Methods Of Treating T Cell Exhaustion By Inhibiting Or Modulating T Cell Receptor Signaling
LYNN; Rachel ; et al.
U.S. patent application number 16/499762 was filed with the patent office on 2020-04-02 for methods of treating t cell exhaustion by inhibiting or modulating t cell receptor signaling. The applicant listed for this patent is THE BOARD OF TRUSTEES OF THE LE-LAND STANDFORD JUNIOR UNIVERSITY. Invention is credited to Rachel LYNN, Crystal MACKALL, Sanjay MALHOTRA, Evan WEBER.
| Application Number | 20200101108 16/499762 |
| Document ID | / |
| Family ID | 63677091 |
| Filed Date | 2020-04-02 |









View All Diagrams
| United States Patent Application | 20200101108 |
| Kind Code | A1 |
| LYNN; Rachel ; et al. | April 2, 2020 |
METHODS OF TREATING T CELL EXHAUSTION BY INHIBITING OR MODULATING T CELL RECEPTOR SIGNALING
Abstract
Provided herein are compositions and methods for preventing or reversing T cell exhaustion. In particular, the present invention relates to methods of preventing or reversing T cell exhaustion by exposing T cells experiencing T cell exhaustion to particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib), or by expanding genetically engineered T cells in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib).
| Inventors: | LYNN; Rachel; (Stanford, CA) ; MACKALL; Crystal; (Stanford, CA) ; WEBER; Evan; (Stanford, CA) ; MALHOTRA; Sanjay; (Stanford, CA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 63677091 | ||||||||||
| Appl. No.: | 16/499762 | ||||||||||
| Filed: | March 30, 2018 | ||||||||||
| PCT Filed: | March 30, 2018 | ||||||||||
| PCT NO: | PCT/US2018/025394 | ||||||||||
| 371 Date: | September 30, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 62479930 | Mar 31, 2017 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61K 31/5025 20130101; A61K 45/06 20130101; A61P 43/00 20180101; A61K 31/497 20130101; A61K 31/506 20130101; C12N 2510/00 20130101; A61K 35/17 20130101; A61K 39/001112 20180801; A61K 2039/5158 20130101; C07K 2317/622 20130101; C12N 2501/727 20130101; A61K 39/001171 20180801; C07K 2319/33 20130101; C07K 2319/03 20130101; C12N 5/0636 20130101; A61K 39/00 20130101; A61K 2039/5156 20130101; A61P 35/02 20180101; C07K 16/3084 20130101 |
| International Class: | A61K 35/17 20060101 A61K035/17; A61K 31/5025 20060101 A61K031/5025; A61K 31/506 20060101 A61K031/506; C07K 16/30 20060101 C07K016/30; C12N 5/0783 20060101 C12N005/0783 |
Claims
1. A method for preventing and/or reversing T cell exhaustion in a subject, the method comprising administering to the subject a therapeutically effective amount of a tyrosine kinase inhibitor.
2. The method of claim 1, wherein the tyrosine kinase inhibitor is capable of inhibiting TCR signaling and/or CAR signaling.
3. The method of claim 1, wherein the tyrosine kinase inhibitor is a Lck inhibitor.
4. The method of claim 1, wherein the tyrosine kinase inhibitor is dasatinib or ponatinib.
5. The method of claim 1, wherein treatment increases secretion of IL-2 by T cells in the subject.
6. The method of claim 1, wherein treatment decreases apoptosis of T cells in the subject.
7. The method of claim 1, wherein treatment decreases expression of at least one T cell exhaustion marker selected from the group consisting of PD-1, TIM-3, and LAG-3.
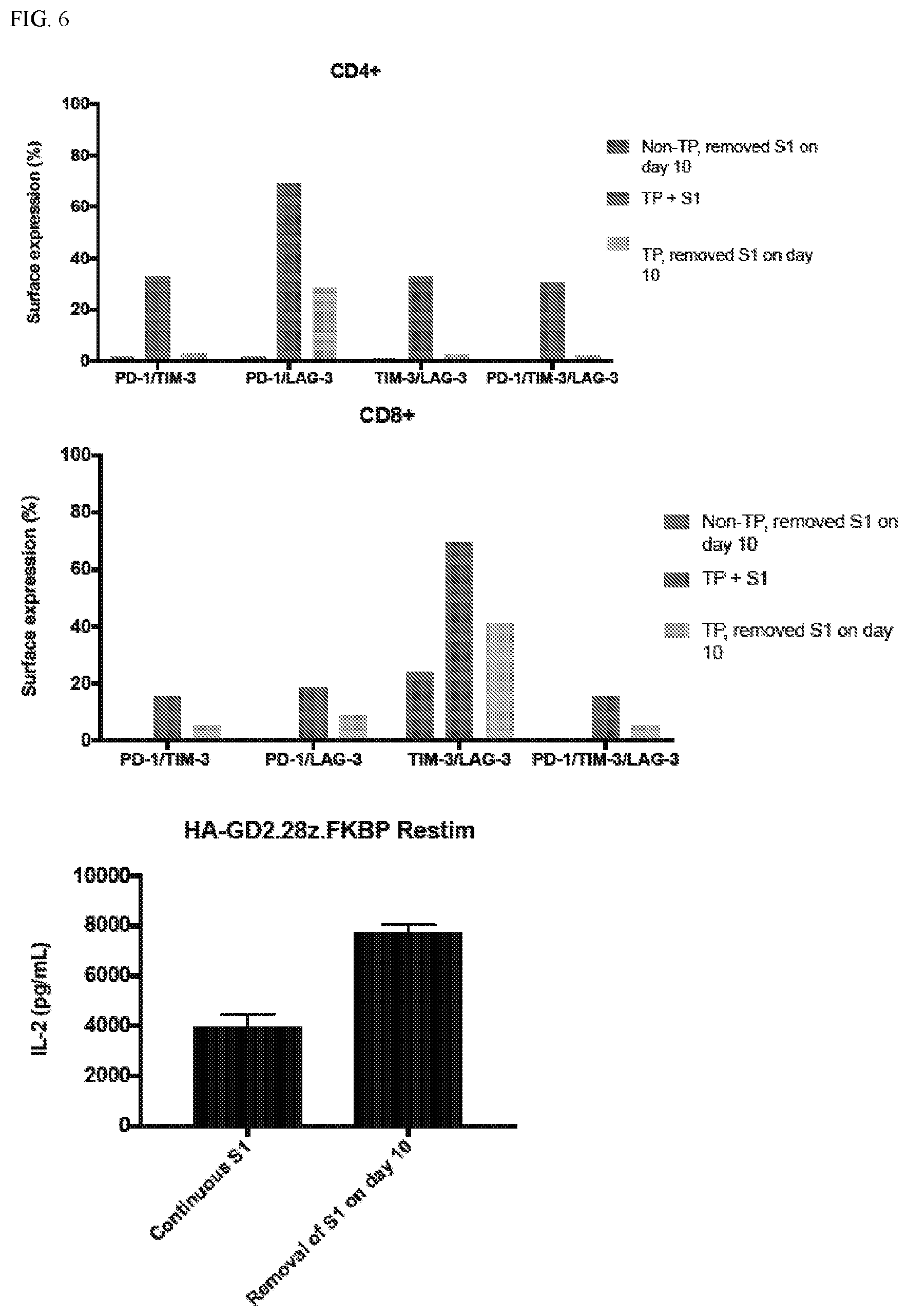
8. The method of claim 1, wherein treatment increases expression of CD62L or CCR7.
9. The method of claim 1, wherein multiple cycles of treatment are administered to the subject.
10. The method of claim 7, wherein the tyrosine kinase inhibitor is administered intermittently.
11. The method of claim 1, wherein the tyrosine kinase inhibitor is administered for a period of time sufficient to restore at least partial T cell function then discontinued.
12. The method of claim 1, wherein the tyrosine kinase inhibitor is administered orally.
13. The method of claim 1, wherein the subject is human.
14. The method of claim 1, wherein the subject has a chronic infection or cancer.
15. The method of claim 1, wherein treatment is prophylactic.
16. A method for treating an immune system related condition or disease in a subject comprising administering to the subject genetically engineered T cells and a therapeutically effective amount of a tyrosine kinase inhibitor.
17. The method of claim 16, wherein the tyrosine kinase inhibitor is capable of inhibiting TCR signaling and/or CAR signaling.
18. The method of claim 16, wherein the tyrosine kinase inhibitor is a Lck inhibitor.
19. The method of claim 16, wherein the tyrosine kinase inhibitor is dasatinib or ponatinib.
20. The method of claim 16, wherein the tyrosine kinase inhibitor and the genetically engineered T cells are administered simultaneously and/or at different time points.
21. The method of claim 16, wherein the immune system related condition or disease is selected from cancer or an autoimmune disease or condition.
22. The method of claim 16, wherein the genetically engineered T cells are selected from CAR T cells, genetically engineered TCR expressing T cells, genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, genetically engineered T cells configured for transduced T-cell therapy, and/or viral specific T cells reengineered with a TCR or CAR.
23. The method of claim 16, further comprising administering to said subject one or more anticancer agents.
24. The method of claim 23, wherein the one or more anticancer agents is selected from a chemotherapeutic agent and radiation therapy.
25. A composition comprising a genetically engineered T cell population, wherein the genetically engineered T cell population was expanded in the presence of a tyrosine kinase inhibitor.
26. The composition of claim 25, wherein the tyrosine kinase inhibitor is capable of inhibiting TCR signaling and/or CAR signaling.
27. The composition of claim 25, wherein the tyrosine kinase inhibitor is a Lck inhibitor.
28. The composition of claim 25, wherein the tyrosine kinase inhibitor is dasatinib or ponatinib.
29. The composition of claim 25, wherein the genetically engineered T cell population is selected from CART cell population, a population of genetically engineered TCR expressing T cells, a population of genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, a population of genetically engineered T cells configured for transduced T-cell therapy, and/or a population of viral specific T cells reengineered with a TCR or CAR.
30. A method of generating a population of genetically engineered T cells resistant to T cell exhaustion, comprising expanding a population of genetically engineered T cells in the presence of a tyrosine kinase inhibitor.
31. The method of claim 30, wherein the tyrosine kinase inhibitor is capable of inhibiting TCR signaling and/or CAR signaling.
32. The method of claim 30, wherein the tyrosine kinase inhibitor is a Lck inhibitor.
33. The method of claim 30, wherein the tyrosine kinase inhibitor is dasatinib or ponatinib.
34. The method of claim 30, wherein the population of genetically engineered T cells is selected from CAR T cell population, a population of genetically engineered TCR expressing T cells, a population of genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, a population of genetically engineered T cells configured for transduced T-cell therapy, and/or a population of viral specific T cells reengineered with a TCR or CAR.
35. A method of treating an immune system related condition or disease, comprising administering to the subject a genetically engineered T cell population that were expanded in the presence of a tyrosine kinase inhibitor.
36. The method of claim 35, wherein the tyrosine kinase inhibitor is capable of inhibiting TCR signaling and/or CAR signaling.
37. The method of claim 35, wherein the tyrosine kinase inhibitor is a Lck inhibitor.
38. The method of claim 35, wherein the tyrosine kinase inhibitor is dasatinib or ponatinib.
39. The method of claim 35, wherein the genetically engineered T cell population is selected from CAR T cell population, a population of genetically engineered TCR expressing T cells, a population of genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, a population of genetically engineered T cells configured for transduced T-cell therapy, and/or a population of viral specific T cells reengineered with a TCR or CAR.
40. The method of claim 35, wherein the subject is undergoing an adoptive T cell therapy.
41. The method of claim 40, wherein the adoptive T cell therapy is a CAR T-cell therapy.
42. The method of claim 40, wherein the adoptive T cell therapy is a transduced T-cell therapy.
43. The method of claim 40, wherein the adoptive T cell therapy is a tumor infiltrating lymphocyte (TIL) therapy.
44. The method of claim 35, wherein the immune system related condition or disease is selected from cancer or an autoimmune disease or condition.
45. The method of claim 35, further comprising administering to said subject one or more anticancer agents.
46. The method of claim 45, wherein the one or more anticancer agents is selected from a chemotherapeutic agent and radiation therapy.
47. A method for preventing and/or reversing toxicity related to genetically engineered T cell administered to a subject, comprising administering to the subject a therapeutically effective amount of a tyrosine kinase inhibitor.
48. The method of claim 47, wherein the tyrosine kinase inhibitor is capable of inhibiting TCR signaling and/or CAR signaling.
49. The method of claim 47, wherein the tyrosine kinase inhibitor is a Lck kinase inhibitor.
50. The method of claim 47, wherein the tyrosine kinase inhibitor is dasatinib or ponatinib.
51. The method of claim 33, wherein the genetically engineered T cells are selected from CAR T cells, genetically engineered TCR expressing T cells, genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, genetically engineered T cells configured for transduced T-cell therapy, and/or viral specific T cells reengineered with a TCR or CAR.
52. The method of claim 47, wherein the subject is undergoing an adoptive T cell therapy.
53. The method of claim 52, wherein the adoptive T cell therapy is a CAR T-cell therapy.
54. The method of claim 52, wherein the adoptive T cell therapy is a transduced T-cell therapy
55. The method of claim 52, wherein the adoptive T cell therapy is a tumor infiltrating lymphocyte (TIL) therapy.
56. The method of claim 47, wherein the toxicity related to genetically engineered T cell administered to a subject is cytokine release syndrome.
57. The method of claim 47, wherein the toxicity related to genetically engineered T cell administered to a subject is on-target off tumor toxicity or off-target off-tumor toxicity.
Description
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of U.S. provisional application Ser. No. 62/479,930, filed Mar. 31, 2017, which is incorporated herein by reference in its entirety.
FIELD OF THE INVENTION
[0002] Provided herein are compositions and methods for preventing or reversing T cell exhaustion. In particular, the present invention relates to methods of preventing or reversing T cell exhaustion by exposing T cells experiencing T cell exhaustion to particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib), or by expanding genetically engineered T cells in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib).
INTRODUCTION
[0003] T cells are immune cells that become activated via T cell receptor (TCR) signaling following engagement with antigen. Physiologic activation through the T cell receptor renders T cells capable of mediating potent antitumor or anti-infective effects. During resolution of an acute inflammatory response, a subset of activated effector T cells differentiate into long-lived memory cells. By contrast, in patients with chronic infections or cancer, T cells not infrequently undergo pathologic differentiation toward a state of dysfunction, which has been termed T cell exhaustion. T cell exhaustion is characterized by marked changes in metabolic function, transcriptional programming, loss of effector function (e.g., cytokine secretion, killing capacity), and co-expression of multiple surface inhibitory receptors. The root cause of T cell exhaustion is persistent antigen exposure leading to continuous TCR signaling. Prevention or reversal of T cell exhaustion has been long sought as a means to enhance T cell effectiveness in patients with cancer or chronic infections.
[0004] The present invention addresses this urgent need.
SUMMARY OF THE INVENTION
[0005] Immune cells respond to the presence of foreign antigens with a wide range of responses, including the secretion of preformed and newly formed mediators, phagocytosis of particles, endocytosis, cytotoxicity against target cells, as well as cell proliferation and/or differentiation. T cells are a subgroup of cells which together with other immune cell types (e.g., polymorphonuclear, eosinophils, basophils, mast cells, B cells, and NK cells), constitute the cellular component of the immune system (see, e.g., U.S. Pat. No. 6,057,294; US Pat. Appl. 20050070478). Under physiological conditions T cells function in immune surveillance and in the elimination of foreign antigen. However, under pathological conditions there is compelling evidence that T cells play a major role in the causation and propagation of disease. In these disorders, breakdown of T cell immunological tolerance, either central or peripheral is a fundamental process in the causation of autoimmune disease.
[0006] It is well established that T cell receptor (TCR) engagement and costimulatory signaling provide the critical signals that regulate T cell activation, proliferation and cytolytic functions. T cells respond to antigen via a polypeptide complex composed of the ligand-binding T cell receptor (TCR) disulfide-linked .alpha. and .beta. subunits (or .gamma. and .delta. subunits in .gamma..delta. T cells) that have single transmembrane (TM) spans per subunit and small intracellular tails and associate non-covalently with hetero- (CD3.gamma..epsilon. and CD3.delta..epsilon.) and homodimeric (.zeta..zeta.) signaling subunits (see, e.g., Cambier J. C. Curr Opin Immunol 1992; 4:257-64). The CD3.epsilon., .delta., and .gamma. chains have single Ig-family extracellular domains, single presumably .alpha.-helical TM spans, and intrinsically disordered intracellular domains of 40-60 residues, whereas each subunit has a small extracellular region (9 residues) carrying the intersubunit disulfide bond, a single presumably .alpha.-helical TM span per subunit, and a large, intrinsically disordered cytoplasmic domain of approximately 110 residues. An understanding of the process of TCR-mediated TM signal transduction and subsequent T cell activation, leading to T cell proliferation and differentiation, is therefore pivotal to both health and disease. Disturbance in TCR signaling can lead to inflammatory and other T cell-related disorders.
[0007] T cells expressing chimeric antigen receptors (CARs) at high levels undergo tonic, antigen independent signaling due to receptor clustering. Such T cells function poorly as a result of T cell exhaustion, as evidenced by high levels of PD-1, TIM-3, LAG-3, diminished antigen induced cytokine production, and excessive programmed cell death. Tonic signaling can be prevented by transiently decreasing CAR associated TCR signaling proteins (e.g., TCR zeta) to levels below the threshold required for tonic signaling.
[0008] Experiments conducted during the course of developing embodiments for the present invention demonstrated that treatment with a particular tyrosine kinase inhibitor that inhibits T cell receptor signaling (e.g., a Lck tyrosine kinase inhibitor (e.g., dasatinib)) (e.g., a Src family tyrosine kinase inhibitor) reduced expression of the T cell exhaustion markers and improved formation of T cell memory. Accordingly, the present invention relates to methods of preventing or reversing T cell exhaustion by transiently inhibiting T cell receptor (TCR) signaling to restore T cell function with particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib).
[0009] Additional experiments determined that CAR T cells co-cultured with tumor cells in the presence of dasatinib or ponatinib exhibit attenuated activation and degranulation, fail to secrete cytokine, and display attenuated killing in response to tumor antigen.
[0010] Additional experiments determined that dasatinib potently inhibits the phosphorylation of CAR CD3z as well as distal signaling proteins after CAR crosslinking.
[0011] Additional experiments determined that tonically signaling CAR T cells expanded in the presence of dasatinib exhibit a reduction in canonical exhaustion marker expression in a dose-dependent manner, retain the capacity to form memory, display augmented cytokine secretion in response to tumor antigen, and display augmented cytotoxicity.
[0012] Additional experiments determined that in vivo dasatinib treatment suppresses exhaustion marker expression, augments memory formation, and facilitates cell survival/proliferation.
[0013] Accordingly, provided herein are compositions and methods for preventing or reversing T cell exhaustion. In particular, the present invention relates to methods of preventing or reversing T cell exhaustion by exposing T cells experiencing T cell exhaustion to particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib), or by expanding genetically engineered T cells in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib).
[0014] In certain embodiments, the present invention provides methods for treating a subject to mitigate T cell exhaustion, the method comprising administering to the subject a therapeutically effective amount of a tyrosine kinase inhibitor. Such embodiments are not limited to a particular tyrosine kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is capable of inhibiting TCR signaling and/or CAR signaling. In some embodiments, the tyrosine kinase inhibitor is a Lck kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is a Fyn kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is a Src family tyrosine kinase inhibitor. In some embodiments, tyrosine kinase inhibitor is dasatinib or ponatinib. In some embodiments, the treatment is prophylactic.
[0015] Such methods are not limited to a particular manner of treating the subject for T cell exhaustion. In some embodiments, the treatment increases secretion of IL-2 by T cells in the subject. In some embodiments, the treatment decreases apoptosis of T cells in the subject. In some embodiments, the treatment decreases expression of at least one T cell exhaustion marker selected from the group consisting of PD-1, TIM-3, and LAG-3. In some embodiments, the treatment increases expression of CD62L or CCR7.
