Treatment Of Cancer Using Anti-cd19 Chimeric Antigen Receptor
June; Carl H. ; et al.
U.S. patent application number 16/435257 was filed with the patent office on 2019-12-26 for treatment of cancer using anti-cd19 chimeric antigen receptor. This patent application is currently assigned to Novartis AG. The applicant listed for this patent is Novartis AG, The Trustees of the University of Pennsylvania. Invention is credited to Jennifer Brogdon, John Byrd, Jason Dubovsky, Joseph A. Fraietta, Saar Gill, David Jonathan Glass, Amy Johnson, Carl H. June, Saad Kenderian, Joan Mannick, Marcela Maus, Leon Murphy, Natarajan Muthusamy, David L. Porter, Marco Ruella, William Raj Sellers, Mariusz Wasik.
| Application Number | 20190388471 16/435257 |
| Document ID | / |
| Family ID | 53008859 |
| Filed Date | 2019-12-26 |










View All Diagrams
| United States Patent Application | 20190388471 |
| Kind Code | A1 |
| June; Carl H. ; et al. | December 26, 2019 |
TREATMENT OF CANCER USING ANTI-CD19 CHIMERIC ANTIGEN RECEPTOR
Abstract
The invention provides compositions and methods for treating diseases associated with expression of CD19, e.g., by administering a recombinant T cell comprising the CD19 CAR as described herein, in combination with a kinase inhibitor, e.g., a kinase inhibitor described herein. The invention also provides kits and compositions described herein.
| Inventors: | June; Carl H.; (Merion Station, PA) ; Porter; David L.; (Springfield, PA) ; Maus; Marcela; (Lexington, MA) ; Wasik; Mariusz; (Ardmore, PA) ; Gill; Saar; (Philadelphia, PA) ; Fraietta; Joseph A.; (Williamstown, NJ) ; Ruella; Marco; (Ardmore, PA) ; Byrd; John; (Columbus, OH) ; Dubovsky; Jason; (Columbus, OH) ; Johnson; Amy; (Dublin, OH) ; Muthusamy; Natarajan; (Galloway, OH) ; Kenderian; Saad; (Philadelphia, PA) ; Mannick; Joan; (Cambridge, MA) ; Glass; David Jonathan; (Cambridge, MA) ; Murphy; Leon; (Cambridge, MA) ; Brogdon; Jennifer; (Sudbury, MA) ; Sellers; William Raj; (Cambridge, MA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Assignee: | Novartis AG Basel PA The Trustees of the University of Pennsylvania Philadelphia PA The Trustees of the University of Pennsylvania Philadelphia |
||||||||||
| Family ID: | 53008859 | ||||||||||
| Appl. No.: | 16/435257 | ||||||||||
| Filed: | June 7, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 14680860 | Apr 7, 2015 | 10357514 | ||
| 16435257 | ||||
| 62097278 | Dec 29, 2014 | |||
| 62087888 | Dec 5, 2014 | |||
| 62076238 | Nov 6, 2014 | |||
| 62036493 | Aug 12, 2014 | |||
| 62007309 | Jun 3, 2014 | |||
| 61976396 | Apr 7, 2014 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61P 35/02 20180101; C12N 2501/599 20130101; A61P 35/00 20180101; A61K 2039/505 20130101; A61K 35/17 20130101; A61K 31/53 20130101; C07K 16/2803 20130101; A61K 45/06 20130101; C12N 5/0636 20130101; A61K 2039/5156 20130101; C07K 2317/24 20130101; A61K 31/436 20130101; A61K 31/519 20130101; C07K 2317/565 20130101; A61K 2039/5158 20130101; C12N 2510/00 20130101; C07K 2317/14 20130101; A61K 39/3955 20130101; C07K 2317/622 20130101; C07K 14/7051 20130101; A61K 39/39558 20130101; A61P 43/00 20180101; C12N 2501/727 20130101; C07K 2319/03 20130101; C07K 2317/73 20130101; A61K 31/436 20130101; A61K 2300/00 20130101; A61K 31/519 20130101; A61K 2300/00 20130101; A61K 31/53 20130101; A61K 2300/00 20130101 |
| International Class: | A61K 35/17 20060101 A61K035/17; A61K 39/395 20060101 A61K039/395; A61K 31/519 20060101 A61K031/519; A61K 45/06 20060101 A61K045/06; C07K 14/725 20060101 C07K014/725; A61K 31/436 20060101 A61K031/436; A61K 31/53 20060101 A61K031/53; C12N 5/0783 20060101 C12N005/0783; C07K 16/28 20060101 C07K016/28 |
Claims
1. A method of treating a mammal having a disease associated with expression of CD19 comprising administering to the mammal an effective amount of a population of cells that expresses a CAR molecule that binds CD19 (a CAR19-expressing cell), in combination with one or more kinase inhibitors chosen from a Bruton's tyrosine kinase (BTK) inhibitor, a cyclin dependent kinase 4 (CDK4) inhibitor, an mTOR inhibitor, or a mitogen activated protein kinase interacting kinase (MNK) inhibitor.
2. (canceled)
3. The method of claim 1, wherein the CAR19-expressing cell is administered to the mammal after administration of the kinase inhibitor.
4. The method of claim 1, wherein the mammal is, or is identified as being, a complete or partial responder to the BTK inhibitor, or a complete or partial responder to the CAR19-expressing cell.
5. The method of claim 1, wherein the BTK inhibitor is chosen from ibrutinib, GDC-0834, RN-486, CGI-560, CGI-1764, HM-71224, CC-292, ONO-4059, CNX-774, or LFM-A13.
6. The method of claim 1, wherein: (i) the CDK4 inhibitor is chosen from: palbociclib, aloisine A, flavopiridol, 2-(2-chlorophenyl)-5,7-dihydroxy-8-[(3S,4R)-3-hydroxy-1-methyl-4-piperidi- nyl]-4-chromenone; crizotinib (PF-02341066, P276-00, RAF265, indisulam, roscovitine, dinaciclib, BMS 387032, MLN8054, AG-024322, AT7519, AZD5438, BMS908662; or ribociclib; (ii) the mTOR inhibitor is chosen from: rapamycin, a rapamycin analog such as everolimus, temsirolimus, ridaforolimus, semapimod, AZD8055, PF04691502, SF1126, XL765, or OSI-027; and (iii) the MNK inhibitor is chosen from: CGP052088, CGP57380, cercosporamide, ETC-1780445-2, or 4-amino-5-(4-fluoroanilino)-pyrazolo [3,4-d] pyrimidine.
7-8. (canceled)
9. The method of claim 1, wherein the kinase inhibitor is ibrutinib and the ibrutinib has a dose of about 250 mg, 300 mg, 350 mg, 400 mg, 420 mg, 440 mg, 460 mg, 480 mg, 500 mg, 520 mg, 540 mg, 560 mg, 580 mg, or 600 mg daily.
10. The method of claim 1, wherein the cell expresses a CAR molecule comprising an anti-CD19 binding domain, a transmembrane domain, and an intracellular signaling domain, optionally, wherein the intracellular signaling domain comprises a costimulatory domain and/or a primary signaling domain, wherein the anti-CD19 binding domain comprises: (i) a light chain complementary determining region 1 (LC CDR1), a light chain complementary determining region 2 (LC CDR2), a light chain complementary determining region 3 (LC CDR3), a heavy chain complementary determining region 1 (HC CDR1), a heavy chain complementary determining region 2 (HC CDR2), and a heavy chain complementary determining region 3 (HC CDR3) of an anti-CD19 binding domain; (ii) a murine light chain variable region of Table 7, a murine heavy chain variable region of Table 7, or both; (iii) a LC CDR1 of SEQ ID NO: 5, a LC CDR2 of SEQ ID NO: 26, and a LC CDR3 of SEQ ID NO: 27; and/or wherein the anti-CD19 binding domain comprises a HC CDR1 of SEQ ID NO: 19, a LC CDR2 of any of SEQ ID NOS: 20-23, and a HC CDR3 of SEQ ID NO: 24; (iv) a sequence of SEQ ID NO:59, or a sequence with 95-99% identify thereof; or (v) a sequence chosen from: SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO: 4, SEQ ID NO:5, SEQ ID NO:6, SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO: 11 and SEQ ID NO: 12, or a sequence with 95-99% identity thereof, and optionally, the anti-CD19 binding domain is a scFv that comprises a light chain variable region attached to a heavy chain variable via a linker, wherein the linker comprises a sequence of SEQ ID NO: 53.
11-19. (canceled)
20. The method of claim 10, wherein the CAR molecule comprises a transmembrane domain of a protein chosen from: the alpha, beta or zeta chain of the T-cell receptor, CD28, CD3 epsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137 or CD154, optionally, wherein the transmembrane domain comprises a sequence of SEQ ID NO: 15.
21. (canceled)
22. The method of claim 20, wherein the anti-CD19 binding domain is connected to the transmembrane domain by a hinge region, wherein the hinge region comprises a sequence of SEQ ID NO:14 or SEQ ID NO:45.
23. The method of claim 10, wherein the CAR molecule comprises a costimulatory domain, wherein the costimulatory domain comprises a sequence of SEQ ID NO: 16 or SEQ ID NO:51.
24. The method of claim 10, wherein the CAR molecule comprises an intracellular signaling domain, wherein the intracellular signaling domain comprises: (i) a functional signaling domain of 4-1BB, a functional signaling domain of CD3 zeta, or both, or wherein the intracellular signaling domain comprises a sequence of CD27, a functional signaling domain of CD3 zeta, or both; (ii) a sequence of SEQ ID NO: 16, a sequence of SEQ ID NO: 17, or both; (iii) a sequence of SEQ ID NO: 16, a sequence of SEQ ID NO: 43, or both; (iv) a sequence of SEQ ID NO: 51, a sequence of SEQ ID NO: 17, or both; or (v) a sequence of SEQ ID NO: 51, a sequence of SEQ ID NO: 43, or both.
25-26. (canceled)
27. The method of claim 10, wherein CAR molecule comprises an amino acid sequence of SEQ ID NO:58, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, SEQ ID NO:38, SEQ ID NO:39, SEQ ID NO:40, SEQ ID NO:41, or SEQ ID NO:42.
28. The method of claim 1, further comprising administration of an agent which inhibits an immune inhibitory molecule chosen from: PD1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 or TGFR beta.
29. The method of claim 1, wherein the disease associated with expression of CD19 is a cancer, optionally, a hematological cancer chosen from a leukemia or lymphoma.
30. (canceled)
31. The method of claim 29, wherein the cancer is chosen from: chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), multiple myeloma, acute lymphoid leukemia (ALL), Hodgkin lymphoma, B-cell acute lymphoid leukemia (BALL), T-cell acute lymphoid leukemia (TALL), small lymphocytic leukemia (SLL), B cell prolymphocytic leukemia, blastic plasmacytoid dendritic cell neoplasm, Burkitt's lymphoma, diffuse large B cell lymphoma (DLBCL), DLBCL associated with chronic inflammation, follicular lymphoma, pediatric follicular lymphoma, hairy cell leukemia, small cell- or a large cell-follicular lymphoma, malignant lymphoproliferative conditions, MALT lymphoma (extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue), Marginal zone lymphoma, myelodysplasia and myelodysplastic syndrome, non-Hodgkin lymphoma, plasmablastic lymphoma, plasmacytoid dendritic cell neoplasm, Waldenstrom macroglobulinemia, splenic marginal zone lymphoma, splenic lymphoma/leukemia, splenic diffuse red pulp small B-cell lymphoma, hairy cell leukemia-variant, lymphoplasmacytic lymphoma, a heavy chain disease, plasma cell myeloma, solitary plasmocytoma of bone, extraosseous plasmocytoma, nodal marginal zone lymphoma, pediatric nodal marginal zone lymphoma, primary cutaneous follicle center lymphoma, lymphomatoid granulomatosis, primary mediastinal (thymic) large B-cell lymphoma, intravascular large B-cell lymphoma, ALK+ large B-cell lymphoma, large B-cell lymphoma arising in HHV8-associated multicentric Castleman disease, primary effusion lymphoma, B-cell lymphoma, or unclassifiable lymphoma.
32. (canceled)
33. The method of claim 1, further comprising administration of a cytokine chosen from IL-7, IL-15, or IL-21.
34. The method of claim 1, wherein the CAR is a regulatable CAR (RCAR), wherein the RCAR comprises: an intracellular signaling member comprising an intracellular signaling domain and a first switch domain, an antigen binding member comprising an antigen binding domain that binds CD19 and a second switch domain; and a transmembrane domain.
35. (canceled)
36. The method of claim 1, wherein the CAR19-expressing cell is administered in combination a second kinase inhibitor, wherein the second kinase inhibitor is other than ibrutinib, when the mammal is, or is identified as being, a non-responder or relapser to ibrutinib, wherein second kinase inhibitor is chosen from one or more of GDC-0834, RN-486, CGI-560, CGI-1764, HM-71224, CC-292, ONO-4059, CNX-774, or LFM-A13, or a combination thereof.
37. The method of claim 1, wherein the mammal is, or is identified as being, a partial responder to the kinase inhibitor, and the mammal is administered the CAR19-expressing cell, alone or in combination with the BTK inhibitor, during the period of partial response.
38. The method of claim 1, wherein the kinase inhibitor is ibrutinib and the ibrutinib is formulated for administration for 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 or more cycles, wherein cycle length is 21 or 28 days.
39. (canceled)
40. The method of claim 1, wherein the mammal has undergone lymphodepletion, wherein the lymphodepletion comprises administration of one or more of melphalan, cytoxan, cyclophosphamide, and fludarabine.
41. (canceled)
42. A method of making a CAR-expressing immune effector population of cells, comprising: contacting the population of cells with a BTK inhibitor; and introducing a nucleic acid encoding a CAR molecule into the population of cells under conditions such that the CAR molecule is expressed.
43. (canceled)
44. The method of claim 42, which comprises contacting the population of cells with the BTK inhibitor for 10-20, 20-30, 30-40, 40-60, or 60-120 minutes and subsequently removing most or all of the BTK inhibitor from the population of cells, wherein the BTK inhibitor is chosen from: ibrutinib, GDC-0834, RN-486, CGI-560, CGI-1764, HM-71224, CC-292, ONO-4059, CNX-774, or LFM-A13.
45. (canceled)
46. A reaction mixture comprising a population of immune effector cells, a BTK inhibitor, and a CAR molecule or a nucleic acid encoding a CAR molecule, wherein the BTK inhibitor is chosen from ibrutinib, GDC-0834, RN-486, CGI-560, CGI-1764, HM-71224, CC-292, ONO-4059, CNX-774, or LFM-A13.
47. A composition comprising a population of cells that expresses a CAR molecule that binds CD19, and one or more kinase inhibitors, wherein the kinase inhibitor is chosen from a Bruton's tyrosine kinase (BTK) inhibitor, a cyclin dependent kinase 4 (CDK4) inhibitor, an mTOR inhibitor, or a mitogen activated protein kinase interacting kinase (MNK) inhibitor, wherein the CAR19-expressing cell and the one or more kinase inhibitors are present in a single dose form, or as two or more dose forms.
Description
[0001] This application is a divisional of U.S. Ser. No. 14/680,860, filed Apr. 7, 2015, which claims priority to U.S. Ser. No. 61/976,396 filed Apr. 7, 2014, U.S. Ser. No. 62/007,309 filed Jun. 3, 2014, U.S. Ser. No. 62/036,493 filed Aug. 12, 2014, U.S. Ser. No. 62/076,238 filed Nov. 6, 2014, U.S. Ser. No. 62/087,888 filed Dec. 5, 2014, and U.S. Ser. No. 62/097,278 filed Dec. 29, 2014, the contents of which are incorporated herein by reference in their entireties. International Application Number PCT/US15/24671, filed Apr. 7, 2015, is also incorporated herein by reference in its entirety.
SEQUENCE LISTING
[0002] The instant application contains a Sequence Listing which has been submitted electronically in ASCII format and is hereby incorporated by reference in its entirety. Said ASCII copy, created on Apr. 6, 2015, is named N2067-7051US_SL.txt and is 249,332 bytes in size.
FIELD OF THE INVENTION
[0003] The present invention relates generally to the use of T cells engineered to express a Chimeric Antigen Receptor (CAR), e.g., in combination with another agent such as, e.g., a kinase inhibitor and/or a cytokine, to treat a disease associated with expression of the Cluster of Differentiation 19 protein (CD19).
BACKGROUND OF THE INVENTION
[0004] Many patients with B cell malignancies are incurable with standard therapy. In addition, traditional treatment options often have serious side effects. Attempts have been made in cancer immunotherapy, however, several obstacles render this a very difficult goal to achieve clinical effectiveness. Although hundreds of so-called tumor antigens have been identified, these are generally derived from self and thus are poorly immunogenic. Furthermore, tumors use several mechanisms to render themselves hostile to the initiation and propagation of immune attack.
[0005] Recent developments using chimeric antigen receptor (CAR) modified autologous T cell (CART) therapy, which relies on redirecting T cells to a suitable cell-surface molecule on cancer cells such as B cell malignancies, show promising results in harnessing the power of the immune system to treat B cell malignancies and other cancers (see, e.g., Sadelain et al., Cancer Discovery 3:388-398 (2013)). The clinical results of the murine derived CART19 (i.e., "CTL019") have shown promise in establishing complete remissions in patients suffering with CLL as well as in childhood ALL (see, e.g., Kalos et al., Sci Transl Med 3:95ra73 (2011), Porter et al., NEJM 365:725-733 (2011), Grupp et al., NEJM 368:1509-1518 (2013)). Besides the ability for the chimeric antigen receptor on the genetically modified T cells to recognize and destroy the targeted cells, a successful therapeutic T cell therapy needs to have the ability to proliferate and persist over time, and to further monitor for leukemic cell escapees. The variable quality of T cells whether it's a result of anergy, suppression or exhaustion will have effects on CAR-transformed T cells' performance but for which skilled practitioners have limited control over at this time. To be effective, CAR transformed patient T cells need to persist and maintain the ability to proliferate in response to the CAR's antigen. It has been shown that ALL patient T cells perform can do this with CART19 comprising a murine scFv (see, e.g., Grupp et al., NEJM 368:1509-1518 (2013)).
SUMMARY OF THE INVENTION
[0006] The disclosure features, at least in part, compositions and methods of treating disorders such as cancer (e.g., hematological cancers or other B-cell malignancies) using immune effector cells (e.g., T cells or NK cells) that express a Chimeric Antigen Receptor (CAR) molecule (e.g., a CAR that binds to a B-cell antigen, e.g., Cluster of Differentiation 19 protein (CD19) (e.g., OMIM Acc. No. 107265, Swiss Prot. Acc No. P15391). The compositions include, and the methods include administering, immune effector cells (e.g., T cells or NK cells) expressing a B cell targeting CAR, in combination with a kinase inhibitor (e.g., one or more of a CDK4/6 inhibitor, a BTK inhibitor, an mTOR inhibitor, a MNK inhibitor, a dual PI3K/mTOR inhibitor, or a combination thereof). In some embodiments, the combination maintains, or has better clinical effectiveness, as compared to either therapy alone. The invention further pertains to the use of engineered cells, e.g., immune effector cells (e.g., T cells or NK cells), to express a CAR molecule that binds to a B-cell antigen, e.g., CD19, in combination with a kinase inhibitor (e.g., a kinase inhibitor chosen from one or more of a cyclin dependent kinase 4 (CDK4) inhibitor, a Bruton's tyrosine kinase (BTK) inhibitor, an mTOR inhibitor, a mitogen activated protein kinase interacting kinase (MNK) inhibitor, a dual phosphatidylinositol 3-kinase (PI3K)/mTOR inhibitor, or a combination thereof) to treat a disorder associated with expression of a B-cell antigen, e.g., CD19 (e.g., a cancer, e.g., a hematological cancer).
[0007] Accordingly, in one aspect, the invention pertains to a method of treating a subject, e.g., a mammal, having a disease associated with expression of a B-cell antigen, e.g., CD19. The method comprises administering to the mammal an effective amount of a cell e.g., an immune effector cell (e.g., a T cell or NK cell) that expresses a CAR molecule that binds the B-cell antigen, in combination with a kinase inhibitor, e.g., a kinase inhibitor described herein. In one embodiment, the CAR molecule binds to CD19, e.g., a CAR molecule that binds CD19 described herein. In other embodiments, the CAR molecule binds to one or more of CD20, CD22 or ROR1.
[0008] In one embodiment, the disease associated with expression of a B-cell antigen (e.g., expression of one or more of CD19, CD20, CD22 or ROR1), is selected from a proliferative disease such as a cancer, a malignancy, or a precancerous condition such as a myelodysplasia, a myelodysplastic syndrome or a preleukemia, or is a non-cancer related indication associated with expression of the B-cell antigen, e.g., one or more of CD19, CD20, CD22 or ROR1. In one embodiment, the disease is a solid or liquid tumor. In one embodiment, the cancer is pancreatic cancer. In one embodiment, the disease is a hematologic cancer. In one embodiment, the hematological cancer is leukemia. In one embodiment, the cancer is selected from the group consisting of one or more acute leukemias including but not limited to B-cell acute lymphoid leukemia (BALL), T-cell acute lymphoid leukemia (TALL), small lymphocytic leukemia (SLL), acute lymphoid leukemia (ALL); one or more chronic leukemias including but not limited to chronic myelogenous leukemia (CML), chronic lymphocytic leukemia (CLL). Additional hematological cancers or hematologic conditions include, but are not limited to, mantle cell lymphoma (MCL), B cell prolymphocytic leukemia, blastic plasmacytoid dendritic cell neoplasm, Burkitt's lymphoma, diffuse large B cell lymphoma (DLBCL), follicular lymphoma, hairy cell leukemia, small cell- or a large cell-follicular lymphoma, malignant lymphoproliferative conditions, MALT lymphoma, Marginal zone lymphoma, multiple myeloma, myelodysplasia and myelodysplastic syndrome, non-Hodgkin lymphoma, Hodgkin lymphoma, plasmablastic lymphoma, plasmacytoid dendritic cell neoplasm, and Waldenstrom macroglobulinemia. In certain embodiments, the disease associated with B-cell antigen (e.g., e.g., one or more of CD19, CD20, CD22 or ROR1) expression is a "preleukemia" which is a diverse collection of hematological conditions united by ineffective production (or dysplasia) of myeloid blood cells. In some embodiments, the disease associated with B-cell antigen (e.g., one or more of CD19, CD20, CD22 or ROR1) expression includes, but is not limited to atypical and/or non-classical cancers, malignancies, precancerous conditions or proliferative diseases expressing the B-cell antigen (e.g., one or more of CD19, CD20, CD22 or ROR1). Any combination of the diseases associated with B-cell antigen (e.g., one or more of CD19, CD20, CD22 or ROR1) expression described herein can be treated with the methods and compositions described herein.
[0009] In one embodiment, the disease associated with expression of the B-cell antigen (e.g., one or more of CD19, CD20, CD22 or ROR1) is a lymphoma, e.g., MCL, Hodgkin lymphoma, or DLBCL. In one embodiment, the disease associated with expression of the B-cell antigen (e.g., one or more of CD19, CD20, CD22 or ROR1) is leukemia, e.g., SLL, CLL and/or ALL. In one embodiment, the disease associated with expression of the B-cell antigen is multiple myeloma (e.g., a multiple myeloma that is CD19-negative, e.g., having a vast majority (99.95%) of the neoplastic plasma cells with a CD19-negative phenotype, e.g., as detected by both flow cytometry and RT-PCR.
[0010] In one embodiment, the kinase inhibitor is a CDK4 inhibitor, e.g., a CDK4 inhibitor described herein, e.g., a CD4/6 inhibitor, such as, e.g., 6-Acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylami- no)-8H-pyrido[2,3-d]pyrimidin-7-one, hydrochloride (also referred to as palbociclib or PD0332991). In one embodiment, the kinase inhibitor is a BTK inhibitor, e.g., a BTK inhibitor described herein, such as, e.g., ibrutinib. In one embodiment, the kinase inhibitor is an mTOR inhibitor, e.g., an mTOR inhibitor described herein, such as, e.g., rapamycin, a rapamycin analog, OSI-027. The mTOR inhibitor can be, e.g., an mTORC1 inhibitor and/or an mTORC2 inhibitor, e.g., an mTORC1 inhibitor and/or mTORC2 inhibitor described herein. In one embodiment, the kinase inhibitor is a MNK inhibitor, e.g., a MNK inhibitor described herein, such as, e.g., 4-amino-5-(4-fluoroanilino)-pyrazolo [3,4-d]pyrimidine. The MNK inhibitor can be, e.g., a MNK1a, MNK1b, MNK2a and/or MNK2b inhibitor. In one embodiment, the inhibitor can be a dual PI3K/mTOR inhibitor, e.g., PF-04695102.
[0011] In one embodiment, the kinase inhibitor is a CDK4 inhibitor selected from aloisine A; flavopiridol or HMR-1275, 2-(2-chlorophenyl)-5,7-dihydroxy-8-[(3S,4R)-3-hydroxy-1-methyl-4-piperidi- nyl]-4-chromenone; crizotinib (PF-02341066); 2-(2-Chlorophenyl)-5,7-dihydroxy-8-[(2R,3S)-2-(hydroxymethyl)-1-methyl-3-- pyrrolidinyl]-4H-1-benzopyran-4-one, hydrochloride (P276-00); 1-methyl-5-[[2-[5-(trifluoromethyl)-1H-imidazol-2-yl]-4-pyridinyl]oxy]-N-- [4-(trifluoromethyl)phenyl]-1H-benzimidazol-2-amine (RAF265); indisulam (E7070); roscovitine (CYC202); palbociclib (PD0332991); dinaciclib (SCH727965); N-[5-[[(5-tert-butyloxazol-2-yl)methyl]thio]thiazol-2-yl]piperidine-4-car- boxamide (BMS 387032); 4-[[9-chloro-7-(2,6-difluorophenyl)-5H-pyrimido[5,4-d][2]benzazepin-2-yl]- amino]-benzoic acid (MLN8054); 5-[3-(4,6-difluoro-1H-benzimidazol-2-yl)-1H-indazol-5-yl]-N-ethyl-4-methy- l-3-pyridinemethanamine (AG-024322); 4-(2,6-dichlorobenzoylamino)-1H-pyrazole-3-carboxylic acid N-(piperidin-4-yl)amide (AT7519); 4-[2-methyl-1-(1-methylethyl)-1H-imidazol-5-yl]-N-[4-(methylsulfonyl)phen- yl]-2-pyrimidinamine (AZD5438); XL281 (BMS908662); and ribociclib.
[0012] In one embodiment, the kinase inhibitor is a CDK4 inhibitor, e.g., palbociclib (PD0332991), and the palbociclib is administered at a dose of about 50 mg, 60 mg, 70 mg, 75 mg, 80 mg, 90 mg, 100 mg, 105 mg, 110 mg, 115 mg, 120 mg, 125 mg, 130 mg, 135 mg (e.g., 75 mg, 100 mg or 125 mg) daily for a period of time, e.g., daily for 14-21 days of a 28 day cycle, or daily for 7-12 days of a 21 day cycle. In one embodiment, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 or more cycles of palbociclib are administered.
[0013] In one embodiment, the kinase inhibitor is a BTK inhibitor selected from ibrutinib (PCI-32765); GDC-0834; RN-486; CGI-560; CGI-1764; HM-71224; CC-292; ONO-4059; CNX-774; and LFM-A13. In a preferred embodiment, the BTK inhibitor does not reduce or inhibit the kinase activity of interleukin-2-inducible kinase (ITK), and is selected from GDC-0834; RN-486; CGI-560; CGI-1764; HM-71224; CC-292; ONO-4059; CNX-774; and LFM-A13.
[0014] In one embodiment, the kinase inhibitor is a BTK inhibitor, e.g., ibrutinib (PCI-32765), and the ibrutinib is administered at a dose of about 250 mg, 300 mg, 350 mg, 400 mg, 420 mg, 440 mg, 460 mg, 480 mg, 500 mg, 520 mg, 540 mg, 560 mg, 580 mg, 600 mg (e.g., 250 mg, 420 mg or 560 mg) daily for a period of time, e.g., daily for 21 day cycle cycle, or daily for 28 day cycle. In one embodiment, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 or more cycles of ibrutinib are administered.
[0015] In one embodiment, the kinase inhibitor is an mTOR inhibitor selected from temsirolimus; ridaforolimus (1R,2R,4S)-4-[(2R)-2 [(1R,9S,12S, 15R, 16E, 18R, 19R,21R,23S,24E,26E,28Z,30S,32S,35R)-1,18-dihydroxy-19,30-dimethoxy-15,17- ,21,23, 29,35-hexamethyl-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatricyclo[3- 0.3.1.0.sup.4,9]hexatriaconta-16,24,26,28-tetraen-12-yl]propyl]-2-methoxyc- yclohexyl dimethylphosphinate, also known as AP23573 and MK8669; everolimus (RAD001); rapamycin (AY22989); semapimod; (5-{2,4-bis[(3S)-3-methylmorpholin-4-yl]pyrido[2,3-d]pyrimidin-7-yl}-2-me- thoxyphenyl)methanol (AZD8055); 2-amino-8-[trans-4-(2-hydroxyethoxy)cyclohexyl]-6-(6-methoxy-3-pyridinyl)- -4-methyl-pyrido[2,3-d]pyrimidin-7(8H)-one (PF04691502); and N.sup.2-[1,4-dioxo-4-[[4-(4-oxo-8-phenyl-4H-1-benzopyran-2-yl)morpholiniu- m-4-yl]methoxy]butyl]-L-arginylglycyl-L-.alpha.-aspartylL-serine-, inner salt (SF1126); and XL765.
[0016] In one embodiment, the kinase inhibitor is an mTOR inhibitor, e.g., rapamycin, and the rapamycin is administered at a dose of about 3 mg, 4 mg, 5 mg, 6 mg, 7 mg, 8 mg, 9 mg, 10 mg (e.g., 6 mg) daily for a period of time, e.g., daily for 21 day cycle cycle, or daily for 28 day cycle. In one embodiment, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 or more cycles of rapamycin are administered. In one embodiment, the kinase inhibitor is an mTOR inhibitor, e.g., everolimus and the everolimus is administered at a dose of about 2 mg, 2.5 mg, 3 mg, 4 mg, 5 mg, 6 mg, 7 mg, 8 mg, 9 mg, 10 mg, 11 mg, 12 mg, 13 mg, 14 mg, 15 mg (e.g., 10 mg) daily for a period of time, e.g., daily for 28 day cycle. In one embodiment, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 or more cycles of everolimus are administered.
[0017] In one embodiment, the kinase inhibitor is an MNK inhibitor selected from CGP052088; 4-amino-3-(p-fluorophenylamino)-pyrazolo [3,4-d]pyrimidine (CGP57380); cercosporamide; ETC-1780445-2; and 4-amino-5-(4-fluoroanilino)-pyrazolo [3,4-d]pyrimidine.
[0018] In one embodiment, the kinase inhibitor is a dual phosphatidylinositol 3-kinase (PI3K) and mTOR inhibitor selected from 2-Amino-8-[trans-4-(2-hydroxyethoxy)cyclohexyl]-6-(6-methoxy-3-pyridinyl)- -4-methyl-pyrido[2,3-d]pyrimidin-7(8H)-one (PF-04691502); N-[4-[[4-(Dimethylamino)-1-piperidinyl]carbonyl]phenyl]-N-[4-(4,6-di-4-mo- rpholinyl-1,3,5-triazin-2-yl)phenyl]urea (PF-05212384, PKI-587); 2-Methyl-2-{4-[3-methyl-2-oxo-8-(quinolin-3-yl)-2,3-dihydro-1H-imidazo[4,- 5-c]quinolin-1-yl]phenyl}propanenitrile (BEZ-235); apitolisib (GDC-0980, RG7422); 2,4-Difluoro-N-{2-(methyloxy)-5-[4-(4-pyridazinyl)-6-quinolinyl]- -3-pyridinyl}benzenesulfonamide (GSK2126458); 8-(6-methoxypyridin-3-yl)-3-methyl-1-(4-(piperazin-1-yl)-3-(trifluorometh- yl)phenyl)-1H-imidazo[4,5-c]quinolin-2(3H)-one Maleic acid (NVP-BGT226); 3-[4-(4-Morpholinylpyrido[3',2':4,5]furo[3,2-d]pyrimidin-2-yl]phenol (PI-103); 5-(9-isopropyl-8-methyl-2-morpholino-9H-purin-6-yl)pyrimidin-2-- amine (VS-5584, SB2343); and N-[2-[(3,5-Dimethoxyphenyl)amino]quinoxalin-3-yl]-4-[(4-methyl-3-methoxyp- henyl)carbonyl]aminophenylsulfonamide (XL765).
[0019] In one embodiment, the cell expresses a CAR molecule comprising an anti-CD19 binding domain (e.g., a murine or humanized antibody or antibody fragment that specifically binds to CD19), a transmembrane domain, and an intracellular signaling domain (e.g., an intracellular signaling domain comprising a costimulatory domain and/or a primary signaling domain). In one embodiment, the CAR comprises an antibody or antibody fragment which includes an anti-CD19 binding domain described herein (e.g., a murine or humanized antibody or antibody fragment that specifically binds to CD19 as described herein), a transmembrane domain described herein, and an intracellular signaling domain described herein (e.g., an intracellular signaling domain comprising a costimulatory domain and/or a primary signaling domain described herein).
[0020] In one embodiment, the CAR molecule is capable of binding CD19 (e.g., wild-type or mutant human CD19). In one embodiment, the CAR molecule comprises an anti-CD19 binding domain comprising one or more (e.g., all three) light chain complementary determining region 1 (LC CDR1), light chain complementary determining region 2 (LC CDR2), and light chain complementary determining region 3 (LC CDR3) of an anti-CD19 binding domain described herein, and one or more (e.g., all three) heavy chain complementary determining region 1 (HC CDR1), heavy chain complementary determining region 2 (HC CDR2), and heavy chain complementary determining region 3 (HC CDR3) of an anti-CD19 binding domain described herein, e.g., an anti-CD19 binding domain comprising one or more, e.g., all three, LC CDRs and one or more, e.g., all three, HC CDRs. In one embodiment, the anti-CD19 binding domain comprises one or more (e.g., all three) heavy chain complementary determining region 1 (HC CDR1), heavy chain complementary determining region 2 (HC CDR2), and heavy chain complementary determining region 3 (HC CDR3) of an anti-CD19 binding domain described herein, e.g., the anti-CD19 binding domain has two variable heavy chain regions, each comprising a HC CDR1, a HC CDR2 and a HC CDR3 described herein. In one embodiment, the anti-CD19 binding domain comprises a murine light chain variable region described herein (e.g., in Table 7) and/or a murine heavy chain variable region described herein (e.g., in Table 7). In one embodiment, the anti-CD19 binding domain is a scFv comprising a murine light chain and a murine heavy chain of an amino acid sequence of Table 7. In an embodiment, the anti-CD19 binding domain (e.g., an scFv) comprises: a light chain variable region comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of an amino acid sequence of a light chain variable region provided in Table 7, or a sequence with 95-99% identity with an amino acid sequence of Table 7; and/or a heavy chain variable region comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of an amino acid sequence of a heavy chain variable region provided in Table 7, or a sequence with 95-99% identity to an amino acid sequence of Table 7. In one embodiment, the anti-CD19 binding domain comprises a sequence of SEQ ID NO:59, or a sequence with 95-99% identify thereof. In one embodiment, the anti-CD19 binding domain is a scFv, and a light chain variable region comprising an amino acid sequence described herein, e.g., in Table 7, is attached to a heavy chain variable region comprising an amino acid sequence described herein, e.g., in Table 7, via a linker, e.g., a linker described herein. In one embodiment, the anti-CD19 binding domain includes a (Gly.sub.4-Ser)n linker, wherein n is 1, 2, 3, 4, 5, or 6, preferably 3 or 4 (SEQ ID NO: 53). The light chain variable region and heavy chain variable region of a scFv can be, e.g., in any of the following orientations: light chain variable region-linker-heavy chain variable region or heavy chain variable region-linker-light chain variable region.
[0021] In one embodiment, the CAR molecule comprises a humanized anti-CD19 binding domain that includes one or more (e.g., all three) light chain complementary determining region 1 (LC CDR1), light chain complementary determining region 2 (LC CDR2), and light chain complementary determining region 3 (LC CDR3) of a humanized anti-CD19 binding domain described herein, and one or more (e.g., all three) heavy chain complementary determining region 1 (HC CDR1), heavy chain complementary determining region 2 (HC CDR2), and heavy chain complementary determining region 3 (HC CDR3) of a humanized anti-CD19 binding domain described herein, e.g., a humanized anti-CD19 binding domain comprising one or more, e.g., all three, LC CDRs and one or more, e.g., all three, HC CDRs. In one embodiment, the humanized anti-CD19 binding domain comprises at least HC CDR2. In one embodiment, the humanized anti-CD19 binding domain comprises one or more (e.g., all three) heavy chain complementary determining region 1 (HC CDR1), heavy chain complementary determining region 2 (HC CDR2), and heavy chain complementary determining region 3 (HC CDR3) of a humanized anti-CD19 binding domain described herein, e.g., the humanized anti-CD19 binding domain has two variable heavy chain regions, each comprising a HC CDR1, a HC CDR2 and a HC CDR3 described herein. In one embodiment, the humanized anti-CD19 binding domain comprises at least HC CDR2. In one embodiment, the light chain variable region comprises one, two, three or all four framework regions of VK3_L25 germline sequence. In one embodiment, the light chain variable region has a modification (e.g., substitution, e.g., a substitution of one or more amino acid found in the corresponding position in the murine light chain variable region of SEQ ID NO: 58, e.g., a substitution at one or more of positions 71 and 87). In one embodiment, the heavy chain variable region comprises one, two, three or all four framework regions of VH4_4-59 germline sequence. In one embodiment, the heavy chain variable region has a modification (e.g., substitution, e.g., a substitution of one or more amino acid found in the corresponding position in the murine heavy chain variable region of SEQ ID NO: 58, e.g., a substitution at one or more of positions 71, 73 and 78). In one embodiment, the humanized anti-CD19 binding domain comprises a light chain variable region described herein (e.g., in Table 3) and/or a heavy chain variable region described herein (e.g., in Table 3). In one embodiment, the humanized anti-CD19 binding domain is a scFv comprising a light chain and a heavy chain of an amino acid sequence of Table 3. In an embodiment, the humanized anti-CD19 binding domain (e.g., an scFv) comprises: a light chain variable region comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of an amino acid sequence of a light chain variable region provided in Table 3, or a sequence with 95-99% identity with an amino acid sequence of Table 3; and/or a heavy chain variable region comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of an amino acid sequence of a heavy chain variable region provided in Table 3, or a sequence with 95-99% identity to an amino acid sequence of Table 3. In one embodiment, the humanized anti-CD19 binding domain comprises a sequence selected from a group consisting of SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO: 4, SEQ ID NO:5, SEQ ID NO:6, SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 and SEQ ID NO: 12, or a sequence with 95-99% identify thereof. In one embodiment, the humanized anti-CD19 binding domain is a scFv, and a light chain variable region comprising an amino acid sequence described herein, e.g., in Table 3, is attached to a heavy chain variable region comprising an amino acid sequence described herein, e.g., in Table 3, via a linker, e.g., a linker described herein. In one embodiment, the humanized anti-CD19 binding domain includes a (Gly.sub.4-Ser)n linker, wherein n is 1, 2, 3, 4, 5, or 6, preferably 3 or 4 (SEQ ID NO: 53). The light chain variable region and heavy chain variable region of a scFv can be, e.g., in any of the following orientations: light chain variable region-linker-heavy chain variable region or heavy chain variable region-linker-light chain variable region.
[0022] In one embodiment, the CAR molecule comprises a transmembrane domain of a protein selected from the group consisting of the alpha, beta or zeta chain of the T-cell receptor, CD28, CD3 epsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137 and CD154. In one embodiment, the transmembrane domain comprises a sequence of SEQ ID NO: 15. In one embodiment, the transmembrane domain comprises an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 20, 10 or 5 modifications (e.g., substitutions) of an amino acid sequence of SEQ ID NO: 15, or a sequence with 95-99% identity to an amino acid sequence of SEQ ID NO: 15.
[0023] In one embodiment, the anti-CD19 binding domain is connected to the transmembrane domain by a hinge region, e.g., a hinge region described herein. In one embodiment, the encoded hinge region comprises SEQ ID NO:14 or SEQ ID NO:45, or a sequence with 95-99% identity thereof.
[0024] In one embodiment, the CAR molecule further comprises a sequence encoding a costimulatory domain, e.g., a costimulatory domain described herein. In one embodiment, the costimulatory domain comprises a functional signaling domain of a protein selected from the group consisting of OX40, CD2, CD27, CD28, CDS, ICAM-1, LFA-1 (CD11a/CD18) and 4-1BB (CD137). In one embodiment, the costimulatory domain comprises a sequence of SEQ ID NO: 16. In one embodiment, the costimulatory domain comprises a sequence of SEQ ID NO:51. In one embodiment, the costimulatory domain comprises an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 20, 10 or 5 modifications (e.g., substitutions) of an amino acid sequence of SEQ ID NO: 16 or SEQ ID NO:51, or a sequence with 95-99% identity to an amino acid sequence of SEQ ID NO: 16 or SEQ ID NO:51.
[0025] In one embodiment, the CAR molecule further comprises a sequence encoding an intracellular signaling domain, e.g., an intracellular signaling domain described herein. In one embodiment, the intracellular signaling domain comprises a functional signaling domain of 4-1BB and/or a functional signaling domain of CD3 zeta. In one embodiment, the intracellular signaling domain comprises the sequence of SEQ ID NO: 16 and/or the sequence of SEQ ID NO: 17. In one embodiment, the intracellular signaling domain comprises the sequence of SEQ ID NO: 16 and/or the sequence of SEQ ID NO:43. In one embodiment, the intracellular signaling domain comprises a functional signaling domain of CD27 and/or a functional signaling domain of CD3 zeta. In one embodiment, the intracellular signaling domain comprises the sequence of SEQ ID NO: 51 and/or the sequence of SEQ ID NO:17. In one embodiment, the intracellular signaling domain comprises the sequence of SEQ ID NO:51 and/or the sequence of SEQ ID NO:43. In one embodiment, the intracellular signaling domain comprises an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 20, 10 or 5 modifications (e.g., substitutions) of an amino acid sequence of SEQ ID NO:16 or SEQ ID NO:51 and/or an amino acid sequence of SEQ ID NO:17 or SEQ ID NO:43, or a sequence with 95-99% identity to an amino acid sequence of SEQ ID NO:16 or SEQ ID NO:51 and/or an amino acid sequence of SEQ ID NO:17 or SEQ ID NO:43. In one embodiment, the intracellular signaling domain comprises the sequence of SEQ ID NO:16 or SEQ ID NO:51 and the sequence of SEQ ID NO: 17 or SEQ ID NO:43, wherein the sequences comprising the intracellular signaling domain are expressed in the same frame and as a single polypeptide chain.
[0026] In one embodiment, the CAR molecule further comprises a leader sequence, e.g., a leader sequence described herein. In one embodiment, the leader sequence comprises an amino acid sequence of SEQ ID NO: 13, or a sequence with 95-99% identity to an amino acid sequence of SEQ ID NO:13.
[0027] In one embodiment, the CAR molecule comprises a leader sequence, e.g., a leader sequence described herein, e.g., a leader sequence of SEQ ID NO: 13, or having 95-99% identity thereof; an anti-CD19 binding domain described herein, e.g., an anti-CD19 binding domain comprising a LC CDR1, a LC CDR2, a LC CDR3, a HC CDR1, a HC CDR2 and a HC CDR3 described herein, e.g., a murine anti-CD19 binding domain described in Table 7, a humanized anti-CD19 binding domain described in Table 3, or a sequence with 95-99% identify thereof; a hinge region, e.g., a hinge region described herein, e.g., a hinge region of SEQ ID NO: 14 or having 95-99% identity thereof; a transmembrane domain, e.g., a transmembrane domain described herein, e.g., a transmembrane domain having a sequence of SEQ ID NO:15 or a sequence having 95-99% identity thereof; an intracellular signaling domain, e.g., an intracellular signaling domain described herein (e.g., an intracellular signaling domain comprising a costimulatory domain and/or a primary signaling domain). In one embodiment, the intracellular signaling domain comprises a costimulatory domain, e.g., a costimulatory domain described herein, e.g., a 4-1BB costimulatory domain having a sequence of SEQ ID NO:16 or SEQ ID NO:51, or having 95-99% identity thereof, and/or a primary signaling domain, e.g., a primary signaling domain described herein, e.g., a CD3 zeta stimulatory domain having a sequence of SEQ ID NO: 17 or SEQ ID NO:43, or having 95-99% identity thereof.
[0028] In one embodiment, the CAR molecule comprises (e.g., consists of) an amino acid sequence of SEQ ID NO:58, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, SEQ ID NO:38, SEQ ID NO:39, SEQ ID NO:40, SEQ ID NO:41 or SEQ ID NO:42, or an amino acid sequence having at least one, two, three, four, five, 10, 15, 20 or 30 modifications (e.g., substitutions) but not more than 60, 50 or 40 modifications (e.g., substitutions) of an amino acid sequence of SEQ ID NO:58, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, SEQ ID NO:38, SEQ ID NO:39, SEQ ID NO:40, SEQ ID NO:41 or SEQ ID NO:42, or an amino acid sequence having 85%, 90%, 95%, 96%, 97%, 98% or 99% identity to an amino acid sequence of SEQ ID NO:58, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, SEQ ID NO:38, SEQ ID NO:39, SEQ ID NO:40, SEQ ID NO:41 or SEQ ID NO:42.
[0029] In one embodiment, the cell expressing the CAR molecule comprises a vector that includes a nucleic acid sequence encoding the CAR molecule. In one embodiment, the vector is selected from the group consisting of a DNA, a RNA, a plasmid, a lentivirus vector, adenoviral vector, or a retrovirus vector. In one embodiment, the vector is a lentivirus vector. In one embodiment, the vector further comprises a promoter. In one embodiment, the promoter is an EF-1 promoter. In one embodiment, the EF-1 promoter comprises a sequence of SEQ ID NO: 100. In one embodiment, the vector is an in vitro transcribed vector, e.g., a vector that transcribes RNA of a nucleic acid molecule described herein. In one embodiment, the nucleic acid sequence in the in vitro vector further comprises a poly(A) tail, e.g., a poly A tail described herein, e.g., comprising about 150 adenosine bases (SEQ ID NO: 104). In one embodiment, the nucleic acid sequence in the in vitro vector further comprises a 3'UTR, e.g., a 3' UTR described herein, e.g., comprising at least one repeat of a 3'UTR derived from human beta-globulin. In one embodiment, the nucleic acid sequence in the in vitro vector further comprises promoter, e.g., a T2A promoter.
[0030] In certain embodiments of the compositions and methods disclosed herein, the cell expressing the CAR molecule (also referred to herein as a "CAR-expressing cell") is a cell or population of cells as described herein, e.g., a human immune effector cell or population of cells (e.g., a human T cell or a human NK cell, e.g., a human T cell described herein or a human NK cell described herein). In one embodiment, the human T cell is a CD8+ T cell. In one embodiment, the cell is an autologous T cell. In one embodiment, the cell is an allogeneic T cell. In one embodiment, the cell is a T cell and the T cell is diaglycerol kinase (DGK) deficient. In one embodiment, the cell is a T cell and the T cell is Ikaros deficient. In one embodiment, the cell is a T cell and the T cell is both DGK and Ikaros deficient. It shall be understood that the compositions and methods disclosed herein reciting the term "cell" encompass compositions and methods comprising one or more cells, e.g., a population of cells.
[0031] In another embodiment, the cell expressing the CAR molecule, e.g., as described herein, can further express another agent, e.g., an agent which enhances the activity of a CAR-expressing cell.
[0032] In one embodiment, the method further includes administering a cell expressing the CAR molecule, as described herein, optionally in combination with a kinase inhibitor, e.g., a BTK inhibitor such as ibrutinib, in combination with an agent which enhances the activity of a CAR-expressing cell. In certain embodiments, the agent is a cytokine, e.g., IL-7, IL-15, IL-21, or a combination thereof. In one embodiment, the method includes administering IL-7 to the subject. The cytokine can be delivered in combination with, e.g., simultaneously or shortly after, administration of the CAR-expressing cell. Alternatively, the cytokine can be delivered after a prolonged period of time after administration of the CAR-expressing cell, e.g., after assessment of the subject's response to the CAR-expressing cell.
[0033] In other embodiments, the agent which enhances the activity of a CAR-expressing cell can be an agent which inhibits an immune inhibitory molecule. Examples of immune inhibitory molecules include PD1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 and TGFR beta. In one embodiment, the agent which inhibits an immune inhibitory molecule comprises a first polypeptide, e.g., an immune inhibitory molecule, associated with a second polypeptide that provides a positive signal to the cell, e.g., an intracellular signaling domain described herein. In one embodiment, the agent comprises a first polypeptide, e.g., of an inhibitory molecule such as PD1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 or TGFR beta, or a fragment of any of these (e.g., at least a portion of the extracellular domain of any of these), and a second polypeptide which is an intracellular signaling domain described herein (e.g., comprising a costimulatory domain (e.g., 41BB, CD27 or CD28, e.g., as described herein) and/or a primary signaling domain (e.g., a CD3 zeta signaling domain described herein). In one embodiment, the agent comprises a first polypeptide of PD1 or a fragment thereof (e.g., at least a portion of the extracellular domain of PD1), and a second polypeptide of an intracellular signaling domain described herein (e.g., a CD28 signaling domain described herein and/or a CD3 zeta signaling domain described herein).
[0034] In one embodiment, lymphocyte infusion, for example allogeneic lymphocyte infusion, is used in the treatment of the cancer, wherein the lymphocyte infusion comprises at least one CAR-expressing cell that binds toa B-cell antigen (e.g., CD19) (also referred to herein as CD19 CAR-expressing cell), as described herein. In one embodiment, autologous lymphocyte infusion is used in the treatment of the cancer, wherein the autologous lymphocyte infusion comprises at least one CD19-expressing cell.
[0035] In one embodiment, the CD19 CAR expressing cell, e.g., T cell, is administered to a subject that has received a previous stem cell transplantation, e.g., autologous stem cell transplantation.
[0036] In one embodiment, the CD19 CAR expressing cell, e.g., T cell, is administered to a subject that has received a previous dose of melphalan.
[0037] In one embodiment, the cell expressing the CAR molecule, e.g., a CAR molecule described herein, is administered in combination with an agent that ameliorates one or more side effect associated with administration of a cell expressing a CAR molecule, e.g., an agent described herein.
[0038] In one embodiment, the kinase inhibitor, is administered in combination with an agent that ameliorates one or more side effect associated with administration of the kinase inhibitor, e.g., an agent described herein.
[0039] In one embodiment, the cell expressing the CAR molecule, e.g., a CAR molecule described herein, and the kinase inhibitor are administered in combination with an additional agent that treats the disease associated with CD19, e.g., an additional agent described herein.
[0040] In one embodiment, the cells expressing a CAR molecule, e.g., a CAR molecule described herein, are administered at a dose and/or dosing schedule described herein.
[0041] In one embodiment, the CAR molecule is introduced into T cells, e.g., using in vitro transcription, and the subject (e.g., human) receives an initial administration of cells comprising a CAR molecule, and one or more subsequent administrations of cells comprising a CAR molecule, wherein the one or more subsequent administrations are administered less than 15 days, e.g., 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, or 2 days after the previous administration. In one embodiment, more than one administration of cells comprising a CAR molecule are administered to the subject (e.g., human) per week, e.g., 2, 3, or 4 administrations of cells comprising a CAR molecule are administered per week. In one embodiment, the subject (e.g., human subject) receives more than one administration of cells comprising a CAR molecule per week (e.g., 2, 3 or 4 administrations per week) (also referred to herein as a cycle), followed by a week of no administration of cells comprising a CAR molecule, and then one or more additional administration of cells comprising a CAR molecule (e.g., more than one administration of the cells comprising a CAR molecule per week) is administered to the subject. In another embodiment, the subject (e.g., human subject) receives more than one cycle of cells comprising a CAR molecule, and the time between each cycle is less than 10, 9, 8, 7, 6, 5, 4, or 3 days. In one embodiment, the cells comprising a CAR molecule are administered every other day for 3 administrations per week. In one embodiment, the cells comprising a CAR molecule are administered for at least two, three, four, five, six, seven, eight or more weeks.
[0042] In one embodiment, the combination of the kinase inhibitor and the cells expressing a CAR molecule, e.g., a CAR molecule described herein, are administered as a first line treatment for the disease, e.g., the cancer, e.g., the cancer described herein. In another embodiment, the combination of the kinase inhibitor and the cells expressing a CAR molecule, e.g., a CAR molecule described herein, are administered as a second, third, fourth line treatment for the disease, e.g., the cancer, e.g., the cancer described herein.
[0043] In one embodiment, a cell (e.g., a population of cells) described herein is administered to the subject.
[0044] In one embodiment, the method includes administering a population of cells, a plurality of which comprise a CAR molecule described herein. In some embodiments, the population of CAR-expressing cells comprises a mixture of cells expressing different CARs. For example, in one embodiment, the population of CAR-expressing cells can include a first cell expressing a CAR having an anti-CD19 binding domain described herein, and a second cell expressing a CAR having a different anti-CD19 binding domain, e.g., an anti-CD19 binding domain described herein that differs from the anti-CD19 binding domain in the CAR expressed by the first cell. As another example, the population of CAR-expressing cells can include a first cell expressing a CAR that includes an anti-CD19 binding domain, e.g., as described herein, and a second cell expressing a CAR that includes an antigen binding domain to a target other than CD19 (e.g., CD123 or mesothelin). In one embodiment, the population of CAR-expressing cells includes, e.g., a first cell expressing a CAR that includes a primary intracellular signaling domain, and a second cell expressing a CAR that includes a secondary signaling domain.
[0045] In one embodiment, the method includes administering a population of cells wherein at least one cell in the population expresses a CAR having an anti-CD19 domain described herein, and an agent which enhances the activity of a CAR-expressing cell, e.g., a second cell expressing the agent which enhances the activity of a CAR-expressing cell. For example, in one embodiment, the agent can be an agent which inhibits an immune inhibitory molecule. Examples of immune inhibitory molecules include PD1, PD-L1, CTLA-4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 and TGFR beta. In one embodiment, the agent which inhibits an immune inhibitory molecule comprises a first polypeptide, e.g., an inhibitory molecule, associated with a second polypeptide that provides a positive signal to the cell, e.g., an intracellular signaling domain described herein. In one embodiment, the agent comprises a first polypeptide, e.g., of an inhibitory molecule such as PD1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 or TGFR beta, or a fragment of any of these (e.g., at least a portion of an extracellular domain of any of these), and a second polypeptide which is an intracellular signaling domain described herein (e.g., comprising a costimulatory domain (e.g., 41BB, CD27 or CD28, e.g., as described herein) and/or a primary signaling domain (e.g., a CD3 zeta signaling domain described herein). In one embodiment, the agent comprises a first polypeptide of PD1 or a fragment thereof (e.g., at least a portion of the extracellular domain of PD1), and a second polypeptide of an intracellular signaling domain described herein (e.g., a CD28 signaling domain described herein and/or a CD3 zeta signaling domain described herein).
[0046] In another aspect, the invention pertains to a cell expressing a CAR molecule described herein for use as a medicament in combination with a kinase inhibitor, e.g., a kinase inhibitor described herein (e.g., a BTK inhibitor such as ibrutinib). In another aspect, the invention pertains to a kinase inhibitor described herein (e.g., a BTK inhibitor such as ibrutinib) for use as a medicament in combination with a cell expressing a CAR molecule described herein.
[0047] In another aspect, the invention pertains to a cell expressing a CAR molecule described herein for use in combination with a kinase inhibitor, e.g., a kinase inhibitor described herein (e.g., a BTK inhibitor such as ibrutinib), in the treatment of a disease expressing the B-cell antigen (e.g., CD19). In another aspect, the invention pertains to a kinase inhibitor described herein (e.g., a BTK inhibitor such as ibrutinib), for use in combination with a cell expressing a CAR molecule described herein, in the treatment of a disease expressing the B-cell antigen (e.g., CD19). The disease may be, e.g., a cancer such as a hematologic cancer. The cancer may be, e.g., a lymphoma, CLL, MCL, ALL, DLBCL, multiple myeloma, or another cancer described herein.
[0048] In another aspect, the invention pertains to a cell expressing a CAR molecule described herein for use as a medicament in combination with a cytokine, e.g., IL-7, IL-15 and/or IL-21 as described herein. In another aspect, the invention pertains to a cytokine described herein for use as a medicament in combination with a cell expressing a CAR molecule described herein.
[0049] In another aspect, the invention pertains to a cell expressing a CAR molecule described herein for use in combination with a cytokine, e.g., IL-7, IL-15 and/or IL-21 as described herein, in the treatment of a disease expressing CD19. In another aspect, the invention pertains to a cytokine described herein for use in combination with a cell expressing a CAR molecule described herein, in the treatment of a disease expressing CD19.
[0050] In another aspect, the invention pertains to a method of treating a mammal having Hodgkin lymphoma, comprising administering to the mammal an effective amount of the cell (e.g., cells) expressing a CAR molecule, e.g., a CAR molecule described herein.
[0051] In one embodiment, the cell expressing a CAR molecule, e.g., a CAR molecule described herein, is administered in combination with an agent that increases the efficacy of a cell expressing a CAR molecule, e.g., an agent described herein.
[0052] In one embodiment, the cell expressing a CAR molecule, e.g., a CAR molecule described herein, is administered in combination with an agent that ameliorates one or more side effect associated with administration of a cell expressing a CAR molecule, e.g., an agent described herein.
[0053] In one embodiment, the cell expressing a CAR molecule, e.g., a CAR molecule described herein, is administered in combination with an agent that treats Hodgkin lymphoma, e.g., an agent described herein.
[0054] In one embodiment, the cell expressing a CAR molecule, e.g., a CAR molecule described herein, is administered in combination with a low, immune enhancing dose of an mTOR inhibitor, e.g., an mTOR inhibitor described herein. While not wishing to be bound by theory, it is believed that treatment with a low, immune enhancing, dose (e.g., a dose that is insufficient to completely suppress the immune system but sufficient to improve immune function) is accompanied by a decrease in PD-1 positive T cells or an increase in PD-1 negative cells. PD-1 positive T cells, but not PD-1 negative T cells, can be exhausted by engagement with cells which express a PD-1 ligand, e.g., PD-L1 or PD-L2.
[0055] In an embodiment this approach can be used to optimize the performance of a CAR cell described herein in the subject. While not wishing to be bound by theory, it is believed that, in an embodiment, the performance of endogenous, non-modified immune effector cells, e.g., T cells, is improved. While not wishing to be bound by theory, it is believed that, in an embodiment, the performance of a CD19 CAR expressing cell is improved. In other embodiments, cells, e.g., T cells, which have, or will be engineered to express a CAR, can be treated ex vivo by contact with an amount of an mTOR inhibitor that increases the number of PD1 negative immune effector cells, e.g., T cells or increases the ratio of PD1 negative immune effector cells, e.g., T cells/PD1 positive immune effector cells, e.g., T cells.
[0056] In an embodiment, administration of a low, immune enhancing, dose of an mTOR inhibitor, e.g., an allosteric inhibitor, e.g., RAD001, or a catalytic inhibitor, is initiated prior to administration of an CAR expressing cell described herein, e.g., T cells. In an embodiment, the mTOR inhibitor is RAD001 or rapamycin. In an embodiment, the CAR cells are administered after a sufficient time, or sufficient dosing, of an mTOR inhibitor, such that the level of PD1 negative immune effector cells, e.g., T cells, or the ratio of PD1 negative immune effector cells, e.g., T cells/PD1 positive immune effector cells, e.g., T cells, has been, at least transiently, increased.
[0057] In an embodiment, the cell, e.g., an immune effector cell (e.g., a T cell or NK cell), to be engineered to express a CAR, is harvested after a sufficient time, or after sufficient dosing of the low, immune enhancing, dose of an mTOR inhibitor, such that the level of PD1 negative immune effector cells, e.g., T cells, or the ratio of PD1 negative immune effector cells, e.g., T cells/PD1 positive immune effector cells, e.g., T cells, in the subject or harvested from the subject has been, at least transiently, increased.
[0058] In embodiments, any of the methods described herein further comprise performing lymphodepletion on a subject, e.g., prior to administering the one or more cells that express a CAR molecule described herein, e.g., a CAR molecule that binds CD19. The lymphodepletion can comprise, e.g., administering one or more of melphalan, cytoxan, cyclophosphamide, and fludarabine.
[0059] In some embodiments, the CAR-expressing cell that is administered comprises a regulatable CAR (RCAR), e.g., an RCAR as described herein. The RCAR may comprise, e.g., an intracellular signaling member comprising an intracellular signaling domain and a first switch domain, an antigen binding member comprising an antigen binding domain that binds CD19 and a second switch domain; and a transmembrane domain. The method may further comprise administering a dimerization molecule, e.g., in an amount sufficient to cause dimerization of the first switch and second switch domains.
[0060] In some embodiments, the CAR-expressing cell and the kinase inhibitor are administered simultaneously or substantially simultaneously, e.g., as a first line of therapy. In some embodiments, the method comprises administering a combination of the BTK inhibitor (e.g., ibrutinib) and the CAR-expressing cell (e.g., a CAR19-expressing cell) to the subject, as a first line therapy.
[0061] In other embodiments, the CAR-expressing cell and the kinase inhibitor are administered sequentially. For example, the kinase inhibitor is administered before the CAR-expressing cell, or the CAR-expressing cell is administered before the kinase inhibitor.
[0062] In some embodiments, the disease associated with expression of CD19 is a hematological cancer (e.g., a hematological cancer described herein such as CLL, MCL, or ALL) and the subject is, or is identified as, a partial responder, non-responder, or relapser to one or more therapies for the hematological cancer, e.g., to a BTK inhibitor such as ibrutinib. In some embodiments, the subject has, or is identified as having, a BTK mutation. The mutation may be, e.g., a point mutation, an insertion, or a deletion. The mutation may be, e.g., a mutation at the binding site for the BTK inhibitor, e.g., at or near the ATP-binding pocket. The mutation may confer a decreased response (e.g., resistance) to the BTK inhibitor.
[0063] In some embodiments of any of the methods disclosed herein, the method comprises administering the BTK inhibitor (e.g., ibrutinib) to the subject, reducing the amount (e.g., ceasing administration) of the BTK inhibitor, and subsequently administering the CAR-expressing cell (e.g., a CAR19-expressing cell) to the subject.
[0064] In some embodiments, the method comprises administering the BTK inhibitor (e.g., ibrutinib) to the subject and subsequently administering a combination of the BTK inhibitor and the CAR-expressing cell (e.g., a CAR19-expressing cell) to the subject.
[0065] In some embodiments, the method comprises administering the BTK inhibitor (e.g., ibrutinib) to the subject, reducing the amount (e.g., ceasing or discontinuing administration) of the BTK inhibitor, and subsequently administering a combination of the CAR-expressing cell (e.g., a CAR19-expressing cell) and a second BTK inhibitor (e.g., a BTK inhibitor other than the first BTK inhibitor, e.g., other than ibrutinib) to the subject. In some embodiments, the second BTK inhibitor is chosen from one or more of GDC-0834, RN-486, CGI-560, CGI-1764, HM-71224, CC-292, ONO-4059, CNX-774, or LFM-A13, or a combination thereof.
[0066] In some embodiments, the disease associated with expression of the B-cell antigen (e.g., CD19) is a hematological cancer (e.g., a hematological cancer described herein, e.g., CLL, MCL, or ALL), and the method delays or decreases resistance to the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib), the the CAR-expressing cell (e.g., a CAR19-expressing cell) to the subject, or both. In some embodiments, the disease associated with expression of CD19 is a hematological cancer (e.g., a hematological cancer described herein, e.g., CLL, MCL, or ALL), and wherein the method prolongs remission or delays relapse of the hematological cancer. For example, remission can be prolonged, relapse can be delayed, resistance can be delayed, or resistance can be decreased, compared to the expected course of disease when treated with a monotherapy of the kinase inhibitor or the CAR-expressing cell.
[0067] Exemplary treatment regimens that can be used in any of the aforesaid methods include one or more of the following:
[0068] In one embodiment, the kinase inhibitor and the CAR-expressing cell (e.g., the CAR19-expressing cell) are administered to the subject, e.g., mammal, as a first line of therapy.
[0069] In another embodiment, the CAR-expressing cell (e.g., the CAR19-expressing cell) is administered to the subject, e.g., mammal, after administration of the kinase inhibitor.
[0070] In other embodiments, the CAR-expressing cell (e.g., the CAR19-expressing cell) is administered after ceasing administration of the kinase inhibitor.
[0071] In other embodiments, administration of the kinase inhibitor is begun prior to administration of the CAR19-expressing cell, and the CAR19-expressing cell is administered in combination with continued administration of the kinase inhibitor.
[0072] In one embodiment, a subject is administered a kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib), e.g., as a first line therapy. After a predetermined time interval, (e.g., 1 or 2 months but also 2 weeks, 3 weeks, 1 month, 1.5 months, 2 months, 3 months, 4 months, 6 months, 9 months, 12 months, 15 months, or 18 months), a CAR-expressing cell (e.g., a CAR19-expressing cell) is administered to the subject alone, or in combination with the kinase inhibitor. In some embodiments, the subject's response to the treatment is assessed at predetermined time intervals, e.g., before or during treatment with the kinase inhibitor and/or CAR-expressing cell. If the assessment shows that the subject is a complete responder, the CAR-expressing cell (e.g., a CAR19-expressing cell) is not administered. If the assessment shows that the subject is a partial responder, or has stable disease in response, to the kinase inhibitor, the CAR-expressing cell (e.g., a CAR19-expressing cell) is administered in combination with the kinase inhibitor e.g., as described herein. If the assessment shows that the subject is a non-responder or relapser, the CAR-expressing cell (e.g., a CAR19-expressing cell) is administered in combination with the kinase inhibitor or a second kinase inhibitor, e.g., a second kinase inhibitor as described herein.
[0073] In other embodiments, the subject, e.g., mammal, is, or is identified as being, a complete or partial responder to the BTK inhibitor (e.g., ibrutinib), or a complete or partial responder to the CAR19-expressing cell.
[0074] In some embodiments, when a subject is (or is identified as being) a complete responder to the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib), the subject is not administered a CAR-expressing cell (e.g., a CAR19-expressing cell) during the period of complete response. In other embodiments, when a subject is (or is identified as being) a complete responder (e.g., a complete responder to ibrutinib) to the kinase inhibitor, the subject is administered a CAR-expressing cell (e.g., a CAR19-expressing cell) during the period of complete response. In an embodiment, after the CAR-expressing cell (e.g., a CAR19-expressing cell), the subject experiences a prolonged response or delayed relapse (e.g., compared to the expected course of disease when treated without the CAR therapy).
[0075] In some embodiments, when a subject is (or is identified as being) a partial responder to the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib), the subject is not administered a CAR-expressing cell (e.g., a CAR19-expressing cell) during the period of partial response. In other embodiments, when a subject is (or is identified as being) a partial responder to the kinase inhibitor, the subject is administered a CAR-expressing cell (e.g., a CAR19-expressing cell) (alone or in combination with the BTK inhibitor) during the period of partial response. In an embodiment, after the CAR therapy, the subject experiences a complete response and/or prolonged response or delayed relapse (e.g., compared to the expected course of disease when treated without CAR therapy).
[0076] In some embodiments, when a subject has (or is identified as having) stable disease after treatment with the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib), the subject is not administered a CAR therapy during the period of stable disease. In other embodiments, when a subject has (or is identified as having) stable disease after treatment with the kinase inhibitor, the subject is administered a CAR therapy during the period of stable disease. In an embodiment, after the CAR therapy, the subject experiences a partial response, a complete response and/or prolonged response or delayed relapse (e.g., compared to the expected course of disease when treated without CAR therapy).
[0077] In some embodiments, when a subject has (or is identified as having) progressive disease after treatment with the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib), the subject is not administered a CAR-expressing cell (e.g., a CAR19-expressing cell) during the period of progressive disease. In other embodiments, when a subject has (or is identified as having) progressive disease after treatment with the kinase inhibitor, the subject is administered a CAR-expressing cell (e.g., a CAR19-expressing cell) during the period of progressive disease. In an embodiment, after the CAR therapy, the subject experiences stable disease, a partial response, a complete response and/or prolonged response or delayed relapse (e.g., compared to the expected course of disease when treated without CAR therapy).
[0078] In other embodiments, the CAR-expressing cell is administered in combination a second kinase inhibitor, wherein the second kinase inhibitor is other than ibrutinib, when the mammal is, or is identified as being, a non-responder or relapser to ibrutinib. The second kinase inhibitor can be chosen from one or more of GDC-0834, RN-486, CGI-560, CGI-1764, HM-71224, CC-292, ONO-4059, CNX-774, or LFM-A13, or a combination thereof.
[0079] In other embodiments, the subject, e.g., the mammal, is (or is identified as being) a partial responder to the kinase inhibitor, and the subject is administered the CAR-expressing cell (e.g., the CAR19-expressing cell), alone or in combination with the BTK inhibitor, during the period of partial response.
[0080] In other embodiments, the subject, e.g., the mammal, is (or has identified as being) a non-responder having progressive or stable disease after treatment with ibrutinib, and the subject is administered the CAR-expressing cell (e.g., the CAR19-expressing cell), alone or in combination with a second BTK inhibitor, during the period of progressive or stable disease, wherein the second kinase inhibitor is other than ibrutinib.
[0081] In another aspect, provided herein is a method of treating a subject, e.g., a mammal, having a disease associated with expression of the B-cell antigen (e.g., CD19). The method comprises administering to the subject an effective amount of a kinase inhibitor as described herein (e.g., a BTK kinase inhibitor described herein, e.g., ibrutinib) and a CAR-expressing cell (e.g., a CAR19-expressing cell) in combination (e.g. simultaneously (or substantially simultaneously), or sequentially).
[0082] In some embodiments, the kinase inhibitor and the CAR-expressing cell (e.g., a CAR19 cell) are administered, in combination, e.g., as a first line of therapy,
[0083] In some embodiments, the kinase inhibitor is administered initially, e.g., a monotherapy or first line of therapy; after reducing the amount (e.g., ceasing or discontinuing administration) of the kinase inhibitor, administering the CAR-expressing cell (e.g., a CAR19-expressing cell) to the subject.
[0084] In other embodiments, the kinase inhibitor is administered initially, e.g., a monotherapy or first line of therapy; and subsequently administering a combination of the kinase inhibitor and the CAR-expressing cell (e.g., a CAR19-expressing cell) to the subject.
[0085] In other embodiments, the kinase inhibitor is administered initially, e.g., a monotherapy or first line of therapy; after reducing the amount (e.g., ceasing or discontinuing administration) of the kinase inhibitor, administering a combination of a second kinase inhibitor and the CAR-expressing cell (e.g., a CAR19-expressing cell) to the subject.
[0086] In some embodiments, the subject's response to the treatment is assessed at predetermined time intervals, e.g., before or during treatment with the kinase inhibitor and/or CAR-expressing cell. If the assessment shows that the subject is a complete responder, the CAR-expressing cell (e.g., a CAR19-expressing cell) is not administered. If the assessment shows that the subject is a partial responder, or has stable disease in response, to the kinase inhibitor, the CAR-expressing cell (e.g., a CAR19-expressing cell) is administered in combination with the kinase inhibitor e.g., as described herein. If the assessment shows that the subject is a non-responder or relapser, the CAR-expressing cell (e.g., a CAR19-expressing cell) is administered in combination with the kinase inhibitor or a second kinase inhibitor, e.g., a second kinase inhibitor as described herein.
[0087] In some embodiments, the disease associated with expression of a B-cell antigen (e.g., CD19) is a hematological cancer, leukemia, lymphoma, MCL, CLL, ALL, Hodgkin lymphoma, or multiple myeloma.
[0088] In some embodiments, the kinase inhibitor is a BTK inhibitor chosen from ibrutinib, GDC-0834, RN-486, CGI-560, CGI-1764, HM-71224, CC-292, ONO-4059, CNX-774, or LFM-A13; a CDK4 inhibitor chosen from palbociclib, aloisine A, flavopiridol, 2-(2-chlorophenyl)-5,7-dihydroxy-8-[(3S,4R)-3-hydroxy-1-methyl-4-piperidi- nyl]-4-chromenone; crizotinib (PF-02341066, P276-00, RAF265, indisulam, roscovitine, dinaciclib, BMS 387032, MLN8054, AG-024322, AT7519, AZD5438, BMS908662; or ribociclib; a mTOR inhibitor chosen from rapamycin, a rapamycin analog such as everolimus, temsirolimus, ridaforolimus, semapimod, AZD8055, PF04691502, SF1126, XL765, or OSI-027; or a MNK inhibitor is chosen from: CGP052088, CGP57380, cercosporamide, or ETC-1780445-2, or 4-amino-5-(4-fluoroanilino)-pyrazolo[3,4-d]pyrimidine.
[0089] In some aspects, the invention features a method of treating or providing an anti-tumor immunity in a subject, e.g., mammal, having Hodgkin lymphoma. The method comprises administering to the subject an effective amount of a cell that expresses a CAR molecule that binds CD19, alone or in combination with a second therapy.
[0090] In another aspect, the invention features a method of treating, or providing anti-tumor immunity to a subject, e.g., a mammal, having a multiple myeloma (e.g., a CD19-positive multiple myeloma, or a CD19-negative myeloma). In one embodiment, the multiple myeloma is CD19-negative, e.g., has a vast majority (99.95%) of the neoplastic plasma cells with a CD19-negative phenotype, e.g., as detected by both flow cytometry and RT-PCR. The method comprises administering to the subject an effective amount of a cell that expresses a CAR molecule that binds CD19, alone or in combination with a second therapy (e.g., a standard of care therapy for multiple myeloma). The method may further comprise administering a kinase inhibitor as described herein.
[0091] In embodiments of the methods related to Hodgkin lymphoma or multiple myeloma, the CAR molecule is a humanized CAR molecule, e.g., as described herein. In embodiments, the CAR molecule is a CAR molecule as described herein. For instance, in embodiments the CAR molecule comprises an anti-CD19 binding domain that comprises a one or more of (e.g., 2, 3, 4, 5, or all of) LC CDR1 of SEQ ID NO: 5, a LC CDR2 of SEQ ID NO: 26, and a LC CDR3 of SEQ ID NO: 27; a HC CDR1 of SEQ ID NO: 19, a LC CDR2 of any of SEQ ID NOS: 20-23, and a HC CDR3 of SEQ ID NO: 24.
[0092] In some embodiments of the methods related to Hodgkin lymphoma or multiple myeloma, the CAR molecule (e.g., CART19 or CTL019) is administered as a monotherapy. In some embodiments, the method further comprises administering a kinase inhibitor, e.g., a BTK inhibitor (such as ibrutinib), a CDK4 inhibitor, an mTOR inhibitor, or a MNK inhibitor.
[0093] In some embodiments of the methods related to multiple myeloma, the CAR molecule (e.g., CART19 or CTL019) is administered in combination a standard of care therapy for multiple myeloma, e.g., with myeloablative chemotherapy and/or autologous stem cell transplant rescue (e.g., after melphalan administration (e.g., high dose melphalan)).
[0094] In another aspect, the invention features a composition comprising a cell that expresses a CAR molecule that binds a B cell antigen (e.g., one or more of CD19, CD20. CD22 or ROR1), and one or more kinase inhibitors, wherein the kinase inhibitor is chosen from a Bruton's tyrosine kinase (BTK) inhibitor, a cyclin dependent kinase 4 (CDK4) inhibitor, an mTOR inhibitor, or a mitogen activated protein kinase interacting kinase (MNK) inhibitor. The CAR-expressing cell and the one or more kinase inhibitors can be present in a single dose form, or as two or more dose forms.
[0095] In embodiments, the compositions disclosed herein are for use as a medicament.
[0096] In embodiments, the compositions disclosed herein are use in the treatment of a disease associated with expression of a B-cell antigen (e.g., CD19).
Methods and Compositions for Producing CAR-Expressing Cells
[0097] The present disclosure also provides, in certain aspects, a method of making a population of immune effector cells (e.g., T cells or NK cells) that can be engineered to express a CAR (e.g., a CAR described herein), the method comprising: providing a population of immune effector cells; and contacting the immune effector cells with a kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib) under conditions sufficient to inhibit a target of the kinase inhibitor (e.g., BTK and/or ITK). The method can further comprise contacting, e.g., transducing, the immune effector cells with a nucleic acid encoding a CAR molecule.
[0098] In some aspects, the disclosure provides a method of making a CAR-expressing cell (e.g., a CAR-expressing immune effector cell or population of cells), comprising: contacting the cell or population of cells with a kinase inhibitor, e.g., a BTK inhibitor such as ibrutinib; and introducing (e.g., transducing) a nucleic acid encoding a CAR molecule into the cell or population of cells under conditions such that the CAR molecule is expressed.
[0099] In certain embodiments of the methods of producing CAR-expressing cells, the CAR molecule encoded by the nucleic acid is a CAR molecule that binds CD19. In embodiments, the method further comprises culturing the cell or cells under conditions that allow the cell or at least a sub-population of the cells to express the CAR molecule. In embodiments, the cell is a T cell or NK cell, or the population of cells includes T cells, NK cells, or both. In embodiments, the method comprises contacting the cell or cells with the kinase inhibitor (e.g., for 10-20, 20-30, 30-40, 40-60, or 60-120 minutes) and subsequently removing most or all of the kinase inhibitor from the cell or cells. In embodiments, the kinase inhibitor is added after the cell or cells are harvested or before the cell or cells are stimulated. In embodiments, the kinase inhibitor is a BTK inhibitor, a CDK4 inhibitor, an mTOR inhibitor, or a MNK inhibitor. In embodiments, the kinase inhibitor is ibrutinib. In embodiments, the population of cells also comprises cancer cells, e.g., leukemia or lymphoma cells. The cancer cells may be, e.g., CLL, MCL, or ALL cells. In embodiments, the kinase inhibitor inhibits a target (e.g., BTK) in the cancer cells, e.g., reduces its activity by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, or 99%. In embodiments, the kinase inhibitor inhibits a target (e.g., ITK) in the immune effector cells, e.g., reduces its activity by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, or 99%.
[0100] In some aspects, the present disclosure also provides a reaction mixture comprising a kinase inhibitor (e.g., a BTK inhibitor) and a CAR molecule or a nucleic acid encoding a CAR molecule. In some embodiments, the reaction mixture further comprises a population of immune effector cells.
[0101] In some embodiments, one or more of the immune effector cells expresses the CAR molecule or comprises the nucleic acid encoding the CAR molecule. In some embodiments, the kinase inhibitor is chosen from a BTK inhibitor, a CDK4 inhibitor, an mTOR inhibitor, or a MNK inhibitor. In some embodiments, the BTK inhibitor is chosen from: ibrutinib, GDC-0834, RN-486, CGI-560, CGI-1764, HM-71224, CC-292, ONO-4059, CNX-774, or LFM-A13. In embodiments, the reaction mixture comprises cancer cells, e.g., haematological cancer cells. The cancer cells may be, e.g., cells that were harvested from the subject when the immune effector cells were harvested from the subject.
[0102] In certain aspects, the present disclosure also provides a reaction mixture comprising a population of immune effector cells, and a CAR molecule or a nucleic acid encoding a CAR molecule, wherein the immune effector cells comprise covalently inactivated ITK. In embodiments, at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, or 99% of ITK is covalently inactivated. In some embodiments, the reaction mixture further comprises cancer cells. In embodiments, the cancer cells comprise covalently inactivated BTK. In embodiments, at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, or 99% of BTK is covalently inactivated. In embodiments, the BTK or ITK forms a covalent bond at or near its ATP binding domain to a small molecule such as ibrutinib. In embodiments, the BTK forms a covalent bond at or near its cysteine-481 to a small molecule such as ibrutinib.
[0103] In embodiments, a reaction mixture as described herein further comprises a buffer or other reagent, e.g., a PBS containing solution. In embodiments, the reaction mixture further comprises an agent that activates and/or expands to cells of the population, e.g., an agent that stimulates a CD3/TCR complex associated signal and/or a ligand that stimulates a costimulatory molecule on the surface of the cells. In embodiments, the agent is a bead conjugated with anti-CD3 antibody, or a fragment thereof, and/or anti-CD28 antibody, or a fragment thereof. In embodiments, the reaction mixture further comprises one or more factors for proliferation and/or viability, including serum (e.g., fetal bovine or human serum), interleukin-2 (IL-2), insulin, IFN-.gamma., IL-4, IL-7, GM-CSF, IL-10, IL-12, IL-15, TGF.beta., and TNF-.alpha. or any other additives for the growth of cells. In embodiments, the reaction mixture further comprises IL-15 and/or IL-7. In embodiments, a plurality of the cells of the population in the reaction mixture comprise a nucleic acid molecule, e.g., a nucleic acid molecule described herein, that comprises a CAR encoding sequence, e.g., a CD19 CAR encoding sequence, e.g., as described herein. In embodiments, a plurality of the cells of the population in the reaction mixture comprise a vector comprising a nucleic acid sequence encoding a CAR, e.g., a CAR described herein, e.g., a CD19 CAR described herein. In embodiments, the vector is a vector described herein, e.g., a vector selected from the group consisting of a DNA, a RNA, a plasmid, a lentivirus vector, adenoviral vector, or a retrovirus vector. In embodiments, the reaction mixture further comprises a cryoprotectant or stabilizer such as, e.g., a saccharide, an oligosaccharide, a polysaccharide and a polyol (e.g., trehalose, mannitol, sorbitol, lactose, sucrose, glucose and dextran), salts and crown ethers. In one embodiment, the cryoprotectant is dextran.
[0104] In some embodiments, the method of making disclosed herein further comprises contacting the population of immune effector cells with a nucleic acid encoding a telomerase subunit, e.g., hTERT. The the nucleic acid encoding the telomerase subunit can be DNA.
[0105] In some embodiments, the method of making disclosed herein further comprises culturing the population of immune effector cells in serum comprising 2% hAB serum.
[0106] Headings, sub-headings or numbered or lettered elements, e.g., (a), (b), (i) etc, are presented merely for ease of reading. The use of headings or numbered or lettered elements in this document does not require the steps or elements be performed in alphabetical order or that the steps or elements are necessarily discrete from one another.
[0107] All publications, patent applications, patents, and other references mentioned herein are incorporated by reference in their entirety.
[0108] Other features, objects, and advantages of the invention will be apparent from the description and drawings, and from the claims.
BRIEF DESCRIPTION OF THE DRAWINGS
[0109] FIG. 1A, 1B and 1C are graphic representations of cytotoxicity as assayed in ND317 (normal donor) T cell transduced with mouse anti-CD19 CAR or the humanized anti-CD19 CARs of the invention and cultured with either control K562 cells that do not express CD19 (K562cc) as shown in FIG. 1A, K562 cells transformed with CD19 (K562.CD19) as shown in FIG. 1B or malignant B cells isolated from a CLL patient (Pt 14 B cell isolate) as shown in FIG. 1C.
[0110] FIGS. 2A and 2B are graphs showing the proliferative response of humanized and mouse anti-CD19 CAR-expressing cells to CD19+ cells, where higher number of viable CAR+ T cells correlates with populations showing maximal CD4+ and CD8+ T cell proliferation to primary CLL cells.
[0111] FIG. 3 is a graphic representation of the deconvoluted HPLC mass spectra for scFvs of the invention, where the top row depicts untreated scFv and the bottom row depicts the cognate deglycosylated scFv.
[0112] FIG. 4 is a graphic representation of the conformation stability as measured by Differential Scanning Fluorimetry. The Tm of mouse scFv was 57.degree. C. (thick line). All humanized scFv variants show higher Tm at around 70.degree. C. as compared to the parental mouse scFv. The residues introduced by humanization have improved the Tm by more than 10.degree. C.
[0113] FIG. 5 is a graphic representation of CD19 CAR transduced T cell proliferation, wherein the CART19 cells are directed either towards (a) a chronic myelogenous leukemia ("CML") cell line that is negative for the expression of CD19, and hence used as a negative control; (b) recombinant K562 cells positive for expression of CD19, and hence used as a positive control; or (c) to Pt14 B cells isolated from a CLL patient and which expresses CD19 on the cell surface.
[0114] FIGS. 6A and 6B are schematics of representative CARs.
[0115] FIG. 7 depicts HALLX5447 primary ALL disease progression in NSG mice after treatment with CD19 transduced CAR T cells. The growth of primary human ALL cells in NSG mice after treatment with CAR T cells specific for CD19 demonstrated control of disease progression. Mean percentage of CD19.sup.+ human ALL cells was an indicator of disease burden in the peripheral blood in NSG mice to day 65 post tumor implant. Black circles: mice treated with 100 ul of PBS via the tail vein; red squares: mice treated with mock transduced T cells; blue triangles: mice treated with murine CD19 CAR transduced T cells; and inverted purple triangles: mice treated with humanized CD19 CAR transduced T cells. Significance calculated by ANOVA; * denotes P<0.01.
[0116] FIG. 8 depicts CD19 expression in a patient's tumor cells. CD138.sup.+ CD45.sup.dim tumor cells were stained for CD19 (x-axis) and CD38 (y-axis). Approximately 1-2% of the tumor cells expressed the CD19 antigen.
[0117] FIGS. 9A and 9B are two graphs showing the cell proliferation and cell size of CART19 cells when treated with increasing concentrations of ibrutinib (10 nM, 100 nM, and 1000 nM).
[0118] FIGS. 10A-1, 10A-2, 10A-3, 10A-4, and 10B shows the proliferation of CART19 cells stimulated with MCL cell lines, while in the presence or absence of ibrutinib. FIGS. 10A-1, 10A-2, 10A-3, and 10A-4 is a series of histograms showing the proliferation of CART19 cells stimulated with tumor cell lines MOLM14, JEKO-1, and RL, in the presence or absence of increasing concentrations of ibrutinib (10 nM, 100 nM, and 1000 nM). Cells were stained by CFSE and analyzed by flow cytometry to determine the percentage of proliferating cells, designated by the bar in each histogram. FIG. 10B is a quantification of representative histograms in FIG. 10A-, 10A-2, 10A-3, and 10A-4.
[0119] FIG. 11A-1, 11A-2, 11A-3, 11A-4, 11A-5, 11A-6, and 11B shows CD107a degranulation of CART19 cells stimulated with MCL cell lines in the presence or absence of ibrutinib. FIGS. 11A-1, 11A-2, 11A-3, 11A-4, 11A-5, and 11A-6 is a series of flow cytometry profiles showing CD107a degranulation of CART19 cells stimulated with tumor cell lines (MOLM14, JEKO-1, and RL) in the presence or absence of increasing concentrations of ibrutinib (10 nM, 100 nM, and 1000 nM). CD107a expression is measured in the y-axis. FIG. 11B is the quantification of the results from FIGS. 11A-1, 11A-2, 11A-3, 11A-4, 11A-5, and 11A-6.
[0120] FIGS. 12-1, 12-1, 12-3, 12-4, 12-5, and 12-6 is a series of flow cytometry profiles showing intra-cytoplasmatic IL-2 production by CART19 cells stimulated with tumor cell lines (MOLM14, JEKO-1, and RL) in the presence or absence of increasing concentrations of ibrutinib (10 nM, 100 nM, and 1000 nM). The y-axis represents IL-2 expression.
[0121] FIGS. 13-1, 13-2, 13-3, 13-4, 13-5, and 13-6 is a series of flow cytometry profiles showing intra-cytoplasmatic TNF-.alpha. production by CART19 cells stimulated with tumor cell lines (MOLM14, JEKO-1, and RL) in the presence or absence of increasing concentrations of ibrutinib (10 nM, 100 nM, and 1000 nM). The y-axis represents TNF-.alpha. expression.
[0122] FIGS. 14-1, 14-2, 14-3, 14-4, 14-5, and 14-6 is a series of flow cytometry profiles showing intra-cytoplasmatic IFN-g production by CART19 cells stimulated with tumor cell lines (MOLM14, JEKO-1, and RL) in the presence or absence of increasing concentrations of ibrutinib (10 nM, 100 nM, and 1000 nM). The y-axis represents IFN-g expression.
[0123] FIG. 15-1, 15-2, 15-3, 15-4, 15-5, 15-6, 15-7, 15-8, 15-9, and 15-10 are a series of graphs showing cytokine secretion from CART19 cells stimulated with tumor cell lines (MOLM14, JEKO-1, and RL) in the presence or absence of increasing concentrations of ibrutinib (10 nM, 100 nM, and 1000 nM).
[0124] FIGS. 16A, 16B, 16C, 16D, 16E, and 16 F are graphs showing CART19 killing of tumor cells, MOLM14 (FIGS. 16A and 16D), JEKO (FIGS. 16B and 16E), and RL (FIGS. 16C and 16F), alone or in the presence of increasing concentrations of ibrutinib. Untransduced (UTD) or CART19 cells were incubated with tumor cells at varying ratios and the total flux of cells (FIGS. 16A, 16B, and 16C) and percentage of dead cells was assessed (16D, 16E, and 16F).
[0125] FIGS. 17A, 17B, and 17C are graphic representations of CART19 killing of tumor cells after 24 hours as measured by flow cytometry to count the total number of cells. Tumor cell lines MOLM14 (FIG. 17A), JEKO (FIG. 17B), and RL (FIG. 17C) were incubated with untransduced (UTD) or CART19 cells alone (ALONE), or in combination with varying concentrations of ibrutinib.
[0126] FIGS. 18A, 18B, 18C, and 18D are graphic representations of CART19 dose finding in the RL MCL mouse model. Tumor burden was monitored by bioluminescence imaging (BLI) over time (FIGS. 18A and 18B). Overall survival was monitored over time (FIG. 18C).
[0127] FIGS. 19A and 19B are graphic representations of CART19 dose finding in the JEKO-1 MCL mouse model. Tumor size is monitored by bioluminescence imaging (BLI) over time (FIG. 19A) and overall survival was also monitored over time (FIG. 19B).
[0128] FIG. 20 is a schematic showing the protocol for administering and assessing CART19 and ibrutinib combination therapy in in vivo mouse models.
[0129] FIGS. 21A, 21B, 21C, and 21D is a graphic representation showing the transduction efficiency of PBMCs for generating CART19 T cells when cells are treated with ibrutinib prior to transduction. Untreated PBMCs were analyzed for CAR19 expression before transduction (FIG. 21A) and after transduction (FIG. 21C). Ibrutinib-treated PBMCs were analyzed for CAR19 expression before transduction (FIG. 21B) and after transduction (FIG. 21D).
[0130] FIGS. 22A, 22B, 22C, 22D, and 22E is a graphic representation of the effect of ibrutinib treatment on CD3/CD28-stimulated T cell proliferation. Increasing concentrations of ibrutinib was assessed: untreated (FIG. 22A); 0.1 .mu.M ibrutinib (FIG. 22B); 0.5 .mu.M ibrutinib (FIG. 22C), 1 .mu.M ibrutinib (FIG. 22D), and 5 .mu.M ibrutinib (FIG. 22E).
[0131] FIG. 23 is a graphic representation demonstrating that ibrutinib does not affect CART19 cytotoxicity.
[0132] FIG. 24 is a series of graphic representations that demonstrate that ibrutinib treatment does not promote skewing of T.sub.H1/T.sub.H2 cytokines in CART19 cells.
[0133] FIGS. 25A and 25B are graphic representations showing that continuous administration of ibrutinib does not affect CART19 function in clearing tumor cells in vivo. FIG. 25A shows the Nalm/6 cells detected in peripheral blood during each treatment regimen. FIG. 25B shows a Kaplan-Meier survival curve comparing the survival of mice receiving CART19 with or without ibrutinib dosing.
[0134] FIGS. 26A, 26B, 26C, 26D, 26E, and 26F are graphic representations that showing the efficiency of CAR19 transduction in CLL patient cells at the indicated timepoints during ibrutinib treatment. Cells were not transduced (FIGS. 26A, 26B, and 26C), or were transduced with CAR19 (FIGS. 26D, 26E, and 26F). Cells stained with GAM express CAR19 and are present in the boxes in each profile.
[0135] FIGS. 27-1, 27-2, and 27-3 are a series of graphic representations depicting the proliferation rate of untransduced cells compared to cells that were transduced with CAR19 at the indicated timepoints during ibrutinib treatment from a panel of patients.
[0136] FIGS. 28A, 28B, and 28C are graphic representations demonstrating that ibrutinib treatment in CLL patients induces lymphocytosis. Cells from the patient were isolated at the indicated timepoints: baseline (FIG. 28A); cycle 2, day 1 (FIG. 28B); and cycle 12, day 1 (FIG. 28C).
[0137] FIGS. 29A, 29B, and 29C are graphic representations demonstrating that ibrutinib treatment in three CLL patients reduces CD200 expression on tumor cells over time in CLL. Each profile contains an overlay of CD200 expression histograms from cells isolated at the indicated timepoints: baseline (screen); cycle 2, day 1; and cycle 12, day 1.
[0138] FIGS. 30A, 30B, and 30C are graphic representations demonstrating that ibrutinib treatment in CLL patients decreases the frequency of PD1+ T cells over time. Cells from the patient were isolated at the indicated timepoints: baseline (FIG. 30A); cycle 2, day 1 (FIG. 30B); and cycle 12, day 1 (FIG. 30C).
[0139] FIGS. 31A and 31B is a graphic representation demonstrating the sensitivity of MCL cell lines RL (FIG. 31A) and JEKO-1 (FIG. 31B) to ibrutinib treatment.
[0140] FIG. 32 is a graphic representation demonstrating the effect of ibrutinib treatment in an in vivo model of MCL.
[0141] FIG. 33 (left panel and right panel) are images of immunohistochemical analysis of a Hogdkin's lymphoma showing CD19 expressing cells present in the tumor. FIG. 33 (left panel) is at 1.times. magnification and FIG. 33 (right panel) is at 20.times. magnification.
[0142] FIG. 34 is a schematic diagram of the experimental set-up for a study to assess the therapeutic efficacy of CART19 treatment in patients with Hodgkin lymphoma.
[0143] FIGS. 35A, 35B, 35C, and 35D show flow cytometry analysis of PD1 and CAR19 expression on T cells. FIGS. 35A and 35B are representative flow cytometry profiles demonstrating the distribution of PD-1 and CAR19 expression on CD4+ T cells from subjects that are complete responders (CR) or non-responders (NR) to CART therapy. FIG. 35C is a graph showing the percent of PD1 cells in the CD4+ T cell population from groups of subjects with different responses to CART therapy. FIG. 35D is a graph showing the percent of PD1 cells in the CD8+ T cell population from groups of subjects with different responses to CART therapy.
[0144] FIGS. 36A and 36B show the distribution of PD1 expression in CD4 and CAR19-expressing cells (FIG. 36A) or CD8 and CAR19-expressing cells (FIG. 36B) from groups of subjects with different responses to CART therapy.
[0145] FIG. 37 shows flow cytometry analysis of PD1, CAR19, LAG3, and TIM3 expression on T cells from subjects that are complete responders (CR) or non-responders (NR) to CART therapy.
[0146] FIGS. 38A and 38B show the distribution of PD1 and LAG3 expression (FIG. 38A) or PD1 and TIM3 expression (FIG. 38B) from groups of subjects with different responses to CART therapy.
[0147] FIG. 39 shows the plasma cell IgA immunophenotyping from a myeloma patient who received CART19, demonstrating the response to CART19 therapy.
[0148] FIGS. 40A and 40B are graphs showing an increase in titers to influenza vaccine strains as compared to placebo. In FIG. 40A, the increase above baseline in influenza geometric mean titers to each of the 3 influenza vaccine strains (H1N1 A/California/07/2009, H3N2 A/Victoria/210/2009, B/Brisbane/60/2008) relative to the increase in the placebo cohort 4 weeks after vaccination is shown for each of the RAD001 dosing cohorts in the intention to treat population. The bold black line indicates the 1.2 fold increase in titers relative to placebo that is required to be met for 2 out of 3 influenza vaccine strains to meet the primary endpoint of the study. The star "*" indicates that the increase in GMT titer relative to placebo exceeds 1 with posterior probability of at least 80%. FIG. 40B is a graph of the same data as in FIG. 40A for the subset of subjects with baseline influenza titers <=1:40.
[0149] FIG. 41 shows a scatter plot of RAD001 concentration versus fold increase in geometric mean titer to each influenza vaccine strain 4 weeks after vaccination. RAD001 concentrations (1 hour post dose) were measured after subjects had been dosed for 4 weeks. All subjects who had pharmacokinetic measurements were included in the analysis set. The fold increase in geometric mean titers at 4 weeks post vaccination relative to baseline is shown on the y axis.
[0150] FIG. 42 is a graphic representation showing increase in titers to heterologous influenza strains as compared to placebo. The increase above baseline in influenza geometric mean titers to 2 heterologous influenza strains (A/H1N1 strain A/New Jersey/8/76 and A/H3N2 strain A/Victoria/361/11) not contained in the influenza vaccine relative to the increase in the placebo cohort 4 weeks after vaccination is shown for each of the RAD001 dosing cohorts in the intention to treat population. * indicates increase in titer relative to placebo exceeds 1 with a posterior probability of at least 80%.
[0151] FIGS. 43A and 43B are graphic representations of IgG and IgM levels before and after influenza vaccination. Levels of anti-A/H1N1/California/07/2009 influenza IgG and IgM were measured in serum obtained from subjects before and 4 weeks post influenza vaccination. No significant difference in the change from baseline to 4 weeks post vaccination in anti-H1N1 influenza IgG and IgM levels were detected between the RAD001 and placebo cohorts (all p values >0.05 by Kruskal-Wallis rank sum test).
[0152] FIGS. 44A, 44B, and 44C are graphic representations of the decrease in percent of PD-1-positive CD4 and CD8 and increase in PD-1-negative CD4 T cells after RAD001 treatment. The percent of PD-1-positive CD4, CD8 and PD-1-negative CD4 T cells was determined by FACS analysis of PBMC samples at baseline, after 6 weeks of study drug treatment (Week 6) and 6 weeks after study drug discontinuation and 4 weeks after influenza vaccination (Week 12). FIG. 44A shows there was a significant decrease (-37.1--28.5%) in PD-1-positive CD4 T cells at week 12 in cohorts receiving RAD001 at dose levels 0.5 mg/Day (n=25), 5 mg/Week (n=29) and 20 mg/Week (n=30) as compared to the placebo cohort (n=25) with p=0.002 (0.02), p=0.003 (q=0.03), and p=0.01 (q=0.05) respectively. FIG. 44B shows there was a significant decrease (-43.3--38.5%) in PD-1-positive CD8 T cells at week 12 in cohorts receiving RAD001 (n=109) at dose levels 0.5 mg/Day (n=25), 5 mg/Week (n=29) and 20 mg/Week (n=30) as compared to the placebo cohort (n=25) with p=0.01 (0.05), p=0.007 (q=0.04), and p=0.01 (q=0.05) respectively. FIG. 44C shows was a significant increase (3.0-4.9%) in PD-1-negative CD4 T cells at week 12 in cohorts receiving RAD001 (n=109) at dose levels 0.5 mg/Day (n=25), 5 mg/Week (n=29) and 20 mg/Week (n=30) as compared to the placebo cohort (n=25) with p=0.0007 (0.02), p=0.03 (q=0.07), and p=0.03 (q=0.08) respectively.
[0153] FIGS. 45A and 45B are graphic representations of the decrease in percent of PD-1-positive CD4 and CD8 and increase in PD-1-negative CD4 T cells after RAD001 treatment adjusted for differences in baseline PD-1 expression. The percent of PD-1-positive CD4, CD8 and PD-1-negative CD4 T cells was determined by FACS analysis of PBMC samples at baseline, after 6 weeks of study drug treatment (Week 6) and 6 weeks after study drug discontinuation and 4 weeks after influenza vaccination (Week 12). FIG. 45A shows a significant decrease of 30.2% in PD-1+CD4 T cells at week 6 in the pooled RAD cohort (n=84) compared to placebo cohort (n=25) with p=0.03 (q=0.13). The decrease in PD-1-positive CD4 T cells at week 12 in the pooled RAD as compared to the placebo cohort is 32.7% with p=0.05 (q=0.19). FIG. 45B shows a significant decrease of 37.4% in PD-1-positive CD8 T cells at week 6 in the pooled RAD001 cohort (n=84) compared to placebo cohort (n=25) with p=0.008 (q=0.07). The decrease in PD-1-positive CD8 T cells at week 12 in the pooled RAD001 as compared to the placebo cohort is 41.4% with p=0.066 (q=0.21). FIGS. 45A and 45B represent the data in FIGS. 44A, 44B, and 44C but with the different RAD001 dosage groups of FIGS. 44A, 44B, and 44C pooled into the single RAD001-treated group in FIGS. 45A and 45B.
[0154] FIG. 46 depicts increases in exercise and energy in elderly subjects in response to RAD001.
[0155] FIGS. 47A and 47B depict the predicted effect of RAD001 on P70 S6K activity in cells. FIG. 47A depicts P70 S6 kinase inhibition with higher doses of weekly and daily RAD001; FIG. 47B depicts P70 S6 kinase inhibition with lower doses of weekly RAD001.
[0156] FIGS. 48A and 48B show IL-7 receptor (CD127) expression on cancer cell lines and CART cells. Expression of CD127 was measured by flow cytometry analysis in three cancer cell lines: RL (mantle cell lymphoma), JEKO (also known as Jeko-1, mantle cell lymphoma), and Nalm-6 (B-ALL) (FIG. 48A). CD127 expression was measured by flow cytometry analysis on CD3 positive (CART) cells that had been infused and circulating in NSG mice (FIG. 48B).
[0157] FIGS. 49A, 49B, and 49C show the anti-tumor response after CART19 treatment and subsequent IL-7 treatment. NSG mice engrafted with a luciferase-expressing mantle lymphoma cell line (RL-luc) at Day 0 were treated with varying dosages of CART19 cells at Day 6, and tumor burden was monitored. Mice were divided into 4 groups and received no CART19 cells, 0.5.times.10.sup.6 CART19 cells (CART19 0.5E6), 1.times.10.sup.6 CART19 cells (CART19 1E6), or 2.times.10.sup.6 CART19 cells (CART19 2E6). Tumor burden after CART treatment was measured by detection of bioluminescence (mean BLI) (FIG. 49A). Mice receiving 0.5.times.10.sup.6 CART19 cells (CART19 0.5E6) or 1.times.10.sup.6 CART19 cells (CART19 1E6) were randomized to receive recominbant human IL-7 (rhIL-7) or not. Tumor burden, represented here by mean bioluminescence (BLI), was monitored for the three mice (#3827, #3829, and #3815, receiving the indicated initial CART19 dose) from FIG. 49A that were treated with IL-7 starting at Day 85 (FIG. 49B). IL-7 was administered through IP injection 3 times weekly. Tumor burden, represented here by mean bioluminescence (BLI) before Day 85 (PRE) and after Day 115 (POST) was compared between mice that did not receive IL-7 (CTRL) and mice that received IL-7 treatment (IL-7) (FIG. 49C).
[0158] FIGS. 50A and 50B show the T cell dynamics after IL-7 treatment. The level of human T cells detected in the blood was monitored for each of the mice receiving IL-7 or control mice (FIG. 50A). The level of CART19 cells (CD3+ cells) detected in the blood was measured before (PRE) and 14 days after (Day 14) initiation of IL-7 treatment (FIG. 50B).
[0159] FIG. 51 depicts the structures of two exemplary RCAR configurations. The antigen binding members comprise an antigen binding domain, a transmembrane domain, and a switch domain. The intracellular binding members comprise a switch domain, a co-stimulatory signaling domain and a primary signaling domain. The two configurations demonstrate that the first and second switch domains described herein can be in different orientations with respect to the antigen binding member and the intracellular binding member. Other RCAR configurations are further described herein.
[0160] FIG. 52A is an image of a RL cell line.
[0161] FIG. 52B is a set of flow cytometry scatterplots showing the expression of CD19 and CD5 in RL primary and RL cell lines.
[0162] FIG. 52C is an image showing t(11;14) translocation by fluorescence in-situ hybridization (FISH).
[0163] FIG. 52D is a graph showing the IC50 (by percentage MTT conversion) of ibrutinib inhibition in different cell lines.
[0164] FIG. 52E is a set of images and graphs showing engraftment of RL cells in NOD-SCID-gamma chain knockout (NSG) mice and the resulting tumor burden.
[0165] FIG. 52F is a set of histological images showing localization of MCL cells to various organs in mice.
[0166] FIG. 52G is a set of histological images of mice that have been injected with MCL-RL cells.
[0167] FIG. 53A is a set of graphs showing the number of CD107a+ CART19 cells when exposed to various MCL cell lines.
[0168] FIG. 53B is a set of graphs showing the amount of IL-2 and TNF-alpha produced by CART19 cells when exposed to various MCL cell lines.
[0169] FIG. 53C is a graph showing the percent killing of various MCL cell lines by CART19 cells at various effector:target cell ratios.
[0170] FIG. 53D is a graph showing the amount of carboxyfluorescein succinimidyl ester (CFSE), a measure of proliferation, in CART19 cells exposed to various MCL cell lines.
[0171] FIG. 53E is a set of graphs showing the percentage of T cells before and after expansion.
[0172] FIG. 53F is a set of graphs showing the percentage of untranduced or CAR-19 transduced T cells that express or produce various biomolecules (e.g., cytokines).
[0173] FIG. 54A is a set of images showing the activation of interleukin-2-inducible T-cell kinase (ITK) when CART19 cells were stimulated specifically or non-specifically.
[0174] FIG. 54B is a set of graphs showing CD107a surface expression (a measure of degranulation), IL-2 production, and TNF-alpha production by CART19 cells with various concentrations with ibrutinib.
[0175] FIG. 54C is a set of histograms showing the amount of CFSE in CART19 cells with various concentrations of ibrutinib and exposed to various MCL cell lines.
[0176] FIGS. 54D-1 and 54D-2 is a set of graphs showing the expression or production of various cytokines and biomarkers as indicators of the Th1 or Th2 state of CART19 cells when combined with different concentrations of ibrutinib.
[0177] FIG. 54E is a set of graphs showing the percentage killing by CART19 cells of various MCL cell lines when combined with different concentrations of ibrutinib.
[0178] FIG. 54F is a bar graph showing the expression of various markers of intrinsic cytotoxic function of CART19 cells when combined with various concentrations of ibrutinib.
[0179] FIG. 55 is a schematic of an in vivo mouse model experimental setup to test the effect of CART19 and/or ibrutinib on MCL-RL-injected mice, with a readout being luminescence (a measure of the number of tumor cells).
[0180] FIG. 56 is a schematic of an in vivo mouse model experimental setup to test the effect of CART19 and/or ibrutinib on MCL-RL-injected mice, with a readout being luminescence (a measure of the number of tumor cells).
[0181] FIG. 57 is a set of graphs showing the luminescence (a measure of the measure of tumor cell number) in mice treated with ibrutinib at different concentrations and their overall survival after treatment.
[0182] FIG. 58 is a set of graphs showing the luminescence (a measure of tumor cell number) in mice treated with ibrutinib or CART19 cells as well as their overall survival after treatment.
[0183] FIG. 59 is a graph showing the luminescence (a measure of tumor cell number) in mice after treatment with ibrutinib, untransduced T cells, ibrutinib with untransduced T cells, CART19 cells, and CART19 cells with ibrutinib.
[0184] FIG. 60 is a graph showing the luminescence (a measure of tumor cell number) in mice after treatment with ibrutinib alone, CART19 cells alone, or the combination of ibrutinib with CART19 cells.
[0185] FIG. 61A is a set of graphs showing the level of Th1 cytokines produced in mice treated with ibrutinib and/or CART19 cells. FIG. 61B is a set of graphs showing the level of Th2 cytokines produced in mice treated with ibrutinib and/or CART19 cells.
[0186] FIG. 61C is a graph showing the percentage of cells expressing the proliferation marker Ki67 in mice treated with CART19 cells or CART19 cells plus ibrutinib.
[0187] FIG. 61D is a graph showing the percentage of cells expressing the anti-apoptotic marker BCL-2 in mice treated with CART19 cells or CART19 cells plus ibrutinib.
DETAILED DESCRIPTION
Definitions
[0188] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which the invention pertains.
[0189] The term "a" and "an" refers to one or to more than one (i.e., to at least one) of the grammatical object of the article. By way of example, "an element" means one element or more than one element.
[0190] The term "about" when referring to a measurable value such as an amount, a temporal duration, and the like, is meant to encompass variations of .+-.20% or in some instances.+-.10%, or in some instances.+-.5%, or in some instances.+-.1%, or in some instances.+-.0.1% from the specified value, as such variations are appropriate to perform the disclosed methods.
[0191] The term "Chimeric Antigen Receptor" or alternatively a "CAR" refers to a set of polypeptides, typically two in the simplest embodiments, which when in an immune effector cell, provides the cell with specificity for a target cell, typically a cancer cell, and with intracellular signal generation. In some embodiments, a CAR comprises at least an extracellular antigen binding domain, a transmembrane domain and a cytoplasmic signaling domain (also referred to herein as "an intracellular signaling domain") comprising a functional signaling domain derived from a stimulatory molecule and/or costimulatory molecule as defined below. In some aspects, the set of polypeptides are contiguous with each other, e.g., are in the same polypeptide chain (e.g., comprise a chimeric fusion protein). In some embodiments, the set of polypeptides are not contiguous with each other, e.g., are in different polypeptide chains. In some embodiments, the set of polypeptides include a dimerization switch that, upon the presence of a dimerization molecule, can couple the polypeptides to one another, e.g., can couple an antigen binding domain to an intracellular signaling domain. In one aspect, the stimulatory molecule is the zeta chain associated with the T cell receptor complex. In one aspect, the cytoplasmic signaling domain further comprises one or more functional signaling domains derived from at least one costimulatory molecule as defined below. In one aspect, the costimulatory molecule is chosen from the costimulatory molecules described herein, e.g., 4-1BB (i.e., CD137), CD27 and/or CD28. In one aspect, the CAR comprises a chimeric fusion protein comprising an extracellular antigen binding domain, a transmembrane domain and an intracellular signaling domain comprising a functional signaling domain derived from a stimulatory molecule. In one aspect, the CAR comprises a chimeric fusion protein comprising an extracellular antigen binding domain, a transmembrane domain and an intracellular signaling domain comprising a functional signaling domain derived from a costimulatory molecule and a functional signaling domain derived from a stimulatory molecule. In one aspect, the CAR comprises a chimeric fusion protein comprising an extracellular antigen binding domain, a transmembrane domain and an intracellular signaling domain comprising two functional signaling domains derived from one or more costimulatory molecule(s) and a functional signaling domain derived from a stimulatory molecule. In one aspect, the CAR comprises a chimeric fusion protein comprising an extracellular antigen binding domain, a transmembrane domain and an intracellular signaling domain comprising at least two functional signaling domains derived from one or more costimulatory molecule(s) and a functional signaling domain derived from a stimulatory molecule. In one aspect the CAR comprises an optional leader sequence at the amino-terminus (N-ter) of the CAR fusion protein. In one aspect, the CAR further comprises a leader sequence at the N-terminus of the extracellular antigen binding domain, wherein the leader sequence is optionally cleaved from the antigen binding domain (e.g., a scFv) during cellular processing and localization of the CAR to the cellular membrane.
[0192] The term "signaling domain" refers to the functional portion of a protein which acts by transmitting information within the cell to regulate cellular activity via defined signaling pathways by generating second messengers or functioning as effectors by responding to such messengers.
[0193] As used herein, the term "CD19" refers to the Cluster of Differentiation 19 protein, which is an antigenic determinant detectable on leukemia precursor cells. The human and murine amino acid and nucleic acid sequences can be found in a public database, such as GenBank, UniProt and Swiss-Prot. For example, the amino acid sequence of human CD19 can be found as UniProt/Swiss-Prot Accession No. P15391 and the nucleotide sequence encoding of the human CD19 can be found at Accession No. NM_001178098. As used herein, "CD19" includes proteins comprising mutations, e.g., point mutations, fragments, insertions, deletions and splice variants of full length wild-type CD19. CD19 is expressed on most B lineage cancers, including, e.g., acute lymphoblastic leukaemia, chronic lymphocyte leukaemia and non-Hodgkin lymphoma. Other cells with express CD19 are provided below in the definition of "disease associated with expression of CD19." It is also an early marker of B cell progenitors. See, e.g., Nicholson et al. Mol. Immun. 34 (16-17): 1157-1165 (1997). In one aspect the antigen-binding portion of the CART recognizes and binds an antigen within the extracellular domain of the CD19 protein. In one aspect, the CD19 protein is expressed on a cancer cell.
[0194] As used herein, the term "CD20" refers to an antigenic determinant known to be detectable on B cells. Human CD20 is also called membrane-spanning 4-domains, subfamily A, member 1 (MS4A1). The human and murine amino acid and nucleic acid sequences can be found in a public database, such as GenBank, UniProt and Swiss-Prot. For example, the amino acid sequence of human CD20 can be found at Accession Nos. NP_690605.1 and NP_068769.2, and the nucleotide sequence encoding transcript variants 1 and 3 of the human CD20 can be found at Accession No. NM_152866.2 and NM_021950.3, respectively. In one aspect the antigen-binding portion of the CAR recognizes and binds an antigen within the extracellular domain of the CD20 protein. In one aspect, the CD20 protein is expressed on a cancer cell.
[0195] As used herein, the term "CD22," refers to an antigenic determinant known to be detectable on leukemia precursor cells. The human and murine amino acid and nucleic acid sequences can be found in a public database, such as GenBank, UniProt and Swiss-Prot. For example, the amino acid sequences of isoforms 1-5 human CD22 can be found at Accession Nos. NP 001762.2, NP 001172028.1, NP 001172029.1, NP 001172030.1, and NP 001265346.1, respectively, and the nucleotide sequence encoding variants 1-5 of the human CD22 can be found at Accession No. NM 001771.3, NM 001185099.1, NM 001185100.1, NM 001185101.1, and NM 001278417.1, respectively. In one aspect the antigen-binding portion of the CAR recognizes and binds an antigen within the extracellular domain of the CD22 protein. In one aspect, the CD22 protein is expressed on a cancer cell.
[0196] As used herein, the term "ROR1" refers to an antigenic determinant known to be detectable on leukemia precursor cells. The human and murine amino acid and nucleic acid sequences can be found in a public database, such as GenBank, UniProt and Swiss-Prot. For example, the amino acid sequences of isoforms land 2 precursors of human ROR1 can be found at Accession Nos. NP_005003.2 and NP_001077061.1, respectively, and the mRNA sequences encoding them can be found at Accession Nos. NM_005012.3 and NM_001083592.1, respectively. In one aspect the antigen-binding portion of the CAR recognizes and binds an antigen within the extracellular domain of the ROR1 protein. In one aspect, the ROR1 protein is expressed on a cancer cell.
[0197] The term "antibody," as used herein, refers to a protein, or polypeptide sequence derived from an immunoglobulin molecule which specifically binds with an antigen. Antibodies can be polyclonal or monoclonal, multiple or single chain, or intact immunoglobulins, and may be derived from natural sources or from recombinant sources. Antibodies can be tetramers of immunoglobulin molecules.
[0198] The term "antibody fragment" refers to at least one portion of an antibody, that retains the ability to specifically interact with (e.g., by binding, steric hinderance, stabilizing/destabilizing, spatial distribution) an epitope of an antigen. Examples of antibody fragments include, but are not limited to, Fab, Fab', F(ab').sub.2, Fv fragments, scFv antibody fragments, disulfide-linked Fvs (sdFv), a Fd fragment consisting of the VH and CH1 domains, linear antibodies, single domain antibodies such as sdAb (either VL or VH), camelid VHH domains, multi-specific antibodies formed from antibody fragments such as a bivalent fragment comprising two Fab fragments linked by a disulfide brudge at the hinge region, and an isolated CDR or other epitope binding fragments of an antibody. An antigen binding fragment can also be incorporated into single domain antibodies, maxibodies, minibodies, nanobodies, intrabodies, diabodies, triabodies, tetrabodies, v-NAR and bis-scFv (see, e.g., Hollinger and Hudson, Nature Biotechnology 23:1126-1136, 2005). Antigen binding fragments can also be grafted into scaffolds based on polypeptides such as a fibronectin type III (Fn3)(see U.S. Pat. No. 6,703,199, which describes fibronectin polypeptide minibodies).
[0199] The term "scFv" refers to a fusion protein comprising at least one antibody fragment comprising a variable region of a light chain and at least one antibody fragment comprising a variable region of a heavy chain, wherein the light and heavy chain variable regions are contiguously linked, e.g., via a synthetic linker, e.g., a short flexible polypeptide linker, and capable of being expressed as a single chain polypeptide, and wherein the scFv retains the specificity of the intact antibody from which it is derived. Unless specified, as used herein an scFv may have the VL and VH variable regions in either order, e.g., with respect to the N-terminal and C-terminal ends of the polypeptide, the scFv may comprise VL-linker-VH or may comprise VH-linker-VL.
[0200] The portion of the CAR of the invention comprising an antibody or antibody fragment thereof may exist in a variety of forms where the antigen binding domain is expressed as part of a contiguous polypeptide chain including, for example, a single domain antibody fragment (sdAb), a single chain antibody (scFv), a humanized antibody or bispecific antibody (Harlow et al., 1999, In: Using Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press, NY; Harlow et al., 1989, In: Antibodies: A Laboratory Manual, Cold Spring Harbor, N.Y.; Houston et al., 1988, Proc. Natl. Acad. Sci. USA 85:5879-5883; Bird et al., 1988, Science 242:423-426). In one aspect, the antigen binding domain of a CAR composition of the invention comprises an antibody fragment. In a further aspect, the CAR comprises an antibody fragment that comprises a scFv. The precise amino acid sequence boundaries of a given CDR can be determined using any of a number of well-known schemes, including those described by Kabat et al. (1991), "Sequences of Proteins of Immunological Interest," 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. ("Kabat" numbering scheme), Al-Lazikani et al., (1997) JMB 273, 927-948 ("Chothia" numbering scheme), or a combination thereof.
[0201] As used herein, the term "binding domain" or "antibody molecule" refers to a protein, e.g., an immunoglobulin chain or fragment thereof, comprising at least one immunoglobulin variable domain sequence. The term "binding domain" or "antibody molecule" encompasses antibodies and antibody fragments. In an embodiment, an antibody molecule is a multispecific antibody molecule, e.g., it comprises a plurality of immunoglobulin variable domain sequences, wherein a first immunoglobulin variable domain sequence of the plurality has binding specificity for a first epitope and a second immunoglobulin variable domain sequence of the plurality has binding specificity for a second epitope. In an embodiment, a multispecific antibody molecule is a bispecific antibody molecule. A bispecific antibody has specificity for no more than two antigens. A bispecific antibody molecule is characterized by a first immunoglobulin variable domain sequence which has binding specificity for a first epitope and a second immunoglobulin variable domain sequence that has binding specificity for a second epitope.
[0202] The portion of the CAR of the invention comprising an antibody or antibody fragment thereof may exist in a variety of forms where the antigen binding domain is expressed as part of a contiguous polypeptide chain including, for example, a single domain antibody fragment (sdAb), a single chain antibody (scFv), a humanized antibody, or bispecific antibody (Harlow et al., 1999, In: Using Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press, NY; Harlow et al., 1989, In: Antibodies: A Laboratory Manual, Cold Spring Harbor, N.Y.; Houston et al., 1988, Proc. Natl. Acad. Sci. USA 85:5879-5883; Bird et al., 1988, Science 242:423-426). In one aspect, the antigen binding domain of a CAR composition of the invention comprises an antibody fragment. In a further aspect, the CAR comprises an antibody fragment that comprises a scFv.
[0203] The term "antibody heavy chain," refers to the larger of the two types of polypeptide chains present in antibody molecules in their naturally occurring conformations, and which normally determines the class to which the antibody belongs.
[0204] The term "antibody light chain," refers to the smaller of the two types of polypeptide chains present in antibody molecules in their naturally occurring conformations. Kappa (K) and lambda (.lamda.) light chains refer to the two major antibody light chain isotypes.
[0205] The term "recombinant antibody" refers to an antibody which is generated using recombinant DNA technology, such as, for example, an antibody expressed by a bacteriophage or yeast expression system. The term should also be construed to mean an antibody which has been generated by the synthesis of a DNA molecule encoding the antibody and which DNA molecule expresses an antibody protein, or an amino acid sequence specifying the antibody, wherein the DNA or amino acid sequence has been obtained using recombinant DNA or amino acid sequence technology which is available and well known in the art.
[0206] The term "antigen" or "Ag" refers to a molecule that provokes an immune response. This immune response may involve either antibody production, or the activation of specific immunologically-competent cells, or both. The skilled artisan will understand that any macromolecule, including virtually all proteins or peptides, can serve as an antigen. Furthermore, antigens can be derived from recombinant or genomic DNA. A skilled artisan will understand that any DNA, which comprises a nucleotide sequences or a partial nucleotide sequence encoding a protein that elicits an immune response therefore encodes an "antigen" as that term is used herein. Furthermore, one skilled in the art will understand that an antigen need not be encoded solely by a full length nucleotide sequence of a gene. It is readily apparent that the present invention includes, but is not limited to, the use of partial nucleotide sequences of more than one gene and that these nucleotide sequences are arranged in various combinations to encode polypeptides that elicit the desired immune response. Moreover, a skilled artisan will understand that an antigen need not be encoded by a "gene" at all. It is readily apparent that an antigen can be generated synthesized or can be derived from a biological sample, or might be macromolecule besides a polypeptide. Such a biological sample can include, but is not limited to a tissue sample, a tumor sample, a cell or a fluid with other biological components.
[0207] The term "anti-cancer effect" refers to a biological effect which can be manifested by various means, including but not limited to, e.g., a decrease in tumor volume, a decrease in the number of cancer cells, a decrease in the number of metastases, an increase in life expectancy, decrease in cancer cell proliferation, decrease in cancer cell survival, or amelioration of various physiological symptoms associated with the cancerous condition. An "anti-cancer effect" can also be manifested by the ability of the peptides, polynucleotides, cells and antibodies in prevention of the occurrence of cancer in the first place. The term "anti-tumor effect" refers to a biological effect which can be manifested by various means, including but not limited to, e.g., a decrease in tumor volume, a decrease in the number of tumor cells, a decrease in tumor cell proliferation, or a decrease in tumor cell survival.
[0208] The term "autologous" refers to any material derived from the same individual to whom it is later to be re-introduced into the individual.
[0209] The term "allogeneic" refers to any material derived from a different animal of the same species as the individual to whom the material is introduced. Two or more individuals are said to be allogeneic to one another when the genes at one or more loci are not identical. In some aspects, allogeneic material from individuals of the same species may be sufficiently unlike genetically to interact antigenically
[0210] The term "xenogeneic" refers to a graft derived from an animal of a different species.
[0211] The term "cancer" refers to a disease characterized by the uncontrolled growth of aberrant cells. Cancer cells can spread locally or through the bloodstream and lymphatic system to other parts of the body. Examples of various cancers are described herein and include but are not limited to, breast cancer, prostate cancer, ovarian cancer, cervical cancer, skin cancer, pancreatic cancer, colorectal cancer, renal cancer, liver cancer, brain cancer, lymphoma, leukemia, lung cancer and the like. The terms "tumor" and "cancer" are used interchangeably herein, e.g., both terms encompass solid and liquid, e.g., diffuse or circulating, tumors. As used herein, the term "cancer" or "tumor" includes premalignant, as well as malignant cancers and tumors.
[0212] The phrase "disease associated with expression of CD19" includes, but is not limited to, a disease associated with expression of CD19 or condition associated with cells which express, or at any time expressed, CD19 including, e.g., proliferative diseases such as a cancer or malignancy or a precancerous condition such as a myelodysplasia, a myelodysplastic syndrome or a preleukemia; or a noncancer related indication associated with cells which express CD19. For the avoidance of doubt, a disease associated with expression of CD19 may include a condition associated with cells which do not presently express CD19, e.g., because CD19 expression has been downregulated, e.g., due to treatment with a molecule targeting CD19, e.g., a CD19 CAR, but which at one time expressed CD19. In one aspect, a cancer associated with expression of CD19 is a hematological cancer. In one aspect, the hematolical cancer is a leukemia or a lymphoma. In one aspect, a cancer associated with expression of CD19 includes cancers and malignancies including, but not limited to, e.g., one or more acute leukemias including but not limited to, e.g., B-cell acute Lymphoid Leukemia (BALL), T-cell acute Lymphoid Leukemia (TALL), acute lymphoid leukemia (ALL); one or more chronic leukemias including but not limited to, e.g., chronic myelogenous leukemia (CML), Chronic Lymphoid Leukemia (CLL). Additional cancers or hematologic conditions associated with expression of CD19 comprise, but are not limited to, e.g., B cell prolymphocytic leukemia, blastic plasmacytoid dendritic cell neoplasm, Burkitt's lymphoma, diffuse large B cell lymphoma, Follicular lymphoma, Hairy cell leukemia, small cell- or a large cell-follicular lymphoma, malignant lymphoproliferative conditions, MALT lymphoma, mantle cell lymphoma (MCL), Marginal zone lymphoma, multiple myeloma, myelodysplasia and myelodysplastic syndrome, non-Hodgkin lymphoma, Hodgkin lymphoma, plasmablastic lymphoma, plasmacytoid dendritic cell neoplasm, Waldenstrom macroglobulinemia, and "preleukemia" which are a diverse collection of hematological conditions united by ineffective production (or dysplasia) of myeloid blood cells, and the like. Further diseases associated with expression of CD19 expression include, but not limited to, e.g., atypical and/or non-classical cancers, malignancies, precancerous conditions or proliferative diseases associated with expression of CD19. Non-cancer related indications associated with expression of CD19 include, but are not limited to, e.g., autoimmune disease, (e.g., lupus), inflammatory disorders (allergy and asthma) and transplantation. In some embodiments, the tumor antigen-expressing cells express, or at any time expressed, mRNA encoding the tumor antigen. In an embodiment, the tumor antigen-expressing cells produce the tumor antigen protein (e.g., wild-type or mutant), and the tumor antigen protein may be present at normal levels or reduced levels. In an embodiment, the tumor antigen-expressing cells produced detectable levels of a tumor antigen protein at one point, and subsequently produced substantially no detectable tumor antigen protein.
