Proteolytically Cleavable Fusion Proteins With High Molar Specific Activity
METZNER; Hubert ; et al.
U.S. patent application number 16/674390 was filed with the patent office on 2020-03-26 for proteolytically cleavable fusion proteins with high molar specific activity. The applicant listed for this patent is CSL Behring GmbH. Invention is credited to Hubert METZNER, Stefan SCHULTE, Thomas WEIMER.
| Application Number | 20200095567 16/674390 |
| Document ID | / |
| Family ID | 46329953 |
| Filed Date | 2020-03-26 |

| United States Patent Application | 20200095567 |
| Kind Code | A1 |
| METZNER; Hubert ; et al. | March 26, 2020 |
PROTEOLYTICALLY CLEAVABLE FUSION PROTEINS WITH HIGH MOLAR SPECIFIC ACTIVITY
Abstract
The invention relates to therapeutic fusion proteins in which a coagulation factor is fused to a half-life enhancing polypeptide, and in which both are connected by a linker peptide that is proteolytically cleavable. The cleavage of such linkers liberates the coagulation factor from activity-compromising steric hindrance caused by the half-life enhancing polypeptide and thereby allows the generation of fusion proteins may show relatively high molar specific activity when tested in coagulation-related assays. Furthermore, the fact that the linker is cleavable can enhance the rates of inactivation and/or elimination after proteolytic cleavage of the peptide linker compared to the rates measured for corresponding therapeutic fusion proteins linked by the non-cleavable linker having the amino acid sequence GGGGGGV.
| Inventors: | METZNER; Hubert; (Marburg, DE) ; WEIMER; Thomas; (Gladenbach, DE) ; SCHULTE; Stefan; (Marburg, DE) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 46329953 | ||||||||||
| Appl. No.: | 16/674390 | ||||||||||
| Filed: | November 5, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 15808453 | Nov 9, 2017 | |||
| 16674390 | ||||
| 14623868 | Feb 17, 2015 | |||
| 15808453 | ||||
| 13074153 | Mar 29, 2011 | |||
| 14623868 | ||||
| 12000739 | Dec 17, 2007 | 7939632 | ||
| 13074153 | ||||
| 11812016 | Jun 14, 2007 | |||
| 12000739 | ||||
| 60819620 | Jul 11, 2006 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | C07K 14/765 20130101; C12N 9/6424 20130101; C07K 14/745 20130101; C12Y 304/21022 20130101; C12N 9/6443 20130101; A61P 7/00 20180101; C07K 2319/50 20130101; A61P 7/04 20180101; C12Y 304/21021 20130101; C07K 14/755 20130101; C12N 9/6472 20130101; C07K 2319/31 20130101; C12N 9/6437 20130101; A61K 38/00 20130101; C07K 14/76 20130101; A61K 48/00 20130101 |
| International Class: | C12N 9/64 20060101 C12N009/64; C07K 14/745 20060101 C07K014/745; C07K 14/755 20060101 C07K014/755; C07K 14/76 20060101 C07K014/76; C07K 14/765 20060101 C07K014/765 |
Foreign Application Data
| Date | Code | Application Number |
|---|---|---|
| Jun 14, 2006 | EP | 06012262.9 |
Claims
1-33. (canceled)
34. A method of administering an effective amount of a coagulation factor fusion protein to a patient in need thereof, wherein the coagulation factor fusion protein comprises: a) a von Willebrand Factor; b) a half-life enhancing polypeptide (HLEP), and c) a peptide linker which joins the von Willebrand Factor and the half-life enhancing polypeptide; wherein the HLEP is an immunoglobulin without an antigen binding domain, wherein the peptide linker is cleavable by one or more proteases involved in coagulation or activated by coagulation enzymes, and wherein the peptide linker is cleaved during a coagulatory event; and wherein the patient suffers from a blood coagulation disorder.
35. The method of claim 34, wherein said fusion protein has at least one of the following properties, in comparison to the respective therapeutic fusion protein linked by a non-cleavable linker having the amino acid sequence GGGGGGV (SEQ ID NO: 94): i) an increased molar specific activity in at least one coagulation-related assay, ii) an increased inactivation rate of the activated coagulation factor after the peptide linker is proteolytically cleaved in a coagulation-related mode, and iii) an increased elimination rate after the peptide linker is proteolytically cleaved in a coagulation-mode.
36. The method of claim 34, wherein said fusion protein has a higher in vivo recovery compared to the in vivo recovery of a von Willebrand Factor when not fused to a half-life enhancing polypeptide.
37. The method of claim 34, wherein said fusion protein has an increased half-life in plasma compared to the half-life in plasma of a von Willebrand Factor when not fused to a half-life enhancing polypeptide.
38. The method of claim 34, wherein the molar specific activity of the fusion protein is increased at least 25% compared to that of the respective fusion protein linked by a non-cleavable linker consisting of the amino acid sequence GGGGGGV (SEQ ID NO: 94) in at least one coagulation-related assays.
39. The method of claim 34, wherein the inactivation rate of the von Willebrand Factor after cleavage of the peptide linker which links the von Willebrand Factor to the half-life enhancing polypeptide is increased at least 10% as compared to the inactivation rate of a von Willebrand Factor in a respective fusion protein linked by a non-cleavable linker consisting of the amino acid sequence GGGGGGV (SEQ ID NO: 94).
40. The method of claim 34, wherein the elimination rate of the von Willebrand Factor after cleavage of the peptide linker which links the von Willebrand Factor to the half-life enhancing polypeptide is increased by at least 10% as compared to the elimination rate of a von Willebrand Factor in a respective fusion protein linked by a non-cleavable linker consisting of the amino acid sequence GGGGGGV (SEQ ID NO: 94).
41. The method of claim 34, wherein the peptide linker is cleavable by a protease that naturally activates FVII or FVIII in vivo.
42. The method of claim 34, wherein the kinetics of linker cleavage by the protease is not delayed by more than a factor of 3 compared to the activation kinetics of the coagulation factor.
43. The method of claim 34, wherein the linker is cleaved by thrombin during coagulation.
44. The method of claim 34, wherein the blood coagulation disorder is hemophilia A.
45. The method of claim 34, wherein the peptide linker is cleavable by a protease that is activated directly or indirectly by a coagulation factor that is activated during a coagulation event.
46. A method of administering an effective amount of a coagulation factor fusion protein to a patient in need thereof, comprising: (a) administering a composition comprising said coagulation factor fusion protein, (b) administering a composition comprising a polynucleotide encoding said coagulation factor fusion protein via a gene therapy protocol, or (c) administering a composition comprising a plasmid or vector comprising a polynucleotide encoding said coagulation factor fusion protein via a gene therapy protocol, wherein the coagulation factor fusion protein comprises: a) a von Willebrand Factor; b) a half-life enhancing polypeptide (HLEP), and c) a peptide linker which joins the von Willebrand Factor and the half-life enhancing polypeptide; wherein the HLEP is an immunoglobulin without an antigen binding domain, wherein the peptide linker is cleavable by one or more proteases involved in coagulation or activated by coagulation enzymes, and wherein the peptide linker is cleaved during coagulation; and wherein the patient suffers from a blood coagulation disorder.
47. The method of claim 46, wherein the administration comprises administering via a gene therapy protocol (a) a composition comprising a polynucleotide encoding said coagulation factor fusion protein or (b) a composition comprising a plasmid or vector comprising a polynucleotide encoding said coagulation factor fusion protein.
48. The method of claim 46, comprising administering a composition comprising said fusion protein.
49. The method of claim 34, wherein the von Willebrand Factor or the immunoglobulin comprises a sequence that is 95% identical to the sequence of a wild-type human von Willebrand Factor or a wild-type human immunoglobulin, respectively.
50. The method of claim 34, wherein the von Willebrand Factor or the immunoglobulin comprises a sequence that is identical to the sequence of a wild-type human von Willebrand Factor or a wild-type human immunoglobulin respectively.
Description
[0001] This application is a divisional of U.S. application Ser. No. 15/808,453, filed Nov. 9, 2017, which is a divisional of U.S. application Ser. No. 14/623,868, filed Feb. 17, 2015, which is a continuation of U.S. application Ser. No. 13/074,153, filed Mar. 29, 2011, which is a division of U.S. application Ser. No. 12/000,739, which was filed Dec. 17, 2007, and issued as U.S. Pat. No. 7,939,632 on May 10, 2011, which is a continuation-in-part of U.S. application Ser. No. 11/812,016, filed Jun. 14, 2007, which claims priority to U.S. Provisional Application No. 60/819,620, filed Jul. 11, 2006, all of which are incorporated herein by reference. The application also claims priority to European Patent Application No. 06012262.9, filed Jun. 14, 2006, which is incorporated herein by reference.
[0002] The present invention relates to the field of modified therapeutic fusion proteins with increased half-life compared to their non-modified parent therapeutic polypeptides. The invention, for example, relates to coagulation factors fused to half-life enhancing polypeptides (HLEPs), which are connected by linker peptides that are proteolytically cleavable. The cleavage of such linkers liberates the therapeutic polypeptide from activity-compromising steric hindrance caused by the HLEP and thereby allows the generation of fusion proteins, which retain a high molar specific activity of the coagulation factor. When the therapeutic fusion proteins are zymogens, one may use linkers that liberate the therapeutic polypeptide essentially simultaneously with its activation in vivo upon exposure to the corresponding protease(s). Embodiments of the present invention may show a faster inactivation rate of a given coagulation factor once the coagulation factor is activated and the peptide linker is proteolytically cleaved in a coagulation-related mode and/or a faster elimination rate of a given coagulation factor once the coagulation factor is activated and the peptide linker is proteolytically cleaved in a coagulation-related mode, compared to the corresponding fusion protein without cleavable linker.
[0003] One aspect of the invention is demonstrated for example by human vitamin K-dependent polypeptides Factor IX, Factor VII, and Factor VIIa. The same concept also may be applied to other coagulation factors. Any half-life enhancing polypeptide (HLEP) may be connected to the therapeutic polypeptide by a cleavable linker peptide. For instance albumin or immunoglobulins or fragments derived therefrom without an antigen binding domain, such as the Fc fragment, may serve as HLEPs. The invention also relates to cDNA sequences coding for the therapeutic polypeptides and derivatives thereof genetically fused to a cDNA coding for HLEPs, such as human serum albumin linked by oligonucleotides that code for cleavable, intervening peptide linkers. Such encoded derivatives may exhibit improved half-life and molar specific activities that are increased in comparison to their non-cleavable counterparts. The invention also relates to recombinant expression vectors containing such cDNA sequences, host cells transformed with such recombinant expression vectors, recombinant polypeptides and derivatives which may have biological activities comparable to the unmodified wild type therapeutic polypeptide, but having improved half-lifes. The invention also relates to processes for the manufacture of such recombinant proteins and their derivatives. The invention also relates to a transfer vector for use in human gene therapy, which comprises such modified DNA sequences, which can be useful to increase half-life in vivo.
[0004] Several recombinant, therapeutic polypeptides are commercially available for therapeutic and prophylactic use in humans. The patients in general may benefit from the specific mode of action of the recombinant active ingredients but a disadvantage may be their limited availability due to expensive and complex manufacturing processes. A reduction of the necessary dose or the frequency of administration of such products could improve this situation. A reduced frequency of administration could improve the convenience for the patient and, therefore, also the acceptance of the therapy. Several ideas have been described to attempt to achieve the goal of an increased in vivo half-life after administration. See, e.g. Ballance et al. (WO 01/79271), Sheffield et al. (Sheffield W. P. et al. (2004), Br. J. Haematol. 126: 565-573), WO 2002/04598, WO 2003/059935, WO 2004/081053, WO 2004/101740, WO 2005/001025, WO 91/09125, and WO 03/068934.
[0005] Fusions of coagulation factors to half-life enhancing polypeptides have been suggested to lengthen the half-life of coagulation factors administered to patients. However, once a coagulation factor is activated during coagulation either by proteolytic cleavage of the zymogen (like FIX) or by contact of an already proteolytically "pre"-activated factor to a second polypeptide (like FVIIa binding to Tissue Factor), it may no longer be desirable to maintain the long half-life of the now activated coagulation factor, as this might lead to thrombotic complications. This is the case for a wild type coagulation factor such as FVIIa (Aledort L. M., J Thromb Haemost 2(10): 1700-1708 (2004)) and may be even more relevant if the activated factor has an increased half-life. It is therefore one objective of some embodiments of the present invention to provide long-lived coagulation factors, which after activation or after availability of a cofactor have a half-life comparable to that of an unmodified coagulation factor.
[0006] Fusions of the coagulation factors to half-life enhancing polypeptides as described in the prior art and as also shown in examples 6 and 7 may suffer in general from a reduced molar specific activity of the fused coagulation factor. Another aspect of the present invention is to provide coagulation factors with enhanced half-life that show increased molar specific activity compared to the corresponding therapeutic fusion protein without a cleavable linker.
[0007] Some embodiments of this invention include therapeutic fusion proteins comprising: [0008] a) a coagulation factor, [0009] b) a half-life enhancing polypeptide (HLEP) chosen from albumin and immunoglobulin, and [0010] c) a peptide linker, which linker joins the coagulation factor and the half-life enhancing polypeptide; wherein the peptide linker is cleavable by proteases involved in coagulation or proteases activated by coagulation enzymes, and wherein the therapeutic fusion protein has, in comparison to the respective therapeutic fusion protein linked by a non-cleavable linker having the amino acid sequence GGGGGGV (SEQ ID NO: 94), at least one of the following properties: [0011] i) an increased molar specific activity in at least one coagulation-related assay, [0012] ii) an increased inactivation rate of the activated coagulation factor after the peptide linker is proteolytically cleaved in a coagulation-related mode, and [0013] iii) an increased elimination rate of the activated coagulation factor after the peptide linker is proteolytically cleaved in a coagulation-related mode.
[0014] A "coagulation factor," as used herein, includes variants or derivatives thereof, such as genetically engineered or chemically modified variants or active fragments thereof. See below for additional description of examples of "coagulation factors."
[0015] "Albumin," as used herein, includes polypeptides of the albumin family of proteins such as human serum albumin and bovine serum albumin, including variants and derivatives thereof, such as genetically engineered or chemically modified albumin variants and fragments of albumin proteins. See below for additional description of examples of "albumin."
[0016] "Immunoglobulin," as used herein, includes variants and derivatives of immunoglobulin proteins, such as genetically engineered or chemically modified immunoglobulin variants and fragments of immunoglobulin, for example, an Fc fragment or other fragment not containing an antigen binding domain. See below for additional description of examples of "immunoglobulin."
[0017] As a consequence of the cleavable linker, after cleavage of the peptide linker in a coagulation-related mode the coagulation factor more closely resembles the behaviour of the native, non-fused factor and does not show an increased half-life of the active factor with potentially prothrombotic effect.
[0018] "Proteolytic cleavage in a coagulation-related mode," as used herein, means any proteolytic cleavage that occurs as a consequence of the activation of at least one coagulation factor or coagulation cofactor.
[0019] The phrase "activated coagulation factor after the peptide linker is proteolytically cleaved in a coagulation-related mode," as used herein means that the coagulation factor is either activated almost in parallel to the proteolytic cleavage of the linker peptide, or that the coagulation factor was already activated before the proteolytic cleavage of the linker peptide. Activation may occur, for example by proteolytic cleavage of the coagulation factor or by binding to a cofactor.
[0020] A further aspect of the present invention is to provide therapeutic fusion proteins comprising: [0021] a) a coagulation factor, [0022] b) a half-life enhancing polypeptide (HLEP) chosen from albumin and immunoglobulin, and [0023] c) a peptide linker which joins the coagulation factor and the half-life enhancing polypeptide; wherein the peptide linker is cleavable by proteases involved in coagulation or activated by coagulation enzymes, and wherein the therapeutic fusion protein has, in comparison to the respective therapeutic fusion protein linked by a non-cleavable linker having the amino acid sequence GGGGGGV (SEQ ID NO: 94), at least one of the following properties: [0024] i) an increased molar specific activity in at least one coagulation-related assay, [0025] ii) an increased inactivation rate of the activated coagulation factor after the peptide linker is proteolytically cleaved in a coagulation-related mode, [0026] iii) an increased elimination rate of the activated coagulation factor after the peptide linker is proteolytically cleaved in a coagulation-related mode, and [0027] iv) an enhanced in vivo recovery as compared to the in vivo recovery of the unmodified coagulation factor.
[0028] Some embodiments include therapeutic fusion proteins which have an enhanced in vivo recovery compared to the unmodified coagulation factor by at least 10%, for example, by at least 25% or by 40% or more.
[0029] Exemplary coagulation factors are vitamin-K dependent coagulation factors, FVIIa, and FIX, and fragments and variants thereof, such as genetically engineered or chemically modified variants or fragments, such as are described in more detail below.
[0030] HLEPs may be albumin and fragments or variants thereof and immunoglobulins including fragments and variants thereof, as described above, and in more detail, below.
