Transposon Nucleic Acids Comprising A Calibration Sequence For Dna Sequencing
KAVANAGH; Ian ; et al.
U.S. patent application number 16/386746 was filed with the patent office on 2019-10-03 for transposon nucleic acids comprising a calibration sequence for dna sequencing. The applicant listed for this patent is THERMO FISHER SCIENTIFIC BALTICS UAB. Invention is credited to Heli HAAKANA, Ian KAVANAGH, Laura-Leena KIISKINEN.
| Application Number | 20190300937 16/386746 |
| Document ID | / |
| Family ID | 47556174 |
| Filed Date | 2019-10-03 |


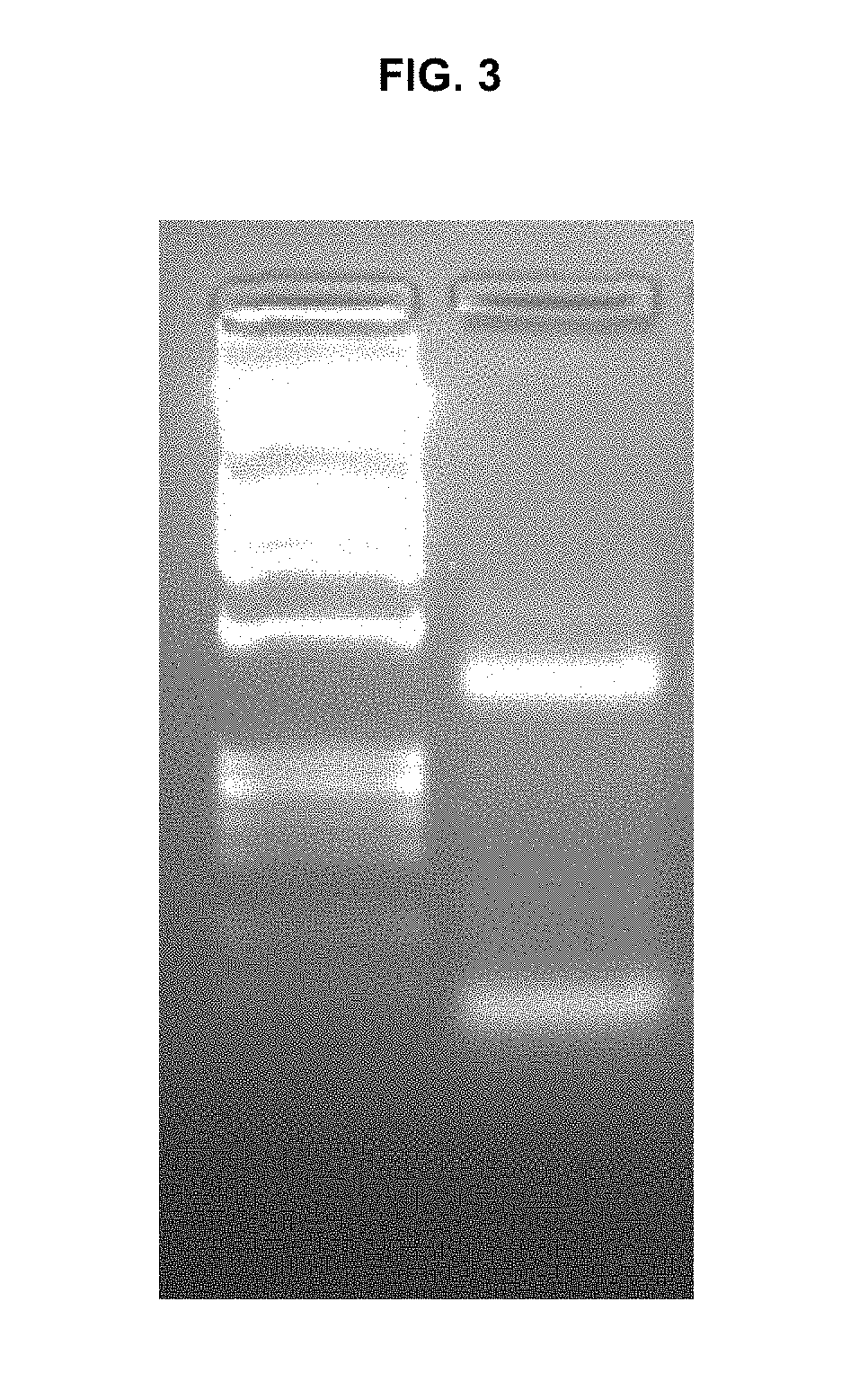





| United States Patent Application | 20190300937 |
| Kind Code | A1 |
| KAVANAGH; Ian ; et al. | October 3, 2019 |
TRANSPOSON NUCLEIC ACIDS COMPRISING A CALIBRATION SEQUENCE FOR DNA SEQUENCING
Abstract
Transposon nucleic acids comprising a transposon end sequence and a calibration sequence for DNA sequencing in the transposon end sequence. In one embodiment, the transposon end sequence is a Mu transposon end. A method for the generation of DNA fragmentation library based on a transposition reaction in the presence of a transposon end with the calibration sequence providing facilitated downstream handling of the produced DNA fragments, e.g., in the generation of sequencing templates.
| Inventors: | KAVANAGH; Ian; (Luzern, CH) ; KIISKINEN; Laura-Leena; (Espoo, FI) ; HAAKANA; Heli; (Espoo, FI) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 47556174 | ||||||||||
| Appl. No.: | 16/386746 | ||||||||||
| Filed: | April 17, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 15791740 | Oct 24, 2017 | 10308978 | ||
| 16386746 | ||||
| 14836248 | Aug 26, 2015 | 9834811 | ||
| 15791740 | ||||
| 13553395 | Jul 19, 2012 | 9145623 | ||
| 14836248 | ||||
| 61509691 | Jul 20, 2011 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | C40B 50/06 20130101; C12N 15/1093 20130101; C12Q 1/6806 20130101; C12Q 2525/131 20130101; C12Q 1/6806 20130101; C40B 40/08 20130101; C12Q 2525/155 20130101 |
| International Class: | C12Q 1/6806 20060101 C12Q001/6806; C40B 50/06 20060101 C40B050/06; C12N 15/10 20060101 C12N015/10; C40B 40/08 20060101 C40B040/08 |
Claims
1. A nucleic acid comprising a modified transposon end sequence, wherein (a) the modified transposon end sequence comprises (i) a first strand having the nucleotide sequence of SEQ ID NO: 1 that is modified to contain a calibration sequence that is four nucleotides in length and (ii) a second strand that is complementary to the first strand, and (b) the calibration sequence contains four different nucleotide bases in any order and is located in the 3' end region of sequence of SEQ ID NO: 1 3' of position 17 of SEQ ID NO: 1.
