Apilimod For Use In The Treatment Of Colorectal Cancer
BEEHARRY; Neil ; et al.
U.S. patent application number 15/524846 was filed with the patent office on 2019-07-11 for apilimod for use in the treatment of colorectal cancer. The applicant listed for this patent is AI Therapeutics, Inc.. Invention is credited to Paul BECKETT, Neil BEEHARRY, Chris CONRAD, Sophia GAYLE, Marylens HERNANDEZ, Sean LANDRETTE, Henri LICHENSTEIN, Jonathan M. ROTHBERG, Tian XU.
| Application Number | 20190209576 15/524846 |
| Document ID | / |
| Family ID | 54548301 |
| Filed Date | 2019-07-11 |











View All Diagrams
| United States Patent Application | 20190209576 |
| Kind Code | A1 |
| BEEHARRY; Neil ; et al. | July 11, 2019 |
APILIMOD FOR USE IN THE TREATMENT OF COLORECTAL CANCER
Abstract
The present invention relates to methods for treating colorectal cancer with apilimod and related compositions and methods.
| Inventors: | BEEHARRY; Neil; (Guilford, CT) ; GAYLE; Sophia; (East Haven, CT) ; LANDRETTE; Sean; (Meriden, CT) ; BECKETT; Paul; (Yorktown Heights, NY) ; CONRAD; Chris; (Guilford, CT) ; XU; Tian; (Cambridge, MA) ; HERNANDEZ; Marylens; (Guilford, CT) ; ROTHBERG; Jonathan M.; (Guilford, CT) ; LICHENSTEIN; Henri; (Guilford, CT) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 54548301 | ||||||||||
| Appl. No.: | 15/524846 | ||||||||||
| Filed: | November 6, 2015 | ||||||||||
| PCT Filed: | November 6, 2015 | ||||||||||
| PCT NO: | PCT/US2015/059526 | ||||||||||
| 371 Date: | May 5, 2017 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 62077127 | Nov 7, 2014 | |||
| 62115228 | Feb 12, 2015 | |||
| 62119540 | Feb 23, 2015 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | G01N 33/57419 20130101; A61K 45/06 20130101; A61K 31/5377 20130101; A61K 9/0019 20130101; A61K 9/0053 20130101; G01N 2800/52 20130101; G01N 33/5743 20130101; A61K 31/437 20130101; A61P 35/04 20180101; A61K 31/44 20130101; A61K 31/5377 20130101; A61K 2300/00 20130101; A61K 31/437 20130101; A61K 2300/00 20130101; A61K 31/44 20130101; A61K 2300/00 20130101 |
| International Class: | A61K 31/5377 20060101 A61K031/5377; A61P 35/04 20060101 A61P035/04; A61K 45/06 20060101 A61K045/06; A61K 9/00 20060101 A61K009/00; A61K 31/437 20060101 A61K031/437 |
Claims
1. A method for treating colorectal cancer in a subject in need thereof, the method comprising administering to the subject a pharmaceutical composition comprising a therapeutically effective amount of apilimod, or a pharmaceutically, acceptable salt thereof.
2. The method of claim 1, wherein the apilimod is apilimod dimesylate.
3. The method of claim 1, wherein the colorectal cancer is refractory or metastatic.
4. The method of claim 1, wherein the composition is an oral dosage form or a dosage form suitable for intravenous administration.
5. The method of claim 1, wherein the colorectal cancer is a stage III or stage IV colorectal cancer, as defined by the TNM staging system of the World Health Organization.
6. The method of claim 1, further comprising administering at least one additional active agent.
7. The method of claim 1, wherein the at least one additional active agent is a therapeutic agent or a non-therapeutic agent, or a combination thereof.
8. The method of claim 7, wherein the therapeutic agent is selected from the group consisting of a protein kinase inhibitor, a PD-1/PDL-1 pathway inhibitor, a platinum based anti-neoplastic agent, a topoisomerase inhibitor, a nucleoside metabolic inhibitor, an alkylating agent, an intercalating agent, a tubulin binding agent, a BRAT inhibitor, and combinations thereof.
9. The method of claim 8, wherein the therapeutic agent is selected from the group consisting of vemurafenib, oxaliplatin, regorafenib, irinotecan, 5-fluorouracil, pembrolizumab, avelumab, atezolizumab (MPDL3280A), nivolumab (BMS-936558), pidilizumab (MK-3475), MSB0010718C, MEDI4736, and combinations thereof.
