Hz, 1H), 4.73 (d, J=5.2 Hz, 1H), 4.48-4.38 (m, 2H), 4.37-4.28 (m, 1H), 4.17 (t, J=5.6 Hz, 1H), 4.08-3.94 (m, 2H), 3.94-3.80 (m, 1H), 1.48 (sep, J=12.0, 6.1 Hz, 1H), 1.34 (p, J=7.3 Hz, 4H), 1.29 (d, J=7.2 Hz, 3H), 0.87 (t, J=7.4 Hz, 6H). .sup.31PNMR (162 MHz, CD.sub.3OD) .delta. 3.71 (s). HPLC (2-98% MeCN--H.sub.2O gradient with 0.1% TFA modifier over 8.5 min, 1.5 mL/min, Column: Phenomenex Kinetex C18, 2.6 um 100 .ANG., 4.6.times.100 mm) t.sub.R=5.585 min.
[0311] Second Eluting Diastereomer is (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cy- ano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)pr- opanoate:
##STR00135##
[0312] .sup.1HNMR (400 MHz, CD.sub.3OD) .delta. 8.08 (s, 1H), 7.36-7.28 (m, 3H), 7.23-7.14 (m, 3H), 7.08 (d, J=4.8 Hz, 1H), 4.71 (d, J=5.3 Hz, 1H), 4.45-4.34 (m, 2H), 4.32-4.24 (m, 1H), 4.14 (t, J=5.8 Hz, 1H), 4.08-3.94 (m, 2H), 3.93-3.85 (m, 1H), 1.47 (sep, J=6.2 Hz, 1H), 1.38-1.26 (m, 7H), 0.87 (t, J=7.5 Hz, 6H). .sup.31PNMR (162 MHz, CD.sub.3OD) .delta. 3.73 (s). HPLC (2-98% MeCN--H.sub.2O gradient with 0.1% TFA modifier over 8.5 min, 1.5 mL/min, Column: Phenomenex Kinetex C18, 2.6 um 100 .ANG., 4.6.times.100 mm) t.sub.R=5.629 min.
Example 13. (2S)-ethyl 2-((((2R,3S,4R,5R)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-5-cyano-3- ,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphorylamino)propanoa- te (Compound 10)
##STR00136##
[0314] The preparation of (2S)-ethyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propan- oate is described below.
Procedure 1. Preparation Via Chloridate A
##STR00137##
[0316] Prepared from Compound 1 and chloridate A using same method as for the preparation of compound 8. .sup.1H NMR (300 MHz, CD.sub.3OD) .delta. 7.95 (m, 1H), 7.32-6.97 (m, 7H), 4.78 (m, 1H), 4.43-4.08 (m, 6H), 3.83 (m, 1H), 1.31-1.18 (m, 6H). .sup.31P NMR (121.4 MHz, CD.sub.3OD) .delta. 3.7. LCMS m/z 547.0 [M+H], 545.0 [M-H].
Procedure 2. Preparation Via Nitro-Benzene Compound L
##STR00138##
[0318] Compound 1 (50 mg, 0.17 mmol) was dissolved in NMP-THF (1:1 mL)) and cooled with ice bath. tBuMgCl (0.257 mL, 0.257 mmol) was then added over about 5 min. The resulting mixture was allowed to warm to RT and was stirred for about 30 min. Then a solution of compound L (Prepared according to US20120009147, 74.6 mg, 0.189 mmol) in THF (2 mL) was added. After about 30 min, the reaction mixture was purified by HPLC (acetonitrile 10 to 80% in water) to give compound 29 as a yellow solid. The solid was further purified with silica gel chromatography (MeOH 0 to 20% DCM) to afford compound 29. .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 7.76 (d, J=6.0 Hz, 1H), 7.25-7.14 (m, 2H), 7.11-6.99 (m, 3H), 6.87-6.72 (m, 2H), 4.70 (d, J=5.4 Hz, 1H), 4.39-4.24 (m, 2H), 4.20 (dddd, J=9.7, 7.9, 5.1, 2.8 Hz, 1H), 4.10 (dt, J=12.8, 5.5 Hz, 1H), 4.06-3.91 (m, 2H), 3.72 (ddq, J=14.3, 9.3, 7.1 Hz, 1H), 1.17 (dd, J=7.1, 1.0 Hz, 1H), 1.14-1.06 (m, 5H). .sup.31P NMR (162 MHz, CD.sub.3OD) .delta. 3.73, 3.68. MS m/z=547 (M+1).sup.+.
Example 14. (2S)-ethyl 2-((((2R,3R,4R,5R)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-5-cyano-4- -fluoro-3-hydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphorylamino)pro- panoate (Compound 11)
##STR00139##
[0320] Compound 11 was prepared from Compound 2 and chloridate A using same method as for the preparation of compound 8. .sup.1H NMR (300 MHz, CD.sub.3OD) .delta. 7.91 (m, 1H), 7.33-7.16 (m, 5H), 6.98-6.90 (m, 2H), 5.59 (m, 1H), 4.50-4.15 (m, 4H), 4.12-3.90 (m, 3H), 1.33-1.18 (m, 6H). .sup.31P NMR (121.4 MHz, CD.sub.3OD) .delta. 3.8. LCMS m/z 549.0 [M+H], 547.1 [M-H].
Example 15. (2S,2'S)-diethyl 2,2'-((((2R,3S,4R,5R)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-5-cyan- o-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)phosphoryl)bis(azanediyl)dipro- panoate (Compound 12)
##STR00140##
[0322] The nucleoside 1 (14.6 mg, 0.05 mmol) was dissolved in anhydrous trimethyl phosphate (0.5 mL) and stirred under N.sub.2(g) at RT. POCl.sub.3 (9.2 .mu.L, 0.1 mmol) was added and the mixture stirred for about 60 min. Alanine ethyl ester hydrochloride (61 mg, 0.4 mmol) and then Et.sub.3N (70 .mu.L, 0.5 mmol) was added. The resultant mixture was stirred for about 15 min. and then additional Et.sub.3N (70 .mu.l, 0.5 mmol) was added to give a solution pH of 9-10. The mixture was stirred for about 2 h. and then diluted with EtOAc, washed with saturated aqueous NaHCO.sub.3 solution followed by saturated aqueous NaCl solution. The organic layer was dried over anhydrous Na.sub.2SO.sub.4 and concentrated under reduced pressure. The residue was subjected to preparative HPLC (C18 column) to yield the product 12. .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 8.13 (s, 1H), 7.41 (d, J=4.8 Hz, 1H), 7.18 (d, J=4.8 Hz, 1H), 4.78 (d, J=5.6 Hz, 1H), 4.36 (m, 1H), 4.25-4.08 (m, 7H), 3.83 (m, 2H), 1.33-1.23 (m, 12H). .sup.31P NMR (121.4 MHz, CD.sub.3OD) .delta. 13.8. LCMS m/z 570.0 [M+H], 568.0 [M-H].
Example 16. (2S,3R,4S,5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-2-ethynyl-5-(h- ydroxymethyl)tetrahydrofuran-3,4-diol (Compound 13)
##STR00141##
[0324] The preparation of (2S,3R,4S,5R)-2-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-2-ethynyl-5-(h- ydroxymethyl)tetrahydrofuran-3,4-diol is described below.
##STR00142##
[0325] The nucleoside alcohol (0.6 g, 1.08 mmol) (prepared as described in Compound 1 synthesis) was dissolved in anhydrous THF (8 mL) and placed under N.sub.2(g). The reaction mixture was stirred and cooled to about 0.degree. C. and then treated with a 0.5N solution of ethynyl magnesium bromide in THF (17.2 mL, 17.2 mmol). The reaction mixture was stirred overnight at RT. AcOH (1.5 mL) was added to quench the reaction. The mixture was concentrated under reduced pressure and the residue redissolved in CH.sub.2Cl.sub.2. The solution subjected to a plug of silica gel eluting with 0 to 80% EtOAc in Hexanes to provide the title product as a crude mixture. LCMS m/z 579 [M+H].
##STR00143##
[0326] The crude ethynyl alcohol (0.624 g, 1.08 mmol) was dissolved in anhydrous CH.sub.2Cl.sub.2 (10 mL) and placed under N.sub.2(g). The mixture was stirred and sulfonic acid (0.2 mL, 2.74 mmol) was added. The reaction mixture was stirred for about 12 h. at RT. When complete by LCMS, Et.sub.3N (0.56 mL) was added to quench the reaction. The reaction was concentrated under reduced pressure and the residue subjected to silica gel chromatography eluting with 0 to 75% EtOAc in Hexanes to yield the ethynyl nucleoside as a mixture of anomers. LCMS m/z 561 [M+H].
##STR00144##
[0327] The tribenzyl nucleoside (0.650 g, 1.16 mmol) was dissolved in anhydrous CH.sub.2Cl.sub.2 (30 mL) and cooled to -78.degree. C. under N.sub.2(g). A solution of boron tribromide (1 N in CH.sub.2Cl.sub.2, 5.5 mL) was added and the reaction mixture stirred for 1 h. at -78.degree. C. A solution of MeOH (10 mL) and pyridine (2 mL) was added to quench the reaction and the mixture was allowed to rise to RT. The mixture was concentrated under reduced pressure and subjected to preparative HPLC to provide the .alpha.-anomer (20 mg) and .beta.-anomer 13 (110 mg). (.beta.-anomer) .sup.1H NMR (300 MHz, DMSO) .delta. 7.81 (s, 1H), 7.76 (br s, 2H), 6.80-6.85 (m, 2H), 5.11 (d, J=7.2 Hz, 1H), 4.90 (d, J=6.0 Hz, 1H), 4.82 (dd, J=7.2, 4.8 Hz, 1H), 4.62 (t, J=6.3 Hz, 1H), 3.95-3.99 (m, 1H), 3.85-3.91 (dd, J=11.4, 5.7 Hz, 1H), 3.61-3.67 (m, 1H), 3.47-3.55 (m, 1H), 3.52 (d, J=0.9 Hz, 1H). (.alpha.-anomer) .sup.1H NMR (300 MHz, DMSO) .delta. 7.80 (s, 1H), 7.59 (bs, 2H), 6.80 (d, J=4.5 Hz, 1H), 6.54 (d, J=4.2 Hz, 1H), 5.00 (d, J=7.2 Hz, 1H), 4.89 (d, J=4.8 Hz, 1H), 4.74 (t, J=5.7 Hz, 1H), 4.58 (t, J=4.5 Hz, 1H), 4.27 (m, 1H), 3.88 (m, 1H), 3.64-3.72 (m, 1H), 3.51-3.59 (m, 1H), 3.48 (d, J=0.6 Hz, 1H). LCMS m/z 291 [M+H].
Example 17. (2R,3R,4R)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-1,3,4-tris(benzyl- oxy)hexane-2,5-diol (Compound 14)
##STR00145##
[0329] The preparation of (2R,3R,4R)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-1,3,4-tris(benzyl- oxy)hexane-2,5-diol is described below.
##STR00146##
[0330] The tribenzyl alcohol from Compound 1 synthesis (0.250 g, 0.453 mmol) was dissolved in anhydrous THF (25 mL) and stirred under N.sub.2(g). The reaction mixture was cooled to 0.degree. C. and then a 3.0 N solution of methyl magnesium chloride in THF (1.2 mL, 3.62 mmol) was added. The reaction mixture was stirred overnight at RT. Acetic acid (1.5 mL) was added to quench the reaction and then the mixture was concentrated under reduced pressure. The residue was redissolved in CH.sub.2Cl.sub.2 and subjected to a plug of silica gel eluting with 0 to 80% EtOAc in hexanes. The crude product (0.452 g) was then used in the next reaction without further purification. LCMS m/z 569 [M+H].
##STR00147##
[0331] The crude methyl nucleoside (0.452 g, 0.796 mmol) was dissolved in anhydrous CH.sub.2Cl.sub.2 (20 mL) and stirred under N.sub.2(g). Methanesulfonic acid (0.2 mL, 2.78 mmol) was added and the reaction stirred for about 12 hr at RT. Et.sub.3N (0.56 mL) was added to quench the reaction and then the mixture concentrated under reduced pressure. The residue was subjected to silica gel chromatography eluting with 0 to 75% EtOAc in Hexanes to yield the product as a mixture of anomers. LCMS m/z 551 [M+H].
##STR00148##
[0332] The tribenzyl nucleoside (0.20 g, 0.364 mmol) was dissolved in AcOH (30 mL). and charged with Pd/C (Degussa) (400 mg). The stirred mixture was flushed with N.sub.2(g) three times and then H.sub.2 (g) was introduced, The reaction was stirred under H.sub.2 (g) for 2 h. and then the catalyst removed by filtration. The solution was concentrated under reduced pressure and under the residue was re-dissolved in H.sub.2O. The solution was subjected to preparative HPLC under neutral conditions to provide the .alpha.-anomer and .beta.-anomer 14. (.alpha.-anomer) .sup.1H NMR (300 MHz, D.sub.2O) .delta. 7.81 (s, 1H), 7.22 (d, 1H), 6.75 (d, 1H), 4.47 (d, 1H), 4.25-4.31 (m, 1H), 3.88-4.95 (m, 1H), 3.58-3.86 (dd, 2H), 1.50 (s, 3H). (.beta.-anomer) .sup.1H NMR (300 MHz, D.sub.2O) .delta. 7.91 (s, 1H), 7.26 (d, 1H), 6.90 (d, 1H), 4.61 (d, 1H), 4.00-4.09 (m, 2H), 3.63-3.82 (dd, 2H), 1.67 (s, 3H). LCMS m/z 281 [M+H].
Example 18. S,S'-2,2'-((((2R,3S,4R,5R)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-5- -cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)phosphoryl)bis(oxy)bis(et- hane-2,1-diyl) bis(2,2-dimethylpropanethioate) (Compound 15)
##STR00149##
[0334] The nucleoside 1 (0.028 g, 0.096 mmol) was dissolved in trimethylphosphate (1 mL). The reaction was stirred under N.sub.2(g) and then treated with 1H-tetrazole (0.021 g, 0.29 mmol). The reaction mixture was cooled to 0.degree. C. and the phosphane (Nucleoside Nucleotides, Nucleic acids; 14; 3-5; 1995; 763-766. Lefebvre, Isabelle; Pompon, Alain; Perigaud, Christian; Girardet, Jean-Luc; Gosselin, Gilles; et al.) (87 mg, 0.192 mmol) was added. The reaction was stirred for 2 h. and then quenched with 30% hydrogen peroxide (0.120 mL). The mixture was stirred for 30 min at RT and then treated with saturated aqueous sodium thiosulfate (1 mL). The mixture was stirred for 10 min. and then concentrated under reduced pressure. The residue was subjected to preparative HPLC to isolate the title product 15. .sup.1H NMR (300 MHz, CD.sub.3CN) .delta. 7.98 (s, 1H), 6.92 (d, 1H), 6.81 (d, 1H), 6.44 (bs, 2H), 4.82 (m, 2H), 4.47 (m, 1H), 4.24 (m, 2H), 4.00 (m, 4H), 3.80 (bs, 1H), 3.11 (m, 4H), 1.24 (s, 9H). .sup.31P NMR (121.4 MHz, CD.sub.3CN) .delta. -1.85 (s). LCMS m/z 661 [M+H].
Example 19. S,S'-2,2'-((((2R, 3S, 4R, 5S)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-5-ethynyl-3,4-dihydroxyt- etrahydrofuran-2-yl)methoxy)phosphoryl)bis(oxy)bis(ethane-2,1-diyl) bis(2,2-dimethylpropanethioate) (Compound 16)
##STR00150##
[0336] Compound 16 was prepared using the same method as compound 15 except substituting compound 13 as the starting nucleoside. .sup.1H NMR (300 MHz, CD.sub.3CN) .delta. 7.91 (s, 1H), 6.86 (d, J=4.8 Hz, 1H), 6.76 (d, J=4.5 Hz, 1H), 6.29 (bs, 2H), 4.69 (t, J=2.7 Hz, 1H), 4.58 (d, J=5.7 Hz, 1H), 4.14-4.33 (m, 5H), 3.99-4.07 (m, 4H), 3.53 (d, J=5.4 Hz, 1H), 3.11 (q, J=5.7 Hz, 4H), 1.22 (s, 18H). LCMS m/z 658.9 [M+]. Tr=2.31
