Compositions And Methods Relating To Universal Glycoforms For Enhanced Antibody Efficacy
WONG; Chi-Huey ; et al.
U.S. patent application number 16/591229 was filed with the patent office on 2020-05-28 for compositions and methods relating to universal glycoforms for enhanced antibody efficacy. This patent application is currently assigned to Academia Sinica. The applicant listed for this patent is Academia Sinica. Invention is credited to Che MA, Chi-Huey WONG, Chung-Yi WU.
| Application Number | 20200165649 16/591229 |
| Document ID | / |
| Family ID | 54699741 |
| Filed Date | 2020-05-28 |









View All Diagrams
| United States Patent Application | 20200165649 |
| Kind Code | A1 |
| WONG; Chi-Huey ; et al. | May 28, 2020 |
COMPOSITIONS AND METHODS RELATING TO UNIVERSAL GLYCOFORMS FOR ENHANCED ANTIBODY EFFICACY
Abstract
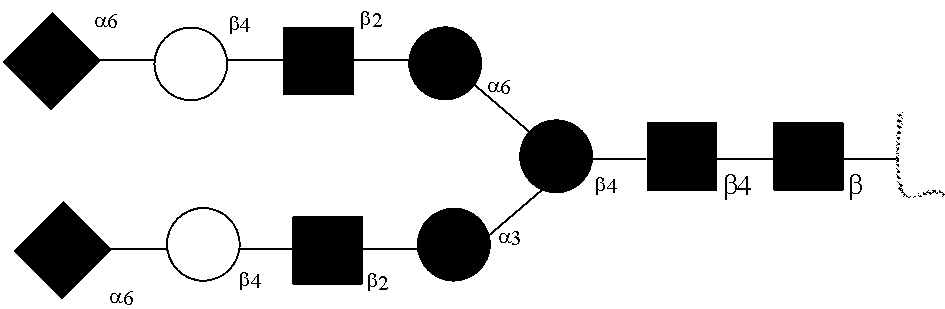
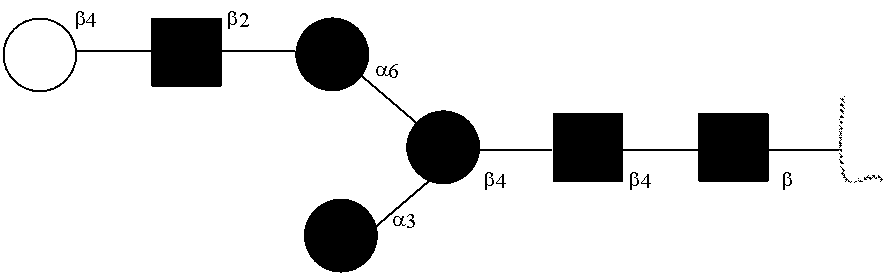
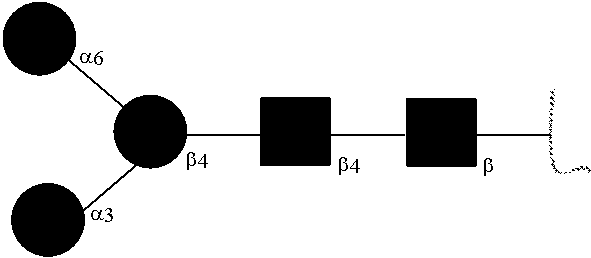
The present disclosure relates to glycoproteins, particularly monoclonal antibodies, comprising a glycoengineered Fc region, wherein said Fc region comprises an optimized N-glycan having the structure of Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2. The glycoengineered Fc region binds Fc.gamma.RIIA or Fc.gamma.RIIIA with a greater affinity, relative to comparable monoclonal antibodies comprising the wild-type Fc region. The monoclonal antibodies of the invention are particularly useful in preventing, treating, or ameliorating one or more symptoms associated with a disease, disorder, or infection where an enhanced efficacy of effector cell function (e.g., ADCC) mediated by Fc.gamma.R is desired, e.g., cancer, autoimmune, infectious disease, and in enhancing the therapeutic efficacy of therapeutic antibodies the effect of which is mediated by ADCC.
| Inventors: | WONG; Chi-Huey; (Rancho Santa Fe, CA) ; WU; Chung-Yi; (New Taipei City, TW) ; MA; Che; (Taipei, TW) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Assignee: | Academia Sinica Taipei TW |
||||||||||
| Family ID: | 54699741 | ||||||||||
| Appl. No.: | 16/591229 | ||||||||||
| Filed: | October 2, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 16018400 | Jun 26, 2018 | |||
| 16591229 | ||||
| 14723297 | May 27, 2015 | 10023892 | ||
| 16018400 | ||||
| 62110338 | Jan 30, 2015 | |||
| 62020199 | Jul 2, 2014 | |||
| 62003908 | May 28, 2014 | |||
| 62003104 | May 27, 2014 | |||
| 62003136 | May 27, 2014 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61K 39/3955 20130101; C07K 16/241 20130101; A61P 35/00 20180101; C07K 2317/41 20130101; A61P 3/10 20180101; C07K 16/30 20130101; C07K 16/32 20130101; Y02A 50/466 20180101; C07K 2317/24 20130101; A61P 1/04 20180101; A61P 31/12 20180101; C07K 16/2887 20130101; C12Y 302/01051 20130101; C12N 9/24 20130101; C07K 16/2896 20130101; A61P 37/02 20180101; A61P 21/04 20180101; C07K 16/1018 20130101; C07K 2317/734 20130101; C07K 16/00 20130101; C07K 2317/72 20130101; A61P 17/06 20180101; A61K 2039/505 20130101; A61P 29/00 20180101; C07K 16/18 20130101; C07K 2317/732 20130101; Y02A 50/412 20180101; A61P 25/00 20180101; A61P 27/02 20180101; C12P 19/14 20130101; A61P 31/14 20180101; Y02A 50/30 20180101; A61P 31/20 20180101; C12Y 302/01 20130101; A61P 31/22 20180101; C07K 2317/92 20130101; A61P 7/00 20180101; A61P 35/02 20180101; A61P 31/18 20180101; A61P 13/12 20180101; A61P 19/02 20180101; A61K 45/06 20130101; A61K 39/42 20130101 |
| International Class: | C12P 19/14 20060101 C12P019/14; C12N 9/24 20060101 C12N009/24; C07K 16/32 20060101 C07K016/32; C07K 16/30 20060101 C07K016/30; C07K 16/28 20060101 C07K016/28; C07K 16/24 20060101 C07K016/24; C07K 16/10 20060101 C07K016/10; C07K 16/00 20060101 C07K016/00; A61K 45/06 20060101 A61K045/06; A61K 39/42 20060101 A61K039/42; A61K 39/395 20060101 A61K039/395; C07K 16/18 20060101 C07K016/18 |
Claims
1. A composition comprising a homogeneous population of monoclonal antibodies or antigen binding fragment thereof, wherein each glycoantibody or antigen binding fragment molecule comprising a single, uniform N-glycan on the Fc region, wherein the N-glycan has the structure of Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2, and wherein the N-glycan is optimized for improving effector cell function.
2. (canceled)
3. (canceled)
4. The composition of claim 1, wherein the monoclonal antibodies are selected from human IgG1, IgG2, IgG3, and IgG4.
5. The composition of claim 1, wherein the monoclonal antibodies bind to at least an antigen associated with cancers, autoimmune or inflammatory diseases, or infectious diseases.
6. The composition of claim 1, wherein the monoclonal antibodies bind to an antigen associated with cancers.
7. The composition of claim 6, wherein the antigen is selected from the group consisting of GD2, GD3, GM2, Globo-H, SSEA-3, SSEA-4, CD16A, CD30, CD32B, CD33, CD52, EpCAM, CEA, gpA33, HER2/neu, A33, CDS, CD11c, CD19, CD20, CD22, CD23, CD27, CD40, CD45, CD79a, CD79b, CD103, CTLA4, ErbB1, ErbB3, ErbB4, VEGF receptor, TNF-.alpha. receptor, TNF-.beta. receptor, or TNF-.gamma. receptor, gpA33, Mucins, TAG-72, CAIX, PSMA, Folate-binding protein, VEGF, VEGFR, Integrin .alpha.V.beta.3, Integrin .alpha.5.beta.1, EGFR, ERBB2, ERBB3, MET, IGF1R, EPHA3, TRAILR1, TRAILR2, RANKL, FAP and Tenascin.
8. The composition of claim 1, wherein the monoclonal antibodies bind to an antigen associated with an autoimmune or inflammatory disease.
9. The composition of claim 1, wherein the antigen is selected from the group consisting of interleukin 5 and its receptor, a tumor necrosis factor and its receptor.
10. The composition of claim 1, wherein the monoclonal antibodies bind to an antigen expressed on a virus infected cell.
11. The composition of claim 1, wherein the antigen is selected from the group consisting of gp120, CXCR4 and Vero toxin.
12. The composition of claim 1, wherein the composition is produced in vitro.
13. A pharmaceutical formulation comprising a composition according to claim 1 and a pharmaceutically acceptable carrier.
14. A method for enhancing antibody-dependent cell mediated cytotoxicity (ADCC) activity, the method comprising administering to a subject in need thereof an amount of a composition according to claim 1.
15. A method for preventing, treating, or ameliorating one or more symptoms associated with a disease, disorder, or infection, the method comprising administering to a subject in need thereof a therapeutically effective amount of the pharmaceutical composition according to claim 13.
16. The method of claim 15, wherein the disease, disorder, or infection is selected from a group consisting of cancer, autoimmune disorder, inflammatory disorder or infectious infection.
17. The method of claim 16, wherein the cancer is selected from the group consisting of brain cancer, lung cancer, breast cancer, oral cancer, esophagus cancer, stomach cancer, liver cancer, bile duct cancer, pancreas cancer, colon cancer, kidney cancer, cervix cancer, ovary cancer and prostate cancer. In some embodiments, the cancer is brain cancer, lung cancer, breast cancer, ovarian cancer, prostate cancer, colon cancer, or pancreas cancer.
18. The method of claim 16, wherein the cancer is selected from the group consisting of B cell lymphomas, NHL, precursor B cell lymphoblastic leukemia/lymphoma and mature B cell neoplasms, B cell chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), B cell prolymphocytic leukemia, lymphoplasmacytic lymphoma, mantle cell lymphoma (MCL), follicular lymphoma (FL), low-grade, intermediate-grade and high-grade (FL), cutaneous follicle center lymphoma, marginal zone B cell lymphoma, MALT type marginal zone B cell lymphoma, nodal marginal zone B cell lymphoma, splenic type marginal zone B cell lymphoma, hairy cell leukemia, diffuse large B cell lymphoma, Burkitt's lymphoma, plasmacytoma, plasma cell myeloma, post-transplant lymphoproliferative disorder, Waldenstrom's macroglobulinemia, and anaplastic large-cell lymphoma (ALCL).
19. The method of claim 16, wherein the autoimmune or inflammatory disease is selected from the group consisting of rheumatoid arthritis, juvenile rheumatoid arthritis, systemic lupus erythematosus (SLE), lupus nephritis, ulcerative colitis, Wegener's disease, inflammatory bowel disease, idiopathic thrombocytopenic purpura (ITP), thrombotic thrombocytopenic purpura (TTP), autoimmune thrombocytopenia, multiple sclerosis, psoriasis, IgA nephropathy, IgM polyneuropathies, myasthenia gravis, vasculitis, diabetes mellitus, Reynaud's syndrome, Sjorgen's syndrome and glomerulonephritis.
20. The method of claim 19, wherein the autoimmune or inflammatory disease is rheumatoid arthritis.
21. (canceled)
22. The method of claim 21, wherein the infectious disease is caused by HIV, HCV, or a combination thereof.
23. The method of claim 15, wherein an enhanced efficacy of effector cell function mediated by Fc.gamma.R is desired for preventing, treating, or ameliorating one or more symptoms associated with the disease, disorder, or infection.
24. (canceled)
25. The method of claim 15, wherein the pharmaceutical composition is administered alone or in conjunction with a second therapeutic agent selected from a group consisting of a second antibody, a chemotherapeutic agent and an immunosuppressive agent.
26. (canceled)
27. (canceled)
28. The composition of claim 1, wherein the monoclonal antibodies comprise a light chain sequence and a heavy chain sequence of Rituximab (Rituxan.RTM.).
29. The composition of claim 1, wherein the monoclonal antibodies comprise a light chain sequence and a heavy chain sequence of Trastuzumab (Herceptin.RTM.).
30. The composition of claim 1, wherein the monoclonal antibodies comprise a light chain sequence and a heavy chain sequence of Adalimumab (Humira).
31. The composition of claim 1, wherein the monoclonal antibodies are F16 monoclonal antibodies.
32. A method for treating a cancer, the method comprising administering to a subject in need thereof a therapeutically effective amount of a pharmaceutical composition comprising an essentially homogeneous population of FDA approved monoclonal antibodies for treatment of cancer, or antigen-binding fragments thereof, wherein the FDA approved monoclonal antibodies or antigen binding fragments thereof have been glycoengineered to have a Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 at each Asn-297 position in the Fc region.
33. The method of claim 32, wherein the FDA approved antibodies or antigen-binding fragments thereof are selected from the group consisting of Rituximab, Ibritumomab tiuxetan, Obinutuzumab, Ofatumumab, and Tositumomab.
34. A method for treating a cancer, the method comprising administering to a subject in need thereof a therapeutically effective amount of a pharmaceutical composition comprising an essentially homogeneous population of glycoengineered monoclonal glycoantibodies or antigen binding fragments thereof, wherein the glycoengineered monoclonal or antigen binding fragments thereof are selected from the group consisting of Alemtuzumab, Belimumab, Bevacizumab, Brentuximab vedotin, Canakinumab, Cetuximab, Denosumab, Ibritumomab tiuxetan, Ipilimumab, Nivolumab, Obinutuzumab, Ofatumumab, Panitumumab, Pembrolizumab, Pertuzumab, Ramucirumab, Rituximab, Siltuximab, Tocilizumab, Tositumomab and Trastuzumab, and wherein the glycoengineered monoclonal glycoantibodies or antigen binding fragments thereof have a Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 at each Asn-297 position in the Fc region.
35. A method for treating a cancer, the method comprising administering to a subject in need thereof a therapeutically effective amount of a pharmaceutical composition comprising an essentially homogeneous population of monoclonal antibodies or antigen binding fragments thereof, wherein the monoclonal antibodies or antigen binding fragments thereof are selected from the group consisting of Alemtuzumab, Belimumab, Bevacizumab, Brentuximab vedotin, Canakinumab, Cetuximab, Denosumab, Ibritumomab tiuxetan, Ipilimumab, Nivolumab, Obinutuzumab, Ofatumumab, Panitumumab, Pembrolizumab, Pertuzumab, Ramucirumab, Rituximab, Siltuximab, Tocilizumab, Tositumomab and Trastuzumab, and wherein the monoclonal antibodies or antigen binding fragments thereof have been glycoengineered to have a Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 at each Asn-297 position in the Fc region.
Description
RELATED APPLICATIONS
[0001] This application claims the benefit of priority of, and is a Continuation of, U.S. application Ser. No. 16/018,400, filed Jun. 26, 2018, which is a continuation of U.S. application Ser. No. 14/723,297, filed May 27, 2015, now issued as U.S. Pat. No. 10,023,892, which claims priority to U.S. provisional applications U.S. Serial No. (USSN) 62/003,136, filed May 27, 2014, U.S. Ser. No. 62/003,104, filed May 27, 2014, U.S. Ser. No. 62/003,908, filed May 28, 2014, U.S. Ser. No. 62/020,199, filed Jul. 2, 2014, and U.S. Ser. No. 62/110,338, filed Jan. 30, 2015. The contents of each of which is hereby incorporated by reference in its entirety.
BACKGROUND OF THE INVENTION
[0002] Antibody-based therapies have a proven record of efficacy against many diseases including inflammatory disorders, cancers, infectious diseases, and solid organ transplant rejection. Currently more than 40 therapeutic monoclonal antibodies (mAbs) are approved for clinical use in USA, EU and several other countries. Most of them are for therapy of cancer and immune diseases. Examples of therapeutic antibodies with anti-tumor activities include anti-CD20, anti-Her2, anti-EGFR, anti-CD40, anti-CTLA-4, and anti-PD-1 antibodies.
[0003] Most of therapeutic antibodies are monoclonal and prepared by the hybridoma technology in which transgenic humanized mice were incorporated to express murine/human chimeric or humanized antibodies to avoid undesired immunological responses derived from species difference. Recently, the development of fully human antibodies has become a major trend and its impressive progress is beneficial from the utilization of phage-displayed antibody libraries or single B cells.
[0004] Like many other mammalian proteins, antibodies are heterogeneously glycosylated, and the glycosylation in the Fc region has been an important issue in the development of efficacious and safe therapeutic monoclonal antibodies because the glycan can significantly affect the antibody's activity through interaction with the Fc receptors. Consequently, there is a need for the development of homogeneous monoclonal antibodies with well-defined Fc-glycan to understand these interactions and to improve the safety and efficacy in medication. Toward this goal, it has been reported that the removal of the core fucose residue would enhance the antibody-dependent cellular cytotoxicity (ADCC) activity of IgGs due to the increased interaction between Fc-glycan and human Fc.gamma.RIIIa receptor. The two FDA approved glyco-engineered antibodies, mogamulizumab (POTELLIGENT.RTM.) and obinutuzuman (GA101), are defucosylated antibodies in which POTELLIGENT.RTM. was produced by the FUT8 knockout CHO cell line and GA101 was from the GnT-III overexpressing system. In addition, more Fc.gamma.IIIa was expressed on the monocytes of long-term RA, and the tendency of more fucosylation was also found in the IgG heavy chain of RA patients, implying the possibility of RA treatment and remission with afucosylated pharmaceutical antibodies, which not only neutralize proinflammatory cytokines but also compete with autologous autoantibodies for Fc.gamma.IIIa.
[0005] Thus, it is of great interest to generate therapeutic monoclonal antibodies with optimized Fc glycoforms.
SUMMARY OF THE INVENTION
[0006] The present disclosure is based on the discovery of glyco-optimized Fc for monoclonal antibodies, specifically a homogeneous population of monoclonal antibodies ("glycoantibodies"). The optimized glycoform exhibits an enhanced efficacy of effector cell function (e.g., ADCC).
[0007] The term "glycoantibodies" was coined by the inventor, Dr. Chi-Huey Wong, to refer to a homogeneous population of monoclonal antibodies (preferably, therapeutic monoclonal antibodies) having a single, uniform N-glycan on Fc. The individual glycoantibodies comprising the homogeneous population are substantially identical, bind to the same epitope, and contain the same Fc glycan with a well-defined glycan structure and sequence.
[0008] "Substantially identical" means the objects being compared have such close resemblance as to be essentially the same--as understood by one having ordinary skill in the art. "Substantially identical" encompasses "identical".
[0009] As used herein, the term "glycoantibodies" ("GAbs") refers to a homogeneous population of IgG molecules having the same N-glycan on Fc. The term "glycoantibody" ("GAb") refers to an individual IgG molecule in the glycoantibodies.
[0010] Accordingly, one aspect of the present disclosure relates to a composition of a homogeneous population of monoclonal antibodies comprising a single, uniform N-glycan on Fc, wherein the structure is an optimized N-glycan structure for enhancing the efficacy of effector cell function.
[0011] In preferred embodiments, the N-glycan is attached to the Asn-297 of the Fc region.
[0012] In preferred embodiments, wherein the N-glycan consists of the structure of Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAC.sub.2.
[0013] The glycoantibodies described herein may be produced in vitro. The glycoantibodies may be generated by Fc glycoengineering. In certain embodiments, the glycoantibodies are enzymatically or chemoenzymatically engineered from the monoclonal antibodies obtained by mammalian cell culturing.
[0014] In some embodiments, the Fc region of the glycoantibodies described herein exhibits an increased binding affinity for Fc.gamma.RIIA or Fc.gamma.RIIIA relative to a wild-type Fc region in the corresponding monoclonal antibodies.
[0015] In some embodiments, the glycoantibodies described herein exhibit an enhanced antibody-dependent cell mediated cytotoxicity (ADCC) activity relative to wild-type immunoglobulins.
[0016] In some embodiments, the glycoantibodies are selected from a group consisting of human IgG1, IgG2, IgG3, and IgG4.
[0017] The monoclonal antibodies may be humanized, human or chimeric.
[0018] The glycoantibodies described herein may bind to an antigen associated with cancers, autoimmune disorders, inflammatory disorders or infectious diseases.
[0019] In some embodiments, the glycoantibody described herein is a glycoengineered anti-CD20. In some examples, the glycoantibody described herein is a glycoengineered Rituximab (Rituxan.RTM.).
[0020] In some embodiments, the glycoantibody described herein is a glycoengineered anti-HER2. In some examples, the glycoantibody described herein is a glycoengineered Trastuzumab (Herceptin.RTM.).
[0021] In some embodiments, the glycoantibody described herein is a glycoengineered anti-TNF.alpha.. In some examples, the glycoantibody described herein is a glycoengineered Adalimumab (Humira.RTM.).
[0022] In some embodiments, the glycoantibody described herein is a glycoengineered F16 antibodies.
[0023] Another aspect of the present disclosure features a pharmaceutical composition comprising a composition of glycoantibodies described herein and a pharmaceutically acceptable carrier. The pharmaceutical composition may be used in therapeutics such as oncology, autoimmune disorders, inflammatory disorders and infectious diseases.
[0024] In some embodiments, the pharmaceutical composition is used for preventing, treating, or ameliorating one or more symptoms associated with a disease, disorder, or infection where an enhanced efficacy of effector cell function (e.g., ADCC) mediated by Fc.gamma.R is desired, e.g., cancer, autoimmune, infectious disease, and in enhancing the therapeutic efficacy of therapeutic antibodies the effect of which is mediated by ADCC.
[0025] Disclosed herein also include methods for enhancing antibody-dependent cell mediated cytotoxicity (ADCC) activity, the method comprising administering to a subject an amount of glycoantibodies described herein.
[0026] Further, disclosed herein include methods for preventing, treating, or ameliorating one or more symptoms associated with a disease, disorder, or infection, the method comprising administering to a subject in need thereof a therapeutically effective amount of the pharmaceutical composition described herein. The disease, disorder, or infection may be selected from a group consisting of cancers, autoimmune disorders, inflammatory disorders and infectious infections.
[0027] Another aspect of the present disclosure features a method for treating a viral disease in a human subject in need thereof, comprising (a) administering to the subject a first compound that blocks an inhibitory receptor of an NK cell, and (b) administering to the subject a therapeutically effective amount of the pharmaceutical composition described herein.
[0028] In these treatment methods described herein, the pharmaceutical composition of glycoantibodies can be administered alone or in conjunction with a second therapeutic agent such as a second antibody, or a chemotherapeutic agent or an immunosuppressive agent.
[0029] This application refers to various issued patent, published patent applications, journal articles, and other publications, all of which are incorporated herein by reference.
[0030] The details of one or more embodiments of the invention are set forth in the description below. Other features or advantages of the present invention will be apparent from the following drawings and detailed description of several embodiments, and also from the appending claims.
BRIEF DESCRIPTION OF THE DRAWINGS
[0031] FIGS. 1a, 1b show (a) general strategy for the preparation of homogeneous antibody through remodeling of the glycan structures on the Fc region of IgG1 (b).
[0032] FIGS. 2a, 2b, 2c, 2d, 2e show that antibody dependent B-cell depletion activity of various glycoengineered Rituximab. The depletion of human B cells was conducted using freshly prepared human PBMC cells and analyzed on FACS, based on the CD19+CD2- B cells. (A) Compared to a series of different glycoengineered Riruximabs, the 2,6-NSCT Rituximab showed higher depletion ability. (B) In the whole blood B-cell depletion activity of 10 donors, the 2,6-sialylated Rituximab was significantly more active than the non-treated Rituximab with a p value of 0.0016, whereas the mono-GlcNAc Rituximab showed the lowest activity. (C) The prepared Rituximab-resistant cells of Ramos and Raji express lower level of CD20 on cell surface. (D, E) The 2,6-NSCT Rituximab showed a remarkable ADCC efficacy towards both normal and resistant cells, whereas non-treated antibody dramatically lost its activity towards resistant strains.
[0033] FIGS. 3a, 3b, 3c, 3d show that EC50 of glycoengineered Herceptin in V158 Fc.gamma.RIIIa mediated ADCC reporter bioassay. Experiments were performed under E/T ratio of 6 to 1 with SKBR3 as target cells and V158 Fc.gamma.RIIIa engineered Jurkat as effector cells. All data shown in the same graph were experiments done in the same microplate and the same batch of effector cells; bars of 95% confidence interval were plotted. (A) afucosylated Herceptin G8 and commercial Herceptin showed a similar ADCC effect, illustrating that the defucosylation advantage of anti-Fc.gamma.RIIIa is lost in the afucosylated Herceptin G8. (B) Bisected and its non-bisected analogue Herceptin, G9 and G4 showed similar EC50 values, indicating that no better bisected glycan mediated ADCC function was observed in this assay. (C) Compared to glycoengineered Herceptin G1 with two galactose terminals, no significant EC50 change in the 2,6-sialylated antibody was observed, whereas the apparent EC50 increase was shown in the 2,3-sialylated Herceptin. The results indicated that the 2,3-sialylation on Fc would lower the effector cell activation but the 2,6-linked one would not. Curves of fold induction were results of induced luminescence divided by induction of no antibody control. (D) Samples with lowest EC50 in graph (A) to (C) were chosen and compared to commercial Herceptin. All samples demonstrated better activity in this ADCC reporter bioassay.
[0034] FIGS. 4a, 4b, 4c show that anti-influenza antibody FI6 with a modified homogeneous SCT glycan attached to its Fc Asn297 (FI6m) significantly showed an enhancement of its ADCC activity and prophylactically protects mice from a lethal dose of H1N1 virus challenge. (a) Cytotoxicity is represented as the percentage of lysed HEK293T cells (target cells) expressed with influenza H1 hemagglutinin (HA) (A/California/07/09) when incubated with PBMCs (effector cells) and various concentrations of antibodies. (b) ADCC activity was shown as fold increases of bioluminescence from a luciferase reporter assay that gave signals when ADCC signaling nuclear factor of activated T-cell pathway was activated. HA expressed HEK293T cells (target cells) were incubated with NK cells with the said luciferase reporter (effector cells) and various amounts of anti-influenza antibody FI6 and FI6m. Curve fitting was done with software GraphPad Prism in 4PL nonlinear regression. (c) Survival of mice was monitored upon lethal dose (10 MLD50) infection of influenza virus A/California/07/09 (H1N1). Two hours before infection, each group of mice (N=9) was intraperitoneally given either 2.5 mg/kg of FI6, FI6m or PBS, respectively. The FI6 and FI6m groups had significant survival difference (p<0.01).
DETAILED DESCRIPTION OF THE INVENTION
Definitions
[0035] The practice of the present invention will employ, unless otherwise indicated, conventional techniques of molecular biology, microbiology, recombinant DNA, and immunology, which are within the skill of the art. Such techniques are explained fully in the literature. See, for example, Molecular Cloning A Laboratory Manual, 2nd Ed., ed. by Sambrook, Fritsch and Maniatis (Cold Spring Harbor Laboratory Press, 1989); DNA Cloning, Volumes I and II (D. N. Glover ed., 1985); Culture Of Animal Cells (R. I. Freshney, Alan R. Liss, Inc., 1987); Immobilized Cells And Enzymes (IRL Press, 1986); B. Perbal, A Practical Guide To Molecular Cloning (1984); the treatise, Methods In Enzymology (Academic Press, Inc., N.Y.); Gene Transfer Vectors For Mammalian Cells (J. H. Miller and M. P. Calos eds., 1987, Cold Spring Harbor Laboratory); Methods In Enzymology, Vols. 154 and 155 (Wu et al. eds.), Immunochemical Methods In Cell And Molecular Biology (Mayer and Walker, eds., Academic Press, London, 1987); Antibodies: A Laboratory Manual, by Harlow and Lane s (Cold Spring Harbor Laboratory Press, 1988); and Handbook Of Experimental Immunology, Volumes I-IV (D. M. Weir and C. C. Blackwell, eds., 1986).
[0036] The term "glycoantibodies" was coined by the inventor, Dr. Chi-Huey Wong, to refer to a homogeneous population of monoclonal antibodies (preferably, therapeutic monoclonal antibodies) having a single, uniformed glycoform bound to the Fc region. The individual glycoantibodies comprising the essentially homogeneous population are identical, bind to the same epitope, and contain the same Fc glycan with a well-defined glycan structure and sequence.
[0037] As used herein, the term "anti-CD20 glycoantibodies" ("anti-CD20 GAbs") refers to a homogeneous population of anti-CD20 IgG molecules having the same glycoform on Fc.
[0038] The term "anti-CD20 glycoantibody" ("anti-CD20 GAb") refers to an individual IgG antibody molecule in the anti-CD20 glycoantibodies. As used herein, "molecule" can also refer to antigen binding fragments.
[0039] As used herein, the term "glycan" refers to a polysaccharide, oligosaccharide or monosaccharide. Glycans can be monomers or polymers of sugar residues and can be linear or branched. A glycan may include natural sugar residues (e.g., glucose, N-acetylglucosamine, N-acetyl neuraminic acid, galactose, mannose, fucose, hexose, arabinose, ribose, xylose, etc.) and/or modified sugars (e.g., 2'-fluororibose, 2'-deoxyribose, phosphomannose, 6' sulfo N-acetylglucosamine, etc). Glycan is also used herein to refer to the carbohydrate portion of a glycoconjugate, such as a glycoprotein, glycolipid, glycopeptide, glycoproteome, peptidoglycan, lipopolysaccharide or a proteoglycan. Glycans usually consist solely of 0-glycosidic linkages between monosaccharides. For example, cellulose is a glycan (or more specifically a glucan) composed of -1,4-linked D-glucose, and chitin is a glycan composed of -1,4-linked N-acetyl-D-glucosamine. Glycans can be homo or heteropolymers of monosaccharide residues, and can be linear or branched. Glycans can be found attached to proteins as in glycoproteins and proteoglycans. They are generally found on the exterior surface of cells. O- and N-linked glycans are very common in eukaryotes but may also be found, although less commonly, in prokaryotes. N-Linked glycans are found attached to the R-group nitrogen (N) of asparagine in the sequon. The sequon is a Asn-X-Ser or Asn-X-Thr sequence, where X is any amino acid except praline.
[0040] As used herein, the terms "fucose", "core fucose" and "core fucose residue" are used interchangeably and refer to a fucose in .alpha.1,6-position linked to the N-acetylglucosamine.
[0041] As used herein, the terms "N-glycan", "N-linked glycan", "N-linked glycosylation", "Fc glycan" and "Fc glycosylation" are used interchangeably and refer to an N-linked oligosaccharide attached by an N-acetylglucosamine (GlcNAc) linked to the amide nitrogen of an asparagine residue in a Fc-containing polypeptide. The term "Fc-containing polypeptide" refers to a polypeptide, such as an antibody, which comprises an Fc region.
[0042] As used herein, the term "glycosylation pattern" and "glycosylation profile" are used interchangeably and refer to the characteristic "fingerprint" of the N-glycan species that have been released from a glycoprotein or antibody, either enzymatically or chemically, and then analyzed for their carbohydrate structure, for example, using LC-HPLC, or MALDI-TOF MS, and the like. See, for example, the review in Current Analytical Chemistry, Vol. 1, No. 1 (2005), pp. 28-57; herein incorporated by reference in its entirety.
[0043] As used herein, the term "glycoengineered Fc" when used herein refers to N-glycan on the Fc region has been altered or engineered either enzymatically or chemically. The term "Fc glycoengineering" as used herein refers to the enzymatic or chemical process used to make the glycoengineered Fc. Exemplary methods of engineering are described in, for example, Wong et al U.S. Ser. No. 12/959,351, the contents of which is hereby incorporated by reference.
[0044] The terms "homogeneous", "uniform", "uniformly" and "homogeneity" in the context of a glycosylation profile of Fc region are used interchangeably and are intended to mean a single glycosylation pattern represented by one desired N-glycan species, with little or no trace amount of precursor N-glycan. In certain embodiments, the trace amount of the precursor N-glycan is less than about 2%.
[0045] "Essentially pure" protein means a composition comprising at least about 90% by weight of the protein, based on total weight of the composition, including, for example, at least about 91%, at least about 92%, at least about 93%, at least about 94%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, or at least about 99% by weight.
[0046] "Essentially homogeneous" protein means a composition comprising at least about 98% by weight of protein, including for example, at least about 98.5%, at least about 99% based on total weight of the composition. In certain embodiments, the protein is an antibody, structural variants, and/or antigen binding fragment thereof.
[0047] As used herein, the terms "IgG", "IgG molecule", "monoclonal antibody", "immunoglobulin", and "immunoglobulin molecule" are used interchangeably. As used herein, "molecule" can also refer to antigen binding fragments.
[0048] As used herein, the term "Fc receptor" or "FcR" describes a receptor that binds to the Fc region of an antibody. The preferred FcR is a native sequence human FcR. Moreover, a preferred FcR is one which binds an IgG antibody (a gamma receptor) and includes receptors of the Fc.gamma.RI (CD64), Fc.gamma.RII (CD32), and Fc.gamma.RIII (CD16) subclasses, including allelic variants and alternatively spliced forms of these receptors. Fc.gamma.RII receptors include Fc.gamma.RIIA (an "activating receptor") and Fc.gamma.RIIB (an "inhibiting receptor"), which have similar amino acid sequences that differ primarily in the cytoplasmic domains thereof. Activating receptor Fc.gamma.RIIA contains an immunoreceptor tyrosine-based activation motif (ITAM) in its cytoplasmic domain. Inhibiting receptor Fc.gamma.RIIB contains an immunoreceptor tyrosine-based inhibition motif (ITIM) in its cytoplasmic domain. (see review M. in Daeron, Annu. Rev. Immunol. 15:203-234 (1997)). FcRs are reviewed in Ravetch and Kinet, Annu. Rev. Immunol 9:457-92 (1991); Capel et al., Immunomethods 4:25-34 (1994); and de Haas et al., J. Lab. Clin. Med. 126:330-41 (1995). Other FcRs, including those to be identified in the future, are encompassed by the term "FcR" herein. The term also includes the neonatal receptor, FcRn, which is responsible for the transfer of maternal IgGs to the fetus (Guyer et al., J. Immunol. 117:587 (1976) and Kim et al., J. Immunol. 24:249 (1994)).
[0049] The term "effector function" as used herein refers to a biochemical event that results from the interaction of an antibody Fc region with an Fc receptor or ligand. Exemplary "effector functions" include C1q binding; complement dependent cytotoxicity; Fc receptor binding; antibody-dependent cell-mediated cytotoxicity (ADCC); phagocytosis; down regulation of cell surface receptors (e.g. B cell receptor; BCR), etc. Such effector functions can be assessed using various assays known in the art.
