Method And Kit For Determining The Genome Integrity And/or The Quality Of A Library Of Dna Sequences Obtained By Deterministic R
Klein; Christoph Andreas ; et al.
U.S. patent application number 16/455470 was filed with the patent office on 2019-10-17 for method and kit for determining the genome integrity and/or the quality of a library of dna sequences obtained by deterministic r. The applicant listed for this patent is Fraunhofer-Gesellschaft Zur Forderung Der Angewandten Forschung E.V., Menarini Silicon Biosystems S.p.A.. Invention is credited to Christoph Andreas Klein, Nicol Manaresi, Bernhard Michael Polzer.
| Application Number | 20190316194 16/455470 |
| Document ID | / |
| Family ID | 49680939 |
| Filed Date | 2019-10-17 |

| United States Patent Application | 20190316194 |
| Kind Code | A1 |
| Klein; Christoph Andreas ; et al. | October 17, 2019 |
METHOD AND KIT FOR DETERMINING THE GENOME INTEGRITY AND/OR THE QUALITY OF A LIBRARY OF DNA SEQUENCES OBTAINED BY DETERMINISTIC RESTRICTION SITE WHOLE GENOME AMPLIFICATION
Abstract
A method for determining the integrity of the genome of a sample and/or the quality of a library of DNA sequences obtained by deterministic restriction site whole genome amplification can include (a) amplifying the library of DNA sequences to produce first, second, and third PCR products each of a different size from 50 bp to 1000 bp, by PCR using at least one first primer pair, one second primer pair and one third primer pair, the primer pairs each hybridizing to a DNA sequence of the library having a length from 1000 bp to 5000 bp and corresponding to a sequence of the genome located respectively on a first, second and third chromosome arm; (b) detecting the first, second and third PCR products; (c) correlating the presence of the first, second and third PCR products with the integrity of the genome of the sample and/or the quality of the library.
| Inventors: | Klein; Christoph Andreas; (Regensburg, DE) ; Polzer; Bernhard Michael; (Muenchen, DE) ; Manaresi; Nicol; (Bologna, IT) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 49680939 | ||||||||||
| Appl. No.: | 16/455470 | ||||||||||
| Filed: | June 27, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 15101299 | Jun 2, 2016 | 10392657 | ||
| PCT/IB2014/066602 | Dec 4, 2014 | |||
| 16455470 | ||||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | C12Q 2535/122 20130101; C12Q 1/686 20130101; C40B 30/00 20130101; C40B 40/06 20130101; C12Q 2545/101 20130101; C12Q 2545/101 20130101; C12Q 1/6869 20130101; C12Q 1/6869 20130101; C12Q 2535/122 20130101 |
| International Class: | C12Q 1/6869 20060101 C12Q001/6869; C40B 40/06 20060101 C40B040/06; C40B 30/00 20060101 C40B030/00; C12Q 1/686 20060101 C12Q001/686 |
Foreign Application Data
| Date | Code | Application Number |
|---|---|---|
| Dec 4, 2013 | EP | 13195770.6 |
Claims
1-15. (canceled)
16. A kit for determining the integrity of the genome of a sample and/or the quality of a library of DNA sequences obtained by deterministic restriction site whole genome amplification (DRS-WGA) of the genome of the sample, the kit comprising: at least one first primer pair which hybridizes to a DNA sequence of the library having a length from 1000 bp to 5000 bp and encompasses the D5S2117 region of chromosome 5q, at least one second primer pair which hybridizes to a DNA sequence of the library having a length from 1000 bp to 5000 bp and encompasses exons 2 and 3 of the TRP53 gene of the genome, and at least one third primer pair which hybridizes to a DNA sequence of the library having a length from 1000 bp to 5000 bp and encompasses the KRT19psuedo-gene 1 of the genome, wherein the first, second and third primer pairs give rise, when amplified by PCR, to PCR products having different size from one another.
17. The kit of claim 16, wherein when amplified by PCR, the first second, and third primer pairs give rise to a first PCR product from 50 bp to 1000 bp, a second PCR product from 50 bp to 1000 bp having a size other than the first PCR product, and a third PCR product from 50 bp to 1000 bp having a size other than the first and second PCR products.
18. The kit of claim 16, wherein a forward primer of the at least one first primer pair is SEQ ID NO:4 and a reverse primer of the at least one first primer pair is SEQ ID NO:3.
19. The kit of claim 16, wherein a forward primer of the at least one second primer pair is SEQ ID NO:6 and a reverse primer of the at least one second primer pair is SEQ ID NO:5.
20. The kit of claim 16, wherein a forward primer of the at least one third primer pair is SEQ ID NO:8 and a reverse primer of the at least one third primer pair is SEQ ID NO:7.
21. The kit of claim 16, further comprising instructions to amplify the library of DNA sequences by PCR using the at least one first primer pair, the at least one second primer pair, and the at least one third primer pair, and to detect the first, second, and third PCR products, wherein the presence of any one or more of the first, second, and third PCR products corresponds to a level of the integrity of the genome of the sample and/or the quality of the library of DNA sequences.
22. The kit of claim 16, further comprising at least one fourth primer pair which hybridizes to a DNA sequence of the library having a length from 80 bp to 300 bp, wherein, when amplified by PCR, a fourth PCR product results having a size other than the first, second, and third PCR product.
23. The kit of claim 22, wherein the fourth PCR product is from 50 bp to 200 bp.
24. The kit of claim 22, wherein the DNA sequence of the library to which the at least one fourth primer pair hybridises encompasses Codon 12/13 of the KRAS gene.
25. The kit of claim 23, wherein a forward primer of the at least one fourth primer pair is SEQ ID NO:17 and a reverse primer of the at least one first primer pair is SEQ ID NO:18.
26. The kit of claim 22, further comprising instructions to amplify the library of DNA sequences by PCR using the at least one first primer pair, the at least one second primer pair, the at least one third primer pair, and the at lea tone fourth primer pair and to detect the first, second, third, and fourth PCR products, wherein the presence of any one or more of the first, second, third, and fourth PCR products corresponds to a level of the integrity of the genome of the sample and/or the quality of the library of DNA sequences.
27. The kit of claim 16, wherein the sample consists of 5 cells or less.
28. The kit of claim 16, wherein the sample is a single cell.
29. A method of predicting the success of a genetic analysis assay after amplification of single cell genomic DNA by DRS-WGA comprising amplifying a library of DNA sequences by PCR using the kit of claim 16 and detecting the first, second, and third PCR products.