[0213] The phrase "disease associated with expression of a B-cell antigen" includes, but is not limited to, a disease associated with expression of one or more of CD19, CD20, CD22 or ROR1, or a condition associated with cells which express, or at any time expressed, one or more of CD19, CD20, CD22 or ROR1, including, e.g., proliferative diseases such as a cancer or malignancy or a precancerous condition such as a myelodysplasia, a myelodysplastic syndrome or a preleukemia; or a noncancer related indication associated with cells which express one or more of CD19, CD20, CD22 or ROR1. For the avoidance of doubt, a disease associated with expression of the B-cell antigen may include a condition associated with cells which do not presently express the B-cell antigen, e.g., because the antigen expression has been downregulated, e.g., due to treatment with a molecule targeting the B-cell antigen, e.g., a B-cell targeting CAR, but which at one time expressed the antigen. The phrase "disease associated with expression of a B-cell antigen" includes a disease associated with expression of CD19, as described herein.
[0214] The term "conservative sequence modifications" refers to amino acid modifications that do not significantly affect or alter the binding characteristics of the antibody or antibody fragment containing the amino acid sequence. Such conservative modifications include amino acid substitutions, additions and deletions. Modifications can be introduced into an antibody or antibody fragment of the invention by standard techniques known in the art, such as site-directed mutagenesis and PCR-mediated mutagenesis. Conservative amino acid substitutions are ones in which the amino acid residue is replaced with an amino acid residue having a similar side chain. Families of amino acid residues having similar side chains have been defined in the art. These families include amino acids with basic side chains (e.g., lysine, arginine, histidine), acidic side chains (e.g., aspartic acid, glutamic acid), uncharged polar side chains (e.g., glycine, asparagine, glutamine, serine, threonine, tyrosine, cysteine, tryptophan), nonpolar side chains (e.g., alanine, valine, leucine, isoleucine, proline, phenylalanine, methionine), beta-branched side chains (e.g., threonine, valine, isoleucine) and aromatic side chains (e.g., tyrosine, phenylalanine, tryptophan, histidine). Thus, one or more amino acid residues within a CAR of the invention can be replaced with other amino acid residues from the same side chain family and the altered CAR can be tested using the functional assays described herein.
[0215] The term "stimulation," refers to a primary response induced by binding of a stimulatory molecule (e.g., a TCR/CD3 complex or CAR) with its cognate ligand (or tumor antigen in the case of a CAR) thereby mediating a signal transduction event, such as, but not limited to, signal transduction via the TCR/CD3 complex or signal transduction via the appropriate NK receptor or signaling domains of the CAR. Stimulation can mediate altered expression of certain molecules.
[0216] The term "stimulatory molecule," refers to a molecule expressed by an immune cell (e.g., T cell, NK cell, B cell) that provides the cytoplasmic signaling sequence(s) that regulate activation of the immune cell in a stimulatory way for at least some aspect of the immune cell signaling pathway. In one aspect, the signal is a primary signal that is initiated by, for instance, binding of a TCR/CD3 complex with an MHC molecule loaded with peptide, and which leads to mediation of a T cell response, including, but not limited to, proliferation, activation, differentiation, and the like. A primary cytoplasmic signaling sequence (also referred to as a "primary signaling domain") that acts in a stimulatory manner may contain a signaling motif which is known as immunoreceptor tyrosine-based activation motif or ITAM. Examples of an ITAM containing cytoplasmic signaling sequence that is of particular use in the invention includes, but is not limited to, those derived from CD3 zeta, common FcR gamma (FCER1G), Fc gamma RIIa, FcR beta (Fc Epsilon R1b), CD3 gamma, CD3 delta, CD3 epsilon, CD79a, CD79b, DAP10, and DAP12. In a specific CAR of the invention, the intracellular signaling domain in any one or more CARS of the invention comprises an intracellular signaling sequence, e.g., a primary signaling sequence of CD3-zeta. In a specific CAR of the invention, the primary signaling sequence of CD3-zeta is the sequence provided as SEQ ID NO: 17, or the equivalent residues from a non-human species, e.g., mouse, rodent, monkey, ape and the like. In a specific CAR of the invention, the primary signaling sequence of CD3-zeta is the sequence as provided in SEQ ID NO: 43, or the equivalent residues from a non-human species, e.g., mouse, rodent, monkey, ape and the like.
[0217] The term "antigen presenting cell" or "APC" refers to an immune system cell such as an accessory cell (e.g., a B-cell, a dendritic cell, and the like) that displays a foreign antigen complexed with major histocompatibility complexes (MHC's) on its surface. T-cells may recognize these complexes using their T-cell receptors (TCRs). APCs process antigens and present them to T-cells.
[0218] An "intracellular signaling domain," as the term is used herein, refers to an intracellular portion of a molecule. The intracellular signaling domain generates a signal that promotes an immune effector function of the CAR containing cell, e.g., a CART cell. Examples of immune effector function, e.g., in a CART cell, include cytolytic activity and helper activity, including the secretion of cytokines.
[0219] In an embodiment, the intracellular signaling domain can comprise a primary intracellular signaling domain. Exemplary primary intracellular signaling domains include those derived from the molecules responsible for primary stimulation, or antigen dependent simulation. In an embodiment, the intracellular signaling domain can comprise a costimulatory intracellular domain. Exemplary costimulatory intracellular signaling domains include those derived from molecules responsible for costimulatory signals, or antigen independent stimulation. For example, in the case of a CART, a primary intracellular signaling domain can comprise a cytoplasmic sequence of a T cell receptor, and a costimulatory intracellular signaling domain can comprise cytoplasmic sequence from co-receptor or costimulatory molecule.
[0220] A primary intracellular signaling domain can comprise a signaling motif which is known as an immunoreceptor tyrosine-based activation motif or ITAM. Examples of ITAM containing primary cytoplasmic signaling sequences include, but are not limited to, those derived from CD3 zeta, common FcR gamma (FCER1G), Fc gamma RIIa, FcR beta (Fc Epsilon R1b), CD3 gamma, CD3 delta, CD3 epsilon, CD79a, CD79b, DAP10, and DAP12.
[0221] The term "zeta" or alternatively "zeta chain", "CD3-zeta" or "TCR-zeta" is defined as the protein provided as GenBan Acc. No. BAG36664.1, or the equivalent residues from a non-human species, e.g., mouse, rodent, monkey, ape and the like, and a "zeta stimulatory domain" or alternatively a "CD3-zeta stimulatory domain" or a "TCR-zeta stimulatory domain" is defined as the amino acid residues from the cytoplasmic domain of the zeta chain, or functional derivatives thereof, that are sufficient to functionally transmit an initial signal necessary for T cell activation. In one aspect the cytoplasmic domain of zeta comprises residues 52 through 164 of GenBank Acc. No. BAG36664.1 or the equivalent residues from a non-human species, e.g., mouse, rodent, monkey, ape and the like, that are functional orthologs thereof. In one aspect, the "zeta stimulatory domain" or a "CD3-zeta stimulatory domain" is the sequence provided as SEQ ID NO: 17. In one aspect, the "zeta stimulatory domain" or a "CD3-zeta stimulatory domain" is the sequence provided as SEQ ID NO:43.
[0222] The term "costimulatory molecule" refers to the cognate binding partner on a T cell that specifically binds with a costimulatory ligand, thereby mediating a costimulatory response by the T cell, such as, but not limited to, proliferation. Costimulatory molecules are cell surface molecules other than antigen receptors or their ligands that are contribute to an efficient immune response. Costimulatory molecules include, but are not limited to an MHC class I molecule, BTLA and a Toll ligand receptor, as well as OX40, CD27, CD28, CDS, ICAM-1, LFA-1 (CD1 1a/CD18), ICOS (CD278), and 4-1BB (CD137). Further examples of such costimulatory molecules include CDS, ICAM-1, GITR, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), NKp44, NKp30, NKp46, CD160, CD19, CD4, CD8alpha, CD8beta, IL2R beta, IL2R gamma, IL7R alpha, ITGA4, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11 d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11 b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, NKG2D, NKG2C, TNFR2, TRANCE/RANKL, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), CD69, SLAMF6 (NTB-A, Ly108), SLAM (SLAMFI, CD150, IPO-3), BLAME (SLAMF8), SELPLG (CD162), LTBR, LAT, GADS, SLP-76, PAG/Cbp, CD19a, and a ligand that specifically binds with CD83.
[0223] A costimulatory intracellular signaling domain can be the intracellular portion of a costimulatory molecule. A costimulatory molecule can be represented in the following protein families: TNF receptor proteins, Immunoglobulin-like proteins, cytokine receptors, integrins, signaling lymphocytic activation molecules (SLAM proteins), and activating NK cell receptors. Examples of such molecules include CD27, CD28, 4-1BB (CD137), OX40, GITR, CD30, CD40, ICOS, BAFFR, HVEM, ICAM-1, lymphocyte function-associated antigen-1 (LFA-1), CD2, CDS, CD7, CD287, LIGHT, NKG2C, NKG2D, SLAMF7, NKp80, NKp30, NKp44, NKp46, CD160, B7-H3, and a ligand that specifically binds with CD83, and the like.
[0224] The intracellular signaling domain can comprise the entire intracellular portion, or the entire native intracellular signaling domain, of the molecule from which it is derived, or a functional fragment or derivative thereof.
[0225] The term "4-1BB" refers to a member of the TNFR superfamily with an amino acid sequence provided as GenBank Acc. No. AAA62478.2, or the equivalent residues from a non-human species, e.g., mouse, rodent, monkey, ape and the like; and a "4-1BB costimulatory domain" is defined as amino acid residues 214-255 of GenBank accno. AAA62478.2, or the equivalent residues from a non-human species, e.g., mouse, rodent, monkey, ape and the like. In one aspect, the "4-1BB costimulatory domain" is the sequence provided as SEQ ID NO: 16 or the equivalent residues from a non-human species, e.g., mouse, rodent, monkey, ape and the like.
[0226] "Immune effector cell," as that term is used herein, refers to a cell that is involved in an immune response, e.g., in the promotion of an immune effector response. Examples of immune effector cells include T cells, e.g., alpha/beta T cells and gamma/delta T cells, B cells, natural killer (NK) cells, natural killer T (NKT) cells, mast cells, and myeloic-derived phagocytes.
[0227] "Immune effector function or immune effector response," as that term is used herein, refers to function or response, e.g., of an immune effector cell, that enhances or promotes an immune attack of a target cell. E.g., an immune effector function or response refers a property of a T or NK cell that promotes killing or the inhibition of growth or proliferation, of a target cell. In the case of a T cell, primary stimulation and co-stimulation are examples of immune effector function or response.
[0228] The term "encoding" refers to the inherent property of specific sequences of nucleotides in a polynucleotide, such as a gene, a cDNA, or an mRNA, to serve as templates for synthesis of other polymers and macromolecules in biological processes having either a defined sequence of nucleotides (e.g., rRNA, tRNA and mRNA) or a defined sequence of amino acids and the biological properties resulting therefrom. Thus, a gene, cDNA, or RNA, encodes a protein if transcription and translation of mRNA corresponding to that gene produces the protein in a cell or other biological system. Both the coding strand, the nucleotide sequence of which is identical to the mRNA sequence and is usually provided in sequence listings, and the non-coding strand, used as the template for transcription of a gene or cDNA, can be referred to as encoding the protein or other product of that gene or cDNA.
[0229] Unless otherwise specified, a "nucleotide sequence encoding an amino acid sequence" includes all nucleotide sequences that are degenerate versions of each other and that encode the same amino acid sequence. The phrase nucleotide sequence that encodes a protein or a RNA may also include introns to the extent that the nucleotide sequence encoding the protein may in some version contain an intron(s).
[0230] The term "effective amount" or "therapeutically effective amount" are used interchangeably herein, and refer to an amount of a compound, formulation, material, or composition, as described herein effective to achieve a particular biological result.
[0231] The term "endogenous" refers to any material from or produced inside an organism, cell, tissue or system.
[0232] The term "exogenous" refers to any material introduced from or produced outside an organism, cell, tissue or system.
[0233] The term "expression" refers to the transcription and/or translation of a particular nucleotide sequence driven by a promoter.
[0234] The term "transfer vector" refers to a composition of matter which comprises an isolated nucleic acid and which can be used to deliver the isolated nucleic acid to the interior of a cell. Numerous vectors are known in the art including, but not limited to, linear polynucleotides, polynucleotides associated with ionic or amphiphilic compounds, plasmids, and viruses. Thus, the term "transfer vector" includes an autonomously replicating plasmid or a virus. The term should also be construed to further include non-plasmid and non-viral compounds which facilitate transfer of nucleic acid into cells, such as, for example, a polylysine compound, liposome, and the like. Examples of viral transfer vectors include, but are not limited to, adenoviral vectors, adeno-associated virus vectors, retroviral vectors, lentiviral vectors, and the like.
[0235] The term "expression vector" refers to a vector comprising a recombinant polynucleotide comprising expression control sequences operatively linked to a nucleotide sequence to be expressed. An expression vector comprises sufficient cis-acting elements for expression; other elements for expression can be supplied by the host cell or in an in vitro expression system. Expression vectors include all those known in the art, including cosmids, plasmids (e.g., naked or contained in liposomes) and viruses (e.g., lentiviruses, retroviruses, adenoviruses, and adeno-associated viruses) that incorporate the recombinant polynucleotide.
[0236] The term "lentivirus" refers to a genus of the Retroviridae family. Lentiviruses are unique among the retroviruses in being able to infect non-dividing cells; they can deliver a significant amount of genetic information into the DNA of the host cell, so they are one of the most efficient methods of a gene delivery vector. HIV, SIV, and FIV are all examples of lentiviruses.
[0237] The term "lentiviral vector" refers to a vector derived from at least a portion of a lentivirus genome, including especially a self-inactivating lentiviral vector as provided in Milone et al., Mol. Ther. 17(8): 1453-1464 (2009). Other examples of lentivirus vectors that may be used in the clinic, include but are not limited to, e.g., the LENTIVECTOR.RTM. gene delivery technology from Oxford BioMedica, the LENTIMAX.TM. vector system from Lentigen and the like. Nonclinical types of lentiviral vectors are also available and would be known to one skilled in the art.
[0238] The term "homologous" or "identity" refers to the subunit sequence identity between two polymeric molecules, e.g., between two nucleic acid molecules, such as, two DNA molecules or two RNA molecules, or between two polypeptide molecules. When a subunit position in both of the two molecules is occupied by the same monomeric subunit; e.g., if a position in each of two DNA molecules is occupied by adenine, then they are homologous or identical at that position. The homology between two sequences is a direct function of the number of matching or homologous positions; e.g., if half (e.g., five positions in a polymer ten subunits in length) of the positions in two sequences are homologous, the two sequences are 50% homologous; if 90% of the positions (e.g., 9 of 10), are matched or homologous, the two sequences are 90% homologous.
[0239] "Humanized" forms of non-human (e.g., murine) antibodies are chimeric immunoglobulins, immunoglobulin chains or fragments thereof (such as Fv, Fab, Fab', F(ab')2 or other antigen-binding subsequences of antibodies) which contain minimal sequence derived from non-human immunoglobulin. For the most part, humanized antibodies and antibody fragments thereof are human immunoglobulins (recipient antibody or antibody fragment) in which residues from a complementary-determining region (CDR) of the recipient are replaced by residues from a CDR of a non-human species (donor antibody) such as mouse, rat or rabbit having the desired specificity, affinity, and capacity. In some instances, Fv framework region (FR) residues of the human immunoglobulin are replaced by corresponding non-human residues. Furthermore, a humanized antibody/antibody fragment can comprise residues which are found neither in the recipient antibody nor in the imported CDR or framework sequences. These modifications can further refine and optimize antibody or antibody fragment performance. In general, the humanized antibody or antibody fragment thereof will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the CDR regions correspond to those of a non-human immunoglobulin and all or a significant portion of the FR regions are those of a human immunoglobulin sequence. The humanized antibody or antibody fragment can also comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin. For further details, see Jones et al., Nature, 321: 522-525, 1986; Reichmann et al., Nature, 332: 323-329, 1988; Presta, Curr. Op. Struct. Biol., 2: 593-596, 1992.
[0240] "Fully human" refers to an immunoglobulin, such as an antibody or antibody fragment, where the whole molecule is of human origin or consists of an amino acid sequence identical to a human form of the antibody or immunoglobulin.
[0241] The term "isolated" means altered or removed from the natural state. For example, a nucleic acid or a peptide naturally present in a living animal is not "isolated," but the same nucleic acid or peptide partially or completely separated from the coexisting materials of its natural state is "isolated." An isolated nucleic acid or protein can exist in substantially purified form, or can exist in a non-native environment such as, for example, a host cell.
[0242] In the context of the present invention, the following abbreviations for the commonly occurring nucleic acid bases are used. "A" refers to adenosine, "C" refers to cytosine, "G" refers to guanosine, "T" refers to thymidine, and "U" refers to uridine.
[0243] The term "operably linked" or "transcriptional control" refers to functional linkage between a regulatory sequence and a heterologous nucleic acid sequence resulting in expression of the latter. For example, a first nucleic acid sequence is operably linked with a second nucleic acid sequence when the first nucleic acid sequence is placed in a functional relationship with the second nucleic acid sequence. For instance, a promoter is operably linked to a coding sequence if the promoter affects the transcription or expression of the coding sequence. Operably linked DNA sequences can be contiguous with each other and, e.g., where necessary to join two protein coding regions, are in the same reading frame.
[0244] The term "parenteral" administration of an immunogenic composition includes, e.g., subcutaneous (s.c.), intravenous (i.v.), intramuscular (i.m.), or intrasternal injection, intratumoral, or infusion techniques.
[0245] The term "nucleic acid" or "polynucleotide" refers to deoxyribonucleic acids (DNA) or ribonucleic acids (RNA) and polymers thereof in either single- or double-stranded form. Unless specifically limited, the term encompasses nucleic acids containing known analogues of natural nucleotides that have similar binding properties as the reference nucleic acid and are metabolized in a manner similar to naturally occurring nucleotides. Unless otherwise indicated, a particular nucleic acid sequence also implicitly encompasses conservatively modified variants thereof (e.g., degenerate codon substitutions), alleles, orthologs, SNPs, and complementary sequences as well as the sequence explicitly indicated. Specifically, degenerate codon substitutions may be achieved by generating sequences in which the third position of one or more selected (or all) codons is substituted with mixed-base and/or deoxyinosine residues (Batzer et al., Nucleic Acid Res. 19:5081 (1991); Ohtsuka et al., J. Biol. Chem. 260:2605-2608 (1985); and Rossolini et al., Mol. Cell. Probes 8:91-98 (1994)).
[0246] The terms "peptide," "polypeptide," and "protein" are used interchangeably, and refer to a compound comprised of amino acid residues covalently linked by peptide bonds. A protein or peptide must contain at least two amino acids, and no limitation is placed on the maximum number of amino acids that can comprise a protein's or peptide's sequence. Polypeptides include any peptide or protein comprising two or more amino acids joined to each other by peptide bonds. As used herein, the term refers to both short chains, which also commonly are referred to in the art as peptides, oligopeptides and oligomers, for example, and to longer chains, which generally are referred to in the art as proteins, of which there are many types. "Polypeptides" include, for example, biologically active fragments, substantially homologous polypeptides, oligopeptides, homodimers, heterodimers, variants of polypeptides, modified polypeptides, derivatives, analogs, fusion proteins, among others. A polypeptide includes a natural peptide, a recombinant peptide, or a combination thereof.
[0247] The term "promoter" refers to a DNA sequence recognized by the synthetic machinery of the cell, or introduced synthetic machinery, required to initiate the specific transcription of a polynucleotide sequence.
[0248] The term "promoter/regulatory sequence" refers to a nucleic acid sequence which is required for expression of a gene product operably linked to the promoter/regulatory sequence. In some instances, this sequence may be the core promoter sequence and in other instances, this sequence may also include an enhancer sequence and other regulatory elements which are required for expression of the gene product. The promoter/regulatory sequence may, for example, be one which expresses the gene product in a tissue specific manner.
[0249] The term "constitutive" promoter refers to a nucleotide sequence which, when operably linked with a polynucleotide which encodes or specifies a gene product, causes the gene product to be produced in a cell under most or all physiological conditions of the cell.
[0250] The term "inducible" promoter refers to a nucleotide sequence which, when operably linked with a polynucleotide which encodes or specifies a gene product, causes the gene product to be produced in a cell substantially only when an inducer which corresponds to the promoter is present in the cell.
[0251] The term "tissue-specific" promoter refers to a nucleotide sequence which, when operably linked with a polynucleotide encodes or specified by a gene, causes the gene product to be produced in a cell substantially only if the cell is a cell of the tissue type corresponding to the promoter.
[0252] The term "flexible polypeptide linker" or "linker" as used in the context of a scFv refers to a peptide linker that consists of amino acids such as glycine and/or serine residues used alone or in combination, to link variable heavy and variable light chain regions together. In one embodiment, the flexible polypeptide linker is a Gly/Ser linker and comprises the amino acid sequence (Gly-Gly-Gly-Ser)n, where n is a positive integer equal to or greater than 1. For example, n=1, n=2, n=3. n=4, n=5 and n=6, n=7, n=8, n=9 and n=10 (SEQ ID NO:105). In one embodiment, the flexible polypeptide linkers include, but are not limited to, (Gly.sub.4 Ser).sub.4 (SEQ ID NO: 106) or (Gly.sub.4 Ser).sub.3 (SEQ ID NO: 107). In another embodiment, the linkers include multiple repeats of (Gly.sub.2Ser), (GlySer) or (Gly.sub.3Ser) (SEQ ID NO: 108). Also included within the scope of the invention are linkers described in WO2012/138475, incorporated herein by reference).
[0253] As used herein, a 5' cap (also termed an RNA cap, an RNA 7-methylguanosine cap or an RNA m.sup.7G cap) is a modified guanine nucleotide that has been added to the "front" or 5' end of a eukaryotic messenger RNA shortly after the start of transcription. The 5' cap consists of a terminal group which is linked to the first transcribed nucleotide. Its presence is critical for recognition by the ribosome and protection from RNases. Cap addition is coupled to transcription, and occurs co-transcriptionally, such that each influences the other. Shortly after the start of transcription, the 5' end of the mRNA being synthesized is bound by a cap-synthesizing complex associated with RNA polymerase. This enzymatic complex catalyzes the chemical reactions that are required for mRNA capping. Synthesis proceeds as a multi-step biochemical reaction. The capping moiety can be modified to modulate functionality of mRNA such as its stability or efficiency of translation.
[0254] As used herein, "in vitro transcribed RNA" refers to RNA, preferably mRNA, that has been synthesized in vitro. Generally, the in vitro transcribed RNA is generated from an in vitro transcription vector. The in vitro transcription vector comprises a template that is used to generate the in vitro transcribed RNA.
[0255] As used herein, a "poly(A)" is a series of adenosines attached by polyadenylation to the mRNA. In the preferred embodiment of a construct for transient expression, the polyA is between 50 and 5000 (SEQ ID NO: 109), preferably greater than 64, more preferably greater than 100, most preferably greater than 300 or 400. poly(A) sequences can be modified chemically or enzymatically to modulate mRNA functionality such as localization, stability or efficiency of translation.
[0256] As used herein, "polyadenylation" refers to the covalent linkage of a polyadenylyl moiety, or its modified variant, to a messenger RNA molecule. In eukaryotic organisms, most messenger RNA (mRNA) molecules are polyadenylated at the 3' end. The 3' poly(A) tail is a long sequence of adenine nucleotides (often several hundred) added to the pre-mRNA through the action of an enzyme, polyadenylate polymerase. In higher eukaryotes, the poly(A) tail is added onto transcripts that contain a specific sequence, the polyadenylation signal. The poly(A) tail and the protein bound to it aid in protecting mRNA from degradation by exonucleases. Polyadenylation is also important for transcription termination, export of the mRNA from the nucleus, and translation. Polyadenylation occurs in the nucleus immediately after transcription of DNA into RNA, but additionally can also occur later in the cytoplasm. After transcription has been terminated, the mRNA chain is cleaved through the action of an endonuclease complex associated with RNA polymerase. The cleavage site is usually characterized by the presence of the base sequence AAUAAA near the cleavage site. After the mRNA has been cleaved, adenosine residues are added to the free 3' end at the cleavage site.
[0257] As used herein, "transient" refers to expression of a non-integrated transgene for a period of hours, days or weeks, wherein the period of time of expression is less than the period of time for expression of the gene if integrated into the genome or contained within a stable plasmid replicon in the host cell.
[0258] The term "signal transduction pathway" refers to the biochemical relationship between a variety of signal transduction molecules that play a role in the transmission of a signal from one portion of a cell to another portion of a cell. The phrase "cell surface receptor" includes molecules and complexes of molecules capable of receiving a signal and transmitting signal across the membrane of a cell.
[0259] The term "subject" is intended to include living organisms in which an immune response can be elicited (e.g., mammals, human).
[0260] The term, a "substantially purified" cell refers to a cell that is essentially free of other cell types. A substantially purified cell also refers to a cell which has been separated from other cell types with which it is normally associated in its naturally occurring state. In some instances, a population of substantially purified cells refers to a homogenous population of cells. In other instances, this term refers simply to cell that have been separated from the cells with which they are naturally associated in their natural state. In some aspects, the cells are cultured in vitro. In other aspects, the cells are not cultured in vitro.
[0261] The term "therapeutic" as used herein means a treatment. A therapeutic effect is obtained by reduction, suppression, remission, or eradication of a disease state.
[0262] The term "prophylaxis" as used herein means the prevention of or protective treatment for a disease or disease state.
[0263] In the context of the present invention, "tumor antigen" or "hyperproliferative disorder antigen" or "antigen associated with a hyperproliferative disorder" refers to antigens that are common to specific hyperproliferative disorders. In certain aspects, the hyperproliferative disorder antigens of the present invention are derived from, cancers including but not limited to primary or metastatic melanoma, thymoma, lymphoma, sarcoma, lung cancer, liver cancer, non-Hodgkin lymphoma, Hodgkin lymphoma, leukemias, uterine cancer, cervical cancer, bladder cancer, kidney cancer and adenocarcinomas such as breast cancer, prostate cancer, ovarian cancer, pancreatic cancer, and the like.
[0264] The term "transfected" or "transformed" or "transduced" refers to a process by which exogenous nucleic acid is transferred or introduced into the host cell. A "transfected" or "transformed" or "transduced" cell is one which has been transfected, transformed or transduced with exogenous nucleic acid. The cell includes the primary subject cell and its progeny.
[0265] The term "specifically binds," refers to an antibody, or a ligand, which recognizes and binds with a binding partner (e.g., a stimulatory tumor antigen) protein present in a sample, but which antibody or ligand does not substantially recognize or bind other molecules in the sample.
[0266] "Regulatable chimeric antigen receptor (RCAR)," as that term is used herein, refers to a set of polypeptides, typically two in the simplest embodiments, which when in a RCARX cell, provides the RCARX cell with specificity for a target cell, typically a cancer cell, and with regulatable intracellular signal generation or proliferation, which can optimize an immune effector property of the RCARX cell. An RCARX cell relies at least in part, on an antigen binding domain to provide specificity to a target cell that comprises the antigen bound by the antigen binding domain. In an embodiment, an RCAR includes a dimerization switch that, upon the presence of a dimerization molecule, can couple an intracellular signaling domain to the antigen binding domain.
[0267] "Membrane anchor" or "membrane tethering domain", as that term is used herein, refers to a polypeptide or moiety, e.g., a myristoyl group, sufficient to anchor an extracellular or intracellular domain to the plasma membrane.
[0268] "Switch domain," as that term is used herein, e.g., when referring to an RCAR, refers to an entity, typically a polypeptide-based entity, that, in the presence of a dimerization molecule, associates with another switch domain. The association results in a functional coupling of a first entity linked to, e.g., fused to, a first switch domain, and a second entity linked to, e.g., fused to, a second switch domain. A first and second switch domain are collectively referred to as a dimerization switch. In embodiments, the first and second switch domains are the same as one another, e.g., they are polypeptides having the same primary amino acid sequence, and are referred to collectively as a homodimerization switch. In embodiments, the first and second switch domains are different from one another, e.g., they are polypeptides having different primary amino acid sequences, and are referred to collectively as a heterodimerization switch. In embodiments, the switch is intracellular. In embodiments, the switch is extracellular. In embodiments, the switch domain is a polypeptide-based entity, e.g., FKBP or FRB-based, and the dimerization molecule is small molecule, e.g., a rapalogue. In embodiments, the switch domain is a polypeptide-based entity, e.g., an scFv that binds a myc peptide, and the dimerization molecule is a polypeptide, a fragment thereof, or a multimer of a polypeptide, e.g., a myc ligand or multimers of a myc ligand that bind to one or more myc scFvs. In embodiments, the switch domain is a polypeptide-based entity, e.g., myc receptor, and the dimerization molecule is an antibody or fragments thereof, e.g., myc antibody.
[0269] "Dimerization molecule," as that term is used herein, e.g., when referring to an RCAR, refers to a molecule that promotes the association of a first switch domain with a second switch domain. In embodiments, the dimerization molecule does not naturally occur in the subject, or does not occur in concentrations that would result in significant dimerization. In embodiments, the dimerization molecule is a small molecule, e.g., rapamycin or a rapalogue, e.g, RAD001.
[0270] The term "bioequivalent" refers to an amount of an agent other than the reference compound (e.g., RAD001), required to produce an effect equivalent to the effect produced by the reference dose or reference amount of the reference compound (e.g., RAD001). In an embodiment the effect is the level of mTOR inhibition, e.g., as measured by P70 S6 kinase inhibition, e.g., as evaluated in an in vivo or in vitro assay, e.g., as measured by an assay described herein, e.g., the Boulay assay, or measurement of phosphorylated S6 levels by western blot. In an embodiment, the effect is alteration of the ratio of PD-1 positive/PD-1 negative T cells, as measured by cell sorting. In an embodiment a bioequivalent amount or dose of an mTOR inhibitor is the amount or dose that achieves the same level of P70 S6 kinase inhibition as does the reference dose or reference amount of a reference compound. In an embodiment, a bioequivalent amount or dose of an mTOR inhibitor is the amount or dose that achieves the same level of alteration in the ratio of PD-1 positive/PD-1 negative T cells as does the reference dose or reference amount of a reference compound.
[0271] The term "low, immune enhancing, dose" when used in conjuction with an mTOR inhibitor, e.g., an allosteric mTOR inhibitor, e.g., RAD001 or rapamycin, or a catalytic mTOR inhibitor, refers to a dose of mTOR inhibitor that partially, but not fully, inhibits mTOR activity, e.g., as measured by the inhibition of P70 S6 kinase activity. Methods for evaluating mTOR activity, e.g., by inhibition of P70 S6 kinase, are discussed herein. The dose is insufficient to result in complete immune suppression but is sufficient to enhance the immune response. In an embodiment, the low, immune enhancing, dose of mTOR inhibitor results in a decrease in the number of PD-1 positive T cells and/or an increase in the number of PD-1 negative T cells, or an increase in the ratio of PD-1 negative T cells/PD-1 positive T cells. In an embodiment, the low, immune enhancing, dose of mTOR inhibitor results in an increase in the number of naive T cells. In an embodiment, the low, immune enhancing, dose of mTOR inhibitor results in one or more of the following:
[0272] an increase in the expression of one or more of the following markers: CD62L.sup.highCD127.sup.high, CD27.sup.+, and BCL2, e.g., on memory T cells, e.g., memory T cell precursors;
[0273] a decrease in the expression of KLRG1, e.g., on memory T cells, e.g., memory T cell precursors; and
[0274] an increase in the number of memory T cell precursors, e.g., cells with any one or combination of the following characteristics: increased CD62L.sup.high, increased CD127.sup.high increased CD27+, decreased KLRG1, and increased BCL2;
wherein any of the changes described above occurs, e.g., at least transiently, e.g., as compared to a non-treated subject.
[0275] "Refractory" as used herein refers to a disease, e.g., cancer, that does not respond to a treatment. In embodiments, a refractory cancer can be resistant to a treatment before or at the beginning of the treatment. In other embodiments, the refractory cancer can become refractory during a treatment.
[0276] A "complete responder" as used herein refers to a subject having a disease, e.g., a cancer, who exhibits a complete response, e.g., a complete remission, to a treatment. A complete response may be identified, e.g., using the Cheson criteria as described herein.
[0277] A "partial responder" as used herein refers to a subject having a disease, e.g., a cancer, who exhibits a partial response, e.g., a partial remission, to a treatment. A partial response may be identified, e.g., using the Cheson criteria.
[0278] A "non-responder" as used herein refers to a subject having a disease, e.g., a cancer, who does not exhibit a response to a treatment, e.g., the patient has stable disease or progressive disease. A non-responder may be identified, e.g., using the Cheson criteria as described herein.
[0279] The term "relapse" as used herein refers to reappearance of a disease (e.g., cancer) after an initial period of responsiveness (e.g., complete response or partial response). The initial period of responsiveness may involve the level of cancer cells falling below a certain threshold, e.g., below 20%, 1%, 10%, 5%, 4%, 3%, 2%, or 1%. The reappearance may involve the level of cancer cells rising above a certain threshold, e.g., above 20%, 1%, 10%, 5%, 4%, 3%, 2%, or 1%. Relapse may be identified, e.g., using the Cheson criteria as described herein.
[0280] Ranges: throughout this disclosure, various aspects of the invention can be presented in a range format. It should be understood that the description in range format is merely for convenience and brevity and should not be construed as an inflexible limitation on the scope of the invention. Accordingly, the description of a range should be considered to have specifically disclosed all the possible subranges as well as individual numerical values within that range. For example, description of a range such as from 1 to 6 should be considered to have specifically disclosed subranges such as from 1 to 3, from 1 to 4, from 1 to 5, from 2 to 4, from 2 to 6, from 3 to 6 etc., as well as individual numbers within that range, for example, 1, 2, 2.7, 3, 4, 5, 5.3, and 6. As another example, a range such as 95-99% identity, includes something with 95%, 96%, 97%, 98% or 99% identity, and includes subranges such as 96-99%, 96-98%, 96-97%, 97-99%, 97-98% and 98-99% identity. This applies regardless of the breadth of the range.
DESCRIPTION
[0281] Provided herein are compositions of matter and methods of use for the treatment of a disease such as cancer (e.g., hematological cancers or other B cell malignancies) using immune effector cells (e.g., T cells or NK cells) that express a chimeric antigen receptor (CAR) (e.g., a CAR that targets a B-cell marker, such as CD19). The methods include, inter alia, administering immune effector cells (e.g., T cells or NK cells) expressing a B cell targeting CAR described herein in combination with another agent such as a kinase inhibitor, e.g., a kinase inhibitor described herein.
[0282] The present invention provides, at least in part, experiments supporting the high efficacy of a combination of a CAR therapy (e.g., a B-cell targeting CAR therapy) and a kinase inhibitor, e.g., a BTK inhibitor such as ibrutinib. The combination of a kinase inhibitor, e.g., a BTK inhibitor such as ibrutinib, with a CAR therapy can increase efficacy of the combination therapy relative to a monotherapy of the kinase inhibitor, or a dose of CAR-expressing cells, or both. These beneficial effects can, for example, allow for a lower dose of the kinase inhibitor or the CAR-expressing cells, or both, while maintaining efficacy. The results herein are applicable to a wide range of cancers, e.g., hematological cancers and other B cell malignancies. For example, ibrutinib inhibits BTK, which is elevated in most lymphomas. An immune effector cell (e.g., T cell or NK cell) that expresses CAR19 targets cancers with CD19 surface expression, which is expressed in most B cell malignancies. Alternatively or in combination with CAR19, any other B-cell targeting CAR (e.g., a CAR targeting one or more of: CD20, CD22, or ROR1) can be used in the combination therapies described herein. Therefore, the combination of a CAR therapy (e.g., one or more of a CD19 CAR, CD20 CAR, CD22 CAR or ROR1 CAR therapy) with a BTK inhibitor (e.g., ibrutinib) is suitable for treating a wide range of cancers involving overproliferation of B cells, including lymphomas (e.g., Hodgkin lymphoma), MCL, CLL, DLBCL, and multiple myeloma.
[0283] According to the present invention, ibrutinib can reduce tumor masses and mobilize neoplastic B cells in the peripheral blood (see e.g., Example 8 herein). Without wishing to be bound by theory, certain lymphomas, such as MCL, are characterized by masses of cancerous cells in proliferation centers in lymph nodes. CAR-expressing immune effector cells sometimes have difficulty penetrating these densely packed masses. Thus, a BTK inhibitor, such as ibrutinib, can reduce tumor masses and mobilize neoplastic B cells in the peripheral blood, making the lymphoma cells more vulnerable to the CAR-expressing cells.
[0284] Alternatively or in combination, BTK inhibitors, such as ibrutinib, can also affect the CAR-expressing cells. The present invention demonstrates that ibrutinib treatment increases the level of circulating CART19 cells (see e.g., data shown in Example 8). Without wishing to be bound by theory, the increase in the level of circulating CART19 cells may be a result of, for example, increased proliferation, alteration of T cell phenotype, or other factors. For example, ibrutinib can inhibit ITK, a kinase with homology to BTK. ITK is expressed in T cells, and its inhibition may alter the T cell phenotype. Treatment with a kinase inhibitor, such as ibrutinib, can alter the T cell phenotype from a Th2 phenotype to a Th1 phenotype, and thus increase the T cell proliferative capacity. Pre-treatment, or co-administration, to a subject, of a BTK inhibitor may increase the T cell proliferative capacity in the subject, thus increasing the level of circulating CAR-expressing cells. In addition, a subject pre-treated with a BTK inhibitor, e.g., ibrutinib, can have a T cell population with a higher proliferative capacity in their apheresis for CAR manufacturing.
[0285] In one aspect, the invention provides a number of chimeric antigen receptors (CAR) comprising an antibody or antibody fragment engineered for specific binding to a B-cell antigen (e.g., chosen from one or more of CD19, CD20, CD22 or ROR1 protein). In one aspect, the invention provides a cell (e.g., T cell) engineered to express a CAR, wherein the CAR T cell ("CART") exhibits an anticancer property. In one aspect a cell is transformed with the CAR and the CAR is expressed on the cell surface. In some embodiments, the cell (e.g., T cell) is transduced with a viral vector encoding a CAR. In some embodiments, the viral vector is a retroviral vector. In some embodiments, the viral vector is a lentiviral vector. In some such embodiments, the cell may stably express the CAR. In another embodiment, the cell (e.g., T cell) is transfected with a nucleic acid, e.g., mRNA, cDNA, DNA, encoding a CAR. In some such embodiments, the cell may transiently express the CAR.
[0286] In one aspect, the anti-CD19 protein binding portion of the CAR is a scFv antibody fragment. In one aspect such antibody fragments are functional in that they retain the equivalent binding affinity, e.g., they bind the same antigen with comparable affinity, as the IgG antibody from which it is derived. In one aspect such antibody fragments are functional in that they provide a biological response that can include, but is not limited to, activation of an immune response, inhibition of signal-transduction origination from its target antigen, inhibition of kinase activity, and the like, as will be understood by a skilled artisan. In one aspect, the anti-CD19 antigen binding domain of the CAR is a scFv antibody fragment that is humanized compared to the murine sequence of the scFv from which it is derived. In one aspect, the parental murine scFv sequence is the CAR19 construct provided in PCT publication WO2012/079000 (incorporated herein by reference) and provided herein as SEQ ID NO:59. In one embodiment, the anti-CD19 binding domain is a scFv described in WO2012/079000 and provided in SEQ ID NO:59.
[0287] In some aspects, the antibodies of the invention are incorporated into a chimeric antigen receptor (CAR). In one aspect, the CAR comprises the polypeptide sequence provided as SEQ ID NO: 12 in PCT publication WO2012/079000, and provided herein as SEQ ID NO: 58, wherein the scFv domain is substituted by one or more sequences selected from SEQ ID NOS: 1-12. In one aspect, the scFv domains of SEQ ID NOS:1-12 are humanized variants of the scFv domain of SEQ ID NO:59, which is an scFv fragment of murine origin that specifically binds to human CD19. Humanization of this mouse scFv may be desired for the clinical setting, where the mouse-specific residues may induce a human-anti-mouse antigen (HAMA) response in patients who receive CART19 treatment, e.g., treatment with T cells transduced with the CAR19 construct.
[0288] In one aspect, the anti-CD19 binding domain, e.g., humanized scFv, portion of a CAR of the invention is encoded by a transgene whose sequence has been codon optimized for expression in a mammalian cell. In one aspect, entire CAR construct of the invention is encoded by a transgene whose entire sequence has been codon optimized for expression in a mammalian cell. Codon optimization refers to the discovery that the frequency of occurrence of synonymous codons (i.e., codons that code for the same amino acid) in coding DNA is biased in different species. Such codon degeneracy allows an identical polypeptide to be encoded by a variety of nucleotide sequences. A variety of codon optimization methods is known in the art, and include, e.g., methods disclosed in at least U.S. Pat. Nos. 5,786,464 and 6,114,148.
[0289] In one aspect, the humanized CAR19 comprises the scFv portion provided in SEQ ID NO: 1. In one aspect, the humanized CAR19 comprises the scFv portion provided in SEQ ID NO:2. In one aspect, the humanized CAR19 comprises the scFv portion provided in SEQ ID NO:3. In one aspect, the humanized CAR19 comprises the scFv portion provided in SEQ ID NO:4. In one aspect, the humanized CAR19 comprises the scFv portion provided in SEQ ID NO:5. In one aspect, the humanized CAR19 comprises the scFv portion provided in SEQ ID NO:6. In one aspect, the humanized CAR19 comprises the scFv portion provided in SEQ ID NO:7. In one aspect, the humanized CAR19 comprises the scFv portion provided in SEQ ID NO:8. In one aspect, the humanized CAR19 comprises the scFv portion provided in SEQ ID NO:9. In one aspect, the humanized CAR19 comprises the scFv portion provided in SEQ ID NO:10. In one aspect, the humanized CAR19 comprises the scFv portion provided in SEQ ID NO:11. In one aspect, the humanized CAR19 comprises the scFv portion provided in SEQ ID NO:12.
[0290] In one aspect, the CARs of the invention combine an antigen binding domain of a specific antibody with an intracellular signaling molecule. For example, in some aspects, the intracellular signaling molecule includes, but is not limited to, CD3-zeta chain, 4-1BB and CD28 signaling modules and combinations thereof. In one aspect, the CD19 CAR comprises a CAR selected from the sequence provided in one or more of SEQ ID NOS: 31-42. In one aspect, the CD19 CAR comprises the sequence provided in SEQ ID NO:31. In one aspect, the CD19 CAR comprises the sequence provided in SEQ ID NO:32. In one aspect, the CD19 CAR comprises the sequence provided in SEQ ID NO:33. In one aspect, the CD19 CAR comprises the sequence provided in SEQ ID NO:34. In one aspect, the CD19 CAR comprises the sequence provided in SEQ ID NO:35. In one aspect, the CD19 CAR comprises the sequence provided in SEQ ID NO:36. In one aspect, the CD19 CAR comprises the sequence provided in SEQ ID NO:37. In one aspect, the CD19 CAR comprises the sequence provided in SEQ ID NO:38. In one aspect, the CD19 CAR comprises the sequence provided in SEQ ID NO:39. In one aspect, the CD19 CAR comprises the sequence provided in SEQ ID NO:40. In one aspect, the CD19 CAR comprises the sequence provided in SEQ ID NO:41. In one aspect, the CD19 CAR comprises the sequence provided in SEQ ID NO:42.
[0291] Furthermore, the present invention provides CD19 CAR compositions and their use in medicaments or methods for treating, among other diseases, cancer or any malignancy or autoimmune diseases involving cells or tissues which express CD19.
[0292] In one aspect, the CAR of the invention can be used to eradicate CD19-expressing normal cells, thereby applicable for use as a cellular conditioning therapy prior to cell transplantation. In one aspect, the CD19-expressing normal cell is a CD19-expressing normal stem cell and the cell transplantation is a stem cell transplantation.
[0293] In one aspect, the invention provides a cell (e.g., T cell) engineered to express a chimeric antigen receptor (CAR), wherein the CAR-expressing cell, e.g., CAR T cell ("CART"), exhibits an anticancer property. A preferred antigen is CD19. In one aspect, the antigen binding domain of the CAR comprises a partially humanized anti-CD19 antibody fragment. In one aspect, the antigen binding domain of the CAR comprises a partially humanized anti-CD19 antibody fragment comprising a scFv. Accordingly, the invention provides a CD19-CAR that comprises a humanized anti-CD19 binding domain and is engineered into an immune effector cell, e.g., a T cell or an NK cell, and methods of their use for adoptive therapy.
[0294] In one aspect, the CD19-CAR comprises at least one intracellular domain selected from the group of a CD137 (4-1BB) signaling domain, a CD28 signaling domain, a CD3zeta signal domain, and any combination thereof. In one aspect, the CD19-CAR comprises at least one intracellular signaling domain is from one or more co-stimulatory molecule(s) other than a CD137 (4-1BB) or CD28.
Chimeric Antigen Receptor (CAR)
[0295] The present invention encompasses a recombinant DNA construct comprising sequences encoding a CAR, wherein the CAR comprises an antibody or antibody fragment that binds specifically to a B-cell antigen (e.g., CD19, e.g., human CD19), wherein the sequence of the antibody fragment is contiguous with and in the same reading frame as a nucleic acid sequence encoding an intracellular signaling domain. The intracellular signaling domain can comprise a costimulatory signaling domain and/or a primary signaling domain, e.g., a zeta chain. The costimulatory signaling domain refers to a portion of the CAR comprising at least a portion of the intracellular domain of a costimulatory molecule. In one embodiment, the antigen binding domain is a murine antibody or antibody fragment described herein. In one embodiment, the antigen binding domain is a humanized antibody or antibody fragment.
[0296] In specific aspects, a CAR construct of the invention comprises a scFv domain selected from the group consisting of SEQ ID NOS:1-12 or an scFV domain of SEQ ID NO:59, wherein the scFv may be preceded by an optional leader sequence such as provided in SEQ ID NO: 13, and followed by an optional hinge sequence such as provided in SEQ ID NO: 14 or SEQ ID NO:45 or SEQ ID NO:47 or SEQ ID NO:49, a transmembrane region such as provided in SEQ ID NO: 15, an intracellular signalling domain that includes SEQ ID NO: 16 or SEQ ID NO:51 and a CD3 zeta sequence that includes SEQ ID NO:17 or SEQ ID NO:43, wherein the domains are contiguous with and in the same reading frame to form a single fusion protein. Also included in the invention is a nucleotide sequence that encodes the polypeptide of each of the scFv fragments selected from the group consisting of SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4, SEQ ID NO:5, SEQ ID NO:6, SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11, SEQ ID NO:12 and SEQ ID NO:59. Also included in the invention is a nucleotide sequence that encodes the polypeptide of each of the scFv fragments selected from the group consisting of SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4, SEQ ID NO:5, SEQ ID NO:6, SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11, SEQ ID NO:12 and SEQ ID NO:59, and each of the domains of SEQ ID NOS: 13-17, plus the encoded CD19CAR fusion protein of the invention. In one aspect an exemplary CD19CAR constructs comprise an optional leader sequence, an extracellular antigen binding domain, a hinge, a transmembrane domain, and an intracellular stimulatory domain. In one aspect an exemplary CD19CAR construct comprises an optional leader sequence, an extracellular antigen binding domain, a hinge, a transmembrane domain, an intracellular costimulatory domain and an intracellular stimulatory domain. Specific CD19 CAR constructs containing humanized scFv domains of the invention are provided as SEQ ID NOS: 31-42, or a murine scFv domain as provided as SEQ ID NO:59.
[0297] Full-length CAR sequences are also provided herein as SEQ ID NOS: 31-42 and 58, as shown in Table 7 and Table 3.
[0298] An exemplary leader sequence is provided as SEQ ID NO: 13. An exemplary hinge/spacer sequence is provided as SEQ ID NO: 14 or SEQ ID NO:45 or SEQ ID NO:47 or SEQ ID NO:49. An exemplary transmembrane domain sequence is provided as SEQ ID NO:15. An exemplary sequence of the intracellular signaling domain of the 4-1BB protein is provided as SEQ ID NO: 16. An exemplary sequence of the intracellular signaling domain of CD27 is provided as SEQ ID NO:51. An exemplary CD3zeta domain sequence is provided as SEQ ID NO: 17 or SEQ ID NO:43.
[0299] In one aspect, the present invention encompasses a recombinant nucleic acid construct comprising a nucleic acid molecule encoding a CAR, wherein the nucleic acid molecule comprises the nucleic acid sequence encoding an anti-CD19 binding domain, e.g., described herein, that is contiguous with and in the same reading frame as a nucleic acid sequence encoding an intracellular signaling domain. In one aspect, the anti-CD19 binding domain is selected from one or more of SEQ ID NOS:1-12 and 58. In one aspect, the anti-CD 19 binding domain is encoded by a nucleotide residues 64 to 813 of the sequence provided in one or more of SEQ ID NOS:61-72 and 59. In one aspect, the anti-CD19 binding domain is encoded by a nucleotide residues 64 to 813 of SEQ ID NO:61. In one aspect, the anti-CD19 binding domain is encoded by a nucleotide residues 64 to 813 of SEQ ID NO:62. In one aspect, the anti-CD19 binding domain is encoded by a nucleotide residues 64 to 813 of SEQ ID NO:63. In one aspect, the anti-CD19 binding domain is encoded by a nucleotide residues 64 to 813 of SEQ ID NO:64. In one aspect, the anti-CD19 binding domain is encoded by a nucleotide residues 64 to 813 of SEQ ID NO:65. In one aspect, the anti-CD19 binding domain is encoded by a nucleotide residues 64 to 813 of SEQ ID NO:66. In one aspect, the anti-CD19 binding domain is encoded by a nucleotide residues 64 to 813 of SEQ ID NO:67. In one aspect, the anti-CD19 binding domain is encoded by a nucleotide residues 64 to 813 of SEQ ID NO:68. In one aspect, the anti-CD19 binding domain is encoded by a nucleotide residues 64 to 813 of SEQ ID NO:69. In one aspect, the anti-CD19 binding domain is encoded by a nucleotide residues 64 to 813 of SEQ ID NO:70. In one aspect, the anti-CD19 binding domain is encoded by a nucleotide residues 64 to 813 of SEQ ID NO:71. In one aspect, the anti-CD19 binding domain is encoded by a nucleotide residues 64 to 813 of SEQ ID NO:72.
[0300] In one aspect, the present invention encompasses a recombinant nucleic acid construct comprising a transgene encoding a CAR, wherein the nucleic acid molecule comprises a nucleic acid sequence encoding an anti-CD19 binding domain selected from one or more of SEQ ID NOS:61-72, wherein the sequence is contiguous with and in the same reading frame as the nucleic acid sequence encoding an intracellular signaling domain. An exemplary intracellular signaling domain that can be used in the CAR includes, but is not limited to, one or more intracellular signaling domains of, e.g., CD3-zeta, CD28, 4-1BB, and the like. In some instances, the CAR can comprise any combination of CD3-zeta, CD28, 4-1BB, and the like. In one aspect the nucleic acid sequence of a CAR construct of the invention is selected from one or more of SEQ ID NOS:85-96. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:85. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:86. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:87. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:88. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:89. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:90. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:91. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:92. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:93. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:94. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:95. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:96. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:97. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:98. In one aspect the nucleic acid sequence of a CAR construct is SEQ ID NO:99.
[0301] The nucleic acid sequences coding for the desired molecules can be obtained using recombinant methods known in the art, such as, for example by screening libraries from cells expressing the gene, by deriving the gene from a vector known to include the same, or by isolating directly from cells and tissues containing the same, using standard techniques. Alternatively, the nucleic acid of interest can be produced synthetically, rather than cloned.
[0302] The present invention includes retroviral and lentiviral vector constructs expressing a CAR that can be directly transduced into a cell.
[0303] The present invention also includes an RNA construct that can be directly transfected into a cell. A method for generating mRNA for use in transfection involves in vitro transcription (IVT) of a template with specially designed primers, followed by polyA addition, to produce a construct containing 3' and 5' untranslated sequence ("UTR"), a 5' cap and/or Internal Ribosome Entry Site (IRES), the nucleic acid to be expressed, and a polyA tail, typically 50-2000 bases in length (SEQ ID NO: 118). RNA so produced can efficiently transfect different kinds of cells. In one embodiment, the template includes sequences for the CAR. In an embodiment, an RNA CAR vector is transduced into a T cell by electroporation.
Antigen Binding Domain
[0304] In one aspect, the CAR of the invention comprises a target-specific binding element otherwise referred to as an antigen binding domain. The choice of moiety depends upon the type and number of ligands that define the surface of a target cell. For example, the antigen binding domain may be chosen to recognize a ligand that acts as a cell surface marker on target cells associated with a particular disease state. Thus examples of cell surface markers that may act as ligands for the antigen binding domain in a CAR of the invention include those associated with viral, bacterial and parasitic infections, autoimmune disease and cancer cells.
[0305] In one aspect, the CAR-mediated T-cell response can be directed to an antigen of interest by way of engineering an antigen binding domain that specifically binds a desired antigen into the CAR.
[0306] In one aspect, the portion of the CAR comprising the antigen binding domain comprises an antigen binding domain that targets CD19. In one aspect, the antigen binding domain targets human CD19. In one aspect, the antigen binding domain of the CAR has the same or a similar binding specificity as the FMC63 scFv fragment described in Nicholson et al. Mol. Immun. 34 (16-17): 1157-1165 (1997). In one embodiment, the antigen binding domain of the CAR includes the scFv fragment described in Nicholson et al. Mol. Immun. 34 (16-17): 1157-1165 (1997).
[0307] The antigen binding domain can be any domain that binds to the antigen including but not limited to a monoclonal antibody, a polyclonal antibody, a recombinant antibody, a murine antibody, a human antibody, a humanized antibody, and a functional fragment thereof, including but not limited to a single-domain antibody such as a heavy chain variable domain (VH), a light chain variable domain (VL) and a variable domain (VHH) of camelid derived nanobody, and to an alternative scaffold known in the art to function as antigen binding domain, such as a recombinant fibronectin domain, and the like.
[0308] In one embodiment, the CAR molecule comprises an anti-CD19 binding domain comprising one or more (e.g., all three) light chain complementary determining region 1 (LC CDR1), light chain complementary determining region 2 (LC CDR2), and light chain complementary determining region 3 (LC CDR3) of an anti-CD19 binding domain described herein, and one or more (e.g., all three) heavy chain complementary determining region 1 (HC CDR1), heavy chain complementary determining region 2 (HC CDR2), and heavy chain complementary determining region 3 (HC CDR3) of an anti-CD19 binding domain described herein, e.g., an anti-CD19 binding domain comprising one or more, e.g., all three, LC CDRs and one or more, e.g., all three, HC CDRs. In one embodiment, the anti-CD19 binding domain comprises one or more (e.g., all three) heavy chain complementary determining region 1 (HC CDR1), heavy chain complementary determining region 2 (HC CDR2), and heavy chain complementary determining region 3 (HC CDR3) of an anti-CD19 binding domain described herein, e.g., the anti-CD19 binding domain has two variable heavy chain regions, each comprising a HC CDR1, a HC CDR2 and a HC CDR3 described herein. In one embodiment, the anti-CD19 binding domain comprises a murine light chain variable region described herein (e.g., in Table 7) and/or a murine heavy chain variable region described herein (e.g., in Table 7). In one embodiment, the anti-CD19 binding domain is a scFv comprising a murine light chain and a murine heavy chain of an amino acid sequence of Table 7. In an embodiment, the anti-CD19 binding domain (e.g., an scFv) comprises: a light chain variable region comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of an amino acid sequence of a light chain variable region provided in Table 7, or a sequence with 95-99% identity with an amino acid sequence of Table 7; and/or a heavy chain variable region comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of an amino acid sequence of a heavy chain variable region provided in Table 7, or a sequence with 95-99% identity to an amino acid sequence of Table 7. In one embodiment, the anti-CD19 binding domain comprises a sequence of SEQ ID NO:59, or a sequence with 95-99% identify thereof. In one embodiment, the anti-CD19 binding domain is a scFv, and a light chain variable region comprising an amino acid sequence described herein, e.g., in Table 7, is attached to a heavy chain variable region comprising an amino acid sequence described herein, e.g., in Table 7, via a linker, e.g., a linker described herein. In one embodiment, the anti-CD19 binding domain includes a (Gly.sub.4-Ser)n linker, wherein n is 1, 2, 3, 4, 5, or 6, preferably 3 or 4 (SEQ ID NO: 53). The light chain variable region and heavy chain variable region of a scFv can be, e.g., in any of the following orientations: light chain variable region-linker-heavy chain variable region or heavy chain variable region-linker-light chain variable region.
[0309] In some instances, it is beneficial for the antigen binding domain to be derived from the same species in which the CAR will ultimately be used in. For example, for use in humans, it may be beneficial for the antigen binding domain of the CAR to comprise human or humanized residues for the antigen binding domain of an antibody or antibody fragment.
[0310] Thus, in one aspect, the antigen binding domain comprises a humanized antibody or an antibody fragment. In one embodiment, the humanized anti-CD19 binding domain comprises one or more (e.g., all three) light chain complementary determining region 1 (LC CDR1), light chain complementary determining region 2 (LC CDR2), and light chain complementary determining region 3 (LC CDR3) of a murine or humanized anti-CD19 binding domain described herein, and/or one or more (e.g., all three) heavy chain complementary determining region 1 (HC CDR1), heavy chain complementary determining region 2 (HC CDR2), and heavy chain complementary determining region 3 (HC CDR3) of a murine or humanized anti-CD19 binding domain described herein, e.g., a humanized anti-CD19 binding domain comprising one or more, e.g., all three, LC CDRs and one or more, e.g., all three, HC CDRs. In one embodiment, the humanized anti-CD19 binding domain comprises one or more (e.g., all three) heavy chain complementary determining region 1 (HC CDR1), heavy chain complementary determining region 2 (HC CDR2), and heavy chain complementary determining region 3 (HC CDR3) of a murine or humanized anti-CD19 binding domain described herein, e.g., the humanized anti-CD19 binding domain has two variable heavy chain regions, each comprising a HC CDR1, a HC CDR2 and a HC CDR3 described herein. In one embodiment, the humanized anti-CD19 binding domain comprises a humanized light chain variable region described herein (e.g., in Table 3) and/or a humanized heavy chain variable region described herein (e.g., in Table 3). In one embodiment, the humanized anti-CD19 binding domain comprises a humanized heavy chain variable region described herein (e.g., in Table 3), e.g., at least two humanized heavy chain variable regions described herein (e.g., in Table 3). In one embodiment, the anti-CD19 binding domain is a scFv comprising a light chain and a heavy chain of an amino acid sequence of Table 3. In an embodiment, the anti-CD19 binding domain (e.g., an scFv) comprises: a light chain variable region comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of an amino acid sequence of a light chain variable region provided in Table 3, or a sequence with 95-99% identity with an amino acid sequence of Table 3; and/or a heavy chain variable region comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of an amino acid sequence of a heavy chain variable region provided in Table 3, or a sequence with 95-99% identity to an amino acid sequence of Table 3. In one embodiment, the humanized anti-CD19 binding domain comprises a sequence selected from a group consisting of SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4, SEQ ID NO:5, SEQ ID NO:6, SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11, and SEQ ID NO:12, or a sequence with 95-99% identify thereof. In one embodiment, the nucleic acid sequence encoding the humanized anti-CD19 binding domain comprises a sequence selected from a group consisting of SEQ ID NO:61, SEQ ID NO:62, SEQ ID NO:63, SEQ ID NO:64, SEQ ID NO:65, SEQ ID NO:66, SEQ ID NO:67, SEQ ID NO:68, SEQ ID NO:70, SEQ ID NO:71 and SEQ ID NO:72, or a sequence with 95-99% identify thereof. In one embodiment, the humanized anti-CD19 binding domain is a scFv, and a light chain variable region comprising an amino acid sequence described herein, e.g., in Table 3, is attached to a heavy chain variable region comprising an amino acid sequence described herein, e.g., in Table 3, via a linker, e.g., a linker described herein. In one embodiment, the humanized anti-CD19 binding domain includes a (Gly.sub.4-Ser)n linker, wherein n is 1, 2, 3, 4, 5, or 6, preferably 3 or 4 (SEQ ID NO:53). The light chain variable region and heavy chain variable region of a scFv can be, e.g., in any of the following orientations: light chain variable region-linker-heavy chain variable region or heavy chain variable region-linker-light chain variable region.
[0311] In one aspect, the antigen binding domain portion comprises one or more sequence selected from SEQ ID NOS:1-12. In one aspect the humanized CAR is selected from one or more sequence selected from SEQ ID NOS: 31-42. In some aspects, a non-human antibody is humanized, where specific sequences or regions of the antibody are modified to increase similarity to an antibody naturally produced in a human or fragment thereof.
[0312] A humanized antibody can be produced using a variety of techniques known in the art, including but not limited to, CDR-grafting (see, e.g., European Patent No. EP 239,400; International Publication No. WO 91/09967; and U.S. Pat. Nos. 5,225,539, 5,530,101, and 5,585,089, each of which is incorporated herein in its entirety by reference), veneering or resurfacing (see, e.g., European Patent Nos. EP 592,106 and EP 519,596; Padlan, 1991, Molecular Immunology, 28(4/5):489-498; Studnicka et al., 1994, Protein Engineering, 7(6):805-814; and Roguska et al., 1994, PNAS, 91:969-973, each of which is incorporated herein by its entirety by reference), chain shuffling (see, e.g., U.S. Pat. No. 5,565,332, which is incorporated herein in its entirety by reference), and techniques disclosed in, e.g., U.S. Patent Application Publication No. US2005/0042664, U.S. Patent Application Publication No. US2005/0048617, U.S. Pat. Nos. 6,407,213, 5,766,886, International Publication No. WO 9317105, Tan et al., J. Immunol., 169:1119-25 (2002), Caldas et al., Protein Eng., 13(5):353-60 (2000), Morea et al., Methods, 20(3):267-79 (2000), Baca et al., J. Biol. Chem., 272(16):10678-84 (1997), Roguska et al., Protein Eng., 9(10):895-904 (1996), Couto et al., Cancer Res., 55 (23 Supp):5973s-5977s (1995), Couto et al., Cancer Res., 55(8):1717-22 (1995), Sandhu J S, Gene, 150(2):409-10 (1994), and Pedersen et al., J. Mol. Biol., 235(3):959-73 (1994), each of which is incorporated herein in its entirety by reference. Often, framework residues in the framework regions will be substituted with the corresponding residue from the CDR donor antibody to alter, for example improve, antigen binding. These framework substitutions are identified by methods well-known in the art, e.g., by modeling of the interactions of the CDR and framework residues to identify framework residues important for antigen binding and sequence comparison to identify unusual framework residues at particular positions. (See, e.g., Queen et al., U.S. Pat. No. 5,585,089; and Riechmann et al., 1988, Nature, 332:323, which are incorporated herein by reference in their entireties.)
[0313] A humanized antibody or antibody fragment has one or more amino acid residues remaining in it from a source which is nonhuman. These nonhuman amino acid residues are often referred to as "import" residues, which are typically taken from an "import" variable domain. As provided herein, humanized antibodies or antibody fragments comprise one or more CDRs from nonhuman immunoglobulin molecules and framework regions wherein the amino acid residues comprising the framework are derived completely or mostly from human germline. Multiple techniques for humanization of antibodies or antibody fragments are well-known in the art and can essentially be performed following the method of Winter and co-workers (Jones et al., Nature, 321:522-525 (1986); Riechmann et al., Nature, 332:323-327 (1988); Verhoeyen et al., Science, 239:1534-1536 (1988)), by substituting rodent CDRs or CDR sequences for the corresponding sequences of a human antibody, i.e., CDR-grafting (EP 239,400; PCT Publication No. WO 91/09967; and U.S. Pat. Nos. 4,816,567; 6,331,415; 5,225,539; 5,530,101; 5,585,089; 6,548,640, the contents of which are incorporated herein by reference herein in their entirety). In such humanized antibodies and antibody fragments, substantially less than an intact human variable domain has been substituted by the corresponding sequence from a nonhuman species. Humanized antibodies are often human antibodies in which some CDR residues and possibly some framework (FR) residues are substituted by residues from analogous sites in rodent antibodies. Humanization of antibodies and antibody fragments can also be achieved by veneering or resurfacing (EP 592,106; EP 519,596; Padlan, 1991, Molecular Immunology, 28(4/5):489-498; Studnicka et al., Protein Engineering, 7(6):805-814 (1994); and Roguska et al., PNAS, 91:969-973 (1994)) or chain shuffling (U.S. Pat. No. 5,565,332), the contents of which are incorporated herein by reference herein in their entirety.
[0314] The choice of human variable domains, both light and heavy, to be used in making the humanized antibodies is to reduce antigenicity. According to the so-called "best-fit" method, the sequence of the variable domain of a rodent antibody is screened against the entire library of known human variable-domain sequences. The human sequence which is closest to that of the rodent is then accepted as the human framework (FR) for the humanized antibody (Sims et al., J. Immunol., 151:2296 (1993); Chothia et al., J. Mol. Biol., 196:901 (1987), the contents of which are incorporated herein by reference herein in their entirety). Another method uses a particular framework derived from the consensus sequence of all human antibodies of a particular subgroup of light or heavy chains. The same framework may be used for several different humanized antibodies (see, e.g., Nicholson et al. Mol. Immun. 34 (16-17): 1157-1165 (1997); Carter et al., Proc. Natl. Acad. Sci. USA, 89:4285 (1992); Presta et al., J. Immunol., 151:2623 (1993), the contents of which are incorporated herein by reference herein in their entirety). In some embodiments, the framework region, e.g., all four framework regions, of the heavy chain variable region are derived from a VH4_4-59 germline sequence. In one embodiment, the framework region can comprise, one, two, three, four or five modifications, e.g., substitutions, e.g., from the amino acid at the corresponding murine sequence (e.g., of SEQ ID NO:59). In one embodiment, the framework region, e.g., all four framework regions of the light chain variable region are derived from a VK3_1.25 germline sequence. In one embodiment, the framework region can comprise, one, two, three, four or five modifications, e.g., substitutions, e.g., from the amino acid at the corresponding murine sequence (e.g., of SEQ ID NO:59).
[0315] In some aspects, the portion of a CAR composition of the invention that comprises an antibody fragment is humanized with retention of high affinity for the target antigen and other favorable biological properties. According to one aspect of the invention, humanized antibodies and antibody fragments are prepared by a process of analysis of the parental sequences and various conceptual humanized products using three-dimensional models of the parental and humanized sequences. Three-dimensional immunoglobulin models are commonly available and are familiar to those skilled in the art. Computer programs are available which illustrate and display probable three-dimensional conformational structures of selected candidate immunoglobulin sequences. Inspection of these displays permits analysis of the likely role of the residues in the functioning of the candidate immunoglobulin sequence, e.g., the analysis of residues that influence the ability of the candidate immunoglobulin to bind the target antigen. In this way, FR residues can be selected and combined from the recipient and import sequences so that the desired antibody or antibody fragment characteristic, such as increased affinity for the target antigen, is achieved. In general, the CDR residues are directly and most substantially involved in influencing antigen binding.
[0316] A humanized antibody or antibody fragment may retain a similar antigenic specificity as the original antibody, e.g., in the present invention, the ability to bind human CD19. In some embodiments, a humanized antibody or antibody fragment may have improved affinity and/or specificity of binding to human CD19.
[0317] In one aspect, the anti-CD19 binding domain is characterized by particular functional features or properties of an antibody or antibody fragment. For example, in one aspect, the portion of a CAR composition of the invention that comprises an antigen binding domain specifically binds human CD19. In one aspect, the antigen binding domain has the same or a similar binding specificity to human CD19 as the FMC63 scFv described in Nicholson et al. Mol. Immun. 34 (16-17): 1157-1165 (1997). In one aspect, the invention relates to an antigen binding domain comprising an antibody or antibody fragment, wherein the antibody binding domain specifically binds to a CD19 protein or fragment thereof, wherein the antibody or antibody fragment comprises a variable light chain and/or a variable heavy chain that includes an amino acid sequence of SEQ ID NO: 1-12 or SEQ ID NO:59. In one aspect, the antigen binding domain comprises an amino acid sequence of an scFv selected from SEQ ID NOs: 1-12 or SEQ ID NO:59. In certain aspects, the scFv is contiguous with and in the same reading frame as a leader sequence. In one aspect the leader sequence is the polypeptide sequence provided as SEQ ID NO:13.
[0318] In one aspect, the anti-CD19 binding domain is a fragment, e.g., a single chain variable fragment (scFv). In one aspect, the anti-CD19 binding domain is a Fv, a Fab, a (Fab')2, or a bi-functional (e.g. bi-specific) hybrid antibody (e.g., Lanzavecchia et al., Eur. J. Immunol. 17, 105 (1987)). In one aspect, the antibodies and fragments thereof of the invention binds a CD19 protein with wild-type or enhanced affinity.
[0319] In some instances, scFvs can be prepared according to method known in the art (see, for example, Bird et al., (1988) Science 242:423-426 and Huston et al., (1988) Proc. Natl. Acad. Sci. USA 85:5879-5883). ScFv molecules can be produced by linking VH and VL regions together using flexible polypeptide linkers. The scFv molecules comprise a linker (e.g., a Ser-Gly linker) with an optimized length and/or amino acid composition. The linker length can greatly affect how the variable regions of a scFv fold and interact. In fact, if a short polypeptide linker is employed (e.g., between 5-10 amino acids) intrachain folding is prevented. Interchain folding is also required to bring the two variable regions together to form a functional epitope binding site. For examples of linker orientation and size see, e.g., Hollinger et al. 1993 Proc Natl Acad. Sci. U.S.A. 90:6444-6448, U.S. Patent Application Publication Nos. 2005/0100543, 2005/0175606, 2007/0014794, and PCT publication Nos.
[0320] WO2006/020258 and WO2007/024715, is incorporated herein by reference.
[0321] A scFv can comprise a linker of at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45, 50, or more amino acid residues between its VL and VH regions. The linker sequence may comprise any naturally occurring amino acid. In some embodiments, the linker sequence comprises amino acids glycine and serine. In another embodiment, the linker sequence comprises sets of glycine and serine repeats such as (Gly4Ser)n, where n is a positive integer equal to or greater than 1 (SEQ ID NO:18). In one embodiment, the linker can be (Gly4Ser).sub.4 (SEQ ID NO: 106) or (Gly4Ser).sub.3 (SEQ ID NO: 107). Variation in the linker length may retain or enhance activity, giving rise to superior efficacy in activity studies.
[0322] In some embodiments, the amino acid sequence of the antigen binding domain (or other portions or the entire CAR) can be modified, e.g., an amino acid sequence described herein can be modified, e.g., by a conservative substitution. Families of amino acid residues having similar side chains have been defined in the art, including basic side chains (e.g., lysine, arginine, histidine), acidic side chains (e.g., aspartic acid, glutamic acid), uncharged polar side chains (e.g., glycine, asparagine, glutamine, serine, threonine, tyrosine, cysteine), nonpolar side chains (e.g., alanine, valine, leucine, isoleucine, proline, phenylalanine, methionine, tryptophan), beta-branched side chains (e.g., threonine, valine, isoleucine) and aromatic side chains (e.g., tyrosine, phenylalanine, tryptophan, histidine).
[0323] Percent identity in the context of two or more nucleic acids or polypeptide sequences, refers to two or more sequences that are the same. Two sequences are "substantially identical" if two sequences have a specified percentage of amino acid residues or nucleotides that are the same (e.g., 60% identity, optionally 70%, 71%. 72%. 73%, 74%, 75%, 76%, 77%, 78%, 79%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% identity over a specified region, or, when not specified, over the entire sequence), when compared and aligned for maximum correspondence over a comparison window, or designated region as measured using one of the following sequence comparison algorithms or by manual alignment and visual inspection. Optionally, the identity exists over a region that is at least about 50 nucleotides (or 10 amino acids) in length, or more preferably over a region that is 100 to 500 or 1000 or more nucleotides (or 20, 50, 200 or more amino acids) in length.
[0324] For sequence comparison, typically one sequence acts as a reference sequence, to which test sequences are compared. When using a sequence comparison algorithm, test and reference sequences are entered into a computer, subsequence coordinates are designated, if necessary, and sequence algorithm program parameters are designated. Default program parameters can be used, or alternative parameters can be designated. The sequence comparison algorithm then calculates the percent sequence identities for the test sequences relative to the reference sequence, based on the program parameters. Methods of alignment of sequences for comparison are well known in the art. Optimal alignment of sequences for comparison can be conducted, e.g., by the local homology algorithm of Smith and Waterman, (1970) Adv. Appl. Math. 2:482c, by the homology alignment algorithm of Needleman and Wunsch, (1970) J. Mol. Biol. 48:443, by the search for similarity method of Pearson and Lipman, (1988) Proc. Nat'l. Acad. Sci. USA 85:2444, by computerized implementations of these algorithms (GAP, BESTFIT, FASTA, and TFASTA in the Wisconsin Genetics Software Package, Genetics Computer Group, 575 Science Dr., Madison, Wis.), or by manual alignment and visual inspection (see, e.g., Brent et al., (2003) Current Protocols in Molecular Biology).
[0325] Two examples of algorithms that are suitable for determining percent sequence identity and sequence similarity are the BLAST and BLAST 2.0 algorithms, which are described in Altschul et al., (1977) Nuc. Acids Res. 25:3389-3402; and Altschul et al., (1990) J. Mol. Biol. 215:403-410, respectively. Software for performing BLAST analyses is publicly available through the National Center for Biotechnology Information.
[0326] The percent identity between two amino acid sequences can also be determined using the algorithm of E. Meyers and W. Miller, (1988) Comput. Appl. Biosci. 4:11-17) which has been incorporated into the ALIGN program (version 2.0), using a PAM120 weight residue table, a gap length penalty of 12 and a gap penalty of 4. In addition, the percent identity between two amino acid sequences can be determined using the Needleman and Wunsch (1970) J. Mol. Biol. 48:444-453) algorithm which has been incorporated into the GAP program in the GCG software package (available at www.gcg.com), using either a Blossom 62 matrix or a PAM250 matrix, and a gap weight of 16, 14, 12, 10, 8, 6, or 4 and a length weight of 1, 2, 3, 4, 5, or 6.
[0327] In one aspect, the present invention contemplates modifications of the starting antibody or fragment (e.g., scFv) amino acid sequence that generate functionally equivalent molecules. For example, the VH or VL of an anti-CD19 binding domain, e.g., scFv, comprised in the CAR can be modified to retain at least about 70%, 71%. 72%. 73%, 74%, 75%, 76%, 77%, 78%, 79%, 80%,81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% identity of the starting VH or VL framework region of the anti-CD19 binding domain, e.g., scFv. The present invention contemplates modifications of the entire CAR construct, e.g., modifications in one or more amino acid sequences of the various domains of the CAR construct in order to generate functionally equivalent molecules. The CAR construct can be modified to retain at least about 70%, 71%. 72%. 73%, 74%, 75%, 76%, 77%, 78%, 79%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% identity of the starting CAR construct.
[0328] Bispecific CARs
[0329] In an embodiment a multispecific antibody molecule is a bispecific antibody molecule. A bispecific antibody has specificity for no more than two antigens. A bispecific antibody molecule is characterized by a first immunoglobulin variable domain sequence which has binding specificity for a first epitope and a second immunoglobulin variable domain sequence that has binding specificity for a second epitope. In an embodiment the first and second epitopes are on the same antigen, e.g., the same protein (or subunit of a multimeric protein). In an embodiment the first and second epitopes overlap. In an embodiment the first and second epitopes do not overlap. In an embodiment the first and second epitopes are on different antigens, e.g., different proteins (or different subunits of a multimeric protein). In an embodiment a bispecific antibody molecule comprises a heavy chain variable domain sequence and a light chain variable domain sequence which have binding specificity for a first epitope and a heavy chain variable domain sequence and a light chain variable domain sequence which have binding specificity for a second epitope. In an embodiment a bispecific antibody molecule comprises a half antibody having binding specificity for a first epitope and a half antibody having binding specificity for a second epitope. In an embodiment a bispecific antibody molecule comprises a half antibody, or fragment thereof, having binding specificity for a first epitope and a half antibody, or fragment thereof, having binding specificity for a second epitope. In an embodiment a bispecific antibody molecule comprises a scFv, or fragment thereof, have binding specificity for a first epitope and a scFv, or fragment thereof, have binding specificity for a second epitope.
Transmembrane Domain
[0330] With respect to the transmembrane domain, in various embodiments, a CAR can be designed to comprise a transmembrane domain that is attached to the extracellular domain of the CAR. A transmembrane domain can include one or more additional amino acids adjacent to the transmembrane region, e.g., one or more amino acid associated with the extracellular region of the protein from which the transmembrane was derived (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 up to 15 amino acids of the extracellular region) and/or one or more additional amino acids associated with the intracellular region of the protein from which the transmembrane protein is derived (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 up to 15 amino acids of the intracellular region). In one aspect, the transmembrane domain is one that is associated with one of the other domains of the CAR, e.g., in one embodiment, the transmembrane domain may be from the same protein that the signaling domain, costimulatory domain or the hinge domain is derived from. In another aspect, the transmembrane domain is not derived from the same protein that any other domain of the CAR is derived from. In some instances, the transmembrane domain can be selected or modified by amino acid substitution to avoid binding of such domains to the transmembrane domains of the same or different surface membrane proteins, e.g., to minimize interactions with other members of the receptor complex. In one aspect, the transmembrane domain is capable of homodimerization with another CAR on the cell surface of a CAR-expressing cell. In a different aspect the amino acid sequence of the transmembrane domain may be modified or substituted so as to minimize interactions with the binding domains of the native binding partner present in the same CAR-expressing cell.
[0331] The transmembrane domain may be derived either from a natural or from a recombinant source. Where the source is natural, the domain may be derived from any membrane-bound or transmembrane protein. In one aspect the transmembrane domain is capable of signaling to the intracellular domain(s) whenever the CAR has bound to a target. A transmembrane domain of particular use in this invention may include at least the transmembrane region(s) of e.g., the alpha, beta or zeta chain of the T-cell receptor, CD28, CD3 epsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137, CD154. In some embodiments, a transmembrane domain may include at least the transmembrane region(s) of, e.g., KIRDS2, OX40, CD2, CD27, LFA-1 (CD11a, CD18), ICOS (CD278), 4-1BB (CD137), GITR, CD40, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), NKp44, NKp30, NKp46, CD160, CD19, IL2R beta, IL2R gamma, IL7R a, ITGA1, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, TNFR2, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), SLAMF6 (NTB-A, Ly108), SLAM (SLAMFI, CD150, IPO-3), BLAME (SLAMF8), SELPLG (CD162), LTBR, PAG/Cbp, NKG2D, NKG2C.
[0332] In some instances, the transmembrane domain can be attached to the extracellular region of the CAR, e.g., the antigen binding domain of the CAR, via a hinge, e.g., a hinge from a human protein. For example, in one embodiment, the hinge can be a human Ig (immunoglobulin) hinge, e.g., an IgG4 hinge, an IgD hinge), a GS linker (e.g., a GS linker described herein), a KIR2DS2 hinge or a CD8a hinge. In one embodiment, the hinge or spacer comprises (e.g., consists of) the amino acid sequence of SEQ ID NO: 14. In one aspect, the transmembrane domain comprises (e.g., consists of) a transmembrane domain of SEQ ID NO: 15.
[0333] In one aspect, the hinge or spacer comprises an IgG4 hinge. For example, in one embodiment, the hinge or spacer comprises a hinge of the amino acid sequence ESKYGPPCPPCPAPEFLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNW YVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPSSIEK TISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYK TTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGKM (SEQ ID NO:45). In some embodiments, the hinge or spacer comprises a hinge encoded by a nucleotide sequence of
TABLE-US-00001 (SEQ ID NO: 46) GAGAGCAAGTACGGCCCTCCCTGCCCCCCTTGCCCTGCCCCCGAGTTC CTGGGCGGACCCAGCGTGTTCCTGTTCCCCCCCAAGCCCAAGGACACC CTGATGATCAGCCGGACCCCCGAGGTGACCTGTGTGGTGGTGGACGTG TCCCAGGAGGACCCCGAGGTCCAGTTCAACTGGTACGTGGACGGCGTG GAGGTGCACAACGCCAAGACCAAGCCCCGGGAGGAGCAGTTCAATAGC ACCTACCGGGTGGTGTCCGTGCTGACCGTGCTGCACCAGGACTGGCTG AACGGCAAGGAATACAAGTGTAAGGTGTCCAACAAGGGCCTGCCCAGC AGCATCGAGAAAACCATCAGCAAGGCCAAGGGCCAGCCTCGGGAGCCC CAGGTGTACACCCTGCCCCCTAGCCAAGAGGAGATGACCAAGAACCAG GTGTCCCTGACCTGCCTGGTGAAGGGCTTCTACCCCAGCGACATCGCC GTGGAGTGGGAGAGCAACGGCCAGCCCGAGAACAACTACAAGACCACC CCCCCTGTGCTGGACAGCGACGGCAGCTTCTTCCTGTACAGCCGGCTG ACCGTGGACAAGAGCCGGTGGCAGGAGGGCAACGTCTTTAGCTGCTCC GTGATGCACGAGGCCCTGCACAACCACTACACCCAGAAGAGCCTGAGC CTGTCCCTGGGCAAGATG.
[0334] In one aspect, the hinge or spacer comprises an IgD hinge. For example, in one embodiment, the hinge or spacer comprises a hinge of the amino acid sequence RWPESPKAQASSVPTAQPQAEGSLAKATTAPATTRNTGRGGEEKKKEKEKEEQEERET KTPECPSHTQPLGVYLLTPAVQDLWLRDKATFTCFVVGSDLKDAHLTWEVAGKVPTG GVEEGLLERHSNGSQSQHSRLTLPRSLWNAGTSVTCTLNHPSLPPQRLMALREPAAQA PVKLSLNLLAS SDPPEAAS WLLCEVS GFSPPNILLMWLEDQREVNTS GFAPARPPPQPG STTFWAWSVLRVPAPPSPQPATYTCVVSHEDSRTLLNASRSLEVSYVTDH (SEQ ID NO:47). In some embodiments, the hinge or spacer comprises a hinge encoded by a nucleotide sequence of
TABLE-US-00002 (SEQ ID NO: 48) AGGTGGCCCGAAAGTCCCAAGGCCCAGGCATCTAGTGTTCCTACTGCACA GCCCCAGGCAGAAGGCAGCCTAGCCAAAGCTACTACTGCACCTGCCACTA CGCGCAATACTGGCCGTGGCGGGGAGGAGAAGAAAAAGGAGAAAGAGAAA GAAGAACAGGAAGAGAGGGAGACCAAGACCCCTGAATGTCCATCCCATAC CCAGCCGCTGGGCGTCTATCTCTTGACTCCCGCAGTACAGGACTTGTGGC TTAGAGATAAGGCCACCTTTACATGTTTCGTCGTGGGCTCTGACCTGAAG GATGCCCATTTGACTTGGGAGGTTGCCGGAAAGGTACCCACAGGGGGGGT TGAGGAAGGGTTGCTGGAGCGCCATTCCAATGGCTCTCAGAGCCAGCACT CAAGACTCACCCTTCCGAGATCCCTGTGGAACGCCGGGACCTCTGTCACA TGTACTCTAAATCATCCTAGCCTGCCCCCACAGCGTCTGATGGCCCTTAG AGAGCCAGCCGCCCAGGCACCAGTTAAGCTTAGCCTGAATCTGCTCGCCA GTAGTGATCCCCCAGAGGCCGCCAGCTGGCTCTTATGCGAAGTGTCCGGC TTTAGCCCGCCCAACATCTTGCTCATGTGGCTGGAGGACCAGCGAGAAGT GAACACCAGCGGCTTCGCTCCAGCCCGGCCCCCACCCCAGCCGGGTTCTA CCACATTCTGGGCCTGGAGTGTCTTAAGGGTCCCAGCACCACCTAGCCCC CAGCCAGCCACATACACCTGTGTTGTGTCCCATGAAGATAGCAGGACCCT GCTAAATGCTTCTAGGAGTCTGGAGGTTTCCTACGTGACTGACCATT.
[0335] In one aspect, the transmembrane domain may be recombinant, in which case it will comprise predominantly hydrophobic residues such as leucine and valine. In one aspect a triplet of phenylalanine, tryptophan and valine can be found at each end of a recombinant transmembrane domain.
[0336] Optionally, a short oligo- or polypeptide linker, between 2 and 10 amino acids in length may form the linkage between the transmembrane domain and the cytoplasmic region of the CAR. A glycine-serine doublet provides a particularly suitable linker. For example, in one aspect, the linker comprises the amino acid sequence of GGGGSGGGGS (SEQ ID NO:49). In some embodiments, the linker is encoded by a nucleotide sequence of
TABLE-US-00003 (SEQ ID NO: 50) GGTGGCGGAGGTTCTGGAGGTGGAGGTTCC.
[0337] In one aspect, the hinge or spacer comprises a KIR2DS2 hinge.
Cytoplasmic Domain
[0338] The cytoplasmic domain or region of the CAR includes an intracellular signaling domain. An intracellular signaling domain is generally responsible for activation of at least one of the normal effector functions of the immune cell in which the CAR has been introduced. The term "effector function" refers to a specialized function of a cell. Effector function of a T cell, for example, may be cytolytic activity or helper activity including the secretion of cytokines. Thus the term "intracellular signaling domain" refers to the portion of a protein which transduces the effector function signal and directs the cell to perform a specialized function. While usually the entire intracellular signaling domain can be employed, in many cases it is not necessary to use the entire chain. To the extent that a truncated portion of the intracellular signaling domain is used, such truncated portion may be used in place of the intact chain as long as it transduces the effector function signal. The term intracellular signaling domain is thus meant to include any truncated portion of the intracellular signaling domain sufficient to transduce the effector function signal.
[0339] Examples of intracellular signaling domains for use in the CAR of the invention include the cytoplasmic sequences of the T cell receptor (TCR) and co-receptors that act in concert to initiate signal transduction following antigen receptor engagement, as well as any derivative or variant of these sequences and any recombinant sequence that has the same functional capability.
[0340] It is known that signals generated through the TCR alone are insufficient for full activation of the T cell and that a secondary and/or costimulatory signal is also required. Thus, T cell activation can be said to be mediated by two distinct classes of cytoplasmic signaling sequences: those that initiate antigen-dependent primary activation through the TCR (primary intracellular signaling domains) and those that act in an antigen-independent manner to provide a secondary or costimulatory signal (secondary cytoplasmic domain, e.g., a costimulatory domain).
[0341] A primary signaling domain regulates primary activation of the TCR complex either in a stimulatory way, or in an inhibitory way. Primary intracellular signaling domains that act in a stimulatory manner may contain signaling motifs which are known as immunoreceptor tyrosine-based activation motifs or ITAMs.
[0342] Examples of ITAM containing primary intracellular signaling domains that are of particular use in the invention include those of CD3 zeta, common FcR gamma (FCER1G), Fc gamma RIIa, FcR beta (Fc Epsilon R1b), CD3 gamma, CD3 delta, CD3 epsilon, CD79a, CD79b, DAP10, and DAP12. In one embodiment, a CAR of the invention comprises an intracellular signaling domain, e.g., a primary signaling domain of CD3-zeta.
[0343] In one embodiment, a primary signaling domain comprises a modified ITAM domain, e.g., a mutated ITAM domain which has altered (e.g., increased or decreased) activity as compared to the native ITAM domain. In one embodiment, a primary signaling domain comprises a modified ITAM-containing primary intracellular signaling domain, e.g., an optimized and/or truncated ITAM-containing primary intracellular signaling domain. In an embodiment, a primary signaling domain comprises one, two, three, four or more ITAM motifs.
[0344] Further examples of molecules containing a primary intracellular signaling domain that are of particular use in the invention include those of DAP10, DAP12, and CD32.
[0345] The intracellular signalling domain of the CAR can comprise the CD3-zeta signaling domain by itself or it can be combined with any other desired intracellular signaling domain(s) useful in the context of a CAR of the invention. For example, the intracellular signaling domain of the CAR can comprise a CD3 zeta chain portion and a costimulatory signaling domain. The costimulatory signaling domain refers to a portion of the CAR comprising the intracellular domain of a costimulatory molecule. A costimulatory molecule is a cell surface molecule other than an antigen receptor or its ligands that is required for an efficient response of lymphocytes to an antigen. Examples of such molecules include CD27, CD28, 4-1BB (CD137), OX40, CD30, CD40, PD1, ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, B7-H3, and a ligand that specifically binds with CD83, and the like. For example, CD27 costimulation has been demonstrated to enhance expansion, effector function, and survival of human CART cells in vitro and augments human T cell persistence and antitumor activity in vivo (Song et al. Blood. 2012; 119(3):696-706). Further examples of such costimulatory molecules include CDS, ICAM-1, GITR, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), NKp44, NKp30, NKp46, CD160, CD19, CD4, CD8alpha, CD8beta, IL2R beta, IL2R gamma, IL7R alpha, ITGA4, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, TNFR2, TRANCE/RANKL, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), NKG2D, CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), CD69, SLAMF6 (NTB-A, Ly108), SLAM (SLAMFI, CD150, IPO-3), BLAME (SLAMF8), SELPLG (CD162), LTBR, LAT, GADS, SLP-76, PAG/Cbp, and CD19a.
[0346] The intracellular signaling sequences within the cytoplasmic portion of the CAR of the invention may be linked to each other in a random or specified order. Optionally, a short oligo- or polypeptide linker, for example, between 2 and 10 amino acids (e.g., 2, 3, 4, 5, 6, 7, 8, 9, or 10 amino acids) in length may form the linkage between intracellular signaling sequence. In one embodiment, a glycine-serine doublet can be used as a suitable linker. In one embodiment, a single amino acid, e.g., an alanine, a glycine, can be used as a suitable linker.
[0347] In one aspect, the intracellular signaling domain is designed to comprise two or more, e.g., 2, 3, 4, 5, or more, costimulatory signaling domains. In an embodiment, the two or more, e.g., 2, 3, 4, 5, or more, costimulatory signaling domains, are separated by a linker molecule, e.g., a linker molecule described herein. In one embodiment, the intracellular signaling domain comprises two costimulatory signaling domains. In some embodiments, the linker molecule is a glycine residue. In some embodiments, the linker is an alanine residue.
[0348] In one aspect, the intracellular signaling domain is designed to comprise the signaling domain of CD3-zeta and the signaling domain of CD28. In one aspect, the intracellular signaling domain is designed to comprise the signaling domain of CD3-zeta and the signaling domain of 4-1BB. In one aspect, the signaling domain of 4-1BB is a signaling domain of SEQ ID NO: 16. In one aspect, the signaling domain of CD3-zeta is a signaling domain of SEQ ID NO: 17.
[0349] In one aspect, the intracellular signaling domain is designed to comprise the signaling domain of CD3-zeta and the signaling domain of CD27. In one aspect, the signaling domain of CD27 comprises an amino acid sequence of QRRKYRSNKGESPVEPAEPCRYSCPREEEGSTIPIQEDYRKPEPACSP (SEQ ID NO:51). In one aspect, the signalling domain of CD27 is encoded by a nucleic acid sequence of
TABLE-US-00004 (SEQ ID NO: 52) AGGAGTAAGAGGAGCAGGCTCCTGCACAGTGACTACATGAACATGACTCC CCGCCGCCCCGGGCCCACCCGCAAGCATTACCAGCCCTATGCCCCACCAC GCGACTTCGCAGCCTATCGCTCC.
[0350] In one aspect, the CAR-expressing cell described herein can further comprise a second CAR, e.g., a second CAR that includes a different antigen binding domain, e.g., to the same target (CD19) or a different target (e.g., CD123 or mesothelin). In one embodiment, when the CAR-expressing cell comprises two or more different CARs, the antigen binding domains of the different CARs can be such that the antigen binding domains do not interact with one another. For example, a cell expressing a first and second CAR can have an antigen binding domain of the first CAR, e.g., as a fragment, e.g., an scFv, that does not form an association with the antigen binding domain of the second CAR, e.g., the antigen binding domain of the second CAR is a VHH.
[0351] In another aspect, the CAR-expressing cell described herein can further express another agent, e.g., an agent which enhances the activity of a CAR-expressing cell. For example, in one embodiment, the agent can be an agent which inhibits an inhibitory molecule. Inhibitory molecules, e.g., PD1, can, in some embodiments, decrease the ability of a CAR-expressing cell to mount an immune effector response. Examples of inhibitory molecules include PD1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 and TGFR beta. In one embodiment, the agent which inhibits an inhibitory molecule comprises a first polypeptide, e.g., an inhibitory molecule, associated with a second polypeptide that provides a positive signal to the cell, e.g., an intracellular signaling domain described herein. In one embodiment, the agent comprises a first polypeptide, e.g., of an inhibitory molecule such as PD1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 or TGFR beta, or a fragment of any of these (e.g., at least a portion of an extracellular domain of any of these), and a second polypeptide which is an intracellular signaling domain described herein (e.g., comprising a costimulatory domain (e.g., 41BB, CD27 or CD28, e.g., as described herein) and/or a primary signaling domain (e.g., a CD3 zeta signaling domain described herein). In one embodiment, the agent comprises a first polypeptide of PD1 or a fragment thereof (e.g., at least a portion of an extracellular domain of PD1), and a second polypeptide of an intracellular signaling domain described herein (e.g., a CD28 signaling domain described herein and/or a CD3 zeta signaling domain described herein). PD1 is an inhibitory member of the CD28 family of receptors that also includes CD28, CTLA-4, ICOS, and BTLA. PD-1 is expressed on activated B cells, T cells and myeloid cells (Agata et al. 1996 Int. Immunol 8:765-75). Two ligands for PD1, PD-L1 and PD-L2 have been shown to downregulate T cell activation upon binding to PD1 (Freeman et a. 2000 J Exp Med 192:1027-34; Latchman et al. 2001 Nat Immunol 2:261-8; Carter et al. 2002 Eur J Immunol 32:634-43). PD-L1 is abundant in human cancers (Dong et al. 2003 J Mol Med 81:281-7; Blank et al. 2005 Cancer Immunol. Immunother 54:307-314; Konishi et al. 2004 Clin Cancer Res 10:5094). Immune suppression can be reversed by inhibiting the local interaction of PD1 with PD-L1.
[0352] In one embodiment, the agent comprises the extracellular domain (ECD) of an inhibitory molecule, e.g., Programmed Death 1 (PD1), can be fused to a transmembrane domain and intracellular signaling domains such as 41BB and CD3 zeta (also referred to herein as a PD1 CAR). In one embodiment, the PD1 CAR, when used in combinations with a CD19 CAR described herein, improves the persistence of the T cell. In one embodiment, the CAR is a PD1 CAR comprising the extracellular domain of PD1 indicated as underlined in SEQ ID NO: 121. In one embodiment, the PD1 CAR comprises the amino acid sequence of SEQ ID NO:121.
TABLE-US-00005 (SEQ ID NO: 121) Malpvtalllplalllhaarppgwfldspdrpwnpptfspallvvtegdn atftcsfsntsesfvlnwyrmspsnqtdklaafpedrsqpgqdcrfrvtq lpngrdfhmsvvrarrndsgtylcgaislapkaqikeslraelrvterra evptahpspsprpagqfqtlvtttpaprpptpaptiasqplslrpeacrp aaggavhtrgldfacdiyiwaplagtcgvlllslvitlyckrgrkkllyi fkqpfmrpvqttqeedgcscrfpeeeeggcelrvkfsrsadapaykqgqn qlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkma eayseigmkgerrrgkghdglyqglstatkdtydalhmqalppr.
[0353] In one embodiment, the PD1 CAR comprises the amino acid sequence provided below (SEQ ID NO:119).
TABLE-US-00006 (SEQ ID NO: 119) pgwfldspdrpwnpptfspallvvtegdnatftcsfsntsesfvlnwyrm spsnqtdklaafpedrsqpgqdcrfrvtqlpngrdfhmsvvrarrndsgt ylcgaislapkaqikeslraelrvterraevptahpspsprpagqfqtlv tttpaprpptpaptiasqplslrpeacrpaaggavhtrgldfacdiyiwa plagtcgvlllslvitlyckrgrkkllyifkqpfmrpvqttqeedgcscr fpeeeeggcelrvkfsrsadapaykqgqnqlynelnlgrreeydvldkrr grdpemggkprrknpqeglynelqkdkmaeayseigmkgerrrgkghdgl yqglstatkdtydalhmqalppr.
[0354] In one embodiment, the agent comprises a nucleic acid sequence encoding the PD1 CAR, e.g., the PD1 CAR described herein. In one embodiment, the nucleic acid sequence for the PD1 CAR is shown below, with the PD1 ECD underlined below in SEQ ID NO: 120
TABLE-US-00007 (SEQ ID NO: 120) atggccctccctgtcactgccctgcttctccccctcgcactcctgctcca cgccgctagaccacccggatggtttctggactctccggatcgcccgtgga atcccccaaccttctcaccggcactcttggttgtgactgagggcgataat gcgaccttcacgtgctcgttctccaacacctccgaatcattcgtgctgaa ctggtaccgcatgagcccgtcaaaccagaccgacaagctcgccgcgtttc cggaagatcggtcgcaaccgggacaggattgtcggttccgcgtgactcaa ctgccgaatggcagagacttccacatgagcgtggtccgcgctaggcgaaa cgactccgggacctacctgtgcggagccatctcgctggcgcctaaggccc aaatcaaagagagcttgagggccgaactgagagtgaccgagcgcagagct gaggtgccaactgcacatccatccccatcgcctcggcctgcggggcagtt tcagaccctggtcacgaccactccggcgccgcgcccaccgactccggccc caactatcgcgagccagcccctgtcgctgaggccggaagcatgccgccct gccgccggaggtgctgtgcatacccggggattggacttcgcatgcgacat ctacatttgggctcctctcgccggaacttgtggcgtgctccttctgtccc tggtcatcaccctgtactgcaagcggggtcggaaaaagcttctgtacatt ttcaagcagcccttcatgaggcccgtgcaaaccacccaggaggaggacgg ttgctcctgccggttccccgaagaggaagaaggaggttgcgagctgcgcg tgaagttctcccggagcgccgacgcccccgcctataagcagggccagaac cagctgtacaacgaactgaacctgggacggcgggaagagtacgatgtgct ggacaagcggcgcggccgggaccccgaaatgggcgggaagcctagaagaa agaaccctcaggaaggcctgtataacgagctgcagaaggacaagatggcc gaggcctactccgaaattgggatgaagggagagcggcggaggggaaaggg gcacgacggcctgtaccaaggactgtccaccgccaccaaggacacatacg atgccctgcacatgcaggcccttccccctcgc.
[0355] In another aspect, the present invention provides a population of CAR-expressing cells, e.g., CART cells. In some embodiments, the population of CAR-expressing cells comprises a mixture of cells expressing different CARs. For example, in one embodiment, the population of CAR-expressing cells can include a first cell expressing a CAR having an anti-CD19 binding domain described herein, and a second cell expressing a CAR having a different anti-CD19 binding domain, e.g., an anti-CD19 binding domain described herein that differs from the anti-CD19 binding domain in the CAR expressed by the first cell. As another example, the population of CAR-expressing cells can include a first cell expressing a CAR that includes an anti-CD19 binding domain, e.g., as described herein, and a second cell expressing a CAR that includes an antigen binding domain to a target other than CD19 (e.g., CD123). In one embodiment, the population of CAR-expressing cells includes, e.g., a first cell expressing a CAR that includes a primary intracellular signaling domain, and a second cell expressing a CAR that includes a secondary signaling domain.
[0356] In another aspect, the present invention provides a population of cells wherein at least one cell in the population expresses a CAR having an anti-CD19 binding domain described herein, and a second cell expressing another agent, e.g., an agent which enhances the activity of a CAR-expressing cell. For example, in one embodiment, the agent can be an agent which inhibits an inhibitory molecule. Inhibitory molecules, e.g., can, in some embodiments, decrease the ability of a CAR-expressing cell to mount an immune effector response. Examples of inhibitory molecules include PD1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 or TGFR beta. In one embodiment, the agent which inhibits an inhibitory molecule comprises a first polypeptide, e.g., an inhibitory molecule, associated with a second polypeptide that provides a positive signal to the cell, e.g., an intracellular signaling domain described herein. In one embodiment, the agent comprises a first polypeptide, e.g., of an inhibitory molecule such as PD1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 or TGFR beta, or a fragment of any of these (e.g., at least a portion of an extracellular domain of any of these), and a second polypeptide which is an intracellular signaling domain described herein (e.g., comprising a costimulatory domain (e.g., 41BB, CD27 or CD28, e.g., as described herein) and/or a primary signaling domain (e.g., a CD3 zeta signaling domain described herein). In one embodiment, the agent comprises a first polypeptide of PD1 or a fragment thereof (e.g., at least a portion of the extracellular domain of PD1), and a second polypeptide of an intracellular signaling domain described herein (e.g., a CD28 signaling domain described herein and/or a CD3 zeta signaling domain described herein).
Regulatable Chimeric Antigen Receptors
[0357] In some embodiments, a regulatable CAR (RCAR) where the CAR activity can be controlled is desirable to optimize the safety and efficacy of a CAR therapy. There are many ways CAR activities can be regulated. For example, inducible apoptosis using, e.g., a caspase fused to a dimerization domain (see, e.g., Di Stasa et al., N Engl. J. Med. 2011 Nov. 3; 365(18):1673-1683), can be used as a safety switch in the CAR therapy of the instant invention. In one embodiment, the cells (e.g., T cells or NK cells) expressing a CAR of the present invention further comprise an inducible apoptosis switch, wherein a human caspase (e.g., caspase 9) or a modified version is fused to a modification of the human FKB protein that allows conditional dimerization. In the presence of a small molecule, such as a rapalog (e.g., AP 1903, AP20187), the inducible caspase (e.g., caspase 9) is activated and leads to the rapid apoptosis and death of the cells (e.g., T cells or NK cells) expressing a CAR of the present invention. Examples of a caspase-based inducible apoptosis switch (or one or more aspects of such a switch) have been described in, e.g., US2004040047; US20110286980; US20140255360; WO1997031899; WO2014151960; WO2014164348; WO2014197638; WO2014197638; all of which are incorporated by reference herein.
[0358] In an aspect, a RCAR comprises a set of polypeptides, typically two in the simplest embodiments, in which the components of a standard CAR described herein, e.g., an antigen binding domain and an intracellular signaling domain, are partitioned on separate polypeptides or members. In some embodiments, the set of polypeptides include a dimerization switch that, upon the presence of a dimerization molecule, can couple the polypeptides to one another, e.g., can couple an antigen binding domain to an intracellular signaling domain. In one embodiment, the CARs of the present invention utilizes a dimerization switch as those described in, e.g., WO2014127261, which is incorporated by reference herein.
[0359] In an aspect, an RCAR comprises two polypeptides or members: 1) an intracellular signaling member comprising an intracellular signaling domain, e.g., a primary intracellular signaling domain described herein, and a first switch domain; 2) an antigen binding member comprising an antigen binding domain, e.g., that targets CD19, as described herein and a second switch domain. Optionally, the RCAR comprises a transmembrane domain described herein. In an embodiment, a transmembrane domain can be disposed on the intracellular signaling member, on the antigen binding member, or on both. (Unless otherwise indicated, when members or elements of an RCAR are described herein, the order can be as provided, but other orders are included as well. In other words, in an embodiment, the order is as set out in the text, but in other embodiments, the order can be different. E.g., the order of elements on one side of a transmembrane region can be different from the example, e.g., the placement of a switch domain relative to a intracellular signaling domain can be different, e.g., reversed).
[0360] In an embodiment, the first and second switch domains can form an intracellular or an extracellular dimerization switch. In an embodiment, the dimerization switch can be a homodimerization switch, e.g., where the first and second switch domain are the same, or a heterodimerization switch, e.g., where the first and second switch domain are different from one another.
[0361] In embodiments, an RCAR can comprise a "multi switch." A multi switch can comprise heterodimerization switch domains or homodimerization switch domains. A multi switch comprises a plurality of, e.g., 2, 3, 4, 5, 6, 7, 8, 9, or 10, switch domains, independently, on a first member, e.g., an antigen binding member, and a second member, e.g., an intracellular signaling member. In an embodiment, the first member can comprise a plurality of first switch domains, e.g., FKBP-based switch domains, and the second member can comprise a plurality of second switch domains, e.g., FRB-based switch domains. In an embodiment, the first member can comprise a first and a second switch domain, e.g., a FKBP-based switch domain and a FRB-based switch domain, and the second member can comprise a first and a second switch domain, e.g., a FKBP-based switch domain and a FRB-based switch domain.
[0362] In an embodiment, the intracellular signaling member comprises one or more intracellular signaling domains, e.g., a primary intracellular signaling domain and one or more costimulatory signaling domains.
[0363] In an embodiment, the antigen binding member may comprise one or more intracellular signaling domains, e.g., one or more costimulatory signaling domains. In an embodiment, the antigen binding member comprises a plurality, e.g., 2 or 3 costimulatory signaling domains described herein, e.g., selected from 41BB, CD28, CD27, ICOS, and OX40, and in embodiments, no primary intracellular signaling domain. In an embodiment, the antigen binding member comprises the following costimulatory signaling domains, from the extracellular to intracellular direction: 41BB-CD27; 41BB-CD27; CD27-41BB; 41BB-CD28; CD28-41BB; OX40-CD28; CD28-OX40; CD28-41BB; or 41BB-CD28. In such embodiments, the intracellular binding member comprises a CD3zeta domain. In one such embodiment the RCAR comprises (1) an antigen binding member comprising, an antigen binding domain, a transmembrane domain, and two costimulatory domains and a first switch domain; and (2) an intracellular signaling domain comprising a transmembrane domain or membrane tethering domain and at least one primary intracellular signaling domain, and a second switch domain.
[0364] An embodiment provides RCARs wherein the antigen binding member is not tethered to the surface of the CAR cell. This allows a cell having an intracellular signaling member to be conveniently paired with one or more antigen binding domains, without transforming the cell with a sequence that encodes the antigen binding member. In such embodiments, the RCAR comprises: 1) an intracellular signaling member comprising: a first switch domain, a transmembrane domain, an intracellular signaling domain, e.g., a primary intracellular signaling domain, and a first switch domain; and 2) an antigen binding member comprising: an antigen binding domain, and a second switch domain, wherein the antigen binding member does not comprise a transmembrane domain or membrane tethering domain, and, optionally, does not comprise an intracellular signaling domain. In some embodiments, the RCAR may further comprise 3) a second antigen binding member comprising: a second antigen binding domain, e.g., a second antigen binding domain that binds a different antigen than is bound by the antigen binding domain; and a second switch domain.
[0365] Also provided herein are RCARs wherein the antigen binding member comprises bispecific activation and targeting capacity. In this embodiment, the antigen binding member can comprise a plurality, e.g., 2, 3, 4, or 5 antigen binding domains, e.g., scFvs, wherein each antigen binding domain binds to a target antigen, e.g. different antigens or the same antigen, e.g., the same or different epitopes on the same antigen. In an embodiment, the plurality of antigen binding domains are in tandem, and optionally, a linker or hinge region is disposed between each of the antigen binding domains. Suitable linkers and hinge regions are described herein.
[0366] An embodiment provides RCARs having a configuration that allows switching of proliferation. In this embodiment, the RCAR comprises: 1) an intracellular signaling member comprising: optionally, a transmembrane domain or membrane tethering domain; one or more co-stimulatory signaling domain, e.g., selected from 41BB, CD28, CD27, ICOS, and OX40, and a switch domain; and 2) an antigen binding member comprising: an antigen binding domain, a transmembrane domain, and a primary intracellular signaling domain, e.g., a CD3zeta domain, wherein the antigen binding member does not comprise a switch domain, or does not comprise a switch domain that dimerizes with a switch domain on the intracellular signaling member. In an embodiment, the antigen binding member does not comprise a co-stimulatory signaling domain. In an embodiment, the intracellular signaling member comprises a switch domain from a homodimerization switch. In an embodiment, the intracellular signaling member comprises a first switch domain of a heterodimerization switch and the RCAR comprises a second intracellular signaling member which comprises a second switch domain of the heterodimerization switch. In such embodiments, the second intracellular signaling member comprises the same intracellular signaling domains as the intracellular signaling member. In an embodiment, the dimerization switch is intracellular. In an embodiment, the dimerization switch is extracellular.
[0367] In any of the RCAR configurations described here, the first and second switch domains comprise a FKBP-FRB based switch as described herein.
[0368] Also provided herein are cells comprising an RCAR described herein. Any cell that is engineered to express a RCAR can be used as a RCARX cell. In an embodiment the RCARX cell is a T cell, and is referred to as a RCART cell. In an embodiment the RCARX cell is an NK cell, and is referred to as a RCARN cell.
[0369] Also provided herein are nucleic acids and vectors comprising RCAR encoding sequences. Sequence encoding various elements of an RCAR can be disposed on the same nucleic acid molecule, e.g., the same plasmid or vector, e.g., viral vector, e.g., lentiviral vector. In an embodiment, (i) sequence encoding an antigen binding member and (ii) sequence encoding an intracellular signaling member, can be present on the same nucleic acid, e.g., vector. Production of the corresponding proteins can be achieved, e.g., by the use of separate promoters, or by the use of a bicistronic transcription product (which can result in the production of two proteins by cleavage of a single translation product or by the translation of two separate protein products). In an embodiment, a sequence encoding a cleavable peptide, e.g., a P2A or F2A sequence, is disposed between (i) and (ii). In an embodiment, a sequence encoding an IRES, e.g., an EMCV or EV71 IRES, is disposed between (i) and (ii). In these embodiments, (i) and (ii) are transcribed as a single RNA. In an embodiment, a first promoter is operably linked to (i) and a second promoter is operably linked to (ii), such that (i) and (ii) are transcribed as separate mRNAs.
[0370] Alternatively, the sequence encoding various elements of an RCAR can be disposed on the different nucleic acid molecules, e.g., different plasmids or vectors, e.g., viral vector, e.g., lentiviral vector. E.g., the (i) sequence encoding an antigen binding member can be present on a first nucleic acid, e.g., a first vector, and the (ii) sequence encoding an intracellular signaling member can be present on the second nucleic acid, e.g., the second vector.
Dimerization Switches
[0371] Dimerization switches can be non-covalent or covalent. In a non-covalent dimerization switch, the dimerization molecule promotes a non-covalent interaction between the switch domains. In a covalent dimerization switch, the dimerization molecule promotes a covalent interaction between the switch domains.
[0372] In an embodiment, the RCAR comprises a FKBP/FRAP, or FKBP/FRB,-based dimerization switch. FKBP12 (FKBP, or FK506 binding protein) is an abundant cytoplasmic protein that serves as the initial intracellular target for the natural product immunosuppressive drug, rapamycin. Rapamycin binds to FKBP and to the large PI3K homolog FRAP (RAFT, mTOR). FRB is a 93 amino acid portion of FRAP, that is sufficient for binding the FKBP-rapamycin complex (Chen, J., Zheng, X. F., Brown, E. J. & Schreiber, S. L. (1995) Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc Natl Acad Sci USA 92: 4947-51.)
[0373] In embodiments, an FKBP/FRAP, e.g., an FKBP/FRB, based switch can use a dimerization molecule, e.g., rapamycin or a rapamycin analog.
[0374] The amino acid sequence of FKBP is as follows:
TABLE-US-00008 (SEQ ID NO: 122) D V P D Y A S L G G P S S P K K K R K V S R G V Q V E T I S P G D G R T F P K R G Q T C V V H Y T G M L E D G K K F D S S R D R N K P F K F M L G K Q E V I R G W E E G V A Q M S V G Q R A K L T I S P D Y A Y G A T G H P G I I P P H A T L V F D V E L L K L E T S Y
[0375] In embodiments, an FKBP switch domain can comprise a fragment of FKBP having the ability to bind with FRB, or a fragment or analog thereof, in the presence of rapamycin or a rapalog, e.g., the underlined portion of SEQ ID NO: 122, which is:
TABLE-US-00009 (SEQ ID NO: 123) V Q V E T I S P G D G R T F P K R G Q T C V V H Y T G M L E D G K K F D S S R D R N K P F K F M L G K Q E V I R G W E E G V A Q M S V G Q R A K L T I S P D Y A Y G A T G H P G I I P P H A T L V F D V E L L K L E T S
[0376] The amino acid sequence of FRB is as follows:
TABLE-US-00010 (SEQ ID NO: 124) ILWHEMWHEG LEEASRLYFG ERNVKGMFEV LEPLHAMMER GPQTLKETSF NQAYGRDLME AQEWCRKYMK SGNVKDLTQA WDLYYHVFRR ISK
[0377] "FKBP/FRAP, e.g., an FKBP/FRB, based switch" as that term is used herein, refers to a dimerization switch comprising: a first switch domain, which comprises an FKBP fragment or analog thereof having the ability to bind with FRB, or a fragment or analog thereof, in the presence of rapamycin or a rapalog, e.g., RAD001, and has at least 70, 75, 80, 85, 90, 95, 96, 97, 98, or 99% identity with, or differs by no more than 30, 25, 20, 15, 10, 5, 4, 3, 2, or 1 amino acid residues from, the FKBP sequence of SEQ ID NO: 122 or 123; and a second switch domain, which comprises an FRB fragment or analog thereof having the ability to bind with FRB, or a fragment or analog thereof, in the presence of rapamycin or a rapalog, and has at least 70, 75, 80, 85, 90, 95, 96, 97, 98, or 99% identity with, or differs by no more than 30, 25, 20, 15, 10, 5, 4, 3, 2, or 1 amino acid residues from, the FRB sequence of SEQ ID NO: 124. In an embodiment, a RCAR described herein comprises one switch domain comprises amino acid residues disclosed in SEQ ID NO: 122 (or SEQ ID NO: 123), and one switch domain comprises amino acid residues disclosed in SEQ ID NO: 124.
[0378] In embodiments, the FKBP/FRB dimerization switch comprises a modified FRB switch domain that exhibits altered, e.g., enhanced, complex formation between an FRB-based switch domain, e.g., the modified FRB switch domain, a FKBP-based switch domain, and the dimerization molecule, e.g., rapamycin or a rapalogue, e.g., RAD001. In an embodiment, the modified FRB switch domain comprises one or more mutations, e.g., 2, 3, 4, 5, 6, 7, 8, 9, 10 or more, selected from mutations at amino acid position(s) L2031, E2032, S2035, R2036, F2039, G2040, T2098, W2101, D2102, Y2105, and F2108, where the wild-type amino acid is mutated to any other naturally-occurring amino acid. In an embodiment, a mutant FRB comprises a mutation at E2032, where E2032 is mutated to phenylalanine (E2032F), methionine (E2032M), arginine (E2032R), valine (E2032V), tyrosine (E2032Y), isoleucine (E2032I), e.g., SEQ ID NO: 125, or leucine (E2032L), e.g., SEQ ID NO: 126. In an embodiment, a mutant FRB comprises a mutation at T2098, where T2098 is mutated to phenylalanine (T2098F) or leucine (T2098L), e.g., SEQ ID NO: 127. In an embodiment, a mutant FRB comprises a mutation at E2032 and at T2098, where E2032 is mutated to any amino acid, and where T2098 is mutated to any amino acid, e.g., SEQ ID NO: 128. In an embodiment, a mutant FRB comprises an E2032I and a T2098L mutation, e.g., SEQ ID NO: 129. In an embodiment, a mutant FRB comprises an E2032L and a T2098L mutation, e.g., SEQ ID NO: 130.
TABLE-US-00011 TABLE 14 Exemplary mutant FRB having increased affinity for a dimerization molecule. SEQ ID FRB mutant Amino Acid Sequence NO: E2032I mutant ILWHEMWHEGLIEASRLYFGERNVKGMFEVLEPLHAMMERGPQTLKETSFNQAYGR 125 DLMEAQEWCRKYMKSGNVKDLTQAWDLYYHVFRRISKTS E2032L mutant ILWHEMWHEGLLEASRLYFGERNVKGMFEVLEPLHAMMERGPQTLKETSFNQAYGR 126 DLMEAQEWCRKYMKSGNVKDLTQAWDLYYHVFRRISKTS 12098L mutant ILWHEMWHEGLEEASRLYFGERNVKGMFEVLEPLHAMMERGPQTLKETSFNQAYGR 127 DLMEAQEWCRKYMKSGNVKDLLQAWDLYYHVFRRISKTS E2032, T2098 ILWHEMWHEGLXEASRLYFGERNVKGMFEVLEPLHAMMERGPQTLKETSFNQAYGR 128 mutant DLMEAQEWCRKYMKSGNVKDLXQAWDLYYHVFRRISKTS E2032I, T2098L ILWHEMWHEGLIEASRLYFGERNVKGMFEVLEPLHAMMERGPQTLKETSFNQAYGR 129 mutant DLMEAQEWCRKYMKSGNVKDLLQAWDLYYHVFRRISKTS E2032L, T2098L ILWHEMWHEGLLEASRLYFGERNVKGMFEVLEPLHAMMERGPQTLKETSFNQAYGR 130 mutant DLMEAQEWCRKYMKSGNVKDLLQAWDLYYHVFRRISKTS
[0379] Other suitable dimerization switches include a GyrB-GyrB based dimerization switch, a Gibberellin-based dimerization switch, a tag/binder dimerization switch, and a halo-tag/snap-tag dimerization switch. Following the guidance provided herein, such switches and relevant dimerization molecules will be apparent to one of ordinary skill.
Dimerization Molecule
[0380] Association between the switch domains is promoted by the dimerization molecule. In the presence of dimerization molecule interaction or association between switch domains allows for signal transduction between a polypeptide associated with, e.g., fused to, a first switch domain, and a polypeptide associated with, e.g., fused to, a second switch domain. In the presence of non-limiting levels of dimerization molecule signal transduction is increased by 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2, 5, 10, 50, 100 fold, e.g., as measured in a system described herein.
[0381] Rapamycin and rapamycin analogs (sometimes referred to as rapalogues), e.g., RAD001, can be used as dimerization molecules in a FKBP/FRB-based dimerization switch described herein. In an embodiment the dimerization molecule can be selected from rapamycin (sirolimus), RAD001 (everolimus), zotarolimus, temsirolimus, AP-23573 (ridaforolimus), biolimus and AP21967. Additional rapamycin analogs suitable for use with FKBP/FRB-based dimerization switches are further described in the section entitled "Combination Therapies", or in the subsection entitled "Exemplary mTOR inhibitors".
Split CAR
[0382] In some embodiments, the CAR-expressing cell uses a split CAR. The split CAR approach is described in more detail in publications WO2014/055442 and WO2014/055657. Briefly, a split CAR system comprises a cell expressing a first CAR having a first antigen binding domain and a costimulatory domain (e.g., 41BB), and the cell also expresses a second CAR having a second antigen binding domain and an intracellular signaling domain (e.g., CD3 zeta). When the cell encounters the first antigen, the costimulatory domain is activated, and the cell proliferates. When the cell encounters the second antigen, the intracellular signaling domain is activated and cell-killing activity begins. Thus, the CAR-expressing cell is only fully activated in the presence of both antigens.
RNA Transfection
