Dosage Of A Gluten Peptide Composition
Anderson; Robert P.
U.S. patent application number 16/660420 was filed with the patent office on 2020-05-14 for dosage of a gluten peptide composition. This patent application is currently assigned to Immusan T, Inc.. The applicant listed for this patent is Immusan T, Inc. Invention is credited to Robert P. Anderson.
| Application Number | 20200147168 16/660420 |
| Document ID | / |
| Family ID | 51582541 |
| Filed Date | 2020-05-14 |








View All Diagrams
| United States Patent Application | 20200147168 |
| Kind Code | A1 |
| Anderson; Robert P. | May 14, 2020 |
DOSAGE OF A GLUTEN PEPTIDE COMPOSITION
Abstract
Provided herein are methods and compositions for treating subjects with Celiac disease.
| Inventors: | Anderson; Robert P.; (Shrewsbury, MA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Assignee: | Immusan T, Inc. Cambridge MA |
||||||||||
| Family ID: | 51582541 | ||||||||||
| Appl. No.: | 16/660420 | ||||||||||
| Filed: | October 22, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 15021059 | Mar 10, 2016 | 10449228 | ||
| PCT/US2014/054959 | Sep 10, 2014 | |||
| 16660420 | ||||
| 62014666 | Jun 19, 2014 | |||
| 61876172 | Sep 10, 2013 | |||
| 61983989 | Apr 24, 2014 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | G01N 2333/57 20130101; G01N 2333/55 20130101; A61K 9/08 20130101; G01N 33/6866 20130101; A61K 38/10 20130101; A61K 9/0021 20130101; A61P 1/00 20180101 |
| International Class: | A61K 38/10 20060101 A61K038/10; G01N 33/68 20060101 G01N033/68; A61K 9/00 20060101 A61K009/00; A61K 9/08 20060101 A61K009/08 |
Claims
1. A method for treating Celiac disease in a subject, the method comprising: administering to the subject: (a) a first peptide comprising the amino acid sequence ELQPFPQPELPYPQPQ (SEQ ID NO: 1), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated; (b) a second peptide comprising the amino acid sequence EQPFPQPEQPFPWQP (SEQ ID NO: 2), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal proline is amidated; and (c) a third peptide comprising the amino acid sequence EPEQPIPEQPQPYPQQ (SEQ ID NO: 3), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated; wherein 50 micrograms of the first peptide and an equimolar amount of each of the second and third peptides are administered once or twice per week to the subject.
2. The method of claim 1, wherein 50 micrograms of the first peptide and the equimolar amount of each of the second and third peptides are administered twice per week to the subject.
3. The method of claim 1, wherein 50 micrograms of the first peptide and the equimolar amount of each of the second and third peptides are administered once per week to the subject.
4. The method of claim 1, wherein the first, second and third peptides are in equimolar amounts in a composition, and the composition is administered to the subject.
5. The method of claim 4, wherein the first, second and third peptides are each in an amount of 50 micrograms in the composition.
6. The method of claim 1, wherein the first, second and third peptides or the composition are/is administered intradermally.
7. The method of claim 6, wherein the first, second and third peptides or the composition are/is administered as a bolus by intradermal injection.
8. The method of claim 1, wherein the first, second and third peptides or the composition are/is formulated as a sterile, injectable solution.
9. The method of claim 8, wherein the sterile, injectable solution is sodium chloride.
10. The method of claim 9, wherein the sodium chloride is sterile sodium chloride 0.9% USP.
11. The method of claim 1, wherein, when the administration is twice a week, the first, second and third peptides or the composition are/is administered for four weeks.
12. The method of claim 1, wherein the first, second and third peptides or the composition are/is administered for three weeks.
13. The method of claim 1, wherein the first, second and third peptides or the composition are/is administered for eight weeks.
14. The method of claim 1, wherein the subject is HLA-DQ2.5 positive.
15. The method of claim 1, wherein the subject is on a gluten-free diet.
16. The method of claim 1, wherein the method further comprises assessing immune tolerance after administration of the first, second and third peptides.
17. The method of claim 16, wherein assessing immune tolerance comprises measuring a T cell response to gluten and/or to the first, second and third peptides in a sample comprising T cells from the subject.
18. The method of claim 17, wherein measuring the T cell response comprises contacting the sample with gluten and/or the first, second and third peptides and measuring the T cell response in the sample after the contacting.
19. The method of claim 18, wherein the T cell response is measured by measuring a level of IFN-.gamma..
20-21. (canceled)
22. A composition, comprising: a. a first peptide comprising the amino acid sequence ELQPFPQPELPYPQPQ (SEQ ID NO: 1), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated; b. a second peptide comprising the amino acid sequence EQPFPQPEQPFPWQP (SEQ ID NO: 2), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal proline is amidated; and c. a third peptide comprising the amino acid sequence EPEQPIPEQPQPYPQQ (SEQ ID NO: 3), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated; wherein 50 micrograms of the first peptide and an equimolar amount of each of the second and third peptides are present in the composition.
23-32. (canceled)
Description
BACKGROUND
[0001] Celiac disease, also known as coeliac disease or Celiac sprue (Coeliac sprue), affects approximately 1% of people in Europe and North America. In many of those affected, Celiac disease is unrecognised, but this clinical oversight is now being rectified with greater clinical awareness. A gluten free diet is the only currently approved treatment for Celiac disease, and because regular ingestion of as little as 50 mg of gluten (equivalent to 1/100.sup.th of a standard slice of bread) can damage the small intestine; chronic inflammation of the small bowel is commonplace in subjects on a gluten free diet. Persistent inflammation of the small intestine has been shown to increase the risk of cancer, osteoporosis and death. As gluten is so widely used, for example, in commercial soups, sauces, ice-creams, etc., maintaining a gluten-free diet is difficult.
[0002] Celiac disease occurs in genetically susceptible individuals who possess either HLA-DQ2.5 (encoded by the genes HLA-DQA1*05 and HLA-DQB1*02) accounting for about 90% of individuals, HLA-DQ2.2 (encoded by the genes HLA-DQA1*02 and HLA-DQB1*02), or HLA-DQ8 (encoded by the genes HLA-DQA1*03 and HLA-DQB1*0302). Without wishing to be bound by theory, it is believed that such individuals mount an inappropriate HLA-DQ2- and/or DQ8-restricted CD4.sup.+ T cell-mediated immune response to peptides derived from the aqueous-insoluble proteins of wheat flour, gluten, and related proteins in rye and barley.
SUMMARY
[0003] Described herein are specific dosages and dosage schedules of a composition for use in treating subjects with Celiac disease. In some aspects, any one of the compositions provided can include a first peptide comprising the amino acid sequence PFPQPELPY (SEQ ID NO: 4) and the amino acid sequence PQPELPYPQ (SEQ ID NO: 5), a second peptide comprising the amino acid sequence PFPQPEQPF (SEQ ID NO: 6) and the amino acid sequence PQPEQPFPW (SEQ ID NO: 7), and a third peptide comprising the amino acid sequence EQPIPEQPQ (SEQ ID NO: 8) and the amino acid sequence PIPEQPQPY (SEQ ID NO: 9), optionally wherein the N-terminus of one or more of the peptides (e.g., the N-terminus of each of the peptides) comprises a pyroglutamate and the C-terminus of one or more of the peptides (e.g., the C-terminus of each of the peptides) comprises an amino acid having an amidated carboxyl group. Without wishing to be bound by theory as above-mentioned SEQ ID NOs: 4-9 are thought to be T-cell epitopes. In some embodiments, the composition includes a first peptide comprising the amino acid sequence ELQPFPQPELPYPQPQ (SEQ ID NO: 1), wherein the N-terminal glutamate is a pyroglutamate and the carboxyl group of the C-terminal glutamine is amidated: a second peptide comprising the amino acid sequence EQPFPQPEQPFPWQP (SEQ ID NO: 2), wherein the N-terminal glutamate is a pyroglutamate and the carboxyl group of the C-terminal proline is amidated; and a third peptide comprising the amino acid sequence EPEQPIPEQPQPYPQQ (SEQ ID NO: 3), wherein the N-terminal glutamate is a pyroglutamate and the carboxyl group of the C-terminal glutamine is amidated. It is believed that administration of the compositions provided herein in the dosages and dosage schedules described herein to a subject with Celiac disease will induce immune tolerance in the subject such that the subject may consume or come into contact with at least wheat, rye, barley and optionally oats without a significant T cell response which would normally lead to symptoms of Celiac disease.
[0004] Accordingly, aspects of the disclosure relate to compositions and methods for treating a subject with Celiac disease.
[0005] In some aspects, the disclosure relates to a method for treating Celiac disease in a subject, the method comprising administering any one of the compositions provided herein to the subject. In some embodiments, the (a) first peptide comprising the amino acid sequence PFPQPELPY (SEQ ID NO: 4) and the amino acid sequence PQPELPYPQ (SEQ ID NO: 5), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal glutamine is amidated); (b) a second peptide comprising the amino acid sequence PFPQPEQPF (SEQ ID NO: 6) and the amino acid sequence PQPEQPFPW (SEQ ID NO: 7), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal proline is amidated); and (c) a third peptide comprising the amino acid sequence EQPIPEQPQ (SEQ ID NO: 8) and the amino acid sequence PIPEQPQPY (SEQ ID NO: 9), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal glutamine is amidated); and wherein 50 micrograms of the first peptide and an equimolar amount of each of the second and third peptides are administered once or twice per week to the subject. In some aspects, the disclosure relates to a method for treating Celiac disease in a subject, the method comprising administering to the subject: (a) first peptide comprising the amino acid sequence ELQPFPQPELPYPQPQ (SEQ ID NO: 1), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated; (b) a second peptide comprising the amino acid sequence EQPFPQPEQPFPWQP (SEQ ID NO: 2), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal proline is amidated; and (c) a third peptide comprising the amino acid sequence EPEQPIPEQPQPYPQQ (SEQ ID NO: 3), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated; wherein 50 micrograms of the first peptide and an equimolar amount of each of the second and third peptides are administered once or twice per week to the subject. In some embodiments, the 50 micrograms of the first peptide and the equimolar amount of each of the second and third peptides are administered twice per week to the subject. In some embodiments, the 50 micrograms of the first peptide and the equimolar amount of each of the second and third peptides are administered once per week to the subject.
[0006] In some embodiments of any one of the methods provided, the first, second and third peptides are in equimolar amounts in a composition, and the composition is administered to the subject. In some embodiments of any one of the methods provided, the first, second and third peptides are each in an amount of 50 micrograms in the composition. In some embodiments of any one of the methods provided, the first, second and third peptides or the composition are/is administered intradermally. In some embodiments of any one of the methods provided, the first, second and third peptides or the composition are/is administered as a bolus by intradermal injection. In some embodiments of any one of the methods provided, the first, second and third peptides or the composition are/is formulated as a sterile, injectable solution. In some embodiments of any one of the methods provided, the sterile, injectable solution is sodium chloride. In some embodiments of any one of the methods provided, the sodium chloride is sterile sodium chloride 0.9% USP. In some embodiments of any one of the methods provided, the first, second and third peptides or the composition are/is administered for eight weeks. In some embodiments of any one of the methods provided, the first, second and third peptides or the composition are/is administered for four weeks. In some of these embodiments of any one of the methods provided, when administration is for four weeks, the first, second and third peptides are administered biweekly for the four weeks. In some embodiments of any one of the methods provided, the first, second and third peptides or the composition are/is administered for three weeks. In some of these embodiments of any one of the methods provided, when administration is for three weeks, the first, second and third peptides are administered weekly for the three weeks. In some embodiments of any one of the methods provided, the subject is HLA-DQ2.5 positive. In some embodiments of any one of the methods provided, the subject is on a gluten-free diet.
[0007] In some aspects, the disclosure relates to a method for treating Celiac disease in a subject, the method comprising administering to the subject: (a) first peptide comprising the amino acid sequence PFPQPELPY (SEQ ID NO: 4) and the amino acid sequence PQPELPYPQ (SEQ ID NO: 5), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal glutamine is amidated); (b) a second peptide comprising the amino acid sequence PFPQPEQPF (SEQ ID NO: 6) and the amino acid sequence PQPEQPFPW (SEQ ID NO: 7), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal proline is amidated); and (c) a third peptide comprising the amino acid sequence EQPIPEQPQ (SEQ ID NO: 8) and the amino acid sequence PIPEQPQPY (SEQ ID NO: 9), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal glutamine is amidated); and wherein 100 micrograms of the first peptide and an equimolar amount of each of the second and third peptides are administered once or twice per week to the subject. In some aspects, the disclosure relates to a method for treating Celiac disease in a subject, the method comprising administering to the subject: (a) first peptide comprising the amino acid sequence ELQPFPQPELPYPQPQ (SEQ ID NO: 1), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated; (b) a second peptide comprising the amino acid sequence EQPFPQPEQPFPWQP (SEQ ID NO: 2), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal proline is amidated; and (c) a third peptide comprising the amino acid sequence EPEQPIPEQPQPYPQQ (SEQ ID NO: 3), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated; wherein 100 micrograms of the first peptide and an equimolar amount of each of the second and third peptides are administered once or twice per week to the subject. In some embodiments, the 100 micrograms of the first peptide and the equimolar amount of each of the second and third peptides are administered twice per week to the subject. In some embodiments, the 100 micrograms of the first peptide and the equimolar amount of each of the second and third peptides are administered once per week to the subject.
[0008] In some embodiments of any one of the methods provided, the first, second and third peptides are in equimolar amounts in a composition, and the composition is administered to the subject. In some embodiments of any one of the methods provided, the first, second and third peptides are each in art amount of 100 micrograms in the composition. In some embodiments of any one of the methods provided, the first, second and third peptides or the composition are/is administered intradermally. In some embodiments of any one of the methods provided, the first, second and third peptides or the composition are/is administered as a bolus by intradermal injection. In some embodiments of any one of the methods provided, the first, second and third peptides or the composition are/is formulated as a sterile, injectable solution. In some embodiments of any one of the methods provided, the sterile, injectable solution is sodium chloride. In some embodiments, the sodium chloride is sterile sodium chloride 0.9% USP. In some embodiments of any one of the methods provided, the first, second and third peptides or the composition are/is administered for eight weeks. In some embodiments of any one of the methods provided, the first, second and third peptides or the composition are/is administered for four weeks. In some of these embodiments of any one of the methods provided, when administration is for four weeks, the first, second and third peptides are administered biweekly for the four weeks. In some embodiments of any one of the methods provided, the subject is HLA-DQ2.5 positive. In some embodiments of any one of the methods provided, the subject is on a gluten-free diet.
