Distinguishing Antagonistic And Agonistic Anti B7-h1 Antibodies
Dong; Haidong
U.S. patent application number 16/407699 was filed with the patent office on 2019-11-28 for distinguishing antagonistic and agonistic anti b7-h1 antibodies. This patent application is currently assigned to Mayo Foundation for Medical Education and Research. The applicant listed for this patent is Mayo Foundation for Medical Education and Research. Invention is credited to Haidong Dong.
| Application Number | 20190361033 16/407699 |
| Document ID | / |
| Family ID | 54554772 |
| Filed Date | 2019-11-28 |









View All Diagrams
| United States Patent Application | 20190361033 |
| Kind Code | A1 |
| Dong; Haidong | November 28, 2019 |
DISTINGUISHING ANTAGONISTIC AND AGONISTIC ANTI B7-H1 ANTIBODIES
Abstract
Materials and methods for distinguishing agonistic anti-B7-H1 antibodies from antagonistic anti-B7-H1 antibodies, and for treating subjects diagnosed with clinical conditions such as cancer, pathogenic infection, or autoimmune disease.
| Inventors: | Dong; Haidong; (Rochester, MN) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Assignee: | Mayo Foundation for Medical
Education and Research Rochester MN |
||||||||||
| Family ID: | 54554772 | ||||||||||
| Appl. No.: | 16/407699 | ||||||||||
| Filed: | May 9, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 15311552 | Nov 16, 2016 | 10302653 | ||
| PCT/US15/31993 | May 21, 2015 | |||
| 16407699 | ||||
| 62001984 | May 22, 2014 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61K 39/3955 20130101; C07K 2317/76 20130101; G01N 33/57484 20130101; C07K 2317/73 20130101; C07K 16/2827 20130101; G01N 33/6854 20130101; G01N 2440/14 20130101; A61K 2039/505 20130101; C07K 2317/74 20130101; C07K 2317/75 20130101; A61K 39/3955 20130101; G01N 2333/912 20130101; A61K 2300/00 20130101; G01N 2333/70532 20130101 |
| International Class: | G01N 33/68 20060101 G01N033/68; C07K 16/28 20060101 C07K016/28; A61K 39/395 20060101 A61K039/395; G01N 33/574 20060101 G01N033/574 |
Claims
1. A composition comprising a pharmaceutically acceptable carrier and an antibody that specifically binds to B7-H1, wherein the antibody is identified as having antagonistic activity but not agonistic activity, agonistic activity but not antagonistic activity, predominantly antagonistic activity, or predominantly agonistic activity.
2. The composition of claim 1, wherein the antibody is identified as having antagonistic activity but not agonistic activity, or as having predominantly antagonistic activity.
3. The composition of claim 2, wherein the antibody was identified as having antagonistic activity based at least in part on its ability to block forward signaling of B7-H1 through PD-1.
4. The composition of claim 1, wherein the antibody is identified as having agonistic activity but not antagonistic activity, or as having predominantly agonistic activity.
5. The composition of claim 4, wherein the antibody was identified as having agonistic activity based at least in part on its ability to trigger signaling through p38 MAPK.
Description
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application is a divisional of U.S. application Ser. No. 15/311,552, filed Nov. 16, 2016 (now U.S. Pat. No. 10,302,653), which is a National Stage application under 35 U.S.C. .sctn. 371 of International Application No. PCT/US2015/031993, having an International Filing Date of May 21, 2015, which claims the benefit of U.S. Provisional Ser. No. 62/001,984, filed on May 22, 2014. The disclosures of the prior applications are considered part of (and are incorporated by reference in) the disclosure of this application.
TECHNICAL FIELD
[0002] This document relates to materials and methods for distinguishing agonistic anti-B7-H1 antibodies from antagonistic anti-B7-H1 antibodies, and for treating subjects diagnosed as having a clinical condition such as cancer or a pathogenic infection with B7-H1 antibodies identified as having or not having particular activities.
BACKGROUND
[0003] Elevated tumor expression of B7-H1 (also known as PD-L1) is predictive of an aggressive disease course, including increased risk of progression and cancer-related death (Thompson et al., Cancer Res 66:3381-3385, 2006; and Zang and Allison, Clin Cancer Res 13:5271-5279, 2007). Retrospective studies suggested that tumors exploit B7-H1 expression to inhibit host T cell function, thereby fostering malignant progression. The concept of tumor B7-H1-mediated immune evasion was an impetus for implementing B7-H1 blockade as a tumor immunotherapy (Zang and Allison, supra; Zou and Chen, Nat Rev Immunol 8:467-477, 2008, Dong and Chen, J Mol Med 81:281-287, 2003; Li et al., Clin Cancer Res 15:1623-1634, 2009; and Webster et al., J Immunol 179:2860-2869, 2007). B7-H1 blockade can dramatically improve tumor immunotherapy by increasing the function of effector CD8 T cells (Strome et al., Cancer Res 63:6501-6505, 2003; Hirano et al., Cancer Res 65:1089-1096, 2005; Blank et al., Int J Cancer 119:317-327, 2006; and Iwai et al., Proc Natl Acad Sci USA 99:12293-12297, 2002). Preclinical studies also provided evidence that B7-H1 blockade may be useful as a treatment for advanced human solid cancers (Dong and Chen, supra; Brahmer et al., New Engl J Med 366:2455-2465, 2012; and Dong and Chen, Cell Mol Immunol 3:179-187, 2006). In these studies, however, only a small portion of treated patients exhibited lasting objective responses (Brahmer et al., supra).
SUMMARY
[0004] This document is based, at least in part, on the discovery that unintentional disruption of a previously unknown function of B7-H1 in T cell survival may seriously counter beneficial effects of B7-H1 blockade, and on the development of assays for distinguishing agonistic B7-H1 antibodies from antagonistic B7-H1 antibodies. As described herein, an anti-B7-H1 antibody given at the early stage of immunization can increase the numbers of effector CD8 T cells, while reducing the numbers of effector CD8 T cells when given at a later stage. The hypotheses that B7-H1 expressed by activated CD8 T cells has an intrinsic pro-survival function required for establishment of T cell immunity, and that ligation of B7-H1 by agonistic antibody may disrupt its pro-survival function in T cells, are new concepts in addressing T cell survival and differentiation. The findings discussed herein challenge the conventional assumption that B7-H1 is singularly an immune inhibitory molecule. By understanding the intrinsic signaling pathways of B7-H1 in T cells, it may be possible to develop new subcellular targets for regulating T cell survival, to screen B7-H1 blockade antibodies to improve T cell immunity against cancer and pathogenic infections, and to provide improved treatment methods for patients.
[0005] In one aspect, this document features a method for identifying an anti B7-H1 antibody as having agonistic activity. The method can include contacting a population of activated T cells with the antibody, performing a quantitative assay to measure the level of p38 mitogen-activated protein kinase (MAPK) activation in the T cells, and identifying the antibody as having agonistic activity when the level of p38 MAPK activation is increased in the activated T cells as compared to a control level of p38 MAPK activation. The level of p38 MAPK activation can be measured using flow cytometry, or measured as an increase in phosphorylation. The method can include contacting the T cell population with the anti B7-H1 antibody for 12-36 hours (e.g., for 24 hours). The control level of p38 MAPK activation can be the level of p38 MAPK activation in a population of activated T cells contacted with control IgG.
[0006] In another aspect, this document features a method for modulating T cell survival or function in a subject. The method can include administering to the subject an antibody that specifically binds to B7-H1, where the antibody is identified as having antagonistic activity but not agonistic activity, agonistic activity but not antagonistic activity, predominantly antagonistic activity, or predominantly agonistic activity, and where the antibody is administered in an amount effective to increase or decrease T cell function or survival in the subject. For example, the subject can be a subject diagnosed as having cancer or a pathogenic infection, and the antibody can be identified as having antagonistic activity but not agonistic activity, or as having predominantly antagonistic activity, where the antibody is administered in an amount effective to increase T cell function or survival in the subject. The antibody can be identified as having antagonistic activity based at least in part on its ability to block forward signaling of B7-H1 through PD-1. In another example, the subject can be a subject diagnosed as having an autoimmune disease, and the antibody can be identified as having agonistic activity but not antagonistic activity, or as having predominantly agonistic activity, where the antibody is administered in an amount effective to decrease T cell function or survival in the subject. The antibody can be identified as having agonistic activity based at least in part on its ability to trigger signaling through p38 mitogen-activated protein kinase (MAPK).
[0007] This document also features a composition comprising a pharmaceutically acceptable carrier and an antibody that specifically binds to B7-H1, where the antibody is identified as having antagonistic activity but not agonistic activity, agonistic activity but not antagonistic activity, predominantly antagonistic activity, or predominantly agonistic activity. The antibody can be identified as having antagonistic activity but not agonistic activity, or as having predominantly antagonistic activity, and can be identified as having antagonistic activity based at least in part on its ability to block forward signaling of B7-H1 through PD-1. The antibody can be identified as having agonistic activity but not antagonistic activity, or as having predominantly agonistic activity, and can be identified as having agonistic activity based at least in part on its ability to trigger signaling through p38 MAPK.
[0008] In still another aspect, this document features an antibody that specifically binds to B7-H1, or a composition containing a pharmaceutically acceptable carrier and an antibody that specifically binds to B7-H1, for use in modulating T cell survival or function in a subject. The antibody can be identified as having antagonistic activity but not agonistic activity, agonistic activity but not antagonistic activity, predominantly antagonistic activity, or predominantly agonistic activity. In use, the antibody can be administered in an amount effective to increase or decrease T cell function or survival in the subject. For example, the subject can be a subject diagnosed as having cancer or a pathogenic infection, and the antibody can be identified as having antagonistic activity but not agonistic activity, or as having predominantly antagonistic activity, where the antibody is to be administered in an amount effective to increase T cell function or survival in the subject. The antibody can be identified as having antagonistic activity based at least in part on its ability to block forward signaling of B7-H1 through PD-1. In another example, the subject can be a subject diagnosed as having an autoimmune disease, and the antibody can be identified as having agonistic activity but not antagonistic activity, or as having predominantly agonistic activity, where the antibody is to be administered in an amount effective to decrease T cell function or survival in the subject. The antibody can be identified as having agonistic activity based at least in part on its ability to trigger signaling through p38 MAPK.
[0009] Unless otherwise defined, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention pertains. Although methods and materials similar or equivalent to those described herein can be used to practice the invention, suitable methods and materials are described below. All publications, patent applications, patents, and other references mentioned herein are incorporated by reference in their entirety. In case of conflict, the present specification, including definitions, will control. In addition, the materials, methods, and examples are illustrative only and not intended to be limiting.
[0010] The details of one or more embodiments of the invention are set forth in the accompanying drawings and the description below. Other features, objects, and advantages of the invention will be apparent from the description and drawings, and from the claims.
DESCRIPTION OF DRAWINGS
[0011] FIG. 1 is a diagram depicting a bi-directional signaling model for B7-H1 in T cells.
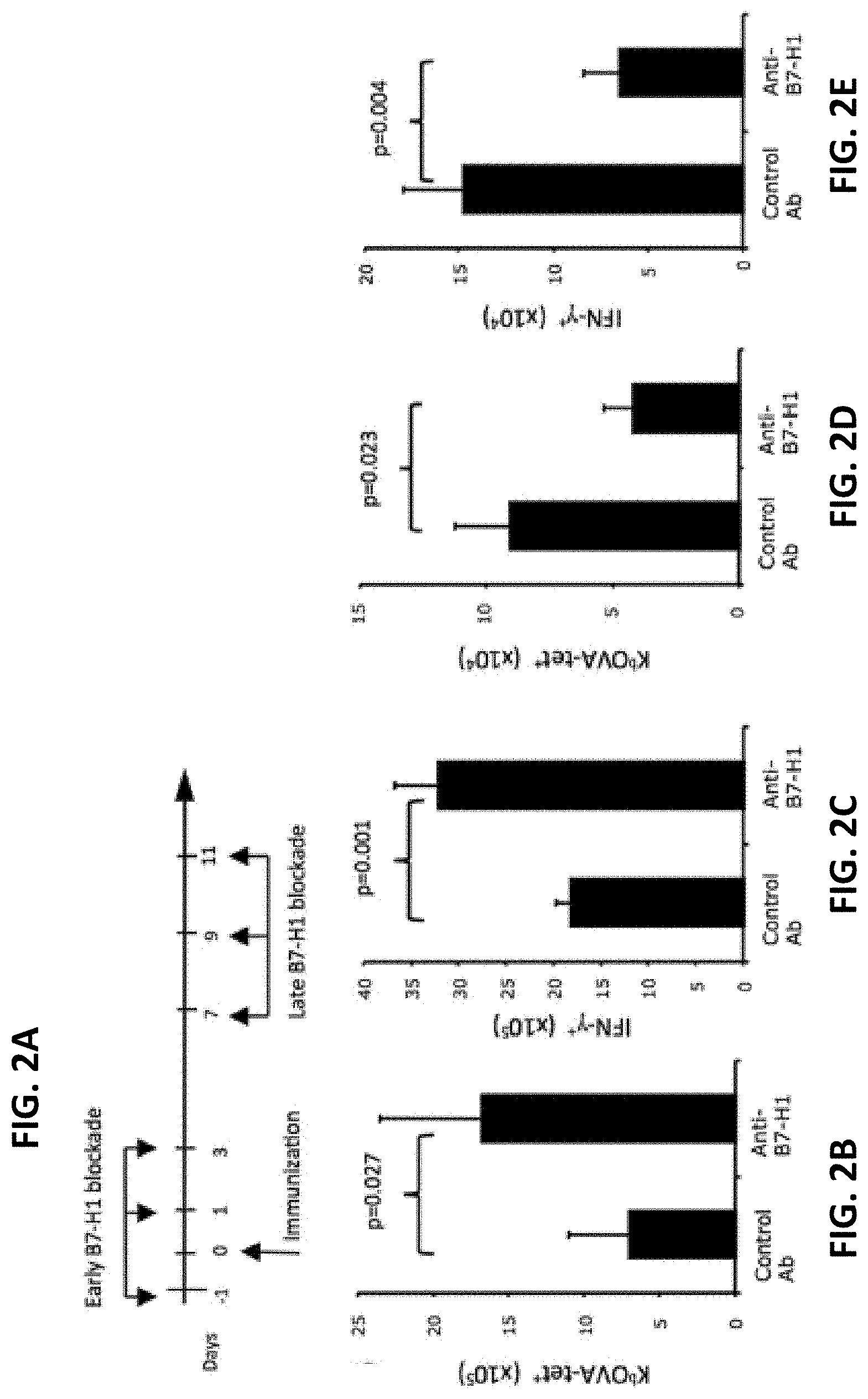
[0012] FIG. 2A is a schematic depicting early vs. late B7-H1 blockade methods. FIGS. 2B-2E are a series of graphs plotting the effects of late B7-H1 blockade on the numbers of effector CD8 T cells. C57BL/6 mice were immunized with OVA protein/poly I:C. Spleen cells were isolated 7 days after the last injection of antibodies and analyzed for KbOVA tetramer (tet) binding and intracellular IFN-.gamma. production. Graphs show the average numbers of tetramer.sup.+ (FIG. 2B) and IFN-.gamma..sup.+ (FIG. 2C) CD8 T cells after early blockade, and the average numbers of tetramer.sup.+ (FIG. 2D) and IFN-.gamma..sup.+ (FIG. 2E) CD8 T cells after late blockade.