2. The nucleic acid of claim 1, wherein the calibration sequence is chosen from among the sequences of 5'-TCAG-3', 5'-GTCA-3' and 5'-TGCA-3'.
3. The nucleic acid of claim 1, wherein the modified transposon end sequence comprises the nucleotide sequence of SEQ ID NO: 3, SEQ ID NO: 4 or SEQ ID NO: 6.
4. The nucleic acid of claim 1, wherein the modified transposon end sequence comprises a cleavage site.
5. An in vitro transpososome assembly reaction mixture comprising a transposase, a nucleic acid comprising a modified transposon end sequence, and reaction mixture components suitable for assembly of transposoomes, wherein (a) the modified transposon end sequence comprises (i) a first strand having the nucleotide sequence of SEQ ID NO: 1 that is modified to contain a calibration sequence that is four nucleotides in length and (ii) a second strand that is complementary to the first strand, (b) the calibration sequence contains four different nucleotide bases in any order and is located in the 3' end region of sequence of SEQ ID NO: 1 3' of position 17 of SEQ ID NO: 1 and (c) the modified transposon end sequence and the transposase are capable of forming a transpososome.
6. The in vitro transpososome assembly reaction mixture of claim 5, wherein the calibration sequence is chosen from among the sequences of 5'-TCAG-3', 5'-GTCA-3' and 5'-TGCA-3'.
7. The in vitro transpososome assembly reaction mixture of claim 5, wherein the modified transposon end sequence comprises the nucleotide sequence of SEQ ID NO: 3, SEQ ID NO: 4, SEQ ID NO: 5 or SEQ ID NO: 6.
8. The in vitro transpososome assembly reaction mixture of claim 5, wherein the modified transposon end sequence comprises the nucleotide sequence of SEQ ID NO: 5.
9. A kit comprising a nucleic acid comprising a modified transposon end sequence of claim 1, a transposase and a buffer for performing a transposition reaction.
10. The kit of claim 9, further comprising a DNA polymerase.
11. A method for in vitro assembly of one or more transpososome complexes, comprising contacting a transposase and a nucleic acid comprising a modified transposon end sequence under a condition for forming a transpososome complex, thereby forming one or more transpososome complexes, wherein (a) the modified transposon end sequence comprises (i) a first strand having the nucleotide sequence of SEQ ID NO: 1 that is modified to contain a calibration sequence that is four nucleotides in length and (ii) a second strand that is complementary to the first strand, and (b) the calibration sequence contains four different nucleotide bases in any order and is located in the 3' end region of sequence of SEQ ID NO: 1 3' of position 17 of SEQ ID NO: 1.
12. The method of claim 11, wherein the calibration sequence is chosen from among the sequences of 5'-TCAG-3', 5'-GTCA-3' and 5'-TGCA-3'.
13. The method of claim 11, wherein the modified transposon end sequence comprises the nucleotide sequence of SEQ ID NO: 3, SEQ ID NO: 4 or SEQ ID NO: 6.
14. The method of claim 11, wherein the modified transposon end sequence comprises a cleavage site.
15. The method of claim 13, further comprising contacting the one or more transpososome complexes with a plurality of target nucleic acids.
16. The method of claim 15, further comprising incubating the one or more transpososome complexes and plurality of target nucleic acids under a condition for performing a transposition reaction, wherein the transposition reaction results in fragmentation of the plurality of target nucleic acids and incorporation of a modified transposon end sequence into the ends of the fragmented target nucleic acids, thereby generating a plurality of target nucleic acid fragments attached at both ends to a modified transposon end sequence.
17. The method of claim 16, further comprising contacting a DNA polymerase having 5'-3' exonuclease or strand displacement activity with the plurality of target nucleic acid fragments to generate fully double-stranded nucleic acid molecules.
18. The method of claim 17, further comprising denaturating the fully double-stranded nucleic acid molecules to produce single-stranded nucleic acids, and amplifying the single-stranded nucleic acids.
19. The method of claim 18, further comprising sequencing the single-stranded nucleic acids.
Description
[0001] This application is a continuation application of U.S. patent application Ser. No. 15/791,740, filed Oct. 24, 2017, which is a divisional application of U.S. patent application Ser. No. 14/836,248, filed Aug. 26, 2015, now issued as U.S. Pat. No. 9,834,811, which is a continuation application of U.S. patent application Ser. No. 13/553,395, filed Jul. 19, 2012, now issued as U.S. Pat. No. 9,145,623, which claims priority to U.S. Provisional Application Ser. No. 61/509,691, filed Jul. 20, 2011, each of which is expressly incorporated by reference herein in its entirety.
[0002] The invention relates to the field of high throughput multiplex DNA sequencing. The invention is directed to transposon nucleic acids comprising a transposon end sequence and a calibration sequence for DNA sequencing in the transposon end sequence. In one embodiment, this transposon end sequence is a Mu transposon end. The invention is also directed to a method for generation of a DNA fragmentation library based on a transposition reaction in the presence of a transposon end with the calibration sequence, providing facilitated downstream handling of the produced DNA fragments, e.g., in the generation of sequencing templates.
BACKGROUND
[0003] "DNA sequencing" generally refers to methodologies aiming to determine the primary sequence information in a given nucleic acid molecule. Traditionally, Maxam-Gilbert and Sanger sequencing methodologies have been applied successfully for several decades, as well as a pyrosequencing method. However, these methodologies have been difficult to multiplex, as they require a wealth of labor and equipment time, and the cost of sequencing is excessive for entire genomes. These methodologies required each nucleic acid target molecule to be individually processed, the steps including, e.g., subcloning and transformation into E. coli bacteria, extraction, purification, amplification, and sequencing reaction preparation and analysis.