[0050] As used herein, the term "Antibody-dependent cell-mediated cytotoxicity" or "ADCC" refers to a form of cytotoxicity in which secreted Ig bound onto Fc receptors (FcRs) present on certain cytotoxic cells (e.g. Natural Killer (NK) cells, neutrophils, and macrophages) enable these cytotoxic effector cells to bind specifically to an antigen-bearing target cell and subsequently kill the target cell with cytotoxins. The antibodies "arm" the cytotoxic cells and are absolutely required for such killing. The primary cells for mediating ADCC, NK cells, express Fc.gamma.RIII only, whereas monocytes express Fc.gamma.RI, Fc.gamma.RII and Fc.gamma.RIII. FcR expression on hematopoietic cells is summarized in Table 3 on page 464 of Ravetch and Kinet, Annu. Rev. Immunol 9:457-92 (1991). To assess ADCC activity of a molecule of interest, an in vitro ADCC assay, such as that described in U.S. Pat. Nos. 5,500,362 or 5,821,337 may be performed. Useful effector cells for such assays include peripheral blood mononuclear cells (PBMC) and Natural Killer (NK) cells. Alternatively, or additionally, ADCC activity of the molecule of interest may be assessed in vivo, e.g., in a animal model such as that disclosed in Clynes et al. PNAS (USA) 95:652-656 (1998).
[0051] The term "Complement dependent cytotoxicity" or "CDC" as used herein refers to the lysis of a target cell in the presence of complement. Activation of the classical complement pathway is initiated by the binding of the first component of the complement system (C1q) to antibodies (of the appropriate subclass) which are bound to their cognate antigen. To assess complement activation, a CDC assay, e.g. as described in Gazzano-Santoro et al., J. Immunol. Methods 202:163 (1996), may be performed.
[0052] "Chimeric" antibodies (immunoglobulins) have a portion of the heavy and/or light chain identical with or homologous to corresponding sequences in antibodies derived from a particular species or belonging to a particular antibody class or subclass, while the remainder of the chain(s) is identical with or homologous to corresponding sequences in antibodies derived from another species or belonging to another antibody class or subclass, as well as fragments of such antibodies, so long as they exhibit the desired biological activity (U.S. Pat. No. 4,816,567; and Morrison et al., Proc. Natl. Acad. Sci. USA 81:6851-6855 (1984)). Humanized antibody as used herein is a subset of chimeric antibodies.
[0053] "Humanized" forms of non-human (e.g., murine) antibodies are chimeric antibodies which contain minimal sequence derived from non-human immunoglobulin. For the most part, humanized antibodies are human immunoglobulins (recipient or acceptor antibody) in which hypervariable region residues of the recipient are replaced by hypervariable region residues from a non-human species (donor antibody) such as mouse, rat, rabbit or nonhuman primate having the desired specificity, affinity, and capacity. In some instances, Fv framework region (FR) residues of the human immunoglobulin are replaced by corresponding non-human residues. Furthermore, humanized antibodies may comprise residues which are not found in the recipient antibody or in the donor antibody. These modifications are made to further refine antibody performance such as binding affinity. Generally, the humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non-human immunoglobulin and all or substantially all of the FR regions are those of a human immunoglobulin sequence although the FR regions may include one or more amino acid substitutions that improve binding affinity. The number of these amino acid substitutions in the FR is typically no more than 6 in the H chain, and in the L chain, no more than 3. The humanized antibody optionally also will comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin. For further details, see Jones et al., Nature 321:522-525 (1986); Reichmann et al., Nature 332:323-329 (1988); and Presta, Curr. Op. Struct. Biol. 2:593-596 (1992). See also the following review articles and references cited therein: Vaswani and Hamilton, Ann. Allergy, Asthma & Immunol. 1:105-115 (1998); Harris, Biochem. Soc. Transactions 23:1035-1038 (1995); Hurle and Gross, Curr. Op. Biotech. 5:428-433 (1994).
[0054] As used herein, the term "antigen" is defined as any substance capable of eliciting an immune response. As used herein, the term "antigen specific" refers to a property of a cell population such that supply of a particular antigen, or a fragment of the antigen, results in specific cell proliferation.
[0055] As used herein, the term "immunogenicity" refers to the ability of an immunogen, antigen, or vaccine to stimulate an immune response.
[0056] As used herein, the term "epitope" is defined as the parts of an antigen molecule which contact the antigen binding site of an antibody or a T cell receptor.
[0057] As used herein, the term "specifically binding," refers to the interaction between binding pairs (e.g., an antibody and an antigen). In various instances, specifically binding can be embodied by an affinity constant of about 10-6 moles/liter, about 10-7 moles/liter, or about 10-8 moles/liter, or less.
[0058] An "isolated" antibody is one which has been identified and separated and/or recovered from a component of its natural environment. Contaminant components of its natural environment are materials which would interfere with research, diagnostic or therapeutic uses for the antibody, and may include enzymes, hormones, and other proteinaceous or nonproteinaceous solutes.
[0059] The phrase "substantially similar," "substantially the same", "equivalent", or "substantially equivalent", as used herein, denotes a sufficiently high degree of similarity between two numeric values (for example, one associated with a molecule and the other associated with a reference/comparator molecule) such that one of skill in the art would consider the difference between the two values to be of little or no biological and/or statistical significance within the context of the biological characteristic measured by said values (e.g., Kd values, anti-viral effects, etc.). The difference between said two values is, for example, less than about 50%, less than about 40%, less than about 30%, less than about 20%, and/or less than about 10% as a function of the value for the reference/comparator molecule.
[0060] The phrase "substantially reduced," or "substantially different", as used herein, denotes a sufficiently high degree of difference between two numeric values (generally one associated with a molecule and the other associated with a reference/comparator molecule) such that one of skill in the art would consider the difference between the two values to be of statistical significance within the context of the biological characteristic measured by said values (e.g., Kd values). The difference between said two values is, for example, greater than about 10%, greater than about 20%, greater than about 30%, greater than about 40%, and/or greater than about 50% as a function of the value for the reference/comparator molecule.
[0061] "Binding affinity" generally refers to the strength of the sum total of noncovalent interactions between a single binding site of a molecule (e.g., an antibody) and its binding partner (e.g., an antigen). Unless indicated otherwise, as used herein, "binding affinity" refers to intrinsic binding affinity which reflects a 1:1 interaction between members of a binding pair (e.g., antibody and antigen). The affinity of a molecule X for its partner Y can generally be represented by the dissociation constant (Kd). Affinity can be measured by common methods known in the art, including those described herein. Low-affinity antibodies generally bind antigen slowly and tend to dissociate readily, whereas high-affinity antibodies generally bind antigen faster and tend to remain bound longer. A variety of methods of measuring binding affinity are known in the art, any of which can be used for purposes of the present invention. Specific illustrative embodiments are described in the following.
[0062] The "variable region" or "variable domain" of an antibody refers to the amino-terminal domains of heavy or light chain of the antibody. These domains are generally the most variable parts of an antibody and contain the antigen-binding sites.
[0063] The term "variable" refers to the fact that certain portions of the variable domains differ extensively in sequence among antibodies and are used in the binding and specificity of each particular antibody for its particular antigen. However, the variability is not evenly distributed throughout the variable domains of antibodies. It is concentrated in three segments called complementarity-determining regions (CDRs) or hypervariable regions both in the light-chain and the heavy-chain variable domains. The more highly conserved portions of variable domains are called the framework (FR). The variable domains of native heavy and light chains each comprise four FR regions, largely adopting a beta-sheet configuration, connected by three CDRs, which form loops connecting, and in some cases forming part of, the beta-sheet structure. The CDRs in each chain are held together in close proximity by the FR regions and, with the CDRs from the other chain, contribute to the formation of the antigen-binding site of antibodies (see Kabat et al., Sequences of Proteins of Immunological Interest, Fifth Edition, National Institute of Health, Bethesda, Md. (1991)). The constant domains are not involved directly in binding an antibody to an antigen, but exhibit various effector functions, such as participation of the antibody in antibody-dependent cellular toxicity.
[0064] Papain digestion of antibodies produces two identical antigen-binding fragments, called "Fab" fragments, each with a single antigen-binding site, and a residual "Fc" fragment, whose name reflects its ability to crystallize readily. Pepsin treatment yields an F(ab')2 fragment that has two antigen-combining sites and is still capable of cross-linking antigen.
[0065] "Fv" is the minimum antibody fragment which contains a complete antigen-recognition and -binding site. In a two-chain Fv species, this region consists of a dimer of one heavy- and one light-chain variable domain in tight, non-covalent association. In a single-chain Fv species, one heavy- and one light-chain variable domain can be covalently linked by a flexible peptide linker such that the light and heavy chains can associate in a "dimeric" structure analogous to that in a two-chain Fv species. It is in this configuration that the three CDRs of each variable domain interact to define an antigen-binding site on the surface of the VH-VL dimer. Collectively, the six CDRs confer antigen-binding specificity to the antibody. However, even a single variable domain (or half of an Fv comprising only three CDRs specific for an antigen) has the ability to recognize and bind antigen, although at a lower affinity than the entire binding site.
[0066] The Fab fragment also contains the constant domain of the light chain and the first constant domain (CH1) of the heavy chain. Fab' fragments differ from Fab fragments by the addition of a few residues at the carboxy terminus of the heavy chain CH1 domain including one or more cysteines from the antibody hinge region. Fab'-SH is the designation herein for Fab' in which the cysteine residue(s) of the constant domains bear a free thiol group. F(ab')2 antibody fragments originally were produced as pairs of Fab' fragments which have hinge cysteines between them. Other chemical couplings of antibody fragments are also known.
[0067] The "light chains" of antibodies (immunoglobulins) from any vertebrate species can be assigned to one of two clearly distinct types, called kappa (.kappa.) and lambda (.lamda.), based on the amino acid sequences of their constant domains.
[0068] Depending on the amino acid sequences of the constant domains of their heavy chains, antibodies (immunoglobulins) can be assigned to different classes. There are five major classes of immunoglobulins: IgA, IgD, IgE, IgG and IgM, and several of these may be further divided into subclasses (isotypes), e.g., IgG1, IgG2, IgG3, IgG4, IgA1, and IgA2. The heavy chain constant domains that correspond to the different classes of immunoglobulins are called .alpha., .delta., .epsilon., .gamma., and .mu., respectively. The subunit structures and three-dimensional configurations of different classes of immunoglobulins are well known and described generally in, for example, Abbas et al. Cellular and Mol. Immunology, 4th ed. (2000). An antibody may be part of a larger fusion molecule, formed by covalent or non-covalent association of the antibody with one or more other proteins or peptides.
[0069] The terms "full length antibody," "intact antibody" and "whole antibody" are used herein interchangeably, to refer to an antibody in its substantially intact form, not antibody fragments as defined below. The terms particularly refer to an antibody with heavy chains that contain the Fc region.
[0070] "Antibody fragments" comprise only a portion of an intact antibody, wherein the portion retains at least one, and as many as most or all, of the functions normally associated with that portion when present in an intact antibody. In one embodiment, an antibody fragment comprises an antigen binding site of the intact antibody and thus retains the ability to bind antigen. In another embodiment, an antibody fragment, for example one that comprises the Fc region, retains at least one of the biological functions normally associated with the Fc region when present in an intact antibody, such as FcRn binding, antibody half life modulation, ADCC function and complement binding. In one embodiment, an antibody fragment is a monovalent antibody that has an in vivo half life substantially similar to an intact antibody. For example, such an antibody fragment may comprise an antigen binding arm linked to an Fc sequence capable of conferring in vivo stability to the fragment.
[0071] The term "monoclonal antibody" as used herein refers to an antibody obtained from a population of substantially homogeneous antibodies, i.e., the individual antibodies comprising the population are identical except for possible naturally occurring mutations that may be present in minor amounts. Thus, the modifier "monoclonal" indicates the character of the antibody as not being a mixture of discrete antibodies. Such monoclonal antibody typically includes an antibody comprising a polypeptide sequence that binds a target, wherein the target-binding polypeptide sequence was obtained by a process that includes the selection of a single target binding polypeptide sequence from a plurality of polypeptide sequences. For example, the selection process can be the selection of a unique clone from a plurality of clones, such as a pool of hybridoma clones, phage clones or recombinant DNA clones. It should be understood that the selected target binding sequence can be further altered, for example, to improve affinity for the target, to humanize the target binding sequence, to improve its production in cell culture, to reduce its immunogenicity in vivo, to create a multispecific antibody, etc., and that an antibody comprising the altered target binding sequence is also a monoclonal antibody of this invention. In contrast to polyclonal antibody preparations which typically include different antibodies directed against different determinants (epitopes), each monoclonal antibody of a monoclonal antibody preparation is directed against a single determinant on an antigen. In addition to their specificity, the monoclonal antibody preparations are advantageous in that they are typically uncontaminated by other immunoglobulins. The modifier "monoclonal" indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies, and is not to be construed as requiring production of the antibody by any particular method. For example, the monoclonal antibodies to be used in accordance with the present invention may be made by a variety of techniques, including, for example, the hybridoma method (e.g., Kohler et al., Nature, 256: 495 (1975); Harlow et al., Antibodies: A Laboratory Manual, (Cold Spring Harbor Laboratory Press, 2nd ed. 1988); Hammerling et al., in: Monoclonal Antibodies and T-Cell hybridomas 563-681 (Elsevier, N.Y., 1981)), recombinant DNA methods (see, e.g., U.S. Pat. No. 4,816,567), phage display technologies (See, e.g., Clackson et al., Nature, 352: 624-628 (1991); Marks et al., J. Mol. Biol. 222: 581-597 (1992); Sidhu et al., J. Mol. Biol. 338(2): 299-310 (2004); Lee et al., J. Mol. Biol. 340(5): 1073-1093 (2004); Fellouse, Proc. Natl. Acad. Sci. USA 101(34): 12467-12472 (2004); and Lee et al., J. Immunol. Methods 284(1-2): 119-132 (2004), and technologies for producing human or human-like antibodies in animals that have parts or all of the human immunoglobulin loci or genes encoding human immunoglobulin sequences (see, e.g., WO98/24893; WO96/34096; WO96/33735; WO91/10741; Jakobovits et al., Proc. Natl. Acad. Sci. USA 90: 2551 (1993); Jakobovits et al., Nature 362: 255-258 (1993); Bruggemann et al., Year in Immunol. 7:33 (1993); U.S. Pat. Nos. 5,545,807; 5,545,806; 5,569,825; 5,625,126; 5,633,425; 5,661,016; Marks et al., Bio. Technology 10: 779-783 (1992); Lonberg et al., Nature 368: 856-859 (1994); Morrison, Nature 368: 812-813 (1994); Fishwild et al., Nature Biotechnol. 14: 845-851 (1996); Neuberger, Nature Biotechnol. 14: 826 (1996) and Lonberg and Huszar, Intern. Rev. Immunol. 13: 65-93 (1995).
[0072] The monoclonal antibodies herein specifically include "chimeric" antibodies in which a portion of the heavy and/or light chain is identical with or homologous to corresponding sequences in antibodies derived from a particular species or belonging to a particular antibody class or subclass, while the remainder of the chain(s) is identical with or homologous to corresponding sequences in antibodies derived from another species or belonging to another antibody class or subclass, as well as fragments of such antibodies, so long as they exhibit the desired biological activity (U.S. Pat. No. 4,816,567; and Morrison et al., Proc. Natl. Acad. Sci. USA 81:6851-6855 (1984)).
[0073] See also the following review articles and references cited therein: Vaswani and Hamilton, Ann. Allergy, Asthma & Immunol. 1:105-115 (1998); Harris, Biochem. Soc. Transactions 23:1035-1038 (1995); Hurle and Gross, Curr. Op. Biotech. 5:428-433 (1994).
[0074] The term "hypervariable region", "HVR", or "HV", when used herein refers to the regions of an antibody variable domain which are hypervariable in sequence and/or form structurally defined loops. Generally, antibodies comprise six hypervariable regions; three in the VH (H1, H2, H3), and three in the VL (L1, L2, L3). A number of hypervariable region delineations are in use and are encompassed herein. The Kabat Complementarity Determining Regions (CDRs) are based on sequence variability and are the most commonly used (Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991)). Chothia refers instead to the location of the structural loops (Chothia and Lesk J. Mol. Biol. 196:901-917 (1987)). The AbM hypervariable regions represent a compromise between the Kabat CDRs and Chothia structural loops, and are used by Oxford Molecular's AbM antibody modeling software. The "contact" hypervariable regions are based on an analysis of the available complex crystal structures. The residues from each of these hypervariable regions are noted below.
Loop Kabat AbM Chothia Contact
L1 L24-L34 L24-L34 L26-L32 L30-L36
L2 L50-L56 L50-L56 L50-L52 L46-L55
L3 L89-L97 L89-L97 L91-L96 L89-L96
H1 H31-H35B H26-H35B H26-H32 H30-H35B
(Kabat Numbering)
H1 H31-H35 H26-H35 H26-H32 H30-H35
(Chothia Numbering)
H2 H50-H65 H50-H58 H53-H55 H47-H58
H3 H95-H102 H95-H102 H96-H101 H93-H101
[0075] Hypervariable regions may comprise "extended hypervariable regions" as follows: 24-36 or 24-34 (L1), 46-56 or 50-56 or 49-56 (L2) and 89-97 or 89-96 (L3) in the VL and 26-35 (H1), 50-65 or 49-65 (H2) and 93-102, 94-102, or 95-102 (H3) in the VH. The variable domain residues are numbered according to Kabat et al., supra, for each of these definitions.
[0076] "Framework" or "FR" residues are those variable domain residues other than the hypervariable region residues as herein defined.
[0077] The term "variable domain residue numbering as in Kabat" or "amino acid position numbering as in Kabat," and variations thereof, refers to the numbering system used for heavy chain variable domains or light chain variable domains of the compilation of antibodies in Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991). Using this numbering system, the actual linear amino acid sequence may contain fewer or additional amino acids corresponding to a shortening of, or insertion into, a FR or HVR of the variable domain. For example, a heavy chain variable domain may include a single amino acid insert (residue 52a according to Kabat) after residue 52 of H2 and inserted residues (e.g. residues 82a, 82b, and 82c, etc. according to Kabat) after heavy chain FR residue 82. The Kabat numbering of residues may be determined for a given antibody by alignment at regions of homology of the sequence of the antibody with a "standard" Kabat numbered sequence.
[0078] "Single-chain Fv" or "scFv" antibody fragments comprise the VH and VL domains of antibody, wherein these domains are present in a single polypeptide chain. Generally, the scFv polypeptide further comprises a polypeptide linker between the VH and VL domains which enables the scFv to form the desired structure for antigen binding. For a review of scFv see Pluckthun, in The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg and Moore eds., Springer-Verlag, New York, pp. 269-315 (1994).
[0079] The term "diabodies" refers to small antibody fragments with two antigen-binding sites, which fragments comprise a heavy-chain variable domain (VH) connected to a light-chain variable domain (VL) in the same polypeptide chain (VH-VL). By using a linker that is too short to allow pairing between the two domains on the same chain, the domains are forced to pair with the complementary domains of another chain and create two antigen-binding sites. Diabodies are described more fully in, for example, EP 404,097; WO93/1161; and Hollinger et al., Proc. Natl. Acad. Sci. USA 90: 6444-6448 (1993).
[0080] A "human antibody" is one which possesses an amino acid sequence which corresponds to that of an antibody produced by a human and/or has been made using any of the techniques for making human antibodies as disclosed herein. This definition of a human antibody specifically excludes a humanized antibody comprising non-human antigen-binding residues.
[0081] An "affinity matured" antibody is one with one or more alterations in one or more HVRs thereof which result in an improvement in the affinity of the antibody for antigen, compared to a parent antibody which does not possess those alteration(s). In one embodiment, an affinity matured antibody has nanomolar or even picomolar affinities for the target antigen. Affinity matured antibodies are produced by procedures known in the art. Marks et al. Bio/Technology 10:779-783 (1992) describes affinity maturation by VH and VL domain shuffling. Random mutagenesis of CDR and/or framework residues is described by: Barbas et al. Proc Nat. Acad. Sci. USA 91:3809-3813 (1994); Schier et al. Gene 169:147-155 (1995); Yelton et al. J. Immunol. 155:1994-2004 (1995); Jackson et al., J. Immunol. 154(7):3310-9 (1995); and Hawkins et al, J. Mol. Biol. 226:889-896 (1992).
[0082] A "blocking" antibody or an "antagonist" antibody is one which inhibits or reduces biological activity of the antigen it binds. Certain blocking antibodies or antagonist antibodies substantially or completely inhibit the biological activity of the antigen.
[0083] An "agonist antibody", as used herein, is an antibody which mimics at least one of the functional activities of a polypeptide of interest.
[0084] A "disorder" is any condition that would benefit from treatment with an antibody of the invention. This includes chronic and acute disorders or diseases including those pathological conditions which predispose the mammal to the disorder in question. Non-limiting examples of disorders to be treated herein include cancer.
[0085] The terms "cell proliferative disorder" and "proliferative disorder" refer to disorders that are associated with some degree of abnormal cell proliferation. In one embodiment, the cell proliferative disorder is cancer.
[0086] "Tumor," as used herein, refers to all neoplastic cell growth and proliferation, whether malignant or benign, and all pre-cancerous and cancerous cells and tissues. The terms "cancer," "cancerous," "cell proliferative disorder," "proliferative disorder" and "tumor" are not mutually exclusive as referred to herein.
[0087] The terms "cancer" and "cancerous" generally refer to or describe the physiological condition in mammals that is typically characterized by unregulated cell growth/proliferation. Examples of cancer include, but are not limited to, carcinoma, lymphoma (e.g., Hodgkin's and non-Hodgkin's lymphoma), blastoma, sarcoma, and leukemia. More particular examples of such cancers include squamous cell cancer, small-cell lung cancer, non-small cell lung cancer, adenocarcinoma of the lung, squamous carcinoma of the lung, cancer of the peritoneum, hepatocellular cancer, gastrointestinal cancer, pancreatic cancer, glioblastoma, cervical cancer, ovarian cancer, liver cancer, bladder cancer, hepatoma, breast cancer, colon cancer, colorectal cancer, endometrial or uterine carcinoma, salivary gland carcinoma, kidney cancer, liver cancer, prostate cancer, vulval cancer, thyroid cancer, hepatic carcinoma, leukemia and other lymphoproliferative disorders, and various types of head and neck cancer.
[0088] As used herein, the term "antigen" is defined as any substance capable of eliciting an immune response.
[0089] As used herein, the term "antigen specific" refers to a property of a cell population such that supply of a particular antigen, or a fragment of the antigen, results in specific cell proliferation.
[0090] The term "CD20 expressing cancer" as used herein refers to all cancers in which the cancer cells show an expression of the CD20 antigen. Preferably CD20 expressing cancer as used herein refers to lymphomas (preferably B-Cell Non-Hodgkin's lymphomas (NHL)) and lymphocytic leukemias. Such lymphomas and lymphocytic leukemias include e.g. a) follicular lymphomas, b) Small Non-Cleaved Cell Lymphomas/Burkitt's lymphoma (including endemic Burkitt's lymphoma, sporadic Burkitt's lymphoma and Non-Burkitt's lymphoma) c) marginal zone lymphomas (including extranodal marginal zone B cell lymphoma (Mucosa-associated lymphatic tissue lymphomas, MALT), nodal marginal zone B cell lymphoma and splenic marginal zone lymphoma), d) Mantle cell lymphoma (MCL), e) Large Cell Lymphoma (including B-cell diffuse large cell lymphoma (DLCL), Diffuse Mixed Cell Lymphoma, Immunoblastic Lymphoma, Primary Mediastinal B-Cell Lymphoma, Angiocentric Lymphoma-Pulmonary B-Cell Lymphoma) f) hairy cell leukemia, g) lymphocytic lymphoma, Waldenstrom's macroglobulinemia, h) acute lymphocytic leukemia (ALL), chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), B-cell prolymphocytic leukemia, i) plasma cell neoplasms, plasma cell myeloma, multiple myeloma, plasmacytoma j) Hodgkin's disease. More preferably the CD20 expressing cancer is a B-Cell Non-Hodgkin's lymphomas (NHL). Especially the CD20 expressing cancer is a Mantle cell lymphoma (MCL), acute lymphocytic leukemia (ALL), chronic lymphocytic leukemia (CLL), B-cell diffuse large cell lymphoma (DLCL), Burkitt's lymphoma, hairy cell leukemia, follicular lymphoma, multiple myeloma, marginal zone lymphoma, post transplant lymphoproliferative disorder (PTLD), HIV associated lymphoma, Waldenstrom's macro globulinemia, or primary CNS lymphoma.
[0091] As used herein, "treatment" refers to clinical intervention in an attempt to alter the natural course of the individual or cell being treated, and can be performed either for prophylaxis or during the course of clinical pathology. Desirable effects of treatment include preventing occurrence or recurrence of disease, alleviation of symptoms, diminishment of any direct or indirect pathological consequences of the disease, preventing or decreasing inflammation and/or tissue/organ damage, decreasing the rate of disease progression, amelioration or palliation of the disease state, and remission or improved prognosis. In some embodiments, antibodies of the invention are used to delay development of a disease or disorder.
[0092] An "individual" or a "subject" is a vertebrate. In certain embodiments, the vertebrate is a mammal. Mammals include, but are not limited to, farm animals (such as cows), sport animals, pets (such as cats, dogs, and horses), primates, mice and rats. In certain embodiments, the vertebrate is a human.
[0093] "Mammal" for purposes of treatment refers to any animal classified as a mammal, including humans, domestic and farm animals, and zoo, sports, or pet animals, such as dogs, horses, cats, cows, etc. In certain embodiments, the mammal is human.
[0094] An "effective amount" refers to an amount effective, at dosages and for periods of time necessary, to achieve the desired therapeutic or prophylactic result.
[0095] A "therapeutically effective amount" of a substance/molecule of the invention may vary according to factors such as the disease state, age, sex, and weight of the individual, and the ability of the substance/molecule, to elicit a desired response in the individual. A therapeutically effective amount is also one in which any toxic or detrimental effects of the substance/molecule are outweighed by the therapeutically beneficial effects. A "prophylactically effective amount" refers to an amount effective, at dosages and for periods of time necessary, to achieve the desired prophylactic result. Typically but not necessarily, since a prophylactic dose is used in subjects prior to or at an earlier stage of disease, the prophylactically effective amount would be less than the therapeutically effective amount.
[0096] The term "cytotoxic agent" as used herein refers to a substance that inhibits or prevents the function of cells and/or causes destruction of cells. The term is intended to include radioactive isotopes (e.g., At211, I131, I125, Y90, Re186, Re188, Sm153, Bi212, P32, Pb212 and radioactive isotopes of Lu), chemotherapeutic agents (e.g., methotrexate, adriamicin, vinca alkaloids (vincristine, vinblastine, etoposide), doxorubicin, melphalan, mitomycin C, chlorambucil, daunorubicin or other intercalating agents, enzymes and fragments thereof such as nucleolyticenzymes, antibiotics, and toxins such as small molecule toxins or enzymatically active toxins of bacterial, fungal, plant or animal origin, including fragments and/or variants thereof, and the various antitumor or anticancer agents disclosed below. Other cytotoxic agents are described below. A tumoricidal agent causes destruction of tumor cells.
[0097] A "chemotherapeutic agent" is a chemical compound useful in the treatment of cancer. Examples of chemotherapeutic agents include alkylating agents such as thiotepa and CYTOXAN.RTM. cyclosphosphamide; alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa; ethylenimines and methylamelamines including altretamine, triethylenemelamine, trietylenephosphoramide, triethiylenethiophosphoramide and trimethylolomelamine; acetogenins (especially bullatacin and bullatacinone); delta-9-tetrahydrocannabinol (dronabinol, MARINOL.RTM.); beta-lapachone; lapachol; colchicines; betulinic acid; a camptothecin (including the synthetic analogue topotecan (HYCAMTIN.RTM.), CPT-11 (irinotecan, CAMPTOSAR.RTM.), acetylcamptothecin, scopolectin, and 9-aminocamptothecin); bryostatin; callystatin; CC-1065 (including its adozelesin, carzelesin and bizelesin synthetic analogues); podophyllotoxin; podophyllinic acid; teniposide; cryptophycins (particularly cryptophycin 1 and cryptophycin 8); dolastatin; duocarmycin (including the synthetic analogues, KW-2189 and CB1-TM1); eleutherobin; pancratistatin; a sarcodictyin; spongistatin; nitrogen mustards such as chlorambucil, chlomaphazine, cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosureas such as carmustine, chlorozotocin, fotemustine, lomustine, nimustine, and ranimnustine; antibiotics such as the enediyne antibiotics (e.g., calicheamicin, especially calicheamicin gamma1I and calicheamicin omegaI1 (see, e.g., Agnew, Chem. Intl. Ed. Engl., 33: 183-186 (1994)); dynemicin, including dynemicin A; an esperamicin; as well as neocarzinostatin chromophore and related chromoprotein enediyne antiobiotic chromophores), aclacinomysins, actinomycin, authramycin, azaserine, bleomycins, cactinomycin, carabicin, caminomycin, carzinophilin, chromomycinis, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L-norleucine, ADRIAMYCIN.RTM. doxorubicin (including morpholino-doxorubicin, cyanomorpholino-doxorubicin, 2-pyrrolino-doxorubicin and deoxydoxorubicin), epirubicin, esorubicin, idarubicin, marcellomycin, mitomycins such as mitomycin C, mycophenolic acid, nogalamycin, olivomycins, peplomycin, potfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin; anti-metabolites such as methotrexate and 5-fluorouracil (5-FU); folic acid analogues such as denopterin, methotrexate, pteropterin, trimetrexate; purine analogs such as fludarabine, 6-mercaptopurine, thiamiprine, thioguanine; pyrimidine analogs such as ancitabine, azacitidine, 6-azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine; androgens such as calusterone, dromostanolone propionate, epitiostanol, mepitiostane, testolactone; anti-adrenals such as aminoglutethimide, mitotane, trilostane; folic acid replenisher such as frolinic acid; aceglatone; aldophosphamide glycoside; aminolevulinic acid; eniluracil; amsacrine; bestrabucil; bisantrene; edatraxate; defofamine; demecolcine; diaziquone; elformithine; elliptinium acetate; an epothilone; etoglucid; gallium nitrate; hydroxyurea; lentinan; lonidainine; maytansinoids such as maytansine and ansamitocins; mitoguazone; mitoxantrone; mopidanmol; nitraerine; pentostatin; phenamet; pirarubicin; losoxantrone; 2-ethylhydrazide; procarbazine; PSK.RTM. polysaccharide complex (JHS Natural Products, Eugene, Oreg.); razoxane; rhizoxin; sizofuran; spirogermanium; tenuazonic acid; triaziquone; 2,2',2''-trichlorotriethylamine; trichothecenes (especially T-2 toxin, verracurin A, roridin A and anguidine); urethan; vindesine (ELDISINE.RTM., FILDESIN.RTM.); dacarbazine; mannomustine; mitobronitol; mitolactol; pipobroman; gacytosine; arabinoside ("Ara-C"); thiotepa; taxoids, e.g., TAXOL.RTM. paclitaxel (Bristol-Myers Squibb Oncology, Princeton, N.J.), ABRAXANE.TM. Cremophor-free, albumin-engineered nanoparticle formulation of paclitaxel (American Pharmaceutical Partners, Schaumberg, Ill.), and TAXOTERE.RTM. doxetaxel (Rhone-Poulenc Rorer, Antony, France); chloranbucil; gemcitabine (GEMZAR.RTM.); 6-thioguanine; mercaptopurine; methotrexate; platinum analogs such as cisplatin and carboplatin; vinblastine (VELBAN.RTM.); platinum; etoposide (VP-16); ifosfamide; mitoxantrone; vincristine (ONCOVIN.RTM.); oxaliplatin; leucovovin; vinorelbine (NAVELBINE.RTM.); novantrone; edatrexate; daunomycin; aminopterin; ibandronate; topoisomerase inhibitor RFS 2000;
