Method And System For Selective Access Of Stored Or Transmitted Bioinformatics Data
Baluch; Mohamed Khoso ; et al.
U.S. patent application number 16/341426 was filed with the patent office on 2020-02-06 for method and system for selective access of stored or transmitted bioinformatics data. This patent application is currently assigned to GENOMSYS SA. The applicant listed for this patent is GENOMSYS SA. Invention is credited to Mohamed Khoso Baluch, Daniele Renzi, Giorgio Zoia.
| Application Number | 20200042735 16/341426 |
| Document ID | / |
| Family ID | 61905752 |
| Filed Date | 2020-02-06 |



View All Diagrams
| United States Patent Application | 20200042735 |
| Kind Code | A1 |
| Baluch; Mohamed Khoso ; et al. | February 6, 2020 |
METHOD AND SYSTEM FOR SELECTIVE ACCESS OF STORED OR TRANSMITTED BIOINFORMATICS DATA
Abstract
The storage or transmission of genomic data is realized by employing a structured compressed genomic dataset in a file or in a stream of genomic data. Selective access to the data, or subsets of the data, corresponding to specific genomic regions is achieved by employing user-defined labels based on data classification and a specific indexing mechanism.
| Inventors: | Baluch; Mohamed Khoso; (Chantilly, VA) ; Zoia; Giorgio; (Lausanne, CH) ; Renzi; Daniele; (Lausanne, CH) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Assignee: | GENOMSYS SA Lausanne CH |
||||||||||
| Family ID: | 61905752 | ||||||||||
| Appl. No.: | 16/341426 | ||||||||||
| Filed: | February 14, 2017 | ||||||||||
| PCT Filed: | February 14, 2017 | ||||||||||
| PCT NO: | PCT/US2017/017841 | ||||||||||
| 371 Date: | April 11, 2019 |
| Current U.S. Class: | 1/1 |
| Current CPC Class: | G16B 30/20 20190201; G16B 45/00 20190201; H03M 7/3086 20130101; G16B 20/10 20190201; G16B 50/00 20190201; G16B 30/10 20190201; G06F 21/602 20130101; G06F 16/285 20190101; G16B 50/30 20190201; G06F 16/2282 20190101; G06F 16/2365 20190101; G16B 30/00 20190201; G06F 7/00 20130101; G16B 50/40 20190201; G16B 40/10 20190201; G16B 99/00 20190201; G06F 3/048 20130101; G16B 20/20 20190201; G16B 50/50 20190201; H03M 7/70 20130101; G06F 21/6245 20130101; G06F 21/6218 20130101; G16B 40/00 20190201; G16B 50/10 20190201 |
| International Class: | G06F 21/62 20060101 G06F021/62; G16B 30/10 20060101 G16B030/10; G16B 50/40 20060101 G16B050/40; G16B 50/30 20060101 G16B050/30; G16B 20/20 20060101 G16B020/20; G06F 16/28 20060101 G06F016/28; G06F 16/22 20060101 G06F016/22; G06F 21/60 20060101 G06F021/60 |
Foreign Application Data
| Date | Code | Application Number |
|---|---|---|
| Oct 11, 2016 | EP | PCT/EP2016/074297 |
| Oct 11, 2016 | EP | PCT/EP2016/074301 |
| Oct 11, 2016 | EP | PCT/EP2016/074307 |
| Oct 11, 2016 | EP | PCT/EP2016/074311 |
Claims
1. A method for selective access of regions of genomic data by employing labels, said labels comprising: an identifier of a reference genomic sequence, an identifier of said genomic regions, and an identifier of the data class of said genomic data, wherein said genomic data are sequences of genomic reads, and wherein said data classes can be of the following type or a subset of them: "Class P" comprising genomic reads which do not present any mismatch with respect to a reference sequence, "Class N" comprising genomic reads including only mismatches in positions where the sequencing machine was not able to call any "base" and the number of said mismatches does not exceed a given threshold, "Class M" comprising genomic reads in which mismatches are constituted by positions where the sequencing machine was not able to call any base, named "n type" mismatches, and/or it called a different base than the reference sequence, named "s type" mismatches, and said numbers of mismatches do not exceed given thresholds for the number of mismatches of "n type", of "s type" and a threshold obtained from a given function (f(n,s)), "Class I" when the genomic reads can possibly have the same type of mismatches of "Class M", and in addition at least one mismatch of type: "insertion" ("i type"), "deletion" ("d type"), soft clips ("c type"), and wherein the numbers of mismatches for each type does not exceed the corresponding given thresholds and a threshold provided by a given function (w(n,s,i,d,c)), "Class U" comprising all reads that do not find any classification in the classes P, N, M, I.
2. (canceled)
3. (canceled)
4. The method of claim 1, further comprising the case of said genomic data being paired sequences of genomic reads.
5. The method of claim 4 wherein said data class of paired reads can be of the following types or a subset of them: "Class P" comprising genomic read pairs which do not present any mismatch with respect to a reference sequence, "Class N" comprising genomic reads pairs including only mismatches in positions where the sequencing machine was not able to call any "base" and said numbers of mismatches for each read do not exceed a given threshold, "Class M" comprising genomic read pairs including only mismatches in positions where the sequencing machine was not able to call any "base" and said numbers of mismatches for each read do not exceed a given threshold, named "n type" mismatches, and/or it called a different base than the reference sequence, named "s type" mismatches, and said numbers of mismatches does not exceed a given thresholds for the number of mismatches of "n type", of "s type" and a threshold obtained from a given function (f(n,s)), "Class I" comprising read pairs which can possibly have the same type of mismatches of "Class M" pairs, and in addition at least one mismatch of type: "insertion" ("i type") "deletion" ("d type") soft clips ("c type"), and wherein the number of mismatches for each type does not exceed the corresponding given threshold and a threshold provided by a given function (w(n,s,i,d,c)), "Class HM" comprising read pairs for which only one read mate does not satisfy the matching rules for being classified in any of the classes P, N, M, I, Class "U" comprising all reads pairs for which both reads do not satisfy the matching rules for being classified in the classes P, N, M, I.
6. The method of claim 1, wherein said identifier of said genomic regions is comprised in a master index table.
7. The method of claim 6 wherein said genomic data and said labels are entropy coded.
8. The method of claim 7 wherein said master index table is comprised in a genomic dataset header.
9. The method of claim 8, wherein said regions of genomic data are dispersed among separate Access Units.
10. The method of claim 9 wherein the location of said regions of genomic data, in a file, is indicated in a local index table.
11. The method of claim 1, wherein said labels are user specified.
12. The method of claim 1, wherein said regions are protected and/or encrypted in a separate manner, without encrypting the whole genomic file.
13. The method of claim 1, wherein said labels are stored in a genomic label list (GLL).
14. A method for encoding genomic data with selective access to regions of genomic data as claimed in claim 1.
15. The method of claim 13, wherein said genomic label list is periodically retransmitted or updated in order to enable multiple synchronization points.
16. A method for decoding a stream or a file of genomic data with selective access to regions of genomic data as claimed in claim 1.
17. An apparatus for encoding genomic data as claimed in claim 14.
18. An apparatus for decoding genomic data as claimed in claim 16.
19. Storing means for storing genomic data encoded according to claim 14.
20. A computer-readable medium comprising instructions that when executed cause at least one processor to perform the encoding method of claim 14.
21. A computer-readable medium comprising instructions that when executed cause at least one processor to perform the decoding method of claim 16.
Description
TECHNICAL FIELD
[0001] The present application provides new methods for the efficient storage, transmission and multiplexing of bioinformatics data, and in particular genomic sequencing data, in compressed form that enable efficient selective access and selective protection of the different data categories composing the genomic datasets.
BACKGROUND
[0002] An appropriate representation of genome sequencing data is fundamental to enable efficient processing, storage and transmission of genomic data to make possible and facilitate analysis applications such as genome variants calling and all analysis performed, with various purposes, by processing the sequencing data and metadata. Today, genome sequencing information is generated by High Throughput Sequencing (HTS) machines in the form of sequences of nucleotides (a. k. a. bases) represented by strings of letters from a defined vocabulary.
[0003] These sequencing machines do not read out an entire genomes or genes, but they produce short random fragments of nucleotide sequences known as sequence reads. A quality score is associated to each nucleotide in a sequence read. Such number represents the confidence level given by the machine to the read of a specific nucleotide at a specific location in the nucleotide sequence. This raw sequencing data generated by NGS machines are commonly stored in FASTQ files (see also FIG. 1).
[0004] The smallest vocabulary to represent sequences of nucleotides obtained by a sequencing process is composed by five symbols: {A, C, G, T, N} representing the four types of nucleotides present in DNA namely Adenine, Cytosine, Guanine, and Thymine plus the symbol N to indicate that the sequencing machine was not able to call any base with a sufficient level of confidence, so the type of base in such position remains undetermined in the reading process. In RNA Thymine is replaced by Uracil (U). The nucleotides sequences produced by sequencing machines are called "reads". In case of paired reads the term "template" is used to designate the original sequence from which the read pair has been extracted. Sequence reads can be composed by a number of nucleotides in a range from a few dozen up to several thousand. Some technologies produce sequence reads in pairs where each read can be originated from one of the two DNA strands.
[0005] In the genome sequencing field the term "coverage" is used to express the level of redundancy of the sequence data with respect to a reference genome. For example, to reach a coverage of 30.times. on a human genome (3.2 billion bases long) a sequencing machine shall produce a total of about 30.times.3.2 billion bases so that in average each position in the reference is "covered" 30 times.
State of the Art Solutions
[0006] The most used genome information representations of sequencing data are based on FASTQ and SAM file formats which are commonly made available in zipped form in the attempt of reducing the original size. The traditional file formats, respectively FASTQ and SAM for non-aligned and aligned sequencing data, are constituted by plain text characters and are thus compressed by using general purpose approaches such as LZ (from Lempel and Ziv) schemes (the well-known zip, gzip etc). When general purpose compressors such as gzip are used, the result of the compression is usually a single blob of binary data. The information in such monolithic form results quite difficult to archive, transfer and elaborate particularly in the case of high throughput sequencing when the volumes of data are extremely large.
[0007] After sequencing, each stage of a genomic information processing pipeline produces data represented by a completely new data structure (file format) despite the fact that in reality only a small fraction of the generated data is new with respect to the previous stage.
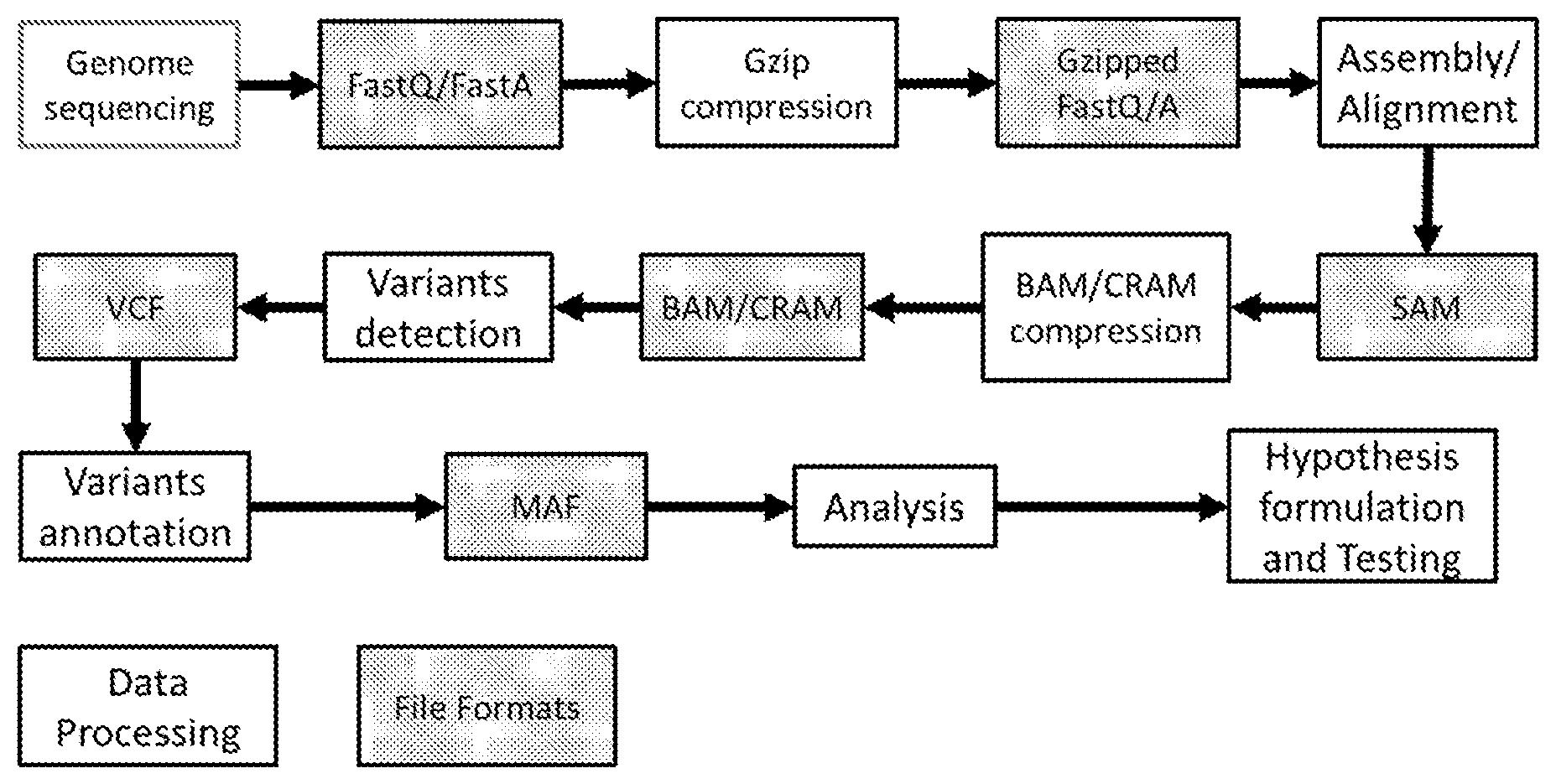
[0008] FIG. 1 shows the main stages of a typical genomic information processing pipeline with the indication of the associated file format representation.
[0009] Commonly used solutions presents several drawbacks: data archival is inefficient for the fact that a different file format is used at each stage of the genomic information processing pipelines which implies the multiple replication of data, with the consequent rapid increase of the required storage space. This is inefficient and unnecessary and it is also becoming not sustainable for the increase of the data volume generated by HTS machines. This has in fact consequences in terms of available storage space and generated costs, and it is also hindering the benefits of genomic analysis in healthcare from reaching a larger portion of the population. The impact of the IT costs generated by the exponential growth of sequence data to be stored and analysed is currently one of the main challenges the scientific community and that the healthcare industry have to face (see Scott D. Kahn "On the future of genomic data"--Science 331, 728 (2011) and Pavlichin, D. S., Weissman, T., and G. Yona. 2013. "The human genome contracts again" Bioinformatics 29(17): 2199-2202). At the same time several are the initiatives attempting to scale genome sequencing from a few selected individuals to large populations (see Josh P. Roberts "Million Veterans Sequenced"--Nature Biotechnology 31, 470 (2013))
[0010] The transfer of genomic data is slow and inefficient because the currently used data formats are organized into monolithic files of up to several hundred Gigabytes of size which need to be entirely transferred at the receiving end in order to be processed. This implies that the analysis of a small segment of the data requires the transfer of the entire file with significant costs in terms of consumed bandwidth and waiting time. Often online transfer is prohibitive for the large volumes of the data to be transferred, and the transport of the data is performed by physically moving storage media such as hard disk drives or storage servers from one location to another.
[0011] These limitations occurring when employing state of the art approaches are overcome by the present invention.
[0012] Processing the data is slow and inefficient for to the fact that the information is not structured in such a way that the portions of the different classes of data and metadata required by commonly used analysis applications cannot be retrieved without the need of accessing the data in its totality. This fact implies that common analysis pipelines can require to run for days or weeks wasting precious and costly processing resources because of the need, at each stage of accessing, of parsing and filtering large volumes of data even if the portions of data relevant for the specific analysis purpose is much smaller.
[0013] These limitations are preventing health care professionals from timely obtaining genomic analysis reports and promptly reacting to diseases outbreaks. The present invention provides a solution to this need.
[0014] There is another technical limitation that is overcome by the present invention.
[0015] In fact the invention aims at providing an appropriate genomic sequencing data and metadata representation by organizing and partitioning the data so that the compression of data and metadata is maximized and several functionality such as selective access and support for incremental updates are efficiently enabled.
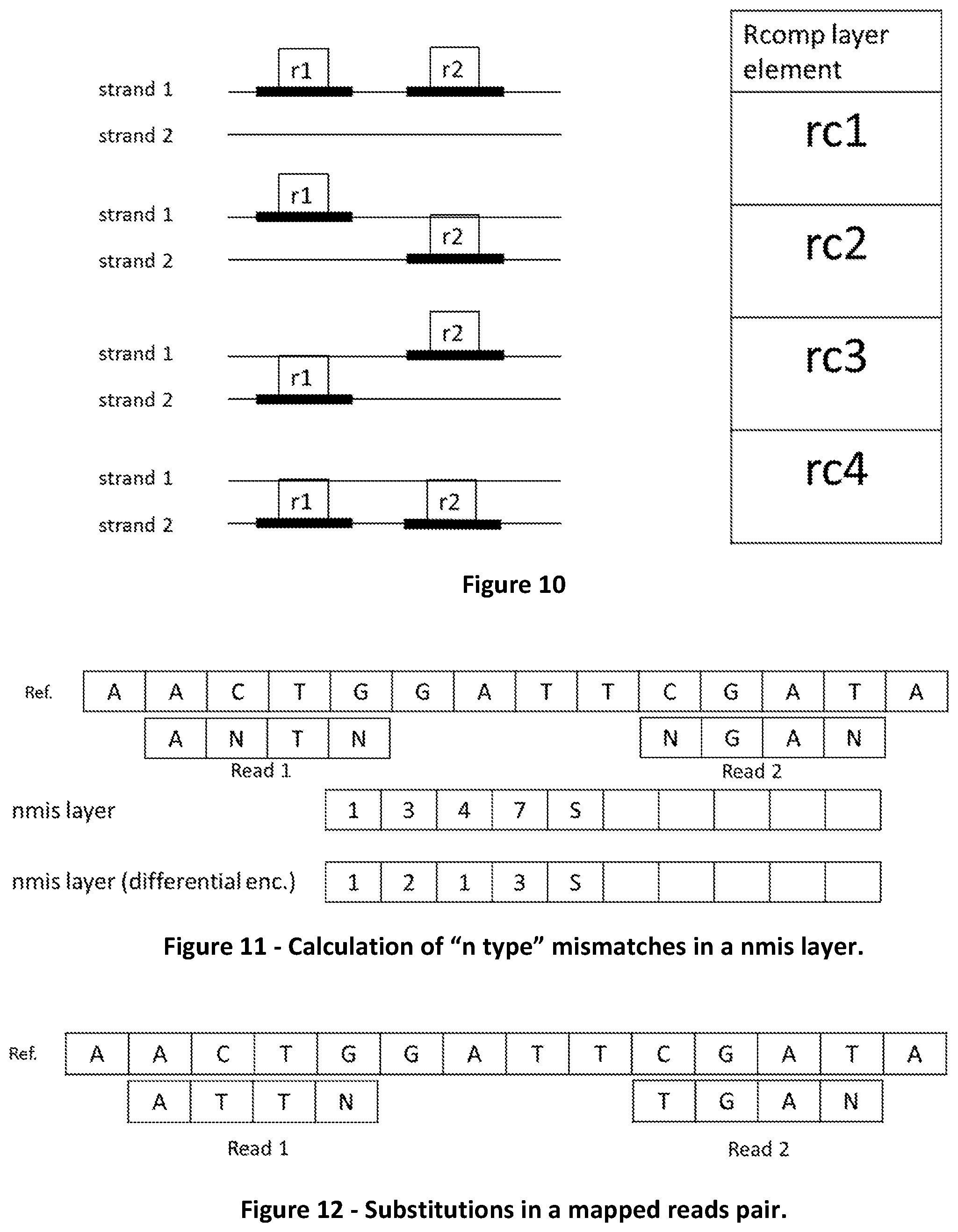
[0016] A key aspect of the invention is a specific definition of classes of data and metadata to be represented by an appropriate source model, coded (i.e. compressed) separately by being structured in specific layers. The most important achievements of this invention with respect to existing state of the art methods consist in: [0017] the increase of compression performance due to the reduction of the information source entropy constituted by providing an efficient model for each class of data or metadata; [0018] the possibility of performing selective accesses to portions of the compressed data and metadata for any further processing purpose directly in the compressed domain; [0019] the possibility of defining user specified "labels" identifying genomic regions or sub-regions or aggregations of regions or sub-regions to enable efficient selective access to the compressed data by means of parsing a "labels list" contained in the genomic file header; [0020] the possibility of implementing access control and protection to the different genomic regions or sub-regions identified by a label; [0021] the possibility of incrementally (without the need of re-encoding) updating and adding encoded data and metadata with new sequencing data and/or metadata and/or new analysis results; [0022] the possibility of efficiently processing data as soon as they are produced by the sequencing machine or alignment tools without the need of waiting the end of the sequencing or alignment process.
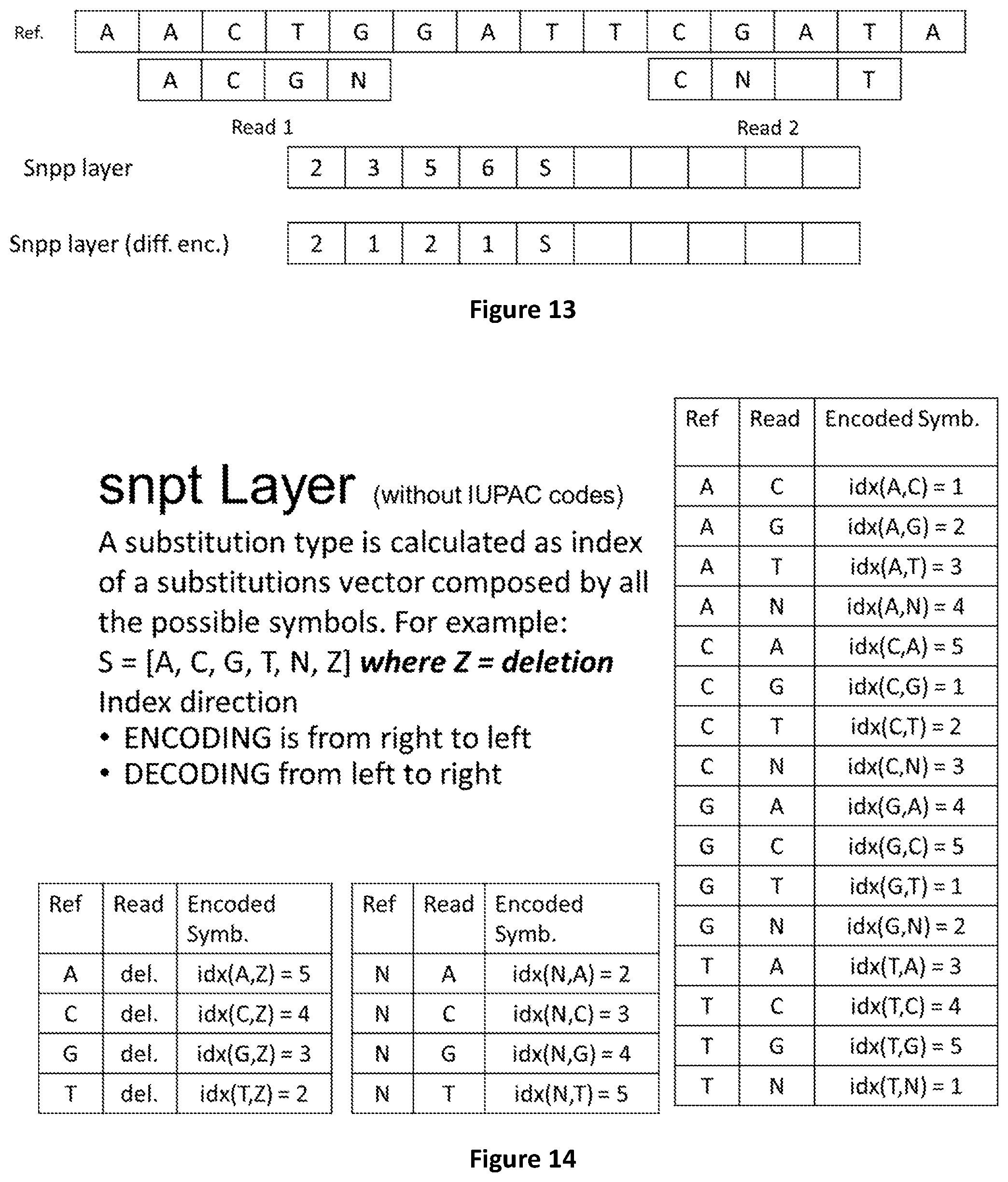
[0023] The present application discloses a method and system addressing the problem of efficient manipulation, storage and transmission of very large amounts of genomic sequencing data, by employing a structured access units approach combined with multiplexing techniques.
[0024] The present application overcomes all the limitations of the prior art approaches related to the functionality of genomic data accessibility, selective data protection, efficient processing of data subsets, transmission and streaming functionality combined with an efficient compression.
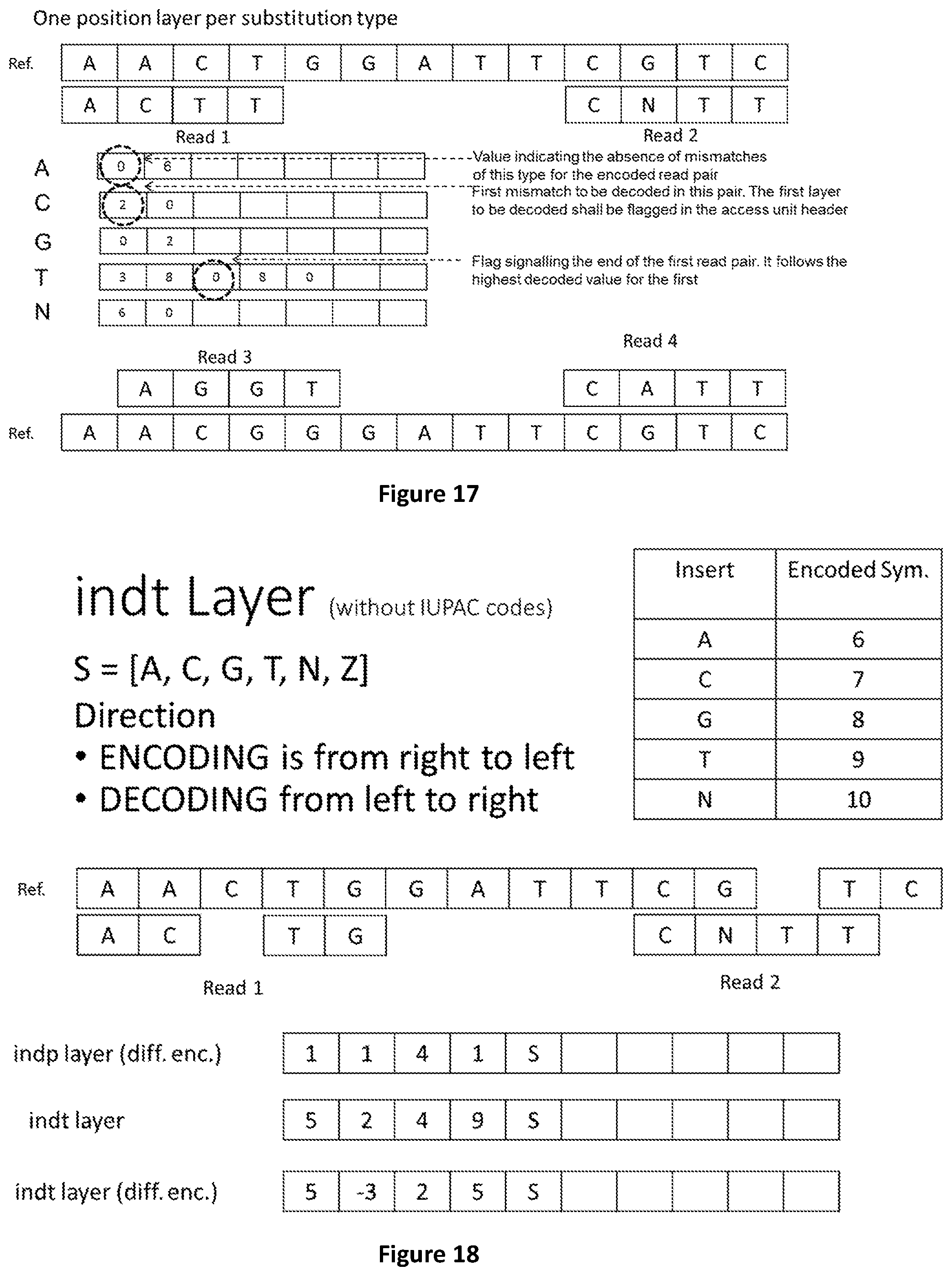
[0025] Today the most used representation format for genomic data is the Sequence Alignment Mapping (SAM) textual format and its binary correspondent BAM. SAM files are human readable ASCII text files whereas BAM adopts a block based variant of gzip. BAM files can be indexed to enable a limited modality of random access. This is supported by the creation of a separate index file.
[0026] The BAM format is characterized by poor compression performance for the following reasons: [0027] 1. It focuses on compressing the inefficient and redundant SAM file format rather than on extracting the actual genomic information conveyed by SAM files and using appropriate models for compressing it. [0028] 2. It employs a general purpose text compression algorithm such as gzip rather than exploiting the specific nature of each data source (the genomic information itself). [0029] 3. It lacks any concept and does not support any functionality related to data classification that would enable the implementation of mechanisms providing selective access to specific classes of genomic data.
[0030] A more sophisticated approach to genomic data compression that is less commonly used, but more efficient than BAM is CRAM (CRAM specification: https://samtools.github.io/hts-specs/CRAMv3.pdf). CRAM provides a more efficient compression for the adoption of differential encoding with respect to an existing reference (it partially exploits the data source redundancy), but it still lacks features such as incremental updates, support for streaming and selective access to specific classes of compressed data.
[0031] CRAM relies on the concept of the CRAM record. Each CRAM record encodes a single mapped or unmapped reads by encoding all the elements necessary to reconstruct it.