Example 20. ((2R, 3S, 4R, 5R)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytet- rahydrofuran-2-yl)methyl Tetrahydrogen Triphosphate (Compound 17)
##STR00151##
[0338] Compound 17 was prepared from compound 1 using a similar procedure to the preparation of compound 6. The product was isolated as the sodium salt. .sup.1H NMR (400 MHz, D.sub.2O) .delta. 7.76 (s, 1H), 6.88 (d, J=4.8 Hz, 1H), 6.73 (d, J=4.4 Hz, 1H), 4.86 (d, J=5.2 Hz, 1H), 4.43 (m, 1H), 4.39 (m, 1H), 4.05 (m, 1H), 3.94 (m, 1H). .sup.31P NMR (121.4 MHz, D.sub.2O) .delta. -5.4 (d, 1P), -10.8 (d, 1P), -21.1 (t, 1P). LCMS m/z 530 [M-H], 531.9 [M+H] Tr=0.22 min. HPLC ion exchange Tr=9.95 min.
Example 21. ((2R, 3S, 4R, 5S)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-5-ethynyl-3,4-dihydroxyt- etrahydrofuran-2-yl)methyl Tetrahydrogen Triphosphate (Compound 18)
##STR00152##
[0340] Compound 18 was prepared from compound 13 using a similar procedure to the preparation of compound 6. The product was isolated as the TEA salt. .sup.1H NMR (300 MHz, D.sub.2O) .delta. 7.85 (s, 1H), 7.09 (d, J=4.6 Hz, 1H), 6.95 (d, J=4.7 Hz, 1H), 4.23 (m, 2H), 4.08 (m, 2H), 3.06 (q, J=7.4 Hz, 20H), 1.14 (t, J=7.3 Hz, 30H). .sup.31P NMR (121.4 MHz, D.sub.2O) .delta. -10.8 (d, 1P), -11.2 (d, 1P), -23.2 (t, 1P). LCMS m/z 530.8 [M+H], Tr=0.46. HPLC ion exchange Tr=9.40 min.
Example 22. ((2R, 3S, 4R, 5S)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-5-methylte- trahydrofuran-2-yl)methyl Tetrahydrogen Triphosphate (Compound 19)
##STR00153##
[0342] Compound 19 was prepared from compound 14 using a similar procedure to the preparation of compound 6. .sup.1H NMR (400 MHz, D.sub.2O) .delta. 7.78 (s, 1H), 6.98 (m, 1H), 6.84 (m, 1H), 4.45 (m, 1H), 4.04 (m, 4H), 1.54 (s, 3H). .sup.31P NMR (161 MHz, D.sub.2O) .delta. -10.6 (m), -23.0 (m). LCMS m/z 521.0 [M+H].
Example 23. ((2R,3R,4R,5R)-5-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)-5-cyano-4-flu- oro-3-hydroxytetrahydrofuran-2-yl)methyl Tetrahydrogen Triphosphate (Compound 20)
##STR00154##
[0344] Compound 20 was prepared from compound 2 using a similar procedure to the preparation of compound 6. .sup.1H NMR (400 MHz, D.sub.2O) .delta. 7.78 (s, 1H), 6.93 (d, J=4.4 Hz, 1H), 6.78 (d, J=4.8 Hz, 1H), 5.45 (dd, J=53, 4.4 Hz, 1H), 4.38-4.50 (m, 2H), 4.13-4.20 (m, 2H). .sup.31P NMR (161 MHz, D.sub.2O) .delta. -5.7 (d, 1P), -11.0 (d, 1P), -21.5 (t, 1P). LCMS m/z 533.9.0 [M+H], 532.0 [M-H] Tr=1.25 min. HPLC ion exchange Tr=11.0 min.
Example 24. (2S)-ethyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-3-phe- nylpropanoate (21)
##STR00155##
[0346] The preparation of (2S)-ethyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-3-phe- nylpropanoate is described below.
Preparation of (S)-ethyl 2-amino-3-phenylpropanoate hydrochloride
##STR00156##
[0348] L-Phenylalanine (5 g, 30 mmol) was taken up in EtOH (30 mL). TMSCl (6.915 mL, 54 mmol) was added to the reaction at RT. The reaction vessel was fitted with a reflux condenser and the reaction was placed in an 80.degree. C. bath. The reaction was stirred overnight. The next day the reaction was cooled to RT, concentrated under reduced pressure and the resulting residue was taken up in Et.sub.2O. The resulting slurry was filtered and the isolate solids were further washed with Et.sub.2O. The washed solids were placed under high vacuum to yield example (S)-ethyl 2-amino-3-phenylpropanoate hydrochloride. .sup.1N NMR (400 MHz, DMSO-d.sub.6) .delta. 8.52 (s, 3H), 7.30 (m, 5H), 4.24 (ABX, J.sub.AX=7.8 Hz, J.sub.BX=6.2 Hz, 1H), 4.11 (m, 2H), 3.17, 3.05 (ABX, J.sub.AB=-14 Hz, J.sub.BX=5.8 Hz, J.sub.AX=7.6 Hz, 2H), 1.09 (t, J=6.8 Hz, 3H).
Preparation of (2S)-ethyl 2-(((4-nitrophenoxy)(phenoxy)phosphoryl)amino)-3-phenylpropanoate (Compound D)
##STR00157##
[0350] (S)-ethyl 2-amino-3-phenylpropanoate hydrochloride (1.01 g, 4.41 mmol) was dissolved in DCM (50 mL). This solution was cooled to about 0.degree. C. and PhOP(O)Cl.sub.2 (0.656 mL, 4.41 mmol) was added, followed by the slow addition of Et.sub.3N (1.62 mL, 11.5 mmol) over 5 min. The cold bath was removed and the reaction was allowed to warm to RT and stir over a period of 80 min. p-NO.sub.2PhOH (0.583 g, 4.19 mmol) was added, followed by more Et.sub.3N (0.3 mL, 2.1 mmol). The reaction progress was monitored by LC/MS. Upon completion of the reaction, it was diluted with Et.sub.2O, and the resulting solids were removed by filtration. The filtrate was concentrated and compound D was isolated by silica gel column chromatography (25 g dry load cartridge, 120 g column; eluent: 100% hexanes ramping to 55% EtOAc in hexanes). .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 8.17 (m, 2H), 7.33 (m, 2H), 7.09-7.25 (m, 10H), 4.17 (m, 1H), 4.07 (m, 2H), 3.08 (m, 1H), 2.84 (m, 1H), 1.14 (m, 3H). .sup.31P NMR (162 MHz, DMSO-d.sub.6) .delta. -1.479 (s), -1.719 (s). MS m/z=471.01 [M+1].
Preparation of (2S)-ethyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-3-phe- nylpropanoate (Compound 21)
##STR00158##
[0352] Compound 1 (0.030 g, 0.103 mmol) was dissolved in DMF (1 mL) and then THF (0.5 mL) was added. t-BuMgCl (1M/THF, 154.5 .mu.L, 0.154 .mu.mol) was added to the reaction in a drop-wise manner with vigorous stirring. The resulting white slurry was stirred at RT for about 30 min. A solution of compound D (0.058 g, 0.124 mmol) in THF (1 mL) was added in a drop-wise manner to the reaction at RT. The reaction progress was monitored by LC/MS. When the reaction progressed to 50% conversion, the reaction was cooled in an ice bath and quenched with glacial acetic acid (70 .mu.L). The reaction was concentrated and compound 21 was isolated from the residue by reverse phase HPLC. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 7.91 (d, J=4 Hz, 1H), 7.90 (brs, 2H), 7.09-7.30 (m, 8H), 7.01, (t, J=8.2 Hz, 2H), 6.89 (d, J=4.4 Hz, 1H), 6.82 (t, J=4.4 Hz, 1H), 6.27 (m, 1H), 6.14 (m, 1H), 5.34 (m, 1H), 4.62 (t, J=5.6 Hz, 1H), 4.15 (m, 1H), 3.78-4.01 (m, 6H), 2.92 (m, 1H), 2.78 (m, 1H), 1.04 (m, 3H). .sup.31P NMR (162 MHz, DMSO-d.sub.6) .delta. 3.69 (s), 3.34 (s). MS m/z=623.0 [M+H].
Example 25. (2S)-ethyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-0)methoxy)(phenoxy)phosphoryl)amino)-3-meth- ylbutanoate (22)
##STR00159##
[0354] The preparation of (2S)-ethyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-3-met- hylbutanoate is described below.
Preparation of (2S)-ethyl 3-methyl-2-(((4-nitrophenoxy)(phenoxy)phosphoryl)amino) butanoate (Compound E)
##STR00160##
[0356] The (S)-ethyl 2-amino-3-methylbutanoate (0.351 g, 1.932 mmol) was dissolved in DCM (17 mL). This solution was cooled in an ice bath and PhOP(O)Cl.sub.2 (0.287 mL, 1.932 mmol) was added, followed by the slow addition of Et.sub.3N (1.62 mL, 11.4 mmol) over about 5 min. The cold bath was removed and the reaction was allowed to warm to RT and stir over a period of 1 h. p-NO.sub.2PhOH (0.255 g, 1.836 mmol) was added, and the reaction progress was monitored by LC/MS. Upon completion of the reaction, the mixture was diluted with Et.sub.2O, and the resulting solids were removed by filtration. The filtrate was concentrated and compound E was isolated by silica gel column chromatography (12 g dry load cartridge, 80 g column; eluent: 100% hexanes ramping to 55% EtOAc in hexanes). .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 8.30 (d, J=9.2 Hz, 2H), 7.48 (t, J=9.6 Hz, 2H), 7.40 (t, J=7.8 Hz, 2H), 7.20-7.27 (m, 3H), 6.60 (quart, J=11.6 Hz, 1H), 4.01 (m, 2H), 3.61 (m, 1H), 1.93 (m, 1H), 1.11 (m, 3H), 0.79 (m, 6H). .sup.31P NMR (162 MHz, DMSO-d.sub.6) .delta. -0.342 (s), -0.578 (s). MS m/z=422.9 [M+H].
Preparation of (2S)-ethyl 2-(((((2R,3 S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydro- xytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-3-methylbutanoat- e (Compound 22)
##STR00161##
[0358] Compound 1 (0.040 g, 0.137 mmol) was dissolved in NMP (1.5 mL) and then THF (0.25 mL) was added. This solution was cooled in an ice bath and t-BuMgCl (1M/THF, 425.7 .mu.L, 0.426 .mu.mol) was added in a drop-wise manner with vigorous stirring. The ice bath was removed and the resulting white slurry was stirred at RT for about 15 min. A solution of compound E (0.081 g, 0.192 mmol) in THF (0.5 mL) was added in a drop-wise manner to the reaction at RT. The reaction progress was monitored by LC/MS. When the reaction progressed to 50% conversion, the reaction was cooled in an ice bath and quenched with glacial acetic acid (70 .mu.L). The reaction was concentrated and compound 22 was semi-purified from the residue by reverse phase HPLC. The semi-pure material was further purified by silica gel column chromatography (12 g dry load cartridge, 40 g column; eluent: 100% EtOAc ramping to 10% MeOH in EtOAc) to yield compound 22. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 7.91 (d, J=1.6 Hz, 1H), 7.88 (brs, 2H), 7.32 (m, 2H), 7.15 (m, 3H), 6.90 (t, J=4.2 Hz, 1H), 6.84 (d, J=4.8 Hz, 1H), 6.26 (dd, J=13.4, 6.2 Hz, 1H), 5.87 (quart. J=11.2 Hz, 1H), 5.35 (m, 1H), 4.64 (m, 1H), 4.25 (m, 2H), 3.93-4.15 (m, 4H), 3.45 (m, 1H), 1.87 (m, 1H), 1.09-1.16 (m, 3H), 0.70-0.83 (m, 6H). .sup.31P NMR (162 MHz, DMSO-d.sub.6) .delta. 4.59 (s), 4.47 (s). MS m/z=575.02 [M+H].
Example 26. (S)-isopropyl 2-(((R)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cy- ano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)pr- opanoate (23)
##STR00162##
[0360] The preparation of (S)-isopropyl 2-(((R)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cy- ano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)pr- opanoate is described below.
##STR00163##
[0361] Compound 1 (60.0 mg, 206 .mu.mop was dissolved in NMP (0.28 mL). THF (0.2 mL) was added followed by tert-butyl magnesium chloride (1.0M solution in tetrahydrofuran, 0.309 mL) at RT under an argon atmosphere. After 20 min, a solution of compound F (Prepared according to Cho, A. et al J. Med. Chem. 2014, 57, 1812-1825., 81 mg, 206 .mu.mop in THF (0.2 mL) was added, and the resulting mixture was warmed to about 50.degree. C. After 3 h, the reaction mixture was allowed to cool to RT and was purified directly by preparatory HPLC (Phenominex Synergi 4u Hydro-RR 80 .ANG. 150.times.30 mm column, 5-100% acetonitrile/water gradient) to afford compound 23. .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 7.86 (s, 1H), 7.34-7.26 (m, 2H), 7.21-7.12 (m, 3H), 6.91 (d, J=4.6 Hz, 1H), 6.87 (d, J=4.6 Hz, 1H), 4.92 (sept, J=6.3 Hz, 1H), 4.80 (d, J=5.4 Hz, 1H), 4.43-4.34 (m, 1H), 4.33-4.24 (m, 1H), 4.18 (t, J=5.6 Hz, 1H), 3.82 (dq, J=9.7, 7.1 Hz, 2H), 1.27 (dd, J=7.1, 1.0 Hz, 3H), 1.18 (dd, J=6.3, 4.8 Hz, 6H). .sup.31P NMR (162 MHz, CD.sub.3OD) .delta. 3.72 (s). LC/MS: t.sub.R=1.39 min, MS m/z=561.11 [M+H]; LC system: Thermo Accela 1250 UHPLC; MS system: Thermo LCQ Fleet; Column: Kinetex 2.6u XB-C18 100A, 50.times.4.6 mm; Solvents: ACN with 0.1% acetic acid, water with 0.1% acetic acid; Gradient: 0 min-2.0 min 2-100% ACN, 2.0 min-3.05 min 100% ACN, 3.05 min-3.2 min 100%-2% ACN, 3.2 min-3.5 min 2% ACN at 2 .mu.l/min. HPLC: t.sub.R=2.523 min; HPLC system: Agilent 1100 series.; Column: Gemini 5.mu. C18 110A, 50.times.4.6 mm; Solvents: ACN with 0.1% TFA, Water with 0.1% TFA; Gradient: 0 min-5.0 min 2-98% ACN, 5.0 min-6.0 min 98% ACN at 2 mL/min.
Example 27. (2S)-cyclobutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl) Amino)propanoate (24)
##STR00164##
[0363] The preparation of (2S)-cyclobutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propan- oate is described below.
Preparation of (2S)-cyclobutyl 2-(((4-nitrophenoxy)(phenoxy)phosphoryl)amino)propanoate (Compound G)
##STR00165##
[0365] Phenyl dichlorophosphate (1.49 mL, 10 mmol) was dissolved in 10 mL of anhydrous DCM and stirred under atmosphere nitrogen in an ice bath. L-Alanine isobutyl ester hydrochloride (0.9 g, 5 mmol) was added in one portion. Triethylamine (765 .mu.L, 5.5 mmol) was then added dropwise. Reaction stirred for about 1 h. More Triethylamine (765 .mu.L, 5.5 mmol) was added dropwise and the reaction was stirred for about 45 min. p-Nitrophenol (1.25 g, 9 mmol) was added in one portion and stirred for about 30 min. Triethylamine (765 .mu.L, 5.5 mmol) was added and the reaction mixture was stirred for about 2 h. Additional p-nitrophenol (1.25 g, 9 mmol) and triethylamine (765 .mu.L, 5.5 mmol) were then added, and the reaction was stirred for another about 2 h. The reaction mixture was concentrated under reduced pressure. The resulting crude was diluted with EtOAc and washed twice with 5% aqueous citric acid solution, followed with saturated aqueous sodium chloride solution. The organic layer was then dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude residue was purified with silica gel column (0-20-50% EtOAc in hexanes) to give compound G. .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 8.33-8.23 (m, 2H), 7.52-7.33 (m, 4H), 7.33-7.17 (m, 3H), 4.96-4.85 (m, 1H), 4.07-3.96 (m, 1H), 2.27 (m, 2H), 2.07-1.91 (m, 2H), 1.83-1.70 (m, 1H), 1.70-1.55 (m, 1H), 1.32 (m, 3H). .sup.31P NMR (162 MHz, CD.sub.3OD) .delta. -1.36, -1.59. MS m/z=420.9 [M+H].