[0009] In some embodiments of any one of the methods above, the method further comprises assessing immune tolerance after administration of the first, second and third peptides. In some embodiments of any one of the methods provided, assessing immune tolerance comprises measuring a T cell response to gluten and/or to any one of the compositions provided herein, such as one that comprises the first, second and third peptides provided herein, in a sample comprising T cells from the subject. In some embodiments of any one of the methods provided, measuring the T cell response comprises contacting the sample with gluten and/or any one of the compositions provided, such as one that comprises the first, second and third peptides provided herein, and measuring the T cell response in the sample after the contacting. In some embodiments of any one of the methods provided, the T cell response is measured by measuring a level of IFN-.gamma.. In some embodiments of any one of the methods provided, a subject is identified as tolerized if IFN-.gamma. levels <7.2 pg/mL or as otherwise provided in the Examples. In some embodiments, measuring the level of IFN-.gamma. comprises an immuno-based assay. In some embodiments, the immuno-based assay comprises an ELISA.
[0010] In other aspects, the disclosure relates to a composition, comprising: (a) first peptide comprising the amino acid sequence PFPQPELPY (SEQ ID NO: 4) and the amino acid sequence PQPELPYPQ (SEQ ID NO: 5), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal glutamine is amidated); (b) a second peptide comprising the amino acid sequence PFPQPEQPF (SEQ ID NO: 6) and the amino acid sequence PQPEQPFPW (SEQ ID NO: 7), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal proline is amidated); and (c) a third peptide comprising the amino acid sequence EQPIPEQPQ (SEQ ID NO: 8) and the amino acid sequence PIPEQPQPY (SEQ ID NO: 9), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal glutamine is amidated); wherein 50 micrograms of the first peptide and an equimolar amount of each of the second and third peptides are present in the composition. In other aspects, the disclosure relates to a composition, comprising: (a) a first peptide comprising the amino acid sequence ELQPFPQPELPYPQPQ (SEQ ID NO: 1), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated; (b) a second peptide comprising the amino acid sequence EQPFPQPEQPFPWQP (SEQ ID NO: 2), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal proline is amidated: and (c) a third peptide comprising the amino acid sequence EPEQPIPEQPQPYPQQ (SEQ ID NO: 3), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated; wherein 50 micrograms of the first peptide and an equimolar amount of each of the second and third peptides are present in the composition. In yet other aspects, the disclosure relates to a composition, comprising: (a) first peptide comprising the amino acid sequence PFPQPELPY (SEQ ID NO: 4) and the amino acid sequence PQPELPYPQ (SEQ ID NO: 5), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal glutamine is amidated); (b) a second peptide comprising the amino acid sequence PFPQPEQPF (SEQ ID NO: 6) and the amino acid sequence PQPEQPFPW (SEQ ID NO: 7), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal proline is amidated); and (c) a third peptide comprising the amino acid sequence EQPIPEQPQ (SEQ ID NO: 8) and the amino acid sequence PIPEQPQPY (SEQ ID NO: 9), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal glutamine is amidated); wherein 100 micrograms of the first peptide and an equimolar amount of each of the second and third peptides are present in the composition. In other aspects of the disclosure, the composition, comprises: (a) a first peptide comprising the amino acid sequence ELQPFPQPELPYPQPQ (SEQ ID NO: 1), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated; (b) a second peptide comprising the amino acid sequence EQPFPQPEQPFPWQP (SEQ ID NO: 2), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal proline is amidated; and (c) a third peptide comprising the amino acid sequence EPEQPIPEQPQPYPQQ (SEQ ID NO: 3), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated; wherein 100 micrograms of the first peptide and an equimolar amount of each of the second and third peptides are present in the composition.
[0011] In some embodiments of any one of the compositions provided, the first, second and third peptides are in equimolar amounts in the composition. In some embodiments of any one of the compositions provided, the first, second and third peptides are each in an amount of 50 micrograms in the composition. In some embodiments of any one of the compositions provided, the first, second and third peptides are each in an amount of 100 micrograms in the composition. In some embodiments of any one of the compositions provided, the composition is formulated for intradermal administration to a subject. In some embodiments of any one of the compositions provided, the composition is formulated as a bolus for intradermal injection to a subject. In some embodiments of any one of the compositions provided, the composition is formulated as a sterile, injectable solution. In some embodiments of any one of the compositions provided, the sterile, injectable solution is sodium chloride. In some embodiments of any one of the compositions provided, the sodium chloride is sterile sodium chloride 0.9% USP. In some embodiments of any one of the compositions provided, the composition is comprised in a kit. In some embodiments, the first, second and third peptides are contained in the same container in the kit. In some embodiments, the first, second and third peptides are contained in separate containers in the kit.
BRIEF DESCRIPTION OF THE DRAWINGS
[0012] The following drawings form part of the present specification and are included to further demonstrate certain aspects of the present disclosure, which can be better understood by reference to one or more of these drawings in combination with the detailed description of specific embodiments presented herein.
[0013] FIG. 1 is a diagram of the study format in Example 3.
[0014] FIG. 2 is a graph showing the pharmacokinetics of peptide 1 (SEQ ID NO: 1, with an N-terminal pyroglutamate and a C-terminal amide group). The x-axis is time in hours after the dose. The y-axis is log(plasma concentration).
[0015] FIG. 3 is a graph showing the pharmacokinetics of peptide 2 (SEQ ID NO: 2, with an N-terminal pyroglutamate and a C-terminal amide group). The x-axis is time in hours after the dose. The y-axis is log(plasma concentration).
[0016] FIG. 4 is a graph showing the pharmacokinetics of peptide 3 (SEQ ID NO: 3, with an N-terminal pyroglutamate and a C-terminal amide group). The x-axis is time in hours after the dose. The y-axis is log(plasma concentration).
[0017] FIG. 5 is a table showing responsiveness and tolerance by ex vivo whole blood cytokine release stimulated by immuno-dominant gluten-derived T cell epitopes before and after celiac disease patients are treated with the peptide composition.
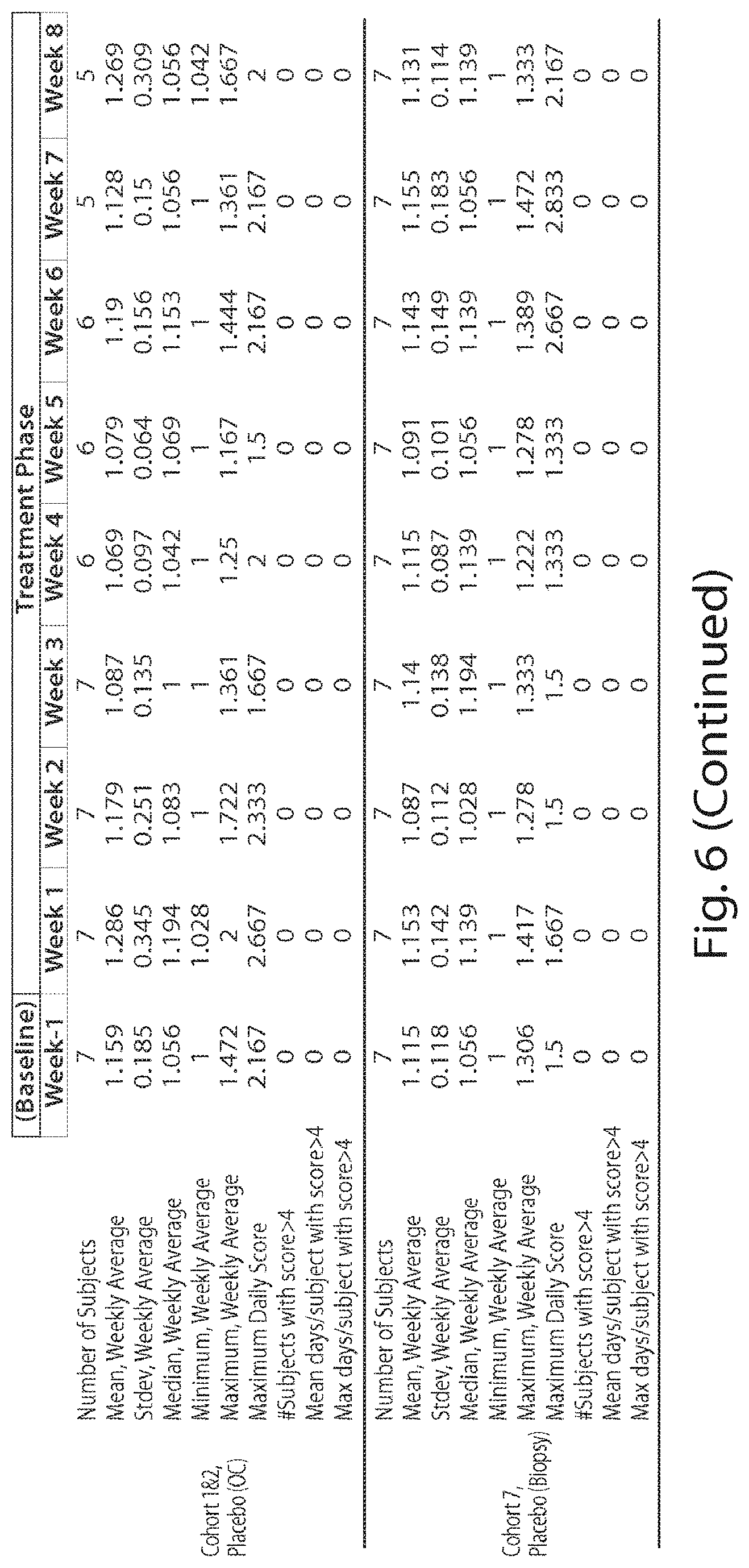
[0018] FIG. 6 is a table showing the symptom scores during dosing.
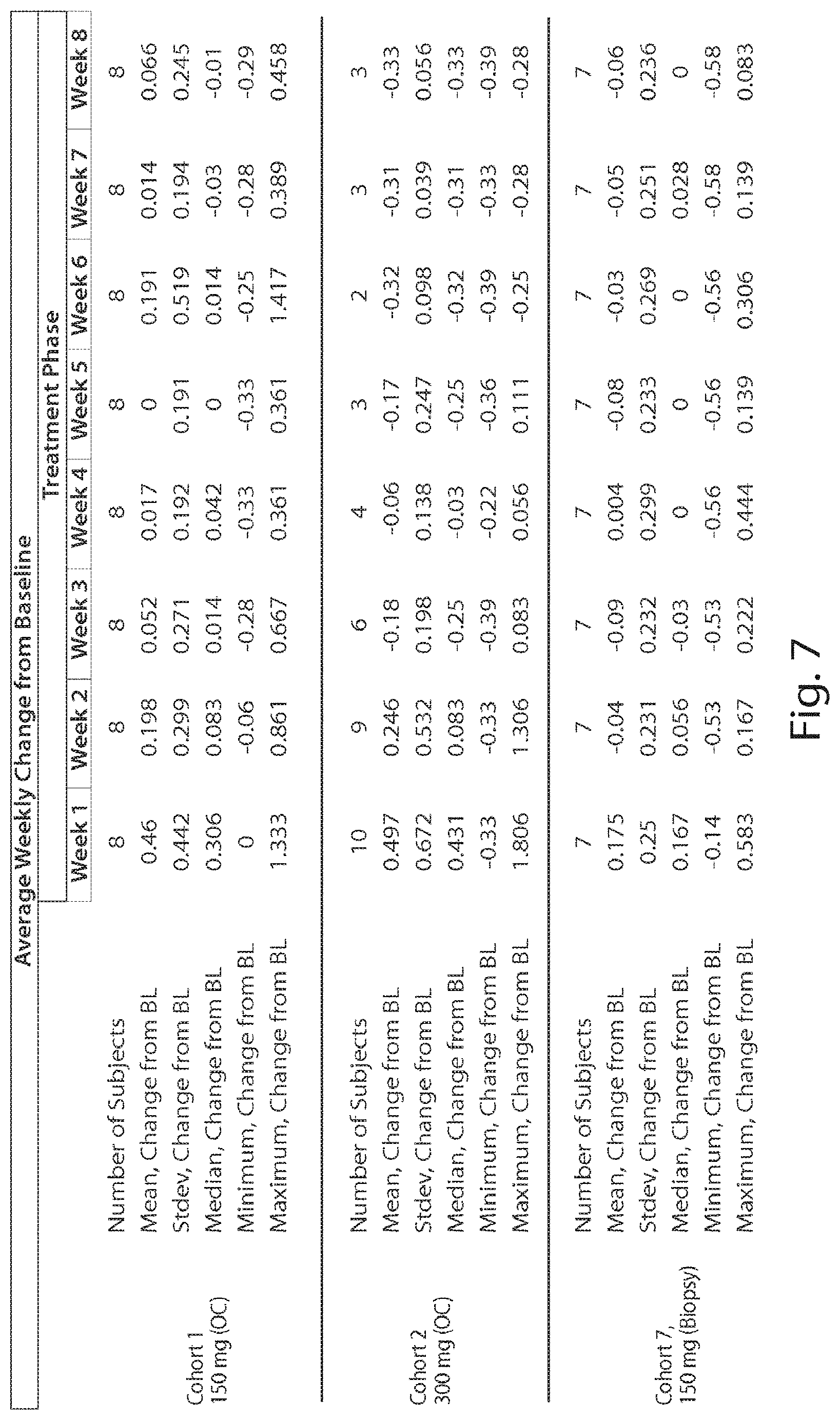
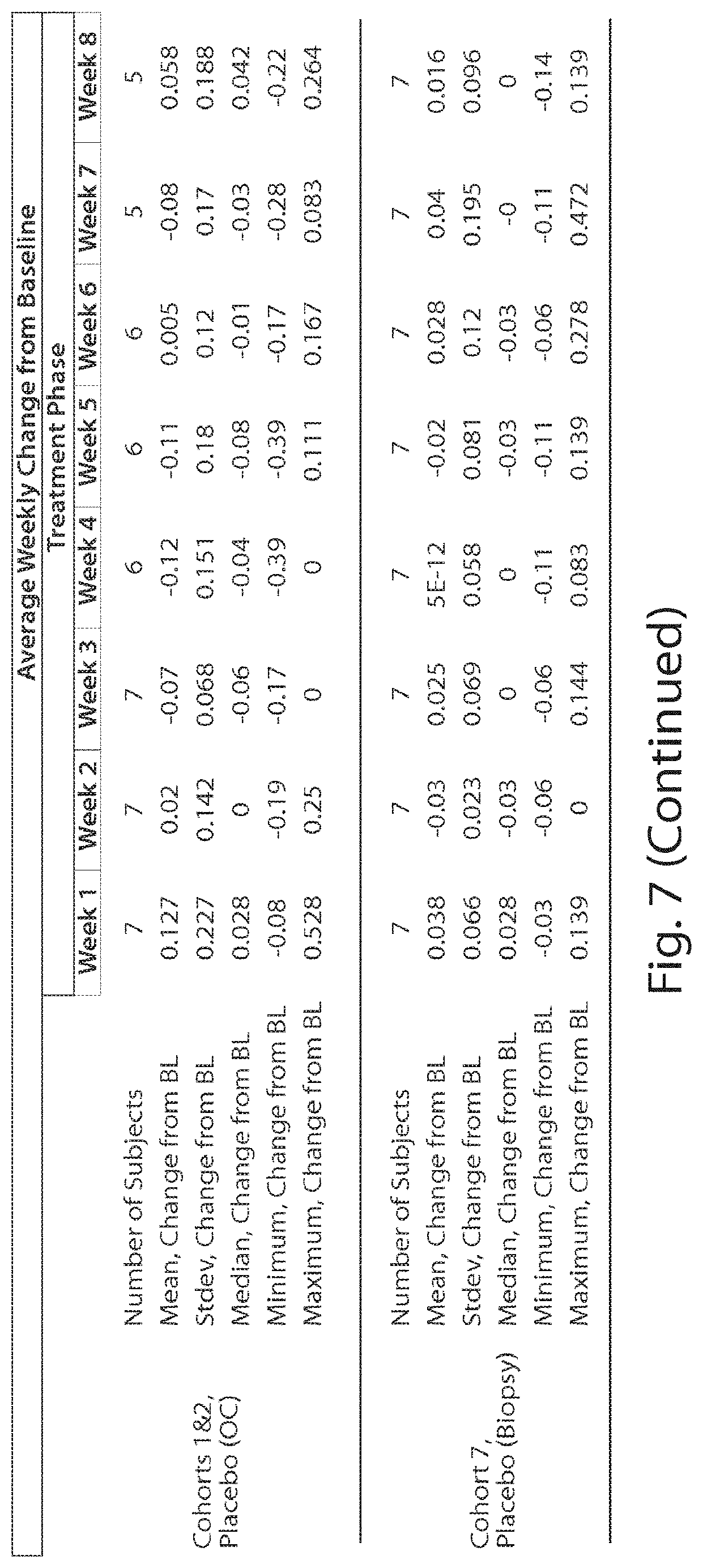
[0019] FIG. 7 is a table showing the symptom scores during dosing as changed from baseline.