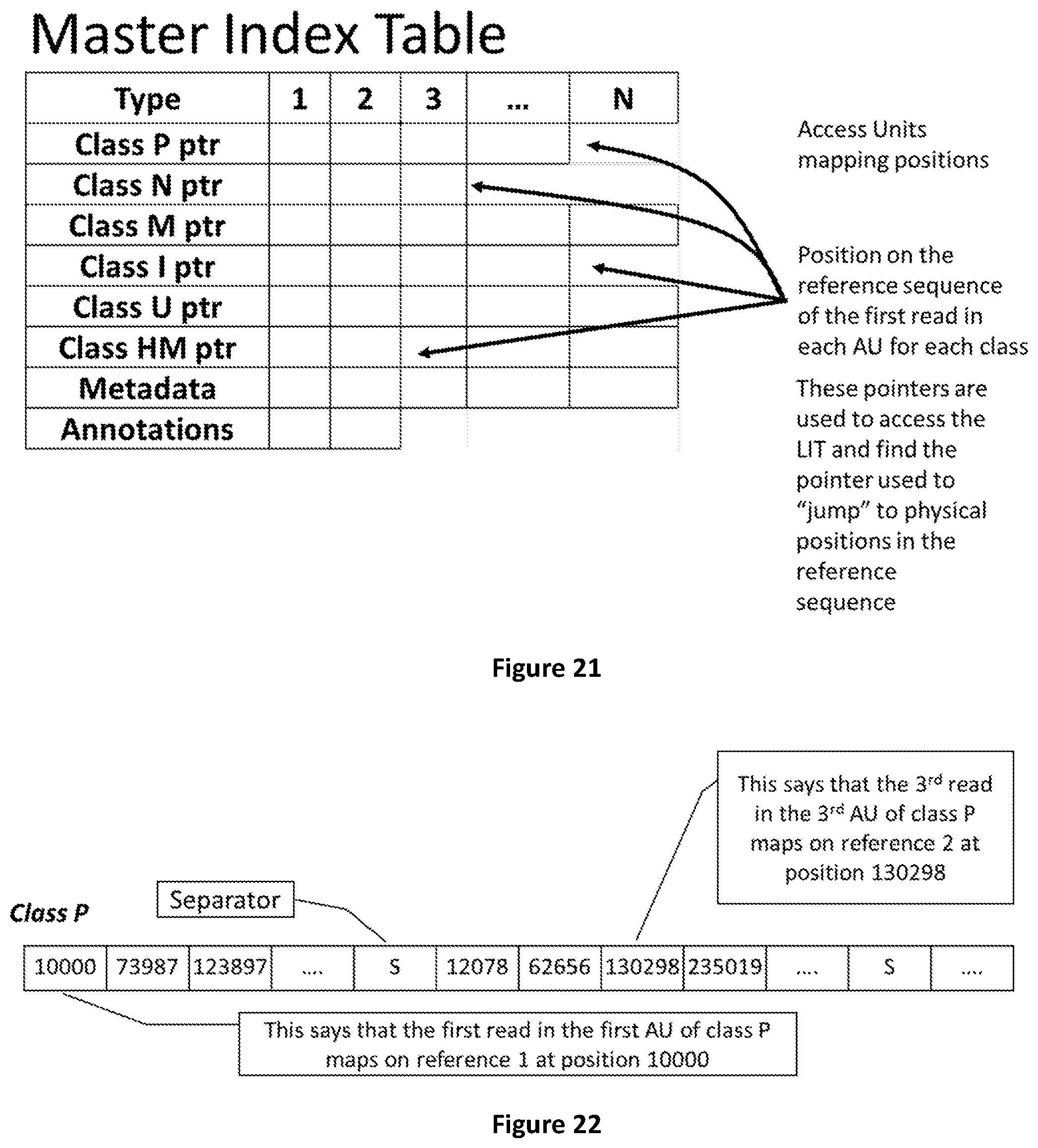
[0032] CRAM presents the following drawbacks and limitations that are solved and removed by the invention described in this document: [0033] 1. CRAM does not support data indexing and random access to data subsets sharing specific features. Data indexing is out of the scope of the specification (see section 12 of CRAM specification v 3.0) and it is implemented as a separate file. Conversely the approach of the invention described in this document employs a data indexing method that is integrated with the encoding process and indexes are embedded in the encoded (i.e. compressed) bit stream. [0034] 2. CRAM does not support the aggregation of the data related to several sequencing runs so that selective access is efficient and segregation of runs (i.e. the process of extracting the genomic information from the actual organic sample) is preserved. CRAM does provide the possibility to label reads as belonging to different groups, but this is provided on a read by read base and reads from different groups are then mixed in the file structure. In the present invention a method is described to structure the data so as to keep segregation among different sequencing runs so that efficient selective access is available. [0035] 3. CRAM is built by core data blocks that can contain any type of mapped reads (perfectly matching reads, reads with substitutions only, reads with insertions or deletions (also referred to as "indels")). There is no notion of data classification and grouping of reads in classes according to the result of mapping with respect to a reference sequence. This means that all data need to be inspected even if only reads with specific features are searched. Such limitation is solved by the invention by classifying and partitioning data in classes before coding. [0036] 4. CRAM is based on the concept of encapsulating each read into a "CRAM record". This implies the need to inspect each complete "record" when reads characterized by specific biological features (e.g. reads with substitutions, but without "indels", or perfectly mapped reads) are searched. Conversely, in the present invention there is the notion of data classes coded separately in separate information layers and there is no notion of record encapsulating each read. This enables more efficient access to set of reads with specific biological characteristics (e.g. reads with substitutions, but without "indels", or perfectly mapped reads) without the need of decoding each (block of) read(s) to inspect its features. [0037] 5. In a CRAM record each field in a record is associated to a specific flag and each flag must always have the same meaning as there is no notion of context since each CRAM record can contain any different type of data. This coding mechanism introduces redundant information and prevents the usage of efficient context based entropy coding. [0038] Conversely in the present invention there is no notion of flag denoting data because this is intrinsically defined by the information "layer" the data belongs to. This implies a largely reduced number of symbols to be used and a consequent reduction of the information source entropy which results into a more efficient compression. Such improvement is possible because the use of different "layers" enables the encoder to reuse the same symbol across each layer with different meanings according to the context. In CRAM each flag must always have the same meaning as there is no notion of contexts and each CRAM record can contain any type of data. [0039] 6. In CRAM, substitutions, insertions and deletions are represented by using different syntax elements, option that increases the size of the information source alphabet and yields a higher source entropy. Conversely the approach of the disclosed invention uses a single alphabet and encoding for substitutions, insertions and deletions. This makes the encoding and decoding process simpler and produces a lower entropy source model which coding yields bitstreams characterized by high compression performance. [0040] 7. CRAM does not provide any mechanism to uniquely identify specific regions or sub regions of the genomic data or aggregations thereof. Apart from the definition of loci in terms of start and end positions on the reference sequence, according to the CRAM specification there is no way to: [0041] label a region and access it using the defined label instead of the genomic start and end position. Start and end positions of the same genomic region may change if a new reference sequence is published, while a defined label would hide such change to any end user. The encoding and decoding system would take care of adapting the actual region identified by the label to the newly published reference sequence [0042] aggregate several regions or sub-regions under the same label so that any end user would be able to select the required data via a single query not involving complex nested queries. The entire aggregation mechanism would be embedded in the encoding and decoding system as described in this document. [0043] 8. CRAM does not provide or support any mechanism to implement selective protection and access control relative to specific regions or sub regions of the genomic data or aggregations thereof, neither when such regions are pre-defined nor when they are specified by the user inserting appropriate "Labels".
[0044] Beside CRAM also the other approaches to genomic data compression and processing present strong limitations to most of the desired functionality and do not support features that are provided by this invention disclosure as described and specified in the following of the document.
[0045] Genomic compression algorithms used in the state of the art can be classified into these categories: [0046] Transform-based [0047] LZ-based [0048] Read reordering [0049] Assembly-based [0050] Statistical modeling
[0051] The first two categories share the disadvantage of not exploiting the specific characteristics of the data source (genomic sequence reads) and process the genomic data as string of text to be compressed without taking into account the specific properties of such kind of information (e.g. redundancy among reads, reference to an existing sample). Two of the most advanced toolkits for genomic data compression, namely CRAM and Goby ("Compression of structured high-throughput sequencing data", F. Campagne, K. C. Dorff, N. Chambwe, J. T. Robinson, J. P. Mesirov, T. D. Wu), make a poor use of arithmetic coding as they implicitly model data as independent and identically distributed by a Geometric distribution. Goby is slightly more sophisticated since it converts all the fields to a list of integers and each list is encoded independently using arithmetic coding without using any context. In the most efficient mode of operation, Goby is able to perform some inter-list modeling over the integer lists to improve compression. These prior art solutions yield poor compression ratios and data structures that are difficult if not impossible to selectively access and manipulate once compressed. Downstream analysis stages can result to be inefficient and very slow due to the necessity of handling large and rigid data structures even to perform simple operation or to access selected regions of the genomic dataset.
[0052] A simplified vision of the relation among the file formats used in genome processing pipelines is depicted in FIG. 1. In this diagram file inclusion does not imply the existence of a nested file structure, but it only represents the type and amount of information that can be encoded for each format (i.e. SAM contains all information in FASTQ, but organized in a different file structure). CRAM contains the same genomic information as SAM/BAM, but it has more flexibility in the type of compression that can be used, therefore it is represented as a superset of SAM/BAM.
[0053] The use of multiple file formats for the storage of genomic information is highly inefficient and costly. Having different file formats at different stages of the genomic information life cycle implies a linear growth of utilized storage space even if the incremental information is minimal. Further disadvantages of prior art solutions are listed below. [0054] 1. Accessing, analysing or adding annotations (metadata) to raw data stored in compressed FastQ files or any combination thereof requires the decompression and recompression of the entire file with extensive usage of computational resources and time. [0055] 2. Retrieving specific subsets of information such as read mapping position, read variant position and type, indels position and types, or any other metadata and annotation contained in aligned data stored in BAM files requires to access the whole data volume associated to each read. Selective access to a single class of metadata is not possible with prior art solutions. [0056] 3. Prior art file formats require that the whole file is received at the end user before processing can start. For example the alignment of reads could start before the sequencing process has been completed relying on an appropriate data representation. Sequencing, alignment and analysis could proceed and run in parallel. [0057] 4. Prior art solution do not support structuring and are not able of distinguishing genomic data obtained by different sequencing processes according to their specific generation semantic (e.g. sequencing obtained at different time of the life of the same individual). The same limitation occurs for sequencing obtained by different types of biological samples of the same individual. [0058] 5. The protection by means of access control mechanisms (e.g. encryption, watermarking, digital signature, hashing) of entire or selected portions of the data is not supported by prior art solutions. For example the protection of: [0059] a. selected DNA regions [0060] b. only those sequences containing variants [0061] c. chimeric sequences only [0062] d. unmapped sequences only [0063] e. regions or sub-regions or aggregations of regions or sub-regions identified by user defined Labels [0064] f. specific metadata (e.g. origin of the sequenced sample, identity of sequenced individual, type of sample) [0065] is not supported in files and data formats of prior art solutions. [0066] 6. The transcoding from sequencing data aligned to a given reference (i.e. a SAM/BAM file) to a new reference requires to process the entire data volume even if the new reference differs only by a single nucleotide position from the previous reference.
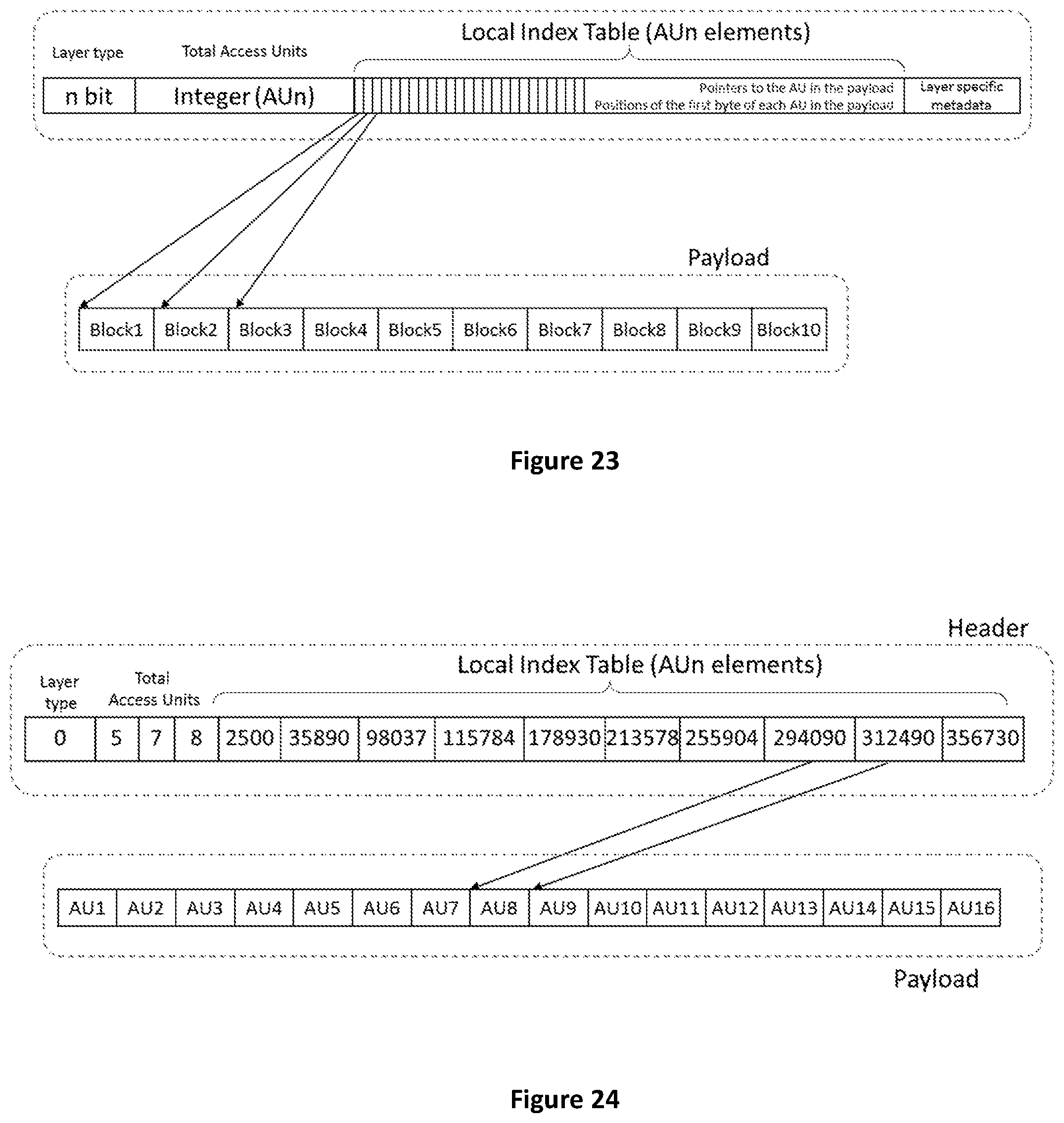
[0067] Therefore there is the clear need of an appropriate Genomic Information Storage Format (Genomic File Format) and Transport Mechanism that enable efficient compression, support selective access and protection functionality in the compressed domain, of local and remotely stored data and support the incremental addition of heterogeneous metadata in the compressed domain at all levels of the different stages of the genomic data processing.
[0068] The present invention provides a solution to the limitations of the state of the art by employing the method, devices and computer programs as claimed in the accompanying set of claims.
LIST OF FIGURES
[0069] FIG. 1 shows the main steps of a typical genomic pipeline and the related file formats.
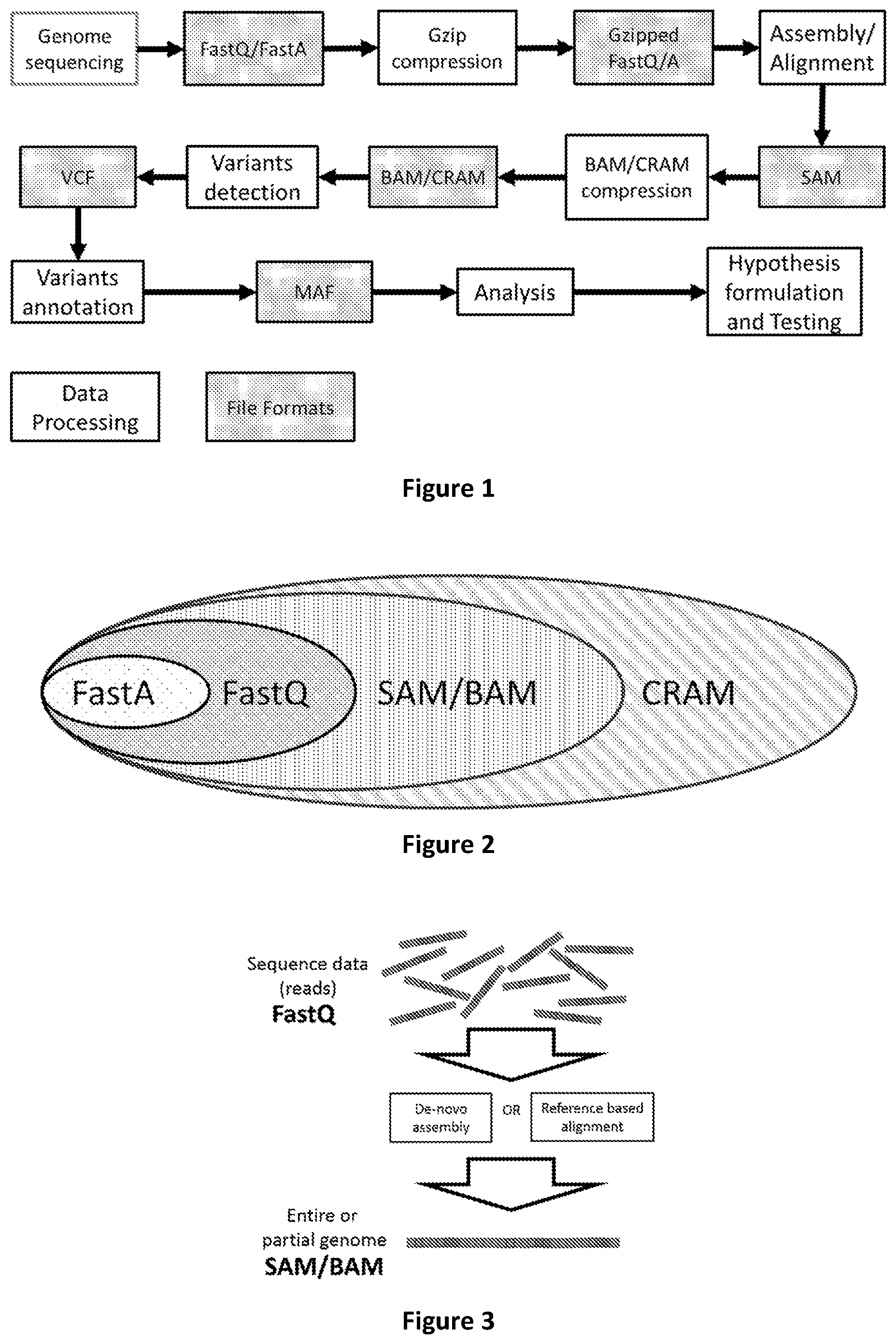
[0070] FIG. 2 shows the mutual relationship among the most used genomic file formats
[0071] FIG. 3 shows how genomic sequence reads are assembled in an entire or partial genome via de-novo assembly or reference based alignment.
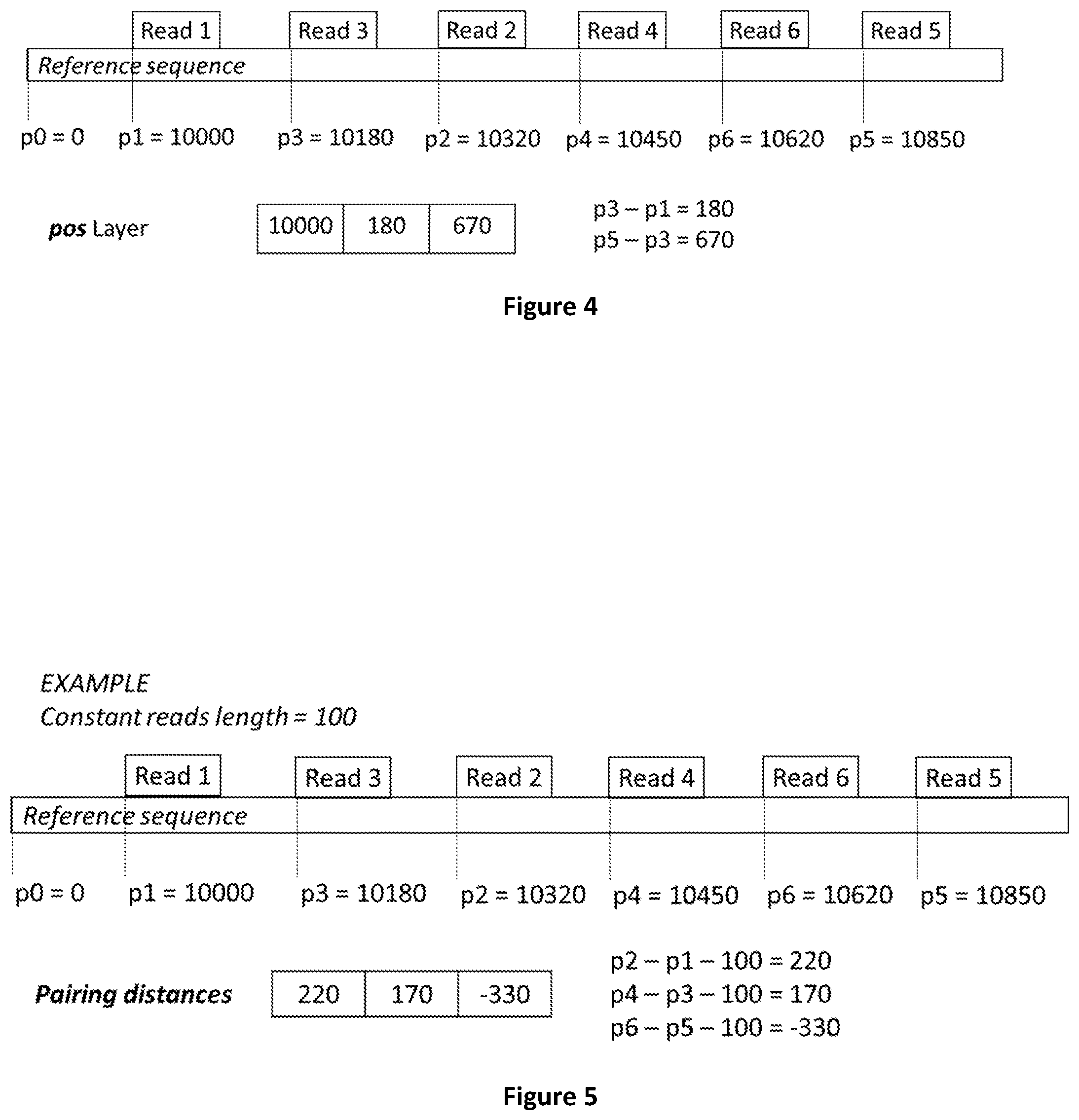
[0072] FIG. 4 shows how reads mapping positions on the reference sequence are calculated.
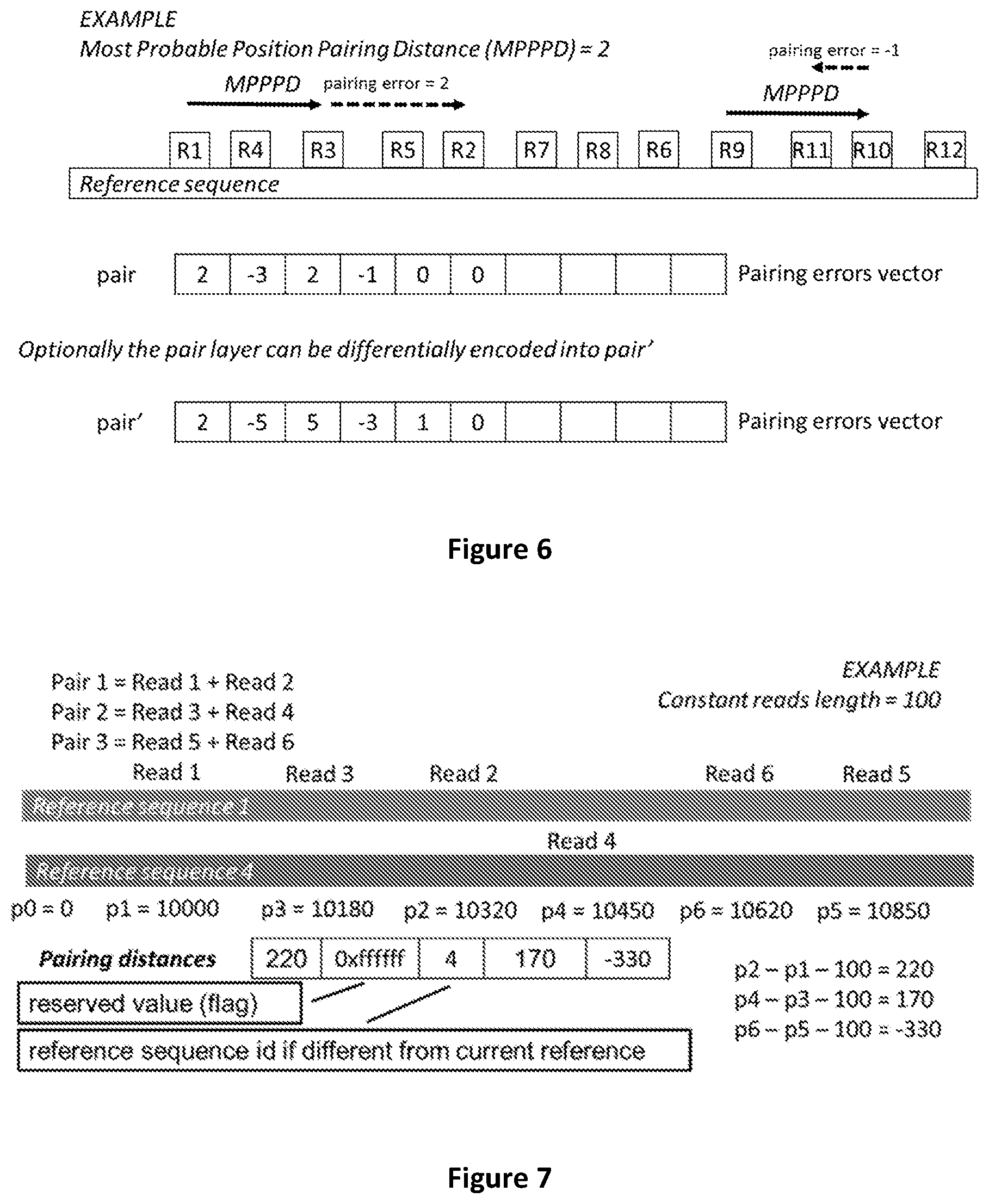
[0073] FIG. 5 shows how reads pairing distances are calculated.
[0074] FIG. 6 shows how pairing errors are calculated.
[0075] FIG. 7 shows how the pairing distance is encoded when a read mate pair is mapped on a different chromosome.
[0076] FIG. 8 shows how sequence reads can be generated from the first or second DNA strand of a genome.
[0077] FIG. 9 shows how a read mapped on strand 2 has a corresponding reverse complemented read on strand 1.
[0078] FIG. 10 shows the four possible combinations of reads composing a reads pair and the respective encoding in the rcomp layer.
[0079] FIG. 11 shows how "n type" mismatches are encoded in a nmis layer.
[0080] FIG. 12 shows an example of substitutions in a mapped read pair.
[0081] FIG. 13 shows how substitutions positions can be calculated either as absolute or differential values.
[0082] FIG. 14 shows how symbols encoding substitutions without IUPAC codes are calculated.
[0083] FIG. 15 shows how substitution types are encoded in the snpt layer.
[0084] FIG. 16 shows how symbols encoding substitutions with IUPAC codes are calculated.
[0085] FIG. 17 shows an alternative source model for substitution where only positions are encoded, but one layer per substitution type is used.
[0086] FIG. 18 shows how to encode substitutions, insertions and deletions in a reads pair of class I when IUPAC codes are not used.
[0087] FIG. 19 shows how to encode substitutions, insertions and deletions in a reads pair of class I when IUPAC codes are used.
[0088] FIG. 20 shows the structure of the Genomic Dataset Header of the genomic information data structure disclosed by this invention.
[0089] FIG. 21 shows how the Master Index Table contains the positions on the reference sequences of the first read in each Access Unit.
[0090] FIG. 22 shows an example of partial MIT showing the mapping positions of the first read in each pos AU of class P.
[0091] FIG. 23 shows how the Local Index Table in the layer header is a vector of pointers to the AUs in the payload.
[0092] FIG. 24 shows an example of Local Index Table.
[0093] FIG. 25 shows the functional relation between Master Index Table and Local Index Tables
[0094] FIG. 26 shows how Access Units are composed by blocks of data belonging to several layers. Layers are composed by Blocks subdivided in Packets.
[0095] FIG. 27 shows how a Genomic Access Unit of type 1 (containing positional, pairing, reverse complement and read length information) is packetized and encapsulated in a Genomic Data Multiplex.
[0096] FIG. 28 shows how Access Units are composed by a header and multiplexed blocks belonging to one or more layers of homogeneous data. Each block can be composed by one or more packets containing the actual descriptors of the genomic information.
[0097] FIG. 29 shows the structure of Access Units of type 0 which do not need to refer to any information coming from other access units to be accessed or decoded and accessed.
[0098] FIG. 30 shows the structure of Access Units of type 1.
[0099] FIG. 31 shows the structure of Access Units of type 2 which contain data that refer to an access unit of type 1. These are the positions of N bases in the encoded reads.
[0100] FIG. 32 shows the structure of Access Units of type 3 which contain data that refer to an access unit of type 1. These are the positions and types of mismatches in the encoded reads.
[0101] FIG. 33 shows the structure of Access Units of type 4 which contain data that refer to an access unit of type 1. These are the positions and types of mismatches in the encoded reads.
[0102] FIG. 34 shows the first five type of Access Units.
[0103] FIG. 35 shows that Access Units of type 1 refer to Access Units of type 0 to be decoded.
[0104] FIG. 36 shows that Access Units of type 2 refer to Access Units of type 0 and 1 to be decoded.
[0105] FIG. 37 shows that Access Units of type 3 refer to Access Units of type 0 and 1 to be decoded.
[0106] FIG. 38 shows that Access Units of type 4 refer to Access Units of type 0 and 1 to be decoded.
[0107] FIG. 39 shows the Access Units required to decode sequence reads with mismatches mapped on the second segment of the reference sequence (AU 0-2).