[0016] Such methods are not limited to particular manner of administration. In some embodiments, multiple cycles of treatment are administered to the subject. In some embodiments, the tyrosine kinase inhibitor is administered intermittently. In some embodiments, the tyrosine kinase inhibitor is administered for a period of time sufficient to restore at least partial T cell function then discontinued. In some embodiments, the tyrosine kinase inhibitor is administered orally.
[0017] Such methods are not limited to a particular type or kind of subject. In some embodiments, the subject is a human. In some embodiments, the subject has a chronic infection or cancer.
[0018] In certain embodiments, the present invention provides for treating an immune system related condition or disease in a subject comprising administering to the subject genetically engineered T cells and a therapeutically effective amount of a tyrosine kinase inhibitor. Such embodiments are not limited to a particular tyrosine kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is capable of inhibiting TCR signaling and/or CAR signaling. In some embodiments, the tyrosine kinase inhibitor is a Lck kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is a Fyn kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is a Src family tyrosine kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is dasatinib or ponatinib. In some embodiments, the treatment is prophylactic. In some embodiments, the tyrosine kinase inhibitor and the genetically engineered T cells are administered simultaneously and/or at different time points.
[0019] Such methods are not limited to a specific type or kind of genetically engineered T cells. In some embodiments, the genetically engineered T cells include, but are not limited to, CAR T cells, genetically engineered TCR expressing T cells, genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, genetically engineered T cells configured for transduced T-cell therapy, and/or viral specific T cells reengineered with a TCR or CAR.
[0020] Such methods are not limited to treating a specific immune system related condition or disease. In some embodiments, the immune system related condition or disease is selected from cancer or an autoimmune disease or condition.
[0021] In certain embodiments, the present invention provides methods for preventing and/or reversing toxicity related to genetically engineered T cell administered to a subject, comprising administering to the subject a therapeutically effective amount of a tyrosine kinase inhibitor. Such embodiments are not limited to a particular tyrosine kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is capable of inhibiting TCR signaling and/or CAR signaling. In some embodiments, the tyrosine kinase inhibitor is a Lck kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is a Fyn kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is a Src family tyrosine kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is dasatinib or ponatinib.
[0022] Such methods are not limited to a specific type or kind of genetically engineered T cells. In some embodiments, the genetically engineered T cells include, but are not limited to, CAR T cells, genetically engineered TCR expressing T cells, genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, genetically engineered T cells configured for transduced T-cell therapy, and/or viral specific T cells reengineered with a TCR or CAR.
[0023] Such methods are not limited to a particular type or kind of adoptive T cell therapy. In some embodiments, the adoptive T cell therapy is a CAR T-cell therapy. In some embodiments, the adoptive T cell therapy is a transduced T-cell therapy. In some embodiments, the adoptive T cell therapy is a tumor infiltrating lymphocyte (TIL) therapy.
[0024] Such methods are not limited to a particular type or kind of toxicity related to genetically engineered T cell administered to a subject. In some embodiments, the toxicity related to genetically engineered T cell administered to a subject is cytokine release syndrome. In some embodiments, the toxicity related to genetically engineered T cell administered to a subject is on-target off tumor toxicity or off-target off-tumor toxicity.
[0025] In certain embodiments, the present invention provides compositions comprising a genetically engineered T cell population, wherein the genetically engineered T cell population was expanded in the presence of tyrosine kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is capable of inhibiting TCR signaling and/or CAR signaling inhibitor. In some embodiments, the tyrosine kinase inhibitor dasatinib or ponatinib.
[0026] In certain embodiments, the present invention provides methods of generating a population of genetically engineered T cells resistant to T cell exhaustion, comprising expanding a population of genetically engineered T cells in the presence of a tyrosine kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is capable of inhibiting TCR signaling and/or CAR signaling inhibitor. In some embodiments, the tyrosine kinase inhibitor is dasatinib or ponatinib. Such methods are not limited to a specific type or kind of genetically engineered T cells. In some embodiments, the genetically engineered T cells include, but are not limited to, CAR T cells, genetically engineered TCR expressing T cells, genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, genetically engineered T cells configured for transduced T-cell therapy, and/or viral specific T cells reengineered with a TCR or CAR. Such methods are not limited to a specific expanding technique as such techniques are well known in the art.
[0027] In certain embodiments, the present invention provides methods of treating an immune system related condition or disease in a subject undergoing an adoptive T cell therapy, comprising administering to the subject a genetically engineered T cell population that were expanded in the presence of a tyrosine kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is capable of inhibiting TCR signaling inhibitor and/or CAR signaling. In some embodiments, the tyrosine kinase inhibitor is a Lck kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is a Fyn kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is a Src family tyrosine kinase inhibitor. In some embodiments, the tyrosine kinase inhibitor is dasatinib or ponatinib. In some embodiments, the immune system related condition or disease is selected from cancer or an autoimmune disease or condition.
[0028] Such methods are not limited to a specific type or kind of genetically engineered T cells. In some embodiments, the genetically engineered T cells include, but are not limited to, CAR T cells, genetically engineered TCR expressing T cells, genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, genetically engineered T cells configured for transduced T-cell therapy, and/or viral specific T cells reengineered with a TCR or CAR.
[0029] Such methods are not limited to a particular type or kind of adoptive T cell therapy. In some embodiments, the adoptive T cell therapy is a CAR T-cell therapy. In some embodiments, the adoptive T cell therapy is a transduced T-cell therapy. In some embodiments, the adoptive T cell therapy is a tumor infiltrating lymphocyte (TIL) therapy.
[0030] The present invention contemplates that exposure of animals (e.g., humans) suffering from cancer (e.g., and/or cancer related disorders) to adoptive T cell therapies (e.g., a CAR T-cell therapy, a transduced T-cell therapy, and a tumor infiltrating lymphocyte (TIL) therapy) with genetically engineered T cell populations and compositions comprising particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) will inhibit the growth of cancer cells or supporting cells outright and/or render such cells as a population more susceptible to the cell death-inducing activity of cancer therapeutic drugs or radiation therapies. In such embodiments, the methods result in improved therapy outcome as such particular tyrosine kinase inhibitors are capable of 1) modulating TCR signaling within the genetically engineered T cell population (e.g., decreasing expression of one or more of PD-1, TIM-3, and LAG-3; increasing expression of memory markers (e.g., CD62L or CCR7); increasing secretion of IL-2 and other cytokines), and 2) preventing and/or reversing T cell exhaustion within the genetically engineered T cell population. Thus, the present invention provides methods for treating cancer (e.g., and/or cancer related disorders) with adoptive T cell therapies (e.g., a CAR T-cell therapy, a transduced T-cell therapy, and a tumor infiltrating lymphocyte (TIL) therapy) in a subject comprising administering to the subject (e.g., simultaneously and/or at different time points) genetically engineered T cells, particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib), and additional cancer therapeutic drugs or radiation therapies.
[0031] The present invention contemplates that exposure of animals (e.g., humans) suffering from cancer (e.g., and/or cancer related disorders) to adoptive T cell therapies (e.g., a CAR T-cell therapy, a transduced T-cell therapy, and a tumor infiltrating lymphocyte (TIL) therapy) with genetically engineered T cell populations that were expanded in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) will inhibit the growth of cancer cells or supporting cells outright and/or render such cells as a population more susceptible to the cell death-inducing activity of cancer therapeutic drugs or radiation therapies. In such embodiments, the methods result in improved therapy outcome as such genetically engineered T cell populations are resistant and/or less prone to T cell exhaustion. Thus, the present invention provides methods for treating cancer (e.g., and/or cancer related disorders) with adoptive T cell therapies (e.g., a CAR T-cell therapy, a transduced T-cell therapy, and a tumor infiltrating lymphocyte (TIL) therapy) in a subject comprising administering to the subject (e.g., simultaneously and/or at different time points) genetically engineered T cell populations that were expanded in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) and additional cancer therapeutic drugs or radiation therapies.
[0032] The present invention contemplates that such methods (e.g., adoptive T cell therapies with genetically engineered T cell populations and compositions comprising particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib)) (e.g., adoptive T cell therapies with genetically engineered T cell populations that were expanded in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib)) satisfy an unmet need for the treatment of multiple cancer types, either when administered as monotherapy or when administered in a temporal relationship with additional agent(s), such as other cell death-inducing or cell cycle disrupting cancer therapeutic drugs or radiation therapies (combination therapies), so as to render a greater proportion of the cancer cells or supportive cells susceptible to executing the apoptosis program compared to the corresponding proportion of cells in an animal treated only with the cancer therapeutic drug or radiation therapy alone.
[0033] In certain embodiments of the invention, combination treatment of animals with such methods (e.g., adoptive T cell therapies with genetically engineered T cell populations and compositions comprising particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib)) (e.g., adoptive T cell therapies with genetically engineered T cell populations that were expanded in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib)) produce a greater tumor response and clinical benefit in such animals compared to those treated with the anticancer drugs/radiation alone. Since the doses for all approved anticancer drugs and radiation treatments are known, the present invention contemplates the various combinations of them with such methods.
[0034] A non-limiting exemplary list of cancer (e.g., and/or cancer related disorders) includes, but is not limited to, pancreatic cancer, breast cancer, prostate cancer, lymphoma, skin cancer, colon cancer, melanoma, malignant melanoma, ovarian cancer, brain cancer, primary brain carcinoma, head and neck cancer, glioma, glioblastoma, liver cancer, bladder cancer, non-small cell lung cancer, head or neck carcinoma, breast carcinoma, ovarian carcinoma, lung carcinoma, small-cell lung carcinoma, Wilms' tumor, cervical carcinoma, testicular carcinoma, bladder carcinoma, pancreatic carcinoma, stomach carcinoma, colon carcinoma, prostatic carcinoma, genitourinary carcinoma, thyroid carcinoma, esophageal carcinoma, myeloma, multiple myeloma, adrenal carcinoma, renal cell carcinoma, endometrial carcinoma, adrenal cortex carcinoma, malignant pancreatic insulinoma, malignant carcinoid carcinoma, choriocarcinoma, mycosis fungoides, malignant hypercalcemia, cervical hyperplasia, leukemia, acute lymphocytic leukemia, chronic lymphocytic leukemia, acute myelogenous leukemia, chronic myelogenous leukemia, chronic granulocytic leukemia, acute granulocytic leukemia, hairy cell leukemia, neuroblastoma, rhabdomyosarcoma, Kaposi's sarcoma, polycythemia vera, essential thrombocytosis, Hodgkin's disease, non-Hodgkin's lymphoma, soft-tissue sarcoma, osteogenic sarcoma, primary macroglobulinemia, and retinoblastoma, and the like, T and B cell mediated autoimmune diseases; inflammatory diseases; infections; hyperproliferative diseases; AIDS; degenerative conditions, vascular diseases, and the like. In some embodiments, the cancer cells being treated are metastatic. In other embodiments, the cancer cells being treated are resistant to anticancer agents.
BRIEF DESCRIPTION OF DRAWINGS
[0035] FIG. 1: Characterization of the GD2.28z.FKBP CAR. T cells were transduced with lentivirus encoding the GD2.28z.FKBP CAR on day 1 after activation and subsequently cultured with various concentrations of shield-1 in the growth medium. On day 7, CAR expression was quantified via FACS.
[0036] FIG. 2: Removal of S1 from culture medium results in reversal of T cell exhaustion marker surface expression.
[0037] FIG. 3: Removal of S1 from culture medium results in maintenance of CD62L expression and prevention of apoptosis.
[0038] FIG. 4: Removal of S1 from culture medium results in reversal of function T cell exhaustion.
[0039] FIG. 5: Removal of surface CAR results in more effective prevention of T cell exhaustion compared PD-1/PDL-1 blockade.
[0040] FIG. 6: Removal of surface CAR rescues exhaustion in PD-1/TIM-3/LAG-3 triple positive CAR T cells after only 4 days.
[0041] FIG. 7: Dasatinib inhibits cytokine secretion of CAR T cells in response to tumor antigen.
[0042] FIG. 8: Dasatinib reverses exhaustion marker expression and co-expression.
[0043] FIG. 9: Dasatinib treatment results in maintenance of CD62L expression.
[0044] FIG. 10: Dasatinib Treatment results in augmented IL-2 and IFN.gamma. secretion in response to tumor antigen.
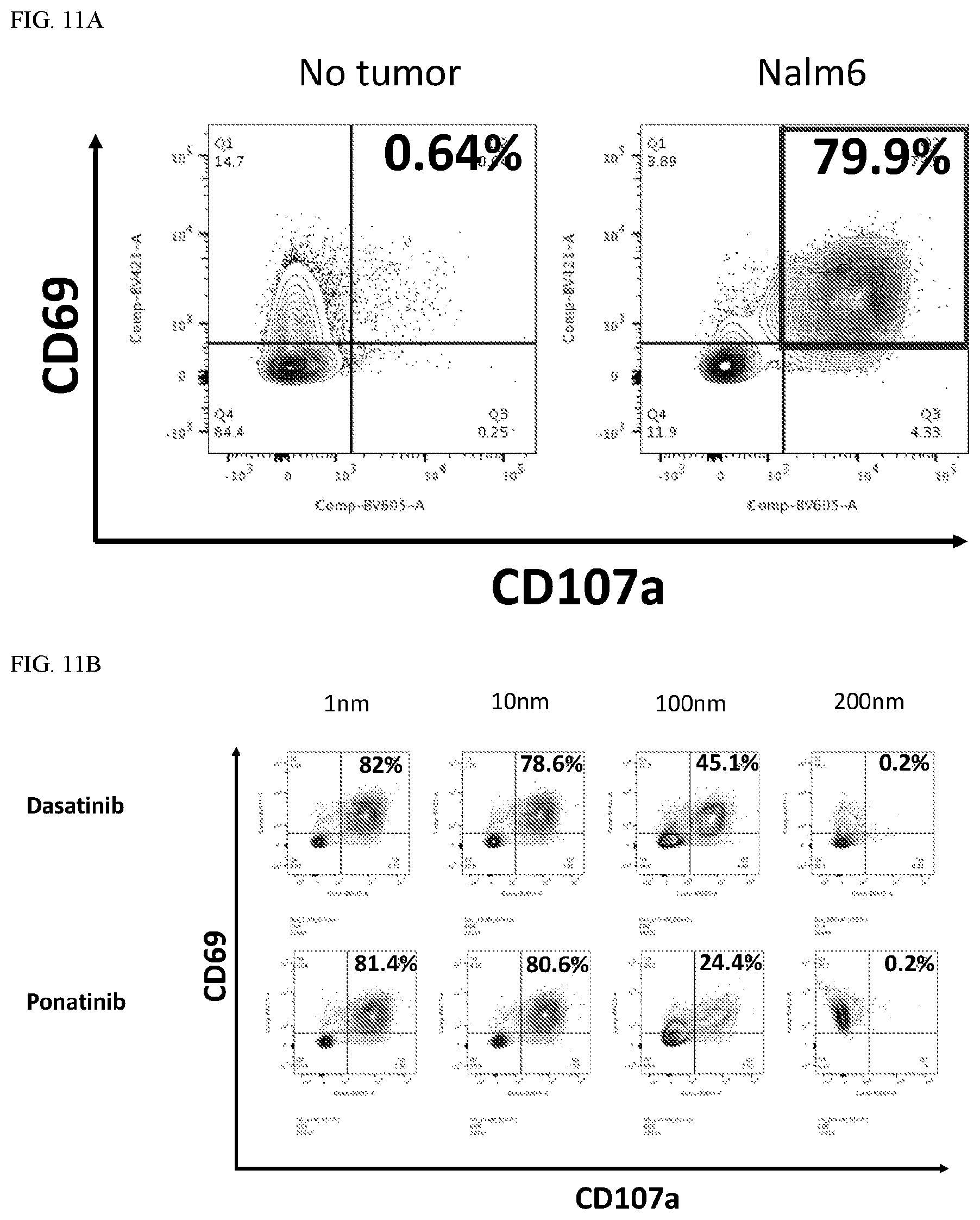
[0045] FIG. 11: CAR T cells co-cultured with tumor cells in the presence of dasatinib or ponatinib exhibit attenuated activation and degranulation. As shown, CD19.28z CAR T cells were cultured in the presence or absence of various concentrations of dasatinib or ponatinib for at least 48 hours. CAR T cells were then co-cultured with CD19-bearing Nalm6 tumor cells for 6 hours. CD69 and CD107a surface expression was subsequently assessed via FACS. Plots display cells gated on the CD8+ CAR+ population. Such results demonstrate that 80% of CD19.28z CART cells become activated (surface CD69 is a surrogate for activation) and degranulate (surface CD107a is a surrogate for degranulation) in response to tumor. However, dasatinib and ponatinib dose-dependently inhibit CAR T cells' ability to respond to tumor in this manner.
[0046] FIG. 12: CAR T cells co-cultured with tumor cells in the presence of dasatinib or ponatinib fail to secrete cytokine. As shown, high affinity GD2.28z (HA-GD2.28z) CAR T cells were co-cultured with GD2-overexpressing nalm6 for 24 hours in the presence of absence of various concentrations of dasatinib or ponatinib. Supernatant was then collected and analyzed for IL-2 and IFN.gamma. via ELISA. These results demonstrate that using the HA-GD2.28z CAR, dasatinib and ponatinib inhibit CART cell secretion of IL-2 and IFN.gamma. in response to tumor.
[0047] FIG. 13: CAR T cells cultured in the presence of dasatinib display attenuated killing in response to tumor antigen. An incucyte assay was conducted in which CD19.BBz CAR T cells were co-cultured with nalm6 tumor cells expressing a GFP reporter for 72 hours in the presence of 1 uM dasatinib or vehicle (DMSO). Tumor GFP fluorescence was measured over time. GFP values were normalized to the fluorescence intensity at the first time point. These results demonstrate that dasatinib blunts the ability of the CD19.28z CAR to kill tumor cells. FIGS. 11, 12 and 13 demonstrate that dasatinib or ponatinib could serve as a rapid and reversible safety "OFF" switch for CAR T cells that are having deleterious effects in a given patient.
[0048] FIG. 14: Dasatinib potently inhibits the phosphorylation of CAR CD3z as well as distal signaling proteins after CAR crosslinking. 2E6 HA-GD2.28z CAR T cells cultured in 1 uM dasatinib or vehicle were removed from culture on day 10 post-activation. Idiotype primary antibody and a crosslinking secondary antibody were then added to the cells at 5 ug/mL to initiate signaling through the CAR. Shown here, dasatinib potently inhibits crosslinking-induced phosphorylation of the CD3z domain on the CAR, as well as phosphorylation of distal signaling kinases Akt and ERK1/2. This is a representative blot of n=3 independent experiments.
[0049] FIG. 15: Tonically signaling CART cells expanded in the presence of dasatinib exhibit a reduction in canonical exhaustion marker expression in a dose-dependent manner. HA-GD2.28z CART cells were expanded in the presence of various concentrations of dasatinib or vehicle (DMSO). On day 14 post-activation, cells were removed from cultured, stained, and their exhaustion phenotype was analyzed via FACS. Representative plots from 3 independent experiments. FIG. 15A: CAR+ T cell canonical exhaustion marker expression.
[0050] FIG. 15B: CAR+ CD4+ (left) or CAR+ CD8+ (right) exhaustion marker co-expression. These results demonstrate that the HA-GD2.28z CAR tonically signals in the absence of antigen, which ultimately induces T cells exhaustion, as defined by expression of multiple inhibitory receptors, lack of memory formation, and decreased effector function. FIG. 14 demonstrates that expanding HA-GD2.28z CAR T cells in the presence of dasatinib dose-dependently attenuates exhaustion marker single expression (a) or co-expression (b).
