Methods For The Identification Of Variant Recognition Sites For Rare-cutting Engineered Double-strand-break-inducing Agents And
DESCHAMPS; STEPHANE ; et al.
U.S. patent application number 16/415179 was filed with the patent office on 2019-12-05 for methods for the identification of variant recognition sites for rare-cutting engineered double-strand-break-inducing agents and . This patent application is currently assigned to E. I. DU PONT DE NEMOURS AND COMPANY. The applicant listed for this patent is E. I. DU PONT DE NEMOURS AND COMPANY, PIONEER HI-BRED INTERNATIONAL, INC.. Invention is credited to STEPHANE DESCHAMPS, JAMES ENGLISH, ZHONGSEN LI, VICTOR LLACA, JOSHUA K. YOUNG.
| Application Number | 20190367934 16/415179 |
| Document ID | / |
| Family ID | 50513431 |
| Filed Date | 2019-12-05 |


View All Diagrams
| United States Patent Application | 20190367934 |
| Kind Code | A1 |
| DESCHAMPS; STEPHANE ; et al. | December 5, 2019 |
METHODS FOR THE IDENTIFICATION OF VARIANT RECOGNITION SITES FOR RARE-CUTTING ENGINEERED DOUBLE-STRAND-BREAK-INDUCING AGENTS AND COMPOSITIONS AND USES THEREOF
Abstract
Methods for the identification of variant recognition sites for rare cutting engineered double strand break inducing agents and compositions thereof are provided. Further provided are nucleic acid constructs, yeast, plants, plant cells, explants, seeds and grain having the of variant recognition sites. Various methods of identifying variant recognition sites with increased substrate activity for a rare cutting engineered double strand break inducing agents are provided.
| Inventors: | DESCHAMPS; STEPHANE; (WEST DES MOINES, IA) ; ENGLISH; JAMES; (SAN RAMON, CA) ; LI; ZHONGSEN; (HOCKESSIN, DE) ; YOUNG; JOSHUA K.; (JOHNSTON, IA) ; LLACA; VICTOR; (NEWARK, DE) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Assignee: | E. I. DU PONT DE NEMOURS AND
COMPANY WILMINGTON DE PIONEER HI-BRED INTERNATIONAL, INC. JOHNSTON IA |
||||||||||
| Family ID: | 50513431 | ||||||||||
| Appl. No.: | 16/415179 | ||||||||||
| Filed: | May 17, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 14775777 | Sep 14, 2015 | 10329574 | ||
| PCT/US14/22500 | Mar 10, 2014 | |||
| 16415179 | ||||
| 61777238 | Mar 12, 2013 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | C12Q 1/6811 20130101; C12N 15/8241 20130101; A01H 6/14 20180501; C12Q 1/6811 20130101; C12Q 1/6811 20130101; C12N 9/22 20130101; A01H 6/46 20180501; C12N 15/8213 20130101; A01H 6/82 20180501; C12Q 2521/313 20130101; C12Q 2525/191 20130101; C12Q 2525/191 20130101; C12Q 2521/507 20130101 |
| International Class: | C12N 15/82 20060101 C12N015/82; C12Q 1/6811 20060101 C12Q001/6811; C12N 9/22 20060101 C12N009/22 |
Claims
1) A method to identify a double-strand break site for a double-strand-break-inducing agent in genomic DNA, said method comprising: a. contacting genomic DNA with a double-strand-break-inducing agent capable of introducing a double-strand break into said genomic DNA, wherein the double-strand break results in a nucleotide overhang; b. ligating a first adapter to said nucleotide overhang of (a), resulting in ligated DNA; c. obtaining fragments of the ligated DNA obtained in step (b) and ligating a second adapter to the DNA fragments to allow for the amplification and sequencing of the DNA fragments that each comprise the first adapter on one end and the second adapter on the other end; d. amplifying and sequencing the DNA fragments of (c); e. aligning nucleotide sequences of the DNA fragments obtained in (d) with a reference genome DNA sequence; and, f. identifying a variant recognition site comprising at least one nucleotide base alteration when compared to the intended recognition site of said engineered double-strand break-inducing agent.
2) The method of claim 1 wherein the double-strand-break-inducing agent is selected from the group consisting of: a meganuclease, a zinc finger nuclease, a TAL effector nuclease, a transposase, a Cas endonuclease, and a site-specific recombinase.
3) The method of claim 1 wherein the nucleotide overhang is a 3' nucleotide overhang.
4) The method of claim 1 wherein the nucleotide overhang is a 5' nucleotide overhang.
5) The method of claim 1 wherein the first adapter ligated to the nucleotide overhang is a non-5' phosphorylated adapter.
6) The method of claim 1 wherein the genomic DNA is selected from the group consisting of a prokaryotic DNA, eukaryotic DNA and synthetic DNA.
7) The method of claim 6 wherein the eukaryotic DNA is isolated from a plant, yeast, or animal.
8) The method of claim 7 wherein the plant is selected from the group consisting of soybean, sunflower, cotton, alfalfa, canola, cotton, tobacco, potato, Arabidopsis, safflower, maize, rice, sorghum, barley, wheat, millet, oats, sugarcane, turfgrass, and switch grass.
9) (canceled)
10) A method to identify a variant double-strand break site with improved cleavage activity for a double-strand-break-inducing agent capable of introducing a double strand break in an intended recognition site, said method comprising: a. contacting genomic DNA with double-strand-break-inducing agent capable of introducing a double strand break into said genomic DNA, wherein the double strand break results in a nucleotide overhang; b. ligating a first adapter to said nucleotide overhang; c. shearing the ligated DNA obtained in step (b) and ligating a second adapter to the sheared nucleotide end to allow for the amplification and sequencing of genomic DNA fragments surrounding the double strand break; d. amplifying and sequencing the DNA fragments of (c); e. aligning nucleotide sequences of the DNA fragments obtained in (d) with a reference genome DNA sequence; and, f. identifying a variant double-strand break site comprising at least one nucleotide base alteration when compared to the intended double-strand break site of said rare-cutting engineered double-strand-break-inducing agent; g. analyzing the a rare-cutting engineered double-strand-break-inducing agent activity at the variant double-strand break site of (e); and, g. identifying a variant double-strand break site that results in an increased activity of the double-strand-break-inducing agent when compared to the activity at the intended double-strand break site.
11) The method of claim 10 wherein the increased activity of the double-strand-break-inducing agent is evidenced by a. a higher percent (%) cleavage of the variant double-strand break site when compared to the percent (%) cleavage of intended double-strand break site, wherein the double-strand break sites are located on genomic DNA; b. a higher percent (%) cleavage of the variant double-strand break site when compared to the percent (%) cleavage of intended double-strand break site, wherein the double-strand break sites are located on plasmid DNA; c. a higher yeast assay score for the variant double-strand break site when compared to the intended double-strand break site; or, d. any combination of (a), (b) and (c)
12) A method for introducing into the genome of a cell a variant double-strand break site for a double-strand-break-inducing agent capable of introducing a double strand break in an intended double-strand break site, said method comprising: a. providing a donor DNA comprising a variant double-strand break site for a double-strand-break-inducing agent capable of introducing a double-strand break in an intended double-strand break site, wherein said a double-strand-break-inducing agent is also capable of introducing a double-strand break in said variant double-strand break site; b. providing a plant cell; c. contacting the plant cell with the donor DNA; and, d. identifying at least one plant cell from (c) comprising in its genome said variant double-strand break site.
13) The method of claim 12 wherein the double-strand-break-inducing agent is selected from the group consisting of: a meganuclease, a zinc finger nuclease, a TAL effector nuclease, a transposase, a Cas endonuclease, and a site-specific recombinase.
14)-42) (canceled)
Description
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application is a continuation of U.S. application Ser. No. 14/775,777 filed 14 Sep. 2015, now allowed, which is a 371 National Stage Entry of PCT/US14/22500 which was filed 10 Mar. 2014, which claims the benefit of U.S. Patent Application Ser. No. 61/777,238, filed Mar. 12, 2013, all of which are herein incorporated by reference in their entireties.
FIELD OF THE INVENTION
[0002] This invention is in the field of molecular biology. More specifically, this invention pertains to methods for identifying and using variant recognition sites for rare-cutting engineered double strand break inducing agents.
REFERENCE TO SEQUENCE LISTING SUBMITTED ELECTRONICALLY
[0003] The official copy of the sequence listing is submitted electronically via EFS-Web as an ASCII formatted sequence listing with a file named BB2223USPCN_SequenceListing_ST25.txt created on 14 May 2019 and having a size of 65,816 bytes and is filed concurrently with the specification. The sequence listing comprised in this ASCII formatted document is part of the specification and is herein incorporated by reference in its entirety.
BACKGROUND OF THE INVENTION
[0004] Recombinant DNA technology has made it possible to insert foreign DNA sequences into the genome of an organism, thus, altering the organism's phenotype. The most commonly used plant transformation methods are Agrobacterium infection and biolistic particle bombardment in which transgenes integrate into a plant genome in a random fashion and in an unpredictable copy number. Thus, efforts are undertaken to control transgene integration in plants.
[0005] Methods for inserting or modifying a DNA sequence into the genome of a variety of organism have been developed and can involve site-specific integration techniques, which rely on homologous recombination (U.S. Pat. No. 7,102,055 issued on Sep. 5, 2006) or designer endonucleases such as meganucleases, zinc finger nucleases or TALENs (US patent publication 2009-0133152 A1, published May 21, 2009).
[0006] While these systems have provided useful techniques for targeted insertion of sequences of interest, there remains a need for identifying more recognition sites for rare cutting double strand break inducing agents and for identifying recognition sites with increased activity towards rare cutting double strand beak inducing agents.
BRIEF SUMMARY OF THE INVENTION
[0007] Compositions and methods are provided which employ variant recognition sites for a rare-cutting engineered double-strand-break-inducing agent capable of introducing a rare double strand break in an intended recognition site.
[0008] Methods for identifying a variant recognition site for a rare-cutting engineered double-strand-break-inducing agent capable of introducing a rare double strand break in an intended recognition site are provided. One method comprises, a) contacting genomic DNA with a rare-cutting engineered double-strand-break-inducing agent capable of introducing a double-strand break into said genomic DNA, wherein the double-strand break results in a nucleotide overhang, b) ligating a first adapter to said nucleotide overhang, c) shearing the ligated DNA obtained in step (b) and ligating at least one second adapter to the sheared nucleotide end to allow for the amplification and sequencing of genomic DNA fragments surrounding the double strand break, d) aligning nucleotide sequences of the DNA fragments obtained in (c) with a reference genome DNA sequence; and e) identifying a variant recognition site comprising at least one nucleotide base alteration when compared to the intended recognition site of said engineered double-strand break-inducing agent. Another method comprises a method to identify a variant recognition site for a rare-cutting engineered double-strand-break-inducing agent capable of introducing a rare double strand break in an intended recognition site, said method comprising: a) contacting genomic DNA with a rare-cutting engineered double-strand-break-inducing agent capable of introducing a double-strand break into said genomic DNA, wherein the double-strand break results in a blunt end; b) creating a nucleotide overhang from the blunt end of (a); c) ligating a first adapter to the nucleotide overhang of (b); d) shearing the ligated DNA obtained in step (c) and ligating at least one second adapter to the sheared nucleotide end to allow for the amplification and sequencing of genomic DNA fragments surrounding the double strand break; e) aligning nucleotide sequences of the DNA fragments obtained in (d) with a reference genome DNA sequence; and, f) identifying a variant recognition site comprising at least one nucleotide base alteration when compared to the intended recognition site of said engineered double-strand break-inducing agent. The rare-cutting engineered double-strand-break-inducing agent can be selected from the group consisting of a meganuclease, a zinc finger nuclease, a TAL effector nuclease, a transposase, a Cas endonuclease and a site-specific recombinase. The nucleotide overhang can be a 3' or 5' nucleotide overhang.
[0009] Further provided are methods to identify a variant recognition site with an improved cleavage activity for a rare-cutting engineered double-strand-break-inducing agent capable of introducing a double strand break in an intended recognition site. The increased activity of the rare-cutting engineered double-strand-break-inducing agent is evidenced by a) a higher percent (%) cleavage of the variant recognition site when compared to the percent (%) cleavage of intended recognition site, wherein the recognition sites are located on genomic DNA; b) a higher percent (%) cleavage of the variant recognition site when compared to the percent (%) cleavage of intended recognition site, wherein the recognition sites are located on plasmid DNA; c) a higher yeast assay score for the variant recognition site when compared to the intended recognition site; or, d) any combination of (a), (b) and (c).
[0010] Further provided are methods for targeting the insertion of a polynucleotide of interest to a specific chromosomal site within a plant genome, said method comprising: a) transforming a plant cell or a plant with a DNA fragment comprising a polynucleotide of interest, wherein said genome of said plant cell or plant comprises at least one variant recognition site selected from the group consisting of SEQ ID NOs: 15, 16, 17, 18, 19, 20 and 21 or SEQ ID NOs: 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 and 35; and, b) providing a meganuclease capable of providing a double strand break into the variable recognition site of (a); and, c) selecting said plant cell or plant comprising said polynucleotide of interest integrated into said variant recognition site.
[0011] Various compositions include a plant, a seed or a plant cell comprising in its genome a variant recognition site for a rare-cutting engineered double-strand-break-inducing agent capable of introducing a rare double strand break in an intended recognition site.
BRIEF DESCRIPTION OF THE DRAWINGS AND SEQUENCE LISTING
[0012] The invention can be more fully understood from the following detailed description and the accompanying drawings and Sequence Listing, which form a part of this application. The sequence descriptions and sequence listing attached hereto comply with the rules governing nucleotide and amino acid sequence disclosures in patent applications as set forth in 37 C.F.R. .sctn..sctn. 1.821 1.825. The sequence descriptions contain the three letter codes for amino acids as defined in 37 C.F.R. .sctn..sctn. 1.821 1.825, which are incorporated herein by reference.
[0013] FIG. 1A Genomic recognition site peak signature from a sample treated with meganuclease. Mapped sequence data originates from and comes back towards the site of cleavage. The directionality of the mapped reads is indicated in the pile-up view and by the arrows. FIG. 1B The mock control contains no enrichment or peak signature as observed for the treated sample.
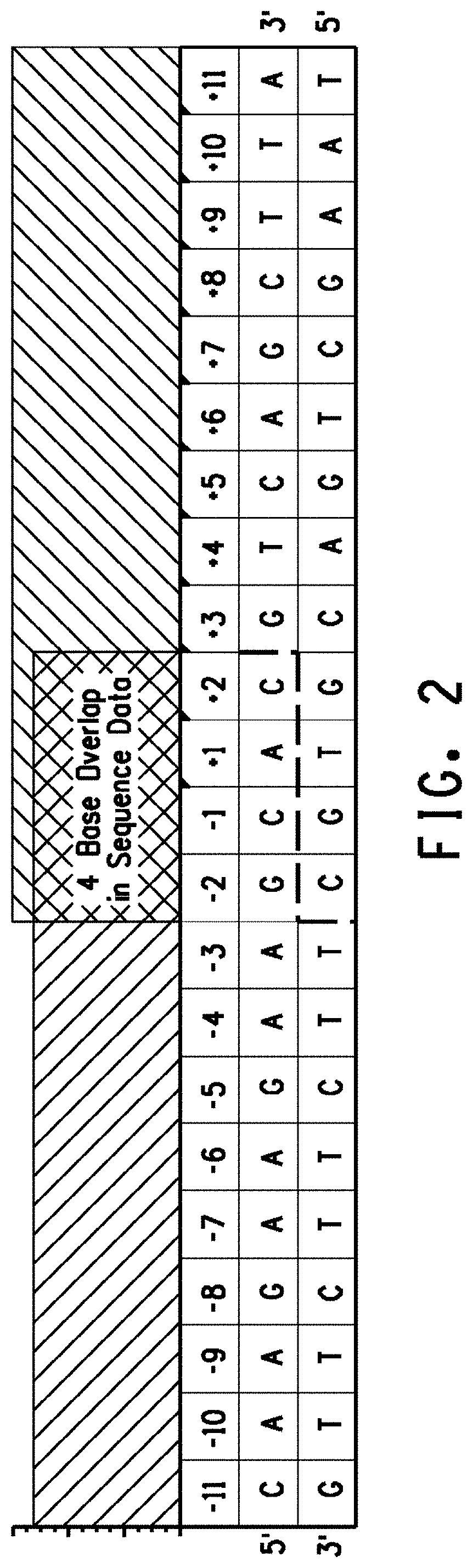
[0014] FIG. 2. The 4 base overlap in the peak recognition site signature corresponds to the overhang generated by the meganuclease and defines the sequence of the genomic variant recognition site. The dashed line defines the overhangs produced by recognition site cleavage.
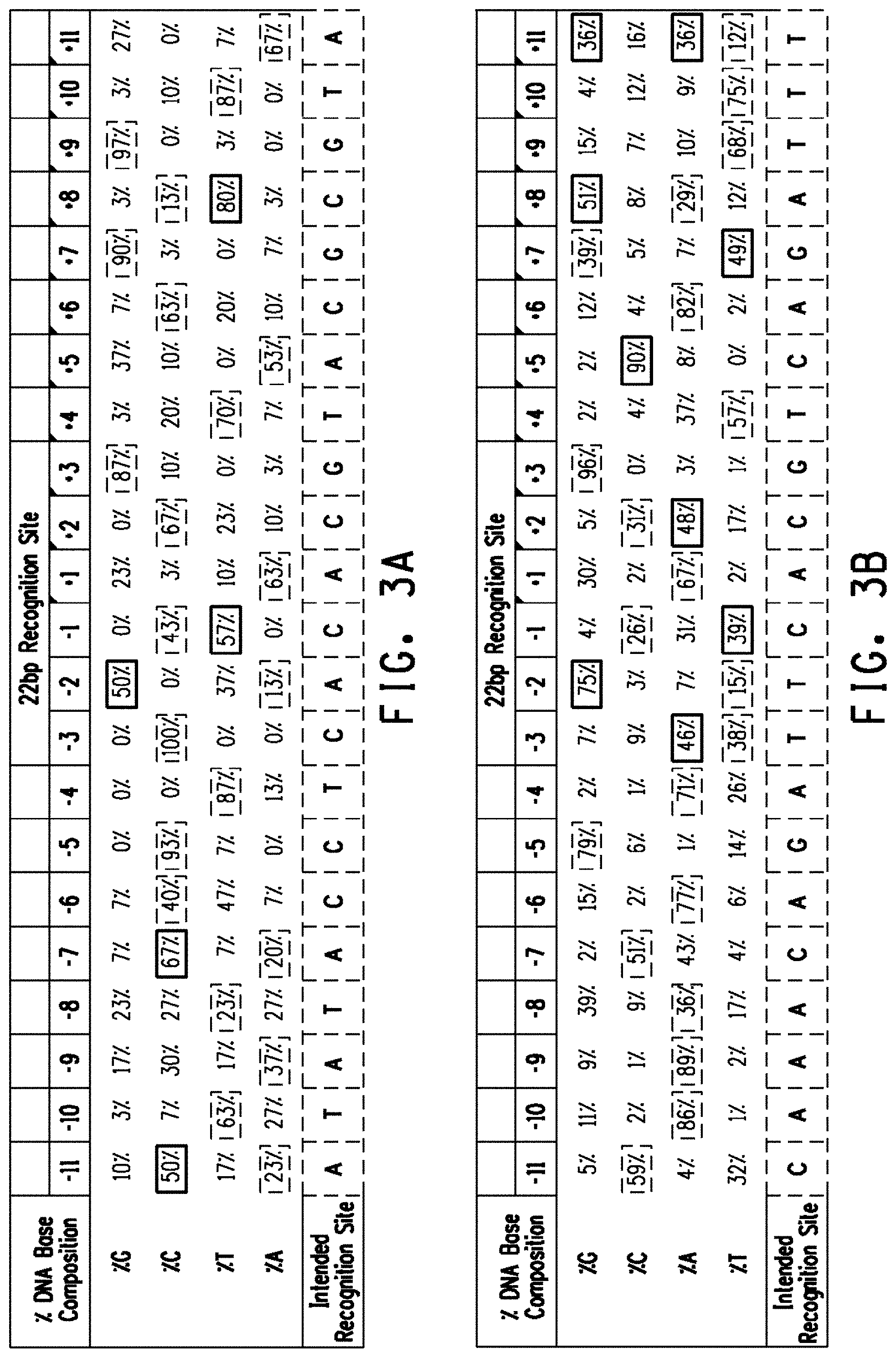
[0015] FIGS. 3A and 3B show the percent DNA base composition of oriented genomic variant recognition sites. Preferred off-nucleotides are outlined while the intended recognition bases are shaded. FIG. 3A Lig3-4 meganuclease DNA base composition of 30 genomic variant recognition sites. The LIG3-4 intended recognition site (SEQ ID NO:13) is shown at the bottom of FIG. 3A. FIG. 3B MHP14+ meganuclease DNA base composition of 254 genomic variant recognition sites. The MHP14+ intended recognition site (SEQ ID NO:14) is shown at the bottom of FIG. 3B.
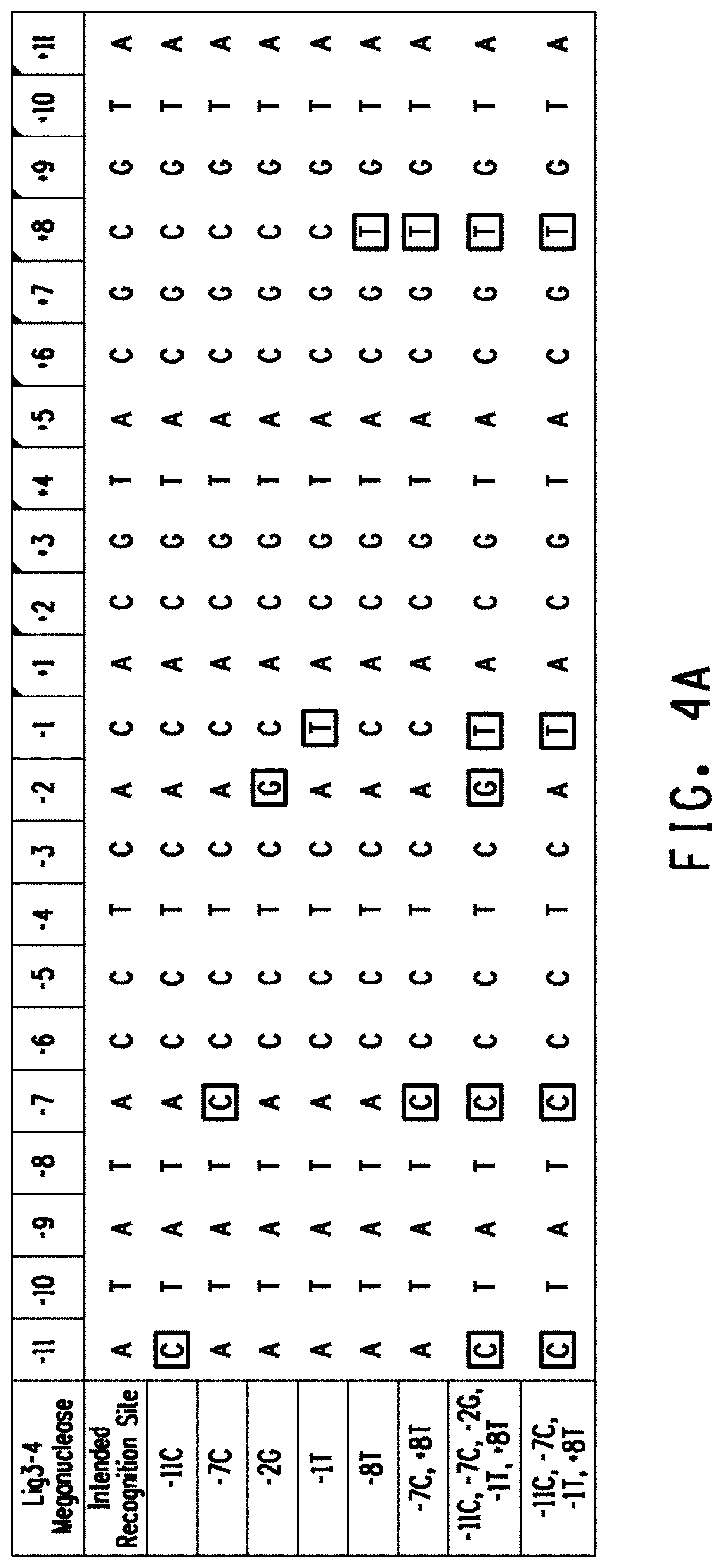
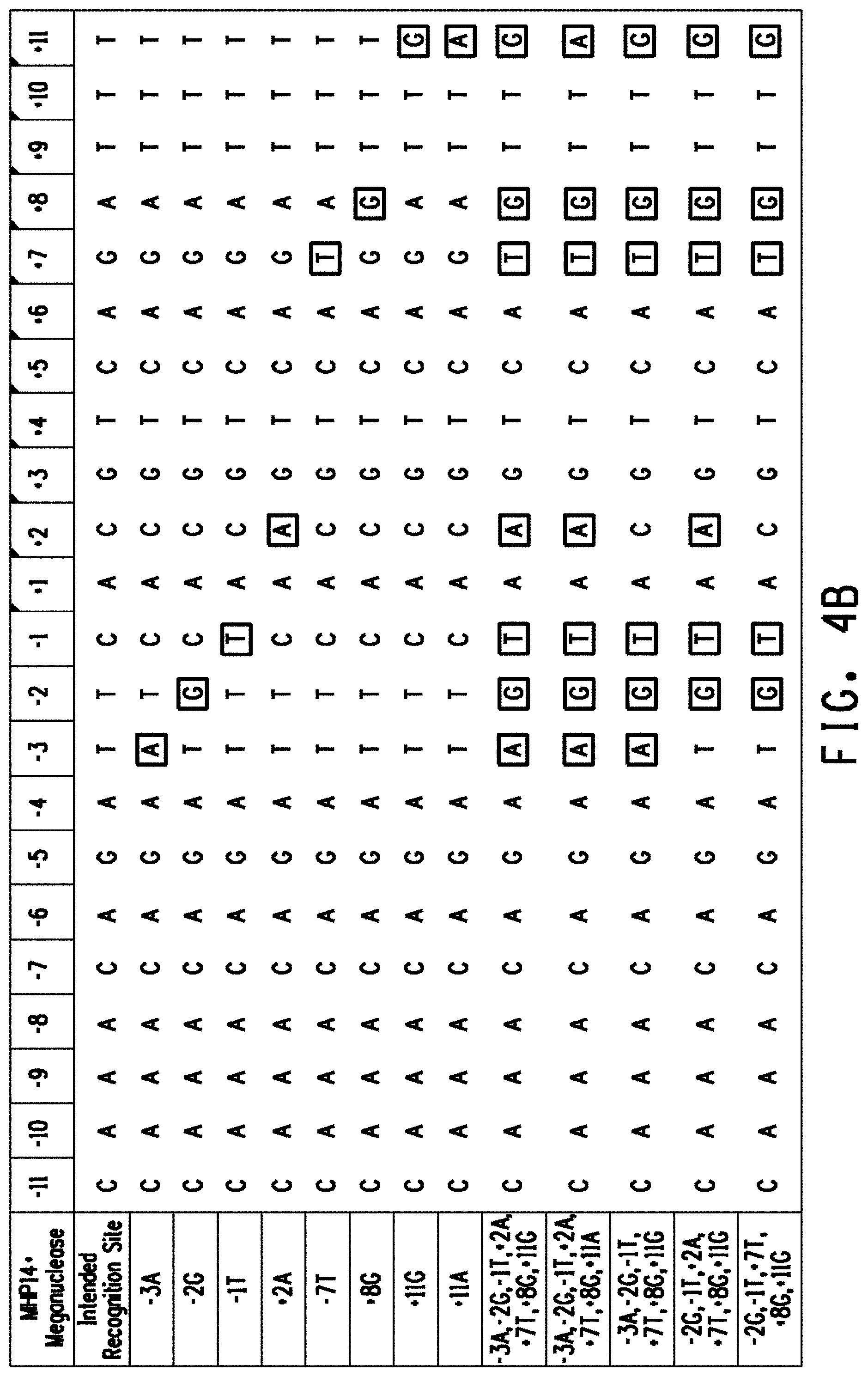
[0016] FIG. 4A Alignment of the intended recognition site for the LIG3-4 meganuclease and LIG3-4 variant recognition sites. Preferred off-nucleotides (outlined) were introduced into the LIG3-4 intended recognition site individually (resulting in the creation of variant recognition sites -11C, -7C, -2G, -1T, +8T corresponding to SEQ ID NOs: 15-19) and in combination (resulting in the creation of variant recognition sites corresponding to SEQ ID NOs:20-22). FIG. 4B Alignment of the intended recognition site for the MHP14+ meganuclease and MHP14+ variant recognition sites. Preferred off-nucleotides (outlined) were introduced into the MHP14+ intended recognition site individually (resulting in the creation of variant recognition sites -3A, -2G, -1T, +2A, +7T, +8G, +11G, +11A corresponding to SEQ ID NOs: 23-30) and in combination (resulting in the creation of variant recognition sites corresponding to SEQ ID NOs: 31-35).
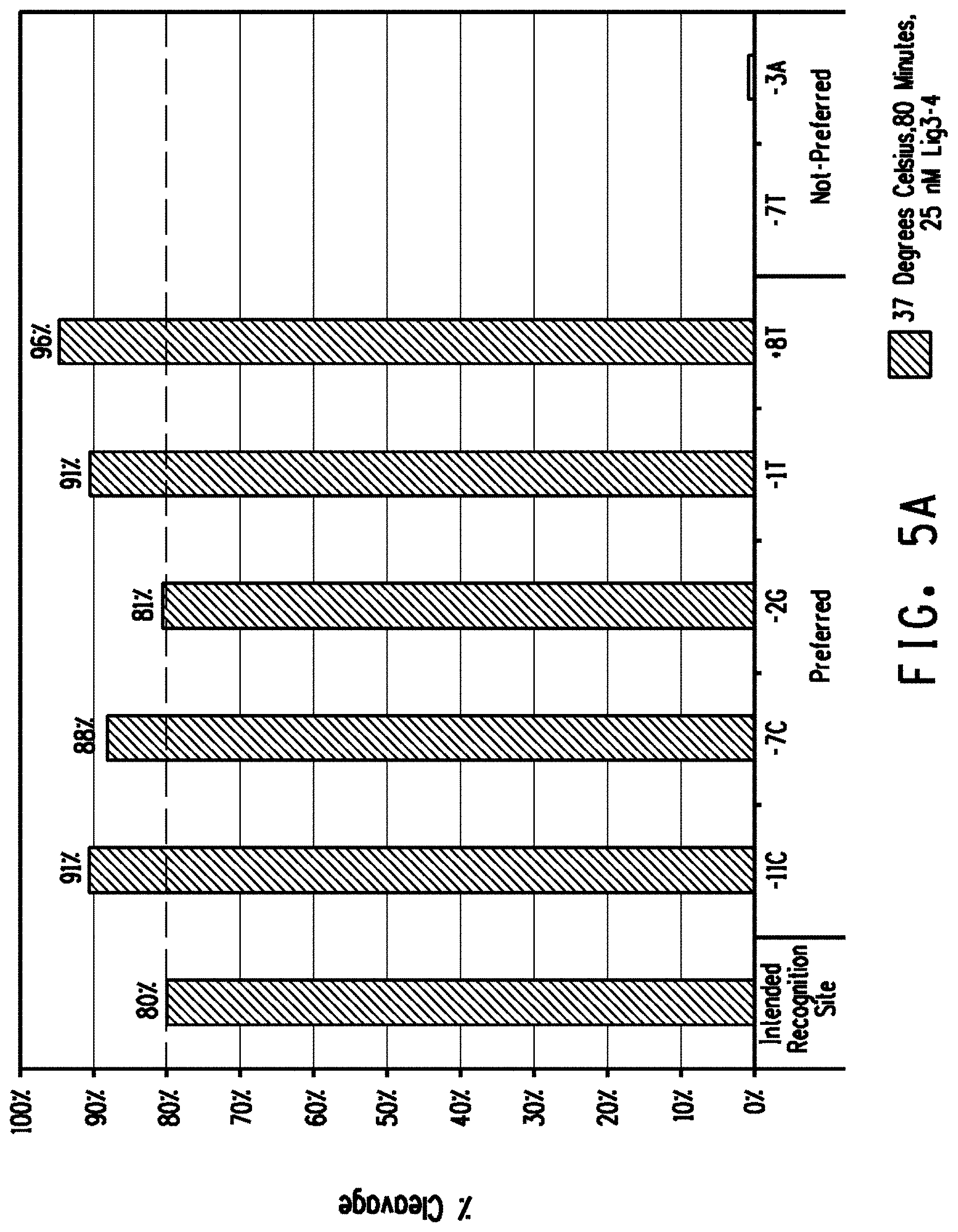
[0017] FIGS. 5A and 5B show the comparison of plasmid DNA cleavage activity between the intended recognition site and preferred off-nucleotides individually placed into the intended recognition site for the FIG. 5A Lig3-4 meganuclease and FIG. 5B MHP14+ meganuclease. The percent cleavage activity of the intended recognition site is marked with a dashed line. As a control, at least two bases not preferred in the percent DNA base composition of genomic variant recognition sites were also assayed.