[0108] FIG. 40 shows how raw genomic sequence data that becomes available can be incrementally added to pre-encoded genomic data.
[0109] FIG. 41 shows how a data structure based on Access Units enables genomic data analysis to start before the sequencing process is completed.
[0110] FIG. 42 shows how new analysis performed on existing data can imply that reads are moved from AUs of type 4 to one of type 3.
[0111] FIG. 43 shows how newly generated analysis data are encapsulated in a new AU of type 8 and a corresponding index is created in the MIT.
[0112] FIG. 44 shows how to transcode data due to the publication of a new reference sequence (genome).
[0113] FIG. 45 shows how reads mapped to a new genomic region with better quality (e.g. no indels) are moved from AU of type 4 to AU of type 3
[0114] FIG. 46 shows how, in case new mapping location is found, (e.g. with less mismatches) the related reads can be moved from one AU to another of the same type.
[0115] FIG. 47 shows how selective encryption can be applied on Access Units of Type 4 only as they contain the sensible information to be protected.
[0116] FIG. 48 shows the data encapsulation in a genomic multiplex where one or more genomic datasets 482-483 contain Genomic streams 484 and streams of Genomic Datasets Mapping Table Lists 481, Genomic Dataset Mapping Tables 485, and Reference Identifiers Mapping Tables 487. Each genomic stream is composed by a Header 488 and Access Units 486. Access Units encapsulate Blocks 489 which are composed by Packets 4810.
[0117] FIG. 49 shows how raw genomic sequence data (499) or aligned genomic data (produced by element 491) are processed to be encapsulated in a Genomic Multiplex. The alignment (491) and reference genome construction (492) stages can be necessary to prepare the data for encoding. Data classes (498) generated by a data classification unit (494) can be further classified with respect to one or more transformed reference generated by a reference transformation unit (4919). The transformed classes (4918) are then sent to layers encoders (495-497). The generated layers (4911) are encoded by entropy coders (4912-4914) which generate Genomic Streams of Access Units (4915) fed to the Genomic Multiplexer (4916).
[0118] FIG. 50 shows how a genomic demultiplexer (500) extracts Genomic Streams (501) from the Genomic Multiplex (5010), one decoder per AU type (502-504) extracts the genomic layers which are then decoded (506-507) into various data classes (5011) which are used by class decoders (509) to reconstruct genomic formats such as for example FASTQ and SAM/BAM. When present in the multiplexed bitstream (5010) a genomic stream containing one or more reference transformations is decoded by an entropy decoder (504) to produce reference transformation descriptors (5012). Reference transformation descriptors are processed by a reference transformation unit (5013) to transform one or more "external" references to generate one or more transformed references (5014) to be used by the class decoders (509).
[0119] FIG. 51 shows the process of encoding sequence reads belonging to class U using a self-generated reference sequence using six layers of descriptors. Four layers are the same used for other classes P, N, M, I while two layers are specific to class U reads.
[0120] FIG. 52 shows how a label is built to aggregate genomic regions belonging to two different references.
[0121] FIG. 53 shows how an existing label can be updated in case new results of analysis require to add an additional region R4 to the existing ones (R1, R2 and R3).
[0122] FIG. 54 shows how the labeling mechanism can be used to implement access control and data protection on specific genomic regions or sub regions. The simple case uses one access control rule (AC) and one protection mechanism (e. g. encryption) for all genomic regions identified by one label.
[0123] FIG. 55 shows how the different genomic regions identified by the same label can be protected by several different access control rules (AC) and several different encryption keys.
[0124] FIG. 56 shows how an alternative encoding of reads of class U where a signed POS descriptor is used to encode the mapping position of a read on the computed reference FIG. 57 shows how half mapped read pairs can help in filling unknown regions of the reference sequence by assembling longer contigs with unmapped reads.
[0125] FIG. 58 shows the hierarchical structure of headers for genomic data stored following the structure described in this invention.
[0126] FIG. 59 shows how a device implementing the labeling mechanism described by this invention enables concurrent access to data related to several genomic regions when they are stored in different records of a database. This can happen either in presence of controlled access or not.
[0127] FIG. 60 shows how vectors of thresholds are used in encoders of classes N, M and I to generate separated subclasses of data
[0128] FIG. 61 provides an example of how reference transformations can change the class reads belong to when all or a subset of mismatches are removed (i.e. the read belonging to class M before transformation is assigned to class P after the transformation of the reference has been applied).
[0129] FIG. 62 shows how reference transformations can be applied to remove mismatches (MMs) from reads. In some cases reference transformations may generate new mismatches or change the type of mismatches found when referring to the reference before the transformation has been applied.
[0130] FIG. 63 The same reference transformation A0 can be used for all classes of data or different transformations AN, A.sub.M, A.sub.I are used for each class N, M, I
SUMMARY
[0131] The features of the claims below solve the problem of existing prior art solutions by providing
a method for selective access of regions of genomic data by employing labels, said labels comprising: an identifier of a reference genomic sequence (521), an identifier of said genomic regions (522), and an identifier of the data class (523) of said genomic data
[0132] In another aspect of the method said genomic data are sequences of genomic reads.
[0133] In another aspect of the method data classes can be of the following type or a subset of them: [0134] "Class P" comprising genomic reads which do not present any mismatch with respect to a reference sequence [0135] "Class N" comprising genomic reads including only mismatches in positions where the sequencing machine was not able to call any "base" and the number of said mismatches does not exceed a given threshold [0136] "Class M" comprising genomic reads in which mismatches are constituted by positions where the sequencing machine was not able to call any base, named "n type" mismatches, and/or it called a different base than the reference sequence, named "s type" mismatches, and said numbers of mismatches do not exceed given thresholds for the number of mismatches of "n type", of "s type" and a threshold obtained from a given function (f(n,s)) [0137] "Class I" when the genomic reads can possibly have the same type of mismatches of "Class M", and in addition at least one mismatch of type: "insertion" ("i type"), "deletion" ("d type"), soft clips ("c type"), and wherein the numbers of mismatches for each type does not exceed the corresponding given thresholds and a threshold provided by a given function (w(n,s,i,d,c)) [0138] "Class U" comprising all reads that do not find any classification in the classes P, N, M, I
[0139] In another aspect of the method said genomic data are paired sequences of genomic reads.
[0140] In another aspect of the method said data class of paired reads can be of the following types or a subset of them: [0141] "Class P" comprising genomic read pairs which do not present any mismatch with respect to a reference sequence [0142] "Class N" comprising genomic reads pairs including only mismatches in positions where the sequencing machine was not able to call any "base" and said numbers of mismatches for each read do not exceed a given threshold [0143] "Class M" comprising genomic read pairs including only mismatches in positions where the sequencing machine was not able to call any "base" and said numbers of mismatches for each read do not exceed a given threshold, named "n type" mismatches, and/or it called a different base than the reference sequence, named "s type" mismatches, and said numbers of mismatches does not exceed a given thresholds for the number of mismatches of "n type", of "s type" and a threshold obtained from a given function (f(n,s)) [0144] "Class I" comprising read pairs which can possibly have the same type of mismatches of "Class M" pairs, and in addition at least one mismatch of type: "insertion" ("i type") "deletion" ("d type") soft clips ("c type"), and wherein the number of mismatches for each type does not exceed the corresponding given threshold and a threshold provided by a given function (w(n,s,i,d,c)) [0145] "Class HM" comprising read pairs for which only one read mate does not satisfy the matching rules for being classified in any of the classes P, N, M, I [0146] Class "U" comprising all reads pairs for which both reads do not satisfy the matching rules for being classified in the classes P, N, M, I
[0147] In another aspect of the method said identifier of said genomic regions is comprised in a master index table.
[0148] In another aspect of the method said genomic data and said labels are entropy coded.
[0149] In another aspect of the method said master index table (4812) is comprised in a genomic dataset header (4813).
[0150] In another aspect of the method said regions of genomic data are dispersed among separate Access Units (524, 486).
[0151] In another aspect of the method the location of said regions of genomic data, in a file, is indicated in a local index table (525).
[0152] In another aspect of the method said labels are user specified.
[0153] In another aspect of the method said regions are protected and/or encrypted in a separate manner, without encrypting the whole genomic file.
[0154] In another aspect of the method said labels are stored in a genomic label list (GLL)
[0155] In another aspect the method further comprises encoding genomic data with selective access to regions of genomic data as previously defined.
[0156] In another aspect of the method said genomic label list is periodically retransmitted or updated in order to enable multiple synchronization points
[0157] In another aspect the method further comprises decoding a stream or a file of genomic data with selective access to regions of genomic data as previously defined.
[0158] The present invention further provides an apparatus for encoding genomic data as previously defined.
[0159] The present invention further provides an apparatus for decoding genomic data as previously defined.
[0160] The present invention further provides a storing mean for storing genomic data encoded as previously defined.
[0161] The present invention further provides a computer-readable medium comprising instructions that when executed cause at least one processor to perform the encoding method previously defined.
[0162] The present invention further provides a computer-readable medium comprising instructions that when executed cause at least one processor to perform the decoding method previously defined.
DETAILED DESCRIPTION
[0163] The present invention describes a labelling mechanism providing selective access and selective access control to genomic regions or sub-regions or aggregations of regions or sub-regions of compressed genomic data stored in a file format and/or the relevant access units to be used to store, transport, access and process genomic or proteomic information in the form of sequences of symbols representing molecules.
[0164] These molecules include, for example, nucleotides, amino acids and proteins. One of the most important pieces of information represented as sequence of symbols are the data generated by high-throughput genome sequencing devices.
[0165] The genome of any living organism is usually represented as a string of symbols expressing the chain of nucleic acids (bases) characterizing that organism. Current state of the art genome sequencing technology is able to produce only a fragmented representation of the genome in the form of several (up to billions) strings of nucleic acids associated to metadata (identifiers, level of accuracy etc.). Such strings are usually called "sequence reads" or "reads".
[0166] The typical steps of the genomic information life cycle comprise Sequence reads extraction, Mapping and Alignment, Variant detection, Variant annotation and Functional and Structural Analysis (see FIG. 1).
[0167] Sequence reads extraction is the process --performed by either a human operator or a machine--of representation of fragments of genetic information in the form of sequences of symbols representing the molecules composing a biological sample. In the case of nucleic acids such molecules are called "nucleotides". The sequences of symbols produced by the extraction are commonly referred to as "reads". This information is usually encoded in prior art as FASTA files including a textual header and a sequence of symbols representing the sequenced molecules.
[0168] When the biological sample is sequenced to extract DNA of a living organism the alphabet is composed by the symbols (A,C,G,T,N).
[0169] When the biological sample is sequenced to extract RNA of a living organism the alphabet is composed by the symbols (A,C,G,U,N).
[0170] In case the IUPAC extended set of symbols, so called "ambiguity codes" are also generated by the sequencing machine, the alphabet used for the symbols composing the reads are (A, C, G, T, U, W, S, M, K, R, Y, B, D, H, V, N or -).
[0171] When the IUPAC ambiguity codes are not used, a sequence of quality score can be associated to each sequence read. In such case prior art solutions encode the resulting information as a FASTQ file. Sequencing devices can introduce errors in the sequence reads such as: [0172] 1. identification of a wrong symbol (i.e. representing a different nucleic acid) to represent the nucleic acid actually present in the sequenced sample; this is usually called "substitution error" (mismatch); [0173] 2. insertion in one sequence read of additional symbols that do not refer to any actually present nucleic acid; this is usually called "insertion error"; [0174] 3. deletion from one sequence read of symbols that represent nucleic acids that are actually present in the sequenced sample; this is usually called "deletion error"; [0175] 4. recombination of one or more fragments into a single fragment which does not reflect the reality of the originating sequence.
[0176] The term "coverage" is used in literature to quantify the extent to which a reference genome or part thereof can be covered by the available sequence reads. Coverage is said to be: [0177] partial (less than 1.times.) when some parts of the reference genome are not mapped by any available sequence read [0178] single (1.times.) when all nucleotides of the reference genome are mapped by one and only one symbol present in the sequence reads [0179] multiple (2.times., 3.times., N.times.) when each nucleotide of the reference genome is mapped multiple times.
[0180] Sequence alignment refers to the process of arranging sequence reads by finding regions of similarity that may be a consequence of functional, structural, or evolutionary relationships among the sequences. When the alignment is performed with reference to a pre-existing nucleotides sequence referred to as "reference genome", the process is called "mapping". Sequence alignment can also be performed without a pre-existing sequence (i.e. reference genome) in such cases the process is known in prior art as "de novo" alignment. Prior art solutions store this information in SAM, BAM or CRAM files. The concept of aligning sequences to reconstruct a partial or complete genome is depicted in FIG. 3.
[0181] Variant detection (a.k.a. variant calling) is the process of translating the aligned output of genome sequencing machines, (sequence reads generated by NGS devices and aligned), to a summary of the unique characteristics of the organism being sequenced that cannot be found in other pre-existing sequence or can be found in a few pre-existing sequences only. These characteristics are called "variants" because they are expressed as differences between the genome of the organism under study and a reference genome. Prior art solutions store this information in a specific file format called VCF file.
[0182] Variant annotation is the process of assigning functional information to the genomic variants identified by the process of variant calling. This implies the classification of variants according to their relationship to coding sequences in the genome and according to their impact on the coding sequence and the gene product. This is in prior art usually stored in a MAF file.
[0183] The process of analysis of DNA (variant, CNV=copy number variation, methylation etc) strands to define their relationship with genes (and proteins) functions and structure is called functional or structural analysis. Several different solutions exist in the prior art for the storage of this data.
[0184] Genomic File Format
[0185] The invention disclosed in this document consists in the definition of a selective and controlled data access applied to a compressed data structure for representing, processing manipulating and transmitting genome sequencing data that differs from prior art solutions for at least the following aspects: [0186] It does not rely on any prior art representation formats of genomic information (i.e. FASTQ, SAM). [0187] It supports efficient handling and selective random access to data produced by multiple sequencing runs structured as multiple genomic datasets. Partitioning data from different sequencing runs into the same data structure enables analysts to simultaneously perform queries on them with great advantage for population genetics studies. [0188] It implements a new original classification of the genomic data and metadata according to their specific characteristics. Sequence reads are mapped to a reference sequence and grouped in distinct classes according to the results of the alignment process. This results in data classes with lower information entropy that can be more efficiently encoded applying different specific compression algorithms such as Huffman coding, arithmetic coding (CABAC, CAVLAC), Asymmetric Numerical Systems, Lempel-Ziv and its derivations. [0189] It implements a new method to associate data classes or subsets of data classes to specific genomic regions, or sub-regions or aggregations of regions or sub-regions, by means of user defined Labels that enable the selective access and protection of said compressed data classes corresponding to specific genomic regions or sub-regions or aggregations of regions or sub-regions. [0190] It defines syntax elements and the related encoding/decoding process conveying the sequence reads and the alignment information into a representation which is more efficient to be processed for downstream analysis applications.
[0191] Classifying the reads according to the result of mapping and coding them using descriptors to be stored in layers (position layer, mate distance layer, mismatch type layer etc, etc, . . . ) present the following advantages: [0192] A reduction of the information entropy when the different syntax elements are modelled by a specific source model which yields higher compression performance. [0193] A more efficient access to data that are already organized in groups/layers that have a specific meaning for the downstream analysis stages and that can be accesses separately and independently directly in the compressed domain. [0194] The presence of a modular data structure that can be updated incrementally by accessing only the required information without the need of decoding (i.e. decompressing) the whole data content. [0195] The genomic information produced by sequencing machines is intrinsically highly redundant due to the nature of the information itself and to the need of mitigating the errors intrinsic in the sequencing process. This implies that the relevant genetic information which needs to be identified and analyzed (the variations with respect to a reference) is only a small fraction of the produced data. Prior art genomic data representation formats are not conceived to "isolate" the meaningful information at a given analysis stage from the rest of the information so as to make it promptly available and understandable by the analysis applications. [0196] The solution brought by the disclosed invention is to represent genomic data in such a way that any relevant portion of data is readily available to the analysis applications without the need of accessing and decompressing the entirety of data and the redundancy of the data is efficiently reduced by efficient compression to minimize the required storage space and transmission bandwidth.
[0197] The key elements of the invention are: [0198] 1. The specification of a file format that "contains" structured and user-defined selectively accessible data elements called Access Units (AU) in compressed form. Such approach can be seen as the opposite of prior art approaches, SAM and BAM for instance, in which data are structured in non-compressed form and then the entire file is compressed. A first clear advantage of the approach is to be able to efficiently and naturally provide various forms of user-defined structured selective access to the data elements in the compressed domain which is impossible or extremely awkward in prior art approaches. [0199] 2. The structuring of the genomic information into specific "layers" of homogeneous data and metadata presents the considerable advantage of enabling the definition of different models of the information sources characterized by low entropy. Such models not only can differ from layer to layer, but can also differ inside each layer when the compressed data within layers are partitioned into Data Blocks included into Access Units. This structuring enables the use of the most appropriate compression for each class of data or metadata and portion of them with significant gains in coding efficiency versus prior art approaches. [0200] 3. The information is structured in Access Units (AU) so that any relevant subset of data used by genomic analysis applications is efficiently and selectively accessible by means of appropriate interfaces. These features enable faster access to data and yield a more efficient processing. [0201] 4. The definition of a Master Index Table and Local Index Tables enabling selective access to the information carried by the layers of encoded (i.e. compressed) data without the need to decode the entire volume of compressed data. [0202] 5. The possibility of accessing only the AUs that correspond to specific user defined genomic regions or sub-regions or aggregations of regions or sub-regions and data classes of interest by parsing a "Label List" present in the file header. [0203] 6. The possibility of providing different types of access control to different AUs and portions of data contained into the AU according to the user defined "Labels" identifying associated genomic regions. [0204] 7. The possibility of performing realignment of already aligned and compressed genomic data sets when they need to be re-aligned versus newly published reference genomes by performing an efficient transcoding of selected data portions in the compressed domain. The frequent release of new reference genomes currently requires resource consuming and time for the transcoding processes to re-align already compressed and stored genomic data with respect to the newly published references because all data volume need to be processed.
[0205] The method described in this document aims at exploiting the available a-priori knowledge on genomic data to define an alphabet of syntax elements with reduced entropy. In genomics the available knowledge is represented by an existing genomic sequence usually --but not necessarily --of the same species as the one to be processed. As an example, human genomes of different individuals differ only of a fraction of 1%. However, such small amount of data contain enough information to enable early diagnosis, personalized medicine, customized drugs synthesis etc. This invention aims at defining a genomic information representation format where the relevant information is efficiently accessible, access can be selectively controlled and data protected, the information is efficiently transportable and all such processing is performed handling compressed data structures.
[0206] The technical features used in the present invention are: [0207] 1. Partitioning genomic information generated by different sequencing runs into different genomic datasets in order to enable efficient data retrieval and processing when querying one or more of the available datasets. [0208] 2. Partition of the genome sequence data and metadata in "classes" sharing common features; [0209] 3. Definition of the structure of the genomic information carried by each data classes in which the genomic data is partitioned, into a sets of "layers" of descriptors in order to reduce the information entropy as much as possible; [0210] 4. Definition of a Master Index Table and Local Index Tables to enable selective access to the data classes and associated information by accessing only the desired layers of coded information (i.e. compressed) without the need to decode the entire coded genomic information; [0211] 5. Usage of different source models and entropy coders to code the syntax elements belonging to different layers of the data classes defined as specified in point 2; [0212] 6. Definition of specific mechanisms establishing a correspondence among dependent layers to enable selective access to the data without the need to decode all the layers if not necessary or desired; [0213] 7. Definition of a mechanism for labelling compressed data corresponding to specific genomic regions or sub-regions or aggregations of regions or sub-regions and corresponding data "classes" or subsets of data classes by "Labels" enabling efficient selective access; [0214] 8. Definition of mechanisms for the selective protection of specific genomic regions or sub-regions or aggregations of regions or sub-regions and corresponding data "classes" or subsets of data classes and any combination thereof. [0215] 9. Coding of the datasets or data "classes" with respect to one or more pre-existing or constructed reference sequences that can be further transformed to reduce the entropy of the sequence data representation.
[0216] In order to solve all the mentioned problems of the prior art in terms of efficient selective access and selective access control to specific data "classes", specific genomic regions or sub-regions or aggregations of regions or sub-regions, while preserving efficient transmission and storing by means of an efficient compressed representation, the present invention application provides a specific data structure specification that implements appropriate data reordering into accessible units of homogeneous and/or semantically significant data enabling seamless access and processing required by state of the art genome data analysis applications.
[0217] In particular the present invention adopts a data structure based on the concept of Access Unit, "Labels" and the multiplexing of the relevant data, concepts which are absent from all state of the art genomic data formats.
[0218] Genomic data are structured and encoded into different Access Units. Hereafter follows a description of the genomic data that are contained into different Access Units and can be identified by "Labels" associating genomic data to specific genomic regions or sub-regions or aggregations of regions or sub-regions versus reference genomes.
[0219] Genomic Data Classification According to Matching Rules
[0220] The sequence reads generated by sequencing machines are classified by the disclosed invention into five different "classes" according to the matching results of the alignment with respect to one or more pre-existing reference sequences.
[0221] When aligning a DNA sequence of nucleotides with respect to a reference sequence the following cases can be identified: [0222] 1. A region in the reference sequence is found to match the sequence read without any error (i.e. perfect mapping). Such sequence of nucleotides is referenced to as "perfectly matching read" or denoted as "Class P". [0223] 2. A region in the reference sequence is found to match the sequence read with a type and a number of mismatches determined only by the number of positions in which the sequencing machine generating the read was not able to call any base (or nucleotide). Such type of mismatches are denoted by an "N" the letter used to indicate an undefined nucleotide base. In this document this type of mismatch referred to as "n type" mismatch. Such sequences is referenced to as "N mismatching reads" or "Class N". Once the read is classified to belong to "Class N" it is useful to limit the degree of matching inaccuracy to a given upper bound and set a boundary between what is considered a valid matching and what it is not. Therefore, the reads assigned to Class N are also constrained by setting a threshold (MAXN) that defines the maximum number of undefined bases (i.e. bases called as "N") that a read can contain. Such classification implicitly defines the required minimum matching accuracy (or maximum degree of mismatch) that all reads belonging to Class N shares when referred to the corresponding reference sequence, which constitute an useful criterion for applying selective data searches to the compressed data. [0224] 3. A region in the reference sequence is found to match the sequence read with types and number of mismatches determined by the number of positions in which the sequencing machine generating the read was not able to call any nucleotide base, if present (i.e. "n type" mismatches), plus the number of mismatches in which a different base, than the one present in the reference, has been called. Such type of mismatch denoted as "substitution" is also called Single Nucleotide Variation (SNV) or Single Nucleotide Polymorphism (SNP). In this document this type of mismatch is also referred to as "s type" mismatch. The sequence read is then referenced to as "M mismatching reads" and assigned to "Class M". Like in the case of "Class N", also for all reads belonging to "Class M" it is useful to limit the degree of matching inaccuracy to a given upper bound, and set a boundary between what is considered a valid matching and what it is not. Therefore, the reads assigned to Class M are also constrained by defining a set of thresholds, one for the number "n" of mismatches of "n type" (MAXN) if present, and another for the number of substitutions "s" (MAXS). A third constraint is a threshold defined by any function of both numbers "n" and "s", f(n,s). Such third constraint enable to generate classes with an upper bound of matching inaccuracy according to any meaningful selective access criterion. For instance, and not as a limitation, f(n,s) can be (n+s)1/2 or (n+s) or any linear or non-linear expression that sets a boundary to the maximum matching inaccuracy level that is admitted for a read belonging to "Class M". Such boundary constitutes a very useful criterion for applying the desired selective data searches to the compressed data when analyzing sequence reads for various purposes because it makes possible to set a further boundary to any possible combination of the numbers "n" of "n type" mismatches and "s" of "s type" mismatches (substitutions) beyond the simple threshold applied to the one type or to the other. [0225] 4. A fourth class is constituted by sequencing reads presenting at least one mismatch of any type among "insertion", "deletion" (a.k.a. indels) and "clipped", plus, if present, any mismatches type belonging to class N or M. Such sequence is referenced to as "I mismatching reads" and assigned to "Class I". Insertions are constituted by an additional sequence of one or more nucleotides not present in the reference, but present in the read sequence. In this document this type of mismatch is referred to as "i type" mismatch. In literature when the inserted sequence is at the edges of the sequence it is also referred to as "soft clipped" (i.e. the nucleotides are not matching the reference but are kept in the aligned reads contrarily to "hard clipped" nucleotides which are discarded). In this document this type of mismatch is referred to as "c type" mismatch. Keeping or discarding nucleotides is a decisions taken by the aligner stage and not by the classifier of reads disclosed in this invention which receives and processes the reads as they are determined by the sequencing machine or by the following alignment stage. Deletion are "holes" (missing nucleotides) in the read with respect to the reference. In this document this type of mismatch is referred to as "d type" mismatch. Like in the case of classes "N" and "M" it is possible and appropriate to define a limit to the matching inaccuracy. The definition of the set of constraints for "Class I" is based on the same principles used for "Class M" and is reported in Table 1 in the last table lines. Beside a threshold for each type of mismatch admissible for class I data, a further constraint is defined by a threshold determined by any function of the number of the mismatches "n", "s", "d", "i" and "c", w(n,s,d,i,c). Such additional constraint make possible to generate classes with an upper bound of matching inaccuracy according to any meaningful user defined selective access criterion. For instance, and not as a limitation, w(n,s,d,i,c) can be (n+s+d+i+c)1/5 or (n+s+d+i+c) or any linear or non-linear expression that sets a boundary to the maximum matching inaccuracy level that is admitted for a read belonging to "Class I". Such boundary constitutes a very useful criterion for applying the desired selective data searches to the compressed data when analyzing sequence reads for various purposes because it enables to set a further boundary to any possible combination of the number of mismatches admissible in "Class I" reads beyond the simple threshold applied to each type of admissible mismatch. [0226] 5. A fifth class includes all reads that do now find any matching considered valid (i.e not satisfying the set of matching rules defining an upper bound to the maximum matching inaccuracy as specified in Table 1) for each data class when referring to the reference sequence. Such sequences are said to be "Unmapped" when referring to the reference sequences and are classified as belonging to the "Class U".
[0227] Classification of Read Pairs According to Matching Rules
[0228] The classification specified in the previous section concerns single sequence reads. In the case of sequencing technologies that generates read in pairs (i.e. Illumina Inc.) in which two reads are known to be separated by an unknown sequence of variable length, it is appropriate to consider the classification of the entire pair to a single data class. A read that is coupled with another is said to be its "mate".
[0229] If both paired reads belong to the same class the assignment to a class of the entire pair is obvious, the entire pair is assigned to the same class for any class (i.e. P, N, M, I, U). In the case the two reads belong to a different class, but none of them belongs to the "Class U", then the entire pair is assigned to the class with the highest priority defined according to the following expression:
P<N<M<I
in which "Class P" has the lowest priority and "Class I" has the highest priority.
[0230] In case only one of the reads belongs to "Class U" and its mate to any of the Classes P, N, M, I a sixth class is defined as "Class HM" which stands for "Half Mapped".
[0231] The definition of such specific class of reads is motivated by the fact that it is used for attempting to determine gaps or unknown regions existing in reference genomes (a.k.a. little known or unknown regions). Such regions are reconstructed by mapping pairs at the edges using the pair read that can be mapped on the known regions. The unmapped mate is then used to build the so called "contigs" of the unknown region as it is shown in FIG. 57. Therefore providing a selective access to only such type of read pairs greatly reduces the associated computation burden enabling much efficient processing of such data originated by large amounts of data sets that using the state of the art solutions would require to be entirely inspected.
[0232] The table below summarizes the matching rules applied to reads in order to define the class of data each read belongs to. The rules are defined in the first five columns of the table in terms of presence or absence of type of mismatches (n, s, d, i and c type mismatches). The sixth column provides rules in terms of maximum threshold for each mismatch type and any function f(n,s) and w(n,s,d,i,c) of the possible mismatch types.
TABLE-US-00001 TABLE 1 Type of mismatches and set of constrains that each sequence reads must satisfy to be classified in the data classes defined in this invention disclosure. Number and types of mismatches found when matching a read with a reference sequence Number of Number of unknown Number of Number of Number of clipped Set of matching Assignement bases ("N") substitutions deletions Insertions bases accuracy constraints Class 0 0 0 0 0 0 P n > 0 0 0 0 0 n .ltoreq. MAXN N n > MAXN U n .gtoreq. 0 s > 0 0 0 0 n .ltoreq. MAXN and M s .ltoreq. MAXS and f(n, s) .ltoreq. MAXM n > MAXN or U s > MAXS or f(n, s) > MAXM n .gtoreq. 0 s .gtoreq. 0 d .gtoreq. 0* i .gtoreq. 0* c .gtoreq. 0* n .ltoreq. MAXN and I *At least one mismatch s .ltoreq. MAXS and of type d, i, c must be resent d .ltoreq. MAXD and (i.e. d > 0 or i > 0 or > 0) i .ltoreq. MAXI and c .ltoreq. MAXC w(n, s, d, i, c) .ltoreq. MAXTOT d .gtoreq. 0 i .gtoreq. 0 c .gtoreq. 0 n > MAXN or U s > MAXS or d > MAXD or i > MAXI or c > MAXC w(n, s, d, i, c) > MAXTOT
[0233] Matching Rules Partition of Sequence Read Data Classes N, M and I into Subclasses with Different Degrees of Matching Accuracy
[0234] The data classes of type N, M and I as defined in the previous sections can be further decomposed into an arbitrary number of distinct sub-classes with different degrees of matching accuracy. Such option is an important technical advantage in providing a finer granularity and as consequence a much more efficient selective access to each data class. As an example and not as a limitation, to partition the Class N into a number k of subclasses (Sub-Class N.sub.1, . . . , Sub-Class N.sub.k) it is necessary to define a vector with the corresponding components MAXN.sub.1, MAXN.sub.2, MAXN.sub.(k-1), MAXN.sub.(k), with the condition that MAXN.sub.1<MAXN.sub.2< . . . <MAXN.sub.(k-1)<MAXN and assign each read to the lowest ranked sub-class that satisfy the constrains specified in Table 1 when evaluated for each element of the vector. This is shown in FIG. 60 where a data classification unit 601 contains Class P, N, M, I U, HM encoder and encoders for annotations and metadata. Class N encoder is configured with a vector of thresholds, MAXN.sub.1 to MAXN.sub.k 602 which generates k subclasses of N data (606).
