Pharmaceutical Composition, Methods For Treating And Uses Thereof
EICKELMANN; Peter ; et al.
U.S. patent application number 16/446086 was filed with the patent office on 2020-03-05 for pharmaceutical composition, methods for treating and uses thereof. The applicant listed for this patent is Boehringer Ingelheim International GmbH. Invention is credited to Uli Christian BROEDL, Peter EICKELMANN, Rolf GREMPLER, Michael MARK, Leo John SEMAN, Leo THOMAS.
| Application Number | 20200069713 16/446086 |
| Document ID | / |
| Family ID | 42008536 |
| Filed Date | 2020-03-05 |










View All Diagrams
| United States Patent Application | 20200069713 |
| Kind Code | A1 |
| EICKELMANN; Peter ; et al. | March 5, 2020 |
PHARMACEUTICAL COMPOSITION, METHODS FOR TREATING AND USES THEREOF
Abstract
The invention relates to the treatment or prevention of one or more conditions selected from type 1 diabetes mellitus, type 2 diabetes mellitus, impaired glucose tolerance and hyperglycemia using a SGLT-2 inhibitor. In addition the present invention relates to methods for preventing or treating of metabolic disorders and related conditions.
| Inventors: | EICKELMANN; Peter; (Mittelbiberach, DE) ; MARK; Michael; (Biberach an der Riss, DE) ; SEMAN; Leo John; (Cheshire, CT) ; THOMAS; Leo; (Biberach an der Riss, DE) ; BROEDL; Uli Christian; (Oakville, CA) ; GREMPLER; Rolf; (Mittelbiberach, DE) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 42008536 | ||||||||||
| Appl. No.: | 16/446086 | ||||||||||
| Filed: | June 19, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 15286017 | Oct 5, 2016 | |||
| 16446086 | ||||
| 15046653 | Feb 18, 2016 | |||
| 15286017 | ||||
| 14046109 | Oct 4, 2013 | |||
| 15046653 | ||||
| 13439324 | Apr 4, 2012 | |||
| 14046109 | ||||
| 12704062 | Feb 11, 2010 | |||
| 13439324 | ||||
| 61152318 | Feb 13, 2009 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61K 9/2018 20130101; A61P 1/18 20180101; A61P 43/00 20180101; A61K 31/7034 20130101; A61K 31/7048 20130101; A61P 3/00 20180101; A61P 37/06 20180101; A61P 19/06 20180101; A61P 9/00 20180101; A61P 9/06 20180101; A61P 9/08 20180101; A61P 3/10 20180101; A61K 9/2027 20130101; A61K 9/4866 20130101; A61K 47/26 20130101; A61P 3/08 20180101; A61P 13/12 20180101; A61P 9/10 20180101; A61K 9/0019 20130101; A61P 11/00 20180101; A61P 9/04 20180101; A61P 25/00 20180101; A61P 3/04 20180101; A61P 27/12 20180101; A61P 27/02 20180101; A61P 5/50 20180101; A61P 3/06 20180101 |
| International Class: | A61K 31/7034 20060101 A61K031/7034; A61K 31/7048 20060101 A61K031/7048 |
Claims
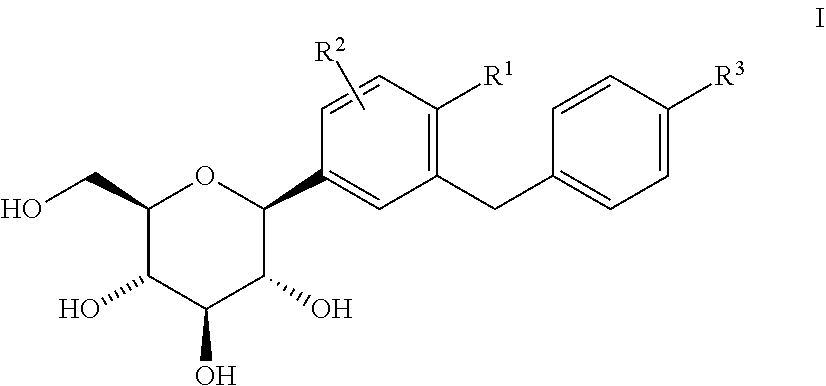
1. Method for preventing, slowing the progression of, delaying or treating a metabolic disorder selected from the group consisting of type 1 diabetes mellitus, type 2 diabetes mellitus, impaired glucose tolerance, impaired fasting blood glucose, hyperglycemia, postprandial hyperglycemia, overweight, obesity, metabolic syndrome, gestational diabetes, new onset diabetes after transplantation (NODAT) and complications associated therewith, and post-transplant metabolic syndrome (PTMS) and complications associated therewith in a patient in need thereof characterized in that a pharmaceutical composition comprising an SGLT2 inhibitor is administered to the patient, wherein the SGLT2 inhibitor is a glucopyranosyl-substituted benzene derivative of the formula (I) ##STR00005## wherein R.sup.1 denotes Cl, methyl or cyano; R.sup.2 denotes H, methyl, methoxy or hydroxy and R.sup.3 denotes ethyl, cyclopropyl, ethynyl, ethoxy, (R)-tetrahydrofuran-3-yloxy or (S)-tetrahydrofuran-3-yloxy; or a prodrug thereof.
2. Method according to claim 1 wherein the patient: (1) is an individual diagnosed of one or more of the conditions selected from the group consisting of overweight, obesity, visceral obesity and abdominal obesity; or (2) is an individual who shows one, two or more of the following conditions: (a) a fasting blood glucose or serum glucose concentration greater than 100 mg/dL, in particular greater than 125 mg/dL; (b) a postprandial plasma glucose equal to or greater than 140 mg/dL; (c) an HbA1c value equal to or greater than 6.5%, in particular equal to or greater than 8.0%; or (3) is an individual wherein one, two, three or more of the following conditions are present: (a) obesity, visceral obesity and/or abdominal obesity, (b) triglyceride blood level .gtoreq.150 mg/dL, (c) HDL-cholesterol blood level <40 mg/dL in female patients and <50 mg/dL in male patients, (d) a systolic blood pressure .gtoreq.130 mm Hg and a diastolic blood pressure .gtoreq.85 mm Hg, (e) a fasting blood glucose level .gtoreq.100 mg/dL.
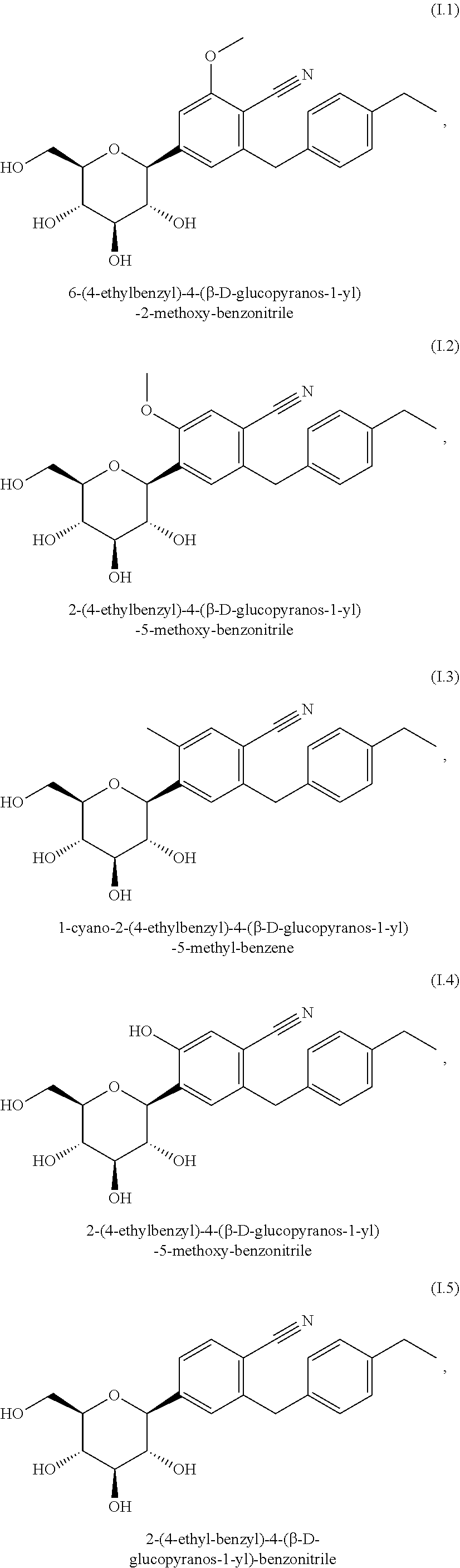
3. Method according to claim 1, wherein the SGLT2 inhibitor is selected from the group consisting of the group of compounds (I.1) to (I.11): (I.1) 6-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-2-methoxy-benzonit- rile, (I.2) 2-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-5-methoxy-benzonitrile, (I.3) 1-cyano-2-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-5-methyl-b- enzene, (I.4) 2-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-5-hydroxy-benzonitrile, (I.5) 2-(4-ethyl-benzyl)-4-(.beta.-D-glucopyranos-1-yl)-benzonitrile, (I.6) 2-(4-cyclopropyl-benzyl)-4-(.beta.-D-glucopyranos-1-yl)-benzonitril- e, (I.7) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-(4-ethynyl-benzyl)-benz- ene, (I.8) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-[4-((R)-tetrahydrofur- an-3-yloxy)-benzyl]-benzene, (I.9) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-[4-((S)-tetrahydrofuran-3-yloxy- )-benzyl]-benzene, (I.10) 1-methyl-2-[4-((R)-tetrahydrofuran-3-yloxy)-benzyl]-4-(.beta.-D-glucopyra- nos-1-yl)-benzene, and (I.11) 1-methyl-2-[4-((S)-tetrahydrofuran-3-yloxy)-benzyl]-4-(.beta.-D-glucopyra- nos-1-yl)-benzene.
4. Method according to claim 1 wherein the pharmaceutical composition additionally comprises one or more pharmaceutically acceptable carriers.
5. Method for improving glycemic control and/or for reducing of fasting plasma glucose, of postprandial plasma glucose and/or of glycosylated hemoglobin HbA1c in a patient in need thereof characterized in that a pharmaceutical composition comprising an SGLT2 is administered to the patient, wherein the SGLT2 inhibitor is a glucopyranosyl-substituted benzene derivative of the formula (I) ##STR00006## wherein R.sup.1 denotes Cl, methyl or cyano; R.sup.2 denotes H, methyl, methoxy or hydroxy and R.sup.3 denotes ethyl, cyclopropyl, ethynyl, ethoxy, (R)-tetrahydrofuran-3-yloxy or (S)-tetrahydrofuran-3-yloxy; or a prodrug thereof.
6. Method according to claim 5 wherein the patient: (1) is an individual diagnosed of one or more of the conditions selected from the group consisting of overweight, obesity, visceral obesity and abdominal obesity; or (2) is an individual who shows one, two or more of the following conditions: (a) a fasting blood glucose or serum glucose concentration greater than 100 mg/dL, in particular greater than 125 mg/dL; (b) a postprandial plasma glucose equal to or greater than 140 mg/dL; (c) an HbA1c value equal to or greater than 6.5%, in particular equal to or greater than 8.0%; or (3) is an individual wherein one, two, three or more of the following conditions are present: (a) obesity, visceral obesity and/or abdominal obesity, (b) triglyceride blood level .gtoreq.150 mg/dL, (c) HDL-cholesterol blood level <40 mg/dL in female patients and <50 mg/dL in male patients, (d) a systolic blood pressure .gtoreq.130 mm Hg and a diastolic blood pressure .gtoreq.85 mm Hg, (e) a fasting blood glucose level .gtoreq.100 mg/dL.
7. Method according to claim 5, wherein the SGLT2 inhibitor is selected from the group consisting of the group of compounds (I.1) to (I.11): (I.1) 6-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-2-methoxy-benzonit- rile, (I.2) 2-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-5-methoxy-benzonitrile, (I.3) 1-cyano-2-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-5-methyl-b- enzene, (I.4) 2-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-5-hydroxy-benzonitrile, (1.5) 2-(4-ethyl-benzyl)-4-(.beta.-D-glucopyranos-1-yl)-benzonitrile, (I.6) 2-(4-cyclopropyl-benzyl)-4-(.beta.-D-glucopyranos-1-yl)-benzonitril- e, (I.7) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-(4-ethynyl-benzyl)-benz- ene, (I.8) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-[4-((R)-tetrahydrofur- an-3-yloxy)-benzyl]-benzene, (I.9) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-[4-((S)-tetrahydrofuran-3-yloxy- )-benzyl]-benzene, (I.10) 1-methyl-2-[4-((R)-tetrahydrofuran-3-yloxy)-benzyl]-4-(.beta.-D-glucopyra- nos-1-yl)-benzene, and (I.11) 1-methyl-2-[4-((S)-tetrahydrofuran-3-yloxy)-benzyl]-4-(.beta.-D-glucopyra- nos-1-yl)-benzene.
8. Method according to claim 5 wherein the pharmaceutical composition additionally comprises one or more pharmaceutically acceptable carriers.
9. Method for preventing, slowing, delaying or reversing progression from impaired glucose tolerance, impaired fasting blood glucose, insulin resistance and/or from metabolic syndrome to type 2 diabetes mellitus in a patient in need thereof characterized in that a pharmaceutical composition comprising an SGLT2 inhibitor is administered to the patient, wherein the SGLT2 inhibitor is a glucopyranosyl-substituted benzene derivative of the formula (I) ##STR00007## wherein R.sup.1 denotes Cl, methyl or cyano; R.sup.2 denotes H, methyl, methoxy or hydroxy and R.sup.3 denotes ethyl, cyclopropyl, ethynyl, ethoxy, (R)-tetrahydrofuran-3-yloxy or (S)-tetrahydrofuran-3-yloxy; or a prodrug thereof.
10. Method according to claim 9 wherein the patient: (1) is an individual diagnosed of one or more of the conditions selected from the group consisting of overweight, obesity, visceral obesity and abdominal obesity; or (2) is an individual who shows one, two or more of the following conditions: (a) a fasting blood glucose or serum glucose concentration greater than 100 mg/dL, in particular greater than 125 mg/dL; (b) a postprandial plasma glucose equal to or greater than 140 mg/dL; (c) an HbA1c value equal to or greater than 6.5%, in particular equal to or greater than 8.0%; or (3) is an individual wherein one, two, three or more of the following conditions are present: (a) obesity, visceral obesity and/or abdominal obesity, (b) triglyceride blood level .gtoreq.150 mg/dL, (c) HDL-cholesterol blood level <40 mg/dL in female patients and <50 mg/dL in male patients, (d) a systolic blood pressure .gtoreq.130 mm Hg and a diastolic blood pressure .gtoreq.85 mm Hg, (e) a fasting blood glucose level .gtoreq.100 mg/dL.
11. Method according to claim 9, wherein the SGLT2 inhibitor is selected from the group consisting of the group of compounds (I.1) to (I.11): (I.1) 6-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-2-methoxy-benzonit- rile, (I.2) 2-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-5-methoxy-benzonitrile, (I.3) 1-cyano-2-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-5 -methyl-benzene, (I.4) 2-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-5-hydroxy-benzonitrile, (I.5) 2-(4-ethyl-benzyl)-4-(.beta.-D-glucopyranos-1-yl)-benzonitrile, (I.6) 2-(4-cyclopropyl-benzyl)-4-(.beta.-D-glucopyranos-1-yl)-benzonitril- e, (I.7) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-(4-ethynyl-benzyl)-benz- ene, (I.8) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-[4-((R)-tetrahydrofur- an-3-yloxy)-benzyl]-benzene, (I.9) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-[4-((S)-tetrahydrofuran-3-yloxy- )-benzyl]-benzene, (I.10) 1-methyl-2-[4-((R)-tetrahydrofuran-3-yloxy)-benzyl]-4-(.beta.-D-glucopyra- nos-1-yl)-benzene, and (I.11) 1-methyl-2-[4-((S)-tetrahydrofuran-3-yloxy)-benzyl]-4-(.beta.-D-glucopyra- nos-1-yl)-benzene.
12. Method according to claim 9 wherein the pharmaceutical composition additionally comprises one or more pharmaceutically acceptable carriers.
13. Method for preventing, slowing the progression of, delaying or treating of a condition or disorder selected from the group consisting of complications of diabetes mellitus such as cataracts and micro- and macrovascular diseases, such as nephropathy, retinopathy, neuropathy, tissue ischaemia, diabetic foot, arteriosclerosis, myocardial infarction, accute coronary syndrome, unstable angina pectoris, stable angina pectoris, stroke, peripheral arterial occlusive disease, cardiomyopathy, heart failure, heart rhythm disorders and vascular restenosis, in a patient in need thereof characterized in that a pharmaceutical composition comprising an SGLT2 inhibitor is administered to the patient, wherein the SGLT2 inhibitor is a glucopyranosyl-substituted benzene derivative of the formula (I) ##STR00008## wherein R.sup.1 denotes Cl, methyl or cyano; R.sup.2 denotes H, methyl, methoxy or hydroxy and R.sup.3 denotes ethyl, cyclopropyl, ethynyl, ethoxy, (R)-tetrahydrofuran-3-yloxy or (S)-tetrahydrofuran-3-yloxy; or a prodrug thereof.
14. Method according to claim 13 wherein the patient: (1) is an individual diagnosed of one or more of the conditions selected from the group consisting of overweight, obesity, visceral obesity and abdominal obesity; or (2) is an individual who shows one, two or more of the following conditions: (a) a fasting blood glucose or serum glucose concentration greater than 100 mg/dL, in particular greater than 125 mg/dL; (b) a postprandial plasma glucose equal to or greater than 140 mg/dL; (c) an HbA1c value equal to or greater than 6.5%, in particular equal to or greater than 8.0%; or (3) is an individual wherein one, two, three or more of the following conditions are present: (a) obesity, visceral obesity and/or abdominal obesity, (b) triglyceride blood level .gtoreq.150 mg/dL, (c) HDL-cholesterol blood level <40 mg/dL in female patients and <50 mg/dL in male patients, (d) a systolic blood pressure .gtoreq.130 mm Hg and a diastolic blood pressure .gtoreq.85 mm Hg, (e) a fasting blood glucose level .gtoreq.100 mg/dL.