30. The method of claim 29, wherein the genetic analysis assay is selected from the group consisting of Sanger sequencing for mutation analysis, assessment of specific copy number chambers, quantitative PCR for gene amplification, metaphase Comparative Genomic Hybridization (CGH), and array Comparative Genomic Hybridization (CGH).
31. A method of determining the integrity of the genome of a sample, comprising amplifying a library of DNA sequences by PCR using the kit of claim 16 and detecting the first, second, and third PCR products.
32. The method of claim 31, wherein determining the integrity of the genome of a sample allows to determine a biological status of the cell/cells of the sample.
Description
[0001] The present invention relates to a method and a kit for determining the integrity of the genome of a sample, in particular a single cell, and/or the quality of a library of DNA sequences obtained by deterministic restriction site whole genome amplification (DRS-WGA) of the genome of the sample.
STATE OF THE ART
[0002] Whole Genome Amplification (WGA) permits detection of somatic mutations and copy alterations in DNA of limited starting material, such as in the case of single circulating tumour cells (CTC) of cancer patients or in preimplantation diagnostics.
[0003] For the diagnostic use of WGA for single cell analysis, quality of genomic DNA (i.e. genome integrity) of the single cell sample of interest plays a major role for successful molecular analysis after WGA.
[0004] In particular, CTCs have been described as being frequently apoptotic (Mehes, G., et al., Circulating breast cancer cells are frequently apoptotic. Am J Pathol, 2001. 159(1): p. 17-20).
[0005] Moreover, during caspase-mediated apoptosis genomic DNA is fragmented into small pieces of 180 bp to 200 bp length (Wyllie, A H., Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature, 1980. 284(5756): p. 555-6).
[0006] It is thus important to assess the Genome Integrity status of a single cell, as this can be linked to the biological status of the cell itself, and give clinically relevant information on the overall status of a cancer patient, which goes beyond the information provided by just counting the CTCs and complements the molecular characterization of those CTCs.
[0007] Besides, DNA crosslinking and/or fragmentation occur with chemical treatment (e.g. fixation) applied on patient-derived cells and tissues for needed sample conservation after biopsy.
[0008] To predict performance of molecular assays for single cell analysis and evaluation of resulting data derived from such samples, assessing the genomic integrity of single cells is of paramount importance.
[0009] Available single cell WGA kits assess quality of whole genome amplification by measuring the concentration of the WGA product only. As protocols for these methods include at least one random step during the procedure of single cell DNA amplification, specific assays to evaluate genome integrity of the input sample (in general a single cell) such as apoptotic or non-apoptotic status, or the quality of the output of the WGA product, such as the suitability for further genetic analysis, are difficult.
[0010] A specific kind of WGA is deterministic restriction site whole genome amplification (hereinafter referred to as DRS-WGA). DRS-WGA, which is known from EP1109938 and is commercialised as Ampli1.TM. by Silicon Biosystems Spa, is based on specific restriction digestion of double stranded DNA at MseI sites (TTAA) and ligation of a universal adaptor for amplification.
[0011] DRS-WGA has been shown to be better for the amplification of single cells (see for example: Lee Y S, et al: Comparison of whole genome amplification methods for further quantitative analysis with microarray-based comparative genomic hybridization. Taiwan J Obstet Gynecol. 2008, 47(1):32-41) and also more tolerant to DNA degradation due to fixative treatment (see for example: Stoecklein N. H. et al: SCOMP is Superior to Degenerated Oligonucleotide Primed-PCR for Global Amplification of Minute Amounts of DNA from Microdissected Archival Samples. American Journal of Pathology 2002, Vol. 161, No. 1; Arneson N. et al.: Comparison of Whole Genome Amplification methods for analysis of DNA extracted from microdissected early breast lesions in formalin-fixed paraffin-embedded tissue. ISRN Oncol. 2012; 2012;710692).
[0012] To date there are no specific assays to evaluate the genome integrity of an input sample from the DRS-WGA product or the quality of the DRS-WGA product obtained.
[0013] A need is therefore felt to develop methods and kits allowing to determine the genome integrity of an input sample and/or the quality of the DRS-WGA product obtained and permitting to predict performance of molecular assays downstream of DRS-WGA for single cell analysis and evaluation of resulting data.
[0014] An object of the present invention is therefore to provide a method for determining the integrity of the genome of a sample and/or the quality of a library of DNA sequences obtained by DRS-WGA that provides robust and reliable results and allows in particular to assess the biological status of a cell/cells of the sample and/or predict the performance of molecular assays downstream of the DRS-WGA.
[0015] This object is achieved by the present invention as it relates to a method as defined in claim 1.
[0016] It is a further object of the present invention to provide a kit as defined in claim 8.
Definitions
[0017] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention pertains.
[0018] By the term "sample", there is intended a sample comprising at least one particle of a biological entity, said particle comprising at least a DNA sequence representing the genome or a substantial subset of the genome of that biological entity. By way of non-limiting example, said entity may be a human, said at least one particle may be a set of 5 cells or less, a single cell, or a single-cell nucleus, or a haploid germ cell, or a chromosome.
[0019] By the term "genome" there is intended the entire genome or said substantial subset of the genome.
[0020] By the term "integrity" of the genome there is intended the absence of DNA damages such as double strand breaks, or nicks or similar conditions which may hamper the replication of the genome or its normal functionality.
[0021] By the term "quality" of a library of DNA sequences there is intended the suitability of the library of DNA sequences to be used for the genetic characterization of certain features such as, by way of non-limiting example, the presence of point mutations, deletions, insertions, copy number variations.
