Treating Sickle Cell Disease With A Pyruvate Kinase R Activating Compound
Ericsson; Anna ; et al.
U.S. patent application number 16/576720 was filed with the patent office on 2020-04-30 for treating sickle cell disease with a pyruvate kinase r activating compound. The applicant listed for this patent is FORMA Therapeutics, Inc.. Invention is credited to Anna Ericsson, Neal Green, Gary Gustafson, David R. Lancia, JR., Gary Marshall, Lorna Mitchell, Madhu Mondal, Schroeder Patricia, Kelly J. Patrick, Maria Ribadeneira, David Richard, Forsyth Sanjeev, Zhongguo Wang.
| Application Number | 20200129485 16/576720 |
| Document ID | / |
| Family ID | 69774634 |
| Filed Date | 2020-04-30 |










View All Diagrams
| United States Patent Application | 20200129485 |
| Kind Code | A1 |
| Ericsson; Anna ; et al. | April 30, 2020 |
TREATING SICKLE CELL DISEASE WITH A PYRUVATE KINASE R ACTIVATING COMPOUND
Abstract
Compounds that activate pyruvate kinase R can be used for the treatment of sickle cell disease (SCD). Methods and compositions for the treatment of SCD are provided herein, including a therapeutic compound designated as Compound 1.
| Inventors: | Ericsson; Anna; (Shrewsbury, MA) ; Green; Neal; (Newton, MA) ; Gustafson; Gary; (Ridgefield, CT) ; Lancia, JR.; David R.; (Boston, MA) ; Marshall; Gary; (Watertown, MA) ; Mitchell; Lorna; (West Beach, AU) ; Richard; David; (Littleton, MA) ; Wang; Zhongguo; (Lexington, MA) ; Sanjeev; Forsyth; (Milton, MA) ; Patrick; Kelly J.; (Concord, MA) ; Mondal; Madhu; (Winchester, MA) ; Ribadeneira; Maria; (Cambridge, MA) ; Patricia; Schroeder; (Somerville, MA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 69774634 | ||||||||||
| Appl. No.: | 16/576720 | ||||||||||
| Filed: | September 19, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 62733558 | Sep 19, 2018 | |||
| 62733562 | Sep 19, 2018 | |||
| 62782933 | Dec 20, 2018 | |||
| 62789641 | Jan 8, 2019 | |||
| 62811904 | Feb 28, 2019 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61K 31/436 20130101; A61P 7/06 20180101; A61K 9/0053 20130101 |
| International Class: | A61K 31/436 20060101 A61K031/436; A61P 7/06 20060101 A61P007/06; A61K 9/00 20060101 A61K009/00 |
Claims
1. A method of reducing levels of 2,3-diphosphoglycerate (2,3-DPG) in a patient's red blood cells, the method comprising orally administering to the patient in need thereof a therapeutically effective amount of the compound (S)-1-(5-((2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-yl)sulfonyl)- -3,4,5,6-tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)-3-hydroxy-2-phenylpropan- -1 in a pharmaceutical composition each day.
2. The method of claim 1, comprising administering a total of 25 mg-1,500 mg of the compound (S)-1-(5-((2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-yl)sulfonyl)-3,4,5,6-- tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)-3-hydroxy-2-phenylpropan-1 to the patient per day.
3. The method of claim 1, wherein the compound is administered in a single dose each day.
4. The method of claim 1, wherein the compound is administered in a divided dose each day.
5. The method of claim 2, wherein the compound is administered in a single dose each day.
6. The method of claim 2, wherein the compound is administered in a divided dose each day.
7. A method of reducing levels of 2,3-diphosphoglycerate (2,3-DPG) in a patient's red blood cells, the method comprising administering to the patient in need thereof a therapeutically effective amount of Compound 1: ##STR00026## in a pharmaceutical composition comprising Compound 1 and a pharmaceutically acceptable carrier.
8. The method of claim 7, comprising administering a total of 25 mg-1,500 mg of the Compound 1 to the patient per day.
9. The method of claim 7, wherein the Compound 1 is administered in a single dose each day.
10. The method of claim 7, wherein the Compound 1 is administered in a divided dose each day.
11. The method of claim 8, wherein the Compound 1 is administered in a single dose each day.
12. The method of claim 8, wherein the Compound 1 is administered in a divided dose each day.
13. A method of reducing levels of 2,3-diphosphoglycerate (2,3-DPG) in a patient's red blood cells, the method comprising orally administering to the patient in need thereof a total of 25 mg-1,500 mg of the Compound 1 to the patient per day in a single or divided dose: ##STR00027## in a pharmaceutical composition comprising Compound 1 and a pharmaceutically acceptable carrier.
14. The method of claim 13, wherein the Compound 1 is administered in a single dose each day.
15. The method of claim 13, wherein the Compound 1 is administered in a divided dose each day.
Description
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of U.S. Provisional Application No. 62/733,558, filed on Sep. 19, 2018, U.S. Provisional Application No. 62/733,562, filed on Sep. 19, 2018, U.S. Provisional Application No. 62/782,933, filed on Dec. 20, 2018, U.S. Provisional Application No. 62/789,641, filed on Jan. 8, 2019, and U.S. Provisional Application No. 62/811,904, filed on Feb. 28, 2019, each of which is incorporated by reference in its entirety.
TECHNICAL FIELD
[0002] This disclosure relates to the treatment of sickle cell disease (SCD), including the treatment of patients diagnosed with SCD by the administration of a compound that activates pyruvate kinase R (PKR).
BACKGROUND
[0003] Sickle cell disease (SCD) is a chronic hemolytic anemia caused by inheritance of a mutated form of hemoglobin (Hgb), sickle Hgb (HgbS). It is the most common inherited hemolytic anemia, affecting 70,000 to 80,000 patients in the United States (US). SCD is characterized by polymerization of HgbS in red blood cells (RBCs) when HgbS is in the deoxygenated state (deoxy-HgbS), resulting in a sickle-shaped deformation. Sickled cells aggregate in capillaries precipitating vaso-occlusive events that generally present as acute and painful crises resulting in tissue ischemia, infarction, and long-term tissue damage. RBCs in patients with SCD tend to be fragile due to sickling and other factors, and the mechanical trauma of circulation causes hemolysis and chronic anemia. Finally, damaged RBCs have abnormal surfaces that adhere to and damage vascular endothelium, provoking a proliferative/inflammatory response that underlies large-vessel stroke and potentially pulmonary-artery hypertension. Collectively, these contribute to the significant morbidity and increased mortality associated with this disease.
[0004] Currently, therapeutic treatment of SCD is inadequate. The only known cure for SCD is hematopoietic stem cell transplantation which has serious risks, is typically recommended for only the most serious cases, and is largely offered only to children with sibling-matched donors. Gene therapy is also under investigation with promising preliminary results; however, there are market access hurdles, mainly high cost and treatment complexities, that are likely to limit its broad use in the near term. There have been few advances in therapies for SCD over the past two decades. Hydroxyurea (HU) induces HgbF which interrupts the polymerization of HgbS, and thereby has activity in decreasing the onset of vaso-occlusive crises and pathological sequelae of SCD. While HU is in wide use as a backbone therapy for SCD, it remains only partially effective, and is associated with toxicity, such as myelosuppression and teratogenicity. Patients receiving HU still experience hemolysis, anemia, and vaso-occlusive crises, suggesting a need for more effective therapies, either as a replacement or in combination with HU. Beyond HU, therapeutic intervention is largely supportive care, aimed at managing the symptoms of SCD. For instance, blood transfusions help with the anemia and other SCD complications by increasing the number of normal RBCs. However, repeated transfusions lead to iron overload and the need for chelation therapies to avoid consequent tissue damage. In addition to these approaches, analgesic medications are used to manage pain.
[0005] Given the current standard of care for SCD, there is a clear medical need for a noninvasive, disease-modifying therapy with appropriate safety and efficacy profiles.
SUMMARY
[0006] One aspect of the disclosure relates to methods of treating SCD comprising the administration of a therapeutically effective amount of a pyruvate kinase R (PKR) activator to a patient in need thereof diagnosed with SCD. Pyruvate kinase R (PKR) is the isoform of pyruvate kinase expressed in RBCs, and is a key enzyme in glycolysis. The invention is based in part on the discovery that the activation of PKR can target both sickling, by reducing deoxy-HgbS, and hemolysis. Targeting hemolysis may be achieved by improving RBC membrane integrity. One aspect of the disclosure is the recognition that activation of PKR can reduce 2,3-diphosphoglycerate (2,3-DPG), which leads to decreased deoxy-HgbS (and, therefore, sickling), as well as can increase ATP, which promotes membrane health and reduces hemolysis. Another aspect of the disclosure is the recognition that activation of PKR can reduce 2,3-diphosphoglycerate (2,3-DPG), which inhibits Hgb deoxygenation/increases oxygen affinity of HgbS and leads to decreased deoxy-HgbS (and, therefore, sickling), as well as can increase ATP, which promotes membrane health and reduces hemolysis. Accordingly, in one embodiment, PKR activation (e.g., by administration of a therapeutically effective amount of a PKR Activating Compound to a patient diagnosed with SCD) reduces RBC sickling via a reduction in levels of 2,3-diphosphoglycerate (2,3-DPG), which in turn reduces the polymerization of sickle Hgb (HgbS) into rigid aggregates that deform the cell. Furthermore, in some embodiments, PKR activation may contribute to overall RBC membrane integrity via increasing levels of adenosine triphosphate (ATP), which is predicted to reduce vaso-occlusive and hemolytic events which cause acute pain crises and anemia in SCD patients.
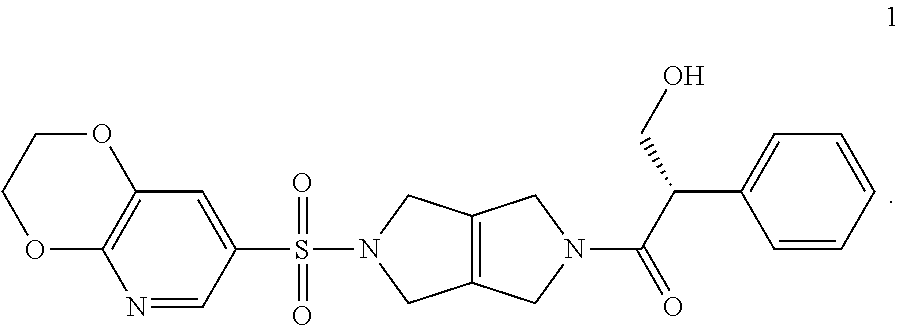
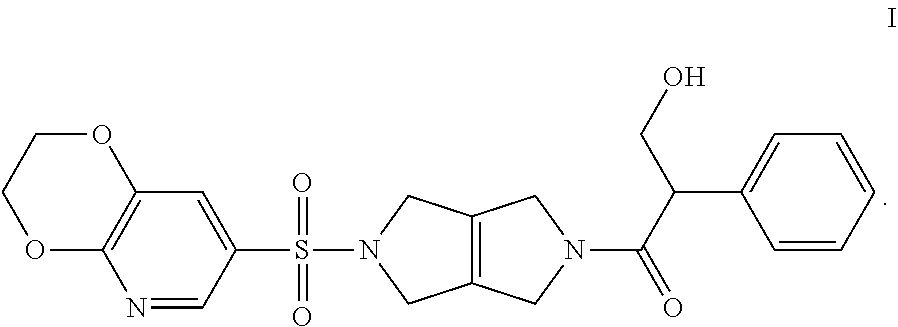
[0007] Preferably, a patient diagnosed with SCD is treated with a compound that is a PKR Activating Compound. The PKR activator can be a compound identified as a PKR Activating Compound or a composition identified as a PKR Activating Composition having an AC.sub.50 value of less than 1 M using the Luminescence Assay described in Example 2, or a pharmaceutically acceptable salt and/or other solid form thereof. For example, the PKR Activating Compound can be the compound (S)-1-(5-((2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-yl)sulfonyl)-3,4,5,6-- tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)-3-hydroxy-2-phenylpropan-1-one (Compound 1):
##STR00001##
or a pharmaceutically acceptable salt thereof. Compound 1 is a selective, orally bioavailable PKR Activating Compound that decreases 2,3-DPG, increases ATP, and has anti-sickling effects in disease models with a wide therapeutic margin relative to preclinical toxicity.
[0008] PKR Activating Compounds can be readily identified as compounds of Formula I:
##STR00002##
or a pharmaceutically acceptable salt thereof, (e.g., Compound 1 and mixtures of Compound 1 with its stereoisomer) having an AC.sub.50 value of less than 1 M using the Luminescence Assay described in Example 2.
[0009] In other embodiments, the PKR Activating Compound can be any of the compounds listed in FIG. 1, or a pharmaceutically acceptable salt thereof.
[0010] PKR Activating Compounds, such as 1-(5-((2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-yl)sulfonyl)-3,4,5,6-tetr- ahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)-3-hydroxy-2-phenylpropan-1-one, or a pharmaceutically acceptable salt thereof, are useful in pharmaceutical compositions for the treatment of patients diagnosed with SCD. PKR Activating Compounds, such as any of the compounds listed in FIG. 1, or a pharmaceutically acceptable salt thereof, are useful in pharmaceutical compositions for the treatment of patients diagnosed with SCD. The compositions comprising a compound of Formula I (e.g., Compound 1), or a pharmaceutically acceptable salt thereof, can be obtained by certain processes also provided herein. The compositions comprising any of the compounds listed in FIG. 1, or a pharmaceutically acceptable salt thereof, can be obtained by certain processes also provided herein.
[0011] The methods of treating SCD provided herein can offer greater protection against vaso-occlusive crises and hemolytic anemia, as compared to existing and emerging therapies. Therefore, use of a PKR Activating Compound, such as Compound 1, provides a novel and improved therapeutic approach either alone or in combination with drugs that act through alternative mechanisms, such as hydroxyurea (HU). In addition, use of a PKR Activating Compound, such as any of the compounds listed in FIG. 1, provides a novel and improved therapeutic approach either alone or in combination with drugs that act through alternative mechanisms, such as hydroxyurea (HU).
BRIEF DESCRIPTION OF THE DRAWINGS
[0012] FIG. 1 is a table of PKR Activating Compounds.
[0013] FIG. 2 is a schematic showing the relationship of PKR activation to the reduction of the clinical consequences of sickle cell disease (SCD).
[0014] FIG. 3 is a graph showing the oxyhemoglobin dissociation curve and modulating factors by plotting the relationship between hemoglobin saturation (percent) vs. partial pressure of oxygen (mmHg).
[0015] FIG. 4A is a chemical synthesis scheme for compounds of Formula I, including a synthesis of Compound 1 (separately provided in FIG. 4B).
[0016] FIG. 4B is a chemical synthesis scheme for Compound 1.
[0017] FIG. 4C is a general chemical synthesis of the compounds listed in FIG. 1.
[0018] FIG. 5 is a graph showing activation of recombinant PKR-R510Q with Compound 1, plotting the normalized rate vs. concentration of phosphoenolpyruvate (PEP) (Example 3).
[0019] FIG. 6 is a graph of data showing activation of recombinant PKR-R510Q by Compound 1 in the enzyme assay of Example 3.
[0020] FIG. 7 is a graph of data showing PKR activation in human red blood cells treated with Compound 1 (Example 4).
[0021] FIG. 8A (Study 1) and FIG. 8B (Study 2) are each graphs showing the observed changes in 2,3-DPG levels in blood from mice following 7 days of once daily (QD) oral treatment with Compound 1 (Example 5).
[0022] FIG. 9 is a graph showing observed changes in 2,3-DPG levels in blood from mice following 7 days of once daily (QD) oral treatment with Compound 1 (Example 5, Study 2).
[0023] FIG. 10A (Study 1) and FIG. 10B (Study 2) are graphs of data measuring ATP concentrations in red blood cells of mice following 7 days of once daily (QD) oral treatment with Compound 1 (Example 5).
[0024] FIG. 11 is a graph of the blood 2,3-DPG levels measured over time in healthy volunteers who received a single dose of Compound 1 or placebo.
[0025] FIG. 12 is a graph of the blood 2,3-DPG levels measured 24 hours post-dose in healthy volunteers who received a single dose of Compound 1 or placebo.
[0026] FIG. 13 is a graph of the blood 2,3-DPG levels measured over time in healthy volunteers who received daily doses of Compound 1 or placebo for 14 days.
[0027] FIG. 14 is a graph of the blood 2,3-DPG levels measured on day 14 in healthy volunteers who received daily doses of Compound 1 or placebo for 14 days.
[0028] FIG. 15 is a graph of the blood ATP levels measured on day 14 in healthy volunteers who received daily doses of Compound 1 or placebo for 14 days.
[0029] FIG. 16 is a graph plotting the blood concentration of Compound 1 (ng/mL) measured in healthy volunteer (HV) patients on a first (left) axis and the concentration of 2,3-DPG (micrograms/mL) measured in these HV patients on a second (right) axis after administration of a single dose of Compound 1 (400 mg).
DETAILED DESCRIPTION
[0030] Methods of treating SCD preferably include administration of a therapeutically effective amount of a compound (e.g., Compound 1) that reduces HgbS polymerization, for example by increasing HgbS affinity for oxygen. Methods of treating SCD also preferably include administration of a therapeutically effective amount of a compound (e.g., any of the compounds listed in FIG. 1) that reduces HgbS polymerization, for example by increasing HgbS affinity for oxygen. Methods of lowering 2,3-DPG and/or increasing ATP levels in human RBCs comprise administering a PKR Activating Compound, such as Compound 1. Methods of lowering 2,3-DPG and/or increasing ATP levels in human RBCs also comprise administering a PKR Activating Compound, such as any of the compounds listed in FIG. 1. Together these effects are consistent with providing therapies to reduce HgbS sickling and to improve RBC membrane health, presenting a unique disease-modifying mechanism for treating SCD.
[0031] A PKR Activating Compound, such as Compound 1, is useful to promote activity in the glycolytic pathway. A PKR Activating Compound, such as any of the compounds listed in FIG. 1, also is useful to promote activity in the glycolytic pathway. As the enzyme that catalyzes the last step of glycolysis, PKR directly impacts the metabolic health and primary functions of RBCs. PKR Activating Compounds (e.g., Compound 1), are useful to decrease 2,3-DPG and increase ATP. PKR Activating Compounds (e.g., any of the compounds listed in FIG. 1), are useful to decrease 2,3-DPG and increase ATP. PKR Activating Compounds (e.g., Compound 1 or any of the compounds listed in FIG. 1, preferably Compound 1) are also useful to increase Hgb oxygen affinity in RBC. The disclosure is based in part on the discovery that PKR activation is a therapeutic modality for SCD, whereby HgbS polymerization and RBC sickling are reduced via decreased 2,3-DPG and increased ATP levels.
[0032] SCD is the most common inherited blood disorder and clinically manifests with potentially severe pathological conditions associated with substantial physical, emotional, and economic burden. For instance, acute vaso-occlusive pain crises can be debilitating and necessitate rapid medical response. Chronic hemolytic anemia causes fatigue and often necessitates blood transfusions and supportive care. Over time, impaired oxygen transport through microvasculature precipitates organ and tissue damage. While there are a number of options available for treating symptoms, overall disease management would benefit from therapies that target upstream processes to prevent vaso-occlusion and hemolysis.