Preparation (2S)-cyclobutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propan- oate (Compound 24)
##STR00166##
[0367] Compound 1 (58 mg, 0.2 mmol) was mixed with compound G (101 mg, 0.24 mmol) in 2 mL of anhydrous DMF. Magnesium chloride (42 mg, 0.44 mmol) was added in one portion. The reaction mixture was heated to about 50.degree. C. DIPEA (87 .mu.L, 0.5 mmol) was added, and the reaction was stirred for about 2 h at about 50.degree. C. The reaction mixture was cooled to room temperature, was diluted with EtOAc and was washed with 5% aqueous citric acid solution followed by saturated aqueous sodium chloride solution. The organic layer was then dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude residue was purified with silica gel column (0-2-5% MeOH in DCM) to afford compound 24. .sup.1H NMR (400 MHz, Methanol-d.sub.4) .delta. 7.85 (m, 1H), 7.34-7.22 (m, 2H), 7.22-7.08 (m, 3H), 6.94-6.84 (m, 2H), 4.95-4.85 (m, 1H), 4.79 (m, 1H), 4.46-4.34 (m, 2H), 4.34-4.24 (m, 1H), 4.19 (m, 1H), 3.81 (m, 1H), 2.27 (m, 2H), 2.01 (m, 2H), 1.84-1.68 (m, 1H), 1.62 (m, 1H), 1.30-1.16 (m, 3H). .sup.31P NMR (162 MHz, cd.sub.3od) .delta. 3.70, 3.65. MS m/z=573.0 [M+H].
Example 28. (2S)-isopropyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-3-phe- nylpropanoate (25)
##STR00167##
[0369] The preparation of (2S)-isopropyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-3-phe- nylpropanoate is described below.
Preparation of (2S)-isopropyl 2-(((4-nitrophenoxy)(phenoxy)phosphoryl)amino)-3-phenylpropanoate (Compound H)
##STR00168##
[0371] Phenyl dichlorophosphate (718 .mu.L, 4.8 mmol) was dissolved in 10 mL of anhydrous DCM and stirred under a nitrogen atmosphere in an ice bath. L-Phenylalanine isopropyl ester hydrochloride (1 g, 4.1 mmol) was added in one portion. Another 10 mL of anhydrous DCM was added. Triethylamine (736 .mu.L, 5.3 mmol) was added dropwise and the reaction mixture was stirred for about 30 min. More triethylamine (736 .mu.L, 5.3 mmol) was then added dropwise and the reaction mixture was stirred for 30 min. Additional triethylamine (736 .mu.L, 5.3 mmol) was then added dropwise and the reaction mixture was stirred for about 15 min. p-Nitrophenol (600 mg, 4.32 mmol) was then added. The ice bath was then removed and the reaction mixture was allowed to warm to room temperature and stirred for about 2 h. More p-nitrophenol (50 mg) and triethylamine (736 .mu.L, 5.3 mmol) were the added and the reaction mixture was stirred for about 1 h.
[0372] The reaction mixture was then concentrated under reduced pressure, and was diluted with EtOAc and washed twice with 5% aqueous citric acid solution, followed with saturated aqueous sodium chloride solution. The organic layer was dried over anhydrous sodium sulfate and was concentrated under reduced pressure. The crude was purified with silica gel column (0-15% EtOAc in hexanes) to give compound H. .sup.1H NMR (400 MHz, CDCl.sub.3) .delta. 8.17 (m, 2H), 7.38-7.13 (m, 10H), 7.13-7.02 (m, 2H), 4.95 (m, 1H), 4.31 (m, 1H), 3.69 (m, 1H), 3.02 (dd, J=6.1, 1.8 Hz, 2H), 1.21-1.08 (m, 6H). .sup.31P NMR (162 MHz, cdcl3) .delta. -2.96, -2.98. MS m/z=485.0 [M+H].
Preparation of (2S)-isopropyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-3-phe- nylpropanoate (Compound 25)
##STR00169##
[0374] Compound 1 (58 mg, 0.2 mmol) and compound H (116 mg, 0.24 mmol) were mixed and 2 mL of anhydrous DMF was added. The reaction mixture was stirred under a nitrogen atmosphere at room temperature. 1M tBuMgCl in THF (300 .mu.L, 0.3 mmol) was added dropwise over 3 minutes and the reaction mixture was then stirred for about 16 h. The reaction mixture was diluted with EtOAc and washed with 5% aqueous citric acid solution, saturated aqueous sodium bicarbonate solution and then saturated aqueous sodium chloride solution. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude residue was purified with silica gel column (0-5% MeOH in DCM) to give compound 25. .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 7.84 (m, 1H), 7.27-7.08 (m, 8H), 7.08-6.97 (m, 2H), 6.88 (m, 2H), 4.91-4.84 (m, 1H), 4.74 (m, 1H), 4.26 (m, 1H), 4.19-4.04 (m, 2H), 4.04-3.91 (m, 2H), 2.97 (m, 1H), 2.82 (m, 1H), 1.14 (m, 3H), 1.06 (m, 3H). .sup.31P NMR (162 MHz, CD.sub.3OD) .delta. 3.63, 3.25. MS m/z=637.0 [M+H].
Example 29. (S)-methyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cy- ano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino) Propanoate (26)
##STR00170##
[0376] The preparation of (S)-methyl 2-(((S)-(((2R,3 S,4R,5R)-5-(4-aminopyrrolo[2,1-f][ 1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(ph- enoxy)phosphoryl)amino)propanoate is described below.
##STR00171##
[0377] Compound 1 (100 mg, 0.34 mmol) was dissolved in THF (2 mL) and cooled with an ice water bath. Then 1M t-BuMgCl (0.52 mL, 0.77 mmol) was added dropwise slowly. The resulting mixture was stirred for about 30 min at room temperature. Then compound I (Prepared according to WO 2012142085, 219 mg, 0.52 mmol) in THF (2 mL) was added over 5 min and the resulting mixture was stirred for about 24 h at room temperature. The reaction mixture was then diluted with EtOAc, cooled under ice-water bath, washed with aq NaHCO.sub.3 (2 mL), washed with brine, dried with sodium sulfate, and concentrated in vacuo. The resulting mixture was purified by silica gel column chromatography (MeOH 0 to 20% in DCM) and prep-HPLC (acetonitrile 10 to 80% in water) to give compound 26. .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 7.86 (s, 1H), 7.29 (dd, J=8.6, 7.2 Hz, 2H), 7.21-7.09 (m, 3H), 6.94-6.81 (m, 2H), 4.79 (d, J=5.4 Hz, 1H), 4.38 (ddq, J=10.8, 5.3, 2.7 Hz, 2H), 4.33-4.23 (m, 1H), 4.18 (t, J=5.5 Hz, 1H), 3.86 (dq, J=9.9, 7.1 Hz, 1H), 3.62 (s, 3H), 1.27 (dd, J=7.2, 1.1 Hz, 3H). MS m/z=533 (M+1).sup.+.
Example 30. (S)-neopentyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cy- ano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino) Propanoate (27)
##STR00172##
[0379] The preparation of (S)-neopentyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cy- ano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)pr- opanoate is described below.
##STR00173##
[0380] Compound 1 (100 mg, 0.34 mmol) was dissolved in THF (2 mL) and cooled under ice water bath. Then 1M t-BuMgCl (0.52 mL, 0.77 mmol) was added dropwise slowly. The resulting mixture was stirred for about 30 min at room temperature. Then compound J (Prepared according to WO2012075140, 248 mg, 0.52 mmol) was added over about 5 min and the resulting mixture was stirred for about 24 h at room temperature, diluted with EtOAc, cooled under ice-water bath, treated with aq NaHCO.sub.3 (2 mL), washed with brine, dried with sodium sulfate, and concentrated in vacuo. The resulting mixture was purified by silica gel column chromatography (MeOH 0 to 20% in DCM) and prep-HPLC (acetonitrile 10 to 80% in water) to give Compound 27. .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 7.86 (s, 1H), 7.36-7.24 (m, 2H), 7.23-7.10 (m, 3H), 6.96-6.85 (m, 2H), 4.78 (d, J=5.4 Hz, 1H), 4.38 (tdd, J=10.0, 4.9, 2.5 Hz, 2H), 4.32-4.24 (m, 1H), 4.17 (t, J=5.6 Hz, 1H), 3.91 (dq, J=9.8, 7.1 Hz, 1H), 3.81 (d, J=10.5 Hz, 1H), 3.69 (d, J=10.5 Hz, 1H), 1.31 (dd, J=7.2, 1.1 Hz, 3H), 0.89 (s, 9H). MS m/z=589 (M+1).sup.+.
Example 31. (2S)-cyclopentyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino) Propanoate (28)
##STR00174##
[0382] The preparation of (2S)-cyclopentyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propan- oate is described below.
##STR00175##
[0383] Compound 1 (100 mg, 0.34 mmol) was dissolved in THF (2 mL) and cooled under ice water bath. Then 1M t-BuMgCl (0.52 mL, 0.77 mmol) was added dropwise slowly. The resulting mixture was stirred for about 30 min at room temperature. Then compound K (Prepared according to WO2012075140, 247 mg, 0.52 mmol) in THF (2 mL) was added over about 5 min and the resulting mixture was stirred for about 24 h at room temperature, diluted with EtOAc, cooled under ice-water bath, treated with aq NaHCO.sub.3 (2 mL), washed with brine, dried with sodium sulfate, and concentrated in vacuo. The resulting mixture was purified by silica gel column chromatography (MeOH 0 to 20% in DCM) and prep-HPLC (acetonitrile 10 to 80% in water) to give example 28. .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 7.85 (s, 1H), 7.33-7.22 (m, 2H), 7.14 (tdd, J=7.6, 2.1, 1.1 Hz, 3H), 6.95-6.87 (m, 2H), 5.13-5.00 (m, 1H), 4.78 (d, J=5.4 Hz, 1H), 4.48-4.35 (m, 2H), 4.30 (ddd, J=10.6, 5.7, 3.6 Hz, 1H), 4.19 (t, J=5.4 Hz, 1H), 3.78 (dq, J=9.2, 7.1 Hz, 1H), 1.81 (dtd, J=12.5, 5.9, 2.4 Hz, 2H), 1.74-1.49 (m, 6H), 1.21 (dd, J=7.1, 1.2 Hz, 3H). MS m/z=587 (M+1).sup.+.
Example 32. (2S)-cyclohexyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino) Propanoate (29)
##STR00176##
##STR00177##
[0385] To a mixture of compound 1 (50 mg, 0.343 mmol), compound M (Prepared according to US20130143835, 93 mg, 0.209 mmol), and MgCl.sub.2 (24.5 mg, 0.257 mmol) in DMF (1 mL) was added diisopropylethylamine (0.075 mL, 0.43 mmol) dropwise over about 5 min at about 0.degree. C. The resulting mixture was stirred at about 50.degree. C. for about 1 h. The reaction mixture was then cooled with an ice-water bath, treated with 1M citric acid (0.5 mL), and was purified directly by prep-HPLC (ACN 0 to 70% in water) to afford compound 29. .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 7.84 (s, 1H), 7.32-7.23 (m, 2H), 7.18-7.10 (m, 3H), 6.93-6.87 (m, 2H), 4.78 (d, J=5.4 Hz, 1H), 4.67 (td, J=8.7, 4.2 Hz, 1H), 4.48-4.35 (m, 2H), 4.30 (ddd, J=10.8, 5.7, 3.7 Hz, 1H), 4.20 (t, J=5.4 Hz, 1H), 3.88-3.71 (m, 1H), 1.83-1.63 (m, 4H), 1.58-1.46 (m, 1H), 1.46-1.24 (m, 5H), 1.24 (s, 3H). .sup.31P NMR (162 MHz, CD.sub.3OD) .delta. 3.75. MS m/z=601 (M+1).sup.+.
Example 33. Ethyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-2-met- hylpropanoate (30)
##STR00178##
[0387] The preparation of ethyl 2-(((((2R,3 S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydro- xytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-2-methylpropanoa- te is described below.
Preparation of Ethyl 2-((tert-butoxycarbonyl)amino)-2-methylpropanoate
##STR00179##
[0389] Take up triphenylphosphine (6.18 g, 25.00 mmol) in THF (30 mL). Next charge DIAD (4.92 mL, 25.00 mmol) and stir at room temperature for 10 min. Dissolve 2-((tert-butoxycarbonyl)amino)-2-methylpropanoic acid (5.08 g, 25.00 mmol) in THF (20 mL) and add to the reaction mixture followed by the addition of ethanol (2.19 mL, 37.49 mmol). Allow the reaction to stir at room temperature for about 1 h. The solvents were removed under reduced pressure and the crude was taken up in 1:1 Et.sub.2O:Hexanes (120 mL). The solid triphenylphosphine oxide was filtered off and the solvent was removed under reduced pressure. The crude was taken up in minimal CH.sub.2Cl.sub.2 and purified by silica gel chromatography 0-50% EtOAc/Hex to afford ethyl 2-((tert-butoxycarbonyl)amino)-2-methylpropanoate. .sup.1H NMR (400 MHz, Chloroform-d) .delta. 4.18 (q, J=7.1 Hz, 2H), 1.49 (s, 6H), 1.43 (s, 9H), 1.27 (t, J=7.1 Hz, 3H).
Preparation of Ethyl 2-amino-2-methylpropanoate Hydrochloride
##STR00180##
[0391] Take up ethyl 2-((tert-butoxycarbonyl)amino)-2-methylpropanoate (2.71 g, 11.72 mmol) in CH.sub.2Cl.sub.2 (25 mL) and slowly add 4N HCl in dioxane (25 mmol) and stir at room temperature. At 1 h, the reaction was determined to be complete by TLC. The solvents were removed under reduced pressure and the crude was coevaporated with Et.sub.2O two times then placed under high vacuum to afford ethyl 2-amino-2-methylpropanoate hydrochloride. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 8.70 (s, 3H), 4.18 (q, J=7.1 Hz, 2H), 1.46 (s, 6H), 1.21 (t, J=7.1 Hz, 3H).