[0235] In the case of the classes of type M and I the same principle is applied by defining a vector with the same properties for MAXM and MAXTOT respectively and use each vector components as threshold for checking if the functions f(n,s) and w(n,s,d,i,c) satisfy the constraint. Like in the case of sub-classes of type N, the assignment is given to the lowest sub-class for which the constraint is satisfied. The number of sub-classes for each class type is independent and any combination of subdivisions is admissible. This is shown in FIG. 60 where a Class M encoder and a Class I encoder are configured respectively with a vector of thresholds MAXM.sub.1 to MAXM.sub.j (603) and MAXTOT.sub.1 to MAXTOT.sub.h (604). The two encoders generate respectively j subclasses of M data (607) and h subclasses of I data (608). When two reads in a pair are classified in the same sub-class, then the pair belongs to the same sub-class.
[0236] When two reads in a pair are classified into sub-classes of different classes, then the pair belongs to the sub-class of the class of higher priority according to the following expression:
N<M<I
where N has the lowest priority and I has the highest priority.
[0237] When two reads belong to different sub-classes of one of classes N or M or I, then the pair belongs to the sub-class with the highest priority according to the following expressions:
N.sub.1<N.sub.2< . . . <N.sub.k
M.sub.1<M.sub.2< . . . <M.sub.j
I.sub.1<I.sub.2< . . . <I.sub.h
where the highest index has the highest priority.
[0238] Transformations of the "External" Reference Sequences
[0239] The mismatches found for the reads classified in the classes N, M and I can be used to create "transformed references" to be used to compress more efficiently the read representation. Reads classified as belonging to the Classes N, M or I (with respect to the pre-existing (i.e. "external") reference sequence denoted as RS.sub.0) can be coded with respect to the "transformed" reference sequence RS.sub.1 according to the occurrence of the actual mismatches with the transformed reference. For example if read.sup.M.sub.in belonging to Class M (denoted as the i.sup.th read of class M) containing mismatches with respect to the reference sequence RS.sub.n, then after "transformation" read.sup.M.sub.in=read.sup.P.sub.i(n+1) can be obtained with A(Ref.sub.n)=Ref.sub.n+1 where A is the transformation from reference sequence RS.sub.n to reference sequence RS.sub.n+1.
[0240] FIG. 61 shows an example on how reads containing mismatches (belonging to Class M) with respect to reference sequence 1 (RS.sub.1) can be transformed into perfectly matching reads with respect to the reference sequence 2 (RS.sub.2) obtained from RS.sub.1 by modifying the bases corresponding to the mismatch positions. They remain classified and they are coded together the other reads in the same data class access unit, but the coding is done using only the descriptors and descriptor values needed for a Class P read. This transformation can be denoted as:
RS.sub.2=A(RS.sub.1)
[0241] When the representation of the transformation A which generates RS.sub.2 when applied to RS.sub.1 plus the representation of the reads versus RS.sub.2 corresponds to a lower entropy than the representation of the reads of class M versus RS.sub.1, it is advantageous to transmit the representation of the transformation A and the corresponding representation of the read versus RS.sub.2 because an higher compression of the data representation is achieved.
[0242] The coding of the transformation A for transmission in the compressed bitstream requires the definition of two additional syntax elements as defined in the table below.
TABLE-US-00002 Syntax elements Semantic Comments rftp Reference position of difference between transformation reference and contig position used for prediction rftt Reference type of difference between reference and transformation contig used for prediction. Same syntax type described for the snpt descriptor defined below.
[0243] FIG. 62 shows an example on how a reference transformation is applied to reduce the number of mismatches to be coded on the mapped reads.
[0244] It has to be observed that, in some cases the transformation applied to the reference: [0245] May introduce mismatches in the representations of the reads that were not present when referring to the reference before applying the transformation. [0246] May modify the types of mismatches, a read may contain A instead of G while all other reads contain C instead of G), but mismatches remain in the same position. [0247] Different data classes and subsets of data of each data class may refer to the same transformed reference sequence or to reference sequences obtained by applying different transformations to the same pre-existing reference sequence.
[0248] FIG. 61 shows an example on how reads can change the type of coding from a data class to another by means of the appropriate set of descriptors (e.g. using the descriptors of a Class P to code a read from Class M) after a reference transformation is applied and the read is represented using the transformed reference. This occurs for example when the transformation changes all bases corresponding to the mismatches of a read in the bases actually present in the read, thus virtually transforming a read belonging to Class M (when referring to the original non transformed reference sequence) into a virtual read of Class P (when referring to the transformed reference). The definition of the set of descriptors used for each class of data is provided in the following sections.
[0249] FIG. 63 shows how the different classes of data can use the same "transformed" reference R.sub.1=A.sub.0(R.sub.0) (630) to re-encode the reads or different transformations A.sub.N (631), A.sub.M (632), A.sub.I (633) can be separately applied to each class of data
[0250] Genomic Data Headers for Global Parameters
[0251] The data structure of said genomic data requires the storage of global parameters and metadata to be used by the decoding engine. These data are organized in the following structures: For file based storage: [0252] Datasets Multiplex Header [0253] Dataset Header [0254] Descriptors Layer Header [0255] Block Header
[0256] The hierarchical relationship among these headers is shown in FIG. 58.
[0257] For transport in a streaming scenario: [0258] Datasets Mapping Tables List [0259] Datasets Mapping Table [0260] Transport Block Header [0261] Packet Header
[0262] A dataset is defined as the ensemble of coding elements needed to reconstruct the genomic information related to a single genomic sequencing run and all the following analysis. If the same genomic sample is sequenced twice in two distinct runs, the obtained data will be encoded in two distinct datasets.
[0263] Datasets Multiplex Header
[0264] This is the data structure prepended to one or more datasets aggregated in a "Multiplex".
TABLE-US-00003 Syntax Description Datasets_ Multiplex _Header { Multiplex_id Label to identify this Datasets Multiplex from any other Datasets Multiplex. Version_number Version number of the Dataset Multiplex. The version number shall be incremented by a unit whenever the definition of the Datasets Multiplex changes. List_number Number of the current datasets list. gd_number Number of datasets composing the datasets Multiplex. for (i=0; i<gd_number;i++) { genomic_dataset_ID Field identifying the dataset. This field shall not take any single value more than once within one version of the Dataset List } Metadata Data structure carrying metadata to be used for application-specific processing such as data analysis and content protection. }
[0265] This is the data structure prepended to an encoded dataset.
TABLE-US-00004 TABLE 2 Genomic Dataset Header structure. Element Type Description Dataset_ID Byte array Unique identifier for the encoded content Major_Brand Byte array Major + Minor Minor_Version Byte array version of the encoding algorithm Header Size Integer Size in bytes of the entire encoded content Reads Length Integer Size of reads in case of constant reads length. A special value (e.g. 0) is reserved for variable reads length Ref count Integer Number of reference sequences used Access Units counters Byte array Total Number of (e.g. encoded Access integers) Units per reference sequence Ref ids Byte array Unique identifiers for reference sequences Ref_count Integer number of references for (i=0; i<Ref_count; i++) { Reference_genome:Ref_ID string:string Unambiguous ID, as a characters string, identifying the reference sequence(s) used in this Dataset } for (i=0; i<Ref_count; i++) { Ref blocks Byte array Number of encoded blocks per each reference } Dataset label size Integer The size of the following element Dataset label String A string of character used to identify the dataset Dataset type Integer The type of data encoded in the dataset (e.g. aligned, not aligned) Master index table Byte array This is a Alignment positions of first read in each block (Access Unit). multidimensional l.e. smaller position of the first read on the reference genome array supporting per each block of the six classes random access to 1 per pos class (six) per reference Access Units. Label List Byte array This is a list of Sub-part of the Genomic Dataset Header indicating (e.g. Labels, each one number of Labels integers) represented as a for each Label: multidimensional the Label ID array in order to the number of reference sequences concerned support selective by the label access to specific for each reference sequence genomic regions the reference identifier or sub-regions or the number of regions covered by the aggregations of label, regions or sub- for each region: regions. the class ID the start position in the genomic range the end position in the genomic range Start position and end position can be replaced by "block numbers", composing, together with reference sequence ID and class ID, a three dimensional vector addressing the coordinates of the Master Index Table. Parameters set Byte array Encoding parameters used to configure the encoding process and sent to the decoder.
[0266] Descriptors Layer Header
[0267] Descriptors (a.k.a. syntax elements) are described in the following sections of this document and are the building blocks of the genomic information representation described by this invention. They are organized in layers (a.k.a. descriptors streams) of homogeneous elements partitioned according to the specific statistical properties of each descriptor. This has the advantage of reducing the entropy of each layer and improving compression efficiency.
[0268] Each layer is prepended by the Descriptors Layer Header described below.
TABLE-US-00005 Syntax Description Descriptors_Layer_Header { Descriptors_Layer_ID Descriptors layer ID, table specified in this specification Num_Of_Blocks Number of Blocks in the Descriptors Layer Label size Size of the human readable label Label (Human-Readable) Label Flag Flag used to interpret the following metadata Local Index Table The Local Index Table structure as described in this invention Metadata Data structure carrying metadata to be used for application- specific processing such as data analysis and content protection. }
[0269] Block Header
[0270] Every Descriptors Layer is composed by one or multiple Genomic Data Blocks. One or more Blocks from different Layers compose an Access Unit, depending on the Class of data.
[0271] An Access Unit is a set of Genomic Blocks that can be decoded either independently from other Access Units by using only globally available data (e.g. decoder configuration) or by using information contained in other Access Units.
TABLE-US-00006 Syntax Semantic Block_Header { Descriptors_Layer_ID Unambiguously identifies the descriptors stream. Same as Descriptors_Layer_ID in Descriptor Layer Header Block size (BS) Number of bytes composing Block, including this header and payload, and excluding padding (total Block size will be BS + padding size). }
[0272] Definition of the Information Necessary to Represent Sequence Reads into Layers of Descriptors
[0273] Once the classification of reads is completed with the definition of the Classes, further processing consists in defining a set of distinct syntax elements which represent the remaining information enabling the reconstruction of the DNA read sequence when represented as being mapped on a given reference sequence.
[0274] A sequence read (e.g. a DNA segment) referred to a given reference sequence can be fully expressed by: [0275] The starting position on the reference sequence pos (292). [0276] A flag signaling if the read has to be considered as a reverse complement versus the reference rcomp (293). [0277] A distance to the mate pair in case of paired reads pair (294). [0278] The value of the read length (295) in case of the sequencing technology produces variable length reads. In case of constant reads length the read length associated to each reads can obviously be omitted and can be stored in the Genomic Dataset Header. [0279] For each mismatch: [0280] Mismatch position nmis (300) for class N, snpp (311) for class M, and indp (321) for class I) [0281] Mismatch type (not present in class N, snpt (312) in class M, indt (322) in class I) [0282] Flags (296) indicating specific characteristics of the sequence read such as: [0283] template having multiple segments in sequencing [0284] each segment properly aligned according to the aligner [0285] unmapped segment [0286] next segment in the template unmapped [0287] signalization of first or last segment [0288] quality control failure [0289] PCR or optical duplicate [0290] secondary alignment [0291] supplementary alignment [0292] Soft clipped nucleotides string (323) when present for class I [0293] Flag indicating the reference used for alignment and compression (e.g. internal reference for class U) if applicable (descriptor rtype). [0294] For class U, descriptor indc identifies those parts of the reads (typically the edges) that do not match, with a specified set of matching accuracy constraints, with the "internal" reference sequences. [0295] Descriptor ureads is used to encode verbatim the reads that cannot be mapped on any available reference being it "external" (i.e pre-existing like an actual reference genome) or a "internal" reference sequence.
[0296] This classification creates groups of descriptors (syntax elements) that can be used to univocally represent genome sequence reads. The table below summarizes the syntax elements needed for each class of reads aligned with "pre-existing" (i.e. "external") or "constructed" (i.e. "internal") references.
TABLE-US-00007 TABLE 3 Defined layers per class of data. P N M I U HM pos X X X X X X pair X X X X X rcomp X X X X X flags X X X X X rlen X X X X X nmis X snpp X X snpt X X indp X X indt X X indc X X ureads X X rtype X
[0297] Reads belonging to class P are characterized and can be perfectly reconstructed by only a position, a reverse complement information and an offset between mates in case they have been obtained by a sequencing technology yielding mated pairs, some flags and a read length.
[0298] The next section details how these descriptors are defined for classes P, N, M and I while for class U they are described in a later section.
[0299] Class HM is applied to read pairs only and it is a special case where one read belongs to class P, N, M or I and the other to class U.
[0300] Position Descriptors Layer
[0301] In each Access Unit, only the mapping position of the first encoded read is stored in the AU header as absolute position on the reference genome. All the other positions are expressed as a difference with respect to the previous position and are stored in a specific layer. This modeling of the information source, defined by the sequence of read positions, is in general characterized by a reduced entropy particularly for sequencing processes generating high coverage results. Once the absolute position of the first alignment has been stored, all positions of other reads are expressed as difference (distance) with respect to the first one.
[0302] For example FIG. 4 shows how after encoding the starting position of the first alignment as position "10000" on the reference sequence, the position of the second read starting at position 10180 is coded as "180". With high coverage data (>50.times.) most of the descriptors of the position vector will show very high occurrences of low values such as 0 and 1 and other small integers. FIG. 10 shows how the positions of three read pairs are encoded in a pos Layer.
[0303] The same source model is used for the positions of reads belonging to classes N, M, P and I. In order to enable any combination of selective access to the data, the positions of reads belonging to the four classes are encoded in separate layers as depicted in Table I.
[0304] Reverse Complement Descriptor Layer
[0305] Each read of the read pairs produced by sequencing technologies can be originated from either genome strands of the sequenced organic sample. However, only one of the two strands is used as reference sequence. FIG. 8 shows how in a reads pair one read (read 1) can be originated from one strand and the other (read 2) can be originated from the other strand.
[0306] When the strand 1 is used as reference sequence, read 2 can be encoded as reverse complement of the corresponding fragment on strand 1. This is shown in FIG. 9.
[0307] In case of coupled reads, four are the possible combinations of direct and reverse complement mate pairs. This is shown in FIG. 10. The rcomp layer codes the four possible combinations.
[0308] The same coding is used for the reverse complement information of reads belonging to classes P, N, M, I. In order to enable enhanced selective access to the data, the reverse complement information of reads belonging to the four classes are coded in different layers as depicted in Table 3.
[0309] Pairing Descriptors Layer
[0310] The pairing descriptor is stored in the pair layer. Such layer stores descriptors encoding the information needed to reconstruct the originating reads pairs, when the employed sequencing technology produces reads by pairs. Although at the date of the disclosure of the invention the vast majority of sequencing data is generated by using a technology generating paired reads, it is not the case of all technologies. This is the reason for which the presence of this layer is not necessary to reconstruct all sequencing data information if the sequencing technology of the genomic data considered does not generate paired reads information.
Definitions
[0311] mate pair: read associated to another read in a read pair (e.g. Read 2 is the mate pair of Read 1 in the example of FIG. 4) [0312] pairing distance: number of nucleotide positions on the reference sequence which separate one position in the first read (pairing anchor, e.g. last nucleotide of first read) from one position of the second read (e.g. the first nucleotide of the second read) [0313] most probable pairing distance (MPPD): this is the most probable pairing distance expressed in number of nucleotide positions. [0314] position pairing distance (PPD): the PPD is a way to express a pairing distance in terms of the number of reads separating one read from its respective mate present in a specific position descriptor layer. [0315] most probable position pairing distance (MPPPD): is the most probable number of reads separating one read from its mate pair present in a specific position descriptor layer. [0316] position pairing error (PPE): is defined as the difference between the MPPD or MPPPD and the actual position of the mate. [0317] pairing anchor: position of first read last nucleotide in a pair used as reference to calculate the distance of the mate pair in terms of number of nucleotide positions or number of read positions.
[0318] FIG. 5 shows how the pairing distance among read pairs is calculated.
[0319] The pair descriptor layer is the vector of pairing errors calculated as number of reads to be skipped to reach the mate pair of the first read of a pair with respect to the defined decoding pairing distance.
[0320] FIG. 6 shows an example of how pairing errors are calculated, both as absolute value and as differential vector (characterized by lower entropy for high coverages).
[0321] The same descriptors are used for the pairing information of reads belonging to classes N, M, P and I. In order to enable the selective access to the different data classes, the pairing information of reads belonging to the four classes are encoded in different layer as depicted in.
[0322] Pairing Information in Case of Reads Mapped on Different References
[0323] In the process of mapping sequence reads on a reference sequence it is not uncommon to have the first read in a pair mapped on one reference (e.g. chromosome 1) and the second on a different reference (e.g. chromosome 4). In this case the pairing information described above has to be integrated by additional information related to the reference sequence used to map one of the reads. This is achieved by coding
1. A reserved value (flag) indicating that the pair is mapped on two different sequences (different values indicate if read1 or read2 are mapped on the sequence that is not currently encoded) 2. a unique reference identifier referring to the reference identifiers encoded in the Genomic Dataset Header structure as described in Table 2. 3. a third element containing the mapping information on the reference identified at point 2 and expressed as offset with respect to the last encoded position.
[0324] FIG. 7 provides an example of this scenario.
[0325] In FIG. 7, since Read 4 is not mapped on the currently encoded reference sequence, the genomic encoder signals this information by crafting additional descriptors in the pair layer. In the example shown in FIG. 7 Read 4 of pair 2 is mapped on reference no. 4 while the currently encoded reference is no. 1. This information is encoded using 3 components:
1) One special reserved value is encoded as pairing distance (in this case 0xffffff) 2) A second descriptor provides a reference ID as listed in the Genomic Dataset Header (in this case 4) 3) The third element contains the mapping information on the concerned reference (170).
[0326] Mismatch Descriptors for Class N Reads
[0327] Class N includes all reads in which only "n type" mismatches are present, at the place of an A, C, G or T base a N is found as called base. All other bases of the read perfectly match the reference sequence.
[0328] FIG. 11 shows how: [0329] the positions of "N" in read 1 are coded as [0330] absolute position in read 1 or [0331] as differential position with respect to the previous "N" in the same read. the positions of "N" in read 2 are coded as [0332] absolute position in read 2+read 1 length or [0333] differential position with respect to the previous N In the nmis layer, the coding of each reads pair is terminated by a special "separator" symbol.
[0334] Encoding Substitutions (Mismatches or SNPs)
[0335] A substitution is defined as the presence, in a mapped read, of a different nucleotide with respect to the one that is present in the reference sequence at the same position (see FIG. 12).
[0336] Each substitution can be encoded as [0337] "position" (snpp layer) and "type" (snpt layer). See FIG. 13, FIG. 14, FIG. 16 and FIG. 15. [0338] OR [0339] "position" only but using one snpp layer per mismatch type. See FIG. 17
[0340] Substitutions Positions
[0341] A substitution position is calculated as for the values of the nmis layer, i.e.: In read 1 substitutions are encoded [0342] as absolute position in read 1 OR [0343] as differential position with respect to the previous substitution in the same read In read 2 substitutions are encoded
[0344] In read 1 substitutions are encoded [0345] as absolute position in read 2+read 1 length OR [0346] as differential position with respect to the previous substitution FIG. 13 shows how substitutions positions are encoded in layer snpp. Substitutions positions can be calculated either as absolute or as differential values.
[0347] In the snpp layer, the encoding of each reads pair is terminated by a special "separator" symbol.
[0348] Substitutions Types Descriptors
[0349] For class M (and I as described in the next sections), mismatches are coded by an index (moving from right to left) from the actual symbol present in the reference to the corresponding substitution symbol present in the read {A, C, G, T, N, Z}. For example if the aligned read presents a C instead of a T which is present at the same position in the reference, the mismatch index will be denoted as "4". The decoding process reads the encoded syntax element, the nucleotide at the given position on the reference and moves from left to right to retrieve the decoded symbol. E.g. a "2" received for a position where a G is present in the reference will be decoded as "N". FIG. 14 shows all the possible substitutions and the respective encoding symbols when IUPAC ambiguity codes are not used and FIG. 15 provides an example of encoding of substitutions types in the snpt layer.
[0350] In case of presence of IUPAC ambiguity codes, the substitution indexes change as shown in FIG. 16.
[0351] In case the encoding of substation types described above presents high information entropy, an alternative method of substitution encoding consists in storing only the mismatches positions in separate layers, one per nucleotide, as depicted in FIG. 17.
[0352] Encoding of Insertions and Deletions
[0353] For class I, mismatches and deletions are coded by an indexes (moving from right to left) from the actual symbol present in the reference to the corresponding substitution symbol present in the read: {A, C, G, T, N, Z}. For example if the aligned read presents a C instead of a T present at the same position in the reference, the mismatch index will be "4". In case the read presents a deletion where a A is present in the reference, the coded symbol will be "5". The decoding process reads the coded syntax element, the nucleotide at the given position on the reference and moves from left to right to retrieve the decoded symbol. E.g. a "3" received for a position where a G is present in the reference will be decoded as "Z" which indicates the presence of a deletion in the sequence read.
[0354] Inserts are coded as 6, 7, 8, 9, 10 respectively for inserted A, C, G, T, N.
[0355] In case of adoption of the IUPAC ambiguity codes the substitution mechanism results to be exactly the same however the substitution vector is extended as: S={A, C, G, T, N, Z, M, R, W, S, Y, K, V, H, D, B} and insertions use different codes: 16, 17, 18, 19, 20.
[0356] FIG. 18 and FIG. 19 show examples of how to encode substitutions, inserts and deletions in a reads pair of class I.
[0357] The following structures of file format, access units and multiplexing are described referring to the coding elements disclosed here above. However, the access units, the file format and the multiplexing produce the same technical advantage also with other and different algorithms of source modeling and genomic data compression.
[0358] Construction of "Internal" References for Unmapped Reads of "Class U" and "Class HM"
[0359] In the case of the reads belonging to Class U or the unmapped pair of "Class HM" since they cannot be mapped to any "external" reference sequence satisfying the specified set of matching accuracy constraints for belonging to any of the classes P, N, M, or I, one or more "internal" reference sequences are constructed and used for the compressed representation of the reads belonging to these data classes.
[0360] Several approaches are possible to construct appropriate "internal" references such as for instance and not as limitation: [0361] the partitioning of the non-mapped reads into clusters containing reads that share a common contiguous genomic sequence of at least a minimal size (signature). Each cluster can be uniquely identified by its signature. [0362] the sorting of reads in any meaningful order (e.g. lexicographic order) and the use of the last N reads as "internal" reference for the encoding of the N+1. This method is shown in FIG. 51. [0363] performing a so called "de-novo assembly" on a subset of the reads of class U so as to be able to align and encode all or a relevant sub-set of the reads belonging to said class according to the specified matching accuracy constraints or a new set of constraints.
[0364] If the read being coded can be mapped on the "internal" reference satisfying the specified set of matching accuracy constraints, the information necessary to reconstruct the read after compression is coded using syntax elements that can be of the following types: [0365] 1. Start position of the matching portion on the internal reference in terms of read number in the internal reference (pos layer). This position can be encoded either as absolute or differential value with respect to the previously encoded read. [0366] 2. Offset of the start position from the beginning of the corresponding read in the internal reference (pair layer). E.g. in case of constant read length the actual position is pos*length+pair. [0367] 3. Possibly present mismatches coded as mismatch position (snpp layer) and type (snpt layer) [0368] 4. Those parts of the reads (typically the edges identified by pair) that do not match with the internal reference (or do so, but with a number of mismatches above a defined threshold) are encoded in the indc layer. A padding operation can be performed to the edges of the part of the internal reference used in order to reduce the entropy of the mismatches encoded in the indc layer, as shown in FIG. 51. The most appropriate padding strategy can be chosen by the encoder according to the statistical properties of the genomic data being processed. Possible padding strategies include: [0369] a. No padding [0370] b. Constant padding pattern chosen according to its frequency in the currently encoded data. [0371] c. Variable padding pattern according to the statistical properties of the current context defined in terms of the latest N encoded reads
[0372] The specific type of padding strategy will be signaled by special values in the indc layer header [0373] 5. A flag that indicates if the read has been encoded using an internal self-generated, external or no-reference (rtype layer) [0374] 6. Reads which are encoded verbatim (ureads).
[0375] FIG. 51 provides an example of such encoding procedure.
[0376] FIG. 56 shows an alternative encoding of unmapped reads on the internal reference where pos+pair syntax elements are replaced by a signed pos. In this case pos would express the distance --in terms of positions on the reference sequence --of the left most nucleotide position of read n with respect of the position of the left most nucleotide of read n-1.
[0377] This coding approach can be extended to support N start positions per read so that reads can be split over two or more reference positions. This can be particularly useful to encode reads generated by those sequencing technology (e.g. from Pacific Bioscience) producing very long reads (50K+bases) which usually present repeated patterns generated by loops in the sequencing methodology. The same approach can be used as well to encode chimeric sequence reads defined as reads that align to two distinct portions of the genome with little or no overlap.
[0378] The approach described above can be clearly applied beyond the simple class U and could be applied to any layer containing syntax elements related to reads positions (pos layers).
[0379] File Format: Selective Access to Regions of Genomic Data by Using the Master Index Table
[0380] In order to support selective access to specific regions of the aligned data, the data structure described in this document implements an indexing tool called Master Index Table (MIT). This is a multi-dimensional array containing the loci at which specific reads map on the used reference sequences. The values contained in the MIT are the mapping positions of the first read in each pos layer so that non-sequential access to each Access Unit is supported. The MIT contains one section per each class of data (P, N, M, I, U and HM) and per each reference sequence. The MIT is contained in the Genomic Dataset Header of the encoded data. FIG. 20 shows the structure of the Genomic Dataset Header, FIG. 21 shows a generic visual representation of the MIT and FIG. 22 shows an example of MIT for the class P of encoded reads.
[0381] The values contained in the MIT depicted in FIG. 22 are used to directly access the region of interest (and the corresponding AU) in the compressed domain.
[0382] For example, with reference to FIG. 22, if it is required to access the region comprised between position 150,000 and 250,000 on reference 2, a decoding application would skip to the second reference in the MIT and would look for the two values k1 and k2 so that k1<150,000 and k2>250,000. Where k1 and k2 are 2 indexes read from the MIT. In the example of FIG. 22 this would result in positions 3 and 4 of the second vector of the MIT. These returned values will then be used by the decoding application to fetch the positions of the appropriate data from the pos layer Local Index Table as described in the next section.
[0383] Together with pointers to the layer containing the data belonging to the four classes of genomic data described above, the MIT can be uses as an index of additional metadata and/or annotations added to the genomic data during its life cycle.
[0384] Local Index Table
[0385] Each data layer described above is prefixed with a data structure referred to as local header. The local header contains a unique identifier of the layer, a vector of Access Units counters per each reference sequence, a Local Index Table (LIT) and optionally some layer specific metadata. The LIT is a vector of pointers to the physical position of the data belonging to each AU in the layer payload. FIG. 23 depicts the generic layer header and payload where the LIT is used to access specific regions of the encoded data in a non-sequential way.
[0386] In the previous example, in order to access region 150,000 to 250,000 of reads aligned on the reference sequence no. 2, the decoding application retrieved positions 3 and 4 from the MIT. These values shall be used by the decoding process to access the 3.sup.rd and 4.sup.th elements of the corresponding section of the LIT. In the example shown in FIG. 24, the Total Access Units counters contained in the layer header are used to skip the LIT indexes related to AUs related to reference 1 (5 in the example). The indexes containing the physical positions of the requested AUs in the encoded stream are therefore calculated as:
position of the data blocks belonging to the requested AU=data blocks belonging to AUs of reference 1 to be skipped+position retrieved using the MIT, i.e. First block position: 5+3=8 Last block position: 5+4=9
[0387] The blocks of data retrieved using the indexing mechanism called Local Index Table, are part of the Access Units requested.
[0388] FIG. 26 shows how the data blocks retrieved using the MIT and the LIT compose one or more Access Units.
[0389] Access Units The genomic data classified in data classes and structured in compressed or uncompressed layers are organized into different Access Units.
[0390] Genomic Access Units (AU) are defined as sections of genome data (in a compressed or uncompressed form) that reconstructs nucleotide sequences and/or the relevant metadata, and/or sequence of DNA/RNA (e.g. the virtual reference) and/or annotation data generated by a genome sequencing machine and/or a genomic processing device or analysis application. An example of Access Unit is provided in FIG. 26.
[0391] An Access Unit is a block of data that can be decoded either independently from other Access Units by using only globally available data (e.g. decoder configuration) or by using information contained in other Access Units.
[0392] Access Units are differentiated by: [0393] type, characterizing the nature of the genomic data and data sets they carry and the way they can be accessed, [0394] order, providing a unique order to Access Units belonging to the same type.
[0395] Access units of any type can be further classified into different "categories".
[0396] Hereafter follows a non-exhaustive list of definition of different types of genomic Access Units: [0397] 1) Access units of type 0 do not need to refer to any information coming from other Access Units to be accessed or decoded and accessed. The entire information carried by the data or data sets they contain can be independently read and processed by a decoding device or processing application. [0398] 2) Access units of type 1 contain data that refer to data carried by Access Units of type 0. Reading or decoding and processing the data contained in Access Units of type 1 requires having access to one or more Access Units of type 0. Access unit of type 1 encode genomic data related to sequence reads of "Class P" [0399] 3) Access Units of type 2 contain data that refer to data carried by Access Units of type 0. Reading or decoding and processing the data contained in Access Units of type 2 requires having access to one or more Access Units of type 0. Access unit of type 2 encode genomic data related to sequence reads of "Class N" [0400] 4) Access Units of type 3 contain data that refer to data carried by Access Units of type 0. Reading or decoding and processing the data contained in Access Units of type 3 requires having access to one or more Access Units of type 0. Access unit of type 3 encode genomic data related to sequence reads of "Class M" [0401] 5) Access Units of type 4 contain data that refer to data carried by Access Units of type 0. Reading or decoding and processing the data contained in Access Units of type 4 requires having access to one or more Access Units of type 0. Access unit of type 4 encode genomic data related to sequence reads of "Class I" [0402] 6) Access Units of type 5 contain reads that cannot be mapped on any available reference sequence ("Class U") and are encoded used an internally constructed reference sequence. Access Units of type 5 contain data that refer to data carried by Access Units of type 0. Reading or decoding and processing the data contained in Access Units of type 5 requires having access to one or more Access Units of type 0. [0403] 7) Access Units of type 6 contain read pairs where one read can belong to any of the four classes P, N, M, I and the other cannot be mapped on any available reference sequence ("Class HM"). Access Units of type 6 contain data that refer to data carried by Access Units of type 0. Reading or decoding and processing the data contained in Access Units of type 6 requires having access to one or more Access Units of type 0. [0404] 8) Access Units of type 7 contain metadata (e.g. quality scores) and/or annotation data associated to the data or data sets contained in the access unit of type 1. Access Units of type 7 may be classified and labelled in different layers. [0405] 9) Access Units of type 8 contain data or data sets classified as annotation data. Access Units of type 8 may be classified and labelled in layers. [0406] 10) Access Units of additional types can extend the structure and mechanisms described here. As an example, but not as a limitation, the results of genomic variant calling, structural and functional analysis can be encoded in Access Units of new types. The data organization in Access Units described herein does not prevent any type of data to be encapsulated in Access Units being the mechanism completely transparent with respect to the nature of encoded data.
