Intergenic Sites Between Conserved Genes in the Genome of Modified Vaccinia Ankara (MVA) Vaccinia Virus
Moss; Bernard ; et al.
U.S. patent application number 16/579276 was filed with the patent office on 2020-03-05 for intergenic sites between conserved genes in the genome of modified vaccinia ankara (mva) vaccinia virus. The applicant listed for this patent is The USA, as represented by the Secretary, Department of Health and Human Services, The USA, as represented by the Secretary, Department of Health and Human Services. Invention is credited to Patricia L. Earl, Bernard Moss, Linda S. Wyatt.
| Application Number | 20200071724 16/579276 |
| Document ID | / |
| Family ID | 40032225 |
| Filed Date | 2020-03-05 |












View All Diagrams
| United States Patent Application | 20200071724 |
| Kind Code | A1 |
| Moss; Bernard ; et al. | March 5, 2020 |
Intergenic Sites Between Conserved Genes in the Genome of Modified Vaccinia Ankara (MVA) Vaccinia Virus
Abstract
The pesent invention relates to new insertion sites useful for the integration exogenous sequence into an intergenic region (IGR) of a vaccinia virus genome, where the IGR is located between or is flanked by two adjacent open reading frames (ORFs) of the vaccinia virus genome, and wherein the ORFs correspond to conserved genes, and to related plasmid vectors useful to insert exogenous DNA into the genome of a vaccinia virus, and further to recombinant vaccinia viruses comprising an exogenous sequence inserted into said new insertion site as a medicine or vaccine.
| Inventors: | Moss; Bernard; (Bethesda, MD) ; Wyatt; Linda S.; (Rockville, MD) ; Earl; Patricia L.; (Chevy Chase, MD) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 40032225 | ||||||||||
| Appl. No.: | 16/579276 | ||||||||||
| Filed: | September 23, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 14833913 | Aug 24, 2015 | 10421978 | ||
| 16579276 | ||||
| 12377847 | Feb 17, 2010 | 9133478 | ||
| PCT/IB07/04575 | Aug 24, 2007 | |||
| 14833913 | ||||
| 60840093 | Aug 25, 2006 | |||
| 60840755 | Aug 28, 2006 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61P 31/18 20180101; A61K 2039/5256 20130101; C12N 2710/24111 20130101; C12N 15/86 20130101; C12N 2710/24141 20130101; C12N 2710/24152 20130101; C12N 2710/24143 20130101; A61P 31/12 20180101; C12N 15/8636 20130101; A61P 37/00 20180101 |
| International Class: | C12N 15/86 20060101 C12N015/86; C12N 15/863 20060101 C12N015/863 |
Claims
1-33. (canceled)
34. A method of stabilizing a heterologous nucleic acid insert in a recombinant virus, the method comprising making a mutation in a run of 4 or more purines or 4 or more pyrimidines, in the nucleic acid insert, so that the length of the run of the 4 or more purines or the 4 or more pyrimidines is reduced.
35. The method of claim 34, wherein the mutation is a substitution mutation.
36. The method of claim 34, wherein the mutation is a silent mutation.
37. The method of claim 34, wherein the mutation is made in a run of 4 or more guanine (G) or cytosine (C) residues.
38. The method of claim 34, wherein the mutation is made in a run of 4 or more adenine (A) or thymine (T) residues.
39. The method of claim 34, wherein the recombinant virus is a recombinant vaccinia virus.
40. The method of claim 39, wherein the heterologous nucleic acid insert is located in an intergenic region (IGR) of the vaccinia virus (VV) genome, wherein the IGR is located between or is flanked by two adjacent open reading frames (ORFs) of the VV genome, and wherein the ORFs correspond to genes that are essential for replication of vaccinia virus.
41. The method of claim 34, wherein the sequence of the heterologous nucleic acid insert is derived from human immunodeficiency virus (HIV).
42. The method of claim 34, wherein the heterologous nucleic acid insert comprises the HIV gagpol gene or the HIV env gene.
43. A recombinant virus comprising a heterologous nucleic acid insert, wherein the presence of the heterologous insert has been stabilized using the method of claim 34.
44. The recombinant virus of claim 43, wherein the virus is a vaccinia virus.
45. The recombinant virus of claim 44, wherein the heterologous nucleic acid insert is located in an intergenic region (IGR) of the vaccinia virus (VV) genome, wherein the IGR is located between or is flanked by two adjacent open reading frames (ORFs) of the VV genome, and wherein the ORFs correspond to genes that are essential for replication of vaccinia virus.
46. The recombinant virus of claim 43, wherein the sequence of the heterologous nucleic acid insert is derived from human immunodeficiency virus (HIV).
47. The recombinant virus of claim 43, wherein the heterologous nucleic acid insert comprises the HIV gagpol gene or the HIV env gene.
Description
RELATED APPLICATIONS
[0001] This application claims the benefit of U.S. Provisional Application No. 60/840,093 filed Aug. 25, 2006, and U.S. Provisional Application No. 60/840,755 filed Aug. 28, 2006, both of which are hereby expressly incorporated by reference in their entireties.
FIELD OF THE INVENTION
[0002] The present invention relates to insertion sites useful for the stable integration of exogenous DNA sequences into the MVA genome.
DESCRIPTION OF THE RELATED ART
[0003] The members of the poxvirus family have large double-stranded DNA genomes encoding several hundred proteins (Moss, B. 2007 "Poxviridae: The Viruses and Their Replication" in Fields Virology, 5.sup.th Ed. (D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus, Eds), Lippincott Williams & Wilkins, Philadelphia, Pa.). The genomic sequence of the highly attenuated vaccinia strain modified vaccinia Ankara (MVA) (Mayr, A. et al. 1978 Zentralbl Bakteriol 167:375-390), which cannot grow in most mammalian cells and which is a good candidate for a recombinant vaccine vector, is known (Sutter, G. and Moss, B. 1992 Proc Natl Acad Sci USA 89:10847-10851; and Sutter, G. et al. 1994 Vaccine 12:1032-1040) has been passaged over 570 times in chicken embryo fibroblasts, during which six major deletions relative to the parental wild-type strain Ankara, accompanied by a severe restriction in host range, have occurred (Meyer, H. et al. 1991 J Gen Virol 72:1031-1038).
SUMMARY OF THE INVENTION
[0004] The present invention relates to new insertion sites useful for the integration of exogenous sequences into an intergenic region (IGR) of a vaccinia virus genome, where the IGR is located between or is flanked by two adjacent open reading frames (ORFs) of the vaccinia virus genome, and where the ORFs correspond to conserved genes, and to related plasmid vectors useful to insert exogenous DNA into the genome of a vaccinia virus, and further to recombinant vaccinia viruses comprising an exogenous sequence inserted into said new insertion site as a medicine or vaccine.
BRIEF DESCRIPTION OF THE DRAWINGS
[0005] FIG. 1. Phylogenetic relationships of HIV-1 and HIV-2 based on identity of pol gene sequences. SIV.sub.cpz and SIV.sub.smm are subhuman primate lentiviruses recovered from a chimpanzee and sooty mangabey monkey, respectively.
[0006] FIG. 2. Phylogenetic relationships of HIV-1 groups M, N and O with four different SIV.sub.cpz isolates based on full-length pol gene sequences. The bar indicates a genetic distance of 0.1 (10% nucleotide divergence) and the asterisk positions group N HIV-1 isolates based on env sequences.
[0007] FIG. 3. Tropic and biologic properties of HIV-1 isolates.
[0008] FIG. 4. HIV-encoded proteins. The location of the HIV genes, the sizes of primary translation products (in some cases polyproteins), and the processed mature viral proteins are indicated.
[0009] FIG. 5. Schematic representation of a mature HIV-1 virion.
[0010] FIG. 6. Linear representation of the HIV-1 Env glycoprotein. The arrow indicates the site of gp160 cleavage to gp120 and gp41. In gp120, cross-hatched areas represent variable domains (V.sub.1 to V.sub.5) and open boxes depict conserved sequences (C.sub.1 to C.sub.5). In the gp41 ectodomain, several domains are indicated: the N-terminal fusion peptide, and the two ectodomain helices (N- and C-helix). The membrane-spanning domain is represented by a black box. In the gp41 cytoplasmic domain, the Tyr-X-X-Leu (YXXL) endocytosis motif (SEQ ID NO: 1) and two predicted helical domains (helix-1 and -2) are shown. Amino acid numbers are indicated.
[0011] FIG. 7. pLW-73 transfer vector.
[0012] FIG. 8-1 through FIG. 8-9. Nucleotide sequence of the pLW-73 transfer vector (top strand, SEQ ID NO: 2; bottom strand, SEQ ID NO: 3).
[0013] FIG. 9. Nucleotide sequence encoding Ugandan clade D Env protein (isolate AO7412) (SEQ ID NO: 4).
[0014] FIG. 10-1 through FIG. 10-2. Codon altered nucleotide sequence encoding Ugandan clade D gagpol protein (isolate AO3349) (SEQ ID NO: 5).
[0015] FIG. 11. Generation of recombinant MVAs and analysis of stability of inserted genes. A) Schematic diagram of insertion of env and gagpol into Del II and Del III sites, respectively. B) Evaluation of stability by immunostaining.
[0016] FIG. 12. Types and frequency of env mutations in MVA/65A/G env.
[0017] FIG. 13. Insertion of Env in I8R/G1L IGR and Gag Pol in Del III.
[0018] FIG. 14. Modifications to A/G constructs to increase stability.
[0019] FIG. 15. Env expression after plaque passages.
[0020] FIG. 16. PCR and Western blot analysis of individual clones.
[0021] FIG. 17. Expression of A/G env by double recombinant MVA.
[0022] FIG. 18. Recombinant viruses expressing env and gagpol from Ugandan HIV-1 isolates.
[0023] FIG. 19. MVA/UGD4a--anlaysis of non-staining env plaques.
[0024] FIG. 20. Modification of UGD env gene in recombinant MVA.
[0025] FIG. 21. MVA/UGD4b--analysis of non-staining gag plaques. *, location of runs of 4-6 G or C residues.
[0026] FIG. 22. Modification of UGD gagpol gene in recombinant MVA.
[0027] FIG. 23. Construction of stable recombinant MVA expressing UGD env and gagpol.
[0028] FIG. 24. Cellular responses elicited by MVA/UGD4d.
[0029] FIG. 25. Antibody responses elicited by MVA/UGD4d.
DEPOSIT OF MICROORGANISM
[0030] The following microorganism has been deposited in accordance with the terms of the Budapest Treaty with the American Type Culture Collection (ATCC), Manassas, Va., on the date indicated:
TABLE-US-00001 Microorganism Accession No. Date MVA 1974/NIH Clone 1 PTA-5095 Mar. 27, 2003
[0031] MVA 1974/NIH Clone 1 was deposited as ATCC Accession No.: PTA-5095 on Mar. 27, 2003 with the American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, Va. 20110-2209, USA. This deposit was made under the provisions of the Budapest Treaty on the International Recognition of the Deposit of Microorganisms for the Purposes of Patent Procedure and the Regulations thereunder (Budapest Treaty). This assures maintenance of a viable culture of the deposit for 30 years from date of deposit. The deposit will be made available by ATCC under the terms of the Budapest Treaty, and subject to an agreement between Applicant and ATCC which assures permanent and unrestricted availability of the progeny of the culture of the deposit to the public upon issuance of the pertinent U.S. patent or upon laying open to the public of any U.S. or foreign patent application, whichever comes first, and assures availability of the progeny to one determined by the U.S. Commissioner of Patents and Trademarks to be entitled thereto according to 35 USC .sctn. 122 and the Commissioner's rules pursuant thereto (including 37 CFR .sctn. 1.14). Availability of the deposited strain is not to be construed as a license to practice the invention in contravention of the rights granted under the authority of any government in accordance with its patent laws.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENT
[0032] Unless defined otherwise, technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. See, e.g., Singleton P and Sainsbury D., Dictionary of Microbiology and Molecular Biology, 3rd ed., J. Wiley & Sons, Chichester, N.Y., 2001 and Fields Virology, 5.sup.th Ed. (D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus, eds), Lippincott Williams & Wilkins, Philadelphia, Pa., 2007. The transitional term "comprising" is synonymous with "including," "containing," or "characterized by," is inclusive or open-ended and does not exclude additional, unrecited elements or method steps.
[0033] The transitional phrase "consisting of" excludes any element, step, or ingedient not specified in the claim, but does not exclude additional components or steps that are unrelated to the invention such as impurities ordinarily associated therewith.
[0034] The transitional phrase "consisting essentially of" limits the scope of a claim to the specified materials or steps and those that do not materially affect the basic and novel characteristic(s) of the claimed invention.
[0035] Poxviruses are divided into the subfamilies Chordopoxvirinae and Entomopoxvirinae, based on vertebrate and insect host range. The subfamily Chordopoxvirinae consists of eight genera: Orthopoxvirus, Parapoxvirus, Avipoxvirus, Capripoxvirus, Leporipoxvirus, Suipoxvirus, Molluscipoxvirus, and Yatapoxvirus. The prototypal member of the genus Orthopoxvirus is vaccinia virus.
[0036] Complete genome sequences have been reported for at least one member of each chordopoxvirus genus and two entomopoxviruses. Nearly 100 genes are conserved in all chordopoxviruses, and about half of these are also present in entomopoxviruses. Based on the above, several generalizations can be made: Genes are largely nonoverlapping, tend to occur in blocks pointing toward the nearer end of the genome, are usually located in the central region if highly conserved and concerned with essential replication functions, and are usually located in the end regions if variable and concerned with host interactions. The arrangement of the central genes is remarkably similar in all chordopoxviruses. A convention for naming vaccinia virus genes or ORFs (open reading frames), originating prior to sequencing the entire genome and subsequently used for the complete sequence of the Copenhagen strain of vaccinia virus, consists of using the HindlIl restriction endonuclease DNA fragment letter, followed by the ORF number (from left to right) within the fragment, and L or R, depending on the direction of the ORF. An exception to this rule was made for the HindIII C fragment; the ORFs were numbered from the right in order to avoid starting at the highly variable left end of the genome. Polypeptide names correspond to gene names, except that L or R is dropped. In most subsequent complete poxvirus genome sequences, ORFs were numbered successively from one end of the genome to the other. Nevertheless, the old letter designations have been retained as common names to provide continuity in the literature. The ORF number of the Western Reserve (WR) strain of vaccinia virus is commonly shown in reference books because this strain has been used for the great majority of biochemical and genetic studies.
[0037] The inventors of an embodiment of the present invention identified new sites for the insertion of exogenous DNA sequences into the genome of modified vaccinia Ankara (MVA) virus. The new insertion sites are located in the intergenic regions (IGRs) of the viral genome, wherein the IGRs are, in turn, located between or are flanked by two adjacent open reading frames (ORFs) of the MVA genome, and wherein the ORFs correspond to conserved genes.
[0038] Accordingly, an embodiment of the invention relates to a recombinant MVA comprising a heterologous DNA sequence inserted into an IGR of the viral genome. According to the present embodiment, one or more exogenous DNA sequences may be inserted into one or more IGRs.
[0039] It was surprisingly found that exogenous DNA sequences remain stable inserted into IGRs of the MVA genome. The genome of MVA is to be considered as being quite unstable. It seems that genes or DNA sequences non-essential for propagation of the virus are deleted or fragmented. Although it was--on the one hand--found that stable recombinant MVAs are obtained when heterologous DNA sequences are inserted into the naturally occurring deletion sites of the MVA genome, it was--on the other hand--found that sometimes these recombinant MVAs are unstable. Therefore, it could be concluded that inserting heterologous DNA sequences non-essential for viral propagation into spaces between ORFs would be expected to be deleted by the virus as well.
[0040] While the nucleotide sequence of an ORF encodes an amino acid sequence forming a peptide, polypeptide or protein, the IGRs between two ORFs have no coding capacity, but may comprise regulatory elements, binding sites, promoter and/or enhancer sequences essential for or involved in the transcriptional control of the viral gene expression. Thus, the IGR may be involved in the regulatory control of the viral life cycle. However, the inventors of the present embodiment have also shown that the new insertion sites have the unexpected advantage that exogenous DNA sequences can be stably inserted into the MVA genome without influencing or changing the typical characteristics and gene expression of MVA. The new insertion sites are especially useful, since no ORF or coding sequence of MVA is altered.
[0041] The nucleotide sequence of an ORF regularly starts with a start codon and ends with a stop codon. Depending on the orientation of the two adjacent ORFs the IGR, the region in between these ORFs, is flanked either by the two stop codons of the two adjacent ORFs, or, by the two start codons of the two adjacent ORFs, or, by the stop codon of the first ORF and the start codon of the second ORF, or, by the start codon of the first ORF and the stop codon of the second ORF.
[0042] Accordingly, the insertion site for the exogenous DNA sequence into the IGR may be downstream or 3' of the stop codon of a first ORF. In case the adjacent ORF, also termed second ORF, has the same orientation as the first ORF, this insertion site downstream of the stop codon of the first ORF lies upstream or 5' of the start codon of the second ORF.
[0043] In case the second ORF has an opposite orientation relative to the first ORF, which means the orientation of the two adjacent ORFs points to each other, then the insertion site lies downstream of the stop codons of both ORFs.