15. Method according to claim 13, wherein the SGLT2 inhibitor is selected from the group consisting of the group of compounds (I.1) to (I.11): (I.1) 6-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-2-methoxy-benzonit- rile, (I.2) 2-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-5-methoxy-benzonitrile, (I.3) 1-cyano-2-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-5-methyl-b- enzene, (I.4) 2-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-5-hydroxy-benzonitrile, (I.5) 2-(4-ethyl-benzyl)-4-(.beta.-D-glucopyranos-1-yl)-benzonitrile, (I.6) 2-(4-cyclopropyl-benzyl)-4-(.beta.-D-glucopyranos-1-yl)-benzonitril- e, (I.7) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-(4-ethynyl-benzyl)-benz- ene, (I.8) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-[4-((R)-tetrahydrofur- an-3-yloxy)-benzyl]-benzene, (I.9) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-[4-((S)-tetrahydrofuran-3-yloxy- )-benzyl]-benzene, (I.10) 1-methyl-2-[4-((R)-tetrahydrofuran-3-yloxy)-benzyl]-4-(.beta.-D-glucopyra- nos-1-yl)-benzene, and (I.11) 1-methyl-2-[4-((S)-tetrahydrofuran-3-yloxy)-benzyl]-4-(.beta.-D-glucopyra- nos-1-yl)-benzene.
16. Method according to claim 13 wherein the pharmaceutical composition additionally comprises one or more pharmaceutically acceptable carriers.
17. Method for reducing body weight and/or body fat, or preventing an increase in body weight and/or body fat, or facilitating a reduction in body weight and/or body fat, in a patient in need thereof characterized in that a pharmaceutical composition comprising an SGLT2 inhibitor is administered to the patient, wherein the SGLT2 inhibitor is a glucopyranosyl-substituted benzene derivative of the formula (I) ##STR00009## wherein R.sup.1 denotes Cl, methyl or cyano; R.sup.2 denotes H, methyl, methoxy or hydroxy and R.sup.3 denotes ethyl, cyclopropyl, ethynyl, ethoxy, (R)-tetrahydrofuran-3-yloxy or (S)-tetrahydrofuran-3-yloxy; or a prodrug thereof.
18. Method according to claim 17 wherein the patient: (1) is an individual diagnosed of one or more of the conditions selected from the group consisting of overweight, obesity, visceral obesity and abdominal obesity; or (2) is an individual who shows one, two or more of the following conditions: (a) a fasting blood glucose or serum glucose concentration greater than 100 mg/dL, in particular greater than 125 mg/dL; (b) a postprandial plasma glucose equal to or greater than 140 mg/dL; (c) an HbA1c value equal to or greater than 6.5%, in particular equal to or greater than 8.0%; or (3) is an individual wherein one, two, three or more of the following conditions are present: (a) obesity, visceral obesity and/or abdominal obesity, (b) triglyceride blood level .gtoreq.150 mg/dL, (c) HDL-cholesterol blood level <40 mg/dL in female patients and <50 mg/dL in male patients, (d) a systolic blood pressure >130 mm Hg and a diastolic blood pressure .gtoreq.85 mm Hg, (e) a fasting blood glucose level .gtoreq.100 mg/dL.
19. Method according to claim 17, wherein the SGLT2 inhibitor is selected from the group consisting of the group of compounds (I.1) to (I.11): (I.1) 6-(4-ethylbenzyl)-4-(.beta.-D-glucopyranos-1-yl)-2-methoxy-benzonit- rile, (I.2) 2-(4-ethylbenzyl)-4-(.beta.D-glucopyranos-1-yl)-5-methoxy-benzonitrile, (I.3) 1-cyano-2-(4-ethylbenzyl)-4-(.beta.D-glucopyranos- 1-yl)-5 -methyl-benzene, (I.4) 2-(4-ethylbenzyl)-4-(.beta.D-glucopyranos-1-yl)-5-hydroxy-benzonitrile, (I.5) 2-(4-ethyl-benzyl)-4-(.beta.D-glucopyranos-1-yl)-benzonitrile, (I.6) 2-(4-cyclopropyl-benzyl)-4-(.beta.-D-glucopyranos-1-yl)-benzonitril- e, (I.7) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-(4-ethynyl-benzyl)-benz- ene, (I.8) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2- [4-((R)-tetrahydrofuran-3-yloxy)-benzyl]-benzene, (I.9) 1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2- [4-((S)-tetrahydrofuran-3-yloxy)-benzyl]-benzene, (I.10) 1-methyl-2-[4-((R)-tetrahydrofuran-3-yloxy)-benzyl]-4-(.beta.-D-glucopyra- nos-1-yl)-benzene, and (I.11) 1-methyl-2-[4-((S)-tetrahydrofuran-3-yloxy)-benzyl]-4-(.beta.-D-glucopyra- nos-1-yl)-benzene.
20. Method according to claim 17 wherein the pharmaceutical composition additionally comprises one or more pharmaceutically acceptable carriers.
21.-36. (canceled)
Description
[0001] This application is a Continuation of U.S. patent application Ser. No. 15/286,017 filed on Oct. 5, 2016, which is a Continuation of U.S. patent application Ser. No. 15/046,653 filed on Feb. 18, 2016, which is a Continuation of U.S. patent application Ser. No. 14/046,109 filed on Oct. 4, 2013, which is a Continuation of U.S. patent application Ser. No. 13/439,324 filed on Apr. 4, 2012, which is a Continuation of U.S. patent application Ser. No. 12/704,062 filed on Feb. 11, 2010, which claims benefit from U.S. Provisional Application No. 61/152,318, filed on Feb. 13, 2009, the content of which are incorporated herein in their entireties.
TECHNICAL FIELD OF THE INVENTION
[0002] The invention relates to a pharmaceutical composition comprising an SGLT2-inhibitor as described hereinafter which is suitable in the treatment or prevention of one or more conditions selected from type 1 diabetes mellitus, type 2 diabetes mellitus, impaired glucose tolerance, impaired fasting blood glucose and hyperglycemia inter alia.
[0003] Furthermore the invention relates to methods [0004] for preventing, slowing progression of, delaying, or treating a metabolic disorder; [0005] for improving glycemic control and/or for reducing of fasting plasma glucose, of postprandial plasma glucose and/or of glycosylated hemoglobin HbA1c; [0006] for preventing, slowing, delaying or reversing progression from impaired glucose tolerance, impaired fasting blood glucose, insulin resistance and/or from metabolic syndrome to type 2 diabetes mellitus; [0007] for preventing, slowing progression of, delaying or treating of a condition or disorder selected from the group consisting of complications of diabetes mellitus; [0008] for reducing body weight and/or body fat, or preventing an increase in body weight and/or body fat, or facilitating a reduction in body weight and/or body fat; [0009] for preventing or treating the degeneration of pancreatic beta cells and/or for improving and/or restoring the functionality of pancreatic beta cells and/or restoring the functionality of pancreatic insulin secretion; [0010] for preventing, slowing, delaying or treating diseases or conditions attributed to an abnormal accumulation of ectopic fat; [0011] maintaining and/or improving the insulin sensitivity and/or for treating or preventing hyperinsulinemia and/or insulin resistance; [0012] for preventing, slowing progression of, delaying, or treating new onset diabetes after transplantation (NODAT) and/or post-transplant metabolic syndrome (PTMS); [0013] for preventing, delaying, or reducing NODAT and/or PTMS associated complications including micro- and macrovascular diseases and events, graft rejection, infection, and death; [0014] for treating hyperuricemia and hyperuricemia associated conditions; [0015] for treating or preventing kidney stones; [0016] for treating hyponatremia; in patients in need thereof characterized in that a pharmaceutical composition comprising an SGLT2 inhibitor as defined hereinafter is administered.
[0017] In addition the present invention relates to the use of an SGLT2 inhibitor for the manufacture of a medicament for use in a method as described hereinbefore and hereinafter.
[0018] The invention also relates to a use of a pharmaceutical composition according to this invention for the manufacture of a medicament for use in a method as described hereinbefore and hereinafter.
BACKGROUND OF THE INVENTION
[0019] Type 2 diabetes is an increasingly prevalent disease that due to a high frequency of complications leads to a significant reduction of life expectancy. Because of diabetes-associated microvascular complications, type 2 diabetes is currently the most frequent cause of adult-onset loss of vision, renal failure, and amputations in the industrialized world. In addition, the presence of type 2 diabetes is associated with a two to five fold increase in cardiovascular disease risk.
[0020] After long duration of disease, most patients with type 2 diabetes will eventually fail on oral therapy and become insulin dependent with the necessity for daily injections and multiple daily glucose measurements.
[0021] The UKPDS (United Kingdom Prospective Diabetes Study) demonstrated that intensive treatment with metformin, sulfonylureas or insulin resulted in only a limited improvement of glycemic control (difference in HbA1c.about.0.9%). In addition, even in patients within the intensive treatment arm glycemic control deteriorated significantly over time and this was attributed to deterioration of .beta.-cell function. Importantly, intensive treatment was not associated with a significant reduction in macrovascular complications, i.e. cardiovascular events. Therefore many patients with type 2 diabetes remain inadequately treated, partly because of limitations in long term efficacy, tolerability and dosing inconvenience of existing antihyperglycemic therapies.
[0022] Oral antidiabetic drugs conventionally used in therapy (such as e.g. first- or second-line, and/or mono- or (initial or add-on) combination therapy) include, without being restricted thereto, metformin, sulphonylureas, thiazolidinediones, glinides and a-glucosidase inhibitors.
[0023] The high incidence of therapeutic failure is a major contributor to the high rate of long-term hyperglycemia-associated complications or chronic damages (including micro- and macrovascular complications such as e.g. diabetic nephrophathy, retinopathy or neuropathy, or cardiovascular complications) in patients with type 2 diabetes.
[0024] Therefore, there is an unmet medical need for methods, medicaments and pharmaceutical compositions with a good efficacy with regard to glycemic control, with regard to disease-modifying properties and with regard to reduction of cardiovascular morbidity and mortality while at the same time showing an improved safety profile.
[0025] SGLT2 inhibitors inhibitors represent a novel class of agents that are being developed for the treatment or improvement in glycemic control in patients with type 2 diabetes. Glucopyranosyl-substituted benzene derivative are described in the prior art as SGLT2 inhibitors, for example in WO 01/27128, WO 03/099836, WO 2005/092877, WO 2006/034489, WO 2006/064033, WO 2006/117359, WO 2006/117360, WO 2007/025943, WO 2007/028814, WO 2007/031548, WO 2007/093610, WO 2007/128749, WO 2008/049923, WO 2008/055870, WO 2008/055940. The glucopyranosyl-substituted benzene derivatives are proposed as inducers of urinary sugar excretion and as medicaments in the treatment of diabetes.
[0026] Renal filtration and reuptake of glucose contributes, among other mechanisms, to the steady state plasma glucose concentration and can therefore serve as an antidiabetic target. Reuptake of filtered glucose across epithelial cells of the kidney proceeds via sodium-dependent glucose cotransporters (SGLTs) located in the brush-border membranes in the tubuli along the sodium gradient. There are at least 3 SGLT isoforms that differ in their expression pattern as well as in their physico-chemical properties. SGLT2 is exclusively expressed in the kidney, whereas SGLT1 is expressed additionally in other tissues like intestine, colon, skeletal and cardiac muscle. SGLT3 has been found to be a glucose sensor in interstitial cells of the intestine without any transport function. Potentially, other related, but not yet characterized genes, may contribute further to renal glucose reuptake. Under normoglycemia, glucose is completely reabsorbed by SGLTs in the kidney, whereas the reuptake capacity of the kidney is saturated at glucose concentrations higher than 10 mM, resulting in glucosuria ("diabetes mellitus"). This threshold concentration can be decreased by SGLT2-inhibition. It has been shown in experiments with the SGLT inhibitor phlorizin that SGLT-inhibition will partially inhibit the reuptake of glucose from the glomerular filtrate into the blood leading to a decrease in blood glucose concentrations and to glucosuria.
Aim of the Present Invention
[0027] The aim of the present invention is to provide a pharmaceutical composition and method for preventing, slowing progression of, delaying or treating a metabolic disorder, in particular of type 2 diabetes mellitus.
[0028] A further aim of the present invention is to provide a pharmaceutical composition and method for improving glycemic control in a patient in need thereof, in particular in patients with type 2 diabetes mellitus.
[0029] Another aim of the present invention is to provide a pharmaceutical composition and method for improving glycemic control in a patient with insufficient glycemic control.
[0030] Another aim of the present invention is to provide a pharmaceutical composition and method for preventing, slowing or delaying progression from impaired glucose tolerance (IGT), impaired fasting blood glucose (IFG), insulin resistance and/or metabolic syndrome to type 2 diabetes mellitus.
[0031] Yet another aim of the present invention is to provide a pharmaceutical composition and method for preventing, slowing progression of, delaying or treating of a condition or disorder from the group consisting of complications of diabetes mellitus.
[0032] A further aim of the present invention is to provide a pharmaceutical composition and method for reducing the weight or preventing an increase of the weight in a patient in need thereof.
[0033] Another aim of the present invention is to provide a pharmaceutical composition with a high efficacy for the treatment of metabolic disorders, in particular of diabetes mellitus, impaired glucose tolerance (IGT), impaired fasting blood glucose (IFG), and/or hyperglycemia, which has good to very good pharmacological and/or pharmacokinetic and/or physicochemical properties.
[0034] Further aims of the present invention become apparent to the one skilled in the art by description hereinbefore and in the following and by the examples.
SUMMARY OF THE INVENTION
[0035] Within the scope of the present invention it has now surprisingly been found that a pharmaceutical composition comprising a SGLT2 inhibitor as defined hereinafter can advantageously be used for preventing, slowing progression of, delaying or treating a metabolic disorder, in particular for improving glycemic control in patients. This opens up new therapeutic possibilities in the treatment and prevention of type 2 diabetes mellitus, overweight, obesity, complications of diabetes mellitus and of neighboring disease states.
[0036] Therefore, in a first aspect the present invention provides a method for preventing, slowing the progression of, delaying or treating a metabolic disorder selected from the group consisting of type 1 diabetes mellitus, type 2 diabetes mellitus, impaired glucose tolerance (IGT), impaired fasting blood glucose (IFG), hyperglycemia, postprandial hyperglycemia, overweight, obesity, metabolic syndrome and gestational diabetes in a patient in need thereof characterized in that an SGLT2 inhibitor as defined hereinbefore and hereinafter is administered to the patient.
[0037] According to another aspect of the invention, there is provided a method for improving glycemic control and/or for reducing of fasting plasma glucose, of postprandial plasma glucose and/or of glycosylated hemoglobin HbA1c in a patient in need thereof characterized in that an SGLT2 inhibitor as defined hereinbefore and hereinafter is administered to the patient.
[0038] The pharmaceutical composition according to this invention may also have valuable disease-modifying properties with respect to diseases or conditions related to impaired glucose tolerance (IGT), impaired fasting blood glucose (IFG), insulin resistance and/or metabolic syndrome.
[0039] According to another aspect of the invention, there is provided a method for preventing, slowing, delaying or reversing progression from impaired glucose tolerance (IGT), impaired fasting blood glucose (IFG), insulin resistance and/or from metabolic syndrome to type 2 diabetes mellitus in a patient in need thereof characterized in that an SGLT2 inhibitor as defined hereinbefore and hereinafter is administered to the patient.
[0040] As by the use of a pharmaceutical composition according to this invention, an improvement of the glycemic control in patients in need thereof is obtainable, also those conditions and/or diseases related to or caused by an increased blood glucose level may be treated.
[0041] According to another aspect of the invention, there is provided a method for preventing, slowing the progression of, delaying or treating of a condition or disorder selected from the group consisting of complications of diabetes mellitus such as cataracts and micro- and macrovascular diseases, such as nephropathy, retinopathy, neuropathy, tissue ischaemia, diabetic foot, arteriosclerosis, myocardial infarction, accute coronary syndrome, unstable angina pectoris, stable angina pectoris, stroke, peripheral arterial occlusive disease, cardiomyopathy, heart failure, heart rhythm disorders and vascular restenosis, in a patient in need thereof characterized in that an SGLT2 inhibitor as defined hereinbefore and hereinafter is administered to the patient. In particular one or more aspects of diabetic nephropathy such as hyperperfusion, proteinuria and albuminuria may be treated, their progression slowed or their onset delayed or prevented. The term "tissue ischaemia" particularly comprises diabetic macroangiopathy, diabetic microangiopathy, impaired wound healing and diabetic ulcer. The terms "micro- and macrovascular diseases" and "micro- and macrovascular complications" are used interchangeably in this application.
[0042] By the administration of a pharmaceutical composition according to this invention and due to the activity of the SGLT2 inhibitor excessive blood glucose levels are not converted to insoluble storage forms, like fat, but excreted through the urine of the patient. In animal models it can be seen that loss of fat accounts for the majority of the observed weight loss whereas no significant changes in body water or protein content are observed. Therefore, no gain in weight or even a reduction in body weight is the result.
[0043] According to another aspect of the invention, there is provided a method for reducing body weight and/or body fat, or preventing an increase in body weight and/or body fat, or facilitating a reduction in body weight and/or body fat, in a patient in need thereof characterized in that an SGLT2 inhibitor as defined hereinbefore and hereinafter is administered to the patient.
[0044] The pharmacological effect of the SGLT2 inhibitor in the pharmaceutical composition according to this invention is independent of insulin. Therefore, an improvement of the glycemic control is possible without an additional strain on the pancreatic beta cells. By an administration of a pharmaceutical composition according to this invention a beta-cell degeneration and a decline of beta-cell functionality such as for example apoptosis or necrosis of pancreatic beta cells can be delayed or prevented. Furthermore, the functionality of pancreatic cells can be improved or restored, and the number and size of pancreatic beta cells increased. It may be shown that the differentiation status and hyperplasia of pancreatic beta-cells disturbed by hyperglycemia can be normalized by treatment with a pharmaceutical composition according to this invention.