[0033] The described clinical symptoms are largely due to perturbations in RBC membrane shape and function resulting from aggregation of HgbS molecules. Unlike normal Hgb, HgbS polymerizes when it is in the deoxygenated state and ultimately causes a deformed, rigid membrane that is unable to pass through small blood vessels, thereby blocking normal blood flow through microvasculature. The loss of membrane elasticity also increases hemolysis, reducing RBC longevity. Furthermore, decreased cellular ATP and oxidative damage contribute to a sickle RBC membrane that is stiffer and weaker than that of normal RBCs. The damaged membrane has a greater propensity for adhering to vasculature, leading to hemolysis, increased aggregation of sickled RBCs, and increased coagulation and inflammation associated with vaso-occlusive crises.
[0034] The underlying cause of sickling is the formation of rigid deoxy-HgbS aggregates that alter the cell shape and consequently impact cellular physiology and membrane elasticity. These aggregates are highly structured polymers of deoxygenated HgbS; the oxygenated form does not polymerize. Polymerization is promoted by a subtle shift in conformation from the oxygen-bound relaxed (R)-state to the unbound tense (T)-state. In the latter, certain residues within the 0-chain of HgbS are able to interact in a specific and repetitive manner, facilitating polymerization.
[0035] The concentration of deoxy-HgbS depends on several factors, but the predominant factor is the partial pressure of oxygen (PO.sub.2). Oxygen reversibly binds to the heme portions of the Hgb molecule. As oxygenated blood flows via capillaries to peripheral tissues and organs that are actively consuming oxygen, PO.sub.2 drops and Hgb releases oxygen. The binding of oxygen to Hgb is cooperative and the relationship to PO.sub.2 levels fits a sigmoidal curve (FIG. 3). This relationship can be impacted by temperature, pH, carbon dioxide, and the glycolytic intermediate 2,3-DPG. 2,3-DPG binds within the central cavity of the Hgb tetramer, causes allosteric changes, and reduces Hgb's affinity for oxygen. Therefore, therapeutic approaches that increase oxygen affinity (i.e., reduce deoxygenation) of HgbS would presumably decrease polymer formation, changes to the cell membrane, and clinical consequences associated with SCD.
[0036] One aspect of this disclosure is targeting PKR activation to reduce 2,3-DPG levels, based on PKR's role in controlling the rate of glycolysis in RBCs. A decrease in 2,3-DPG with PKR activation has been demonstrated in preclinical studies and in healthy volunteers and patients with pyruvate kinase deficiency. Additionally, PKR activation would be expected to increase ATP, and has been observed to do so in these same studies. Given the role of ATP in the maintenance of a healthy RBC membrane and protection from oxidative stress, elevating its levels is likely to have broad beneficial effects. Therefore, activation of PKR offers the potential for a 2,3-DPG effect (i.e., reduced cell membrane damage from HgbS polymerization) that is augmented by ATP support for membrane integrity. It is via these changes that a PKR activator is could positively impact physiological changes that lead to the clinical pathologies of SCD (FIG. 2). In another aspect, the disclosure relates to a method of improving the anemia and the complications associated with anemia in SCD patients (e.g., .gtoreq.12 years of age) with Hgb SS or Hgb SB.sup.0-thalassemia.
[0037] Compound 1 is a selective, orally bioavailable PKR activator that has been shown to decrease 2,3-DPG, increase ATP, and have anti-sickling effects in disease models with a wide therapeutic margin relative to preclinical toxicity.
[0038] Methods of treatment can comprise administering to a subject in need thereof a therapeutically effective amount of (i) a PKR Activating Compound (e.g., a compound disclosed herein), or a pharmaceutically acceptable salt thereof; or (ii) a PKR Activating Composition (e.g., a pharmaceutical composition comprising a compound disclosed herein, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier). The pharmaceutical composition may be orally administered in any orally acceptable dosage form. In some embodiments, to increase the lifetime of red blood cells, a compound, composition, or pharmaceutical composition described herein is added directly to whole blood or packed cells extracorporeally or provided to the subject (e.g., the patient) directly. A patient and/or subject can be selected for treatment using a compound described herein by first evaluating the patient and/or subject to determine whether the subject is in need of activation of PKR, and if the subject is determined to be in need of activation of PKR, then administering to the subject a PKR Activating Compound in a pharmaceutically acceptable composition. A patient and/or subject can be selected for treatment using a compound described herein by first evaluating the patient and/or subject to determine whether the subject is diagnosed with SCD, and if the subject is diagnosed with SCD, then administering to the subject a PKR Activating Compound in a pharmaceutically acceptable composition. For example, administration of a therapeutically effective amount of a PKR Activating Compound can include administration of a total of about 25 mg-1,500 mg of Compound 1 each day, in single or divided doses. In some embodiments, Compound 1 is administered to patients diagnosed with SCD in total once daily (QD) doses of 25 mg, 50 mg, 75 mg, 100 mg, 125 mg, 150 mg, and/or higher if tolerated (e.g., 250 mg, 300 mg, 500 mg, 600 mg, 1000 mg, and/or 1500 mg). In some embodiments, a human dose of 80 to 130 mg of Compound 1 is administered once daily (QD) to a patient in need thereof (e.g., a patient diagnosed with SCD). In some embodiments, a PKR Activating Compound is administered in an amount of 400 mg per day (e.g., 400 mg QD or 200 mg BID). In some embodiments, Compound 1 or a pharmaceutically acceptable salt thereof is administered in an amount of 400 mg per day (e.g., 400 mg QD or 200 mg BID). In some embodiments, any of the compounds listed in FIG. 1 or a pharmaceutically acceptable salt thereof is administered in an amount of 400 mg per day (e.g., 400 mg QD or 200 mg BID). In some embodiments, a PKR Activating Compound is administered in an amount of 700 mg per day (e.g., 700 mg QD or 350 mg BID). In some embodiments, Compound 1 or a pharmaceutically acceptable salt thereof is administered in an amount of 700 mg per day (e.g., 700 mg QD or 350 mg BID). In some embodiments, any of the compounds listed in FIG. 1 or a pharmaceutically acceptable salt thereof is administered in an amount of 700 mg per day (e.g., 700 mg QD or 350 mg BID). In some embodiments, a PKR Activating Compound is administered in an amount of 100 mg, 200 mg, 400 mg, 600 mg, 700 mg, 1100 mg, or 1500 mg per day, in single or divided doses. In some embodiments, Compound 1 or a pharmaceutically acceptable salt thereof is administered in an amount of 100 mg, 200 mg, 400 mg, 600 mg, 700 mg, 1100 mg, or 1500 mg per day, in single or divided doses. In some embodiments, any of the compounds listed in FIG. 1 or a pharmaceutically acceptable salt thereof is administered in an amount of 100 mg, 200 mg, 400 mg, 600 mg, 700 mg, 1100 mg, or 1500 mg per day, in single or divided doses.
[0039] Methods of treating a patient diagnosed with SCD can include administering to the patient in need thereof a therapeutic compound targeting reduction of deoxy-HgbS, which may or may not directly improve RBC membrane integrity. Compound 1 has been shown to decrease 2,3-DPG and increase ATP, and reduced cell sickling has been demonstrated in disease models. Accordingly, in some embodiments, the methods of treatment can address not only sickling, but also hemolysis and anemia.
[0040] Methods of treating a patient diagnosed with sickle cell disease, and PKR Activating Compounds for use in such methods, can include administering to the patient the PKR Activating Compound (e.g., a composition comprising one or more compounds of Formula I, such as Compound 1 or a mixture of Compound 1 and Compound 2) in an amount sufficient to reduce 2,3-DPG levels in the patient's red blood cells. In some embodiments, the amount is sufficient to reduce 2,3-DPG levels by at least 30% after 24 hours, or greater (e.g., reducing 2,3-DPG levels in the patient's red blood cells by at least 40% after 24 hours). In some embodiments, the amount is sufficient to reduce 2,3-DPG levels by 30-50% after 24 hours. In some embodiments, the amount is sufficient to reduce 2,3-DPG levels by 40-50% after 24 hours. In some embodiments, the amount is sufficient to reduce 2,3-DPG levels by at least 25% after 12 hours. In some embodiments, the amount is sufficient to reduce 2,3-DPG levels by 25-45% after 12 hours. In some embodiments, the amount is sufficient to reduce 2,3-DPG levels by at least 15% after 6 hours. In some embodiments, the amount is sufficient to reduce 2,3-DPG levels by 15-30% after 6 hours. In some embodiments, the amount is sufficient to reduce 2,3-DPG levels by at least 40% on day 14 of treatment. In some embodiments, the amount is sufficient to reduce 2,3-DPG levels by 40-60% on day 14 of treatment. In some embodiments, the amount is sufficient to reduce 2,3-DPG levels by at least 50% on day 14 of treatment. In some embodiments, the amount is sufficient to reduce 2,3-DPG levels by 50-60% on day 14 of treatment.
[0041] Methods of treating a patient diagnosed with sickle cell disease, and PKR Activating Compounds for use in such methods, can also include administering to the patient the PKR Activating Compound (e.g., a composition comprising one or more compounds of Formula I, such as Compound 1 or a mixture of Compound 1 and Compound 2) in a daily amount sufficient to increase the patient's ATP blood levels. In some embodiments, the amount is sufficient to increase ATP blood levels by at least 40% on day 14 of treatment, or greater (e.g., t least 50% on day 14 of treatment). In some embodiments, the amount is sufficient to increase ATP blood levels by 40-65% on day 14 of treatment. In some embodiments, the amount is sufficient to increase ATP blood levels by at least 50% on day 14 of treatment, or greater (e.g., at least 50% on day 14 of treatment). In some embodiments, the amount is sufficient to increase ATP blood levels by 50-65% on day 14 of treatment.
[0042] In some examples, a pharmaceutical composition comprising Compound 1 can be used in a method of treating a patient diagnosed with sickle cell disease, the method comprising administering to the patient 400 mg of Compound 1 once per day (QD)
##STR00003##
In some examples, a pharmaceutical composition comprising Compound 1 can be used in a method of treating a patient diagnosed with sickle cell disease, the method comprising administering to the patient 200 mg of Compound 1 twice per day (BID)
##STR00004##
[0043] In some embodiments, the present disclosure provides PKR Activating Compounds of Formula I:
##STR00005##
or a pharmaceutically acceptable salt thereof. In some embodiments, a PKR Activating Compound is 1-(5-((2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-yl)sulfonyl)-3,4,5,6-tetr- ahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)-3-hydroxy-2-phenylpropan-1-one.
[0044] The compound of Formula I is preferably Compound 1:
##STR00006##
or a pharmaceutically acceptable salt thereof. In some embodiments, a compound of Formula I is (S)-1-(5-((2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-yl)sulfonyl)-3,4,5,6-- tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)-3-hydroxy-2-phenylpropan-1-one.
[0045] In some embodiments, the present disclosure provides a PKR Activating Compound that is any of the compounds listed in FIG. 1, or a pharmaceutically acceptable salt thereof.
[0046] The present disclosure also provides compositions (e.g. pharmaceutical compositions) comprising a compound of Formula I. In some embodiments, a provided composition containing a compound of Formula I comprises a mixture of Compound 1 and Compound 2:
##STR00007##
or a pharmaceutically acceptable salt thereof. The present disclosure also provides compositions (e.g. pharmaceutical compositions) comprising any of the compounds listed in FIG. 1, or a pharmaceutically acceptable salt thereof.
[0047] Pharmaceutical compositions comprising a PKR Activating Composition containing a compound of Formula (I) can be formulated for oral administration (e.g., as a capsule or tablet). For example, Compound 1 can be combined with suitable compendial excipients to form an oral unit dosage form, such as a capsule or tablet, containing a target dose of Compound 1. The drug product can be prepared by first manufacturing Compound 1 as an active pharmaceutical ingredient (API), followed by roller compaction/milling with intragranular excipients and blending with extra granular excipients. A Drug Product can contain the Compound 1 API and excipient components in Table 1 in a tablet in a desired dosage strength of Compound 1 (e.g., a 25 mg or 100 mg tablet formed from a Pharmaceutical Composition in Table 1). The blended material can be compressed to form tablets and then film coated.
[0048] The pharmaceutical composition preferably comprises about 30-70% by weight of (S)-1-(5-((2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-yl)sulfonyl- )-3,4,5,6-tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)-3-hydroxy-2-phenylpropa- n-1-one, and a pharmaceutically acceptable excipient in an oral dosage form.
TABLE-US-00001 TABLE 1 Exemplary Pharmaceutical Compositions of Compound 1 for Oral Administration % Formulation Function (weight) Examplary Component API 30-70% Compound 1 Filler 15-40% Microcrystalline Cellulose Dry binder 2-10% Crospovidone Kollidon CL Glidant 0.25-1.25% Colloidal Silicon Dioxide Lubricant 0.25-1.00% Magnesium Stearate, Hyqual
[0049] In some embodiments, a provided composition containing a compound of Formula I comprises a mixture of (S)-1-(5-((2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-yl)sulfonyl)-3,4,5,6-- tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)-3-hydroxy-2-phenylpropan-1-one and (R)-1-(5-((2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-yl)sulfonyl)-3,4,- 5,6-tetrahydropyrro[3,4-c]pyrrol-2(1H)-yl)-3-hydroxy-2-phenylpropan-1-one. In some embodiments, a provided composition containing a compound of Formula I is a mixture of Compound 1 and Compound 2 as part of a PKR Activating Composition. In some embodiments, a compound of Formula I is racemic. In some embodiments, a compound of Formula I consists of about 50% of Compound 1 and about 50% of Compound 2. In some embodiments, a compound of Formula I is not racemic. In some embodiments, a compound of Formula I does not consist of about 50% of Compound 1 and about 50% of Compound 2. In some embodiments, a compound of Formula I comprises about 99-95%, about 95-90%, about 90-80%, about 80-70%, or about 70-60% of Compound 1. In some embodiments, a compound of Formula I comprises about 99%, 98%, 95%, 90%, 80%, 70%, or 60% of Compound 1.
[0050] In some embodiments, a PKR Activating Composition comprises a mixture of Compound 1 and Compound 2. In some embodiments, a PKR Activating Composition comprises a mixture of Compound 1 and Compound 2, wherein the PKR Activating Composition comprises a therapeutically effective amount of Compound 1.
[0051] Compositions comprising a compound of Formula I can be prepared as shown in FIG. 4A and FIG. 4B. Compounds of Formula I can be obtained by the general chemical synthesis scheme of FIG. 4A. Compound 1 can be obtained by the chemical synthesis route of FIG. 4A or FIG. 4B. In brief, compounds of Formula I (FIG. 4A) and/or Compound 1 (FIG. 4B) can be obtained from a series of four reaction steps from commercially available starting materials. Commercially available 7-bromo-2H,3H-[1,4]dioxino[2,3-b]pyridine was treated with a mixture of n-butyl lithium and dibutylmagnesium followed by sulfuryl chloride to give sulfonyl chloride 3. Treatment of 3 with tert-butyl 1H,2H,3H,4H,5H,6H-pyrrolo[3,4-c]pyrrole-2-carboxylate in the presence of triethylamine (TEA) afforded Boc-protected monosulfonamide 4. Compound 4 was then de-protected in the presence of trifluoroacetic acid (TFA) to give 5, the free base of the monosulfonamide. The last step to generate Compound 1 (FIG. 4B) or Compound 1 and Compound 2 (FIG. 4A) was an amide coupling of 5 and tropic acid in the presence of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluoro-phosphate (HATU).
[0052] The compounds listed in FIG. 1 can be prepared as shown in FIG. 4C and as described in International Publication No. WO 2018/175474, published Sep. 27, 2018. Generally, the compounds listed in FIG. 1 can be prepared by acylation and sulfonylation of the secondary amine groups of hexahydropyrrolopyrrole 6. For example, sulfonylation of 6 with a suitable sulfonyl chloride 7 affords sulfonyl hexahydropyrrolopyrrole 8, which is then treated with a suitable carboxylic acid 9 in the presence of an amide coupling reagent (e.g., HATU) to afford compound 10 (Path 1). Alternatively, acylation of 6 with a suitable carboxylic acid 9 in the presence of an amide coupling reagent affords acyl hexahydropyrrolopyrrole 11, which is then treated with a suitable sulfonyl chloride 7 to afford compound 10 (Path 2). As a person of ordinary skill would understand, well-known protecting groups, functionalization reactions, and separation techniques can be used in conjunction with Paths 1 and 2 to obtain the specific compounds listed in FIG. 1.
[0053] Methods of treating SCD also include administration of a therapeutically effective amount of a bioactive compound (e.g., a small molecule, nucleic acid, or antibody or other therapy) that reduces HgbS polymerization, for example by increasing HgbS affinity for oxygen.
[0054] In other embodiments, the disclosure relates to each of the following numbered embodiments:
1. A composition comprising a PKR Activating Compound of Formula I, or a pharmaceutically acceptable salt thereof:
##STR00008##
2. The composition of embodiment 1, wherein the compound of Formula I is Compound 1, or a pharmaceutically acceptable salt thereof:
##STR00009##
3. The composition of embodiment 2, wherein the composition comprises a mixture of Compound 1 and Compound 2, or a pharmaceutically acceptable salt thereof:
##STR00010##
