Methods For Generating A Glucose Permease Library And Uses Thereof
Manchester; Shawn ; et al.
U.S. patent application number 16/722658 was filed with the patent office on 2020-04-23 for methods for generating a glucose permease library and uses thereof. The applicant listed for this patent is Zymergen Inc.. Invention is credited to Shawn Manchester, Jeffrey Mellin.
| Application Number | 20200123529 16/722658 |
| Document ID | / |
| Family ID | 60785478 |
| Filed Date | 2020-04-23 |




| United States Patent Application | 20200123529 |
| Kind Code | A1 |
| Manchester; Shawn ; et al. | April 23, 2020 |
METHODS FOR GENERATING A GLUCOSE PERMEASE LIBRARY AND USES THEREOF
Abstract
The present disclosure describes methods for generating microbial strains expressing a heterologous bacterial glucose permease gene that produce biomolecules of interest. In aspects, the disclosure provides novel bacterial strains, which express a heterologous bacterial glucose permease gene whose expression is controlled by a native Corynebacterium glutamicum promoter or a mutant promoter derived therefrom. Also provided herein are methods for producing a library of bacterial glucose permease genes using a promoter ladder comprising a plurality of promoters derived from Corynebacterium glutamicum.
| Inventors: | Manchester; Shawn; (Oakland, CA) ; Mellin; Jeffrey; (Oakland, CA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 60785478 | ||||||||||
| Appl. No.: | 16/722658 | ||||||||||
| Filed: | December 20, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 16313613 | Dec 27, 2018 | 10544411 | ||
| PCT/US2017/039997 | Jun 29, 2017 | |||
| 16722658 | ||||
| 62356924 | Jun 30, 2016 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | C12P 1/04 20130101; C12P 21/02 20130101; C12Y 207/01001 20130101; C12Y 207/01069 20130101; C12P 19/02 20130101; C12N 15/1037 20130101; C07K 1/047 20130101; C12N 9/1205 20130101 |
| International Class: | C12N 15/10 20060101 C12N015/10; C12P 1/04 20060101 C12P001/04; C12N 9/12 20060101 C12N009/12; C12P 21/02 20060101 C12P021/02; C07K 1/04 20060101 C07K001/04; C12P 19/02 20060101 C12P019/02 |
Claims
1.-66. (canceled)
67. A host cell comprising a heterologous glucose permease gene functionally linked to a first promoter polynucleotide sequence, wherein the first promoter polynucleotide sequence is derived from Corynebacterium glutamicum, is less than 100 base pairs in length, is able to constitutively express genes across different growth conditions, and is able to form a ladder of promoters comprising a plurality of promoters with incrementally increasing levels of promoter activity.
68. The host cell of claim 67, wherein the glucose permease gene is a gene that encodes a polypeptide with an amino acid sequence selected from SEQ ID NO: 13, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 9 and SEQ ID NO: 14.
69. The host cell of claim 67, wherein the glucose permease gene is a gene with a nucleotide sequence selected from SEQ ID NO: 23, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, SEQ ID NO: 19 and SEQ ID NO: 24.
70. The host cell of claim 67, further comprising a hexokinase gene functionally linked to a second promoter polynucleotide sequence, wherein the second promoter polynucleotide sequence is derived from Corynebacterium glutamicum, is less than 100 base pairs in length, is able to constitutively express genes across different growth conditions, and is able to form a ladder of promoters comprising a plurality of promoters with incrementally increasing levels of promoter activity.
71. The host cell of claim 70, wherein the hexokinase gene is a gene that encodes a polypeptide sequence selected from SEQ ID NO: 15 and SEQ ID NO: 16.
72. The host cell of claim 70, wherein the hexokinase gene is a gene with a nucleotide sequence selected from SEQ ID NO: 25 and SEQ ID NO: 26.
73. A method for generating a microorganism capable of increased production of a biomolecule from glucose, the method comprising: (a) genetically modifying a host microorganism, wherein the modifying comprises introducing a glucose permease gene from a library of glucose permease genes into the genome of the host microorganism, wherein each glucose permease gene from the library of glucose permease genes is functionally linked to a promoter polynucleotide sequence, wherein the promoter polynucleotide sequence is derived from Corynebacterium glutamicum, is less than 100 base pairs in length, is able to constitutively express genes across different growth conditions, and is able to form a ladder of promoters comprising a plurality of promoters with incrementally increasing levels of promoter activity, wherein the modification generates a strain of the host microorganism expressing the glucose permease gene; (b) repeating step (a) for a plurality of rounds until a plurality of strains of the host microorganism are generated, wherein each strain of the plurality of strains of the host microorganism expresses a separate glucose permease gene from the library of glucose permease genes; (c) contacting each strain of the plurality of strains of the host microorganism with a carbon source comprising glucose under fermentative conditions; and (d) selecting each strain of the host microorganism that produces an increased amount of a biomolecule from glucose as compared to the amount of the biomolecule produce from glucose from a control microorganism, wherein the control microorganism does not express a glucose permease gene from the library of glucose permease genes.
74. The method of claim 73, wherein the library of glucose permease genes comprises genes that encode polypeptide sequences of SEQ ID NO: 13, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 9, SEQ ID NO: 14 or a combination thereof.
75. The method of claim 73, wherein the library of glucose permease genes comprises genes with a nucleotide sequence of SEQ ID NO: 23, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, SEQ ID NO: 19, SEQ ID NO: 24 or a combination thereof.
76. The method of claim 73, further comprising introducing a hexokinase gene from a library of hexokinase genes, wherein each hexokinase gene from the library of hexokinase genes is functionally linked to a promoter polynucleotide sequence, wherein the promoter polynucleotide sequence is derived from Corynebacterium glutamicum, is less than 100 base pairs in length, is able to constitutively express genes across different growth conditions, and is able to form a ladder of promoters comprising a plurality of promoters with incrementally increasing levels of promoter activity.
77. The method of claim 76, wherein the library of hexokinase genes comprises genes that encode polypeptide sequences of SEQ ID NO: 15 and/or SEQ ID NO: 16.
78. The method of claim 76, wherein the library of hexokinase genes comprises genes with nucleotide sequences of SEQ ID NO: 25 and/or SEQ ID NO: 26.
79. A library of glucose permease genes, wherein each glucose permease gene in the library of glucose permease genes is functionally linked to a promoter polynucleotide sequence, wherein the promoter polynucleotide sequence is derived from Corynebacterium glutamicum, is less than 100 base pairs in length, is able to constitutively express genes across different growth conditions, and is able to form a ladder of promoters comprising a plurality of promoters with incrementally increasing levels of promoter activity.
80. The library of claim 79, wherein the library of glucose permease genes comprises genes that encode polypeptide sequences of SEQ ID NO: 13, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 9, SEQ ID NO: 14 or a combination thereof.
81. The library of claim 79, wherein the library of glucose permease genes comprises genes with nucleotide sequences of SEQ ID NO: 23, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, SEQ ID NO: 19, SEQ ID NO: 24 or a combination thereof.
82. The library of claim 79, wherein each glucose permease gene in the library of glucose permease genes is a first portion of a chimeric construct, wherein the chimeric construct comprises a second portion, wherein the second portion is a hexokinase gene.
83. The library of claim 82, wherein the hexokinase gene is functionally linked to a promoter polynucleotide sequence, wherein the promoter polynucleotide sequence is derived from Corynebacterium glutamicum, is less than 100 base pairs in length, is able to constitutively express genes across different growth conditions, and is able to form a ladder of promoters comprising a plurality of promoters with incrementally increasing levels of promoter activity.
84. The library of claim 83, wherein the library of hexokinase-genes comprises genes that encode polypeptide sequences of SEQ ID NO: 15 and/or SEQ ID NO: 16.
85. The library of claim 83, wherein the library of hexokinase genes comprises genes with nucleotide sequences of SEQ ID NO: 25 and/or SEQ ID NO: 26.
86. A method of producing a biomolecule comprising introducing a glucose permease gene from the library of claim 79 into a host cell and culturing the host cell under conditions suitable for producing the biomolecule.
Description
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of priority from U.S. Provisional Application Ser. No. 62/356,924, filed Jun. 30, 2016, which is hereby incorporated by reference in its entirety for all purposes.
STATEMENT REGARDING SEQUENCE LISTING
[0002] The Sequence Listing associated with this application is provided in text format in lieu of a paper copy, and is hereby incorporated by reference into the specification. The name of the text file containing the Sequence Listing is ZYMR_005_01WO_SeqList_ST25.txt. The text file is 49 KB, was created on Jun. 28, 2017, and is being submitted electronically via EFS-Web.
FIELD
[0003] The present disclosure is directed to microbial genomic engineering. The disclosed genomic engineering method entails the generation of a library of glucose permease genes and/or glucokinase genes and introducing said library into microbial hosts in order to produce strains with a desired phenotype (e.g. microbial production of commercial products).
BACKGROUND
[0004] Glucose transport in some microorganisms such as, for example, Corynebacterium. glutamicum is natively accomplished using the phosphotransferase transport system (PTS). In this system, phosphorylation of glucose is carried out simultaneously to transport. The phospho donor is phosphoenolpyruvate (PEP), therefore linking transport directly to glycolytic flux. In addition, the PTS system is natively regulated by a number of transcriptional processes in ways that are not always ideal for the production of commercial products.
[0005] Microbial processes for the production of various commercial products from glucose strive to maximize the efficiency with which the carbon skeleton of glucose is converted into the desired product. Control of glucose flux is critical for the production of products in ways that are dependent on the fermentation process, strain of microbial host being used (e.g., C. glutamicum), and small molecule being produced. If there is too much flux through glycolysis under high concentrations of glucose, glycolytic by-products (usually organic acids) are produced which decrease yield of product. If there is too little transport of glucose into the cell, then it is difficult to produce product at high rates. The genotypes of strains which are engineered in various ways to produce specific products interact with process conditions to lead to situations in which more or less glucose transport occurs than would be ideal to maximize yield or productivity.
[0006] Microbial strain improvement has been attempted by the expression of different glucose permeases and glucokinases which may alter glucose transport in such a way as to increase yield or productivity of commercial products. This has been demonstrated in a number of cases. For example, deletion of the native PTS system for glucose transport and overexpression of a native C. glutamicum permease along with a native C. glutamicum kinase led to the increased yield of lysine production from glucose (see Linder et al. Appl. Environ. Microbiol. June 2011 vol. 77, no. 11 pp 3571-3581, the contents of which are hereby incorporated by reference in their entirety). In another example, overexpression of the glucose permease and glucokinase from Z. mobilis in C. glutamicum was used for the production of small molecules (see U.S. Pat. No. 5,602,030, the contents of which are hereby incorporated by reference in its entirety).
[0007] However, the selection of a particular glucose permease to create the ideal level of glucose transport for a given metabolic process to produce a specific commercial product relies on a good understanding of a number of interacting factors, including the interaction of the genotype of a strain with the process environment in which fermentation takes place. Further, the correct expression, affinity, and transport rate, in combination with glucose and other carbon source concentrations may be required to deliver a balanced flux of glucose into the cell to match the flux through the pathway of interest. Understanding these parameters a priori and then choosing a single permease which embodies them can be difficult or impossible.
[0008] Thus, there is a great need in the art for new methods of engineering industrial microbes for producing specific commercial products, which do not suffer from the aforementioned drawbacks inherent with traditional strain improvement programs.
SUMMARY OF THE DISCLOSURE
[0009] In one aspect, provided herein is a host cell comprising a heterologous glucose permease gene functionally linked to a first promoter polynucleotide, wherein the first promoter polynucleotide comprises a nucleotide sequence selected from SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, and SEQ ID NO: 8. In some cases, the glucose permease gene is a bacterial glucose permease gene. In some cases, the bacterial glucose permease gene is a gene that encodes a polypeptide sequence selected from SEQ ID NO: 13, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 9 and SEQ ID NO: 14. In some cases, the bacterial glucose permease gene is a gene with a nucleotide sequence selected from SEQ ID NO: 23, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, SEQ ID NO: 19 and SEQ ID NO: 24. In some cases, the host cell further comprises a hexokinase gene functionally linked to a second promoter polynucleotide, wherein the second promoter polynucleotide comprises a nucleotide sequence selected from SEQ ID NO: 2, SEQ ID NO: 1, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, and SEQ ID NO: 8. In some cases, the hexokinase gene is a glucokinase gene. In some cases, the glucokinase gene is a bacterial glucokinase gene. In some cases, the bacterial glucokinase gene is a gene that encodes a polypeptide with an amino acid sequence selected from SEQ ID NO: 15 and SEQ ID NO: 16. In some cases, the bacterial glucokinase gene is a gene with a nucleotide sequence selected from SEQ ID NO: 25 and SEQ ID NO: 26. In some cases, the first promoter polynucleotide and the second promoter polynucleotide are different. In some cases, the first promoter polynucleotide and the second promoter polynucleotide are identical. In some cases, the host cell belongs to the genus Corynebacterium. In some cases, the host cell is Corynebacterium glutamicum. In some cases, the host cell is used in a method of producing a biomolecule from glucose comprising culturing the host cell under conditions suitable for producing the biomolecule. In some cases, the biomolecule is a small molecule, a nucleotide, an amino acid, an organic acid, or an alcohol. In some cases, the amino acid is tyrosine, phenylalanine, tryptophan, aspartic acid, asparagine, threonine, isoleucine, methionine, or lysine. In some cases, the organic acid is succinate, lactate or pyruvate. In some cases, the alcohol is ethanol or isobutanol.
[0010] In another aspect, provided herein is a method for generating a microorganism capable of increased production of a biomolecule from glucose, the method comprising: a) genetically modifying a host microorganism, wherein the modifying comprises introducing a glucose permease gene from a library of glucose permease genes into the genome of the host microorganism, wherein each glucose permease gene from the library of glucose permease genes is functionally linked to a promoter comprising a nucleotide sequence selected from SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, and SEQ ID NO: 8 and wherein the modification generates a strain of the host microorganism expressing the glucose permease gene; b) repeating step a) for a plurality of rounds until a plurality of strains of the host microorganism are generated, wherein each strain of the plurality of strains of the host microorganism expresses a separate glucose permease gene from the library of glucose permease genes; c) contacting each strain of the plurality of strains of the host microorganism with a carbon source comprising glucose under fermentative conditions; and d) selecting each strain of the host microorganism that produces an increased amount of a biomolecule from glucose as compared to the amount of the biomolecule produce from glucose from a control microorganism, wherein the control microorganism does not express a glucose permease gene from the library of glucose permease genes. In some cases, each of the glucose permease genes in the library of glucose permease genes is a bacterial glucose permease gene. In some cases, the library of bacterial glucose permease genes comprises genes that encode polypeptide sequences of SEQ ID NO: 13, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 9, SEQ ID NO: 14 or a combination thereof. In some cases, the library of bacterial glucose permease genes comprises genes with a nucleotide sequence of SEQ ID NO: 23, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, SEQ ID NO: 19, SEQ ID NO: 24 or a combination thereof. In some cases, the method further comprises introducing a hexokinase gene from a library of hexokinase genes, wherein each hexokinase gene from the library of hexokinase genes is functionally linked to a promoter polynucleotide, wherein the promoter polynucleotide comprises a sequence selected from SEQ ID NO: 2, SEQ ID NO: 1, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, and SEQ ID NO: 8. In some cases, the introduction of each hexokinase gene from the library of hexokinase genes is concurrent with the introduction of each glucose permease gene from the library of glucose permease genes. In some cases, each hexokinase gene from the library of hexokinase genes is present in a chimeric construct comprising a glucose permease gene from the library of glucose permease genes. In some cases, the hexokinase gene is a glucokinase gene. In some cases, the glucokinase gene is a bacterial glucokinase gene. In some cases, the library of bacterial glucokinase genes comprises genes that encode polypeptide sequences of SEQ ID NO: 15 and/or SEQ ID NO: 16. In some cases, the library of bacterial glucokinase genes comprises genes with nucleotide sequences of SEQ ID NO: 25 and/or SEQ ID NO: 26. In some cases, the promoter polynucleotide functionally linked to the glucose permease gene and the promoter polynucleotide functionally linked to the hexokinase gene are different. In some cases, the promoter polynucleotide functionally linked to the glucose permease gene and the promoter polynucleotide functionally linked to the hexokinase gene are identical. In some cases, the host microorganism belongs to the genus Corynebacterium. In some cases, the host microorganism is Corynebacterium glutamicum. In some cases, the introducing is performed by transformation, transduction or electroporation. In some cases, the biomolecule is a small molecule, an amino acid, a nucleotide, an organic acid, or an alcohol. In some cases, the amino acid is tyrosine, phenylalanine, tryptophan, aspartic acid, asparagine, threonine, isoleucine, methionine, or lysine. In some cases, the organic acid is succinate, lactate or pyruvate. In some cases, the alcohol is ethanol or isobutanol.
[0011] In yet another aspect, provided herein is a library of glucose permease genes, wherein each glucose permease gene in the library of glucose permease genes is functionally linked to a promoter comprising a nucleotide sequence selected from SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, and SEQ ID NO: 8. In some cases, each glucose permease gene is a bacterial glucose permease gene. In some cases, the library of bacterial glucose permease genes comprises genes that encode polypeptide sequences of SEQ ID NO: 13, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 9, SEQ ID NO: 14 or a combination thereof. In some cases, the library of bacterial glucose permease genes comprises genes with nucleotide sequences of SEQ ID NO: 23, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, SEQ ID NO: 19, SEQ ID NO: 24 or a combination thereof. In some cases, each glucose permease gene in the library of glucose permease genes is a first portion of a chimeric construct, wherein the chimeric construct comprises a second portion, wherein the second portion is a hexokinase gene. In some cases, the hexokinase gene is functionally linked to a promoter polynucleotide, wherein the promoter polynucleotide comprises a sequence selected from SEQ ID NO: 2, SEQ ID NO: 1, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, and SEQ ID NO: 8. In some cases, the hexokinase gene is a glucokinase gene. In some cases, the glucokinase gene is a bacterial glucokinase gene. In some cases, the library of bacterial glucokinase genes comprises genes that encode polypeptide sequences of SEQ ID NO: 15 and/or SEQ ID NO: 16. In some cases, the library of bacterial glucokinase genes comprises genes with nucleotides sequences of SEQ ID NO: 25 and/or SEQ ID NO: 26. In some cases, the promoter polynucleotide functionally linked to the glucose permease gene and the promoter polynucleotide functionally linked to the hexokinase gene are different. In some cases, the promoter polynucleotide functionally linked to the glucose permease gene and the promoter polynucleotide functionally linked to the hexokinase gene are identical. In some cases, the library is used in a method of producing a biomolecule comprising introducing a glucose permease gene from the library into a host cell and culturing the host cell under conditions suitable for producing the biomolecule. In some cases, the biomolecule is an amino acid, a nucleotide, an organic acid, or an alcohol. In some cases, the amino acid is tyrosine, phenylalanine, tryptophan, aspartic acid, asparagine, threonine, isoleucine, methionine, or lysine. In some cases, the organic acid is succinate, lactate or pyruvate. In some cases, the alcohol is ethanol or isobutanol. In some cases, the host cell belongs to the genus Corynebacterium. In some cases, the host cell is Corynebacterium glutamicum. In some cases, the introducing is performed by transformation, transduction or electroporation.
[0012] In another aspect, provided herein is an isolated, synthetic or recombinant polynucleotide comprising a codon optimized polynucleotide selected from SEQ ID NO: 23, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, SEQ ID NO: 19 and SEQ ID NO: 24, wherein the polynucleotide is codon optimized for expression in a host cell. In some cases, the host cell is E. coli and/or C. glutamicum.
[0013] In a further aspect, provided herein is an isolated, synthetic or recombinant polynucleotide comprising a codon optimized polynucleotide selected from SEQ ID NO: 25 and SEQ ID NO: 26, wherein the polynucleotide is codon optimized for expression in a host cell. In some cases, the host cell is E. coli and/or C. glutamicum.
[0014] In yet another aspect, provided herein is an isolated, synthetic or recombinant polynucleotide comprising a first codon optimized polynucleotide and a second codon optimized polynucleotide, wherein the first polynucleotide and the second polynucleotide are each codon optimized for expression in a host cell, and wherein the first codon optimized polynucleotide encodes a polypeptide with glucose permease activity and the second codon optimized polynucleotide encodes a polypeptide with glucokinase activity. In some cases, the first codon optimized polynucleotide is selected from SEQ ID NO: 23, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, SEQ ID NO: 19 and SEQ ID NO: 24. In some cases, the second codon optimized polynucleotide is selected from SEQ ID NO: 25 and SEQ ID NO: 26. In some cases, the polypeptide with glucose permease activity comprises a sequence selected from SEQ ID NO: 13, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 9 and SEQ ID NO: 14. In some cases, the polypeptide with glucokinase activity comprises a sequence selected from SEQ ID NO: 15 and SEQ ID NO: 16. In some cases, the host cell is E. coli and/or C. glutamicum.
BRIEF DESCRIPTION OF THE FIGURES
[0015] FIG. 1 illustrates performance of glucose permeases in evaluation method as described in Example 1.
[0016] FIG. 2 illustrates performance of glucose permeases in desired fermentation conditions as described in Example 1.