[0020] FIG. 8 is a table showing the symptom scores during gluten challenge.
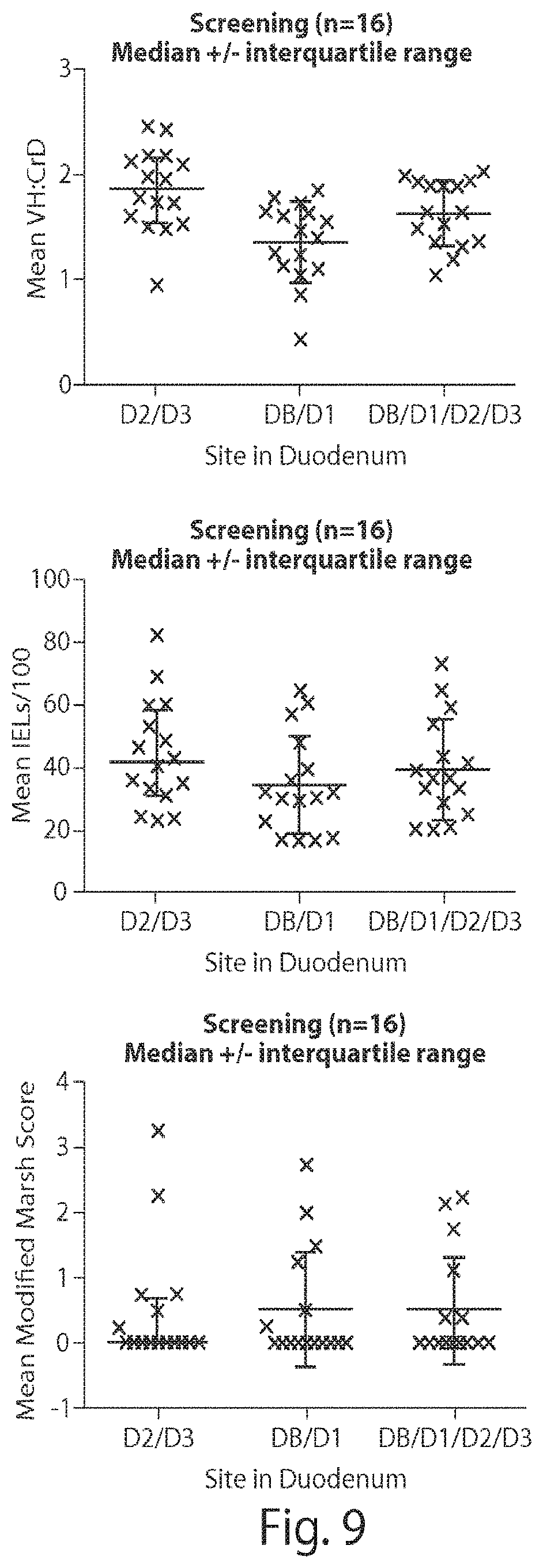
[0021] FIG. 9 is a series of graphs showing the mean villous height to crypt depth (VH:CrD) ratio at different sites in the duodenum.
[0022] FIG. 10 is a series of graphs showing VH:CrD before and after treatment with peptide composition or placebo.
[0023] FIG. 11 is a series of graphs showing intraepithelial lymphocyte (IEL) count before and after treatment with peptide composition or placebo.
[0024] FIG. 12 is a series of graphs showing the modified marsh score before and after treatment with peptide composition or placebo.
[0025] FIG. 13 is a table showing ELISA and MAGPIX data from whole blood contacted with peptide composition or controls in samples collected from cohort 1 (150 micrograms peptide composition) after gluten oral challenge.
[0026] FIG. 14 is a table showing ELISA and MAGPIX data from whole blood contacted with peptide composition or controls in samples collected from cohort 2 (300 micrograms peptide composition) after gluten oral challenge.
[0027] FIG. 15 is a table showing ELISA and MAGPIX data from whole blood contacted with peptide composition or controls in samples collected from placebo cohort (cohorts 1 and 2 placebo) after gluten oral challenge.
[0028] FIG. 16 is a diagram showing an exemplary time course for cohorts 1 and 2 (150 and 300 micrograms peptide composition, respectively).
DETAILED DESCRIPTION
General Techniques and Definitions
[0029] Unless specifically defined otherwise, all technical and scientific terms used herein shall be taken to have the same meaning as commonly understood by one of ordinary skill in the art (e.g., in cell culture, molecular genetics, immunology, immunohistochemistry, protein chemistry, and biochemistry).
[0030] Unless otherwise indicated, the recombinant protein, cell culture, and immunological techniques utilized in the present disclosure are standard procedures, well known to those skilled in the art. Such techniques are described and explained throughout the literature in sources such as, J. Perbal, A Practical Guide to Molecular Cloning, John Wiley and Sons (1984); J. Sambrook et. al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbour Laboratory Press (1989); T. A. Brown (editor), Essential Molecular Biology: A Practical Approach, Volumes 1 and 2, IRL Press (1991); D. M. Glover and B. D. Hames (editors), DNA Cloning: A Practical Approach, Volumes 1-4, IRL Press (1995 and 1996); F. M. Ausubel et al. (editors), Current Protocols in Molecular Biology, Greene Pub. Associates and Wiley-Interscience (1988, including all updates until present); Ed Harlow and David Lane (editors) Antibodies: A Laboratory Manual, Cold Spring Harbour Laboratory, (1988); and J. E. Coligan et al. (editors), Current Protocols in Immunology, John Wiley & Sons (including all updates until present).
[0031] The term "Celiac disease" refers to an immune-mediated systemic disorder elicited by gluten and related prolamines in genetically susceptible individuals, characterized by the presence of a variable combination of gluten-dependent clinical manifestations, celiac disease-specific antibodies, human leukocyte antigen (HLA)-DQ2 and HLA-DQ8 haplotypes, and enteropathy. The disease encompasses a spectrum of conditions characterised by an inappropriate CD4.sup.+ T cell response to gluten, or a peptide thereof. The severe form of celiac disease is characterised by a flat small intestinal mucosa (hyperplastic villous atrophy) and other forms are characterised by milder histological abnormalities in the small intestine, such as intra-epithelial lymphocytosis without villous atrophy. Serological abnormalities associated with celiac disease include the presence of autoantibodies specific for tissue transglutaminase-2, and antibodies specific for deamidated gluten-derived peptides. The clinical manifestations associated with celiac disease can include fatigue, chronic diarrhoea, malabsorption of nutrients, weight loss, abdominal distension, anaemia as well as a substantially enhanced risk for the development of osteoporosis and intestinal malignancies (lymphoma and carcinoma). A central feature in the current definitive diagnosis of celiac disease is that intestinal histology, celiac disease-specific serology and clinical abnormalities resolve or improve with exclusion of dietary gluten.
[0032] The terms "human leukocyte antigen" and "HLA" are here defined as a genetic fingerprint expressed on human white blood cells, composed of proteins that play a critical role in activating the body's immune system to respond to foreign organisms. In humans and other animals, the HLA is also collectively referred to as the "major histocompatibility complex" (MHC).
[0033] The term "subject" includes inter alia an individual, patient, target, host or recipient regardless of whether the subject is a human or non-human animal including mammalian species and also avian species. The term "subject", therefore, includes a human, non-human primate (for example, gorilla, marmoset, African Green Monkey), livestock animal (for example, sheep, cow, pig, horse, donkey, goat), laboratory test animal (for example, rat, mouse, rabbit, guinea pig, hamster), companion animal (for example, dog, cat), captive wild animal (for example, fox, deer, game animals) and avian species including poultry birds (for example, chickens, ducks, geese, turkeys). The preferred subject, however, is a human. In some embodiments, the subject is a human on a gluten-free diet. In some embodiments, the subject is a human who is HLA-DQ2.5 positive. In some embodiments, the subject is a human who is HLA-DQ2.5 positive and HLA-DQ8 negative. In some embodiments, the subject is human who is HLA-DQ2.5 positive and HLA-DQ8 positive.
Peptides
[0034] The terms "peptide", "polypeptide", and "protein" can generally be used interchangeably and also encompass pharmaceutical salts thereof. However, the term "peptide" is typically used to refer to relatively short molecules comprising less than 50, more preferably less than 25, amino acids.
[0035] The overall length of each peptide defined herein may be, for example, 7 to 50 amino acids, such as 7, 8, 9 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 30, 35, 40, 45, or 50 amino acids, or any integer in between. It is contemplated that shorter peptides may prove useful, particularly those that are 20 or fewer amino acids in length, in therapeutics to reduce the likelihood of anaphylaxis but longer peptides with multiple epitopes are likely to be as effective as multiple short peptides, for example, in functional T cell-based diagnostics in vitro.
[0036] It is believed that the peptides of the disclosure, such as those that comprise SEQ ID NOs: 1, 2, and 3, are capable of generating a T cell response in a subject having Celiac disease. Without wishing to be bound by theory, T cell responses in a subject with Celiac disease are thought to be caused by T-cell receptor ligation of the minimal T cell epitopes present in SEQ ID NOs: 1, 2, and 3 that are presented by HLA-DQ2.5 on the surface of antigen presenting cells.
[0037] In some embodiments, a peptide is modified during or after translation or synthesis (for example, by farnesylation, prenylation, myristoylation, glycosylation, palmitoylation, acetylation, phosphorylation [such as phosphotyrosine, phosphoserine or phosphothreonine], amidation, derivatisation by known protecting/blocking groups, proteolytic cleavage, linkage to an antibody molecule or other cellular ligand, and the like). Any of the numerous chemical modification methods known within the art may be utilised including, but not limited to, specific chemical cleavage by cyanogen bromide, trypsin, chymotrypsin, papain, V8 protease, NaBH.sub.4, acetylation, formylation, oxidation, reduction, metabolic synthesis in the presence of tunicamycin, etc.
[0038] The phrases "protecting group" and "blocking group" as used herein, refers to modifications to the peptide, which protect it from undesirable chemical reactions, particularly in vivo. Examples of such protecting groups include esters of carboxylic acids and boronic acids, ethers of alcohols and acetals, and ketals of aldehydes and ketones. Examples of suitable groups include acyl protecting groups such as, for example, furoyl, formyl, adipyl, azelayl, suberyl, dansyl, acetyl, theyl, benzoyl, trifluoroacetyl, succinyl and methoxysuccinyl; aromatic urethane protecting groups such as, for example, benzyloxycarbonyl (Cbz); aliphatic urethane protecting groups such as, for example, t-butoxycarbonyl (Boc) or 9-fluorenylmethoxy-carbonyl (FMOC); pyroglutamate and amidation. Many other modifications providing increased potency, prolonged activity, ease of purification, and/or increased half-life will be known to the person skilled in the art.
[0039] The peptides may comprise one or more modifications, which may be natural post-translation modifications or artificial modifications. The modification may provide a chemical moiety (typically by substitution of a hydrogen, for example, of a C--H bond), such as an amino, acetyl, acyl, amide, carboxy, hydroxy or halogen (for example, fluorine) group, or a carbohydrate group. Typically, the modification may be present on the N- and/or C-terminus. Furthermore, one or more of the peptides may be PEGylated, where the PEG (polyethyleneoxy group) provides for enhanced lifetime in the blood stream. One or more of the peptides may also be combined as a fusion or chimeric protein with other proteins, or with specific binding agents that allow targeting to specific moieties on a target cell. The peptide may also be chemically modified at the level of amino acid side chains, of amino acid chirality, and/or of the peptide backbone.
[0040] Particular changes may be made to the peptides to improve resistance to degradation or optimise solubility properties or otherwise improve bioavailability compared to the parent peptide, thereby providing peptides having similar or improved therapeutic, diagnostic and/or pharmacokinetic properties. A preferred such modification includes the use of an N-terminal pyroglutamate and/or a C-terminal amide (such as the respective N-terminal pyroglutamate and C-terminal glutamine of SEQ ID NOs: 1, 2, and 3). Such modifications significantly increase the half-life and bioavailability of the peptides compared to the parent peptides having a free N- and C-terminus.