[0031] The peptide linker in some embodiments may comprise a sequence of the therapeutic polypeptide to be administered or a variant thereof, which should result in a decreased risk of neoantigenic properties (formation of a novel potentially immunogenic epitope due to the occurrence of a peptide within the therapeutic antigen which does not exist in human proteins) of the expressed fusion protein. Also in case the therapeutic protein is a zymogen (e.g. needs to be proteolytically activated) the kinetics of the peptide linker cleavage may more closely reflect the coagulation-related activation kinetics of the zymogen. Thus, in some embodiments, a zymogen and a corresponding peptide linker are activated and respectively cleaved, with comparable kinetics. For this reason, embodiments of the present invention also relate to fusion proteins of a zymogen and a HLEP, where the kinetics of the linker cleavage by relevant proteases are not delayed by more than a factor of 3, such as not by more than a factor of 2, compared to the kinetics of the zymogen activation.
[0032] In another embodiment, the peptide linker comprises cleavage sites for more than one protease. This can be achieved, for example, by a peptide linker that can be cleaved at the same position by different proteases or by a peptide linker that provides two or more different cleavage sites. This may be advantageous circumstances where the therapeutic fusion protein must be activated by proteolytic cleavage to achieve enzymatic activity and where different proteases may contribute to this activation step. This is the case, for example, upon activation of FIX, which can either be achieved by FXIa or by FVIIa/Tissue Factor (TF).
[0033] Some embodiments of the invention are therapeutic fusion proteins wherein the peptide linker is cleavable by the protease that normally activates the coagulation factor in vivo, thereby ensuring that the cleavage of the linker is linked to the activation of the coagulation factor at a site at which coagulation occurs.
[0034] Other exemplary therapeutic fusion proteins according to the invention are those wherein the linker is cleavable by the coagulation factor which is part of the therapeutic fusion protein once it is activated, thus also ensuring that cleavage of the fusion protein is connected with a coagulatory event.
[0035] Other exemplary therapeutic fusion proteins according to the invention are those wherein the linker is cleavable by a protease, which itself is activated directly or indirectly by the activity of the coagulation factor which is part of the therapeutic fusion protein, thus also ensuring that cleavage of the fusion protein is connected with a coagulatory event.
[0036] One class of therapeutic fusion proteins included in this invention comprises those wherein the linker is cleavable by FXIa and/or by FVIIa/TF and the coagulation factor is FIX.
[0037] For example, embodiments of the invention include fusion proteins comprising the vitamin K-dependent polypeptide Factor IX, cleavable linkers, and albumin as the HLEP, as well as their corresponding cDNA sequences. The invention also relates to cDNA sequences coding for any other coagulation factors which can be proteolytically activated or that are involved in the activation of other zymogens or polypeptides. In some embodiments, these cDNAs are genetically fused to cDNA sequences coding for human serum albumin or other HLEPs, and are linked by oligonucleotides that code for intervening, cleavable peptide linkers. The expressed therapeutic fusion proteins may exhibit molar specific activities which are increased in comparison to their non-cleavable counterparts. The invention also relates to recombinant expression vectors containing such fused cDNA sequences, host cells transformed with such recombinant expression vectors, recombinant therapeutic fusion proteins and derivatives that may have biological activities almost comparable to the unmodified wild type therapeutic polypeptides but having improved in vivo half-life. The invention also relates to processes for the manufacture of the recombinant polypeptides of the invention and their derivatives. The invention also relates to a transfer vector for use in human gene therapy, which comprises such modified DNA sequences useful to increase product levels in vivo.
[0038] Some therapeutic fusion proteins according to the invention have a molar specific activity, in particular a molar specific activity in at least one coagulation-related assay, that is at least 25% increased compared to that of the corresponding therapeutic fusion protein without a cleavable linker. Other therapeutic fusion proteins have a molar specific activity that is increased by at least 50%, or by at least 100%, in at least one of the different coagulation-related assays available, compared to that of the corresponding therapeutic fusion protein without a cleavable linker, such as one with the non-cleavable sequence GGGGGGV (SEQ ID NO: 94).
[0039] Additional embodiments of the present invention are therapeutic fusion proteins wherein the inactivation rate of the activated coagulation factor after cleavage of the peptide linker which links the coagulation factor to the half-life enhancing polypeptide is increased by at least 10% as compared to the inactivation rate of the activated coagulation factor in a corresponding therapeutic fusion protein without a cleavable linker. Other embodiments are therapeutic fusion proteins in which the inactivation rate is increased by at least 25%, or by at least 50% as compared to the inactivation rate of the activated coagulation factor in a corresponding therapeutic fusion protein without a cleavable linker, such as one with the non-cleavable sequence GGGGGGV (SEQ ID NO: 94).
[0040] Additional embodiments of the present invention are therapeutic fusion proteins wherein the elimination rate of the coagulation factor after cleavage of the peptide linker that links the coagulation factor to the half-life enhancing polypeptide is increased by at least 10% as compared to the elimination rate of the coagulation factor in a corresponding therapeutic fusion protein without a cleavable linker. Other embodiments are therapeutic fusion proteins in which the elimination rate is increased by at least 25%, or by at least 50% as compared to the elimination rate of the coagulation factor in a corresponding therapeutic fusion protein without a cleavable linker, such as one with the non-cleavable sequence GGGGGGV (SEQ ID NO: 94).
[0041] Vitamin K-Dependent Polypeptides
[0042] Vitamin K-dependent polypeptides as one group of the therapeutic polypeptides are polypeptides that are .gamma.-carboxylated enzymatically in the liver using vitamin K as a cofactor. Such vitamin K-dependent polypeptides e.g. include Factors II, VII, IX, X, Protein C, Protein S, GAS6, and Protein Z.
[0043] Human FIX
[0044] Human FIX, one member of the group of vitamin K-dependent polypeptides, is a single-chain glycoprotein with a molecular weight of 57 kDa, which is secreted by liver cells into the blood stream as an inactive zymogen of 415 amino acids. It contains 12 .gamma.-carboxy-glutamic acid residues localized in the N-terminal Gla-domain of the polypeptide. The Gla residues require vitamin K for their biosynthesis. Following the Gla domain there are two epidermal growth factor domains, an activation peptide, and a trypsin-type serine protease domain. Further posttranslational modifications of FIX encompass hydroxylation (Asp 64), N-(Asn157 and Asn167) as well as 0-type glycosylation (Ser53, Ser61, Thr159, Thr169, and Thr172), sulfation (Tyr155), and phosphorylation (Ser158).
[0045] FIX is converted to its active form, Factor IXa, by proteolysis of the activation peptide at Arg145-Ala146 and Arg180-Val181 leading to the formation of two polypeptide chains, an N-terminal light chain (18 kDa) and a C-terminal heavy chain (28 kDa), which are held together by one disulfide bridge. Activation cleavage of Factor IX can be achieved in vitro e.g. by Factor XIa or Factor VIIa/TF. Factor IX is present in human plasma in a concentration of 5-10/.mu.g/ml. Terminal plasma half-life of Factor IX in humans was found to be about 15 to 18 hours (White G C et al. 1997. Recombinant factor IX. Thromb Haemost. 78: 261-265; Ewenstein B M et al. 2002. Pharmacokinetic analysis of plasma-derived and recombinant F IX concentrates in previously treated patients with moderate or severe hemophilia B. Transfusion 42:190-197).
[0046] Hemophilia B is caused by non-functional or missing Factor IX and is treated with Factor IX concentrates from plasma or a recombinant form of Factor IX. As haemophilia B patients often receive at least biweekly prophylactic administrations of Factor IX to avoid spontaneous bleedings, it is desirable to increase the intervals of time between administrations by increasing the half-life of the Factor IX product applied. An improvement in plasma half-life may bring significant benefit to the patient. Up to now no pharmaceutical preparation of a Factor IX with improved plasma half-life is commercially available nor have any data been published showing F IX variants with prolonged in vivo half-life and almost unchanged molar specific activity in coagulation-related assays. Therefore, a great medical need still exists to develop forms of Factor IX which have a longer functional half-life in vivo.
[0047] Factor VII and Factor VIIa
[0048] FVII is a single-chain glycoprotein with a molecular weight of 50 kDa, which is secreted by liver cells into the blood stream as an inactive zymogen of 406 amino acids. FVII is converted to its active form Factor VIIa, by proteolysis of the single peptide bond at Arg152-Ile153 leading to the formation of two polypeptide chains, a N-terminal light chain (24 kDa) and a C-terminal heavy chain (28 kDa), which are held together by one disulfide bridge. In contrast to other vitamin K-dependent coagulation factors, no activation peptide is cleaved off during activation. Activation cleavage of Factor VII can be achieved in vitro, for example, by Factor Xa, Factor IXa, Factor VIIa, Factor XIIa, Factor Seven Activating Protease (FSAP), and thrombin. Mollerup et al. (Biotechnol. Bioeng. (1995) 48: 501-505) reported that some cleavage also occurs in the heavy chain at Arg290 and/or Arg315.
[0049] Factor VII is present in plasma in a concentration of 500 ng/ml. About 1% or 5 ng/ml of Factor VII is present as activated Factor VIIa. The terminal plasma half-life of Factor VII was found to be about 4 hours and that of Factor VIIa about 2 hours.
[0050] By administering supraphysiological concentrations of Factor VIIa hemostasis can be achieved bypassing the need for Factor Villa and Factor IXa. The cloning of the cDNA for Factor VII (U.S. Pat. No. 4,784,950) made it possible to develop activated Factor VII as a pharmaceutical. Factor VIIa was successfully administered for the first time in 1988. Ever since the number of indications of Factor VIIa has grown steadily showing a potential to become an universal hemostatic agent to stop bleeding (Erhardtsen, 2002). However, the short terminal half-life of Factor VIIa of approximately 2 hours and reduced in vivo recovery may be limiting its application. Therefore, a great medical need still exists to develop forms of Factor VIIa which have an improved half-life but otherwise almost uncompromised molar specific activity, inactivation kinetics, and/or elimination kinetics after start of coagulation.
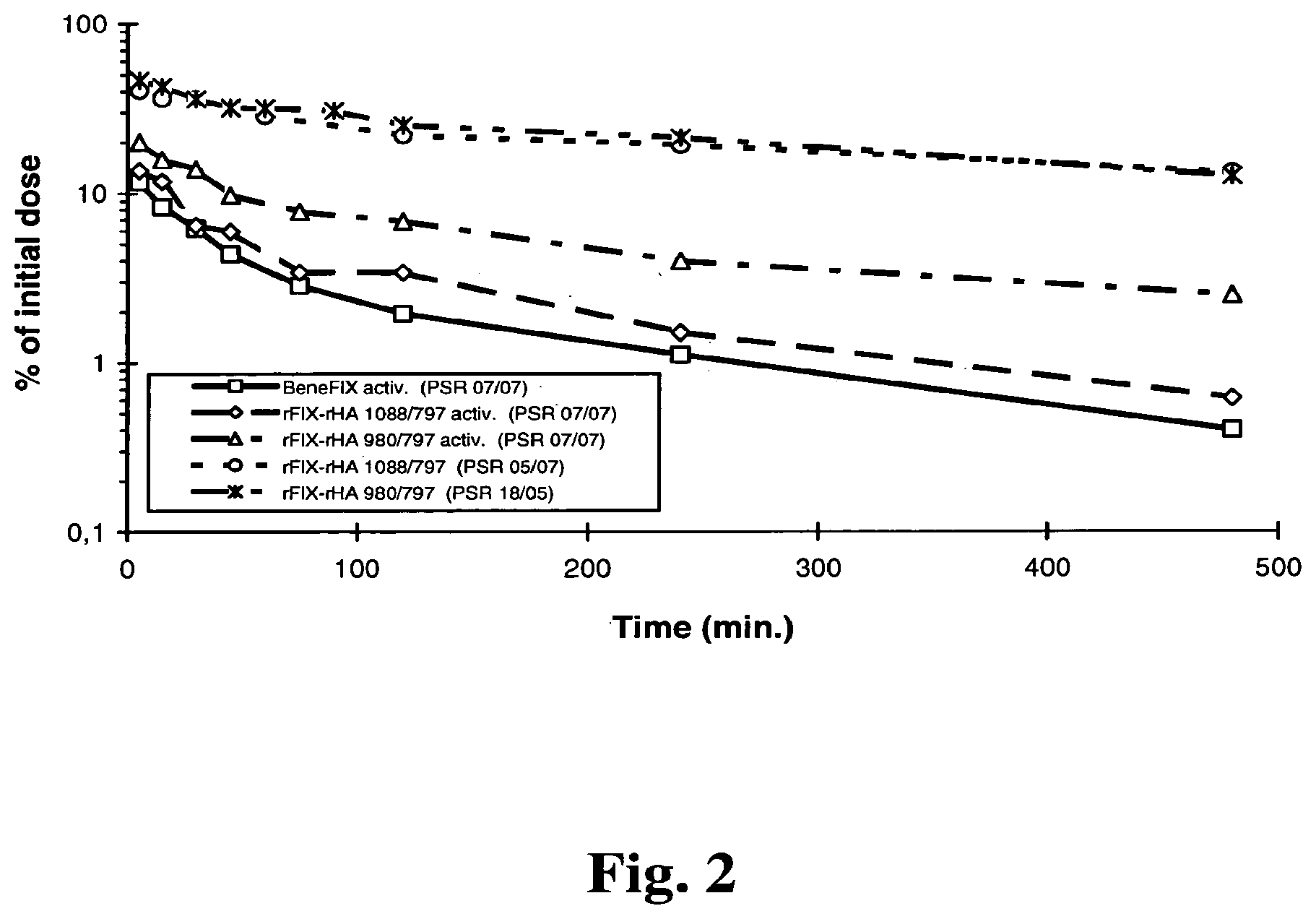
[0051] Therapeutic Fusion Proteins
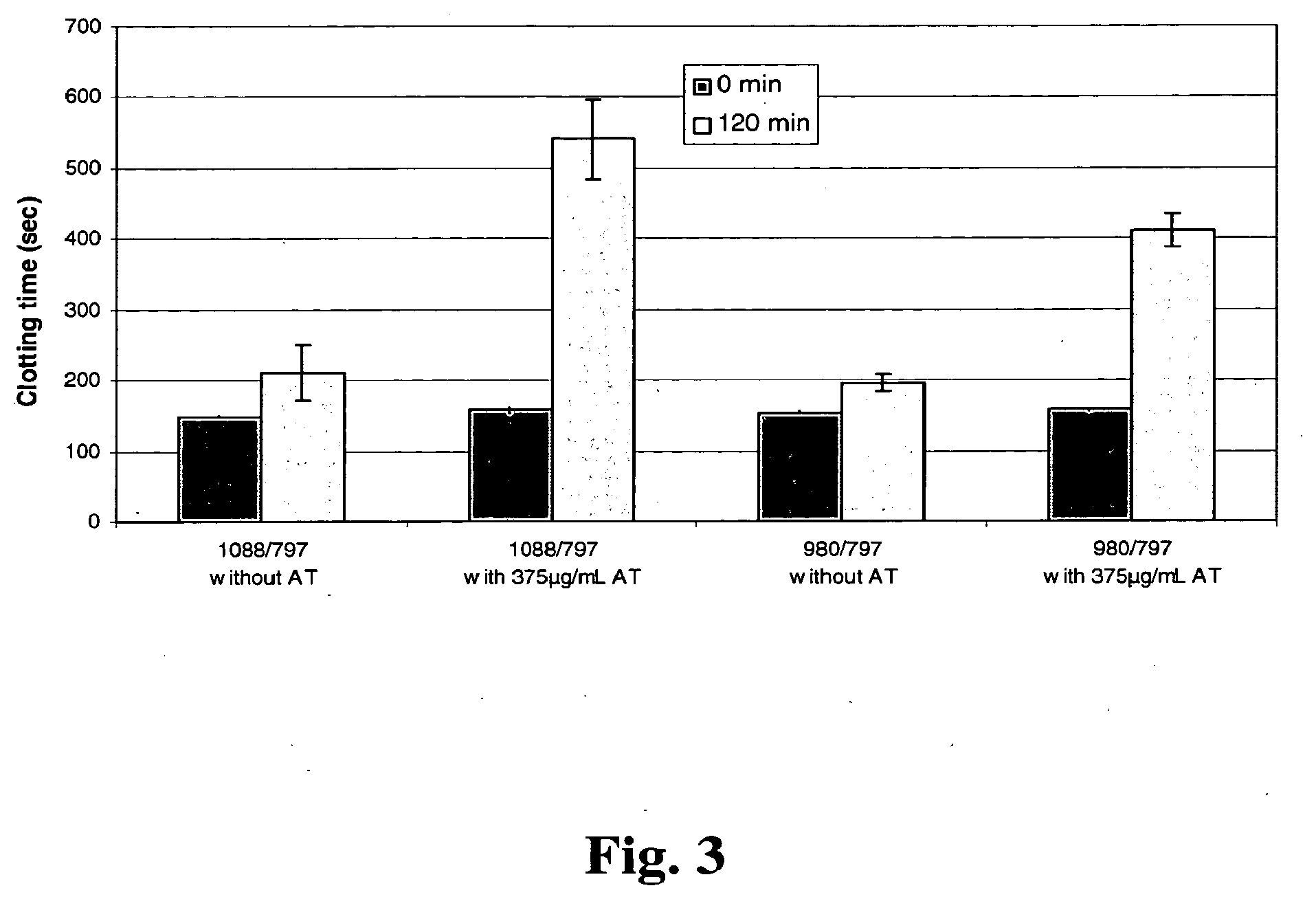
[0052] "Therapeutic fusion proteins" or "fusion proteins" as used herein are coagulation factors fused to a half-life enhancing polypeptide such that, upon administration to a human or other animal, may produce a prophylactic or therapeutic effect. These therapeutic fusion proteins may be administered to a human or other animal via, for example, intravenous, intramuscular, subcutaneous, oral, topical, parenteral or other routes. In addition, gene therapy protocols may be used which involve administration of a polynucleotide encoding the fusion protein or a composition comprising that polynucleotide, such as a plasmid or vector or host cell. Specific classes of therapeutic fusion proteins described, i.e. by the examples below, are coagulation factors, such as vitamin K-dependent polypeptides, linked to half-life enhancing polypeptides, such as albumin and immunoglobulin. The expression "therapeutic fusion protein" is used interchangeably with "fusion protein".