Preparation of Ethyl 2-methyl-2-(((4-nitrophenoxy)(phenoxy)phosphoryl)amino)propanoate (Compound N)
##STR00181##
[0393] Take up phenyl dichlorophosphate (0.97 mL, 6.50 mmol) and ethyl 2-amino-2-methylpropanoate hydrochloride (1.09 g, 6.50 mmol) in CH.sub.2Cl.sub.2 (50 mL). Cool the reaction mixture to about 0.degree. C. and slowly add TEA (1.75 mL, 12.45 mmol). Remove the cold bath and allow the reaction mixture to stir at room temperature. After about 2 h, the addition of the amino acid was determined to be complete by .sup.31P NMR. Charge p-nitrophenol (0.860 g, 6.17 mmol) followed by the addition of TEA (0.87 g, 7.69 mmol). Allow the reaction to stir at room temperature. After about 2 h, the reaction was determined to be complete by LCMS. The reaction was diluted with Et.sub.2O and the TEA.HCl salts were filtered off The crude was concentrated and purified by silica gel chromatography (0-50% EtOAc/Hex) to afford compound N. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 8.37-8.21 (m, 2H), 7.55-7.44 (m, 2H), 7.43-7.33 (m, 2H), 7.30-7.09 (m, 3H), 6.57 (d, J=10.1 Hz, 1H), 3.99 (q, J=7.1 Hz, 2H), 1.39 (s, 6H), 1.08 (t, J=7.1 Hz, 3H). .sup.31P NMR (162 MHz, DMSO-d.sub.6) .delta. -2.87. LC/MS: t.sub.R=1.65 min, MS m/z=408.97 [M+1].; LC system: Thermo Accela 1250 UHPLC; MS system: Thermo LCQ Fleet; Column: Kinetex 2.6.mu. XB-C18 100A, 50.times.3.00 mm; Solvents: Acetonitrile with 0.1% formic acid, Water with 0.1% formic acid; Gradient: 0 min-2.4 min 2-100% ACN, 2.4 min-2.80 min 100% ACN, 2.8 min-2.85 min 100%-2% ACN, 2.85 min-3.0 min 2% ACN at 1.8 mL/min.
Preparation of Ethyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-2-met- hylpropanoate (Compound 30)
##STR00182##
[0395] Take up compound 1 (66 mg, 0.23 mmol) in NMP (2.0 mL). Cool the mixture to about 0.degree. C. and slowly add tBuMgCl (1.0M in THF, 0.34 mL, 0.34 mmol). Allow the reaction to stir at about 0.degree. C. for about 30 min, then add a solution of compound N (139 mg, 0.34 mmol) dissolved in THF (1.0 mL). Remove the cold bath and place the reaction in about 50.degree. C. preheated oil bath. After about 2 h, the reaction was cooled to room temperature and quenched with acetic acid and methanol. The crude was concentrated and purified by reverse phase HPLC without modifier to afford compound 30. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 7.89 (m, 3H), 7.31 (q, J=8.1 Hz, 2H), 7.22-7.05 (m, 3H), 6.87 (d, J=4.5, 1H), 6.80 (d, J=4.5 Hz, 1H), 6.27 (d, J=11.7, 1H), 5.81 (d, J=9.7, 1H), 5.35 (d, J=5.6 Hz, 1H), 4.64 (dt, J=9.0, 5.6 Hz, 1H), 4.24 (m, 2H), 4.11 (m, 1H), 4.04-3.90 (m, 3H), 1.39-1.23 (m, 6H), 1.10 (t, J=7.1, 3H). .sup.31P NMR (162 MHz, DMSO-d.sub.6) .delta. 2.45, 2.41. LC/MS: t.sub.R=1.03 min, MS m/z=561.03 [M+1]; LC system: Thermo Accela 1250 UHPLC; MS system: Thermo LCQ Fleet; Column: Kinetex 2.6.mu. XB-C18 100A, 50.times.3.00 mm; Solvents: Acetonitrile with 0.1% formic acid, Water with 0.1% formic acid; Gradient: 0 min-2.4 min 2-100% ACN, 2.4 min-2.80 min 100% ACN, 2.8 min-2.85 min 100%-2% ACN, 2.85 min-3.0 min 2% ACN at 1.8 mL/min.
Example 34. Isopropyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-2-met- hylpropanoate (31)
##STR00183##
[0397] The preparation of Isopropyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-2-met- hylpropanoate is described below.
Preparation of Isopropyl 2-((tert-butoxycarbonyl)amino)-2-methylpropanoate
##STR00184##
[0399] Take up triphenylphosphine (6.17 g, 25.00 mmol) in THF (30 mL). Next charge DIAD (4.92 mL, 25.00 mmol) and stir at room temperature for about 10 min. Dissolve 2-((tert-butoxycarbonyl)amino)-2-methylpropanoic acid (5.07 g, 25.00 mmol) dissolved in THF (20 mL) and add to the reaction mixture followed by the addition of isopropanol (1.91 mL, 25.00 mmol). Allow the reaction to stir at room temperature for about 1 h. The solvents were removed under reduced pressure and the crude was taken up in 1:1 Et.sub.2O:Hexanes (120 mL). The solid triphenylphosphine oxide was filtered off and the solvent was removed under reduced pressure. The crude was taken up in minimal CH.sub.2Cl.sub.2 and purified by silica gel chromatography (0-50% EtOAc/Hex) to afford isopropyl 2-((tert-butoxycarbonyl)amino)-2-methylpropanoate. .sup.1H NMR (400 MHz, Chloroform-d) .delta. 5.03 (p, J=6.2 Hz, 1H), 1.48 (s, 6H), 1.40 (d, J=6.2 Hz, 9H), 1.24 (d, J=6.3 Hz, 6H).
Preparation of Isopropyl 2-amino-2-methylpropanoate Hydrochloride
##STR00185##
[0401] Take up isopropyl 2-((tert-butoxycarbonyl)amino)-2-methylpropanoate (4.09 g, 16.67 mmol) in CH.sub.2Cl.sub.2 (50 mL) and slowly add 4N HCl in dioxane (50 mmol) and stir at room temperature. At about 1 h, the reaction was determined to be complete by TLC. The solvents were removed under reduced pressure and the crude was coevaporated with Et.sub.2O two times then placed under high vacuum to afford isopropyl 2-amino-2-methylpropanoate hydrochloride. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 8.61 (s, 3H), 4.96 (p, J=6.2 Hz, 1H), 1.44 (s, 6H), 1.22 (d, J=6.2 Hz, 6H).
Preparation of Isopropyl 2-methyl-2-(((4-nitrophenoxy)(phenoxy)phosphoryl)amino) propanoate (Compound 0)
##STR00186##
[0403] Take up phenyl dichlorophosphate (0.83 mL, 5.58 mmol) and isopropyl 2-amino-2-methylpropanoate hydrochloride (1.01 g, 5.58 mmol) in CH.sub.2Cl.sub.2 (50 mL). Cool the reaction mixture to 0.degree. C. and slowly add TEA (1.61 mL, 11.45 mmol). Remove the cold bath and allow the reaction mixture to stir at room temperature. After about 2 h, the addition of the amino acid was determined to be complete by .sup.31P NMR. Charge p-nitrophenol (0.74 g, 5.30 mmol) followed by the addition of TEA (0.81, 5.84 mmol). Allow the reaction to stir at room temperature. After about 2 h, the reaction was determined to be complete by LCMS. The reaction was diluted with Et.sub.2O and the TEA.HCl salts were filtered off. The crude was concentrated and purified by silica gel chromatography (0-50% EtOAc/Hex) to afford compound O. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 8.42-8.19 (m, 2H), 7.55-7.43 (m, 2H), 7.39 (dd, J=8.6, 7.2 Hz, 2H), 7.30-7.12 (m, 3H), 6.53 (d, J=10.1 Hz, 1H), 4.82 (hept, J=6.3 Hz, 1H), 1.38 (s, 6H), 1.09 (d, J=6.3, 6H). .sup.31P NMR (162 MHz, DMSO-d.sub.6) .delta. -2.84. LC/MS: t.sub.R=1.73 min, MS m/z=422.92 [M+1]; LC system: Thermo Accela 1250 UHPLC; MS system: Thermo LCQ Fleet; Column: Kinetex XB-C18 100A, 50.times.3.00 mm; Solvents: Acetonitrile with 0.1% formic acid, Water with 0.1% formic acid; Gradient: 0 min-2.4 min 2-100% ACN, 2.4 min-2.80 min 100% ACN, 2.8 min-2.85 min 100%-2% ACN, 2.85 min-3.0 min 2% ACN at 1.8 mL/min.
Preparation of Isopropyl 2-(((((2R,3 S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydro- xytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)-2-methylpropanoa- te (Compound 31)
##STR00187##
[0405] Take up compound 1 (66 mg, 0.23 mmol) in NMP (2.0 mL). Cool the mixture to about 0.degree. C. and slowly add tBuMgCl (1.0M in THF, 0.57 mL, 0.57 mmol). Allow the reaction to stir at about 0.degree. C. for about 30 min, then add a solution of compound 0 (143 mg, 0.34 mmol) dissolved in THF (1.0 mL). Remove the cold bath and place the reaction in an about 50.degree. C. preheated oil bath. After about 2 h, the reaction was cooled to room temperature and was quenched with acetic acid and methanol. The crude was concentrated and purified by reverse phase HPLC without modifier to afford compound 31. .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 7.88 (m, 3H), 7.30 (td, J=8.5, 7.0 Hz, 2H), 7.20-7.04 (m, 3H), 6.87 (d, J=4.5, 1H), 6.80 (d, J=4.5 Hz, 1H), 6.27 (d, 6.1 Hz, 1H), 5.75 (t, J=9.1 Hz, 1H), 5.34 (d, J=5.7 Hz, 1H), 4.81 (p, J=6.3 Hz, 1H), 4.71-4.50 (m, 1H), 4.23 (m, 2H), 4.11 (m, 1H), 4.03-3.83 (m, 1H), 1.37-1.23 (m, 6H), 1.18-1.04 (m, 6H). .sup.31P NMR (162 MHz, DMSO) .delta. 2.47, 2.43. LC/MS: t.sub.R=1.08 min, MS m/z=575.06 [M+1]; LC system: Thermo Accela 1250 UHPLC; MS system: Thermo LCQ Fleet; Column: Kinetex 2.6.mu. XB-C18 100A, 50.times.3.00 mm; Solvents: Acetonitrile with 0.1% formic acid, Water with 0.1% formic acid; Gradient: 0 min-2.4 min 2-100% ACN, 2.4 min-2.80 min 100% ACN, 2.8 min-2.85 min 100%-2% ACN, 2.85 min-3.0 min 2% ACN at 1.8 mL/min.
Example 35. (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cy- ano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl) amino)propanoate (32)
##STR00188##
[0407] The preparation of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cy- ano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)pr- opanoate is described below.
Preparation of (3R,4R,5R)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl)dihydrofuran-2(3H)-one
##STR00189##
[0409] (3R,4R,5R)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-- 2-ol (15.0 g) was combined with MTBE (60.0 mL), KBr (424.5 mg), aqueous K.sub.2HPO.sub.4 solution (2.5M, 14.3 mL), and TEMPO (56 mg). This mixture was cooled to about 1.degree. C. Aqueous bleach solution (7.9% wt.) was slowly charged in portions until complete consumption of starting material as indicated through a starch/iodide test. The layers were separated, and the aqueous layer was extracted with MTBE. The combined organic phase was dried over MgSO.sub.4 and concentrated under reduced pressure to yield the product as a solid.
Preparation (4-amino-7-iodopyrrolo[2,1-f][1,2,4]triazine)
##STR00190##
[0411] To a cold solution of 4-aminopyrrolo[2,1-f][1,2,4]-triazine (10.03 g; 74.8 mmol) in N,N-dimethylformamide (70.27 g), N-iodosuccinimide (17.01 g; 75.6 mmol) was charged in portions, while keeping the contents at about 0.degree. C. Upon reaction completion (about 3 h at about 0.degree. C.), the reaction mixture was transferred into a 1 M sodium hydroxide aqueous solution (11 g NaOH and 276 mL water) while keeping the contents at about 20-30.degree. C. The resulting slurry was agitated at about 22.degree. C. for 1.5 h and then filtered. The solids are rinsed with water (50 mL) and dried at about 50.degree. C. under vacuum to yield 4-amino-7-iodopyrrolo[2,1-f][1,2,4]triazine as a solid. .sup.1H NMR (400 MHz, DMSO-d6) .delta. 7.90 (s, 1H), 7.78 (br s, 2H), 6.98 (d, J=4.4 Hz, 1H), 6.82 (d, J=4.4 Hz, 1H). .sup.13C NMR (101 MHz, DMSO-d6) .delta. 155.7, 149.1, 118.8, 118.1, 104.4, 71.9. MS m/z=260.97 [M+H].
Preparation (3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyloxy- )-5-((benzyloxy)methyl)tetrahydrofuran-2-ol via (4-amino-7-iodopyrrolo[2,1-f][1,2,4]triazine)
##STR00191##
[0413] To a reactor under a nitrogen atmosphere was charged iodobase 2 (81 g) and THF (1.6 L). The resulting solution was cooled to about 5.degree. C., and TMSCl (68 g) was charged. PhMgCl (345 mL, 1.8 M in THF) was then charged slowly while maintaining an internal temperature at about .ltoreq.5.degree. C. The reaction mixture was stirred at about 0.degree. C. for 30 min, and then cooled to about -15.degree. C. iPrMgCl--LiCl (311 mL, 1.1 M in THF) was charged slowly while maintaining an internal temperature below about -12.degree. C. After about 10 minutes of stirring at about -15.degree. C., the reaction mixture was cooled to about -20.degree. C., and a solution of lactone 1 (130 g) in THF (400 mL) was charged. The reaction mixture was then agitated at about -20.degree. C. for about 1 h and quenched with AcOH (57 mL). The reaction mixture was warmed to about 0.degree. C. and adjusted to pH 7-8 with aqueous NaHCO.sub.3 (5 wt %, 1300 mL). The reaction mixture was then diluted with EtOAc (1300 mL), and the organic and aqueous layers were separated. The organic layer was washed with 1N HCl (1300 mL), aqueous NaHCO.sub.3 (5 wt %, 1300 mL), and brine (1300 mL), and then dried over anhydrous Na.sub.2SO.sub.4 and concentrated to dryness. Purification by silica gel column chromatography using a gradient consisting of a mixture of MeOH and EtOAc afforded the product.
Preparation ((2S)-2-ethylbutyl 2-(((perfluorophenoxy)(phenoxy)phosphoryl)amino)propanoate) (Mixture of Sp and Rp)
##STR00192##