[0041] In a particular embodiment, a composition comprising a first peptide comprising the amino acid sequence ELQPFPQPELPYPQPQ (SEQ ID NO: 1), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated (i.e., the free C-terminal COO is amidated); a second peptide comprising the amino acid sequence EQPFPQPEQPFPWQP (SEQ ID NO: 2), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal proline is amidated (i.e., the free C-terminal COO is amidated); and a third peptide comprising the amino acid sequence EPEQPIPEQPQPYPQQ (SEQ ID NO: 3), wherein the N-terminal glutamate is a pyroglutamate and the C-terminal glutamine is amidated (i.e., the free C-terminal COO is amidated) is contemplated. In some embodiments, the first, second and/or third peptides consist essentially of or consist of the amino acid sequence of SEQ ID NO: 1, 2, or 3, respectively. Compositions are further described herein.
[0042] In another embodiment, a composition comprising first peptide comprising the amino acid sequence PFPQPELPY (SEQ ID NO: 4) and the amino acid sequence PQPELPYPQ (SEQ ID NO: 5), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal glutamine is amidated); a second peptide comprising the amino acid sequence PFPQPEQPF (SEQ ID NO: 6) and the amino acid sequence PQPEQPFPW (SEQ ID NO: 7), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal proline is amidated); and a third peptide comprising the amino acid sequence EQPIPEQPQ (SEQ ID NO: 8) and the amino acid sequence PIPEQPQPY (SEQ ID NO: 9), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal proline is amidated) is contemplated.
[0043] Certain peptides described herein may exist in particular geometric or stereoisomeric forms. The present disclosure contemplates all such forms, including cis-(Z) and trans-(E) isomers, R- and S-enantiomers, diastereomers, (D)-isomers, (L)-isomers, the racemic mixtures thereof, and other mixtures thereof, as, falling within the scope of the disclosure. Additional asymmetric carbon atoms may be present in a substituent, such as an alkyl group. All such isomers, as well as mixtures thereof, are intended to be included in this disclosure. In another example, to prevent cleavage by peptidases, any one or more of the peptides may include a non-cleavable peptide bond in place of a particularly sensitive peptide bond to provide a more stable peptide. Such non-cleavable peptide bonds may include beta amino acids.
[0044] In certain embodiments, any one or more of the peptides may include a functional group, for example, in place of the scissile peptide bond, which facilitates inhibition of a serine-, cysteine- or aspartate-type protease, as appropriate. For example, the disclosure includes a peptidyl diketone or a peptidyl keto ester, a peptide haloalkylketone, a peptide sulfonyl fluoride, a peptidyl boronate, a peptide epoxide, a peptidyl diazomethane, a peptidyl phosphonate, isocoumarins, benzoxazin-4-ones, carbamates, isocyantes, isatoic anhydrides or the like. Such functional groups have been provided in other peptide molecules, and general routes for their synthesis are known.
[0045] The peptides may be in a salt form, preferably, a pharmaceutically acceptable salt form. "A pharmaceutically acceptable salt form" includes the conventional non-toxic salts or quaternary ammonium salts of a peptide, for example, from non-toxic organic or inorganic acids. Conventional non-toxic salts include, for example, those derived from inorganic acids such as hydrochloride, hydrobromic, sulphuric, sulfonic, phosphoric, nitric, and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, palmitic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicyclic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isothionic, and the like.
Peptide Production
[0046] The peptides can be prepared in any suitable manner. For example, the peptides can be recombinantly and/or synthetically produced.
[0047] The peptides may be synthesised by standard chemistry techniques, including synthesis by an automated procedure using a commercially available peptide synthesiser. In general, peptides may be prepared by solid-phase peptide synthesis methodologies which may involve coupling each protected amino acid residue to a resin support, preferably a 4-methylbenzhydrylamine resin, by activation with dicyclohexylcarbodiimide to yield a peptide with a C-terminal amide. Alternatively, a chloromethyl resin (Merrifield resin) may be used to yield a peptide with a free carboxylic acid at the C-terminal. After the last residue has been attached, the protected peptide-resin is treated with hydrogen fluoride to cleave the peptide from the resin, as well as deprotect the side chain functional groups. Crude product can be further purified by gel filtration, high pressure liquid chromatography (HPLC), partition chromatography, or ion-exchange chromatography.
[0048] If desired, and as outlined above, various groups may be introduced into the peptide of the composition during synthesis or during expression, which allow for linking to other molecules or to a surface. For example, cysteines can be used to make thioethers, histidines for linking to a metal ion complex, carboxyl groups for forming amides or esters, amino groups for forming amides, and the like.
[0049] The peptides may also be produced using cell-free translation systems. Standard translation systems, such as reticulocyte lysates and wheat germ extracts, use RNA as a template; whereas "coupled" and "linked" systems start with DNA templates, which are transcribed into RNA then translated.
[0050] Alternatively, the peptides may be produced by transfecting host cells with expression vectors that comprise a polynucleotide(s) that encodes one or more peptides. For recombinant production, a recombinant construct comprising a sequence which encodes one or more of the peptides is introduced into host cells by conventional methods such as calcium phosphate transfection, DEAE-dextran mediated transfection, microinjection, cationic lipid-mediated transfection, electroporation, transduction, scrape lading, ballistic introduction or infection.
[0051] One or more of the peptides may be expressed in suitable host cells, such as, for example, mammalian cells (for example, COS, CHO, BHK, 293 HEK, VERO, HeLa, HepG2, MDCK, W138, or NIH 3T3 cells), yeast (for example, Saccharomyces or Pichia), bacteria (for example, E. coli, P. pastoris, or B. subtilis), insect cells (for example, baculovirus in Sf9 cells) or other cells under the control of appropriate promoters using conventional techniques. Following transformation of the suitable host strain and growth of the host strain to an appropriate cell density, the cells are harvested by centrifugation, disrupted by physical or chemical means, and the resulting crude extract retained for further purification of the peptide or variant thereof.
[0052] Suitable expression vectors include, for example, chromosomal, non-chromosomal and synthetic polynucleotides, for example, derivatives of SV40, bacterial plasmids, phage DNAs, yeast plasmids, vectors derived from combinations of plasmids and phage DNAs, viral DNA such as vaccinia viruses, adenovirus, adeno-associated virus, lentivirus, canary pox virus, fowl pox virus, pseudorabies, baculovirus, herpes virus and retrovirus. The polynucleotide may be introduced into the expression vector by conventional procedures known in the art.
[0053] The polynucleotide which encodes one or more peptides may be operatively linked to an expression control sequence, i.e., a promoter, which directs mRNA synthesis. Representative examples of such promoters include the LTR or SV40 promoter, the E. coli lac or trp, the phage lambda PL promoter and other promoters known to control expression of genes in prokaryotic or eukaryotic cells or in viruses. The expression vector may also contain a ribosome binding site for translation initiation and a transcription terminator. The expression vectors may also include an origin of replication and a selectable marker, such as the ampicillin resistance gene of E. coli to permit selection of transformed cells, i.e., cells that are expressing the heterologous polynucleotide. The nucleic acid molecule encoding one or more of the peptides may be incorporated into the vector in frame with translation initiation and termination sequences.
[0054] One or more of the peptides can be recovered and purified from recombinant cell cultures (i.e., from the cells or culture medium) by well-known methods including ammonium sulphate or ethanol precipitation, acid extraction, anion or cation exchange chromatography, phosphocellulose chromatography, hydrophobic interaction chromatography, affinity chromatography, hydroxyapatite chromatography, lectin chromatography, and HPLC. Well known techniques for refolding proteins may be employed to regenerate active conformation when the peptide is denatured during isolation and or purification.
[0055] To produce a glycosylated peptide, it is preferred in some embodiments that recombinant techniques be used. To produce a glycosylated peptide, it is preferred in some embodiments that mammalian cells such as, COS-7 and Hep-G2 cells be employed in the recombinant techniques.
[0056] The peptides can also be prepared by cleavage of longer peptides, especially from food extracts.
[0057] Pharmaceutically acceptable salts of the peptides can be synthesised from the peptides which contain a basic or acid moiety by conventional chemical methods. Generally, the salts are prepared by reacting the free base or acid with stoichiometric amounts or with an excess of the desired salt-forming inorganic or organic acid or base in a suitable solvent.
Compositions, Vaccine Compositions, and Administration
Compositions and Vaccine Compositions
[0058] The disclosure also provides a composition comprising a first peptide comprising the amino acid sequence ELQPFPQPELPYPQPQ (SEQ ID NO: 1), wherein the N-terminal glutamate is a pyroglutamate and the carboxyl group of the C-terminal glutamine is amidated; a second peptide comprising the amino acid sequence EQPFPQPEQPFPWQP (SEQ ID NO: 2), wherein the N-terminal glutamate is a pyroglutamate and the carboxyl group of the C-terminal proline is amidated; and a third peptide comprising the amino acid sequence EPEQPIPEQPQPYPQQ (SEQ ID NO: 3), wherein the N-terminal glutamate is a pyroglutamate and the carboxyl group of the C-terminal glutamine is amidated. In some embodiments, the composition is a vaccine composition.
[0059] The disclosure additionally provides a composition comprising a first peptide comprising the amino acid sequence PFPQPELPY (SEQ ID NO: 4) and the amino acid sequence PQPELPYPQ (SEQ ID NO: 5), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal glutamine is amidated); a second peptide comprising the amino acid sequence PFPQPEQPF (SEQ ID NO: 6) and the amino acid sequence PQPEQPFPW (SEQ ID NO: 7), optionally wherein the N-terminus comprises a pyroglutamate (e.g., any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal proline is amidated); and a third peptide comprising the amino acid sequence EQPIPEQPQ (SEQ ID NO: 8) and the amino acid sequence PIPEQPQPY (SEQ ID NO: 9), optionally wherein the N-terminus comprises a pyroglutamate (e.g, any N-terminal glutamate is a pyroglutamate) and the C-terminus is amidated (e.g., any C-terminal proline is amidated). In some embodiments, the composition is a vaccine composition.
[0060] As used herein, the term "vaccine" refers to a composition comprising peptide(s) that can be administered to a subject having Celiac disease to modulate the subject's response to gluten. The vaccine may reduce the immunological reactivity of a subject towards gluten. Preferably, the vaccine induces tolerance to gluten.
[0061] Without being bound by any theory, administration of the vaccine composition to a subject may induce tolerance by clonal deletion of gluten-specific effector T cell populations, for example, gluten-specific CD4.sup.+ T cells, or by inactivation (anergy) of said T cells such that they become less responsive, preferably, unresponsive to subsequent exposure to gluten (or peptides thereof). Deletion or inactivation of said T cells can be measured, for example, by contacting ex vivo a sample comprising said T cells with gluten or a peptide thereof and measuring the response of said T cells to the gluten or peptide thereof. An exemplary T cell response measurement is measurement of the level of interferon-gamma (IFN-.gamma., see, e.g., NCBI Gene ID 3458 and Protein ID NP_000610.2) in the sample after contact with the gluten or peptide thereof. A decreased level of IFN-.gamma.may indicate deletion or inactivation of said T cells. The level of IFN-.gamma. can be measured using any method known to those of skill in the art, e.g., using immuno-based detection methods such as Western blot or enzyme-linked immunosorbent assay (ELISA).
[0062] Alternatively, or in addition, administration of the vaccine composition may modify the cytokine secretion profile of the subject (for example, result in decreased IL-4, IL-2, TNF-.alpha. and/or IFN-.gamma., and/or increased IL-10). The vaccine composition may induce suppressor T cell subpopulations, for example Treg cells, to produce IL-10 and/or TGF-.beta. and thereby suppress gluten-specific effector T cells. The cytokine secretion profile of the subject can be measured using any method known to those of skill in the art, e.g., using immuno-based detection methods such as Western blot or enzyme-linked immunosorbent assay (ELISA).
[0063] The vaccine composition of the disclosure can be used for prophylactic treatment of a subject capable of developing Celiac disease and/or used in ongoing treatment of a subject who has Celiac disease. In some embodiments, the composition is for use in treating Celiac disease in a subject. In some embodiments, the subject is HLA-DQ2.5 positive. In some embodiments, the subject is HLA-DQ2.5 positive and HLA-DQ8 negative.
Effective Amount
[0064] The amount of a composition to be administered is referred to as the "effective amount". The term "effective amount" means the amount sufficient to provide the desired therapeutic or physiological effect when administered under appropriate or sufficient conditions. In some embodiments, the effective amount is 150 micrograms of the peptides provided herein (i.e., 50 micrograms of the first peptide and an equimolar amount of each of the second and third peptides). In some embodiments, the effective amount is 26.5 nmol of each of the first, second, and third peptides. Methods for producing equimolar peptide compositions are known in the art and provided herein (see, e.g., Example 1 and Muller et al. Successful immunotherapy with T-cell epitope peptides of bee venom phospholipase A2 induces specific T-cell anergy in patient allergic to bee venom. J. Allergy Clin. Immunol. Vol. 101, Number 6, Part 1: 747-754 (1998)). In some embodiments, the effective amount is 300 micrograms of the peptides provided herein (i.e., 100 micrograms of the first peptide and an equimolar amount of each of the second and third peptides). In some embodiments, this effective amount of the peptides is administered in sterile sodium chloride 0.9% USP as a bolus intradermal injection.
[0065] The effective amounts provided herein are believed to modify the T cell response, e.g., by inducing immune tolerance, to wheat, barley and rye in the subject, and preferably wheat, barley, rye and oats. Thus, a subject treated according to the disclosure preferably is able to eat at least wheat, rye, barley and optionally oats without a significant T cell response which would normally lead to clinical manifestations of active Celiac disease.
Pharmaceutically Acceptable Carriers
[0066] The composition may include a pharmaceutically acceptable carrier. The term "pharmaceutically acceptable carrier" refers to molecular entities and compositions that do not produce an allergic, toxic or otherwise adverse reaction when administered to a subject, particularly a mammal, and more particularly a human. The pharmaceutically acceptable carrier may be solid or liquid. Useful examples of pharmaceutically acceptable carriers include, but are not limited to, diluents, excipients, solvents, surfactants, suspending agents, buffering agents, lubricating agents, adjuvants, vehicles, emulsifiers, absorbants, dispersion media, coatings, stabilizers, protective colloids, adhesives, thickeners, thixotropic agents, penetration agents, sequestering agents, isotonic and absorption delaying agents that do not affect the activity of the active agents of the disclosure. In sortie embodiments, the pharmaceutically acceptable carrier is a sodium chloride solution (e.g., sodium chloride 0.9% USP).
[0067] The carrier can be any of those conventionally used and is limited only by chemico-physical considerations, such as solubility and lack of reactivity with the active agent, and by the route of administration. Suitable carriers for this disclosure include those conventionally used, for example, water, saline, aqueous dextrose, lactose, Ringer's solution, a buffered solution, hyaluronan, glycols, starch, cellulose, glucose, lactose, sucrose, gelatin, malt, rice, flour, chalk, silica gel, magnesium stearate, sodium stearate, glycerol monostearate, sodium chloride, glycerol, propylene glycol, water, ethanol, and the like. Liposomes may also be used as carriers.