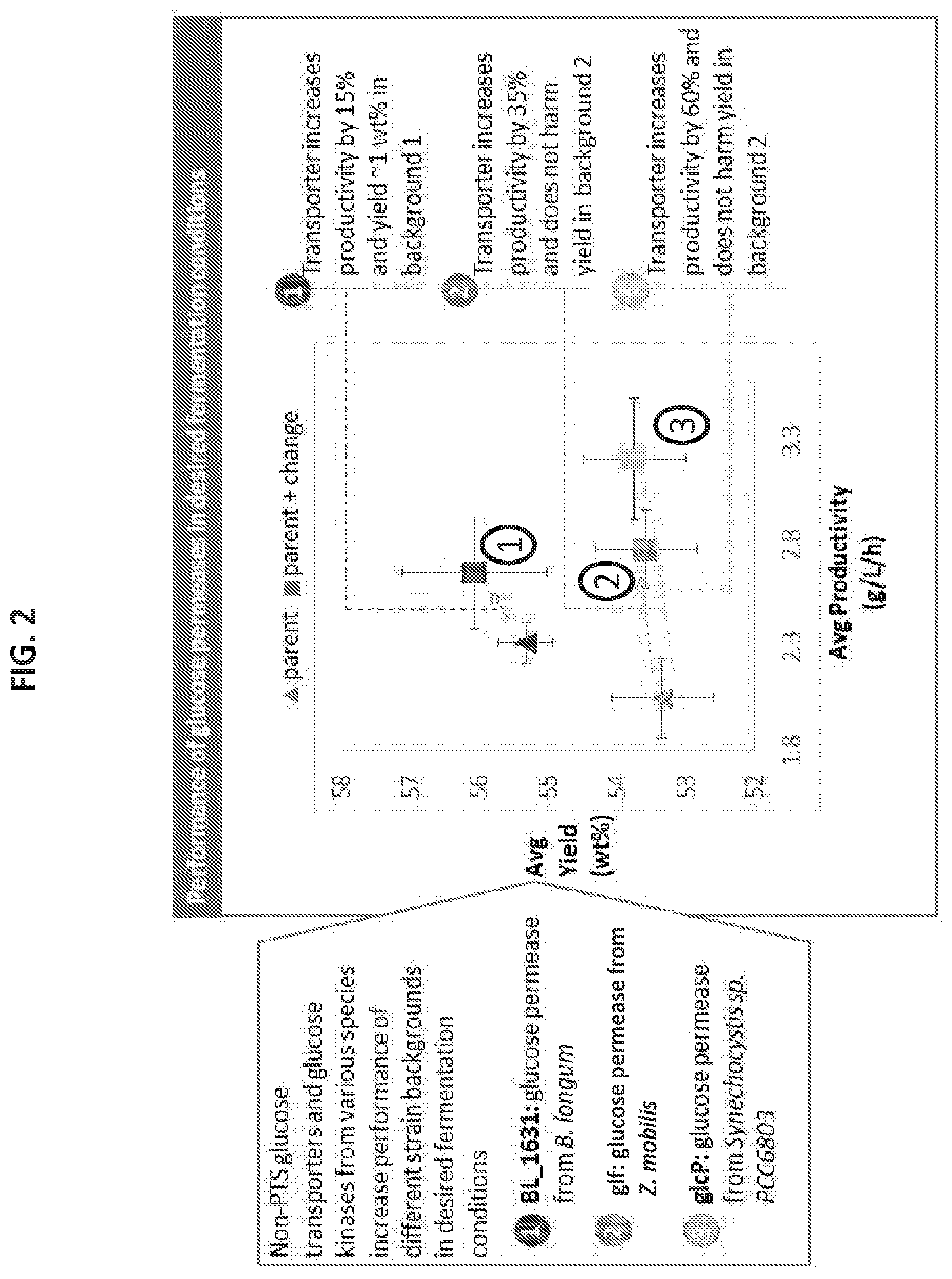
[0017] FIG. 3 illustrates assembly of transformation plasmids of the present disclosure, and their integration into host organisms. The insert sequence insert DNA is generated by combining one or more synthesized oligonucleotides in an assembly reaction. DNA inserts contain desired promoter sequence flanked by direct repeat region (i.e., homology arms) designed for looping out DNA in subsequent steps. Assembled plasmids contain the insert DNA (permease gene and/or glucokinase gene functionally linked to promoters provided herein), and optionally, one or more selection markers.
[0018] FIG. 4 illustrates a procedure for looping-out selected regions of DNA from host strains. Direct repeat (DR) regions of the inserted DNA form a loop with corresponding sequences in the host strain's genome. Cells counter selected for selection marker exhibit DNA deletion of loop DNA.
DETAILED DESCRIPTION
Definitions
[0019] While the following terms are believed to be well understood by one of ordinary skill in the art, the following definitions are set forth to facilitate explanation of the presently disclosed subject matter.
[0020] The term "a" or "an" refers to one or more of that entity, i.e. can refer to a plural referents. As such, the terms "a" or "an", "one or more" and "at least one" are used interchangeably herein. In addition, reference to "an element" by the indefinite article "a" or "an" does not exclude the possibility that more than one of the elements is present, unless the context clearly requires that there is one and only one of the elements.
[0021] Unless the context requires otherwise, throughout the present specification and claims, the word "comprise" and variations thereof, such as, "comprises" and "comprising" are to be construed in an open, inclusive sense that is as "including, but not limited to".
[0022] Reference throughout this specification to "one embodiment" or "an embodiment" means that a particular feature, structure or characteristic described in connection with the embodiment may be included in at least one embodiment of the present disclosure. Thus, the appearances of the phrases "in one embodiment" or "in an embodiment" in various places throughout this specification may not necessarily all refer to the same embodiment. It is appreciated that certain features of the invention, which are, for clarity, described in the context of separate embodiments, may also be provided in combination in a single embodiment. Conversely, various features of the invention, which are, for brevity, described in the context of a single embodiment, may also be provided separately or in any suitable sub-combination.
[0023] As used herein the terms "cellular organism" "microorganism" or "microbe" should be taken broadly. These terms can be used interchangeably and include, but may not be limited to, the two prokaryotic domains, Bacteria and Archaea, as well as certain eukaryotic fungi and protists. In some embodiments, the disclosure refers to the "microorganisms" or "cellular organisms" or "microbes" of lists/tables and figures present in the disclosure. This characterization can refer to not only the identified taxonomic genera of the tables and figures, but also the identified taxonomic species, as well as the various novel and newly identified or designed strains of any organism in said tables or figures. The same characterization holds true for the recitation of these terms in other parts of the Specification, such as in the Examples.
[0024] The term "prokaryotes" is art recognized and refers to cells which contain no nucleus or other cell organelles. The prokaryotes are generally classified in one of two domains, the Bacteria and the Archaea. The definitive difference between organisms of the Archaea and Bacteria domains is based on fundamental differences in the nucleotide base sequence in the 16S ribosomal RNA.
[0025] The term "Archaea" refers to a categorization of organisms of the division Mendosicutes, typically found in unusual environments and distinguished from the rest of the prokaryotes by several criteria, including the number of ribosomal proteins and the lack of muramic acid in cell walls. On the basis of ssrRNA analysis, the Archaea consist of two phylogenetically-distinct groups: Crenarchaeota and Euryarchaeota. On the basis of their physiology, the Archaea can be organized into three types: methanogens (prokaryotes that produce methane); extreme halophiles (prokaryotes that live at very high concentrations of salt (NaCl); and extreme (hyper) thermophilus (prokaryotes that live at very high temperatures). Besides the unifying archaeal features that distinguish them from Bacteria (i.e., no murein in cell wall, ester-linked membrane lipids, etc.), these prokaryotes exhibit unique structural or biochemical attributes which adapt them to their particular habitats. The Crenarchaeota consists mainly of hyperthermophilic sulfur-dependent prokaryotes and the Euryarchaeota contains the methanogens and extreme halophiles.
[0026] "Bacteria" or "eubacteria" refers to a domain of prokaryotic organisms. Bacteria include at least 11 distinct groups as follows: (1) Gram-positive (gram+) bacteria, of which there are two major subdivisions: (1) high G+C group (Actinomycetes, Mycobacteria, Micrococcus, others) (2) low G+C group (Bacillus, Clostridia, Lactobacillus, Staphylococci, Streptococci, Mycoplasmas); (2) Proteobacteria, e.g., Purple photosynthetic+non-photosynthetic Gram-negative bacteria (includes most "common" Gram-negative bacteria); (3) Cyanobacteria, e.g., oxygenic phototrophs; (4) Spirochetes and related species; (5) Planctomyces; (6) Bacteroides, Flavobacteria; (7) Chlamydia; (8) Green sulfur bacteria; (9) Green non-sulfur bacteria (also anaerobic phototrophs); (10) Radioresistant micrococci and relatives; (11) Thermotoga and Thermosipho thermophiles.
[0027] A "eukaryote" is any organism whose cells contain a nucleus and other organelles enclosed within membranes. Eukaryotes belong to the taxon Eukarya or Eukaryota. The defining feature that sets eukaryotic cells apart from prokaryotic cells (the aforementioned Bacteria and Archaea) is that they have membrane-bound organelles, especially the nucleus, which contains the genetic material, and is enclosed by the nuclear envelope.
[0028] The terms "genetically modified microorganism," "recombinant microorganism," "recombinant host cell," and "recombinant strain" can be used interchangeably herein and can refer to microorganisms that have been genetically modified. Thus, the terms include a microorganism (e.g., bacteria, yeast cell, fungal cell, etc.) that has been genetically altered, modified, or engineered, such that it exhibits an altered, modified, or different genotype and/or phenotype (e.g., when the genetic modification affects coding nucleic acid sequences of the microorganism), as compared to the naturally-occurring microorganism from which it was derived. It is understood that the terms refer not only to the particular recombinant microorganism in question, but also to the progeny or potential progeny of such a microorganism.
[0029] The term "wild-type microorganism" can describe a cell that occurs in nature, i.e. a cell that has not been genetically modified.
[0030] The term "genetically engineered" may refer to any manipulation of a microorganism's genome (e.g. by insertion or deletion of nucleic acids).
[0031] As used herein, the term "allele(s)" can mean any of one or more alternative forms of a gene, all of which alleles relate to at least one trait or characteristic. In a diploid cell, the two alleles of a given gene can occupy corresponding loci on a pair of homologous chromosomes. Since the present disclosure, in embodiments, relates to QTLs, i.e. genomic regions that may comprise one or more genes or regulatory sequences, it is in some instances more accurate to refer to "haplotype" (i.e. an allele of a chromosomal segment) instead of "allele", however, in those instances, the term "allele" should be understood to comprise the term "haplotype".
[0032] As used herein, the term "locus" (loci plural) can mean a specific place or places or a site on a chromosome where for example a gene or genetic marker is found.
[0033] As used herein, the term "genetically linked" can refer to two or more traits that are co-inherited at a high rate during breeding such that they are difficult to separate through crossing.
[0034] A "recombination" or "recombination event" as used herein can refer to a chromosomal crossing over or independent assortment. The term "recombinant" can refer to an organism having a new genetic makeup arising as a result of a recombination event.
[0035] As used herein, the term "phenotype" can refer to the observable characteristics of an individual cell, cell culture, organism, or group of organisms which results from the interaction between that individual's genetic makeup (i.e., genotype) and the environment.
[0036] As used herein, the term "chimeric" or "recombinant" when describing a nucleic acid sequence or a protein sequence can refer to a nucleic acid, or a protein sequence, that links at least two heterologous polynucleotides, or two heterologous polypeptides, into a single macromolecule, or that can re-arrange one or more elements of at least one natural nucleic acid or protein sequence. For example, the term "recombinant" can refer to an artificial combination of two otherwise separated segments of sequence, e.g., by chemical synthesis or by the manipulation of isolated segments of nucleic acids by genetic engineering techniques.
[0037] As used herein, a "synthetic nucleotide sequence" or "synthetic polynucleotide sequence" can be a nucleotide sequence that is not known to occur in nature or that is not naturally occurring. Generally, such a synthetic nucleotide sequence will comprise at least one nucleotide difference when compared to any other naturally occurring nucleotide sequence.
[0038] As used herein, the term "nucleic acid" can refer to a polymeric form of nucleotides of any length, either ribonucleotides or deoxyribonucleotides, or analogs thereof. This term can refer to the primary structure of the molecule, and thus includes double- and single-stranded DNA, as well as double- and single-stranded RNA. It can also include modified nucleic acids such as methylated and/or capped nucleic acids, nucleic acids containing modified bases, backbone modifications, and the like. The terms "nucleic acid" and "nucleotide sequence" can be used interchangeably.
[0039] As used herein, the term "gene" can refer to any segment of DNA associated with a biological function. Thus, genes can include, but are not limited to, coding sequences and/or the regulatory sequences required for their expression. Genes can also include non-expressed DNA segments that, for example, form recognition sequences for other proteins. Genes can be obtained from a variety of sources, including cloning from a source of interest or synthesizing from known or predicted sequence information, and may include sequences designed to have desired parameters.
[0040] As used herein, the term "homologous" or "homologue" or "ortholog" is known in the art and can refer to related sequences that share a common ancestor or family member and are determined based on the degree of sequence identity. The terms "homology," "homologous," "substantially similar" and "corresponding substantially" can be used interchangeably herein. They can refer to nucleic acid fragments wherein changes in one or more nucleotide bases do not affect the ability of the nucleic acid fragment to mediate gene expression or produce a certain phenotype. These terms can also refer to modifications of the nucleic acid fragments of the instant disclosure such as deletion or insertion of one or more nucleotides that do not substantially alter the functional properties of the resulting nucleic acid fragment relative to the initial, unmodified fragment. It is therefore understood, as those skilled in the art will appreciate, that the disclosure can encompass more than the specific exemplary sequences. These terms can describe the relationship between a gene found in one species, subspecies, variety, cultivar or strain and the corresponding or equivalent gene in another species, subspecies, variety, cultivar or strain. For purposes of this disclosure homologous sequences can be compared. "Homologous sequences" or "homologues" or "orthologs" can be thought, believed, or known to be functionally related. A functional relationship may be indicated in any one of a number of ways, including, but not limited to: (a) degree of sequence identity and/or (b) the same or similar biological function. Preferably, both (a) and (b) are indicated. Homology can be determined using software programs readily available in the art, such as those discussed in Current Protocols in Molecular Biology (F. M. Ausubel et al., eds., 1987) Supplement 30, section 7.718, Table 7.71. Some alignment programs are MacVector (Oxford Molecular Ltd, Oxford, U.K.), ALIGN Plus (Scientific and Educational Software, Pennsylvania) and AlignX (Vector NTI, Invitrogen, Carlsbad, Calif.). Another alignment program is Sequencher (Gene Codes, Ann Arbor, Mich.), using default parameters.
[0041] As used herein, the term "nucleotide change" can refer to, e.g., nucleotide substitution, deletion, and/or insertion, as is well understood in the art. For example, mutations contain alterations that produce silent substitutions, additions, or deletions, but do not alter the properties or activities of the encoded protein or how the proteins are made.
[0042] As used herein, the term "protein modification" can refer to, e.g., amino acid substitution, amino acid modification, deletion, and/or insertion, as is well understood in the art.
[0043] As used herein, the term "at least a portion" or "fragment" of a nucleic acid or polypeptide can mean a portion having the minimal size characteristics of such sequences, or any larger fragment of the full length molecule, up to and including the full length molecule. A fragment of a polynucleotide of the disclosure may encode a biologically active portion of a genetic regulatory element. A biologically active portion of a genetic regulatory element can be prepared by isolating a portion of one of the polynucleotides of the disclosure that comprises the genetic regulatory element and assessing activity as described herein. Similarly, a portion of a polypeptide may be 4 amino acids, 5 amino acids, 6 amino acids, 7 amino acids, and so on, going up to the full length polypeptide. The length of the portion to be used can depend on the particular application. A portion of a nucleic acid useful as a hybridization probe may be as short as 12 nucleotides; in some embodiments, it is 20 nucleotides. A portion of a polypeptide useful as an epitope may be as short as 4 amino acids. A portion of a polypeptide that performs the function of the full-length polypeptide can generally be longer than 4 amino acids.
[0044] Variant polynucleotides also encompass sequences that can be derived from a mutagenic and recombinogenic procedure such as DNA shuffling. Strategies for such DNA shuffling are known in the art. See, for example, Stemmer (1994) PNAS 91:10747-10751; Stemmer (1994) Nature 370:389-391; Crameri et al. (1997) Nature Biotech. 15:436-438; Moore et al. (1997) J. Mol. Biol. 272:336-347; Zhang et al. (1997) PNAS 94:4504-4509; Crameri et al. (1998) Nature 391:288-291; and U.S. Pat. Nos. 5,605,793 and 5,837,458.
[0045] For PCR amplifications of the polynucleotides disclosed herein, oligonucleotide primers can be designed for use in PCR reactions to amplify corresponding DNA sequences from cDNA or genomic DNA extracted from any organism of interest. Methods for designing PCR primers and PCR cloning are generally known in the art and are disclosed in Sambrook et al. (2001) Molecular Cloning: A Laboratory Manual (3.sup.rd ed., Cold Spring Harbor Laboratory Press, Plainview, N.Y.). See also Innis et al., eds. (1990) PCR Protocols: A Guide to Methods and Applications (Academic Press, New York); Innis and Gelfand, eds. (1995) PCR Strategies (Academic Press, New York); and Innis and Gelfand, eds. (1999) PCR Methods Manual (Academic Press, New York). Known methods of PCR can include, but are not limited to, methods using paired primers, nested primers, single specific primers, degenerate primers, gene-specific primers, vector-specific primers, partially-mismatched primers, and the like.
[0046] The term "primer" as used herein can refer to an oligonucleotide which is capable of annealing to the amplification target allowing a DNA polymerase to attach, thereby serving as a point of initiation of DNA synthesis when placed under conditions in which synthesis of primer extension product is induced, i.e., in the presence of nucleotides and an agent for polymerization such as DNA polymerase and at a suitable temperature and pH. The (amplification) primer is preferably single stranded for maximum efficiency in amplification. Preferably, the primer is an oligodeoxyribonucleotide. The primer must be sufficiently long to prime the synthesis of extension products in the presence of the agent for polymerization. The exact lengths of the primers will depend on many factors, including temperature and composition (A/T vs. G/C content) of primer. A pair of bi-directional primers consists of one forward and one reverse primer as commonly used in the art of DNA amplification such as in PCR amplification.
[0047] The terms "stringency" or "stringent hybridization conditions" can refer to hybridization conditions that affect the stability of hybrids, e.g., temperature, salt concentration, pH, formamide concentration and the like. These conditions can be empirically optimized to maximize specific binding and minimize non-specific binding of primer or probe to its target nucleic acid sequence. The terms as used can include reference to conditions under which a probe or primer will hybridize to its target sequence, to a detectably greater degree than other sequences (e.g. at least 2-fold over background). Stringent conditions can be sequence dependent and will be different in different circumstances. Longer sequences can hybridize specifically at higher temperatures. Generally, stringent conditions can be selected to be about 5.degree. C. lower than the thermal melting point (Tm) for the specific sequence at a defined ionic strength and pH. The Tm can be the temperature (under defined ionic strength and pH) at which 50% of a complementary target sequence hybridizes to a perfectly matched probe or primer. Typically, stringent conditions may be those in which the salt concentration is less than about 1.0 M Na+ ion, typically about 0.01 to 1.0 M Na+ion concentration (or other salts) at pH 7.0 to 8.3 and the temperature is at least about 30.degree. C. for short probes or primers (e.g. 10 to 50 nucleotides) and at least about 60.degree. C. for long probes or primers (e.g. greater than 50 nucleotides). Stringent conditions may also be achieved with the addition of destabilizing agents such as formamide. Exemplary low stringent conditions or "conditions of reduced stringency" can include hybridization with a buffer solution of 30% formamide, 1 M NaCl, 1% SDS at 37.degree. C. and a wash in 2.times.SSC at 40.degree. C. Exemplary high stringency conditions include hybridization in 50% formamide, 1M NaCl, 1% SDS at 37.degree. C., and a wash in 0.1.times.SSC at 60.degree. C. Hybridization procedures are well known in the art and are described by e.g. Ausubel et al., 1998 and Sambrook et al., 2001. In some embodiments, stringent conditions are hybridization in 0.25 M Na2HPO4 buffer (pH 7.2) containing 1 mM Na2EDTA, 0.5-20% sodium dodecyl sulfate at 45.degree. C., such as 0.5%, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19% or 20%, followed by a wash in 5.times.SSC, containing 0.1% (w/v) sodium dodecyl sulfate, at 55.degree. C. to 65.degree. C.
[0048] As used herein, "promoter" or "promoter polynucleotide" can refer to a DNA sequence capable of controlling the expression of a coding sequence or functional RNA. The promoter sequence consists of proximal and more distal upstream elements, the latter elements can often be referred to as enhancers. Accordingly, an "enhancer" can be a DNA sequence that can stimulate promoter activity, and may be an innate element of the promoter or a heterologous element inserted to enhance the level or tissue specificity of a promoter. Promoters may be derived in their entirety from a native gene, or be composed of different elements derived from different promoters found in nature, or even comprise synthetic DNA segments. It is understood by those skilled in the art that different promoters may direct the expression of a gene in different tissues or cell types, or at different stages of development, or in response to different environmental conditions. It is further recognized that since in most cases the exact boundaries of regulatory sequences have not been completely defined, DNA fragments of some variation may have identical promoter activity.
[0049] As used herein, the phrases "recombinant construct", "expression construct", "chimeric construct", "construct", and "recombinant DNA construct" can be used interchangeably herein. A recombinant construct comprises an artificial combination of nucleic acid fragments, e.g., regulatory and coding sequences that are not found together in nature. For example, a chimeric construct may comprise regulatory sequences and coding sequences that are derived from different sources, or regulatory sequences and coding sequences derived from the same source, but arranged in a manner different than that found in nature. In some cases, a chimeric construct can be a recombinant construct comprising a plurality of regulatory (e.g., promoter) and coding sequences (e.g., glucose permease gene and hexokinase gene (glucokinase gene)). Each coding sequence in a chimeric construct comprising a plurality of coding sequences can be controlled by or functionally linked to a separate regulatory sequence). Such constructs described herein may be used by itself or may be used in conjunction with a vector. If a vector is used then the choice of vector can be dependent upon the method that will be used to transform host cells as is well known to those skilled in the art. For example, a plasmid vector can be used. The skilled artisan is well aware of the genetic elements that must be present on the vector in order to successfully transform, select and propagate host cells comprising any of the isolated nucleic acid fragments of the disclosure. The skilled artisan will also recognize that different independent transformation events will result in different levels and patterns of expression (Jones et al., (1985) EMBO J. 4:2411-2418; De Almeida et al., (1989) Mol. Gen. Genetics 218:78-86), and thus that multiple events must be screened in order to obtain lines displaying the desired expression level and pattern. Such screening may be accomplished by Southern analysis of DNA, Northern analysis of mRNA expression, immunoblotting analysis of protein expression, or phenotypic analysis, among others. Vectors can be plasmids, viruses, bacteriophages, pro-viruses, phagemids, transposons, artificial chromosomes, and the like, that replicate autonomously or can integrate into a chromosome of a host cell. A vector can also be a naked RNA polynucleotide, a naked DNA polynucleotide, a polynucleotide composed of both DNA and RNA within the same strand, a poly-lysine-conjugated DNA or RNA, a peptide-conjugated DNA or RNA, a liposome-conjugated DNA, or the like, that is not autonomously replicating. As used herein, the term "expression" refers to the production of a functional end-product e.g., an mRNA or a protein (precursor or mature).
[0050] "Operably linked" or "functionally linked" can mean in this context the sequential arrangement of the promoter polynucleotide according to the disclosure with a further oligo- or polynucleotide (e.g., glucose permease gene and/or glucokinase gene), resulting in transcription of said further polynucleotide (e.g., glucose permease gene and/or glucokinase gene). In other words, "operably linked" or "functionally linked" can mean the promoter controls the transcription of the gene (e.g. glucose permease gene and/or glucokinase gene) adjacent or downstream or 3' to said promoter.
[0051] The term "carbon source" generally can refer to a substance suitable to be used as a source of carbon for cell growth. Carbon sources can include, but are not limited to, biomass hydrolysates, starch, sucrose, cellulose, hemicellulose, xylose, and lignin, as well as monomeric components of these substrates. Carbon sources can comprise various organic compounds in various forms, including, but not limited to polymers, carbohydrates, acids, alcohols, aldehydes, ketones, amino acids, peptides, etc. These can include, for example, various monosaccharides such as glucose, dextrose (D-glucose), maltose, oligosaccharides, polysaccharides, saturated or unsaturated fatty acids, succinate, lactate, acetate, ethanol, etc., or mixtures thereof. Photosynthetic organisms can additionally produce a carbon source as a product of photosynthesis. In some embodiments, carbon sources may be selected from biomass hydrolysates and glucose.
[0052] The term "feedstock" can be defined as a raw material or mixture of raw materials supplied to a microorganism or fermentation process from which other products can be made. For example, a carbon source, such as biomass or the carbon compounds derived from biomass can be a feedstock for a microorganism that produces a product of interest (e.g. small molecule, peptide, synthetic compound, fuel, alcohol, etc.) in a fermentation process. However, a feedstock may contain nutrients other than a carbon source.