[0407] Access Units of type 0 are ordered (e.g. numbered), but they do not need to be stored and/or transmitted in an ordered manner (technical advantage: parallel processing/parallel streaming, multiplexing)
[0408] Access Units of type 1, 2, 3, 4, 5 and 6 do not need to be ordered and do not need to be stored and/or transmitted in an ordered manner (technical advantage: parallel processing/parallel streaming).
[0409] FIG. 26 shows how Access Units are composed by a header and one or more layers of homogeneous data. Each layer can be composed by one or more blocks. Each block contains several packets and the packets are a structured sequence of the descriptors introduced above to represent e.g. reads positions, pairing information, reverse complement information, mismatches positions and types etc.
[0410] Each Access unit can have a different number of packets in each block, but within an Access Unit all blocks have the same number of packets.
[0411] Each data packet can be identified by the combination of 3 identifiers X Y Z where: [0412] X identifies the access unit it belongs to [0413] Y identifies the block it belongs to (i.e. the data type it encapsulates) [0414] Z is an identifier expressing the packet order with respect to other packets in the same block
[0415] FIG. 28 shows an example of Access Units and packets labelling where AU T N is an access unit of type T with identifier N which may or may not imply a notion of order according to the Access Unit Type. Identifiers are used to uniquely associate Access Units of one type with those of other types required to completely decode the carried genomic data.
[0416] Access Units of any type can be further classified and labelled in different "categories" according to different sequencing processes. For example, but not as a limitation, classification and labelling can take place when [0417] 1. sequencing the same organism at different times (Access Units contain genomic information with a "temporal" connotation), [0418] 2. sequencing organic samples of different nature of the same organisms (e.g. skin, blood, hair for human samples). These are Access Units with "biological" connotation.
[0419] The access units of type 1, 2, 3, 4, 5 and 6 are built according to the result of a matching function applied on genome sequence fragments (a.k.a. reads) with respect to the reference sequence encoded in Access Units of type 0 they refer to.
[0420] For example access units (AUs) of type 1 (see FIG. 30) may contain the positions and the reverse complement flags of those reads which result in a perfect match (or maximum possible score corresponding to the selected matching function) when a matching function is applied to specific regions of the reference sequence encoded in AUs of type 0. Together with the data contained in AUs of type 0, such matching function information is sufficient to completely reconstruct all genome sequence reads represented by the data set carried by the access units of type 1.
[0421] With reference to the genomic data classification previously described in this document, the Access Units of type 1 described above would contain information related to genomic sequence reads of class P (perfect matches).
[0422] In case of variable reads length and paired reads the data contained in AUs of type 1 mentioned in the previous example, have to be integrated with the data representing the information about reads pairing and reads length in order to be able to completely reconstruct the genomic data including the reads pairs association. With respect to the data classification previously introduced in the present document, pair and rlen layers would be encoded in AU of type 1.
[0423] The matching functions applied with respect to access units of type 1 to classify the content of AU for the type 2, 3 and 4 can provide results such as: [0424] 1. each sequence contained in the AU of type 1 perfectly matches sequences contained in the AU of type 0 in correspondence to the specified position; [0425] 2. each sequence contained in the AU of type 2 perfectly matches a sequence contained in the AU of type 0 in correspondence to the specified position, except for the "N" symbols present (base not called by the sequencing device) in the sequence in the AU of type 2; [0426] 3. each sequence contained in the AU of type 3 includes variants in the form of substituted symbols (variants) with respect to the sequence contained in the AU of type 0 in correspondence to the specified position; [0427] 4. each sequence contained in the AU of type 4 includes variants in the form of substituted symbols (variants), insertions and/or deletions with respect to the sequence contained in the AU of type 0 in correspondence to the specified position. [0428] 5. each sequence contained in the AU of type 5 do not map any sequence contained in the AU of type 0. [0429] 6. each sequence pair contained in the AU of type 6 presents one sequence that can belong to any class P, N, M and I (points 1 to 4 above) while the other sequence does not map any sequence contained in the AU of type 0.
[0430] Access units of type 0 are ordered (e.g. numbered), but they do not need to be stored and/or transmitted in an ordered manner (technical advantage: parallel processing/parallel streaming, multiplexing)
[0431] Access units of type 1, 2, 3, 4, 5 and 6 do not need to be ordered and do not need to be stored and/or transmitted in an ordered manner (technical advantage: parallel processing/parallel streaming).
[0432] Identifying Access Units by Using "Labels" Associated to Specific Genomic Regions
[0433] An additional mechanism is provided by the disclosed invention enabling user-defined selective access to data classes referring to specific genomic regions or sub-regions or aggregations of regions or sub-regions.
[0434] A "Label" is an identifier which is assigned to a specific genomic region or sub-region or aggregations of regions or sub-regions. Labels identify genomic regions by specifying: the reference sequence id ("Ref ids"), the index of the MIT corresponding to the desired region of the reference sequence, and the data classes. An example is provided in FIG. 52.
[0435] A single, a subset, or all data classes can be referenced by a Label, enabling selective access to only a sub-set of the data associated to a specific genomic region or sub-regions or aggregations of regions or sub-regions.
[0436] A Label list should be created by a Genomic Labels Generator (4917 FIG. 49), in a storage scenario, and/or in a streaming scenario to make available the available Labels to the analysis applications applying a selective access to the stored or streamed data.
[0437] A Label List might include the following elements: [0438] the number of Labels [0439] for each Label in the list: [0440] the Label ID [0441] the number of reference sequences concerned by the label [0442] for each reference sequence [0443] the reference identifier [0444] the number of regions covered by the label, [0445] for each region: [0446] the class ID [0447] the start position in the genomic range [0448] the end position in the genomic range
[0449] The table below reports a pseudo-syntax for a generic "Label List".
TABLE-US-00008 TABLE 4 Syntax of the generic "Label List" data format. Syntax Description Label_list( ) { num_Labels total number of labels in the list for (i=0; i<num_Labels;i++) { Label_id label identifier num_ref number of references concerned by the current label for (j = 0; j < num_ref; j++) { ref_id current reference num_regions number of different regions of this reference identified by the label for (k = 0; k < num_regions; k++) { class_id type of class, start and end position of start_pos this region end_pos } } } }
[0450] In case Genomic Data are compressed and streamed, one or more Access Units can be identified using a specific "Label" by means of a Block Header field ("Label ID"), which serves as an identifier for the "Label" in the "Label List" which the current block belongs to. Such field enables a dynamic mapping of blocks to "Labels", typical for streaming scenarios.
[0451] In the Genomic File Format, the "start_pos" and "end_pos" fields can be replaced by the block numbers referring to all "blocks" belonging to a specific "Label", as follows:
TABLE-US-00009 TABLE 5 Efficient implementation of the "Label List" Syntax data format in the case of a compressed file. Syntax Data type Description num_Labels Bitstring number of labels in the genomic dataset for (i=0; i<num_Labels;i++) { Label_id Bitstring label identifier Label_length_in_blocks Bitstring number of data blocks identified by one label for (j = 0; j < Label_length_in_blocks;j++) { ref_id Bitstring reference id for this block class_id Bitstring class id for this block block_num Bitstring block number in the Master Index Table } }
[0452] The use of block numbers instead of "start_pos" and "end_pos" presents a relevant technical advantage because it enables a direct access to the "Master Index Table" (MIT), considering that the three dimensional vector consisting of: "ref_num", "class_id" and "block_num" can be used as coordinates to directly address the MIT itself.
[0453] In storage scenarios, the "Label List" is created by a Genomic Labels Generator (4917) and sent to the genomic multiplexer (see also FIG. 49). The demultiplexer parses the Label List syntax and exposes the available Labels to the data access application, which according to the specific data access required selects the Access Units corresponding to the subset of "Labels".
[0454] The possibility of using "Labels" to identify Access Units associated to specific genomic regions does not prevent using the indexing tools such as MIT and LIT without "Labels" to achieve random data access functionality. Generic random access can be achieved by specifying a three dimensional vector determining the MIT and LIT coordinates of interest (reference id, position range and classes) and ignoring the information carried by the Label List.
[0455] FIG. 51 shows how labels are used to aggregate and uniquely identify several genomic regions by using indexes contained in the MIT.
[0456] FIG. 59 shows how a device (592) implementing the labelling mechanism disclosed by this invention can enable concurrent access to several records of data (596) stored in a database (595). Selective protection of one or more regions identified by the same label is supported as well by means of a dedicated module (591) in charge of parsing the query (591) and dispatching the required metadata to the security module (594) in charge of enforcing access control. The labels decoder (593) is in charge of translating the label syntax into object identifiers that can be protected (and therefore access is controlled by the security module 594) or not.
Technical Effects
[0457] The technical effect of structuring genomic information in Access Units or Access Units identified by Labels as described here is that the genomic data:
1. can be selectively queried in order to access: [0458] specific "categories" of data (e.g. with a specific temporal or biological connotation) without having to decompress the entire genomic data or data sets and/or the related metadata. [0459] specific regions of the genome for all "categories", a subset of "categories", a single "category" (with or without the associated metadata) without the need to decompress other regions of the genome [0460] specific genomic regions or sub-regions or aggregations of regions or sub-regions identified by user defined "Labels" by only parsing the "Label List" main header and accessing (i.e. retrieving and decompressing) only the corresponding Access Units 2. can be incrementally updated with new data that can be available when: [0461] new analysis is performed on the genomic data or data sets [0462] new genomic data or data sets are generated by sequencing the same organisms (different biological samples, different biological sample of the same type, e.g. blood sample, but acquired at a different time, etc.) 3. can be efficiently transcoded to a new data format in case of [0463] new genomic data or data sets to be used as new reference (e.g. new reference genome carried by AU of type 0) [0464] update of the encoding format specification 4. can be protected with different levels of granularity in terms of both access control (e.g. encryption) and permissions enforcement. For example these scenarios are enabled: [0465] the same access control rule and encryption keys can be applied to all the genomic regions or sub-regions identified by one label (see FIG. 54 for an example); [0466] different access control rules and different encryption keys can be used to protect each single region or sub-regions aggregated under the same label (see FIG. 55 for an example).
[0467] With respect to prior art solutions such as SAM/BAM, the described technical features address the issues of requiring data filtering to happen at the application level when the entire data has been retrieved and decompressed from the encoded format.
[0468] Hereafter follows examples of application scenario where the association of access unit structure, file format and Labelling mechanism becomes instrumental for a technological advantage.
[0469] Selective Access
[0470] In particular the disclosed data structure based on Access Units of different types possibly including user defined "Labels" enables to: [0471] extract only the read information (data or data sets) of the whole sequencing of all "categories" or a subset (i.e. one or more layers) or a single "category" without having to decompress also the associated metadata information (limitation of current state of the art: SAM/BAM that cannot even support distinction between different categories or layers); [0472] extract all the reads aligned on specific regions of the assumed reference sequence for all categories, subsets of the categories, a single category (with or without the associated metadata) without the need of decompressing also other regions of the genome (limitation of current state of the art: SAM/BAM); [0473] extract all the reads belonging to a single, a subset or all data "classes" aligned on specific genomic regions or sub-regions or aggregations of regions or sub-regions identified by user specified "Labels" for all categories, subsets of the categories, a single category (with or without the associated metadata) without the need of decompressing also other data associated to other regions of the genome (limitation of current state of the art: SAM/BAM).
[0474] FIG. 39 shows how the access to the genomic information mapped on the second segment of the reference sequence (AU 0-2) with mismatches only requires the decoding of AUs 0-2, 1-2 and 3-2 only. This is an example of selective access according to both a criteria related to a mapping region (i.e. position on the reference sequence) and a criteria related to the matching function applied to the encoded sequence reads with respect to the reference sequence (e.g. mismatches only in this example).
[0475] A further technical advantage is that the querying on the data is much more efficient in terms of data accessibility and execution speed because it can be based on accessing and decoding only selected "categories", specific regions of longer genomic sequences and only specific layers for access units of type 1, 2, 3, 4 that match the criteria of the applied queries and any combination thereof.
[0476] The organization of access units of type 1, 2, 3, 4 into layers allow for efficient extraction of nucleotides sequences [0477] with specific variations (e.g. mismatches, insertions, deletions) with respect to one or more reference genomes; [0478] that do not map to any of the considered reference genomes; [0479] that perfectly map on one or more reference genomes; [0480] that map with one or more accuracy levels.
[0481] FIG. 52 shows how the access to the genomic information associated only to specific genomic regions or sub-regions or aggregations of regions or sub-regions associated to user defined "Labels". The syntax of a label is based on a three coordinates system where each region or sub-region associated to a label can be uniquely identified by: [0482] 1. reference ID, [0483] 2. data type (class) [0484] 3. block number in the MIT (corresponding to a genomic region).
[0485] These three coordinates can be used to identify [0486] the MIT location containing the genomic position of the region on the corresponding reference and [0487] the LIT location containing the physical location of the data representing the respective genomic region or sub-region
[0488] Like in the case of accessing data related to a specified genomic region, a further technical advantage is that the querying on the data results to be much more efficient in terms of data accessibility and execution speed because it can be based on accessing and decoding only selected "categories", of the labelled specific regions and only specific layers for access units of type 1, 2, 3, 4 that corresponds to the "Labels" of the applied queries and any combination thereof.
[0489] Another technical advantage of this labelling mechanism is the possibility of efficiently retrieving encoded genomic information that has been scattered among several Access Units due to its characteristics such as position on the reference genome, type of mismatches with respect to the reference (524).
[0490] Filtering genomic data according to the characteristics of the mapped reads (e.g. perfectly matching, substitutions only, etc.) today can take hours when using the traditional formats such as BAM and CRAM. This is due to the fact that the data are sparse within the compressed format and require decompression and filtering using pipelines of commands. The present invention describes a data structure that enables data filtering in a matter of seconds. Memory usage can be as well reduced by a factor that is proportional with the file size (from 10.times. to 100.times.) since the present invention does not require the decoding (i.e. memory allocation) of the entire file.
[0491] Selective Access to Specific Genomic Regions Identified by User Specified "Labels" in "Storage" and "Streaming" Scenarios.
[0492] For example let's suppose that sequencing data is compressed and selective access to "GeneXY" and "GeneWZ" is required. The two genomic regions corresponding to "GeneXY" and "GeneWZ" in the compressed file format or in the compressed stream must be labelled. Depending if a compressed data file is generated for storage or a compressed data stream is generated for streaming, two methods are used.
[0493] In the case of a compressed data file, the multiplexer creates a "Label List" which includes two Labels with: "Label_ID"=GeneXY and "Label_ID"=GeneWZ. The Label parameter "Label_lenght_in_blocks" and for each block the parameters: "ref_num", "class_ID", "block_num" are determined by the multiplexer based on the position on the reference of the "GeneXY" and "GeneWZ" regions and the class of data for which the selective access is desired. The complete syntax is reported in Table 5.
[0494] In the case of a compressed stream, the multiplexer creates a "Label List" which includes two Labels with: "Label_ID"=GeneXY and "Label_ID"=GeneWZ. The Label parameters "ref ID", "class_ID", "start_pos" and "end_pos" are determined by the multiplexer based on the position on the reference of the "GeneXY" and "GeneWZ" regions and the class of data for which the selective access is desired. The complete syntax is reported in Table 4.
[0495] The method used in the case of a compressed stream is generic and could be used also in the case of a compressed file for storage, but the corresponding implementation would result less efficient because the use of block numbers, as described in the case of compressed file, enables a direct access to the "Master Index Table" (MIT).
[0496] In both cases mentioned above (streaming and storage), the mechanism of retrieval of the genomic data identified by the labels follows is the same.
[0497] When parsing a label a decoding device will: [0498] 1. Identify the reference sequence from the first element of the label [0499] 2. Identify the class of data from the second element of the label [0500] 3. Identify the block of the MIT (corresponding to a genomic region) from the third element of the label [0501] 4. The two coordinates parsed in 1 and 2 enable the decoder to identify the required Genomic Streams (484), [0502] 5. Each Genomic Stream starts with a header containing a LIT (525) containing pointers to the descriptors encoding data mapped to each genomic region. The third coordinate parsed in 3 is used to access the correct pointer in the LIT of each Genomic Stream. [0503] 6. The decoder can efficiently retrieve all the descriptors to decode the genomic data identified by the decoded Genomic Label even if they are scattered among different Access Units (524).
[0504] Incremental Update
[0505] The access units of type 7 and 8 allow for easy insertion of annotations without the need of depacketizing/decoding/decompressing the whole file thereby adding to the efficient handling of the file which is a limitation of prior art approaches. Existing compression solutions may have to access and process a large amount of compressed data before the desired genomic data can be accessed. This will cause inefficient RAM bandwidth utilization and more power consumption also in hardware implementations. Power consumption and memory access issues may be alleviated by using the approach based on Access Units described here.
[0506] The data indexing mechanism described in the Master Index Table (see FIG. 21) together with the utilization of Access Unites and the possibility of identifying Access Units with user-defined "Labels" associated to specific genomic regions or sub-regions or aggregations of regions or sub-regions enables incremental update of the encoded content as described below. This mechanism is shown with an example in FIG. 53.
[0507] Insertion of Additional Data
[0508] New genomic information can be periodically added to existing genomic data for several reasons. For example when: [0509] An organism is sequenced at different moments in time; [0510] Several different samples of the same individual are sequenced at the same time; [0511] New data generated by a sequencing process (streaming).
[0512] In the above mentioned situations, structuring data using the Access Units described here and the data structure described in the file format section enables the incremental integration of the newly generated data without the need to re-encode the existing data. The incremental update process can be implemented as follows: [0513] 1. The newly generated AUs can simply be concatenated in the file with the pre-existing AUs and [0514] 2. the indexing of the newly generated data or data sets are included in the Master Index Table described in the file format section of this document. One index shall position the newly generated AU on the existing reference sequence, other indexes consist in pointers of the newly generated AUs in the physical file to enable direct and selective access to them. [0515] 3. The existing and/or newly generated AU can be identified with user defined "Labels" corresponding to specific genomic regions or sub-regions or aggregations of regions or sub-regions and a "Label List" can be included or updated.
[0516] This mechanism is illustrated in FIG. 40 where pre-existing data encoded in 3 AUs of type 1 and 4 AUs per each type from 2 to 4 are updated with 3 AUs per type with encoding data coming for example from a new sequence run for the same individual.
[0517] The mechanism of creating or updating "Labels" and the "Label List" are illustrated in FIG. 52 and FIG. 53.
[0518] In the specific use case of streaming genomic data and data sets in compressed form, the incremental update of a pre-existing data set may be useful when analyzing data as soon as they are generated by a sequencing machine and before the actual sequencing is completed. An encoding engine (compressor) can assemble several AUs in parallel by "clustering" sequence reads that map on the same region of the selected reference sequence. Once the first AU contains a number of reads above a pre-configured threshold/parameter, the AU is ready to be sent to the analysis application. Together with the newly encoded Access Unit, the encoding engine (the compressor) shall make sure that all Access Units the new AU depends on have already been sent to the receiving end or is sent together with it. For example an AU of type 3 will require the appropriate AU of type 0 and type 1 to be present at the receiving end in order to be properly decoded.
[0519] By means of the described mechanism, a receiving variant calling application would be able to start calling variants on the AU received before the sequencing process has been completed at the transmitting side. A schematic of this process is depicted in FIG. 41.
[0520] New Analysis of Results.
[0521] During the genome processing life cycle several iterations of genome analysis can be applied on the same data (e.g. different variant calling using different processing algorithm). The use of AUs as defined in this document and the data structure described in the file format section of this document enable incremental update of existing compressed data with the results of new analysis. For example, new analysis performed on existing compressed data can produce new data in these cases: [0522] 1. A new analysis can modify existing results already associated with the encoded data. This use case is depicted in FIG. 42 and it is implemented by moving entirely or partially the content of one Access Unit from one type to another. In case new AUs need to be created (due to a pre-defined maximum size per AU), the related indexes in the Master Index Table must be created and the related vector is sorted when needed. [0523] 2. New data are produced from new analysis and have to be associated to existing encoded data. In this case new AUs of type 7 can be produced and concatenated with the existing vector of AUs of the same type. This and the related update of the Master Index Table are depicted in FIG. 43
[0524] The use cases described above and depicted in FIG. 42 and FIG. 43 are enabled by: [0525] 1. The possibility to have direct access only to data with poor mapping quality (e.g. AUs of type 4); [0526] 2. The possibility to remap reads to a new genomic region by simply creating a new Access Unit possibly belonging to a new type (e.g. reads included in a Type 4 AU can be remapped to a new region with less (type 2-3) mismatches and included in a newly created AU); [0527] 3. The possibility to create AU of type 8 (433) containing only the newly created analysis results and/or related annotations. In this case the newly created AUs only require to contain "pointers" to the existing AUs to which they refer to. [0528] 4. The possibility of performing in a single run new analysis on several genomic regions and sub-regions identified by the same Label without the need to repeat the analysis on each single genomic region or sub-region. Labels as described in this document would enable users to manipulate non-contiguous genomic segments as if they were a single genomic sequence. [0529] 5. The possibility of updating with new analysis results several genomic regions or sub regions identified by a single Label. The new results (usually expressed in the form of metadata) would be linked to the label identifying the aggregation of potentially several genomic regions and sub regions without the need of creating several links from the results to each genomic region or sub region.
[0530] Transcoding
[0531] Compressed genomic data can require transcoding, for example, in the following situations: [0532] Publication of new reference sequences; [0533] Use of a different mapping algorithm (re-mapping).
[0534] When genomic data is mapped on an existing public reference genome, the publication of a new version of said reference sequence or the desire to map the data using a different processing algorithm, today requires a process of re-mapping. When remapping compressed data using prior art file formats such as SAM or CRAM the entire compressed data has to be decompressed into its "raw" form to be mapped again with reference to the newly available reference sequence or using a different mapping algorithm. This is true even if the newly published reference is only slightly different from the previous or the different mapping algorithm used produces a mapping that is very close (or identical) to the previous mapping.
[0535] The advantage of transcoding genomic data structured using Access Units described here is that: [0536] 1. Mapping versus a new reference genome only requires re-encoding (decompressing and compressing) the data of AUs that map on the genome regions that have changes. Additionally the user may select those compressed reads that for any reason might need to be re-mapped even if they originally do not map on the changed region (this may happen if the user believes that the previous mapping is of poor quality). This use case is depicted in FIG. 44. [0537] 2. In case the newly published reference genome differs from the previous only in terms of entire regions shifted to different genomic locations ("loci"), the transcoding operation results particularly simple and efficient. In fact in order to move all the reads mapped to the "shifted" region it is sufficient to change only the value of the absolute position contained in the related (set of) AU(s) header. Each AU header contain the absolute position the first read contained in the AU is mapped to on the reference sequence, while all other reads positions are encoded differentially with respect to the first. Therefore, by simply updating the value of the absolute position of the first read, all the reads in the AU are moved accordingly. This mechanism cannot be implemented by state of the art approaches such as CRAM and BAM because genome data positions are encoded in the compressed payload, thus requiring complete decompression and re-compression of all genome data sets. [0538] 3. When a different mapping algorithm is used, it is possible to apply it only on a portion of compressed reads that was deemed mapped with poor quality. For example it can be appropriate to apply the new mapping algorithm only on reads which did not perfectly match on the reference genome. With existing formats today it is not possible (or it's only partially possible with some limitations) to extract reads according to their mapping quality (i.e. presence and number of mismatches). If new mapping results are returned by the new mapping tools the related reads can be transcoded from one AU from another of the same type (FIG. 46) or from one AU of one type to an AU of another type (FIG. 45).
[0539] Moreover, prior art compression solutions may have to access and process a large amount of compressed data before the desired genomic data can be accessed. This will cause inefficient RAM bandwidth utilization and more power consumption and in hardware implementations. Power consumption and memory access issues may be alleviated by using the approach based on Access Units described here.
[0540] A further advantage of the adoption of the genomic access units described here is the facilitation of parallel processing and suitability for hardware implementations. Current solutions such as SAM/BAM and CRAM are conceived for single-threaded software implementation.
[0541] Selective Protection
[0542] The approach based on Access Units organized in several types an layers as described in this document enables the implementation of content protection mechanisms otherwise not possible with state of the art monolithic solutions.
[0543] A person skilled in the art knows that the majority of genomic information related to an organism's genetic profile relies in the differences (variants) with respect to a known sequence (e.g. a reference genome or a population of genomes). An individual genetic profile to be protected from unauthorized access will therefore be encoded in Access Units of type 3 and 4 as described in this document. The implementation of controlled access to the most sensible genomic information produced by a sequencing and analysis process can therefore be realized by encrypting only the payload of AUs of type 3 and 4 (see FIG. 47 for an example). This will generate significant savings in terms of both processing power and bandwidth since the resources consuming encryption process shall be applied on a subset of data only.
[0544] Selective Protection of Specific Genomic Regions Identified by "Labels"
[0545] The labelling mechanism enables different mechanisms of data protection and access control. For example FIG. 54 shows how one protection mechanism (e.g. encryption) and one access control rule (AC) can be applied to several genomic regions identified by the same label. In a more sophisticated scenario, data protection can be implemented by applying a different access control rule and a different protection mechanism (encryption) to each region identified by a label. This is shown in FIG. 55.
[0546] Additionally, selective encryption of genomic regions or sub-regions or aggregations of regions or sub-regions identified by different "Labels" can be easily implemented by applying encryption only to compressed data corresponding to a "Label" for both file and streamed scenarios. For instance two genomic regions labelled as "GeneXY" and "GeneWZ" like in the example of section "Selective Access to Specific Genomic Regions identified by User Specified "Labels" in "storage" and "streaming" scenarios" can be differentiated by only encrypting data labelled by "GeneXY" and leaving in clear the compressed data labelled as "GeneWZ". Encryption rules can be carried by the metadata fields (in both storage and streaming scenarios) and associated to each element of the "Label List"
[0547] Transport of Genomic Access Units
[0548] Genomic Data Multiplex
[0549] Genomic Access Units can be transported over a communication network within a Genomic Data Multiplex. A Genomic Data Multiplex is defined as a sequence of packetized genomic data and metadata represented according to the data classification disclosed as part of this invention, transmitted in network environments where errors, such as packet losses, may occur.
[0550] The Genomic Data Multiplex is conceived to ease and render more efficient the transport of genomic coded data over different environments (typically network environments) and has the following advantages not present in state of the art solutions: [0551] 1. it enables encapsulation of either a stream or a sequence of genomic data (described below) or Genomic File Format generated by an encoding tool into one or more Genomic Data Multiplex, in order to carry it over a network environment, and then recover a valid and identical stream or file format in order to render the transmission and access to information more efficient [0552] 2. It enables selective retrieval of encoded genomic data from the encapsulated Genomic Data Streams, for decoding and presentation. [0553] 3. It enables multiplexing several Genomic Datasets into a single container of information for transport and it enables de-multiplexing a subset of the carried information into a new Genomic Data Multiplex. [0554] 4. It enables the multiplexing of data and metadata produced by different sources (with the consequent separate access) and/or sequencing/analysis processes and transmit the resulting Genomic Data Multiplex over a network environment. [0555] 5. It supports identification of errors such as packet losses. [0556] 6. It supports proper reorder data which may arrive out of order due to network delays, therefore rendering more efficient the transmission of genomic data when compared with the state of the art solutions
[0557] An Example of Genomic Data Multiplexing is Shown in FIG. 49.
[0558] Genomic Dataset
[0559] In the context of the present invention a Genomic Dataset is defined as a structured set of Genomic Data including, for example, genomic data of a living organism, one or more sequences and metadata generated by several steps of genomic data processing, or the result of the genomic sequencing of a living organism. One Genomic Data Multiplex may include multiple Genomic Datasets (as in a multi-channel scenario) where each dataset refers to a different organism. The multiplexing mechanism of the several datasets into a single Genomic Data Multiplex is governed by information contained in data structures called Genomic Datasets List (GDL), Genomic Dataset Mapping Tables List (GDMTL) and Genomic Dataset Mapping Table (GDMT).
[0560] Genomic Dataset List
[0561] A Genomic Dataset List (GDL) is defined as a data structure listing all Genomic Datasets available in a Genomic Data Multiplex. Each of the listed Genomic Datasets is identified by a unique value called Genomic Dataset ID (GID).
[0562] Each Genomic Dataset listed in the GDL is associated to: [0563] one Genomic Data Stream carrying one Genomic Dataset Mapping Table (GDMT) and identified by a specific value of Stream ID (genomic_dataset_map_SID); [0564] one Genomic Data Stream carrying one Reference ID Mapping Table (RIDMT) and identified by a specific value of Stream ID (reference_id_map_SID).
[0565] The GDL is sent as payload of a single Transport Packet at the beginning of a Genomic Data Stream transmission; it can then be periodically re-transmitted in order to enable random access to the Stream.
[0566] The syntax of the GDL data structure is provided in the table below with an indication of the data type associated to each syntax element.
TABLE-US-00010 Syntax Data type genomic_dataset_list( ) { list_length bitstring multiplex_id bitstring version_number bitstring applicable_section_flag bit list_ID bitstring for (i = 0; i < N; i++) { N = number of Genomic Datasets in this Genomic Multiplex genomic_dataset_ID bitstring genomic_dataset_map_SID bitstring reference_id_map_SID bitstring genomic_Label_list_SID bitstring } Checksum bitstring }
[0567] The syntax elements composing the GDL described above have the following meaning and function.