[0051] FIG. 16: Tonically signaling CAR T cells expanded in the presence of dasatinib retain the capacity to form memory. CD19.28z or HA-GD2.28z were expanded in the presence or absence of 1 uM dasatinib or vehicle (DMSO). On Day 14 post-activation, cells were removed from cultured for FACS analysis. This representative plot shows CAR+ T cells. The red box highlights the CD45RA low, CCR7 high population, which corresponds to central memory-like T cells. These results demonstrate expanding tonically signaling HA-GD2.28z CAR T cells in dasatinib also augments memory formation, here demonstrated by a marked increase in the CD45RA low, CCR7 high population, which corresponds to a central memory-like phenotype.
[0052] FIG. 17: Tonically signaling CAR T cells expanded in the presence of dasatinib display augmented cytokine secretion in response to tumor antigen. HA-GD2.28z CAR T cells were expanded in the presence or absence of various concentrations of dasatinib or ponatinib. Drug was removed from the T cells 24 hours prior to co-culture with GD2-overexpressing nalm6 tumor cells in order to allow the T cells to regain the ability to signal in response to tumor. After 24 hours, supernatants were collected and IL-2 and IFN.gamma. secretion was assessed via ELISA. FIGS. 11, 12, 13, 15 and 16 demonstrate that dasatinib and ponatinib can inhibit CART cell signaling and function. FIG. 17 shows that expansion of tonically signaling HA-GD2.28z CART cells in the presence of these drugs followed by remove of the drugs prior to co-culturing with tumor cells results in augmentation of IL-2 and IFN.gamma..
[0053] FIG. 18: Tonically signaling CART cells expanded in the presence of dasatinib display augmented cytotoxicity. HA-GD2.28z CAR T cells were expanded in the presence or absence of dasatinib or vehicle (DMSO) for 96 hours. On day 14 post-activation, dasatinib was removed from the T cells 24 hours prior to an incucyte assay in which T cells were co-cultured at a 1:8 E:T ratio with GD2-overexpressing nalm6 tumor. Tumor GFP fluorescence was measured over time. GFP values were normalized to the fluorescence intensity at the first time point. These results demonstrate that inhibiting tonical signaling of HA-GD2.28z CAR T cells by including dasatinib in the culture medium during expansion followed by removal of dasatinib prior to co-culture rescues the ability of these CAR T cells to kill tumor.
[0054] FIG. 19: GD2-overexpressing Nalm6 in the presence and absence of dasatinib. 0.5E6 143B tumor cells were engrafted intramuscularly in the legs of mice. On day 3 post-engraftment, 10E6 GD2.BBz CART cells expanded in the presence of dasatinib or vehicle (DMSO) were infused into mice intravenously. The left plot displays the mean leg area+/-SEM (n=5 mice). FIGS. 19 and 20 recapitulate the findings from FIGS. 14, 15, 16 and 17 in an in vivo setting. Culturing different types of CARs (GD2.BBz, HA-GD2.28z) in dasatinib and then infusing them in vivo augments their anti-tumor function.
[0055] FIG. 20A: 0.5E6 143B tumor cells were engrafted intramuscularly in the legs of mice. On day 3 post-engraftment, 10E6 HA-GD2.28z CAR T cells expanded in the presence of dasatinib or vehicle (DMSO) were infused into mice intravenously. The top plot displays the mean leg area+/-SEM (n=5 mice). FIGS. 19 and 20 recapitulate the findings from FIGS. 14, 15, 16 and 17 in an in vivo setting. Culturing different types of CARs (GD2.BBz, HA-GD2.28z) in dasatinib and then infusing them in vivo augments their anti-tumor function.
[0056] FIG. 20B: 1E6 GD2-overexpressing nalm6 tumor cells were engrafted intravenously in mice. On day 3 post-engraftment, 2E6 CAR+ HA-GD2.28z CAR T cells expanded in the presence of dasatinib or vehicle (DMSO) were infused into mice intravenously. The top plot displays the mean tumor luminescence+/-SEM (n=5 mice). FIGS. 19 and 20 recapitulate the findings from FIGS. 14, 15, 16 and 17 in an in vivo setting. Culturing different types of CARs (GD2.BBz, HA-GD2.28z) in dasatinib and then infusing them in vivo augments their anti-tumor function.
[0057] FIG. 21: 1E6 GD2-overexpressing nalm6 tumor cells were engrafted intravenously in mice. On day 3 post-engraftment, 2E6 HA-GD2.28z CAR T cells expanded in the presence of dasatinib or vehicle (DMSO) were infused into mice intravenously. On day 17 post-engraftment, blood samples were taken from each mouse and mixed with counting beads. FACS analysis was performed, and the number of CD4+ and CD8+ cells for each mouse was calculated. This plot displays the mean CD4+ or CD8+ cells per mouse+/-SEM (n=5 mice). FIG. 21 demonstrates one of the mechanisms by which dasatinib augments function. After infusing dasatinib-treated CAR T cells into mice, blood samples were taken and the number of circulating CAR T cells analyzed, a typical readout for in vivo CAR T cell proliferation in response to tumor. The vehicle HA-GD2.28z CAR T cells did not exhibit significantly more in vivo proliferation than mock T cells, as these cells were likely exhausted when they were initially infused into the mice. However, CAR T cells that were expanded in dasatinib retained their anti-tumor function and thus proliferated robustly in vivo.
[0058] FIG. 22A,B,C,D,E: In vivo dasatinib treatment suppresses exhaustion marker expression, augments memory formation, and facilitates cell survival/proliferation. Mice were engrafted with 1E6 GD2-overexpressing nalm6 tumor cells via intravenous injection. On day 4 post-engraftment, 2E6 HA-GD2.28z CAR T cells were infused into mice intravenously. Mice were dosed with 50 mg/kg dasatinib via intraperitoneal injection on days 21-23 post-tumor engraftment. 5 hours after dasatinib dosing on day 23, 1 mouse receiving vehicle and 1 mouse receiving dasatinib were sacrificed, and spleens/blood were harvested, surface stained, and phenotyped via FACS. A and C) CAR+ T cells constituted a higher percentage of total circulating cells (A) or total splenic cells (C) in the mouse treated with dasatinib versus the vehicle control. B and D) In contrast to mice treated with vehicle (red), circulating or splenic CD8+ CAR+ T cells in dasatinib-treated mice (blue) exhibited a phenotype consistent with a non-activated or resting T cell, indicating that dasatinib suppressed CAR T cell activation and induced memory formation (i.e., higher CD62L expression) in vivo. E) On days 27-29 post-tumor engraftment, 1 mouse received 50 mg/kg dasatinib each day and a different mouse received vehicle control. On days 30-32, mice were untreated. On day 32, tumor luminescence was assessed. The 3 days of dasatinib dosing were sufficient to induce a robust reinvigoration of the anti-tumor response (blue). These data indicate that iterative dosing of dasatinib may reinvigorate exhausted T cells in vivo.
[0059] FIG. 23: The nucleic acid and amino acid sequence for CD19.28z (FMC63 scFv).
[0060] FIG. 24: The nucleic acid and amino acid sequence for CD19.BBz (FMC63 scFv).
[0061] FIG. 25: The nucleic acid and amino acid sequence for GD2.BBz (14G2a scFv).
[0062] FIG. 26: The nucleic acid and amino acid sequence for HA-GD2.28z (High affinity 14G2a scFv).
DEFINITIONS
[0063] It must be noted that, as used in this specification and the appended claims, the singular forms "a," "an" and "the" include plural referents unless the content clearly dictates otherwise. Thus, for example, reference to "a T cell" includes two or more T cells, and the like.
[0064] The term "about," particularly in reference to a given quantity, is meant to encompass deviations of plus or minus five percent.
[0065] The term "chimeric antigen receptor" or "CAR," as used herein, refers to an artificial T cell receptor that is engineered to be expressed on an immune effector cell and specifically bind an antigen. CARs may be used as a therapy with adoptive cell transfer. T cells are removed from a patient and modified so that they express the receptors specific to a particular form of antigen. In some embodiments, the CARs have been expressed with specificity to a tumor associated antigen, for example. CARs may also comprise an intracellular activation domain, a transmembrane domain and an extracellular domain comprising a tumor associated antigen binding region. The specificity of CAR designs may be derived from ligands of receptors (e.g., peptides). In some embodiments, a CAR can target cancers by redirecting the specificity of a T cell expressing the CAR specific for tumor associated antigens.
[0066] "Pharmaceutically acceptable excipient or carrier" refers to an excipient that may optionally be included in the compositions of the invention and that causes no significant adverse toxicological effects to the patient.
[0067] "Pharmaceutically acceptable salt" includes, but is not limited to, amino acid salts, salts prepared with inorganic acids, such as chloride, sulfate, phosphate, diphosphate, bromide, and nitrate salts, or salts prepared from the corresponding inorganic acid form of any of the preceding, e.g., hydrochloride, etc., or salts prepared with an organic acid, such as malate, maleate, fumarate, tartrate, succinate, ethylsuccinate, citrate, acetate, lactate, methanesulfonate, benzoate, ascorbate, para-toluenesulfonate, palmoate, salicylate and stearate, as well as estolate, gluceptate and lactobionate salts. Similarly, salts containing pharmaceutically acceptable cations include, but are not limited to, sodium, potassium, calcium, aluminum, lithium, and ammonium (including substituted ammonium).
[0068] The term "T cell" refers to T lymphocytes as defined in the art and is intended to include thymocytes, immature T lymphocytes, mature T lymphocytes, resting T lymphocytes, or activated T lymphocytes. The T cells can be CD4.sup.+ T cells, CD8.sup.+ T cells, CD4.sup.+CD8.sup.+ T cells, or CD4.sup.-CD8.sup.- cells. The T cells can also be T helper cells, such as T helper 1 (TH1), or T helper 2 (TH2) cells, or TH17 cells, as well as cytotoxic T cells, regulatory T cells, natural killer T cells, naive T cells, memory T cells, or gamma delta T cells.
[0069] The T cells can be a purified population of T cells, or alternatively the T cells can be in a population with cells of a different type, such as B cells and/or other peripheral blood cells. The T cells can be a purified population of a subset of T cells, such as CD4.sup.+ T cells, or they can be a population of T cells comprising different subsets of T cells. In another embodiment of the invention, the T cells are T cell clones that have been maintained in culture for extended periods of time. T cell clones can be transformed to different degrees. In a specific embodiment, the T cells are a T cell clone that proliferates indefinitely in culture.
[0070] In some embodiments, the T cells are primary T cells. The term "primary T cells" is intended to include T cells obtained from an individual, as opposed to T cells that have been maintained in culture for extended periods of time. Thus, primary T cells are particularly peripheral blood T cells obtained from a subject. A population of primary T cells can be composed of mostly one subset of T cells. Alternatively, the population of primary T cells can be composed of different subsets of T cells.
[0071] The T cells can be from previously stored blood samples, from a healthy individual, or alternatively from an individual affected with a condition. The condition can be an infectious disease, such as a condition resulting from a viral infection, a bacterial infection or an infection by any other microorganism, or a hyperproliferative disease, such as cancer like melanoma. In yet another embodiment of the invention, the T cells are from a subject suffering from or susceptible to an autoimmune disease or T-cell pathologies. The T cells can be of human origin, murine origin or any other mammalian species.
[0072] "T cell exhaustion" refers to loss of T cell function, which may occur as a result of an infection or a disease. T cell exhaustion is associated with increased expression of PD-1, TIM-3, and LAG-3, apoptosis, and reduced cytokine secretion.
[0073] By "therapeutically effective dose or amount" of an inhibitor of TCR signaling (e.g., dasatinib) is intended an amount that, when administered as described herein, brings about a positive therapeutic response in treatment of T cell exhaustion, such as restored T cell function. Improved T cell function may include decreased expression of PD-1, TIM-3, and LAG-3, maintenance of memory markers (e.g., CD62L or CCR7), prevention of apoptosis, and increased secretion of IL-2 and other cytokines. The exact amount required will vary from subject to subject, depending on the species, age, and general condition of the subject, the severity of the condition being treated, the particular drug or drugs employed, mode of administration, and the like. An appropriate "effective" amount in any individual case may be determined by one of ordinary skill in the art using routine experimentation, based upon the information provided herein.
[0074] The terms "subject," "individual," and "patient," are used interchangeably herein and refer to any vertebrate subject, including, without limitation, humans and other primates, including non-human primates such as chimpanzees and other apes and monkey species; farm animals such as cattle, sheep, pigs, goats and horses; domestic mammals such as dogs and cats; laboratory animals including rodents such as mice, rats and guinea pigs; birds, including domestic, wild and game birds such as chickens, turkeys and other gallinaceous birds, ducks, geese, and the like. The term does not denote a particular age. Thus, both adult and newborn individuals are intended to be covered.
DETAILED DESCRIPTION OF THE INVENTION
[0075] The invention is based on the discovery that transient inhibition or modulation of TCR signaling and/or CAR signaling in human T cells can prevent or reverse T cell exhaustion and restore T cell function. The inventors have shown that GD2-CAR expressing T cells develop functional exhaustion, exhibited by expression of PD-1, TIM-3, and LAG-3 exhaustion markers. Cessation of tonic signaling restores the ability of T cells to secrete IL-2 in response to tumor antigen. The inventors further showed that treatment with dasatinib, a Lck tyrosine kinase inhibitor that inhibits T cell receptor signaling, reduced expression of the T cell exhaustion markers and improved preservation of T cell memory.
[0076] Protein tyrosine kinases are a family of enzymes catalysing the transfer of the terminal phosphate of adenosine triphosphate to tyrosine residues in protein substrates. Phosphorylation of tyrosine residues on protein substrates leads to transduction of intracellular signals which regulate a wide variety of intracellular processes such as growth and activation of cells of the immune system, e.g. T-cells. As T-cell activation is implicated in a number of inflammatory conditions and other disorders of the immune system (e.g. autoimmune diseases), modulation of the activity of protein tyrosine kinases appears to be an attractive route to the management of inflammatory diseases. A large number of protein tyrosine kinases have been identified which may be receptor protein tyrosine kinases, e.g. the insulin receptor, or non-receptor protein tyrosine kinases.
[0077] Protein tyrosine kinases of the Src family have been found to be particularly important for intracellular signal transduction related to inflammatory responses (see, e.g., D. Okutani et al., Am. J. Physiol. Lung Cell Mol. Physiol. 291, 2006, pp. L129-L141; CA. Lowell, Mol. Immunol. 41, 2004, pp. 631-643). While some of Src family protein tyrosine kinases, e.g. Src, Yes and Fyn, are expressed in a variety of cell types and tissues, the expression of others is restricted to specific cell types, e.g. hematopoietic cells. Thus, the protein tyrosine kinase Lck is expressed almost exclusively in T-cells as the first signalling molecule to be activated downstream of the T-cell receptor, and its activity is essential for T-cell signal transduction. Expression of Hck, Lyn and Fgr is increased by inflammatory stimuli such as LPS in mature monocytes and macrophages. Also, if gene expression of the main B-cell Src family kinases, namely Lyn, Fyn and BIk, is disrupted, immature B-cells are prevented from developing into mature B-cells. Src family kinases have also been identified as essential for the recruitment and activation of monocytes, macrophages and neutrophils as well as being involved in the inflammatory response of tissue cells.
[0078] As noted, receptor tyrosine kinases are essential components of signal transduction pathways that mediate cell-to-cell communication and their function as relay points for signaling pathways. They have a key role in numerous processes that control cellular proliferation and differentiation, regulate cell growth and cellular metabolism, and promote cell survival and apoptosis. Lck (p56.sup.lck or lymphocyte specific kinase) is a cytoplasmic tyrosine kinase of the Src family expressed in T cells and natural killer (NK) cells. Genetic evidence from knockout mice and human mutations demonstrates that Lck kinase activity is critical for T cell receptor (TCR)-mediated signaling, leading to normal T-cell development and activation. As such, selective inhibition of Lck is useful in the treatment of T-cell-mediated autoimmune and inflammatory disorders and/or organ transplant rejection.
[0079] The invention is further based on the discovery that the Lck kinase inhibitor dasatinib and the receptor tyrosine kinase inhibitor ponatinib have the potential to address several important challenges currently facing the field of adoptive T cell therapies (e.g., CART cell therapies). First, these drugs were shown to potently inhibit CAR signaling, which provides a method to regulate CAR activity and thus mitigate CAR T cell toxicity while preserving the option to continue therapy once the toxicity has resolved, as the inhibitory effect of dasatinib and ponatinib on CAR T cell function is reversible. Second, expansion of CAR T cells in the presence of dasatinib or ponatinib was shown to prevent CAR tonic signaling and in turn enhance the functional capacity of CAR T cells. Lastly, providing short periods of CAR T cell "rest" in vivo via iterative drug dosing was shown to be one method by which CAR T cell exhaustion could be prevented or reversed and/or memory could be induced.
[0080] Accordingly, provided herein are compositions and methods for preventing or reversing T cell exhaustion. In particular, the present invention relates to methods of preventing or reversing T cell exhaustion by exposing T cells experiencing T cell exhaustion to particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib), or by expanding genetically engineered T cells in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib).
[0081] As such, the present invention contemplates that exposure of animals (e.g., humans) undergoing adoptive T cell therapies (e.g., a CAR T-cell therapy, a transduced T-cell therapy, and a tumor infiltrating lymphocyte (TIL) therapy) with genetically engineered T cell populations to compositions comprising particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) will result in improved therapy outcome as such particular tyrosine kinase inhibitors are capable of 1) modulating TCR signaling within the genetically engineered T cell population (e.g., decreasing expression of one or more of PD-1, TIM-3, and LAG-3; increasing expression of memory markers (e.g., CD62L or CCR7); increasing secretion of IL-2 and other cytokines), and 2) preventing and/or reversing T cell exhaustion within the genetically engineered T cell population. Indeed, the present invention contemplates that the use of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) (e.g., Src family kinase inhibitors) (e.g., Lck inhibitors) within adoptive T cell therapies satisfies an unmet need as the effectiveness of such therapies are frequently compromised by such T cell populations experiencing T cell exhaustion. Thus, the present invention provides methods for treating an immune system related condition or disease (e.g., cancer) in a subject comprising administering to the subject (e.g., simultaneously and/or at different time points) genetically engineered T cells and particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib). Such methods are not limited to a specific type or kind of genetically engineered T cells. In some embodiments, the genetically engineered T cells include, but are not limited to, CAR T cells, genetically engineered TCR expressing T cells, genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, genetically engineered T cells configured for transduced T-cell therapy, and/or viral specific T cells reengineered with a TCR or CAR.
[0082] Such tyrosine kinase inhibitors may be administered by any suitable mode of administration, but is typically administered orally. Multiple cycles of treatment may be administered to a subject. In certain embodiments, the tyrosine kinase inhibitors are administered according to a daily dosing regimen or intermittently. In another embodiment, the tyrosine kinase inhibitors are administered for a period of time sufficient to restore at least partial T cell function, then discontinued.
[0083] The present invention contemplates that ex vivo expansion of a population of T cells with particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) will result in a population T cells that are resistant and/or less prone to T cell exhaustion. Thus, the present invention provides compositions comprising a population of T cells that were expanded in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) (e.g., Src family kinase inhibitors) (e.g., Lck inhibitors). Thus, the present invention provides methods of expanding a population of T cells to generate T cell populations that are resistant and/or less prone to T cell exhaustion through expanding such T cells in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib). Thus, the present invention provides kits comprising T cell populations that were expanded in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) and additional agents (e.g., additional agents useful in expanding T cells) (e.g., additional agents useful in adoptive T cell therapies (e.g., a CAR T-cell therapy, a transduced T-cell therapy, and a tumor infiltrating lymphocyte (TIL) therapy). Such methods are not limited to a specific type or kind of genetically engineered T cells. In some embodiments, the genetically engineered T cells include, but are not limited to, CAR T cells, genetically engineered TCR expressing T cells, genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, genetically engineered T cells configured for transduced T-cell therapy, and/or viral specific T cells reengineered with a TCR or CAR.