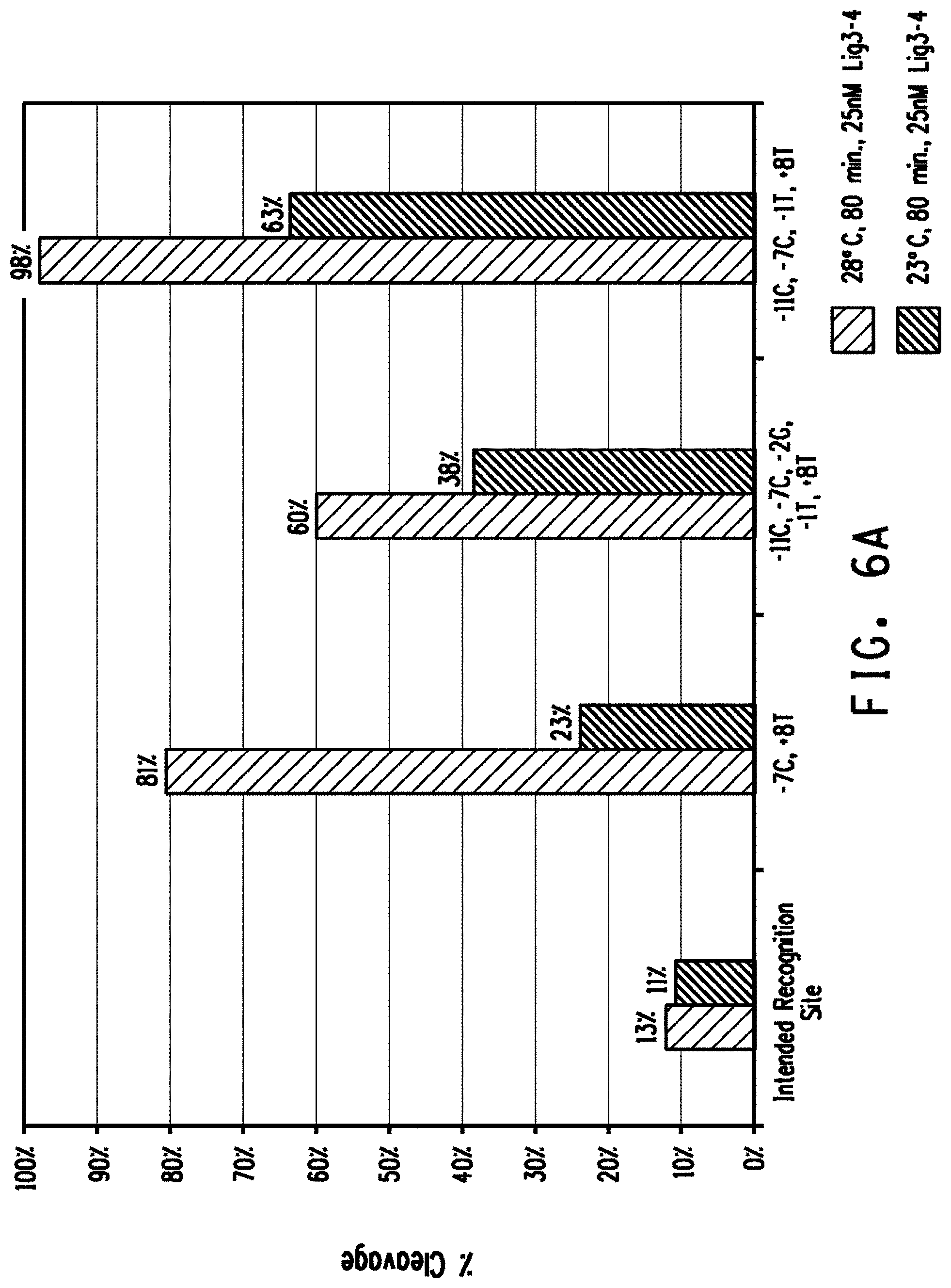
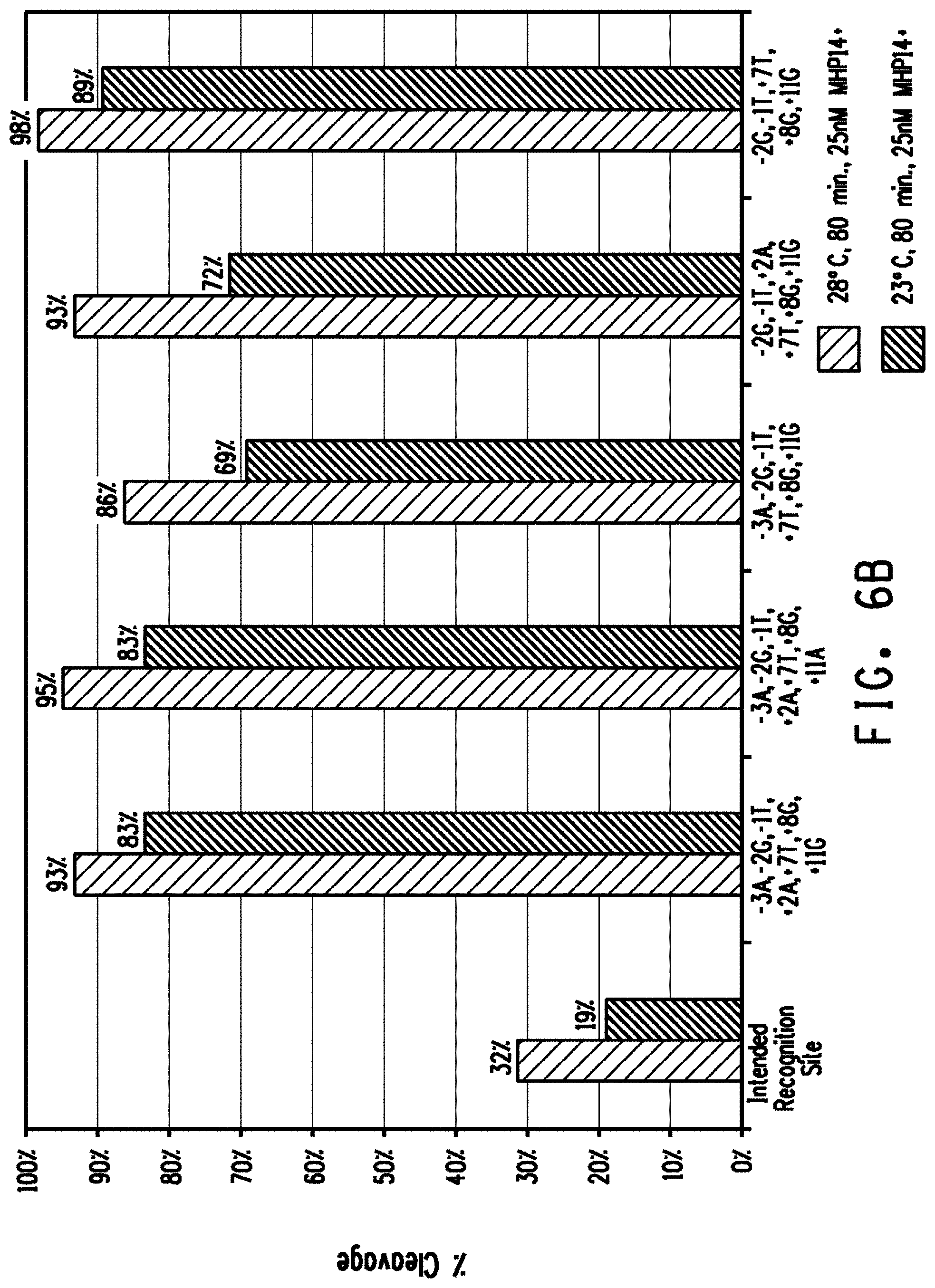
[0018] FIGS. 6A and 6B show a comparison of plasmid DNA cleavage activity between the intended recognition site and preferred off-nucleotides placed in combination into the intended recognition site for the FIG. 6A Lig3-4 meganuclease and FIG. 6B MHP14+ meganuclease.
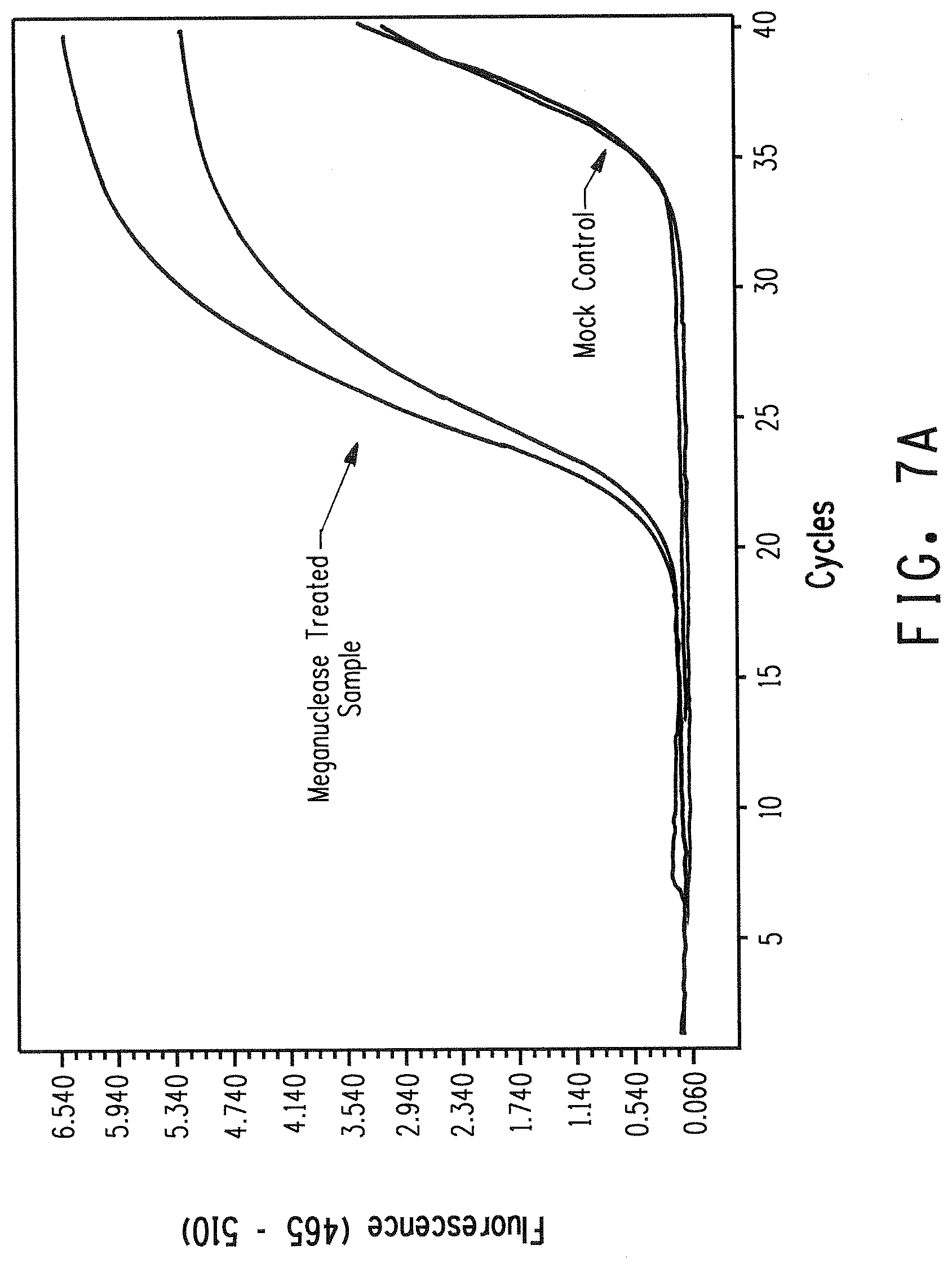
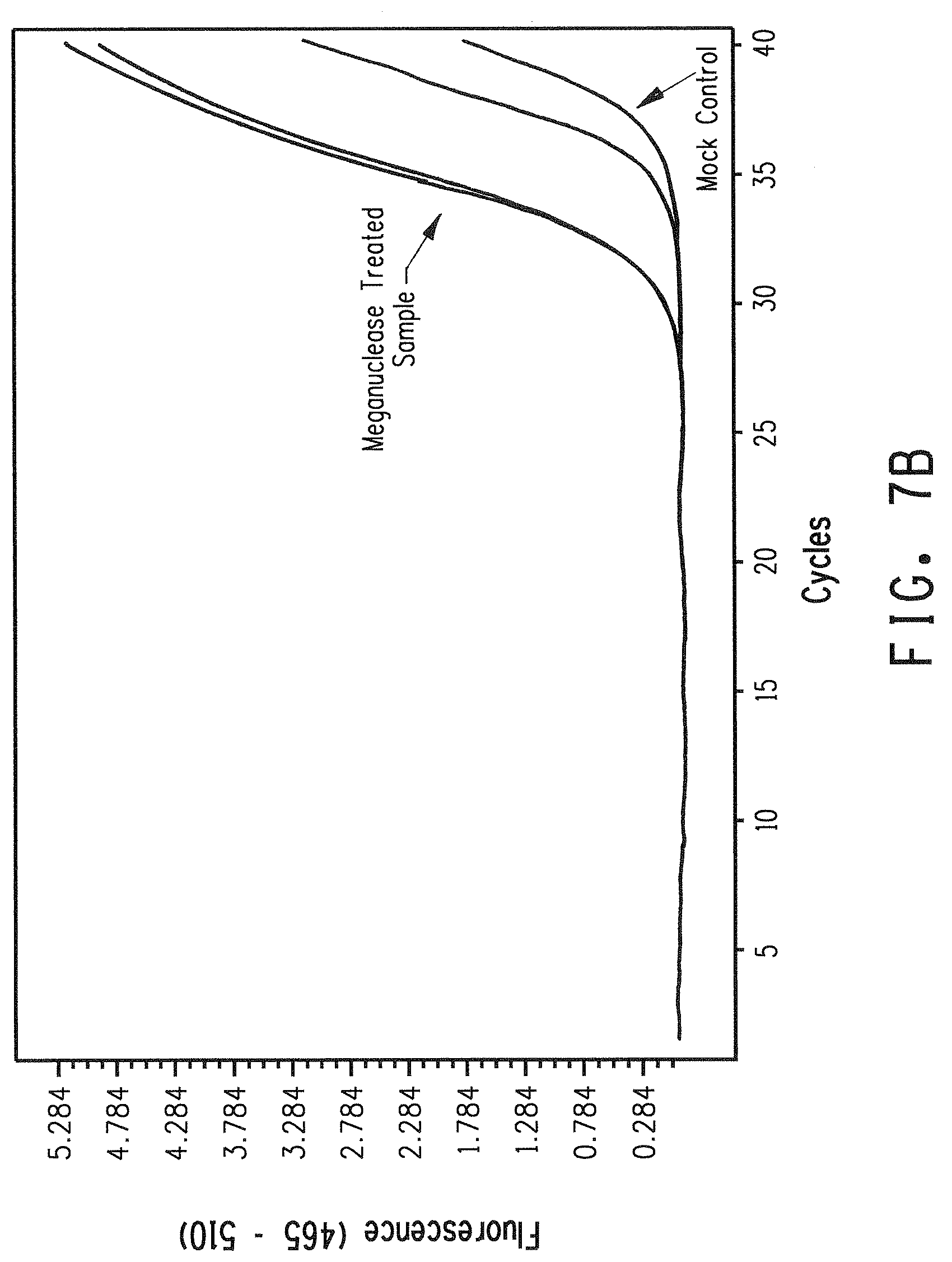
[0019] FIG. 7 shows a comparison of real-time PCR amplification plots from genomic variant meganuclease recognition site libraries created with either phosphorylated or non-phosphorylated biotinylated adapters containing a fully degenerate 4 nucleotide 3' overhang. FIG. 7A shows results for the non-phosphorylated adapter. FIG. 7B shows results for the phosphorylated adapter.
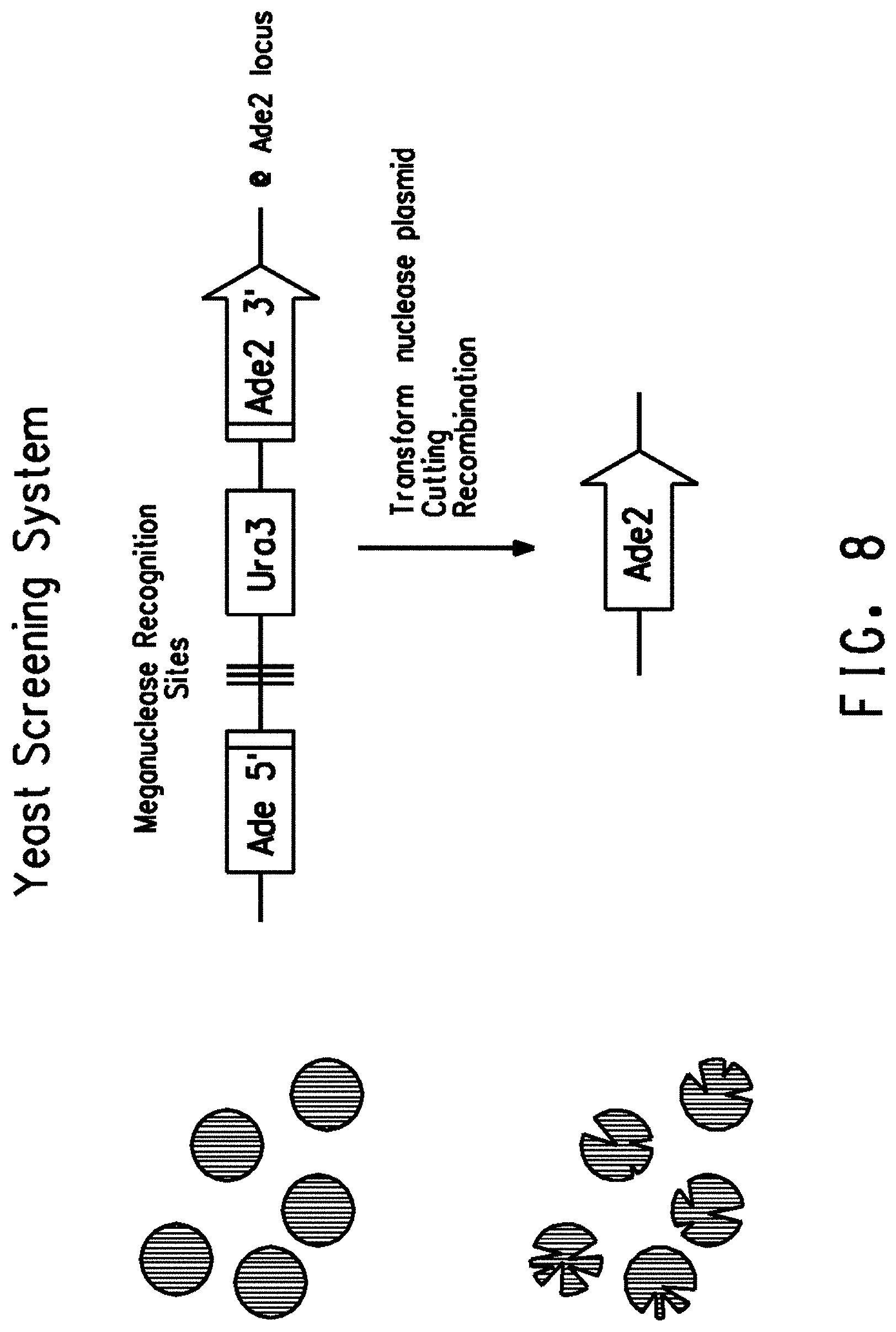
[0020] FIG. 8 shows a diagram representing the yeast screening system used to determine the meganuclease activity in yeast. Gene fragments corresponding to the first 1000 nucleotides of the yeast Ade2 coding sequence (Ade2 5' fragment) and the last 1011 nucleotides of the yeast Ade2 coding sequence (Ade2 3' fragment) were disrupted by a fragment including the yeast ura3 gene (Ura3) and meganuclease recognition sites for I-SceI.
[0021] FIG. 9 shows the numerical scale and corresponding white sectoring of yeast colonies used to quantify meganuclease activity. Since the sectoring phenotype is a qualitative measure of meganuclease activity, a 0-4 numerical scoring system was implemented. A score of 0 indicates that no white sectors (no meganuclease cutting) were observed; a score of 4 indicates completely white colonies (complete cutting of the recognition site); scores of 1-3 indicate intermediate white sectoring phenotypes (and intermediate degrees of recognition site cutting)
[0022] FIG. 10A shows a comparison of plasmid DNA cleavage activity between the MHP14+ intended recognition site (SEQ ID NO.: 14), a MHP14+ variant recognition site (SEQ ID NO: 11) that occurs naturally in the maize genome (labeled maize variant recognition site), and MHP14+ variant recognition sites (SEQ ID NOs: 31-35) that are not endogenous to the maize genome. FIG. 10B shows a comparison of the relative copy number of the MHP14+ variant recognition site of SEQ ID NO:11 and the MHP14+ intended recognition site (SEQ ID NO: 14) in mature maize embryos.
[0023] FIG. 11A is the map of plasmid PHP57712, and FIG. 11B is the map of plasmid PHP62552.
SEQUENCES
[0024] SEQ ID NO: 1 is the nucleotide sequence encoding the single chain LIG3-4 meganuclease fusion polypeptide.
[0025] SEQ ID NO: 2 is the amino acid sequence of the LIG3-4 meganuclease fusion polypeptide.
[0026] SEQ ID NO: 3 is the nucleotide sequence encoding the single chain MHP14+ meganuclease.
[0027] SEQ ID NO: 4 is the amino acid sequence of the MHP14+ meganuclease.
[0028] SEQ ID NO: 5 is the nucleotide sequence of a biotinylated, dephosphorylated adapter designed with a fully-degenerated 4 bp 3' overhang.
[0029] SEQ ID NO: 6 is the nucleotide sequence of recovery primer A.
[0030] SEQ ID NO: 7 is the nucleotide sequence of recovery primer B.
[0031] SEQ ID NO: 8 is the nucleotide sequence of an Illumina-compatibe adapter.
[0032] SEQ ID NO: 9 is the nucleotide sequence of a sequence tag.
[0033] SEQ ID NO: 10 is the nucleotide sequence of the complement sequence tag of SEQ ID NO: 9.
[0034] SEQ ID NO: 11 is the nucleotide sequence of the 5'-3' sequence shown in FIG. 2.
[0035] SEQ ID NO: 12 is the nucleotide sequence of the 3'-5' sequence shown in FIG. 2.
[0036] SEQ ID NO: 13 is the nucleotide sequence of the intended recognition site for the LIG3-4 meganuclease (also shown in FIG. 3 A and FIG. 4).
[0037] SEQ ID NO: 14 is the nucleotide sequence of the intended recognition site for the MHP14+ meganuclease (also shown in FIG. 3 B and FIG. 4).
[0038] SEQ ID NOs:15-22 are nucleotide sequences of variant recognition sites for the LIG3-4 meganuclease.
[0039] SEQ ID NOs:23-36 are nucleotide sequences of variant recognition sites for the MHP14+ meganuclease.
[0040] SEQ ID NO: 36 is the nucleotide sequence of the Ade2 yeast gene.
[0041] SEQ ID NO: 37 is the nucleotide sequence of the intended recognition site for the MS26 meganuclease
[0042] SEQ ID NO: 38 is the nucleotide sequence of plasmid PHP57712
[0043] SEQ ID NO: 39 is the nucleotide sequence of plasmid PHP62552
DETAILED DESCRIPTION OF THE INVENTION
[0044] The present inventions now will be described more fully hereinafter with reference to the accompanying drawings, in which some, but not all embodiments of the inventions are shown. Indeed, these inventions may be embodied in many different forms and should not be construed as limited to the embodiments set forth herein; rather, these embodiments are provided so that this disclosure will satisfy applicable legal requirements. Like numbers refer to like elements throughout.
[0045] Many modifications and other embodiments of the inventions set forth herein will come to mind to one skilled in the art to which these inventions pertain having the benefit of the teachings presented in the foregoing descriptions and the associated drawings. Therefore, it is to be understood that the inventions are not to be limited to the specific embodiments disclosed and that modifications and other embodiments are intended to be included within the scope of the appended claims. Although specific terms are employed herein, they are used in a generic and descriptive sense only and not for purposes of limitation.
[0046] All publications and patent applications mentioned in the specification are indicative of the level of those skilled in the art to which this invention pertains. All publications and patent applications are herein incorporated by reference to the same extent as if each individual publication or patent application was specifically and individually indicated to be incorporated by reference.
[0047] As used herein and in the appended claims, the singular forms "a", "an", and "the" include plural reference unless the context clearly dictates otherwise. Thus, for example, reference to "a plant" includes a plurality of such plants; reference to "a cell" includes one or more cells and equivalents thereof known to those skilled in the art, and so forth.
[0048] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which the invention pertains. Although any methods and materials similar or equivalent to those described herein can be used in the practice for testing of the present invention, specific examples of appropriate materials and methods are described herein.
[0049] In the context of this disclosure, a number of terms and abbreviations are used. The following definitions are provided.
[0050] As used herein, the terms "target site", "target sequence", "genomic target site" and "genomic target sequence" are used interchangeably herein and refer to a polynucleotide sequence in the genome of a plant cell or yeast cell that comprises a recognition site for a double-strand-break-inducing agent.
[0051] An "artificial target site" is a target sequence that has been introduced into the genome of an organism such as a plant or yeast. Such an artificial target sequence can be identical in sequence to an endogenous or native target sequence in the genome of the organism but can be located in a different position (i.e., a non-endogenous or non-native position) in the genome of the organism.
[0052] The terms "endogenous target sequence" and "native target sequence" are used interchangeable herein to refer to a target sequence that is endogenous or native to the genome of a host (such as a plant or yeast) and is at the endogenous or native position of that target sequence in the genome of the host (such as a plant or yeast).
[0053] The term "double-strand-break-inducing agent" as used herein refers to any nuclease which produces a double-strand break in the target sequence. Producing the double-strand break in a target sequence or other DNA can be referred to herein as "cutting" or "cleaving" the target sequence or other DNA.
[0054] The term "rare-cutting double-strand-break-inducing agent" as used herein refers to any nuclease which produces a double-strand break in a target sequence, but cuts at rare occasions (in contrast to restriction enzymes, for example) in the genome of an organism. Rare-cutting double-strand-break-inducing agents include but are not limited to endonucleases such as meganucleases, (US patent application 2332 and BB1990), zinc finger nucleases (Kim, Y. G., J. Cha, et al. (1996). "Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage); Cas endonucleases (WO2007/025097 application published Mar. 1, 2007) and TALENs (Christian, M., T. Cermak, et al. 2010. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186(2): 757-61). Cleavage by rare cutting endonucleases usually generates cohesive ends, with 3' overhangs for LAGLIDADG meganucleases (Chevalier, B. S. and B. L. Stoddard. 2001. Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res 29(18): 3757-74) and 5' overhangs for Zinc Finger nucleases (Smith, J., M. Bibikova, et al. 2000, Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res 28(17): 3361-9). FokI-based TALE-nucleases (TALENs) have a similar functional layout than Zinc-Finger Nucleases, with the Zinc-finger DNA binding domain being replaced by the TALE domain (Li, T., S. Huang, et al. 2011. TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res 39(1): 359-72; Christian, M., T. Cermak, et al. 2010). Cleavage with Cas endonucleases such as Cas9 endonucleases can result in blunt ends.
[0055] An "endonuclease" refers to an enzyme that cleaves the phosphodiester bond within a polynucleotide chain.
[0056] Endonucleases include restriction endonucleases that cleave DNA at specific sites without damaging the bases. Restriction endonucleases include Type I, Type II, Type III, and Type IV endonucleases, which further include subtypes. In the Type I and Type III systems, both the methylase and restriction activities are contained in a single complex.
[0057] Type I and Type III restriction endonucleases recognize specific recognition sites, but typically cleave at a variable position from the recognition site, which can be hundreds of base pairs away from the recognition site. In Type II systems the restriction activity is independent of any methylase activity, and cleavage typically occurs at specific sites within or near to the recognition site. Most Type II enzymes cut palindromic sequences, however Type IIa enzymes recognize non-palindromic recognition sites and cleave outside of the recognition site, Type IIb enzymes cut sequences twice with both sites outside of the recognition site, and Type IIs enzymes recognize an asymmetric recognition site and cleave on one side and at a defined distance of about 1-20 nucleotides from the recognition site. Type IV restriction enzymes target methylated DNA. Restriction enzymes are further described and classified, for example in the REBASE database (webpage at rebase.neb.com; Roberts et al., (2003) Nucleic Acids Res 31:418-20), Roberts et al., (2003) Nucleic Acids Res 31:1805-12, and Belfort et al., (2002) in Mobile DNA II, pp. 761-783, Eds. Craigie et al., (ASM Press, Washington, D.C.).
[0058] An "engineered rare-cutting double-strand-break-inducing agent" refers to any rare-cutting double-strand-break-inducing agent that is engineered (modified or derived) from its native form to specifically recognize and induce a double-strand break in the desired recognition site. Thus, an engineered rare-cutting double-strand-break-inducing agent can be derived from a native, naturally-occurring nuclease or it could be artificially created or synthesized. The modification of the nuclease can be as little as one nucleotide. In some embodiments, the engineered rare-cutting double-strand-break-inducing agent induces a double-strand break in a recognition site, wherein the recognition site was not a sequence that would have been recognized by a native (non-engineered or non-modified) rare-cutting double-strand-break-inducing agent. Producing a double-strand break in a recognition site or other DNA can be referred to herein as "cutting" or "cleaving" the recognition site or other DNA.
[0059] A "meganuclease" refers to a homing endonuclease, which like restriction endonucleases, bind and cut at a specific recognition site, however the recognition sites for meganucleases are typically longer, about 18 bp or more. In some embodiments of the invention, the meganuclease has been engineered (or modified) to cut a specific endogenous recognition sequence, wherein the endogenous target sequence prior to being cut by the engineered double-strand-break-inducing agent was not a sequence that would have been recognized by a native (non-engineered or non-modified) endonuclease.
[0060] A "meganuclease polypeptide" refers to a polypeptide having meganuclease activity and thus capable of producing a double-strand break in the recognition sequence.
[0061] Meganucleases have been classified into four families based on conserved sequence motifs, the families are the LAGLIDADG, GIY-YIG, H-N-H, and His-Cys box families. These motifs participate in the coordination of metal ions and hydrolysis of phosphodiester bonds. HEases are notable for their long recognition sites, and for tolerating some sequence polymorphisms in their DNA substrates. The naming convention for meganuclease is similar to the convention for other restriction endonuclease. Meganucleases are also characterized by prefix F-, I-, or PI- for enzymes encoded by free-standing open reading frames, introns, and inteins, respectively. For example, intron-, intein-, and freestanding gene encoded meganuclease from Saccharomyces cerevisiae are denoted I-SceI, PI-SceI, and F-SceII, respectively. Meganuclease domains, structure and function are known, see for example, Guhan and Muniyappa (2003) Crit Rev Biochem Mol Biol 38:199-248; Lucas et al., (2001) Nucleic Acids Res 29:960-9; Jurica and Stoddard, (1999) Cell Mol Life Sci 55:1304-26; Stoddard, (2006) Q Rev Biophys 38:49-95; and Moure et al., (2002) Nat Struct Biol 9:764. In some examples a naturally occurring variant, and/or engineered derivative meganuclease is used. Methods for modifying the kinetics, cofactor interactions, expression, optimal conditions, and/or recognition site specificity, and screening for activity are known, see for example, Epinat et al., (2003) Nucleic Acids Res 31:2952-62; Chevalier et al., (2002) Mol Cell 10:895-905; Gimble et al., (2003) Mol Biol 334:993-1008; Seligman et al., (2002) Nucleic Acids Res 30:3870-9; Sussman et al., (2004) J Mol Biol 342:31-41; Rosen et al., (2006) Nucleic Acids Res 34:4791-800; Chames et al., (2005) Nucleic Acids Res 33:e178; Smith et al., (2006) Nucleic Acids Res 34:e149; Gruen et al., (2002) Nucleic Acids Res 30:e29; Chen and Zhao, (2005) Nucleic Acids Res 33:e154; WO2005105989; WO2003078619; WO2006097854; WO2006097853; WO2006097784; and WO2004031346.
[0062] Any meganuclease can be used herein, including, but not limited to, I-SceI, I-SceII, I-SceIII, I-SceIV, I-SceV, I-SceVI, I-SceVII, I-CeuI, I-CeuAIIP, I-CreI, I-CrepsbIP, I-CrepsbIIP, I-CrepsbIIIP, I-CrepsbIVP, I-TliI, I-PpoI, PI-PspI, F-SceI, F-SceII, F-SuvI, F-TevI, F-TevII, I-AmaI, I-AniI, I-ChuI, I-CmoeI, I-CpaI, I-CpaII, I-CsmI, I-CvuI, I-CvuAIP, I-DdiI, I-DdiII, I-DirI, I-DmoI, I-HmuI, I-HmuII, I-HsNIP, I-LlaI, I-Msol, I-NaaI, I-NanI, I-NclIP, I-NgrIP, I-NitI, I-NjaI, I-Nsp236IP, I-PakI, I-PboIP, I-PcuIP, I-PcuAI, I-PcuVI, I-PgrIP, I-PobIP, I-PorI, I-PorIIP, I-PbpIP, I-SpBetaIP, I-ScaI, I-SexIP, I-SneIP, I-SpomI, I-SpomCP, I-SpomIP, I-SpomIIP, I-SquIP, I-Ssp6803I, I-SthPhiJP, I-SthPhiST3P, I-SthPhiSTe3bP, I-TdeIP, I-TevI, I-TevII, I-TevIII, I-UarAP, I-UarHGPAIP, I-UarHGPA13P, I-VinIP, I-ZbiIP, PI-MtuI, PI-MtuHIP PI-MtuHIIP, PI-PfuI, PI-PfuII, PI-PkoI, PI-PkoII, PI-Rma438121P, PI-SpBetaIP, PI-SceI, PI-TfuI, PI-TfuII, PI-ThyI, PI-TliI, PI-TliII, or any active variants or fragments thereof. In a specific embodiment, the engineered endonuclease is derived from I-Cre-I having the sequence set forth in SEQ ID NO: 15, 21 or 26 or an active variant or fragment thereof.