[0044] As a third alternative, in case the two adjacent ORFs read in opposite directions, but the orientation of the two adjacent ORFs points away from each other, which is synonymous with a positioning that is characterized in that the start codons of the two ORFs are adjacent to each other, then the exogenous DNA is inserted upstream relative to both start codons.
[0045] ORFs in the MVA genome occur in two coding directions. Consequently, mRNA synthesis activity occurs from left to right, i.e., forward direction and, correspondingly, from right to left (reverse direction). It is common practice in poxvirology and it became a standard classification for vaccinia viruses to identify ORFs by their orientation and their position on the different Hindlll restriction digest fragments of the genome. For the nomenclature, the different Hindlll fragments are named by descending capital letters corresponding with their descending size. The ORF are numbered from left to right on each HindIII fragment and the orientation of the ORF is indicated by a capital L (standing for transcription from right to Left) or R (standing for transcription from left to Right). Additionally, there is a more recent publication of the MVA genome structure, which uses a different nomenclature, simply numbering the ORF from the left to the right end of the genome and indicating their orientation with a capital L or R (Antoine, G. et al. 1998 Virology 244:365-396). As an example the I8R ORF, according to the old nomenclature, corresponds to the 069R ORF according to Antoine et al.
[0046] In their efforts to make recombinants of modified vaccinia virus Ankara (MVA) expressing HIV genes as candidate vaccines, the inventors have observed instability in the expression of the HIV genes. They have determined that one of the causes of instability is due to deletions of the foreign gene and flanking MVA. To overcome this problem they set out to insert foreign genes between conserved genes in order to prevent viable deletions from occurring in recombinant MVAs. Viruses with such deletions have a growth advantage and will thus overgrow rMVA virus populations. If one inserts foreign genes between conserved genes in the vaccinia genome (these genes are considered to be required for vaccinia virus replication and are therefore "essential genes"), any deletion of an essential gene would inhibit virus replication, and, therefore, not overgrow the recombinant MVAs. Thus, the stable expression of the rMVA population is maintained. The strain of MVA that the inventors have been using to make their recombinants was provided by them to the Centers for Disease Control and Prevention (CDC) and was subsequently sequenced by Acambis (Genbank Accession number AY603355). The strain of MVA that Bavarian Nordic has based their WO03/097845 publication on is vaccinia virus strain modified vaccinia Ankara (Genbank Accession number U94848) sequenced by Antoine, G. et al. 1998 Virology 244:365-396. (Please note that the gene numbers in these two sequences for a given gene are different.)
[0047] The inventors initially looked at genes conserved in the Poxviridae family as well as those genes conserved in subfamily Chordopoxvirinae (the vertebrate poxviruses) (Upton, C. et al. 2003 Journal of Virology 77:7590-7600). These genes are listed in the nomenclature of Copenhagen vaccinia virus (Genbank Accession number M35027) given on the Poxvirus Bioinformatics Resource Center found on the world wide web at poxvirus.org. These genes total 49 conserved genes in the Poxvirus family and 41 additional genes conserved in chordopoxviruses, making a total of 90 conserved genes. From these 90 conserved genes, the inventors listed intergenic sites between conserved gene pairs. These gene pairs are listed in Table 1. (Please note that genes are marked that have not been included in the Bavarian Nordic WO03/097845 publication). The orientations of these genes are variable, with some being transcribed to the right, some to the left. This means that some of the intergenic sites contain promoters that would have to be preserved in the construction of the insertion vector. In addition, for overlapping conserved genes, during vector construction the genes would have to be reconstructed using alternative codons to minimize the repeating sequences.
[0048] In a preferred embodiment, the inventors focused on conserved genes whose orientation is "end to end" such that the 3' stop codon of the genes are in close proximity to one another. The construction of transfer vectors used in these sites are facilitated by the fact that there would be no promoter in this region between the stop codons. If there are intergenic nucleotides separating the stop codons, then construction of the insertion vector is straightforward. If the stop codon of one gene is within the 3' end of the other gene, then during construction of the plasmid transfer vector, the gene can be reconstructed using alternative codons to minimize repeating sequences, or, depending on the size of the overlap, simply corrected in the PCR of the flanks so as not to overlap. Table 2 gives the intergenic sites that meet the requirement of the orientation of the conserved genes being "end to end". Those intergenic sites highlighted in bolded text have no overlapping ends and therefore are simplest to construct.
[0049] The inventors specifically focused on the six intergenic sites that have no overlapping ends. In a working example, of these six, they chose the intergenic site, 071-072 (I8R-G1L), to insert their foreign gene.
[0050] Besides using the requirement of conserved genes as listed above in the Upton publication, for any gene that has been experimentally deleted and virus replication is reduced by 10 fold in the mutant, this gene could be considered as an "essential gene". If this gene lies adjacent to another essential or conserved gene, the intergenic site between the two genes could be considered as a different site of insertion for a foreign gene.
TABLE-US-00002 TABLE 1 Intergenic Sites between Conserved Genes Listed in Genes/ CDC/Acambis Antoine et WO03/097845 Copenhagen Genes al. Genes publ? N = No F9L-F10L 040-041 038L-039L F12L-F13L 044-045 042L-043L N F17R-E1L 049-050 047R-048L N E1L-E2L 050-051 048L-049L N E8R-E9L 057-058 055R-056L E9L-E10R 058-059 056L-057L N I1L-I2L 064-065 062L-063L N I2L-I3L 065-066 063L-064L N I5L-I6L 068-069 066L-067L I6L-I7L 069-070 067L-068L N I7L-I8R 070-071 068L-069R N I8R-G1L 071-072 069R-070L N G1L-G3L 072-073 070L-071L N G3L-G2R 073-074 071L-072R N G2R-G4L 074-075 072R-073L N G4L-G5R 075-076 073L-074R N G5R-G5.5R 076-077 074R-075R N G5.5R-G6R 077-078 075R-076R N G6R-G7L 078-079 076R-077L N G7L-G8R 079-080 077L-078R G8R-G9R 080-081 078R-079R G9R-L1R 081-082 079R-080R N L1R-L2R 082-083 080R-081R L2R-L3L 083-084 081R-082L L3L-L4R 084-085 082L-083R L4R-L5R 085-086 083R-084R N L5R-J1R 086-087 084R-085R N J3R-J4R 089-090 087R-088R N J4R-J5L 090-091 088R-089L J5L-J6R 091-092 089L-090R J6R-H1L 092-093 090R-091L N H1L-H2R 093-094 091L-092R N H2R-H3L 094-095 092R-093L H3L-H4L 095-096 093L-094L N H4L-H5R 096-097 094L-095R H5R-H6R 097-098 095R-096R N H6R-H7R 098-099 096R-097R H7R-D1R 099-100 097R-098R D1R-D2L 100-101 098R-099L N D2L-D3R 101-102 099L-100R N D3R-D4R 102-103 100R-101R N D4R-D5R 103-104 101R-102R D5R-D6R 104-105 102R-103R N D6R-D7R 105-106 103R-104R D9R-D10R 108-109 106R-107R N D10R-D11L 109-110 107R-108L D11L-D12L 110-111 108L-109L D12L-D13L 111-112 109L-110L D13L-A1L 112-113 110L-111L A1L-A2L 113-114 111L-112L N A2L-A2.5L 114-115 112L-113L N A2.5L-A3L 115-116 113L-114L A3L-A4L 116-117 114L-115L A4L-A5R 117-118 115L-116R A5R-A6L 118-119 116R-117L N A6L-A7L 119-120 117L-118L A7L-A8R 120-121 118L-119R A8R-A9L 121-122 119R-120L N A9L-A10L 122-123 120L-121L N A10L-A11R 123-124 121L-122R N A11R-A12L 124-125 122R-123L A12L-A13L 125-126 123L-124L A13L-A14L 126-127 124L-125L A14L-A14.5L 127-128 .sup. 125L-125.5L N A14.5L-A15L 128-129 125.5L-126L.sup. N A15L-A16L 129-130 126L-127L N A16L-A17L 130-131 127L-128L N A17L-A18R 131-132 128L-129R N A18R-A19L 132-133 129R-130L N A19L-A21L 133-134 130L-131L N A21L-A20R 134-135 131L-132R N A20R-A22R 135-136 132R-133R N A22R-A23R 136-137 133R-134R A23R-A24R 137-138 134R-135R A28L-A29L 141-142 139L-140L N A29L-A30L 142-143 140L-141L N
TABLE-US-00003 TABLE 2 Conserved genes with "end to end" orientation Overlapping CDC/Acambis Antoine Genes end to end ends genes genes F17R-E1L Yes 049-050 047R-048L E8R-E9L No 057-058 055R-056L I8R-G1L No 071-072 069R-070L G2R-G4L Yes 074-075 072R-073L G6R-G7L Yes 078-079 076R-077L L2R-L3L Yes 083-084 081R-082L J4R-J5L No 090-091 088R-089L J6R-H1L Yes 092-093 090R-091L H2R-H3L No 094-095 092R-093L D1R-D2L Yes 100-101 098R-099L D10R-D11L No 109-110 107R-108L A5R-A6L Yes 118-119 116R-117L A8R-A9L Yes 121-122 119R-120L A11R-A12L No 124-125 122R-123L A18R-A19L Yes 132-133 129R-130L Genes in bolded text have no overlapping ends and thus are simplest to use as intergenic sites.
[0051] According to the present invention, heterologous DNA sequences can be inserted into one or more IGRs in between two adjacent ORFs selected from the group consisting of (using the nomenclature according to CDC/Acambis):
[0052] 044-045, 049-050, 050-051, 058-059, 064-065, 065-066, 069-070, 070-071, 071-072, 072-073, 073-074, 074-075, 075-076, 076-077, 077-078, 078-079, 081-082, 085-086, 086-087, 089-090, 092-093, 093-094, 095-096, 097-098, 100-101, 101-102, 102-103, 104-105, 108-109, 113-114, 114-115, 118-119, 121-122, 122-123, 123-124, 127-128, 128-129, 129-130, 130-131, 131-132, 132-133, 133-134, 134-135, 135-136, 141-142, and 142-143, in an exemplary manner or corresponding thereto in other strains of vaccinia virus.
[0053] In a preferred embodiment, the heterologous sequence is inserted into an IGR flanked by two adjacent ORFs with "end to end" orientation selected from the group consisting .of 049-050, 071-072, 074-075, 078-079, 092-093, 100-101, 118-119, 121-122, and 132-133.
[0054] In a working example, the heterologous DNA sequence is inserted into an IGR in which the conserved genes have no overlapping ends 071-072.
[0055] Heterologous or exogenous DNA sequences are sequences which, in nature, are not normally found associated with the poxvirus as used according to the present invention. According to a further embodiment of the present invention, the exogenous DNA sequence comprises at least one coding sequence. The coding sequence is operatively linked to a transcription control element, preferably to a poxviral transcription control element. Additionally, also combinations between poxviral transcription control element and, e.g., internal ribosomal entry sites can be used.
[0056] According to a further embodiment, the exogenous DNA sequence can also comprise two or more coding sequences linked to one or several transcription control elements. Preferably, the coding sequence encodes one or more proteins, polypeptides, peptides, foreign antigens or antigenic epitopes, especially those of therapeutically interesting genes.
[0057] Therapeutically interesting genes according to the present invention may be genes derived from or homologous to genes of pathogenous or infectious microorganisms which are disease causing. Accordingly, in the context of the present invention such therapeutically interesting genes are presented to the immune system of an organism in order to affect, preferably induce a specific immune response and, thereby, vaccinate or prophylactically protect the organism against an infection with the microorganism. In further preferred embodiments of the present invention the therapeutically interesting genes are selected from genes of infectious viruses, e.g.,--but not limited to--dengue virus, hepatitis virus B or C, or human immunodeficiency viruses such as HIV.
[0058] According to a preferred embodiment of the present invention the heterologous DNA sequence is derived from HIV and encodes HIV env, wherein the HIV env gene is preferably inserted into the IGR between the ORFs 071-072 (I8R-G1L).
[0059] Furthermore, therapeutically interesting genes according to the present invention also comprise disease related genes, which have a therapeutic effect on proliferative disorder, cancer or metabolic diseases. For example, a therapeutically interesting gene regarding cancer could be a cancer antigen that has the capacity to induce a specific anti-cancer immune reaction.
[0060] According to a further embodiment of the present invention, the coding sequence comprises at least one marker or selection gene.
[0061] Selection genes transduce a particular resistance to a cell, whereby a certain selection method becomes possible. The skilled practitioner is familiar with a variety of selection genes, which can be used in a poxviral system. Among these are, e.g., neomycin resistance gene (NPT) or phosphoribosyl transferase gene (gpt).
[0062] Marker genes induce a color reaction in transduced cells, which can be used to identify transduced cells. The skilled practitioner is familiar with a variety of marker genes, which can be used in a poxviral system. Among these are the gene encoding, e.g., .beta.-galactosidase (.beta.-gal), .beta.-glucosidase (.beta.-glu), green fluorescence protein (EGFP) or blue fluorescence protein.
[0063] According to still a further embodiment of the present invention the exogenous
[0064] DNA sequence comprises a spacing sequence, which separates poxviral transcription control element and/or coding sequence in the exogenous DNA sequence from the stop codon and/or the start codon of the adjacent ORFs. This spacer sequence between the stop/start codon of the adjacent ORF and the inserted coding sequence in the exogenous DNA has the advantage to stabilize the inserted exogenous DNA and, thus, any resulting recombinant virus. The size of the spacer sequence is variable as long as the sequence is without its own coding or regulatory function.
[0065] According to a further embodiment, the spacer sequence separating the poxviral transcription control element and/or the coding sequence in the exogenous DNA sequence from the stop codon of the adjacent ORF is at least one nucleotide long.
[0066] According to another embodiment of the present invention, the spacing sequence separating the poxviral transcription control element and/or the coding sequence in the exogenous DNA sequence from the start codon of the adjacent ORF is at least 30 nucleotides. Particularly, in cases where a typical vaccinia virus promoter element is identified upstream of a start codon the insertion of exogenous DNA may not separate the promoter element from the start codon of the adjacent ORF. A typical vaccinia promoter element can be identified by scanning for e.g., the sequence "TAAAT" for late promoters (Davison & Moss 1989 J. Mol. Biol.; 210:771-784) and an A/T rich domain for early promoters. A spacing sequence of about 30 nucleotides is the preferred distance to secure that a poxviral promoter located upstream of the start codon of the ORF is not influenced. Additionally, according to a further preferred embodiment, the distance between the inserted exogenous DNA and the start codon of the adjacent ORF is around 50 nucleotides and more preferably around 100 nucleotides.
[0067] According to a further preferred embodiment of the present invention, the spacing sequence comprises an additional poxviral transcription control element which is capable to control the transcription of the adjacent ORF.
[0068] A typical MVA strain which can be used according to the present invention for generating a recombinant MVA is MVA 1974/NIH Clone 1 that has been deposited as ATCC Accession No.: PTA-5095 on Mar. 27, 2003 with the American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, Va. 20110-2209, USA.
[0069] The term "derivatives" of a virus according to the present invention refers to progeny viruses showing the same characteristic features as the parent virus but showing differences in one or more parts of its genome. The term "derivative of MVA" describes a virus, which has the same functional characteristics compared to MVA. For example, a derivative of MVA 1974/NIH Clone 1 has the characteristic features of MVA 1974/NIH Clone 1. One of these characteristics of MVA 1974/NIH Clone for derivatives thereof is its attenuation and severe restriction in host range.
[0070] The recombinant MVA according to the present invention is useful as a medicament or vaccine. It is, according to a further embodiment, used for the introduction of the exogenous coding sequence into a target cell, said sequence being either homologous or heterologous to the genome of the target cell.
[0071] The introduction of an exogenous coding sequence into a target cell may be done in vitro to produce proteins, polypeptides, peptides, antigens or antigenic epitopes. This method comprises the infection of a host cell with the recombinant MVA according to the invention, cultivation of the infected host cell under suitable conditions, and isolation and/or enrichment of the polypeptide, peptide, protein, antigen, epitope and/or virus produced by said host cell.
[0072] Furthermore, the method for introduction of one or more homologous or one or more heterologous sequence into cells may be applied for in vitro and in vivo therapy. For in vitro therapy, isolated cells that have been previously (ex vivo) infected with the recombinant MVA according to the invention are administered to the living animal body for affecting, preferably inducing an immune response. For in vivo therapy, the recombinant poxvirus according to the invention is directly administered to the living animal body for affecting, preferably inducing an immune response. In this case, the cells surrounding the site of inoculation, but also cells where the virus is transported to via, e.g., the blood stream, are directly infected in vivo by the recombinant MVA according to the invention. After infection, these cells synthesize the proteins, peptides or antigenic epitopes of the therapeutic genes, which are encoded by the exogenous coding sequences and, subsequently, present them or parts thereof on the cellular surface. Specialized cells of the immune system recognize the presentation of such heterologous proteins, peptides or epitopes and launch a specific immune response.
[0073] Since the MVA is highly growth restricted and, thus, highly attenuated, it is useful for the treatment of a wide range of mammals including humans, including immune-compromised animals or humans. The present invention also provides pharmaceutical compositions and vaccines for inducing an immune response in a living animal body, including a human.