TABLE-US-00011 section_length bitstring field, specifying the number of bytes composing the section, starting immediately following the section_length field, and including the CRC. multiplex_id bitstring field which serves as a label to identify this multiplexed stream from any other multiplex within a network. version_number bitstring field indicating the version number of the whole Genomic Dataset List Section. The version number shall be incremented by 1 whenever the definition of the Genomic Dataset Mapping Table changes. When the applicable_section_flag is set to `1`, then the version_number shall be that of the currently applicable Genomic Dataset List. When the applicable_section_flag is set to `0`, then the version_number shall be that of the next applicable Genomic Dataset List. applicable_section_flag A 1 bit indicator, which when set to `1` indicates that the Genomic Dataset Mapping Table sent is currently applicable. When the bit is set to `0`, it indicates that the table sent is not yet applicable and shall be the next table to become valid. list_ID This is a bitstring field identifying the current genomic dataset list. genomic_dataset_ID genomic_dataset_ID is a bitstring field which specifies the genomic dataset to which the genomic_dataset_map_SID is applicable. This field shall not take any single value more than once within one version of the Genomic Dataset Mapping Table. genomic_dataset_map_SID genomic_dataset_map_SID is a bitstring field identifying the Genomic Data Stream carrying the Genomic Dataset Mapping Table (GDMT) associated to this Genomic Dataset. No genomic_dataset_ID shall have more than one genomic_dataset_map_SID associated. The value of the genomic_dataset_map_SID is defined by the user. reference_id_map_SID reference_id_map_SID is a bitstring field identifying the Genomic Data Stream carrying the Reference ID Mapping Table (RIDMT) associated to this Genomic Dataset. No genomic_dataset_ID shall have more than one reference_id_map_SID associated. The value of the reference_id_map_SID is defined by the user. genomic_Label_list_SID genomic_Label_list_SID is a bitstring field identifying the Genomic Data Stream carrying the Genomic Label List (GLL) associated to this Genomic Dataset. No genomic_dataset_ID shall have more than one genomic_Label_list_SID associated. The value of the genomic_Label_list_SID is defined by the user. Chacksum This is a bitstring field that contains an integrity check value for the entire GDL. One typical algorithm used for this purpose function is the CRC32 algorithm producing a 32 bit value other algorithms include the hashing functions MD5 and SHA-256.
[0568] Genomic Dataset Mapping Table
[0569] The Genomic Dataset Mapping Table (GDMT) is produced and transmitted at the beginning of a streaming process (and possibly periodically re-transmitted, updated or identical in order to enable the update of correspondence points and the relevant dependencies in the streamed data). The GDMT is carried by a single Packet following the Genomic Dataset List and lists the SIDs identifying the Genomic Data Streams composing one Genomic Dataset. The GDMT is the complete collection of all identifiers of Genomic Data Streams (e.g., the genomic sequence, reference genome, metadata, etc) composing one Genomic Dataset carried by a Genomic Multiplex. A genomic dataset mapping table is instrumental in enabling random access to genomic sequences by providing the identifier of the stream of genomic data associated to each genomic dataset.
[0570] The syntax of the GDMT data structure is provided in the table below with an indication of the data type associated to each syntax element.
TABLE-US-00012 genomic_dataset_mapping_table( ) { table_length bitstring genomic_dataset_ID bitstring version_number bitstring applicable_section_flag bit mapping_table_ID bitstring genomic_dataset_ef_length bitstring for (i=0; i<N; i++) { N = number of extension fields associated to this Genomic Dataset extension_field( ) data structure } for (i = 0;i < M ; i++) { M = number of Genomic Data Streams associated to this specific Dataset data_type bitstring genomic_data_SID bitstring gd_component_ef_length bitstring for (I = 0; I < K; i++) { K = number of extension fields associated to each Genomic Data Stream extension_field ( ) data structure } } Chaecksum bitstring }
[0571] The syntax elements composing the GDMT described above have the following meaning and function.
TABLE-US-00013 version_number, These elements have the same meaning as for the GDL applicable_section_flag table_length, bitstring field specifying the number of bytes composing the table, starting after the table_length field, and including the Checksum field. genomic_dataset_ID bitstring field identifying a Genomic Dataset mapping_table_ID bitstring bit field identifying the current Genomic Dataset Mapping Table genomic_dataset_ef_length bitstring field specifying the number of bytes of the optional extension_field associated with this Genomic Dataset data_type bitstring field specifying the type of genomic data carried by the packets identified by the genomic_data_SID. genomic_data_SID bitstring bit field specifying the Stream ID of the packets carrying the encoded genomic data associated with one component of this Genomic Dataset (e.g. read p positions, read p pairing information etc. as defined in this invention) gd_component_ef_length bitstring field specifying the number of bytes of the optional extension_field associated with the genomic Stream identified by genomic_data_SID. Checksum This is a bitstring field that contains an integrity check value for the entire GDMT. One typical algorithm used for this purpose function is the CRC32 algorithm producing a 32 bit value or hashing functions such as MD5 and SHA-256.
[0572] extension_fields are optional descriptors that might be used to further describe either a Genomic Dataset or one Genomic Dataset component.
[0573] The data_type field can have the following values
TABLE-US-00014 data_type Description 0 Dataset Header 1 Layer Header 2 to 15 User-defined extensions 16 to N 16 + Descriptors_Layer_ID
[0574] Genomic Datasets Mapping Tables List
[0575] This structure carries information about all the datasets mapping tables related to a Genomic Datasets Multiplex.
TABLE-US-00015 Syntax Description Datasets_mapping_tables_list{ Multiplex_id Datasets Multiplex ID, as in Datasets Multiplex Header. for (i=0; i<gd_number;i++) { Note: gd_number as in Datasets Multiplex Header. dataset_mapping_table_SID Stream ID of Dataset Mapping Table of i-th Dataset. } }
[0576] Reference ID Mapping Table
[0577] The Reference ID Mapping Table (RIDMT) is produced and transmitted at the beginning of a streaming process. The RIDMT is carried by a single Packet following the Genomic Dataset List. The RIDMT specifies a mapping between the numeric identifiers of reference sequences (REFID) contained in the Block header of an access unit and the (typically literal) reference identifiers contained in the Genomic Dataset Header specified in Table 2.
[0578] The RIDMT can be periodically re-transmitted in order to: [0579] enable the update of correspondence points and the relevant dependencies in the streamed data, [0580] support the integration of new reference sequences added to the pre-existing ones (e.g. synthetic references created by de-novo assembly processes)