[0063] TAL effector nucleases are a new class of sequence-specific nucleases that can be used to make double-strand breaks at specific target sequences in the genome of a plant or other organism. TAL effector nucleases are created by fusing a native or engineered transcription activator-like (TAL) effector, or functional part thereof, to the catalytic domain of an endonuclease, such as, for example, FokI. The unique, modular TAL effector DNA binding domain allows for the design of proteins with potentially any given DNA recognition specificity. Thus, the DNA binding domains of the TAL effector nucleases can be engineered to recognize specific DNA target sites and thus, used to make double-strand breaks at desired target sequences. See, WO 2010/079430; Morbitzer et al. (2010) PNAS 10.1073/pnas.1013133107; Scholze & Boch (2010) Virulence 1:428-432; Christian et al. Genetics (2010) 186:757-761; Li et al. (2010) Nuc. Acids Res. (2010) doi:10.1093/nar/gkq704; and Miller et al. (2011) Nature Biotechnology 29:143-148; all of which are herein incorporated by reference.
[0064] As used herein, the term "Cas gene" refers to a gene that is generally coupled, associated or close to or in the vicinity of flanking CRISPR loci.
[0065] CRISPR loci (Clustered Regularly Interspaced Short Palindromic Repeats) (also known as SPIDRs--SPacer Interspersed Direct Repeats) constitute a family of recently described DNA loci. CRISPR loci consist of short and highly conserved DNA repeats (typically 24 to 40 bps, repeated from 1 to 140 times--also referred to as CRISPR-repeats) which are partially palindromic. The repeated sequences (usually specific to a species) are interspaced by variable sequences of constant length (typically 20 to 58 by depending on the CRISPR locus (WO2007/024097 published Mar. 1, 2007).
[0066] CRISPR loci were first recognized in E. coli (Ishino et al. (1987) J. Bacterial. 169:5429-5433; Nakata et al. (1989) J. Bacterial. 171:3553-3556). Similar interspersed short sequence repeats have been identified in Haloferax mediterranei, Streptococcus pyogenes, Anabaena, and Mycobacterium tuberculosis (Groenen et al. (1993) Mol. Microbiol. 10:1057-1065; Hoe et al. (1999) Emerg. Infect. Dis. 5:254-263; Masepohl et al. (1996) Biochim. Biophys. Acta 1307:26-30; Mojica et al. (1995) Mol. Microbiol. 17:85-93). The CRISPR loci differ from other SSRs by the structure of the repeats, which have been termed short regularly spaced repeats (SRSRs) (Janssen et al. (2002) OMICS J. Integ. Biol. 6:23-33; Mojica et al. (2000) Mol. Microbiol. 36:244-246). The repeats are short elements that occur in clusters, that are always regularly spaced by variable sequences of constant length (Mojica et al. (2000) Mol. Microbiol. 36:244-246).
[0067] The terms "Cas gene", "CRISPR-associated (Cas) gene" are used interchangeably herein. A comprehensive review of the Cas protein family is presented in Haft et al. (2005) Computational Biology, PLoS Comput Biol 1(6): e60. doi:10.1371/journal.pcbi.0010060. As described therein, 41 CRISPR-associated (Cas) gene families are described, in addition to the four previously known gene families. It shows that CRISPR systems belong to different classes, with different repeat patterns, sets of genes, and species ranges. The number of Cas genes at a given CRISPR locus can vary between species.
[0068] As used herein, the term "Cas endonuclease" refers to a Cas protein encoded by a Cas gene, wherein said Cas protein is capable of introducing a double strand break into a DNA target sequence. The Cas endonuclease unwinds the DNA duplex in close proximity of the genomic target site and cleaves both DNA strands upon recognition of a target sequence by a guide RNA, but only if the correct protospacer-adjacent motif (PAM) is approximately oriented at the 3' end of the target sequence.
[0069] As used herein, the term "guide RNA" refers to a synthetic fusion of two RNA molecules, a crRNA (CRISPR RNA) comprising a variable targeting domain, and a tracrRNA. In one embodiment, the guide RNA comprises a variable targeting domain of 12 to 30 nucleotide sequences and a RNA fragment that can interact with a Cas endonuclease.
[0070] The term "variable targeting domain" refers to a nucleotide sequence 5-prime of the GUUUU sequence motif in the guide RNA, that is complementary to one strand of a double strand DNA target site in the genome of a plant cell, plant or seed. In one embodiment, the variable targeting domain is 12 to 30 nucleotides in length.
[0071] In one embodiment, the guide RNA and Cas endonuclease are capable of forming a complex that enables the Cas endonuclease to introduce a double strand break at a DNA target site.
[0072] As used herein, the term "recognition site" refers to a DNA sequence at which a double-strand break is induced in a cell genome by a rare-cutting double-strand-break-inducing agent. The terms "recognition site", "recognition sequence" are used interchangeably herein. The recognition site can be an endogenous site in a host (such as a yeast or plant) genome, or alternatively, the recognition site can be heterologous to the host (yeast or plant) and thereby not be naturally occurring in the genome, or the recognition site can be found in a heterologous genomic location compared to where it occurs in nature.
[0073] As used herein, the term "endogenous recognition site" refers to a rare-cutting double-strand-break-inducing agent recognition site that is endogenous or native to the genome of a host (such as a plant or yeast) and is located at the endogenous or native position of that recognition site in the genome of the host (such as a plant or yeast). The length of the recognition site can vary, and includes, for example, recognition sites that are at least 4, 6, 8, 10, 12, 14, 16, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70 or more nucleotides in length. It is further possible that the recognition site could be palindromic, that is, the sequence on one strand reads the same in the opposite direction on the complementary strand. The nick/cleavage site could be within the recognition sequence or the nick/cleavage site could be outside of the recognition sequence. In another variation, the cleavage could occur at nucleotide positions immediately opposite each other to produce a blunt end cut or, in other cases, the incisions could be staggered to produce single-stranded overhangs, also called "sticky ends", which can be either 5' overhangs, or 3' overhangs.
[0074] As used herein, the term "intended recognition site" refers to the recognition sequence to which an engineered rare-cutting double-strand-break-inducing agent, such as an engineered meganuclease, was directed to specifically recognize and induce a double-strand break. In one embodiment, the rare-cutting double-strand-break-inducing agent is a LIG3-4 engineered meganuclease (SEQ ID NO: 2) which was designed to recognize the intended recognition sequence of SEQ ID NO: 13 (US patent publication 2009-0133152 A1, published May 21, 2009). In another embodiment, the rare-cutting double-strand-break-inducing agent is a MHP14+ engineered meganuclease (SEQ ID NO: 4) which was designed to recognize the intended recognition sequence of SEQ ID NO: 14 (in U.S. patent application Ser. No. 13/427,138 filed on Mar. 22, 2012).
[0075] As used herein, the term "variant recognition site" refers to a variant nucleotide sequence that comprises at least one base nucleotide alteration when compared to the intended recognition site to which an engineered rare-cutting double-strand-break-inducing agent such as a meganuclease, was directed to specifically recognize and induce a double-strand break. Such "alteration" include, for example: (i) replacement of at least one nucleotide, (ii) a deletion of at least one nucleotide, (iii) an insertion of at least one nucleotide, or (iv) any combination of (i)-(iii). Active variants and fragments of the recognition can comprise at least 65%, 70%, 75%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence identity to the given recognition sequence, wherein the active variants retain biological activity and hence are capable of being recognized and cleaved by an endonuclease. Variant recognition sites can comprise at least one (1) and up to 2, 3, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 off-nucleotide preferences. In one embodiment, variant recognition sites are non-endogenous to the host genome, such variant recognition sites include, but are not limited to, the maize variant recognition sites shown in FIG. 4 (SEQ ID NOs: 15-22 and SEQ ID NOs: 23-35). In another embodiment, the variant recognition sites are present in the host genome (referred to as genomic variant recognition sites) or endogenous to the host genome, such as plant or yeast genomes. In some embodiments, the variant recognition sites can be introduced into a plant genome by the mutagenesis of an endogenous genomic sequence. Methods for the site-specific mutagenesis of genomic DNA are known in the art, and include those described, for example in U.S. Pat. Nos. 5,565,350, 5,731,181, and 6,870,075. Other methods include the use of zinc finger nucleases, such as those methods described in U.S. Patent Publication 20050208489.
[0076] A "genomic variant recognition site" refers to a variant recognition site of a rare-cutting double-strand-break-inducing agent, such as a meganuclease, that is endogenous to the genome of an organism (such as a plant or yeast). One example of a variant recognition site that is endogenous to the maize genome is SEQ ID NO: 11.
[0077] The term "preferred off-nucleotides" or "off-nucleotide preferences" can be used interchangeably and refers to nucleotides that are located at the same position relative to the nucleotides of the intended recognition site, but are more prevalent in the identified genomic variant recognition sites (see for example the prevalence for a +8T (80%) compared with an intended recognition site +8C (13%) in FIG. 3A). In most instances, the preferred off-nucleotide when placed into the intended recognition site is cleaved at a higher percentage than the intended recognition site (see for example +8T (96% cleavage) compared to +8C (80% cleavage in FIG. 5A).
[0078] In one embodiment, the intended recognition sequence of the LIG3-4 engineered meganuclease comprises SEQ ID NO: 13, whereas the variant recognition site of the LIG3-4 engineered meganuclease comprises SEQ ID NO: 15, 16, 17, 18, 19, 20, 21, or 22. In another embodiment, the intended recognition sequence of the MHP14+ engineered meganuclease comprises SEQ ID NO: 14, whereas the variant recognition site of the MHP14+ engineered meganuclease comprises SEQ ID NO: 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, or 35.
[0079] A "variant recognition site locus" is the position on a chromosome comprising the variable recognition site. Preferably, the variant recognition site locus is within 0, 50, 100, 200, 300, 400, 500, 600, 700, 800, 900 or 1000 base pairs of the variant recognition site.
[0080] The term "meganuclease activity" as used herein refers to the ability of a meganuclease to produce a double-strand break at a desired recognition sequence and thus retain double-strand-break-inducing activity. Producing the double-strand break in a recognition sequence or other DNA can be referred to herein as "cutting" or "cleaving" the recognition sequence or other DNA.
[0081] Assays for meganuclease activity are known and generally measure the overall activity and specificity of the meganuclease on DNA substrates containing the recognition site. These DNA substrates include but are not limited to genomic DNA and plasmid DNA. For example the meganuclease activity can be measured in-vitro as described herein in Example 3 and Example 9. In short, time-course digestions can be carried out at 37.degree. C., 28.degree. C., and 23.degree. C. (or any temperature ranging between 37.degree. C., 36.degree. C., 35.degree. C., 34.degree. C., 33.degree. C., 32.degree. C., 31.degree. C., 30.degree. C., 29.degree. C., 28.degree. C., 27.degree. C., 26.degree. C., 25.degree. C., 24.degree. C. and 23.degree. C.), on plasmid or genomic DNA containing a meganuclease recognition site and the % digestion of each sample (also referred to as % cleavage or to as % loss of meganuclease recognition sites) (indicative of meganuclease activity) can be determined by real-time PCR.
[0082] Meganuclease activity can also be measured using a yeast screening assay as described herein (FIGS. 8 and 9 and Example 16). In short, yeast cells with a functional Ade2 gene are white, whereas those lacking Ade2 function exhibit red pigmentation due to accumulation of a metabolite earlier in the adenine biosynthetic pathway resulting in red colonies with white sectors. The degree of white sectoring, sometimes extending to entire colonies, indicates the amount of meganuclease cutting activity. Since the sectoring phenotype is a qualitative measure of meganuclease activity, a 0-4 numerical scoring system was implemented. As shown in FIG. 3, a score of 0 indicates that no white sectors (no meganuclease cutting) were observed; a score of 4 indicates completely white colonies (complete cutting of the recognition site); scores of 1-3 indicate intermediate white sectoring phenotypes (and intermediate degrees of recognition site cutting.
[0083] Furthermore, meganuclease activity can be measured in-planta by determining the Target Site (TS) mutation rate. Target site mutation rate is defined as: (number of events with target site modification/total number events)*100%.
[0084] An "increased" or an "increased" activity are used interchangeably herein. An "increased" or "increased" meganuclease activity comprises any statistically significant increase in the activity of the parental meganuclease polypeptide as determined through any activity assays described herein.
[0085] The meganuclease can be provided via a polynucleotide encoding the endonuclease. Such a polynucleotide encoding an endonuclease can be modified to substitute codons having a higher frequency of usage in a plant, as compared to the naturally occurring polynucleotide sequence. For example the polynucleotide encoding the meganuclease can be modified to substitute codons having a higher frequency of usage in a maize or soybean plant, as compared to the naturally occurring polynucleotide sequence.
[0086] A "control meganuclease" or "reference meganuclease" can be used interchangeably and refers to any meganuclease to which a variant meganuclease is compared to. Control meganucleases can include, but are not limited to, parental or corresponding meganucleases or any wild-type I-Cre1 type meganucleases.
[0087] Numbering of an amino acid or nucleotide polymer, such any one of the meganucleases of the invention, corresponds to numbering of a selected amino acid polymer or nucleic acid when the position of a given monomer component (amino acid residue, incorporated nucleotide, etc.) of the polymer corresponds to the same residue position in a selected reference polypeptide or polynucleotide.
[0088] As used herein, a "genomic region of interest" is a segment of a chromosome in the genome of a plant that is desirable for introducing a polynucleotide of interest or trait of interest. The genomic region of interest can include, for example, one or more polynucleotides of interest. Generally, a genomic region of interest of the present invention comprises a segment of chromosome that is 0-15 centi-morgan (cM).
[0089] As used herein, a "polynucleotide of interest" within a genomic region of interest is any coding and/or non-coding portion of the genomic region of interest including, but not limited to, a transgene, a native gene, a mutated gene, and a genetic marker such as, for example, a single nucleotide polymorphism (SNP) marker and a simple sequence repeat (SSR) marker.
[0090] As used herein, "physically linked," "in physical linkage", and "genetically linked" are used to refer to any two or more genes, transgenes, native genes, mutated genes, alterations, target sites, markers, and the like that are part of the same DNA molecule or chromosome.
[0091] As used herein, an "isolated" polynucleotide or polypeptide, or biologically active portion thereof, is substantially or essentially free from components that normally accompany or interact with the polynucleotide or polypeptide as found in its naturally occurring environment. Thus, an isolated or purified polynucleotide or polypeptide is substantially free of other cellular material or culture medium when produced by recombinant techniques, or substantially free of chemical precursors or other chemicals when chemically synthesized. Optimally, an "isolated" polynucleotide is free of sequences (optimally protein encoding sequences) that naturally flank the polynucleotide (i.e., sequences located at the 5' and 3' ends of the polynucleotide) in the genomic DNA of the organism from which the polynucleotide is derived. For example, in various embodiments, the isolated polynucleotide can contain less than about 5 kb, 4 kb, 3 kb, 2 kb, 1 kb, 0.5 kb, or 0.1 kb of nucleotide sequence that naturally flank the polynucleotide in genomic DNA of the cell from which the polynucleotide is derived. A polypeptide that is substantially free of cellular material includes preparations of polypeptides having less than about 30%, 20%, 10%, 5%, or 1% (by dry weight) of contaminating protein. When the polypeptide of the invention or biologically active portion thereof is recombinantly produced, optimally culture medium represents less than about 30%, 20%, 10%, 5%, or 1% (by dry weight) of chemical precursors or non-protein-of-interest chemicals.
[0092] As used herein, polynucleotide or polypeptide is "recombinant" when it is artificial or engineered, or derived from an artificial or engineered protein or nucleic acid. For example, a polynucleotide that is inserted into a vector or any other heterologous location, e.g., in a genome of a recombinant organism, such that it is not associated with nucleotide sequences that normally flank the polynucleotide as it is found in nature is a recombinant polynucleotide. A polypeptide expressed in vitro or in vivo from a recombinant polynucleotide is an example of a recombinant polypeptide. Likewise, a polynucleotide sequence that does not appear in nature, for example, a variant of a naturally occurring gene is recombinant.
[0093] A "subsequence" or "fragment" is any portion of an entire sequence.
[0094] Sequence Comparisons
[0095] The following terms are used to describe the sequence relationships between two or more polynucleotides or polypeptides: (a) "reference sequence", (b) "comparison window", (c) "sequence identity", and, (d) "percent sequence identity."
[0096] (a) As used herein, "reference sequence" is a defined sequence used as a basis for sequence comparison. A reference sequence may be a subset or the entirety of a specified sequence; for example, as a segment of a full-length cDNA or gene sequence, or the complete cDNA or gene sequence or protein sequence.
[0097] (b) As used herein, "comparison window" makes reference to a contiguous and specified segment of a polypeptide sequence, wherein the polypeptide sequence in the comparison window may comprise additions or deletions (i.e., gaps) compared to the reference sequence (which does not comprise additions or deletions) for optimal alignment of the two polypeptides. Generally, the comparison window is at least 5, 10, 15, or 20 contiguous amino acid in length, or it can be 30, 40, 50, 100, or longer. Those of skill in the art understand that to avoid a high similarity to a reference sequence due to inclusion of gaps in the polypeptide sequence a gap penalty is typically introduced and is subtracted from the number of matches.
[0098] Methods of alignment of sequences for comparison are well known in the art. Thus, the determination of percent sequence identity between any two sequences can be accomplished using a mathematical algorithm. Non-limiting examples of such mathematical algorithms are the algorithm of Myers and Miller (1988) CABIOS 4:11-17; the local alignment algorithm of Smith et al. (1981) Adv. Appl. Math. 2:482; the global alignment algorithm of Needleman and Wunsch (1970) J. Mol. Biol. 48:443-453; the search-for-local alignment method of Pearson and Lipman (1988) Proc. Natl. Acad. Sci. 85:2444-2448; the algorithm of Karlin and Altschul (1990) Proc. Natl. Acad. Sci. USA 872264, modified as in Karlin and Altschul (1993) Proc. Natl. Acad. Sci. USA 90:5873-5877. Computer implementations of these mathematical algorithms can be utilized for comparison of sequences to determine sequence identity. Such implementations include, but are not limited to: CLUSTAL in the PC/Gene program (available from Intelligenetics, Mountain View, Calif.); the ALIGN program (Version 2.0) and GAP, BESTFIT, BLAST, FASTA, and TFASTA in the GCG Wisconsin Genetics Software Package, Version 10 (available from Accelrys Inc., 9685 Scranton Road, San Diego, Calif., USA). Alignments using these programs can be performed using the default parameters. The CLUSTAL program is well described by Higgins et al. (1988) Gene 73:237-244 (1988); Higgins et al. (1989) CABIOS 5:151-153; Corpet et al. (1988) Nucleic Acids Res. 16:10881-90; Huang et al. (1992) CABIOS 8:155-65; and Pearson et al. (1994) Meth. Mol. Biol. 24:307-331. The ALIGN program is based on the algorithm of Myers and Miller (1988) supra. A PAM120 weight residue table, a gap length penalty of 12, and a gap penalty of 4 can be used with the ALIGN program when comparing amino acid sequences. The BLAST programs of Altschul et al (1990) J. Mol. Biol. 215:403 are based on the algorithm of Karlin and Altschul (1990) supra. BLAST nucleotide searches can be performed with the BLASTN program, score=100, wordlength=12, to obtain nucleotide sequences homologous to a nucleotide sequence encoding a protein of the invention. BLAST protein searches can be performed with the BLASTX program, score=50, wordlength=3, to obtain amino acid sequences homologous to a protein or polypeptide of the invention. BLASTP protein searches can be performed using default parameters. See, blast.ncbi.nlm.nih.gov/Blast.cgi.
[0099] Sequence alignments and percent similarity calculations may be determined using the Megalign program of the LASARGENE bioinformatics computing suite (DNASTAR Inc., Madison, Wis.) or using the AlignX program of the Vector NTI bioinformatics computing suite (Invitrogen, Carlsbad, Calif.). Multiple alignment of the sequences are performed using the Clustal method of alignment (Higgins and Sharp, CABIOS 5:151-153 (1989)) with the default parameters (GAP PENALTY=10, GAP LENGTH PENALTY=10). Default parameters for pairwise alignments and calculation of percent identity of protein sequences using the Clustal method are KTUPLE=1, GAP PENALTY=3, WINDOW=5 and DIAGONALS SAVED=5. For nucleic acids these parameters are GAP PENALTY=10, GAP LENGTH PENALTY=10, KTUPLE=2, GAP PENALTY=5, WINDOW=4 and DIAGONALS SAVED=4. A "substantial portion" of an amino acid or nucleotide sequence comprises enough of the amino acid sequence of a polypeptide or the nucleotide sequence of a gene to afford putative identification of that polypeptide or gene, either by manual evaluation of the sequence by one skilled in the art, or by computer-automated sequence comparison and identification using algorithms such as BLAST (Altschul, S. F. et al., J. Mol. Biol. 215:403-410 (1993)) and Gapped Blast (Altschul, S. F. et al., Nucleic Acids Res. 25:3389-3402 (1997)). BLASTN refers to a BLAST program that compares a nucleotide query sequence against a nucleotide sequence database.
[0100] "Gene" refers to a nucleic acid fragment that expresses a specific protein, including regulatory sequences preceding (5' non-coding sequences) and following (3' non-coding sequences) the coding sequence. "Native gene" refers to a gene as found in nature with its own regulatory sequences. "Chimeric gene" or "recombinant expression construct", which are used interchangeably, refers to any gene that is not a native gene, comprising regulatory and coding sequences that are not found together in nature. Accordingly, a chimeric gene may comprise regulatory sequences and coding sequences that are derived from different sources, or regulatory sequences and coding sequences derived from the same source, but arranged in a manner different than that found in nature. "Endogenous gene" refers to a native gene in its natural location in the genome of an organism. A "foreign" gene refers to a gene not normally found in the host organism, but that is introduced into the host organism by gene transfer. Foreign genes can comprise native genes inserted into a non-native organism, or chimeric genes. A "transgene" is a gene that has been introduced into the genome by a transformation procedure.
[0101] "Coding sequence" refers to a DNA sequence which codes for a specific amino acid sequence. "Regulatory sequences" refer to nucleotide sequences located upstream (5' non-coding sequences), within, or downstream (3' non-coding sequences) of a coding sequence, and which influence the transcription, RNA processing or stability, or translation of the associated coding sequence. Regulatory sequences may include, but are not limited to, promoters, translation leader sequences, introns, and polyadenylation recognition sequences.
[0102] "Codon degeneracy" refers to divergence in the genetic code permitting variation of the nucleotide sequence without affecting the amino acid sequence of an encoded polypeptide. Accordingly, the instant invention relates to any nucleic acid fragment comprising a nucleotide sequence that encodes all or a substantial portion of the amino acid sequences set forth herein. The skilled artisan is well aware of the "codon-bias" exhibited by a specific host cell in usage of nucleotide codons to specify a given amino acid. Therefore, when synthesizing a nucleic acid fragment for increased expression in a host cell, it is desirable to design the nucleic acid fragment such that its frequency of codon usage approaches the frequency of preferred codon usage of the host cell.
[0103] As used herein, "sequence identity" or "identity" in the context of two polynucleotides or polypeptide sequences makes reference to the residues in the two sequences that are the same when aligned for maximum correspondence over a specified comparison window. When percentage of sequence identity is used in reference to proteins it is recognized that residue positions which are not identical often differ by conservative amino acid substitutions, where amino acid residues are substituted for other amino acid residues with similar chemical properties (e.g., charge or hydrophobicity). When sequences differ in conservative substitutions, the percent sequence identity may be adjusted upwards to correct for the conservative nature of the substitution. Sequences that differ by such conservative substitutions are said to have "sequence similarity" or "similarity". Means for making this adjustment are well known to those of skill in the art. Typically this involves scoring a conservative substitution as a partial rather than a full mismatch, thereby increasing the percent sequence identity. Thus, for example, where an identical amino acid is given a score of 1 and a non-conservative substitution is given a score of zero, a conservative substitution is given a score between zero and 1. The scoring of conservative substitutions is calculated, e.g., as implemented in the program PC/GENE (Intelligenetics, Mountain View, Calif.).
[0104] As used herein, "percent sequence identity" means the value determined by comparing two aligned sequences over a comparison window, wherein the portion of the polynucleotide sequence in the comparison window may comprise additions or deletions (i.e., gaps) as compared to the reference sequence (which does not comprise additions or deletions) for optimal alignment of the two sequences. The percentage is calculated by determining the number of positions at which the identical nucleic acid base or amino acid residue occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the window of comparison, and multiplying the result by 100 to yield the percent sequence identity.
Polynucleotide Constructs
[0105] Provided herein are polynucleotides or nucleic acid molecules comprising the variant recognition sites for rare-cutting double-strand-break-inducing agents or any active variants or fragments thereof. The terms "polynucleotide," "polynucleotide sequence," "nucleic acid sequence," and "nucleic acid fragment" are used interchangeably herein. These terms encompass nucleotide sequences and the like. The use of the term "polynucleotide" is not intended to limit the present invention to polynucleotides comprising DNA. Those of ordinary skill in the art will recognize that polynucleotides can comprise ribonucleotides and combinations of ribonucleotides and deoxyribonucleotides. Such deoxyribonucleotides and ribonucleotides include both naturally occurring molecules and synthetic analogues. The polynucleotides of the invention also encompass all forms of sequences including, but not limited to, single-stranded forms, double-stranded forms, hairpins, stem-and-loop structures, and the like.
[0106] Further provided are recombinant polynucleotides comprising the various rare-cutting double-strand-break-inducing agents such as engineered meganucleases. The terms "recombinant polynucleotide", "recombinant nucleotide", "recombinant DNA" and "recombinant DNA construct" are used interchangeably herein. A recombinant construct comprises an artificial or heterologous combination of nucleic acid sequences, e.g., regulatory and coding sequences that are not found together in nature. For example, a transfer cassette can comprise restriction sites and a heterologous polynucleotide of interest. In other embodiments, a recombinant construct may comprise regulatory sequences and coding sequences that are derived from different sources, or regulatory sequences and coding sequences derived from the same source, but arranged in a manner different than that found in nature. Such a construct may be used by itself or may be used in conjunction with a vector. If a vector is used, then the choice of vector is dependent upon the method that will be used to transform host cells as is well known to those skilled in the art. For example, a plasmid vector can be used. The skilled artisan is well aware of the genetic elements that must be present on the vector in order to successfully transform, select and propagate host cells comprising any of the isolated nucleic acid fragments provided herein. The skilled artisan will also recognize that different independent transformation events will result in different levels and patterns of expression (Jones et al., EMBO J. 4:2411-2418 (1985); De Almeida et al., Mol. Gen. Genetics 218:78-86 (1989)), and thus that multiple events must be screened in order to obtain lines displaying the desired expression level and pattern. Such screening may be accomplished by Southern analysis of DNA, Northern analysis of mRNA expression, immunoblotting analysis of protein expression, or phenotypic analysis, among others.
[0107] The meganuclease polynucleotides disclosed herein can be provided in expression cassettes for expression in the plant of interest. The cassette can include 5' and 3' regulatory sequences operably linked to a meganuclease polynucleotide or active variant or fragment thereof. "Operably linked" is intended to mean a functional linkage between two or more elements. For example, an operable linkage between a polynucleotide of interest and a regulatory sequence (i.e., a promoter) is a functional link that allows for expression of the polynucleotide of interest. Operably linked elements may be contiguous or non-contiguous. When used to refer to the joining of two protein coding regions, by operably linked is intended that the coding regions are in the same reading frame. The cassette may additionally contain at least one additional gene to be cotransformed into the organism. Alternatively, the additional gene(s) can be provided on multiple expression cassettes. Such an expression cassette is provided with a plurality of restriction sites and/or recombination sites for insertion of the meganuclease polynucleotide or active variant or fragment thereof to be under the transcriptional regulation of the regulatory regions. The expression cassette may additionally contain selectable marker genes.
[0108] The expression cassette can include in the 5'-3' direction of transcription, a transcriptional and translational initiation region (i.e., a promoter), a meganuclease polynucleotide or active variant or fragment thereof, and a transcriptional and translational termination region (i.e., termination region) functional in plants. The regulatory regions (i.e., promoters, transcriptional regulatory regions, and translational termination regions) and/or the meganuclease polynucleotide or active variant or fragment thereof may be native/analogous to the host cell or to each other. Alternatively, the regulatory regions and/or the meganuclease polynucleotide of or active variant or fragment thereof may be heterologous to the host cell or to each other.
[0109] As used herein, "heterologous" in reference to a sequence is a sequence that originates from a foreign species, or, if from the same species, is substantially modified from its native form in composition and/or genomic locus by deliberate human intervention. For example, a promoter operably linked to a heterologous polynucleotide is from a species different from the species from which the polynucleotide was derived, or, if from the same/analogous species, one or both are substantially modified from their original form and/or genomic locus, or the promoter is not the native promoter for the operably linked polynucleotide.
[0110] While it may be optimal to express the sequences using heterologous promoters, the native promoter sequences may be used. Such constructs can change expression levels of the meganuclease polynucleotide in the plant or plant cell. Thus, the phenotype of the plant or plant cell can be altered.
[0111] The termination region may be native with the transcriptional initiation region, may be native with the operably linked meganuclease polynucleotide or active variant or fragment thereof, may be native with the plant host, or may be derived from another source (i.e., foreign or heterologous) to the promoter, the meganuclease polynucleotide or active fragment or variant thereof, the plant host, or any combination thereof. Convenient termination regions are available from the Ti-plasmid of A. tumefaciens, such as the octopine synthase and nopaline synthase termination regions. See also Guerineau et al. (1991) Mol. Gen. Genet. 262:141-144; Proudfoot (1991) Cell 64:671-674; Sanfacon et al. (1991) Genes Dev. 5:141-149; Mogen et al. (1990) Plant Cell 2:1261-1272; Munroe et al. (1990) Gene 91:151-158; Ballas et al. (1989) Nucleic Acids Res. 17:7891-7903; and Joshi et al. (1987) Nucleic Acids Res. 15:9627-9639.
[0112] Where appropriate, the polynucleotides may be optimized for increased expression in the transformed plant. That is, the polynucleotides can be synthesized using plant-preferred codons for improved expression. See, for example, Campbell and Gowri (1990) Plant Physiol. 92:1-11 for a discussion of host-preferred codon usage. Methods are available in the art for synthesizing plant-preferred genes. See, for example, U.S. Pat. Nos. 5,380,831, and 5,436,391, and Murray et al. (1989) Nucleic Acids Res. 17:477-498, herein incorporated by reference.
[0113] Additional sequence modifications are known to enhance gene expression in a cellular host. These include elimination of sequences encoding spurious polyadenylation signals, exon-intron splice site signals, transposon-like repeats, and other such well-characterized sequences that may be deleterious to gene expression. The G-C content of the sequence may be adjusted to levels average for a given cellular host, as calculated by reference to known genes expressed in the host cell. When possible, the sequence is modified to avoid predicted hairpin secondary mRNA structures.
[0114] The expression cassettes may additionally contain 5' leader sequences. Such leader sequences can act to enhance translation. Translation leaders are known in the art and include: picornavirus leaders, for example, EMCV leader (Encephalomyocarditis 5' noncoding region) (Elroy-Stein et al. (1989) Proc. Natl. Acad. Sci. USA 86:6126-6130); potyvirus leaders, for example, TEV leader (Tobacco Etch Virus) (Gallie et al. (1995) Gene 165(2):233-238), MDMV leader (Maize Dwarf Mosaic Virus) (Virology 154:9-20), and human immunoglobulin heavy-chain binding protein (BiP) (Macejak et al. (1991) Nature 353:90-94); untranslated leader from the coat protein mRNA of alfalfa mosaic virus (AMV RNA 4) (Jobling et al. (1987) Nature 325:622-625); tobacco mosaic virus leader (TMV) (Gallie et al. (1989) in Molecular Biology of RNA, ed. Cech (Liss, New York), pp. 237-256); and maize chlorotic mottle virus leader (MCMV) (Lommel et al. (1991) Virology 81:382-385. See also, Della-Cioppa et al. (1987) Plant Physiol. 84:965-968.
[0115] In preparing the expression cassette, the various DNA fragments may be manipulated, so as to provide for the DNA sequences in the proper orientation and, as appropriate, in the proper reading frame. Toward this end, adapters or linkers may be employed to join the DNA fragments or other manipulations may be involved to provide for convenient restriction sites, removal of superfluous DNA, removal of restriction sites, or the like. For this purpose, in vitro mutagenesis, primer repair, restriction, annealing, resubstitutions, e.g., transitions and transversions, may be involved.
[0116] A number of promoters can be used to express the various meganuclease sequence disclosed herein, including the native promoter of the polynucleotide sequence of interest. The promoters can be selected based on the desired outcome. Such promoters include, for example, constitutive, tissue-preferred, or other promoters for expression in plants.
[0117] Constitutive promoters include, for example, the core promoter of the Rsyn7 promoter and other constitutive promoters disclosed in WO 99/43838 and U.S. Pat. No. 6,072,050; the core CaMV 35S promoter (Odell et al. (1985) Nature 313:810-812); rice actin (McElroy et al. (1990) Plant Cell 2:163-171); ubiquitin (Christensen et al. (1989) Plant Mol. Biol. 12:619-632 and Christensen et al. (1992) Plant Mol. Biol. 18:675-689); pEMU (Last et al. (1991) Theor. Appl. Genet. 81:581-588); MAS (Velten et al. (1984) EMBO J. 3:2723-2730); ALS promoter (U.S. Pat. No. 5,659,026), and the like. Other constitutive promoters include, for example, U.S. Pat. Nos. 5,608,149; 5,608,144; 5,604,121; 5,569,597; 5,466,785; 5,399,680; 5,268,463; 5,608,142; and 6,177,611.