4. The composition of embodiment 1, comprising the compound: 1-(5-((2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-yl)sulfony)-3,4,5,6-tetra- hydrpyrrolo[3,4-c]pyrrol-2(1H)-yl)-3-hydroxy-2-phenylpropan-1-one. 5. The composition of any one of embodiments 1-4, formulated as an oral unit dosage form. 6. A method of treating a patient diagnosed with a sickle cell disease (SCD), the method comprising administering to the patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising (S)-1-(5-((2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-yl)sulfonyl)-3,4,5,6-- tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)-3-hydroxy-2-phenylpropan-1-one, or a pharmaceutically acceptable salt thereof. 7. The method of embodiment 6, wherein the method comprises oral administration of the pharmaceutical composition comprising (S)-1-(5-((2,3-dihydro-[1,4]dioxino[2,3-b]pyridin-7-yl)sulfonyl)-3,4,5,6-- tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)-3-hydroxy-2-phenylpropan-1-one, as the only PKR Activating Compound in the pharmaceutical composition. 8. A method of treating a patient diagnosed with a sickle cell disease (SCD), the method comprising administering to the patient in need thereof a therapeutically effective amount of a pharmaceutical composition comprising Compound 1:
##STR00011##
or a pharmaceutically acceptable salt thereof. 9. A composition comprising a compound of Formula I obtainable by a process comprising the step of converting compound 5 into a compound of Formula I in a reaction described as Step 4:
##STR00012##
10. The composition of embodiment 9, wherein the process further comprises first obtaining the compound 5 from a compound 4 by a process comprising Step 3:
##STR00013##
11. The composition of embodiment 10, wherein the process further comprises first obtaining the compound 4 from a compound 3 by a process comprising Step 2:
##STR00014##
12. The composition of embodiment 11, wherein the process further comprises first obtaining the compound 3 from a process comprising Step 1:
##STR00015##
13. A method of treating a patient diagnosed with sickle cell disease (SCD), the method comprising administering to the patient in need thereof a therapeutically effective amount of a PKR Activating Compound having an AC.sub.50 value of less than 1 M using the Luminescence Assay described in Example 2. 14. The method of embodiment 13, wherein the PKR Activating Compound is Compound 1. 15. The method of any one of embodiments 13-14, wherein the PKR Activating Compound is orally administered to the patient in need thereof.
16. The use of Compound 1:
##STR00016##
[0055] or a pharmaceutically acceptable salt thereof, for the treatment of patients diagnosed with sickle cell disease (SCD). 17. The use of a PKR Activating Compound having an AC.sub.50 value of less than 1 M using the Luminescence Assay described in Example 2, in the treatment of patients diagnosed with sickle cell disease. 18. The method of any one of embodiments 6-8 or 13-15, comprising the administration of Compound 1 once per day. 19. The method of any one of embodiments 6-8 or 13-15, comprising the administration of a total of 25 mg-1,500 mg of Compound 1 each day. 20. The method of any one of embodiments 18-19, comprising the administration of a total of 25 mg-130 mg of Compound 1 each day.
[0056] In other embodiments, the disclosure relates to each of the following numbered embodiments:
1. A method for reducing 2,3-diphosphoglycerate (2,3-DPG) levels in a patient's red blood cells, comprising administering to the patient a PKR Activating Compound in a therapeutically effective amount, wherein the PKR Activating Compound is a compound of Formula I:
##STR00017##
or a pharmaceutically acceptable salt thereof, having an AC.sub.50 value of less than 1 M using the Luminescence Assay described in Example 2. 2. The method of embodiment 1, wherein the PKR Activating Compound is Compound 1:
##STR00018##
or a pharmaceutically acceptable salt thereof. 3. The method of embodiment 1, wherein the PKR Activating Compound is Compound 1:
##STR00019##
4. The method of embodiment 3, wherein the PKR Activating Compound is administered in an amount of 25-1500 mg per day. 5. The method of embodiment 3, wherein the PKR Activating Compound is administered once daily in an amount of 250 mg, 300 mg, 500 mg, 600 mg, 1000 mg, or 1500 mg per day. 6. The method of embodiment 3, wherein the PKR Activating Compound is administered once daily in an amount of 100 mg per day. 7. The method of embodiment 3, wherein the PKR Activating Compound is administered once daily in an amount of 600 mg per day. 8. The method of embodiment 3, wherein the PKR Activating Compound is administered once per day. 9. The method of embodiment 3, wherein the PKR Activating Compound is orally administered to the patient. 10. The method of embodiment 3, wherein Compound 1 is the only PKR Activating Compound administered to the patient.
[0057] In other embodiments, the disclosure relates to each of the following numbered embodiments:
1. A method for reducing 2,3-diphosphoglycerate (2,3-DPG) levels in a patient's red blood cells, comprising administering to the patient the PKR Activating Compound in an amount sufficient to reduce 2,3-DPG levels in the patient's red blood cells by at least 30% after 24 hours, wherein the PKR Activating Compound is a compound of Formula I:
##STR00020##
or a pharmaceutically acceptable salt thereof, having an AC.sub.50 value of less than 1 M using the Luminescence Assay described in Example 2. 2. The method of embodiment 1, wherein the PKR Activating Compound is Compound 1:
##STR00021##
or a pharmaceutically acceptable salt thereof. 3. The method of embodiment 1, wherein the PKR Activating Compound is Compound 1:
##STR00022##
4. The method of embodiment 1, wherein Compound 1 is the only PKR Activating Compound administered to the patient. 5. The method of any one of embodiments 1-4, wherein the PKR Activating Compound is orally administered to the patient. 6. The method of any one of embodiments 1-5, wherein the PKR Activating Compound is administered once per day. 7. The method of any one of embodiments 1-6, wherein the PKR Activating Compound is administered in an amount sufficient to reduce 2,3-DPG levels in the patient's red blood cells by at least 40% after 24 hours. 8. The method of any one of embodiments 1-7, wherein the PKR Activating Compound is administered in a daily amount sufficient to increase the patient's ATP blood levels by at least 40% on day 14 of treatment. 9. The method of any one of embodiments 1-5, wherein the PKR Activating Compound is administered in an amount of 100 mg, 200 mg, 400 mg, 600 mg, 700 mg, 1100 mg, or 1500 mg per day. 10. The method of any one of embodiments 1-5, wherein the PKR Activating Compound is administered in an amount of 200 mg per day. 11. The method of embodiment 10, wherein the PKR Activating Compound is administered in an amount of 200 mg per day once per day (QD). 12. The method of embodiment 10, wherein the PKR Activating Compound is administered in an amount of 100 mg per day twice per day (BID). 13. The method of any one of embodiments 1-5, wherein the PKR Activating Compound is administered in an amount of 400 mg per day. 14. The method of embodiment 13, wherein the PKR Activating Compound is administered in an amount of 400 mg once per day (QD). 15. The method of embodiment 13, wherein the PKR Activating Compound is administered in an amount of 200 mg twice per day (BID). 16. The method of any one of embodiments 1-5, wherein the PKR Activating Compound is administered in an amount of 600 mg per day. 17. The method of embodiment 16, wherein the PKR Activating Compound is administered in an amount of 300 mg twice per day (BID). 18. The method of any one of embodiments 1-5, wherein the PKR Activating Compound is administered in an amount of 700 mg per day. 19. The method of embodiment 18, wherein the PKR Activating Compound is administered in an amount of 700 mg once per day (QD). 30. The method of embodiment 18, wherein the PKR Activating Compound is administered in an amount of 350 mg twice per day (BID).
[0058] The present disclosure enables one of skill in the relevant art to make and use the inventions provided herein in accordance with multiple and varied embodiments. Various alterations, modifications, and improvements of the present disclosure that readily occur to those skilled in the art, including certain alterations, modifications, substitutions, and improvements are also part of this disclosure. Accordingly, the foregoing description and drawings are by way of example to illustrate the discoveries provided herein.
EXAMPLES
[0059] As the enzyme that catalyzes the last step of glycolysis, PKR underlies reactions that directly impact the metabolic health and primary functions of RBCs. The following Examples demonstrate how PKR activation by Compound 1 impacts RBCs. The primary effect of Compound 1 on RBCs is a decrease in 2,3-DPG that is proposed to reduce Hgb sickling and its consequences on RBCs and oxygen delivery to tissues. Compound 1 also increases ATP, which may provide metabolic resources to support cell membrane integrity and protect against loss of deformability and increased levels of hemolysis in SCD. With the combination of effects Compound 1 has on RBCs, it is likely to reduce the clinical sequelae of sickle Hgb and provide therapeutic benefits for patients with SCD.
[0060] The PKR Activating Compound designated Compound 1 was prepared as described in Example 1, and tested for PKR activating activity in the biochemical assay of Example 2.
[0061] The biological enzymatic activity of PKR (i.e., formation of ATP and/or pyruvate) was evaluated in enzyme and cell assays with Compound 1, as described in Example 3 and Example 4, respectively. Results from enzyme assays show that Compound 1 is an activator of recombinant wt-PKR and mutant PKR, (e.g., R510Q), which is one of the most prevalent PKR mutations in North America. PKR exists in both a dimeric and tetrameric state, but functions most efficiently as a tetramer. Compound 1 is an allosteric activator of PKR and is shown to stabilize the tetrameric form of PKR, thereby lowering the K.sub.m (the Michaelis-Menten constant) for PEP.
[0062] In vivo testing in mice (Examples 5) demonstrated PKR activation in wt mice, and provided an evaluation of effects on RBCs and Hgb in a murine model of SCD. Compound 1 was well tolerated up to the highest dose tested, and exposures increased in a dose-proportional manner. Levels of 2,3-DPG were reduced by >30% for doses 2120 mg/kg Compound 1 (AUC from 0 to 24 hours (AUC.sub.0-24>5200 hrng/mL) and levels of ATP were increased by >40% for 260 mg/kg Compound 1 (AUC.sub.0-24>4000 hrng/mL).
[0063] In some embodiments, a daily dose of between 100 mg to 1500 mg of a PKR Activating Compound is administered to humans. In some embodiments, a daily dose of between 100 mg to 1500 mg of Compound 1 is administered to humans. In some embodiments, a daily dose of between 100 mg to 1500 mg of any of the compounds listed in FIG. 1 is administered to humans. In particular, a total daily dose of 100 mg-600 mg of a PKR Activating Compound can be administered to humans (including, e.g., a dose of 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, or 600 mg, per day, in single or divided doses). In particular, a total daily dose of 100 mg-600 mg of Compound 1 can be administered to humans (including, e.g., a dose of 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, or 600 mg, per day, in single or divided doses). In particular, a total daily dose of 100 mg-600 mg of any of the compounds listed in FIG. 1 can be administered to humans (including, e.g., a dose of 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, or 600 mg, per day, in single or divided doses). In some embodiments, a daily dose of 400 mg (e.g., 400 mg QD or 200 mg BID) of a PKR Activating Compound is administered to humans. In some embodiments, a daily dose of 400 mg (e.g., 400 mg QD or 200 mg BID) of Compound 1, or a pharmaceutically acceptable salt thereof, is administered to humans. In some embodiments, a daily dose of 400 mg (e.g., 400 mg QD or 200 mg BID) of any of the compounds listed in FIG. 1 is administered to humans.
Example 1: Synthesis of Compounds of Formula I
[0064] The PKR Activating Compound 1 was obtained by the method described herein and the reaction scheme shown in FIG. 4A and/or FIG. 4B. Compound 1 has a molecular weight of 457.50 Da.
Step 1. 2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl chloride (3)
[0065] Into a 100 mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen was placed a solution of n-BuLi in hexane (2.5 M, 2 mL, 5.0 mmol, 0.54 equiv) and a solution of n-Bu.sub.2Mg in heptanes (1.0 M, 4.8 mL, 4.8 mmol, 0.53 equiv). The resulting solution was stirred for 10 min at RT (20.degree. C.). This was followed by the dropwise addition of a solution of 7-bromo-2H,3H-[1,4]dioxino[2,3-b]pyridine (2 g, 9.26 mmol, 1.00 equiv) in tetrahydrofuran (16 mL) with stirring at -10.degree. C. in 10 min. The resulting mixture was stirred for 1 h at -10.degree. C. The reaction mixture was slowly added to a solution of sulfuryl chloride (16 mL) at -10.degree. C. The resulting mixture was stirred for 0.5 h at -10.degree. C. The reaction was then quenched by the careful addition of 30 mL of saturated ammonium chloride solution at 0.degree. C. The resulting mixture was extracted with 3.times.50 mL of dichloromethane. The organic layers were combined, dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. The residue was purified by silica gel column chromatography, eluting with ethyl acetate/petroleum ether (1:3). This provided 1.3 g (60%) of 2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl chloride as a white solid. LCMS m/z: calculated for C.sub.7H.sub.6ClNO.sub.4S: 235.64; found: 236 [M+H].sup.+.
Step 2. tert-Butyl 5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H-pyrrol- o[3,4-c]pyrrole-2-carboxylate (4)
[0066] Into a 100-mL round-bottom flask was placed 2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl chloride (1.3 g, 5.52 mmol, 1.00 equiv), tert-butyl 1H,2H,3H,4H,5H,6H-pyrrolo[3,4-c]pyrrole-2-carboxylate (1.16 g, 5.52 mmol), dichloromethane (40 mL), and triethylamine (1.39 g, 13.74 mmol, 2.49 equiv). The solution was stirred for 2 h at 20.degree. C., then diluted with 40 mL of water. The resulting mixture was extracted with 3.times.30 mL of dichloromethane. The organic layers were combined, dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. The residue was purified by silica gel column chromatography, eluting with dichloromethane/methanol (10:1). This provided 1.2 g (53%) of tert-butyl 5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H-pyrrol- o[3,4-c]pyrrole-2-carboxylate as a yellow solid. LCMS m/z: calculated for C.sub.18H.sub.23N.sub.3O.sub.6S: 409.46; found: 410 [M+H].sup.+.
Step 3. 2-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H- -pyrrolo[3,4-c]pyrrole (5)
[0067] Into a 100-mL round-bottom flask was placed tert-butyl 5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H-pyrrol- o[3,4-c]pyrrole-2-carboxylate (1.2 g, 2.93 mmol, 1.00 equiv), dichloromethane (30 mL), and trifluoroacetic acid (6 mL). The solution was stirred for 1 h at 20.degree. C. The resulting mixture was concentrated under vacuum. The residue was dissolved in 10 mL of methanol and the pH was adjusted to 8 with sodium bicarbonate (2 mol/L). The resulting solution was extracted with 3.times.10 mL of dichloromethane. The organic layers were combined, dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. The crude product was purified by silica gel column chromatography, eluting with dichloromethane/methanol (10:1). This provided 650 mg (72%) of 2-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H-pyrrol- o[3,4-c]pyrrole as a yellow solid. LCMS m/z: calculated for C.sub.13H.sub.15N.sub.3O.sub.4S: 309.34; found: 310 [M+H].sup.+.
Step 4. (S)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4- H,5H,6H-pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one (1) and (R)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H- -pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one (2)
[0068] Into a 100 mL round-bottom flask was placed 2-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H-pyrrol- o[3,4-c]pyrrole (150 mg, 0.48 mmol, 1.00 equiv), 3-hydroxy-2-phenylpropanoic acid (97 mg, 0.58 mmol, 1.20 equiv), dichloromethane (10 mL), HATU (369 mg, 0.97 mmol, 2.00 equiv) and DIEA (188 mg, 1.46 mmol, 3.00 equiv). The resulting solution was stirred overnight at 20.degree. C. The reaction mixture was diluted with 20 mL of water and was then extracted with 3.times.20 mL of dichloromethane. The organic layers were combined, dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. The residue was purified by prep-TLC eluted with dichloromethane/methanol (20:1) and further purified by prep-HPLC (Column: XBridge C18 OBD Prep Column, 100 .ANG., 5 m, 19 mm.times.250 mm; Mobile Phase A: water (10 mmol/L NH.sub.4HCO.sub.3), Mobile Phase B: MeCN; Gradient: 15% B to 45% B over 8 min; Flow rate: 20 mL/min; UV Detector: 254 nm). The two enantiomers were separated by prep-Chiral HPLC (Column, Daicel CHIRALPAK.RTM. IF, 2.0 cm.times.25 cm, 5 m; mobile phase A: DCM, phase B: MeOH (hold 60% MeOH over 15 min); Flow rate: 16 mL/min; Detector, UV 254 & 220 nm). This resulted in peak 1 (2, Rt: 8.47 min) 9.0 mg (4%) of (R)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H- -pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one as a yellow solid; and peak 2 (1, Rt: 11.83 min) 10.6 mg (5%) of (S)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6H- -pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one as a yellow solid.
[0069] (1): .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 8.13 (d, J=2.0 Hz, 1H), 7.61 (d, J=2.0 Hz, 1H), 7.31-7.20 (m, 5H), 4.75 (t, J=5.2 Hz, 1H), 4.50-4.47 (m, 2H), 4.40-4.36 (m, 1H), 4.32-4.29 (m, 2H), 4.11-3.87 (m, 8H), 3.80-3.77 (m, 1H), 3.44-3.41 (m, 1H). LC-MS (ESI) m/z: calculated for C.sub.22H.sub.23N.sub.3O.sub.6S: 457.13; found: 458.0 [M+H].sup.+.
[0070] (2): .sup.1H NMR (400 MHz, DMSO-d.sub.6) .delta. 8.13 (d, J=2.0 Hz, 1H), 7.60 (d, J=2.0 Hz, 1H), 7.31-7.18 (m, 5H), 4.75 (t, J=5.2 Hz, 1H), 4.52-4.45 (m, 2H), 4.40-4.36 (m, 1H), 4.34-4.26 (m, 2H), 4.11-3.87 (m, 8H), 3.80-3.78 (m, 1H), 3.44-3.43 (m, 1H). LC-MS (ESI) m/z: calculated for C.sub.22H.sub.23N.sub.3O.sub.6S: 457.13; found: 458.0 [M+H].sup.+.
Step 5. (S)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4- H,5H,6H-pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one (1)