[0415] L-Alanine 2-ethylbutyl ester hydrochloride (5.0 g, 23.84 mmol) was combined with methylene chloride (40 mL), cooled to about -78.degree. C., and phenyl dichlorophosphate (3.65 mL, 23.84 mmol) was added. Triethylamine (6.6 mL, 47.68 mmol) was added over about 60 min at about -78.degree. C. and the resulting mixture was stirred at ambient temperature for 3 h. The reaction mixture was cooled to about 0.degree. C. and pentafluorophenol (4.4 g, 23.84 mmol) was added. Triethylamine (3.3 mL, 23.84 mmol) was added over about 60 min. The mixture was stirred for about 3 h at ambient temperature and concentrated under reduced pressure. The residue was dissolved in EtOAc, washed with an aqueous sodium carbonate solution several times, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using a gradient of EtOAc and hexanes (0 to 30%). Product containing fractions were concentrated under reduced pressure to give (2S)-2-ethylbutyl 2-(((perfluorophenoxy)(phenoxy)phosphoryl)amino)propanoate as a solid. .sup.1H NMR (400 MHz, Chloroform-d) .delta. 7.41-7.32 (m, 4H), 7.30-7.17 (m, 6H), 4.24-4.16 (m, 1H), 4.13-4.03 (m, 4H), 4.01-3.89 (m, 1H), 1.59-1.42 (m, 8H), 1.40-1.31 (m, 8H), 0.88 (t, J=7.5 Hz, 12H). .sup.31P NMR (162 MHz, Chloroform-d) .delta. -1.52. .sup.19F NMR (377 MHz, Chloroform-d) .delta. -153.63,-153.93 (m), -160.05 (td, J=21.9, 3.6 Hz), -162.65 (qd, J=22.4, 20.5, 4.5 Hz). MS m/z=496 [M+H].
Preparation ((2S)-2-ethylbutyl 2-(((perfluorophenoxy)(phenoxy)phosphoryl)amino)propanoate)
##STR00193##
[0417] L-alanine-2-ethylbutylester hydrochloride (40.10 g, 0.191 mmol) was dissolved in dichloromethane (533 g) and the solution was cooled with stirring to about -15.degree. C. under N.sub.2(g). Phenyl dichlorophosphate (40.32 g, 0.191 mol) was added followed by slow addition of triethylamine (41.58 g, 0.411 mmol) and the reaction mixture was stirred at about -15.degree. C. for about 1.5 h. Pentafluorophenol (35.14 g, 0.191 mol) was added, followed by triethylamine (19.23 g, 0.190 mol) and the reaction mixture was stirred for about 2 h. The reaction mixture was warmed to about 0.degree. C. and 0.5 M HCl (279.19 g) was added. The mixture was warmed to about 22.degree. C. and the organic layer was separated and washed with 5% KHCO.sub.3 aqueous solution (281 g), then water (281 g). An aliquot of the organic layer (453.10 g of the 604.30 g solution) was concentrated to about 120 mL volume, isopropyl acetate (157 g) was added and the solution was concentrated to dryness. The residue was dissolved in isopropyl acetate (158 g). The resulting solution was concentrated to about 120 mL volume and the temperature was adjusted to about 45.degree. C. n-Heptane (165 g) was added and the mixture was cooled to 22.degree. C. over about 1 h. n-Heptane (167 g) was added and the mixture was cooled to about 0.degree. C. Triethylamine (2.90 g, 0.0287 mol) was added and the mixture was stirred at 0.degree. C. for about 17 h. The mixture was filtered, the solids were rinsed with n-heptane (145 g) and the solids were dried under vacuum at about 40.degree. C. for about 15 h to provide 2-ethylbutyl ((S)-(penthafluorophenoxy)(phenoxy)phosphoryl)-L-alaninate.
Preparation 2-ethylbutyl ((S)-(4-nitrophenoxy)(phenoxy)phosphoryl)-L-alaninate
##STR00194##
[0419] A slurry of L-alanine-2-ethylbutylester hydrochloride (20.08 g, 95.8 mmol) and isopropyl acetate (174 g) was cooled with stirring to about -20.degree. C.). Phenyl dichlorophosphate (20.37 g, 96.5 mmol) was added, followed by slow addition of triethyl amine (20.97 g, 207.2 mmol) and the mixture was stirred at about -20.degree. C. for about 1 h. 4-Nitrophenol (13.23 g, 95.1 mmol) was added, followed by slow addition of triethylamine (10.01 g, 98.8 mmol) and the reaction mixture was stirred for about 1.5 h. The reaction mixture was warmed to about 0.degree. C. and 0.5 M HCl (140 g) was added. The organic layer was separated and washed with 5% Na.sub.2CO.sub.3 (2.times.100 g) and 10% NaCl (2.times.100 g). The organic layer was then concentrated to about 80 mL volume and isopropylacetate (4 g) was added, followed by n-heptane (110 g). Product seed crystals (0.100 g) were added followed by a second portion of n-heptane (110 g) and the mixture was cooled to about 0.degree. C. 1,8-Diazabicycloundec-7-ene (1.49 g, 9.79 mmol) was added and the mixture was stirred at about 0.degree. C. for about 21 h. The resultant solids were filtered and washed first with n-heptane (61 g) and then with H.sub.2O (2.times.100 g). The solids were stirred with H.sub.2O (200 g) for about 1.5 h, filtered, and rinsed with H.sub.2O (3.times.100 g), then n-heptane (61 g). The obtained solids were dried under vacuum at about 40.degree. C. for about 19 h to provide 2-ethylbutyl ((S)-(4-nitrophenoxy)(phenoxy)phosphoryl)-L-alaninate.
Preparation of Title Compound (Mixture of Sp and Rp)
##STR00195##
[0421] The nucleoside (29 mg, 0.1 mmol) and the phosphonamide (60 mg, 0.12 mmol) and N,N-dimethylformamide (2 mL) were combined at ambient temperature. Tert-Butyl magnesium chloride (1M in THF, 0.15 mL) was slowly added. After about 1 h, the reaction was diluted with ethyl acetate, washed with aqueous citric acid solution (5% wt.), aqueous saturated NaHCO.sub.3 solution and saturated brine solution. The organic phase was dried over Na.sub.2SO.sub.4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using a gradient of methanol and CH.sub.2Cl.sub.2 (0 to 5%). Product containing fractions were concentrated under reduced pressure to provide the product.
Preparation of (3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxyme- thyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile
##STR00196##
[0423] To a mixture of (2R,3R,4S,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-- 5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile (5.8 g, 0.02 mol), 2,2-dimethoxypropane (11.59 mL, 0.09 mol) and acetone (145 mL) at ambient temperature was added sulfuric acid (18M, 1.44 mL). The mixture was warmed to about 45.degree. C. After about 30 min, the mixture was cooled to ambient temperature and sodium bicarbonate (5.8 g) and water 5.8 mL) were added. After 15 min, the mixture was concentrated under reduced pressure. The residue was taken up in ethyl acetate (150 mL) and water (50 mL). The aqueous layer was extracted with ethyl acetate (2.times.50 mL). The combined organic phase was dried over sodium sulfate and concentrated under reduced pressure to give crude (2R,3R,4S,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-- 5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile. .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 7.84 (s, 1H), 6.93 (d, J=4.6 Hz, 1H), 6.89 (d, J=4.6 Hz, 1H), 5.40 (d, J=6.7 Hz, 1H), 5.00 (dd, J=6.7, 3.3 Hz, 1H), 4.48-4.40 (m, 1H), 3.81-3.72 (m, 2H), 1.71 (s, 3H), 1.40 (s, 3H). MS m/z=332.23 [M+1].
Preparation of (3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxyme- thyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile TsOH Salt
##STR00197##
[0425] To a mixture of (2R,3R,4S,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-- 5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile (5.0 g, 17.2 mmol, 1.0 equiv.), 2,2-dimethoxypropane (10.5 mL, 86 mmol, 5.0 equiv.) and acetone (25 mL) at ambient temperature was added p-tolylsulfonic acid (3.59 g, 1.1 equiv.). The mixture was stirred at ambient temperature. After about 30 min, isopropyl acetate (25 mL) was added over about one hour. The resulting slurry was filtered and rinsed with 2:1 heptane:isopropyl acetate (25 ml). The product was dried under vacuum at about 40.degree. C.
Preparation of (3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxyme- thyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile
##STR00198##
[0427] To a mixture of (2R,3R,4S,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-- 5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile (5 g, 17.2 mmol, 1.0 equiv.), 2,2-dimethoxypropane (10.5 mL, 86 mmol, 5.0 equiv.) and acetone (25 mL) at ambient temperature was added p-tolylsulfonic acide (3.59 g, 1.1 equiv.). The mixture was stirred at ambient temperature. After 30 min, isopropyl acetate (25 mL) was added over one hour. The resulting slurry was filtered and rinsed with 2:1 heptane:isopropyl acetate (25 ml). The product was dried under vacuum at 40.degree. C. The isolated solid was added to a reactor and 5% K.sub.2CO.sub.3 solution (50 ml) and ethyl acetate (50 mL) were added. The layers were separated, and the aqueous layer washed with ethyl acetate (25 ml). The combined organic layers were washed with water (25 ml), then concentrated to ca.25 ml. The reactor was refilled with isopropyl acetate (25 ml) and concentrated to ca. 25 ml. The reactor was again refilled with isopropyl acetate (25 ml) and concentrated to 25 ml. The resulting solution was seeded, producing a thick slurry. To this was added heptane (25 ml) over one hour. The resulting slurry was filtered and rinsed with 2:1 heptane:isopropyl acetate (25 ml). The product was dried under vacuum at 40.degree. C. 0 (2R,3R,4S,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-- 5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile. .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 7.84 (s, 1H), 6.93 (d, J=4.6 Hz, 1H), 6.89 (d, J=4.6 Hz, 1H), 5.40 (d, J=6.7 Hz, 1H), 5.00 (dd, J=6.7, 3.3 Hz, 1H), 4.48-4.40 (m, 1H), 3.81-3.72 (m, 2H), 1.71 (s, 3H), 1.40 (s, 3H). MS m/z=332.23 [M+1].
Preparation of (2S)-2-ethylbutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-- 3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propan- oate
##STR00199##
[0429] Acetonitrile (100 mL) was combined with (2S)-2-ethylbutyl nitrophenoxy)(phenoxy)phosphoryl)-amino)propanoate (9.6 g, 21.31 mmol), the substrate alcohol (6.6 g, 0.02 mol),), magnesium chloride ((1.9 g, 19.91 mmol) at ambient temperature. The mixture was agitated for about 15 min and N,N-diisopropylethylamine (8.67 mL, 49.78 mmol) was added. After about 4 h, the reaction was diluted with ethyl acetate (100 mL), cooled to about 0.degree. C. and combined with aqueous citric acid solution (5% wt., 100 mL). The organic phase was washed with aqueous citric acid solution (5% wt., 100 mL) and aqueous saturated ammonium chloride solution (40 mL), aqueous potassium carbonate solution (10% wt., 2.times.100 mL), and aqueous saturated brine solution (100 mL). The organic phase was dried with sodium sulfate and concentrated under reduced pressure to provide crude product. .sup.1H NMR (400 MHz, CD.sub.3OD) .delta. 7.86 (s, 1H), 7.31-7.22 (m, 2H), 7.17-7.09 (m, 3H), 6.93-6.84 (m, 2H), 5.34 (d, J=6.7 Hz, 1H), 4.98 (dd, J=6.6, 3.5 Hz, 1H), 4.59-4.50 (m, 1H), 4.36-4.22 (m, 2H), 4.02 (dd, J=10.9, 5.7 Hz, 1H), 3.91 (dd, J=10.9, 5.7 Hz, 1H), 3.83 (dq, J=9.7, 7.1 Hz, 1H), 1.70 (s, 3H), 1.50-1.41 (m, 1H), 1.39 (s, 3H), 1.36-1.21 (m, 7H), 0.86 (t, J=7.4 Hz, 6H). MS m/z=643.21 [M+1].
Preparation of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cy- ano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)pr- opanoate (Compound 32)
##STR00200##
[0431] The crude acetonide (12.85 g) was combined with tetrahydrofuran (50 mL) and concentrated under reduced pressure. The residue was taken up in tetrahydrofuran (100 mL), cooled to about 0.degree. C. and concentrated HCl (20 mL) was slowly added. The mixture was allowed to warm to ambient temperature. After consumption of the starting acetonide as indicated by HPLC analysis, water (100 mL) was added followed by aqueous saturated sodium bicarbonate solution (200 mL). The mixture was extracted with ethyl acetate (100 mL), the organic phase washed with aqueous saturated brine solution (50 mL), dried over sodium sulfated and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using a gradient of methanol and ethyl acetate (0 to 20%). Product containing fractions were concentrated under reduced pressure to provide the product.
Preparation of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cy- ano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)pr- opanoate (Compound 32)
##STR00201##
[0433] To a vial containing (S)-2-ethylbutyl 2-(((S)-(((3aR,4R,6R,6aR)-6-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-- cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methoxy)(phenoxy)p- hosphoryl)amino)propanoate (30 mg, 0.05 mmol) was added an 80% aqueous formic acid solution (1.5 mL). After 18 h at about 20.degree. C. complete conversion was confirmed by HPLC and LC-MS. MS

