[0004] So called "next-generation" technologies or "massive parallel sequencing" platforms allow millions of nucleic acid molecules to be sequenced simultaneously. The methods rely on sequencing-by-synthesis approach, while certain other platforms are based on sequencing-by-ligation technology. Although very efficient, all of these new technologies rely on multiplication of the sequencing templates. Thus, for each application, a pool of sequencing templates needs to be produced. A major advancement for template generation was the use of in vitro transposition technology. The earliest in vitro transposition-assisted sequencing template generation methodology (Tenkanen U.S. Pat. No. 6,593,113) discloses a method in which the transposition reaction results in fragmentation of the target DNA, and the subsequent amplification reaction is carried out in the presence of a fixed primer complementary to the known sequence of the target DNA and a selective primer having a complementary sequence to the end of a transposon DNA.
[0005] In vitro transposition methodology has also been applied to "next generation" sequencing platforms. Grunenwald (U.S. Patent Application 20100120098) disclose methods using a transposase and a transposon end for generating extensive fragmentation and 5'-tagging of double-stranded target DNA in vitro. The method is based on the use of a DNA polymerase for generating 5'- and 3'-tagged single-stranded DNA fragments after fragmentation without performing a PCR amplification reaction.
[0006] Many "next-generation" sequencing instruments require a specific calibration sequence to be read first as a part of the sequence to be analyzed (e.g. ion torrent PGM and Roche 454 Genome Sequencer FLX System). This calibration sequence has known bases in particular order and it calibrates the instrument so that it is capable of differentiating the signal generated from different bases during the DNA sequencing reaction. It is necessary that each of the sequencing templates comprises this calibration sequence.
[0007] Methods that facilitate the downstream handling of the fragmented DNA obtained from the transposition step are needed.