10. The method of claim 9, wherein the therapeutic agent s vemurafenib.
11. The method of claim 9, wherein the therapeutic agent s regorafenib.
12. The method of claim 9, wherein the cancer is refractory or metastatic colorectal cancer.
13. The method of claim 1 further comprising administering a non-therapeutic agent selected to ameliorate one or more side effects of the apilimod.
14. (canceled)
15. The method of claim 7, wherein the non-therapeutic agent is selected from the group consisting of ondansetron, granisetron, dolasetron and palonosetron.
16. The method of claim 7, wherein the non-therapeutic agent is selected from the group consisting of pindolol and risperidone.
17. (canceled)
18. (canceled)
19. (canceled)
20. (canceled)
21. (canceled)
22. (canceled)
23. (canceled)
24. (canceled)
25. A method for inducing or potentiating autophagy or apoptosis in a colorectal cancer cell, the method comprising contacting the cell with a composition comprising apilimod dimesylate.
26. A method for identifying a human colorectal cancer patient for treatment with a combination therapy comprising apilimod and vemurafenib, the method comprising assaying a biological sample of the subject's cancer for one or more of the V600E BRAF protein mutation, the V600K BRAF protein mutation, or the genetic equivalents thereof, wherein a subject having either of these mutations is identified as a patient for treatment with a combination therapy comprising apilimod and vemurafenib.
27. (canceled)
28. (canceled)
29. A method for identifying a colorectal cancer as sensitive to apilimod treatment, the method comprising assaying the expression of the SNX10 gene in a sample of the cancer, such as a biopsy sample, wherein high SNX10 gene expression indicates that the colorectal cancer is sensitive to apilimod treatment.
Description
RELATED APPLICATIONS
[0001] This application is a national stage entry, filed under 35 U.S.C. .sctn. 371, of International Application No. PCT/US2015/059526, filed on Nov. 6, 2015, which claims priority to U.S. Pat. App. Ser. No. 62/077,127, filed on Nov. 7, 2014, U.S. Pat. App. Ser. No. 62/115,228, filed on Feb. 12, 2015, and U.S. Pat. App. Ser. No. 62/119,540, filed on Feb. 23, 2015, the contents of which are hereby fully incorporated by reference.
FIELD OF THE INVENTION
[0002] The present invention relates to compositions comprising apilimod and methods of using same in the treatment of colorectal cancer.
BACKGROUND OF THE INVENTION
[0003] Colorectal cancer remains a serious health concern. Globally it is the third most common cancer, making up 10% of all cases. In 2012 there were 14 million new cases and 694,000 deaths from colorectal cancer. Five year survival rates in the United States are around 65%, highlighting the need for more effective therapies.
[0004] Treatments for colorectal cancer may include some combination of surgery, radiation therapy, chemotherapy, and targeted therapy. Cancers that are confined within the wall of the colon may be curable with surgery. But in many cases, it is not possible to completely eliminate or cure the cancer once it has metastasized. Depending upon where and how big the metastases are, treatment may involve chemotherapy, surgery, gene therapy, immunotherapy, radiation therapy, and combinations of these. There remains a need for more effective treatment options for colorectal cancer. The present invention addresses that need.
SUMMARY OF THE INVENTION
[0005] The present invention is based in part on the surprising discovery that apilimod is a highly cytotoxic agent in colorectal cancer cells, including colorectal cancer cells that are resistant to other therapies, and further in combination with certain chemotherapy agents.
[0006] In one aspect, the disclosure provides a composition for treating colorectal cancer in a subject in need thereof, the composition comprising a therapeutically effective amount of apilimod, or a pharmaceutically acceptable salt, solvate, clathrate, hydrate, polymorph, prodrug, analog or derivative thereof. In embodiments, the apilimod is apilimod dimesylate. In embodiments, the colorectal cancer is refractory or metastatic. In embodiments, the colorectal cancer is a stage III or stage IV colorectal cancer, as defined by the TNM staging system of the World Health Organization. In embodiments, the composition is an oral dosage form or a dosage form suitable for intravenous administration.