[0383] Disclosed herein are methods for producing an in vitro transcribed RNA CAR. The present invention also includes a CAR encoding RNA construct that can be directly transfected into a cell. A method for generating mRNA for use in transfection can involve in vitro transcription (IVT) of a template with specially designed primers, followed by polyA addition, to produce a construct containing 3' and 5' untranslated sequence ("UTR"), a 5' cap and/or Internal Ribosome Entry Site (IRES), the nucleic acid to be expressed, and a polyA tail, typically 50-2000 bases in length (SEQ ID NO: 118). RNA so produced can efficiently transfect different kinds of cells. In one aspect, the template includes sequences for the CAR.
[0384] In one aspect the anti-CD19 CAR is encoded by a messenger RNA (mRNA). In one aspect, the mRNA encoding the anti-CD19 CAR is introduced into an immune effector cell, e.g., a T cell or a NK cell, for production of a CAR-expressing cell, e.g., a CART cell or a CAR NK cell.
[0385] In one embodiment, the in vitro transcribed RNA CAR can be introduced to a cell as a form of transient transfection. The RNA is produced by in vitro transcription using a polymerase chain reaction (PCR)-generated template. DNA of interest from any source can be directly converted by PCR into a template for in vitro mRNA synthesis using appropriate primers and RNA polymerase. The source of the DNA can be, for example, genomic DNA, plasmid DNA, phage DNA, cDNA, synthetic DNA sequence or any other appropriate source of DNA. The desired temple for in vitro transcription is a CAR of the present invention. For example, the template for the RNA CAR comprises an extracellular region comprising a single chain variable domain of an anti-tumor antibody; a hinge region, a transmembrane domain (e.g., a transmembrane domain of CD8a); and a cytoplasmic region that includes an intracellular signaling domain, e.g., comprising the signaling domain of CD3-zeta and the signaling domain of 4-1BB.
[0386] In one embodiment, the DNA to be used for PCR contains an open reading frame. The DNA can be from a naturally occurring DNA sequence from the genome of an organism. In one embodiment, the nucleic acid can include some or all of the 5' and/or 3' untranslated regions (UTRs). The nucleic acid can include exons and introns. In one embodiment, the DNA to be used for PCR is a human nucleic acid sequence. In another embodiment, the DNA to be used for PCR is a human nucleic acid sequence including the 5' and 3' UTRs. The DNA can alternatively be an artificial DNA sequence that is not normally expressed in a naturally occurring organism. An exemplary artificial DNA sequence is one that contains portions of genes that are ligated together to form an open reading frame that encodes a fusion protein. The portions of DNA that are ligated together can be from a single organism or from more than one organism.
[0387] PCR is used to generate a template for in vitro transcription of mRNA which is used for transfection. Methods for performing PCR are well known in the art. Primers for use in PCR are designed to have regions that are substantially complementary to regions of the DNA to be used as a template for the PCR. "Substantially complementary," as used herein, refers to sequences of nucleotides where a majority or all of the bases in the primer sequence are complementary, or one or more bases are non-complementary, or mismatched. Substantially complementary sequences are able to anneal or hybridize with the intended DNA target under annealing conditions used for PCR. The primers can be designed to be substantially complementary to any portion of the DNA template. For example, the primers can be designed to amplify the portion of a nucleic acid that is normally transcribed in cells (the open reading frame), including 5' and 3' UTRs. The primers can also be designed to amplify a portion of a nucleic acid that encodes a particular domain of interest. In one embodiment, the primers are designed to amplify the coding region of a human cDNA, including all or portions of the 5' and 3' UTRs. Primers useful for PCR can be generated by synthetic methods that are well known in the art. "Forward primers" are primers that contain a region of nucleotides that are substantially complementary to nucleotides on the DNA template that are upstream of the DNA sequence that is to be amplified. "Upstream" is used herein to refer to a location 5, to the DNA sequence to be amplified relative to the coding strand. "Reverse primers" are primers that contain a region of nucleotides that are substantially complementary to a double-stranded DNA template that are downstream of the DNA sequence that is to be amplified. "Downstream" is used herein to refer to a location 3' to the DNA sequence to be amplified relative to the coding strand.
[0388] Any DNA polymerase useful for PCR can be used in the methods disclosed herein. The reagents and polymerase are commercially available from a number of sources.
[0389] Chemical structures with the ability to promote stability and/or translation efficiency may also be used. The RNA preferably has 5' and 3' UTRs. In one embodiment, the 5' UTR is between one and 3000 nucleotides in length. The length of 5' and 3' UTR sequences to be added to the coding region can be altered by different methods, including, but not limited to, designing primers for PCR that anneal to different regions of the UTRs. Using this approach, one of ordinary skill in the art can modify the 5' and 3' UTR lengths required to achieve optimal translation efficiency following transfection of the transcribed RNA.
[0390] The 5' and 3' UTRs can be the naturally occurring, endogenous 5' and 3' UTRs for the nucleic acid of interest. Alternatively, UTR sequences that are not endogenous to the nucleic acid of interest can be added by incorporating the UTR sequences into the forward and reverse primers or by any other modifications of the template. The use of UTR sequences that are not endogenous to the nucleic acid of interest can be useful for modifying the stability and/or translation efficiency of the RNA. For example, it is known that AU-rich elements in 3' UTR sequences can decrease the stability of mRNA. Therefore, 3' UTRs can be selected or designed to increase the stability of the transcribed RNA based on properties of UTRs that are well known in the art.
[0391] In one embodiment, the 5' UTR can contain the Kozak sequence of the endogenous nucleic acid. Alternatively, when a 5' UTR that is not endogenous to the nucleic acid of interest is being added by PCR as described above, a consensus Kozak sequence can be redesigned by adding the 5' UTR sequence. Kozak sequences can increase the efficiency of translation of some RNA transcripts, but does not appear to be required for all RNAs to enable efficient translation. The requirement for Kozak sequences for many mRNAs is known in the art. In other embodiments the 5' UTR can be 5'UTR of an RNA virus whose RNA genome is stable in cells. In other embodiments various nucleotide analogues can be used in the 3' or 5' UTR to impede exonuclease degradation of the mRNA.
[0392] To enable synthesis of RNA from a DNA template without the need for gene cloning, a promoter of transcription should be attached to the DNA template upstream of the sequence to be transcribed. When a sequence that functions as a promoter for an RNA polymerase is added to the 5' end of the forward primer, the RNA polymerase promoter becomes incorporated into the PCR product upstream of the open reading frame that is to be transcribed. In one preferred embodiment, the promoter is a T7 polymerase promoter, as described elsewhere herein. Other useful promoters include, but are not limited to, T3 and SP6 RNA polymerase promoters. Consensus nucleotide sequences for T7, T3 and SP6 promoters are known in the art.
[0393] In a preferred embodiment, the mRNA has both a cap on the 5' end and a 3' poly(A) tail which determine ribosome binding, initiation of translation and stability mRNA in the cell. On a circular DNA template, for instance, plasmid DNA, RNA polymerase produces a long concatameric product which is not suitable for expression in eukaryotic cells. The transcription of plasmid DNA linearized at the end of the 3' UTR results in normal sized mRNA which is not effective in eukaryotic transfection even if it is polyadenylated after transcription.
[0394] On a linear DNA template, phage T7 RNA polymerase can extend the 3' end of the transcript beyond the last base of the template (Schenborn and Mierendorf, Nuc Acids Res., 13:6223-36 (1985); Nacheva and Berzal-Herranz, Eur. J. Biochem., 270:1485-65 (2003).
[0395] The conventional method of integration of polyA/T stretches into a DNA template is molecular cloning. However polyA/T sequence integrated into plasmid DNA can cause plasmid instability, which is why plasmid DNA templates obtained from bacterial cells are often highly contaminated with deletions and other aberrations. This makes cloning procedures not only laborious and time consuming but often not reliable. That is why a method which allows construction of DNA templates with polyA/T 3' stretch without cloning highly desirable.
[0396] The polyA/T segment of the transcriptional DNA template can be produced during PCR by using a reverse primer containing a polyT tail, such as 100T tail (SEQ ID NO: 110) (size can be 50-5000 T (SEQ ID NO: 111)), or after PCR by any other method, including, but not limited to, DNA ligation or in vitro recombination. Poly(A) tails also provide stability to RNAs and reduce their degradation. Generally, the length of a poly(A) tail positively correlates with the stability of the transcribed RNA. In one embodiment, the poly(A) tail is between 100 and 5000 adenosines (SEQ ID NO: 112).
[0397] Poly(A) tails of RNAs can be further extended following in vitro transcription with the use of a poly(A) polymerase, such as E. coli polyA polymerase (E-PAP). In one embodiment, increasing the length of a poly(A) tail from 100 nucleotides to between 300 and 400 nucleotides (SEQ ID NO: 113) results in about a two-fold increase in the translation efficiency of the RNA. Additionally, the attachment of different chemical groups to the 3' end can increase mRNA stability. Such attachment can contain modified/artificial nucleotides, aptamers and other compounds. For example, ATP analogs can be incorporated into the poly(A) tail using poly(A) polymerase. ATP analogs can further increase the stability of the RNA.
[0398] 5' caps on also provide stability to RNA molecules. In a preferred embodiment, RNAs produced by the methods disclosed herein include a 5' cap. The 5' cap is provided using techniques known in the art and described herein (Cougot, et al., Trends in Biochem. Sci., 29:436-444 (2001); Stepinski, et al., RNA, 7:1468-95 (2001); Elango, et al., Biochim. Biophys. Res. Commun., 330:958-966 (2005)).
[0399] The RNAs produced by the methods disclosed herein can also contain an internal ribosome entry site (IRES) sequence. The IRES sequence may be any viral, chromosomal or artificially designed sequence which initiates cap-independent ribosome binding to mRNA and facilitates the initiation of translation. Any solutes suitable for cell electroporation, which can contain factors facilitating cellular permeability and viability such as sugars, peptides, lipids, proteins, antioxidants, and surfactants can be included.
[0400] RNA can be introduced into target cells using any of a number of different methods, for instance, commercially available methods which include, but are not limited to, electroporation (Amaxa Nucleofector-II (Amaxa Biosystems, Cologne, Germany)), (ECM 830 (BTX) (Harvard Instruments, Boston, Mass.) or the Gene Pulser II (BioRad, Denver, Colo.), Multiporator (Eppendort, Hamburg Germany), cationic liposome mediated transfection using lipofection, polymer encapsulation, peptide mediated transfection, or biolistic particle delivery systems such as "gene guns" (see, for example, Nishikawa, et al. Hum Gene Ther., 12(8):861-70 (2001).
Non-Viral Delivery Methods
[0401] In some aspects, non-viral methods can be used to deliver a nucleic acid encoding a CAR described herein into a cell or tissue or a subject.
[0402] In some embodiments, the non-viral method includes the use of a transposon (also called a transposable element). In some embodiments, a transposon is a piece of DNA that can insert itself at a location in a genome, for example, a piece of DNA that is capable of self-replicating and inserting its copy into a genome, or a piece of DNA that can be spliced out of a longer nucleic acid and inserted into another place in a genome. For example, a transposon comprises a DNA sequence made up of inverted repeats flanking genes for transposition.
[0403] Exemplary methods of nucleic acid delivery using a transposon include a Sleeping Beauty transposon system (SBTS) and a piggyBac (PB) transposon system. See, e.g., Aronovich et al. Hum. Mol. Genet. 20.R1 (2011):R14-20; Singh et al. Cancer Res. 15(2008):2961-2971; Huang et al. Mol. Ther. 16(2008):580-589; Grabundzija et al. Mol. Ther. 18(2010):1200-1209; Kebriaei et al. Blood. 122.21(2013):166; Williams. Molecular Therapy 16.9(2008):1515-16; Bell et al. Nat. Protoc. 2.12(2007):3153-65; and Ding et al. Cell. 122.3(2005):473-83, all of which are incorporated herein by reference.
[0404] The SBTS includes two components: 1) a transposon containing a transgene and 2) a source of transposase enzyme. The transposase can transpose the transposon from a carrier plasmid (or other donor DNA) to a target DNA, such as a host cell chromosome/genome. For example, the transposase binds to the carrier plasmid/donor DNA, cuts the transposon (including transgene(s)) out of the plasmid, and inserts it into the genome of the host cell. See, e.g., Aronovich et al. supra.
[0405] Exemplary transposons include a pT2-based transposon. See, e.g., Grabundzija et al. Nucleic Acids Res. 41.3(2013):1829-47; and Singh et al. Cancer Res. 68.8(2008): 2961-2971, all of which are incorporated herein by reference. Exemplary transposases include a Tc1/mariner-type transposase, e.g., the SB10 transposase or the SB11 transposase (a hyperactive transposase which can be expressed, e.g., from a cytomegalovirus promoter). See, e.g., Aronovich et al.; Kebriaei et al.; and Grabundzija et al., all of which are incorporated herein by reference.
[0406] Use of the SBTS permits efficient integration and expression of a transgene, e.g., a nucleic acid encoding a CAR described herein. Provided herein are methods of generating a cell, e.g., T cell or NK cell, that stably expresses a CAR described herein, e.g., using a transposon system such as SBTS.
[0407] In accordance with methods described herein, in some embodiments, one or more nucleic acids, e.g., plasmids, containing the SBTS components are delivered to a cell (e.g., T or NK cell). For example, the nucleic acid(s) are delivered by standard methods of nucleic acid (e.g., plasmid DNA) delivery, e.g., methods described herein, e.g., electroporation, transfection, or lipofection. In some embodiments, the nucleic acid contains a transposon comprising a transgene, e.g., a nucleic acid encoding a CAR described herein. In some embodiments, the nucleic acid contains a transposon comprising a transgene (e.g., a nucleic acid encoding a CAR described herein) as well as a nucleic acid sequence encoding a transposase enzyme. In other embodiments, a system with two nucleic acids is provided, e.g., a dual-plasmid system, e.g., where a first plasmid contains a transposon comprising a transgene, and a second plasmid contains a nucleic acid sequence encoding a transposase enzyme. For example, the first and the second nucleic acids are co-delivered into a host cell.
[0408] In some embodiments, cells, e.g., T or NK cells, are generated that express a CAR described herein by using a combination of gene insertion using the SBTS and genetic editing using a nuclease (e.g., Zinc finger nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), the CRISPR/Cas system, or engineered meganuclease re-engineered homing endonucleases).
[0409] In some embodiments, use of a non-viral method of delivery permits reprogramming of cells, e.g., T or NK cells, and direct infusion of the cells into a subject. Advantages of non-viral vectors include but are not limited to the ease and relatively low cost of producing sufficient amounts required to meet a patient population, stability during storage, and lack of immunogenicity.
Nucleic Acid Constructs Encoding a CAR
[0410] The present invention also provides nucleic acid molecules encoding one or more CAR constructs described herein. In one aspect, the nucleic acid molecule is provided as a messenger RNA transcript. In one aspect, the nucleic acid molecule is provided as a DNA construct.
[0411] Accordingly, in one aspect, the invention pertains to an isolated nucleic acid molecule encoding a chimeric antigen receptor (CAR), wherein the CAR comprises a anti-CD19 binding domain (e.g., a humanized anti-CD19 binding domain), a transmembrane domain, and an intracellular signaling domain comprising a stimulatory domain, e.g., a costimulatory signaling domain and/or a primary signaling domain, e.g., zeta chain. In one embodiment, the anti-CD19 binding domain is an anti-CD19 binding domain described herein, e.g., an anti-CD19 binding domain which comprises a sequence selected from a group consisting of SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4, SEQ ID NO:5, SEQ ID NO:6, SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11, SEQ ID NO:12 and SEQ ID NO:59, or a sequence with 95-99% identify thereof. In one embodiment, the transmembrane domain is transmembrane domain of a protein selected from the group consisting of the alpha, beta or zeta chain of the T-cell receptor, CD28, CD3 epsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137 and CD154. In one embodiment, the transmembrane domain comprises a sequence of SEQ ID NO: 15, or a sequence with 95-99% identity thereof. In one embodiment, the anti-CD19 binding domain is connected to the transmembrane domain by a hinge region, e.g., a hinge described herein. In one embodiment, the hinge region comprises SEQ ID NO: 14 or SEQ ID NO:45 or SEQ ID NO:47 or SEQ ID NO:49, or a sequence with 95-99% identity thereof. In one embodiment, the isolated nucleic acid molecule further comprises a sequence encoding a costimulatory domain. In one embodiment, the costimulatory domain is a functional signaling domain of a protein selected from the group consisting of OX40, CD27, CD28, CDS, ICAM-1, LFA-1 (CD11a/CD18), ICOS (CD278), and 4-1BB (CD137). In one embodiment, the costimulatory domain comprises a sequence of SEQ ID NO: 16, or a sequence with 95-99% identity thereof. In one embodiment, the intracellular signaling domain comprises a functional signaling domain of 4-1BB and a functional signaling domain of CD3 zeta. In one embodiment, the intracellular signaling domain comprises the sequence of SEQ ID NO: 16 or SEQ ID NO:51, or a sequence with 95-99% identity thereof, and the sequence of SEQ ID NO: 17 or SEQ ID NO:43, or a sequence with 95-99% identity thereof, wherein the sequences comprising the intracellular signaling domain are expressed in the same frame and as a single polypeptide chain.
[0412] In another aspect, the invention pertains to an isolated nucleic acid molecule encoding a CAR construct comprising a leader sequence of SEQ ID NO: 13, a scFv domain having a sequence selected from the group consisting of SEQ ID NO: 1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4, SEQ ID NO:5, SEQ ID NO:6, SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11, SEQ ID NO:12, and SEQ ID NO:59, (or a sequence with 95-99% identify thereof), a hinge region of SEQ ID NO: 14 or SEQ ID NO:45 or SEQ ID NO:47 or SEQ ID NO:49 (or a sequence with 95-99% identity thereof), a transmembrane domain having a sequence of SEQ ID NO: 15 (or a sequence with 95-99% identity thereof), a 4-1BB costimulatory domain having a sequence of SEQ ID NO: 16 or a CD27 costimulatory domain having a sequence of SEQ ID NO:51 (or a sequence with 95-99% identity thereof), and a CD3 zeta stimulatory domain having a sequence of SEQ ID NO: 17 or SEQ ID NO:43 (or a sequence with 95-99% identity thereof).
[0413] In another aspect, the invention pertains to an isolated polypeptide molecule encoded by the nucleic acid molecule. In one embodiment, the isolated polypeptide molecule comprises a sequence selected from the group consisting of SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, SEQ ID NO:38, SEQ ID NO:39, SEQ ID NO:40, SEQ ID NO:41, SEQ ID NO:42, SEQ ID NO:59 or a sequence with 95-99% identify thereof.
[0414] In another aspect, the invention pertains to a nucleic acid molecule encoding a chimeric antigen receptor (CAR) molecule that comprises an anti-CD19 binding domain, a transmembrane domain, and an intracellular signaling domain comprising a stimulatory domain, and wherein said anti-CD19 binding domain comprises a sequence selected from the group consisting of SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4, SEQ ID NO:5, SEQ ID NO:6, SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11, SEQ ID NO:12 and SEQ ID NO:59, or a sequence with 95-99% identify thereof.
[0415] In one embodiment, the encoded CAR molecule further comprises a sequence encoding a costimulatory domain. In one embodiment, the costimulatory domain is a functional signaling domain of a protein selected from the group consisting of OX40, CD27, CD28, CDS, ICAM-1, LFA-1 (CD11a/CD18) and 4-1BB (CD137). In one embodiment, the costimulatory domain comprises a sequence of SEQ ID NO: 16. In one embodiment, the transmembrane domain is a transmembrane domain of a protein selected from the group consisting of the alpha, beta or zeta chain of the T-cell receptor, CD28, CD3 epsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137 and CD154. In one embodiment, the transmembrane domain comprises a sequence of SEQ ID NO: 15. In one embodiment, the intracellular signaling domain comprises a functional signaling domain of 4-1BB and a functional signaling domain of zeta. In one embodiment, the intracellular signaling domain comprises the sequence of SEQ ID NO: 16 and the sequence of SEQ ID NO: 17, wherein the sequences comprising the intracellular signaling domain are expressed in the same frame and as a single polypeptide chain. In one embodiment, the anti-CD19 binding domain is connected to the transmembrane domain by a hinge region. In one embodiment, the hinge region comprises SEQ ID NO: 14. In one embodiment, the hinge region comprises SEQ ID NO:45 or SEQ ID NO:47 or SEQ ID NO:49.
[0416] In another aspect, the invention pertains to an encoded CAR molecule comprising a leader sequence of SEQ ID NO: 13, a scFv domain having a sequence selected from the group consisting of SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4, SEQ ID NO:5, SEQ ID NO:6, SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11, SEQ ID NO: 12, and SEQ ID NO:59, or a sequence with 95-99% identify thereof, a hinge region of SEQ ID NO:14 or SEQ ID NO:45 or SEQ ID NO:47 or SEQ ID NO:49, a transmembrane domain having a sequence of SEQ ID NO: 15, a 4-1BB costimulatory domain having a sequence of SEQ ID NO: 16 or a CD27 costimulatory domain having a sequence of SEQ ID NO:51, and a CD3 zeta stimulatory domain having a sequence of SEQ ID NO:17 or SEQ ID NO:43. In one embodiment, the encoded CAR molecule comprises a sequence selected from a group consisting of SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, SEQ ID NO:38, SEQ ID NO:39, SEQ ID NO:40, SEQ ID NO:41, SEQ ID NO:42, and SEQ ID NO:59, or a sequence with 95-99% identify thereof.
[0417] The nucleic acid sequences coding for the desired molecules can be obtained using recombinant methods known in the art, such as, for example by screening libraries from cells expressing the gene, by deriving the gene from a vector known to include the same, or by isolating directly from cells and tissues containing the same, using standard techniques. Alternatively, the gene of interest can be produced synthetically, rather than cloned.
[0418] The present invention also provides vectors in which a DNA of the present invention is inserted. Vectors derived from retroviruses such as the lentivirus are suitable tools to achieve long-term gene transfer since they allow long-term, stable integration of a transgene and its propagation in daughter cells. Lentiviral vectors have the added advantage over vectors derived from onco-retroviruses such as murine leukemia viruses in that they can transduce non-proliferating cells, such as hepatocytes. They also have the added advantage of low immunogenicity. A retroviral vector may also be, e.g., a gammaretroviral vector. A gammaretroviral vector may include, e.g., a promoter, a packaging signal (W), a primer binding site (PBS), one or more (e.g., two) long terminal repeats (LTR), and a transgene of interest, e.g., a gene encoding a CAR. A gammaretroviral vector may lack viral structural gens such as gag, pol, and env. Exemplary gammaretroviral vectors include Murine Leukemia Virus (MLV), Spleen-Focus Forming Virus (SFFV), and Myeloproliferative Sarcoma Virus (MPSV), and vectors derived therefrom. Other gammaretroviral vectors are described, e.g., in Tobias Maetzig et al., "Gammaretroviral Vectors: Biology, Technology and Application" Viruses. 2011 June; 3(6): 677-713.
[0419] In another embodiment, the vector comprising the nucleic acid encoding the desired CAR of the invention is an adenoviral vector (A5/35). In another embodiment, the expression of nucleic acids encoding CARs can be accomplished using of transposons such as sleeping beauty, crisper, CAS9, and zinc finger nucleases. See below June et al. 2009Nature Reviews Immunology 9.10: 704-716, is incorporated herein by reference.
[0420] In brief summary, the expression of natural or synthetic nucleic acids encoding CARs is typically achieved by operably linking a nucleic acid encoding the CAR polypeptide or portions thereof to a promoter, and incorporating the construct into an expression vector. The vectors can be suitable for replication and integration eukaryotes. Typical cloning vectors contain transcription and translation terminators, initiation sequences, and promoters useful for regulation of the expression of the desired nucleic acid sequence.
[0421] The expression constructs of the present invention may also be used for nucleic acid immunization and gene therapy, using standard gene delivery protocols. Methods for gene delivery are known in the art. See, e.g., U.S. Pat. Nos. 5,399,346, 5,580,859, 5,589,466, incorporated by reference herein in their entireties. In another embodiment, the invention provides a gene therapy vector.
[0422] The nucleic acid can be cloned into a number of types of vectors. For example, the nucleic acid can be cloned into a vector including, but not limited to a plasmid, a phagemid, a phage derivative, an animal virus, and a cosmid. Vectors of particular interest include expression vectors, replication vectors, probe generation vectors, and sequencing vectors.
[0423] Further, the expression vector may be provided to a cell in the form of a viral vector. Viral vector technology is well known in the art and is described, for example, in Sambrook et al., 2012, MOLECULAR CLONING: A LABORATORY MANUAL, volumes 1-4, Cold Spring Harbor Press, NY), and in other virology and molecular biology manuals. Viruses, which are useful as vectors include, but are not limited to, retroviruses, adenoviruses, adeno-associated viruses, herpes viruses, and lentiviruses. In general, a suitable vector contains an origin of replication functional in at least one organism, a promoter sequence, convenient restriction endonuclease sites, and one or more selectable markers, (e.g., WO 01/96584; WO 01/29058; and U.S. Pat. No. 6,326,193).
[0424] A number of viral based systems have been developed for gene transfer into mammalian cells. For example, retroviruses provide a convenient platform for gene delivery systems. A selected gene can be inserted into a vector and packaged in retroviral particles using techniques known in the art. The recombinant virus can then be isolated and delivered to cells of the subject either in vivo or ex vivo. A number of retroviral systems are known in the art. In some embodiments, adenovirus vectors are used. A number of adenovirus vectors are known in the art. In one embodiment, lentivirus vectors are used.
[0425] Additional promoter elements, e.g., enhancers, regulate the frequency of transcriptional initiation. Typically, these are located in the region 30-110 bp upstream of the start site, although a number of promoters have been shown to contain functional elements downstream of the start site as well. The spacing between promoter elements frequently is flexible, so that promoter function is preserved when elements are inverted or moved relative to one another. In the thymidine kinase (tk) promoter, the spacing between promoter elements can be increased to 50 bp apart before activity begins to decline. Depending on the promoter, it appears that individual elements can function either cooperatively or independently to activate transcription. Exemplary promoters include the CMV IE gene, EF-1.alpha., ubiquitin C, or phosphoglycerokinase (PGK) promoters.
[0426] An example of a promoter that is capable of expressing a CAR transgene in a mammalian T cell is the EF1a promoter. The native EF1a promoter drives expression of the alpha subunit of the elongation factor-1 complex, which is responsible for the enzymatic delivery of aminoacyl tRNAs to the ribosome. The EF1a promoter has been extensively used in mammalian expression plasmids and has been shown to be effective in driving CAR expression from transgenes cloned into a lentiviral vector. See, e.g., Milone et al., Mol. Ther. 17(8): 1453-1464 (2009). In one aspect, the EF1a promoter comprises the sequence provided as SEQ ID NO: 100.
[0427] Another example of a promoter is the immediate early cytomegalovirus (CMV) promoter sequence. This promoter sequence is a strong constitutive promoter sequence capable of driving high levels of expression of any polynucleotide sequence operatively linked thereto. However, other constitutive promoter sequences may also be used, including, but not limited to the simian virus 40 (SV40) early promoter, mouse mammary tumor virus (MMTV), human immunodeficiency virus (HIV) long terminal repeat (LTR) promoter, MoMuLV promoter, an avian leukemia virus promoter, an Epstein-Barr virus immediate early promoter, a Rous sarcoma virus promoter, as well as human gene promoters such as, but not limited to, the actin promoter, the myosin promoter, the elongation factor-1.alpha. promoter, the hemoglobin promoter, and the creatine kinase promoter. Further, the invention should not be limited to the use of constitutive promoters. Inducible promoters are also contemplated as part of the invention. The use of an inducible promoter provides a molecular switch capable of turning on expression of the polynucleotide sequence which it is operatively linked when such expression is desired, or turning off the expression when expression is not desired. Examples of inducible promoters include, but are not limited to a metallothionine promoter, a glucocorticoid promoter, a progesterone promoter, and a tetracycline promoter.
[0428] A vector may also include, e.g., a signal sequence to facilitate secretion, a polyadenylation signal and transcription terminator (e.g., from Bovine Growth Hormone (BGH) gene), an element allowing episomal replication and replication in prokaryotes (e.g. SV40 origin and ColE1 or others known in the art) and/or elements to allow selection (e.g., ampicillin resistance gene and/or zeocin marker).
[0429] In order to assess the expression of a CAR polypeptide or portions thereof, the expression vector to be introduced into a cell can also contain either a selectable marker gene or a reporter gene or both to facilitate identification and selection of expressing cells from the population of cells sought to be transfected or infected through viral vectors. In other aspects, the selectable marker may be carried on a separate piece of DNA and used in a co-transfection procedure. Both selectable markers and reporter genes may be flanked with appropriate regulatory sequences to enable expression in the host cells. Useful selectable markers include, for example, antibiotic-resistance genes, such as neo and the like.
[0430] Reporter genes are used for identifying potentially transfected cells and for evaluating the functionality of regulatory sequences. In general, a reporter gene is a gene that is not present in or expressed by the recipient organism or tissue and that encodes a polypeptide whose expression is manifested by some easily detectable property, e.g., enzymatic activity. Expression of the reporter gene is assayed at a suitable time after the DNA has been introduced into the recipient cells. Suitable reporter genes may include genes encoding luciferase, beta-galactosidase, chloramphenicol acetyl transferase, secreted alkaline phosphatase, or the green fluorescent protein gene (e.g., Ui-Tei et al., 2000 FEBS Letters 479: 79-82). Suitable expression systems are well known and may be prepared using known techniques or obtained commercially. In general, the construct with the minimal 5' flanking region showing the highest level of expression of reporter gene is identified as the promoter. Such promoter regions may be linked to a reporter gene and used to evaluate agents for the ability to modulate promoter-driven transcription.
[0431] Methods of introducing and expressing genes into a cell are known in the art. In the context of an expression vector, the vector can be readily introduced into a host cell, e.g., mammalian, bacterial, yeast, or insect cell by any method in the art. For example, the expression vector can be transferred into a host cell by physical, chemical, or biological means.
[0432] Physical methods for introducing a polynucleotide into a host cell include calcium phosphate precipitation, lipofection, particle bombardment, microinjection, electroporation, and the like. Methods for producing cells comprising vectors and/or exogenous nucleic acids are well-known in the art. See, for example, Sambrook et al., 2012, MOLECULAR CLONING: A LABORATORY MANUAL, volumes 1-4, Cold Spring Harbor Press, NY). A preferred method for the introduction of a polynucleotide into a host cell is calcium phosphate transfection
[0433] Biological methods for introducing a polynucleotide of interest into a host cell include the use of DNA and RNA vectors. Viral vectors, and especially retroviral vectors, have become the most widely used method for inserting genes into mammalian, e.g., human cells. Other viral vectors can be derived from lentivirus, poxviruses, herpes simplex virus I, adenoviruses and adeno-associated viruses, and the like. See, for example, U.S. Pat. Nos. 5,350,674 and 5,585,362.
[0434] Chemical means for introducing a polynucleotide into a host cell include colloidal dispersion systems, such as macromolecule complexes, nanocapsules, microspheres, beads, and lipid-based systems including oil-in-water emulsions, micelles, mixed micelles, and liposomes. An exemplary colloidal system for use as a delivery vehicle in vitro and in vivo is a liposome (e.g., an artificial membrane vesicle). Other methods of state-of-the-art targeted delivery of nucleic acids are available, such as delivery of polynucleotides with targeted nanoparticles or other suitable sub-micron sized delivery system.
[0435] In the case where a non-viral delivery system is utilized, an exemplary delivery vehicle is a liposome. The use of lipid formulations is contemplated for the introduction of the nucleic acids into a host cell (in vitro, ex vivo or in vivo). In another aspect, the nucleic acid may be associated with a lipid. The nucleic acid associated with a lipid may be encapsulated in the aqueous interior of a liposome, interspersed within the lipid bilayer of a liposome, attached to a liposome via a linking molecule that is associated with both the liposome and the oligonucleotide, entrapped in a liposome, complexed with a liposome, dispersed in a solution containing a lipid, mixed with a lipid, combined with a lipid, contained as a suspension in a lipid, contained or complexed with a micelle, or otherwise associated with a lipid. Lipid, lipid/DNA or lipid/expression vector associated compositions are not limited to any particular structure in solution. For example, they may be present in a bilayer structure, as micelles, or with a "collapsed" structure. They may also simply be interspersed in a solution, possibly forming aggregates that are not uniform in size or shape. Lipids are fatty substances which may be naturally occurring or synthetic lipids. For example, lipids include the fatty droplets that naturally occur in the cytoplasm as well as the class of compounds which contain long-chain aliphatic hydrocarbons and their derivatives, such as fatty acids, alcohols, amines, amino alcohols, and aldehydes.
[0436] Lipids suitable for use can be obtained from commercial sources. For example, dimyristyl phosphatidylcholine ("DMPC") can be obtained from Sigma, St. Louis, Mo.; dicetyl phosphate ("DCP") can be obtained from K & K Laboratories (Plainview, N.Y.); cholesterol ("Choi") can be obtained from Calbiochem-Behring; dimyristyl phosphatidylglycerol ("DMPG") and other lipids may be obtained from Avanti Polar Lipids, Inc. (Birmingham, Ala.). Stock solutions of lipids in chloroform or chloroform/methanol can be stored at about -20.degree. C. Chloroform is used as the only solvent since it is more readily evaporated than methanol. "Liposome" is a generic term encompassing a variety of single and multilamellar lipid vehicles formed by the generation of enclosed lipid bilayers or aggregates. Liposomes can be characterized as having vesicular structures with a phospholipid bilayer membrane and an inner aqueous medium. Multilamellar liposomes have multiple lipid layers separated by aqueous medium. They form spontaneously when phospholipids are suspended in an excess of aqueous solution. The lipid components undergo self-rearrangement before the formation of closed structures and entrap water and dissolved solutes between the lipid bilayers (Ghosh et al., 1991 Glycobiology 5: 505-10). However, compositions that have different structures in solution than the normal vesicular structure are also encompassed. For example, the lipids may assume a micellar structure or merely exist as nonuniform aggregates of lipid molecules. Also contemplated are lipofectamine-nucleic acid complexes.
[0437] Regardless of the method used to introduce exogenous nucleic acids into a host cell or otherwise expose a cell to the inhibitor of the present invention, in order to confirm the presence of the recombinant DNA sequence in the host cell, a variety of assays may be performed. Such assays include, for example, "molecular biological" assays well known to those of skill in the art, such as Southern and Northern blotting, RT-PCR and PCR; "biochemical" assays, such as detecting the presence or absence of a particular peptide, e.g., by immunological means (ELISAs and Western blots) or by assays described herein to identify agents falling within the scope of the invention.
[0438] The present invention further provides a vector comprising a CAR encoding nucleic acid molecule. In one aspect, a CAR vector can be directly transduced into a cell, e.g., a T cell. In one aspect, the vector is a cloning or expression vector, e.g., a vector including, but not limited to, one or more plasmids (e.g., expression plasmids, cloning vectors, minicircles, minivectors, double minute chromosomes), retroviral and lentiviral vector constructs. In one aspect, the vector is capable of expressing the CAR construct in mammalian T cells. In one aspect, the mammalian T cell is a human T cell.
Sources of Cells
[0439] Prior to expansion and genetic modification or other modification, a source of cells, e.g., T cells or natural killer (NK) cells, can be obtained from a subject. The term "subject" is intended to include living organisms in which an immune response can be elicited (e.g., mammals). Examples of subjects include humans, monkeys, chimpanzees, dogs, cats, mice, rats, and transgenic species thereof. T cells can be obtained from a number of sources, including peripheral blood mononuclear cells, bone marrow, lymph node tissue, cord blood, thymus tissue, tissue from a site of infection, ascites, pleural effusion, spleen tissue, and tumors.
[0440] In certain aspects of the present disclosure, immune effector cells, e.g., T cells, can be obtained from a unit of blood collected from a subject using any number of techniques known to the skilled artisan, such as Ficoll.TM. separation. In one preferred aspect, cells from the circulating blood of an individual are obtained by apheresis. The apheresis product typically contains lymphocytes, including T cells, monocytes, granulocytes, B cells, other nucleated white blood cells, red blood cells, and platelets. In one aspect, the cells collected by apheresis may be washed to remove the plasma fraction and, optionally, to place the cells in an appropriate buffer or media for subsequent processing steps. In one embodiment, the cells are washed with phosphate buffered saline (PBS). In an alternative embodiment, the wash solution lacks calcium and may lack magnesium or may lack many if not all divalent cations.
[0441] Initial activation steps in the absence of calcium can lead to magnified activation. As those of ordinary skill in the art would readily appreciate a washing step may be accomplished by methods known to those in the art, such as by using a semi-automated "flow-through" centrifuge (for example, the Cobe 2991 cell processor, the Baxter CytoMate, or the Haemonetics Cell Saver 5) according to the manufacturer's instructions. After washing, the cells may be resuspended in a variety of biocompatible buffers, such as, for example, Ca-free, Mg-free PBS, PlasmaLyte A, or other saline solution with or without buffer. Alternatively, the undesirable components of the apheresis sample may be removed and the cells directly resuspended in culture media.
[0442] In one aspect, T cells are isolated from peripheral blood lymphocytes by lysing the red blood cells and depleting the monocytes, for example, by centrifugation through a PERCOLL.TM. gradient or by counterflow centrifugal elutriation.
[0443] The methods described herein can include, e.g., selection of a specific subpopulation of immune effector cells, e.g., T cells, that are a T regulatory cell-depleted population, CD25+ depleted cells, using, e.g., a negative selection technique, e.g., described herein. Preferably, the population of T regulatory depleted cells contains less than 30%, 25%, 20%, 15%, 10%, 5%, 4%, 3%, 2%, 1% of CD25+ cells.
[0444] In one embodiment, T regulatory cells, e.g., CD25+ T cells, are removed from the population using an anti-CD25 antibody, or fragment thereof, or a CD25-binding ligand, IL-2. In one embodiment, the anti-CD25 antibody, or fragment thereof, or CD25-binding ligand is conjugated to a substrate, e.g., a bead, or is otherwise coated on a substrate, e.g., a bead. In one embodiment, the anti-CD25 antibody, or fragment thereof, is conjugated to a substrate as described herein.
[0445] In one embodiment, the T regulatory cells, e.g., CD25+ T cells, are removed from the population using CD25 depletion reagent from Miltenyi.TM.. In one embodiment, the ratio of cells to CD25 depletion reagent is 1e7 cells to 20 uL, or 1e7 cells to 15 uL, or 1e7 cells to 10 uL, or 1e7 cells to 5 uL, or 1e7 cells to 2.5 uL, or 1e7 cells to 1.25 uL. In one embodiment, e.g., for T regulatory cells, e.g., CD25+ depletion, greater than 500 million cells/ml is used. In a further aspect, a concentration of cells of 600, 700, 800, or 900 million cells/ml is used.
[0446] In one embodiment, the population of immune effector cells to be depleted includes about 6.times.10.sup.9 CD25+ T cells. In other aspects, the population of immune effector cells to be depleted include about 1.times.10.sup.9 to 1.times.10.sup.10 CD25+ T cell, and any integer value in between. In one embodiment, the resulting population T regulatory depleted cells has 2.times.10.sup.9 T regulatory cells, e.g., CD25+ cells, or less (e.g., 1.times.10.sup.9, 5.times.10.sup.8, 1.times.10.sup.8, 5.times.10.sup.7, 1.times.10.sup.7, or less CD25+ cells).
[0447] In one embodiment, the T regulatory cells, e.g., CD25+ cells, are removed from the population using the CliniMAC system with a depletion tubing set, such as, e.g., tubing 162-01. In one embodiment, the CliniMAC system is run on a depletion setting such as, e.g., DEPLETION2.1.
[0448] Without wishing to be bound by a particular theory, decreasing the level of negative regulators of immune cells (e.g., decreasing the number of unwanted immune cells, e.g., T.sub.REG cells), in a subject prior to apheresis or during manufacturing of a CAR-expressing cell product can reduce the risk of subject relapse. For example, methods of depleting T.sub.REG cells are known in the art. Methods of decreasing T.sub.REG cells include, but are not limited to, cyclophosphamide, anti-GITR antibody (an anti-GITR antibody described herein), CD25-depletion, and combinations thereof.
[0449] In some embodiments, the manufacturing methods comprise reducing the number of (e.g., depleting) T.sub.REG cells prior to manufacturing of the CAR-expressing cell. For example, manufacturing methods comprise contacting the sample, e.g., the apheresis sample, with an anti-GITR antibody and/or an anti-CD25 antibody (or fragment thereof, or a CD25-binding ligand), e.g., to deplete T.sub.REG cells prior to manufacturing of the CAR-expressing cell (e.g., T cell, NK cell) product.
[0450] In an embodiment, a subject is pre-treated with one or more therapies that reduce T.sub.REG cells prior to collection of cells for CAR-expressing cell product manufacturing, thereby reducing the risk of subject relapse to CAR-expressing cell treatment. In an embodiment, methods of decreasing T.sub.REG cells include, but are not limited to, administration to the subject of one or more of cyclophosphamide, anti-GITR antibody, CD25-depletion, or a combination thereof. Administration of one or more of cyclophosphamide, anti-GITR antibody, CD25-depletion, or a combination thereof, can occur before, during or after an infusion of the CAR-expressing cell product.
[0451] In an embodiment, a subject is pre-treated with cyclophosphamide prior to collection of cells for CAR-expressing cell product manufacturing, thereby reducing the risk of subject relapse to CAR-expressing cell treatment. In an embodiment, a subject is pre-treated with an anti-GITR antibody prior to collection of cells for CAR-expressing cell product manufacturing, thereby reducing the risk of subject relapse to CAR-expressing cell treatment.
[0452] In one embodiment, the population of cells to be removed are neither the regulatory T cells or tumor cells, but cells that otherwise negatively affect the expansion and/or function of CART cells, e.g. cells expressing CD14, CD11b, CD33, CD15, or other markers expressed by potentially immune suppressive cells. In one embodiment, such cells are envisioned to be removed concurrently with regulatory T cells and/or tumor cells, or following said depletion, or in another order.
[0453] The methods described herein can include more than one selection step, e.g., more than one depletion step. Enrichment of a T cell population by negative selection can be accomplished, e.g., with a combination of antibodies directed to surface markers unique to the negatively selected cells. One method is cell sorting and/or selection via negative magnetic immunoadherence or flow cytometry that uses a cocktail of monoclonal antibodies directed to cell surface markers present on the cells negatively selected. For example, to enrich for CD4+ cells by negative selection, a monoclonal antibody cocktail can include antibodies to CD14, CD20, CD11b, CD16, HLA-DR, and CD8.
[0454] The methods described herein can further include removing cells from the population which express a tumor antigen, e.g., a tumor antigen that does not comprise CD25, e.g., CD19, CD30, CD38, CD123, CD20, CD14 or CD11b, to thereby provide a population of T regulatory depleted, e.g., CD25+ depleted, and tumor antigen depleted cells that are suitable for expression of a CAR, e.g., a CAR described herein. In one embodiment, tumor antigen expressing cells are removed simultaneously with the T regulatory, e.g., CD25+ cells. For example, an anti-CD25 antibody, or fragment thereof, and an anti-tumor antigen antibody, or fragment thereof, can be attached to the same substrate, e.g., bead, which can be used to remove the cells or an anti-CD25 antibody, or fragment thereof, or the anti-tumor antigen antibody, or fragment thereof, can be attached to separate beads, a mixture of which can be used to remove the cells. In other embodiments, the removal of T regulatory cells, e.g., CD25+ cells, and the removal of the tumor antigen expressing cells is sequential, and can occur, e.g., in either order.
[0455] Also provided are methods that include removing cells from the population which express a check point inhibitor, e.g., a check point inhibitor described herein, e.g., one or more of PD1+ cells, LAG3+ cells, and TIM3+ cells, to thereby provide a population of T regulatory depleted, e.g., CD25+ depleted cells, and check point inhibitor depleted cells, e.g., PD1+, LAG3+ and/or TIM3+ depleted cells. Exemplary check point inhibitors include B7-H1, B7-1, CD160, P1H, 2B4, PD1, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, TIGIT, CTLA-4, BTLA and LAIR1. In one embodiment, check point inhibitor expressing cells are removed simultaneously with the T regulatory, e.g., CD25+ cells. For example, an anti-CD25 antibody, or fragment thereof, and an anti-check point inhibitor antibody, or fragment thereof, can be attached to the same bead which can be used to remove the cells, or an anti-CD25 antibody, or fragment thereof, and the anti-check point inhibitor antibody, or fragment there, can be attached to separate beads, a mixture of which can be used to remove the cells. In other embodiments, the removal of T regulatory cells, e.g., CD25+ cells, and the removal of the check point inhibitor expressing cells is sequential, and can occur, e.g., in either order.
[0456] Methods described herein can include a positive selection step. For example, T cells can isolated by incubation with anti-CD3/anti-CD28 (e.g., 3.times.28)-conjugated beads, such as DYNABEADS.RTM. M-450 CD3/CD28 T, for a time period sufficient for positive selection of the desired T cells. In one embodiment, the time period is about 30 minutes. In a further embodiment, the time period ranges from 30 minutes to 36 hours or longer and all integer values there between. In a further embodiment, the time period is at least 1, 2, 3, 4, 5, or 6 hours. In yet another embodiment, the time period is 10 to 24 hours, e.g., 24 hours. Longer incubation times may be used to isolate T cells in any situation where there are few T cells as compared to other cell types, such in isolating tumor infiltrating lymphocytes (TIL) from tumor tissue or from immunocompromised individuals. Further, use of longer incubation times can increase the efficiency of capture of CD8+ T cells. Thus, by simply shortening or lengthening the time T cells are allowed to bind to the CD3/CD28 beads and/or by increasing or decreasing the ratio of beads to T cells (as described further herein), subpopulations of T cells can be preferentially selected for or against at culture initiation or at other time points during the process. Additionally, by increasing or decreasing the ratio of anti-CD3 and/or anti-CD28 antibodies on the beads or other surface, subpopulations of T cells can be preferentially selected for or against at culture initiation or at other desired time points.
[0457] In one embodiment, a T cell population can be selected that expresses one or more of IFN-.sup..gamma., TNF.alpha., IL-17A, IL-2, IL-3, IL-4, GM-CSF, IL-10, IL-13, granzyme B, and perforin, or other appropriate molecules, e.g., other cytokines. Methods for screening for cell expression can be determined, e.g., by the methods described in PCT Publication No.: WO 2013/126712.
[0458] For isolation of a desired population of cells by positive or negative selection, the concentration of cells and surface (e.g., particles such as beads) can be varied. In certain aspects, it may be desirable to significantly decrease the volume in which beads and cells are mixed together (e.g., increase the concentration of cells), to ensure maximum contact of cells and beads. For example, in one aspect, a concentration of 10 billion cells/ml, 9 billion/ml, 8 billion/ml, 7 billion/ml, 6 billion/ml, or 5 billion/ml is used. In one aspect, a concentration of 1 billion cells/ml is used. In yet one aspect, a concentration of cells from 75, 80, 85, 90, 95, or 100 million cells/ml is used. In further aspects, concentrations of 125 or 150 million cells/ml can be used.
[0459] Using high concentrations can result in increased cell yield, cell activation, and cell expansion. Further, use of high cell concentrations allows more efficient capture of cells that may weakly express target antigens of interest, such as CD28-negative T cells, or from samples where there are many tumor cells present (e.g., leukemic blood, tumor tissue, etc.). Such populations of cells may have therapeutic value and would be desirable to obtain. For example, using high concentration of cells allows more efficient selection of CD8+ T cells that normally have weaker CD28 expression.
[0460] In a related aspect, it may be desirable to use lower concentrations of cells. By significantly diluting the mixture of T cells and surface (e.g., particles such as beads), interactions between the particles and cells is minimized. This selects for cells that express high amounts of desired antigens to be bound to the particles. For example, CD4+ T cells express higher levels of CD28 and are more efficiently captured than CD8+ T cells in dilute concentrations. In one aspect, the concentration of cells used is 5.times.10.sup.6/ml. In other aspects, the concentration used can be from about 1.times.10.sup.5/ml to 1.times.10.sup.6/ml, and any integer value in between.
[0461] In other aspects, the cells may be incubated on a rotator for varying lengths of time at varying speeds at either 2-10.degree. C. or at room temperature.
[0462] T cells for stimulation can also be frozen after a washing step. Wishing not to be bound by theory, the freeze and subsequent thaw step provides a more uniform product by removing granulocytes and to some extent monocytes in the cell population. After the washing step that removes plasma and platelets, the cells may be suspended in a freezing solution. While many freezing solutions and parameters are known in the art and will be useful in this context, one method involves using PBS containing 20% DMSO and 8% human serum albumin, or culture media containing 10% Dextran 40 and 5% Dextrose, 20% Human Serum Albumin and 7.5% DMSO, or 31.25% Plasmalyte-A, 31.25% Dextrose 5%, 0.45% NaCl, 10% Dextran 40 and 5% Dextrose, 20% Human Serum Albumin, and 7.5% DMSO or other suitable cell freezing media containing for example, Hespan and PlasmaLyte A, the cells then are frozen to -80.degree. C. at a rate of 10 per minute and stored in the vapor phase of a liquid nitrogen storage tank. Other methods of controlled freezing may be used as well as uncontrolled freezing immediately at -20.degree. C. or in liquid nitrogen.
[0463] In certain aspects, cryopreserved cells are thawed and washed as described herein and allowed to rest for one hour at room temperature prior to activation using the methods of the present invention.
[0464] Also contemplated in the context of the invention is the collection of blood samples or apheresis product from a subject at a time period prior to when the expanded cells as described herein might be needed. As such, the source of the cells to be expanded can be collected at any time point necessary, and desired cells, such as T cells, isolated and frozen for later use in immune effector cell therapy for any number of diseases or conditions that would benefit from immune effector cell therapy, such as those described herein. In one aspect a blood sample or an apheresis is taken from a generally healthy subject. In certain aspects, a blood sample or an apheresis is taken from a generally healthy subject who is at risk of developing a disease, but who has not yet developed a disease, and the cells of interest are isolated and frozen for later use. In certain aspects, the T cells may be expanded, frozen, and used at a later time. In certain aspects, samples are collected from a patient shortly after diagnosis of a particular disease as described herein but prior to any treatments. In a further aspect, the cells are isolated from a blood sample or an apheresis from a subject prior to any number of relevant treatment modalities, including but not limited to treatment with agents such as natalizumab, efalizumab, antiviral agents, chemotherapy, radiation, immunosuppressive agents, such as cyclosporin, azathioprine, methotrexate, mycophenolate, and FK506, antibodies, or other immunoablative agents such as CAMPATH, anti-CD3 antibodies, cytoxan, fludarabine, cyclosporin, FK506, rapamycin, mycophenolic acid, steroids, FR901228, and irradiation.
[0465] In a further aspect of the present invention, T cells are obtained from a patient directly following treatment that leaves the subject with functional T cells. In this regard, it has been observed that following certain cancer treatments, in particular treatments with drugs that damage the immune system, shortly after treatment during the period when patients would normally be recovering from the treatment, the quality of T cells obtained may be optimal or improved for their ability to expand ex vivo. Likewise, following ex vivo manipulation using the methods described herein, these cells may be in a preferred state for enhanced engraftment and in vivo expansion. Thus, it is contemplated within the context of the present invention to collect blood cells, including T cells, dendritic cells, or other cells of the hematopoietic lineage, during this recovery phase. Further, in certain aspects, mobilization (for example, mobilization with GM-CSF) and conditioning regimens can be used to create a condition in a subject wherein repopulation, recirculation, regeneration, and/or expansion of particular cell types is favored, especially during a defined window of time following therapy. Illustrative cell types include T cells, B cells, dendritic cells, and other cells of the immune system.
[0466] In one embodiment, the immune effector cells expressing a CAR molecule, e.g., a CAR molecule described herein, are obtained from a subject that has received a low, immune enhancing dose of an mTOR inhibitor. In an embodiment, the population of immune effector cells, e.g., T cells, to be engineered to express a CAR, are harvested after a sufficient time, or after sufficient dosing of the low, immune enhancing, dose of an mTOR inhibitor, such that the level of PD1 negative immune effector cells, e.g., T cells, or the ratio of PD1 negative immune effector cells, e.g., T cells/PD1 positive immune effector cells, e.g., T cells, in the subject or harvested from the subject has been, at least transiently, increased.
[0467] In other embodiments, population of immune effector cells, e.g., T cells, which have, or will be engineered to express a CAR, can be treated ex vivo by contact with an amount of an mTOR inhibitor that increases the number of PD1 negative immune effector cells, e.g., T cells or increases the ratio of PD1 negative immune effector cells, e.g., T cells/PD1 positive immune effector cells, e.g., T cells.
[0468] In one embodiment, a T cell population is diaglycerol kinase (DGK)-deficient. DGK-deficient cells include cells that do not express DGK RNA or protein, or have reduced or inhibited DGK activity. DGK-deficient cells can be generated by genetic approaches, e.g., administering RNA-interfering agents, e.g., siRNA, shRNA, miRNA, to reduce or prevent DGK expression. Alternatively, DGK-deficient cells can be generated by treatment with DGK inhibitors described herein.
[0469] In one embodiment, a T cell population is Ikaros-deficient. Ikaros-deficient cells include cells that do not express Ikaros RNA or protein, or have reduced or inhibited Ikaros activity, Ikaros-deficient cells can be generated by genetic approaches, e.g., administering RNA-interfering agents, e.g., siRNA, shRNA, miRNA, to reduce or prevent Ikaros expression. Alternatively, Ikaros-deficient cells can be generated by treatment with Ikaros inhibitors, e.g., lenalidomide.
[0470] In embodiments, a T cell population is DGK-deficient and Ikaros-deficient, e.g., does not express DGK and Ikaros, or has reduced or inhibited DGK and Ikaros activity. Such DGK and Ikaros-deficient cells can be generated by any of the methods described herein.
[0471] In an embodiment, the NK cells are obtained from the subject. In another embodiment, the NK cells are an NK cell line, e.g., NK-92 cell line (Conkwest).
Allogeneic CAR
[0472] In embodiments described herein, the immune effector cell can be an allogeneic immune effector cell, e.g., T cell or NK cell. For example, the cell can be an allogeneic T cell, e.g., an allogeneic T cell lacking expression of a functional T cell receptor (TCR) and/or human leukocyte antigen (HLA), e.g., HLA class I and/or HLA class II.
[0473] A T cell lacking a functional TCR can be, e.g., engineered such that it does not express any functional TCR on its surface, engineered such that it does not express one or more subunits that comprise a functional TCR or engineered such that it produces very little functional TCR on its surface. Alternatively, the T cell can express a substantially impaired TCR, e.g., by expression of mutated or truncated forms of one or more of the subunits of the TCR. The term "substantially impaired TCR" means that this TCR will not elicit an adverse immune reaction in a host.
[0474] A T cell described herein can be, e.g., engineered such that it does not express a functional HLA on its surface. For example, a T cell described herein, can be engineered such that cell surface expression HLA, e.g., HLA class 1 and/or HLA class II, is downregulated.
[0475] In some embodiments, the T cell can lack a functional TCR and a functional HLA, e.g., HLA class I and/or HLA class II.
[0476] Modified T cells that lack expression of a functional TCR and/or HLA can be obtained by any suitable means, including a knock out or knock down of one or more subunit of TCR or HLA. For example, the T cell can include a knock down of TCR and/or HLA using siRNA, shRNA, clustered regularly interspaced short palindromic repeats (CRISPR) transcription-activator like effector nuclease (TALEN), or zinc finger endonuclease (ZFN).
[0477] In some embodiments, the allogeneic cell can be a cell which does not express or expresses at low levels an inhibitory molecule, e.g. by any method described herein. For example, the cell can be a cell that does not express or expresses at low levels an inhibitory molecule, e.g., that can decrease the ability of a CAR-expressing cell to mount an immune effector response. Examples of inhibitory molecules include PD1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 and TGFR beta. Inhibition of an inhibitory molecule, e.g., by inhibition at the DNA, RNA or protein level, can optimize a CAR-expressing cell performance. In embodiments, an inhibitory nucleic acid, e.g., an inhibitory nucleic acid, e.g., a dsRNA, e.g., an siRNA or shRNA, a clustered regularly interspaced short palindromic repeats (CRISPR), a transcription-activator like effector nuclease (TALEN), or a zinc finger endonuclease (ZFN), e.g., as described herein, can be used.
siRNA and shRNA to Inhibit TCR or HLA
[0478] In some embodiments, TCR expression and/or HLA expression can be inhibited using siRNA or shRNA that targets a nucleic acid encoding a TCR and/or HLA in a T cell.
[0479] Expression of siRNA and shRNAs in T cells can be achieved using any conventional expression system, e.g., such as a lentiviral expression system.
[0480] Exemplary shRNAs that downregulate expression of components of the TCR are described, e.g., in US Publication No.: 2012/0321667. Exemplary siRNA and shRNA that downregulate expression of HLA class I and/or HLA class II genes are described, e.g., in U.S. publication No.: US 2007/0036773.
CRISPR to Inhibit TCR or HLA
[0481] "CRISPR" or "CRISPR to TCR and/or HLA" or "CRISPR to inhibit TCR and/or HLA" as used herein refers to a set of clustered regularly interspaced short palindromic repeats, or a system comprising such a set of repeats. "Cas", as used herein, refers to a CRISPR-associated protein. A "CRISPR/Cas" system refers to a system derived from CRISPR and Cas which can be used to silence or mutate a TCR and/or HLA gene.
[0482] Naturally-occurring CRISPR/Cas systems are found in approximately 40% of sequenced eubacteria genomes and 90% of sequenced archaea. Grissa et al. (2007) BMC Bioinformatics 8: 172. This system is a type of prokaryotic immune system that confers resistance to foreign genetic elements such as plasmids and phages and provides a form of acquired immunity. Barrangou et al. (2007) Science 315: 1709-1712; Marragini et al. (2008) Science 322: 1843-1845.
[0483] The CRISPR/Cas system has been modified for use in gene editing (silencing, enhancing or changing specific genes) in eukaryotes such as mice or primates. Wiedenheft et al. (2012) Nature 482: 331-8. This is accomplished by introducing into the eukaryotic cell a plasmid containing a specifically designed CRISPR and one or more appropriate Cas.
[0484] The CRISPR sequence, sometimes called a CRISPR locus, comprises alternating repeats and spacers. In a naturally-occurring CRISPR, the spacers usually comprise sequences foreign to the bacterium such as a plasmid or phage sequence; in the TCR and/or HLA CRISPR/Cas system, the spacers are derived from the TCR or HLA gene sequence.
[0485] RNA from the CRISPR locus is constitutively expressed and processed by Cas proteins into small RNAs. These comprise a spacer flanked by a repeat sequence. The RNAs guide other Cas proteins to silence exogenous genetic elements at the RNA or DNA level. Horvath et al. (2010) Science 327: 167-170; Makarova et al. (2006) Biology Direct 1: 7. The spacers thus serve as templates for RNA molecules, analogously to siRNAs. Pennisi (2013) Science 341: 833-836.
[0486] As these naturally occur in many different types of bacteria, the exact arrangements of the CRISPR and structure, function and number of Cas genes and their product differ somewhat from species to species. Haft et al. (2005) PLoS Comput. Biol. 1: e60; Kunin et al. (2007) Genome Biol. 8: R61; Mojica et al. (2005) J. Mol. Evol. 60: 174-182; Bolotin et al. (2005) Microbiol. 151: 2551-2561; Pourcel et al. (2005) Microbiol. 151: 653-663; and Stern et al. (2010) Trends. Genet. 28: 335-340. For example, the Cse (Cas subtype, E. coli) proteins (e.g., CasA) form a functional complex, Cascade, that processes CRISPR RNA transcripts into spacer-repeat units that Cascade retains. Brouns et al. (2008) Science 321: 960-964. In other prokaryotes, Cas6 processes the CRISPR transcript. The CRISPR-based phage inactivation in E. coli requires Cascade and Cas3, but not Cas 1 or Cas2. The Cmr (Cas RAMP module) proteins in Pyrococcus furiosus and other prokaryotes form a functional complex with small CRISPR RNAs that recognizes and cleaves complementary target RNAs. A simpler CRISPR system relies on the protein Cas9, which is a nuclease with two active cutting sites, one for each strand of the double helix. Combining Cas9 and modified CRISPR locus RNA can be used in a system for gene editing. Pennisi (2013) Science 341: 833-836.
[0487] The CRISPR/Cas system can thus be used to edit a TCR and/or HLA gene (adding or deleting a basepair), or introducing a premature stop which thus decreases expression of a TCR and/or HLA. The CRISPR/Cas system can alternatively be used like RNA interference, turning off TCR and/or HLA gene in a reversible fashion. In a mammalian cell, for example, the RNA can guide the Cas protein to a TCR and/or HLA promoter, sterically blocking RNA polymerases.
[0488] Artificial CRISPR/Cas systems can be generated which inhibit TCR and/or HLA, using technology known in the art, e.g., that described in U.S. Publication No. 20140068797, and Cong (2013) Science 339: 819-823. Other artificial CRISPR/Cas systems that are known in the art may also be generated which inhibit TCR and/or HLA, e.g., that described in Tsai (2014) Nature Biotechnol., 32:6 569-576, U.S. Pat. Nos. 8,871,445; 8,865,406; 8,795,965; 8,771,945; and 8,697,359.
TALEN to Inhibit TCR and/or HLA
[0489] "TALEN" or "TALEN to HLA and/or TCR" or "TALEN to inhibit HLA and/or TCR" refers to a transcription activator-like effector nuclease, an artificial nuclease which can be used to edit the HLA and/or TCR gene.
[0490] TALENs are produced artificially by fusing a TAL effector DNA binding domain to a DNA cleavage domain. Transcription activator-like effects (TALEs) can be engineered to bind any desired DNA sequence, including a portion of the HLA or TCR gene. By combining an engineered TALE with a DNA cleavage domain, a restriction enzyme can be produced which is specific to any desired DNA sequence, including a HLA or TCR sequence. These can then be introduced into a cell, wherein they can be used for genome editing. Boch (2011) Nature Biotech. 29: 135-6; and Boch et al. (2009) Science 326: 1509-12; Moscou et al. (2009) Science 326: 3501.
[0491] TALEs are proteins secreted by Xanthomonas bacteria. The DNA binding domain contains a repeated, highly conserved 33-34 amino acid sequence, with the exception of the 12th and 13th amino acids. These two positions are highly variable, showing a strong correlation with specific nucleotide recognition. They can thus be engineered to bind to a desired DNA sequence.
[0492] To produce a TALEN, a TALE protein is fused to a nuclease (N), which is a wild-type or mutated FokI endonuclease. Several mutations to FokI have been made for its use in TALENs; these, for example, improve cleavage specificity or activity. Cermak et al. (2011) Nucl. Acids Res. 39: e82; Miller et al. (2011) Nature Biotech. 29: 143-8; Hockemeyer et al. (2011) Nature Biotech. 29: 731-734; Wood et al. (2011) Science 333: 307; Doyon et al. (2010) Nature Methods 8: 74-79; Szczepek et al. (2007) Nature Biotech. 25: 786-793; and Guo et al. (2010) J. Mol. Biol. 200: 96.
[0493] The FokI domain functions as a dimer, requiring two constructs with unique DNA binding domains for sites in the target genome with proper orientation and spacing. Both the number of amino acid residues between the TALE DNA binding domain and the FokI cleavage domain and the number of bases between the two individual TALEN binding sites appear to be important parameters for achieving high levels of activity. Miller et al. (2011) Nature Biotech. 29: 143-8.
[0494] A HLA or TCR TALEN can be used inside a cell to produce a double-stranded break (DSB). A mutation can be introduced at the break site if the repair mechanisms improperly repair the break via non-homologous end joining. For example, improper repair may introduce a frame shift mutation. Alternatively, foreign DNA can be introduced into the cell along with the TALEN; depending on the sequences of the foreign DNA and chromosomal sequence, this process can be used to correct a defect in the HLA or TCR gene or introduce such a defect into a wt HLA or TCR gene, thus decreasing expression of HLA or TCR.
[0495] TALENs specific to sequences in HLA or TCR can be constructed using any method known in the art, including various schemes using modular components. Zhang et al. (2011) Nature Biotech. 29: 149-53; Geibler et al. (2011) PLoS ONE 6: e19509.
Zinc Finger Nuclease to Inhibit HLA and/or TCR
[0496] "ZFN" or "Zinc Finger Nuclease" or "ZFN to HLA and/or TCR" or "ZFN to inhibit HLA and/or TCR" refer to a zinc finger nuclease, an artificial nuclease which can be used to edit the HLA and/or TCR gene.
[0497] Like a TALEN, a ZFN comprises a FokI nuclease domain (or derivative thereof) fused to a DNA-binding domain. In the case of a ZFN, the DNA-binding domain comprises one or more zinc fingers. Carroll et al. (2011) Genetics Society of America 188: 773-782; and Kim et al. (1996) Proc. Natl. Acad. Sci. USA 93: 1156-1160.
[0498] A zinc finger is a small protein structural motif stabilized by one or more zinc ions. A zinc finger can comprise, for example, Cys2His2, and can recognize an approximately 3-bp sequence. Various zinc fingers of known specificity can be combined to produce multi-finger polypeptides which recognize about 6, 9, 12, 15 or 18-bp sequences. Various selection and modular assembly techniques are available to generate zinc fingers (and combinations thereof) recognizing specific sequences, including phage display, yeast one-hybrid systems, bacterial one-hybrid and two-hybrid systems, and mammalian cells.
[0499] Like a TALEN, a ZFN must dimerize to cleave DNA. Thus, a pair of ZFNs are required to target non-palindromic DNA sites. The two individual ZFNs must bind opposite strands of the DNA with their nucleases properly spaced apart. Bitinaite et al. (1998) Proc. Natl. Acad. Sci. USA 95: 10570-5.
[0500] Also like a TALEN, a ZFN can create a double-stranded break in the DNA, which can create a frame-shift mutation if improperly repaired, leading to a decrease in the expression and amount of HLA and/or TCR in a cell. ZFNs can also be used with homologous recombination to mutate in the HLA or TCR gene.
[0501] ZFNs specific to sequences in HLA AND/OR TCR can be constructed using any method known in the art. See, e.g., Provasi (2011) Nature Med. 18: 807-815; Torikai (2013) Blood 122: 1341-1349; Cathomen et al. (2008) Mol. Ther. 16: 1200-7; and Guo et al. (2010) J. Mol. Biol. 400: 96; U.S. Patent Publication 2011/0158957; and U.S. Patent Publication 2012/0060230.
Telomerase Expression
[0502] While not wishing to be bound by any particular theory, in some embodiments, a therapeutic T cell has short term persistence in a patient, due to shortened telomeres in the T cell; accordingly, transfection with a telomerase gene can lengthen the telomeres of the T cell and improve persistence of the T cell in the patient. See Carl June, "Adoptive T cell therapy for cancer in the clinic", Journal of Clinical Investigation, 117:1466-1476 (2007). Thus, in an embodiment, an immune effector cell, e.g., a T cell, ectopically expresses a telomerase subunit, e.g., the catalytic subunit of telomerase, e.g., TERT, e.g., hTERT. In some aspects, this disclosure provides a method of producing a CAR-expressing cell, comprising contacting a cell with a nucleic acid encoding a telomerase subunit, e.g., the catalytic subunit of telomerase, e.g., TERT, e.g., hTERT. The cell may be contacted with the nucleic acid before, simultaneous with, or after being contacted with a construct encoding a CAR.
[0503] In one aspect, the disclosure features a method of making a population of immune effector cells (e.g., T cells or NK cells). In an embodiment, the method comprises: providing a population of immune effector cells (e.g., T cells or NK cells), contacting the population of immune effector cells with a nucleic acid encoding a CAR; and contacting the population of immune effector cells with a nucleic acid encoding a telomerase subunit, e.g., hTERT, under conditions that allow for CAR and telomerase expression.
[0504] In an embodiment, the nucleic acid encoding the telomerase subunit is DNA. In an embodiment, the nucleic acid encoding the telomerase subunit comprises a promoter capable of driving expression of the telomerase subunit.
[0505] In an embodiment, hTERT has the amino acid sequence of GenBank Protein ID AAC51724.1 (Meyerson et al., "hEST2, the Putative Human Telomerase Catalytic Subunit Gene, Is Up-Regulated in Tumor Cells and during Immortalization" Cell Volume 90, Issue 4, 22 Aug. 1997, Pages 785-795) as follows:
TABLE-US-00012 (SEQ ID NO: 131) MPRAPRCRAVRSLLRSHYREVLPLATFVRRLGPQGWRLVQRGDPAAFRAL VAQCLVCVPWDARPPPAAPSFRQVSCLKELVARVLQRLCERGAKNVLAFG FALLDGARGGPPEAFTTSVRSYLPNTVTDALRGSGAWGLLLRRVGDDVLV HLLARCALFVLVAPSCAYQVCGPPLYQLGAATQARPPPHASGPRRRLGCE RAWNHSVREAGVPLGLPAPGARRRGGSASRSLPLPKRPRRGAAPEPERTP VGQGSWAHPGRTRGPSDRGFCVVSPARPAEEATSLEGALSGTRHSHPSVG RQHHAGPPSTSRPPRPWDTPCPPVYAETKHFLYSSGDKEQLRPSFLLSSL RPSLTGARRLVETIFLGSRPWMPGTPRRLPRLPQRYWQMRPLFLELLGNH AQCPYGVLLKTHCPLRAAVTPAAGVCAREKPQGSVAAPEEEDTDPRRLVQ LLRQHSSPWQVYGFVRACLRRLVPPGLWGSRHNERRFLRNTKKFISLGKH AKLSLQELTWKMSVRGCAWLRRSPGVGCVPAAEHRLREEILAKFLHWLMS VYVVELLRSFFYVTETTFQKNRLFFYRKSVWSKLQSIGIRQHLKRVQLRE LSEAEVRQHREARPALLTSRLRFIPKPDGLRPIVNMDYVVGARTFRREKR AERLTSRVKALFSVLNYERARRPGLLGASVLGLDDIHRAWRTFVLRVRAQ DPPPELYFVKVDVTGAYDTIPQDRLTEVIASIIKPQNTYCVRRYAVVQKA AHGHVRKAFKSHVSTLTDLQPYMRQFVAHLQETSPLRDAVVIEQSSSLNE ASSGLFDVFLRFMCHHAVRIRGKSYVQCQGIPQGSILSTLLCSLCYGDME NKLFAGIRRDGLLLRLVDDFLLVTPHLTHAKTFLRTLVRGVPEYGCVVNL RKTVVNFPVEDEALGGTAFVQMPAHGLFPWCGLLLDTRTLEVQSDYSSYA RTSIRASLTFNRGFKAGRNMRRKLFGVLRLKCHSLFLDLQVNSLQTVCTN IYKILLLQAYRFHACVLQLPFHQQVWKNPTFFLRVISDTASLCYSILKAK NAGMSLGAKGAAGPLPSEAVQWLCHQAFLLKLTRHRVTYVPLLGSLRTAQ TQLSRKLPGTTLTALEAAANPALPSDFKTILD
[0506] In an embodiment, the hTERT has a sequence at least 80%, 85%, 90%, 95%, 96{circumflex over ( )}, 97%, 98%, or 99% identical to the sequence of SEQ ID NO: 131. In an embodiment, the hTERT has a sequence of SEQ ID NO: 131. In an embodiment, the hTERT comprises a deletion (e.g., of no more than 5, 10, 15, 20, or 30 amino acids) at the N-terminus, the C-terminus, or both. In an embodiment, the hTERT comprises a transgenic amino acid sequence (e.g., of no more than 5, 10, 15, 20, or 30 amino acids) at the N-terminus, the C-terminus, or both.
[0507] In an embodiment, the hTERT is encoded by the nucleic acid sequence of GenBank Accession No. AF018167 (Meyerson et al., "hEST2, the Putative Human Telomerase Catalytic Subunit Gene, Is Up-Regulated in Tumor Cells and during Immortalization" Cell Volume 90, Issue 4, 22 Aug. 1997, Pages 785-795):
TABLE-US-00013 (SEQ ID NO: 132) 1 caggcagcgt ggtcctgctg cgcacgtggg aagccctggc cccggccacc cccgcgatgc 61 cgcgcgctcc ccgctgccga gccgtgcgct ccctgctgcg cagccactac cgcgaggtgc 121 tgccgctggc cacgttcgtg cggcgcctgg ggccccaggg ctggcggctg gtgcagcgcg 181 gggacccggc ggctttccgc gcgctggtgg cccagtgcct ggtgtgcgtg ccctgggacg 241 cacggccgcc ccccgccgcc ccctccttcc gccaggtgtc ctgcctgaag gagctggtgg 301 cccgagtgct gcagaggctg tgcgagcgcg gcgcgaagaa cgtgctggcc ttcggcttcg 361 cgctgctgga cggggcccgc gggggccccc ccgaggcctt caccaccagc gtgcgcagct 421 acctgcccaa cacggtgacc gacgcactgc gggggagcgg ggcgtggggg ctgctgttgc 481 gccgcgtggg cgacgacgtg ctggttcacc tgctggcacg ctgcgcgctc tttgtgctgg 541 tggctcccag ctgcgcctac caggtgtgcg ggccgccgct gtaccagctc ggcgctgcca 601 ctcaggcccg gcccccgcca cacgctagtg gaccccgaag gcgtctggga tgcgaacggg 661 cctggaacca tagcgtcagg gaggccgggg tccccctggg cctgccagcc ccgggtgcga 721 ggaggcgcgg gggcagtgcc agccgaagtc tgccgttgcc caagaggccc aggcgtggcg 781 ctgcccctga gccggagcgg acgcccgttg ggcaggggtc ctgggcccac ccgggcagga 841 cgcgtggacc gagtgaccgt ggtttctgtg tggtgtcacc tgccagaccc gccgaagaag 901 ccacctcttt ggagggtgcg ctctctggca cgcgccactc ccacccatcc gtgggccgcc 961 agcaccacgc gggcccccca tccacatcgc ggccaccacg tccctgggac acgccttgtc 1021 ccccggtgta cgccgagacc aagcacttcc tctactcctc aggcgacaag gagcagctgc 1081 ggccctcctt cctactcagc tctctgaggc ccagcctgac tggcgctcgg aggctcgtgg 1141 agaccatctt tctgggttcc aggccctgga tgccagggac tccccgcagg ttgccccgcc 1201 tgccccagcg ctactggcaa atgcggcccc tgtttctgga gctgcttggg aaccacgcgc 1261 agtgccccta cggggtgctc ctcaagacgc actgcccgct gcgagctgcg gtcaccccag 1321 cagccggtgt ctgtgcccgg gagaagcccc agggctctgt ggcggccccc gaggaggagg 1381 acacagaccc ccgtcgcctg gtgcagctgc tccgccagca cagcagcccc tggcaggtgt 1441 acggcttcgt gcgggcctgc ctgcgccggc tggtgccccc aggcctctgg ggctccaggc 1501 acaacgaacg ccgcttcctc aggaacacca agaagttcat ctccctgggg aagcatgcca 1561 agctctcgct gcaggagctg acgtggaaga tgagcgtgcg gggctgcgct tggctgcgca 1621 ggagcccagg ggttggctgt gttccggccg cagagcaccg tctgcgtgag gagatcctgg 1681 ccaagttcct gcactggctg atgagtgtgt acgtcgtcga gctgctcagg tctttctttt 1741 atgtcacgga gaccacgttt caaaagaaca ggctcttttt ctaccggaag agtgtctgga 1801 gcaagttgca aagcattgga atcagacagc acttgaagag ggtgcagctg cgggagctgt 1861 cggaagcaga ggtcaggcag catcgggaag ccaggcccgc cctgctgacg tccagactcc 1921 gcttcatccc caagcctgac gggctgcggc cgattgtgaa catggactac gtcgtgggag 1981 ccagaacgtt ccgcagagaa aagagggccg agcgtctcac ctcgagggtg aaggcactgt 2041 tcagcgtgct caactacgag cgggcgcggc gccccggcct cctgggcgcc tctgtgctgg 2101 gcctggacga tatccacagg gcctggcgca ccttcgtgct gcgtgtgcgg gcccaggacc 2161 cgccgcctga gctgtacttt gtcaaggtgg atgtgacggg cgcgtacgac accatccccc 2221 aggacaggct cacggaggtc atcgccagca tcatcaaacc ccagaacacg tactgcgtgc 2281 gtcggtatgc cgtggtccag aaggccgccc atgggcacgt ccgcaaggcc ttcaagagcc 2341 acgtctctac cttgacagac ctccagccgt acatgcgaca gttcgtggct cacctgcagg 2401 agaccagccc gctgagggat gccgtcgtca tcgagcagag ctcctccctg aatgaggcca 2461 gcagtggcct cttcgacgtc ttcctacgct tcatgtgcca ccacgccgtg cgcatcaggg 2521 gcaagtccta cgtccagtgc caggggatcc cgcagggctc catcctctcc acgctgctct 2581 gcagcctgtg ctacggcgac atggagaaca agctgtttgc ggggattcgg cgggacgggc 2641 tgctcctgcg tttggtggat gatttcttgt tggtgacacc tcacctcacc cacgcgaaaa 2701 ccttcctcag gaccctggtc cgaggtgtcc ctgagtatgg ctgcgtggtg aacttgcgga 2761 agacagtggt gaacttccct gtagaagacg aggccctggg tggcacggct tttgttcaga 2821 tgccggccca cggcctattc ccctggtgcg gcctgctgct ggatacccgg accctggagg 2881 tgcagagcga ctactccagc tatgcccgga cctccatcag agccagtctc accttcaacc 2941 gcggcttcaa ggctgggagg aacatgcgtc gcaaactctt tggggtcttg cggctgaagt 3001 gtcacagcct gtttctggat ttgcaggtga acagcctcca gacggtgtgc accaacatct 3061 acaagatcct cctgctgcag gcgtacaggt ttcacgcatg tgtgctgcag ctcccatttc 3121 atcagcaagt ttggaagaac cccacatttt tcctgcgcgt catctctgac acggcctccc 3181 tctgctactc catcctgaaa gccaagaacg cagggatgtc gctgggggcc aagggcgccg 3241 ccggccctct gccctccgag gccgtgcagt ggctgtgcca ccaagcattc ctgctcaagc 3301 tgactcgaca ccgtgtcacc tacgtgccac tcctggggtc actcaggaca gcccagacgc 3361 agctgagtcg gaagctcccg gggacgacgc tgactgccct ggaggccgca gccaacccgg 3421 cactgccctc agacttcaag accatcctgg actgatggcc acccgcccac agccaggccg 3481 agagcagaca ccagcagccc tgtcacgccg ggctctacgt cccagggagg gaggggcggc 3541 ccacacccag gcccgcaccg ctgggagtct gaggcctgag tgagtgtttg gccgaggcct 3601 gcatgtccgg ctgaaggctg agtgtccggc tgaggcctga gcgagtgtcc agccaagggc 3661 tgagtgtcca gcacacctgc cgtcttcact tccccacagg ctggcgctcg gctccacccc 3721 agggccagct tttcctcacc aggagcccgg cttccactcc ccacatagga atagtccatc 3781 cccagattcg ccattgttca cccctcgccc tgccctcctt tgccttccac ccccaccatc 3841 caggtggaga ccctgagaag gaccctggga gctctgggaa tttggagtga ccaaaggtgt 3901 gccctgtaca caggcgagga ccctgcacct ggatgggggt ccctgtgggt caaattgggg 3961 ggaggtgctg tgggagtaaa atactgaata tatgagtttt tcagttttga aaaaaaaaaa 4021 aaaaaaa
[0508] In an embodiment, the hTERT is encoded by a nucleic acid having a sequence at least 80%, 85%, 90%, 95%, 96, 97%, 98%, or 99% identical to the sequence of SEQ ID NO: 132. In an embodiment, the hTERT is encoded by a nucleic acid of SEQ ID NO: 132.
Activation and Expansion of Immune Effector Cells (e.g., T Cells)
[0509] Immune effector cells such as T cells may be activated and expanded generally using methods as described, for example, in U.S. Pat. Nos. 6,352,694; 6,534,055; 6,905,680; 6,692,964; 5,858,358; 6,887,466; 6,905,681; 7,144,575; 7,067,318; 7,172,869; 7,232,566; 7,175,843; 5,883,223; 6,905,874; 6,797,514; 6,867,041; and U.S. Patent Application Publication No. 20060121005.
[0510] The procedure for ex vivo expansion of hematopoietic stem and progenitor cells is described in U.S. Pat. No. 5,199,942, incorporated herein by reference, can be applied to the cells of the present invention. Other suitable methods are known in the art, therefore the present invention is not limited to any particular method of ex vivo expansion of the cells. Briefly, ex vivo culture and expansion of T cells can comprise: (1) collecting CD34+ hematopoietic stem and progenitor cells from a mammal from peripheral blood harvest or bone marrow explants; and (2) expanding such cells ex vivo. In addition to the cellular growth factors described in U.S. Pat. No. 5,199,942, other factors such as flt3-L, IL-1, IL-3 and c-kit ligand, can be used for culturing and expansion of the cells.
[0511] Generally, a population of immune effector cells e.g., T regulatory cell depleted cells, may be expanded by contact with a surface having attached thereto an agent that stimulates a CD3/TCR complex associated signal and a ligand that stimulates a costimulatory molecule on the surface of the T cells. In particular, T cell populations may be stimulated as described herein, such as by contact with an anti-CD3 antibody, or antigen-binding fragment thereof, or an anti-CD2 antibody immobilized on a surface, or by contact with a protein kinase C activator (e.g., bryostatin) in conjunction with a calcium ionophore. For co-stimulation of an accessory molecule on the surface of the T cells, a ligand that binds the accessory molecule is used. For example, a population of T cells can be contacted with an anti-CD3 antibody and an anti-CD28 antibody, under conditions appropriate for stimulating proliferation of the T cells. To stimulate proliferation of either CD4+ T cells or CD8+ T cells, an anti-CD3 antibody and an anti-CD28 antibody can be used. Examples of an anti-CD28 antibody include 9.3, B-T3, XR-CD28 (Diaclone, Besancon, France) can be used as can other methods commonly known in the art (Berg et al., Transplant Proc. 30(8):3975-3977, 1998; Haanen et al., J. Exp. Med. 190(9):13191328, 1999; Garland et al., J. Immunol Meth. 227(1-2):53-63, 1999).
[0512] In certain aspects, the primary stimulatory signal and the costimulatory signal for the T cell may be provided by different protocols. For example, the agents providing each signal may be in solution or coupled to a surface. When coupled to a surface, the agents may be coupled to the same surface (i.e., in "cis" formation) or to separate surfaces (i.e., in "trans" formation). Alternatively, one agent may be coupled to a surface and the other agent in solution. In one aspect, the agent providing the costimulatory signal is bound to a cell surface and the agent providing the primary activation signal is in solution or coupled to a surface. In certain aspects, both agents can be in solution. In one aspect, the agents may be in soluble form, and then cross-linked to a surface, such as a cell expressing Fc receptors or an antibody or other binding agent which will bind to the agents. In this regard, see for example, U.S. Patent Application Publication Nos. 20040101519 and 20060034810 for artificial antigen presenting cells (aAPCs) that are contemplated for use in activating and expanding T cells in the present invention.
[0513] In one aspect, the two agents are immobilized on beads, either on the same bead, i.e., "cis," or to separate beads, i.e., "trans." By way of example, the agent providing the primary activation signal is an anti-CD3 antibody or an antigen-binding fragment thereof and the agent providing the costimulatory signal is an anti-CD28 antibody or antigen-binding fragment thereof; and both agents are co-immobilized to the same bead in equivalent molecular amounts. In one aspect, a 1:1 ratio of each antibody bound to the beads for CD4+ T cell expansion and T cell growth is used. In certain aspects of the present invention, a ratio of anti CD3:CD28 antibodies bound to the beads is used such that an increase in T cell expansion is observed as compared to the expansion observed using a ratio of 1:1. In one particular aspect an increase of from about 1 to about 3 fold is observed as compared to the expansion observed using a ratio of 1:1. In one aspect, the ratio of CD3:CD28 antibody bound to the beads ranges from 100:1 to 1:100 and all integer values there between. In one aspect, more anti-CD28 antibody is bound to the particles than anti-CD3 antibody, i.e., the ratio of CD3:CD28 is less than one. In certain aspects, the ratio of anti CD28 antibody to anti CD3 antibody bound to the beads is greater than 2:1. In one particular aspect, a 1:100 CD3:CD28 ratio of antibody bound to beads is used. In one aspect, a 1:75 CD3:CD28 ratio of antibody bound to beads is used. In a further aspect, a 1:50 CD3:CD28 ratio of antibody bound to beads is used. In one aspect, a 1:30 CD3:CD28 ratio of antibody bound to beads is used. In one preferred aspect, a 1:10 CD3:CD28 ratio of antibody bound to beads is used. In one aspect, a 1:3 CD3:CD28 ratio of antibody bound to the beads is used. In yet one aspect, a 3:1 CD3:CD28 ratio of antibody bound to the beads is used.
[0514] Ratios of particles to cells from 1:500 to 500:1 and any integer values in between may be used to stimulate T cells or other target cells. As those of ordinary skill in the art can readily appreciate, the ratio of particles to cells may depend on particle size relative to the target cell. For example, small sized beads could only bind a few cells, while larger beads could bind many. In certain aspects the ratio of cells to particles ranges from 1:100 to 100:1 and any integer values in-between and in further aspects the ratio comprises 1:9 to 9:1 and any integer values in between, can also be used to stimulate T cells. The ratio of anti-CD3- and anti-CD28-coupled particles to T cells that result in T cell stimulation can vary as noted above, however certain preferred values include 1:100, 1:50, 1:40, 1:30, 1:20, 1:10, 1:9, 1:8, 1:7, 1:6, 1:5, 1:4, 1:3, 1:2, 1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1, 10:1, and 15:1 with one preferred ratio being at least 1:1 particles per T cell. In one aspect, a ratio of particles to cells of 1:1 or less is used. In one particular aspect, a preferred particle: cell ratio is 1:5. In further aspects, the ratio of particles to cells can be varied depending on the day of stimulation. For example, in one aspect, the ratio of particles to cells is from 1:1 to 10:1 on the first day and additional particles are added to the cells every day or every other day thereafter for up to 10 days, at final ratios of from 1:1 to 1:10 (based on cell counts on the day of addition). In one particular aspect, the ratio of particles to cells is 1:1 on the first day of stimulation and adjusted to 1:5 on the third and fifth days of stimulation. In one aspect, particles are added on a daily or every other day basis to a final ratio of 1:1 on the first day, and 1:5 on the third and fifth days of stimulation. In one aspect, the ratio of particles to cells is 2:1 on the first day of stimulation and adjusted to 1:10 on the third and fifth days of stimulation. In one aspect, particles are added on a daily or every other day basis to a final ratio of 1:1 on the first day, and 1:10 on the third and fifth days of stimulation. One of skill in the art will appreciate that a variety of other ratios may be suitable for use in the present invention. In particular, ratios will vary depending on particle size and on cell size and type. In one aspect, the most typical ratios for use are in the neighborhood of 1:1, 2:1 and 3:1 on the first day.
[0515] In further aspects, the cells, such as T cells, are combined with agent-coated beads, the beads and the cells are subsequently separated, and then the cells are cultured. In an alternative aspect, prior to culture, the agent-coated beads and cells are not separated but are cultured together. In a further aspect, the beads and cells are first concentrated by application of a force, such as a magnetic force, resulting in increased ligation of cell surface markers, thereby inducing cell stimulation.
[0516] By way of example, cell surface proteins may be ligated by allowing paramagnetic beads to which anti-CD3 and anti-CD28 are attached (3.times.28 beads) to contact the T cells. In one aspect the cells (for example, 10.sup.4 to 10.sup.9 T cells) and beads (for example, DYNABEADS.RTM. M-450 CD3/CD28 T paramagnetic beads at a ratio of 1:1) are combined in a buffer, for example PBS (without divalent cations such as, calcium and magnesium). Again, those of ordinary skill in the art can readily appreciate any cell concentration may be used. For example, the target cell may be very rare in the sample and comprise only 0.01% of the sample or the entire sample (i.e., 100%) may comprise the target cell of interest. Accordingly, any cell number is within the context of the present invention. In certain aspects, it may be desirable to significantly decrease the volume in which particles and cells are mixed together (i.e., increase the concentration of cells), to ensure maximum contact of cells and particles. For example, in one aspect, a concentration of about 10 billion cells/ml, 9 billion/ml, 8 billion/ml, 7 billion/ml, 6 billion/ml, 5 billion/ml, or 2 billion cells/ml is used. In one aspect, greater than 100 million cells/ml is used. In a further aspect, a concentration of cells of 10, 15, 20, 25, 30, 35, 40, 45, or 50 million cells/ml is used. In yet one aspect, a concentration of cells from 75, 80, 85, 90, 95, or 100 million cells/ml is used. In further aspects, concentrations of 125 or 150 million cells/ml can be used. Using high concentrations can result in increased cell yield, cell activation, and cell expansion. Further, use of high cell concentrations allows more efficient capture of cells that may weakly express target antigens of interest, such as CD28-negative T cells. Such populations of cells may have therapeutic value and would be desirable to obtain in certain aspects. For example, using high concentration of cells allows more efficient selection of CD8+ T cells that normally have weaker CD28 expression.
[0517] In one embodiment, cells transduced with a nucleic acid encoding a CAR, e.g., a CAR described herein, are expanded, e.g., by a method described herein. In one embodiment, the cells are expanded in culture for a period of several hours (e.g., about 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 18, 21 hours) to about 14 days (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 or 14 days). In one embodiment, the cells are expanded for a period of 4 to 9 days. In one embodiment, the cells are expanded for a period of 8 days or less, e.g., 7, 6 or 5 days. In one embodiment, the cells, e.g., a CD19 CAR cell described herein, are expanded in culture for 5 days, and the resulting cells are more potent than the same cells expanded in culture for 9 days under the same culture conditions. Potency can be defined, e.g., by various T cell functions, e.g. proliferation, target cell killing, cytokine production, activation, migration, or combinations thereof. In one embodiment, the cells, e.g., a CD19 CAR cell described herein, expanded for 5 days show at least a one, two, three or four fold increase in cells doublings upon antigen stimulation as compared to the same cells expanded in culture for 9 days under the same culture conditions. In one embodiment, the cells, e.g., the cells expressing a CD19 CAR described herein, are expanded in culture for 5 days, and the resulting cells exhibit higher proinflammatory cytokine production, e.g., IFN-.gamma. and/or GM-CSF levels, as compared to the same cells expanded in culture for 9 days under the same culture conditions. In one embodiment, the cells, e.g., a CD19 CAR cell described herein, expanded for 5 days show at least a one, two, three, four, five, ten fold or more increase in pg/ml of proinflammatory cytokine production, e.g., IFN-.gamma. and/or GM-CSF levels, as compared to the same cells expanded in culture for 9 days under the same culture conditions.
[0518] Several cycles of stimulation may also be desired such that culture time of T cells can be 60 days or more. Conditions appropriate for T cell culture include an appropriate media (e.g., Minimal Essential Media or RPMI Media 1640 or, X-vivo 15, (Lonza)) that may contain factors necessary for proliferation and viability, including serum (e.g., fetal bovine or human serum), interleukin-2 (IL-2), insulin, IFN-.gamma., IL-4, IL-7, GM-CSF, IL-10, IL-12, IL-15, TGF.beta., and TNF-.alpha. or any other additives for the growth of cells known to the skilled artisan. Other additives for the growth of cells include, but are not limited to, surfactant, plasmanate, and reducing agents such as N-acetyl-cysteine and 2-mercaptoethanol. Media can include RPMI 1640, AIM-V, DMEM, MEM, .alpha.-MEM, F-12, X-Vivo 15, and X-Vivo 20, Optimizer, with added amino acids, sodium pyruvate, and vitamins, either serum-free or supplemented with an appropriate amount of serum (or plasma) or a defined set of hormones, and/or an amount of cytokine(s) sufficient for the growth and expansion of T cells. Antibiotics, e.g., penicillin and streptomycin, are included only in experimental cultures, not in cultures of cells that are to be infused into a subject. The target cells are maintained under conditions necessary to support growth, for example, an appropriate temperature (e.g., 37.degree. C.) and atmosphere (e.g., air plus 5% CO.sub.2).
[0519] In one embodiment, the cells are expanded in an appropriate media (e.g., media described herein) that includes one or more interleukin that result in at least a 200-fold (e.g., 200-fold, 250-fold, 300-fold, 350-fold) increase in cells over a 14 day expansion period, e.g., as measured by a method described herein such as flow cytometry. In one embodiment, the cells are expanded in the presence of IL-15 and/or IL-7 (e.g., IL-15 and IL-7).
[0520] In embodiments, methods described herein, e.g., CAR-expressing cell manufacturing methods, comprise removing T regulatory cells, e.g., CD25+ T cells, from a cell population, e.g., using an anti-CD25 antibody, or fragment thereof, or a CD25-binding ligand, IL-2. Methods of removing T regulatory cells, e.g., CD25+ T cells, from a cell population are described herein. In embodiments, the methods, e.g., manufacturing methods, further comprise contacting a cell population (e.g., a cell population in which T regulatory cells, such as CD25+ T cells, have been depleted; or a cell population that has previously contacted an anti-CD25 antibody, fragment thereof, or CD25-binding ligand) with IL-15 and/or IL-7. For example, the cell population (e.g., that has previously contacted an anti-CD25 antibody, fragment thereof, or CD25-binding ligand) is expanded in the presence of IL-15 and/or IL-7.
[0521] In some embodiments a CAR-expressing cell described herein is contacted with a composition comprising a interleukin-15 (IL-15) polypeptide, a interleukin-15 receptor alpha (IL-15Ra) polypeptide, or a combination of both a IL-15 polypeptide and a IL-15Ra polypeptide e.g., hetIL-15, during the manufacturing of the CAR-expressing cell, e.g., ex vivo. In embodiments, a CAR-expressing cell described herein is contacted with a composition comprising a IL-15 polypeptide during the manufacturing of the CAR-expressing cell, e.g., ex vivo. In embodiments, a CAR-expressing cell described herein is contacted with a composition comprising a combination of both a IL-15 polypeptide and a IL-15 Ra polypeptide during the manufacturing of the CAR-expressing cell, e.g., ex vivo. In embodiments, a CAR-expressing cell described herein is contacted with a composition comprising hetIL-15 during the manufacturing of the CAR-expressing cell, e.g., ex vivo.
[0522] In one embodiment the CAR-expressing cell described herein is contacted with a composition comprising hetIL-15 during ex vivo expansion. In an embodiment, the CAR-expressing cell described herein is contacted with a composition comprising an IL-15 polypeptide during ex vivo expansion. In an embodiment, the CAR-expressing cell described herein is contacted with a composition comprising both an IL-15 polypeptide and an IL-15Ra polypeptide during ex vivo expansion. In one embodiment the contacting results in the survival and proliferation of a lymphocyte subpopulation, e.g., CD8+ T cells.
[0523] T cells that have been exposed to varied stimulation times may exhibit different characteristics. For example, typical blood or apheresed peripheral blood mononuclear cell products have a helper T cell population (TH, CD4+) that is greater than the cytotoxic or suppressor T cell population (TC, CD8+). Ex vivo expansion of T cells by stimulating CD3 and CD28 receptors produces a population of T cells that prior to about days 8-9 consists predominately of TH cells, while after about days 8-9, the population of T cells comprises an increasingly greater population of TC cells. Accordingly, depending on the purpose of treatment, infusing a subject with a T cell population comprising predominately of TH cells may be advantageous. Similarly, if an antigen-specific subset of TC cells has been isolated it may be beneficial to expand this subset to a greater degree.
[0524] Further, in addition to CD4 and CD8 markers, other phenotypic markers vary significantly, but in large part, reproducibly during the course of the cell expansion process. Thus, such reproducibility enables the ability to tailor an activated T cell product for specific purposes.
[0525] In other embodiments, the method of making disclosed herein further comprises contacting the population of immune effector cells with a nucleic acid encoding a telomerase subunit, e.g., hTERT. The the nucleic acid encoding the telomerase subunit can be DNA.
[0526] In some embodiments, a kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib) is added during the CAR cell manufacturing process. According to the non-limiting theory herein, the kinase inhibitor can improve the quality of the population of cells produced. For instance, CAR-expressing cells are often produced from a cancer patient's own plasma apheresis sample, which can contain cancer cells, and the kinase inhibitor can alter signalling in those cancer cells (e.g., a BTK-expressing cancer such as CLL or MCL), e.g., reducing their proliferation or increasing levels of apoptosis. As another example, the kinase inhibitor may alter signalling in the CAR-expressing cells (or immune effector cells before they express CAR), e.g., by inhibiting ITK in T cells. The kinase inhibitor may shift the balance of T cells from TH2 cells towards TH1 cells.
[0527] The kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib) can be added to the reaction mixture in a level sufficient to inhibit its target, e.g., BTK. In some embodiments, the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib) is added at a concentration of about 0.1-0.2, 0.2-0.5, 0.5-1, 1-2, 2-5, or 5-10 .mu.M. In some embodiments, the kinase inhibitor is a covalent inhibitor (such as ibrutinib) and a short pulse is sufficient to irreversibly inactivate the target while avoiding nonspecific toxicity. Consequently, the kinase inhibitor may be added for, e.g., 10-20, 20-30, 30-40, 40-60, or 60-120 minutes. The kinase inhibitor may also be added for longer periods of time, for instance if the kinase inhibitor has a noncovalent mode of action. Thus, the kinase inhibitor may be added for, e.g., 2-4, 4-6, 6-8, 8-12, 12-18, or 18-24 hours, or for 1-2, 2-3, 3-4, 4-6, 6-8, 8-10 days, or for the entire length of time the cells are being cultured. The kinase inhibitor may be added at various points during the manufacturing process, for example, after harvesting the cells, before stimulating with beads, after stimulating with beads, before transduction, after transduction, or before administration of the cells to the patient. In some embodiments, the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib) is added after harvesting the cells or before stimulating, e.g., with beads. Before and after, in this context, can refer to, e.g., about 1, 5, 15, 30, 45, or 60 minutes before or after, or 1, 2, 3, 4, 5, or 6 hours before or after.
[0528] Once a CD19 CAR is constructed, various assays can be used to evaluate the activity of the molecule, such as but not limited to, the ability to expand T cells following antigen stimulation, sustain T cell expansion in the absence of re-stimulation, and anti-cancer activities in appropriate in vitro and animal models. Assays to evaluate the effects of a CD19 CAR are described in further detail below
[0529] Western blot analysis of CAR expression in primary T cells can be used to detect the presence of monomers and dimers. See, e.g., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Very briefly, T cells (1:1 mixture of CD4.sup.+ and CD8.sup.+ T cells) expressing the CARs are expanded in vitro for more than 10 days followed by lysis and SDS-PAGE under reducing conditions. CARs containing the full length TCR-.zeta. cytoplasmic domain and the endogenous TCR-.zeta. chain are detected by western blotting using an antibody to the TCR-.zeta. chain. The same T cell subsets are used for SDS-PAGE analysis under non-reducing conditions to permit evaluation of covalent dimer formation.
[0530] In vitro expansion of CAR.sup.+ T cells following antigen stimulation can be measured by flow cytometry. For example, a mixture of CD4.sup.+ and CD8.sup.+ T cells are stimulated with .alpha.CD3/.alpha.CD28 beads followed by transduction with lentiviral vectors expressing GFP under the control of the promoters to be analyzed. Exemplary promoters include the CMV IE gene, EF-1.alpha., ubiquitin C, or phosphoglycerokinase (PGK) promoters. GFP fluorescence is evaluated on day 6 of culture in the CD4.sup.+ and/or CD8.sup.+ T cell subsets by flow cytometry. See, e.g., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Alternatively, a mixture of CD4.sup.+ and CD8.sup.+ T cells are stimulated with .alpha.CD3/.alpha.CD28 coated magnetic beads on day 0, and transduced with CAR on day 1 using a bicistronic lentiviral vector expressing CAR along with eGFP using a 2A ribosomal skipping sequence. Cultures are re-stimulated with either CD19.sup.+ K562 cells (K562-CD19), wild-type K562 cells (K562 wild type) or K562 cells expressing hCD32 and 4-1BBL in the presence of anti-CD3 and anti-CD28 antibody (K562-BBL-3/28) following washing. Exogenous IL-2 is added to the cultures every other day at 100 IU/ml. GFP.sup.+ T cells are enumerated by flow cytometry using bead-based counting. See, e.g., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009).
[0531] Sustained CAR.sup.+ T cell expansion in the absence of re-stimulation can also be measured. See, e.g., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Briefly, mean T cell volume (fl) is measured on day 8 of culture using a Coulter Multisizer particle counter, a Nexcelom Cellometer Vision or Millipore Scepter, following stimulation with .alpha.CD3/.alpha.CD28 coated magnetic beads on day 0, and transduction with the indicated CAR on day 1.
[0532] Animal models can also be used to measure a CART activity. For example, xenograft model using human CD19-specific CAR.sup.+ T cells to treat a primary human pre-B ALL in immunodeficient mice can be used. See, e.g., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Very briefly, after establishment of ALL, mice are randomized as to treatment groups. Different numbers of .alpha.CD19-.zeta. and .alpha.CD19-BB-.zeta. engineered T cells are coinjected at a 1:1 ratio into NOD-SCID-.gamma..sup.-/- mice bearing B-ALL. The number of copies of .alpha.CD19-.zeta. and .alpha.CD19-BB-.zeta. vector in spleen DNA from mice is evaluated at various times following T cell injection. Animals are assessed for leukemia at weekly intervals. Peripheral blood CD19.sup.+ B-ALL blast cell counts are measured in mice that are injected with .alpha.CD19-.zeta. CAR.sup.+ T cells or mock-transduced T cells. Survival curves for the groups are compared using the log-rank test. In addition, absolute peripheral blood CD4.sup.+ and CD8.sup.+ T cell counts 4 weeks following T cell injection in NOD-SCID-.gamma..sup.-/- mice can also be analyzed. Mice are injected with leukemic cells and 3 weeks later are injected with T cells engineered to express CAR by a bicistronic lentiviral vector that encodes the CAR linked to eGFP. T cells are normalized to 45-50% input GFP.sup.+ T cells by mixing with mock-transduced cells prior to injection, and confirmed by flow cytometry. Animals are assessed for leukemia at 1-week intervals. Survival curves for the CAR.sup.+ T cell groups are compared using the log-rank test.
[0533] Dose dependent CAR treatment response can be evaluated. See, e.g., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). For example, peripheral blood is obtained 35-70 days after establishing leukemia in mice injected on day 21 with CAR T cells, an equivalent number of mock-transduced T cells, or no T cells. Mice from each group are randomly bled for determination of peripheral blood CD19.sup.+ ALL blast counts and then killed on days 35 and 49. The remaining animals are evaluated on days 57 and 70.
[0534] Assessment of cell proliferation and cytokine production has been previously described, e.g., at Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Briefly, assessment of CAR-mediated proliferation is performed in microtiter plates by mixing washed T cells with K562 cells expressing CD19 (K19) or CD32 and CD137 (KT32-BBL) for a final T-cell:K562 ratio of 2:1. K562 cells are irradiated with gamma-radiation prior to use. Anti-CD3 (clone OKT3) and anti-CD28 (clone 9.3) monoclonal antibodies are added to cultures with KT32-BBL cells to serve as a positive control for stimulating T-cell proliferation since these signals support long-term CD8.sup.+ T cell expansion ex vivo. T cells are enumerated in cultures using CountBright.TM. fluorescent beads (Invitrogen, Carlsbad, Calif.) and flow cytometry as described by the manufacturer. CAR.sup.+ T cells are identified by GFP expression using T cells that are engineered with eGFP-2A linked CAR-expressing lentiviral vectors. For CAR+ T cells not expressing GFP, the CAR+ T cells are detected with biotinylated recombinant CD19 protein and a secondary avidin-PE conjugate. CD4+ and CD8.sup.+ expression on T cells are also simultaneously detected with specific monoclonal antibodies (BD Biosciences). Cytokine measurements are performed on supernatants collected 24 hours following re-stimulation using the human TH1/TH2 cytokine cytometric bead array kit (BD Biosciences, San Diego, Calif.) according the manufacturer's instructions. Fluorescence is assessed using a FACScalibur flow cytometer, and data is analyzed according to the manufacturer's instructions.
[0535] Cytotoxicity can be assessed by a standard .sup.51Cr-release assay. See, e.g., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Briefly, target cells (K562 lines and primary pro-B-ALL cells) are loaded with .sup.51Cr (as NaCrO.sub.4, New England Nuclear, Boston, Mass.) at 37.degree. C. for 2 hours with frequent agitation, washed twice in complete RPMI and plated into microtiter plates. Effector T cells are mixed with target cells in the wells in complete RPMI at varying ratios of effector cell:target cell (E:T). Additional wells containing media only (spontaneous release, SR) or a 1% solution of triton-X 100 detergent (total release, TR) are also prepared. After 4 hours of incubation at 37.degree. C., supernatant from each well is harvested. Released 51Cr is then measured using a gamma particle counter (Packard Instrument Co., Waltham, Mass.). Each condition is performed in at least triplicate, and the percentage of lysis is calculated using the formula: % Lysis=(ER-SR)/(TR-SR), where ER represents the average 51Cr released for each experimental condition.
[0536] Imaging technologies can be used to evaluate specific trafficking and proliferation of CARs in tumor-bearing animal models. Such assays have been described, for example, in Barrett et al., Human Gene Therapy 22:1575-1586 (2011). Briefly, NOD/SCID/.gamma.c.sup.-/- (NSG) mice are injected IV with Nalm-6 cells followed 7 days later with T cells 4 hour after electroporation with the CAR constructs. The T cells are stably transfected with a lentiviral construct to express firefly luciferase, and mice are imaged for bioluminescence. Alternatively, therapeutic efficacy and specificity of a single injection of CAR.sup.+ T cells in Nalm-6 xenograft model can be measured as the following: NSG mice are injected with Nalm-6 transduced to stably express firefly luciferase, followed by a single tail-vein injection of T cells electroporated with CD19 CAR 7 days later. Animals are imaged at various time points post injection. For example, photon-density heat maps of firefly luciferasepositive leukemia in representative mice at day 5 (2 days before treatment) and day 8 (24 hr post CAR.sup.+ PBLs) can be generated.
[0537] Other assays, including those described in the Example section herein as well as those that are known in the art can also be used to evaluate the CD19 CAR constructs of the invention.
Kinase Inhibitor
[0538] In one embodiment, the kinase inhibitor is a CDK4 inhibitor, e.g., a CDK4 inhibitor described herein, e.g., a CDK4/6 inhibitor, such as, e.g., 6-Acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylami- no)-8H-pyrido[2,3-d]pyrimidin-7-one, hydrochloride (also referred to as palbociclib or PD0332991). In one embodiment, the kinase inhibitor is a BTK inhibitor, e.g., a BTK inhibitor described herein, such as, e.g., ibrutinib. In one embodiment, the kinase inhibitor is an mTOR inhibitor, e.g., an mTOR inhibitor described herein, such as, e.g., rapamycin, a rapamycin analog, OSI-027. The mTOR inhibitor can be, e.g., an mTORC1 inhibitor and/or an mTORC2 inhibitor, e.g., an mTORC1 inhibitor and/or mTORC2 inhibitor described herein. In one embodiment, the kinase inhibitor is a MNK inhibitor, e.g., a MNK inhibitor described herein, such as, e.g., 4-amino-5-(4-fluoroanilino)-pyrazolo [3,4-d]pyrimidine. The MNK inhibitor can be, e.g., a MNK1a, MNK1b, MNK2a and/or MNK2b inhibitor.
[0539] In one embodiment, the kinase inhibitor is a CDK4 inhibitor selected from aloisine A; flavopiridol or HMR-1275, 2-(2-chlorophenyl)-5,7-dihydroxy-8-[(3S,4R)-3-hydroxy-1-methyl-4-piperidi- nyl]-4-chromenone; crizotinib (PF-02341066; 2-(2-Chlorophenyl)-5,7-dihydroxy-8-[(2R,3S)-2-(hydroxymethyl)-1-methyl-3-- pyrrolidinyl]-4H-1-benzopyran-4-one, hydrochloride (P276-00); 1-methyl-5-[[2-[5-(trifluoromethyl)-1H-imidazol-2-yl]-4-pyridinyl]oxy]-N-- [4-(trifluoromethyl)phenyl]-1H-benzimidazol-2-amine (RAF265); indisulam (E7070); roscovitine (CYC202); palbociclib (PD0332991); dinaciclib (SCH727965); N-[5-[[(5-tert-butyloxazol-2-yl)methyl]thio]thiazol-2-yl]piperidine-4-car- boxamide (BMS 387032); 4-[[9-chloro-7-(2,6-difluorophenyl)-5H-pyrimido[5,4-d][2]benzazepin-2-yl]- amino]-benzoic acid (MLN8054); 5-[3-(4,6-difluoro-1H-benzimidazol-2-yl)-1H-indazol-5-yl]-N-ethyl-4-methy- l-3-pyridinemethanamine (AG-024322); 4-(2,6-dichlorobenzoylamino)-1H-pyrazole-3-carboxylic acid N-(piperidin-4-yl)amide (AT7519); 4-[2-methyl-1-(1-methylethyl)-1H-imidazol-5-yl]-N-[4-(methylsulfonyl)phen- yl]-2-pyrimidinamine (AZD5438); and XL281 (BMS908662).
[0540] In one embodiment, the kinase inhibitor is a CDK4 inhibitor, e.g., palbociclib (PD0332991), and the palbociclib is administered at a dose of about 50 mg, 60 mg, 70 mg, 75 mg, 80 mg, 90 mg, 100 mg, 105 mg, 110 mg, 115 mg, 120 mg, 125 mg, 130 mg, 135 mg (e.g., 75 mg, 100 mg or 125 mg) daily for a period of time, e.g., daily for 14-21 days of a 28 day cycle, or daily for 7-12 days of a 21 day cycle. In one embodiment, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 or more cycles of palbociclib are administered.
[0541] An exemplary CDK4/6 inhibitor is LEE011 (also called ribociclib), the structure of which is shown below.
##STR00001##
Without being bound by theory, it is believed that administration of a CAR-expressing cell described herein with a CDK4/6 inhibitor (e.g., LEE011 or other CDK4/6 inhibitor described herein) can achieve higher responsiveness, e.g., with higher remission rates and/or lower relapse rates, e.g., compared to a CDK4/6 inhibitor alone.
[0542] While not wishing to be bound by theory, in some embodiments the CDK4/6 inhibitor acts to reduce cyclin D1 activity in cancer cells, e.g., MCL cells. Some cancer cells are characterized by elevated cyclin D1 levels due to a translocation. CDK4 complexes with cyclin D to promote cell cycle progression. Accordingly, in some embodiments, administration of the CK4/6 inhibitor reduces cancer cell proliferation. See, e.g., Marzec et al., "Mantle cell lymphoma cells express predominantly cyclin D1a isoform and are highly sensitive to selective inhibition of CDK4 kinase activity." Blood. 2006 Sep. 1;108(5):1744-50. Epub 2006 May 11.
[0543] In one embodiment, the kinase inhibitor is a BTK inhibitor selected from ibrutinib (PCI-32765); GDC-0834; RN-486; CGI-560; CGI-1764; HM-71224; CC-292; ONO-4059; CNX-774; and LFM-A13. In an embodiment, the BTK inhibitor does not reduce or inhibit the kinase activity of interleukin-2-inducible kinase (ITK), and is selected from GDC-0834; RN-486; CGI-560; CGI-1764; HM-71224; CC-292; ONO-4059; CNX-774; and LFM-A13.
[0544] In one embodiment, the kinase inhibitor is a BTK inhibitor, e.g., ibrutinib (PCI-32765), and the ibrutinib is administered at a dose of about 250 mg, 300 mg, 350 mg, 400 mg, 420 mg, 440 mg, 460 mg, 480 mg, 500 mg, 520 mg, 540 mg, 560 mg, 580 mg, 600 mg (e.g., 250 mg, 420 mg or 560 mg) daily for a period of time, e.g., daily for 21 day cycle cycle, or daily for 28 day cycle. In one embodiment, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 or more cycles of ibrutinib are administered.
[0545] In embodiments, the BTK inhibitor (e.g., ibrutinib) is administered to a subject that has CLL, mantle cell lymphoma (MCL), or small lymphocytic lymphoma (SLL). For example, the subject to whom the BTK inhibitor is administered has a deletion in the short arm of chromosome 17 (del(17p), e.g., in a leukemic cell). In other examples, the subject to whom the BTK inhibitor is administered does not have a del(17p). In embodiments, the subject to whom the BTK inhibitor is administered has relapsed CLL or SLL, e.g., the subject has previously been administered a cancer therapy (e.g., previously been administered one, two, three, or four prior cancer therapies). In embodiments, the subject to whom the BTK inhibitor is administered has refractory CLL or SLL. In other embodiments, the subject to whom the BTK inhibitor is administered has follicular lymphoma, e.g., relapse or refractory follicular lymphoma.
[0546] The structure of ibrutinib (1-[(3R)-3-[4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl]- piperidin-1-yl]prop-2-en-1-one) is shown below.
##STR00002##
[0547] Concentrations of ibrutinib exceeding approximately 400 nM are not clinically relevant. In the study by Advani et al (J Clin Oncol 2012; 31:88), the mean peak serum concentration of ibrutinib was about 130 ng/ml, which is equivalent to 295 nM (based on the molecular weight of ibrutinib of 440.5). Therefore, data showing the effect of ibrutinib on T cells at a concentration of 1 uM significantly exceeds the clinically relevant concentration in vivo.
[0548] In one embodiment, the kinase inhibitor is an mTOR inhibitor selected from temsirolimus; ridaforolimus (1R,2R,4S)-4-[(2R)-2 [(1R,9S,12S,15R,16E, 18R, 19R,21R,23S,24E,26E,28Z,30S,32S,35R)-1,18-dihydroxy-19,30-dimethoxy-15,17- ,21,23, 29,35-hexamethyl-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatricyclo[3- 0.3.1.0.sup.4,9]hexatriaconta-16,24,26,28-tetraen-12-yl]propyl]-2-methoxyc- yclohexyl dimethylphosphinate, also known as AP23573 and MK8669; everolimus (RAD001); rapamycin (AY22989); simapimod; (5-{2,4-bis[(3S)-3-methylmorpholin-4-yl]pyrido[2,3-d]pyrimidin-7-yl}-2-me- thoxyphenyl)methanol (AZD8055); 2-amino-8-[trans-4-(2-hydroxyethoxy)cyclohexyl]-6-(6-methoxy-3-pyridinyl)- -4-methyl-pyrido[2,3-d]pyrimidin-7(8H)-one (PF04691502); and N.sup.2-[1,4-dioxo-4-[[4-(4-oxo-8-phenyl-4H-1-benzopyran-2-yl)morpholiniu- m-4-yl]methoxy]butyl]-L-arginylglycyl-L-.alpha.-aspartylL-serine-, inner salt (SF1126); and XL765.
[0549] In one embodiment, the kinase inhibitor is an mTOR inhibitor, e.g., rapamycin, and the rapamycin is administered at a dose of about 3 mg, 4 mg, 5 mg, 6 mg, 7 mg, 8 mg, 9 mg, 10 mg (e.g., 6 mg) daily for a period of time, e.g., daily for 21 day cycle cycle, or daily for 28 day cycle. In one embodiment, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 or more cycles of rapamycin are administered. In one embodiment, the kinase inhibitor is an mTOR inhibitor, e.g., everolimus and the everolimus is administered at a dose of about 2 mg, 2.5 mg, 3 mg, 4 mg, 5 mg, 6 mg, 7 mg, 8 mg, 9 mg, 10 mg, 11 mg, 12 mg, 13 mg, 14 mg, 15 mg (e.g., 10 mg) daily for a period of time, e.g., daily for 28 day cycle. In one embodiment, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 or more cycles of everolimus are administered.
[0550] In one embodiment, the kinase inhibitor is an MNK inhibitor selected from CGP052088; 4-amino-3-(p-fluorophenylamino)-pyrazolo [3,4-d]pyrimidine (CGP57380); cercosporamide; ETC-1780445-2; and 4-amino-5-(4-fluoroanilino)-pyrazolo [3,4-d]pyrimidine.
[0551] In embodiments, a CAR-expressing cell described herein, optionally in combination with a kinase inhibitor, e.g., a BTK inhibitor such as ibrutinib, is administered to a subject in combination with a phosphoinositide 3-kinase (PI3K) inhibitor (e.g., a PI3K inhibitor described herein, e.g., idelalisib or duvelisib) and/or rituximab. In embodiments, a CAR-expressing cell described herein is administered to a subject in combination with idelalisib and rituximab. In embodiments, a CAR-expressing cell described herein is administered to a subject in combination with duvelisib and rituximab. Idelalisib (also called GS-1101 or CAL-101; Gilead) is a small molecule that blocks the delta isoform of PI3K. The structure of idelalisib (5-Fluoro-3-phenyl-2-[(1S)-1-(7H-purin-6-ylamino)propyl]-4(3H)-quinazolin- one) is shown below.
##STR00003##
Duvelisib is a small molecule that blocks PI3K-.delta.,.gamma.. The structure of duvelisib (8-Chloro-2-phenyl-3-[(1S)-1-(9H-purin-6-ylamino)ethyl]-1(2H)-isoquinolin- one) is shown below.
##STR00004##
In embodiments, the subject has CLL. In embodiments, the subject has relapsed CLL, e.g., the subject has previously been administered a cancer therapy (e.g., previously been administered an anti-CD20 antibody or previously been administered ibrutinib). For example, the subject has a deletion in the short arm of chromosome 17 (del(17p), e.g., in a leukemic cell). In other examples, the subject does not have a del(17p). In embodiments, the subject comprises a leukemic cell comprising a mutation in the immunoglobulin heavy-chain variable-region (IgV.sub.H) gene. In other embodiments, the subject does not comprise a leukemic cell comprising a mutation in the immunoglobulin heavy-chain variable-region (IgV.sub.H) gene. In embodiments, the subject has a deletion in the long arm of chromosome 11 (del(11q)). In other embodiments, the subject does not have a del(11q). In embodiments, idelalisib is administered at a dosage of about 100-400 mg (e.g., 100-125, 125-150, 150-175, 175-200, 200-225, 225-250, 250-275, 275-300, 325-350, 350-375, or 375-400 mg), e.g., BID. In embodiments, duvelisib is administered at a dosage of about 15-100 mg (e.g., about 15-25, 25-50, 50-75, or 75-100 mg), e.g., twice a day. In embodiments, rituximab is administered at a dosage of about 350-550 mg/m.sup.2 (e.g., 350-375, 375-400, 400-425, 425-450, 450-475, or 475-500 mg/m.sup.2), e.g., intravenously.
[0552] In one embodiment, the kinase inhibitor is a dual phosphatidylinositol 3-kinase (PI3K) and mTOR inhibitor selected from 2-Amino-8-[trans-4-(2-hydroxyethoxy)cyclohexyl]-6-(6-methoxy-3-pyridinyl)- -4-methyl-pyrido[2,3-d]pyrimidin-7(8H)-one (PF-04691502); N-[4-[[4-(Dimethylamino)-1-piperidinyl]carbonyl]phenyl]-N'-[4-(4,6-di-4-m- orpholinyl-1,3,5-triazin-2-yl)phenyl]urea (PF-05212384, PKI-587); 2-Methyl-2-{4-[3-methyl-2-oxo-8-(quinolin-3-yl)-2,3-dihydro-1H-imidazo[4,- 5-c]quinolin-1-yl]phenyl}propanenitrile (BEZ-235); apitolisib (GDC-0980, RG7422); 2,4-Difluoro-N-{2-(methyloxy)-5-[4-(4-pyridazinyl)-6-quinolinyl]- -3-pyridinyl}benzenesulfonamide (GSK2126458); 8-(6-methoxypyridin-3-yl)-3-methyl-1-(4-(piperazin-1-yl)-3-(trifluorometh- yl)phenyl)-1H-imidazo[4,5-c]quinolin-2(3H)-one Maleic acid (NVP-BGT226); 3-[4-(4-Morpholinylpyrido[3',2':4,5]furo[3,2-d]pyrimidin-2-yl]phenol (PI-103); 5-(9-isopropyl-8-methyl-2-morpholino-9H-purin-6-yl)pyrimidin-2-- amine (VS-5584, SB2343); and N-[2-[(3,5-Dimethoxyphenyl)amino]quinoxalin-3-yl]-4-[(4-methyl-3-methoxyp- henyl)carbonyl]aminophenylsulfonamide (XL765).
[0553] In embodiments, a CAR-expressing cell described herein, optionally in combination with a kinase inhibitor, e.g., a BTK inhibitor such as ibrutinib, is administered to a subject in combination with an anaplastic lymphoma kinase (ALK) inhibitor. Exemplary ALK kinase inhibitors include but are not limited to crizotinib (Pfizer), ceritinib (Novartis), alectinib (Chugai), brigatinib (also called AP26113; Ariad), entrectinib (Ignyta), PF-06463922 (Pfizer), TSR-O11 (Tesaro) (see, e.g., Clinical Trial Identifier No. NCT02048488), CEP-37440 (Teva), and X-396 (Xcovery). In some embodiments, the subject has a solid cancer, e.g., a solid cancer described herein, e.g., lung cancer.
[0554] The chemical name of crizotinib is 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol- -4-yl)pyridin-2-amine. The chemical name of ceritinib is 5-Chloro-N.sup.2-[2-isopropoxy-5-methyl-4-(4-piperidinyl)phenyl]-N.sup.4-- [2-(isopropylsulfonyl)phenyl]-2,4-pyrimidinediamine. The chemical name of alectinib is 9-ethyl-6,6-dimethyl-8-(4-morpholinopiperidin-1-yl)-11-oxo-6,11-dihydro-5- H-benzo[b]carbazole-3-carbonitrile. The chemical name of brigatinib is 5-Chloro-N.sup.2-{4-[4-(dimethylamino)-1-piperidinyl]-2-methoxyphenyl}-N.- sup.4-[2-(dimethylphosphoryl)phenyl]-2,4-pyrimidinediamine. The chemical name of entrectinib is N-(5-(3,5-difluorobenzyl)-1H-indazol-3-yl)-4-(4-methylpiperazin-1-yl)-2-(- (tetrahydro-2H-pyran-4-yl)amino)benzamide. The chemical name of PF-06463922 is (10R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2- H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carb- onitrile. The chemical structure of CEP-37440 is (S)-2-((5-chloro-2-((6-(4-(2-hydroxyethyl)piperazin-1-yl)-1-methoxy-6,7,8- ,9-tetrahydro-5H-benzo[7]annulen-2-yl)amino)pyrimidin-4-yl)amino)-N-methyl- benzamide. The chemical name of X-396 is (R)-6-amino-5-(1-(2,6-dichloro-3-fluorophenyl)ethoxy)-N-(4-(4-methylpiper- azine-1-carbonyl)phenyl)pyridazine-3-carboxamide.
[0555] In embodiments, a CAR-expressing cell described herein, optionally in combination with a kinase inhibitor, e.g., a BTK inhibitor such as ibrutinib, is administered to a subject in combination with an indoleamine 2,3-dioxygenase (IDO) inhibitor. IDO is an enzyme that catalyzes the degradation of the amino acid, L-tryptophan, to kynurenine. Many cancers overexpress IDO, e.g., prostatic, colorectal, pancreatic, cervical, gastric, ovarian, head, and lung cancer. pDCs, macrophages, and dendritic cells (DCs) can express IDO. Without being bound by theory, it is thought that a decrease in L-tryptophan (e.g., catalyzed by IDO) results in an immunosuppressive milieu by inducing T-cell anergy and apoptosis. Thus, without being bound by theory, it is thought that an IDO inhibitor can enhance the efficacy of a CAR-expressing cell described herein, e.g., by decreasing the suppression or death of a CAR-expressing immune cell. In embodiments, the subject has a solid tumor, e.g., a solid tumor described herein, e.g., prostatic, colorectal, pancreatic, cervical, gastric, ovarian, head, or lung cancer. Exemplary inhibitors of IDO include but are not limited to 1-methyl-tryptophan, indoximod (NewLink Genetics) (see, e.g., Clinical Trial Identifier Nos. NCT01191216; NCT01792050), and INCB024360 (Incyte Corp.) (see, e.g., Clinical Trial Identifier Nos. NCT01604889; NCT01685255)
[0556] In embodiments, a CAR-expressing cell described herein, optionally in combination with a kinase inhibitor, e.g., a BTK inhibitor such as ibrutinib, is administered to a subject in combination with a modulator of myeloid-derived suppressor cells (MDSCs). MDSCs accumulate in the periphery and at the tumor site of many solid tumors. These cells suppress T cell responses, thereby hindering the efficacy of CAR-expressing cell therapy. Without being bound by theory, it is thought that administration of a MDSC modulator enhances the efficacy of a CAR-expressing cell described herein. In an embodiment, the subject has a solid tumor, e.g., a solid tumor described herein, e.g., glioblastoma. Exemplary modulators of MDSCs include but are not limited to MCS110 and BLZ945. MCS110 is a monoclonal antibody (mAb) against macrophage colony-stimulating factor (M-CSF). See, e.g., Clinical Trial Identifier No. NCT00757757. BLZ945 is a small molecule inhibitor of colony stimulating factor 1 receptor (CSF1R). See, e.g., Pyonteck et al. Nat. Med. 19(2013):1264-72. The structure of BLZ945 is shown below.
##STR00005##
[0557] In some embodiments, a CAR-expressing cell described herein, optionally in combination with a kinase inhibitor, e.g., a BTK inhibitor such as ibrutinib, is administered to a subject in combination with a interleukin-15 (IL-15) polypeptide, a interleukin-15 receptor alpha (IL-15Ra) polypeptide, or a combination of both a IL-15 polypeptide and a IL-15Ra polypeptide e.g., hetIL-15 (Admune Therapeutics, LLC). hetIL-15 is a heterodimeric non-covalent complex of IL-15 and IL-15Ra. hetIL-15 is described in, e.g., U.S. Pat. No. 8,124,084, U.S. 2012/0177598, U.S. 2009/0082299, U.S. 2012/0141413, and U.S. 2011/0081311, incorporated herein by reference. In embodiments, het-IL-15 is administered subcutaneously. In embodiments, the subject has a cancer, e.g., solid cancer, e.g., melanoma or colon cancer. In embodiments, the subject has a metastatic cancer.
Therapeutic Application
[0558] CD19 Associated Diseases and/or Disorders
[0559] In one aspect, the invention provides methods for treating a disease associated with CD19 expression. In one aspect, the invention provides methods for treating a disease wherein part of the tumor is negative for CD19 and part of the tumor is positive for CD19. For example, the CAR of the invention is useful for treating subjects that have undergone treatment for a disease associated with elevated expression of CD19, wherein the subject that has undergone treatment for elevated levels of CD19 exhibits a disease associated with elevated levels of CD19.
[0560] The therapies described herein can be used to treat, e.g., subjects who respond to a kinase inhibitor such as ibrutinib (e.g., partial response or complete response) or subjects who do not (e.g., non-responders or relapsers). Without wishing to be bound by theory, a number of patients undergoing treatment with kinase inhibitors (e.g., BTK inhibitors such as ibrutinib) may show a reduced response to the treatment (e.g., are partial or non-responders to the treatment, or relapse during treatment). According, administration of the CAR-therapies disclosed herein, in combination with the kinase inhibitors can result in beneficial effects.
[0561] Exemplary therapeutic regimens for these subjects are described below.