[0071] Alternatively, Compound 1 can be synthesized using the procedure described here as Step 5. A solution of 7-((3,4,5,6-tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)sulfonyl)-2,3-dihydro- -[1,4]dioxino[2,3-b]pyridine (130.9 mg, 0.423 mmol) in DMF (2.5 ml) was cooled on an ice bath, then treated with (S)-3-hydroxy-2-phenylpropanoic acid (84.8 mg, 0.510 mmol), HATU (195.5 mg, 0.514 mmol), and DIEA (0.30 mL, 1.718 mmol) and stirred at ambient temperature overnight. The solution was diluted with EtOAc (20 mL), washed sequentially with water (20 mL) and brine (2.times.20 mL), dried (MgSO.sub.4), filtered, treated with silica gel, and evaporated under reduced pressure. The material was chromatographed by Biotage MPLC (10 g silica gel column, 0 to 5% MeOH in DCM) to provide a white, slightly sticky solid. The sample was readsorbed onto silica gel and chromatographed (10 g silica gel column, 0 to 100% EtOAc in hexanes) to provide (2S)-1-(5-[2H,3H-[1,4]dioxino[2,3-b]pyridine-7-sulfonyl]-1H,2H,3H,4H,5H,6- H-pyrrolo[3,4-c]pyrrol-2-yl)-3-hydroxy-2-phenylpropan-1-one (106.5 mg, 0.233 mmol, 55% yield) as a white solid.
Example 2: Biochemical Assay for Identification of PKR Activating Activity
[0072] PKR Activating Compounds can be identified with the biochemical Luminescence Assay of Example 2. The PKR activating activity of a series of chemical compounds was evaluated using the Luminescence Assay below, including compounds designated Compound 1, Compound 2, and Compounds 6, 7, and 8 below, and the compounds listed in FIG. 1.
[0073] For each tested compound, the ability to activate PKR was determined using the following Luminescence Assay. The effect of phosphorylation of adenosine-5'-diphosphate (ADP) by PKR is determined by the Kinase Glo Plus Assay (Promega) in the presence or absence of FBP (D-fructose-1,6-diphosphate; BOC Sciences, CAS: 81028-91-3) as follows. Unless otherwise indicated, all reagents are purchased from Sigma-Aldrich. All reagents are prepared in buffer containing 50 mM Tris-HCl, 100 mM KCl, 5 mM MgCl.sub.2, and 0.01% Triton X100, 0.03% BSA, and 1 mM DTT. Enzyme and PEP (phosphoenolpyruvate) are added at 2.times. to all wells of an assay-ready plate containing serial dilutions of test compounds or DMSO vehicle. Final enzyme concentrations for PKR(wt), PKR(R510Q), and PKR(G332S) are 0.8 nM, 0.8 nM, and 10 nM respectively. Final PEP concentration is 100 M. The Enzyme/PEP mixture is incubated with compounds for 30 minutes at RT before the assay is initiated with the addition of 2.times.ADP and KinaseGloPlus. Final concentration of ADP is 100 M. Final concentration of KinaseGloPlus is 12.5%. For assays containing FBP, that reagent is added at 30 .mu.M upon reaction initiation. Reactions are allowed to progress for 45 minutes at RT until luminescence is recorded by the BMG PHERAstar FS Multilabel Reader. The compound is tested in triplicate at concentrations ranging from 42.5 .mu.M to 2.2 nM in 0.83% DMSO. AC.sub.50 measurements were obtained by the standard four parameter fit algorithm of ActivityBase XE Runner (max, min, slope and AC.sub.50). The AC.sub.50 value for a compound is the concentration (.mu.M) at which the activity along the four parameter logistic curve fit is halfway between minimum and maximum activity.
[0074] As set forth in Tables 2 and 3 below and in FIG. 1, AC.sub.50 values are defined as follows: .ltoreq.0.1 .mu.M (+++); >0.1 .mu.M and .ltoreq.1.0 .mu.M (++); >1.0 .mu.M and .ltoreq.40 .mu.M (+); >40 .mu.M (0).
TABLE-US-00002 TABLE 2 Luminescence Assay Data AC.sub.50 AC.sub.50 AC.sub.50 Compound (PKRG332S) (PKRR510Q) (WT) 1 ++ +++ +++ 2 + + +
TABLE-US-00003 TABLE 3 Additional Luminescence Assay Data AC.sub.50 AC.sub.50 Compound Structure (PKRG332S) (PKRR510Q) 6 ##STR00023## ++ + 7 ##STR00024## 0 0 8 ##STR00025## 0 0
[0075] Compounds and compositions described herein are activators of wild type PKR and certain PKR mutants having lower activities compared to the wild type. Such mutations in PKR can affect enzyme activity (catalytic efficiency), regulatory properties, and/or thermostability of the enzyme. One example of a PKR mutation is G332S. Another example of a PKR mutation is R510Q.
Example 3: Enzyme Assays of a PKR Activating Compound
[0076] The effect of 2 .mu.M Compound 1 on maximum velocity (V.sub.max) and PEP K.sub.m (Michaelis-Menten constant, i.e., the concentration of PEP at which v=1/2v.sub.max) was evaluated for wt-PKR and PKR-R510Q. Tests were conducted in the presence and absence of fructose-1,6-bisphosphate (FBP), a known allosteric activator of PKR. Assessments were made up to 60 min at RT, and V.sub.max and PEP K.sub.m were calculated. The effect of Compound 1 on V.sub.max ranged from no effect to a modest increase (see FIG. 5 for a representative curve). Compound 1 consistently reduced the PEP K.sub.m, typically by .about.2 fold, for wt-PKR and PKR-R510Q in the presence or absence of FBP (Table 4), demonstrating that Compound 1 can enhance the rate of PKR at physiological concentrations of PEP.
TABLE-US-00004 TABLE 4 Effect of Compound 1 on PKR Enzyme Kinetic Parameters No FBP 30 .mu.M FBP Kinetic 2 .mu.M 2 .mu.M Enzyme Parameter.sup.a DMSO Compound 1 DMSO Compound 1 WT- V.sub.max 1.00 1.14 1.19 1.16 PKR PEP K.sub.m 4.84 2.44 1.98 1.00 PKR V.sub.max 1.54 1.56 1.00 1.29 R510Q PEP K.sub.m 6.20 1.70 2.01 1.00 .sup.aAll values in Table 4 are normalized to 1.00, relative to the other values in the same row.
[0077] Activation of wt-PKR and PKR-R510Q by different concentrations of Compound 1 was evaluated for PEP concentrations at or below K.sub.m. Compound 1 increased the rate of ATP formation, with AC.sub.50 values ranging from <0.05 to <0.10 .mu.M and a range of <2.0 to <3.0 maximum-fold activation (i.e., <200% to <300%) (Table 5). Representative data from PKR-R510Q showed that the effect was concentration dependent (FIG. 6).
TABLE-US-00005 TABLE 5 Activation of PKR Wild and Mutant Types by Compound 1 PK Enzyme Maximum-fold Activation AC.sub.50 (.mu.M) WT-PKR <2.0 <0.05 PKR R510Q <3.0 <0.10
Example 4: Cell Assays of a PKR Activating Compound
[0078] The activation of wt-PKR by Compound 1 in mature human erythrocytes ex vivo was evaluated in purified RBCs purchased from Research Blood Components. Cells treated with Compound 1 for 3 hr in glucose-containing media were washed, lysed, and assayed using a Biovision Pyruvate Kinase Assay (K709-100). The assay was repeated multiple times to account for donor-to-donor variability and the relatively narrow dynamic range. Mean maximum activation increase (Max-Min) was <100% and mean 50% effective concentration (EC.sub.50) was <125 nM (Table 6). wt-PKR was activated in a concentration-dependent manner (FIG. 7).
TABLE-US-00006 TABLE 6 Wild Type PKR Activation in Human Red Blood Cells Treated with Compound 1 Replicate Max-Min (%) EC.sub.50 (nM) 1 <125 <250 2 <150 <150 3 <100 <50 4 <50 <50 Mean <100 <125
[0079] Mouse RBCs were isolated fresh from whole blood using a Ficoll gradient and assayed with methods similar to those used in the human RBCs assays. Maximum activation increase, and EC.sub.50 values were comparable to the effects in human RBCs (Table 7).
TABLE-US-00007 TABLE 7 Effect of Compound 1 on PKR Activation in Mouse Red Blood Cells Replicate Max-Min (%) EC.sub.50 (nM) 1 <50 <125 2 <100 <125 Mean <100 <125
Example 5: Pharmacokinetic/Pharmacodynamic Studies of Compound 1 in Wild Type Mice
[0080] Two pharmacokinetic (PK)/phamacodynamic (PD) studies were conducted in Balb/c mice that were administered Compound 1 once daily by oral gavage (formulated in 10% Cremophor EL/10% PG/80% DI water) for 7 days (QD.times.7) at doses of 0 (vehicle), 3.75, 7.5, 15, 30, 60 mg/kg (Study 1); 0 (vehicle), 7.5, 15, 30, 60, 120, or 240 mg/kg (Study 2). On the 7th day, whole blood was collected 24 hours after dosing and snap frozen. Samples were later thawed and analyzed by LC/MS for 2,3-DPG and ATP levels. In both studies, Compound 1 was well tolerated. No adverse clinical signs were observed and there were no differences in body weight change compared with the vehicle group.
[0081] The levels of 2,3-DPG decreased with Compound 1 treatment (FIGS. 8A and 8B (Studies 1 and 2) and FIG. 9 (Study 2)). In general, reductions were >20% at .gtoreq.15 mg/kg Compound 1, and >30% for 120 and 240 mg/kg Compound 1. Together, the results from the highest doses provide in vivo evidence that 2,3-DPG decreases with PKR activation.
[0082] Evaluation of ATP levels in these studies showed that treatment with Compound 1 increased levels of ATP. In Study 1, ATP increased 21% and 79% with 30 and 60 mg/kg Compound 1, respectively, compared to vehicle, and in Study 2, ATP levels increased with exposure with doses up to 120 mg/kg Compound 1 with a maximum increase of .about.110% compared to vehicle (FIG. 10A and FIG. 10B). At the highest dose, 240 mg/kg Compound 1, ATP levels increased by 45%. Levels of ATP correlated with Compound 1 exposure in a manner similar across both studies.
Example 6: A SAD/MAD Study to Assess the Safety, Pharmacokinetics, and Pharmacodynamics of Compound 1 in Healthy Volunteers and Sickle Cell Disease Patients
[0083] Compound 1 will be evaluated in a randomized, placebo-controlled, double blind, single ascending and multiple ascending dose study to assess the safety, pharmacokinetics, and pharmacodynamics of Compound 1 in healthy volunteers and sickle cell disease patients. The use of Compound 1 is disclosed herein for treatment of sickle cell disease in humans.
[0084] Compound 1 is an oral small-molecule agonist of pyruvate kinase red blood cell isozyme (PKR) being developed for the treatment of hemolytic anemias. This human clinical trial study will characterize the safety, tolerability and the pharmacokinetics/pharmacodynamics (PK/PD) of a single ascending dose and multiple ascending doses of Compound 1 in the context of phase 1 studies in healthy volunteers and sickle cell disease patients. The effects of food on the absorption of Compound 1 will also be evaluated, in healthy volunteers.
[0085] The objectives of the study include the following: [0086] 1. To evaluate the safety and tolerability of a single ascending dose and multiple ascending doses of Compound 1 in healthy volunteers and sickle cell disease (SCD) patients. [0087] 2. To characterize the pharmacokinetics (PK) of Compound 1. [0088] 3. To evaluate the levels of 2,3-diphosphoglycerate (DPG) and adenosine triphosphate (ATP) in the red blood cells (RBCs) of healthy volunteers and SCD patients after single and multiple doses of Compound 1. [0089] 4. To evaluate the relationship between Compound 1 plasma concentration and potential effects on the QT interval in healthy volunteers. [0090] 5. To evaluate the effect of single ascending doses of Compound 1 on other electrocardiogram (ECG) parameters (heart rate, PR and QRS interval and T-wave morphology) in healthy volunteers. [0091] 6. To explore food effects on the PK of Compound 1 in healthy volunteers. [0092] 7. To explore the association of Compound 1 exposure and response variables (such as safety, pharmacodynamics (PD), hematologic parameters as appropriate). [0093] 8. To explore effects of Compound 1 after single and multiple doses on RBC function. [0094] 9. To explore effects of Compound 1 after multiple doses in SCD patients on RBC metabolism, inflammation and coagulation.
[0095] This is a first-in-human (FIH), Phase 1 study of Compound 1 that will characterize the safety, PK, and PD of Compound 1 after a single dose and after repeated dosing first in healthy adult volunteers and then in adolescents or adults with sickle cell disease. The study arms and assigned interventions to be employed in the study are summarized in Table 8. Initially, a dose range of Compound 1 in single ascending dose (SAD) escalation cohorts will be explored in healthy subjects. Enrollment of healthy subjects into 2-week multiple ascending dose (MAD) escalation cohorts will be initiated once the safety and PK from at least two SAD cohorts is available to inform the doses for the 2-week MAD portion of the study. The MAD cohorts will then run in parallel to the single dose cohorts. A single dose cohort is planned to understand food effects (FE) on the PK of Compound 1. After the SAD and FE studies in healthy subjects are completed, the safety, PK and PD of a single dose of Compound 1 that was found to be safe in healthy subjects will then be evaluated in sickle cell disease (SCD) subjects. Multiple dose studies in SCD subjects will then be initiated upon completion of MAD studies in healthy volunteers. Compound 1 will be administered in 25 mg and 100 mg tablets delivered orally.
TABLE-US-00008 TABLE 8 Arms Assigned Interventions Experimental: Single ascending dose cohorts Drug: Compound 1/Placebo in healthy subjects Healthy volunteer subjects will receive Healthy volunteer subject cohorts Compound 1/placebo and be monitored randomized 6:2 receiving a single dose of for side effects while undergoing Compound 1 or placebo. The first cohort pharmacokinetics and pharmacodynamic will receive 200 mg of Compound 1 or studies placebo. Dose escalation will occur if Compound 1 or placebo is tolerated. The maximum dose of Compound 1 or placebo will be 1500 mg. Planned doses for the SAD cohorts are listed in Table 9. Experimental: Multiple ascending dose Drug: Compound 1/Placebo cohorts in healthy subjects Healthy volunteer subjects will receive Healthy volunteer subject cohorts Compound 1/placebo and be monitored randomized 9:3 to receive Compound 1 or for side effects while undergoing placebo for 14 days continuous dosing. pharmacokinetics and pharmacodynamic The first cohort will receive 100 mg of studies Compound 1 or placebo daily X 14 days. Alternatively, the first cohort will receive 200 mg (e.g., 100 mg BID or 200 mg QD) of Compound 1 or placebo daily X 14 days. The maximum dose of Compound 1/placebo will be 600 mg Compound 1/placebo daily for 14 days. Planned doses for the MAD cohorts are listed in Table 10. Experimental: Food Effect Cohort in healthy Drug: Compound 1 subjects Healthy subjects will receive Compound Healthy Volunteer subject cohort of 10 1 with or without food and undergo subjects who will receive a single dose of pharmacokinetic studies Compound 1 with food and without food. Dose will be administered per the protocol defined dose. Healthy Volunteer subject cohort of 10 subjects who will receive a single dose of Compound 1 with food and without food. Dose will be 500 mg of Compound 1, but is subject to change based on the pharmacokinetic profile of Compound 1 observed in the initial SAD cohorts and the safety profile of Compound 1 observed in prior SAD and MAD cohorts. Experimental: Single ascending dose cohorts Drug: Compound 1/Placebo in SCD subjects SCD subjects will receive Compound Sickle cell disease subject cohort 1/placebo and be monitored for side randomized 6:2 receiving a single dose of effects while undergoing Compound 1 or placebo. The dose of pharmacokinetic and pharmacodynamics Compound 1/placebo administered will be studies a dose that was found to be safe in healthy subjects. The dose of Compound 1/placebo administered also will be a dose that was found to be pharmacodynamically active (e.g., results in a reduction in 2,3-DPG) in healthy subjects. Experimental: Multiple ascending dose Drug: Compound 1/Placebo cohorts in SCD subjects SCD subjects will receive Compound Sickle cell disease subject cohorts 1/placebo and be monitored for side randomized 9:3 to receive Compound 1 or effects while undergoing placebo for 14 days continuous dosing. pharmacokinetic and pharmacodynamics The dose of Compound 1/placebo studies administered will be a dose less than maximum tolerable dose evaluated in MAD healthy volunteers. The dose of Compound 1/placebo also will be a dose that was found to be pharmacodynamically active (e.g., results in a reduction in RBC 2,3-DPG and increase in RBC ATP) in MAD healthy volunteers.
TABLE-US-00009 TABLE 9 Dose Level/Cohort Dose Tablet Strength (#/day) SAD 1 200 mg 100 mg (2/day) SAD 2 400 mg 100 mg (4/day) SAD 3 700 mg 100 mg (7/day) SAD 4 1100 mg 100 mg (11/day) SAD 5 1500 mg 100 mg (15/day)
TABLE-US-00010 TABLE 10 Dose Level/Cohort Dose Tablet Strength (#/day) MAD 1 100 mg 100 mg (1/day) or 25 mg (4/day) MAD 2 200 mg 100 mg (2/day) MAD 3 400 mg 100 mg (4/day) MAD 4 600 mg 100 mg (6/day)
[0096] Outcome Measures
[0097] Primary Outcome Measures.
1. Incidence, frequency, and severity of adverse events (AEs) per CTCAE v5.0 of a single ascending dose and multiple ascending doses of Compound 1 in adult healthy volunteers and SCD patients.
[0098] [Time Frame: Up to 3 weeks of monitoring]
2. Maximum observed plasma concentration (Cmax)
[0099] [Time Frame: Up to 3 weeks of testing]
3. Time to maximum observed plasma concentration (Tmax)
[0100] [Time Frame: Up to 3 weeks of testing]
4. Area under the plasma concentration-time curve from time zero until the 24-hour time point (AUC0-24)
[0101] [Time Frame: Up to 3 weeks of testing]
5. Area under the plasma concentration-time curve from time zero until last quantifiable time point (AUC0-last)
[0102] [Time Frame: Up to 3 weeks of testing]
6. Area under the plasma concentration-time curve from time zero to infinity (AUC0-inf)
[0103] [Time Frame: Up to 3 weeks of testing]
7. Terminal elimination half-life (t1/2)
[0104] [Time Frame: Up to 3 weeks of testing]
8. Apparent clearance (CL/F)
[0105] [Time Frame: Up to 3 weeks of testing]
9. Apparent volume of distribution (Vd/F)
[0106] [Time Frame: Up to 3 weeks of testing]
10. Terminal disposition rate constant (Lz)
[0107] [Time Frame: Up to 3 weeks of testing]
11. Renal clearance (CIR)
[0108] [Time Frame: Up to 3 weeks of testing]
[0109] Secondary Outcome Measures:
12. Change from baseline in the levels of 2,3-diphosphoglycerate (DPG) and adenosine triphosphate (ATP) in the red blood cells (RBCs) of healthy volunteers and SCD patients after single and multiple doses of Compound 1.
[0110] [Time Frame: Up to 3 weeks of testing]
13. Model-based estimate of change from baseline QT interval corrected using Fridericia's correction formula (QTcF) and 90% confidence interval at the estimated Cmax after a single dose of Compound 1 in healthy volunteers.
[0111] [Time Frame: up to 7 days]
14. Change from baseline heart rate after a single dose of Compound 1 in healthy volunteers
[0112] [Time Frame: up to 7 days]
15. Change from baseline PR after a single dose of Compound 1 in healthy volunteers
[0113] [Time Frame: up to 7 days]
16. Change from baseline QRS after a single dose of Compound 1 in healthy volunteers
[0114] [Time Frame: up to 7 days]