[0074] The pharmaceutical composition may generally include one or more pharmaceutical acceptable and/or approved carriers, additives, antibiotics, preservatives, adjuvants, diluents and/or stabilizers. Such auxiliary substances can be water, saline, glycerol, ethanol, wetting or emulsifying agents, pH buffering substances, or the like. Suitable carriers are typically large, slowly metabolized molecules such as proteins, polysaccharides, polylactic acids, polyglycollic acids, polymeric amino acids, amino acid copolymers, lipid aggregates, or the like.
[0075] For the preparation of vaccines, the recombinant poxvirus according to the invention is converted into a physiologically acceptable form. This can be done based on the experience in the preparation of poxvirus vaccines used for vaccination against smallpox (as described by Stickl, H. et al. 1974 Dtsch Med Wochenschr. 99:2386-2392). For example, the purified virus is stored at -80.degree. C. with a titer of 5.times.10E8 TCID.sub.50/ml formulated in about 10 mM Tris, 140 mM NaCl pH 7.4. For the preparation of vaccine shots, e.g., 10E2-10E8 particles of the virus are lyophilized in 100 ml of phosphate-buffered saline (PBS) in the presence of 2% peptone and 1% human albumin in an ampoule, preferably a glass ampoule. Alternatively, the vaccine shots can be produced by stepwise freeze-drying of the virus in a formulation. This formulation can contain additional additives such as mannitol, dextran, sugar, glycine, lactose or polyvinylpyrrolidone or other aids such as antioxidants or inert gas, stabilizers or recombinant proteins (e.g., human serum albumin) suitable for in vivo administration. The glass ampoule is then sealed and can be stored between 4.degree. C. and room temperature for several months. However, as long as no need exists the ampoule is stored preferably at temperatures below -20.degree. C.
[0076] For vaccination or therapy the lyophilisate can be dissolved in 0.1 to 0.5 ml of an aqueous solution, preferably physiological saline or Tris buffer, and administered either systemically or locally, i.e., parenterally, subcutaneous, intramuscularly, by scarification or any other path of administration know to the skilled practitioner. The mode of administration, the dose and the number of administrations can be optimized by those skilled in the art in a known manner. However, most commonly a patient is vaccinated with a second shot about one month to six weeks after the first vaccination shot.
[0077] The present invention further relates to plasmid vectors, which can be used to generate recombinant MVA according to the present invention, and also relates to certain DNA sequences.
[0078] Regularly, the IGR located between or flanked by two adjacent ORFs comprises nucleotide sequences in which the exogenous DNA sequence of interest can be inserted. Accordingly, the plasmid vector according to the present invention comprises a DNA sequence derived from or homologous to the genome of MVA, wherein said DNA sequence comprises a complete or partial fragment of an IGR sequence located between or flanked by two adjacent ORFs of the viral genome. Preferably, the plasmid vector comprises inserted into said IGR-derived sequence at least one cloning site for the insertion of an exogenous DNA sequence of interest and, preferably, for the insertion of a poxviral transcription control element operatively linked to said heterologous DNA sequence. Optionally, the plasmid vector comprises a reporter- and/or selection gene cassette. The plasmid vector preferably also comprises sequences of the two adjacent ORFs flanking said complete or partial fragment of the IGR sequence.
[0079] Some IGRs have been identified which do not include nucleotide sequences. In these cases, the plasmid vector comprises DNA sequences of the IGR flanking sequences, i.e., DNA sequences of the two adjacent ORFs. Preferably, the cloning site for the insertion of the heterologous DNA sequence is inserted into the IGR. The DNA of the IGR flanking sequences is used to direct the insertion of exogenous DNA sequences into the corresponding IGR in the MVA genome. Such a plasmid vector may additionally include a complete or partial fragment of an IGR sequence which comprises the cloning site for the insertion of the heterologous DNA sequence and, optionally, of the, reporter- and/or selection gene cassette.
[0080] IGR-DNA sequences as well as IGR flanking sequences of the two adjacent ORFs are preferably selected from IGRs and ORFs, respectively, selected from the group consisting of (using the nomenclature according to CDC/Acambis):
[0081] 044-045, 049-050, 050-051, 058-059, 064-065, 065-066, 069-070, 070-071, 071-072, 072-073, 073-074, 074-075, 075-076, 076-077, 077-078, 078-079, 081-082, 085-086, 086-087, 089-090, 092-093, 093-094, 095-096, 097-098, 100-101, 101-102, 102-103, 104-105, 108-109, 113-114, 114-115, 118-119, 121-122, 122-123, 123-124, 127-128, 128-129, 129-130, 130-131, 131-132, 132-133, 133-134, 134-135, 135-136, 141-142, and 142-143, in an exemplary manner or corresponding thereto in other strains of vaccinia virus.
[0082] The sequences are, more preferably, selected from IGRs and ORFs, respectively, selected from the group consisting of 049-050, 071-072, 074-075, 078-079, 092-093, 100-101, 118-119, 121-122, and 132-133.
[0083] In a working example, the IGR derived sequence is selected as 071-072.
[0084] The DNA sequences are preferably derived from or homologous to the genome of the MVA deposited as ATCC Accession No.: PTA-5095 on Mar. 27, 2003 with the American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, Va. 20110-2209, USA.
[0085] To generate a plasmid vector according to the present invention the sequences are isolated and cloned into a standard cloning vector, such as pBluescript (Stratagene), wherein they flank the exogenous DNA to be inserted into the MVA genome. Optionally, such a plasmid vector comprises a selection- or reporter gene cassette, which can be deleted from the final recombinant virus, due to a repetitive sequence included into said cassette.
[0086] Methods to introduce exogenous DNA sequences by a plasmid vector into an MVA genome and methods to obtain recombinant MVA are well known to the person skilled in the art and, additionally, can be deduced can be deduced from Molecular Cloning, A Laboratory Manual, Second Edition, J. Sambrook, E. F. Fritsch and T. Maniatis, Cold Spring Harbor Laboratory Press, 1989 and Current Protocols in Molecular Biology, John Wiley and Son Inc. 1998, Chapter 16, section TV, "Expression of proteins in mammalian cells using vaccinia viral vectors".
[0087] The MVA according to the present invention may be produced by transfecting a cell with a plasmid vector according to the present invention, infecting the transfected cell with an MVA and, subsequently, identifying, isolating and, optionally, purifying the MVA according to the invention.
[0088] The DNA sequences according to the invention can be used to identify or isolate the MVA or its derivatives according to the invention and cells or individuals infected with an MVA according to the present invention. The DNA sequences are, e.g., used to generate PCR-primers, hybridization probes or are used in array technologies.
HIVs and Their Replication
[0089] The etiological agent of acquired immune deficiency syndrome (AIDS) is recognized to be a retrovirus exhibiting characteristics typical of the lentivirus genus, referred to as human immunodeficiency virus (HIV). The phylogenetic relationships of the human lentiviruses are shown in FIG. 1. HIV-2 is more closely related to SIV.sub.smm, a virus isolated from sooty mangabey monkeys in the wild, than to HIV-1. It is currently believed that HIV-2 represents a zoonotic transmission of SIV.sub.smm to man. A series of lentiviral isolates from captive chimpanzees, designated SIV.sub.cpz, are close genetic relatives of HIV-1.
[0090] The earliest phylogenetic analyses of HIV-1 isolates focused on samples from Europe/North America and Africa; discrete clusters of viruses were identified from these two areas of the world. Distinct genetic subtypes or clades of HIV-1 were subsequently defined and classified into three groups: M (major); O (outlier); and N (non-M or O) (FIG. 2). The M group of HIV-1, which includes over 95% of the global virus isolates, consists of at least eight discrete clades (A, B, C, D, F, G, H, and J), based on the sequence of complete viral genomes. Members of HIV-1 group O have been recovered from individuals living in Cameroon, Gabon, and Equatorial Guinea; their genomes share less than 50% identity in nucleotide sequence with group M viruses. The more recently discovered group N HIV-I strains have been identified in infected Cameroonians, fail to react serologically in standard whole-virus enzyme-linked immunosorbent assay (ELISA), yet are readily detectable by conventional Western blot analysis.
[0091] Most current knowledge about HIV-1 genetic variation comes from studies of group M viruses of diverse geographic origin. Data collected during the past decade indicate that the HIV-1 population present within an infected individual can vary from 6% to 10% in nucleotide sequence. HIV-1 isolates within a clade may exhibit nucleotide distances of 15% in gag and up to 30% in gp120 coding sequences. Interclade genetic variation may range between 30% and 40% depending on the gene analyzed.
[0092] All of the HIV-1 group M subtypes can be found in Africa. Clade A viruses are genetically the most divergent and were the most common HIV-1 subtype in Africa early in the epidemic. With the rapid spread of HIV-1 to southern Africa during the mid to late 1990s, clade C viruses have become the dominant subtype and now account for 48% of HIV-1 infections worldwide. Clade B viruses, the most intensively studied HIV-1 subtype, remain the most prevalent isolates in Europe and North America.
[0093] High rates of genetic recombination are a hallmark of retroviruses. It was initially believed that simultaneous infections by genetically diverse virus strains were not likely to be established in individuals at risk for HIV-1. By 1995, however, it became apparent that a significant fraction of the HIV-1 group M global diversity included interclade viral recombinants. It is now appreciated that HIV-1 recombinants will be found in geographic areas such as Africa, South America, and Southeast Asia, where multiple HIV-1 subtypes coexist and may account for more than 10% of circulating HIV-1 strains. Molecularly; the genomes of these recombinant viruses resemble patchwork mosaics, with juxtaposed diverse HIV-1 subtype segments, reflecting the multiple crossover events contributing to their generation. Most HIV-1 recombinants have arisen in Africa and a majority contains segments originally derived from clade A viruses. In Thailand, for example, the composition of the predominant circulating strain consists of a clade A gag plus pol gene segment and a clade E env gene. Because the clade E env gene in Thai HIV-1 strains is closely related to the clade E env present in virus isolates from the Central African Republic, it is believed that the original recombination event occurred in Africa, with the subsequent introduction of a descendent virus into Thailand. Interestingly, no full-length HIV-1 subtype E isolate (i.e., with subtype E gag, pol, and env genes) has been reported to date.
[0094] The discovery that a and p chemokine receptors function as coreceptors for virus fusion and entry into susceptible CD4.sup.+cells has led to a revised classification scheme for HIV-1 (FIG. 3). Isolates can now be grouped on the basis of chemokine receptor utilization in fusion assays in which HIV-1 gp120 and CD4.sup.+ coreceptor proteins are expressed in separate cells. As indicated in FIG. 3, HIV-1 isolates using the CXCR4 receptor (now designated X4 viruses) are usually T cell line (TCL)-tropic syncytium inducing (SI) strains, whereas those exclusively utilizing the CCR5 receptor (R5 viruses) are predominantly macrophage (M)-tropic and non-syncytium inducing (NSI). The dual-tropic R5/X4 strains, which may comprise the majority of patient isolates and exhibit a continuum of tropic phenotypes, are frequently SI.
[0095] As is the case for all replication-competent retroviruses, the three primary HIV-1 translation products, all encoding structural proteins, are initially synthesized as polyprotein precursors, which are subsequently processed by viral or cellular proteases into mature particle-associated proteins (FIG. 4). The 55-kd Gag precursor Pr55.sup.Gag is cleaved into the matrix (MA), capsid (CA), nucleocapsid (NC), and p6 proteins. Autocatalysis of the 160-kd Gag-Pol polyprotein, Pr160.sup.Gag-Pol, gives rise to the protease (PR), the heterodimeric reverse transcriptase (RT), and the integrase (IN) proteins, whereas proteolytic digestion by a cellular enzyme(s) converts the glycosylated 160-kd Env precursor gpl 60 to the gp120 surface (SU) and gp41 transmembrane (TM) cleavage products. The remaining six HIV-1-encoded proteins (Vif, Vpr, Tat, Rev, Vpu, and Nef) are the primary translation products of spliced mRNAs.
Gag
[0096] The Gag proteins of HIV, like those of other retroviruses, are necessary and sufficient for the formation of noninfectious, virus-like particles. Retroviral Gag proteins are generally synthesized as polyprotein precursors; the HIV-1 Gag precursor has been named, based on its apparent molecular mass, Pr55.sup.Gag. As noted previously, the mRNA for Pr55.sup.Gag is the unspliced 9.2-kb transcript (FIG. 4) that requires Rev for its expression in the cytoplasm. When the pol ORF is present, the viral protease (PR) cleaves Pr55.sup.Gag during or shortly after budding from the cell to generate the mature Gag proteins p17 (MA), p24 (CA), p7 (NC), and p6 (see FIG. 4). In the virion, MA is localized immediately inside the lipid bilayer of the viral envelope, CA forms the outer portion of the cone-shaped core structure in the center of the particle, and NC is present in the core in a ribonucleoprotein complex with the viral RNA genome (FIG. 5).
[0097] The HIV Pr55.sup.Gag precursor oligomerizes following its translation and is targeted to the plasma membrane, where particles of sufficient size and density to be visible by EM are assembled. Formation of virus-like particles by Pr55.sup.Gag is a self-assembly process, with critical Gag-Gag interactions taking place between multiple domains along the Gag precursor. The assembly of virus-like particles does not require the participation of genomic RNA (although the presence of nucleic acid appears to be essential), pol-encoded enzymes, or Env glycoproteins, but the production of infectious virions requires the encapsidation of the viral RNA genome and the incorporation of the Env glycoproteins and the Gag-Pol polyprotein precursor Prl 60.sup.Gag-Pol .
Pol
[0098] Downstream of gag lies the most highly conserved region of the HIV genome, the pol gene, which encodes three enzymes: PR, RT, and IN (see FIG. 4). RT and IN are required, respectively, for reverse transcription of the viral RNA genome to a double-stranded DNA copy, and for the integration of the viral DNA into the host cell chromosome. PR plays a critical role late in the life cycle by mediating the production of mature, infectious virions. The pol gene products are derived by enzymatic cleavage of a 160-kd Gag-Pol fusion protein, referred to as Pr160.sup.Gag-Pol. This fusion protein is produced by ribosomal frameshifting during translation of Pr55.sup.Gag (see FIG. 4). The frame-shifting mechanism for Gag-Pol expression, also utilized by many other retroviruses, ensures that the pol-derived proteins are expressed at a low level, approximately 5% to 10% that of Gag. Like Pr55.sup.Gag, the N-terminus of Pr160.sup.Gag-Pol is myristylated and targeted to the plasma membrane.
Protease
[0099] Early pulse-chase studies performed with avian retroviruses clearly indicated that retroviral Gag proteins are initially synthesized as polyprotein precursors that are cleaved to generate smaller products. Subsequent studies demonstrated that the processing function is provided by a viral rather than a cellular enzyme, and that proteolytic digestion of the Gag and Gag-Pol precursors is essential for virus infectivity. Sequence analysis of retroviral PRs indicated that they are related to cellular "aspartic" proteases such as pepsin and renin. Like these cellular enzymes, retroviral PRs use two apposed Asp residues at the active site to coordinate a water molecule that catalyzes the hydrolysis of a peptide bond in the target protein. Unlike the cellular aspartic proteases, which function as pseudodimers (using two folds within the same molecule to generate the active site), retroviral PRs function as true dimers. X-ray crystallographic data from HIV-1 PR indicate that the two monomers are held together in part by a four-stranded antiparallel (3-sheet derived from both N- and C-terminal ends of each monomer. The substrate-binding site is located within a cleft formed between the two monomers. Like their cellular homologs, the HIV PR dimer contains flexible "flaps" that overhang the binding site and may stabilize the substrate within the cleft; the active-site Asp residues lie in the center of the dimer. Interestingly, although some limited amino acid homology is observed surrounding active-site residues, the primary sequences of retroviral PRs are highly divergent, yet their structures are remarkably similar.
Reverse Transcriptase
[0100] By definition, retroviruses possess the ability to convert their single-stranded RNA genomes into double-stranded DNA during the early stages of the infection process. The enzyme that catalyzes this reaction is RT, in conjunction with its associated RNaseH activity. Retroviral RTs have three enzymatic activities: (a) RNA-directed DNA polymerization (for minus-strand DNA synthesis), (b) RNaseH activity (for the degradation of the tRNA primer and genomic RNA present in DNA-RNA hybrid intermediates), and (c) DNA-directed DNA polymerization (for second- or plus-strand DNA synthesis).