[0562] In some cases, when the subject is a non-responder or relapser to a kinase inhibitor (e.g. a BTK inhibitor such as ibrutinib), the kinase inhibitor is withdrawn and CAR therapy is administered. In other cases, when the subjects does not respond to a kinase inhibitor (e.g. a BTK inhibitor such as ibrutinib), the kinase inhibitor therapy is continued and CAR therapy is added to the regimen. This use is supported, e.g., by experiments in Example 8 herein which indicate that CAR therapy is effective as a monotherapy in ibrutinib-resistant cells. Without wishing to be bound by theory, continuing kinase inhibitor therapy can improve the efficacy of the CAR therapy, e.g., by increasing the number of CAR-expressing cells in the bloodstream (see Example 8 herein).
[0563] Without being bound by theory, a subject who is a non-responder or relapser to a kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib) can be non-responsive for at least two reasons: the subjects may have a mutation in the drug target (e.g., BTK, e.g., a C481S mutation) that prevents target inhibition, or can have alterations in other pathways that can drive proliferation even when the target is adequately inhibited (e.g., a mutation in PLC.gamma., such as an activating mutation in PLC.gamma. resulting in constitutive BTK-independent cell signaling). The treatment can be altered depending on the reason for non-responsiveness. For instance, in the first situation (in some embodiments), if the subjects has (or is identified as having) a mutation that prevents the kinase inhibitor from inhibiting its target, a second kinase inhibitor (e.g., directed against the same target) can be substituted for (or administered in combination with) the kinase inhibitor. More specifically, in some embodiments where the patient has (or is identified as having) a mutation that prevents ibrutinib from inhibiting BTK, a second BTK inhibitor, e.g., a BTK inhibitor described herein such as GDC-0834, RN-486, CGI-560, CGI-1764, HM-71224, CC-292, ONO-4059, CNX-774, or LFM-A13) can be substituted for ibrutinib. Without wishing to be bound by theory, the second kinase inhibitor may act on a region of the target that is not disrupted by the mutation, and therefore the subject is sensitive to the second kinase inhibitor. In other embodiments, the original kinase inhibitor (e.g. a BTK inhibitor such as ibrutinib) is maintained. According to the non-limiting theory here, the original kinase inhibitor may have useful activity on the CAR-expressing cells, e.g., promoting a TH1 phenotype, promoting proliferation, or otherwise increasing levels or activity of the cells.
[0564] As noted above, in some cases a subject is non-responsive because the subject has an alteration (e.g., a mutation) in another pathway that can drive proliferation even when the target is adequately inhibited. Accordingly, if the subject has (or is identified has having) an alteration in a pathway that makes the kinase inhibitor's activity ineffectual, the kinase inhibitor therapy can be maintained. Without wishing to be bound by theory, the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib) activity can promote useful biological changes in the cancer cells even if the kinase inhibitor alone is not sufficient to slow proliferation. For instance, the kinase inhibitor can be sufficient to mobilize cancer cells out of the lymph nodes, making them more vulnerable to the CAR therapy.
[0565] Turning now to subjects who respond to a kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib), various therapeutic regimens are now described. In some embodiments, when a subject is (or is identified as being) a complete responder to the kinase inhibitor, the subject is not administered a CAR therapy during the period of complete response. In other embodiments, when a subject is (or is identified as being) a complete responder to the kinase inhibitor, the subject is administered a CAR therapy during the period of complete response. In an embodiment, after the CAR therapy, the subject experiences a prolonged response or delayed relapse (e.g., compared to the expected course of disease when treated without CAR therapy). For instance, MCL treated with ibrutinib monotherapy has a median duration of response of about 17.5 months.
[0566] In some embodiments, when a subject is (or is identified as being) a partial responder to the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib), the subject is not administered a CAR therapy during the period of partial response. In other embodiments, when a subject is (or is identified as being) a partial responder to the kinase inhibitor, the subject is administered a CAR therapy during the period of partial response. In an embodiment, after the CAR therapy, the subject experiences a complete response and/or prolonged response or delayed relapse (e.g., compared to the expected course of disease when treated without CAR therapy).
[0567] In some embodiments, when a subject has (or is identified as having) stable disease after the beginning of treatment with the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib), the subject is not administered a CAR therapy during the period of stable disease. In other embodiments, when a subject has (or is identified as having) stable disease after the beginning of treatment with the kinase inhibitor, the subject is administered a CAR therapy during the period of stable disease. In an embodiment, after the CAR therapy, the subject experiences a partial response, a complete response and/or prolonged response or delayed relapse (e.g., compared to the expected course of disease when treated without CAR therapy).
[0568] In some embodiments, when a subject has (or is identified as having) progressive disease after the beginning of treatment with the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib), the subject is not administered a CAR therapy during the period of progressive disease. In other embodiments, when a subject has (or is identified as having) progressive disease after the beginning of treatment with the kinase inhibitor, the subject is administered a CAR therapy during the period of progressive disease. In an embodiment, after the CAR therapy, the subject experiences stable disease, a partial response, a complete response and/or prolonged response or delayed relapse (e.g., compared to the expected course of disease when treated without CAR therapy).
[0569] Thus, one or more disease assessment steps can be performed before or during treatment, to determine which course of treatment is suitable for a given patient. For instance, a subject can be administered a kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib) as a first line therapy. Then, after a period of time (e.g., 1 or 2 months but also 2 weeks, 3 weeks, 1 month, 1.5 months, 2 months, 3 months, 4 months, 6 months, 9 months, 12 months, 15 months, or 18 months) the patient's response can be assessed. If the assessment shows that the subject is a complete responder, in some embodiments CAR therapy is not administered, e.g., as described above. If the assessment shows that the subject is a partial responder or has stable disease, in some embodiments CAR therapy is administered in combination with the kinase inhibitor e.g., as described above. If the assessment shows that the subject is a non-responder or relapser, in some embodiments CAR therapy is administered in combination with the kinase inhibitor or a second kinase inhibitor, e.g., as described above. In some embodiments, the kinase inhibitor controls the disease while a CAR-expressing cell is being manufactured, e.g., while the patient's own T cells are being engineered to express a CAR and/or other factors.
[0570] Clinical standards for classifying a patient's responder status or relapser status are known in the art. As an example, for malignant lymphoma, standardized response criteria are described in Cheson et al, J Clin Oncol 17:1244 (1999) and Cheson et al., "Revised Response Criteria for Malignant Lymphoma", J Clin Oncol 25:579-586 (2007) (both of which are incorporated by reference herein in their entireties). Accordingly, in some embodiments, a subject is considered a complete responder, partial responder, having stable disease, a non-responder, or a relapser according to Cheson criteria or modified Cheson criteria. Criteria for classifying other hematological malignancies are known in the art.
[0571] According to the criteria in Table 2 of Cheson 2007, a complete responder has disappearance of all evidence of disease; a partial responder has regression of measurable disease and no new sites; a patient with stable disease has a failure to attain CR/PR or PD; and a patient with relapsed disease or progressive disease has any new lesion or increase by greater than or equal to 50% of previously involved sites from nadir. The assessment can involve a determination of whether the disease is FDG-avid, PET positive or negative, whether nodules are present e.g., palpable in the liver or spleen, and whether bone marrow is cleared or shows involvement.
[0572] The CAR therapy and the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib) can be administered, e.g., simultaneously or sequentially. In some embodiments, the CAR therapy is begun at substantially the same time as kinase inhibitor therapy begins. In some embodiments, the CAR therapy is begun before the kinase inhibitor therapy begins. In some embodiments, the CAR therapy is begun after the kinase inhibitor therapy begins. For instance, the CAR therapy can be begun, e.g., at least 1, 2, 3, or 4 weeks, or 1, 2, 3, 4, 6, 9, 12, 15, 18, or 24 months after the kinase inhibitor therapy begins. In some embodiments, the CAR therapy is begun while a patient has physiologically relevant levels of the kinase inhibitor in their body.
[0573] When administered in combination, the CAR therapy and the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib), or both, can be administered in an amount or dose that is higher, lower or the same than the amount or dosage of each agent used individually, e.g., as a monotherapy. In certain embodiments, the administered amount or dosage of the CAR therapy, the kinase inhibitor, or both, is lower (e.g., at least 20%, at least 30%, at least 40%, or at least 50%) than the amount or dosage of each agent used individually, e.g., as a monotherapy. In other embodiments, the amount or dosage of the CAR therapy, the kinase inhibitor, or both, that results in a desired effect (e.g., treatment of cancer) is lower (e.g., at least 20%, at least 30%, at least 40%, or at least 50% lower) than the amount or dosage of each agent used individually, e.g., as a monotherapy, required to achieve the same therapeutic effect.
[0574] When administered in combination, the CAR therapy and the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib), or both, can be administered with a duration that is longer, shorter, or the same than the duration of each agent used individually, e.g., as a monotherapy. In certain embodiments, the duration of administration of the CAR therapy, the kinase inhibitor, or both, is shorter (e.g., at least 20%, at least 30%, at least 40%, or at least 50%) than the duration of each agent used individually, e.g., as a monotherapy. In other embodiments, the duration of administration of the CAR therapy, the kinase inhibitor, or both, that results in a desired effect (e.g., treatment of cancer) is shorter (e.g., at least 20%, at least 30%, at least 40%, or at least 50% shorter) than the duration of each agent used individually, e.g., as a monotherapy, required to achieve the same therapeutic effect. In some embodiment, the patient is administered an abbreviated course of the kinase inhibitor (e.g., a BTK inhibitor such as ibrutinib). For instance, the abbreviated course of the kinase inhibitor may last about 0-2, 2-4, 4-6, 6-8, 8-10, 10-12, 12-15, 15-18, 18-21, or 21-24 months total or may last about 0-2, 2-4, 4-6, 6-8, 8-10, 10-12, 12-15, 15-18, 18-21, or 21-24 months after administration of the CAR therapy. In embodiments, the abbreviated course of the kinase inhibitor ends before relapse. In embodiments, the kinase inhibitor is administered at normal (e.g., monotherapy) levels during the abbreviated course.
[0575] In embodiments, a single dose of CAR-expressing cells comprises about 5.times.10.sup.8 CD19 CART cells. A dose of CAR-expressing cells may also comprise about 5.times.10.sup.6, 1.times.10.sup.7, 2.times.10.sup.7, 5.times.10.sup.7, 1.times.10.sup.8, 2.times.10.sup.8, 5.times.10.sup.8, 1.times.10.sup.9, 2.times.10.sup.9, or 5.times.10.sup.9 cells, e.g., CD19 CAR cells, e.g., CD19 CART cells.
[0576] In one aspect, the invention pertains to a vector comprising CD19 CAR operably linked to promoter for expression in mammalian cells, e.g., T cells. In one aspect, the invention provides a recombinant cell, e.g., a T cell, expressing the CD19 CAR for use in treating CD19-expressing tumors, wherein the recombinant T cell expressing the CD19 CAR is termed a CD19 CART. In one aspect, the CD19 CART described herein, is capable of contacting a tumor cell with at least one CD19 CAR expressed on its surface such that the CART targets the tumor cell and growth of the tumor is inhibited.
[0577] In one aspect, the invention pertains to a method of inhibiting growth of a CD19-expressing tumor cell, comprising contacting the tumor cell with a CD19 CAR expressing cell, e.g., a CD19 CART cell, described herein such that the CART is activated in response to the antigen and targets the cancer cell, wherein the growth of the tumor is inhibited. The CD19 CAR-expressing cell, e.g., T cell, is administered in combination with a kinase inhibitor, e.g., a kinase inhibitor described herein.
[0578] Administered "in combination", as used herein, means that two (or more) different treatments are delivered to the subject during the course of the subject's affliction with the disorder, e.g., the two or more treatments are delivered after the subject has been diagnosed with the disorder and before the disorder has been cured or eliminated or treatment has ceased for other reasons. In some embodiments, the delivery of one treatment is still occurring when the delivery of the second begins, so that there is overlap in terms of administration. This is sometimes referred to herein as "simultaneous" or "concurrent delivery". In other embodiments, the delivery of one treatment ends before the delivery of the other treatment begins. In some embodiments of either case, the treatment is more effective because of combined administration. For example, the second treatment is more effective, e.g., an equivalent effect is seen with less of the second treatment, or the second treatment reduces symptoms to a greater extent, than would be seen if the second treatment were administered in the absence of the first treatment, or the analogous situation is seen with the first treatment. In some embodiments, delivery is such that the reduction in a symptom, or other parameter related to the disorder is greater than what would be observed with one treatment delivered in the absence of the other. The effect of the two treatments can be partially additive, wholly additive, or greater than additive. The delivery can be such that an effect of the first treatment delivered is still detectable when the second is delivered. In one embodiment, the CAR-expressing cell is administered at a dose and/or dosing schedule described herein, and the kinase inhibitor or agent that enhances the activity of the CAR-expressing cell is administered at a dose and/or dosing schedule described herein.
[0579] The invention includes a type of cellular therapy where T cells are genetically modified to express a chimeric antigen receptor (CAR) and the CAR T cell is infused to a recipient in need thereof. The infused cell is able to kill tumor cells in the recipient. Unlike antibody therapies, CAR-modified T cells are able to replicate in vivo resulting in long-term persistence that can lead to sustained tumor control. In various aspects, the T cells administered to the patient, or their progeny, persist in the patient for at least four months, five months, six months, seven months, eight months, nine months, ten months, eleven months, twelve months, thirteen months, fourteen month, fifteen months, sixteen months, seventeen months, eighteen months, nineteen months, twenty months, twenty-one months, twenty-two months, twenty-three months, two years, three years, four years, or five years after administration of the T cell to the patient.
[0580] The invention also includes a type of cellular therapy where T cells are modified, e.g., by in vitro transcribed RNA, to transiently express a chimeric antigen receptor (CAR) and the CAR T cell is infused to a recipient in need thereof. The infused cell is able to kill tumor cells in the recipient. Thus, in various aspects, the T cells administered to the patient, is present for less than one month, e.g., three weeks, two weeks, one week, after administration of the T cell to the patient.
[0581] Without wishing to be bound by any particular theory, the anti-tumor immunity response elicited by the CAR-modified T cells may be an active or a passive immune response, or alternatively may be due to a direct vs indirect immune response. In one aspect, the CAR transduced T cells exhibit specific proinflammatory cytokine secretion and potent cytolytic activity in response to human cancer cells expressing the CD19, resist soluble CD19 inhibition, mediate bystander killing and mediate regression of an established human tumor. For example, antigen-less tumor cells within a heterogeneous field of CD19-expressing tumor may be susceptible to indirect destruction by CD19-redirected T cells that has previously reacted against adjacent antigen-positive cancer cells.
[0582] In one aspect, the fully-human CAR-modified T cells of the invention may be a type of vaccine for ex vivo immunization and/or in vivo therapy in a mammal. In one aspect, the mammal is a human.
[0583] With respect to ex vivo immunization, at least one of the following occurs in vitro prior to administering the cell into a mammal: i) expansion of the cells, ii) introducing a nucleic acid encoding a CAR to the cells or iii) cryopreservation of the cells.
[0584] Ex vivo procedures are well known in the art and are discussed more fully below. Briefly, cells are isolated from a mammal (e.g., a human) and genetically modified (i.e., transduced or transfected in vitro) with a vector expressing a CAR disclosed herein. The CAR-modified cell can be administered to a mammalian recipient to provide a therapeutic benefit. The mammalian recipient may be a human and the CAR-modified cell can be autologous with respect to the recipient. Alternatively, the cells can be allogeneic, syngeneic or xenogeneic with respect to the recipient. In addition to using a cell-based vaccine in terms of ex vivo immunization, also included in the methods described herein are compositions and methods for in vivo immunization to elicit an immune response directed against an antigen in a patient.
[0585] Generally, the cells activated and expanded as described herein may be utilized in the treatment and prevention of diseases that arise in individuals who are immunocompromised. In particular, the CAR-expressing cells described herein are used in the treatment of diseases, disorders and conditions associated with expression of CD19. In certain aspects, the cells are used in the treatment of patients at risk for developing diseases, disorders and conditions associated with expression of CD19. Thus, the present invention provides methods for the treatment or prevention of diseases, disorders and conditions associated with expression of CD19 comprising administering to a subject in need thereof, a therapeutically effective amount of the CAR-expressing cells described herein, in combination with a kinase inhibitor, e.g., a kinase inhibitor described herein.
[0586] The present invention also provides methods for inhibiting the proliferation or reducing a CD19-expressing cell population, the methods comprising contacting a population of cells comprising a CD19-expressing cell with an anti-CD19 CAR-expressing cell described herein that binds to the CD19-expressing cell, and contacting the population of CD19-expressing cells with a kinase inhibitor, e.g., a kinase inhibitor described herein. In a specific aspect, the present invention provides methods for inhibiting the proliferation or reducing the population of cancer cells expressing CD19, the methods comprising contacting the CD19-expressing cancer cell population with an anti-CD19 CAR-expressing cell described herein that binds to the CD19-expressing cell, and contacting the CD19-expressing cell with a kinase inhibitor, e.g., a kinase inhibitor described herein. In one aspect, the present invention provides methods for inhibiting the proliferation or reducing the population of cancer cells expressing CD19, the methods comprising contacting the CD19-expressing cancer cell population with an anti-CD19 CAR-expressing cell described herein that binds to the CD19-expressing cell and contacting the CD19-expressing cell with a kinase inhibitor, e.g., a kinase inhibitor described herein. In certain aspects, the combination of the anti-CD19 CAR-expressing cell described herein and the kinase inhibitor, e.g., a kinase inhibitor described herein, reduces the quantity, number, amount or percentage of cells and/or cancer cells by at least 25%, at least 30%, at least 40%, at least 50%, at least 65%, at least 75%, at least 85%, at least 95%, or at least 99% in a subject with or animal model for a hematological cancer or another cancer associated with CD19-expressing cells relative to a negative control. In one aspect, the subject is a human.
[0587] The present invention also provides methods for preventing, treating and/or managing a disease associated with CD19-expressing cells (e.g., a hematologic cancer or atypical cancer expressing CD19), the methods comprising administering to a subject in need an anti-CD19 CAR-expressing cell that binds to the CD19-expressing cell and administering a kinase inhibitor, e.g., a kinase inhibitor described herein. In one aspect, the subject is a human. Non-limiting examples of disorders associated with CD19-expressing cells include autoimmune disorders (such as lupus), inflammatory disorders (such as allergies and asthma) and cancers (such as hematological cancers or atypical cancers expressing CD19).
[0588] The present invention also provides methods for preventing, treating and/or managing a disease associated with CD19-expressing cells, the methods comprising administering to a subject in need an anti-CD19 CART cell of the invention that binds to the CD19-expressing cell. In one aspect, the subject is a human.
[0589] The present invention provides methods for preventing relapse of cancer associated with CD19-expressing cells, the methods comprising administering to a subject in need thereof an anti-CD19 expressing cell (such as an anti-CD19 CART cell) of the invention that binds to the CD19-expressing cell. In one aspect, the methods comprise administering to the subject in need thereof an effective amount of an anti-CD19 expressing cell (such as an anti-CD19 CART cell) described herein that binds to the CD19-expressing cell in combination with an effective amount of another therapy.
[0590] In one aspect, the invention pertains to a method of treating cancer in a subject. The method comprises administering to the subject a cell (e.g., an immune effector cell) expressing a B-cell targeting CAR, e.g., a T cell or NK cell, described herein, in combination with a kinase inhibitor, e.g., a kinase inhibitor described herein, such that the cancer is treated in the subject. An example of a cancer that is treatable by the methods described herein is a cancer associated with expression of the B-cell antigen, e.g., CD19. In one embodiment, the disease is a solid or liquid tumor. In one embodiment, the disease is a hematologic cancer. In one embodiment, the hematologic cancer is leukemia. In one embodiment, the hematologic cancer is a mature B cell neoplasm, e.g., according to WHO classification. In one embodiment, the hematologic cancer is a CD19+B-lymphocyte-derived malignancy. In one embodiment, the cancer is selected from the group consisting of one or more acute leukemias including but not limited to B-cell acute lymphoid leukemia (BALL), T-cell acute lymphoid leukemia (TALL), small lymphocytic leukemia (SLL), acute lymphoid leukemia (ALL); one or more chronic leukemias including but not limited to chronic myelogenous leukemia (CML), chronic lymphocytic leukemia (CLL); additional hematologic cancers or hematologic conditions including, but not limited to mantle cell lymphoma (MCL), B cell prolymphocytic leukemia, blastic plasmacytoid dendritic cell neoplasm, Burkitt's lymphoma, diffuse large B cell lymphoma (DLBCL) (e.g., T-cell/histiocyte rich large B-cell lymphoma, primary DLCBL of the CNS, primary cutaneous DLBCL leg type, or EBV+ DLBCL of the elderly), DLBCL associated with chronic inflammation, follicular lymphoma, pediatric follicular lymphoma, hairy cell leukemia, small cell- or a large cell-follicular lymphoma, malignant lymphoproliferative conditions, MALT lymphoma (extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue), Marginal zone lymphoma, multiple myeloma, myelodysplasia and myelodysplastic syndrome, non-Hodgkin lymphoma, Hodgkin lymphoma, plasmablastic lymphoma, plasmacytoid dendritic cell neoplasm, Waldenstrom macroglobulinemia, splenic marginal zone lymphoma, splenic lymphoma/leukemia (e.g., unclassifiable), splenic diffuse red pulp small B-cell lymphoma, hairy cell leukemia-variant, lymphoplasmacytic lymphoma, a heavy chain disease (e.g., alpha heavy chain disease, gamma heavy chain disease, or mu heavy chain disease), plasma cell myeloma, solitary plasmocytoma of bone, extraosseous plasmocytoma, nodal marginal zone lymphoma, pediatric nodal marginal zone lymphoma, primary cutaneous follicle center lymphoma, lymphomatoid granulomatosis, primary mediastinal (theymic) large B-cell lymphoma, intravascular large B-cell lymphoma, ALK+ large B-cell lymphoma, large B-cell lymphoma arising in HHV8-associated multicenric Castleman disease, primary effusion lymphoma, B-cell lymphoma, unclassifiable (e.g., with features intermediate between DLBCL and Burkitt lymphoma or intermediate between DLBCL and classical Hodgkin lymphoma), and "preleukemia" which are a diverse collection of hematological conditions united by ineffective production (or dysplasia) of myeloid blood cells, and to disease associated with B-cell antigen- (e.g., CD19-) expression include, but not limited to atypical and/or non-classical cancers, malignancies, precancerous conditions or proliferative diseases expressing B-cell antigen (e.g., CD19); and any combination thereof.
[0591] In some embodiments, the cancer is Hodgkin lymphoma, and the patient is treated with CAR expressing cells, e.g., as a monotherapy, or in combination with one or more additional therapeutics. In embodiments, the Hodgkin lymphoma is stage I, II, III, or IV. The additional therapeutic may comprise, e.g., a kinase inhibitor such as a BTK inhibitor like ibrutinib. The additional therapeutic may comprise a treatment for Hodgkin lymphoma. The additional therapeutic may comprise, e.g., radiation therapy, MOPP (Mustargen, Oncovin, Prednisone, and Procarbazine), ABVD (Adriamycin, bleomycin, vinblastine, and dacarbazine), Stanford V (a regimen with chemotherapy and radiation treatment), or BEACOPP (Bleomycin, Etoposide, Adriamycin, Cyclophosphamide, Oncovin, Procarbazine, Prednisone). In some embodiments, the subject has previously been treated with, or is resistant to, or is refractory to, one or more of radiation therapy, MOPP, Stanford V, or BEACOPP.
[0592] Non-cancer related indications associated with expression of B-cell antigen, e.g., one or more of CD19, CD20, CD22 or ROR1, include, but are not limited to, e.g., autoimmune disease, (e.g., lupus), inflammatory disorders (allergy and asthma) and transplantation.
[0593] In some embodiments, a cancer that can be treated with the combination described herein is multiple myeloma. Multiple myeloma is a cancer of the blood, characterized by accumulation of a plasma cell clone in the bone marrow. Current therapies for multiple myeloma include, but are not limited to, treatment with lenalidomide, which is an analog of thalidomide. Lenalidomide has activities which include anti-tumor activity, angiogenesis inhibition, and immunomodulation. In some embodiments, a CD19 CAR, e.g., as described herein, may be used to target myeloma cells. In some embodiments, the combination described herein can be used with one or more additional therapies, e.g., lenalidomide treatment.
[0594] The CAR-expressing cells described herein may be administered either alone, or as a pharmaceutical composition in combination with diluents and/or with other components such as IL-2 or other cytokines or cell populations.
[0595] In embodiments, a lymphodepleting chemotherapy is administered to the subject prior to, concurrently with, or after administration (e.g., infusion) of CAR cells, e.g., CAR-expressing cells described herein. In an example, the lymphodepleting chemotherapy is administered to the subject prior to administration of CAR cells. For example, the lymphodepleting chemotherapy ends 1-4 days (e.g., 1, 2, 3, or 4 days) prior to CAR cell infusion. In embodiments, multiple doses of CAR cells are administered, e.g., as described herein. For example, a single dose comprises about 5.times.10.sup.8 CAR cells. In embodiments, a lymphodepleting chemotherapy is administered to the subject prior to, concurrently with, or after administration (e.g., infusion) of a CAR-expressing cell described herein.
Hematologic Cancer
[0596] Hematological cancer conditions are the types of cancer such as leukemia, lymphoma and malignant lymphoproliferative conditions that affect blood, bone marrow and the lymphatic system.
[0597] Leukemia can be classified as acute leukemia and chronic leukemia. Acute leukemia can be further classified as acute myelogenous leukemia (AML) and acute lymphoid leukemia (ALL). Chronic leukemia includes chronic myelogenous leukemia (CML) and chronic lymphoid leukemia (CLL). Other related conditions include myelodysplastic syndromes (MDS, formerly known as "preleukemia") which are a diverse collection of hematological conditions united by ineffective production (or dysplasia) of myeloid blood cells and risk of transformation to AML.
[0598] Lymphoma is a group of blood cell tumors that develop from lymphocytes. Exemplary lymphomas include non-Hodgkin lymphoma and Hodgkin lymphoma.
Combination Therapies
[0599] The combination of a CAR-expressing cell described herein (e.g., and a kinase inhibitor described herein) may be used in combination with other known agents and therapies.
[0600] A CAR-expressing cell described herein, the kinase inhibitor and/or the at least one additional therapeutic agent can be administered simultaneously, in the same or in separate compositions, or sequentially. For sequential administration, the CAR-expressing cell described herein can be administered first, and the additional agent can be administered second, or the order of administration can be reversed.
[0601] The CAR therapy and/or other therapeutic agents, procedures or modalities can be administered during periods of active disorder, or during a period of remission or less active disease. The CAR therapy can be administered before another treatment, concurrently with the treatment, post-treatment, or during remission of the disorder.
[0602] When administered in combination, the CAR therapy and one or more additional agent (e.g., kinase inhibitor and/or a third agent), or all, can be administered in an amount or dose that is higher, lower or the same than the amount or dosage of each agent used individually, e.g., as a monotherapy. In certain embodiments, the administered amount or dosage of the CAR therapy, the additional agent (e.g., kinase inhibitor and/or third agent), or all, is lower (e.g., at least 20%, at least 30%, at least 40%, or at least 50%) than the amount or dosage of each agent used individually, e.g., as a monotherapy. In other embodiments, the amount or dosage of the CAR therapy, the additional agent (e.g., kinase inhibitor and/or third agent), or all, that results in a desired effect (e.g., treatment of cancer) is lower (e.g., at least 20%, at least 30%, at least 40%, or at least 50% lower) than the amount or dosage of each agent used individually, e.g., as a monotherapy, required to achieve the same therapeutic effect.
[0603] In further aspects, the combination of the CAR-expressing cell described herein (e.g., and the kinase inhibitor) may be used in a treatment regimen in combination with surgery, chemotherapy, radiation, immunosuppressive agents, such as cyclosporin, azathioprine, methotrexate, mycophenolate, and FK506, antibodies, or other immunoablative agents such as CAMPATH, anti-CD3 antibodies or other antibody therapies, cytoxin, fludarabine, cyclosporin, FK506, rapamycin, mycophenolic acid, steroids, FR901228, cytokines, and irradiation. peptide vaccine, such as that described in Izumoto et al. 2008 J Neurosurg 108:963-971.
[0604] In one embodiment, the combination of a CAR-expressing cell described herein (e.g., and a kinase inhibitor described herein) can be used in combination with another chemotherapeutic agent. Exemplary chemotherapeutic agents include an anthracycline (e.g., doxorubicin (e.g., liposomal doxorubicin)); a vinca alkaloid (e.g., vinblastine, vincristine, vindesine, vinorelbine); an alkylating agent (e.g., cyclophosphamide, decarbazine, melphalan, ifosfamide, temozolomide); an immune cell antibody (e.g., alemtuzamab, gemtuzumab, rituximab, ofatumumab, tositumomab, brentuximab); an antimetabolite (including, e.g., folic acid antagonists, pyrimidine analogs, purine analogs and adenosine deaminase inhibitors (e.g., fludarabine)); a TNFR glucocorticoid induced TNFR related protein (GITR) agonist; a proteasome inhibitor (e.g., aclacinomycin A, gliotoxin or bortezomib); an immunomodulator such as thalidomide or a thalidomide derivative (e.g., lenalidomide).
[0605] General Chemotherapeutic agents considered for use in combination therapies include anastrozole (Arimidex.RTM.), bicalutamide (Casodex.RTM.), bleomycin sulfate (Blenoxane.RTM.), busulfan (Myleran.RTM.), busulfan injection (Busulfex.RTM.), capecitabine (Xeloda.RTM.), N4-pentoxycarbonyl-5-deoxy-5-fluorocytidine, carboplatin (Paraplatin.RTM.), carmustine (BiCNU.RTM.), chlorambucil (Leukeran.RTM.), cisplatin (Platinol.RTM.), cladribine (Leustatin.RTM.), cyclophosphamide (Cytoxan.RTM. or Neosar.RTM.), cytarabine, cytosine arabinoside (Cytosar-U.RTM.), cytarabine liposome injection (DepoCyt.RTM.), dacarbazine (DTIC-Dome.RTM.), dactinomycin (Actinomycin D, Cosmegan), daunorubicin hydrochloride (Cerubidine.RTM.), daunorubicin citrate liposome injection (DaunoXome.RTM.), dexamethasone, docetaxel (Taxotere.RTM.), doxorubicin hydrochloride (Adriamycin.RTM., Rubex.RTM.), etoposide (Vepesid.RTM.), fludarabine phosphate (Fludara.RTM.), 5-fluorouracil (Adrucil.RTM., Efudex.RTM.), flutamide (Eulexin.RTM.), tezacitibine, gemcitabine (difluorodeoxycitidine), hydroxyurea (Hydrea.RTM.), Idarubicin (Idamycin.RTM.), ifosfamide (IFEX.RTM.), irinotecan (Camptosar.RTM.), L-asparaginase (ELSPAR.RTM.), leucovorin calcium, melphalan (Alkeran.RTM.), 6-mercaptopurine (Purinethol.RTM.), methotrexate (Folex.RTM.), mitoxantrone (Novantrone.RTM.), mylotarg, paclitaxel (Taxol.RTM.), phoenix (Yttrium90/MX-DTPA), pentostatin, polifeprosan 20 with carmustine implant (Gliadel.RTM.), tamoxifen citrate (Nolvadex.RTM.), teniposide (Vumon.RTM.), 6-thioguanine, thiotepa, tirapazamine (Tirazone.RTM.), topotecan hydrochloride for injection (Hycamptin.RTM.), vinblastine (Velban.RTM.), vincristine (Oncovin.RTM.), and vinorelbine (Navelbine.RTM.).
[0606] Exemplary alkylating agents include, without limitation, nitrogen mustards, ethylenimine derivatives, alkyl sulfonates, nitrosoureas and triazenes): uracil mustard (Aminouracil Mustard.RTM., Chlorethaminacil.RTM., Demethyldopan.RTM., Desmethyldopan.RTM., Haemanthamine.RTM., Nordopan.RTM., Uracil nitrogen Mustard.RTM., Uracillost.RTM., Uracilmostaza.RTM., Uramustin.RTM., Uramustine.RTM.), chlormethine (Mustargen.RTM.), cyclophosphamide (Cytoxan.RTM., Neosar.RTM., Clafen.RTM., Endoxan.RTM., Procytox.RTM., Revimmune.TM.), ifosfamide (Mitoxana.RTM.), melphalan (Alkeran.RTM.), Chlorambucil (Leukeran.RTM.), pipobroman (Amedel.RTM., Vercyte.RTM.), triethylenemelamine (Hemel.RTM., Hexalen.RTM., Hexastat.RTM.), triethylenethiophosphoramine, Temozolomide (Temodar.RTM.), thiotepa (Thioplex.RTM.), busulfan (Busilvex.RTM., Myleran.RTM.), carmustine (BiCNU.RTM.), lomustine (CeeNU.RTM.), streptozocin (Zanosar.RTM.), and Dacarbazine (DTIC-Dome.RTM.). Additional exemplary alkylating agents include, without limitation, Oxaliplatin (Eloxatin.RTM.); Temozolomide (Temodar.RTM. and Temodal.RTM.); Dactinomycin (also known as actinomycin-D, Cosmegen.RTM.); Melphalan (also known as L-PAM, L-sarcolysin, and phenylalanine mustard, Alkeran.RTM.); Altretamine (also known as hexamethylmelamine (HMM), Hexalen.RTM.); Carmustine (BiCNU.RTM.); Bendamustine (Treanda.RTM.); Busulfan (Busulfex.RTM. and Myleran.RTM.); Carboplatin (Paraplatin.RTM.); Lomustine (also known as CCNU, CeeNU.RTM.); Cisplatin (also known as CDDP, Platinol.RTM. and Platinol.RTM.-AQ); Chlorambucil (Leukeran.RTM.); Cyclophosphamide (Cytoxan.RTM. and Neosar.RTM.); Dacarbazine (also known as DTIC, DIC and imidazole carboxamide, DTIC-Dome.RTM.); Altretamine (also known as hexamethylmelamine (HMM), Hexalen.RTM.); Ifosfamide (Ifex.RTM.); Prednumustine; Procarbazine (Matulane.RTM.); Mechlorethamine (also known as nitrogen mustard, mustine and mechloroethamine hydrochloride, Mustargen.RTM.); Streptozocin (Zanosar.RTM.); Thiotepa (also known as thiophosphoamide, TESPA and TSPA, Thioplex.RTM.); Cyclophosphamide (Endoxan.RTM., Cytoxan.RTM., Neosar.RTM., Procytox.RTM., Revimmune.RTM.); and Bendamustine HCl (Treanda.RTM.).
[0607] In embodiments, a CAR-expressing cell described herein, optionally in combination with a kinase inhibitor e.g., a BTK inhibitor such as ibrutinib, is administered to a subject in combination with fludarabine, cyclophosphamide, and/or rituximab. In embodiments, a CAR-expressing cell described herein is administered to a subject in combination with fludarabine, cyclophosphamide, and rituximab (FCR). In embodiments, the subject has CLL. For example, the subject has a deletion in the short arm of chromosome 17 (del(17p), e.g., in a leukemic cell). In other examples, the subject does not have a del(17p). In embodiments, the subject comprises a leukemic cell comprising a mutation in the immunoglobulin heavy-chain variable-region (IgV.sub.H) gene. In other embodiments, the subject does not comprise a leukemic cell comprising a mutation in the immunoglobulin heavy-chain variable-region (IgV.sub.H) gene. In embodiments, the fludarabine is administered at a dosage of about 10-50 mg/m.sup.2 (e.g., about 10-15, 15-20, 20-25, 25-30, 30-35, 35-40, 40-45, or 45-50 mg/m.sup.2), e.g., intravenously. In embodiments, the cyclophosphamide is administered at a dosage of about 200-300 mg/m.sup.2 (e.g., about 200-225, 225-250, 250-275, or 275-300 mg/m.sup.2), e.g., intravenously. In embodiments, the rituximab is administered at a dosage of about 400-600 mg/m.sup.2 (e.g., 400-450, 450-500, 500-550, or 550-600 mg/m.sup.2), e.g., intravenously.
[0608] In embodiments, a CAR-expressing cell described herein, optionally in combination with a kinase inhibitor e.g., a BTK inhibitor such as ibrutinib, is administered to a subject in combination with bendamustine and rituximab. In embodiments, the subject has CLL. For example, the subject has a deletion in the short arm of chromosome 17 (del(17p), e.g., in a leukemic cell). In other examples, the subject does not have a del(17p). In embodiments, the subject comprises a leukemic cell comprising a mutation in the immunoglobulin heavy-chain variable-region (IgV.sub.H) gene. In other embodiments, the subject does not comprise a leukemic cell comprising a mutation in the immunoglobulin heavy-chain variable-region (IgV.sub.H) gene. In embodiments, the bendamustine is administered at a dosage of about 70-110 mg/m.sup.2 (e.g., 70-80, 80-90, 90-100, or 100-110 mg/m.sup.2), e.g., intravenously. In embodiments, the rituximab is administered at a dosage of about 400-600 mg/m.sup.2 (e.g., 400-450, 450-500, 500-550, or 550-600 mg/m.sup.2), e.g., intravenously.
[0609] In embodiments, a CAR-expressing cell described herein, optionally in combination with a kinase inhibitor e.g., a BTK inhibitor such as ibrutinib, is administered to a subject in combination with rituximab, cyclophosphamide, doxorubicine, vincristine, and/or a corticosteroid (e.g., prednisone). In embodiments, a CAR-expressing cell described herein is administered to a subject in combination with rituximab, cyclophosphamide, doxorubicine, vincristine, and prednisone (R-CHOP). In embodiments, the subject has diffuse large B-cell lymphoma (DLBCL). In embodiments, the subject has nonbulky limited-stage DLBCL (e.g., comprises a tumor having a size/diameter of less than 7 cm). In embodiments, the subject is treated with radiation in combination with the R-CHOP. For example, the subject is administered R-CHOP (e.g., 1-6 cycles, e.g., 1, 2, 3, 4, 5, or 6 cycles of R-CHOP), followed by radiation. In some cases, the subject is administered R-CHOP (e.g., 1-6 cycles, e.g., 1, 2, 3, 4, 5, or 6 cycles of R-CHOP) following radiation.
[0610] In embodiments, a CAR-expressing cell described herein, optionally in combination with a kinase inhibitor e.g., a BTK inhibitor such as ibrutinib, is administered to a subject in combination with etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and/or rituximab. In embodiments, a CAR-expressing cell described herein is administered to a subject in combination with etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab (EPOCH-R). In embodiments, a CAR-expressing cell described herein is administered to a subject in combination with dose-adjusted EPOCH-R (DA-EPOCH-R). In embodiments, the subject has a B cell lymphoma, e.g., a Myc-rearranged aggressive B cell lymphoma.
[0611] In embodiments, a CAR-expressing cell described herein, optionally in combination with a kinase inhibitor e.g., a BTK inhibitor such as ibrutinib, is administered to a subject in combination with rituximab and/or lenalidomide. Lenalidomide ((RS)-3-(4-Amino-1-oxo 1,3-dihydro-2H-isoindol-2-yl)piperidine-2,6-dione) is an immunomodulator. In embodiments, a CAR-expressing cell described herein is administered to a subject in combination with rituximab and lenalidomide. In embodiments, the subject has follicular lymphoma (FL) or mantle cell lymphoma (MCL). In embodiments, the subject has FL and has not previously been treated with a cancer therapy. In embodiments, lenalidomide is administered at a dosage of about 10-20 mg (e.g., 10-15 or 15-20 mg), e.g., daily. In embodiments, rituximab is administered at a dosage of about 350-550 mg/m.sup.2 (e.g., 350-375, 375-400, 400-425, 425-450, 450-475, or 475-500 mg/m.sup.2), e.g., intravenously.
[0612] Exemplary immunomodulators include, e.g., afutuzumab (available from Roche.RTM.); pegfilgrastim (Neulasta.RTM.); lenalidomide (CC-5013, Revlimid.RTM.); thalidomide (Thalomid.RTM.), pomelidomide, actimid (CC4047); and IRX-2 (mixture of human cytokines including interleukin 1, interleukin 2, and interferon .gamma., CAS 951209-71-5, available from IRX Therapeutics).
[0613] Exemplary anthracyclines include, e.g., doxorubicin (Adriamycin.RTM. and Rubex.RTM.); bleomycin (Lenoxane.RTM.); daunorubicin (dauorubicin hydrochloride, daunomycin, and rubidomycin hydrochloride, Cerubidine.RTM.); daunorubicin liposomal (daunorubicin citrate liposome, DaunoXome.RTM.); mitoxantrone (DHAD, Novantrone.RTM.); epirubicin (Ellence.TM.); idarubicin (Idamycin.RTM., Idamycin PFS.RTM.); mitomycin C (Mutamycin.RTM.); geldanamycin; herbimycin; ravidomycin; and desacetylravidomycin.
[0614] Exemplary vinca alkaloids include, e.g., vinorelbine tartrate (Navelbine.RTM.), Vincristine (Oncovin.RTM.), and Vindesine (Eldisine.RTM.)); vinblastine (also known as vinblastine sulfate, vincaleukoblastine and VLB, Alkaban-AQ.RTM. and Velban.RTM.); and vinorelbine (Navelbine.RTM.).
[0615] Exemplary proteosome inhibitors include bortezomib (Velcade.RTM.); carfilzomib (PX-171-007, (S)-4-Methyl-N--((S)-1-(((S)-4-methyl-1-((R)-2-methyloxiran-2-yl)-1-oxope- ntan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)-2-((S)-2-(2-morpholinoacetamid- o)-4-phenylbutanamido)-pentanamide); marizomib (NPI-0052); ixazomib citrate (MLN-9708); delanzomib (CEP-18770); and O-Methyl-N-[(2-methyl-5-thiazolyl)carbonyl]-L-seryl-O-methyl-N-[(1 S)-2-[(2R)-2-methyl-2-oxiranyl]-2-oxo-1-(phenylmethyl)ethyl]-L-serinamide (ONX-0912).
[0616] In embodiments, a CAR-expressing cell described herein, optionally in combination with a kinase inhibitor e.g., a BTK inhibitor such as ibrutinib, is administered to a subject in combination with brentuximab. Brentuximab is an antibody-drug conjugate of anti-CD30 antibody and monomethyl auristatin E. In embodiments, the subject has Hodgkin lymphoma (HL), e.g., relapsed or refractory HL. In embodiments, the subject comprises CD30+HL. In embodiments, the subject has undergone an autologous stem cell transplant (ASCT). In embodiments, the subject has not undergone an ASCT. In embodiments, brentuximab is administered at a dosage of about 1-3 mg/kg (e.g., about 1-1.5, 1.5-2, 2-2.5, or 2.5-3 mg/kg), e.g., intravenously, e.g., every 3 weeks.
[0617] In embodiments, a CAR-expressing cell described herein, optionally in combination with a kinase inhibitor e.g., a BTK inhibitor such as ibrutinib, is administered to a subject in combination with brentuximab and dacarbazine or in combination with brentuximab and bendamustine. Dacarbazine is an alkylating agent with a chemical name of 5-(3,3-Dimethyl-1-triazenyl)imidazole-4-carboxamide. Bendamustine is an alkylating agent with a chemical name of 4-[5-[Bis(2-chloroethyl)amino]-1-methylbenzimidazol-2-yl]butanoic acid. In embodiments, the subject has Hodgkin lymphoma (HL). In embodiments, the subject has not previously been treated with a cancer therapy. In embodiments, the subject is at least 60 years of age, e.g., 60, 65, 70, 75, 80, 85, or older. In embodiments, dacarbazine is administered at a dosage of about 300-450 mg/m.sup.2 (e.g., about 300-325, 325-350, 350-375, 375-400, 400-425, or 425-450 mg/m.sup.2), e.g., intravenously. In embodiments, bendamustine is administered at a dosage of about 75-125 mg/m.sup.2 (e.g., 75-100 or 100-125 mg/m.sup.2, e.g., about 90 mg/m.sup.2), e.g., intravenously. In embodiments, brentuximab is administered at a dosage of about 1-3 mg/kg (e.g., about 1-1.5, 1.5-2, 2-2.5, or 2.5-3 mg/kg), e.g., intravenously, e.g., every 3 weeks.
[0618] In some embodiments, a CAR-expressing cell described herein is administered to a subject in combination with a CD20 inhibitor, e.g., an anti-CD20 antibody (e.g., an anti-CD20 mono- or bispecific antibody) or a fragment thereof. Exemplary anti-CD20 antibodies include but are not limited to rituximab, ofatumumab, ocrelizumab, veltuzumab, obinutuzumab, TRU-015 (Trubion Pharmaceuticals), ocaratuzumab, and Pro131921 (Genentech). See, e.g., Lim et al. Haematologica. 95.1(2010):135-43.
[0619] In some embodiments, the anti-CD20 antibody comprises rituximab. Rituximab is a chimeric mouse/human monoclonal antibody IgG1 kappa that binds to CD20 and causes cytolysis of a CD20 expressing cell, e.g., as described in www.accessdata.fda.gov/drugsatfda_docs/label/2010/103705s5311lbl.pdf. In embodiments, a CAR-expressing cell described herein is administered to a subject in combination with rituximab. In embodiments, the subject has CLL or SLL.
[0620] In some embodiments, rituximab is administered intravenously, e.g., as an intravenous infusion. For example, each infusion provides about 500-2000 mg (e.g., about 500-550, 550-600, 600-650, 650-700, 700-750, 750-800, 800-850, 850-900, 900-950, 950-1000, 1000-1100, 1100-1200, 1200-1300, 1300-1400, 1400-1500, 1500-1600, 1600-1700, 1700-1800, 1800-1900, or 1900-2000 mg) of rituximab. In some embodiments, rituximab is administered at a dose of 150 mg/m.sup.2 to 750 mg/m.sup.2, e.g., about 150-175 mg/m.sup.2, 175-200 mg/m.sup.2, 200-225 mg/m.sup.2, 225-250 mg/m.sup.2, 250-300 mg/m.sup.2, 300-325 mg/m.sup.2, 325-350 mg/m.sup.2, 350-375 mg/m.sup.2, 375-400 mg/m.sup.2, 400-425 mg/m.sup.2, 425-450 mg/m.sup.2, 450-475 mg/m.sup.2, 475-500 mg/m.sup.2, 500-525 mg/m.sup.2, 525-550 mg/m.sup.2, 550-575 mg/m.sup.2, 575-600 mg/m.sup.2, 600-625 mg/m.sup.2, 625-650 mg/m.sup.2, 650-675 mg/m.sup.2, or 675-700 mg/m.sup.2, where m.sup.2 indicates the body surface area of the subject. In some embodiments, rituximab is administered at a dosing interval of at least 4 days, e.g., 4, 7, 14, 21, 28, 35 days, or more. For example, rituximab is administered at a dosing interval of at least 0.5 weeks, e.g., 0.5, 1, 2, 3, 4, 5, 6, 7, 8 weeks, or more. In some embodiments, rituximab is administered at a dose and dosing interval described herein for a period of time, e.g., at least 2 weeks, e.g., at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 weeks, or greater. For example, rituximab is administered at a dose and dosing interval described herein for a total of at least 4 doses per treatment cycle (e.g., at least 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, or more doses per treatment cycle).
[0621] In some embodiments, the anti-CD20 antibody comprises ofatumumab. Ofatumumab is an anti-CD20 IgG1.kappa. human monoclonal antibody with a molecular weight of approximately 149 kDa. For example, ofatumumab is generated using transgenic mouse and hybridoma technology and is expressed and purified from a recombinant murine cell line (NS0). See, e.g., www.accessdata.fda.gov/drugsatfda_docs/label/2009/125326lbl.pdf; and Clinical Trial Identifier number NCT01363128, NCT01515176, NCT01626352, and NCT01397591. In embodiments, a CAR-expressing cell described herein is administered to a subject in combination with ofatumumab. In embodiments, the subject has CLL or SLL.
[0622] In some embodiments, ofatumumab is administered as an intravenous infusion. For example, each infusion provides about 150-3000 mg (e.g., about 150-200, 200-250, 250-300, 300-350, 350-400, 400-450, 450-500, 500-550, 550-600, 600-650, 650-700, 700-750, 750-800, 800-850, 850-900, 900-950, 950-1000, 1000-1200, 1200-1400, 1400-1600, 1600-1800, 1800-2000, 2000-2200, 2200-2400, 2400-2600, 2600-2800, or 2800-3000 mg) of ofatumumab. In embodiments, ofatumumab is administered at a starting dosage of about 300 mg, followed by 2000 mg, e.g., for about 11 doses, e.g., for 24 weeks. In some embodiments, ofatumumab is administered at a dosing interval of at least 4 days, e.g., 4, 7, 14, 21, 28, 35 days, or more. For example, ofatumumab is administered at a dosing interval of at least 1 week, e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 24, 26, 28, 20, 22, 24, 26, 28, 30 weeks, or more. In some embodiments, ofatumumab is administered at a dose and dosing interval described herein for a period of time, e.g., at least 1 week, e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 22, 24, 26, 28, 30, 40, 50, 60 weeks or greater, or 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 months or greater, or 1, 2, 3, 4, 5 years or greater. For example, ofatumumab is administered at a dose and dosing interval described herein for a total of at least 2 doses per treatment cycle (e.g., at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 18, 20, or more doses per treatment cycle).
[0623] In some cases, the anti-CD20 antibody comprises ocrelizumab. Ocrelizumab is a humanized anti-CD20 monoclonal antibody, e.g., as described in Clinical Trials Identifier Nos. NCT00077870, NCT01412333, NCT00779220, NCT00673920, NCT01194570, and Kappos et al. Lancet. 19.378(2011):1779-87.
[0624] In some cases, the anti-CD20 antibody comprises veltuzumab. Veltuzumab is a humanized monoclonal antibody against CD20. See, e.g., Clinical Trial Identifier No. NCT00547066, NCT00546793, NCT01101581, and Goldenberg et al. Leuk Lymphoma. 51(5)(2010):747-55.
[0625] In some cases, the anti-CD20 antibody comprises GA101. GA101 (also called obinutuzumab or R05072759) is a humanized and glyco-engineered anti-CD20 monoclonal antibody. See, e.g., Robak. Curr. Opin. Investig. Drugs. 10.6(2009):588-96; Clinical Trial Identifier Numbers: NCT01995669, NCT01889797, NCT02229422, and NCT01414205; and www.accessdata.fda.gov/drugsatfda_docs/label/2013/125486s000lbl.pdf.
[0626] In some cases, the anti-CD20 antibody comprises AME-133v. AME-133v (also called LY2469298 or ocaratuzumab) is a humanized IgG1 monoclonal antibody against CD20 with increased affinity for the Fc.gamma.RIIIa receptor and an enhanced antibody dependent cellular cytotoxicity (ADCC) activity compared with rituximab. See, e.g., Robak et al. BioDrugs 25.1(2011):13-25; and Forero-Torres et al. Clin Cancer Res. 18.5(2012):1395-403.
[0627] In some cases, the anti-CD20 antibody comprises PRO131921. PRO131921 is a humanized anti-CD20 monoclonal antibody engineered to have better binding to Fc.gamma.RIIIa and enhanced ADCC compared with rituximab. See, e.g., Robak et al. BioDrugs 25.1(2011):13-25; and Casulo et al. Clin Immunol. 154.1(2014):37-46; and Clinical Trial Identifier No. NCT00452127.
[0628] In some cases, the anti-CD20 antibody comprises TRU-015. TRU-015 is an anti-CD20 fusion protein derived from domains of an antibody against CD20. TRU-015 is smaller than monoclonal antibodies, but retains Fc-mediated effector functions. See, e.g., Robak et al. BioDrugs 25.1(2011):13-25. TRU-015 contains an anti-CD20 single-chain variable fragment (scFv) linked to human IgG1 hinge, CH2, and CH3 domains but lacks CH1 and CL domains.
[0629] In some embodiments, an anti-CD20 antibody described herein is conjugated or otherwise bound to a therapeutic agent, e.g., a chemotherapeutic agent (e.g., cytoxan, fludarabine, histone deacetylase inhibitor, demethylating agent, peptide vaccine, anti-tumor antibiotic, tyrosine kinase inhibitor, alkylating agent, anti-microtubule or anti-mitotic agent), anti-allergic agent, anti-nausea agent (or anti-emetic), pain reliever, or cytoprotective agent described herein.
[0630] In embodiments, a CAR-expressing cell described herein is administered to a subject in combination with a B-cell lymphoma 2 (BCL-2) inhibitor (e.g., venetoclax, also called ABT-199 or GDC-0199) and/or rituximab. In embodiments, a CAR-expressing cell described herein is administered to a subject in combination with venetoclax and rituximab. Venetoclax is a small molecule that inhibits the anti-apoptotic protein, BCL-2. The structure of venetoclax (4-(4-{[2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-en-1-yl]methyl}piperazi- n-1-yl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfon- yl)-2-(1H-pyrrololfonyl)-2-([2,3-b]pyridin-5-yloxy)benzamide) is shown below.
##STR00006##
[0631] In embodiments, the subject has CLL. In embodiments, the subject has relapsed CLL, e.g., the subject has previously been administered a cancer therapy. In embodiments, venetoclax is administered at a dosage of about 15-600 mg (e.g., 15-20, 20-50, 50-75, 75-100, 100-200, 200-300, 300-400, 400-500, or 500-600 mg), e.g., daily. In embodiments, rituximab is administered at a dosage of about 350-550 mg/m2 (e.g., 350-375, 375-400, 400-425, 425-450, 450-475, or 475-500 mg/m2), e.g., intravenously, e.g., monthly.
[0632] In an embodiment, cells expressing a CAR described herein, optionally in combination with a kinase inhibitor, e.g., a BTK inhibitor such as ibrutinib, are administered to a subject in combination with a molecule that decreases the Treg cell population. Methods that decrease the number of (e.g., deplete) Treg cells are known in the art and include, e.g., CD25 depletion, cyclophosphamide administration, modulating GITR function. Without wishing to be bound by theory, it is believed that reducing the number of Treg cells in a subject prior to apheresis or prior to administration of a CAR-expressing cell described herein reduces the number of unwanted immune cells (e.g., Tregs) in the tumor microenvironment and reduces the subject's risk of relapse. In one embodiment, cells expressing a CAR described herein, optionally in combination with a kinase inhibitor, e.g., a BTK inhibitor such as ibrutinib, are administered to a subject in combination with a molecule targeting GITR and/or modulating GITR functions, such as a GITR agonist and/or a GITR antibody that depletes regulatory T cells (Tregs). In embodiments, cells expressing a CAR described herein, optionally in combination with a kinase inhibitor, e.g., a BTK inhibitor such as ibrutinib, are administered to a subject in combination with cyclophosphamide. In one embodiment, the GITR binding molecules and/or molecules modulating GITR functions (e.g., GITR agonist and/or Treg depleting GITR antibodies) are administered prior to administration of the CAR-expressing cell. For example, in one embodiment, the GITR agonist can be administered prior to apheresis of the cells. In embodiments, cyclophosphamide is administered to the subject prior to administration (e.g., infusion or re-infusion) of the CAR-expressing cell or prior to aphersis of the cells. In embodiments, cyclophosphamide and an anti-GITR antibody are administered to the subject prior to administration (e.g., infusion or re-infusion) of the CAR-expressing cell or prior to apheresis of the cells. In one embodiment, the subject has cancer (e.g., a solid cancer or a hematological cancer such as ALL or CLL). In an embodiment, the subject has CLL. In embodiments, the subject has ALL. In embodiments, the subject has a solid cancer, e.g., a solid cancer described herein.
[0633] Exemplary GITR agonists include, e.g., GITR fusion proteins and anti-GITR antibodies (e.g., bivalent anti-GITR antibodies) such as, e.g., a GITR fusion protein described in U.S. Pat. No. 6,111,090, European Patent No.: 090505B1, U.S. Pat. No. 8,586,023, PCT Publication Nos.: WO 2010/003118 and 2011/090754, or an anti-GITR antibody described, e.g., in U.S. Pat. No. 7,025,962, European Patent No.: 1947183B1, U.S. Pat. Nos. 7,812,135, 8,388,967, 8,591,886, European Patent No.: EP 1866339, PCT Publication No.: WO 2011/028683, PCT Publication No.: WO 2013/039954, PCT Publication No.: WO2005/007190, PCT Publication No.: WO 2007/133822, PCT Publication No.: WO2005/055808, PCT Publication No.: WO 99/40196, PCT Publication No.: WO 2001/03720, PCT Publication No.: WO99/20758, PCT Publication No.: WO2006/083289, PCT Publication No.: WO 2005/115451, U.S. Pat. No. 7,618,632, and PCT Publication No.: WO 2011/051726.
[0634] In one embodiment, the combination of a CAR expressing cell described herein and a kinase inhibitor described herein is administered to a subject in combination with a GITR agonist, e.g., a GITR agonist described herein. In one embodiment, the GITR agonist is administered prior to the CAR-expressing cell. For example, in one embodiment, the GITR agonist can be administered prior to apheresis of the cells. In one embodiment, the subject has CLL.
[0635] Drugs that inhibit either the calcium dependent phosphatase calcineurin (cyclosporine and FK506) or inhibit the p70S6 kinase that is important for growth factor induced signaling (rapamycin). (Liu et al., Cell 66:807-815, 1991; Henderson et al., Immun. 73:316-321, 1991; Bierer et al., Curr. Opin. Immun. 5:763-773, 1993) can also be used. In a further aspect, the cell compositions of the present invention may be administered to a patient in conjunction with (e.g., before, simultaneously or following) bone marrow transplantation, T cell ablative therapy using chemotherapy agents such as, fludarabine, external-beam radiation therapy (XRT), cyclophosphamide, and/or antibodies such as OKT3 or CAMPATH. In one aspect, the cell compositions of the present invention are administered following B-cell ablative therapy such as agents that react with CD20, e.g., Rituxan. For example, in one embodiment, subjects may undergo standard treatment with high dose chemotherapy followed by peripheral blood stem cell transplantation. In certain embodiments, following the transplant, subjects receive an infusion of the expanded immune cells of the present invention. In an additional embodiment, expanded cells are administered before or following surgery.
[0636] In one embodiment, the subject can be administered an agent which reduces or ameliorates a side effect associated with the administration of a CAR-expressing cell. Side effects associated with the administration of a CAR-expressing cell include, but are not limited to CRS, and hemophagocytic lymphohistiocytosis (HLH), also termed Macrophage Activation Syndrome (MAS). Symptoms of CRS include high fevers, nausea, transient hypotension, hypoxia, and the like. Accordingly, the methods described herein can comprise administering a CAR-expressing cell described herein to a subject and further administering an agent to manage elevated levels of a soluble factor resulting from treatment with a CAR-expressing cell. In one embodiment, the soluble factor elevated in the subject is one or more of IFN-.gamma., TNF.alpha., IL-2 receptor and IL-6. Therefore, an agent administered to treat this side effect can be an agent that neutralizes one or more of these soluble factors. Examples of such agents include, but are not limited to a steroid (e.g., corticosteroid), an inhibitor of TNF.alpha., and an inhibitor of IL-6. An example of a TNF.alpha. inhibitor is an anti-TNF.alpha. antibody molecule such as, infliximab, adalimumab, certolizumab pegol, and golimumab. Another example of a TNF.alpha. inhibitor is a fusion protein such as entanercept. Small molecule inhibitor of TNF.alpha. include, but are not limited to, xanthine derivatives (e.g. pentoxifylline) and bupropion. An example of an IL-6 inhibitor is an anti-IL-6 antibody molecule or anti-IL-6 receptor antibody molecule such as tocilizumab (toc), sarilumab, elsilimomab, CNTO 328, ALD518/BMS-945429, CNTO 136, CPSI-2364, CDP6038, VX30, ARGX-109, FE301, and FM101. In one embodiment, the anti-IL-6 receptor antibody molecule is tocilizumab. An example of an IL-1R based inhibitor is anakinra.
[0637] In one embodiment, the subject can be administered an agent which enhances the activity of a CAR-expressing cell. For example, in one embodiment, the agent can be an agent which inhibits an inhibitory molecule. Inhibitory molecules, e.g., Programmed Death 1 (PD1), can, in some embodiments, decrease the ability of a CAR-expressing cell to mount an immune effector response. Examples of inhibitory molecules include PD1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 and TGFR beta. Inhibition of an inhibitory molecule, e.g., by inhibition at the DNA, RNA or protein level, can optimize a CAR-expressing cell performance. In embodiments, an inhibitory nucleic acid, e.g., an inhibitory nucleic acid, e.g., a dsRNA, e.g., an siRNA or shRNA, a clustered regularly interspaced short palindromic repeats (CRISPR), a transcription-activator like effector nuclease (TALEN), or a zinc finger endonuclease (ZFN), can be used to inhibit expression of an inhibitory molecule in the CAR-expressing cell. In an embodiment the inhibitor is an shRNA. In an embodiment, the inhibitory molecule is inhibited within a CAR-expressing cell. In these embodiments, a dsRNA molecule that inhibits expression of the inhibitory molecule is linked to the nucleic acid that encodes a component, e.g., all of the components, of the CAR. In one embodiment, the inhibitor of an inhibitory signal can be, e.g., an antibody or antibody fragment that binds to an inhibitory molecule. For example, the agent can be an antibody or antibody fragment that binds to PD1, PD-L1, PD-L2 or CTLA4 (e.g., ipilimumab (also referred to as MDX-010 and MDX-101, and marketed as Yervoy.RTM.; Bristol-Myers Squibb; Tremelimumab (IgG2 monoclonal antibody available from Pfizer, formerly known as ticilimumab, CP-675,206).). In an embodiment, the agent is an antibody or antibody fragment that binds to TIM3. In an embodiment, the agent is an antibody or antibody fragment that binds to LAG3. In an embodiment, the agent is an antibody or antibody fragment that binds to CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5).
[0638] PD1 is an inhibitory member of the CD28 family of receptors that also includes CD28, CTLA-4, ICOS, and BTLA. PD1 is expressed on activated B cells, T cells and myeloid cells (Agata et al. 1996 Int. Immunol 8:765-75). Two ligands for PD1, PD-L1 and PD-L2 have been shown to downregulate T cell activation upon binding to PD1 (Freeman et al. 2000 J Exp Med 192:1027-34; Latchman et al. 2001 Nat Immunol 2:261-8; Carter et al. 2002 Eur J Immunol 32:634-43). PD-L1 is abundant in human cancers (Dong et al. 2003 J Mol Med 81:281-7; Blank et al. 2005 Cancer Immunol. Immunother 54:307-314; Konishi et al. 2004 Clin Cancer Res 10:5094). Immune suppression can be reversed by inhibiting the local interaction of PD1 with PD-L. Antibodies, antibody fragments, and other inhibitors of PD1, PD-L1 and PD-L2 are known and may be used combination with a CD19 CAR described herein. For example, nivolumab (also referred to as BMS-936558 or MDX1106; Bristol-Myers Squibb) is a fully human IgG4 monoclonal antibody which specifically blocks PD1. Nivolumab (clone 5C4) and other human monoclonal antibodies that specifically bind to PD1 are disclosed in U.S. Pat. No. 8,008,449 and WO2006/121168. Pidilizumab (CT-011; Cure Tech) is a humanized IgG1k monoclonal antibody that binds to PD1. Pidilizumab and other humanized anti-PD1 monoclonal antibodies are disclosed in WO2009/101611. Pembrolizumab (formerly known as lambrolizumab, and also referred to as Keytruda, MK03475; Merck) is a humanized IgG4 monoclonal antibody that binds to PD1. Pembrolizumab and other humanized anti-PD1 antibodies are disclosed in U.S. Pat. No. 8,354,509 and WO2009/114335. MEDI4736 (Medimmune) is a human monoclonal antibody that binds to PDL1, and inhibits interaction of the ligand with PD1. MDPL3280A (Genentech/Roche) is a human Fc optimized IgG1 monoclonal antibody that binds to PD-L. MDPL3280A and other human monoclonal antibodies to PD-L1 are disclosed in U.S. Pat. No. 7,943,743 and U.S Publication No.: 20120039906. Other anti-PD-L1 binding agents include YW243.55.570 (heavy and light chain variable regions are shown in SEQ ID NOs 20 and 21 in WO2010/077634) and MDX-1 105 (also referred to as BMS-936559, and, e.g., anti-PD-L1 binding agents disclosed in WO2007/005874). AMP-224 (B7-DCIg; Amplimmune; e.g., disclosed in WO2010/027827 and WO2011/066342), is a PD-L2 Fc fusion soluble receptor that blocks the interaction between PD1 and B7-H1. Other anti-PD1 antibodies include AMP 514 (Amplimmune), among others, e.g., anti-PD1 antibodies disclosed in U.S. Pat. No. 8,609,089, US 2010028330, and/or US 20120114649.
[0639] TIM3 (T cell immunoglobulin-3) also negatively regulates T cell function, particularly in IFN-g-secreting CD4+T helper 1 and CD8+T cytotoxic 1 cells, and plays a critical role in T cell exhaustion. Inhibition of the interaction between TIM3 and its ligands, e.g., galectin-9 (Gal9), phosphotidylserine (PS), and HMGB1, can increase immune response. Antibodies, antibody fragments, and other inhibitors of TIM3 and its ligands are available in the art and may be used combination with a CD19 CAR described herein. For example, antibodies, antibody fragments, small molecules, or peptide inhibitors that target TIM3 binds to the IgV domain of TIM3 to inhibit interaction with its ligands. Antibodies and peptides that inhibit TIM3 are disclosed in WO2013/006490 and US20100247521. Other anti-TIM3 antibodies include humanized versions of RMT3-23 (disclosed in Ngiow et al., 2011, Cancer Res, 71:3540-3551), and clone 8B. 2C12 (disclosed in Monney et al., 2002, Nature, 415:536-541). Bi-specific antibodies that inhibit TIM3 and PD-1 are disclosed in US20130156774.
[0640] In other embodiments, the agent which enhances the activity of a CAR-expressing cell is a CEACAM inhibitor (e.g., CEACAM-1, CEACAM-3, and/or CEACAM-5 inhibitor). In one embodiment, the inhibitor of CEACAM is an anti-CEACAM antibody molecule. Exemplary anti-CEACAM-1 antibodies are described in WO 2010/125571, WO 2013/082366 WO 2014/059251 and WO 2014/022332, e.g., a monoclonal antibody 34B1, 26H7, and 5F4; or a recombinant form thereof, as described in, e.g., US 2004/0047858, U.S. Pat. No. 7,132,255 and WO 99/052552. In other embodiments, the anti-CEACAM antibody binds to CEACAM-5 as described in, e.g., Zheng et al. PLoS One. 2010 Sep. 2;5(9). pii: e12529 (DOI:10:1371/journal.pone.0021146), or crossreacts with CEACAM-1 and CEACAM-5 as described in, e.g., WO 2013/054331 and US 2014/0271618.
[0641] Without wishing to be bound by theory, carcinoembryonic antigen cell adhesion molecules (CEACAM), such as CEACAM-1 and CEACAM-5, are believed to mediate, at least in part, inhibition of an anti-tumor immune response (see e.g., Markel et al. J Immunol. 2002 Mar. 15;168(6):2803-10; Markel et al. J Immunol. 2006 Nov. 1;177(9):6062-71; Markel et al. Immunology. 2009 February; 126(2):186-200; Markel et al. Cancer Immunol Immunother. 2010 February; 59(2):215-30; Ortenberg et al. Mol Cancer Ther. 2012 June; 11(6):1300-10; Stern et al. J Immunol. 2005 Jun. 1;174(11):6692-701; Zheng et al. PLoS One. 2010 Sep. 2;5(9). pii: e12529). For example, CEACAM-1 has been described as a heterophilic ligand for TIM-3 and as playing a role in TIM-3-mediated T cell tolerance and exhaustion (see e.g., WO 2014/022332; Huang, et al. (2014) Nature doi:10.1038/nature13848). In embodiments, co-blockade of CEACAM-1 and TIM-3 has been shown to enhance an anti-tumor immune response in xenograft colorectal cancer models (see e.g., WO 2014/022332; Huang, et al. (2014), supra). In other embodiments, co-blockade of CEACAM-1 and PD-1 reduce T cell tolerance as described, e.g., in WO 2014/059251. Thus, CEACAM inhibitors can be used with the other immunomodulators described herein (e.g., anti-PD-1 and/or anti-TIM-3 inhibitors) to enhance an immune response against a cancer, e.g., a melanoma, a lung cancer (e.g., NSCLC), a bladder cancer, a colon cancer an ovarian cancer, and other cancers as described herein.
[0642] LAG3 (lymphocyte activation gene-3 or CD223) is a cell surface molecule expressed on activated T cells and B cells that has been shown to play a role in CD8+ T cell exhaustion. Antibodies, antibody fragments, and other inhibitors of LAG3 and its ligands are available in the art and may be used combination with a CD19 CAR described herein. For example, 3BMS-986016 (Bristol-Myers Squib) is a monoclonal antibody that targets LAG3. IMP701 (Immutep) is an antagonist LAG3 antibody and IMP731 (Immutep and GlaxoSmithKline) is a depleting LAG3 antibody. Other LAG3 inhibitors include IMP321 (Immutep), which is a recombinant fusion protein of a soluble portion of LAG3 and Ig that binds to MHC class II molecules and activates antigen presenting cells (APC). Other antibodies are disclosed, e.g., in WO2010/019570.
[0643] In some embodiments, the CAR therapy and kinase inhibitor are administered in combination with a toll like receptor (TLR) agonist. The TLR agonist can be a TLR9 agonist. In some embodiments, the TLR agonist is an oligodeoxynucleotide, e.g., a CG-enriched oligodeoxynucleotide, e.g., an unmethylated CG-enriched oligodeoxynucleotide. See, e.g., Sagiv-Barfi et al., "Ibrutinib enhances the antitumor immune response induced by intratumoral injection of a TLR9 ligand in syngeneic mouse lymphoma model." Blood. 2015 Feb. 6. pii: blood-2014-08-593137, which is incorporated herein by reference in its entirety. In some embodiments, the TLR agonist is administered in combination with a CAR-expressing NK cell. Without being bound by theory, the TLR agonist may promote activation of NK cells such as CAR-expressing NK cells. In some embodiments, the TLR agonist is administered by injection, e.g., intrarumoral injection.
[0644] In some embodiments, the agent which enhances the activity of a CAR-expressing cell can be, e.g., a fusion protein comprising a first domain and a second domain, wherein the first domain is an inhibitory molecule, or fragment thereof, and the second domain is a polypeptide that is associated with a positive signal, e.g., a polypeptide comprising an intracellular signaling domain as described herein. In some embodiments, the polypeptide that is associated with a positive signal can include a costimulatory domain of CD28, CD27, ICOS, e.g., an intracellular signaling domain of CD28, CD27 and/or ICOS, and/or a primary signaling domain, e.g., of CD3 zeta, e.g., described herein. In one embodiment, the fusion protein is expressed by the same cell that expressed the CAR. In another embodiment, the fusion protein is expressed by a cell, e.g., a T cell that does not express an anti-CD19 CAR.
[0645] In one embodiment, the agent which enhances activity of a CAR-expressing cell described herein is miR-17-92.
[0646] In one embodiment, the agent which enhances activity of a CAR-described herein is a cytokine. Cytokines have important functions related to T cell expansion, differentiation, survival, and homeostatis. Cytokines that can be administered to the subject receiving a CAR-expressing cell described herein include: IL-2, IL-4, IL-7, IL-9, IL-15, IL-18, and IL-21, or a combination thereof. In preferred embodiments, the cytokine administered is IL-7, IL-15, or IL-21, or a combination thereof. The cytokine can be administered once a day or more than once a day, e.g., twice a day, three times a day, or four times a day. The cytokine can be administered for more than one day, e.g. the cytokine is administered for 2 days, 3 days, 4 days, 5 days, 6 days, 1 week, 2 weeks, 3 weeks, or 4 weeks. For example, the cytokine is administered once a day for 7 days.
[0647] In embodiments, the cytokine is administered in combination with CAR-expressing cells. The cytokine can be administered simultaneously or concurrently with the CAR-expressing cells, e.g., administered on the same day. The cytokine may be prepared in the same pharmaceutical composition as the CAR-expressing cells, or may be prepared in a separate pharmaceutical composition. Alternatively, the cytokine can be administered shortly after administration of the CAR-expressing T cells, e.g., 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, or 7 days after administration of the CAR-expressing cells. In embodiments where the cytokine is administered in a dosing regimen that occurs over more than one day, the first day of the cytokine dosing regimen can be on the same day as administration with the CAR-expressing cells, or the first day of the cytokine dosing regimen can be 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, or 7 days after administration of the CAR-expressing T cells. In one embodiment, on the first day, the CAR-expressing cells are administered to the subject, and on the second day, a cytokine is administered once a day for the next 7 days. In a preferred embodiment, the cytokine to be administered in combination with the CAR-expressing cells is IL-7, IL-15, and/or IL-21.
[0648] In other embodiments, the cytokine is administered a sufficient period of time after administration of the CAR-expressing cells, e.g., at least 2 weeks, 3 weeks, 4 weeks, 6 weeks, 8 weeks, 10 weeks, 12 weeks, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, or 1 year or more after administration of CAR-expressing cells. In one embodiment, the cytokine is administered after assessment of the subject's response to the CAR-expressing cells. For example, the subject is administered CAR-expressing cells according to the dosage and regimens described herein. The response of the subject to CART therapy is assessed at 2 weeks, 3 weeks, 4 weeks, 6 weeks, 8 weeks, 10 weeks, 12 weeks, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, or 1 year or more after administration of CAR-expressing cells, using any of the methods described herein, including inhibition of tumor growth, reduction of circulating tumor cells, or tumor regression. Subjects that do not exhibit a sufficient response to CART therapy can be administered a cytokine. Administration of the cytokine to the subject that has sub-optimal response to the CART therapy improves CART efficacy and/or anti-tumor activity. In a preferred embodiment, the cytokine administered after administration of CAR-expressing cells is IL-7.
[0649] Combination with a Low Dose of an mTOR Inhibitor
[0650] In one embodiment, the cells expressing a CAR molecule, e.g., a CAR molecule described herein, are administered in combination with a low, immune enhancing dose of an mTOR inhibitor.
[0651] In an embodiment, a dose of an mTOR inhibitor is associated with, or provides, mTOR inhibition of at least 5 but no more than 90%, at least 10 but no more than 90%, at least 15, but no more than 90%, at least 20 but no more than 90%, at least 30 but no more than 90%, at least 40 but no more than 90%, at least 50 but no more than 90%, at least 60 but no more than 90%, or at least 70 but no more than 90%.
[0652] In an embodiment, a dose of an mTOR inhibitor is associated with, or provides, mTOR inhibition of at least 5 but no more than 80%, at least 10 but no more than 80%, at least 15, but no more than 80%, at least 20 but no more than 80%, at least 30 but no more than 80%, at least 40 but no more than 80%, at least 50 but no more than 80%, or at least 60 but no more than 80%.
[0653] In an embodiment, a dose of an mTOR inhibitor is associated with, or provides, mTOR inhibition of at least 5 but no more than 70%, at least 10 but no more than 70%, at least 15, but no more than 70%, at least 20 but no more than 70%, at least 30 but no more than 70%, at least 40 but no more than 70%, or at least 50 but no more than 70%.
[0654] In an embodiment, a dose of an mTOR inhibitor is associated with, or provides, mTOR inhibition of at least 5 but no more than 60%, at least 10 but no more than 60%, at least 15, but no more than 60%, at least 20 but no more than 60%, at least 30 but no more than 60%, or at least 40 but no more than 60%.
[0655] In an embodiment, a dose of an mTOR inhibitor is associated with, or provides, mTOR inhibition of at least 5 but no more than 50%, at least 10 but no more than 50%, at least 15, but no more than 50%, at least 20 but no more than 50%, at least 30 but no more than 50%, or at least 40 but no more than 50%.
[0656] In an embodiment, a dose of an mTOR inhibitor is associated with, or provides, mTOR inhibition of at least 5 but no more than 40%, at least 10 but no more than 40%, at least 15, but no more than 40%, at least 20 but no more than 40%, at least 30 but no more than 40%, or at least 35 but no more than 40%.
[0657] In an embodiment, a dose of an mTOR inhibitor is associated with, or provides, mTOR inhibition of at least 5 but no more than 30%, at least 10 but no more than 30%, at least 15, but no more than 30%, at least 20 but no more than 30%, or at least 25 but no more than 30%.
[0658] In an embodiment, a dose of an mTOR inhibitor is associated with, or provides, mTOR inhibition of at least 1, 2, 3, 4 or 5 but no more than 20%, at least 1, 2, 3, 4 or 5 but no more than 30%, at least 1, 2, 3, 4 or 5, but no more than 35, at least 1, 2, 3, 4 or 5 but no more than 40%, or at least 1, 2, 3, 4 or 5 but no more than 45%.
[0659] In an embodiment, a dose of an mTOR inhibitor is associated with, or provides, mTOR inhibition of at least 1, 2, 3, 4 or 5 but no more than 90%.
[0660] As is discussed herein, the extent of mTOR inhibition can be expressed as the extent of P70 S6 kinase inhibition, e.g., the extent of mTOR inhibition can be determined by the level of decrease in P70 S6 kinase activity, e.g., by the decrease in phosphorylation of a P70 S6 kinase substrate. The level of mTOR inhibition can be evaluated by a method described herein, e.g. by the Boulay assay, or measurement of phosphorylated S6 levels by western blot.
Exemplary mTOR Inhibitors
[0661] As used herein, the term "mTOR inhibitor" refers to a compound or ligand, or a pharmaceutically acceptable salt thereof, which inhibits the mTOR kinase in a cell. In an embodiment an mTOR inhibitor is an allosteric inhibitor. In an embodiment an mTOR inhibitor is a catalytic inhibitor.
[0662] Allosteric mTOR inhibitors include the neutral tricyclic compound rapamycin (sirolimus), rapamycin-related compounds, that is compounds having structural and functional similarity to rapamycin including, e.g., rapamycin derivatives, rapamycin analogs (also referred to as rapalogs) and other macrolide compounds that inhibit mTOR activity.
[0663] Rapamycin is a known macrolide antibiotic produced by Streptomyces hygroscopicus having the structure shown in Formula A.
##STR00007##
[0664] See, e.g., McAlpine, J. B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S. L., et al., J. Am. Chem. Soc. (1991) 113: 7433; U.S. Pat. No. 3,929,992. There are various numbering schemes proposed for rapamycin. To avoid confusion, when specific rapamycin analogs are named herein, the names are given with reference to rapamycin using the numbering scheme of formula A.
[0665] Rapamycin analogs useful in the invention are, for example, O-substituted analogs in which the hydroxyl group on the cyclohexyl ring of rapamycin is replaced by OR.sub.1 in which R.sub.1 is hydroxyalkyl, hydroxyalkoxyalkyl, acylaminoalkyl, or aminoalkyl; e.g. RAD001, also known as, everolimus as described in U.S. Pat. No. 5,665,772 and WO94/09010 the contents of which are incorporated by reference. Other suitable rapamycin analogs include those substituted at the 26- or 28-position. The rapamycin analog may be an epimer of an analog mentioned above, particularly an epimer of an analog substituted in position 40, 28 or 26, and may optionally be further hydrogenated, e.g. as described in U.S. Pat. No. 6,015,815, WO95/14023 and WO99/15530 the contents of which are incorporated by reference, e.g. ABT578 also known as zotarolimus or a rapamycin analog described in U.S. Pat. No. 7,091,213, WO98/02441 and WO01/14387 the contents of which are incorporated by reference, e.g. AP23573 also known as ridaforolimus.
[0666] Examples of rapamycin analogs suitable for use in the present invention from U.S. Pat. No. 5,665,772 include, but are not limited to, 40-O-benzyl-rapamycin, 40-O-(4'-hydroxymethyl)benzyl-rapamycin, 40-O-[4'-(1,2-dihydroxyethyl)]benzyl-rapamycin, 40-O-allyl-rapamycin, 40-O-[3'-(2,2-dimethyl-1,3-dioxolan-4(S)-yl)-prop-2'-en-1'-yl]-rapamycin, (2'E,4'S)-40-O-(4',5'-dihydroxypent-2'-en-1'-yl)-rapamycin, 40-O-(2-hydroxy)ethoxycarbonylmethyl-rapamycin, 40-O-(2-hydroxy)ethyl-rapamycin, 40-O-(3-hydroxy)propyl-rapamycin, 40-O-(6-hydroxy)hexyl-rapamycin, 40-O-[2-(2-hydroxy)ethoxy]ethyl-rapamycin, 40-O-[(3S)-2,2-dimethyldioxolan-3-yl]methyl-rapamycin, 40-O-[(2S)-2,3-dihydroxyprop-1-yl]-rapamycin, 40-O-(2-acetoxy)ethyl-rapamycin, 40-O-(2-nicotinoyloxy)ethyl-rapamycin, 40-O-[2-(N-morpholino)acetoxy]ethyl-rapamycin, 40-O-(2-N-imidazolylacetoxy)ethyl-rapamycin, 40-O-[2-(N-methyl-N'-piperazinyl)acetoxy]ethyl-rapamycin, 39-O-desmethyl-39,40-O,O-ethylene-rapamycin, (26R)-26-dihydro-40-O-(2-hydroxy)ethyl-rapamycin, 40-O-(2-aminoethyl)-rapamycin, 40-O-(2-acetaminoethyl)-rapamycin, 40-O-(2-nicotinamidoethyl)-rapamycin, 40-O-(2-(N-methyl-imidazo-2'-ylcarbethoxamido)ethyl)-rapamycin, 40-O-(2-ethoxycarbonylaminoethyl)-rapamycin, 40-O-(2-tolylsulfonamidoethyl)-rapamycin and 40-O-[2-(4',5'-dicarboethoxy-1',2',3'-triazol-1'-yl)-ethyl]-rapamycin.
[0667] Other rapamycin analogs useful in the present invention are analogs where the hydroxyl group on the cyclohexyl ring of rapamycin and/or the hydroxy group at the 28 position is replaced with an hydroxyester group are known, for example, rapamycin analogs found in U.S. RE44,768, e.g. temsirolimus.
[0668] Other rapamycin analogs useful in the preset invention include those wherein the methoxy group at the 16 position is replaced with another substituent, preferably (optionally hydroxy-substituted) alkynyloxy, benzyl, orthomethoxybenzyl or chlorobenzyl and/or wherein the methoxy group at the 39 position is deleted together with the 39 carbon so that the cyclohexyl ring of rapamycin becomes a cyclopentyl ring lacking the 39 position methyoxy group; e.g. as described in WO95/16691 and WO96/41807 the contents of which are incorporated by reference. The analogs can be further modified such that the hydroxy at the 40-position of rapamycin is alkylated and/or the 32-carbonyl is reduced.
[0669] Rapamycin analogs from WO95/16691 include, but are not limited to, 16-demethoxy-16-(pent-2-ynyl)oxy-rapamycin, 16-demethoxy-16-(but-2-ynyl)oxy-rapamycin, 16-demethoxy-16-(propargyl)oxy-rapamycin, 16-demethoxy-16-(4-hydroxy-but-2-ynyl)oxy-rapamycin, 16-demethoxy-16-benzyloxy-40-O-(2-hydroxyethyl)-rapamycin, 16-demethoxy-16-benzyloxy-rapamycin, 16-demethoxy-16-ortho-methoxybenzyl-rapamycin, 16-demethoxy-40-O-(2-methoxyethyl)-16-pent-2-ynyl)oxy-rapamycin, 39-demethoxy-40-desoxy-39-formyl-42-nor-rapamycin, 39-demethoxy-40-desoxy-39-hydroxymethyl-42-nor-rapamycin, 39-demethoxy-40-desoxy-39-carboxy-42-nor-rapamycin, 39-demethoxy-40-desoxy-39-(4-methyl-piperazin-1-yl)carbonyl-42-nor-rapamy- cin, 39-demethoxy-40-desoxy-39-(morpholin-4-yl)carbonyl-42-nor-rapamycin, 39-demethoxy-40-desoxy-39-[N-methyl, N-(2-pyridin-2-yl-ethyl)]carbamoyl-42-nor-rapamycin and 39-demethoxy-40-desoxy-39-(p-toluenesulfonylhydrazonomethyl)-42-nor-rapam- ycin.
[0670] Rapamycin analogs from WO96/41807 include, but are not limited to, 32-deoxo-rapamycin, 16-O-pent-2-ynyl-32-deoxo-rapamycin, 16-O-pent-2-ynyl-32-deoxo-40-O-(2-hydroxy-ethyl)-rapamycin, 16-O-pent-2-ynyl-32-(S)-dihydro-40-O-(2-hydroxyethyl)-rapamycin, 32(S)-dihydro-40-O-(2-methoxy)ethyl-rapamycin and 32(S)-dihydro-40-O-(2-hydroxyethyl)-rapamycin.
[0671] Another suitable rapamycin analog is umirolimus as described in US2005/0101624 the contents of which are incorporated by reference.
[0672] RAD001, otherwise known as everolimus (Afinitor.RTM.), has the chemical name (1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-1,18-dihydrox- y-12-{(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methyl- ethyl}-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-11,36-dioxa-4-aza-tric- yclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraene-2,3,10,14,20-pentaone
[0673] Further examples of allosteric mTOR inhibitors include sirolimus (rapamycin, AY-22989), 40-[3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate]-rapamycin (also called temsirolimus or CCI-779) and ridaforolimus (AP-23573/MK-8669). Other examples of allosteric mTor inhibitors include zotarolimus (ABT578) and umirolimus.
[0674] Alternatively or additionally, catalytic, ATP-competitive mTOR inhibitors have been found to target the mTOR kinase domain directly and target both mTORC1 and mTORC2. These are also more effective inhibitors of mTORC1 than such allosteric mTOR inhibitors as rapamycin, because they modulate rapamycin-resistant mTORC1 outputs such as 4EBP1-T37/46 phosphorylation and cap-dependent translation.
[0675] Catalytic inhibitors include: BEZ235 or 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]q- uinolin-1-yl)-phenyl]-propionitrile, or the monotosylate salt form. the synthesis of BEZ235 is described in WO2006/122806; CCG168 (otherwise known as AZD-8055, Chresta, C. M., et al., Cancer Res, 2010, 70(1), 288-298) which has the chemical name {5-[2,4-bis-((S)-3-methyl-morpholin-4-yl)-pyrido[2,3d]pyrimidin-7-yl]-2-m- ethoxy-phenyl}-methanol; 3-[2,4-bis[(3S)-3-methylmorpholin-4-yl]pyrido[2,3-d]pyrimidin-7-yl]-N-met- hylbenzamide (WO09104019); 3-(2-aminobenzo[d]oxazol-5-yl)-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-4-- amine (WO10051043 and WO2013023184); A N-(3-(N-(3-((3,5-dimethoxyphenyl)amino)quinoxaline-2-yl) sulfamoyl)phenyl)-3-methoxy-4-methylbenzamide (WO07044729 and WO12006552); PKI-587 (Venkatesan, A. M., J. Med. Chem., 2010, 53, 2636-2645) which has the chemical name 1-[4-[4-(dimethylamino)piperidine-1-carbonyl]phenyl]-3-[4-(4,6-dimorpholi- no-1,3,5-triazin-2-yl)phenyl]urea; GSK-2126458 (ACS Med. Chem. Lett., 2010, 1, 39-43) which has the chemical name 2,4-difluoro-N-{2-methoxy-5-[4-(4-pyridazinyl)-6-quinolinyl]-3-pyridinyl}- benzenesulfonamide; 5-(9-isopropyl-8-methyl-2-morpholino-9H-purin-6-yl)pyrimidin-2-amine (WO10114484); (E)-N-(8-(6-amino-5-(trifluoromethyl)pyridin-3-yl)-1-(6-(2-cyanopropan-2-- yl)pyridin-3-yl)-3-methyl-1H-imidazo[4,5-c]quinolin-2(3H)-ylidene)cyanamid- e (WO12007926).
[0676] Further examples of catalytic mTOR inhibitors include 8-(6-methoxy-pyridin-3-yl)-3-methyl-1-(4-piperazin-1-yl-3-trifluoromethyl- -phenyl)-1,3-dihydro-imidazo[4,5-c]quinolin-2-one (WO2006/122806) and Ku-0063794 (Garcia-Martinez J M, et al., Biochem J., 2009, 421(1), 29-42. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR).) WYE-354 is another example of a catalytic mTor inhibitor (Yu K, et al. (2009). Biochemical, Cellular, and In vivo Activity of Novel ATP-Competitive and Selective Inhibitors of the Mammalian Target of Rapamycin. Cancer Res. 69(15): 6232-6240).
[0677] mTOR inhibitors useful according to the present invention also include prodrugs, derivatives, pharmaceutically acceptable salts, or analogs thereof of any of the foregoing.
[0678] mTOR inhibitors, such as RAD001, may be formulated for delivery based on well-established methods in the art based on the particular dosages described herein. In particular, U.S. Pat. No. 6,004,973 (incorporated herein by reference) provides examples of formulations useable with the mTOR inhibitors described herein.
Evaluation of mTOR Inhibition
[0679] mTOR phosphorylates the kinase P70 S6, thereby activating P70 S6 kinase and allowing it to phosphorylate its substrate. The extent of mTOR inhibition can be expressed as the extent of P70 S6 kinase inhibition, e.g., the extent of mTOR inhibition can be determined by the level of decrease in P70 S6 kinase activity, e.g., by the decrease in phosphorylation of a P70 S6 kinase substrate. One can determine the level of mTOR inhibition, by measuring P70 S6 kinase activity (the ability of P70 S6 kinase to phosphorylate a substrate), in the absence of inhibitor, e.g., prior to administration of inhibitor, and in the presences of inhibitor, or after the administration of inhibitor. The level of inhibition of P70 S6 kinase gives the level of mTOR inhibition. Thus, if P70 S6 kinase is inhibited by 40%, mTOR activity, as measured by P70 S6 kinase activity, is inhibited by 40%. The extent or level of inhibition referred to herein is the average level of inhibition over the dosage interval. By way of example, if the inhibitor is given once per week, the level of inhibition is given by the average level of inhibition over that interval, namely a week.
[0680] Boulay et al., Cancer Res, 2004, 64:252-61, hereby incorporated by reference, teaches an assay that can be used to assess the level of mTOR inhibition (referred to herein as the Boulay assay). In an embodiment, the assay relies on the measurement of P70 S6 kinase activity from biological samples before and after administration of an mTOR inhibitor, e.g., RAD001. Samples can be taken at preselected times after treatment with an mTOR inhibitor, e.g., 24, 48, and 72 hours after treatment. Biological samples, e.g., from skin or peripheral blood mononuclear cells (PBMCs) can be used. Total protein extracts are prepared from the samples. P70 S6 kinase is isolated from the protein extracts by immunoprecipitation using an antibody that specifically recognizes the P70 S6 kinase. Activity of the isolated P70 S6 kinase can be measured in an in vitro kinase assay. The isolated kinase can be incubated with 40S ribosomal subunit substrates (which is an endogenous substrate of P70 S6 kinase) and gamma-.sup.32P under conditions that allow phosphorylation of the substrate. Then the reaction mixture can be resolved on an SDS-PAGE gel, and .sup.32P signal analyzed using a PhosphorImager. A .sup.32P signal corresponding to the size of the 40S ribosomal subunit indicates phosphorylated substrate and the activity of P70 S6 kinase. Increases and decreases in kinase activity can be calculated by quantifying the area and intensity of the .sup.32P signal of the phosphorylated substrate (e.g., using ImageQuant, Molecular Dynamics), assigning arbitrary unit values to the quantified signal, and comparing the values from after administration with values from before administration or with a reference value. For example, percent inhibition of kinase activity can be calculated with the following formula: 1-(value obtained after administration/value obtained before administration).times.100. As described above, the extent or level of inhibition referred to herein is the average level of inhibition over the dosage interval.
[0681] Methods for the evaluation of kinase activity, e.g., P70 S6 kinase activity, are also provided in U.S. Pat. No. 7,727,950, hereby incorporated by reference.
[0682] The level of mTOR inhibition can also be evaluated by a change in the ration of PD1 negative to PD1 positive T cells. T cells from peripheral blood can be identified as PD1 negative or positive by art-known methods.
Low-Dose mTOR Inhibitors
[0683] Methods described herein use low, immune enhancing, dose mTOR inhibitors, doses of mTOR inhibitors, e.g., allosteric mTOR inhibitors, including rapalogs such as RAD001. In contrast, levels of inhibitor that fully or near fully inhibit the mTOR pathway are immunosuppressive and are used, e.g., to prevent organ transplant rejection. In addition, high doses of rapalogs that fully inhibit mTOR also inhibit tumor cell growth and are used to treat a variety of cancers (See, e.g., Antineoplastic effects of mammalian target of rapamycine inhibitors. Salvadori M. World J Transplant. 2012 Oct. 24;2(5):74-83; Current and Future Treatment Strategies for Patients with Advanced Hepatocellular Carcinoma: Role of mTOR Inhibition. Finn R S. Liver Cancer. 2012 November; 1(3-4):247-256; Emerging Signaling Pathways in Hepatocellular Carcinoma. Moeini A, Cornella H, Villanueva A. Liver Cancer. 2012 September; 1(2):83-93; Targeted cancer therapy--Are the days of systemic chemotherapy numbered?Joo W D, Visintin I, Mor G. Maturitas. 2013 Sep. 20; Role of natural and adaptive immunity in renal cell carcinoma response to VEGFR-TKIs and mTOR inhibitor. Santoni M, Berardi R, Amantini C, Burattini L, Santini D, Santoni G, Cascinu S. Int J Cancer. 2013 Oct. 2).
[0684] The present invention is based, at least in part, on the surprising finding that doses of mTOR inhibitors well below those used in current clinical settings had a superior effect in increasing an immune response in a subject and increasing the ratio of PD-1 negative T cells/PD-1 positive T cells. It was surprising that low doses of mTOR inhibitors, producing only partial inhibition of mTOR activity, were able to effectively improve immune responses in human human subjects and increase the ratio of PD-1 negative T cells/PD-1 positive T cells.
[0685] Alternatively, or in addition, without wishing to be bound by any theory, it is believed that low, a low, immune enhancing, dose of an mTOR inhibitor can increase naive T cell numbers, e.g., at least transiently, e.g., as compared to a non-treated subject. Alternatively or additionally, again while not wishing to be bound by theory, it is believed that treatment with an mTOR inhibitor after a sufficient amount of time or sufficient dosing results in one or more of the following:
[0686] an increase in the expression of one or more of the following markers: CD62L.sup.highCD.sub.127.sup.high, CD27.sup.+, and BCL2, e.g., on memory T cells, e.g., memory T cell precursors;
[0687] a decrease in the expression of KLRG1, e.g., on memory T cells, e.g., memory T cell precursors; and
[0688] an increase in the number of memory T cell precursors, e.g., cells with any one or combination of the following characteristics: increased CD62L.sup.high, increased CD127.sup.high, increased CD27.sup.+, decreased KLRG1, and increased BCL2;
and wherein any of the changes described above occurs, e.g., at least transiently, e.g., as compared to a non-treated subject (Araki, K et al. (2009) Nature 460:108-112). Memory T cell precursors are memory T cells that are early in the differentiation program. For example, memory T cells have one or more of the following characteristics: increased CD62L.sup.high, increased CD127high, increased CD27.sup.+, decreased KLRG1, and/or increased BCL2.
[0689] In an embodiment, the invention relates to a composition, or dosage form, of an mTOR inhibitor, e.g., an allosteric mTOR inhibitor, e.g., a rapalog, rapamycin, or RAD001, or a catalytic mTOR inhibitor, which, when administered on a selected dosing regimen, e.g., once daily or once weekly, is associated with: a level of mTOR inhibition that is not associated with complete, or significant immune suppression, but is associated with enhancement of the immune response.
[0690] An mTOR inhibitor, e.g., an allosteric mTOR inhibitor, e.g., a rapalog, rapamycin, or RAD001, or a catalytic mTOR inhibitor, can be provided in a sustained release formulation. Any of the compositions or unit dosage forms described herein can be provided in a sustained release formulation. In some embodiments, a sustained release formulation will have lower bioavailability than an immediate release formulation. E.g., in embodiments, to attain a similar therapeutic effect of an immediate release forlation a sustained release formulation will have from about 2 to about 5, about 2.5 to about 3.5, or about 3 times the amount of inhibitor provided in the immediate release formulation.
[0691] In an embodiment, immediate release forms, e.g., of RAD001, typically used for one administration per week, having 0.1 to 20, 0.5 to 10, 2.5 to 7.5, 3 to 6, or about 5, mgs per unit dosage form, are provided. For once per week administrations, these immediate release formulations correspond to sustained release forms, having, respectively, 0.3 to 60, 1.5 to 30, 7.5 to 22.5, 9 to 18, or about 15 mgs of an mTOR inhibitor, e.g., an allosteric mTOR inhibitor, e.g., rapamycin or RAD001. In embodiments both forms are administered on a once/week basis.
[0692] In an embodiment, immediate release forms, e.g., of RAD001, typically used for one administration per day, having having 0.005 to 1.5, 0.01 to 1.5, 0.1 to 1.5, 0.2 to 1.5, 0.3 to 1.5, 0.4 to 1.5, 0.5 to 1.5, 0.6 to 1.5, 0.7 to 1.5, 0.8 to 1.5, 1.0 to 1.5, 0.3 to 0.6, or about 0.5 mgs per unit dosage form, are provided. For once per day administrations, these immediate release forms correspond to sustained release forms, having, respectively, 0.015 to 4.5, 0.03 to 4.5, 0.3 to 4.5, 0.6 to 4.5, 0.9 to 4.5, 1.2 to 4.5, 1.5 to 4.5, 1.8 to 4.5, 2.1 to 4.5, 2.4 to 4.5, 3.0 to 4.5, 0.9 to 1.8, or about 1.5 mgs of an mTOR inhibitor, e.g., an allosteric mTOR inhibitor, e.g., rapamycin or RAD001. For once per week administrations, these immediate release forms correspond to sustained release forms, having, respectively, 0.1 to 30, 0.2 to 30, 2 to 30, 4 to 30, 6 to 30, 8 to 30, 10 to 30, 1.2 to 30, 14 to 30, 16 to 30, 20 to 30, 6 to 12, or about 10 mgs of an mTOR inhibitor, e.g., an allosteric mTOR inhibitor, e.g., rapamycin or RAD001.
[0693] In an embodiment, immediate release forms, e.g., of RAD001, typically used for one administration per day, having having 0.01 to 1.0 mgs per unit dosage form, are provided. For once per day administrations, these immediate release forms correspond to sustained release forms, having, respectively, 0.03 to 3 mgs of an mTOR inhibitor, e.g., an allosteric mTOR inhibitor, e.g., rapamycin or RAD001. For once per week administrations, these immediate release forms correspond to sustained release forms, having, respectively, 0.2 to 20 mgs of an mTOR inhibitor, e.g., an allosteric mTOR inhibitor, e.g., rapamycin or RAD001.
[0694] In an embodiment, immediate release forms, e.g., of RAD001, typically used for one administration per week, having having 0.5 to 5.0 mgs per unit dosage form, are provided. For once per week administrations, these immediate release forms correspond to sustained release forms, having, respectively, 1.5 to 15 mgs of an mTOR inhibitor, e.g., an allosteric mTOR inhibitor, e.g., rapamycin or RAD001.
[0695] As described above, one target of the mTOR pathway is the P70 S6 kinase. Thus, doses of mTOR inhibitors which are useful in the methods and compositions described herein are those which are sufficient to achieve no greater than 80% inhibition of P70 S6 kinase activity relative to the activity of the P70 S6 kinase in the absence of an mTOR inhibitor, e.g., as measured by an assay described herein, e.g., the Boulay assay. In a further aspect, the invention provides an amount of an mTOR inhibitor sufficient to achieve no greater than 38% inhibition of P70 S6 kinase activity relative to P70 S6 kinase activity in the absence of an mTOR inhibitor.
[0696] In one aspect the dose of mTOR inhibitor useful in the methods and compositions of the invention is sufficient to achieve, e.g., when administered to a human subject, 90+/-5% (i.e., 85-95%), 89+/-5%, 88+/-5%, 87+/-5%, 86+/-5%, 85+/-5%, 84+/-5%, 83+/-5%, 82+/-5%, 81+/-5%, 80+/-5%, 79+/-5%, 78+/-5%, 77+/-5%, 76+/-5%, 75+/-5%, 74+/-5%, 73+/-5%, 72+/-5%, 71+/-5%, 70+/-5%, 69+/-5%, 68+/-5%, 67+/-5%, 66+/-5%, 65+/-5%, 64+/-5%, 63+/-5%, 62+/-5%, 61+/-5%, 60+/-5%, 59+/-5%, 58+/-5%, 57+/-5%, 56+/-5%, 55+/-5%, 54+/-5%, 54+/-5%, 53+/-5%, 52+/-5%, 51+/-5%, 50+/-5%, 49+/-5%, 48+/-5%, 47+/-5%, 46+/-5%, 45+/-5%, 44+/-5%, 43+/-5%, 42+/-5%, 41+/-5%, 40+/-5%, 39+/-5%, 38+/-5%, 37+/-5%, 36+/-5%, 35+/-5%, 34+/-5%, 33+/-5%, 32+/-5%, 31+/-5%, 30+/-5%, 29+/-5%, 28+/-5%, 27+/-5%, 26+/-5%, 25+/-5%, 24+/-5%, 23+/-5%, 22+/-5%, 21+/-5%, 20+/-5%, 19+/-5%, 18+/-5%, 17+/-5%, 16+/-5%, 15+/-5%, 14+/-5%, 13+/-5%, 12+/-5%, 11+/-5%, or 10+/-5%, inhibition of P70 S6 kinase activity, e.g., as measured by an assay described herein, e.g., the Boulay assay.
[0697] P70 S6 kinase activity in a subject may be measured using methods known in the art, such as, for example, according to the methods described in U.S. Pat. No. 7,727,950, by immunoblot analysis of phosphoP70 S6K levels and/or phosphoP70 S6 levels or by in vitro kinase activity assays.
[0698] As used herein, the term "about" in reference to a dose of mTOR inhibitor refers to up to a +/-10% variability in the amount of mTOR inhibitor, but can include no variability around the stated dose.
[0699] In some embodiments, the invention provides methods comprising administering to a subject an mTOR inhibitor, e.g., an allosteric inhibitor, e.g., RAD001, at a dosage within a target trough level. In some embodiments, the trough level is significantly lower than trough levels associated with dosing regimens used in organ transplant and cancer patients. In an embodiment mTOR inhibitor, e.g., RAD001, or rapamycin, is administered to result in a trough level that is less than/2, 1/4, 1/10, or 1/20 of the trough level that results in immunosuppression or an anticancer effect. In an embodiment mTOR inhibitor, e.g., RAD001, or rapamycin, is administered to result in a trough level that is less than/2, 1/4, 1/10, or 1/20 of the trough level provided on the FDA approved packaging insert for use in immunosuppression or an anticancer indications.
[0700] In an embodiment a method disclosed herein comprises administering to a subject an mTOR inhibitor, e.g., an allosteric inhibitor, e.g., RAD001, at a dosage that provides a target trough level of 0.1 to 10 ng/ml, 0.1 to 5 ng/ml, 0.1 to 3 ng/ml, 0.1 to 2 ng/ml, or 0.1 to 1 ng/ml.
[0701] In an embodiment a method disclosed herein comprises administering to a subject an mTOR inhibitor, e.g., an allosteric inhibitor, e.g., RAD001, at a dosage that provides a target trough level of 0.2 to 10 ng/ml, 0.2 to 5 ng/ml, 0.2 to 3 ng/ml, 0.2 to 2 ng/ml, or 0.2 to 1 ng/ml.
[0702] In an embodiment a method disclosed herein comprises administering to a subject an mTOR inhibitor, e.g. an, allosteric inhibitor, e.g., RAD001, at a dosage that provides a target trough level of 0.3 to 10 ng/ml, 0.3 to 5 ng/ml, 0.3 to 3 ng/ml, 0.3 to 2 ng/ml, or 0.3 to 1 ng/ml.
[0703] In an embodiment a method disclosed herein comprises administering to a subject an mTOR inhibitor, e.g., an allosteric inhibitor, e.g., RAD001, at a dosage that provides a target trough level of 0.4 to 10 ng/ml, 0.4 to 5 ng/ml, 0.4 to 3 ng/ml, 0.4 to 2 ng/ml, or 0.4 to 1 ng/ml.
[0704] In an embodiment a method disclosed herein comprises administering to a subject an mTOR inhibitor, e.g., an allosteric inhibitor, e.g., RAD001, at a dosage that provides a target trough level of 0.5 to 10 ng/ml, 0.5 to 5 ng/ml, 0.5 to 3 ng/ml, 0.5 to 2 ng/ml, or 0.5 to 1 ng/ml.
[0705] In an embodiment a method disclosed herein comprises administering to a subject an mTOR inhibitor, e.g., an allosteric inhibitor, e.g., RAD001, at a dosage that provides a target trough level of 1 to 10 ng/ml, 1 to 5 ng/ml, 1 to 3 ng/ml, or 1 to 2 ng/ml.
[0706] As used herein, the term "trough level" refers to the concentration of a drug in plasma just before the next dose, or the minimum drug concentration between two doses.
[0707] In some embodiments, a target trough level of RAD001 is in a range of between about 0.1 and 4.9 ng/ml. In an embodiment, the target trough level is below 3 ng/ml, e.g., is between 0.3 or less and 3 ng/ml. In an embodiment, the target trough level is below 3 ng/ml, e.g., is between 0.3 or less and 1 ng/ml.
[0708] In a further aspect, the invention can utilize an mTOR inhibitor other than RAD001 in an amount that is associated with a target trough level that is bioequivalent to the specified target trough level for RAD001. In an embodiment, the target trough level for an mTOR inhibitor other than RAD001, is a level that gives the same level of mTOR inhibition (e.g., as measured by a method described herein, e.g., the inhibition of P70 S6 kinase) as does a trough level of RAD001 described herein.
Pharmaceutical Compositions: mTOR Inhibitors
[0709] In one aspect, the present invention relates to pharmaceutical compositions comprising an mTOR inhibitor, e.g., an mTOR inhibitor as described herein, formulated for use in combination with CAR cells described herein.
[0710] In some embodiments, the mTOR inhibitor is formulated for administration in combination with an additional, e.g., as described herein.
[0711] In general, compounds of the invention will be administered in therapeutically effective amounts as described above via any of the usual and acceptable modes known in the art, either singly or in combination with one or more therapeutic agents.
[0712] The pharmaceutical formulations may be prepared using conventional dissolution and mixing procedures. For example, the bulk drug substance (e.g., an mTOR inhibitor or stabilized form of the compound (e.g., complex with a cyclodextrin derivative or other known complexation agent) is dissolved in a suitable solvent in the presence of one or more of the excipients described herein. The mTOR inhibitor is typically formulated into pharmaceutical dosage forms to provide an easily controllable dosage of the drug and to give the patient an elegant and easily handleable product.
[0713] Compounds of the invention can be administered as pharmaceutical compositions by any conventional route, in particular enterally, e.g., orally, e.g., in the form of tablets or capsules, or parenterally, e.g., in the form of injectable solutions or suspensions, topically, e.g., in the form of lotions, gels, ointments or creams, or in a nasal or suppository form. Where an mTOR inhibitor is administered in combination with (either simultaneously with or separately from) another agent as described herein, in one aspect, both components can be administered by the same route (e.g., parenterally). Alternatively, another agent may be administered by a different route relative to the mTOR inhibitor. For example, an mTOR inhibitor may be administered orally and the other agent may be administered parenterally.
Sustained Release
[0714] mTOR inhibitors, e.g., allosteric mTOR inhibitors or catalytic mTOR inhibitors, disclosed herein can be provided as pharmaceutical formulations in form of oral solid dosage forms comprising an mTOR inhibitor disclosed herein, e.g., rapamycin or RAD001, which satisfy product stability requirements and/or have favorable pharmacokinetic properties over the immediate release (IR) tablets, such as reduced average plasma peak concentrations, reduced inter- and intra-patient variability in the extent of drug absorption and in the plasma peak concentration, reduced C.sub.max/C.sub.min ratio and/or reduced food effects. Provided pharmaceutical formulations may allow for more precise dose adjustment and/or reduce frequency of adverse events thus providing safer treatments for patients with an mTOR inhibitor disclosed herein, e.g., rapamycin or RAD001.
[0715] In some embodiments, the present disclosure provides stable extended release formulations of an mTOR inhibitor disclosed herein, e.g., rapamycin or RAD001, which are multi-particulate systems and may have functional layers and coatings.
[0716] The term "extended release, multi-particulate formulation as used herein refers to a formulation which enables release of an mTOR inhibitor disclosed herein, e.g., rapamycin or RAD001, over an extended period of time e.g. over at least 1, 2, 3, 4, 5 or 6 hours. The extended release formulation may contain matrices and coatings made of special excipients, e.g., as described herein, which are formulated in a manner as to make the active ingredient available over an extended period of time following ingestion.
[0717] The term "extended release" can be interchangeably used with the terms "sustained release" (SR) or "prolonged release". The term "extended release" relates to a pharmaceutical formulation that does not release active drug substance immediately after oral dosing but over an extended in accordance with the definition in the pharmacopoeias Ph. Eur. (7.sup.th edition) monograph for tablets and capsules and USP general chapter <1151> for pharmaceutical dosage forms. The term "Immediate Release" (IR) as used herein refers to a pharmaceutical formulation which releases 85% of the active drug substance within less than 60 minutes in accordance with the definition of "Guidance for Industry: "Dissolution Testing of Immediate Release Solid Oral Dosage Forms" (FDA CDER, 1997). In some embodiments, the term "immediate release" means release of everolimus from tablets within the time of 30 minutes, e.g., as measured in the dissolution assay described herein.
[0718] Stable extended release formulations of an mTOR inhibitor disclosed herein, e.g., rapamycin or RAD001, can be characterized by an in-vitro release profile using assays known in the art, such as a dissolution assay as described herein: a dissolution vessel filled with 900 mL phosphate buffer pH 6.8 containing sodium dodecyl sulfate 0.2% at 37.degree. C. and the dissolution is performed using a paddle method at 75 rpm according to USP by according to USP testing monograph 711, and Ph.Eur. testing monograph 2.9.3. respectively.
[0719] In some embodiments, stable extended release formulations of an mTOR inhibitor disclosed herein, e.g., rapamycin or RAD001, release the mTOR inhibitor in the in-vitro release assay according to following release specifications:
[0720] 0.5 h: <45%, or <40, e.g., <30%
[0721] 1 h: 20-80%, e.g., 30-60%
[0722] 2 h: >50%, or >70%, e.g., >75%
[0723] 3 h: >60%, or >65%, e.g., >85%, e.g., >90%.
[0724] In some embodiments, stable extended release formulations of an mTOR inhibitor disclosed herein, e.g., rapamycin or RAD001, release 50% of the mTOR inhibitor not earlier than 45, 60, 75, 90, 105 min or 120 min in the in-vitro dissolution assay.
Biopolymer Delivery Methods
[0725] In some embodiments, one or more CAR-expressing cells as disclosed herein, optionally in combination with a kinase inhibitor, e.g., a BTK inhibitor such as ibrutinib, can be administered or delivered to the subject via a biopolymer scaffold, e.g., a biopolymer implant. Biopolymer scaffolds can support or enhance the delivery, expansion, and/or dispersion of the CAR-expressing cells described herein. A biopolymer scaffold comprises a biocompatible (e.g., does not substantially induce an inflammatory or immune response) and/or a biodegradable polymer that can be naturally occurring or synthetic.
[0726] Examples of suitable biopolymers include, but are not limited to, agar, agarose, alginate, alginate/calcium phosphate cement (CPC), beta-galactosidase (.beta.-GAL), (1,2,3,4,6-pentaacetyl a-D-galactose), cellulose, chitin, chitosan, collagen, elastin, gelatin, hyaluronic acid collagen, hydroxyapatite, poly(3-hydroxybutyrate-co-3-hydroxy-hexanoate) (PHBHHx), poly(lactide), poly(caprolactone) (PCL), poly(lactide-co-glycolide) (PLG), polyethylene oxide (PEO), poly(lactic-co-glycolic acid) (PLGA), polypropylene oxide (PPO), polyvinyl alcohol) (PVA), silk, soy protein, and soy protein isolate, alone or in combination with any other polymer composition, in any concentration and in any ratio. The biopolymer can be augmented or modified with adhesion- or migration-promoting molecules, e.g., collagen-mimetic peptides that bind to the collagen receptor of lymphocytes, and/or stimulatory molecules to enhance the delivery, expansion, or function, e.g., anti-cancer activity, of the cells to be delivered. The biopolymer scaffold can be an injectable, e.g., a gel or a semi-solid, or a solid composition.
[0727] In some embodiments, CAR-expressing cells described herein are seeded onto the biopolymer scaffold prior to delivery to the subject. In embodiments, the biopolymer scaffold further comprises one or more additional therapeutic agents described herein (e.g., another CAR-expressing cell, an antibody, or a small molecule) or agents that enhance the activity of a CAR-expressing cell, e.g., incorporated or conjugated to the biopolymers of the scaffold. In embodiments, the biopolymer scaffold is injected, e.g., intratumorally, or surgically implanted at the tumor or within a proximity of the tumor sufficient to mediate an anti-tumor effect. Additional examples of biopolymer compositions and methods for their delivery are described in Stephan et al., Nature Biotechnology, 2015, 33:97-101; and WO2014/110591.
Pharmaceutical Compositions and Treatments
[0728] Pharmaceutical compositions of the present invention may comprise a CAR-expressing cell, e.g., a plurality of CAR-expressing cells, as described herein, in combination with one or more pharmaceutically or physiologically acceptable carriers, diluents or excipients. Such compositions may comprise buffers such as neutral buffered saline, phosphate buffered saline and the like; carbohydrates such as glucose, mannose, sucrose or dextrans, mannitol; proteins; polypeptides or amino acids such as glycine; antioxidants; chelating agents such as EDTA or glutathione; adjuvants (e.g., aluminum hydroxide); and preservatives. Compositions of the present invention are in one aspect formulated for intravenous administration.
[0729] Pharmaceutical compositions of the present invention may be administered in a manner appropriate to the disease to be treated (or prevented). The quantity and frequency of administration will be determined by such factors as the condition of the patient, and the type and severity of the patient's disease, although appropriate dosages may be determined by clinical trials.
[0730] In one embodiment, the pharmaceutical composition is substantially free of, e.g., there are no detectable levels of a contaminant, e.g., selected from the group consisting of endotoxin, mycoplasma, replication competent lentivirus (RCL), p24, VSV-G nucleic acid, HIV gag, residual anti-CD3/anti-CD28 coated beads, mouse antibodies, pooled human serum, bovine serum albumin, bovine serum, culture media components, vector packaging cell or plasmid components, a bacterium and a fungus. In one embodiment, the bacterium is at least one selected from the group consisting of Alcaligenes faecalis, Candida albicans, Escherichia coli, Haemophilus influenza, Neisseria meningitides, Pseudomonas aeruginosa, Staphylococcus aureus, Streptococcus pneumonia, and Streptococcus pyogenes group A.
[0731] When "an immunologically effective amount," "an anti-tumor effective amount," "a tumor-inhibiting effective amount," or "therapeutic amount" is indicated, the precise amount of the compositions of the present invention to be administered can be determined by a physician with consideration of individual differences in age, weight, tumor size, extent of infection or metastasis, and condition of the patient (subject). It can generally be stated that a pharmaceutical composition comprising the T cells described herein may be administered at a dosage of 10.sup.4 to 10.sup.9 cells/kg body weight, in some instances 10.sup.5 to 10.sup.6 cells/kg body weight, including all integer values within those ranges. T cell compositions may also be administered multiple times at these dosages. The cells can be administered by using infusion techniques that are commonly known in immunotherapy (see, e.g., Rosenberg et al., New Eng. J. of Med. 319:1676, 1988).
[0732] In certain aspects, it may be desired to administer activated T cells to a subject and then subsequently redraw blood (or have an apheresis performed), activate T cells therefrom according to the present invention, and reinfuse the patient with these activated and expanded T cells. This process can be carried out multiple times every few weeks. In certain aspects, T cells can be activated from blood draws of from 10cc to 400cc. In certain aspects, T cells are activated from blood draws of 20cc, 30cc, 40cc, 50cc, 60cc, 70cc, 80cc, 90cc, or 100cc.
[0733] The administration of the subject compositions may be carried out in any convenient manner, including by aerosol inhalation, injection, ingestion, transfusion, implantation or transplantation. The compositions described herein may be administered to a patient trans arterially, subcutaneously, intradermally, intratumorally, intranodally, intramedullary, intramuscularly, by intravenous (i.v.) injection, or intraperitoneally. In one aspect, the T cell compositions of the present invention are administered to a patient by intradermal or subcutaneous injection. In one aspect, the T cell compositions of the present invention are administered by i.v. injection. The compositions of T cells may be injected directly into a tumor, lymph node, or site of infection.
[0734] In a particular exemplary aspect, subjects may undergo leukapheresis, wherein leukocytes are collected, enriched, or depleted ex vivo to select and/or isolate the cells of interest, e.g., T cells. These T cell isolates may be expanded by methods known in the art and treated such that one or more CAR constructs of the invention may be introduced, thereby creating a CAR T cell of the invention. Subjects in need thereof may subsequently undergo standard treatment with high dose chemotherapy followed by peripheral blood stem cell transplantation. In certain aspects, following or concurrent with the transplant, subjects receive an infusion of the expanded CAR T cells of the present invention. In an additional aspect, expanded cells are administered before or following surgery.
[0735] The dosage of the above treatments to be administered to a patient will vary with the precise nature of the condition being treated and the recipient of the treatment. The scaling of dosages for human administration can be performed according to art-accepted practices. The dose for CAMPATH, for example, will generally be in the range 1 to about 100 mg for an adult patient, usually administered daily for a period between 1 and 30 days. The preferred daily dose is 1 to 10 mg per day although in some instances larger doses of up to 40 mg per day may be used (described in U.S. Pat. No. 6,120,766).
[0736] In one embodiment, the CAR is introduced into T cells, e.g., using in vitro transcription, and the subject (e.g., human) receives an initial administration of CAR T cells of the invention, and one or more subsequent administrations of the CAR T cells of the invention, wherein the one or more subsequent administrations are administered less than 15 days, e.g., 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, or 2 days after the previous administration. In one embodiment, more than one administration of the CAR T cells of the invention are administered to the subject (e.g., human) per week, e.g., 2, 3, or 4 administrations of the CAR T cells of the invention are administered per week. In one embodiment, the subject (e.g., human subject) receives more than one administration of the CAR T cells per week (e.g., 2, 3 or 4 administrations per week) (also referred to herein as a cycle), followed by a week of no CAR T cells administrations, and then one or more additional administration of the CAR T cells (e.g., more than one administration of the CAR T cells per week) is administered to the subject. In another embodiment, the subject (e.g., human subject) receives more than one cycle of CAR T cells, and the time between each cycle is less than 10, 9, 8, 7, 6, 5, 4, or 3 days. In one embodiment, the CAR T cells are administered every other day for 3 administrations per week. In one embodiment, the CAR T cells of the invention are administered for at least two, three, four, five, six, seven, eight or more weeks.
[0737] In one aspect, CAR-expressing cells are generated using lentiviral viral vectors, such as lentivirus. Cells, e.g., CARTs generated that way will have stable CAR expression.
[0738] In one aspect, CAR-expressing cells, e.g., CARTs, are generated using a viral vector such as a gammaretroviral vector, e.g., a gammaretroviral vector described herein. CARTs generated using these vectors can have stable CAR expression.
[0739] In one aspect, CARTs transiently express CAR vectors for 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 days after transduction. Transient expression of CARs can be effected by RNA CAR vector delivery. In one aspect, the CAR RNA is transduced into the T cell by electroporation.
[0740] A potential issue that can arise in patients being treated using transiently expressing CAR T cells (particularly with murine scFv bearing CARTs) is anaphylaxis after multiple treatments.
[0741] Without being bound by this theory, it is believed that such an anaphylactic response might be caused by a patient developing humoral anti-CAR response, i.e., anti-CAR antibodies having an anti-IgE isotype. It is thought that a patient's antibody producing cells undergo a class switch from IgG isotype (that does not cause anaphylaxis) to IgE isotype when there is a ten to fourteen day break in exposure to antigen.
[0742] If a patient is at high risk of generating an anti-CAR antibody response during the course of transient CAR therapy (such as those generated by RNA transductions), CART infusion breaks should not last more than ten to fourteen days.
EXAMPLES
[0743] The invention is further described in detail by reference to the following experimental examples. These examples are provided for purposes of illustration only, and are not intended to be limiting unless otherwise specified. Thus, the invention should in no way be construed as being limited to the following examples, but rather, should be construed to encompass any and all variations which become evident as a result of the teaching provided herein.
[0744] Without further description, it is believed that one of ordinary skill in the art can, using the preceding description and the following illustrative examples, make and utilize the compounds of the present invention and practice the claimed methods. The following working examples specifically point out various aspects of the present invention, and are not to be construed as limiting in any way the remainder of the disclosure.
Example 1: Humanization of Murine Anti-CD19 Antibody
[0745] A CD19 antibody molecule can be, e.g., an antibody molecule (e.g., a humanized anti-CD19 antibody molecule) described in WO2014/153270, which is incorporated herein by reference in its entirety. Humanization of murine CD19 antibody is desired for the clinical setting, where the mouse-specific residues may induce a human-anti-mouse antigen (HAMA) response in patients who receive CART19 treatment, i.e., treatment with T cells transduced with the CAR19 construct. VH and VL sequences of hybridoma derived murine CD19 antibody were extracted from published literature (Nicholson et al, 1997, supra). Humanization was accomplished by grafting CDR regions from murine CD19 antibody onto human germline acceptor frameworks VH4_4-59 and VK3_L25 (vBASE database). In addition to the CDR regions, five framework residues, i.e. VH #71, #73, #78 and VL #71 #87, thought to support the structural integrity of the CDR regions were retained from the murine sequence. Further, the human J elements JH4 and JK2 were used for the heavy and light chain, respectively. The resulting amino acid sequences of the humanized antibody were designated FMC63_VL_hz and FMC63_VH_hz1, respectively, and are shown below in Table 1. The residue numbering follows Kabat (Kabat E. A. et al, 1991, supra). For CDR definitions, both Kabat as well as Chothia et al, 1987 supra) were used. Residues coming from mouse CD19 are shown in bold/italic. Positions #60/61/62 boxed indicate potential post-translational modification (PTM) site in CDR H2, also termed HCDR2.
TABLE-US-00014 TABLE 1 Amino acid sequences of humanized CD19 variable domains (SEQ ID NOs: 114-117, respectively, in order of appearance). ##STR00008##
[0746] These humanized CD19 IgGs were used to generate soluble scFvs to test for expression and scFvs for the full CART CD19 constructs (See Examples below). Of interest was that during humanization, position 62 in the CDRH2 region prefers to be a serine residue rather than the alanine present in the murine CDRH2. The murine sequence lacks a post-translational modification (PTM), and has asparagine-serine-alanine at positions 60/61/62, respectively in CDRH2. This generates potential PTM motifs (indicated as the boxed cite in CDRH2) during the course of humanization. Whether the PTM site generated during humanization process was actually a "true" PTM site or merely a theoretical one was tested. It was hypothesized that the amino acid motif asparagine followed by serine (NS) may be susceptible to post-translational deamidation but not something that was readily apparent. It was also hypothesized that asparagine followed by any amino acid except proline and then followed by serine (N.times.S, x.noteq.P) may be susceptible to post-translational N-glycosylation. To test this hypothesis, two IgG variants, were generated in which the asparagine at position 60 (known to be a glycosylation site) was mutated to serine, or glutamine and designated FMC63_VH_hz2 (N60S) and FMC63_VH_hz2 (N60Q), respectively. These constructs were generated in order to eliminate the potential post-translational modification site (PTM) and test for retained activity (See Example 2 below).
Cloning:
[0747] DNA sequences coding for mouse and humanized VL and VH domains were obtained, and the codons for the constructs were optimized for expression in cells from Homo sapiens.
[0748] Sequences coding for VL and VH domain were subcloned from the cloning vectors into expression vectors suitable for secretion in mammalian cells. The heavy and light chains were cloned into individual expression vectors to allow co-transfection. Elements of the expression vector include a promoter (Cytomegalovirus (CMV) enhancer-promoter), a signal sequence to facilitate secretion, a polyadenylation signal and transcription terminator (Bovine Growth Hormone (BGH) gene), an element allowing episomal replication and replication in prokaryotes (e.g. SV40 origin and ColE1 or others known in the art) and elements to allow selection (ampicillin resistance gene and zeocin marker).
Expression:
[0749] Chimera and humanized IgG candidates were expressed in HEK293F mammalian cells at 1 ml scale. Cleared supernatants were used for FACS binding studies. More precisely, HEK293F cells were diluted to 5E5 cells/ml in FreeStyle medium supplemented with Pen/Strep and 1 ml transferred into 24 round bottom deep well plate. 0.5 .mu.g of light and 0.5 .mu.g of heavy chain mammalian expression plasmids were diluted in the same medium together with 4 .mu.l of FuGENE HD (Roche REF 04709705001). After 15 min RT incubation, DNA/Fugene mix was added drop-wise to the cells and placed in a 5% CO.sub.2 incubator at 250 rpm, 37.degree. C. for five days. Supernatant were then separated from the cells by centrifugation. To measure IgG content, aliquots of 200 .mu.L were placed in the wells of 96-well microtiter plates. All samples and standards were measured in duplicate using Protein A Dip and read biosensors (Fortebio Cat No 18-5010). The plate was placed in an Octet instrument (ForteBio) and allowed to equilibrate to 27.degree. C. in the thermostated chamber. Data were processed automatically using the Octet User Software version 3.0 and concentration determined by comparing to an IgG standard curve.
Binding Analysis by FACS:
[0750] Humanized and chimera antibodies were evaluated with a flow cytometry binding assay using cell line 300.19-hsCD19FL. This cell line was generated by transfecting the mouse preB cell line 300.19 with a vector (hCD19 FL/pEF4-myc-His A) encoding the full length human CD19 encoding sequence and natural promoter as well as a Zeocin resistance gene. In brief, 300.19 cells were electroporated with the linearized plasmid and then cells expressing high levels of hsCD19 were identified using an APC-conjugated anti-human CD19 Ab (clone HIB19 from BD 555415) and subsequently sorted using a FACS Aria flow cytometer. The sorted hsCD19+ cells were cultured and confirmed to stably express high levels of hsCD19.
[0751] The binding assay could be performed directly with the serum free culture media containing the expressed IgG. All evaluated IgGs were normalized to the same concentration (85 nM), before to be diluted by a 3 fold serial dilution down to 1.4 pM. Then, in a 96-well plate, aliquots of 5.times.10.sup.5 cells/well were incubated for 30 min at 4.degree. C. with diluted IgGs. Cells were washed twice with FACS buffer (0.5% BSA in PBS) before addition of the detection antibody, an APC conjugated goat anti-hu IgG, Fc fragment specific (Dianova #109-136-098), diluted 1:1000 in FACS buffer. Cells were incubated a further 30 min at 4.degree. C., then washed twice in FACS buffer and assayed using FACS Calibur (BD Bioscience). Binding curves plotting (median of fluorescence intensity versus IgG concentration) and EC.sub.50 determination were performed with GraphPad Prism.TM. 3.0 software with nonlinear regression analysis, sigmoidal dose response (variable slope).
[0752] The FACS analyses show that apparent binding for all evaluated IgGs can vary widely, with some constructs exhibiting a 5 to 10 fold shift in EC50 as an IgG versus a scFv. Based on EC.sub.50 values, lead candidates are chosen that have a binding affinity within a factor of 2 or better compared to the chimeric reference.