17. Change from baseline T-wave morphology after a single dose of Compound 1 in healthy volunteers
[0115] [Time Frame: up to 7 days]
[0116] Exploratory Outcome Measures:
18. Effect of food on C.sub.max, AUC.sub.0-24/AUC.sub.last 19. Effect of AUC.sub.last/AUC.sub.0-24, C.sub.max, minimum plasma concentration (C.sub.min), peak-to trough ratio, dose linearity, accumulation ratio on safety, PD, and hematologic parameters of interest, as assessed by exposure-response analyses 20. Effect of chronic Compound 1 dosing on SCD RBC response to oxidative stress in SCD Patients (including evaluation of glutathione, glutathione peroxidase and superoxide dismutase levels) 21. Effect of chronic Compound 1 dosing on measurable markers of inflammation in SCD Patients (C-reactive protein, ferritin, interleukin [IL]-113, IL-6, IL-8, and tumor necrosis factor-.alpha.) 22. Effects of chronic Compound 1 dosing on measurable markers of hypercoagulation in SCD patients (D-dimer, prothrombin 1.2, and thrombin-antithrombin [TAT] complexes)
[0117] Eligibility
[0118] Minimum age: 18 Years (healthy volunteers); 12 Years (SCD subjects)
[0119] Maximum age: 60 Years
[0120] Sex: All
[0121] Gender Based: No
[0122] Accepts Healthy Volunteers: Yes
[0123] Inclusion Criteria: [0124] Healthy volunteer: subjects must be between 18 and 60 years of age; SCD: subjects must be between 12 and 50 years of age [0125] Subjects must have the ability to understand and sign written informed consent, which must be obtained prior to any study-related procedures being completed. [0126] Subjects must be in general good health, based upon the results of medical history, a physical examination, vital signs, laboratory profile, and a 12-lead ECG. [0127] Subjects must have a body mass index (BMI) within the range of 18 kg/m2 to 33 kg/m.sup.2 (inclusive) and a minimum body weight of 50 kg (healthy volunteer subjects) or 40 kg (SCD subjects) [0128] For SCD subjects, sickle cell disease previously confirmed by hemoglobin electrophoresis or genotyping indicating one of the following hemoglobin genotypes: Hgb SS, Hgb S.beta..sup.+-thalassemia, Hgb S.beta..sup.0-thalassemia, or Hgb SC [0129] All males and females of child bearing potential must agree to use medically accepted contraceptive regimen during study participation and up to 90 days after. [0130] Subjects must be willing to abide by all study requirements and restrictions.
[0131] Exclusion Criteria. [0132] Evidence of clinically significant medical condition or other condition that might significantly interfere with the absorption, distribution, metabolism, or excretion of study drug, or place the subject at an unacceptable risk as a participant in this study [0133] History of clinically significant cardiac diseases including condition disturbances [0134] Abnormal hematologic, renal and liver function studies [0135] History of drug or alcohol abuse
[0136] Results (Healthy Volunteers)
[0137] Four healthy SAD cohorts were evaluated at doses of 200, 400, 700, and 1000 mg, and four healthy MAD cohorts received 200 to 600 mg total daily doses for 14 days at QD or BID dosing (100 mg BID, 200 mg BID, 300 mg BID, and 400 mg QD). In the food effect (FE) cohort, 10 healthy subjects received 200 mg of Compound 1 QD with and without food.
[0138] No serious adverse events (SAEs) or AEs leading to withdrawal were reported in the SAD and MAD cohorts of healthy volunteers. In PK assessments, Compound 1 was rapidly absorbed with a median T.sub.max of 1 hr postdose. Single dose exposure increased in greater than dose-proportional manner at doses .gtoreq.700 mg. In multiple-doses delivered BID or QD, linear PK was observed across all dose levels (100-300 mg BID, 400 mg QD), and exposure remained steady up to day 14, without cumulative effect. Compound 1 exposure under fed/fasted conditions was similar.
[0139] PD activity was demonstrated at all dose levels evaluated in Compound 1-treated subjects (Table 11). Table 11 reports the mean maximum percentage change in 2,3-DPG and ATP across all doses and timepoints in the SAD and MAD cohorts. As shown in Table 11, a mean decrease in 2,3-DPG, and a mean increase in ATP, relative to baseline, was observed in both the SAD and MAD cohorts. Within 24 hr of a single dose of Compound 1, a decrease in 2,3-DPG was observed. After 14 days of Compound 1 dosing these PD effects were maintained along with an increase in ATP over baseline. Accordingly, the mean maximum reduction in the concentration of 2,3-DPG was at least about 40% in patients receiving Compound 1 in the SAD study and at least about 50% in patients receiving Compound 1 in the MAD study.
TABLE-US-00011 TABLE 11 Summary of Mean Maximum Percent Change in Key PD Measures from Baseline SAD MAD Placebo Compound 1 Placebo Compound 1 PD Marker Statistics (N = 8) (N = 24) (N = 12 (N = 36) 2,3-DPG Mean -19.5 -46.8 -17.0 -56.3 (95% CI) (-25.0, -14.0) (-50.3, -43.2) (-22.9, -11.1) (-58.9, -53.7) P-value <0.0001 <0.0001 ATP Mean 9.2 24.4 7.2 68.5 (95% CI) (0.5, 18.0) (18.4, 30.3) (-0.3, 14.7) (63.6, 73.3) P-value 0.0094 <0.0001
[0140] In the SAD cohorts, the subjects' blood 2,3-DPG levels were measured periodically after dosing by a qualified LC-MS/MS method for the quantitation of 2,3-DPG in blood. Decreased 2,3-DPG blood levels were observed 6 hours following a single dose of Compound 1 at all dose levels (earlier timepoints were not collected). Maximum decreases in 2,3-DPG levels generally occurred .about.24 hours after the first dose with the reduction sustained .about.48-72 hr postdose. Table 12 reports the median percentage change in 2,3-DPG blood levels, relative to baseline, measured over time in healthy volunteers after a single dose of Compound 1 (200 mg, 400 mg, 700 mg, or 1000 mg) or placebo. Accordingly, the median reduction in the concentration of 2,3-DPG, relative to baseline, was at least about 30% at all dose levels tested 24 hours after administration of the single dose.
TABLE-US-00012 TABLE 12 Median Percentage Change in 2,3-DPG Levels Time After Dose Dose Placebo 200 mg 400 mg 700 mg 1000 mg 0 0.0 0.0 0.0 0.0 0.0 6 -7.8 -18 -23 -29 -20 8 -7.6 -17 -29 -28 -31 12 -4.0 -25 -40 -41 -44 16 -6.0 -33 -35 -46 -50 24 -2.0 -31 -39 -49 -48 36 -6.9 -33 -38 -46 -47 48 -15 -29 -31 -48 -47 72 -6.9 -18 -30 -33 -21
[0141] FIG. 11 is a graph of the blood 2,3-DPG levels measured over time in healthy volunteers who received a single dose of Compound 1 (200 mg, 400 mg, 700 mg, or 1000 mg) or placebo. As shown in FIG. 11, healthy volunteers who received Compound 1 experienced a decrease in blood 2,3-DPG levels, relative subjects who received the placebo. FIG. 12 is a graph of the blood 2,3-DPG levels measured 24 hours post-dose in healthy volunteers who received a single dose of Compound 1 (200 mg, 400 mg, 700 mg, or 1000 mg) or placebo. As shown in FIG. 12, healthy volunteers who received Compound 1 experienced a decrease in blood 2,3-DPG levels at 24 hours post-dose, relative to subjects who received the placebo.
[0142] In the MAD cohorts, the subjects' blood 2,3-DPG levels were measured periodically after dosing by a qualified LC-MS/MS method for the quantitation of 2,3-DPG in blood. The maximum decrease in 2,3-DPG on Day 14 was 55% from baseline (median). 2,3-DPG levels reached a nadir and plateaued on Day 1 and had not returned to baseline levels 72 hours after the final dose on Day 14. Table 13 reports the median percentage change in 2,3-DPG blood levels, relative to baseline, measured over time after the first dose on days 1 and 14 in healthy volunteers who received daily doses of Compound 1 (100 mg BID, 200 mg BID, or 300 mg BID) or placebo for 14 days. Accordingly, the median reduction in the concentration of 2,3-DPG, relative to baseline, was at least about 25% at all dose levels tested 24 hours after administration of the first dose on day 1 and at least about 40% at all dose levels tested 24 hours after administration of the first dose on day 14.
TABLE-US-00013 TABLE 13 Median Percentage Change in 2,3-DPG Levels (Days 1 and 14) Time After Dose First 100 mg BID 200 mg BID 300 mg BID Placebo Daily Day Day Day Day Dose 1 14 1 14 1 14 1 14 0 0.0 -42.0 0.0 -48.2 0.0 -59.4 0.0 -7.6 6 -16.1 -44.3 -13.1 -48.5 -18.8 -53.0 -2.9 -10.9 8 -12.1 -44.7 -22.3 -44.3 -23.8 -54.2 -0.6 -1.6 12 -18.1 -43.6 -23.1 -42.2 -31.6 -55.3 -7.1 -1.6 16 -18.4 -43.9 -33.9 -42.9 -40.7 -52.4 -6.7 -5.3 24 -27.8 -44.1 -43.5 -44.3 -50.8 -52.1 1.1 -10.7 48 -34.7 -38.7 -44.5 -1.0 72 -20.2 -20.2 -32.9 -7.0
[0143] FIG. 13 is a graph of the blood 2,3-DPG levels measured over time in healthy volunteers who received daily doses of Compound 1 (100 mg BID, 200 mg BID, 300 mg BID, or 400 mg QD) or placebo for 14 days. As shown in FIG. 13, healthy volunteers who received Compound 1 experienced a decrease in blood 2,3-DPG levels, relative subjects who received the placebo. FIG. 14 is a graph of the blood 2,3-DPG levels measured on day 14 in healthy volunteers who received daily doses of Compound 1 (100 mg BID, 200 mg BID, 300 mg BID, or 400 mg QD) or placebo for 14 days. As shown in FIG. 14, healthy volunteers who received Compound 1 experienced a decrease in blood 2,3-DPG levels, relative to subjects who received the placebo.
[0144] In the MAD cohorts, the subjects' blood ATP levels were measured on day 14 by a qualified LC-MS/MS method for the quantitation of ATP in blood. ATP levels were elevated, relative to baseline, on day 14, and remained elevated 60 hours after the last dose. Table 14 reports the median percentage change in blood ATP levels, relative to baseline, measured over time after the first dose on day 14 in healthy volunteers who received daily doses of Compound 1 (100 mg BID, or 200 mg BID) or placebo for 14 days.
TABLE-US-00014 TABLE 14 Median Percentage Change in ATP Levels (Day 14) Time After First Dose Daily Dose 100 mg BID 200 mg BID Placebo 0 41.5 55.3 -0.5 6 43.8 48.1 2.8 8 47.8 58.4 -4.1 12 45.4 56.2 2.3 16 44.8 57.0 -6.8 24 55.0 64.0 2.9 48 52.2 58.9 4.7 72 49.2 54.0 2.2
[0145] FIG. 15 is a graph of the blood ATP levels measured on day 14 in healthy volunteers who received daily doses of Compound 1 (100 mg BID, 200 mg BID, 300 mg BID, or 400 mg QD) or placebo for 14 days. As shown in FIG. 15, healthy volunteers who received Compound 1 experienced an increase in blood ATP levels, relative to subjects who received the placebo.
[0146] FIG. 16 is a graph plotting the blood concentration of Compound 1 (ng/mL) measured in healthy volunteer (HV) patients on a first (left) axis and the concentration of 2,3-DPG (micrograms/mL) measured in these HV patients on a second (right) axis after administration of a single dose of Compound 1 (400 mg). Solid symbols represent geometric means and Standard errors of the observed Compound 1 plasma and 2,3 DPG concentrations. As shown in the figure, the observed 2,3 DPG modulation does not track directly plasma pharmacokinetics (blood concentration of Compound 1) where the pharmacodynamic maximum (i.e., the minimum of the 2,3-DPG concentration, at time .about.24 h) occurred nearly 24 h after the pharmacokinetic maximum (i.e., maximum of the PK curve, at time .about.1-2 h). The observed pharmacodynamic response in HVs was durable, with a calculated PD half-life of .about.20 h, where 2,3-DPG depression was observed long after plasma levels were undetectable. Taken together, this suggests that identifying the pharmacologically active dose cannot be adequately performed using pharmacokinetic parameters (C.sub.max/C.sub.min/AUC) in isolation, but rather support an approach that includes integrating the temporal pharmacokinetic/pharmacodynamic relationship to provide the platform of evidence that QD dosing may be feasible in sickle cell disease patients.
Example 7: Analysis of ATP and 2,3 DPG in K2EDTA Whole Blood by LC-MS/MS
[0147] The following procedures are employed for the analysis of ATP and 2,3-DPG in human whole blood K2EDTA using a protein precipitation extraction procedure and analysis by LC-MS/MS.
[0148] This bioanalytical method applies to the parameters described below:
TABLE-US-00015 Assay Range 25,000-1,500,000 ng/mL Extraction Volume 15.0 uL Species/Matrix/ Water as a surrogate for Human Anticoagulant Whole Blood K2EDTA Extraction type Protein Precipitation Sample Storage 80.degree. C. Mass Spectrometer API-5500 Acquisition software Analyst/ Aria System
[0149] The following precautions are followed:
[0150] 1. Standard and QC samples are prepared on ice and stored in plastic containers.
[0151] 2. Study samples and QC samples are thawed on ice.
[0152] 3. Extraction is performed on ice.
[0153] The following definitions and abbreviations are employed:
TABLE-US-00016 CRB Carryover remediation blanks FT Freeze-thaw MPA Mobile phase A MPB Mobile phase B NA Not applicable NR Needle rinse RT Retention time SIP Stability in progress TBD To be determined
[0154] The following chemicals, matrix, and reagents are used:
TABLE-US-00017 K2EDTA Human Whole Blood, BioreclamationIVT or equivalent (Note: BioReclamationIVT and BioIVT are considered equivalent) Acetonitrile (ACN), HPLC Grade or better Ammonium Acetate (NH.sub.4OAc), HPLC grade or equivalent Ammonium Hydroxide (NH.sub.4OH, 28-30%), ACS grade or better Dimethylsulfoxide (DMSO), ACS grade or better Formic Acid (FA), 88% ACS grade Isopropanol (IPA), HPLC Grade or better Methanol (MeOH), HPLC Grade or better Water (H.sub.2O), Milli-Q or HPLC Grade ATP - Analyte, Sponsor or supplier ATP-IS- IS, Sponsor or supplier 2,3-DPG - Analyte, Sponsor or supplier 2,3-DPG-IS- IS, Sponsor or supplier
[0155] The following procedures are used for reagent preparation. Any applicable weights and volumes listed are nominal and may be proportionally adjusted as long as the targeted composition is achieved:
TABLE-US-00018 Nominal Volumes Final Solution for Solution Storage Solution Composition Preparation Conditions Mobile Phase A 10 mM Weigh Ambient (MPA) Ammoniumn approximately Temperature Acetate in 770.8 mg of water pH 8.5 Ammonium Acetate; add to a bottle with 1000 mL of water. Adjust pH to 8.3-8.7 using Ammonium Hydroxide. Mobile Phase B 5:95 MPA:ACN Add 50.0 mL of Ambient (MPB) MPA to 950 mL of Temperature CAN. Mix. Needle Rinse 1 25:25:25:25:0.1 Add 500 mL of (NR1) (v:v:v:v:v) MeOH, 500 mL of Ambient MeOH:ACN: ACN, 500 mL of H2O:IPA:NH.sub.4OH H.sub.2O, 500 mL of Temperature IPA, and 2 mL of NH.sub.4OH. Mix. Needle Rings 2 90:10:0.1 (v:v:v) Add 2 mL of FA to Ambient (NR2) H.sub.20:MeOH:FA 200 mL of MeOH Temperature and 1800 mL of H.sub.20. Mix.
[0156] Calibration standards are prepared using water as the matrix according to the table presented below. The indicated standard is prepared by diluting the indicated spiking volume of stock solution with the indicated matrix volume.
TABLE-US-00019 Stock Spiking Matrix Final Final Calibration Stock Conc. Vol. Vol. Vol. Conc. Standard Solution (ng/mL) (mL) (mL) (mL) (ng/mL) STD-6 ATP Stock 60,000,000 0.0100 0.380 0.400 1,500,000 2,3-DPG Stock 60,000,000 0.0100 STD-5 STD-6 1,500,000 0.100 0.200 0.300 500,000 STD-4 STD-6 1,500,000 0.0500 0.325 0.375 200,000 STD-3 STD-6 1,500,000 0.0250 0.350 0.375 100,000 STD-2 STD-5 500,000 0.0500 0.450 0.500 50,000 STD-1 STD-5 500,000 0.0250 0.475 0.500 25,000 Cond. STD-5 500,000 0.0250 0.975 1.00 12,500
[0157] Quality control standards are prepared using water as the matrix according to the table presented below. The indicated quality control standard is prepared by diluting the indicated spiking volume of stock solution with the indicated matrix volume.
TABLE-US-00020 Quality Stock Spiking Matrix Final Final Control Stock Conc. Vol. Vol. Vol. Conc. Standard Solution (ng/mL) (mL) (mL) (mL) (ng/mL) QC-High ATP Stock 60,000,000 0.160 7.68 8.00 1,200,000 2,3-DPG 60,000,000 0.160 Stock QC-Mid QC-High 1,200,000 1.50 4.50 6.00 300,000 QC-Low QC-Mid 300,000 1.50 4.50 6.00 75,000
[0158] An internal standard spiking solution is prepared with a final concentration of 12,500 ng/mL ATP and 2,3-DPG by diluting stock solutions of ATP and 2,3-DPG at concentrations of 1,000,000 ng/mL with water. 0.200 mL each of the ATP and 2,3-DPG stock solutions are diluted with 15.6 mL of water to produce a final volume of 16.0 mL at a final concentration of 12,500 ng/mL of ATP and 2,3-DPG.
[0159] The following procedures are used for sample extraction prior to analysis via LC-MS/MS. 15.0 .mu.L of the calibration standards, quality controls, matrix blanks, and samples are aliquoted into a 96-well plate. 50.0 .mu.L of the internal standard spiking solution is added to all samples on the plate, with the exception of the matrix blank samples; 50.0 .mu.L of water is added to the matrix blank samples. Subsequently, 150 .mu.L of water is added to all samples on the plate. The plate is then covered and agitated by vortex at high speed for ten minutes, after which 750 .mu.L of methanol is added to all samples on the plate. The plate is covered and agitated by vortex for approximately 1 minute. The plate is then centrifuged at approximately 3500 RPM at approximately 4.degree. C. for five minutes. After centrifugation, a liquid handler is used to transfer 50 L of each sample to a new 96-well plate, and 200 .mu.L of acetonitrile is added to all samples on the plate. The newly prepared plate is covered and agitated by vortex for approximately 1 minute. The plate is then centrifuged at approximately 3500 RPM at approximately 4.degree. C. for 2 minutes.
[0160] The following LC parameters and gradient conditions are used for analysis of the extracted samples:
TABLE-US-00021 LC Parameters Analytical Column Vendor: SeQuant Description: ZIC-pHILIC Dimensions: 50 mm .times. 2.1 mm Column Heater Temperature: 40.degree. C. Plate Rack Position: Cold Stack Cold Stack Set Point: 5.degree. C. Mobile Phase Mobile Phase A 10 mM Ammoniumn (MPA) Acetate in water pH 8.5 Mobile Phase B 5:95 MPA:ACN (MPB) Injection Volume 5 .mu.L
TABLE-US-00022 LC Gradient Time Flow Gradient Step (s) (mL/min) Setting % MPB 1 50 0.400 Step 5 2 30 0.400 Ramp 95 3 70 0.400 Step 5
Data is collected starting at 0.08 min and is collected over a data window length of 0.70 min.
[0161] The following MS parameters are used for analysis of the extracted samples using an API-5500 Mass Spectrometer:
TABLE-US-00023 Interface: Turbo Ion Spray Ionization, positive-ion mode Scan Mode: Multiple Reaction Monitoring (MRM) Scan Parameters: Parent/Product: Dwell Time (ms): 506.0/159.0 50 521.0/159.0 25 265.0/166.8 50 268.0/169.8 25 Source Temperature: 400.degree. C.
[0162] The collected MS data is analyzed and sample concentrations are quantified using peak area ratios with a linear 1/x.sup.2 regression type.
* * * * *