[0053] The term "volumetric productivity" or "production rate" can be defined as the amount of product formed per volume of medium per unit of time. Volumetric productivity can be reported in gram per liter per hour (g/L/h).
[0054] The term "specific productivity" can defined as the rate of formation of the product. To describe productivity as an inherent parameter of the microorganism and not of the fermentation process, productivity can herein further be defined as the specific productivity in gram product per gram of cell dry weight (CDW) per hour (g/g CDW/h). Using the relation of CDW to OD.sub.600 for the given microorganism specific productivity can also be expressed as gram product per liter culture medium per optical density of the culture broth at 600 nm (OD) per hour (g/L/h/OD)
[0055] The term "yield" can be defined as the amount of product obtained per unit weight of raw material and may be expressed as g product per g substrate (g/g). Yield may be expressed as a percentage of the theoretical yield. "Theoretical yield" is defined as the maximum amount of product that can be generated per a given amount of substrate as dictated by the stoichiometry of the metabolic pathway used to make the product.
[0056] The term "titre" or "titer" can be defined as the strength of a solution or the concentration of a substance in solution. For example, the titre of a product of interest (e.g. small molecule, peptide, synthetic compound, fuel, alcohol, etc.) in a fermentation broth can be described as g of product of interest in solution per liter of fermentation broth (g/L).
[0057] The term "total titer" can be defined as the sum of all product of interest produced in a process, including but not limited to the product of interest in solution, the product of interest in gas phase if applicable, and any product of interest removed from the process and recovered relative to the initial volume in the process or the operating volume in the process.
[0058] As used herein, the term "glucose permease" can refer to any transporter (e.g., myo-inositol transporter and/or glucose permease) that exhibits an affinity for glucose and subsequently facilitates its transport across the cell membrane of a host cell. The glucose permease can be a transmembrane protein. The transport can be passive transport whereby glucose diffuses in or out of the host cell as facilitated by the glucose permease. The glucose permease can be derived from a prokaryotic cell (i.e., Bacteria or Archaea) or a eukaryotic cell (e.g., a fungal cell). The prokaryotic glucose permease protein can be from any genus and species of bacteria or Archaea known in the art. The eukaryotic glucose permease protein can be, for example, from any genus and species of fungus known in the art. The term "bacterial glucose permease" as used herein can refer to a glucose permease as described herein derived from a bacteria.
[0059] As used herein, the term "glucose permease gene" can refer to any nucleic acid (e.g., genomic DNA, cDNA and/or mRNA) that when transcribed and/or translated encodes a glucose permease protein as described herein. The term "bacterial glucose permease gene" as used herein can refer to a bacterial glucose permease protein as described herein derived from a bacteria.
[0060] As used herein, the term "hexokinase" can refer to any protein derived from a prokaryotic cell (i.e., Bacteria or Archaea) or a eukaryotic cell (e.g., a fungal cell) that is an enzyme that can facilitate the phosphorylation of a hexose (six-carbon sugar). As provided herein, a hexokinase can be a glucokinase. As used herein, the term "glucokinase" can refer to any protein derived from a prokaryotic cell (i.e., Bacteria or Archaea) or a eukaryotic cell (e.g., a fungal cell) that is an enzyme that can facilitate the phosphorylation of glucose to glucose-6-phosphate. The prokaryotic hexokinase or glucokinase protein can be from any genus and species of bacteria or Archaea known in the art. The eukaryotic hexokinase or glucokinase protein can be, for example, from any genus and species of fungus known in the art. The term "bacterial hexokinase" as used herein can refer to a hexokinase as described herein derived from a bacteria. The term "bacterial glucokinase" as used herein can refer to a glucokinase as described herein derived from a bacteria.
[0061] As used herein, the "hexokinase gene" can refer to any nucleic acid (e.g., genomic DNA, cDNA and/or mRNA) that when transcribed and/or translated encodes a hexokinase protein as described herein. As used herein, the "glucokinase gene" can refer to any nucleic acid (e.g., genomic DNA, cDNA and/or mRNA) that when transcribed and/or translated encodes a glucokinase protein as described herein. The term "bacterial hexokinase gene" as used herein can refer to a bacterial hexokinase protein as described herein derived from a bacteria. The term "bacterial glucokinase gene" as used herein can refer to a bacterial glucokinase protein as described herein derived from a bacteria.
[0062] The term "product of interest" or "biomolecule" as used herein refers to any product produced by microbes from feedstock. In some cases, the product of interest may be a small molecule, enzyme, peptide, amino acid, organic acid, synthetic compound, fuel, alcohol, etc. For example, the product of interest or biomolecule may be any primary or secondary extracellular metabolite. The primary metabolite may be, inter alia, ethanol, citric acid, lactic acid, glutamic acid, glutamate, lysine, threonine, tryptophan and other amino acids, vitamins, polysaccharides, etc. The secondary metabolite may be, inter alia, an antibiotic compound like penicillin, or an immunosuppressant like cyclosporin A, a plant hormone like gibberellin, a statin drug like lovastatin, a fungicide like griseofulvin, etc. The product of interest or biomolecule may also be any intracellular component produced by a microbe, such as: a microbial enzyme, including: catalase, amylase, protease, pectinase, glucose isomerase, cellulase, hemicellulase, lipase, lactase, streptokinase, and many others. The intracellular component may also include recombinant proteins, such as: insulin, hepatitis B vaccine, interferon, granulocyte colony-stimulating factor, streptokinase and others.
Overview
[0063] The present disclosure provides a microbial genomic engineering method that does not suffer from the myriad of problems associated with traditional microbial strain improvement programs.
[0064] One aspect provided herein is a method for generating a microorganism (e.g., bacteria) that is capable of increased production of a biomolecule or product of interest. In general, the methods for generating a microorganism for use in producing any biomolecule as provided herein can entail genetically modifying a host microorganism by introducing a member of a library of target genes into said host microorganism to generate a genomically engineered strain of said microorganism, culturing said engineered strain under conditions suitable to produce the biomolecule or product of interest, and selecting said engineered strain if said engineered strain produces an increased amount of the biomolecule or product of interest. The increased amount can be as compared to a wild-type strain of the host microorganism. The increased amount can be as compared to a strain of the host microorganism that does not contain a member of the library of target genes. The library of target genes can comprise a plurality of vectors, wherein each vector in the library comprises a chimeric construct comprising at least one promoter polynucleotide functionally linked or coupled to a target gene.
[0065] An exemplary workflow of one of the embodiments of the invention entails selecting a target gene, acquiring or synthesizing nucleic acid (e.g., DNA) for the target gene, and cloning said acquired or synthesized target gene into a suitable vector. Any method known in the art and/or provided herein can be used to assemble or clone the target gene or target genes into a suitable vector. The vector can be any vector known in the art and/or provided herein that is compatible with the host microorganism to be utilized. Once the vector comprising the target gene(s) is assembled, it can be introduced into the host microorganism. The introduction of the vector can be using any method known in the art and/or provided herein. The host microorganism can be any host microorganism provided herein. Once introduced into the host microorganism, genetically modified hosts can be selected and the insertion of the target gene(s) can be evaluated. The target gene(s) can be engineered to be inserted into specific locations of the host microorganism's genome. In some cases, the target gene(s) is inserted into a neutral site of the genome that facilitates expression of the target gene(s) without perturbing unintended pathways/processes within the host microorganism. In some cases, the target gene(s) replace specific gene(s) within the host microorganism. The specific gene can be the homologous target gene normally present in the host microorganism. The integration site, such as, for example, the neutral integration site can be determined empirically such that various sites can tested and a site that permits expression of the integrated target gene(s) without being detrimental to the host cell can be chosen. Integration into a desired site (e.g., neutral site) can be facilitated by cloning the target gene(s) into a vector comprising portions of sequence homologous to the desired integration site (i.e., homologous arms) and subsequently performing a recombination event in the host cell. The target gene(s) can be inserted between the portions of homologous sequence. In one embodiment, the vector comprises about 2 kb of sequence homologous to the desired integration site. The sequence homologous to the desired site can flank a glucose permease gene insert and/or glucose permease-glucokinase gene insert such that a first portion of the sequence is upstream (i.e., 5') of the gene insert and a second portion of the sequence is downstream (i.e., 3') of the gene insert. In another embodiment, the vector comprises about 4 kb of sequence homologous to the desired integration site. In this embodiment, the vector comprises about 2 kb of sequence homologous to the desired integration site upstream (i.e., 5') to a glucose permease gene insert and/or glucose permease-glucokinase gene insert and about 2 kb of sequence homologous to the desired integration site downstream (i.e., 3') to a glucose permease gene insert and/or glucose permease-glucokinase gene insert. In one embodiment, integration is performed by a single-cross-over integration and subsequent loop out of the plasmid backbone facilitated by counter-selection on a marker present in the vector backbone. In one embodiment, the target gene is any bacterial glucose permease gene known in the art and/or provided herein. In one embodiment, the target gene is any bacterial glucokinase gene known in the art and/or provided herein. In one embodiment, target genes are any bacterial glucose permease gene known in the art and/or provided herein and any bacterial glucokinase gene known in the art and/or provided herein.
[0066] Evaluation of the insertion can be performed using any method know in the art such as, for example, amplifying and/or sequencing of the genetically modified microorganism's genome or portions thereof. In some cases, the methods provided herein also entail the removal or looping out of selection markers through counter selection as described herein. The looping out can be performed using any of the methods provided herein.
[0067] Following the evaluation of the insertion of the target gene(s) and, optional, removal of selection markers, the genetically modified strain can be evaluated for its ability to produce a biomolecule or product of interest. Prior to evaluation an optional step can be expanding the strain. Expansion can entail culturing the genetically modified strain on plates or in wells in a multi-well plate in growth media suitable for expansion. The evaluation step can entail culturing the genetically modified strain on plates or in wells in a multi-well plate comprising growth media/conditions designed to mimic actual conditions for producing a biomolecule or product of interest. In some cases, the growth media in this step is suitable for the production of biomolecules or products of interest derived from the metabolic processing of glucose. If the genetically modified strain possesses or is predicted to produce a desired or threshold rate of production or yield of the biomolecule or product of interest as determined from the evaluation step, the strain can be selected and placed in cold storage. The prediction can be based on measuring the amount of product of interest and biomass formed at various time points during culturing of the strain and using said measurements to predict how said strain will perform under expanded or larger scale conditions (e.g., fermentation conditions). In one embodiment, the prediction is based on a linear regression analysis of the performance of the strain during the evaluation method.
[0068] In some cases, a genetically modified strain possessing or predicted to produce a desired or threshold rate of production or yield of the biomolecule or product of interest is transferred to or grown in a larger culture under conditions for producing the biomolecule or product of interest (e.g., fermentation conditions). This step can be used in order to determine if the selected strain can perform as predicted under actual conditions for the production of the biomolecule or product of interest. In some cases, the steps provided herein for the introduction and evaluation of each target gene from a library of target genes such as those provided herein are repeated for each target gene from the library in order to select one or more strains of genetically modified microorganisms that produce a desired or threshold yield and/or productivity rate of a biomolecule or product of interest.
[0069] In one embodiment, the biomolecule or product of interest is derived from glucose and the metabolic processing thereof by the microorganism such that the methods provided herein entail the generation of a strain or strains of microorganisms that produce an increased amount of a biomolecule or product of interest derived from the metabolic processing of glucose by the strain or strains. In one embodiment, the methods provided herein entail the introduction of one or more target genes involved in glucose transport and/or metabolism. In one embodiment, the one or more target genes are utilized in a phosphotransferase system (PTS). In one embodiment, the target gene is a glucose permease gene such that a glucose permease gene is introduced into the host microorganism in the methods provided herein. The glucose permease gene can be a heterologous gene in the host microorganism. In one embodiment, the target gene is a hexokinase gene such that a hexokinase gene is introduced into the host microorganism in the methods provided herein. In one embodiment, both a glucose permease gene and a hexokinase gene are introduced into the host microorganism in the methods provided herein. In one embodiment, the introduction of a glucose permease gene and/or hexokinase gene into the host microorganism produces a non-PTS recombinant glucose uptake system in the host microorganism. The recombinant glucose uptake system can serve to uncouple glucose transport from phosphoenolpyruvate (PEP) utilization, thereby producing more PEP for the synthesis of biomolecules or products of interest. The biomolecules or products of interest produced by the methods provided herein can be any commercial product produced from glucose. In some cases, the biomolecule or product of interest is a small molecule, an amino acid, an organic acid, or an alcohol. The amino acid can be tyrosine, phenylalanine, tryptophan, aspartic acid, asparagine, threonine, isoleucine, methionine, or lysine. The organic acid can be succinate, lactate or pyruvate. The alcohol can be ethanol or isobutanol.
[0070] In one embodiment, the disclosed microbial genomic engineering method utilizes a library of glucose permease genes and/or hexokinase genes. A glucose permease gene can be selected based on the glucose permeases affinity for glucose and/or glucose transport rate. In some cases, the microbes are engineered utilizing a glucose permease library, a hexokinase (e.g., glucokinase) library or a combination of glucose permease and hexokinase (e.g., glucokinase) libraries. In one embodiment, the library contains a plurality of chimeric construct inserts such that each insert in the library comprises a glucose permease gene and a hexokinase (e.g., glucokinase) gene. Following engineering, the microbes can be efficiently screened or evaluated for resultant outcome, e.g. production of a product from glucose as provided herein. This process of utilizing the libraries provided herein to define particular genomic alterations and then testing/screening host microbial genomes harboring the alterations can be implemented in an efficient and iterative manner and can be used to identify specific combinations of glucose permease/hexokinase genes (e.g., glucokinase genes) whose expression in a host cell produces a desired or threshold level of a biomolecule or product of interest form glucose.
[0071] In one embodiment, each glucose permease gene or hexokinase gene (glucokinase gene) as provided herein for use in the methods provided herein is under the control of or functionally linked to a native promoter or any of the promoter polynucleotides provided herein. A "promoter polynucleotide" or a "promoter" or a "polynucleotide having promoter activity" can mean a polynucleotide, preferably deoxyribopolynucleotide, or a nucleic acid, preferably deoxyribonucleic acid (DNA), which when functionally linked to a polynucleotide to be transcribed determines the point and frequency of initiation of transcription of the coding polynucleotide (e.g., glucose permease gene or glucokinase gene), thereby enabling the strength of expression of the controlled polynucleotide to be influenced. In one embodiment, each glucose permease gene and/or hexokinase gene (e.g., glucokinase gene) in a library comprising glucose permease genes and/or hexokinase genes (e.g., glucokinase genes) is under the control of the same or an identical promoter. In one embodiment, each glucose permease gene and/or hexokinase gene (e.g., glucokinase gene) in a library comprising glucose permease genes and/or hexokinase genes (e.g., glucokinase genes) is under the control of separate or different promoter. In yet another embodiment, each target gene in a chimeric construct in a library of chimeric constructs comprising the target genes are under the control of the same or an identical promoter. In a further embodiment, each target gene in a chimeric construct in a library of chimeric constructs comprising the target genes are under the control of a separate or different promoter.
[0072] In one embodiment, provided herein is a promoter ladder for use in generating a library of glucose permease genes or hexokinase genes or glucokinase genes. The term "promoter ladder" as used herein refers to a plurality of promoters with incrementally increasing levels of promoter activity. The term "promoter activity" as used herein refers to the ability of the promoter to initiate transcription of a polynucleotide sequence into mRNA. Methods of assessing promoter activity are well known to those of skill in the art and can include, for example the methods described in Example 2 of U.S. 62/264,232, filed on Dec. 7, 2015 and PCT/US16/65464 (i.e., PCT Publication No. WO2017/100376), each of which is herein incorporated by references in its entirety. The term "constitutive promoter" as used herein can refer to a promoter that directs the transcription of its associated genes at a constant rate regardless of the internal or external cellular conditions.
Promoters
[0073] In some embodiments, the present disclosure teaches methods of selecting promoters with optimal expression properties to modulate RNA degradation function and produce beneficial effects on overall-host strain productivity.
[0074] Promoters regulate the rate at which genes are transcribed and can influence transcription in a variety of ways. Constitutive promoters, for example, direct the transcription of their associated genes at a constant rate regardless of the internal or external cellular conditions, while regulatable promoters increase or decrease the rate at which a gene is transcribed depending on the internal and/or the external cellular conditions, e.g. growth rate, temperature, responses to specific environmental chemicals, and the like. Promoters can be isolated from their normal cellular contexts and engineered to regulate the expression of virtually any gene, enabling the effective modification of cellular growth, product yield and/or other phenotypes of interest.
[0075] In some embodiments, the present disclosure teaches methods of identifying one or more promoters and/or generating variants of one or more promoters within a host cell, which exhibit a range of expression strengths (e.g. promoter ladders discussed infra), or superior regulatory properties (i.e., tighter regulatory control for selected genes). A particular combination of these identified and/or generated promoters can be grouped together as a promoter ladder for use in the RNA degradation perturbation experiments explained in more detail below.
[0076] In some embodiments, promoter ladders are created by identifying natural, native, or wild-type promoters associated with a target gene of interest that have a range of expression strengths. These identified promoters can be grouped together as a promoter ladder.
[0077] In some embodiments, promoter ladders are created by: identifying natural, native, or wild-type promoters associated with a target gene of interest and then mutating said promoter to derive multiple mutated promoter sequences. Each of these mutated promoters is tested for effect on target gene expression. In some embodiments, the edited promoters are tested for expression activity across a variety of conditions, such that each promoter variant's activity is documented/characterized/annotated and stored in a database. The resulting edited promoter variants are subsequently organized into promoter ladders arranged based on the strength of their expression (e.g., with highly expressing variants near the top, and attenuated expression near the bottom, therefore leading to the term "ladder").
[0078] In some embodiments, the present disclosure teaches promoter ladders that are a combination of identified naturally occurring promoters and mutated variant promoters.
[0079] In some embodiments, the present disclosure teaches methods of identifying natural, native, or wild-type promoters that satisfied both of the following criteria: 1) represented a ladder of constitutive promoters; and 2) could be encoded by short DNA sequences, ideally less than 100 base pairs. In some embodiments, constitutive promoters of the present disclosure exhibit constant gene expression across two selected growth conditions (typically compared among conditions experienced during industrial cultivation). In some embodiments, the promoters of the present disclosure will consist of a .about.60 base pair core promoter, and a 5' UTR between 26- and 40 base pairs in length.
[0080] In some embodiments, one or more of the aforementioned identified naturally occurring promoter sequences are chosen for gene editing. In some embodiments, the natural promoters are edited via any known genetic mutation methods. In other embodiments, the promoters of the present disclosure are edited by synthesizing new promoter variants with the desired sequence.
[0081] The entire disclosures of U.S. Patent Application No. 62/264,232, filed on Dec. 7, 2015, and PCT/US16/65464 (PCT Publication No. WO2017/100376), filed Dec. 7, 2016 are each hereby incorporated by reference in its entirety for all purposes.
[0082] A non-exhaustive list of the promoters of the present disclosure is provided in Table 1 below. Each of the promoter sequences in Table 1 can be referred to as a heterologous promoter or heterologous promoter polynucleotide.
TABLE-US-00001 TABLE 1 Selected promoter sequences of the present disclosure. SEQ ID Promoter Short No. Name 1 P1 2 P2 3 P3 4 P4 5 P5 6 P6 7 P7 8 P8
[0083] In some embodiments, the promoters of the present invention exhibit at least 100%, 99%, 98%, 97%, 96%, 95%, 94%, 93%, 92%, 91%, 90%, 89%, 88%, 87%, 86%, 85%, 84%, 83%, 82%, 81%, 80%, 79%, 78%, 77%, 76%, or 75% sequence identity with a promoter sequences from Table 1.
Glucose Permeases
[0084] Provided herein is a library of glucose permease genes for use in the methods provided herein. The library of glucose permease genes can comprise one or more glucose permease genes. Each glucose permease gene in the library can be a native form of the glucose permease gene or a mutated form. The mutated form can comprise one or more mutations selected from an insertion, deletion, single nucleotide polymorphism (SNP), or translocation. Each glucose permease gene in the library can be a bacterial glucose permease gene. The glucose permease gene can be any glucose permease gene from a prokaryotic cell (i.e., Bacteria and/or Archaea) known in the art. The glucose permease gene can be any glucose permease gene from a eukaryotic cell (e.g., fungal) known in the art. A glucose permease can be considered any protein comprising glucose permease activity. For example, a glucose permease for use herein can be any transporter (e.g., myo-inositol transporter) that exhibits an affinity for glucose and subsequently facilitates its transport across the cell membrane of a host cell. The host cell can be any host cell provided herein. In one embodiment, the library of glucose permease genes comprises glucose permease genes from any strain/species/sub-species of Mycobacterium (e.g., Mycobacterium smegmatis), Streptomyces (e.g., Streptomyces coelicolor), Zymomonas (e.g., Zymomonas mobilis), Synechocystis (e.g., Synechocystis sp. PCC6803), Bifidobacterium (e.g., Bifidobacterium longum), Escherichia (e.g., Escherichia coli), Bacillus (e.g., Bacillus subtilis), Corynebacterium (e.g., Corynebacterium glutamicum), Saccharomyces (e.g., S. cerevisiae) or a combination thereof. In one embodiment, the library of glucose permease genes comprises glucose permease genes that encode polypeptide sequences selected from SEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 13, SEQ ID NO: 14 or a combination thereof.
[0085] In some embodiments, the permeases of the present invention exhibit at least 100%, 99%, 98%, 97%, 96%, 95%, 94%, 93%, 92%, 91%, 90%, 89%, 88%, 87%, 86%, 85%, 84%, 83%, 82%, 81%, 80%, 79%, 78%, 77%, 76%, or 75% sequence identity with a permease provided herein.
[0086] In one embodiment, the library of glucose permease genes comprises glucose permease genes selected from SEQ ID NO: 19, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, SEQ ID NO: 23, SEQ ID NO: 24 or a combination thereof.
[0087] In some embodiments, the permease genes of the present invention exhibit at least 100%, 99%, 98%, 97%, 96%, 95%, 94%, 93%, 92%, 91%, 90%, 89%, 88%, 87%, 86%, 85%, 84%, 83%, 82%, 81%, 80%, 79%, 78%, 77%, 76%, or 75% sequence identity with a permease gene provided herein.
[0088] Each glucose permease in the library can be functionally linked or under the control of its native promoter or a mutated form of its native promoter. Each glucose permease gene in the library can be functionally linked to or controlled by any promoter provided herein. Each glucose permease gene in the library can be controlled by a promoter polynucleotide sequence that comprises a sequence selected from SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 7, and SEQ ID NO: 8. Each glucose permease gene in the library can be controlled by a promoter polynucleotide sequence that contains a sequence selected from SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, and SEQ ID NO: 8. In one embodiment, each glucose permease gene in the library is present as a set of glucose permease genes, wherein each set has one glucose permease gene functionally linked to SEQ ID NO. 1, one glucose permease gene functionally linked to SEQ ID NO. 2, one glucose permease gene functionally linked to SEQ ID NO. 3, one glucose permease gene functionally linked to SEQ ID NO. 4, one glucose permease gene functionally linked to SEQ ID NO. 5, one glucose permease gene functionally linked to SEQ ID NO. 6, one glucose permease gene functionally linked to SEQ ID NO. 7 and one glucose permease gene functionally linked to SEQ ID NO. 8 or a combination thereof. Each glucose permease gene in a library of glucose permease genes can be present in a chimeric construct such that the gene can be flanked by one or more regulatory sequences and/or sequence homologous to sequence present in the genome of a host cell. The sequence homologous to sequence present in the host cell can facilitate integration of the glucose permease gene into a site or locus of the host cell genome that comprises complementary sequence. Integration can be via a recombination event. The regulatory sequence can be any regulatory sequence known in the art or provided herein such as, for example, a promoter, start, stop, signal, secretion and/or termination sequence used by the genetic machinery of the host cell.