Example 2: Characterization of Anti-CD19 Soluble scFv Fragments Derived from Humanized CD19 IgG Antibodies
[0753] Soluble scFv fragments were generated from the humanized CD19 IgGs described in Example 1 using standard molecule biology techniques. These soluble scFvs were used in characterization studies to examine the stability, cell surface expression, and binding properties of the scFvs. Additionally, experiments were also conducted to investigate the impact of the potential PTM introduced during the humanization process.
scFv Expression and Purification
[0754] For transfection of each scFv construct, around 3e8 293F cells were transfected with 100 .mu.g of plasmid using PEI as the transfection reagent at the ratio of 3:1 (PEI:DNA). The cells were grown in 100 ml EXPi293 Expression media (Invitrogen) in a shaker flask at 37.degree. C., 125 rpm, 8% CO.sub.2. The culture was harvested after six days and used for protein purification.
[0755] 293F cells were harvested by spinning down at 3500 g for 20 minutes. The supernatant was collected and filtered through VacuCap90 PF Filter Unit (w/0.8/0.2 .mu.m Super Membrane, PALL). Around 400 .mu.l 400 ul of Ni-NTA agarose beads (Qiagen) were added to the supernatant. The mixture was rotated and incubated for 4 hrs at 4.degree. C. It was loaded onto a purification column and washed with washing buffer with 20 mM Histidine. The protein was eluted with 500 .mu.l elution buffer with 300 mM Histidine. The samples were dialyzed against PBS buffer at 4C overnight. Protein samples were quantified using nanodrop 2000c.
scFv Conformation and Colloidal Stability Analysis
[0756] Thermostability of the scFv was determined by DSF: mix 10-20 .mu.l of protein sample with the dye Sypro Orange (Invitrogen Cat#S6650) of a final dilution at 1:1000, in a total volume of 25 .mu.l in PBS, run BioRad CFX1000 (25 C for 2 min, then increment 0.5.degree. C. for 30 second, 25 to 95.degree. C.).
[0757] For analytical SEC experiment, around 15-20 .mu.g of scFv protein sample in 20 .mu.l PBS was injected onto TSKgel Super SW2000 at 0.3 ml/min flow rate on n Agilent 1100 series.
EC50 by FACS Binding
[0758] Mouse cell line 300.CD19 were grown in RPMI 1640 with 0.5 mg/ml Zeocin. Around 5e5 cells/per well were transferred to the BD Falcon 96 well plate. The cells were spin down at 900 rpm (Sorval Legend XT centrifuge) for 3 minutes. The supernatant were removed. Anti-CD19 scFv protein samples were diluted in DPBS with 5% FBS. The samples were added into the wells, mixed well with the cells and incubated for 1 hour. The cells were washed twice in the DPBS with 5% FBS. The cells were incubated with antipoly His PE (R&D) for 1 hour, washed twice before FACS analysis (LSRII from BD Biosciences).
Kinetic Analysis by Proteon
[0759] Kinetics were determined using Bio-Rad Proteon. Immobilization was performed using standard amine coupling on a GLC sensor chip. The scFv samples were diluted to 0.03 mg/mL in acetate pH 4.5 and applied to the chip at a flow rate of 30 .mu.L/min for 300 seconds. The CD19 ligand was then serial diluted in PBS-Tween and injected at a flow rate of 50 .mu.L/min for 120 seconds with a dissociation time of 480 seconds. The chip surface was regenerated with glycine pH 2.5. Data was fitted using a 1:1 Langmuir model.
Surface Expression of CART19 Constructs and Staining by FACS
[0760] HEK293F suspension cells transiently transfected with different anti-hCD19 CARTs were harvested 2 days after the transfection. Around 1e6 cells were placed into each well of a V-shape 96 well plate (Greiner Bio-One, Germany) and washed three times with 0.2 ml FACS buffer (1.times.PBS containing 4% bovine serum albumin (BSA) (BSA fraction V, Roche Diagnostics, Indianapolis, Ind.). Cells were resuspended in 0.2 ml of the FCAS buffer with either 0.2 .mu.g of biotinylated protein L (GenScript, Piscataway, N.J.) or 100 nM of hCD19(AA 1-291)-hIgG1 Fc (Generated in NIBRI) and incubated at 4.degree. C. for 30 minutes. Cells were then washed with 0.2 ml of FACS buffer three times, and incubated with 1 .mu.l Streptavidin Alexa Fluor 488 (Life Technologies, Grand Island, N.Y.) in 0.2 ml of FACS buffer for samples with protein L, or 2 .mu.l of PE anti-human Fcx (Jackson ImmunoResearch Laboratories, West Grove, Pa.) in 0.2 ml of FACS buffer for samples with hCD19-hIgG1 Fc for 30 minutes at 4.degree. C. in the dark. After washing with 0.2 ml of FACS buffer three times, cells were analyzed on a LSRII (BD Biosciences, San Jose, Calif.) machine using the FACSDiva software (BD Biosciences, San Jose, Calif.). Immunofluorescence staining was analyzed as the relative log fluorescence of live cells, and the percentage of the Alexa Fluor 488 positive or PE positive cells were measured.
Analysis of Potential PMTs Generated During the Humanization Process
[0761] Of interest was that during humanization, position 62 in the CDRH2 region prefers to be a serine residue rather than the alanine present in the murine CDRH2 as described in Example 1. Whether the PTM site generated during humanization process was actually a "true" PTM site or merely a theoretical one was tested. Two IgG variants were generated in which the asparagine at position 60 (known to be a glycosylation site) was mutated to serine, or glutamine and designated FMC63_VH_hz2 (N60S) and FMC63_VH_hz2 (N60Q), respectively. These constructs were generated in order to eliminate the potential post-translational modification site (PTM) and test for retained activity.
Results
[0762] Anti-CD19 humanized scFvs and mouse scFv were expressed in 293F cells and purified through His tag. The expression and yield of all humanized scFvs was much higher than the original mouse scFv (data not shown).
[0763] To confirm identity and assess integrity, the scFV constructs are analyzed with or without incubation with N-glycanase F (PNGaseF) followed by both high-performance liquid chromatography mass spectrometry (HPLC-MS) (See FIG. 3) and SDS-PAGE (data not shown). PNGaseF is an enzyme specific for the removal of N-linked glycan structures from the consensus sequence N--X-S/T/C where X is any amino acid except proline. Briefly, the samples are diluted in water to 0.1 .mu.g/L and either left untreated or incubated with PNGaseF at a 1:2 (w/w) PNGaseF: scFV ratio for 3 hours at 37.degree. C.
[0764] SDS-PAGE analysis is performed using a NuPAGE 4-12% Bis-Tris gel from Novex. Approximately 2 .mu.g scFV are loaded into each lane and the electrophoresis is conducted at 200 V constant for 40 minutes. Following electrophoresis, the gel is stained using PhastGel Blue R 250 stain (Amersham Pharmacia) and destained with 10% acetic acid, 30% methanol.
[0765] HPLC-MS analysis is performed on the Water's Acquity UPLC system coupled to a Xevo-Tof mass spectrometer. Approximately 1 .mu.g of each sample is loaded onto a R 1/10 2.1 .times.100 mm 10 am POROS column (Applied Biosciences) set to 60.degree. C. at a flow rate of 0.5 mL/min. Mobile phases are composed of 0.1% formic acid (A) and 0.1% formic acid, 75% isopropanol, 25% acetonitrile (B). Protein is eluted from the column with a reverse phase gradient from 25%-90% B in 12 minutes. The acquisition is performed using electrospray positive scan at the m/z range of 600-4000 Da with a source cone voltage ramp 20-50V. The resulting spectra are deconvoluted using MaxEnt1.
[0766] The glycosylation site was introduced during the process of humanization. The non-PTM variants (VH: N60S or N60Q) were without this additional form. The construct was the only one with a consensus site of N-linked glycosylation in HC CDR2. From the SDS-PAGE analysis, the untreated samples migrated as single bands consistent with the approximate molecular weights of the sequences for all constructs except 103101-WT (S/N) for which doublet is observed. This construct is the only one with a consensus site of N-linked glycosylation in H-CDR2. When treated with PNGaseF, the higher molecular weight band of the doublet is no longer present suggesting partial occupancy of the site. Similarly, the observed molecular weights from the deconvoluted mass spectra are consistent with those predicted from the amino acid sequences. However, while the other constructs demonstrated a single primary molecular species, 103101-WT (S/N) also had a population 1217 Daltons higher than that predicted from the sequence which is no longer present after treatment with PNGaseF. This is consistent with the presence of a single predominant N-linked glycoform, likely oligomannose 5 based upon mass. The presence of the glycosylated form was confirmed by the MS analysis as shown in FIG. 3.
[0767] The conformation stability was measured by Differential Scanning Fluorimetry (DSF). As shown in FIG. 4, the Tm of mouse scFv was 57.degree. C., while the human variants showed higher Tm at around 70.degree. C. The Tm for all the humanized scFv is much better than the murine scFv, clearly showing that all the humanized scFv are more stable than the murine scFv. This stability will likely translate to the CART19 construct, likely leading to improved therapeutic properties.
[0768] The activity of the purified scFv was measure by binding to hCD19 expression cells as well as by binding to hCD19 antigen using SPR based detection method. Mouse cell line 300 was used to determine the binding of scFvs. The EC.sub.50 of mouse scFv for hCD19 was around 06-1.6 nM. The humanized variants showed EC.sub.50 of the same range in the low or sub nM EC.sub.50s range.
Example 3: CD19 CAR Constructs
[0769] ScFv to be used in the final CAR construct were derived from the humanized IgG described in Example 1. The order in which the VL and VH domains appear in the scFv was varied (i.e., VL-VH, or VH-VL orientation), and where either three or four copies of the "G4S" (SEQ ID NO: 18) subunit, in which each subunit comprises the sequence GGGGS (SEQ ID NO:18) (e.g., (G4S).sub.3 (SEQ ID NO:107) or (G4S).sub.4 (SEQ ID NO:106)), connect the variable domains to create the entirety of the scFv domain, as shown in Table 2.
TABLE-US-00015 TABLE 2 Humanized CD19 scFv constructs showing VH and VL orientation and linker length ("3G4S" is disclosed as SEQ ID NO: 107 and "4G4S" is disclosed as SEQ ID NO: 106). construct ID Length aa annotation Vh change mscFvCTL019 486 VL-VH, 3G4S 104879 491 VL-VH, 4G4S N/S 104880 491 VL-VH, 4G4S N/Q 104881 491 VH-VL, 4G4S N/S 104882 491 VH-VL, 4G4S N/Q 104875 486 VL-VH, 3G4S N/S 104876 486 VL-VH, 3G4S N/Q 104877 486 VH-VL, 3G4S N/S 104878 486 VH-VL, 3G4S N/Q 105974 491 VL-VH, 4G4S S/N 105975 491 VH-VL, 4G4S S/N 105976 486 VL-VH, 3G4S S/N 105977 486 VH-VL, 3G4S S/N
[0770] The sequences of the humanized scFv fragments (SEQ ID NOS: 1-12) are provided below in Table 3. Full CAR constructs were generated using SEQ ID NOs: 1-12 with additional sequences, SEQ ID NOs: 13-17, shown below, to generate full CAR constructs with SEQ ID NOs: 31-42.
TABLE-US-00016 leader (amino acid sequence) (SEQ ID NO: 13) MALPVTALLLPLALLLHAARP leader (nucleic acid sequence) (SEQ ID NO: 54) ATGGCCCTGCCTGTGACAGCCCTGCTGCTGCCTCTGGCTCTGCTGCTGCA TGCCGCTAGACCC CD8 hinge (amino acid sequence) (SEQ ID NO: 14) TTTPAPRPPTPAPTIASQPLSLRPEACRPAAGGAVHTRGLDFACD CD8 hinge (nucleic acid sequence) (SEQ ID NO: 55) ACCACGACGCCAGCGCCGCGACCACCAACACCGGCGCCCACCATCGCGTC GCAGCCCCTGTCCCTGCGCCCAGAGGCGTGCCGGCCAGCGGCGGGGGGCG CAGTGCACACGAGGGGGCTGGACTTCGCCTGTGAT CD8 transmembrane (amino acid sequence) (SEQ ID NO: 15) IYIWAPLAGTCGVLLLSLVITLYC transmembrane (nucleic acid sequence) (SEQ ID NO: 56) ATCTACATCTGGGCGCCCTTGGCCGGGACTTGTGGGGTCCTTCTCCTGTC ACTGGTTATCACCCTTTACTGC 4-1BB Intracellular domain (amino acid sequence) (SEQ ID NO: 16) KRGRKKLLYIFKQPFMRPVQTTQEEDGCSCRFPEEEEGGCEL 4-1BB Intracellular domain (nucleic acid sequence) (SEQ ID NO: 60) AAACGGGGCAGAAAGAAACTCCTGTATATATTCAAACAACCATTTATGAG ACCAGTACAAACTACTCAAGAGGAAGATGGCTGTAGCTGCCGATTTCCAG AAGAAGAAGAAGGAGGATGTGAACTG CD3 zeta domain (amino acid sequence) (SEQ ID NO: 17) RVKFSRSADAPAYKQGQNQLYNELNLGRREEYDVLDKRRGRDPEMGGKPR RKNPQEGLYNELQKDKMAEAYSEIGMKGERRRGKGHDGLYQGLSTATKDT YDALHMQALPPR CD3 zeta (nucleic acid sequence) (SEQ ID NO: 101) AGAGTGAAGTTCAGCAGGAGCGCAGACGCCCCCGCGTACAAGCAGGGCCA GAACCAGCTCTATAACGAGCTCAATCTAGGACGAAGAGAGGAGTACGATG TTTTGGACAAGAGACGTGGCCGGGACCCTGAGATGGGGGGAAAGCCGAGA AGGAAGAACCCTCAGGAAGGCCTGTACAATGAACTGCAGAAAGATAAGAT GGCGGAGGCCTACAGTGAGATTGGGATGAAAGGCGAGCGCCGGAGGGGCA AGGGGCACGATGGCCTTTACCAGGGTCTCAGTACAGCCACCAAGGACACC TACGACGCCCTTCACATGCAGGCCCTGCCCCCTCGC CD3 zeta domain (amino acid sequence; NCBI Reference Sequence NM_000734.3) (SEQ ID NO: 43) RVKFSRSADAPAYQQGQNQLYNELNLGRREEYDVLDKRRGRDPEMGGKPR RKNPQEGLYNELQKDKMAEAYSEIGMKGERRRGKGHDGLYQGLSTATKDT YDALHMQALPPR CD3 zeta (nucleic acid sequence; NCBI Reference Sequence NM_000734.3); (SEQ ID NO: 44) AGAGTGAAGTTCAGCAGGAGCGCAGACGCCCCCGCGTACCAGCAGGGCCA GAACCAGCTCTATAACGAGCTCAATCTAGGACGAAGAGAGGAGTACGATG TTTTGGACAAGAGACGTGGCCGGGACCCTGAGATGGGGGGAAAGCCGAGA AGGAAGAACCCTCAGGAAGGCCTGTACAATGAACTGCAGAAAGATAAGAT GGCGGAGGCCTACAGTGAGATTGGGATGAAAGGCGAGCGCCGGAGGGGCA AGGGGCACGATGGCCTTTACCAGGGTCTCAGTACAGCCACCAAGGACACC TACGACGCCCTTCACATGCAGGCCCTGCCCCCTCGC IgG4 Hinge (amino acid sequence) (SEQ ID NO: 102) ESKYGPPCPPCPAPEFLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQ EDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKE YKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCL VKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQ EGNVFSCSVMHEALHNHYTQKSLSLSLGKM IgG4 Hinge (nucleotide sequence) (SEQ ID NO: 103) GAGAGCAAGTACGGCCCTCCCTGCCCCCCTTGCCCTGCCCCCGAGTTCCT GGGCGGACCCAGCGTGTTCCTGTTCCCCCCCAAGCCCAAGGACACCCTGA TGATCAGCCGGACCCCCGAGGTGACCTGTGTGGTGGTGGACGTGTCCCAG GAGGACCCCGAGGTCCAGTTCAACTGGTACGTGGACGGCGTGGAGGTGCA CAACGCCAAGACCAAGCCCCGGGAGGAGCAGTTCAATAGCACCTACCGGG TGGTGTCCGTGCTGACCGTGCTGCACCAGGACTGGCTGAACGGCAAGGAA TACAAGTGTAAGGTGTCCAACAAGGGCCTGCCCAGCAGCATCGAGAAAAC CATCAGCAAGGCCAAGGGCCAGCCTCGGGAGCCCCAGGTGTACACCCTGC CCCCTAGCCAAGAGGAGATGACCAAGAACCAGGTGTCCCTGACCTGCCTG GTGAAGGGCTTCTACCCCAGCGACATCGCCGTGGAGTGGGAGAGCAACGG CCAGCCCGAGAACAACTACAAGACCACCCCCCCTGTGCTGGACAGCGACG GCAGCTTCTTCCTGTACAGCCGGCTGACCGTGGACAAGAGCCGGTGGCAG GAGGGCAACGTCTTTAGCTGCTCCGTGATGCACGAGGCCCTGCACAACCA CTACACCCAGAAGAGCCTGAGCCTGTCCCTGGGCAAGATG
[0771] These clones all contained a Q/K residue change in the signal domain of the co-stimulatory domain derived from 4-1BB.
TABLE-US-00017 TABLE 3 Humanized CD19 CAR Constructs Name SEQ ID Sequence CAR 1 CAR1 scFv 1 EIVMTQSPATLSLSPGERATLSCRASQDISKYLNWYQQKPGQAPRLLIYHT domain SRLHSGIPARFSGSGSGTDYTLTISSLQPEDFAVYFCQQGNTLPYTFGQGT KLEIKGGGGSGGGGSGGGGSQVQLQESGPGLVKPSETLSLTCTVSGVSLPD YGVSWIRQPPGKGLEWIGVIWGSETTYYSSSLKSRVTISKDNSKNQVSLKL SSVTAADTAVYYCAKHYYYGGSYAMDYWGQGTLVTVSS 103101 61 atggccctccctgtcaccgccctgctgcttccgctggctcttctgctccacgccgc CAR1 tcggcccgaaattgtgatgacccagtcacccgccactcttagcctttcacccggtg Soluble agcgcgcaaccctgtcttgcagagcctcccaagacatctcaaaataccttaattgg scFv-nt tatcaacagaagcccggacaggctcctcgccttctgatctaccacaccagccggct ccattctggaatccctgccaggttcagcggtagcggatctgggaccgactacaccc tcactatcagctcactgcagccagaggacttcgctgtctatttctgtcagcaaggg aacaccctgccctacacctttggacagggcaccaagctcgagattaaaggtggagg tggcagcggaggaggtgggtccggcggtggaggaagccaggtccaactccaagaaa gcggaccgggtcttgtgaagccatcagaaactctttcactgacttgtactgtgagc ggagtgtctctccccgattacggggtgtcttggatcagacagccaccggggaaggg tctggaatggattggagtgatttggggctctgagactacttactactcttcatccc tcaagtcacgcgtcaccatctcaaaggacaactctaagaatcaggtgtcactgaaa ctgtcatctgtgaccgcagccgacaccgccgtgtactattgcgctaagcattacta ttatggcgggagctacgcaatggattactggggacagggtactctggtcaccgtgt ccagccaccaccatcatcaccatcaccat 103101 73 MALPVTALLLPLALLLHAARPeivmtqspatlslspgeratlscrasqdiskylnw CAR1 yqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcqqg Soluble ntlpytfgqgtkleikggggsggggsggggsqvqlqesgpglvkpsetlsltctvs scFv-aa gvslpdygvswirqppgkglewigviwgsettyyssslksrvtiskdnsknqvslk lssvtaadtavyycakhyyyggsyamdywgqgtlvtvsshhhhhhhh 104875 85 atggccctccctgtcaccgccctgctgcttccgctggctcttctgctccacgccgc CAR 1- tcggcccgaaattgtgatgacccagtcacccgccactcttagcctttcacccggtg Full-nt agcgcgcaaccctgtcttgcagagcctcccaagacatctcaaaataccttaattgg tatcaacagaagcccggacaggctcctcgccttctgatctaccacaccagccggct ccattctggaatccctgccaggttcagcggtagcggatctgggaccgactacaccc tcactatcagctcactgcagccagaggacttcgctgtctatttctgtcagcaaggg aacaccctgccctacacctttggacagggcaccaagctcgagattaaaggtggagg tggcagcggaggaggtgggtccggcggtggaggaagccaggtccaactccaagaaa gcggaccgggtcttgtgaagccatcagaaactctttcactgacttgtactgtgagc ggagtgtctctccccgattacggggtgtcttggatcagacagccaccggggaaggg tctggaatggattggagtgatttggggctctgagactacttactactcttcatccc tcaagtcacgcgtcaccatctcaaaggacaactctaagaatcaggtgtcactgaaa ctgtcatctgtgaccgcagccgacaccgccgtgtactattgcgctaagcattacta ttatggcgggagctacgcaatggattactggggacagggtactctggtcaccgtgt ccagcaccactaccccagcaccgaggccacccaccccggctcctaccatcgcctcc cagcctctgtccctgcgtccggaggcatgtagacccgcagctggtggggccgtgca tacccggggtcttgacttcgcctgcgatatctacatttgggcccctctggctggta cttgcggggtcctgctgctttcactcgtgatcactctttactgtaagcgcggtcgg aagaagctgctgtacatctttaagcaacccttcatgaggcctgtgcagactactca agaggaggacggctgttcatgccggttcccagaggaggaggaaggcggctgcgaac tgcgcgtgaaattcagccgcagcgcagatgctccagcctacaagcaggggcagaac cagctctacaacgaactcaatcttggtcggagagaggagtacgacgtgctggacaa gcggagaggacgggacccagaaatgggcgggaagccgcgcagaaagaatccccaag agggcctgtacaacgagctccaaaaggataagatggcagaagcctatagcgagatt ggtatgaaaggggaacgcagaagaggcaaaggccacgacggactgtaccagggact cagcaccgccaccaaggacacctatgacgctcttcacatgcaggccctgccgcctc gg 104875 31 MALPVTALLLPLALLLHAARPeivmtqspatlslspgeratlscrasqdiskylnw CAR 1- yqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcqqg Full-aa ntlpytfgqgtkleikggggsggggsggggsqvqlqesgpglvkpsetlsltctvs gvslpdygvswirqppgkglewigviwgsettyyssslksrvtiskdnsknqvslk lssvtaadtavyycakhyyyggsyamdywgqgtlvtvsstttpaprpptpaptias qplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitlyckrgr kkllyifkqpfmrpvqttgeedgcscrfpeeeeggcelrvkfsrsadapaykqgqn qlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkmaeaysei gmkgerrrgkghdglyqglstatkdtydalhmqalppr CAR 2 CAR2 scFv 2 eivmtqspatlslspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhs domain giparfsgsgsgtdytltisslqpedfavyfcqqgntlpytfgqgtkleikggggs ggggsggggsqvqlqesgpglvkpsetlsltctvsgvslpdygvswirqppgkgle wigviwgsettyyqsslksrvtiskdnsknqvslklssvtaadtavyycakhyyyg gsyamdywgqgtlvtvss 103102 62 atggccctccctgtcaccgccctgctgcttccgctggctcttctgctccacgccgc CAR2- tcggcccgaaattgtgatgacccagtcacccgccactcttagcctttcacccggtg Soluble agcgcgcaaccctgtcttgcagagcctcccaagacatctcaaaataccttaattgg scFv-nt tatcaacagaagcccggacaggctcctcgccttctgatctaccacaccagccggct ccattctggaatccctgccaggttcagcggtagcggatctgggaccgactacaccc tcactatcagctcactgcagccagaggacttcgctgtctatttctgtcagcaaggg aacaccctgccctacacctttggacagggcaccaagctcgagattaaaggtggagg tggcagcggaggaggtgggtccggcggtggaggaagccaggtccaactccaagaaa gcggaccgggtcttgtgaagccatcagaaactctttcactgacttgtactgtgagc ggagtgtctctccccgattacggggtgtcttggatcagacagccaccggggaaggg tctggaatggattggagtgatttggggctctgagactacttactaccaatcatccc tcaagtcacgcgtcaccatctcaaaggacaactctaagaatcaggtgtcactgaaa ctgtcatctgtgaccgcagccgacaccgccgtgtactattgcgctaagcattacta ttatggcgggagctacgcaatggattactggggacagggtactctggtcaccgtgt ccagccaccaccatcatcaccatcaccat 103102 74 MALPVTALLLPLALLLHAARPeivmtqspatlslspgeratlscrasqdiskylnw CAR2- yqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcqqg Soluble ntlpytfgqgtkleikggggsggggsggggsqvqlqesgpglvkpsetlsltctvs scFv-aa gvslpdygvswirqppgkglewigviwgsettyyqsslksrvtiskdnsknqvslk lssvtaadtavyycakhyyyggsyamdywgqgtlvtvsshhhhhhhh 104876 86 atggccctccctgtcaccgccctgctgcttccgctggctcttctgctccacgccgc CAR 2- tcggcccgaaattgtgatgacccagtcacccgccactcttagcctttcacccggtg Full-nt agcgcgcaaccctgtcttgcagagcctcccaagacatctcaaaataccttaattgg tatcaacagaagcccggacaggctcctcgccttctgatctaccacaccagccggct ccattctggaatccctgccaggttcagcggtagcggatctgggaccgactacaccc tcactatcagctcactgcagccagaggacttcgctgtctatttctgtcagcaaggg aacaccctgccctacacctttggacagggcaccaagctcgagattaaaggtggagg tggcagcggaggaggtgggtccggcggtggaggaagccaggtccaactccaagaaa gcggaccgggtcttgtgaagccatcagaaactctttcactgacttgtactgtgagc ggagtgtctctccccgattacggggtgtcttggatcagacagccaccggggaaggg tctggaatggattggagtgatttggggctctgagactacttactaccaatcatccc tcaagtcacgcgtcaccatctcaaaggacaactctaagaatcaggtgtcactgaaa ctgtcatctgtgaccgcagccgacaccgccgtgtactattgcgctaagcattacta ttatggcgggagctacgcaatggattactggggacagggtactctggtcaccgtgt ccagcaccactaccccagcaccgaggccacccaccccggctcctaccatcgcctcc cagcctctgtccctgcgtccggaggcatgtagacccgcagctggtggggccgtgca tacccggggtcttgacttcgcctgcgatatctacatttgggcccctctggctggta cttgcggggtcctgctgctttcactcgtgatcactctttactgtaagcgcggtcgg aagaagctgctgtacatctttaagcaacccttcatgaggcctgtgcagactactca agaggaggacggctgttcatgccggttcccagaggaggaggaaggcggctgcgaac tgcgcgtgaaattcagccgcagcgcagatgctccagcctacaagcaggggcagaac cagctctacaacgaactcaatcttggtcggagagaggagtacgacgtgctggacaa gcggagaggacgggacccagaaatgggcgggaagccgcgcagaaagaatccccaag agggcctgtacaacgagctccaaaaggataagatggcagaagcctatagcgagatt ggtatgaaaggggaacgcagaagaggcaaaggccacgacggactgtaccagggact cagcaccgccaccaaggacacctatgacgctcttcacatgcaggccctgccgcctc gg 104876 32 MALPVTALLLPLALLLHAARPeivmtqspatlslspgeratlscrasqdiskylnw CAR 2- yqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcqqg Full-aa ntlpytfgqgtkleikggggsggggsggggsqvqlqesgpglvkpsetlsltctvs gvslpdygvswirqppgkglewigviwgsettyyqsslksrvtiskdnsksqvslk lssvtaadtavyycakhyyyggsyamdywgqgtlvtvsstttpaprpptpaptias qplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitlyckrgr kkllyifkqpfmrpvqttqeedgcscrfpeeeeggcelrvkfsrsadapaykqgqn qlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkmaeaysei gmkgerrrgkghdglyqglstatkdtydalhmqalppr CAR 3 CAR3 scFv 3 qvqlqesgpglvkpsetlsltctvsgvslpdygvswirqppgkglewigviwgset domain tyyssslksrvtiskdnsknqvslklssvtaadtavyycakhyyyggsyamdywgq gtivtvssggggsggggsggggseivmtqspatlslspgeratlscrasqdiskyl nwyqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcq qgntlpytfgqgtkleik 103104 63 atggctctgcccgtgaccgcactcctcctgccactggctctgctgcttcacgccgc CAR 3- tcgcccacaagtccagcttcaagaatcagggcctggtctggtgaagccatctgaga Soluble ctctgtccctcacttgcaccgtgagcggagtgtccctcccagactacggagtgagc scFv-nt tggattagacagcctcccggaaagggactggagtggatcggagtgatttggggtag cgaaaccacttactattcatcttccctgaagtcacgggtcaccatttcaaaggata actcaaagaatcaagtgagcctcaagctctcatcagtcaccgccgctgacaccgcc gtgtattactgtgccaagcattactactatggagggtcctacgccatggactactg gggccagggaactctggtcactgtgtcatctggtggaggaggtagcggaggaggcg ggagcggtggaggtggctccgaaatcgtgatgacccagagccctgcaaccctgtcc ctttctcccggggaacgggctaccctttcttgtcgggcatcacaagatatctcaaa atacctcaattggtatcaacagaagccgggacaggcccctaggcttcttatctacc acacctctcgcctgcatagcgggattcccgcacgctttagcgggtctggaagcggg accgactacactctgaccatctcatctctccagcccgaggacttcgccgtctactt ctgccagcagggtaacaccctgccgtacaccttcggccagggcaccaagcttgaga tcaaacatcaccaccatcatcaccatcac 103104 75 MALPVTALLLPLALLLHAARPqvqlqesgpglvkpsetlsltctvsgvslpdygvs CAR 3- wirqppgkglewigviwgsettyyssslksrvtiskdnsknqvslklssvtaadta Soluble vyycakhyyyggsyamdywgqgtlvtvssggggsggggsggggseivmtqspatls scFv-aa lspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhsgiparfsgsgsg tdytltisslqpedfavyfcqqgntlpytfgqgtkleikhhhhhhhh 104877 87 atggctctgcccgtgaccgcactcctcctgccactggctctgctgcttcacgccgc CAR 3- tcgcccacaagtccagcttcaagaatcagggcctggtctggtgaagccatctgaga Full-nt ctctgtccctcacttgcaccgtgagcggagtgtccctcccagactacggagtgagc tggattagacagcctcccggaaagggactggagtggatcggagtgatttggggtag cgaaaccacttactattcatcttccctgaagtcacgggtcaccatttcaaaggata actcaaagaatcaagtgagcctcaagctctcatcagtcaccgccgctgacaccgcc gtgtattactgtgccaagcattactactatggagggtcctacgccatggactactg gggccagggaactctggtcactgtgtcatctggtggaggaggtagcggaggaggcg ggagcggtggaggtggctccgaaatcgtgatgacccagagccctgcaaccctgtcc ctttctcccggggaacgggctaccctttcttgtcgggcatcacaagatatctcaaa atacctcaattggtatcaacagaagccgggacaggcccctaggcttcttatctacc acacctctcgcctgcatagcgggattcccgcacgctttagcgggtctggaagcggg accgactacactctgaccatctcatctctccagcccgaggacttcgccgtctactt ctgccagcagggtaacaccctgccgtacaccttcggccagggcaccaagcttgaga tcaaaaccactactcccgctccaaggccacccacccctgccccgaccatcgcctct cagccgctttccctgcgtccggaggcatgtagacccgcagctggtggggccgtgca tacccggggtcttgacttcgcctgcgatatctacatttgggcccctctggctggta cttgcggggtcctgctgctttcactcgtgatcactctttactgtaagcgcggtcgg aagaagctgctgtacatctttaagcaacccttcatgaggcctgtgcagactactca agaggaggacggctgttcatgccggttcccagaggaggaggaaggcggctgcgaac tgcgcgtgaaattcagccgcagcgcagatgctccagcctacaagcaggggcagaac cagctctacaacgaactcaatcttggtcggagagaggagtacgacgtgctggacaa gcggagaggacgggacccagaaatgggcgggaagccgcgcagaaagaatccccaag agggcctgtacaacgagctccaaaaggataagatggcagaagcctatagcgagatt ggtatgaaaggggaacgcagaagaggcaaaggccacgacggactgtaccagggact cagcaccgccaccaaggacacctatgacgctcttcacatgcaggccctgccgcctc gg 104877 33 MALPVTALLLPLALLLHAARPqvqlqesgpglvkpsetlsltctvsgvslpdygvs CAR 3- wirqppgkglewigviwgsettyyssslksrvtiskdnsknqvslklssvtaadta Full-aa vyycakhyyyggsyamdywgqgtlvtvssggggsggggsggggseivmtqspatls lspgeratlscrasgdiskylnwyqqkpgqaprlliyhtsrlhsgiparfsgsgsg tdytltisslqpedfavyfcqqgntlpytfgqgtkleiktttpaprpptpaptias qplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitlyckrgr kkllyifkqpfmrpvqttgeedgcscrfpeeeeggcelrvkfsrsadapaykqgqn qlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkmaeaysei gmkgerrrgkghdglyqglstatkdtydalhmqalppr CAR4 CAR4 scFv 4 qvqlqesgpglvkpsetlsltctvsgvslpdygvswirqppgkglewigviwgset domain tyyqsslksrvtiskdnsknqvslklssvtaadtavyycakhyyyggsyamdywgq gtlvtvssggggsggggsggggseivmtqspatlslspgeratlscrasqdiskyl nwyqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcq qgntlpytfgqgtkleik 103106 64 atggctctgcccgtgaccgcactcctcctgccactggctctgctgcttcacgccgc CAR4- tcgcccacaagtccagcttcaagaatcagggcctggtctggtgaagccatctgaga Soluble ctctgtccctcacttgcaccgtgagcggagtgtccctcccagactacggagtgagc scFv-nt tggattagacagcctcccggaaagggactggagtggatcggagtgatttggggtag cgaaaccacttactatcaatcttccctgaagtcacgggtcaccatttcaaaggata actcaaagaatcaagtgagcctcaagctctcatcagtcaccgccgctgacaccgcc gtgtattactgtgccaagcattactactatggagggtcctacgccatggactactg gggccagggaactctggtcactgtgtcatctggtggaggaggtagcggaggaggcg ggagcggtggaggtggctccgaaatcgtgatgacccagagccctgcaaccctgtcc ctttctcccggggaacgggctaccctttcttgtcgggcatcacaagatatctcaaa atacctcaattggtatcaacagaagccgggacaggcccctaggcttcttatctacc acacctctcgcctgcatagcgggattcccgcacgctttagcgggtctggaagcggg accgactacactctgaccatctcatctctccagcccgaggacttcgccgtctactt ctgccagcagggtaacaccctgccgtacaccttcggccagggcaccaagcttgaga tcaaacatcaccaccatcatcaccatcac 103106 76 MALPVTALLLPLALLLHAARPqvqlqesgpglvkpsetlsltctvsgvslpdygvs CAR4- wirqppgkglewigviwgsettyyqsslksrvtiskdnsknqvslklssvtaadta Soluble vyycakhyyyggsyamdywgqgtlvtvssggggsggggsggggseivmtqspatls scFv-aa lspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhsgiparfsgsgsg tdytltisslqpedfavyfcqqgntlpytfgqgtkleikhhhhhhhh 104878 88 atggctctgcccgtgaccgcactcctcctgccactggctctgctgcttcacgccgc CAR 4- tcgcccacaagtccagcttcaagaatcagggcctggtctggtgaagccatctgaga Full-nt ctctgtccctcacttgcaccgtgagcggagtgtccctcccagactacggagtgagc tggattagacagcctcccggaaagggactggagtggatcggagtgatttggggtag cgaaaccacttactatcaatcttccctgaagtcacgggtcaccatttcaaaggata actcaaagaatcaagtgagcctcaagctctcatcagtcaccgccgctgacaccgcc gtgtattactgtgccaagcattactactatggagggtcctacgccatggactactg gggccagggaactctggtcactgtgtcatctggtggaggaggtagcggaggaggcg ggagcggtggaggtggctccgaaatcgtgatgacccagagccctgcaaccctgtcc ctttctcccggggaacgggctaccctttcttgtcgggcatcacaagatatctcaaa atacctcaattggtatcaacagaagccgggacaggcccctaggcttcttatctacc acacctctcgcctgcatagcgggattcccgcacgctttagcgggtctggaagcggg accgactacactctgaccatctcatctctccagcccgaggacttcgccgtctactt ctgccagcagggtaacaccctgccgtacaccttcggccagggcaccaagcttgaga tcaaaaccactactcccgctccaaggccacccacccctgccccgaccatcgcctct
cagccgctttccctgcgtccggaggcatgtagacccgcagctggtggggccgtgca tacccggggtcttgacttcgcctgcgatatctacatttgggcccctctggctggta cttgcggggtcctgctgctttcactcgtgatcactctttactgtaagcgcggtcgg aagaagctgctgtacatctttaagcaacccttcatgaggcctgtgcagactactca agaggaggacggctgttcatgccggttcccagaggaggaggaaggcggctgcgaac tgcgcgtgaaattcagccgcagcgcagatgctccagcctacaagcaggggcagaac cagctctacaacgaactcaatcttggtcggagagaggagtacgacgtgctggacaa gcggagaggacgggacccagaaatgggcgggaagccgcgcagaaagaatccccaag agggcctgtacaacgagctccaaaaggataagatggcagaagcctatagcgagatt ggtatgaaaggggaacgcagaagaggcaaaggccacgacggactgtaccagggact cagcaccgccaccaaggacacctatgacgctcttcacatgcaggccctgccgcctc gg 104878 34 MALPVTALLLPLALLLHAARPqvqlqesgpglvkpsetlsltctvsgvslpdygvs CAR 4- wirqppgkglewigviwgsettyyqsslksrvtiskdnsknqvslklssvtaadta Full-aa vyycakhyyyggsyamdywgqgtlvtvssggggsggggsggggseivmtqspatls lspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhsgiparfsgsgsg tdytltisslqpedfavyfcqqgntlpytfgqgtkleiktttpaprpptpaptias qplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitlyckrgr kkllyifkqpfmrpvqttgeedgcscrfpeeeeggcelrvkfsrsadapaykqgqn qlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkmaeaysei gmkgerrrgkghdglyqglstatkdtydalhmqalppr CAR 5 CAR5 scFv 5 eivmtqspatlslspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhs domain giparfsgsgsgtdytltisslqpedfavyfcqqgntlpytfgqgtkleikggggs ggggsggggsggggsqvqlqesgpglvkpsetlsltctvsgvslpdygvswirqpp gkglewigviwgsettyyssslksrvtiskdnsknqvslklssvtaadtavyycak hyyyggsyamdywgqgtlvtvss 99789 65 atggccctcccagtgaccgctctgctgctgcctctcgcacttcttctccatgccgc CAR5- tcggcctgagatcgtcatgacccaaagccccgctaccctgtccctgtcacccggcg Soluble agagggcaaccctttcatgcagggccagccaggacatttctaagtacctcaactgg scFv-nt tatcagcagaagccagggcaggctcctcgcctgctgatctaccacaccagccgcct ccacagcggtatccccgccagattttccgggagcgggtctggaaccgactacaccc tcaccatctcttctctgcagcccgaggatttcgccgtctatttctgccagcagggg aatactctgccgtacaccttcggtcaaggtaccaagctggaaatcaagggaggcgg aggatcaggcggtggcggaagcggaggaggtggctccggaggaggaggttcccaag tgcagcttcaagaatcaggacccggacttgtgaagccatcagaaaccctctccctg acttgtaccgtgtccggtgtgagcctccccgactacggagtctcttggattcgcca gcctccggggaagggtcttgaatggattggggtgatttggggatcagagactactt actactcttcatcacttaagtcacgggtcaccatcagcaaagataatagcaagaac caagtgtcacttaagctgtcatctgtgaccgccgctgacaccgccgtgtactattg tgccaaacattactattacggagggtcttatgctatggactactggggacagggga ccctggtgactgtctctagccatcaccatcaccaccatcatcac 99789 77 MALPVTALLLPLALLLHAARPeivmtqspatlslspgeratlscrasqdiskylnw CAR5- yqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcqqg Soluble ntlpytfgqgtkleikggggsggggsggggsggggsqvqlqesgpglvkpsetlsl scFV-aa tctvsgvslpdygvswirqppgkglewigviwgsettyyssslksrvtiskdnskn qvslklssvtaadtavyycakhyyyggsyamdywgqgtlvtvsshhhhhhhh 104879 89 atggccctccctgtcaccgccctgctgcttccgctggctcttctgctccacgccgc CAR5- tcggcccgaaattgtgatgacccagtcacccgccactcttagcctttcacccggtg Full-nt agcgcgcaaccctgtcttgcagagcctcccaagacatctcaaaataccttaattgg tatcaacagaagcccggacaggctcctcgccttctgatctaccacaccagccggct ccattctggaatccctgccaggttcagcggtagcggatctgggaccgactacaccc tcactatcagctcactgcagccagaggacttcgctgtctatttctgtcagcaaggg aacaccctgccctacacctttggacagggcaccaagctcgagattaaaggtggagg tggcagcggaggaggtgggtccggcggtggaggaagcggcggaggcgggagccagg tccaactccaagaaagcggaccgggtcttgtgaagccatcagaaactctttcactg acttgtactgtgagcggagtgtctctccccgattacggggtgtcttggatcagaca gccaccggggaagggtctggaatggattggagtgatttggggctctgagactactt actactcttcatccctcaagtcacgcgtcaccatctcaaaggacaactctaagaat caggtgtcactgaaactgtcatctgtgaccgcagccgacaccgccgtgtactattg cgctaagcattactattatggcgggagctacgcaatggattactggggacagggta ctctggtcaccgtgtccagcaccactaccccagcaccgaggccacccaccccggct cctaccatcgcctcccagcctctgtccctgcgtccggaggcatgtagacccgcagc tggtggggccgtgcatacccggggtcttgacttcgcctgcgatatctacatttggg cccctctggctggtacttgcggggtcctgctgctttcactcgtgatcactctttac tgtaagcgcggtcggaagaagctgctgtacatctttaagcaacccttcatgaggcc tgtgcagactactcaagaggaggacggctgttcatgccggttcccagaggaggagg aaggcggctgcgaactgcgcgtgaaattcagccgcagcgcagatgctccagcctac aagcaggggcagaaccagctctacaacgaactcaatcttggtcggagagaggagta cgacgtgctggacaagcggagaggacgggacccagaaatgggcgggaagccgcgca gaaagaatccccaagagggcctgtacaacgagctccaaaaggataagatggcagaa gcctatagcgagattggtatgaaaggggaacgcagaagaggcaaaggccacgacgg actgtaccagggactcagcaccgccaccaaggacacctatgacgctcttcacatgc aggccctgccgcctcgg 104879 35 MALPVTALLLPLALLLHAARPeivmtqspatlslspgeratlscrasqdiskylnw CAR5- yqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcqqg Full-aa ntlpytfgqgtkleikggggsggggsggggsggggsqvqlqesgpglvkpsetlsl tctvsgvslpdygvswirqppgkglewigviwgsettyyssslksrvtiskdnskn qvslklssvtaadtavyycakhyyyggsyamdywgqgtivtvsstttpaprpptpa ptiasqplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitly ckrgrkkllyifkqpfmrpvqttgeedgcscrfpeeeeggcelrvkfsrsadapay kqgqnqlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkmae ayseigmkgerrrgkghdglyqglstatkdtydalhmqalppr CAR 6 CAR6 6 eivmtqspatlslspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhs scFv giparfsgsgsgtdytltisslqpedfavyfcqqgntlpytfgqgtkleikggggs domain ggggsggggsggggsqvqlqesgpglvkpsetlsltctvsgvslpdygvswirqpp gkglewigviwgsettyyqsslksrvtiskdnsknqvslklssvtaadtavyycak hyyyggsyamdywgqgtlvtvss 99790 66 atggccctcccagtgaccgctctgctgctgcctctcgcacttcttctccatgccgc CAR6- tcggcctgagatcgtcatgacccaaagccccgctaccctgtccctgtcacccggcg Soluble agagggcaaccctttcatgcagggccagccaggacatttctaagtacctcaactgg scFv-nt tatcagcagaagccagggcaggctcctcgcctgctgatctaccacaccagccgcct ccacagcggtatccccgccagattttccgggagcgggtctggaaccgactacaccc tcaccatctcttctctgcagcccgaggatttcgccgtctatttctgccagcagggg aatactctgccgtacaccttcggtcaaggtaccaagctggaaatcaagggaggcgg aggatcaggcggtggcggaagcggaggaggtggctccggaggaggaggttcccaag tgcagcttcaagaatcaggacccggacttgtgaagccatcagaaaccctctccctg acttgtaccgtgtccggtgtgagcctccccgactacggagtctcttggattcgcca gcctccggggaagggtcttgaatggattggggtgatttggggatcagagactactt actaccagtcatcacttaagtcacgggtcaccatcagcaaagataatagcaagaac caagtgtcacttaagctgtcatctgtgaccgccgctgacaccgccgtgtactattg tgccaaacattactattacggagggtcttatgctatggactactggggacagggga ccctggtgactgtctctagccatcaccatcaccaccatcatcac 99790 78 MALPVTALLLPLALLLHAARPeivmtqspatlslspgeratlscrasqdiskylnw CAR6- yqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcqqg Soluble ntlpytfgqgtkleikggggsggggsggggsggggsqvqlqesgpglvkpsetlsl scFv-aa tctvsgvslpdygvswirqppgkglewigviwgsettyyqsslksrvtiskdnskn qvslklssvtaadtavyycakhyyyggsyamdywgqgtlvtvsshhhhhhhh 104880 90 atggccctccctgtcaccgccctgctgcttccgctggctcttctgctccacgccgc CAR6- tcggcccgaaattgtgatgacccagtcacccgccactcttagcctttcacccggtg Full-nt agcgcgcaaccctgtcttgcagagcctcccaagacatctcaaaataccttaattgg tatcaacagaagcccggacaggctcctcgccttctgatctaccacaccagccggct ccattctggaatccctgccaggttcagcggtagcggatctgggaccgactacaccc tcactatcagctcactgcagccagaggacttcgctgtctatttctgtcagcaaggg aacaccctgccctacacctttggacagggcaccaagctcgagattaaaggtggagg tggcagcggaggaggtgggtccggcggtggaggaagcggaggcggagggagccagg tccaactccaagaaagcggaccgggtcttgtgaagccatcagaaactctttcactg acttgtactgtgagcggagtgtctctccccgattacggggtgtcttggatcagaca gccaccggggaagggtctggaatggattggagtgatttggggctctgagactactt actaccaatcatccctcaagtcacgcgtcaccatctcaaaggacaactctaagaat caggtgtcactgaaactgtcatctgtgaccgcagccgacaccgccgtgtactattg cgctaagcattactattatggcgggagctacgcaatggattactggggacagggta ctctggtcaccgtgtccagcaccactaccccagcaccgaggccacccaccccggct cctaccatcgcctcccagcctctgtccctgcgtccggaggcatgtagacccgcagc tggtggggccgtgcatacccggggtcttgacttcgcctgcgatatctacatttggg cccctctggctggtacttgcggggtcctgctgctttcactcgtgatcactctttac tgtaagcgcggtcggaagaagctgctgtacatctttaagcaacccttcatgaggcc tgtgcagactactcaagaggaggacggctgttcatgccggttcccagaggaggagg aaggcggctgcgaactgcgcgtgaaattcagccgcagcgcagatgctccagcctac aagcaggggcagaaccagctctacaacgaactcaatcttggtcggagagaggagta cgacgtgctggacaagcggagaggacgggacccagaaatgggcgggaagccgcgca gaaagaatccccaagagggcctgtacaacgagctccaaaaggataagatggcagaa gcctatagcgagattggtatgaaaggggaacgcagaagaggcaaaggccacgacgg actgtaccagggactcagcaccgccaccaaggacacctatgacgctcttcacatgc aggccctgccgcctcgg 104880 36 MALPVTALLLPLALLLHAARPeivmtqspatlslspgeratlscrasgdiskylnw CAR6- yqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcqqg Full-aa ntlpytfgqgtkleikggggsggggsggggsggggsqvqlqesgpglvkpsetlsl tctvsgvslpdygvswirqppgkglewigviwgsettyygsslksrvtiskdnskn qvslklssvtaadtavyycakhyyyggsyamdywgqgtlvtvsstttpaprpptpa ptiasqplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitly ckrgrkkllyifkqpfmrpvqttqeedgcscrfpeeeeggcelrvkfsrsadapay kqgqnqlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkmae ayseigmkgerrrgkghdglyqglstatkdtydalhmqalppr CAR 7 CAR7 scFv 7 qvqlqesgpglvkpsetlsltctvsgvslpdygvswirqppgkglewigviwgset domain tyyssslksrvtiskdnsknqvslklssvtaadtavyycakhyyyggsyamdywgq gtivtvssggggsggggsggggsggggseivmtqspatlslspgeratlscrasqd iskylnwyqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfa vyfcqqgntlpytfgqgtkleik 100796 67 atggcactgcctgtcactgccctcctgctgcctctggccctccttctgcatgccgc CAR7- caggccccaagtccagctgcaagagtcaggacccggactggtgaagccgtctgaga Soluble ctctctcactgacttgtaccgtcagcggcgtgtccctccccgactacggagtgtca scFv-nt tggatccgccaacctcccgggaaagggcttgaatggattggtgtcatctggggttc tgaaaccacctactactcatcttccctgaagtccagggtgaccatcagcaaggata attccaagaaccaggtcagccttaagctgtcatctgtgaccgctgctgacaccgcc gtgtattactgcgccaagcactactattacggaggaagctacgctatggactattg gggacagggcactctcgtgactgtgagcagcggcggtggagggtctggaggtggag gatccggtggtggtgggtcaggcggaggagggagcgagattgtgatgactcagtca ccagccaccctttctctttcacccggcgagagagcaaccctgagctgtagagccag ccaggacatttctaagtacctcaactggtatcagcaaaaaccggggcaggcccctc gcctcctgatctaccatacctcacgccttcactctggtatccccgctcggtttagc ggatcaggatctggtaccgactacactctgaccatttccagcctgcagccagaaga tttcgcagtgtatttctgccagcagggcaatacccttccttacaccttcggtcagg gaaccaagctcgaaatcaagcaccatcaccatcatcaccaccat 100796 79 MALPVTALLLPLALLLHAARPqvqlqesgpglvkpsetlsltctvsgvslpdygvs CAR7- wirqppgkglewigviwgsettyyssslksrvtiskdnsknqvslklssvtaadta Soluble vyycakhyyyggsyamdywgqgtlvtvssggggsggggsggggsggggseivmtqs scFv-aa patlslspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhsgiparfs gsgsgtdytltisslqpedfavyfcqqgntlpytfgqgtkleikhhhhhhhh 104881 91 atggctctgcccgtgaccgcactcctcctgccactggctctgctgcttcacgccgc CAR 7 tcgcccacaagtccagcttcaagaatcagggcctggtctggtgaagccatctgaga Full-nt ctctgtccctcacttgcaccgtgagcggagtgtccctcccagactacggagtgagc tggattagacagcctcccggaaagggactggagtggatcggagtgatttggggtag cgaaaccacttactattcatcttccctgaagtcacgggtcaccatttcaaaggata actcaaagaatcaagtgagcctcaagctctcatcagtcaccgccgctgacaccgcc gtgtattactgtgccaagcattactactatggagggtcctacgccatggactactg gggccagggaactctggtcactgtgtcatctggtggaggaggtagcggaggaggcg ggagcggtggaggtggctccggaggtggcggaagcgaaatcgtgatgacccagagc cctgcaaccctgtccctttctcccggggaacgggctaccctttcttgtcgggcatc acaagatatctcaaaatacctcaattggtatcaacagaagccgggacaggccccta ggcttcttatctaccacacctctcgcctgcatagcgggattcccgcacgctttagc gggtctggaagcgggaccgactacactctgaccatctcatctctccagcccgagga cttcgccgtctacttctgccagcagggtaacaccctgccgtacaccttcggccagg gcaccaagcttgagatcaaaaccactactcccgctccaaggccacccacccctgcc ccgaccatcgcctctcagccgctttccctgcgtccggaggcatgtagacccgcagc tggtggggccgtgcatacccggggtcttgacttcgcctgcgatatctacatttggg cccctctggctggtacttgcggggtcctgctgctttcactcgtgatcactctttac tgtaagcgcggtcggaagaagctgctgtacatctttaagcaacccttcatgaggcc tgtgcagactactcaagaggaggacggctgttcatgccggttcccagaggaggagg aaggcggctgcgaactgcgcgtgaaattcagccgcagcgcagatgctccagcctac aagcaggggcagaaccagctctacaacgaactcaatcttggtcggagagaggagta cgacgtgctggacaagcggagaggacgggacccagaaatgggcgggaagccgcgca gaaagaatccccaagagggcctgtacaacgagctccaaaaggataagatggcagaa gcctatagcgagattggtatgaaaggggaacgcagaagaggcaaaggccacgacgg actgtaccagggactcagcaccgccaccaaggacacctatgacgctcttcacatgc aggccctgccgcctcgg 104881 37 MALPVTALLLPLALLLHAARPqvqlqesgpglvkpsetlsltctvsgvslpdygvs CAR 7 wirqppgkglewigviwgsettyyssslksrvtiskdnsknqvslklssvtaadta Full-aa vyycakhyyyggsyamdywgqgtivtvssggggsggggsggggsggggseivmtqs patlslspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhsgiparfs gsgsgtdytltisslqpedfavyfcqqgntlpytfgqgtkleiktttpaprpptpa ptiasqplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitly ckrgrkkllyifkqpfmrpvqttgeedgcscrfpeeeeggcelrvkfsrsadapay kqgqnqlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkmae ayseigmkgerrrgkghdglyqglstatkdtydalhmqalppr CAR 8 CAR8 scFv 8 qvqlqesgpglvkpsetlsltctvsgvslpdygvswirqppgkglewigviwgset domain tyyqsslksrvtiskdnsknqvslklssvtaadtavyycakhyyyggsyamdywgq gtivtvssggggsggggsggggsggggseivmtqspatlslspgeratlscrasqd iskylnwyqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfa vyfcqqgntlpytfgqgtkleik 100798 68 atggcactgcctgtcactgccctcctgctgcctctggccctccttctgcatgccgc CAR8- caggccccaagtccagctgcaagagtcaggacccggactggtgaagccgtctgaga Soluble ctctctcactgacttgtaccgtcagcggcgtgtccctccccgactacggagtgtca scFv-nt tggatccgccaacctcccgggaaagggcttgaatggattggtgtcatctggggttc tgaaaccacctactaccagtcttccctgaagtccagggtgaccatcagcaaggata attccaagaaccaggtcagccttaagctgtcatctgtgaccgctgctgacaccgcc gtgtattactgcgccaagcactactattacggaggaagctacgctatggactattg gggacagggcactctcgtgactgtgagcagcggcggtggagggtctggaggtggag gatccggtggtggtgggtcaggcggaggagggagcgagattgtgatgactcagtca ccagccaccctttctctttcacccggcgagagagcaaccctgagctgtagagccag ccaggacatttctaagtacctcaactggtatcagcaaaaaccggggcaggcccctc gcctcctgatctaccatacctcacgccttcactctggtatccccgctcggtttagc ggatcaggatctggtaccgactacactctgaccatttccagcctgcagccagaaga tttcgcagtgtatttctgccagcagggcaatacccttccttacaccttcggtcagg gaaccaagctcgaaatcaagcaccatcaccatcatcatcaccac 100798 80 MALPVTALLLPLALLLHAARPqvqlqesgpglvkpsetlsltctvsgvslpdygvs CAR8- wirqppgkglewigviwgsettyyqsslksrvtiskdnsknqvslklssvtaadta Soluble vyycakhyyyggsyamdywgqgtivtvssggggsggggsggggsggggseivmtqs scFv-aa patlslspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhsgiparfs
gsgsgtdytltisslqpedfavyfcqqgntlpytfgqgtkleikhhhhhhhh 104882 92 atggctctgcccgtgaccgcactcctcctgccactggctctgctgcttcacgccgc CAR 8- tcgcccacaagtccagcttcaagaatcagggcctggtctggtgaagccatctgaga Full-nt ctctgtccctcacttgcaccgtgagcggagtgtccctcccagactacggagtgagc tggattagacagcctcccggaaagggactggagtggatcggagtgatttggggtag cgaaaccacttactatcaatcttccctgaagtcacgggtcaccatttcaaaggata actcaaagaatcaagtgagcctcaagctctcatcagtcaccgccgctgacaccgcc gtgtattactgtgccaagcattactactatggagggtcctacgccatggactactg gggccagggaactctggtcactgtgtcatctggtggaggaggtagcggaggaggcg ggagcggtggaggtggctccggaggcggtgggtcagaaatcgtgatgacccagagc cctgcaaccctgtccctttctcccggggaacgggctaccctttcttgtcgggcatc acaagatatctcaaaatacctcaattggtatcaacagaagccgggacaggccccta ggcttcttatctaccacacctctcgcctgcatagcgggattcccgcacgctttagc gggtctggaagcgggaccgactacactctgaccatctcatctctccagcccgagga cttcgccgtctacttctgccagcagggtaacaccctgccgtacaccttcggccagg gcaccaagcttgagatcaaaaccactactcccgctccaaggccacccacccctgcc ccgaccatcgcctctcagccgctttccctgcgtccggaggcatgtagacccgcagc tggtggggccgtgcatacccggggtcttgacttcgcctgcgatatctacatttggg cccctctggctggtacttgcggggtcctgctgctttcactcgtgatcactctttac tgtaagcgcggtcggaagaagctgctgtacatctttaagcaacccttcatgaggcc tgtgcagactactcaagaggaggacggctgttcatgccggttcccagaggaggagg aaggcggctgcgaactgcgcgtgaaattcagccgcagcgcagatgctccagcctac aagcaggggcagaaccagctctacaacgaactcaatcttggtcggagagaggagta cgacgtgctggacaagcggagaggacgggacccagaaatgggcgggaagccgcgca gaaagaatccccaagagggcctgtacaacgagctccaaaaggataagatggcagaa gcctatagcgagattggtatgaaaggggaacgcagaagaggcaaaggccacgacgg actgtaccagggactcagcaccgccaccaaggacacctatgacgctcttcacatgc aggccctgccgcctcgg 104882 38 MALPVTALLLPLALLLHAARPqvqlqesgpglvkpsetlsltctvsgvslpdygvs CAR 8- wirqppgkglewigviwgsettyygsslksrvtiskdnsknqvslklssvtaadta Full-aa vyycakhyyyggsyamdywgqgtlvtvssggggsggggsggggsggggseivmtqs patlslspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhsgiparfs gsgsgtdytltisslqpedfavyfcqqgntlpytfgqgtkleiktttpaprpptpa ptiasqplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitly ckrgrkkllyifkqpfmrpvqttgeedgcscrfpeeeeggcelrvkfsrsadapay kqgqnqlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkmae ayseigmkgerrrgkghdglyqglstatkdtydalhmqalppr CAR 9 CAR9 scFv 9 eivmtqspatlslspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhs domain giparfsgsgsgtdytltisslqpedfavyfcqqgntlpytfgqgtkleikggggs ggggsggggsggggsqvqlqesgpglvkpsetlsltctvsgvslpdygvswirqpp gkglewigviwgsettyynsslksrvtiskdnsknqvslklssvtaadtavyycak hyyyggsyamdywgqgtlvtvss 99789 69 atggccctcccagtgaccgctctgctgctgcctctcgcacttcttctccatgccgc CAR9- tcggcctgagatcgtcatgacccaaagccccgctaccctgtccctgtcacccggcg Soluble agagggcaaccctttcatgcagggccagccaggacatttctaagtacctcaactgg scFv-nt tatcagcagaagccagggcaggctcctcgcctgctgatctaccacaccagccgcct ccacagcggtatccccgccagattttccgggagcgggtctggaaccgactacaccc tcaccatctcttctctgcagcccgaggatttcgccgtctatttctgccagcagggg aatactctgccgtacaccttcggtcaaggtaccaagctggaaatcaagggaggcgg aggatcaggcggtggcggaagcggaggaggtggctccggaggaggaggttcccaag tgcagcttcaagaatcaggacccggacttgtgaagccatcagaaaccctctccctg acttgtaccgtgtccggtgtgagcctccccgactacggagtctcttggattcgcca gcctccggggaagggtcttgaatggattggggtgatttggggatcagagactactt actacaattcatcacttaagtcacgggtcaccatcagcaaagataatagcaagaac caagtgtcacttaagctgtcatctgtgaccgccgctgacaccgccgtgtactattg tgccaaacattactattacggagggtcttatgctatggactactggggacagggga ccctggtgactgtctctagccatcaccatcaccaccatcatcac 99789 81 MALPVTALLLPLALLLHAARPeivmtqspatlslspgeratlscrasqdiskylnw CAR9- yqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcqqg Soluble ntlpytfgqgtkleikggggsggggsggggsggggsqvqlqesgpglvkpsetlsl scFv-aa tctvsgvslpdygvswirqppgkglewigviwgsettyynsslksrvtiskdnskn qvslklssvtaadtavyycakhyyyggsyamdywgqgtlvtvsshhhhhhhh 105974 93 atggccctccctgtcaccgccctgctgcttccgctggctcttctgctccacgccgc CAR 9- tcggcccgaaattgtgatgacccagtcacccgccactcttagcctttcacccggtg Full-nt agcgcgcaaccctgtcttgcagagcctcccaagacatctcaaaataccttaattgg tatcaacagaagcccggacaggctcctcgccttctgatctaccacaccagccggct ccattctggaatccctgccaggttcagcggtagcggatctgggaccgactacaccc tcactatcagctcactgcagccagaggacttcgctgtctatttctgtcagcaaggg aacaccctgccctacacctttggacagggcaccaagctcgagattaaaggtggagg tggcagcggaggaggtgggtccggcggtggaggaagcggaggcggtgggagccagg tccaactccaagaaagcggaccgggtcttgtgaagccatcagaaactctttcactg acttgtactgtgagcggagtgtctctccccgattacggggtgtcttggatcagaca gccaccggggaagggtctggaatggattggagtgatttggggctctgagactactt actacaactcatccctcaagtcacgcgtcaccatctcaaaggacaactctaagaat caggtgtcactgaaactgtcatctgtgaccgcagccgacaccgccgtgtactattg cgctaagcattactattatggcgggagctacgcaatggattactggggacagggta ctctggtcaccgtgtccagcaccactaccccagcaccgaggccacccaccccggct cctaccatcgcctcccagcctctgtccctgcgtccggaggcatgtagacccgcagc tggtggggccgtgcatacccggggtcttgacttcgcctgcgatatctacatttggg cccctctggctggtacttgcggggtcctgctgctttcactcgtgatcactctttac tgtaagcgcggtcggaagaagctgctgtacatctttaagcaacccttcatgaggcc tgtgcagactactcaagaggaggacggctgttcatgccggttcccagaggaggagg aaggcggctgcgaactgcgcgtgaaattcagccgcagcgcagatgctccagcctac aagcaggggcagaaccagctctacaacgaactcaatcttggtcggagagaggagta cgacgtgctggacaagcggagaggacgggacccagaaatgggcgggaagccgcgca gaaagaatccccaagagggcctgtacaacgagctccaaaaggataagatggcagaa gcctatagcgagattggtatgaaaggggaacgcagaagaggcaaaggccacgacgg actgtaccagggactcagcaccgccaccaaggacacctatgacgctcttcacatgc aggccctgccgcctcgg 105974 39 MALPVTALLLPLALLLHAARPeivmtqspatlslspgeratlscrasqdiskylnw CAR 9- yqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcqqg Full-aa ntlpytfgqgtkleikggggsggggsggggsggggsqvqlqesgpglvkpsetlsl tctvsgvslpdygvswirqppgkglewigviwgsettyynsslksrvtiskdnskn qvslklssvtaadtavyycakhyyyggsyamdywgqgtivtvsstttpaprpptpa ptiasqplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitly ckrgrkkllyifkqpfmrpvqttgeedgcscrfpeeeeggcelrvkfsrsadapay kqgqnqlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkmae ayseigmkgerrrgkghdglyqglstatkdtydalhmqalppr CAR10 CAR10 10 qvqlqesgpglvkpsetlsltctvsgvslpdygvswirqppgkglewigviwgset scFv tyynsslksrvtiskdnsknqvslklssvtaadtavyycakhyyyggsyamdywgq domain gtlvtvssggggsggggsggggsggggseivmtqspatlslspgeratlscrasqd iskylnwyqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfa vyfcqqgntlpytfgqgtkleik 100796 70 atggcactgcctgtcactgccctcctgctgcctctggccctccttctgcatgccgc CAR10- caggccccaagtccagctgcaagagtcaggacccggactggtgaagccgtctgaga Soluble ctctctcactgacttgtaccgtcagcggcgtgtccctccccgactacggagtgtca scFv-nt tggatccgccaacctcccgggaaagggcttgaatggattggtgtcatctggggttc tgaaaccacctactacaactcttccctgaagtccagggtgaccatcagcaaggata attccaagaaccaggtcagccttaagctgtcatctgtgaccgctgctgacaccgcc gtgtattactgcgccaagcactactattacggaggaagctacgctatggactattg gggacagggcactctcgtgactgtgagcagcggcggtggagggtctggaggtggag gatccggtggtggtgggtcaggcggaggagggagcgagattgtgatgactcagtca ccagccaccctttctctttcacccggcgagagagcaaccctgagctgtagagccag ccaggacatttctaagtacctcaactggtatcagcaaaaaccggggcaggcccctc gcctcctgatctaccatacctcacgccttcactctggtatccccgctcggtttagc ggatcaggatctggtaccgactacactctgaccatttccagcctgcagccagaaga tttcgcagtgtatttctgccagcagggcaatacccttccttacaccttcggtcagg gaaccaagctcgaaatcaagcaccatcaccatcatcaccaccat 100796 82 MALPVTALLLPLALLLHAARPqvqlqesgpglvkpsetlsltctvsgvslpdygvs CAR10- wirqppgkglewigviwgsettyynsslksrvtiskdnsknqvslklssvtaadta Soluble vyycakhyyyggsyamdywgqgtlvtvssggggsggggsggggsggggseivmtqs scFv-aa patlslspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhsgiparfs gsgsgtdytltisslqpedfavyfcqqgntlpytfgqgtkleikhhhhhhhh 105975 94 atggccctccctgtcaccgccctgctgcttccgctggctcttctgctccacgccgc CAR 10 tcggcccgaaattgtgatgacccagtcacccgccactcttagcctttcacccggtg Full-nt agcgcgcaaccctgtcttgcagagcctcccaagacatctcaaaataccttaattgg tatcaacagaagcccggacaggctcctcgccttctgatctaccacaccagccggct ccattctggaatccctgccaggttcagcggtagcggatctgggaccgactacaccc tcactatcagctcactgcagccagaggacttcgctgtctatttctgtcagcaaggg aacaccctgccctacacctttggacagggcaccaagctcgagattaaaggtggagg tggcagcggaggaggtgggtccggcggtggaggaagcggaggcggtgggagccagg tccaactccaagaaagcggaccgggtcttgtgaagccatcagaaactctttcactg acttgtactgtgagcggagtgtctctccccgattacggggtgtcttggatcagaca gccaccggggaagggtctggaatggattggagtgatttggggctctgagactactt actacaactcatccctcaagtcacgcgtcaccatctcaaaggacaactctaagaat caggtgtcactgaaactgtcatctgtgaccgcagccgacaccgccgtgtactattg cgctaagcattactattatggcgggagctacgcaatggattactggggacagggta ctctggtcaccgtgtccagcaccactaccccagcaccgaggccacccaccccggct cctaccatcgcctcccagcctctgtccctgcgtccggaggcatgtagacccgcagc tggtggggccgtgcatacccggggtcttgacttcgcctgcgatatctacatttggg cccctctggctggtacttgcggggtcctgctgctttcactcgtgatcactctttac tgtaagcgcggtcggaagaagctgctgtacatctttaagcaacccttcatgaggcc tgtgcagactactcaagaggaggacggctgttcatgccggttcccagaggaggagg aaggcggctgcgaactgcgcgtgaaattcagccgcagcgcagatgctccagcctac aagcaggggcagaaccagctctacaacgaactcaatcttggtcggagagaggagta cgacgtgctggacaagcggagaggacgggacccagaaatgggcgggaagccgcgca gaaagaatccccaagagggcctgtacaacgagctccaaaaggataagatggcagaa gcctatagcgagattggtatgaaaggggaacgcagaagaggcaaaggccacgacgg actgtaccagggactcagcaccgccaccaaggacacctatgacgctcttcacatgc aggccctgccgcctcgg 105975 40 MALPVTALLLPLALLLHAARPEIVMTQSPATLSLSPGERATLSCRASQDISKYLNW CAR 10 YQQKPGQAPRLLIYHTSRLHSGIPARFSGSGSGTDYTLTISSLQPEDFAVYFCQQG Full-aa NTLPYTFGQGTKLEIKGGGGSGGGGSGGGGSGGGGSQVQLQESGPGLVKPSETLSL TCTVSGVSLPDYGVSWIRQPPGKGLEWIGVIWGSETTYYNSSLKSRVTISKDNSKN QVSLKLSSVTAADTAVYYCAKHYYYGGSYAMDYWGQGTLVTVSSTTTPAPRPPTPA PTIASQPLSLRPEACRPAAGGAVHTRGLDFACDIYIWAPLAGTCGVLLLSLVITLY CKRGRKKLLYIFKQPFMRPVQTTQEEDGCSCRFPEEEEGGCELRVKFSRSADAPAY KQGQNQLYNELNLGRREEYDVLDKRRGRDPEMGGKPRRKNPQEGLYNELQKDKMAE AYSEIGMKGERRRGKGHDGLYQGLSTATKDTYDALHMQALPPR CAR11 CAR11 11 eivmtqspatlslspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhs scFv giparfsgsgsgtdytltisslqpedfavyfcqqgntlpytfgqgtkleikggggs domain ggggsggggsqvqlqesgpglvkpsetlsltctvsgvslpdygvswirqppgkgle wigviwgsettyynsslksrvtiskdnsknqvslklssvtaadtavyycakhyyyg gsyamdywgqgtlvtvss 103101 71 Atggccctccctgtcaccgccctgctgcttccgctggctcttctgctccacgccgc CAR11- tcggcccgaaattgtgatgacccagtcacccgccactcttagcctttcacccggtg Soluble agcgcgcaaccctgtcttgcagagcctcccaagacatctcaaaataccttaattgg scFv-nt tatcaacagaagcccggacaggctcctcgccttctgatctaccacaccagccggct ccattctggaatccctgccaggttcagcggtagcggatctgggaccgactacaccc tcactatcagctcactgcagccagaggacttcgctgtctatttctgtcagcaaggg aacaccctgccctacacctttggacagggcaccaagctcgagattaaaggtggagg tggcagcggaggaggtgggtccggcggtggaggaagccaggtccaactccaagaaa gcggaccgggtcttgtgaagccatcagaaactctttcactgacttgtactgtgagc ggagtgtctctccccgattacggggtgtcttggatcagacagccaccggggaaggg tctggaatggattggagtgatttggggctctgagactacttactacaattcatccc tcaagtcacgcgtcaccatctcaaaggacaactctaagaatcaggtgtcactgaaa ctgtcatctgtgaccgcagccgacaccgccgtgtactattgcgctaagcattacta ttatggcgggagctacgcaatggattactggggacagggtactctggtcaccgtgt ccagccaccaccatcatcaccatcaccat 103101 83 MALPVTALLLPLALLLHAARPeivmtqspatlslspgeratlscrasqdiskylnw CAR11- yqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcqqg Soluble ntlpytfgqgtkleikggggsggggsggggsqvqlqesgpglvkpsetlsltctvs scFv-aa gvslpdygvswirqppgkglewigviwgsettyynsslksrvtiskdnsknqvslk lssvtaadtavyycakhyyyggsyamdywgqgtlvtvsshhhhhhhh 105976 95 atggctctgcccgtgaccgcactcctcctgccactggctctgctgcttcacgccgc CAR 11 tcgcccacaagtccagcttcaagaatcagggcctggtctggtgaagccatctgaga Full-nt ctctgtccctcacttgcaccgtgagcggagtgtccctcccagactacggagtgagc tggattagacagcctcccggaaagggactggagtggatcggagtgatttggggtag cgaaaccacttactataactcttccctgaagtcacgggtcaccatttcaaaggata actcaaagaatcaagtgagcctcaagctctcatcagtcaccgccgctgacaccgcc gtgtattactgtgccaagcattactactatggagggtcctacgccatggactactg gggccagggaactctggtcactgtgtcatctggtggaggaggtagcggaggaggcg ggagcggtggaggtggctccggaggtggcggaagcgaaatcgtgatgacccagagc cctgcaaccctgtccctttctcccggggaacgggctaccctttcttgtcgggcatc acaagatatctcaaaatacctcaattggtatcaacagaagccgggacaggccccta ggcttcttatctaccacacctctcgcctgcatagcgggattcccgcacgctttagc gggtctggaagcgggaccgactacactctgaccatctcatctctccagcccgagga cttcgccgtctacttctgccagcagggtaacaccctgccgtacaccttcggccagg gcaccaagcttgagatcaaaaccactactcccgctccaaggccacccacccctgcc ccgaccatcgcctctcagccgctttccctgcgtccggaggcatgtagacccgcagc tggtggggccgtgcatacccggggtcttgacttcgcctgcgatatctacatttggg cccctctggctggtacttgcggggtcctgctgctttcactcgtgatcactctttac tgtaagcgcggtcggaagaagctgctgtacatctttaagcaacccttcatgaggcc tgtgcagactactcaagaggaggacggctgttcatgccggttcccagaggaggagg aaggcggctgcgaactgcgcgtgaaattcagccgcagcgcagatgctccagcctac aagcaggggcagaaccagctctacaacgaactcaatcttggtcggagagaggagta cgacgtgctggacaagcggagaggacgggacccagaaatgggcgggaagccgcgca gaaagaatccccaagagggcctgtacaacgagctccaaaaggataagatggcagaa gcctatagcgagattggtatgaaaggggaacgcagaagaggcaaaggccacgacgg actgtaccagggactcagcaccgccaccaaggacacctatgacgctcttcacatgc aggccctgccgcctcgg 105976 41 MALPVTALLLPLALLLHAARPQVQLQESGPGLVKPSETLSLTCTVSGVSLPDYGVS CAR 11 WIRQPPGKGLEWIGVIWGSETTYYNSSLKSRVTISKDNSKNQVSLKLSSVTAADTA Full-aa VYYCAKHYYYGGSYAMDYWGQGTLVTVSSGGGGSGGGGSGGGGSGGGGSEIVMTQS PATLSLSPGERATLSCRASQDISKYLNWYQQKPGQAPRLLIYHTSRLHSGIPARFS GSGSGTDYTLTISSLQPEDFAVYFCQQGNTLPYTFGQGTKLEIKTTTPAPRPPTPA PTIASQPLSLRPEACRPAAGGAVHTRGLDFACDIYIWAPLAGTCGVLLLSLVITLY CKRGRKKLLYIFKQPFMRPVQTTQEEDGCSCRFPEEEEGGCELRVKFSRSADAPAY KQGQNQLYNELNLGRREEYDVLDKRRGRDPEMGGKPRRKNPQEGLYNELQKDKMAE AYSEIGMKGERRRGKGHDGLYQGLSTATKDTYDALHMQALPPR CAR12 CAR12 12 qvqlqesgpglvkpsetlsltctvsgvslpdygvswirqppgkglewigviwgset scFv tyynsslksrvtiskdnsknqvslklssvtaadtavyycakhyyyggsyamdywgq domain gtlvtvssggggsggggsggggseivmtqspatlslspgeratlscrasqdiskyl nwyqqkpgqaprlliyhtsrlhsgiparfsgsgsgtdytltisslqpedfavyfcq qgntlpytfgqgtkleik 103104 72 atggctctgcccgtgaccgcactcctcctgccactggctctgctgcttcacgccgc CAR12- tcgcccacaagtccagcttcaagaatcagggcctggtctggtgaagccatctgaga Soluble ctctgtccctcacttgcaccgtgagcggagtgtccctcccagactacggagtgagc
scFv-nt tggattagacagcctcccggaaagggactggagtggatcggagtgatttggggtag cgaaaccacttactataactcttccctgaagtcacgggtcaccatttcaaaggata actcaaagaatcaagtgagcctcaagctctcatcagtcaccgccgctgacaccgcc gtgtattactgtgccaagcattactactatggagggtcctacgccatggactactg gggccagggaactctggtcactgtgtcatctggtggaggaggtagcggaggaggcg ggagcggtggaggtggctccgaaatcgtgatgacccagagccctgcaaccctgtcc ctttctcccggggaacgggctaccctttcttgtcgggcatcacaagatatctcaaa atacctcaattggtatcaacagaagccgggacaggcccctaggcttcttatctacc acacctctcgcctgcatagcgggattcccgcacgctttagcgggtctggaagcggg accgactacactctgaccatctcatctctccagcccgaggacttcgccgtctactt ctgccagcagggtaacaccctgccgtacaccttcggccagggcaccaagcttgaga tcaaacatcaccaccatcatcaccatcac 103104 84 MALPVTALLLPLALLLHAARPqvqlqesgpglvkpsetlsltctvsgvslpdygvs CAR12- wirqppgkglewigviwgsettyynsslksrvtiskdnsknqvslklssvtaadta Soluble vyycakhyyyggsyamdywgqgtlvtvssggggsggggsggggseivmtqspatls scFv-aa lspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhsgiparfsgsgsg tdytltisslqpedfavyfcqqgntlpytfgqgtkleikhhhhhhhh 105977 96 atggccctccctgtcaccgccctgctgcttccgctggctcttctgctccacgccgc CAR 12- tcggcccgaaattgtgatgacccagtcacccgccactcttagcctttcacccggtg Full-nt agcgcgcaaccctgtcttgcagagcctcccaagacatctcaaaataccttaattgg tatcaacagaagcccggacaggctcctcgccttctgatctaccacaccagccggct ccattctggaatccctgccaggttcagcggtagcggatctgggaccgactacaccc tcactatcagctcactgcagccagaggacttcgctgtctatttctgtcagcaaggg aacaccctgccctacacctttggacagggcaccaagctcgagattaaaggtggagg tggcagcggaggaggtgggtccggcggtggaggaagccaggtccaactccaagaaa gcggaccgggtcttgtgaagccatcagaaactctttcactgacttgtactgtgagc ggagtgtctctccccgattacggggtgtcttggatcagacagccaccggggaaggg tctggaatggattggagtgatttggggctctgagactacttactacaactcatccc tcaagtcacgcgtcaccatctcaaaggacaactctaagaatcaggtgtcactgaaa ctgtcatctgtgaccgcagccgacaccgccgtgtactattgcgctaagcattacta ttatggcgggagctacgcaatggattactggggacagggtactctggtcaccgtgt ccagcaccactaccccagcaccgaggccacccaccccggctcctaccatcgcctcc cagcctctgtccctgcgtccggaggcatgtagacccgcagctggtggggccgtgca tacccggggtcttgacttcgcctgcgatatctacatttgggcccctctggctggta cttgcggggtcctgctgctttcactcgtgatcactctttactgtaagcgcggtcgg aagaagctgctgtacatctttaagcaacccttcatgaggcctgtgcagactactca agaggaggacggctgttcatgccggttcccagaggaggaggaaggcggctgcgaac tgcgcgtgaaattcagccgcagcgcagatgctccagcctacaagcaggggcagaac cagctctacaacgaactcaatcttggtcggagagaggagtacgacgtgctggacaa gcggagaggacgggacccagaaatgggcgggaagccgcgcagaaagaatccccaag agggcctgtacaacgagctccaaaaggataagatggcagaagcctatagcgagatt ggtatgaaaggggaacgcagaagaggcaaaggccacgacggactgtaccagggact cagcaccgccaccaaggacacctatgacgctcttcacatgcaggccctgccgcctc gg 105977 42 MALPVTALLLPLALLLHAARPEIVMTQSPATLSLSPGERATLSCRASQDISKYLNW CAR 12- YQQKPGQAPRLLIYHTSRLHSGIPARFSGSGSGTDYTLTISSLQPEDFAVYFCQQG Full-aa NTLPYTFGQGTKLEIKGGGGSGGGGSGGGGSQVQLQESGPGLVKPSETLSLTCTVS GVSLPDYGVSWIRQPPGKGLEWIGVIWGSETTYYNSSLKSRVTISKDMSKMQVSLK LSSVTAADTAVYYCAKHYYYGGSYAMDYWGQGTLVTVSSTTTPAPRPPTPAPTIAS QPLSLRPEACRPAAGGAVHTRGLDFACDIYIWAPLAGTCGVLLLSLVITLYCKRGR KKLLYIFKQPFMRPVQTTQEEDGCSCREPEEEEGGCELRVKFSRSADAPAYKQGQN QLYNELNLGRREEYDVLDKRRGRDPEMGGKPRRKMPQEGLYNELQKDKMAEAYSEI GMKGERRRGKGHDGLYQGLSTATKDTYDALHMQALPPR
TABLE-US-00018 TABLE 7 Murine CD19 CAR Constructs CTL019 CTL019- 97 atggccctgcccgtcaccgctctgctgctgccccttgctctgcttcttcatgcagc Soluble aaggccggacatccagatgacccaaaccacctcatccctctctgcctctcttggag scFv-Histag- acagggtgaccatttcttgtcgcgccagccaggacatcagcaagtatctgaactgg nt tatcagcagaagccggacggaaccgtgaagctcctgatctaccatacctctcgcct gcatagcggcgtgccctcacgcttctctggaagcggatcaggaaccgattattctc tcactatttcaaatcttgagcaggaagatattgccacctatttctgccagcagggt aataccctgccctacaccttcggaggagggaccaagctcgaaatcaccggtggagg aggcagcggcggtggagggtctggtggaggtggttctgaggtgaagctgcaagaat caggccctggacttgtggccccttcacagtccctgagcgtgacttgcaccgtgtcc ggagtctccctgcccgactacggagtgtcatggatcagacaacctccacggaaagg actggaatggctcggtgtcatctggggtagcgaaactacttactacaattcagccc tcaaaagcaggctgactattatcaaggacaacagcaagtcccaagtctttcttaag atgaactcactccagactgacgacaccgcaatctactattgtgctaagcactacta ctacggaggatcctacgctatggattactggggacaaggtacttccgtcactgtct cttcacaccatcatcaccatcaccatcac CTL019- 98 MALPVTALLLPLALLLHAARPdiqmtqttsslsaslgdrvtiscrasqdiskylnw Soluble yqqkpdgtvklliyhtsrlhsgvpsrfsgsgsgtdysltisnleqediatyfcqqg scFv-Histag- ntlpytfgggtkleitggggsggggsggggsevklqesgpglvapsqslsvtctvs aa gvslpdygvswirqpprkglewlgviwgsettyynsalksrltiikdnsksqvflk mnslqtddtaiyycakhyyyggsyamdywgqgtsvtvsshhhhhhhh CTL019 99 atggccttaccagtgaccgccttgctcctgccgctggccttgctgctccacgccgc Full-nt caggccggacatccagatgacacagactacatcctccctgtctgcctctctgggag acagagtcaccatcagttgcagggcaagtcaggacattagtaaatatttaaattgg tatcagcagaaaccagatggaactgttaaactcctgatctaccatacatcaagatt acactcaggagtcccatcaaggttcagtggcagtgggtctggaacagattattctc tcaccattagcaacctggagcaagaagatattgccacttacttttgccaacagggt aatacgcttccgtacacgttcggaggggggaccaagctggagatcacaggtggcgg tggctcgggcggtggtgggtcgggtggcggcggatctgaggtgaaactgcaggagt caggacctggcctggtggcgccctcacagagcctgtccgtcacatgcactgtctca ggggtctcattacccgactatggtgtaagctggattcgccagcctccacgaaaggg tctggagtggctgggagtaatatggggtagtgaaaccacatactataattcagctc tcaaatccagactgaccatcatcaaggacaactccaagagccaagttttcttaaaa atgaacagtctgcaaactgatgacacagccatttactactgtgccaaacattatta ctacggtggtagctatgctatggactactggggccaaggaacctcagtcaccgtct cctcaaccacgacgccagcgccgcgaccaccaacaccggcgcccaccatcgcgtcg cagcccctgtccctgcgcccagaggcgtgccggccagcggcggggggcgcagtgca cacgagggggctggacttcgcctgtgatatctacatctgggcgcccttggccggga cttgtggggtccttctcctgtcactggttatcaccctttactgcaaacggggcaga aagaaactcctgtatatattcaaacaaccatttatgagaccagtacaaactactca agaggaagatggctgtagctgccgatttccagaagaagaagaaggaggatgtgaac tgagagtgaagttcagcaggagcgcagacgcccccgcgtacaagcagggccagaac cagctctataacgagctcaatctaggacgaagagaggagtacgatgttttggacaa gagacgtggccgggaccctgagatggggggaaagccgagaaggaagaaccctcagg aaggcctgtacaatgaactgcagaaagataagatggcggaggcctacagtgagatt gggatgaaaggcgagcgccggaggggcaaggggcacgatggcctttaccagggtct cagtacagccaccaaggacacctacgacgcccttcacatgcaggccctgccccctc gc CTL019 58 MALPVTALLLPLALLLHAARPdiqmtqttsslsaslgdrvtiscrasqdiskylnw Full-aa yqqkpdgtvklliyhtsrlhsgvpsrfsgsgsgtdysltisnleqediatyfcqqg ntlpytfgggtkleitggggsggggsggggsevklqesgpglvapsqslsvtctvs gvslpdygvswirqpprkglewlgviwgsettyynsalksrltiikdnsksqvflk mnslqtddtaiyycakhyyyggsyamdywgqgtsvtvsstttpaprpptpaptias qplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitlyckrgr kkllyifkqpfmrpvqttqeedgcscrfpeeeeggcelrvkfsrsadapaykqgqn qlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkmaeaysei gmkgerrrgkghdglyqglstatkdtydalhmqalppr CTL019 59 diqmtqttsslsaslgdrvtiscrasqdiskylnwyqqkpdgtvklliyhtsrlhs scFv gvpsrfsgsgsgtdysltisnleqediatyfcqqgntlpytfgggtkleitggggs domain ggggsggggsevklqesgpglvapsqslsvtctvsgvslpdygvswirqpprkgle wlgviwgsettyynsalksrltiikdnsksqvflkmnslqtddtaiyycakhyyyg gsyamdywgqgtsvtvss
[0772] The sequences of humanized CDR sequences of the scFv domains are shown in Table 4 for the heavy chain variable domains and in Table 5 for the light chain variable domains. "ID" stands for the respective SEQ ID NO for each CDR.
TABLE-US-00019 TABLE 4 Heavy Chain Variable Domain CDRs (Kabat) Candidate FW HCDR1 ID HCDR2 ID HCDR3 ID murine_CART19 GVSLPDYGVS 19 VIWGSETTYYNSALKS 20 HYYYGGSYAMDY 24 humanized_CART19 a VH4 GVSLPDYGVS 19 VIWGSETTYY S LKS 21 HYYYGGSYAMDY 24 humanized_CART19 b VH4 GVSLPDYGVS 19 VIWGSETTYY S LKS 22 HYYYGGSYAMDY 24 humanized_CART19 c VH4 GVSLPDYGVS 19 VIWGSETTYYNS LKS 23 HYYYGGSYAMDY 24
TABLE-US-00020 TABLE 5 Light Chain Variable Domain CDRs Candidate FW LCDR1 ID LCDR2 ID LCDR3 ID murine_CART19 RASQDISKYLN 25 HTSRLHS 26 QQGNTLPYT 27 humanized_CART19 a VK3 RASQDISKYLN 25 HTSRLHS 26 QQGNTLPYT 27 humanized_CART19 b VK3 RASQDISKYLN 25 HTSRLHS 26 QQGNTLPYT 27 humanized_CART19 c VK3 RASQDISKYLN 25 HTSRLHS 26 QQGNTLPYT 27
[0773] Table 6 is an identification key correlating the CD19 constructs numerical names to the specific orientation of the light and heavy chains of the scFv, the number of linker units (i.e., (G4S).sub.3 (SEQ ID NO: 107) or (G4S).sub.4 (SEQ ID NO: 106)), separating the heavy and light chains, and the distinguishing amino acid sequences in the heavy chain CDR2.
TABLE-US-00021 TABLE 6 CD19 CAR designations. Clone Alt. Chain Site of Heavy SEQ ID ID/CAR# Clone ID Orientation Linkers CDR2 mutation NO 104875 C2136 L2H 3x YSSSL 28 (CAR1) 104876 C2137 L2H 3x YQSSL 29 (CAR2) 104877 C2138 H2L 3x YSSSL 28 (CAR3) 104878 C2139 H2L 3x YQSSL 29 (CAR4) 104879 C2140 L2H 4x YSSSL 28 (CAR5) 104880 C2141 L2H 4x YQSSL 29 (CAR6) 104881 C2142 H2L 4x YSSSL 28 (CAR7) 104882 C2143 H2L 4x YQSSL 29 (CAR8) 105974 C2144 L2H 4x YNSSL 30 (CAR9) 105975 C2145 H2L 4x YNSSL 30 (CAR10) 105976 C2146 L2H 3x YNSSL 30 (CAR11) 105977 C2147 H2L 3x YNSSL 30 (CAR12) CTL019 muCART19 L2H 3x YNSAL 57
[0774] The CAR scFv fragments were then cloned into lentiviral vectors to create a full length CAR construct in a single coding frame, and using the EF1 alpha promoter for expression (SEQ ID NO: 100).
TABLE-US-00022 EF1 alpha promoter (SEQ ID NO: 100) CGTGAGGCTCCGGTGCCCGTCAGTGGGCAGAGCGCACATCGCCCACAGTC CCCGAGAAGTTGGGGGGAGGGGTCGGCAATTGAACCGGTGCCTAGAGAAG GTGGCGCGGGGTAAACTGGGAAAGTGATGTCGTGTACTGGCTCCGCCTTT TTCCCGAGGGTGGGGGAGAACCGTATATAAGTGCAGTAGTCGCCGTGAAC GTTCTTTTTCGCAACGGGTTTGCCGCCAGAACACAGGTAAGTGCCGTGTG TGGTTCCCGCGGGCCTGGCCTCTTTACGGGTTATGGCCCTTGCGTGCCTT GAATTACTTCCACCTGGCTGCAGTACGTGATTCTTGATCCCGAGCTTCGG GTTGGAAGTGGGTGGGAGAGTTCGAGGCCTTGCGCTTAAGGAGCCCCTTC GCCTCGTGCTTGAGTTGAGGCCTGGCCTGGGCGCTGGGGCCGCCGCGTGC GAATCTGGTGGCACCTTCGCGCCTGTCTCGCTGCTTTCGATAAGTCTCTA GCCATTTAAAATTTTTGATGACCTGCTGCGACGCTTTTTTTCTGGCAAGA TAGTCTTGTAAATGCGGGCCAAGATCTGCACACTGGTATTTCGGTTTTTG GGGCCGCGGGCGGCGACGGGGCCCGTGCGTCCCAGCGCACATGTTCGGCG AGGCGGGGCCTGCGAGCGCGGCCACCGAGAATCGGACGGGGGTAGTCTCA AGCTGGCCGGCCTGCTCTGGTGCCTGGCCTCGCGCCGCCGTGTATCGCCC CGCCCTGGGCGGCAAGGCTGGCCCGGTCGGCACCAGTTGCGTGAGCGGAA AGATGGCCGCTTCCCGGCCCTGCTGCAGGGAGCTCAAAATGGAGGACGCG GCGCTCGGGAGAGCGGGCGGGTGAGTCACCCACACAAAGGAAAAGGGCCT TTCCGTCCTCAGCCGTCGCTTCATGTGACTCCACGGAGTACCGGGCGCCG TCCAGGCACCTCGATTAGTTCTCGAGCTTTTGGAGTACGTCGTCTTTAGG TTGGGGGGAGGGGTTTTATGCGATGGAGTTTCCCCACACTGAGTGGGTGG AGACTGAAGTTAGGCCAGCTTGGCACTTGATGTAATTCTCCTTGGAATTT GCCCTTTTTGAGTTTGGATCTTGGTTCATTCTCAAGCCTCAGACAGTGGT TCAAAGTTTTTTTCTTCCATTTCAGGTGTCGTGA.
Analysis of the humanized CAR constructs was conducted as described in Example 4.
Example 4: Analysis of Humanized CD19 Constructs in CART
[0775] To evaluate the feasibility of targeting CD19 via a CAR technology, the single chain variable fragments for an anti-CD19 antibody is cloned into a lentiviral CAR expression vector with the CD3zeta chain and the 4-1BB costimulatory molecule in four different configurations and the optimal construct is selected based on the quantity and quality of the effector T cell response of CD19 CAR transduced T cells ("CART19" or "CART19 T cells") in response to CD19+ targets. Effector T cell responses include, but are not limited to, cellular expansion, proliferation, doubling, cytokine production and target cell killing or cytolytic activity (degranulation).
Materials and Methods
Generation of Redirected Humanized CART19 T Cells
[0776] The humanized CART19 lentiviral transfer vectors are used to produce the genomic material packaged into the VSVg psuedotyped lentiviral particles. Lentiviral transfer vector DNA is mixed with the three packaging components of VSVg, gag/pol and rev in combination with lipofectamine reagent to transfect them together in to 293T cells. After 24 and 48 hr, the media is collected, filtered and concentrated by ultracentrifugation. The resulting viral preparation is stored at -80 C. The number of transducing units is determined by titration on SupT1 cells. Redirected CART19 T cells are produced by activating fresh naive T cells by engaging with CD3.times.28 beads for 24 hrs and then adding the appropriate number of transducing units to obtain the desired percentage of transduced T cells. These modified T cells are allowed to expand until they become rested and come down in size at which point they are cryopreserved for later analysis. The cell numbers and sizes are measured using a coulter multisizer III. Before cryopreserving, percentage of cells transduced (expressing the CART19 on the cell surface) and their relative fluorescence intensity of that expression are determined by flow cytometric analysis on an LSRII. From the histogram plots, the relative expression levels of the CARs can be examined by comparing percentage transduced with their relative fluorescent intensity.
Evaluating Cytolytic Activity, Proliferation Capabilities and Cytokine Secretion of Humanized CART19 Redirected T Cells.
[0777] To evaluate the functional abilities of humanized CAR19 T cells to kill, proliferate and secrete cytokines, the cells are thawed and allowed to recover overnight. In addition to the humanized CART19, the murine CART19 was used for comparative purposes while SS1-BBz was used as non-targeting expressed CAR for background CAR/T cell effect. The "control" gold standard (GS) CART19 was used in all assays to compare assay variation. Importantly, the GS CART19 are cells produced in research grade (i.e., not clinical grade) manufacturing conditions and include the addition of IL-2 to the growth culture. This likely impacts the overall viability and functionality of these cells and should not be evaluated as a direct comparison to the research grade production of the other transduced T cell populations. The T cell killing was directed towards K562, a chronic myelogenous leukemia cell line expressing or not expressing CD19 or Pt14, B cells isolated from CLL patients. For this flow based cytotoxicity assay, the target cells are stained with CSFE to quantitate their presence. The target cells were stained for CD19 expression to confirm similar target antigens levels. The cytolytic activities of CAR19 T cells are measured at a titration of effector:target cell ratios of 10:1, 3:1, 1:1, 0.3:1 and 0:1 where effectors were defined as T cells expressing the anti-CD19 chimeric receptor. Assays were initiated by mixing an appropriate number of T cells with a constant number of targets cells. After 16 hrs, total volume of each mixture was removed and each well washed combining appropriately. The T cells were stained for CD2 and all cells stained with live/dead marker 7AAD. After the final wash, the pelleted cells were re-suspended in a specific volume with a predetermined number of counting beads. Cell staining data was collected by LSRII flow cytometry and analyzed with FloJo software using beads to quantitate results.
[0778] For measuring cell proliferation and cytokine production of humanized CAR19 T cells, cells are thawed and allowed to recover overnight. In addition to the humanized CART19, the murine CART19 was used for comparative purposes while SS1-BBz was used as a non-targeting expressed CAR for background CAR/T cell effect. The "control" gold standard (GS) CART19 was used in all assays to compare assay variation. The T cells were directed either towards K562, a chronic myelogenous leukemia cell line expressing or not expressing CD19 or Pt14, B cells isolated from CLL patients. In addition, CD3.times.28 beads were used to evaluate the potential of T cells to respond to the endogenous immunological signals. To analyze proliferation, T cells were stained with CSFE. The proliferation is the dilution of the CSFE stain reflecting the separation of the parental markings now into two daughter cells. The assay tests only an effector:target ratios of 1:1 and 1:0 where effectors were defined as T cells expressing the anti-CD19 chimeric receptor. The assay is done in duplicate and 24 hrs after mixing of the cells, 50% of the media is removed/replaced for cytokine analysis using the Luminex 10-plex panel of human cytokines detection. After 5 days, T cells were stained for CAR expression, phenotyped as either CD4 or CD8 cells and stained for live/dead with 7AAD. After the final wash, the pelleted cells were re-suspended in a specific volume with a predetermined number of BD counting beads. Cell staining data was collected by LSRII flow cytometry and analyzed with FloJo software using beads to quantitate results. Total cell counts were determined by number of cells counted relative to a specific number of beads multiplied by the fraction of beads yet to be counted.
[0779] To evaluate the potential for the humanized CART19 cells to function similarly to the currently successful murine CART19, we wanted to assess in vitro their ability to kill targeted cells, to proliferate in response to the targeted antigen and to show signs of persistence. By packaging each of the humanized CART19 lentiviral constructs and titering them on SupT1 cells, we are able to determine the amount of virus to normalize transductions to be around 50%. This allows for more direct comparisons of activity starting with similar average intergration sites per cell.
[0780] The therapeutic CAR19 T cells are generated by starting with the blood from a normal apheresed donor whose naive T cells are obtained by negative selection for T cells, CD4+ and CD8+ lymphocytes. These cells are activated by CD3.times.28 beads in 10% RPMI at 37C, 5% CO.sub.2.
[0781] After 24 hrs, the T cells are blasting and the normalized amount of virus is added. The T cells begin to divide into a logarithmic growth pattern which is monitored by measuring the cell counts per ml and cell size. As the T cells begin to rest down, the logarithmic growth wanes and the cell size shrinks. The combination of slowing growth rate and T cell size approaching .about.300 fl determines the state for T cells to be cryopreserved or restimulated.
[0782] There is a very similar trend of T cells resting down as seen by size. The almost overlapping pattern between the humanized CART cells with the current murine CART19 and UTD population indicates no unusual effect of the humanized CAR19 on the normal T cell expansion following activation. As a control, SS1-BBz is used to define unwanted antigen independent CAR activity. The expansion profile in total cell numbers shows the differences in the actual numbers in the individual expansions are likely due mainly to different starting number of cells. By normalizing starting T cell numbers, a tight cluster is seen for all the CART19 cells. In addition, the unwanted effect of antigen independent CAR activation is detected in the line running lower and away from the group.
[0783] The level of surface expression for each of these CAR19 expressing cells was determined. The titered virus normalized for transduction show comparable expression levels correlating with transduction efficiency, percent cells transduced. Some CARs had their titers extrapolated from earlier packagings, and though their percentages transduced are lower, their MFI are also reduced as expected. The results indicate that there is no detectable negative effect of the humanized CAR19 on the cells ability to expand normally when compared to the UTD and murine CAR19 T cells.
[0784] The ability of the humanized CART19 cells to selectively discern a cell surface specific epitope expressed on cells and destroy them is analyzed. Wild type K562 cells do not express CD19 but can be transduced to express CD19. Comparing these killing curves, titrating the amount of effector cells shows that those cells expressing CD19 are destroyed. Redirected T cells from the same donor and modified with either humanized CART19 cells or current clinical murine CART19 cells indicate no difference in their ability to kill. The killing curves show that a very similar killing capacity is found among humanized CART19 cells targeting CD19+ CLL cells from patient 14. Interestingly, there is a decrease in overall cytolytic activity, in particularly GS CART19, suggesting these cells may possess specific inhibitory properties. The similar level of CD19 expressed on the targets cells indicates the expression level is not the reason for differences in cell killing.
[0785] The necessary property of the humanized CART19 cells to proliferate after seeing target cells is found in all constructs after being stimulated by the control CD3.times.28 beads and the CD19 expressing targets. Targeting Pt14 CLL cells appear to indicate a slightly greater proliferation rate with scFvs with a light to heavy chain orientation with no bias seen when having a 3.times. or 4.times.GGGGS linkage (SEQ ID NOS 107 and 106, respectively). The proliferative results reflect the total number of cells accumulated over the 5 days, indicating that the humanized CART19s, 2146, 2144, 2136, 2141 and 2137 drive a more proliferative signal to the T cells. Impressively, this was detected in the humanized CART19 cells targeting Pt14 CLL cells.
[0786] Overall, the humanized CART19 constructs exhibit very similar characteristics to the current murine CART19 in cytolytic activity, proliferative response and cytokine secretion to antigen specific targets. The potential of humanized CART19 cells, (2146, 2144, 2136, 2141 and 2137), to drive a more proliferative signal to the T cells upon target activation would seem to be an extra benefit of these new constructs to potentially enhance therapeutic response.
Results
[0787] Using both degranulation and cytokine production assays, it is demonstrated that the engineered CART19 T cells specifically target CD19+ cells.
[0788] ND317 cells transduced with humanized CD19CAR constructs (a.k.a. "huCART19") of the invention were analyzed. There was a tight similarity in size of the T cells during their expansions after CD3.times.28 activation and transduction with the humanized CART19 candidates relative to the murine CART19 and unmodified (UTD) T cells.
[0789] Experiments showed little difference in the number of T cells that accumulated during their expansions after CD3.times.28 activation and transduction with the different humanized CART19 candidates relative to the murine CART19 and unmodified (UTD) T cells.
[0790] Cell surface expressions of humanized CART19 are comparable and their expression level very similar to murine CART19. The overlay of histograms plotting the cell surface expression staining pattern of each humanized CART19 transduced T cells and the mean fluorescent intensity (MFI) calculated from these profiles correlates well with the percentage of cells transduced.
[0791] Furthermore the humanized CART19 have similar specific cytotoxic activities in targeting CD19 expressing target cells and comparable to murine CART19. Plots from 16 hr-flow-based killing assays using titrating Effector to Target (E:T) ratios with effector humanized CART19 cells targeting CSFE labeled K562cc (FIG. 1A. non-expressing CD19 controls), K562.CD19 (FIG. 1B, K562 cells transduced to express CD19) or Pt14 (FIG. 1C, B cells from CLL patient). The cytolytic activities of all the humanized CART19 cells are similar and comparable to the murine CART19. The differences in the cytolytic activity between different targets is similar and comparable indicating the murine CART19's activity is preserved in the humanized form of CART19.
[0792] Histogram overlays of CFSE marked humanized CART19 cells 6 days after being mixed with target cells show their proliferative capacity (FIG. 5). The proliferative response delivered from the CAR19 is a necessary response after engagement with and killing of target cells to develop a positive clinical response. The dilution of SS1-BBz CSFE staining, an indicator of dividing daughter cells diluting out the parental cell's stain, is a result of unrested T cells maintaining divisions in a targeting independent mechanism.
[0793] The cell populations overall ability to proliferate is evaluated with CD3.times.28 beads which mimics the endogenous engagement of the TCR and the co-stimulator CD28. Data indicates each cell population has a comparable proliferation potential. All humanized and murine CART19 cells proliferate strongly and comparably upon engagement with K562 cells expressing CD19. Humanized CART19 cells also responded well to B cells obtained from a CLL patient though some seem to respond slightly less. As shown in FIGS. 2A and 2B, the humanized CART19 cells 2136, 2137, 2140, 2141, 2144 and 2146 can be seen to have a slightly more robust proliferation as evidenced by the greater dilution of CSFE staining. These constructs all have the same variable chain orientation of light to heavy, indicating that this is the orientation of choice. A closer look at the amino acid changes in the heavy CDR2 site (Table 1) reveals that each of the three variations YSSSL, YQSSL and YNSSL (SEQ ID NOS:28, 29 and 30, respectively) are represented in the constructs that appeared to have the more robust proliferations after seeing targets. In addition, these observed constructs have both the G4S linker containing 3 copies of the subunit (3G4S) (SEQ ID NO: 107) and the G4S linker containing 4 copies of the subunit (4G4S) (SEQ ID NO: 106), indicating the linker size did not influence function.
[0794] From the proliferative expansions described above, the total cell numbers after 5 days post tumor engagement is determined. The cells show a decline in numbers than were initially seeded, indicating activation is required to maintain survival. An endogenous activation control is analyzed to show that the total cell count at the end of 6 days was similar. Humanized CART19 cells targeting K562 cells expressing CD19 show that the two murine CART19 cells both end up with the higher cell numbers, with 2146 slightly above all the other constructs with similar values. Total cell numbers were also analyzed 6 days after exposure to B cells from Patient 14 (pt14), and interestingly shows that the previously selected out humanized CART19 constructs 2146, 2144, 2136, 2141 and 2137, all of which have the light to heavy chain orientation and represent the three amino acid variations YSSSL, YQSSL and YNSSL (SEQ ID NOS: 28, 29 and 30, respectively), resulted in higher total cell numbers, higher than the murine CART19s. This unexpected differentiation between the various humanized anti-CD19CAR clones may translate to better clinical efficacy of CART cells transduced with these constructs.
[0795] Background levels of cytokine produced from humanized CART19 cells after exposure to the control K562 cells not expressing CD19 were analyzed. 24 hr supernatants were analyzed using a luminex 30-plex panel. The potential cytokine profile from stimulation of the endogenous immune system with the CD3.times.28 beads indicate each of the cell populations have a comparable cytokine profile.
[0796] Data also shows that the humanized CART19 and murine CART19 produce similar cytokine profiles at similar levels when responding to the same targets. The cytokine profile was lower but similar when targeting the Pt14 target cells.
Example 5: Humanized CD19 CAR T Cell Treatment in an In Vivo ALL Model
[0797] Primary human ALL cells can be grown in immune compromised mice without having to culture them in vitro. These mice can be used to test the efficacy of chimeric antigen receptor (CAR) T cells in a model that represents the patient population that will be found in the clinic. The model used here, HALLX5447, was passaged twice in NOD.Cg-Prkdc.sup.scidIl2rg.sup.tm1WjlSzJ (NSG) mice, prior to use in studies testing the efficacy of CAR T cells.
[0798] Murine CD19 CAR T cells have previously been shown to target and kill leukemia cells in an NSG mouse model of primary human ALL. The CD19 scFv (single chain Fc variable fragment) has been humanized and the present example compares the ability of T cells expressing a humanized CD19 CAR (CAR 2) to eliminate ALL tumor cells in vivo to that of the murine CD19 CAR T cells. Here, the efficacy of these cells has been directly compared in mice with established primary human ALL, as assayed by peripheral blood FACS analysis of human CD19 cells. Following an implant of 1.5.times.10.sup.6 primary ALL cells intravenously, a disease burden of 2.5-4% CD19 human cells in the blood was achieved by 2 weeks post-tumor implantation. This CD19 percentage is of total cells in the blood of the mice. 100% of human cells in the mice prior to treatment with CAR T cells are tumor cells. Percentages above 2% CD19 human cells in the peripheral blood are considered to be established human ALL disease in this model. The leukemia-bearing mice were treated with the CAR T cells once the leukemia is established in the mice, approximately two to three weeks after tumor implantation. Mice in each group were treated with 5.times.10.sup.6 total human T cells. The transduction efficiencies of the donor human T cells with the CAR expressing lentivirus were between 40-60%. Following treatment with the T cells, mice were bled weekly for analysis of the percentage of CD19 human cells in the blood as a biomarker for disease progression.
Materials and Methods:
[0799] Primary Human ALL Cells:
[0800] Primary cells were not cultured in vitro prior to implantation. These cells were harvested from a patient with ALL and then transferred into mice for establishment and expansion. After the tumor cells were expanded in the mice, the bone marrow and splenocytes were harvested and viably frozen in separate batches for re-implantation. The cells were frozen in 90% FBS and 10% DMSO at a minimum concentration of 5.times.10.sup.6 cells per milliliter. For re-implantation, the frozen ALL cells were thawed and then injected intravenously in to NSG mice, in order to generate mice with ALL that will be used to compare the anti-tumor efficacy of the humanized CD19 CAR T cells and the murine CD19 CAR T cells.
[0801] Mice:
[0802] 6 week old NSG (NOD.Cg-Prkdc.sup.scidIl2rg.sup.tm1Wjl/SzJ) mice were received from the Jackson Laboratory (stock number 005557). Animals were allowed to acclimate to the Novartis NIBRI animal facility for at least 3 days prior to experimentation. Animals were handled in accordance with Novartis ACUC regulations and guidelines.
[0803] Tumor Implantation:
[0804] In vivo serially passaged primary human ALL cells, model HALLX5447, were thawed in a 37.degree. C. water bath. The cells were then transferred to a 15 ml conical tube and washed twice with cold sterile PBS. The primary ALL cells were then counted and resuspended at a concentration of 15.times.10.sup.6 cells per milliliter of PBS. The cells were placed on ice and immediately (within one hour) implanted in the mice. The ALL cells were injected intravenously via the tail vein in a 100 .mu.l volume, for a total of 1.5.times.10.sup.6 cells per mouse.
[0805] CAR T Cell Dosing:
[0806] Mice were administered 5.times.10.sup.6 T cells 16 days after tumor implantation. Cells were partially thawed in a 37 degree Celsius water bath and then completely thawed by the addition of 1 ml of cold sterile PBS to the tube containing the cells. The thawed cells were transferred to a 15 ml falcon tube and adjusted to a final volume of 10 mls with PBS. The cells were washed twice at 1000 rpm for 10 minutes each time and then counted on a hemocytometer. T cells were then resuspended at a concentration of 50.times.10.sup.6 cells per ml of cold PBS and kept on ice until the mice were dosed. The mice were injected intravenously via the tail vein with 100 .mu.l of the CAR T cells for a dose of 5.times.10.sup.6 T cells per mouse. Five mice per group were treated either with 100 .mu.l of PBS alone (PBS), untransduced T cells (Mock), murine CD19 CAR T cells (muCTL019), or humanized CD19 CAR T cells (huCTL019). The untransduced T cells, muCTL019 T cells, and huCTL019 T cells were all prepared from the same human donor in parallel.
[0807] Animal Monitoring:
[0808] The health status of the mice was monitored daily, including twice weekly body weight measurements. The percent change in body weight was calculated as (BW.sub.current-BW.sub.initial)/(BW.sub.initial).times.100%. Tumor burden was monitored weekly by peripheral blood FACS analysis. Mice were bled weekly via the tail vein into EDTA coated tubes that were kept on ice. 10-20 .mu.l of blood was plated from the tubes into 96 well plates on ice. Red blood cells were lysed with ACK red blood cell lysis buffer (Life Technologies, catalog number A10492-01) and then washed twice with cold PBS. The cells were incubated with an Fc blocking mix of human and mouse Fc block (Miltenyi Biotec, catalog numbers 130-059-901 and 130-092-575) for 30 minutes and then incubated with an anti-human CD19 antibody for 30 minutes. The cells were fixed with a 2% paraformaldehyde solution for 20 minutes, washed and stored in PBS+2% FBS overnight prior to analysis on a BD Canto or Fortessa, followed by further analysis using the FlowJo FACS analysis software. The cells were analyzed to determine the percent of human CD19.sup.+ cells in the blood of the human HALLX5447 ALL tumor-bearing NSG mice. CD19 percentages in the blood are reported as the mean.+-.standard error of the mean (SEM).
[0809] Percent treatment/control (T/C) values were calculated using the following formula:
% T/C=100 .times..DELTA.T/.DELTA.C if .DELTA.T.gtoreq.0;
% Regression=100 .times..DELTA.T/T.sub.initial if .DELTA.T<0;
where T=mean peripheral blood CD19 percentage of the drug-treated group on the final day of the study; T.sub.initial=peripheral blood CD19 percentage of the drug-treated group on initial day of dosing; .DELTA.T=mean peripheral blood CD19 percentage of the drug-treated group on the final day of the study-mean peripheral blood CD19 percentage of the drug treated group on the initial day of dosing; C=mean peripheral blood CD19 percentage of the control group on the final day of the study; and .DELTA.C=mean peripheral blood CD19 percentage of the control group on the final day of the study -mean peripheral blood CD19 percentage of the control group on the initial day of dosing.
[0810] T/C values in the range of 100% to 42% are interpreted to have no or minimal anti-tumor activity; T/C values that are .ltoreq.42% and >10% are interpreted to have anti-tumor activity or tumor growth inhibition. T/C values .ltoreq.10% or regression values .gtoreq.-10% are interpreted to be tumor stasis. Regression values <-10% are reported as regression.
Results:
[0811] The anti-tumor activity of murine and humanized CD19 CAR T cells were evaluated and directly compared in a primary model of human ALL. Following tumor implantation on day 0, mice were randomized into treatment groups and treated with 5.times.10.sup.6 T cells intravenously on day 16. ALL disease burden and animal health were monitored until animals achieved endpoint. The mice in all the groups were euthanized on day 65 post-tumor implantation when disease burden in the control groups was above 80% human CD19.sup.+ cells in the peripheral blood.
[0812] A clear difference in disease burden was seen between the control groups and the groups treated with either the murine or the humanized CD19 CAR T cells with P<0.01 from day 24 after tumor implantation, and continuing to the end of the study at day 65. The murine and human CD19 CAR T cells demonstrate a similar ability to control human HALLX5447 ALL tumor cell growth in NSG mice. Both groups showed a peak peripheral blood disease level of 12-15% human CD19.sup.+ cells at day 21 post HALLX5447 implantation. 42 days after tumor cell implantation, no human CD19.sup.+ cells were detectable in the huCTL019 group, while the percentage of human CD19.sup.+ cells in the muCTL019 group dropped to about 1%. Both the murine and the humanized CD19 CAR T cells resulted in a comparable ability to control the expansion of primary human ALL cells in this model (P>0.05). The % T/C values for the mock transduced T cell group was 94.40%, demonstrating that the mock transduced T cells had no anti-tumor activity. The percent regression of the muCTL019 group was -89.75% and the huCTL019 group was -90.46%, demonstrating that both of these treatments were able to cause a regression of the HALLX5447 tumor model. The peripheral blood human CD19.sup.+ cell percentages as a measure of the disease burden in these mice is shown in FIG. 7. The PBS treatment group, which did not receive any T cells, demonstrated baseline primary ALL tumor growth kinetics in intravenously implanted NSG mice. The Mock treatment group received untransduced T cells that underwent the same in vitro expansion process as the CAR T cells. These cells serve as a T cell control to show the non-specific response of the T cells in this tumor model. Both the PBS and Mock transduced T cell treatment groups demonstrated continuous tumor progression throughout the experiment. Both the murine and the humanized CD19 CAR T cells control the progression of disease within one week of the 5.times.10.sup.6 T cell injections and demonstrate a similar ability to sustain disease control over the course of this 65 day study.
[0813] The anti-tumor activity of murine and humanized CD19 CAR transduced T cells was assessed in an efficacy study in NSG mice bearing a primary human ALL model, HALLX5447. This study demonstrated that both the murine and humanized CD19 CAR T cells (muCTL019 and huCTL019) are capable of mounting an anti-tumor response in a primary model of human ALL. In addition, this response, as assayed by peripheral blood disease burden is the same for the muCTL019 and huCTL019 cells. Both the murine and humanized CD19 CAR T cells control primary ALL growth within a week of the mice being dosed with the T cells. Initially after treatment, the disease burden continued to increase before decreasing to virtually undetectable levels. One treatment with either the murine or humanized CAR T cells resulted in a sustained anti-tumor response over the course of the 65 day disease progression in control treated mice. The humanized CD19 CAR T cells demonstrated a similar ability to mount an efficacious anti-CD19 tumor response and control ALL disease burden as was seen with the murine CD19 CAR T cells.
Example 6: CD19 CAR T Cells for Use in Treating Multiple Myeloma
[0814] Even with current regimens of chemotherapy, targeted therapies, and autologous stem cell transplant, myeloma is considered an incurable disease. The present example describes treating multiple myeloma (MM) with autologous T cells directed to CD19 with a chimeric antigen receptor (lentivirus/CD19:4-1BB:CD3zeta; also known as "CART19" or CTL019). This example demonstrates that CD19-directed CAR therapies have the potential to establish deep, long-term durable remissions based on targeting the myeloma stem cell and/or tumor cells that express very low (undetectable by most methods) levels of CD19.
[0815] In treating a patient with an aggressive secondary plasma cell leukemia, we found that CART19 administered two days after a salvage autologous stem cell transplant resulted in rapid clearance of plasma cell leukemia and a very good partial response in a patient who had progressed through multiple lines of chemotherapy. This patient was transfusion-dependent for months prior to the treatment; at two months after the treatment, she has recovered her blood counts (with normal-range platelet counts and white blood cell counts) and has not required transfusions since she was discharged from the hospital from her treatment.
[0816] Because myeloma cells do not naturally express CD19, the finding that CART19 treatment induced a rapid and significant tumor response in this tumor was surprising. Without wishing to be bound by a particular theory, it was reasoned that CART19 could be used to treat myeloma because: (1) while myeloma cells are traditionally thought to be negative for CD19 expression by flow cytometry, there are data indicating that myeloma cells may express very low levels of CD19, such that expression is detectable by RNA but not by flow cytometry or immunohistochemistry; and (2) the concept of targeting the clonotypic B cell, which is thought to be the cancerous stem cell that gives rise to multiple myeloma, and is particularly resistant to chemotherapy. There is a clonal relationship between B cells and myeloma tumor cells, but traditional myeloma therapy is aimed at the malignant plasma cells rather than B cells. CART19 for treating myeloma therefore targets a different cell population than most myeloma therapies.
[0817] In our single patient experience, the patient had circulating plasma cells, and we were able to test her tumor cells for the expression of CD19. Approximately 1-2% of her tumor cells expressed the CD19 antigen. (FIG. 8). Thus, it was reasoned that CART19 may have a direct effect on a very small population of her tumor cells; a very good partial response, though would not have been predicted based on targeting only the very small population of CD19+ tumor cells.
[0818] In this case, CART19 was administered following autologous stem cell transplant rescue after high-dose melphalan. Although this is a standard therapy in myeloma, it is not curative. Furthermore, this patient had previously undergone tandem autologous stem cell transplants and relapsed early (<6 months) after transplant. Without wishing to be bound by a particular theory, use of CART19 cells as described in the present example may have a non-overlapping mechanism in the treatment of myeloma when combined with a salvage autologous stem cell transplant.
[0819] A patient with refractory multiple myeloma was treated with CTL019 after myeloablative chemotherapy and ASCT. Remission was maintained despite loss of detectable CTL019 and reconstitution of normal CD19-positive B cells, indicating that this response did not require sustained CTL019 activity. Moreover, this patient's response was realized even though the vast majority (99.95%) of the neoplastic plasma cells were CD19-negative by both flow cytometry and RT-PCR.
[0820] The absence of detectable CD19 expression in this patient's dominant neoplastic plasma cell population suggests that the clinically relevant target of CTL019 resided outside this dominant CD19-negative population. Neoplastic plasma cells in multiple myeloma patients exhibit genetic, immunophenotypic, and functional heterogeneity. Particular subpopulations may be required for survival of the clone through anti-myeloma therapy. In the patient reported here, for example, the small CD19-expressing subset of plasma cells might have been relatively melphalan-resistant but sensitive to CTL019. This finding suggests that therapeutically targeting a small subset of the clone can lead to durable clinical benefit when coupled with conventional anti-myeloma therapy.
[0821] Alternatively, the clinically relevant target of CTL019 in this patient may have resided outside the neoplastic plasma cell population. For instance, the CTL019 may target a stem cell population that is relatively small but gives rise to neoplastic plasma cells. Multiple myeloma may therefore be a disease of multiple late B-lineage cell types, not just terminally differentiated plasma cells, such that therapies like CTL019 that target B lymphocytes might be useful adjuncts to therapies that directly target plasma cells.
[0822] Ten additional multiple myeloma patients will be treated with CART19 in a Phase I trial, at least three patients have been treated to date.
Dose Rationale and Risks/Benefits
[0823] We have chosen to use flat dosing via the intravenous route of administration for this protocol. The primary objective of this protocol was to test the safety and feasibility of administering CART-19 cells to patients with multiple myeloma. The primary toxicities that were anticipated are (I) cytokine release when the CARs encounter their surrogate CD19 antigen on malignant or normal B cells; (2) depletion of normal B cells, similar to rituximab therapy; (3) steroid-responsive skin and gastrointestinal syndromes resembling graft-versus-host disease as has been seen previously when expanded/costimulated autologous T-cells have been coupled with ASCT for MM. A theoretical concern was whether transformation or uncontrolled proliferation of the CART-19 T cells might occur in response to high levels of CD19. This was less a concern in this application compared to another study of CLL patients, as the burden of clonotypic B-cells in MM is expected to be far lower than the burden of malignant B-cells in the refractory CLL patients treated on that study.
Dose Rationale
[0824] With the first 3 patients, we have observed clinical activity at doses ranging from 1.4.times.10.sup.7 to 1.1.times.10.sup.9 CART-19 cells. This observation demonstrates, at least in the first 3 patients treated, that there is not an obvious dose response relationship. A complete response was observed in patients administered with two log fold difference in dose. Thus, unlike standard drugs that are metabolized, CAR T cells can have a wide dose response range. This is most likely because the CAR T cells are able to proliferate extensively in the patients. We therefore set a dose range of 1-5.times.10.sup.8 CART-19 cells for infusion. In this single-patient study offered on a compassionate use basis, the patient was offered up to 5.times.10.sup.8 CART19 cells, with no lower dose limit. For the ten patient trial, patients will be offered 1-5.times.10.sup.7 CART-19 cells.
General Design
[0825] This was single patient-study offered on a compassionate use basis; it was modeled after a Phase I study to determine if the infusion of autologous T cells transduced to express CART-19 is safe. The primary goals of the study were to determine the safety, tolerability and engraftment potential of CART-19 T cells in patients undergoing salvage ASCT after early relapse following first ASCT. The protocol consists of an open label pilot study.
[0826] At entry subjects will undergo a bone marrow biopsy and routine laboratory and imaging assessment of their MM. Eligible subjects will undergo steady-state apheresis to obtain large numbers of peripheral blood mononuclear cells (PBMC) for CART-19 manufacturing. The T cells will be purified from the PBMC, transduced with TCR.zeta./4-1BB lentiviral vector, expanded in vitro and then frozen for future administration. The number of patients who have inadequate T cell collections, expansion or manufacturing compared to the number of patients who have T cells successfully manufactured will be recorded; feasibility of product manufacturing is not expected to be problematic in this patient population.
[0827] Subjects will generally have had adequate peripheral blood stem cells remaining stored from the mobilization/collection performed in preparation for their first ASCT to conduct two additional ASCT. Those who do not will undergo a second mobilization/collection procedure either before or after their steady-state apheresis with a regimen according to the treating physician's preference. Approximately two weeks after the initial leukapheresis, subjects will be admitted to the hospital and receive high-dose melphalan (day -2) followed by infusion of autologous stem cells two days later (day 0), and all subjects will receive infusion of CART-19 cells twelve to fourteen days later (day +12-14). Up to 10 patients will be enrolled.
[0828] All subjects will have blood tests to assess safety, and engraftment and persistence of the CART-19 cells at regular intervals through week 4 of the study. At day +42 and day +100, subjects will undergo bone marrow aspirates/biopsies to assess the bone marrow plasma cell burden and trafficking of CART-19 cells to the bone marrow. A formal response assessment will be made at day 100 according to International Myeloma Working Group (IMWG) criteria136, and TTP will be monitored according to routine clinical practice for patients with multiple myeloma. The main efficacy outcome measured in this study will be a comparison of TTP after a patient's initial ASCT to TTP after the ASCT on this study.
[0829] As the primary endpoint of this study is safety and feasibility of infusion of CART-19 cells with ASCT, the study will employ an early stopping rule. Briefly, if less than 2 severe, unexpected adverse events occur among the first five subjects treated, the study will then accrue an additional five subjects towards a target enrollment of 10. We will observe treated subjects for 40 days after CART-19 infusion (i.e., through the first official response assessment at day 42) before enrolling a subsequent subject until five subjects have been enrolled and so observed. For treatment of the second group of five patients, no waiting period will be required between subjects.
[0830] Following the 6 months of intensive follow-up, subjects will be evaluated at least quarterly for two years with a medical history, physical examination, and blood tests. Following this evaluation, subjects will enter a roll-over study for annual follow-up by phone and questionnaire for up to additional thirteen years to assess for the diagnosis of long-term health problems, such as development of new malignancy.
Primary Study Endpoints
[0831] This pilot trial is designed to test the safety and feasibility of the autologous T cells transduced with the CD19 TCR/4-1BB in patients undergoing salvage ASCT for MM following early relapse after first ASCT.
Primary Safety and Feasibility Endpoints Include:
[0832] Occurrence of study-related adverse events, defined as NCJ CTC 2: grade 3 signs/symptoms, laboratory toxicities and clinical events that are possibly, likely or definitely related to study treatment at any time from the infusion until week 24. This will include infusional toxicity and any toxicity possibly related to the CART-19 cells including but not limited to:
a. Fevers b. Rash c. Neutropenia, thrombocytopenia, anemia, marrow aplasia d. Hepatic dysfunction e. Pulmonary infiltrates or other pulmonary toxicity f. GVHD-like syndromes affecting gastrointestinal tract or skin.
[0833] Feasibility to manufacture CART-19 cells from patient apheresis products. The number of manufactured products that do not meet release criteria for vector transduction efficiency, T cell purity, viability, sterility and tumor contamination will be determined.
[0834] The depth and duration of response following autologous stem cell transplant with CART19 will be compared to the depth and duration of response that each patient initially achieved following standard autologous stem cell transplant.
Subject Selection and Withdrawal
Inclusion Criteria
[0835] Subjects must have undergone a prior ASCT for MM and have progressed within 365 days of stem cell infusion. Subjects who have undergone two prior ASCTs as part of a planned tandem ASCT consolidation regimen are eligible. Progression will be defined according to IMWG criteria for progressive disease or, for patients who attained CR or sCR after initial ASCT, criteria for relapse from CR (Durie et al. Leukemia 2006; 20(9):1467-1473). N.B.: There is no requirement that patients must enroll within 365 days of prior ASCT, and patients may be treated with other agents, including experimental agents, following relapse/progression after prior ASCT before enrollment on this study.
[0836] Subjects must have signed written, informed consent.
[0837] Subjects must have adequate vital organ function to receive high-dose melphalan as defined by the following criteria, measured within 12 weeks prior to the date of melphalan infusion: a. Serum creatinine .ltoreq.2.5 or estimated creatinine clearance .gtoreq.30 ml/min and not dialysis-dependent. b. SGOT .ltoreq.3.times. the upper limit of normal and total bilirubin .ltoreq.2.0 mg/dl (except for patients in whom hyperbilirubinemia is attributed to Gilbert's syndrome). c. Left ventricular ejection fraction (LVEF) .gtoreq.45% or, if LVEF is <45%, a formal evaluation by a cardiologist identifying no clinically significant cardiovascular function impairment. LVEF assessment must have been performed within six weeks of enrollment. d. Adequate pulmonary function with FEV1, FVC, TLC, DLCO (after appropriate adjustment for lung volume and hemoglobin concentration) .gtoreq.40% of predicted values. Pulmonary function testing must have been performed within six weeks of enrollment.
[0838] Subjects must have an ECOG performance status of 0-2, unless a higher performance status is due solely to bone pain.
Exclusion Criteria
[0839] Subjects must not:
[0840] Have any active and uncontrolled infection.
[0841] Have active hepatitis B, hepatitis C, or HIV infection.
[0842] Any uncontrolled medical disorder that would preclude participation as outlined.
Treatment Regimen
[0843] Therapy for Relapsed/Progressive Multiple Myeloma
[0844] Patients may receive, prior to enrollment, therapy for relapsed/progressive multiple myeloma according to the preference of their treating physicians. Therapy may continue upon enrollment.
[0845] Patients must stop all therapy for two weeks prior to apheresis and for two weeks prior to high-dose melphalan. If more than two weeks are expected to lapse between apheresis and high-dose melphalan, patients may resume therapy after apheresis at the discretion of their treating physicians.
[0846] High-Dose Melphalan (Day -2)
[0847] Patients will be admitted to the hospital on day -3 or -2 and will undergo examination by the attending physician and routine laboratory tests, which will include monitoring parameters for tumor lysis syndrome, prior to commencement of the treatment protocol. Blood for MM monitoring laboratory tests (SPEP, quantitative immunoglobulins, and serum free light chain analysis), will be drawn prior to initiation of therapy if such tests had not been drawn within 7 days of admission.
[0848] High-dose therapy will consist of melphalan at a dose of 200 mg/m.sup.2 administered intravenously over approximately 20 minutes on day -2. The dose of melphalan will be reduced to 140 mg/m.sup.2 for patients >70 years of age or for patients of any age whom, at the discretion of the treating physician, may not tolerate a dose of 200 mg/m.sup.2 All patients will receive standard anti-emetic prophylaxis, which may include dexamethasone, and standard antibiotic prophylaxis.
[0849] Stem-Cell Re-Infusion (Day 0)
[0850] Stem cell infusion will take place on day 0, at least 18 hours after the administration of the high-dose melphalan. Stem cells will be infused intravenously over approximately 20-60 minutes following premedication according to standard institutional practice. At least 2.times.10.sup.6 CD34+ progenitors/kg body weight should be infused. In addition, at least 1.times.10.sup.6 CD34+ progenitors/kg body weight should be available as a back-up stem-cell product to be infused in the event of delayed engraftment or late graft failure. G-CSF should be administered SQ beginning on day +5, dosed according to standard institutional practice. Other supportive care measures such as transfusion support will be done in accordance with standard institutional guidelines.
[0851] CART19 Cell Infusion (Day +12-14)
[0852] A single dose of CART-19 transduced T cells will be given consisting of up to 5.times.10.sup.7 CART-19 cells. The minimal acceptable dose for infusion of cells transduced with the CD19 TCR 4-1BB vector is 1.times.10.sup.7. CART-19 cells will be given as a single dose by rapid i.v. infusion on day +12-14 after stem cell infusion. If patient fails to meet any of the inclusion criteria described herein in the 12-14 day window, the CART-19 infusion may be delayed beyond day +12-14 until the criteria is satisfied.
[0853] Maintenance Lenalidomide
[0854] Subjects who received and tolerated maintenance lenalidomide after their first ASCT will re-initiate lenalidomide maintenance therapy at approximately day +100, assuming there are no contraindications in the judgment of the treating physician. The starting dose will be 10 mg daily unless prior experience dictates an alternative starting dose for a particular patient. Maintenance therapy will continue until disease progression or intolerance.
[0855] Preparation and Administration of Study Drug
[0856] The CART-19 T cells are prepared in the CVPF and are not released from the CVPF until FDA approved release criteria for the infused cells (e.g., cell dose, cell purity, sterility, average copy number of vectors/cell, etc.) are met. Upon release, the cells are taken to the bedside for administration.
[0857] Cell thawing. The frozen cells will be transported in dry ice to the subject's bedside. The cells will be thawed at the bedside using a water bath maintained at 36.degree. C. to 38.degree. C. The bag will be gently massaged until the cells have just thawed. There should be no frozen clumps left in the container. If the CART-19 cell product appears to have a damaged or the bag to be leaking, or otherwise appears to be compromised, it should not be infused and should be returned to the CVPF as specified below.
[0858] Premedication. Side effects following T cell infusions include transient fever, chills, and/or nausea; see Cruz et al. for review (Cytotherapy 2010; 12(6):743-749). It is recommended that the subject be pre-medicated with acetaminophen and diphenhydramine hydrochloride prior to the infusion of CART-19 cells. These medications may be repeated every six hours as needed. A course of non-steroidal anti-inflammatory medication may be prescribed if the patient continues to have fever not relieved by acetaminophen. It is recommended that patients not receive systemic corticosteroids such as hydrocortisone, prednisone, methylprednisolone or dexamethasone at any time, except in the case of a life-threatening emergency, since this may have an adverse effect on T cells.
[0859] Febrile reaction. In the unlikely event that the subject develops sepsis or systemic bacteremia following CAR T cell infusion, appropriate cultures and medical management should be initiated. If a contaminated CART-19 T cell product is suspected, the product can be retested for sterility using archived samples that are stored in the CVPF.
[0860] Administration. The infusion will take place in an isolated room in Rhoads, using precautions for immunosuppressed patients. The transduced T cells will be administered by rapid intravenous infusion at a flow rate of approximately 10 mL to 20 ml per minute through an 18-gauge latex free Y-type blood set with a 3-way stopcock. The duration of the infusion will be based on the total volume to be infused and the recommended infusion rate. Each infusion bag will have affixed to it a label containing the following: "FOR AUTOLOGOUS USE ONLY." In addition the label will have at least two unique identifiers such as the subject's initials, birth date, and study number. Prior to the infusion, two individuals will independently verify all this information in the presence of the subject and so confirm that the information is correctly matched to the participant.
[0861] Emergency medical equipment (i.e., emergency trolley) will be available during the infusion in case the subject has an allergic response, or severe hypotensive crisis, or any other reaction to the infusion. Vital signs (temperature, respiration rate, pulse, and blood pressure) will be taken before and after infusion, then every 15 minutes for at least one hour and until these signs are satisfactory and stable. The subject will be asked not to leave until the physician considers it is safe for him or her to do so.
Packaging
[0862] Infusion will be comprised of a single dose of 1-5.times.10.sup.7 CA T19-transduced cells, with a minimal acceptable dose of 1.times.10.sup.7 CART-19 cells for infusion. Each bag will contain an aliquot (volume dependent upon dose) of cryomedia containing the following infusible grade reagents (% v/v): 31.25% plasmalyte-A, 31.25% dextrose (5%), 0.45% NaCl, up to 7.5% DMSO, 1% dextran 40, 5% human serum albumin.
Apheresis
[0863] A large volume (12-15 liters or 4-6 blood volumes) apheresis procedure is carried out at the apheresis center. PBMC are obtained for CART-19 during this procedure. From a single leukapheresis, the intention is to harvest at least 5.times.10.sup.9 white blood cells to manufacture CART-19 T cells. Baseline blood leukocytes for FDA look-back requirements and for research are also obtained and cryopreserved. The cell product is expected to be ready for release approximately 2-4 weeks later. Flow cytometry lymphocyte subset quantitation, including CD19 and CD20 B cell determination. Baseline assessment is made for human anti-VSV-G and anti-murine antibody (HAMA). If a subject has previously had an adequate apberesis collection banked according to current Good Manufacturing Practices at the Clinical Cell and Vaccine Production Facility these cells may be used as the source of cells for CART-19 manufacturing. Using a banked apheresis product would avert the expense, time, and risk to the subject of undergoing an additional apheresis collection.
Cytoreductive Chemotherapy
[0864] The lymphodepleting chemotherapy will be high-dose melphalan as described herein.
CART-19 Infusion
[0865] Infusion will begin on day +12-14 after stem-cell reinfusion.
[0866] On day +12-14 prior to the first infusion, patients will have a CBC with differential, and assessment of CD3, CD4 and CD8 counts since chemotherapy is given in part to induce lymphopenia.
[0867] The first dose will be administered using a single dose. The cells are thawed at the patient's bedside. The thawed cells will be given at as rapid an infusion rate as tolerated such that the duration of the infusion will be approximately 10-15 minutes. In order to facilitate mixing, the cells will be administered simultaneously using a Y-adapter. Subjects will be infused and premedicated as described herein. Subjects' vital signs will be assessed and pulse oxymetry done prior to dosing, at the end of the infusion, and every 15 minutes thereafter for 1 hour and until these are stable and satisfactory. A blood sample for determination of a baseline CART-19 level is obtained any time prior to the first infusion and 20 minutes to 4 hours after each infusion (and sent to TCSL).
[0868] Patients experiencing toxicities related to high-dose melphalan will have their infusion schedule delayed until these toxicities have resolved. The specific toxicities warranting delay of T cell infusions include: 1) Pulmonary: Requirement for supplemental oxygen to keep saturation greater than 95% or presence of radiographic abnormalities on chest x-ray that are progressive; 2) Cardiac: New cardiac arrhythmia not controlled with medical management 3) Hypotension requiring vasopressor support. 4) Active Infection: Positive blood cultures for bacteria, fungus, or virus within 48-hours of T cell infusion.
Management of Toxicity
[0869] Uncontrolled T cell proliferation. Toxicity associated with allogeneic or autologous T cell infusions has been managed with a course of pharmacologic immunosuppression. T body associated toxicity has been reported to respond to systemic corticosteroids. If uncontrolled T cell proliferation occurs (grade 3 or 4 toxicity related to CART-19 cells), subjects may be treated with corticosteroids. Subjects will be treated with pulse methylprednisolone (2 mg/kg i.v. divided q8 hr.times.2 days), followed by a rapid taper.
[0870] In addition, based on the observations of subjects treated on another protocol, there is some concern for macrophage activation syndrome (MAS), though the CD19+ tumor burden is expected to be much lower in patients with myeloma than in patients with CLL. Treatment and timing of treatment of this toxicity will be at the discretion of the patient's physician and the study investigator. Suggested management might include: if the subject has a fever greater than 101.degree. F. that lasts more than 2 consecutive days and there is no evidence of infection (negative blood cultures, CXR or other source), tocilizumab 4 mg/kg can be considered. The addition of corticosteroids and anti-TNF therapy can be considered at the physician's discretion.
[0871] B cell depletion. It is possible that B cell depletion and hypogammaglobulinemia will occur. This is common with anti-CD20 directed therapies. In the event of clinically significant hypogammaglobulinemia (i.e. systemic infections), subjects will be given intravenous immunoglobulin (IVIG) by established clinical dosing guidelines to restore normal levels of serum immunoglobulin levels, as has been done with Rituximab.
[0872] Primary graft failure. Primary graft failure (i.e., non-engraftment) may be more common after second ASCT compared to first ASCT. Eligibility criteria stipulate that sufficient stem cells must be available for rescue reinfusion at the discretion of the treating physician in the event of primary graft failure.
[0873] Results
[0874] Three treatment-refractory, advanced multiple myeloma patients have now been treated with CTL019 in this ongoing trial. Results for two of these patients show that both have had substantial anti-tumor effects from the CTL019 therapy based on the primary efficacy assessment at the three-month time-point. The third patient has not yet reached the three-month time point. The results for the two patients are described in more detail below.
[0875] The first myeloma patient has completed her +100 day response assessment and she had a very good response to the CART19 therapy. The following tests were performed with the following results: [0876] SPEP/immunofixation: negative [0877] urine immunofixation: faint unmeasurable kappa light chain band on her immunofixation (also present at day 38, so not new) Otherwise, the patient meet the criteria for stringent complete remission including: [0878] serum free light chain ratio: normal [0879] bone marrow biopsy: negative [0880] IgA immunophenotyping: IgA is below the limit of detection
[0881] Other than the faint unmeasurable kappa light chain result from urine immunofixation, the patient met all criteria for "stringent complete remission". The summary of the plasma cell immunophenotyping at 3 time points (day -2, day +38, day +103) is shown in FIG. 39, and demonstrates that the patient's IgA is below the limit of detection. The summary shows heavy myeloma burden at day -2 and none detectable at day +38 and +103, which classifies the patient as "MRD negative" by flow analysis. At day +103, the summary shows recovery of normal, polyclonal, CD19+ plasma cells and B cells. The patient had no symptoms of disease or therapy and is functioning like a normal person.
[0882] The second patient treated has not yet reached the +100 day time point. However, at this time point, she is doing well but it is too early to determine the effect of the CTL019 infusion.
Example 7: Kinase Inhibitor/CAR19 T-Cell Combined Therapy for Mantle Cell Lymphoma
[0883] Adoptive T-cell therapy holds considerable promise for the treatment of lymphoid malignancies. Promising clinical responses in small lymphocytic lymphoma/chronic lymphocytic leukemia (SLL/CLL) and acute lymphocytic leukemia (ALL), using adoptive transfer of autologous T cells transduced with chimeric antigen receptors (CAR) against the B-cell specific CD19 antigen (CAR19 T cells/CART19 cells) using CTL109. We have recently reported initial data on 3 patients with chemotherapy-refractive SLL/CLL enrolled in a phase I trial to treat CD19-positive malignancies using CD19-specific CAR (CAR19 T cells/CART19 cells). The approach used involved the genetic modification of patient-derived bulk T cells using a lentivirus to express a CD19-targeting CAR that contains signaling domains derived from CD137 and TcRz. For this study cells were expanded using our anti-CD3 and -CD28 bead expansion methodology, and cells were infused early post lymphodepletion without cytokine support (Kalos M, et al. (2011) Sci Transl Med. 3: 95ra73; and Porter D L, et al. (2011) N Engl J Med. 365: 725-733). These early results were extremely promising: (i) following a single course of treatment 2/3 patients achieved complete remissions and remain disease free now at 15+ months post treatment, while the third patient, who was treated with corticosteroids soon after the T cell infusion demonstrated a strong partial response. (ii) In these patients we were able to recapitulate the elements thought to be required for ultimate efficacy of adoptive T cell therapy-based strategies, namely robust in vivo T cell expansion, disease eradication, T cell contraction, and long-term functional persistence. To date, 10 SLL/CLL patients have been treated with 2 remaining in the complete clinical and molecular remission, 5 experiencing partial remission, and 3 displaying lack of measurable response. In another study, two patients with B cell ALL achieved complete remission with 1 relapsing with leukemic cells lacking CD19 expression.
[0884] Mantle cell lymphoma (MCL), both before and after large cell transformation, will also likely to benefit from the CART19-based adoptive therapy, in particular when combined with kinase inhibitors such as those that directly affect MCL cells.
[0885] To further analyze the combination of CART19-based adoptive therapy in combination with kinase inhibitors, high throughput screens will be used to evaluate several inhibitors targeting the kinases critical for MCL pathogenesis: CDK4/6, BTK, and mTOR in combination with CART19 cells. The most promising combinations will be evaluated in greater detail, both in vitro and in vivo, in MCL xenotransplant mouse model, which ultimately may guide the development of a clinical protocol to evaluate combination of small molecule kinase inhibitor and the CART cell immunotherapy in MCL patients.
[0886] In this study, preclinical studies will be performed to determine potential clinical efficacy of this approach in the various subtypes of MCL and to evaluate the ability to therapeutically target MCL cells using CART19 cells either alone or in will combination with small molecule inhibitors of selected proteins from the kinase family expression and activity of which is critical for survival and growth of MCL cells.
Research Plan
[0887] In a pre-clinical setting, the ability to therapeutically target MCL cells, both cultured and primary-type cells, using inhibitors of kinases with documented pathogenic relevance in MCL and CART19 cells will be evaluated. A high throughput MTT assay will be used to determine the effect of these agents to identify potential optimal combinations, dosing and timing of the agent application. The most promising 2-3 combinations will be evaluated in the greater detail in regard to cell function, phosphorylation-based cell signaling, and gene expression first in vitro and later in vivo in the MCL xenotransplant model.
In-Vitro Studies to Characterize the Ability of Kinase Inhibitor/CART-19 Cell Combinations to Effectively Target MCL Cells
[0888] In this aim, detailed functional, phenotypic, biochemical, and molecular assays listed above to study in-vitro the impact of the small molecule kinase inhibitors on MCL cells as well as to examine interactions between CART19 cells and MCL cells and the impact of the inhibitors on these interactions will be examined.
[0889] The benchmarks for accomplishing this aim will be to generate a comprehensive data set to: [0890] i. document that CART19 cells are activated by and lyse the cultured and primary MCL cells; [0891] ii. demonstrate that the selected kinase inhibitors enhance the ability of each other and/or CART19 cells to eliminate MCL cells without negatively impacting CART19 cell function when appropriately applied in regard to the dose and, for some, timing of the inhibitor vs. CART19 cell administration; and [0892] iii. establish a regimen for the schedule and dosing for the BTK inhibitor to be used in the in vivo MCL xenotransplant experiments using the NSG mice.
[0893] The goals of this study are, e.g., to evaluate whether identify the optimal therapeutic combinations of small molecule inhibitors targeting kinases critical for MCL pathobiology: CDK4, BTK, and mTOR together with CART19 cells, monitor CART19 activity, and characterize the functional, biochemical, and molecular effects of the therapy on MCL.
[0894] These studies should to establish a rational schema for schedule for the timing and dose of BTK treatment kinase inhibitor in conjunction with CART19 therapy to be evaluated in-vivo in aim 2.
In-Vivo Studies to Evaluate the Ability of CART19 Cells to Target Follicular Lymphoma, Alone and in Combination with BTK Inhibitor
[0895] In this aim, we will test in animal models the ability of the selected inhibitor/CART19 cell combination(s) to affect growth of established and primary MCL cells.
[0896] The benchmarks for accomplishing this aim will be to generate a data set to: [0897] i. demonstrate that the selected inhibitor/CART19 cell combination markedly enhances the survival of animals engrafted with MCL as compared to the controls (single and mock agent treated animals); [0898] ii. establish a regimen for the schedule and dosing for the selected kinase inhibitor/CART19 combination to be used as the basis for a future clinical trial.
[0899] The goals of this study include evaluating the treatment and dose schedule defined in aim 1 for the identified kinase inhibitor/CART19 plus BTK inhibitor combination, and to test whether BTK treatment synergizes with CART19 to target MCL in NSG mice xenotransplanted with MCL cells, both cultured and primary.
[0900] The following cell types, compounds, animals and experimental methodologies will be used to accomplish the proposed aims:
[0901] MCL Cells:
[0902] Four MCL cell lines (Jeko-1, Mino, SP-49, and SP-53) and viably frozen samples from 15 primary MCL (12 typical and 3 blastoid). While the cell lines grow well spontaneously, the primary cells will be cultured alone as well as in the presence of conditioned medium collected from HS5 bone marrow stromal cells to improve their viability.
[0903] CART19 Cells:
[0904] Primary human T cells engineered to express CAR19 will be generated using lentivirus transduction and using the established protocols ((Kalos M, et al. (2011) Sci Transl Med. 3: 95ra73; and Porter D L, et al. (2011) N Engl J Med. 365: 725-733). Following a single transduction event T cells typically express CAR19 at frequencies exceeding 30%.
[0905] Our studies will use CART19 populations from five SLL/CLL patients (50-100 vials/patient at with 1.times.10.sup.7 cells/vial are already available). CART19 cells will be identified using an anti-CAR19-specific idiotype antibody (STM). CART19 activity will be controlled both in vitro and in vivo in NSG mice in the standardized manner using CD19+ NALM-6, CD19-negative K562, and CD19-transduced K562 cell lines. Although CART19 cell function is not MHC restricted, CART19 cell from at least 5 MCL patients will also be used.
[0906] Kinase Inhibitors.
[0907] Inhibitors of the following kinases will be tested: CDK4/6 (PD0332991), BTK (PCI-32765), mTORC1 (rapamycin), MNK (4-Amino-5-(4-fluoroanilino)-pyrazolo[3,4-d]pyrimidine (Marzec M, et al. PLoS One 6:e24849); and a novel compound from Eli Lilly (Gupta M, et al. (2012) Blood 119:476-487); mTOR (OSI-027), and dual PI3K/mTOR (PF-04691502).
[0908] The compounds will be evaluated first at the pre-determined spectrum of effective doses, including the non-toxic concentrations reached in patients' sera, to assure optimal kinase inhibition.
[0909] Animals:
[0910] The in-vivo experiments will be performed using NOD-SCID-IL-2Rgc null (NSG) mice which are bred and available from the Stem Cell and Xenograft Core using breeders obtained from Jackson Laboratory (Bar Harbor). Mice will be housed in sterile conditions using HEPAfiltered microisolators and fed with irradiated food and acidified water.
[0911] Transplanted mice are treated with antibiotics (neomycin and polymixin) for the duration of the experiment. Six to eight week-old animals, equal mixes of males and females, will be utilized for all studies in accordance with protocols approved by the Institutional Animal Care and Use Committee.
[0912] We have used NSG animals in previous T cell adoptive transfer studies specifically to evaluate differential activity of CART19 cells (Witzig T E, et al. (2010) Hematology Am Soc Hematol Educ Program. 2010:265-270, MTT assay). The high throughput MTT assay to evaluate MCL cell growth will be performed first in response to the kinase inhibitors applied either alone or in various combinations. This assay is able to simultaneously determine cell proliferation rate and viability, allowing efficient evaluation of many possible combinations of small molecule inhibitors in the presence or absence of CART19 cells. The key aspects of this analysis will be to characterize the drug effect in regard to potential synergistic, additive, or antagonistic effect. In addition, the effect of the small molecule inhibitors on CART19 cells will be evaluated. While BTK inhibition should be B-cell specific, mTOR and CDK4/6 inhibition will affect CART19 cells. Establishing the proper timing of the drug application to minimize their potential effect on CART19 cells will be one of the aims of these experiments.
[0913] To perform the test, MCL cells will be seeded in 96-well plates at 1.times.10.sup.4 cells/well, in triplicates, and exposed to medium or kinase inhibitors in various combinations and various concentrations of CART-19 cells. After 48 and 74 hrs, the relative number of metabolically active cells will be determined by the use of MTT reduction colorimetric assay (Promega).
[0914] The significance of difference between the mean values (+/-S.D.) of the controls and different treatment conditions will be evaluated using Student's t-test with the P value of <0.05 considered to be statistically significant.
[0915] Cell Proliferation and Apoptosis Assays:
[0916] The most promising drug combinations will be next evaluated in the CFSE labeling and terminal dUTP nick-end labeling (tunel) assays to determine both cytostatic and cytotoxic components of MCL cell growth inhibition, respectively. In the former assay, MCL cells will be labeled with CFSE addition of the BTK inhibitor and/or unlabeled CART-19 cells. After 48 hrs, the cultured cells will be the analyzed by FACS for the CFSE labeling pattern of the MCL-type cells. The tunel assay will be done using the ApoAlert DNA Fragmentation Assay Kit from BD Biosciences according to the manufacturer's protocol.
[0917] In brief, MCL cells will be cultured with the inhibitors and/or CART19 cells for 48 or 72 hours. After being washed, cells will be stained with labeled anti-CD20 antibody and permeabilized, washed, and incubated in TdT buffer for 1 hour at 37.degree. C. The reaction will be stopped, the cells washed, resuspended, and analyzed by flow cytometry using the CellQuest PRO software.
[0918] CART19 Functional Assays:
[0919] We will measure effector activity of CART19 cells against MCL cell lines using CD107 degranulation, Intracellular Cytokine Secretion (ICS) assays, proliferationcytolysis assays, and multiplex cytokine detection assays (32). For degranulation and ICS assays, effector (T cells) and Target (tumor cells) will be co-incubated in the presence of anti-CD107 antibody for 4 hours at E:T of 0.2:1 followed by staining for surface (CAR19, CD3, CD8, CD4) and intracellular cytokine markers as per established protocols. Cytolysis of MCL cells will be assessed using flow-cytometry-based cytolysis assays. For proliferation assays, effector cells will be pre-loaded with CFSE (Carboxyfluorescein succinimidyl estercarboxy-fluoroscein-succinil esterase), mixed with target cells at E:T of 0.2:1, co-incubated at 37.degree. C. for 4 days, stained for surface markers (CAR19, CD3, CD8, CD4) and analyzed for dilution of CFSE by flow-cytometry.
[0920] Multiplex Cytokine Assays.
[0921] We will measure production of cytokines by CART19 cells in response to MCL targets using Luminex-based bead assays as described in (STM32). For these analyses we will employ the Invitrogen 30-plex kit that simultaneously measures IL-1.beta., IL-1RA, IL-2, IL-2R, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12 (p40/p70), IL-13, IL-15, IL-17, TNF-.alpha., IFN-.alpha., IFN-.gamma., GM-CSF, MIP-1.alpha., MIP-1.beta., IP-10, MIG, Eotaxin, RANTES, MCP-1, VEGF, G-CSF, EGF, FGF-basic, and HGF in serum, plasma, or tissue culture supernatant.
[0922] Multiparametric Flow Cytometry Analysis of CART19T Cells:
[0923] We will measure the modulation of surface markers associated with functional activation and suppression on CART19 cells following co-incubation with tumor cells using four color flow cytometry and a custom BD LSR II equipped with 4 lasers (blue (488 nM), violet (405 nM), Green (532 nM), and Red (633 nM) available through the University of Pennsylvania Abramson Cancer Center Flow Cytometry Core. All flow cytometry data will be analyzed using FlowJo software (TreeStar, San Carlos, Calif.). These analyses will be performed essentially as described in (STM42), using a dump channel to exclude dead cells and target cells (CD19+), and a CAR19 idiotype-specific reagent to detect CART19 cells (STM). We will evaluate the following markers on CART19-positive and -negative cells (CD3+/CD8+ and CD3+/CD4+) post co-incubation with tumor cells, on either intact or permeabilized cells as needed. We have established multi-parametric panels for these markers: [0924] activation/effector function: CD25, CD154, CD134, CD137, CD69, CD57, CD28, T-bet [0925] inhibition: CD152 (CTLA4), PD1, LAG3, CD200 [0926] suppression (Treg) CD4+/CD25++/CD127-, Fox-P3+
[0927] Simultaneously, MCL cells identified by CD19 and CD5 staining, will be examined for expression of the immunosuppressive proteins: CD174 (PD-L1) CD173 (PD-L2) and CD152.
[0928] Inhibitor Impact on Cell Signaling:
[0929] This part of the study will focus only on the selected compounds; the ones that proved to be the most effective in the functional assays (cell growth, proliferation, and apoptosis) described above. The effect will be studied separately for each drug and for the selected combinations and the studies will be adjusted to the specific compounds. For example, while the mTORC1 and MNK inhibitor combination will evaluate mTORC1 signaling, in particular the eIF-4E phosphorylation, BTK inhibition will focus on the PI3K-AKT and MEK-ERK pathways, and CDK4/6 inhibition on Rb phosphorylation. These studies will be performed by Western blotting using phospho-specific antibodies as described (Marzec M, et al. (2006) Blood. 108:1744-1750; Marzec M, et al. (2008) Blood 111: 2181-2189; Zhang Q, et al. (2011) Proc Natl Acad Sci USA 108: 11977-11982). In brief, the MCL cells will be lysed and the protein extracts will be assayed using the Lowry method (Bio-Rad) and loaded into the polyacrylamide gel. To examine protein phosphorylation, the blotted membranes will incubated with the phosphor-specific antibodies, for example the ones specific for S6rp S235/236, eIF4E S209, 4E-BP1 T37/46, 4E-BP1 T70 (Cell Signaling) to evaluate the mTORC1 and MNK activity and their inhibition. Next, the membranes will be incubated with the appropriate secondary, peroxidase-conjugated antibodies. The blots will be developed using the ECL Plus System from Amersham.
[0930] Genome-Scale Gene Expression Analysis:
[0931] Inhibition of cell signaling typically leads to changes in gene transcription. To determine the effects of the selected inhibitor, or a few inhibitors on gene transcription in MCL, a genome-scale gene expression analysis will be performed as done as described in Marzec M, et al. (2008) Blood 111: 2181-2189; Zhang Q, et al. (2011) Proc Natl Acad Sci USA 108: 11977-11982. In brief, the cells will be treated in triplicate cultures with the selected inhibitor or its diluent for 0, 4, and 8 hours. The total RNA will be further purified to enrich for mRNA which will be reverse transcribed, labeled and examined by hybridization to the Affimetrix microchip against all known gene exons. The microarray data will be normalized and summarized using RMA as implemented in GeneSpring and MASS algorithm. The resulting p values will be corrected for multiple testing using false discovery rate (FDR) by the Benjamini-Hochberg step-up method. Differential expression testing will be accomplished using a variety of tools including SAM and PartekPro. The emerging genes of interest will then be clustered based on expression patterns (GeneSpring or Spotfire), and clusters will be analyzed for functional groups and pathways in KEGG, Ingenuity Pathway Analysis, and Gene Ontology databases using the NIH-David as the search tool. For the genes identified based on the data, the independent expression conformation by the quantitative RTPCR will be performed on a larger pool of samples (at least 20) of various types of MCL (standard vs. blastoid and SOX11-positive vs. SOX11-negative).
[0932] Whole-Exome DNA Sequence Analysis:
[0933] To better characterize the MCL cases in regard to their pathogenesis and, to the extent possible, response to the proposed here combination therapies, the sequence of exomic DNA will be examined. Whole-exome capture and next generation sequencing of the MCL and normal peripheral blood DNA samples will performed using the NimbleGen Sequence Capture 2.1M Human Exome Array and the HiSeq 2000/1000 Illumina instrument.
[0934] Evaluation of the Treatment Effect in the Xenotransplanted Tumors:
[0935] The NSG mice will carry the MCL tumors tumors (derived from both MCL cell lines: Jeko and Mino and primary cells implanted as either tissue fragments or, less preferably, cell suspensions). The tumors will be propagated by subcutaneous implantation of the small tumor fragments. The therapy will be initiated once the tumors reach 0.2-0.3 cm in the diameter. The kinase inhibitor(s) will administered by gavage at dose and timing preselected in vitro (for example, we expect to apply BTK inhibitor simultaneously with CART19 cells, given its B-cell specificity and expected lack of any inhibitory effect on CART19 cells). CART-19 cells will be injected into the tail vein of the tumor-bearing mice at a dose of 1.times.10.sup.7/animal, the kinase inhibitor(s) will administered by gavage at dose and timing preselected in vitro. a dose we have established to be sufficient to reproducibly eradicate malignant cells and, at the same time, not to induce xeno-graft versus host disease. A large master stock of CART19 cells (lx 10.sup.10) will be generated and frozen to minimize variability associated with effector cell differences. The primary measure from these experiments will be survival, which we will assess using Kaplan/Meier curves. As a secondary measure we will evaluate differential expansion of CART19 cells in animals following T cell infusion. This will be made possible by the fact that the infused T cell product will be composed of CART19-positive and -negative cells at a defined ratio. For these analyses, animals will be bled weekly by tail vein bleed (25 microliters each time), followed by red blood cell lysis and staining for human CD3, CD4, CD8, and CART19. Preferential expansion of CART19 cells (at least a 2-fold increase in the CART19+/CART19-ratio will be evidence for selective MCL-driven CART19 cell expansion. To assess the treatment results, volumes of the implanted subcutaneous tumors will be measured determined as follows, according to the formula: volume=0.4ab2, where a and b designate respectively long and short diameters of the tumor. Tumor volumes differences between the treated and untreated groups of mice will be statistically analyzed using a standard t-test. Mice will be sacrificed at either the end-point of the experiments (>30 days), or if tumors reach >1.2 cm in diameter, or when any evidence of the animal distress noted. Tumor volumes differences between the treated and untreated groups of mice will be statistically analyzed using a standard t-test. The tumors as well as the internal organs will be harvested, processed and analyzed by histology and, for selected tissues, by immuno-histochemistry using the battery of antibodies against B cells (CD20, CD79a, Pax-5, CD10, BCL-6) and T cells (CD2, CD3, CD4, CD5, CD7, CD8, TIA-1), and the proliferation marker Ki-67.
[0936] Statistical Analysis:
[0937] In the in vitro functional studies, the significance of difference between the mean values (+/-S.D.) of the controls and different treatment conditions will be evaluated using Student's t-test with the P value of <0.05 considered to be statistically significant. Based on our previous experiences, the differences between the experimental mouse groups are expected to be large. Thus, 10 NSG mice will be used for each treatment group, which will ensure at least 90% power at 0.05 type 1 error level with a two-sided two sample/-test, given the ratio between the difference in treatment means and the standard deviation is at least 3, which is expected. Data will be presented as mean.+-.SEM. Comparison among groups will be made using the two sample t-test. A value of p<0.05 is considered to be significant. For tumor-free survival studies, groups of 10 mice will be used for survival comparison, and the disease status (tumor vs. no tumor) and tumor-free time for each mouse will be recorded. The Kaplan-Meier survival curve will be plotted and the log-rank test will be performed to compare the survival curves. The significance level is controlled at 0.05.
Example 8: Ibrutinib/CAR19 T-Cell Combined Therapy for Mantle Cell Lymphoma
[0938] The experiments described in this example characterize CART19 activity in combination with ibrutinib treatment for treating mantle cell lymphoma in vitro and in vivo. Ibrutinib is a small molecule inhibitor of BTK often used for treatment of some hematological cancers. The in vitro experiments described herein include assessment of proliferation, cytokine production, CD107a degranulation, and cytotoxicity. Xenoplant mouse models were utilized to investigate the efficacy and optimal dosage of CART19 with ibrutinib treatment in vivo. Although ibrutinib displays considerable activity in MCL, about 30% of patients do not respond, and among the responders, only 21% to about one-third experience complete remission (Wang et al. NEJM 369.6(2013):507-16). Achievement of a complete remission is associated with improved progression-free survival. Furthermore, therapy can lead to drug resistance with the duration of median response of 17.5 months. In some settings, mutations in BTK binding sites or immediately downstream have been observed after ibrutinib therapy, highlighting a mechanism of drug resistance that may become increasingly frequent. See, e.g., Woyach et al. NEJM. 370.24(2014):2286-94. Also, blockade of BTK function leads to inhibition of B cell receptor (BCR) signaling and is not directly cytotoxic. See, e.g., Ponader et al. Blood. 119.5(2012): 1182-89. Lack of cytotoxicity and failure to eradicate malignant clones predispose to clonal evolution under a selection pressure. Also, preliminary findings of increased transformation to aggressive disease in patients treated with ibrutinib for CLL are concerning. See, e.g., Byrd et al. NEJM. 369.1(2013):32-42; and Parikh et al. Blood. 123.11(2014):1647-57.
[0939] Infusion of autologous T cells transduced with chimeric antigen receptors (CAR) against the B-cell specific CD19 antigen (CTL019, CART19) leads to dramatic clinical responses in the majority of patients with various B-cell neoplasms, foremost acute lymphoblastic leukemia (ALL). See, e.g., Maude et al. NEJM. 371.16(2014):1507-17; and Ruella et al. Expert Opin. Biol. Ther. (2015):1-6. The presence of lymph node masses or bulky disease may lead to decreased T cell infiltration and consequent reduced anti-tumor activity. Bulky lymphadenopathy does not appear to impair the response to ibrutinib. Wang et al. NEJM. 369.6(2013):507-16. Also, ibrutinib has shown particular efficacy in reducing tumor masses and mobilizing neoplastic B cells in the peripheral blood.
Methods
Cell Lines and Primary Samples.
[0940] MCL cell lines were obtained from ATCC (Mino, Jeko-1, SP-49) while MCL-RL was generated from a progressive pleural effusion of a MCL patient. For in vitro experiments, cell lines were maintained in culture with RPMI media supplemented with 10% fetal calf serum, penicillin, and streptomycin. For some experiments, MCL-RL and Jeko-1 cells were transduced with click beetle green luciferase/eGFP and then sorted to obtain a >99% positive population. The acute leukemia cell lines MOLM-14, K562 or NALM-6 and the T-ALL cell line JURKAT were used as controls. These cell lines were originally obtained from the ATCC. De-identified primary human MCL bone marrow (BM) and peripheral blood (PB) specimens were obtained from the clinical practices of University of Pennsylvania. For all functional studies, primary cells were thawed at least 12 hours before experiment and rested at 37.degree. C.
Generation of CAR Constructs and CAR T Cells.
[0941] The murine anti-CD19 Chimeric antigen receptor (containing a CD8 hinge, 41BB costimulatory domain and CD3 zeta signaling domain) was generated as previously described. See, e.g., Milone et al. Molecular Therapy: the Journal of the American Society of Gene Therapy. 17.8(2009):1453-64. Production of CAR-expressing T cells was performed as previously described. See, e.g., Gill et al. Blood. 123.15(2014):2343-54. Normal donor CD4 and CD8 T cells or PB mononuclear cells (PBMC) were obtained from the Human Immunology Core of the University of Pennsylvania. T cells were plated at 1.times.10.sup.6/ml, with a CD4:CD8 ratio of 1:1 and expanded in X-vivo 15 media (Lonza, 04-418Q), human serum AB 5% (Gemini, 100-512), penicillin/streptomycin (Gibco, 15070063) and Glutamax (Gibco, 35050061) using anti-CD3/CD28 Dynabeads (Life Technologies, 11161D) added on the day 1 of culture and removed on day 6. T cells were transduced with lentivirus on day 2. T cells were expanded in culture for 8-15 days and harvested when the median cell volume was below 300 fl. T cells were then cryopreserved in FBS 10% DMSO for future experiments. Prior to all experiments, T cells were thawed and rested overnight at 37.degree. C.
Ibrutinib.
[0942] Ibrutinib (PCI-32765) was purchased from MedKoo (#202171) or Selleck Biochemicals (#S2680) as a powder or DMSO solution. For in vitro experiments, ibrutinib was diluted to the concentrations of 10, 100 and 1000 nM. For in vivo experiments, ibrutinib powder was dissolved in a 10% HP-beta-cyclodextrin solution (1.6 mg/ml) and administered to mice in the drinking water.
Multiparametric Flow Cytometry Analysis.
[0943] Anti-human antibodies were purchased from Biolegend, eBioscience, or Becton Dickinson. Cells were isolated from in vitro culture or from animals, washed once in PBS supplemented with 2% fetal calf serum, and stained for 15 minutes at room temperature. For cell number quantitation, Countbright (Invitrogen) beads were used according to the manufacturer's instructions. In all analyses, the population of interest was gated based on forward vs. side scatter characteristics followed by singlet gating, and live cells were gated using Live Dead Aqua (Invitrogen). Time gating was included for quality control. Surface expression of CAR19 was detected as previously described. See, e.g., Kalos et al. Science Translational Medicine. 3.95(2011):95ra73. Flow cytometry was performed on a four-laser Fortessa-LSR cytometer (Becton-Dickinson) and analyzed with FlowJo X 10.0.7r2 (Tree Star).
Degranulation Assay.
[0944] Degranulation assay was performed as previously described. See, e.g., Kalos et al. Science Translational Medicine. 3.95(2011):95ra73. T cells were incubated with target cells at a 1:5 ratio in T cell media. Anti-CD107a-PECY7 (Biolegend), anti-CD28 (BD Biosciences), anti-CD49d (BD Biosciences) antibodies and monensin (BD Biosciences) were added to the co-culture. After 4 hours, cells were harvested and stained for CAR expression, CD3, CD8 and Live Dead aqua staining (Invitrogen). Cells were fixed and permeabilized (Invitrogen Fix/Perm buffers) and intracellular staining was then performed to detect multiple cytokines (IFN, TNF.alpha., IL-2, GM-CSF, MIP1b).
Proliferation Assay.
[0945] T cells were washed and resuspended at 1.times.10.sup.7/ml in 100 ul of PBS and stained with 100 ul of CFSE 2.5 uM (Invitrogen) for 5 minutes at 37.degree. C. The reaction was then quenched with cold media, and cells were washed three times. Targets were irradiated at a dose of 100 Gy. T cells were incubated at a 1:1 ratio with irradiated target cells for 120 hours, adding media at 24 hours. Cells were then harvested, stained for CD3, CAR and Live Dead aqua (Invitrogen), and Countbright beads (Invitrogen) were added prior to flow cytometric analysis for absolute quantification.
Cytotoxicity Assays.
[0946] Luciferase/eGFP+ NALM-6 or RL cells were used for cytotoxicity assay as previously described. See, e.g., Gill et al. Blood. 123.15(2014):2343-54. Targets were incubated at the indicated ratios with effector T cells for 4 or 16 hours. Killing was calculated by bioluminescence imaging on a Xenogen IVIS-200 Spectrum camera.
[0947] Cytokine measurements. Effector and target cells were co-incubated at a 1:1 ratio in T cell media for 24 h. Supernatant was harvested and analyzed by 30-plex Luminex array (Luminex Corp, FLEXMAP 3D) according to the manufacturer's protocol (Invitrogen). See, e.g., Kalos et al. Science Translational Medicine. 3.95(2011):95ra73.
In Vivo Experiments.
[0948] NOD-SCID-.gamma. chain-/- (NSG) mice originally obtained from Jackson Laboratories were purchased from the Stem Cell and Xenograft Core of the University of Pennsylvania. All experiments were performed on protocols approved by the Institutional Animal Care and Use Committee (IACUC). Schematics of the utilized xenograft models are discussed herein. Cells (MCL cell lines or T cells) were injected in 200 ul of PBS at the indicated concentration into the tail veins of mice. Bioluminescent imaging was performed using a Xenogen IVIS-200 Spectrum camera and analyzed with LivingImage software v. 4.3.1 (Caliper LifeSciences). Animals were euthanized at the end of the experiment or when they met pre-specified endpoints according to the IACUC protocols.
Immunohistochemistry.
[0949] Immuno-histochemical (IHC) staining of formalin fixed paraffin embedded tissues was performed on a Leica Bond-III instrument using the Bond Polymer Refine Detection System. Antibodies against CD3, CD4, CD8, Pax5 and CyclinD1 were used undiluted. Heat-induced epitope retrieval was done for 20 minutes with ER2 solution (Leica Microsystems AR9640). Images were digitally acquired using the Aperio ScanScope.TM..
Statistical Analysis.
[0950] All statistics were performed using GraphPad Prism 6 for windows, version 6.04.
Mantle Cell Lymphoma Cell Lines
[0951] Most mantle cell lymphoma (MCL) lines in existence have been immortalized and propagated for many generations in vitro, thus losing their dependence on B cell receptor signaling. Consequently, they are poorly sensitive to ibrutinib. In vitro experiments were performed to determine the sensitivity of MCL cell lines on ibrutinib treatment. These cell lines were also used to assess the efficacy of ibrutinib/CART19 combination treatment in experiments discussed further in this example. MCL cells were harvested from the pleural effusion of a patient with multiply relapsed MCL. Both the original cells (RL.sup.primary) and a cell line derived from them (RL) had a blastoid morphology, typical MCL immunophenotype and were positive for the classical t(11;14) translocation by fluorescence in-situ hybridization (FISH) (FIG. 52A, 52B, 52C).
[0952] RL and JEKO-1 cells were cultured with different doses of ibrutinib (0.1 nM, 1 nM, 10 nM, 100 nM, 1 .mu.M, and 10 .mu.M) and sensitivity to ibrutinib was determined by measuring the reduction of bioluminescence (BLI). As shown in FIG. 31A, RL cells were sensitive to ibrutinib treatment in a dose-dependent manner. However, JEKO-1 cells were resistant to ibrutinib treatment (no demonstration of reduced bioluminescence as a function of increased ibrutinib dosage).
[0953] Sensitivity of RL, Jeko-1, and Mino cells to ibrutinib were also assayed using a MTT assay. Exposure of RL to increasing concentrations of ibrutinib in vitro led to a dose-dependent inhibition of proliferation and of downstream mediator phosphorylation, indicating an on-target effect of ibrutinib, with an IC50 of 10 nM. In contrast, the established MCL cell lines Jeko-1 and Mino were relatively resistant to ibrutinib, with IC50 results up to 10 .mu.M (FIG. 52D). To confirm RL as a viable model for in vivo experiments, immunodeficient NOD-SCID-.gamma. chain knockout (NSG) mice were engrafted with 1.times.10.sup.6 luciferase-expressing RL cells (FIG. 52E). Their tumor burden and survival were assessed. After intravenous injection, MCL engrafted in all mice and localized to the spleen and liver, followed by dissemination to bone marrow, blood and lymph nodes (FIG. 52F). Histology of affected spleen and liver is consistent with parenchymal infiltration of MCL. (FIG. 52G).
[0954] These results showed that these different cell lines could therefore be used to model both ibrutinib-sensitive and ibrutinib-resistant MCL.
Mantle Cell Lymphoma Cells are Sensitive to Killing by CART19
[0955] Most preclinical work showing the efficacy of CART19 has been done using B-ALL cell lines, which are not sensitive to ibrutinib. Furthermore, the best clinical responses to date have been reported in patients with B-ALL, whereas patients with indolent B-cell malignancies have reportedly lower responses.
[0956] To show that MCL is sensitive to killing by CART19 in this model, healthy donor T cells were transduced with an anti-CD19 CAR construct that has been used in clinical trials. See, e.g., Porter, NEJM 2011. A series of in vitro experiments were performed to show that the ibrutinib-sensitive cell line RL and the ibrutinib-resistant cell line Jeko-1 lead to equivalent CART-19 degranulation, cytokine production, killing and proliferation (FIG. 53A, 53B, 53C, 53D). In addition, peripheral blood or bone marrow was obtained from two patients with MCL in leukemic phase, permitting the expansion and transduction of autologous T cells with anti-CD19 CAR. Autologous patient-derived CART19 cells were reactive to MCL whereas the untransduced T cells were unreactive to their respective autologous MCL (FIG. 53E, 53F). These results indicate that MCL are sensitive to the effector functions of CART19.
Assessment of Ibrutinib/CART19 Treatment In Vitro
[0957] An effect of ibrutinib on T cells was previously discounted based on short-term activity assays, as described in Honigberg et al. Proc. Natl. Acad. Sci. USA. 107(2010):13075-80. Later, an analysis of the effect of ibrutinib on the T cell kinase ITK was reported to support an immunomodulatory role of ibrutinib on CD4 T cells by inhibiting Th2-type polarization. See Dubovsky et al. Blood 122.15(2013):2539-49. Cytokine analysis of patients treated with CART19 by several groups indicates that CART19 therapy is associated with both Th1 (IL2, IFN.gamma., TNF), Th2 (IL-4, IL-5, IL-10) and other cytokines (see, e.g., Kalos et al. Science Translational Medicine 3.95(2011):95ra73). In this example, the effect of CART19 function was evaluated with ibrutinib at, above, and below the ibrutinib concentrations that would be expected in patients (mean peak concentration in serum 100-150 ng/ml) See Advani et al. J. Clin. Oncol. 2013; 31:88.
[0958] CART19 cells were found to contain ITK. Non-specific stimulation of CART19 cells via the TCR in the presence of ibrutinib led to a reduction in phosphorylated ITK (pITK-Y.sub.180) as previously reported for CD4+ T cells. See Dubovsky et al. Blood 122.15(2013):2539-49. In contrast, specific stimulation of CART19 cells via the CAR did not lead to diminished ITK activation (FIG. 54A). This observation indicated that CART19 function would likely not be adversely affected by exposure to ibrutinib.
[0959] In addition, the short- and long-term in vitro function of CART19 cells in the presence of ibrutinib was determined. Ibrutinib at clinically relevant concentrations did not impair CART19 cell proliferation, degranulation or cytokine production; although at supra-physiologic concentrations, there was inhibition of CART19 cell functions, likely representing non-specific toxicity (FIG. 54B, 54C, 10B).
[0960] In particular, PBMCs isolated from a normal healthy donor was transduced with a lentiviral anti-CD19 CAR construct as described above. The resulting CAR19-expressing T cells (CART19) were cultured and passaged to determine proliferation and expansion capacity. A 1:1 ratio of CD4:CD8 expressing T cells were cultured culture with or without different concentrations of ibrutinib (10 nM, 100 nM, and 1000 nM ibrutinib). Ibrutinib was added at each cell passage. The number of cells was counted at day 0, day 5, day 6, day 7, day 9, and day 10 (FIG. 9A). Cell volume was also monitored (FIG. 9B).
[0961] CFSE-staining and flow cytometry analysis was also used to assess proliferation of the CART19 cells in the presence of ibrutinib after stimulation by tumor cell lines MOLM14, JEKO-1, and RL. MOLM14 is an AML cell line, and JEKO-1 and RL are mantle cell lymphoma cell lines. Specifically, the RL cells are a novel MCL cell line derived from neoplastic B-cells obtained from a pleural effusion of a relapsed MCL patient. CART19 cells and tumor cells were mixed in a 1:1 ratio and proliferation was assessed over 5 days. Percentage of proliferating cells are designated in each histogram in FIGS. 10A-1, 10A-2, 10A-3, and 10A-4. Quantification of proliferation as shown in FIG. 10B shows that high doses of ibrutinib might inhibit CART19 cell proliferation during co-culture with MCL cell lines.
[0962] Degranulation of T cells indicates the activation of cytolytic T cells and the ability to initiate antigen-specific cytotoxicity. CD107a is a functional marker of degranulation of T cells transiently expressed on the cell surface after T cell stimulation. Flow cytometry analysis was used to quantify CD107a-expressing CART19 T cells after stimulation with tumor cell lines MOLM14, JEKO-1, and RL. CD107a-expressing cells were present in Q2 (quadrant 2) of the cell profiles shown in FIGS. 11A-1, 11A-2, 11A-3, 11A-4, 11A-5, and 11A-6. Quantification of the results obtained from the profiles of FIGS. 11A-1, 11A-2, 11A-3, 11A-4, 11A-5, and 11A-6 is shown in FIG. 11B. These results indicate that co-culture of CART19 cells with either MCL-RL or JEKO-1 led to massive CAR-specific CD107a degranulation.
[0963] Cytokine production by CART19 T cells in the presence of ibrutinib was also quantified after stimulation by different tumor cell lines. IL-2, TNF-.alpha., and IFN-.gamma. production was assessed by flow cytometry. Cells producing the cytokines are present in quadrant 2 of the profiles shown in FIGS. 12-1, 12-1, 12-3, 12-4, 12-5, and 12-6, FIGS. 13-1, 13-2, 13-3, 13-4, 13-5, and 13-6, and FIGS. 14-1, 14-2, 14-3, 14-4, 14-5, and 14-6. CART19 T cells stimulated by JEKO-1 and RL showed an increase in cytokine expression. Also, increasing concentrations of ibrutinib treatment did not affect the percentage of the CART19 cytokine-producing cells.
[0964] Cytokine secretion from CART19 cells after stimulation by tumor cells and in the presence of varying concentration of ibrutinib was analyzed by 30-plex LUMINEX assay. Cytokines secreted by T.sub.H1 cells, such as IL-2, IFN-.gamma., and TNF-.alpha., was assayed. Cytokines secreted by T.sub.H2 cells, including IL-4, IL-5, IL-6, IL-10, IL-13, IL-15, IL-17, MIP1a, GM-CSF, MIP-1b, CP-1, IL-1Ra, IL-7, IP-10, IL-1b, VEGF, G-CSF, EGF, HGF, IFNa, IL-12, RANTES, Eotaxin, IL-2R, MIG, and IL-8, was assayed. As shown in FIGS. 15-1 through 15-10, T.sub.H1 and T.sub.H2 cytokines were secreted by the CART19 T cells.
[0965] Also, experiments using two different techniques indicated that there were no differences in Th1/Th2 polarization between ibrutinib-exposed and ibrutinib-unexposed CART19 cells (FIGS. 54D-1 and 54D-2).
[0966] Killing of MCL cells by CART19 cells was augmented in the presence of ibrutinib, suggesting an at least additive effect from the combination (FIG. 54E). However, intrinsic cytotoxic function of CART19 cells was not augmented in the presence of ibrutinib. (FIG. 54F).
[0967] Additional bioluminescence assays were performed to assess CART19 cell killing of tumor cells. CART19 cells were plated with tumor cells MOLM14, JEKO-1, and RL carrying a luciferase reporter in varying ratios, such as 1:1; 1:0.5; 1:0.25; and 1:0 in a 96 well plate, in duplicate. After 24 hours, the bioluminescence was detected and quantified. Results indicated that bioluminescence in MOLM14 samples did not decrease after incubation with CART19 cells. However, for JEKO-1 and RL cells, the bioluminescence decreased in the presence of CART19 cells, indicating that CART19 cells mediated JEKO-1 and RL cell killing (FIGS. 16A, 16B, 16C, 16D, 16E, and 16F). There was a further decrease in bioluminescence when treated with ibrutinib, indicating that the combination of ibrutinib and CART19 caused increased JEKO-1 and RL cell killing than with CART19 treatment alone. Consistent with these results, calculation of total cells after each treatment showed that CART19 treatment of JEKO-1 and RL cells caused reduction in cell number and a further reduction when treated with CART19 and ibrutinib (FIGS. 17B and 17C), suggesting that the combination therapy was not only efficacious for killing MCL cells, but led to efficient killing of MCL cell lines.
Assessment of Ibrutinib/CART19 Treatment In Vivo
[0968] In these experiments, mouse models of mantle cell lymphoma were used to assess CART19 and ibrutinib combination therapy in vivo.
[0969] Schematics such as those shown in FIGS. 20, 55, and 5A were used in this example. FIG. 20 shows a schematic of testing CART19/ibrutinib combination therapy in the in vivo mouse models of MCL. 1.times.10.sup.6 cells from RL (MCL-3), JEKO-1 (MCL-4), and NALM6 (MCL-5) cell lines are injected in NSG mice (20 mice for each experiment). After 1 week to allow engraftment, treatment is initiated at Day 0, in which the treatment is: 0.5-1.times.10.sup.6 CART19 cells (via injection), 25 mg/kg/day ibrutinib (oral gavage), or CART19+ ibrutinib treatment. At Day 7, Day 14, Day 21, and Day 28, the bioluminescence is imaged to monitor tumor size. Mice that are receiving ibrutinib treatment are continuously treated with ibrutinib. Survival of the mice is also monitored. FIGS. 55 and 56 show additional schematics of testing CART19/ibrutinib combination therapy in the in vivo mouse models of MCL. 2.times.106 MCL-RL cells are injected into NSG mice. After a week to allow engraftment (engraftment confirmed by bioluminescence imaging), treatment is initiated at Day 7 (after MCL-RL cell injection), in which treatment is: vehicle control, CART19 cells (2.times.10.sup.6 cells), and/or ibrutinib (125 mg/kg/day). At days 14, 21, 28, and 35 (after injection of MCL-RL cells), the bioluminescence is imaged to monitor tumor size.
[0970] The effect of ibrutinib treatment alone on an in vivo mouse model of MCL was examined. RL cells transfected with the GFP/luciferase gene were intravenously injected into immunodeficient NSG mice, resulting in 100% MCL engraftment in liver and spleen, with eventual spread into lymph nodes and bone marrow. Mice were treated with varying doses of ibrutinib, 25 mg/kg/day and 250 mg/kg/day. Mean bioluminescence, representing tumor growth, was assessed at various timepoints. As shown in FIG. 32, RL-derived tumors demonstrated dose-related sensitivity. Additional experiments titrating ibrutinib doses in RL cell-containing mice were performed and are shown in FIG. 57. A higher dose led to a better antitumor activity without increasing toxicity, in line with the higher dose of ibrutinib used in the clinic for MCL (Wang et al. NEJM 369.6(2013):507-16).
[0971] CART19 dose finding was also performed. Two MCL cell lines, RL (MCL-1) and JEKO-1 (MCL-2), carrying a GFP-luciferase reporter were injected into NSG mice at day 0. CART19 T cells were injected at day 7 at varying dosages, for example at 0.5.times.e6 cells, 1.times.e6 cells, or 2.times.e6 cells. Mice were monitored, for example, for 100 days. At various timepoints, the mice were monitored for tumor size (e.g., bioluminescence imaging) (FIGS. 18A and 18B, and FIG. 19A), and for overall survival (e.g., Kaplan-Meier survival curve) (FIG. 18C and FIG. 19B). Different doses of CART19 cells showing a dose-dependent anti-tumor efficacy, with 2.times.10.sup.6 CART19cells/mouse being the most effective dose (FIG. 19A).
[0972] These studies provided an opportunity to conduct a head-to-head comparison of the two of therapies for MCL. As shown in FIG. 58, long-term survival was achieved only in mice treated with CART-19 cells. There was no difference in anti-tumor effect when comparing untransduced T cells plus ibrutinib with ibrutinib alone. Therefore, in all subsequent experiments, the control groups were vehicle and ibrutinib alone (FIG. 59).
[0973] The addition of CART19 to ibrutinib was also tested, as detailed in the schematic in FIG. 56. Evaluation of the effect of ibrutinib on tumor burden indicated modestly delayed tumor growth at early time points. In contrast, CART19 cell therapy led to a clear decrease in tumor burden for several weeks. In mice receiving CART19 cells alone, this was followed by an indolent relapse beginning at Day 40, whereas mice that were treated with CART19 cells as well as ibrutinib had no detectable disease until Day 80 (FIG. 60). Histopathology of organs harvested at the end of the experiment showed persistence of disease in all control and ibrutinib treated mice with foci of tumor necrosis in the ibrutinib-treated. Most of the mice treated with CART19 alone showed indolent relapse at long term that was accompanied by the persistence of CART19 cells, while mice treated with CART19-ibrutinib showed clearance of the tumor and disappearance of CART19 from involved organs (data not shown).
Mechanism of Combined Effect of CART19 and Ibrutinib
[0974] The in vitro experiments herein indicated that ibrutinib neither impaired nor clearly augmented short-term CART19 effector functions. The in vivo studies showed that ibrutinib monotherapy had a modest anti-tumor effect. The results indicated that ibrutinib could significantly enhance the anti-tumor function of CART19 cells (FIG. 60). Therefore, experiments were performed to determine the mechanism for this effect.
[0975] Inhibition of ITK has been shown to inhibit Th2 polarization and skew towards a Th1 phenotype (Dubovsky et al. Blood 122.15(2013):2539-49). In mice treated with CART19 cells and ibrutinib, an increase in Th1 cells when compared with CART19 cell monotherapy was not observed using this assay (FIG. 61A, 61B). However, exposure of mice to ibrutinib led to an increase in peripheral CART19 cells. There was no difference in the proliferation marker Ki67 between the treatment group and a control group (FIG. 61C), so this assay did not detect a difference in proliferation. Similarly, there was no difference in the anti-apoptotic marker Bcl2 or the apoptosis marker phosphotidyl serine, suggesting that the difference in CART cell numbers was not related to an impairment of apoptosis (FIG. 61D). As ibrutinib has been associated with peripheral lymphocytosis in patients, experiments were done to determine whether this was also found in mice treated with ibrutinib alone. One week after beginning ibrutinib, there were more circulating MCL cells and fewer nodal/organ MCL cells in ibrutinib-treated mice. Interestingly, this increase was observed in both ibrutinib-sensitive and -resistant in vivo model, as it was also observed in NSG mice engrafted with the acute leukemia cell line NALM-6 (data not shown).
[0976] In order to understand the role of ibrutinib in T cell expansion in vivo we engrafted NSG mice with MCL-RL WT cells and treated them with luciferase-positive T cells. Both CTL019 and CTL019-ibrutinib treated mice showed intense T cell expansion compared to UTD or UTD-ibrutinib (data not shown). We then investigated the frequency of different T cells subsets in vivo and did not see differences in PB T cells 1 week after T cells infusion. (data not shown). Since CXCR4 is involved in ibrutinib-driven B cell mobilization in humans, we checked the expression of CXCR4 in vivo in PB T cells of mice treated with CTL019 or CTL019-ibrutinib: CXCR4 level were similar in the 2 groups (data not shown). Lastly, expression of inhibitory/costimulatory receptor on PB of T cells of mice treated with CART19 and CART19-ibrutinib was analized. No difference in expression of TIM3, LAG3, CD137 or CTLA4 was evident, however a trend to a reduced PD-1 expression was noted in mice treated with CTL019 and UTD combined with ibrutinib.
Conclusions
[0977] Therapies for B-cell malignancies include small molecule inhibitors of BCR signaling and CD19-directed T cell based therapies. In the setting of relapsed MCL, the BTK inhibitor ibrutinib is now approved by the FDA and engenders high initial response rates. Unfortunately, these responses tend to be transient and require higher drug doses than those used for CLL. CART-19 leads to durable responses in patients with high-risk B-ALL, and it may be efficacious in other B-cell malignancies as well. Preliminary data suggest that the responses of mature B-cell malignancies to CART-19 may be lower than those of B-ALL, but the mechanism of this disparity has not yet been ascertained. This example investigated the impact of adding ibrutinib to CART19 in the treatment of MCL.
[0978] Different MCL cell lines with variable sensitivities to ibrutinib (IC50 ranging from 10 nM to 10 pM) were used for in vitro experiments. These different cell lines were used to model both ibrutinib-sensitive and ibrutinib-resistant MCL. At all but the highest doses of ibrutinib, CART19 cell function was unimpaired, with intact T cell expansion kinetics, tumor recognition and killing, and cytokine production. Furthermore, the results did not reveal a T helper polarization upon ibrutinib exposure. This finding may be due to a combination of factors, including the use of a mixed culture of CD4 and CD8 cells, in contrast to the model of CD4-only experimentation performed by Dubovksy et al. Blood 122.15(2013):2539-49. Both ibrutinib-sensitive and ibrutinib-resistant cell lines strongly activated CART19 cells and induced killing, cytokine production and proliferation. Combination of CART19 and ibrutinib in vitro led to at least additive tumor killing. The results in this example show a superiority of CART19 over ibrutinib when each was used as monotherapy at clinically relevant doses and schedules of administration (single dose for CART19, continuous administration for ibrutinib).
[0979] A systemic xenograft MCL model was also generated in this example, using the MCL-RL cell line generated in a laboratory. Treatment of these mice with different doses of allogeneic CAR19 T cells led to a dose dependent anti-tumor effect. A similar dose response to CART-19 was also observed in the ibrutinib resistant JEKO-1 cell line. MCL-RL was treated in vivo with different doses (e.g., 0, 25 and 125 mg/kg/day) of Ibrutinib, leading to a median overall survival respectively of 70, 81 and 100 days (p<0.001). A direct in vivo comparison of the ibrutinib 125 mg/kg and CART19 showed a significantly improved tumor control for CART19 treated mice. Also, MCL-RL engrafted mice were treated with vehicle, ibrutinib, CART19 or the combination of CART19 and ibrutinib (iCART19). At clinically relevant doses, monotherapy of MCL with CART19 was superior over monotherapy with ibrutinib, and the combination of ibrutinib with CART19 led to an augmented anti-tumor effect. In particular, the iCART19 combination in vivo led to initially higher circulating levels of CART19 cells, followed by deep tumor responses, and relapses were significantly delayed when ibrutinib was added to CART19. The iCART19 combination resulted in an improved tumor control with 80% of mice reaching complete remission and long-term disease-free survival. Mechanistically, mice treated with ibrutinib had higher numbers of circulating CART19 cells without changes in Th1/Th2 or memory phenotype. Thus, the results herein show that ibrutinib can be combined with CART19 in a rational manner and suggest that the properties of each of these therapies may compensate for deficiencies of the other, thus leading to enhanced long-term anti-tumor effect. The experiments and results of combining BCR signaling inhibition with anti-CD19 directed T cell therapy pave the way to rational combinations of non-crossresistant therapies for B cell malignancies.
[0980] The kinetics of tumor response and relapse suggest that ibrutinib serves either to deepen the initial response achieved by CTL019 alone, or to enhance the long-term immunosurveillance capacity of CTL019 cells.
Example 9: Ibrutinib/CAR19 T-Cell Combined Therapy for Chronic Lymphocytic Leukemia
[0981] Ibrutinib is also utilized for treatment of chronic lymphocytic leukemia (CLL). Ibrutinib has not demonstrated cytotoxic effects on T cells or NK cells. However, in CLL cells, ibrutinib promotes programmed cell death and inhibits tumor cell migration and adhesion. In this example, experiments were performed to examine: 1) the effect of ibrutinib on CART19 production from patients undergoing ibrutinib therapy; and 2) the optimal timing of treatment with ibrutinib in combination with CART19 for optimal in vivo function.
Effect of Ibrutinib on CART19 Production and Function
[0982] Normal donor PBMCs were obtained using apheresis as described herein, for example in Example 6. The PBMCs were incubated with 5 .mu.M of ibrutinib for 30 minutes or were left untreated (for control), and then the cells were washed twice. The cells were then transduced with lentiviral constructs containing CAR19 to generate CART19 T cells using the methods described herein, for example, in Example 4. The number of CART19 T cells generated from ibrutinib-treated PBMCs FACs analysis was performed to determine the number of CART19 T cells generated from ibrutinib-treated PBMCs compared to untreated PBMCs. As shown in FIG. 21, 15% of the transduced ibrutinib-treated PBMCs expressed CAR19 and 12% of the transduced untreated PBMCs expressed CAR19. These results demonstrate that ibrutinib treatment does not affect lentiviral transduction efficiency or CART19 T cell production. Therefore, CART19 T cells can be manufactured from CLL patients undergoing ibrutinib treatment.
[0983] Further in vitro analysis was performed to determine the effect of ibrutinib on CART19 T cell proliferation, CART19 cytotoxicity, and the ratio of T.sub.H1:T.sub.H2 cytokine production.
[0984] For measuring cell proliferation, cells were stained with CFSE for detection of proliferating cells and analyzed by FACS, as described in Example 4. CART19 T cells were incubated with varying concentrations of ibrutinib (0.1 .mu.M, 0.5 .mu.M, 1 .mu.M, and 5 .mu.M), and were either stimulated with CD3/CD28 beads or were left unstimulated. In FIG. 22, histograms were overlayed to compare the unstimulated to the CD3/CD28 stimulated CART19 T cells at each concentration of ibrutinib. For each concentration of ibrutinib, CD3/CD28-stimulated T cell proliferation was observed, thereby demonstrating that ibrutinib treatment did not affect CART19 cell proliferation.
[0985] The effect of ibrutinib on cytotoxicity of CART19 T cells was also assessed, using methods described in Example 4. Untransduced and CAR19-transduced T cells were treated with media, DMSO, or 1.mu.M ibrutinib. A flow-based killing assay was performed using titrating effector to target (E;T) ratios with effector CART19 cells (media, DMSO, or ibrutinib-treated) to determined specific cytotoxic activity against CD19-expressing target cells. As shown in FIG. 23, CART19 T cells treated with ibrutinib demonstrated the same percentage of specific cytotoxic activity against CD19-expressing target cells as controls (CART19 T cells treated with media or DMSO). Thus, ibrutinib treatment does not affect CART19 cytotoxicity.
[0986] Ibrutinib treatment can limit T.sub.H2 activation, and therefore may promote T.sub.H1 selective pressure in T cells and skew T.sub.H1/T.sub.H2 cytokines in human CLL patients. Assays were performed to measure T.sub.H1 and T.sub.H2 cytokine production in the presence or absence of ibrutinib or DMSO (control). As shown in FIG. 24, ibrutinib does not promote skewing of T.sub.H1 and T.sub.H2 cytokines in CART19 cells.
Efficacy of Ibrutinib/CART19 Combination Treatment In Vivo
[0987] CART19 function was assessed in an in vivo mouse model. Nalm/6 cells (human acute lymphoblastic leukemia cell line) were implanted into NSG mice, and the mice were monitored daily for at least 50 days. The Nalm/6 tumor model produces tumors that are not sensitive to ibrutinib, and therefore allow analysis of CART19 function in vivo and efficacy in reducing tumor volume/treating cancer. Starting on day 7, mice were administered either DMSO (control) or ibrutinib daily by oral gavage. CART19 was administered on day 7 or day 9, or mice were left untreated (control). At days 4, 11, 18, 25 and 32, the number of Nalm/6 cells circulating in the mice were measured, e.g., by peripheral blood FACS analysis, to determine the efficacy of the CART19 for clearing Nalm/6 cells in the presence or absence of ibrutinib. For example, the cells were stained with anti-human CD19 antibody to determine the percent of human CD19+(Nalm/6) cells in the blood of the tumor-bearing NSG mice.
[0988] As shown in FIG. 25A, mice that did not receive CART19 injections exhibited an increase in tumor Nalm/6 cells. However, mice that received CART19 injections in combination with daily administration of DMSO showed successful clearing (reduction) of Nalm/6 cells. Mice that received CART19 injections in combination with daily administration of ibrutinib also showed successful clearing of the Nalm/6 cells with the same kinetics and efficacy as the mice that received DMSO treatment. Therefore, these results demonstrate that ibrutinib treatment does not impair CART19 function in clearing Nalm/6 cells from this tumor model.
[0989] Health status of the mice were monitored for at least 50 days after injection of the Nalm/6 cells. The Kaplan-Meier survival curve of FIG. 25B shows that none of the mice that were untreated (did not receive CART19 T cells) and received either DMSO or ibrutinib survived greater than 30 days after injection of the Nalm/6 cells. However, treatment with CART19 T cells, with DMSO or with ibrutinib increased the survival of the mice. Thus, these results taken together with the tumor burden results from FIGS. 13-1, 13-2, 13-3, 13-4, 13-5, and 13-6 demonstrate that ibrutinib treatment does not impair CART19 function in clearing tumor cells from the in vivo NSG-Nalm/6 tumor model.
Optimal Timing of Ibrutinib/CART19 Combination Treatment in CLL Patients
[0990] The optimal time during ibrutinib treatment of CLL for administration of CART19 was assessed using samples from CLL patients who were undergoing ibrutinib treatment for one year. PBMC samples from 9 CLL patients (Patient 111330026, Patient 111330030, Patient 111330039, Patient 111330056, Patient 111330073, Patient 111330074, Patient 111330081, Patient 111330086, and Patient 111330111) were isolated at different cycles of the ibrutinib treatment and were used to manufacture CART19 T cells. The PBMC samples were collected before ibrutinib treatment to establish a baseline, and then collected during ibrutinib treatment at cycle 2, day 1, and cycle 12, day 1. Several different parameters of CART19 manufacturing were assessed, such as transduction, proliferation, cyotoxicity, and cytokine production. Other evaluations can include ex vivo immunophenotyping, such as assessment of memory, inhibitor molecules, and exhaustion.
[0991] FIG. 26 shows the results from flow cytometry analysis from CAR19 transduction of T cells from Patient 111330030, which was representative of the results obtained from the other 8 patients after CAR transduction. PBMCs were collected from the patients at the indicated times (baseline, e.g., before treatment; cycle 2 at day 1; and cycle 12 at day 1) and were transduced with lentiviral vectors containing CAR19, using methods as described, for example, in Example 4. The transduced PBMCs were then stained for annexin, CD3, CD4, and GAM (to detect CAR), and then analyzed by FACS analysis. The boxed region in the graphs in FIGS. 14-1, 14-2, 14-3, 14-4, 14-5, and 14-6 shows the percentage of cells that were GAM positive, and successfully transduced to CART19 T cells. The lower three graphs show that CAR19 was successfully transduced in about 23% of cells at baseline, 28% of cells at cycle 2, day 1, and 44% of cells at cycle 12, day 1.
[0992] Next, the proliferation rate (or population doublings) of the CART19 cells or the untransduced cells (control) was assessed at each time point (baseline, cycle 2 at day 1, and cycle 12 at day 1) for 12 days. FIG. 27-1 through 27-3 shows the graphic representations of the population doublings over 12 days for three patients, Patient 111330039 (#39), Patient 111330026 (#26), and Patient 111330030 (#30). These results indicate that with regard to proliferation rate, PBMCs isolated between or at cycle 2, day 1 and cycle 12, day 1 are preferred for CAR transduction.
Potential Mechanisms for Ibrutinib Treatment Affecting CART19 Function
[0993] Various mechanisms of ibrutinib treatment that may affect CART19 function was assessed from CLL patient samples.
[0994] Analysis of CD19-expressing (CD19+) cells was assessed by FACS analysis. PBMCs from a CLL patient undergoing ibrutinib treatment were isolated at baseline (before ibrutinib treatment), at cycle 2, day 1, and at cycle 12, day 1, and were subsequently stained for CD19. FACS analysis showed that ibrutinib causes a decrease in CD19-expressing cells (FIG. 28A, FIG. 28B, and FIG. 28C). Thus, these results indicate that ibrutinib treatment induces lymphocytosis.
[0995] Additional analysis was performed to examine CD200 expression on tumor cells over time during ibrutinib treatment. The immune-suppressive molecule CD200 is up-regulated on primary B cell CLL tumor cells. CD200 binds to its receptor, CD200R, which is expressed on cells of the monocyte/macrophage lineage and on T lymphocytes. Interaction of CD200 with its receptor delivers an inhibitory signal to the macrophage lineage altering cytokine profiles from T.sub.H1 to T.sub.H2 and results in the induction of regulatory T cells. Samples from Patients 111330030, 111330026, and 111330039 at baseline (screen), cycle 2, day 1, and cycle 12, day 1 were stained for annexin, CD19 (to sort for CD19-expressing tumor cells), and CD200 and analyzed by FACS. The histograms detecting CD200 expression on tumor cells from each timepoint was overlaid for each patient (FIGS. 29A, 29B, and 29C). Generally, CD200 expression on tumor cells decreased over time during ibrutinib treatment.
[0996] The frequency of PD1-expressing T cells during ibrutinib treatment was also assessed. Samples from patients were obtained at baseline, cycle 2, day 1, and cycle 12, day 1 and stained with annexin, CD3, CD8, and PD1. Cells that were negative for annexin and positive for CD3 were analyzed for CD8 and PD1 expression. The cells that express CD8 and PD1 are designated by the box in FIGS. 30A, 30B, and 30C. Comparison of FACS profiles of cells at baseline (FIG. 30A), cycle 2, day 1 (FIG. 30B), and cycle 12, day 1 (FIG. 30C), indicates that ibrutinib treatment decreases the frequency of PD1-expressing cells over time.
[0997] The data obtained from the experiments described above indicate that the optimal time for administering CART19 therapy to CLL patients receiving ibrutinib is between cycle 2 and cycle 12, or at cycle 12.
Example 10: CAR19 T Cell Therapy for Hodgkin Lymphoma
[0998] CAR19 T cell therapy can also be used to treat Hodgkin lymphoma (HL). Hodgkin lymphoma is characterized by the presence of malignant Hodgkin Reed-Sternberg (HRS) cells that are derived from clonal germinal center B cells. There are several factors that indicate the therapeutic efficacy of CAR19 T cell therapy for HL. CD19 staining of HL tumors shows CD19-expressing (CD19.sup.+) cells within the tumor and tumor microenvironment (FIG. 33). A study has shown that a clonal B cell population (CD20+CD27+ ALDH.sup.+) that expresses CD19 is responsible for the generation and maintenance of Hodgkin lymphoma cell lines, and also circulates in the blood of most HL patients (Jones et al., Blood, 2009, 113(23):5920-5926). This clonal B cell population has also been suggested to give rise to or contribute to the generation of the malignant HRS cells. Thus, CART19 therapy would deplete this B cell population that contributes to tumorigenesis or maintenance of tumor cells. Another study showed that B cell depletion retards solid tumor growth in multiple murine models (Kim et al., J Immunotherapy, 2008, 31(5):446-57). In support of the idea that depletion of B cells in the HL tumormicroenvironment results in some anti-tumor effect, current therapies, such as rituxan, are being clinically tested for targeting and depletion of tumoral B cells in HL (Younes et al., Blood, 2012, 119(18):4123-8). De novo carcinogenesis related to chronic inflammation has also been shown to be B-cell dependent (de Visser, et al., Cancer Cell, 2005, 7(5):411-23). The results from these studies indicate that targeting of the B cell population, particularly in the HL tumor microenvironment, would be useful for treating HL, by reducing or inhibiting disease progression or tumor growth.
[0999] In addition, normal CD19-expressing B cells also infiltrate the tumor microenvironment in HL. Previous studies with CART19 therapy in CLL and ALL (e.g., described in Examples 4 and 5) show that CART19 exposure to CD19+ targets leads to cytokine production and macrophage production. Thus, modulation of the HL tumor microenvironment from a pro-tumor microenvironment to an anti-tumor microenvironment can be achieved by infusing CART19 to interact with normal CD19+B cells present in the HL. For example, CART19 exposure to CD19-expressing targets causes cytokine production, e.g., inflammatory cytokines, that promote anti-tumor activity through the expansion of cytotoxic T cells, activation of macrophages, and recruitment of other immune effector cells with various functions that inhibit tumor growth, such as leukocytes, macrophages, and antigen-presenting cells. Because the target CD19+B cells may not be malignant (e.g., normally circulating B cells), a transient rather than protracted CART19 effect may be preferred for modulation of the tumor microenvironment.
[1000] A study to examine the therapeutic efficacy of CART19 therapy in HL patients can be performed as described below (FIG. 34). The study will also assess the safety and tolerability of CART19 in HL subjects, and determine the effect of CART19 cells on the HL tumor microenvironment.
[1001] 8 patients with classical HL are treated in this study. Patients are of all ages, though separate protocols for drug delivery can be established for pediatric and adult patients. Patients in this study have no available potentially curative treatment options (such as autologous (ASCT) or allogeneic stem cell transplantation), or are not suitable for such curative treatment options. For example, patients can be any of the following: PET+ after salvage chemotherapy, PET+ after treatment with brentuximab, or PET+ after ASCT with or without prior brentuximab exposure. The patients will have a limited prognosis (several months to less than or equal to 2 year expected survival) with currently available therapies. And finally, the patients will not have received anti-CD20 antibody therapy. Patients are excluded due to lack of feasibility, e.g., if the patient has insufficient numbers of T cells for 6 infusions of CART19.
[1002] An mRNA CAR19 is produced by in vitro transcription. The CAR19 mRNA is electroporated into donor T cells, and the resulting cells are expanded and stimulated by incubation with CD3/CD28 beads. Dosages containing 1.times.10.sup.8-5.times.10.sup.8 RNA-electroporated CAR19 T cells are delivered to the patient three times a week for two weeks (e.g., at day 0, 2, 4, 7, 9 and 11). The overall response rate will be assessed by clinical, CT, and PET scanning at 1 month after treatment. Response and survival will be monitored monthly for the first 6 months, then every 3 months until 2 years after the first CART19 infusion (day 0). Monitoring techniques include biopsy of the tumor or lymph node (e.g., for immunohistochemical analysis and/or RNA for gene expression profiling) and PET scanning before and after CART19 treatment. For example, the effect of the CART19 cells on the HL tumor microenvironment are analyzed by comparing the results of gene expression profiling performed on accessible lymph node biopsies from selected patients before treatment and approximately one week after treatment (or the appropriate time after treatment to allow for alteration of cellular phenotype). To assess the safety and tolerability of CART19 treatment, the frequency and severity of adverse events are reported, including the frequency of cytokine release syndrome (CRS) and macrophage activation syndrome (MAS).
[1003] Chemotherapy may be administered concurrently with CART19 treatment. The first dose of CART19 can be preceded by lymphodepleting chemotherapy, e.g., cytoxan.
Example 11: Non-Responder Subset of CLL Patients Exhibit Increased Expression of Immune Checkpoint Inhibitor Molecules
[1004] In this study, CART19 cells from clinical manufacture from 34 CLL patients were assessed for expression of immune checkpoint inhibitor molecules, such as PD-1, LAG3, and TIM3. The response of this cohort to CART19 was known and hence a correlation between response and biomarker expression patterns could be assessed.
[1005] Manufactured CART19 cells from CLL patients with different responses to CART therapy were analyzed by flow cytometry to determine the expression of CAR and the immune checkpoint inhibitor molecules PD-1, LAG3, and TIM3. The CART19 cells were from: healthy donors (HD) (n=2); CLL patients that responded to CART therapy (CR) (n=5); CLL patients that partially responded to CART therapy (PR) (n=8); CLL patients that did not respond to CART therapy (NR) (n=21). Cells were stained with fluorescently labeled antibodies that specifically recognize CD3, CD4, CD8, CD27, CD45RO, the CAR19 molecule, and immune checkpoint molecules PD-1, LAG3, and TIM3, according to standard methods for flow cytometry analysis known in the art. Expression of each marker, e.g., CD4+, CD8+, etc., was determined by flow cytometry analysis software, and subpopulations (e.g., CD4+ T cells, CD8+ T cells, or CAR19-expressing T cells) were further analyzed for the expression of immune checkpoint molecules PD-1, LAG3, and TIM3.
[1006] An example of the flow cytometry profiles analysis used to determine surface marker expression is shown in FIGS. 35A and 35B. T cells expressing CD4 were determined using flow cytometry, and were further analyzed for CAR19 and PD-1 expression, such that the x-axis of the profiles indicate CAR19 expression (the top left (Q5) and bottom left (Q8) quadrants show the CAR19-negative CD4+ cells, while the top right (Q6) and bottom right (Q7) quadrants show the CAR19-expressing CD4+ cells) and the y-axis shows PD-1 expression (the bottom left (Q8) and right (Q7) quadrants show the PD-1 negative CD4+ cells and the top left (Q5) and right (Q6) quadrants show the PD-1-expressing CD4+ cells). In the CD4+ population from a CART responder, 44.7% of the CD4+ cells overall expressed PD-1, and about 22.3% of the CAR19-expressing cells were PD-1 positive, while 27.2% of CAR19-expressing cells were PD-1 negative (FIG. 35A). In contrast, in the CD4+ population from a non-responder, there was a significant decrease in CAR19-expressing cells overall (about 15.3% compared to the 49.5% in CR), with 14.7% of the CAR19-expressing cells being PD-1 positive while only 0.64% were PD-1 negative (FIG. 35B). Comparison between the profiles in FIG. 35A and FIG. 35B shows that a much higher percentage of the CD4+ cells from a non-responder express PD-1 (about 92.9%) compared to the CART responder (about 44.7%).
[1007] Using the methods and analysis described above, the percentage of PD-1 expressing (PD-1+) cells of the CD4+ population and the CD8+ population was determined for each patient in each response group. Non-responders were shown to have a greater percentage of PD-1+ cells in both the CD4+(FIG. 35C) and CD8+(FIG. 35D) populations compared to those that responded to CAR therapy (CR); the increase of average PD-1 percentage was statistically significant for both CD4+ and CD8+ populations. Partial responders (PR) exhibited higher percentages of PD-1+ cells than responders (CR) in both CD4+(FIG. 35C) and CD8+(FIG. 35D) populations.
[1008] Next, the percentage of PD-1 expressing (PD-1+) cells of the CAR19-expressing CD4+ population and the CAR19-expressing CD8+ population was determined for each patient in each response group. Similar analysis was performed as above, with the additional step of analyzing the CD4+ and CD8+ cells for CAR19-expression, and after identification of the CAR19-expressing cells, determining the percentage of cells with PD-1 expression from the populations of CAR19-expressing cells. A similar trend as that observed in the CD4+ and CD8+ overall populations was observed for the CAR19 expressing CD4+ and CD8+ populations: non-responders were shown to have a greater percentage of PD-1+ cells in both the CD4+(FIG. 36A) and CD8+(FIG. 36B) populations compared to those that responded to CAR therapy (CR); the increase of average PD-1 percentage was statistically significant for both CD4+ and CD8+ populations. Partial responders (PR) exhibited higher percentages of PD-1+ cells than responders (CR) in both CD4+(FIG. 36A) and CD8+(FIG. 36B) populations.
[1009] Further analysis was performed to determine the distribution of cells expressing PD-1, LAG3, and TIM3 from patients with different responses to CAR therapy. Representative cell profile analysis for PD-1, LAG3, and TIM3expression in the CD4+ population is shown in FIG. 37. The cell populations were first analyzed for CD4+ and CD8+ expression. The CD4+ population (or CD8+ population, not shown) was then analyzed for PD-1 and CAR19 expression (FIG. 37, left profiles). As described previously, non-responders (NR) had a significantly increased percentage of cells that were PD-1+ overall compared to CART responders (CR) (about 92.9% PD-1 positive for NR compared to 44.7% PD-1 positive for CR). Moreover, in non-responders, CAR19-expressing cells were mostly PD-1 positive (14.7% PD-1 positive and CAR+ compared to 0.64% PD-1 negative and CAR+). Then the populations were analyzed for PD-1 and LAG3 co-expression (FIG. 37, middle profiles). Cells that expressed both PD-1 and LAG3 are shown in the top right quadrant (Q2). Non-responders had a significantly increased percentage of cells that expressed both immune checkpoint inhibitors, PD-1 and LAG3, compared to CART responders (67.3% compared to 7.31%). PD-1 expression was also analyzed with TIM3 expression. In FIG. 37, right profiles, the box indicates the cells that express both PD-1 and TIM3. Similar to the results obtained with PD-1 and LAG3, the non-responders had a significantly higher percentage of cells that expressed both immune checkpoint inhibitors, PD-1 and TIM3, compared to CART responders (83.3% compared to 28.5%). The percentage of PD-1 expressing cells (PD1+), PD-1 and LAG3-expressing cells (PD1+ LAG3+), and PD-1 and TIM3-expressing cells (PD1+TIM3+) was determined for each patient in each response group using the flow cytometry analysis as described above. Non-responders were shown to have an increased percentage of PD1+ LAG3+ cells (FIG. 38A) and PD1+TIM3+ cells (FIG. 38B) compared to CART responders that was statistically significant for both cell populations. Partial responders also showed an increased percentage of both cell populations compared to CART responders, with the averages being decreased compared to the non-responders.
[1010] These results indicate that patients that do not respond to CAR therapy exhibit increased expression of immune checkpoint inhibitors (e.g., PD-1, LAG3, and TIM3) compared to patients that respond or partially respond to CAR therapy. Thus, these results show that agents that inhibit or decrease expression of immune checkpoint inhibitors, e.g., PD-1, LAG3, or TIM3, may be useful for administration to patients receiving CAR therapy to prevent immune suppression through immune checkpoint pathways (e.g., mediated by PD-1, LAG3, or TIM3), thereby increasing the efficacy of the CAR-expressing cells.
Example 12: Effects of mTOR Inhibition on Immunosenescence in the Elderly
[1011] One of the pathways most clearly linked to aging is the mTOR pathway. The mTOR inhibitor rapamycin has been shown to extend lifespan in mice and improve a variety of aging-related conditions in old mice (Harrison, D E et al. (2009) Nature 460:392-395; Wilkinson J E et al. (2012) Aging Cell 11:675-682; and Flynn, J M et al. (2013) Aging Cell 12:851-862). Thus, these findings indicate that mTOR inhibitors may have beneficial effects on aging and aging-related conditions in humans.
[1012] An age-related phenotype that can be studied in a short clinical trial timeframe is immunosenescence. Immunosenescence is the decline in immune function that occurs in the elderly, leading to an increased susceptibility to infection and a decreased response to vaccination, including influenza vaccination. The decline in immune function with age is due to an accumulation of immune defects, including a decrease in the ability of hematopoietic stem cells (HSCs) to generate naive lymphocytes, and an increase in the numbers of exhausted PD-1 positive lymphocytes that have defective responses to antigenic stimulation (Boraschi, D et al. (2013) Sci. Transl. Med. 5:185ps8; Lages, C S et al. (2010) Aging Cell 9:785-798; and Shimatani, K et al., (2009) Proc. Natl. Acad. Sci. USA 106:15807-15812). Studies in elderly mice showed that 6 weeks of treatment with the mTOR inhibitor rapamycin rejuvenated HSC function leading to increased production of naive lymphocytes, improved response to influenza vaccination, and extended lifespan (Chen, C et al. (2009) Sci. Signal. 2:ra75).
[1013] To assess the effects of mTOR inhibition on human aging-related phenotypes and whether the mTOR inhibitor RAD001 ameliorates immunosenescence, the response to influenza vaccine in elderly volunteers receiving RAD001 or placebo was evaluated. The findings presented herein suggest that RAD001 enhanced the response to influenza vaccine in elderly volunteers at doses that were well tolerated. RAD001 also reduced the percentage of programmed death (PD)-1 positive CD4 and CD8 T lymphocytes that accumulate with age. These results show that mTOR inhibition has beneficial effects on immunosenescence in elderly volunteers.
[1014] As described herein, a 6 week treatment with the mTOR inhibitor RAD001, an analog of rapamycin, improved the response to influenza vaccination in elderly human volunteers.
Methods
Study Population
[1015] Elderly volunteers >=65 years of age without unstable underlying medical diseases were enrolled at 9 sites in New Zealand and Australia. Exclusion criteria at screening included hemoglobin <9.0 g/dL, white blood cell count <3,500/mm.sup.3, neutrophil count <2,000/mm.sup.3, or platelet count <125,000/mm.sup.3, uncontrolled diabetes, unstable ischemic heart disease, clinically significant underlying pulmonary disease, history of an immunodeficiency or receiving immunosuppressive therapy, history of coagulopathy or medical condition requiring long-term anticoagulation, estimated glomerular filtration rate <30 ml/min, presence of severe uncontrolled hypercholesterolemia (>350 mg/dL, 9.1 mmol/L) or hypertriglyceridemia (>500 mg/dL, 5.6 mmol/L).
[1016] Baseline demographics between the treatment arms were similar (Table 7). Of the 218 subjects enrolled, 211 completed the study. Seven subjects withdrew from the study. Five subjects withdrew due to adverse events (AEs), one subject withdrew consent, and one subject left the study as a result of a protocol violation.
TABLE-US-00023 TABLE 8 Demographic and Baseline characteristics of the Study Patients RAD001 RAD001 RAD001 0.5 mg 5 mg 20 mg Placebo daily weekly weekly pooled Total Population N = 53 N = 53 N = 53 N = 59 N = 218 Age (Years) Mean (SD) 70.8 (5.0) 72.0 (5.3) 71.4 (5.2) 71.1 (5.1) 71.3 (5.2) Gender Male - n 34 (64%) 27 (51%) 32 (60%) 31 (53%) 124 (57%) (%) BMI* Mean (SD) 27.4 (4.2) 28.8 (5.0) 28.0 (4.1) 28.0 (4.2) 28.0 (4.4) (kg/m2) Race - n (%) Caucasian 48 (91%) 50 (94%) 46 (87%) 54 (92%) 198 (91%) Other 5 (9%) 3 (6%) 7 (13%) 5 (8%) 20 (9%) *The body-mass index is weight in kilograms divided by the square of the height in meters Study Design and Conduct
[1017] From December 2011 to April 2012, 218 elderly volunteers were enrolled in a randomized, observer-blind, placebo-controlled trial. The subjects were randomized to treatment arms using a validated automated randomization system with a ratio of RAD001 to placebo of 5:2 in each treatment arm. The treatment arms were:
[1018] RAD001 0.5 mg daily or placebo
[1019] RAD001 5 mg weekly or placebo
[1020] RAD001 20 mg weekly or placebo
[1021] The trial was observer-blind because the placebo in the RAD001 0.5 mg daily and 20 mg weekly cohorts differed slightly from the RAD001 tablets in those cohorts. The study personnel evaluating the subjects did not see the study medication and therefore were fully blinded. The treatment duration for all cohorts was 6 weeks during which time subjects underwent safety evaluations in the clinic every 2 weeks. After subjects had been dosed for 4 weeks, RAD001 steady state levels were measured pre-dose and at one hour post dose. After completing the 6 week course of study drug, subjects were given a 2 week drug free break to reverse any possible RAD001-induced immunosuppression, and then were given a 2012 seasonal influenza vaccination (Agrippal.RTM., Novartis Vaccines and Diagnostics, Siena, Italy) containing the strains H1N1 A/California/07/2009, H3N2 A/Victoria/210/2009, B/Brisbane/60/2008. Four weeks after influenza vaccination, subjects had serum collected for influenza titer measurements. Antibody titers to the 3 influenza vaccine strains as well as to 2 heterologous strains (A/H1N1 strain A/New Jersy/8/76 and A/H3N2 strain A/Victoria/361/11) were measured by standard hemagglutination inhibition assay (Kendal, A P et al. (1982) Concepts and procedures for laboratory-based influenza surveillance. Atlanta: Centers for Disease Control and Prevention B17-B35). Levels of IgG and IgM specific for the A/H1N1/California/07/2009 were measured in serum samples taken before and 4 weeks after influenza vaccination as described previously (Spensieri, F. et al. (2013) Proc. Natl. Acad. Sci. USA 110:14330-14335). Results were expressed as fluorescence intensity.
[1022] All subjects provided written informed consent. The study was conducted in accordance with the principals of Good Clinical Practice and was approved by the appropriate ethics committees and regulatory agencies.
Safety
[1023] Adverse event assessment and blood collection for hematologic and biochemical safety assessments were performed during study visits. Adverse event information was also collected in diaries that subjects filled out at home during the 6 weeks they were on study drug. Data on all adverse events were collected from the time of informed consent until 30 days after the last study visit. Events were classified by the investigators as mild, moderate or severe.
Statistical Analysis
[1024] The primary analysis of geometric mean titer ratios was done using a normal Bayesian regression model with non-informative priors. This model was fitted to each antibody titer on the log scale. The primary outcome in each model was the Day 84 measurement. The Day 63 measurement was included in the outcome vector. The model fitted using SAS 9.2 proc mixed with the prior statement. The covariance structure of the matrix was considered as unstructured (option type=UN). A flat prior was used. For the secondary analysis of seroconversion rates, logistic regression was used.
[1025] The intention to treat population was defined as all subjects who received at least one full dose of study drug and who had no major protocol deviations impacting efficacy data. 199 out of the total of 218 subjects enrolled in the study were in the intention to treat population.
Immunophenotyping
[1026] Peripheral blood mononuclear cells were isolated from whole blood collected at 3 time points: baseline; after 6 weeks of study drug treatment; and at the end of study when subjects had been off study drug for 6 weeks and 4 weeks after influenza vaccination. Seventy-six PBMC subsets were analyzed by flow cytometry using 8-color immunophenotyping panels at the Human Immune Monitoring Center at Stanford University, CA, USA as described previously (Maecker, H T et al. (2012) Nat Rev Immunol. 12:191-200). Seventy-six PBMC subsets were analyzed by flow cytometry using 8-color lyophilized immunophenotyping panels (BD Lyoplate, BD Biosciences, San Diego, Calif.). PBMC samples with viability >80% and yield of 2.times.10.sup.6 cells or greater were included in the analysis.
[1027] Relative changes of the immunophenotypes from baseline to Week 6 of study drug treatment and from baseline to the end of study (Week 12) were calculated for each of the RAD001 dosing cohorts. Student T test was conducted to examine if the relative change of the immunophenotypes from baseline to the two blood sampling time points was significantly different from zero, respectively, within each dosing group after adjusting for placebo effect. Missing data imputation in treatment effect analysis was not conducted. Therefore if a patient has a missing phenotype data at baseline, this patient was not be included in the analysis for this phenotype. If a patient had a missing phenotype data at 6 or 12 weeks, then this patient did not contribute to the analysis of this phenotype for the affected timepoint.
[1028] 608 tests in 76 phenotypes under 3 dosing groups were conducted to compare the treatment effect against the placebo effect. Stratified false discovery rate (FDR) control methodology was implemented to control the occurrence of false positives associated with multiple testing yet provide considerably better power. The cell type group was taken as the stratification factor and conducted FDR (q-value) calculation within each stratum respectively. All null-hypotheses were rejected at 0.05 significance level with corresponding q-value .ltoreq.0.1. The multiple testing adjustment strategy with rejecting at 0.05 significance level and corresponding q<0.1 ensured that less than 10% of the findings are false.
[1029] In a second analysis, the immunophenotype changes between pooled treatment and placebo groups, where all three RAD001 dosing groups were combined. To determine which immunophenotype changes differed between the treated and placebo groups, within-patient cell count ratios for each measured phenotype were calculated between baseline and Week 6 of study drug treatment and between baseline and the end of study (Week 12). The ratios were log transformed, and analyzed by analysis of covariance at each time point in order to detect a difference between the pooled treatment and placebo groups. 152 tests in 76 phenotypes were performed to compare the pooled treatment effect against the placebo effect. Stratified false discovery rate (FDR) control methodology was implemented to control the occurrence of false positives associated with multiple testing yet provide considerably better power (Benjamini, Y. et al. (1995) J. Roy. Statist. 57:289-300; and Sun, L. et al. (2006) Genet. Epidemiol. 30:519-530). The cell type group was taken as the stratification factor and FDR (q-value) calculation was conducted within each stratum respectively. All null-hypotheses at 0.05 significance level and q-value less than 20% were rejected. This can be interpreted as rejecting only those hypotheses with P values less than 0.05 and less than 20% probability that the each observed significant result is due to multiple testing.
Results
[1030] In general, RAD001 was well tolerated, particularly the 0.5 mg daily and 5 mg weekly dosing regimens. No deaths occurred during the study. Three subjects experienced four serious adverse events (SAEs) that were assessed as unrelated to RAD001. The 4 SAEs were retinal hemorrhage of the left eye with subsequent blindness in a subject with normal platelet counts who had completed a 6 week course of 5 mg weekly RAD001 6 weeks previously; severe back pain in a subject treated with placebo and severe gastroenteritis in a subject treated with placebo. A list of treatment-related adverse events (AEs) with an incidence >2% in any treatment group is provided in Table 9. The most common RAD001-related AE was mouth ulcer that, in the majority of cases, was of mild severity. Overall, subjects who received RAD001 had a similar incidence of severe AEs as those treated with placebo. Only one severe AE was assessed as related to RAD001 mouth ulcers in a subject treated with 20 mg weekly RAD001.
TABLE-US-00024 TABLE 9 Incidence of treatment-related AEs >2% in any treatment group by preferred term RAD001 RAD001 RAD001 Placebo, 0.5 mg daily 5 mg weekly 20 mg weekly pooled Total N = 53 N = 53 N = 53 N = 59 N = 218 n (%) n (%) n (%) n (%) n (%) Total AE(s) 35 46 109 21 211 Patients with AE(s) 22 (41.5%) 20 (37.7%) 27 (50.9%) 12 (20.3%) 81 (37.2%) Mouth ulceration 6 (11.3%) 2 (3.8%) 9 (17.0%) 3 (5.1%) 20 (9.2%) Headache 0 2 (3.8%) 9 (17.0%) 1 (1.7%) 12 (5.5%) Blood cholesterol 2 (3.8%) 2 (3.8%) 2 (3.8%) 0 6 (2.8%) increased Diarrhea 1 (1.9%) 4 (7.5%) 1 (1.9%) 0 6 (2.8%) Dyspepsia 0 3 (5.7%) 2 (3.8%) 1 (1.7%) 6 (2.8%) Fatigue 0 2 (3.8%) 4 (7.5%) 0 6 (2.8%) Low density lipoprotein 2 (3.8%) 1 (1.9%) 2 (3.8%) 0 5 (2.3%) increased Tongue ulceration 3 (5.7%) 1 (1.9%) 0 1 (1.7%) 5 (2.3%) Insomnia 1 (1.9%) 2 (3.8%) 1 (1.9%) 0 4 (1.8%) Dry mouth 0 0 2 (3.8%) 1 (1.7%) 3 (1.4%) Neutropenia 0 0 3 (5.7%) 0 3 (1.4%) Oral pain 0 2 (3.8%) 1 (1.9%) 0 3 (1.4%) Pruritus 0 2 (3.8%) 1 (1.9%) 0 3 (1.4%) Conjunctivitis 0 2 (3.8%) 0 0 2 (0.9%) Erythema 0 2 (3.8%) 0 0 2 (0.9%) Limb discomfort 0 2 (3.8%) 0 0 2 (0.9%) Mucosal inflammation 0 0 2 (3.8%) 0 2 (0.9%) Paresthesia oral 2 (3.8%) 0 0 0 2 (0.9%) Stomatitis 0 0 2 (3.8%) 0 2 (0.9%) Thrombocytopenia 0 0 2 (3.8%) 0 2 (0.9%) Urinary tract infection 0 0 2 (3.8%) 0 2 (0.9%)
[1031] The ability of RAD001 to improve immune function in elderly volunteers was evaluated by measuring the serologic response to the 2012 seasonal influenza vaccine. The hemagglutination inhibition (HI) geometric mean titers (GMT) to each of the 3 influenza vaccine strains at baseline and 4 weeks after influenza vaccination are provided in Table 10. The primary analysis variable was the HI GMT ratio (4 weeks post vaccination/baseline). The study was powered to be able to demonstrate that in at least 2 out of 3 influenza vaccine strains there was 1) a .gtoreq.1.2-fold GMT increase relative to placebo; and 2) a posterior probability no lower than 80% that the placebo-corrected GMT ratio exceeded 1. This endpoint was chosen because a 1.2-fold increase in the influenza GMT ratio induced by the MF-59 vaccine adjuvant was associated with a decrease in influenza illness (Iob, A et al. (2005) Epidemiol Infect 133:687-693).
TABLE-US-00025 TABLE 10 HI GMTs for each influenza vaccine strain at baseline and at 4 weeks after influenza vaccination Influenza RAD001 RAD001 5 mg RAD001 Vaccine 0.5 mg daily weekly 20 mg weekly Placebo Strain Time N = 50 N = 49 N = 49 N = 55 A/H1N1 GMT (CV %) Baseline 102.8 (186.9) 84.2 (236.4) 90.1 (188.4) 103.2 (219.7) Week 4 190.2 (236.9) 198.73 (195.6) 129.7 (175.9) 169.4 (259.8) GMT ratio 2.6 (302.5) 2.5 (214.3) 1.8 (201.5) 2.0 (132.7) (CV %) A/H3N2 GMT (CV %) Baseline 106.8 (168.2) 126.04 (162.6) 137.1 (211.5) 131.7 (162.3) Week 4 194.4 (129.1) 223.0 (118.8) 223.0 (163.6) 184.3 (153.2) GMT ratio 2.1 (152.6) 2.0 (189.2) 2.1 (277.3) 1.6 (153.6) (CV %) B GMT (CV %) Baseline 44.2 (96.6) 64.8 (87.3) 58.0 (156.0) 57.0 (112.6) Week 4 98.4 (94.8) 117.3 (99.9) 99.2 (124.1) 114.6 (136.7) GMT ratio 2.5 (111.2) 2.2 (112.8) 2.1 (126.5) 2.2 (109.2) (CV %) Baseline indicates 2 weeks prior to influenza vaccination Week 4 indicates 4 weeks after influenza vaccination N is number of subjects per cohort GMT is geometric mean titer GMT ratio is the GMT at week 4 post vaccination/GMT at baseline CV % indicates coefficient of variation
[1032] In the intent-to-treat (ITT) population, the low, immune enhancing, dose RAD001 (0.5 mg daily or 5 mg weekly) cohorts but not higher dose (20 mg weekly) cohort met the primary endpoint of the study (FIG. 40A). This demonstrates that there is a distinct immunomodulatory mechanism of RAD001 at the lower doses, and that at the higher dose the known immunosuppressive effects of mTOR inhibition may come into play. Furthermore, the results suggest a trend toward improved immune function in the elderly after low, immune enhancing, dose RAD001 treatment.
[1033] In a subgroup analysis, the subset of subjects with low baseline influenza titers (.ltoreq.1:40) experienced a greater RAD001-associated increase in titers than did the ITT population (FIG. 40B). These data show that RAD001 is particularly effective at enhancing the influenza vaccine response of subjects who did not have protective (>1:40) titers at baseline, and therefore were at highest risk of influenza illness.
[1034] Scatter plots of RAD001 concentration versus increase in titer to each influenza vaccine strain show an inverse exposure/response relationship (FIG. 41). Modeling and simulation based on mTOR mediated phosphorylation of S6 kinase (S6K) predicts that the 20 mg weekly dosing regimen inhibits mTOR-mediated S6K activity almost completely, the 5 mg weekly dosing regimen inhibits S6K activity by over 50%, and the 0.5 mg daily dosing regiment inhibits S6K phosphorylation by approximately 38% during the dosing interval (Tanaka, C et al. (2008) J. Clin. Oncol 26:1596-1602). Thus, partial mTOR inhibition, e.g., mTOR-mediated S6K phosphorylation, with low, immune enhancing, dose RAD001 may be as, if not more effective, than near complete mTOR inhibition with high dose RAD001 at enhancing the immune response of the elderly.
[1035] Rates of seroconversion 4 weeks after influenza vaccination were also evaluated. Seroconversion was defined as the change from a negative pre-vaccination titer (i.e., HI titer <1:10) to post-vaccination HI titer .gtoreq.1:40 or at least 4-fold increase from a non-negative (.gtoreq.1:10) pre-vaccination HI titer. In the intention-to-treat population, seroconversion rates for the H3N2 and B strains were increased in the RAD001 as compared to the placebo cohorts although the increases did not meet statistical significance (Table 11). In the subpopulation of subjects with baseline influenza titers <=1:40, RAD001 treatment also increased the rates of seroconversion to the H3N2 and B strains, and these results reached statistical significance for the B strain in the 0.5 mg daily dosing cohort. These data further show that RAD001 enhanced the serologic response to influenza vaccination in the elderly.
TABLE-US-00026 TABLE 11 Percent of subjects with seroconversion to influenza 4 weeks after vaccination Placebo 0.5 mg 5 mg 20 mg N = 54 N = 48 N = 49 N = 48 Intention to Treat Population H1N1 24 27 27 17 H3N2 17 27 24 25 B 17 27 22 19 Subjects with Baseline Titers <= 40 H1N1 40 42 45 36 H3N2 42 64 53 71 B 16 40* 33 28 *Odds ratio for seroconversion between RAD001 and Placebo significantly different than 1 (two-sided p-value < 0.05 obtained by logistic regression with treatment as fixed effect)
[1036] Current seasonal influenza vaccines often provide inadequate protection against continuously emerging strains of influenza that present as variants of previously circulating viruses. However, mice vaccinated against influenza in the presence of the mTOR inhibitor rapamycin, as compared to placebo, developed a broader serologic response to influenza. The broader serologic response included antibodies to conserved epitopes expressed by multiple subtypes of influenza that provided protection against infection with heterologous strains of influenza not contained in the vaccine (Keating, R et al. (2013) Nat Immunology 14:2166-2178). To determine if RAD001 broadened the serologic response to influenza in the elderly volunteers, HI titers to 2 heterologous strains of influenza not contained in the influenza vaccine (A/H1N1 strain A/New Jersey/8/76 and A/H3N2 strain A/Victoria/361/11) were measured. The increase in the HI GMT ratios for the heterologous strains was higher in the RAD001 as compared to placebo cohorts (FIG. 42). In addition, seroconversion rates for the heterologous strains were higher in the RAD001 as compared to placebo cohorts. The increase in seroconversion rates in the 5 and 20 mg weekly RAD001 dosing cohorts was statistically significant for the H3N2 heterologous strain (Table 12). The H3N2 seroconversion rate for the pooled RAD001 cohorts was 39% versus 20% for the placebo cohort (p=0.007). The results presented herein suggest that mTOR inhibition broadens the serologic response of elderly volunteers to influenza vaccination, and increases antibody titers to heterologous strains of influenza not contained in the seasonal influenza vaccine.
[1037] Broadened serologic response to heterologous strains of influenza in mice treated with rapamycin has been associated with an inhibition of class switching in B cells and an increase in anti-influenza IgM levels (Keating, R. et al. (2013) Nat Immunol 14:2166-2178). However, inhibition of class switching may not be involved in the broadened serologic response in humans treated with RAD001 because the post-vaccination anti-influenza IgM and IgG levels did not differ between RAD001 and placebo treated cohorts (FIG. 43).
TABLE-US-00027 TABLE 12 Percentage of subjects who seroconvert to heterologous strains of influenza 4 weeks after seasonal influenza vaccination Placebo, RAD001 RAD001 RAD001 pooled 0.5 mg daily 5 mg weekly 20 mg weekly A/H1N1 strain: 7% 17% 16% 8% A/NewJersey/8/76 A/H3N2 strain: 20% 38% 39%* 40% * A/Victoria/361/11 * Odds ratio for seroconversion between RAD001 and Placebo significantly different than 1 (two-sided p-value < 0.05 obtained by logistic regression with treatment as fixed effect)
[1038] To address the mechanism by which RAD001 enhanced immune function in elderly volunteers, immunophenotyping was performed on PBMC samples obtained from subjects at baseline, after 6 weeks of study drug treatment and 4 weeks after influenza vaccination (6 weeks after study drug discontinuation). Although the percentage of most PBMC subsets did not differ between the RAD001 and placebo cohorts, the percentage of PD-1 positive CD4 and CD8 cells was lower in the RAD001 as compared to placebo cohorts (FIG. 44). PD-1 positive CD4 and CD8 cells accumulate with age and have defective responses to antigen stimulation because PD-1 inhibits T cell receptor-induced T cell proliferation, cytokine production and cytolytic function (Lages, C S et al. (2010) Aging Cell 9:785-798). There was an increase in percentage of PD-1 positive T cells over time in the placebo cohort. At week 12 (4 weeks post-vaccination) this increase may have been due to influenza vaccination since influenza virus has been shown to increase PD-1 positive T cells (Erikson, J J et al. (2012) JCI 122:2967-2982). However the percentage of CD4 PD-1 positive T cells decreased from baseline at week 6 and 12 in all RAD001 cohorts (FIG. 44A). The percentage of CD8 PD-1 positive cells also decreased from baseline at both week 6 and 12 in the two lower dose RAD001 cohorts (FIG. 44B). The percentage of PD-1 negative CD4 T cells was evaluated and increased in the RAD001 cohorts as compared to the placebo cohorts (FIG. 44C).
[1039] Under more stringent statistical analysis, where the results from the RAD001 cohorts were pooled and adjusted for differences in baseline PD-1 expression, there was a statistically significant decrease of 30.2% in PD-1 positive CD4 T cells at week 6 in the pooled RAD cohort (n=84) compared to placebo cohort (n=25) with p=0.03 (q=0.13) (FIG. 45A). The decrease in PD-1 positive CD4 T cells at week 12 in the pooled RAD as compared to the placebo cohort is 32.7% with p=0.05 (q=0.19). FIG. 45B shows a statistically significant decrease of 37.4% in PD-1 positive CD8 T cells at week 6 in the pooled RAD001 cohort (n=84) compared to placebo cohort (n=25) with p=0.008 (q=0.07). The decrease in PD-1 positive CD8 T cells at week 12 in the pooled RAD001 as compared to the placebo cohort is 41.4% with p=0.066 (q=0.21). Thus, the results from FIGS. 44 and 45 together suggest that the RAD001-associated decrease in the percentage of PD-1 positive CD4 and CD8 T cells may contribute to enhanced immune function.
Conclusion
[1040] In conclusion, the data presented herein show that the mTOR inhibitor RAD001 ameliorates the age-related decline in immunological function of the human elderly as assessed by response to influenza vaccination, and that this amelioration is obtained with an acceptable risk/benefit balance. In a study of elderly mice, 6 weeks treatment with the mTOR inhibitor rapamycin not only enhanced the response to influenza vaccination but also extended lifespan, suggesting that amelioration of immunosenescence may be a marker of a more broad effect on aging-related phenotypes.
[1041] Since RAD001 dosing was discontinued 2 weeks prior to vaccination, the immune enhancing effects of RAD001 may be mediated by changes in a relevant cell population that persists after discontinuation of drug treatment. The results presented herein show that RAD001 decreased the percentage of exhausted PD-1 positive CD4 and CD8 T cells as compared to placebo. PD-1 expression is induced by TCR signaling and remains high in the setting of persistent antigen stimulation including chronic viral infection. While not wishing to be bound by theory, is possible that RAD001 reduced chronic immune activation in elderly volunteers and thereby led to a decrease in PD-1 expression. RAD001 may also directly inhibit PD-1 expression as has been reported for the immunophilin cyclosporine A (Oestreich, K J et al. (2008) J Immunol. 181:4832-4839). A RAD001-induced reduction in the percentage of PD-1 positive T cells is likely to improve the quality of T cell responses. This is consistent with previous studies showing that mTOR inhibition improved the quality of memory CD8 T cell response to vaccination in mice and primates (Araki, K et al. (2009) Nature 460:108-112). In aged mice, mTOR inhibition has also been shown to increase the number of hematopoietic stem cells, leading to increased production of naive lymphocytes (Chen, C et al. (2009) Sci Signal 2:ra75). Although significant differences in the percentages of naive lymphocytes in the RAD001 versus placebo cohorts were not detected in this example, this possible mechanism may be further investigated.
[1042] The mechanism by which RAD001 broadened the serologic response to heterologous strains of influenza may be further investigated. Rapamycin has also been shown to inhibit class switching in B cells after influenza vaccination. As a result, a unique repertoire of anti-influenza antibodies was generated that promoted cross-strain protection against lethal infection with influenza virus subtypes not contained in the influenza vaccine (Keating, R et al. (2013) Nat Immunol. 14:2166-2178). The results described herein did not show that RAD001 altered B cell class switching in the elderly subjects who had discontinued RAD001 2 weeks prior to influenza vaccination. Although the underlying mechanism requires further elucidation, the increased serologic response to heterologous influenza strains described herein may confer enhanced protection to influenza illness in years when there is a poor match between the seasonal vaccine and circulating strains of influenza in the community.
[1043] The effect of RAD001 on influenza antibody titers was comparable to the effect of the MF59 vaccine adjuvant that is approved to enhance the response of the elderly to influenza vaccination (Podda, A (2001) Vaccine 19:2673-2680). Therefore, RAD001-driven enhancement of the antibody response to influenza vaccination may translate into clinical benefit as demonstrated with MF59-adjuvanted influenza vaccine in the elderly (Iob, A et al. (2005) Epidemiol Infect. 133:687-693). However, RAD001 is also used to suppress the immune response of organ transplant patients. These seemingly paradoxical findings raise the possibility that the immunomodulatory effects of mTOR inhibitors may be dose and/or antigen-dependent (Ferrer, I R et al. (2010) J Immunol. 185:2004-2008). A trend toward an inverse RAD001 exposure/vaccination response relationship was seen herein. It is possible that complete mTOR inhibition suppresses immune function through the normal cyclophilin-rapamycin mechanism, whereas partial mTOR inhibition, at least in the elderly, enhances immune function due to a distinct aging-related phenotype inhibition. Of interest, mTOR activity is increased in a variety of tissues including hematopoietic stem cells in aging animal models (Chen C. et al. (2009) Sci Signal 2:ra75 and Barns, M. et al. (2014) Int J Biochem Cell Biol. 53:174-185). Thus, turning down mTOR activity to levels seen in young tissue, as opposed to more complete suppression of mTOR activity, may be of clinical benefit in aging indications.
[1044] The safety profile of mTOR inhibitors such as RAD001 in the treatment of aging-related indications has been of concern. The toxicity of RAD001 at doses used in oncology or organ transplant indications includes rates of stomatitis, diarrhea, nausea, cytopenias, hyperlipidemia, and hyperglycemia that would be unacceptable for many aging-related indications. However, these AEs are related to the trough levels of RAD001 in blood. Therefore the RAD001 dosing regimens used in this study were chosen to minimize trough levels. The average RAD001 trough levels of the 0.5 mg daily, 5 mg weekly and 20 mg weekly dosing cohorts were 0.9 ng/ml, below 0.3 ng/ml (the lower limit of quantification), and 0.7 ng/ml, respectively. These trough levels are significantly lower than the trough levels associated with dosing regimens used in organ transplant and cancer patients. In addition, the limited 6 week course of treatment decreased the risk of adverse events. These findings suggest that the dosing regimens used in this study may have an acceptable risk/benefit for some conditions of the elderly. Nonetheless, significant numbers of subjects in the experiments described herein developed mouth ulcers even when dosed as low as 0.5 mg daily. Therefore the safety profile of low, immune enhancing, dose RAD001 warrants further study. Development of mTOR inhibitors with cleaner safety profiles than currently available rapalogs may provide better therapeutic options in the future for aging-associated conditions.
Example 13: Enhancement of Immune Response to Vaccine in Elderly Subjects
[1045] Immune function declines in the elderly, leading to an increase incidence of infection and a decreased response to vaccination. As a first step in determining if mTOR inhibition has anti-aging effects in humans, a randomized placebo-controlled trial was conducted to determine if the mTOR inhibitor RAD001 reverses the aging-related decline in immune function as assessed by response to vaccination in elderly volunteers. In all cases, appropriate patent consents were obtained and the study was approved by national health authorities.
[1046] The following 3 dosing regimens of RAD001 were used in the study:
20 mg weekly (trough level: 0.7 ng/ml) 5 mg weekly (trough level was below detection limits) 0.5 mg daily (trough level: 0.9 ng/ml)
[1047] These dosing regimens were chosen because they have lower trough levels than the doses of RAD001 approved for transplant and oncology indications. Trough level is the lowest level of a drug in the body. The trough level of RAD001 associated with the 10 mg daily oncology dosing regimen is approximately 20 ng/ml. The trough level associated with the 0.75-1.5 mg bid transplant dosing regimen is approximately 3 ng/ml. In contrast, the trough level associated with the dosing regimens used in our immunization study were 3-20 fold lower.
[1048] Since RAD001-related AEs are associated with trough levels, the 3 dosing regimens were predicted to have adequate safety for normal volunteers. In addition, the 3 doses were predicted to give a range of mTOR inhibition. P70 S6 Kinase (P70 S6K) is a downstream target that is phosphorylated by mTOR. Levels of P70 S6K phosphorylation serve as a measure of mTOR activity. Based on modeling and simulation of P70 S6K phosphorylation data obtained in preclinical and clinical studies of RAD001, 20 mg weekly was predicted to almost fully inhibit mTOR activity for a full week, whereas 5 mg weekly and 0.5 mg daily were predicted to partially inhibit mTOR activity.
[1049] Elderly volunteers >=65 years of age were randomized to one of the 3 RAD001 treatment groups (50 subjects per arm) or placebo (20 subjects per arm). Subjects were treated with study drug for 6 weeks, given a 2 week break, and then received influenza (Aggrippal, Novartis) and pneumoccal (Pneumovax 23, Merck), vaccinations. Response to influenza vaccination was assessed by measuring the geometric mean titers (GMTs) by hemagglutination inhibition assay to the 3 influenza strains (H1N1, H3N2 and B influenza subtypes) in the influenza vaccine 4 weeks after vaccination. The primary endpoints of the study were (1) safety and tolerability and (2) a 1.2 fold increase in influenza titers as compared to placebo in 2/3 of the influenza vaccine strains 4 weeks after vaccination. This endpoint was chosen because a 1.2 fold increase in influenza titers is associated with a decrease in influenza illness post vaccination, and therefore is clinically relevant. The 5 mg weekly and 0.5 mg daily doses were well tolerated and unlike the 20 mg weekly dose, met the GMT primary endpoint (FIG. 40A). Not only did RAD001 improve the response to influenza vaccination, it also improved the response to pneumococcal vaccination as compared to placebo in elderly volunteers. The pneumococcal vaccine contains antigens from 23 pneumococcal serotypes. Antibody titers to 7 of the serotypes were measured in our subjects. Antibody titers to 6/7 serotypes were increased in all 3 RAD cohorts compared to placebo.
[1050] The combined influenza and pneumococcal titer data suggest that partial (less than 80-100%) mTOR inhibition is more effective at reversing the aging-related decline in immune function than more complete mTOR inhibition.
Example 14: Low Dose mTOR Inhibition Increases Energy and Exercise
[1051] In preclinical models, mTOR inhibition with the rapalog rapamycin increases spontaneous physical activity in old mice (Wilkinson et al. Rapamycin slows aging in mice. (2012) Aging Cell; 11:675-82). Of interest, subjects in the 0.5 mg daily dosing cohort described in Example 13 also reported increased energy and exercise ability as compared to placebo in questionnaires administered one year after dosing (FIG. 46). These data suggest that partial mTOR inhibition with rapalogs may have beneficial effects on aging-related morbidity beyond just immune function.
Example 15: P70 S6 Kinase Inhibition with RAD001
[1052] Modeling and simulation were performed to predict daily and weekly dose ranges of RAD001 that are predicted to partially inhibit mTOR activity. As noted above, P70 S6K is phosphorylated by mTOR and is the downstream target of mTOR that is most closely linked to aging because knockout of P70 S6K increases lifespan. Therefore modeling was done of doses of RAD001 that partially inhibit P70 S6K activity. Weekly dosing in the range of >=0.1 mg and <20 mg are predicted to achieve partial inhibition of P70 S6K activity (FIG. 47).
[1053] For daily dosing, concentrations of RAD001 from 30 pM to 4 nM partially inhibited P70 S6K activity in cell lines (Table 13). These serum concentrations are predicted to be achieved with doses of RAD001 >=0.005 mg to <1.5 mg daily.
TABLE-US-00028 TABLE 13 Percent inhibition of P70 S6K activity in HeLa cells in vitro RAD001 6 32 160 800 4 20 concentration 0 pM pM pM pM nM nM % P70 S6K 0 0 18 16 62 90 95 inhibition
Conclusion
[1054] Methods of treating aging-related morbidity, or generally enhancing an immune response, with doses of mTOR inhibitors that only partially inhibit P70 S6K. The efficacy of partial mTOR inhibition with low doses of RAD001 in aging indications is an unexpected finding. RAD001 dose ranges between >=0.1 mg to <20 mg weekly and >=0.005 mg to <1.5 mg daily will achieve partial mTOR inhibition and therefore are expected to have efficacy in aging-related morbidity or in the enhancement of the immune response.
Example 16: Exogenous IL-7 Enhances the Function of CAR T Cells
[1055] After adoptive transfer of CAR T cells, some patients experience limited persistence of the CAR T cells, which can result in suboptimal levels of anti-tumor activity. In this example, the effects of administration of exogenous human IL-7 is assessed in mouse xenograft models where an initial suboptimal response to CAR T cells has been observed.
[1056] Expression of the IL-7 receptor CD127 was first assessed in different cancer cell lines and in CAR-expressing cells. Two mantle cell lymphoma cell lines (RL and Jeko-1) and one B-ALL cell line (Nalm-6) were analyzed by flow cytometry for CD127 expression. As shown in FIG. 48A, out of the three cancer cell lines tested, RL was shown to have the highest expression of CD127, followed by Jeko-1 and Nalm-6. CART19 cells were infused into NSG mice and CD127 expression was assessed on the circulating CART19 cells by flow cytometry. As shown in FIG. 48B, CD127 is uniformly expressed on all circulating CART19 cells.
[1057] Next, the effect of exogenous IL-7 treatment on anti-tumor activity of CART19 cells was assessed in a lymphoma animal model. NSG mice were engrafted with a luciferase-expressing mantle cell line (RL luc) on Day 0 (D0), followed by treatment of CART19 cells on Day 6. The NSG mice were divided into groups, where one group received no CART19 cells, a second group received 0.5.times.10.sup.6 CART19 cells, a third group received 1.times.10.sup.6 CART19 cells, and a fourth group received 2.times.10.sup.6 CART19 cells. Tumor size was monitored by measuring the mean bioluminescence of the engrafted tumors over more than 80 days. Only mice receiving 2.times.10.sup.6 CART19 cells demonstrated rejection of the tumor and inhibition of tumor growth (FIG. 49A). Mice from the two groups receiving 0.5.times.10.sup.6 CART19 cells or 1.times.10.sup.6 CART19 cells were shown to s a suboptimal anti-tumor response. Mice from these two groups were then randomized, where three mice (mouse #3827 and #3829 which received 0.5.times.10.sup.6 CART19 cells, and mouse #3815 which received 1.times.10.sup.6 CART19 cells) received exogenous recombinant human IL-7 at a dosage of 200 ng/mouse by intraperitoneal injection three times weekly starting at Day 85, and two mice did not. The tumor burden of mice receiving exogenous IL-7 from Day 85-125, as detected by mean bioluminescence, is shown in FIG. 49B. All mice receiving IL-7 showed a dramatic response of 1-3 log reduction in tumor burden. Mice that originally received a higher dose of CART19 cells (mouse #3815 which received 1.times.10.sup.6 CART19 cells) showed a more profound response. When comparing the tumor burden of mice that received IL-7 treatment to control, before and after IL-7 treatment, tumor reduction in tumor burden was only seen in the mice that had received IL-7 treatment (FIG. 49C).
[1058] T cell dynamics following IL-7 treatment in the lymphoma animal model was also examined. Human CART19 cells were not detectable in the blood prior to IL-7 treatment. Upon treatment of IL-7, there was rapid, but variable increase in the numbers of T cells in the treated mice (FIG. 50A). The extent of T cell expansion observed in mice receiving the IL-7 also correlated with tumor response. The mouse with the highest number of T cells detected in the blood at peak expansion during IL-7 treatment (mouse #3815) had the most robust reduction in tumor burden (see FIG. 49B). Moreover, the time of peak expansion correlated with the T cell dose injected as baseline. The number/level CD3-expressing cells in the blood were also measured before and after IL-7 treatment. In control mice, very few CD3-expressing cells were detected, while IL-7-treated mice showed a significant increase in CD3+ cells after IL-7 treatment (FIG. 50B).
[1059] Together, the results in this example demonstrate that exogenous IL-7 treatment increases T cell proliferation and anti-tumor activity in vivo, indicating that use of IL-7 in patients with suboptimal results after CAR therapy can improve anti-tumor response in these patients.
EQUIVALENTS
[1060] The disclosures of each and every patent, patent application, and publication cited herein are hereby incorporated herein by reference in their entirety. While this invention has been disclosed with reference to specific aspects, it is apparent that other aspects and variations of this invention may be devised by others skilled in the art without departing from the true spirit and scope of the invention. The appended claims are intended to be construed to include all such aspects and equivalent variations.
Sequence CWU 1
1
1321242PRTArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic polypeptide" 1Glu Ile Val Met Thr Gln Ser Pro
Ala Thr Leu Ser Leu Ser Pro Gly1 5 10 15Glu Arg Ala Thr Leu Ser Cys
Arg Ala Ser Gln Asp Ile Ser Lys Tyr 20 25 30Leu Asn Trp Tyr Gln Gln
Lys Pro Gly Gln Ala Pro Arg Leu Leu Ile 35 40 45Tyr His Thr Ser Arg
Leu His Ser Gly Ile Pro Ala Arg Phe Ser Gly 50 55 60Ser Gly Ser Gly
Thr Asp Tyr Thr Leu Thr Ile Ser Ser Leu Gln Pro65 70 75 80Glu Asp
Phe Ala Val Tyr Phe Cys Gln Gln Gly Asn Thr Leu Pro Tyr 85 90 95Thr
Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys Gly Gly Gly Gly Ser 100 105
110Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gln Val Gln Leu Gln Glu
115 120 125Ser Gly Pro Gly Leu Val Lys Pro Ser Glu Thr Leu Ser Leu
Thr Cys 130 135 140Thr Val Ser Gly Val Ser Leu Pro Asp Tyr Gly Val
Ser Trp Ile Arg145 150 155 160Gln Pro Pro Gly Lys Gly Leu Glu Trp
Ile Gly Val Ile Trp Gly Ser 165 170 175Glu Thr Thr Tyr Tyr Ser Ser
Ser Leu Lys Ser Arg Val Thr Ile Ser 180 185 190Lys Asp Asn Ser Lys
Asn Gln Val Ser Leu Lys Leu Ser Ser Val Thr 195 200 205Ala Ala Asp
Thr Ala Val Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly 210 215 220Gly
Ser Tyr Ala Met Asp Tyr Trp Gly Gln Gly Thr Leu Val Thr Val225 230
235 240Ser Ser2242PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 2Glu Ile Val Met Thr Gln
Ser Pro Ala Thr Leu Ser Leu Ser Pro Gly1 5 10 15Glu Arg Ala Thr Leu
Ser Cys Arg Ala Ser Gln Asp Ile Ser Lys Tyr 20 25 30Leu Asn Trp Tyr
Gln Gln Lys Pro Gly Gln Ala Pro Arg Leu Leu Ile 35 40 45Tyr His Thr
Ser Arg Leu His Ser Gly Ile Pro Ala Arg Phe Ser Gly 50 55 60Ser Gly
Ser Gly Thr Asp Tyr Thr Leu Thr Ile Ser Ser Leu Gln Pro65 70 75
80Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln Gly Asn Thr Leu Pro Tyr
85 90 95Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys Gly Gly Gly Gly
Ser 100 105 110Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gln Val Gln
Leu Gln Glu 115 120 125Ser Gly Pro Gly Leu Val Lys Pro Ser Glu Thr
Leu Ser Leu Thr Cys 130 135 140Thr Val Ser Gly Val Ser Leu Pro Asp
Tyr Gly Val Ser Trp Ile Arg145 150 155 160Gln Pro Pro Gly Lys Gly
Leu Glu Trp Ile Gly Val Ile Trp Gly Ser 165 170 175Glu Thr Thr Tyr
Tyr Gln Ser Ser Leu Lys Ser Arg Val Thr Ile Ser 180 185 190Lys Asp
Asn Ser Lys Asn Gln Val Ser Leu Lys Leu Ser Ser Val Thr 195 200
205Ala Ala Asp Thr Ala Val Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly
210 215 220Gly Ser Tyr Ala Met Asp Tyr Trp Gly Gln Gly Thr Leu Val
Thr Val225 230 235 240Ser Ser3242PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 3Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu Val Lys
Pro Ser Glu1 5 10 15Thr Leu Ser Leu Thr Cys Thr Val Ser Gly Val Ser
Leu Pro Asp Tyr 20 25 30Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys
Gly Leu Glu Trp Ile 35 40 45Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr
Tyr Ser Ser Ser Leu Lys 50 55 60Ser Arg Val Thr Ile Ser Lys Asp Asn
Ser Lys Asn Gln Val Ser Leu65 70 75 80Lys Leu Ser Ser Val Thr Ala
Ala Asp Thr Ala Val Tyr Tyr Cys Ala 85 90 95Lys His Tyr Tyr Tyr Gly
Gly Ser Tyr Ala Met Asp Tyr Trp Gly Gln 100 105 110Gly Thr Leu Val
Thr Val Ser Ser Gly Gly Gly Gly Ser Gly Gly Gly 115 120 125Gly Ser
Gly Gly Gly Gly Ser Glu Ile Val Met Thr Gln Ser Pro Ala 130 135
140Thr Leu Ser Leu Ser Pro Gly Glu Arg Ala Thr Leu Ser Cys Arg
Ala145 150 155 160Ser Gln Asp Ile Ser Lys Tyr Leu Asn Trp Tyr Gln
Gln Lys Pro Gly 165 170 175Gln Ala Pro Arg Leu Leu Ile Tyr His Thr
Ser Arg Leu His Ser Gly 180 185 190Ile Pro Ala Arg Phe Ser Gly Ser
Gly Ser Gly Thr Asp Tyr Thr Leu 195 200 205Thr Ile Ser Ser Leu Gln
Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln 210 215 220Gln Gly Asn Thr
Leu Pro Tyr Thr Phe Gly Gln Gly Thr Lys Leu Glu225 230 235 240Ile
Lys4242PRTArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic polypeptide" 4Gln Val Gln Leu Gln Glu Ser Gly
Pro Gly Leu Val Lys Pro Ser Glu1 5 10 15Thr Leu Ser Leu Thr Cys Thr
Val Ser Gly Val Ser Leu Pro Asp Tyr 20 25 30Gly Val Ser Trp Ile Arg
Gln Pro Pro Gly Lys Gly Leu Glu Trp Ile 35 40 45Gly Val Ile Trp Gly
Ser Glu Thr Thr Tyr Tyr Gln Ser Ser Leu Lys 50 55 60Ser Arg Val Thr
Ile Ser Lys Asp Asn Ser Lys Asn Gln Val Ser Leu65 70 75 80Lys Leu
Ser Ser Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys Ala 85 90 95Lys
His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp Tyr Trp Gly Gln 100 105
110Gly Thr Leu Val Thr Val Ser Ser Gly Gly Gly Gly Ser Gly Gly Gly
115 120 125Gly Ser Gly Gly Gly Gly Ser Glu Ile Val Met Thr Gln Ser
Pro Ala 130 135 140Thr Leu Ser Leu Ser Pro Gly Glu Arg Ala Thr Leu
Ser Cys Arg Ala145 150 155 160Ser Gln Asp Ile Ser Lys Tyr Leu Asn
Trp Tyr Gln Gln Lys Pro Gly 165 170 175Gln Ala Pro Arg Leu Leu Ile
Tyr His Thr Ser Arg Leu His Ser Gly 180 185 190Ile Pro Ala Arg Phe
Ser Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu 195 200 205Thr Ile Ser
Ser Leu Gln Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln 210 215 220Gln
Gly Asn Thr Leu Pro Tyr Thr Phe Gly Gln Gly Thr Lys Leu Glu225 230
235 240Ile Lys5247PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 5Glu Ile Val Met Thr Gln
Ser Pro Ala Thr Leu Ser Leu Ser Pro Gly1 5 10 15Glu Arg Ala Thr Leu
Ser Cys Arg Ala Ser Gln Asp Ile Ser Lys Tyr 20 25 30Leu Asn Trp Tyr
Gln Gln Lys Pro Gly Gln Ala Pro Arg Leu Leu Ile 35 40 45Tyr His Thr
Ser Arg Leu His Ser Gly Ile Pro Ala Arg Phe Ser Gly 50 55 60Ser Gly
Ser Gly Thr Asp Tyr Thr Leu Thr Ile Ser Ser Leu Gln Pro65 70 75
80Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln Gly Asn Thr Leu Pro Tyr
85 90 95Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys Gly Gly Gly Gly
Ser 100 105 110Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly
Gly Ser Gln 115 120 125Val Gln Leu Gln Glu Ser Gly Pro Gly Leu Val
Lys Pro Ser Glu Thr 130 135 140Leu Ser Leu Thr Cys Thr Val Ser Gly
Val Ser Leu Pro Asp Tyr Gly145 150 155 160Val Ser Trp Ile Arg Gln
Pro Pro Gly Lys Gly Leu Glu Trp Ile Gly 165 170 175Val Ile Trp Gly
Ser Glu Thr Thr Tyr Tyr Ser Ser Ser Leu Lys Ser 180 185 190Arg Val
Thr Ile Ser Lys Asp Asn Ser Lys Asn Gln Val Ser Leu Lys 195 200
205Leu Ser Ser Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys Ala Lys
210 215 220His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp Tyr Trp Gly
Gln Gly225 230 235 240Thr Leu Val Thr Val Ser Ser
2456247PRTArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic polypeptide" 6Glu Ile Val Met Thr Gln Ser Pro
Ala Thr Leu Ser Leu Ser Pro Gly1 5 10 15Glu Arg Ala Thr Leu Ser Cys
Arg Ala Ser Gln Asp Ile Ser Lys Tyr 20 25 30Leu Asn Trp Tyr Gln Gln
Lys Pro Gly Gln Ala Pro Arg Leu Leu Ile 35 40 45Tyr His Thr Ser Arg
Leu His Ser Gly Ile Pro Ala Arg Phe Ser Gly 50 55 60Ser Gly Ser Gly
Thr Asp Tyr Thr Leu Thr Ile Ser Ser Leu Gln Pro65 70 75 80Glu Asp
Phe Ala Val Tyr Phe Cys Gln Gln Gly Asn Thr Leu Pro Tyr 85 90 95Thr
Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys Gly Gly Gly Gly Ser 100 105
110Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gln
115 120 125Val Gln Leu Gln Glu Ser Gly Pro Gly Leu Val Lys Pro Ser
Glu Thr 130 135 140Leu Ser Leu Thr Cys Thr Val Ser Gly Val Ser Leu
Pro Asp Tyr Gly145 150 155 160Val Ser Trp Ile Arg Gln Pro Pro Gly
Lys Gly Leu Glu Trp Ile Gly 165 170 175Val Ile Trp Gly Ser Glu Thr
Thr Tyr Tyr Gln Ser Ser Leu Lys Ser 180 185 190Arg Val Thr Ile Ser
Lys Asp Asn Ser Lys Asn Gln Val Ser Leu Lys 195 200 205Leu Ser Ser
Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys Ala Lys 210 215 220His
Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp Tyr Trp Gly Gln Gly225 230
235 240Thr Leu Val Thr Val Ser Ser 2457247PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 7Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu Val Lys
Pro Ser Glu1 5 10 15Thr Leu Ser Leu Thr Cys Thr Val Ser Gly Val Ser
Leu Pro Asp Tyr 20 25 30Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys
Gly Leu Glu Trp Ile 35 40 45Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr
Tyr Ser Ser Ser Leu Lys 50 55 60Ser Arg Val Thr Ile Ser Lys Asp Asn
Ser Lys Asn Gln Val Ser Leu65 70 75 80Lys Leu Ser Ser Val Thr Ala
Ala Asp Thr Ala Val Tyr Tyr Cys Ala 85 90 95Lys His Tyr Tyr Tyr Gly
Gly Ser Tyr Ala Met Asp Tyr Trp Gly Gln 100 105 110Gly Thr Leu Val
Thr Val Ser Ser Gly Gly Gly Gly Ser Gly Gly Gly 115 120 125Gly Ser
Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Glu Ile Val Met 130 135
140Thr Gln Ser Pro Ala Thr Leu Ser Leu Ser Pro Gly Glu Arg Ala
Thr145 150 155 160Leu Ser Cys Arg Ala Ser Gln Asp Ile Ser Lys Tyr
Leu Asn Trp Tyr 165 170 175Gln Gln Lys Pro Gly Gln Ala Pro Arg Leu
Leu Ile Tyr His Thr Ser 180 185 190Arg Leu His Ser Gly Ile Pro Ala
Arg Phe Ser Gly Ser Gly Ser Gly 195 200 205Thr Asp Tyr Thr Leu Thr
Ile Ser Ser Leu Gln Pro Glu Asp Phe Ala 210 215 220Val Tyr Phe Cys
Gln Gln Gly Asn Thr Leu Pro Tyr Thr Phe Gly Gln225 230 235 240Gly
Thr Lys Leu Glu Ile Lys 2458247PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 8Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu Val Lys
Pro Ser Glu1 5 10 15Thr Leu Ser Leu Thr Cys Thr Val Ser Gly Val Ser
Leu Pro Asp Tyr 20 25 30Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys
Gly Leu Glu Trp Ile 35 40 45Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr
Tyr Gln Ser Ser Leu Lys 50 55 60Ser Arg Val Thr Ile Ser Lys Asp Asn
Ser Lys Asn Gln Val Ser Leu65 70 75 80Lys Leu Ser Ser Val Thr Ala
Ala Asp Thr Ala Val Tyr Tyr Cys Ala 85 90 95Lys His Tyr Tyr Tyr Gly
Gly Ser Tyr Ala Met Asp Tyr Trp Gly Gln 100 105 110Gly Thr Leu Val
Thr Val Ser Ser Gly Gly Gly Gly Ser Gly Gly Gly 115 120 125Gly Ser
Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Glu Ile Val Met 130 135
140Thr Gln Ser Pro Ala Thr Leu Ser Leu Ser Pro Gly Glu Arg Ala
Thr145 150 155 160Leu Ser Cys Arg Ala Ser Gln Asp Ile Ser Lys Tyr
Leu Asn Trp Tyr 165 170 175Gln Gln Lys Pro Gly Gln Ala Pro Arg Leu
Leu Ile Tyr His Thr Ser 180 185 190Arg Leu His Ser Gly Ile Pro Ala
Arg Phe Ser Gly Ser Gly Ser Gly 195 200 205Thr Asp Tyr Thr Leu Thr
Ile Ser Ser Leu Gln Pro Glu Asp Phe Ala 210 215 220Val Tyr Phe Cys
Gln Gln Gly Asn Thr Leu Pro Tyr Thr Phe Gly Gln225 230 235 240Gly
Thr Lys Leu Glu Ile Lys 2459247PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 9Glu Ile Val Met Thr Gln Ser Pro Ala Thr Leu Ser Leu
Ser Pro Gly1 5 10 15Glu Arg Ala Thr Leu Ser Cys Arg Ala Ser Gln Asp
Ile Ser Lys Tyr 20 25 30Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala
Pro Arg Leu Leu Ile 35 40 45Tyr His Thr Ser Arg Leu His Ser Gly Ile
Pro Ala Arg Phe Ser Gly 50 55 60Ser Gly Ser Gly Thr Asp Tyr Thr Leu
Thr Ile Ser Ser Leu Gln Pro65 70 75 80Glu Asp Phe Ala Val Tyr Phe
Cys Gln Gln Gly Asn Thr Leu Pro Tyr 85 90 95Thr Phe Gly Gln Gly Thr
Lys Leu Glu Ile Lys Gly Gly Gly Gly Ser 100 105 110Gly Gly Gly Gly
Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gln 115 120 125Val Gln
Leu Gln Glu Ser Gly Pro Gly Leu Val Lys Pro Ser Glu Thr 130 135
140Leu Ser Leu Thr Cys Thr Val Ser Gly Val Ser Leu Pro Asp Tyr
Gly145 150 155 160Val Ser Trp Ile Arg Gln Pro Pro Gly Lys Gly Leu
Glu Trp Ile Gly 165 170 175Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr
Asn Ser Ser Leu Lys Ser 180 185 190Arg Val Thr Ile Ser Lys Asp Asn
Ser Lys Asn Gln Val Ser Leu Lys 195 200 205Leu Ser Ser Val Thr Ala
Ala Asp Thr Ala Val Tyr Tyr Cys Ala Lys 210 215 220His Tyr Tyr Tyr
Gly Gly Ser Tyr Ala Met Asp Tyr Trp Gly Gln Gly225 230 235 240Thr
Leu Val Thr Val Ser Ser 24510247PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 10Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu Val Lys
Pro Ser Glu1 5 10 15Thr Leu Ser Leu Thr Cys Thr Val Ser Gly Val Ser
Leu Pro Asp Tyr 20 25 30Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys
Gly Leu Glu Trp Ile 35 40 45Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr
Tyr Asn Ser Ser Leu Lys 50 55 60Ser Arg Val Thr Ile Ser Lys Asp Asn
Ser Lys Asn Gln Val Ser Leu65 70 75 80Lys Leu Ser Ser Val Thr Ala
Ala Asp Thr Ala Val Tyr Tyr Cys Ala 85 90 95Lys His Tyr Tyr Tyr Gly
Gly Ser Tyr Ala Met Asp Tyr Trp Gly Gln 100 105 110Gly Thr Leu Val
Thr Val Ser Ser Gly Gly Gly Gly Ser Gly Gly Gly 115 120
125Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Glu Ile Val Met
130 135 140Thr Gln Ser Pro Ala Thr Leu Ser Leu Ser Pro Gly Glu Arg
Ala Thr145 150 155 160Leu Ser Cys Arg Ala Ser Gln Asp Ile Ser Lys
Tyr Leu Asn Trp Tyr 165 170 175Gln Gln Lys Pro Gly Gln Ala Pro Arg
Leu Leu Ile Tyr His Thr Ser 180 185 190Arg Leu His Ser Gly Ile Pro
Ala Arg Phe Ser Gly Ser Gly Ser Gly 195 200 205Thr Asp Tyr Thr Leu
Thr Ile Ser Ser Leu Gln Pro Glu Asp Phe Ala 210 215 220Val Tyr Phe
Cys Gln Gln Gly Asn Thr Leu Pro Tyr Thr Phe Gly Gln225 230 235
240Gly Thr Lys Leu Glu Ile Lys 24511242PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 11Glu Ile Val Met Thr Gln Ser Pro Ala Thr Leu Ser Leu
Ser Pro Gly1 5 10 15Glu Arg Ala Thr Leu Ser Cys Arg Ala Ser Gln Asp
Ile Ser Lys Tyr 20 25 30Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala
Pro Arg Leu Leu Ile 35 40 45Tyr His Thr Ser Arg Leu His Ser Gly Ile
Pro Ala Arg Phe Ser Gly 50 55 60Ser Gly Ser Gly Thr Asp Tyr Thr Leu
Thr Ile Ser Ser Leu Gln Pro65 70 75 80Glu Asp Phe Ala Val Tyr Phe
Cys Gln Gln Gly Asn Thr Leu Pro Tyr 85 90 95Thr Phe Gly Gln Gly Thr
Lys Leu Glu Ile Lys Gly Gly Gly Gly Ser 100 105 110Gly Gly Gly Gly
Ser Gly Gly Gly Gly Ser Gln Val Gln Leu Gln Glu 115 120 125Ser Gly
Pro Gly Leu Val Lys Pro Ser Glu Thr Leu Ser Leu Thr Cys 130 135
140Thr Val Ser Gly Val Ser Leu Pro Asp Tyr Gly Val Ser Trp Ile
Arg145 150 155 160Gln Pro Pro Gly Lys Gly Leu Glu Trp Ile Gly Val
Ile Trp Gly Ser 165 170 175Glu Thr Thr Tyr Tyr Asn Ser Ser Leu Lys
Ser Arg Val Thr Ile Ser 180 185 190Lys Asp Asn Ser Lys Asn Gln Val
Ser Leu Lys Leu Ser Ser Val Thr 195 200 205Ala Ala Asp Thr Ala Val
Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly 210 215 220Gly Ser Tyr Ala
Met Asp Tyr Trp Gly Gln Gly Thr Leu Val Thr Val225 230 235 240Ser
Ser12242PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 12Gln Val Gln Leu Gln
Glu Ser Gly Pro Gly Leu Val Lys Pro Ser Glu1 5 10 15Thr Leu Ser Leu
Thr Cys Thr Val Ser Gly Val Ser Leu Pro Asp Tyr 20 25 30Gly Val Ser
Trp Ile Arg Gln Pro Pro Gly Lys Gly Leu Glu Trp Ile 35 40 45Gly Val
Ile Trp Gly Ser Glu Thr Thr Tyr Tyr Asn Ser Ser Leu Lys 50 55 60Ser
Arg Val Thr Ile Ser Lys Asp Asn Ser Lys Asn Gln Val Ser Leu65 70 75
80Lys Leu Ser Ser Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys Ala
85 90 95Lys His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp Tyr Trp Gly
Gln 100 105 110Gly Thr Leu Val Thr Val Ser Ser Gly Gly Gly Gly Ser
Gly Gly Gly 115 120 125Gly Ser Gly Gly Gly Gly Ser Glu Ile Val Met
Thr Gln Ser Pro Ala 130 135 140Thr Leu Ser Leu Ser Pro Gly Glu Arg
Ala Thr Leu Ser Cys Arg Ala145 150 155 160Ser Gln Asp Ile Ser Lys
Tyr Leu Asn Trp Tyr Gln Gln Lys Pro Gly 165 170 175Gln Ala Pro Arg
Leu Leu Ile Tyr His Thr Ser Arg Leu His Ser Gly 180 185 190Ile Pro
Ala Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu 195 200
205Thr Ile Ser Ser Leu Gln Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln
210 215 220Gln Gly Asn Thr Leu Pro Tyr Thr Phe Gly Gln Gly Thr Lys
Leu Glu225 230 235 240Ile Lys1321PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
peptide" 13Met Ala Leu Pro Val Thr Ala Leu Leu Leu Pro Leu Ala Leu
Leu Leu1 5 10 15His Ala Ala Arg Pro 201445PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 14Thr Thr Thr Pro Ala Pro Arg Pro Pro Thr Pro Ala Pro
Thr Ile Ala1 5 10 15Ser Gln Pro Leu Ser Leu Arg Pro Glu Ala Cys Arg
Pro Ala Ala Gly 20 25 30Gly Ala Val His Thr Arg Gly Leu Asp Phe Ala
Cys Asp 35 40 451524PRTArtificial Sequencesource/note="Description
of Artificial Sequence Synthetic peptide" 15Ile Tyr Ile Trp Ala Pro
Leu Ala Gly Thr Cys Gly Val Leu Leu Leu1 5 10 15Ser Leu Val Ile Thr
Leu Tyr Cys 201642PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 16Lys Arg Gly Arg Lys
Lys Leu Leu Tyr Ile Phe Lys Gln Pro Phe Met1 5 10 15Arg Pro Val Gln
Thr Thr Gln Glu Glu Asp Gly Cys Ser Cys Arg Phe 20 25 30Pro Glu Glu
Glu Glu Gly Gly Cys Glu Leu 35 4017112PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 17Arg Val Lys Phe Ser Arg Ser Ala Asp Ala Pro Ala Tyr
Lys Gln Gly1 5 10 15Gln Asn Gln Leu Tyr Asn Glu Leu Asn Leu Gly Arg
Arg Glu Glu Tyr 20 25 30Asp Val Leu Asp Lys Arg Arg Gly Arg Asp Pro
Glu Met Gly Gly Lys 35 40 45Pro Arg Arg Lys Asn Pro Gln Glu Gly Leu
Tyr Asn Glu Leu Gln Lys 50 55 60Asp Lys Met Ala Glu Ala Tyr Ser Glu
Ile Gly Met Lys Gly Glu Arg65 70 75 80Arg Arg Gly Lys Gly His Asp
Gly Leu Tyr Gln Gly Leu Ser Thr Ala 85 90 95Thr Lys Asp Thr Tyr Asp
Ala Leu His Met Gln Ala Leu Pro Pro Arg 100 105 110185PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
peptide" 18Gly Gly Gly Gly Ser1 51910PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
peptide" 19Gly Val Ser Leu Pro Asp Tyr Gly Val Ser1 5
102016PRTArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic peptide" 20Val Ile Trp Gly Ser Glu Thr Thr Tyr
Tyr Asn Ser Ala Leu Lys Ser1 5 10 152116PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
peptide" 21Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr Ser Ser Ser Leu
Lys Ser1 5 10 152216PRTArtificial Sequencesource/note="Description
of Artificial Sequence Synthetic peptide" 22Val Ile Trp Gly Ser Glu
Thr Thr Tyr Tyr Gln Ser Ser Leu Lys Ser1 5 10 152316PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
peptide" 23Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr Asn Ser Ser Leu
Lys Ser1 5 10 152412PRTArtificial Sequencesource/note="Description
of Artificial Sequence Synthetic peptide" 24His Tyr Tyr Tyr Gly Gly
Ser Tyr Ala Met Asp Tyr1 5 102511PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
peptide" 25Arg Ala Ser Gln Asp Ile Ser Lys Tyr Leu Asn1 5
10267PRTArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic peptide" 26His Thr Ser Arg Leu His Ser1
5279PRTArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic peptide" 27Gln Gln Gly Asn Thr Leu Pro Tyr Thr1
5285PRTArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic peptide" 28Tyr Ser Ser Ser Leu1
5295PRTArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic peptide" 29Tyr Gln Ser Ser Leu1
5305PRTArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic peptide" 30Tyr Asn Ser Ser Leu1
531486PRTArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic polypeptide" 31Met Ala Leu Pro Val Thr Ala Leu
Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg Pro Glu Ile
Val Met Thr Gln Ser Pro Ala Thr Leu 20 25 30Ser Leu Ser Pro Gly Glu
Arg Ala Thr Leu Ser Cys Arg Ala Ser Gln 35 40 45Asp Ile Ser Lys Tyr
Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala 50 55 60Pro Arg Leu Leu
Ile Tyr His Thr Ser Arg Leu His Ser Gly Ile Pro65 70 75 80Ala Arg
Phe Ser Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile 85 90 95Ser
Ser Leu Gln Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln Gly 100 105
110Asn Thr Leu Pro Tyr Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys
115 120 125Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly
Ser Gln 130 135 140Val Gln Leu Gln Glu Ser Gly Pro Gly Leu Val Lys
Pro Ser Glu Thr145 150 155 160Leu Ser Leu Thr Cys Thr Val Ser Gly
Val Ser Leu Pro Asp Tyr Gly 165 170 175Val Ser Trp Ile Arg Gln Pro
Pro Gly Lys Gly Leu Glu Trp Ile Gly 180 185 190Val Ile Trp Gly Ser
Glu Thr Thr Tyr Tyr Ser Ser Ser Leu Lys Ser 195 200 205Arg Val Thr
Ile Ser Lys Asp Asn Ser Lys Asn Gln Val Ser Leu Lys 210 215 220Leu
Ser Ser Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys Ala Lys225 230
235 240His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp Tyr Trp Gly Gln
Gly 245 250 255Thr Leu Val Thr Val Ser Ser Thr Thr Thr Pro Ala Pro
Arg Pro Pro 260 265 270Thr Pro Ala Pro Thr Ile Ala Ser Gln Pro Leu
Ser Leu Arg Pro Glu 275 280 285Ala Cys Arg Pro Ala Ala Gly Gly Ala
Val His Thr Arg Gly Leu Asp 290 295 300Phe Ala Cys Asp Ile Tyr Ile
Trp Ala Pro Leu Ala Gly Thr Cys Gly305 310 315 320Val Leu Leu Leu
Ser Leu Val Ile Thr Leu Tyr Cys Lys Arg Gly Arg 325 330 335Lys Lys
Leu Leu Tyr Ile Phe Lys Gln Pro Phe Met Arg Pro Val Gln 340 345
350Thr Thr Gln Glu Glu Asp Gly Cys Ser Cys Arg Phe Pro Glu Glu Glu
355 360 365Glu Gly Gly Cys Glu Leu Arg Val Lys Phe Ser Arg Ser Ala
Asp Ala 370 375 380Pro Ala Tyr Lys Gln Gly Gln Asn Gln Leu Tyr Asn
Glu Leu Asn Leu385 390 395 400Gly Arg Arg Glu Glu Tyr Asp Val Leu
Asp Lys Arg Arg Gly Arg Asp 405 410 415Pro Glu Met Gly Gly Lys Pro
Arg Arg Lys Asn Pro Gln Glu Gly Leu 420 425 430Tyr Asn Glu Leu Gln
Lys Asp Lys Met Ala Glu Ala Tyr Ser Glu Ile 435 440 445Gly Met Lys
Gly Glu Arg Arg Arg Gly Lys Gly His Asp Gly Leu Tyr 450 455 460Gln
Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr Asp Ala Leu His Met465 470
475 480Gln Ala Leu Pro Pro Arg 48532486PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 32Met Ala Leu Pro Val Thr Ala Leu Leu Leu Pro Leu Ala
Leu Leu Leu1 5 10 15His Ala Ala Arg Pro Glu Ile Val Met Thr Gln Ser
Pro Ala Thr Leu 20 25 30Ser Leu Ser Pro Gly Glu Arg Ala Thr Leu Ser
Cys Arg Ala Ser Gln 35 40 45Asp Ile Ser Lys Tyr Leu Asn Trp Tyr Gln
Gln Lys Pro Gly Gln Ala 50 55 60Pro Arg Leu Leu Ile Tyr His Thr Ser
Arg Leu His Ser Gly Ile Pro65 70 75 80Ala Arg Phe Ser Gly Ser Gly
Ser Gly Thr Asp Tyr Thr Leu Thr Ile 85 90 95Ser Ser Leu Gln Pro Glu
Asp Phe Ala Val Tyr Phe Cys Gln Gln Gly 100 105 110Asn Thr Leu Pro
Tyr Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys 115 120 125Gly Gly
Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gln 130 135
140Val Gln Leu Gln Glu Ser Gly Pro Gly Leu Val Lys Pro Ser Glu
Thr145 150 155 160Leu Ser Leu Thr Cys Thr Val Ser Gly Val Ser Leu
Pro Asp Tyr Gly 165 170 175Val Ser Trp Ile Arg Gln Pro Pro Gly Lys
Gly Leu Glu Trp Ile Gly 180 185 190Val Ile Trp Gly Ser Glu Thr Thr
Tyr Tyr Gln Ser Ser Leu Lys Ser 195 200 205Arg Val Thr Ile Ser Lys
Asp Asn Ser Lys Asn Gln Val Ser Leu Lys 210 215 220Leu Ser Ser Val
Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys Ala Lys225 230 235 240His
Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp Tyr Trp Gly Gln Gly 245 250
255Thr Leu Val Thr Val Ser Ser Thr Thr Thr Pro Ala Pro Arg Pro Pro
260 265 270Thr Pro Ala Pro Thr Ile Ala Ser Gln Pro Leu Ser Leu Arg
Pro Glu 275 280 285Ala Cys Arg Pro Ala Ala Gly Gly Ala Val His Thr
Arg Gly Leu Asp 290 295 300Phe Ala Cys Asp Ile Tyr Ile Trp Ala Pro
Leu Ala Gly Thr Cys Gly305 310 315 320Val Leu Leu Leu Ser Leu Val
Ile Thr Leu Tyr Cys Lys Arg Gly Arg 325 330 335Lys Lys Leu Leu Tyr
Ile Phe Lys Gln Pro Phe Met Arg Pro Val Gln 340 345 350Thr Thr Gln
Glu Glu Asp Gly Cys Ser Cys Arg Phe Pro Glu Glu Glu 355 360 365Glu
Gly Gly Cys Glu Leu Arg Val Lys Phe Ser Arg Ser Ala Asp Ala 370 375
380Pro Ala Tyr Lys Gln Gly Gln Asn Gln Leu Tyr Asn Glu Leu Asn
Leu385 390 395 400Gly Arg Arg Glu Glu Tyr Asp Val Leu Asp Lys Arg
Arg Gly Arg Asp 405 410 415Pro Glu Met Gly Gly Lys Pro Arg Arg Lys
Asn Pro Gln Glu Gly Leu 420 425 430Tyr Asn Glu Leu Gln Lys Asp Lys
Met Ala Glu Ala Tyr Ser Glu Ile 435 440 445Gly Met Lys Gly Glu Arg
Arg Arg Gly Lys Gly His Asp Gly Leu Tyr 450 455 460Gln Gly Leu Ser
Thr Ala Thr Lys Asp Thr Tyr Asp Ala Leu His Met465 470 475 480Gln
Ala Leu Pro Pro Arg 48533486PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 33Met Ala Leu Pro Val Thr Ala Leu Leu Leu Pro Leu Ala
Leu Leu Leu1 5 10 15His Ala Ala Arg Pro Gln Val Gln Leu Gln Glu Ser
Gly Pro Gly Leu 20 25 30Val Lys Pro Ser Glu Thr Leu Ser Leu Thr Cys
Thr Val Ser Gly Val 35 40 45Ser Leu Pro Asp Tyr Gly Val Ser Trp Ile
Arg Gln Pro Pro Gly Lys 50 55 60Gly Leu Glu Trp Ile Gly Val Ile Trp
Gly Ser Glu Thr Thr Tyr Tyr65 70 75 80Ser Ser Ser Leu Lys Ser Arg
Val Thr Ile Ser Lys Asp Asn Ser Lys 85 90 95Asn Gln Val Ser Leu Lys
Leu Ser Ser Val Thr Ala Ala Asp Thr Ala 100 105 110Val Tyr Tyr Cys
Ala Lys His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met 115 120 125Asp Tyr
Trp Gly Gln Gly Thr Leu Val Thr Val Ser Ser Gly Gly Gly 130 135
140Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Glu Ile Val
Met145 150 155 160Thr Gln Ser Pro Ala Thr Leu Ser Leu Ser Pro Gly
Glu Arg Ala Thr 165 170 175Leu Ser Cys Arg Ala Ser Gln Asp Ile Ser
Lys Tyr Leu Asn Trp Tyr
180 185 190Gln Gln Lys Pro Gly Gln Ala Pro Arg Leu Leu Ile Tyr His
Thr Ser 195 200 205Arg Leu His Ser Gly Ile Pro Ala Arg Phe Ser Gly
Ser Gly Ser Gly 210 215 220Thr Asp Tyr Thr Leu Thr Ile Ser Ser Leu
Gln Pro Glu Asp Phe Ala225 230 235 240Val Tyr Phe Cys Gln Gln Gly
Asn Thr Leu Pro Tyr Thr Phe Gly Gln 245 250 255Gly Thr Lys Leu Glu
Ile Lys Thr Thr Thr Pro Ala Pro Arg Pro Pro 260 265 270Thr Pro Ala
Pro Thr Ile Ala Ser Gln Pro Leu Ser Leu Arg Pro Glu 275 280 285Ala
Cys Arg Pro Ala Ala Gly Gly Ala Val His Thr Arg Gly Leu Asp 290 295
300Phe Ala Cys Asp Ile Tyr Ile Trp Ala Pro Leu Ala Gly Thr Cys
Gly305 310 315 320Val Leu Leu Leu Ser Leu Val Ile Thr Leu Tyr Cys