[0007] In embodiments, the composition further comprises at least one additional active agent. In embodiments, the at least one additional active agent is a therapeutic agent or a non-therapeutic agent, or a combination thereof. In embodiments, the at least one additional active agent is a therapeutic agent selected from the group consisting of a protein kinase inhibitor, a platinum based anti-neoplastic agent, a topoisomerase inhibitor, a nucleoside metabolic inhibitor, an alkylating agent, an intercalating agent, a tubulin binding agent, a BRAF inhibitor, and combinations thereof. In embodiments, the therapeutic agent is selected from the group consisting of vemurafenib, oxaliplatin, regorafenib, irinotecan, 5-fluorouracil, and combinations thereof. In one embodiment, the therapeutic agent is vemurafenib or regorafenib.
[0008] In embodiments, the composition further comprises a non-therapeutic agent selected to ameliorate one or more side effects of the apilimod. In embodiments, the non-therapeutic agent is selected from the group consisting of ondansetron, granisetron, dolasetron and palonosetron or from pindolol and risperidone.
[0009] In one aspect, the disclosure provides a method for treating colorectal cancer in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of apilimod, or a pharmaceutically acceptable salt, solvate, clathrate, hydrate, polymorph, prodrug, analog or derivative thereof. In embodiments, the apilimod is apilimod dimesylate.
[0010] In embodiments, the method further comprises administering at least one additional active agent to the subject. The at least one additional active agent may be a therapeutic agent or a non-therapeutic agent, or a combination thereof. The at least one additional active agent may be administered in a single dosage form with the apilimod or in a separate dosage form.
[0011] In embodiments, the at least one additional active agent is a therapeutic agent. In embodiments, the therapeutic agent is a B-Raf enzyme inhibitor. In embodiments, the therapeutic agent is vemurafenib, oxaliplatin, regorafenib, irinotecan, 5-fluorouracil.
[0012] In embodiments, the method comprises administering apilimod and vemurafenib together in a combination therapy regimen for the treatment of colorectal cancer. In embodiments, the colorectal cancer is a late-stage colorectal cancer. In one embodiment, the late-stage colorectal cancer is resistant to vemurafenib alone.
[0013] In embodiments, the at least one additional active agent is selected from an alkylating agent, an intercalating agent, a tubulin binding agent, a PD-1/PDL-1 pathway inhibitor, and combinations thereof. In embodiments, the at least one additional active agent is a therapeutic agent selected from vemurafenib, 5-fluorouracil (Fluoroplex.RTM., irinotecan (Camptostar.RTM.), capecitabine (Xeloda.RTM.), oxaliplatin (Eloxatin.RTM.) and regorafenib (Stivarga.RTM.), and combinations thereof. In embodiments, the at least one additional active agent is a therapeutic agent selected from bevacizumab (Avastin.RTM.), ramucirumab (Cyramza.RTM.), cetuximab (Erbitex.RTM.), panitumumab (Vecibix.RTM.), ipilimumab (Yervoy.RTM.), pembrolizumab (Keytruda.TM.), dabrafenib (Tafinlar.TM.), vemurafenib (Zelboraf.TM.), trametinib (Mekinist.TM.), Zviv-Aflibercept (Zaltrap.RTM.), nivolumab (Opdivo.RTM.) and combinations thereof. In embodiments, the at least one additional active agent is a PD-1/PDL-1 pathway inhibitor. In embodiments, the PD-1/PDL-1 pathway inhibitor is selected from pembrolizumab (Keytruda), avelumab, atezolizumab (MPDL3280A), nivolumab (BMS-936558), pidilizumab (MK-3475), MSB0010718C, and MEDI4736.
[0014] In embodiments, the at least one additional active agent is a non-therapeutic agent selected to ameliorate one or more side effects of apilimod. In one embodiment, the non-therapeutic agent is selected from the group consisting of ondansetron, granisetron, dolasetron and palonosetron. In one embodiment, the non-therapeutic agent is selected from the group consisting of pindolol and risperidone.
[0015] In embodiments, the apilimod may be administered in any suitable dosage form. In one embodiment, the apilimod is administered in an oral dosage form. In another embodiment, the dosage form is suitable for intravenous administration. In one embodiment, where the dosage form is suitable for intravenous administration, administration is by a single injection or by a drip bag.
[0016] In embodiments of the methods described here, the subject is a human colorectal cancer patient. In one embodiment, the human colorectal cancer patient in need of treatment according to the methods described here is one having late-stage, malignant or metastatic colorectal cancer. In embodiments, the human colorectal cancer patient in need of treatment is one whose cancer is refractory to a standard chemotherapy regimen. In embodiments, the human colorectal cancer patient in need of treatment is one whose colorectal cancer has recurred following treatment with a standard chemotherapy regimen.