Hexokinases
[0089] Provided herein is a library of hexokinase genes for use in the methods provided herein. The library of hexokinase genes can comprise one or more hexokinase genes. Each hexokinase gene in the library can be a native form or a mutated form of the gene. The mutated form can comprise one or more mutations selected from an insertion, deletion, single nucleotide polymorphism (SNP), or translocation. Each hexokinase gene can be a glucokinase gene. Each glucokinase gene in the library can be a bacterial glucokinase gene. The glucokinase gene can be any glucokinase gene from a prokaryotic cell (i.e., Bacteria and/or Archaea) known in the art. The glucokinase gene can be any glucokinase gene from a eukaryotic cell (e.g., fungal) known in the art. A glucokinase can be considered any kinase known in the art that can utilize glucose as a substrate and phosphorylate glucose to produce glucose-6-phosphate. In one embodiment, the library of glucokinase genes comprises glucokinase genes from any strain/species/sub-species of Corynebactium (e.g., C. glutamicum), Zymomonas (e.g., Zymomonas mobilis), Staphylococcus (e.g., S. aureus glkA), Enterococcus (e.g., E. faecalis), Escherichia (e.g., E. coli), Clostridium (e.g., C. difficile), Streptococcus (e.g., S. pneumonia), Bacillus (e.g., B. anthracis), Renibacterium (e.g., R. salmoninarium), Saccharomyces (e.g., S. cerevisiae) or a combination thereof. In one embodiment, the library of glucokinase genes comprises glucokinase genes that encode polypeptide sequences selected from SEQ ID NO: 15 and/or SEQ ID NO: 16.
[0090] In some embodiments, the hexokinases (e.g., glucokinases) of the present invention exhibit at least 100%, 99%, 98%, 97%, 96%, 95%, 94%, 93%, 92%, 91%, 90%, 89%, 88%, 87%, 86%, 85%, 84%, 83%, 82%, 81%, 80%, 79%, 78%, 77%, 76%, or 75% sequence identity with a hexokinase (e.g., glucokinase) provided herein.
[0091] In one embodiment, the library of glucokinase genes comprises glucokinase genes selected from SEQ ID NO: 25 and/or SEQ ID NO: 26.
[0092] In some embodiments, the hexokinase genes (e.g., glucokinase genes) of the present invention exhibit at least 100%, 99%, 98%, 97%, 96%, 95%, 94%, 93%, 92%, 91%, 90%, 89%, 88%, 87%, 86%, 85%, 84%, 83%, 82%, 81%, 80%, 79%, 78%, 77%, 76%, or 75% sequence identity with a hexokinase gene (e.g., glucokinase gene) provided herein.
[0093] Each hexokinase gene (e.g., glucokinase gene) in the library can be functionally linked or under the control of its native promoter or a mutated form of its native promoter. Each hexokinase gene (e.g., glucokinase gene) in the library can be functionally linked to or controlled by any promoter provided herein. Each hexokinase gene (e.g., glucokinase gene) in the library can be controlled by a promoter polynucleotide sequence that comprises a sequence selected from SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 7, and SEQ ID NO: 8. Each hexokinase gene (e.g., glucokinase gene) in the library can be controlled by a promoter polynucleotide sequence that contains a sequence selected from SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, and SEQ ID NO: 8. In one embodiment, each hexokinase gene (e.g., glucokinase gene) in the library is present as a set of hexokinase genes (e.g., glucokinase genes), wherein each set has one hexokinase gene (e.g., glucokinase gene) functionally linked to SEQ ID NO. 1, one hexokinase gene (e.g., glucokinase gene) functionally linked to SEQ ID NO. 2, one hexokinase gene (e.g., glucokinase gene) functionally linked to SEQ ID NO. 3, one hexokinase gene (e.g., glucokinase gene) functionally linked to SEQ ID NO. 4, one hexokinase gene (e.g., glucokinase gene) functionally linked to SEQ ID NO. 5, one hexokinase gene (e.g., glucokinase gene) functionally linked to SEQ ID NO. 6, one hexokinase gene (e.g., glucokinase gene) functionally linked to SEQ ID NO. 7 and one hexokinase gene (e.g., glucokinase gene) functionally linked to SEQ ID NO. 8 or a combination thereof. Each hexokinase gene in a library of hexokinase genes can be present in a chimeric construct such that the gene can be flanked by one or more regulatory sequences and/or sequence homologous to sequence present in the genome of a host cell. The sequence homologous to sequence present in the host cell can facilitate integration of the hexokinase gene into a site or locus of the host cell genome that comprises complementary sequence. Integration can be via a recombination event. The regulatory sequence can be any regulatory sequence known in the art or provided herein such as, for example, a promoter, start, stop, signal, secretion and/or termination sequence used by the genetic machinery of the host cell.
[0094] Provided herein is a library comprising glucose permease genes and hexokinase genes for use in the methods provided herein. In one embodiment, the glucose permease genes and the hexokinase genes are present in a single chimeric insert. Each glucose permease gene or hexokinase gene (e.g., glucokinase gene) in a chimeric construct in the library can be a native form or a mutated form of either gene. A mutated form of either gene can comprise one or more mutations selected from an insertion, deletion, single nucleotide polymorphism (SNP), or translocation. The glucose permease gene can be a bacterial glucose permease gene. The glucose permease gene can be any bacterial glucose permease gene known in the art. In one embodiment, a glucose permease gene in a chimeric construct comprises a glucose permease gene from any strain/species/sub-species of Mycobacterium (e.g., Mycobacterium smegmatis), Streptomyces (e.g., Streptomyces coelicolor), Zymomonas (e.g., Zymomonas mobilis), Synechocystis (e.g., Synechocystis sp. PCC6803), or Bifidobacterium (e.g., Bifidobacterium longum) Escherichia (e.g., Escherichia coli), Bacillus (e.g., Bacillus subtilis) or Corynebacterium (e.g., Corynebacterium glutamicum). In one embodiment, the glucose permease gene in a chimeric construct is a gene that encodes a polypeptide sequence selected from SEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 13 or SEQ ID NO: 14. In one embodiment, the glucose permease gene in a chimeric construct is selected from SEQ ID NO: 19, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, SEQ ID NO: 23, SEQ ID NO: 24 or a combination thereof. The hexokinase gene in each chimeric construct can be a glucokinase gene. Each glucokinase gene in each chimeric construct can be a bacterial glucokinase gene. The glucokinase gene can be any bacterial glucokinase gene known in the art. In one embodiment, a glucokinase gene in a chimeric construct comprises a glucokinase gene from any strain/species/sub-species of Corynebactium (e.g., C. glutamicum), Zymomonas (e.g., Zymomonas mobilis), Staphylococcus (e.g., S. aureus glkA), Enterococcus (e.g., E. faecalis), Escherichia (e.g., E. coli), Clostridium (e.g., C. difficile), Streptococcus (e.g., S. pneumonia), Bacillus (e.g., B. anthracis) or Renibacterium (e.g., R. salmoninarium). In one embodiment, the glucokinase gene in the chimeric construct comprises a glucokinase gene that encodes a polypeptide sequence selected from SEQ ID NO: 15 and/or SEQ ID NO: 16. In one embodiment, the glucokinase gene in the chimeric construct is selected from SEQ ID NO: 25 and/or SEQ ID NO: 26. In the chimeric construct as provided herein the glucose permease gene can be any glucose permease gene provided herein, while the glucokinase gene can be any glucokinase gene provided herein. In one embodiment, a library comprising chimeric glucose permease gene and glucokinase gene constructs comprises a plurality of constructs, whereby the plurality comprises each possible combination of glucose permease genes and glucokinase genes provided herein.
[0095] Each glucose permease gene and/or hexokinase gene (e.g., glucokinase gene) in a chimeric construct as provided herein can be functionally linked or under the control of its native promoter or a mutated form of its native promoter. Each glucose permease gene and/or hexokinase gene (e.g., glucokinase gene) in a chimeric construct as provided herein can be functionally linked to or controlled by any promoter provided herein. Each glucose permease genes in a chimeric construct as provided herein can be controlled by a promoter polynucleotide sequence that comprises or contains a sequence selected from SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 7, and SEQ ID NO: 8. Each hexokinase gene (e.g., glucokinase gene) in a chimeric construct as provided herein can be controlled by a promoter polynucleotide sequence that comprises or contains a sequence selected from SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, and SEQ ID NO: 8. The glucose permease gene and the hexokinase gene (e.g., glucokinase gene) in a chimeric construct as provided herein can each be functionally linked to a promoter that comprises or contains the same sequence. The glucose permease gene and the hexokinase gene (e.g., glucokinase gene) in a chimeric construct as provided herein can each be functionally linked to a promoter that comprises or contains different sequence.
Generating Mutated forms of Glucose Permease and/or Hexokinase Genes
[0096] As provided herein, a glucose permease gene and/or a hexokinase gene (e.g., glucokinase gene) for use in the methods provided herein can be a mutated form of the gene from which it is derived. The mutated gene can be mutated in any way known in the art or provided herein.
[0097] In some embodiments, the present disclosure teaches mutating cell populations by introducing, deleting, or replacing selected portions of genomic DNA. Thus, in some embodiments, the present disclosure teaches methods for targeting mutations to a specific locus (e.g., glucose permease or glucokinase). In other embodiments, the present disclosure teaches the use of gene editing technologies such as ZFNs, TALENS, or CRISPR, to selectively edit target DNA regions. Following mutation of the cell populations, the targeted mutations can be isolated from the cells and subsequently used for generating a library of glucose permease and/or hexokinase genes as described herein.
[0098] In some embodiments, the present disclosure teaches mutating selected DNA regions (e.g., glucose permease genes or glucokinase genes) outside of the host organism. For example, in some embodiments, the present disclosure teaches mutating native glucose permease genes or hexokinase genes (e.g., glucokinase gene).
[0099] In some embodiments, the selected regions of DNA are produced in vitro via gene shuffling of natural variants, or shuffling with synthetic oligos, plasmid-plasmid recombination, virus plasmid recombination, or virus-virus recombination. In other embodiments, the genomic regions are produced via error-prone PCR or site-directed mutagenesis.
[0100] In some embodiments, generating mutations in selected genetic regions containing a glucose permease or hexokinase gene is accomplished by "reassembly PCR." Briefly, oligonucleotide primers (oligos) are synthesized for PCR amplification of segments of a nucleic acid sequence of interest (e.g., glucose permease gene or glucokinase gene), such that the sequences of the oligonucleotides overlap the junctions of two segments. The overlap region is typically about 10 to 100 nucleotides in length. Each of the segments is amplified with a set of such primers. The PCR products are then "reassembled" according to assembly protocols. In brief, in an assembly protocol, the PCR products are first purified away from the primers, by, for example, gel electrophoresis or size exclusion chromatography. Purified products are mixed together and subjected to about 1-10 cycles of denaturing, reannealing, and extension in the presence of polymerase and deoxynucleoside triphosphates (dNTP's) and appropriate buffer salts in the absence of additional primers ("self-priming"). Subsequent PCR with primers flanking the gene are used to amplify the yield of the fully reassembled and shuffled genes.
[0101] In some embodiments of the disclosure, mutated permease or hexokinase DNA regions, such as those discussed above, are enriched for mutant sequences so that the multiple mutant spectrum, i.e. possible combinations of mutations, is more efficiently sampled. In some embodiments, mutated sequences are identified via a mutS protein affinity matrix (Wagner et al., Nucleic Acids Res. 23(19):3944-3948 (1995); Su et al., Proc. Natl. Acad. Sci. (U.S.A.), 83:5057-5061 (1986)) with a preferred step of amplifying the affinity-purified material in vitro prior to an assembly reaction. This amplified material is then put into an assembly or reassembly PCR reaction.
Generation of Libraries Comprising Glucose Permease and/or Hexokinase Genes
[0102] In some embodiments, the present disclosure teaches inserting and/or replacing and/or deleting a DNA segment comprising a glucose permease and/or glucokinase gene of the host organism (e.g.). In some aspects, the methods taught herein involve building an oligonucleotide of interest (i.e. a glucose permease or glucose permease-hexokinase segment), which can be incorporated into the genome of a host organism. In some embodiments, the glucose permease or glucose permease-hexokinase DNA segments of the present disclosure can be obtained via any method known in the art, including, copying or cutting from a known template, mutation, or DNA synthesis. In some embodiments, the present disclosure is compatible with commercially available gene synthesis products for producing DNA sequences (e.g., GeneArt.TM., GeneMaker.TM., GenScript.TM., Anagen.TM., Blue Heron.TM., Entelechon.TM., GeNOsys, Inc., or Qiagen.TM.).
[0103] In some embodiments, the glucose permease or glucose permease-hexokinase DNA segment is designed to incorporate the glucose permease or glucose permease-hexokinase DNA segment into a selected DNA region of the host organism (e.g., adding a beneficial non-PTS glucose transport system). The selected DNA region can be a neutral integration site. In other embodiments, the glucose permease or glucose permease-hexokinase DNA segment is designed to remove the native permease and/or hexokinase gene from the DNA of the host organisms (e.g., removing a native PTS glucose transport system).
[0104] In some embodiments, the glucose permease gene, hexokinase gene or glucose permease-hexokinase genes used in the inventive methods can be synthesized in stages as oligonucleotides using any of the methods of enzymatic or chemical synthesis known in the art. The oligonucleotides may be synthesized on solid supports such as controlled pore glass (CPG), polystyrene beads, or membranes composed of thermoplastic polymers that may contain CPG. Oligonucleotides can also be synthesized on arrays, on a parallel microscale using microfluidics (Tian et al., Mol. BioSyst., 5, 714-722 (2009)), or known technologies that offer combinations of both (see Jacobsen et al., U.S. Pat. App. No. 2011/0172127).
[0105] Synthesis on arrays or through microfluidics offers an advantage over conventional solid support synthesis by reducing costs through lower reagent use. The scale required for gene synthesis is low, so the scale of oligonucleotide product synthesized from arrays or through microfluidics is acceptable. However, the synthesized oligonucleotides are of lesser quality than when using solid support synthesis (See Tian infra.; see also Staehler et al., U.S. Pat. App. No. 2010/0216648).
[0106] A great number of advances have been achieved in the traditional four-step phosphoramidite chemistry since it was first described in the 1980's (see for example, Sierzchala, et al. J. Am. Chem. Soc., 125, 13427-13441 (2003) using peroxy anion deprotection; Hayakawa et al., U.S. Pat. No. 6,040,439 for alternative protecting groups; Azhayev et al, Tetrahedron 57, 4977-4986 (2001) for universal supports; Kozlov et al., Nucleosides, Nucleotides, and Nucleic Acids, 24 (5-7), 1037-1041 (2005) for improved synthesis of longer oligonucleotides through the use of large-pore CPG; and Damha et al., NAR, 18, 3813-3821 (1990) for improved derivatization).
[0107] Regardless of the type of synthesis, the resulting oligonucleotides may then form the smaller building blocks for longer polynucleotides (i.e., glucose permease gene, hexokinase gene or glucose permease-hexokinase genes). In some embodiments, smaller oligonucleotides can be joined together using protocols known in the art, such as polymerase chain assembly (PCA), ligase chain reaction (LCR), and thermodynamically balanced inside-out synthesis (TBIO) (see Czar et al. Trends in Biotechnology, 27, 63-71 (2009)). In PCA, oligonucleotides spanning the entire length of the desired longer product are annealed and extended in multiple cycles (typically about 55 cycles) to eventually achieve full-length product. LCR uses ligase enzyme to join two oligonucleotides that are both annealed to a third oligonucleotide. TBIO synthesis starts at the center of the desired product and is progressively extended in both directions by using overlapping oligonucleotides that are homologous to the forward strand at the 5' end of the gene and against the reverse strand at the 3' end of the gene.
[0108] Another method of synthesizing a larger double stranded DNA fragment is to combine smaller oligonucleotides through top-strand PCR (TSP). In this method, a plurality of oligonucleotides spans the entire length of a desired product and contain overlapping regions to the adjacent oligonucleotide(s). Amplification can be performed with universal forward and reverse primers, and through multiple cycles of amplification a full-length double stranded DNA product is formed. This product can then undergo optional error correction and further amplification that results in the desired double stranded DNA fragment end product.
[0109] In one method of TSP, the set of smaller oligonucleotides that will be combined to form the full-length desired product are between 40-200 bases long and overlap each other by at least about 15-20 bases. For practical purposes, the overlap region should be at a minimum long enough to ensure specific annealing of oligonucleotides and have a high enough melting temperature (T.sub.m) to anneal at the reaction temperature employed. The overlap can extend to the point where a given oligonucleotide is completely overlapped by adjacent oligonucleotides. The amount of overlap does not seem to have any effect on the quality of the final product. The first and last oligonucleotide building block in the assembly should contain binding sites for forward and reverse amplification primers. In one embodiment, the terminal end sequence of the first and last oligonucleotide contain the same sequence of complementarity to allow for the use of universal primers.
Assembling/Cloning Plasmids
[0110] In some embodiments, the present disclosure teaches methods for constructing vectors capable of inserting desired glucose permease genes and/or glucokinase genes DNA sections into the genome of host organisms. In some embodiments, the present disclosure teaches methods of cloning vectors comprising the insert DNA (e.g., glucose permease gene and/or glucokinase gene), homology arms, and at least one selection marker. (see FIG. 3).
[0111] In some embodiments, the present disclosure is compatible with any vector suited for transformation into the host organism. In some embodiments, the present disclosure teaches use of shuttle vectors compatible with a host cell. In one embodiment, a shuttle vector for use in the methods provided herein is a shuttle vector compatible with an E. coli and/or Corynebacterium host cell. Shuttle vectors for use in the methods provided herein can comprise markers for selection and/or counter-selection as described herein. The markers can be any markers known in the an and/or provided herein. The shuttle vectors can further comprise any regulatory sequence(s) and/or sequences useful in the assembly of said shuttle vectors as known in the art. The shuttle vectors can further comprise any origins of replication that may be needed for propagation in a host cell as provided herein such as, for example, E. coli or C. glutamicum. The regulatory sequence can be any regulatory sequence known in the art or provided herein such as, for example, a promoter, start, stop, signal, secretion and/or termination sequence used by the genetic machinery of the host cell. The termination sequence can be SEQ ID NO: 17 or 18. In certain instances, the target DNA can be inserted into vectors, constructs or plasmids obtainable from any repository or catalogue product, such as a commercial vector (see e.g., DNA2.0 custom or GATEWAY.RTM. vectors).
[0112] In some embodiments, the assembly/cloning methods of the present disclosure may employ at least one of the following assembly strategies: i) type II conventional cloning, ii) type II S-mediated or "Golden Gate" cloning (see, e.g., Engler, C., R. Kandzia, and S. Marillonnet. 2008 "A one pot, one step, precision cloning method with high throughput capability". PLos One 3:e3647; Kotera, I., and T. Nagai. 2008 "A high-throughput and single-tube recombination of crude PCR products using a DNA polymerase inhibitor and type IIS restriction enzyme." J Biotechnol 137:1-7; Weber, E., R. Gruetzner, S. Werner, C. Engler, and S. Marillonnet. 2011 Assembly of Designer TAL Effectors by Golden Gate Cloning. PloS One 6:e19722), iii) GATEWAY.RTM. recombination, iv) TOPO.RTM. cloning, exonuclease-mediated assembly (Aslanidis and de Jong 1990. "Ligation-independent cloning of PCR products (LIC-PCR)." Nucleic Acids Research, Vol. 18, No. 20 6069), v) homologous recombination, vi) non-homologous end joining, or a combination thereof. Modular type IIS based assembly strategies are disclosed in PCT Publication WO 2011/154147, the disclosure of which is included herein by reference.
[0113] In some embodiments, the present disclosure teaches cloning vectors with at least one selection marker. Various selection marker genes are known in the art often encoding antibiotic resistance function for selection in prokaryotic (e.g., against ampicillin, kanamycin, tetracycline, chloramphenycol, zeocin, spectinomycin/streptomycin) or eukaryotic cells (e.g. geneticin, neomycin, hygromycin, puromycin, blasticidin, zeocin) under selective pressure. Other marker systems allow for screening and identification of wanted or unwanted cells such as the well-known blue/white screening system used in bacteria to select positive clones in the presence of X-gal or fluorescent reporters such as green or red fluorescent proteins expressed in successfully transduced host cells. Another class of selection markers most of which are only functional in prokaryotic systems relates to counter selectable marker genes often also referred to as "death genes" which express toxic gene products that kill producer cells. Examples of such genes include sacB, rpsL(strA), tetAR, pheS, thyA, gata-1, or ccdB, the function of which is described in (Reyrat et al. 1998 "Counterselectable Markers: Untapped Tools for Bacterial Genetics and Pathogenesis." Infect Immun. 66(9): 4011-4017).
[0114] In one embodiment, the vector into which the target DNA segment is cloned into comprises a promoter polynucleotide from a promoter ladder or library as provided herein. In one embodiment, provided herein is promoter ladder comprising or containing a sequence selected from SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, or SEQ ID NO: 8.
[0115] In one embodiment, the vector comprises a first promoter polynucleotide and a second promoter polynucleotide. The first and/or second promoter polynucleotide can comprise or contain a sequence selected from SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7, or SEQ ID NO: 8. The promoter polynucleotide can be used in each case for over-expressing or under-expressing a glucose permease and/or hexokinase in a host microorganism.
[0116] In some embodiments, each generated strain comprising a heterologous glucose permease gene or glucose permease gene-glucokinase gene is cultured and analyzed under one or more criteria of the present disclosure (e.g., productivity of a biomolecule or product of interest). Data from each of the analyzed host strains is associated/correlated with a particular glucose permease gene or glucose permease gene/glucokinase gene combination, and is recorded for future use. Thus, the present disclosure enables the creation of large and highly annotated genetic diversity libraries/depositories that identify the effect of a glucose permease gene or combination of glucose permease gene/glucokinase gene on any number of microbial genetic or phenotypic traits of interest.
[0117] In some embodiments, the present disclosure teaches the use of vectors for cloning the glucose permease gene and/or hexokinase gene with start and/or stop codon variants such that the cloned gene utilizes the start and/or stop codon variant. For example, typical stop codons for S. cerevisiae and mammals are UAA and UGA, respectively. The typical stop codon for monocotyledonous plants is UGA, whereas insects and E. coli commonly use UAA as the stop codon (Dalphin et al. (1996) Nucl. Acids Res. 24: 216-218).