BRIEF DESCRIPTION OF THE DRAWINGS
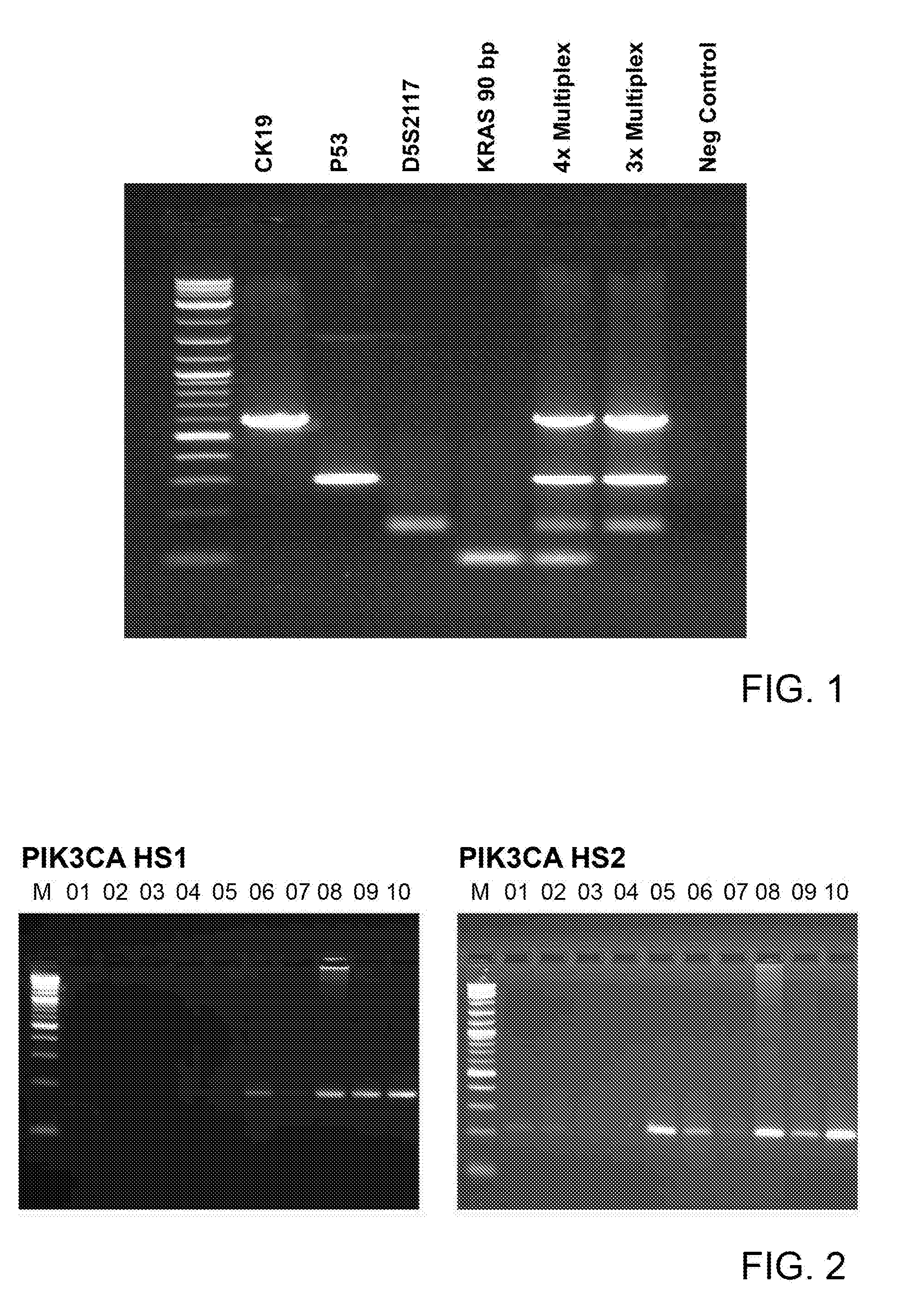
[0022] FIG. 1 shows an agarose gel picture of single marker PCRs, and of a 4-multiplex assay and a 3-multiplex assay according to preferred embodiments of the present invention;
[0023] FIG. 2 shows an agarose gel picture of PCRs on PIK3CA hotspot 1 and hotspot2 (M=size marker, 0132 breast13, 02=breast15, 03=breast17, 04=breast20, 05=prostate14, 06=prostate16, 07=prostate24, 0832 melanoma13, 09=melanoma14, 10=melanoma16);
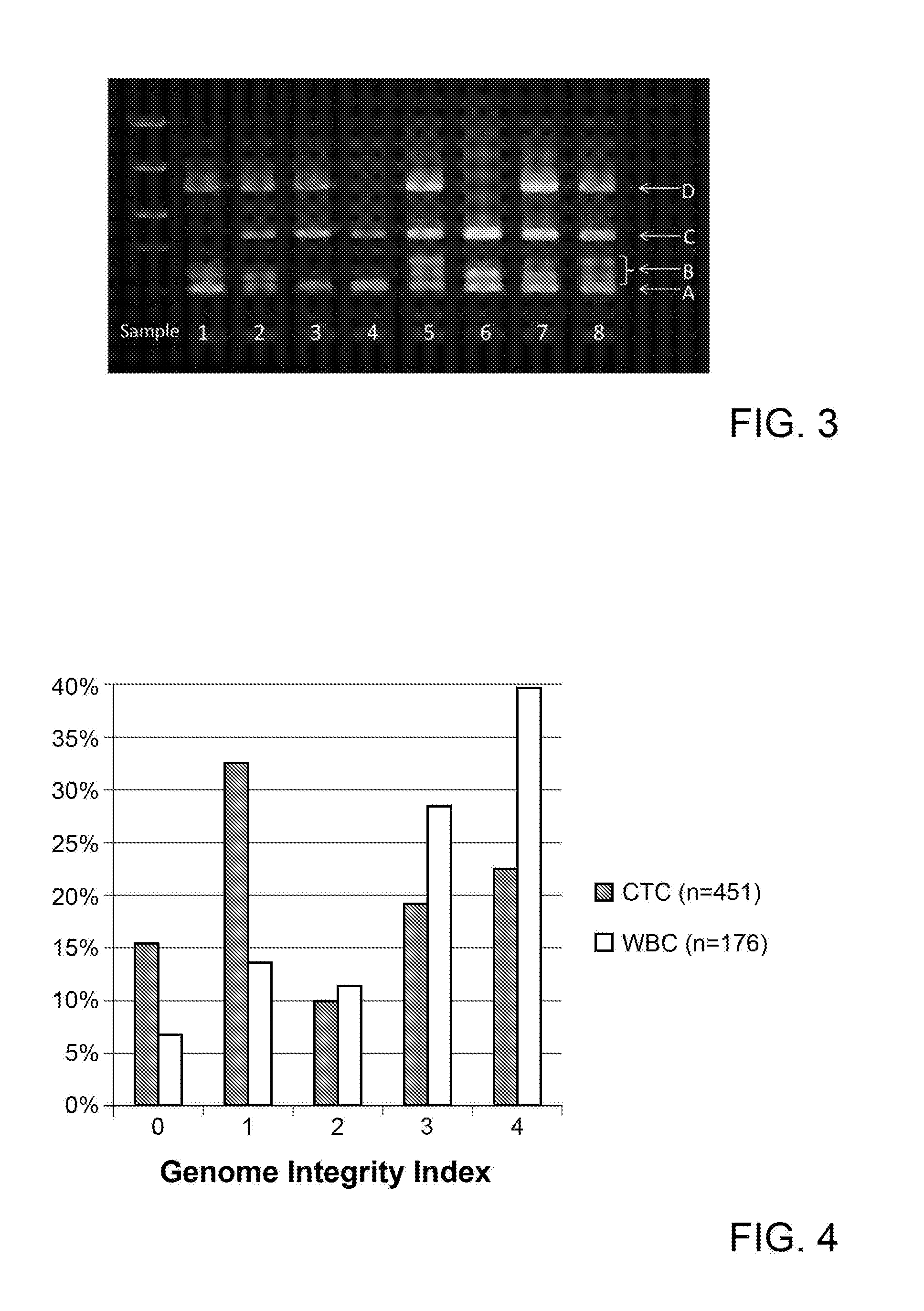
[0024] FIG. 3 shows an agarose gel picture of 8 samples tested by the preferred 4-multiplex assay of FIG. 1.