[0013] FIGS. 3A-3C show that B7-H1 deficient CD8 T cells exhibited impaired protective immunity against tumor challenge. FIG. 3A is a graph plotting B16-OVA tumor growth in mice transferred with activated CD8 T cells. Tumor sizes are shown as mean.+-.SD of five mice per group. *p<0.05. FIG. 3B shows the accumulation of transferred OT-1 CD8 T cells (KbOVA-tetramer, tet.sup.+) at the tumor site and spleen. Numbers show the percentage of tet.sup.+ CD8 T cells. FIG. 3C is a graph plotting cytolytic activity in the spleens of recipient mice. EL-4 cells that were pulsed with OVA peptide (solid lines) or control peptide (dotted lines) were used as target cells in a 4 hour calcein release assay. Data are representative of three independent experiments with three mice per group.
[0014] FIGS. 4A-4D demonstrate increased apoptosis of B7-H1 deficient CD8 T cells following antigen stimulation. FIGS. 4A and 4B show apoptosis of activated CD8 T cells. Numbers are average percentages of apoptotic (Annexin V.sup.+ TMRE.sup.low or active caspase-3.sup.+) CD8 T cells. *p<0.05, **p<0.01. FIG. 4C is a graph plotting the numbers of viable T cells (n=3). *p<0.05. FIG. 4D is a series of graphs plotting proliferation (based on the dilution of CFSE). Numbers are the percentages of proliferating T cells that have undergone three or more times of division, *p<0.05.
[0015] FIGS. 5A and 5B indicate a T cell intrinsic function of B7-H1 in T cell contraction. FIG. 5A is a pair of graphs showing the effects of separate transfer of WT or B7-H1 KO T cells (Thy1.2.sup.+) into Thy1.1.sup.+ host model. Similar expansion was observed (N.S., no significant difference) at day 4 (top panel), while a bit more contraction of B7-H1 KO T cells was observed on day 6 following immunization (*p<0.05 compared to WT T cells) (bottom panel). FIG. 5B is a pair of graphs showing the effects of co-transfer of pre-activated WT (Thy1.2.sup.+CD45.1.sup.+) and B7-H1 KO (Thy1.2.sup.+CD45.1-) into a Thy1.1.sup.+ host model. Injection of anti-PD-1 antibody (bottom panel) did not interfere with T cell contraction on day 2 post transfer.
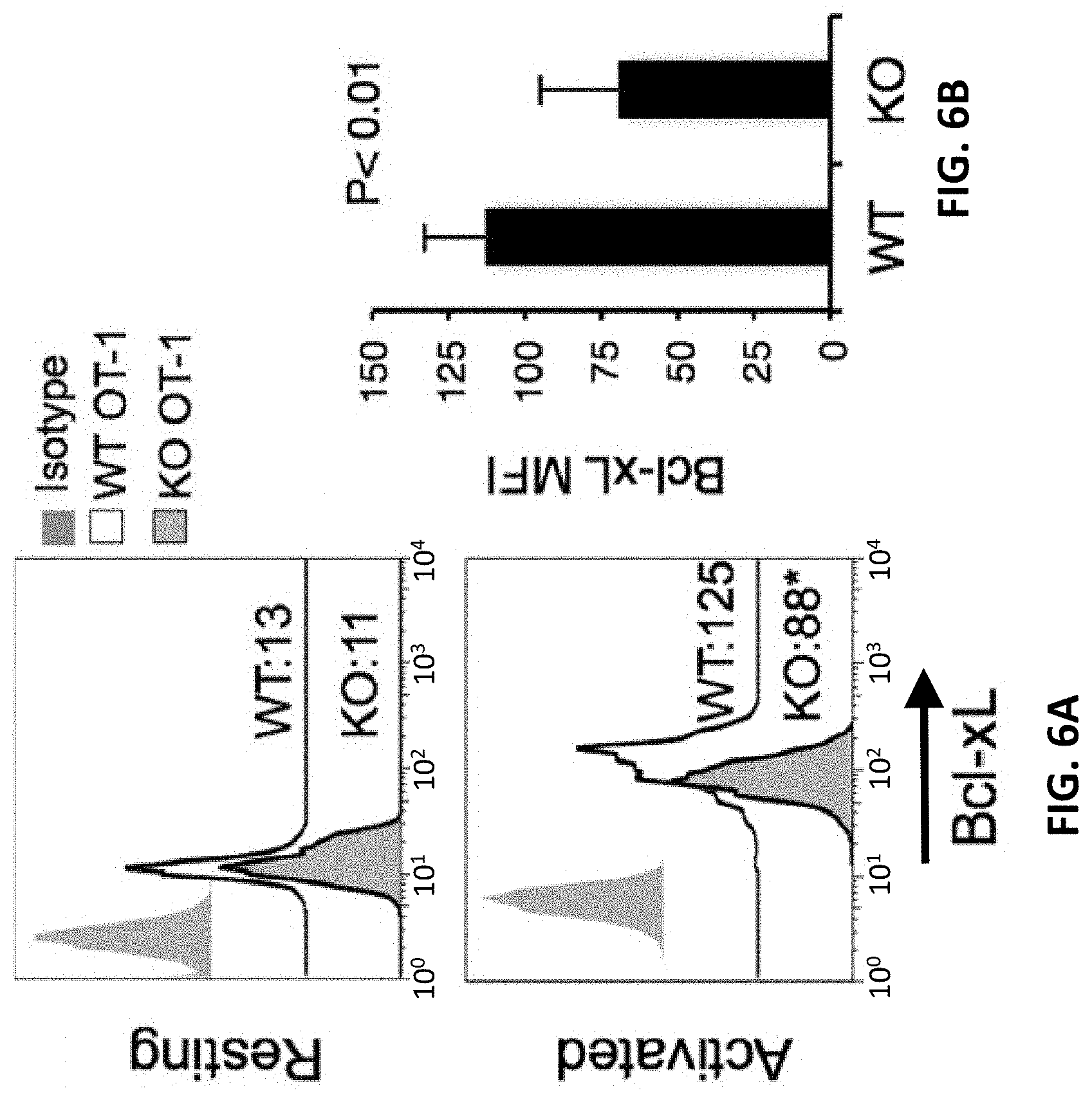
[0016] FIGS. 6A and 6B demonstrate lower Bcl-xL expression by B7-H1 KO T cells. FIG. 6A shows intracellular staining for Bcl-xL, Bcl-2, and Bim in resting (top panel) and activated (bottom panel) WT and B7-H1 KO CD8 T cells. MFI: mean fluorescence intensity. *p<0.01 compared with WT cells. FIG. 6B is a bar graph plotting the average MFI of Bcl-xL expressed by activated WT and B7-H1 KO T cells (mean.+-.SD, n=3).
[0017] FIG. 7 is a graph plotting p38 MAPK activation, which was increased in the absence of B7-H1. T cells isolated from WT or B7-H1 KO mice were activated with anti-CD3/CD28 beads for 48 hours. Data show the histogram of phosphor-p38 MAPK expression.
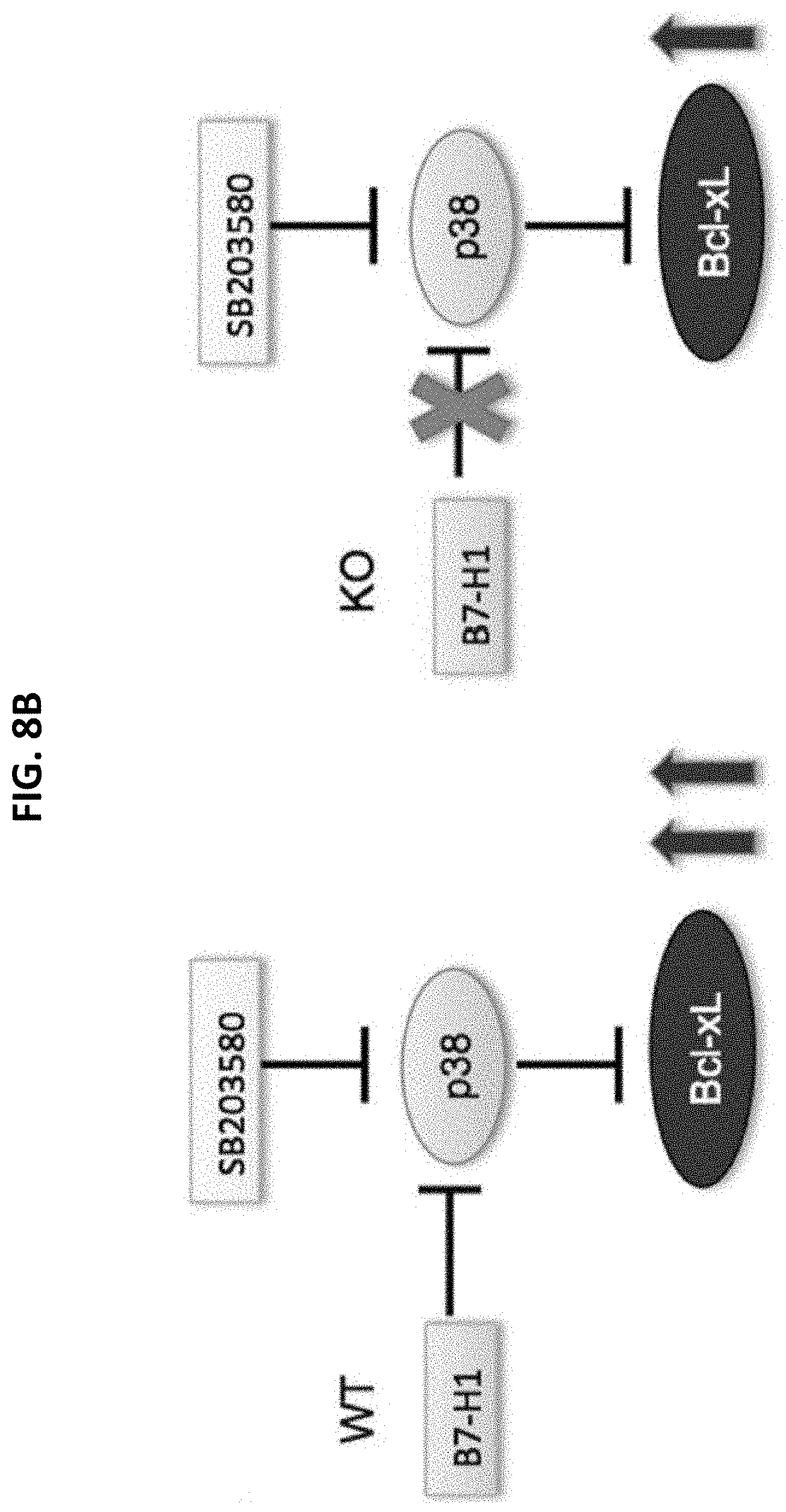
[0018] FIGS. 8A and 8B show regulation of Bcl-xL by B7-H1 via p38 MAPK. Pre-activated WT and B7-H1 KO CD8 T cells were incubated with SB203580 (10 uM) or solvent DMSO for 48 hours. FIG. 8A is a pair of histograms plotting intracellular staining for Bcl-xL in WT T cells (left panel) and in KO T cells (center panel). The percent increase in Bcl-xL was higher in WT T cells than in KO T cells (right panel). FIG. 8B is a diagram of a potential mechanism of regulation of Bcl-xL by B7-H1 via p38 MAPK.
[0019] FIGS. 9A and 9B are pictures of Western blots showing B7-H1 associated protein DNA-PKcs in T cells. For FIG. 9A, immunoprecipitation (IP) with cell lysate of Kaspas299 was performed with anti-B7-H1 mAb (5H1) or control Ab (Ct). For FIG. 9B, IP with anti-B7-H1 or anti-DNA-PK (H106) antibody was followed by Western blotting (WB) with either anti-B7-H1 or anti-DNAPK. Whole cell lysate was used as input.
[0020] FIG. 10 is a picture of a Western blot showing that B7-H1 is associated with DNA-PK in activated human primary T cells. T cells were activated by PHA for 48 hours.