[0053] Half-Life Enhancing Polypeptide (HLEP)
[0054] Albumin and immunoglobulin have been described above as examples of half-life enhancing polypeptides (HLEPs). The terms "human serum albumin" (HSA) and "human albumin" (HA) are used interchangeably in this application. The terms "albumin" and "serum albumin" are broader, and encompass human serum albumin as well as albumin from other species, and fragments and variants thereof.
[0055] "Albumin," as used herein, includes polypeptides of the albumin family of proteins. As used herein, "albumin" refers collectively to albumin polypeptide or amino acid sequence, or an albumin fragment or variant having one or more functional activities (e.g., biological activities) of albumin. Examples include human serum albumin and bovine serum albumin, including variants and derivatives thereof, such as genetically engineered or chemically modified albumin variants and fragments of albumin proteins. For example, "albumin" refers to human albumin or fragments thereof, such as the mature form of human albumin as shown in SEQ ID NO:1 herein or albumin from other vertebrates or fragments thereof, or analogs or variants of these molecules or fragments thereof.
[0056] The albumin portion of the albumin fusion proteins may comprise the full length of, for instance, the HA sequence as described above, or may include one or more fragments thereof that are capable of stabilizing or prolonging the therapeutic activity. Such fragments may be of 10 or more amino acids in length or may include about 15, 20, 25, 30, 50, or more contiguous amino acids from the HA sequence or may include part or all of specific domains of HA.
[0057] The albumin portion of the albumin fusion proteins of the invention may comprise a variant or derivative or analog of normal HA, either natural or artificial. The therapeutic polypeptide portion of the fusion proteins of the invention may also comprise variants of the corresponding therapeutic polypeptides as described herein.
[0058] The terms "variants" "derivatives" and "analogs", throughout this application, when applied to any protein disclosed herein, each include, for example, insertions, deletions, and substitutions, either conservative or non-conservative, either natural or artificial (i.e. engineered), where such changes do not substantially alter the active site, or a fragment, such as an active domain that confers the therapeutic activities of the therapeutic polypeptides, or chemical modifications that also allow for therapeutic activity. Examples include, for instance coagulation factors, albumin, or immunoglobulins that are 80%, 85%, 90% and 95% identical in sequence to a wild-type, human coagulation factor, albumin, or immunoglobulin sequence.
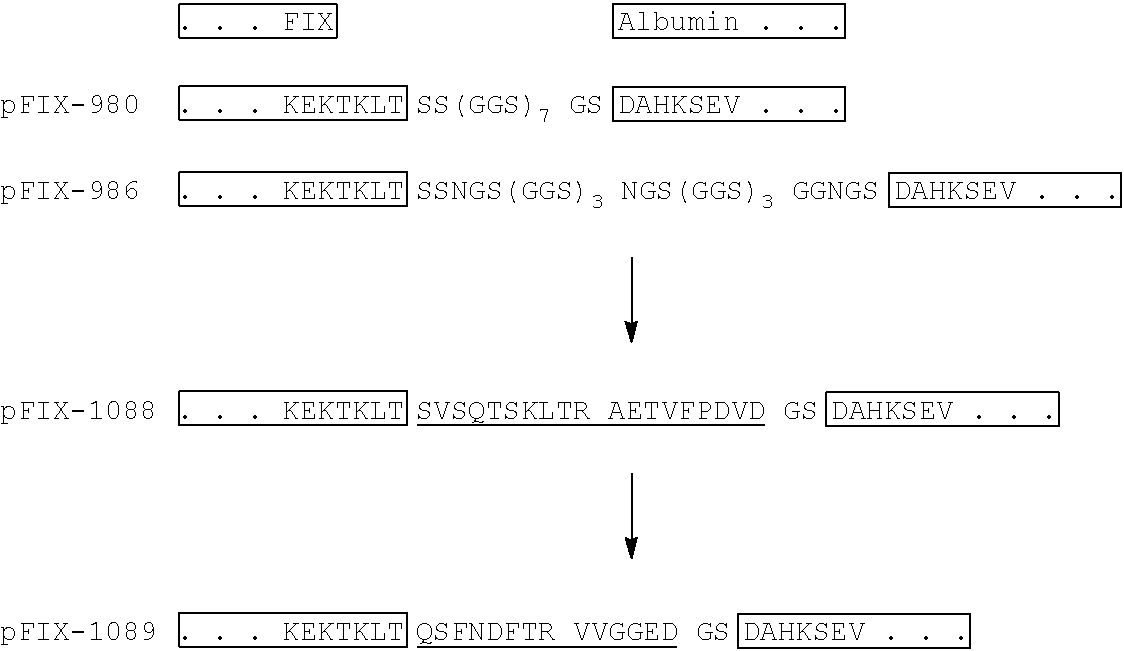
[0059] In particular, the albumin fusion proteins of the invention may include naturally occurring polymorphic variants of human albumin and fragments of human albumin. The albumin may be derived from any vertebrate, especially any mammal, for example human, cow, sheep, or pig. Non-mammalian albumins include, but are not limited to, hen and salmon. The albumin portion of the albumin-linked polypeptide may be from a different animal than the therapeutic polypeptide portion.
[0060] Generally speaking, an albumin fragment or variant encompassed within the term "albumin" will be at least 10, for example at least 40, or more than 70 amino acids long. That albumin variant may comprise at least one whole domain of albumin or fragments of said domains, for example domains 1 (amino acids 1-194 of SEQ ID NO:1), 2 (amino acids 195-387 of SEQ ID NO:1), 3 (amino acids 388-585 of SEQ ID NO:1), 1+2 (1-387 of SEQ ID NO:1), 2+3 (195-585 of SEQ ID NO:1) or 1+3 (amino acids 1-194 of SEQ ID NO:1+amino acids 388-585 of SEQ ID NO:1). Each domain is itself made up of two homologous subdomains namely 1-105, 120-194, 195-291, 316-387, 388-491 and 512-585, with flexible inter-subdomain linker regions comprising residues Lys106 to Glu119, Glu292 to Val315, and Glu492 to Ala511.
[0061] The albumin portion of an albumin fusion protein of the invention may comprise at least one subdomain or domain of HA or conservative modifications thereof.
[0062] All fragments and variants of albumin are encompassed by the invention as fusion partners of a coagulation factor as long as they lead to a half-life extension of the therapeutic fusion protein in plasma of at least 25% as compared to the non-fused coagulation factor.
[0063] The albumin family of proteins, included within the term "albumin" used herein, comprise evolutionarily related serum transport proteins, for example, albumin, alpha-fetoprotein (AFP; Beattie & Dugaiczyk 1982. Gene 20:415-422), afamin (AFM; Lichenstein et al. 1994. J. Biol. Chem. 269:18149-18154) and vitamin D binding protein (DBP; Cooke & David 1985. J. Clin. Invest. 76:2420-2424). Alpha-fetoprotein has been claimed to enhance the half-life of an attached therapeutic polypeptide in vivo (WO 2005/024044). Their genes represent a multigene cluster with structural and functional similarities mapping to the same chromosomal region in humans, mice and rat. The structural similarity of those albumin family members suggests their usability as HLEPs. Some embodiments of the invention, therefore, may use such albumin family members, or fragments and variants thereof as defined above, as HLEPs.
[0064] Albumin family members encompassed within the term "albumin" herein also comprise the full length of the respective proteins AFP, AFM and DBP, or may include one or more fragments thereof that are capable of stabilizing or prolonging the therapeutic activity. Such fragments may be of 10 or more amino acids in length or may include about 15, 20, 25, 30, 50, or more contiguous amino acids of the respective protein sequence or may include part or all of specific domains of the respective protein, as long as the HLEP fragments provide a half-life extension of at least 25% as compared to the non-fused coagulation factor. Albumin family members of the therapeutic fusion proteins of the invention may also include naturally occurring polymorphic variants of AFP, AFM and DBP.
[0065] The term "immunoglobulin" as used herein, encompasses, for instance, IgG and IgG-fragments, which may also be used as HLEPs, as long as the corresponding HLEP fragments provide a half-life extension of at least 25% as compared to the non-fused coagulation factor. The therapeutic polypeptide portion may be connected to the IgG or the IgG fragments via a cleavable linker that allows high molar specific activities of the fusion protein. For example, fusion proteins comprising FVII or VIIa and IgG or fragments thereof may be prepared. A linker sequence of the present invention liberating FVII (FVIIa) molecules upon cleavage by a protease of the coagulation cascade such as, e.g., FXIa, FXa, or FIXa could be able to elevate the clotting activity of the constructs to an activity level comparable to the monomer/dimer or even higher. A FIX-Fc fusion protein with cleavable linker is exemplarily shown in SEQ ID NO:93. Cleavable linkers such as those shown in table 3a and 3b may also be applied in such embodiments.
[0066] The invention also relates to fusion proteins comprising linking a coagulation factor, including, for example, a fragment or variant thereof, to the N- or C-terminus of a HLEP, including a fragment or variant thereof, such that an intervening cleavable peptide linker is introduced between the therapeutic polypeptide and the HLEP such that the fusion protein formed has an increased in vivo half-life compared to the coagulation factor which has not been linked to a HLEP and that the fusion protein has an at least 25% higher molar specific activity compared to the corresponding fusion protein with non-cleavable linker in at least one of the different coagulation-related assays available.
[0067] "Coagulation factor" as used in this application includes, but is not limited to, polypeptides consisting of Factor IX, Factor VII, Factor VIII, von Willebrand Factor, Factor V, Factor X, Factor XI, Factor XII, Factor XIII, Factor I, Factor II (Prothrombin), Protein C, Protein S, GAS6, or Protein Z as well as their activated forms. A "coagulation factor," as used herein, also includes variants or derivatives thereof, such as genetically engineered or chemically modified variants or active fragments thereof. For instance, useful therapeutic polypeptides may be wild-type polypeptides or may contain mutations. Degree and location of glycosylation or other post-translation modifications may vary depending on the chosen host cells and the nature of the host cellular environment. When referring to specific amino acid sequences, posttranslational modifications of such sequences are encompassed in this application.
[0068] "Coagulation factor" within the above definition includes polypeptides that have the natural amino acid sequence including any natural polymorphisms. It also includes polypeptides with a slightly modified amino acid sequence, for instance, a modified N-terminal or C-terminal end including terminal amino acid deletions or additions, as long as those polypeptides substantially retain the activity of the respective therapeutic polypeptide. Variants included may differ in one or more amino acid residues from the wild type sequence. Examples of such differences may include truncation of the N- and/or C-terminus by one or more amino acid residues (e.g. 1 to 30 amino acid residues), or addition of one or more extra residues at the N- and/or C-terminus, as well as conservative amino acid substitutions, i.e. substitutions performed within groups of amino acids with similar characteristics, e.g. (1) small amino acids, (2) acidic amino acids, (3) polar amino acids, (4) basic amino acids, (5) hydrophobic amino acids, and (6) aromatic amino acids. Examples of such conservative substitutions are shown in the following table.
TABLE-US-00001 TABLE 1 (1) Alanine Glycine (2) Aspartic acid Glutamic acid (3a) Asparagine Glutamine (3b) Serine Threonine (4) Arginine Histidine Lysine (5) Isoleucine Leucine Methionine Valine (6) Phenylalanine Tyrosine Tryptophane
[0069] The in vivo half-life of the fusion proteins of the invention, in general determined as terminal half-life or .beta.-half-life, may be at least about 25%, at least about 50%, or more than 100% higher than the in vivo half-life of the non-fused polypeptide.
[0070] The fusion proteins of the present invention also have at least a 25%, and may have at least a 50% or at least 100% increased molar specific activity compared to the corresponding fusion proteins without cleavable linkers.
[0071] The "molar specific activity" (or molar specific coagulation-related activity as considered here in particular) in this regard is defined for purposes herein as the activity expressed per mole (or e.g. nmole) of the therapeutic polypeptide or therapeutic fusion protein of interest. Calculation of the molar specific activity allows a direct comparison of the activity of the different constructs that is not affected by the different molecular weights or optical densities of the polypeptides studied. The molar specific activity may be calculated as exemplified in table 2 below for FIX and a FIX-HSA fusion protein.
TABLE-US-00002 TABLE 2 Calculation of molar specific activity as shown for a purified FIX-HSA fusion protein Molar optical Calculation of molar Activity/Vol/OD.sub.280 density (OD.sub.(280) specific activity Product OD.sub.(280nm, 1%) MW (IU/L/OD.sub.280) at 1 mol/L) (IU/mol) FIX 13.3 .sup.1) 57 000 determined for 75810 (= MW .times. = (Activity/Vol/OD.sub.280) .times. product OD.sub.(280, 1%)/10) (OD.sub.280 at 1 mol/L) HSA 5.7 .sup.2) 66 300 37791 (= MW .times. OD.sub.(280, 1%)/10) FIX-HSA determined for 113601 (= sum = (Activity/Vol/OD.sub.280) .times. product of molar optical (OD.sub.280 at 1 mol/L) density of FIX and HSA) .sup.1)R. G. DiScipio et al., Biochem. 16: 698-706 (1977) .sup.2)C. Chaudhury et al, J. Exp. Med. 197(3): 315-322 (2003)
[0072] In order to determine a molar specific coagulation-related activity, any assay may be used that determines enzymatic or cofactor activities that are relevant to the coagulation process.
[0073] Therefore "coagulation-related assays" in the sense of the invention comprise any assay which determines enzymatic or cofactor activities that are of relevance in the coagulation process or that are able to determine that either the intrinsic or the extrinsic coagulation cascade has been activated. The "coagulation-related" assay thus may be direct coagulation assays like aPTT, PT, or the thrombin generation assays. However, other assays such as chromogenic assays applied for specific coagulation factors are also included. Examples of such assays or corresponding reagents are Pathromtin.RTM. SL (aPTT assay, Dade Behring) or Thromborel.RTM. S (Prothrombin time assay, Dade Behring) with corresponding coagulation factor deficient plasma (Dade Behring), Thrombin generation assay kits (Technoclone.TM., Thrombinoscope.TM.) using e.g. coagulation factor deficient plasma, chromogenic assays like Biophen.TM. Factor IX (Hyphen BioMed), Staclot.RTM. FVIIa-rTF (Roche Diagnostics GmbH), Coatest.RTM. Factor VIII:C/4 (Chromogenix), or others.
[0074] For purposes of this invention, an increase in any one of the above assays or an equivalent coagulation-related assay is considered to show an increase in molar specific activity. For example, a 25% increase refers to a 25% increase in any of the above or an equivalent assay.
[0075] To determine whether therapeutic fusion proteins fall within the scope of the present invention, the standard against which the molar specific activity of these proteins is compared is a construct in which the respective coagulation factor and the respective HLEP are linked by a non-cleavable linker having the amino acid sequence GGGGGGV (SEQ ID NO: 94).
[0076] In the case of FIX, aPTT assays are often used for determination of coagulation activity. Such a coagulation assay (aPTT assay) is described in example 5 in more detail. However, other coagulation-related assays or assay principles may be applied to determine molar specific activity for FIX.
[0077] Recombinant therapeutic polypeptide drugs are usually expensive and not all countries can afford costly therapies based on such drugs. Increasing the in vivo recovery of such drugs could make the use of these products cheaper and subsequently more patients would benefit from them. In the case of the fusion proteins of the present invention an increased in vivo recovery would also be a desirable advantage. "In vivo recovery" as used herein in the sense of the invention means the amount of product found in blood or plasma shortly after administration of the product. For detection of the in vivo recovery in general the plasma content is determined a few minutes (e.g. 5 or 15 min) after administration of the product.