[0101] The mature HIV-1 RT holoenzyme is a heterodimer of 66 and 51 kd subunits. The 51-kd subunit (p51) is derived from the 66-kd (p66) subunit by proteolytic removal of the C-terminal 15-kd RNaseH domain of p66 by PR (see FIG. 4). The crystal structure of HIV-1 RT reveals a highly asymmetric folding in which the orientations of the p66 and p51 subunits differ substantially. The p66 subunit can be visualized as a right hand, with the polymerase active site within the palm, and a deep template-binding cleft formed by the palm, fingers, and thumb subdomains. The polymerase domain is linked to RNaseH by the connection subdomain. The active site, located in the palm, contains three critical Asp residues (110, 185, and 186) in close proximity, and two coordinated Mg.sup.2+ ions. Mutation of these Asp residues abolishes RT polymerizing activity. The orientation of the three active-site Asp residues is similar to that observed in other DNA polymerases (e.g., the Klenow fragment of E. coli DNA poll). The p51 subunit appears to be rigid and does not form a polymerizing cleft; Asp 110, 185, and 186 of this subunit are buried within the molecule. Approximately 18 base pairs of the primer-template duplex lie in the nucleic acid binding cleft, stretching from the polymerase active site to the RNaseH domain.
[0102] In the RT-primer-template-dNTP structure, the presence of a dideoxynucleotide at the 3' end of the primer allows visualization of the catalytic complex trapped just prior to attack on the incoming dNTP. Comparison with previously obtained structures suggests a model whereby the fingers close in to trap the template and dNTP prior to nucleophilic attack of the 3'-OH of the primer on the incoming dNTP. After the addition of the incoming dNTP to the growing chain, it has been proposed that the fingers adopt a more open configuration, thereby releasing the pyrophosphate and enabling RT to bind the next dNTP. The structure of the HIV-1 RNaseH has also been determined by x-ray crystallography; this domain displays a global folding similar to that of E. coli RNaseH.
Integrase
[0103] A distinguishing feature of retrovirus replication is the insertion of a DNA copy of the viral genome into the host cell chromosome following reverse transcription. The integrated viral DNA (the provirus) serves as the template for the synthesis of viral RNAs and is maintained as part of the host cell genome for the lifetime of the infected cell. Retroviral mutants deficient in the ability to integrate generally fail to establish a productive infection.
[0104] The integration of viral DNA is catalyzed by integrase, a 32-kd protein generated by PR-mediated cleavage of the C-terminal portion of the HIV-1 Gag-Pol polyprotein (see FIG. 4).
[0105] Retroviral IN proteins are composed of three structurally and functionally distinct domains: an N-terminal, zinc-finger-containing domain, a core domain, and a relatively nonconserved C-terminal domain. Because of its low solubility, it has not yet been possible to crystallize the entire 288-amino-acid HIV-1 IN protein. However, the structure of all three domains has been solved independently by x-ray crystallography or NMR methods. The crystal structure of the core domain of the avian sarcoma virus IN has also been determined. The N-terminal domain (residues 1 to 55), whose structure was solved by NMR spectroscopy, is composed of four helices with a zinc coordinated by amino acids His-12, His-16, Cys-40, and Cys-43. The structure of the N-terminal domain is reminiscent of helical DNA binding proteins that contain a so-called helix-turn-helix motif; however, in the HIV-1 structure this motif contributes to dimer formation. Initially, poor solubility hampered efforts to solve the structure of the core domain. However, attempts at crystallography were successful when it was observed that a Phe-to-Lys change at IN residue 185 greatly increased solubility without disrupting in vitro catalytic activity. Each monomer of the HIV-1 IN core domain (IN residues 50 to 212) is composed of a five-stranded n-sheet flanked by helices; this structure bears striking resemblance to other polynucleotidyl transferases including RNaseH and the bacteriophage MuA transposase. Three highly conserved residues are found in analogous positions in other polynucleotidyl transferases; in HIV-1 IN these are Asp-64, Asp-116 and Glu-152, the so-called D,D-35-E motif. Mutations at these positions block HIV IN function both in vivo and in vitro. The close proximity of these three amino acids in the crystal structure of both avian sarcoma virus and HIV-1 core domains supports the hypothesis that these residues play a central role in catalysis of the polynucleotidyl transfer reaction that is at the heart of the integration process. The C-terminal domain, whose structure has been solved by NMR methods, adopts a five-stranded .beta.-barrel folding topology reminiscent of a Src homology 3 (SH3) domain. Recently, the x-ray structures of SIV and Rous sarcoma virus IN protein fragments encompassing both the core and C-terminal domains have been solved.
Env
[0106] The HIV Env glycoproteins play a major role in the virus life cycle. They contain the determinants that interact with the CD4 receptor and coreceptor, and they catalyze the fusion reaction between the lipid bilayer of the viral envelope and the host cell plasma membrane. In addition, the HIV Env glycoproteins contain epitopes that elicit immune responses that are important from both diagnostic and vaccine development perspectives.
[0107] The HIV Env glycoprotein is synthesized from the singly spliced 4.3-kb Vpu/Env bicistronic mRNA (see FIG. 4); translation occurs on ribosomes associated with the rough endoplasmic reticulum (ER). The 160-kd polyprotein precursor (gp160) is an integral membrane protein that is anchored to cell membranes by a hydropholkc stop-transfer signal in the domain destined to be the mature TM Env glycoprotein, gp41 (FIG. 6). The gp160 is cotranslationally glycosylated, forms disulfide bonds, and undergoes oligomerization in the ER. The predominant oligomeric form appears to be a trimer, although dimers and tetramers are also observed. The gp160 is transported to the Golgi, where, like other retroviral envelope precursor proteins, it is proteolytically cleaved by cellular enzymes to the mature SU glycoprotein gp120 and TM glycoprotein gp41 (see FIG. 6). The cellular enzyme responsible for cleavage of retroviral Env precursors following a highly conserved Lys/Arg-X-Lys/Arg-Arg motif is furin or a furin-like protease, although other enzymes may also catalyze gp160 processing. Cleavage of gp160 is required for Env-induced fusion activity and virus infectivity. Subsequent to gp160 cleavage, gp120 and gp41 form a noncovalent association that is critical for transport of the Env complex from the Golgi to the cell surface. The gp120-gp41 interaction is fairly weak, and a substantial amount of gp120 is shed from the surface of Env-expressing cells.
[0108] The HIV Env glycoprotein complex, in particular the SU (gp120) domain, is very heavily glycosylated; approximately half the molecular mass of gp160 is composed of oligosaccharide side chains. During transport of Env from its site of synthesis in the ER to the plasma membrane, many of the side chains are modified by the addition of complex sugars. The numerous oligosaccharide side chains form what could be imagined as a sugar cloud obscuring much of gp120 from host immune recognition. As shown in FIG. 6, gp120 contains interspersed conserved (C.sub.1 to C.sub.5) and variable (V.sub.1 to V.sub.5) domains. The Cys residues present in the gp120s of different isolates are highly conserved and form disulfide bonds that link the first four variable regions in large loops.
[0109] A primary function of viral Env glycoproteins is to promote a membrane fusion reaction between the lipid bilayers of the viral envelope and host cell membranes. This membrane fusion event enables the viral core to gain entry into the host cell cytoplasm. A number of regions in both gp120 and gp41 have been implicated, directly or indirectly, in Env-mediated membrane fusion. Studies of the HA.sub.2 hemagglutinin protein of the orthomyxoviruses and the F protein of the paramyxoviruses indicated that a highly hydrophobic domain at the N-terminus of these proteins, referred to as the fusion peptide, plays a critical role in membrane fusion. Mutational analyses demonstrated that an analogous domain was located at the N-terminus of the HIV-1, HIV-2, and SIV TM glycoproteins (see FIG. 6). Nonhydrophobic substitutions within this region of gp41 greatly reduced or blocked syncytium formation and resulted in the production of noninfectious progeny virions.
[0110] C-terminal to the gp41 fusion peptide are two amphipathic helical domains (see FIG. 6) which play a central role in membrane fusion. Mutations in the N-terminal helix (referred to as the N-helix), which contains a Leu zipper-like heptad repeat motif, impair infectivity and membrane fusion activity, and peptides derived from these sequences exhibit potent antiviral activity in culture. The structure of the ectodomain of HIV-1 and SIV gp41, the two helical motifs in particular, has been the focus of structural analyses in recent years. Structures were determined by x-ray crystallography or NMR spectroscopy either for fusion proteins containing the helical domains, a mixture of peptides derived from the N- and C-helices, or in the case of the SIV structure, the intact gp41 ectodomain sequence from residue 27 to 149. These studies obtained fundamentally similar trimeric structures, in which the two helical domains pack in an antiparallel fashion to generate a six-helix bundle. The N-helices form a coiled-coil in the center of the bundle, with the C-helices packing into hydrophobic grooves on the outside.
[0111] In the steps leading to membrane fusion CD4 binding induces conformation changes in Env that facilitate coreceptor binding. Following the formation of a ternary gp120/CD4/coreceptor complex, gp41 adopts a hypothetical conformation that allows the fusion peptide to insert into the target lipid bilayer. The formation of the gp41 six-helix bundle (which involves antiparallel interactions between the gp41 N- and C-helices) brings the viral and cellular membranes together and membrane fusion takes place.
Use of Recombinant MVA Virus To Boost CD+8 Cell Immune Response
[0112] The present invention relates to generation of a CD8.sup.+ T cell immune response against an antigen and also eliciting an antibody response. More particularly, the present invention relates to "prime and boost" immunization regimes in which the immune response induced by administration of a priming composition is boosted by administration of a boosting composition. The present invention is based on prior experimental demonstration that effective boosting can be achieved using modified vaccinia Ankara (MVA) vectors, following priming with any of a variety of different types of priming compositions including recombinant MVA itself.
[0113] A major protective component of the immune response against a number of pathogens is mediated by T lymphocytes of the CD8.sup.+ type, also known as cytotoxic T lymphocytes (CTL). An important function of CD8.sup.+ cells is secretion of gamma interferon (IFN.gamma.), and this provides a measure of CD8.sup.+ T cell immune response. A second component of the immune response is antibody directed to the proteins of the pathogen.
[0114] The present invention employs MVA which, as prior experiments show, has been found to be an effective means for providing a boost to a CD8.sup.+ T cell immune response primed to antigen using any of a variety of different priming compositions and also eliciting an antibody response.
[0115] Notably, prior experimental work demonstrates that use of predecessors of the present invention allows for recombinant MVA virus expressing an HIV antigen to boost a CD8.sup.+ T cell immune response primed by a DNA vaccine and also eliciting an antibody response. The MVA may be found to induce a CD8.sup.+ T cell response after immunization. Recombinant MVA may also be shown to prime an immune response that is boosted by one or more inoculations of recombinant MVA.
[0116] Non-human primates immunized with plasmid DNA and boosted with the MVA were effectively protected against intramucosal challenge with live virus (Amara et al 2001 Science 292:69-74). Advantageously, the inventors contemplate that a vaccination regime using intradermal, intramuscular or mucosal immunization for both prime and boost can be employed, constituting a general immunization regime suitable for inducing CD8.sup.+ T cells and also eliciting an antibody response, e.g., in humans.
[0117] The present invention in various aspects and embodiments employs an MVA vector encoding an HIV antigen for boosting a CD8.sup.+ T cell immune response to the antigen primed by previous administration of nucleic acid encoding the antigen and also eliciting an antibody response.
[0118] A general aspect of the present invention provides for the use of an MVA vector for boosting a CD8.sup.+ T cell immune response to an HIV antigen and also eliciting an antibody response.
[0119] One aspect of the present invention provides a method of boosting a CD8.sup.+ T cell immune response to an HIV antigen in an individual, and also eliciting an antibody response, the method including provision in the individual of an MVA vector including nucleic acid encoding the antigen operably linked to regulatory sequences for production of antigen in the individual by expression from the nucleic acid, whereby a CD8.sup.+ T cell immune response to the antigen previously primed in the individual is boosted.
[0120] An immune response to an HIV antigen may be primed by immunization with plasmid DNA or by infection with an infectious agent.
[0121] A further aspect of the invention provides a method of inducing a CD8.sup.+ T cell immune response to an HIV antigen in an individual, and also eliciting an antibody response, the method comprising administering to the individual a priming composition comprising nucleic acid encoding the antigen and then administering a boosting composition which comprises an MVA vector including nucleic acid encoding the antigen operably linked to regulatory sequences for production of antigen in the individual by expression from the nucleic acid.
[0122] A further aspect provides for use of an MVA vector, as disclosed, in the manufacture of a medicament for administration to a mammal to boost a CD8.sup.+ T cell immune response to an HIV antigen, and also eliciting an antibody response. Such a medicament is generally for administration following prior administration of a priming composition comprising nucleic acid encoding the antigen.
[0123] The priming composition may comprise DNA encoding the antigen, such DNA preferably being in the form of a circular plasmid that is not capable of replicating in mammalian cells. Any selectable marker should not be resistance to an antibiotic used clinically, so for example Kanamycin resistance is preferred to Ampicillin resistance. Antigen expression should be driven by a promoter which is active in mammalian cells, for instance the cytomegalovirus immediate early (CMV IE) promoter.
[0124] In particular embodiments of the various aspects of the present invention, administration of a priming composition is followed by boosting with a boosting composition, or first and second boosting compositions, the first and second boosting compositions being the same or different from one another. Still further boosting compositions may be employed without departing from the present invention. In one embodiment, a triple immunization regime employs DNA, then adenovirus as a first boosting composition, then MVA as a second boosting composition, optionally followed by a further (third) boosting composition or subsequent boosting administration of one or other or both of the same or different vectors. Another option is DNA then MVA then adenovirus, optionally followed by subsequent boosting administration of one or other or both of the same or different vectors.
[0125] The antigen to be encoded in respective priming and boosting compositions (however many boosting compositions are employed) need not be identical, but should share at least one CD8.sup.+ T cell epitope. The antigen may correspond to a complete antigen, or a fragment thereof. Peptide epitopes or artificial strings of epitopes may be employed, more efficiently cutting out unnecessary protein sequence in the antigen and encoding sequence in the vector or vectors. One or more additional epitopes may be included, for instance epitopes which are recognized by T helper cells, especially epitopes recognized in individuals of different HLA types.
[0126] An HIV antigen of the invention to be encoded by a recombinant MVA virus includes polypeptides having immunogenic activity elicited by an amino acid sequence of an HIV Env, Gag, Pol, Vif, Vpr, Tat, Rev, Vpu, or Nef amino acid sequence as at least one CD8.sup.+T cell epitope. This amino acid sequence substantially corresponds to at least one 10-900 amino acid fragment and/or consensus sequence of a known HIV Env or Pol; or at least one 10-450 amino acid fragment and/or consensus sequence of a known HIV Gag; or at least one 10-100 amino acid fragment and/or consensus sequence of a known HIV Vif, Vpr, Tat, Rev, Vpu, or Nef.
[0127] Although a full length Env precursor sequence is presented for use in the present invention, Env is optionally deleted of subsequences. For example, regions of the gp120 surface and gp41 transmembrane cleavage products can be deleted.
[0128] Although a full length Gag precursor sequence is presented for use in the present invention, Gag is optionally deleted of subsequences. For example, regions of the matrix protein (p17), regions of the capsid protein (p24), regions of the nucleocapsid protein (p7), and regions of p6 (the C-terminal peptide of the Gag polyprotein) can be deleted.
[0129] Although a full length Pol precursor sequence is presented for use in the present invention, Pol is optionally deleted of subsequences. For example, regions of the protease protein (p10), regions of the reverse transcriptase protein (p66/p51), and regions of the integrase protein (p32) can be deleted.
[0130] Such an HIV Env, Gag, or Pol can have overall identity of at least 50% to a known Env, Gag, or Pol protein amino acid sequence, such as 50-99% identity, or any range or value therein, while eliciting an immunogenic response against at least one strain of an HIV.