[0021] FIG. 11A is a diagram depicting the domains of the B7-H1 protein. FIG. 11B shows a representative amino acid sequence for B7-H1 (SEQ ID NO:1). The intracellular domain (ICD) of B7-H1 is underlined and in bold. Serine and threonine residues are circled.
[0022] FIG. 12 is an image showing co-localization of B7-H1 and DNA-PKcs in cells from a human breast tumor cell line (MDA-MB-231) treated with topo I inhibitor for 2 hours to induce translocation of B7-H1. Cells were stained for DNA-PKcs (Red), B7-H1 (Green) and nuclei (Blue).
[0023] FIG. 13A show the percentages of WT (Thy1.1.sup.+ CD45.2.sup.+) and B7-H1 KO (Thy1.1.sup.-CD45.2.sup.+) CD8 T cells detected in the spleen on day 15 post transfer. FIG. 13B is a graph plotting the numbers of transferred T cells (n=3). *p<0.05, **p<0.01.
[0024] FIG. 14 is a diagram showing the proposed role of B7-H1 expressed by T cells in memory T cell generation. In the absence of B7-H1, some effector T cells may undergo more apoptosis during the contraction phase, and fewer of them become memory cells.
[0025] FIG. 15 is a diagram depicting the inducible B7-H1 expression in T cells. Injection of Dox induces B7-H1 expression in T cells by activating transcription of B7-H1 via rtTA driven by a CD3.delta. promoter, and releasing the repressor (tTS) from tetO.
[0026] FIGS. 16A and 16B show that ligation of B7-H1 in human T cells incubated with various anti-B7-H1 mAbs or control Ab in the presence of anti-CD3/CD28 beads led to increased activation of p38 MAPK. FIG. 16A is a graph plotting the average levels of phosphor-p38 MAPK. MFI: mean fluorescence intensity. FIG. 16B is a histogram of phosphor-p38 MAPK expression.
[0027] FIGS. 17A and 17B show that T cell apoptosis was reduced when pre-activated CD8 T cells were incubated with SB203580 to inhibit p38 MAPK. FIG. 17A is a graph plotting the percent of apoptotic T cells (TMRE.sup.low Annexin V.sup.+). FIG. 17B shows a representative staining of TMRE and Annexin V in CD8 T cells.
[0028] FIG. 18 is a list of candidate proteins in the MAPK/ERK pathway that will be screened and compared in B7-H1 and mock transfected tumor cells.
[0029] FIGS. 19A and 19B show that B7-H1.sup.+ CD8 T cells are increased in RCC patients. FIG. 19A is a picture showing cells stained for B7-H1 (brown, arrows) and CD8 (red). FIG. 19B is a graph plotting the percentages of B7-H1.sup.+ CD8 T cells in tumor infiltrating lymphocytes (TIL) and peripheral blood of RCC patients. *p=0.017, **p=0.043 vs. normal donors (n=17).
[0030] FIGS. 20A and 20B are a pair of graphs showing the kinetics of tumor-reactive CD8 T cells and their expression of B7-H1, after B16-OVA tumor cells were subcutaneously injected into C57BL/6 mice. FIG. 20A is a graph plotting the kinetics of CD11a.sup.high CD8 T cells (dashed line) and tumor growth (solid line). FIG. 20B is a graph plotting B7-H1 levels (MFI) on CD11a.sup.high CD8 T cells from spleen and tumor infiltrating lymphocytes (TIL) of tumor mice, or on naive CD8 T cells (baseline).
[0031] FIG. 21 is a graph plotting the average levels of phosphorp38 MAPK in mouse T cells incubated with plate bound anti-B7-H1 mAb or control Ab in the presence of anti-CD3/CD28 beads, and showing that ligation of B7-H1 increased activation of p38 MAPK. MFI: mean fluorescence intensity. *p<0.05.
[0032] FIG. 22A is a series of histograms showing the levels of phosphorylated AKT in freshly purified human peripheral blood CD8 T cells after incubation with anti-CD3, anti-B7-H1 (H1A and 5H1), or an isotype control antibody to B7-H1. AKT phosphorylation was assessed by intracellular staining with an anti-phosphor-AKT (S473) antibody. FIG. 22B is a graph plotting the level of phosphorylated AKT in the cells, showing pooled data from three donors.
DETAILED DESCRIPTION
[0033] Some B7-H1 antibodies may disrupt a previously unknown function of B7-H1 in T cells, as indicated by the apparent ability of anti-B7-H1 antibodies to reduce CD8 T cell responses (Xu et al., supra; and Pulko et al., J Immunol 187:5606-5614, 2011). A bi-directional signaling model of B7-H1 is proposed in FIG. 1. In this model, the extrinsic signaling of B7-H1 is mediated by PD-1, which impairs T cell function and survival via reduction of AKT activation (Patsoukis et al., Science Signaling 5:ra46, 2012) and up-regulation of Bim (Gibbons et al., Oncoimmunology 1:1061-1073, 2012). As a result, T cell function and survival are compromised. Current B7-H1 therapies using blocking antibodies are aimed at blocking this extrinsic effect of B7-H1, thereby enhancing antitumor T cell immunity. The intrinsic signaling of B7-H1 is mediated by an unknown mechanism that leads to stabilizing pro-survival molecules in T cells. The data presented herein reveal that B7-H1 per se is required for the survival of activated T cells (Pulko et al., supra), and that ligation of B7-H1 by antibody triggers proapoptosis signals in T cells (Dong et al., supra). Therefore, if the intrinsic prosurvival function of B7-H1 is disrupted (for example, by an agonistic antibody), CD8 T cells that express B7-H1 would undergo apoptosis, resulting in compromised immunity.
[0034] To achieve maximal therapeutic effects of B7-H1 blockade therapy in cancer treatment, it is imperative that the new function of B7-H1 expressed by T cells be fully characterized. However, no methodology has been reported for evaluating whether the use of anti-B7-H1 antibodies in treatment of cancer has a potential to impart unfavorable effects on T cell survival, and whether such unfavorable effects could be avoided by screening anti-B7-H1 antibodies before administration to patients. This document provides materials and methods focused on defining the nature of intrinsic signals of B7-H1 in T cell apoptosis/differentiation, and on evaluating the impact of agonistic anti-B7-H1 antibodies on antitumor T cell immunity. The studies discussed herein provide new approaches (i.e., screening antibodies) for advancing checkpoint blockade immunologically and therapeutically.
[0035] A representative example of a human B7-H1 polypeptide has the sequence set forth in GENBANK.RTM. Accession No. AAF25807 (GI No. 6708119) (SEQ ID NO:1; FIG. 11B); the corresponding human B7-H1 nucleic acid has the sequence set forth in GENBANK.RTM. Accession No. AF177937 (GI No. 6708118) (SEQ ID NO:2).
[0036] The term "antibody" includes monoclonal antibodies, polyclonal antibodies, recombinant antibodies, humanized antibodies (Jones et al. (1986) Nature 321:522-525; Riechmann et al. (1988) Nature 332:323-329; and Presta (1992) Curr Op Struct Biol 2:593-596), chimeric antibodies (Morrison et al. (1984) Proc Natl Acad Sci USA 81:6851-6855), multispecific antibodies (e.g., bispecific antibodies) formed from at least two antibodies, and antibody fragments. The term "antibody fragment" comprises any portion of the afore-mentioned antibodies, such as their antigen binding or variable regions. Examples of antibody fragments include Fab fragments, Fab' fragments, F(ab')2 fragments, Fv fragments, diabodies (Hollinger et al. (1993) Proc Natl Acad Sci USA 90:6444-6448), single chain antibody molecules (Pluckthun in: The Pharmacology of Monoclonal Antibodies 113, Rosenburg and Moore, eds., Springer Verlag, N.Y. (1994), 269-315) and other fragments as long as they exhibit the desired capability of binding to B7-H1.
[0037] Examples of anti-human B7-H1 antibodies include, without limitation, anti-human B7-H1 antibodies commercially available from Biolegend (e.g., Catalog No. 329701 or 329702; San Diego, Calif.) or eBioscience (e.g., Catalog No. 14-5983-80 or 14-5983-82).
[0038] The term "antibody," as used herein, also includes antibody-like molecules that contain engineered sub-domains of antibodies or naturally occurring antibody variants. These antibody-like molecules may be single-domain antibodies such as V.sub.H-only or V.sub.L-only domains derived either from natural sources such as camelids (Muyldermans et al. (2001) Rev Mol Biotechnol 74:277-302) or through in vitro display of libraries from humans, camelids or other species (Holt et al. (2003) Trends Biotechnol 21:484-90). In certain embodiments, the polypeptide structure of the antigen binding proteins can be based on antibodies, including, but not limited to, minibodies, synthetic antibodies (sometimes referred to as "antibody mimetics"), human antibodies, antibody fusions (sometimes referred to as "antibody conjugates"), and fragments thereof, respectively.
[0039] An "Fv fragment" is the minimum antibody fragment that contains a complete antigen-recognition and -binding site. This region consists of a dimer of one heavy chain variable domain and one light chain variable domain in tight, non-covalent association. It is in this configuration that the three CDR's of each variable domain interact to define an antigen-binding site on the surface of the VH-VL dimer. Collectively, the six CDR's confer antigen-binding specificity to the antibody. However, even a single variable domain (or half of an Fv comprising only three CDR's specific for an antigen) has the ability to recognize and bind the antigen, although usually at a lower affinity than the entire binding site. The "Fab fragment" also contains the constant domain of the light chain and the first constant domain (C.sub.H1) of the heavy chain. The "Fab fragment" differs from the "Fab' fragment" by the addition of a few residues at the carboxy terminus of the heavy chain C.sub.H1 domain, including one or more cysteines from the antibody hinge region. The "F(ab')2 fragment" originally is produced as a pair of "Fab' fragments" which have hinge cysteines between them. Methods of preparing such antibody fragments, such as papain or pepsin digestion, are known to those skilled in the art.
[0040] An antibody can be of the IgA-, IgD-, IgE-, IgG- or IgM-type, including IgG- or IgM-types such as, without limitation, IgG1-, IgG2-, IgG3-, IgG4-, IgM1- and IgM2-types. For example, in some cases, the antibody is of the IgG1-, IgG2- or IgG4-type.
[0041] In some embodiments, antibodies as used in the methods described herein can be fully human or humanized antibodies. Human antibodies can avoid certain problems associated with xenogeneic antibodies, such as antibodies that possess murine or rat variable and/or constant regions. First, because the effector portion is human, it can interact better with other parts of the human immune system, e.g., to destroy target cells more efficiently by complement-dependent cytotoxicity or antibody-dependent cellular cytotoxicity. Second, the human immune system should not recognize the antibody as foreign. Third, half-life in human circulation will be similar to naturally occurring human antibodies, allowing smaller and less frequent doses to be given. Methods for preparing human antibodies are known in the art.
[0042] In addition to human antibodies, "humanized" antibodies can have many advantages. Humanized antibodies generally are chimeric or mutant monoclonal antibodies from mouse, rat, hamster, rabbit or other species, bearing human constant and/or variable region domains or specific changes. Techniques for generating humanized antibodies are well known to those of skill in the art. For example, controlled rearrangement of antibody domains joined through protein disulfide bonds to form new, artificial protein molecules or "chimeric" antibodies can be utilized (Konieczny et al. (1981) Haematologia (Budap.) 14:95). Recombinant DNA technology can be used to construct gene fusions between DNA sequences encoding mouse antibody variable light and heavy chain domains and human antibody light and heavy chain constant domains (Morrison et al. (1984) Proc Natl Acad Sci USA 81:6851).
[0043] DNA sequences encoding antigen binding portions or complementarity determining regions (CDR's) of murine monoclonal antibodies can be grafted by molecular means into DNA sequences encoding frameworks of human antibody heavy and light chains (Jones et al. (1986) Nature 321:522; Riechmann et al. (1988) Nature 332:323). Expressed recombinant products are called "reshaped" or humanized antibodies, and comprise the framework of a human antibody light or heavy chain and antigen recognition portions, CDR's, of a murine monoclonal antibody.
[0044] Other methods for designing heavy and light chains and for producing humanized antibodies are described in, for example, U.S. Pat. Nos. 5,530,101; 5,565,332; 5,585,089; 5,639,641; 5,693,761; 5,693,762; and 5,733,743. Yet additional methods for humanizing antibodies are described in U.S. Pat. Nos. 4,816,567; 4,935,496; 5,502,167; 5,558,864; 5,693,493; 5,698,417; 5,705,154; 5,750,078; and 5,770,403, for example.
[0045] The methods provided herein can include determining whether an antibody against B7-H1 has antagonistic function, such that it has the ability to block forward signaling of B7-H1 through PD-1, and/or determining whether an antibody against B7-H1 has agonistic function, such that it has the ability to trigger signaling through p38 MAPK. The antagonistic ability of an antibody to block forward signaling of B7-H1 through PD-1 can result in increased T cell function, while the agonistic ability of an antibody to trigger signaling through p38 MAPK can result in decreased T cell function and survival. Thus, an anti B7-H1 antibody that has the ability to block forward signaling through PD-1 but has low ability or lacks the ability to trigger signaling through p38 MAPK, may be particularly useful for treating cancer and other disorders (e.g., pathogenic infections) in which it can be advantageous to increase T cell function. In contrast, a dual function anti B7-H1 antibody that also has the ability to trigger signaling through p38 MAPK may have the opposite effect on T cell function and survival, and thus may not have a significant clinical benefit in treatment of cancer patients. Such antibodies may, however, be useful for treating conditions (e.g., autoimmune disorders) in which decreased T cell function is desired.