[0581] The syntax of the RIDMT data structure is provided in the table below with an indication of the data type associated to each syntax element.
TABLE-US-00016 Syntax Data type reference_id_mapping_table( ) { table_length bitstring genomic_dataset_ID bitstring version_number bitstring applicable_section_flag bit reference_id_mapping_table_ ID bitstring for (i = 0; i < N; i++) { N = number of reference sequences associated with the Genomic Dataset identified by genomic_dataset_ID ref_string_length bitstring for (i=0;i<ref_string_length;i++){ ref_string[i] byte } REFID bitstring } Checksum bitstring (e.g. CRC-32 or MD5 hash) }
[0582] The syntax elements composing the RIDMT described above have the following meaning and function.
TABLE-US-00017 table_length, genomic_dataset_ID, These elements have the same meaning as for the version_number, applicable_section_flag GDMT reference_id_mapping_table_ID bitstring field identifying the current Reference ID Mapping Table ref_string_length bitstring field specifying the number of characters (bytes) composing ref_string, excluding the end of string (`\0`) character. ref_string[i] byte field encoding each character of the string representation of a reference sequence (e.g. "chr1" for chromosome 1). The end of string (`\0`) character is not necessary, as it is implicitly inferred from the ref_string_length field REFID This is a bitstring field uniquely identifying a reference sequence. This is encoded in the data Block header as REFID field. Checksum This is a bitstring field that contains an integrity check value for the entire RIDMT. One typical algorithm used for this purpose function is the CRC32 algorithm producing a 32 bit value or any hash function producing longer strings of bits.
[0583] Genomic Label List
[0584] As described above, a label is an identifier which is assigned to a specific genomic regions or sub-regions or aggregations of regions or sub-regions.
[0585] Labels identify genomic regions by specifying the reference sequence id, the position range with respect to the reference sequence and the data classes that they identify.
[0586] For such purpose, the Genomic Label List (GLL) is created during the packetization process by the multiplexer and transmitted.
[0587] The depacketizer of the demultiplexer parses the GLL syntax and exposes the available "Labels" to the data access application, which has the possibility to select and access the desired sub-set of data.
[0588] The GLL is (optionally) produced and transmittedat the beginning of a stream and typically transmitted periodically in order to enable multiple synchronization points (4811), and provides the list of "Labels" associated to the Multiplex and Dataset identified by the multiplex_id and dataset_id fields.
[0589] The syntax of the GLL data structure is provided in the table below with an indication of the data type associated to each syntax element.
TABLE-US-00018 TABLE 6 Complete syntax of "Label List" data format for the streamed compressed data scenario. Syntax Description genomic_label_list( ) { table_length multiplex_id dataset_id num_labels total number of labels in the list for (i=0; i<num_labels;i++) { Label_id label identifier num_ref number of references concerned by the current label for (j = 0; j < num_ref; j++) { ref_id current reference num_regions number of different regions of this reference identified by the label for (k = 0; k < num_regions; k++) { class_id type of class, start and end position of start_pos this region end_pos } } } Checksum e.g. CRC-32 or MD5 hash }
[0590] The syntax elements composing the GLL described above have the following meaning and function.
TABLE-US-00019 TABLE 7 Description of syntax elements of Table 6. table_length Bitstring field specifying the number of bytes composing the list, starting after the table_length field, and including the Checksum field multiplex_ID Byte which serves as a label to identify the Genomic Multiplex from any other multiplex within a network dataset_ID Byte which serves as a label to identify the Genomic Dataset from any other dataset within the multiplex identified by multiplex_id num_Labels Bitstring representing the total number of Labels in this GLL Label_id Bitstring identifying the i-th Label num_ref Bitstring identifying the number of references concerned by the current label ref_id Bitstring identifying the j-th reference sequence the i-th Label refers to num_regions Bistring identifying the number of regions conveyed by the i-th Label class_id Bitstring identifying the class of the k-th region in the j-th reference in the i-th Label start_pos Bitstring indicating the position in the j-th reference sequence of the first read of the k-th region in the i-th Label end_pos indicating the position in the j-th reference sequence of the last read of the k-th region in the i-th Label Checksum Bitstring field that contains an integrity check value for the entire GLL. One typical algorithm used for this purpose function is the CRC32 algorithm producing a 32 bit value or hashing functions producing longer strings of bits (e.g. MD5, SHA-256).
[0591] Genomic Data Stream
[0592] A Genomic Data Multiplex contains one or several Genomic Data Streams where each stream can transport [0593] data structures containing transport information (e.g. Genomic Dataset List, Genomic Dataset Mapping Table etc.) [0594] data belonging to one of the Genomic Data Layers described in this invention. [0595] Metadata related to the genomic data [0596] Any other data
[0597] A Genomic Data Stream containing genomic data is essentially a packetized version of a Genomic Data Layer where each packet is prepended with a header describing the packet content and how it is related to other elements of the Multiplex.
[0598] The Genomic Data Stream format described in this document and the File Format described in this document are mutually convertible. Whereas a full file format can be reconstructed in full only after all data have been received, in case of streaming a decoding tool can reconstruct and access, and start processing the partial data at any time.
[0599] A Genomic Data Stream is composed by several Genomic Data Blocks each containing one or more Genomic Data Packets. Genomic Data Blocks (GDBs) are containers of genomic information composing one genomic AU. GDB can be split into several Genomic Data Packets, according to the communication channel requirements.
[0600] Genomic access units are composed by one or more Genomic Data Blocks belonging to different Genomic Data Streams.
[0601] Genomic Data Packets (GDPs) are transmission units composing one GDB. Packet size is typically set according to the communication channel requirements.
[0602] FIG. 27 shows the relationship among Genomic Multiplex, Streams, Access Units, Blocks and Packets when encoding data belonging to the P class as defined in this invention. In this example three Genomic Streams encapsulate information on position, pairing and reverse complement of sequence reads.
[0603] Genomic Data Blocks are composed by a header, a payload of compressed data and padding information.
[0604] The table below provides an example of implementation of a GDB header with a description of each field and a typical data type.