[0017] In embodiments, the standard chemotherapy regimen comprises an immunotherapy regimen or a targeted therapy regimen. In embodiments, the immunotherapy regimen comprises one or more therapeutic agents selected from the group consisting of anti-CTLA4 antibodies (e.g., ipilimumab), PD-1/PDL-1 pathway inhibitors (e.g., pembrolizumab (Keytruda), MSB0010718C, MEDI4736, MPDL3280A, BMS-936559), nivolumab (Opdivo), pidilizumab, AMP-224, and Interleukin-2 (IL-2, aldesleukin, Proleukin). In embodiments, the targeted therapy regimen comprises one or more therapeutic agents selected from the group consisting of BRAF inhibitors (e.g., dabrafenib (Tafinlar), sorafenib (Nexavar) and vemurafenib (Zelboraf), MEK inhibitors (e.g., trametinib (Mekinist)) and KIT inhibitors (e.g., dasatinib (Sprycel), imatinib (Gleevec), and nilotinib (Tasigna)). In embodiments, the PD-1/PDL-1 pathway inhibitor is selected from pembrolizumab (Keytruda), avelumab, atezolizumab (MPDL3280A), nivolumab (BMS-936558), pidilizumab (MK-3475), MSB0010718C, and MEDI4736.
[0018] In embodiments, the method is a method of treating a colorectal cancer using a combination therapy comprising apilimod and a chemotherapy regimen for the treatment of the colorectal cancer. In one embodiment, the apilimod is administered as an adjunctive therapy to the chemotherapy regimen. In embodiments, the chemotherapy regimen comprises one or more of dacarbazine, temozolomide, Nab-paclitaxel, carmustine, cisplatin, carboplatin, and vinblastine for the treatment of malignant or metastatic colorectal cancer. In embodiments, the chemotherapy regimen comprises one or more of vemurafenib, dabrafenib and trametinib for metastatic colorectal cancer. In one embodiment, the chemotherapy regimen comprises one or more of high dose interleukin-2 and ipilimumab.
[0019] In one embodiment, the disclosure provides methods for inducing or potentiating autophagy or apoptosis in a colorectal cancer cell. In accordance with this embodiment, the colorectal cancer cell may be in vitro or in vivo. In one embodiment, the colorectal cancer cell is in vitro. In one embodiment, the colorectal cancer cell is in vivo in a mammalian subject. In one embodiment, the colorectal cancer cell is a late stage colorectal cancer. In one embodiment, the colorectal cancer cell is a metastatic cell or a cell that has metastasized. In embodiments, the colorectal cancer cell has a BRAF mutant phenotype. In embodiments, the BRAF mutant phenotype is characterized by the V600E or V600K mutation of human BRAF.
[0020] The disclosure also provides a method for inducing or potentiating autophagy or apoptosis in a colorectal cancer cell, the method comprising contacting the cell with a composition comprising apilimod, or a pharmaceutically acceptable salt, solvate, clathrate, hydrate, polymorph, prodrug, analog or derivative thereof. In embodiments, the apilimod is apilimod dimesylate.
[0021] The disclosure also provides a method for identifying a human colorectal cancer patient for treatment with a combination therapy comprising an apilimod composition and vemurafenib, the method comprising assaying a biological sample of the subject's cancer for one or more of the V600E BRAF protein mutation, the V600K BRAF protein mutation, or the genetic equivalents thereof, wherein a subject having either of these mutations is identified as a patient for treatment with a combination therapy comprising an apilimod composition and vemurafenib.
[0022] In embodiments, the disclosure also provides a method for identifying a colorectal cancer that is sensitive to apilimod, the method comprising assaying a sample of cancer cells from the cancer, for example a biopsy, for expression of the SNX10 gene, wherein high expression levels of SNX10 in the sample of cancer cells indicates that the cells are sensitive to apilimod. In embodiments, the SNX10 gene expression level is called as `high` relative to the SNX10 gene expression level of a reference cell line. For example, an SNX10 gene expression level in a biopsy sample that is higher than that of COL0205 cells, may be considered "high" for purposes of indicating sensitivity to apilimod.
BRIEF DESCRIPTION OF THE DRAWINGS
[0023] FIG. 1: LAM-002 (apilimod dimesylate)+oxaliplatin combination in HCT116 cells (5 day assay). A, bar graph showing cell viability (%). B, combination index (CI) vs fractional effect graph showing synergy determinations at ED.sub.50, ED.sub.75 and ED.sub.90.