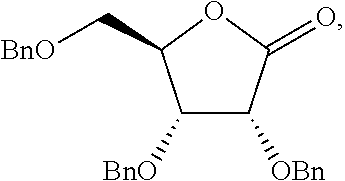






















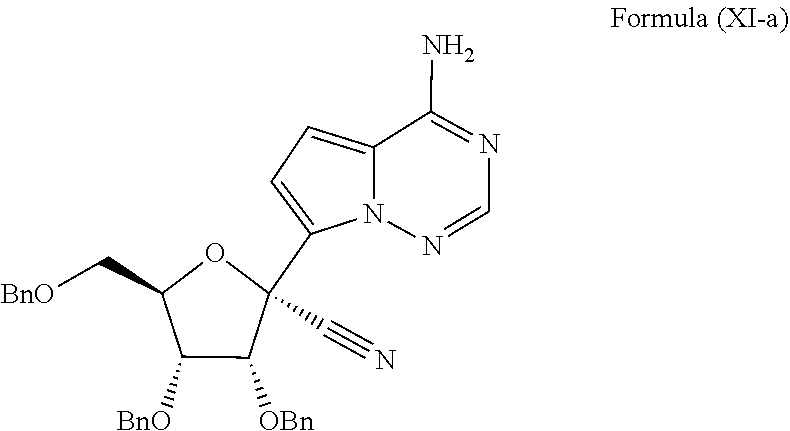




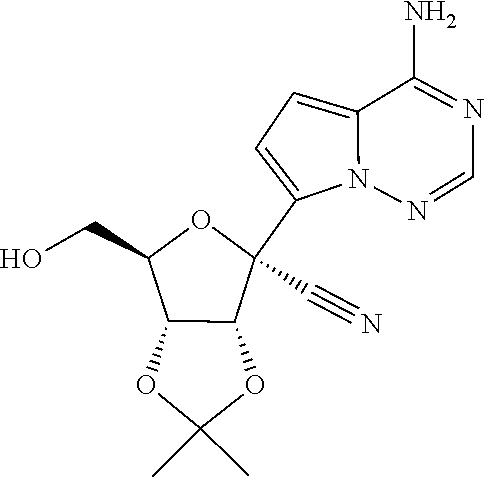



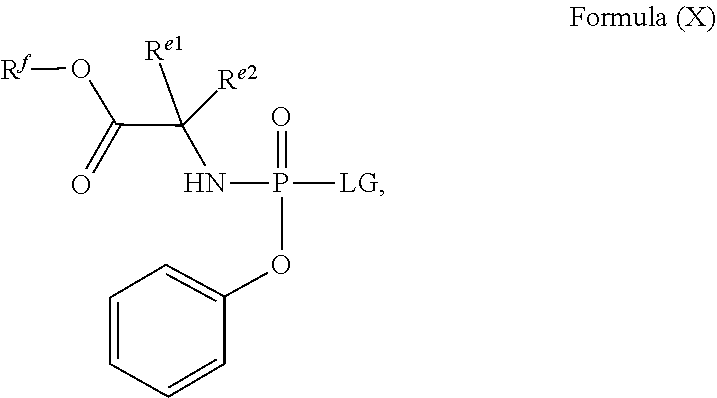



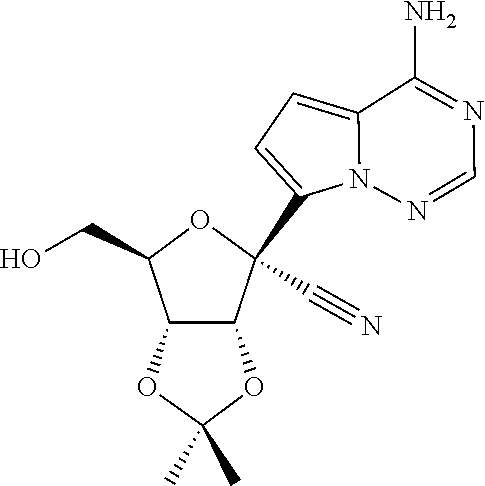







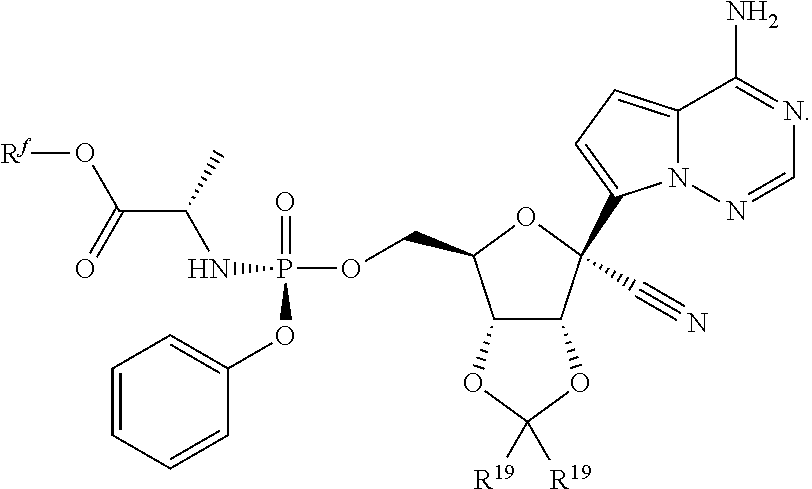




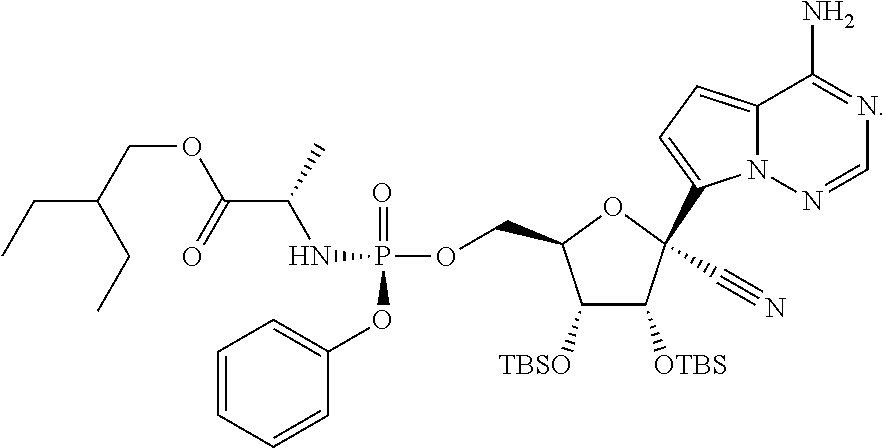




















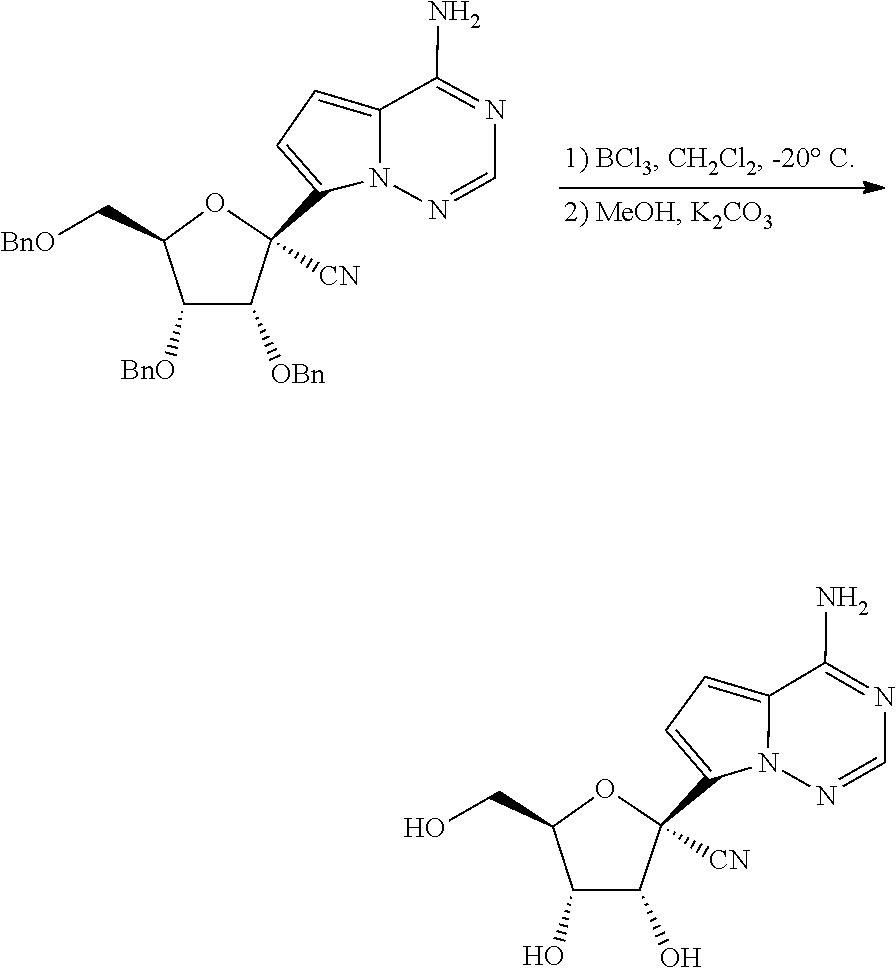




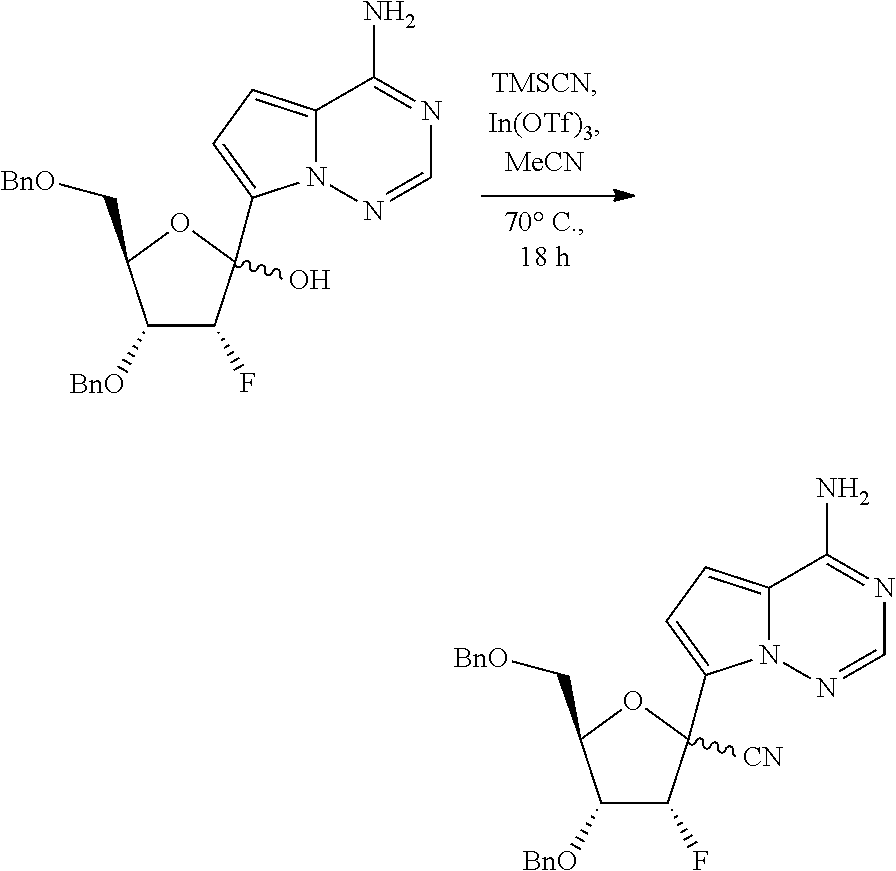






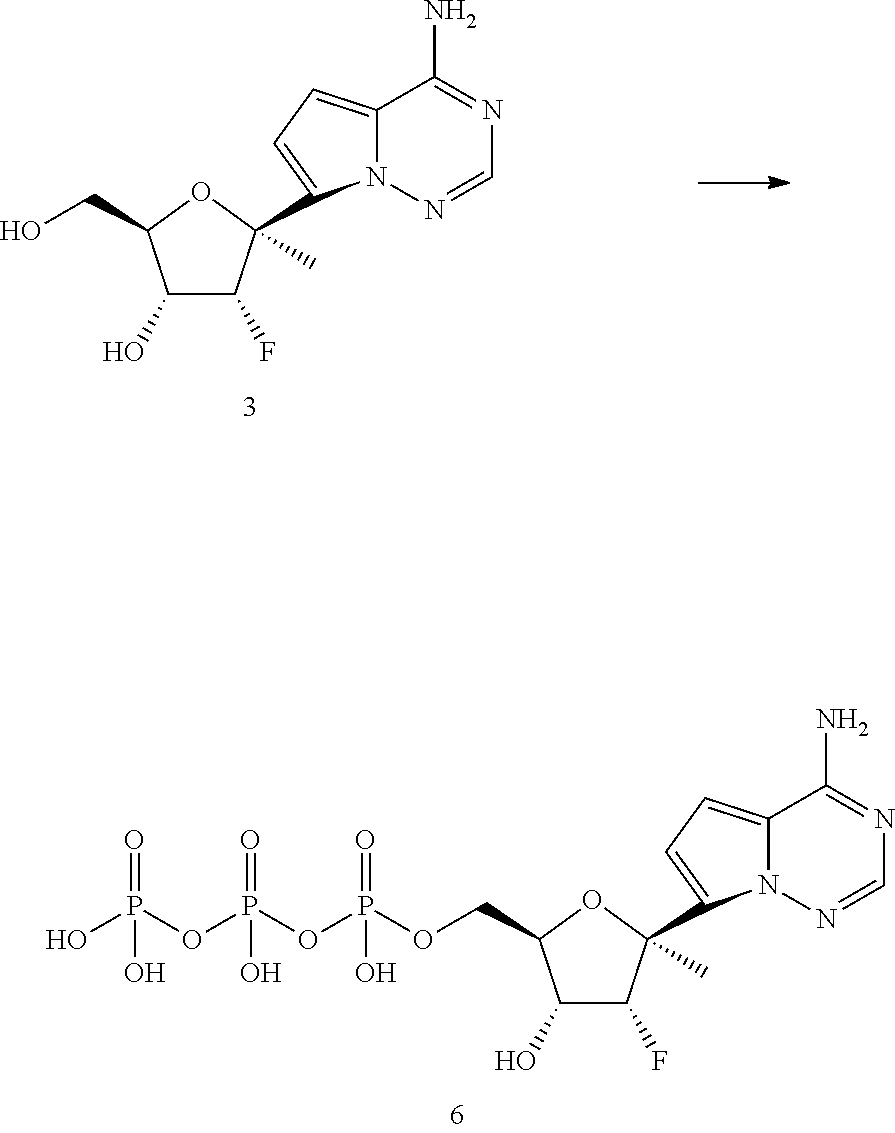
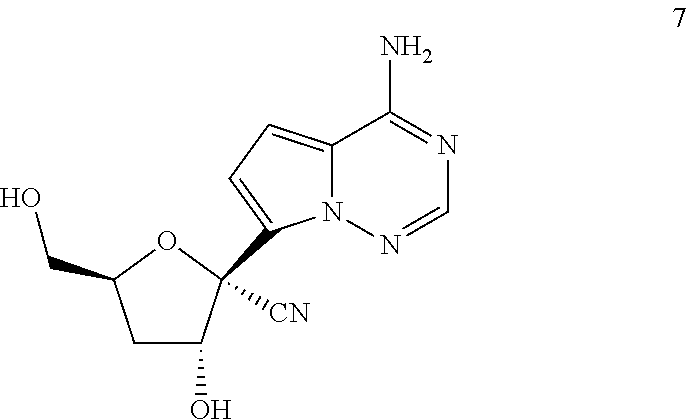