TABLE-US-00020 TABLE 8 Description of Genomic Data Block syntax elements. Data type Description Data type Block Start Code Reserved value used to unambiguously identify the beginning bitstring Prefix (BSCP) of a Genomic Data Block. Block Header Block Header as defined in this document bitstring POS Flag (PSF) If the POS Flag is set, the block contains the 40 bit POS field at bit the end of the block header and before the optional fields. Padding Flag (PDF) If the Padding Flag is set, the block contains additional padding bit bytes after the payload which are not part of the payload. Block size (BS) Number of bytes composing the block, including this header bitstring and payload, and excluding padding (total block size will be BS + padding size). Access Unit ID (AUID) Unambiguous ID, linearly increasing (not necessarily by 1, even bitstring though recommended). Needed to implement proper random access, as described in the Master Index Table defined in this invention. Label ID (LID) Unambiguous ID, linearly increasing by 1, identifying the bitstring genomic region/classes (Label) this block belongs to. It corresponds to the i-th index in the main for loop in the Genomic Label List described above. (Optional) Reference Unambiguous ID, identifying the reference sequence the AU bitstring ID (REFID) containing this block refers to. This is needed, along with POS field, to have proper random access, as described in the Master Index Table. (Optional) POS (POS) Present if PSF is 1. Position on the reference sequence of the bitstring first read in the block. (Extra optional fields) Additional optional fields, presence signaled by BS. bytestring (Optional) Padding (Optional, presence signaled by PDF) Fixed bitstring value that bitstring can be inserted in order to meet the channel requirements. If present, the first byte indicates how many bytes compose the padding. It is discarded by the decoder.
[0605] The use of AUID, POS and BS enables the decoder to reconstruct the data indexing mechanisms referred to as Master Index Table (MIT) and Local Index Table (LIT) in this invention. In a data streaming scenario the use of AUID and BS enables the receiving end to dynamically re-create a LIT locally, without the need to send extra-data. The use of AUID, BS and POS will enable to recreate a MIT locally without the need to send additional data.
[0606] This has the technical advantage to [0607] reduce the encoding overhead which might be large if the entire LIT is transmitted; [0608] avoid the need of a complete mapping between genomic positions and Access Units which is not normally available in a streaming scenario
[0609] A Genomic Data Block can be split into one or more Genomic Data Packets, depending on network layer constraints such as maximum packet size, packet loss rate, etc. A Genomic Data Packet is composed by a header and a payload of encoded or encrypted genomic data as described in the table below.
TABLE-US-00021 TABLE 9 Description of Genomic Data Packet syntax elements. Data type Description Data size Stream ID (SID) Unambiguously identifies data type carried by this bitstring packet. A Genomic Dataset Mapping Table is needed at the beginning of the stream in order to map Stream IDs to data types. Used also for updating correspondence points and relevant dependencies. Access Unit Marker Bit Set for the last packet of the access unit. Allows to bit (MB) identify the last packet of an AU. Packet Counter Counter associated to each Stream ID linearly increasing bitstring Number (SN) by 1. Needed to identify gaps/packet losses. Wrap around at 255. Packet Size (PS) Number of bytes composing the packet, including bitstring header, optional fields and payload. Extension Flag (EF) Set if extension fields are present. bit Extension Fields Optional fields, presence signaled by PS. bytestring Payload Block data (entire block or fragment) bytestring
[0610] The Genomic Multiplex can be properly decoded only when at least one Genomic Dataset List, one Genomic Dataset Mapping Table and one Reference ID Mapping Table have been received, allowing to map every packet to a specific Genomic Dataset component.
[0611] Genomic Packet Header
[0612] Every Genomic Data Block may be split in fragments, which may be transmitted in the payload of Genomic Data Packets, depending on channel requirements, such as packet loss rate, protocol maximum packet size, etc.
[0613] A Genomic Data Packet is defined as follows.
TABLE-US-00022 Syntax Description Packet_header( ) { Layer ID (LID) Unambiguously identifies data type carried by this Packet. Unique for each sub-stream/data type. Mapping Table needed at beginning of stream in order to map Layer IDs to data types. Reserved To maintain byte-alignment Access Unit Marker Bit (MB) Set for the last Packet of the Access Unit. Allows identifying the end of an AU as a set of Blocks. Sequence Number (SN) Packet counter, linearly increasing by 1. Needed to identify packet losses as gaps in SNs for each individual sub-stream. Associated to LID, i.e., different SN for every LID. Packet Size (PS) Number of bytes composing Packet, including header, optional fields and payload. Extension Flag (EF) Set if extension field is present. [optional] Extension field Optional field, present if EF is set. }
[0614] Multiplex Encoding Process
[0615] FIG. 49 shows how before being transformed in the data structures presented in this invention, raw genomic sequence data need to be mapped (491) on one or more reference sequence known a-priori (4920). In case a reference sequence is not available a "constructed" reference can be built from the raw sequence data (492). Already aligned data can be re-aligned in order to reduce the information entropy. After alignment, a genomic classifier (494) creates the data classes according to the matching functions described in Table land separates metadata (e.g. quality values) and annotation data from the genomic sequences. A reference transformation (4919) can be applied on the external reference (4920) in order to further reduce the entropy of the generated classes of data (498). The transformed data classes (4918) are fed to layers encoders (495-497) to produce genomic layers (491) which are then encoded by entropy encoders (4912-4914). The genomic streams generated by the entropy encoders are then sent to Genomic Multiplexer (4916) which generates the Genomic Multiplex. Genomic labels generated by a Genomic Labels Generator (4917) can be associated to the genomic streams (4915) by the Multiplexer (4916).
* * * * *
References
D00000
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
D00012
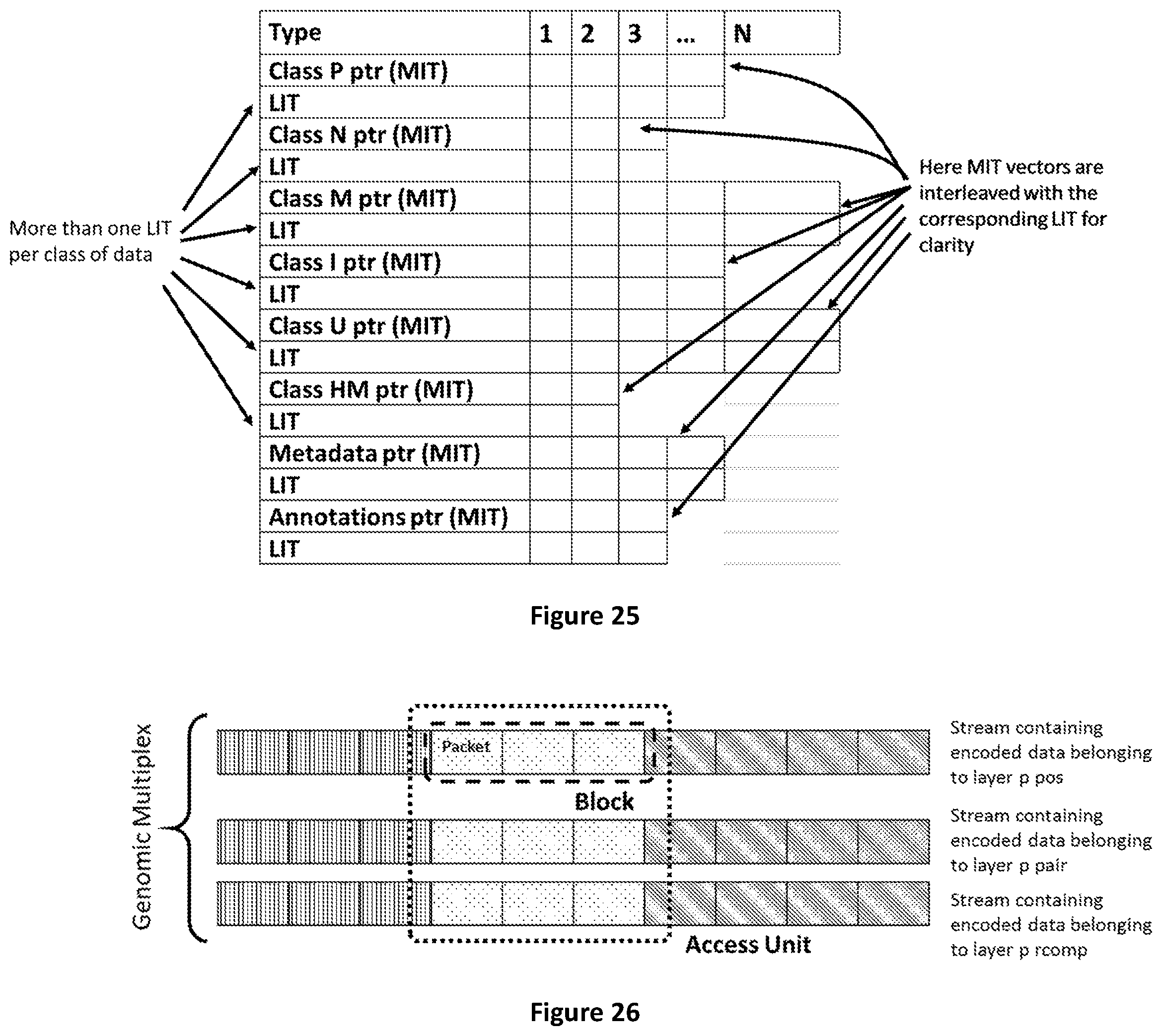
D00013
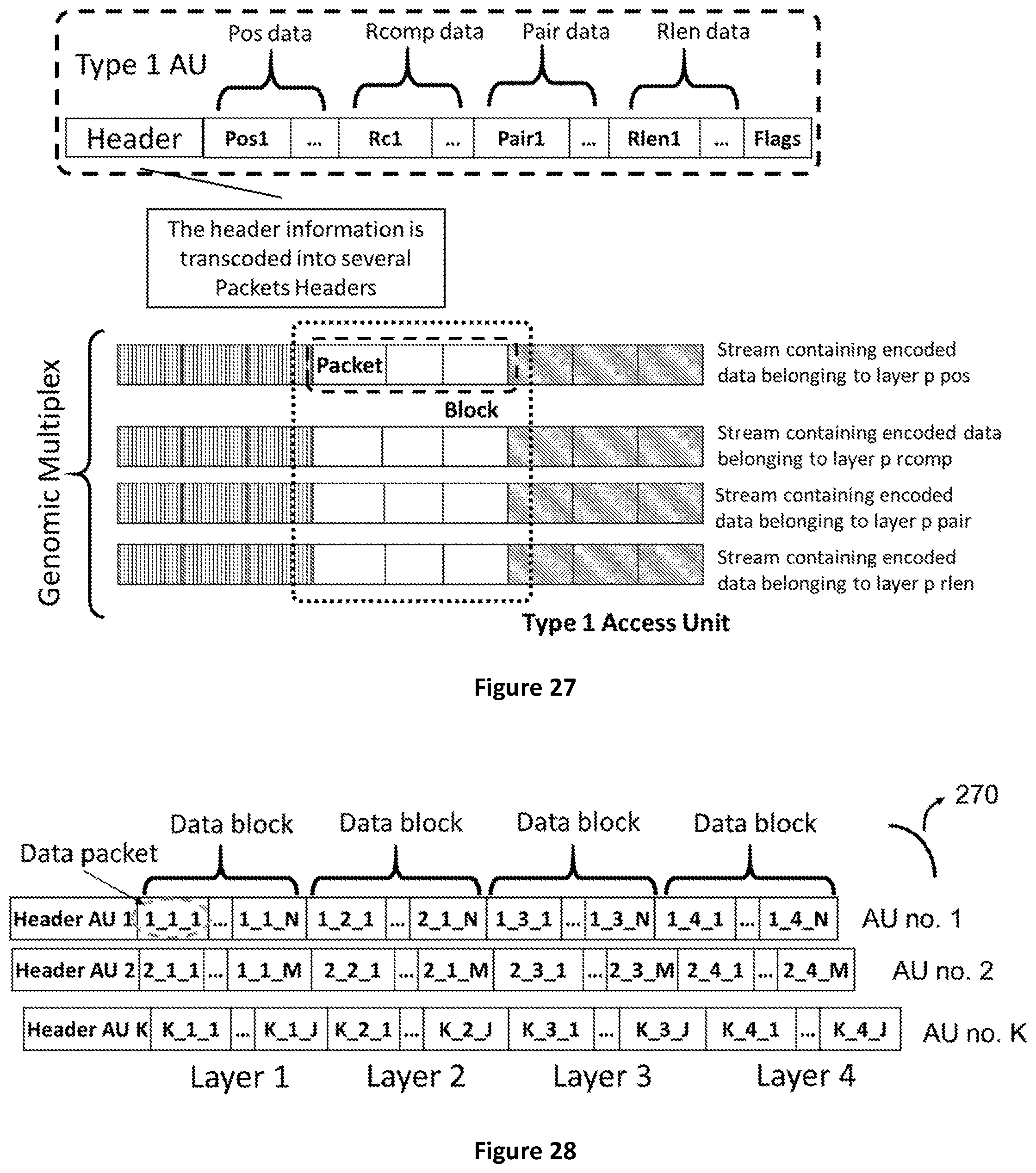
D00014

D00015
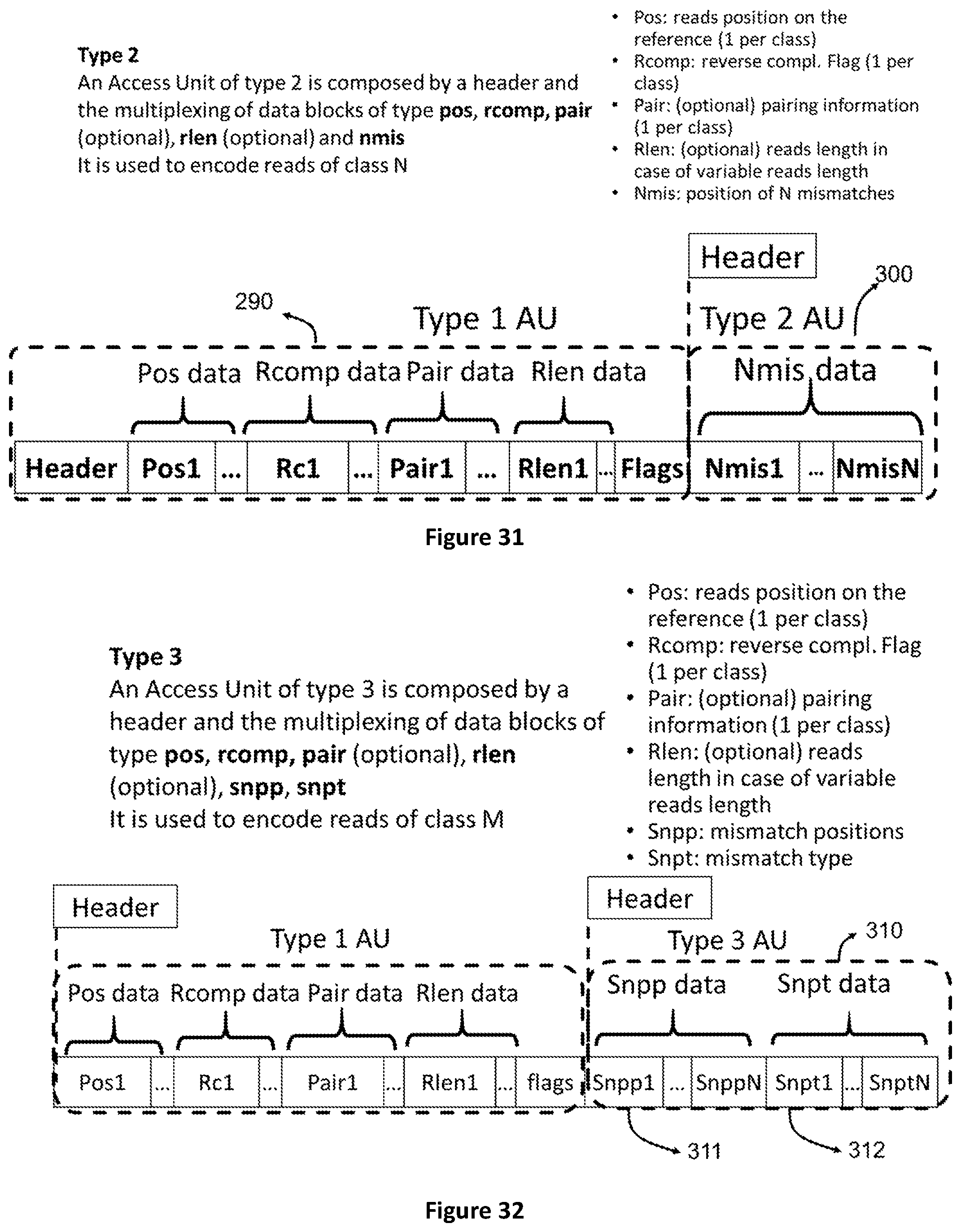
D00016
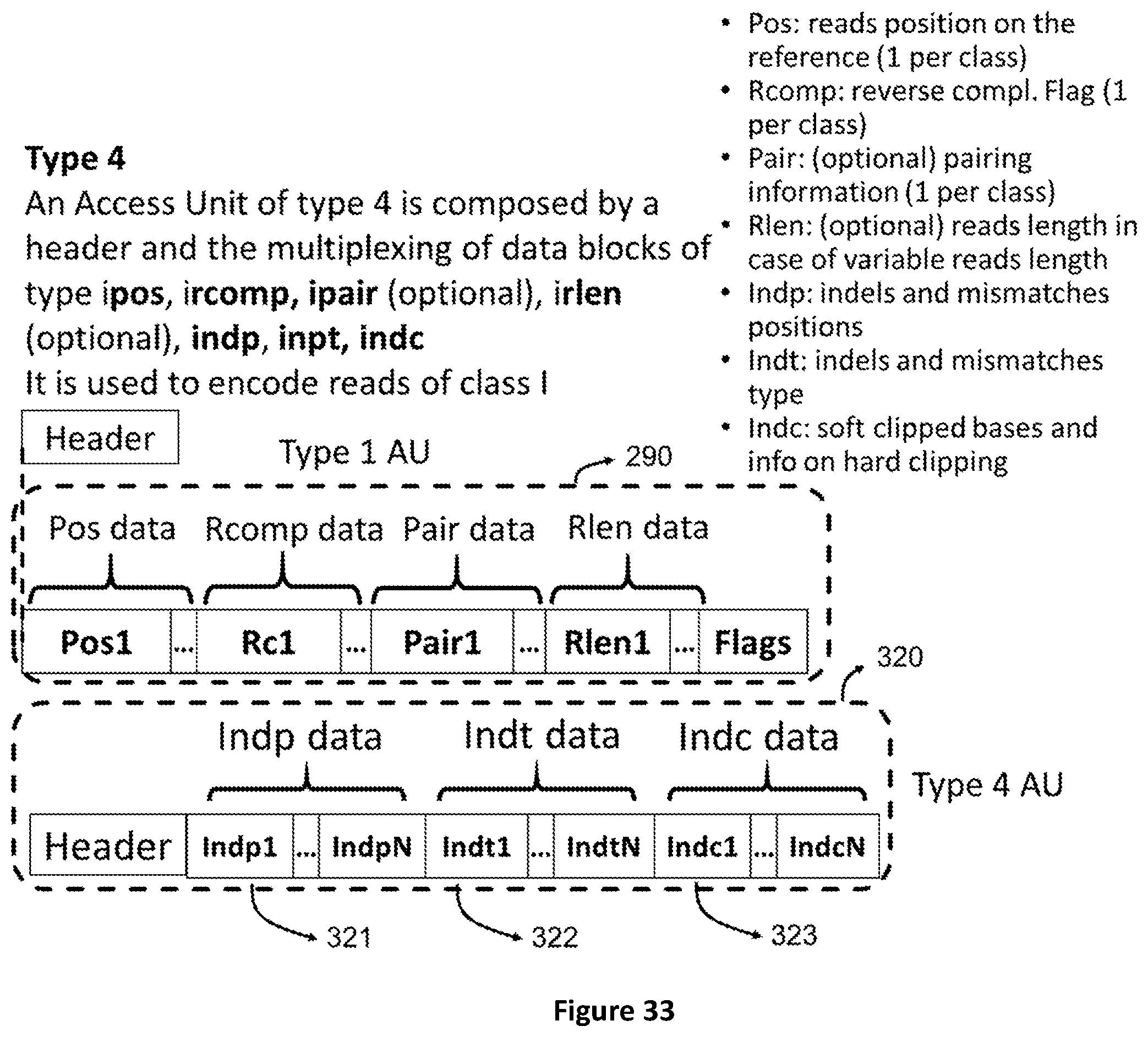
D00017
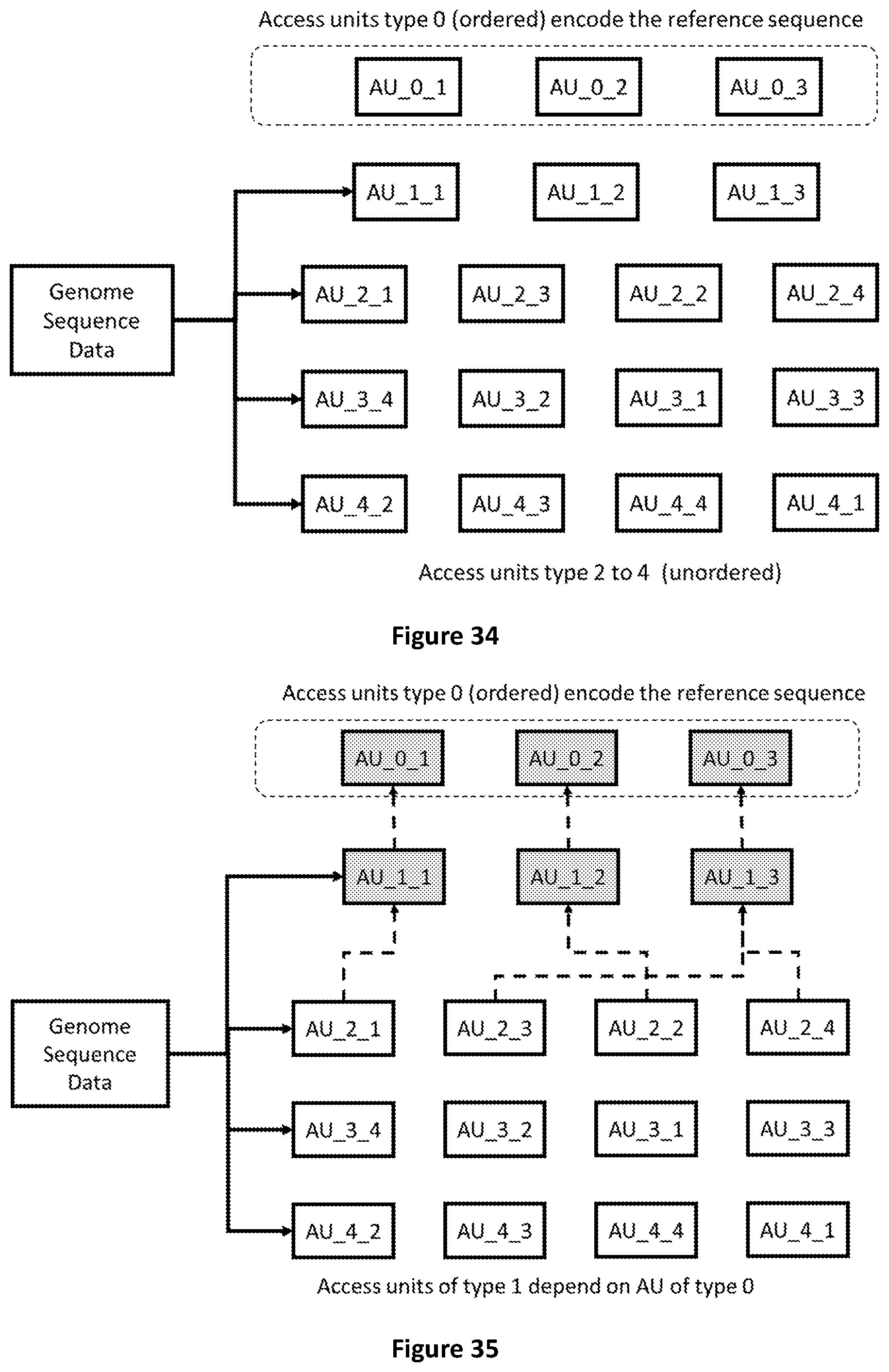
D00018
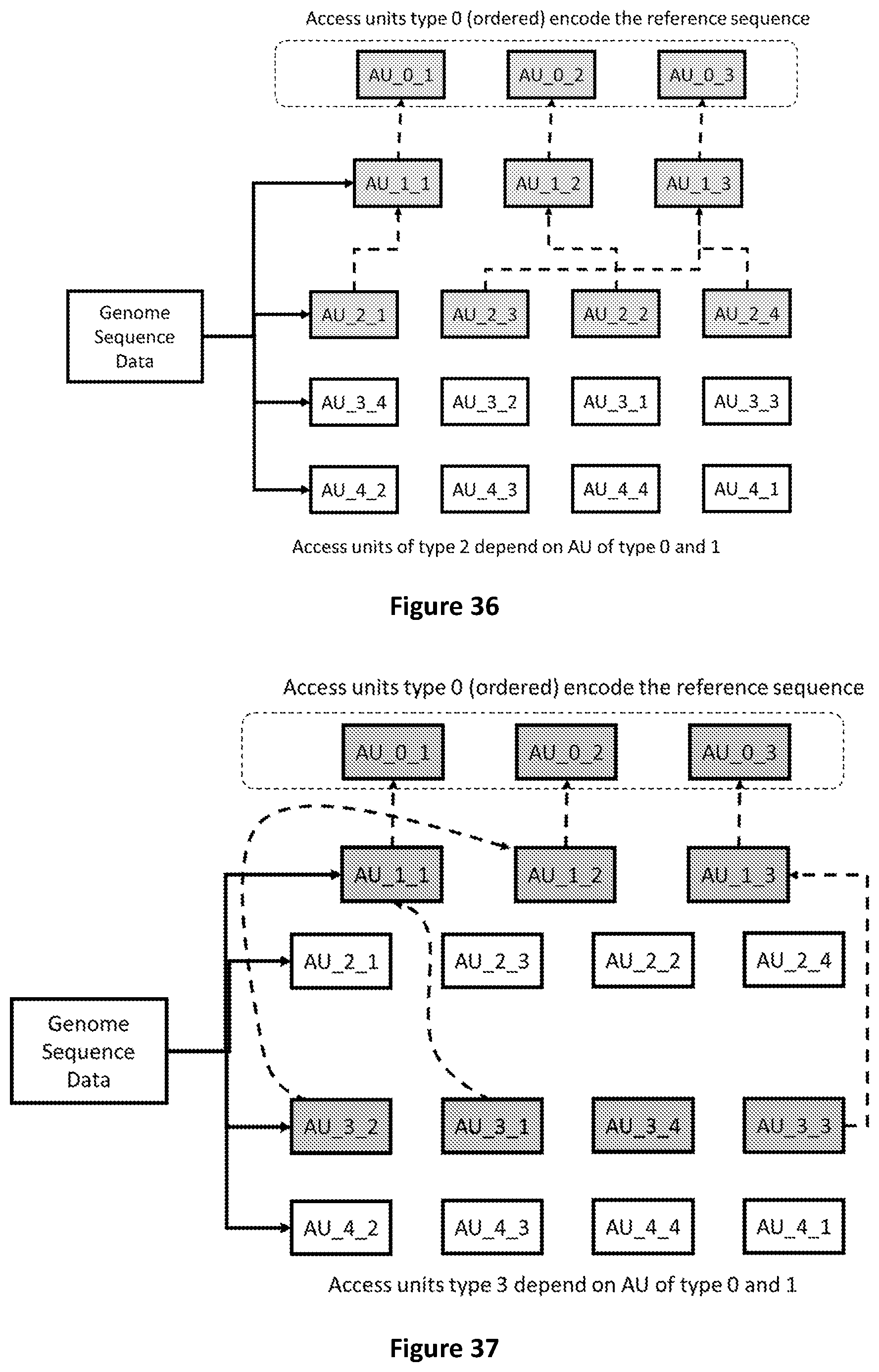
D00019
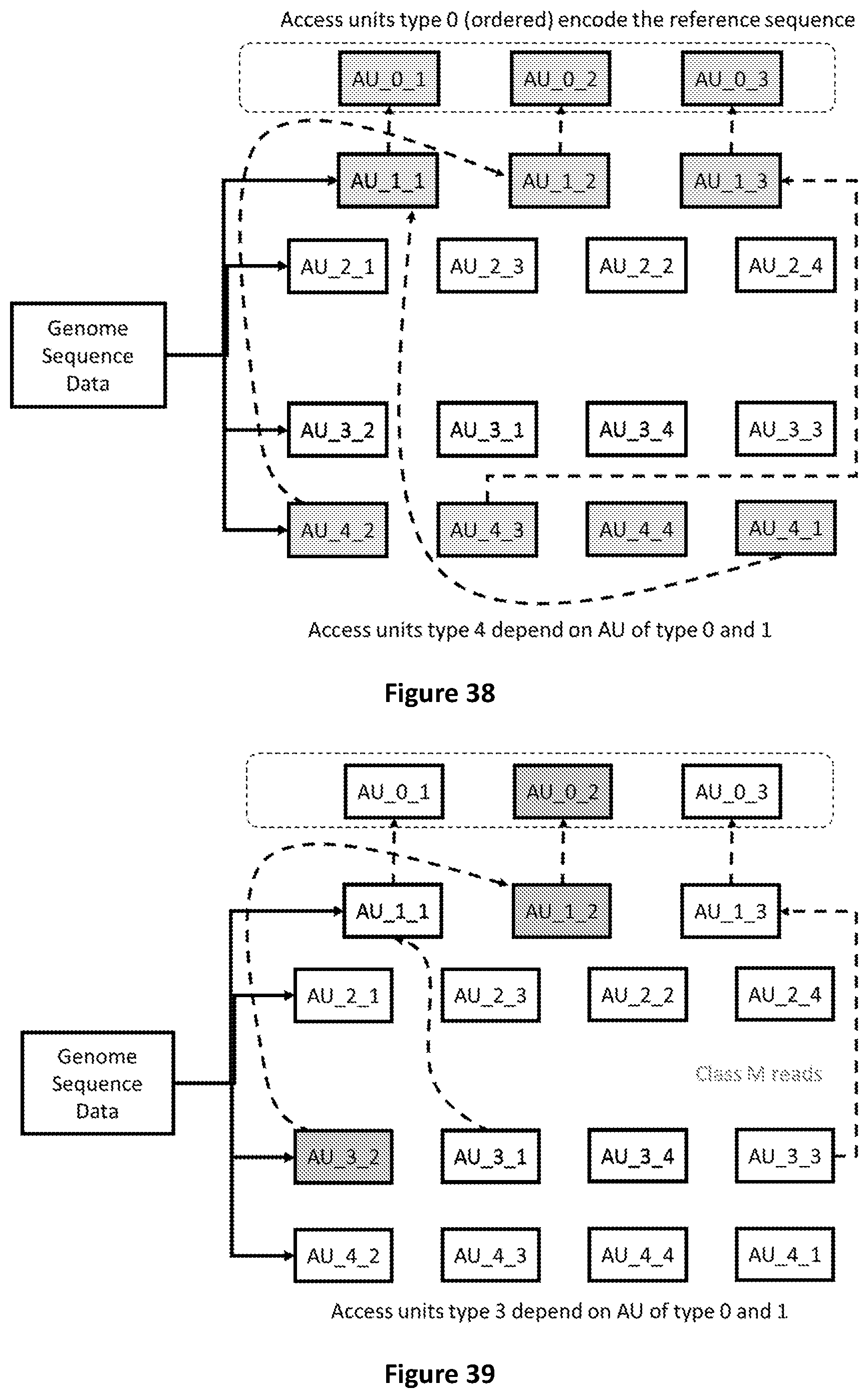
D00020
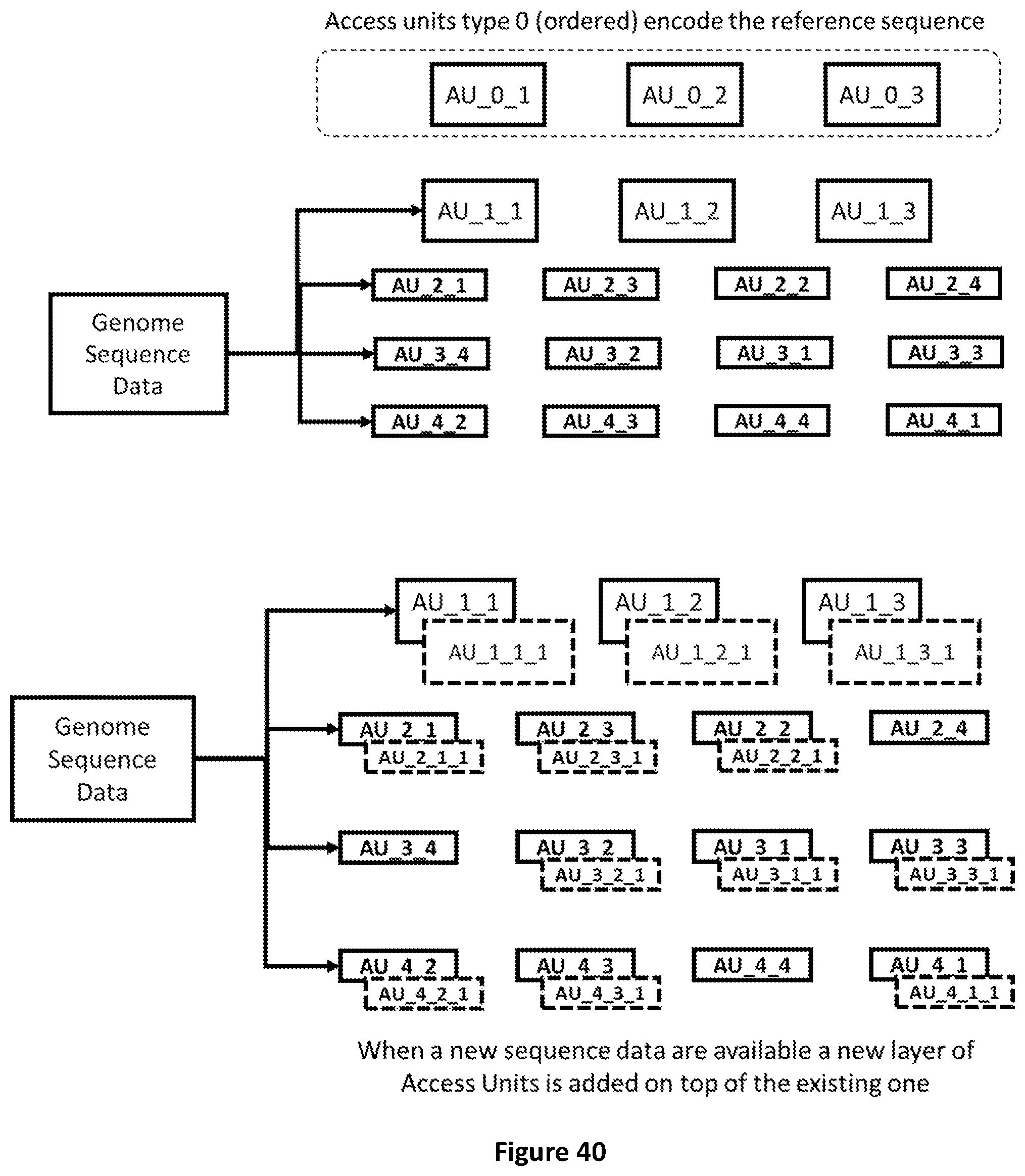
D00021
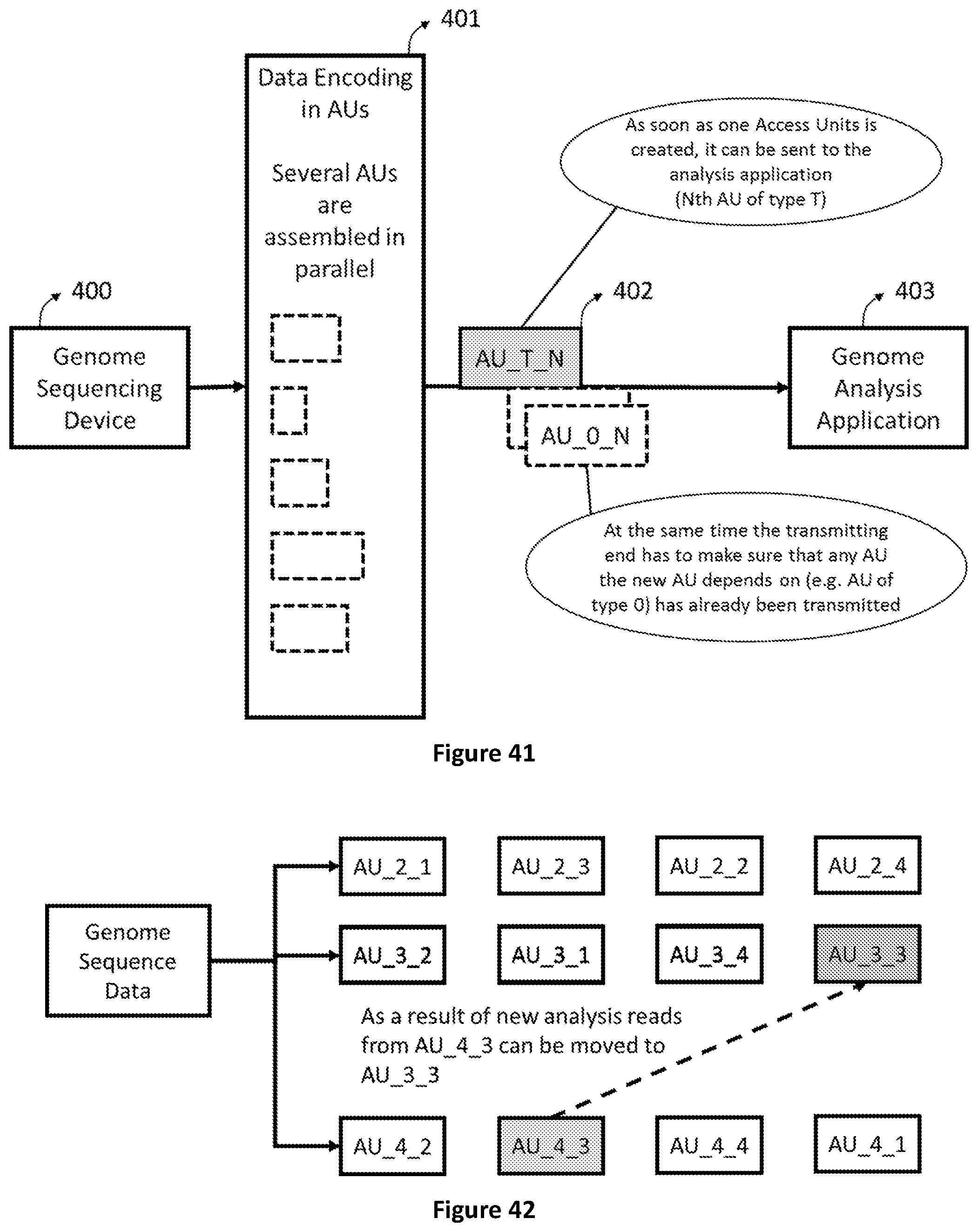
D00022
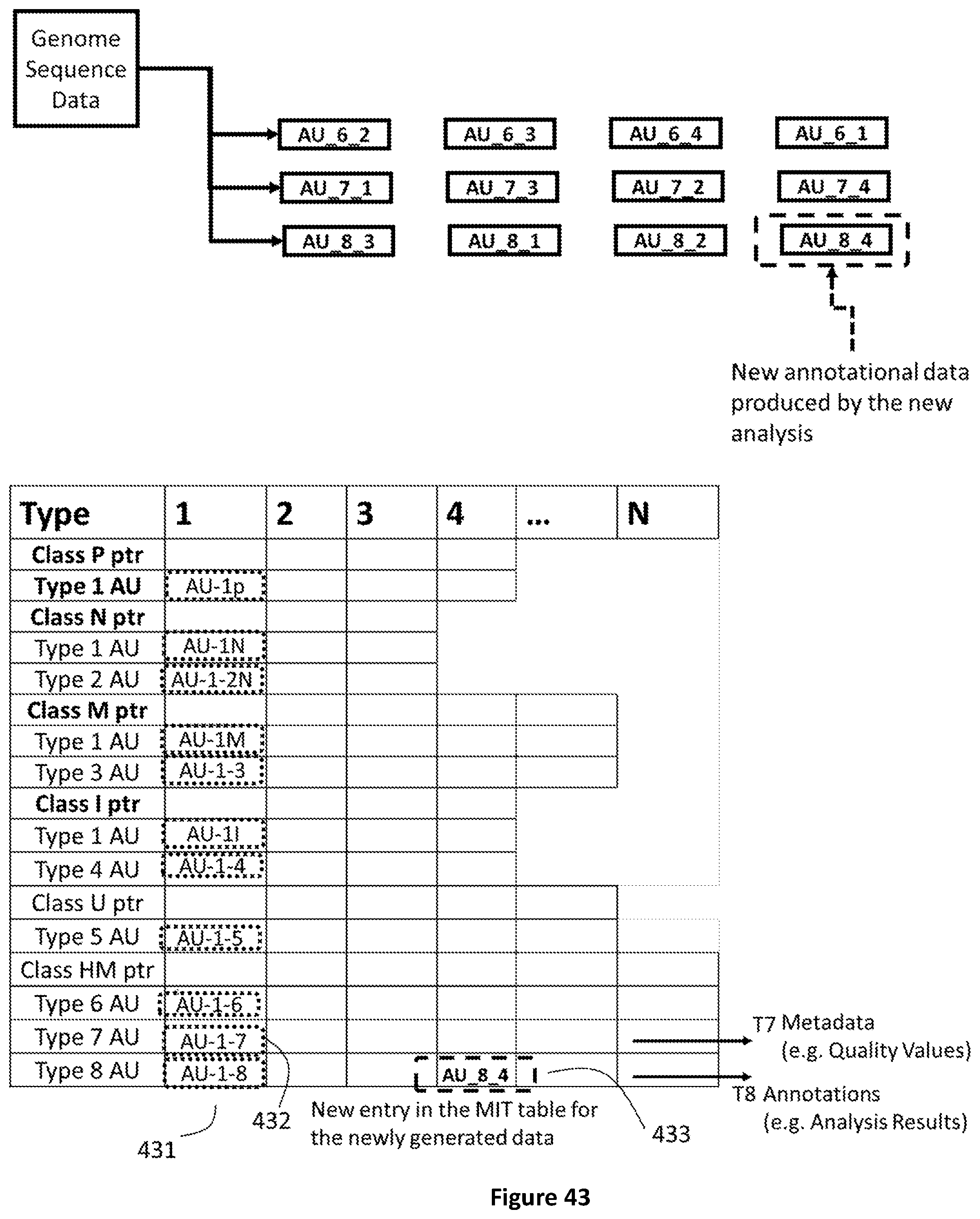
D00023
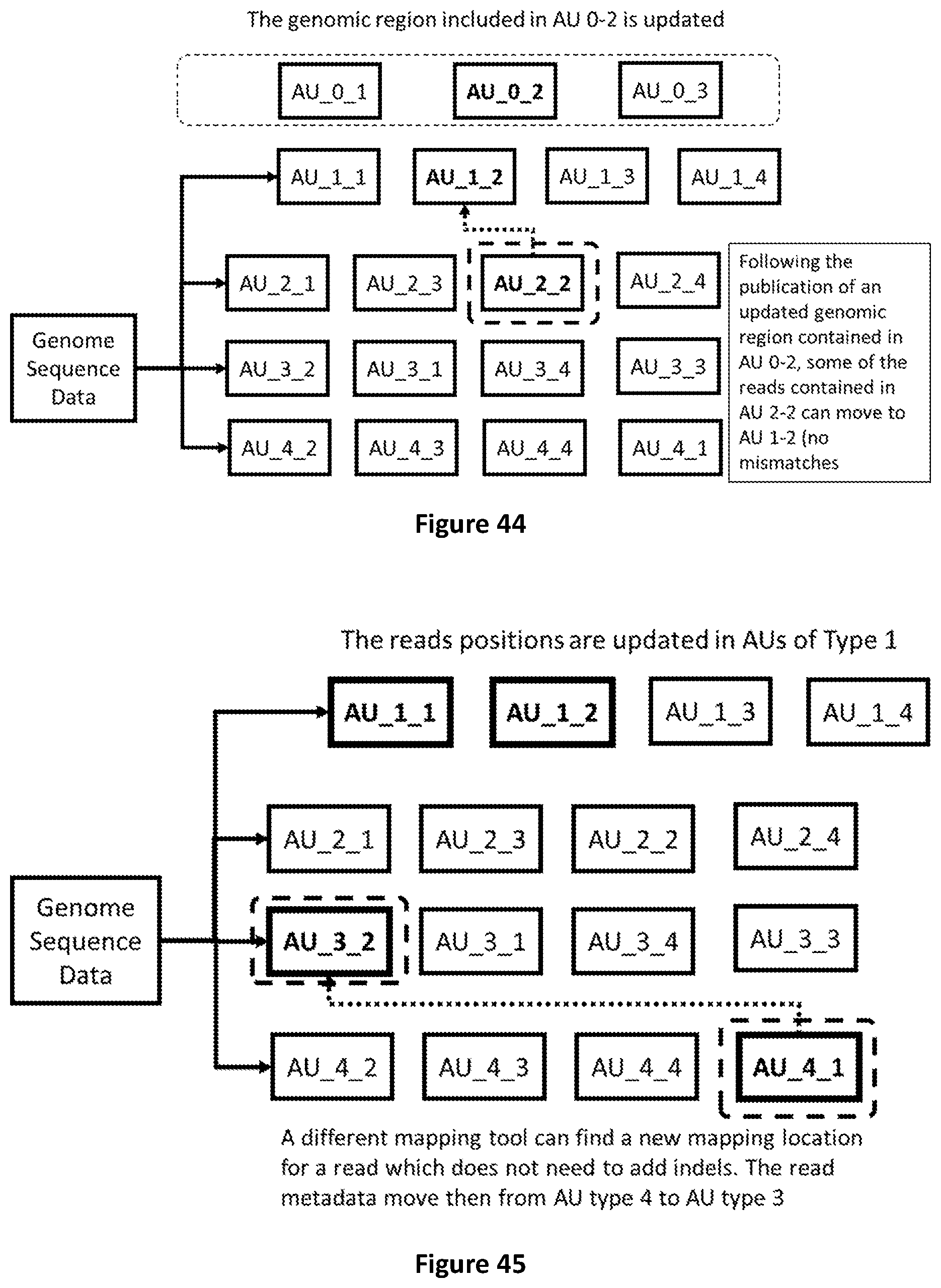
D00024
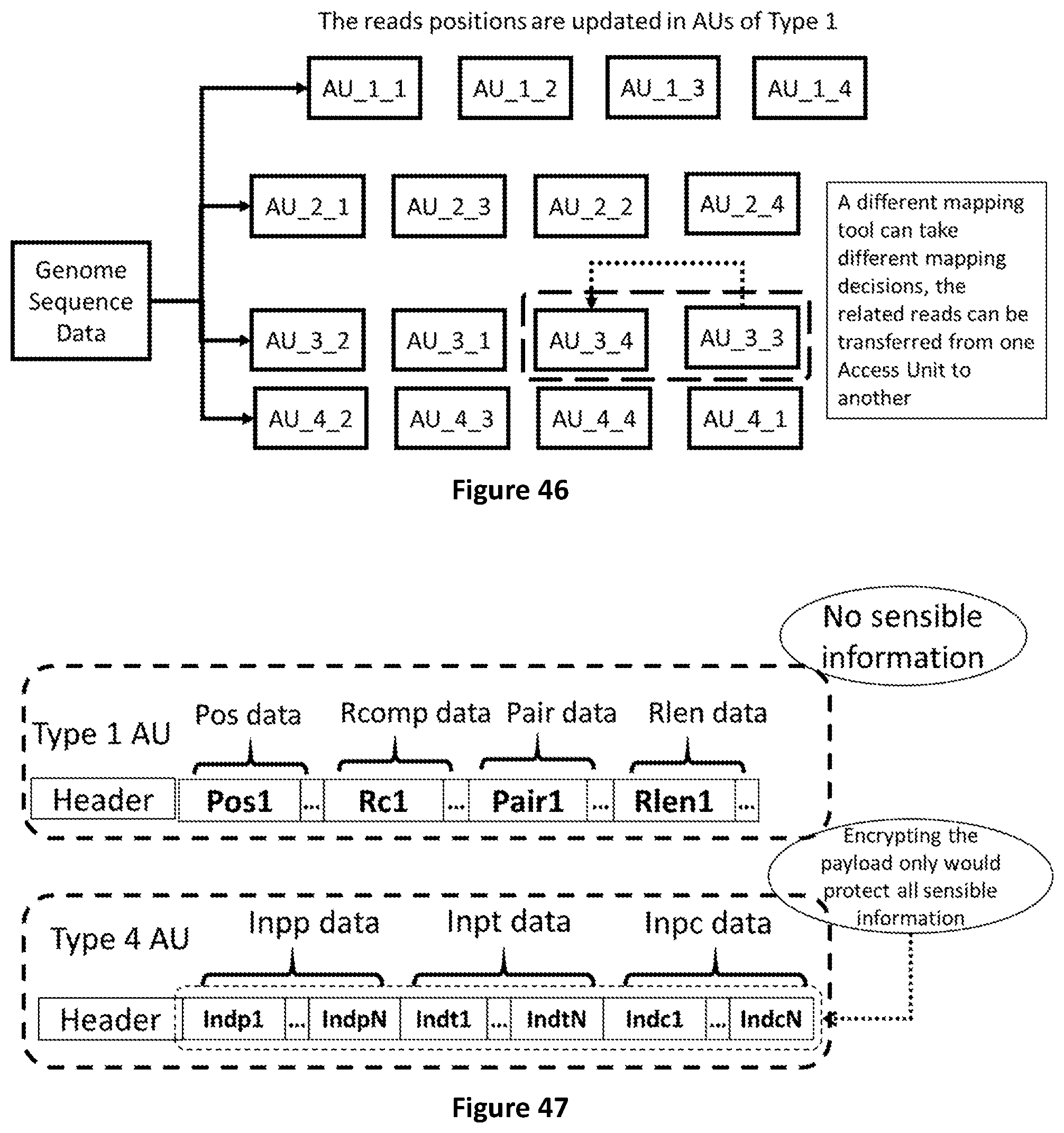
D00025
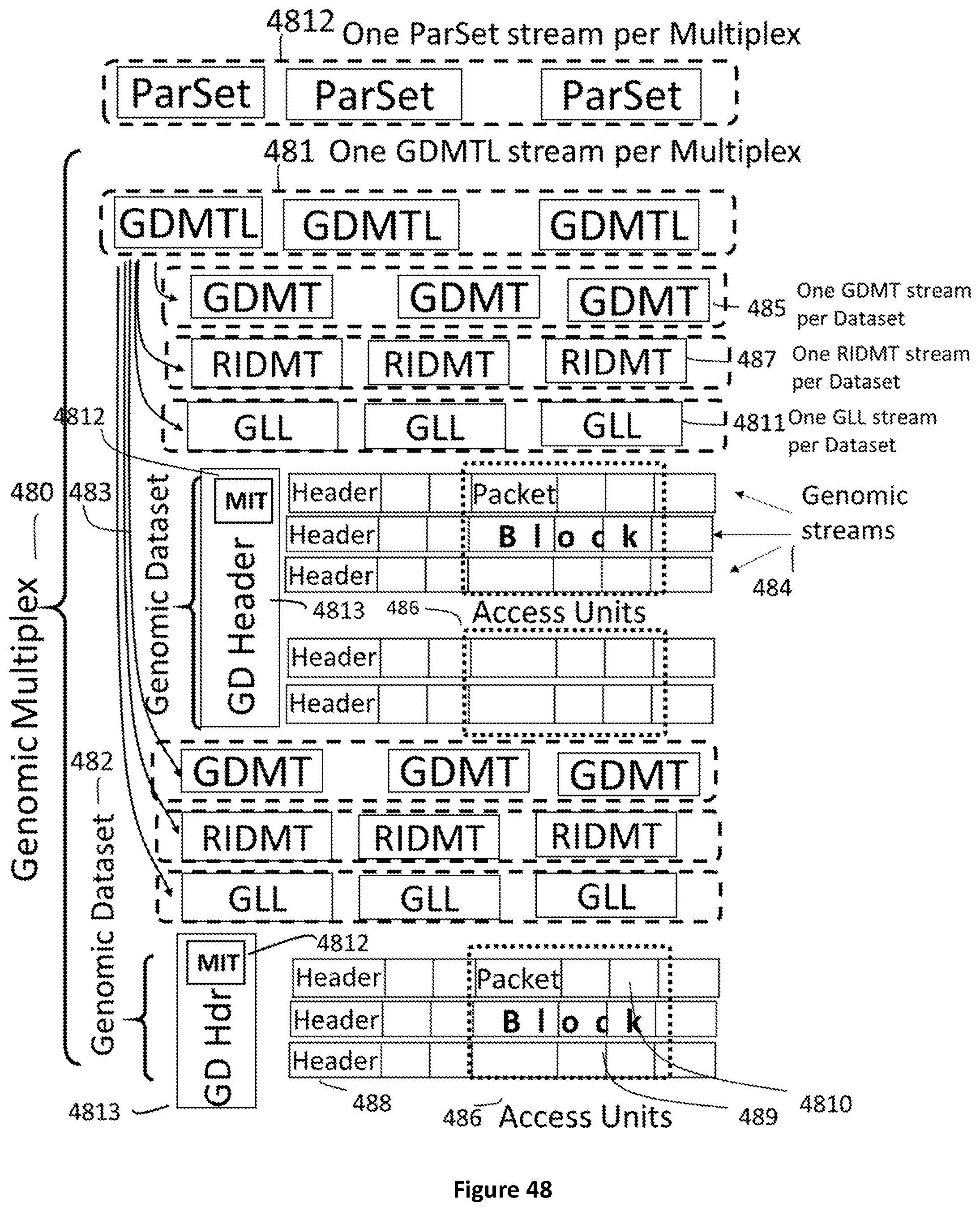
D00026
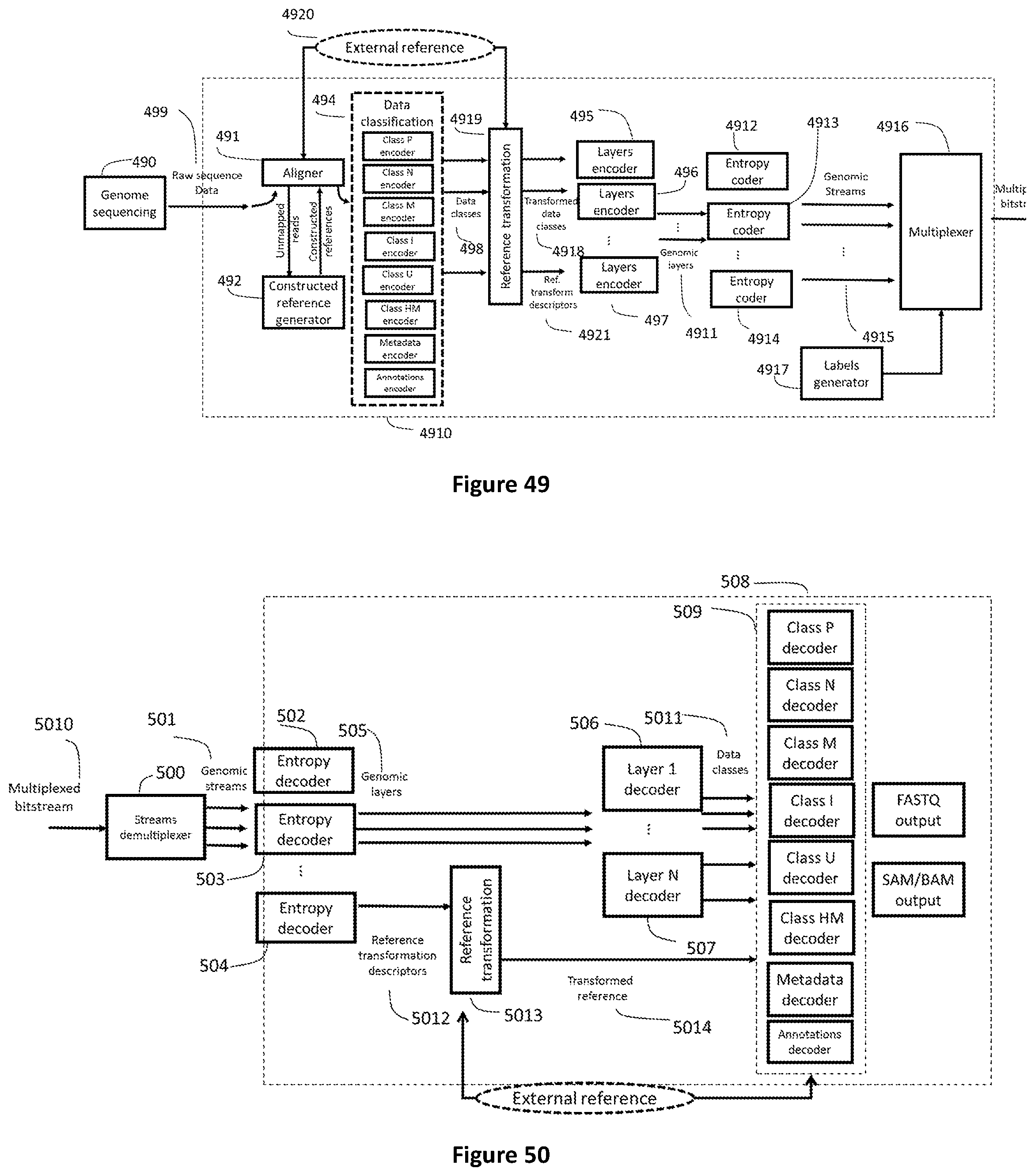
D00027
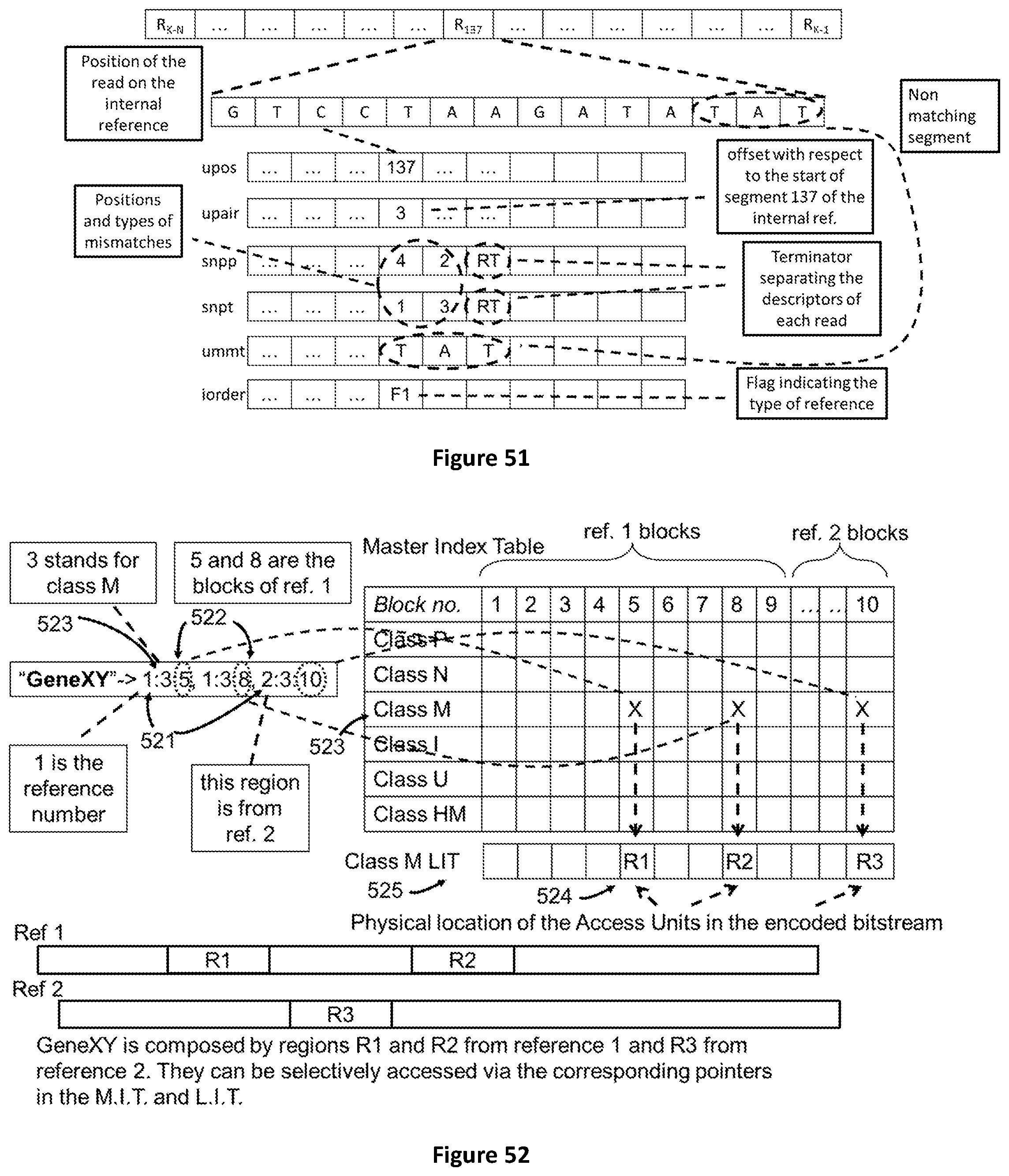
D00028
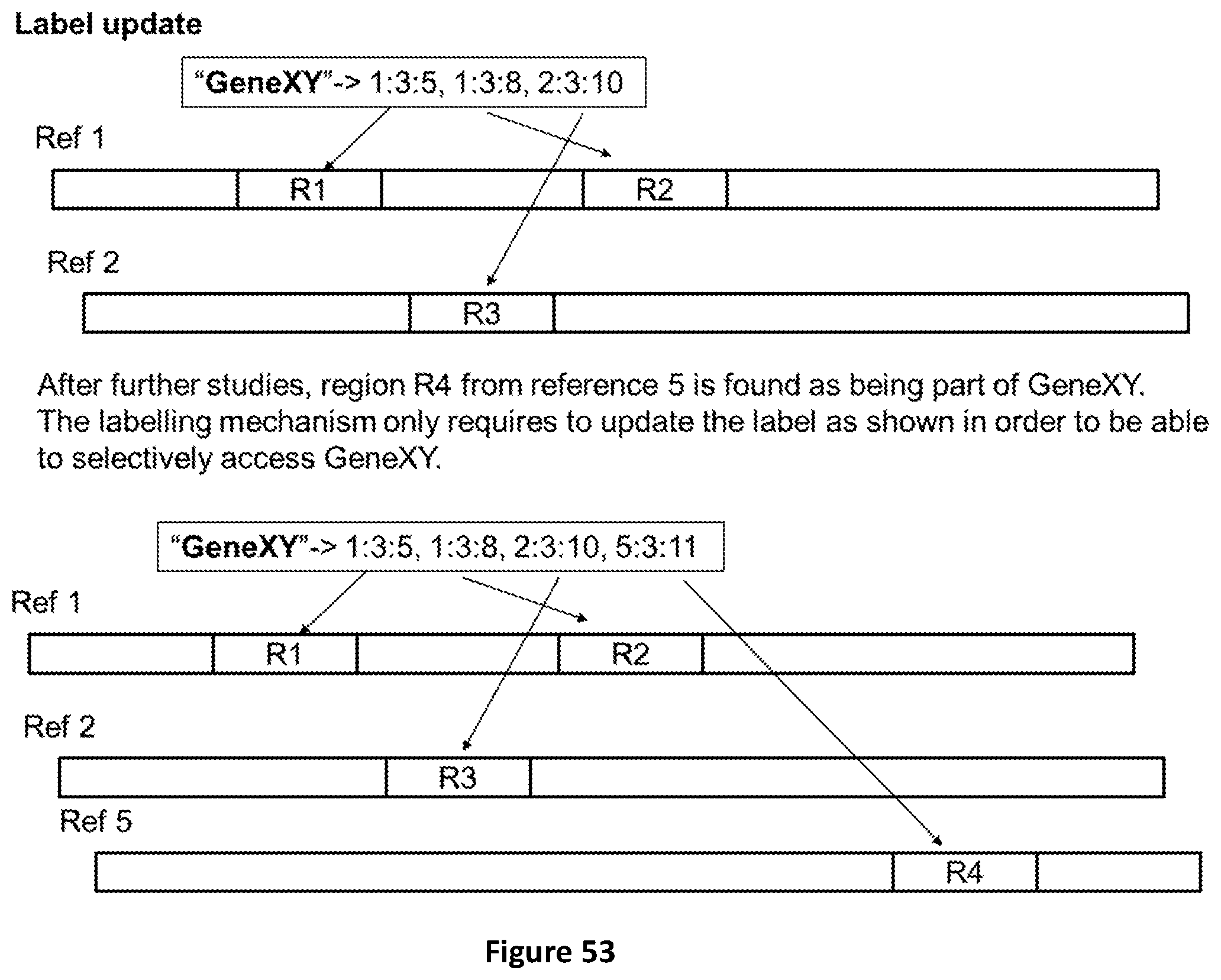
D00029
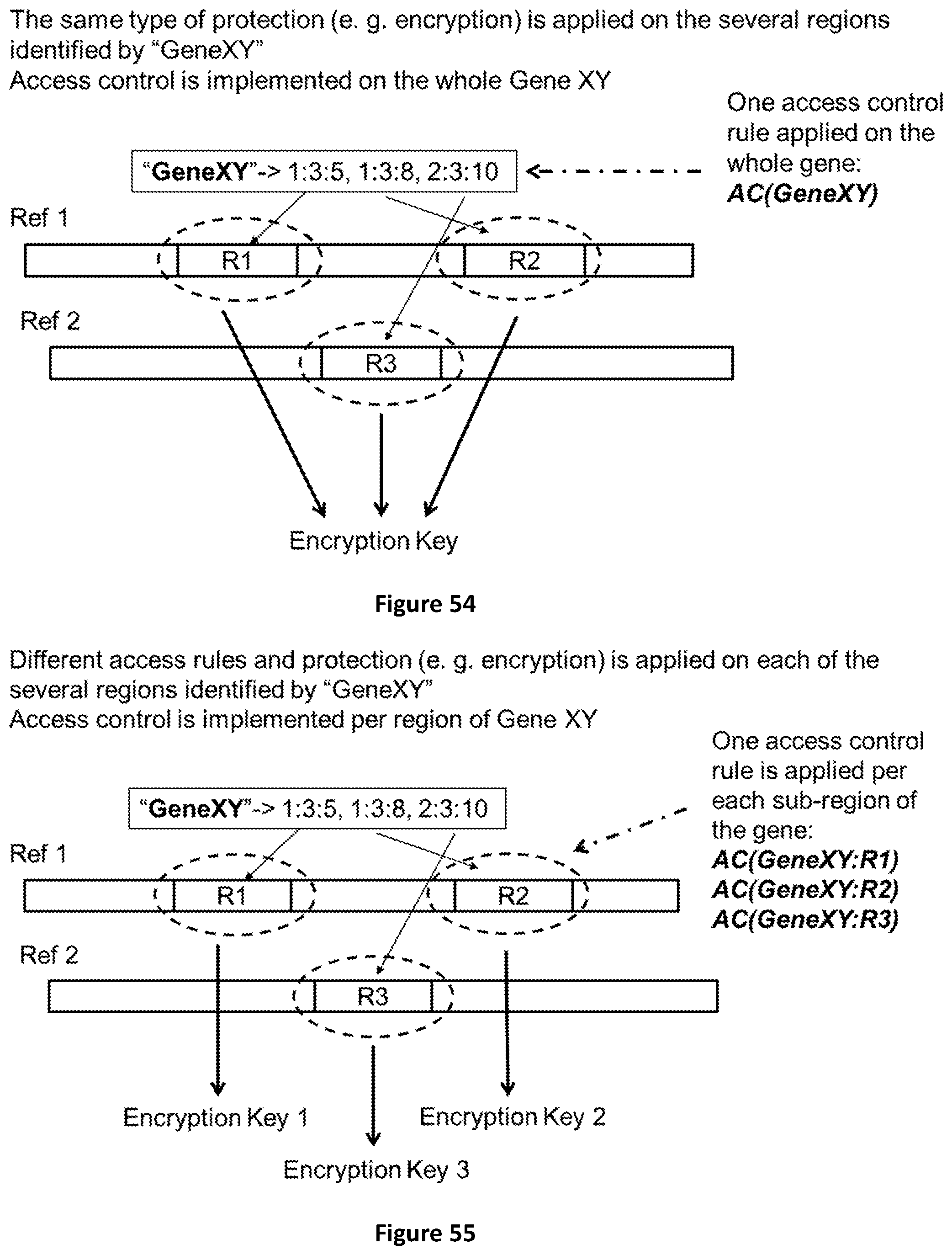
D00030

D00031

D00032
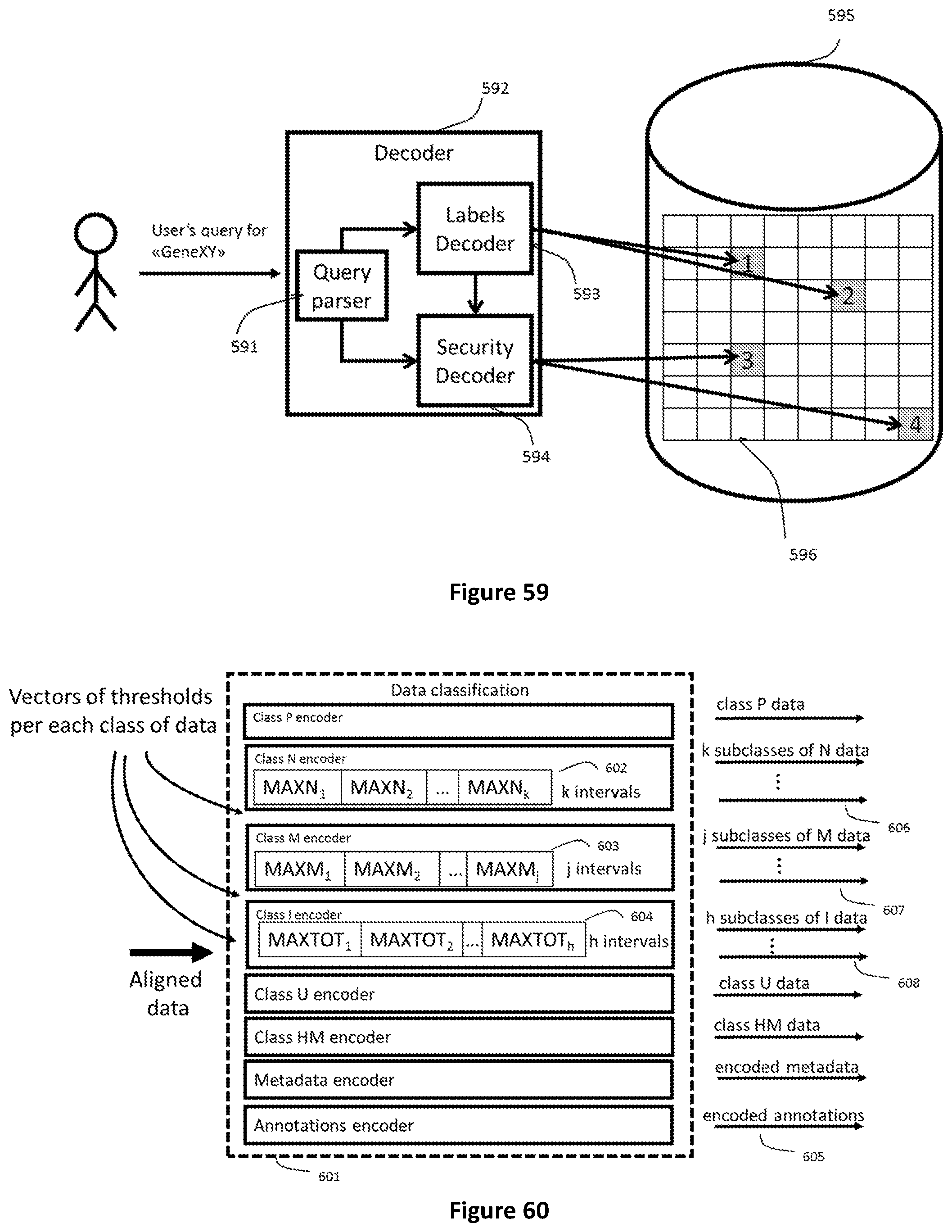
D00033
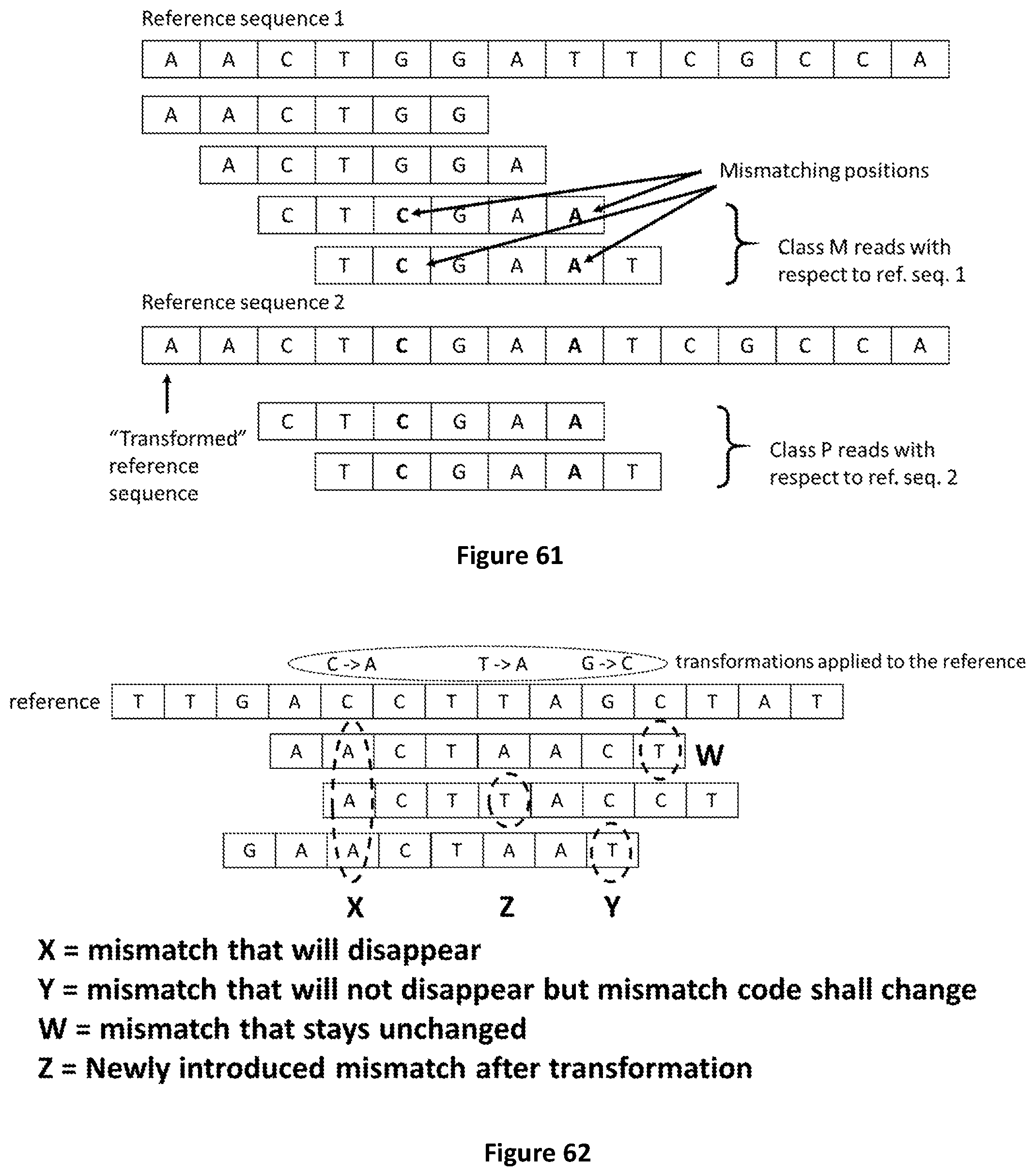
D00034
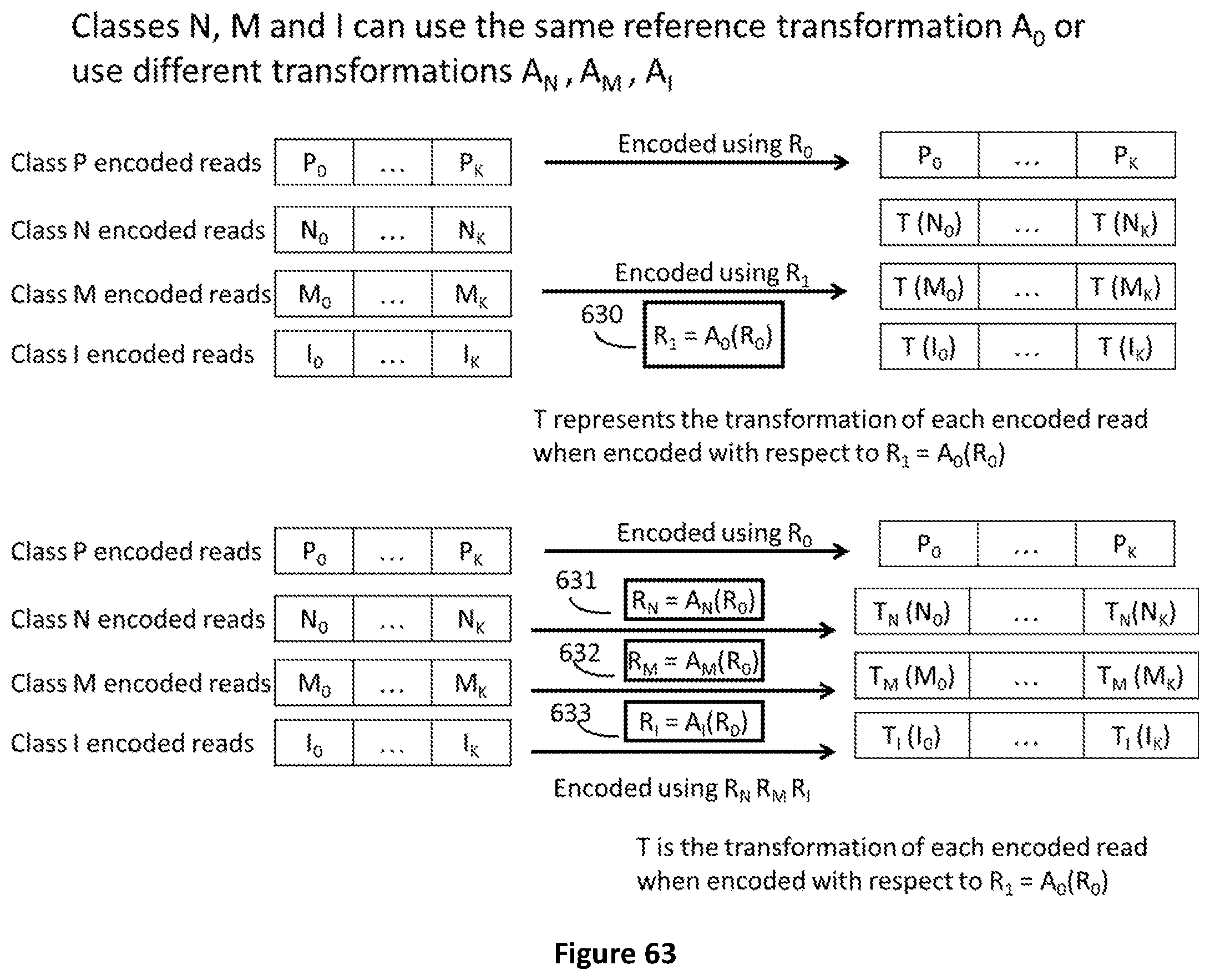
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.