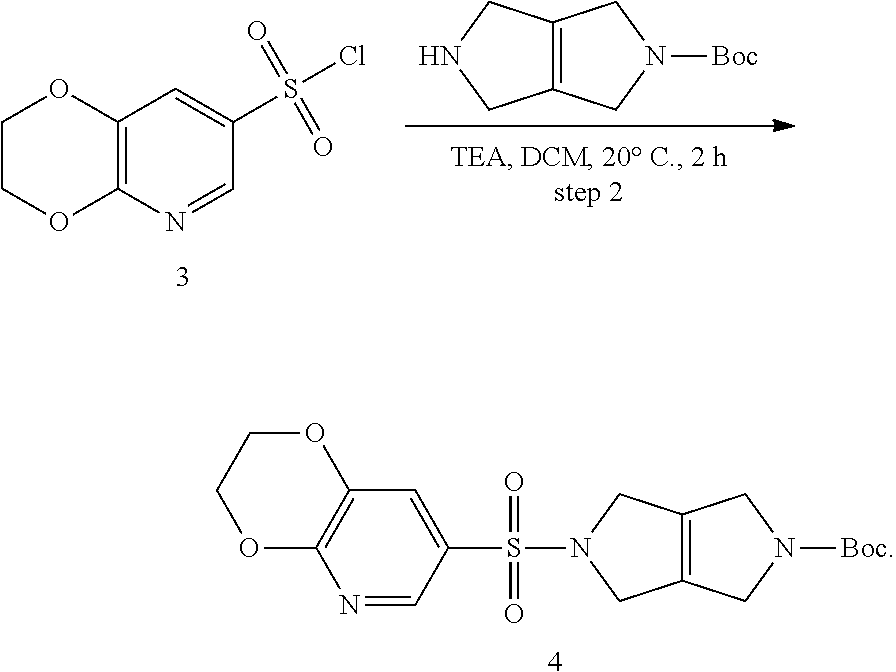



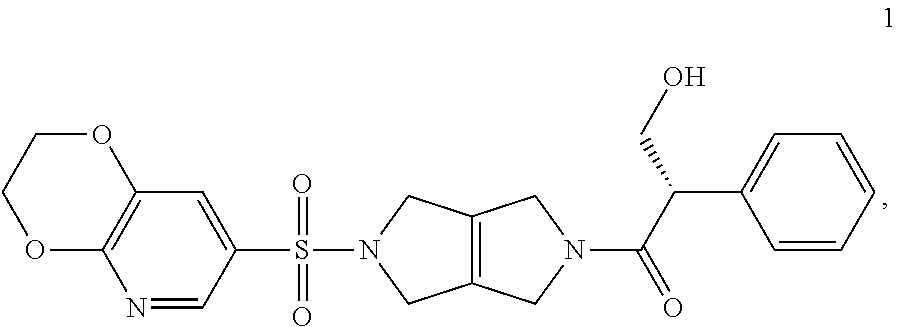



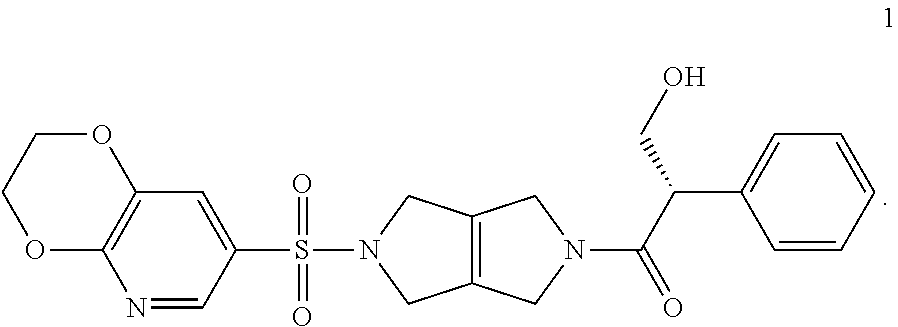
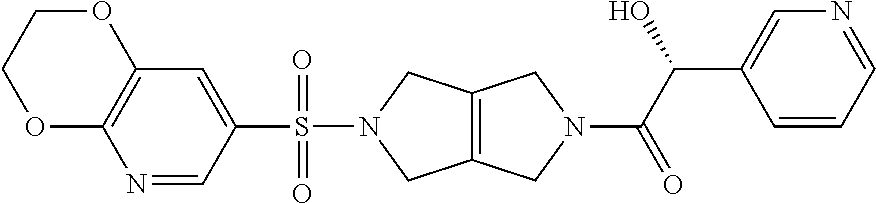