[0118] Tissue-preferred promoters can be utilized to target enhanced meganuclease expression within a particular plant tissue. Tissue-preferred promoters include those described in Yamamoto et al. (1997) Plant J. 12(2):255-265; Kawamata et al. (1997) Plant Cell Physiol. 38(7):792-803; Hansen et al. (1997) Mol. Gen Genet. 254(3):337-343; Russell et al. (1997) Transgenic Res. 6(2):157-168; Rinehart et al. (1996) Plant Physiol. 112(3):1331-1341; Van Camp et al. (1996) Plant Physiol. 112(2):525-535; Canevascini et al. (1996) Plant Physiol. 112(2):513-524; Yamamoto et al. (1994) Plant Cell Physiol. 35(5):773-778; Lam (1994) Results Probl. Cell Differ. 20:181-196; Orozco et al. (1993) Plant Mol Biol. 23(6):1129-1138; Matsuoka et al. (1993) Proc Natl. Acad. Sci. USA 90(20):9586-9590; and Guevara-Garcia et al. (1993) Plant J. 4(3):495-505. Such promoters can be modified, if necessary, for weak expression.
[0119] Leaf-preferred promoters are known in the art. See, for example, Yamamoto et al. (1997) Plant J. 12(2):255-265; Kwon et al. (1994) Plant Physiol. 105:357-67; Yamamoto et al. (1994) Plant Cell Physiol. 35(5):773-778; Gotor et al. (1993) Plant J. 3:509-18; Orozco et al. (1993) Plant Mol. Biol. 23(6):1129-1138; and Matsuoka et al. (1993) Proc. Natl. Acad. Sci. USA 90(20):9586-9590.
[0120] Synthetic promoters can be used to express meganuclease sequences or biologically active variants and fragments thereof.
[0121] The expression cassette can also comprise a selectable marker gene for the selection of transformed cells. Selectable marker genes are utilized for the selection of transformed cells or tissues. Marker genes include genes encoding antibiotic resistance, such as those encoding neomycin phosphotransferase II (NEO) and hygromycin phosphotransferase (HPT), as well as genes conferring resistance to herbicidal compounds, such as glyphosate, glufosinate ammonium, bromoxynil, sulfonylureas, dicamba, and 2,4-dichlorophenoxyacetate (2,4-D). Additional selectable markers include phenotypic markers such as .beta.-galactosidase and fluorescent proteins such as green fluorescent protein (GFP) (Su et al. (2004) Biotechnol Bioeng 85:610-9 and Fetter et al. (2004) Plant Cell 16:215-28), cyan florescent protein (CYP) (Bolte et al. (2004) J. Cell Science 117:943-54 and Kato et al. (2002) Plant Physiol 129:913-42), and yellow florescent protein (PhiYFP.TM. from Evrogen, see, Bolte et al. (2004) J. Cell Science 117:943-54). For additional selectable markers, see generally, Yarranton (1992) Curr. Opin. Biotech. 3:506-511; Christopherson et al. (1992) Proc. Natl. Acad. Sci. USA 89:6314-6318; Yao et al. (1992) Cell 71:63-72; Reznikoff (1992) Mol. Microbiol. 6:2419-2422; Barkley et al. (1980) in The Operon, pp. 177-220; Hu et al. (1987) Cell 48:555-566; Brown et al. (1987) Cell 49:603-612; Figge et al. (1988) Cell 52:713-722; Deuschle et al. (1989) Proc. Natl. Acad. Aci. USA 86:5400-5404; Fuerst et al. (1989) Proc. Natl. Acad. Sci. USA 86:2549-2553; Deuschle et al. (1990) Science 248:480-483; Gossen (1993) Ph.D. Thesis, University of Heidelberg; Reines et al. (1993) Proc. Natl. Acad. Sci. USA 90:1917-1921; Labow et al. (1990) Mol. Cell. Biol. 10:3343-3356; Zambretti et al. (1992) Proc. Natl. Acad. Sci. USA 89:3952-3956; Baim et al. (1991) Proc. Natl. Acad. Sci. USA 88:5072-5076; Wyborski et al. (1991) Nucleic Acids Res. 19:4647-4653; Hillenand-Wissman (1989) Topics Mol. Struc. Biol. 10:143-162; Degenkolb et al. (1991) Antimicrob. Agents Chemother. 35:1591-1595; Kleinschnidt et al. (1988) Biochemistry 27:1094-1104; Bonin (1993) Ph.D. Thesis, University of Heidelberg; Gossen et al. (1992) Proc. Natl. Acad. Sci. USA 89:5547-5551; Oliva et al. (1992) Antimicrob. Agents Chemother. 36:913-919; Hlavka et al. (1985) Handbook of Experimental Pharmacology, Vol. 78 (Springer-Verlag, Berlin); Gill et al. (1988) Nature 334:721-724. Such disclosures are herein incorporated by reference. The above list of selectable marker genes is not meant to be limiting. Any selectable marker gene can be used in the present invention.
Method of Introducing
[0122] The rare-cutting double-strand-break-inducing agent, such as a meganuclease may be introduced by any means known in the art. For example, a cell, yeast or plant having the intended or variant recognition site in its genome is provided. The meganuclease may be transiently expressed or the polypeptide itself can be directly provided to the cell. Alternatively, a nucleotide sequence capable of expressing the meganuclease may be stably integrated into the genome of the plant. In the presence of the corresponding intended or variant recognition site and the meganuclease, a donor DNA can be inserted into the transformed plant's genome. Alternatively, the different components may be brought together by sexually crossing transformed plants. Thus a sequence encoding a meganuclease and/or intended or variant recognition site can be sexually crossed to one another to allow each component of the system to be present in a single plant. The meganuclease may be under the control of a constitutive or inducible promoter. Such promoters of interest are discussed in further detail elsewhere herein.
[0123] Various methods can be used to introduce a sequence of interest such as, any of the rare-cutting double-strand-break-inducing agents into a plant or plant part. "Introducing" is intended to mean presenting to the plant, plant cell or plant part the polynucleotide or polypeptide in such a manner that the sequence gains access to the interior of a cell of the plant. The methods of the invention do not depend on a particular method for introducing a sequence into a plant or plant part, only that the polynucleotide or polypeptides gains access to the interior of at least one cell of the plant. Methods for introducing polynucleotide or polypeptides into plants are known in the art including, but not limited to, stable transformation methods, transient transformation methods, and virus-mediated methods.
[0124] "Stable transformation" is intended to mean that the nucleotide construct introduced into a plant integrates into the genome of the plant and is capable of being inherited by the progeny thereof. "Transient transformation" is intended to mean that a polynucleotide is introduced into the plant and does not integrate into the genome of the plant or a polypeptide is introduced into a plant.
[0125] Transformation protocols as well as protocols for introducing polypeptides or polynucleotide sequences into plants may vary depending on the type of plant or plant cell, i.e., monocot or dicot, targeted for transformation. Suitable methods of introducing polypeptides and polynucleotides into plant cells include microinjection (Crossway et al. (1986) Biotechniques 4:320-334), electroporation (Riggs et al. (1986) Proc. Natl. Acad. Sci. USA 83:5602-5606, Agrobacterium-mediated transformation (U.S. Pat. Nos. 5,563,055 and 5,981,840), direct gene transfer (Paszkowski et al. (1984) EMBO J. 3:2717-2722), and ballistic particle acceleration (see, for example, U.S. Pat. Nos. 4,945,050; 5,879,918; 5,886,244; and, 5,932,782; Tomes et al. (1995) in Plant Cell, Tissue, and Organ Culture: Fundamental Methods, ed. Gamborg and Phillips (Springer-Verlag, Berlin); McCabe et al. (1988) Biotechnology 6:923-926); and Lec1 transformation (WO 00/28058). Also see Weissinger et al. (1988) Ann. Rev. Genet. 22:421-477; Sanford et al. (1987) Particulate Science and Technology 5:27-37 (onion); Christou et al. (1988) Plant Physiol. 87:671-674 (soybean); McCabe et al. (1988) Bio/Technology 6:923-926 (soybean); Finer and McMullen (1991) In Vitro Cell Dev. Biol. 27P:175-182 (soybean); Singh et al. (1998) Theor. Appl. Genet. 96:319-324 (soybean); Datta et al. (1990) Biotechnology 8:736-740 (rice); Klein et al. (1988) Proc. Natl. Acad. Sci. USA 85:4305-4309 (maize); Klein et al. (1988) Biotechnology 6:559-563 (maize); U.S. Pat. Nos. 5,240,855; 5,322,783; and, 5,324,646; Klein et al. (1988) Plant Physiol. 91:440-444 (maize); Fromm et al. (1990) Biotechnology 8:833-839 (maize); Hooykaas-Van Slogteren et al. (1984) Nature (London) 311:763-764; U.S. Pat. No. 5,736,369 (cereals); Bytebier et al. (1987) Proc. Natl. Acad. Sci. USA 84:5345-5349 (Liliaceae); De Wet et al. (1985) in The Experimental Manipulation of Ovule Tissues, ed. Chapman et al. (Longman, N.Y.), pp. 197-209 (pollen); Kaeppler et al. (1990) Plant Cell Reports 9:415-418 and Kaeppler et al. (1992) Theor. Appl. Genet. 84:560-566 (whisker-mediated transformation); D'Halluin et al. (1992) Plant Cell 4:1495-1505 (electroporation); Li et al. (1993) Plant Cell Reports 12:250-255 and Christou and Ford (1995) Annals of Botany 75:407-413 (rice); Osjoda et al. (1996) Nature Biotechnology 14:745-750 (maize via Agrobacterium tumefaciens); all of which are herein incorporated by reference.
[0126] In specific embodiments, the rare-cutting double-strand-break-inducing agent sequence, such as a meganuclease sequence, or active variant or fragments thereof can be provided to a yeast cell or plant using a variety of transient transformation methods. Such transient transformation methods include, but are not limited to, the introduction of the meganuclease protein or active variants and fragments thereof directly into a yeast cell or plant. Such methods include, for example, microinjection or particle bombardment. See, for example, Crossway et al. (1986) Mol Gen. Genet. 202:179-185; Nomura et al. (1986) Plant Sci. 44:53-58; Hepler et al. (1994) Proc. Natl. Acad. Sci. 91: 2176-2180 and Hush et al. (1994) The Journal of Cell Science 107:775-784, all of which are herein incorporated by reference.
[0127] Generally, such methods involve incorporating a nucleotide construct of the invention within a DNA or RNA molecule. It is recognized that the an meganuclease sequence may be initially synthesized as part of a viral polyprotein, which later may be processed by proteolysis in vivo or in vitro to produce the desired recombinant protein. Further, it is recognized that promoters of the invention also encompass promoters utilized for transcription by viral RNA polymerases. Methods for introducing polynucleotides into plants and expressing a protein encoded therein, involving viral DNA or RNA molecules, are known in the art. See, for example, U.S. Pat. Nos. 5,889,191, 5,889,190, 5,866,785, 5,589,367, 5,316,931, and Porta et al. (1996) Molecular Biotechnology 5:209-221; herein incorporated by reference.
[0128] Methods are known in the art for the targeted insertion of a polynucleotide at a specific location in the plant genome. In one embodiment, the insertion of the polynucleotide at a desired genomic location is achieved using a site-specific recombination system. See, for example, WO99/25821, WO99/25854, WO99/25840, WO99/25855, and WO99/25853, all of which are herein incorporated by reference. Briefly, the polynucleotide of the invention can be contained in transfer cassette flanked by two non-recombinogenic recombination sites. The transfer cassette is introduced into a plant having stably incorporated into its genome a target site which is flanked by two non-recombinogenic recombination sites that correspond to the sites of the transfer cassette. An appropriate recombinase is provided and the transfer cassette is integrated at the target site. The polynucleotide of interest is thereby integrated at a specific chromosomal position in the plant genome. Other methods to target polynucleotides are set forth in WO 2009/114321 (herein incorporated by reference), which describes "custom" meganucleases produced to modify plant genomes, in particular the genome of maize. See, also, Gao et al. (2010) Plant Journal 1:176-187.
[0129] The cells that have been transformed may be grown into plants in accordance with conventional ways. See, for example, McCormick et al. (1986) Plant Cell Reports 5:81-84. These plants may then be grown, and either pollinated with the same transformed strain or different strains, and the resulting progeny having constitutive expression of the desired phenotypic characteristic identified. Two or more generations may be grown to ensure that expression of the desired phenotypic characteristic is stably maintained and inherited and then seeds harvested to ensure expression of the desired phenotypic characteristic has been achieved. In this manner, the present invention provides transformed seed (also referred to as "transgenic seed") having a polynucleotide of the invention, for example, an expression cassette of the invention, stably incorporated into their genome.
[0130] As used herein, "primers" are isolated polynucleotides that are annealed to a complementary target DNA strand by nucleic acid hybridization to form a hybrid between the primer and the target DNA strand, then extended along the target DNA strand by a polymerase, e.g., a DNA polymerase. Primer pairs of the invention refer to their use for amplification of a target polynucleotide, e.g., by the polymerase chain reaction (PCR) or other conventional nucleic-acid amplification methods. "PCR" or "polymerase chain reaction" is a technique used for the amplification of specific DNA segments (see, U.S. Pat. Nos. 4,683,195 and 4,800,159; herein incorporated by reference.
[0131] Probes and primers are of sufficient nucleotide length to bind to the target DNA sequence and specifically detect and/or identify a polynucleotide encoding a meganuclease polypeptide or active variant or fragment thereof as describe elsewhere herein. It is recognized that the hybridization conditions or reaction conditions can be determined by the operator to achieve this result. This length may be of any length that is of sufficient length to be useful in a detection method of choice. Such probes and primers can hybridize specifically to a target sequence under high stringency hybridization conditions. Probes and primers according to embodiments of the present invention may have complete DNA sequence identity of contiguous nucleotides with the target sequence, although probes differing from the target DNA sequence and that retain the ability to specifically detect and/or identify a target DNA sequence may be designed by conventional methods. Accordingly, probes and primers can share about 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or greater sequence identity or complementarity to the target polynucleotide.
[0132] Methods for preparing and using probes and primers are described, for example, in Molecular Cloning: A Laboratory Manual, 2.sup.nd ed, vol. 1-3, ed. Sambrook et al., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. 1989 (hereinafter, "Sambrook et al., 1989"); Current Protocols in Molecular Biology, ed. Ausubel et al., Greene Publishing and Wiley-Interscience, New York, 1992 (with periodic updates) (hereinafter, "Ausubel et al., 1992"); and Innis et al., PCR Protocols: A Guide to Methods and Applications, Academic Press: San Diego, 1990. PCR primer pairs can be derived from a known sequence, for example, by using computer programs intended for that purpose such as the PCR primer analysis tool in Vector NTI version 10 (Invitrogen); PrimerSelect (DNASTAR Inc., Madison, Wis.); and Primer (Version 0.5.COPYRGT., 1991, Whitehead Institute for Biomedical Research, Cambridge, Mass.). Additionally, the sequence can be visually scanned and primers manually identified using guidelines known to one of skill in the art.
Yeast and Plants
[0133] Yeast, plants, plant cells, plant parts and seeds, and grain having the variant recognition sequences for rare-cutting double-strand-break-inducing agents, such as meganucleases disclosed herein, are provided. In specific embodiments, the yeast, plants and/or plant parts have stably incorporated at least one heterologous variant recognition sequence disclosed herein or an active variant or fragment thereof. Thus, yeast, plants, plant cells, plant parts and seed are provided which comprise at least one variant recognition sequence of any one of SEQ ID NOs: 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 or any combination thereof, or a biologically active fragment and/or variant thereof. In specific embodiments, the variant recognition sequences expresses increased cleavage activity towards the rare-cutting double-strand-break-inducing agent.
[0134] As used herein, the term plant includes plant cells, plant protoplasts, plant cell tissue cultures from which plants can be regenerated, plant calli, plant clumps, and plant cells that are intact in plants or parts of plants such as embryos, pollen, ovules, seeds, leaves, flowers, branches, fruit, kernels, ears, cobs, husks, stalks, roots, root tips, anthers, and the like. Grain is intended to mean the mature seed produced by commercial growers for purposes other than growing or reproducing the species. Progeny, variants, and mutants of the regenerated plants are also included within the scope of the invention, provided that these parts comprise the introduced polynucleotides.
[0135] A transformed plant or transformed plant cell provided herein is one in which genetic alteration, such as transformation, has been affected as to a gene of interest, or is a plant or plant cell which is descended from a plant or cell so altered and which comprises the alteration. A "transgene" is a gene that has been introduced into the genome by a transformation procedure. Accordingly, a "transgenic plant" is a plant that contains a transgene, whether the transgene was introduced into that particular plant by transformation or by breeding; thus, descendants of an originally-transformed plant are encompassed by the definition. A "control" or "control plant" or "control plant cell" provides a reference point for measuring changes in phenotype of the subject plant or plant cell. A control plant or plant cell may comprise, for example: (a) a wild-type plant or cell, i.e., of the same genotype as the starting material for the genetic alteration which resulted in the subject plant or cell; (b) a plant or plant cell of the same genotype as the starting material but which has been transformed with a null construct (i.e., with a construct which does not express the transgene, such as a construct comprising a marker gene); (c) a plant or plant cell which is a non-transformed segregant among progeny of a subject plant or plant cell; (d) a plant or plant cell genetically identical to the subject plant or plant cell but which is not exposed to conditions or stimuli that would induce expression of the transgene; or (e) the subject plant or plant cell itself, under conditions in which the construct is not expressed.
[0136] Plant cells that have been transformed to express a meganuclease provided herein can be grown into whole plants. The regeneration, development, and cultivation of plants from single plant protoplast transformants or from various transformed explants is well known in the art. See, for example, McCormick et al. (1986) Plant Cell Reports 5:81-84; Weissbach and Weissbach, In: Methods for Plant Molecular Biology, (Eds.), Academic Press, Inc. San Diego, Calif., (1988). This regeneration and growth process typically includes the steps of selection of transformed cells, culturing those individualized cells through the usual stages of embryonic development through the rooted plantlet stage. Transgenic embryos and seeds are similarly regenerated. The resulting transgenic rooted shoots are thereafter planted in an appropriate plant growth medium such as soil. Preferably, the regenerated plants are self-pollinated to provide homozygous transgenic plants. Otherwise, pollen obtained from the regenerated plants is crossed to seed-grown plants of agronomically important lines. Conversely, pollen from plants of these important lines is used to pollinate regenerated plants. Two or more generations may be grown to ensure that expression of the desired phenotypic characteristic is stably maintained and inherited and then seeds harvested to ensure expression of the desired phenotypic characteristic has been achieved. In this manner, the compositions presented herein provide transformed seed (also referred to as "transgenic seed") having a polynucleotide provided herein, for example, a target site, stably incorporated into their genome.
[0137] The variant recognition sequences and active variant and fragments thereof disclosed herein may be used for transformation of any plant species, including, but not limited to, monocots and dicots. Examples of plant species of interest include, but are not limited to, corn (Zea mays), Brassica sp. (e.g., B. napus, B. raga, B. juncea), particularly those Brassica species useful as sources of seed oil, alfalfa (Medicago sativa), rice (Oryza sativa), rye (Secale cereale), sorghum (Sorghum bicolor, Sorghum vulgare), millet (e.g., pearl millet (Pennisetum glaucum), proso millet (Panicum miliaceum), foxtail millet (Setaria italica), finger millet (Eleusine coracana)), sunflower (Helianthus annuus), safflower (Carthamus tinctorius), wheat (Triticum aestivum), soybean (Glycine max), tobacco (Nicotiana tabacum), potato (Solanum tuberosum), peanuts (Arachis hypogaea), cotton (Gossypium barbadense, Gossypium hirsutum), sweet potato (Ipomoea batatus), cassava (Manihot esculenta), coffee (Coffea spp.), coconut (Cocos nucifera), pineapple (Ananas comosus), citrus trees (Citrus spp.), cocoa (Theobroma cacao), tea (Camellia sinensis), banana (Musa spp.), avocado (Persea americana), fig (Ficus casica), guava (Psidium guajava), mango (Mangifera indica), olive (Olea europaea), papaya (Carica papaya), cashew (Anacardium occidentale), macadamia (Macadamia integrifolia), almond (Prunus amygdalus), sugar beets (Beta vulgaris), sugarcane (Saccharum spp.), oats, barley, vegetables, ornamentals, and conifers.
[0138] Vegetables include tomatoes (Lycopersicon esculentum), lettuce (e.g., Lactuca sativa), green beans (Phaseolus vulgaris), lima beans (Phaseolus limensis), peas (Lathyrus spp.), and members of the genus Cucumis such as cucumber (C. sativus), cantaloupe (C. cantalupensis), and musk melon (C. melo). Ornamentals include azalea (Rhododendron spp.), hydrangea (Macrophylla hydrangea), hibiscus (Hibiscus rosasanensis), roses (Rosa spp.), tulips (Tulipa spp.), daffodils (Narcissus spp.), petunias (Petunia hybrida), carnation (Dianthus caryophyllus), poinsettia (Euphorbia pulcherrima), and chrysanthemum.
[0139] Conifers that may be employed in practicing the present invention include, for example, pines such as loblolly pine (Pinus taeda), slash pine (Pinus elliotii), ponderosa pine (Pinus ponderosa), lodgepole pine (Pinus contorta), and Monterey pine (Pinus radiata); Douglas-fir (Pseudotsuga menziesii); Western hemlock (Tsuga canadensis); Sitka spruce (Picea glauca); redwood (Sequoia sempervirens); true firs such as silver fir (Abies amabilis) and balsam fir (Abies balsamea); and cedars such as Western red cedar (Thuja plicata) and Alaska yellow-cedar (Chamaecyparis nootkatensis), and Poplar and Eucalyptus. In specific embodiments, plants of the present invention are crop plants (for example, corn, alfalfa, sunflower, Brassica, soybean, cotton, safflower, peanut, sorghum, wheat, millet, tobacco, etc.). In other embodiments, corn and soybean plants are optimal, and in yet other embodiments corn plants are optimal.
[0140] Other plants of interest include grain plants that provide seeds of interest, oil-seed plants, and leguminous plants. Seeds of interest include grain seeds, such as corn, wheat, barley, rice, sorghum, rye, etc. Oil-seed plants include cotton, soybean, safflower, sunflower, Brassica, maize, alfalfa, palm, coconut, etc. Leguminous plants include beans and peas. Beans include guar, locust bean, fenugreek, soybean, garden beans, cowpea, mungbean, lima bean, fava bean, lentils, chickpea, etc.
Non-limiting examples of compositions and methods disclosed herein are as follows: [0141] 1) A method to identify a variant recognition site for a rare-cutting engineered double-strand-break-inducing agent capable of introducing a rare double strand break in an intended recognition site, said method comprising: [0142] a) contacting genomic DNA with a rare-cutting engineered double-strand-break-inducing agent capable of introducing a double-strand break into said genomic DNA, wherein the double-strand break results in a nucleotide overhang; [0143] b) ligating a first adapter to said nucleotide overhang; [0144] c) shearing the ligated DNA obtained in step (b) and ligating at least one second adapter to the sheared nucleotide end to allow for the amplification and sequencing of genomic DNA fragments surrounding the double strand break; [0145] d) aligning nucleotide sequences of the DNA fragments obtained in (c) with a reference genome DNA sequence; and, [0146] e) identifying a variant recognition site comprising at least one nucleotide base alteration when compared to the intended recognition site of said engineered double-strand break-inducing agent. [0147] 2) The method of embodiment 1 wherein the rare-cutting engineered double-strand-break-inducing agent is selected from the group consisting of a meganuclease, a zinc finger nuclease, a TAL effector nuclease, a transposase, and a site-specific recombinase. [0148] 3) The method of embodiment 1 wherein the nucleotide overhang is a 3' nucleotide overhang. [0149] 4) The method of embodiment 1 wherein the nucleotide overhang is a 5' nucleotide overhang. [0150] 5) The method of embodiment 1 wherein the first adapter ligated to the nucleotide overhang is a non-5' phosphorylated adapter. [0151] 6) The method of embodiment 1 wherein the genomic DNA is selected from the group consisting of a prokaryotic DNA, eukaryotic DNA and synthetic DNA. 7) The method of embodiment 6 wherein the eukaryotic DNA is isolated from a plant, yeast or animal. [0152] 8) The method of embodiment 7 wherein the plant is selected from the group consisting of soybean, sunflower, cotton, alfalfa, canola, cotton, tobacco, potato, Arabidopsis, safflower, maize, rice, sorghum, barley, wheat, millet, oats, sugarcane, turfgrass, and switch grass. [0153] 9) The method of embodiment 1, wherein the double-strand-break-inducing agent is derived from I-CreI. [0154] 10) A method to identify a variant recognition site with an improved cleavage activity for a rare-cutting engineered double-strand-break-inducing agent capable of introducing a rare double strand break in an intended recognition site, said method comprising: [0155] a) contacting genomic DNA with rare-cutting engineered double-strand-break-inducing agent capable of introducing a double strand break into said genomic DNA, wherein the double strand break results in a nucleotide overhang; [0156] b) ligating a first adapter to said nucleotide overhang; [0157] c) shearing the ligated DNA obtained in step (b) and ligating a second adapter to the sheared nucleotide end to allow for the amplification and sequencing of genomic DNA fragments surrounding the double strand break; [0158] d) aligning nucleotide sequences of the DNA fragments obtained in (c) with a reference genome DNA sequence; and, [0159] e) identifying a variant recognition site comprising at least one nucleotide base alteration when compared to the intended recognition site of said rare-cutting engineered double-strand-break-inducing agent; [0160] f) analyzing the a rare-cutting engineered double-strand-break-inducing agent activity at the variant recognition sites of d); [0161] g) identifying a variant recognition site that results in an increased activity of the rare-cutting engineered double-strand-break-inducing agent when compared to the activity at the intended recognition site. [0162] 11) The method of embodiment 10 wherein the increased activity of the rare-cutting engineered double-strand-break-inducing agent is evidenced by [0163] a) a higher percent (%) cleavage of the variant recognition site when compared to the percent (%) cleavage of intended recognition site, wherein the recognition sites are located on genomic DNA; [0164] b) a higher percent (%) cleavage of the variant recognition site when compared to the percent (%) cleavage of intended recognition site, wherein the recognition sites are located on plasmid DNA; [0165] c) a higher yeast assay score for the variant recognition site when compared to the intended recognition site; or, [0166] d) any combination of (a), (b) and (c) [0167] 12) A method for introducing into the genome of a cell a variant recognition site for a rare-cutting engineered double-strand-break-inducing agent capable of introducing a rare double strand break in an intended recognition site, said method comprising: [0168] a) providing a donor DNA comprising a variant recognition site for a rare-cutting engineered double-strand-break-inducing agent capable of introducing a double-strand break in an intended recognition site, wherein said a rare-cutting engineered double-strand-break-inducing agent is also capable of introducing a double-strand break in said variant recognition site; [0169] b) providing a plant cell; [0170] c) contacting the plant cell with the donor DNA; and, [0171] d) identifying at least one plant cell from (c) comprising in its genome said variant recognition site. [0172] 13) The method of embodiment 12 wherein the rare-cutting engineered double-strand-break-inducing agent is selected from the group consisting of a meganuclease, a zinc finger nuclease, a TAL effector nuclease, a transposase, a Cas endonuclease and a site-specific recombinase. [0173] 14) An isolated polynucleotide comprising a variant recognition site with an improved cleavage activity for an engineered meganuclease capable of introducing a double strand break in an intended recognition site, wherein said variant recognition site comprises a nucleotide sequence with at least 1 nucleotide base substitution when compared to the intended recognition site of SEQ ID NO: 14. [0174] 15) The isolated polynucleotide of embodiment 1, wherein said variant recognition site comprises a sequence with at least 2, 3, 4, 5, 6 or 7 base pair alterations when compared to SEQ ID NO: 14. [0175] 16) The isolated polynucleotide of embodiment 14, wherein said variant recognition site comprises: [0176] a) an adenine (A) at a position corresponding to the nucleotide position 9 in SEQ ID NO: 14; [0177] b) a guanine (G) at a position corresponding to the nucleotide position 10 in SEQ ID NO: 14; [0178] c) a thymine (T) at a position corresponding to the nucleotide position 11 in SEQ ID NO: 14; [0179] d) an adenine (A) at a position corresponding to the nucleotide position 13 in SEQ ID NO: 14; [0180] e) a thymine (T) at a position corresponding to the nucleotide position 18 in SEQ ID NO: 14; [0181] f) a guanine (G) at a position corresponding to the nucleotide position 19 in SEQ ID NO: 14; [0182] g) a guanine (G) or an adenine (A) at a position corresponding to the nucleotide position 22 in SEQ ID NO: 14; or, [0183] h) any combination of a) to g). [0184] 17) The isolated polynucleotide of embodiment 14, wherein said variant recognition sequence is selected from the group consisting of SEQ ID NOs: 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 and 35. [0185] 18) The isolated polynucleotide of embodiment 14, wherein the improved cleavage activity is evidenced by: [0186] a) a higher percent (%) cleavage of the variant recognition site when compared to the percent (%) cleavage of intended recognition site of SEQ ID NO:14, wherein the recognition sites are located on genomic DNA; [0187] b) a higher percent (%) cleavage of the variant recognition site when compared to the percent (%) cleavage of intended recognition site, wherein the recognition sites are located on plasmid DNA; [0188] c) a higher yeast assay score for the variant recognition site when compared to the intended recognition site; or, [0189] d) any combination of (a), (b) and (c) [0190] 19) A recombinant DNA fragment comprising the isolated polynucleotide of embodiment 14. [0191] 20) A cell comprising the recombinant DNA fragment of embodiment 19. [0192] 21) The cell of embodiment 20, wherein the cell is a yeast or plant cell. [0193] 22) A transgenic plant or seed comprising the plant cell of embodiment 21. [0194] 23) The transgenic plant of embodiment 22 wherein said plant is selected from the group consisting of maize, wheat, rice, barley, sugarcane, sorghum, rye, switch grass, soybean, Brassica, sunflower, cotton, or alfalfa. [0195] 24) An isolated polynucleotide comprising a variant recognition site with an improved cleavage activity for an engineered meganuclease capable of introducing a double strand break in an intended recognition site, wherein said variant recognition site comprises a nucleotide sequence with at least 1 nucleotide base substitution when compared to the intended recognition site of SEQ ID NO: 13. [0196] 25) The isolated polynucleotide of embodiment 24, wherein said variant recognition site comprises a sequence with at least 2, 3, 4, or 5 base pair alterations when compared to SEQ ID NO: 13. [0197] 26) The isolated polynucleotide of embodiment 24, wherein said variant recognition site comprises: [0198] a) a cytosine (C) at a position corresponding to the nucleotide position 1 in SEQ ID NO: 13 [0199] b) a cytosine (C)) at a position corresponding to the nucleotide position 5 in SEQ ID NO: 13; [0200] c) a guanine (G) at a position corresponding to the nucleotide position 10 in SEQ ID NO: 13; [0201] d) a thymine (T) an adenine (A) at a position corresponding to the nucleotide position 11 in SEQ ID NO: 13; [0202] e) a thymine (T) at a position corresponding to the nucleotide position 19 in SEQ ID NO: 13; [0203] f) any combination of a) to e). [0204] 27) The isolated polynucleotide of embodiment 24, wherein said variant recognition sequence is selected from the group consisting of SEQ ID NOs: 15, 16, 17, 18, 19, 20 and 21. [0205] 28) The isolated polynucleotide of embodiment 24, wherein the improved cleavage activity is evidenced by: [0206] a) a higher percent (%) cleavage of the variant recognition site when compared to the percent (%) cleavage of intended recognition site of SEQ ID NO:13, wherein the recognition sites are located on genomic DNA; [0207] b) a higher percent (%) cleavage of the variant recognition site when compared to the percent (%) cleavage of intended recognition site, wherein the recognition sites are located on plasmid DNA; [0208] c) a higher yeast assay score for the variant recognition site when compared to the intended recognition site; or, [0209] d) any combination of (a), (b) and (c). [0210] 29) A recombinant DNA fragment comprising the isolated polynucleotide of embodiment 24. [0211] 30) A cell comprising the recombinant DNA fragment of embodiment 29. [0212] 31) The cell of embodiment 30, wherein the cell is a yeast or plant cell. [0213] 32) A transgenic plant or seed comprising the plant cell of embodiment 31. [0214] 33) The transgenic plant of embodiment 32 wherein said plant is selected from the group consisting of maize, wheat, rice, barley, sugarcane, sorghum, rye, switch grass, soybean, Brassica, sunflower, cotton, or alfalfa. [0215] 34) A method for targeting the insertion of a polynucleotide of interest to a specific chromosomal site within a plant genome, said method comprising: [0216] a) transforming a plant cell or a plant with a DNA fragment comprising a polynucleotide of interest, wherein said genome of said plant cell or plant comprises at least one variant recognition site selected from the group consisting of SEQ ID NOs: 15, 16, 17, 18, 19, 20 and 21; and, [0217] b) providing a meganuclease capable of providing a double strand break into the variable recognition site of (a); and, [0218] c) selecting said plant cell or plant comprising said polynucleotide of interest integrated into said variant recognition site. [0219] 35) The method of embodiment 34 wherein providing said meganuclease comprises integrating in the genome of said plant cell or plant a nucleotide sequence encoding the meganuclease of SEQ ID NO: 1. [0220] 36) A plant or plant cell obtained by the method of embodiment 34. [0221] 37) A method for targeting the insertion of a polynucleotide of interest to a specific chromosomal site within a plant genome, said method comprising: [0222] a) transforming a plant cell or a plant with a DNA fragment comprising a polynucleotide of interest, wherein said genome of said plant cell or plant comprises at least one variant recognition site selected from the group consisting of SEQ ID NOs: 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 and 35; [0223] b) providing a meganuclease capable of providing a double strand break into the variable recognition site of (a); and [0224] c) selecting said plant cell or plant comprising said polynucleotide of interest integrated into said variant recognition site. [0225] 38) The method of embodiment 37 wherein providing the meganuclease comprises integrating in the genome of said plant cells a nucleotide sequence encoding the meganuclease of SEQ ID NO: 3 [0226] 39) A plant or plant cell obtained by the method of embodiment 37. [0227] 40) A method to identify a variant recognition site for a rare-cutting engineered double-strand-break-inducing agent capable of introducing a rare double strand break in an intended recognition site, said method comprising: [0228] a) contacting genomic DNA with a rare-cutting engineered double-strand-break-inducing agent capable of introducing a double-strand break into said genomic DNA, wherein the double-strand break results in a blunt end; [0229] b) creating a nucleotide overhang from the blunt end of (a); [0230] c) ligating a first adapter to the nucleotide overhang of (b); [0231] d) shearing the ligated DNA obtained in step (c) and ligating at least one second adapter to the sheared nucleotide end to allow for the amplification and sequencing of genomic DNA fragments surrounding the double strand break; [0232] e) aligning nucleotide sequences of the DNA fragments obtained in (d) with a reference genome DNA sequence; and, [0233] f) identifying a variant recognition site comprising at least one nucleotide base alteration when compared to the intended recognition site of said engineered double-strand break-inducing agent. [0234] 41) The method of embodiment 40 wherein the rare-cutting engineered double-strand-break-inducing agent is a Cas endonuclease [0235] 42) The method of embodiment 41 wherein the Cas endonuclease is capable of forming a complex with a guide RNA, wherein said complex enables the Cas endonuclease to introduce a double strand break into said genomic DNA. [0236] 43) The method of embodiment 40 wherein the genomic DNA is selected from the group consisting of a prokaryotic DNA, eukaryotic DNA and synthetic DNA.