Lys Arg Gly Arg 325 330 335Lys Lys Leu Leu Tyr Ile Phe Lys Gln Pro
Phe Met Arg Pro Val Gln 340 345 350Thr Thr Gln Glu Glu Asp Gly Cys
Ser Cys Arg Phe Pro Glu Glu Glu 355 360 365Glu Gly Gly Cys Glu Leu
Arg Val Lys Phe Ser Arg Ser Ala Asp Ala 370 375 380Pro Ala Tyr Lys
Gln Gly Gln Asn Gln Leu Tyr Asn Glu Leu Asn Leu385 390 395 400Gly
Arg Arg Glu Glu Tyr Asp Val Leu Asp Lys Arg Arg Gly Arg Asp 405 410
415Pro Glu Met Gly Gly Lys Pro Arg Arg Lys Asn Pro Gln Glu Gly Leu
420 425 430Tyr Asn Glu Leu Gln Lys Asp Lys Met Ala Glu Ala Tyr Ser
Glu Ile 435 440 445Gly Met Lys Gly Glu Arg Arg Arg Gly Lys Gly His
Asp Gly Leu Tyr 450 455 460Gln Gly Leu Ser Thr Ala Thr Lys Asp Thr
Tyr Asp Ala Leu His Met465 470 475 480Gln Ala Leu Pro Pro Arg
48534486PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 34Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu 20 25 30Val Lys Pro
Ser Glu Thr Leu Ser Leu Thr Cys Thr Val Ser Gly Val 35 40 45Ser Leu
Pro Asp Tyr Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys 50 55 60Gly
Leu Glu Trp Ile Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr65 70 75
80Gln Ser Ser Leu Lys Ser Arg Val Thr Ile Ser Lys Asp Asn Ser Lys
85 90 95Asn Gln Val Ser Leu Lys Leu Ser Ser Val Thr Ala Ala Asp Thr
Ala 100 105 110Val Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly Gly Ser
Tyr Ala Met 115 120 125Asp Tyr Trp Gly Gln Gly Thr Leu Val Thr Val
Ser Ser Gly Gly Gly 130 135 140Gly Ser Gly Gly Gly Gly Ser Gly Gly
Gly Gly Ser Glu Ile Val Met145 150 155 160Thr Gln Ser Pro Ala Thr
Leu Ser Leu Ser Pro Gly Glu Arg Ala Thr 165 170 175Leu Ser Cys Arg
Ala Ser Gln Asp Ile Ser Lys Tyr Leu Asn Trp Tyr 180 185 190Gln Gln
Lys Pro Gly Gln Ala Pro Arg Leu Leu Ile Tyr His Thr Ser 195 200
205Arg Leu His Ser Gly Ile Pro Ala Arg Phe Ser Gly Ser Gly Ser Gly
210 215 220Thr Asp Tyr Thr Leu Thr Ile Ser Ser Leu Gln Pro Glu Asp
Phe Ala225 230 235 240Val Tyr Phe Cys Gln Gln Gly Asn Thr Leu Pro
Tyr Thr Phe Gly Gln 245 250 255Gly Thr Lys Leu Glu Ile Lys Thr Thr
Thr Pro Ala Pro Arg Pro Pro 260 265 270Thr Pro Ala Pro Thr Ile Ala
Ser Gln Pro Leu Ser Leu Arg Pro Glu 275 280 285Ala Cys Arg Pro Ala
Ala Gly Gly Ala Val His Thr Arg Gly Leu Asp 290 295 300Phe Ala Cys
Asp Ile Tyr Ile Trp Ala Pro Leu Ala Gly Thr Cys Gly305 310 315
320Val Leu Leu Leu Ser Leu Val Ile Thr Leu Tyr Cys Lys Arg Gly Arg
325 330 335Lys Lys Leu Leu Tyr Ile Phe Lys Gln Pro Phe Met Arg Pro
Val Gln 340 345 350Thr Thr Gln Glu Glu Asp Gly Cys Ser Cys Arg Phe
Pro Glu Glu Glu 355 360 365Glu Gly Gly Cys Glu Leu Arg Val Lys Phe
Ser Arg Ser Ala Asp Ala 370 375 380Pro Ala Tyr Lys Gln Gly Gln Asn
Gln Leu Tyr Asn Glu Leu Asn Leu385 390 395 400Gly Arg Arg Glu Glu
Tyr Asp Val Leu Asp Lys Arg Arg Gly Arg Asp 405 410 415Pro Glu Met
Gly Gly Lys Pro Arg Arg Lys Asn Pro Gln Glu Gly Leu 420 425 430Tyr
Asn Glu Leu Gln Lys Asp Lys Met Ala Glu Ala Tyr Ser Glu Ile 435 440
445Gly Met Lys Gly Glu Arg Arg Arg Gly Lys Gly His Asp Gly Leu Tyr
450 455 460Gln Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr Asp Ala Leu
His Met465 470 475 480Gln Ala Leu Pro Pro Arg 48535491PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 35Met Ala Leu Pro Val Thr Ala Leu Leu Leu Pro Leu Ala
Leu Leu Leu1 5 10 15His Ala Ala Arg Pro Glu Ile Val Met Thr Gln Ser
Pro Ala Thr Leu 20 25 30Ser Leu Ser Pro Gly Glu Arg Ala Thr Leu Ser
Cys Arg Ala Ser Gln 35 40 45Asp Ile Ser Lys Tyr Leu Asn Trp Tyr Gln
Gln Lys Pro Gly Gln Ala 50 55 60Pro Arg Leu Leu Ile Tyr His Thr Ser
Arg Leu His Ser Gly Ile Pro65 70 75 80Ala Arg Phe Ser Gly Ser Gly
Ser Gly Thr Asp Tyr Thr Leu Thr Ile 85 90 95Ser Ser Leu Gln Pro Glu
Asp Phe Ala Val Tyr Phe Cys Gln Gln Gly 100 105 110Asn Thr Leu Pro
Tyr Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys 115 120 125Gly Gly
Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly 130 135
140Gly Gly Gly Ser Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu
Val145 150 155 160Lys Pro Ser Glu Thr Leu Ser Leu Thr Cys Thr Val
Ser Gly Val Ser 165 170 175Leu Pro Asp Tyr Gly Val Ser Trp Ile Arg
Gln Pro Pro Gly Lys Gly 180 185 190Leu Glu Trp Ile Gly Val Ile Trp
Gly Ser Glu Thr Thr Tyr Tyr Ser 195 200 205Ser Ser Leu Lys Ser Arg
Val Thr Ile Ser Lys Asp Asn Ser Lys Asn 210 215 220Gln Val Ser Leu
Lys Leu Ser Ser Val Thr Ala Ala Asp Thr Ala Val225 230 235 240Tyr
Tyr Cys Ala Lys His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp 245 250
255Tyr Trp Gly Gln Gly Thr Leu Val Thr Val Ser Ser Thr Thr Thr Pro
260 265 270Ala Pro Arg Pro Pro Thr Pro Ala Pro Thr Ile Ala Ser Gln
Pro Leu 275 280 285Ser Leu Arg Pro Glu Ala Cys Arg Pro Ala Ala Gly
Gly Ala Val His 290 295 300Thr Arg Gly Leu Asp Phe Ala Cys Asp Ile
Tyr Ile Trp Ala Pro Leu305 310 315 320Ala Gly Thr Cys Gly Val Leu
Leu Leu Ser Leu Val Ile Thr Leu Tyr 325 330 335Cys Lys Arg Gly Arg
Lys Lys Leu Leu Tyr Ile Phe Lys Gln Pro Phe 340 345 350Met Arg Pro
Val Gln Thr Thr Gln Glu Glu Asp Gly Cys Ser Cys Arg 355 360 365Phe
Pro Glu Glu Glu Glu Gly Gly Cys Glu Leu Arg Val Lys Phe Ser 370 375
380Arg Ser Ala Asp Ala Pro Ala Tyr Lys Gln Gly Gln Asn Gln Leu
Tyr385 390 395 400Asn Glu Leu Asn Leu Gly Arg Arg Glu Glu Tyr Asp
Val Leu Asp Lys 405 410 415Arg Arg Gly Arg Asp Pro Glu Met Gly Gly
Lys Pro Arg Arg Lys Asn 420 425 430Pro Gln Glu Gly Leu Tyr Asn Glu
Leu Gln Lys Asp Lys Met Ala Glu 435 440 445Ala Tyr Ser Glu Ile Gly
Met Lys Gly Glu Arg Arg Arg Gly Lys Gly 450 455 460His Asp Gly Leu
Tyr Gln Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr465 470 475 480Asp
Ala Leu His Met Gln Ala Leu Pro Pro Arg 485 49036491PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 36Met Ala Leu Pro Val Thr Ala Leu Leu Leu Pro Leu Ala
Leu Leu Leu1 5 10 15His Ala Ala Arg Pro Glu Ile Val Met Thr Gln Ser
Pro Ala Thr Leu 20 25 30Ser Leu Ser Pro Gly Glu Arg Ala Thr Leu Ser
Cys Arg Ala Ser Gln 35 40 45Asp Ile Ser Lys Tyr Leu Asn Trp Tyr Gln
Gln Lys Pro Gly Gln Ala 50 55 60Pro Arg Leu Leu Ile Tyr His Thr Ser
Arg Leu His Ser Gly Ile Pro65 70 75 80Ala Arg Phe Ser Gly Ser Gly
Ser Gly Thr Asp Tyr Thr Leu Thr Ile 85 90 95Ser Ser Leu Gln Pro Glu
Asp Phe Ala Val Tyr Phe Cys Gln Gln Gly 100 105 110Asn Thr Leu Pro
Tyr Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys 115 120 125Gly Gly
Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly 130 135
140Gly Gly Gly Ser Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu
Val145 150 155 160Lys Pro Ser Glu Thr Leu Ser Leu Thr Cys Thr Val
Ser Gly Val Ser 165 170 175Leu Pro Asp Tyr Gly Val Ser Trp Ile Arg
Gln Pro Pro Gly Lys Gly 180 185 190Leu Glu Trp Ile Gly Val Ile Trp
Gly Ser Glu Thr Thr Tyr Tyr Gln 195 200 205Ser Ser Leu Lys Ser Arg
Val Thr Ile Ser Lys Asp Asn Ser Lys Asn 210 215 220Gln Val Ser Leu
Lys Leu Ser Ser Val Thr Ala Ala Asp Thr Ala Val225 230 235 240Tyr
Tyr Cys Ala Lys His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp 245 250
255Tyr Trp Gly Gln Gly Thr Leu Val Thr Val Ser Ser Thr Thr Thr Pro
260 265 270Ala Pro Arg Pro Pro Thr Pro Ala Pro Thr Ile Ala Ser Gln
Pro Leu 275 280 285Ser Leu Arg Pro Glu Ala Cys Arg Pro Ala Ala Gly
Gly Ala Val His 290 295 300Thr Arg Gly Leu Asp Phe Ala Cys Asp Ile
Tyr Ile Trp Ala Pro Leu305 310 315 320Ala Gly Thr Cys Gly Val Leu
Leu Leu Ser Leu Val Ile Thr Leu Tyr 325 330 335Cys Lys Arg Gly Arg
Lys Lys Leu Leu Tyr Ile Phe Lys Gln Pro Phe 340 345 350Met Arg Pro
Val Gln Thr Thr Gln Glu Glu Asp Gly Cys Ser Cys Arg 355 360 365Phe
Pro Glu Glu Glu Glu Gly Gly Cys Glu Leu Arg Val Lys Phe Ser 370 375
380Arg Ser Ala Asp Ala Pro Ala Tyr Lys Gln Gly Gln Asn Gln Leu
Tyr385 390 395 400Asn Glu Leu Asn Leu Gly Arg Arg Glu Glu Tyr Asp
Val Leu Asp Lys 405 410 415Arg Arg Gly Arg Asp Pro Glu Met Gly Gly
Lys Pro Arg Arg Lys Asn 420 425 430Pro Gln Glu Gly Leu Tyr Asn Glu
Leu Gln Lys Asp Lys Met Ala Glu 435 440 445Ala Tyr Ser Glu Ile Gly
Met Lys Gly Glu Arg Arg Arg Gly Lys Gly 450 455 460His Asp Gly Leu
Tyr Gln Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr465 470 475 480Asp
Ala Leu His Met Gln Ala Leu Pro Pro Arg 485 49037491PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 37Met Ala Leu Pro Val Thr Ala Leu Leu Leu Pro Leu Ala
Leu Leu Leu1 5 10 15His Ala Ala Arg Pro Gln Val Gln Leu Gln Glu Ser
Gly Pro Gly Leu 20 25 30Val Lys Pro Ser Glu Thr Leu Ser Leu Thr Cys
Thr Val Ser Gly Val 35 40 45Ser Leu Pro Asp Tyr Gly Val Ser Trp Ile
Arg Gln Pro Pro Gly Lys 50 55 60Gly Leu Glu Trp Ile Gly Val Ile Trp
Gly Ser Glu Thr Thr Tyr Tyr65 70 75 80Ser Ser Ser Leu Lys Ser Arg
Val Thr Ile Ser Lys Asp Asn Ser Lys 85 90 95Asn Gln Val Ser Leu Lys
Leu Ser Ser Val Thr Ala Ala Asp Thr Ala 100 105 110Val Tyr Tyr Cys
Ala Lys His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met 115 120 125Asp Tyr
Trp Gly Gln Gly Thr Leu Val Thr Val Ser Ser Gly Gly Gly 130 135
140Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly
Gly145 150 155 160Ser Glu Ile Val Met Thr Gln Ser Pro Ala Thr Leu
Ser Leu Ser Pro 165 170 175Gly Glu Arg Ala Thr Leu Ser Cys Arg Ala
Ser Gln Asp Ile Ser Lys 180 185 190Tyr Leu Asn Trp Tyr Gln Gln Lys
Pro Gly Gln Ala Pro Arg Leu Leu 195 200 205Ile Tyr His Thr Ser Arg
Leu His Ser Gly Ile Pro Ala Arg Phe Ser 210 215 220Gly Ser Gly Ser
Gly Thr Asp Tyr Thr Leu Thr Ile Ser Ser Leu Gln225 230 235 240Pro
Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln Gly Asn Thr Leu Pro 245 250
255Tyr Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys Thr Thr Thr Pro
260 265 270Ala Pro Arg Pro Pro Thr Pro Ala Pro Thr Ile Ala Ser Gln
Pro Leu 275 280 285Ser Leu Arg Pro Glu Ala Cys Arg Pro Ala Ala Gly
Gly Ala Val His 290 295 300Thr Arg Gly Leu Asp Phe Ala Cys Asp Ile
Tyr Ile Trp Ala Pro Leu305 310 315 320Ala Gly Thr Cys Gly Val Leu
Leu Leu Ser Leu Val Ile Thr Leu Tyr 325 330 335Cys Lys Arg Gly Arg
Lys Lys Leu Leu Tyr Ile Phe Lys Gln Pro Phe 340 345 350Met Arg Pro
Val Gln Thr Thr Gln Glu Glu Asp Gly Cys Ser Cys Arg 355 360 365Phe
Pro Glu Glu Glu Glu Gly Gly Cys Glu Leu Arg Val Lys Phe Ser 370 375
380Arg Ser Ala Asp Ala Pro Ala Tyr Lys Gln Gly Gln Asn Gln Leu
Tyr385 390 395 400Asn Glu Leu Asn Leu Gly Arg Arg Glu Glu Tyr Asp
Val Leu Asp Lys 405 410 415Arg Arg Gly Arg Asp Pro Glu Met Gly Gly
Lys Pro Arg Arg Lys Asn 420 425 430Pro Gln Glu Gly Leu Tyr Asn Glu
Leu Gln Lys Asp Lys Met Ala Glu 435 440 445Ala Tyr Ser Glu Ile Gly
Met Lys Gly Glu Arg Arg Arg Gly Lys Gly 450 455 460His Asp Gly Leu
Tyr Gln Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr465 470 475 480Asp
Ala Leu His Met Gln Ala Leu Pro Pro Arg 485 49038491PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 38Met Ala Leu Pro Val Thr Ala Leu Leu Leu Pro Leu Ala
Leu Leu Leu1 5 10 15His Ala Ala Arg Pro Gln Val Gln Leu Gln Glu Ser
Gly Pro Gly Leu 20 25 30Val Lys Pro Ser Glu Thr Leu Ser Leu Thr Cys
Thr Val Ser Gly Val 35 40 45Ser Leu Pro Asp Tyr Gly Val Ser Trp Ile
Arg Gln Pro Pro Gly Lys 50 55 60Gly Leu Glu Trp Ile Gly Val Ile Trp
Gly Ser Glu Thr Thr Tyr Tyr65 70 75 80Gln Ser Ser Leu Lys Ser Arg
Val Thr Ile Ser Lys Asp Asn Ser Lys 85 90 95Asn Gln Val Ser Leu Lys
Leu Ser Ser Val Thr Ala Ala Asp Thr Ala 100 105 110Val Tyr Tyr Cys
Ala Lys His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met 115 120 125Asp Tyr
Trp Gly Gln Gly Thr Leu Val Thr Val Ser Ser Gly Gly Gly
130 135 140Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly
Gly Gly145 150 155 160Ser Glu Ile Val Met Thr Gln Ser Pro Ala Thr
Leu Ser Leu Ser Pro 165 170 175Gly Glu Arg Ala Thr Leu Ser Cys Arg
Ala Ser Gln Asp Ile Ser Lys 180 185 190Tyr Leu Asn Trp Tyr Gln Gln
Lys Pro Gly Gln Ala Pro Arg Leu Leu 195 200 205Ile Tyr His Thr Ser
Arg Leu His Ser Gly Ile Pro Ala Arg Phe Ser 210 215 220Gly Ser Gly
Ser Gly Thr Asp Tyr Thr Leu Thr Ile Ser Ser Leu Gln225 230 235
240Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln Gly Asn Thr Leu Pro
245 250 255Tyr Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys Thr Thr
Thr Pro 260 265 270Ala Pro Arg Pro Pro Thr Pro Ala Pro Thr Ile Ala
Ser Gln Pro Leu 275 280 285Ser Leu Arg Pro Glu Ala Cys Arg Pro Ala
Ala Gly Gly Ala Val His 290 295 300Thr Arg Gly Leu Asp Phe Ala Cys
Asp Ile Tyr Ile Trp Ala Pro Leu305 310 315 320Ala Gly Thr Cys Gly
Val Leu Leu Leu Ser Leu Val Ile Thr Leu Tyr 325 330 335Cys Lys Arg
Gly Arg Lys Lys Leu Leu Tyr Ile Phe Lys Gln Pro Phe 340 345 350Met
Arg Pro Val Gln Thr Thr Gln Glu Glu Asp Gly Cys Ser Cys Arg 355 360
365Phe Pro Glu Glu Glu Glu Gly Gly Cys Glu Leu Arg Val Lys Phe Ser
370 375 380Arg Ser Ala Asp Ala Pro Ala Tyr Lys Gln Gly Gln Asn Gln
Leu Tyr385 390 395 400Asn Glu Leu Asn Leu Gly Arg Arg Glu Glu Tyr
Asp Val Leu Asp Lys 405 410 415Arg Arg Gly Arg Asp Pro Glu Met Gly
Gly Lys Pro Arg Arg Lys Asn 420 425 430Pro Gln Glu Gly Leu Tyr Asn
Glu Leu Gln Lys Asp Lys Met Ala Glu 435 440 445Ala Tyr Ser Glu Ile
Gly Met Lys Gly Glu Arg Arg Arg Gly Lys Gly 450 455 460His Asp Gly
Leu Tyr Gln Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr465 470 475
480Asp Ala Leu His Met Gln Ala Leu Pro Pro Arg 485
49039491PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 39Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Glu Ile Val Met Thr Gln Ser Pro Ala Thr Leu 20 25 30Ser Leu Ser
Pro Gly Glu Arg Ala Thr Leu Ser Cys Arg Ala Ser Gln 35 40 45Asp Ile
Ser Lys Tyr Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala 50 55 60Pro
Arg Leu Leu Ile Tyr His Thr Ser Arg Leu His Ser Gly Ile Pro65 70 75
80Ala Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile
85 90 95Ser Ser Leu Gln Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln
Gly 100 105 110Asn Thr Leu Pro Tyr Thr Phe Gly Gln Gly Thr Lys Leu
Glu Ile Lys 115 120 125Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly
Gly Gly Gly Ser Gly 130 135 140Gly Gly Gly Ser Gln Val Gln Leu Gln
Glu Ser Gly Pro Gly Leu Val145 150 155 160Lys Pro Ser Glu Thr Leu
Ser Leu Thr Cys Thr Val Ser Gly Val Ser 165 170 175Leu Pro Asp Tyr
Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys Gly 180 185 190Leu Glu
Trp Ile Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr Asn 195 200
205Ser Ser Leu Lys Ser Arg Val Thr Ile Ser Lys Asp Asn Ser Lys Asn
210 215 220Gln Val Ser Leu Lys Leu Ser Ser Val Thr Ala Ala Asp Thr
Ala Val225 230 235 240Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly Gly
Ser Tyr Ala Met Asp 245 250 255Tyr Trp Gly Gln Gly Thr Leu Val Thr
Val Ser Ser Thr Thr Thr Pro 260 265 270Ala Pro Arg Pro Pro Thr Pro
Ala Pro Thr Ile Ala Ser Gln Pro Leu 275 280 285Ser Leu Arg Pro Glu
Ala Cys Arg Pro Ala Ala Gly Gly Ala Val His 290 295 300Thr Arg Gly
Leu Asp Phe Ala Cys Asp Ile Tyr Ile Trp Ala Pro Leu305 310 315
320Ala Gly Thr Cys Gly Val Leu Leu Leu Ser Leu Val Ile Thr Leu Tyr
325 330 335Cys Lys Arg Gly Arg Lys Lys Leu Leu Tyr Ile Phe Lys Gln
Pro Phe 340 345 350Met Arg Pro Val Gln Thr Thr Gln Glu Glu Asp Gly
Cys Ser Cys Arg 355 360 365Phe Pro Glu Glu Glu Glu Gly Gly Cys Glu
Leu Arg Val Lys Phe Ser 370 375 380Arg Ser Ala Asp Ala Pro Ala Tyr
Lys Gln Gly Gln Asn Gln Leu Tyr385 390 395 400Asn Glu Leu Asn Leu
Gly Arg Arg Glu Glu Tyr Asp Val Leu Asp Lys 405 410 415Arg Arg Gly
Arg Asp Pro Glu Met Gly Gly Lys Pro Arg Arg Lys Asn 420 425 430Pro
Gln Glu Gly Leu Tyr Asn Glu Leu Gln Lys Asp Lys Met Ala Glu 435 440
445Ala Tyr Ser Glu Ile Gly Met Lys Gly Glu Arg Arg Arg Gly Lys Gly
450 455 460His Asp Gly Leu Tyr Gln Gly Leu Ser Thr Ala Thr Lys Asp
Thr Tyr465 470 475 480Asp Ala Leu His Met Gln Ala Leu Pro Pro Arg
485 49040491PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 40Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Glu Ile Val Met Thr Gln Ser Pro Ala Thr Leu 20 25 30Ser Leu Ser
Pro Gly Glu Arg Ala Thr Leu Ser Cys Arg Ala Ser Gln 35 40 45Asp Ile
Ser Lys Tyr Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala 50 55 60Pro
Arg Leu Leu Ile Tyr His Thr Ser Arg Leu His Ser Gly Ile Pro65 70 75
80Ala Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile
85 90 95Ser Ser Leu Gln Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln
Gly 100 105 110Asn Thr Leu Pro Tyr Thr Phe Gly Gln Gly Thr Lys Leu
Glu Ile Lys 115 120 125Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly
Gly Gly Gly Ser Gly 130 135 140Gly Gly Gly Ser Gln Val Gln Leu Gln
Glu Ser Gly Pro Gly Leu Val145 150 155 160Lys Pro Ser Glu Thr Leu
Ser Leu Thr Cys Thr Val Ser Gly Val Ser 165 170 175Leu Pro Asp Tyr
Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys Gly 180 185 190Leu Glu
Trp Ile Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr Asn 195 200
205Ser Ser Leu Lys Ser Arg Val Thr Ile Ser Lys Asp Asn Ser Lys Asn
210 215 220Gln Val Ser Leu Lys Leu Ser Ser Val Thr Ala Ala Asp Thr
Ala Val225 230 235 240Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly Gly
Ser Tyr Ala Met Asp 245 250 255Tyr Trp Gly Gln Gly Thr Leu Val Thr
Val Ser Ser Thr Thr Thr Pro 260 265 270Ala Pro Arg Pro Pro Thr Pro
Ala Pro Thr Ile Ala Ser Gln Pro Leu 275 280 285Ser Leu Arg Pro Glu
Ala Cys Arg Pro Ala Ala Gly Gly Ala Val His 290 295 300Thr Arg Gly
Leu Asp Phe Ala Cys Asp Ile Tyr Ile Trp Ala Pro Leu305 310 315
320Ala Gly Thr Cys Gly Val Leu Leu Leu Ser Leu Val Ile Thr Leu Tyr
325 330 335Cys Lys Arg Gly Arg Lys Lys Leu Leu Tyr Ile Phe Lys Gln
Pro Phe 340 345 350Met Arg Pro Val Gln Thr Thr Gln Glu Glu Asp Gly
Cys Ser Cys Arg 355 360 365Phe Pro Glu Glu Glu Glu Gly Gly Cys Glu
Leu Arg Val Lys Phe Ser 370 375 380Arg Ser Ala Asp Ala Pro Ala Tyr
Lys Gln Gly Gln Asn Gln Leu Tyr385 390 395 400Asn Glu Leu Asn Leu
Gly Arg Arg Glu Glu Tyr Asp Val Leu Asp Lys 405 410 415Arg Arg Gly
Arg Asp Pro Glu Met Gly Gly Lys Pro Arg Arg Lys Asn 420 425 430Pro
Gln Glu Gly Leu Tyr Asn Glu Leu Gln Lys Asp Lys Met Ala Glu 435 440
445Ala Tyr Ser Glu Ile Gly Met Lys Gly Glu Arg Arg Arg Gly Lys Gly
450 455 460His Asp Gly Leu Tyr Gln Gly Leu Ser Thr Ala Thr Lys Asp
Thr Tyr465 470 475 480Asp Ala Leu His Met Gln Ala Leu Pro Pro Arg
485 49041491PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 41Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu 20 25 30Val Lys Pro
Ser Glu Thr Leu Ser Leu Thr Cys Thr Val Ser Gly Val 35 40 45Ser Leu
Pro Asp Tyr Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys 50 55 60Gly
Leu Glu Trp Ile Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr65 70 75
80Asn Ser Ser Leu Lys Ser Arg Val Thr Ile Ser Lys Asp Asn Ser Lys
85 90 95Asn Gln Val Ser Leu Lys Leu Ser Ser Val Thr Ala Ala Asp Thr
Ala 100 105 110Val Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly Gly Ser
Tyr Ala Met 115 120 125Asp Tyr Trp Gly Gln Gly Thr Leu Val Thr Val
Ser Ser Gly Gly Gly 130 135 140Gly Ser Gly Gly Gly Gly Ser Gly Gly
Gly Gly Ser Gly Gly Gly Gly145 150 155 160Ser Glu Ile Val Met Thr
Gln Ser Pro Ala Thr Leu Ser Leu Ser Pro 165 170 175Gly Glu Arg Ala
Thr Leu Ser Cys Arg Ala Ser Gln Asp Ile Ser Lys 180 185 190Tyr Leu
Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala Pro Arg Leu Leu 195 200
205Ile Tyr His Thr Ser Arg Leu His Ser Gly Ile Pro Ala Arg Phe Ser
210 215 220Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile Ser Ser
Leu Gln225 230 235 240Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln
Gly Asn Thr Leu Pro 245 250 255Tyr Thr Phe Gly Gln Gly Thr Lys Leu
Glu Ile Lys Thr Thr Thr Pro 260 265 270Ala Pro Arg Pro Pro Thr Pro
Ala Pro Thr Ile Ala Ser Gln Pro Leu 275 280 285Ser Leu Arg Pro Glu
Ala Cys Arg Pro Ala Ala Gly Gly Ala Val His 290 295 300Thr Arg Gly
Leu Asp Phe Ala Cys Asp Ile Tyr Ile Trp Ala Pro Leu305 310 315
320Ala Gly Thr Cys Gly Val Leu Leu Leu Ser Leu Val Ile Thr Leu Tyr
325 330 335Cys Lys Arg Gly Arg Lys Lys Leu Leu Tyr Ile Phe Lys Gln
Pro Phe 340 345 350Met Arg Pro Val Gln Thr Thr Gln Glu Glu Asp Gly
Cys Ser Cys Arg 355 360 365Phe Pro Glu Glu Glu Glu Gly Gly Cys Glu
Leu Arg Val Lys Phe Ser 370 375 380Arg Ser Ala Asp Ala Pro Ala Tyr
Lys Gln Gly Gln Asn Gln Leu Tyr385 390 395 400Asn Glu Leu Asn Leu
Gly Arg Arg Glu Glu Tyr Asp Val Leu Asp Lys 405 410 415Arg Arg Gly
Arg Asp Pro Glu Met Gly Gly Lys Pro Arg Arg Lys Asn 420 425 430Pro
Gln Glu Gly Leu Tyr Asn Glu Leu Gln Lys Asp Lys Met Ala Glu 435 440
445Ala Tyr Ser Glu Ile Gly Met Lys Gly Glu Arg Arg Arg Gly Lys Gly
450 455 460His Asp Gly Leu Tyr Gln Gly Leu Ser Thr Ala Thr Lys Asp
Thr Tyr465 470 475 480Asp Ala Leu His Met Gln Ala Leu Pro Pro Arg
485 49042486PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 42Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Glu Ile Val Met Thr Gln Ser Pro Ala Thr Leu 20 25 30Ser Leu Ser
Pro Gly Glu Arg Ala Thr Leu Ser Cys Arg Ala Ser Gln 35 40 45Asp Ile
Ser Lys Tyr Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala 50 55 60Pro
Arg Leu Leu Ile Tyr His Thr Ser Arg Leu His Ser Gly Ile Pro65 70 75
80Ala Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile
85 90 95Ser Ser Leu Gln Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln
Gly 100 105 110Asn Thr Leu Pro Tyr Thr Phe Gly Gln Gly Thr Lys Leu
Glu Ile Lys 115 120 125Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly
Gly Gly Gly Ser Gln 130 135 140Val Gln Leu Gln Glu Ser Gly Pro Gly
Leu Val Lys Pro Ser Glu Thr145 150 155 160Leu Ser Leu Thr Cys Thr
Val Ser Gly Val Ser Leu Pro Asp Tyr Gly 165 170 175Val Ser Trp Ile
Arg Gln Pro Pro Gly Lys Gly Leu Glu Trp Ile Gly 180 185 190Val Ile
Trp Gly Ser Glu Thr Thr Tyr Tyr Asn Ser Ser Leu Lys Ser 195 200
205Arg Val Thr Ile Ser Lys Asp Asn Ser Lys Asn Gln Val Ser Leu Lys
210 215 220Leu Ser Ser Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys
Ala Lys225 230 235 240His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp
Tyr Trp Gly Gln Gly 245 250 255Thr Leu Val Thr Val Ser Ser Thr Thr
Thr Pro Ala Pro Arg Pro Pro 260 265 270Thr Pro Ala Pro Thr Ile Ala
Ser Gln Pro Leu Ser Leu Arg Pro Glu 275 280 285Ala Cys Arg Pro Ala
Ala Gly Gly Ala Val His Thr Arg Gly Leu Asp 290 295 300Phe Ala Cys
Asp Ile Tyr Ile Trp Ala Pro Leu Ala Gly Thr Cys Gly305 310 315
320Val Leu Leu Leu Ser Leu Val Ile Thr Leu Tyr Cys Lys Arg Gly Arg
325 330 335Lys Lys Leu Leu Tyr Ile Phe Lys Gln Pro Phe Met Arg Pro
Val Gln 340 345 350Thr Thr Gln Glu Glu Asp Gly Cys Ser Cys Arg Phe
Pro Glu Glu Glu 355 360 365Glu Gly Gly Cys Glu Leu Arg Val Lys Phe
Ser Arg Ser Ala Asp Ala 370 375 380Pro Ala Tyr Lys Gln Gly Gln Asn
Gln Leu Tyr Asn Glu Leu Asn Leu385 390 395 400Gly Arg Arg Glu Glu
Tyr Asp Val Leu Asp Lys Arg Arg Gly Arg Asp 405 410 415Pro Glu Met
Gly Gly Lys Pro Arg Arg Lys Asn Pro Gln Glu Gly Leu 420 425 430Tyr
Asn Glu Leu Gln Lys Asp Lys Met Ala Glu Ala Tyr Ser Glu Ile 435 440
445Gly Met Lys Gly Glu Arg Arg Arg Gly Lys Gly His Asp Gly Leu Tyr
450 455 460Gln Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr Asp Ala Leu
His Met465 470 475 480Gln Ala Leu Pro Pro Arg 48543112PRTHomo
sapiens 43Arg Val Lys Phe Ser Arg Ser Ala Asp Ala Pro Ala Tyr Gln
Gln Gly1 5 10 15Gln Asn Gln Leu Tyr Asn Glu Leu Asn Leu Gly Arg Arg
Glu Glu Tyr 20 25 30Asp Val Leu Asp Lys Arg Arg Gly Arg Asp Pro Glu
Met Gly Gly Lys 35 40 45Pro Arg Arg Lys Asn Pro Gln Glu Gly Leu Tyr
Asn Glu Leu Gln Lys 50 55 60Asp Lys Met Ala Glu Ala Tyr Ser Glu Ile
Gly Met Lys Gly Glu Arg65 70 75 80Arg Arg Gly Lys Gly His Asp Gly
Leu Tyr Gln Gly Leu Ser Thr Ala 85 90 95Thr Lys Asp Thr Tyr Asp
Ala Leu His Met Gln Ala Leu Pro Pro Arg 100 105 11044336DNAHomo
sapiens 44agagtgaagt tcagcaggag cgcagacgcc cccgcgtacc agcagggcca
gaaccagctc 60tataacgagc tcaatctagg acgaagagag gagtacgatg ttttggacaa
gagacgtggc 120cgggaccctg agatgggggg aaagccgaga aggaagaacc
ctcaggaagg cctgtacaat 180gaactgcaga aagataagat ggcggaggcc
tacagtgaga ttgggatgaa aggcgagcgc 240cggaggggca aggggcacga
tggcctttac cagggtctca gtacagccac caaggacacc 300tacgacgccc
ttcacatgca ggccctgccc cctcgc 33645230PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 45Glu Ser Lys Tyr Gly Pro Pro Cys Pro Pro Cys Pro Ala
Pro Glu Phe1 5 10 15Leu Gly Gly Pro Ser Val Phe Leu Phe Pro Pro Lys
Pro Lys Asp Thr 20 25 30Leu Met Ile Ser Arg Thr Pro Glu Val Thr Cys
Val Val Val Asp Val 35 40 45Ser Gln Glu Asp Pro Glu Val Gln Phe Asn
Trp Tyr Val Asp Gly Val 50 55 60Glu Val His Asn Ala Lys Thr Lys Pro
Arg Glu Glu Gln Phe Asn Ser65 70 75 80Thr Tyr Arg Val Val Ser Val
Leu Thr Val Leu His Gln Asp Trp Leu 85 90 95Asn Gly Lys Glu Tyr Lys
Cys Lys Val Ser Asn Lys Gly Leu Pro Ser 100 105 110Ser Ile Glu Lys
Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro 115 120 125Gln Val
Tyr Thr Leu Pro Pro Ser Gln Glu Glu Met Thr Lys Asn Gln 130 135
140Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile
Ala145 150 155 160Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn
Tyr Lys Thr Thr 165 170 175Pro Pro Val Leu Asp Ser Asp Gly Ser Phe
Phe Leu Tyr Ser Arg Leu 180 185 190Thr Val Asp Lys Ser Arg Trp Gln
Glu Gly Asn Val Phe Ser Cys Ser 195 200 205Val Met His Glu Ala Leu
His Asn His Tyr Thr Gln Lys Ser Leu Ser 210 215 220Leu Ser Leu Gly
Lys Met225 23046690DNAArtificial Sequencesource/note="Description
of Artificial Sequence Synthetic polynucleotide" 46gagagcaagt
acggccctcc ctgcccccct tgccctgccc ccgagttcct gggcggaccc 60agcgtgttcc
tgttcccccc caagcccaag gacaccctga tgatcagccg gacccccgag
120gtgacctgtg tggtggtgga cgtgtcccag gaggaccccg aggtccagtt
caactggtac 180gtggacggcg tggaggtgca caacgccaag accaagcccc
gggaggagca gttcaatagc 240acctaccggg tggtgtccgt gctgaccgtg
ctgcaccagg actggctgaa cggcaaggaa 300tacaagtgta aggtgtccaa
caagggcctg cccagcagca tcgagaaaac catcagcaag 360gccaagggcc
agcctcggga gccccaggtg tacaccctgc cccctagcca agaggagatg
420accaagaacc aggtgtccct gacctgcctg gtgaagggct tctaccccag
cgacatcgcc 480gtggagtggg agagcaacgg ccagcccgag aacaactaca
agaccacccc ccctgtgctg 540gacagcgacg gcagcttctt cctgtacagc
cggctgaccg tggacaagag ccggtggcag 600gagggcaacg tctttagctg
ctccgtgatg cacgaggccc tgcacaacca ctacacccag 660aagagcctga
gcctgtccct gggcaagatg 69047282PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 47Arg Trp Pro Glu Ser Pro Lys Ala Gln Ala Ser Ser Val
Pro Thr Ala1 5 10 15Gln Pro Gln Ala Glu Gly Ser Leu Ala Lys Ala Thr
Thr Ala Pro Ala 20 25 30Thr Thr Arg Asn Thr Gly Arg Gly Gly Glu Glu
Lys Lys Lys Glu Lys 35 40 45Glu Lys Glu Glu Gln Glu Glu Arg Glu Thr
Lys Thr Pro Glu Cys Pro 50 55 60Ser His Thr Gln Pro Leu Gly Val Tyr
Leu Leu Thr Pro Ala Val Gln65 70 75 80Asp Leu Trp Leu Arg Asp Lys
Ala Thr Phe Thr Cys Phe Val Val Gly 85 90 95Ser Asp Leu Lys Asp Ala
His Leu Thr Trp Glu Val Ala Gly Lys Val 100 105 110Pro Thr Gly Gly
Val Glu Glu Gly Leu Leu Glu Arg His Ser Asn Gly 115 120 125Ser Gln
Ser Gln His Ser Arg Leu Thr Leu Pro Arg Ser Leu Trp Asn 130 135
140Ala Gly Thr Ser Val Thr Cys Thr Leu Asn His Pro Ser Leu Pro
Pro145 150 155 160Gln Arg Leu Met Ala Leu Arg Glu Pro Ala Ala Gln
Ala Pro Val Lys 165 170 175Leu Ser Leu Asn Leu Leu Ala Ser Ser Asp
Pro Pro Glu Ala Ala Ser 180 185 190Trp Leu Leu Cys Glu Val Ser Gly
Phe Ser Pro Pro Asn Ile Leu Leu 195 200 205Met Trp Leu Glu Asp Gln
Arg Glu Val Asn Thr Ser Gly Phe Ala Pro 210 215 220Ala Arg Pro Pro
Pro Gln Pro Gly Ser Thr Thr Phe Trp Ala Trp Ser225 230 235 240Val
Leu Arg Val Pro Ala Pro Pro Ser Pro Gln Pro Ala Thr Tyr Thr 245 250
255Cys Val Val Ser His Glu Asp Ser Arg Thr Leu Leu Asn Ala Ser Arg
260 265 270Ser Leu Glu Val Ser Tyr Val Thr Asp His 275
28048847DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 48aggtggcccg
aaagtcccaa ggcccaggca tctagtgttc ctactgcaca gccccaggca 60gaaggcagcc
tagccaaagc tactactgca cctgccacta cgcgcaatac tggccgtggc
120ggggaggaga agaaaaagga gaaagagaaa gaagaacagg aagagaggga
gaccaagacc 180cctgaatgtc catcccatac ccagccgctg ggcgtctatc
tcttgactcc cgcagtacag 240gacttgtggc ttagagataa ggccaccttt
acatgtttcg tcgtgggctc tgacctgaag 300gatgcccatt tgacttggga
ggttgccgga aaggtaccca cagggggggt tgaggaaggg 360ttgctggagc
gccattccaa tggctctcag agccagcact caagactcac ccttccgaga
420tccctgtgga acgccgggac ctctgtcaca tgtactctaa atcatcctag
cctgccccca 480cagcgtctga tggcccttag agagccagcc gcccaggcac
cagttaagct tagcctgaat 540ctgctcgcca gtagtgatcc cccagaggcc
gccagctggc tcttatgcga agtgtccggc 600tttagcccgc ccaacatctt
gctcatgtgg ctggaggacc agcgagaagt gaacaccagc 660ggcttcgctc
cagcccggcc cccaccccag ccgggttcta ccacattctg ggcctggagt
720gtcttaaggg tcccagcacc acctagcccc cagccagcca catacacctg
tgttgtgtcc 780catgaagata gcaggaccct gctaaatgct tctaggagtc
tggaggtttc ctacgtgact 840gaccatt 8474910PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
peptide" 49Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser1 5
105030PRTArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic polypeptide" 50Gly Gly Thr Gly Gly Cys Gly Gly
Ala Gly Gly Thr Thr Cys Thr Gly1 5 10 15Gly Ala Gly Gly Thr Gly Gly
Ala Gly Gly Thr Thr Cys Cys 20 25 305148PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 51Gln Arg Arg Lys Tyr Arg Ser Asn Lys Gly Glu Ser Pro
Val Glu Pro1 5 10 15Ala Glu Pro Cys Arg Tyr Ser Cys Pro Arg Glu Glu
Glu Gly Ser Thr 20 25 30Ile Pro Ile Gln Glu Asp Tyr Arg Lys Pro Glu
Pro Ala Cys Ser Pro 35 40 4552123DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 52aggagtaaga ggagcaggct cctgcacagt gactacatga
acatgactcc ccgccgcccc 60gggcccaccc gcaagcatta ccagccctat gccccaccac
gcgacttcgc agcctatcgc 120tcc 1235330PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide"misc_feature(1)..(30)/note="This sequence may encompass
1-6 repeating "Gly Gly Gly Gly Ser" repeating units" 53Gly Gly Gly
Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly1 5 10 15Gly Gly
Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser 20 25
305463DNAArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic oligonucleotide" 54atggccctgc ctgtgacagc
cctgctgctg cctctggctc tgctgctgca tgccgctaga 60ccc
6355135DNAArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic polynucleotide" 55accacgacgc cagcgccgcg
accaccaaca ccggcgccca ccatcgcgtc gcagcccctg 60tccctgcgcc cagaggcgtg
ccggccagcg gcggggggcg cagtgcacac gagggggctg 120gacttcgcct gtgat
1355672DNAArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic oligonucleotide" 56atctacatct gggcgccctt
ggccgggact tgtggggtcc ttctcctgtc actggttatc 60accctttact gc
72575PRTArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic peptide" 57Tyr Asn Ser Ala Leu1
558486PRTArtificial Sequencesource/note="Description of Artificial
Sequence Synthetic polypeptide" 58Met Ala Leu Pro Val Thr Ala Leu
Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg Pro Asp Ile
Gln Met Thr Gln Thr Thr Ser Ser Leu 20 25 30Ser Ala Ser Leu Gly Asp
Arg Val Thr Ile Ser Cys Arg Ala Ser Gln 35 40 45Asp Ile Ser Lys Tyr
Leu Asn Trp Tyr Gln Gln Lys Pro Asp Gly Thr 50 55 60Val Lys Leu Leu
Ile Tyr His Thr Ser Arg Leu His Ser Gly Val Pro65 70 75 80Ser Arg
Phe Ser Gly Ser Gly Ser Gly Thr Asp Tyr Ser Leu Thr Ile 85 90 95Ser
Asn Leu Glu Gln Glu Asp Ile Ala Thr Tyr Phe Cys Gln Gln Gly 100 105
110Asn Thr Leu Pro Tyr Thr Phe Gly Gly Gly Thr Lys Leu Glu Ile Thr
115 120 125Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly
Ser Glu 130 135 140Val Lys Leu Gln Glu Ser Gly Pro Gly Leu Val Ala
Pro Ser Gln Ser145 150 155 160Leu Ser Val Thr Cys Thr Val Ser Gly
Val Ser Leu Pro Asp Tyr Gly 165 170 175Val Ser Trp Ile Arg Gln Pro
Pro Arg Lys Gly Leu Glu Trp Leu Gly 180 185 190Val Ile Trp Gly Ser
Glu Thr Thr Tyr Tyr Asn Ser Ala Leu Lys Ser 195 200 205Arg Leu Thr
Ile Ile Lys Asp Asn Ser Lys Ser Gln Val Phe Leu Lys 210 215 220Met
Asn Ser Leu Gln Thr Asp Asp Thr Ala Ile Tyr Tyr Cys Ala Lys225 230
235 240His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp Tyr Trp Gly Gln
Gly 245 250 255Thr Ser Val Thr Val Ser Ser Thr Thr Thr Pro Ala Pro
Arg Pro Pro 260 265 270Thr Pro Ala Pro Thr Ile Ala Ser Gln Pro Leu
Ser Leu Arg Pro Glu 275 280 285Ala Cys Arg Pro Ala Ala Gly Gly Ala
Val His Thr Arg Gly Leu Asp 290 295 300Phe Ala Cys Asp Ile Tyr Ile
Trp Ala Pro Leu Ala Gly Thr Cys Gly305 310 315 320Val Leu Leu Leu
Ser Leu Val Ile Thr Leu Tyr Cys Lys Arg Gly Arg 325 330 335Lys Lys
Leu Leu Tyr Ile Phe Lys Gln Pro Phe Met Arg Pro Val Gln 340 345
350Thr Thr Gln Glu Glu Asp Gly Cys Ser Cys Arg Phe Pro Glu Glu Glu
355 360 365Glu Gly Gly Cys Glu Leu Arg Val Lys Phe Ser Arg Ser Ala
Asp Ala 370 375 380Pro Ala Tyr Lys Gln Gly Gln Asn Gln Leu Tyr Asn
Glu Leu Asn Leu385 390 395 400Gly Arg Arg Glu Glu Tyr Asp Val Leu
Asp Lys Arg Arg Gly Arg Asp 405 410 415Pro Glu Met Gly Gly Lys Pro
Arg Arg Lys Asn Pro Gln Glu Gly Leu 420 425 430Tyr Asn Glu Leu Gln
Lys Asp Lys Met Ala Glu Ala Tyr Ser Glu Ile 435 440 445Gly Met Lys
Gly Glu Arg Arg Arg Gly Lys Gly His Asp Gly Leu Tyr 450 455 460Gln
Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr Asp Ala Leu His Met465 470
475 480Gln Ala Leu Pro Pro Arg 48559242PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 59Asp Ile Gln Met Thr Gln Thr Thr Ser Ser Leu Ser Ala
Ser Leu Gly1 5 10 15Asp Arg Val Thr Ile Ser Cys Arg Ala Ser Gln Asp
Ile Ser Lys Tyr 20 25 30Leu Asn Trp Tyr Gln Gln Lys Pro Asp Gly Thr
Val Lys Leu Leu Ile 35 40 45Tyr His Thr Ser Arg Leu His Ser Gly Val
Pro Ser Arg Phe Ser Gly 50 55 60Ser Gly Ser Gly Thr Asp Tyr Ser Leu
Thr Ile Ser Asn Leu Glu Gln65 70 75 80Glu Asp Ile Ala Thr Tyr Phe
Cys Gln Gln Gly Asn Thr Leu Pro Tyr 85 90 95Thr Phe Gly Gly Gly Thr
Lys Leu Glu Ile Thr Gly Gly Gly Gly Ser 100 105 110Gly Gly Gly Gly
Ser Gly Gly Gly Gly Ser Glu Val Lys Leu Gln Glu 115 120 125Ser Gly
Pro Gly Leu Val Ala Pro Ser Gln Ser Leu Ser Val Thr Cys 130 135
140Thr Val Ser Gly Val Ser Leu Pro Asp Tyr Gly Val Ser Trp Ile
Arg145 150 155 160Gln Pro Pro Arg Lys Gly Leu Glu Trp Leu Gly Val
Ile Trp Gly Ser 165 170 175Glu Thr Thr Tyr Tyr Asn Ser Ala Leu Lys
Ser Arg Leu Thr Ile Ile 180 185 190Lys Asp Asn Ser Lys Ser Gln Val
Phe Leu Lys Met Asn Ser Leu Gln 195 200 205Thr Asp Asp Thr Ala Ile
Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly 210 215 220Gly Ser Tyr Ala
Met Asp Tyr Trp Gly Gln Gly Thr Ser Val Thr Val225 230 235 240Ser
Ser60126DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 60aaacggggca
gaaagaaact cctgtatata ttcaaacaac catttatgag accagtacaa 60actactcaag
aggaagatgg ctgtagctgc cgatttccag aagaagaaga aggaggatgt 120gaactg
12661813DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 61atggccctcc
ctgtcaccgc cctgctgctt ccgctggctc ttctgctcca cgccgctcgg 60cccgaaattg
tgatgaccca gtcacccgcc actcttagcc tttcacccgg tgagcgcgca
120accctgtctt gcagagcctc ccaagacatc tcaaaatacc ttaattggta
tcaacagaag 180cccggacagg ctcctcgcct tctgatctac cacaccagcc
ggctccattc tggaatccct 240gccaggttca gcggtagcgg atctgggacc
gactacaccc tcactatcag ctcactgcag 300ccagaggact tcgctgtcta
tttctgtcag caagggaaca ccctgcccta cacctttgga 360cagggcacca
agctcgagat taaaggtgga ggtggcagcg gaggaggtgg gtccggcggt
420ggaggaagcc aggtccaact ccaagaaagc ggaccgggtc ttgtgaagcc
atcagaaact 480ctttcactga cttgtactgt gagcggagtg tctctccccg
attacggggt gtcttggatc 540agacagccac cggggaaggg tctggaatgg
attggagtga tttggggctc tgagactact 600tactactctt catccctcaa
gtcacgcgtc accatctcaa aggacaactc taagaatcag 660gtgtcactga
aactgtcatc tgtgaccgca gccgacaccg ccgtgtacta ttgcgctaag
720cattactatt atggcgggag ctacgcaatg gattactggg gacagggtac
tctggtcacc 780gtgtccagcc accaccatca tcaccatcac cat
81362813DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 62atggccctcc
ctgtcaccgc cctgctgctt ccgctggctc ttctgctcca cgccgctcgg 60cccgaaattg
tgatgaccca gtcacccgcc actcttagcc tttcacccgg tgagcgcgca
120accctgtctt gcagagcctc ccaagacatc tcaaaatacc ttaattggta
tcaacagaag 180cccggacagg ctcctcgcct tctgatctac cacaccagcc
ggctccattc tggaatccct 240gccaggttca gcggtagcgg atctgggacc
gactacaccc tcactatcag ctcactgcag 300ccagaggact tcgctgtcta
tttctgtcag caagggaaca ccctgcccta cacctttgga 360cagggcacca
agctcgagat taaaggtgga ggtggcagcg gaggaggtgg gtccggcggt
420ggaggaagcc aggtccaact ccaagaaagc ggaccgggtc ttgtgaagcc
atcagaaact 480ctttcactga cttgtactgt gagcggagtg tctctccccg
attacggggt gtcttggatc 540agacagccac cggggaaggg tctggaatgg
attggagtga tttggggctc tgagactact 600tactaccaat catccctcaa
gtcacgcgtc accatctcaa aggacaactc taagaatcag 660gtgtcactga
aactgtcatc tgtgaccgca gccgacaccg ccgtgtacta ttgcgctaag
720cattactatt atggcgggag ctacgcaatg gattactggg gacagggtac
tctggtcacc 780gtgtccagcc accaccatca tcaccatcac cat
81363813DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 63atggctctgc
ccgtgaccgc actcctcctg ccactggctc tgctgcttca cgccgctcgc 60ccacaagtcc
agcttcaaga atcagggcct ggtctggtga agccatctga gactctgtcc
120ctcacttgca ccgtgagcgg agtgtccctc ccagactacg gagtgagctg
gattagacag 180cctcccggaa agggactgga gtggatcgga gtgatttggg
gtagcgaaac cacttactat 240tcatcttccc tgaagtcacg ggtcaccatt
tcaaaggata actcaaagaa
tcaagtgagc 300ctcaagctct catcagtcac cgccgctgac accgccgtgt
attactgtgc caagcattac 360tactatggag ggtcctacgc catggactac
tggggccagg gaactctggt cactgtgtca 420tctggtggag gaggtagcgg
aggaggcggg agcggtggag gtggctccga aatcgtgatg 480acccagagcc
ctgcaaccct gtccctttct cccggggaac gggctaccct ttcttgtcgg
540gcatcacaag atatctcaaa atacctcaat tggtatcaac agaagccggg
acaggcccct 600aggcttctta tctaccacac ctctcgcctg catagcggga
ttcccgcacg ctttagcggg 660tctggaagcg ggaccgacta cactctgacc
atctcatctc tccagcccga ggacttcgcc 720gtctacttct gccagcaggg
taacaccctg ccgtacacct tcggccaggg caccaagctt 780gagatcaaac
atcaccacca tcatcaccat cac 81364813DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 64atggctctgc ccgtgaccgc actcctcctg ccactggctc
tgctgcttca cgccgctcgc 60ccacaagtcc agcttcaaga atcagggcct ggtctggtga
agccatctga gactctgtcc 120ctcacttgca ccgtgagcgg agtgtccctc
ccagactacg gagtgagctg gattagacag 180cctcccggaa agggactgga
gtggatcgga gtgatttggg gtagcgaaac cacttactat 240caatcttccc
tgaagtcacg ggtcaccatt tcaaaggata actcaaagaa tcaagtgagc
300ctcaagctct catcagtcac cgccgctgac accgccgtgt attactgtgc
caagcattac 360tactatggag ggtcctacgc catggactac tggggccagg
gaactctggt cactgtgtca 420tctggtggag gaggtagcgg aggaggcggg
agcggtggag gtggctccga aatcgtgatg 480acccagagcc ctgcaaccct
gtccctttct cccggggaac gggctaccct ttcttgtcgg 540gcatcacaag
atatctcaaa atacctcaat tggtatcaac agaagccggg acaggcccct
600aggcttctta tctaccacac ctctcgcctg catagcggga ttcccgcacg
ctttagcggg 660tctggaagcg ggaccgacta cactctgacc atctcatctc
tccagcccga ggacttcgcc 720gtctacttct gccagcaggg taacaccctg
ccgtacacct tcggccaggg caccaagctt 780gagatcaaac atcaccacca
tcatcaccat cac 81365828DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 65atggccctcc cagtgaccgc tctgctgctg cctctcgcac
ttcttctcca tgccgctcgg 60cctgagatcg tcatgaccca aagccccgct accctgtccc
tgtcacccgg cgagagggca 120accctttcat gcagggccag ccaggacatt
tctaagtacc tcaactggta tcagcagaag 180ccagggcagg ctcctcgcct
gctgatctac cacaccagcc gcctccacag cggtatcccc 240gccagatttt
ccgggagcgg gtctggaacc gactacaccc tcaccatctc ttctctgcag
300cccgaggatt tcgccgtcta tttctgccag caggggaata ctctgccgta
caccttcggt 360caaggtacca agctggaaat caagggaggc ggaggatcag
gcggtggcgg aagcggagga 420ggtggctccg gaggaggagg ttcccaagtg
cagcttcaag aatcaggacc cggacttgtg 480aagccatcag aaaccctctc
cctgacttgt accgtgtccg gtgtgagcct ccccgactac 540ggagtctctt
ggattcgcca gcctccgggg aagggtcttg aatggattgg ggtgatttgg
600ggatcagaga ctacttacta ctcttcatca cttaagtcac gggtcaccat
cagcaaagat 660aatagcaaga accaagtgtc acttaagctg tcatctgtga
ccgccgctga caccgccgtg 720tactattgtg ccaaacatta ctattacgga
gggtcttatg ctatggacta ctggggacag 780gggaccctgg tgactgtctc
tagccatcac catcaccacc atcatcac 82866828DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 66atggccctcc cagtgaccgc tctgctgctg cctctcgcac
ttcttctcca tgccgctcgg 60cctgagatcg tcatgaccca aagccccgct accctgtccc
tgtcacccgg cgagagggca 120accctttcat gcagggccag ccaggacatt
tctaagtacc tcaactggta tcagcagaag 180ccagggcagg ctcctcgcct
gctgatctac cacaccagcc gcctccacag cggtatcccc 240gccagatttt
ccgggagcgg gtctggaacc gactacaccc tcaccatctc ttctctgcag
300cccgaggatt tcgccgtcta tttctgccag caggggaata ctctgccgta
caccttcggt 360caaggtacca agctggaaat caagggaggc ggaggatcag
gcggtggcgg aagcggagga 420ggtggctccg gaggaggagg ttcccaagtg
cagcttcaag aatcaggacc cggacttgtg 480aagccatcag aaaccctctc
cctgacttgt accgtgtccg gtgtgagcct ccccgactac 540ggagtctctt
ggattcgcca gcctccgggg aagggtcttg aatggattgg ggtgatttgg
600ggatcagaga ctacttacta ccagtcatca cttaagtcac gggtcaccat
cagcaaagat 660aatagcaaga accaagtgtc acttaagctg tcatctgtga
ccgccgctga caccgccgtg 720tactattgtg ccaaacatta ctattacgga
gggtcttatg ctatggacta ctggggacag 780gggaccctgg tgactgtctc
tagccatcac catcaccacc atcatcac 82867828DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 67atggcactgc ctgtcactgc cctcctgctg cctctggccc
tccttctgca tgccgccagg 60ccccaagtcc agctgcaaga gtcaggaccc ggactggtga
agccgtctga gactctctca 120ctgacttgta ccgtcagcgg cgtgtccctc
cccgactacg gagtgtcatg gatccgccaa 180cctcccggga aagggcttga
atggattggt gtcatctggg gttctgaaac cacctactac 240tcatcttccc
tgaagtccag ggtgaccatc agcaaggata attccaagaa ccaggtcagc
300cttaagctgt catctgtgac cgctgctgac accgccgtgt attactgcgc
caagcactac 360tattacggag gaagctacgc tatggactat tggggacagg
gcactctcgt gactgtgagc 420agcggcggtg gagggtctgg aggtggagga
tccggtggtg gtgggtcagg cggaggaggg 480agcgagattg tgatgactca
gtcaccagcc accctttctc tttcacccgg cgagagagca 540accctgagct
gtagagccag ccaggacatt tctaagtacc tcaactggta tcagcaaaaa
600ccggggcagg cccctcgcct cctgatctac catacctcac gccttcactc
tggtatcccc 660gctcggttta gcggatcagg atctggtacc gactacactc
tgaccatttc cagcctgcag 720ccagaagatt tcgcagtgta tttctgccag
cagggcaata cccttcctta caccttcggt 780cagggaacca agctcgaaat
caagcaccat caccatcatc accaccat 82868828DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 68atggcactgc ctgtcactgc cctcctgctg cctctggccc
tccttctgca tgccgccagg 60ccccaagtcc agctgcaaga gtcaggaccc ggactggtga
agccgtctga gactctctca 120ctgacttgta ccgtcagcgg cgtgtccctc
cccgactacg gagtgtcatg gatccgccaa 180cctcccggga aagggcttga
atggattggt gtcatctggg gttctgaaac cacctactac 240cagtcttccc
tgaagtccag ggtgaccatc agcaaggata attccaagaa ccaggtcagc
300cttaagctgt catctgtgac cgctgctgac accgccgtgt attactgcgc
caagcactac 360tattacggag gaagctacgc tatggactat tggggacagg
gcactctcgt gactgtgagc 420agcggcggtg gagggtctgg aggtggagga
tccggtggtg gtgggtcagg cggaggaggg 480agcgagattg tgatgactca
gtcaccagcc accctttctc tttcacccgg cgagagagca 540accctgagct
gtagagccag ccaggacatt tctaagtacc tcaactggta tcagcaaaaa
600ccggggcagg cccctcgcct cctgatctac catacctcac gccttcactc
tggtatcccc 660gctcggttta gcggatcagg atctggtacc gactacactc
tgaccatttc cagcctgcag 720ccagaagatt tcgcagtgta tttctgccag
cagggcaata cccttcctta caccttcggt 780cagggaacca agctcgaaat
caagcaccat caccatcatc atcaccac 82869828DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 69atggccctcc cagtgaccgc tctgctgctg cctctcgcac
ttcttctcca tgccgctcgg 60cctgagatcg tcatgaccca aagccccgct accctgtccc
tgtcacccgg cgagagggca 120accctttcat gcagggccag ccaggacatt
tctaagtacc tcaactggta tcagcagaag 180ccagggcagg ctcctcgcct
gctgatctac cacaccagcc gcctccacag cggtatcccc 240gccagatttt
ccgggagcgg gtctggaacc gactacaccc tcaccatctc ttctctgcag
300cccgaggatt tcgccgtcta tttctgccag caggggaata ctctgccgta
caccttcggt 360caaggtacca agctggaaat caagggaggc ggaggatcag
gcggtggcgg aagcggagga 420ggtggctccg gaggaggagg ttcccaagtg
cagcttcaag aatcaggacc cggacttgtg 480aagccatcag aaaccctctc
cctgacttgt accgtgtccg gtgtgagcct ccccgactac 540ggagtctctt
ggattcgcca gcctccgggg aagggtcttg aatggattgg ggtgatttgg
600ggatcagaga ctacttacta caattcatca cttaagtcac gggtcaccat
cagcaaagat 660aatagcaaga accaagtgtc acttaagctg tcatctgtga
ccgccgctga caccgccgtg 720tactattgtg ccaaacatta ctattacgga
gggtcttatg ctatggacta ctggggacag 780gggaccctgg tgactgtctc
tagccatcac catcaccacc atcatcac 82870828DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 70atggcactgc ctgtcactgc cctcctgctg cctctggccc
tccttctgca tgccgccagg 60ccccaagtcc agctgcaaga gtcaggaccc ggactggtga
agccgtctga gactctctca 120ctgacttgta ccgtcagcgg cgtgtccctc
cccgactacg gagtgtcatg gatccgccaa 180cctcccggga aagggcttga
atggattggt gtcatctggg gttctgaaac cacctactac 240aactcttccc
tgaagtccag ggtgaccatc agcaaggata attccaagaa ccaggtcagc
300cttaagctgt catctgtgac cgctgctgac accgccgtgt attactgcgc
caagcactac 360tattacggag gaagctacgc tatggactat tggggacagg
gcactctcgt gactgtgagc 420agcggcggtg gagggtctgg aggtggagga
tccggtggtg gtgggtcagg cggaggaggg 480agcgagattg tgatgactca
gtcaccagcc accctttctc tttcacccgg cgagagagca 540accctgagct
gtagagccag ccaggacatt tctaagtacc tcaactggta tcagcaaaaa
600ccggggcagg cccctcgcct cctgatctac catacctcac gccttcactc
tggtatcccc 660gctcggttta gcggatcagg atctggtacc gactacactc
tgaccatttc cagcctgcag 720ccagaagatt tcgcagtgta tttctgccag
cagggcaata cccttcctta caccttcggt 780cagggaacca agctcgaaat
caagcaccat caccatcatc accaccat 82871813DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 71atggccctcc ctgtcaccgc cctgctgctt ccgctggctc
ttctgctcca cgccgctcgg 60cccgaaattg tgatgaccca gtcacccgcc actcttagcc
tttcacccgg tgagcgcgca 120accctgtctt gcagagcctc ccaagacatc
tcaaaatacc ttaattggta tcaacagaag 180cccggacagg ctcctcgcct
tctgatctac cacaccagcc ggctccattc tggaatccct 240gccaggttca
gcggtagcgg atctgggacc gactacaccc tcactatcag ctcactgcag
300ccagaggact tcgctgtcta tttctgtcag caagggaaca ccctgcccta
cacctttgga 360cagggcacca agctcgagat taaaggtgga ggtggcagcg
gaggaggtgg gtccggcggt 420ggaggaagcc aggtccaact ccaagaaagc
ggaccgggtc ttgtgaagcc atcagaaact 480ctttcactga cttgtactgt
gagcggagtg tctctccccg attacggggt gtcttggatc 540agacagccac
cggggaaggg tctggaatgg attggagtga tttggggctc tgagactact
600tactacaatt catccctcaa gtcacgcgtc accatctcaa aggacaactc
taagaatcag 660gtgtcactga aactgtcatc tgtgaccgca gccgacaccg
ccgtgtacta ttgcgctaag 720cattactatt atggcgggag ctacgcaatg
gattactggg gacagggtac tctggtcacc 780gtgtccagcc accaccatca
tcaccatcac cat 81372813DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 72atggctctgc ccgtgaccgc actcctcctg ccactggctc
tgctgcttca cgccgctcgc 60ccacaagtcc agcttcaaga atcagggcct ggtctggtga
agccatctga gactctgtcc 120ctcacttgca ccgtgagcgg agtgtccctc
ccagactacg gagtgagctg gattagacag 180cctcccggaa agggactgga
gtggatcgga gtgatttggg gtagcgaaac cacttactat 240aactcttccc
tgaagtcacg ggtcaccatt tcaaaggata actcaaagaa tcaagtgagc
300ctcaagctct catcagtcac cgccgctgac accgccgtgt attactgtgc
caagcattac 360tactatggag ggtcctacgc catggactac tggggccagg
gaactctggt cactgtgtca 420tctggtggag gaggtagcgg aggaggcggg
agcggtggag gtggctccga aatcgtgatg 480acccagagcc ctgcaaccct
gtccctttct cccggggaac gggctaccct ttcttgtcgg 540gcatcacaag
atatctcaaa atacctcaat tggtatcaac agaagccggg acaggcccct
600aggcttctta tctaccacac ctctcgcctg catagcggga ttcccgcacg
ctttagcggg 660tctggaagcg ggaccgacta cactctgacc atctcatctc
tccagcccga ggacttcgcc 720gtctacttct gccagcaggg taacaccctg
ccgtacacct tcggccaggg caccaagctt 780gagatcaaac atcaccacca
tcatcaccat cac 81373271PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 73Met Ala Leu Pro Val Thr Ala Leu Leu Leu Pro Leu Ala
Leu Leu Leu1 5 10 15His Ala Ala Arg Pro Glu Ile Val Met Thr Gln Ser
Pro Ala Thr Leu 20 25 30Ser Leu Ser Pro Gly Glu Arg Ala Thr Leu Ser
Cys Arg Ala Ser Gln 35 40 45Asp Ile Ser Lys Tyr Leu Asn Trp Tyr Gln
Gln Lys Pro Gly Gln Ala 50 55 60Pro Arg Leu Leu Ile Tyr His Thr Ser
Arg Leu His Ser Gly Ile Pro65 70 75 80Ala Arg Phe Ser Gly Ser Gly
Ser Gly Thr Asp Tyr Thr Leu Thr Ile 85 90 95Ser Ser Leu Gln Pro Glu
Asp Phe Ala Val Tyr Phe Cys Gln Gln Gly 100 105 110Asn Thr Leu Pro
Tyr Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys 115 120 125Gly Gly
Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gln 130 135
140Val Gln Leu Gln Glu Ser Gly Pro Gly Leu Val Lys Pro Ser Glu
Thr145 150 155 160Leu Ser Leu Thr Cys Thr Val Ser Gly Val Ser Leu
Pro Asp Tyr Gly 165 170 175Val Ser Trp Ile Arg Gln Pro Pro Gly Lys
Gly Leu Glu Trp Ile Gly 180 185 190Val Ile Trp Gly Ser Glu Thr Thr
Tyr Tyr Ser Ser Ser Leu Lys Ser 195 200 205Arg Val Thr Ile Ser Lys
Asp Asn Ser Lys Asn Gln Val Ser Leu Lys 210 215 220Leu Ser Ser Val
Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys Ala Lys225 230 235 240His
Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp Tyr Trp Gly Gln Gly 245 250
255Thr Leu Val Thr Val Ser Ser His His His His His His His His 260
265 27074271PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 74Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Glu Ile Val Met Thr Gln Ser Pro Ala Thr Leu 20 25 30Ser Leu Ser
Pro Gly Glu Arg Ala Thr Leu Ser Cys Arg Ala Ser Gln 35 40 45Asp Ile
Ser Lys Tyr Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala 50 55 60Pro
Arg Leu Leu Ile Tyr His Thr Ser Arg Leu His Ser Gly Ile Pro65 70 75
80Ala Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile
85 90 95Ser Ser Leu Gln Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln
Gly 100 105 110Asn Thr Leu Pro Tyr Thr Phe Gly Gln Gly Thr Lys Leu
Glu Ile Lys 115 120 125Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly
Gly Gly Gly Ser Gln 130 135 140Val Gln Leu Gln Glu Ser Gly Pro Gly
Leu Val Lys Pro Ser Glu Thr145 150 155 160Leu Ser Leu Thr Cys Thr
Val Ser Gly Val Ser Leu Pro Asp Tyr Gly 165 170 175Val Ser Trp Ile
Arg Gln Pro Pro Gly Lys Gly Leu Glu Trp Ile Gly 180 185 190Val Ile
Trp Gly Ser Glu Thr Thr Tyr Tyr Gln Ser Ser Leu Lys Ser 195 200
205Arg Val Thr Ile Ser Lys Asp Asn Ser Lys Asn Gln Val Ser Leu Lys
210 215 220Leu Ser Ser Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys
Ala Lys225 230 235 240His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp
Tyr Trp Gly Gln Gly 245 250 255Thr Leu Val Thr Val Ser Ser His His
His His His His His His 260 265 27075271PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 75Met Ala Leu Pro Val Thr Ala Leu Leu Leu Pro Leu Ala
Leu Leu Leu1 5 10 15His Ala Ala Arg Pro Gln Val Gln Leu Gln Glu Ser
Gly Pro Gly Leu 20 25 30Val Lys Pro Ser Glu Thr Leu Ser Leu Thr Cys
Thr Val Ser Gly Val 35 40 45Ser Leu Pro Asp Tyr Gly Val Ser Trp Ile
Arg Gln Pro Pro Gly Lys 50 55 60Gly Leu Glu Trp Ile Gly Val Ile Trp
Gly Ser Glu Thr Thr Tyr Tyr65 70 75 80Ser Ser Ser Leu Lys Ser Arg
Val Thr Ile Ser Lys Asp Asn Ser Lys 85 90 95Asn Gln Val Ser Leu Lys
Leu Ser Ser Val Thr Ala Ala Asp Thr Ala 100 105 110Val Tyr Tyr Cys
Ala Lys His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met 115 120 125Asp Tyr
Trp Gly Gln Gly Thr Leu Val Thr Val Ser Ser Gly Gly Gly 130 135
140Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Glu Ile Val
Met145 150 155 160Thr Gln Ser Pro Ala Thr Leu Ser Leu Ser Pro Gly
Glu Arg Ala Thr 165 170 175Leu Ser Cys Arg Ala Ser Gln Asp Ile Ser
Lys Tyr Leu Asn Trp Tyr 180 185 190Gln Gln Lys Pro Gly Gln Ala Pro
Arg Leu Leu Ile Tyr His Thr Ser 195 200 205Arg Leu His Ser Gly Ile
Pro Ala Arg Phe Ser Gly Ser Gly Ser Gly 210 215 220Thr Asp Tyr Thr
Leu Thr Ile Ser Ser Leu Gln Pro Glu Asp Phe Ala225 230 235 240Val
Tyr Phe Cys Gln Gln Gly Asn Thr Leu Pro Tyr Thr Phe Gly Gln 245 250
255Gly Thr Lys Leu Glu Ile Lys His His His His His His His His 260
265 27076271PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 76Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu 20 25 30Val Lys Pro
Ser Glu Thr Leu Ser Leu Thr Cys Thr Val Ser Gly Val 35 40 45Ser Leu
Pro Asp Tyr Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys 50 55 60Gly
Leu Glu Trp Ile Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr65 70 75
80Gln Ser Ser Leu Lys Ser Arg Val Thr Ile Ser Lys Asp Asn Ser Lys
85 90 95Asn Gln Val Ser Leu Lys Leu Ser Ser Val Thr Ala Ala Asp Thr
Ala 100 105 110Val Tyr Tyr Cys Ala Lys His Tyr Tyr
Tyr Gly Gly Ser Tyr Ala Met 115 120 125Asp Tyr Trp Gly Gln Gly Thr
Leu Val Thr Val Ser Ser Gly Gly Gly 130 135 140Gly Ser Gly Gly Gly
Gly Ser Gly Gly Gly Gly Ser Glu Ile Val Met145 150 155 160Thr Gln
Ser Pro Ala Thr Leu Ser Leu Ser Pro Gly Glu Arg Ala Thr 165 170
175Leu Ser Cys Arg Ala Ser Gln Asp Ile Ser Lys Tyr Leu Asn Trp Tyr
180 185 190Gln Gln Lys Pro Gly Gln Ala Pro Arg Leu Leu Ile Tyr His
Thr Ser 195 200 205Arg Leu His Ser Gly Ile Pro Ala Arg Phe Ser Gly
Ser Gly Ser Gly 210 215 220Thr Asp Tyr Thr Leu Thr Ile Ser Ser Leu
Gln Pro Glu Asp Phe Ala225 230 235 240Val Tyr Phe Cys Gln Gln Gly
Asn Thr Leu Pro Tyr Thr Phe Gly Gln 245 250 255Gly Thr Lys Leu Glu
Ile Lys His His His His His His His His 260 265
27077276PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 77Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Glu Ile Val Met Thr Gln Ser Pro Ala Thr Leu 20 25 30Ser Leu Ser
Pro Gly Glu Arg Ala Thr Leu Ser Cys Arg Ala Ser Gln 35 40 45Asp Ile
Ser Lys Tyr Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala 50 55 60Pro
Arg Leu Leu Ile Tyr His Thr Ser Arg Leu His Ser Gly Ile Pro65 70 75
80Ala Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile
85 90 95Ser Ser Leu Gln Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln
Gly 100 105 110Asn Thr Leu Pro Tyr Thr Phe Gly Gln Gly Thr Lys Leu
Glu Ile Lys 115 120 125Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly
Gly Gly Gly Ser Gly 130 135 140Gly Gly Gly Ser Gln Val Gln Leu Gln
Glu Ser Gly Pro Gly Leu Val145 150 155 160Lys Pro Ser Glu Thr Leu
Ser Leu Thr Cys Thr Val Ser Gly Val Ser 165 170 175Leu Pro Asp Tyr
Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys Gly 180 185 190Leu Glu
Trp Ile Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr Ser 195 200
205Ser Ser Leu Lys Ser Arg Val Thr Ile Ser Lys Asp Asn Ser Lys Asn
210 215 220Gln Val Ser Leu Lys Leu Ser Ser Val Thr Ala Ala Asp Thr
Ala Val225 230 235 240Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly Gly
Ser Tyr Ala Met Asp 245 250 255Tyr Trp Gly Gln Gly Thr Leu Val Thr
Val Ser Ser His His His His 260 265 270His His His His
27578276PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 78Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Glu Ile Val Met Thr Gln Ser Pro Ala Thr Leu 20 25 30Ser Leu Ser
Pro Gly Glu Arg Ala Thr Leu Ser Cys Arg Ala Ser Gln 35 40 45Asp Ile
Ser Lys Tyr Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala 50 55 60Pro
Arg Leu Leu Ile Tyr His Thr Ser Arg Leu His Ser Gly Ile Pro65 70 75
80Ala Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile
85 90 95Ser Ser Leu Gln Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln
Gly 100 105 110Asn Thr Leu Pro Tyr Thr Phe Gly Gln Gly Thr Lys Leu
Glu Ile Lys 115 120 125Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly
Gly Gly Gly Ser Gly 130 135 140Gly Gly Gly Ser Gln Val Gln Leu Gln
Glu Ser Gly Pro Gly Leu Val145 150 155 160Lys Pro Ser Glu Thr Leu
Ser Leu Thr Cys Thr Val Ser Gly Val Ser 165 170 175Leu Pro Asp Tyr
Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys Gly 180 185 190Leu Glu
Trp Ile Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr Gln 195 200
205Ser Ser Leu Lys Ser Arg Val Thr Ile Ser Lys Asp Asn Ser Lys Asn
210 215 220Gln Val Ser Leu Lys Leu Ser Ser Val Thr Ala Ala Asp Thr
Ala Val225 230 235 240Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly Gly
Ser Tyr Ala Met Asp 245 250 255Tyr Trp Gly Gln Gly Thr Leu Val Thr
Val Ser Ser His His His His 260 265 270His His His His
27579276PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 79Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu 20 25 30Val Lys Pro
Ser Glu Thr Leu Ser Leu Thr Cys Thr Val Ser Gly Val 35 40 45Ser Leu
Pro Asp Tyr Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys 50 55 60Gly
Leu Glu Trp Ile Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr65 70 75
80Ser Ser Ser Leu Lys Ser Arg Val Thr Ile Ser Lys Asp Asn Ser Lys
85 90 95Asn Gln Val Ser Leu Lys Leu Ser Ser Val Thr Ala Ala Asp Thr
Ala 100 105 110Val Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly Gly Ser
Tyr Ala Met 115 120 125Asp Tyr Trp Gly Gln Gly Thr Leu Val Thr Val
Ser Ser Gly Gly Gly 130 135 140Gly Ser Gly Gly Gly Gly Ser Gly Gly
Gly Gly Ser Gly Gly Gly Gly145 150 155 160Ser Glu Ile Val Met Thr
Gln Ser Pro Ala Thr Leu Ser Leu Ser Pro 165 170 175Gly Glu Arg Ala
Thr Leu Ser Cys Arg Ala Ser Gln Asp Ile Ser Lys 180 185 190Tyr Leu
Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala Pro Arg Leu Leu 195 200
205Ile Tyr His Thr Ser Arg Leu His Ser Gly Ile Pro Ala Arg Phe Ser
210 215 220Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile Ser Ser
Leu Gln225 230 235 240Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln
Gly Asn Thr Leu Pro 245 250 255Tyr Thr Phe Gly Gln Gly Thr Lys Leu
Glu Ile Lys His His His His 260 265 270His His His His
27580276PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 80Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu 20 25 30Val Lys Pro
Ser Glu Thr Leu Ser Leu Thr Cys Thr Val Ser Gly Val 35 40 45Ser Leu
Pro Asp Tyr Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys 50 55 60Gly
Leu Glu Trp Ile Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr65 70 75
80Gln Ser Ser Leu Lys Ser Arg Val Thr Ile Ser Lys Asp Asn Ser Lys
85 90 95Asn Gln Val Ser Leu Lys Leu Ser Ser Val Thr Ala Ala Asp Thr
Ala 100 105 110Val Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly Gly Ser
Tyr Ala Met 115 120 125Asp Tyr Trp Gly Gln Gly Thr Leu Val Thr Val
Ser Ser Gly Gly Gly 130 135 140Gly Ser Gly Gly Gly Gly Ser Gly Gly
Gly Gly Ser Gly Gly Gly Gly145 150 155 160Ser Glu Ile Val Met Thr
Gln Ser Pro Ala Thr Leu Ser Leu Ser Pro 165 170 175Gly Glu Arg Ala
Thr Leu Ser Cys Arg Ala Ser Gln Asp Ile Ser Lys 180 185 190Tyr Leu
Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala Pro Arg Leu Leu 195 200
205Ile Tyr His Thr Ser Arg Leu His Ser Gly Ile Pro Ala Arg Phe Ser
210 215 220Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile Ser Ser
Leu Gln225 230 235 240Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln
Gly Asn Thr Leu Pro 245 250 255Tyr Thr Phe Gly Gln Gly Thr Lys Leu
Glu Ile Lys His His His His 260 265 270His His His His
27581276PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 81Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Glu Ile Val Met Thr Gln Ser Pro Ala Thr Leu 20 25 30Ser Leu Ser
Pro Gly Glu Arg Ala Thr Leu Ser Cys Arg Ala Ser Gln 35 40 45Asp Ile
Ser Lys Tyr Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala 50 55 60Pro
Arg Leu Leu Ile Tyr His Thr Ser Arg Leu His Ser Gly Ile Pro65 70 75
80Ala Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile
85 90 95Ser Ser Leu Gln Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln
Gly 100 105 110Asn Thr Leu Pro Tyr Thr Phe Gly Gln Gly Thr Lys Leu
Glu Ile Lys 115 120 125Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly
Gly Gly Gly Ser Gly 130 135 140Gly Gly Gly Ser Gln Val Gln Leu Gln
Glu Ser Gly Pro Gly Leu Val145 150 155 160Lys Pro Ser Glu Thr Leu
Ser Leu Thr Cys Thr Val Ser Gly Val Ser 165 170 175Leu Pro Asp Tyr
Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys Gly 180 185 190Leu Glu
Trp Ile Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr Asn 195 200
205Ser Ser Leu Lys Ser Arg Val Thr Ile Ser Lys Asp Asn Ser Lys Asn
210 215 220Gln Val Ser Leu Lys Leu Ser Ser Val Thr Ala Ala Asp Thr
Ala Val225 230 235 240Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly Gly
Ser Tyr Ala Met Asp 245 250 255Tyr Trp Gly Gln Gly Thr Leu Val Thr
Val Ser Ser His His His His 260 265 270His His His His
27582276PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 82Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu 20 25 30Val Lys Pro
Ser Glu Thr Leu Ser Leu Thr Cys Thr Val Ser Gly Val 35 40 45Ser Leu
Pro Asp Tyr Gly Val Ser Trp Ile Arg Gln Pro Pro Gly Lys 50 55 60Gly
Leu Glu Trp Ile Gly Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr65 70 75
80Asn Ser Ser Leu Lys Ser Arg Val Thr Ile Ser Lys Asp Asn Ser Lys
85 90 95Asn Gln Val Ser Leu Lys Leu Ser Ser Val Thr Ala Ala Asp Thr
Ala 100 105 110Val Tyr Tyr Cys Ala Lys His Tyr Tyr Tyr Gly Gly Ser
Tyr Ala Met 115 120 125Asp Tyr Trp Gly Gln Gly Thr Leu Val Thr Val
Ser Ser Gly Gly Gly 130 135 140Gly Ser Gly Gly Gly Gly Ser Gly Gly
Gly Gly Ser Gly Gly Gly Gly145 150 155 160Ser Glu Ile Val Met Thr
Gln Ser Pro Ala Thr Leu Ser Leu Ser Pro 165 170 175Gly Glu Arg Ala
Thr Leu Ser Cys Arg Ala Ser Gln Asp Ile Ser Lys 180 185 190Tyr Leu
Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala Pro Arg Leu Leu 195 200
205Ile Tyr His Thr Ser Arg Leu His Ser Gly Ile Pro Ala Arg Phe Ser
210 215 220Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile Ser Ser
Leu Gln225 230 235 240Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln
Gly Asn Thr Leu Pro 245 250 255Tyr Thr Phe Gly Gln Gly Thr Lys Leu
Glu Ile Lys His His His His 260 265 270His His His His
27583271PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 83Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Glu Ile Val Met Thr Gln Ser Pro Ala Thr Leu 20 25 30Ser Leu Ser
Pro Gly Glu Arg Ala Thr Leu Ser Cys Arg Ala Ser Gln 35 40 45Asp Ile
Ser Lys Tyr Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Ala 50 55 60Pro
Arg Leu Leu Ile Tyr His Thr Ser Arg Leu His Ser Gly Ile Pro65 70 75
80Ala Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile
85 90 95Ser Ser Leu Gln Pro Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln
Gly 100 105 110Asn Thr Leu Pro Tyr Thr Phe Gly Gln Gly Thr Lys Leu
Glu Ile Lys 115 120 125Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly
Gly Gly Gly Ser Gln 130 135 140Val Gln Leu Gln Glu Ser Gly Pro Gly
Leu Val Lys Pro Ser Glu Thr145 150 155 160Leu Ser Leu Thr Cys Thr
Val Ser Gly Val Ser Leu Pro Asp Tyr Gly 165 170 175Val Ser Trp Ile
Arg Gln Pro Pro Gly Lys Gly Leu Glu Trp Ile Gly 180 185 190Val Ile
Trp Gly Ser Glu Thr Thr Tyr Tyr Asn Ser Ser Leu Lys Ser 195 200
205Arg Val Thr Ile Ser Lys Asp Asn Ser Lys Asn Gln Val Ser Leu Lys
210 215 220Leu Ser Ser Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys
Ala Lys225 230 235 240His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp
Tyr Trp Gly Gln Gly 245 250 255Thr Leu Val Thr Val Ser Ser His His
His His His His His His 260 265 27084271PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 84Met Ala Leu Pro Val Thr Ala Leu Leu Leu Pro Leu Ala
Leu Leu Leu1 5 10 15His Ala Ala Arg Pro Gln Val Gln Leu Gln Glu Ser
Gly Pro Gly Leu 20 25 30Val Lys Pro Ser Glu Thr Leu Ser Leu Thr Cys
Thr Val Ser Gly Val 35 40 45Ser Leu Pro Asp Tyr Gly Val Ser Trp Ile
Arg Gln Pro Pro Gly Lys 50 55 60Gly Leu Glu Trp Ile Gly Val Ile Trp
Gly Ser Glu Thr Thr Tyr Tyr65 70 75 80Asn Ser Ser Leu Lys Ser Arg
Val Thr Ile Ser Lys Asp Asn Ser Lys 85 90 95Asn Gln Val Ser Leu Lys
Leu Ser Ser Val Thr Ala Ala Asp Thr Ala 100 105 110Val Tyr Tyr Cys
Ala Lys His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met 115 120 125Asp Tyr
Trp Gly Gln Gly Thr Leu Val Thr Val Ser Ser Gly Gly Gly 130 135
140Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Glu Ile Val
Met145 150 155 160Thr Gln Ser Pro Ala Thr Leu Ser Leu Ser Pro Gly
Glu Arg Ala Thr 165 170 175Leu Ser Cys Arg Ala Ser Gln Asp Ile Ser
Lys Tyr Leu Asn Trp Tyr 180 185 190Gln Gln Lys Pro Gly Gln Ala Pro
Arg Leu Leu Ile Tyr His Thr Ser 195 200 205Arg Leu His Ser Gly Ile
Pro Ala Arg Phe Ser Gly Ser Gly Ser Gly 210 215 220Thr Asp Tyr Thr
Leu Thr Ile Ser Ser Leu Gln Pro Glu Asp Phe Ala225 230 235 240Val
Tyr Phe Cys Gln Gln Gly Asn Thr Leu Pro Tyr Thr Phe Gly Gln 245 250
255Gly Thr Lys Leu Glu Ile Lys His His His His His His His His 260
265 270851458DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 85atggccctcc ctgtcaccgc cctgctgctt ccgctggctc
ttctgctcca cgccgctcgg 60cccgaaattg tgatgaccca gtcacccgcc actcttagcc
tttcacccgg tgagcgcgca 120accctgtctt gcagagcctc ccaagacatc
tcaaaatacc ttaattggta tcaacagaag 180cccggacagg ctcctcgcct
tctgatctac cacaccagcc ggctccattc tggaatccct 240gccaggttca
gcggtagcgg atctgggacc gactacaccc tcactatcag ctcactgcag
300ccagaggact tcgctgtcta tttctgtcag caagggaaca ccctgcccta
cacctttgga 360cagggcacca agctcgagat taaaggtgga ggtggcagcg
gaggaggtgg gtccggcggt 420ggaggaagcc aggtccaact ccaagaaagc
ggaccgggtc ttgtgaagcc atcagaaact 480ctttcactga cttgtactgt
gagcggagtg tctctccccg attacggggt gtcttggatc 540agacagccac
cggggaaggg tctggaatgg attggagtga tttggggctc tgagactact
600tactactctt catccctcaa gtcacgcgtc accatctcaa aggacaactc
taagaatcag 660gtgtcactga aactgtcatc tgtgaccgca gccgacaccg
ccgtgtacta ttgcgctaag 720cattactatt atggcgggag ctacgcaatg
gattactggg gacagggtac tctggtcacc 780gtgtccagca ccactacccc
agcaccgagg ccacccaccc cggctcctac catcgcctcc 840cagcctctgt
ccctgcgtcc ggaggcatgt agacccgcag ctggtggggc cgtgcatacc
900cggggtcttg acttcgcctg cgatatctac atttgggccc ctctggctgg
tacttgcggg 960gtcctgctgc tttcactcgt gatcactctt tactgtaagc
gcggtcggaa gaagctgctg 1020tacatcttta agcaaccctt catgaggcct
gtgcagacta ctcaagagga ggacggctgt 1080tcatgccggt tcccagagga
ggaggaaggc ggctgcgaac tgcgcgtgaa attcagccgc 1140agcgcagatg
ctccagccta caagcagggg cagaaccagc tctacaacga actcaatctt
1200ggtcggagag aggagtacga cgtgctggac aagcggagag gacgggaccc
agaaatgggc 1260gggaagccgc gcagaaagaa tccccaagag ggcctgtaca
acgagctcca aaaggataag 1320atggcagaag cctatagcga gattggtatg
aaaggggaac gcagaagagg caaaggccac 1380gacggactgt accagggact
cagcaccgcc accaaggaca cctatgacgc tcttcacatg 1440caggccctgc cgcctcgg
1458861458DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 86atggccctcc
ctgtcaccgc cctgctgctt ccgctggctc ttctgctcca cgccgctcgg 60cccgaaattg
tgatgaccca gtcacccgcc actcttagcc tttcacccgg tgagcgcgca
120accctgtctt gcagagcctc ccaagacatc tcaaaatacc ttaattggta
tcaacagaag 180cccggacagg ctcctcgcct tctgatctac cacaccagcc
ggctccattc tggaatccct 240gccaggttca gcggtagcgg atctgggacc
gactacaccc tcactatcag ctcactgcag 300ccagaggact tcgctgtcta
tttctgtcag caagggaaca ccctgcccta cacctttgga 360cagggcacca
agctcgagat taaaggtgga ggtggcagcg gaggaggtgg gtccggcggt
420ggaggaagcc aggtccaact ccaagaaagc ggaccgggtc ttgtgaagcc
atcagaaact 480ctttcactga cttgtactgt gagcggagtg tctctccccg
attacggggt gtcttggatc 540agacagccac cggggaaggg tctggaatgg
attggagtga tttggggctc tgagactact 600tactaccaat catccctcaa
gtcacgcgtc accatctcaa aggacaactc taagaatcag 660gtgtcactga
aactgtcatc tgtgaccgca gccgacaccg ccgtgtacta ttgcgctaag
720cattactatt atggcgggag ctacgcaatg gattactggg gacagggtac
tctggtcacc 780gtgtccagca ccactacccc agcaccgagg ccacccaccc
cggctcctac catcgcctcc 840cagcctctgt ccctgcgtcc ggaggcatgt
agacccgcag ctggtggggc cgtgcatacc 900cggggtcttg acttcgcctg
cgatatctac atttgggccc ctctggctgg tacttgcggg 960gtcctgctgc
tttcactcgt gatcactctt tactgtaagc gcggtcggaa gaagctgctg
1020tacatcttta agcaaccctt catgaggcct gtgcagacta ctcaagagga
ggacggctgt 1080tcatgccggt tcccagagga ggaggaaggc ggctgcgaac
tgcgcgtgaa attcagccgc 1140agcgcagatg ctccagccta caagcagggg
cagaaccagc tctacaacga actcaatctt 1200ggtcggagag aggagtacga
cgtgctggac aagcggagag gacgggaccc agaaatgggc 1260gggaagccgc
gcagaaagaa tccccaagag ggcctgtaca acgagctcca aaaggataag
1320atggcagaag cctatagcga gattggtatg aaaggggaac gcagaagagg
caaaggccac 1380gacggactgt accagggact cagcaccgcc accaaggaca
cctatgacgc tcttcacatg 1440caggccctgc cgcctcgg
1458871458DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 87atggctctgc
ccgtgaccgc actcctcctg ccactggctc tgctgcttca cgccgctcgc 60ccacaagtcc
agcttcaaga atcagggcct ggtctggtga agccatctga gactctgtcc
120ctcacttgca ccgtgagcgg agtgtccctc ccagactacg gagtgagctg
gattagacag 180cctcccggaa agggactgga gtggatcgga gtgatttggg
gtagcgaaac cacttactat 240tcatcttccc tgaagtcacg ggtcaccatt
tcaaaggata actcaaagaa tcaagtgagc 300ctcaagctct catcagtcac
cgccgctgac accgccgtgt attactgtgc caagcattac 360tactatggag
ggtcctacgc catggactac tggggccagg gaactctggt cactgtgtca
420tctggtggag gaggtagcgg aggaggcggg agcggtggag gtggctccga
aatcgtgatg 480acccagagcc ctgcaaccct gtccctttct cccggggaac
gggctaccct ttcttgtcgg 540gcatcacaag atatctcaaa atacctcaat
tggtatcaac agaagccggg acaggcccct 600aggcttctta tctaccacac
ctctcgcctg catagcggga ttcccgcacg ctttagcggg 660tctggaagcg
ggaccgacta cactctgacc atctcatctc tccagcccga ggacttcgcc
720gtctacttct gccagcaggg taacaccctg ccgtacacct tcggccaggg
caccaagctt 780gagatcaaaa ccactactcc cgctccaagg ccacccaccc
ctgccccgac catcgcctct 840cagccgcttt ccctgcgtcc ggaggcatgt
agacccgcag ctggtggggc cgtgcatacc 900cggggtcttg acttcgcctg
cgatatctac atttgggccc ctctggctgg tacttgcggg 960gtcctgctgc
tttcactcgt gatcactctt tactgtaagc gcggtcggaa gaagctgctg
1020tacatcttta agcaaccctt catgaggcct gtgcagacta ctcaagagga
ggacggctgt 1080tcatgccggt tcccagagga ggaggaaggc ggctgcgaac
tgcgcgtgaa attcagccgc 1140agcgcagatg ctccagccta caagcagggg
cagaaccagc tctacaacga actcaatctt 1200ggtcggagag aggagtacga
cgtgctggac aagcggagag gacgggaccc agaaatgggc 1260gggaagccgc
gcagaaagaa tccccaagag ggcctgtaca acgagctcca aaaggataag
1320atggcagaag cctatagcga gattggtatg aaaggggaac gcagaagagg
caaaggccac 1380gacggactgt accagggact cagcaccgcc accaaggaca
cctatgacgc tcttcacatg 1440caggccctgc cgcctcgg
1458881458DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 88atggctctgc
ccgtgaccgc actcctcctg ccactggctc tgctgcttca cgccgctcgc 60ccacaagtcc
agcttcaaga atcagggcct ggtctggtga agccatctga gactctgtcc
120ctcacttgca ccgtgagcgg agtgtccctc ccagactacg gagtgagctg
gattagacag 180cctcccggaa agggactgga gtggatcgga gtgatttggg
gtagcgaaac cacttactat 240caatcttccc tgaagtcacg ggtcaccatt
tcaaaggata actcaaagaa tcaagtgagc 300ctcaagctct catcagtcac
cgccgctgac accgccgtgt attactgtgc caagcattac 360tactatggag
ggtcctacgc catggactac tggggccagg gaactctggt cactgtgtca
420tctggtggag gaggtagcgg aggaggcggg agcggtggag gtggctccga
aatcgtgatg 480acccagagcc ctgcaaccct gtccctttct cccggggaac
gggctaccct ttcttgtcgg 540gcatcacaag atatctcaaa atacctcaat
tggtatcaac agaagccggg acaggcccct 600aggcttctta tctaccacac
ctctcgcctg catagcggga ttcccgcacg ctttagcggg 660tctggaagcg
ggaccgacta cactctgacc atctcatctc tccagcccga ggacttcgcc
720gtctacttct gccagcaggg taacaccctg ccgtacacct tcggccaggg
caccaagctt 780gagatcaaaa ccactactcc cgctccaagg ccacccaccc
ctgccccgac catcgcctct 840cagccgcttt ccctgcgtcc ggaggcatgt
agacccgcag ctggtggggc cgtgcatacc 900cggggtcttg acttcgcctg
cgatatctac atttgggccc ctctggctgg tacttgcggg 960gtcctgctgc
tttcactcgt gatcactctt tactgtaagc gcggtcggaa gaagctgctg
1020tacatcttta agcaaccctt catgaggcct gtgcagacta ctcaagagga
ggacggctgt 1080tcatgccggt tcccagagga ggaggaaggc ggctgcgaac
tgcgcgtgaa attcagccgc 1140agcgcagatg ctccagccta caagcagggg
cagaaccagc tctacaacga actcaatctt 1200ggtcggagag aggagtacga
cgtgctggac aagcggagag gacgggaccc agaaatgggc 1260gggaagccgc
gcagaaagaa tccccaagag ggcctgtaca acgagctcca aaaggataag
1320atggcagaag cctatagcga gattggtatg aaaggggaac gcagaagagg
caaaggccac 1380gacggactgt accagggact cagcaccgcc accaaggaca
cctatgacgc tcttcacatg 1440caggccctgc cgcctcgg
1458891473DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 89atggccctcc
ctgtcaccgc cctgctgctt ccgctggctc ttctgctcca cgccgctcgg 60cccgaaattg
tgatgaccca gtcacccgcc actcttagcc tttcacccgg tgagcgcgca
120accctgtctt gcagagcctc ccaagacatc tcaaaatacc ttaattggta
tcaacagaag 180cccggacagg ctcctcgcct tctgatctac cacaccagcc
ggctccattc tggaatccct 240gccaggttca gcggtagcgg atctgggacc
gactacaccc tcactatcag ctcactgcag 300ccagaggact tcgctgtcta
tttctgtcag caagggaaca ccctgcccta cacctttgga 360cagggcacca
agctcgagat taaaggtgga ggtggcagcg gaggaggtgg gtccggcggt
420ggaggaagcg gcggaggcgg gagccaggtc caactccaag aaagcggacc
gggtcttgtg 480aagccatcag aaactctttc actgacttgt actgtgagcg
gagtgtctct ccccgattac 540ggggtgtctt ggatcagaca gccaccgggg
aagggtctgg aatggattgg agtgatttgg 600ggctctgaga ctacttacta
ctcttcatcc ctcaagtcac gcgtcaccat ctcaaaggac 660aactctaaga
atcaggtgtc actgaaactg tcatctgtga ccgcagccga caccgccgtg
720tactattgcg ctaagcatta ctattatggc gggagctacg caatggatta
ctggggacag 780ggtactctgg tcaccgtgtc cagcaccact accccagcac
cgaggccacc caccccggct 840cctaccatcg cctcccagcc tctgtccctg
cgtccggagg catgtagacc cgcagctggt 900ggggccgtgc atacccgggg
tcttgacttc gcctgcgata tctacatttg ggcccctctg 960gctggtactt
gcggggtcct gctgctttca ctcgtgatca ctctttactg taagcgcggt
1020cggaagaagc tgctgtacat ctttaagcaa cccttcatga ggcctgtgca
gactactcaa 1080gaggaggacg gctgttcatg ccggttccca gaggaggagg
aaggcggctg cgaactgcgc 1140gtgaaattca gccgcagcgc agatgctcca
gcctacaagc aggggcagaa ccagctctac 1200aacgaactca atcttggtcg
gagagaggag tacgacgtgc tggacaagcg gagaggacgg 1260gacccagaaa
tgggcgggaa gccgcgcaga aagaatcccc aagagggcct gtacaacgag
1320ctccaaaagg ataagatggc agaagcctat agcgagattg gtatgaaagg
ggaacgcaga 1380agaggcaaag gccacgacgg actgtaccag ggactcagca
ccgccaccaa ggacacctat 1440gacgctcttc acatgcaggc cctgccgcct cgg
1473901473DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 90atggccctcc
ctgtcaccgc cctgctgctt ccgctggctc ttctgctcca cgccgctcgg 60cccgaaattg
tgatgaccca gtcacccgcc actcttagcc tttcacccgg tgagcgcgca
120accctgtctt gcagagcctc ccaagacatc tcaaaatacc ttaattggta
tcaacagaag 180cccggacagg ctcctcgcct tctgatctac cacaccagcc
ggctccattc tggaatccct 240gccaggttca gcggtagcgg atctgggacc
gactacaccc tcactatcag ctcactgcag 300ccagaggact tcgctgtcta
tttctgtcag caagggaaca ccctgcccta cacctttgga 360cagggcacca
agctcgagat taaaggtgga ggtggcagcg gaggaggtgg gtccggcggt
420ggaggaagcg gaggcggagg gagccaggtc caactccaag aaagcggacc
gggtcttgtg 480aagccatcag aaactctttc actgacttgt actgtgagcg
gagtgtctct ccccgattac 540ggggtgtctt ggatcagaca gccaccgggg
aagggtctgg aatggattgg agtgatttgg 600ggctctgaga ctacttacta
ccaatcatcc ctcaagtcac gcgtcaccat ctcaaaggac 660aactctaaga
atcaggtgtc actgaaactg tcatctgtga ccgcagccga caccgccgtg
720tactattgcg ctaagcatta ctattatggc gggagctacg caatggatta
ctggggacag 780ggtactctgg tcaccgtgtc cagcaccact accccagcac
cgaggccacc caccccggct 840cctaccatcg cctcccagcc tctgtccctg
cgtccggagg catgtagacc cgcagctggt 900ggggccgtgc atacccgggg
tcttgacttc gcctgcgata tctacatttg ggcccctctg 960gctggtactt
gcggggtcct gctgctttca ctcgtgatca ctctttactg taagcgcggt
1020cggaagaagc tgctgtacat ctttaagcaa cccttcatga ggcctgtgca
gactactcaa 1080gaggaggacg gctgttcatg ccggttccca gaggaggagg
aaggcggctg cgaactgcgc 1140gtgaaattca gccgcagcgc agatgctcca
gcctacaagc aggggcagaa ccagctctac 1200aacgaactca atcttggtcg
gagagaggag tacgacgtgc tggacaagcg gagaggacgg 1260gacccagaaa
tgggcgggaa gccgcgcaga aagaatcccc aagagggcct gtacaacgag
1320ctccaaaagg ataagatggc agaagcctat agcgagattg gtatgaaagg
ggaacgcaga 1380agaggcaaag gccacgacgg actgtaccag ggactcagca
ccgccaccaa ggacacctat 1440gacgctcttc acatgcaggc cctgccgcct cgg
1473911473DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 91atggctctgc
ccgtgaccgc actcctcctg ccactggctc tgctgcttca cgccgctcgc 60ccacaagtcc
agcttcaaga atcagggcct ggtctggtga agccatctga gactctgtcc
120ctcacttgca ccgtgagcgg agtgtccctc ccagactacg gagtgagctg
gattagacag 180cctcccggaa agggactgga gtggatcgga gtgatttggg
gtagcgaaac cacttactat 240tcatcttccc tgaagtcacg ggtcaccatt
tcaaaggata actcaaagaa tcaagtgagc 300ctcaagctct catcagtcac
cgccgctgac accgccgtgt attactgtgc caagcattac 360tactatggag
ggtcctacgc catggactac tggggccagg gaactctggt cactgtgtca
420tctggtggag gaggtagcgg aggaggcggg agcggtggag gtggctccgg
aggtggcgga 480agcgaaatcg tgatgaccca gagccctgca accctgtccc
tttctcccgg ggaacgggct 540accctttctt gtcgggcatc acaagatatc
tcaaaatacc tcaattggta tcaacagaag 600ccgggacagg cccctaggct
tcttatctac cacacctctc gcctgcatag cgggattccc 660gcacgcttta
gcgggtctgg aagcgggacc gactacactc tgaccatctc atctctccag
720cccgaggact tcgccgtcta cttctgccag cagggtaaca ccctgccgta
caccttcggc 780cagggcacca agcttgagat caaaaccact actcccgctc
caaggccacc cacccctgcc 840ccgaccatcg cctctcagcc gctttccctg
cgtccggagg catgtagacc cgcagctggt 900ggggccgtgc atacccgggg
tcttgacttc gcctgcgata tctacatttg ggcccctctg 960gctggtactt
gcggggtcct gctgctttca ctcgtgatca ctctttactg taagcgcggt
1020cggaagaagc tgctgtacat ctttaagcaa cccttcatga ggcctgtgca
gactactcaa 1080gaggaggacg gctgttcatg ccggttccca gaggaggagg
aaggcggctg cgaactgcgc 1140gtgaaattca gccgcagcgc agatgctcca
gcctacaagc aggggcagaa ccagctctac 1200aacgaactca atcttggtcg
gagagaggag tacgacgtgc tggacaagcg gagaggacgg 1260gacccagaaa
tgggcgggaa gccgcgcaga aagaatcccc aagagggcct gtacaacgag
1320ctccaaaagg ataagatggc agaagcctat agcgagattg gtatgaaagg
ggaacgcaga 1380agaggcaaag gccacgacgg actgtaccag ggactcagca
ccgccaccaa ggacacctat 1440gacgctcttc acatgcaggc cctgccgcct cgg
1473921473DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 92atggctctgc
ccgtgaccgc actcctcctg ccactggctc tgctgcttca cgccgctcgc 60ccacaagtcc
agcttcaaga atcagggcct ggtctggtga agccatctga gactctgtcc
120ctcacttgca ccgtgagcgg agtgtccctc ccagactacg gagtgagctg
gattagacag 180cctcccggaa agggactgga gtggatcgga gtgatttggg
gtagcgaaac cacttactat 240caatcttccc tgaagtcacg ggtcaccatt
tcaaaggata actcaaagaa tcaagtgagc 300ctcaagctct catcagtcac
cgccgctgac accgccgtgt attactgtgc caagcattac 360tactatggag
ggtcctacgc catggactac tggggccagg gaactctggt cactgtgtca
420tctggtggag gaggtagcgg aggaggcggg agcggtggag gtggctccgg
aggcggtggg 480tcagaaatcg tgatgaccca gagccctgca accctgtccc
tttctcccgg ggaacgggct 540accctttctt gtcgggcatc acaagatatc
tcaaaatacc tcaattggta tcaacagaag 600ccgggacagg cccctaggct
tcttatctac cacacctctc gcctgcatag cgggattccc 660gcacgcttta
gcgggtctgg aagcgggacc gactacactc tgaccatctc atctctccag
720cccgaggact tcgccgtcta cttctgccag cagggtaaca ccctgccgta
caccttcggc 780cagggcacca agcttgagat caaaaccact actcccgctc
caaggccacc cacccctgcc 840ccgaccatcg cctctcagcc gctttccctg
cgtccggagg catgtagacc cgcagctggt 900ggggccgtgc atacccgggg
tcttgacttc gcctgcgata tctacatttg ggcccctctg 960gctggtactt
gcggggtcct gctgctttca ctcgtgatca ctctttactg taagcgcggt
1020cggaagaagc tgctgtacat ctttaagcaa cccttcatga ggcctgtgca
gactactcaa 1080gaggaggacg gctgttcatg ccggttccca gaggaggagg
aaggcggctg cgaactgcgc 1140gtgaaattca gccgcagcgc agatgctcca
gcctacaagc aggggcagaa ccagctctac 1200aacgaactca atcttggtcg
gagagaggag tacgacgtgc tggacaagcg gagaggacgg 1260gacccagaaa
tgggcgggaa gccgcgcaga aagaatcccc aagagggcct gtacaacgag
1320ctccaaaagg ataagatggc agaagcctat agcgagattg gtatgaaagg
ggaacgcaga 1380agaggcaaag gccacgacgg actgtaccag ggactcagca
ccgccaccaa ggacacctat 1440gacgctcttc acatgcaggc cctgccgcct cgg
1473931473DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 93atggccctcc
ctgtcaccgc cctgctgctt ccgctggctc ttctgctcca cgccgctcgg 60cccgaaattg
tgatgaccca gtcacccgcc actcttagcc tttcacccgg tgagcgcgca
120accctgtctt gcagagcctc ccaagacatc tcaaaatacc ttaattggta
tcaacagaag 180cccggacagg ctcctcgcct tctgatctac cacaccagcc
ggctccattc tggaatccct 240gccaggttca gcggtagcgg atctgggacc
gactacaccc tcactatcag ctcactgcag 300ccagaggact tcgctgtcta
tttctgtcag caagggaaca ccctgcccta cacctttgga 360cagggcacca
agctcgagat taaaggtgga ggtggcagcg gaggaggtgg gtccggcggt
420ggaggaagcg gaggcggtgg gagccaggtc caactccaag aaagcggacc
gggtcttgtg 480aagccatcag aaactctttc actgacttgt actgtgagcg
gagtgtctct ccccgattac 540ggggtgtctt ggatcagaca gccaccgggg
aagggtctgg aatggattgg agtgatttgg 600ggctctgaga ctacttacta
caactcatcc ctcaagtcac gcgtcaccat ctcaaaggac 660aactctaaga
atcaggtgtc actgaaactg tcatctgtga ccgcagccga caccgccgtg
720tactattgcg ctaagcatta ctattatggc gggagctacg caatggatta
ctggggacag 780ggtactctgg tcaccgtgtc cagcaccact accccagcac
cgaggccacc caccccggct 840cctaccatcg cctcccagcc tctgtccctg
cgtccggagg catgtagacc cgcagctggt 900ggggccgtgc atacccgggg
tcttgacttc gcctgcgata tctacatttg ggcccctctg 960gctggtactt
gcggggtcct gctgctttca ctcgtgatca ctctttactg taagcgcggt
1020cggaagaagc tgctgtacat ctttaagcaa cccttcatga ggcctgtgca
gactactcaa 1080gaggaggacg gctgttcatg ccggttccca gaggaggagg
aaggcggctg cgaactgcgc 1140gtgaaattca gccgcagcgc agatgctcca
gcctacaagc aggggcagaa ccagctctac 1200aacgaactca atcttggtcg
gagagaggag tacgacgtgc tggacaagcg gagaggacgg 1260gacccagaaa
tgggcgggaa gccgcgcaga aagaatcccc aagagggcct gtacaacgag
1320ctccaaaagg ataagatggc agaagcctat agcgagattg gtatgaaagg
ggaacgcaga 1380agaggcaaag gccacgacgg actgtaccag ggactcagca
ccgccaccaa ggacacctat 1440gacgctcttc acatgcaggc cctgccgcct cgg
1473941473DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 94atggccctcc
ctgtcaccgc cctgctgctt ccgctggctc ttctgctcca cgccgctcgg 60cccgaaattg
tgatgaccca gtcacccgcc actcttagcc tttcacccgg tgagcgcgca
120accctgtctt gcagagcctc ccaagacatc tcaaaatacc ttaattggta
tcaacagaag 180cccggacagg ctcctcgcct tctgatctac cacaccagcc
ggctccattc tggaatccct 240gccaggttca gcggtagcgg atctgggacc
gactacaccc tcactatcag ctcactgcag 300ccagaggact tcgctgtcta
tttctgtcag caagggaaca ccctgcccta cacctttgga 360cagggcacca
agctcgagat taaaggtgga ggtggcagcg gaggaggtgg gtccggcggt
420ggaggaagcg gaggcggtgg gagccaggtc caactccaag aaagcggacc
gggtcttgtg 480aagccatcag aaactctttc actgacttgt actgtgagcg
gagtgtctct ccccgattac 540ggggtgtctt ggatcagaca gccaccgggg
aagggtctgg aatggattgg
agtgatttgg 600ggctctgaga ctacttacta caactcatcc ctcaagtcac
gcgtcaccat ctcaaaggac 660aactctaaga atcaggtgtc actgaaactg
tcatctgtga ccgcagccga caccgccgtg 720tactattgcg ctaagcatta
ctattatggc gggagctacg caatggatta ctggggacag 780ggtactctgg
tcaccgtgtc cagcaccact accccagcac cgaggccacc caccccggct
840cctaccatcg cctcccagcc tctgtccctg cgtccggagg catgtagacc
cgcagctggt 900ggggccgtgc atacccgggg tcttgacttc gcctgcgata
tctacatttg ggcccctctg 960gctggtactt gcggggtcct gctgctttca
ctcgtgatca ctctttactg taagcgcggt 1020cggaagaagc tgctgtacat
ctttaagcaa cccttcatga ggcctgtgca gactactcaa 1080gaggaggacg
gctgttcatg ccggttccca gaggaggagg aaggcggctg cgaactgcgc
1140gtgaaattca gccgcagcgc agatgctcca gcctacaagc aggggcagaa
ccagctctac 1200aacgaactca atcttggtcg gagagaggag tacgacgtgc
tggacaagcg gagaggacgg 1260gacccagaaa tgggcgggaa gccgcgcaga
aagaatcccc aagagggcct gtacaacgag 1320ctccaaaagg ataagatggc
agaagcctat agcgagattg gtatgaaagg ggaacgcaga 1380agaggcaaag
gccacgacgg actgtaccag ggactcagca ccgccaccaa ggacacctat
1440gacgctcttc acatgcaggc cctgccgcct cgg 1473951473DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 95atggctctgc ccgtgaccgc actcctcctg ccactggctc
tgctgcttca cgccgctcgc 60ccacaagtcc agcttcaaga atcagggcct ggtctggtga
agccatctga gactctgtcc 120ctcacttgca ccgtgagcgg agtgtccctc
ccagactacg gagtgagctg gattagacag 180cctcccggaa agggactgga
gtggatcgga gtgatttggg gtagcgaaac cacttactat 240aactcttccc
tgaagtcacg ggtcaccatt tcaaaggata actcaaagaa tcaagtgagc
300ctcaagctct catcagtcac cgccgctgac accgccgtgt attactgtgc
caagcattac 360tactatggag ggtcctacgc catggactac tggggccagg
gaactctggt cactgtgtca 420tctggtggag gaggtagcgg aggaggcggg
agcggtggag gtggctccgg aggtggcgga 480agcgaaatcg tgatgaccca
gagccctgca accctgtccc tttctcccgg ggaacgggct 540accctttctt
gtcgggcatc acaagatatc tcaaaatacc tcaattggta tcaacagaag
600ccgggacagg cccctaggct tcttatctac cacacctctc gcctgcatag
cgggattccc 660gcacgcttta gcgggtctgg aagcgggacc gactacactc
tgaccatctc atctctccag 720cccgaggact tcgccgtcta cttctgccag
cagggtaaca ccctgccgta caccttcggc 780cagggcacca agcttgagat
caaaaccact actcccgctc caaggccacc cacccctgcc 840ccgaccatcg
cctctcagcc gctttccctg cgtccggagg catgtagacc cgcagctggt
900ggggccgtgc atacccgggg tcttgacttc gcctgcgata tctacatttg
ggcccctctg 960gctggtactt gcggggtcct gctgctttca ctcgtgatca
ctctttactg taagcgcggt 1020cggaagaagc tgctgtacat ctttaagcaa
cccttcatga ggcctgtgca gactactcaa 1080gaggaggacg gctgttcatg
ccggttccca gaggaggagg aaggcggctg cgaactgcgc 1140gtgaaattca
gccgcagcgc agatgctcca gcctacaagc aggggcagaa ccagctctac
1200aacgaactca atcttggtcg gagagaggag tacgacgtgc tggacaagcg
gagaggacgg 1260gacccagaaa tgggcgggaa gccgcgcaga aagaatcccc
aagagggcct gtacaacgag 1320ctccaaaagg ataagatggc agaagcctat
agcgagattg gtatgaaagg ggaacgcaga 1380agaggcaaag gccacgacgg
actgtaccag ggactcagca ccgccaccaa ggacacctat 1440gacgctcttc
acatgcaggc cctgccgcct cgg 1473961458DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 96atggccctcc ctgtcaccgc cctgctgctt ccgctggctc
ttctgctcca cgccgctcgg 60cccgaaattg tgatgaccca gtcacccgcc actcttagcc
tttcacccgg tgagcgcgca 120accctgtctt gcagagcctc ccaagacatc
tcaaaatacc ttaattggta tcaacagaag 180cccggacagg ctcctcgcct
tctgatctac cacaccagcc ggctccattc tggaatccct 240gccaggttca
gcggtagcgg atctgggacc gactacaccc tcactatcag ctcactgcag
300ccagaggact tcgctgtcta tttctgtcag caagggaaca ccctgcccta
cacctttgga 360cagggcacca agctcgagat taaaggtgga ggtggcagcg
gaggaggtgg gtccggcggt 420ggaggaagcc aggtccaact ccaagaaagc
ggaccgggtc ttgtgaagcc atcagaaact 480ctttcactga cttgtactgt
gagcggagtg tctctccccg attacggggt gtcttggatc 540agacagccac
cggggaaggg tctggaatgg attggagtga tttggggctc tgagactact
600tactacaact catccctcaa gtcacgcgtc accatctcaa aggacaactc
taagaatcag 660gtgtcactga aactgtcatc tgtgaccgca gccgacaccg
ccgtgtacta ttgcgctaag 720cattactatt atggcgggag ctacgcaatg
gattactggg gacagggtac tctggtcacc 780gtgtccagca ccactacccc
agcaccgagg ccacccaccc cggctcctac catcgcctcc 840cagcctctgt
ccctgcgtcc ggaggcatgt agacccgcag ctggtggggc cgtgcatacc
900cggggtcttg acttcgcctg cgatatctac atttgggccc ctctggctgg
tacttgcggg 960gtcctgctgc tttcactcgt gatcactctt tactgtaagc
gcggtcggaa gaagctgctg 1020tacatcttta agcaaccctt catgaggcct
gtgcagacta ctcaagagga ggacggctgt 1080tcatgccggt tcccagagga
ggaggaaggc ggctgcgaac tgcgcgtgaa attcagccgc 1140agcgcagatg
ctccagccta caagcagggg cagaaccagc tctacaacga actcaatctt
1200ggtcggagag aggagtacga cgtgctggac aagcggagag gacgggaccc
agaaatgggc 1260gggaagccgc gcagaaagaa tccccaagag ggcctgtaca
acgagctcca aaaggataag 1320atggcagaag cctatagcga gattggtatg
aaaggggaac gcagaagagg caaaggccac 1380gacggactgt accagggact
cagcaccgcc accaaggaca cctatgacgc tcttcacatg 1440caggccctgc cgcctcgg
145897813DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 97atggccctgc
ccgtcaccgc tctgctgctg ccccttgctc tgcttcttca tgcagcaagg 60ccggacatcc
agatgaccca aaccacctca tccctctctg cctctcttgg agacagggtg
120accatttctt gtcgcgccag ccaggacatc agcaagtatc tgaactggta
tcagcagaag 180ccggacggaa ccgtgaagct cctgatctac catacctctc
gcctgcatag cggcgtgccc 240tcacgcttct ctggaagcgg atcaggaacc
gattattctc tcactatttc aaatcttgag 300caggaagata ttgccaccta
tttctgccag cagggtaata ccctgcccta caccttcgga 360ggagggacca
agctcgaaat caccggtgga ggaggcagcg gcggtggagg gtctggtgga
420ggtggttctg aggtgaagct gcaagaatca ggccctggac ttgtggcccc
ttcacagtcc 480ctgagcgtga cttgcaccgt gtccggagtc tccctgcccg
actacggagt gtcatggatc 540agacaacctc cacggaaagg actggaatgg
ctcggtgtca tctggggtag cgaaactact 600tactacaatt cagccctcaa
aagcaggctg actattatca aggacaacag caagtcccaa 660gtctttctta
agatgaactc actccagact gacgacaccg caatctacta ttgtgctaag
720cactactact acggaggatc ctacgctatg gattactggg gacaaggtac
ttccgtcact 780gtctcttcac accatcatca ccatcaccat cac
81398271PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 98Met Ala Leu Pro Val
Thr Ala Leu Leu Leu Pro Leu Ala Leu Leu Leu1 5 10 15His Ala Ala Arg
Pro Asp Ile Gln Met Thr Gln Thr Thr Ser Ser Leu 20 25 30Ser Ala Ser
Leu Gly Asp Arg Val Thr Ile Ser Cys Arg Ala Ser Gln 35 40 45Asp Ile
Ser Lys Tyr Leu Asn Trp Tyr Gln Gln Lys Pro Asp Gly Thr 50 55 60Val
Lys Leu Leu Ile Tyr His Thr Ser Arg Leu His Ser Gly Val Pro65 70 75
80Ser Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Tyr Ser Leu Thr Ile
85 90 95Ser Asn Leu Glu Gln Glu Asp Ile Ala Thr Tyr Phe Cys Gln Gln
Gly 100 105 110Asn Thr Leu Pro Tyr Thr Phe Gly Gly Gly Thr Lys Leu
Glu Ile Thr 115 120 125Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly
Gly Gly Gly Ser Glu 130 135 140Val Lys Leu Gln Glu Ser Gly Pro Gly
Leu Val Ala Pro Ser Gln Ser145 150 155 160Leu Ser Val Thr Cys Thr
Val Ser Gly Val Ser Leu Pro Asp Tyr Gly 165 170 175Val Ser Trp Ile
Arg Gln Pro Pro Arg Lys Gly Leu Glu Trp Leu Gly 180 185 190Val Ile
Trp Gly Ser Glu Thr Thr Tyr Tyr Asn Ser Ala Leu Lys Ser 195 200
205Arg Leu Thr Ile Ile Lys Asp Asn Ser Lys Ser Gln Val Phe Leu Lys
210 215 220Met Asn Ser Leu Gln Thr Asp Asp Thr Ala Ile Tyr Tyr Cys
Ala Lys225 230 235 240His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp
Tyr Trp Gly Gln Gly 245 250 255Thr Ser Val Thr Val Ser Ser His His
His His His His His His 260 265 270991458DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 99atggccttac cagtgaccgc cttgctcctg ccgctggcct
tgctgctcca cgccgccagg 60ccggacatcc agatgacaca gactacatcc tccctgtctg
cctctctggg agacagagtc 120accatcagtt gcagggcaag tcaggacatt
agtaaatatt taaattggta tcagcagaaa 180ccagatggaa ctgttaaact
cctgatctac catacatcaa gattacactc aggagtccca 240tcaaggttca
gtggcagtgg gtctggaaca gattattctc tcaccattag caacctggag
300caagaagata ttgccactta cttttgccaa cagggtaata cgcttccgta
cacgttcgga 360ggggggacca agctggagat cacaggtggc ggtggctcgg
gcggtggtgg gtcgggtggc 420ggcggatctg aggtgaaact gcaggagtca
ggacctggcc tggtggcgcc ctcacagagc 480ctgtccgtca catgcactgt
ctcaggggtc tcattacccg actatggtgt aagctggatt 540cgccagcctc
cacgaaaggg tctggagtgg ctgggagtaa tatggggtag tgaaaccaca
600tactataatt cagctctcaa atccagactg accatcatca aggacaactc
caagagccaa 660gttttcttaa aaatgaacag tctgcaaact gatgacacag
ccatttacta ctgtgccaaa 720cattattact acggtggtag ctatgctatg
gactactggg gccaaggaac ctcagtcacc 780gtctcctcaa ccacgacgcc
agcgccgcga ccaccaacac cggcgcccac catcgcgtcg 840cagcccctgt
ccctgcgccc agaggcgtgc cggccagcgg cggggggcgc agtgcacacg
900agggggctgg acttcgcctg tgatatctac atctgggcgc ccttggccgg
gacttgtggg 960gtccttctcc tgtcactggt tatcaccctt tactgcaaac
ggggcagaaa gaaactcctg 1020tatatattca aacaaccatt tatgagacca
gtacaaacta ctcaagagga agatggctgt 1080agctgccgat ttccagaaga
agaagaagga ggatgtgaac tgagagtgaa gttcagcagg 1140agcgcagacg
cccccgcgta caagcagggc cagaaccagc tctataacga gctcaatcta
1200ggacgaagag aggagtacga tgttttggac aagagacgtg gccgggaccc
tgagatgggg 1260ggaaagccga gaaggaagaa ccctcaggaa ggcctgtaca
atgaactgca gaaagataag 1320atggcggagg cctacagtga gattgggatg
aaaggcgagc gccggagggg caaggggcac 1380gatggccttt accagggtct
cagtacagcc accaaggaca cctacgacgc ccttcacatg 1440caggccctgc cccctcgc
14581001184DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 100cgtgaggctc
cggtgcccgt cagtgggcag agcgcacatc gcccacagtc cccgagaagt 60tggggggagg
ggtcggcaat tgaaccggtg cctagagaag gtggcgcggg gtaaactggg
120aaagtgatgt cgtgtactgg ctccgccttt ttcccgaggg tgggggagaa
ccgtatataa 180gtgcagtagt cgccgtgaac gttctttttc gcaacgggtt
tgccgccaga acacaggtaa 240gtgccgtgtg tggttcccgc gggcctggcc
tctttacggg ttatggccct tgcgtgcctt 300gaattacttc cacctggctg
cagtacgtga ttcttgatcc cgagcttcgg gttggaagtg 360ggtgggagag
ttcgaggcct tgcgcttaag gagccccttc gcctcgtgct tgagttgagg
420cctggcctgg gcgctggggc cgccgcgtgc gaatctggtg gcaccttcgc
gcctgtctcg 480ctgctttcga taagtctcta gccatttaaa atttttgatg
acctgctgcg acgctttttt 540tctggcaaga tagtcttgta aatgcgggcc
aagatctgca cactggtatt tcggtttttg 600gggccgcggg cggcgacggg
gcccgtgcgt cccagcgcac atgttcggcg aggcggggcc 660tgcgagcgcg
gccaccgaga atcggacggg ggtagtctca agctggccgg cctgctctgg
720tgcctggcct cgcgccgccg tgtatcgccc cgccctgggc ggcaaggctg
gcccggtcgg 780caccagttgc gtgagcggaa agatggccgc ttcccggccc
tgctgcaggg agctcaaaat 840ggaggacgcg gcgctcggga gagcgggcgg
gtgagtcacc cacacaaagg aaaagggcct 900ttccgtcctc agccgtcgct
tcatgtgact ccacggagta ccgggcgccg tccaggcacc 960tcgattagtt
ctcgagcttt tggagtacgt cgtctttagg ttggggggag gggttttatg
1020cgatggagtt tccccacact gagtgggtgg agactgaagt taggccagct
tggcacttga 1080tgtaattctc cttggaattt gccctttttg agtttggatc
ttggttcatt ctcaagcctc 1140agacagtggt tcaaagtttt tttcttccat
ttcaggtgtc gtga 1184101336DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 101agagtgaagt tcagcaggag cgcagacgcc cccgcgtaca
agcagggcca gaaccagctc 60tataacgagc tcaatctagg acgaagagag gagtacgatg
ttttggacaa gagacgtggc 120cgggaccctg agatgggggg aaagccgaga
aggaagaacc ctcaggaagg cctgtacaat 180gaactgcaga aagataagat
ggcggaggcc tacagtgaga ttgggatgaa aggcgagcgc 240cggaggggca
aggggcacga tggcctttac cagggtctca gtacagccac caaggacacc
300tacgacgccc ttcacatgca ggccctgccc cctcgc 336102230PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 102Glu Ser Lys Tyr Gly Pro Pro Cys Pro Pro Cys Pro Ala
Pro Glu Phe1 5 10 15Leu Gly Gly Pro Ser Val Phe Leu Phe Pro Pro Lys
Pro Lys Asp Thr 20 25 30Leu Met Ile Ser Arg Thr Pro Glu Val Thr Cys
Val Val Val Asp Val 35 40 45Ser Gln Glu Asp Pro Glu Val Gln Phe Asn
Trp Tyr Val Asp Gly Val 50 55 60Glu Val His Asn Ala Lys Thr Lys Pro
Arg Glu Glu Gln Phe Asn Ser65 70 75 80Thr Tyr Arg Val Val Ser Val
Leu Thr Val Leu His Gln Asp Trp Leu 85 90 95Asn Gly Lys Glu Tyr Lys
Cys Lys Val Ser Asn Lys Gly Leu Pro Ser 100 105 110Ser Ile Glu Lys
Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro 115 120 125Gln Val
Tyr Thr Leu Pro Pro Ser Gln Glu Glu Met Thr Lys Asn Gln 130 135
140Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile
Ala145 150 155 160Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn
Tyr Lys Thr Thr 165 170 175Pro Pro Val Leu Asp Ser Asp Gly Ser Phe
Phe Leu Tyr Ser Arg Leu 180 185 190Thr Val Asp Lys Ser Arg Trp Gln
Glu Gly Asn Val Phe Ser Cys Ser 195 200 205Val Met His Glu Ala Leu
His Asn His Tyr Thr Gln Lys Ser Leu Ser 210 215 220Leu Ser Leu Gly
Lys Met225 230103690DNAArtificial Sequencesource/note="Description
of Artificial Sequence Synthetic polynucleotide" 103gagagcaagt
acggccctcc ctgcccccct tgccctgccc ccgagttcct gggcggaccc 60agcgtgttcc
tgttcccccc caagcccaag gacaccctga tgatcagccg gacccccgag
120gtgacctgtg tggtggtgga cgtgtcccag gaggaccccg aggtccagtt
caactggtac 180gtggacggcg tggaggtgca caacgccaag accaagcccc
gggaggagca gttcaatagc 240acctaccggg tggtgtccgt gctgaccgtg
ctgcaccagg actggctgaa cggcaaggaa 300tacaagtgta aggtgtccaa
caagggcctg cccagcagca tcgagaaaac catcagcaag 360gccaagggcc
agcctcggga gccccaggtg tacaccctgc cccctagcca agaggagatg
420accaagaacc aggtgtccct gacctgcctg gtgaagggct tctaccccag
cgacatcgcc 480gtggagtggg agagcaacgg ccagcccgag aacaactaca
agaccacccc ccctgtgctg 540gacagcgacg gcagcttctt cctgtacagc
cggctgaccg tggacaagag ccggtggcag 600gagggcaacg tctttagctg
ctccgtgatg cacgaggccc tgcacaacca ctacacccag 660aagagcctga
gcctgtccct gggcaagatg 690104150DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 104aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 60aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 120aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
15010540PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic
polypeptide"misc_feature(1)..(40)/note="This sequence may encompass
1-10 repeating "Gly Gly Gly Ser" repeating units" 105Gly Gly Gly
Ser Gly Gly Gly Ser Gly Gly Gly Ser Gly Gly Gly Ser1 5 10 15Gly Gly
Gly Ser Gly Gly Gly Ser Gly Gly Gly Ser Gly Gly Gly Ser 20 25 30Gly
Gly Gly Ser Gly Gly Gly Ser 35 4010620PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
peptide" 106Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly
Ser Gly1 5 10 15Gly Gly Gly Ser 2010715PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
peptide" 107Gly Gly Gly Gly Ser Gly Gly Gly Gly Ser Gly Gly Gly Gly
Ser1 5 10 151084PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic peptide" 108Gly Gly Gly
Ser11095000DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic
polynucleotide"misc_feature(1)..(5000)/note="This sequence may
encompass 50-5000 nucleotides" 109aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 60aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 120aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 180aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
240aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 300aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 360aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 420aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 480aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
540aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 600aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 660aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 720aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 780aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
840aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 900aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 960aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1020aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1080aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
1140aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 1200aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1260aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1320aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
1380aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 1440aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 1500aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1560aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1620aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
1680aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 1740aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 1800aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1860aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1920aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
1980aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 2040aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 2100aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 2160aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 2220aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
2280aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 2340aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 2400aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 2460aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 2520aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
2580aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 2640aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 2700aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 2760aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 2820aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
2880aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 2940aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 3000aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 3060aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 3120aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
3180aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 3240aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 3300aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 3360aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 3420aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
3480aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 3540aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 3600aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 3660aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 3720aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
3780aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 3840aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 3900aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 3960aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4020aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
4080aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 4140aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 4200aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4260aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4320aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
4380aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 4440aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 4500aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4560aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4620aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
4680aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 4740aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 4800aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4860aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4920aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
4980aaaaaaaaaa aaaaaaaaaa 5000110100DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide" 110tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 60tttttttttt tttttttttt tttttttttt tttttttttt
1001115000DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic
polynucleotide"misc_feature(1)..(5000)/note="This sequence may
encompass 50-5000 nucleotides" 111tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 60tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 120tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 180tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
240tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 300tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 360tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 420tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 480tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
540tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 600tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 660tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 720tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 780tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
840tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 900tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 960tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 1020tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 1080tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
1140tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 1200tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 1260tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 1320tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 1380tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
1440tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 1500tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 1560tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 1620tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 1680tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
1740tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 1800tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 1860tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 1920tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 1980tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
2040tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 2100tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 2160tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 2220tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 2280tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
2340tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 2400tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 2460tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 2520tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 2580tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
2640tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 2700tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 2760tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 2820tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 2880tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
2940tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 3000tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 3060tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 3120tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 3180tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
3240tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 3300tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 3360tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 3420tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 3480tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
3540tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 3600tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 3660tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 3720tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 3780tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
3840tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 3900tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 3960tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 4020tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 4080tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
4140tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 4200tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 4260tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 4320tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 4380tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
4440tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 4500tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 4560tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 4620tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 4680tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
4740tttttttttt tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt 4800tttttttttt tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt 4860tttttttttt tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt 4920tttttttttt tttttttttt
tttttttttt tttttttttt tttttttttt tttttttttt 4980tttttttttt
tttttttttt 50001125000DNAArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polynucleotide"misc_feature(1)..(5000)/note="This sequence may
encompass 100-5000 nucleotides" 112aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 60aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 120aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 180aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
240aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 300aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 360aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 420aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 480aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
540aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 600aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 660aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 720aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 780aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
840aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 900aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 960aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1020aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1080aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
1140aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 1200aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 1260aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1320aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1380aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
1440aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 1500aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 1560aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1620aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1680aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
1740aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 1800aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 1860aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1920aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1980aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
2040aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 2100aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 2160aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 2220aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 2280aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
2340aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 2400aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 2460aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 2520aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 2580aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
2640aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 2700aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 2760aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 2820aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 2880aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
2940aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 3000aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 3060aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 3120aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 3180aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
3240aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 3300aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 3360aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 3420aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 3480aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
3540aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 3600aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 3660aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 3720aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 3780aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
3840aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 3900aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 3960aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4020aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4080aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
4140aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 4200aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 4260aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4320aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4380aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
4440aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 4500aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 4560aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4620aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4680aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
4740aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 4800aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 4860aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4920aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 4980aaaaaaaaaa
aaaaaaaaaa 5000113400DNAArtificial Sequencesource/note="Description
of Artificial Sequence Synthetic
polynucleotide"misc_feature(1)..(400)/note="This sequence may
encompass 100-400 nucleotides" 113aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 60aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 120aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 180aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
240aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 300aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 360aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 400114120PRTArtificial Sequencesource/note="Description
of Artificial Sequence Synthetic polypeptide" 114Gln Val Gln Leu
Gln Glu Ser Gly Pro Gly Leu Val Lys Pro Ser Glu1 5 10 15Thr Leu Ser
Leu Thr Cys Thr Val Ser Gly Val Ser Leu Pro Asp Tyr 20 25 30Gly Val
Ser Trp Ile Arg Gln Pro Pro Gly Lys Gly Leu Glu Trp Ile 35 40 45Gly
Val Ile Trp Gly Ser Glu Thr Thr Tyr Tyr Asn Ser Ser Leu Lys 50 55
60Ser Arg Val Thr Ile Ser Lys Asp Asn Ser Lys Asn Gln Val Ser Leu65
70 75 80Lys Leu Ser Ser Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys
Ala 85 90 95Lys His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp Tyr Trp
Gly Gln 100 105 110Gly Thr Leu Val Thr Val Ser Ser 115
120115120PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 115Gln Val Gln Leu Gln
Glu Ser Gly Pro Gly Leu Val Lys Pro Ser Glu1 5 10 15Thr Leu Ser Leu
Thr Cys Thr Val Ser Gly Val Ser Leu Pro Asp Tyr 20 25 30Gly Val Ser
Trp Ile Arg Gln Pro Pro Gly Lys Gly Leu Glu Trp Ile 35 40 45Gly Val
Ile Trp Gly Ser Glu Thr Thr Tyr Tyr Ser Ser Ser Leu Lys 50 55 60Ser
Arg Val Thr Ile Ser Lys Asp Asn Ser Lys Asn Gln Val Ser Leu65 70 75
80Lys Leu Ser Ser Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys Ala
85 90 95Lys His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp Tyr Trp Gly
Gln 100 105 110Gly Thr Leu Val Thr Val Ser Ser 115
120116120PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 116Gln Val Gln Leu Gln
Glu Ser Gly Pro Gly Leu Val Lys Pro Ser Glu1 5 10 15Thr Leu Ser Leu
Thr Cys Thr Val Ser Gly Val Ser Leu Pro Asp Tyr 20 25 30Gly Val Ser
Trp Ile Arg Gln Pro Pro Gly Lys Gly Leu Glu Trp Ile 35 40 45Gly Val
Ile Trp Gly Ser Glu Thr Thr Tyr Tyr Gln Ser Ser Leu Lys 50 55 60Ser
Arg Val Thr Ile Ser Lys Asp Asn Ser Lys Asn Gln Val Ser Leu65 70 75
80Lys Leu Ser Ser Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys Ala
85 90 95Lys His Tyr Tyr Tyr Gly Gly Ser Tyr Ala Met Asp Tyr Trp Gly
Gln 100 105 110Gly Thr Leu Val Thr Val Ser Ser 115
120117107PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 117Glu Ile Val Met Thr
Gln Ser Pro Ala Thr Leu Ser Leu Ser Pro Gly1 5 10 15Glu Arg Ala Thr
Leu Ser Cys Arg Ala Ser Gln Asp Ile Ser Lys Tyr 20 25 30Leu Asn Trp
Tyr Gln Gln Lys Pro Gly Gln Ala Pro Arg Leu Leu Ile 35 40 45Tyr His
Thr Ser Arg Leu His Ser Gly Ile Pro Ala Arg Phe Ser Gly 50 55 60Ser
Gly Ser Gly Thr Asp Tyr Thr Leu Thr Ile Ser Ser Leu Gln Pro65 70 75
80Glu Asp Phe Ala Val Tyr Phe Cys Gln Gln Gly Asn Thr Leu Pro Tyr
85 90 95Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys 100
1051182000DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic
polynucleotide"misc_feature(1)..(2000)/note="This sequence may
encompass 50-2000 nucleotides" 118aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 60aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 120aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 180aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
240aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 300aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 360aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 420aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 480aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
540aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 600aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 660aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 720aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 780aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
840aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 900aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 960aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1020aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1080aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
1140aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 1200aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 1260aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1320aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1380aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
1440aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 1500aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 1560aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1620aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1680aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
1740aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa 1800aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa 1860aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1920aaaaaaaaaa aaaaaaaaaa
aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa aaaaaaaaaa 1980aaaaaaaaaa
aaaaaaaaaa 2000119373PRTArtificial Sequencesource/note="Description
of Artificial Sequence Synthetic polypeptide" 119Pro Gly Trp Phe
Leu Asp Ser Pro Asp Arg Pro Trp Asn Pro Pro Thr1 5 10 15Phe Ser Pro
Ala Leu Leu Val Val Thr Glu Gly Asp Asn Ala Thr Phe 20 25 30Thr Cys
Ser Phe Ser Asn Thr Ser Glu Ser Phe Val Leu Asn Trp Tyr 35 40 45Arg
Met Ser Pro Ser Asn Gln Thr Asp Lys Leu Ala Ala Phe Pro Glu 50 55
60Asp Arg Ser Gln Pro Gly Gln Asp Cys Arg Phe Arg Val Thr Gln Leu65
70 75 80Pro Asn Gly Arg Asp Phe His Met Ser Val Val Arg Ala Arg Arg
Asn 85 90 95Asp Ser Gly Thr Tyr Leu Cys Gly Ala Ile Ser Leu Ala Pro
Lys Ala 100 105 110Gln Ile Lys Glu Ser Leu Arg Ala Glu Leu Arg Val
Thr Glu Arg Arg 115 120 125Ala Glu Val Pro Thr Ala His Pro Ser Pro
Ser Pro Arg Pro Ala Gly 130 135 140Gln Phe Gln Thr Leu Val Thr Thr
Thr Pro Ala Pro Arg Pro Pro Thr145 150 155 160Pro Ala Pro Thr Ile
Ala Ser Gln Pro Leu Ser Leu Arg Pro Glu Ala 165 170 175Cys Arg Pro
Ala Ala Gly Gly Ala Val His Thr Arg Gly Leu Asp Phe 180 185 190Ala
Cys Asp Ile Tyr Ile Trp Ala Pro Leu Ala Gly Thr Cys Gly Val 195 200
205Leu Leu Leu Ser Leu Val Ile Thr Leu Tyr Cys Lys Arg Gly Arg Lys
210 215 220Lys Leu Leu Tyr Ile Phe Lys Gln Pro Phe Met Arg Pro Val
Gln Thr225 230 235 240Thr Gln Glu Glu Asp Gly Cys Ser Cys Arg Phe
Pro Glu Glu Glu Glu 245 250 255Gly Gly Cys Glu Leu Arg Val Lys Phe
Ser Arg Ser Ala Asp Ala Pro 260 265 270Ala Tyr Lys Gln Gly Gln Asn
Gln Leu Tyr Asn Glu Leu Asn Leu Gly 275 280 285Arg Arg Glu Glu Tyr
Asp Val Leu Asp Lys Arg Arg Gly Arg Asp Pro 290 295 300Glu Met Gly
Gly Lys Pro Arg Arg Lys Asn Pro Gln Glu Gly Leu Tyr305 310 315
320Asn Glu Leu Gln Lys Asp Lys Met Ala Glu Ala Tyr Ser Glu Ile Gly
325 330 335Met Lys Gly Glu Arg Arg Arg Gly Lys Gly His Asp Gly Leu
Tyr Gln 340 345 350Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr Asp Ala
Leu His Met Gln 355 360 365Ala Leu Pro Pro Arg
3701201182DNAArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polynucleotide" 120atggccctcc
ctgtcactgc cctgcttctc cccctcgcac tcctgctcca cgccgctaga 60ccacccggat
ggtttctgga ctctccggat cgcccgtgga atcccccaac cttctcaccg
120gcactcttgg ttgtgactga gggcgataat gcgaccttca cgtgctcgtt
ctccaacacc 180tccgaatcat tcgtgctgaa ctggtaccgc atgagcccgt
caaaccagac cgacaagctc 240gccgcgtttc cggaagatcg gtcgcaaccg
ggacaggatt gtcggttccg cgtgactcaa 300ctgccgaatg gcagagactt
ccacatgagc gtggtccgcg ctaggcgaaa cgactccggg 360acctacctgt
gcggagccat ctcgctggcg cctaaggccc aaatcaaaga gagcttgagg
420gccgaactga gagtgaccga gcgcagagct gaggtgccaa ctgcacatcc
atccccatcg 480cctcggcctg cggggcagtt tcagaccctg gtcacgacca
ctccggcgcc gcgcccaccg 540actccggccc caactatcgc gagccagccc
ctgtcgctga ggccggaagc atgccgccct 600gccgccggag gtgctgtgca
tacccgggga ttggacttcg catgcgacat ctacatttgg 660gctcctctcg
ccggaacttg tggcgtgctc cttctgtccc tggtcatcac cctgtactgc
720aagcggggtc ggaaaaagct tctgtacatt ttcaagcagc ccttcatgag
gcccgtgcaa 780accacccagg aggaggacgg ttgctcctgc cggttccccg
aagaggaaga aggaggttgc 840gagctgcgcg tgaagttctc ccggagcgcc
gacgcccccg cctataagca gggccagaac 900cagctgtaca acgaactgaa
cctgggacgg cgggaagagt acgatgtgct ggacaagcgg 960cgcggccggg
accccgaaat gggcgggaag cctagaagaa agaaccctca ggaaggcctg
1020tataacgagc tgcagaagga caagatggcc gaggcctact ccgaaattgg
gatgaaggga 1080gagcggcgga ggggaaaggg gcacgacggc ctgtaccaag
gactgtccac cgccaccaag 1140gacacatacg atgccctgca catgcaggcc
cttccccctc gc 1182121394PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 121Met Ala Leu Pro Val Thr Ala Leu Leu Leu Pro Leu Ala
Leu Leu Leu1 5 10 15His Ala Ala Arg Pro Pro Gly Trp Phe Leu Asp Ser
Pro Asp Arg Pro 20 25 30Trp Asn Pro Pro Thr Phe Ser Pro Ala Leu Leu
Val Val Thr Glu Gly 35 40 45Asp Asn Ala Thr Phe Thr Cys Ser Phe Ser
Asn Thr Ser Glu Ser Phe 50 55 60Val Leu Asn Trp Tyr Arg Met Ser Pro
Ser Asn Gln Thr Asp Lys Leu65 70 75 80Ala Ala Phe Pro Glu Asp Arg
Ser Gln Pro Gly Gln Asp Cys Arg Phe 85 90 95Arg Val Thr Gln Leu Pro
Asn Gly Arg Asp Phe His Met Ser Val Val 100 105 110Arg Ala Arg Arg
Asn Asp Ser Gly Thr Tyr Leu Cys Gly Ala Ile Ser 115 120 125Leu Ala
Pro Lys Ala Gln Ile Lys Glu Ser Leu Arg Ala Glu Leu Arg 130 135
140Val Thr Glu Arg Arg Ala Glu Val Pro Thr Ala His Pro Ser Pro
Ser145 150 155 160Pro Arg Pro Ala Gly Gln Phe Gln Thr Leu Val Thr
Thr Thr Pro Ala 165 170 175Pro Arg Pro Pro Thr Pro Ala Pro Thr Ile
Ala Ser Gln Pro Leu Ser 180 185 190Leu Arg Pro Glu Ala Cys Arg Pro
Ala Ala Gly Gly Ala Val His Thr 195 200 205Arg Gly Leu Asp Phe Ala
Cys Asp Ile Tyr Ile Trp Ala Pro Leu Ala 210 215 220Gly Thr Cys Gly
Val Leu Leu Leu Ser Leu Val Ile Thr Leu Tyr Cys225 230 235 240Lys
Arg Gly Arg Lys Lys Leu Leu Tyr Ile Phe Lys Gln Pro Phe Met 245 250
255Arg Pro Val Gln Thr Thr Gln Glu Glu Asp Gly Cys Ser Cys Arg Phe
260 265 270Pro Glu Glu Glu Glu Gly Gly Cys Glu Leu Arg Val Lys Phe
Ser Arg 275 280 285Ser Ala Asp Ala Pro Ala Tyr Lys Gln Gly Gln Asn
Gln Leu Tyr Asn 290 295 300Glu Leu Asn Leu Gly Arg Arg Glu Glu Tyr
Asp Val Leu Asp Lys Arg305 310 315 320Arg Gly Arg Asp Pro Glu Met
Gly Gly Lys Pro Arg Arg Lys Asn Pro 325 330 335Gln Glu Gly Leu Tyr
Asn Glu Leu Gln Lys Asp Lys Met Ala Glu Ala 340 345 350Tyr Ser Glu
Ile Gly Met Lys Gly Glu Arg Arg Arg Gly Lys Gly His 355 360 365Asp
Gly Leu Tyr Gln Gly Leu Ser Thr Ala Thr Lys Asp Thr Tyr Asp 370 375
380Ala Leu His Met Gln Ala Leu Pro Pro Arg385
390122132PRTArtificial Sequencesource/note="Description of
Artificial Sequence Synthetic polypeptide" 122Asp Val Pro Asp Tyr
Ala Ser Leu Gly Gly Pro Ser Ser Pro Lys Lys1 5 10 15Lys Arg Lys Val
Ser Arg Gly Val Gln Val Glu Thr Ile Ser Pro Gly 20 25 30Asp Gly Arg
Thr Phe Pro Lys Arg Gly Gln Thr Cys Val Val His Tyr 35 40 45Thr Gly
Met Leu Glu Asp Gly Lys Lys Phe Asp Ser Ser Arg Asp Arg 50 55 60Asn
Lys Pro Phe Lys Phe Met Leu Gly Lys Gln Glu Val Ile Arg Gly65 70 75
80Trp Glu Glu Gly Val Ala Gln Met Ser Val Gly Gln Arg Ala Lys Leu
85 90 95Thr Ile Ser Pro Asp Tyr Ala Tyr Gly Ala Thr Gly His Pro Gly
Ile 100 105 110Ile Pro Pro His Ala Thr Leu Val Phe Asp Val Glu Leu
Leu Lys Leu 115 120 125Glu Thr Ser Tyr 130123108PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 123Val Gln Val Glu Thr Ile Ser Pro Gly Asp Gly Arg Thr
Phe Pro Lys1 5 10 15Arg Gly Gln Thr Cys Val Val His Tyr Thr Gly Met
Leu Glu Asp Gly 20 25 30Lys Lys Phe Asp Ser Ser Arg Asp Arg Asn Lys
Pro Phe Lys Phe Met 35 40 45Leu Gly Lys Gln Glu Val Ile Arg Gly Trp
Glu Glu Gly Val Ala Gln 50 55 60Met Ser Val Gly Gln Arg Ala Lys Leu
Thr Ile Ser Pro Asp Tyr Ala65 70 75 80Tyr Gly Ala Thr Gly His Pro
Gly Ile Ile Pro Pro His Ala Thr Leu 85 90 95Val Phe Asp Val Glu Leu
Leu Lys Leu Glu Thr Ser 100 10512493PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 124Ile Leu Trp His Glu Met Trp His Glu Gly Leu Glu Glu
Ala Ser Arg1 5 10 15Leu Tyr Phe Gly Glu Arg Asn Val Lys Gly Met Phe
Glu Val Leu Glu 20 25 30Pro Leu His Ala Met Met Glu Arg Gly Pro Gln
Thr Leu Lys Glu Thr 35 40 45Ser Phe Asn Gln Ala Tyr Gly Arg Asp Leu
Met Glu Ala Gln Glu Trp 50 55 60Cys Arg Lys Tyr Met Lys Ser Gly Asn
Val Lys Asp Leu Thr Gln Ala65 70 75 80Trp Asp Leu Tyr Tyr His Val
Phe Arg Arg Ile Ser Lys 85 9012595PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 125Ile Leu Trp His Glu Met Trp His Glu Gly Leu Ile Glu
Ala Ser Arg1 5 10 15Leu Tyr Phe Gly Glu Arg Asn Val Lys Gly Met Phe
Glu Val Leu Glu 20 25 30Pro Leu His Ala Met Met Glu Arg Gly Pro Gln
Thr Leu Lys Glu Thr 35 40 45Ser Phe Asn Gln Ala Tyr Gly Arg Asp Leu
Met Glu Ala Gln Glu Trp 50 55 60Cys Arg Lys Tyr Met Lys Ser Gly Asn
Val Lys Asp Leu Thr Gln Ala65 70 75 80Trp Asp Leu Tyr Tyr His Val
Phe Arg Arg Ile Ser Lys Thr Ser 85 90 9512695PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 126Ile Leu Trp His Glu Met Trp His Glu Gly Leu Leu Glu
Ala Ser Arg1 5 10 15Leu Tyr Phe Gly Glu Arg Asn Val Lys Gly Met Phe
Glu Val Leu Glu 20 25 30Pro Leu His Ala Met Met Glu Arg Gly Pro Gln
Thr Leu Lys Glu Thr 35 40 45Ser Phe Asn Gln Ala Tyr Gly Arg
Asp Leu Met Glu Ala Gln Glu Trp 50 55 60Cys Arg Lys Tyr Met Lys Ser
Gly Asn Val Lys Asp Leu Thr Gln Ala65 70 75 80Trp Asp Leu Tyr Tyr
His Val Phe Arg Arg Ile Ser Lys Thr Ser 85 90 9512795PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 127Ile Leu Trp His Glu Met Trp His Glu Gly Leu Glu Glu
Ala Ser Arg1 5 10 15Leu Tyr Phe Gly Glu Arg Asn Val Lys Gly Met Phe
Glu Val Leu Glu 20 25 30Pro Leu His Ala Met Met Glu Arg Gly Pro Gln
Thr Leu Lys Glu Thr 35 40 45Ser Phe Asn Gln Ala Tyr Gly Arg Asp Leu
Met Glu Ala Gln Glu Trp 50 55 60Cys Arg Lys Tyr Met Lys Ser Gly Asn
Val Lys Asp Leu Leu Gln Ala65 70 75 80Trp Asp Leu Tyr Tyr His Val
Phe Arg Arg Ile Ser Lys Thr Ser 85 90 9512895PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide"MOD_RES(12)..(12)Any amino acidMOD_RES(78)..(78)Any
amino acid 128Ile Leu Trp His Glu Met Trp His Glu Gly Leu Xaa Glu
Ala Ser Arg1 5 10 15Leu Tyr Phe Gly Glu Arg Asn Val Lys Gly Met Phe
Glu Val Leu Glu 20 25 30Pro Leu His Ala Met Met Glu Arg Gly Pro Gln
Thr Leu Lys Glu Thr 35 40 45Ser Phe Asn Gln Ala Tyr Gly Arg Asp Leu
Met Glu Ala Gln Glu Trp 50 55 60Cys Arg Lys Tyr Met Lys Ser Gly Asn
Val Lys Asp Leu Xaa Gln Ala65 70 75 80Trp Asp Leu Tyr Tyr His Val
Phe Arg Arg Ile Ser Lys Thr Ser 85 90 9512995PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 129Ile Leu Trp His Glu Met Trp His Glu Gly Leu Ile Glu
Ala Ser Arg1 5 10 15Leu Tyr Phe Gly Glu Arg Asn Val Lys Gly Met Phe
Glu Val Leu Glu 20 25 30Pro Leu His Ala Met Met Glu Arg Gly Pro Gln
Thr Leu Lys Glu Thr 35 40 45Ser Phe Asn Gln Ala Tyr Gly Arg Asp Leu
Met Glu Ala Gln Glu Trp 50 55 60Cys Arg Lys Tyr Met Lys Ser Gly Asn
Val Lys Asp Leu Leu Gln Ala65 70 75 80Trp Asp Leu Tyr Tyr His Val
Phe Arg Arg Ile Ser Lys Thr Ser 85 90 9513095PRTArtificial
Sequencesource/note="Description of Artificial Sequence Synthetic
polypeptide" 130Ile Leu Trp His Glu Met Trp His Glu Gly Leu Leu Glu
Ala Ser Arg1 5 10 15Leu Tyr Phe Gly Glu Arg Asn Val Lys Gly Met Phe
Glu Val Leu Glu 20 25 30Pro Leu His Ala Met Met Glu Arg Gly Pro Gln
Thr Leu Lys Glu Thr 35 40 45Ser Phe Asn Gln Ala Tyr Gly Arg Asp Leu
Met Glu Ala Gln Glu Trp 50 55 60Cys Arg Lys Tyr Met Lys Ser Gly Asn
Val Lys Asp Leu Leu Gln Ala65 70 75 80Trp Asp Leu Tyr Tyr His Val
Phe Arg Arg Ile Ser Lys Thr Ser 85 90 951311132PRTHomo sapiens
131Met Pro Arg Ala Pro Arg Cys Arg Ala Val Arg Ser Leu Leu Arg Ser1
5 10 15His Tyr Arg Glu Val Leu Pro Leu Ala Thr Phe Val Arg Arg Leu
Gly 20 25 30Pro Gln Gly Trp Arg Leu Val Gln Arg Gly Asp Pro Ala Ala
Phe Arg 35 40 45Ala Leu Val Ala Gln Cys Leu Val Cys Val Pro Trp Asp
Ala Arg Pro 50 55 60Pro Pro Ala Ala Pro Ser Phe Arg Gln Val Ser Cys
Leu Lys Glu Leu65 70 75 80Val Ala Arg Val Leu Gln Arg Leu Cys Glu
Arg Gly Ala Lys Asn Val 85 90 95Leu Ala Phe Gly Phe Ala Leu Leu Asp
Gly Ala Arg Gly Gly Pro Pro 100 105 110Glu Ala Phe Thr Thr Ser Val
Arg Ser Tyr Leu Pro Asn Thr Val Thr 115 120 125Asp Ala Leu Arg Gly
Ser Gly Ala Trp Gly Leu Leu Leu Arg Arg Val 130 135 140Gly Asp Asp
Val Leu Val His Leu Leu Ala Arg Cys Ala Leu Phe Val145 150 155
160Leu Val Ala Pro Ser Cys Ala Tyr Gln Val Cys Gly Pro Pro Leu Tyr
165 170 175Gln Leu Gly Ala Ala Thr Gln Ala Arg Pro Pro Pro His Ala
Ser Gly 180 185 190Pro Arg Arg Arg Leu Gly Cys Glu Arg Ala Trp Asn
His Ser Val Arg 195 200 205Glu Ala Gly Val Pro Leu Gly Leu Pro Ala
Pro Gly Ala Arg Arg Arg 210 215 220Gly Gly Ser Ala Ser Arg Ser Leu
Pro Leu Pro Lys Arg Pro Arg Arg225 230 235 240Gly Ala Ala Pro Glu
Pro Glu Arg Thr Pro Val Gly Gln Gly Ser Trp 245 250 255Ala His Pro
Gly Arg Thr Arg Gly Pro Ser Asp Arg Gly Phe Cys Val 260 265 270Val
Ser Pro Ala Arg Pro Ala Glu Glu Ala Thr Ser Leu Glu Gly Ala 275 280
285Leu Ser Gly Thr Arg His Ser His Pro Ser Val Gly Arg Gln His His
290 295 300Ala Gly Pro Pro Ser Thr Ser Arg Pro Pro Arg Pro Trp Asp
Thr Pro305 310 315 320Cys Pro Pro Val Tyr Ala Glu Thr Lys His Phe
Leu Tyr Ser Ser Gly 325 330 335Asp Lys Glu Gln Leu Arg Pro Ser Phe
Leu Leu Ser Ser Leu Arg Pro 340 345 350Ser Leu Thr Gly Ala Arg Arg
Leu Val Glu Thr Ile Phe Leu Gly Ser 355 360 365Arg Pro Trp Met Pro
Gly Thr Pro Arg Arg Leu Pro Arg Leu Pro Gln 370 375 380Arg Tyr Trp
Gln Met Arg Pro Leu Phe Leu Glu Leu Leu Gly Asn His385 390 395
400Ala Gln Cys Pro Tyr Gly Val Leu Leu Lys Thr His Cys Pro Leu Arg
405 410 415Ala Ala Val Thr Pro Ala Ala Gly Val Cys Ala Arg Glu Lys
Pro Gln 420 425 430Gly Ser Val Ala Ala Pro Glu Glu Glu Asp Thr Asp
Pro Arg Arg Leu 435 440 445Val Gln Leu Leu Arg Gln His Ser Ser Pro
Trp Gln Val Tyr Gly Phe 450 455 460Val Arg Ala Cys Leu Arg Arg Leu
Val Pro Pro Gly Leu Trp Gly Ser465 470 475 480Arg His Asn Glu Arg
Arg Phe Leu Arg Asn Thr Lys Lys Phe Ile Ser 485 490 495Leu Gly Lys
His Ala Lys Leu Ser Leu Gln Glu Leu Thr Trp Lys Met 500 505 510Ser
Val Arg Gly Cys Ala Trp Leu Arg Arg Ser Pro Gly Val Gly Cys 515 520
525Val Pro Ala Ala Glu His Arg Leu Arg Glu Glu Ile Leu Ala Lys Phe
530 535 540Leu His Trp Leu Met Ser Val Tyr Val Val Glu Leu Leu Arg
Ser Phe545 550 555 560Phe Tyr Val Thr Glu Thr Thr Phe Gln Lys Asn
Arg Leu Phe Phe Tyr 565 570 575Arg Lys Ser Val Trp Ser Lys Leu Gln
Ser Ile Gly Ile Arg Gln His 580 585 590Leu Lys Arg Val Gln Leu Arg
Glu Leu Ser Glu Ala Glu Val Arg Gln 595 600 605His Arg Glu Ala Arg
Pro Ala Leu Leu Thr Ser Arg Leu Arg Phe Ile 610 615 620Pro Lys Pro
Asp Gly Leu Arg Pro Ile Val Asn Met Asp Tyr Val Val625 630 635
640Gly Ala Arg Thr Phe Arg Arg Glu Lys Arg Ala Glu Arg Leu Thr Ser
645 650 655Arg Val Lys Ala Leu Phe Ser Val Leu Asn Tyr Glu Arg Ala
Arg Arg 660 665 670Pro Gly Leu Leu Gly Ala Ser Val Leu Gly Leu Asp
Asp Ile His Arg 675 680 685Ala Trp Arg Thr Phe Val Leu Arg Val Arg
Ala Gln Asp Pro Pro Pro 690 695 700Glu Leu Tyr Phe Val Lys Val Asp
Val Thr Gly Ala Tyr Asp Thr Ile705 710 715 720Pro Gln Asp Arg Leu
Thr Glu Val Ile Ala Ser Ile Ile Lys Pro Gln 725 730 735Asn Thr Tyr
Cys Val Arg Arg Tyr Ala Val Val Gln Lys Ala Ala His 740 745 750Gly
His Val Arg Lys Ala Phe Lys Ser His Val Ser Thr Leu Thr Asp 755 760
765Leu Gln Pro Tyr Met Arg Gln Phe Val Ala His Leu Gln Glu Thr Ser
770 775 780Pro Leu Arg Asp Ala Val Val Ile Glu Gln Ser Ser Ser Leu
Asn Glu785 790 795 800Ala Ser Ser Gly Leu Phe Asp Val Phe Leu Arg
Phe Met Cys His His 805 810 815Ala Val Arg Ile Arg Gly Lys Ser Tyr
Val Gln Cys Gln Gly Ile Pro 820 825 830Gln Gly Ser Ile Leu Ser Thr
Leu Leu Cys Ser Leu Cys Tyr Gly Asp 835 840 845Met Glu Asn Lys Leu
Phe Ala Gly Ile Arg Arg Asp Gly Leu Leu Leu 850 855 860Arg Leu Val
Asp Asp Phe Leu Leu Val Thr Pro His Leu Thr His Ala865 870 875
880Lys Thr Phe Leu Arg Thr Leu Val Arg Gly Val Pro Glu Tyr Gly Cys
885 890 895Val Val Asn Leu Arg Lys Thr Val Val Asn Phe Pro Val Glu
Asp Glu 900 905 910Ala Leu Gly Gly Thr Ala Phe Val Gln Met Pro Ala
His Gly Leu Phe 915 920 925Pro Trp Cys Gly Leu Leu Leu Asp Thr Arg
Thr Leu Glu Val Gln Ser 930 935 940Asp Tyr Ser Ser Tyr Ala Arg Thr
Ser Ile Arg Ala Ser Leu Thr Phe945 950 955 960Asn Arg Gly Phe Lys
Ala Gly Arg Asn Met Arg Arg Lys Leu Phe Gly 965 970 975Val Leu Arg
Leu Lys Cys His Ser Leu Phe Leu Asp Leu Gln Val Asn 980 985 990Ser
Leu Gln Thr Val Cys Thr Asn Ile Tyr Lys Ile Leu Leu Leu Gln 995
1000 1005Ala Tyr Arg Phe His Ala Cys Val Leu Gln Leu Pro Phe His
Gln 1010 1015 1020Gln Val Trp Lys Asn Pro Thr Phe Phe Leu Arg Val
Ile Ser Asp 1025 1030 1035Thr Ala Ser Leu Cys Tyr Ser Ile Leu Lys
Ala Lys Asn Ala Gly 1040 1045 1050Met Ser Leu Gly Ala Lys Gly Ala
Ala Gly Pro Leu Pro Ser Glu 1055 1060 1065Ala Val Gln Trp Leu Cys
His Gln Ala Phe Leu Leu Lys Leu Thr 1070 1075 1080Arg His Arg Val
Thr Tyr Val Pro Leu Leu Gly Ser Leu Arg Thr 1085 1090 1095Ala Gln
Thr Gln Leu Ser Arg Lys Leu Pro Gly Thr Thr Leu Thr 1100 1105
1110Ala Leu Glu Ala Ala Ala Asn Pro Ala Leu Pro Ser Asp Phe Lys
1115 1120 1125Thr Ile Leu Asp 11301324027DNAHomo sapiens
132caggcagcgt ggtcctgctg cgcacgtggg aagccctggc cccggccacc
cccgcgatgc 60cgcgcgctcc ccgctgccga gccgtgcgct ccctgctgcg cagccactac
cgcgaggtgc 120tgccgctggc cacgttcgtg cggcgcctgg ggccccaggg
ctggcggctg gtgcagcgcg 180gggacccggc ggctttccgc gcgctggtgg
cccagtgcct ggtgtgcgtg ccctgggacg 240cacggccgcc ccccgccgcc
ccctccttcc gccaggtgtc ctgcctgaag gagctggtgg 300cccgagtgct
gcagaggctg tgcgagcgcg gcgcgaagaa cgtgctggcc ttcggcttcg
360cgctgctgga cggggcccgc gggggccccc ccgaggcctt caccaccagc
gtgcgcagct 420acctgcccaa cacggtgacc gacgcactgc gggggagcgg
ggcgtggggg ctgctgttgc 480gccgcgtggg cgacgacgtg ctggttcacc
tgctggcacg ctgcgcgctc tttgtgctgg 540tggctcccag ctgcgcctac
caggtgtgcg ggccgccgct gtaccagctc ggcgctgcca 600ctcaggcccg
gcccccgcca cacgctagtg gaccccgaag gcgtctggga tgcgaacggg
660cctggaacca tagcgtcagg gaggccgggg tccccctggg cctgccagcc
ccgggtgcga 720ggaggcgcgg gggcagtgcc agccgaagtc tgccgttgcc
caagaggccc aggcgtggcg 780ctgcccctga gccggagcgg acgcccgttg
ggcaggggtc ctgggcccac ccgggcagga 840cgcgtggacc gagtgaccgt
ggtttctgtg tggtgtcacc tgccagaccc gccgaagaag 900ccacctcttt
ggagggtgcg ctctctggca cgcgccactc ccacccatcc gtgggccgcc
960agcaccacgc gggcccccca tccacatcgc ggccaccacg tccctgggac
acgccttgtc 1020ccccggtgta cgccgagacc aagcacttcc tctactcctc
aggcgacaag gagcagctgc 1080ggccctcctt cctactcagc tctctgaggc
ccagcctgac tggcgctcgg aggctcgtgg 1140agaccatctt tctgggttcc
aggccctgga tgccagggac tccccgcagg ttgccccgcc 1200tgccccagcg
ctactggcaa atgcggcccc tgtttctgga gctgcttggg aaccacgcgc
1260agtgccccta cggggtgctc ctcaagacgc actgcccgct gcgagctgcg
gtcaccccag 1320cagccggtgt ctgtgcccgg gagaagcccc agggctctgt
ggcggccccc gaggaggagg 1380acacagaccc ccgtcgcctg gtgcagctgc
tccgccagca cagcagcccc tggcaggtgt 1440acggcttcgt gcgggcctgc
ctgcgccggc tggtgccccc aggcctctgg ggctccaggc 1500acaacgaacg
ccgcttcctc aggaacacca agaagttcat ctccctgggg aagcatgcca
1560agctctcgct gcaggagctg acgtggaaga tgagcgtgcg gggctgcgct
tggctgcgca 1620ggagcccagg ggttggctgt gttccggccg cagagcaccg
tctgcgtgag gagatcctgg 1680ccaagttcct gcactggctg atgagtgtgt
acgtcgtcga gctgctcagg tctttctttt 1740atgtcacgga gaccacgttt
caaaagaaca ggctcttttt ctaccggaag agtgtctgga 1800gcaagttgca
aagcattgga atcagacagc acttgaagag ggtgcagctg cgggagctgt
1860cggaagcaga ggtcaggcag catcgggaag ccaggcccgc cctgctgacg
tccagactcc 1920gcttcatccc caagcctgac gggctgcggc cgattgtgaa
catggactac gtcgtgggag 1980ccagaacgtt ccgcagagaa aagagggccg
agcgtctcac ctcgagggtg aaggcactgt 2040tcagcgtgct caactacgag
cgggcgcggc gccccggcct cctgggcgcc tctgtgctgg 2100gcctggacga
tatccacagg gcctggcgca ccttcgtgct gcgtgtgcgg gcccaggacc
2160cgccgcctga gctgtacttt gtcaaggtgg atgtgacggg cgcgtacgac
accatccccc 2220aggacaggct cacggaggtc atcgccagca tcatcaaacc
ccagaacacg tactgcgtgc 2280gtcggtatgc cgtggtccag aaggccgccc
atgggcacgt ccgcaaggcc ttcaagagcc 2340acgtctctac cttgacagac
ctccagccgt acatgcgaca gttcgtggct cacctgcagg 2400agaccagccc
gctgagggat gccgtcgtca tcgagcagag ctcctccctg aatgaggcca
2460gcagtggcct cttcgacgtc ttcctacgct tcatgtgcca ccacgccgtg
cgcatcaggg 2520gcaagtccta cgtccagtgc caggggatcc cgcagggctc
catcctctcc acgctgctct 2580gcagcctgtg ctacggcgac atggagaaca
agctgtttgc ggggattcgg cgggacgggc 2640tgctcctgcg tttggtggat
gatttcttgt tggtgacacc tcacctcacc cacgcgaaaa 2700ccttcctcag
gaccctggtc cgaggtgtcc ctgagtatgg ctgcgtggtg aacttgcgga
2760agacagtggt gaacttccct gtagaagacg aggccctggg tggcacggct
tttgttcaga 2820tgccggccca cggcctattc ccctggtgcg gcctgctgct
ggatacccgg accctggagg 2880tgcagagcga ctactccagc tatgcccgga
cctccatcag agccagtctc accttcaacc 2940gcggcttcaa ggctgggagg
aacatgcgtc gcaaactctt tggggtcttg cggctgaagt 3000gtcacagcct
gtttctggat ttgcaggtga acagcctcca gacggtgtgc accaacatct
3060acaagatcct cctgctgcag gcgtacaggt ttcacgcatg tgtgctgcag
ctcccatttc 3120atcagcaagt ttggaagaac cccacatttt tcctgcgcgt
catctctgac acggcctccc 3180tctgctactc catcctgaaa gccaagaacg
cagggatgtc gctgggggcc aagggcgccg 3240ccggccctct gccctccgag
gccgtgcagt ggctgtgcca ccaagcattc ctgctcaagc 3300tgactcgaca
ccgtgtcacc tacgtgccac tcctggggtc actcaggaca gcccagacgc
3360agctgagtcg gaagctcccg gggacgacgc tgactgccct ggaggccgca
gccaacccgg 3420cactgccctc agacttcaag accatcctgg actgatggcc
acccgcccac agccaggccg 3480agagcagaca ccagcagccc tgtcacgccg
ggctctacgt cccagggagg gaggggcggc 3540ccacacccag gcccgcaccg
ctgggagtct gaggcctgag tgagtgtttg gccgaggcct 3600gcatgtccgg
ctgaaggctg agtgtccggc tgaggcctga gcgagtgtcc agccaagggc
3660tgagtgtcca gcacacctgc cgtcttcact tccccacagg ctggcgctcg
gctccacccc 3720agggccagct tttcctcacc aggagcccgg cttccactcc
ccacatagga atagtccatc 3780cccagattcg ccattgttca cccctcgccc
tgccctcctt tgccttccac ccccaccatc 3840caggtggaga ccctgagaag
gaccctggga gctctgggaa tttggagtga ccaaaggtgt 3900gccctgtaca
caggcgagga ccctgcacct ggatgggggt ccctgtgggt caaattgggg
3960ggaggtgctg tgggagtaaa atactgaata tatgagtttt tcagttttga
aaaaaaaaaa 4020aaaaaaa 4027
References
D00001
D00002
D00003
D00004
D00005