[0025] FIG. 4 shows a histogram of the distribution of the Genome Integrity Index among single Circulating Tumour Cells (CTCs) and single leukocytes (WBCs) from breast cancer patients.
DETAILED DESCRIPTION OF THE INVENTION
[0026] The method according to the present invention for determining the integrity of the genome of a sample and/or the quality of a library of DNA sequences obtained by deterministic restriction site whole genome amplification (DRS-WGA) of the genome of the sample comprises steps (a) to (d).
[0027] By allowing the determination of the integrity of the genome of a sample and/or the quality of a library of sequences obtained by DRS-WGA of the genome of the sample, the method also allows to assess the biological status of of a cell/cells of the sample and/or predict the success rate of genetic analysis assays on the library of DNA sequences.
[0028] The sample preferably consists of 5 cells or less, more preferably the sample is a single cell. The single cell is preferably a circulating tumour cell (CTC), a circulating fetal cell, a circulating endothelial cell (CEC), an oocyte, a cumulus cell, a sperm, a blastomere, or a trophectoderm cell.
[0029] In step (a), the library of DNA sequences is provided.
[0030] In step (b), the library of DNA sequences is amplified by PCR using at least one first primer pair which hybridises to a DNA sequence of the library having a length from 1000 bp to 5000 bp, preferably from 1000 bp to 2000 bp, and corresponding to a sequence of the genome located on a first chromosome arm, the step of amplifying giving rise to a first PCR product from 50 bp to 1000 bp.
[0031] Preferably, the DNA sequence of the library to which the first primer pair hybridises encompasses the D5S2117 region of chromosome 5q.
[0032] More preferably, the forward primer of the first primer pair is SEQ ID NO:4 and the reverse primer of the first primer pair is SEQ ID NO:3.
[0033] In step (c) the first PCR product is detected. Agarose gel electrophoresis may be used to separate and detect the PCR product as well as other methods known in the art.
[0034] In step (d) the presence of the first PCR product is correlated with the integrity of the genome of the sample and/or the quality of the library of DNA sequences.
[0035] Advantageously, in step (b) at least one second primer pair is used which hybridises to a DNA sequence of the library having a length from 1000 bp to 5000 bp, more preferably from 1000 bp to 2000 bp, and corresponding to a sequence of the genome located on a second chromosome arm other than the first chromosome arm, the step of amplifying giving rise to a second PCR product from 50 bp to 1000 bp having a size other than the first PCR product.
[0036] The second PCR product is also detected in step (c) and the presence of the second PCR product is also correlated with the integrity of the genome of the sample and/or the quality of the library of DNA sequences in step (d).
[0037] Preferably, the DNA sequence of the library to which the second primer pair hybridises encompasses exons 2 and 3 of the TRP53 gene.
[0038] More preferably, the forward primer of the second primer pair is SEQ ID NO:6 and the reverse primer of second primer pair is SEQ ID NO:5.
[0039] Advantageously, in step (b) at least one third primer pair is used which hybridises to a DNA sequence of the library having a length from 1000 bp to 5000 bp, more preferably from 1000 bp to 2000 bp, and corresponding to a sequence of the genome located on a third chromosome arm other than the first and second chromosome arms, the step of amplifying giving rise to a third PCR product from 50 bp to 1000 bp having a size other than the first and second PCR products.
[0040] The third PCR product is also detected in step (c) and the presence of the third PCR product is also correlated with the integrity of the genome of the sample and/or the quality of the library of DNA sequences in step (d).
[0041] Preferably, the DNA sequence of the library to which the third primer pair hybridises encompasses the KRT19 pseudo-gene 1 (indicated for short as CK19).
[0042] More preferably, the forward primer of the third primer pair is SEQ ID NO:8 and the reverse primer of the third primer pair is SEQ ID NO:7.
[0043] Even more preferably, in step (b) at least one fourth primer pair is used which hybridises to a DNA sequence of the library having a length from 80 bp to 300 bp, the step of amplifying giving rise to a fourth PCR product from 50 bp to 200 bp having a size other than the first, the second and the third PCR products.
[0044] When step (b) uses at least one fourth primer pair, the fourth PCR product is also detected in step (c) and the presence of the fourth PCR product is also correlated with the integrity of the genome of the sample and/or the quality of the library of DNA sequences in step (d).
[0045] Preferably, the DNA sequence of the library to which the fourth primer pair hybridises encompasses codons 12 and 13 of the KRAS gene.
[0046] Even more preferably, the forward primer of the fourth primer pair is SEQ ID NO:17 and the reverse primer of the fourth primer pair is SEQ ID NO:18.
[0047] According to the present invention there is also provided a kit for determining the integrity of the genome of a sample and/or the quality of a library of DNA sequences obtained by deterministic restriction site whole genome amplification (DRS-WGA) of the genome of the sample comprising: [0048] at least one first primer pair which hybridises to a DNA sequence of the library having a length from 1000 bp to 5000 bp and corresponding to a sequence of the genome located on a first chromosome arm, [0049] at least one second primer pair which hybridises to a DNA sequence of the library having a length from 1000 bp to 5000 bp and corresponding to a sequence of the genome located on a second chromosome arm other than the first chromosome arm, [0050] at least one third primer pair which hybridises to a DNA sequence of the library having a length from 1000 bp to 5000 bp and corresponding to a sequence of the genome located on a third chromosome arm other than the first and second chromosome arms, wherein the first, second and third primer pairs give rise, when amplified by PCR, to PCR products having different size from one another.