[0237] 44) The method of embodiment 43 wherein the eukaryotic DNA is isolated from a plant, yeast or animal. [0238] 45) A method to identify a variant recognition site with an improved cleavage activity for a rare-cutting engineered double-strand-break-inducing agent capable of introducing a rare double strand break in an intended recognition site, said method comprising: [0239] a) contacting genomic DNA with rare-cutting engineered double-strand-break-inducing agent capable of introducing a double strand break into said genomic DNA, wherein the double strand break results in a blunt end [0240] b) creating a nucleotide overhang from the blunt end of (a); [0241] c) ligating a first adapter the nucleotide overhang of (b); [0242] d) shearing the ligated DNA obtained in step (c) and ligating a second adapter to the sheared nucleotide end to allow for the amplification and sequencing of genomic DNA fragments surrounding the double strand break; [0243] e) aligning nucleotide sequences of the DNA fragments obtained in (d) with a reference genome DNA sequence; and, [0244] f) identifying a variant recognition site comprising at least one nucleotide base alteration when compared to the intended recognition site of said rare-cutting engineered double-strand-break-inducing agent; [0245] g) analyzing the rare-cutting engineered double-strand-break-inducing agent activity at the variant recognition sites of f); and, [0246] h) identifying a variant recognition site that results in an increased activity of the rare-cutting engineered double-strand-break-inducing agent when compared to the activity at the intended recognition site. [0247] 46) The method of embodiment 45 wherein the rare-cutting engineered double-strand-break-inducing agent is a Cas endonuclease [0248] 47) The method of embodiment 46 wherein the Cas endonuclease is capable of forming a complex with a guide RNA, wherein said complex enables the Cas endonuclease to introduce a double strand break into said genomic DNA.
EXPERIMENTAL
Example 1
Creation of Rare Cutting Engineered Meganucleases
A. LIG3-4 Meganuclease and LIG3-4 Intended Recognition Sequence
[0249] An endogenous maize genomic target site comprising the LIG3-4 intended recognition sequence (SEQ ID NO: 13) was selected for design of a rare-cutting double-strand break inducing agent (SEQ ID NO: 1) as described in US patent publication 2009-0133152 A1 (published May 21, 2009). The LIG3-4 intended recognition sequence is a 22 bp polynucleotide having the following sequence:
TABLE-US-00001 (SEQ ID NO: 13) ATATACCTCACACGTACGCGTA.
B. MHP14+ Meganucleases and MHP14 Recognition Site
[0250] An endogenous maize genomic target site comprising the MHP14+ intended recognition site (SEQ ID NO: 14) was selected for design of a rare-cutting double-strand break inducing agent (SEQ ID NO: 3) as described in U.S. patent application Ser. No. 13/427,138 filed on Mar. 22, 2012). The MHP14+ intended recognition site is a 22 bp polynucleotide located and having the following sequence:
TABLE-US-00002 (SEQ ID NO: 14) CAAACAGATTCACGTCAGATTT.
Example 2
Meganuclease Protein Production in E. coli
[0251] In order to produce purified protein for in vitro genomic and plasmid based meganuclease cleavage activity assays, DNA fragments corresponding to the open-reading-frames of Lig3-4 meganuclease (SEQ ID NO: 2) and MHP14+ meganuclease (SEQ ID NO: 4) were placed into a pQE80 (Qiagen) expression vector, transformed into BL21-Gold (Agilent Technologies) E. coli cells, and grown overnight on solid LB media containing 100 ppm of carbenicillin. Colonies were resuspended in 2 ml of 2XYT media and 250 .mu.l of the cell suspension was used to inoculate a 50 ml culture of 2XYT supplemented with 100 ppm of carbenicillin. Cultures were grown at 37.degree. C. for 1 to 1.5 hrs or until the OD600 reached 0.8 and then protein expression was induced by the addition of 0.5 ml of 100 mM IPTG. Cultures were cooled to room temperature and allowed to express protein for 2 hrs. Cells were pelleted by centrifuging for 10 minutes at 5,000 rcf. The supernant was decanted, the pellet resuspended in 1 ml of Buffer 1 (50 mM Tris-HCl (pH8.0), 500 mM NaCl, 10 mM imidizole), and transferred to a 1.5 ml microfuge tube. Cells were disrupted by sonication with a two-step 1/8'' microtip with 20 pulses (duty cycle 50, power 4) on a Branson 450 Analog Sonifer and centrifuged at 20,000 rcf for 15 minutes at 4.degree. C. The supernant was diluted with 4 ml of Buffer 1 and loaded onto a disposable column containing 0.3 ml of Nickel-NTA Superflow resin (Qiagen). The column was washed with 5 ml of Buffer 2 (50 mM Tris-HCl (pH8.0), 500 mM NaCl, 60 mM imidizole) and the protein eluted with 0.6 ml of Buffer 4 (50 mM Tris-HCl (pH8.0), 500 mM NaCl, 250 mM imidizole) into a Vivaspin column (GE). To concentrate the samples, the vivaspin columns were centrifuged at 14,800 rcf for approximately 6 minutes or until the meniscus was between 75 and 50. A buffer exchange was performed using a Zeba Spin Desalting Column (Pierce) pre-equilibrated with storage buffer (25 mM Tris-HCl (pH8.0), 100 mM NaCl, 10 mM MgCl.sub.2, 5 mM EDTA, 50% Glycerol). After the buffer exchange, Bovine Serum Albumin was added to a final concentration of 100 ng/.mu.l and purified protein was stored at -20.degree. C. until use.
Example 3
In Vitro Genomic DNA Cleavage Assays
[0252] To generate material for the capture of genomic variant recognition sites, in vitro assays were carried out with 114 nM of purified meganuclease protein isolated as described in Example 2 and 6.07 .mu.g of purified maize genomic DNA at 32.degree. C. for 80 minutes in a final volume of 80 .mu.l in the presence of digestion buffer (50 mM Tris-HCl (pH 7.9), 100 mM NaCl, 10 mM MgCl.sub.2, 1 mM DTT, 5 mM EDTA). After 80 minutes, the entire reaction was stopped with an equal volume of stop buffer (100 mM Tris-HCl (pH 8.0), 600 mM NaCl, 2% SDS, 100 mM EDTA, 1 mg of proteinase K per ml) and incubated at 50.degree. C. for 30-45 minutes. Stopped reactions were purified by phenol/chloroform extraction and ethanol precipitated in the presence of 0.2 M NaCl. Precipitated genomic DNA was washed twice with 70% ethanol, dried, and resuspended in 34 .mu.l of water.
[0253] Meganuclease protein concentration was determined visually on Nu-PAGE gels (Life Technologies) by calculating and then comparing band intensity with serially diluted samples of known concentration and genomic DNA concentration was determined using a Hoechst dye fluorometric assay.
[0254] To confirm cleavage (representing the % loss of meganuclease recognition sites) at the intended genomic recognition site, real-time PCR was performed on 1 .mu.l of purified genomic DNA with a TaqMan assay spanning the meganuclease recognition site. The % cleavage or loss of meganuclease recognition sites was calculated via the .DELTA..DELTA.Ct method relative to an internal control TaqMan assay using the mock control as a calibrator.
Example 4
Capture of Genomic Variant Recognition Sites & Generation of Libraries for Illumina Deep Sequencing
[0255] Our method utilizes a novel adapter approach specifically tailored for the capture of cleaved I-CreI or engineered I-CreI homing endonuclease genomic variant recognition sites whose sequence is unknown and different in composition from the intended recognition site which is different from methods using restriction enzymes to perform reduced representation sequencing, restriction associated DNA (RAD-tag or RADseq) deep sequencing, whole genome sequencing (WGS), or genotype by sequencing (GBS).
[0256] Since the I-CreI homing endonuclease generates a 3' 4 nucleotide overhang at the center of its 22 bp recognition site (+2, +1, -1, -2) upon cleavage (Thompson et al. (1992) Gene 119:247-51 and Durrenberger et al. (1993) Mol. Gen. Genet. 236:409-14) and has been demonstrated to cleave its recognition site in the context of different combinations of center 4 base pairs (+2, +1, -1, -2) (Molina et al. (2012) Nucleic Acids Res. 40:6936-45), adapters were generated containing a 3' 4 nucleotide overhang containing all possible DNA nucleotide combinations (G, T, A, or C) of the overhang in an equimolar distribution. Thus, allowing for the efficient ligation and perfect complementation to all possible overhangs generated by recognition site cleavage in the genome subjected to I-CreI or engineered I-CreI homing endonuclease cleavage.
[0257] Similar strategies can be employed for other rare cutting double strand break inducing agents such as Zinc Finger and TALEN nucleases that cleave DNA with the non-specific catalytic domain of FokI generating overhangs of variable length and nucleotide composition within the intervening spacer region (Smith et al. (2000) Nucleic Acids Res. 28:3361-69 and Li et al. (2011) Nucleic Acids Res. 39:359-72). To capture genomic variant recognition sites, non-phosphorylated biotinylated adapters were synthesized and purified by HPLC (Integrated DNA Technologies, Inc.) containing a fully degenerate 4 nucleotide 3' nucleotide overhang complementary to the 4 nucleotide 3' overhang generated by meganuclease recognition site cleavage (SEQ ID NO: 5) and ligated to approximately 2 .mu.g of meganuclease-cleaved genomic DNA (prepared as described in Example 3) in a 100 .mu.l T4 ligase reaction (NEB) (representing a first adapter of the method to identify a variant recognition site for a rare-cutting engineered double-strand-break-inducing agent). Samples comprising the ligated DNA were then loaded in sonication microtubes and randomly sheared to an average peak size of 300 bps by sonication in a Covaris E220 system. The settings were 10% duty cycle, 140 peak incident power, and 200 cycles per burst. Fragments ranging from 200 to 500 bp were fractionated by electrophoresis in an agarose gel followed by gel extraction using the Qiagen Gel Extraction Kit according to manufacturer's recommendations.
[0258] Non-biotinylated ends were repaired using the End-It End repair kit (Epicentre) in a 75 .mu.l reaction and column-purified (Qiagen). Single 3' A overhang extension was performed by incubating the repaired DNA at 37.degree. C. for 30 minutes in a 50 .mu.l reaction containing ATP, 1.times. Klenow buffer (NEBnext), and 15 units Klenow (exo-). Samples were later purified by column (Qiagen) and ligated to indexed IIlumina TruSeq-compatible adapters (representing a second set of adapters of the method to identify a variant recognition site for a rare-cutting engineered double-strand-break-inducing agent) in a 50 .mu.l reaction containing 0.3 mM indexed adapter, 1.times. Quick ligation buffer and 5 units T4 DNA ligase (NEB) at room temperature. After ligation, samples were incubated at 65.degree. C. for 15 minutes and the volume adjusted to 100 .mu.l. Streptavidin magnetic capture was performed using Dynabeads M-280 streptavidin beads (Invitrogen). A total of 100 .mu.l resuspended Streptavidin-Dynabeads (M-280) were washed twice in TE and resuspended in 100 .mu.l 2.times. B&W buffer (10 mM Tris-HCL, 1 mM EDTA, 100 .mu.l 0.5M EDTA, 2M NaCl). Samples were incubated at 30.degree. C. for 30 minutes, the supernatant removed, and beads washed 4 times with 1 ml of 1.times.B&W buffer. The final enriched sample was resuspended in 30 .mu.l EB buffer.
[0259] Fragments were recovered from the beads by 12-cycle PCR using Phusion master mix (NEB), in a 50 .mu.l reaction in the presence of 0.4 pmol of recovery primer A (5'GTTGACATGCTGGATTGAGACTTC; SEQ ID NO: 6) and primer B (5'CAAGCAGAAGACGGCATACGA; SEQ ID NO: 7) according to manufacturer, except that an annealing temperature of 66.degree. C. and extension time of 30 seconds were used. Recovered DNA was digested with SbfI (NEB) and purified twice with Agencourt AMPure XP Beads (SPRI) according to manufacturer instructions. The sample was ligated to an Illumina-compatibe adapter with a SbfI compatible overhang (SEQ ID NO: 8). The supernatant was cleaned-up twice using Agencourt AMPure XP Beads, first using a 1:1.8 and then a 1:1 sample-to-bead ratio. The final samples were resuspended in 50 .mu.l of EB buffer. A second amplification with the standard TruSeq PCR primer cocktail (Illumina) was performed, using a 60.degree. C. annealing temperature, followed by clean-up twice using Agencourt AMPure XP Beads in a 1:1.8 and then a 1:1 sample-to-bead ratio. The final sample was resuspended in 20 .mu.l. Samples were evaluated on a bioanalyzer, relatively quantified using qPCR with Illumina qPCR primers, and pooled. Prior to sequencing, the pools were size selected using the lab Xchip (Caliper), according to manufacturer's instructions.
Example 5
Non-Phosphorylated Adapters Enhance Enrichment of Meganuclease Cleaved Genomic Recognition Sites
[0260] To examine the effect that phosphorylation has on the ability of a first adapter (as described in Example 4) to capture and enrich for meganuclease cleaved genomic recognition sites, libraries were made with both phosphorylated and non-phosphorylated adapters. After normalizing the DNA concentration of the libraries, they were examined for enrichment of the cleaved intended recognition site by real-time PCR with a TaqMan assay immediately adjacent to the intended recognition site. As shown in FIG. 7, both libraries demonstrated enrichment relative to the mock control but the amplification plots from the library generated with the non-phosphorylated adapter reached logarithmic amplification at a much earlier amplification cycle than the library generated with the phosphorylated adapter. Using standard curves derived from maize genomic DNA, the library generated with the non-phosphorylated adapter was estimated to be approximately 900 times more enriched for the left half of the intended recognition site than the library generated with the phosphorylated adapter.
Example 6
Illumina Deep Sequencing & Post-Run Trim
[0261] After the capture and enrichment for genomic variant recognition sites and DNA preparation as described in Example 4, cluster generation and paired-end read sequencing were performed on an Illumina cBot and Genome Analyzer IIx, respectively, according to the manufacturer's instructions. Approximately 30% (v/v) phiX DNA control (Illumina) was added to the DNA library solution prior to clustering. The random base composition of the phiX DNA fragments was expected to offset any base composition bias present at the vicinity of the meganuclease site. 100 cycle paired-end recipes were used on the Illumina Genome Analyzer. Sequences and quality scores were generated by the Illumina pipeline version 2.9 software for image analysis and base calling. During base calling, the phiX control data were used to obtain error estimates and re-calibrate raw quality scores for the other samples. After initial base calling, additional filtering was performed by the Illumina software where reads are excluded if the noise estimate exceeds thresholds defined by the Illumina base calling pipeline. Base call conversion to FASTQ format (using the Illumina CASSAVA software) was followed by additional filtering where reads were trimmed and filtered according to each base's respective quality score (where bases with a quality score below 10 are trimmed from the 3' end of the read).
Example 7
In Silico Enrichment & Physical Mapping of Sequence Data to the Genomic Reference
[0262] To further enrich for meganuclease cleaved and adapter-ligated genomic DNA fragments, the set of reads from a sequencing experiment was filtered using a custom script that searches for read pairs or singletons wherein at least one member of the pair (or singleton) containing a sequence tag, GCAGGACGT (SEQ ID NO: 9), at the beginning of the read or its complement ACGTCCTGC (SEQ ID NO: 10) at the end of the read. Pairs or singleton reads matching this sequence are written to a new file for use in the mapping phase, while the rest are discarded.
[0263] To reunite both halves of the cleaved genomic recognition site, the enriched read set was physically mapped to the target reference genome using bowtie version 0.12.7 (Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25.) to identify the locations of the genome homologous to the reads. Alignment settings for specificity were adjusted on a case by case basis depending on the characteristics of the target genome and that genome's similarity to the source genotype of the data. The resulting alignments were then used for peak detection and identification of variant recognition sequences present in the genome.
Example 8
Peak Detection and Identification of Genomic Variant Recognition Site, and Composition
[0264] Using the genomic alignments generated in Example 7, peak detection was carried out using the MACs (Model-based analysis of ChiP-Seq) peak detection algorithm in Genedata Expressionist Refiner 7.5 (Genedata) with the parameters listed in Table 1.
TABLE-US-00003 TABLE 1 MACs (Model-based analysis of ChiP-Seq) peak detection algorithm settings in Genedata Expressionist Refiner 7.5 (Genedata). Experiment Setup: ChIP Samples with Control Method: MACS Minimum Fold Enrichment 10 Maximum Fold Enrichment 1.00E+09 Bandwidth 200 bp p-Value Threshold 1.00E-05
The chromosomal regions with significant enrichment relative to the mock control were exported to excel. Regions with the greatest difference between the treated and mock control samples were prioritized and confirmed as being enriched relative to the mock control and having a peak signature resulting from genomic DNA cleavage in the Genome Browser functionality of Genedata Expressionist Refiner 7.5. As shown in FIG. 1, recognition site peak signatures contain sequence data originating and diverging from the site of cleavage with the overlapping center corresponding to the overhang generated by the double-strand break inducing reagent. Based on the overhang defined by the peak signature, the precise variant recognition site sequence present in the genomic DNA could be identified as shown in FIG. 2. To determine the correct orientation of genomic variant recognition site sequences, they were aligned to the intended target site in both sense and anti-sense orientations with the best fitting orientation used as the recognition site sequence. A small proportion of genomic variant recognition sites fitted equally well in both orientations. These were left in the sense orientation. The oriented genomic variant recognition sites were aligned and the percent DNA nucleotide composition was calculated for each individual position of the recognition site and compared with the intended recognition site. As shown in FIG. 3, some positions in recognition sites exhibited an off-nucleotide preference; a preference for a nucleotide other than what was targeted in the intended recognition site.
Example 9
In Vitro Plasmid DNA Cleavage Assays
[0265] In vitro meganuclease cleavage activity can also be assayed using plasmid DNA, To compare meganuclease cleavage activity at intended and variant recognition sites, annealed oligonucleotides (synthesized by Integrated DNA Technologies, Inc.) containing the intended or variant recognition site with EcoRI and HindIII overhangs were cloned into the HindIII and EcoRI restriction endonucleases sites of the pBluescript SK+ plasmid (Stratagene, now an Agilent Technologies company), and in vitro DNA cleavage activity assayed as described in Example 3 with the following modifications. Timed digestions were carried out with 0.25 nM of linearized plasmid substrate containing a single intended or variant recognition site with 25 nM of purified meganuclease protein. In vitro assays were carried out at 37.degree. C., 28.degree. C., and 23.degree. C. to best examine the cleavage activity at a given variant recognition site. Stopped reactions were purified with a Qiagen PCR purification column per the manufacturer's instruction and purified DNA was diluted 200-fold prior to the quantification of cleavage activity activity or % loss of recognition sites by qPCR.
Example 10
Identifying Variant Recognition Sites and Effect of Off-Nucleotide Preferences on Meganuclease Cleavage Activity
[0266] To assess the effect that the off-nucleotide preferences had on meganuclease cleavage activity, the off-nucleotide preferences were introduced into the intended recognition site individually and in combination (see FIG. 4), hereby generating variant recognition sites. Examples of such variant recognition sites are shown in Table 2 for the LIG3-4 meganuclease and Table 3 for the MHP14+ meganuclease.
TABLE-US-00004 TABLE 2 List of variant recognition sites for the LIG3-4 meganuclease. Nucleotides with an asterisk (*) indicate a modification compared to the nucleotide at the corresponding location of the intended recognition site (SEQ ID NO: 13). name of SEQ variable ID recognition nucleotide position of reference nucleotide (SEQ ID NO: 13) NO: site 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 13 intended A T A T A C C T C A C A C G T A C G C G T A LIG3-4 15 -11C C* T A T A C C T C A C A C G T A C G C G T A 16 -7C A T A T C* C C T C A C A C G T A C G C G T A 17 -2G A T A T A C C T C G* C A C G T A C G C G T A 18 -1T A T A T A C C T C A T* A C G T A C G C G T A 19 +8T A T A T A C C T C A C A C G T A C G T* G T A 20 -70, +8T A T A T C* C C T C A C A C G T A C G T* G T A 21 -11C, -7C, C* T A T C* C C T C G* T* A C G T A C G T* G T A -2G, -1T, +8T 22 -11C, -7C, C* T A T C* C C T C A T* A C G T A C G T* G T A -1T, +8T
TABLE-US-00005 TABLE 3 List of variant recognition sites for the MHP14+ meganuclease. Nucleotides with an asterisk (*) indicate a modification compared to the nucleotide at the corresponding location of the intended recognition site (SEQ ID NO: 14). name of variable SEQ recogni- nucleotide position of reference nucleotide ID tion (SEQ ID NO13 or SEQ ID NO: 14) NO: site 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 14 MHP14+ C A A A C A G A T T C A C G T C A G A T T T 23 -3A C A A A C A G A A* T C A C G T C A G A T T T 24 -2G C A A A C A G A T G* C A C G T C A G A T T T 25 -1T C A A A C A G A T T T* A C G T C A G A T T T 26 +2A C A A A C A G A T T C A A* G T C A G A T T T 27 +7T C A A A C A G A T T C A C G T C A T* A T T T 28 +8G C A A A C A G A T T C A C G T C A G G* T T T 29 +11G C A A A C A G A T T C A C G T C A G A T T G* 30 +11A C A A A C A G A T T C A C G T C A G A T T A* 31 -3A, -2G, C A A A C A G A A* G* T* A A* G T C A T* G* T T G* -1T, +2A, +7T, +8G, +11G 32 -3A, -2G, C A A A C A G A A* G* T* A A* G T C A T* G* T T A* -1T, +2A, +7T, +8G, +11A 33 -3A, -2G, C A A A C A G A A* G* T* A C G T C A T* G* T T G* -1T, +7T, +8G, +11G 34 -2G, -1T, C A A A C A G A T G* T* A A* G T C A T* G* T T G* +2A, +7T, +8G, +11G 35 -2G, -1T, C A A A C A G A T G* T* A C G T C A T* G* T T G* +7T, +8G, +11G
[0267] The variant recognition sites were then cloned into the HindIII and EcoRI restriction endonucleases sites of a pBluescript SK+ vector and meganuclease activity was assayed by determining the % cleavage or % loss of recognition sites on plasmid DNA substrates as described in Example 9.
[0268] When individually introduced into their corresponding Lig3-4 and MHP14+ intended recognition sites, the off-nucleotide preferences conferred plasmid DNA cleavage activity equal to or greater than the intended recognition site (see FIGS. 5A and 5B). While nucleotides not preferred were not cleaved well (see FIGS. 5A and 5B). Interestingly, even off-nucleotide preferences within the center 4 bases (+2, +1, -1, -2) reported as not being directly contacted by the I-CreI homing endonuclease (Jurica et al. (1998) Mol. Cell 2:469-76; Grizot et al. (2011) Nucleic Acids Res. 39:6124-36, and Ulge et al. (2011) Nucleic Acids Res. 39:4330-9)) enhanced cleavage activity. The one exception to this was the poor cleavage activity for an adenine at position +2 for the MHP14+ meganuclease. However, when all the preferred nucleotides for MHP14+ were examined in combination as shown in FIG. 6B (recognition sites -3A, -2G, -1T, +2A, +7T, +8G, +11G and -3A, -2G, -1T, +2A, +7T, +8G, +11A), cleavage activity far greater than that of the intended recognition site was observed. Also if +2A was removed as in the -3A, -2G, -1T, +7T, +8G, +11G recognition site, there is a slight decrease in cleavage activity (see FIG. 6B) indicating that cleavage of the adenine at position+2 is context specific and dependent on the recognition or conformation of nucleotide bases adjacent to it.
[0269] In combination, off-nucleotide preferences had an additive effect on cleavage efficiency with the best combinations being cleaved approximately 5 to 6 times more efficiently than the intended recognition site (FIG. 6). Many of the variant recognition sites could even be cleaved at temperatures as low as 23.degree. C. where only slight cleavage activity was observed at the intended recognition site (FIGS. 6A and B). Some combinations of preferred off-nucleotides in the center 4 bases (+2,+1,-1,-2) and in positions immediately adjacent (+3,-3) influenced the magnitude of cleavage activity gained relative to the intended recognition site.
[0270] Taken together; our data indicates that the methods described herein can be used to infer the preferred DNA base contacts made by a meganuclease at individual positions across its DNA binding interface allowing a thorough evaluation of cleavage specificity providing a novel approach to examining meganuclease specificity within a genomic DNA context. Our methods also allow the identification of variant recognition sites that are cleaved more efficiently than the intended recognition site.
Example 11
Uses of Variant Recognition Sites in Plant or Animals
[0271] The variant recognition sites identified in Example 10 with improved cleavage activity over the intended recognition site (-7C, 8T (Lig3-4); -11C, -7C, -2G, -1T, +8T (Lig3-4); -11C, -7C, -1T, +8T (Lig3-4); -3A, -2G, -1T, +2A, +7T, +8G, +11G (MHP14+); -3A, -2G, -1T, +2A, +7T, +8G, +11A (MHP14+); -3A, -2G, -1T, +7T, +8G, +11G (MHP14+); -2G, -1T, +2A, +7T, +8G, +11G (MHP14+); -2G, -1T, +7T, +8G, +11G (MHP14+), corresponding to SEQ ID NOs: 13-35), or any other variant recognition site identified by the method described herein, can be transformed into any plant or animal genome and targeted for mutagenesis or gene insertion. Since the cleavage activity at these recognition sites is enhanced relative to the intended recognition site, site modification rates including deletion, insertion, or any combination of the two may also be enhanced. The variant recognition sites may also be placed individually or in combination on transgenic expression cassettes allowing for the alteration, excision, or insertion of transgenic pieces.
Example 12
Application to Other Rare-Cutting Double-Strand Break Inducing Reagents
[0272] Since oligonucleotides with both 5' and 3' degenerate ends may be synthesized in a wide range of user-specified configurations and lengths (Integrated DNA Technologies, Inc) and annealed to form double stranded DNA adapters with either 5' or 3' degenerate overhang, the methods we have established here would be applicable to any rare-cutting double-strand break reagent that creates a DNA base overhang upon cleavage. This would include but not be limited to other homing endonucleases, Zinc-Finger nucleases and TALENs.
Example 13
Transformation of Maize Immature Embryos
[0273] Transformation can be accomplished by various methods known to be effective in plants, including particle-mediated delivery, Agrobacterium-mediated transformation, PEG-mediated delivery, and electroporation.
[0274] a. Particle-Mediated Delivery
[0275] Transformation of maize immature embryos using particle delivery is performed as follows. Media recipes follow below.
[0276] The ears are husked and surface sterilized in 30% Clorox bleach plus 0.5% Micro detergent for 20 minutes, and rinsed two times with sterile water. The immature embryos are isolated and placed embryo axis side down (scutellum side up), 25 embryos per plate, on 560Y medium for 4 hours and then aligned within the 2.5-cm target zone in preparation for bombardment. Alternatively, isolated embryos are placed on 560L (Initiation medium) and placed in the dark at temperatures ranging from 26.degree. C. to 37.degree. C. for 8 to 24 hours prior to placing on 560Y for 4 hours at 26.degree. C. prior to bombardment as described above.
[0277] Plasmids containing the double strand brake inducing agent and donor DNA are constructed using standard molecular biology techniques and co-bombarded with plasmids containing the developmental genes ODP2 (AP2 domain transcription factor ODP2 (Ovule development protein 2); US20090328252 A1) and Wushel (US2011/0167516).
[0278] The plasmids and DNA of interest are precipitated onto 0.6 .mu.m (average diameter) gold pellets using a water-soluble cationic lipid Tfx.TM.-50 (Cat# E1811, Promega, Madison, Wis., USA) as follows. DNA solution is prepared on ice using 1 .mu.g of plasmid DNA and optionally other constructs for co-bombardment such as 50 ng (0.5 .mu.l) of each plasmid containing the developmental genes ODP2 (AP2 domain transcription factor ODP2 (Ovule development protein 2); US20090328252 A1) and Wushel. To the pre-mixed DNA, 20 .mu.l of prepared gold particles (15 mg/ml) and 5 .mu.l Tfx-50 is added in water and mixed carefully. Gold particles are pelleted in a microfuge at 10,000 rpm for 1 min and supernatant is removed. The resulting pellet is carefully rinsed with 100 ml of 100% EtOH without resuspending the pellet and the EtOH rinse is carefully removed. 105 .mu.l of 100% EtOH is added and the particles are resuspended by brief sonication. Then, 10 .mu.l is spotted onto the center of each macrocarrier and allowed to dry about 2 minutes before bombardment.
[0279] Alternatively, the plasmids and DNA of interest are precipitated onto 1.1 .mu.m (average diameter) tungsten pellets using a calcium chloride (CaCl.sub.2)) precipitation procedure by mixing 100 .mu.l prepared tungsten particles in water, 10 .mu.l (1 .mu.g) DNA in Tris EDTA buffer (1 .mu.g total DNA), 100 .mu.l 2.5 M CaCl2, and 10 .mu.l 0.1 M spermidine. Each reagent is added sequentially to the tungsten particle suspension, with mixing. The final mixture is sonicated briefly and allowed to incubate under constant vortexing for 10 minutes. After the precipitation period, the tubes are centrifuged briefly, liquid is removed, and the particles are washed with 500 ml 100% ethanol, followed by a 30 second centrifugation. Again, the liquid is removed, and 105 .mu.l 100% ethanol is added to the final tungsten particle pellet. For particle gun bombardment, the tungsten/DNA particles are briefly sonicated. 10 .mu.l of the tungsten/DNA particles is spotted onto the center of each macrocarrier, after which the spotted particles are allowed to dry about 2 minutes before bombardment.
[0280] The sample plates are bombarded at level #4 with a Biorad Helium Gun. All samples receive a single shot at 450 PSI, with a total of ten aliquots taken from each tube of prepared particles/DNA.
[0281] Following bombardment, the embryos are incubated on 560P (maintenance medium) for 12 to 48 hours at temperatures ranging from 26 C to 37 C, and then placed at 26 C. After 5 to 7 days the embryos are transferred to 560R selection medium containing 3 mg/liter Bialaphos, and subcultured every 2 weeks at 26 C. After approximately 10 weeks of selection, selection-resistant callus clones are transferred to 288J medium to initiate plant regeneration. Following somatic embryo maturation (2-4 weeks), well-developed somatic embryos are transferred to medium for germination and transferred to a lighted culture room. Approximately 7-10 days later, developing plantlets are transferred to 272V hormone-free medium in tubes for 7-10 days until plantlets are well established. Plants are then transferred to inserts in flats (equivalent to a 2.5'' pot) containing potting soil and grown for 1 week in a growth chamber, subsequently grown an additional 1-2 weeks in the greenhouse, then transferred to Classic 600 pots (1.6 gallon) and grown to maturity. Plants are monitored and scored for transformation efficiency, and/or modification of regenerative capabilities.
[0282] Initiation medium (560 L) comprises 4.0 g/l N6 basal salts (SIGMA C-1416), 1.0 ml/l Eriksson's Vitamin Mix (1000.times. SIGMA-1511), 0.5 mg/l thiamine HCl, 20.0 g/l sucrose, 1.0 mg/l 2,4-D, and 2.88 g/l L-proline (brought to volume with D-I H.sub.2O following adjustment to pH 5.8 with KOH); 2.0 g/l Gelrite (added after bringing to volume with D-I H2O); and 8.5 mg/l silver nitrate (added after sterilizing the medium and cooling to room temperature).
[0283] Maintenance medium (560P) comprises 4.0 g/l N6 basal salts (SIGMA C-1416), 1.0 ml/l Eriksson's Vitamin Mix (1000.times. SIGMA-1511), 0.5 mg/l thiamine HCl, 30.0 g/l sucrose, 2.0 mg/l 2,4-D, and 0.69 g/l L-proline (brought to volume with D-I H2O following adjustment to pH 5.8 with KOH); 3.0 g/l Gelrite (added after bringing to volume with D-I H2O); and 0.85 mg/l silver nitrate (added after sterilizing the medium and cooling to room temperature).