[0131] Percent identity can be determined, for example, by comparing sequence information using the GAP computer program, version 6.0, available from the University of Wisconsin Genetics Computer Group (UWGCG). The GAP program utilizes the alignment method of Needleman and Wunsch (J Mol Biol 1970 48:443), as revised by Smith and Waterman (Adv Appl Math 1981 2:482). Briefly, the GAP program defines identity as the number of aligned symbols (i.e., nucleotides or amino acids) which are identical, divided by the total number of symbols in the shorter of the two sequences. The preferred default parameters for the GAP program include: (1) a unitary comparison matrix (containing a value of 1 for identities and 0 for non-identities) and the weighted comparison matrix of Gribskov and Burgess (Nucl Acids Res 1986 14:6745), as described by Schwartz and Dayhoff (eds., Atlas of Protein Sequence and Structure, National Biomedical Research Foundation, Washington, D.C. 1979, pp. 353-358); (2) a penalty of 3.0 for each gap and an additional 0.10 penalty for each symbol in each gap; and (3) no penalty for end gaps.
[0132] In a preferred embodiment, an Env of the present invention is a variant form of at least one HIV envelope protein. Preferably, the Env is composed of gp120 and the membrane-spanning and ectodomain of gp41 but lacks part or all of the cytoplasmic domain of gp41.
[0133] Known HIV sequences are readily available from commercial and institutional HIV sequence databases, such as GENBANK, or as published compilations, such as Myers et al. eds., Human Retroviruses and AIDS, A Compilation and Analysis of Nucleic Acid and Amino Acid Sequences, Vol. I and II, Theoretical Biology and Biophysics, Los Alamos, N. Mex. (1993), or on the world wide web at hiv-web.lanl.gov/.
[0134] Substitutions or insertions of an HIV Env, Gag, or Pol to obtain an additional HIV Env, Gag, or Pol, encoded by a nucleic acid for use in a recombinant MVA virus of the present invention, can include substitutions or insertions of at least one amino acid residue (e.g., 1-25 amino acids). Alternatively, at least one amino acid (e.g., 1-25 amino acids) can be deleted from an HIV Env, Gag, or Pol sequence. Preferably, such substitutions, insertions or deletions are identified based on safety features, expression levels, immunogenicity and compatibility with high replication rates of MVA.
[0135] Amino acid sequence variations in an HIV Env, Gag, or Pol of the present invention can be prepared e.g., by mutations in the DNA. Such HIV Env, Gag, or Pol include, for example, deletions, insertions or substitutions of nucleotides coding for different amino acid residues within the amino acid sequence. Obviously, mutations that will be made in nucleic acid encoding an HIV Env, Gag, or Pol must not place the sequence out of reading frame and preferably will not create complementary domains that could produce secondary mRNA structures.
[0136] HIV Env, Gag, or Pol-encoding nucleic acid of the present invention can also be prepared by amplification or site-directed mutagenesis of nucleotides in DNA or RNA encoding an HIV Env, Gag, or Pol and thereafter synthesizing or reverse transcribing the encoding DNA to produce DNA or RNA encoding an HIV Env, Gag, or Pol, based on the teaching and guidance presented herein.
[0137] Recombinant MVA viruses expressing HIV Env, Gag, or Pol of the present invention, include a finite set of HIV Env, Gag, or Pol-encoding sequences as substitution nucleotides that can be routinely obtained by one of ordinary skill in the art, without undue experimentation, based on the teachings and guidance presented herein. For a detailed description of protein chemistry and structure, see Schulz, G. E. et al., 1978 Principles of Protein Structure, Springer-Verlag, New York, N.Y., and Creighton, T. E., 1983 Proteins: Structure and Molecular Properties, W. H. Freeman & Co., San Francisco, Calif. For a presentation of nucleotide sequence substitutions, such as codon preferences, see Ausubel et al. eds. Current Protocols in Molecular Biology, Greene Publishing Assoc., New York, N.Y. 1994 at .sctn..sctn. A.1.1-A.1.24, and Sambrook, J. et al. 1989 Molecular Cloning: A Laboratory Manual, Second Edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. at Appendices C and D.
[0138] Thus, one of ordinary skill in the art, given the teachings and guidance presented herein, will know how to substitute other amino acid residues in other positions of an HIV env, gag, or pol DNA or RNA to obtain alternative HIV Env, Gag, or Pol, including substitutional, deletional or insertional variants.
[0139] Within the MVA vector, regulatory sequences for expression of the encoded antigen will include a promoter. By "promoter" is meant a sequence of nucleotides from which transcription may be initiated of DNA operably linked downstream (i.e., in the 3' direction on the sense strand of double-stranded DNA). "Operably linked" means joined as part of the same nucleic acid molecule, suitably positioned and oriented for transcription to be initiated from the promoter. DNA operably linked to a promoter is "under transcriptional initiation regulation" of the promoter. Other regulatory sequences including terminator fragments, polyadenylation sequences, marker genes and other sequences may be included as appropriate, in accordance with the knowledge and practice of the ordinary person skilled in the art: see, for example, Moss, B. (2001). Poxviridae: the viruses and their replication. In Fields Virology, D. M. Knipe, and P. M. Howley, eds. (Philadelphia, Lippincott Williams & Wilkins), pp. 2849-2883. Many known techniques and protocols for manipulation of nucleic acid, for example in preparation of nucleic acid constructs, mutagenesis, sequencing, introduction of DNA into cells and gene expression, and analysis of proteins, are described in detail in Current Protocols in Molecular Biology, 1998 Ausubel et al. eds., John Wiley & Sons.
[0140] Promoters for use in aspects and embodiments of the present invention may be compatible with poxvirus expression systems and include natural, modified and synthetic sequences.
[0141] Either or both of the priming and boosting compositions may include an adjuvant, such as granulocyte macrophage-colony stimulating factor (GM-CSF) or encoding nucleic acid therefor.
[0142] Administration of the boosting composition is generally about 1 to 6 months after administration of the priming composition, preferably about 1 to 3 months.
[0143] Preferably, administration of priming composition, boosting composition, or both priming and boosting compositions, is intradermal, intramuscular or mucosal immunization.
[0144] Administration of MVA vaccines may be achieved by using a needle to inject a suspension of the virus. An alternative is the use of a needleless injection device to administer a virus suspension (using, e.g., Biojector.TM. needleless injector) or a resuspended freeze-dried powder containing the vaccine, providing for manufacturing individually prepared doses that do not need cold storage. This would be a great advantage for a vaccine that is needed in rural areas of Africa.
[0145] MVA is a virus with an excellent safety record in human immunizations. The generation of recombinant viruses can be accomplished simply, and they can be manufactured reproducibly in large quantities. Intradermal, intramuscular or mucosal administration of recombinant MVA virus is therefore highly suitable for prophylactic or therapeutic vaccination of humans against AIDS which can be controlled by a CD8.sup.+ T cell response.
[0146] The individual may have AIDS such that delivery of the antigen and generation of a CD8.sup.+ T cell immune response to the antigen is of benefit or has a therapeutically beneficial effect.
[0147] Most likely, administration will have prophylactic aim to generate an immune response against HIV or AIDS before infection or development of symptoms.
[0148] Components to be administered in accordance with the present invention may be formulated in pharmaceutical compositions. These compositions may comprise a pharmaceutically acceptable excipient, carrier, buffer, stabilizer or other materials well known to those skilled in the art. Such materials should be non-toxic and should not interfere with the efficacy of the active ingredient. The precise nature of the carrier or other material may depend on the route of administration, e.g., intravenous, cutaneous or subcutaneous, nasal, intramuscular, intraperitoneal routes.
[0149] As noted, administration is preferably intradermal, intramuscular or mucosal.
[0150] Physiological saline solution, dextrose or other saccharide solution or glycols such as ethylene glycol, propylene glycol or polyethylene glycol may be included.
[0151] For intravenous, cutaneous, subcutaneous, intramuscular or mucosal injection, or injection at the site of affliction, the active ingredient will be in the form of a parenterally acceptable aqueous solution which is pyrogen-free and has suitable pH, isotonicity and stability. Those of relevant skill in the art are well able to prepare suitable solutions using, for example, isotonic vehicles such as Sodium Chloride Injection, Ringer's Injection, Lactated Ringer's Injection. Preservatives, stabilizers, buffers, antioxidants and/or other additives may be included as required.
[0152] A slow-release formulation may be employed.
[0153] Following production of MVA particles and optional formulation of such particles into compositions, the particles may be administered to an individual, particularly human or other primate. Administration may be to another mammal, e.g., rodent such as mouse, rat or hamster, guinea pig, rabbit, sheep, goat, pig, horse, cow, donkey, dog or cat.
[0154] Administration is preferably in a "prophylactically effective amount" or a "therapeutically effective amount" (as the case may be, although prophylaxis may be considered therapy), this being sufficient to show benefit to the individual. The actual amount administered, and rate and time-course of administration, will depend on the nature and severity of what is being treated. Prescription of treatment, e.g., decisions on dosage etc, is within the responsibility of general practitioners and other medical doctors, or in a veterinary context a veterinarian, and typically takes account of the disorder to be treated, the condition of the individual patient, the site of delivery, the method of administration and other factors known to practitioners. Examples of the techniques and protocols mentioned above can be found in Remington's Pharmaceutical Sciences, 16th edition, 1980, Osol, A. (ed.).
[0155] In one preferred regimen, DNA is administered at a dose of 300 .mu.g to 3 mg/injection, followed by MVA at a dose of 10.sup.6 to 10.sup.9 infectious virus particles/injection.
[0156] A composition may be administered alone or in combination with other treatments, either simultaneously or sequentially dependent upon the condition to be treated.
[0157] Delivery to a non-human mammal need not be for a therapeutic purpose, but may be for use in an experimental context, for instance in investigation of mechanisms of immune responses to an antigen of interest, e.g., protection against HIV or AIDS.
A shuttle Plasmid, Recombinant MVA/HIV1 Clinical Vaccine Construct and Mechanism for Retention of Intact Foreign Gene Inserts in Recombinant MVA by Codon Alteration of the Foreign Gene and Insertion of the Foreign Gene Between Two Vaccinia Virus Essential Genes
[0158] The invention provides mechanisms for: [0159] retention of intact foreign genes by inserting them between two vaccinia virus genes that are essential for MVA replication. Deletion of the foreign gene can provide a significant growth advantage for the recombinant MVA allowing it to compete with MVA containing the intact foreign gene upon repeated passage. However, most deletions of a foreign gene include loss of some part of the flanking vaccinia virus DNA. If that vaccinia virus DNA is essential, then those viruses with deletions will not replicate and compete with the MVA containing the intact foreign gene. This methodology will be useful in production of recombinant vaccinia viruses that must be amplified to large scale such as for use in clinical trials, and [0160] stabilizing foreign gene inserts by alteration of specific "hot spots" that otherwise readily undergo mutation after repeated passage of the recombinant virus. This methodology is useful in production of recombinant viruses that must be amplified to large scale such as for use in clinical trials.
[0161] And describes: [0162] the shuttle plasmid, pLW-73, used for insertion of a foreign gene between 2 essential vaccinia virus genes; and [0163] the recombinant MVA/HIV-1 clinical vaccine construct MVAJUGD4d, a material that embodies use of these two mechanisms.
Novel Methods for Generation of Stable Recombinant MVA Viruses
[0164] The inventors have made modified vaccinia virus Ankara (MVA) recombinants expressing env and gagpol genes from HIV-1 isolates from different geographical locations. The foreign genes were inserted into 2 sites, Deletion II and Deletion III of MVA. The stability of these genes after repeated passage of recombinant MVA in tissue culture has proven to be variable. The inventors demonstrated that the instability was due to either deletion of the entire foreign gene and some flanking DNA or specific point mutations resulting in propagation of progeny virions that have a growth advantage because they do not express the foreign gene. Here the inventors describe two novel methods of retaining the intact foreign gene recombinant MVA. First, the inventors constructed a transfer vector that directs insertion of a foreign gene between two essential vaccinia virus genes in the conserved central region of the genome. Use of this site for insertion of genes prevents the outgrowth of variants containing large deletions that include the essential vaccinia virus DNA. In addition, this plasmid can be used for insertion of additional genes into recombinant viruses. Second, analysis of isolates with point mutations revealed certain "hot spots" with a propensity for insertion or deletion of a single base that causes premature termination during translation. The inventors showed that generation of silent mutations in these sites resulted in stabilization of the inserted gene.
I. Novel Transfer Vector Construction and Application
[0165] Construction of Novel Transfer Vector, pLW-73
[0166] 1: The central region of the MVA genome, K7R-A24R, was examined for 1) pairs of genes conserved in the poxvirus family or chordopoxvirus subfamily and 2) genes that are in opposite orientation such that their 3' ends are in close proximity, thereby providing an insertion site that would not disrupt a vaccina promoter. The site chosen as the new insertion site was between two essential genes, I8R and G1L.
[0167] 2. The left flank of the new vector was constructed in the following way: Plasmid LAS-1 was cut with restriction enzymes EcoRI and XhoI to remove the del III MVA flank, GFP, and direct repeat of MVA flank. This insert was cut with Ascl and SacI and the GFP fragment was isolated. Five hundred thirty one base pairs at the end of the I8R gene (including the TAA stop codon) was PCR amplified with EcoRI and AscI restriction sites on the ends of the PCR product. PCR amplification of 229 base pairs of the direct repeat (from the end of the I8R gene including the TAA stop codon) was performed with oligonucleotides containing SacI and XhoI restriction sites. All four pieces of DNA, 1) the vector backbone with EcoRI and Xho 1 ends, 2) new left flank containing end of I8R with EcoRI and Asci ends, 3) GFP with Acsl and Sacl ends and the 4) direct repeat of the I8R flank with SacI and XhoI ends were ligated together to make plasmid pLW-72.
[0168] 3. The right flank was made as follows: pLW-72 was cut with restriction enzymes PstI and HindIII to release del III flank of the MVA in the plasmid. Seven hundred and two base pairs at the end of the G1L gene was PCR amplified with Pstl and HindIII restriction enzyme sites on the ends and ligated into the pLW-72 vector to make pLW-73 (FIG. 7). The sequence of pLW-73 is given in FIG. 8.
[0169] 4. The salient features of pLW-73 are: 1) the vector was designed for insertion of foreign genes between essential genes in MVA genome. The left flank consists of end of I8R gene and right flank consists of end of G1L gene. 2) the GFP gene is included for easy initial selection of recombinant virus 3) the GFP is flanked by direct repeats of the I8R gene which allows for transient expression of GFP as the GFP will be lost upon repeated passage of the recombinant virus. Referring to WO 2004/087201, features 2 and 3 were also contained in earlier plasmids used for making MVA/HIV recombinants, pLAS-1 and pLAS-2.
Application of pLW-73
[0170] 1. The env gene from the clade B ADA isolate of HIV-1 was cloned into pLW-73 and a recombinant MVA virus was made. DNA sequencing confirmed the location and integrity of the env gene.
[0171] 2. A recombinant MVA virus expressing the Ugandan clade D (isolate AO7412) env gene (FIG. 9) in the Deletion II site of MVA proved to be unstable, i.e., after repeated serial passage in culture, the gene was deleted from a significant portion of the virus progeny. The same gene was then cloned into pLW-73 and a recombinant MVA virus was made and characterized. The env gene insert was stable after repeated serial passage (8.times.) in culture i.e., no deletions of the inserted gene or the MVA flanking region were found. In addition, no other mutations arose when the gene was inserted into this site.
IIH. Point Mutation of "Hot Spots"
[0172] Analysis of Point Mutations
[0173] A recombinant MVA virus expressing the Ugandan Clade D (isolate AO3349) gagpol gene in the Deletion III site of MVA proved to be unstable. The major genetic alteration was the generation of single point mutations in runs of 4-6 G or C residues (Table 3). In addition, similar point mutations were found in non-staining plaques from similar recombinant viruses expressing the gagpol genes from a Kenyan clade A isolate and a Tanzanian clade C isolate of HIV-1.
[0174] Mutagenesis of Hot Spots and Analysis of Stability in Recombinant Virus