[0045] According to another aspect of the invention, there is provided a method for preventing, slowing, delaying or treating the degeneration of pancreatic beta cells and/or the decline of the functionality of pancreatic beta cells and/or for improving and/or restoring the functionality of pancreatic beta cells and/or restoring the functionality of pancreatic insulin secretion in a patient in need thereof characterized in that an SGLT2 inhibitor as defined hereinbefore and hereinafter is administered to the patient.
[0046] By the administration of a pharmaceutical composition according to the present invention, an abnormal accumulation of ectopic fat, in particular of the liver, may be reduced or inhibited. Therefore, according to another aspect of the present invention, there is provided a method for preventing, slowing, delaying or treating diseases or conditions attributed to an abnormal accumulation of ectopic fat, in particular liver fat, in a patient in need thereof characterized in that an SGLT2 inhibitor as defined hereinbefore and hereinafter is administered to the patient. Diseases or conditions which are attributed to an abnormal accumulation of liver fat are particularly selected from the group consisting of general fatty liver, non-alcoholic fatty liver (NAFL), non-alcoholic steatohepatitis (NASH), hyperalimentation-induced fatty liver, diabetic fatty liver, alcoholic-induced fatty liver or toxic fatty liver.
[0047] As a result thereof, another aspect of the invention provides a method for maintaining and/or improving the insulin sensitivity and/or for treating or preventing hyperinsulinemia and/or insulin resistance in a patient in need thereof characterized in that an SGLT2 inhibitor as defined hereinbefore and hereinafter is administered to the patient.
[0048] According to another aspect of the invention, there is provided a method for preventing, slowing progression of, delaying, or treating new onset diabetes after transplantation (NODAT) and/or post-transplant metabolic syndrome (PTMS).
[0049] According to a further aspect of the invention, there is provided a method for preventing, delaying, or reducing NODAT and/or PTMS associated complications including micro- and macrovascular diseases and events, graft rejection, infection, and death.
[0050] The pharmaceutical composition according to the invention is capable of facilitating the lowering of serum total urate levels in the patient. Therefore according to another aspect of the invention, there is provided a method for treating hyperuricemia and hyperuricemia-associated conditions, such as for example gout, hypertension and renal failure, in a patient in need thereof. The patient may be a diabetic or non-diabetic patient.
[0051] The administration of a pharmaceutical composition increases the urine excretion of glucose. This increase in osmotic excretion and water release and the lowering of urate levels are beneficial as a treatment or prevention for kidney stones. Therefore in a further aspect of the invention, there is provided a method for treating or preventing kidney stones.
[0052] According to a further aspect of the invention, there is provided a method for treating hyponatremia, water retention and water intoxication. By the administration of the pharmaceutical composition according to this invention it may be possible to reverse the effects of hyponatremia, water retention and water intoxication by acting on the kidney to reverse water retention and electrolyte imbalances associated with these diseases and disorders.
[0053] According to another aspect of the invention there is provided the use of an SGLT2 inhibitor for the manufacture of a medicament for [0054] preventing, slowing the progression of, delaying or treating a metabolic disorder selected from the group consisting of type 1 diabetes mellitus, type 2 diabetes mellitus, impaired glucose tolerance (IGT), impaired fasting blood glucose (IFG), hyperglycemia, postprandial hyperglycemia, overweight, obesity, metabolic syndrome and gestational diabetes; or [0055] improving glycemic control and/or for reducing of fasting plasma glucose, of postprandial plasma glucose and/or of glycosylated hemoglobin HbA1c; or [0056] preventing, slowing, delaying or reversing progression from impaired glucose tolerance (IGT), impaired fasting blood glucose (IFG), insulin resistance and/or from metabolic syndrome to type 2 diabetes mellitus; or [0057] preventing, slowing the progression of, delaying or treating of a condition or disorder selected from the group consisting of complications of diabetes mellitus such as cataracts and micro- and macrovascular diseases, such as nephropathy, retinopathy, neuropathy, tissue ischaemia, diabetic foot, arteriosclerosis, myocardial infarction, accute coronary syndrome, unstable angina pectoris, stable angina pectoris, stroke, peripheral arterial occlusive disease, cardiomyopathy, heart failure, heart rhythm disorders and vascular restenosis,; or [0058] reducing body weight and/or body fat, or preventing an increase in body weight and/or body fat, or facilitating a reduction in body weight and/or body fat; or [0059] preventing, slowing, delaying or treating the degeneration of pancreatic beta cells and/or the decline of the functionality of pancreatic beta cells and/or for improving and/or restoring the functionality of pancreatic beta cells and/or restoring the functionality of pancreatic insulin secretion; or [0060] preventing, slowing, delaying or treating diseases or conditions attributed to an abnormal accumulation of ectopic fat; or [0061] maintaining and/or improving the insulin sensitivity and/or for treating or preventing hyperinsulinemia and/or insulin resistance; [0062] preventing, slowing progression of, delaying, or treating new onset diabetes after transplantation (NODAT) and/or post-transplant metabolic syndrome (PTMS); [0063] preventing, delaying, or reducing NODAT and/or PTMS associated complications including micro- and macrovascular diseases and events, graft rejection, infection, and death; [0064] treating hyperuricemia and hyperuricemia associated conditions; [0065] treating or prevention kidney stones; [0066] treating hyponatremia; in a patient in need thereof characterized in that the SGLT2 inhibitor is administered, as defined hereinbefore and hereinafter.
[0067] According to another aspect of the invention, there is provided the use of a pharmaceutical composition according to the present invention for the manufacture of a medicament for a therapeutic and preventive method as described hereinbefore and hereinafter.
[0068] Definitions
[0069] The term "active ingredient" of a pharmaceutical composition according to the present invention means the SGLT2 inhibitor according to the present invention. An "active ingredient is also sometimes referred to herein as an "active substance".
[0070] The term "body mass index" or "BMI" of a human patient is defined as the weight in kilograms divided by the square of the height in meters, such that BMI has units of kg/m.sup.2.
[0071] The term "overweight" is defined as the condition wherein the individual has a BMI greater than or 25 kg/m.sup.2 and less than 30 kg/m.sup.2. The terms "overweight" and "pre-obese" are used interchangeably.
[0072] The term "obesity" is defined as the condition wherein the individual has a BMI equal to or greater than 30 kg/m.sup.2. According to a WHO definition the term obesity may be categorized as follows: the term "class I obesity" is the condition wherein the BMI is equal to or greater than 30 kg/m.sup.2 but lower than 35 kg/m.sup.2; the term "class II obesity" is the condition wherein the BMI is equal to or greater than 35 kg/m.sup.2 but lower than 40 kg/m.sup.2; the term "class III obesity" is the condition wherein the BMI is equal to or greater than 40 kg/m.sup.2.
[0073] The term "visceral obesity" is defined as the condition wherein a waist-to-hip ratio of greater than or equal to 1.0 in men and 0.8 in women is measured. It defines the risk for insulin resistance and the development of pre-diabetes.
[0074] The term "abdominal obesity" is usually defined as the condition wherein the waist circumference is >40 inches or 102 cm in men, and is >35 inches or 94 cm in women. With regard to a Japanese ethnicity or Japanese patients abdominal obesity may be defined as waist circumference 85 cm in men and 90 cm in women (see e.g. investigating committee for the diagnosis of metabolic syndrome in Japan).
[0075] The term "euglycemia" is defined as the condition in which a subject has a fasting blood glucose concentration within the normal range, greater than 70 mg/dL (3.89 mmol/L) and less than 100 mg/dL (5.6 mmol/L). The word "fasting" has the usual meaning as a medical term.
[0076] The term "hyperglycemia" is defined as the condition in which a subject has a fasting blood glucose concentration above the normal range, greater than 100 mg/dL (5.6 mmol/L). The word "fasting" has the usual meaning as a medical term.
[0077] The term "hypoglycemia" is defined as the condition in which a subject has a blood glucose concentration below the normal range, in particular below 70 mg/dL (3.89 mmol/L).
[0078] The term "postprandial hyperglycemia" is defined as the condition in which a subject has a 2 hour postprandial blood glucose or serum glucose concentration greater than 200 mg/dL (11.11 mmol/L).
[0079] The term "impaired fasting blood glucose" or "IFG" is defined as the condition in which a subject has a fasting blood glucose concentration or fasting serum glucose concentration in a range from 100 to 125 mg/dl (i.e. from 5.6 to 6.9 mmol/l), in particular greater than 110 mg/dL and less than 126 mg/dl (7.00 mmol/L). A subject with "normal fasting glucose" has a fasting glucose concentration smaller than 100 mg/dl, i.e. smaller than 5.6 mmol/l.
[0080] The term "impaired glucose tolerance" or "IGT" is defined as the condition in which a subject has a 2 hour postprandial blood glucose or serum glucose concentration greater than 140 mg/dl (7.78 mmol/L) and less than 200 mg/dL (11.11 mmol/L). The abnormal glucose tolerance, i.e. the 2 hour postprandial blood glucose or serum glucose concentration can be measured as the blood sugar level in mg of glucose per dL of plasma 2 hours after taking 75 g of glucose after a fast. A subject with "normal glucose tolerance" has a 2 hour postprandial blood glucose or serum glucose concentration smaller than 140 mg/dl (7.78 mmol/L).
[0081] The term "hyperinsulinemia" is defined as the condition in which a subject with insulin resistance, with or without euglycemia, has fasting or postprandial serum or plasma insulin concentration elevated above that of normal, lean individuals without insulin resistance, having a waist-to-hip ratio <1.0 (for men) or <0.8 (for women).
[0082] The terms "insulin-sensitizing", "insulin resistance-improving" or "insulin resistance-lowering" are synonymous and used interchangeably.
[0083] The term "insulin resistance" is defined as a state in which circulating insulin levels in excess of the normal response to a glucose load are required to maintain the euglycemic state (Ford E S, et al. JAMA. (2002) 287:356-9). A method of determining insulin resistance is the euglycaemic-hyperinsulinaemic clamp test. The ratio of insulin to glucose is determined within the scope of a combined insulin-glucose infusion technique. There is found to be insulin resistance if the glucose absorption is below the 25th percentile of the background population investigated (WHO definition). Rather less laborious than the clamp test are so called minimal models in which, during an intravenous glucose tolerance test, the insulin and glucose concentrations in the blood are measured at fixed time intervals and from these the insulin resistance is calculated. With this method, it is not possible to distinguish between hepatic and peripheral insulin resistance.
[0084] Furthermore, insulin resistance, the response of a patient with insulin resistance to therapy, insulin sensitivity and hyperinsulinemia may be quantified by assessing the "homeostasis model assessment to insulin resistance (HOMA-IR)" score, a reliable indicator of insulin resistance (Katsuki A, et al. Diabetes Care 2001; 24: 362-5). Further reference is made to methods for the determination of the HOMA-index for insulin sensitivity (Matthews et al., Diabetologia 1985, 28: 412-19), of the ratio of intact proinsulin to insulin (Forst et al., Diabetes 2003, 52(Suppl.1): A459) and to an euglycemic clamp study. In addition, plasma adiponectin levels can be monitored as a potential surrogate of insulin sensitivity. The estimate of insulin resistance by the homeostasis assessment model (HOMA)-IR score is calculated with the formula (Galvin P, et al. Diabet Med 1992;9:921-8):
[0085] HOMA-IR=[fasting serum insulin (.mu.U/mL)].times.[fasting plasma glucose(mmol/L)/22.5]
[0086] As a rule, other parameters are used in everyday clinical practice to assess insulin resistance. Preferably, the patient's triglyceride concentration is used, for example, as increased triglyceride levels correlate significantly with the presence of insulin resistance.
[0087] Patients with a predisposition for the development of IGT or IFG or type 2 diabetes are those having euglycemia with hyperinsulinemia and are by definition, insulin resistant. A typical patient with insulin resistance is usually overweight or obese. If insulin resistance can be detected, this is a particularly strong indication of the presence of pre-diabetes. Thus, it may be that in order to maintain glucose homoeostasis a person needs 2-3 times as much insulin as a healthy person, without this resulting in any clinical symptoms.
[0088] The methods to investigate the function of pancreatic beta-cells are similar to the above methods with regard to insulin sensitivity, hyperinsulinemia or insulin resistance: An improvement of beta-cell function can be measured for example by determining a HOMA-index for beta-cell function (Matthews et al., Diabetologia 1985, 28: 412-19), the ratio of intact proinsulin to insulin (Forst et al., Diabetes 2003, 52(SuppL1): A459), the insulin/C-peptide secretion after an oral glucose tolerance test or a meal tolerance test, or by employing a hyperglycemic clamp study and/or minimal modeling after a frequently sampled intravenous glucose tolerance test (Stumvoll et al., Eur J Clin Invest 2001, 31: 380-81).
[0089] The term "pre-diabetes" is the condition wherein an individual is pre-disposed to the development of type 2 diabetes. Pre-diabetes extends the definition of impaired glucose tolerance to include individuals with a fasting blood glucose within the high normal range 100 mg/dL (J. B. Meigs, et al. Diabetes 2003; 52:1475-1484) and fasting hyperinsulinemia (elevated plasma insulin concentration). The scientific and medical basis for identifying pre-diabetes as a serious health threat is laid out in a Position Statement entitled "The Prevention or Delay of Type 2 Diabetes" issued jointly by the American Diabetes Association and the National Institute of Diabetes and Digestive and Kidney Diseases (Diabetes Care 2002; 25:742-749).
[0090] Individuals likely to have insulin resistance are those who have two or more of the following attributes: 1) overweight or obese, 2) high blood pressure, 3) hyperlipidemia, 4) one or more 1.sup.st degree relative with a diagnosis of IGT or IFG or type 2 diabetes. Insulin resistance can be confirmed in these individuals by calculating the HOMA-IR score. For the purpose of this invention, insulin resistance is defined as the clinical condition in which an individual has a HOMA-IR score >4.0 or a HOMA-IR score above the upper limit of normal as defined for the laboratory performing the glucose and insulin assays.
[0091] The term "type 2 diabetes" is defined as the condition in which a subject has a fasting blood glucose or serum glucose concentration greater than 125 mg/dL (6.94 mmol/L). The measurement of blood glucose values is a standard procedure in routine medical analysis. If a glucose tolerance test is carried out, the blood sugar level of a diabetic will be in excess of 200 mg of glucose per dL (11.1 mmol/l) of plasma 2 hours after 75 g of glucose have been taken on an empty stomach. In a glucose tolerance test 75 g of glucose are administered orally to the patient being tested after 10-12 hours of fasting and the blood sugar level is recorded immediately before taking the glucose and 1 and 2 hours after taking it. In a healthy subject, the blood sugar level before taking the glucose will be between 60 and 110 mg per dL of plasma, less than 200 mg per dL 1 hour after taking the glucose and less than 140 mg per dL after 2 hours. If after 2 hours the value is between 140 and 200 mg, this is regarded as abnormal glucose tolerance.
[0092] The term "late stage type 2 diabetes mellitus" includes patients with a secondary drug failure, indication for insulin therapy and progression to micro- and macrovascular complications e.g. diabetic nephropathy, or coronary heart disease (CHD).
[0093] The term "HbA1c" refers to the product of a non-enzymatic glycation of the haemoglobin B chain. Its determination is well known to one skilled in the art. In monitoring the treatment of diabetes mellitus the HbA1c value is of exceptional importance. As its production depends essentially on the blood sugar level and the life of the erythrocytes, the HbA1c in the sense of a "blood sugar memory" reflects the average blood sugar levels of the preceding 4-6 weeks. Diabetic patients whose HbA1c value is consistently well adjusted by intensive diabetes treatment (i.e. <6.5% of the total haemoglobin in the sample), are significantly better protected against diabetic microangiopathy. For example, metformin on its own achieves an average improvement in the HbA1c value in the diabetic of the order of 1.0-1.5%. This reduction of the HbA1C value is not sufficient in all diabetics to achieve the desired target range of <6.5% and preferably <6% HbA1c.
[0094] The term "insufficient glycemic control" or "inadequate glycemic control" in the scope of the present invention means a condition wherein patients show HbA1c values above 6.5%, in particular above 7.0%, even more preferably above 7.5%, especially above 8%.
[0095] The "metabolic syndrome", also called "syndrome X" (when used in the context of a metabolic disorder), also called the "dysmetabolic syndrome" is a syndrome complex with the cardinal feature being insulin resistance (Laaksonen D E, et al. Am J Epidemiol 2002; 156:1070-7). According to the ATP III/NCEP guidelines (Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) JAMA: Journal of the American Medical Association (2001) 285:2486-2497), diagnosis of the metabolic syndrome is made when three or more of the following risk factors are present: [0096] 1. Abdominal obesity, defined as waist circumference >40 inches or 102 cm in men, and >35 inches or 94 cm in women; or with regard to a Japanese ethnicity or Japanese patients defined as waist circumference 85 cm in men and .gtoreq.90 cm in women; [0097] 2. Triglycerides: .gtoreq.150 mg/dL [0098] 3. HDL-cholesterol <40 mg/dL in men [0099] 4. Blood pressure .gtoreq.130/85 mm Hg (SBP.gtoreq.130 or DBP.gtoreq.85) [0100] 5. Fasting blood glucose.gtoreq.100 mg/dL
[0101] The NCEP definitions have been validated (Laaksonen D E, et al. Am J Epidemiol. (2002) 156:1070-7). Triglycerides and HDL cholesterol in the blood can also be determined by standard methods in medical analysis and are described for example in Thomas L (Editor): "Labor and Diagnose", TH-Books Verlagsgesellschaft mbH, Frankfurt/Main, 2000.
[0102] According to a commonly used definition, hypertension is diagnosed if the systolic blood pressure (SBP) exceeds a value of 140 mm Hg and diastolic blood pressure (DBP) exceeds a value of 90 mm Hg. If a patient is suffering from manifest diabetes it is currently recommended that the systolic blood pressure be reduced to a level below 130 mm Hg and the diastolic blood pressure be lowered to below 80 mm Hg.