[0118] In one embodiment, the methods of the provided disclosure comprise codon optimizing one or more genes expressed by the host organism. Methods for optimizing codons to improve expression in various hosts are known in the art and are described in the literature (see U.S. Pat. App. Pub. No. 2007/0292918, incorporated herein by reference in its entirety). Optimized coding sequences containing codons preferred by a particular prokaryotic or eukaryotic host (see also, Murray et al. (1989) Nucl. Acids Res. 17:477-508) can be prepared, for example, to increase the rate of translation or to produce recombinant RNA transcripts having desirable properties, such as a longer half-life, as compared with transcripts produced from a non-optimized sequence.
[0119] In some embodiments, a glucose permease gene or polynucleotide provided herein comprises a molecule codon optimized for translation in a host cell provided herein, such as, for example, E. coli and/or C. glutamicum. The gene or polynucleotide can be an isolated, synthetic or recombinant nucleic acid. The codon optimized glucose permease gene or polynucleotide can be selected from SEQ ID NO: 19, SEQ ID NO: 20, SEQ ID NO: 21, SEQ ID NO: 22, SEQ ID NO: 23, SEQ ID NO: 24. In some cases, provided herein is a permease gene or polynucleotide that is codon optimized to encode a polypeptide sequence selected from SEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 13 or SEQ ID NO: 14. The codon optimized glucose permease gene or polynucleotide provided herein can be generated using a method known in the art for generating codon optimized polynucleotides such as, for example, GenScript's OptimumGene.TM. gene design system or DNA2.0 GeneGPS.RTM. Expression Optimization technology.
[0120] In some embodiments, a hexokinase (e.g., glucokinase) gene or polynucleotide provided herein comprises a molecule codon optimized for translation in a host cell provided herein, such as, for example, E. coli and/or C. glutamicum. The gene or polynucleotide can be an isolated, synthetic or recombinant nucleic acid. The codon optimized hexokinase gene (e.g., glucokinase gene) can be selected from SEQ ID NO: 25 or SEQ ID NO: 26. In some cases, provided herein is a hexokinase (e.g., glucokinase) gene or polynucleotide that is codon optimized to encode a polypeptide sequence selected from SEQ ID NO: 15 or SEQ ID NO: 16. The codon optimized hexokinase (e.g., glucokinase) gene or polynucleotide provided herein can be generated using a method known in the art for generating codon optimized polynucleotides such as, for example, GenScript's OptimumGene.TM. gene design system or DNA2.0 GeneGPS.RTM. Expression Optimization technology.
[0121] Protein expression is governed by a host of factors including those that affect transcription, mRNA processing, and stability and initiation of translation. Optimization can thus address any of a number of sequence features of any particular gene. As a specific example, a rare codon induced translational pause can result in reduced protein expression. A rare codon induced translational pause includes the presence of codons in the polynucleotide of interest that are rarely used in the host organism may have a negative effect on protein translation due to their scarcity in the available tRNA pool.
[0122] Alternate translational initiation also can result in reduced heterologous protein expression. Alternate translational initiation can include a synthetic polynucleotide sequence inadvertently containing motifs capable of functioning as a ribosome binding site (RBS). These sites can result in initiating translation of a truncated protein from a gene-internal site. One method of reducing the possibility of producing a truncated protein, which can be difficult to remove during purification, includes eliminating putative internal RBS sequences from an optimized polynucleotide sequence.
[0123] Repeat-induced polymerase slippage can result in reduced heterologous protein expression. Repeat-induced polymerase slippage involves nucleotide sequence repeats that have been shown to cause slippage or stuttering of DNA polymerase which can result in frameshift mutations. Such repeats can also cause slippage of RNA polymerase. In an organism with a high G+C content bias, there can be a higher degree of repeats composed of G or C nucleotide repeats. Therefore, one method of reducing the possibility of inducing RNA polymerase slippage, includes altering extended repeats of G or C nucleotides.
[0124] Interfering secondary structures also can result in reduced heterologous protein expression. Secondary structures can sequester the RBS sequence or initiation codon and have been correlated to a reduction in protein expression. Stemloop structures can also be involved in transcriptional pausing and attenuation. An optimized polynucleotide sequence can contain minimal secondary structures in the RBS and gene coding regions of the nucleotide sequence to allow for improved transcription and translation.
[0125] For example, the optimization process can begin by identifying the desired amino acid sequence to be expressed by the host. From the amino acid sequence a candidate polynucleotide or DNA sequence can be designed. During the design of the synthetic DNA sequence, the frequency of codon usage can be compared to the codon usage of the host expression organism and rare host codons can be removed from the synthetic sequence. Additionally, the synthetic candidate DNA sequence can be modified in order to remove undesirable enzyme restriction sites and add or remove any desired signal sequences, linkers or untranslated regions. The synthetic DNA sequence can be analyzed for the presence of secondary structure that may interfere with the translation process, such as G/C repeats and stem-loop structures.
Transformation of Host Cells
[0126] In some embodiments, the vectors of the present disclosure may be introduced into the host cells using any of a variety of techniques, including transformation, transfection, transduction, viral infection, gene guns, or Ti-mediated gene transfer. Particular methods include calcium phosphate transfection, DEAE-Dextran mediated transfection, lipofection, or electroporation (Davis, L., Dibner, M., Battey, I., 1986 "Basic Methods in Molecular Biology"). Other methods of transformation include for example, lithium acetate transformation and electroporation See, e.g., Gietz et al., Nucleic Acids Res. 27:69-74 (1992); Ito et al., J. Bacterol. 153:163-168 (1983); and Becker and Guarente, Methods in Enzymology 194:182-187 (1991). In some embodiments, transformed host cells are referred to as recombinant host strains.
[0127] In some embodiments, the present disclosure teaches high throughput transformation of cells using 96-well plate robotics platform and liquid handling machines known in the art.
[0128] In some embodiments, the present disclosure teaches screening transformed cells with one or more selection markers. In one such embodiment, cells transformed with a vector comprising a kanamycin resistance marker (KanR) are plated on media containing effective amounts of the kanamycin antibiotic. Colony forming units visible on kanamycin-laced media are presumed to have incorporated the vector cassette into their genome. Insertion of the desired sequences can be confirmed via PCR, restriction enzyme analysis, and/or sequencing of the relevant insertion site.
Looping Out of Selected Sequences
[0129] In some embodiments, the present disclosure teaches methods of looping out selected regions of DNA from the host organisms. The looping out method can be as described in Nakashima et al. 2014 "Bacterial Cellular Engineering by Genome Editing and Gene Silencing." Int. J. Mol. Sci. 15(2), 2773-2793. In some embodiments, the present disclosure teaches looping out selection markers from positive transformants. Looping out deletion techniques are known in the art, and are described in (Tear et al. 2014 "Excision of Unstable Artificial Gene-Specific inverted Repeats Mediates Scar-Free Gene Deletions in Escherichia coli." Appl. Biochem. Biotech. 175:1858-1867). The looping out methods used in the methods provided herein can be performed using single-crossover homologous recombination or double-crossover homologous recombination. In one embodiment, looping out of selected regions as described herein can entail using single-crossover homologous recombination as described herein.
[0130] First, loop out vectors are inserted into selected target regions within the genome of the host organism (e.g., via homologous recombination, CRISPR, or other gene editing technique). In one embodiment, single-crossover homologous recombination is used between a circular plasmid or vector and the host cell genome in order to loop-in the circular plasmid or vector such as depicted in FIG. 3. The inserted vector can be designed with a sequence which is a direct repeat of an existing or introduced nearby host sequence, such that the direct repeats flank the region of DNA slated for looping and deletion. Once inserted, cells containing the loop out plasmid or vector can be counter selected for deletion of the selection region (e.g., see FIG. 4; lack of resistance to the selection gene).
Host Microorganisms
[0131] The genomic engineering methods provided herein are exemplified with industrial microbial cell cultures, but can be applicable to any organism where desired traits can be identified in a population of genetic mutants.
[0132] Thus, as used herein, the term "microorganism" should be taken broadly. It includes, but is not limited to, the two prokaryotic domains, Bacteria and Archaea, as well as certain eukaryotic fungi and protists. However, in certain aspects, "higher" eukaryotic organisms such as insects, plants, and animals can be utilized in the methods taught herein.
[0133] Suitable host cells include, but are not limited to: bacterial cells, algal cells, plant cells, fungal cells, insect cells, and mammalian cells. In one illustrative embodiment, suitable host cells include E. coli (e.g., SHuffle.TM. competent E. coli available from New England BioLabs in Ipswich, Mass.).
[0134] Other suitable host organisms of the present disclosure include microorganisms of the genus Corynebacterium. In some embodiments, preferred Corynebacterium strains/species include: C. efficiens, with the deposited type strain being DSM44549, C. glutamicum, with the deposited type strain being ATCC13032, and C. ammoniagenes, with the deposited type strain being ATCC6871. In some embodiments, the preferred host of the present disclosure is C. glutamicum.
[0135] Suitable host strains of the genus Corynebacterium, in particular of the species Corynebacterium glutamicum, are in particular the known wild-type strains: Corynebacterium glutamicum ATCC13032, Corynebacterium acetoglutamicum ATCC15806, Corynebacterium acetoacidophilum ATCC13870, Corynebacterium melassecola ATCC17965, Corynebacterium thermoaminogenes FERM BP-1539, Brevibacterium flavum ATCC14067, Brevibacterium lactofermentum ATCC13869, and Brevibacterium divaricatum ATCC14020; and L-amino acid-producing mutants, or strains, prepared therefrom, such as, for example, the L-lysine-producing strains: Corynebacterium glutamicum FERM-P 1709, Brevibacterium flavum FERM-P 1708, Brevibacterium lactofermentum FERM-P 1712, Corynebacterium glutamicum FERM-P 6463, Corynebacterium glutamicum FERM-P 6464, Corynebacterium glutamicum DM58-1, Corynebacterium glutamicum DG52-5, Corynebacterium glutamicum DSM5714, and Corynebacterium glutamicum DSM12866.
[0136] The term "Micrococcus glutamicus" has also been in use for C. glutamicum. Some representatives of the species C. efficiens have also been referred to as C. thermoaminogenes in the prior art, such as the strain FERM BP-1539, for example.
[0137] In some embodiments, the host cell of the present disclosure is a eukaryotic cell. Suitable eukaryotic host cells include, but are not limited to: fungal cells, algal cells, insect cells, animal cells, and plant cells. Suitable fungal host cells include, but are not limited to: Ascomycota, Basidiomycota, Deuteromycota, Zygomycota, Fungi imperfecti. Certain preferred fungal host cells include yeast cells and filamentous fungal cells. Suitable filamentous fungi host cells include, for example, any filamentous forms of the subdivision Eumycotina and Oomycota. (see, e.g., Hawksworth et al., In Ainsworth and Bisby's Dictionary of The Fungi, 8.sup.th edition, 1995, CAB International, University Press, Cambridge, UK, which is incorporated herein by reference). Filamentous fungi are characterized by a vegetative mycelium with a cell wall composed of chitin, cellulose and other complex polysaccharides. The filamentous fungi host cells are morphologically distinct from yeast.
[0138] In certain illustrative, but non-limiting embodiments, the filamentous fungal host cell may be a cell of a species of: Achlya, Acremonium, Aspergillus, Aureobasidium, Bjerkandera, Ceriporiopsis, Cephalosporium, Chrysosporium, Cochliobolus, Corynascus, Cryphonectria, Cryptococcus, Coprinus, Coriolus, Diplodia, Endothis, Fusarium, Gibberella, Gliocladium, Humicola, Hypocrea, Myceliophthora (e.g., Myceliophthora thermophila), Mucor, Neurospora, Penicillium, Podospora, Phlebia, Piromyces, Pyricularia, Rhizomucor, Rhizopus, Schizophyllum, Sporotrichum, Talaromyces, Thermoascus, Thielavia, Tramates, Tolypocladium, Trichoderma, Verticillium, Volvariella, or teleomorphs, or anamorphs, and synonyms or taxonomic equivalents thereof.
[0139] Suitable yeast host cells include, but are not limited to: Candida, Hansenula, Saccharomyces, Schizosaccharomyces, Pichia, Kluyveromyces, and Yarrowia. In some embodiments, the yeast cell is Hansenula polymorpha, Saccharomyces cerevisiae, Saccharomyces carlsbergensis, Saccharomyces diastaticus, Saccharomyces norbensis, Saccharomyces kluyveri, Schizosaccharomyces pombe, Pichia pastoris, Pichia finlandica, Pichia trehalophila, Pichia kodamae, Pichia membranaefaciens, Pichia opuntiae, Pichia thermotolerans, Pichia salictaria, Pichia quercuum, Pichia pijperi, Pichia stipitis, Pichia methanolica, Pichia angusta, Kluyveromyces lactis, Candida albicans, or Yarrowia lipolytica.
[0140] In certain embodiments, the host cell is an algal such as, Chlamydomonas (e.g., C. Reinhardtii) and Phormidium (P. sp. ATCC29409).
[0141] In other embodiments, the host cell is a prokaryotic cell. Suitable prokaryotic cells include gram positive, gram negative, and gram-variable bacterial cells. The host cell may be a species of, but not limited to: Agrohacterium, Alicyclobacillus, Anabaena, Anacystis, Acinetobacter, Acidothermus, Arthrohacter, Azohacter, Bacillus, Bifidohacterium, Brevihacterium, Butyrivihrio, Buchnera, Campestris, Camplyohacter, Clostridium, Corynebacterium, Chromatium, Coprococcus, Escherichia, Enterococcus, Enterobacter, Erwinia, Fusobacterium, Faecalibacterium, Francisella, Flavobacterium, Geobacillus, Haemophilus, Helicobacter, Klebsiella, Lactobacillus, Laclococcus, Ilyobacler, Micrococcus, Microbacterium, Mesorhizobium, Methylobacterium, Methylobacterium, Mycobacterium, Neisseria, Pantoea, Pseudomonas, Prochlorococcus, Rhodobacter, Rhodopseudomonas, Rhodopseudomonas, Roseburia, Rhodospirillum, Rhodococcus, Scenedesmus, Streptomyces, Streptococcus, Synecoccus, Saccharomonospora, Staphylococcus, Serratia, Salmonella, Shigella, Thermoanaerobacterium, Tropheryma, Tularensis, Temecula, Thermosynechococcus, Thermococcus, Ureaplasma, Xanthomonas, Xylella, Yersinia, and Zymomonas. In some embodiments, the host cell is Corynebacterium glutamicum.
[0142] In some embodiments, the bacterial host strain is an industrial strain. Numerous bacterial industrial strains are known and suitable in the methods and compositions described herein.
[0143] In some embodiments, the bacterial host cell is of the Agrobacterium species (e.g., A. radiobacter, A. rhizogenes, A. rubi), the Arthrobacter species (e.g., A. aurescens, A. citreus, A. globformis, A. hydrocarboglutamicus, A. mysorens, A. nicotianae, A. paraffineus, A. protophonniae, A. roseoparaffinus, A. sulfureus, A. ureafaciens), the Bacillus species (e.g., B. thuringiensis, B. anthracis, B. megaterium, B. subtilis, B. lentils, B. circulars, B. pumilus, B. lautus, B. coagulans, B. brevis, B. firmus, B. alkaophius, B. licheniformis, B. clausii, B. stearothermophilus, B. halodurans and B. amyloliquefaciens. In particular embodiments, the host cell will be an industrial Bacillus strain including but not limited to B. subtilis, B. pumilus, B. licheniformis, B. megaterium, B. clausii, B. stearothermophilus and B. amyloliquefaciens. In some embodiments, the host cell will be an industrial Clostridium species (e.g., C. acetobutylicum, C. tetani E88, C. lituseburense, C. saccharobutylicum, C. perfringens, C. beijerinckii). In some embodiments, the host cell will be an industrial Corynebacterium species (e.g., C. glutamicum, C. acetoacidophilum). In some embodiments, the host cell will be an industrial Escherichia species (e.g., E. coli). In some embodiments, the host cell will be an industrial Erwinia species (e.g., E. uredovora, E. carotovora, E. ananas, E. herbicola, E. punctata, E. terreus). In some embodiments, the host cell will be an industrial Pantoea species (e.g., P. citrea, P. agglomerans). In some embodiments, the host cell will be an industrial Pseudomonas species, (e.g., P. putida, P. aeruginosa, P. mevalonii). In some embodiments, the host cell will be an industrial Streptococcus species (e.g., S. equisimiles, S. pyogenes, S. uberis). In some embodiments, the host cell will be an industrial Streptomyces species (e.g., S. ambofaciens, S. achromogenes, S. avermitilis, S. coelicolor, S. aureofaciens, S. aureus, S. fungicidicus, S. griseus, S. lividans). In some embodiments, the host cell will be an industrial Zymomonas species (e.g., Z. mobilis, Z. lipolytica), and the like.
[0144] In various embodiments, strains that may be used in the practice of the disclosure including both prokaryotic and eukaryotic strains, are readily accessible to the public from a number of culture collections such as American Type Culture Collection (ATCC), Deutsche Sammlung von Mikroorganismen and Zellkulturen GmbH (DSM), Centraalbureau Voor Schimmelcultures (CBS), and Agricultural Research Service Patent Culture Collection, Northern Regional Research Center (NRRL).
[0145] In some embodiments, the methods of the present disclosure are also applicable to multi-cellular organisms. For example, the platform could be used for improving the performance of crops. The organisms can comprise a plurality of plants such as Gramineae, Fetucoideae, Poacoideae, Agrostis, Phleum, Dactylis, Sorgum, Setaria, Zea, Oryza, Triticum, Secale, Avena, Hordeum, Saccharum, Poa, Festuca, Stenotaphrum, Cynodon, Coix, Olyreae, Phareae, Compositae or Leguminosae. For example, the plants can be corn, rice, soybean, cotton, wheat, rye, oats, barley, pea, beans, lentil, peanut, yam bean, cowpeas, velvet beans, clover, alfalfa, lupine, vetch, lotus, sweet clover, wisteria, sweet pea, sorghum, millet, sunflower, canola or the like. Similarly, the organisms can include a plurality of animals such as non-human mammals, fish, insects, or the like.
Cell Fermentation and Culture
[0146] Microorganisms of the present disclosure including those genetically engineered as described herein can be cultured in conventional nutrient media modified as appropriate for any desired biosynthetic reactions or selections. In some embodiments, the present disclosure teaches culture in inducing media for activating promoters. In some embodiments, the present disclosure teaches media with selection agents, including selection agents of transformants (e.g., antibiotics), or selection of organisms suited to grow under inhibiting conditions (e.g., high ethanol conditions). In some embodiments, the present disclosure teaches growing cell cultures in media optimized for cell growth. In other embodiments, the present disclosure teaches growing cell cultures in media optimized for product yield such as, for example, products or biomolecules of interest derived from metabolic processing of glucose. In some embodiments, the present disclosure teaches growing cultures in media capable of inducing cell growth and also contains the necessary precursors for final product production (e.g., high levels of sugars for ethanol production). The biomolecules or products of interest produced by the methods provided herein can be any commercial product produced from glucose. In some cases, the biomolecule or product of interest is a small molecule, an amino acid, an organic acid, or an alcohol. The amino acid can be tyrosine, phenylalanine, tryptophan, aspartic acid, asparagine, threonine, isoleucine, methionine, or lysine. The organic acid can be succinate, lactate or pyruvate. The alcohol can be ethanol or isobutanol.
[0147] Culture conditions, such as temperature, pH and the like, are those suitable for use with the host cell selected for expression, and will be apparent to those skilled in the art. As noted, many references are available for the culture and production of many cells, including cells of bacterial, plant, animal (including mammalian) and archebacterial origin. See e.g., Sambrook, Ausubel (all supra), as well as Berger, Guide to Molecular Cloning Techniques, Methods in Enzymology volume 152 Academic Press, Inc., San Diego, Calif.; and Freshney (1994) Culture of Animal Cells, a Manual of Basic Technique, third edition, Wiley-Liss, New York and the references cited therein; Doyle and Griffiths (1997) Mammalian Cell Culture: Essential Techniques John Wiley and Sons, NY; Humason (1979) Animal Tissue Techniques, fourth edition W.H. Freeman and Company; and Ricciardelle et al., (1989) In Vitro Cell Dev. Biol. 25:1016-1024, all of which are incorporated herein by reference. For plant cell culture and regeneration, Payne et al. (1992) Plant Cell and Tissue Culture in Liquid Systems John Wiley & Sons, Inc. New York, N.Y.; Gamborg and Phillips (eds) (1995) Plant Cell, Tissue and Organ Culture; Fundamental Methods Springer Lab Manual, Springer-Verlag (Berlin Heidelberg N.Y.); Jones, ed. (1984) Plant Gene Transfer and Expression Protocols, Humana Press, Totowa, N.J. and Plant Molecular Biology (1993) R. R. D. Croy, Ed. Bios Scientific Publishers, Oxford, U.K. ISBN 0 12 198370 6, all of which are incorporated herein by reference. Cell culture media in general are set forth in Atlas and Parks (eds.) The Handbook of Microbiological Media (1993) CRC Press, Boca Raton, Fla., which is incorporated herein by reference. Additional information for cell culture is found in available commercial literature such as the Life Science Research Cell Culture Catalogue from Sigma-Aldrich, Inc (St Louis, Mo.) ("Sigma-LSRCCC") and, for example, The Plant Culture Catalogue and supplement also from Sigma-Aldrich, Inc (St Louis, Mo.) ("Sigma-PCCS"), all of which are incorporated herein by reference.
[0148] The culture medium or fermentation medium to be used must in a suitable manner satisfy the demands of the respective strains. Descriptions of culture media for various microorganisms are present in the "Manual of Methods for General Bacteriology" of the American Society for Bacteriology (Washington D.C., USA, 1981). The terms culture medium and fermentation medium are interchangeable.
[0149] In some embodiments, the present disclosure teaches that the microorganisms produced may be cultured continuously--as described, for example, in WO 05/021772--or discontinuously in a batch process (batch cultivation) or in a fed-batch or repeated fed-batch process for the purpose of producing the desired organic-chemical compound. A summary of a general nature about known cultivation methods is available in the textbook by Chmiel (Bioproze technik. 1: Einfuhrung in die Bioverfahrenstechnik (Gustav Fischer Verlag, Stuttgart, 1991)) or in the textbook by Storhas (Bioreaktoren and periphere Einrichtungen (Vieweg Verlag, Braunschweig/Wiesbaden, 1994)).
[0150] In some embodiments, the cells of the present disclosure are grown under batch or continuous fermentations conditions. Classical batch fermentation is a closed system, wherein the compositions of the medium is set at the beginning of the fermentation and is not subject to artificial alternations during the fermentation. A variation of the batch system is a fed-batch fermentation which also finds use in the present disclosure. In this variation, the substrate is added in increments as the fermentation progresses. Fed-batch systems are useful when catabolite repression is likely to inhibit the metabolism of the cells and where it is desirable to have limited amounts of substrate in the medium. Batch and fed-batch fermentations are common and well known in the art. Continuous fermentation is a system where a defined fermentation medium is added continuously to a bioreactor and an equal amount of conditioned medium is removed simultaneously for processing and harvesting of desired proteins. In some embodiments, continuous fermentation generally maintains the cultures at a constant high density where cells are primarily in log phase growth. In some embodiments, continuous fermentation generally maintains the cultures at a stationary or late log/stationary, phase growth. Continuous fermentation systems strive to maintain steady state growth conditions.