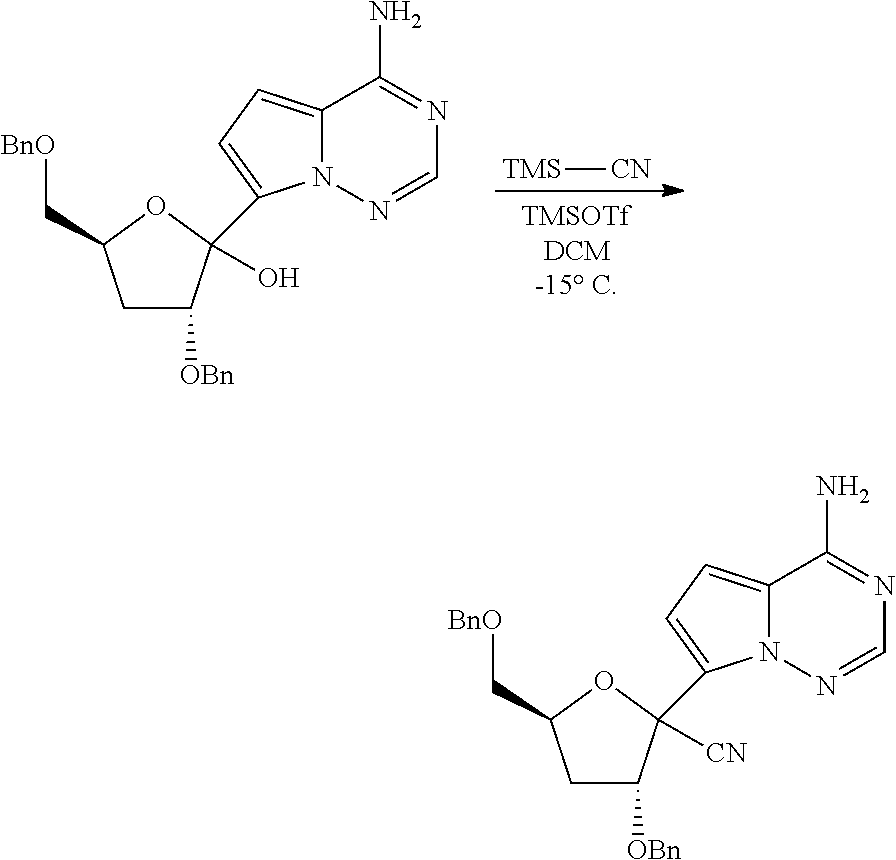








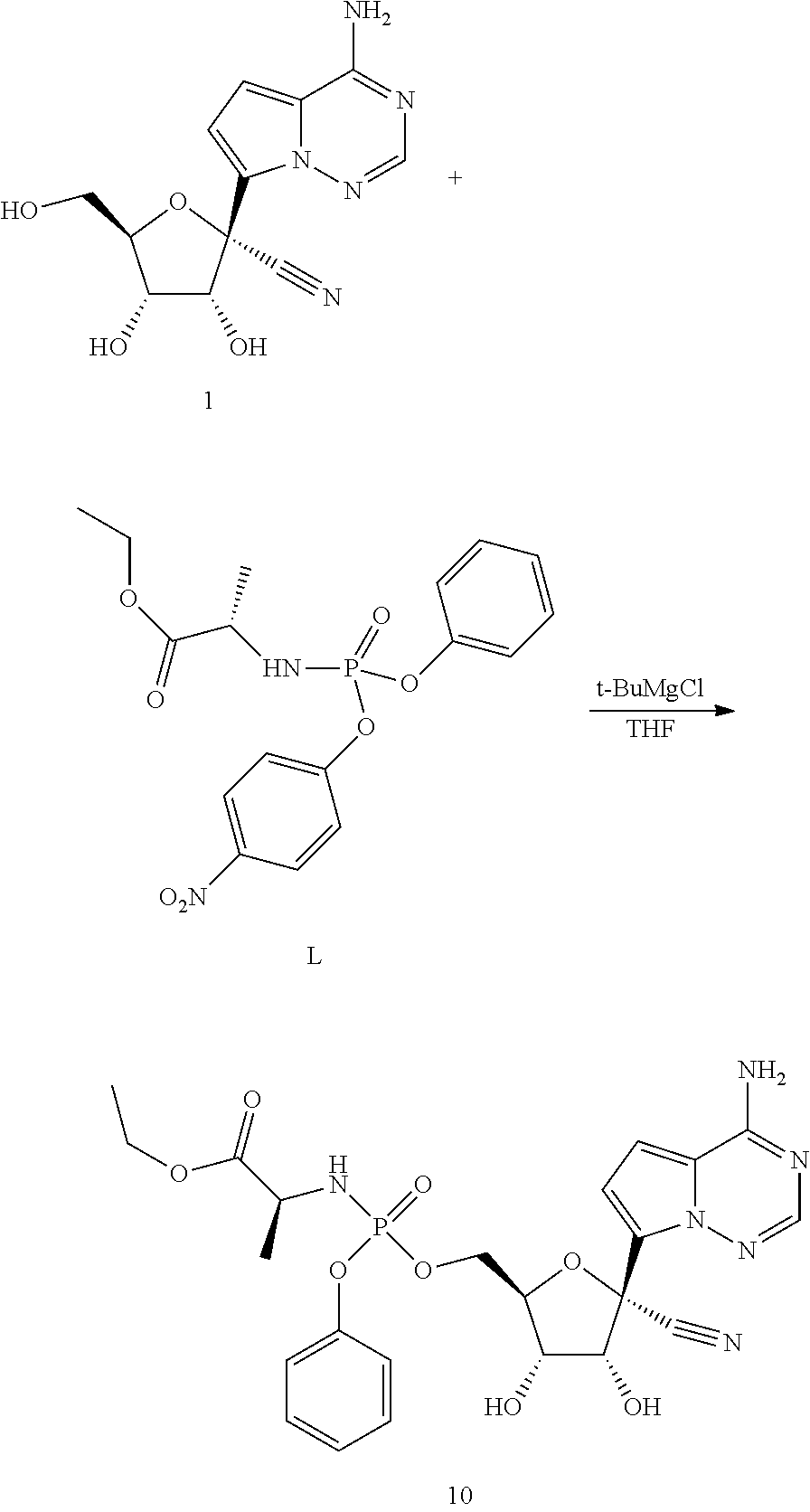









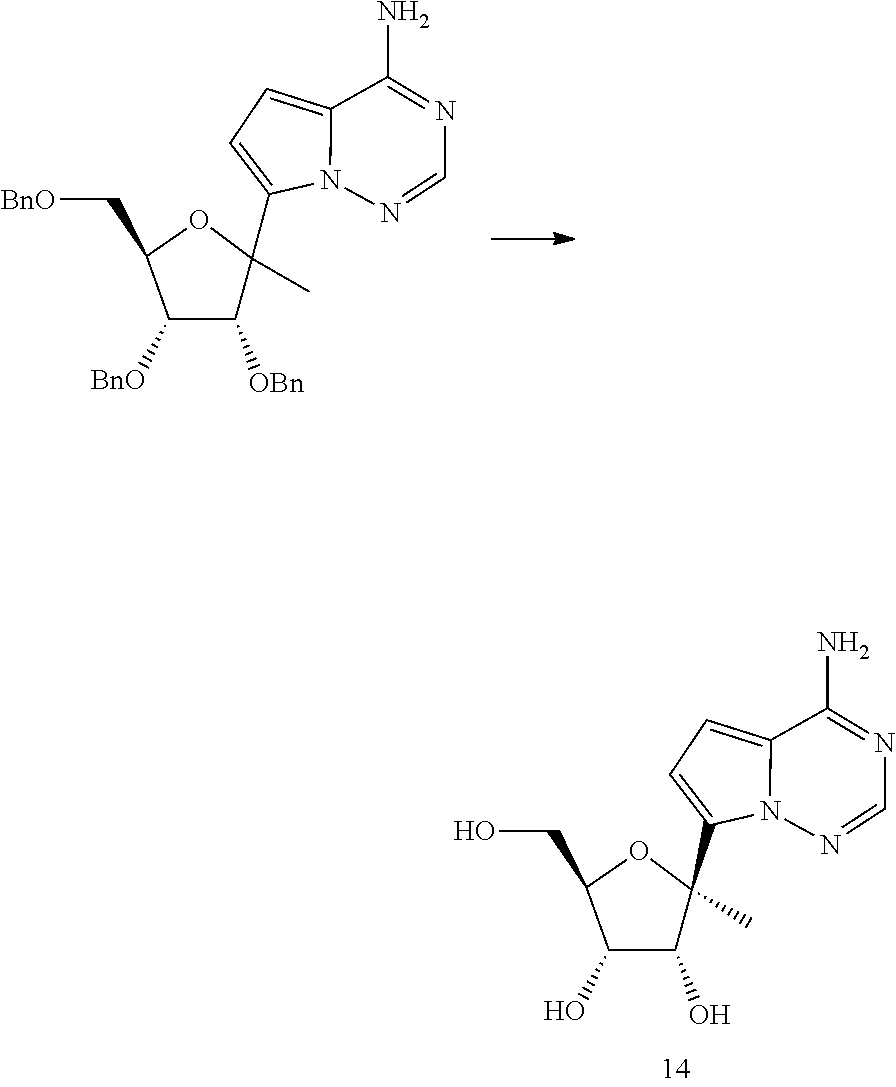



















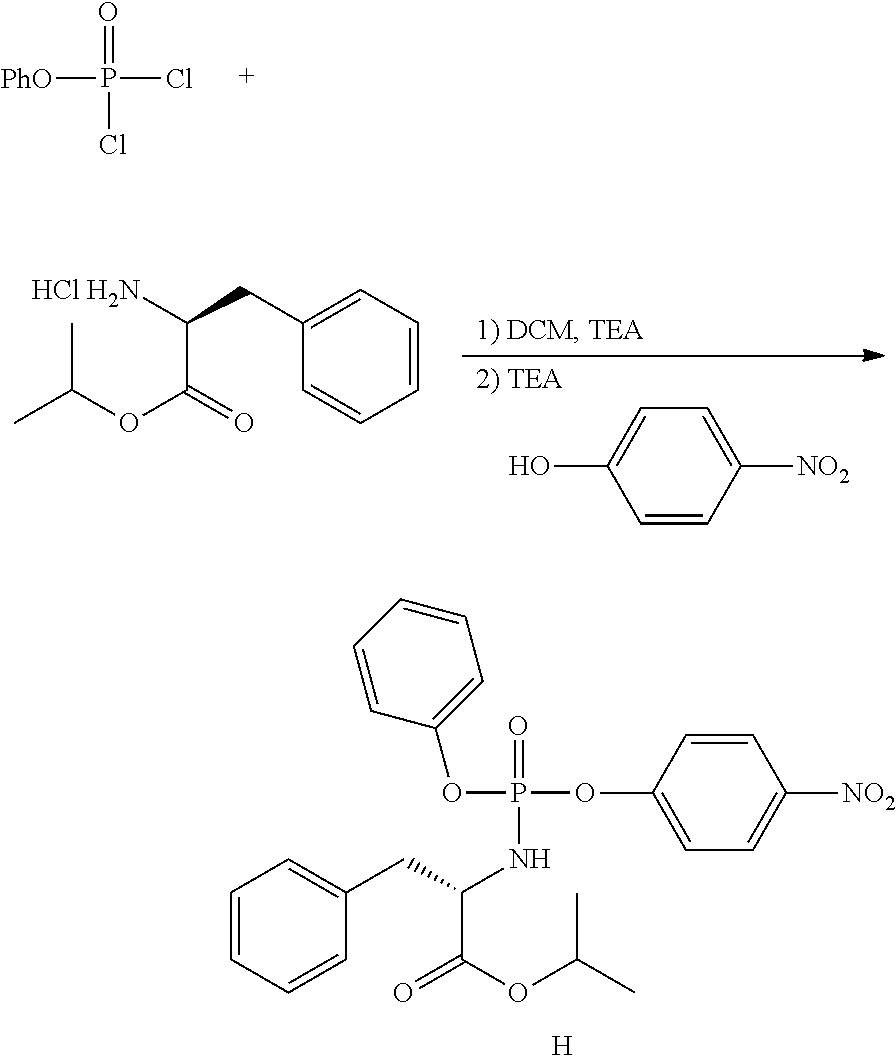



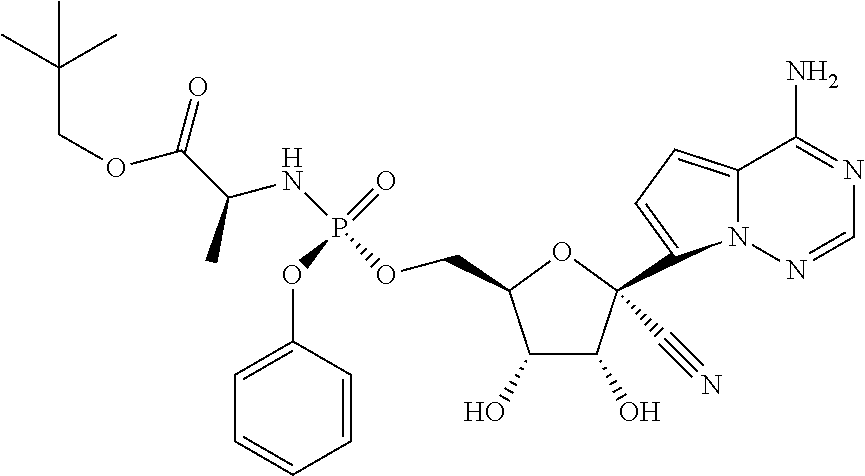



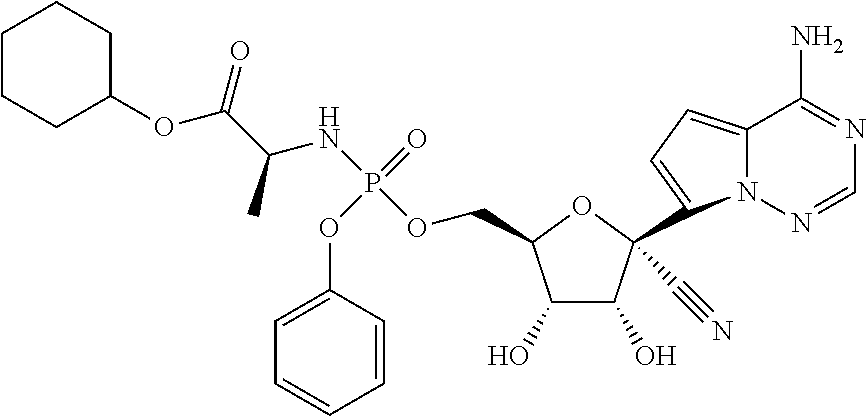