difluoromethylornithine (DMFO); retinoids such as retinoic acid; capecitabine (XELODA.RTM.); pharmaceutically acceptable salts, acids or derivatives of any of the above; as well as combinations of two or more of the above such as CHOP, an abbreviation for a combined therapy of cyclophosphamide, doxorubicin, vincristine, and prednisolone, and FOLFOX, an abbreviation for a treatment regimen with oxaliplatin (ELOXATIN.TM.) combined with 5-FU and leucovovin.
[0098] As used herein, "treatment" refers to clinical intervention in an attempt to alter the natural course of the individual or cell being treated, and can be performed either for prophylaxis or during the course of clinical pathology. Desirable effects of treatment include preventing occurrence or recurrence of disease, alleviation of symptoms, diminishment of any direct or indirect pathological consequences of the disease, preventing or decreasing inflammation and/or tissue/organ damage, decreasing the rate of disease progression, amelioration or palliation of the disease state, and remission or improved prognosis. In some embodiments, antibodies of the invention are used to delay development of a disease or disorder.
[0099] An "individual" or a "subject" is a vertebrate. In certain embodiments, the vertebrate is a mammal. Mammals include, but are not limited to, farm animals (such as cows), sport animals, pets (such as cats, dogs, and horses), primates, mice and rats. In certain embodiments, the vertebrate is a human.
[0100] "Mammal" for purposes of treatment refers to any animal classified as a mammal, including humans, domestic and farm animals, and zoo, sports, or pet animals, such as dogs, horses, cats, cows, etc. In certain embodiments, the mammal is human.
[0101] An "effective amount" refers to an amount effective, at dosages and for periods of time necessary, to achieve the desired therapeutic or prophylactic result.
[0102] A "therapeutically effective amount" of a substance/molecule of the invention may vary according to factors such as the disease state, age, sex, and weight of the individual, and the ability of the substance/molecule, to elicit a desired response in the individual. A therapeutically effective amount is also one in which any toxic or detrimental effects of the substance/molecule are outweighed by the therapeutically beneficial effects. A "prophylactically effective amount" refers to an amount effective, at dosages and for periods of time necessary, to achieve the desired prophylactic result. Typically but not necessarily, since a prophylactic dose is used in subjects prior to or at an earlier stage of disease, the prophylactically effective amount would be less than the therapeutically effective amount.
[0103] The term "cytotoxic agent" as used herein refers to a substance that inhibits or prevents the function of cells and/or causes destruction of cells. The term is intended to include radioactive isotopes (e.g., At211, I131, I125, Y90, Re186, Re188, Sm153, Bi212, P32, Pb212 and radioactive isotopes of Lu), chemotherapeutic agents (e.g., methotrexate, adriamicin, vinca alkaloids (vincristine, vinblastine, etoposide), doxorubicin, melphalan, mitomycin C, chlorambucil, daunorubicin or other intercalating agents, enzymes and fragments thereof such as nucleolyticenzymes, antibiotics, and toxins such as small molecule toxins or enzymatically active toxins of bacterial, fungal, plant or animal origin, including fragments and/or variants thereof, and the various antitumor or anticancer agents disclosed below. Other cytotoxic agents are described below. A tumoricidal agent causes destruction of tumor cells.
[0104] "Treating" or "treatment" or "alleviation" refers to both therapeutic treatment and prophylactic or preventative measures; wherein the object is to prevent or slow down (lessen) the targeted pathologic condition or disorder. Those in need of treatment include those already with the disorder as well as those prone to have the disorder or those in whom the disorder is to be prevented. A subject or mammal is successfully "treated" for an infection if, after receiving a therapeutic amount of an antibody according to the methods of the present invention, the patient shows observable and/or measurable reduction in or absence of one or more of the following: reduction in the number of infected cells or absence of the infected cells; reduction in the percent of total cells that are infected; and/or relief to some extent, one or more of the symptoms associated with the specific infection; reduced morbidity and mortality, and improvement in quality of life issues. The above parameters for assessing successful treatment and improvement in the disease are readily measurable by routine procedures familiar to a physician.
[0105] The term "therapeutically effective amount" refers to an amount of an antibody or a drug effective to "treat" a disease or disorder in a subject or mammal See preceding definition of "treating."
[0106] Administration "in combination with" one or more further therapeutic agents includes simultaneous (concurrent) and consecutive administration in any order.
[0107] "Carriers" as used herein include pharmaceutically acceptable carriers, excipients, or stabilizers that are nontoxic to the cell or mammal being exposed thereto at the dosages and concentrations employed. Often the physiologically acceptable carrier is an aqueous pH buffered solution. Examples of physiologically acceptable carriers include buffers such as phosphate, citrate, and other organic acids; antioxidants including ascorbic acid; low molecular weight (less than about 10 residues) polypeptide; proteins, such as serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine, arginine or lysine; monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrins; chelating agents such as EDTA; sugar alcohols such as mannitol or sorbitol; salt-forming counterions such as sodium; and/or nonionic surfactants such as TWEEN.TM. polyethylene glycol (PEG), and PLURONICS.TM.
Glycoantibodies
[0108] The glycosylation of recombinant proteins produced from mammalian cells in culture is an important process in ensuring the effective use of therapeutic antibodies (Goochee et al., 1991; Jenkins and Curling, 1994). Mammalian cell culture delivers a heterogeneous mixture of glycosylation patterns which do not all have the same properties. Properties like safety, efficacy and the serum half-life of therapeutic proteins can be affected by these glycosylation patterns. We have successfully addressed the glycoform heterogeneity problem by the development of a novel class of monoclonal antibodies, named "glycoantibodies".
[0109] The term "glycoantibodies" was coined by the inventor, Dr. Chi-Huey Wong, to refer to a homogeneous population of monoclonal antibodies (preferably, therapeutic monoclonal antibodies) having a single, uniformed glycoform on Fc. The individual glycoantibodies comprising the homogeneous population are identical, bind to the same epitope, and contain the same Fc glycan with a well-defined glycan structure and sequence.
[0110] Glycoantibodies may be generated from monoclonal antibodies (preferably, therapeutic monoclonal antibodies) commercially available or in the development. Monoclonal antibodies for therapeutic use can be humanized, human or chimeric.
[0111] The term "parental antibody" as used herein refers to the monoclonal antibody used to produce a glycoantibody. The parental antibodies can be obtained by cell culturing such as mammalian cell culture, Pichia pastoris or insect cell lines. Preferrably, the parental antibodies are produced in mammalian cell culture. The parental antibodies may be FDA approved or in development.
[0112] Monoclonal antibodies can be prepared using a wide variety of techniques known in the art including the use of hybridoma, recombinant, and phage display technologies, or a combination thereof. For example, monoclonal antibodies can be produced using hybridoma techniques including those known in the art and taught, for example, in Harlow et al., Antibodies: A Laboratory Manual, (Cold Spring Harbor Laboratory Press, 2nd ed. 1988); Hammerling, et al., in: Monoclonal Antibodies and T-Cell Hybridomas 563-681 (Elsevier, N.Y., 1981); each of which is incorporated herein by reference in its entirety. The term "monoclonal antibody" (abbreviated as "mAb") as used herein is not limited to antibodies produced through hybridoma technology. The term "monoclonal antibody" refers to an antibody that is derived from a single clone, including any eukaryotic, prokaryotic, or phage clone, and not the method by which it is produced. A "monoclonal antibody" may comprise, or alternatively consist of, two proteins, i.e., a heavy and a light chain.
[0113] Described herein are the functionally active glycoantibodies derived from therapeutic monoclonal antibodies by Fc glycoengineering. The glycoantibodies with optimized glycoforms exhibit more potent biological activities compared to the therapeutic monoclonal antibodies. It is contemplated that the glycoantibodies with optimized glycoforms may provide an alternative for therapeutic use.
[0114] Glycoantibodies of the invention consist of a single, uniformed glycoform (N-glycan) on Fc. In some embodiments, the N-glycan is attached to the Asn-297 of the Fc region.
[0115] The N-glycans according to the invention have a common pentasaccharide core of Man.sub.3GlcNAc.sub.2 which is also referred to as "trimannose core" or "pentasaccharide core", wherein "Man" refers to mannose, "Glc" refers to glucose, "NAc" refers to N-acetyl, and GlcNAc refers to N-acetylglucosamine.
[0116] In some embodiments, the N-glycan has a biantennary structure.
[0117] The N-glycan described herein may have intrachain substitutions comprising "bisecting" GlcNAc. When a glycan comprises a bisecting GlcNAc on the trimannose core, the structure is represented as Man.sub.3GlcNAc.sub.3. When a glycan comprises a core fucose attached to the trimannose core, the structure is represented as Man.sub.3GlcNAc.sub.2(F). The N-glycan may comprise one or more terminal sialic acids (e.g. N-acetylneuraminic acid). The structure represented as "Sia" refers to a terminal sialic acid. Sialylation may occur on either the .alpha.1-3 or .alpha.1-6 arm of the biantennary structures.
[0118] In some embodiments, the N-glycan described herein comprises at least one .alpha.2-6 terminal sialic acid. In certain embodiments, the N-glycan comprises one .alpha.2-6 terminal sialic acid. In a preferred embodiment, the N-glycan comprises two .alpha.2-6 terminal sialic acids.
[0119] In some embodiments, the N-glycan described herein comprises at least one .alpha.2-3 terminal sialic acid. In certain embodiments, the N-glycan comprises one .alpha.2-3 terminal sialic acid. In a preferred embodiment, the N-glycan comprises two .alpha.2-3 terminal sialic acids.
[0120] In some embodiments, the N-glycan described herein comprises at least one galactose. In certain embodiments, the N-glycan comprises one galactose. In a preferred embodiment, the N-glycan comprises two galactoses.
[0121] Preferrably, the N-glycan according to the disclosure is free of core fucose.
[0122] Table 1 lists exemplary N-glycans in glycoantibodies.
TABLE-US-00001 TABLE 1 GAb Glycan structure Glycan sequence 1-101 ##STR00001## Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-102 ##STR00002## Sia(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-103 ##STR00003## Sia(.alpha.2-6)GalGlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-104 ##STR00004## Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-105 ##STR00005## GalGlcNAcMan.sub.3GlcNAc.sub.2 1-106 ##STR00006## GalGlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-107 ##STR00007## GlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-108 ##STR00008## GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-109 ##STR00009## GlcNAcMan.sub.3GlcNAc.sub.2 1-110 ##STR00010## GlcNAcMan.sub.3GlcNAc.sub.2 1-111 ##STR00011## Man.sub.3GlcNAc.sub.2 1-112 ##STR00012## Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GLcNAc.sub.2 1-113 ##STR00013## Sia(.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-114 ##STR00014## Sia(.alpha.2-6)GalGlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-115 ##STR00015## Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-116 ##STR00016## GalGlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-117 ##STR00017## Sia.sub.2(.alpha.2-3)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-118 ##STR00018## Sia(.alpha.2-3)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-119 ##STR00019## Sia.sub.2(.alpha.2-3)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-120 ##STR00020## Sia(.alpha.2-3)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-121 ##STR00021## Sia.sub.2(.alpha.2-3/.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.- 2 1-122 ##STR00022## Sia.sub.2(.alpha.2-6/.alpha.2-3)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.- 2 1-123 ##STR00023## Sia.sub.2(.alpha.2-3/.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.- 2 1-124 ##STR00024## Sia.sub.2(.alpha.2-6/.alpha.2-3)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.- 2 1-125 ##STR00025## Sia(.alpha.2-3)GalGlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-126 ##STR00026## Sia(.alpha.2-3)GalGlcNAc.sub.3Man.sub.3GlcNAc.sub.2
Glycosylation on Fc can affect a variety of immunoglobulin effector-mediated functions, including ADCC, CDC and circulating half-life. ADCC enhancement is a key strategy for improving therapeutic antibody drug efficacy. It has the potential of lowering effective drug dosage for benefits of lower drug cost. The glycoantibodies described herein can be characterized by functional properties.
(I) Glycoantibodies for Cancers
[0123] Glycoantibodies described herein may be useful for treating a cancer. The FDA has approved multiple therapeutic monoclonal antibodies for cancer therapies, and many more are being studied in clinical trials either alone or in combination with other treatments. These monoclonal antibodies ("parental antibodies") can be used to produce glycoantibodies.
[0124] Exemplary monoclonal antibodies for cancers include, but are not limited to, Ado-trastuzumab emtansine (Kadcyla), Alemtuzumab (Campath), Belimumab (Benlysta), Bevacizumab (Avastin), Brentuximab vedotin (Adcetris), Cabozantinib (Cometriq), Canakinumab (Ilaris), Cetuximab (Erbitux), Denosumab (Xgeva), Ibritumomab tiuxetan (Zevalin), Ipilimumab (Yervoy), Nivolumab (Opdivo), Obinutuzumab (Gazyva), Ofatumumab (Arzerra, HuMax-CD20), Panitumumab (Vectibix), Pembrolizumab (Keytruda), Pertuzumab (Perjeta), Ramucirumab (Cyramza), Rituximab (Rituxan, Mabthera), Siltuximab (Sylvant), Tocilizumab, Tositumomab (Bexxar) and Trastuzumab (Herceptin).
Anti-CD20 Glycoantibodies (Anti-CD20 GAb)
[0125] The "CD20" antigen is a non-glycosylated, transmembrane phosphoprotein with a molecular weight of approximately 35 kD that is found on the surface of greater than 90% of B cells from peripheral blood or lymphoid organs. CD20 is expressed during early pre-B cell development and remains until plasma cell differentiation; it is not found on human stem cells, lymphoid progenitor cells or normal plasma cells. CD20 is present on both normal B cells as well as malignant B cells. Other names for CD20 in the literature include "B-lymphocyte-restricted differentiation antigen" and "Bp35". The CD20 antigen is described in, for example, Clark and Ledbetter, Adv. Can Res. 52:81-149 (1989) and Valentine et al. J. Biol. Chem. 264(19):11282-11287 (1989).
[0126] The present disclosure features a novel class of anti-CD20 antibodies, termed "anti-CD20 glycoantibodies" ("anti-CD20 GAb"). The anti-CD20 glycoantibodies can be generated from anti-CD20 monoclonal antibodies by Fc glycoengineering. The individual anti-CD20 glycoantibodies comprising the homogeneous population are identical and contain the same Fc glycan with a well-defined glycan structure and sequence. The anti-CD20 GAb according to the present invention specifically binds to the same epitope of a human CD20 antigen on a cell membrane as its patent antibody.
[0127] The term "parental antibody" as used herein refers to the anti-CD20 monoclonal antibody used to produce an anti-CD20 glycoantibody.
[0128] The parental antibodies can be obtained by cell culturing such as mammalian cell culture, Pichia pastoris or insect cell lines. Preferrably, the parental antibodies are produced in mammalian cell culture. The parental antibodies may be FDA approved or in development. Exemplary parental antibodies include, but not limited to, Rituximab, Ofatumumab, Tositumomab, Ocrelizumab, 11B8 or 7D8 (disclosed in WO2004/035607), an anti-CD20 antibody disclosed in WO 2005/103081 such as C6, an anti-CD antibody disclosed in W02003/68821 such as IMMU-106 (from Immunomedics), an anti-CD20 antibody disclosed in W02004/103404 such as AME-133 (from Applied Molecular Evolution/Lilly), and anti-CD20 antibody disclosed in US 2003/0118592 such as TRU-015 (from Trubion Pharmaceuticals Inc), 90Y-labeled 2B8 murine antibody designated "Y2B8" (ZEVALIN.RTM.) (Biogen-Idec, Inc.) (e.g., U.S. Pat. No. 5,736,137, Anderson et al.; ATCC deposit HB11388); murine and chimeric 2H7 antibody (e.g., U.S. Pat. No. 5,677,180, Robinson et al.); humanized 2H7 antibodies such as rhuMAb2H7 and other versions (Genentech, Inc.) (e.g., WO 2004/056312, Adams et al., and other references noted below); human monoclonal antibodies against CD20 (GenMab A/S/Medarex, Inc.) (e.g., WO 2004/035607 and WO 2005/103081, Teeling et al.); a chimerized or humanized monoclonal antibody binding to an extracellular epitope of CD20 (Biomedics Inc.) (e.g., WO 2006/106959, Numazaki et al.); humanized LL2 and similar antibodies (Immunomedics, Inc.) (e.g., U.S. Pat. No. 7,151,164 and US 2005/0106108, Hansen); A20 antibodies (Immunomedics, Inc.) such as chimeric A20 (cA20) or humanized A20 antibody (hA20, IMMUN-106T, veltuzumab) (e.g., US 2003/0219433, Hansen et al.); fully human antibodies against CD20 (Amgen/AstraZeneca) (e.g., WO 2006/130458, Gazit et al.); antibodies against CD20 (Avestha Gengraine Technologies Pvt Ltd.) (e.g., WO 2006/126069, Morawala); and chimeric or humanized B-Ly1 antibodies to CD20 (Roche/GlycArt Biotechnology AG) such as GA101 (e.g., WO 2005/044859; US 2005/0123546; US 2004/0072290; and US 2003/0175884, Umana et al.).
[0129] In some embodiments, the exemplary anti-CD20 GAb described herein comprise a heavy chain having the amino acid sequence set forth in SEQ ID NO: 1, and a light chain having the amino acid sequence set forth in SEQ ID NO: 2. In a preferred embodiment, the anti-CD20 GAb comprises a light chain sequence and a heavy chain sequence of Rituximab.
Table 2 below shows the heavy chain and the light chain sequences of Rituximab.
TABLE-US-00002 TABLE 2 >Rituximab Accession Number: DB00073 Source: http://www.drugbank.ca/drugs/DB00073 >Rituximab heavy chain QVQLQQPGAELVKPGASVKMSCKASGYTFTSYNMHWVKQTPGRGLEW IGAIYPGNGDTSYNQKFKGKATLTADKSSSTAYMQLSSLTSEDSAVY YCARSTYYGGDWYFNVWGAGTTVTVSAASTKGPSVFPLAPSSKSTSG GTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSS VVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPA PELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWY VDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSN KALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGF YPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQ GNVFSCSVMHEALHNHYTQKSLSLSPGK (SEQ ID: 2) >Rituximab light chain QIVLSQSPAILSASPGEKVTMTCRASSSVSYIHWFQQKPGSSPKPWI YATSNLASGVPVRFSGSGSGTSYSLTISRVEAEDAATYYCQQWTSNP PTFGGGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPR EAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKH KVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID: 1)
[0130] In some embodiments, the N-glycan is attached to the Asn-297 of the Fc region.
[0131] The N-glycans according to the invention have a common pentasaccharide core of Man.sub.3GlcNAc.sub.2 which is also referred to as "trimannose core" or "pentasaccharide core", wherein "Man" refers to mannose, "Glc" refers to glucose, "NAc" refers to N-acetyl, and GlcNAc refers to N-acetylglucosamine.
[0132] In some embodiments, the N-glycan has a biantennary structure.
[0133] The N-glycan described herein may have intrachain substitutions comprising "bisecting" GlcNAc. When a glycan comprises a bisecting GlcNAc on the trimannose core, the structure is represented as Man.sub.3GlcNAc.sub.3. When a glycan comprises a core fucose attached to the trimannose core, the structure is represented as Man.sub.3GlcNAc.sub.2(F). The N-glycan may comprise one or more terminal sialic acids (e.g. N-acetylneuraminic acid). The structure represented as "Sia" refers to a terminal sialic acid. Sialylation may occur on either the .alpha.1-3 or .alpha.1-6 arm of the biantennary structures.
[0134] In some embodiments, the N-glycan described herein comprises at least one .alpha.2-6 terminal sialic acid. In certain embodiments, the N-glycan comprises one .alpha.2-6 terminal sialic acid. In a preferred embodiment, the N-glycan comprises two .alpha.2-6 terminal sialic acids.
[0135] In some embodiments, the N-glycan described herein comprises at least one .alpha.2-3 terminal sialic acid. In certain embodiments, the N-glycan comprises one .alpha.2-3 terminal sialic acid. In a preferred embodiment, the N-glycan comprises two .alpha.2-3 terminal sialic acids.
[0136] In some embodiments, the N-glycan described herein comprises at least one galactose. In certain embodiments, the N-glycan comprises one galactose. In a preferred embodiment, the N-glycan comprises two galactoses.
[0137] Preferrably, the N-glycan according to the disclosure is free of core fucose.
[0138] Table 3 lists exemplary N-glycans in anti-CD20 glycoantibodies. Embodiments of the present disclosure may include or exclude any of the N-glycans listed herein.
TABLE-US-00003 TABLE 3 GAb Glycan structure Glycan sequence 1-101 ##STR00027## Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-102 ##STR00028## Sia(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-103 ##STR00029## Sia(.alpha.2-6)GalGlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-104 ##STR00030## Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-105 ##STR00031## GalGlcNAcMan.sub.3GlcNAc.sub.2 1-106 ##STR00032## GalGlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-107 ##STR00033## GlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-108 ##STR00034## GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-109 ##STR00035## GlcNAcMan.sub.3GlcNAc.sub.2 1-110 ##STR00036## GlcNAcMan.sub.3GlcNAc.sub.2 1-111 ##STR00037## Man.sub.3GlcNAc.sub.2 1-112 ##STR00038## Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-113 ##STR00039## Sia(.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-114 ##STR00040## Sia(.alpha.2-6)GalGlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-115 ##STR00041## Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-116 ##STR00042## GalGlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-117 ##STR00043## Sia.sub.2(.alpha.2-3)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-118 ##STR00044## Sia(.alpha.2-3)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-119 ##STR00045## Sia2(.alpha.2-3)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-120 ##STR00046## Sia(.alpha.2-3)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2 1-121 ##STR00047## Sia.sub.2(.alpha.2-3/.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.- 2 1-122 ##STR00048## Sia.sub.2(.alpha.2-6/.alpha.2-3)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.- 2 1-123 ##STR00049## Sia.sub.2(.alpha.2-3/.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.- 2 1-124 ##STR00050## Sia.sub.2(.alpha.2-6/.alpha.2-3)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.- 2 1-125 ##STR00051## Sia(.alpha.2-3)GalGlcNAc.sub.2Man.sub.3GlcNAc.sub.2 1-126 ##STR00052## Sia(.alpha.2-3)GalGlcNAc.sub.3Man.sub.3GlcNAc.sub.2
Biological Characteristic of Anti-CD20 Glycoantibodies
[0139] Glycosylation on Fc can affect a variety of immunoglobulin effector-mediated functions, including ADCC, CDC and circulating half-life. ADCC enhancement is a key strategy for improving therapeutic antibody drug efficacy. It has the potential of lowering effective drug dosage for benefits of lower drug cost. The anti-CD20 glycoantibodies described herein can be characterized by functional properties. The anti-CD20 GAb has cell growth inhibitory activities including apoptosis against human CD20 expressing cells. In some embodiments, the anti-CD20 GAb exhibits more potent cell growth inhibitory activities as compared to its patent antibody.
ADCC Activities of Anti-CD20 Glycoantibodies
[0140] The increased ADCC activity of the glycoantibody according to the invention is at least about 5 fold, including but not limited to, at least about 6 fold, about 7 fold, about 8 fold, about 9 fold about 10 fold, about 15 fold, about 20 fold, about 25 fold, about 30 fold, about 35 fold, about 40 fold, about 50 fold, about 60 fold, and about 80 fold or at least about a value in the range between any of the two numbers listed herein compared to the ADCC activity of the parental antibody.
[0141] Table 4 lists exemplary enhanced ADCC activities of anti-CD20 GAbs as compared to Rituximab. Exemplary assays are described in the examples.
TABLE-US-00004 TABLE 4 Anti-CD20 Rituximab GAb101 GAb104 GAb105 GAb107 GAb108 GAb111 ADCC 1 >50 >50 30~50 >50 10~30 5~10 (fold)
[0142] A number of anti-CD20 GAbs described herein, in particular GAb101, and GAb104, exhibit enhanced ADCC activity compared to it parental antibody, Rituximab. It is contemplated that the glycoantibodies of the invention may exhibit superior effect as therapeutic agents for B cell-mediated malignant tumors and immunological diseases in which B cells or antibodies produced by B cells are involved, and an object of the present invention is to use the anti-CD20 GAb in development of therapeutic agents.
CDC Activities of Anti-CD20 Glycoantibodies
[0143] The glycoantibody described herein is surprisingly able to provide improved ADCC without affecting CDC. Exemplary CDC assays are described in the examples. In exemplary embodiments, ADCC of the glycoantibody is increased but other immunoglobulin-type effector functions such as complement-dependent cytoxicity (CDC) remain similar or are not significantly affected.
Binding Between Fc.gamma.RIII and Anti-CD20 Glycoantibodies
[0144] Table 5 lists exemplary Fc.gamma.RIIIA binding of anti-CD20 GAbs and Rituximab.
[0145] Fc.gamma.RIIIA binding may be measured using assays known in the art. Exemplary assays are described in the examples. The Fc receptor binding may be determined as the relative ratio of anti-CD20 GAb vs Rituximab. Fc receptor binding in exemplary embodiments is increased by at least 1.2-fold, 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 15-fold or 20-fold, 30-fold, 40-fold, 50-fold, 100-fold or higher.
TABLE-US-00005 TABLE 5 Binding Constant of FcRIIIA to variable glycoantibodies by SPR Curve KD (nM) Rmax (RU) Fold Note Rittman 100~300 49.29 ##STR00053## GAb101 1~25 90.48 A Category A: increase>30X Category B: increase 15~30X Category C: increase 5~10X ##STR00054## GAb104 1~25 93.4 A ##STR00055## GAb111 40~130 56.28 C ##STR00056## GAb108 40~130 67.01 C ##STR00057## GAb107 7~30 76.02 B ##STR00058## GAb109 40~130 51.03 C ##STR00059## GAb110 40~130 38.43 C ##STR00060## GAb105 1~25 72.12 A ##STR00061## GAb106 7~30 70.8 B ##STR00062## GAb102 1~25 67.52 A
[0146] As compared to Rituximab, the binding data showed that the anti-CD20 GAbs, in particular GAb101 and GAb104, exhibit stronger binding affinity for the target molecule CD20.
[0147] Taken together, anti-CD20 Gabs, exhibit enhanced ADCC activity and stronger Fc.gamma.RIIIA binding affinity as compared to Rituximab. It is contemplated that the glycoantibodies of the invention may provide a superior clinical response either alone or, in a composition comprising two or more such antibodies, and optionally in combination with other treatments such as chemotherapy. It is contemplated that the ADCC-enhanced anti-CD20 glycoantibody may provide an alternative therapeutic for B-cell lymphoma and other diseases. The glycoantibodies of the present invention advantageously can be used to alter current routes of administration and current therapeutic regimens, as their increased effector function means they can be dosed at lower concentrations and with less frequency, thereby reducing the potential for antibody toxicity and/or development of antibody tolerance. Furthermore, the improved effector function yields new approaches to treating clinical indications that have previously been resistant or refractory to treatment with the corresponding anti-CD20 monoclonal antibody produced in recombinant host systems.
Preparation of Anti-CD20 GAb
[0148] The anti-CD20 glycoantibodies of the invention can be produced by Fc glycoengineering from anti-CD20 monoclonal antibodies ("parental antibodies") commercially available or in the preclinical or clinical development. Preferrably, the monoclonal antibodies are therapeutic monoclonal antibodies. Fc glycoengineering may be performed enzymatically or chemoenzymatically. In a preferred embodiment, the parental antibody is Rituximab.
[0149] The N-glycans in the glycoantibodies of the invention are preferrably defucosylated.
[0150] Defucosylation of N-glycans is a process to remove core fucoses in N-glycans of the Fc domains. Defucosylation can be employed enzymatically. Since N-glycans are embedded between two Fc domains, the enzymatic defucosylation efficiency is much lower due to steric hindrance, i.e., access of fucosidase to fucose residues is blocked by potions of the Fc domains.
[0151] Many .alpha.-fucosidases are known in the art. Examples include .alpha.-fucosidases from Turbo cornutus, Charonia lampas, Bacillus fulminans, Aspergillus niger, Clostridium perfringens, Bovine kidney (Glyko), chicken liver (Tyagarajan et al., 1996, Glycobiology 6:83-93) and .alpha.-fucosidase II from Xanthomonas manihotis (Glyko, PROzyme). Many varieties of fucosidase are also commercially available (Glyko, Novato, Calif.; PROzyme, San Leandro, Calif.; Calbiochem-Novabiochem Corp., San Diego, Calif.; among others). However, none of .alpha.-fucosidases are known to efficiently remove the core fucose from N-linked glycans.
[0152] WO 2013/12066 disclosed the defucosylation of (Fuc.alpha.1,6)GlcNAc-Rituximab by an .alpha.-fucosidase from bovine kidney. As described in WO 2013/12066, a reaction mixture of (Fuc al, 6)GlcNAc-Rituximab was incubated with .alpha.-fucosidase from bovine kidney (commercially available from Prozyme) at 37.degree. C. for 20 days to completely remove the fucose in (Fuc.alpha.1,6)GlcNAc-Rituximab.
[0153] Thermal instability of immunoglobulin has been reported (Vermeer et al., Biophys J. January 78: 394-404 (2000)). The Fab fragment is most sensitive to heat treatment, whereas the Fc fragment is most sensitive to decreasing pH. To examine the thermal stability and functional activity of the antibody, we performed the same experiment as described in WO 2013/12066, and found the antibody lost about 10% binding affinity to CD20 after thermal treatment at 37.degree. C. for 3 days. Furthermore, we found the antibody lost about 20% binding affinity to CD20 after thermal treatment at 37.degree. C. for 7 days. It is contemplated that the antibody will significantly lose the binding affinity to CD20 after prolonged thermal treatment, such as at 37.degree. C. for 20 days, as described in WO 2013/12066.