[0084] The present invention contemplates that ex vivo expansion of a population of genetically engineered T cells (e.g., genetically engineered for use within adoptive T cell therapies (e.g., a CAR T-cell therapy, a transduced T-cell therapy, and a tumor infiltrating lymphocyte (TIL) therapy)) with particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) (e.g., Src family kinase inhibitors) (e.g., Lck inhibitors) will result in genetically engineered T cells that are resistant and/or less prone to T cell exhaustion. Thus, the present invention provides compositions comprising a population of genetically engineered T cells that were expanded in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib). Thus, the present invention provides methods of expanding a population of genetically engineered T cells to generate genetically engineered T cell populations that are resistant and/or less prone to T cell exhaustion through expanding such T cells in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib). Thus, the present invention provides kits comprising genetically engineered T cell populations that were expanded in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib). Such methods are not limited to a specific type or kind of genetically engineered T cells. In some embodiments, the genetically engineered T cells include, but are not limited to, CAR T cells, genetically engineered TCR expressing T cells, genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, genetically engineered T cells configured for transduced T-cell therapy, and/or viral specific T cells reengineered with a TCR or CAR.
[0085] The present invention contemplates that exposure of animals (e.g., humans) undergoing adoptive T cell therapies (e.g., a CAR T-cell therapy, a transduced T-cell therapy, and a tumor infiltrating lymphocyte (TIL) therapy) with genetically engineered T cell populations that were expanded in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) will result in improved therapy outcome as such genetically engineered T cell populations are resistant and/or less prone to T cell exhaustion. Thus, the present invention provides methods of treating an immune system related condition or disease (e.g., cancer) in a subject comprising administering a population of genetically engineered T cells expanded in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) (e.g., Src family kinase inhibitors) (e.g., Lck inhibitors). Such methods are not limited to a specific type or kind of genetically engineered T cells. In some embodiments, the genetically engineered T cells include, but are not limited to, CAR T cells, genetically engineered TCR expressing T cells, genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, genetically engineered T cells configured for transduced T-cell therapy, and/or viral specific T cells reengineered with a TCR or CAR.
[0086] Such embodiments are not limited to a particular type or kind of an immune system related condition or disease.
[0087] For example, in some embodiments, the immune system related condition or disease is an autoimmune disease or condition (e.g., Acquired Immunodeficiency Syndrome (AIDS), alopecia areata, ankylosing spondylitis, antiphospholipid syndrome, autoimmune Addison's disease, autoimmune hemolytic anemia, autoimmune hepatitis, autoimmune inner ear disease (AIED), autoimmune lymphoproliferative syndrome (ALPS), autoimmune thrombocytopenic purpura (ATP), Behcet's disease, cardiomyopathy, celiac sprue-dermatitis hepetiformis; chronic fatigue immune dysfunction syndrome (CFIDS), chronic inflammatory demyelinating polyneuropathy (CIPD), cicatricial pemphigold, cold agglutinin disease, crest syndrome, Crohn's disease, Degos' disease, dermatomyositis-juvenile, discoid lupus, essential mixed cryoglobulinemia, fibromyalgia-fibromyositis, Graves' disease, Guillain-Barre syndrome, Hashimoto's thyroiditis, idiopathic pulmonary fibrosis, idiopathic thrombocytopenia purpura (ITP), IgA nephropathy, insulin-dependent diabetes mellitus, juvenile chronic arthritis (Still's disease), juvenile rheumatoid arthritis, Meniere's disease, mixed connective tissue disease, multiple sclerosis, myasthenia gravis, pernacious anemia, polyarteritis nodosa, polychondritis, polyglandular syndromes, polymyalgia rheumatica, polymyositis and dermatomyositis, primary agammaglobulinemia, primary biliary cirrhosis, psoriasis, psoriatic arthritis, Raynaud's phenomena, Reiter's syndrome, rheumatic fever, rheumatoid arthritis, sarcoidosis, scleroderma (progressive systemic sclerosis (PSS), also known as systemic sclerosis (SS)), Sjogren's syndrome, stiff-man syndrome, systemic lupus erythematosus, Takayasu arteritis, temporal arteritis/giant cell arteritis, ulcerative colitis, uveitis, vitiligo, Wegener's granulomatosis, and any combination thereof).
[0088] For example, in some embodiments, the immune system related condition or disease is cancer (e.g., breast cancer, prostate cancer, ovarian cancer, cervical cancer, skin cancer, pancreatic cancer, colorectal cancer, renal cancer, liver cancer, brain cancer, lymphoma, leukemia, lung cancer, and thyroid carcinoma).
[0089] The present invention contemplates that the use of genetically engineered T cell populations that were expanded in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) within adoptive T cell therapies (e.g., a CAR T-cell therapy, a transduced T-cell therapy, and a tumor infiltrating lymphocyte (TIL) therapy) satisfies an unmet need as such therapies are frequently compromised by such T cell populations experiencing T cell exhaustion. Such methods are not limited to a specific type or kind of genetically engineered T cells. In some embodiments, the genetically engineered T cells include, but are not limited to, CAR T cells, genetically engineered TCR expressing T cells, genetically engineered T cells configured for tumor infiltrating lymphocyte (TIL) therapy, genetically engineered T cells configured for transduced T-cell therapy, and/or viral specific T cells reengineered with a TCR or CAR.
[0090] The embodiments of the present invention are not limited to specific types of tyrosine kinase inhibitors. In some embodiments, the tyrosine kinase inhibitors are a Lck tyrosine kinase inhibitors. In some embodiments, the tyrosine kinase inhibitor is a Src family kinase inhibitor (e.g., Src kinase inhibitor, Yes kinase inhibitor, Fyn kinase inhibitor, Fgr kinase inhibitor, Lck kinase inhibitor, Hck kinase inhibitor, Blk kinase inhibitor, Lyn kinase inhibitor). In some embodiments, the tyrosine kinase inhibitor is dasatinib
##STR00001##
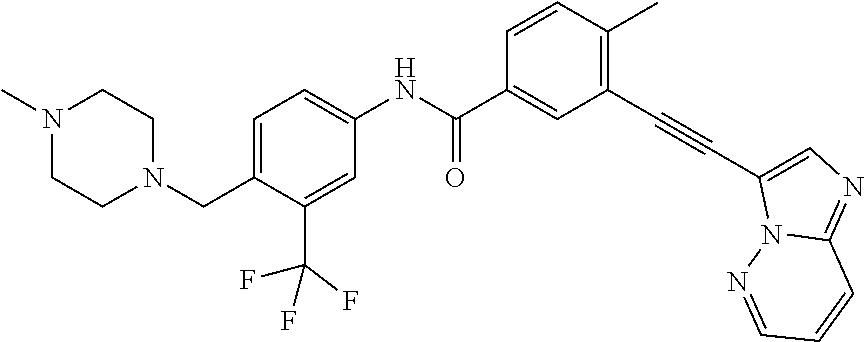
(N-(2-chloro-6-methylphenyl)-2-(6-(4-(2-hydroxyethyl)piperazin-1-yl)-2-me- thylpyrimidin-4-ylamino)thiazole-5-carboxamide), or a pharmaceutically acceptable salt, solvate, or prodrug thereof. In some embodiments, the tyrosine kinase inhibitor is ponatinib
##STR00002##
(3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methyl-N-(4-((4-methylpiperazi- n-1-yl)methyl)-3-(trifluoromethyl)phenyl)benzamide), or a pharmaceutically acceptable salt, solvate, or prodrug thereof.
[0091] Some embodiments of the present invention provide for administering such methods (e.g., adoptive T cell therapies with genetically engineered T cell populations and compositions comprising particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib)) (e.g., adoptive T cell therapies with genetically engineered T cell populations that were expanded in the presence of particular tyrosine kinase inhibitors (e.g., dasatinib, ponatinib)) in combination with an effective amount of at least one additional therapeutic agent (including, but not limited to, chemotherapeutic antineoplastics, apoptosis-modulating agents, antimicrobials, antivirals, antifungals, and anti-inflammatory agents) and/or therapeutic technique (e.g., surgical intervention, and/or radiotherapies). In a particular embodiment, the additional therapeutic agent(s) is an anticancer agent.
[0092] Tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) can be formulated into pharmaceutical compositions optionally comprising one or more pharmaceutically acceptable excipients. Exemplary excipients include, without limitation, carbohydrates, inorganic salts, antimicrobial agents, antioxidants, surfactants, buffers, acids, bases, and combinations thereof. Excipients suitable for injectable compositions include water, alcohols, polyols, glycerine, vegetable oils, phospholipids, and surfactants. A carbohydrate such as a sugar, a derivatized sugar such as an alditol, aldonic acid, an esterified sugar, and/or a sugar polymer may be present as an excipient. Specific carbohydrate excipients include, for example: monosaccharides, such as fructose, maltose, galactose, glucose, D-mannose, sorbose, and the like; disaccharides, such as lactose, sucrose, trehalose, cellobiose, and the like; polysaccharides, such as raffinose, melezitose, maltodextrins, dextrans, starches, and the like; and alditols, such as mannitol, xylitol, maltitol, lactitol, xylitol, sorbitol (glucitol), pyranosyl sorbitol, myoinositol, and the like. The excipient can also include an inorganic salt or buffer such as citric acid, sodium chloride, potassium chloride, sodium sulfate, potassium nitrate, sodium phosphate monobasic, sodium phosphate dibasic, and combinations thereof.
[0093] A surfactant can be present as an excipient. Exemplary surfactants include: polysorbates, such as "Tween 20" and "Tween 80," and pluronics such as F68 and F88 (BASF, Mount Olive, N.J.); sorbitan esters; lipids, such as phospholipids such as lecithin and other phosphatidylcholines, phosphatidylethanolamines (although preferably not in liposomal form), fatty acids and fatty esters; steroids, such as cholesterol; chelating agents, such as EDTA; and zinc and other such suitable cations.
[0094] Acids or bases can be present as an excipient in the pharmaceutical composition. Nonlimiting examples of acids that can be used include those acids selected from the group consisting of hydrochloric acid, acetic acid, phosphoric acid, citric acid, malic acid, lactic acid, formic acid, trichloroacetic acid, nitric acid, perchloric acid, phosphoric acid, sulfuric acid, fumaric acid, and combinations thereof. Examples of suitable bases include, without limitation, bases selected from the group consisting of sodium hydroxide, sodium acetate, ammonium hydroxide, potassium hydroxide, ammonium acetate, potassium acetate, sodium phosphate, potassium phosphate, sodium citrate, sodium formate, sodium sulfate, potassium sulfate, potassium fumerate, and combinations thereof.
[0095] The amount of the tyrosine kinase inhibitor (e.g., dasatinib, ponatinib) (e.g., when contained in a drug delivery system) in the pharmaceutical composition will vary depending on a number of factors, but will optimally be a therapeutically effective dose when the composition is in a unit dosage form or container (e.g., a vial). A therapeutically effective dose can be determined experimentally by repeated administration of increasing amounts of the composition in order to determine which amount produces a clinically desired endpoint.
[0096] The amount of any individual excipient in the pharmaceutical composition will vary depending on the nature and function of the excipient and particular needs of the composition. Typically, the optimal amount of any individual excipient is determined through routine experimentation, i.e., by preparing compositions containing varying amounts of the excipient (ranging from low to high), examining the stability and other parameters, and then determining the range at which optimal performance is attained with no significant adverse effects. Generally, however, the excipient(s) will be present in the composition in an amount of about 1% to about 99% by weight, preferably from about 5% to about 98% by weight, more preferably from about 15 to about 95% by weight of the excipient, with concentrations less than 30% by weight most preferred. These foregoing pharmaceutical excipients along with other excipients are described in "Remington: The Science & Practice of Pharmacy", 19.sup.th ed., Williams & Williams, (1995), the "Physician's Desk Reference", 52.sup.nd ed., Medical Economics, Montvale, N.J. (1998), and Kibbe, A. H., Handbook of Pharmaceutical Excipients, 3.sup.rd Edition, American Pharmaceutical Association, Washington, D.C., 2000.
[0097] The pharmaceutical compositions encompass all types of formulations and in particular those that are suited for injection, e.g., powders or lyophilates that can be reconstituted with a solvent prior to use, as well as ready for injection solutions or suspensions, dry insoluble compositions for combination with a vehicle prior to use, and emulsions and liquid concentrates for dilution prior to administration. Examples of suitable diluents for reconstituting solid compositions prior to injection include bacteriostatic water for injection, dextrose 5% in water, phosphate buffered saline, Ringer's solution, saline, sterile water, deionized water, and combinations thereof. With respect to liquid pharmaceutical compositions, solutions and suspensions are envisioned. Additional preferred compositions include those for oral, ocular, or localized delivery.
[0098] The pharmaceutical preparations herein can also be housed in a syringe, an implantation device, or the like, depending upon the intended mode of delivery and use. Preferably, the pharmaceutical compositions comprising one or more tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) described herein are in unit dosage form, meaning an amount of a conjugate or composition of the invention appropriate for a single dose, in a premeasured or pre-packaged form.
[0099] The pharmaceutical compositions herein may optionally include one or more additional agents, or may be combined with one or more additional agents, such as other drugs for treating T cell exhaustion (e.g., anti-PD-1 checkpoint inhibitor, such as nivolumab), or other medications used to treat a subject for an infection or disease associated with T cell exhaustion (e.g., antiviral, antibiotic, or anti-cancer drugs and therapies, including adoptive T cell therapies). Compounded preparations may be used including at least one tyrosine kinase inhibitor (e.g., dasatinib, ponatinib) and one or more other agents, such as other drugs for treating T cell exhaustion or an infection or disease associated with T cell exhaustion. Alternatively, such agents can be contained in a separate composition from the composition comprising a tyrosine kinase inhibitor (e.g., dasatinib, ponatinib) and co-administered concurrently, before, or after the composition comprising a tyrosine kinase inhibitor (e.g., dasatinib, ponatinib).
[0100] At least one therapeutically effective cycle of treatment with a tyrosine kinase inhibitor (e.g., a tyrosine kinase inhibitor (e.g., dasatinib, ponatinib)) will be administered to a subject for treatment of T cell exhaustion. By "therapeutically effective cycle of treatment" is intended a cycle of treatment that when administered, brings about a positive therapeutic response with respect to treatment of an individual for T cell exhaustion. Of particular interest is a cycle of treatment with a tyrosine kinase inhibitor (e.g., dasatinib, ponatinib) that, when administered transiently as described herein, restores T cell function. For example, a therapeutically effective dose or amount of a tyrosine kinase inhibitor may decrease expression of PD-1, TIM-3, and LAG-3, improve maintenance of memory markers (e.g., CD62L or CCR7), prevent apoptosis, and increase secretion of IL-2 and other cytokines.
[0101] In certain embodiments, multiple therapeutically effective doses of pharmaceutical compositions comprising one or more tyrosine kinase inhibitors (e.g., dasatinib, ponatinib), and/or one or more other therapeutic agents, such as other drugs for treating T cell exhaustion (e.g., anti-PD-1 checkpoint inhibitor, such as nivolumab), or other medications used to treat a subject for an infection or disease associated with T cell exhaustion (e.g., antiviral, antibiotic, or anti-cancer drugs and therapies, including adoptive T cell therapies) will be administered. The pharmaceutical compositions of the present invention are typically, although not necessarily, administered orally, via injection (subcutaneously, intravenously, or intramuscularly), by infusion, or locally. Additional modes of administration are also contemplated, such as topical, intralesion, intracerebral, intracerebroventricular, intraparenchymatous, pulmonary, rectal, transdermal, transmucosal, intrathecal, pericardial, intra-arterial, intraocular, intraperitoneal, and so forth.
[0102] The pharmaceutical preparation can be in the form of a liquid solution or suspension immediately prior to administration, but may also take another form such as a syrup, cream, ointment, tablet, capsule, powder, gel, matrix, suppository, or the like. The pharmaceutical compositions comprising one or more tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) and other agents may be administered using the same or different routes of administration in accordance with any medically acceptable method known in the art.
[0103] In another embodiment, the pharmaceutical compositions comprising one or more tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) and/or other agents are administered prophylactically, e.g., to prevent T cell exhaustion. Such prophylactic uses will be of particular value for subjects with a chronic infection or cancer, who are at risk of developing T cell exhaustion.
[0104] In another embodiment of the invention, the pharmaceutical compositions comprising one or more tyrosine kinase inhibitors (e.g., dasatinib, ponatinib) and/or other agents are in a sustained-release formulation, or a formulation that is administered using a sustained-release device. Such devices are well known in the art, and include, for example, transdermal patches, and miniature implantable pumps that can provide for drug delivery over time in a continuous, steady-state fashion at a variety of doses to achieve a sustained-release effect with a non-sustained-release pharmaceutical composition.
[0105] The invention also provides a method for administering a conjugate comprising a tyrosine kinase inhibitor (e.g., dasatinib, ponatinib) as provided herein to a patient suffering from a condition that is responsive to treatment with a tyrosine kinase inhibitor (e.g., dasatinib, ponatinib) contained in the conjugate or composition. The method comprises administering, via any of the herein described modes, a therapeutically effective amount of the conjugate or drug delivery system, preferably provided as part of a pharmaceutical composition. The method of administering may be used to treat any condition that is responsive to treatment with a tyrosine kinase inhibitor (e.g., dasatinib, ponatinib). More specifically, the pharmaceutical compositions herein are effective in treating T cell exhaustion.
[0106] Those of ordinary skill in the art will appreciate which conditions a tyrosine kinase inhibitor (e.g., dasatinib, ponatinib) can effectively treat. The actual dose to be administered will vary depending upon the age, weight, and general condition of the subject as well as the severity of the condition being treated, the judgment of the health care professional, and conjugate being administered. Therapeutically effective amounts can be determined by those skilled in the art, and will be adjusted to the particular requirements of each particular case.
[0107] Generally, a therapeutically effective amount will range from about 0.50 mg to 5 grams of a tyrosine kinase inhibitor daily, more preferably from about 5 mg to 2 grams daily, even more preferably from about 7 mg to 1.5 grams daily. Preferably, such doses are in the range of 10-600 mg four times a day (QID), 200-500 mg QID, 25-600 mg three times a day (TID), 25-50 mg TID, 50-100 mg TID, 50-200 mg TID, 300-600 mg TID, 200-400 mg TID, 200-600 mg TID, 100 to 700 mg twice daily (BID), 100-600 mg BID, 200-500 mg BID, or 200-300 mg BID. The amount of compound administered will depend on the potency of the tyrosine kinase inhibitor and the magnitude or effect desired and the route of administration.
[0108] A purified tyrosine kinase inhibitor (again, preferably provided as part of a pharmaceutical preparation) can be administered alone or in combination with one or more other therapeutic agents, such as other drugs for treating T cell exhaustion (e.g., anti-PD-1 checkpoint inhibitor, such as nivolumab), or other medications used to treat a subject for an infection or disease associated with T cell exhaustion (e.g., antiviral, antibiotic, or anti-cancer drugs); or adoptive T cell therapies (e.g., a CAR T-cell therapy, a transduced T-cell therapy, and a tumor infiltrating lymphocyte (TIL) therapy); or other medications used to treat a particular condition or disease according to a variety of dosing schedules depending on the judgment of the clinician, needs of the patient, and so forth. The specific dosing schedule will be known by those of ordinary skill in the art or can be determined experimentally using routine methods. Exemplary dosing schedules include, without limitation, administration five times a day, four times a day, three times a day, twice daily, once daily, three times weekly, twice weekly, once weekly, twice monthly, once monthly, and any combination thereof. Preferred compositions are those requiring dosing no more than once a day.
[0109] A tyrosine kinase inhibitor can be administered prior to, concurrent with, or subsequent to other agents or therapies. If provided at the same time as other agents or therapies, one or tyrosine kinase inhibitors can be provided in the same or in a different composition. Thus, one or more tyrosine kinase inhibitors and other agents can be presented to the individual by way of concurrent therapy. By "concurrent therapy" is intended administration to a subject such that the therapeutic effect of the combination of the substances is caused in the subject undergoing therapy. For example, concurrent therapy may be achieved by administering a dose of a pharmaceutical composition comprising a tyrosine kinase inhibitor and a dose of a pharmaceutical composition comprising at least one other agent, such as another drug for treating T cell exhaustion, which in combination comprise a therapeutically effective dose, according to a particular dosing regimen. Similarly, one or more tyrosine kinase inhibitors and one or more other therapeutic agents can be administered in at least one therapeutic dose. Administration of the separate pharmaceutical compositions or therapies can be performed simultaneously or at different times (i.e., sequentially, in either order, on the same day, or on different days), as long as the therapeutic effect of the combination of these substances is caused in the subject undergoing therapy.