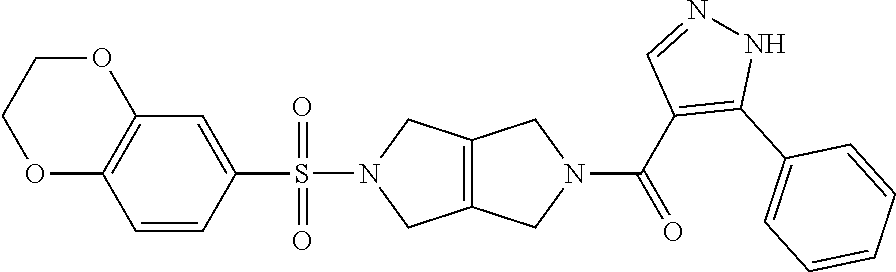


D00000

D00001
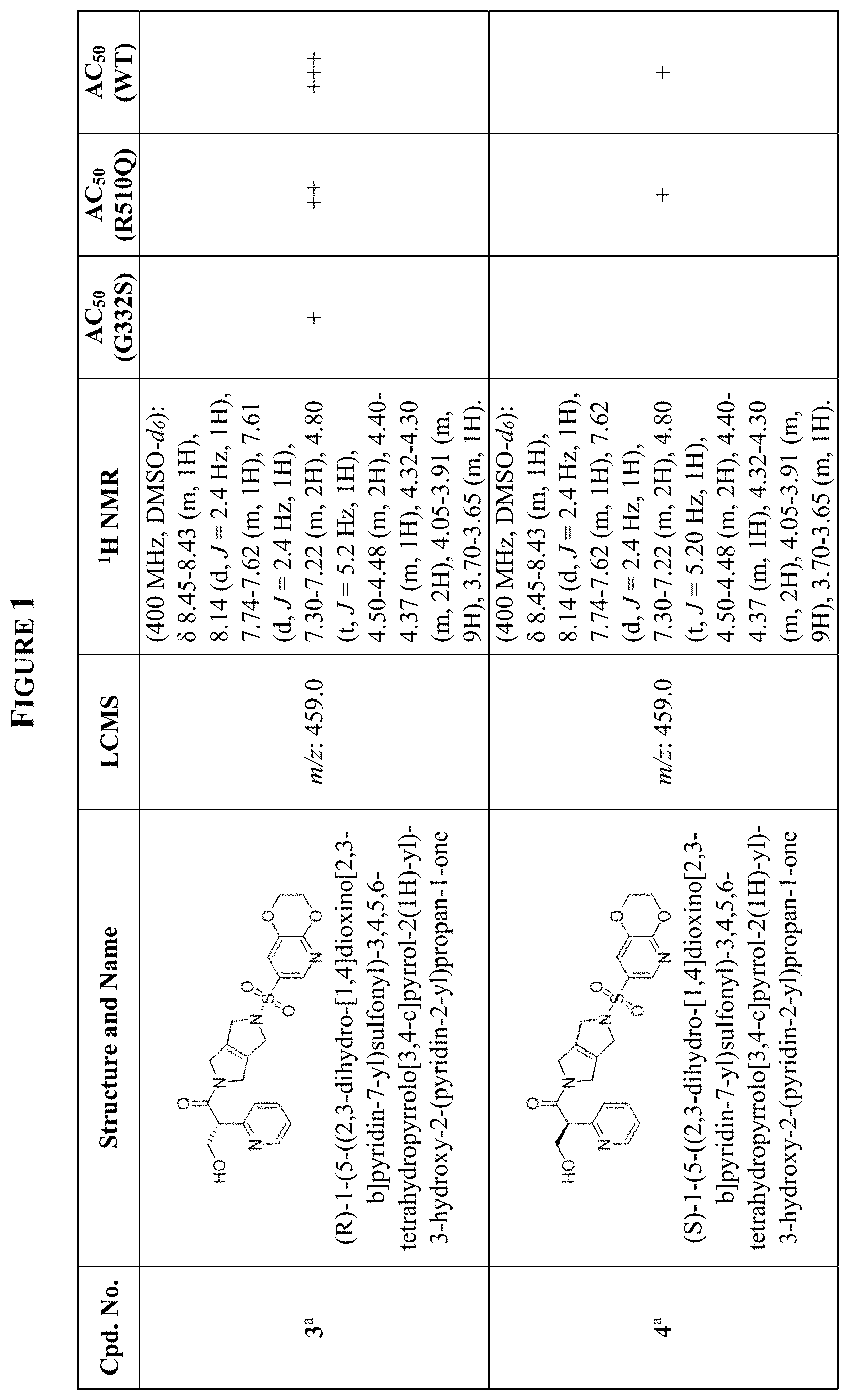
D00002

D00003
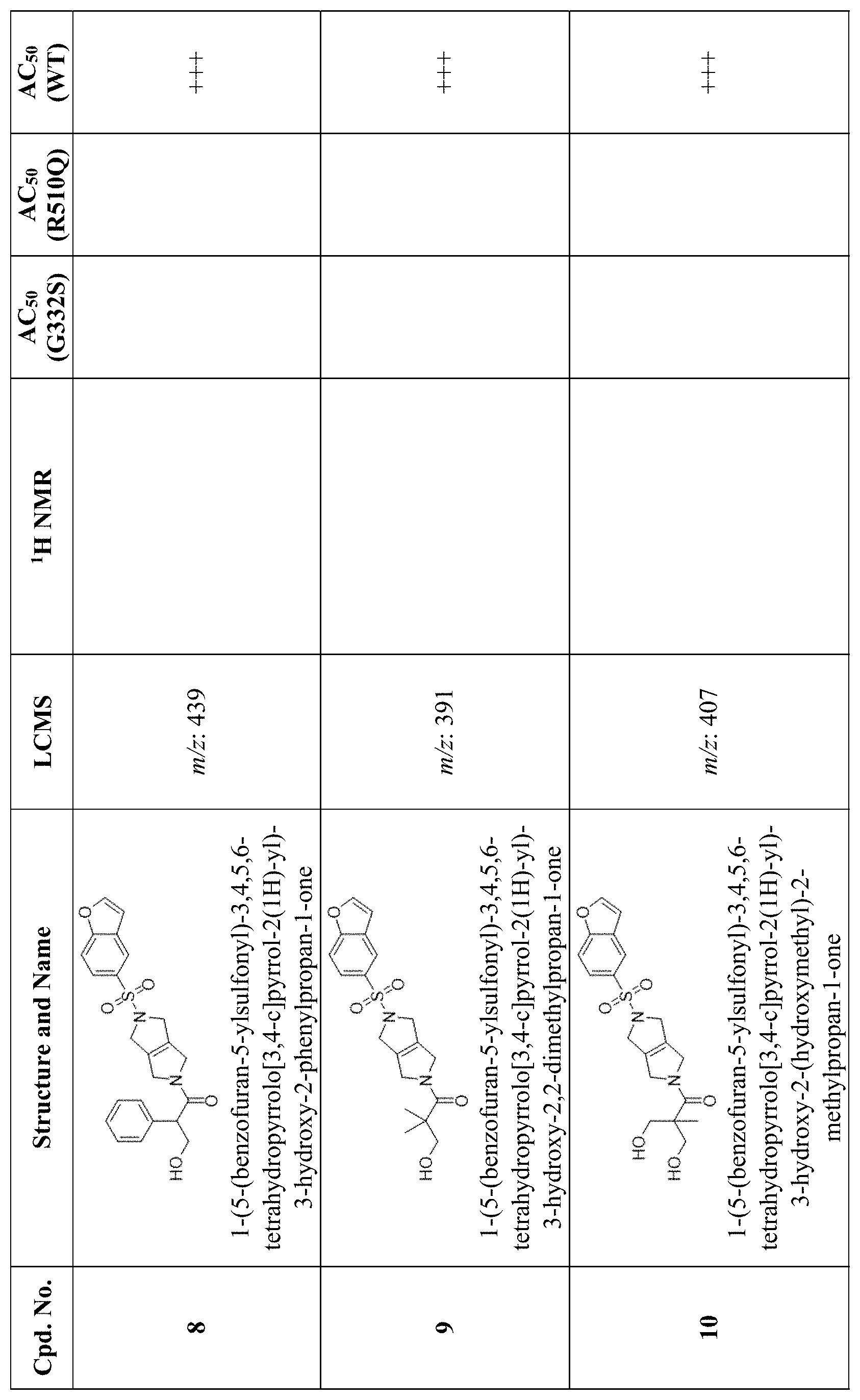
D00004
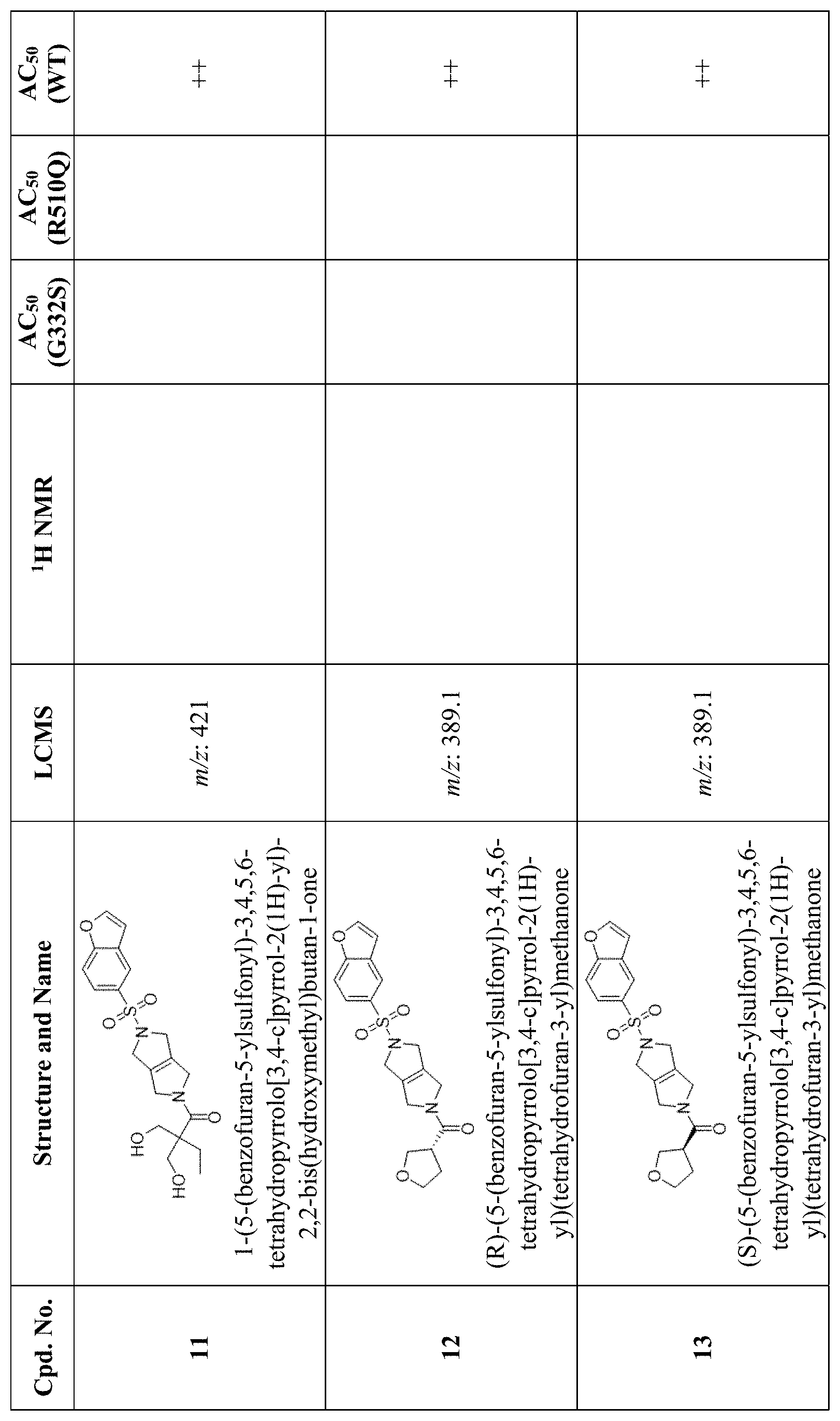
D00005