[0103] The definitions of NODAT (new onset diabetes after transplantation) and PTMS (post-transplant metabolic syndrome) follow closely that of the American Diabetes Association diagnostic criteria for type 2 diabetes, and that of the International Diabetes Federation (IDF) and the American Heart Association/National Heart, Lung, and Blood Institute, for the metabolic syndrome. NODAT and/or PTMS are associated with an increased risk of micro- and macrovascular disease and events, graft rejection, infection, and death. A number of predictors have been identified as potential risk factors related to NODAT and/or PTMS including a higher age at transplant, male gender, the pre-transplant body mass index, pre-transplant diabetes, and immunosuppression.
[0104] The term "gestational diabetes" (diabetes of pregnancy) denotes a form of the diabetes which develops during pregnancy and usually ceases again immediately after the birth.
[0105] Gestational diabetes is diagnosed by a screening test which is carried out between the 24th and 28th weeks of pregnancy. It is usually a simple test in which the blood sugar level is measured one hour after the administration of 50 g of glucose solution. If this 1 h level is above 140 mg/dl, gestational diabetes is suspected. Final confirmation may be obtained by a standard glucose tolerance test, for example with 75 g of glucose.
[0106] The term "hyperuricemia" denotes a condition of high serum total urate levels. In human blood, uric acid concentrations between 3.6 mg/dL (ca. 214 .mu.mol/L) and 8.3 mg/dL (ca. 494 .mu.mol/L) are considered normal by the American Medical Association. High serum total urate levels, or hyperuricemia, are often associated with several maladies. For example, high serum total urate levels can lead to a type of arthritis in the joints kown as gout. Gout is a condition created by a build up of monosodium urate or uric acid crystals on the articular cartilage of joints, tendons and surrounding tissues due to elevated concentrations of total urate levels in the blood stream. The build up of urate or uric acid on these tissues provokes an inflammatory reaction of these tissues. Saturation levels of uric acid in urine may result in kidney stone formation when the uric acid or urate crystallizes in the kidney. Additionally, high serum total urate levels are often associated with the so-called metabolic syndrome, including cardiovascular disease and hypertension.
[0107] The term "hyponatremia" denotes a condition of a positive balance of water with or without a deficit of sodium, which is recognized when the plasma sodium falls below the level of 135 mml/L. Hyponatremia is a condition which can occur in isolation in individuals that over-consume water; however, more often hyponatremia is a complication of medication or other underlying medical condition that leas to a diminished excretion of water. Hyponatremia may lead to water intoxication, which occurs when the normal tonicity of extracellular fluid falls below the safe limit, due to retention of excess water. Water intoxication is a potentially fatal disturbance in brain function. Typical symptoms of water intoxication include nausea, vomiting, headache and malaise.
[0108] The term "SGLT2 inhibitor" in the scope of the present invention relates to compounds, in particular to glucopyranosyl-derivatives, i.e. compounds having a glucopyranosyl-moiety, which show an inhibitory effect on the sodium-glucose transporter 2 (SGLT2), in particular the human SGLT2. The inhibitory effect on hSGLT2 measured as 1050 is prerably below 1000 nM, even more preferably below 100 nM, most preferably below 50 nM. The inhibitory effect on hSGLT2 can be determined by methods known in the literature, in particular as described in the application WO 2005/092877 or WO 2007/093610 (pages 23/24), which are incorporated herein by reference in its entirety. The term "SGLT2 inhibitor" also comprises any pharmaceutically acceptable salts thereof, hydrates and solvates thereof, including the respective crystalline forms.
[0109] The terms "treatment" and "treating" comprise therapeutic treatment of patients having already developed said condition, in particular in manifest form. Therapeutic treatment may be symptomatic treatment in order to relieve the symptoms of the specific indication or causal treatment in order to reverse or partially reverse the conditions of the indication or to stop or slow down progression of the disease. Thus the compositions and methods of the present invention may be used for instance as therapeutic treatment over a period of time as well as for chronic therapy.
[0110] The terms "prophylactically treating", "preventivally treating" and "preventing" are used interchangeably and comprise a treatment of patients at risk to develop a condition mentioned hereinbefore, thus reducing said risk.
[0111] The term "tablet" comprises tablets without a coating and tablets with one or more coatings. Furthermore the "term" tablet comprises tablets having one, two, three or even more layers and press-coated tablets, wherein each of the beforementioned types of tablets may be without or with one or more coatings. The term "tablet" also comprises mini, melt, chewable, effervescent and orally disintegrating tablets.
[0112] The terms "pharmacopoe" and "pharmacopoeias" refer to standard pharmacopoeias such as the "USP 31-NF 26 through Second Supplement" (United States Pharmacopeial Convention) or the "European Pharmacopoeia 6.3" (European Directorate for the Quality of Medicines and Health Care, 2000-2009).
BRIEF DESCRIPTION OF THE FIGURES
[0113] FIG. 1 shows an X-ray powder diffractogram of the crystalline form (I.9X) of the compound (1.9).
[0114] FIG. 2 shows the thermoanalysis and determination of the melting point via DSC of the crystalline form (I9.X) of the compound (1.9).
[0115] FIGS. 3A and 3B show the blood glucose level and blood glucose AUC results of the administration of a compound of the invention to ZDF rats.
[0116] FIG. 4A shows the results of the body weight analysis in the administration of a compound of the invention to Wistar rats.
[0117] FIG. 4B shows the results of the body fat content analysis in the administration of a compound of the invention to Wistar rats.
DETAILED DESCRIPTION
[0118] The aspects according to the present invention, in particular the pharmaceutical compositions, methods and uses, refer to SGLT2 inhibitors as defined hereinbefore and hereinafter.
[0119] Preferably the SGLT2 inhibitor is selected from a glucopyranosyl-substituted benzene derivative of the formula (I)
##STR00001##
wherein R.sup.1 denotes Cl, methyl or cyano; R.sup.2 denotes H, methyl, methoxy or hydroxy and R.sup.3 denotes ethyl, cyclopropyl, ethynyl, ethoxy, (R)-tetrahydrofuran-3-yloxy or (S)-tetrahydrofuran-3-yloxy; or a prodrug of one of the beforementioned SGLT2 inhibitors.
[0120] Compounds of the formula (I) and methods of their synthesis are described for example in the following patent applications: WO 2005/092877, WO 2006/117360, WO 2006/117359, WO 2006/120208, WO 2006/064033, WO 2007/031548, WO 2007/093610, WO 2008/020011, WO 2008/055870.
[0121] In the above glucopyranosyl-substituted benzene derivatives of the formula (I) the following definitions of the substituents are preferred.
[0122] Preferably R.sup.1 denotes chloro or cyano; in particular chloro.
[0123] Preferably R.sup.2 denotes H.
[0124] Preferably R.sup.3 denotes ethyl, cyclopropyl, ethynyl, (R)-tetrahydrofuran-3-yloxy or (S)-tetrahydrofuran-3-yloxy. Even more preferably R.sup.3 denotes cyclopropyl, ethynyl, (R)-tetrahydrofuran-3-yloxy or (S)-tetrahydrofuran-3-yloxy. Most preferably R.sup.3 denotes ethynyl, (R)-tetrahydrofuran-3-yloxy or (S)-tetrahydrofuran-3-yloxy.
[0125] Preferred glucopyranosyl-substituted benzene derivatives of the formula (I) are selected from the group of compounds (I.1) to (I.11):
##STR00002## ##STR00003## ##STR00004##
[0126] Even more preferred glucopyranosyl-substituted benzene derivatives of the formula (I) are selected from the compounds (I.6), (I.7), (I.8), (I.9) and (I.11). Even more preferred glucopyranosyl-substituted benzene derivatives of the formula (I) are selected from the compounds (I.8) and (I.9).
[0127] According to this invention, it is to be understood that the definitions of the above listed glucopyranosyl-substituted benzene derivatives of the formula (I) also comprise their hydrates, solvates and polymorphic forms thereof, and prodrugs thereof. With regard to the preferred compound (I.7) an advantageous crystalline form is described in the international patent application WO 2007/028814 which hereby is incorporated herein in its entirety. With regard to the preferred compound (I.8), an advantageous crystalline form is described in the international patent application WO 2006/117360 which hereby is incorporated herein in its entirety. With regard to the preferred compound (I.9) an advantageous crystalline form is described in the international patent applciation WO 2006/117359 which hereby is incorporated herein in its entirety. With regard to the preferred compound (I.11) an advantageous crystalline form is described in the international patent applciation WO 2008/049923 which hereby is incorporated herein in its entirety. These crystalline forms possess good solubility properties which enable a good bioavailability of the SGLT2 inhibitor. Furthermore, the crystalline forms are physico-chemically stable and thus provide a good shelf-life stability of the pharmaceutical composition.
[0128] For avoidance of any doubt, the disclosure of each of the foregoing documents cited above in connection with the specified SGLT2 inhibitors is specifically incorporated herein by reference in its entirety.
[0129] A preferred crystalline form (I.9X) of the compound (I.9) can be characterized by an X-ray powder diffraction pattern that comprises peaks at 18.84, 20.36 and 25.21 degrees 20 (.+-.0.1 degrees 2.THETA.), wherein said X-ray powder diffraction pattern (XRPD) is made using CuK.sub..alpha.1 radiation.
[0130] In particular said X-ray powder diffraction pattern comprises peaks at 14.69, 18.84, 19.16, 19.50, 20.36 and 25.21 degrees 2.THETA. (.+-.0.1 degrees 2.THETA.), wherein said X-ray powder diffraction pattern is made using CuK.sub..alpha.1 radiation.
[0131] In particular said X-ray powder diffraction pattern comprises peaks at 14.69, 17.95, 18.43, 18.84, 19.16, 19.50, 20.36, 22.71, 23.44, 24.81, 25.21 and 25.65 degrees 2.THETA. (.+-.0.1 degrees 2.THETA.), wherein said X-ray powder diffraction pattern is made using CuK.sub..alpha.1 radiation.
[0132] More specifically, the crystalline form (I.9X) is characterised by an X-ray powder diffraction pattern, made using CuK.sub..alpha.1 radiation, which comprises peaks at degrees 2.THETA. (.+-.0.1 degrees 2.THETA.) as contained in Table 1.
TABLE-US-00001 TABLE 1 X-ray powder diffraction pattern of the crystalline form (I.9X) (only peaks up to 30.degree. in 2.THETA. are listed): 2.THETA. d-value Intensity I/I.sub.0 [.degree.] [.ANG.] [%] 4.46 19.80 8 9.83 8.99 4 11.68 7.57 4 13.35 6.63 14 14.69 6.03 42 15.73 5.63 16 16.20 5.47 8 17.95 4.94 30 18.31 4.84 22 18.43 4.81 23 18.84 4.71 100 19.16 4.63 42 19.50 4.55 31 20.36 4.36 74 20.55 4.32 13 21.18 4.19 11 21.46 4.14 13 22.09 4.02 19 22.22 4.00 4 22.71 3.91 28 23.44 3.79 27 23.72 3.75 3 24.09 3.69 3 24.33 3.66 7 24.81 3.59 24 25.21 3.53 46 25.65 3.47 23 26.40 3.37 2 26.85 3.32 8 27.26 3.27 17 27.89 3.20 2 28.24 3.16 3 29.01 3.08 4 29.41 3.03 18
[0133] Even more specifically, the crystalline form (I.9X) is characterised by an X-ray powder diffraction pattern, made using CuK.sub..alpha.1 radiation, which comprises peaks at degrees 2.THETA. (.+-.0.1 degrees 2.THETA.) as shown in FIG. 1.
[0134] Furthermore the crystalline form (I.9X) is characterised by a melting point of about 149.degree. C..+-.3.degree. C. (determined via DSC; evaluated as onset-temperature; heating rate 10 K/min).The obtained DSC curve is shown in FIG. 2.
[0135] The X-ray powder diffraction patterns are recorded, within the scope of the present invention, using a STOE--STADI P-diffractometer in transmission mode fitted with a location-sensitive detector .quadrature.(OED) and a Cu-anode as X-ray source (CuK.alpha.1 radiation, .quadrature..lamda.=1,54056 .ANG., 40 kV, 40 mA). In the Table 1 above the values "2.THETA. [.degree.]" denote the angle of diffraction in degrees and the values "d [.ANG.]" denote the specified distances in .ANG. between the lattice planes. The intensity shown in the FIG. 1 is given in units of cps (counts per second).
[0136] In order to allow for experimental error, the above described 2.THETA. values should be considered accurate to .+-.0.1 degrees 2.THETA., in particular .+-.0.05 degrees 2.THETA.. That is to say, when assessing whether a given sample of crystals of the compound (I.9) is the crystalline form in accordance with the invention, a 2.THETA. value which is experimentally observed for the sample should be considered identical with a characteristic value described above if it falls within .+-.0.1 degrees 2.THETA. of the characteristic value, in particular if it falls within .+-.0.05 degrees 2.THETA. of the characteristic value.
[0137] The melting point is determined by DSC (Differential Scanning calorimetry) using a DSC 821 (Mettler Toledo).
[0138] In one embodiment, a pharmaceutical composition or dosage form according to the present invention comprises the compound (I.9), wherein at least 50% by weight of the compound (I.9) is in the form of its crystalline form (I.9X) as defined hereinbefore. Preferably in said composition or dosage form at least 80% by weight, more preferably at least 90% by weight of the compound (I.9) is in the form of its crystalline form (I.9X) as defined hereinbefore.
[0139] Regarding the active pharmaceutical ingredients it can be found that the dissolution properties of the pharmaceutical composition and dosage form is affected inter alia by the particle size and particle size distribution of the respective active pharmaceutical ingredient. In the pharmaceutical composition and pharmaceutical dosage form according to the invention the active pharmaceutical ingredients preferably have a particle size distribution such that at least 90% of the respective active pharmaceutical ingredient particles, with regard to the distribution by volume, has a particle size smaller than 200 .mu.m, i.e. X90<200 .mu.m.
[0140] In particular, with regard to the glucopyranosyl-substituted benzene derivative of the formula (I), in particular the compound (I.9) or its crystalline form (I.9X), it was found that the particle size influence the manufacturability, in particular that too small particles influence the manufacturability by sticking or filming. On the other hand too large particles negatively affect the dissolution properties of the pharmaceutical composition and dosage form and thus the bioavailability. In the following preferred ranges of the particle size distribution are described.
[0141] Therefore, in one aspect, in the pharmaceutical composition and pharmaceutical dosage form according to the invention the glucopyranosyl-substituted benzene derivative of the formula (I), in particular the compound (I.9), preferably its crystalline form (I9.X), preferably has a particle size distribution (by volume) such that at least 90% of the respective active pharmaceutical ingredient has a particle size smaller than 200 .mu.m, i.e. X90<200 .mu.m, preferably X90 150 .mu.m. More preferably the particle size distribution is such that X90.ltoreq.100 .mu.m, even more preferably X90.ltoreq.90 .mu.m. In addition the particle size distribution is preferably such that X90.gtoreq.1 .mu.m, more preferably X90.gtoreq.5 .mu.m, even more preferably X90.gtoreq.10 .mu.m. Therefore preferred particle size distributions are such that 1 .mu.m.ltoreq.X90.ltoreq.200 .mu.m, particularly 1 .mu.m.ltoreq.X90.ltoreq.150 .mu.m, more preferably 5 .mu.m.ltoreq.X90.ltoreq.150 .mu.m, even more preferably 5 .mu.m.ltoreq.X90.ltoreq.100 .mu.m, even more preferably 10 .mu.m.ltoreq.X90.ltoreq.100 .mu.m. A preferred example X90.ltoreq.75 .mu.m. Another preferred example is 20 .mu.m.ltoreq.X90.ltoreq.50 .mu.m.
[0142] Furthermore in the pharmaceutical composition and pharmaceutical dosage form according to the invention the glucopyranosyl-substituted benzene derivative of the formula (I), in particular the compound (I.9), preferably its crystalline form (I9.X), preferably has a particle size distribution (by volume) such that X50.ltoreq.90 .mu.m, more preferably X50.ltoreq.75 .mu.m, even more preferably X50.ltoreq.50 .mu.m, most preferably X50.ltoreq.40 .mu.m. In addition the particle size distribution is preferably such that X50.gtoreq.1 .mu.m, more preferably X50.gtoreq.5 .mu.m, even more preferably X50.gtoreq.8 .mu.m. Therefore preferred particle size distributions are such that 1 .mu.m.ltoreq.X50.ltoreq.90 .mu.m, particularly 1 .mu.m.ltoreq.X50.ltoreq.75 .mu.m, more preferably 5 .mu.m.ltoreq.X50.ltoreq.75 .mu.m, even more preferably 5 .mu.m.ltoreq.X50.ltoreq.50 .mu.m. A preferred example is 8 .mu.m.ltoreq.X50.ltoreq.40 .mu.m.
[0143] Furthermore in the pharmaceutical composition and pharmaceutical dosage form according to the invention the glucopyranosyl-substituted benzene derivative of the formula (I), in particular the compound (I.9), preferably its crystalline form (I9.X), preferably has a particle size distribution (by volume) such that X10.ltoreq.0.1 .mu.m, more preferably X10.ltoreq.0.5 .mu.m, even more preferably X10.ltoreq.1 .mu.m, in particular X10.ltoreq.2 .mu.m.