SUMMARY
[0008] The invention is related to the modification of a transposon end sequence so that it includes a calibration sequence for DNA sequencing. When the transposon end sequence is inserted into the target DNA in the fragmentation reaction, the calibration sequence is simultaneously incorporated into the target sequence.
[0009] A modified transposon nucleic acid comprising a transposon end sequence and an engineered calibration sequence for DNA sequencing in the transposon end sequence, and a kit for DNA sequencing containing the modified transposon nucleic acid.
[0010] An in vitro method for generating a DNA library by incubating a transposon complex comprising a transposon nucleic acid and a transposase with a target DNA of interest under conditions for carrying out a transposition reaction, where the transposon nucleic acid comprises a transposon end sequence recognizable by the transposase, where the transposon end sequence comprises a calibration sequence for DNA sequencing, and where the transposition reaction results in fragmentation of the target DNA and incorporation of the transposon end into the 5' end of the fragmented target DNA.
BRIEF DESCRIPTION OF THE DRAWINGS
[0011] The patent or application file contains at least one drawing executed in color. A Petition under 37 C.F.R. .sctn. 1.84 requesting acceptance of the color drawing is being filed separately. Copies of this patent or patent application publication with color drawing(s) will be provided by the Office upon request and payment of the necessary fee.
[0012] FIG. 1 is a gel showing a complex formation with MuA transposase and varying transposon end sequences.
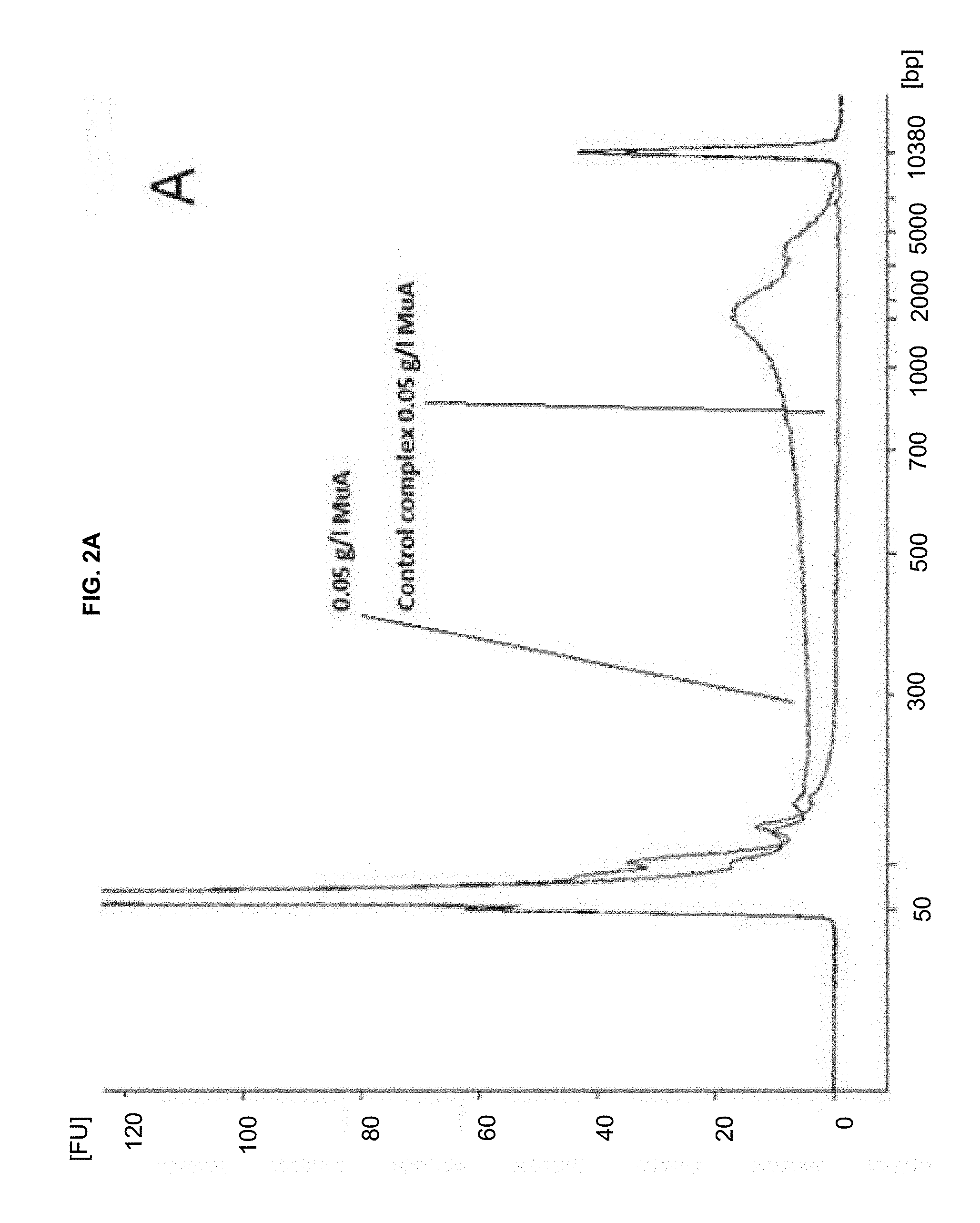
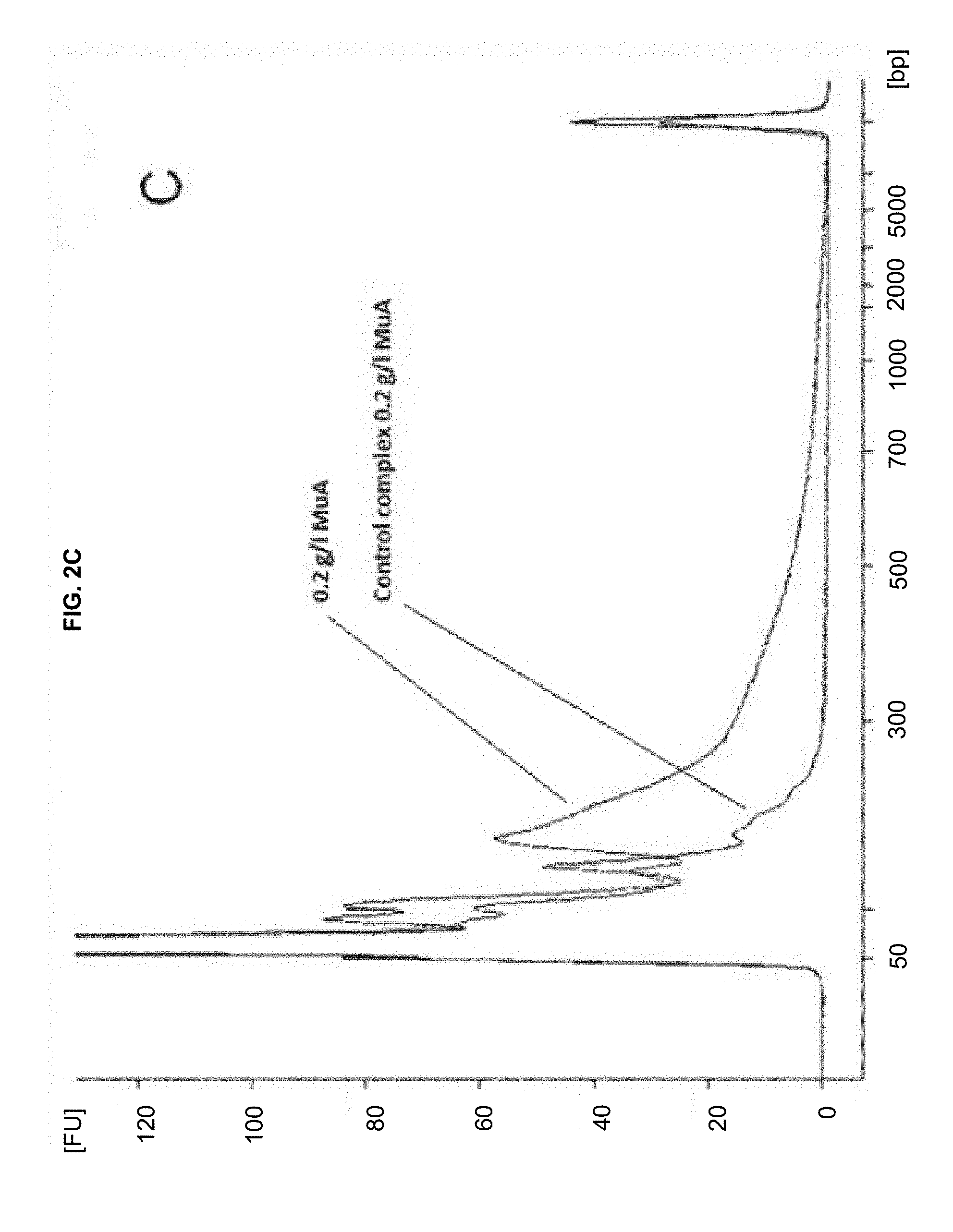
[0013] FIGS. 2A-C show human genomic DNA fragmented with different amounts of transpososome complexes.
[0014] FIG. 3 is a gel showing a complex formation similar to FIG. 1, but with transposon end of SEQ ID NO: 5.
[0015] FIGS. 4A-B are similar to FIGS. 2A-C, but with transposon end of SEQ ID NO: 5.