[0024] FIG. 2: LAM-002+regorafenib combination in HCT116 cells (5 day assay). A, bar graph showing cell viability (%). B, combination index (CI) vs fractional effect graph showing synergy determinations at ED.sub.50, ED.sub.75 and ED.sub.90.
[0025] FIG. 3: LAM-002+regorafenib combination in RKO cells (5 day assay). A, bar graph showing cell viability (%). B, combination index (CI) vs fractional effect graph showing synergy determinations at ED.sub.50, ED.sub.75 and ED.sub.90.
[0026] FIG. 4: LAM-002+irinotecan combination in HCT116 cells (5 day assay). A, bar graph showing cell viability (%). B, combination index (CI) vs fractional effect graph showing synergy determinations at ED.sub.50, ED.sub.75 and ED.sub.90.
[0027] FIG. 5: LAM-002+5-fluorouracil combination in HCT116 cells (5 day assay). A, bar graph showing cell viability (%). B, combination index (CI) vs fractional effect graph showing synergy determinations at ED.sub.50, ED.sub.75 and ED.sub.90.
[0028] FIG. 6: LAM-002+vemurafenib combination in HCT116 cells (5 day assay). A, bar graph showing cell viability (%). B, combination index (CI) vs fractional effect graph showing synergy determinations at ED.sub.50, ED.sub.75 and ED.sub.90.
[0029] FIG. 7: LAM-002+vemurafenib combination in RKO cells (5 day assay). A, bar graph showing cell viability (%). B, combination index (CI) vs fractional effect graph showing synergy determinations at ED.sub.50, ED.sub.75 and ED.sub.90.
[0030] FIG. 8: LAM-002+vemurafenib combination in HT-29 cells (5 day assay). A, bar graph showing cell viability (%). B, combination index (CI) vs fractional effect graph showing synergy determinations at ED.sub.50, ED.sub.75 and ED.sub.90.
[0031] FIG. 9: LAM-002+vemurafenib combination in HCT-15 cells (5 day assay). A, bar graph showing cell viability (%). B, combination index (CI) vs fractional effect graph showing synergy determinations at ED.sub.50, ED.sub.75 and ED.sub.90.
[0032] FIG. 10: LAM-002+vemurafenib combination in SW1116 cells (5 day assay). A, bar graph showing cell viability (%). B, combination index (CI) vs fractional effect graph showing synergy determinations at ED.sub.50, ED.sub.75 and ED.sub.90.
[0033] FIG. 11: LAM-002+vemurafenib combination in SW480 cells (5 day assay). A, bar graph showing cell viability (%). B, combination index (CI) vs fractional effect graph showing synergy determinations at ED.sub.50, ED.sub.75 and ED.sub.90.
[0034] FIG. 12: LAM-002+vemurafenib combination in SW620 cells (5 day assay). A, bar graph showing cell viability (%). B, combination index (CI) vs fractional effect graph showing synergy determinations at ED.sub.50, ED.sub.75 and ED.sub.90.
DETAILED DESCRIPTION OF THE INVENTION
[0035] The present invention provides compositions and methods related to the use of apilimod for treating colorectal cancer in a subject, preferably a human subject, in need of such treatment. The invention generally relates to new uses of apilimod based upon the surprising discovery of apilimod's cytotoxic activity against a range of cancer cells including numerous colorectal cancer cell lines. In addition, the present invention provides novel therapeutic approaches to colorectal cancer treatment based upon combination therapy utilizing apilimod and at least one additional therapeutic agent. The combination therapies described herein exploit the unique cytotoxic activity of apilimod which can provide a synergistic effect when combined with other therapeutic agents, including for example, anti-cancer agents.