[0144] Therefore a pharmaceutical composition or pharmaceutical dosage form according to this invention may preferably be characterized by the above specified particle size distributions X90, X50 and/or X10 or one of the following embodiments:
TABLE-US-00002 Glucopyranosyl-substituted benzene derivative, in particular of Embodiment the compound (I.9) 1 X90 < 200 .mu.m 2 1 .mu.m .ltoreq. X90 .ltoreq. 150 .mu.m 3 5 .mu.m .ltoreq. X90 .ltoreq. 150 .mu.m 4 10 .mu.m .ltoreq. X90 .ltoreq. 100 .mu.m 5 X90 .ltoreq.150 .mu.m 1 .mu.m .ltoreq. X50 .ltoreq. 75 .mu.m 6 X90 .ltoreq. 150 .mu.m 5 .mu.m .ltoreq. X50 .ltoreq. 50 .mu.m 7 X90 .ltoreq. 150 .mu.m 1 .mu.m .ltoreq. X50 .ltoreq. 75 .mu.m X10 .gtoreq.0.1 .mu.m 8 X90 .ltoreq.150 .mu.m 5 .mu.m .ltoreq. X50 .ltoreq. 50 .mu.m X10 .gtoreq. 0.5 .mu.m
[0145] The value X90 refers to the 90% value of the volume distribution measured using a laser diffractometer. In other words, for the purposes of the present invention, the X90 value denotes the particle size below which 90% of the quantity of particles is found based on the volume distribution. Analogously the value X50 refers to the 50% value (median) of the volume distribution measured using a laser diffractometer. In other words, for the purposes of the present invention, the X50 value denotes the particle size below which 50% of the quantity of particles is found based on the volume distribution. Analogously the value X10 refers to the 10% value of the volume distribution measured using a laser diffractometer. In other words, for the purposes of the present invention, the X10 value denotes the particle size below which 10% of the quantity of particles is found based on the volume distribution.
[0146] Preferably all X90, X50, X10 values hereinbefore and hereinafter are by volume and determined by laser-diffraction method, in particular low angle laser light scattering, i.e. Fraunhofer diffraction. A preferred test is described in the experimental section. The laser diffraction method is sensitive to the volume of a particle and provides a volume-average particle size, which is equivalent to the weight-average particle size if the density is constant. The skilled artesian knows that the results of the particle size distribution determination by one technique can be correlated with that from another technique, for example on an empirical basis by routine experimentation. Alternatively the particle size distribution in the pharmaceutical composition or dosage form can be determined by microscopy, in particular electron microscopy or scanning electron microscopy.
[0147] In the following the suitable excipients and carriers in the pharmaceutical compositions according to the invention are described in further detail.
[0148] A pharmaceutical composition according to the invention typically comprises one or more diluents, one or more disintegrants and optionally one or more binders. Some of the excipients may have two or more functions at the same time, e.g. act as a filler and a binder.
[0149] Suitable diluents according to the invention are for example, lactose, in particular lactose monohydrate, cellulose and derivatives, such as powdered cellulose, microcrystalline or silicified microcrystalline cellulose, cellulose acetate, starches and derivatives such as pregelatinized starch, corn starch, wheat starch, rice starch, potato starch, sterilizable maize, sodium chloride, calcium carbonate, calcium phosphate, particularly dibasic calcium phosphate, calcium sulphate, dicalcium or tricalcium phosphate, magnesium carbonate, magnesium oxide, sugars and derivatives such as confectioner's sugar, fructose, sucrose, dextrates, dextrin, D-sorbitol sulfobutylether B-cyclodextrin, dextrose, polydextrose, trehalose, maltose, maltitol, mannitol, maltodextrin, sorbitol, inulin, xylitol, erythritol, isomalt, kaolin and lactitol. Preferred diluents are lactose monohydrate and microcrystalline cellulose.
[0150] Suitable disintegrants according to the invention are for example powdered cellulose, crospovidone, croscarmellose sodium, docusate sodium, low-substituted hydroxypropyl cellulose, magnesium aluminum silicate, microcrystalline cellulose, polacrilin potassium, sodium starch glycolate, starch, particularly pregelatinized starch and corn starch. A preferred disintegrant is croscarmellose sodium.
[0151] Any binder usually employed in pharmaceutical compositions may be used in the context of the instant invention. Binders are for example naturally occurring or partially or totally synthetic polymers selected from acacia, agar, alginic acid, carbomers, carmellose sodium, carrageenan, cellulose acetate phthalate, ceratonia, chitosan, confectionar's sugar, copovidone, povidone, cottonseed oil, dextrate, dextrin, dextrose, polydextrose, maltodextrin, maltose, cellulose and derivatives thereof such as microcrystalline cellulose, methylcellulose, ethylcellulose, hydroxyethyl cellulose, hydroxyethyl methylcellulose, hydroxypropyl celluloses, carboxymethylcelluloses, hypromelloses (cellulose hydroxypropyl methyl ether), starch and derivatives thereof, such as pregelatinized starch, hydroxypropylstarch, corn starch, gelatin, glyceryl behenate, tragacanth, guar gum, hydrogenated vegetable oils, inulin, poloxamer, polycarbophils, polyethylene oxide, polyvinylpyrrolidone, copolymers of N-vinylpyrrolidone and vinyl acetate, polymethacrylates, polyethylene glycols, alginates such as sodium alginate, gelatin, sucrose, sunflower oil, zein as well as derivatives and mixtures thereof. Preferred binders are microcrystalline cellulose and hydroxypropyl cellulose.
[0152] The pharmaceutical composition according to the present invention may also comprise one or more lubricants. Suitable lubricants according to the invention are stearic acid as well as salts thereof including talc, sodium stearate, calcium stearate, zinc stearate, magnesium stearate, sodium stearyl fumarate, glyceryl monostearate, particularly magnesium stearate, polyethylene glycols, in particular polyethylene glycol with a molecular weight in a range from about 4400 to about 9000, hydrogenated castor oil, fatty acid, for example fumaric acid, and salts of fatty acids, in particular the calcium, magnesium, sodium or pottasium salts thereof, for example calcium behenate, calcium stearate, sodium stearyl fumarate or magnesium stearate (for example (e.g. HyQual.RTM., Mallinckrodt), glycerides such as glyceryl behenate (Compritol.RTM. 888), Dynasan.RTM. 118 or Boeson.RTM. VP.
[0153] The pharmaceutical composition according to the present invention may also comprise one or more glidants. Suitable glidants according to the invention are silicon dioxide, particularly colloidal silicon dioxide (e.g. Aerosil.RTM., Cab-O-Sil.RTM.), stearic acid as well as salts thereof including sodium stearate, calcium stearate, zinc stearate, magnesium stearate, magnesium silicate, calcium silicate, magnesium trisilicate and talc. Preferred glidants are colloidal silicon dioxide and talc.
[0154] In another embodiment, a pharmaceutical composition according to the instant invention comprises
TABLE-US-00003 Amount (% by weight) Active ingredient 0.5-25 One or more diluents 65-93 One or more binders 1-5 One or more disintegrants 1-4 Optionally additional additives ad 100%
[0155] In one aspect, the active ingredient is a compound of the formula (I), for example of the formula (I.9) or its crystalline form (I.9X).
[0156] In another embodiment, a pharmaceutical composition according to the instant invention comprises
TABLE-US-00004 Amount (% by weight) Active ingredient 0.5-25 One or more diluents 65-90 One or more binders 1-5 One or more disintegrants 1-3 Optionally additional additives ad 100%
[0157] The active ingredient is a compound of the formula (I), for example of the formula (I.9) or its crystalline form (I.9X).
[0158] In another embodiment, a pharmaceutical composition according to the instant invention comprises
TABLE-US-00005 Amount (% by weight) Active ingredient 0.5-25 Lactose monohydrate 28-70 Microcrystalline cellulose 20-50 Hydroxypropyl cellulose 1-5 Croscarmellose sodium 1-4 Optionally additional additives ad 100%
[0159] In one aspect, the active ingredient is a compound of the formula (I), for example of the formula (I.9) or its crystalline form (I.9X).
[0160] In another embodiment, a pharmaceutical composition according to the instant invention comprises
TABLE-US-00006 Amount (% by weight) Active ingredient 0.5-25 Lactose monohydrate 35-90 Microcrystalline cellulose 0-30 Hydroxypropyl cellulose 1-5 Croscarmellose sodium 1-3 Additional additives ad 100%
[0161] The active ingredient is a compound of the formula (I), for example of the formula (I.9) or its crystalline form (I.9X).
[0162] In one embodiment, the ratio of said disintegrant to said binder in a pharmaceutical composition of the present invention is between 1.5:3.5 and 1:1.
[0163] In one embodiment, the active ingredient represents 25% or less of the weight of the pharmaceutical composition. Preferably, the active ingredient represents 0.5% to 25% of the weight of the pharmaceutical composition. More preferably, the active ingredient represents 1.0% to 20% of the weight of the pharmaceutical composition. Even more preferably, the active ingredient represents 2.0% to 15% of the weight of the pharmaceutical composition.
[0164] In the following, preferred ranges of the amount of the glucopyranosyl-substituted benzene derivative to be employed in the pharmaceutical dosage form according to this invention are described. These ranges refer to the amounts to be administered per day with respect to an adult patient, in particular to a human being, for example of approximately 70 kg body weight, and can be adapted accordingly with regard to an administration 2, 3, 4 or more times daily and with regard to other routes of administration and with regard to the age of the patient. The ranges of the dosage and amounts are calculated for the active ingredient.
[0165] A preferred amount of the glucopyranosyl-substituted benzene derivative, in particular the compound (I.9) or its crystalline form (I.9X) is in a range from 0.5 to 100 mg, preferably from 0.5 to 50 mg, even more preferably from 1 to 25 mg, even more preferably 5 to 25 mg. Preferred dosages of the glucopyranosyl-substituted benzene derivative are for example 1 mg, 2 mg, 2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg, 20 mg, 25 mg and 50 mg.
[0166] A pharmaceutical composition according to the present invention may be comprised in a tablet, a capsule or a film-coated tablet,
[0167] In one embodiment, a tablet comprising a pharmaceutical composition according to the present invention comprises a lubricant, such as magnesium stearate. Such lubricant may be present in a concentration of 0.25-2% in said tablet.
[0168] In one embodiment, a tablet comprising a pharmaceutical composition according to the present invention comprises a glidant, such as colloidal silicon dioxide. Such glidant may be present in a concentration of 0.25-2% in said tablet.
[0169] A tablet according to the invention may be film-coated. Typically a film coat represents 2-5% by weight of the total composition and comprises preferably a film-forming agent, a plasticizer, a glidant and optionally one or more pigments. An exemplary coat composition may comprise hydroxypropylmethyl-cellulose (HPMC), polyethylene glycol (PEG), talc, titanium dioxide and optionally iron oxide, including iron oxide red and/or yellow.
[0170] In one embodiment, the pharmaceutical dosage form according to the invention has dissolution properties such that after 45 minutes at least 75%, preferably at least 90% by weight of the pharmaceutical active ingredient is dissolved. In another embodiment after 30 minutes at least 75%, preferably at least 90% by weight of the pharmaceutical active ingredien is dissolved. In another embodiment after 15 minutes at least 75%, preferably at least 90% by weight of the pharmaceutical active ingredient is dissolved. The dissolution properties can be determined in a standard dissolution test, for example as described in pharmacopoeias, such as the USP31-NF26 S2, chapter 711 (dissolution).
[0171] In one embodiment, the pharmaceutical dosage form according to the invention has disintegration properties such that within 40 minutes, alternatively within 30 minutes, preferably within 20 minutes, more preferably within 15 minutes the pharmaceutical dosage form is disintegrated. The disintegration properties can be determined in a standard disintegration test, for example as described in pharmacopoeias, such as the USP31-NF26 S2, chapter 701 (disintegration).
[0172] In one embodiment, the pharmaceutical dosage form according to the invention has a high content uniformity, preferably within a range from 85 to 115%, more preferably from 90 to 110%, even more preferably from 95 to 105% by weight with regard to the pharmaceutical ingredient. The content uniformity can be determined in a standard test using for example randomly 10 selected pharmaceutical dosage forms, for example as described in pharmacopoeias.
[0173] A dosage form according to this invention, such as a tablet, capsule or film-coated tablet, may be prepared by methods well-known to the one skilled in the art.
[0174] Suitable methods of manufacturing a tablet include compression of the pharmaceutical composition in the form of a powder, i.e. direct compression, or compression of the pharmaceutical composition in the form of granules, and if needed with additional excipients.
[0175] Granules of the pharmaceutical composition according to the invention may be prepared by methods well-known to the one skilled in the art. Preferred methods for the granulation of the active ingredients together with the excipients include wet granulation, for example high shear wet granulation and fluidized bed wet granulation, dry granulation, also called roller compaction.
[0176] In the wet granulation process the granulation liquid are the solvent alone or a preparation of one or more binders in a solvent or mixture of solvents. Suitable binders are described hereinbefore. Examples are hypromellose, hydroxypropyl cellulose, povidone and copovidone. Suitable solvents are for example purified water, ethanol, methanol, isopropanol, acetone, preferably purified water, including mixtures thereof. The solvent is a volatile component, which does not remain in the final product. The one or more active ingredients and the other excipients, in particular the one or more diluents and the one or more disintegrants, usually with exception of the lubricant, are premixed and granulated with the granulation liquid, for example using a high shear granulator. The wet granulation step is usually followed by one or more drying and sieving steps. For example a drying oven or a fluid bed dryer can then be used for drying.
[0177] The dried granules are sieved through an appropriate sieve. After optional addition of the other excipients, in particular disintegrant, binder, filler and/or glidant, with exception of the lubricant the mixture is blended in a suitable blender, for example a free fall blender, followed by addition of the one or more lubricants, for example magnesium stearate, and final blending in the blender.
[0178] An exemplary wet granulation process for making a pharmaceutical composition according to the instant invention comprises the steps of: [0179] (1) Premixing the active ingredient and the main portion of the excipients including the binder in a mixer to obtain a pre-mixture; [0180] (2) granulating the pre-mixture of step (1) by adding the granulation liquid, preferably purified water; [0181] (3) drying the granules of step (2) in a fluidized bed dryer or a drying oven; [0182] (4) optionally dry sieving of the dried granules of step (3); [0183] (5) mixing the dried granules of step (4) with the remaining excipients like filler, binder, disintegrant and/or glidant in a mixer to obtain the main mixture; [0184] (6) mixing the main mixture of step (5) with the lubricnat in a mixer to obtain the final mixture; [0185] (7) tableting the final mixture of step (6) by compressing it on a suitable tablet press to produce tablets cores; [0186] (8) optionally film-coating of the tablet cores of step (7) with a non-functional coat.
[0187] The present invention also provides a pharmaceutical composition obtainable by the above process.
[0188] An exemplary direct compression process according to the present invention for making a pharmaceutical composition comprises the steps of: [0189] (1) Premixing the active ingredient and the main portion of the excipients in a mixer to obtain a pre-mixture; [0190] (2) optionally dry screening the pre-mixture through a screen in order to segregate cohesive particles and to improve content uniformity; [0191] (3) mixing the pre-mixture of step (1) or (2) in a mixer, optionally by adding remaining excipients to the mixture and continuing mixing; [0192] (4) tableting the final mixture of step (3) by compressing it on a suitable tablet press to produce the tablet cores; [0193] (5) optionally film-coating of the tablet cores of step (4) with a non-functional coat.
[0194] The present invention also provides a pharmaceutical composition obtainable by the above process.
[0195] An exemplary dry granulation process according to the present invention for making a pharmaceutical composition comprises the steps of: [0196] (1) mixing the active ingredient or a pharmaceutically acceptable salt thereof with either all or a portion of the excipients in a mixer; [0197] (2) compaction of the mixture of step (1) on a suitable roller compactor; [0198] (3) reducing the ribbons obtained during step (2) to small granules by suitable milling or sieving steps; [0199] (4) optionally mixing the granules of step (3) with the remaining excipients in a mixer to obtain the final mixture; [0200] (5) tabletting the granules of step (3) or the final mixture of step (4) by compressing it on a suitable tablet press to produce the tablet cores; [0201] (6) optionally film-coating of the tablet cores of step (5) with a non-functional coat.
[0202] In one embodiment, the size of the granules according to the present invention is in the range from 25 to 800 .mu.m, for example from 40 .mu.m to 500 .mu.m. The size of the granules may be measured via sieve analysis, for example with a sonic sifter. In one embodiment, at least 80%, at least 90%, or at least 95% by weight of the granules is in the given range.
[0203] When this invention refers to patients requiring treatment or prevention, it relates primarily to treatment and prevention in humans, but the pharmaceutical composition may also be used accordingly in veterinary medicine in mammals. In the scope of this invention adult patients are preferably humans of the age of 18 years or older. Also in the scope of this invention, patients are adolescent humans, i.e. humans of age 10 to 17 years, preferably of age 13 to 17 years. It is assumed that in a adolescent population the administration of the pharmaceutical composition according to the invention a very good HbA1c lowering and a very good lowering of the fasting plasma glucose can be seen. In addition it is assumed that in an adolescent population, in particular in overweight and/or obese patients, a pronounced weight loss can be observed.
[0204] As described hereinbefore by the administration of the pharmaceutical composition according to this invention and in particular in view of the high SGLT2 inhibitory activity of the SGLT2 inhibitors therein, excessive blood glucose is excreted through the urine of the patient, so that no gain in weight or even a reduction in body weight may result. Therefore, a treatment or prophylaxis according to this invention is advantageously suitable in those patients in need of such treatment or prophylaxis who are diagnosed of one or more of the conditions selected from the group consisting of overweight and obesity, in particular class I obesity, class II obesity, class III obesity, visceral obesity and abdominal obesity. In addition a treatment or prophylaxis according to this invention is advantageously suitable in those patients in which a weight increase is contraindicated. The pharmaceutical composition as well as the methods according to the present invention allow a reduction of the HbA1c value to a desired target range, for example <7% and preferably <6.5%, for a higher number of patients and for a longer time of therapeutic treatment compared with a corresponding monotherapy or a therapy using only two of the combination partners.
[0205] The pharmaceutical composition according to this invention and in particular the SGLT2 inhibitor therein exhibits a very good efficacy with regard to glycemic control, in particular in view of a reduction of fasting plasma glucose, postprandial plasma glucose and/or glycosylated hemoglobin (HbA1c). By administering a pharmaceutical composition according to this invention, a reduction of HbA1c equal to or greater than preferably 0.5%, even more preferably equal to or greater than 1.0% can be achieved and the reduction is particularly in the range from 1.0% to 2.0%.