[0046] Methods for determining whether an anti B7-H1 antibody has antagonistic and/or agonistic function include those described herein (see, e.g., the Examples below). For example, the effect of an antibody on T cell survival can be tested in vivo using an animal model, by administering the antibody to an immunized animal and then examining the number and/or percentage of antigen specific and functional CD8 T cells in the animal's spleen.
[0047] In some cases, the potential agonistic function of a particular anti B7-H1 antibody can be evaluated by assaying the effect of the antibody on p38 MAPK activation. For example, the activity of an anti B7-H1 antibody can be assessed by contacting activated T cells with the antibody (e.g., for 2-48 hours, such as 6-36 hours, 12-36 hours, or about 24 hours), and measuring the level of p38 MAPK activation in the T cells. An antibody can be identified as having agonistic activity when the level of p38 MAPK activation is increased in the activated T cells as compared to a control level of p38 MAPK activation (e.g., the level of p38 MAPK activation in activated T cells contacted with a control IgG rather than with the anti B7-H1 antibody). The activation of p38 MAPK can be indicated by an increase in the level of p38 MAPK phosphorylation, for example, and any suitable method can be used to assess the level of p38 MAPK activation. In some embodiments, flow cytometry can be used.
[0048] In some cases, the potential antagonist function of an anti B7-H1 antibody can be tested by determining whether the antibody can block the binding of PD-1 protein to B7-H1 expressed by tumor cells in vitro, or whether it can block B7-H1-mediated T cell apoptosis or apoptotic signaling in vitro. See, e.g., Dong et al., (2002) Nature Med 8(8):793-800; and Gibbons et al. (2012) Oncoimmunol 1(7):1061-1073.
[0049] As discussed below, for example, three anti-mouse B7-H1 antibodies (10B5, 9G2 and MIH5) were identified as having antagonistic function, and two of those (9G2 and MIH5) also were identified as having agonistic function. See, Example 9 and FIG. 21. In contrast, 10B5 was found to have only antagonistic function, and no agonistic activity. Three anti-human B7-H1 antibodies (5H1, H1A, and 2.2B) also were evaluated. Of these, 5H1 had only antagonistic function, while H1A and 2.2B had only agonistic activity. See, Example 9 and FIGS. 16A and 16B.
[0050] Antibodies against B7-H1 can be incorporated into pharmaceutical compositions for treatment of cancer or other diseases (e.g., autoimmune disorders or pathogenic infections). Thus, this document also provides for the use of such molecules in the manufacture of medicaments for treating clinical conditions such as cancer, pathogenic infections, or autoimmune disorders. The compositions can further include one or more pharmaceutically acceptable carriers, diluents and/or adjuvants. The potency of the pharmaceutical compositions provided herein typically is based on the binding of the antibody to B7-H1.
[0051] A "pharmaceutically acceptable carrier" (also referred to as an "excipient" or a "carrier") is a pharmaceutically acceptable solvent, suspending agent, stabilizing agent, or any other pharmacologically inert vehicle for delivering one or more therapeutic compounds to a subject, which is nontoxic to the cell or mammal being exposed thereto at the dosages and concentrations employed. Pharmaceutically acceptable carriers can be liquid or solid, and can be selected with the planned manner of administration in mind so as to provide for the desired bulk, consistency, and other pertinent transport and chemical properties, when combined with one or more of therapeutic compounds and any other components of a given pharmaceutical composition. Typical pharmaceutically acceptable carriers that do not deleteriously react with amino acids include, by way of example and not limitation: water, saline solution, binding agents (e.g., polyvinylpyrrolidone or hydroxypropyl methylcellulose), fillers (e.g., lactose and other sugars, gelatin, or calcium sulfate), lubricants (e.g., starch, polyethylene glycol, or sodium acetate), disintegrates (e.g., starch or sodium starch glycolate), and wetting agents (e.g., sodium lauryl sulfate). Pharmaceutically acceptable carriers also include aqueous pH buffered solutions or liposomes (small vesicles composed of various types of lipids, phospholipids and/or surfactants which are useful for delivery of a drug to a mammal). Further examples of pharmaceutically acceptable carriers include buffers such as phosphate, citrate, and other organic acids, antioxidants such as ascorbic acid, low molecular weight (less than about 10 residues) polypeptides, proteins such as serum albumin, gelatin, or immunoglobulins, hydrophilic polymers such as polyvinylpyrrolidone, amino acids such as glycine, glutamine, asparagine, arginine or lysine, monosaccharides, disaccharides, and other carbohydrates including glucose, mannose or dextrins, chelating agents such as EDTA, sugar alcohols such as mannitol or sorbitol, salt-forming counterions such as sodium, and/or nonionic surfactants such as TWEEN.TM., polyethylene glycol (PEG), and PLURONICS.TM..
[0052] Pharmaceutical compositions can be formulated by mixing one or more active agents with one or more physiologically acceptable carriers, diluents, and/or adjuvants, and optionally other agents that are usually incorporated into formulations to provide improved transfer, delivery, tolerance, and the like. A pharmaceutical composition can be formulated, e.g., in lyophilized formulations, aqueous solutions, dispersions, or solid preparations, such as tablets, dragees or capsules. A multitude of appropriate formulations can be found in the formulary known to all pharmaceutical chemists: Remington's Pharmaceutical Sciences (18th ed, Mack Publishing Company, Easton, Pa. (1990)), particularly Chapter 87 by Block, Lawrence, therein. These formulations include, for example, powders, pastes, ointments, jellies, waxes, oils, lipids, lipid (cationic or anionic) containing vesicles (such as LIPOFECTIN.TM.), DNA conjugates, anhydrous absorption pastes, oil-in-water and water-in-oil emulsions, emulsions carbowax (polyethylene glycols of various molecular weights), semi-solid gels, and semi-solid mixtures containing carbowax. Any of the foregoing mixtures may be appropriate in treatments and therapies as described herein, provided that the active agent in the formulation is not inactivated by the formulation and the formulation is physiologically compatible and tolerable with the route of administration. See, also, Baldrick (2000) Regul Toxicol Pharmacol 32:210-218; Wang (2000) Int J Pharm 203:1-60; Charman (2000) J Pharm Sci 89:967-978; and Powell et al. (1998) PDA J Pharm Sci Technol 52:238-311), and the citations therein for additional information related to formulations, excipients and carriers well known to pharmaceutical chemists.
[0053] Pharmaceutical compositions include, without limitation, solutions, emulsions, aqueous suspensions, and liposome-containing formulations. These compositions can be generated from a variety of components that include, for example, preformed liquids, self-emulsifying solids and self-emulsifying semisolids. Emulsions are often biphasic systems comprising of two immiscible liquid phases intimately mixed and dispersed with each other; in general, emulsions are either of the water-in-oil (w/o) or oil-in-water (o/w) variety. Emulsion formulations have been widely used for oral delivery of therapeutics due to their ease of formulation and efficacy of solubilization, absorption, and bioavailability.
[0054] Compositions and formulations can include sterile aqueous solutions, which also can contain buffers, diluents and other suitable additives (e.g., penetration enhancers, carrier compounds and other pharmaceutically acceptable carriers). Compositions additionally can contain other adjunct components conventionally found in pharmaceutical compositions. Thus, the compositions also can include compatible, pharmaceutically active materials such as, for example, antipruritics, astringents, local anesthetics or anti-inflammatory agents, or additional materials useful in physically formulating various dosage forms of the compositions provided herein, such as dyes, flavoring agents, preservatives, antioxidants, opacifiers, thickening agents and stabilizers. Furthermore, the composition can be mixed with auxiliary agents, e.g., lubricants, preservatives, stabilizers, wetting agents, emulsifiers, salts for influencing osmotic pressure, buffers, colorings, flavorings, and aromatic substances. When added, however, such materials should not unduly interfere with the biological activities of the polypeptide components within the compositions provided herein. The formulations can be sterilized if desired.
[0055] In some embodiments, a composition containing an antibody against B7-H7 can be in the form of a solution or powder with or without a diluent to make an injectable suspension. The composition may contain additional ingredients including, without limitation, pharmaceutically acceptable vehicles, such as saline, water, lactic acid, mannitol, or combinations thereof, for example.
[0056] Methods for using an anti B7-H1 antibody or a composition containing an anti B7-H1 antibody to treat a clinical condition in a subject also are provided herein. The methods can include, for example, administering an anti B7-H1 antibody to a subject identified as being in need thereof, where the subject has a clinical condition (e.g., cancer, a pathogenic infection, or an autoimmune disorder) in which modulation of T cell survival or activity may be beneficial, and where the antibody is identified as having antagonistic and/or agonistic activity with regard to B7-H1. For example, an anti B7-H1 antibody with antagonistic but not agonistic activity (or predominantly antagonistic activity) can be useful for treating a clinical condition (e.g., cancer or a pathogenic infection) in which it is desired to reduce or inhibit B7-H1-mediated inhibition of T cell function and survival. Alternatively, an anti B7-H1 antibody with agonistic but not antagonistic activity (or predominantly agonistic activity) can be useful for treating a clinical condition (e.g., an autoimmune disorder) in which it is desired to increase B7-H1/PD-1-mediated inhibition of T cell function and survival. It is to be noted that antibodies with both agonistic and antagonistic effects also can be useful in the methods of treatment provided herein, particularly where an antibody has predominantly antagonistic or predominantly agonistic effects. An antibody can be considered to have "predominantly" antagonistic effects when it acts more strongly as an antagonist than an agonist, so that the overriding effect is an inhibition of B7-H1-mediated inhibition of T cell function and survival. Conversely, an antibody can be considered to have "predominantly" agonistic effects when it acts more strongly as an agonist than an antagonist, so that the overriding effect is a decrease of B7-H1-mediated T cell survival.
[0057] Any appropriate method can be used to administer an anti-B7-H1 antibody or a composition as described herein to a mammal. Administration can be, for example, parenteral (e.g., by subcutaneous, intrathecal, intraventricular, intramuscular, or intraperitoneal injection, or by intravenous drip). Administration can be rapid (e.g., by injection) or can occur over a period of time (e.g., by slow infusion or administration of slow release formulations). In some embodiments, administration can be topical (e.g., transdermal, sublingual, ophthalmic, or intranasal), pulmonary (e.g., by inhalation or insufflation of powders or aerosols), or oral. In addition, a composition containing an antibody or fusion protein as described herein can be administered prior to, after, or in lieu of surgical resection of a tumor.
[0058] A composition containing an antibody against B7-H1 can be administered to a mammal in any appropriate amount, at any appropriate frequency, and for any appropriate duration effective to achieve a desired outcome (e.g., to increase progression-free survival or to increase the number of naturally-occurring tumor-reactive CD8+ T cells in a cancer patient). In some embodiments, for example, a composition containing an anti-B7-H1 antibody can be administered to a mammal having cancer to reduce the progression rate of the cancer by 5, 10, 25, 50, 75, 100, or more percent. For example, the progression rate can be reduced such that no additional cancer progression is detected. In some embodiments, a composition containing an anti B7-H1 antibody can be administered to a mammal having cancer under conditions where progression-free survival is increased (e.g., by 5, 10, 25, 50, 75, 100, or more percent) as compared to the median progression-free survival of corresponding mammals having untreated cancer or the median progression-free survival of corresponding mammals having cancer and treated with other therapies (e.g., chemotherapeutic agents). Progression-free survival can be measured over any length of time (e.g., one month, two months, three months, four months, five months, six months, or longer). Any appropriate method can be used to determine whether or not the progression rate of cancer is reduced. For skin cancer (e.g., melanoma), for example, the progression rate can be assessed by imaging tissue at different time points and determining the amount of cancer cells present. The amounts of cancer cells determined within tissue at different times can be compared to determine the progression rate. After treatment as described herein, the progression rate can be determined again over another time interval. In some cases, the stage of cancer after treatment can be determined and compared to the stage before treatment to determine whether or not the progression rate has been reduced.
[0059] An effective amount of a composition containing an antibody as provided herein can be any amount that reduces a symptom of the condition being treated, without significant toxicity. With cancer, for example, an effective amount can reduce the progression rate of the cancer, increase the progression-free survival rate, or increase the median time to progression. Optimum dosages can vary depending on the relative potency of individual polypeptides (e.g., antibodies and fusion proteins), and can generally be estimated based on EC.sub.50 found to be effective in in vitro and in vivo animal models. Typically, dosage is from 0.01 .mu.g to 100 g per kg of body weight. For example, an effective amount of an antibody or fusion protein can be from about 1 mg/kg to about 100 mg/kg (e.g., about 5 mg/kg, about 10 mg/kg, about 20 mg/kg, about 50 mg/kg, or about 75 mg/kg). If a particular subject fails to respond to a particular amount, then the amount of the antibody can be increased by, for example, two fold. After receiving this higher concentration, the subject can be monitored for both responsiveness to the treatment and toxicity symptoms, and adjustments made accordingly. The effective amount can remain constant or can be adjusted as a sliding scale or variable dose depending on the mammal's response to treatment. Various factors can influence the actual effective amount used for a particular application. For example, the frequency of administration, duration of treatment, use of multiple treatment agents, route of administration, and severity of the clinical condition may require an increase or decrease in the actual effective amount administered.
[0060] The frequency of administration can be, for example, once or more daily, biweekly, weekly, monthly, or even less. The frequency of administration can remain constant or can be variable during the duration of treatment. A course of treatment can include rest periods. For example, a composition containing an antibody as provided herein can be administered over a first period of time, followed by a rest period, and such a regimen can be repeated one or more times. As with the effective amount, various factors can influence the actual frequency of administration used for a particular application. For example, the effective amount, duration of treatment, use of multiple treatment agents, route of administration, and severity of the clinical condition may require an increase or decrease in administration frequency.
[0061] After administering an anti B7-H1 antibody to a subject, the subject can be monitored to determine whether or not the clinical condition has improved. For example, a cancer patient can be assessed after treatment to determine whether or not the progression of the cancer has been reduced (e.g., stopped). Any method, including those that are standard in the art, can be used to assess progression and survival rates.