[0154] In our attempts to synthesize the glycoantibodies with improved therapeutic values, we unexpectedly discovered a Bacteroides fragilis .alpha.-fucosidase (GenBank accession no. YP_212855.1) that is capable of efficiently removing fucose residues from N-linked glycans. Efficient defucosylation has been successfully achieved using the specific enzyme. Importantly, the efficiency of making the glycoantibodies of the invention has been valuably improved by the use of the specific .alpha.-fucosidase that yields a facile defucosylation of N-glycans, as illustrated in FIG. 1.
[0155] Accordingly, the present invention provides a composition of the .alpha.-fucosidase, and an improved method for removing core fucoses of N-glycans using the .alpha.-fucosidase. The .alpha.-fucosidase comprises a polypeptide having an amino acid sequence having at least 80%, 85% 90%, 95%, 98% or 99% identity to the sequences of SEQ ID NO: 5 or variants thereof. The improved method of defucosylation comprises contacting an antibody with an .alpha.-fucosidase, and in which the .alpha.-fucosidase comprises a polypeptide having an amino acid sequence having at least 80%, 85%, 90%, 95%, 98% or 99% identity to the sequences of SEQ ID NO: 5, a variant or a fragment thereof.
[0156] Described herein includes an improved method for making an anti-CD20 glycoantibody, the method comprising the steps of (a) contacting an anti-CD20 monoclonal antibody with an .alpha.-fucosidase and at least one endoglycosidase, thereby yielding a defucosylated antibody having a single N-acetylglucosamine (GlcNAc), and (b) adding a carbohydrate moiety to GlcNAc under suitable conditions.
[0157] In some embodiments, the anti-CD20 monoclonal antibody according to the method of the invention is Rituximab.
[0158] Endoglycosidase is used to trim off the variable portions of an oligosaccharide in N-glycan. Examples of endoglycosidases used herein include, but not limited to, EndoA, EndoF, EndoF1, EndoF2, EndoF3, EndoH, EndoM, EndoS, EndoS2 and variants thereof.
[0159] The .alpha.-fucosidase according to the method of the invention comprises a polypeptide having an amino acid sequence having at least 85% identity to the sequences of SEQ ID NO: 5, a functional variant thereof.
[0160] In some embodiments, the .alpha.-fucosidase comprises a polypeptide having an amino acid sequence having at least 90% or 95% identity to the sequences of SEQ ID NO: 5, a variant or a fragment thereof.
[0161] In certain embodiments, the .alpha.-fucosidase is a recombinant Bacteroides .alpha.-fucosidase.
TABLE-US-00006 TABLE 6 QQKYQPTEANLKARSEFQDNKFGIFLHWGLYAMLATGEWTMTNNNLN YKEYAKLAGGFYPSKFDADKWVAAIKASGAKYICFTTRHHEGFSMFD TKYSDYNIVKATPFKRDVVKELADACAKHGIKLHFYYSHIDWYREDA PQGRTGRRTGRPNPKGDWKSYYQFMNNQLTELLTNYGPIGAIWFDGW WDQDINPDFDWELPEQYALIHRLQPACLVGNNHHQTPFAGEDIQIFE RDLPGENTAGLSGQSVSHLPLETCETMNGMWGYKITDQNYKSTKTLI HYLVKAAGKDANLLMNIGPQPDGELPEVAVQRLKEVGEWMSKYGETI YGTRGGLVAPHDWGVTTQKGNKLYVHILNLQDKALFLPIVDKKVKKA VVFADKTPVRFTKNKEGIVLELAKVPTDVDYVVELTID (SEQ ID: 5)
[0162] Step (a) in the method of the invention leads to a defucosylated antibody having a single N-acetylglucosamine (GlcNAc). Subsequent enzyme-mediated glycosylation using a transglycosylase is performed to add a designated carbohydrate moiety to GlcNAc and extend the sugar chain. A homogenous population of glycoantibodies can therefore be produced. Examples of transglycosylases as described herein include, but not limited to, EndoA, EndoF, EndoF1, EndoF2, Endo F3, EndoH, EndoM, EndoS, Endo S2 and variants thereof.
[0163] In some embodiments, the carbohydrate moiety according to the method the invention is selected from the group consisting of Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2, Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2, Sia.sub.2(.alpha.2-3)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2, Sia.sub.2(.alpha.2-3)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2, Sia.sub.2(.alpha.2-3/.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.- 2, Sia.sub.2(.alpha.2-6/.alpha.2-3)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.su- b.2, Sia.sub.2(.alpha.2-3/.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.- sub.2, Sia.sub.2(.alpha.2-6/.alpha.2-3)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNA- c.sub.2, Sia(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-3)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-3)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-6)GalGlcNAc.sub.2Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-3)GalGlcNAc.sub.2Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-6)GalGlcNAc.sub.3Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-3)GalGlcNAc.sub.3Man.sub.3GlcNAc.sub.2, Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2, GalGlcNAcMan.sub.3GlcNAc.sub.2, Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2, GalGlcNAc.sub.2Man.sub.3GlcNAc.sub.2, GalGlcNAc.sub.3Man.sub.3GlcNAc.sub.2, GlcNAc.sub.3Man.sub.3GlcNAc.sub.2, GlcNAc.sub.2Man.sub.3GlcNAc.sub.2, GlcNAcMan.sub.3GlcNAc.sub.2 and Man.sub.3GlcNAc.sub.2.
[0164] In preferred embodiments, the carbohydrate moiety is selected from the group consisting of Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2, Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2, Sia.sub.2(.alpha.2-3)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2, Sia.sub.2(.alpha.2-3)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2, Sia.sub.2(.alpha.2-3/.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.- 2, Sia.sub.2(.alpha.2-6/.alpha.2-3)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.su- b.2, Sia.sub.2(.alpha.2-3/.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.- sub.2, Sia.sub.2(.alpha.2-6/.alpha.2-3)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNA- c.sub.2, Sia(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-3)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-6)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-3)Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-6)GalGlcNAc.sub.2Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-3)GalGlcNAc.sub.2Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-6)GalGlcNAc.sub.3Man.sub.3GlcNAc.sub.2, Sia(.alpha.2-3)GalGlcNAc.sub.3Man.sub.3GlcNAc.sub.2, Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.sub.2, GalGlcNAcMan.sub.3GlcNAc.sub.2 and Gal.sub.2GlcNAc.sub.3Man.sub.3GlcNAc.sub.2.
[0165] Step (b) in the method of the invention leads to sugar chain extension. One method for sugar chain extension is through an enzyme-catalyzed glycosylation reaction. It is well known in the art that glycosylation using a sugar oxazoline as the sugar donor among the enzyme-catalyzed glycosylation reactions is useful for synthesizing oligosaccharides because the glycosylation reaction is an addition reaction and advances without any accompanying elimination of acid, water, or the like. (Fujita, et al., Biochim. Biophys. Acta 2001, 1528, 9-14)
[0166] In some embodiments, the carbohydrate moiety is a sugar oxazoline.
[0167] Suitable conditions also include incubation of the reaction mixture for at least 20 minutes, 30 minutes, 40 minutes, 50 minutes, 60 minutes, 70 minutes, 80 minutes, 90 minutes or 100 minutes, preferably less than 60 minutes. Incubation preferably takes place at room temperature, more preferably at approximately 20.degree. C., 25.degree. C., 30.degree. C., 35.degree. C., 40.degree. C. or 45.degree. C., and most preferably at approximately 37.degree. C.
[0168] It will be understood that the polypeptide of the .alpha.-fucosidase of the invention may be derivatized or modified to assist with their isolation or purification. Thus, in one embodiment of the invention, the polypeptide for use in the invention is derivatized or modified by addition of a ligand which is capable of binding directly and specifically to a separation means. Alternatively, the polypeptide is derivatized or modified by addition of one member of a binding pair and the separation means comprises a reagent that is derivatized or modified by addition of the other member of a binding pair. Any suitable binding pair can be used. In a preferred embodiment where the polypeptide for use in the invention is derivatized or modified by addition of one member of a binding pair, the polypeptide is preferably histidine-tagged or biotin-tagged. Typically the amino acid coding sequence of the histidine or biotin tag is included at the gene level and the proteins are expressed recombinantly in E. coli. The histidine or biotin tag is typically present at one end of the polypeptide, either at the N-terminus or at the C-terminus. The histidine tag typically consists of six histidine residues (SEQ ID NO: 23), although it can be longer than this, typically up to 7, 8, 9, 10 or 20 amino acids or shorter, for example 5, 4, 3, 2 or 1 amino acids. Furthermore, the histidine tag may contain one or more amino acid substitutions, preferably conservative substitutions as defined above.
[0169] Variant polypeptide as described herein are those for which the amino acid sequence varies from that in SEQ ID NO: 5, but exhibit the same or similar function of the enzyme comprising the polypeptide having an amino acid sequence of SEQ ID NO: 5.
[0170] As used herein percent (%) sequence identity with respect to a sequence is defined as the percentage of amino acid residues in a candidate polypeptide sequence that are identical with the amino acid residues in the reference polypeptide sequence, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity. Alignment for purposes of determining percent sequence identity can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST, ALIGN or Megalign (DNASTAR) software. Those skilled in the art can determine appropriate parameters for measuring alignment, including any algorithms needed to achieve maximal alignment over the full length of the sequences being compared.
[0171] Some preferred embodiments of the invention are demonstrated in the examples.
[0172] Methods for humanizing non-human antibodies are well known in the art. Generally, a humanized antibody has one or more amino acid residues introduced into it from a source which is non-human. These non-human amino acid residues are often referred to as "import" residues, which are typically taken from an "import" variable domain. Humanization can be essentially performed following the method of Winter and co-workers (Jones et al., Nature, 321:522-525 (1986); Riechmann et al., Nature, 332:323-327 (1988); Verhoeyen et al., Science, 239:1534-1536 (1988)), by substituting rodent CDRs or CDR sequences for the corresponding sequences of a human antibody. Accordingly, such "humanized" antibodies are chimeric antibodies (U.S. Pat. No. 4,816,567), wherein substantially less than an intact human variable domain has been substituted by the corresponding sequence from a non-human species. In practice, humanized antibodies are typically human antibodies in which some CDR residues and possibly some FR residues are substituted by residues from analogous sites in rodent antibodies.
[0173] The choice of human variable domains, both light and heavy, to be used in making the humanized antibodies is very important to reduce antigenicity. According to the so-called "best-fit" method, the sequence of the variable domain of a rodent antibody is screened against the entire library of known human variable-domain sequences. The human sequence which is closest to that of the rodent is then accepted as the human framework (FR) for the humanized antibody (Sims et al., J. Immunol., 151:2296 (1993); Chothia et al., J. Mol. Biol., 196:901 (1987)). Another method uses a particular framework derived from the consensus sequence of all human antibodies of a particular subgroup of light or heavy chains. The same framework may be used for several different humanized antibodies (Carter et al., Proc. Natl. Acad Sci. USA, 89:4285 (1992); Prestaetal, J. Immnol., 151:2623 (1993)).
[0174] It is further important that antibodies be humanized with retention of high affinity for the antigen and other favorable biological properties. To achieve this goal, according to a preferred method, humanized antibodies are prepared by a process of analysis of the parental sequences and various conceptual humanized products using three-dimensional models of the parental and humanized sequences. Three-dimensional immunoglobulin models are commonly available and are familiar to those skilled in the art. Computer programs are available which illustrate and display probable three-dimensional conformational structures of selected candidate immunoglobulin sequences. Inspection of these displays permits analysis of the likely role of the residues in the functioning of the candidate immunoglobulin sequence, i. e., the analysis of residues that influence the ability of the candidate immunoglobulin to bind its antigen. In this way, FR residues can be selected and combined from the recipient and import sequences so that the desired antibody characteristic, such as increased affinity for the target antigen(s), is achieved. In general, the CDR residues are directly and most substantially involved in influencing antigen binding.
[0175] Alternatively, it is now possible to produce transgenic animals (e.g., mice) that are capable, upon immunization, of producing a full repertoire of human antibodies in the absence of endogenous immunoglobulin production. For example, it has been described that the homozygous deletion of the antibody heavy-chain joining region (JH) gene in chimeric and germ-line mutant mice results in complete inhibition of endogenous antibody production. Transfer of the human germ-line immunoglobulin gene array in such germ-line mutant mice will result in the production of human antibodies upon antigen challenge. See, e.g., Jakobovits et al., Proc. Natl. Acad. Sci. USA, 90:2551 (1993); Jakobovits et al., Nature, 362:255-258 (1993); Bruggermann et al., Year in Immuno., 7:33 (1993). Human antibodies can also be derived from phage-display libraries (Hoogenboom et al., J. Mol. Biol., 227:381 (1991); Marks et al., J. Mol. Biol., 222:581-597 (1991)).
Anti-HER2 Glycoantibodies (Anti-HER2 GAb)
[0176] The HER2 gene is overexpressed or amplified in approximately 30% of breast cancers. Breast cancer patients with HER2 overexpression or amplification have shortened disease-free and overall survivals. The HER2 protein is thought to be a unique and useful target for antibody therapy of cancers overexpressing the HER2 gene. A monoclonal antibody anti-HER2, Trastuzumab (Herceptin.RTM.), has been successfully used in therapy for malignant cancers relating to this target, which was approved by FDA in 1998 for the treatment of HER2 overexpressing breast cancer. A need remains for improved therapeutic antibodies against HER2 which are more effective in preventing and/or treating a range of diseases involving cells expressing HER2, including but not limited breast cancer.
[0177] The present disclosure features a novel class of anti-HER2 antibodies, termed "anti-HER2 glycoantibodies" ("anti-HER2 GAb"). The anti-HER2 glycoantibodies can be generated from anti-HER2 monoclonal antibodies by Fc glycoengineering. The individual anti-HER2 glycoantibodies comprising the homogeneous population are identical and contain the same Fc glycan with a well-defined glycan structure and sequence. The anti-HER2 GAb according to the present invention specifically binds to the same epitope of a human HER2 antigen as its patent antibody.
[0178] The term "parental antibody" as used herein refers to the anti-HER2 monoclonal antibody used to produce an anti-HER2 glycoantibody.
[0179] The parental antibodies can be obtained by cell culturing such as mammalian cell culture, Pichia pastoris or insect cell lines. Preferrably, the parental antibodies are produced in mammalian cell culture. The parental antibodies may be FDA approved or in development. FDA approved anti-HER2 therapeutic antibodies include Trastuzumab (Herceptin), Lapatinib (Tykerb), Pertuzumab (Perjeta), Ado-trastuzumab emtansine (Kadcyla, Genentech).
[0180] In some embodiments, the anti-HER2 GAb described herein comprise a heavy chain having the amino acid sequence set forth in SEQ ID NO: 11, and a light chain having the amino acid sequence set forth in SEQ ID NO: 12. In a preferred embodiment, the anti-HER2 GAb comprises a light chain sequence and a heavy chain sequence of Trastuzumab.
Table 7 below shows the heavy chain and the light chain sequences of Trastuzumab.
TABLE-US-00007 TABLE 7 Trastuzumab Accession Number: DB00072 Source: http://www.drugbank.ca/drugs/DB00072 >Amino acid sequence for Trastuzumab light chain DIQMTQSPSSLSASVGDRVTITCRASQDVNTAVAWYQQKPGKAPK LLIYSASFLYSGVPSRFSGSRSGTDFTLTISSLQPEDFATYYCQQ HYTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCL LNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID: 12) >Amino acid sequence for Trastuzumab heavy chain EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGL EWVARIYPTNGYTRTADSVKGRFTISADTSKNTAYLQMNSLRAED TAVYYCSRWGGDGFYAMDYWGQGTLVTVSSASTKGPSVFPLAPSS KSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSS GLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDK THTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVS HEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQD WLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRE EMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDG SFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK (SEQ ID: 11)
[0181] Glycosylation on Fc can affect a variety of immunoglobulin effector-mediated functions, including ADCC, CDC and circulating half-life. ADCC enhancement is a key strategy for improving therapeutic antibody drug efficacy. It has the potential of lowering effective drug dosage for benefits of lower drug cost. The anti-HER2 glycoantibodies described herein can be characterized by functional properties. The anti-HER2 GAb has cell growth inhibitory activities including apoptosis against human HER2 expressing cells. In some embodiments, the anti-HER2 GAb exhibits more potent cell growth inhibitory activities as compared to its patent antibody.
[0182] The ADCC activity of the glycoantibody according to the invention is at least 3 fold increased, preferably at least 9 fold, more preferably at least 10 fold increased ADCC activity, preferably at least 12 fold increased ADCC activity, preferably at least 20 fold increased ADCC activity, most preferred at least 30 fold increased ADCC activity compared to the ADCC activity of the parental antibody.
[0183] The ADCC lysis activity of the inventive glycoantibody can be measured in comparison to the parental antibody using target cancer cell lines such as SKBR5, SKBR3, LoVo, MCF7, OVCAR3 and/or Kato III.
[0184] Table 8 lists exemplary enhanced ADCC activities of anti-HER2 GAbs as compared to Trastuzumab. Exemplary assays are described in the examples.
TABLE-US-00008 TABLE 8 Anti-HER2 Trastuzumab GAb101 GAb104 GAb105 GAb107 GAb108 GAb111 ADCC 1 >30 >30 20~30 >10 5~10 1~5 (fold)
[0185] A number of anti-HER2 GAbs described herein, in particular GAb101, and GAb104, exhibit enhanced ADCC activity compared to it parental antibody, Rituximab. It is contemplated that the glycoantibodies of the invention may exhibit superior effect as therapeutic agents for HER2-positive diseases, and an object of the present invention is to use the anti-HER2 GAb in development of therapeutic agents.
[0186] The glycoantibody described herein is surprisingly able to provide improved ADCC without affecting CDC. Exemplary CDC assays are described in the examples. In exemplary embodiments, ADCC of the glycoantibody is increased but other immunoglobulin-type effector functions such as complement-dependent cytoxicity (CDC) remain similar or are not significantly affected.
[0187] Table 9 lists exemplary Fc.gamma.RIIIA binding of anti-HER2 GAbs and Herceptin.
TABLE-US-00009 TABLE 9 Sample KD (M) Rmax (RU) Fold Herceptin 80~200 30.01 1-fold 101 1~25 44.98 >10X 104 1~25 55.68 >10X 111 35~100 41.54 1~5X 108 25~100 53.98 1~5X 107 20~90 39.88 3~10X 109 25~80 48.19 2~10X 110 70~150 18.15 1~5X 106 25~80 52.82 1~10X 103 15~70 59.89 4~10X 117 1~50 26.95 1~5X
[0188] Fc.gamma.RIIIA binding may be measured using assays known in the art, Exemplary assays are described in the examples. The Fc receptor binding may be determined as the relative ratio of anti-HER2 GAb vs Trastuzumab. Fc receptor binding in exemplary embodiments is increased by at least 2.5-fold, 3-fold, 4-fold, 5-fold 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 15-fold or 20-fold, 30-fold, 40-fold, 50-fold or higher.
[0189] As compared to Trastuzumab, the binding data showed that the anti-HER2 GAbs, in particular GAb101 and GAb104, exhibit stronger binding affinity for the target molecule HER2.
[0190] Taken together, anti-HER2 GAbs, in particular GAb101, and GAb104, exhibit enhanced ADCC activity and stronger Fc.gamma.RIIIA binding affinity as compared to Trastuzumab. It is contemplated that the glycoantibodies of the invention may provide a superior clinical response either alone or, preferably, in a composition comprising two or more such antibodies, and optionally in combination with other treatments such as chemotherapy. It is contemplated that the ADCC-enhanced anti-HER2 glycoantibody may provide an alternative therapeutic for HER2-positive diseases. The glycoantibodies of the present invention advantageously can be used to alter current routes of administration and current therapeutic regimens, as their increased effector function means they can be dosed at lower concentrations and with less frequency, thereby reducing the potential for antibody toxicity and/or development of antibody tolerance. Furthermore, their improved effector function yields new approaches to treating clinical indications that have previously been resistant or refractory to treatment with the corresponding anti-HER2 monoclonal antibody produced in recombinant host systems.
[0191] The anti-HER2 glycoantibodies of the invention can be produced by Fc glycoengineering from anti-HER2 monoclonal antibodies ("parental antibodies") commercially available or in the preclinical or clinical development. Preferrably, the monoclonal antibodies are therapeutic monoclonal antibodies. Fc glycoengineering may be performed enzymatically or chemoenzymatically. In a preferred embodiment, the parental antibody is Trastuzumab.
[0192] The N-glycans in the glycoantibodies of the invention are preferrably defucosylated.
[0193] The method for making an anti-HER2 glycoantibody is similar to the methods described herein for making an anti-CD20 glycoantibody. Briefly, the method comprises the steps of (a) contacting an anti-HER2 monoclonal antibody with an .alpha.-fucosidase and at least one endoglycosidase, thereby yielding a defucosylated antibody having a single N-acetylglucosamine (GlcNAc), and (b) adding a desired carbohydrate moiety to GlcNAc under suitable conditions.
[0194] In preferred embodiments, the carbohydrate moiety is Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.
(II) Glycoantibodies for Autoimmunity and/or Inflammation
[0195] Glycoantibodies described herein may be useful for treating an autoimmunity and/or inflammation. Exemplary monoclonal antibodies for autoimmunity and inflammation include, but are not limited to, Natalizumab (Tysabri; Biogen Idec/Elan), Vedolizumab (MLN2; Millennium Pharmaceuticals/Takeda), Belimumab (Benlysta; Human Genome Sciences/GlaxoSmithKline), Atacicept (TACI-Ig; Merck/Serono), Alefacept (Amevive; Astellas), Otelixizumab (TRX4; Tolerx/GlaxoSmithKline), Teplizumab (MGA031; MacroGenics/Eli Lilly), Rituximab (Rituxan/Mabthera; Genentech/Roche/Biogen Idec), Ofatumumab (Arzerra; Genmab/GlaxoSmithKline), Ocrelizumab (2H7; Genentech/Roche/Biogen Idec), Epratuzumab (hLL2; Immunomedics/UCB), Alemtuzumab (Campath/MabCampath; Genzyme/Bayer), Abatacept (Orencia; Bristol-Myers Squibb), Eculizumab (Soliris; Alexion pharmaceuticals), Omalizumab (Xolair; Genentech/Roche/Novartis), Canakinumab (Ilaris; Novartis), Mepolizumab (Bosatria; GlaxoSmithKline), Reslizumab (SCH55700; Ception Therapeutics), Tocilizumab (Actemra/RoActemra; Chugai/Roche), Ustekinumab (Stelara; Centocor), Briakinumab (ABT-874; Abbott), Etanercept (Enbrel; Amgen/Pfizer), Infliximab (Remicade; Centocor/Merck), Adalimumab (Humira/Trudexa; Abbott), Certolizumab pegol (Cimzia; UCB), and Golimumab (Simponi; Centocor).
Anti-TNF.alpha. Glycoantibodies (Anti-TNF.alpha. GAb)
[0196] Monocytes and macrophages secrete cytokines known as tumor necrosis factor-.alpha. (TNF.alpha.) and tumor necrosis factor-.beta. (TNF.beta.) in response to endotoxin or other stimuli. TNF.alpha. is a soluble homotrimer of 17 kD protein subunits (Smith, et al., J. Biol. Chem. 262:6951-6954 (1987)). A membrane-bound 26 kD precursor form of TNF also exists (Kriegler, et al., Cell 53:45-53 (1988)). TNF-.alpha. is a potent inducer of the inflammatory response, a key regulator of innate immunity and plays an important role in the regulation of Th1 immune responses against intracellular bacteria and certain viral infections. However, dysregulated TNF can also contribute to numerous pathological situations. These include immune-mediated inflammatory diseases (IMIDs) including rheumatoid arthritis, Crohn's disease, psoriatic arthritis, ankylosing spondylitis, ulcerative colitis and severe chronic plaque psoriasis.
[0197] The present disclosure features a novel class of anti-TNF.alpha. monoclonal antibodies, termed "anti-TNF.alpha. glycoantibodies" ("anti-TNF.alpha. GAbs"). Anti-TNF.alpha. glycoantibodies can be generated from anti-TNF.alpha. monoclonal antibodies ("parental antibodies") by Fc glycoengineering. The term "parental antibodies" as used herein refers to the anti-TNF.alpha. monoclonal antibodies used to produce anti-TNF.alpha. glycoantibodies. The individual anti-TNF.alpha. glycoantibodies comprising the homogeneous population are identical and contain the same Fc glycan with a well-defined glycan structure and sequence. Anti-TNF.alpha. glycoantibodies of the invention may bind to the same epitope of a human TNF.alpha. antigen as its patental antibodies do.
[0198] The parental antibodies may be produced in cells such as mammalian cells, Pichia pastoris or insect cells. Preferrably, the parental antibodies are produced in mammalian cells. The parental antibodies may be FDA approved or in development. Anti-TNF.alpha. monoclonal antibodies approved or in development include Infliximab, Adalimumab, Golimumab, CDP870 (certolizumab), TNF-TeAb and CDP571.
[0199] An anti-TNF.alpha. glycoantibody of the invention may comprise a heavy chain having the amino acid sequence set forth in SEQ ID NO: 21, and a light chain having the amino acid sequence set forth in SEQ ID NO: 22. An anti-TNF.alpha. glycoantibody of the invention may comprise a light chain sequence and a heavy chain sequence of Adalimumab (Humira.RTM.).
[0200] Table 10 below shows the heavy chain and the light chain sequences of Adalimumab.
TABLE-US-00010 TABLE 10 Adalimumab Accession Number: DB00051 Source: http://www.drugbank.ca/drugs/DB00051 >Light chain: DIQMTQSPSSLSASVGDRVTITCRASQGIRNYLAWYQQKPGKAPKLL IYAASTLQSGVPSRFSGSGSGTDFTLTISSLQPEDVATYYCQRYNRA PYTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYP REAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEK HKVYACEVTHQGLSSPVTKSFNRGECLSKADYEKHKVYACEVTHQGL SSPVTKSFNRGEC (SEQ ID: 22) >Heavy chain: EVQLVESGGGLVQPGRSLRLSCAASGFTFDDYAMHWVRQAPGKGLEW VSAITWNSGHIDYADSVEGRFTISRDNAKNSLYLQMNSLRAEDTAVY YCAKVSYLSTASSLDYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSG GTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSS VVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSC (SEQ ID: 21)
[0201] An anti-TNF.alpha. glycoantibody of the invention can be produced by Fc glycoengineering from an anti-TNF.alpha. monoclonal antibody ("parental antibody"). In some embodiments, the parental antibody is Adalimumab (Humira.RTM.).
[0202] The method for making an anti-TNF.alpha. glycoantibody is similar to the methods described herein for making an anti-CD20 glycoantibody. Briefly, the method comprises the steps of (a) contacting an anti-TNF.alpha. monoclonal antibody with an .alpha.-fucosidase and at least one endoglycosidase, thereby yielding a defucosylated antibody having a single N-acetylglucosamine (GlcNAc), and (b) adding a desired carbohydrate moiety to GlcNAc under suitable conditions.
[0203] In preferred embodiments, the carbohydrate moiety is Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.
(III) Glycoantibodies for Infectious Diseases
[0204] In some embodiments, glycoantibodies described herein are useful for treating an infectious disease.
[0205] Exemplary monoclonal antibodies for infectious disease include, but are not limited to, anti-Ebola antibodies such as MB-003 (c13C6, h13F6 and c6D8), ZMab (m1H3, m2G4 and m4G7) and ZMapp (c13C6, c2G4, c4G7), anti-HIV antibodies such as_VRC01, VRC02, VRC03, VRC06, b12, HJ16, 8ANC131, 8ANC134, CH103, NIH45, NIH46, NIH45G54W, NIH46G54W, 3BNC117, 3BNC60, VRC-PG04, 1NC9, 12A12, 12A21, VRC23, PG9, PGT145, PGDM1400, PG16, 2G12, PGT121, PGT128, PGT135, 4E10, 10E8, Z13 and 2F5, and anti-influenza antibodies such as C179, CR6261, F10, FI6, CR8020, CH65, C05, TCN-032, D005, CR9114 and S139/1.
Anti-Viral Glycoantibodies
[0206] In some embodiments, the present disclosure features a novel class of glycoengineered FI6 monoclonal antibodies. F16 monoclonal antibodies are neutralizing anti-influenza A virus antibodies. The neutralizing antibodies response to Influenza A virus. Amino acid sequences of a heavy chain and a lights of the antibodies are as those described in PCT publication WO 2013011347.
[0207] The method for making an FI6 glycoantibody is similar to the methods described herein for making an anti-CD20 glycoantibody. Briefly, the method comprises the steps of (a) contacting an FI6 monoclonal antibody with an .alpha.-fucosidase and at least one endoglycosidase, thereby yielding a defucosylated antibody having a single N-acetylglucosamine (GlcNAc), and (b) adding a desired carbohydrate moiety to GlcNAc under suitable conditions.
[0208] In preferred embodiments, the carbohydrate moiety is Sia.sub.2(.alpha.2-6)Gal.sub.2GlcNAc.sub.2Man.sub.3GlcNAc.
Pharmaceutical Compositions
[0209] The pharmaceutical composition according to the disclosure may be used in therapeutics. For example, the pharmaceutical composition can be used for preventing, treating, or ameliorating one or more symptoms associated with a disease, disorder, or infection where an enhanced efficacy of effector cell function (e.g., ADCC) mediated by Fc.gamma.R is desired, e.g., cancer, autoimmune, infectious disease, and in enhancing the therapeutic efficacy of therapeutic antibodies the effect of which is mediated by ADCC.
[0210] After preparation of the antibodies as described herein, a "pre-lyophilized formulation" can be produced. The antibody for preparing the formulation is preferably essentially pure and desirably essentially homogeneous (i.e. free from contaminating proteins etc). "Essentially pure" protein means a composition comprising at least about 90% by weight of the protein, based on total weight of the composition, preferably at least about 95% by weight. "Essentially homogeneous" protein means a composition comprising at least about 99% by weight of protein, based on total weight of the composition. In certain embodiments, the protein is an antibody.
[0211] The amount of antibody in the pre-lyophilized formulation is determined taking into account the desired dose volumes, mode(s) of administration etc. Where the protein of choice is an intact antibody (a full-length antibody), from about 2 mg/mL to about 50 mg/mL, preferably from about 5 mg/mL to about 40 mg/mL and most preferably from about 20-30 mg/mL is an exemplary starting protein concentration. The protein is generally present in solution. For example, the protein may be present in a pH-buffered solution at a pH from about 4-8, and preferably from about 5-7. Exemplary buffers include histidine, phosphate, Tris, citrate, succinate and other organic acids. The buffer concentration can be from about 1 mM to about 20 mM, or from about 3 mM to about 15 mM, depending, for example, on the buffer and the desired isotonicity of the formulation (e.g. of the reconstituted formulation). The preferred buffer is histidine in that, as demonstrated below, this can have lyoprotective properties. Succinate was shown to be another useful buffer.
[0212] The lyoprotectant is added to the pre-lyophilized formulation. In preferred embodiments, the lyoprotectant is a non-reducing sugar such as sucrose or trehalose. The amount of lyoprotectant in the pre-lyophilized formulation is generally such that, upon reconstitution, the resulting formulation will be isotonic. However, hypertonic reconstituted formulations may also be suitable. In addition, the amount of lyoprotectant must not be too low such that an unacceptable amount of degradation/aggregation of the protein occurs upon lyophilization. Where the lyoprotectant is a sugar (such as sucrose or trehalose) and the protein is an antibody, exemplary lyoprotectant concentrations in the pre-lyophilized formulation are from about 10 mM to about 400 mM, and preferably from about 30 mM to about 300 mM, and most preferably from about 50 mM to about 100 mM.
[0213] The ratio of protein to lyoprotectant is selected for each protein and lyoprotectant combination. In the case of an antibody as the protein of choice and a sugar (e.g., sucrose or trehalose) as the lyoprotectant for generating an isotonic reconstituted formulation with a high protein concentration, the molar ratio of lyoprotectant to antibody may be from about 100 to about 1500 moles lyoprotectant to 1 mole antibody, and preferably from about 200 to about 1000 moles of lyoprotectant to 1 mole antibody, for example from about 200 to about 600 moles of lyoprotectant to 1 mole antibody.