[0110] The invention also provides kits comprising one or more containers holding compositions comprising at least one tyrosine kinase inhibitor (e.g., dasatinib, ponatinib) and optionally one or more other agents for treating T cell exhaustion. Compositions can be in liquid form or can be lyophilized. Suitable containers for the compositions include, for example, bottles, vials, syringes, and test tubes. Containers can be formed from a variety of materials, including glass or plastic. A container may have a sterile access port (for example, the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle).
[0111] The kit can further comprise a second container comprising a pharmaceutically-acceptable buffer, such as phosphate-buffered saline, Ringer's solution, or dextrose solution. It can also contain other materials useful to the end-user, including other pharmaceutically acceptable formulating solutions such as buffers, diluents, filters, needles, and syringes or other delivery devices. The delivery device may be pre-filled with the compositions.
[0112] The kit can also comprise a package insert containing written instructions for methods of using the compositions comprising at least one tyrosine kinase inhibitor (e.g., dasatinib, ponatinib) for treating a subject for T cell exhaustion. The package insert can be an unapproved draft package insert or can be a package insert approved by the Food and Drug Administration (FDA) or other regulatory body.
[0113] One of ordinary skill in the art will readily recognize that the foregoing represents merely a detailed description of certain preferred embodiments of the present invention. Various modifications and alterations of the compositions and methods described above can readily be achieved using expertise available in the art and are within the scope of the invention.
EXAMPLES
[0114] The following examples are illustrative, but not limiting, of the compounds, compositions, and methods of the present invention. Other suitable modifications and adaptations of the variety of conditions and parameters normally encountered in clinical therapy and which are obvious to those skilled in the art are within the spirit and scope of the invention.
Example I. A Method of Preventing or Reversing T Cell Exhaustion by Inhibiting or Modulating TCR Signaling
Introduction
[0115] We previously reported that GD2-CAR expressing T cells develop functional exhaustion within 10 days in culture and are characterized by co-expression of inhibitory receptors, failure to secrete cytokines in response to tumor antigen, and aberrant metabolic function (Long et. al, Nat Med 2015). Control cultures included untransduced T cells (mock) and those expressing CD19-CAR, which does not manifest tonic signaling or develop exhaustion in vitro. Previous work also demonstrated that the zeta chain was required for exhaustion in this system, with CD28 signaling enhancing the potency of the signaling stimulus in inducing exhaustion. Using this model system, we have now optimized a robust, manipulatable, and reproducible in vitro human model of T cell exhaustion to evaluate approaches to prevent or reverse T cell exhaustion.
Results
[0116] We engineered a GD2.28z CAR fused to an FKBP12 mutant destabilization domain (Banaszynski et. al, Cell 2006) (GD2.28z.FKBP) which confers its instability to the CAR and induces rapid protein degradation. We observed that surface expression could be rapidly and dose-dependently regulated by adding or subtracting the stabilizing rapalog shield-1 (S1) in culture medium (FIG. 1). Similar regulatability of CAR expression was also accomplished using an E. coli DHFR mutant (GD2.28z.DHFR, not shown), which could be regulated by trimethoprim, an antibiotic that is commonly used clinically.
[0117] Since tonic signaling is highly dependent upon GD2-CAR receptor levels, precise control of CAR expression levels also precisely regulates levels of tonic signaling. Drug regulated control of levels of CAR expression therefore also allowed modulation of the duration and intensity of GD2.28z tonic signaling. Using this system, we demonstrated that phenotypic and functional changes associated with exhaustion were reversed upon cessation of CAR signaling. As shown in FIG. 2, removal of S1 drug from the culture medium and consequent removal of surface CAR on day 7 post-activation reverses canonical exhaustion marker expression to control levels by day 10 (FIG. 2, n=3). This is most well illustrated by measuring levels of PD-1/TIM-3/LAG-3 triple expressing cell which is highly specific for dysfunctional, exhausted T cells. We demonstrate that Day 10 clear induces increases in levels of triple expressing exhausted cells, but that removal of S1 on Day 7 results in normalization of these levels by Day 10. Similar results were obtained on day 14 for cells in which S1 was removed from culture medium on day 7 or day 10 (not shown).
[0118] Additionally, removal of S1 on day 7 or 10, allow transient degradation of CAR proteins results in maintenance of memory markers (ex. CD62L) and prevention of apoptosis (i.e., annexin V staining) by day 14 compared to T cells that received S1 for the entire duration of the culture (S1) (FIG. 3).
[0119] Because phenotypic markers may not be entirely predictive of T cell function, we also performed functional experiments on CAR T cells provided transient drug exposure in culture. CAR T cells were washed, resuspended in media containing S1, and mixed at a 1:1 ratio with Nalm6 leukemic cells stably expressing surface GD2. Culture supernatants were harvested approximately 24 hours later and cytokine levels were evaluated via ELISA. Similar to GD2.28z CAR that lacks a destabilization domain and therefore have persistent high levels of CAR signaling, cells expressing the GD2.28z.FKBP CAR that experienced continuous drug treatment (FIG. 4, grey bars) secreted minimal amounts of IL-2 on both day 10 and day 14 post-activation, consistent with T cell exhaustion. Alternatively, CART cells that were not exposed to drug during culture (black bars) and therefore did not experience tonic signaling demonstrated significant bioactivity as measured by IL-2 production. Finally, CAR T cells that were exposed to drug during the initial 7 or 10 days of culture and therefore acquired phenotypic and functional evidence of T cell exhaustion, but had drug removed from the culture medium on day 7 or day 10 (blue and red bars, respectively) displayed a restored capacity to secrete IL-2 in response to tumor antigen. Remarkably, exhausted T cells on day 10 (grey bar, day 10 ELISA) could be reinvigorated by removing S1 from the culture medium and "rested" for only 4 days (red bar, day 14 ELISA). Similar, but less dramatic augmentation of IFN.gamma. secretion in conditions in which S1 was removed from culture medium was also observed. These functional data cannot be attributed to differential CAR surface expression, as all groups exhibited similar levels of surface CAR at the conclusion of this co-culture assay (not shown).
[0120] We then compared whether prevention or reversal of T cell exhaustion by removal of surface CAR was more or less potent than treatment with well-characterized anti-PD-1 checkpoint inhibitor, nivolumab (Nivo). CART cells were either treated with continuous S1 (and thus exhibit continuous tonic signaling), continuous S1+nivolumab, or no S1 until the time of the co-culture assay. Interestingly, nivolumab treatment resulted in only modest augmentation of IL-2 secretion at day 10, which was sustained until day 14, suggesting that nivolumab only partially prevented the onset of T cell exhaustion in this system (FIG. 5). Conversely, culturing CART cells without S1, then adding it back to the medium just prior to the co-culture assay (left chart, blue bars), resulted in a far superior prevention of exhaustion, as IL-2 secretion was augmented 5-10 fold compared to CART cells that experienced continuous S1 (black bars). Further, removing tonic signaling on day 7 by removing S1 from the culture medium also resulted in superior IL-2 secretion compared to CAR T cells that experienced continuous S1, and those that experienced continuous S1 and were simultaneously treated with S1. Collectively, these data demonstrate that modulating tonic signaling exhibits more potent effects on prevention or reversal of exhaustion compared to PD-1 blockade.
[0121] Functional studies by several groups, including our lab have verified that co-expression of PD-1, TIM-3, and LAG-3 (triple positive, TP) denotes an exhausted cell subset that is highly dysfunctional. We thus sought to analyze whether cessation of tonic signaling in this cell subset could reverse their phenotype and restore their ability to secrete IL-2 in response to tumor antigen. A high affinity version of our GD2.28z CAR (HA-GD2.28z), which exhibits an even more dramatic exhausted phenotype, was fused to the FKBP12 mutant destabilization domain in order to control its surface expression. On day 10 post-activation, HA-GD2.28z.FKBP CAR T cells that had experienced continuous S1 treatment were sorted in order to isolate a pure PD-1/TIM-3/LAG-3 exhausted population. "Triple positive" exhausted cells were then re-cultured either with or without S1 to test whether removal of tonic signaling could restore their function. FACS and co-culture assays were conducted 4 days later.
[0122] Removal of S1 resulted in a dramatic reversal of the exhausted phenotype. After only 4 days without S1 in the medium, pre-sorted triple positive cells exhibited far less expression of exhaustion markers in both CD4+ and CD8+ CAR T cells (FIG. 6). Importantly, these phenotypic changes also conferred functional augmentation in IL-2 secretion, as removal of S1 resulted in a 2-fold increase in IL-2 secretion compared to triple positive cells that received continuous S1 treatment from days 10-14 (FIG. 6).
[0123] We hypothesized that we could recapitulate the effects of removing surface CAR, and thus tonic signaling, by simply inhibiting kinases in the TCR signaling pathway that are also integral to CAR signaling. One such kinase is Lck, which acts to phosphorylate CD3 zeta in response to TCR or CAR ligation. Dasatinib, a potent receptor tyrosine kinase inhibitor and BCR/ABL antagonist, has also been shown to inhibit T cell activation, proliferation, and cytokine secretion by binding to and inhibiting Lck at low concentrations (Schade et. al, Blood, 2008 and Lee et. al, Leukemia, 2010).
[0124] At 100 nM and 1 .mu.M concentrations, dasatinib potently inhibits CD19.28z CART cell cytokine secretion in response to tumor antigen on day 14 post-activation (FIG. 7), proving that dasatinib disrupts CAR signaling.
[0125] We then asked whether transient dasatinib exposure could reverse T cell exhaustion by treating HA-GD2.28z CART cells with dasatinib on days 10-14 post-activation. Cells were treated with dasatinib for 4 days, then drug was extensively washed from the media, and cells were re-cultured for an additional 24 hours before examining their phenotype and function via FACS and tumor co-culture assays. Interestingly, 4-day treatment with dasatinib reversed exhaustion marker expression and co-expression in a dose-dependent manner (FIG. 8).
[0126] Furthermore, dasatinib treatment resulted in preservation of T cell memory via maintenance of CD62L expression in a dose-dependent manner (FIG. 9.).
[0127] Finally, similar to removal of surface CAR, dasatinib treatment reinvigorated exhausted T cells in a functionally significant manner, as dasatinib-treated CAR T cells secreted more IL-2 (and to a lesser extent, IFN.gamma.) in response to tumor antigen compared to those that never received dasatinib (FIG. 10).
[0128] Collectively, these data demonstrate that selective inhibition or modulation of TCR signaling can substantially enhance the function of exhausted T cells that experience continuous antigen exposure in the context of cancer or chronic infection. In future studies, we will conduct in vivo studies to assess the feasibility of exhaustion reversal in this setting and whether such reversal can enhance antitumor effects in murine models.
Example II
[0129] Chimeric antigen receptors (CARs) are synthetic receptors that combine an extracellular tumor-targeting domain with intracellular domains that mimic endogenous TCR signaling (e.g., 1-2 costimulatory domains, like CD28 or 4-1BB, and a CD3 zeta domain) (see, e.g., Lim & June. Cell 168, 724-740 (2017)). When CAR-expressing T cells encounter antigen-expressing tumor cells, CAR T cells form an immune synapse and initiate downstream signaling through the CAR, resulting in potent T cell activation, degranulation of cytotoxic soluble factors, cytokine release, and proliferation. While CAR T cell therapy has experienced unprecedented clinical success in many patients with hematological malignancies, there are several key challenges that must be addressed before this therapy can be expanded to other tumor types or offered as first-line therapy.
[0130] One challenge is CAR toxicity, which typically manifests in the form of cytokine release syndrome (CRS) or on-target off-tumor activity, both of which have been observed in clinical trials and, in some instances, resulted in patient death (see, e.g., Gust et al. Cancer discovery (2017). doi:10.1158/2159-8290.cd-17-0698; Xu & Tang. Cancer Letters 343, 172-178 (2014); D'Aloia, et al. Cell Death & Disease 9, 282 (2018)). Current methods to counteract CAR toxicity are largely limited to a drug-inducible suicide switches (i.e., inducible Caspase 9) that mediate CAR T cell apoptosis (see, e.g., Gargett & Brown. Frontiers in Pharmacology 5, 235 (2014)). While generally regarded as an effective safety mechanism, utilizing this method eliminates the option to continue therapy after the toxicity event resolves, as the CAR T cells are no longer viable.
[0131] A second key challenge to improving the efficacy of CAR T cell therapy is the prevention of CAR T cell exhaustion. T cell exhaustion results from continuous antigen exposure in the context of chronic viral infection or cancer and is characterized by a hierarchical loss of effector function, sustained co-expression of multiple inhibitory receptors (ex., PD-1, TIM-3, LAG-3), attenuated proliferative capacity, and increase apoptosis (see, e.g., Wherry & Kurachi. Nature Reviews Immunology 15, nri3862 (2015)). There is strong evidence for T cell exhaustion in CART cell therapy. Nearly all CD19.28z CART cells administered disappear by day 60 post-infusion (see, e.g., Lee et al. Long-term outcomes following CD19 CART cell therapy for B-ALL are superior in patients receiving a fludarabine/cyclophosphamide preparative regimen and post-CAR hematopoietic stem cell transplantation. (2016)). CD19.BBz CART cells, which are thought to be more resistant to T cell exhaustion, also exhibit features of exhaustion and are undetectable in approximately 30% of patients who receive this therapy, consequently increasing the risk of CD19 positive relapse (see, e.g., Turtle et al. Journal of Clinical Investigation 126, 2123-2138 (2016); Maude et al. The New England Journal of Medicine 371, 1507-1517 (2014)). Lastly, exhaustion has also been observed in T cells constitutively expressing CARs that manifest scFv aggregation-induced tonic signaling, which occurs both in the absence of tumor antigen. This unintended consequence of high CAR expression ultimately limits their effectiveness by exhausting CART cells in vitro and in vivo (see, e.g., Long et al. Nature Medicine 21, 581-590 (2015); Gomes-Silva et al. Cell Reports 21, 17-26 (2017)).
[0132] Experiments conducted during the course of developing embodiments for the present invention addressed both of these challenges by utilizing FDA-approved small molecule tyrosine kinase inhibitors to modulate CAR T cell activity. Several BCR-Abl inhibitors have been shown to have cross-reactivity with signaling kinases required for T cell activation (see, e.g., Banaszynski, et al. Nature medicine 14, 1123-7 (2008); Banaszynski, et al. Cell 126, 995-1004 (2006); Iwamoto, et al. Chemistry & Biology 17, 981-988 (2010)). Dasatinib potently inhibits T cell activation and effector functions by inhibiting both Lck and Fyn. Similarly, ponatinib can bind to and inhibit Lck, but does not affect the function of Fyn or Src kinases, suggesting that this drug can also inhibit T cell effector function, and likewise, CAR T cell function.
[0133] To test this hypothesis, T cells expressing the CD19-targeting CAR (CD19.28z) were incubated with various concentrations of dasatinib and ponatinib for at least 24 hours. CAR T cells were next co-cultured with antigen-bearing tumor cells for 6 hours in the presence or absence of dasatinib/ponatinib, and subsequently assessed CAR T cell activation and degranulation via CD69 and CD107a co-expression. Nearly 80% of control CART cells were CD69+/CD107a+ upon co-culture with tumor (FIG. 11a). Conversely, nanomolar levels of dasatinib and ponatinib potently inhibited activation and degranulation of CAR T cells in a dose-dependent manner (FIG. 11b). These drugs also potently inhibited CAR T cell IL-2 and IFN.gamma. secretion in response to tumor (FIG. 12). Finally, we observed potent inhibition of CAR T cell cytotoxicity when CAR T cells were co-cultured with tumor cells in the presence of 1 uM dasatinib (FIG. 13).
[0134] To assess whether dasatinib was inhibiting CAR T cell effector function by disrupting CAR signaling, experiments were conducted in which surface CAR were transiently crosslinked on CAR T cells in order to transiently initiate CAR downstream signaling. Under control conditions, cross-linking CAR for 5 minutes induced phosphorylation of the CAR CD3 zeta domain, as well as phosphorylation of distal signaling kinases Akt and Erk1/2 (FIG. 14). Conversely, when CAR T cells were crosslinked in the presence of dasatinib, they resembled non-crosslinked controls, indicating that dasatinib potently disrupted CAR-specific intracellular signaling. Collectively, these experiments indicate that both dasatinib and ponatinib inhibit CAR T cell activity and provide indirect evidence that Lck and/or Fyn are critical for CAR signaling. These experiments also indicate that dasatinib or ponatinib could be utilized clinically to disrupt CAR T cell activity in order to mitigate CAR T cell toxicity.
[0135] As previously mentioned, many constitutively expressed CARs exhibit tonic signaling in the absence of antigen during in vitro expansion, consequently driving them towards T cell exhaustion (see, e.g., Long et al. Nature Medicine 21, 581-590 (2015)). Additional experiments hypothesized that expanding CART cells in the presence of dasatinib or ponatinib would alleviate tonic signaling and consequently yield a healthier, more potent CAR T cell. To test this, T cells expressing a tonically signaling, high-affinity GD2.28z CAR (HA-GD2.28z) were expanded in the presence of various concentrations of dasatinib. Cells were then removed from culture for phenotypic analysis via FACS. Control HA-GD2.28z CART cells exhibited robust single marker expression and co-expression of multiple canonical exhaustion markers (FIG. 15). Conversely, expansion in dasatinib reduced both the frequency of exhaustion-marker co-expressing cells as well as the extent to which these exhaustion markers were expressed in a dose-dependent manner (FIG. 15). CART cell expansion in the presence of dasatinib also augmented T cell memory formation, as a nearly 6-fold increase in the frequency of central-memory-like T cells (CD45RA low, CCR7 high) and a greater-than 2-fold reduction in the frequency of effector-memory-like T cells (CD45RA low, CCR7 low) was observed compared to exhausted CAR T cells cultured in the absence of dasatinib.
[0136] Experiments next hypothesized that the dramatic phenotypic changes observed when tonic signaling was mitigated by dasatinib or ponatinib may coincide with an augmentation in T cell function. To test this, experiments first expanded tonically signaling CAR T cells in the presence or absence of various concentrations of dasatinib or ponatinib. Experiments next removed the drugs from culture in order to allow CAR T cells to regain the capacity to function. 18-24 hours after removal of drug, the differentially expanded CAR T cells were co-cultured with antigen-bearing tumor cells for 24 hours and subsequently assessed cytokine release via ELISA, or co-cultured for 72 hours and assessed cytotoxicity via incucyte assay. Tonically signaling CAR T cells cultured in the absence of dasatinib or ponatinib secreted low levels of cytokine in response to tumor (FIG. 17) and exhibited impaired cytotoxicity (FIG. 18), indicating that these cells were functionally exhausted. Alternatively, expansion of CAR T cells in the presence of dasatinib or ponatinib dose-dependently augmented CAR T cell cytokine secretion (FIG. 17) and also allowed for a more potent cytotoxic response (FIG. 18), confirming that the mitigation of tonic signaling during CAR T cell expansion with these drugs confers profound functional benefits.
[0137] Experiments were conducted that tested whether prevention of CAR tonic signaling in vitro augmented the anti-tumor response in vivo. CAR T cells were expanded with or without 1 uM dasatinib and subsequently infused into NSG mice engrafted with antigen-bearing tumor. In a solid tumor model using the 143B osteosarcoma cell line, both GD2.BBz and HA-GD2.28z CAR T cells grown in the absence of dasatinib failed to control tumor growth (FIGS. 19 and 20, respectively). However, CAR T cells expanded in dasatinib allowed for a near complete and lasting eradication of the tumor (FIGS. 19 and 20). The same effect was observed in a GD2-overexpressing NALM6 leukemia model in which the tumor burden was more established at the time of CART cell infusion (FIG. 21).