[0284] Bombardment medium (560Y) comprises 4.0 g/l N6 basal salts (SIGMA C-1416), 1.0 ml/l Eriksson's Vitamin Mix (1000.times. SIGMA-1511), 0.5 mg/l thiamine HCl, 120.0 g/l sucrose, 1.0 mg/l 2,4-D, and 2.88 g/l L-proline (brought to volume with D-I H2O following adjustment to pH 5.8 with KOH); 2.0 g/l Gelrite (added after bringing to volume with D-I H2O); and 8.5 mg/l silver nitrate (added after sterilizing the medium and cooling to room temperature).
[0285] Selection medium (560R) comprises 4.0 g/l N6 basal salts (SIGMA C-1416), 1.0 ml/l Eriksson's Vitamin Mix (1000.times. SIGMA-1511), 0.5 mg/l thiamine HCl, 30.0 g/l sucrose, and 2.0 mg/l 2,4-D (brought to volume with D-I H2O following adjustment to pH 5.8 with KOH); 3.0 g/l Gelrite (added after bringing to volume with D-I H2O); and 0.85 mg/l silver nitrate and 3.0 mg/l bialaphos (both added after sterilizing the medium and cooling to room temperature).
[0286] Plant regeneration medium (288J) comprises 4.3 g/l MS salts (GIBCO 11117-074), 5.0 ml/l MS vitamins stock solution (0.100 g nicotinic acid, 0.02 g/l thiamine HCL, 0.10 g/l pyridoxine HCL, and 0.40 g/l glycine brought to volume with polished D-I H2O) (Murashige and Skoog (1962) Physiol. Plant. 15:473), 100 mg/l myo-inositol, 0.5 mg/l zeatin, 60 g/l sucrose, and 1.0 ml/l of 0.1 mM abscisic acid (brought to volume with polished D-I H2O after adjusting to pH 5.6); 3.0 g/l Gelrite (added after bringing to volume with D-I H2O); and 1.0 mg/l indoleacetic acid and 3.0 mg/l bialaphos (added after sterilizing the medium and cooling to 60.degree. C.). Hormone-free medium (272V) comprises 4.3 g/l MS salts (GIBCO 11117-074), 5.0 ml/l MS vitamins stock solution (0.100 g/l nicotinic acid, 0.02 g/l thiamine HCL, 0.10 g/l pyridoxine HCL, and 0.40 g/l glycine brought to volume with polished D-I H2O), 0.1 g/l myo-inositol, and 40.0 g/l sucrose (brought to volume with polished D-I H2O after adjusting pH to 5.6); and 6 g/l bacto-agar (added after bringing to volume with polished D-I H2O), sterilized and cooled to 60.degree. C.
[0287] b. Agrobacterium-Mediated Transformation
[0288] Agrobacterium-mediated transformation was performed essentially as described in Djukanovic et al. (2006) Plant Biotech J 4:345-57. Briefly, 10-12 day old immature embryos (0.8-2.5 mm in size) were dissected from sterilized kernels and placed into liquid medium (4.0 g/L N6 Basal Salts (Sigma C-1416), 1.0 ml/L Eriksson's Vitamin Mix (Sigma E-1511), 1.0 mg/L thiamine HCl, 1.5 mg/L 2,4-D, 0.690 g/L L-proline, 68.5 g/L sucrose, 36.0 g/L glucose, pH 5.2). After embryo collection, the medium was replaced with 1 ml Agrobacterium at a concentration of 0.35-0.45 OD550. Maize embryos were incubated with Agrobacterium for 5 min at room temperature, then the mixture was poured onto a media plate containing 4.0 g/L N6 Basal Salts (Sigma C-1416), 1.0 ml/L Eriksson's Vitamin Mix (Sigma E-1511), 1.0 mg/L thiamine HCl, 1.5 mg/L 2,4-D, 0.690 g/L L-proline, 30.0 g/L sucrose, 0.85 mg/L silver nitrate, 0.1 nM acetosyringone, and 3.0 g/L Gelrite, pH 5.8. Embryos were incubated axis down, in the dark for 3 days at 20.degree. C., then incubated 4 days in the dark at 28.degree. C., then transferred onto new media plates containing 4.0 g/L N6 Basal Salts (Sigma C-1416), 1.0 ml/L Eriksson's Vitamin Mix (Sigma E-1511), 1.0 mg/L thiamine HCl, 1.5 mg/L 2,4-D, 0.69 g/L L-proline, 30.0 g/L sucrose, 0.5 g/L MES buffer, 0.85 mg/L silver nitrate, 3.0 mg/L Bialaphos, 100 mg/L carbenicillin, and 6.0 g/L agar, pH 5.8. Embryos were subcultured every three weeks until transgenic events were identified. Somatic embryogenesis was induced by transferring a small amount of tissue onto regeneration medium (4.3 g/L MS salts (Gibco 11117), 5.0 ml/L MS Vitamins Stock Solution, 100 mg/L myo-inositol, 0.1 .mu.M ABA, 1 mg/L IAA, 0.5 mg/L zeatin, 60.0 g/L sucrose, 1.5 mg/L Bialaphos, 100 mg/L carbenicillin, 3.0 g/L Gelrite, pH 5.6) and incubation in the dark for two weeks at 28.degree. C. All material with visible shoots and roots were transferred onto media containing 4.3 g/L MS salts (Gibco 11117), 5.0 ml/L MS Vitamins Stock Solution, 100 mg/L myo-inositol, 40.0 g/L sucrose, 1.5 g/L Gelrite, pH 5.6, and incubated under artificial light at 28.degree. C. One week later, plantlets were moved into glass tubes containing the same medium and grown until they were sampled and/or transplanted into soil.
Example 14
Transient Expression of BBM Enhances Transformation
[0289] Parameters of the transformation protocol can be modified to ensure that the BBM activity is transient. One such method involves precipitating the BBM-containing plasmid in a manner that allows for transcription and expression, but precludes subsequent release of the DNA, for example, by using the chemical PEI. In one example, the BBM plasmid is precipitated onto gold particles with PEI, while the transgenic expression cassette (UBI::moPAT.about.GFPm::PinII; moPAT is the maize optimized PAT gene) to be integrated is precipitated onto gold particles using the standard calcium chloride method.
[0290] Briefly, gold particles were coated with PEI as follows. First, the gold particles were washed. Thirty-five mg of gold particles, 1.0 in average diameter (A.S.I. #162-0010), were weighed out in a microcentrifuge tube, and 1.2 ml absolute EtOH was added and vortexed for one minute. The tube was incubated for 15 minutes at room temperature and then centrifuged at high speed using a microfuge for 15 minutes at 4.degree. C. The supernatant was discarded and a fresh 1.2 ml aliquot of ethanol (EtOH) was added, vortexed for one minute, centrifuged for one minute, and the supernatant again discarded (this is repeated twice). A fresh 1.2 ml aliquot of EtOH was added, and this suspension (gold particles in EtOH) was stored at -20.degree. C. for weeks. To coat particles with polyethylimine (PEI; Sigma #P3143), 250 .mu.l of the washed gold particle/EtOH mix was centrifuged and the EtOH discarded. The particles were washed once in 100 .mu.l ddH2O to remove residual ethanol, 250 .mu.l of 0.25 mM PEI was added, followed by a pulse-sonication to suspend the particles and then the tube was plunged into a dry ice/EtOH bath to flash-freeze the suspension, which was then lyophilized overnight. At this point, dry, coated particles could be stored at -80.degree. C. for at least 3 weeks. Before use, the particles were rinsed 3 times with 250 .mu.l aliquots of 2.5 mM HEPES buffer, pH 7.1, with 1.times. pulse-sonication, and then a quick vortex before each centrifugation. The particles were then suspended in a final volume of 250 .mu.l HEPES buffer. A 25 .mu.l aliquot of the particles was added to fresh tubes before attaching DNA. To attach uncoated DNA, the particles were pulse-sonicated, then 1 .mu.g of DNA (in 5 .mu.l water) was added, followed by mixing by pipetting up and down a few times with a Pipetteman and incubated for 10 minutes. The particles were spun briefly (i.e. 10 seconds), the supernatant removed, and 60 .mu.l EtOH added. The particles with PEI-precipitated DNA-1 were washed twice in 60 .mu.l of EtOH. The particles were centrifuged, the supernatant discarded, and the particles were resuspended in 45 .mu.l water. To attach the second DNA (DNA-2), precipitation using TFX-50 was used. The 45 .mu.l of particles/DNA-1 suspension was briefly sonicated, and then 5 .mu.l of 100 ng/.mu.l of DNA-2 and 2.5 .mu.l of TFX-50 were added. The solution was placed on a rotary shaker for 10 minutes, centrifuged at 10,000 g for 1 minute. The supernatant was removed, and the particles resuspended in 60 .mu.l of EtOH. The solution was spotted onto macrocarriers and the gold particles onto which DNA-1 and DNA-2 had been sequentially attached were delivered into scutellar cells of 10 DAP Hi-II immature embryos using a standard protocol for the PDS-1000. For this experiment, the DNA-1 plasmid contained a UBI::RFP::pinII expression cassette, and DNA-2 contained a UBI::CFP::pinII expression cassette. Two days after bombardment, transient expression of both the CFP and RFP fluorescent markers was observed as numerous red & blue cells on the surface of the immature embryo. The embryos were then placed on non-selective culture medium and allowed to grow for 3 weeks before scoring for stable colonies. After this 3-week period, 10 multicellular, stably-expressing blue colonies were observed, in comparison to only one red colony. This demonstrated that PEI-precipitation could be used to effectively introduce DNA for transient expression while dramatically reducing integration of the PEI-introduced DNA and thus reducing the recovery of RFP-expressing transgenic events. In this manner, PEI-precipitation can be used to deliver transient expression of BBM and/or WUS2.
[0291] For example, the particles are first coated with UBI::BBM::pinII using PEI, then coated with UBI::moPAT-YFP using TFX-50, and then bombarded into scutellar cells on the surface of immature embryos. PEI-mediated precipitation results in a high frequency of transiently expressing cells on the surface of the immature embryo and extremely low frequencies of recovery of stable transformants (relative to the TFX-50 method). Thus, it is expected that the PEI-precipitated BBM cassette expresses transiently and stimulates a burst of embryogenic growth on the bombarded surface of the tissue (i.e. the scutellar surface), but this plasmid will not integrate. The PAT.about.GFP plasmid released from the Ca++/gold particles is expected to integrate and express the selectable marker at a frequency that results in substantially improved recovery of transgenic events. As a control treatment, PEI-precipitated particles containing a UBI::GUS::pinII (instead of BBM) are mixed with the PAT.about.GFP/Ca++ particles. Immature embryos from both treatments are moved onto culture medium containing 3 mg/l bialaphos. After 6-8 weeks, it is expected that GFP+, bialaphos-resistant calli will be observed in the PEI/BBM treatment at a much higher frequency relative to the control treatment (PEI/GUS).
[0292] As an alternative method, the BBM plasmid is precipitated onto gold particles with PEI, and then introduced into scutellar cells on the surface of immature embryos, and subsequent transient expression of the BBM gene elicits a rapid proliferation of embryogenic growth. During this period of induced growth, the explants are treated with Agrobacterium using standard methods for maize (see Example 1), with T-DNA delivery into the cell introducing a transgenic expression cassette such as UBI::moPAT.about.GFPm::pinII. After co-cultivation, explants are allowed to recover on normal culture medium, and then are moved onto culture medium containing 3 mg/l bialaphos. After 6-8 weeks, it is expected that GFP+, bialaphos-resistant calli will be observed in the PEI/BBM treatment at a much higher frequency relative to the control treatment (PEI/GUS).
[0293] It may be desirable to "kick start" callus growth by transiently expressing the BBM and/or WUS2 polynucleotide products. This can be done by delivering BBM and WUS2 5'-capped polyadenylated RNA, expression cassettes containing BBM and WUS2 DNA, or BBM and/or WUS2 proteins. All of these molecules can be delivered using a biolistics particle gun. For example 5'-capped polyadenylated BBM and/or WUS2 RNA can easily be made in vitro using Ambion's mMessage mMachine kit. RNA is co-delivered along with DNA containing a polynucleotide of interest and a marker used for selection/screening such as Ubi::moPAT.about.GFPm::PinII. It is expected that the cells receiving the RNA will immediately begin dividing more rapidly and a large portion of these will have integrated the agronomic gene. These events can further be validated as being transgenic clonal colonies because they will also express the PAT.about.GFP fusion protein (and thus will display green fluorescence under appropriate illumination). Plants regenerated from these embryos can then be screened for the presence of the polynucleotide of interest.
Example 15
Production and Model System Transformation of Somatic Soybean Embryo Cultures with Soybean Expression Vectors and Plant Regeneration
Culture Conditions:
[0294] Soybean embryogenic suspension cultures (cv. Jack) are maintained in 35 mL liquid medium SB196 (infra) on a rotary shaker, 150 rpm, 26.degree. C. with cool white fluorescent lights on 16:8 hr day/night photoperiod at light intensity of 60-85 .mu.E/m2/s. Cultures are subcultured every 7 days to two weeks by inoculating approximately 35 mg of tissue into 35 mL of fresh liquid SB196 (the preferred subculture interval is every 7 days).
[0295] Soybean embryogenic suspension cultures are transformed with the soybean expression plasmids by the method of particle gun bombardment (Klein et al., Nature 327:70 (1987)) using a DuPont Biolistic PDS1000/HE instrument (helium retrofit) for all transformations.
Soybean Embryogenic Suspension Culture Initiation:
[0296] Soybean cultures are initiated twice each month with 5-7 days between each initiation. Pods with immature seeds from available soybean plants are picked 45-55 days after planting. Seeds are removed from the pods and placed into a sterilized magenta box. The soybean seeds are sterilized by shaking them for 15 min in a 5% Clorox solution with 1 drop of Ivory soap (i.e., 95 mL of autoclaved distilled water plus 5 mL Clorox and 1 drop of soap, mixed well). Seeds are rinsed using 2 1-liter bottles of sterile distilled water and those less than 4 mm are placed on individual microscope slides. The small end of the seed is cut and the cotyledons pressed out of the seed coat. When cultures are being prepared for production transformation, cotyledons are transferred to plates containing SB1 medium (25-30 cotyledons per plate). Plates are wrapped with fiber tape and are maintained at 26.degree. C. with cool white fluorescent lights on 16:8 h day/night photoperiod at light intensity of 60-80 .mu.E/m2/s for eight weeks, with a media change after 4 weeks. When cultures are being prepared for model system experiments, cotyledons are transferred to plates containing SB199 medium (25-30 cotyledons per plate) for 2 weeks, and then transferred to SB1 for 2-4 weeks. Light and temperature conditions are the same as described above. After incubation on SB1 medium, secondary embryos are cut and placed into SB196 liquid media for 7 days.
Preparation of DNA for Bombardment:
[0297] Either an intact plasmid or a DNA plasmid fragment containing the genes of interest and the selectable marker gene are used for bombardment. Fragments from soybean expression plasmids are obtained by gel isolation of digested plasmids. In each case, 100 .mu.g of plasmid DNA is used in 0.5 mL of the specific enzyme mix described below. Plasmids are digested with AscI (100 units) in NEBuffer 4 (20 mM Tris-acetate, 10 mM magnesium acetate, 50 mM potassium acetate, 1 mM dithiothreitol, pH 7.9), 100 .mu.g/mL BSA, and 5 mM beta-mercaptoethanol at 37.degree. C. for 1.5 h. The resulting DNA fragments are separated by gel electrophoresis on 1% SeaPlaque GTG agarose (BioWhitaker Molecular Applications) and the DNA fragments containing gene cassettes are cut from the agarose gel. DNA is purified from the agarose using the GELase digesting enzyme following the manufacturer's protocol.
[0298] A 50 .mu.L aliquot of sterile distilled water containing 3 mg of gold particles (3 mg gold) is added to 30 .mu.L of a 10 ng/.mu.L DNA solution (either intact plasmid or DNA fragment prepared as described herein), 25 .mu.L 5M CaCl.sub.2) and 20 .mu.L of 0.1 M spermidine. The mixture is shaken 3 min on level 3 of a vortex shaker and spun for 10 sec in a bench microfuge. The supernatant is removed, followed by a wash with 400 .mu.L 100% ethanol and another brief centrifugation. The 400 .mu.L ethanol is removed and the pellet is resuspended in 40 .mu.L of 100% ethanol. Five .mu.L of DNA suspension is dispensed to each flying disk of the Biolistic PDS1000/HE instrument disk. Each 5 .mu.L aliquot contains approximately 0.375 mg gold per bombardment (e.g., per disk).
[0299] For model system transformations, the protocol is identical except for a few minor changes (i.e., 1 mg of gold particles is added to 5 .mu.L of a 1 .mu.g/.mu.L DNA solution, 50 .mu.L of a 2.5M CaCl.sub.2) is used and the pellet is ultimately resuspended in 85 .mu.L of 100% ethanol thus providing 0.058 mg of gold particles per bombardment).
Tissue Preparation and Bombardment with DNA:
[0300] Approximately 150-200 mg of seven day old embryogenic suspension cultures is placed in an empty, sterile 60.times.15 mm petri dish and the dish is covered with plastic mesh. The chamber is evacuated to a vacuum of 27-28 inches of mercury, and tissue is bombarded one or two shots per plate with membrane rupture pressure set at 1100 PSI. Tissue is placed approximately 3.5 inches from the retaining/stopping screen. Model system transformation conditions are identical except 100-150 mg of embryogenic tissue is used, rupture pressure is set at 650 PSI and tissue is place approximately 2.5 inches from the retaining screen.
Selection of Transformed Embryos:
[0301] Transformed embryos are selected either using hygromycin (when the hygromycin B phosphotransferase (HPT) gene is used as the selectable marker) or chlorsulfuron (when the acetolactate synthase (ALS) gene is used as the selectable marker).
[0302] Following bombardment, the tissue is placed into fresh SB196 media and cultured as described above. Six to eight days post-bombardment, the SB196 is exchanged with fresh SB196 containing either 30 mg/L hygromycin or 100 ng/mL chlorsulfuron, depending on the selectable marker used. The selection media is refreshed weekly. Four to six weeks post-selection, green, transformed tissue is observed growing from untransformed, necrotic embryogenic clusters.
Embryo Maturation:
[0303] For production transformations, isolated, green tissue is removed and inoculated into multiwell plates to generate new, clonally propagated, transformed embryogenic suspension cultures. Transformed embryogenic clusters are cultured for four-six weeks in multiwell plates at 26.degree. C. in SB196 under cool white fluorescent (Phillips cool white Econowatt F40/CW/RS/EW) and Agro (Phillips F40 Agro) bulbs (40 watt) on a 16:8 hr photoperiod with light intensity of 90-120 .mu.E/m.sup.2s. After this time embryo clusters are removed to a solid agar media, SB166, for one-two weeks and then subcultured to SB103 medium for 3-4 weeks to mature embryos. After maturation on plates in SB103, individual embryos are removed from the clusters, dried and screened for alterations in their fatty acid compositions as described in Example 7.
[0304] For model system transformations, embryos are matured in soybean histodifferentiation and maturation liquid medium (SHaM liquid media; Schmidt et al., Cell Biology and Morphogenesis 24:393 (2005)) using a modified procedure. Briefly, after 4 weeks of selection in SB196 as described above, embryo clusters are removed to 35 mL of SB228 (SHaM liquid media) in a 250 mL Erlenmeyer flask. Tissue is maintained in SHaM liquid media on a rotary shaker at 130 rpm and 26.degree. C. with cool white fluorescent lights on a 16:8 hr day/night photoperiod at a light intensity of 60-85 .mu.E/m2/s for 2 weeks as embryos mature. Embryos grown for 2 weeks in SHaM liquid media are equivalent in size and fatty acid content to embryos cultured on SB166/SB103 for 5-8 weeks.
Media Recipes:
TABLE-US-00006 [0305] SB 196 - FN Lite Liquid Proliferation Medium (per liter) MS FeEDTA - 100x Stock 1 10 mL MS Sulfate - 100x Stock 2 10 mL FN Lite Halides - 100x Stock 3 10 mL FN Lite P, B, Mo - 100x Stock 4 10 mL B5 vitamins (1 mL/L) 1.0 mL 2,4-D (10 mg/L final concentration) 1.0 mL KNO.sub.3 2.83 gm (NH.sub.4).sub.2SO.sub.4 0.463 gm asparagine 1.0 gm sucrose (1%) 10 gm pH 5.8
FN Lite Stock Solutions
TABLE-US-00007 [0306] Stock Number 1000 mL 500 mL 1 MS Fe EDTA 100x Stock Na.sub.2 EDTA* 3.724 g 1.862 g FeSO.sub.4--7H.sub.2O 2.784 g 1.392 g 2 MS Sulfate 100x stock MgSO.sub.4--7H.sub.2O 37.0 g 18.5 g MnSO.sub.4--H.sub.2O 1.69 g 0.845 g ZnSO.sub.4--7H.sub.2O 0.86 g 0.43 g CuSO.sub.4--5H.sub.2O 0.0025 g 0.00125 g 3 FN Lite Halides 100x Stock CaCl.sub.2--2H.sub.2O 30.0 g 15.0 g KI 0.083 g 0.0715 g CoCl.sub.2--6H.sub.2O 0.0025 g 0.00125 g 4 FN Lite P, B, Mo 100x Stock KH.sub.2PO.sub.4 18.5 g 9.25 g H.sub.3BO.sub.3 0.62 g 0.31 g Na.sub.2MoO.sub.4--2H.sub.2O 0.025 g 0.0125 g *Add first, dissolve in dark bottle while stirring
SB1 Solid Medium (Per Liter)
[0307] 1 package MS salts (Gibco/BRL--Cat. No. 11117-066)
[0308] 1 mL B5 vitamins 1000.times. stock
[0309] 31.5 g glucose
[0310] 2 mL 2,4-D (20 mg/L final concentration)
[0311] pH 5.7
[0312] 8 g TC agar
SB199 Solid Medium (Per Liter)
[0313] 1 package MS salts (Gibco/BRL--Cat. No. 11117-066)
[0314] 1 mL B5 vitamins 1000.times. stock
[0315] 30 g Sucrose
[0316] 4 ml 2,4-D (40 mg/L final concentration)
[0317] pH 7.0
[0318] 2 gm Gelrite
SB 166 Solid Medium (Per Liter)
[0319] 1 package MS salts (Gibco/BRL--Cat. No. 11117-066)
[0320] 1 mL B5 vitamins 1000.times. stock
[0321] 60 g maltose
[0322] 750 mg MgCl.sub.2 hexahydrate
[0323] 5 g activated charcoal
[0324] pH 5.7
[0325] 2 g gelrite
SB 103 Solid Medium (Per Liter)
[0326] 1 package MS salts (Gibco/BRL--Cat. No. 11117-066)
[0327] 1 mL B5 vitamins 1000.times. stock
[0328] 60 g maltose
[0329] 750 mg MgCl2 hexahydrate
[0330] pH 5.7
[0331] 2 g gelrite
SB 71-4 Solid Medium (Per Liter)
[0332] 1 bottle Gamborg's B5 salts w/sucrose (Gibco/BRL--Cat. No. 21153-036)
[0333] pH 5.7
[0334] 5 g TC agar
2,4-D Stock
[0335] Obtain premade from Phytotech Cat. No. D 295--concentration 1 mg/mL
B5 Vitamins Stock (Per 100 mL)
[0336] Store aliquots at -20.degree. C.
[0337] 10 g myo-inositol
[0338] 100 mg nicotinic acid
[0339] 100 mg pyridoxine HCl
[0340] 1 g thiamine
If the solution does not dissolve quickly enough, apply a low level of heat via the hot stir plate.
SB 228--Soybean Histodifferentiation & Maturation (SHaM) (Per Liter)
TABLE-US-00008 [0341] DDI H.sub.2O 600 mL FN-Lite Macro Salts for SHaM 10X 100 mL MS Micro Salts 1000x 1 mL MS FeEDTA 100x 10 mL CaCl 100x 6.82 mL B5 Vitamins 1000x 1 mL L-Methionine 0.149 g Sucrose 30 g Sorbitol 30 g Adjust volume to 900 mL pH 5.8 Autoclave Add to cooled media (.ltoreq.30.degree. C.): *Glutamine (final concentration 30 mM) 4% 110 mL *Note: Final volume will be 1010 mL after glutamine addition.
Since glutamine degrades relatively rapidly, it may be preferable to add immediately prior to using media. Expiration 2 weeks after glutamine is added; base media can be kept longer w/o glutamine.
FN-Lite Macro for SHAM 10.times.--Stock #1 (Per Liter)
TABLE-US-00009 [0342] (NH.sub.4)2SO.sub.4 (ammonium sulfate) 4.63 g KNO.sub.3 (potassium nitrate) 28.3 g MgSO.sub.4*7H.sub.20 (magnesium sulfate heptahydrate) 3.7 g KH.sub.2PO.sub.4 (potassium phosphate, monobasic) 1.85 g Bring to volume Autoclave
MS Micro 1000.times.--Stock #2 (Per 1 Liter)
TABLE-US-00010 [0343] H.sub.3BO.sub.3 (boric acid) 6.2 g MnSO.sub.4*H.sub.2O (manganese sulfate monohydrate) 16.9 g ZnSO.sub.4*7H20 (zinc sulfate heptahydrate) 8.6 g Na.sub.2MoO.sub.4*2H20 (sodium molybdate dihydrate) 0.25 g CuSO.sub.4*5H.sub.20 (copper sulfate pentahydrate) 0.025 g CoCl.sub.2*6H.sub.20 (cobalt chloride hexahydrate) 0.025 g KI (potassium iodide) 0.8300 g Bring to volume Autoclave
FeEDTA 100.times.--Stock #3 (Per Liter)
TABLE-US-00011 [0344] Na.sub.2EDTA* (sodium EDTA) 3.73 g FeSO.sub.4*7H.sub.20 (iron sulfate heptahydrate) 2.78 g *EDTA must be completely dissolved before adding iron. Bring to Volume Solution is photosensitive. Bottle(s) should be wrapped in foil to omit light. Autoclave
Ca 100.times.--Stock #4 (Per Liter)
TABLE-US-00012 [0345] CaCl2*2H.sub.20 (calcium chloride dihydrate) 44 g Bring to Volume Autoclave
B5 Vitamin 1000.times.--Stock #5 (Per Liter)
TABLE-US-00013 [0346] Thiamine*HCl 10 g Nicotinic Acid 1 g Pyridoxine*HCl 1 g Myo-Inositol 100 g Bring to Volume Store frozen
4% Glutamine--Stock #6 (Per Liter)
TABLE-US-00014 [0347] DDI water heated to 30.degree. C. 900 mL L-Glutamine 40 g Gradually add while stirring and applying low heat. Do not exceed 35.degree. C. Bring to Volume Filter Sterilize Store frozen* * Note: Warm thawed stock in 31.degree. C. bath to fully dissolve crystals.
Regeneration of Soybean Somatic Embryos into Plants:
[0348] In order to obtain whole plants from embryogenic suspension cultures, the tissue must be regenerated. Embyros are matured as described in above. After subculturing on medium SB103 for 3 weeks, individual embryos can be removed from the clusters and screened for alterations in their fatty acid compositions as described in Example 7. It should be noted that any detectable phenotype, resulting from the expression of the genes of interest, could be screened at this stage. This would include, but not be limited to, alterations in fatty acid profile, protein profile and content, carbohydrate content, growth rate, viability, or the ability to develop normally into a soybean plant.
[0349] Matured individual embryos are desiccated by placing them into an empty, small petri dish (35.times.10 mm) for approximately 4 to 7 days. The plates are sealed with fiber tape (creating a small humidity chamber). Desiccated embryos are planted into SB71-4 medium where they are left to germinate under the same culture conditions described above. Germinated plantlets are removed from germination medium and rinsed thoroughly with water and then are planted in Redi-Earth in 24-cell pack tray, covered with clear plastic dome. After 2 weeks the dome is removed and plants hardened off for a further week. If plantlets looked hardy they are transplanted to 10'' pot of Redi-Earth with up to 3 plantlets per pot. After 10 to 16 weeks, mature seeds are harvested, chipped and analyzed for fatty acids.
Example 16
Yeast Screening System for Meganuclease Activity
[0350] Yeast screening strains were generated as hosts for the screening of meganuclease activity. The yeast Ade2 gene (Genetika 1987 Jul. 23(7):1141-8) (SEQ ID NO: 36) was used as a visible marker as well as a selection in the scheme depicted in FIG. 7. Gene fragments corresponding to the first 1000 nucleotides of the Ade2 coding sequence (Ade2 5' fragment) and the last 1011 nucleotides of the Ade2 coding sequence (Ade2 3' fragment) were disrupted by a
[0351] There are 305 nucleotides of sequence duplication between the Ade2 5' fragment and the Ade2 3' fragment. The resulting constructs were used to replace the Ade2 gene (chromosome 15 nucleotide position 566193-564480) of yeast strain BY4247. The resulting yeast screening strains VER8145, VER8189 and HD1327 can be characterized as BY4742 MATa his3delta1 leu2delta0 lys2delta0 ura3delta0 Gal2+). If meganuclease cutting occurs between the duplicated sequences, homologous recombination can occur, resulting in a functional Ade2 gene.
[0352] The generation of a functional Ade2 gene can be used as a selection: when yeast cells are grown on media lacking adenine, only those with a functional Ade2 gene are able to grow.
[0353] The generation of a functional Ade2 gene can also be used as a screen. Yeast cells with a functional Ade2 gene are white, whereas those lacking Ade2 function exhibit red pigmentation due to accumulation of a metabolite earlier in the adenine biosynthetic pathway resulting in red colonies with white sectors as shown in FIGS. 8 and 9. The degree of white sectoring, sometimes extending to entire colonies, indicates the amount of meganuclease cutting activity. Since the sectoring phenotype is a qualitative measure of meganuclease activity, a 0-4 numerical scoring system was implemented. As shown in FIG. 9, a score of 0 indicates that no white sectors (no meganuclease cutting) were observed; a score of 4 indicates completely white colonies (complete cutting of the recognition site); scores of 1-3 indicate intermediate white sectoring phenotypes (and intermediate degrees of recognition site cutting).
Example 17
Analysis of Variant Recognition Sequences for LIG3-4 and MHP14+ Meganuclease in Maize
[0354] To demonstrate the cleavage activity of the variant recognition sites identified herein in planta, a naturally occurring variant recognition site (SEQ ID NO.: 11) for the MHP14+ meganuclease was identified in maize and its in planta cleavage activity (as measure by the frequency of mutagenesis of the recognition site) and in vitro cleavage activity was compared with the cleavage activity of the MHP14+ intended recognition site.
[0355] To determine the cleavage activity of the recognition sites in planta, plasmid DNA containing the MHP14+ meganuclease expression cassette was delivered to maize embryos via particle bombardment to allow for double strand break to occur, followed by realtime PCR that was carried out with TaqMan assays spanning the recognition sites. The relative copy number was calculated via the .DELTA..DELTA.Ct method relative to an internal control TaqMan assay using untransformed embryos as a calibrator. Embryos with a relative copy number less than 0.8 were considered to be cleaved and/or mutagenized. Meganuclease in vitro cleavage activity was assayed as described in Example 9 on plasmid DNA.
[0356] The plasmid DNA in vitro cleavage activity at the variant recognition site of SEQ ID NO:11 was approximately 3 times as efficient as the intended recognition site of SEQ ID NO: 14, depending on the reaction temperature (see FIG. 10A). A similar trend was observed in planta with 35% of the mature embryos demonstrating cleavage at the endogenous variant recognition site, as evidenced by the mutagenesis, while only 15% exhibited cleavage (mutagenesis) at the intended recognition site (see FIG. 10B).
[0357] Similar analysis can be performed for the LIG3-4 variant recognitions sites described herein or for any other variant recognition sites identified. It is expected that site modification rates including deletion, insertion, or any combination of the two would be enhanced for the other variant recognition sites with improved cleavage activity identified in Example 10 (-7C, 8T (Lig3-4); -11C, -7C, -2G, -1T, +8T (Lig3-4); -11C, -7C, -1T, +8T (Lig3-4); -3A, -2G, -1T, +2A, +7T, +8G, +11G (MHP14+); -3A, -2G, -1T, +2A, +7T, +8G, +11A (MHP14+); -3A, -2G, -1T, +7T, +8G, +11G (MHP14+); -2G, -1T, +2A, +7T, +8G, +11G (MHP14+); -2G, -1T, +7T, +8G, +11G (MHP14+), corresponding to SEQ ID NOs: 13-35) or any other variant recognition site identified by the method described herein were to be artificially introduced into the genome.
Example 18
Analysis of Variant Recognition Sequences for LIG3-4 and MHP14+ Meganuclease in Soybean
[0358] To test the intended and variant recognition sites for LIG3-4 and MHP14+ meganucleases in a dicotyledonous plant like soybean, the maize recognition sites sequences can be cloned into transformation DNA constructs and introduced in soybean by biolistic transformation as described in Example 15.
Comparing Multiple Recognition Sites Located at Same Locus
[0359] In order to compare the cleavage activity (cutting efficiencies) of different recognition sequences, one can arrange several recognition sequences together in one DNA construct and insert different constructs containing the multiple recognitions sites preferably at the same genomic locus to eliminate position effects. The FLP/FRT mediated site-specific integration transformation system is a valuable tool to achieve the above purpose by placing different donor DNA constructs at previously characterized targets sites (Plant Physiology, Li et al., 2009; U.S. application Ser. No. 12/634,775). Once the recognitions sites are integrated in the soybean genome, single copy transgenic events can be identified, characterized and selected as new materials for subsequent transformation with corresponding meganucleases to evaluate the cleavage activity (cutting efficiency) of each recognition sites by its corresponding meganuclease. Since several recognition sites are inserted at the same genomic site, the cutting efficiency of the corresponding meganucleases can be compared.