[0062] The invention will be further described in the following examples, which do not limit the scope of the invention described in the claims.
EXAMPLES
Example 1--Late Blockade of B7-H1 Reduces the Numbers of Effector CD8 T Cells Following Immunization
[0063] In an attempt to identify the optimal timing of B7-H1 blockade to improve T cell responses, anti-B7-H1 blocking antibody was administered either during an early (days 0-3) or a late (days 7-10) stage following immunization (FIG. 2A). These time periods were set according to the kinetics of T cell response following ovalbumin (OVA) and poly (I:C) immunization (Ahonen et al., J Exp Med 199:775-784, 2004). Early B7-H1 blockade greatly increased the expansion of OVA antigen specific (K.sup.bOVA tetramer.sup.+) and functional IFN-.gamma..sup.+ CD8 T cells in spleens of immunized mice (FIGS. 2B and 2C). Unexpectedly, late B7-H1 blockade decreased the percentages and numbers of antigen specific (tetramer.sup.+) and effector (IFN-.gamma..sup.+) CD8 T cells in the spleens of mice (FIGS. 2D and 2E). Taken together, the results of early blockade of B7-H1 are consistent with an inhibitory role of B7-H1 expressed by antigen presenting cells (dendritic cells) during the early stage of T cell priming (Pulko et al., J Immunol 183:3634-3641, 2009; and Farley et al., Mol Cell Biol 26:2118-2129, 2006). However, the opposite effects of late B7-H1 blockade indicate an unknown function of B7-H1 expressed by activated/effector T cells during the late stage of T cell responses (Pulko 2011, supra).
Example 2--B7-H1 Deficient Effector CD8 T Cells Fail to Mount a Protective Immunity
[0064] The ability of B7-H1-deficient effector CD8 T cells to mount protective immunity against tumor challenge was examined. WT and B7-H1 KO effector OT-1 CD8 T cells (with TCR specific for OVA antigen) were transferred (i.v.) into recipient mice one day before injection of B16-OVA tumor cells. While B16-OVA tumors progressively grew in the control group of mice without effector T cells transfer, they did not grow out in the mice that received WT effector CD8 T cells (FIG. 3A). However, the growth of B16-OVA tumors could not be completely suppressed in mice transferred with B7-H1 KO effector CD8 T cells (FIG. 3A, p<0.05), suggesting that B7-H1 deficient effector CD8 T cells may have compromised protective function. It also was observed that the frequency and numbers of K.sup.bOVA-tet.sup.+ CD8 T cells decreased by 2-5-fold at the tumor sites and spleens of recipients of B7-H1 KO CD8 T cells compared to recipients of WT CD8 T cells (FIG. 3B, p<0.05). In addition, the cytolytic activity in the spleens of recipients of B7-H1 KO CD8 T cells decreased by 4-7 fold compared to recipients of WT CD8 T cells (FIG. 3C). Taken together, these results suggested that B7-H1 deficient effector CD8 T cells could not mount protective immunity due to compromised cytolytic activity resulting from their reduced accumulation.
Example 3--More Apoptosis by B7-H1 Deficient T Cells Following Initial Proliferation
[0065] To directly identify the function of B7-H1 expressed by CD8 T cells, B7-H1 deficient, OT-1 TCR transgenic mice were produced. These mice had CD8 T cells carrying OVA-specific TCR, but did not express B7-H1 (Pulko 2011, supra). Proliferation to antigen stimulation was examined in vitro and in vivo. Naive B7-H1 KO and WT CD8 OT-1 T cells were found to undergo similar antigen-stimulated proliferation in vitro.
[0066] Next, studies were conducted to ascertain whether they differed in spontaneous apoptosis. Freshly isolated WT and B7-H1 KO CD8 T cells underwent comparably low levels of spontaneous apoptosis, as demonstrated by similar levels of Annexin V binding, TMRE staining (a barometer of mitochondrial transmembrane potential, which decreases during apoptosis; Veuger et al., Cancer Res 63:6008-6015, 2003), and active caspase-3 levels (Pulko 2011, supra). However, when stimulated with antigen (OVA), B7-H1 KO CD8 T cells underwent more apoptosis than WT CD8 T cells, as demonstrated by annexin V.sup.+ and TMRE.sup.low staining (FIG. 4A) and increased levels of active caspase-3 (FIG. 4B). Accordingly, the numbers of viable B7-H1 KO CD8 T cells had about a 2-fold decrease between days 3-5 after activation (FIG. 4C).
[0067] To examine whether increased death of B7-H1 KO CD8 T cells was due to impaired proliferation, CD8 T cells were labeled with carboxyfluorescein succinimidyl ester (CFSE; an intracellular dye for tracking cell division). On day 3 post antigen stimulation, B7-H1 KO and WT OT-1 CD8 T cells underwent similar proliferation (up to 6 divisions), but the percentage of B7-H1 KO CD8 T cells that underwent 3 or more divisions decreased by about 2-fold compared to WT CD8 T cells (FIG. 4D). These results suggested that B7-H1 deficient CD8 T cells undergo normal initial proliferation, but there is not a net increase in the population due to increased apoptosis.
Example 4--Fewer B7-H1 Deficient T Cells Survived the Contraction Phase In Vivo
[0068] To further investigate the role of B7-H1 expressed by antigen-specific CD8 T cells in vivo, WT or B7-H1 KO OT-1 CD8 (Thy1.2.sup.+) cells were transferred into congenic (Thy1.1.sup.+) B6 mice, followed by OVA/poly (I:C) immunization. Similar primary expansion of WT and B7-H1 KO CD8 T cells was observed in the spleens of immunized mice on day 4, but on day 6, B7-H1 KO OT-1 CD8 T cells exhibited more contraction than WT OT-1 CD8 T cells in both frequency and total numbers (FIG. 5A). To confirm this was a T cell intrinsic effect of B7-H1, the same numbers of preactivated WT and B7-H1 KO T cells (1:1) were co-transferred into the same host and anti-PD-1 blocking antibody or control antibody was injected with T cell transfer. If the prosurvival function of B7-H1 requires its ligation with PD-1, an anti-PD-1 antibody that blocks PD-1/B7-H1 ligation would cause a reduction in WT T cells but not in B7-H1 KO T cells (internal control). As shown in FIG. 5B, anti-PD-1 did not dramatically change the percent of WT T cells compared with control antibody, nor did it change the ratio with B7-H1 KO T cells, suggesting that B7-H1 does not need ligation with PD-1 to provide pro-survival function for T cells during contraction. These data thus indicated that T cell intrinsic B7-H1 is required for T cell survival during contraction.
[0069] Taken together, the above studies suggested a previously unknown function for B7-H1 expressed by T cells, and support the central hypothesis that B7-H1 expressed by activated CD8 T cells has an intrinsic pro-survival function that is required for establishment of T cell immunity, and ligation of B7-H1 by agonistic antibody may disrupt its pro-survival function in T cells. The studies discussed below are conducted to identify the mechanisms for B7-H1's function as an intrinsic pro-survival factor for activated T cells, to investigate the role of B7-H1 in T cell differentiation, and to find ways to evaluate the impact of agonistic B7-H1 antibodies in T cell function. By understanding the downstream signaling pathways of B7-H1, new subcellular targets for regulating T cell survival can be developed, and optimal B7-H1 antibodies can be selected to improve protective T cell immunity against cancers and pathogen infections.
Example 5--Defining the T Cell-Intrinsic Role of B7-H1 in T Cell Survival
[0070] The idea that B7-H1 has a T cell-intrinsic pro-survival function is a new concept in the field of B7-H1 biology, distinct from traditional studies into B7-H1-PD1 receptor interactions that promote apoptosis of PD-1.sup.+ T cells (Gibbons et al., supra; and Keir et al., Annu Rev Immunol 26:677-704, 2008). Thus, B7-H1 might use a previously unknown signaling pathway to mediate its T cell pro-survival function. The experiments in this example are carried out to investigate how B7-H1 regulates pro-survival molecule Bcl-xL via the p38 MAPK pathway, and to investigate the role of DNA-PKcs (which was more recently identified as a B7-H1-associated protein) in T cell survival. T cell apoptosis can be triggered by intrinsic (mitochondria-based) and extrinsic (receptor-based) stimuli (Bouillet and O'Reilly, Nat Rev Immunol 9:514-519, 2009). As B7-H1 deficiency does not affect the expression of Fas or Fas ligand in T cells (Pulko 2011, supra), these studies focus on defining how B7-H1 affects intrinsic or mitochondria-based apoptosis.
[0071] The Bcl-2 family is a group of proteins that coordinately control apoptotic cell death by regulating mitochondrial cytochrome c. This family includes both pro-apoptotic and anti-apoptotic members. Preliminary studies examined the levels of Bcl-2 family members (Bcl-2, Bcl-xL, and Bim) in both resting and activated T cells. Intracellular staining revealed similar levels of Bcl-2, Bcl-xL, and Bim in resting WT and B7-H1 KO CD8 T cells (FIG. 6A). In activated T cells, however, Bcl-xL levels were significantly lower in B7-H1 KO CD8 T cells than in WT CD8 T cells (FIGS. 6A and 6B, p<0.01).
[0072] The finding that the loss of B7-H1 is correlated with lower levels of Bcl-xL was both novel and unexpected, because it had been believed that B7-H1 functions as a suppressive regulator for T cells (Keir et al., supra). Since B7-H1 ligation by PD-1 did not affect Bcl-xL expression (Pulko 2011, supra), it is possible that B7-H1 regulates Bcl-xL in an intrinsic manner. The experiments described in this section will (1) examine the expression, function, and (2) phosphorylation of Bcl-xL in T cells in the absence of B7-H1 signaling.
[0073] Unlike Bcl-2 protein, which is constitutively expressed by T cells, Bcl-xL protein levels vary with levels of T cell activation (Boise et al., Immunity 3:87-98, 1995). Its expression is induced by TCR stimulation and up-regulated by CD28 signals. Bcl-xL expression is not stable, however, and it begins to decline at 48 h after activation. Bcl-xL loses its pro-survival function through phosphorylation by p38 MAPK (Farley et al., supra; and Kharbanda et al., J Biol Chem 275:322-327, 2000). In fact, activation of p38 MAPK prevents mitochondria accumulation of Bcl-xL and induces apoptosis of CD8 T cells in vivo (Farley et al., supra; and Merritt et al., Mol Cell Biol 20:936-946, 2000). Thus, it is possible that the decrease of Bcl-xL in B7-H1 KO T cells could be due to increased activation of p38 MAPK.
[0074] To test this possibility, activation of p38 MAPK was measured in B7-H1 KO T cells. The data of FIG. 7 show that the activation of p38 MAPK increased in B7-H1 deficient T cells compared to WT T cells, suggesting a potential regulatory role of B7-H1 in activation of p38. The degree p38 MAPK to which contributes to Bcl-xL levels was tested in WT and B7-H1 KO T cells. Since activation of p38 MAPK led to degradation of Bcl-xL (Farley et al., supra; and Kharbanda et al., supra), p38 MAPK inhibitor SB203580 (a specific pharmacological inhibitor of p38 MAPK) was used to test whether inhibition of p38 MAPK would increase Bcl-xL levels in activated T cells. The results of FIG. 8A show that inhibition of p38 MAPK increased the levels of Bcl-xL in WT T cells. Interestingly, the percent of increase of Bcl-xL was higher in WT T cells compared with B7-H1 KO T cells (p<0.01). These data suggested that B7-H1 could be a negative regulator of p38 MAPK activation. As diagrammed in FIG. 8B, in activated WT T cells, p38 MAPK was under the negative regulation of B7-H1 while Bcl-xL was under the negative control of p38 MAPK. When SB203580 was added, p38 MAPK was negatively regulated by at least two factors: B7-H1 and SB203580. Thus, Bcl-xL was released from p38 MAPK suppression and increased dramatically in activated WT T cells. However, in the absence of B7-H1, as in B7-H1 KO T cells, inhibition of p38 MAPK by SB203580 did not increase as much as in WT T cells.
Example 6--Determining Whether B7-H1 Stabilizes Bcl-xL Levels Via p38 MAPK
[0075] Based on the above data, it was hypothesized that B7-H1 stabilizes Bcl-xL levels by regulating activation of p38 MAPK pathway. The following experiments are performed to test this hypothesis.
[0076] First, degradation of Bcl-xL in the absence of B7-H1 is examined. Since phosphorylation of Bcl-xL leads to its degradation, experiments are conducted to test whether phosphorylation of Bcl-xL increases in the absence of B7-H1. The phosphorylation of Bcl-xL in WT and B7-H1 KO activated CD8 T cells is compared. Intracellular staining and Western blotting are performed to assess phospho-Bcl-xL expression (Millipore AB3116 specific for the Bcl-xL phosphorylated on serine 62) following CD8 T cell activation by anti-CD3/CD28 antibody for 5, 10, 30, and 60 minutes.
[0077] Second, p38 activity in the absence of B7-H1 is examined. In particular, the function of p38 MAPK from B7-H1 WT and KO T cells in phosphorylation of Bcl-xL is evaluated. To directly measure whether p38 MAPK activation increases in B7-H1 KO T cells and results in elevated phosphorylation of Bcl-xL, in vitro kinase assays are performed using recombinant Bcl-xL (ProSpec, East Brunswick, N.J.) as a substrate. Total p38 MAPK is immunoprecipitated from whole-cell lysates of B7-H1 WT or KO CD8 T cells (naive or activated), and then incubated with recombinant Bcl-xL in vitro. To confirm that phosphorylation of Bcl-xL in vitro is directly mediated by activated p38 MAPK, SB203580 is used in this system.