[0138] Experiments were next conducted to determine whether greater CAR T cell proliferation and/or persistence were a few of the key mechanisms by which in vitro expansion in dasatinib augments the anti-tumor response. To test this, CAR T cells were infused into mice that had been engrafted with antigen-bearing tumor. On day 14 post-infusion, blood samples were taken from the mice and the number of circulating CAR T cells was assessed via FACS counting beads. Tonically signaling HA-GD2.28z CAR T cells expanded in the absence of dasatinib did not expand and/or persist at levels greater than mice infused with mock untransduced T cells (FIG. 22). Conversely, both CD4+ and CD8+ CAR T cells that were grown in the presence of 1 uM dasatinib underwent a profound expansion in vivo and persisted (FIG. 22). Collectively, these data indicate that limiting CAR tonic signaling by expanding CAR T cells in dasatinib in vitro augments the in vivo anti-tumor response by increasing the capacity for CAR T cells to expand and persist.
[0139] Experiments were next conducted that questioned whether in vivo administration of dasatinib could alter CAR T cell phenotype and function. As a proof-of-concept experiment, CAR T cells were infused into mice that were engrafted with antigen-bearing tumor and subsequently dosed with dasatinib for 3 consecutive days. The mice were then sacrificed and CAR T cell frequency and phenotype assessed in both the blood and the spleen. The dasatinib-treated mouse exhibited a higher frequency of CAR T cells in both tissues (FIG. 22a, c) compared to the vehicle-treated mouse, indicating that in vivo dasatinib treatment induced in situ proliferation or persistence. Furthermore, CAR T cells that were recovered from the dasatinib-treated mouse exhibited reduced expression of exhaustion markers PD-1 and LAG-3, reduced expression of CD69 (i.e. lower activation state), and higher expression of the memory marker CD62L (FIG. 22b, d) compared to the vehicle-treated mouse, all of which are consistent with the phenotypic changes observed upon in vitro treatment with dasatinib (FIG. 15, 16). These results demonstrate that in vivo administration of dasatinib mitigates CAR T cell exhaustion phenotype and improves memory formation, and indicates that in vivo dasatinib dosing provides a functional benefit in vivo.
[0140] Experiments hypothesized that selective in vivo administration of dasatinib will prevent CAR T cell exhaustion by transiently "resting" CAR T cells that are experiencing chronic antigen stimulation. To test this, CAR T cells were infused into mice that exhibited high tumor burden. On day 27 post-tumor engraftment, the mice were dosed with dasatinib or vehicle for 3 consecutive days, then were not treated for a period of 3 additional days. Subsequent to this treatment regimen, tumor burden was assessed and a profound decrease in tumor size in the dasatinib-treated mouse was observed (FIG. 22e). Conversely, the tumor burden in vehicle-treated mouse continued to increase, indicating that the augmentation in anti-tumor response was specific to dasatinib treatment.
[0141] In summary, dasatinib and ponatinib have the potential to address several important challenges currently facing the field of adoptive T cell therapies (e.g., CART cell therapies). First, these drugs were shown potently inhibit CAR signaling, which provides a method to regulate CAR activity and thus mitigate CAR T cell toxicity while preserving the option to continue therapy once the toxicity has resolved, as the inhibitory effect of dasatinib and ponatinib on CAR T cell function is reversible. Second, expansion of CAR T cells in the presence of dasatinib or ponatinib was shown to prevent CAR tonic signaling and in turn enhance the functional capacity of CAR T cells. Lastly, providing short periods of CAR T cell "rest" in vivo via iterative drug dosing was shown to be one method by which CAR T cell exhaustion could be prevented or reversed and/or memory could be induced.
Example III
[0142] This example describes the materials and methods for Example II.
Cells and Culture Conditions
[0143] NALM6-GL (acute lymphoblastic leukemia line, stably transfected with GFP and luciferase) and NALM6-GL-GD2 (stably transfected to overexpress GD2 synthetase) cell lines were cultured in RPMI-1640. 293T and 143B cell lines were cultured in DMEM (Life Technologies). DMEM and RPMI-1640 were supplemented with 10% heat-inactivated FBS (Gibco, Life Technologies), 10 mM HEPES, 100 U/mL penicillin, 100 .mu.g/ml streptomycin and 2 mM L-glutamine (Gibco, Life Technologies).
[0144] Primary human T cells were obtained from healthy donor buffy coats using a Pan T cell negative selection kit (Miltenyi Biotec). Donor T cells were then aliquoted and stored in Cryostor (StemCell Technologies) in liquid nitrogen. T cells were cultured in AimV (Gibco, Life Technologies) supplemented with 5% heat-inactivated FBS, 10 mM HEPES, 1% glutamax (Gibco, Life Technologies), and 100 u/uL recombinant human IL-2 (Peprotech). Dasatinib (Sigma Aldrich and Adooq Biosciences) or ponatinib (SelleckChem) were cultured at 1 uM unless otherwise specified.
Retroviral Production and T Cell Transduction
[0145] All retroviral supernatants were produced via transient transfection of the 293GP cell line. Briefly, 293GP cells were transfected via Lipofectamine 2000 (Life Technologies) with the plasmids encoding the CARs and RD114 envelope protein. Supernatants were collected at 48 and 72 hours post-transfection, aliquoted and stored at -80 C.
[0146] Upon thawing, T cells were activated at a 3:1 bead:cell ratio using anti-CD3/anti-CD28-coated magnetic beads (Dynabeads, Thermo Fisher) at a concentration of 1.times.10.sup.6 cells/mL. On days 2 and 3 post-activation, T cells were transduced with retrovirus encoding the CAR. Briefly, retrovirus was first spun onto retronectin-coated plates at 3000 rpm for 2 hours, after which T cells were transferred to the plates. On day 4 post-activation, magnetic beads were removed from culture, and T cells were cultured at 0.5.times.10.sup.6 cells/mL every day thereafter. Media supplemented with IL-2 and drug was changed every two days. Transduction efficiencies were routinely 70-90% for all CARs.
Flow Cytometry
[0147] All samples were analyzed with an LSR Fortessa (BD Bioscience) or a Cytoflex (Beckman Coulter) and data were analyzed using FlowJo. Cells were washed twice with PBS and labelled with stained at 1.times.10.sup.6 cells/mL in PBS, followed by two washes with FACS buffer (PBS supplemented with 2% FBS and 0.4% 0.5M EDTA). GD2 CARs were detected with the 14g2a anti-idiotype antibody 1A7. CD19 CARs were detected with the FMC63 anti-idiotype antibody 136.20.1. T cell phenotype was evaluated via: CD4 (OKT4, Biolegend), CD8 (SK1, Biolegend), PD-1 (eBioJ105, eBioscience), TIM-3 (F38-2E2, Biolegend), LAG-3 (3DS223H, eBioscience), CD45RA (L48, BD Biosciences), CCR7 (150503, BD Biosciences), CD62L (DREG-56, BD Biosciences), CD69 (FN50, Biolegend), and CD107a (H4A3, eBioscience). For co-culture assays in which CD107a was assessed, tumor cells and CAR T cells were co-cultured in the presence of 1:1000 monensin (eBioscience) and anti-CD107a for at least 6 hours. All FACS plots displaying CART cell phenotype data were pre-gated on CAR+ cells. For mock-transduced T cells, whole T cell populations were used for analysis.
Incucyte Assay
[0148] 50,000 NALM6-GL or NALM6-GL-GD2 tumor cells were co-cultured with T cells at a 1:8 E:T ratio in 200 uL of complete AimV medium without IL-2 supplementation in each well of a 96-well plate. Plates were loaded into the incucyte and 488 nm fluorescent images were acquired every 2 hours for 48-72 hours. GFP+ tumor cells were identified by size and fluorescence intensity masks, and the total integrated GFP intensity of all counted tumor cells was quantified for each individual well. Values were normalized to t=0, and replicate wells were averaged for data display.
[0149] For experiments in which HA-GD2.28z T cells were expanded in the presence of dasatinib, in some instances, drug was removed from the media 18-24 hours prior to the assay to allow CAR T cells to function in the presence of tumor antigen.
Cytokine Release Assay
[0150] 50,000 NALM6-GL-GD2 tumor cells were co-cultured with T cells at a 1:1 E:T ratio in 200 uL of complete AimV medium without IL-2 supplementation in each well of a 96-well plate. After 24 hours, supernatants were removed and stored at -20 C. IL-2 and IFN.gamma. secretion was assessed via ELISA (Biolegend).
[0151] For experiments in which HA-GD2.28z T cells were expanded in the presence of dasatinib, in some instances, drug was removed from the media 18-24 hours prior to the assay to allow CAR T cells to function in the presence of tumor antigen.
Western Blot
[0152] 2.times.10.sup.6 CAR' cells were removed from culture, pelleted, and resuspended in 100 uL of RITA lysis buffer (10 mM Tris-Cl pH 8.0, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl) supplemented with phosphatase and protease inhibitors (Thermo Fisher), After incubating for 30 minutes at 4 C, supernatants were cleared by centrifugation at 14,000 RPM for 20 minutes at 4 C. Protein concentration in the cleared lysates was measured by a colorimetric reaction (BioRad).
[0153] 1.5 ug of protein lysate was mixed with 6.times. loading buffer and loaded onto 10% SDS-PAGE gels assembled into a mini-protean electrophoresis systems (BioRad). Electrophoresis was performed in tris-glycine-SDS buffer (BioRad) at 100V for 20 minutes and later increased to 150V for 50 minutes. Protein transfer into Immbilon-FL PVDF membranes was performed at 100V for 1 hour in tris-glycine buffer (BioRad #1610771). Primary antibodies targeting CD3-zeta (Cell signaling), pY142-CD3-zeta (Cell Signaling), p44/42 MAPK (Erk1/2, Cell Signaling), p-p44/42 MAPK (p-ERK1/2, Cell Signaling), pSer473-Akt (D9E, Cell Signaling), and pan Akt (40D4, Cell Signaling) were used. The Odyssey (LI-COR) imaging system. LI-COR buffers, and LI-COR secondary antibodies (Goat Anti-Mouse IgG Antibody-800CW-Conjugated and Goat Anti-Rabbit IgG Antibody-680LT-Conjugated) were used for protein detection.
[0154] For CAR crosslinking, CAR T cells were incubated in 5 ug/mL anti-idiotype (clone 1A7) plus 5 ug/mL goat anti-mouse Fab secondary (Jackson Immunoresearch) or secondary alone for 5 minutes at 37 C. Cells were then quenched in ice cold PBS, pelleted at 4 C for 5 minutes, then lysed for western blot analysis.
[0155] In vivo experiments 6-8 week old NSG mice were engrafted with 1.times.10.sup.6 NALM6-GL-GD2 leukemia cells via intravenous injection. At day 4 post-engraftment, 2.times.10.sup.6 HA-GD2.28z CAR+ T cells were infused intravenously. NALM6-GL-GD2 tumor burden was evaluated using the Xenogen IVIS Lumina (Caliper Life Sciences). Mice were first injected intraperitoneally with 3 mg D-luciferin (Caliper Life Sciences) and then imaged 4 minutes later with an exposure time of 30 seconds, or, in cases where 30 seconds resulted in signal saturation, "auto" exposure was selected. Luminescence images were analyzed using Living Image software (Caliper Life Sciences).