[0151] Methods for modulating nutrients and growth factors for continuous fermentation processes as well as techniques for maximizing the rate of product formation are well known in the art of industrial microbiology.
[0152] For example, a non-limiting list of carbon sources for the cultures of the present disclosure include, sugars and carbohydrates such as, for example, glucose, sucrose, lactose, fructose, maltose, molasses, sucrose-containing solutions from sugar beet or sugar cane processing, starch, starch hydrolysate, and cellulose; oils and fats such as, for example, soybean oil, sunflower oil, groundnut oil and coconut fat; fatty acids such as, for example, palmitic acid, stearic acid, and linoleic acid; alcohols such as, for example, glycerol, methanol, and ethanol; and organic acids such as, for example, acetic acid or lactic acid.
[0153] A non-limiting list of the nitrogen sources for the cultures of the present disclosure include, organic nitrogen-containing compounds such as peptones, yeast extract, meat extract, malt extract, corn steep liquor, soybean flour, and urea; or inorganic compounds such as ammonium sulfate, ammonium chloride, ammonium phosphate, ammonium carbonate, and ammonium nitrate. The nitrogen sources can be used individually or as a mixture.
[0154] A non-limiting list of the possible phosphorus sources for the cultures of the present disclosure include, phosphoric acid, potassium dihydrogen phosphate or dipotassium hydrogen phosphate or the corresponding sodium-containing salts. The culture medium may additionally comprise salts, for example in the form of chlorides or sulfates of metals such as, for example, sodium, potassium, magnesium, calcium and iron, such as, for example, magnesium sulfate or iron sulfate, which are necessary for growth. Finally, essential growth factors such as amino acids, for example homoserine and vitamins, for example thiamine, biotin or pantothenic acid, may be employed in addition to the abovementioned substances.
[0155] In some embodiments, the pH of the culture can be controlled by any acid or base, or buffer salt, including, but not limited to sodium hydroxide, potassium hydroxide, ammonia, or aqueous ammonia; or acidic compounds such as phosphoric acid or sulfuric acid in a suitable manner. In some embodiments, the pH is generally adjusted to a value of from 6.0 to 8.5, preferably 6.5 to 8.
[0156] In some embodiments, the cultures of the present disclosure may include an anti-foaming agent such as, for example, fatty acid polyglycol esters. In some embodiments the cultures of the present disclosure are modified to stabilize the plasmids of the cultures by adding suitable selective substances such as, for example, antibiotics.
[0157] In some embodiments, the culture is carried out under aerobic conditions. In order to maintain these conditions, oxygen or oxygen-containing gas mixtures such as, for example, air are introduced into the culture. It is likewise possible to use liquids enriched with hydrogen peroxide. The fermentation is carried out, where appropriate, at elevated pressure, for example at an elevated pressure of from 0.03 to 0.2 MPa. The temperature of the culture is normally from 20.degree. C. to 45.degree. C. and preferably from 25.degree. C. to 40.degree. C., particularly preferably from 30.degree. C. to 37.degree. C. In batch or fed-batch processes, the cultivation is preferably continued until an amount of the desired organic-chemical compound sufficient for being recovered has formed. In some embodiments, the culture is carried out under anaerobic conditions.
Product Recovery and Quantification
[0158] Methods for screening for the production of products of interest are known to those of skill in the art and are discussed throughout the present specification. Such methods may be employed when screening the strains of the disclosure. The biomolecules or products of interest produced by the methods provided herein can be any commercial product produced from glucose. In some cases, the biomolecule or product of interest is an amino acid, an organic acid, or an alcohol. The amino acid can be tyrosine, phenylalanine, tryptophan, aspartic acid, asparagine, threonine, isoleucine, methionine, or lysine. The organic acid can be succinate, lactate or pyruvate. The alcohol can be ethanol or isobutanol.
[0159] In some embodiments, the present disclosure teaches methods of improving strains designed to produce non-secreted intracellular products. For example, the present disclosure teaches methods of improving the robustness, yield, efficiency, or overall desirability of cell cultures producing intracellular enzymes, oils, pharmaceuticals, or other valuable small molecules or peptides. The recovery or isolation of non-secreted intracellular products can be achieved by lysis and recovery techniques that are well known in the art, including those described herein.
[0160] For example, in some embodiments, cells of the present disclosure can be harvested by centrifugation, filtration, settling, or other method. Harvested cells are then disrupted by any convenient method, including freeze-thaw cycling, sonication, mechanical disruption, or use of cell lysing agents, or other methods, which are well known to those skilled in the art.
[0161] The resulting product of interest, e.g. a polypeptide, may be recovered/isolated and optionally purified by any of a number of methods known in the art. For example, a product polypeptide may be isolated from the nutrient medium by conventional procedures including, but not limited to: centrifugation, filtration, extraction, spray-drying, evaporation, chromatography (e.g., ion exchange, affinity, hydrophobic interaction, chromatofocusing, and size exclusion), or precipitation. Finally, high performance liquid chromatography (HPLC) can be employed in the final purification steps. (See for example Purification of intracellular protein as described in Parry et al., 2001, Biochem. J. 353:117, and Hong et al., 2007, Appl. Microbiol. Biotechnol. 73:1331, both incorporated herein by reference).
[0162] In addition to the references noted supra, a variety of purification methods are well known in the art, including, for example, those set forth in: Sandana (1997) Bioseparation of Proteins, Academic Press, Inc.; Bollag et al. (1996) Protein Methods, 2.sup.nd Edition, Wiley-Liss, NY; Walker (1996) The Protein Protocols Handbook Humana Press, NJ; Harris and Angal (1990) Protein Purification Applications: A Practical Approach, IRL Press at Oxford, Oxford, England; Harris and Angal Protein Purification Methods: A Practical Approach, IRL Press at Oxford, Oxford, England; Scopes (1993) Protein Purification: Principles and Practice 3.sup.rd Edition, Springer Verlag, NY; Janson and Ryden (1998) Protein Purification: Principles, High Resolution Methods and Applications, Second Edition, Wiley-VCH, NY and Walker (1998) Protein Protocols on CD-ROM, Humana Press, NJ, all of which are incorporated herein by reference.
[0163] In some embodiments, the present disclosure teaches the methods of improving strains designed to produce secreted products. For example, the present disclosure teaches methods of improving the robustness, yield, efficiency, or overall desirability of cell cultures producing valuable small molecules or peptides.
[0164] In some embodiments, immunological methods may be used to detect and/or purify secreted or non-secreted products produced by the cells of the present disclosure. In one example approach, antibody raised against a product molecule (e.g., against an insulin polypeptide or an immunogenic fragment thereof) using conventional methods is immobilized on beads, mixed with cell culture media under conditions in which the endoglucanase is bound, and precipitated. In some embodiments, the present disclosure teaches the use of enzyme-linked immunosorbent assays (ELISA).
[0165] In other related embodiments, immunochromatography is used, as disclosed in U.S. Pat. Nos. 5,591,645, 4,855,240, 4,435,504, 4,980,298, and Se-Hwan Paek, et al., "Development of rapid One-Step Immunochromatographic assay, Methods", 22, 53-60, 2000), each of which are incorporated by reference herein. A general immunochromatography detects a specimen by using two antibodies. A first antibody exists in a test solution or at a portion at an end of a test piece in an approximately rectangular shape made from a porous membrane, where the test solution is dropped. This antibody is labeled with latex particles or gold colloidal particles (this antibody will be called as a labeled antibody hereinafter). When the dropped test solution includes a specimen to be detected, the labeled antibody recognizes the specimen so as to be bonded with the specimen. A complex of the specimen and labeled antibody flows by capillarity toward an absorber, which is made from a filter paper and attached to an end opposite to the end having included the labeled antibody. During the flow, the complex of the specimen and labeled antibody is recognized and caught by a second antibody (it will be called as a tapping antibody hereinafter) existing at the middle of the porous membrane and, as a result of this, the complex appears at a detection part on the porous membrane as a visible signal and is detected.
[0166] In some embodiments, the screening methods of the present disclosure are based on photometric detection techniques (absorption, fluorescence). For example, in some embodiments, detection may be based on the presence of a fluorophore detector such as GFP bound to an antibody. In other embodiments, the photometric detection may be based on the accumulation on the desired product from the cell culture. In some embodiments, the product may be detectable via UV of the culture or extracts from said culture.
[0167] In some embodiments, the product recovery methods allow for the quantitative determination of the effect on performance of each candidate glucose permease gene and/or glucokinase gene. In some embodiments, the product recovery methods allow for the quantitative determination of the effect on performance of each candidate glucose permease gene/glucokinase gene combination, allowing for comparison of each and selection for the optimal combination.
Selection Criteria and Goals
[0168] The selection of a particular strain of host cell expressing a heterologous glucose permease or glucose permease and glucokinase can be based on specific goals. For example, in some embodiments, the program goal may be to maximize single batch yields of reactions with no immediate time limits. In other embodiments, the program goal may be to rebalance biosynthetic yields to produce a specific product, or to produce a particular ratio of products. In some embodiments, the program goal may be to improve performance characteristics such as yield, titer, productivity, by-product elimination, tolerance to process excursions, optimal growth temperature and growth rate. In some embodiments, the program goal is improved host performance as measured by volumetric productivity, specific productivity, yield or titre, of a product of interest produced by a microbe.
[0169] In other embodiments, the program goal may be to optimize synthesis efficiency of a commercial strain in terms of final product yield per quantity of inputs (e.g., total amount of ethanol produced per pound of sucrose). In other embodiments, the program goal may be to optimize synthesis speed, as measured for example in terms of batch completion rates, or yield rates in continuous culturing systems. In one embodiment, the program goal is to optimize final product yield and/or production rate of a biomolecule or product of interest. The biomolecules or products of interest produced by the methods provided herein can be any commercial product produced from glucose. In some cases, the biomolecule or product of interest is a small molecule, an amino acid, an organic acid, or an alcohol. The amino acid can be tyrosine, phenylalanine, tryptophan, aspartic acid, asparagine, threonine, isoleucine, methionine, or lysine. The organic acid can be succinate, lactate or pyruvate. The alcohol can be ethanol or isobutanol.
[0170] Persons having ordinary skill in the art will recognize how to tailor strain selection criteria to meet the particular project goal. For example, selections of a strain's single batch max yield at reaction saturation may be appropriate for identifying strains with high single batch yields. Selection based on consistency in yield across a range of temperatures and conditions may be appropriate for identifying strains with increased robustness and reliability.
[0171] In some embodiments, the selection criteria for the initial phase and the tank-based validation will be identical. In other embodiments, tank-based selection may operate under additional and/or different selection criteria.
EXAMPLES
[0172] The present invention is further illustrated by reference to the following Examples. However, it should be noted that these Examples, like the embodiments described above, is illustrative and is not to be construed as restricting the scope of the invention in any way.
Example 1: Transformation of Corynebacterium with Glucose Permease and Glucokinase Library
Generation of Glucose Permease Libraries
[0173] A number of glucose permeases from various bacteria were selected for generation of a glucose permease library based on their affinity and transport rates for glucose as reported in the literature. The glucose permease selected for inclusion in the library were the glucose permease genes from Mycobacterium smegmatis that encodes SEQ ID NO: 9; from Bifidobacterium longum (BL_1631 in FIG. 1) that encodes SEQ ID NO: 11; from Zymomonas mobilis (glf in FIG. 1) that encodes SEQ ID NO: 13; from Synechocystis sp. PCC6803 (glcP in FIG. 1) that encodes SEQ ID NO: 12; from Streptomyces coelicolor (SC05578 in FIG. 1) that encodes SEQ ID NO: 10; and the myo-inositol transporter gene from Corynebacterium glutamicum that encodes SEQ ID NO: 14. Additionally, two glucokinases were selected for use in the generation of the glucose permease library based on their compatibility with the host cell. The glucokinases chosen were the ppgK glucokinase gene from Corynebacterium glutamicum that encodes SEQ ID NO: 16 and the glk kinase gene from Z. mobilis that encodes SEQ ID NO: 15.
[0174] For generation of the glucose permease library, each glucose permease described above was paired with a glucokinase described above such that each gene was cloned into a single C. glutamicum/Escherichia coli compatible expression vector using type IIs restriction and ligation cloning techniques. More specifically, genes that encode the M. smegmatis glucose permease (SEQ ID NO: 9), the B. longum glucose permease (BL_1631; SEQ ID NO: 11), the Synechocystis sp. PCC6803 glucose permease (glcP; SEQ ID NO: 12), the C. glutamicum myo-inositol transporter (iolT1; SEQ ID NO: 14) and the S. coelicolor glucose permease (SCO5578; SEQ ID NO: 10) were all individually paired with the gene that encodes C. glutamicum ppgK glucokinase (SEQ ID NO: 16), while the gene that encodes the Z. mobilis glucose permease (glf; SEQ ID NO: 10) was paired with the gene that encodes the Z. mobilis glk kinase (SEQ ID NO: 15) and separately paired with the gene that encodes C. glutamicum ppgK glucokinase (SEQ ID NO: 16). In addition, within each glucose permease-glucokinase construct, a P1 promoter (SEQ ID NO: 1) was cloned in front of the respective glucose permease gene, while a P2 promoter (SEQ ID NO: 2) was cloned in front of the respective glucokinase gene such that each respective permease or glucokinase gene was functionally linked to the preceding or upstream promoter. Finally, each permease gene in a construct ended with a T1 termination sequence (SEQ ID NO: 17), while each glucokinase gene in a construct ended with a T2 termination sequence (SEQ ID NO: 18).
Transformation of Assembled Clones into E. coli
[0175] Vectors containing the glucose permease-glucokinase genes were each individually transformed into E. coli in order to identify correctly assembled clones, and to amplify vector DNA for Corynebacterium transformation. Amplified DNA was validated via PCR. Positive clones were saved at -20.degree. C. fridge for future use.
Transformation of Assembled Clones into Corynebacterium
[0176] Validated clones were then individually transformed into Corynebacterium glutamicum host cells via electroporation. In order to test the effect of strain background on construct performance, two different strain backgrounds (i.e., Parent 1/background 2 and parent 2/background 1 in FIGS. 1 and 2) of C. glutamicum were used with each construct being transformed into each background. Each vector was designed to integrate into a neutral integration site within the C. glutamicum genome that was empirically determined to permit expression of the heterologous glucose permease and glucokinase genes but not be detrimental to the host cell. To facilitate integration, the expression vector further comprised about 2 kb of sequence homologous (i.e., homology arms) to the desired integration site whereby each glucose permease-glucokinase gene cassette described above was inserted between. Integration into the genome occurred by single-crossover integration and then loop-out of the plasmid backbone facilitated by counter-selection on a second marker included in the plasmid backbone.
[0177] Transformed bacteria were then tested for assembly success (correct integration into the genome). Colonies from each Corynebacterium transformation plate were cultured and tested for correct integration via PCR. This process was repeated for each of the transformations conducted for each glucose permease-glucokinase construct. Genomic integration of each transformation was also analyzed with respect to the targeted genome location for each plasmid.
Evaluation of Individual Glucose Permease-Glucokinase Constructs in Corynebacterium
[0178] The phenotype of each transformant was then tested in an evaluation method designed to mimic or simulate a specific fermentation process for producing a desired fermentation end product in order to determine the effects the expression of each construct in each host cell background had on the desired phenotype (i.e., improved ability to produce a desired fermentation end product). Briefly, the evaluation method was an experiment where the transformants were cultured in a 96 well plate format under conditions that were meant to mimic fermentation conditions. The amount of product and biomass formed at various time points was measured and used to predict how each strain would perform under fermentation conditions. This prediction was a linear regression generated from testing strains with various fermentation performance in the evaluation method and determining the correlation of measurements to performance.
[0179] The rate of production and yield of the desired fermentation end product was determined for each permease-glucokinase transformant, some examples of which are shown in FIG. 1. As shown in FIG. 1, for the specific permease-glucokinase inserts shown, the productivity (top) in a fermentation process was predicted to increase in each host background for each permease-glucokinase insert shown vs. the respective control host cell, while the yield (bottom) was predicted to be similar (glcP; BL1631), increased (SCO5578), or decreased (glf) vs. the respective control host cell. Please note that the AU units in FIG. 1 are the output of a linear regression that takes as inputs various measurements made on cultures at small scale and predicts the performance of strains under fermentation conditions.
Assessment of Individual Glucose-Permease-Glucokinase Constructs Under Fermentation Conditions
[0180] Following evaluation as described above, transformants with heterologous glucose permease-glucokinase genes with predicted increased performance (i.e., increased predicted productivity and/or predicted yield) were selected and subsequently grown in medium containing glucose under conditions designed to facilitate fermentation and the production of desired fermentation end products. Following growth of each transformant for a predetermined length of time under fermentation conditions designed to produce a desired end-product, the yield and volumetric productivity of the end-product for each transformant was then determined. Briefly, high-performance liquid chromatography (HPLC) was used to determine the amount of product (i.e., avg yield) produced for a certain amount of substrate fed. Productivity (i.e., avg productivity) was similarly determined with the addition of time and volume data.
[0181] As shown in FIG. 2, the BL_1631 glucose permease-C. glutamicum ppgK kinase construct increased productivity of the host cell with background 1 by 15% as well as increased the yield by about 1%. In addition, in host cell with background 2, both the glk permease-glk kinase construct and the glcP permease-C. glutamicum ppgK kinase construct increased productivity by more than 30%, but did not affect yield. Accordingly, this example shows that the methods provided herein can be used to increase the performance of microbial strains in terms of producing fermentation end products.
INCORPORATION BY REFERENCE
[0182] The following applications are hereby incorporated by reference in their entirety, including all descriptions, references, figures, and claims for all purposes: U.S. application Ser. No. 15/396,230, filed on Dec. 30, 2016; International Application No. PCT/US2016/065465, filed on Dec. 7, 2016; U.S. application Ser. No. 15/140,296, filed on Apr. 27, 2016; U.S. Provisional Application No. 62/368,786, filed on Jul. 29, 2016; and U.S. Provisional Application No. 62/264,232, filed on Dec. 7, 2015.
[0183] All references, articles, publications, patents, patent publications, and patent applications cited herein are incorporated by reference in their entireties for all purposes.
[0184] However, mention of any reference, article, publication, patent, patent publication, and patent application cited herein is not, and should not be taken as an acknowledgment or any form of suggestion that they constitute valid prior art or form part of the common general knowledge in any country in the world.