[0068] Techniques for preparing pharmaceutical compositions are generally known in the art as exemplified by Remington's Pharmaceutical Sciences, 16th Ed. Mack Publishing Company, 1980.
[0069] Administration preferably is intradermal administration. Thus, the composition of the disclosure may be in a form suitable for intradermal injection. In some embodiments, the composition of the disclosure is in the form of a bolus for intradermal injection.
Injectables
[0070] The pharmaceutical composition(s) may be in the form of a sterile injectable aqueous or oleagenous suspension. In some embodiments, the composition is formulated as a sterile, injectable solution. This suspension or solution may be formulated according to known methods using those suitable dispersing or wetting agents and suspending agents which have been mentioned above. The sterile injectable preparation may be a suspension in a non-toxic parenterally-acceptable diluent or solvent, for example as a solution in 1,3-butanediol. Among the acceptable carriers that may be employed are water, Ringer's solution and isotonic sodium chloride solution. In some embodiments, the composition is formulated as a sterile, injectable solution, wherein the solution is a sodium chloride solution (e.g., sodium chloride 0.9% USP). In some embodiments, the composition is formulated as a bolus for intradermal injection.
[0071] Examples of appropriate delivery mechanisms for intradermal administration include, but are not limited to, implants, depots, needles, capsules, and osmotic pumps,
Dosage
[0072] It is especially advantageous to formulate the active in a dosage unit form for ease of administration and uniformity of dosage. "Dosage unit form" as used herein refers to physically discrete units suited as unitary dosages for the subject to be treated; each unit containing a predetermined quantity of active agent calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier. The specification for the dosage unit forms are dictated by and directly dependent on the unique characteristics of the active agent and the particular therapeutic effect to be achieved, and the limitations inherent in the art of compounding such an active agent for the treatment of subjects. Examples of dosage units include sealed ampoules and vials and may be stored in a freeze-dried condition requiring only the addition of the sterile liquid carrier immediately prior to use.
[0073] The composition may also be included in a container, pack, or dispenser together with instructions for administration.
[0074] The actual amount administered (or dose or dosage) and the rate and time-course of administration are as provided herein.
[0075] The administration may occur at least once, e.g., once or twice a week. In some embodiments, a composition described herein is administered once or twice a week. In some embodiments, a composition described herein is administered for 3, 4 or 8 weeks. In some embodiments, a composition described herein is administered once a week for 8 weeks. In some embodiments, a composition described herein is administered once a week for 3 weeks. In some embodiments, a composition described herein is administered twice a week for 4 weeks. In some embodiments, a composition described herein is administered twice a week for 8 weeks. In some embodiments, the administration occurs 3, 8 or 16 times.
Kits
[0076] Another aspect of the disclosure relates to kits. In some embodiments, the kit comprises a composition comprising the peptides as described herein. The peptides can be contained within the same container or separate containers. In some embodiments, the kit can further comprise a placebo. In some embodiments, the peptide or peptides may be contained within the container(s) (e.g., dried onto the wall of the container(s)). In some embodiments, the peptides are contained within a solution separate from the container, such that the peptides may be added to the container at a subsequent time. In some embodiments, the peptides are in lyophilized form in a separate container, such that the peptides may be reconstituted and added to the container at a subsequent time.
[0077] In some embodiments, the kit further comprises instructions for reconstitution, mixing, administration, etc. In some embodiments, the instructions include the methods described herein. Instructions can be in any suitable form, e.g., as a printed insert or a label.
Methods of Treatment
[0078] Aspects of the disclosure relate to use of the compositions described herein for treating a subject having, suspected of having or at risk of having Celiac disease.
[0079] As used herein, the terms "treat", "treating", and "treatment" include abrogating, inhibiting, slowing, or reversing the progression of a disease or condition, or ameliorating or preventing a clinical symptom of the disease (for example, Celiac disease). Treatment may include induction of immune tolerance (for example, to gluten or peptide(s) thereof), modification of the cytokine secretion profile of the subject and/or induction of suppressor T cell subpopulations to secrete cytokines. Thus, a subject treated according to the disclosure preferably is able to eat at least wheat, rye, barley and optionally oats without a significant T cell response which would normally lead to symptoms of Celiac disease.
Identifying Subjects for Treatment
[0080] In some embodiments, methods described herein comprise treating a subject who has Celiac disease. Thus, it may be desirable to identify subjects, such as subjects with Celiac disease, who are likely to benefit from administration of a composition described herein. In any one of the methods provided, the method may comprise a step of identifying a subject likely to benefit from such administration. Any diagnostic method or combinations thereof for Celiac disease is contemplated for identifying such a subject. Exemplary methods include, but is not limited to, intestinal biopsy, serology (measuring the levels of one or more antibodies present in the serum), and genotyping (see, e.g., Husby S, Koletzko S. Korponay-Szabo I R, Mearin M L, Phillips A, Shamir R, Troncone R, Giersiepen K, Branski D, Catassi C et al: European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr 2012, 54(1):136-160. AND/OR Rubio-Tapia A, Hill I D, Kelly C P, Calderwood A H, Murray J A. ACG clinical guidelines: diagnosis and management of celiac disease. Am J Gastroenterol 2013; 108:656-76. AND/OR Ludvigsson J F, Leffler D A, Bai J C, Biagi F, Fasano A, Green P H, Hadjivassiliou M, Kaukinen K, Kelly C P, Leonard J N, Lundin K E, Murray J A, Sanders D S, Walker M M, Zingone F, Ciacci C. The Oslo definitions for coeliac disease and related terms. Gut 2012; 62:43-52.).
[0081] The presence of serum antibodies can be detected using methods known to those of skill in the art, e.g., by ELISA, histology, cytology, immunofluorescence or western blotting. Such antibodies include, but are not limited to: IgA anti-endomysial antibody (IgA EMA), IgA anti-tissue transglutaminase 2 antibody (IgA tTG), IgA anti-deamidated gliadin peptide antibody (IgA DGP), and IgG anti-deamidated gliadin peptide antibody (IgG DGP). Deamidated gliadin peptide-IgA (DGP-IgA) and deamidated gliadin peptide-IgG (DGP-IgG) can be evaluated with commercial kits (e.g. INV 708760, 704525, and 704520, INOVA Diagnostics, San Diego, Calif.).
[0082] Subjects can be tested for the presence of the HLA-DQA and HLA-DQB susceptibility alleles encoding HLA-DQ2.5 (DQA1*05 and DQB1*02), DQ2.2 (DQA1*02 and DQB1*02) or DQ8 (DQA1*03 and DQB1*0302). Exemplary sequences that encode the DQA and DQB to susceptibility alleles include HLA-DQA1*0501 (Genbank accession number: AF515813.1) HLA-DQA1*0505 (AH013295.2), HLA-DQB1*0201 (AY375842.1) or HLA-DQB1*0202 (AY375844.1). Methods of genetic testing are well known in the art (see, e.g., Bunce M, et al. Phototyping: comprehensive DNA typing for HLA-A, B, C, DRB1, DRB3, DRB4, DRB5 & DQB1 by PCR with 144 primer mixes utilizing sequence-specific primers (PCR-SSP). Tissue Antigens 46, 355-367 (1995); Olerup O, Aldener A, Fogdell A. HLA-DQB1 and DQA1 typing by PCR amplification with sequence-specific primers in 2 hours. Tissue antigens 41, 119-134 (1993); Mullighan C G, Bunce M, Welsh K I. High-resolution HLA-DQB1 typing using the polymerase chain reaction and sequence-specific primers. Tissue-Antigens. 50, 688-92 (1997); Koskinen L, Romanos J, Kaukinen K, Mustalahti K, Korponay-Szabo I, et al. (2009) Cost-effective HLA typing with tagging SNPs predicts celiac disease risk haplotypes in the Finnish, Hungarian, and Italian populations. Immunogenetics 61: 247-256.; and Monsuur A J, de Bakker P I, Zhernakova A, Pinto D, Verduijn W, et al. (2008) Effective detection of human leukocyte antigen risk alleles in celiac disease using tag single nucleotide polymorphisms. PLoS ONE 3: e2270). Subjects that have one or more copies of a susceptibility allele are considered to be positive for that allele. Detection of the presence of susceptibility alleles can be accomplished by any nucleic acid assay known in the art, e.g., by polymerase chain reaction (PCR) amplification of DNA extracted from the patient followed by hybridization with sequence-specific oligonucleotide probes or using leukocyte-derived DNA (Koskinen L, Romanos J, Kaukinen K, Mustalahti K, Korponay-Szabo I, Barisani D, Bardella M T, Ziberna F, Vatta S, Szeles G et al: Cost-effective HLA typing with tagging SNPs predicts Celiac disease risk haplotypes in the Finnish, Hungarian, and Italian populations. Immunogenetics 2009, 61(4):247-256; Monsuur A J, de Bakker P I, Zhernakova A, Pinto D, Verduijn W, Romanos J, Auricchio R, Lopez A, van Heel D A, Crusius J B et al: Effective detection of human leukocyte antigen risk alleles in Celiac disease using tag single nucleotide polymorphisms. PLoS ONE 2008, 3(5):e2270).
EXAMPLES
Example 1: Preparation of a 150 Microgram Dosage Composition of the First, Second, and Third Peptide
[0083] A dose of 150 .mu.g the peptide composition was defined by there being 50 .mu.g (26.5 nmol) of pure peptide 1, and an equimolar amount of peptide 2 and peptide 3. The molar equivalent of 50 .mu.g peptide 1 was given by 50 .mu.g/1889.3 g/mol=26.5 nmol. When preparing a solution containing 150 .mu.g of the peptide composition, for the constituent peptides, the weight of each peptide was adjusted according to peptide purity and peptide content of the lyophilized stock material. For example, if the peptide 1 stock material had peptide purity of 98% and its peptide content was 90%, the weight of stock material yielding 50 .mu.g, peptide 1 was 50 .mu.g/(peptide purity.times.peptide content)=50 ug/(0.98.times.0.90)=56.7 ug.
[0084] The molar amount of peptide 1 in 150 .mu.g of the peptide composition was 26.5 nmol, and the weight of lyophilized peptide 2 stock material was therefore given by 26.5 nmol.times.1833.2 g/mol/(peptide purity.times.peptide content). For example, if peptide 2 peptide purity was 99%, and peptide content of 95%, the mass of stock required was 51.7 ug.
[0085] The molar amount of peptide 3 in 150 ug of the peptide composition was 26.5 nmol, and the weight of lyophilized peptide 3 stock material was therefore given by 26.5 nmol.times.1833.2 g/mol/(peptide purity.times.peptide content). For example, if peptide 3 peptide purity was 98%, and peptide content of 92%, the mass of stock required was 55.4 ug.
Example 2: A Phase I Randomized, Double-Blind, Placebo-Controlled, Multiple Ascending Dose Study in Patients with Celiac disease
Primary Objective
[0086] To determine the safety and tolerability of an equimolar composition of peptides comprising SEQ ID NOs: 1, 2, and 3 (each peptide comprising an N-terminal pyroglutamate and C-terminal amidated amino acid) when administered intradermally to human leukocyte antigen (HLA)-DQ2.5+ patients with Celiac disease on a gluten-free diet (GFD) (patient has HLA-DQ2.5 genotype (both HLA-DQA1*05 and HLA-DQB1*02, homozygous or heterozygous) but does not possesses the genes encoding HLA DQ8 (either HLA-DQA1*03 or HLA-DQB1*0302))
Secondary Objectives
[0086] [0087] To assess the pharmacokinetics (PK) of the equimolar composition when administered intradermally to patients with Celiac disease on a gluten-free diet (GFD) [0088] To assess the effect of the equimolar composition on the immunological response and on clinical tolerance to gluten when administered intradermally to patients with Celiac disease on a GFD
Exploratory Objective
[0088] [0089] To assess the effect of the equimolar composition on small bowel mucosal histology in patients with Celiac disease on a GFD
Study Design
[0090] The study included a dose treatment cohort (150 .mu.g per dose, i.e., 50 micrograms of the first peptide and an equimolar amount of each of the second and third peptides) that received the composition provided herein or placebo given intradermally on a twice weekly schedule for eight weeks in a 2:1 ratio.
Drug Dosage, and Route of Administration
[0091] The composition was an equimolar mixture of three peptides. The composition dose: 150 .mu.g Placebo: Sodium chloride 0.9% USP (same as vehicle/diluent for the composition) Dose frequency: twice weekly Route of administration: intradermal injection All study injections were prepared by an unblinded pharmacist at the study center while remaining double-blind to the patient and investigator.
Safety Assessments
[0092] A medical history, physical examination, vital sign measurements, ECG, and laboratory assessment (including hematology, biochemistry, and urinalysis) occurred at certain time points. Adverse events (AE) and concomitant medications were assessed at each visit. A daily gastrointestinal (GI) symptom diary and weekly gastrointestinal symptom rating scale (GSRS) were used to record gastrointestinal symptoms throughout the trial. Cytokine, chemokine, and T-cell measurements were assessed. Presence of antitherapeutic antibodies were assessed. An independent data safety monitoring board (DSMB), whose charter was documented prior to randomization of the first patient, assessed the progress of the clinical trial, including the safety data, and recommended whether to continue, modify, or stop the trial at any time.
Pharmacokinetic Assessments
[0093] For each cohort, serial blood samples were collected for assessment of pharmacokinetics.
Pharmacodynamic Assessments
Specific immune responses were assessed throughout the trial.
Exploratory Assessment
[0094] An upper endoscopy and small bowel biopsy for histological assessment was performed on patients in the biopsy cohort at screening and following the last dose of the composition.
Statistical Methods
[0095] No formal hypothesis testing was performed. Data were summarized appropriately and all data were listed. Adverse events (AE) were summarized, presenting the numbers and percent of patients having any AE and having AEs in each system organ class and preferred term.
Example 3: Results of the Phase I Randomized, Double-Blind, Placebo-Controlled, Multiple Ascending Dose Study in Patients with Celiac disease
[0096] 3 cohorts of subjects with HLA-DQ2.5+ biopsy-proven Celiac disease on a gluten-free diet for at least 1 year were included in the study. The first cohort (Cohort 1) contained 12 subjects who were dosed with 150 mcg of a peptide composition (an equimolar composition in sodium chloride 0.9% USP of 3 peptides, peptide 1, peptide 2, and peptide 3, comprising SEQ ID NOs: 1, 2, and 3, respectively, each peptide comprising an N-terminal pyroglutamate and C-terminal amidated amino acid) or a placebo (sodium chloride 0.9% USP) intradermally, twice a week for 8 weeks total. The second cohort (Cohort 2) contained 12 subjects who were dosed with 300 mcg of the peptide composition or the placebo intradermally, twice a week for 8 weeks total. The peptide composition to placebo ratio for each of Cohorts 1 and 2 were 2:1. Both Cohorts 1 and 2 received an oral gluten challenge and were assessed for gamma-interferron (gIFN) release and then returned to baseline prior to starting the treatment regimen. The third cohort (Cohort 7) contained 14 subjects who were dosed with 150 mcg of the peptide composition or the placebo intradermally, twice a week for 8 weeks total. The peptide composition to placebo ratio for Cohort 7 was 1:1. The subjects in Cohort 7 did not undergo an oral gluten challenge or a gIFN release assay before starting the dosage regimen.