[0051] Preferably, the kit further comprises at least one fourth primer pair which hybridises to a DNA sequence of the library having a length from 80 bp to 300 bp, wherein the first, second, third and fourth primer pairs give rise, when amplified by PCR, to PCR products having different size from one another.
[0052] More preferably, the kit comprises the first, second, third and fourth primer pairs.
[0053] The kit may be used for predicting the success of a genetic analysis assay after amplification of single cell genomic DNA by DRS-WGA. Preferably the genetic analysis assay is Sanger sequencing for mutation analysis, assessment of specific copy number changes, quantitative PCR for gene amplification, metaphase Comparative Genomic Hybridisation (CGH), or array Comparative Genomic Hybridisation (CGH).
[0054] For gene specific assays, such as Sanger sequencing for mutation analysis, samples with at least 1-2 out of 4 PCR products can be used. Positivity to at least 3 out of 4 PCR products is predictive of successful genome-wide analysis with metaphase CGH. For array CGH it is advisable to use samples with 4 out of 4 positive PCR products.
[0055] The kit may also be used to determine the integrity of the genome of a sample and thus determine the biological status of the cell/cells of the sample. The sample preferably consists of 5 cells or less, more preferably the sample is a single cell.
[0056] As a matter of fact, if there is a large number of cells in the sample, there will be, overall, enough template copies for the first, second, third (and optionally fourth) primer pairs to amplify respectively the first, second, third (and optionally fourth) PCR products even if the DNA of the cells of the sample is highly fragmented. The result is the loss of the discrimination power of the method.
[0057] On the other hand, it has been experimentally determined that, even in the case of very damaged DNA cell samples, 5 cells is an amount that allows reliable results with the method, i.e allows to obtain the PCR products of step (d) without them being a results of excess template copies.
[0058] Further, for the analysis of very damaged DNA cell samples (for which single cells could result in PCR products not being amplified), the use of a number of cells greater than one, i.e. two to five, allows to quantify genome integrity thus increasing the power of resolution of the method.
EXAMPLES
[0059] In brief, several primer pairs designed on MseI fragments located on different chromosomal locations and with varying fragment lengths were tested. Three primer pairs that predict successful whole genome analysis of single cell products with high specificity and sensitivity were selected (Example 1). A fourth primer pair of a shorter MseI fragment was added to indicate successful DRS-WGA of low quality cells, e.g. apoptotic CTCs (Example 2). In the examples, PCR products are also referred to as "markers" and PCP amplification reactions including several primer pairs are also referred to as "multiplex assays". In particular, PCR amplification reactions including several primer pairs designed to determine the integrity of the genome of a sample and/or the quality of the library of DNA sequences obtained by deterministic restriction site whole genome amplification (DRS-WGA) of the genome of the sample are also referred to as "quality control assays" or "QC assays".
Example 1
[0060] Two alternative marker combinations were shown to predict the success of metaphase comparative genomic hybridization (CGH) after amplification of single cell genomic DNA with DRS-WGA (Resolution 10-20 Mb) on both cancer cell samples and samples of diploid cells with normal karyotype.
A. Characteristics of the 8 Tested PCR Markers
[0061] PCRs on 8 different MseI-fragments covering an MseI-fragment length from 239-1936 bp were tested (Table 1). Sequences located on 7 different chromosome arms were selected to minimize the chance of a negative assay result because of genomic DNA loss in a single cancer cell.
TABLE-US-00001 Pipetting 1.0 .mu.l Buffer + dNTPs scheme (1x) (10 mM MgCl, 100 mM Tris (pH 8.5), 500 mM KCl, 1 mM dNTPs) 0.5 .mu.l Primer 3' (8 .mu.M) 0.5 .mu.l Primer 5' (8 .mu.M) 0.25 .mu.l BSA (for molecular biology) 7.25 .mu.l PCR-H.sub.2O 0.1 .mu.l Taq polymerase (5 U/.mu.l) 0.5 .mu.l Ampli1 product (test sample)
TABLE-US-00002 Thermal profile Step 1 94.0.degree. C. 2 min Step 2 Annealing Temp 30 s Step 3 72.0.degree. C. 2 min Step 4 94.0.degree. C. 15 s Step 5 Annealing Temp 30 s Step 6 72.0.degree. C. 20 s 14 additional cycles (steps 4-6) Step 7 94.0.degree. C. 15 s Step 8 Annealing Temp 30 s Step 9 72.0.degree. C. 30 s 24 additional cycles (steps 7-9) Step 10 72.0.degree. C. 2 min Step 11 4.degree. C. forever
TABLE-US-00003 TABLE 1 Features of the 8 selected PCR primer pairs tested for the QC assay to assess DRS-WGA quality Annealing Mse-fragment PCR-fragment Primer name SEQ ID NO Sequence temperature Chromosome length length BCR-TT-R 1 TCAGCCTCAGGACTCTTGTG 61.degree. C. 22q 1936 bp 323 bp BCR-TT-F 2 CGTGGACAACTACGGAGTTG 61.degree. C. 22q 1936 bp 323 bp D5S2117-R 3 ACTGAGTCCTCCAACCATGG 58.degree. C. 5q 1376 bp 140 bp* D5S2117-F 4 CCAGGTGAGAACCTAGTCAG 58.degree. C. 5q 1376 bp 140 bp* TRP53- 5 CAGCCCAACCCTTGTCCTTA 58.degree. C. 17p 1374 bp 299 bp Ex2/3-R TRP53- 6 GAAGCGTCTCATGCTGGATC 58.degree. C. 17p 1374 bp 299 bp Ex2/3-F CK19-R 7 TTCATGCTCAGCTGTGACTG 58.degree. C. 6q 1146 bp 614 bp CK19-F 8 GAAGATCCGCGACTGGTAC 58.degree. C. 6q 1146 bp 614 bp IGF2R-R 9 GGATCTTGGTACCACTCATG 58.degree. C. 6q 647 bp 217 bp IGF2R-F 10 GCCACTGTCGAAGTCTGCA 58.degree. C. 6q 647 bp 217 bp RUFY2-R 11 CAGCTAGGAACTCCAGGAAT 64.degree. C. 10q 458 bp 104 bp CA RUFY2-F 12 GTTGAGGGCTTCATCAACAC 64.degree. C. 10q 458 bp 104 bp CCA SMYD1-R 13 CTTTTCCCTGAAGGTCTTAG 55.degree. C. 2p 287 bp 163 bp SMYD1-F 14 GGGTGACCTGCTTGACATC 55.degree. C. 2p 287 bp 163 bp PHACTR2-R 15 TGTGAGAAAGACTTGGAGTT 58.degree. C. 6q 239 bp 205 bp PHACTR2-F 16 ACTGAACAGAGCAGGTCTAC 58.degree. C. 6q 239 bp 205 bp *This primer pair amplifies a microsatellite sequence. Therefore the actual fragment length can vary slightly between alleles of one patient and between different patients.