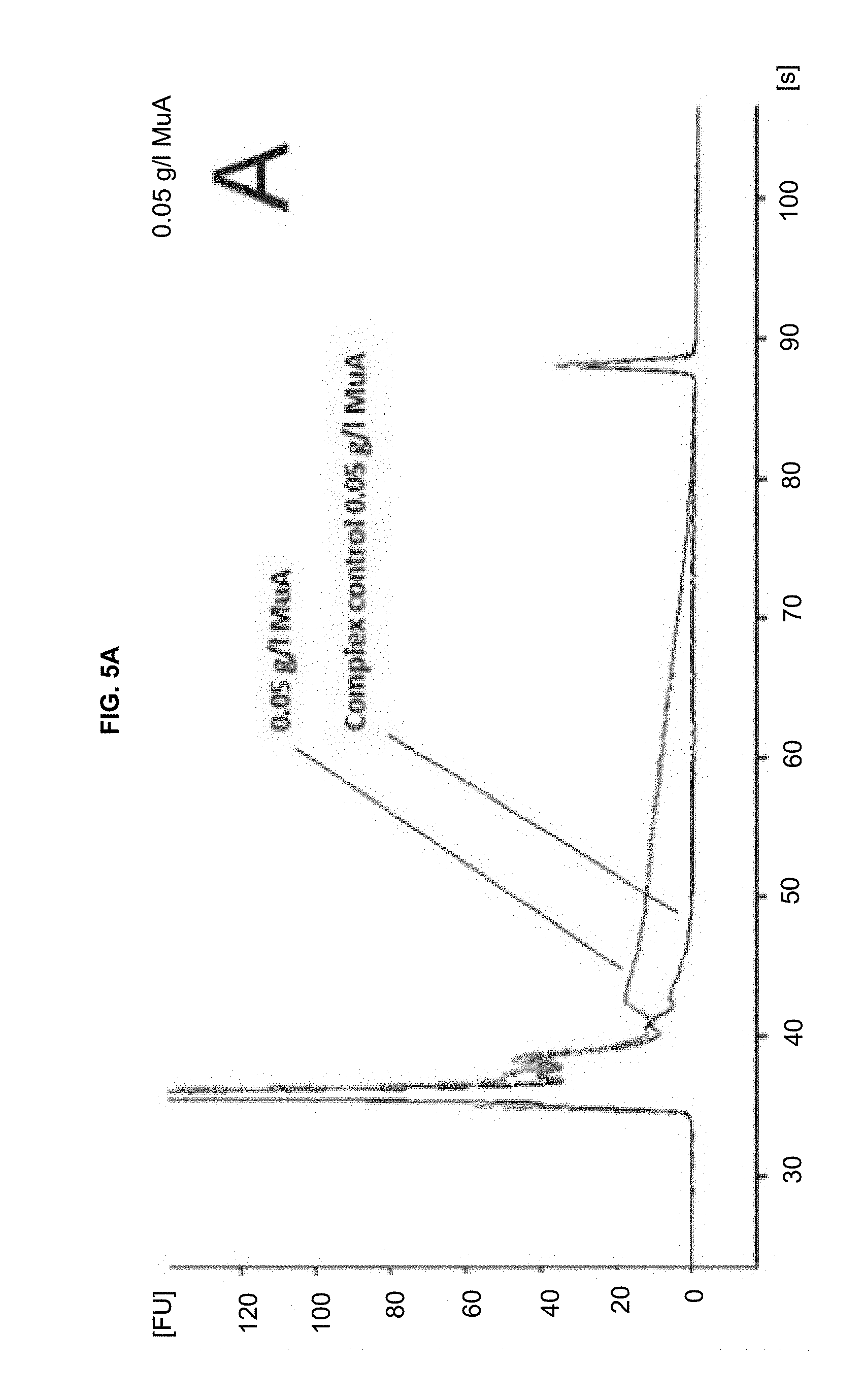
[0016] FIGS. 5A-B are similar to FIGS. 2A-C, but with transposon end of SEQ ID NO: 6.
[0017] FIG. 6 shows a transposition reaction with Mu transposon end sequences on target DNA.
[0018] FIG. 7 shows downstream sequencing reaction with target DNA comprising an incorporated transposon end with calibration sequence.
DETAILED DESCRIPTION
[0019] The terms "calibration sequence" or "key sequence" as used herein generally refer to a nucleic acid sequence that can be used to calibrate a DNA sequencing system. Thus, in embodiments, the particular bases, the order of the bases, and the number of bases that are present in a calibration sequence depends on the requirements of a particular DNA sequencing system.
[0020] In one embodiment, the calibration sequence is a four-nucleotide-long nucleic acid sequence of the four known bases (A, T, C and G) in particular order incorporated into target DNA to be sequenced. The calibration sequence calibrates the sequencing instrument for each sample so that it is capable of differentiating the signal generated from different bases during the DNA sequencing reaction. For example, sequences TCAG, GTCA and TGCA can be used as calibration sequences. However, the four bases could be presented in the calibration sequence in any possible order. In embodiments, the calibration sequence may be longer than four nucleotides, e.g., the calibration sequence may be five, six, seven, eight, nine, ten, or more nucleotides long. The calibration sequence may also comprise bases in addition to, or in place of, the four known bases A, T, G, and C. For example, the calibration sequence may contain derivatized and/or artificial nucleotide bases; such modified bases are known to one skilled in the art.
[0021] The term "transposon", as used herein, refers to a nucleic acid segment that is recognized by a transposase or an integrase enzyme and is an essential component of a functional nucleic acid-protein complex (i.e., a transpososome) capable of transposition. In one embodiment, a minimal nucleic acid-protein complex capable of transposition in a Mu transposition system comprises four MuA transposase protein molecules and a pair of Mu transposon end sequences that are able to interact with MuA (FIG. 6) where the DNA sequences of the fragments from the transposition reaction are, e.g.,
TABLE-US-00001 SEQ ID NO: 1 Insert from Target DNA gap SEQ ID NO: 7 SEQ ID NO: 8 gap SEQ ID NO: 9
and showing the product after gap-filling by a DNA polymerase.
[0022] The term "transposase" as used herein refers to an enzyme that is a component of a functional nucleic acid-protein complex capable of transposition and which is mediating transposition. The term "transposase" also refers to integrases from retrotransposons or of retroviral origin.
[0023] A "transposition reaction" as used herein refers to a reaction where a transposon inserts into a target nucleic acid. Primary components in a transposition reaction are a transposon and a transposase or an integrase enzyme. The method and materials of the invention are exemplified by employing in vitro Mu transposition (Haapa et al. 1999 and Savilahti et al. 1995). Other transposition systems can be used, e.g., Tyl (Devine and Boeke, 1994, and WO 95/23875), Tn7 (Craig, 1996), Tn 10 and IS 10 (Kleckner et al. 1996), Mariner transposase (Lampe et al., 1996), Tcl (Vos et al., 1996, 10(6), 755-61), Tn5 (Park et al., 1992), P element (Kaufman and Rio, 1992) and Tn3 (Ichikawa and Ohtsubo, 1990), bacterial insertion sequences (Ohtsubo and Sekine, 1996), retroviruses (Varmus and Brown 1989), and retrotransposon of yeast (Boeke, 1989).
[0024] A "transposon end sequence" as used herein refers to the nucleotide sequences at the distal ends of a transposon. The transposon end sequences are responsible for identifying the transposon for transposition; they are the DNA sequences the transpose enzyme requires to form a transpososome complex and to perform a transposition reaction. For MuA transposase, this sequence is 50 bp long (SEQ ID NO. 1) described by Goldhaber-Gordon et al., J Biol Chem. 277 (2002) 7703-7712, which is hereby incorporated by reference in its entirety. A transposable DNA of the present invention may comprise only one transposon end sequence. The transposon end sequence in the transposable DNA sequence is thus not linked to another transposon end sequence by a nucleotide sequence, i.e., the transposable DNA contains only one transposase binding sequence. Thus, the transposable DNA comprises a "transposon end" (e.g., Savilahti et al., 1995).