[0097] The progress of each subject before, during and after the trial was assessed using multiple tests including serology (tTG-IgA, DGP-IgG, DGP-IgA, and EMA-IgA), histology, and IFNg whole blood release assay, and cytokine/chemokines in plasma (measured by MAGPIX.RTM. multiplex platform).
[0098] Subject disposition is summarized in Table 1.
TABLE-US-00001 TABLE 1 Subject disposition. Placebo (from Placebo All All Cohort 1 Cohort 2 Cohort 7 Cohorts 1 (from Subjects Subjects Completion (150 mg) (300 mg) (150 mg) and 2) Cohort 7) Dosed Screened Status (N = 8) (N = 8) (N = 7) (N = 7) (N = 7) (N = 39) (N = 67) Screened 67 (100%) Enrolled 8 (100%) 10 (100%) 7 (100%) 7 (100%) 7 (100%) 39 (100%) 39 (58%) Completed the 8 (100%) 6 (60%) 7 (100%) 6 (86%) 7 (100%) 34 (87%) study as required Completed 8 (100%) 2 (20%) 7 (100%) 5 (71%) 7 (100%) 29 (74%) study treatment per protocol (received at least 15 of 16 doses) Received all 7 (88%) 2 (20%) 5 (71%) 4 (57%) 6 (86%) 24 (62%) 16 doses of study treatment Discontinued 8 (80%) 2 (29%) 10 (26%) the study prior to completion
[0099] Subject demographics are summarized in Table 2. The extent of exposure for each subject is summarized in Table 3.
TABLE-US-00002 TABLE 2 Subject Demographics All Cohort 1 Cohort 2 Cohort 7 Placebo subjects (150 mg) (300 mg) (150 mg) (pooled) dosed Parameter Statistic (N = 8) (N = 10) (N = 7) (N = 14) (N = 39) Age (years) N 8 10 7 14 39 Mean 52.0 50.0 42.6 39.1 45.2 SD 11.9 10.1 5.4 15.5 13.0 Median 52.5 52.0 45.0 34.0 47.0 Min 31 28 33 18 18 Max 66 64 47 64 66 Race White n (%) 8 10 7 14 39 (100%) Sex Female n (%) 7 7 5 10 29 (74%) Male n (%) 1 3 2 4 10 (26%) Height (cm) N 8 10 7 14 39 Mean 167.7 170.1 168.4 170.6 169.5 SD 10.0 9.8 8.3 10.0 9.4 Median 168.7 167.0 173.0 170.5 169.0 Min 154 158 156 156 154 Max 186 186 179 186 186 Weight (kg) N 8 10 7 14 39 Mean 70.66 85.34 74.40 66.55 73.62 SD 11.17 13.02 11.58 12.91 14.07 Median 69.20 85.05 73.00 64.10 70.50 Min 60.2 66.0 58.5 48.5 48.5 Max 95.1 105.5 92.5 92.3 105.5 BMI (kg/m{circumflex over ( )}2) N 8 10 7 14 39 Mean 25.24 29.55 26.13 22.81 25.63 SD 4.28 4.54 2.63 3.72 4.60 Median 23.91 28.91 25.23 22.64 25.23 Min 20.7 25.2 23.3 17.2 17.2 Max 33.2 40.2 30.9 32.3 40.2
TABLE-US-00003 TABLE 3 Summary of subject exposure Number of Number of Cohort Treatment Dose level Doses Total Dose Subjects 1 peptide 150 16 2400 7 composition 1 peptide 150 15 2250 1 composition 1 Placebo 0 16 0 4 2 peptide 300 16 4800 2 composition 2 peptide 300 5 1500 1 composition 2 peptide 300 4 1200 2 composition 2 peptide 300 3 900 1 composition 2 peptide 300 2 600 1 composition 2 peptide 300 1 300 3 composition 2 Placebo 0 15 0 1 2 Placebo 0 10 0 1 2 Placebo 0 5 0 1 7 peptide 150 16 2400 5 composition 7 peptide 150 15 2250 2 composition 7 Placebo 0 16 0 6 7 Placebo 0 15 0 1
Immune Tolerance
[0100] Immune tolerance induced by the peptide composition was measured using two types of assays: gIFN release to gluten and cytokine/chemokine plasma assays.
[0101] An ex vivo whole blood cytokine release assay was performed pre-and post-treatment with the peptide composition. Blood was collected 6 days after commencing 3-day oral challenge with gluten (approximately 9 g/day). The MAGPIX.RTM. assay was used to confirm elevated IFN-.gamma. plasma levels in blood incubated with the three constituent peptides present in the peptide composition (0.05 mg/mL/peptide), and also to show that levels of interleukin-2 and IFN-.gamma.-inducible protein (IP-10) correlated with elevated concentrations of IFN-.gamma.. Pretreatment gluten challenge was 4-5 weeks prior to commencing dosing with the peptide composition. Post-treatment 3-day gluten challenge was commenced the day after last dose of the peptide composition.
[0102] Subjects were determined to be responsive to gluten if the subject had detectable gIFN released after gluten challenge before the first dose of the peptide composition. By this criteria, 7 of 8 subjects at 150 mcg and 3 of 4 subjects at 300 mcg were responsive to gluten. Subjects were then determined to be tolerant to gluten after the last dose of the peptide composition if the subject had significantly less gIFN released after the second gluten challenge after the last dose of the peptide composition. By this criteria 5 of 7 subjects at 150 mcg and 1 of 1 at 300 mcg originally responsive to gluten were tolerant to gluten after treatment (2 of the 300 mcg subjects did not finish the treatment and were not included in the tolerance analysis).
[0103] This assessment was also performed using a level of 7.2 pg/mL of IFN-.gamma. in the cytokine release assay as a cut-off level for reactivity v. non-reactivity to the peptide composition. As shown in FIG. 5, most subjects that were reactive to the peptides before treatment (see pre-treatment column in FIG. 5) became non-reactive to the peptides after treatment (see post-treatment column in FIG. 5), indicating that treatment induced immune tolerance to the peptides.
[0104] The second measure of immune tolerance was a plasma cytokine/chemokine assay. Plasma cytokines and chemokines were measured at several timepoints pre and post first and last dose (visits 6 and 21). For a subject to be responsive they needed to have a greater than 2 fold increase after the first dose for IL-8 and MCP-1. For a subject to be tolerant they had to have less than 2 fold increase after the last dose for IL-8 and MCP-1. By this criteria, 8 of 8 subjects in Cohort 1 (150 micrograms), and 3 of 4 subjects in Cohort 2 (300 micrograms) and 6 of 7 in Cohort 7 (150 micrograms) were responsive to the first dose. Of the responsive subjects, 8 of 8, 1 of 1 and 6 of 6 were tolerant in the three cohorts, respectively. These results indicate that immune tolerance to gluten and to the peptide composition was induced in several subjects treated with the peptide composition. The assay results for an exemplary subject from Cohort 1 (150 micrograms the peptide composition) who was both responsive and tolerant to gluten and the peptide composition are shown in Table 4.
TABLE-US-00004 TABLE 4 Tolerance Assay results for representative subject from Cohort 1 (tolerant to both gluten and the peptide composition) Assay Measurement Visit 6 (First dose) Visit 21 (last dose) GI symptoms 18 6 gIFN release, fold increase 345 0.31 gIFN, pg/mL 3,461 -4.6 IL-2, fold increase 10 1 IL-8, fold increase 20 0.9 IL-10, fold increase 8.4 0.75 MCP-1, fold increase 18 1.06 Peptide 1, pharmacokinetics 1.67 1.48
Other Parameters of the Study
[0105] Lymphocyte subpopulations were analyzed to identify whether there was systemic changes during treatment with the peptide composition. FACS analysis showed that there was no change in CD4+, CD8+, NK or B-cell compartments as expected for antigen-specific immunotherapy. There were some fluctuations noted in CD19, CD56 cells. Anti-Therapeutic Antibodies (ATA, also called anti-drug antibodies, ADA) were not detected in any subjects. There was no difference between the pharmacokinetics of the peptides at the first and last dose (FIGS. 2-4). The lack of activation of peptide composition-specific B-cells was consistent with the PK showing no change between the first and last dose. No subjects converted from baseline negative to positive after therapy as measured using standard serology (tissue-transglutaminase-IgA (tTG-IgA), anti-deamidated gliadin peptide-IgG or IgA (DGP-IgG, DGP-IgA), and anti-endomysial-IgA (EMA-IgA)). The biopsy Modified Marsh Scores for the treatment vs. placebo group are shown in Tables 5 and 6.
TABLE-US-00005 TABLE 5 Biopsy Modified Marsh Scores - treated Pre-treatment scores Post-treatment scores Bulb + Bulb + Bulb + Part 2 + Parts (Parts 2, 3) - Bulb + Part 2 + Parts (Parts 2, 3) - Part 1 Part 3 1, 2, 3 (Bulb + Part1) Part 1 Part 3 1, 2, 3 (Bulb + Part1) Subject Number 1 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 4 0 0 0 0 0 0 0 0 5 5 13 18 8 10 13 23 3 6 1 2 3 1 4 4 8 0 7 6 3 9 -3 4 2 6 -2 Parameters N 7 7 7 7 7 7 7 7 Mean 1.7 2.6 4.3 0.9 2.6 2.7 5.3 0.1 Std Dev 2.6 4.8 6.9 3.4 3.8 4.8 8.5 1.5 Median 0 0 0 0 0 0 0 0 Min 0 0 0 -3 0 0 0 -2 Max 6 13 18 8 10 13 23 3 p-value 0.750 1.000
TABLE-US-00006 TABLE 6 Biopsy Modified Marsh Scores - Placebo Pre-treatment scores Post-treatment scores Bulb + Bulb + Bulb + Part 2 + Parts (Parts 2, 3) - Bulb + Part 2 + Parts (Parts 2, 3) - Part 1 Part 3 1, 2, 3 (Bulb + Part1) Part 1 Part 3 1, 2, 3 (Bulb + Part1) Subject Number 1 0 0 0 0 2 1 3 -1 2 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 4 0 0 0 0 0 0 0 0 5 0 0 0 0 4 0 4 -4 6 0 0 0 0 0 0 0 0 7 2 1 3 -1 7 11 18 4 Parameters N 7 7 7 7 7 7 7 7 Mean 0.3 0.1 0.4 -0.1 1.9 1.7 3.6 -0.1 Std Dev 0.8 0.4 1.1 0.4 2.7 4.1 6.6 2.3 Median 0 0 0 0 0 0 0 0 Min 0 0 0 -1 0 0 0 -4 Max 2 1 3 0 7 11 18 4 p-value 1.000 1.000
Adverse Events
[0106] Treatment emergent adverse events are summarized in Tables 7-9. The adverse events were mainly gastrointestinal symptoms and headaches, and were worse early on in the study. The GI adverse events recapitulated oral gluten exposure symptoms. Generally, adverse events occurred about 2 hours post dosing and resolved within 24 hours. Adverse events were primarily noted in week 1 of dosing. Adverse events were more severe in Cohort 1 (150 mg with oral challenge) than in Cohort 7 (150 mg without oral challenge). Interestingly, the gastrointestinal symptoms were worse on active drug vs. placebo early in the study but not late in study. The adverse events that occurred after the last dose (dose 16) were similar to placebo. Three subjects from Cohort 2 (300 micrograms were withdrawn from the study due to gastrointestinal-related adverse events.