[0078] Although it is desirable to have a high in vivo recovery and a long half-life for a non-activated coagulation factor, it is advantageous to limit the half-life of a coagulation factor after its activation or the activation of its co-factor in order to avoid a prothrombotic risk. Therefore, after the coagulation process has been initiated, the half-life of the active coagulation factor should again be reduced. This can either be achieved by enhancing inactivation in a coagulation-related mode or by elimination of the coagulation factor.
[0079] "Inactivation" according to the present invention means the decrease of activity of the therapeutic polypeptide which can be caused, for example, by a complex formation of a coagulation factor and an inhibitor of the corresponding coagulation factor or by further proteolytic cleavage as known, e.g., in the case of FVIII and FV.
[0080] The "inactivation rate" of an activated therapeutic fusion protein is defined as the rate the activity is declining, e.g., by reaction with inhibitors or by proteolytic inactivation. The inactivation rate may be measured by following the molar specific activity of the activated coagulation factor over time in the presence of physiologic amounts of inhibitors of this coagulation factor. Alternatively, the inactivation rate may be determined after administration of the activated product to an animal followed by testing of plasma samples at an appropriate time frame using activity and antigen assays.
[0081] To determine whether therapeutic fusion proteins fall within the scope of the present invention, the standard against which the molar specific activity of these proteins is compared is a construct in which the respective coagulation factor and the respective HLEP are linked by a non-cleavable linker having the amino acid sequence GGGGGGV (SEQ ID NO: 94).
[0082] The "elimination rate" of an activated therapeutic fusion protein is defined as the rate the polypeptide is eliminated from the circulation of humans or other animals. The elimination rate may be determined by measuring the pharmacokinetics of the activated, therapeutic fusion protein after intravenous administration. Using an antigen assay, the elimination by direct removal from the circulation can be determined. Using an activity assay in addition, a specific removal and inactivation rate may be determined.
[0083] To determine whether therapeutic fusion proteins fall within the scope of the present invention, the standard against which the molar specific activity of these proteins is compared is a construct in which the respective coagulation factor and the respective HLEP are linked by a non-cleavable linker having the amino acid sequence GGGGGGV (SEQ ID NO: 94).
[0084] According to some embodiments of this invention, the therapeutic polypeptide moiety is coupled to the HLEP moiety by a cleavable peptide linker. The linker should be non-immunogenic and should be flexible enough to allow cleavage by proteases. The cleavage of the linker should proceed comparably fast as the activation of the therapeutic polypeptide within the fusion protein, if the fusion protein is a zymogen.
[0085] The cleavable linker may comprise a sequence derived from [0086] a) the therapeutic polypeptide to be administered itself if it contains proteolytic cleavage sites that are proteolytically cleaved during activation of the therapeutic polypeptide, [0087] b) a substrate polypeptide of this therapeutic polypeptide, or [0088] c) substrate polypeptide cleaved by a protease which is activated or formed by the direct or indirect involvement of the therapeutic polypeptide.
[0089] The peptide linker in some embodiments comprises a sequence of the therapeutic polypeptide to be applied, which should result in a decreased risk of neoantigenic properties of the expressed fusion protein. Also in case the therapeutic protein is a zymogen (e.g. needs to be proteolytically activated) the kinetics of the peptide linker cleavage will more closely reflect the coagulation-related activation kinetics of the zymogen in some embodiments.
[0090] In some embodiments, the therapeutic polypeptide is FIX zymogen and the HLEP is albumin. In some of those embodiments the linker sequence is either derived from the sequences of the activation regions of FIX, from the cleavage region of any substrate of FIX like FX or FVII or from the cleavage region of any substrate polypeptide that is cleaved by a protease in whose activation FIXa is involved.
[0091] In yet other embodiments, the linker peptide is derived from FIX itself. In other embodiments the linker peptide is derived from FX or FVII. In other embodiments the linker sequence comprises two cleavage sequences that can be cleaved by FXIa or FVIIa/TF, two physiologically relevant activators of FIX.
[0092] Exemplary combinations of therapeutic polypeptide, peptide linker, and HLEP include the constructs listed in tables 3a and 3b, but are not limited to them:
TABLE-US-00003 TABLE 3a Examples of possible constructs Linker derived from (with Coagulation modifications, SEQ ID factor Linker HLEP if applicable) NO: Linker not cleavable or not sufficiently rapidly cleavable FIX HSA FIX R 1 HSA FIX GGGGGGV(Sheffield et al.) HSA 94 FIX (GGS)nGS HSA FIX SS(GGS).sub.7GS HSA 30 FIX SSN GS(GGS).sub.3NGS(GGS).sub.3GGNGS HSA 31 Linker with one cleavage site FIX (1-412) SVSQTSKLTR AETVfPDVD HSA FIX 36 FIX (1-412) S'vSQTSKLTRAETVfPDVD GS HSA FIX 37 FIX SVSQTSKLTR AETVFPDVO HSA FIX 38 FIX SVSQTSKLTR AETVFPDVO GS GGS HSA FIX 95 FIX SVSQTSKLTR AETVFPDVO GS HSA FIX 39 FIX SVSQTSKLTR AETVFPDVO NGS HSA FIX 40 FIX SVSQTSKLTR AETVFPDV HSA FIX 96 FIX QTSKLTR AETVFPDV HSA FIX 97 FIX SKLTR AETVFPDV HSA FIX 98 FIX SVSQTSKLTR AETVFP HSA FIX 99 FIX Sv/SQTSKLTRAETW HSA FIX 100 FIX QTSKLTR AETVF HSA FIX 101 FIX SKLTR AETVF HSA FIX 102 FIX SVSQTSKLTR AFT HSA FIX 103 FIX QTSKLTR AET HSA FIX 104 FIX SKLTR AET HSA FIX 105 FIX SVSQTSKLTR GETWPDVO HSA FIX 41 FIX SVSQTSKLTR TETVFPDVD HSA FIX 42 FIX SVSQTSKLTR SETVFPDVO HSA FIX 43 FIX SVSQTSKLTR LETVFPDVO HSA FIX 44 FIX SVSQTSKLTR TEAVFPDVO HSA FIX 45 FIX SVSQTSKLTR GEAVFPDVD HSA FIX 46 FIX QTSKLTR AETVFPDVO GS HSA FIX 106 FIX SKLTR AETVFPDVO GS HSA FIX 107 FIX SKLTR AETVFPDVO HSA FIX 47 FIX QSFNDFTR WGGED HSA FIX 48 FIX QSFNDFTR WGGED GS HSA FIX 49 FIX QSFNDFTR WGGE HSA FIX 108 FIX QSFNDFTRTVGGED HSA FIX 50 FIX QSFNDFTRLVGGED HSA FIX 51 FIX QSFNDFTR GVGGED HSA FIX 52 FIX QSFNDFTR WGGED NGS HSA FIX 53 FIX QSFNDFTR WGGED N HSA FIX 54 FIX PERGDNNLTRIVGGQE GS HSA FX 109 FIX PERGDNNLTR IVGGQE HSA FX 61 FIX PERGDN NLTR IVGGQ HSA FX 110 FIX DNNLTRM3GQ HSA FX 111 FIX SVSQTSKLTR AETVFPDVD Fc FIX 62 FIX QSFNDFTRWGGED N Fc FIX 63 FIX (1-412) SVSQTSKLTR AETWPDVO Fc FIX 64 FIX ASKPQGR IVGG HSAdelDAJH FVII 112 FIX KRNASKPQGR IVGGKV HSA FVII 65 FIX PEEPQLR MKIMN EEAED HSA FVIII 66 FIX DNSPSFIQIRSVAKKHPKT HSA FVIII 67 FIX LSKNNAJEPR SFSQNSRHPS HSA FVIII 68 FIX DED ENQSPR SFQKKTRH YFIA HSA FVIII 69 FIX SPHVtRNRAQSGSVPQ HSA FVIII 70 FVII or FVIIa PEEPQLR MKNNEEAED YDDDLTDS HSA FVIII 71 FVII or FVIIa DDD NSPSFIQIR SVAKKHPKTWVH YAAEEED HSA FVIII 72 FVII or FVIIa LSKNNAJEPR SFSQN SRHPSTR QKQFN A HSA FVIII 73 FVII or FVIIa DEDENQSPR SFQKKTR H YFIAA HSA FVIII 74 FVII or FVIIa D YGMSSSPHVLRN R AGSGSVPGFKKWFQEFT HSA FVIII 75 FVIII Derived from cleavage sites of FVIII, HSA FVIII, FIX or Fqn FIX, or Fibrinogen WVF Derived from cleavage sites of VWF, HSA FIX, FVIII, VWF FVIII, or FIX vw DIYDEDENQSPR SFQKKTRH YFIA HSA FVIII 76 VWF DNSPSFIQIR SVAKKHP HSA FVIII 77 VWF LSKNNAJEPR SFSQNSRHPS HSA FVIII 78 FIX PVSQTSKLTR AETVFPDV HSA FIX 113 FIX PSVSQTSKLTR AETVFPDV HSA FIX 114
[0093] In the case of linkers derived from the N-terminal region of the FIX activation peptide, according to the natural polymorphism T148-A148 the sequences may also contain A instead of T at this position.
TABLE-US-00004 TABLE 3b Examples of possible constructs with two or more cleavage sites Linker derived from Coagulation (partially incl. SEQ ID factor Linker HLEP Modifications) NO: Linker with two cleavage sites FIX SVSQTSKLTR AETVFPDV TQPERGDN NLTR IVGGQE HSA FIX, FX 79 FIX SKLTR AETVFPDN NLTR IVGGQE HSA FIX, FX 80 FIX R AETVFPDV TQ PERGD NNLTR IVGGQE HSA FIX, FX 81 FIX R AETVFPERGDNNLTR IVGGQE HSA FIX, FX 82 FIX SVSQTSKLTR AETVFPD VDYV NNLTR IVGGQE HSA FIX, FX 83 FIX SVSQTSKLTR AETVFPDVD NNLTR IVGGQE HSA FIX, FX 84 FIX SVSQTSKLTR AETVFPDVD NNLTR IVGGQE HSA FIX, FX 85 FIX SVSQTSKLTRAETVFPDVDYVNSTE AETILDNITQSTQSFN HSA FIX 86 DFTRVVGGEDA FIX SVSQTSKLTR AETVFPDV QSFNDFTR VVGGED HSA FIX 87 FIX SVSQTSKLTR AETVFPDVD SFNDFTR VVGGED HSA FIX 88 FIX SVSQTSKLTR AETVFPD VNASKPQGR IVGGKV HSA FIX and FVII 89 FIX SVSQTSKLTR AETVFPDVN ASKPQGRLVGGKV HSA FIX and FVII 90 FIX SVSQTSKLTR AETVFPDVN ASKPQGRTVGGKV HSA FIX and FVII 91 FIX SVSQTSKLTR AETVFPDVD Fc 92
[0094] Variants and fragments of the linkers described in tables 3a and 3b are also encompassed in the present invention as long as the linkers can still be cleaved by the protease or the proteases that cleave the linkers of tables 3a and 3b or by the type of proteases defined above.
[0095] Other combinations of the cleavage sequences described above and their variants shall be included in the present invention.
[0096] In another embodiment, amino acid substitutions are included that change the post-translational modification pattern of the peptide linker. These can be, for example, substitutions of amino acids that are glycosylated, sulphated, or phosphorylated.
[0097] In another embodiment of the invention the peptide linker between the therapeutic polypeptide and the HLEP moiety contains consensus sites for the addition of posttranslational modifications. Such modifications may comprise glycosylation sites. For example, such modifications may comprise at least one N-glycosylation site of the structure Asn-X-Ser/Thr, wherein X denotes any amino acid except proline. Furthermore, such N-glycosylation sites may be inserted close to the amino and/or carboxy terminus of the peptide linker such that they are capable of shielding potential neo-epitopes which might develop at the sequences where the therapeutic polypeptide moiety is transitioning into the peptide linker or where the peptide linker is transitioning into, for example, an albumin moiety sequence.
BRIEF DESCRIPTION OF THE FIGURES
[0098] FIG. 1: In vitro activation of FIX-albumin fusion proteins by FXIa at 37.degree. C. at a molar ratio of FXIa to fusion protein of about 1:500. One fusion protein with non-cleavable linker (1478/797) and two fusion proteins with cleavable linker (1088/797 and 1089/797) were used. Samples were analyzed by SDS-PAGE under reducing conditions followed by Coomassie blue staining
[0099] FIG. 2: Pharmacokinetics of activated rec FIX and FIX-albumin fusion proteins with and without cleavable linker in comparison to non-activated fusion proteins.
[0100] FIG. 3: Inactivation of activated rec FIX or FIX-albumin fusion protein by AT. Residual FIX activity was determined after 120 min using a non-activated partial thromboplastin time assay.
EXAMPLES
Example 1: Generation of cDNAs Encoding FIX and FIX-Albumin Fusion Proteins
[0101] Factor IX coding sequence was amplified by PCR from a human liver cDNA library (ProQuest, Invitrogen) using primers We1403 and We1404 (SEQ ID NOS:5 and 6). After a second round of PCR using primers We1405 and We1406 (SEQ ID NOS:7 and 8) the resulting fragment was cloned into pCR4TOPO (Invitrogen). From there the FIX cDNA was transferred as an EcoRI Fragment into the EcoRI site of pIRESpuro3 (BD Biosciences) wherein an internal XhoI site had been deleted previously. The resulting plasmid was designated pFIX-496 and was the expression vector for factor IX wild-type.
[0102] For the generation of albumin fusion constructs the FIX cDNA was reamplified by PCR under standard conditions using primers We2610 and We2611 (SEQ ID NOS:9 and 10) deleting the stop codon and introducing an XhoI site instead. The resulting FIX fragment was digested with restriction endonucleases EcoRI and XhoI and ligated into an EcoRI/BamH1 digested pIRESpuro3 together with one XhoI/BamH1 digested linker fragment as described below.
[0103] Two different glycine/serine linker fragments without internal cleavage sites were generated: Oligonucleotides We2148 and We2150 (SEQ ID NOS: 11 and 12) were annealed in equimolar concentrations (10 pmol) under standard PCR conditions, filled up and amplified using a PCR protocol of a 2 min. initial denaturation at 94.degree. C. followed by 7 cycles of 15 sec. of denaturation at 94.degree. C., 15 sec. of annealing at 55.degree. C. and 15 sec. of elongation at 72.degree. C., and finalized by an extension step of 5 min at 72.degree. C. The same procedure was performed using oligonucleotides We2156 and We2157 (SEQ ID NOS: 13 and 14). The resulting linker fragments were digested with restriction endonucleases XhoI and BamH1 and used separately in the above described ligation reaction. The resulting plasmids therefore contained the coding sequence for FIX and a C-terminal extension of a glycine/serine linker.
[0104] Two different cleavable linker fragments derived from the activation sites of FIX were generated: Oligonucleotides We2335 and We2336 (SEQ ID NOS:15 and 16), containing the activation cleavage site of the FIX light chain/activation peptide border region, were annealed, filled, and amplified as described above. The resulting linker fragment was digested with restriction endonucleases XhoI and BamH1 and used in the above described ligation reaction. The resulting plasmid therefore contained the coding sequence for FIX and a C-terminal extension of a cleavable FIX sequence (amino acids 136 to 154 of SEQ ID NO:2). In a subsequent site directed mutagenesis reaction with a commercially available mutagenesis kit (QuickChange XL Site Directed Mutagenesis Kit, Stratagene) using oligonucleotides We2636 and We2637 (SEQ ID NOS:17 and 18) the XhoI site was deleted.
[0105] For generation of the second cleavable linker fragment derived from FIX, the same procedure was performed using oligonucleotides We2337 and We2338 (SEQ ID NOS: 19 and 20) for linker construction. The resulting linker fragment was digested with restriction endonucleases XhoI and BamH1 and used in the above described ligation reaction. The resulting plasmid now contained the coding sequence for FIX and a C-terminal extension of a cleavable FIX sequence derived from the activation cleavage site of the FIX activation peptide/heavy chain border region (amino acids 173 to 186 of SEQ ID NO:2). Oligonucleotides We2638 and We 2639 (SEQ ID NOS:21 and 22) were used for deletion of the XhoI site as described above.
[0106] In the next cloning step the above generated plasmids were digested with BamH1 and a BamH1 fragment containing the cDNA of mature human albumin was inserted. This fragment had been generated by PCR on an albumin cDNA sequence using primers We1862 and We1902 (SEQ ID NOS:23 and 24) under standard conditions.
[0107] The final plasmids with non-cleavable glycine/serine linkers were designated pFIX-980 (SEQ ID NO:30) and pFIX-986 (SEQ ID NO:31), respectively. The final plasmids with cleavable linkers derived from FIX sequences were designated pFIX-1088 (SEQ ID NO:40) and pFIX-1089 (SEQ ID NO:49), respectively. Their linker sequences and the C-terminal FIX and N-terminal albumin sequences are outlined below. Proteolytic cleavage sites within the linkers are indicated with arrows, the FIX derived linker sequences are underlined.