[0025] A "transposase binding sequence" or "transposase binding site" as used herein refers to the nucleotide sequences that are always within the transposon end sequence where a transposase specifically binds when mediating transposition. The transposase binding sequence may comprise more than one site for binding transposase subunits.
[0026] A "transposon joining strand" or "joining end" as used herein means the end of that strand of the double-stranded transposon DNA that is joined by the transposase to the target DNA at the insertion site.
[0027] The term "adaptor" or "adaptor tail" as used herein refers to a non-target nucleic acid component, generally DNA, that provides a means of addressing a nucleic acid fragment to which it is joined. For example, in embodiments, an adaptor comprises a nucleotide sequence that permits identification, recognition, and/or molecular or biochemical manipulation of the DNA to which the adaptor is attached (e.g., by providing a site for annealing an oligonucleotide, such as a primer for extension by a DNA polymerase, or an oligonucleotide for capture or for a ligation reaction).
[0028] Many "next-generation" sequencing instruments (e.g. ion torrent PGM and Roche 454 Genome Sequencer FLX System) require a specific calibration sequence to be read first as a part of the sequence to be analyzed. Because the in vitro transposition technology is already used to fragment target DNA for sequencing, the disclosed method provides transposon end sequences that include the calibration sequence. In this way, the calibration sequence would be incorporated to the target DNA during the fragmentation step. To reduce unusable sequence reads, this calibration sequence may be designed as close to the 3' inserted end of the transposon end (i.e., the joining end) as possible.
[0029] The MuA transposase recognizes a certain transposon end sequence of 50 bp (SEQ ID NO:1) but tolerates some variation at certain positions (Goldhaber-Gordon et al., J Biol Chem. 277 (2002) 7703-7712). Various options for including a calibrator sequence into the transposon end were designed.
[0030] In one embodiment, a modified transposon nucleic acid comprising a transposon end sequence and an engineered calibration sequence for DNA sequencing in the transposon end sequence is provided. The transposon end sequence may be a Mu transposon end sequence, and the Mu transposon end sequence may be any of SEQ ID NOS: 3-6. In one embodiment, the Mu transposon end sequence is SEQ ID NO: 5.
[0031] In one embodiment, a modified transposon nucleic acid comprising a transposon end sequence and an engineered calibration sequence for DNA sequencing in the transposon end sequence is provided, where the transposon end sequence further contains an engineered cleavage site. An engineered cleavage site in the transposon end sequence can be useful for removing parts of the transposon end sequence from the fragmented DNA, which improves downstream amplification (e.g., by reducing intramolecular loop structures, as a result of less complementary sequence) or reduces the amount of transposon end sequence that would be read during sequencing (e.g., single molecule sequencing). In embodiments, the engineered cleavage site may be the incorporation of a uracil base or a restriction site. Modified transposon end sequences comprising an engineered calibration sequence for DNA sequencing and optionally an uracil base or an additional restriction site can be produced, e.g., by regular oligonucleotide synthesis.
[0032] In one embodiment, an in vitro method for generating a DNA library is provided. The method incubates a transposon complex comprising a transposon nucleic acid and a transposase with a target DNA of interest under conditions for carrying out a transposition reaction. Transposon nucleic acid comprises a transposon end sequence that is recognizable by the transposase, and where the transposon end sequence comprises a calibration sequence for DNA sequencing. The transposition reaction results in fragmentation of the target DNA, and incorporates the transposon end into the 5' end of the fragmented target DNA.
[0033] In one embodiment, the method further comprises the step of amplifying the fragmented target DNA in an amplification reaction using a first and second oligonucleotide primer complementary to the transposon end in the 5' ends of the fragmented target DNA. The first and second primer optionally comprise 5' adaptor tails.
[0034] In one embodiment, the method further comprises the step of contacting the fragments of target DNA comprising the transposon end at the 5' ends of the fragmented target DNA with DNA polymerase having 5'-3' exonuclease or strand displacement activity, so that fully double-stranded DNA molecules are produced from the fragments of target DNA. This step is used to fill the gaps generated in the transposition products in the transposition reaction. The length of the gap is characteristic to a certain transposition enzyme, e.g., for MuA the gap length is 5 nucleotides.
[0035] To prepare the transposition products for downstream steps, such as polymerase chain reaction (PCR), the method may comprise the further step of denaturating the fully double-stranded DNA molecules to produce single stranded DNA for use in the amplification reaction.
[0036] In one embodiment, the transposition system used in the method is based on MuA transposase enzyme. For the method, one can assemble in vitro stable but catalytically inactive Mu transposition complexes in conditions devoid of Mg.sup.2+ as disclosed in Savilahti et al., 1995 and Savilahti and Mizuuchi, 1996. In principle, any standard physiological buffer not containing Mg.sup.2+ is suitable for assembly of the inactive Mu transposition complexes. In one embodiment, the in vitro transpososome assembly reaction may contain 150 mM Tris-HCl pH 6.0, 50% (v/v) glycerol, 0.025% (w/v) Triton X-100, 150 mM NaCl, 0.1 mM EDTA, 55 nM transposon DNA fragment, and 245 nM MuA. The reaction volume may range from about 20 .mu.l to about 80 .mu.l. The reaction is incubated at about 30.degree. C. for about 0.5 hours to about four hours. In one embodiment, the assembly reaction is incubated for two hours at about 30.degree. C. Mg.sup.2+ is added for activation.
[0037] In case the transposon end sequence comprises an engineered cleavage site, the method can comprise a further step of incubating the fragmented target DNA with an enzyme specific to the cleavage site so that the transposon ends incorporated to the fragmented target DNA are cleaved at the cleavage site. The cleaving enzyme may be an N-glycosylase or a restriction enzyme, such as uracil-N-glycosylase or a methylation specific restriction enzyme, respectively.
[0038] In one embodiment, the 5' adaptor tail of the first and/or second PCR primer(s) used in the method comprise one or more of the following groups: an amplification tag, a sequencing tag, and/or a detection tag.