[0078] In addition, studies are conducted to determine whether enhanced expression of Bcl-xL rescues T cell apoptosis and contraction of B7-H1 KO T cells. B7-H1 KO mice are bred into Bcl-xL transgenic mice (provided by Dr. Shapiro of Mayo Clinic Rochester). T cell apoptosis and contraction are compared in vitro and in vivo between Bcl-xL Tg and non-Bcl-xLTg B7-H1 KO T cells using models as in the preliminary studies.
[0079] Further studies are conducted to provide insights into mechanisms by which B7-H1 regulates T cell survival, and particularly to determine whether inhibition of p38 MAPK affects T cell apoptosis and contraction of B7-H1 KO T cells. In particular, T cell apoptosis and contraction are compared in vitro and in vivo between B7-H1 KO T cells pre-incubated with SB203580 using models as in the preliminary experiments. Given the pro-survival function of B7-H1 expressed by activated T cells, B7-H1 may stabilize protein levels of Bcl-xL by preventing phosphorylation of Bcl-xL via inhibition of p38 MAPK activation. Accordingly, increased phosphorylation of BclxL and increased activity of p38 MAPK may be observed in B7-H1 KO T cells compared with WT T cells, and introduction of Bcl-xL transgene or inhibition of p38 MAPK may rescue B7-H1 KO T cells from apoptosis and contraction.
[0080] An alternative pathway of Bcl-xL degradation could involve ubiquitination of Bcl-xL (Niture and Jaiswal, J Blot Chem 286:44542-44556, 2011). If B7-H1 signaling data do not support a role for regulation of phosphorylation of Bcl-xL, the extent of ubiquitination of Bcl-xL is examined in B7-H1 KO T cells and WT T cells. Taken together, the results of these studies provide knowledge about how T cell survival and contraction are regulated by the B7-H1/p38 MAPK/Bcl-xL pathway, and facilitate the design of new immune adjuvants to promote T cell survival following antigen stimulation.
Example 7--Defining the Role of DNA-PKcs in B7-H1-Mediated T Cell Survival
[0081] B7-H1 is a transmembrane protein consisting of extracellular, transmembrane, and intracellular domains (FIG. 11A). The extracellular domain (ECD) of B7-H1 interacts with receptors PD-1 and CD80 expressed by activated T cells (Wang et al., J Exp Med 197:1083-1091, 2003). The intracellular domain (ICD) has the potential to deliver intrinsic anti-apoptotic signals (Azuma et al., Blood 111:3635-3643, 2008). It is not clear, however, how ICD mediates B7-H1's pro-survival function. To define the downstream signaling pathway of B7-H1, intracellular binding protein(s) of B7-H1 are identified.
[0082] In pilot studies, a single 450 kDa band was identified in the lysate from a human T cell line (Kaspas299, B7-H1 positive) (Frigola et al., Clin Cancer Res: an official journal of the Am Assoc Cancer Res 17:1915-1923, 2011) using an anti-B7-H1 antibody (5H1) in immunoprecipitation (FIG. 9A). Mass spectrometry analyses indicated that the most abundant protein in the 450 kDa band is DNA-PKcs (DNAdependent protein kinase, catalytic subunit). Western blotting was then performed to confirm that DNA-PK is associated with B7-H1. As shown in FIG. 9B, anti-B7-H1 pulled down a protein from the lysate of Kaspas299 cells that was identified as DNA-PK by Western blot (top panel of FIG. 9B). The presence of B7-H1 in the precipitation was confirmed by Western blotting with an anti-B7-H1 antibody (middle panel of FIG. 9B). In addition, B7-H1 was identified in association with DNA-PK when using anti-DNA-PK in immunoprecipitation with Kaspas299 cell lysate (lower panel of FIG. 9B). In addition, the association of B7-H1 with DNA-PK was identified in activated human T cells, but not in resting T cells (FIG. 10). Since only activated human T cells express B7-H1 protein 1, the association of B7-H1 and DNA-PK in activated T cells suggested a potential functional relationship between B7-H1 and DNA-PK.
[0083] Experiments are conducted to identify the binding sites and intracellular location of B7-H1 association with DNA-PKcs in T cells. The intracellular domain (ICD) of B7-H1 does not contain a tyrosine that could be phosphorylated by a tyrosine kinase, but it does contain serine and threonine residues that could be targets of DNA-PKcs, as DNA-PKcs is a serine/threonine protein kinase. To determine whether serine or threonine residues are required for the association of B7-H1 with DNA-PKcs, mutations are made at these residues in the ICD of B7-H1, and experiments are conducted to test whether these mutants affect the association of B7-H1 and DNA-PKcs. Briefly, B7-H1 mutants in which individual serine or threonine residues are replaced with alanine (FIG. 11B) are produced. B7-H1 negative T cells (Jurkat) are transfected with a mutant B7-H1 and then used in immunoprecipitation assays to test the association of mutant B7-H1 with DNA-PKcs, and to identify a binding site for DNA-PKcs based on the individual serine or threonine mutations. Multiple mutants are produced as needed if the individual mutants are not sufficient to abolish the association of B7-H1 and DNA-PKcs.
[0084] The identification of DNA-PK as a binding protein of B7-H1 was unexpected, as DNA-PK is a nuclear protein involved in DNA repair (Collis et al., Oncogene 24:949-961, 2005), while B7-H1 is an immunoregulatory molecules mainly expressed on the cell surface. Nevertheless, increased expression of DNA-PK has been reported among apoptotic T cells from patients with rheumatoid arthritis (Shao et al., J Exp Med 206:1435-1449, 2009; and Shao et al., EMBO Mol Med 2:415-427, 2010), suggesting that DNA-PK mediated DNA repair may be involved in T cell survival. It is likely that the T cell-intrinsic pro-survival function of B7-H1 is mediated by binding with DNA-PK in the nucleus, where DNA-PK promotes the DNA repair that is needed for T cell survival. The up-regulation of B7-H1 in T cells during the contraction phase following expansion suggests a possible translocation of B7-H1 from the T cell surface into the nucleus, where B7-H1 binds to DNA-PK to promote DNA repair for DNA damage accumulated after intensive T cell expansion (Doering et al., Immunity 37:1130-1144, 2012; and Baitsch et al., J Clin Invest 121:2350-2360, 2011).
[0085] It has been reported that B7-H1 undergoes redistribution from the cell surface into the nucleus in tumor cells upon treatment with chemotherapy drugs (Ghebeh et al., Breast Cancer Res 12:R48, 2010). Using this model, experiments were conducted to determine whether translocation of B7-H1 results in close association with DNA-PKcs in the nucleus. The data of FIG. 12 show that DNA-PKcs mainly localized in the nucleus, while B7-H1 had both cytoplasmic and nuclear distribution. In the nucleus, B7-H1 was identified in association with DNA-PKcs. It is possible that activated T cells recapitulate the pro-survival function of B7-H1 when B7-H1 translocates to nuclei, a potential target of B7-H1 in nuclei could be DNA-PK, as implied by the association of B7-H1 and DNA-PK in activated T cells (FIG. 10). To test this possibility, the translocation of B7-H1 in naive and activated T cells (1-3 days) is examined after TCR stimulation in vitro. The co-localization and intracellular distribution of B7-H1 with DNAPK in resting and activated T cells is analyzed using confocal microscopy. T cell activation may cause B7-H1 distribution into nuclei, where B7-H1 would be closely associated with DNA-PKcs. The strength of TCR stimulation (dose of anti-CD3) and costimulation (CD28) could affect the association and co-localization of B7-H1 and DNA-PKcs. To address this, the dose and anti-CD3 are titrated in the presence or absence of CD28 in these experiments.
[0086] Further experiments are conducted to determine the role of DNA-PKcs in activation of p38 MAPK. DNA-PKcs is a serine/threonine protein kinase (450 KDa) and is a member of the phosphatidylinositol kinase (PIK)-related family. Although DNAPK is believed to play a major role in repairing double strand DNA breaks and V(D)J recombination, DNAPKcs also has signaling functions. It has been reported that DNA-PKcs is required for ERK activation in mouse macrophages (Panta et al., Mol Cell Biol 24:1823-1835, 2004; and Yotsumoto et al., J Immunol 180:809-816, 2008), how DNA-PKcs affects the activation of p38 is not clear. Since p38 activation was increased in B7-H1 KO T cells, the degree to which DNA-PKcs contributes to these changes is tested.
[0087] NU7026 (2-(morpholin-4-yl)-benzo[h]chomen-4-one) is a DNA-PKcs inhibitor. This compound is selective for DNA-PKcs, and 10 .mu.M NU7026 can completely inhibit activity of purified DNA-PK (Veuger et al., supra). NU7026 is added into cultures with pre-activated WT and B7-H1 KO T cells and after 24-72 hours of incubation, the activation of p38 is measured in the cells. As a consequence of p38 MAPK activation regulated by NU7026, the level of Bcl-xL also is measured in the T cells after treatment with NU7026. If p38 activation is regulated by DNA-PKcs, changes in p38 activation in the presence of NU7026 would be observed in WT T cells. If B7-H1 requires DNA-PKcs to regulate activation of p38 MAPK, NU7026 would induce significant changes in p38 MAPK activation in WT T cells, but not in B7-H1 KO T cells. NU7026 increased p38 MAPK activation in WT, but not in B7-H1 KO T cells, suggesting that DNA-PK in association with B7-H1 negatively regulates p38 MAPK activation. Bcl-xL levels would change accordingly with the changes in p38 MAPK activation. In addition to inhibition of DNA-PKcs activity by NU7026, the impact of total DNA-PKcs protein levels on p38 MAPK activation is evaluated. siRNA-mediated knockdown of DNA-PKcs is known to result in reduced ERK activation in mouse macrophages (Yotsumoto et al., supra). Using a similar approach, experiments are conducted to test whether down-regulation of DNA-PKcs in T cells affects p38 MAPK activation in WT and B7-H1 KO T cells. Preliminary data predicts a potential link between DNA-PK and activation of p38 MAPK. DNA double strand breaks induced G2/M cell cycle checkpoint, dependent on activation of p38 MAPK (Pedraza-Alva et al., EMBO J25:763-773, 2006). On the other hand, the involvement of DNA-PK in activation of the MAPK signal cascade has been proposed (Panta et al., supra). Thus, association with DNA-PK would recruit B7-H1 in the regulation of p38 MAPK. Studies to investigate how DNA-PK is involved in the B7-H1 signaling pathway (e.g., in regulation of p38 MAPK activation) in T cells are conducted. A potential link is Akt activation, as studies have identified DNA-PK as a kinase that activates Akt (Feng et al., J Biol Chem 279:41189-41196, 2004; and Dragoi et al. EMBO J24:779-789, 2005). DNA-PKcs colocalizes with Akt at the plasma membrane and phosphorylates Akt on Ser473, resulting in about a 10-fold enhancement of activity. A decrease in activation of Akt was observed in B7-H1 KO T cells. Taken together, results from these studies collectively provide new insight into regulation of T cell survival by a previously unknown B7-H1/p38 MAPK/Bcl-xL pathway.
Example 8--Defining the Role of T Cell Intrinsic B7-H1 in T Cell Differentiation
[0088] To establish protective T cell immunity, primed T cells need to acquire long term survival characteristics and to mount rapid and effective secondary responses to pathogen, traits shared with memory T cells (Pulko 2011, supra; Collis, supra; and Ghebeh et al., supra). Protecting T cells from contraction is a new function of T cell intrinsic B7-H1, suggesting that up-regulation of B7-H1 by effector T cells would give them selective advantage in differentiating into memory T cells. To test this possibility, pre-activated WT and B7-H1 KO CD8 T cells were co-transferred at the same numbers into naive CD45.1+B6.SJL mice to monitor their survival in an antigen-free host (a model for memory cell differentiation) (Pulko 2011, supra). On day 15 after transfer, the transferred WT and B7-H1 KO effector T cells were easily identified by their congenic markers (FIG. 13A). As expected, fewer transferred B7-H1 KO T cells than WT T cells were found in the spleen. The dramatic reduction of B7-H1 KO T cells implied that most effector T cells require B7-H1 to survive and become memory T cells.
[0089] In the linear differentiation model, memory T cells are believed to be generated from effector T cells that survive the contraction phase (Opferman et al., Science 283:1745-1748, 1999; and Wherry et al., Nat Immunol 4:225-234, 2003). Since fewer B7-H1 KO T cells survived at the end of the contraction phase (FIG. 5A) and after 15 days following transfer in vivo (FIG. 13B), it was hypothesized that T cell-intrinsic B7-H1 helps effector T cells to survive the contraction phase and become memory cells. This hypothesis is diagramed in FIG. 14. Both B7-H1 deficient and transgenic T cell models are used to test this hypothesis and define a new mechanism underlying memory T cell differentiation by dissecting the role of T cell intrinsic B7-H1.
[0090] Theiler's murine encephalomyelitis virus (TMEV) is an endogenous pathogen in mice. Intracranial infection of TMEV causes acute encephalitis. Resistant strains of mice (such as C57BL/6 mice H-2.sup.b) effectively clear the TMEV infection and generate a T cell response against the viral protein (Borson et al., J Virol 71:5244-5250, 1997). To easily track viral antigen specific T cell responses, the TMEV strain that includes the H-2K.sup.b restricted OVA epitope SIINFEKL (SEQ ID NO:2; Pavelko et al., Mol Therapy 21:1087-1095, 2013) is used. To evaluate the role of T cell intrinsic B7-H1 in T cell differentiation and mounting an anti-viral immunity, the TMEV-OVA infection model is used.