TABLE-US-00005 ##STR00001##
[0108] For expression in CHO cells the coding sequences for the FIX albumin fusion protein were transferred into vectors pIRESneo3 (BD Biosciences) or pcDNA3.1 (Invitrogen), respectively.
[0109] Using the above protocols and plasmids and by applying molecular biology techniques known to those skilled in the art (and as described e.g. in Current Protocols in Molecular Biology, Ausubel F M et al. (eds.), including Supplement 80, published October 2007, John Wiley & Sons, Inc.) other constructs can be made with insertions of different linker sequences, e.g. as described in tables 3a and 3b.
[0110] For efficient processing of the propeptide in cells expressing FIX in high amounts coexpression of furin is required (Wasley L C et al. 1993. PACE/Furin can process the vitamin K-dependent pro-factor IX precursor within the secretory pathway. J. Biol. Chem. 268:8458-8465). Furin was amplified from a liver cDNA library (Ambion) using primers We1791 and We1792 (SEQ ID NOS:25 and 26). A second round of PCR using primers We1808 and We1809 (SEQ ID NOS:27 and 28) yielded a furin fragment where the carboxyterminal transmembrane domain (TM) was deleted and a stop codon introduced; this fragment was cloned into pCR4TOPO (Invitrogen). From there the furin.DELTA.TM cDNA was transferred as an EcoRI/NotI Fragment into the EcoRI/NotI sites of pIRESpuro3 (BD Biosciences) wherein an internal XhoI site had been deleted previously. The resulting plasmid was designated pFu-797. This plasmid was cotransfected with all FIX constructs in a 1:5 (pFu-797:pFIX-xxx) molar ratio. The amino acid sequence of the secreted furin encoded by pFu-797 is given as SEQ-ID NO:29.
Example 2: Transfection and Expression of FIX and FIX-Albumin Fusion Proteins
[0111] Plasmids were grown up in E. coli TOP10 (Invitrogen) and purified using standard protocols (Qiagen). HEK-293 cells were transfected using the Lipofectamine 2000 reagent (Invitrogen) and grown up in serum-free medium (Invitrogen 293 Express) in the presence of 50 ng/ml Vitamin K and 4 .mu.g/ml Puromycin. Transfected cell populations were spread through T-flasks into roller bottles or small-scale fermenters from which supernatants were harvested for purification.
[0112] Alternatively, CHO K1 or DG44 cells (Invitrogen) were transfected using the Lipofectamine 2000 reagent (Invitrogen) and grown up in serum-free medium (Invitrogen CD-CHO) in the presence of 50 ng/ml Vitamin K and 500-750 ng/ml Geneticin. High expressing clones were selected and spread through T-flasks into roller bottles or small-scale fermenters from which supernatants were harvested for purification.
Example 3: Purification of FIX and FIX-Albumin Fusion Proteins
[0113] Cell culture harvest containing FIX or FIX albumin fusion protein was applied on a Q-Sepharose FF column previously equilibrated with 50 mM TrisxHCl/100 mM NaCl buffer pH 8.0. Subsequently, the column was washed with equilibration buffer containing 200 mM NaCl. Elution of the bound FIX or FIX fusion protein was achieved by a salt gradient using 50 mM TrisxHCl/200 mM NaCl buffer pH 8.0 as a basis. The eluate was further purified by column chromatography on a hydroxylapatite resin. For this purpose, the eluate of the Q-Sepharose FF column was loaded on a hydroxylapatite chromatography column equilibrated with 50 mM TrisxHCl/100 mM NaCl buffer pH 7.2. The column was washed with the same buffer and FIX or FIX-HSA were eluted using a potassium phosphate gradient at pH 7.2. The eluate was dialyzed to reduce the salt concentration and used for biochemical analysis as well as for determination of the pharmacokinetic parameters. FIX antigen and activity were determined as described in example 5.
Example 4: Alternative Purification Scheme of FIX and FIX-Albumin Fusion Proteins
[0114] As described in example 3, cell culture harvest containing FIX or FIX albumin fusion protein was purified by chromatography on Q-Sepharose FF. The Q-Sepharose eluate was further purified by chromatography on a Heparin-Fractogel column. For this purpose, the Heparin-Fractogel column was equilibrated using 50 mM Tris x HCl, 50 mM NaCl pH 8.0 buffer (EP), the Q-Sepharose FF eluate was applied and the column was washed with equilibration buffer containing 75 mM NaCl. FIX or FIX albumin fusion protein, respectively, was eluted using EP adjusted to 300 mM NaCl.
[0115] The Heparin-Fractogel eluate was further purified by chromatography on a hydroxylapatite chromatography column as described in example 3. The purified FIX resp. FIX albumin fusion protein concentrate was subjected to FIX activity and antigen determination according to example 5 and characterized by further in vitro and in vivo investigations.
Example 5: Determination of FIX Activity and Antigen
[0116] FIX activity was determined as clotting or coagulation activity (FIX:C) using commercially available aPTT reagents (Pathromtin SL and FIX depleted plasma, Dade Behring). An internal substandard calibrated against the WHO International FIX concentrate Standard (96/854) was used as a reference.
[0117] FIX antigen (FIX:Ag) was determined by an ELISA acc. to standard protocols known to those skilled in the art. Briefly, microtiter plates were incubated with 100 .mu.L per well of the capture antibody (Paired antibodies for FIX ELISA 1:200, Cedarlane, but other sources of appropriate antibodies may also be applied) overnight at ambient temperature. After washing plates three times with washing buffer B (Sigma P3563), each well was incubated with 200 .mu.L blocking buffer C (Sigma P3688) for one hour at ambient temperature. After another three wash steps with buffer B, serial dilutions of the test sample in buffer B as well as serial dilutions of a substandard (SHP) in buffer B (volumes per well: 100 .mu.L) were incubated for two hours at ambient temperature. After three wash steps with buffer B, 100 .mu.L of a 1:200 dilution of the detection antibody (Paired antibodies for FIX ELISA, peroxidase labelled, Cedarlane) in buffer B were added to each well and incubated for another two hours at ambient temperature. After three wash steps with buffer B, 100 .mu.L of substrate solution (TMB, Dade Behring, OUVF) were added per well and incubated for 30 minutes at ambient temperature in the dark. Addition of 100 .mu.L undiluted stop solution (Dade Behring, OSFA) prepared the samples for reading in a suitable microplate reader at 450 nm wavelength. Concentrations of test samples were then calculated using the standard curve with standard human plasma as reference.
Example 6: Comparison of FIX-Activity/FIX-Antigen Ratio of Different FIX-Albumin Fusion Proteins in Cell Culture Supernatant
[0118] Cell culture supernatants of HEK cells transfected with DNA constructs coding for FIX-albumin fusion proteins that contained different linker peptides were subjected to FIX activity and antigen testing as described above (see example 5). The ratio of FIX:C to FIX:Ag was calculated representing a measure directly proportional to molar specific activity of the different constructs.
[0119] The results shown in table 4 indicate that there is an increase in activity/antigen ratio upon introduction of cleavable linkers into the FIX-HSA molecule. It also shows that the cleavable linker peptide should have a length of more than two amino acids in order to provide clearly increased activity/antigen ratios.
TABLE-US-00006 TABLE 4 FIX:C/FIX:Ag ratios of FIX-albumin fusion proteins containing different linker peptides Fold increase compared to fusion protein 980/797 with non-cleavable linker FIX-HSA (GGGGGGV) construct Linker FIX:C/FIX:Ag (SEQ ID NO: 94) 1182/797 None <0.031 1366/797 RI <0.068 1478/863 GGGGGGV 0.041 -- (Sheffield et al.) (SEQ ID NO: 94) 980/797 SS(GGS).sub.7GS 0.070 1.7 (SEQ ID NO: 30) 986/797 SSNGS(GGS)3NGS 0.076 1.9 (GGS)3GGNGS (SEQ ID NO: 31) 1483/863 SVSQTSKLTR AETVFPDVD 0.688 16.8 GSGGS (SEQ ID NO: 95) 1088/797 SVSQTSKLTR AETVFPDVD GS 0.832 20.3 (SEQ ID NO: 39) 1365/797 SVSQTSKLTR AETVFPDVD 0.630 15.4 (SEQ ID NO: 36) 1482/863 SVSQTSKLTR AETVFP 0.482 11.8 (SEQ ID NO: 99) 1087/797 SVSQTSKLTR AETVFPDVD GS 0.472 11.5 (SEQ ID NO: 39) (FIX deltaKLT) 1089/797 QSFNDFTR VVGGED GS 0.532 13.0 (SEQ ID NO: 49) 1091/797 PERGDNNLTR IVGGQE GS 0.111 2.7 (SEQ ID NO: 109)
Example 7: Comparison of FIX and FIX-Albumin Fusion Proteins in Respect to Molar Specific Activity, Terminal In Vivo Half-Life and In Vivo Recovery in Rats or Rabbits
[0120] Purified recombinant wild type FIX (rFIX 496/797) and FIX-albumin fusion proteins (rFIX 980/797, rFIX 986/797, rFIX-1088/797 and rFIX 1089/797) were tested for FIX activity in a clotting assay as described above. In parallel, the difference of the optical density at 280 and 320 nm was determined as a measure for protein concentration (OD280-320). The ratios of activity per OD280-320 were calculated and based on the molar optical densities the molar specific activities were calculated. In the following table 5 the results are summarized.
TABLE-US-00007 TABLE 5 Molar specific activities of wt FIX compared to FIX-albumin fusions (linker sequences correspond to SEQ ID NOS 94, 30, & 31, respectively, in order of appearance) FIX Molar Optical clotting Activity/ specific density activity Vol/OD activity* Linker (OD280-320) (IU/mL) (IU/mL/OD) (IU/nmol) rFIX, wt (496/797) -- 0.3798 21.2 55.8 4.23 rFIX-HSA GGGGGGV 2.9189 5.8 2.0 0.23 (non-cleavable, 1478/863 (Sheffield et al.) rFIX-HSA SS (GGS).sub.7 GS 1.1122 3.4 3.0 0.35 (non-cleavable, 980/797) rFIX-HSA SS IMGS (GGS)3 0.8107 3.2 4.0 0.45 (non-cleavable, 986/797) NGS (GGS)3 GGN G rFIX-HSA FXIa cleavable 0.3421 11.9 34.8 3.95 (cleavable, 1088/797) rFIX-HSA FXIa cleavable 0.4512 11.3 25.0 2.84 (cleavable, 1089/797) *Molar specific activity based on activity, optical density and the following molar optical densities: Molar optical density of FIX: OD(280 nm, 1 mol/L) = 75 810 Molar optical density of albumin: OD(280 nm, 1 mol/L) = 37 791 Molar optical density of FIX-albumin fusion protein: OD(280 nm, 1 mol/L) = 113 601
[0121] Taking the results summarized in Table 5 into account, two constructs that were generated according to the present invention show highly increased molar specific activities compared to the fusion proteins with non-cleavable linkers. In addition, the molar specific activity of these constructs was only moderately decreased compared to wild type rFIX.
[0122] In vitro investigations of the proteolytic cleavage reactions by Factor XIa (FXIa) confirmed that FIX-albumin fusion proteins containing a cleavable linker such as construct no. 1088/797 or 1089/797 are activated and in parallel the linker is cleaved resulting in release of the albumin moiety (FIG. 1). The fusion protein with non-cleavable linker did not show a corresponding release of the albumin moiety.
[0123] In the case of FVIIa as cleaving protease in the presence of tissue factor, the FIX-albumin fusion proteins 1088/797 or 1089/797 containing a cleavable linker also showed release of the albumin moiety in parallel to release of the FIX activation peptide (Data not shown).
[0124] In addition to determination of molar specific coagulation activity, the polypeptides no. 496/797, 980/797, 986/797, 1088/797 and 1089/797 described above were administered intravenously to narcotized CD/Lewis rats (6 rats per substance) and/or rabbits (4 rabbits per substance) with a dose of 50 IU/kg body weight. Blood samples were drawn prior to test substance administration and at appropriate intervals starting at 5 minutes after administration of the test substances. FIX antigen content was subsequently quantified by an ELISA assay specific for human Factor IX (see above). The mean values of the respective groups were used to calculate in vivo recovery after 5 min. Half-lives for each protein were calculated using the time points of the beta phase of elimination (terminal half-life) according to the formula t.sub.1/2=ln 2/k, whereas k is the slope of the regression line obtained upon plotting FIX:Ag levels in logarithmic scale and time in linear scale.
[0125] Calculated in vivo half-lives are summarized in table 6. In rats as well as in rabbits the in vivo half-lives of the FIX-albumin fusion proteins were found to be significantly increased in comparison to non-fused wild-type recombinant FIX prepared in-house or in comparison to the commercially available recombinant FIX product BeneFIX.RTM.. The in vivo half-lives of the albumin fusion proteins compared to BeneFIX.RTM. were increased to about 200-400%, depending on the animal species or construct used (Table 6).
[0126] To evaluate the in vivo recovery, the FIX antigen levels measured per mL of plasma at their maximum concentrations after intravenous administration (t=5 min) were related to the amount of product applied per kg. Alternatively, a percentage was calculated by relating the determined antigen level (IU/mL) 5 min post infusion to the theoretical product level expected at 100% recovery (product applied per kg divided by an assumed plasma volume of 40 mL per kg). The in vivo recoveries (IVR) of the FIX-albumin fusion proteins were significantly higher than the in vitro recoveries of rFIX (496/797) or BeneFIX.RTM. (Table 7).
TABLE-US-00008 TABLE 6 Terminal in vivo half-lives of FIX preparations derived from recombinant expression (BeneFIX .RTM., rFIX 496/797) and FIX albumin fusion proteins (rFIX 980/797, rFIX 986/797, rFIX 1088/797, and rFIX 1089/797) after intravenous administration of 50 IU/kg into rats and/or 50 IU/kg into rabbits, respectively. Rat experiments PSR18-05, PSRC06-05, Rabbit experiment PSR02-05 PSK11-05 Terminal relative to Terminal relative to half-life BeneFIX half-life BeneFIX (h) [%] (h) [%] rFIX 496/797 4.5* 91 n.t. n.t. rFIX 980/797 11.6* 234 36.9.degree. 410 29.3.degree. 326 (2.sup.nd exp.) rFIX 986/797 10.5* 212 n.t. n.t. rFIX 1088/797 8.3* 168 30.3.degree. 337 rFIX 1089/797 10.5* 212 n.t. n.t. BeneFIX 4.95* (mean of 100 9.0.degree. 100 5.3 and 4.6) *Determined between 120 and 1440 min .degree.Determined between 4 and 96 h
TABLE-US-00009 TABLE 7 In vivo recoveries (amount of substance 5 minutes post administration) of recombinant FIX preparations (BeneFIX, rFIX 496/797) and FIX albumin fusion proteins (rFIX 1088/797, rFIX 1089/797) after intravenous administration of 50 IU/kg into rats. The percentage of in vivo recovery was calculated based on an assumed plasma volume of 40 mL/kg. rat expriment in vivo recovery relative to IU/dL per BeneFIX IU/kg/[%]* [%] rFIX 0.462/18.5 74.6 496/797 rFIX 1.034/41.4 166.5 1088/797 rFIX 1.063/42.5 171.2 1089/797 BeneFIX 0.621/24.8 100 *Calculated based on a plasma volume of 40 mL/kg
Example 8: In Vitro Activation of FIX Albumin Fusion Proteins with/without Cleavable Linker (1088/797 and 980/797) and Determination of Pharmacokinetics in Rats
[0127] FIX-albumin fusion proteins and rec FIX were activated in vitro using commercially available Factor XIa (Kordia). Briefly, identical molar amounts of FIX or FIX-albumin fusion protein (3.0.times.10.sup.-6 mol/L) were activated at 37.degree. C. in solution in the presence of FXIa (1.9.times.10.sup.-8 mol/L) and CaCl.sub.2) (1.5 mmol/L) buffered at pH 6.8. After complete activation as shown by SDS-PAGE the reaction was stopped by addition of a 5.times. molar excess of C1-Inhibitor (Berinert.RTM. P) based on the amount of FXIa. The samples were stored frozen below -70.degree. C. until start of pharmacokinetic investigation.
[0128] A pharmacokinetic investigation of the activated FIX and the FIX-albumin fusion proteins was performed in rats as described in example 7 and the results were compared to a pharmacokinetic results covering non-activated fusion proteins. It turned out that the activated fusion proteins demonstrated significantly reduced half-lives as well as AUC's compared to the non-activated molecules (FIG. 2). Upon activation the FIX-fusion protein with cleavable linker (1088/797) showed a pharmacokinetic behaviour very similar to activated rec FIX (BeneFIX.RTM.) whereas the activated fusion protein with non-cleavable linker (980/797) resulted in a higher initial as well as terminal half-life compared to activated fusion protein 1088/797 with cleavable linker. Therefore, in this example, the cleavable linker results in increased elimination of the coagulation factor after activation and, therefore, avoids accumulation of potentially thrombogenic, activated fusion proteins with extended half-lives.