[0039] The amplification tag is a nucleic acid sequence providing specific sequence complementary to an oligonucleotide primer to be used in the subsequent rounds of amplification. For example, the sequence may be used for facilitating amplification of the nucleic acid obtained. Examples of detection tags are fluorescent and chemiluminescent dyes, a green fluorescent protein, and enzymes that are detectable in the presence of a substrate, e.g., an alkaline phosphatase with NBT plus BCIP, or a peroxidase with a suitable substrate. By using different detection tags, i.e. barcodes, sequences from multiple samples can be sequenced in the same instrument run and identified by the sequence of the detection tag. Examples are Illumina's index sequences in TruSeq DNA Sample Prep Kits, or Molecular barcodes in Life Technologies' SOLiD.TM. DNA Barcoding Kits.
[0040] The sequencing tag provides a nucleic acid sequence permitting the use of the amplified DNA fragments obtained from step c) as templates for next-generation sequencing. For example, the sequencing tag may provide annealing sites for sequencing by hybridization on a solid phase. Such sequencing tag may be Roche 454A and 454B sequencing tags, Applied Biosystems' SOLiD.TM. sequencing tags, ILLUMINA.TM. SOLEXA.TM. sequencing tags, the Pacific Biosciences' SMRT.TM. sequencing tags, Pollonator Polony sequencing tags, and the Complete Genomics sequencing tags.
[0041] The detection tag comprises a sequence or a detectable chemical or biochemical moiety for facilitating detection of the nucleic acid obtained from the amplification step.
[0042] In one embodiment, a kit for use in DNA sequencing is provided. The kit comprises at least a tranposon nucleic acid comprising transposon end sequence and an engineered calibration sequence for DNA sequencing in the transposon end sequence. In one embodiment, the tranposon nucleic acid is a Mu transposon end sequence. In one embodiment, the Mu transposon end sequence is selected from SEQ ID NOS: 3-6. In one embodiment, the Mu transposon end sequence is SEQ ID NO:5. In one embodiment, the tranposon nucleic acid further comprises an engineered cleavage site. The kit may also comprise additional components, e.g., buffers for performing transposition reaction, buffers for DNA sequencing, control DNA, transposase enzyme, and DNA polymerase. The kit can be packaged in a suitable container with instructions for using the kit.
[0043] The publications and other materials used herein to illuminate the background of the invention, and in particular, to provide additional details with respect to its practice, are incorporated herein by reference. The present invention is further described in the following examples, which are not intended to limit the scope of the invention.
Example 1
Native Sequence of the Inserted Strand of MuA Recognition Transposon End
[0044] In various embodiments, a calibration sequence is four bases long and contains each of the four bases (A, T, G, C) sequentially in a row but in any order. The native MuA recognition sequence only contains these at underlined positions which are far from the 3'-end.
TABLE-US-00002 (SEQ ID NO: 1) GTTTTCGCATTTATCGTGAAACGCTTTCGCGTTTTTCGTGCGCCGCTTCA
If these sequences were used as calibrators, a minimum of 34 bases of the inserted transposon sequence would have to be read in every target fragment before reaching the sequence of interest, wasting sequencing reagents and instrument run time. The accuracy of sequence reading also gradually decreases after each cycle, so the best possible accuracy would always be "wasted" on reading the transposon end sequence. In addition, the bases in the "native calibrator sequences" are not necessarily in the preferred order, as defined by the instruments' requirements. That is why the MuA recognition sequence needs to be modified near the 3'-end in order to be used in library generation for instruments that require a calibration sequence. Other transposons, such as the native Tn5 recognition transposon sequence, do not contain the four bases in a row anywhere in its sequence.
[0045] When the transposon end sequence and MuA transposase were incubated together in a suitable buffer, they formed transpososome complexes (FIG. 1). The higher molecular weight DNA band of about 300 bp represents transposon end DNA bound to transpososome complexes (mobility in the gel electrophoresis is retarded due to protein binding) and the smaller DNA band (50 bp) is the free transposon DNA, i.e. not complexed by MuA. M: Molecular weight marker. wt: Native MuA transposon end +MuA transposase. 1: Transposon end of SEQ ID NO:4. 2: Transposon end of SEQ ID NO:2. 3: Transposon end of SEQ ID NO:3. -MuA: Control reaction with native MuA transposon end DNA sequence (SEQ ID NO: 1) without MuA transposase.
[0046] When these complexes (formed using wild-type MuA transposon end DNA and MuA transposase) were incubated with target DNA, the transposon sequences were inserted into DNA and the target DNA was fragmented, as shown in FIGS. 2A-C. FIG. 2A: 0.05 g/L MuA in the fragmentation reaction. FIG. 2B: 0.15 g/l MuA. FIG. 2C: 0.2 g/l MuA, gDNA fragmented with MuA. Control contains MuA complexes but no gDNA.
TABLE-US-00003 TABLE 1 Composition of the complex formation reaction in FIG. 1. The following components of the complex formation reaction were incubated for one hour at 30.degree. C. MuA transposase 0.44 g/l Transposon DNA 3.0 .mu.M Tris-HCl pH 8 120.42 mM HEPES pH 7.6 2.6 mM EDTA 1.05 mM DTT 0.10 mM NaCl 102.1 mM KCl 52 mM Triton X-100 0.05 % glycerol 12.08 % DMSO 10 %
Different amounts of the complex were incubated with human genomic DNA at 30.degree. C. for one hour (Table 2), and each reaction was replicated eight times. The replicates were combined after the fragmentation, DNA was purified with QIAGEN MinElute PCR Purification Kit, and analyzed with Agilent 2100 Bioanalyzer instrument (FIG. 2).
TABLE-US-00004 TABLE 2 Example of final composition of the fragmentation reaction. For the fragmentation reaction, the MuA transposon and transposon DNA concentrations were varied in different experiments, whereas the concentration of other components was kept constant. MuA transposon 0.05 g/l Transposon DNA 0.341 .mu.M gDNA 100 ng Tris-HCl pH 8 40 mM EDTA 0.33 mM NaCl 100 mM MgCl.sub.2 10 mM Triton X-100 0.05 % glycerol 10 % DMSO 3.3 %
Example 2
Changing of the Mu 3'-End to Include the Current Ion Torrent Key Sequence (TCAG)
TABLE-US-00005 [0047] (SEQ ID NO: 2) GTTTTCGCATTTATCGTGAAACGCTTTCGCGTTTTTCGTGCGCCGCTCAG
This transposon end sequence was tested. MuA transposase did not form complexes with this sequence and thus there was no transposition activity (see FIG. 1).