[0175] Using site-directed mutagenesis, silent mutations were made in 6 such regions of the gag gene from the Ugandan HIV-1 isolate. This altered gene, UGD 4d gagpol orf (FIG. 10), was cloned into pLAS-1 and recombined into the same Deletion III site of MVA as was done in construction of the unstable virus. After repeated serial passage (8.times.) in culture, no non-expressing plaques were found. DNA sequencing of the passage 8 virus stock verified that the integrity of the gagpol gene was maintained.
III. Double Recombinant Construction
[0176] MVA/UGD4d Virus
[0177] MVA/UGD4d virus, a recombinant virus that expresses the Ugandan subtype D AO7412 envelope and the AO3349 gagpol, was constructed in the following way: The envelope and gagpol genes were inserted into MVA 1974/NIH Clone 1 by homologous recombination utilizing shuttle plasmids pLW-73 and pLAS-1, respectively. MVA/UGD4d was isolated by 6 rounds of plaque purification in chicken embryo fibroblast cells and subsequently amplified and characterized.
Summary
[0178] 1. A plasmid transfer vector was constructed that directs recombination of a foreign gene between two essential genes, I8R and G1L, in the conserved central region of the MVA genome. The use of this site was shown to inhibit selection of mutant viruses with deletions of inserted gene/MVA flanks.
[0179] 2. Highly mutable runs of G and C residues were altered by site-directed mutagenesis and silent mutations in the coding sequence were generated. This change was shown to stabilize the gene when inserted into Deletion III of MVA.
[0180] 3. Utilizing these two methods above, UGD4d double MVA recombinant that stably expresses both the env and gagpol of Ugandan Clade D was constructed.
EXAMPLE 1
[0181] Recombinant MVAs expressing HIV-1 env and gagpol genes from many different isolates have been made. The stability of inserted genes after repeated passage in tissue culture has proven to be variable. Here the inventors (1) demonstrate that the instability represents a combination of spontaneous mutation or deletion of the inserted gene and selection for non-expressing mutants and (2) describe novel methods for reducing instability.
Overview
[0182] Recombinant MVAs expressing env and gagpol from many different isolates were constructed. Each virus was subjected to repeated passages in chicken embryo fibroblast cells to mimic the large-scale amplification required for production of virus for clinical trials. Insert stability was monitored by env and gag immunostaining of individual plaques. For some recombinant viruses, env and/or gag expression was found to be rapidly lost in a significant fraction of the virus population. To identify the mechanism(s) of loss of expression, individual plaques were isolated and the nature of the mutations was characterized. In some cases, specific DNA sequences with propensity to mutate by addition or deletion of a single nucleotide were identified. Generation of such mutations could be avoided by altering codons without changing the predicted translation product. In other cases, loss of expression was caused by large deletions that frequently extended into flanking non-essential MVA genes. To prevent this from occurring, a new shuttle plasmid was constructed that was designed to direct insertion of foreign genes between two essential MVA genes. Recombination into this site reduced deletions of the foreign DNA. In one case, however, the toxicity associated with high-level HIV env expression was so severe that the selection of rare mutants still resulted in an unstable population. In this case, only truncation of the transmembrane domain of env allowed the construction of a stable recombinant MVA.
Generation of Recombinant MVAs and Analysis of Stability of Inserted Genes
[0183] Env and gagpol genes were cloned into MVA shuttle vectors. Expression and function were analyzed by transient expression assays. Gagpol was recombined into MVA 1974/NIH Clone 1. Recombinant MVA were plaque purified with 6-8 rounds followed by amplification of virus. Env was recombined into the MVA/gagpol isolate and double-recombinant MVA (FIG. 11A) were plaque purified with 6-8 rounds and were amplified. To assess the stability of inserts, virus was serially passaged in CEF cells using a multiplicity of infection (m.o.i.) of .about.1 pfu/cell to mimic large-scale production. Stability was evaluated by determining the percentage of cells expressing env or gag, as determined by immunostaining with monoclonal antibodies (FIG. 11B).
Stability of Recombinant MVAs
[0184] Recombinant MVAs expressing genes from HIV-1 isolates from different geographical locations were constructed. The env and gagpol genes were inserted into deletions II and III of MVA, respectively; both under control of the modified H5 promoter. The stability of env and gagpol genes from seven recombinant MVAs is shown in Table 4. Varying degrees of instability were observed in the seven viruses. In MVA/65A/G, expression of env was rapidly lost with only 25% of virions expressing env by passage 6. In MVA/UGD4a, both env and gagpol expression were increasingly lost with successive virus passages. Since at least 6-7 passages are required for production of a lot of virus for a Phase I trial, these two viruses were deemed unsuitable.
Analysis of Expression of MVA/65A/G
[0185] Referring to FIG. 12, thirteen plaques were randomly picked from P3 and P5 of MVA/65A/G and analyzed by immunostaining with T-24 mAb (binding site shown on a), Western blotting, PCR, and sequencing. Five types of plaques were found and the number of these plaques obtained for each type are given at right of FIG. 12. Plaques a, b, and c stained, but b and c were truncated versions due to base substitution (causing stop codon) (b) and deletion of the end of the env gene and part of MVA flank (c). Nonstaining plaques d and e resulted from addition of G to a 5G run causing a frameshift (d) and large deletion of entire env gene and parts of MVA flanks (e). Thus, base pair addition, substitution, and deletions all contributed to unstable expression of the env gene in MVA/65A/G. This A/G env, the most unstable example worked with, was picked to study modifications that might enhance stability.
Modifications to A/G Constructs to Increase Stability
[0186] 1. Synthetic envelope was made by removing 4 and 5 G and C runs by silent mutations to prevent point mutations.
[0187] 2. Vector 18/G1, i.e., pLW-73. was constructed with an insertion site between essential genes I8R and G1L to prevent deletions of genes and MVA flanks from being viable. The ends of the I8R (500 bp) and GIL (750 bp) genes of MVA were amplified by PCR and inserted into a vector containing vaccinia virus early/late mH5 promoter controlling foreign gene expression. This I8/G1 vector was used to insert foreign genes into MVA by homologous recombination (FIG. 13). Deletions of inserted genes and MVA flanking the inserted gene would not be viable because parts of essential genes would be deleted. Therefore, viruses with these mutations would not be able to overgrow the population with their normal growth advantage.
[0188] 3. A/G gp140 envelope was mutated by deleting the transmembrane domain and the cytoplasmic tail of gp41, resulting in a secreted protein.
Testing Modifications to Increase Stability
[0189] Seven single recombinant viruses were made with env modifications and/or use of new vector as shown in FIG. 14. Five plaques of each virus were isolated and passaged independently in CEF to determine if modifications enhanced envelope stable expression. Passaged plaques were analyzed by immunostaining with mAb T-43 (binding site mapped to 101-125aa of env), Western blotting, PCR, and sequencing.
Env Expression after Plaque Passages
[0190] Referring to FIG. 15, five independently passaged plaque isolates of each of the 7 recombinants listed above, were characterized at passages 1, 3, 5, and 7 by immunostaining with mAb T-43 (binds between 101-125a.a. in gp120). Four of 7 viruses (FIG. 15, a, b, c, e) had unstable protein expression in each of the 5 passaged plaques; two plaque passages of (FIG. 151) also had unstable env expression. These included viruses with the synthetic env in both del II (FIG. 15c) and in the essential gene site (FIG. 15f) of MVA genome. Only recombinant viruses containing the envelope as truncated, secreted gp140 remained stably expressing envelope (FIG. 15, d and g).
Western Blotting, PCR and Sequence Analyses
[0191] From selected plaque passages, clones were picked to analyze protein expression by Western blotting, PCR, and sequence analysis (FIG. 16). For Western blot analysis, T-24 and T-32 binding at the beginning and end of the clade A envelope, respectively, were used in order to determine if only partial or full length envelope was being made. Control viruses, marked c, are at the right of each blot. For the three viruses made in deletion II of MVA (FIG. 16a, b, and c), only in FIG. 16c (i.e., gp140 clones), were all the clones expressing detectable protein in Western. This protein (as measured by T-32) was not truncated. When envelope was inserted into the essential gene site by vector I8/G1 (FIG. 16d, e and f), again, only the gp140 envelope was being expressed in all clones and was not truncated. Although use of I8/G1 vector did not prevent mutations to the env sequence, it did prevent deletions which had been seen in envelope inserted into del II. (Note positive PCR products from all clones tested from I8/G1 vector, but negative PCR products from clones tested using del II vector.)
Expression of Env in Clade A/G Double Recombinant
[0192] Based on previous results with single env analysis, double recombinants expressing gagpol with either gp140 or the synthetic gp160 gene were made and tested for stability of env expression (FIG. 17). Five plaques were isolated from each as previously described, and passaged 7 times to analyze stability of env expression. At passage 7, the passaged plaques were immunostained with both T-43 and T-32 mAbs (which bind to gp120 and gp41, respectively). With T-43 mAb, one of five clones of recombinant expressing synthetic envelope consisted of only non-staining plaques. Subsequent T-32 staining of these plaques showed another plaque had truncated envelope expression. All passaged plaques from double recombinant containing gp140 envelope appeared stable by both T-43 and T-32 immunostaining. Titers were also 2 logs higher than with the other double recombinant. Thus a clade A/G double recombinant stably expressing envelope could only be made with gp140 envelope.
Recombinant Viruses Expressing env and gagpol from Ugandan HIV-1 Isolates
[0193] Recombinant MVA viruses expressing HIV-1 env and gagpol genes from Ugandan isolates AO7412 and AO3349 were constructed as shown in FIG. 18. Four to six independent isolates of each were serially passaged and both genes were found to be unstable whether expressed alone or in combination (Table 5). In contrast, expression of gp140 instead of membrane bound gp160 resulted in stability of the env gene after serial passage (FIG. 18 and Table 5).
MVA/UGD4a--Analysis of Non-Staining Env Plaques To determine the mechanism of instability, 24 individual non-staining plaques (using Mab T-43) were isolated from passage 6 of MVA/UGD4a, amplified, and characterized. Two small deletions (1.2 and 0.3 kb) were identified by PCR amplification and DNA sequencing (FIG. 19). All other isolates contained very large deletions that extended into the flanking MVA. The approximate break-points for these deletions were identified using primer pairs from within the env gene or flanking MVA regions.
Modification of UGD Env Gene in Recombinant MVA
[0194] To ameliorate the problem of instability of the UGD env gene, the AO7412 env gene was inserted into MVA using the new vector, I8/G1, which directs recombination of a foreign gene between 2 essential vaccinia virus genes, I8 and G1 and uses the modified H5 promoter (FIG. 20). Four independent plaques were serially passaged and analyzed for env expression by immunostaining with Mabs T-43 and T-32 at passage 5. In all isolates, the gene was stable (Table 6).
MVA/UGD4b--Analysis of Non-Staining gag Plaques
[0195] To determine the mechanism of instability of the gag gene, 8 individual non-staining plaques (using Mab 183-H12-5C--NIAID AIDS Repository) were picked from passage 6 of MVA/UGD4b, amplified, and the gagpol insert was sequenced (Table 7). In 7 isolates, an insertion or deletion of a single G residue at position 564-569 was found. In one isolate, a C residue was deleted from the sequence CCCC at position 530-534. Furthermore, non-staining plaques from high-passage stocks of MVA/KEA and MVA/TZC revealed, a similar hot-spot for mutation, i.e., position 564-569. Examination of the full sequence of the UGD AO7412 gagpol gene demonstrated 22 runs of 4 or more G or C residues (FIG. 21).
Modification of UGD Gagpol Gene in Recombinant MVA
[0196] Since the mechanism of instability of the gagpol gene was primarily insertion or deletion of a single nucleotide within a run of 4-6 G or C residues, the strategy to improve the stability of this gene was to generate silent mutations at such sites. Thus, site-directed mutagenesis at 6 sites in p17 and p24 gag (Table 3) was employed. The resulting codon altered (c.a.) gene inserted into MVA at the same location, i.e., Deletion III, proved to be stable upon serial passage (FIG. 22 and Table 8).
Construction of Stable, Recombinant MVA Expressing UGD Env and Gagpol
[0197] A recombinant virus expressing the UGD env gene in the I8/G1 locus and the codon altered gagpol gene in Deletion III of MVA was constructed (FIG. 23). Serial passage demonstrated no instability of either gene. Furthermore, the level of protein expression and DNA sequence were unaltered during passage (Table 9).
Conclusions
[0198] Instability of env and gagpol inserts is attributed to the generation of point mutations and deletions and the growth advantage of non-expressing MVA mutants. Instability can generally be reduced by codon alteration and/or insertion into an essential region of the MVA genome (MVA/UGD4d) but env had to be altered in one case (MVA/65A/G).
EXAMPLE 2
Immunogenicity of MVA/UGD4d in BALB/c Mice
[0199] Groups of 10 mice each were immunized by the intraperitoneal route with either 10.sup.6 or 10.sup.7 infectious units of MVA/UGD4d. Groups of 5 mice each were similarly immunized with parental MVA-1974. Mice were immunized at weeks 0 and 3 and bled at weeks 0, 3, and 5. Spleens were harvested at week 5.
[0200] Cellular responses were measured in fresh splenocytes by intracellular cytokine staining. Splenocytes were separately stimulated with the following: 1) immunodominant gag peptide (AMQMLKETI (SEQ ID NO: 6)), 2) env peptides (DTEVHNVWATHACVP (SEQ ID NO: 7) and QQQSNLLRAIEAQQH (SEQ ID NO: 8)), 3) pol peptides (8 peptides with single amino acid variants of ELRQHLLRWGLTT (SEQ ID NO: 9) and HGVYYDPSKDLIAE (SEQ ID NO: 10)), and 4) MVA.
[0201] Cells were stained for surface expression of CD4 and CD8 and then for intracellular expression of IFN-.gamma. and either IL2 or TNF. As shown in FIG. 24, MVA/UGD4d elicited CD8/IFN-.gamma. responses to the gag peptide, pol peptides, and MVA. The gag peptide responses were multifunctional, expressing both IFN-.gamma. and either IL2 or TNF. Also, CD4/IFN-.gamma. responses were elicited to the pool of env peptides.
[0202] Humoral responses were measured by ELISA (FIG. 25). Strong responses to UGD env were demonstrated at 3 weeks after one immunization and were boosted by the second immunization. In addition, strong vaccinia virus responses were elicited after one and two immunizations.
TABLE-US-00004 TABLE 3 MVA/UGD Nucleotide Changes Made to Eliminate Runs of G and C (HIV-1 isolate AO3349) Nucleotide # starting with ATG Original Sequence Modified Sequence 28-32 GGGGG GGAGG 70-74 GGGGG GGAGG 408-411 GGGG GGGA 530-533 CCCC CACC 564-569 GGGGGG AGGAGG 686-689 GGGG GAGG
TABLE-US-00005 TABLE 4 Stability of Recombinant MVAs Percent non-staining plaques LVD passage passage passage passage vaccine Geographic seed 3/4 6/7 9/9 10-13 lot Virus Clade origin env gag env gag env gag env gag env gag env gag KEA5b A Kenya <1 <1 0.13 0.33 0.34 0.36 0.54 2.4 0.64 0.77 65A/G A/G Ivory Coast <2 <1 28 1 75 62B B US <1 <1 <1 <1 6 <1 10 1 TZCa C Tanzania <1 <1 <1 <1 1.7 2.8 3.6 3.7 71C C India <1 <1 <1 1 <1 2 12 14 UGD4a D Uganda <1 <1 3 0.28 6.7 6 12.2 17.4 CMDR E/A Thailand <1 <1 <1 <1 <1 <1 <1 <1
TABLE-US-00006 TABLE 5 Recombinant Viruses Expressing env and gagpol from Ugandan HIV-1 isolates % non-staining passage env gag UGD4a 9 12.2 17.4 5 5.8 2.6 5 2.7 17.6 5 8.4 7.2 5 11.4 8.0 UGD4b 6 1.5 17.0 5 3.3 9.3 5 3.7 8.3 5 7.9 4.4 5 15.2 5.0 UGD1a 4 nd 18.8 4 nd 46.7 4 nd 64.9 4 nd 38.1 5 7.9 44.8 UGD gag3349 8 36.6 8 25.4 6 22.9 6 33.1 UGD env 8 9.0 8 2.9 8 13.3 8 12.5 8 14.3 UGDgag/gp140 5 1.2 18.9 5 2.3 17.6
TABLE-US-00007 TABLE 6 Modification of UGD env Gene in Recombinant MVA % non-staining passage env gag UGD9 5 0.5 5 0.4 5 0.0 5 0.5
TABLE-US-00008 TABLE 7 MVA/UGD4b- Analysis of Non-Staining gag Plaques # individual plaques with mutation gene base # sequence MVA/UGD MVA/KEA MVA/TZC p17 28 GGGGG 70 GGGGG n = 1 p24 408 GGGG 530 CCCC n = 1 564 GGGGGG n = 7 n = 16 n = 21 686 GGGG 1050 GGGGGG P7 1133 GGGG p1 1320 GGGG p6 1361 CCCC 1387 GGGG 1419 GGGG 1473 CCCC Protease 1494 GGGGG RT 1590 GGGGG 1599 GGGGG 2362 GGGG 2380 GGGG 2528 GGGGG 2596 GGGG 2893 GGGG 3001 CCCC
TABLE-US-00009 TABLE 8 Modification of UGD gagpol Gene in Recombinant MVA % non-staining Passage env gag UGD gag (c.a.) 6 0.9 6 0.0 6 0.5
TABLE-US-00010 TABLE 9 Construction of Stable Recombinant MVA Expressing UGD env and gagpol % non-staining Passage env gag UGD4d 11 0.0 0.7
[0203] While the present invention has been described in some detail for purposes of clarity and understanding, one skilled in the art will appreciate that various changes in form and detail can be made without departing from the true scope of the invention. All figures, tables, and appendices, as well as patents, applications, and publications, referred to above, are hereby incorporated by reference.
Sequence CWU 1
1
1014PRTHuman immunodeficiency virus type 1MISC_FEATURE(2)..(3)Xaa
at positions 2 and 3 = any naturally occurring amino acid 1Tyr Xaa
Xaa Leu125044DNAArtificial SequenceSynthetic (pLW-73 Plasmid DNA,
top strand) 2gaattccctg ggacatacgt atatttctat gatctgtctt atatgaagtc
tatacagcga 60atagattcag aatttctaca taattatata ttgtacgcta ataagtttaa