[0091] The same numbers (2.times.10.sup.3) of naive CD8 T cells isolated from WT (Thy1.1) or B7-H1 KO (CD45.2) C57BL/6 mice are co-transferred separately into CD45.1 B6.SJL mice. On the same day of T cell transfer, mice are inoculated intracranially with 2.times.10.sup.6 PFU of the Daniel strain of TMEV (Mendez-Fernandez et al., Eur J Immunol 33:2501-2510, 2003). The function and phenotype of the transferred T cells are analyzed on days 7 (effector phase) and 30 (memory phase) after infection in the brain and draining lymph nodes. H-2K.sup.b/OVA tetramer staining and congenic markers are used to define the transferred WT and B7-H1 KO T cell responses to TMEV infection. The effector or memory phenotype of the transferred T cells is determined by the expression of CD43 (1B11) for effector T cells (Harrington et al., J Exp Med 191:1241-1246, 2000), and CD44 and CD62L for memory T cells. The function of effector/memory T cells is analyzed by ex vivo assays to evaluated degranulation (CD107a expression) and intracellular production of cytokines (IFN-.gamma., IL-2, and TNF-.alpha.) (Webster et al., supra). On day 7 and day 30, a CTL assay is performed to analyze the function of effector (day 7) or memory (day 30) T cells in vivo as previously reported (Pulko 2009, supra). The anti-viral immunity is evaluated by detecting the persistence of TMEV in the brain using TCR and viral plaque assay (Zhang et al., J Neuroimmunol 116:178-187, 2001).
[0092] A sample size of 10 mice per group provides at least 90% power to detect the significant difference at alpha=0.05 based on previous studies (Pavelko et al., supra). By comparing the frequency/number and function of the persistence of transferred B7-H1 WT and KO T cells, the extent to which B7-H1 deficiency affects the generation of functional memory CD8 T cells is determined. Analysis and comparison of their memory phenotype (T effector CD43/1B11.sup.high, T effector memory CD44.sup.highCD62L.sup.low or T central memory CD44.sup.highCD62L.sup.high) allows determination of what subset(s) of CD8 T cells require B7-H1 for their differentiation.
[0093] In addition to using B7-H1 KO mice as models, an inducible B7-H1 transgenic mouse model in which B7-H1 expression can be specifically and temporally induced on T cells (FIG. 15) is used. In this model, a CD3.delta. (T cell-specific) promoter is used to drive the expression of a tetracycline-controlled transcriptional silencer (tTS) and a reverse tetracycline-controlled transcriptional activator (rtTA) in T cells. In the absence of doxycycline (Dox), a derivative of tetracycline, tTS actively suppresses transcription of B7-H1 gene (driven by tetO), preventing leaky expression of B7-H1. In the presence of Dox, tTS dissociates from tetO, whereas rtTA binds tetO with high affinity and induces expression of B7-H1. This approach has been used to induce CD4 T cell-specific gene expression in transgenic mice (Huai et al., Genesis 45:427-431, 2007), demonstrating this system's efficiency and specificity.
[0094] Using this model, T cell expression of B7-H1 is induced on days 0-6 (early stage) or days 8-14 (late stage) respectively by injection of doxycycline, during TMEV-OVA infection. Wild type mice are used as controls for base line T cell responses. The accumulation and function of effector cells and memory T cells in the brain and draining lymph nodes are measured on days 7, 15, and 30 following infection. To assess their protective immunity, immunized mice are challenged with B16-OVA tumor cells on day 21 after immunization, and tumor size is compared between mice with different kinetics/timing of B7-H1 expression by T cells (early vs. late). These experiments permit determination of the optimal timing for turning on B7-H1 expression by T cells during T cell responses to infection in order to enhance protective immunity. Phenotype and functional assays determine how enhanced B7-H1 expression helps memory T cell differentiation.
[0095] As both WT and B7-H1 KO CD8 T cells are from C57BL/6 mice that can effectively respond to OVA epitope in H-2K.sup.b hosts, both types of cells may have comparable primary responses to TMEV-OVA infection on day 7. If B7-H1 is required by effector CD8 T cells to survive during the contraction phase, B7-H1 KO CD8 T cells may have more contraction than WT T cells, and consequently, fewer effector/memory B7-H1 KO T cells are accumulated in the infected brain or lymph nodes on day 30. As a result, high amounts of TMEV remain in the brain tissues of mice transferred with B7-H1 KO T cells compared to mice receiving WT T cells. These results determine whether T cell intrinsic B7-H1 is required to generate a protective T cell immunity.
[0096] The induced T cell B7-H1 expression model is used to determine when T cell B7-H1 is required for survival of activated T cells and subsequent differentiation. The induced B7-H1 model is similar to T cell-specific expression constructs that have been previously established. It is acknowledged that induced B7-H1 might not undergo a degradation process as occurs with natural B7-H1 in T cells, and would overlap or compete with endogenous B7-H1 in T cells. To exclude the overlapping or competitive effects, this model is generated in a B7-H1 KO background. Such a model may be a valuable tool applicable to other immune systems, including evaluating the impact of T cell B7-H1 on the efficacy of tumor vaccines in combination with and B7-H1 blockade as an approach for treating solid tumors.
[0097] Although T cell survival is critical for mounting a protective immunity, T cell trafficking to tumor or infection site also is important. To know whether B7-H1 regulates T cell migration, congenic markers are used to track transferred effector T cells in tumor-bearing or infected mice to examine the migration of WT and B7-H1 KO T cells in vivo. The cytolytic activity of CD8 T cells also may impact the protective immunity mediated by CTLs. If B7-H1 KO T cells do not mount a protective immunity in vivo, experiments are done to test whether it is because of any defects in killing of target cells by B7-H1 KO CD8 T cells. To evaluate this possibility, cytolytic activity is determined using calcein-labeled tumor or infected target cells in vitro.
Example 9--Defining the Agonistic Function of Anti-B7-H1 Antibody on T Cell Function
[0098] To block the B7-H1/PD-1 signaling pathway, antibodies against B7-H1 have been aggressively pursued as an immunotherapeutic option for treating human solid cancers (Brahmer et al., supra). However, the potential agonist functions of such blocking antibodies have not been addressed in the context of T cell biology. Given the pro-survival function of B7-H1 expressed by T cells (Pulko 2011, supra), it is possible that B7-H1 antibody administered in the course of blockade therapy may have agonistic effects on T cell-associated B7-H1, and may disrupt the intrinsic pro-survival function of B7-H1 for T cells. Ligation of B7-H1 by antibody resulted in enhanced apoptosis of fully activated human T cells, with a dramatic increase in transcription of TNF-related apoptosis-inducing ligand (TRAIL) (Dong et al., supra). Interestingly, an autoantibody to B7-H1 was identified in the sera of patients with active rheumatoid arthritis that have ongoing T cell apoptosis (Dong et al., supra; Shao et al. 2009, supra; and Shao et al. 2010, supra). It has been observed that injection of B7-H1 antibody reduced the numbers of effector CD8 T cells but not CD4 T cells at late stages of T cell activation (FIG. 1) (Rowe et al., supra; Xu et al., supra; and Seo et al., Immunology 123:90-99, 2008). Although the reduced outcome of CD8 T cell responses could be explained by a potential co-stimulatory role of B7-H1 (e.g., in promoting T cell expansion) that could be blocked by anti-B7-H1 antibodies, another possibility is that some anti-B7-H1 antibodies might have agonistic function (e.g., disrupting B7-H1's intrinsic pro-survival function, and triggering T cell death). This raises several questions, including how to identify and screen agonistic B7-H1 antibodies for T cells, what is the signaling pathway triggered by agonistic B7-H1 antibodies in T cells, and what is the impact of agonistic B7-H1 antibodies on tumor immunity. This knowledge may be critical for selecting therapeutic anti-B7-H1 antibodies. Identification of agonistic B7-H1 antibodies will help to reduce unwanted negative effects on T cell survival. Further, the selection of B7-H1 antibody with blocking effects but not agonistic effects can maximize the therapeutic effects of anti-B7-H1 antibody in treatment of human cancers.
[0099] To being answering these questions, studies are conducted to select agonistic antibodies to B7-H1 based on their intracellular signaling profile. As noted above, B7-H1 ligation increased transcription of TRAIL in activated human T cells (Dong et al., supra). The TRAIL gene is tightly regulated, potentially due to its considerable apoptosis inducing potential. Activation of p38 MAPK selectively induces apoptosis of CD8 T cells, but not CD4 T cells (Merritt et al., supra; and Rincon and Pedraza-Alva, Immunol Rev 192:131-142, 2003). In addition, activation of p38 MAPK has been correlated with induction of TRAIL expression (Zula et al. Proc Natl Acad Sci USA 108:19689-19694, 2011). Since B7-H1 ligation by antibody induced the up-regulation of TRAIL, B7-H1 ligation may lead to activation of p38 MAPK, which in turn induces TRAIL expression. To test this possibility, the activation (phosphorylation) of p38 MAPK was measured in activated T cells following ligation of B7-H1 by a panel of anti-B7-H1 monoclonal antibodies (5H1, H1A, MDX, and 2.2B). The results shown in FIGS. 16A and 16B demonstrate that H1A and 2.2B significantly increased the phosphorylation of p38 MAPK in activated human T cells. It is noted that antibodies H1A and 2.2B share the same IgG type (mouse IgG1) but have different binding sites from 5H1, suggesting that the epitope on B7-H1 rather than the isotype of antibody determines the activation of p38 MAPK. As described below, measuring the activation of p38 MAPK in T cells is used to screen for B7-H1 antibodies with agonistic function. Based on preliminary data, it is hypothesized that agonistic antibodies against B7-H1 affect CD8 T cell survival via activation of the p38 MAPK pathway.
[0100] To test this hypothesis, studies are conducted to determine whether an anti-B7-H1 antibody with the potential for activating p38 also can enhance T cell apoptosis. CD8 T cells maintain a 2-3 fold higher level of p38 MAPK activity than CD4 T cells, and activation of p38 MAPK in vivo caused a specific loss of CD8 T cells in peripheral lymphoid organs (Merritt et al., supra). The loss of CD8 T cells was attributed to increased apoptosis of CD8 T cells mediated by caspases following p38 MAPK activation. Experiments to test whether inhibition of p38 MAPK reduces apoptosis of human CD8 T cells showed that addition of a specific p38 MAPK inhibitor (SB203580) dramatically reduced apoptosis of activated CD8 T cells in vitro (FIGS. 17A and 17B). Thus, the activation of p38 MAPK appears to induce CD8 T cell apoptosis.
[0101] Using the same model as in FIGS. 17A and 17B, the degree of apoptosis of T cells induced by antibodies is compared, based on their ability to activate p38 MARK. Purified naive (CD45RA+ CCR7+) CD8 T cells from human PBMC are incubated with anti-CD3/CD28 beads in the presence of plate-bound anti-B7-H1 mAb that activates p38 (H1A or 2.2 B) or does not activate p38 (5H1 or MDX); a control group uses isotype control mIgG1. After 48 hours of incubation, the apoptosis of T cells is measured by staining with TMRE (for mitochondria potential) and Annexin V. The number of live T cells is counted to confirm a net loss of cells due to apoptosis. If activation of p38 MAPK enhances CD8 T cell apoptosis, more apoptosis or loss of viable CD8 T cells in culture with H1A or 2.2B anti-B7-H1 mAb compared with 5H1 mAb or control group would be expected. To test whether activation of p38 MAPK is required in T cell apoptosis induced by agonistic B7-H1 antibody, p38 specific inhibitor SB203580 is used in culture with agonist antibody. If activation of p38 MAPK is required for T cell apoptosis, enhanced CD8 T cell apoptosis caused by an agonistic antibody against B7-H1 would be blocked by inhibition of p38 MAPK.
[0102] Studies also are conducted to assess how ligation of B7-H1 by antibody leads to activation of p38 MAPK. A recent report implies that downstream genes involved in the MAPK/ERK pathway are the targets of B7-H1 mediated modulation in tumor cells (Cao et al., Cancer Res 71:1235-1243, 2011). Skin tumors induced in B7-H1 Tg mice show up-regulated Snail and Slug and down regulated Ecadherin, all of which are dependent on the activation of MAPK/ERK signaling (Pece and Gutkind, J Biol Chem 275:41227-41233, 2000; Bonni et al., Science 286:1358-1362, 1999; and Conacci-Sorrell et al., J Cell Biol 163:847-857, 2003). To test whether ligation of B7-H1 might affect the MAPK/ERK activity, the relative phosphorylation levels of 26 proteins (FIG. 18) involved in the MAPK/ERK pathway in T cells are measured and compared after incubation with B7-H1 antibody with or without ability to activate p38 MAPK. Preactivated T cells are incubated with either plate-bound or soluble anti-B7-H1 antibody for different period of times (5, 10, 30, 60, or 120 minutes) followed with a quick cell lysate preparation. Cell lysates are used in Array Assay of Phosphorylation of p38 MAPK pathway, according to manufacturer's directions (R&D Systems, Minneapolis, Minn.). The levels of phosphorylation of individual proteins in this pathway are compared between T cells with and without antibody B7-H1 ligation. Agonistic B7-H1 antibody may alter the activation of some upstream proteins in the MAPK/ERK pathway that could accordingly lead to regulation of p38 MAPK. The regulatory function of these candidate proteins is confirmed by their specific inhibitors or knockdown their protein levels.
[0103] The signaling pathway of B7-H1 following antibody ligation is identified. A panel of anti-human B7-H1 mAbs is used to screen and compare agonistic functions. This information is critical for selecting B7-H1 antibodies having desired blocking properties to be used to treat human solid cancers (Brahmer et al., supra), while agonistic properties are filtered from the B7-H1 antibody inventory to avoid causing undesired effects on T cell survival. Assays and potential molecular signaling pathways developed and identified in these studies may be translated into a screening platform for selecting candidate therapeutic anti-B7-H1 monoclonal antibodies for clinical use. Although these studies focus on the potential impact of anti-B7-H1 antibody on T cell survival and apoptosis, as predicted by previous studies (Dong et al., supra), ligation of B7-H1 also may impact functions of activated T cells (such as cytokine production).