[0360] Towards this end, the intended recognition site for LIG34 (SEQ ID NO:13) as well as the intended recognition sites for MHP14+(SEQ ID NO:14) and MS26 (SEQ ID NO: 37) were cloned into a SSI donor construct PHP57712 (SEQ ID NO: 38) between the selectable marker gene GM-ALS and trait gene cassettes DGAT2 (diacylglycerol acyltransferase) over-expression for high oil, FAD3 (.omega.-3 desaturase) artificial microRNA co-supression for high unsaturated fatty acids, and GAS (galactinol synthase) hairpin co-suppression for high available energy (FIG. 11 A). Transgenic events with the PHP57712 donor DNA integrated at several previously characterized genomic sites were obtained by biolistic particle soybean SSI transformation as described above. Transgenic events with clean insertions of the trait genes and meganuclease recognition sequences were selected and will be used for next round transformation with LIG34, MS26, and MHP14+ meganucleases to test the cleavage activity of the three intended recognition sites.
Comparing Intended Versus Variant Recognition Sites Located at Same Locus
[0361] A variant recognition site for LIG3-4 (SEQ ID NO:22) and a variant recognition site for MHP14+ (SEQ ID NO:35) as well as the intended recognition site for MS26 (SEQ ID NO: 37) were cloned into another SSI donor construct, PHP62252 (SEQ ID NO: 39) (FIG. 11 B) and are being transformed to some of the same soybean genomic sites by SSI transformation. Transgenic events with clean insertions of the trait genes and meganuclease recognition sequences will be selected and used for another round transformation with MS26, MHP14+, and LIG34 meganucleases to test the cleavage activities of the recognition sites. Since all the recognition sites are inserted at the same genomic sites, the cutting efficiencies of the various recognition sites can be more meaningfully compared. The cleavage activity of the variant recognition sites of LIG3-4 and MHP14+(SEQ ID NOs: 22 and 35) can be compared to the cleavage activity of their intended recognition sites (SEQ ID NO: 13 and 14) as described in Example 17.
Example 19
[0362] A Method for Targeting the Insertion of a Polynucleotide of Interest to a Specific Chromosomal Site within a Plant Genome.
[0363] A nucleotide sequence comprising a variable recognition sequence for a double strand break inducing agent is introduced into the target organism's genome establishing a target site (comprising the variable recognition sequence) for insertion of a nucleotide sequences of interest. A library of stable plants or cultured tissues can then be established comprising a variable recognition site at different locations throughout the plant genome.
[0364] One example of such variant recognition sites are SEQ ID NOs: 15, 16, 17, 18, 19, 20 and 21 which can be cleaved by the LIG3-4 meganuclease encoded by SEQ ID NO: 1. Another example of variant recognition sites are SEQ ID NOs: 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 and 35 which can be cleaved by the MHP14+ meganuclease encoded by SEQ ID NO: 3. In one embodiment, the SEQ ID NOs: 15, 16, 17, 18, 19, 20 and 21 and are SEQ ID NOs: 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 and 35 are not endogenous to the maize or plant genome.
[0365] Once a stable plant or cultured tissue is established, a DNA fragment comprising a polynucleotide of interest is introduced into the stably transformed plant or tissues in the presence of a double strand break inducing protein such as a meganuclease protein. This process results in the insertion of the polynucleotide of interest into the variable recognition sequence.
[0366] It is recognized that the transformed plant may comprise multiple target sites, for example, but not limited to, multiple recognition sites capable of being cleaved by a double strand break inducing agent, as well as recombination sites such as FRT sites or LOX sites. Examples of recombination sites are known in the art and include FRT sites (See, for example, Schlake and Bode (1994) Biochemistry 33:12746-12751; Huang et al. (1991) Nucleic Acids Research 19:443-448).
Example 20
Capture of Cas Endonuclease Genomic Variant Recognition Sites & Generation of Libraries for Illumina Deep Sequencing
[0367] To capture genomic DNA variant recognition sites for rare cutting DNA double-strand-break inducing agents where most of the cleaved products result in blunt-ended termini such as for the Cas endonucleases (Gasiunas et al. (2012) Proc. Natl. Acad. Sci. USA 109:E2579-86, Jinek et al. (2012) Science 337:816-21), the addition of an adenine to the 3' termini of cleaved genomic DNA variant recognition site(s) may be utilized. Adapters containing a complementary 3' thymine overhang may then be used to selectively ligate to and enrich for the blunt-ended termini resulting from cleavage by the Cas endonuclease.
[0368] To generate material for the capture of genomic variant recognition sites, in vitro digestion assays would be carried-out and purified essentially as described in Example 3 except purified Cas endonuclease protein and the nucleic acid component(s) need to form a functional Cas endonuclease complex capable of cleaving a DNA target site would be used instead of a meganuclease protein. In vitro reactions may be carried-out in a different buffer, at different temperatures and or length(s) of incubation to foster ideal Cas endonuclease cleavage conditions.
[0369] A single 3' adenine overhang will then be added to the Cas endonuclease cleaved blunt-ended termini by incubating the in vitro digested genomic DNA at 37.degree. C. for 30 minutes in a 50 .mu.l reaction containing ATP, 1.times. Klenow buffer (NEBnext), and 15 units Klenow (exo-) and purified. Non-phosphorylated or phosphoryalted biotinylated adapters synthesized and purified by HPLC containing a 3' thymine nucleotide overhang complementary to the adenine 3' nucleotide overhang may then be ligated to approximately 2 .mu.g of the Cas endonuclease digested 3' adenine extended genomic DNA in a 100 .mu.l T4 ligase reaction (NEB). The resulting adapter ligated Cas endonuclease intended and variant recognition sites may then be enriched for, sequenced and identified similar to that described in Examples 4, 6, 7 and 8.
Sequence CWU 1
1
3911053DNAartificial sequenceLIG3-4 meganuclease 1atgaacacca
agtacaacaa ggagttcctg ctctacctgg ccggcttcgt ggacggcgac 60ggctccatca
aggcgcagat caagccgaac cagtcctgca agttcaagca ccagctctcc
120ctgaccttcc aggtgaccca gaagacgcag aggcgctggt tcctcgacaa
gctggtcgac 180gagatcgggg tgggctacgt ctacgaccgc gggtcggtgt
ccgactacga gctctcccag 240atcaagcccc tgcacaactt cctcacccag
ctccagccgt tcctcaagct gaagcagaag 300caggcgaacc tcgtcctgaa
gatcatcgag cagctcccct cggccaagga gtccccggac 360aagttcctgg
aggtgtgcac gtgggtcgac cagatcgcgg ccctcaacga cagcaagacc
420cgcaagacga cctcggagac ggtgcgggcg gtcctggact ccctcccagg
atccgtggga 480ggtctatcgc catctcaggc atccagcgcc gcatcctcgg
cttcctcaag cccgggttca 540gggatctccg aagcactcag agctggagca
actaagtcca aggaattcct gctctacctg 600gccggcttcg tggacggcga
cggctccatc atcgcgtcca tcaagccgcg ccagtgctac 660aagttcaagc
acgagctccg cctggagttc accgtgaccc agaagacgca gaggcgctgg
720ttcctcgaca agctggtcga cgagatcggg gtgggctacg tctacgaccg
cgggtcggtg 780tccgactacc gcctctccca gatcaagccc ctgcacaact
tcctcaccca gctccagccg 840ttcctcaagc tgaagcagaa gcaggcgaac
ctcgtcctga agatcatcga gcagctcccc 900tcggccaagg agtccccgga
caagttcctg gaggtgtgca cgtgggtcga ccagatcgcg 960gccctcaacg
acagcaagac ccgcaagacg acctcggaga cggtgcgggc ggtcctggac
1020tccctcagcg agaagaagaa gtcgtccccc tga 10532350PRTartificial
sequenceLIG3-4 meganuclease 2Met Asn Thr Lys Tyr Asn Lys Glu Phe
Leu Leu Tyr Leu Ala Gly Phe1 5 10 15Val Asp Gly Asp Gly Ser Ile Lys
Ala Gln Ile Lys Pro Asn Gln Ser 20 25 30Cys Lys Phe Lys His Gln Leu
Ser Leu Thr Phe Gln Val Thr Gln Lys 35 40 45Thr Gln Arg Arg Trp Phe
Leu Asp Lys Leu Val Asp Glu Ile Gly Val 50 55 60Gly Tyr Val Tyr Asp
Arg Gly Ser Val Ser Asp Tyr Glu Leu Ser Gln65 70 75 80Ile Lys Pro
Leu His Asn Phe Leu Thr Gln Leu Gln Pro Phe Leu Lys 85 90 95Leu Lys
Gln Lys Gln Ala Asn Leu Val Leu Lys Ile Ile Glu Gln Leu 100 105
110Pro Ser Ala Lys Glu Ser Pro Asp Lys Phe Leu Glu Val Cys Thr Trp
115 120 125Val Asp Gln Ile Ala Ala Leu Asn Asp Ser Lys Thr Arg Lys
Thr Thr 130 135 140Ser Glu Thr Val Arg Ala Val Leu Asp Ser Leu Pro
Gly Ser Val Gly145 150 155 160Gly Leu Ser Pro Ser Gln Ala Ser Ser
Ala Ala Ser Ser Ala Ser Ser 165 170 175Ser Pro Gly Ser Gly Ile Ser
Glu Ala Leu Arg Ala Gly Ala Thr Lys 180 185 190Ser Lys Glu Phe Leu
Leu Tyr Leu Ala Gly Phe Val Asp Gly Asp Gly 195 200 205Ser Ile Ile
Ala Ser Ile Lys Pro Arg Gln Cys Tyr Lys Phe Lys His 210 215 220Glu
Leu Arg Leu Glu Phe Thr Val Thr Gln Lys Thr Gln Arg Arg Trp225 230
235 240Phe Leu Asp Lys Leu Val Asp Glu Ile Gly Val Gly Tyr Val Tyr
Asp 245 250 255Arg Gly Ser Val Ser Asp Tyr Arg Leu Ser Gln Ile Lys
Pro Leu His 260 265 270Asn Phe Leu Thr Gln Leu Gln Pro Phe Leu Lys
Leu Lys Gln Lys Gln 275 280 285Ala Asn Leu Val Leu Lys Ile Ile Glu
Gln Leu Pro Ser Ala Lys Glu 290 295 300Ser Pro Asp Lys Phe Leu Glu
Val Cys Thr Trp Val Asp Gln Ile Ala305 310 315 320Ala Leu Asn Asp
Ser Lys Thr Arg Lys Thr Thr Ser Glu Thr Val Arg 325 330 335Ala Val
Leu Asp Ser Leu Ser Glu Lys Lys Lys Ser Ser Pro 340 345
35031053DNAartificial sequenceMHP14+ meganuclease 3atgaacacca
agtacaacaa ggagttcctg ctctacctgg ccggcttcgt ggacggcgac 60ggctccatca
tcgcgcagat caagccgaac cagtcctaca agttcaagca ccagctcatg
120ctgaccttca ccgtgaccca gaagacgcag aggcgctggt tcctcgacaa
gctggtcgac 180gagatcgggg tgggcaaggt ccgcgaccgc gggtcggtgt
ccgactacat cctctcccag 240atcaagcccc tgcacaactt cctcacccag
ctccagccgt tcctcaagct gaagcagaag 300caggcgaacc tcgtcctgaa
gatcatcgag cagctcccct cggccaagga gtccccggac 360aagttcctgg
aggtgtgcac gtgggtcgac cagatcgcgg ccctcaacga cagcaagacc
420cgcaagacga cctcggagac ggtgcgggcg gtcctggact ccctcccagg
atccgtggga 480ggtctatcgc catctcaggc atccagcgcc gcatcctcgg
cttcctcaag cccgggttca 540gggatctccg aagcactcag agctggagca
actaagtcca aggaattcct gctctacctg 600gccggcttcg tggacggcga
cggctccatc atcgcggcga tcaagccgaa ccagtcctac 660aagttcaagc
accagctctc cctgaccttc accgtgaccc agaagacgca gaggcgctgg
720ttcctcgaca agctggtcga cgagatcggg gtgggctacg tccgcgacca
ggggtcggtg 780tcccactacc agctctccca gatcaagccc ctgcacaact
tcctcaccca gctccagccg 840ttcctcaagc tgaagcagaa gcaggcgaac
ctcgtcctga agatcatcga gcagctcccc 900tcggccaagg agtccccgga
caagttcctg gaggtgtgca cgtgggtcga ccagatcgcg 960gccctcaacg
acagcaagac ccgcaagacg acctcggaga cggtgcgggc ggttctagac
1020tccctcagcg agaagaagaa gtcgtccccc tga 10534350PRTartificial
sequenceMHP14+ meganuclease 4Met Asn Thr Lys Tyr Asn Lys Glu Phe
Leu Leu Tyr Leu Ala Gly Phe1 5 10 15Val Asp Gly Asp Gly Ser Ile Ile
Ala Gln Ile Lys Pro Asn Gln Ser 20 25 30Tyr Lys Phe Lys His Gln Leu
Met Leu Thr Phe Thr Val Thr Gln Lys 35 40 45Thr Gln Arg Arg Trp Phe
Leu Asp Lys Leu Val Asp Glu Ile Gly Val 50 55 60Gly Lys Val Arg Asp
Arg Gly Ser Val Ser Asp Tyr Ile Leu Ser Gln65 70 75 80Ile Lys Pro
Leu His Asn Phe Leu Thr Gln Leu Gln Pro Phe Leu Lys 85 90 95Leu Lys
Gln Lys Gln Ala Asn Leu Val Leu Lys Ile Ile Glu Gln Leu 100 105
110Pro Ser Ala Lys Glu Ser Pro Asp Lys Phe Leu Glu Val Cys Thr Trp
115 120 125Val Asp Gln Ile Ala Ala Leu Asn Asp Ser Lys Thr Arg Lys
Thr Thr 130 135 140Ser Glu Thr Val Arg Ala Val Leu Asp Ser Leu Pro
Gly Ser Val Gly145 150 155 160Gly Leu Ser Pro Ser Gln Ala Ser Ser
Ala Ala Ser Ser Ala Ser Ser 165 170 175Ser Pro Gly Ser Gly Ile Ser
Glu Ala Leu Arg Ala Gly Ala Thr Lys 180 185 190Ser Lys Glu Phe Leu
Leu Tyr Leu Ala Gly Phe Val Asp Gly Asp Gly 195 200 205Ser Ile Ile
Ala Ala Ile Lys Pro Asn Gln Ser Tyr Lys Phe Lys His 210 215 220Gln
Leu Ser Leu Thr Phe Thr Val Thr Gln Lys Thr Gln Arg Arg Trp225 230
235 240Phe Leu Asp Lys Leu Val Asp Glu Ile Gly Val Gly Tyr Val Arg
Asp 245 250 255Gln Gly Ser Val Ser His Tyr Gln Leu Ser Gln Ile Lys
Pro Leu His 260 265 270Asn Phe Leu Thr Gln Leu Gln Pro Phe Leu Lys
Leu Lys Gln Lys Gln 275 280 285Ala Asn Leu Val Leu Lys Ile Ile Glu
Gln Leu Pro Ser Ala Lys Glu 290 295 300Ser Pro Asp Lys Phe Leu Glu
Val Cys Thr Trp Val Asp Gln Ile Ala305 310 315 320Ala Leu Asn Asp
Ser Lys Thr Arg Lys Thr Thr Ser Glu Thr Val Arg 325 330 335Ala Val
Leu Asp Ser Leu Ser Glu Lys Lys Lys Ser Ser Pro 340 345
350541DNAArtificial sequencedephosphorylated
adaptermisc_feature(38)..(41)n is a, c, g, or t 5ggttgacatg
ctggattgag acttccctgc aggacgtnnn n 41624DNAArtificial
sequenceprimer A 6gttgacatgc tggattgaga cttc 24721DNAArtificial
sequenceprimer B 7caagcagaag acggcatacg a 21861DNAArtificial
sequenceIllumina-compatibe adapter 8aatgatacgg cgaccaccga
gatctacact ctttccctac acgacgctct tccgatctgc 60a 6199DNAArtificial
sequencesequence tag 9gcaggacgt 9109DNAArtificial
sequencecomplement of sequence tag 10acgtcctgc 91122DNAArtificial
sequenceFigure 2 5'-3' SEQUENCE 11caagaagaag cacgtcagct ta
221222DNAArtificial sequenceFigure 2 3'-5' SEQUENCE 12gttcttcttc
gtgcagtcga at 221322DNAArtificial sequenceLIG3-4 Intended
Recognition Sequence 13atatacctca cacgtacgcg ta 221422DNAArtificial
sequenceMHP14+ Intended Recognition Sequence 14caaacagatt
cacgtcagat tt 221522DNAArtificial sequenceLIG3-4 variant
recognition sequence, -11 C 15ctatacctca cacgtacgcg ta
221622DNAArtificial sequenceLIG3-4 variant recognition sequence,
-7C 16atatccctca cacgtacgcg ta 221722DNAArtificial sequenceLIG3-4
variant recognition sequence, -2G 17atatacctcg cacgtacgcg ta
221822DNAArtificial sequenceLIG3-4 variant recognition sequence,
-1T 18atatacctca tacgtacgcg ta 221922DNAArtificial sequenceLIG3-4
variant recognition sequence, +8T 19atatacctca cacgtacgtg ta
222022DNAArtificial sequenceLIG3-4 variant recognition
sequence,-7C, +8T 20atatccctca cacgtacgtg ta 222122DNAArtificial
sequenceLIG3-4 variant recognition sequence, -11C, -7C, -2G, -1T,
+8T 21ctatccctcg tacgtacgtg ta 222222DNAArtificial sequenceLIG3-4
variant recognition sequence, -11C, -7C, -1T, +8T 22ctatccctca
tacgtacgtg ta 222322DNAArtificial sequenceMHP14+ variant
recognition sequence, -3A 23caaacagaat cacgtcagat tt
222422DNAArtificial sequenceMHP14+ variant recognition sequence,
-2G 24caaacagatg cacgtcagat tt 222522DNAArtificial sequenceMHP14+
variant recognition sequence, -1T 25caaacagatt tacgtcagat tt
222622DNAArtificial sequenceMHP14+ variant recognition sequence,
+2A 26caaacagatt caagtcagat tt 222722DNAArtificial sequenceMHP14+
variant recognition sequence, +7T 27caaacagatt cacgtcatat tt
222822DNAArtificial sequenceMHP14+ variant recognition sequence,
+8G 28caaacagatt cacgtcaggt tt 222922DNAArtificial sequenceMHP14+
variant recognition sequence, +11G 29caaacagatt cacgtcagat tg
223022DNAArtificial sequenceMHP14+ variant recognition sequence,
+11A 30caaacagatt cacgtcagat ta 223122DNAArtificial sequenceMHP14+
variant recognition sequence, -3A, -2G, -1T, +2A, +7T, +8G, +11G
31caaacagaag taagtcatgt tg 223222DNAArtificial sequenceMHP14+
variant recognition sequence, -3A, -2G, -1T, +2A, +7T, +8G, +11A
32caaacagaag taagtcatgt ta 223322DNAArtificial sequenceMHP14+
variant recognition sequence, 3A, -2G, -1T, +7T, +8G, +11G
33caaacagaag tacgtcatgt tg 223422DNAArtificial sequenceMHP14+
variant recognition sequence, -2G, -1T, +2A, +7T, +8G, +11G
34caaacagatg taagtcatgt tg 223522DNAArtificial sequenceMHP14+
variant recognition sequence,-2G, -1T, +7T, +8G, +11G 35caaacagatg
tacgtcatgt tg 22363716DNASaccharomyces cerevisiae 36ctactagaaa
gaagggcgtc aaaaatctgc tatgctatta cgaaattact ctataggtta 60gaatgtcatc
ccataggtat ggccctttta aagaagtatc tccacaatca attgcgagaa
120gcctatctag aatcaaaacg acactttatt tccaaaaagg gagattcgac
caacacttcc 180tctaccattg catcatcatc tttcgctggc gcatctgttc
ctctatcttc aaacgaatca 240ggaatgctaa acggcttgaa gcaaattaac
gaacaacaag aatctacatt agaaaccact 300caaaaggaag actagtaacg
ccgtatcgtg attaacgtat tacataagtt acaggattca 360tgcttatggg
ttagctattt cgcccaatgt gtccatctga cattactatt ttgcatttta
420atttaattag aacttgacta gcgcactacc agtatatcat ctcatttccg
taaataccaa 480atgtattata tattgaaagc ttttgaccag gttattataa
aagaaacttc atgctcgaaa 540aagatcattt cgaaaagttg cctagtttca
tgaaatttta aagcagttta tataaatttt 600accttttgat gcggaattga
ctttttcttg aataatacat aacttttctt aaaagaatca 660aagacagata
aaatttaaga gatattaaat attagtgaga agccgagaat tttgtaacac
720caacataaca ctgacatctt taacaacttt taattatgat acatttctta
cgtcatgatt 780gattattaca gctatgctga caaatgactc ttgttgcatg
gctacgaacc gggtaatact 840aagtgattga ctcttgctga ccttttatta
agaactaaat ggacaatatt atggagcatt 900tcatgtataa attggtgcgt
aaaatcgttg gatctctctt ctaagtacat cctactataa 960caatcaagaa
aaacaagaaa atcggacaaa acaatcaagt atggattcta gaacagttgg
1020tatattagga gggggacaat tgggacgtat gattgttgag gcagcaaaca
ggctcaacat 1080taagacggta atactagatg ctgaaaattc tcctgccaaa
caaataagca actccaatga 1140ccacgttaat ggctcctttt ccaatcctct
tgatatcgaa aaactagctg aaaaatgtga 1200tgtgctaacg attgagattg
agcatgttga tgttcctaca ctaaagaatc ttcaagtaaa 1260acatcccaaa
ttaaaaattt acccttctcc agaaacaatc agattgatac aagacaaata
1320tattcaaaaa gagcatttaa tcaaaaatgg tatagcagtt acccaaagtg
ttcctgtgga 1380acaagccagt gagacgtccc tattgaatgt tggaagagat
ttgggttttc cattcgtctt 1440gaagtcgagg actttggcat acgatggaag
aggtaacttc gttgtaaaga ataaggaaat 1500gattccggaa gctttggaag
tactgaagga tcgtcctttg tacgccgaaa aatgggcacc 1560atttactaaa
gaattagcag tcatgattgt gagatctgtt aacggtttag tgttttctta
1620cccaattgta gagactatcc acaaggacaa tatttgtgac ttatgttatg
cgcctgctag 1680agttccggac tccgttcaac ttaaggcgaa gttgttggca
gaaaatgcaa tcaaatcttt 1740tcccggttgt ggtatatttg gtgtggaaat
gttctattta gaaacagggg aattgcttat 1800taacgaaatt gccccaaggc
ctcacaactc tggacattat accattgatg cttgcgtcac 1860ttctcaattt
gaagctcatt tgagatcaat attggatttg ccaatgccaa agaatttcac
1920atctttctcc accattacaa cgaacgccat tatgctaaat gttcttggag
acaaacatac 1980aaaagataaa gagctagaaa cttgcgaaag agcattggcg
actccaggtt cctcagtgta 2040cttatatgga aaagagtcta gacctaacag
aaaagtaggt cacataaata ttattgcctc 2100cagtatggcg gaatgtgaac
aaaggctgaa ctacattaca ggtagaactg atattccaat 2160caaaatctct
gtcgctcaaa agttggactt ggaagcaatg gtcaaaccat tggttggaat
2220catcatggga tcagactctg acttgccggt aatgtctgcc gcatgtgcgg
ttttaaaaga 2280ttttggcgtt ccatttgaag tgacaatagt ctctgctcat
agaactccac ataggatgtc 2340agcatatgct atttccgcaa gcaagcgtgg
aattaaaaca attatcgctg gagctggtgg 2400ggctgctcac ttgccaggta
tggtggctgc aatgacacca cttcctgtca tcggtgtgcc 2460cgtaaaaggt
tcttgtctag atggagtaga ttctttacat tcaattgtgc aaatgcctag
2520aggtgttcca gtagctaccg tcgctattaa taatagtacg aacgctgcgc
tgttggctgt 2580cagactgctt ggcgcttatg attcaagtta tacaacgaaa
atggaacagt ttttattaaa 2640gcaagaagaa gaagttcttg tcaaagcaca
aaagttagaa actgtcggtt acgaagctta 2700tctagaaaac aagtaatata
taagtttatt gatatacttg tacagcaaat aattataaaa 2760tgatatacct
attttttagg ctttgttatg attacatcaa atgtggactt catacataga
2820aatcaacgct tacaggtgtc cttttttaag aatttcatac ataagatcac
ttattataca 2880tacatacata tccagtaaca agaagcaagg aataattacc
tgcttaagtc tgcgattaaa 2940aaaataacgt ttcgatacag ttcatataag
gcggctcaat gcagaaccga ggatagcgct 3000acgtcaggat atctttgtag
ttcccaaata taaatgcgac aatatagttt ctttctttca 3060tatcaataat
atccttttct ccactgaaat cacgaatcaa acctggagca aaaactaaag
3120ccaaattata aagcgtcatt cgattccagt gactgtaccg tgtaaccttt
tctatatgtt 3180cactcagtac tcttaacacc ctataatgtt cccttggaag
gtcttccaat atgtttttta 3240aagcgctctt gctcgacata taagtgtccg
aattctttgc ttctaaggac aactttcctc 3300caacaaatgg caagttttcc
atcatttttt tagatttaac taacctcatc aacggctcgt 3360atatttgaaa
ggtaaagata gggttgggga gctttcttaa gtatcgcttc aacacaccag
3420taacaacgtt gagatcttgt tccgttaaaa tatttggcgt ttcggtattt
tgttgtactt 3480tccatgcaga aaattgcttt tctatttctt ctatgactag
ctgggaacct gattttctat 3540aaatgccctc cgatctcata ttttcttcgt
ctgattcaat aaaatctatg cagacagata 3600gtatcatcgg tatttcattg
ttttcataat tgcacctagc aacgagactt gaaccataca 3660aattgcttcc
atccaaatat tcttctccat ctttactttg tcccatattt gcatcg
37163722DNAArtificial sequenceMS26 intended recognition sequence
37gatggtgacg tacgtgccct ac 223816824DNAArtificial sequenceplasmid
PHP57712misc_feature(12027)..(12027)n is a, c, g, or t 38ggagatccaa
gcttggcgcg ccggcctctg cctgcgttct gctgtggaag ttcctattcc 60gaagttccta
ttctccagaa agtataggaa cttcacatgc tgcctcgtgc aagtcacgat