tctaacactc 120cccgaagatt tgtttataat ccctacaaat ttggatattc
tatggcgtac aaaggaatat 180atagactcgt tcgatattag tacagaaaca
tggaataaat tattatccaa ttattatatg 240aagatgatag agtatgctaa
actttatgta ctaagtccta ttctcgctga ggagttggat 300aattttgaga
ggacgggaga attaactagt attgtacaag aagccatttt atctctaaat
360ttacgaatta agattttaaa ttttaaacat aaagatgatg atacgtatat
acacttttgt 420aaaatattat tcggtgtcta taacggaaca aacgctacta
tatattatca tagacctcta 480acgggatata tgaatatgat ttcagatact
atatttgttc ctgtagataa taactaaggc 540gcgcctttca ttttgttttt
ttctatgcta taaatggtga gcaagggcga ggagctgttc 600accggggtgg
tgcccatcct ggtcgagctg gacggcgacg taaacggcca caagttcagc
660gtgtccggcg agggcgaggg cgatgccacc tacggcaagc tgaccctgaa
gttcatctgc 720accaccggca agctgcccgt gccctggccc accctcgtga
ccaccctgac ctacggcgtg 780cagtgcttca gccgctaccc cgaccacatg
aagcagcacg acttcttcaa gtccgccatg 840cccgaaggct acgtccagga
gcgcaccatc ttcttcaagg acgacggcaa ctacaagacc 900cgcgccgagg
tgaagttcga gggcgacacc ctggtgaacc gcatcgagct gaagggcatc
960gacttcaagg aggacggcaa catcctgggg cacaagctgg agtacaacta
caacagccac 1020aacgtctata tcatggccga caagcagaag aacggcatca
aggtgaactt caagatccgc 1080cacaacatcg aggacggcag cgtgcagctc
gccgaccact accagcagaa cacccccatc 1140ggcgacggcc ccgtgctgct
gcccgacaac cactacctga gcacccagtc cgccctgagc 1200aaagacccca
acgagaagcg cgatcacatg gtcctgctgg agttcgtgac cgccgccggg
1260atcactctcg gcatgcacga gctgtacaag taagagctcg aggacgggag
aattaactag 1320tattgtacaa gaagccattt tatctctaaa tttacgaatt
aagattttaa attttaaaca 1380taaagatgat gatacgtata tacacttttg
taaaatatta ttcggtgtct ataacggaac 1440aaacgctact atatattatc
atagacctct aacgggatat atgaatatga tttcagatac 1500tatatttgtt
cctgtagata ataactaact cgaggccgct ggtacccaac ctaaaaattg
1560aaaataaata caaaggttct tgagggttgt gttaaattga aagcgagaaa
taatcataaa 1620taagcccggg gatcctctag agtcgacctg cagtcaaact
ctaatgacca catctttttt 1680tagagatgaa aaattttcca catctccttt
tgtagacacg actaaacatt ttgcagaaaa 1740aagtttatta gtgtttagat
aatcgtatac ttcatcagtg tagatagtaa atgtgaacag 1800ataaaaggta
ttcttgctca atagattggt aaattccata gaatatatta atcctttctt
1860cttgagatcc cacatcattt caaccagaga cgttttatcc aatgatttac
ctcgtactat 1920accacataca aaactagatt ttgcagtgac gtcgtatctg
gtattcctac caaacaaaat 1980tttactttta gttcttttag aaaattctaa
ggtagaatct ctatttgcca atatgtcatc 2040tatggaatta ccactagcaa
aaaatgatag aaatatatat tgatacatcg cagctggttt 2100tgatctacta
tactttaaaa acgaatcaga ttccataatt gcctgtatat catcagctga
2160aaaactatgt tttacacgta ttccttcggc atttcttttt aatgatatat
cttgtttaga 2220caatgataaa gttatcatgt ccatgagaga cgcgtctccg
tatcgtataa atatttcatt 2280agatgttaga cgcttcatta ggggtatact
tctataaggt ttcttaatca gtccatcatt 2340ggttgcgtca agaacaagct
tgtctcccta tagtgagtcg tattagagct tggcgtaatc 2400atggtcatag
ctgtttcctg tgtgaaattg ttatccgctc acaattccac acaacatacg
2460agccggaagc ataaagtgta aagcctgggg tgcctaatga gtgagctaac
tcacattaat 2520tgcgttgcgc tcactgcccg ctttcgagtc gggaaacctg
tcgtgccagc tgcattaatg 2580aatcggccaa cgcgcgggga gaggcggttt
gcgtattggg cgctcttccg cttcctcgct 2640cactgactcg ctgcgctcgg
tcgttcggct gcggcgagcg gtatcagctc actcaaaggc 2700ggtaatacgg
ttatccacag aatcagggga taacgcagga aagaacatgt gagcaaaagg
2760ccagcaaaag gccaggaacc gtaaaaaggc cgcgttgctg gcgtttttcg
ataggctccg 2820cccccctgac gagcatcaca aaaatcgacg ctcaagtcag
aggtggcgaa acccgacagg 2880actataaaga taccaggcgt ttccccctgg
aagctccctc gtgcgctctc ctgttccgac 2940cctgccgctt accggatacc
tgtccgcctt tctcccttcg ggaagcgtgg cgctttctca 3000tagctcacgc
tgtaggtatc tcagttcggt gtaggtcgtt cgctccaagc tgggctgtgt
3060gcacgaaccc cccgttcagc ccgaccgctg cgccttatcc ggtaactatc
gtcttgagtc 3120caacccggta agacacgact tatcgccact ggcagcagcc
actggtaaca ggattagcag 3180agcgaggtat gtaggcggtg ctacagagtt
cttgaagtgg tggcctaact acggctacac 3240tagaaggaca gtatttggta
tctgcgctct gctgaagcca gttaccttcg gaaaaagagt 3300tggtagctct
tgatccggca aacaaaccac cgctggtagc ggtggttttt ttgtttgcaa
3360gcagcagatt acgcgcagaa aaaaaggatc tcaagaagat cctttgatct
tttctacggg 3420gtctgacgct cagtggaacg aaaactcacg ttaagggatt
ttggtcatga gattatcaaa 3480aaggatcttc acctagatcc ttttaaatta
aaaatgaagt tttaaatcaa tctaaagtat 3540atatgagtaa acttggtctg
acagttacca atgcttaatc agtgaggcac ctatctcagc 3600gatctgtcta
tttcgttcat ccatagttgc ctgactcccc gtcgtgtaga taactacgat
3660acgggagggc ttaccatctg gccccagtgc tgcaatgata ccgcgagacc
cacgctcacc 3720ggctccagat ttatcagcaa taaaccagcc agccggaagg
gccgagcgca gaagtggtcc 3780tgcaacttta tccgcctcca tccagtctat
taattgttgc cgggaagcta gagtaagtag 3840ttcgccagtt aatagtttgc
gcaacgttgt tggcattgct acaggcatcg tggtgtcacg 3900ctcgtcgttt
ggtatggctt cattcagctc cggttcccaa cgatcaaggc gagttacatg
3960atcccccatg ttgtgcaaaa aagcggttag ctccttcggt cctccgatcg
ttgtcagaag 4020taagttggcc gcagtgttat cactcatggt tatggcagca
ctgcataatt ctcttactgt 4080catgccatcc gtaagatgct tttctgtgac
tggtgagtac tcaaccaagt cattctgaga 4140atagtgtatg cggcgaccga
gttgctcttg cccggcgtca atacgggata ataccgcgcc 4200acatagcaga
actttaaaag tgctcatcat tggaaaacgt tcttcggggc gaaaactctc
4260aaggatctta ccgctgttga gatccagttc gatgtaaccc actcgtgcac
ccaactgatc 4320ttcagcatct tttactttca ccagcgtttc tgggtgagca
aaaacaggaa ggcaaaatgc 4380cgcaaaaaag ggaataaggg cgacacggaa
atgttgaata ctcatactct tcctttttca 4440atattattga agcatttatc
agggttattg tctcatgagc ggatacatat ttgaatgtat 4500ttagaaaaat
aaacaaatag gggttccgcg cacatttccc cgaaaagtgc cacctgacgt
4560ctaagaaacc attattatca tgacattaac ctataaaaat aggcgtatca
cgaggccctt 4620tcgtctcgcg cgtttcggtg atgacggtga aaacctctga
cacatgcagc tcccggagac 4680ggtcacagct tgtctgtaag cggatgccgg
gagcagacaa gcccgtcagg gcgcgtcagc 4740gggtgttggc gggtgtcggg
gctggcttaa ctatgcggca tcagagcaga ttgtactgag 4800agtgcaccat
atgcggtgtg aaataccgca cagatgcgta aggagaaaat accgcatcag
4860gcgccattcg ccattcaggc tgcgcaactg ttgggaaggg cgatcggtgc
gggcctcttc 4920gctattacgc cagctggcga aagggggatg tgctgcaagg
cgattaagtt gggtaacgcc 4980agggttttcc cagtcacgac gttgtaaaac
gacggccagt gaattggatt taggtgacac 5040tata 504435044DNAArtificial
SequenceSynthetic (pLW-73 Plasmid DNA, bottom strand, 5'-3')
3tatagtgtca cctaaatcca attcactggc cgtcgtttta caacgtcgtg actgggaaaa
60ccctggcgtt acccaactta atcgccttgc agcacatccc cctttcgcca gctggcgtaa
120tagcgaagag gcccgcaccg atcgcccttc ccaacagttg cgcagcctga
atggcgaatg 180gcgcctgatg cggtattttc tccttacgca tctgtgcggt
atttcacacc gcatatggtg 240cactctcagt acaatctgct ctgatgccgc
atagttaagc cagccccgac acccgccaac 300acccgctgac gcgccctgac
gggcttgtct gctcccggca tccgcttaca gacaagctgt 360gaccgtctcc
gggagctgca tgtgtcagag gttttcaccg tcatcaccga aacgcgcgag
420acgaaagggc ctcgtgatac gcctattttt ataggttaat gtcatgataa
taatggtttc 480ttagacgtca ggtggcactt ttcggggaaa tgtgcgcgga
acccctattt gtttattttt 540ctaaatacat tcaaatatgt atccgctcat
gagacaataa ccctgataaa tgcttcaata 600atattgaaaa aggaagagta
tgagtattca acatttccgt gtcgccctta ttcccttttt 660tgcggcattt
tgccttcctg tttttgctca cccagaaacg ctggtgaaag taaaagatgc
720tgaagatcag ttgggtgcac gagtgggtta catcgaactg gatctcaaca
gcggtaagat 780ccttgagagt tttcgccccg aagaacgttt tccaatgatg
agcactttta aagttctgct 840atgtggcgcg gtattatccc gtattgacgc
cgggcaagag caactcggtc gccgcataca 900ctattctcag aatgacttgg
ttgagtactc accagtcaca gaaaagcatc ttacggatgg 960catgacagta
agagaattat gcagtgctgc cataaccatg agtgataaca ctgcggccaa
1020cttacttctg acaacgatcg gaggaccgaa ggagctaacc gcttttttgc
acaacatggg 1080ggatcatgta actcgccttg atcgttggga accggagctg
aatgaagcca taccaaacga 1140cgagcgtgac accacgatgc ctgtagcaat
gccaacaacg ttgcgcaaac tattaactgg 1200cgaactactt actctagctt
cccggcaaca attaatagac tggatggagg cggataaagt 1260tgcaggacca
cttctgcgct cggcccttcc ggctggctgg tttattgctg ataaatctgg
1320agccggtgag cgtgggtctc gcggtatcat tgcagcactg gggccagatg
gtaagccctc 1380ccgtatcgta gttatctaca cgacggggag tcaggcaact
atggatgaac gaaatagaca 1440gatcgctgag ataggtgcct cactgattaa
gcattggtaa ctgtcagacc aagtttactc 1500atatatactt tagattgatt
taaaacttca tttttaattt aaaaggatct aggtgaagat 1560cctttttgat
aatctcatga ccaaaatccc ttaacgtgag ttttcgttcc actgagcgtc
1620agaccccgta gaaaagatca aaggatcttc ttgagatcct ttttttctgc
gcgtaatctg 1680ctgcttgcaa acaaaaaaac caccgctacc agcggtggtt
tgtttgccgg atcaagagct 1740accaactctt tttccgaagg taactggctt
cagcagagcg cagataccaa atactgtcct 1800tctagtgtag ccgtagttag
gccaccactt caagaactct gtagcaccgc ctacatacct 1860cgctctgcta
atcctgttac cagtggctgc tgccagtggc gataagtcgt gtcttaccgg
1920gttggactca agacgatagt taccggataa ggcgcagcgg tcgggctgaa
cggggggttc 1980gtgcacacag cccagcttgg agcgaacgac ctacaccgaa
ctgagatacc tacagcgtga 2040gctatgagaa agcgccacgc ttcccgaagg
gagaaaggcg gacaggtatc cggtaagcgg 2100cagggtcgga acaggagagc
gcacgaggga gcttccaggg ggaaacgcct ggtatcttta 2160tagtcctgtc
gggtttcgcc acctctgact tgagcgtcga tttttgtgat gctcgtcagg
2220ggggcggagc ctatcgaaaa acgccagcaa cgcggccttt ttacggttcc
tggccttttg 2280ctggcctttt gctcacatgt tctttcctgc gttatcccct
gattctgtgg ataaccgtat 2340taccgccttt gagtgagctg ataccgctcg
ccgcagccga acgaccgagc gcagcgagtc 2400agtgagcgag gaagcggaag
agcgcccaat acgcaaaccg cctctccccg cgcgttggcc 2460gattcattaa
tgcagctggc acgacaggtt tcccgactcg aaagcgggca gtgagcgcaa
2520cgcaattaat gtgagttagc tcactcatta ggcaccccag gctttacact
ttatgcttcc 2580ggctcgtatg ttgtgtggaa ttgtgagcgg ataacaattt
cacacaggaa acagctatga 2640ccatgattac gccaagctct aatacgactc
actataggga gacaagcttg ttcttgacgc 2700aaccaatgat ggactgatta
agaaacctta tagaagtata cccctaatga agcgtctaac 2760atctaatgaa
atatttatac gatacggaga cgcgtctctc atggacatga taactttatc
2820attgtctaaa caagatatat cattaaaaag aaatgccgaa ggaatacgtg
taaaacatag 2880tttttcagct gatgatatac aggcaattat ggaatctgat
tcgtttttaa agtatagtag 2940atcaaaacca gctgcgatgt atcaatatat
atttctatca ttttttgcta gtggtaattc 3000catagatgac atattggcaa
atagagattc taccttagaa ttttctaaaa gaactaaaag 3060taaaattttg
tttggtagga ataccagata cgacgtcact gcaaaatcta gttttgtatg
3120tggtatagta cgaggtaaat cattggataa aacgtctctg gttgaaatga
tgtgggatct 3180caagaagaaa ggattaatat attctatgga atttaccaat
ctattgagca agaatacctt 3240ttatctgttc acatttacta tctacactga
tgaagtatac gattatctaa acactaataa 3300acttttttct gcaaaatgtt
tagtcgtgtc tacaaaagga gatgtggaaa atttttcatc 3360tctaaaaaaa
gatgtggtca ttagagtttg actgcaggtc gactctagag gatccccggg
3420cttatttatg attatttctc gctttcaatt taacacaacc ctcaagaacc
tttgtattta 3480ttttcaattt ttaggttggg taccagcggc ctcgagttag
ttattatcta caggaacaaa 3540tatagtatct gaaatcatat tcatatatcc
cgttagaggt ctatgataat atatagtagc 3600gtttgttccg ttatagacac
cgaataatat tttacaaaag tgtatatacg tatcatcatc 3660tttatgttta
aaatttaaaa tcttaattcg taaatttaga gataaaatgg cttcttgtac
3720aatactagtt aattctcccg tcctcgagct cttacttgta cagctcgtgc
atgccgagag 3780tgatcccggc ggcggtcacg aactccagca ggaccatgtg
atcgcgcttc tcgttggggt 3840ctttgctcag ggcggactgg gtgctcaggt
agtggttgtc gggcagcagc acggggccgt 3900cgccgatggg ggtgttctgc
tggtagtggt cggcgagctg cacgctgccg tcctcgatgt 3960tgtggcggat
cttgaagttc accttgatgc cgttcttctg cttgtcggcc atgatataga
4020cgttgtggct gttgtagttg tactccagct tgtgccccag gatgttgccg
tcctccttga 4080agtcgatgcc cttcagctcg atgcggttca ccagggtgtc
gccctcgaac ttcacctcgg 4140cgcgggtctt gtagttgccg tcgtccttga
agaagatggt gcgctcctgg acgtagcctt 4200cgggcatggc ggacttgaag
aagtcgtgct gcttcatgtg gtcggggtag cggctgaagc 4260actgcacgcc
gtaggtcagg gtggtcacga gggtgggcca gggcacgggc agcttgccgg
4320tggtgcagat gaacttcagg gtcagcttgc cgtaggtggc atcgccctcg
ccctcgccgg 4380acacgctgaa cttgtggccg tttacgtcgc cgtccagctc
gaccaggatg ggcaccaccc 4440cggtgaacag ctcctcgccc ttgctcacca
tttatagcat agaaaaaaac aaaatgaaag 4500gcgcgcctta gttattatct
acaggaacaa atatagtatc tgaaatcata ttcatatatc 4560ccgttagagg
tctatgataa tatatagtag cgtttgttcc gttatagaca ccgaataata
4620ttttacaaaa gtgtatatac gtatcatcat ctttatgttt aaaatttaaa
atcttaattc 4680gtaaatttag agataaaatg gcttcttgta caatactagt
taattctccc gtcctctcaa 4740aattatccaa ctcctcagcg agaataggac
ttagtacata aagtttagca tactctatca 4800tcttcatata ataattggat
aataatttat tccatgtttc tgtactaata tcgaacgagt 4860ctatatattc
ctttgtacgc catagaatat ccaaatttgt agggattata aacaaatctt
4920cggggagtgt tagattaaac ttattagcgt acaatatata attatgtaga
aattctgaat 4980ctattcgctg tatagacttc atataagaca gatcatagaa
atatacgtat gtcccaggga 5040attc 504442214DNAHuman immunodeficiency
virus type 1 4atgagagtga gggagacagt gaggaattat cagcacttgt
ggagatgggg catcatgctc 60cttgggatgt taatgatatg tagtgctgca gaccagctgt
gggtcacagt gtattatggg 120gtacctgtgt ggaaagaagc aaccactact
ctattttgtg catcagatgc taaagcacat 180aaagcagagg cacataatat
ctgggctaca catgcctgtg taccaacaga ccccaatcca 240cgagaaataa
tactaggaaa tgtcacagaa aactttaaca tgtggaagaa taacatggta
300gagcagatgc atgaggatat aatcagttta tgggatcaaa gtctaaaacc
atgtgtaaaa 360ttaaccccac tctgtgttac tttaaactgc actacatatt
ggaatggaac tttacagggg 420aatgaaacta aagggaagaa tagaagtgac
ataatgacat gctctttcaa tataaccaca 480gaaataagag gtagaaagaa
gcaagaaact gcacttttct ataaacttga tgtggtacca 540ctagaggata
aggatagtaa taagactacc aactatagca gctatagatt aataaattgc
600aatacctcag tcgtgacaca ggcgtgtcca aaagtaacct ttgagccaat
tcccatacat 660tattgtgccc cagctggatt tgcgattctg aaatgtaata
ataagacgtt caatggaacg 720ggtccatgca aaaatgtcag cacagtacag
tgtacacatg gaattaggcc agtagtgtca 780actcaactgt tgttgaatgg
cagtctagca gaagaagaga taataattag atctgaaaat 840atcacaaata
atgcaaaaac cataatagta cagcttaatg agtctgtaac aattgattgc
900ataaggccca acaacaatac aagaaaaagt atacgcatag gaccagggca
agcactctat 960acaacagaca taatagggaa tataagacaa gcacattgta
atgttagtaa agtaaaatgg 1020ggaagaatgt taaaaagggt agctgaaaaa
ttaaaagacc ttcttaacca gacaaagaac 1080ataacttttg aaccatcctc
aggaggggac ccagaaatta caacacacag ctttaattgt 1140ggaggggaat
tcttctactg caatacatca ggactattta atgggagtct gcttaatgag
1200cagtttaatg agacatcaaa tgatactctc acactccaat gcagaataaa
acaaattata 1260aacatgtggc aaggagtagg aaaagcaatg tatgcccctc
ccattgcagg accaatcagc 1320tgttcatcaa atattacagg actattgttg
acaagagatg gtggtaatac tggtaatgat 1380tcagagatct tcagacctgg
agggggagat atgagagaca attggagaag tgaattatac 1440aaatataaag
tagtaagaat tgaaccaatg ggtctagcac ccaccagggc aaaaagaaga
1500gtggtggaaa gagaaaaaag agcaatagga ctgggagcta tgttccttgg
gttcttggga 1560gcggcaggaa gcacgatggg cgcagcgtca ctgacgctga
cggtacaggc cagacagtta 1620ttgtctggta tagtgcaaca gcaaaacaat
ttgctgagag ctatagaggc gcaacagcat 1680ctgttgcaac tcacagtctg
gggcattaaa cagctccagg caagagtcct ggctatggaa 1740agctacctaa
aggatcaaca gctcctagga atttggggtt gctctggaaa acacatttgc
1800accactactg tgccctggaa ctctacctgg agtaatagat ctgtagagga
gatttggaat 1860aatatgacct ggatgcagtg ggaaagagaa attgagaatt
acacaggttt aatatacacc 1920ttaattgaag aatcgcaaac ccagcaagaa
aagaatgaac aagaactatt gcaattggat 1980aaatgggcaa gtttgtggaa
ttggtttagt ataacaaaat ggctgtggta tataaaaata 2040ttcataatga
tagtaggagg cttaataggt ttaagaatag tttttgctgt gctttcttta
2100gtaaatagag ttaggcaggg atattcacct ctgtcttttc agaccctcct
cccagccccg 2160aggggacccg acaggcccga aggaatagaa gaagaaggtg
gagagcaagg ctaa 221453068DNAHuman immunodeficiency virus type 1
5atgggtgcga gagcgtcagt attaagcgga ggaaaattag atgaatggga aaaaattcgg
60ttacggccag gaggaaacaa aaaatataga ttaaaacatt tagtatgggc aagcagggag
120ctagaacgat ttgcacttaa tcctggtctt ttagaaacat cagaaggctg
tagacaaata 180atagaacagc tacaaccatc tattcagaca ggatcagagg
aacttaaatc attacataat 240acagtagtaa ccctctattg tgtacatgaa
aggataaagg tagcagatac caaggaagct 300ttagataaga taaaggaaga
acaaaccaaa agtaagaaaa aagcacagca agcaacagct 360gacagcagcc
aggtcagcca aaattatcct atagtacaaa acctacaggg acaaatggta
420caccagtcct tatcacctag gactttgaat gcatgggtaa aagtaataga
agagaaggct 480ttcagcccag aagtaatacc catgttttca gcattatcag
aaggagccac accaacagat 540ttaaacacca tgctaaacac agtaggagga
catcaagcag ccatgcaaat gttaaaagag 600actatcaatg aggaagctgc
agaatgggat aggctacatc cagtgcctgc agggcctgtt 660gcaccaggcc
aaatgagaga accaagagga agtgatatag caggaactac cagtaccctt
720caggaacaaa taggatggat gacaagcaat ccacctatcc cagtaggaga
aatctataaa 780agatggataa tcctaggatt aaataaaata gtaagaatgt
atagccctgt cagcattttg 840gacataagac aaggaccaaa ggaacccttt
agagactatg tagatcggtt ctataaaact 900ctacgagccg agcaagcttc
acaggatgta aaaaattgga tgactgaaac cttgttagtc 960caaaatgcga
atccagattg taaaactatc ttaaaagcat tgggaccagc ggctacatta
1020gaagaaatga tgacagcatg tcagggagtg gggggaccca gtcataaagc
aagagttttg 1080gctgaggcaa tgagccaagc atcaaacaca aatgctgtta
taatgatgca gaggggcaat 1140ttcaagggca agaaaatcat taagtgtttc
aactgtggca aagaaggaca cctagcaaaa 1200aattgtaggg ctcctaggaa
aagaggctgt tggaaatgtg gaaaggaagg gcaccaaatg 1260aaagattgta
atgaaagaca ggctaatttt ttagggagaa tttggccttc ccacaagggg
1320aggccaggga atttccttca gagcagacca gagccaacag ccccaccagc
agagagcttc 1380gggtttgggg aagagataac accctcccag aaacaggagg
ggaaagagga gctgtatcct 1440tcagcctccc tcaaatcact ctttggcaac
gacccctagt cacaataaaa atagggggac 1500agctaaagga agctctatta
gatacaggag cagatgatac agtagtagaa gaaatgaatt 1560tgccaggaaa
atggaaacca aaaatgatag ggggaattgg gggctttatc aaagtaagac
1620agtatgatca aatactcgta gaaatctatg gatataaggc tacaggtaca
gtattagtag 1680gacctacacc tgtcaacata attggaagaa atttgttgac
tcagattggt tgcactttaa 1740attttccaat tagtcctatt gaaactgtac
cagtaaaatt aaagtcaggg atggatggtc 1800caagagttaa acaatggcca
ttgacagaag agaaaataaa agcactaata gaaatttgta 1860cagaaatgga
aaaggaagga aaactttcaa gaattggacc tgaaaatcca tacaatactc
1920caatatttgc cataaagaaa aaagacagta ctaagtggag aaaattagta
gatttcagag 1980aacttaataa gagaactcaa gatttctggg aagttcaact
aggaatacca catcctgcag 2040ggctaaaaaa gaaaaaatca gtaacagtac
tggaggtggg tgatgcatat ttttcagttc 2100ccttatatga agactttaga
aaatacactg cattcaccat acctagtata aacaatgaga 2160caccaggaat
tagatatcag tacaatgtgc ttccacaagg atggaaagga tcaccggcaa
2220tattccaaag tagcatgaca aaaattttag aaccttttag aaaacaaaat
ccagaagtgg 2280ttatctacca atacatgcac
gatttgtatg taggatctga cttagaaata gggcagcata 2340gaataaaaat
agaggaatta aggggacacc tattgaagtg gggatttacc acaccagaca
2400aaaatcatca gaaggaacct ccatttcttt ggatgggtta tgaactccat
cctgataaat 2460ggacagtaca gcctataaaa ctgccagaaa aagaaagctg
gactgtcaat gatctgcaga 2520agttagtggg gaaattaaat tgggcaagtc
aaatttattc aggaattaaa gtaagacaat 2580tatgcaaatg ccttagggga
accaaagcac tgacagaagt agtaccactg acagaagaag 2640cagaattaga
actggcagaa aacagggaac ttctaaaaga aacagtacat ggagtgtatt
2700atgacccatc aaaagactta atagcagaaa tacagaaaca agggcaagac
caatggacat 2760atcaaattta tcaagaacaa tataaaaatt tgaaaacagg
aaagtatgca aagaggagga 2820gtacccacac taatgatgta aaacaattaa
cagaggcagt gcaaaaaata gcccaagaat 2880gtatagtgat atggggaaag
actcctaaat tcagactacc catacaaaag gaaacatggg 2940aaacatggtg
gacagagtat tggcaggcca cctggattcc tgagtgggag tttgtcaata
3000cccctccctt ggttaaatta tggtaccagt tagagaagga acccatagta
ggagcagaaa 3060ccttctaa 306869PRTHuman immunodeficiency virus type
1 6Ala Met Gln Met Leu Lys Glu Thr Ile1 5715PRTHuman
immunodeficiency virus type 1 7Asp Thr Glu Val His Asn Val Trp Ala
Thr His Ala Cys Val Pro1 5 10 15815PRTHuman immunodeficiency virus
type 1 8Gln Gln Gln Ser Asn Leu Leu Arg Ala Ile Glu Ala Gln Gln
His1 5 10 15913PRTHuman immunodeficiency virus type 1 9Glu Leu Arg
Gln His Leu Leu Arg Trp Gly Leu Thr Thr1 5 101014PRTHuman
immunodeficiency virus type 1 10His Gly Val Tyr Tyr Asp Pro Ser Lys
Asp Leu Ile Ala Glu1 5 10
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
D00012
D00013