[0214] In preferred embodiments of the invention, it has been found to be desirable to add a surfactant to the pre-lyophilized formulation. Alternatively, or in addition, the surfactant may be added to the lyophilized formulation and/or the reconstituted formulation. Exemplary surfactants include nonionic surfactants such as polysorbates (e.g. polysorbates 20 or 80); poloxamers (e.g. poloxamer 188); Triton; sodium dodecyl sulfate (SDS); sodium laurel sulfate; sodium octyl glycoside; lauryl-, myristyl-, linoleyl-, or stearyl-sulfobetaine; lauryl-, myristyl-, linoleyl- or stearyl-sarcosine; linoleyl-, myristyl-, or cetyl-betaine; lauroamidopropyl-, cocamidopropyl-, linoleamidopropyl-, myristamidopropyl-, palnidopropyl-, or isostearamidopropyl-betaine (e.g lauroamidopropyl); myristamidopropyl-, palmidopropyl-, or isostearamidopropyl-dimethylamine; sodium methyl cocoyl-, or disodium methyl oleyl-taurate; and the MONAQUAT.TM. series (Mona Industries, Inc., Paterson, N.J.), polyethyl glycol, polypropyl glycol, and copolymers of ethylene and propylene glycol (e.g. Pluronics, PF68 etc). The amount of surfactant added is such that it reduces aggregation of the reconstituted protein and minimizes the formation of particulates after reconstitution. For example, the surfactant may be present in the pre-lyophilized formulation in an amount from about 0.001-0.5%, and preferably from about 0.005-0.05%.
[0215] In certain embodiments of the invention, a mixture of the lyoprotectant (such as sucrose or trehalose) and a bulking agent (e.g. mannitol or glycine) is used in the preparation of the pre-lyophilization formulation. The bulking agent may allow for the production of a uniform lyophilized cake without excessive pockets therein etc.
[0216] Other pharmaceutically acceptable carriers, excipients or stabilizers such as those described in Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980) may be included in the pre-lyophilized formulation (and/or the lyophilized formulation and/or the reconstituted formulation) provided that they do not adversely affect the desired characteristics of the formulation. Acceptable carriers, excipients or stabilizers are nontoxic to recipients at the dosages and concentrations employed and include; additional buffering agents; preservatives; co-solvents; antioxidants including ascorbic acid and methionine; chelating agents such as EDTA; metal complexes (e.g. Zn-protein complexes); biodegradable polymers such as polyesters; and/or salt-forming counterions such as sodium.
[0217] The pharmaceutical compositions and formulations described herein are preferably stable. A "stable" formulation/composition is one in which the antibody therein essentially retains its physical and chemical stability and integrity upon storage. Various analytical techniques for measuring protein stability are available in the art and are reviewed in Peptide and Protein Drug Delivery, 247-301, Vincent Lee Ed., Marcel Dekker, Inc., New York, N.Y., Pubs. (1991) and Jones, A. Adv. Drug Delivery Rev. 10: 29-90 (1993). Stability can be measured at a selected temperature for a selected time period.
[0218] The formulations to be used for in vivo administration must be sterile. This is readily accomplished by filtration through sterile filtration membranes, prior to, or following, lyophilization and reconstitution. Alternatively, sterility of the entire mixture may be accomplished by autoclaving the ingredients, except for protein, at about 120.degree. C. for about 30 minutes, for example.
[0219] After the protein, lyoprotectant and other optional components are mixed together, the formulation is lyophilized. Many different freeze-dryers are available for this purpose such as Hull50.RTM. (Hull, USA) or GT200 (Leybold-Heraeus, Germany) freeze-dryers. Freeze-drying is accomplished by freezing the formulation and subsequently subliming ice from the frozen content at a temperature suitable for primary drying. Under this condition, the product temperature is below the eutectic point or the collapse temperature of the formulation. Typically, the shelf temperature for the primary drying will range from about -30 to 25.degree. C. (provided the product remains frozen during primary drying) at a suitable pressure, ranging typically from about 50 to 250 mTorr. The formulation, size and type of the container holding the sample (e.g., glass vial) and the volume of liquid will mainly dictate the time required for drying, which can range from a few hours to several days (e.g. 40-60 hrs). A secondary drying stage may be carried out at about 0-40.degree. C., depending primarily on the type and size of container and the type of protein employed. However, it was found herein that a secondary drying step may not be necessary. For example, the shelf temperature throughout the entire water removal phase of lyophilization may be from about 15-30.degree. C. (e.g., about 20.degree. C.). The time and pressure required for secondary drying will be that which produces a suitable lyophilized cake, dependent, e.g., on the temperature and other parameters. The secondary drying time is dictated by the desired residual moisture level in the product and typically takes at least about 5 hours (e.g. 10-15 hours). The pressure may be the same as that employed during the primary drying step. Freeze-drying conditions can be varied depending on the formulation and vial size.
[0220] In some instances, it may be desirable to lyophilize the protein formulation in the container in which reconstitution of the protein is to be carried out in order to avoid a transfer step. The container in this instance may, for example, be a 3, 5, 10, 20, 50 or 100 cc vial. As a general proposition, lyophilization will result in a lyophilized formulation in which the moisture content thereof is less than about 5%, and preferably less than about 3%.
[0221] At the desired stage, typically when it is time to administer the protein to the patient, the lyophilized formulation may be reconstituted with a diluent such that the protein concentration in the reconstituted formulation is at least 50 mg/mL, for example from about 50 mg/mL to about 400 mg/mL, more preferably from about 80 mg/mL to about 300 mg/mL, and most preferably from about 90 mg/mL to about 150 mg/mL. Such high protein concentrations in the reconstituted formulation are considered to be particularly useful where subcutaneous delivery of the reconstituted formulation is intended. However, for other routes of administration, such as intravenous administration, lower concentrations of the protein in the reconstituted formulation may be desired (for example from about 5-50 mg/mL, or from about 10-40 mg/mL protein in the reconstituted formulation). In certain embodiments, the protein concentration in the reconstituted formulation is significantly higher than that in the pre-lyophilized formulation. For example, the protein concentration in the reconstituted formulation may be about 2-40 times, preferably 3-10 times and most preferably 3-6 times (e.g. at least three fold or at least four fold) that of the pre-lyophilized formulation.
[0222] Reconstitution generally takes place at a temperature of about 25.degree. C. to ensure complete hydration, although other temperatures may be employed as desired. The time required for reconstitution will depend, e.g., on the type of diluent, amount of excipient(s) and protein. Exemplary diluents include sterile water, bacteriostatic water for injection (BWFI), a pH buffered solution (e.g. phosphate-buffered saline), sterile saline solution, Ringer's solution or dextrose solution. The diluent optionally contains a preservative. Exemplary preservatives have been described above, with aromatic alcohols such as benzyl or phenol alcohol being the preferred preservatives. The amount of preservative employed is determined by assessing different preservative concentrations for compatibility with the protein and preservative efficacy testing. For example, if the preservative is an aromatic alcohol (such as benzyl alcohol), it can be present in an amount from about 0.1-2.0% and preferably from about 0.5-1.5%, but most preferably about 1.0-1.2%. Preferably, the reconstituted formulation has less than 6000 particles per vial which are >10 .mu.m m size.
Therapeutic Applications
[0223] Disclosed herein include methods for preventing, treating, or ameliorating one or more symptoms associated with a disease, disorder, or infection, the method comprising administering to a subject in need thereof a therapeutically effective amount of the pharmaceutical composition described herein. The diseases, disorders, or infections include, but not limited to, cancers, autoimmune disorders, inflammatory disorders and infectious infections.
(I) Treatment of Cancers
[0224] The pharmaceutical composition according to the disclosure may be used in cancers. Disclosed herein include methods for the treatment of cancer in a patient, the method comprising administering to the patient an effective amount of a pharmaceutical composition described herein.
[0225] Examples of cancers include, but not limited to, acoustic neuroma, adenocarcinoma, adrenal gland cancer, anal cancer, angiosarcoma (e.g., lymphangiosarcoma, lymphangioendotheliosarcoma, hemangiosarcoma), appendix cancer, benign monoclonal gammopathy, biliary cancer (e.g., cholangiocarcinoma), bladder cancer, breast cancer (e.g., adenocarcinoma of the breast, papillary carcinoma of the breast, mammary cancer, medullary carcinoma of the breast), brain cancer (e.g., meningioma; glioma, e.g., astrocytoma, oligodendroglioma; medulloblastoma), bronchus cancer, carcinoid tumor, cervical cancer (e.g., cervical adenocarcinoma), choriocarcinoma, chordoma, craniopharyngioma, colorectal cancer (e.g., colon cancer, rectal cancer, colorectal adenocarcinoma), epithelial carcinoma, ependymoma, endotheliosarcoma (e.g., Kaposi's sarcoma, multiple idiopathic hemorrhagic sarcoma), endometrial cancer (e.g., uterine cancer, uterine sarcoma), esophageal cancer (e.g., adenocarcinoma of the esophagus, Barrett's adenocarinoma), Ewing sarcoma, eye cancer (e.g., intraocular melanoma, retinoblastoma), familiar hypereosinophilia, gall bladder cancer, gastric cancer (e.g., stomach adenocarcinoma), gastrointestinal stromal tumor (GIST), head and neck cancer (e.g., head and neck squamous cell carcinoma, oral cancer (e.g., oral squamous cell carcinoma (OSCC), throat cancer (e.g., laryngeal cancer, pharyngeal cancer, nasopharyngeal cancer, oropharyngeal cancer)), hematopoietic cancers (e.g., leukemia such as acute lymphocytic leukemia (ALL) (e.g., B-cell ALL, T-cell ALL), acute myelocytic leukemia (AML) (e.g., B-cell AML, T-cell AML), chronic myelocytic leukemia (CML) (e.g., B-cell CML, T-cell CML), and chronic lymphocytic leukemia (CLL) (e.g., B-cell CLL, T-cell CLL); lymphoma such as Hodgkin lymphoma (HL) (e.g., B-cell HL, T-cell HL) and non-Hodgkin lymphoma (NHL) (e.g., B-cell NHL such as diffuse large cell lymphoma (DLCL) (e.g., diffuse large B-cell lymphoma (DLBCL)), follicular lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), mantle cell lymphoma (MCL), marginal zone B-cell lymphomas (e.g., mucosa-associated lymphoid tissue (MALT) lymphomas, nodal marginal zone B-cell lymphoma, splenic marginal zone B-cell lymphoma), primary mediastinal B-cell lymphoma, Burkitt lymphoma, lymphoplasmacytic lymphoma (i.e., "Waldenstrom's macroglobulinemia"), hairy cell leukemia (HCL), immunoblastic large cell lymphoma, precursor B-lymphoblastic lymphoma and primary central nervous system (CNS) lymphoma; and T-cell NHL such as precursor T-lymphoblastic lymphoma/leukemia, peripheral T-cell lymphoma (PTCL) (e.g., cutaneous T-cell lymphoma (CTCL) (e.g., mycosis fungiodes, Sezary syndrome), angioimmunoblastic T-cell lymphoma, extranodal natural killer T-cell lymphoma, enteropathy type T-cell lymphoma, subcutaneous panniculitis-like T-cell lymphoma, anaplastic large cell lymphoma); a mixture of one or more leukemia/lymphoma as described above; and multiple myeloma (MM)), heavy chain disease (e.g., alpha chain disease, gamma chain disease, mu chain disease), hemangioblastoma, inflammatory myofibroblastic tumors, immunocytic amyloidosis, kidney cancer (e.g., nephroblastoma a.k.a. Wilms' tumor, renal cell carcinoma), liver cancer (e.g., hepatocellular cancer (HCC), malignant hepatoma), lung cancer (e.g., bronchogenic carcinoma, small cell lung cancer (SCLC), non-small cell lung cancer (NSCLC), adenocarcinoma of the lung), leiomyosarcoma (LMS), mastocytosis (e.g., systemic mastocytosis), myelodysplastic syndrome (MDS), mesothelioma, myeloproliferative disorder (MPD) (e.g., polycythemia Vera (PV), essential thrombocytosis (ET), agnogenic myeloid metaplasia (AMM), a.k.a myelofibrosis (MF), chronic idiopathic myelofibrosis, chronic myelocytic leukemia (CML), chronic neutrophilic leukemia (CNL), hypereosinophilic syndrome (HES)), neuroblastoma, neurofibroma (e.g., neurofibromatosis (NF) type 1 or type 2, schwannomatosis), neuroendocrine cancer (e.g., gastroenteropancreatic neuroendoctrine tumor (GEP-NET), carcinoid tumor), osteosarcoma, ovarian cancer (e.g., cystadenocarcinoma, ovarian embryonal carcinoma, ovarian adenocarcinoma), papillary adenocarcinoma, pancreatic cancer (e.g., pancreatic andenocarcinoma, intraductal papillary mucinous neoplasm (IPMN), islet cell tumors), penile cancer (e.g., Paget's disease of the penis and scrotum), pinealoma, primitive neuroectodermal tumor (PNT), prostate cancer (e.g., prostate adenocarcinoma), rectal cancer, rhabdomyosarcoma, salivary gland cancer, skin cancer (e.g., squamous cell carcinoma (SCC), keratoacanthoma (KA), melanoma, basal cell carcinoma (BCC)), small bowel cancer (e.g., appendix cancer), soft tissue sarcoma (e.g., malignant fibrous histiocytoma (MFH), liposarcoma, malignant peripheral nerve sheath tumor (MPNST), chondrosarcoma, fibrosarcoma, myxosarcoma), sebaceous gland carcinoma, sweat gland carcinoma, synovioma, testicular cancer (e.g., seminoma, testicular embryonal carcinoma), thyroid cancer (e.g., papillary carcinoma of the thyroid, papillary thyroid carcinoma (PTC), medullary thyroid cancer), urethral cancer, vaginal cancer and vulvar cancer (e.g., Paget's disease of the vulva).
[0226] In some embodiments, provided glycoantibodies are useful in treating lung cancer. In some embodiments, a provided compound is useful in treating small lung cancer. In some embodiments, a provided compound is useful in treating non-small lung cancer. In some embodiments, a provided compound is useful in treating large bowel cancer. In some embodiments, a provided compound is useful in treating pancreas cancer. In some embodiments, a provided compound is useful in treating biliary tract cancer or endometrial cancer.
Treatment Using Anti-CD20 Glycoantibodies
[0227] In some embodiments, the present disclosure features a method for treating a cancer in a human subject in need thereof, comprising administering to the subject a therapeutically effective amount of anti-CD20 glycoantibodies and a pharmaceutically acceptable carrier.
[0228] Examples of cancers include, but not limited to, B cell lymphomas, NHL, precursor B cell lymphoblastic leukemia/lymphoma and mature B cell neoplasms, B cell chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), B cell prolymphocytic leukemia, lymphoplasmacytic lymphoma, mantle cell lymphoma (MCL), follicular lymphoma (FL), low-grade, intermediate-grade and high-grade (FL), cutaneous follicle center lymphoma, marginal zone B cell lymphoma, MALT type marginal zone B cell lymphoma, nodal marginal zone B cell lymphoma, splenic type marginal zone B cell lymphoma, hairy cell leukemia, diffuse large B cell lymphoma, Burkitt's lymphoma, plasmacytoma, plasma cell myeloma, post-transplant lymphoproliferative disorder, Waldenstrom's macroglobulinemia, and anaplastic large-cell lymphoma (ALCL).
[0229] In certain embodiments, the cancer is B-cell lymphoma such as non-Hodgkin's lymphoma.
Treatment Using Anti-HER2 Glycoantibodies
[0230] In some embodiments, the present disclosure features a method for treating a cancer in a human subject in need thereof, comprising administering to the subject a therapeutically effective amount of anti-HER2 glycoantibodies and a pharmaceutically acceptable carrier.
[0231] Examples of cancers include, but not limited to, breast cancer, brain cancer, lung cancer, oral cancer, esophagus cancer, stomach cancer, liver cancer, bile duct cancer, pancreas cancer, colon cancer, kidney cancer, cervix cancer, ovary cancer and prostate cancer. In some embodiments, the cancer is brain cancer, lung cancer, breast cancer, ovarian cancer, prostate cancer, colon cancer, or pancreas cancer.
[0232] In these treatment methods described herein, the pharmaceutical composition of glycoantibodies can be administered alone or in conjunction with a second therapeutic agents such as a second antibody, or a chemotherapeutic agent or an immunosuppressive agent.
[0233] In certain embodiments, the second therapeutic agent is an anti-cancer agent. Anti-cancer agents encompass biotherapeutic anti-cancer agents as well as chemotherapeutic agents. Exemplary biotherapeutic anti-cancer agents include, but are not limited to, interferons, cytokines (e.g., tumor necrosis factor, interferon .alpha., interferon .gamma.), vaccines, hematopoietic growth factors, monoclonal serotherapy, immunostimulants and/or immunodulatory agents (e.g., IL-1, 2, 4, 6, or 12), immune cell growth factors (e.g., GM-CSF) and antibodies (e.g. HERCEPTIN (trastuzumab), T-DM1, AVASTIN (bevacizumab), ERBITUX (cetuximab), VECTIBIX (panitumumab), RITUXAN (rituximab), BEXXAR (tositumomab)). Exemplary chemotherapeutic agents include, but are not limited to, anti-estrogens (e.g. tamoxifen, raloxifene, and megestrol), LHRH agonists (e.g. goscrclin and leuprolide), anti-androgens (e.g. flutamide and bicalutamide), photodynamic therapies (e.g. vertoporfin (BPD-MA), phthalocyanine, photosensitizer Pc4, and demethoxy-hypocrellin A (2BA-2-DMHA)), nitrogen mustards (e.g. cyclophosphamide, ifosfamide, trofosfamide, chlorambucil, estramustine, and melphalan), nitrosoureas (e.g. carmustine (BCNU) and lomustine (CCNU)), alkylsulphonates (e.g. busulfan and treosulfan), triazenes (e.g. dacarbazine, temozolomide), platinum containing compounds (e.g. cisplatin, carboplatin, oxaliplatin), vinca alkaloids (e.g. vincristine, vinblastine, vindesine, and vinorelbine), taxoids (e.g. paclitaxel or a paclitaxel equivalent such as nanoparticle albumin-bound paclitaxel (ABRAXANE), docosahexaenoic acid bound-paclitaxel (DHA-paclitaxel, Taxoprexin), polyglutamate bound-paclitaxel (PG-paclitaxel, paclitaxel poliglumex, CT-2103, XYOTAX), the tumor-activated prodrug (TAP) ANG1005 (Angiopep-2 bound to three molecules of paclitaxel), paclitaxel-EC-1 (paclitaxel bound to the erbB2-recognizing peptide EC-1), and glucose-conjugated paclitaxel, e.g., 2'-paclitaxel methyl 2-glucopyranosyl succinate; docetaxel, taxol), epipodophyllins (e.g. etoposide, etoposide phosphate, teniposide, topotecan, 9-aminocamptothecin, camptoirinotecan, irinotecan, crisnatol, mytomycin C), anti-metabolites, DHFR inhibitors (e.g. methotrexate, dichloromethotrexate, trimetrexate, edatrexate), IMP dehydrogenase inhibitors (e.g. mycophenolic acid, tiazofurin, ribavirin, and EICAR), ribonuclotide reductase inhibitors (e.g. hydroxyurea and deferoxamine), uracil analogs (e.g. 5-fluorouracil (5-FU), floxuridine, doxifluridine, ratitrexed, tegafur-uracil, capecitabine), cytosine analogs (e.g. cytarabine (ara C), cytosine arabinoside, and fludarabine), purine analogs (e.g. mercaptopurine and Thioguanine), Vitamin D3 analogs (e.g. EB 1089, CB 1093, and KH 1060), isoprenylation inhibitors (e.g. lovastatin), dopaminergic neurotoxins (e.g. 1-methyl-4-phenylpyridinium ion), cell cycle inhibitors (e.g. staurosporine), actinomycin (e.g. actinomycin D, dactinomycin), bleomycin (e.g. bleomycin A2, bleomycin B2, peplomycin), anthracycline (e.g. daunorubicin, doxorubicin, pegylated liposomal doxorubicin, idarubicin, epirubicin, pirarubicin, zorubicin, mitoxantrone), MDR inhibitors (e.g. verapamil), Ca.sup.2+ ATPase inhibitors (e.g. thapsigargin), imatinib, thalidomide, lenalidomide, tyrosine kinase inhibitors (e.g., axitinib (AG013736), bosutinib (SKI-606), cediranib (RECENTIN.TM., AZD2171), dasatinib (SPRYCEL.RTM., BMS-354825), erlotinib (TARCEVA.RTM.), gefitinib (IRESSA.RTM.), imatinib (Gleevec.RTM., CGP57148B, STI-571), lapatinib (TYKERB.RTM., TYVERB.RTM.), lestaurtinib (CEP-701), neratinib (HKI-272), nilotinib (TASIGNA.RTM.), semaxanib (semaxinib, SU5416), sunitinib (SUTENT.RTM., SU11248), toceranib (PALLADIA.RTM.), vandetanib (ZACTIMA.RTM., ZD6474), vatalanib (PTK787, PTK/ZK), trastuzumab (HERCEPTIN.RTM.), bevacizumab (AVASTIN.RTM.), rituximab (RITUXAN.RTM.), cetuximab (ERBITUX.RTM.), panitumumab (VECTIBIX.RTM.), ranibizumab (Lucentis.RTM.), nilotinib (TASIGNA.RTM.), sorafenib (NEXAVAR.RTM.), everolimus (AFINITOR.RTM.), alemtuzumab (CAMPATH.RTM.), gemtuzumab ozogamicin (MYLOTARG.RTM.), temsirolimus (TORISEL.RTM.), ENMD-2076, PCI-32765, AC220, dovitinib lactate (TKI258, CHIR-258), BIBW 2992 (TOVOK.TM.), SGX523, PF-04217903, PF-02341066, PF-299804, BMS-777607, ABT-869, MP470, BIBF 1120 (VARGATEF.RTM.), AP24534, JNJ-26483327, MGCD265, DCC-2036, BMS-690154, CEP-11981, tivozanib (AV-951), OSI-930, MM-121, XL-184, XL-647, and/or XL228), proteasome inhibitors (e.g., bortezomib (VELCADE)), mTOR inhibitors (e.g., rapamycin, temsirolimus (CCI-779), everolimus (RAD-001), ridaforolimus, AP23573 (Ariad), AZD8055 (AstraZeneca), BEZ235 (Novartis), BGT226 (Norvartis), XL765 (Sanofi Aventis), PF-4691502 (Pfizer), GDC0980 (Genetech), SF1126 (Semafoe) and OSI-027 (OSI)), oblimersen, gemcitabine, carminomycin, leucovorin, pemetrexed, cyclophosphamide, dacarbazine, procarbizine, prednisolone, dexamethasone, campathecin, plicamycin, asparaginase, aminopterin, methopterin, porfiromycin, melphalan, leurosidine, leurosine, chlorambucil, trabectedin, procarbazine, discodermolide, carminomycin, aminopterin, and hexamethyl melamine.
(II) Treatment of Autoimmune and/or Inflammatory Diseases
[0234] In some embodiments, glycoantibodies described herein are useful for treating a autoimmune and/or inflammatory diseases.
Treatment Using Anti-CD20 Glycoantibodies
[0235] In some embodiments, the present disclosure features a method for treating a autoimmune or inflammatory disease in a human subject in need thereof, comprising administering to the subject a therapeutically effective amount of anti-CD20 glycoantibodies and a pharmaceutically acceptable carrier.
[0236] Examples of autoimmune or inflammatory diseases include, but not limited to, including, but not limited to, rheumatoid arthritis, juvenile rheumatoid arthritis, systemic lupus erythematosus (SLE), Wegener's disease, inflammatory bowel disease, idiopathic thrombocytopenic purpura (ITP), thrombotic thrombocytopenic purpura (TTP), autoimmune thrombocytopenia, multiple sclerosis, psoriasis, IgA nephropathy, IgM polyneuropathies, myasthenia gravis, vasculitis, diabetes mellitus, Reynaud's syndrome, Crohn's disease, ulcerative colitis, gastritis, Hashimoto's thyroiditis, ankylosing spondylitis, hepatitis C-associated cryoglobulinemic vasculitis, chronic focal encephalitis, bullous pemphigoid, hemophilia A, membranoproliferative glomerulnephritis, adult and juvenile dermatomyositis, adult polymyositis, chronic urticaria, primary biliary cirrhosis, neuromyelitis optica, Graves' dysthyroid disease, bullous pemphigoid, membranoproliferative glonerulonephritis, Churg-Strauss syndrome, asthma, psoriatic arthritis, dermatitis, respiratory distress syndrome, meningitis, encephalitits, uveitis, eczema, atherosclerosis, leukocyte adhesion deficiency, juvenile onset diabetes, Reiter's disease, Behcet's disease, hemolytic anemia, atopic dermatitis, Wegener's granulomatosis, Omenn's syndrome, chronic renal failure, acute infectious mononucleosis, HIV and herpes-associated disease, systemic sclerosis, Sjorgen's syndrome and glomerulonephritis, dermatomyositis, ANCA, aplastic anemia, autoimmune hemolytic anemia (AIHA), factor VIII deficiency, hemophilia A, autoimmune neutropenia, Castleman's syndrome, Goodpasture's syndrome, solid organ transplant rejection, graft versus host disease (GVHD), autoimmune hepatitis, lymphoid interstitial pneumonitis (HIV), bronchiolitis obliterans (non-transplant), Guillain-Barre Syndrome, large vessel vasculitis, giant cell (Takayasu's) arteritis, medium vessel vasculitis, Kawasaki's Disease, and polyarteritis nodosa. In certain embodiments, the autoimmune or inflammatory disease is rheumatoid arthritis.
Treatment Using Anti-TNF.alpha. Glycoantibodies
[0237] In some embodiments, the present disclosure features a method for treating a autoimmune or inflammatory disease in a human subject in need thereof, comprising administering to the subject a therapeutically effective amount of anti-TNF.alpha. glycoantibodies and a pharmaceutically acceptable carrier.
(III) Treatment of Infectious Diseases
[0238] In some embodiments, glycoantibodies described herein are useful for treating a infectious diseases caused by bacterial or vial infections.
[0239] Examples of infectious diseases include, but not limited to, Human Immunodeficiency Virus (HIV), Respiratory syncytial virus (RSV), Cytomegalovirus (CMV), Ebola virus, Hepatitis A virus, Hepatitis B virus, Hepatitis C virus (HCV), Epstein-Barr virus, varicella zoster virus (VZV), Hantaan virus, influenza virus, Herpes simplex virus (HSV), Human herpes virus 6 (HHV-6), human herpes virus 8 (HHV-8), Human papilloma virus, or Parvovirus. SARS virus, measles virus; mumps virus; rubella virus; rabies virus; papillomavirus; vaccinia virus; varicella-zoster virus; variola virus; polio virus; rhino virus; respiratory syncytial virus; P. falciparum; P. vivax; P. malariae; P. ovale; Corynebacterium diphtheriae; Clostridium tetani; Clostridium botulinum; Bordetella pertussis; Haemophilus influenzae; Neisseria meningitidis, serogroup A, B, C, W135 and/or Y; Streptococcus pneumoniae; Streptococcus agalactiae; Streptococcus pyogenes; Staphylococcus aureus; Bacillus anthracis; Moraxella catarrhalis; Chlaymdia trachomatis; Chlamydia pneumoniae; Yersinia pestis; Francisella tularensis; Salmonella species; Vibrio cholerae; toxic E. coli; a human endogenous retrovirus; other microbial pathogens; other microbial toxins, allergens, tumor antigens, autoantigens and alloantigens, chemicals or toxins. In certain embodiments, the infectious disease is caused by HIV, HCV, or a combination thereof.
Treatment Using F16 Glycoantibodies
[0240] In some embodiments, the present disclosure features a method for treating a viral disease in a human subject in need thereof, comprising administering to the subject a therapeutically effective amount of FI6 glycoantibodies and a pharmaceutically acceptable carrier.
[0241] The viral disease may be caused by HIV (Human Immunodeficiency Virus), RSV (Respiratory syncytial virus), CMV (Cytomegalovirus), Ebola virus, Hepatitis A virus, Hepatitis B virus, Hepatitis C virus, Epstein-Barr virus, varicella zos-ter virus (VZV), Hantaan virus, influenza virus, Herpes simplex virus (HSV), Human herpes virus 6 (HHV-6), human herpes virus 8 (HHV-8), Human papilloma virus, or Parvovirus. In separate particular embodiments, the viral disease is caused by HIV or by Hepatitis C virus.
[0242] In some embodiments, the present disclosure features a method for treating a viral disease in a human subject in need thereof, comprising (a) administering to the subject a first compound that blocks an inhibitory receptor of an NK cell, and (b) administering to the subject a therapeutically effective amount of the pharmaceutical composition described herein.
[0243] "Treating" or "treatment" or "alleviation" refers to both therapeutic treatment and prophylactic or preventative measures; wherein the object is to prevent or slow down (lessen) the targeted pathologic condition or disorder. Those in need of treatment include those already with the disorder as well as those prone to have the disorder or those in whom the disorder is to be prevented. A subject or mammal is successfully "treated" for an infection if, after receiving a therapeutic amount of an antibody according to the methods of the present invention, the patient shows observable and/or measurable reduction in or absence of one or more of the following: reduction in the number of infected cells or absence of the infected cells; reduction in the percent of total cells that are infected; and/or relief to some extent, one or more of the symptoms associated with the specific infection; reduced morbidity and mortality, and improvement in quality of life issues. The above parameters for assessing successful treatment and improvement in the disease are readily measurable by routine procedures familiar to a physician.
[0244] The term "therapeutically effective amount" refers to an amount of an antibody or a drug effective to "treat" a disease or disorder in a subject or mammal See preceding definition of "treating."
[0245] Administration "in combination with" one or more further therapeutic agents includes simultaneous (concurrent) and consecutive administration in any order.
[0246] "Carriers" as used herein include pharmaceutically acceptable carriers, excipients, or stabilizers that are nontoxic to the cell or mammal being exposed thereto at the dosages and concentrations employed. Often the physiologically acceptable carrier is an aqueous pH buffered solution. Examples of physiologically acceptable carriers include buffers such as phosphate, citrate, and other organic acids; antioxidants including ascorbic acid; low molecular weight (less than about 10 residues) polypeptide; proteins, such as serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine, arginine or lysine; monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrins; chelating agents such as EDTA; sugar alcohols such as mannitol or sorbitol; salt-forming counterions such as sodium; and/or nonionic surfactants such as TWEEN.TM. polyethylene glycol (PEG), and PLURONICS.TM.
[0247] The following examples are included to demonstrate preferred embodiments of the invention. It should be appreciated by those of skill in the art that the techniques disclosed in the examples which follow represent techniques discovered by the inventor to function well in the practice of the invention, and thus can be considered to constitute preferred modes for its practice. However, those of skill in the art should, in light of the present disclosure, appreciate that many changes can be made in the specific embodiments which are disclosed and still obtain a like or similar result without departing from the spirit and scope of the invention.
Examples
[0248] Exemplary General Procedures
Method A: Glycosylation by Thio-Glycan Donor
[0249] To activate molecular sieves MS-4 .ANG. for glycosylation, it was connected to vacuum system and heated for 1 hour. After the activated molecular sieves was cooled to room temperature, it was added to a flask containing Donor (1.5.about.2.0 eq. for one position glycosylation) and Acceptor (1.0 eq.). Dichloromethane was added to the mixture, and then the solution was stirred at room temperature for 3 h. N-iodosuccinimide (NIS, 1.7.about.2.2 eq.) and trimethylsilyl trifluoromethanesulfonate (TMSOTf, 0.1 eq.) were added to the solution on -78.degree. C., and then the solution was stirred at -20.degree. C. Reaction was monitored by thin-layer chromatography (TLC) analysis, which was carried out on glass-backed silica gel plates (Merck DC Kieselgel 60F254) and visualized by UV light (254 nm) and acidic ceric ammonium molybdate. After the acceptor was consumed completely, the reaction was quenched with sat. NaHCO.sub.3(aq), and 20% Na.sub.2S.sub.2O.sub.3, and then the mixture was filtered through a pad of celite. After the aqueous layer was extracted with two portions of dichloromethane, the combined organic layers were washed with brine, dried over MgSO.sub.4, and concentrated. The crude was purified by silica gel column chromatography (toluene/ethyl acetate as elution system) to give product (the yield was shown on the scheme).
Method B: Glycosylation by Fluoride-Glycan Donor
[0250] A mixture of silver triflate (5 eq.), bis (cyclopentadienyl) hafnium dichloride (3.5 eq.) and 4 .ANG. activated molecular sieves in dry toluene was stirred at room temperature for 1 h. The reaction mixture was then cooled to -50.degree. C., a solution of acceptor (1.0 eq.) and donor (1.2.about.1.5 eq.) in toluene was added. The mixture was stirred at -10.degree. C. for 2-8 h. After TLC indicated complete consumption of acceptor, the reaction was quenched with Et.sub.3N, diluted with EtOAc and filtered through Celite. The filtrate was washed with aqueous NaHCO.sub.3, and a brine solution. The organic layers was dried over Na.sub.2SO.sub.4 and concentrated in vacuo. The crude was purified by silica gel column chromatography (toluene/ethyl acetate as elution system) to give product (the yield was shown on the scheme).
Method C: Deprotection of O-Acetyl
[0251] NaOMe (0.25 eq.) was added to solution of starting material (1.0 eq.) in THF/Methanol (2/3). Reaction was stirred at room temperature and monitored by TLC analysis. After the acetyl group was de-protected completely, the solution was neutralized by IR-120, filtered, and concentrated. The crude was purified by silica gel column chromatography (hexanes/ethyl acetate as elution system) to give product (the yield was shown on the scheme).
Method D: Deprotection of O-Troc