Sequence CWU 1
1
26197DNACorynebacterium glutamicummisc_feature(1)..(97)Pcg0007_39
1tgccgtttct cgcgttgtgt gtggtactac gtggggacct aagcgtgtat tatggaaacg
60tctgtatcgg ataagtagcg aggagtgttc gttaaaa 97297DNACorynebacterium
glutamicummisc_feature(1)..(97)Pcg0007 2tgccgtttct cgcgttgtgt
gtggtactac gtggggacct aagcgtgtaa gatggaaacg 60tctgtatcgg ataagtagcg
aggagtgttc gttaaaa 97393DNACorynebacterium
glutamicummisc_feature(1)..(93)Pcg1860 3cttagctttg acctgcacaa
atagttgcaa attgtcccac atacacataa agtagcttgc 60gtatttaaaa ttatgaacct
aaggggttta gca 93498DNACorynebacterium
glutamicummisc_feature(1)..(98)Pcg0755 4aataaattta taccacacag
tctattgcaa tagaccaagc tgttcagtag ggtgcatggg 60agaagaattt cctaataaaa
actcttaagg acctccaa 98597DNACorynebacterium
glutamicummisc_feature(1)..(97)Pcg0007_265 5tgccgtttct cgcgttgtgt
gtggtactac gtggggacct aagcgtgtac gctggaaacg 60tctgtatcgg ataagtagcg
aggagtgttc gttaaaa 97686DNACorynebacterium
glutamicummisc_feature(1)..(86)Pcg3381 6cgccggataa atgaattgat
tattttaggc tcccagggat taagtctagg gtggaatgca 60gaaatatttc ctacggaagg
tccgtt 86797DNACorynebacterium
glutamicummisc_feature(1)..(97)Pcg0007_119 7tgccgtttct cgcgttgtgt
gtggtactac gtggggacct aagcgtgttg catggaaacg 60tctgtatcgg ataagtagcg
aggagtgttc gttaaaa 97887DNACorynebacterium
glutamicummisc_feature(1)..(87)Pcg3121 8gtggctaaaa cttttggaaa
cttaagttac ctttaatcgg aaacttattg aattcgggtg 60aggcaactgc aactctggac
ttaaagc 879485PRTMycobacterium
smegmatisMISC_FEATURE(1)..(485)MSMEG_4187 9Met Arg Gln Thr Gly Ser
Leu Arg Pro Ile Leu Val Pro Val Trp Ile1 5 10 15Leu Val Leu Val Ala
Ala Leu Ala Gly Cys Ala Thr Arg Thr Asp Asp 20 25 30Gln Pro Thr Glu
Ser Ala Pro Pro Pro Ala Gln Gln Ala Pro Pro Thr 35 40 45Pro Ala Glu
Ile Arg Ala Ile Ala Lys Asp Ala Tyr Ile Trp Gly Phe 50 55 60Pro Leu
Val Asp Asn Tyr Arg Val Gln Tyr Ser Tyr Phe Val Asp Lys65 70 75
80Thr Asp Pro Glu Tyr Lys Gly Gly Phe Asn Glu Val His Asn Thr Ala
85 90 95Arg Leu Tyr Thr Pro Ala Asp Lys Ala Ile Gln Thr Pro Asn Ala
Asp 100 105 110Thr Pro Tyr Ser Phe Val Gly Ala Asp Leu Arg Thr Glu
Pro Leu Val 115 120 125Phe Thr Val Pro Pro Ile Glu Gln Asn Arg Tyr
Phe Ser Leu Gln Phe 130 135 140Val Asp Gly Tyr Thr Tyr Asn Val Ala
Tyr Val Gly Ser Arg Thr Thr145 150 155 160Gly Asn Gly Gly Gly Arg
Tyr Leu Leu Ala Gly Pro Gly Trp Glu Gly 165 170 175Glu Lys Pro Glu
Gly Val Asp Glu Ile Ile Arg Ser Asp Thr Asp Leu 180 185 190Ala Phe
Val Leu Tyr Arg Thr Gln Leu Phe Gly Pro Arg Asp Leu Asp 195 200
205Asn Ile Lys Lys Ile Gln Ala Gly Tyr Gln Val Ala Pro Leu Ser Val
210 215 220Tyr Leu Lys Gln Pro Ser Pro Pro Pro Ala Pro Pro Ile Asp
Phe Thr225 230 235 240Pro Pro Leu Thr Pro Glu Ala Gln Lys Thr Ser
Pro Gln Phe Phe Glu 245 250 255Ile Leu Asn Ala Ala Leu Arg Tyr Ala
Pro Val Lys Pro Glu Glu Gln 260 265 270Glu Met Arg Glu Arg Phe Ala
Arg Ile Gly Ile Gly Pro Asp Gly Asp 275 280 285Phe Asp Ala Asp Lys
Leu Ser Pro Glu Thr Arg Glu Ala Ile Glu Asp 290 295 300Gly Met Ala
Asn Ala Trp Val Glu Phe Asp Arg Phe Lys Gln Asp Lys305 310 315
320Val Asp Thr Gly Glu Val Gly Ser Ala Gln Leu Phe Gly Thr Ala Asp
325 330 335Asp Leu Lys Gly Asn Tyr Leu Tyr Arg Met Ala Gly Ala Val
Leu Gly 340 345 350Ile Tyr Gly Asn Thr Ala Ala Glu Ala Leu Tyr Pro
Ser Ala Met Leu 355 360 365Asp Ala Asp Gly Gln Pro Leu Thr Gly Thr
Asn Ser Tyr Thr Tyr Arg 370 375 380Phe Ala Pro Asp Gln Leu Pro Pro
Val Asn Ala Phe Trp Ser Leu Thr385 390 395 400Ile Tyr Glu Leu Pro
Ser Ser Gln Leu Val Asp Asn Pro Ile Asp Arg 405 410 415Tyr Leu Ile
Asn Ser Glu Met Leu Pro Ser Leu Val Pro Asp Pro Asp 420 425 430Gly
Ala Tyr Thr Leu Arg Ile Gln Asn Thr Gln Pro Pro Glu Asn Glu 435 440
445Ala Asn Trp Leu Pro Ala Pro Lys Gly Pro Phe Thr Leu Val Leu Arg
450 455 460Leu Tyr Trp Pro Lys Pro Asp Ala Leu Asn Gly Thr Trp Gln
Ala Pro465 470 475 480Lys Pro Glu Lys Ile 48510471PRTStreptomyces
coelicolorMISC_FEATURE(1)..(471)SCO5578 10Met Ala Ser Thr Ser Gln
Ala Pro Ser Pro Gly Ala Gly Thr Ala His1 5 10 15Pro Asp His Leu Gly
His Val Ile Phe Ile Ala Ala Ala Ala Ala Met 20 25 30Gly Gly Phe Leu
Phe Gly Tyr Asp Ser Ser Val Ile Asn Gly Ala Val 35 40 45Glu Ala Ile
Arg Asp Arg Tyr Asp Val Gly Ser Ala Val Leu Ala Gln 50 55 60Val Ile
Ala Val Ala Leu Ile Gly Cys Ala Ile Gly Ala Ala Thr Ala65 70 75
80Gly Arg Ile Ala Asp Arg Ile Gly Arg Ile Arg Cys Met Gln Ile Ala
85 90 95Ala Val Leu Phe Thr Val Ser Ala Val Gly Ser Ala Leu Pro Phe
Ala 100 105 110Leu Trp Asp Leu Ala Met Trp Arg Ile Ile Gly Gly Phe
Ala Ile Gly 115 120 125Met Ala Ser Val Ile Gly Pro Ala Tyr Ile Ala
Glu Val Ser Pro Pro 130 135 140Ala Tyr Arg Gly Arg Leu Gly Ser Phe
Gln Gln Ala Ala Ile Val Ile145 150 155 160Gly Ile Ala Val Ser Gln
Leu Val Asn Trp Gly Leu Leu Asn Ala Ala 165 170 175Gly Gly Asp Gln
Arg Gly Glu Leu Met Gly Leu Glu Ala Trp Gln Val 180 185 190Met Leu
Gly Val Met Val Ile Pro Ala Val Leu Tyr Gly Leu Leu Ser 195 200
205Phe Ala Ile Pro Glu Ser Pro Arg Phe Leu Ile Ser Val Gly Lys Arg
210 215 220Glu Arg Ala Lys Lys Ile Leu Glu Glu Val Glu Gly Lys Asp
Val Asp225 230 235 240Phe Asp Ala Arg Val Thr Glu Ile Glu His Ala
Met His Arg Glu Glu 245 250 255Lys Ser Ser Phe Lys Asp Leu Leu Gly
Gly Ser Phe Phe Phe Lys Pro 260 265 270Ile Val Trp Ile Gly Ile Gly
Leu Ser Val Phe Gln Gln Phe Gly Ile 275 280 285Asn Val Ala Phe Tyr
Tyr Ser Ser Thr Leu Trp Gln Ser Val Gly Val 290 295 300Asp Pro Ala
Asp Ser Phe Phe Tyr Ser Phe Thr Thr Ser Ile Ile Asn305 310 315
320Ile Val Gly Thr Val Ile Ala Met Ile Phe Val Asp Arg Val Gly Arg
325 330 335Lys Pro Leu Ala Leu Ile Gly Ser Val Gly Met Val Ile Gly
Leu Ala 340 345 350Leu Glu Ala Trp Ala Phe Ser Phe Asp Leu Val Asp
Gly Lys Leu Pro 355 360 365Ala Thr Gln Gly Trp Val Ala Leu Ile Ala
Ala His Val Phe Val Leu 370 375 380Phe Phe Ala Leu Ser Trp Gly Val
Val Val Trp Val Phe Leu Gly Glu385 390 395 400Met Phe Pro Asn Arg
Ile Arg Ala Ala Ala Leu Gly Val Ala Ala Ser 405 410 415Ala Gln Trp
Ile Ala Asn Trp Ala Ile Thr Ala Ser Phe Pro Ser Leu 420 425 430Ala
Asp Trp Asn Leu Ser Gly Thr Tyr Val Ile Tyr Thr Ile Phe Ala 435 440
445Ala Leu Ser Ile Pro Phe Val Leu Lys Phe Val Lys Glu Thr Lys Gly
450 455 460Lys Ala Leu Glu Glu Met Gly465
47011517PRTBifidobacterium longumMISC_FEATURE(1)..(517)BL1631 11Met
Thr Thr Thr Thr Ala Ser Pro Val Ser Lys Gln Thr Ala Ser Ala1 5 10
15Ala Gln Glu Thr Ser Ala Thr Gly Ala Ala Ala Thr Ala Ile Glu Thr
20 25 30Ile Glu Thr Gly Val Ala Gly Val Ala Gly Ala Ala Thr Asn Ala
Ala 35 40 45Ala Asn Ala Ile Glu Asp Leu Glu Ala Ala Glu Ser His Gly
Phe Ser 50 55 60Thr Arg Phe Pro Leu Asn Ser Ala Phe Ile Phe Thr Phe
Gly Ala Leu65 70 75 80Gly Gly Met Leu Phe Gly Phe Asp Thr Gly Ile
Ile Ser Gly Ala Ser 85 90 95Pro Leu Ile Glu Ser Asp Phe Gly Leu Ser
Val Ser Gln Thr Gly Phe 100 105 110Ile Thr Ser Ser Val Leu Ile Gly
Ser Cys Ala Gly Ala Leu Ser Ile 115 120 125Gly Ala Leu Ser Asp Arg
Phe Gly Arg Lys Lys Leu Leu Ile Val Ser 130 135 140Ala Leu Leu Phe
Leu Leu Gly Ser Gly Leu Cys Ala Ser Ser Thr Gly145 150 155 160Phe
Ala Met Met Val Cys Ala Arg Ile Ile Leu Gly Leu Ala Val Gly 165 170
175Ala Ala Ser Ala Leu Thr Pro Ala Tyr Leu Ala Glu Leu Ala Pro Lys
180 185 190Glu Arg Arg Gly Ser Leu Ser Thr Leu Phe Gln Leu Met Val
Thr Phe 195 200 205Gly Ile Leu Leu Ala Tyr Ala Ser Asn Leu Gly Phe
Leu Asn His Asn 210 215 220Leu Phe Gly Ile Arg Asp Trp Arg Trp Met
Leu Gly Ser Ala Leu Val225 230 235 240Pro Ala Ala Leu Leu Leu Leu
Gly Gly Leu Leu Leu Pro Glu Ser Pro 245 250 255Arg Tyr Leu Val Asn
Lys Gly Asp Thr Arg Asn Ala Phe Lys Val Leu 260 265 270Thr Leu Ile
Arg Lys Asp Val Asp Gln Thr Gln Val Gln Ile Glu Leu 275 280 285Asp
Glu Ile Lys Ala Val Ala Ala Gln Asp Thr Lys Gly Gly Val Arg 290 295
300Glu Leu Phe Arg Ile Ala Arg Pro Ala Leu Val Ala Ala Ile Gly
Ile305 310 315 320Met Leu Phe Gln Gln Leu Val Gly Ile Asn Ser Val
Ile Tyr Phe Leu 325 330 335Pro Gln Val Phe Ile Lys Gly Phe Gly Phe
Pro Glu Gly Asp Ala Ile 340 345 350Trp Val Ser Val Gly Ile Gly Val
Val Asn Phe Val Ser Thr Ile Val 355 360 365Ala Thr Leu Ile Met Asp
Arg Phe Pro Arg Lys Gly Met Leu Ile Phe 370 375 380Gly Ser Ile Val
Met Thr Val Ser Leu Ala Val Leu Ala Val Met Asn385 390 395 400Phe
Val Gly Asp Val Ala Val Leu Ala Val Pro Thr Met Ile Leu Ile 405 410
415Ala Phe Tyr Ile Leu Gly Phe Ala Val Ser Trp Gly Pro Ile Ala Trp
420 425 430Val Leu Ile Gly Glu Ile Phe Pro Leu Ser Val Arg Gly Ile
Gly Ser 435 440 445Ser Phe Gly Ser Ala Ala Asn Trp Leu Gly Asn Phe
Ile Val Ser Gln 450 455 460Phe Phe Leu Val Leu Leu Asp Ala Phe Gly
Asn Asn Val Gly Gly Pro465 470 475 480Phe Ala Ile Phe Gly Val Phe
Ser Ala Leu Ser Ile Pro Phe Val Leu 485 490 495Arg Leu Val Pro Glu
Thr Lys Gly Lys Ser Leu Glu Glu Ile Glu Lys 500 505 510Glu Met Thr
Lys Arg 51512468PRTSynechocystis sp.
PCC6803MISC_FEATURE(1)..(468)glcp (CAA34119.1) 12Met Asn Pro Ser
Ser Ser Pro Ser Gln Ser Thr Ala Asn Val Lys Phe1 5 10 15Val Leu Leu
Ile Ser Gly Val Ala Ala Leu Gly Gly Phe Leu Phe Gly 20 25 30Phe Asp
Thr Ala Val Ile Asn Gly Ala Val Ala Ala Leu Gln Lys His 35 40 45Phe
Gln Thr Asp Ser Leu Leu Thr Gly Leu Ser Val Ser Leu Ala Leu 50 55
60Leu Gly Ser Ala Leu Gly Ala Phe Gly Ala Gly Pro Ile Ala Asp Arg65
70 75 80His Gly Arg Ile Lys Thr Met Ile Leu Ala Ala Val Leu Phe Thr
Leu 85 90 95Ser Ser Ile Gly Ser Gly Leu Pro Phe Thr Ile Trp Asp Phe
Ile Phe 100 105 110Trp Arg Val Leu Gly Gly Ile Gly Val Gly Ala Ala
Ser Val Ile Ala 115 120 125Pro Ala Tyr Ile Ala Glu Val Ser Pro Ala
His Leu Arg Gly Arg Leu 130 135 140Gly Ser Leu Gln Gln Leu Ala Ile
Val Ser Gly Ile Phe Ile Ala Leu145 150 155 160Leu Ser Asn Trp Phe
Ile Ala Leu Met Ala Gly Gly Ser Ala Gln Asn 165 170 175Pro Trp Leu
Phe Gly Ala Ala Ala Trp Arg Trp Met Phe Trp Thr Glu 180 185 190Leu
Ile Pro Ala Leu Leu Tyr Gly Val Cys Ala Phe Leu Ile Pro Glu 195 200
205Ser Pro Arg Tyr Leu Val Ala Gln Gly Gln Gly Glu Lys Ala Ala Ala
210 215 220Ile Leu Trp Lys Val Glu Gly Gly Asp Val Pro Ser Arg Ile
Glu Glu225 230 235 240Ile Gln Ala Thr Val Ser Leu Asp His Lys Pro
Arg Phe Ser Asp Leu 245 250 255Leu Ser Arg Arg Gly Gly Leu Leu Pro
Ile Val Trp Ile Gly Met Gly 260 265 270Leu Ser Ala Leu Gln Gln Phe
Val Gly Ile Asn Val Ile Phe Tyr Tyr 275 280 285Ser Ser Val Leu Trp
Arg Ser Val Gly Phe Thr Glu Glu Lys Ser Leu 290 295 300Leu Ile Thr
Val Ile Thr Gly Phe Ile Asn Ile Leu Thr Thr Ile Val305 310 315
320Ala Ile Ala Phe Val Asp Lys Phe Gly Arg Lys Pro Leu Leu Leu Met
325 330 335Gly Ser Ile Gly Met Thr Ile Thr Leu Gly Ile Leu Ser Val
Val Phe 340 345 350Gly Gly Ala Thr Val Val Asn Gly Gln Pro Thr Leu
Thr Gly Ala Ala 355 360 365Gly Ile Ile Ala Leu Val Thr Ala Asn Leu
Tyr Val Phe Ser Phe Gly 370 375 380Phe Ser Trp Gly Pro Ile Val Trp
Val Leu Leu Gly Glu Met Phe Asn385 390 395 400Asn Lys Ile Arg Ala
Ala Ala Leu Ser Val Ala Ala Gly Val Gln Trp 405 410 415Ile Ala Asn
Phe Ile Ile Ser Thr Thr Phe Pro Pro Leu Leu Asp Thr 420 425 430Val
Gly Leu Gly Pro Ala Tyr Gly Leu Tyr Ala Thr Ser Ala Ala Ile 435 440
445Ser Ile Phe Phe Ile Trp Phe Phe Val Lys Glu Thr Lys Gly Lys Thr
450 455 460Leu Glu Gln Met46513473PRTZymomonas
mobilisMISC_FEATURE(1)..(473)glf permease 13Met Ser Ser Glu Ser Ser
Gln Gly Leu Val Thr Arg Leu Ala Leu Ile1 5 10 15Ala Ala Ile Gly Gly
Leu Leu Phe Gly Tyr Asp Ser Ala Val Ile Ala 20 25 30Ala Ile Gly Thr
Pro Val Asp Ile His Phe Ile Ala Pro Arg His Leu 35 40 45Ser Ala Thr
Ala Ala Ala Ser Leu Ser Gly Met Val Val Val Ala Val 50 55 60Leu Val
Gly Cys Val Thr Gly Ser Leu Leu Ser Gly Trp Ile Gly Ile65 70 75
80Arg Phe Gly Arg Arg Gly Gly Leu Leu Met Ser Ser Ile Cys Phe Val
85 90 95Ala Ala Gly Phe Gly Ala Ala Leu Thr Glu Lys Leu Phe Gly Thr
Gly 100 105 110Gly Ser Ala Leu Gln Ile Phe Cys Phe Phe Arg Phe Leu
Ala Gly Leu 115 120 125Gly Ile Gly Val Val Ser Thr Leu Thr Pro Thr
Tyr Ile Ala Glu Ile 130 135 140Ala Pro Pro Asp Lys Arg Gly Gln Met
Val Ser Gly Gln Gln Met Ala145 150 155 160Ile Val Thr Gly Ala Leu
Thr Gly Tyr Ile Phe Thr Trp Leu Leu Ala 165 170 175His Phe Gly Ser
Ile Asp Trp Val Asn Ala Ser Gly Trp Cys Trp Ser 180 185 190Pro Ala
Ser Glu Gly Leu Ile Gly Ile Ala Phe Leu Leu Leu Leu Leu 195 200
205Thr Ala Pro Asp Thr Pro His Trp Leu Val Met Lys Gly Arg His Ser
210 215 220Glu Ala Ser Lys Ile Leu Ala Arg Leu Glu Pro Gln Ala Asp
Pro Asn225 230 235 240Leu Thr Ile Gln Lys Ile Lys Ala Gly Phe Asp
Lys Ala Met Asp Lys
245 250 255Ser Ser Ala Gly Leu Phe Ala Phe Gly Ile Thr Val Val Phe
Ala Gly 260 265 270Val Ser Val Ala Ala Phe Gln Gln Leu Val Gly Ile
Asn Ala Val Leu 275 280 285Tyr Tyr Ala Pro Gln Met Phe Gln Asn Leu
Gly Phe Gly Ala Asp Thr 290 295 300Ala Leu Leu Gln Thr Ile Ser Ile
Gly Val Val Asn Phe Ile Phe Thr305 310 315 320Met Ile Ala Ser Arg
Val Val Asp Arg Phe Gly Arg Lys Pro Leu Leu 325 330 335Ile Trp Gly
Ala Leu Gly Met Ala Ala Met Met Ala Val Leu Gly Cys 340 345 350Cys
Phe Trp Phe Lys Val Gly Gly Val Leu Pro Leu Ala Ser Val Leu 355 360
365Leu Tyr Ile Ala Val Phe Gly Met Ser Trp Gly Pro Val Cys Trp Val
370 375 380Val Leu Ser Glu Met Phe Pro Ser Ser Ile Lys Gly Ala Ala
Met Pro385 390 395 400Ile Ala Val Thr Gly Gln Trp Leu Ala Asn Ile
Leu Val Asn Phe Leu 405 410 415Phe Lys Val Ala Asp Gly Ser Pro Ala
Leu Asn Gln Thr Phe Asn His 420 425 430Gly Phe Ser Tyr Leu Val Phe
Ala Ala Leu Ser Ile Leu Gly Gly Leu 435 440 445Ile Val Ala Arg Phe
Val Pro Glu Thr Lys Gly Arg Ser Leu Asp Glu 450 455 460Ile Glu Glu
Met Trp Arg Ser Gln Lys465 47014491PRTCorynebacterium
glutamicumMISC_FEATURE(1)..(491)iolT1 14Met Ala Ser Thr Phe Ile Gln
Ala Asp Ser Pro Glu Lys Ser Lys Lys1 5 10 15Leu Pro Pro Leu Thr Glu
Gly Pro Tyr Arg Lys Arg Leu Phe Tyr Val 20 25 30Ala Leu Val Ala Thr
Phe Gly Gly Leu Leu Phe Gly Tyr Asp Thr Gly 35 40 45Val Ile Asn Gly
Ala Leu Asn Pro Met Thr Arg Glu Leu Gly Leu Thr 50 55 60Ala Phe Thr
Glu Gly Val Val Thr Ser Ser Leu Leu Phe Gly Ala Ala65 70 75 80Ala
Gly Ala Met Phe Phe Gly Arg Ile Ser Asp Asn Trp Gly Arg Arg 85 90
95Lys Thr Ile Ile Ser Leu Ala Val Ala Phe Phe Val Gly Thr Met Ile
100 105 110Cys Val Phe Ala Pro Ser Phe Ala Val Met Val Val Gly Arg
Val Leu 115 120 125Leu Gly Leu Ala Val Gly Gly Ala Ser Thr Val Val
Pro Val Tyr Leu 130 135 140Ala Glu Leu Ala Pro Phe Glu Ile Arg Gly
Ser Leu Ala Gly Arg Asn145 150 155 160Glu Leu Met Ile Val Val Gly
Gln Leu Ala Ala Phe Val Ile Asn Ala 165 170 175Ile Ile Gly Asn Val
Phe Gly His His Asp Gly Val Trp Arg Tyr Met 180 185 190Leu Ala Ile
Ala Ala Ile Pro Ala Ile Ala Leu Phe Phe Gly Met Leu 195 200 205Arg
Val Pro Glu Ser Pro Arg Trp Leu Val Glu Arg Gly Arg Ile Asp 210 215
220Glu Ala Arg Ala Val Leu Glu Thr Ile Arg Pro Leu Glu Arg Ala
His225 230 235 240Ala Glu Val Ala Asp Val Glu His Leu Ala Arg Glu
Glu His Ala Val 245 250 255Ser Glu Lys Ser Met Gly Leu Arg Glu Ile
Leu Ser Ser Lys Trp Leu 260 265 270Val Arg Ile Leu Leu Val Gly Ile
Gly Leu Gly Val Ala Gln Gln Leu 275 280 285Thr Gly Ile Asn Ser Ile
Met Tyr Tyr Gly Gln Val Val Leu Ile Glu 290 295 300Ala Gly Phe Ser
Glu Asn Ala Ala Leu Ile Ala Asn Val Ala Pro Gly305 310 315 320Val
Ile Ala Val Val Gly Ala Phe Ile Ala Leu Trp Met Met Asp Arg 325 330
335Ile Asn Arg Arg Thr Thr Leu Ile Thr Gly Tyr Ser Leu Thr Thr Ile
340 345 350Ser His Val Leu Ile Gly Ile Ala Ser Val Ala Phe Pro Val
Gly Asp 355 360 365Pro Leu Arg Pro Tyr Val Ile Leu Thr Leu Val Val
Val Phe Val Gly 370 375 380Ser Met Gln Thr Phe Leu Asn Val Ala Thr
Trp Val Met Leu Ser Glu385 390 395 400Leu Phe Pro Leu Ala Met Arg
Gly Phe Ala Ile Gly Ile Ser Val Phe 405 410 415Phe Leu Trp Ile Ala
Asn Ala Phe Leu Gly Leu Phe Phe Pro Thr Ile 420 425 430Met Glu Ala
Val Gly Leu Thr Gly Thr Phe Phe Met Phe Ala Gly Ile 435 440 445Gly
Val Val Ala Leu Ile Phe Ile Tyr Thr Gln Val Pro Glu Thr Arg 450 455
460Gly Arg Thr Leu Glu Glu Ile Asp Glu Asp Val Thr Ser Gly Val
Ile465 470 475 480Phe Asn Lys Asp Ile Arg Lys Gly Lys Val His 485
49015324PRTZymomonas mobilisMISC_FEATURE(1)..(324)glk kinase 15Met
Glu Ile Val Ala Ile Asp Ile Gly Gly Thr His Ala Arg Phe Ser1 5 10
15Ile Ala Glu Val Ser Asn Gly Arg Val Leu Ser Leu Gly Glu Glu Thr
20 25 30Thr Phe Lys Thr Ala Glu His Ala Ser Leu Gln Leu Ala Trp Glu