[0206] Furthermore, the method and/or use according to this invention is advantageously applicable in those patients who show one, two or more of the following conditions: [0207] (a) a fasting blood glucose or serum glucose concentration greater than 100 mg/dL, in particular greater than 125 mg/dL; [0208] (b) a postprandial plasma glucose equal to or greater than 140 mg/dL; [0209] (c) an HbA1c value equal to or greater than 6.5%, in particular equal to or greater than 7.0%, especially equal to or greater than 7.5%, even more particularly equal to or greater than 8.0%.
[0210] The present invention also discloses the use of the pharmaceutical composition for improving glycemic control in patients having type 2 diabetes or showing first signs of pre-diabetes. Thus, the invention also includes diabetes prevention. If therefore a pharmaceutical composition according to this invention is used to improve the glycemic control as soon as one of the above-mentioned signs of pre-diabetes is present, the onset of manifest type 2 diabetes mellitus can be delayed or prevented.
[0211] Furthermore, the pharmaceutical composition according to this invention is particularly suitable in the treatment of patients with insulin dependency, i.e. in patients who are treated or otherwise would be treated or need treatment with an insulin or a derivative of insulin or a substitute of insulin or a formulation comprising an insulin or a derivative or substitute thereof. These patients include patients with diabetes type 2 and patients with diabetes type 1.
[0212] Therefore, according to a preferred embodiment of the present invention, there is provided a method for improving glycemic control and/or for reducing of fasting plasma glucose, of postprandial plasma glucose and/or of glycosylated hemoglobin HbA1c in a patient in need thereof who is diagnosed with impaired glucose tolerance (IGT), impaired fasting blood glucose (IFG) with insulin resistance, with metabolic syndrome and/or with type 2 or type 1 diabetes mellitus characterized in that an SGLT2 inhibitor as defined hereinbefore and hereinafter is administered to the patient.
[0213] According to another preferred embodiment of the present invention, there is provided a method for improving gycemic control in patients, in particular in adult patients, with type 2 diabetes mellitus as an adjunct to diet and exercise.
[0214] It can be found that by using a pharmaceutical composition according to this invention, an improvement of the glycemic control can be achieved even in those patients who have insufficient glycemic control in particular despite treatment with an antidiabetic drug, for example despite maximal recommended or tolerated dose of oral monotherapy with metformin. A maximal recommended dose with regard to metformin is for example 2000 mg per day or 850 mg three times a day or any equivalent thereof.
[0215] Therefore, the method and/or use according to this invention is advantageously applicable in those patients who show one, two or more of the following conditions:
[0216] (a) insufficient glycemic control with diet and exercise alone;
[0217] (b) insufficient glycemic control despite oral monotherapy with metformin, in particular despite oral monotherapy at a maximal tolerated dose of metformin;
[0218] (c) insufficient glycemic control despite oral monotherapy with another antidiabetic agent, in particular despite oral monotherapy at a maximal tolerated dose of the other antidiabetic agent.
[0219] The lowering of the blood glucose level by the administration of an SGLT2 inhibitor according to this invention is insulin-independent. Therefore, a pharmaceutical composition according to this invention is particularly suitable in the treatment of patients who are diagnosed having one or more of the following conditions [0220] insulin resistance, [0221] hyperinsulinemia, [0222] pre-diabetes, [0223] type 2 diabetes mellitus, particular having a late stage type 2 diabetes mellitus, [0224] type 1 diabetes mellitus.
[0225] Furthermore, a pharmaceutical composition according to this invention is particularly suitable in the treatment of patients who are diagnosed having one or more of the following conditions
[0226] (a) obesity (including class I, II and/or III obesity), visceral obesity and/or abdominal obesity,
[0227] (b) triglyceride blood level .gtoreq.150 mg/dL,
[0228] (c) HDL-cholesterol blood level <40 mg/dL in female patients and <50 mg/dL in male patients,
[0229] (d) a systolic blood pressure .gtoreq.130 mm Hg and a diastolic blood pressure .gtoreq.85 mm Hg,
[0230] (e) a fasting blood glucose level .gtoreq.100 mg/dL.
[0231] It is assumed that patients diagnosed with impaired glucose tolerance (IGT), impaired fasting blood glucose (IFG), with insulin resistance and/or with metabolic syndrome suffer from an increased risk of developing a cardiovascular disease, such as for example myocardial infarction, coronary heart disease, heart insufficiency, thromboembolic events. A glycemic control according to this invention may result in a reduction of the cardiovascular risks.
[0232] Furthermore, a pharmaceutical composition according to this invention is particularly suitable in the treatment of patients after organ transplantation, in particular those patients who are diagnosed having one or more of the following conditions
[0233] (a) a higher age, in particular above 50 years,
[0234] (b) male gender;
[0235] (c) overweight, obesity (including class I, II and/or III obesity), visceral obesity and/or abdominal obesity,
[0236] (d) pre-transplant diabetes,
[0237] (e) immunosuppression therapy.
[0238] Furthermore, a pharmaceutical composition according to this invention is particularly suitable in the treatment of patients who are diagnosed having one or more of the following conditions:
[0239] (a) hyponatremia, in particular chronical hyponatremia;
[0240] (b) water intoxication;
[0241] (c) water retention;
[0242] (d) plasma sodium concentration below 135 mmol/L.
[0243] The patient may be a diabetic or non-diabetic mammal, in particular human.
[0244] Furthermore, a pharmaceutical composition according to this invention is particularly suitable in the treatment of patients who are diagnosed having one or more of the following conditions:
[0245] (a) high serum uric acid levels, in particular greater than 6.0 mg/dL (357 .mu.mol/L);
[0246] (b) a history of gouty arthritis, in particular recurrent gouty arthritis;
[0247] (c) kidney stones, in particular recurrent kidney stones;
[0248] (d) a high propensity for kidney stone formation.
[0249] A pharmaceutical composition according to this invention exhibits a good safety profile. Therefore, a treatment or prophylaxis according to this invention is advantageously possible in those patients for which the mono-therapy with another antidiabetic drug, such as for example metformin, is contraindicated and/or who have an intolerance against such drugs at therapeutic doses. In particular, a treatment or prophylaxis according to this invention may be advantageously possible in those patients showing or having an increased risk for one or more of the following disorders: renal insufficiency or diseases, cardiac diseases, cardiac failure, hepatic diseases, pulmonal diseases, catabolytic states and/or danger of lactate acidosis, or female patients being pregnant or during lactation.
[0250] Furthermore, it can be found that the administration of a pharmaceutical composition according to this invention results in no risk or in a low risk of hypoglycemia. Therefore, a treatment or prophylaxis according to this invention is also advantageously possible in those patients showing or having an increased risk for hypoglycemia.
[0251] A pharmaceutical composition according to this invention is particularly suitable in the long term treatment or prophylaxis of the diseases and/or conditions as described hereinbefore and hereinafter, in particular in the long term glycemic control in patients with type 2 diabetes mellitus.
[0252] The term "long term" as used hereinbefore and hereinafter indicates a treatment of or administration in a patient within a period of time longer than 12 weeks, preferably longer than 25 weeks, even more preferably longer than 1 year.
[0253] Therefore, a particularly preferred embodiment of the present invention provides a method for therapy, preferably oral therapy, for improvement, especially long term improvement, of glycemic control in patients with type 2 diabetes mellitus, especially in patients with late stage type 2 diabetes mellitus, in particular in patients additionally diagnosed of overweight, obesity (including class I, class II and/or class III obesity), visceral obesity and/or abdominal obesity.
[0254] It will be appreciated that the amount of the pharmaceutical composition according to this invention to be administered to the patient and required for use in treatment or prophylaxis according to the present invention will vary with the route of administration, the nature and severity of the condition for which treatment or prophylaxis is required, the age, weight and condition of the patient, concomitant medication and will be ultimately at the discretion of the attendant physician. In general, however, the SGLT2 inhibitor according to this invention is included in the pharmaceutical composition or dosage form in an amount sufficient that by its administration the glycemic control in the patient to be treated is improved.
[0255] For the treatment of hyperuricemia or hyperuricemia associated conditions the SGLT2 inhibitor according to this invention is included in the pharmaceutical composition or dosage form in an amount sufficient that is sufficient to treat hyperuricemia without disturbing the patient's plasma glucose homeostasis, in particular without inducing hypoglycemia.
[0256] For the treatment or prevention of kidney stones the SGLT2 inhibitor according to this invention is included in the pharmaceutical composition or dosage form in an amount sufficient that is sufficient to treat or prevent kidney stones without disturbing the patient's plasma glucose homeostasis, in particular without inducing hypoglycemia.
[0257] For the treatment of hyponatremia and associated conditions the SGLT2 inhibitor according to this invention is included in the pharmaceutical composition or dosage form in an amount sufficient that is sufficient to treat hyponatremia or the associated conditions without disturbing the patient's plasma glucose homeostasis, in particular without inducing hypoglycemia.
[0258] In the following preferred ranges of the amount of the SGLT2 inhibitor to be employed in the pharmaceutical composition and the methods and uses according to this invention are described. These ranges refer to the amounts to be administered per day with respect to an adult patient, in particular to a human being, for example of approximately 70 kg body weight, and can be adapted accordingly with regard to an administration 2, 3, 4 or more times daily and with regard to other routes of administration and with regard to the age of the patient.
[0259] Within the scope of the present invention, the pharmaceutical composition is preferably administered orally. Other forms of administration are possible and described hereinafter. Preferably the one or more dosage forms comprising the SGLT2 inhibitor is oral or usually well known.
[0260] In general, the amount of the SGLT2 inhibitor in the pharmaceutical composition and methods according to this invention is preferably the amount usually recommended for a monotherapy using said SGLT2 inhibitor.
[0261] The preferred dosage range of the SGLT2 inhibitor is in the range from 0.5 mg to 200 mg, even more preferably from 1 to 100 mg, most preferably from 1 to 50 mg per day. The oral administration is preferred. Therefore, a pharmaceutical composition may comprise the hereinbefore mentioned amounts, in particular from 1 to 50 mg or 1 to 25 mg. Particular dosage strengths (e.g. per tablet or capsule) are for example 1, 2.5, 5, 7.5, 10, 12.5, 15, 20, 25 or 50 mg of the SGLT2 inhibitor, such as a compound of the formula (I), in particular of the compound (1.9) or its crystalline form (I.9X). The application of the active ingredient may occur up to three times a day, preferably one or two times a day, most preferably once a day.
[0262] A pharmaceutical composition which is present as a separate or multiple dosage form, preferably as a kit of parts, is useful in combination therapy to flexibly suit the individual therapeutic needs of the patient.
[0263] According to a first embodiment a preferred kit of parts comprises a containment containing a dosage form comprising the SGLT2 inhibitor and at least one pharmaceutically acceptable carrier.
[0264] A further aspect of the present invention is a manufacture comprising the pharmaceutical composition being present as separate dosage forms according to the present invention and a label or package insert comprising instructions that the separate dosage forms are to be administered in combination or alternation.
[0265] According to a first embodiment a manufacture comprises (a) a pharmaceutical composition comprising a SGLT2 inhibitor according to the present invention and (b) a label or package insert which comprises instructions that the medicament is to be administered.
[0266] The desired dose of the pharmaceutical composition according to this invention may conveniently be presented in a once daily or as divided dose administered at appropriate intervals, for example as two, three or more doses per day.
[0267] The pharmaceutical composition may be formulated for oral, rectal, nasal, topical (including buccal and sublingual), transdermal, vaginal or parenteral (including intramuscular, sub-cutaneous and intravenous) administration in liquid or solid form or in a form suitable for administration by inhalation or insufflation. Oral administration is preferred. The formulations may, where appropriate, be conveniently presented in discrete dosage units and may be prepared by any of the methods well known in the art of pharmacy. All methods include the step of bringing into association the active ingredient with one or more pharmaceutically acceptable carriers, like liquid carriers or finely divided solid carriers or both, and then, if necessary, shaping the product into the desired formulation.
[0268] The pharmaceutical composition may be formulated in the form of tablets, granules, fine granules, powders, capsules, caplets, soft capsules, pills, oral solutions, syrups, dry syrups, chewable tablets, troches, effervescent tablets, drops, suspension, fast dissolving tablets, oral fast-dispersing tablets, etc..
[0269] The pharmaceutical composition and the dosage forms preferably comprises one or more pharmaceutical acceptable carriers which must be "acceptable" in the sense of being compatible with the other ingredients of the formulation and not deleterious to the recipient thereof. Examples of pharmaceutically acceptable carriers are known to the one skilled in the art.
[0270] Pharmaceutical compositions suitable for oral administration may conveniently be presented as discrete units such as capsules, including soft gelatin capsules, cachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution, a suspension or as an emulsion, for example as syrups, elixirs or self-emulsifying delivery systems (SEDDS). The active ingredients may also be presented as a bolus, electuary or paste. Tablets and capsules for oral administration may contain conventional excipients such as binding agents, fillers, lubricants, disintegrants, or wetting agents. The tablets may be coated according to methods well known in the art. Oral liquid preparations may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, or may be presented as a dry product for constitution with water or other suitable vehicle before use. Such liquid preparations may contain conventional additives such as suspending agents, emulsifying agents, non-aqueous vehicles (which may include edible oils), or preservatives.
[0271] The pharmaceutical composition according to the invention may also be formulated for parenteral administration (e.g. by injection, for example bolus injection or continuous infusion) and may be presented in unit dose form in ampoules, pre-filled syringes, small volume infusion or in multi-dose containers with an added preservative. The compositions may take such forms as suspensions, solutions, or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents. Alternatively, the active ingredients may be in powder form, obtained by aseptic isolation of sterile solid or by lyophilisation from solution, for constitution with a suitable vehicle, e.g. sterile, pyrogen-free water, before use.
[0272] Pharmaceutical compositions suitable for rectal administration wherein the carrier is a solid are most preferably presented as unit dose suppositories. Suitable carriers include cocoa butter and other materials commonly used in the art, and the suppositories may be conveniently formed by admixture of the active compound(s) with the softened or melted carrier(s) followed by chilling and shaping in moulds.
[0273] The pharmaceutical compositions and methods according to this invention show advantageous effects in the treatment and prevention of those diseases and conditions as described hereinbefore. Advantageous effects may be seen for example with respect to efficacy, dosage strength, dosage frequency, pharmacodynamic properties, pharmacokinetic properties, fewer adverse effects, convenience, compliance, etc..
[0274] Methods for the manufacture of SGLT2 inhibitors according to this invention and of prodrugs thereof are known to the one skilled in the art. Advantageously, the compounds according to this invention can be prepared using synthetic methods as described in the literature, including patent applications as cited hereinbefore. Preferred methods of manufacture are described in the WO 2006/120208 and WO 2007/031548. With regard to compound (I.9) an advantageous crystalline form is described in the international patent application WO 2006/117359 which hereby is incorporated herein in its entirety.
[0275] The active ingredients may be present in the form of a pharmaceutically acceptable salt. Pharmaceutically acceptable salts include, without being restricted thereto, such as salts of inorganic acid like hydrochloric acid, sulfuric acid and phosphoric acid; salts of organic carboxylic acid like oxalic acid, acetic acid, citric acid, malic acid, benzoic acid, maleic acid, fumaric acid, tartaric acid, succinic acid and glutamic acid and salts of organic sulfonic acid like methanesulfonic acid and p-toluenesulfonic acid. The salts can be formed by combining the compound and an acid in the appropriate amount and ratio in a solvent and decomposer. They can be also obtained by the cation or anion exchange from the form of other salts.
[0276] The active ingredients or a pharmaceutically acceptable salt thereof may be present in the form of a solvate such as a hydrate or alcohol adduct.
[0277] Any of the above mentioned pharmaceutical compositions and methods within the scope of the invention may be tested by animal models known in the art. In the following, in vivo experiments are described which are suitable to evaluate pharmacologically relevant properties of pharmaceutical compositions and methods according to this invention.
[0278] Pharmaceutical compositions and methods according to this invention can be tested in genetically hyperinsulinemic or diabetic animals like db/db mice, ob/ob mice, Zucker Fatty (fa/fa) rats or Zucker Diabetic Fatty (ZDF) rats. In addition, they can be tested in animals with experimentally induced diabetes like HanWistar or Sprague Dawley rats pretreated with streptozotocin.
[0279] The effect on glycemic control according to this invention can be tested after single dosing of the SGLT2 inhibitor in an oral glucose tolerance test in the animal models described hereinbefore. The time course of blood glucose is followed after an oral glucose challenge in overnight fasted animals. The pharmaceutical compositions according to the present invention significantly improve glucose excursion, for example compared to another monotherapy, as measured by reduction of peak glucose concentrations or reduction of glucose AUC. In addition, after multiple dosing of the SGLT2 inhibitor in the animal models described hereinbefore, the effect on glycemic control can be determined by measuring the HbA1c value in blood. The pharmaceutical compositions according to this invention significantly reduce HbA1c, for example compared to another monotherapy or compared to a dual-combination therapy.
[0280] The improved independence from insulin of the treatment according to this invention can be shown after single dosing in oral glucose tolerance tests in the animal models described hereinbefore. The time course of plasma insulin is followed after a glucose challenge in overnight fasted animals.
[0281] The increase in active GLP-1 levels by treatment according to this invention after single or multiple dosing can be determined by measuring those levels in the plasma of animal models described hereinbefore in either the fasting or postprandial state. Likewise, a reduction in glucagon levels in plasma can be measured under the same conditions.
[0282] The effect of a SGLT2 inhibitor according to the present invention on beta-cell regeneration and neogenesis can be determined after multiple dosing in the animal models described hereinbefore by measuring the increase in pancreatic insulin content, or by measuring increased beta-cell mass by morphometric analysis after immunhistochemical staining of pancreatic sections, or by measuring increased glucose-stimulated insulin secretion in isolated pancreatic islets.
PHARMACOLOGICAL EXAMPLES
[0283] The following examples show the beneficial effect on glycemic control of the pharmaceutical compositions according to the present invention.