[0252] Zn powder (20 eq.) and acetic acid (0.2 eq.) were added to solution of starting material (1.0 eq.) in THF. Reaction was stirred at room temperature and monitored by thin-layer chromatography (TLC) analysis. After the Troc group was de-protected completely, the solution was filtered, and concentrated. The crude was purified by silica gel column chromatography (hexanes/ethyl acetate as elution system) to give product (the yield was shown on the scheme).
Method E: Deprotection of Benzylidene
[0253] p-Toluenesulfonic acid (pTSA, 1.5 eq.) was added to solution of starting material (1.0 eq.) in ACN/MeOH (2/1). Reaction was stirred at room temperature and monitored by thin-layer chromatography (TLC) analysis. After the benzylidene group was removed completely, the reaction was quenched by trimethylamine and then concentrated. The crude was purified by silica gel column chromatography (hexanes/ethyl acetate as elution system) to give product (the yield was shown on the scheme).
Method F: Global Deprotection
[0254] A mixture of protected oligosaccharides (50 mmol) and 10 mL of ethylene diamine: nBuOH (1/4) were stirred at 90.degree. C. overnight. Volatiles were evaporated, and crude was reacted with 10 mL Ac.sub.2O/pyridine (1/2) overnight. The solvents were removed using high vacuum, and the product was purified by flash column chromatography (acetone/toluene as elute system). The products were de-acetylated using sodium methoxide in MeOH (10 mL) overnight. Reactions were neutralized by using IR-120, then, filtered and concentrated in vacuum. The residues were purified by flash column chromatography (acetone/toluene as elute system). The products were dissolved in 10 mL MeOH: H.sub.2O: HCOOH (6/3/1), Pd(OH)2 (50% by weight) was added, and the reactions were hydrogenated overnight. The reaction mixtures were filtered through celite and concentrated in vacuo. The residues were purified by G-15 gel column chromatography using water as eluent. The products were lypholysed to get white color powders (the yield was shown on the scheme).
Method G: Enzymatic (2,6)-Sialylation
[0255] Starting materials (5 .mu.mol), CTP (1 .mu.mol), Neu5Ac (9.5 .mu.mol), PEP (10 .mu.mol), .quadrature.-2,6 sialyltransferase (200 .mu.L, estimated concentration of 2 mg/L), CMK (80 units), PK (40 units), and PPA (40 units) were dissolved in 50 .mu.mol sodium cacodylate (pH 7.4) containing 1% BSA (130 .mu.L). The reactions were incubated at 37.degree. C. with gentle agitation for 2 d. The products were purified by using G-15 gel chromatography (eluent H.sub.2O) to afford the desired products as white solid after lyophilization.
Building Blocks:
##STR00063##
[0256] Experimental Procedures for Synthesizing Asymmetric N-Glycans
##STR00064##
[0257] Experimental Procedures for Synthesizing Asymmetric N-Glycans
##STR00065##
[0258] Experimental Procedures for Synthesizing Asymmetric N-Glycans
##STR00066##
[0259] Experimental Procedures for Synthesizing Asymmetric N-Glycans
##STR00067##
[0260] Example 1: Glycoengineering of IgG1 Antibody
[0261] The goal of this study is to prepare homogenous antibodies with optimized activities in both anti-cancer and anti-inflammatory functions. Therefore, the commercially available Rituximab IgG1 is selected as a model because it has been used for the treatment of both cancer and autoimmune diseases. The strategy of glycoprotein remodeling was used to first obtain the homogeneous antibody with mono-GlcNAc at the Fc region, then a pure synthetic glycan was ligated with the mono-GlcNAc antibody to obtain the homogeneous antibody for activity assay (FIG. 1a). The fucosidase BfFucH from E. coli was used in combination with an endoglycosidase, either the EndoS from Streptococcus pyogene alone or mixtures of Endo F1/F3 or Endo F1/S, to prepare the homogeneous mono-GlcNAc glycosylated antibody in one-pot within one day. This fucosidase was more efficient than the one from bovine kidney which required 20 days of incubation (23). It was found that incubation at 37.degree. C. for one week would cause the Rituximab structure to deteriorate and leads to a loss of .about.15% binding affinity towards its antigen (supporting information). Then, by using the EndoS mutant (23), a series of synthetic glycan oxazolines were successfully transferred to the mono-GlcNAc Rituximab to form the homogeneous Rituximab with different glycans at the Fc region for subsequent binding and functional assays.
Example 2: Characteristics Between 2, 3- and 2, 6-Sialylated Rituximab
[0262] Although Ravetch's group reported that the 2, 6-sialylated IVIG was the major structure responsible for the anti-inflammation activity comparing to the 2, 3-sialylated IVIG, their detailed interactions with different Fc.gamma.Rs have not been studied (11). Moreover, Raju's studies showed that high levels of sialylation in antibody would deteriorate ADCC (12), but it was not clear whether both 2, 6- and 2, 3-sialylated antibody would have a similar effect on the cytotoxicity. To study the differences of these sialylation linkages, we prepared 2, 6- and 2, 3-sialylated antibodies (denoted as 2,6NSCT-Rituximab and 2,3NSCT-Rituximab) from mono-GlcNAc Rituximab. Compared to the non-modified Rituximab, the mono-GlcNAc Rituximab showed a complete loss or substantially reduced binding affinity towards Fc.gamma.RIIIa, Fc.gamma.RIIa, Fc.gamma.RI and C1q except toward Fc.gamma.RIIb. However, after elongation of the glycan to form the structure of 2,6-NSCT-Rituximab, its binding affinity towards Fc.gamma.RIIa, Fc.gamma.RIIb and especially Fc.gamma.RIIIa increased while no significant change was observed toward C1q (Table 11A). Differently, for the 2,3-NSCT-Rituximab, only the interaction with Fc.gamma.RIIIa was partially increased but its recognition to Fc.gamma.RIIb and Fc.gamma.RIIa was unchanged or even decreased (Table 11A). Corresponding to the comparable binding affinity of C1q to Rituximab and 2,6-NSCT-Rituximab, the FACS results showed that both antibodies displayed a parallel trend in CDC (Table 11B). However, the cytotoxicity of 2,3-NSCT-Rituximab was lower than that of 2,6-NSCT-Rituximab, as shown by the higher value of the half maximal effective concentration (EC50) (Table 11B).
[0263] In addition to CDC, ADCC is also a key issue in considering cytotoxicity relevant to antibody. Defucosylation of IgG1 was reported to effectively raise the ADCC effect via increasing the interaction between the afucosylated Fc-glycan and Fc.gamma.RIIIa (5, 33). We monitored PI-stained dead cells in the PBMC mediated ADCC assay, which was induced by the non-treated Rituximab and treated mAb, 2,3-NSCT- and 2,6-NSCT-Rituximab on flow cytometry using three different CFSE-labeled B lymphoma cells, Raji, Ramos and SKW6.4. Indeed, compared to the commercial Rituximab, both the 2,6-NSCT- and 2,3-NSCT-Rituximab showed a stronger interaction with Fc.gamma.RIIIa and smaller EC50 in ADCC (Table 11A and 11C). Interestingly, the 2,6-sialyl linkage showed an excellent affinity and effect towards Fc.gamma.RIIIa and ADCC, whereas the 2,3-linkage had weaker activities. The ADCC results of the same antibody are comparable among different target cells, including Raji, Ramos and SKW6.4 (Table 11C).
Example 3: Binding Affinity and the B Cell Depletion Activity of Various Afucosylated Rituximab
[0264] In order to study whether the cytotoxicity was affected by the 2,6-sialylation, we prepared other homogeneous afucosylated Rituximabs, including those containing the glycans of bisected modification, mono-sialylation in the 3'-arm, tri-mannose core, terminal GlcNAc endings, galactose tails and other asymmetric glycans. In the surface plasma resonance analysis, none of the modified afucosylated Rituximabs displayed a stronger binding affinity towards Fc.gamma.RIIIa than the 2,6-NSCT-Rituximab, although some Kd variations among different glycoforms were observed (Table 12). Then, we performed the cytotoxicity induction study of engineered antibodies in PBMC mediated depletion of human B cells by analyzing CD19.sup.+ CD3.sup.- B cells on flow cytometry. Corresponding to the SPR data, the B cell depletion efficacy of the 2,6-NSCT-Rituximab was superb when the antibody concentration was 10 ng/mL or larger (FIG. 2A). Moreover, the activity of the 2,6-NSCT-Rituximab was also significantly higher than the non-modified Rituximab with a p value of 0.0016 in the whole blood B cell depletion tests of 10 donors, whereas the mono-GlcNAc Rituximab shows the lowest activity (FIG. 2B). These data indicated that the 2,3- and 2,6-sialylation on immunoglobulin G1 had different activities towards its functions and the 2,6-NSCT is beneficial to Rituximab for B cell depletion. Such results are hardly validated in previous studies because many of the samples were from CHO cells, which expressed proteins with various glycans containing 2,3-sialylation but scarce 2,6-linkage (34).
Example 4: ADCC Efficacy of the 2,6-NSCT Rituximab Towards Resistant Cell Lines
[0265] Like many pharmaceuticals, Rituximab has encountered resistance due to high dosages and long-term medication (35, 36). To understand whether the 2,6-NSCT-Rituximad is effective against drug-resistant cells, we prepared the Rituximab-resistant cell lines of Ramos and Raji to evaluate their PBMC mediating ADCC under different concentrations of the 2, 6-NSCT modified Rituximab (FIG. 2C-E). After co-cultured with Rituximab for a long period of time, both Ramos and Raji B cells evolved into those with less CD20 expression on surface (FIG. 2C). As a result, it's not surprising that the non-modified Rituximab dramatically lost its activity against resistant strains (FIGS. 2D and 2E). However, the 2, 6-NSCT Rituximab showed a superb ADCC activity against both non-resistant and resistant cells.
Example 5: Fc.gamma.RIIIa Binding Affinity of Various Afucosylated Herceptins
[0266] To further evaluate whether the impressive cytotoxicity derived from the 2,6-NSCT glycan modification can be applied to other antibodies, another antibody, Herceptins, were modified with different glycan structures and evaluated.
[0267] The kinetic binding analysis of glycoengineered Herceptins and Fc.gamma.RIIIa were listed in Table 4. Similar to the affinity difference of the 2,3- and 2,6-NSCT-Rituximab in ELISA analysis, the 2,6-NSCT-Herceptin showed a stronger interaction with Fc.gamma.RIIIa, while a detrimental effect was observed with the 2,3-NSCT-Herceptin. Meanwhile, the effect of Fc afucosylation was more significant than the effect of the sialylation with both 2,6- or 2,3-linkage. Moreover, the corresponding Kds of all the glycoengineered Herceptins showed a similar tendency to the cases in Rituximab (Table 13). The antibodies, such as G1, G2, and 2,6-NSCT, had a more than nine-fold increase in affinity for Fc.gamma.RIIIa, compare to the others like G3, G4, G5, G6, G7, G9 and 2,3-NSCT. Specially, in both cases of Rituximab and Herceptin, the afucosylayed glycoengineered G8 almost lost its defucosylation advantage for the ADCC activity. The antibody with bisected glycan, G9 showed a slight but not significant increase in affinity towards Fc.gamma.RIIIa in both Rituximab and Herceptin when it is compared with the non-bisected analogue, G4. Overall, the 2,6-NSCT-Herceptin indeed also showed a superb Fc.gamma.RIIIa binding affinity among these afucosylated analogous in the SPR analysis.
[0268] To further understand the Fc glycosylation effect on the Fc.gamma.RIIIa mediated ADCC of Herceptins, we conducted an ADCC reporter bioassay, which utilized the signaling nuclear factor of the activated T-cell (NFAT) pathway of the V158 Fc.gamma.RIIIa engineered Jurkat effector cells and took SKBR3 as target cells with the E/T ratio of 6. Consistent with the kinetic data, the EC50 of the afucosylated G8 Herceptin showed a loss of Fc.gamma.RIIIa activity and displayed a similar ADCC effect to the fucosylated Herceptin (FIG. 3A). Interestingly, previous study showed that more bisected glycans on antibody caused by the increased level of .beta.(1,4)-N acetylglucosaminyltransferase III correlate with its stronger ADCC (37). On the contrary, our study showed that no significant Kd difference was observed between the bisected and non-bisected antibody of Herceptin and Rituximab, G9 and G4, and the EC50 values of Herceptin glycoforms showed a similar cytotoxicity profile in Fc.gamma.RIIIa cell-mediated assay (FIG. 3B). Thus, we conclude that the bisected IgG1 does not perform better in Fc.gamma.RIIIa dominating ADCC.
[0269] In addition, compared to the non-sialylated G1-Herceptin, the ADCC of 2,3-sialylated Herceptin was obviously reduced, whereas the 2,6-NSCT Herceptin still maintained its activity (FIG. 3C), indicating that sialylation mediated ADCC reduction is caused by the 2,3-linkage. To explore the potential utility of 2,6-NSCT in antibody medication, we selected glycoengineed afucosylated Herceptin samples with the lowest EC50 in each plate for further activity studies (FIG. 3D), and found that all samples exhibited good cytotoxicity and were capable of killing one half of cancer cells under low concentrations.
Example 6: ADCC Effect of the 2,6-NSCT Glycan Modification in Anti-Viral Antibodies
[0270] To explore the utilization of 2,6-NSCT glycan in Fc modification, we evaluated whether the homogenous 2,6-NSCT glycan modification of antibody can increase the ADCC effect of anti-viral antibodies to remove virus infected cells. We prepared an anti-influenza broadly neutralizing antibody, FI6, which was known to bind to the stem region of hemagglutinins (HA) of various subtypes of influenza and its neutralizing activity was linked to ADCC (38). The Fc glycan of FI6 antibody was modified to the homogeneous 2,6-NSCT glycan and mixed with human HEK293T cells, which express HA on cell surface to mimic influenza-infected cells; then, the ADCC effects were measured by both the PBMC-mediated killing in target cells and the activation of ADCC signaling nuclear factor of activated T-cell (NFAT) pathway of the effector cells. The cytotoxicity results showed that the homogeneous 2,6-NSCT glycan modified FI6 (FI6m) indeed exhibits a significantly higher (2- to 3-fold increase) ADCC activity than the ordinary unmodified FI6 antibody (FIG. 4A). In addition, the activation of ADCC signaling NFAT pathway of the effector NK cells was also observed with 2-fold enhancement when the homogeneous FI6m is used (FIG. 4B). Our observation indicated that the homogeneous 2,6-NSCT glycan modification of anti-viral antibodies can be a general strategy to enhance the effector function of ADCC on virus-infected cells.
[0271] Next we tested whether the in vitro ADCC enhancement by homogeneous 2,6-NSCT modification of FI6 can be translated into protection in a mouse model that is given a lethal dose infection of influenza H1N1. The passive transfer of FI6 monoclonal has been shown to protect H1N1 infection previously (39). Indeed, with the homogeneous 2,6-NSCT glycan modification, FI6m employs significantly better protection when mice were challenged with A/California/07/2009 H1N1 virus (FIG. 4C). The survival rate was 66% for FI6m versus 11% for the ordinary FI6 with mixture of complex-type glycans. In conclusion, we have demonstrated that in an influenza virus infection mouse model, the in vitro ADCC enhancement by the homogeneous 2,6-NSCT glycan modification of antibody is consistent with the in vivo protection from viral infection.
[0272] Table 11. Fc.gamma.Rs binding characteristics and the functional assays of the commercial Rituximab and the glycoengineered 2,3-NSCT- and 2,6-NSCT-Rituxmab.
[0273] (A) The binding experiments of the mono-GlcNAc, 2,3-NSCT- and 2,6-NSCT-Rituxmab towards Fc.gamma.Rs and C1q were performed in ELISA. Deglycosylation rendered mono-GlcNAc Rituximab to lose its binding affinity towards Fc.gamma.RIIIa, Fc.gamma.RIIa, Fc.gamma.RI and C1q, whereas the 2,3- and 2,6-sialylated antibodies restored their affinity, and the 2,6-sialylated Rituximab showed enhanced interactions with Fc.gamma.RIIa, Fc.gamma.RIIb and Fc.gamma.RIIIa. (B) The CDC assay performed in FACS. The 2,6-NSCT-Rituximab showed a similar CDC activity to the non-treated antibody, but the results of the 2,3-NSCT-Rituximab showed a reduced CDC efficacy with higher value of EC50. (C) Fresh PBMC mediated ADCC assay. Assay experiments were conducted with 3 different B cells, Raji, Ramos and SKW6.4. The results showed that the activity measured by the EC50 value was significantly increased from the unmodified Rituximab to the glycoengineered afucosylated 2,3-NSCT-Rituximab, with the 2,6-NSCT-Rituximab the highest.
[0274] Table 12. Binding affinity of glycoengineered Rituximab IgG1 to Fc.gamma.RIIIa measured by surface plasma resonance analysis.
[0275] Analyzed antibodies were captured by means of the Human Fab capture kit and detected with the single cycle kinetic method.
[0276] Table 13. Binding affinity of glycoengineered Herceptin IgG1 to Fc.gamma.RIIIa using surface plasma resonance analysis.
[0277] Analyzed antibodies were captured by the F(ab')2 fragment of goat anti-human F(ab').sub.2 and detected by the single cycle kinetic method with double referencing. Data shown are represents of 2 replicates.
TABLE-US-00011 TABLE 11 (A) Roche- 2,6-N S CT- 2,3-NSCT- N- EC50 (nM) Rituximab Rituximab Rituximab Rituximab Fc.gamma.RIIIa 6.2~9.0 0.24~0.27 (30x) 1.2 36.3 Fc.gamma.RIIa 5.4 3.0 (2.1x) 16.5 >110 Fc.gamma.RI 0.19 0.23 0.85 Fc.gamma.RIIb 0.51 0.28 (1.8x) 0.53 0.32 C1q 6.5 8.0 >>50 (B) EC.sub.50 (ug/ml) Ramos Rituximab (Roche) 0.033 2,3-NSCT-Rituximab 0.086 (0.45X) 2,6-NSCT-Rituximab 0.039 (C) EC.sub.50 (ug/ml) Raji Ramos SKW6.4 Rituximab (Roche) 0.319 4.379 2.074 2,3-NSCT-Rituximab 0.086(3.7X) 1.45(3.02X) 1.218(1.7X) 2,6-NSCT-Rituximab 0.022(14.5X) 0.377(11.6X) 0.276(7.5X)
TABLE-US-00012 TABLE 12 Sample ka (1/Ms) kd (1/s) KD (M) Rmax (RU) Fold Rituximab 2.07E+05 0.03344 1.62E-07 49.29 1-fold 2,6-NSCT 6.86E+05 0.005681 8.28E-09 90.48 19.6-fold G1 6.55E+05 0.006116 9.33E-09 93.4 17.4-fold G7 2.22E+05 0.01391 6.27E-08 56.28 2.6-fold G4 3.56E+05 0.01338 3.75E-08 67.01 4.3-fold G9 2.67E+05 0.006993 2.62E-08 76.02 6.2-fold G3 2.39E+05 0.01996 8.36E-08 51.03 1.9-fold G8 4.44E+05 0.05322 1.20E-07 38.43 1.4-fold G2 3.25E+05 0.004263 1.31E-08 72.12 12.4-fold G6 3.67E+05 0.01 2.72E-08 70.8 6.0-fold G5 3.33E+05 0.006284 1.89E-08 67.52 8.6-fold *The fold number was calculated with the K.sub.D value of the commercial Rituximab divided by the K.sub.D value of the glycoengineered Rituximab
TABLE-US-00013 TABLE 13 Sample ka (1/Ms) kd (1/s) KD (M) Rmax (RU) Fold Herceptin 1.45E+05 0.0131 9.09E-08 30.01 1-fold 2,6-NSCT 2.14E+05 0.00209 9.76E-09 44.98 9.3-fold G1 2.04E+05 0.00192 9.37E-09 55.68 9.7-fold G7 1.68E+05 0.0071 4.22E-08 41.54 2.2-fold G4 1.59E+05 0.00447 2.81E-08 53.98 3.2-fold G9 1.74E+05 0.00406 2.33E-08 39.88 3.9-fold G3 1.61E+05 0.00498 3.08E-08 48.19 3.0-fold G8 2.03E+05 0.0156 7.68E-08 18.15 1.2-fold G2 2.15E+05 0.00207 9.61E-09 70.48 9.5-fold G6 1.23E+05 0.00465 2.72E-08 52.82 2.4-fold G5 1.67E+05 0.00318 1.89E-08 59.89 4.8-fold 2,3-NSCT 1.83E+05 0.00473 2.58E-09 26.95 3.5-fold *The fold number was calculated with the K.sub.D value of the commercial Herceptin divided by the K.sub.D value of the glycoengineered Herceptin
Sequence CWU 1 SEQUENCE LISTING <160> NUMBER OF SEQ ID
NOS: 23 <210> SEQ ID NO 1 <211> LENGTH: 213 <212>
TYPE: PRT <213> ORGANISM: Artificial Sequence <220>
FEATURE: <223> OTHER INFORMATION: Description of Artificial
Sequence: Synthetic polypeptide <400> SEQUENCE: 1 Gln Ile Val
Leu Ser Gln Ser Pro Ala Ile Leu Ser Ala Ser Pro Gly 1 5 10 15 Glu
Lys Val Thr Met Thr Cys Arg Ala Ser Ser Ser Val Ser Tyr Ile 20 25
30 His Trp Phe Gln Gln Lys Pro Gly Ser Ser Pro Lys Pro Trp Ile Tyr
35 40 45 Ala Thr Ser Asn Leu Ala Ser Gly Val Pro Val Arg Phe Ser
Gly Ser 50 55 60 Gly Ser Gly Thr Ser Tyr Ser Leu Thr Ile Ser Arg
Val Glu Ala Glu 65 70 75 80 Asp Ala Ala Thr Tyr Tyr Cys Gln Gln Trp
Thr Ser Asn Pro Pro Thr 85 90 95 Phe Gly Gly Gly Thr Lys Leu Glu
Ile Lys Arg Thr Val Ala Ala Pro 100 105 110 Ser Val Phe Ile Phe Pro
Pro Ser Asp Glu Gln Leu Lys Ser Gly Thr 115 120 125 Ala Ser Val Val
Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala Lys 130 135 140 Val Gln
Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln Glu 145 150 155
160 Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser Ser
165 170 175 Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val
Tyr Ala 180 185 190 Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val
Thr Lys Ser Phe 195 200 205 Asn Arg Gly Glu Cys 210 <210> SEQ
ID NO 2 <211> LENGTH: 451 <212> TYPE: PRT <213>
ORGANISM: Artificial Sequence <220> FEATURE: <223>
OTHER INFORMATION: Description of Artificial Sequence: Synthetic
polypeptide <400> SEQUENCE: 2 Gln Val Gln Leu Gln Gln Pro Gly
Ala Glu Leu Val Lys Pro Gly Ala 1 5 10 15 Ser Val Lys Met Ser Cys
Lys Ala Ser Gly Tyr Thr Phe Thr Ser Tyr 20 25 30 Asn Met His Trp
Val Lys Gln Thr Pro Gly Arg Gly Leu Glu Trp Ile 35 40 45 Gly Ala
Ile Tyr Pro Gly Asn Gly Asp Thr Ser Tyr Asn Gln Lys Phe 50 55 60
Lys Gly Lys Ala Thr Leu Thr Ala Asp Lys Ser Ser Ser Thr Ala Tyr 65
70 75 80 Met Gln Leu Ser Ser Leu Thr Ser Glu Asp Ser Ala Val Tyr
Tyr Cys 85 90 95 Ala Arg Ser Thr Tyr Tyr Gly Gly Asp Trp Tyr Phe
Asn Val Trp Gly 100 105 110 Ala Gly Thr Thr Val Thr Val Ser Ala Ala
Ser Thr Lys Gly Pro Ser 115 120 125 Val Phe Pro Leu Ala Pro Ser Ser
Lys Ser Thr Ser Gly Gly Thr Ala 130 135 140 Ala Leu Gly Cys Leu Val
Lys Asp Tyr Phe Pro Glu Pro Val Thr Val 145 150 155 160 Ser Trp Asn
Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala 165 170 175 Val
Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val 180 185
190 Pro Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His
195 200 205 Lys Pro Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys
Ser Cys 210 215 220 Asp Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro
Glu Leu Leu Gly 225 230 235 240 Gly Pro Ser Val Phe Leu Phe Pro Pro
Lys Pro Lys Asp Thr Leu Met 245 250 255 Ile Ser Arg Thr Pro Glu Val
Thr Cys Val Val Val Asp Val Ser His 260 265 270 Glu Asp Pro Glu Val
Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val 275 280 285 His Asn Ala
Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr 290 295 300 Arg
Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly 305 310
315 320 Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro
Ile 325 330 335 Glu Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu
Pro Gln Val 340 345 350 Tyr Thr Leu Pro Pro Ser Arg Asp Glu Leu Thr
Lys Asn Gln Val Ser 355 360 365 Leu Thr Cys Leu Val Lys Gly Phe Tyr
Pro Ser Asp Ile Ala Val Glu 370 375 380 Trp Glu Ser Asn Gly Gln Pro
Glu Asn Asn Tyr Lys Thr Thr Pro Pro 385 390 395 400 Val Leu Asp Ser
Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val 405 410 415 Asp Lys
Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met 420 425 430
His Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser 435
440 445 Pro Gly Lys 450 <210> SEQ ID NO 3 <400>
SEQUENCE: 3 000 <210> SEQ ID NO 4 <400> SEQUENCE: 4 000
<210> SEQ ID NO 5 <211> LENGTH: 414 <212> TYPE:
PRT <213> ORGANISM: Artificial Sequence <220> FEATURE:
<223> OTHER INFORMATION: Description of Artificial Sequence:
Synthetic polypeptide <400> SEQUENCE: 5 Gln Gln Lys Tyr Gln
Pro Thr Glu Ala Asn Leu Lys Ala Arg Ser Glu 1 5 10 15 Phe Gln Asp
Asn Lys Phe Gly Ile Phe Leu His Trp Gly Leu Tyr Ala 20 25 30 Met
Leu Ala Thr Gly Glu Trp Thr Met Thr Asn Asn Asn Leu Asn Tyr 35 40
45 Lys Glu Tyr Ala Lys Leu Ala Gly Gly Phe Tyr Pro Ser Lys Phe Asp
50 55 60 Ala Asp Lys Trp Val Ala Ala Ile Lys Ala Ser Gly Ala Lys
Tyr Ile 65 70 75 80 Cys Phe Thr Thr Arg His His Glu Gly Phe Ser Met
Phe Asp Thr Lys 85 90 95 Tyr Ser Asp Tyr Asn Ile Val Lys Ala Thr
Pro Phe Lys Arg Asp Val 100 105 110 Val Lys Glu Leu Ala Asp Ala Cys
Ala Lys His Gly Ile Lys Leu His 115 120 125 Phe Tyr Tyr Ser His Ile
Asp Trp Tyr Arg Glu Asp Ala Pro Gln Gly 130 135 140 Arg Thr Gly Arg
Arg Thr Gly Arg Pro Asn Pro Lys Gly Asp Trp Lys 145 150 155 160 Ser
Tyr Tyr Gln Phe Met Asn Asn Gln Leu Thr Glu Leu Leu Thr Asn 165 170
175 Tyr Gly Pro Ile Gly Ala Ile Trp Phe Asp Gly Trp Trp Asp Gln Asp
180 185 190 Ile Asn Pro Asp Phe Asp Trp Glu Leu Pro Glu Gln Tyr Ala
Leu Ile 195 200 205 His Arg Leu Gln Pro Ala Cys Leu Val Gly Asn Asn
His His Gln Thr 210 215 220 Pro Phe Ala Gly Glu Asp Ile Gln Ile Phe
Glu Arg Asp Leu Pro Gly 225 230 235 240 Glu Asn Thr Ala Gly Leu Ser
Gly Gln Ser Val Ser His Leu Pro Leu 245 250 255 Glu Thr Cys Glu Thr
Met Asn Gly Met Trp Gly Tyr Lys Ile Thr Asp 260 265 270 Gln Asn Tyr
Lys Ser Thr Lys Thr Leu Ile His Tyr Leu Val Lys Ala 275 280 285 Ala
Gly Lys Asp Ala Asn Leu Leu Met Asn Ile Gly Pro Gln Pro Asp 290 295
300 Gly Glu Leu Pro Glu Val Ala Val Gln Arg Leu Lys Glu Val Gly Glu
305 310 315 320 Trp Met Ser Lys Tyr Gly Glu Thr Ile Tyr Gly Thr Arg
Gly Gly Leu 325 330 335 Val Ala Pro His Asp Trp Gly Val Thr Thr Gln
Lys Gly Asn Lys Leu 340 345 350 Tyr Val His Ile Leu Asn Leu Gln Asp
Lys Ala Leu Phe Leu Pro Ile 355 360 365 Val Asp Lys Lys Val Lys Lys
Ala Val Val Phe Ala Asp Lys Thr Pro 370 375 380 Val Arg Phe Thr Lys
Asn Lys Glu Gly Ile Val Leu Glu Leu Ala Lys 385 390 395 400 Val Pro
Thr Asp Val Asp Tyr Val Val Glu Leu Thr Ile Asp 405 410 <210>
SEQ ID NO 6 <400> SEQUENCE: 6 000 <210> SEQ ID NO 7
<400> SEQUENCE: 7 000 <210> SEQ ID NO 8 <400>
SEQUENCE: 8 000 <210> SEQ ID NO 9 <400> SEQUENCE: 9 000
<210> SEQ ID NO 10 <400> SEQUENCE: 10 000 <210>
SEQ ID NO 11 <211> LENGTH: 450 <212> TYPE: PRT
<213> ORGANISM: Artificial Sequence <220> FEATURE:
<223> OTHER INFORMATION: Description of Artificial Sequence:
Synthetic polypeptide <400> SEQUENCE: 11 Glu Val Gln Leu Val
Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly 1 5 10 15 Ser Leu Arg
Leu Ser Cys Ala Ala Ser Gly Phe Asn Ile Lys Asp Thr 20 25 30 Tyr
Ile His Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val 35 40
45 Ala Arg Ile Tyr Pro Thr Asn Gly Tyr Thr Arg Tyr Ala Asp Ser Val
50 55 60 Lys Gly Arg Phe Thr Ile Ser Ala Asp Thr Ser Lys Asn Thr
Ala Tyr 65 70 75 80 Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala
Val Tyr Tyr Cys 85 90 95 Ser Arg Trp Gly Gly Asp Gly Phe Tyr Ala
Met Asp Tyr Trp Gly Gln 100 105 110 Gly Thr Leu Val Thr Val Ser Ser
Ala Ser Thr Lys Gly Pro Ser Val 115 120 125 Phe Pro Leu Ala Pro Ser
Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala 130 135 140 Leu Gly Cys Leu
Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser 145 150 155 160 Trp
Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val 165 170
175 Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro
180 185 190 Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn
His Lys 195 200 205 Pro Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro
Lys Ser Cys Asp 210 215 220 Lys Thr His Thr Cys Pro Pro Cys Pro Ala
Pro Glu Leu Leu Gly Gly 225 230 235 240 Pro Ser Val Phe Leu Phe Pro
Pro Lys Pro Lys Asp Thr Leu Met Ile 245 250 255 Ser Arg Thr Pro Glu
Val Thr Cys Val Val Val Asp Val Ser His Glu 260 265 270 Asp Pro Glu
Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His 275 280 285 Asn
Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg 290 295
300 Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys
305 310 315 320 Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala
Pro Ile Glu 325 330 335 Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg
Glu Pro Gln Val Tyr 340 345 350 Thr Leu Pro Pro Ser Arg Glu Glu Met
Thr Lys Asn Gln Val Ser Leu 355 360 365 Thr Cys Leu Val Lys Gly Phe
Tyr Pro Ser Asp Ile Ala Val Glu Trp 370 375 380 Glu Ser Asn Gly Gln
Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val 385 390 395 400 Leu Asp
Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp 405 410 415
Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His 420
425 430 Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser
Pro 435 440 445 Gly Lys 450 <210> SEQ ID NO 12 <211>
LENGTH: 214 <212> TYPE: PRT <213> ORGANISM: Artificial
Sequence <220> FEATURE: <223> OTHER INFORMATION:
Description of Artificial Sequence: Synthetic polypeptide
<400> SEQUENCE: 12 Asp Ile Gln Met Thr Gln Ser Pro Ser Ser
Leu Ser Ala Ser Val Gly 1 5 10 15 Asp Arg Val Thr Ile Thr Cys Arg
Ala Ser Gln Asp Val Asn Thr Ala 20 25 30 Val Ala Trp Tyr Gln Gln
Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile 35 40 45 Tyr Ser Ala Ser
Phe Leu Tyr Ser Gly Val Pro Ser Arg Phe Ser Gly 50 55 60 Ser Arg
Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro 65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Gln His Tyr Thr Thr Pro Pro 85
90 95 Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala
Ala 100 105 110 Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu
Lys Ser Gly 115 120 125 Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe
Tyr Pro Arg Glu Ala 130 135 140 Lys Val Gln Trp Lys Val Asp Asn Ala
Leu Gln Ser Gly Asn Ser Gln 145 150 155 160 Glu Ser Val Thr Glu Gln
Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser 165 170 175 Ser Thr Leu Thr
Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr 180 185 190 Ala Cys
Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser 195 200 205
Phe Asn Arg Gly Glu Cys 210 <210> SEQ ID NO 13 <400>
SEQUENCE: 13 000 <210> SEQ ID NO 14 <400> SEQUENCE: 14
000 <210> SEQ ID NO 15 <400> SEQUENCE: 15 000
<210> SEQ ID NO 16 <400> SEQUENCE: 16 000 <210>
SEQ ID NO 17 <400> SEQUENCE: 17 000 <210> SEQ ID NO 18
<400> SEQUENCE: 18 000 <210> SEQ ID NO 19 <400>
SEQUENCE: 19 000 <210> SEQ ID NO 20 <400> SEQUENCE: 20
000 <210> SEQ ID NO 21 <211> LENGTH: 224 <212>
TYPE: PRT <213> ORGANISM: Artificial Sequence <220>
FEATURE: <223> OTHER INFORMATION: Description of Artificial
Sequence: Synthetic polypeptide <400> SEQUENCE: 21 Glu Val
Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Arg 1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Asp Asp Tyr 20
25 30 Ala Met His Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp
Val 35 40 45 Ser Ala Ile Thr Trp Asn Ser Gly His Ile Asp Tyr Ala
Asp Ser Val 50 55 60 Glu Gly Arg Phe Thr Ile Ser Arg Asp Asn Ala
Lys Asn Ser Leu Tyr 65 70 75 80 Leu Gln Met Asn Ser Leu Arg Ala Glu
Asp Thr Ala Val Tyr Tyr Cys 85 90 95 Ala Lys Val Ser Tyr Leu Ser
Thr Ala Ser Ser Leu Asp Tyr Trp Gly 100 105 110 Gln Gly Thr Leu Val
Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser 115 120 125 Val Phe Pro
Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala 130 135 140 Ala
Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val 145 150
155 160 Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro
Ala 165 170 175 Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val
Val Thr Val 180 185 190 Pro Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile
Cys Asn Val Asn His 195 200 205 Lys Pro Ser Asn Thr Lys Val Asp Lys
Lys Val Glu Pro Lys Ser Cys 210 215 220 <210> SEQ ID NO 22
<211> LENGTH: 248 <212> TYPE: PRT <213> ORGANISM:
Artificial Sequence <220> FEATURE: <223> OTHER
INFORMATION: Description of Artificial Sequence: Synthetic
polypeptide <400> SEQUENCE: 22 Asp Ile Gln Met Thr Gln Ser
Pro Ser Ser Leu Ser Ala Ser Val Gly 1 5 10 15 Asp Arg Val Thr Ile
Thr Cys Arg Ala Ser Gln Gly Ile Arg Asn Tyr 20 25 30 Leu Ala Trp
Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile 35 40 45 Tyr
Ala Ala Ser Thr Leu Gln Ser Gly Val Pro Ser Arg Phe Ser Gly 50 55
60 Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80 Glu Asp Val Ala Thr Tyr Tyr Cys Gln Arg Tyr Asn Arg Ala
Pro Tyr 85 90 95 Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg
Thr Val Ala Ala 100 105 110 Pro Ser Val Phe Ile Phe Pro Pro Ser Asp
Glu Gln Leu Lys Ser Gly 115 120 125 Thr Ala Ser Val Val Cys Leu Leu
Asn Asn Phe Tyr Pro Arg Glu Ala 130 135 140 Lys Val Gln Trp Lys Val
Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln 145 150 155 160 Glu Ser Val
Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser 165 170 175 Ser
Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr 180 185
190 Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205 Phe Asn Arg Gly Glu Cys Leu Ser Lys Ala Asp Tyr Glu Lys
His Lys 210 215 220 Val Tyr Ala Cys Glu Val Thr His Gln Gly Leu Ser
Ser Pro Val Thr 225 230 235 240 Lys Ser Phe Asn Arg Gly Glu Cys 245
<210> SEQ ID NO 23 <211> LENGTH: 6 <212> TYPE:
PRT <213> ORGANISM: Artificial Sequence <220> FEATURE:
<223> OTHER INFORMATION: Description of Artificial Sequence:
Synthetic 6xHis tag <220> FEATURE: <223> OTHER
INFORMATION: See specification as filed for detailed description of
substitutions and preferred embodiments <400> SEQUENCE: 23
His His His His His His 1 5
1 SEQUENCE LISTING <160> NUMBER OF SEQ ID NOS: 23 <210>
SEQ ID NO 1 <211> LENGTH: 213 <212> TYPE: PRT
<213> ORGANISM: Artificial Sequence <220> FEATURE:
<223> OTHER INFORMATION: Description of Artificial Sequence:
Synthetic polypeptide <400> SEQUENCE: 1 Gln Ile Val Leu Ser
Gln Ser Pro Ala Ile Leu Ser Ala Ser Pro Gly 1 5 10 15 Glu Lys Val
Thr Met Thr Cys Arg Ala Ser Ser Ser Val Ser Tyr Ile 20 25 30 His
Trp Phe Gln Gln Lys Pro Gly Ser Ser Pro Lys Pro Trp Ile Tyr 35 40
45 Ala Thr Ser Asn Leu Ala Ser Gly Val Pro Val Arg Phe Ser Gly Ser
50 55 60 Gly Ser Gly Thr Ser Tyr Ser Leu Thr Ile Ser Arg Val Glu
Ala Glu 65 70 75 80 Asp Ala Ala Thr Tyr Tyr Cys Gln Gln Trp Thr Ser
Asn Pro Pro Thr 85 90 95 Phe Gly Gly Gly Thr Lys Leu Glu Ile Lys
Arg Thr Val Ala Ala Pro 100 105 110 Ser Val Phe Ile Phe Pro Pro Ser
Asp Glu Gln Leu Lys Ser Gly Thr 115 120 125 Ala Ser Val Val Cys Leu
Leu Asn Asn Phe Tyr Pro Arg Glu Ala Lys 130 135 140 Val Gln Trp Lys
Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln Glu 145 150 155 160 Ser
Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser Ser 165 170
175 Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr Ala
180 185 190 Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys
Ser Phe 195 200 205 Asn Arg Gly Glu Cys 210 <210> SEQ ID NO 2
<211> LENGTH: 451 <212> TYPE: PRT <213> ORGANISM:
Artificial Sequence &l
References