Arg 35 40 45Phe Gly Glu Lys Leu Gly Arg Pro Leu Pro Arg Ala Ala Ala
Ile Ala 50 55 60Trp Ala Gly Pro Val His Gly Glu Val Leu Lys Leu Thr
Asn Asn Pro65 70 75 80Trp Val Leu Arg Pro Ala Thr Leu Asn Glu Lys
Leu Asp Ile Asp Thr 85 90 95His Val Leu Ile Asn Asp Phe Gly Ala Val
Ala His Ala Val Ala His 100 105 110Met Asp Ser Ser Tyr Leu Asp His
Ile Cys Gly Pro Asp Glu Ala Leu 115 120 125Pro Ser Asp Gly Val Ile
Thr Ile Leu Gly Pro Gly Thr Gly Leu Gly 130 135 140Val Ala His Leu
Leu Arg Thr Glu Gly Arg Tyr Phe Val Ile Glu Thr145 150 155 160Glu
Gly Gly His Ile Asp Phe Ala Pro Leu Asp Arg Leu Glu Asp Lys 165 170
175Ile Leu Ala Arg Leu Arg Glu Arg Phe Arg Arg Val Ser Ile Glu Arg
180 185 190Ile Ile Ser Gly Pro Gly Leu Gly Asn Ile Tyr Glu Ala Leu
Ala Ala 195 200 205Ile Glu Gly Val Pro Phe Ser Leu Leu Asp Asp Ile
Lys Leu Trp Gln 210 215 220Met Ala Leu Glu Gly Lys Asp Asn Leu Ala
Glu Ala Ala Leu Asp Arg225 230 235 240Phe Cys Leu Ser Leu Gly Ala
Ile Ala Gly Asp Leu Ala Leu Ala Gln 245 250 255Gly Ala Thr Ser Val
Val Ile Gly Gly Gly Val Gly Leu Arg Ile Ala 260 265 270Ser His Leu
Pro Glu Ser Gly Phe Arg Gln Arg Phe Val Ser Lys Gly 275 280 285Arg
Phe Glu Arg Val Met Ser Lys Ile Pro Val Lys Leu Ile Thr Tyr 290 295
300Pro Gln Pro Gly Leu Leu Gly Ala Ala Ala Ala Tyr Ala Asn Lys
Tyr305 310 315 320Ser Glu Val Glu16250PRTCorynebacterium
glutamicumMISC_FEATURE(1)..(250)ppgK kinase 16Met Thr Glu Thr Gly
Phe Gly Ile Asp Ile Gly Gly Ser Gly Ile Lys1 5 10 15Gly Ala Arg Val
Asn Leu Lys Thr Gly Glu Phe Ile Asp Glu Arg Ile 20 25 30Lys Ile Ala
Thr Pro Lys Pro Ala Thr Pro Glu Ala Val Ala Glu Val 35 40 45Val Ala
Glu Ile Ile Ser Gln Ala Glu Trp Glu Gly Pro Val Gly Ile 50 55 60Thr
Leu Pro Ser Val Val Arg Gly Gln Ile Ala Leu Ser Ala Ala Asn65 70 75
80Ile Asp Lys Ser Trp Ile Gly Thr Asp Val His Glu Leu Phe Asp Arg
85 90 95His Leu Asn Gly Arg Glu Ile Thr Val Leu Asn Asp Ala Asp Ala
Ala 100 105 110Gly Ile Ala Glu Ala Thr Phe Gly Asn Pro Ala Ala Arg
Glu Gly Ala 115 120 125Val Ile Leu Leu Thr Leu Gly Thr Gly Ile Gly
Ser Ala Phe Leu Val 130 135 140Asp Gly Gln Leu Phe Pro Asn Thr Glu
Leu Gly His Met Ile Val Asp145 150 155 160Gly Glu Glu Ala Glu His
Leu Ala Ala Ala Ser Val Lys Glu Asn Glu 165 170 175Asp Leu Ser Trp
Lys Lys Trp Ala Lys His Leu Asn Lys Val Leu Ser 180 185 190Glu Tyr
Glu Lys Leu Phe Ser Pro Ser Val Phe Ile Ile Gly Gly Gly 195 200
205Ile Ser Arg Lys His Glu Lys Trp Leu Pro Leu Met Glu Leu Asp Thr
210 215 220Asp Ile Val Pro Ala Glu Leu Arg Asn Arg Ala Gly Ile Val
Gly Ala225 230 235 240Ala Met Ala Val Asn Gln His Leu Thr Pro 245
2501774DNACorynebacterium glutamicumT1 termination sequence
17acaatagtaa aaggaaccct cacgaactgt gagggttcct tttttgggtt tcgccggagg
60agacgtcgaa aagc 741877DNACorynebacterium glutamicumT2 termination
sequence 18gcatttttag tacgtgcaat aaccactctg gtttttccag ggtggttttt
tgatgccctt 60tttggagtct tcaactg 77191419DNAZymomonas
mobilismisc_feature(1)..(1419)glf permease 19atgtcctcag aatcttccca
aggtctagtt acccgcttgg cgcttattgc tgcaattggt 60ggacttctat tcggatacga
tagtgctgta attgctgcga ttggcactcc agttgacatt 120catttcattg
ctccgcgtca tcttagtgct acagcggcgg cttcacttag tggtatggtt
180gtagtggctg ttcttgtcgg gtgtgtgact ggttctcttc tctcaggttg
gattggcatc 240cgttttggtc gccggggcgg attgcttatg agtagtattt
gtttcgtcgc ggctggattc 300ggagcggcgt tgaccgagaa acttttcggc
actggtggat ctgcacttca gattttctgt 360ttcttccgtt tccttgcggg
actcggaatt ggtgtcgtta gtacacttac tccgacttac 420attgctgaaa
ttgctcctcc ggataagcgt ggacaaatgg tatccggtca acagatggcg
480attgttactg gggctcttac tggatatatt ttcacctggt tgcttgcgca
tttcggatct 540atcgactggg tgaatgcgtc tggatggtgt tggtcaccag
cttccgaagg gctgatcggt 600atcgcgtttc tgcttcttct tttgacggcg
ccagatacgc cgcattggtt ggtgatgaag 660ggccggcatt ctgaagcgtc
caaaattctg gcacgacttg agcctcaggc tgatccaaat 720ctcacgattc
aaaagattaa agctggcttc gataaagcaa tggataaatc ctctgcagga
780ctcttcgcat ttggaatcac cgtagttttt gctggtgtta gtgttgcagc
atttcaacaa 840ttggtaggaa tcaatgctgt actgtattat gctccgcaga
tgtttcagaa tcttggattt 900ggtgctgata cggctctgct tcagactatt
agtattggcg tagtgaactt tattttcacg 960atgattgctt cacgtgtggt
tgaccgcttc ggtcgcaaac cgcttttgat ctggggtgcg 1020ctgggtatgg
cagcgatgat ggcagttctt ggttgctgtt tctggtttaa agtcggcgga
1080gtgcttcctt tggcgtctgt tctactttat atcgctgtat ttggtatgtc
ctggggtccc 1140gtgtgttggg tagtactttc tgaaatgttt ccatcttcta
tcaaaggtgc agctatgcct 1200attgcagtga cgggacagtg gttggctaac
atccttgtca attttctttt caaagtcgct 1260gacggctcac cggcgttgaa
tcagacgttt aaccatggtt tttcttactt ggtattcgcg 1320gctctttcta
ttcttggcgg actaattgtt gctcgctttg tcccggaaac aaagggtcgc
1380tctctcgatg aaattgaaga gatgtggcgt tcacaaaaa
1419201419DNAStreptomyces coelicolormisc_feature(1)..(1419)SCO5578
20atggcttcaa cttcacaggc accatcacca ggggcaggaa ccgcgcatcc agatcatcta
60ggtcatgtca tcttcattgc tgctgctgcg gcgatgggtg gattcctgtt cggatatgat
120tcctctgtaa tcaatggcgc tgttgaagcg attcgggatc gttatgatgt
tggatctgca 180gttcttgcgc aagtaattgc ggtagctctt attggctgtg
caattggcgc tgcgactgcg 240ggacgcatcg cggaccgcat cggacgcatt
cgttgtatgc agattgcagc ggtgcttttt 300accgtaagtg ctgtaggatc
cgcattgcca ttcgctcttt gggatcttgc tatgtggcgt 360attatcggtg
gattcgcgat cggtatggcg agtgtaatcg gcccagctta cattgctgaa
420gtgtctcccc cggcgtatcg tggtcgcctt ggttcttttc agcaagctgc
aattgtcatt 480ggtattgcag tatctcagct agtaaactgg ggtcttttga
atgctgctgg cggggatcag 540cgcggtgagc ttatggggct tgaagcttgg
caagtaatgc ttggtgtcat ggtaattcct 600gcagtcttgt acggactgtt
gtcctttgct attccagagt ctccgcgttt tctcatttca 660gttggcaaac
gtgagcgagc taaaaagatc cttgaagaag tcgaagggaa agacgtcgat
720ttcgacgcgc gtgttaccga aattgagcat gctatgcata gagaagaaaa
atctagtttt 780aaagaccttt tgggtggctc ttttttcttc aagccaattg
tatggatcgg tatcggactt 840agtgtttttc agcaattcgt aggcattaat
gtcgcgttct attactcctc tactctttgg 900cagagtgtgg gtgtggatcc
ggctgactct ttcttctatt cttttactac aagtattatt 960aacatcgttg
gaacggtcat cgctatgatt ttcgtcgatc gggtgggacg caagccgttg
1020gcgctcattg gttccgttgg catggttatt ggactggctt tggaagcttg
ggcgttctca 1080ttcgatctag ttgatggcaa acttcctgca acacagggtt
gggtggcgct tattgctgct 1140catgtttttg tgctgttctt tgcgctttct
tggggtgttg tggtttgggt gttcttggga 1200gaaatgtttc cgaatcgtat
tcgtgctgct gcattgggag tcgcggcatc cgcacaatgg 1260attgcgaatt
gggctatcac cgcgagtttt ccgagtctag ctgactggaa cctttccggt
1320acgtatgtaa tctacacgat ctttgctgcg ctttctattc cttttgtgct
caaatttgtt 1380aaagaaacta agggaaaagc gttggaagaa atgggttag
1419211476DNACorynebacterium glutamicummisc_feature(1)..(1476)iolT1
21atggcttcta cttttattca agctgattca cctgaaaagt caaaaaagct gccacctcta
60actgaaggcc catatcgtaa gcgattgttc tacgttgcgc ttgttgcgac ttttggtggc
120ttgctttttg gatatgatac gggcgtcatt aatggtgctc ttaatccaat
gactcgcgag 180cttggattga cggcttttac tgaaggcgta gttacttctt
ctctcctatt cggtgcggct 240gctggcgcta tgttcttcgg acgcatctct
gataattggg gacgccgtaa gactatcatt 300tctctggctg ttgcattttt
cgttggtact atgatttgtg tattcgcgcc atccttcgcg 360gttatggtag
ttggaagagt ccttttggga ttggctgtgg ggggagcatc aactgttgtg
420cctgtatatc tcgcagaact tgctccgttc gagatccgtg gttctttggc
tggtcgtaac 480gaactcatga ttgtcgtagg ccagttggct gcgtttgtta
ttaatgcaat cattggtaac 540gtgttcggac atcatgatgg tgtctggcgt
tacatgctag cgattgcagc gatcccagca 600attgcgctgt ttttcggcat
gttgcgggta ccggagtccc cacgctggct tgtagagcgg 660gggcgcattg
acgaagctcg tgcggtactt gaaaccattc gtccgttgga acgcgcgcat
720gctgaagtgg ctgatgttga acatcttgcg cgtgaagaac atgctgtaag
tgagaaatca 780atgggtctgc gtgaaatctt gtccagtaaa tggcttgtgc
gcattcttct tgtgggaatt 840gggcttggag tagcacagca acttactggt
atcaatagta ttatgtatta tggccaagtc 900gttctcattg aagcgggttt
cagtgaaaac gcagcgctta ttgctaatgt agctcctggt 960gtgatcgcag
tggttggtgc tttcattgct ctttggatga tggatcgtat caatcgacgc
1020accacgctta ttacgggcta ctcccttacg accatctctc atgttctgat
tggtatcgct 1080tctgttgcgt ttccggttgg tgatccacta cgtccttatg
tgattcttac acttgtagtt 1140gtttttgtgg gatctatgca gacgtttttg
aatgtagcta cgtgggtcat gctttccgag 1200ttgtttccat tggctatgcg
cggattcgca atcggaatta gtgttttctt tttgtggatt 1260gcaaacgcgt
tccttggact tttctttccg acaattatgg aagctgttgg gcttacggga
1320actttcttca tgttcgcggg tattggtgtc gtagcgctga tttttattta
cactcaggtg 1380cccgagactc ggggacgcac ccttgaagaa atcgacgaag
atgttacttc tggagtcatc 1440tttaacaaag acattcgtaa aggtaaagtc cattag
1476221458DNASynechocystis sp. PCC6803misc_feature(1)..(1458)glcp
(CAA34119.1) 22atgcgtcaaa ccggttcttt gcgtcctatt cttgtccctg
tgtggatcct tgtacttgta 60gcggcacttg cgggttgtgc aacacgtact gatgatcagc
cgactgaatc cgctccaccc 120ccagcgcaac aagctccacc aacgccggca
gaaattcgtg cgatcgctaa agatgcttat 180atttggggat tcccgttggt
tgataattac cgtgttcagt atagttactt tgtagacaag 240accgaccccg
agtataaggg tggattcaat gaagtacata atactgctcg gctttatact
300ccggctgata aagctattca gacccctaac gcagatactc cgtattcttt
tgtaggcgca 360gacttgcgta ctgagcctct ggtctttact gtgccgccga
ttgagcagaa tcgttacttc 420tctctgcagt tcgtggatgg ttatacctat
aatgtagctt atgttggttc tcgtacgaca 480ggaaacggcg ggggacgcta
cttgctcgct ggtccaggtt gggagggtga aaagcccgag 540ggtgtagacg
aaatcattcg ctcagacacg gaccttgcgt tcgtccttta tcggacgcag
600ctgtttgggc ctagagattt ggataatatc aagaaaattc aggctggata
tcaagttgcg 660ccgctttccg tttatcttaa acaaccaagt ccacctccgg
ctcctccgat tgattttacg 720cctccactta cgccagaagc gcagaaaacc
tctccacaat tcttcgagat tttgaacgca 780gcgcttcgct atgcgccggt
taaaccagaa gaacaagaaa tgcgagagcg ctttgcacgt 840attggtattg
gccctgatgg cgacttcgat gctgataaac tttctcctga gactcgcgaa
900gcgattgaag atggaatggc gaatgcgtgg gttgaatttg atcgtttcaa
acaagataaa 960gttgacacgg gtgaagtcgg cagtgctcaa ctatttggta
ccgcggatga tctaaaggga 1020aactaccttt accgcatggc tggtgctgtt
ctgggcattt atggaaatac tgcggctgaa 1080gcgctttatc caagtgctat
gcttgatgca gacggccagc ctctcactgg aactaactca 1140tacacgtacc
gatttgcgcc agaccaactt ccgccggtga atgctttctg gtcacttacg
1200atctatgaat tgccttcctc tcaacttgtg gacaatccga ttgatcgcta
tttgattaat 1260agcgaaatgc tcccatcttt ggtcccggat ccagatgggg
cttatacact tcgcatccag 1320aatactcagc cgccagagaa tgaagcaaac
tggctgcctg ctccgaaggg accgtttacg 1380cttgtattgc gcctttactg
gccgaaacct gatgcactta atggaacttg gcaggctcca 1440aaaccagaaa agatttag
1458231458DNAMycobacterium
smegmatismisc_feature(1)..(1458)MSMEG_4187 23atgcgtcaaa ccggttcttt
gcgtcctatt cttgtccctg tgtggatcct tgtacttgta 60gcggcacttg cgggttgtgc
aacacgtact gatgatcagc cgactgaatc cgctccaccc 120ccagcgcaac
aagctccacc aacgccggca gaaattcgtg cgatcgctaa agatgcttat
180atttggggat tcccgttggt tgataattac cgtgttcagt atagttactt
tgtagacaag 240accgaccccg agtataaggg tggattcaat gaagtacata
atactgctcg gctttatact 300ccggctgata aagctattca gacccctaac
gcagatactc cgtattcttt tgtaggcgca 360gacttgcgta ctgagcctct
ggtctttact gtgccgccga ttgagcagaa tcgttacttc 420tctctgcagt
tcgtggatgg ttatacctat aatgtagctt atgttggttc tcgtacgaca
480ggaaacggcg ggggacgcta
cttgctcgct ggtccaggtt gggagggtga aaagcccgag 540ggtgtagacg
aaatcattcg ctcagacacg gaccttgcgt tcgtccttta tcggacgcag
600ctgtttgggc ctagagattt ggataatatc aagaaaattc aggctggata
tcaagttgcg 660ccgctttccg tttatcttaa acaaccaagt ccacctccgg
ctcctccgat tgattttacg 720cctccactta cgccagaagc gcagaaaacc
tctccacaat tcttcgagat tttgaacgca 780gcgcttcgct atgcgccggt
taaaccagaa gaacaagaaa tgcgagagcg ctttgcacgt 840attggtattg
gccctgatgg cgacttcgat gctgataaac tttctcctga gactcgcgaa
900gcgattgaag atggaatggc gaatgcgtgg gttgaatttg atcgtttcaa
acaagataaa 960gttgacacgg gtgaagtcgg cagtgctcaa ctatttggta
ccgcggatga tctaaaggga 1020aactaccttt accgcatggc tggtgctgtt
ctgggcattt atggaaatac tgcggctgaa 1080gcgctttatc caagtgctat
gcttgatgca gacggccagc ctctcactgg aactaactca 1140tacacgtacc
gatttgcgcc agaccaactt ccgccggtga atgctttctg gtcacttacg
1200atctatgaat tgccttcctc tcaacttgtg gacaatccga ttgatcgcta
tttgattaat 1260agcgaaatgc tcccatcttt ggtcccggat ccagatgggg
cttatacact tcgcatccag 1320aatactcagc cgccagagaa tgaagcaaac
tggctgcctg ctccgaaggg accgtttacg 1380cttgtattgc gcctttactg
gccgaaacct gatgcactta atggaacttg gcaggctcca 1440aaaccagaaa agatttag
1458241554DNABifidobacterium longummisc_feature(1)..(1554)BL1631
24atgactacaa caacggcttc accagtatca aaacagaccg cttccgctgc tcaggaaact
60agtgctaccg gtgctgcggc aacggcgatt gaaacgattg agactggtgt tgctggtgtg
120gctggtgcag cgactaatgc agcggcgaat gctatcgaag atctagaggc
agcggaatct 180catggattta gtacgcgctt tcccctgaac tctgcgttta
tcttcacctt cggagcgctt 240ggcggaatgc tgtttggatt cgatacgggc
attatttcag gtgcaagtcc tttgattgag 300tctgactttg gtttgtctgt
atcacagact ggtttcatta cgtctagtgt tctcatcggt 360tcatgtgctg
gcgctttgtc cattggagca ctctctgatc ggttcggtcg caaaaagcta
420cttattgtga gtgcgcttct tttcttgctg ggatccggtt tgtgtgcgtc
ctctactggt 480ttcgcgatga tggtctgtgc tcgtatcatt cttgggctcg
ctgtcggcgc agcgtctgca 540cttactccgg cttaccttgc tgaattggcg
ccgaaagagc gtcgtggatc tctttccacc 600ctttttcagc ttatggttac
tttcggaatt ttgctggctt atgcatctaa cctgggattt 660cttaaccata
atcttttcgg tattcgtgat tggcgctgga tgcttggctc tgcgttggtg
720ccagcggcgc tgctacttct tggtgggttg ttgcttcctg aaagtccgcg
gtacctggtc 780aataaaggtg acactcgcaa tgcttttaag gttcttaccc
ttatccgcaa agacgttgat 840caaacacaag tacagatcga acttgatgaa
atcaaagctg tagctgctca agatacgaaa 900ggcggagtaa gagaactgtt
tcgaatcgca cgcccagcgc ttgtggcagc tatcggaatc 960atgttgttcc
agcaacttgt tggaattaac tctgtcatct atttcctacc tcaggtattc
1020attaaaggct tcggctttcc tgagggtgat gctatttggg tttccgtcgg
tattggtgta 1080gtgaatttcg tttctacaat tgttgcaact cttatcatgg
accgttttcc acgcaagggt 1140atgttgattt ttggtagtat tgtaatgact
gtaagtcttg ctgttttggc tgtgatgaat 1200tttgtgggtg atgttgctgt
acttgcggta ccgactatga ttctgattgc attctatatt 1260ctaggtttcg
ctgtctcctg gggacctatt gcttgggtcc ttattggcga aatctttcca
1320ctttctgtac gtggcattgg atcctccttc ggatctgcgg cgaattggct
aggaaacttt 1380atcgtgagtc aattctttct tgtccttctt gatgcttttg
ggaataatgt tggcggaccg 1440tttgcaattt tcggtgtttt tagtgcgttg
tcaattccgt ttgtcttgcg tcttgtacca 1500gagactaagg gtaaatccct
ggaagaaatc gagaaagaaa tgactaaacg ttag 155425750DNACorynebacterium
glutamicummisc_feature(1)..(750)ppgK kinase 25atgacagaaa ctggtttcgg
aattgatatt ggtggcagtg gaattaaggg tgcgcgtgta 60aatcttaaaa ccggagagtt
tatcgatgaa cggatcaaaa ttgcgacgcc aaagccagcg 120actcccgaag
ctgtagctga agttgtagca gaaatcattt ctcaagctga gtgggaaggc
180cctgtcggaa ttactctgcc atcagttgtt cgcggtcaga ttgcgctttc
cgcggctaat 240atcgataagt cttggattgg tactgacgtc catgaattgt
ttgatcgtca tctcaatggt 300agagagatta cggttcttaa cgacgcggat
gctgctggga ttgcagaagc gacgttcggt 360aatccggcag cacgcgaggg
cgctgttatt cttttgaccc tgggtactgg tattggatct 420gcgttccttg
tggatggtca actatttccg aatactgagc ttggacacat gattgtggac
480ggtgaagagg ctgaacatct ggctgcagct tccgtgaaag aaaatgaaga
tctctcttgg 540aagaaatggg cgaaacatct taacaaagta ttgagtgaat
atgaaaaact attttcccct 600tcagtattca ttatcggggg cggaattagt
cgaaaacatg agaaatggct tccgcttatg 660gaacttgaca ctgatatcgt
gccagcggaa cttcgcaatc gtgctggaat cgttggagcg 720gcgatggctg
tcaatcagca tttgacgcct 75026972DNAZymomonas
mobilismisc_feature(1)..(972)glk kinase 26atggaaattg tcgctattga
tattgggggt acgcatgcac gtttttctat tgctgaagtc 60tccaacggtc gagtacttag
tcttggagaa gagacaacgt ttaaaactgc ggagcatgca 120agtttgcaac
tggcttggga acgttttgga gagaaattgg gtcgcccact gccacgtgcg
180gctgctattg cttgggcggg accagtccat ggtgaagttt tgaagctaac
taataatccg 240tgggtacttc gtccagctac acttaatgaa aaacttgata
ttgatacgca tgttttgatt 300aatgactttg gtgcggtcgc acatgctgtg
gctcacatgg attcttctta tcttgaccat 360atttgtggac ctgacgaagc
gctgccctcc gatggtgtga tcaccatttt gggtccggga 420accggacttg
gtgttgcgca tctacttcgc accgaaggcc gctatttcgt cattgagact
480gaaggcggac atattgattt cgcgccattg gatcgtcttg aagataagat
tcttgcacgc 540ctgcgcgaac gttttcgtcg ggtgagtatc gagagaatca
tctcaggtcc gggattgggt 600aacatttacg aagcgctggc ggctattgaa
ggcgtacctt tctctcttct tgatgacatt 660aagctttggc agatggcgtt
ggaaggcaaa gataatcttg ctgaggctgc actagaccgc 720ttttgtttgt
ccctcggggc aatcgctggt gatcttgcgc ttgcgcaggg tgcaacttct
780gtagtgatcg gtgggggcgt tggtttgcgg attgcatccc atcttccgga
gtcaggtttt 840cgtcaacgct tcgtttctaa gggccgtttc gaacgtgtta
tgagtaaaat cccggtaaaa 900cttattactt atcctcagcc aggactcctt
ggagcggcag ctgcgtatgc taataaatac 960tctgaagttg aa 972
D00000
D00001
D00002
D00003
D00004
S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.