Example 9: Comparison of FIX-Albumin Fusion Proteins with/without Cleavable Linker in Respect to Inactivation Rate of the Activated Coagulation Factors by Antithrombin III (AT)
[0129] FIX fusion proteins with (1088/797) and without (980/797) cleavable linker were activated by incubation with FXIa as described in example 8. The activated factors were incubated with AT for 120 min and residual FIXa activity was determined using a manual FIX clotting assay method without activation (naPTT, see below) acc. to Schnitger and Gross. As control samples the activated FIX-albumin fusion proteins were used in presence of the same amount of AT but without incubation.
[0130] The FIX activity was determined with the aid of a non-activated partial thromboplastin time assay (naPTT) using FIX deficient plasma from Dade Behring. The samples were prediluted in a buffer of pH 6.8 containing His, Gly, Sucrose, and Tween.RTM. 80. The whole determination was performed using coagulometers acc. to Schnitger & Gross. A mixture of 0.1 ml F IX deficient plasma, 0.1 ml sample, and 0.1 ml of 0.1% Phospholipids (Rhone-Poulenc-Nattermann, 1:3 prediluted in imidazole buffer supplemented with 1% HSA) was incubated for 2 minutes at 37.degree. C. The coagulation reaction was initiated by adding 0.1 ml 0.025 mol/l CaCl.sub.2) solution and the clotting time was determined.
[0131] FIG. 3 shows the results of a corresponding inactivation experiment. In the case of the fusion protein with cleavable linker (1088/797) an increase in clotting time from 210 to 540 sec (factor of 2.57.times.) demonstrated an accelerated inactivation process of FIXa activity by AT compared to a fusion protein with non-cleavable linker (980/797) that only showed an increase from 196 to 411 sec (factor of 2.10.times.). Most probably, the albumin residue sterically affects the AT dependent inactivation process in the case of the fusion protein with non-cleavable linker whereas in the case of the fusion protein with cleavable linker the albumin residue is cleaved off resulting in an accelerated inactivation by AT.
Example 10: Design of FIX-HSA Fusion Proteins with Reduced Immunogenicity
[0132] As there is with any fusion between two proteins a slight risk associated that a neoepitope is created around the fusion point it was investigated whether the linker region as described in table 3a and 3b could be modified in order to decrease this risk.
[0133] In the course of this investigation all proposed linker sequences and the adjacent regions of FIX and HSA were analyzed for potential T-cell epitopes by way of prediction of binding capabability to multiple MHC-II alleles. One of these approaches involved the PreDeFT analysis offered by the company EpiVax (146 Clifford St., Providence, R.I. 02903, USA) in which the input sequences were parsed into overlapping 9-mer frames where each frame overlaps the last by 8 amino acids. Each frame was then assessed for its ability to bind with a set of common HLA. These detailed findings were then summarized producing regional and overall assessments of immunogenic potential. Finally, any epitope clusters identified were screened against the non-redundant protein database at GenBank and EpiVax's own database of known MHC ligands and T-cell epitopes.
[0134] As a result of these in silico predictions the following FIX fusion proteins were cloned, expressed and purified:
TABLE-US-00010 SEQ ID NO: 113 FIX- PVSQTSKLTRAETVFPDV-HSA SEQ ID NO: 114 FIX-PSVSQTSKLTRAETVFPDV-HSA
Example 11: Neoantigenicity Test
[0135] FIX-HSA fusion proteins comprising linkers SEQ ID NO:113 or 114 can be shown to display a reduced immunogenicity compared to fusion proteins comprising different linkers or to display a comparable immunogenicity as compared to wild type factor FIX by the following neoantigenicity test.
[0136] Products to be compared are administered subcutaneously into rabbits with or in the absence of Freund's adjuvant. The endpoint assay is a native Western blot.
[0137] A suitable dose of either the FIX-HSA fusion with a linker with reduced immunogenicity or a FIX-HSA fusion with a linker of enhanced immunogenicity or FIX wild type can be administered as a slow bolus.
[0138] A sample can be taken about 30 to 40 days after the start of the immunization and be assayed by a Western blot method to ensure that the rabbits developed antibodies against each of the respective immunogens.
[0139] Antibodies against the test sample for which a potentially increased immunogenicity is to be measured are blocked by an excess of a control sample (e.g. wild type FIX) By doing so all of the antibodies present which formed against native epitopes are unable to react when that antibody is used as a probe in a Western blot. The test and the control sample are run on the Western blot membrane and the blocked antibody is used as a probe.
[0140] If there were epitopes on the test samples (here FIX-HSA fusion protein with a linker) but not in the control sample (wild type FIX) that caused antibody formation in the rabbits, these antibodies would be detected after blocking as residual antibodies which would react with the test sample, but not the control sample.
[0141] Likewise FIX-HSA with a linker having predicted reduced immunogenicity can be used as a control versus a FIX-HSA with a linker having predicted increased immunogenicity as a test sample.
[0142] Preferably the same Western blot is assayed with non-blocked antibodies raised against the test sample as a positive control. Here it is expected that test sample as well as control sample are detected in the Western blot assay.
[0143] If with blocked antibodies raised against the test sample both test and control sample are not detected it can be concluded that the test sample has no neoepitopes as compared to the control sample.
[0144] If with blocked antibodies raised against the test sample only the test sample but not the control sample is detected it can be concluded that the test sample has neoepitopes as compared to the control sample.
[0145] For doing the analysis the IgG fraction of the hyper immune pooled serum can be purified using a protein A column from Pierce or other suppliers with a bed volume for example greater than 1 ml according to the instructions of the supplier. Preferably the rabbit sera are delipidated for example with trichlorotrifluoroethane.
[0146] Blocking is done for example by mixing 0.1-1.00 mg of the purified antiserum with 0.1 to 100 mg of the control sample in for example a 0.5 to 5 ml centrifuge cup, bringing the volume up to 0.5 to 3 ml. The tubes are then rotated slowly at room temperature of a minimum of 2 hours.
[0147] Nonblocked control antibodies to be used as a positive control can be prepared in the same way except that no control sample is used for blocking.
[0148] All blocked and nonblocked antibodies can be added to 3 to 4.5 ml of 5% dry milk solution in TBS+0.1% Tween-20 before incubation with their respective membrane. Preferably Western blots are performed as native Western blots.
BRIEF DESCRIPTION OF THE FIGURES
[0149] FIG. 1: In vitro activation of FIX-albumin fusion proteins by FXIa at 37.degree. C. at a molar ratio of FXIa to fusion protein of about 1:500. One fusion protein with non-cleavable linker (1478/797) and two fusion proteins with cleavable linker (1088/797 and 1089/797) were used. Samples were analyzed by SDS-PAGE under reducing conditions followed by Coomassie blue staining.
[0150] FIG. 2: Pharmacokinetics of activated rec FIX and FIX-albumin fusion proteins with and without cleavable linker in comparison to non-activated fusion proteins.
[0151] FIG. 3: Inactivation of activated rec FIX or FIX-albumin fusion protein by AT. Residual FIX activity was determined after 120 min using a non-activated partial thromboplastin time assay.
Sequence CWU 1
1
1141585PRTHomo sapiens 1Asp Ala His Lys Ser Glu Val Ala His Arg Phe
Lys Asp Leu Gly Glu1 5 10 15Glu Asn Phe Lys Ala Leu Val Leu Ile Ala
Phe Ala Gln Tyr Leu Gln 20 25 30Gln Cys Pro Phe Glu Asp His Val Lys
Leu Val Asn Glu Val Thr Glu 35 40 45Phe Ala Lys Thr Cys Val Ala Asp
Glu Ser Ala Glu Asn Cys Asp Lys 50 55 60Ser Leu His Thr Leu Phe Gly
Asp Lys Leu Cys Thr Val Ala Thr Leu65 70 75 80Arg Glu Thr Tyr Gly
Glu Met Ala Asp Cys Cys Ala Lys Gln Glu Pro 85 90 95Glu Arg Asn Glu
Cys Phe Leu Gln His Lys Asp Asp Asn Pro Asn Leu 100 105 110Pro Arg
Leu Val Arg Pro Glu Val Asp Val Met Cys Thr Ala Phe His 115 120
125Asp Asn Glu Glu Thr Phe Leu Lys Lys Tyr Leu Tyr Glu Ile Ala Arg
130 135 140Arg His Pro Tyr Phe Tyr Ala Pro Glu Leu Leu Phe Phe Ala
Lys Arg145 150 155 160Tyr Lys Ala Ala Phe Thr Glu Cys Cys Gln Ala
Ala Asp Lys Ala Ala 165 170 175Cys Leu Leu Pro Lys Leu Asp Glu Leu
Arg Asp Glu Gly Lys Ala Ser 180 185 190Ser Ala Lys Gln Arg Leu Lys
Cys Ala Ser Leu Gln Lys Phe Gly Glu 195 200 205Arg Ala Phe Lys Ala
Trp Ala Val Ala Arg Leu Ser Gln Arg Phe Pro 210 215 220Lys Ala Glu
Phe Ala Glu Val Ser Lys Leu Val Thr Asp Leu Thr Lys225 230 235
240Val His Thr Glu Cys Cys His Gly Asp Leu Leu Glu Cys Ala Asp Asp
245 250 255Arg Ala Asp Leu Ala Lys Tyr Ile Cys Glu Asn Gln Asp Ser
Ile Ser 260 265 270Ser Lys Leu Lys Glu Cys Cys Glu Lys Pro Leu Leu
Glu Lys Ser His 275 280 285Cys Ile Ala Glu Val Glu Asn Asp Glu Met
Pro Ala Asp Leu Pro Ser 290 295 300Leu Ala Ala Asp Phe Val Glu Ser
Lys Asp Val Cys Lys Asn Tyr Ala305 310 315 320Glu Ala Lys Asp Val
Phe Leu Gly Met Phe Leu Tyr Glu Tyr Ala Arg 325 330 335Arg His Pro
Asp Tyr Ser Val Val Leu Leu Leu Arg Leu Ala Lys Thr 340 345 350Tyr
Glu Thr Thr Leu Glu Lys Cys Cys Ala Ala Ala Asp Pro His Glu 355 360
365Cys Tyr Ala Lys Val Phe Asp Glu Phe Lys Pro Leu Val Glu Glu Pro
370 375 380Gln Asn Leu Ile Lys Gln Asn Cys Glu Leu Phe Glu Gln Leu
Gly Glu385 390 395 400Tyr Lys Phe Gln Asn Ala Leu Leu Val Arg Tyr
Thr Lys Lys Val Pro 405 410 415Gln Val Ser Thr Pro Thr Leu Val Glu
Val Ser Arg Asn Leu Gly Lys 420 425 430Val Gly Ser Lys Cys Cys Lys
His Pro Glu Ala Lys Arg Met Pro Cys 435 440 445Ala Glu Asp Tyr Leu
Ser Val Val Leu Asn Gln Leu Cys Val Leu His 450 455 460Glu Lys Thr
Pro Val Ser Asp Arg Val Thr Lys Cys Cys Thr Glu Ser465 470 475
480Leu Val Asn Arg Arg Pro Cys Phe Ser Ala Leu Glu Val Asp Glu Thr
485 490 495Tyr Val Pro Lys Glu Phe Asn Ala Glu Thr Phe Thr Phe His
Ala Asp 500 505 510Ile Cys Thr Leu Ser Glu Lys Glu Arg Gln Ile Lys
Lys Gln Thr Ala 515 520 525Leu Val Glu Leu Val Lys His Lys Pro Lys
Ala Thr Lys Glu Gln Leu 530 535 540Lys Ala Val Met Asp Asp Phe Ala
Ala Phe Val Glu Lys Cys Cys Lys545 550 555 560Ala Asp Asp Lys Glu
Thr Cys Phe Ala Glu Glu Gly Lys Lys Leu Val 565 570 575Ala Ala Ser
Gln Ala Ala Leu Gly Leu 580 5852415PRTHomo sapiens 2Tyr Asn Ser Gly
Lys Leu Glu Glu Phe Val Gln Gly Asn Leu Glu Arg1 5 10 15Glu Cys Met
Glu Glu Lys Cys Ser Phe Glu Glu Ala Arg Glu Val Phe 20 25 30Glu Asn
Thr Glu Arg Thr Thr Glu Phe Trp Lys Gln Tyr Val Asp Gly 35 40 45Asp
Gln Cys Glu Ser Asn Pro Cys Leu Asn Gly Gly Ser Cys Lys Asp 50 55
60Asp Ile Asn Ser Tyr Glu Cys Trp Cys Pro Phe Gly Phe Glu Gly Lys65
70 75 80Asn Cys Glu Leu Asp Val Thr Cys Asn Ile Lys Asn Gly Arg Cys
Glu 85 90 95Gln Phe Cys Lys Asn Ser Ala Asp Asn Lys Val Val Cys Ser
Cys Thr 100 105 110Glu Gly Tyr Arg Leu Ala Glu Asn Gln Lys Ser Cys
Glu Pro Ala Val 115 120 125Pro Phe Pro Cys Gly Arg Val Ser Val Ser
Gln Thr Ser Lys Leu Thr 130 135 140Arg Ala Glu Thr Val Phe Pro Asp
Val Asp Tyr Val Asn Ser Thr Glu145 150 155 160Ala Glu Thr Ile Leu