120ctcgagttct atagtgtcac ctaaatcgta tgtgtatgat acataaggtt
atgtattaat 180tgtagccgcg ttctaacgac aatatgtcca tatggtgcac
tctcagtaca atctgctctg 240atgccgcata gttaagccag ccccgacacc
cgccaacacc cgctgacgcg ccctgacggg 300cttgtctgct cccggcatcc
gcttacagac aagctgtgac cgtctccggg agctgcatgt 360gtcagaggtt
ttcaccgtca tcaccgaaac gcgcgagacg aaagggcctc gtgatacgcc
420tatttttata ggttaatgtc atgaccaaaa tcccttaacg tgagttttcg
ttccactgag 480cgtcagaccc cgtagaaaag atcaaaggat cttcttgaga
tccttttttt ctgcgcgtaa 540tctgctgctt gcaaacaaaa aaaccaccgc
taccagcggt ggtttgtttg ccggatcaag 600agctaccaac tctttttccg
aaggtaactg gcttcagcag agcgcagata ccaaatactg 660tccttctagt
gtagccgtag ttaggccacc acttcaagaa ctctgtagca ccgcctacat
720acctcgctct gctaatcctg ttaccagtgg ctgctgccag tggcgataag
tcgtgtctta 780ccgggttgga ctcaagacga tagttaccgg ataaggcgca
gcggtcgggc tgaacggggg 840gttcgtgcac acagcccagc ttggagcgaa
cgacctacac cgaactgaga tacctacagc 900gtgagcattg agaaagcgcc
acgcttcccg aagggagaaa ggcggacagg tatccggtaa 960gcggcagggt
cggaacagga gagcgcacga gggagcttcc agggggaaac gcctggtatc
1020tttatagtcc tgtcgggttt cgccacctct gacttgagcg tcgatttttg
tgatgctcgt 1080caggggggcg gagcctatgg aaaaacgcca gcaacgcggc
ctttttacgg ttcctggcct 1140tttgctggcc ttttgctcac atgttctttc
ctgcgttatc ccctgattct gtggataacc 1200gtattaccgc ctttgagtga
gctgataccg ctcgccgcag ccgaacgacc gagcgcagcg 1260agtcagtgag
cgaggaagcg gaagagcgcc caatacgcaa accgcctctc cccgcgcgtt
1320ggccgattca ttaatgcagg ttgatcagat ctcgatcccg cgaaattaat
acgactcact 1380atagggagac cacaacggtt tccctctaga aataattttg
tttaacttta agaaggagat 1440atacccatgg aaaagcctga actcaccgcg
acgtctgtcg agaagtttct gatcgaaaag 1500ttcgacagcg tctccgacct
gatgcagctc tcggagggcg aagaatctcg tgctttcagc 1560ttcgatgtag
gagggcgtgg atatgtcctg cgggtaaata gctgcgccga tggtttctac
1620aaagatcgtt atgtttatcg gcactttgca tcggccgcgc tcccgattcc
ggaagtgctt 1680gacattgggg aattcagcga gagcctgacc tattgcatct
cccgccgtgc acagggtgtc 1740acgttgcaag acctgcctga aaccgaactg
cccgctgttc tgcagccggt cgcggaggct 1800atggatgcga tcgctgcggc
cgatcttagc cagacgagcg ggttcggccc attcggaccg 1860caaggaatcg
gtcaatacac tacatggcgt gatttcatat gcgcgattgc tgatccccat
1920gtgtatcact ggcaaactgt gatggacgac accgtcagtg cgtccgtcgc
gcaggctctc 1980gatgagctga tgctttgggc cgaggactgc cccgaagtcc
ggcacctcgt gcacgcggat 2040ttcggctcca acaatgtcct gacggacaat
ggccgcataa cagcggtcat tgactggagc 2100gaggcgatgt tcggggattc
ccaatacgag gtcgccaaca tcttcttctg gaggccgtgg 2160ttggcttgta
tggagcagca gacgcgctac ttcgagcgga ggcatccgga gcttgcagga
2220tcgccgcggc tccgggcgta tatgctccgc attggtcttg accaactcta
tcagagcttg 2280gttgacggca atttcgatga tgcagcttgg gcgcagggtc
gatgcgacgc aatcgtccga 2340tccggagccg ggactgtcgg gcgtacacaa
atcgcccgca gaagcgcggc cgtctggacc 2400gatggctgtg tagaagtact
cgccgatagt ggaaaccgac gccccagcac tcgtccgagg 2460gcaaaggaat
agtgaggtac agcttggatc gatccggctg ctaacaaagc ccgaaaggaa
2520gctgagttgg ctgctgccac cgctgagcaa taactagcat aaccccttgg
ggcctctaaa 2580cgggtcttga ggggtttttt gctgaaagga ggaactatat
ccggatgatc gtcgaggcct 2640cacgtgttaa cagaagttcc tattccgaag
ttcctattct ctagaaagta taggaacttc 2700caccacacaa cacaatggcg
gccaccgctt ccagaaccac ccgattctct tcttcctctt 2760cacaccccac
cttccccaaa cgcattacta gatccaccct ccctctctct catcaaaccc
2820tcaccaaacc caaccacgct ctcaaaatca aatgttccat ctccaaaccc
cccacggcgg 2880cgcccttcac caaggaagcg ccgaccacgg agcccttcgt
gtcacggttc gcctccggcg 2940aacctcgcaa gggcgcggac atccttgtgg
aggcgctgga gaggcagggc gtgacgacgg 3000tgttcgcgta ccccggcggt
gcgtcgatgg agatccacca ggcgctcacg cgctccgccg 3060ccatccgcaa
cgtgctcccg cgccacgagc agggcggcgt cttcgccgcc gaaggctacg
3120cgcgttcctc cggcctcccc ggcgtctgca ttgccacctc cggccccggc
gccaccaacc 3180tcgtgagcgg cctcgccgac gctttaatgg acagcgtccc
agtcgtcgcc atcaccggcc 3240aggtcagccg ccggatgatc ggcaccgacg
ccttccaaga aaccccgatc gtggaggtga 3300gcagatccat cacgaagcac
aactacctca tcctcgacgt cgacgacatc ccccgcgtcg 3360tcgccgaggc
tttcttcgtc gccacctccg gccgccccgg tccggtcctc atcgacattc
3420ccaaagacgt tcagcagcaa ctcgccgtgc ctaattggga cgagcccgtt
aacctccccg 3480gttacctcgc caggctgccc aggccccccg ccgaggccca
attggaacac attgtcagac 3540tcatcatgga ggcccaaaag cccgttctct
acgtcggcgg tggcagtttg aattccagtg 3600ctgaattgag gcgctttgtt
gaactcactg gtattcccgt tgctagcact ttaatgggtc 3660ttggaacttt
tcctattggt gatgaatatt cccttcagat gctgggtatg catggtactg
3720tttatgctaa ctatgctgtt gacaatagtg atttgttgct tgcctttggg
gtaaggtttg 3780atgaccgtgt tactgggaag cttgaggctt ttgctagtag
ggctaagatt gttcacattg 3840atattgattc tgccgagatt gggaagaaca
agcaggcgca cgtgtcggtt tgcgcggatt 3900tgaagttggc cttgaaggga
attaatatga ttttggagga gaaaggagtg gagggtaagt 3960ttgatcttgg
aggttggaga gaagagatta atgtgcagaa acacaagttt ccattgggtt
4020acaagacatt ccaggacgcg atttctccgc agcatgctat cgaggttctt
gatgagttga 4080ctaatggaga tgctattgtt agtactgggg ttgggcagca
tcaaatgtgg gctgcgcagt 4140tttacaagta caagagaccg aggcagtggt
tgacctcagg gggtcttgga gccatgggtt 4200ttggattgcc tgcggctatt
ggtgctgctg ttgctaaccc tggggctgtt gtggttgaca 4260ttgatgggga
tggtagtttc atcatgaatg ttcaggagtt ggccactata agagtggaga
4320atctcccagt taagatattg ttgttgaaca atcagcattt gggtatggtg
gttcagtggg 4380aggataggtt ctacaagtcc aatagagctc acacctatct
tggagatccg tctagcgaga 4440gcgagatatt cccaaacatg ctcaagtttg
ctgatgcttg tgggataccg gcagcgcgag 4500tgacgaagaa ggaagagctt
agagcggcaa ttcagagaat gttggacacc cctggcccct 4560accttcttga
tgtcattgtg ccccatcagg agcatgtgtt gccgatgatt cccagtaatg
4620gatccttcaa ggatgtgata actgagggtg atggtagaac gaggtactga
ttgcctagac 4680caaatgttcc ttgatgcttg ttttgtacaa tatatataag
ataatgctgt cctagttgca 4740ggatttggcc tgtggtgagc atcatagtct
gtagtagttt tggtagcaag acattttatt 4800ttccttttat ttaacttact
acatgcagta gcatctatct atctctgtag tctgatatct 4860cctgttgtct
gtattgtgcc gttggatttt ttgctgtagt gagactgaaa atgatgtgct
4920agtaataata tttctgttag aaatctaagt agagaatctg ttgaagaagt
caaaagctaa 4980tggaatcagg ttacatattc aatgtttttc tttttttagc
ggttggtaga cgtgtagatt 5040caacttctct tggagctcac ctaggcaatc
agtaaaatgc atattccttt tttaacttgc 5100catttattta cttttagtgg
aaattgtgac caatttgttc atgtagaacg gatttggacc 5160attgcgtcca
caaaacgtct cttttgctcg atcttcacaa agcgataccg aaatccagag
5220atagttttca aaagtcagaa atggcaaagt tataaatagt aaaacagaat
agatgctgta 5280atcgacttca ataacaagtg gcatcacgtt tctagttcta
gacccatcag gggcagatct 5340aggcgcgcgc catatacctc acacgtacgc
gtagatggtg acgtacgtgc cctaccaaac 5400agattcacgt cagatttgaa
gttcctattc cgaagttcct attctacata gagtatagga 5460acttccgata
tcactgcagt ggccggcggc gcgccgtcga cggatccgta cgatccatgc
5520ccttcatttg ccgcttatta attaatttgg taacagtccg tactaatcag
ttacttatcc 5580ttcccccatc ataattaatc ttggtagtct cgaatgccac
aacactgact agtctcttgg 5640atcataagaa aaagccaagg aacaaaagaa
gacaaaacac aatgagagta tcctttgcat 5700agcaatgtct aagttcataa
aattcaaaca aaaacgcaat cacacacagt ggacatcact 5760tatccactag
ctgatcagga tcgccgcgtc aagaaaaaaa aactggaccc caaaagccat
5820gcacaacaac acgtactcac aaaggtgtca atcgagcagc ccaaaacatt
caccaactca 5880acccatcatg agccctcaca tttgttgttt ctaacccaac
ctcaaactcg tattctcttc 5940cgccacctca tttttgttta tttcaacacc
cgtcaaactg catgccaccc cgtggccaaa 6000tgtccatgca tgttaacaag
acctatgact ataaatagct gcaatctcgg cccaggtttt 6060catcatcaag
aaccagttca atatcctagt acaccgtatt aaagaattta agatatactg
6120cggccgcatg actatcgact cacaatacta caagtcgcga gacaaaaacg
acacggcacc 6180caaaatcgcg ggaatccgat atgccccgct atcgacacca
ttactcaacc gatgtgagac 6240cttctctctg gtctggcaca ttttcagcat
tcccactttc ctcacaattt tcatgctatg 6300ctgcgcaatt ccactgctct
ggccatttgt gattgcgtat gtagtgtacg ctgttaaaga 6360cgactccccg
tccaacggag gagtggtcaa gcgatactcg cctatttcaa gaaacttctt
6420catctggaag ctctttggcc gctacttccc cataactctg cacaagacgg
tggatctgga 6480gcccacgcac acatactacc ctctggacgt ccaggagtat
cacctgattg ctgagagata 6540ctggccgcag aacaagtacc tccgagcaat
catcaccacc atcgagtact ttctgcccgc 6600cttcatgaaa cggtctcttt
ctatcaacga gcaggagcag cctgccgagc gagatcctct 6660cctgtctccc
gtttctccca gctctccggg ttctcaacct gacaagtgga ttaaccacga
6720cagcagatat agccgtggag aatcatctgg ctccaacggc cacgcctcgg
gctccgaact 6780taacggcaac ggcaacaacg gcaccactaa ccgacgacct
ttgtcgtccg cctctgctgg 6840ctccactgca tctgattcca cgcttcttaa
cgggtccctc aactcctacg ccaaccagat 6900cattggcgaa aacgacccac
agctgtcgcc cacaaaactc aagcccactg gcagaaaata 6960catcttcggc
taccaccccc acggcattat cggcatggga gcctttggtg gaattgccac
7020cgagggagct ggatggtcca agctctttcc gggcatccct gtttctctta
tgactctcac 7080caacaacttc cgagtgcctc tctacagaga gtacctcatg
agtctgggag tcgcttctgt 7140ctccaagaag tcctgcaagg ccctcctcaa
gcgaaaccag tctatctgca ttgtcgttgg 7200tggagcacag gaaagtcttc
tggccagacc cggtgtcatg gacctggtgc tactcaagcg 7260aaagggtttt
gttcgacttg gtatggaggt cggaaatgtc gcccttgttc ccatcatggc
7320ctttggtgag aacgacctct atgaccaggt tagcaacgac aagtcgtcca
agctgtaccg 7380attccagcag tttgtcaaga acttccttgg attcaccctt
cctttgatgc atgcccgagg 7440cgtcttcaac tacgatgtcg gtcttgtccc
ctacaggcga cccgtcaaca ttgtggttgg 7500ttcccccatt gacttgcctt
atctcccaca ccccaccgac gaagaagtgt ccgaatacca 7560cgaccgatac
atcgccgagc tgcagcgaat ctacaacgag cacaaggatg aatatttcat
7620cgattggacc gaggagggca aaggagcccc agagttccga atgattgagt
aagcggccgc 7680aagtatgaac taaaatgcat gtaggtgtaa gagctcatgg
agagcatgga atattgtatc 7740cgaccatgta acagtataat aactgagctc
catctcactt cttctatgaa taaacaaagg 7800atgttatgat atattaacac
tctatctatg caccttattg ttctatgata aatttcctct 7860tattattata
aatcatctga atcgtgacgg cttatggaat gcttcaaata gtacaaaaac
7920aaatgtgtac tataagactt tctaaacaat tctaacctta gcattgtgaa
cgagacataa 7980gtgttaagaa gacataacaa ttataatgga agaagtttgt
ctccatttat atattatata 8040ttacccactt atgtattata ttaggatgtt
aaggagacat aacaattata aagagagaag 8100tttgtatcca tttatatatt
atatactacc catttatata ttatacttat ccacttattt 8160aatgtcttta
taaggtttga tccatgatat ttctaatatt ttagttgata tgtatatgaa
8220aaggtactat ttgaactctc ttactctgta taaaggttgg atcatcctta
aagtgggtct 8280atttaatttt attgcttctt acagataaaa aaaaaattat
gagttggttt gataaaatat 8340tgaaggattt aaaataataa taaataacat
ataatatatg tatataaatt tattataata 8400taacatttat ctataaaaaa
gtaaatattg tcataaatct atacaatcgt ttagccttgc 8460tggaacgaat
ctcaattatt taaacgagag taaacatatt tgactttttg gttatttaac
8520aaattattat ttaacactat atgaaatttt tttttttatc agcaaagaat
aaaattaaat 8580taagaaggac aatggtgtcc caatccttat acaaccaact
tccacaagaa agtcaagtca 8640gagacaacaa aaaaacaagc aaaggaaatt
ttttaatttg agttgtcttg tttgctgcat 8700aatttatgca gtaaaacact
acacataacc cttttagcag tagagcaatg gttgaccgtg 8760tgcttagctt
cttttatttt atttttttat cagcaaagaa taaataaaat aaaatgagac
8820acttcaggga tgtttcaacg tactttctag acgtacgtct ttccacaata
cataactatt 8880aattaatctt aaataaataa aggataaaat attttttttt
cttcataaag ttaaaatatg 8940ttattttttg tttagatgta tattcgaata
aatctaaata tatgataatg attttttata 9000ttgattaaac atataatcaa
tattaaatat gatatttttt tatataggtt gtacacataa 9060ttttataagg
ataaaaaata tgataaaaat aaattttaaa tatttttata tttacgagaa
9120aaaaaaatat tttagccata aataaatgac cagcatattt tacaacctta
gtaattcata 9180aattcctata tgtatatttg aaattaaaaa cagataatcg
ttaagggaag gaatcctacg 9240tcatctcttg ccatttgttt ttcatgcaaa
cagaaaggga cgaaaaacca cctcaccatg 9300aatcactctt cacaccattt
ttactagcaa acaagtctca acaactgaag ccagctctct 9360ttccgtttct
ttttacaaca ctttctttga aatagtagta ttttttttca catgatttat
9420taacgtgcca aaagatgctt attgaataga gtgcacattt gtaatgtact
actaattaga 9480acatgaaaaa gcattgttct aacacgataa tcctgtgaag
gcgttaactc caaagatcca 9540atttcactat ataaattgtg acgaaagcaa
aatgaattca catagctgag agagaaagga 9600aaggttaact aagaagcaat
acttcagcgg ccgcttctag ctagctaggg tttgggtagt 9660gagtgtaata
aagttgcaaa gtttttggtt aggttacgtt ttgaccttat tattatagtt
9720caaagggaaa cattaattaa aggggattat gaagtgggct ctcttgattc
ttggatgagg 9780atcttactgg gtgaattgag ctgcttagct atggatccca
cagttctacc catcaataag 9840tgcttttgtg gtagtcttgt ggcttccata
tctggggagc ttcatttgcc tttatagtat 9900taaccttctc caagaacaaa
gagagcccac acccttctct tcttttctct cataataatt 9960taaatttgtt
atagactcta aactttaaat gttttttttg aagtttttcc gtttttctct
10020tttgccatga tcccgttctt gctgtggagt aaccttgtcc gaggtatgtg
catgattaga 10080tccatactta atttgtgtgc atcacgaagg tgaggttgaa
atgaactttg cttttttgac 10140cttttaggaa agttcttttg ttgcagtaat
caattttaat tagttttaat tgacactatt 10200acttttattg tcatctttgt
tagttttatt gttgaattga gtgcatattt cctaggaaat 10260tctcttacct
aacatttttt atacagatct atgctcttgg ctcttgccct tactcttggc
10320cttgtgttgg ttatttgtct acatatttat tgactggtcg atgagacatg
tcacaattct 10380tgggcttatt tgttggtcta ataaaaggag tgcttattga
aagatcaaga cggagattcg 10440gttttatata aataaactaa agatgacata
ttagtgtgtt gatgtctctt caggataatt 10500tttgtttgaa ataatatggt
aatgtcttgt ctaaatttgt gtacataatt cttactgatt 10560ttttggattg
ttggattttt ataaacaaat ctgcggccgc atgagccgta aaggttcaat
10620acaacgagtg cttgttttct tagggacaag cattgtactt atgtatgatt
ctgtgtaacc 10680atgagtcttc cacgttgtac taatgtgaag ggcaaaaata
aaacacagaa caagttcgtt 10740tttctcaaat aatgtgaagg tagaaaatgg
aaccatgcct cctctcttgc atgtgattta 10800aaatattagc agatggtacg
tcgagtcgac ctgcaggtcg actcgacgta cgtcctcgaa 10860gagaagggtt
aataacacat tttttaacat ttttaacaca aattttagtt atttaaaaat
10920ttattaaaaa atttaaaata agaagaggaa ctctttaaat aaatctaact
tacaaaattt 10980atgattttta ataagttttc accaataaaa aatgtcataa
aaatatgtta aaaagtatat 11040tatcaatatt ctctttatga taaataaaaa
gaaaaaaaaa ataaaagtta agtgaaaatg 11100agattgaagt gactttaggt
gtgtataaat atatcaaccc cgccaacaat ttatttaatc 11160caaatatatt
gaagtatatt attccatagc ctttatttat ttatatattt attatataaa
11220agctttattt gttctaggtt gttcatgaaa tatttttttg gttttatctc
cgttgtaaga 11280aaatcatgtg ctttgtgtcg ccactcacta ttgcagcttt
ttcatgcatt ggtcagattg 11340acggttgatt gtatttttgt tttttatggt
tttgtgttat gacttaagtc ttcatctctt 11400tatctcttca tcaggtttga
tggttaccta atatggtcca tgggtacatg catggttaaa 11460ttaggtggcc
aactttgttg tgaacgatag aatttttttt atattaagta aactattttt
11520atattatgaa ataataataa aaaaaatatt ttatcattat taacaaaatc
atattagtta 11580atttgttaac tctataataa aagaaatact gtaacattca
cattacatgg taacatcttt 11640ccaccctttc atttgttttt tgtttgatga
ctttttttct tgtttaaatt tatttccctt 11700cttttaaatt tggaatacat
tatcatcata tataaactaa aatactaaaa acaggattac 11760acaaatgata
aataataaca caaatattta taaatctagc tgcaatatat ttaaactagc
11820tatatcgata ttgtaaaata aaactagctg cattgatact gataaaaaaa
tatcatgtgc 11880tttctggact gatgatgcag tatacttttg acattgcctt
tattttattt ttcagaaaag 11940ctttcttagt tctgggttct tcattatttg
tttcccatct ccattgtgaa ttgaatcatt 12000tgcttcgtgt cacaaataca
atttagntag gtacatgcat tggtcagatt cacggtttat 12060tatgtcatga
cttaagttca tggtagtaca ttacctgcca cgcatgcatt atattggtta
12120gatttgatag gcaaatttgg ttgtcaacaa tataaatata aataatgttt
ttatattacg 12180aaataacagt gatcaaaaca aacagtttta tctttattaa
caagattttg tttttgtttg 12240atgacgtttt ttaatgttta cgctttcccc
cttcttttga atttagaaca ctttatcatc 12300ataaaatcaa atactaaaaa
aattacatat ttcataaata ataacacaaa tatttttaaa 12360aaatctgaaa
taataatgaa caatattaca tattatcacg aaaattcatt aataaaaata
12420ttatataaat aaaatgtaat agtagttata tgtaggaaaa aagtactgca
cgcataatat 12480atacaaaaag attaaaatga actattataa ataataacac
taaattaatg gtgaatcata 12540tcaaaataat gaaaaagtaa ataaaatttg
taattaactt ctatatgtat tacacacaca 12600aataataaat aatagtaaaa
aaaattatga taaatattta ccatctcata agatatttaa 12660aataatgata
aaaatataga ttatttttta tgcaactagc tagccaaaaa gagaacacgg
12720gtatatataa aaagagtacc tttaaattct actgtacttc ctttattcct
gacgttttta 12780tatcaagtgg acatacgtga agattttaat tatcagtcta
aatatttcat tagcacttaa 12840tacttttctg ttttattcct atcctataag
tagtcccgat tctcccaaca ttgcttattc 12900acacaactaa ctaagaaagt
cttccatagc cccccaagcg gccgctagtc gactaagtca 12960tcaactattc
caagctacgt atttgggagt ttgtggagta cagcaagatg atatacctag
13020acggtgatat ccaagttttt gacaacattg accacttgtt tgacttgcct
gataactact 13080tctatgcggt gatggactgt ttctgtgagc caacttgggg
ccacactaaa caatatcaga 13140tcggttactg ccagcagtgc ccccataagg
ttcagtggcc cactcacttt gggcccaaac 13200ctcctctcta tttcaatgct
ggcatgtttg tgtatgagcc caatttggct acttaccgtg 13260acctccttca
aacagtccaa gtcacccagc ccacttcctt tgctgaacag gattttttga
13320acatgtactt caaggacaaa tataggccaa ttcctaatgt ctacaatctt
gtgctggcca 13380tgctgtggcg tcaccctgag aacgttgagc ttgacaaagt
taaagtggtt cactactgtg 13440ctgctgggtc taagccttgg aggtacactg
ggaagtgact cgaggtcatc aattactcca 13500agctacgtat ttgggagttc
gtggagtaca agaagacgat atacctagac ggtgacatcc 13560aagtatttgg
aaacatagac cacttgtttg atctgcctga taattatttc tatgcggtga
13620tggattgttt ctgcgagaag acttggagcc acacccctca gttccagatt
gggtactgcc 13680aacagtgccc tgataaggtt caatggccct ctcactttgg
ttccaaacct cctctatatt 13740tcaatgctgg catgtttgtt tatgagccta
atctcgacac ctaccgtgat cttctccaaa 13800ctgtccaact caccaagccc
acttcttttg ctgagcagga ctttctcaac atgtacttca 13860aggacaagta
caagccaata ccgaacatgt acaaccttgt gctggccatg ttgtggcgtc
13920accctgaaaa tgttgaactt gataaagttc aagtggttca ttactgtgct
gctgggtcta 13980agccttggag gttcactggg aagtaactgc aggtcatcaa
ctactccaag ctccgtatat 14040gggagtttgt ggagtacagc aagatgatat
acttggacgg agacattgag gtatatgaga 14100acatagacca cctatttgac
ctacctgatg gtaactttta cgctgtgatg gattgtttct 14160gcgagaagac
atggagtcac acccctcagt acaaggtggg ttactgccag caatgcccgg
14220agaaggtgcg gtggcccacc gaattgggtc agcccccttc tctttacttc
aacgctggca 14280tgttcgtgtt cgaacccaac atcgccacct atcatgacct
attgaaaacg gtgcaagtca 14340ccactcccac ctcgttcgct gaacaagatt
tcttgaacat gtacttcaag gacatttaca 14400agccaatccc tttaaattac
aatcttgtcc tcgccatgct gtggcgccac ccggaaaacg 14460ttaaattaga
ccaagtcaag gttgttcact attgcgcagc ggggtccaag ccatggagat
14520atacggggaa gtagcctagg cgtacgcagg taagtttctg cttctacctt
tgatatatat 14580ataataatta tcattaatta gtagtaatat aatatttcaa
atattttttt caaaataaaa 14640gaatgtagta tatagcaatt gcttttctgt
agtttataag tgtgtatatt ttaatttata 14700acttttctaa tatatgacca
aaacatggtg atgtgcaggt cctaggctac ttccccgtat 14760atctccatgg
cttggacccc gctgcgcaat agtgaacaac cttgacttgg tctaatttaa
14820cgttttccgg gtggcgccac agcatggcga ggacaagatt gtaatttaaa
gggattggct 14880tgtaaatgtc cttgaagtac atgttcaaga aatcttgttc
agcgaacgag gtgggagtgg 14940tgacttgcac cgttttcaat aggtcatgat
aggtggcgat gttgggttcg aacacgaaca 15000tgccagcgtt gaagtaaaga
gaagggggct gacccaattc ggtgggccac cgcaccttct 15060ccgggcattg
ctggcagtaa cccaccttgt actgaggggt gtgactccat gtcttctcgc
15120agaaacaatc catcacagcg taaaagttac catcaggtag gtcaaatagg
tggtctatgt 15180tctcatatac ctcaatgtct ccgtccaagt atatcatctt
gctgtactcc acaaactccc 15240atatacggag cttggagtag ttgatgacct
gcagttactt cccagtgaac ctccaaggct 15300tagacccagc agcacagtaa
tgaaccactt gaactttatc aagttcaaca ttttcagggt 15360gacgccacaa
catggccagc acaaggttgt acatgttcgg tattggcttg tacttgtcct
15420tgaagtacat gttgagaaag tcctgctcag caaaagaagt gggcttggtg
agttggacag 15480tttggagaag atcacggtag gtgtcgagat taggctcata
aacaaacatg ccagcattga 15540aatatagagg aggtttggaa ccaaagtgag
agggccattg aaccttatca gggcactgtt 15600ggcagtaccc aatctggaac
tgaggggtgt ggctccaagt cttctcgcag aaacaatcca 15660tcaccgcata
gaaataatta tcaggcagat caaacaagtg gtctatgttt ccaaatactt
15720ggatgtcacc
gtctaggtat atcgtcttct tgtactccac gaactcccaa atacgtagct
15780tggagtaatt gatgacctcg agtcacttcc cagtgtacct ccaaggctta
gacccagcag 15840cacagtagtg aaccacttta actttgtcaa gctcaacgtt
ctcagggtga cgccacagca 15900tggccagcac aagattgtag acattaggaa
ttggcctata tttgtccttg aagtacatgt 15960tcaaaaaatc ctgttcagca
aaggaagtgg gctgggtgac ttggactgtt tgaaggaggt 16020cacggtaagt
agccaaattg ggctcataca caaacatgcc agcattgaaa tagagaggag
16080gtttgggccc aaagtgagtg ggccactgaa ccttatgggg gcactgctgg
cagtaaccga 16140tctgatattg tttagtgtgg ccccaagttg gctcacagaa
acagtccatc accgcataga 16200agtagttatc aggcaagtca aacaagtggt
caatgttgtc aaaaacttgg atatcaccgt 16260ctaggtatat catcttgctg
tactccacaa actcccaaat acgtagcttg gaatagttga 16320tgacttagtc
gactagcggc cgcgacacaa gtgtgagagt actaaataaa tgctttggtt
16380gtacgaaatc attacactaa ataaaataat caaagcttat atatgccttc
cgctaaggcc 16440gaatgcaaag aaattggttc tttctcgtta tcttttgcca
cttttactag tacgtattaa 16500ttactactta atcatctttg tttacggctc
attatatccg gtctaggcca aggccgcgaa 16560gttaaaagca atgttgtcac
ttgtacgtac taacacatga tgtgatagtt tatgctagct 16620agctataaca
taagctgtct ctgagtgtgt tgtatattaa taaagatcat cactggtgaa
16680tggtgatcgt gtacgtaccc tacttagtag gcaatggaag cacttagagt
gtgctttgtg 16740catggccttg cctctgtttt gagacttttg taatgttttc
gagtttaaat ctttgccttt 16800gcgtacgtgg gcgga
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
D00012
D00013
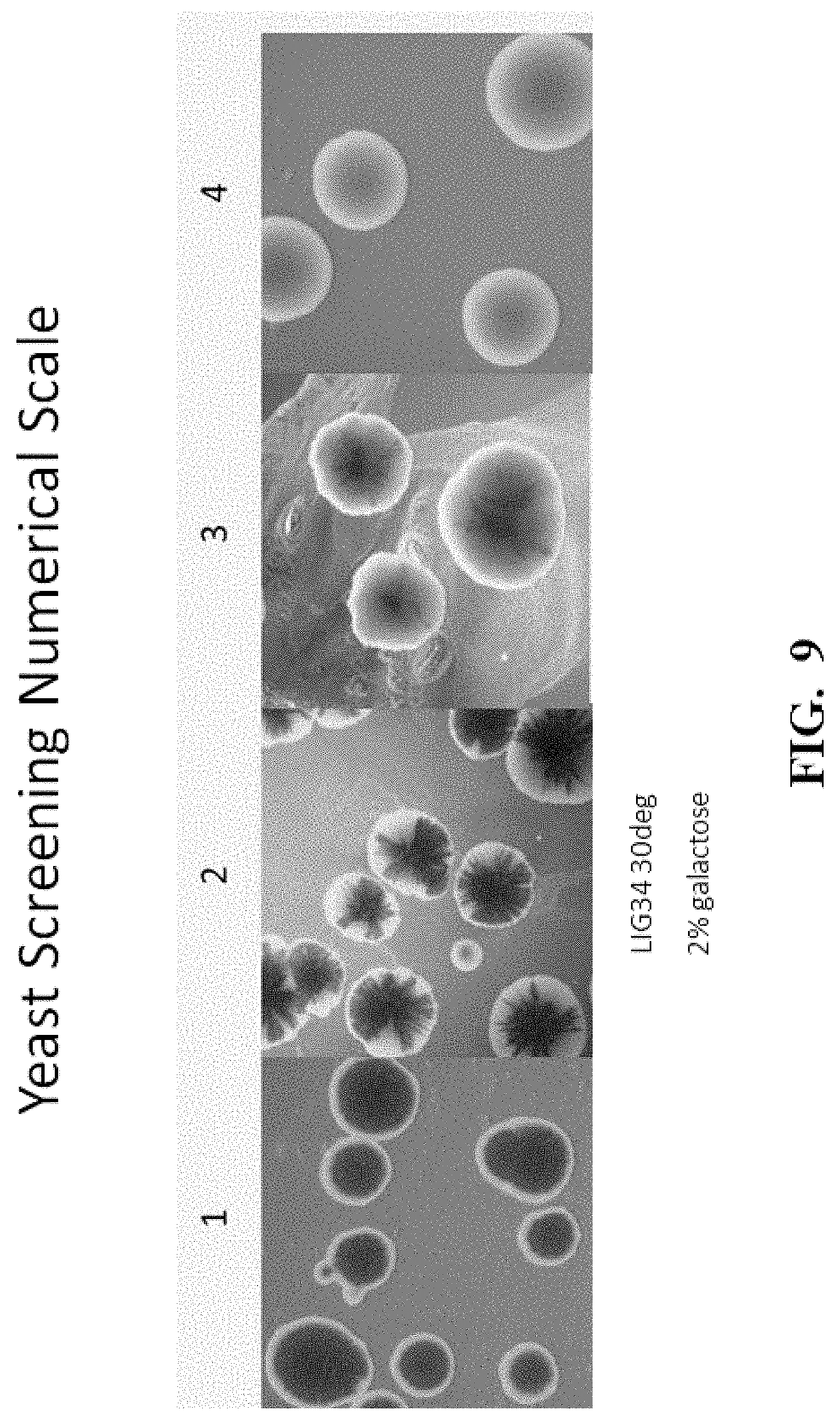
D00014
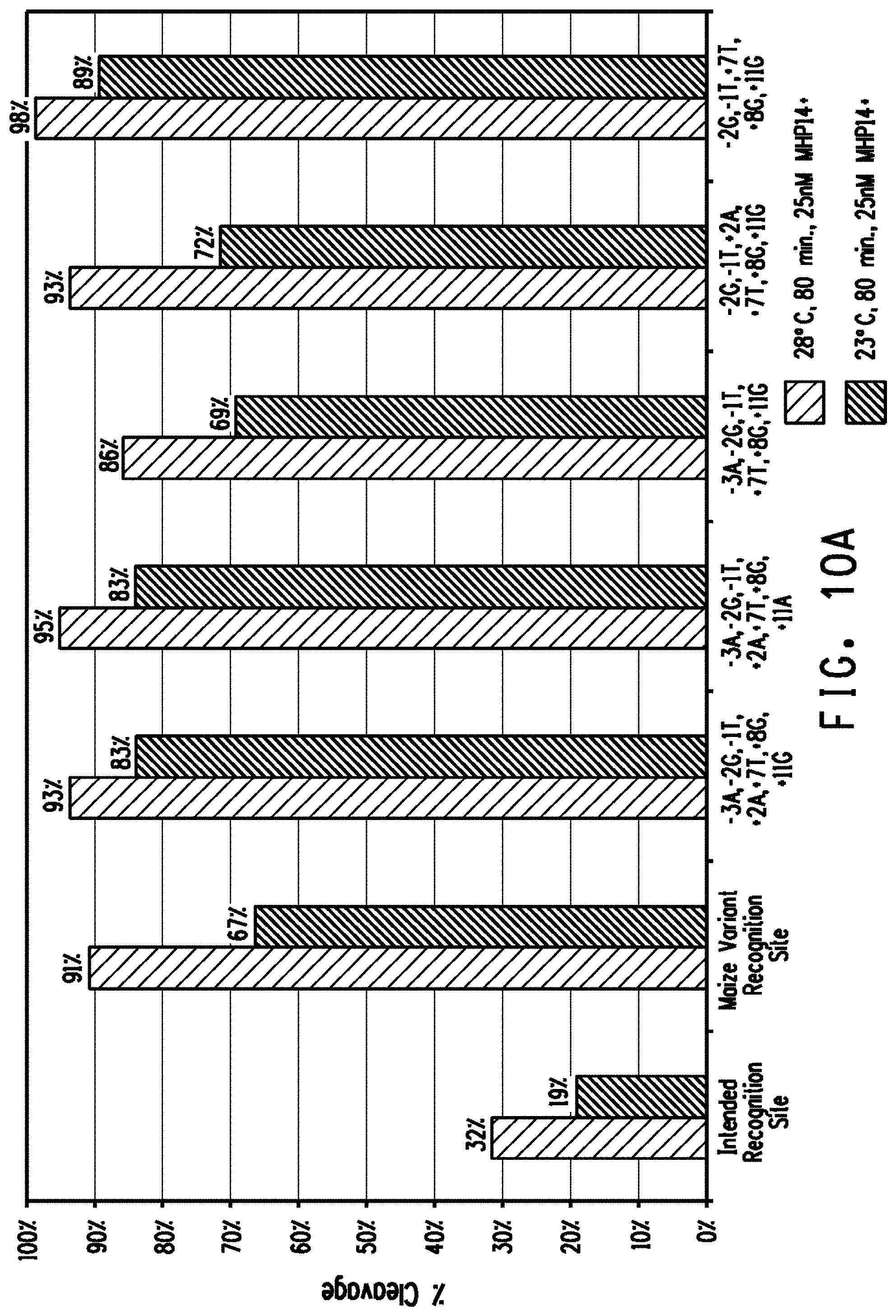
D00015
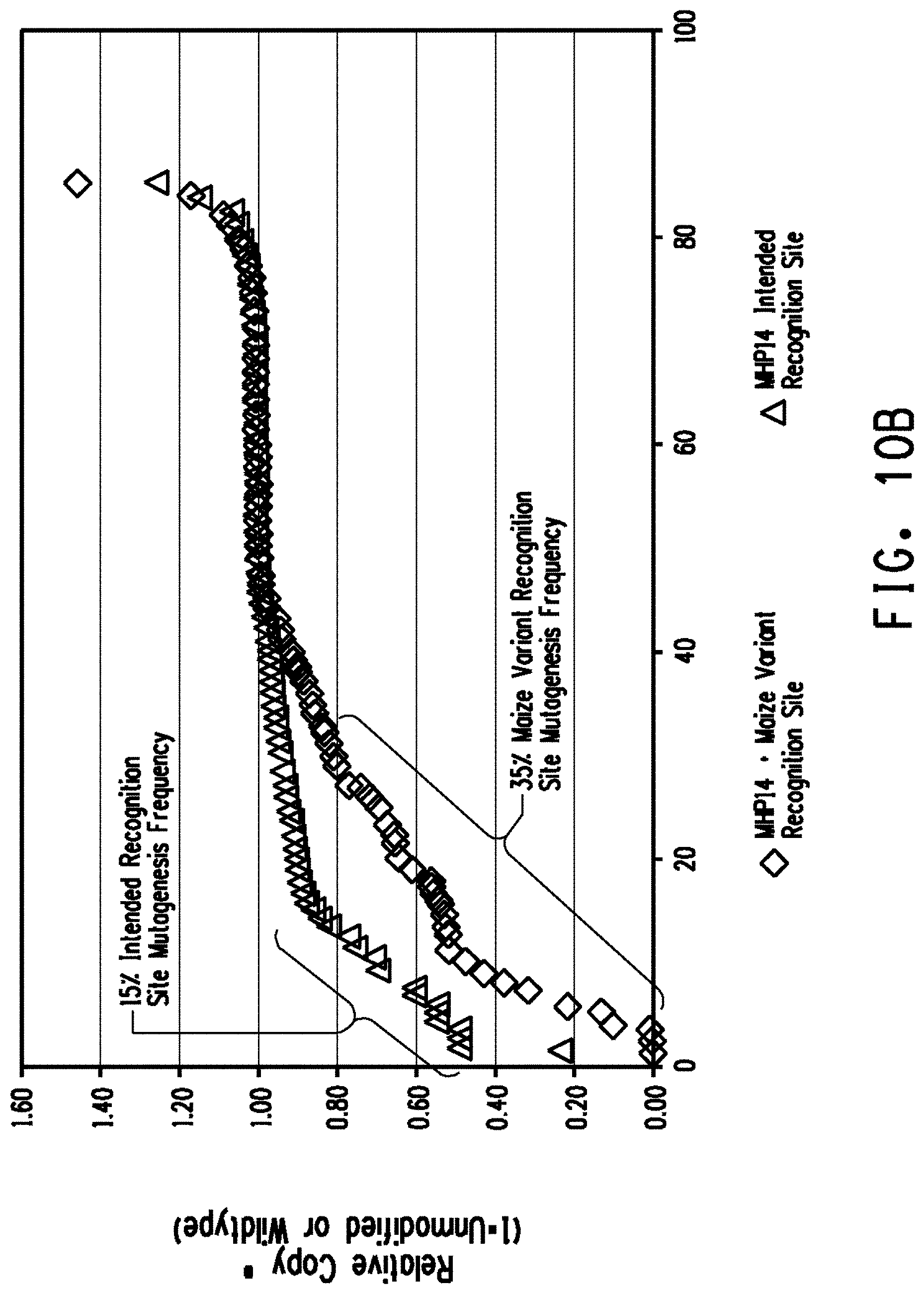
D00016
D00017
S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.