D00006

D00007

D00008

D00009

D00010

D00011

D00012

D00013

D00014
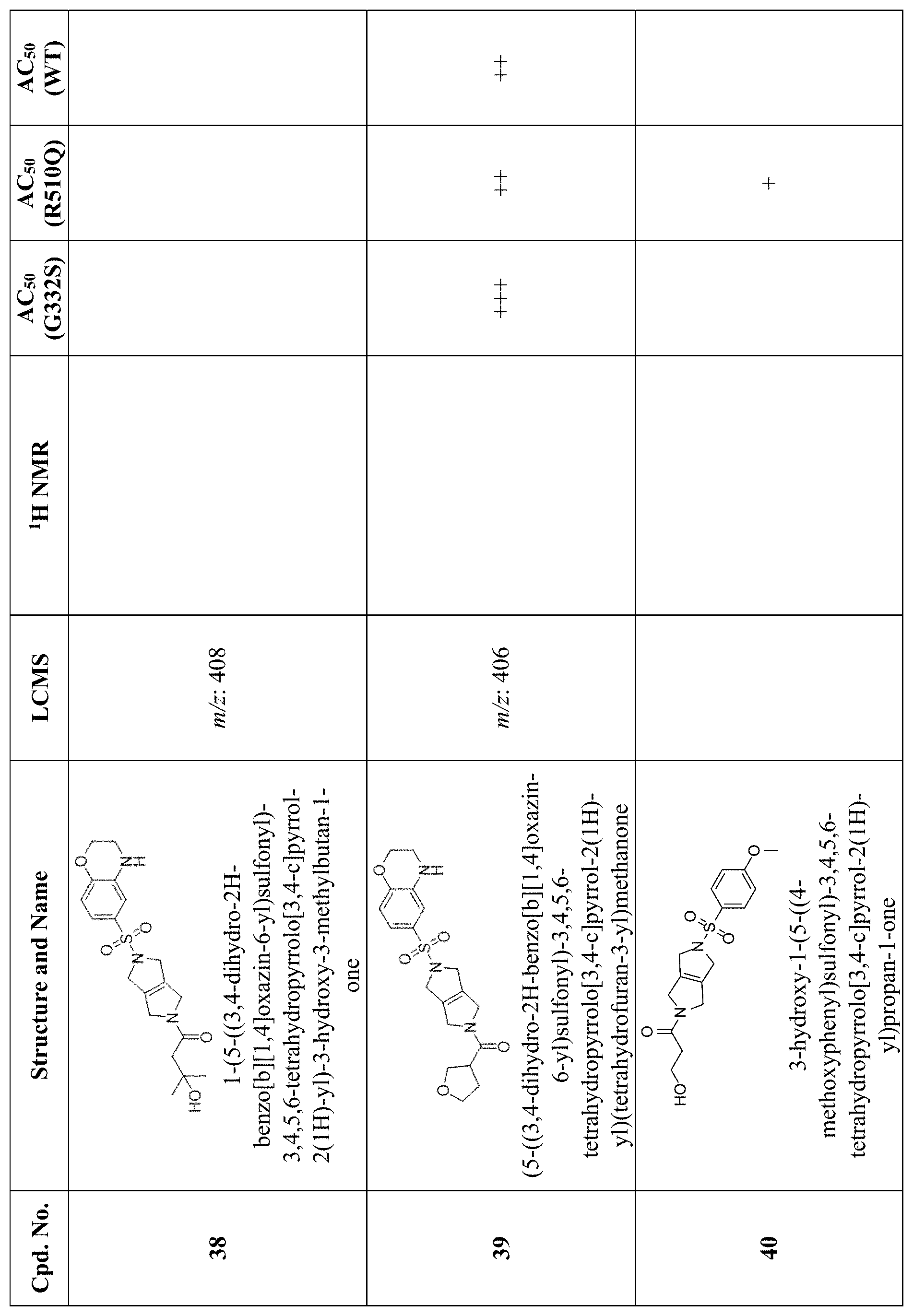
D00015

D00016

D00017

D00018

D00019
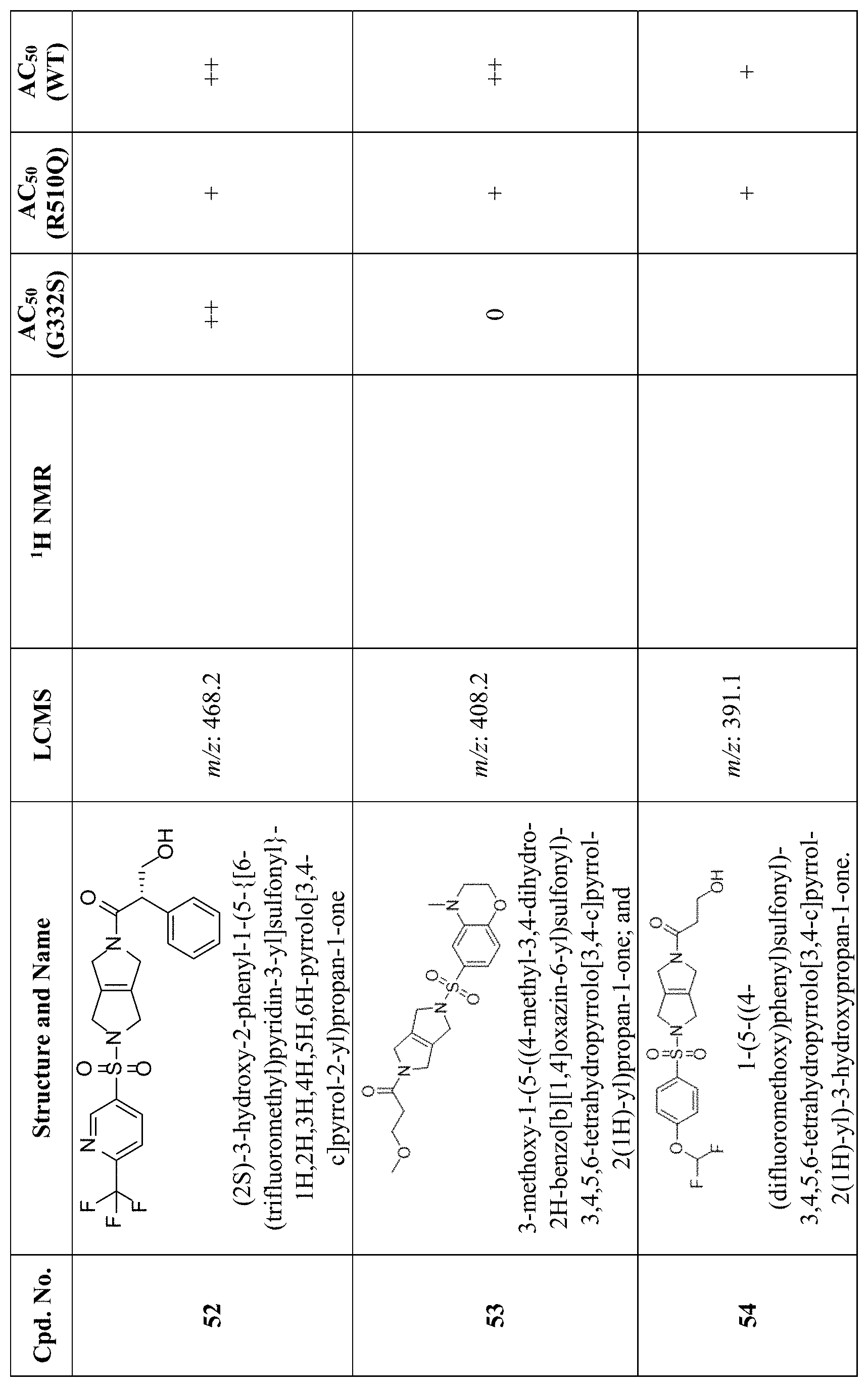
D00020

D00021

D00022

D00023

D00024

D00025

D00026

D00027
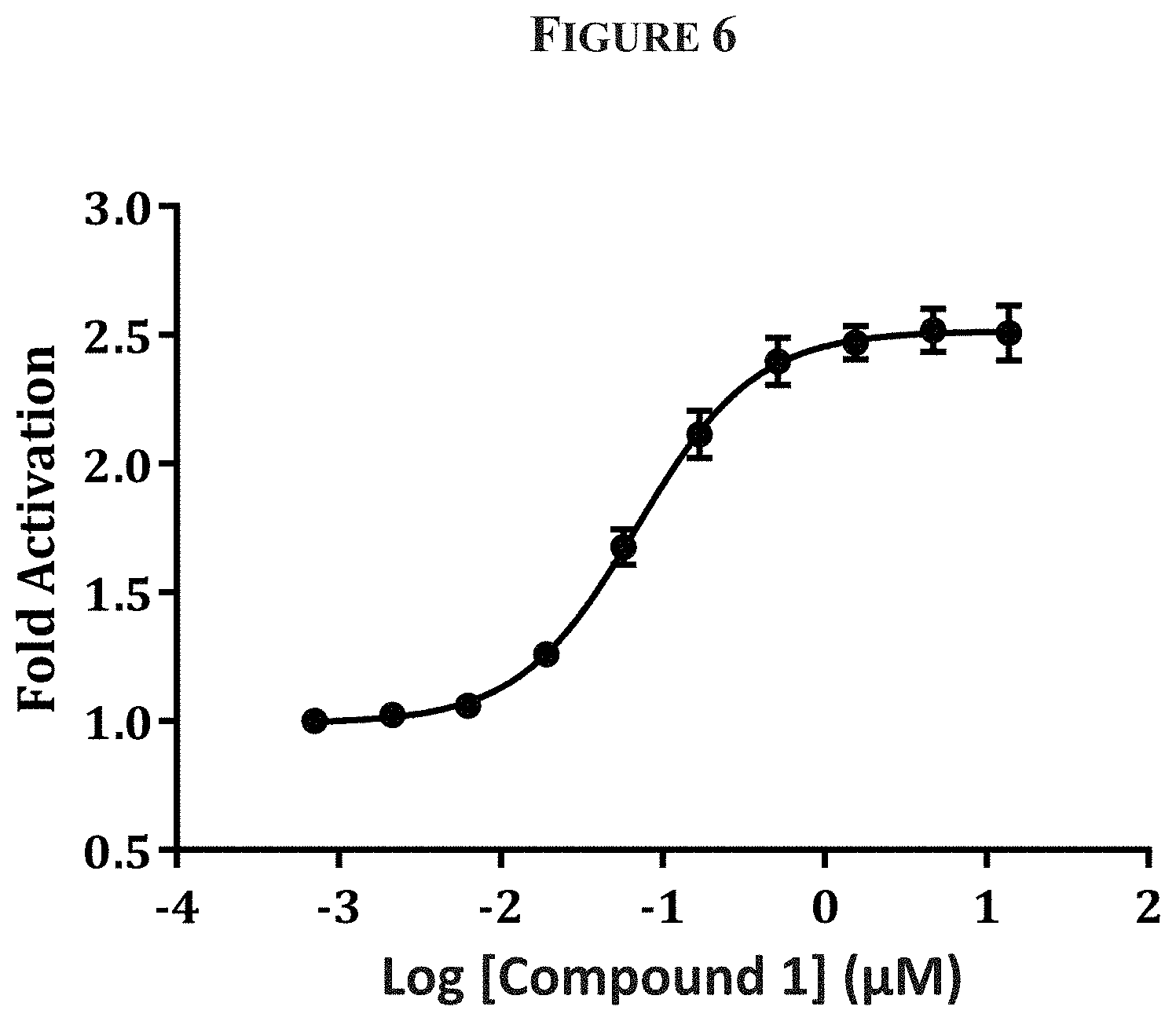
D00028

D00029

D00030
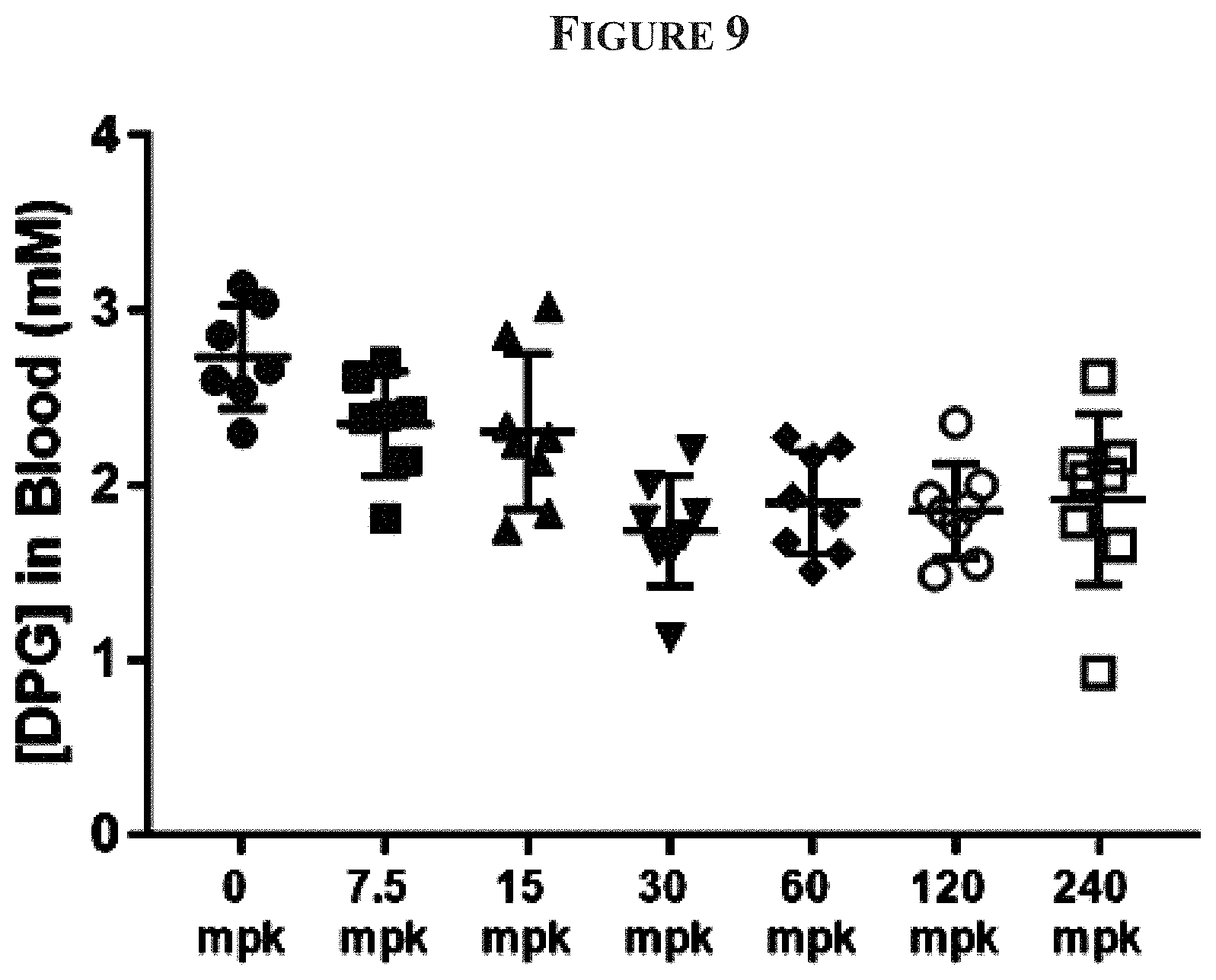
D00031

D00032

D00033

D00034

D00035
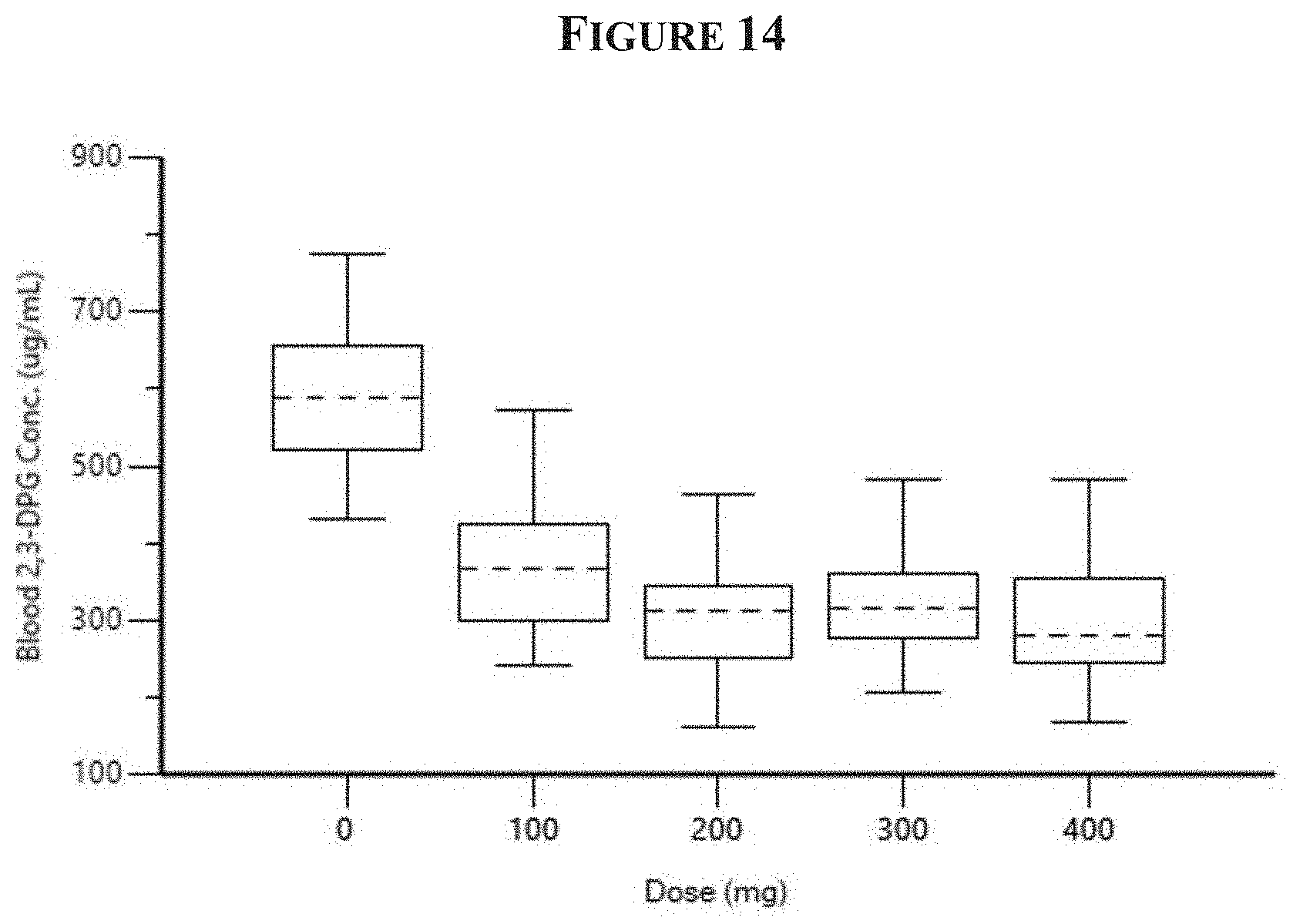
D00036
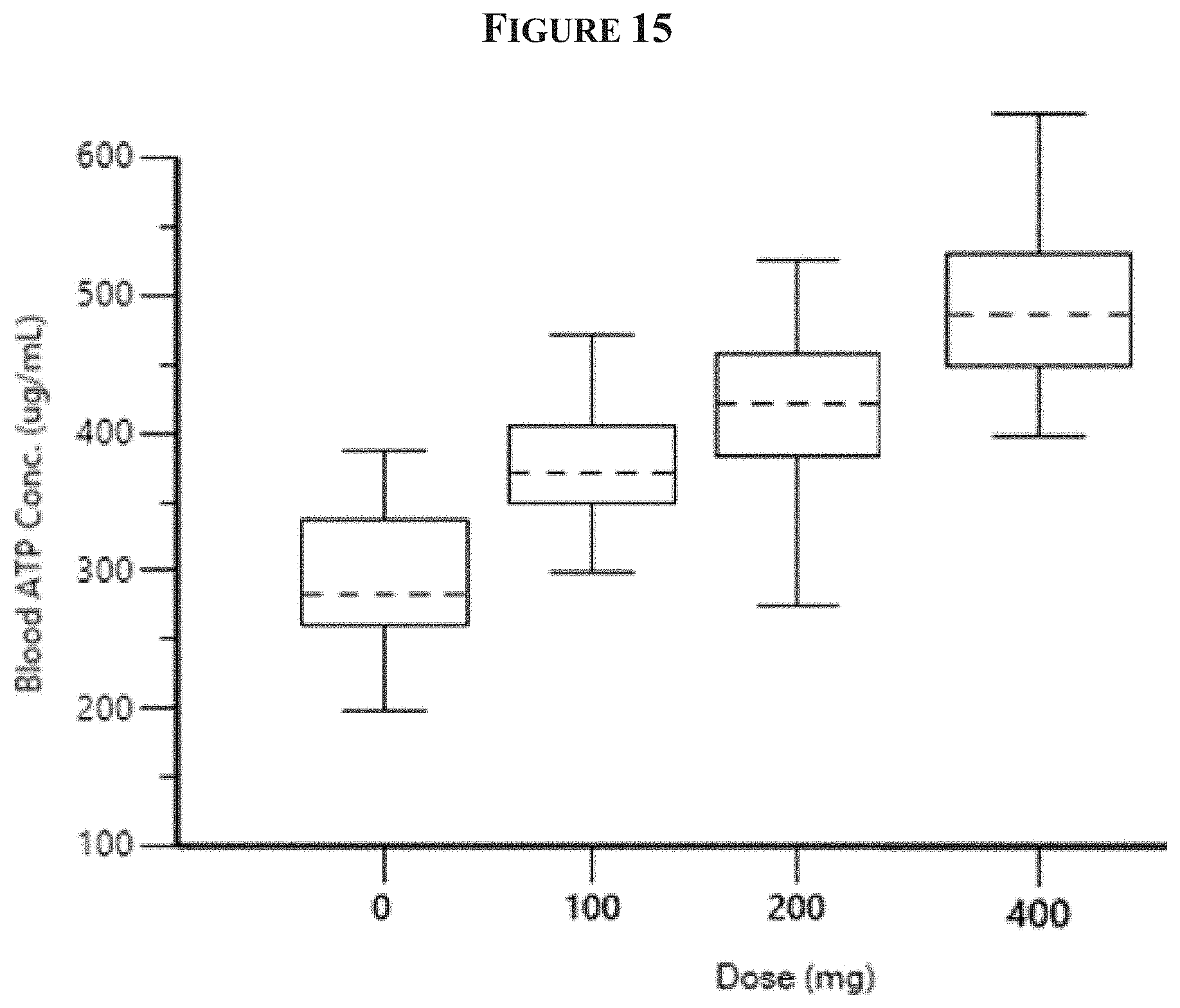
D00037

XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.