TABLE-US-00007 TABLE 7 Treatment emergent adverse events (TEAEs) Placebo (from All Cohort 1 Cohort 2 Cohort 7 Cohorts 1 Placebo Subjects (150 mg) (300 mg) (150 mg) and 2) (Cohort 7) Dosed (N = 8) (N = 10) (N = 7) (N = 7) (N = 7) (N = 39) Number of Subjects 7 (88%) 10 (100%) 6 (86%) 5 (71%) 7 (100%) 35 (90%) with TEAEs Number of Subjects 6 (75%) 9 (90%) 4 (57%) 3 (43%) 3 (43%) 25 (64%) with Study Drug Related TEAEs Number of Subjects 6 (75%) 8 (80%) 3 (43%) 2 (29%) 19 (49%) with Moderate or Severe TEAEs Number of Subjects 5 (63%) 8 (80%) 1 (14%) 14 (36%) with Study Drug Related, Moderate or Severe TEAEs Number of Subjects 1 (10%) 1 (3%) with SAE Number of Treatment- 26 26 22 14 27 115 Emergent Adverse Events Number of Study Drug 17 16 7 5 5 50 Related TEAEs Number of Moderate 11 12 4 2 29 or Severe TEAEs Number of Study Drug 7 8 2 17 Related, Moderate or Severe TEAEs Number of SAEs 1 1
TABLE-US-00008 TABLE 8 AEs by Severity and System Organ Class Placebo (from All Cohort 1 Cohort 2 Cohort 7 Cohorts 1 Placebo Subjects System Organ Class, (150 mg) (300 mg) (150 mg) and 2) (Cohort 7) Dosed Preferred term Severity (N = 8) (N = 10) (N = 7) (N = 7) (N = 7) (N = 39) Nervous system Moderate 2 (25%) 3 (30%) 1 (14%) 6 (15%) disorders [2] [3] [1] [6] Mild 2 (25%) 3 (30%) 4 (57%) 5 (71%) 4 (57%) 18 (46%) [8] [6] [4] [7] [8] [33] Dizziness Mild 2 (29%) 2 (29%) 4 (10%) [4] [2] [6] Headache Moderate 1 (13%) 2 (20%) 1 (14%) 4 (10%) [1] [2] [1] [4] Mild 2 (25%) 3 (30%) 2 (29%) 3 (43%) 3 (43%) 13 (33%) [8] [6] [2] [3] [4] [23] Lethargy Mild 1 (14%) 1 (14%) 2 (5%) [1] [1] [2] Migraine Moderate 1 (13%) 1 (10%) 2 (5%) [1] [1] [2] Mild 1 (14%) 1 (3%) [1] [1] Presyncope Mild 1 (14%) 1 (3%) [1] [1] Gastrointestinal Severe 1 (13%) 1 (10%) 2 (5%) disorders [1] [2] [3] Moderate 4 (50%) 6 (60%) 10 (26%) [5] [6] [11] Mild 2 (25%) 2 (20%) 3 (43%) 1 (14%) 3 (43%) 11 (28%) [2] [2] [4] [1] [5] [14] Abdominal pain Severe 1 (10%) 1 (3%) [2] [2] Mild 1 (14%) 1 (3%) [1] [1] Aphthous Mild 1 (14%) 1 (3%) stomatitis [1] [1] Change of bowel Mild 1 (10%) 1 (3%) habit [1] [1] Diarrhoea Mild 1 (14%) 1 (14%) 2 (5%) [1] [1] [2] Dry mouth Mild 1 (14%) 1 (14%) 2 (5%) [1] [1] [2] Gastrointestinal Moderate 2 (20%) 2 (5%) disorder [2] [2] Gastrointestinal Mild 1 (14%) 1 (3%) sounds abnormal [1] [1] Gastrooesophageal Mild 1 (14%) 1 (3%) reflux disease [1] [1] Lip dry Mild 1 (14%) 1 (3%) [1] [1] Vomiting Severe 1 (13%) 1 (3%) [1] [1]
TABLE-US-00009 TABLE 9 Time course of gastrointestinal/headache adverse events Count of TEAE_YN Week Week Week Week Week Week Grand SI_type Treatment 1 2 3-4 5-6 7-8 9 Total Gastro System peptide 4 0 1 0 3 0 8 Organ Class composition (Cohort 1) peptide 8 0 0 2 0 0 10 composition (Cohort 2) peptide 2 1 0 0 0 1 4 composition (Cohort 7) Placebo 0 0 0 1 0 0 1 (Cohort 1 and 2) Placebo 1 0 2 0 2 0 5 (Cohort 7) Gastro System 15 1 3 3 5 1 28 Organ Class Total Headache peptide 1 3 3 1 1 0 9 composition (Cohort 1) peptide 5 2 0 1 0 0 8 composition (Cohort 2) peptide 1 1 0 0 0 1 3 composition (Cohort 7) Placebo 1 1 0 1 0 0 3 (Cohort 1 and 2) Placebo 2 2 0 0 0 0 4 (Cohort 7) Headache Total 10 9 3 3 1 1 27 Grand Total 25 10 6 6 6 2 55
CONCLUSIONS
[0107] The maximum tolerated dose in this study was 150 micrograms. Tolerance to the peptide composition was induced in 14 of 14 responsive subjects. Tolerance to gluten was induced in 6 of 8 responsive subjects. There was no conversion of negative to positive tTG serology and biopsy was not worsened by exposure to the peptide composition.
[0108] A summary of the study as well as the assessments and further assessments performed during the study in Example 3 are shown in Table 10 below (which is split up into multiple tables). An exemplary time course for cohorts 1 and 2 is shown in FIG. 16.
TABLE-US-00010 TABLE 10 Study parameters and assessments Visit 1-5 Double Blind Oral Challenge Visit V1 V2 V3 Day (S = screening, T = treatment) Parameter Notes Sample S-35 S-34 S-33 S-30 S-28 Treatment: Peptide composition/ Placebo Oral challenge: x x x x Gluten/Placebo cookies IFN-.gamma. Whole Peptide composition- IFN-.gamma. WBRA x x x Blood Release NIL pg/mL Plasma-heparin Assay FACS In PBMC PBMC x ADA Peptide composition Serum x IgG & IgA titer, reflex peptide 1-3 IgG & IgA CD-serology tTG-IgA (4 kits), DGP Serum x IgG & IgA (2 kits), & EmA PK 10 time points 0-6 Plasma-EDTA hours. Only trough levels at V17 Histology-Score Bulb, D1, D2 & D3 Biopsy (Biopsy cohort) duplicate biopsies: Mod-Marsh Score Histology- Bulb, D1, D2 & D3 Biopsy Quantitative duplicate biopsies: (Biopsy cohort) IEL density/100 Histology- Bulb, D1, D2 & D3 Biopsy Quantitative duplicate biopsies: (Biopsy cohort) VH:CrD MAGPIX: In 38 cytokines and Plasma-EDTA x plasma chemokines MAGPIX: In 38 cytokines and PK Plasma-EDTA each of 10 PK chemokines x10 time course plasmas MAGPIX: 38 cytokines and IFN-.gamma. WBRA x x Peptide chemokines Plasma-heparin composition-NIL response (150 mcg cohort only) Induction Phase Visit 1-5 Double Blind Oral Challenge (8 weeks of dosing) V4 V5 V5.1 V5.2 Endoscopy V6 V7 Parameter S-27 S-26 S-23 S-07 S-07 S-07 Screening T + 01 T + 04 Treatment: x x Peptide composition/ Placebo Oral challenge: x x Gluten/Placebo cookies IFN-.gamma. Whole x x x x x Blood Release Assay FACS x x ADA x CD-serology x PK x Histology-Score x (Biopsy cohort) Histology- x Quantitative (Biopsy cohort) Histology- x Quantitative (Biopsy cohort) MAGPIX: In x x plasma MAGPIX: In x each of 10 PK time course plasmas MAGPIX: x Peptide composition-NIL response (150 mcg cohort only) Induction Phase (8 weeks of dosing) V8 V9 V10 V11 V12 V13 V14 V15 Parameter T + 8 T + 11 T + 15 T + 18 T + 22 T + 25 T + 29 T + 32 Treatment: x x x x x x x x Peptide composition/ Placebo Oral challenge: Gluten/Placebo cookies IFN-.gamma. Whole x x Blood Release Assay FACS x x ADA x x CD-serology PK Histology-Score (Biopsy cohort) Histology- Quantitative (Biopsy cohort) Histology- Quantitative (Biopsy cohort) MAGPIX: In x x plasma MAGPIX: In each of 10 PK time course plasmas MAGPIX: Peptide composition-NIL response (150 mcg cohort only) Induction Phase (8 weeks of dosing) V21 V16 V17 V18 V19 V20 (EOT) Endoscopy Parameter T + 36 T + 39 T + 43 T + 46 T + 50 T + 53 Follow-up Treatment: x x x x x x Peptide composition/ Placebo Oral challenge: Gluten/Placebo cookies IFN-.gamma. Whole x x Blood Release Assay FACS x x ADA x CD-serology PK x x Histology-Score x (Biopsy cohort) Histology- x Quantitative (Biopsy cohort) Histology- x Quantitative (Biopsy cohort) MAGPIX: In x x plasma MAGPIX: In x each of 10 PK time course plasmas MAGPIX: Peptide composition-NIL response (150 mcg cohort only) Post-treatment Double Blind Oral Challenge V26 V22 V23 V24 V25 (EOS) Parameter T + 54 T + 55 T + 56 T + 59 T + 61 T + 62 T + 63 T + 66 T + 88 Treatment: Peptide composition/ Placebo Oral challenge: X X X X X X Gluten/Placebo cookies IFN-.gamma. Whole X X X X X Blood Release Assay FACS X X ADA X X CD-serology X X PK Histology-Score (Biopsy cohort) Histology- Quantitative (Biopsy cohort) Histology- Quantitative (Biopsy cohort) MAGPIX: In X X plasma MAGPIX: In each of 10 PK time course plasmas MAGPIX: X X Peptide composition-NIL response (150 mcg cohort only)
Example 4: Gastrointestinal Symptom Rating Seale (GSRS) Data
[0109] The GSRS is a validated diary tool used in Gastroesophageal Reflux Disease (GERD) studies. The GSRS scale is as follows:
[0110] 1=no discomfort at all
[0111] 2=minor discomfort
[0112] 3=mild discomfort
[0113] 4=moderate discomfort
[0114] 5=moderately severe discomfort
[0115] 6=severe discomfort
[0116] 7=very severe discomfort
[0117] The GSRS was used for all subjects described in Example 3 to keep track of symptoms. There were 6 questions on: bloating, diarrhea, hunger pain, nausea, pain, and rumbling. The GSRS was conducted daily (looking at past 24 hours) throughout the trial described in Example 3. The daily values were averaged for 6 days and the following were calculated: cohort means for gluten challenges, dosing period, and change from baseline is dosing period.
[0118] The symptom scores during dosing are shown in FIG. 6. The symptom scores during dosing as changed from baseline are shown in FIG. 7. The symptom scores during gluten challenge are shown in FIG. 8.
[0119] Generally, during dosing, symptoms increased from week -1 to week 1 with Cohorts 1 and 2 (150 micrograms and 300 micrograms with oral challenge, respectively) increasing more than Cohort 7 (150 microgram biopsy). During the course of dosing, the symptoms in all peptide composition cohorts (Cohorts 1, 2, and 7) decreased to where at week 8 there was no change from the week -1 baseline. For the gluten challenge, the symptom scores were higher for active gluten than placebo gluten both pre-dosing and post-dosing with the peptide composition or placebo. However, the mean scores were between 1 ("No symptoms at all") and 2 ("Minor symptoms").
Example 5: Biopsy Data
[0120] Biopsy data from the study in Example 3 is summarized below.
[0121] Small bowel biopsies were conducted at duodenal bulb and parts 1, 2, and 3 of jejunum. 2 biopsies were taken per area=8 pre-dose samples and 8 post-dose samples. Screening biopsies were all required to be Marsh-Oberhuber 0 or 1 for enrollment into the study. After collection of the post-treatment biopsy, H+E stained sections were re-assessed for Marsh-Oberhuber score. Villous height to crypt depth (VH/CrD) ratios were determined in well oriented sections. Intra-epithelial lymphocytes (IEL) per 100 epithelial cells were assessed in anti-CD3-stained slides.
[0122] The Marsh-Oberhuber classification (Oberhuber 1999) is shown in Table 11 below.
TABLE-US-00011 TABLE 11 Marsh-Oberhuber classification Type 0 Type 1 Type 2 Type 3a Type 3b Type 3c Score 0 1 2 3 4 5 IEL* <40 >40 >40 >40 >40 >40 Crypts Normal Normal Hypertrophic Hypertrophic Hypertrophic Hypertrophic Villi Normal Normal Normal Mild atrophy Marked atrophy Absent *Numbers are given as IEL per 100 epithelial cells (counted from H + E stained biopsy sections
[0123] The mean villous height to crypt depth (VH:CrD) is shown in FIG. 9. Two of the 16 screenees excluded as initial read of histology was scored as modified marsh 3 (villous atrophy and increased IEL count) indicating "active disease". At the end of the study, modified marsh score, VH:CrD (N>3) and IEL count per 100 epithelial cells was evaluated by observers blind to the subject number and timing of biopsy collection.
[0124] It was found that there was no change in VH:CrD before and after treatment with peptide composition or placebo (FIG. 10, normal mucosal morphology shows VH:CrD as greater than 3). It was also found that there was no change in IEL count before and after treatment with peptide composition or placebo (FIG. 11, normal IEL count shows fewer than 20 per 100 epithelial cells). It was also found that there was no change in modified marsh score before and after treatment with peptide composition or placebo (FIG. 12, normal mucosa is scored 0, raised IELs 1, crypt hyperplasia 2, villous atrophy 3-5). The quantitative histology is summarized in Table 12 below.
TABLE-US-00012 TABLE 12 VH:CrD Marsh VH:CrD VH:CrD AVG IEL/100 IEL/100 IEL/100 Marsh Marsh AVG AVG AVG DB/D1/ AVG AVG DB/D1/ AVG AVG DB/D1/ D2/D3 DB/D1 D2/D3 D2/D3 DB/D1 D2/D3 D2/D3 DB/D1 D2/D3 Peptide 1.74 1.34 1.57 47.39 33.89 40.64 0.64 0.43 0.54 composition screen mean Placebo 2.07 1.46 1.79 34.89 27.21 31.05 0.04 0.07 0.05 screen mean Peptide 1.65 1.63 1.68 46.50 36.71 41.61 0.68 0.64 0.66 composition follow-up mean Placebo 1.92 1.76 1.84 36.82 30.68 33.75 0.43 0.46 0.45 Followup Mean Peptide -0.09 0.29 0.11 -0.89 2.82 0.96 0.04 0.21 0.13 composition Followup Screen Placebo -0.15 0.30 0.05 1.93 3.46 2.70 0.39 0.39 0.39 Followup Screen
[0125] The purpose of assessing biopsies was because tissue damage defines reactivation and results in complications. It was hypothesized that biopsies from part 2 and 3 of duodenum were probably cleanest since they do not contain Brunners glands as do bulb and part 1. In summary, the peptide composition cohort had a mean VH:CrD of 1.74 before treatment and 1.65 after treatment, an IEL of 47 before treatment and 47 after treatment, and a Marsh score of 0.64 before treatment and 0.68 after treatment. The placebo cohort had a mean VH:CrD of 2.07 before treatment and 1.92 after treatment, an IEL of 35 before treatment and 37 after treatment, and a Marsh score of 0.04 before treatment and 0.43 after treatment. It was determined that exposure to the peptide composition did not worsen the modified marsh score.
Example 6: Further Tolerance Assessments
[0126] The cohorts from Example 3 were analyzed using different ex vivo whole blood cytokine release assays to assess reactivity and tolerance to the peptide composition after treatment with the peptide composition or placebo. The subjects underwent a double-blind placebo-controlled gluten challenge pre- and post-treatment. To be reactive to the peptide composition, the subjects had to have an IFN-.gamma. ELISA after contact with the peptide composition of >7.2 pg/mL above negative control (not contacted with the peptide composition) and a stimulation index of >1.25 or a MAGPIX ratio of peptide composition to negative control of >2. To be tolerant to the peptide composition, the subjects had to have net IFN-.gamma. levels <7.2 pg/mL or stimulation index of <1.25 by ELISA and <2-fold elevation of IFN-.gamma., IL-2 or IP-10 by MAGPIX at visit 23. According to these criteria, it was found that 5 of 7 responsive subjects in Cohort 1 (150 micrograms) became non-reactive to the peptide composition (FIG. 13). It was found that 3 of 3 responsive subjects in Cohort 2 (300 micrograms) became non-reactive to the peptide composition (FIG. 14). It was found that 1 of 7 responsive subjects in Placebo Cohort (Cohorts 1 and 2 placebo) became non-reactive to the peptide composition (FIG. 15).
[0127] The cohorts were also analyzed using plasma cytokine levels. The plasma cytokine levels were measured at 37 time points pre- and post-first and last dose of peptide composition. For a subject to be tolerant to the peptide composition, they had to have less than a 2-fold increase after the last of peptide composition of IL-8 and MCP-1. It was found that 16 of 16 Cytokine Reactive Patients were tolerized to intradermal peptide composition, and peptide composition reactivity was abolished in 8 of 10 Patients treated with the peptide composition after oral gluten challenge. A summary of the change in tolerance/reactivity to the peptide composition is shown in Table 13.