Example 1
[0284] According to a first example an oral glucose tolerance test is performed in overnight fasted 9-weeks old male Zucker Diabetic Fatty (ZDF) rats (ZDF/Crl-Lepr.sup.fa). A pre-dose blood sample is obtained by tail bleed. Blood glucose is measured with a glucometer, and the animals are randomized for blood glucose (n=5/group). Subsequently, the groups receive a single oral administration of either vehicle alone (0.5% aqueous hydroxyethylcellulose containing 3 mM HCl and 0.015% Polysorbat 80) or vehicle containing the SGLT2 inhibitor. The animals receive an oral glucose load (2 g/kg) 30 min after compound administration. Blood glucose is measured in tail blood 30 min, 60 min, 90 min, 120 min, and 180 min after the glucose challenge. Glucose excursion is quantified by calculating the reactive glucose AUC. The data are presented as mean.+-.SEM. The two-sided unpaired Student t-test is used for statistical comparison of the control group and the active groups.
[0285] A representative experiment is shown in FIGS. 3A and 3B. Compound (1.9) (1-chloro-4-(.beta.-D-glucopyranos-1-yl)-2-[4-((S)-tetrahydrofuran-- 3-yloxy)-benzyl]-benzene) was orally administered to ZDF rats at doses of 0.3 mg/kg, 3 mg/kg or 30 mg/kg body weight. The animals then received an oral glucose bolus and the resulting glucose-time profile is shown in FIG. 3A. The baseline-corrected area under the glucose-time curves are shown in FIG. 3B. Compound (1.9) reduced glucose excursion by 15% at 0.3 mg/kg (not significant), by 62% at 3 mg/kg (p<0.001) and by 89% at 30 mg/kg (p<0.001).
Example 2
[0286] According to a second example an oral glucose tolerance test is performed in overnight fasted male Sprague Dawley rats (Crl:CD(SD)) with a body weight of about 200 g. A pre-dose blood sample is obtained by tail bleed. Blood glucose is measured with a glucometer, and the animals are randomized for blood glucose (n=5/group). Subsequently, the groups receive a single oral administration of either vehicle alone (0.5% aqueous hydroxyethylcellulose containing 0.015% Polysorbat 80) or vehicle containing the SGLT2 inhibitor. The animals receive an oral glucose load (2 g/kg) 30 min after compound administration. Blood glucose is measured in tail blood 30 min, 60 min, 90 min, and 120 min after the glucose challenge. Glucose excursion is quantified by calculating the reactive glucose AUC. The data are presented as mean.+-.S.E.M. Statistical comparisons are conducted by Student's t test.
Example 3
Treatment of Pre-Diabetes
[0287] The efficacy of a pharmaceutical composition according to the invention in the treatment of pre-diabetes characterised by pathological fasting glucose and/or impaired glucose tolerance can be tested using clinical studies. In studies over a shorter period (e.g. 2-4 weeks) the success of the treatment is examined by determining the fasting glucose values and/or the glucose values after a meal or after a loading test (oral glucose tolerance test or food tolerance test after a defined meal) after the end of the period of therapy for the study and comparing them with the values before the start of the study and/or with those of a placebo group. In addition, the fructosamine value can be determined before and after therapy and compared with the initial value and/or the placebo value. A significant drop in the fasting or non-fasting glucose levels demonstrates the efficacy of the treatment. In studies over a longer period (12 weeks or more) the success of the treatment is tested by determining the HbA1c value, by comparison with the initial value and/or with the value of the placebo group. A significant change in the HbA1c value compared with the initial value and/or the placebo value demonstrates the efficacy of the pharmaceutical composition according to the invention for treating pre-diabetes.
Example 4
Preventing Manifest Type 2 Diabetes
[0288] Treating patients with pathological fasting glucose and/or impaired glucose tolerance (pre-diabetes) is also in pursuit of the goal of preventing the transition to manifest type 2 diabetes. The efficacy of a treatment can be investigated in a comparative clinical study in which pre-diabetes patients are treated over a lengthy period (e.g. 1-5 years) with either a pharmaceutical composition according to this invention or with placebo or with a non-drug therapy or other medicaments. During and at the end of the therapy, by determining the fasting glucose and/or a loading test (e.g. oGTT), a check is made to determine how many patients exhibit manifest type 2 diabetes, i.e. a fasting glucose level of >125 mg/dl and/or a 2h value according to oGTT of >199 mg/dl. A significant reduction in the number of patients who exhibit manifest type 2 diabetes when treated with a pharmaceutical composition according to this invention as compared to one of the other forms of treatment, demonstrates the efficacy in preventing a transition from pre-diabetes to manifest diabetes.
Example 5
Treatment of Type 2 Diabetes
[0289] Treating patients with type 2 diabetes with the pharmaceutical composition according to the invention, in addition to producing an acute improvement in the glucose metabolic situation, prevents a deterioration in the metabolic situation in the long term. This can be observed is patients are treated for a longer period, e.g. 3 months to 1 year or even 1 to 6 years, with the pharmaceutical composition according to the invention and are compared with patients who have been treated with other antidiabetic medicaments. There is evidence of therapeutic success compared with patients treated with other antidiabetic medicaments if no or only a slight increase in the fasting glucose and/or HbA1c value is observed. Further evidence of therapeutic success is obtained if a significantly smaller percentage of the patients treated with a pharmaceutical composition according to the invention, compared with patients who have been treated with other medicaments, undergo a deterioration in the glucose metabolic position (e.g. an increase in the HbA1c value to >6.5% or >7%) to the point where treatment with an additional oral antidiabetic medicament or with insulin or with an insulin analogue is indicated.
Example 6
Treatment of Insulin Resistance
[0290] In clinical studies running for different lengths of time (e.g. 2 weeks to 12 months) the success of the treatment is checked using a hyperinsulinaemic euglycaemic glucose clamp study. A significant rise in the glucose infusion rate at the end of the study, compared with the initial value or compared with a placebo group, or a group given a different therapy, proves the efficacy of a pharmaceutical composition according to the invention in the treatment of insulin resistance.
Example 7
Treatment of Hyperglycaemia
[0291] In clinical studies running for different lengths of time (e.g. 1 day to 24 months) the success of the treatment in patients with hyperglycaemia is checked by determining the fasting glucose or non-fasting glucose (e.g. after a meal or a loading test with oGTT or a defined meal). A significant fall in these glucose values during or at the end of the study, compared with the initial value or compared with a placebo group, or a group given a different therapy, proves the efficacy of a pharmaceutical composition according to the invention in the treatment of hyperglycaemia.
Example 8
Prevention of Micro- or Macrovascular Complications
[0292] The treatment of type 2 diabetes or pre-diabetes patients with a pharmaceutical composition according to the invention prevents or reduces or reduces the risk of developing microvascular complications (e.g. diabetic neuropathy, diabetic retinopathy, diabetic nephropathy, diabetic foot, diabetic ulcer) or macrovascular complications (e.g. myocardial infarct, acute coronary syndrome, unstable angina pectoris, stable angina pectoris, stroke, peripheral arterial occlusive disease, cardiomyopathy, heart failure, heart rhythm disorders, vascular restenosis). Type 2 diabetes or patients with pre-diabetes are treated long-term, e.g. for 1-6 years, with a pharmaceutical composition according to the invention and compared with patients who have been treated with other antidiabetic medicaments or with placebo. Evidence of the therapeutic success compared with patients who have been treated with other antidiabetic medicaments or with placebo can be found in the smaller number of single or multiple complications. In the case of macrovascular events, diabetic foot and/or diabetic ulcer, the numbers are counted by anamnesis and various test methods. In the case of diabetic retinopathy the success of the treatment is determined by computer-controlled illumination and evaluation of the background to the eye or other ophthalmic methods. In the case of diabetic neuropathy, in addition to anamnesis and clinical examination, the nerve conduction rate can be measured using a calibrated tuning fork, for example. With regard to diabetic nephropathy the following parameters may be investigated before the start, during and at the end of the study: secretion of albumin, creatinin clearance, serum creatinin values, time taken for the serum creatinin values to double, time taken until dialysis becomes necessary.
Example 9
Treatment of Metabolic Syndrome
[0293] The efficacy of a pharmaceutical composition according to the invention can be tested in clinical studies with varying run times (e.g. 12 weeks to 6 years) by determining the fasting glucose or non-fasting glucose (e.g. after a meal or a loading test with oGTT or a defined meal) or the HbA1c value. A significant fall in these glucose values or HbA1c values during or at the end of the study, compared with the initial value or compared with a placebo group, or a group given a different therapy, proves the efficacy of an active substance in the treatment of Metabolic Syndrome. Examples of this are a reduction in systolic and/or diastolic blood pressure, a lowering of the plasma triglycerides, a reduction in total or LDL cholesterol, an increase in HDL cholesterol or a reduction in weight, either compared with the starting value at the beginning of the study or in comparison with a group of patients treated with placebo or a different therapy.
Example 10a
Prevention of NODAT and/or PTMS, and NODAT/PTMS Associated Complications
[0294] Treatment of patients after organ transplantation with the pharmaceutical composition according to the invention prevents the development of NODAT and/or PTMS, and associated complications. The efficacy of the treatment can be investigated in a comparative clinical study in which patients before or immediately after transplantation are treated over a lengthy period (e.g. 1-5 years) with either a pharmaceutical composition according to this intervention or with a placebo or with a non-drug therapy or other medicaments. During and at the end of the therapy, the incidence of NODAT, PTMS, micro- and macrovascular complications, graft rejection, infection and death will be assessed. A significant reduction in the number of patients experiencing these complications demonstrates the efficacy in preventing development of NODAT, PTMS, and associated complications.
Example 10b
Treatment of NODAT and/or PTMS with Prevention, Delay or Reduction of Associated Complications
[0295] Treatment of patients with NODAT and/or PTMS with the pharmaceutical composition according to the invention prevents, delays or reduces the development of NODAT/PTMS associated complications. The efficacy of the treatment can be investigated in a comparative clinical study in which patients with NODAT and/or PTMS are treated over a lengthy period (e.g. 1-5 years) with either a pharmaceutical composition according to this intervention or with a placebo or with a non-drug therapy or other medicaments. During and at the end of the therapy, the incidence of micro- and macrovascular complications, graft rejection, infection and death will be assessed. A significant reduction in the number of patients experiencing these complications demonstrates the efficacy in preventing, delaying or reducing the development of NODAT and/or PTMS associated complications.
Example 11a
Treatment of Gestational Diabetes
[0296] In clinical studies running for a shorter period (e.g. 2-4 weeks) the success of the treatment is checked by determining the fasting glucose values and/or the glucose values after a meal or after a loading test (oral glucose tolerance test or food tolerance test after a defined meal) at the end of the therapeutic period of the study and comparing them with the values before the start of the study and/or with those of a placebo group. In addition, the fructosamine value can be determined before and after treatment and compared with the initial value and/or a placebo value. A significant fall in the fasting or non-fasting glucose levels demonstrates the pharmaceutical compositon according to the invention.
[0297] In longer-running studies (12 weeks or more) the success of the treatment is checked by determining the HbA1c value (compared with initial value and placebo group). A significant change in the HbA1c value compared with the starting value and/or placebo value demonstrates the efficacy of the pharmaceutical composition according to the invention in the treatment of gestational diabetes.
Example 11b
Treatment of Women who have had Gestational Diabetes
[0298] Patients with gestational diabetes have a significantly increased risk of contracting manifest type 2 diabetes after the pregnancy. Therapy may be provided with the objective of preventing the transition to manifest type 2. For this purpose, women with a history of gestational diabetes are treated either with a pharmaceutical composition according to the invention or with placebo or with a non-drug therapy or with other medicaments, over a lengthy period (e.g. 1-4 years). During and at the end of the treatment a check is carried out by determining the fasting glucose and/or by a loading test (e.g. oGTT) to see how many patients have developed manifest type 2 diabetes (fasting glucose level>125 mg/dl and/or 2 h value after oGTT>199 mg/dl). A significant reduction in the number of patients who develop manifest type 2 diabetes when treated with a pharmaceutical composition according to the invention compared with a different type of therapy, is proof of the efficacy of a pharmaceutical composition in preventing manifest diabetes in women with a history of gestational diabetes.
Example 12
Treatment of Hyperuricemia
[0299] Patients with elevated levels of uric acid above the normal range (above 8.3 mg/dL or 494 pmol/L) or patients with a history of gout or gouty arthritis with a uric acid level greater than 6.0 mg/dL or 357 .mu.mol/L have a significant risk of future episodes of gout or gouty arthritis as well as having an increased risk of cardiovascular disease. Therapy may be provided with the objective of lowering serum levels of uric acid as a means of preventing future episodes or flare-ups of gout or gouty arthritis. Additionally, lowering serum uric acid levels may reduce the risk of cardiovascular disease. For this purpose patients with an elevated uric acid level or a history of gout or gouty arthritis are treated either with a pharmaceutical composition according to the invention or with placebo or with a non-drug therapy or with other medicaments, over a lengthy period (e.g. 6 months to 4 years). During and at the end of the treatment a check is carried out by determining the serum uric acid level and the number of episodes of gout or gouty arthritis occurences. A reduction in uric acid below 6.0 mg/dL and/or fewer episodes of gout or gouty arthritis occurrence when treated with a pharmaceutical composition according to the invention compared with a different type of therapy, is proof of the efficacy of a pharmaceutical composition in preventing episodic gout or gouty arthritis or treating hyperuricemia.
[0300] In a twelve week study of patients with manifest type 2 diabetes mellitus serum uric acid levels were measured at baseline and every 4 weeks in patients randomized to an administration of the compound (1.9) of 5 mg, 10 mg, or 25 mg or placebo or metformin 2000 mg, daily for 12 weeks. When compared to baseline, patients receiving all doses of the compound (1.9) had a reduction in their serum uric acid levels of 0.5 to 0.7 mg/dL when compared to baseline, while serum uric acid levels increased in patients randomized to either metformin or placebo.
TABLE-US-00007 Cpd. (I.9) Cpd. (I.9) Cpd. (I.9) Metformin 5 mg 10 mg 25 mg 2000 mg placebo baseline 5.5 mg/dL 5.3 mg/dL 5.4 mg/dL 5.6 mg/dL 5.4 mg/dL 12 weeks 5.0 mg/dL 4.7 mg/dL 4.7 mg/dL 6.2 mg/dL 5.6 mg/dL
Example 13
Treatment of Hyponatremia
[0301] Patients with hyponatremia and water intoxication whether due to an increase in water resorption or an increase in water intake, are at risk of central nervous system abnormalities and possibly death. Therapy may be provided with the objective of increasing the amount of free water to be excreted in the renal filtrate without disturbing sodium balance with the objective of increasing the overall sodium concentration of the interstitial fluids. For this purpose, patients with a history of hyponatremia are treated either with a pharmaceutical composition according to the invention or with placebo or with a non-drug therapy or with other medicaments, over a short period (e.g. 3 to 6 months), with periodic assessment of serum sodium levels. An increase in sodium levels into the normal range reported during this time period when treated with a pharmaceutical composition according to the invention compared with a different type of therapy, is proof of the efficacy of a pharmaceutical composition in treating hyponatremia.
Example 14
Treatment/Prevention of Kidney Stones
[0302] Patients with a history of kidney stones, particularly calcium, mixed calcium, and uric acid stones frequently have a history of hyperuricemia. These renal stones may relate to small urate crystals forming a nidus in the renal filtrate upon which further crystalization of urate or other crystalizing substances in the solute can induce renal stone formation. These stones are not related to renal stones caused by certain kidney infections (such as staghorn--type stones). Therapy may be provided with the objective of increasing the neutral solutes (for example glucose) and free water content of the renal filtrate, making it difficult for a urate nidus to form, despite a possible increase in the absolute amounts of urate in the renal filtrate. These neutral solutes and free water will also reduce the formation of stones other than uric acid stones. For this purpose patients with a history of kidney stones particularly calcium, mixed calcium, and uric acid stones are treated either with a pharmaceutical composition according to the invention or with placebo or with a non-drug therapy or with other medicaments, over a lengthy period (e.g. 6 months to 4 years). A reduction in the number of kidney stones stones particularly calcium, mixed calcium, and uric acid stones reported during this time period when treated with a pharmaceutical composition according to the invention compared with a different type of therapy, is proof of the efficacy of a pharmaceutical composition in preventing kidney stones particularly calcium, mixed calcium, and uric acid stones.
Example 15
Body Weight and Body Fat Reduction
[0303] The following example shows the beneficial effect of the compound (I.9) on body weight and total body fat content. All experimental procedures concerning the use of laboratory animals were carried out under a Home Office Certificate of Designation. An animal model of obesity was used to study the effect of the compound (I.9) on body weight and total body fat content. For this, female Wistar rats were made obese by exposure to a simplified cafeteria diet containing high fat chow, chocolate and ground peanuts for approximately 24 weeks. Following the induction of obesity, rats were given vehicle (0.5% aqueous hydroxyethyl-cellulose) for 7 days and then dosed orally once daily with either vehicle or 3 mg/kg or 10 mg/kg compound (I.9) for 28 days. For the duration of the study rats were maintained on the cafeteria diet. Body weight was monitored daily and the final body weight after 28 day treatment is given in the FIG. 4A. Therein "Cpd A" denotes the glucopyranosyl-substituted benzene derivative (I.9) at a dose of 3 mg/kg or 10 mg/kg. Results are means (adjusted for differences between the body weights of the different treatment groups at baseline (Day 1)).+-.SEM (calculated from the residuals of the statistical model), n=10. After 28 day daily oral treatment with the compound (I.9) a reduced body weight compared to the vehicle-treated control group was observed. Body weight data was analysed by analysis of covariance with body weights on Day 1 as covariate. P values versus vehicle control are indicated by symbols above the bars (*, p<0.05) Multiple comparisons against the vehicle control group were performed by Williams' test for the two freely-feeding "Cpd A" groups. The glucopyranosyl-substituted benzene derivative (1.9) reduced the body weight by 4.1% at 3 mg/kg and significantly by 6.9% at 10 mg/kg.