B. Selection of PCR Markers for the QC Assay
[0062] To select the best possible combination of PCR markers, 72 single cell genomes from different types of human cancers (24 breast disseminated cancer cells (DCCs) 24 prostate DCCs, 24 melanoma DCCs) were re-amplified. For each of the three groups, 12 cells were included which resulted in successful metaphase hybridization in a previous CGH experiment and 12 cells which resulted in a failed metaphase hybridization in a previous CGH experiment. Specific PCRs for all selected markers were performed and the assay was evaluated for accuracy in predicting the outcome of metaphase CGH.
[0063] As single markers, PCRs on the long Mse-fragments D5S2117, TRP53-Ex2/3 and KRT19 pseudogene 1 (hereinafter also referred to as CK19) provided best separation between amplified genomes with successful and failed metaphase CGH. An assay of these three fragments showed high assay accuracy in predicting the success of metaphase CGH. The addition of the PCR on the shortest Mse-fragment PHACTR2 slightly increased the assay accuracy in the collective of 72 DCC genomes.
[0064] In summary, two alternative QC assays for the QC kit for Ampli1 were developed (Table 2).
[0065] Alternative 1: QC assay with 3 markers (D5S2117, TRP53-Ex2/3 and CK19).
[0066] Alternative 2: QC assay with 4 markers (D5S2117, TRP53-Ex2/3, CK19 and PHACTR2).
TABLE-US-00004 TABLE 2 Assay accuracy of two alternative marker combinations for the QC assay (72 DCC samples) Statistical Alternative 1 Alternative 2 measurement 2/3 PCRs+ 3/3 PCRs+ 3/4 PCRs+ 4/4 PCRs+ True+ 35 29 35 29 False- 1 7 1 7 True- 34 36 35 36 False+ 2 0 1 0 Sensitivity 0.97 0.81 0.97 0.81 Specificity 0.94 1.0 0.97 1.0 Positive 0.95 1.0 0.97 1.0 predictive value Negative 0.97 0.84 0.97 0.84 predictive value
[0067] If possible, only samples that are of the highest quality and that are positive for all selected markers (for both alternative assays 100% specificity) should be used. However, the trade-off for applying this high standard of quality control is a high rate of false negatives (7/36=19.4%). If the number of test samples with the highest quality standard is limited, DRS-WGA amplified genomes with 2/3 or 3/4 positive PCRs still predict a high success rate for metaphase CGH (specificity 0.94 for assay with 3 markers and 0.97 for assay with 4 markers, respectively).
C. Testing of PCR Markers on a set of 100 Diploid Cells with Normal Karyotype
[0068] To further test the set of PCR markers for the QC assay, 100 single cell genomes from diploid cells with normal karyotypes were re-amplified. All samples were tested for the 8 different PCR markers. Additionally, metaphase CGH success was checked for 22 genomes with predicted good quality and 10 genomes with predicted bad quality. The results of the statistical evaluation of assay accuracy are shown in Table 3.
TABLE-US-00005 TABLE 3 Assay accuracy of two alternative marker combinations for the Ampli1 QC kit (32 normal samples) Statistical Alternative 1 Alternative 2 measurement 2/3 PCRs+ 3/3 PCRs+ 3/4 PCRs+ 4/4 PCRs+ True+ 22 21 22 21 False- 0 1 0 1 True- 10 10 10 10 False+ 0 0 0 0 Sensitivity 1.0 0.95 1.0 0.95 Specificity 1.0 1.0 1.0 1.0 Positive 1.0 1.0 1.0 1.0 predictive value Negative 1.0 0.91 1.0 0.91 predictive value
D. Further Validation of Selected QC Markers on DRS-WGA Amplified Samples
[0069] As the tested samples in B and C were taken from an existing biobank of single cell genomes, they were amplified with DRS-WGA customized reagents of the related labs (provided by different suppliers). To validate the performance of the proposed QC assay with samples amplified by Ampli1 kit (as provided by Silicon Biosystems), single mononuclear PBLs and cell pools of a healthy donor and single cells and cell pools of the breast cancer cell line SKBR3 were isolated. The markers proposed in B and C for the QC assay were used to predict quality of the amplified genomes. Then, metaphase CGH experiments for a set of samples (5 single cells, 1 cell pool and 1 Ampli1 negative control) for diploid blood cells as well as SKBR3 cells were performed to validate the predictive accuracy of the QC assay.
SKBR: 10/11 single cells and both cell pools showed 4/4 positive bands marker PCRs [0070] 1/11 cells was negative for all tested markers negative control clean in all tested PCRs PBL: 11/11 single cells and both cell pools showed 4/4 positive bands marker PCRs [0071] negative control clean in all tested PCRs
Example 2
[0072] As other downstream analyses, e.g. Sanger sequencing for specific fragments, are not dependent on a quantitatively and qualitatively very high amplification of the single cell DNA, a fourth fragment was included. Experiments were carried out to test whether a successful amplification of this sequence only is enough to predict success of Sanger sequencing of comparable Mse fragments (here PIK3CA hotspots 1 and 2).
A. Protocol for 4-Multiplex Assay
[0073] In order to have compatibility with the already established 3-multiplex assay, new primers were designed for the KRAS Mse-fragment encompassing the frequently mutated nucleotides encoding for codons 12 and 13. These primers amplify a PCR fragment of 91 bp length clearly distinguishable from the other three bands (D5S2117, CK19 and TP53-Exon2/3, see FIG. 1).
[0074] The following pipetting scheme and thermal profile were used adding the following KRAS primers in a concentration of 4 .mu.M to the primer mix.