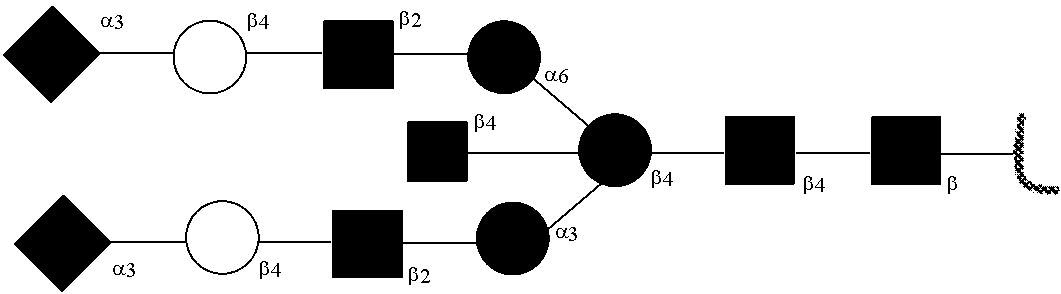
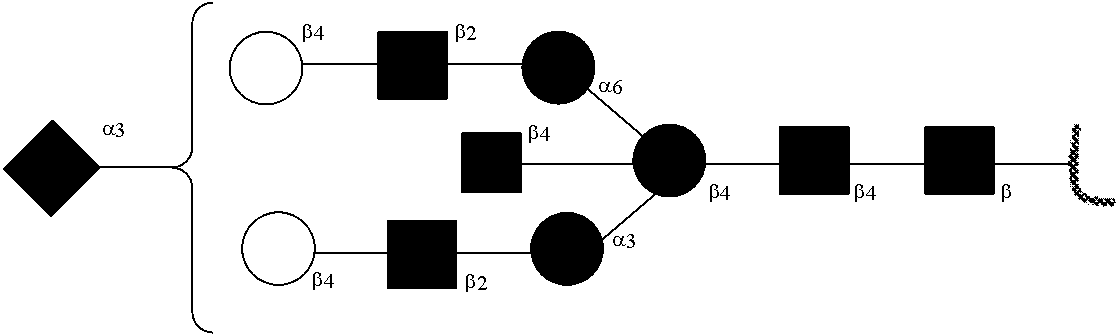