[0304] At the end of the study on Day 34 (24 hours after the last treatment on Day 33) all rats were terminated, the body exsanguinated and the following tissues removed: the caudate liver lobe, the pancreas, the left kidney and one soleus muscle. Body composition (body fat, protein and water) was determined using the FoodScan NIR (near infra-red) meat analyser (Foss UK). This machine has AOAC (Association of Official Analytical Chemists) approval as reference method for the analysis of moisture, fat and protein in meat. The carcasses were milled under liquid nitrogen and a portion of the milled carcass was analysed in the FoodScan Analyser. The results of the determination of body fat content are given in FIG. 4B. Therein "Cpd A" denotes the glucopyranosyl-substituted benzene derivative (I.9) at a dose of 3 mg/kg or 10 mg/kg. After 33 day daily oral treatment with the compound (I.9) a reduced body fat content compared to the control group was observed. Means (n=9-10) are adjusted for differences between treatment groups in body weight at baseline (Day 1). Carcasses from all animals were analysed less terminal bleeds (exsanguination), pancreas and caudate lobe of the liver whilst 4-5 animals per group additionally had the left kidney and one soleus muscle removed. Statistical analysis was by robust regression and included Day 1 body weight as a covariate. Standard errors of the mean (SEM) are calculated from the residuals of the statistical model. Comparisons against the vehicle-treated control group on the cafeteria diet were by Williams' tests for the freely feeding "Cpd A" animals (3 mg/kg and 10 mg/kg). Significant differences are denoted by *p<0.05. The total body fat content (expressed as weight per rat) was significantly lower after treatment with 10 mg/kg of the compound (1.9) in comparison to vehicle-treated animals.
Examples of Formulations
[0305] The following examples of formulations, which may be obtained analogously to methods known in the art, serve to illustrate the present invention more fully without restricting it to the contents of these examples. The term "active substance" denotes a SGLT-2 inhibitor according to this invention, especially a compound of the formula (I), for example a compound of the formula (I.9) or its crystalline form (I.9X).
[0306] The active pharmaceutical ingredient or active sustance, i.e. the compound (I.9), preferably in the crystalline form (I9.X), is milled with a suitable mill like pin- or jet-mill in order to obtain the desired particle size distribution before manufacturing of the pharmaceutical composition or dosage form.
[0307] Examples of typical particle size distribution values X90, X50 and X10 for the preferred active pharmaceutical ingredient according to the invention are shown in the table below.
[0308] Typical particle size distribution results
TABLE-US-00008 Active Active substance substance Batch 1 Batch 2 X10 1.8 .mu.m 1.7 .mu.m X50 18.9 .mu.m 12.1 .mu.m X90 45.3 .mu.m 25.9 .mu.m
Example 1
Dry Ampoule Containing 50 mg of Active Substance Per 10 ml Composition
TABLE-US-00009 [0309] Active substance 50.0 mg Mannitol 50.0 mg water for injections ad 10.0 ml
[0310] Preparation:
[0311] Active substance and mannitol are dissolved in water. After packaging the solution is freeze-dried. To produce the solution ready for use, the product is dissolved in water for injections.
Example 2
Dry Ampoule Containing 25 mg of Active Substance Per 2 ml Composition
TABLE-US-00010 [0312] Active substance 25.0 mg Mannitol 100.0 mg water for injections ad 2.0 ml
[0313] Preparation:
[0314] Active substance and mannitol are dissolved in water. After packaging, the solution is freeze-dried. To produce the solution ready for use, the product is dissolved in water for injections.
Example 3
Tablet Containing 50 mg of Active Substance Composition
TABLE-US-00011 [0315] (1) Active substance 50.0 mg (2) Mannitol 98.0 mg (3) Maize starch 50.0 mg (4) Polyvinylpyrrolidone 15.0 mg (5) Magnesium stearate 2.0 mg 215.0 mg
[0316] Preparation:
[0317] (1), (2) and (3) are mixed together and granulated with an aqueous solution of (4). (5) is added to the dried granulated material. From this mixture tablets are pressed, biplanar, faceted on both sides and with a dividing notch on one side.
[0318] Diameter of the tablets: 9 mm.
Example 4
Capsules Containing 50 mg of Active Substance Composition
TABLE-US-00012 [0319] (1) Active substance 50.0 mg (2) Dried maize starch 58.0 mg (3) Mannitol 50.0 mg (4) Magnesium stearate 2.0 mg 160.0 mg
[0320] Preparation:
[0321] (1) is triturated with (3). This trituration is added to the mixture of (2) and (4) with vigorous mixing. This powder mixture is packed into size 3 hard gelatin capsules in a capsule filling machine.
Example 5
Tablets Containing 2.5mg, 5mg, 10mg, 25mg, 50mg of Active Substance
TABLE-US-00013 [0322] 2.5 mg 5 mg 10 mg 25 mg 50 mg Mg/per Mg/per Mg/per Mg/per Mg/per Active substance tablet tablet tablet tablet tablet Wet granulation active substance 2.5000 5.000 10.00 25.00 50.00 Lactose 40.6250 81.250 162.50 113.00 226.00 Monohydrate Microcrystalline 12.5000 25.000 50.00 40.00 80.00 Cellulose Hydroxypropyl 1.8750 3.750 7.50 6.00 12.00 Cellulose Croscarmellose 1.2500 2.500 5.00 4.00 8.00 Sodium Purified Water q.s. q.s. q.s. q.s. q.s. Dry Adds Microcrystalline 3.1250 6.250 12.50 10.00 20.00 Cellulose Colloidal silicon 0.3125 0.625 1.25 1.00 2.00 dioxide Magnesium stearate 0.3125 0.625 1.25 1.00 2.00 Total core 62.5000 125.000 250.00 200.00 400.00 Film Coating Film coating system 2.5000 4.000 7.00 6.00 9.00 Purified Water q.s. q.s. q.s. q.s. q.s. Total 65.000 129.000 257.00 206.00 409.00
Example 6
[0323] Manufacturing Process for Tablets
Example 7
Pharmaceutical Composition Containing Other Fillers
[0324] Copovidone is dissolved in purified water at ambient temperature to produce a granulation liquid. A glucopyranosyl-substituted benzene derivative according to the present invention, mannitol, pregelatinized starch and corn starch are blended in a suitable mixer, to produce a pre-mix. The pre-mix is moistened with the granulation liquid and subsequently granulated. The moist granulate is sieved through a suitable sieve. The granulate is dried at about 60.degree. C. inlet air temperature in a fluid bed dryer until a loss on drying value of 1-4% is obtained. The dried granulate is sieved through a sieve with a mesh size of 1.0 mm.
[0325] Magnesium stearate is passed through a sieve for delumping and added to the granulate. Subsequently the final blend is produced by final blending in a suitable blender for three minutes and compressed into tablet cores.
[0326] Hydroxypropyl methylcellulose, polyethylene glycol, talc, titanium dioxide and iron oxide are suspended in purified water in a suitable mixer at ambient temperature to produce a coating suspension. The tablet cores are coated with the coating suspension to a weight gain of about 3% to produce film-coated tablets. The following formulation variants can be obtained:
TABLE-US-00014 mg/ mg/ mg/ mg/ mg/ Ingredient tablet tablet tablet tablet tablet Active substance 2.5 5.0 10.0 25.0 50.0 Mannitol 133.4 130.9 125.9 110.9 221.8 Pregelatinised starch 18.0 18.0 18.0 18.0 36.0 Maize starch 18.0 18.0 18.0 18.0 36.0 Copovidone 5.4 5.4 5.4 5.4 10.8 Magnesium stearate 2.7 2.7 2.7 2.7 5.4 Film coat 5.0 5.0 5.0 5.0 10.0 Total 185.0 185.0 185.0 185.0 370.0
Example 8
Pharmaceutical Composition Containg Other Disintegrant
[0327] Copovidone is dissolved in purified water at ambient temperature to produce a granulation liquid. An glucopyranosyl-substituted benzene derivative according to the present invention, mannitol, pregelatinized starch and corn starch are blended in a suitable mixer, to produce a pre-mix. The pre-mix is moistened with the granulation liquid and subsequently granulated. The moist granulate is sieved through a suitable sieve. The granulate is dried at about 60.degree. C. inlet air temperature in a fluid bed dryer until a loss on drying value of 1-4% is obtained. The dried granulate is sieved through a sieve with a mesh size of 1.0 mm.
[0328] Crospovidone is added to the dried granulate and mixed for 5 minutes to produce the main blend. Magnesium stearate is passed through a sieve for delumping and added to main blend. Subsequently the final blend is produced by final blending in a suitable blender for three minutes and compressed into 8 mm round tablet cores with a compression force of 16 kN.
[0329] Hydroxypropyl methylcellulose, polyethylene glycol, talc, titanium dioxide and iron oxide are suspended in purified water in a suitable mixer at ambient temperature to produce a coating suspension. The tablet cores are coated with the coating suspension to a weight gain of about 3% to produce film-coated tablets. The following formulation variants can be obtained:
TABLE-US-00015 mg/ mg/ mg/ mg/ mg/ Ingredient tablet tablet tablet tablet tablet Active substance 2.5 5.0 10.0 25.0 50.0 Mannitol 127.5 125.0 120.0 105.0 210.0 Microcrystalline Cellulose 39.0 39.0 39.0 39.0 78.0 Crospovidone 2.0 2.0 2.0 2.0 4.0 Copovidone 5.4 5.4 5.4 5.4 10.8 Magnesium stearate 3.6 3.6 3.6 3.6 7.2 Film coat 5.0 5.0 5.0 5.0 10.0 Total 185.0 185.0 185.0 185.0 370.0
[0330] The tablet hardness, the friability, the content uniformity, the disintegration time and the dissolution properties are determined as described hereinbefore.
Example 9
Direct Compression Formulation
[0331] 1. Screen the active ingredient, microcrystalline cellulose, croscarmellose.sodium and either hydroxypropyl cellulose or polyethylene glycol powder through a 20 mesh hand screen.
[0332] 2. Add the above items into the high shear mixer and mix for two minutes.
[0333] 3. Make a premix (.about.1/1) of the lactose and colloidal silicon dioxide.
[0334] 4. Screen the premix through a 20 mesh hand screen and add to the mixer.
[0335] 5. Screen the remaining lactose through a 20 mesh hand screen and add to the mixer.
[0336] 6. Mix in components in the mixer for 2 minutes.
[0337] 7. Screen the magnesium stearate through a 30 mesh hand screen and add to the mixer.
[0338] 8. Mix for 1 minute 30 seconds to obtain the final blend.
[0339] 9 Tabletting of the final blend on a suitable tabletting press.
[0340] 10. Optionally film coating of the tablet cores.
TABLE-US-00016 mg/ mg/ mg/ mg/ mg/ Ingredient tablet tablet tablet tablet tablet Active substance 2.5000 5.000 10.00 25.0 50.0 Lactose Monohydrate 43.7500 87.500 175.00 74.0 148.0 Microcrystalline 12.5000 25.000 50.00 80.0 160.0 Cellulose Polyethylene glycol -- -- -- 10.0 20.0 Croscarmellose sodium 1.2500 2.500 5.00 8.0 16.0 Hydroxypropyl cellulose 1.8750 3.750 7.50 -- -- Colloidal Silicon dioxide 0.3125 0.625 1.25 1.0 2.0 Magnesium stearate 0.3125 0.625 1.25 2.0 4.0 Film coat 2.5000 4.000 7.00 6.00 9.00 Purified water q.s. q.s. q.s. q.s. q.s. Total 65.000 129.000 257.00 206.00 409.00
Example 10
Tablets Containing 0.5mg, 5mg, 25mg, 100mg of Active Substance
TABLE-US-00017 [0341] 100 mg 0.5 mg 5 mg 25 mg mg/per Active substance mg/per tablet mg/per tablet mg/per tablet tablet Wet granulation active substance 2.5000 5.000 25.00 100.00 Lactose 60.00 55.00 42.00 168.00 Monohydrate Microcrystalline 20.00 20.00 38.00 152.00 Cellulose Hydroxypropyl 5.00 5.00 7.50 30.00 Cellulose Croscarmellose 4.00 4.00 6.00 24.00 Sodium Purified Water q.s. q.s. q.s. q.s. Dry Adds Microcrystalline 10.00 10.00 30.00 120.00 Cellulose Colloidal silicon -- 0.50 0.75 3.00 dioxide Magnesium stearate 0.50 0.50 0.75 3.00 Total 100.00 100.00 150.00 600.00
[0342] The active substance, e.g. the compound (I.9), preferably in the crystalline form (I.9X), hydroxypropyl cellulose, and croscarmellose sodium are mixed in a blender. This premix is mixed with lactose monohydrate and a portion of microcrystalline cellulose. The resulting blend is granulated with purified water. Multiple granulation subparts may be produced for an individual tablet batch, as needed, depending on the batch size and equipment used. The granulation is discharged onto dryer trays and dried. The granulation is then milled. The remainder of the microcrystalline cellulose is added (as a premix with the colloidal silicon dioxide for all strengths other than the 0.5 mg) to the milled granulation, and mixed. The magnesium stearate is premixed with a portion of the blend, screened into the remainder of the granulation, and mixed.
[0343] The final tablet blend is compressed into tablets using a tablet press. The finished tablets are packaged using a suitable container closure system.
Example 11
Tablets Containing 1 mg, 5mg, 25mg of Active Substance
TABLE-US-00018 [0344] 1 mg 5 mg 25 mg Active substance mg/per tablet mg/per tablet mg/per tablet Wet granulation active substance 1.00 5.00 25.00 Lactose 63.00 59.00 39.00 Monohydrate Microcrystalline 20.00 20.00 20.00 Cellulose Hydroxypropyl 3.00 3.00 3.00 Cellulose Croscarmellose 2.00 2.00 2.00 Sodium Purified Water q.s. q.s. q.s. Dry Adds Microcrystalline 10.00 10.00 10.00 Cellulose Colloidal silicon 0.50 0.50 0.50 dioxide Magnesium stearate 0.50 0.50 0.50 Total 100.00 100.00 100.00
[0345] The active substance, e.g. the compound (I.9), preferably in the crystalline form (I.9X), is passed through a screen and added to a blender or a high shear granulator. The hydroxypropyl cellulose and croscarmellose
D00001
D00002
D00003
P00001
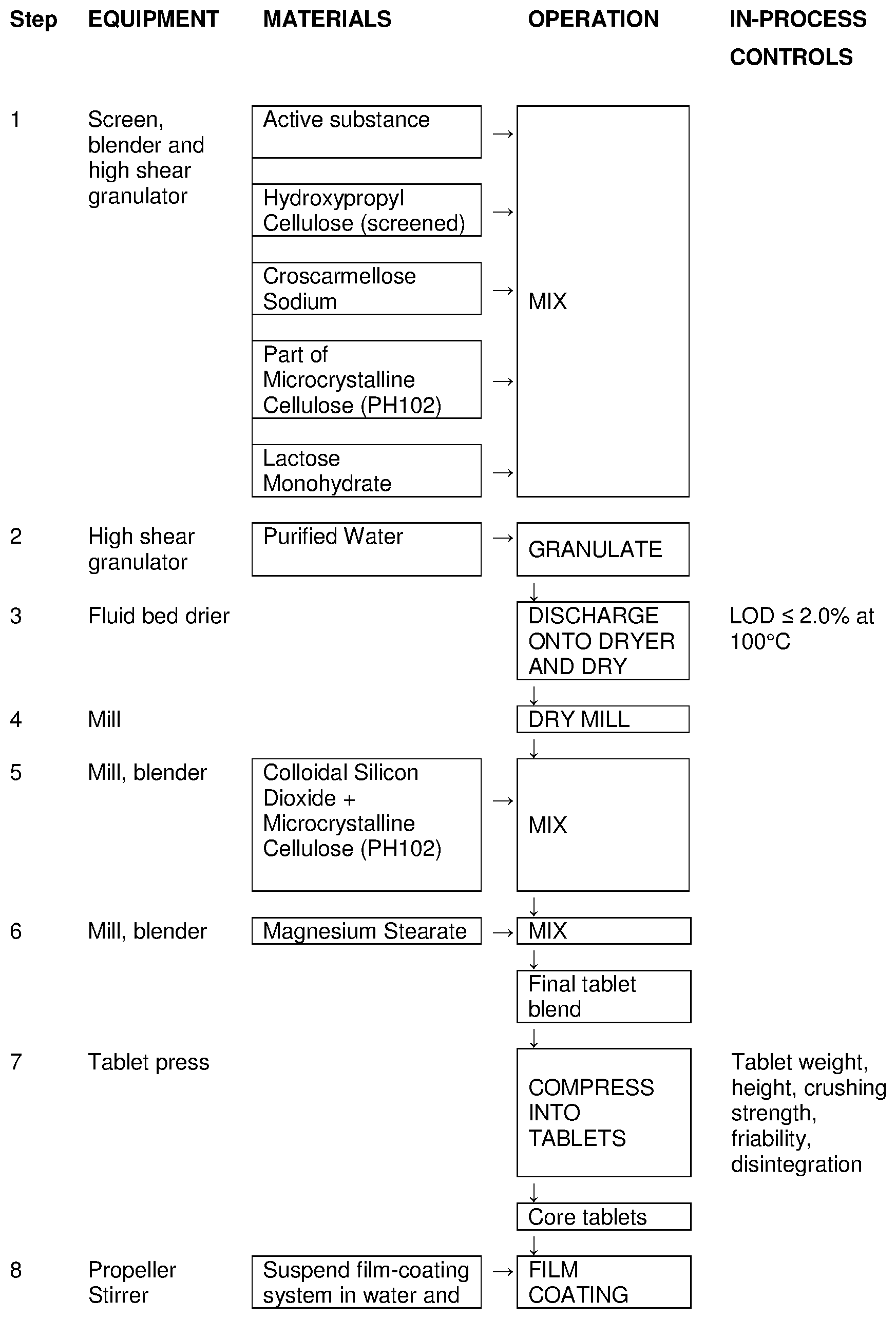
P00002
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.