TABLE-US-00013 TABLE 13 Tolerance/reactivity to the peptide composition Peptide composition Peptide composition reactive first dose reactive last dose Cytokines cohort 1, 2, 7 cohort 1, 2, 7 IL-8 & MCP-1 16/17 0/16 (plasma) (vs. 0/7 in placebo arms) (vs. 0/7 in placebo arms) Gluten Reactive Gluten Reactive Pre-treatment (V2/4) Post-treatment (V23) Cytokines Cohort 1, 2 Cohort 1, 2 IFN-.gamma./IL-2/IP-10 10/12 2/10 ex vivo release (vs. 7/7 in placebo arms) (vs. 6/7 in placebo arms) (6 days after oral challenge) Peptide composition reactive T-cells in vivo =>2 fold plasma cytokine increase Peptide composition reactive T-cells ex vivo =>7.2 pg/ml IFN-.gamma. & SI >1.25 OR >2-fold increase in IFN-.gamma., IP-10 or IL-2
EQUIVALENTS
[0128] While several inventive embodiments have been described and illustrated herein, those of ordinary skill in the art will readily envision a variety of other means and/or structures for performing the function and/or obtaining the results and/or one or more of the advantages described herein, and each of such variations and/or modifications is deemed to be within the scope of the inventive embodiments described herein. More generally, those skilled in the art will readily appreciate that all parameters, dimensions, materials, and configurations described herein are meant to be exemplary and that the actual parameters, dimensions, materials, and/or configurations will depend upon the specific application or applications for which the inventive teachings is/are used. Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents to the specific inventive embodiments described herein. It is, therefore, to be understood that the foregoing embodiments are presented by way of example only and that, within the scope of the appended claims and equivalents thereto, inventive embodiments may be practiced otherwise than as specifically described and claimed. Inventive embodiments of the present disclosure are directed to each individual feature, system, article, material, kit, and/or method described herein. In addition, any combination of two or more such features, systems, articles, materials, kits, and/or methods, if such features, systems, articles, materials, kits, and/or methods are not mutually inconsistent, is included within the inventive scope of the present disclosure.
[0129] All definitions, as defined and used herein, should be understood to control over dictionary definitions, definitions in documents incorporated by reference, and/or ordinary meanings of the defined terms.
[0130] All references, patents and patent applications disclosed herein are incorporated by reference with respect to the subject matter for which each is cited, which in some cases may encompass the entirety of the document.
[0131] The indefinite articles "a" and "an," as used herein in the specification and in the claims, unless clearly indicated to the contrary, should be understood to mean "at least one."
[0132] The phrase "and/or," as used herein in the specification and in the claims, should be understood to mean "either or both" of the elements so conjoined, i.e., elements that are conjunctively present in some cases and disjunctively present in other cases. Multiple elements listed with "and/or" should be construed in the same fashion, i.e., "one or more" of the elements so conjoined. Other elements may optionally be present other than the elements specifically identified by the "and/or" clause, whether related or unrelated to those elements specifically identified. Thus, as a non-limiting example, a reference to "A and/or B", when used in conjunction with open-ended language such as "comprising" can refer, in one embodiment, to A only (optionally including elements other than B); in another embodiment, to B only (optionally including elements other than A); in yet another embodiment, to both A and B (optionally including other elements); etc.
[0133] As used herein in the specification and in the claims, "or" should be understood to have the same meaning as "and/or" as defined above. For example, when separating items in a list, "or" or "and/or" shall be interpreted as being inclusive, i.e., the inclusion of at least one, but also including more than one, of a number or list of elements, and, optionally, additional unlisted items. Only terms clearly indicated to the contrary, such as "only one of" or "exactly one of," or, when used in the claims, "consisting of," will refer to the inclusion of exactly one element of a number or list of elements. In general, the term "or" as used herein shall only be interpreted as indicating exclusive alternatives (i.e. "one or the other but not both") when preceded by terms of exclusivity, such as "either," "one of," "only one of," or "exactly one of." "Consisting essentially of," when used in the claims, shall have its ordinary meaning as used in the field of patent law.
[0134] As used herein in the specification and in the claims, the phrase "at least one," in reference to a list of one or more elements, should be understood to mean at least one element selected from any one or more of the elements in the list of elements, but not necessarily including at least one of each and every element specifically listed within the list of elements and not excluding any combinations of elements in the list of elements. This definition also allows that elements may optionally be present other than the elements specifically identified within the list of elements to which the phrase "at least one" refers, whether related or unrelated to those elements specifically identified. Thus, as a non-limiting example, "at least one of A and B" (or, equivalently, "at least one of A or B," or, equivalently "at least one of A and: or B") can refer, in one embodiment, to at least one, optionally including more than one, A, with no B present (and optionally including elements other than B); in another embodiment, to at least one, optionally including more than one, B, with no A present (and optionally including elements other than A); in yet another embodiment, to at least one, optionally including more than one, A, and at least one, optionally including more than one, B (and optionally including other elements); etc.
[0135] It should also be understood that, unless clearly indicated to the contrary, in any methods claimed herein that include more than one step or act, the order of the steps or acts of the method is not necessarily limited to the order in which the steps or acts of the method are recited.
[0136] In the claims, as well as in the specification above, all transitional phrases such as "comprising," "including," "carrying," "having," "containing," "involving," "holding," "composed of," and the like are to be understood to be open-ended, i.e., to mean including but not limited to. Only the transitional phrases "consisting of" and "consisting essentially of" shall be closed or semi-closed transitional phrases, respectively, as set forth in the United States Patent Office Manual of Patent Examining Procedures, Section 2111.03.
Sequence CWU 1
1
9116PRTArtificial SequenceSynthetic polypeptide 1Glu Leu Gln Pro
Phe Pro Gln Pro Glu Leu Pro Tyr Pro Gln Pro Gln1 5 10
15215PRTArtificial SequenceSynthetic polypeptide 2Glu Gln Pro Phe
Pro Gln Pro Glu Gln Pro Phe Pro Trp Gln Pro1 5 10
15316PRTArtificial SequenceSynthetic polypeptide 3Glu Pro Glu Gln
Pro Ile Pro Glu Gln Pro Gln Pro Tyr Pro Gln Gln1 5 10
1549PRTArtificial SequenceSynthetic polypeptide 4Pro Phe Pro Gln
Pro Glu Leu Pro Tyr1 559PRTArtificial SequenceSynthetic polypeptide
5Pro Gln Pro Glu Leu Pro Tyr Pro Gln1 569PRTArtificial
SequenceSynthetic polypeptide 6Pro Phe Pro Gln Pro Glu Gln Pro Phe1
579PRTArtificial SequenceSynthetic polypeptide 7Pro Gln Pro Glu Gln
Pro Phe Pro Trp1 589PRTArtificial SequenceSynthetic polypeptide
8Glu Gln Pro Ile Pro Glu Gln Pro Gln1 599PRTArtificial
SequenceSynthetic polypeptide 9Pro Ile Pro Glu Gln Pro Gln Pro Tyr1
5
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
D00012
D00013

D00014

D00015
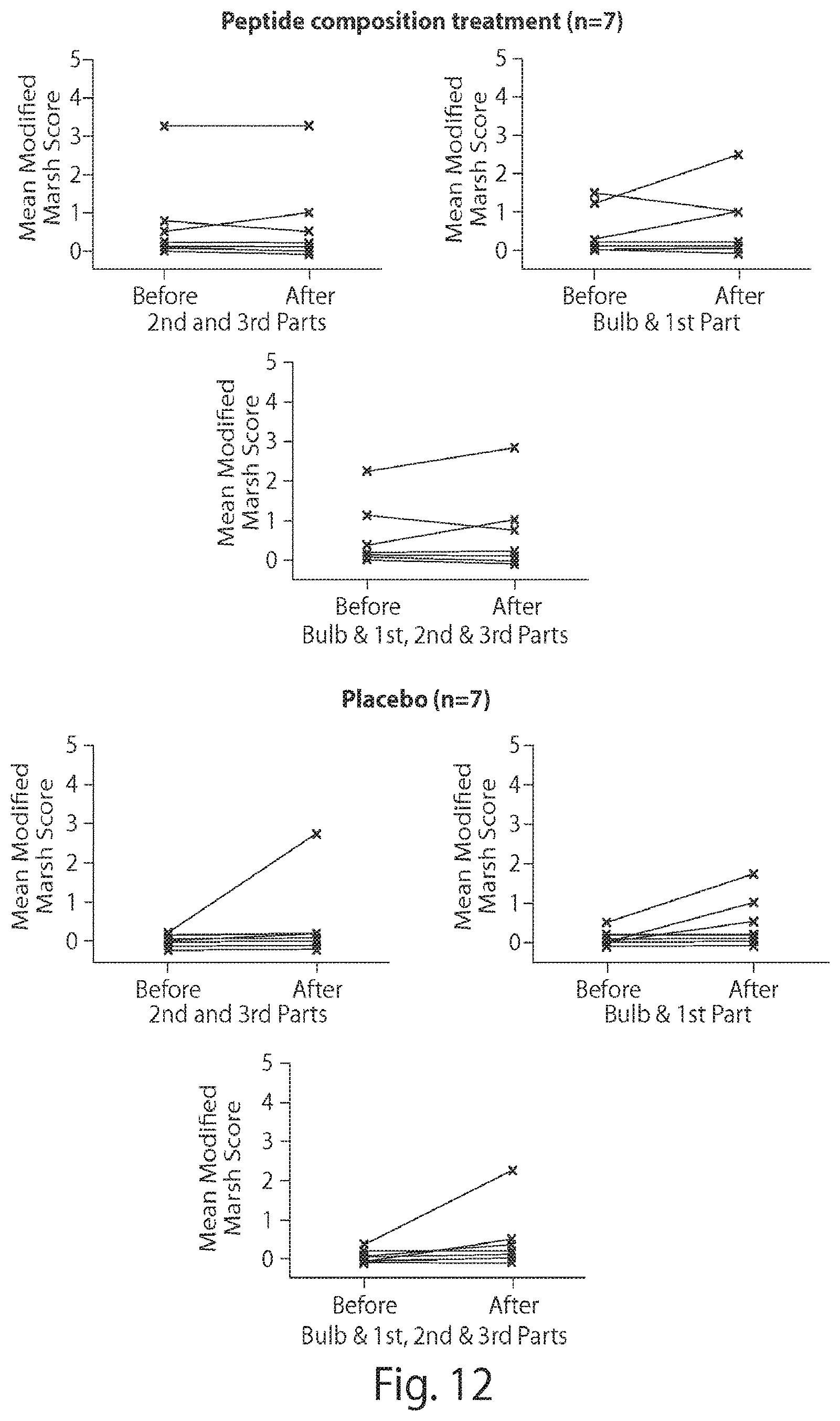
D00016

D00017
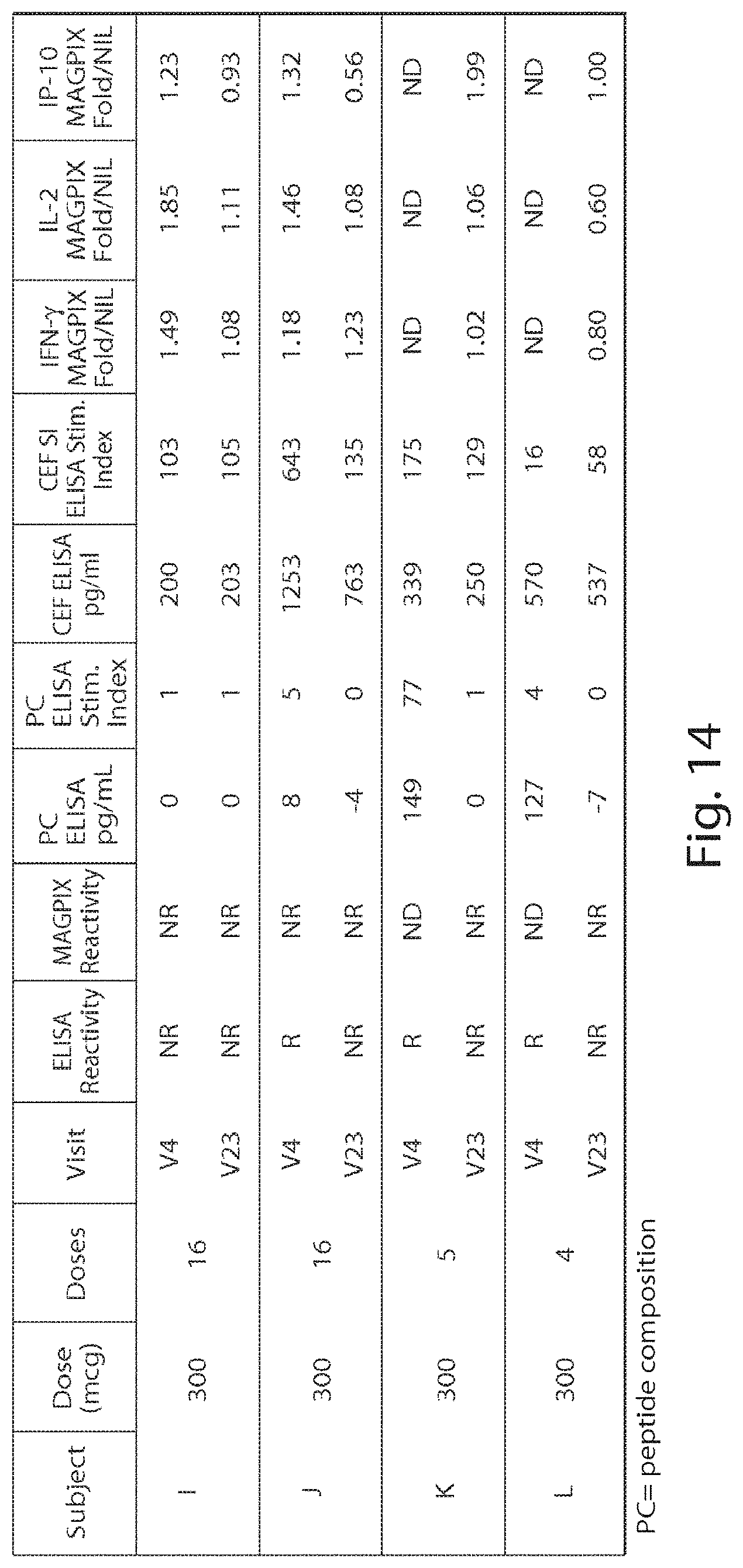
D00018

D00019

S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.