Example 3
Converting the Fourth T of Mu End Sequence to G (Counting from the 3'-End) to Yield GTCA Key
TABLE-US-00006 [0048] (SEQ ID NO: 3) GTTTTCGCATTTATCGTGAAACGCTTTCGCGTTTTTCGTGCGCCGCGTCA
This was tested for transposition activity and MuA transposase formed complexes with this sequence and was also active (FIG. 1).
Example 4
Converting the Third T of Mu End Sequence to G to Yield TGCA Key
TABLE-US-00007 [0049] (SEQ ID NO: 4) GTTTTCGCATTTATCGTGAAACGCTTTCGCGTTTTTCGTGCGCCGCTGCA
This was tested for transposition activity and MuA transposase formed complexes with this sequence and was also active (FIG. 1).
Example 5
Modification of the 5th, 6th, and 8th Bases of Mu End Sequence into G, a, and T, Respectively, to Yield the Current Ion Torrent Key
TABLE-US-00008 [0050] (SEQ ID NO: 5) GTTTTCGCATTTATCGTGAAACGCTTTCGCGTTTTTCGTGCGTCAGTTCA
This transposon end sequence worked well with MuA transposase in complex formation, as shown in FIG. 3, which shows complex formation similar to FIG. 1, but with transposon end of SEQ ID NO: 5, and in fragmentation, as shown in FIG. 4, which is similar to FIG. 2, but with transposon end of SEQ ID NO: 5, i.e. showing human genomic DNA fragmented with different amounts of transpososome complexes. FIG. 4A: 0.05 g/L MuA in the fragmentation reaction. FIG. 4B: 0.15 g/l MuA, with gDNA fragmented with MuA and control that contains MuA complexes but no gDNA.
Example 6
Modification of the 10th, and 11th Bases of Mu End Sequence into a and C, Respectively, to Yield
TABLE-US-00009 [0051] (SEQ ID NO: 6) GTTTTCGCATTTATCGTGAAACGCTTTCGCGTTTTTCGTCAGCCGCTTCA
This transposon end sequence worked well with MuA transposase in complex formation and in fragmentation, as shown in FIG. 5, which is similar to FIG. 2, but with transposon end of SEQ ID NO: 6, i.e. showing human genomic DNA fragmented with different amounts of transpososome complexes. FIG. 5A: 0.05 g/L MuA in the fragmentation reaction. FIG. 5B: 0.15 g/l MuA, with gDNA fragmented with MuA and control that contains MuA complexes but no gDNA.
REFERENCES
[0052] Boeke J. D. 1989. Transposable elements in Saccharomyces cerevisiae in Mobile DNA. [0053] Craig N. L. 1996. Transposon Tn7. Curr. Top. Microbiol. Immunol. 204: 27-48. [0054] Devine, S. E. and Boeke, J. D., Nucleic Acids Research, 1994, 22(18): 3765-3772. [0055] Goldhaber-Gordon et al., J Biol Chem. 277 (2002) 7703-7712 [0056] Haapa, S. et al., Nucleic Acids Research, vol. 27, No. 13, 1999, pp. 2777-2784 [0057] Ichikawa H. and Ohtsubo E., J. Biol. Chem., 1990, 265(31): 18829-32. [0058] Kaufman P. and Rio D. C. 1992. Cell, 69(1): 27-39. [0059] Kleckner N., Chalmers R. M., Kwon D., Sakai J. and Bolland S. TnlO and IS10 Transposition and chromosome rearrangements: mechanism and regulation in vivo and in vitro. Curr. Top. Microbiol. Immunol., 1996, 204: 49-82. [0060] Lampe D. J., Churchill M. E. A. and Robertson H. M., EMBO J., 1996, 15(19): 5470-5479. [0061] Ohtsubo E. & Sekine Y. Bacterial insertion sequences. Curr. Top. Microbiol. Immunol., 1996, 204:1-26. [0062] Park B. T., Jeong M. H. and Kim B. H., Taehan Misaengmul Hakhoechi, 1992, 27(4): 381-9. [0063] Savilahti, H. and K. Mizuuchi. 1996. Mu transpositional recombination: donor DNA cleavage and strand transfer in trans by the Mu transposase. Cell 85:271-280. [0064] Savilahti, H., P. A. Rice, and K. Mizuuchi. 1995. The phage Mu transpososome core: DNA requirements for assembly and function. EMBO J. 14:4893-4903. [0065] Varmus H and Brown. P. A. 1989. Retroviruses, in Mobile DNA. Berg D. E. and Howe M. eds. American Society for Microbiology, Washington D. C. pp. 53-108. [0066] Vos J. C., Baere I. And Plasterk R. H. A., Genes Dev., 1996, 10(6): 755-61.
[0067] Applicants incorporate by reference the material contained in the accompanying computer readable Sequence Listing identified as Sequence Listing_ST25.txt, having a file creation date of Jul. 17, 2012 1:49 P.M. and file size of 1.98 KB.
Sequence CWU 1
1
9150DNAArtificial SequenceBacteriophage Mu 1gttttcgcat ttatcgtgaa
acgctttcgc gtttttcgtg cgccgcttca 50250DNAArtificial
SequenceModified Mu end sequence 2gttttcgcat ttatcgtgaa acgctttcgc
gtttttcgtg cgccgctcag 50350DNAArtificial SequenceModified Mu end
sequence 3gttttcgcat ttatcgtgaa acgctttcgc gtttttcgtg cgccgcgtca
50450DNAArtificial SequenceModified Mu end sequence 4gttttcgcat
ttatcgtgaa acgctttcgc gtttttcgtg cgccgctgca 50550DNAArtificial
SequenceModified Mu end sequence 5gttttcgcat ttatcgtgaa acgctttcgc
gtttttcgtg cgtcagttca 50650DNAArtificial SequenceModified Mu end
sequence 6gttttcgcat ttatcgtgaa acgctttcgc gtttttcgtc agccgcttca
50750DNAArtificial Sequencetransposon 7tgaagcggcg cacgaaaaac
gcgaaagcgt ttcacgataa atgcgaaaac 50850DNAArtificial
Sequencetransposon 8caaaagcgta aatagcactt tgcgaaagcg caaaaagcac
gaggcgaagt 50950DNAArtificial Sequencetransposon 9acttcgccgc
gtgctttttg cgctttcgca aagtgctatt tacgcttttg 50
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.