Asp Asn Ile Thr Gln Ser Thr Gln Ser Phe Asn 165 170 175Asp Phe Thr
Arg Val Val Gly Gly Glu Asp Ala Lys Pro Gly Gln Phe 180 185 190Pro
Trp Gln Val Val Leu Asn Gly Lys Val Asp Ala Phe Cys Gly Gly 195 200
205Ser Ile Val Asn Glu Lys Trp Ile Val Thr Ala Ala His Cys Val Glu
210 215 220Thr Gly Val Lys Ile Thr Val Val Ala Gly Glu His Asn Ile
Glu Glu225 230 235 240Thr Glu His Thr Glu Gln Lys Arg Asn Val Ile
Arg Ile Ile Pro His 245 250 255His Asn Tyr Asn Ala Ala Ile Asn Lys
Tyr Asn His Asp Ile Ala Leu 260 265 270Leu Glu Leu Asp Glu Pro Leu
Val Leu Asn Ser Tyr Val Thr Pro Ile 275 280 285Cys Ile Ala Asp Lys
Glu Tyr Thr Asn Ile Phe Leu Lys Phe Gly Ser 290 295 300Gly Tyr Val
Ser Gly Trp Gly Arg Val Phe His Lys Gly Arg Ser Ala305 310 315
320Leu Val Leu Gln Tyr Leu Arg Val Pro Leu Val Asp Arg Ala Thr Cys
325 330 335Leu Arg Ser Thr Lys Phe Thr Ile Tyr Asn Asn Met Phe Cys
Ala Gly 340 345 350Phe His Glu Gly Gly Arg Asp Ser Cys Gln Gly Asp
Ser Gly Gly Pro 355 360 365His Val Thr Glu Val Glu Gly Thr Ser Phe
Leu Thr Gly Ile Ile Ser 370 375 380Trp Gly Glu Glu Cys Ala Met Lys
Gly Lys Tyr Gly Ile Tyr Thr Lys385 390 395 400Val Ser Arg Tyr Val
Asn Trp Ile Lys Glu Lys Thr Lys Leu Thr 405 410 41531386DNAHomo
sapiens 3atgcagcgcg tgaacatgat catggcagaa tcaccaggcc tcatcaccat
ctgcctttta 60ggatatctac tcagtgctga atgtacagtt tttcttgatc atgaaaacgc
caacaaaatt 120ctgaatcggc caaagaggta taattcaggt aaattggaag
agtttgttca agggaacctt 180gagagagaat gtatggaaga aaagtgtagt
tttgaagaag cacgagaagt ttttgaaaac 240actgaaagaa caactgaatt
ttggaagcag tatgttgatg gagatcagtg tgagtccaat 300ccatgtttaa
atggcggcag ttgcaaggat gacattaatt cctatgaatg ttggtgtccc
360tttggatttg aaggaaagaa ctgtgaatta gatgtaacat gtaacattaa
gaatggcaga 420tgcgagcagt tttgtaaaaa tagtgctgat aacaaggtgg
tttgctcctg tactgaggga 480tatcgacttg cagaaaacca gaagtcctgt
gaaccagcag tgccatttcc atgtggaaga 540gtttctgttt cacaaacttc
taagctcacc cgtgctgaga ctgtttttcc tgatgtggac 600tatgtaaatt
ctactgaagc tgaaaccatt ttggataaca tcactcaaag cacccaatca
660tttaatgact tcactcgggt tgttggtgga gaagatgcca aaccaggtca
attcccttgg 720caggttgttt tgaatggtaa agttgatgca ttctgtggag
gctctatcgt taatgaaaaa 780tggattgtaa ctgctgccca ctgtgttgaa
actggtgtta aaattacagt tgtcgcaggt 840gaacataata ttgaggagac
agaacataca gagcaaaagc gaaatgtgat tcgaattatt 900cctcaccaca
actacaatgc agctattaat aagtacaacc atgacattgc ccttctggaa
960ctggacgaac ccttagtgct aaacagctac gttacaccta tttgcattgc
tgacaaggaa 1020tacacgaaca tcttcctcaa atttggatct ggctatgtaa
gtggctgggg aagagtcttc 1080cacaaaggga gatcagcttt agttcttcag
taccttagag ttccacttgt tgaccgagcc 1140acatgtcttc gatctacaaa
gttcaccatc tataacaaca tgttctgtgc tggcttccat 1200gaaggaggta
gagattcatg tcaaggagat agtgggggac cccatgttac tgaagtggaa
1260gggaccagtt tcttaactgg aattattagc tggggtgaag agtgtgcaat
gaaaggcaaa 1320tatggaatat ataccaaggt atcccggtat gtcaactgga
ttaaggaaaa aacaaagctc 1380acttaa 138641830DNAHomo sapiens
4atgaagtggg taacctttat ttcccttctt tttctcttta gctcggctta ttccaggggt
60gtgtttcgtc gagatgcaca caagagtgag gttgctcatc ggtttaaaga tttgggagaa
120gaaaatttca aagccttggt gttgattgcc tttgctcagt atcttcagca
gtgtccattt 180gaagatcatg taaaattagt gaatgaagta actgaatttg
caaaaacatg tgttgctgat 240gagtcagctg aaaattgtga caaatcactt
catacccttt ttggagacaa attatgcaca 300gttgcaactc ttcgtgaaac
ctatggtgaa atggctgact gctgtgcaaa acaagaacct 360gagagaaatg
aatgcttctt gcaacacaaa gatgacaacc caaacctccc ccgattggtg
420agaccagagg ttgatgtgat gtgcactgct tttcatgaca atgaagagac
atttttgaaa 480aaatacttat atgaaattgc cagaagacat ccttactttt
atgccccgga actccttttc 540tttgctaaaa ggtataaagc tgcttttaca
gaatgttgcc aagctgctga taaagctgcc 600tgcctgttgc caaagctcga
tgaacttcgg gatgaaggga aggcttcgtc tgccaaacag 660agactcaagt
gtgccagtct ccaaaaattt ggagaaagag ctttcaaagc atgggcagta
720gctcgcctga gccagagatt tcccaaagct gagtttgcag aagtttccaa
gttagtgaca 780gatcttacca aagtccacac ggaatgctgc catggagatc
tgcttgaatg tgctgatgac 840agggcggacc ttgccaagta tatctgtgaa
aatcaagatt cgatctccag taaactgaag 900gaatgctgtg aaaaacctct
gttggaaaaa tcccactgca ttgccgaagt ggaaaatgat 960gagatgcctg
ctgacttgcc ttcattagct gctgattttg ttgaaagtaa ggatgtttgc
1020aaaaactatg ctgaggcaaa ggatgtcttc ctgggcatgt ttttgtatga
atatgcaaga 1080aggcatcctg attactctgt cgtgctgctg ctgagacttg
ccaagacata tgaaaccact 1140ctagagaagt gctgtgccgc tgcagatcct
catgaatgct atgccaaagt gttcgatgaa 1200tttaaacctc ttgtggaaga
gcctcagaat ttaatcaaac aaaattgtga gctttttgag 1260cagcttggag
agtacaaatt ccagaatgcg ctattagttc gttacaccaa gaaagtaccc
1320caagtgtcaa ctccaactct tgtagaggtc tcaagaaacc taggaaaagt
gggcagcaaa 1380tgttgtaaac atcctgaagc aaaaagaatg ccctgtgcag
aagactatct atccgtggtc 1440ctgaaccagt tatgtgtgtt gcatgagaaa
acgccagtaa gtgacagagt caccaaatgc 1500tgcacagaat ccttggtgaa
caggcgacca tgcttttcag ctctggaagt cgatgaaaca 1560tacgttccca
aagagtttaa tgctgaaaca ttcaccttcc atgcagatat atgcacactt
1620tctgagaagg agagacaaat caagaaacaa actgcacttg ttgagctcgt
gaaacacaag 1680cccaaggcaa caaaagagca actgaaagct gttatggatg
atttcgcagc ttttgtagag 1740aagtgctgca aggctgacga taaggagacc
tgctttgccg aggagggtaa aaaacttgtt 1800gctgcaagtc aagctgcctt
aggcttataa 1830521DNAArtificial SequenceSynthetic 5ccactttcac
aatctgctag c 21623DNAArtificial SequenceSynthetic 6caattccaat
gaattaacct tgg 23721DNAArtificial SequenceSynthetic 7atgcagcgcg
tgaacatgat c 21825DNAArtificial SequenceSynthetic 8tcattaagtg
agctttgttt tttcc 25921DNAArtificial SequenceSynthetic 9gattcgaatt
cgcccttatg c 211032DNAArtificial SequenceSynthetic 10cgctcgaggt
gagctttgtt ttttccttaa tc 321152DNAArtificial SequenceSynthetic
11ctcgagcggg ggatctggcg ggtctggagg ctctggaggg tcgggaggct ct
521246DNAArtificial SequenceSynthetic 12ggatccagat cccccagagc
ctccagagcc tcccgaccct ccagag 461356DNAArtificial SequenceSynthetic
13ctcgagcaat ggatctggcg ggtctggagg ctctggaggg tcgaatggct ctggag
561464DNAArtificial SequenceSynthetic 14ggatccgttc cctccagacc
cgccagatcc cccagagcct ccagagccat tcgaccctcc 60agag
641546DNAArtificial SequenceSynthetic 15cctcgagctc tgtgagccag
acctccaagc tcaccagggc cgagac 461644DNAArtificial SequenceSynthetic
16gggatccgtc cacatcaggg aagacagtct cggccctggt gagc
441733DNAArtificial SequenceSynthetic 17ggaaaaaaca aagctcactt
ctgtgagcca gac 331833DNAArtificial SequenceSynthetic 18gtctggctca
cagaagtgag ctttgttttt tcc 331938DNAArtificial SequenceSynthetic
19cctcgagcag agcttcaatg acttcacccg ggtggtgg 382041DNAArtificial
SequenceSynthetic 20gggatccatc ctccccgccc accacccggg tgaagtcatt g
412134DNAArtificial SequenceSynthetic 21ggaaaaaaca aagctcactc
agagcttcaa tgac 342234DNAArtificial SequenceSynthetic 22gtcattgaag
ctctgagtga gctttgtttt ttcc 342331DNAArtificial SequenceSynthetic
23gtgggatccg atgcacacaa gagtgaggtt g 312435DNAArtificial
SequenceSynthetic 24cacggatccc tataagccta aggcagcttg acttg
352518DNAArtificial SequenceSynthetic 25caaggagacg ggcgctcc
182619DNAArtificial SequenceSynthetic 26gcccaaggag gggattggc
192730DNAArtificial SequenceSynthetic 27gtggaattca tggagctgag
gccctggttg 302838DNAArtificial SequenceSynthetic 28cacgcggccg
ctcactacag ccgttgcccc gcctccac 3829704PRTHomo Sapiens 29Met Glu Leu
Arg Pro Trp Leu Leu Trp Val Val Ala Ala Thr Gly Thr1 5 10 15Leu Val
Leu Leu Ala Ala Asp Ala Gln Gly Gln Lys Val Phe Thr Asn 20 25 30Thr
Trp Ala Val Arg Ile Pro Gly Gly Pro Ala Val Ala Asn Ser Val 35 40
45Ala Arg Lys His Gly Phe Leu Asn Leu Gly Gln Ile Phe Gly Asp Tyr
50 55 60Tyr His Phe Trp His Arg Gly Val Thr Lys Arg Ser Leu Ser Pro
His65 70 75 80Arg Pro Arg His Ser Arg Leu Gln Arg Glu Pro Gln Val
Gln Trp Leu 85 90 95Glu Gln Gln Val Ala Lys Arg Arg Thr Lys Arg Asp
Val Tyr Gln Glu 100 105 110Pro Thr Asp Pro Lys Phe Pro Gln Gln Trp
Tyr Leu Ser Gly Val Thr 115 120 125Gln Arg Asp Leu Asn Val Lys Ala
Ala Trp Ala Gln Gly Tyr Thr Gly 130 135 140His Gly Ile Val Val Ser
Ile Leu Asp Asp Gly Ile Glu Lys Asn His145 150 155 160Pro Asp Leu
Ala Gly Asn Tyr Asp Pro Gly Ala Ser Phe Asp Val Asn 165 170 175Asp
Gln Asp Pro Asp Pro Gln Pro Arg Tyr Thr Gln Met Asn Asp Asn 180 185
190Arg His Gly Thr Arg Cys Ala Gly Glu Val Ala Ala Val Ala Asn Asn
195 200 205Gly Val Cys Gly Val Gly Val Ala Tyr Asn Ala Arg Ile Gly
Gly Val 210 215 220Arg Met Leu Asp Gly Glu Val Thr Asp Ala Val Glu
Ala Arg Ser Leu225 230 235 240Gly Leu Asn Pro Asn His Ile His Ile
Tyr Ser Ala Ser Trp Gly Pro 245 250 255Glu Asp Asp Gly Lys Thr Val
Asp Gly Pro Ala Arg Leu Ala Glu Glu 260 265 270Ala Phe Phe Arg Gly
Val Ser Gln Gly Arg Gly Gly Leu Gly Ser Ile 275 280 285Phe Val Trp
Ala Ser Gly Asn Gly Gly Arg Glu His Asp Ser Cys Asn 290 295 300Cys
Asp Gly Tyr Thr Asn Ser Ile Tyr Thr Leu Ser Ile Ser Ser Ala305 310
315 320Thr Gln Phe Gly Asn Val Pro Trp Tyr Ser Glu Ala Cys Ser Ser
Thr 325 330 335Leu Ala Thr Thr Tyr Ser Ser Gly Asn Gln Asn Glu Lys
Gln Ile Val 340 345 350Thr Thr Asp Leu Arg Gln Lys Cys Thr Glu Ser
His Thr Gly Thr Ser 355 360 365Ala Ser Ala Pro Leu Ala Ala Gly Ile
Ile Ala Leu Thr Leu Glu Ala 370 375 380Asn Lys Asn Leu Thr Trp Arg
Asp Met Gln His Leu Val Val Gln Thr385 390 395 400Ser Lys Pro Ala
His Leu Asn Ala Asn Asp Trp Ala Thr Asn Gly Val 405 410 415Gly Arg
Lys Val Ser His Ser Tyr Gly Tyr Gly Leu Leu Asp Ala Gly 420 425
430Ala Met Val Ala Leu Ala Gln Asn Trp Thr Thr Val Ala Pro Gln Arg
435 440 445Lys Cys Ile Ile Asp Ile Leu Thr Glu Pro Lys Asp Ile Gly
Lys Arg 450 455 460Leu Glu Val Arg Lys Thr Val Thr Ala Cys Leu Gly
Glu Pro Asn His465 470 475 480Ile Thr Arg Leu Glu His Ala Gln Ala
Arg Leu Thr Leu Ser Tyr Asn 485 490 495Arg Arg Gly Asp Leu Ala Ile
His Leu Val Ser Pro Met Gly Thr Arg 500 505 510Ser Thr Leu Leu Ala
Ala Arg Pro His Asp Tyr Ser Ala Asp Gly Phe 515 520 525Asn Asp
Trp Ala Phe Thr Thr Thr His Ser Trp Asp Glu Asp Pro Ser 530 535
540Gly Glu Trp Val Leu Glu Ile Glu Asn Thr Ser Glu Ala Asn Asn
Tyr545 550 555 560Gly Thr Leu Thr Lys Phe Thr Leu Val Leu Tyr Gly
Thr Ala Pro Glu 565 570 575Gly Leu Pro Val Pro Pro Glu Ser Ser Gly
Cys Lys Thr Leu Thr Ser 580 585 590Ser Gln Ala Cys Val Val Cys Glu
Glu Gly Phe Ser Leu His Gln Lys 595 600 605Ser Cys Val Gln His Cys
Pro Pro Gly Phe Ala Pro Gln Val Leu Asp 610 615 620Thr His Tyr Ser
Thr Glu Asn Asp Val Glu Thr Ile Arg Ala Ser Val625 630 635 640Cys
Ala Pro Cys His Ala Ser Cys Ala Thr Cys Gln Gly Pro Ala Leu 645 650
655Thr Asp Cys Leu Ser Cys Pro Ser His Ala Ser Leu Asp Pro Val Glu
660 665 670Gln Thr Cys Ser Arg Gln Ser Gln Ser Ser Arg Glu Ser Pro
Pro Gln 675 680 685Gln Gln Pro Pro Arg Leu Pro Pro Glu Val Glu Ala
Gly Gln Arg Leu 690 695 7003025PRTArtificialLinker 30Ser Ser Gly
Gly Ser Gly Gly Ser Gly Gly Ser Gly Gly Ser Gly Gly1 5 10 15Ser Gly
Gly Ser Gly Gly Ser Gly Ser 20 253131PRTArtificialLinker 31Ser Ser
Asn Gly Ser Gly Gly Ser Gly Gly Ser Gly Gly Ser Asn Gly1 5 10 15Ser
Gly Gly Ser Gly Gly Ser Gly Gly Ser Gly Gly Asn Gly Ser 20 25
303217PRTArtificialLinker 32Arg Ala Glu Thr Val Phe Pro Asp Val Asp
Tyr Val Asn Ser Thr Glu1 5 10 15Ala3314PRTArtificialLinker 33Arg
Ala Glu Thr Val Phe Pro Asp Val Asp Tyr Val Asn Ser1 5
103417PRTArtificialLinker 34Arg Ala Glu Ala Val Phe Pro Asp Val Asp
Tyr Val Asn Ser Thr Glu1 5 10 15Ala3519PRTArtificialLinker 35Arg
Ala Glu Ala Val Phe Pro Asp Val Asp Tyr Val Asn Ser Thr Glu1 5 10
15Ala Gly Ser3619PRTArtificialLinker 36Ser Val Ser Gln Thr Ser Lys
Leu Thr Arg Ala Glu Thr Val Phe Pro1 5 10 15Asp Val
Asp3721PRTArtificialLinker 37Ser Val Ser Gln Thr Ser Lys Leu Thr
Arg Ala Glu Thr Val Phe Pro1 5 10 15Asp Val Asp Gly Ser
203819PRTArtificialLinker 38Ser Val Ser Gln Thr Ser Lys Leu Thr Arg
Ala Glu Thr Val Phe Pro1 5 10 15Asp Val Asp3921PRTArtificialLinker
39Ser Val Ser Gln Thr Ser Lys Leu Thr Arg Ala Glu Thr Val Phe Pro1
5 10 15Asp Val Asp Gly Ser 204022PRTArtificialLinker 40Ser Val Ser