TABLE-US-00006 KRAS91bp-F (SEQ ID NO: 17) ATAAGGCCTGCTGAAAATGAC KRAS91bp-R (SEQ ID NO: 18) CTGAATTAGCTGTATCGTCAAGG
TABLE-US-00007 Pipetting 1.0 .mu.l Ampli1 .TM. PCR Reaction Buffer scheme (1x) (20 mM MgCl.sub.2 included) 0.2 .mu.l dNTPs (10 mM) 1.0 .mu.l Primer mix (8 primers, each 4 .mu.M) 0.2 .mu.l BSA (20 mg/ml) 6.5 .mu.l PCR-H.sub.2O 0.1 .mu.l Ampli1 .TM. Taq polymerase (5 U/.mu.l) 1.0 .mu.l Ampli1 .TM. product (test sample)
TABLE-US-00008 Thermal profile Step 1 95.0.degree. C. 4 min Step 2 95.0.degree. C. 30 s Step 3 58.0.degree. C. 30 s Step 4 72.0.degree. C. 90 s 32 cycles (steps 2-4) Step 5 72.0.degree. C. 7 min Step 6 4.0.degree. C. forever
[0075] 5 .mu.l of each PCR product were loaded on a 1.2% agarose gel. The results were checked by comparing the obtained amplicons bp length with the expected ones, as shown in table 4.
TABLE-US-00009 TABLE 4 PCR product identification Target Primer marker Chromosome Amplicon length (bp) A KRAS 12p 91 B D5S2117 5q 108-166 C TRP53 17p 299 D KRT19 pseudo- 6q 614 gene 1 (CK19)
[0076] As already noted above, marker B maps on a polymorphic region, therefore the PCR products may be:
[0077] homozygous, and show one band of a bp comprised in the described range (108-166);
[0078] heterozygous and show two bands of different bp comprised in the described range (108-166).
[0079] FIG. 3 shows an example of an agarose gel electrophoresis of PCR products obtained from different samples by using the above disclosed 4 multiplex assay.
[0080] Sample 1 in FIG. 3 for example displays 3/4 positive markers. Marker B heterozygosity or homozigosity (one or two bands) must be counted as one in the evaluation of the positivity for marker B. Samples 2 and 8 for example display 4/4 positive markers.
B. Comparison of 4-Multiplex Assay with Results of Single Fragment PCR Reactions and Established 3-Multiplex
[0081] 72 DRS-WGA libraries of breast, prostate and melanoma DCC that had already been tested by the 3-multiplex assay of Example 1 were selected. Unfortunately the sample Breast was lost due to a fissure in the reaction tube. All other 71 DRS-WGA products were additionally tested with KRAS 91 bp single PCR and the new 4-multiplex assay. In summary, 53/71 samples showed the expected band at 91 bp in single marker PCR, and all of them but one (Breast 24) showed the same band in the 4-multiplex assay. All the other bands for D5S2117, CK19 and TP53-Exon2/3 were detected in the same samples than with 3-multiplex assay. Moreover, the KRAS 91 bp amplicon was detected in all 38 samples that showed one or more bands in the currently used 3-multiplex. Additionally, 15 samples were identified that were negative for D5S2117, CK19 and TP53-Exon2/3 but positive for KRAS91 bp, only. Finally 18 samples were negative for all four tested amplicons.
C. PIK3CA Sequencing of Ten Selected Samples
[0082] Out of the 15 samples which showed the KRAS 91 bp amplicon only, ten samples were selected for PIK3CA sequence analysis (4 breast DCC samples, 3 prostate DCC samples, and 3 melanoma DCC samples). PIK3CA PCRs were performed for HS1 and HS2 fragments, using Ampli1.TM. PIK3CA Seq Kit (Silicon Biosystems SpA, Italy), according to the manufacturer instructions. After PCR all samples were loaded on a 1.5% agarose gel and amplicons visualized after electrophoresis. Strong bands were obtained only for HS1 in four DCC samples (prostate DCC 16, melanoma DCCs 13, 14, and 16) and for HS2 in five DCC samples (prostate DCCs 14, and 16, melanoma DCCs 13, 14, and 16). Additionally, weak but visible bands were detected for an additional two (breast DCC 13, prostate DCC 24) and three samples (breast DCCs 13, and 15, prostate DCC 24), respectively (FIG. 2). One sample (melanoma DCC 16) showed a strong smear for DNA fragment .gtoreq.1 kb, which was also visible in all previous gels of this sample. Nevertheless, the PCR amplicon was purified for all samples and sequenced.
[0083] Sanger sequencing for PIK3CA mutational hotspots resulted in very strong and clean sequences for the samples showing strong amplification in gel electrophoresis (see FIG. 2), including sample melanoma DCC 16. Most of the other samples with lower amplicon concentration showed high background noise, although sequences could easily be edited for two additional samples for HS1 (breast DCC 13, prostate DCC 24) and four additional samples for HS2 (breast DCCs 13, 15, and 20, prostate DCC 24). For the remaining samples background signal was too high for a secure editing of the complete sequence (breast DCCs 15, 17, and 20, prostate DCC 14 for HS1; breast DCC 15 for HS2).
[0084] On a collective of CTCs and White Blood Cells (WBCs) harvested with DEPArray.TM. (Silicon Biosystems SpA) from breast cancer patients peripheral blood enriched with CellSearch.RTM. (Jannsen Diagnostics LLC), the Genome Integrity was assessed by analysing the quality of Ampli1.TM. WGA product according to the invention. The resulting number of PCR products detected following the multiplex PCR reaction (Genome Integrity Index or GII) was found to be significantly skewed, as shown in FIG. 4, toward lower values of GII in CTCs with respect to normal WBCs collected from the same patients, undergoing the same process of enrichment sorting and Ampli1.TM. WGA.
[0085] By way of explanation, a GII with a value of 0 corresponds to a situation in which none of the first, second, third or fourth PCR products is amplified, a GII with a value of 1 corresponds to a situation in which only the fourth PCR product is amplified, a GII with a value of 2 corresponds to a situation in which one among the first, second and third PCR products and the fourth PCR product are amplified, a GII with a value of 3 corresponds to a situation in which two among the first, second and third PCR products and the fourth PCR product are amplified, and a GII with a value of 4 corresponds to a situation in which the first, second, third and fourth PCR products are amplified.
[0086] The GII was further used to assess the success rate of targeted Sanger sequencing for PIK3CA exon 9 and exon 20 mutation hotspots, of a custom qPCR assay to determine the amplification of ERBB2 gene, and of array CGH. Table 5 shows a synopsis of the results.