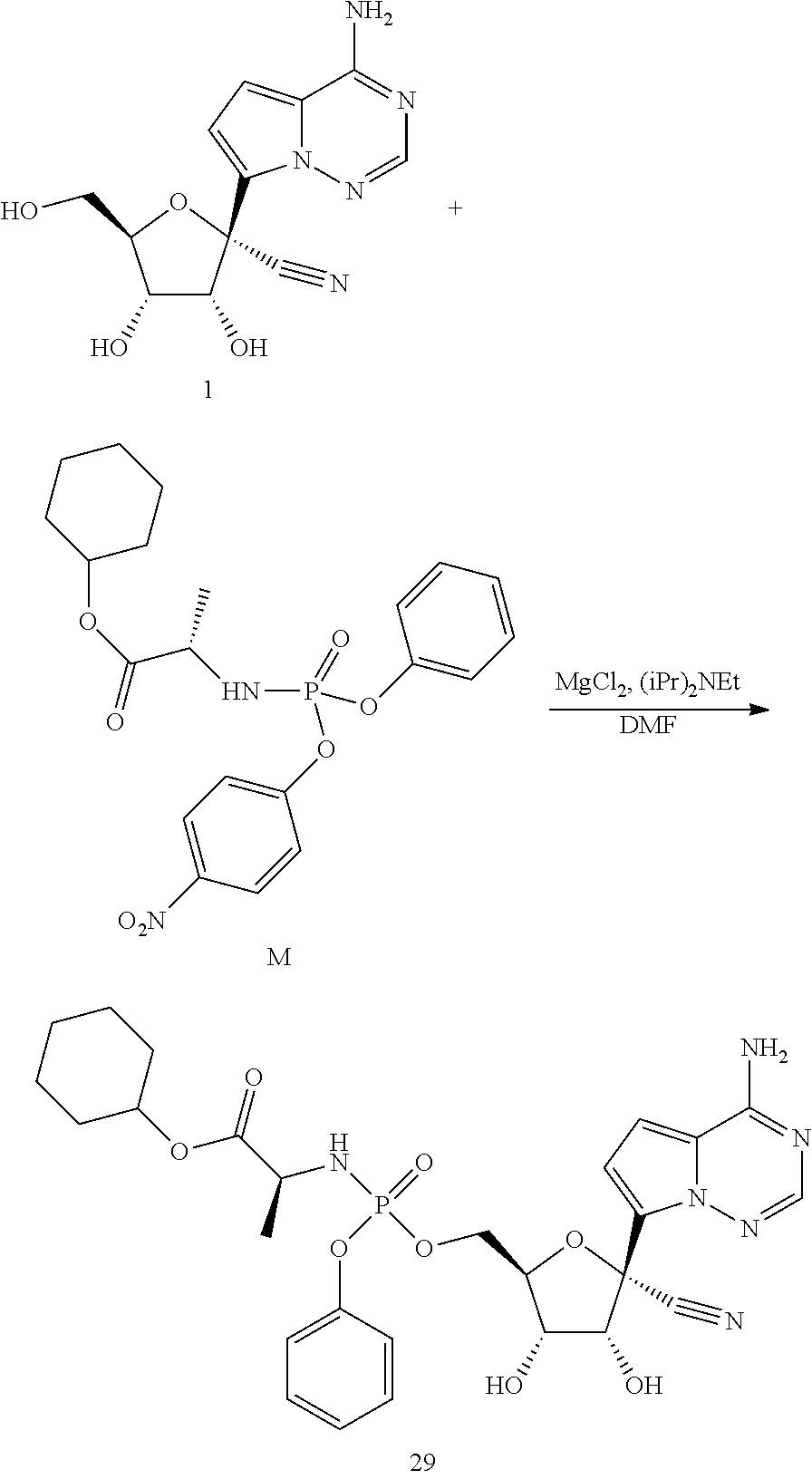







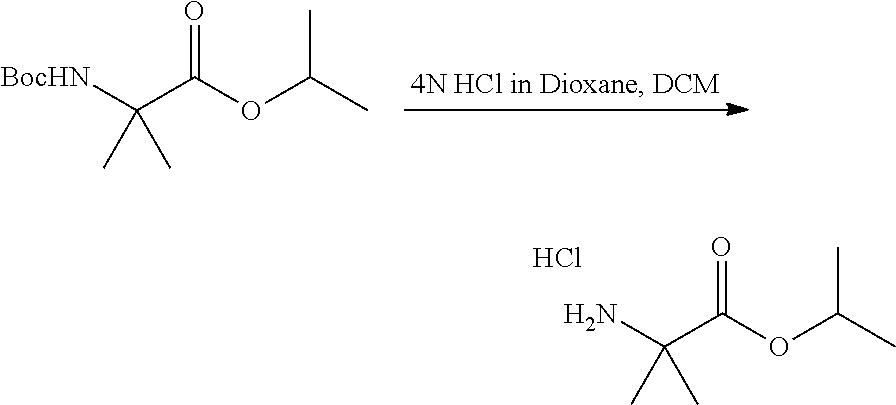






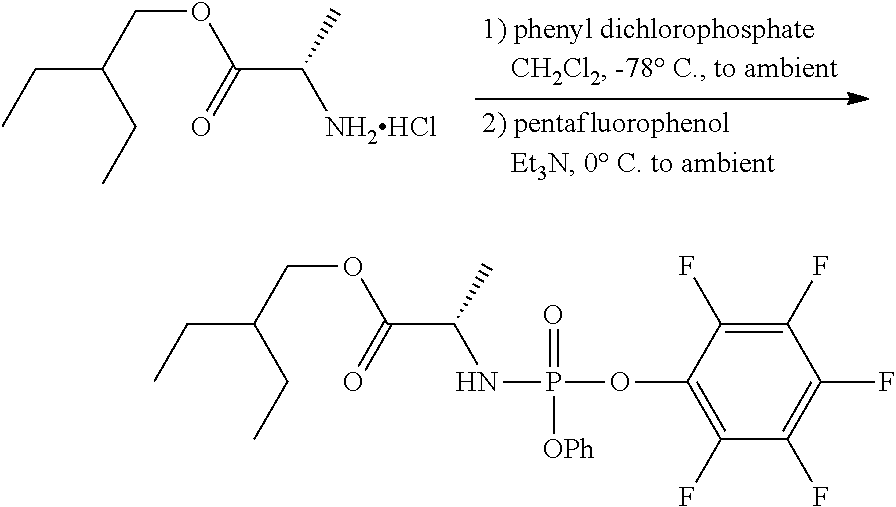























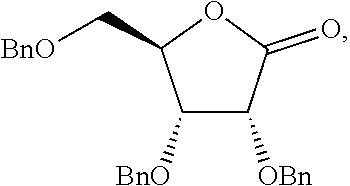











XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.