Gln Thr Ser Lys Leu Thr Arg Ala Glu Thr Val Phe Pro1 5 10 15Asp Val
Asp Asn Gly Ser 204119PRTArtificialLinker 41Ser Val Ser Gln Thr Ser
Lys Leu Thr Arg Gly Glu Thr Val Phe Pro1 5 10 15Asp Val
Asp4219PRTArtificialLinker 42Ser Val Ser Gln Thr Ser Lys Leu Thr
Arg Thr Glu Thr Val Phe Pro1 5 10 15Asp Val
Asp4319PRTArtificialLinker 43Ser Val Ser Gln Thr Ser Lys Leu Thr
Arg Ser Glu Thr Val Phe Pro1 5 10 15Asp Val
Asp4419PRTArtificialLinker 44Ser Val Ser Gln Thr Ser Lys Leu Thr
Arg Leu Glu Thr Val Phe Pro1 5 10 15Asp Val
Asp4519PRTArtificialLinker 45Ser Val Ser Gln Thr Ser Lys Leu Thr
Arg Thr Glu Ala Val Phe Pro1 5 10 15Asp Val
Asp4619PRTArtificialLinker 46Ser Val Ser Gln Thr Ser Lys Leu Thr
Arg Gly Glu Ala Val Phe Pro1 5 10 15Asp Val
Asp4714PRTArtificialLinker 47Ser Lys Leu Thr Arg Ala Glu Thr Val
Phe Pro Asp Val Asp1 5 104814PRTArtificialLinker 48Gln Ser Phe Asn
Asp Phe Thr Arg Val Val Gly Gly Glu Asp1 5
104916PRTArtificialLinker 49Gln Ser Phe Asn Asp Phe Thr Arg Val Val
Gly Gly Glu Asp Gly Ser1 5 10 155014PRTArtificialLinker 50Gln Ser
Phe Asn Asp Phe Thr Arg Thr Val Gly Gly Glu Asp1 5
105114PRTArtificialLinker 51Gln Ser Phe Asn Asp Phe Thr Arg Leu Val
Gly Gly Glu Asp1 5 105214PRTArtificialLinker 52Gln Ser Phe Asn Asp
Phe Thr Arg Gly Val Gly Gly Glu Asp1 5 105317PRTArtificialLinker
53Gln Ser Phe Asn Asp Phe Thr Arg Val Val Gly Gly Glu Asp Asn Gly1
5 10 15Ser5415PRTArtificialLinker 54Gln Ser Phe Asn Asp Phe Thr Arg
Val Val Gly Gly Glu Asp Asn1 5 10 15557PRTArtificialLinker 55Arg
Ile Val Gly Gly Gln Glu1 5567PRTArtificialLinker 56Arg Leu Val Gly
Gly Gln Glu1 5577PRTArtificialLinker 57Arg Thr Val Gly Gly Gln Glu1
5587PRTArtificialLinker 58Arg Val Val Gly Gly Gln Glu1
5597PRTArtificialLinker 59Arg Ala Val Gly Gly Gln Glu1
5607PRTArtificialLinker 60Arg Gly Val Gly Gly Gln Glu1
56116PRTArtificialLinker 61Pro Glu Arg Gly Asp Asn Asn Leu Thr Arg
Ile Val Gly Gly Gln Glu1 5 10 156219PRTArtificialLinker 62Ser Val
Ser Gln Thr Ser Lys Leu Thr Arg Ala Glu Thr Val Phe Pro1 5 10 15Asp
Val Asp6315PRTArtificialLinker 63Gln Ser Phe Asn Asp Phe Thr Arg
Val Val Gly Gly Glu Asp Asn1 5 10 156419PRTArtificialLinker 64Ser
Val Ser Gln Thr Ser Lys Leu Thr Arg Ala Glu Thr Val Phe Pro1 5 10
15Asp Val Asp6516PRTArtificialLinker 65Lys Arg Asn Ala Ser Lys Pro
Gln Gly Arg Ile Val Gly Gly Lys Val1 5 10 156616PRTArtificialLinker
66Pro Glu Glu Pro Gln Leu Arg Met Lys Asn Asn Glu Glu Ala Glu Asp1
5 10 156719PRTArtificialLinker 67Asp Asn Ser Pro Ser Phe Ile Gln
Ile Arg Ser Val Ala Lys Lys His1 5 10 15Pro Lys
Thr6820PRTArtificialLinker 68Leu Ser Lys Asn Asn Ala Ile Glu Pro
Arg Ser Phe Ser Gln Asn Ser1 5 10 15Arg His Pro Ser
206921PRTArtificialLinker 69Asp Glu Asp Glu Asn Gln Ser Pro Arg Ser
Phe Gln Lys Lys Thr Arg1 5 10 15His Tyr Phe Ile Ala
207016PRTArtificialLinker 70Ser Pro His Val Leu Arg Asn Arg Ala Gln
Ser Gly Ser Val Pro Gln1 5 10 157124PRTArtificialLinker 71Pro Glu
Glu Pro Gln Leu Arg Met Lys Asn Asn Glu Glu Ala Glu Asp1 5 10 15Tyr
Asp Asp Asp Leu Thr Asp Ser 207231PRTArtificialLinker 72Asp Asp Asp
Asn Ser Pro Ser Phe Ile Gln Ile Arg Ser Val Ala Lys1 5 10 15Lys His
Pro Lys Thr Trp Val His Tyr Ala Ala Glu Glu Glu Asp 20 25
307328PRTArtificialLinker 73Leu Ser Lys Asn Asn Ala Ile Glu Pro Arg
Ser Phe Ser Gln Asn Ser1 5 10 15Arg His Pro Ser Thr Arg Gln Lys Gln
Phe Asn Ala 20 257422PRTArtificialLinker 74Asp Glu Asp Glu Asn Gln
Ser Pro Arg Ser Phe Gln Lys Lys Thr Arg1 5 10 15His Tyr Phe Ile Ala
Ala 207532PRTArtificialLinker 75Asp Tyr Gly Met Ser Ser Ser Pro His
Val Leu Arg Asn Arg Ala Gln1 5 10 15Ser Gly Ser Val Pro Gln Phe Lys
Lys Val Val Phe Gln Glu Phe Thr 20 25 307624PRTArtificialLinker
76Asp Ile Tyr Asp Glu Asp Glu Asn Gln Ser Pro Arg Ser Phe Gln Lys1
5 10 15Lys Thr Arg His Tyr Phe Ile Ala 207717PRTArtificialLinker
77Asp Asn Ser Pro Ser Phe Ile Gln Ile Arg Ser Val Ala Lys Lys His1
5 10 15Pro7820PRTArtificialLinker 78Leu Ser Lys Asn Asn Ala Ile Glu
Pro Arg Ser Phe Ser Gln Asn Ser1 5 10 15Arg His Pro Ser
207936PRTArtificialLinker 79Ser Val Ser Gln Thr Ser Lys Leu Thr Arg
Ala Glu Thr Val Phe Pro1 5 10 15Asp Val Thr Gln Pro Glu Arg Gly Asp
Asn Asn Leu Thr Arg Ile Val 20 25 30Gly Gly Gln Glu
358023PRTArtificialLinker 80Ser Lys Leu Thr Arg Ala Glu Thr Val Phe
Pro Asp Asn Asn Leu Thr1 5 10 15Arg Ile Val Gly Gly Gln Glu
208127PRTArtificialLinker 81Arg Ala Glu Thr Val Phe Pro Asp Val Thr
Gln Pro Glu Arg Gly Asp1 5 10 15Asn Asn Leu Thr Arg Ile Val Gly Gly
Gln Glu 20 258222PRTArtificialLinker 82Arg Ala Glu Thr Val Phe Pro
Glu Arg Gly Asp Asn Asn Leu Thr Arg1 5 10 15Ile Val Gly Gly Gln Glu
208332PRTArtificialLinker 83Ser Val Ser Gln Thr Ser Lys Leu Thr Arg
Ala Glu Thr Val Phe Pro1 5 10 15Asp Val Asp Tyr Val Asn Asn Leu Thr
Arg Ile Val Gly Gly Gln Glu 20 25 308430PRTArtificialLinker 84Ser
Val Ser Gln Thr Ser Lys Leu Thr Arg Ala Glu Thr Val Phe Pro1 5 10
15Asp Val Asp Asn Asn Leu Thr Arg Ile Val Gly Gly Gln Glu 20 25
308530PRTArtificialLinker 85Ser Val Ser Gln Thr Ser Lys Leu Thr Arg
Ala Glu Thr Val Phe Pro1 5 10 15Asp Val Asp Asn Asn Leu Thr Arg Ile
Val Gly Gly Gln Glu 20 25 308652PRTArtificialLinker 86Ser Val Ser
Gln Thr Ser Lys Leu Thr Arg Ala Glu Thr Val Phe Pro1 5 10 15Asp Val
Asp Tyr Val Asn Ser Thr Glu Ala Glu Thr Ile Leu Asp Asn 20 25 30Ile
Thr Gln Ser Thr Gln Ser Phe Asn Asp Phe Thr Arg Val Val Gly 35 40
45Gly Glu Asp Ala 508732PRTArtificialLinker 87Ser Val Ser Gln Thr
Ser Lys Leu Thr Arg Ala Glu Thr Val Phe Pro1 5 10 15Asp Val Gln Ser
Phe Asn Asp Phe Thr Arg Val Val Gly Gly Glu Asp 20 25
308832PRTArtificialLinker 88Ser Val Ser Gln Thr Ser Lys Leu Thr Arg
Ala Glu Thr Val Phe Pro1 5 10 15Asp Val Asp Ser Phe Asn Asp Phe Thr
Arg Val Val Gly Gly Glu Asp 20 25 308932PRTArtificialLinker 89Ser
Val Ser Gln Thr Ser Lys Leu Thr Arg Ala Glu Thr Val Phe Pro1 5 10
15Asp Val Asn Ala Ser Lys Pro Gln Gly Arg Ile Val Gly Gly Lys Val
20 25 309032PRTArtificialLinker 90Ser Val Ser Gln Thr Ser Lys Leu
Thr Arg Ala Glu Thr Val Phe Pro1 5 10 15Asp Val Asn Ala Ser Lys Pro
Gln Gly Arg Leu Val Gly Gly Lys Val 20 25 309132PRTArtificialLinker
91Ser Val Ser Gln Thr Ser Lys Leu Thr Arg Ala Glu Thr Val Phe Pro1
5 10 15Asp Val Asn Ala Ser Lys Pro Gln Gly Arg Thr Val Gly Gly Lys
Val 20 25 309219PRTArtificialLinker 92Ser Val Ser Gln Thr Ser Lys
Leu Thr Arg Ala Glu Thr Val Phe Pro1 5 10 15Asp Val Asp93666PRTHomo
Sapiens 93Tyr Asn Ser Gly Lys Leu Glu Glu Phe Val Gln Gly Asn Leu
Glu Arg1 5 10 15Glu Cys Met Glu Glu Lys Cys Ser Phe Glu Glu Ala Arg
Glu Val Phe 20 25 30Glu Asn Thr Glu Arg Thr Thr Glu Phe Trp Lys Gln
Tyr Val Asp Gly 35 40 45Asp Gln Cys Glu Ser Asn Pro Cys Leu Asn Gly
Gly Ser Cys Lys Asp 50 55 60Asp Ile Asn Ser Tyr Glu Cys Trp Cys Pro
Phe Gly Phe Glu Gly Lys65 70 75 80Asn Cys Glu Leu Asp Val Thr Cys
Asn Ile Lys Asn Gly Arg Cys Glu 85 90 95Gln Phe Cys Lys Asn Ser Ala
Asp Asn Lys Val Val Cys Ser Cys Thr 100 105 110Glu Gly Tyr Arg Leu
Ala Glu Asn Gln Lys Ser Cys Glu Pro Ala Val 115 120 125Pro Phe Pro
Cys Gly Arg Val Ser Val Ser Gln Thr Ser Lys Leu Thr 130 135 140Arg
Ala Glu Thr Val Phe Pro Asp Val Asp Tyr Val Asn Ser Thr Glu145 150
155 160Ala Glu Thr Ile Leu Asp Asn Ile Thr Gln Ser Thr Gln Ser Phe
Asn 165 170 175Asp Phe Thr Arg Val Val Gly Gly Glu Asp Ala Lys Pro
Gly Gln Phe 180 185 190Pro Trp Gln Val Val Leu Asn Gly Lys Val Asp
Ala Phe Cys Gly Gly 195 200 205Ser Ile Val Asn Glu Lys Trp Ile Val
Thr Ala Ala His Cys Val Glu 210 215 220Thr Gly Val Lys Ile Thr Val
Val Ala Gly Glu His Asn Ile Glu Glu225 230 235 240Thr Glu His Thr
Glu Gln Lys Arg Asn Val Ile Arg Ile Ile Pro His 245 250 255His Asn
Tyr Asn Ala Ala Ile Asn Lys Tyr Asn His Asp Ile Ala Leu 260 265
270Leu Glu Leu Asp Glu Pro Leu Val Leu Asn Ser Tyr Val Thr Pro Ile
275 280 285Cys Ile Ala Asp Lys Glu Tyr Thr Asn Ile Phe Leu Lys Phe
Gly Ser 290 295 300Gly Tyr Val Ser Gly Trp Gly Arg Val Phe His Lys
Gly Arg Ser Ala305 310 315 320Leu Val Leu Gln Tyr Leu Arg Val Pro
Leu Val Asp Arg Ala Thr Cys 325 330 335Leu Arg Ser Thr Lys Phe Thr
Ile Tyr Asn Asn Met Phe Cys Ala Gly 340 345 350Phe His Glu Gly Gly
Arg Asp Ser Cys Gln Gly Asp Ser Gly Gly Pro 355 360 365His Val Thr
Glu Val Glu Gly Thr Ser Phe Leu Thr Gly Ile Ile Ser 370 375 380Trp
Gly Glu Glu Cys Ala Met Lys Gly Lys Tyr Gly Ile Tyr Thr Lys385 390
395 400Val Ser Arg Tyr Val Asn Trp Ile Lys Glu Lys Thr Lys Leu Thr
Ser 405 410 415Val Ser Gln Thr Ser Lys Leu Thr Arg Ala Glu Thr Val
Phe Pro Asp 420 425 430Val Asp Glu Pro Lys Ser Cys Asp Lys Thr His
Thr Cys Pro Pro Cys 435 440 445Pro Ala Pro Glu Leu Leu Gly Gly Pro
Ser Val Phe Leu Phe Pro Pro 450 455 460Lys Pro Lys Asp Thr Leu Met
Ile Ser Arg Thr Pro Glu Val Thr Cys465 470 475 480Val Val Val Asp
Val Ser His Glu Asp Pro Glu Val Lys Phe Asn Trp 485 490 495Tyr Val
Asp Gly Val Glu Val His Asn Ala Lys Thr Lys Pro Arg Glu 500 505
510Glu Gln Tyr Asn Ser Thr Tyr Arg Val Val Ser Val Leu Thr Val Leu
515 520 525His Gln Asp Trp Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val
Ser Asn 530 535 540Lys Ala Leu Pro Ala Pro Ile Glu Lys Thr Ile Ser
Lys Ala Lys Gly545 550 555 560Gln Pro Arg Glu Pro Gln Val Tyr Thr
Leu Pro Pro Ser Arg Glu Glu 565 570 575Met Thr Lys Asn Gln Val Ser
Leu Thr Cys Leu Val Lys Gly Phe Tyr 580 585 590Pro Ser Asp Ile Ala
Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn 595 600 605Asn Tyr Lys
Thr Thr Pro Pro Val Leu Asp Ser Asp Gly Ser Phe Phe 610 615 620Leu
Tyr Ser Lys Leu Thr Val Asp Lys Ser Arg Trp Gln Gln Gly Asn625 630
635 640Val Phe Ser Cys Ser Val Met His Glu Ala Leu His Asn His Tyr
Thr 645 650 655Gln Lys Ser Leu Ser Leu Ser Pro Gly Lys 660
665947PRTArtificialLinker 94Gly Gly Gly Gly Gly Gly Val1
59524PRTArtificialLinker 95Ser Val Ser Gln Thr Ser Lys Leu Thr Arg
Ala Glu Thr Val Phe Pro1 5 10 15Asp Val Asp Gly Ser Gly Gly Ser
209618PRTArtificialLinker 96Ser Val Ser Gln Thr Ser Lys Leu Thr Arg
Ala Glu Thr Val Phe Pro1 5 10 15Asp Val9715PRTArtificialLinker
97Gln Thr Ser Lys Leu Thr Arg Ala Glu Thr Val Phe Pro Asp Val1 5 10
159813PRTArtificialLinker 98Ser Lys Leu Thr Arg Ala Glu Thr Val Phe
Pro Asp Val1 5 109916PRTArtificialLinker 99Ser Val Ser Gln Thr Ser
Lys Leu Thr Arg Ala Glu Thr Val Phe Pro1 5 10
1510015PRTArtificialLinker
100Ser Val Ser Gln Thr Ser Lys Leu Thr Arg Ala Glu Thr Val Phe1 5
10 1510112PRTArtificialLinker 101Gln Thr Ser Lys Leu Thr Arg Ala
Glu Thr Val Phe1 5 1010210PRTArtificialLinker 102Ser Lys Leu Thr
Arg Ala Glu Thr Val Phe1 5 1010313PRTArtificialLinker 103Ser Val
Ser Gln Thr Ser Lys Leu Thr Arg Ala Glu Thr1 5
1010410PRTArtificialLinker 104Gln Thr Ser Lys Leu Thr Arg Ala Glu
Thr1 5 101058PRTArtificialLinker 105Ser Lys Leu Thr Arg Ala Glu
Thr1 510618PRTArtificialLinker 106Gln Thr Ser Lys Leu Thr Arg Ala
Glu Thr Val Phe Pro Asp Val Asp1 5 10 15Gly
Ser10716PRTArtificialLinker 107Ser Lys Leu Thr Arg Ala Glu Thr Val
Phe Pro Asp Val Asp Gly Ser1 5 10 1510813PRTArtificialLinker 108Gln
Ser Phe Asn Asp Phe Thr Arg Val Val Gly Gly Glu1 5
1010918PRTArtificialLinker 109Pro Glu Arg Gly Asp Asn Asn Leu Thr
Arg Ile Val Gly Gly Gln Glu1 5 10 15Gly Ser11015PRTArtificialLinker
110Pro Glu Arg Gly Asp Asn Asn Leu Thr Arg Ile Val Gly Gly Gln1 5
10 1511111PRTArtificialLinker 111Asp Asn Asn Leu Thr Arg Ile Val
Gly Gly Gln1 5 1011211PRTArtificialLinker 112Ala Ser Lys Pro Gln
Gly Arg Ile Val Gly Gly1 5 1011318PRTArtificialLinker 113Pro Val
Ser Gln Thr Ser Lys Leu Thr Arg Ala Glu Thr Val Phe Pro1 5 10 15Asp
Val11419PRTArtificialLinker 114Pro Ser Val Ser Gln Thr Ser Lys Leu
Thr Arg Ala Glu Thr Val Phe1 5 10 15Pro Asp Val
D00001
D00002
D00003
S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.