[0104] Further experiments are carried out to define the impact of agonistic B7-H1 antibody on tumor immunity. B7-H1 expressed by tumor cells may not be the only target of administered therapeutic anti-B7-H1 antibody. B7-H1 positive CD8 T cells are frequently observed in human renal cell carcinoma tissues and peripheral blood (FIGS. 19A and 19B). However, the impact of anti-B7-H1 on the function of tumor-reactive T cells has not been evaluated. Ligation of B7-H1 expression by T cells could affect the outcome of B7-H1 blockade therapy using anti-B7-H1, as anti-B7-H1 antibody may have agonistic effects on T cells in regarding to their survival. Thus, this information is critical.
[0105] To identify the timing for targeting B7-H1 expressed by T cells within tumors, the kinetics of tumor-reactive CD8 T cells within growing tumors were determined, and the levels of B7-H1 expression by tumor-reactive CD8 T cells were measured. CD11 a.sup.high was used as a surrogate marker to identify tumor-reactive CD8 T cells (Gibbons et al., supra). As shown in FIG. 20A, CD11a.sup.high CD8 T cells gradually increased in growing tumors and peaked at day 15, a turning point for rapid growth of tumors. Accordingly, B7-H1 expression increased in CD11a.sup.high CD8 T cells up to day 12, and declined thereafter (FIG. 20B). These results demonstrated that up regulation of B7-H1 in tumor-reactive CD8 T cells accompanies T cell expansion within tumors, and down regulation of B7-H1 advances T cell contraction within tumors, suggesting B7-H1 may be required for T cell accumulation at tumor sites.
[0106] To examine the impact of B7-H1 antibody on the accumulation and function of tumor-infiltrating T cells, a panel of anti-B7-H1 mAbs is compared based on their different agonistic functions. The anti-mouse B7-H1 monoclonal antibodies 10B561 ("10B5"), 10F.9G262 ("9G2"), and MIH563 ("MIH5") are all dual blockers of B7-H1/PD-1 and B7-H1/B7-1 binding. As shown in FIG. 21, 9G2 and MIH5, but not 10B5, increased the activation of p38 MAPK as compared to control IgG (rat IgG), suggesting that 9G2 and MIH5 may have agonistic effects on B7-H1 expressed by T cells. Interestingly, 9G2 may reduce CD8 T cell responses in vivo (Rowe et al., supra; and Xu et al., supra). It is likely that 10F.9G2 has agonistic function, such that when it engages B7-H1 expressed by activated CD8 T cells in vivo, it may induce T cell apoptosis by activation of p38 MAPK.
[0107] Using the tumor model of FIG. 20A, the impacts of anti-B7-H1 mAb with (9G2) or without (10B5) agonistic function are compared. Anti-B7-H1 mAbs are injected during days 6-12, when B7-H1 has higher expression on T cells (FIG. 20B). Antibody or isotype control IgG (200 .mu.g) is injected i.p. every three days (on days 6, 9, and 12). The impact of each antibody is determined by its effects on T cell function and tumor growth. On days 14, 20, 27 after tumor inoculation, CD8 T cells are isolated from tumor tissues, and their frequency and function are analyzed. In addition, tumor growth is measured after antibody injection. Each group includes 5 mice, and all experiments are performed independently at least 3 times. Two-sided, unpaired Student's t-tests are used to evaluate differences in T cell frequency and tumor growth between groups of mice. P-values <0.05 is considered statistically significant.
[0108] These studies are used to investigate the hypothesis that agonistic B7-H1 antibodies may compromise antitumor immunity mediated by CD8 T cells due to activation of p38 MAPK. Since accumulation of sufficient tumor-reactive CD8 T cells at tumor sites is critical for controlling tumors, a low frequency of functional tumor-reactive CD8 T cells would represent a deficiency in antitumor activity. Mice treated with anti-B7-H1 blocking antibody without agonistic function would have a survival advantage compared with mice treated with agonistic B7-H1 antibody. It is acknowledged that p38 MAPK activation may not be the only readout for determining the agonistic function of anti-B7-H1 mAb in vitro, as some antibodies that do not have agonistic effects in vitro might have agonistic effects in vivo. A careful comparison of the frequency and numbers of tumor-reactive CD8 T cells is used to identify the final impact of B7-H1 antibodies on T cells. On the other hand, the potential effects of agonistic B7-H1 antibodies on tumor cells should be considered in interpreting tumor growth data. The anti-apoptosis function of B7-H1 was identified in tumor cells, but the impact of B7-H1 ligation on tumor growth and survival is not clear. The studies discussed herein are used to reveal new mechanisms for B7-H1 in T cell survival. These studies provide knowledge necessary for selecting optimal B7-H1 antibodies to improve the efficacy of checkpoint blockade therapy for human cancers.
Example 10--Ligation of B7-H1 Reduced Phosphorylation of AKT in CD8 T Cells
[0109] Freshly purified human peripheral blood CD8 T cells were incubated with anti-CD3 and H1A or 5H1 antibodies to B7-H1 for 24 hours. The control was an isotype of anti-B7-H1 antibody. AKT phosphorylation was analyzed by intracellular staining with anti-phosphor-AKT (S473) antibody. FIG. 22A contains representative histograms of phosphor-AKT expression, while FIG. 22B is a graph plotting the pooled data from three donors. Ligation of B7-H1 by the H1A antibody significantly reduced AKT phosphorylation (p<0.05).
OTHER EMBODIMENTS
[0110] It is to be understood that while the invention has been described in conjunction with the detailed description thereof, the foregoing description is intended to illustrate and not limit the scope of the invention, which is defined by the scope of the appended claims. Other aspects, advantages, and modifications are within the scope of the following claims.
Sequence CWU 1
1
21290PRTHomo sapiens 1Met Arg Ile Phe Ala Val Phe Ile Phe Met Thr
Tyr Trp His Leu Leu1 5 10 15Asn Ala Phe Thr Val Thr Val Pro Lys Asp
Leu Tyr Val Val Glu Tyr 20 25 30Gly Ser Asn Met Thr Ile Glu Cys Lys
Phe Pro Val Glu Lys Gln Leu 35 40 45Asp Leu Ala Ala Leu Ile Val Tyr
Trp Glu Met Glu Asp Lys Asn Ile 50 55 60Ile Gln Phe Val His Gly Glu
Glu Asp Leu Lys Val Gln His Ser Ser65 70 75 80Tyr Arg Gln Arg Ala
Arg Leu Leu Lys Asp Gln Leu Ser Leu Gly Asn 85 90 95Ala Ala Leu Gln
Ile Thr Asp Val Lys Leu Gln Asp Ala Gly Val Tyr 100 105 110Arg Cys
Met Ile Ser Tyr Gly Gly Ala Asp Tyr Lys Arg Ile Thr Val 115 120
125Lys Val Asn Ala Pro Tyr Asn Lys Ile Asn Gln Arg Ile Leu Val Val
130 135 140Asp Pro Val Thr Ser Glu His Glu Leu Thr Cys Gln Ala Glu
Gly Tyr145 150 155 160Pro Lys Ala Glu Val Ile Trp Thr Ser Ser Asp
His Gln Val Leu Ser 165 170 175Gly Lys Thr Thr Thr Thr Asn Ser Lys
Arg Glu Glu Lys Leu Phe Asn 180 185 190Val Thr Ser Thr Leu Arg Ile
Asn Thr Thr Thr Asn Glu Ile Phe Tyr 195 200 205Cys Thr Phe Arg Arg
Leu Asp Pro Glu Glu Asn His Thr Ala Glu Leu 210 215 220Val Ile Pro
Glu Leu Pro Leu Ala His Pro Pro Asn Glu Arg Thr His225 230 235
240Leu Val Ile Leu Gly Ala Ile Leu Leu Cys Leu Gly Val Ala Leu Thr
245 250 255Phe Ile Phe Arg Leu Arg Lys Gly Arg Met Met Asp Val Lys
Lys Cys 260 265 270Gly Ile Gln Asp Thr Asn Ser Lys Lys Gln Ser Asp
Thr His Leu Glu 275 280 285Glu Thr 2902873DNAHomo sapiens
2atgaggatat ttgctgtctt tatattcatg acctactggc atttgctgaa cgcatttact
60gtcacggttc ccaaggacct atatgtggta gagtatggta gcaatatgac aattgaatgc
120aaattcccag tagaaaaaca attagacctg gctgcactaa ttgtctattg
ggaaatggag 180gataagaaca ttattcaatt tgtgcatgga gaggaagacc
tgaaggttca gcatagtagc 240tacagacaga gggcccggct gttgaaggac
cagctctccc tgggaaatgc tgcacttcag 300atcacagatg tgaaattgca
ggatgcaggg gtgtaccgct gcatgatcag ctatggtggt 360gccgactaca
agcgaattac tgtgaaagtc aatgccccat acaacaaaat caaccaaaga
420attttggttg tggatccagt cacctctgaa catgaactga catgtcaggc
tgagggctac 480cccaaggccg aagtcatctg gacaagcagt gaccatcaag
tcctgagtgg taagaccacc 540accaccaatt ccaagagaga ggagaagctt
ttcaatgtga ccagcacact gagaatcaac 600acaacaacta atgagatttt
ctactgcact tttaggagat tagatcctga ggaaaaccat 660acagctgaat
tggtcatccc agaactacct ctggcacatc ctccaaatga aaggactcac
720ttggtaattc tgggagccat cttattatgc cttggtgtag cactgacatt
catcttccgt 780ttaagaaaag ggagaatgat ggatgtgaaa aaatgtggca
tccaagatac aaactcaaag 840aagcaaagtg atacacattt ggaggagacg taa
873
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
D00012
D00013

D00014

D00015

D00016
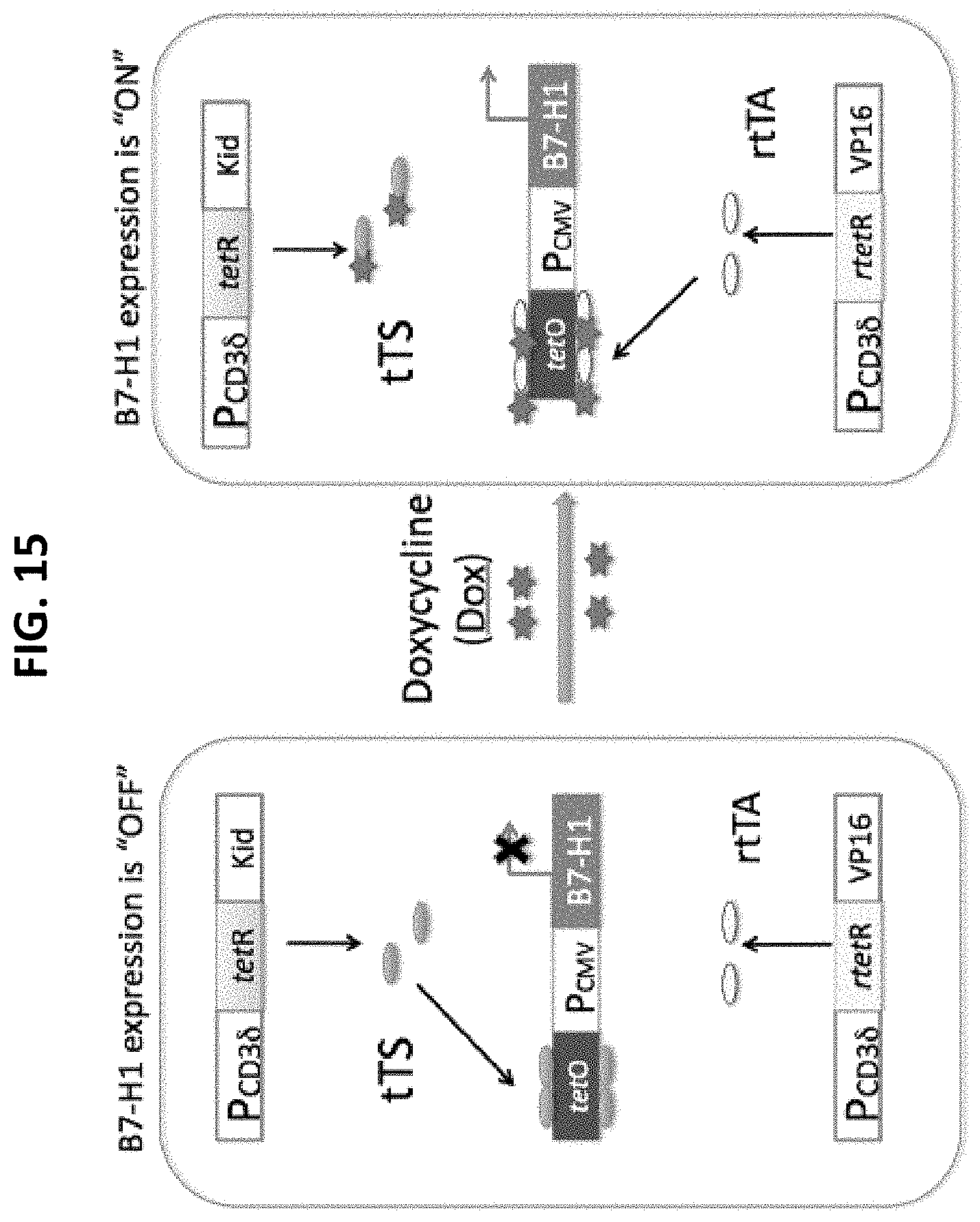
D00017

D00018

D00019

D00020

D00021

D00022

D00023

D00024

S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.