D00006

D00007

D00008

D00009

D00010
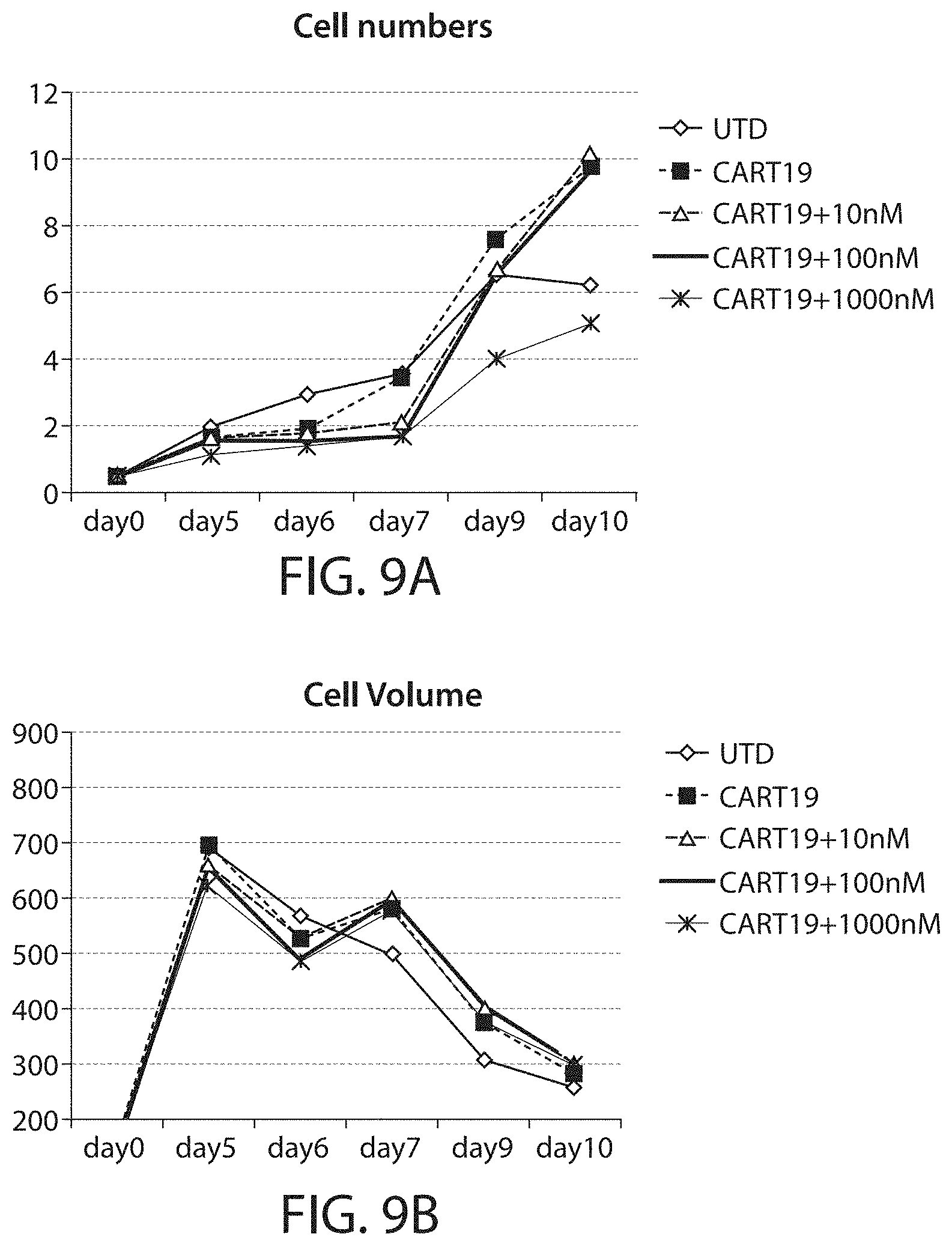
D00011

D00012

D00013

D00014
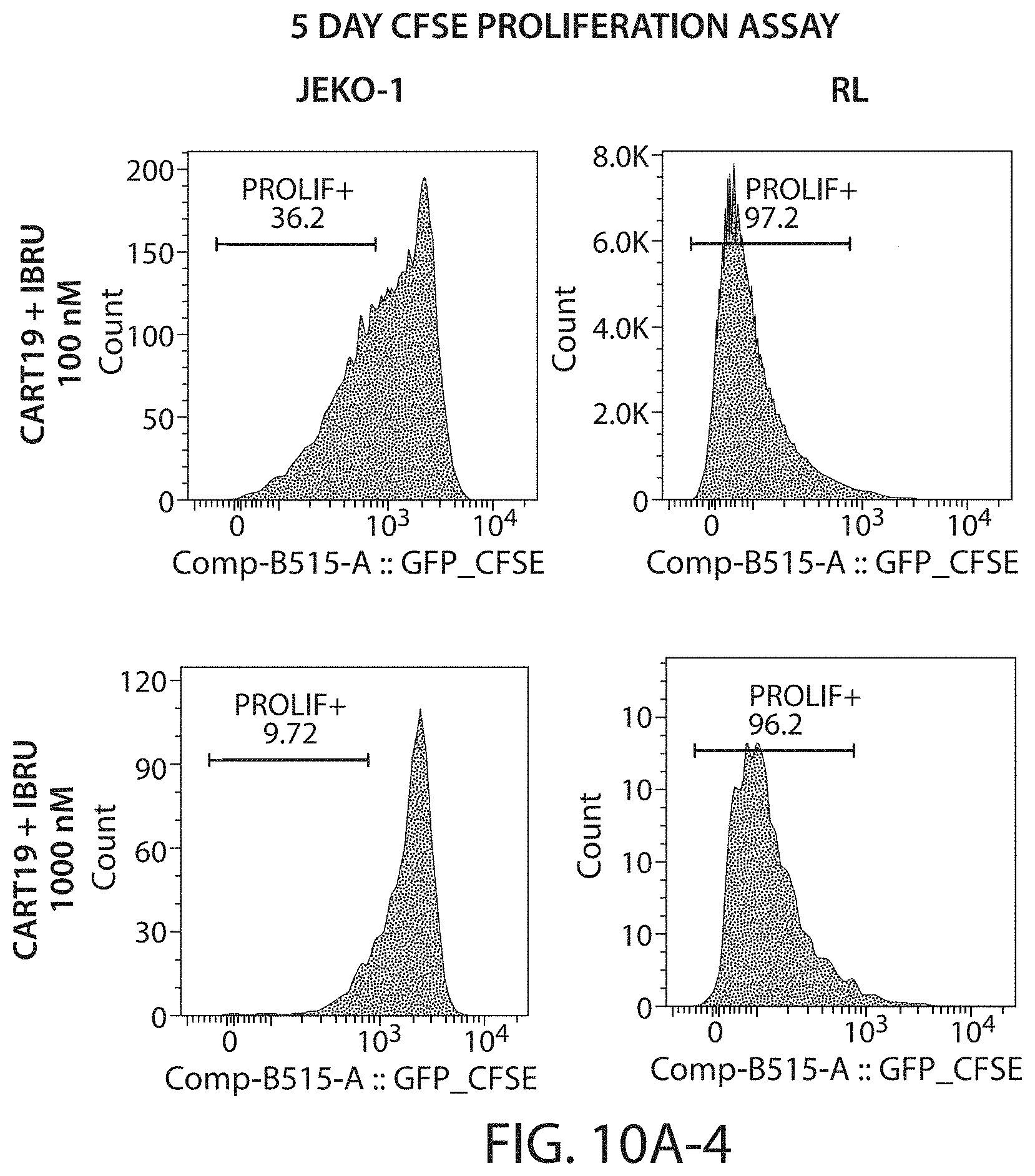
D00015

D00016

D00017

D00018

D00019

D00020

D00021

D00022

D00023
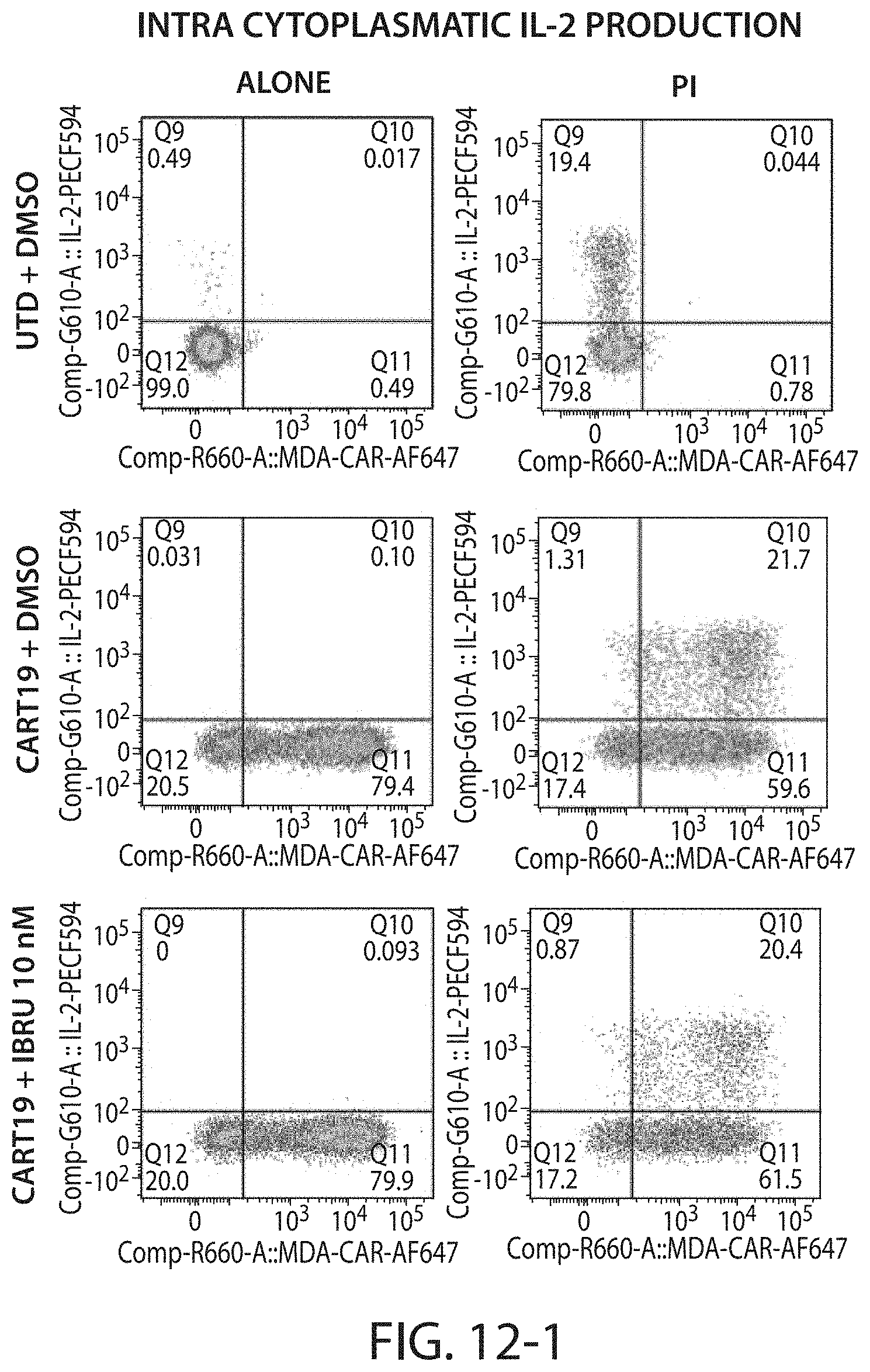
D00024

D00025

D00026

D00027

D00028

D00029

D00030

D00031

D00032

D00033

D00034
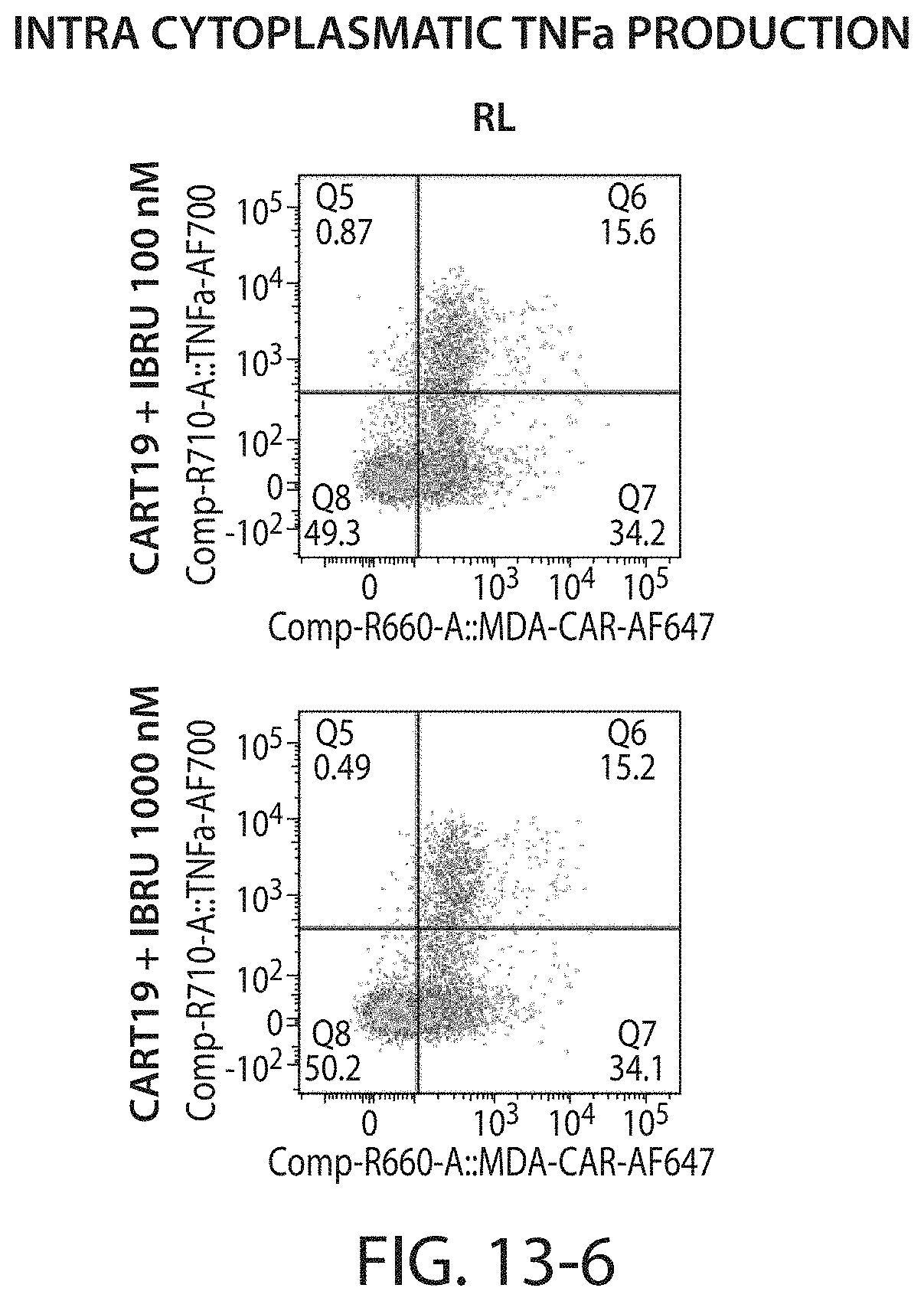
D00035

D00036

D00037

D00038

D00039

D00040

D00041

D00042

D00043

D00044

D00045

D00046

D00047

D00048

D00049

D00050

D00051

D00052

D00053

D00054

D00055

D00056

D00057

D00058

D00059

D00060

D00061

D00062
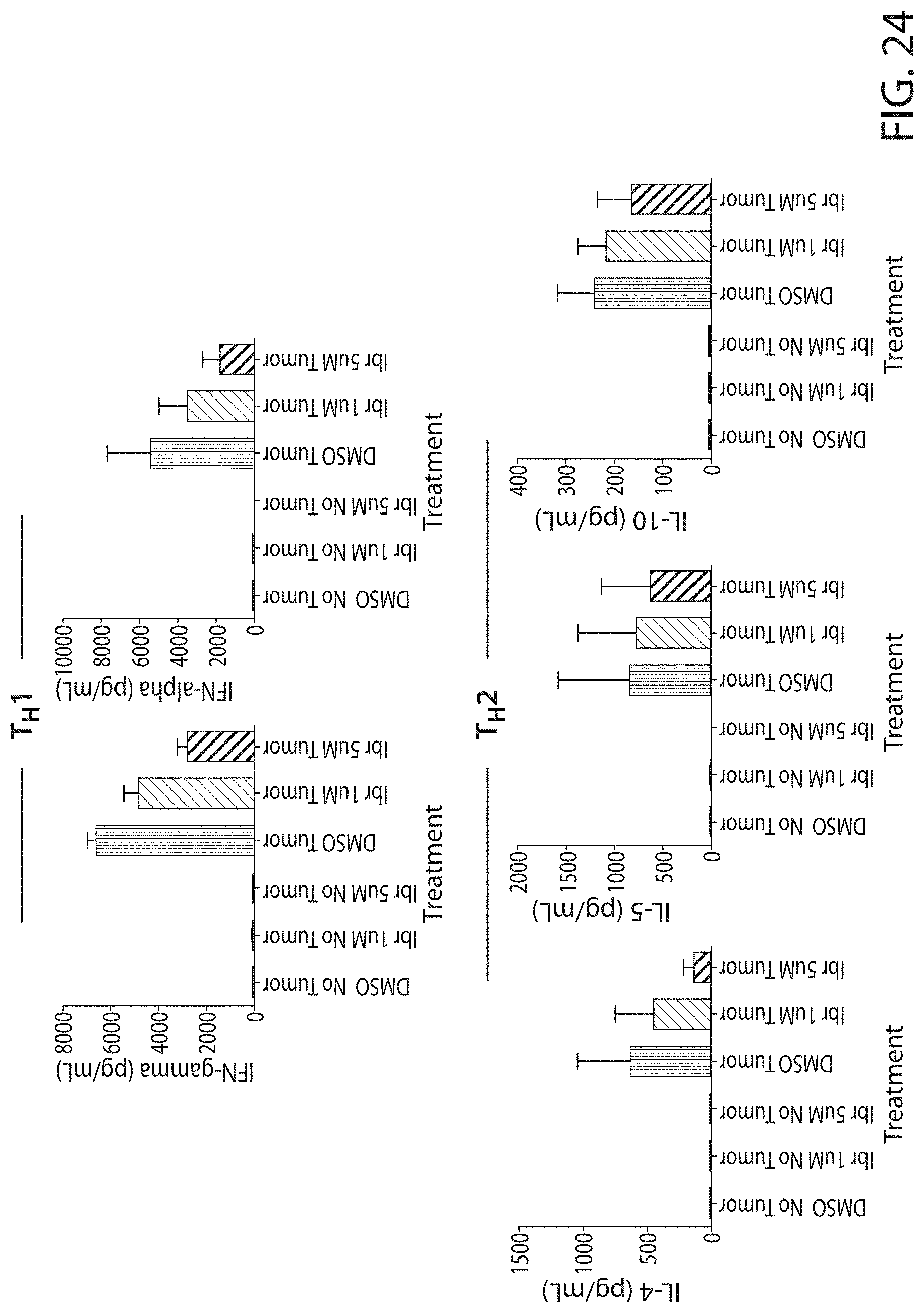
D00063

D00064

D00065

D00066

D00067

D00068

D00069

D00070

D00071

D00072

D00073

D00074

D00075

D00076

D00077

D00078

D00079
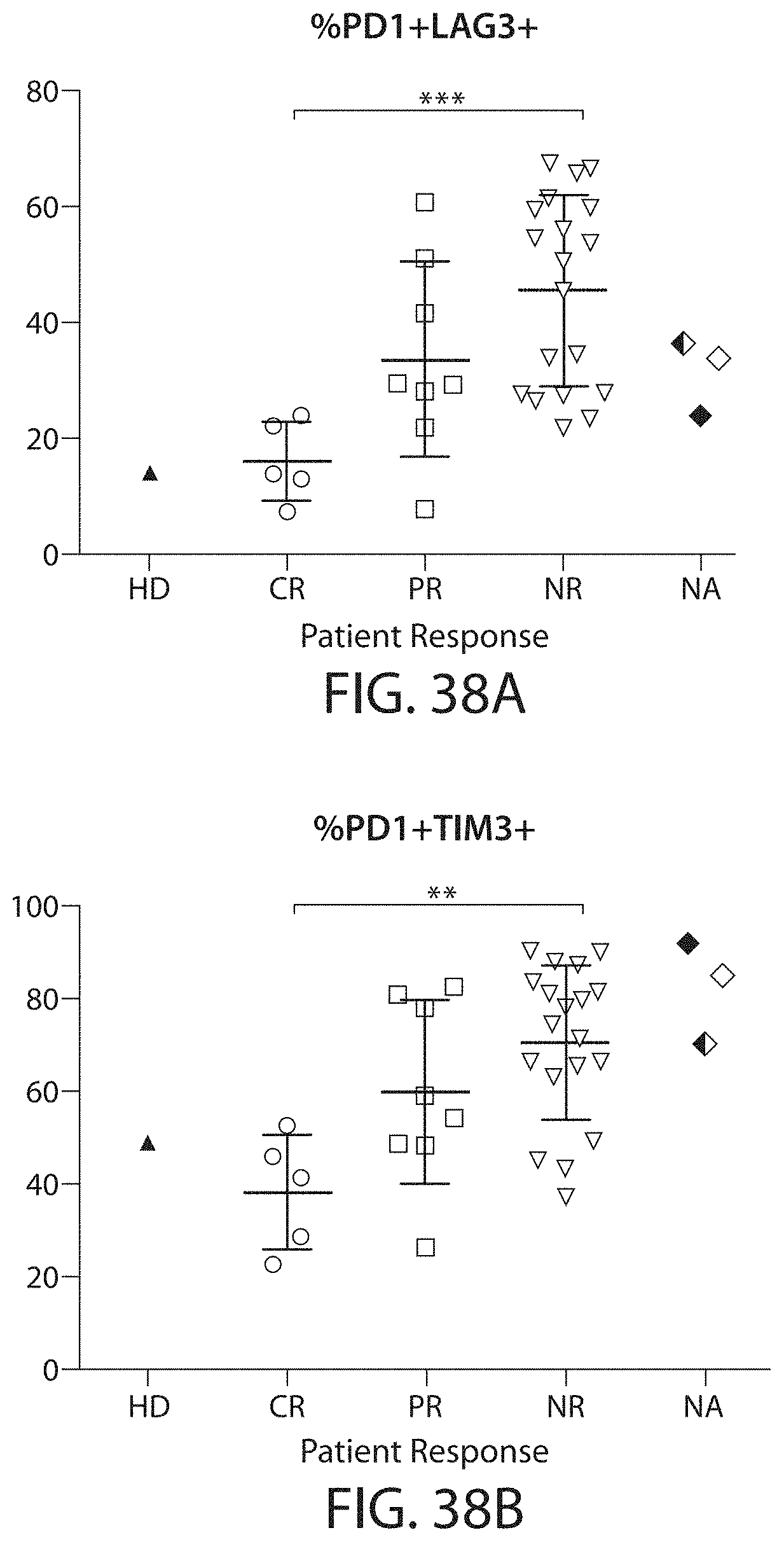
D00080

D00081

D00082

D00083

D00084

D00085

D00086

D00087

D00088

D00089

D00090

D00091

D00092

D00093

D00094

D00095

D00096

D00097

D00098

D00099

D00100

D00101

D00102

D00103
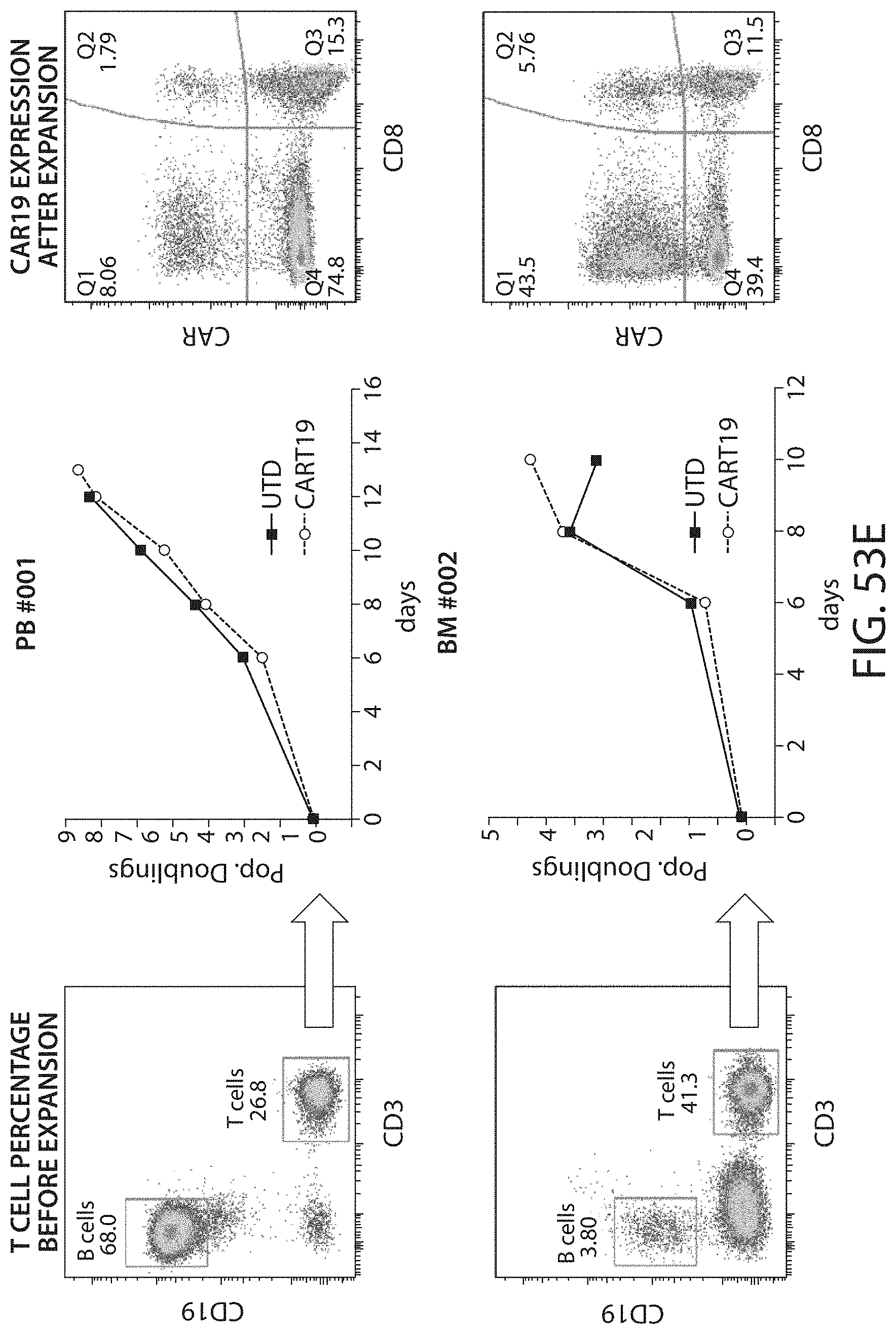
D00104

D00105

D00106

D00107

D00108

D00109

D00110

D00111

D00112

D00113

D00114

D00115

D00116

D00117

D00118

D00119

D00120

P00001

P00002
S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.