D00014

D00015

D00016

D00017

D00018

D00019

D00020

D00021
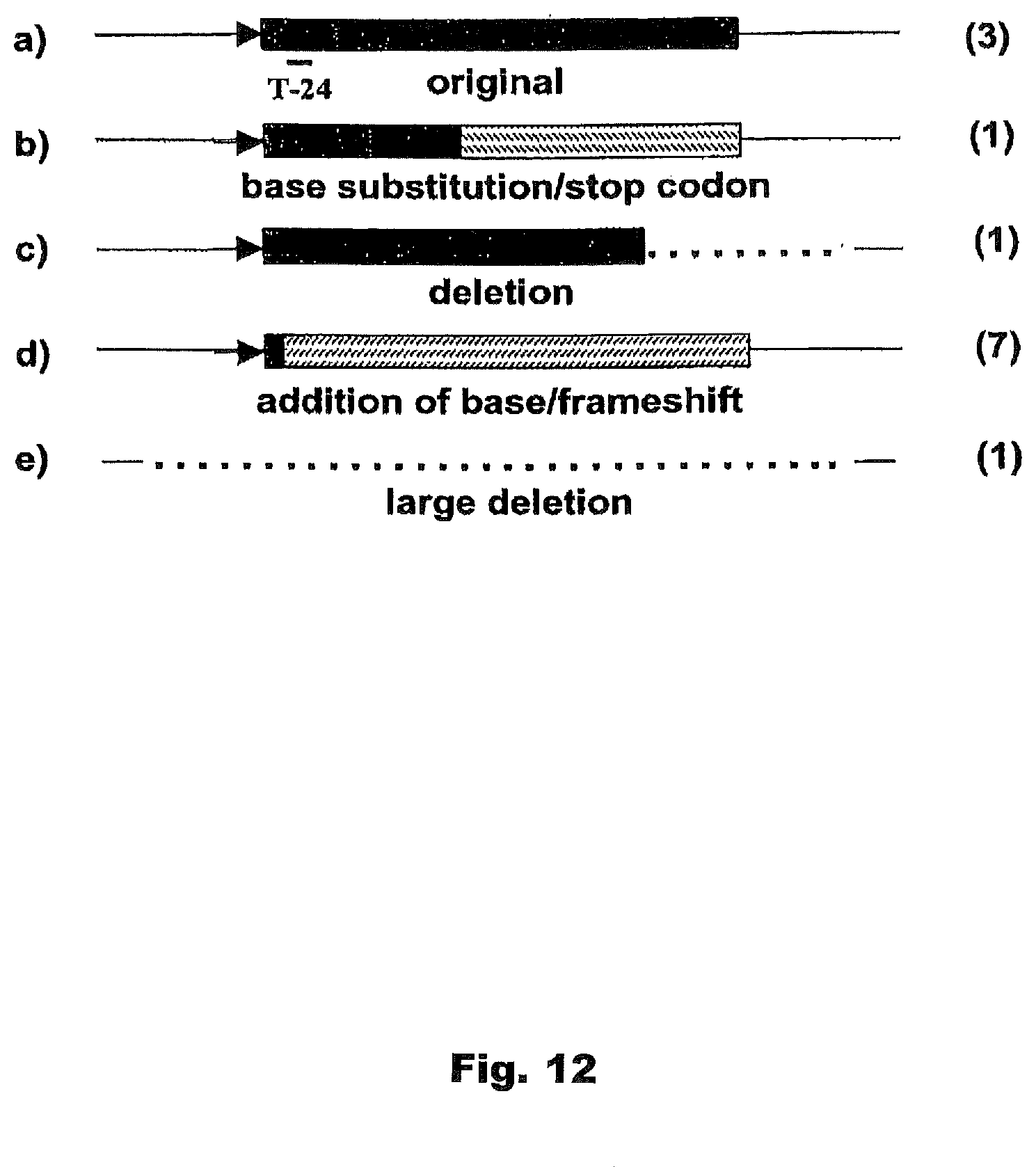
D00022

D00023

D00024

D00025

D00026

D00027

D00028

D00029

D00030

D00031

D00032

D00033

D00034

S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.