[0156] 6-8 week old NSG mice were engrafted with 0.5.times.10.sup.6 143 B osteosarcoma cells intramuscularly. On day 3 post-engraftment, 10.times.10.sup.6 GD2.BBz or HA-GD2.28z were infused intravenously. Osteosarcoma burden was quantified via two-dimensional leg area measurements.
[0157] Mice treated with dasatinib (Adooq Biosciences) were injected intraperitoneally at a concentration of 50 mg/kg in water+10% Kolliphor HS 15 (Sigma Aldrich). Mice treated with vehicle were injected with an equivalent volume of water+10% Kolliphor HS 15.
[0158] Blood samples were taken via retro-orbital bleed and briefly stored in EDTA-coated microvettes (Kent Scientific). Spleens were mechanically disaggregated by passage through a 70-.mu.m filter (BD Biosciences). Both blood and spleen were lysed in ACK lysis buffer (Fisher Scientific) for 5 minutes and subsequently stained with surface marker antibodies for FACS analysis.
Construction of CAR Vectors
[0159] All CAR sequences were inserted into the MSGV retroviral backbone. Each CAR includes a signal peptide, single chain variable fragment (scFv), extracellular hinge region, transmembrane domain, intracellular co-stimulatory domain, and intracellular CD3 zeta domain.
Sequences
[0160] The nucleic acid and amino acid sequence for CD19.28z (FMC63 scFv) is provided at FIG. 23.
[0161] The nucleic acid and amino acid sequence for CD19.BBz (FMC63 scFv) is provided at FIG. 24.
[0162] The nucleic acid and amino acid sequence for GD2.BBz (14G2a scFv) is provided at FIG. 25.
[0163] The nucleic acid and amino acid sequence for HA-GD2.28z (High affinity 14G2a scFv) is provided at FIG. 26.
[0164] Having now fully described the invention, it will be understood by those of skill in the art that the same can be performed within a wide and equivalent range of conditions, formulations, and other parameters without affecting the scope of the invention or any embodiment thereof. All patents, patent applications and publications cited herein are fully incorporated by reference herein in their entirety.
INCORPORATION BY REFERENCE
[0165] The entire disclosure of each of the patent documents and scientific articles referred to herein is incorporated by reference for all purposes.
EQUIVALENTS
[0166] The invention may be embodied in other specific forms without departing from the spirit or essential characteristics thereof. The foregoing embodiments are therefore to be considered in all respects illustrative rather than limiting the invention described herein. Scope of the invention is thus indicated by the appended claims rather than by the foregoing description, and all changes that come within the meaning and range of equivalency of the claims are intended to be embraced therein.
Sequence CWU 1
1
811794DNAArtificial Sequencesynthetic 1atgacaagag ttactaacag
cccctctctc caagctcact tacaggctct ctacttagtc 60cagcacgaag tctggagacc
tctggcggca gcctaccaag aacaactgga ccgaccggtg 120gtacctcacc
cttaccgagt cggcgacaca gtgtgggtcc gccgacacca gactaagaac
180ctagaacctc gctggaaagg accttacaca gtcctgctga ccacccccac
cgccctcaaa 240gtagacggca tcgcagcttg gatacacgcc gcccacgtga
aggctgccga ccccgggggt 300ggaccatcct ctagactgct cgagatgctt
ctcctggtga caagccttct gctctgtgag 360ttaccacacc cagcattcct
cctgatccca gacatccaga tgacacagac tacatcctcc 420ctgtctgcct
ctctgggaga cagagtcacc atcagttgca gggcaagtca ggacattagt
480aaatatttaa attggtatca gcagaaacca gatggaactg ttaaactcct
gatctaccat 540acatcaagat tacactcagg agtcccatca aggttcagtg
gcagtgggtc tggaacagat 600tattctctca ccattagcaa cctggagcaa
gaagatattg ccacttactt ttgccaacag 660ggtaatacgc ttccgtacac
gttcggaggg gggactaagt tggaaataac aggctccacc 720tctggatccg
gcaagcccgg atctggcgag ggatccacca agggcgaggt gaaactgcag
780gagtcaggac ctggcctggt ggcgccctca cagagcctgt ccgtcacatg
cactgtctca 840ggggtctcat tacccgacta tggtgtaagc tggattcgcc
agcctccacg aaagggtctg 900gagtggctgg gagtaatatg gggtagtgaa
accacatact ataattcagc tctcaaatcc 960agactgacca tcatcaagga
caactccaag agccaagttt tcttaaaaat gaacagtctg 1020caaactgatg
acacagccat ttactactgt gccaaacatt attactacgg tggtagctat
1080gctatggact actggggtca aggaacctca gtcaccgtct cctcagcggc
cgcaattgaa 1140gttatgtatc ctcctcctta cctagacaat gagaagagca
atggaaccat tatccatgtg 1200aaagggaaac acctttgtcc aagtccccta
tttcccggac cttctaagcc cttttgggtg 1260ctggtggtgg ttgggggagt
cctggcttgc tatagcttgc tagtaacagt ggcctttatt 1320attttctggg
tgaggagtaa gaggagcagg ctcctgcaca gtgactacat gaacatgact
1380ccccgccgcc ccgggcccac ccgcaagcat taccagccct atgccccacc
acgcgacttc 1440gcagcctatc gctccagagt gaagttcagc aggagcgcag
acgcccccgc gtaccagcag 1500ggccagaacc agctctataa cgagctcaat
ctaggacgaa gagaggagta cgatgttttg 1560gacaagagac gtggccggga
ccctgagatg gggggaaagc cgagaaggaa gaaccctcag 1620gaaggcctgt
acaatgaact gcagaaagat aagatggcgg aggcctacag tgagattggg
1680atgaaaggcg agcgccggag gggcaagggg cacgatggcc tttaccaggg
tctcagtaca 1740gccaccaagg acacctacga cgcccttcac atgcaggccc
tgccccctcg ctaa 17942597PRTArtificial Sequencesynthetic 2Met Thr
Arg Val Thr Asn Ser Pro Ser Leu Gln Ala His Leu Gln Ala1 5 10 15Leu
Tyr Leu Val Gln His Glu Val Trp Arg Pro Leu Ala Ala Ala Tyr 20 25
30Gln Glu Gln Leu Asp Arg Pro Val Val Pro His Pro Tyr Arg Val Gly
35 40 45Asp Thr Val Trp Val Arg Arg His Gln Thr Lys Asn Leu Glu Pro
Arg 50 55 60Trp Lys Gly Pro Tyr Thr Val Leu Leu Thr Thr Pro Thr Ala
Leu Lys65 70 75 80Val Asp Gly Ile Ala Ala Trp Ile His Ala Ala His
Val Lys Ala Ala 85 90 95Asp Pro Gly Gly Gly Pro Ser Ser Arg Leu Leu
Glu Met Leu Leu Leu 100 105 110Val Thr Ser Leu Leu Leu Cys Glu Leu
Pro His Pro Ala Phe Leu Leu 115 120 125Ile Pro Asp Ile Gln Met Thr
Gln Thr Thr Ser Ser Leu Ser Ala Ser 130 135 140Leu Gly Asp Arg Val
Thr Ile Ser Cys Arg Ala Ser Gln Asp Ile Ser145 150 155 160Lys Tyr
Leu Asn Trp Tyr Gln Gln Lys Pro Asp Gly Thr Val Lys Leu 165 170
175Leu Ile Tyr His Thr Ser Arg Leu His Ser Gly Val Pro Ser Arg Phe
180 185 190Ser Gly Ser Gly Ser Gly Thr Asp Tyr Ser Leu Thr Ile Ser
Asn Leu 195 200 205Glu Gln Glu Asp Ile Ala Thr Tyr Phe Cys Gln Gln
Gly Asn Thr Leu 210 215 220Pro Tyr Thr Phe Gly Gly Gly Thr Lys Leu
Glu Ile Thr Gly Ser Thr225 230 235 240Ser Gly Ser Gly Lys Pro Gly
Ser Gly Glu Gly Ser Thr Lys Gly Glu 245 250 255Val Lys Leu Gln Glu
Ser Gly Pro Gly Leu Val Ala Pro Ser Gln Ser 260 265 270Leu Ser Val
Thr Cys Thr Val Ser Gly Val Ser Leu Pro Asp Tyr Gly 275 280 285Val
Ser Trp Ile Arg Gln Pro Pro Arg Lys Gly Leu Glu Trp Leu Gly 290 295
300Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr Asn Ser Ala Leu Lys
Ser305 310 315 320Arg Leu Thr Ile Ile Lys Asp Asn Ser Lys Ser Gln
Val Phe Leu Lys 325 330 335Met Asn Ser Leu Gln Thr Asp Asp Thr Ala
Ile Tyr Tyr Cys Ala Lys 340 345 350His Tyr Tyr Tyr Gly Gly Ser Tyr
Ala Met Asp Tyr Trp Gly Gln Gly 355 360 365Thr Ser Val Thr Val Ser
Ser Ala Ala Ala Ile Glu Val Met Tyr Pro 370 375 380Pro Pro Tyr Leu
Asp Asn Glu Lys Ser Asn Gly Thr Ile Ile His Val385 390 395 400Lys
Gly Lys His Leu Cys Pro Ser Pro Leu Phe Pro Gly Pro Ser Lys 405 410
415Pro Phe Trp Val Leu Val Val Val Gly Gly Val Leu Ala Cys Tyr Ser
420 425 430Leu Leu Val Thr Val Ala Phe Ile Ile Phe Trp Val Arg Ser
Lys Arg 435 440 445Ser Arg Leu Leu His Ser Asp Tyr Met Asn Met Thr
Pro Arg Arg Pro 450 455 460Gly Pro Thr Arg Lys His Tyr Gln Pro Tyr
Ala Pro Pro Arg Asp Phe465 470 475 480Ala Ala Tyr Arg Ser Arg Val
Lys Phe Ser Arg Ser Ala Asp Ala Pro 485 490 495Ala Tyr Gln Gln Gly
Gln Asn Gln Leu Tyr Asn Glu Leu Asn Leu Gly 500 505 510Arg Arg Glu
Glu Tyr Asp Val Leu Asp Lys Arg Arg Gly Arg Asp Pro 515 520 525Glu
Met Gly Gly Lys Pro Arg Arg Lys Asn Pro Gln Glu Gly Leu Tyr 530 535
540Asn Glu Leu Gln Lys Asp Lys Met Ala Glu Ala Tyr Ser Glu Ile
Gly545 550 555 560Met Lys Gly Glu Arg Arg Arg Gly Lys Gly His Asp
Gly Leu Tyr Gln 565 570 575Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr
Asp Ala Leu His Met Gln 580 585 590Ala Leu Pro Pro Arg
59531482DNAArtificial Sequencesynthetic 3atgcttctcc tggtgacaag
ccttctgctc tgtgagttac cacacccagc attcctcctg 60atcccagaca tccagatgac
acagactaca tcctccctgt ctgcctctct gggagacaga 120gtcaccatca
gttgcagggc aagtcaggac attagtaaat atttaaattg gtatcagcag
180aaaccagatg gaactgttaa actcctgatc taccatacat caagattaca
ctcaggagtc 240ccatcaaggt tcagtggcag tgggtctgga acagattatt
ctctcaccat tagcaacctg 300gagcaagaag atattgccac ttacttttgc
caacagggta atacgcttcc gtacacgttc 360ggagggggga ctaagttgga
aataacaggc tccacctctg gatccggcaa gcccggatct 420ggcgagggat
ccaccaaggg cgaggtgaaa ctgcaggagt caggacctgg cctggtggcg
480ccctcacaga gcctgtccgt cacatgcact gtctcagggg tctcattacc
cgactatggt 540gtaagctgga ttcgccagcc tccacgaaag ggtctggagt
ggctgggagt aatatggggt 600agtgaaacca catactataa ttcagctctc
aaatccagac tgaccatcat caaggacaac 660tccaagagcc aagttttctt
aaaaatgaac agtctgcaaa ctgatgacac agccatttac 720tactgtgcca
aacattatta ctacggtggt agctatgcta tggactactg gggtcaagga
780acctcagtca ccgtctcctc agcggccgca accacgacgc cagcgccgcg
accaccaaca 840ccggcgccca ccatcgcgtc gcagcccctg tccctgcgcc
cagaggcgtg ccggccagcg 900gcggggggcg cagtgcacac gagggggctg
gacttcgcct gtgatatcta catctgggcg 960cccttggccg ggacttgtgg
ggtccttctc ctgtcactgg ttatcaccct ttactgcaaa 1020cggggcagaa
agaaactcct gtatatattc aaacaaccat ttatgagacc agtacaaact
1080actcaagagg aagatggctg tagctgccga tttccagaag aagaagaagg
aggatgtgaa 1140ctgagagtga agttcagcag gagcgcagac gcccccgcgt
acaagcaggg ccagaaccag 1200ctctataacg agctcaatct aggacgaaga
gaggagtacg atgttttgga caagagacgt 1260ggccgggacc ctgagatggg
gggaaagccg agaaggaaga accctcagga aggcctgtac 1320aatgaactgc
agaaagataa gatggcggag gcctacagtg agattgggat gaaaggcgag
1380cgccggaggg gcaaggggca cgatggcctt taccagggtc tcagtacagc
caccaaggac 1440acctacgacg cccttcacat gcaggccctg ccccctcgct aa
14824493PRTArtificial Sequencesynthetic 4Met Leu Leu Leu Val Thr
Ser Leu Leu Leu Cys Glu Leu Pro His Pro1 5 10 15Ala Phe Leu Leu Ile
Pro Asp Ile Gln Met Thr Gln Thr Thr Ser Ser 20 25 30Leu Ser Ala Ser
Leu Gly Asp Arg Val Thr Ile Ser Cys Arg Ala Ser 35 40 45Gln Asp Ile
Ser Lys Tyr Leu Asn Trp Tyr Gln Gln Lys Pro Asp Gly 50 55 60Thr Val
Lys Leu Leu Ile Tyr His Thr Ser Arg Leu His Ser Gly Val65 70 75
80Pro Ser Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Tyr Ser Leu Thr
85 90 95Ile Ser Asn Leu Glu Gln Glu Asp Ile Ala Thr Tyr Phe Cys Gln
Gln 100 105 110Gly Asn Thr Leu Pro Tyr Thr Phe Gly Gly Gly Thr Lys
Leu Glu Ile 115 120 125Thr Gly Ser Thr Ser Gly Ser Gly Lys Pro Gly
Ser Gly Glu Gly Ser 130 135 140Thr Lys Gly Glu Val Lys Leu Gln Glu
Ser Gly Pro Gly Leu Val Ala145 150 155 160Pro Ser Gln Ser Leu Ser
Val Thr Cys Thr Val Ser Gly Val Ser Leu 165 170 175Pro Asp Tyr Gly
Val Ser Trp Ile Arg Gln Pro Pro Arg Lys Gly Leu 180 185 190Glu Trp
Leu Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr Asn Ser 195 200
205Ala Leu Lys Ser Arg Leu Thr Ile Ile Lys Asp Asn Ser Lys Ser Gln
210 215 220Val Phe Leu Lys Met Asn Ser Leu Gln Thr Asp Asp Thr Ala
Ile Tyr225 230 235 240Tyr Cys Ala Lys His Tyr Tyr Tyr Gly Gly Ser
Tyr Ala Met Asp Tyr 245 250 255Trp Gly Gln Gly Thr Ser Val Thr Val
Ser Ser Ala Ala Ala Thr Thr 260 265 270Thr Pro Ala Pro Arg Pro Pro
Thr Pro Ala Pro Thr Ile Ala Ser Gln 275 280 285Pro Leu Ser Leu Arg
Pro Glu Ala Cys Arg Pro Ala Ala Gly Gly Ala 290 295 300Val His Thr
Arg Gly Leu Asp Phe Ala Cys Asp Ile Tyr Ile Trp Ala305 310 315
320Pro Leu Ala Gly Thr Cys Gly Val Leu Leu Leu Ser Leu Val Ile Thr
325 330 335Leu Tyr Cys Lys Arg Gly Arg Lys Lys Leu Leu Tyr Ile Phe
Lys Gln 340 345 350Pro Phe Met Arg Pro Val Gln Thr Thr Gln Glu Glu
Asp Gly Cys Ser 355 360 365Cys Arg Phe Pro Glu Glu Glu Glu Gly Gly