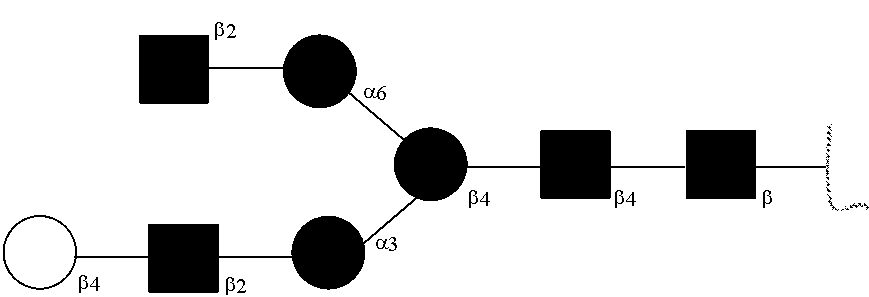



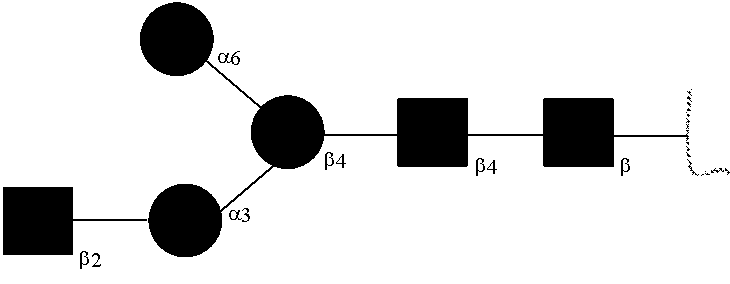



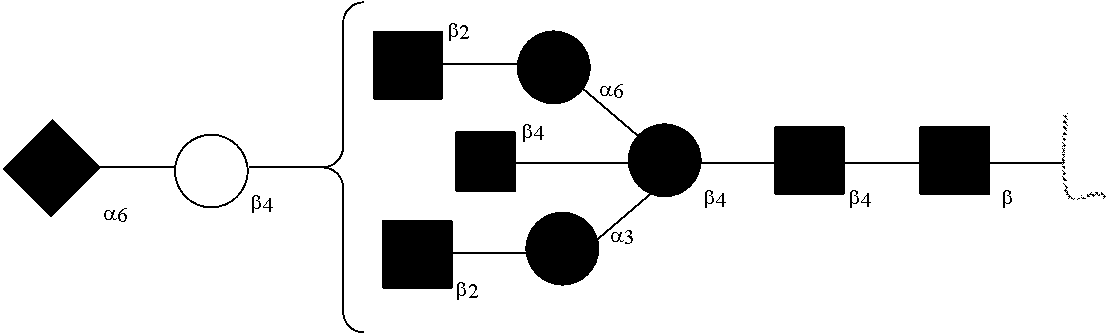



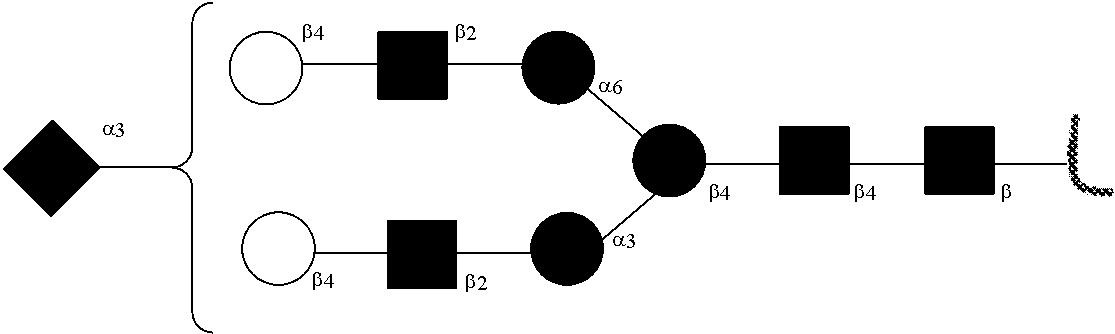

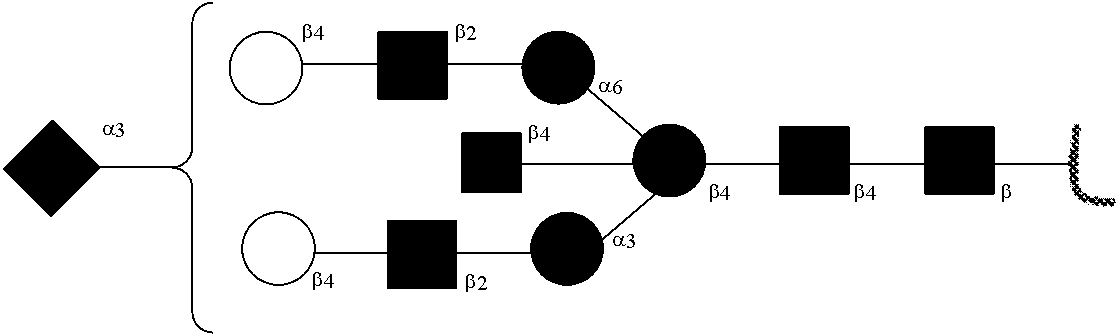



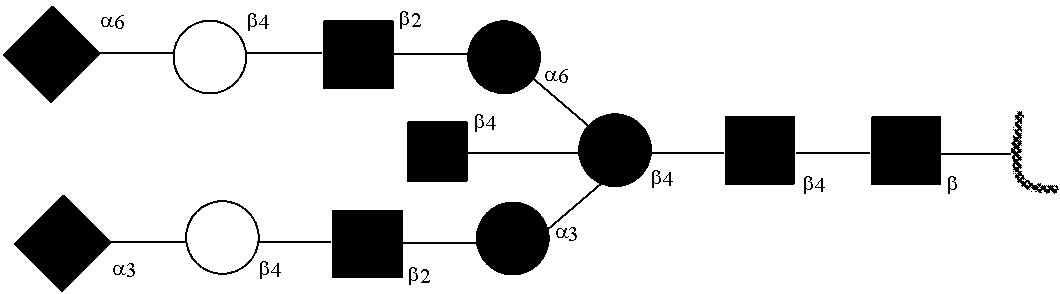










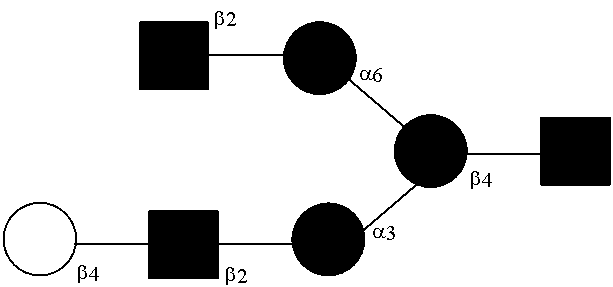
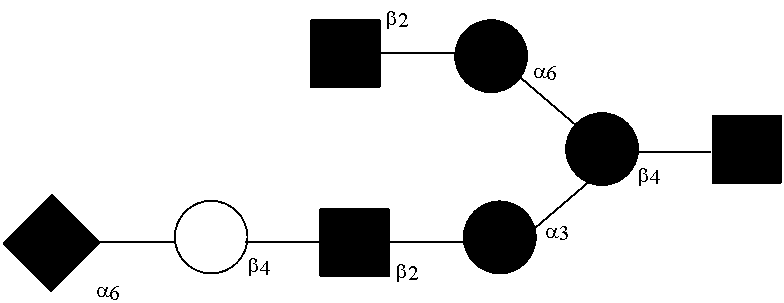



D00000
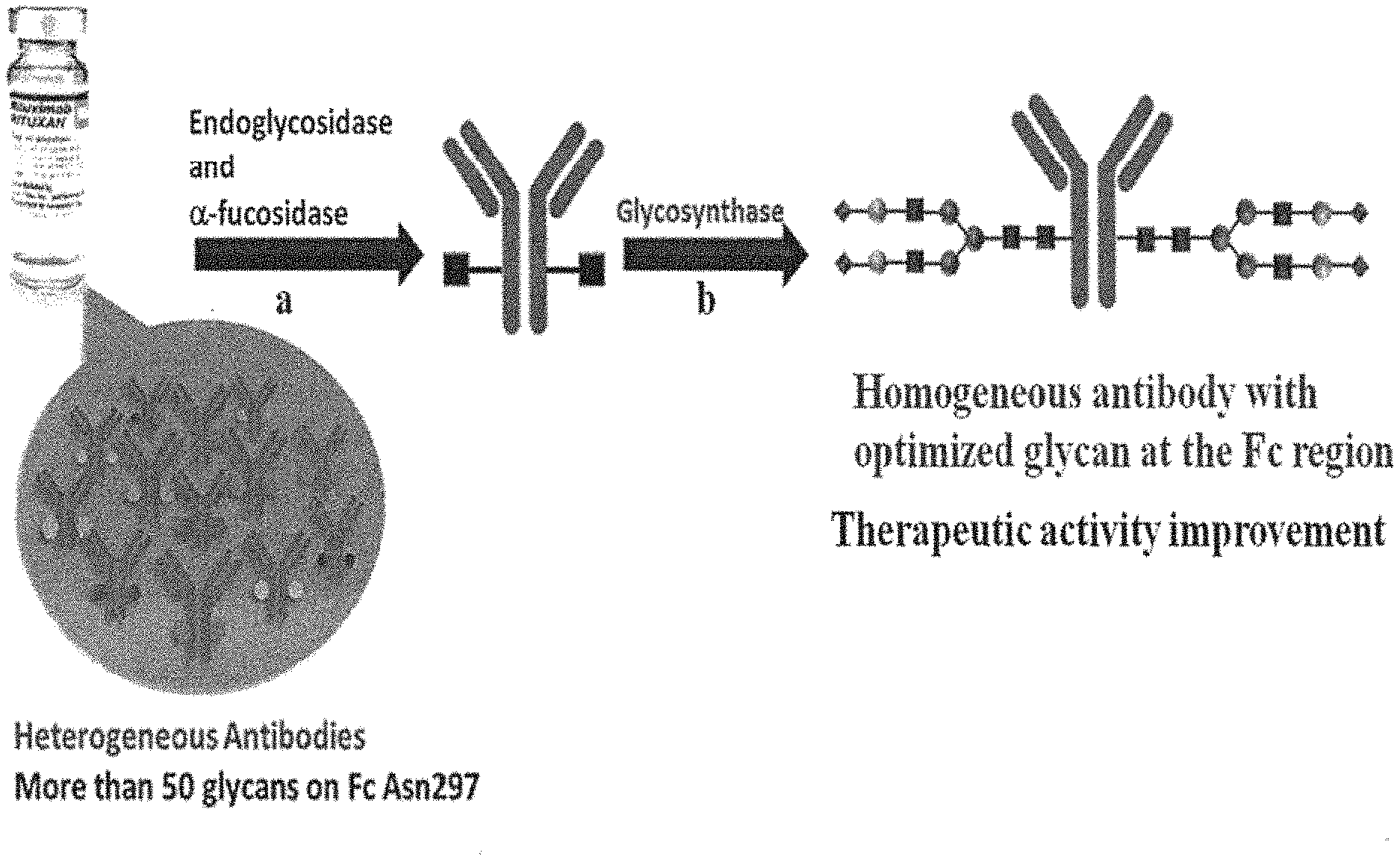
D00001

D00002

D00003

D00004

D00005

D00006

D00007

D00008

D00009

D00010

D00011
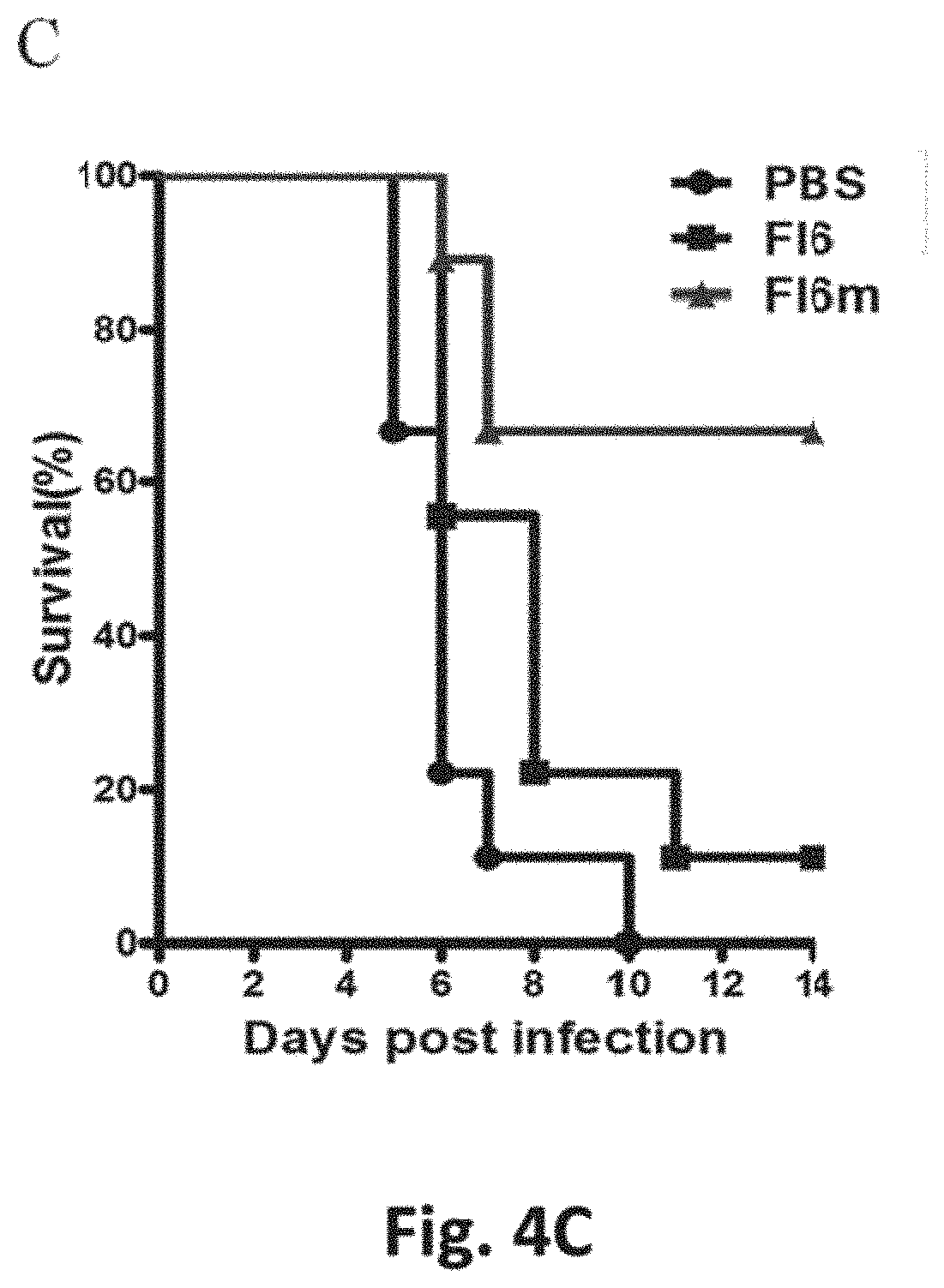
S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.