TABLE-US-00010 TABLE 5 P Value Molecular Analyzed Genome Integrity Index (GII) Chi- assay cells GII 0 GII 1 GII 2 GII 3 GII 4 square PIK3CA HS1 n = 383 7/23 14/25 48/62 102/117 146/156 <0.0001 (30.4%) (56.0%) (77.4%) (87.2%) (93.6%) PIK3CA HS2 n = 383 8/23 18/25 55/62 109/117 149/156 <0.0001 (34.8%) (72.0%) (88.7%) (93.2%) (95.5%) PIK3CA n = 383 4/23 12/25 45/62 97/117 141/156 <0.0001 complete (17.4%) (48.0%) (72.6%) (82.9%) (90.4%) HER2 qPCR n = 351 3/12 8/18 41/61 95/112 136/148 <0.0001 (25.0%) (50.0%) (67.2%) (84.8%) (91.9%) aCGH n = 50 Not Not 4/5 7/9 36/36 0.016 assessed assessed (80.0%) (77.8%) (100%)
[0087] The above examples show that the method according to the present invention allows to determine the integrity of the genome of a sample and the quality of a library of DNA sequences obtained by DRS-WGA of the genome of the sample with robust and reliable results and allows in particular to predict the performance of molecular assays, such as metaphase and array comparative genomic hybridization (CGH), Sanger sequencing and qPCR, downstream of the DRS-WGA.
[0088] From an analysis of the features of the method and kit of the present invention, the resulting advantages are apparent.
[0089] In particular, in virtue of the fact that DRS-WGA is based on specific restriction digestion of double stranded DNA at MseI site (TTAA) and ligation of one universal adaptor for amplification, in principle all fragments generated during amplification that represent the WGA library are known. Thus, by the identification of specific MseI fragments having a length of >1000 bp in a single cell WGA library, the genomic integrity of the DNA of an isolated cell can be measured, and thus the quality of the sample for different kinds of successful molecular analysis determined.
[0090] Since the first, second and third primer pairs have been specifically selected in target regions of the genome corresponding to long amplicons of the DRS-WGA DNA digestion enzyme, MseI, which are more difficult to amplify in case of DNA fragmentation, their amplification is indicative of good overall success of the DRS-WGA on a given sample.
[0091] Further, in virtue of the fact that the first, second and third primer pairs are designed on different chromosome arms, the method allows to assess the integrity of the genome of a sample and/or the quality of the library of DNA sequences obtained over the broadest region of the genome.
[0092] Further, in virtue of the fact that the first, second, third and fourth PCR products have different size, it is possible to use the first, second, third and fourth primer pairs together in a multiplex reaction.
[0093] Moreover, in virtue of the fact that the fourth primer pair hybridises to a DNA sequence of the library having a length from 80 bp to 300 bp, it is possible to predict the performance of molecular assays downstream of the DRS-WGA also for low quality cells, for example apoptotic CTCs. In fact there is a significant difference in performance between the success rate in e.g. targeted sanger sequencing of the PIK3CA exon 9 (30%) and exon 20 (34%) or qPCR for Her2 CNV analysis (25%) of cells for which not even this fourth PCR product is amplified and those cells for which at least this is amplifiable, for which the success rate roughly doubles (56%, 72%, 50% respectively). The two populations would otherwise be indistinguishable when using only the 3 primer pairs hybridizing to long amplicons of the DRS-WGA.
[0094] Finally, it is clear that modifications and variants to the method and kit disclosed and shown may be made without because of this departing from the scope of protection of the appended claims.
[0095] In particular, the method may be multiplexed by using further pairs of primers which do not interfere with the PCR amplification with the first, second, third and possibly fourth primer.
Sequence CWU 1
1
18120DNAHomo sapienssource(1)..(20)/mol_type="DNA" /note="BCR-TT-3"
/organism="Homo sapiens" 1tcagcctcag gactcttgtg 20220DNAHomo
sapienssource(1)..(20)/mol_type="DNA" /note="BCR-TT-5"
/organism="Homo sapiens" 2cgtggacaac tacggagttg 20320DNAHomo
sapienssource(1)..(20)/mol_type="DNA" /note="D5S2117-3"
/organism="Homo sapiens" 3actgagtcct ccaaccatgg 20420DNAHomo
sapienssource(1)..(20)/mol_type="DNA" /note="D5S2117-5"
/organism="Homo sapiens" 4ccaggtgaga acctagtcag 20520DNAHomo
sapienssource(1)..(20)/mol_type="DNA" /note="TRP53-Ex2/3-3"
/organism="Homo sapiens" 5cagcccaacc cttgtcctta 20620DNAHomo
sapienssource(1)..(20)/mol_type="DNA" /note="TRP53-Ex2/3-5"
/organism="Homo sapiens" 6gaagcgtctc atgctggatc 20720DNAHomo
sapienssource(1)..(20)/mol_type="DNA" /note="CK12-3"
/organism="Homo sapiens" 7ttcatgctca gctgtgactg 20819DNAHomo
sapienssource(1)..(19)/mol_type="DNA" /note="CK12-5"
/organism="Homo sapiens" 8gaagatccgc gactggtac 19920DNAHomo
sapienssource(1)..(20)/mol_type="DNA" /note="IGF2R-3"
/organism="Homo sapiens" 9ggatcttggt accactcatg 201019DNAHomo
sapienssource(1)..(19)/mol_type="DNA" /note="IGF2R-5"
/organism="Homo sapiens" 10gccactgtcg aagtctgca 191122DNAHomo
sapienssource(1)..(22)/mol_type="DNA" /note="RUFY2-3"
/organism="Homo sapiens" 11cagctaggaa ctccaggaat ca 221223DNAHomo
sapienssource(1)..(23)/mol_type="DNA" /note="RUFY2-5"
/organism="Homo sapiens" 12gttgagggct tcatcaacac cca 231320DNAHomo
sapienssource(1)..(20)/mol_type="DNA" /note="SMYD1-3"
/organism="Homo sapiens" 13cttttccctg aaggtcttag 201419DNAHomo
sapienssource(1)..(19)/mol_type="DNA" /note="SMYD1-5"
/organism="Homo sapiens" 14gggtgacctg cttgacatc 191520DNAHomo
sapienssource(1)..(20)/mol_type="DNA" /note="PHACTR2-3"
/organism="Homo sapiens" 15tgtgagaaag acttggagtt 201620DNAHomo
sapienssource(1)..(20)/mol_type="DNA" /note="PHACTR2-5"
/organism="Homo sapiens" 16actgaacaga gcaggtctac 201721DNAHomo
sapienssource(1)..(21)/mol_type="DNA" /note="KRAS 91bp 5 "
/organism="Homo sapiens" 17ataaggcctg ctgaaaatga c 211823DNAHomo
sapienssource(1)..(23)/mol_type="DNA" /note="KRAS 91bp 3 "
/organism="Homo sapiens" 18ctgaattagc tgtatcgtca agg 23
D00000
D00001
D00002
S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.