Cys Glu Leu Arg Val Lys 370 375 380Phe Ser Arg Ser Ala Asp Ala Pro
Ala Tyr Lys Gln Gly Gln Asn Gln385 390 395 400Leu Tyr Asn Glu Leu
Asn Leu Gly Arg Arg Glu Glu Tyr Asp Val Leu 405 410 415Asp Lys Arg
Arg Gly Arg Asp Pro Glu Met Gly Gly Lys Pro Arg Arg 420 425 430Lys
Asn Pro Gln Glu Gly Leu Tyr Asn Glu Leu Gln Lys Asp Lys Met 435 440
445Ala Glu Ala Tyr Ser Glu Ile Gly Met Lys Gly Glu Arg Arg Arg Gly
450 455 460Lys Gly His Asp Gly Leu Tyr Gln Gly Leu Ser Thr Ala Thr
Lys Asp465 470 475 480Thr Tyr Asp Ala Leu His Met Gln Ala Leu Pro
Pro Arg 485 49051479DNAArtificial Sequencesynthetic 5atgctgctgc
tcgtgacatc tctgctgctg tgcgagctgc cccaccccgc ctttctgctg 60atccccgata
tcctgctgac ccagacccct ctgagcctgc ctgtgtctct gggcgatcag
120gccagcatca gctgcagatc cagccagagc ctggtgcacc ggaacggcaa
cacctacctg 180cactggtatc tgcagaagcc cggccagagc cccaagctgc
tgattcacaa ggtgtccaac 240cggttcagcg gcgtgcccga cagattttct
ggcagcggct ccggcaccga cttcaccctg 300aagatcagcc gggtggaagc
cgaggacctg ggcgtgtact tctgcagcca gtccacccac 360gtgccccccc
tgacatttgg cgccggaaca aagctggaac tgaagggcag cacaagcggc
420agcggcaagc ctggatctgg cgagggaagc accaagggcg aagtgaagct
gcagcagagc 480ggcccctctc tggtggaacc tggcgcctct gtgatgatct
cctgcaaggc cagcggcagc 540tccttcaccg gctacaacat gaactgggtg
cgccagaaca tcggcaagag cctggaatgg 600atcggcgcca tcgaccccta
ctacggcggc accagctaca accagaagtt caagggcaga 660gccaccctga
ccgtggacaa gagcagctcc accgcctaca tgcacctgaa gtccctgacc
720agcgaggaca gcgccgtgta ctactgcgtg tccggcatgg aatactgggg
ccagggcaca 780agcgtgaccg tgtcctctgc ggccgcaacc acgacgccag
cgccgcgacc accaacaccg 840gcgcccacca tcgcgtcgca gcccctgtcc
ctgcgcccag aggcgtgccg gccagcggcg 900gggggcgcag tgcacacgag
ggggctggac ttcgcctgtg atatctacat ctgggcgccc 960ttggccggga
cttgtggggt ccttctcctg tcactggtta tcacccttta ctgcaaacgg
1020ggcagaaaga aactcctgta tatattcaaa caaccattta tgagaccagt
acaaactact 1080caagaggaag atggctgtag ctgccgattt ccagaagaag
aagaaggagg atgtgaactg 1140agagtgaagt tcagcaggag cgcagacgcc
cccgcgtaca agcagggcca gaaccagctc 1200tataacgagc tcaatctagg
acgaagagag gagtacgatg ttttggacaa gagacgtggc 1260cgggaccctg
agatgggggg aaagccgaga aggaagaacc ctcaggaagg cctgtacaat
1320gaactgcaga aagataagat ggcggaggcc tacagtgaga ttgggatgaa
aggcgagcgc 1380cggaggggca aggggcacga tggcctttac cagggtctca
gtacagccac caaggacacc 1440tacgacgccc ttcacatgca ggccctgccc
cctcgctaa 14796492PRTArtificial Sequencesynthetic 6Met Leu Leu Leu
Val Thr Ser Leu Leu Leu Cys Glu Leu Pro His Pro1 5 10 15Ala Phe Leu
Leu Ile Pro Asp Ile Leu Leu Thr Gln Thr Pro Leu Ser 20 25 30Leu Pro
Val Ser Leu Gly Asp Gln Ala Ser Ile Ser Cys Arg Ser Ser 35 40 45Gln
Ser Leu Val His Arg Asn Gly Asn Thr Tyr Leu His Trp Tyr Leu 50 55
60Gln Lys Pro Gly Gln Ser Pro Lys Leu Leu Ile His Lys Val Ser Asn65
70 75 80Arg Phe Ser Gly Val Pro Asp Arg Phe Ser Gly Ser Gly Ser Gly
Thr 85 90 95Asp Phe Thr Leu Lys Ile Ser Arg Val Glu Ala Glu Asp Leu
Gly Val 100 105 110Tyr Phe Cys Ser Gln Ser Thr His Val Pro Pro Leu
Thr Phe Gly Ala 115 120 125Gly Thr Lys Leu Glu Leu Lys Gly Ser Thr
Ser Gly Ser Gly Lys Pro 130 135 140Gly Ser Gly Glu Gly Ser Thr Lys
Gly Glu Val Lys Leu Gln Gln Ser145 150 155 160Gly Pro Ser Leu Val
Glu Pro Gly Ala Ser Val Met Ile Ser Cys Lys 165 170 175Ala Ser Gly
Ser Ser Phe Thr Gly Tyr Asn Met Asn Trp Val Arg Gln 180 185 190Asn
Ile Gly Lys Ser Leu Glu Trp Ile Gly Ala Ile Asp Pro Tyr Tyr 195 200
205Gly Gly Thr Ser Tyr Asn Gln Lys Phe Lys Gly Arg Ala Thr Leu Thr
210 215 220Val Asp Lys Ser Ser Ser Thr Ala Tyr Met His Leu Lys Ser
Leu Thr225 230 235 240Ser Glu Asp Ser Ala Val Tyr Tyr Cys Val Ser
Gly Met Glu Tyr Trp 245 250 255Gly Gln Gly Thr Ser Val Thr Val Ser
Ser Ala Ala Ala Thr Thr Thr 260 265 270Pro Ala Pro Arg Pro Pro Thr
Pro Ala Pro Thr Ile Ala Ser Gln Pro 275 280 285Leu Ser Leu Arg Pro
Glu Ala Cys Arg Pro Ala Ala Gly Gly Ala Val 290 295 300His Thr Arg
Gly Leu Asp Phe Ala Cys Asp Ile Tyr Ile Trp Ala Pro305 310 315
320Leu Ala Gly Thr Cys Gly Val Leu Leu Leu Ser Leu Val Ile Thr Leu
325 330 335Tyr Cys Lys Arg Gly Arg Lys Lys Leu Leu Tyr Ile Phe Lys
Gln Pro 340 345 350Phe Met Arg Pro Val Gln Thr Thr Gln Glu Glu Asp
Gly Cys Ser Cys 355 360 365Arg Phe Pro Glu Glu Glu Glu Gly Gly Cys
Glu Leu Arg Val Lys Phe 370 375 380Ser Arg Ser Ala Asp Ala Pro Ala
Tyr Lys Gln Gly Gln Asn Gln Leu385 390 395 400Tyr Asn Glu Leu Asn
Leu Gly Arg Arg Glu Glu Tyr Asp Val Leu Asp 405 410 415Lys Arg Arg
Gly Arg Asp Pro Glu Met Gly Gly Lys Pro Arg Arg Lys 420 425 430Asn
Pro Gln Glu Gly Leu Tyr Asn Glu Leu Gln Lys Asp Lys Met Ala 435 440
445Glu Ala Tyr Ser Glu Ile Gly Met Lys Gly Glu Arg Arg Arg Gly Lys
450 455 460Gly His Asp Gly Leu Tyr Gln Gly Leu Ser Thr Ala Thr Lys
Asp Thr465 470 475 480Tyr Asp Ala Leu His Met Gln Ala Leu Pro Pro
Arg 485 49072217DNAArtificial Sequencesynthetic 7atggaattcg
gcctgagctg gctgttcctg gtggccatcc tgaagggcgt gcagtgcagc 60agagatatcc
tgctgaccca gacccctctg agcctgcctg tgtctctggg cgatcaggcc
120agcatcagct gcagatccag ccagagcctg gtgcaccgga acggcaacac
ctacctgcac 180tggtatctgc agaagcccgg ccagagcccc aagctgctga
tccacaaggt gtccaaccgg 240ttcagcggcg tgcccgacag attttctggc
agcggctccg gcaccgactt caccctgaag 300atcagccggg tggaagccga
ggacctgggc gtgtacttct gcagccagtc cacccacgtg 360ccccccctga
catttggcgc cggaacaaag ctggaactga aggggggagg cggatctggc
420ggcggaggaa gtggcggagg gggatctgaa gtgaagctgc agcagtccgg
ccccagcctg 480gtggaacctg gcgcctctgt gatgatctcc tgcaaggcca
gcggcagctc cttcaccggc 540tacaacatga actgggtgcg ccagaacatc
ggcaagagcc tggaatggat cggcgccatc 600gacccctact acggcggcac
cagctacaac cagaagttca agggcagagc caccctgacc 660gtggacaaga
gcagcagcac cgcctacatg cacctgaagt ccctgaccag cgaggacagc
720gccgtgtact actgcgtgtc cggcatgaag tactggggcc agggcacaag
cgtgaccgtg 780tctagcgcca agaccacccc ccctagcgtg tacggaagag
tgacagtgtc ctctgccgag 840cccaagagct gcgacaagac ccacacctgt
cccccttgtc ctgcccctga gctgctggga 900ggcccttccg tgttcctgtt
ccccccaaag cccaaggaca cactgatgat cagcagaacc 960cccgaagtga
cctgcgtggt ggtggacgtg tcccacgagg acccagaagt gaagttcaat
1020tggtacgtgg acggcgtgga agtgcacaac gccaagacaa agcccagaga
ggaacagtac 1080aacagcacct accgggtggt gtccgtgctg accgtgctgc
atcaggattg gctgaacggc 1140aaagagtaca agtgcaaagt gtccaacaag
gccctgcctg cccccatcga gaaaaccatc 1200agcaaggcca agggccagcc
ccgcgaaccc caggtgtaca cactgccccc tagcagggac 1260gagctgacca
agaaccaggt gtccctgaca tgcctcgtga agggcttcta cccctccgat
1320atcgccgtgg aatgggagag caacggccag cccgagaaca actacaagac
aacccctccc 1380gtgctggaca gcgacggctc attcttcctg tacagcaagc
tgacagtgga taagtcccgg 1440tggcagcagg gcaacgtgtt cagctgctcc
gtgatgcacg aggccctgca caaccactac 1500acccagaaaa gcctgtccct
gagccccggc aagaaggacc ccaaagctag cttcgaaatt 1560gaagttatgt
atcctcctcc ttacctagac aatgagaaga gcaatggaac cattatccat
1620gtgaaaggga aacacctttg tccaagtccc ctatttcccg gaccttctaa
gcccttttgg 1680gtgctggtgg tggttggggg agtcctggct tgctatagct
tgctagtaac agtggccttt 1740attattttct gggtgaggag taagaggagc
aggctcctgc acagtgacta catgaacatg 1800actccccgcc gccccgggcc
cacccgcaag cattaccagc cctatgcccc accacgcgac 1860ttcgcagcct
atcgctccag agtgaagttc agcaggagcg cagacgcccc cgcgtacaag
1920cagggccaga accagctcta taacgagctc aatctaggac gaagagagga
gtacgatgtt 1980ttggacaaga gacgtggccg ggaccctgag atggggggaa
agccgagaag gaagaaccct 2040caggaaggcc tgtacaatga actgcagaaa
gataagatgg cggaggccta cagtgagatt 2100gggatgaaag gcgagcgccg
gaggggcaag gggcacgatg gcctttacca gggtctcagt 2160acagccacca
aggacaccta cgacgccctt cacatgcagg ccctgccccc tcgctaa
22178738PRTArtificial Sequencesynthetic 8Met Glu Phe Gly Leu Ser
Trp Leu Phe Leu Val Ala Ile Leu Lys Gly1 5 10 15Val Gln Cys Ser Arg
Asp Ile Leu Leu Thr Gln Thr Pro Leu Ser Leu 20 25 30Pro Val Ser Leu
Gly Asp Gln Ala Ser Ile Ser Cys Arg Ser Ser Gln 35 40 45Ser Leu Val
His Arg Asn Gly Asn Thr Tyr Leu His Trp Tyr Leu Gln 50 55 60Lys Pro
Gly Gln Ser Pro Lys Leu Leu Ile His Lys Val Ser Asn Arg65 70 75
80Phe Ser Gly Val Pro Asp Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp
85 90 95Phe Thr Leu Lys Ile Ser Arg Val Glu Ala Glu Asp Leu Gly Val
Tyr 100 105 110Phe Cys Ser Gln Ser Thr His Val Pro Pro Leu Thr Phe
Gly Ala Gly 115 120 125Thr Lys Leu Glu Leu Lys Gly Gly Gly Gly Ser
Gly Gly Gly Gly Ser 130 135 140Gly Gly Gly Gly Ser Glu Val Lys Leu
Gln Gln Ser Gly Pro Ser Leu145 150 155 160Val Glu Pro Gly Ala Ser
Val Met Ile Ser Cys Lys Ala Ser Gly Ser 165 170 175Ser Phe Thr Gly
Tyr Asn Met Asn Trp Val Arg Gln Asn Ile Gly Lys 180 185 190Ser Leu
Glu Trp Ile Gly Ala Ile Asp Pro Tyr Tyr Gly Gly Thr Ser 195 200
205Tyr Asn Gln Lys Phe Lys Gly Arg Ala Thr Leu Thr Val Asp Lys Ser
210 215 220Ser Ser Thr Ala Tyr Met His Leu Lys Ser Leu Thr Ser Glu
Asp Ser225 230 235 240Ala Val Tyr Tyr Cys Val Ser Gly Met Lys Tyr
Trp Gly Gln Gly Thr 245 250 255Ser Val Thr Val Ser Ser Ala Lys Thr
Thr Pro Pro Ser Val Tyr Gly 260 265 270Arg Val Thr Val Ser Ser Ala
Glu Pro Lys Ser Cys Asp Lys Thr His 275 280 285Thr Cys Pro Pro Cys
Pro Ala Pro Glu Leu Leu Gly Gly Pro Ser Val 290 295 300Phe Leu Phe
Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser Arg Thr305 310 315
320Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu Asp Pro Glu
325 330 335Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn
Ala Lys 340 345 350Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr
Arg Val Val Ser 355 360 365Val Leu Thr Val Leu His Gln Asp Trp Leu
Asn Gly Lys Glu Tyr Lys 370 375 380Cys Lys Val Ser Asn Lys Ala Leu
Pro Ala Pro Ile Glu Lys Thr Ile385 390 395 400Ser Lys Ala Lys Gly
Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu Pro 405 410 415Pro Ser Arg
Asp Glu Leu Thr Lys Asn Gln Val Ser Leu Thr Cys Leu 420 425 430Val
Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser Asn 435 440
445Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp Ser
450 455 460Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys
Ser Arg465 470 475 480Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val
Met His Glu Ala Leu 485 490 495His Asn His Tyr Thr Gln Lys Ser Leu
Ser Leu Ser Pro Gly Lys Lys 500 505 510Asp Pro Lys Ala Ser Phe Glu
Ile Glu Val Met Tyr Pro Pro Pro Tyr 515 520 525Leu Asp Asn Glu Lys
Ser Asn Gly Thr Ile Ile His Val Lys Gly Lys 530 535 540His Leu Cys
Pro Ser Pro Leu Phe Pro Gly Pro Ser Lys Pro Phe Trp545 550 555
560Val Leu Val Val Val Gly Gly Val Leu Ala Cys Tyr Ser Leu Leu Val
565 570 575Thr Val Ala Phe Ile Ile Phe Trp Val Arg Ser Lys Arg Ser
Arg Leu 580 585 590Leu His Ser Asp Tyr Met Asn Met Thr Pro Arg Arg
Pro Gly Pro Thr 595 600 605Arg Lys His Tyr Gln Pro Tyr Ala Pro Pro
Arg Asp Phe Ala Ala Tyr 610 615 620Arg Ser Arg Val Lys Phe Ser Arg
Ser Ala Asp Ala Pro Ala Tyr Lys625 630 635 640Gln Gly Gln Asn Gln
Leu Tyr Asn Glu Leu Asn Leu Gly Arg Arg Glu 645 650 655Glu Tyr Asp
Val Leu Asp Lys Arg Arg Gly Arg Asp Pro Glu Met Gly 660 665 670Gly
Lys Pro Arg Arg Lys Asn Pro Gln Glu Gly Leu Tyr Asn Glu Leu 675 680
685Gln Lys Asp Lys Met Ala Glu Ala Tyr Ser Glu Ile Gly Met Lys Gly
690 695 700Glu Arg Arg Arg Gly Lys Gly His Asp Gly Leu Tyr Gln Gly
Leu Ser705 710 715 720Thr Ala Thr Lys Asp Thr Tyr Asp Ala Leu His
Met Gln Ala Leu Pro 725 730 735Pro Arg
D00000
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010

D00011
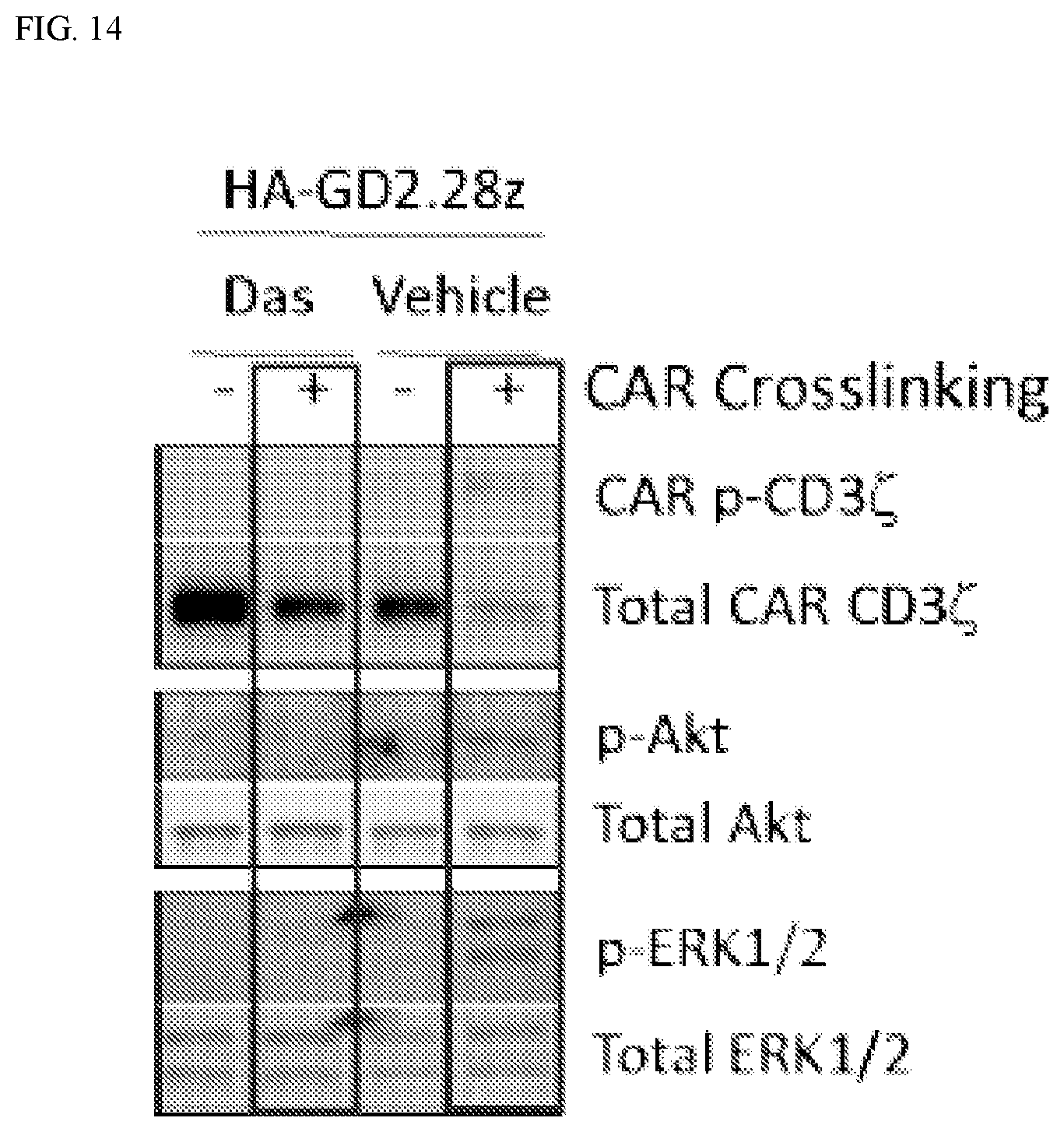
D00012

D00013
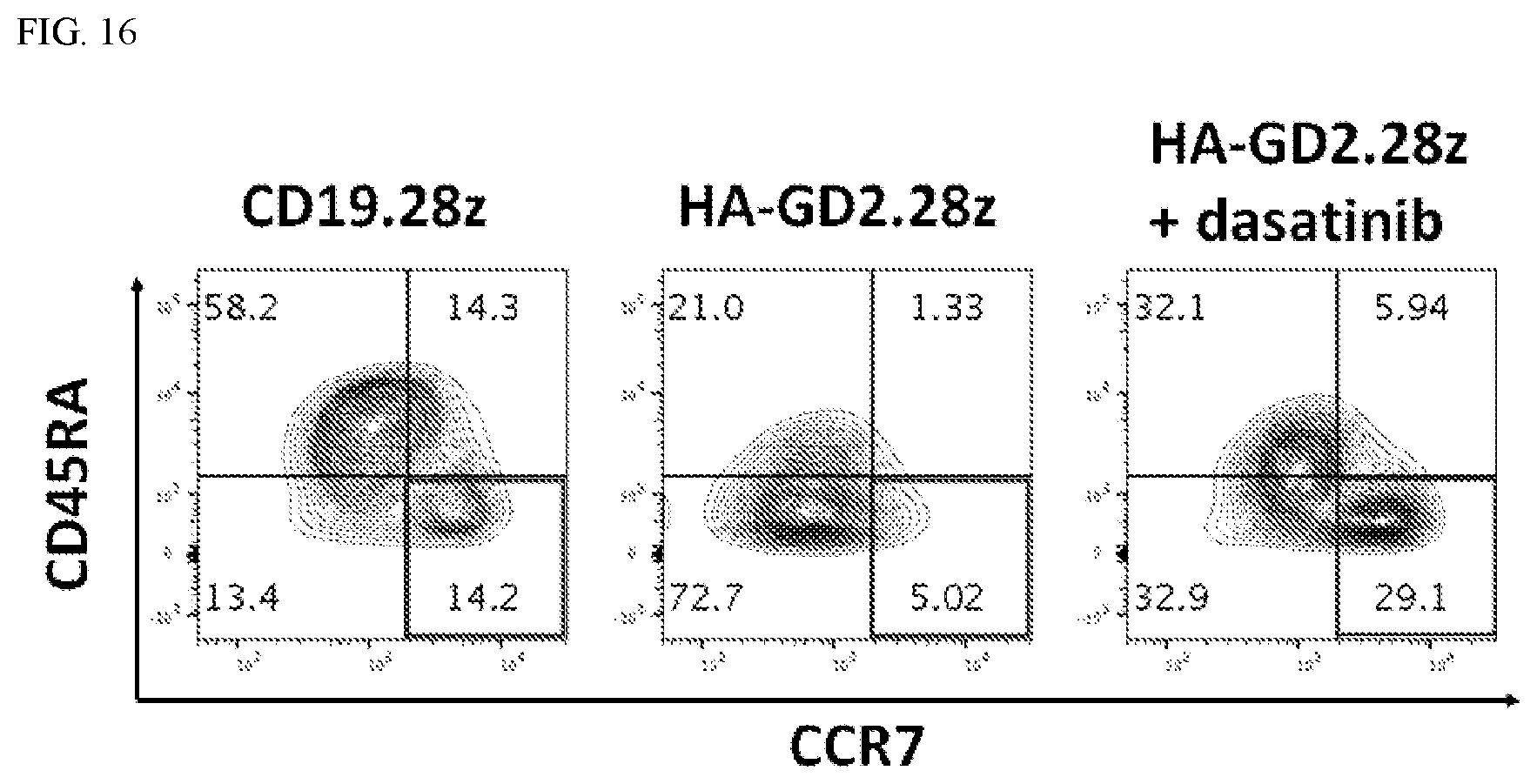
D00014

D00015
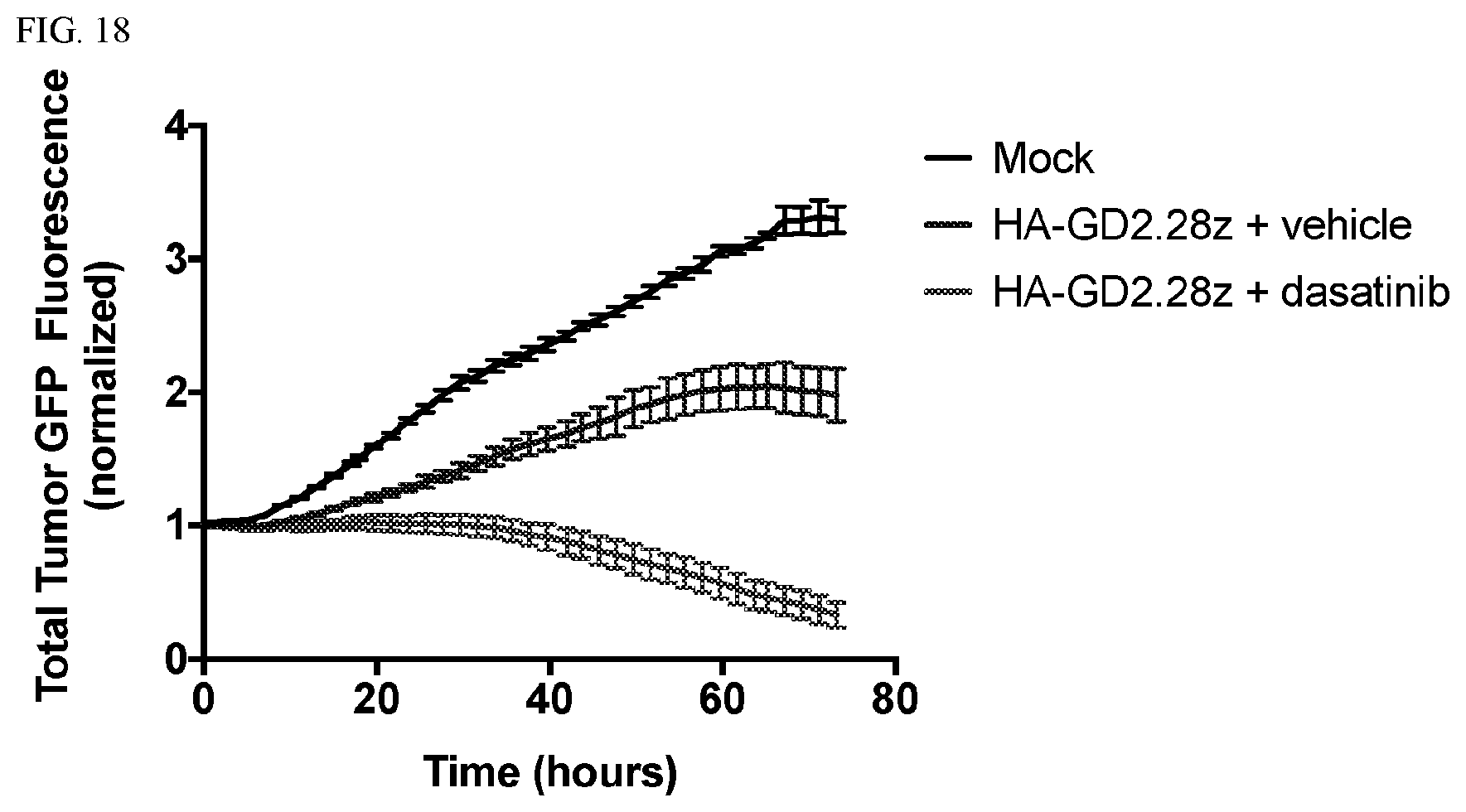
D00016
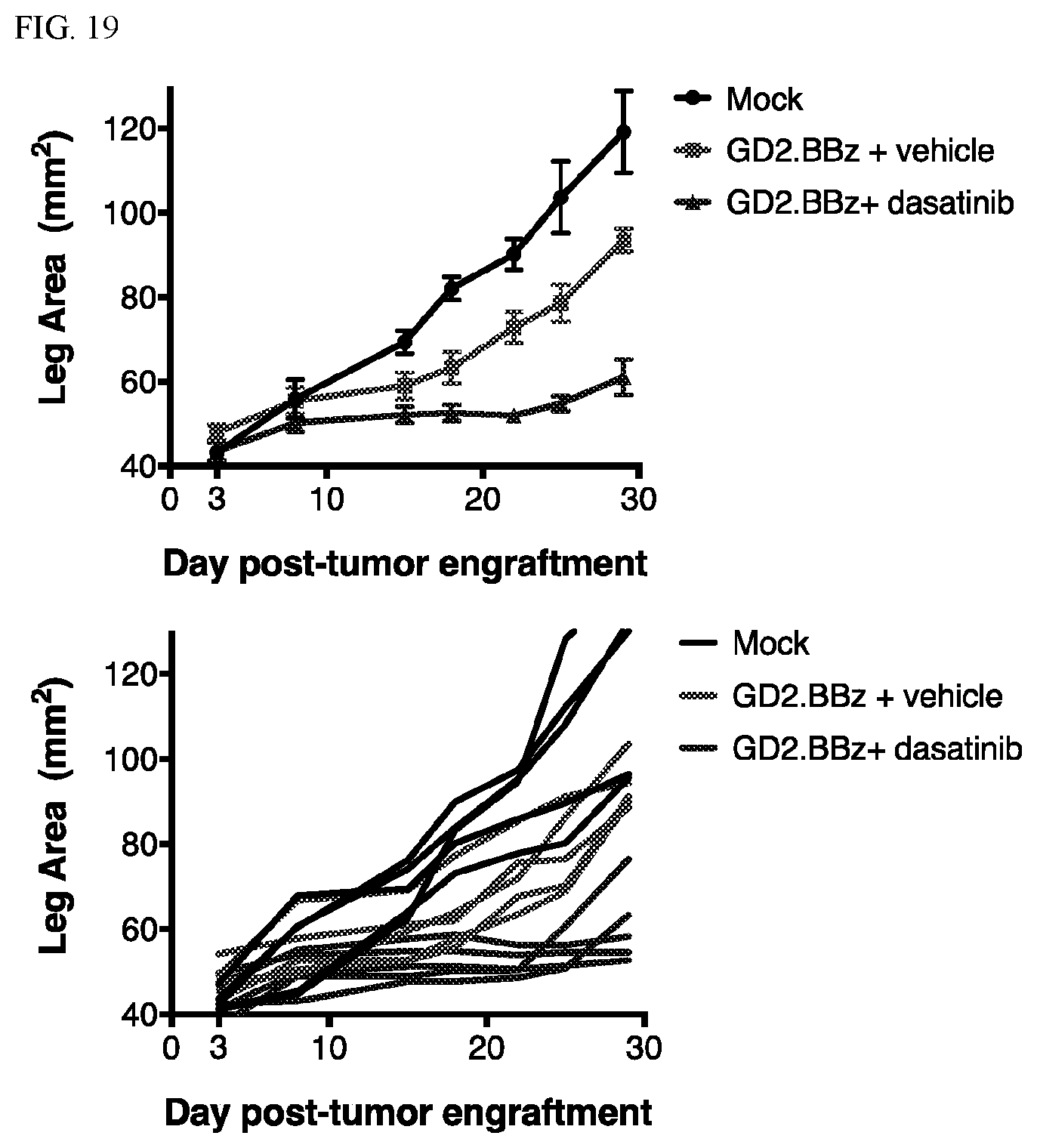
D00017

D00018

D00019

D00020

D00021

D00022

D00023

D00024

D00025

S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.