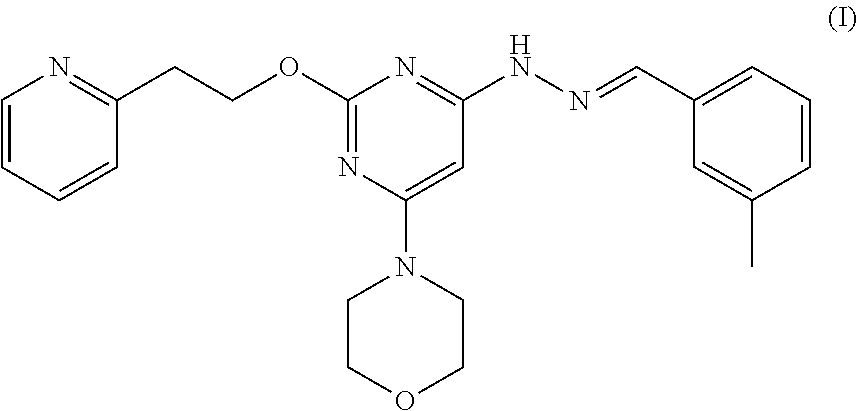
[0036] As used throughout the present disclosure, the term "apilimod" may refer to apilimod itself (free base), or may encompass pharmaceutically acceptable salts, solvates, clathrates, hydrates, polymorphs, prodrugs, analogs or derivatives of apilimod, as described below. In embodiments of the methods for treating colorectal cancer described here, the apilimod is apilimod dimesylate. The structure of apilimod free base is shown in Formula I:
##STR00001##
[0037] The IUPAC name of apilimod is: (E)-4-(6-(2-(3-methylbenzylidene)hydrazinyl)-2-(2-(pyridin-2-yl)ethoxy)py- rimidin-4-yl)morpholine) and the CAS number is 541550-19-0.
[0038] Apilimod can be prepared, for example, according to the methods described in U.S. Pat. Nos. 7,923,557, and 7,863,270, and WO 2006/128129.
[0039] As used herein, the term "pharmaceutically acceptable salt," is a salt formed from, for example, an acid and a basic group of an apilimod composition. Illustrative salts include, but are not limited, to sulfate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, bisulfate, phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate, besylate, gentisinate, fumarate, gluconate, glucaronate, saccharate, formate, benzoate, glutamate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, and pamoate (e.g., 1,1'-methylene-bis-(2-hydroxy-3-naphthoate)) salts.
[0040] The term "pharmaceutically acceptable salt" also refers to a salt prepared from an apilimod composition having an acidic functional group, such as a carboxylic acid functional group, and a pharmaceutically acceptable inorganic or organic base.
[0041] The term "pharmaceutically acceptable salt" also refers to a salt prepared from an apilimod composition having a basic functional group, such as an amino functional group, and a pharmaceutically acceptable inorganic or organic acid.
[0042] The salts of the compounds described herein can be synthesized from the parent compound by conventional chemical methods such as methods described in Pharmaceutical Salts: Properties, Selection, and Use, P. Hemrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, August 2002. Generally, such salts can be prepared by reacting the parent compound with the appropriate acid in water or in an organic solvent, or in a mixture of the two.
[0043] One salt form of a compound described herein can be converted to the free base and optionally to another salt form by methods well known to the skilled person. For example, the free base can be formed by passing the salt solution through a column containing an amine stationary phase (e.g. a Strata-NH.sub.2 column). Alternatively, a solution of the salt in water can be treated with sodium bicarbonate to decompose the salt and precipitate out the free base. The free base may then be combined with another acid using routine methods.
[0044] As used herein, the term "polymorph" means solid crystalline forms of a compound of the present invention (e.g., apilimod) or complex thereof. Different polymorphs of the same compound can exhibit different physical, chemical and/or spectroscopic properties. Different physical properties include, but are not limited to stability (e.g., to heat or light), compressibility and density (important in formulation and product manufacturing), and dissolution rates (which can affect bioavailability). Differences in stability can result from changes in chemical reactivity (e.g., differential oxidation, such that a dosage form discolors more rapidly when comprised of one polymorph than when comprised of another polymorph) or mechanical characteristics (e.g., tablets crumble on storage as a kinetically favored polymorph converts to thermodynamically more stable polymorph) or both (e.g., tablets of one polymorph are more susceptible to breakdown at high humidity). Different physical properties of polymorphs can affect their processing. For example, one polymorph might be more likely to form solvates or might be more difficult to filter or wash free of impurities than another due to, for example, the shape or size distribution of particles of it.
[0045] As used herein, the term "hydrate" means a compound of the present invention (e.g., apilimod) or a salt thereof, which further includes a stoichiometric or non-stoichiometric amount of water bound by non-covalent intermolecular forces.
[0046] As used herein, the term "clathrate" means a compound of the present invention (e.g., apilimod) or a salt thereof in the form of a crystal lattice that contains spaces (e.g., channels) that have a guest molecule (e.g., a solvent or water) trapped within.
[0047] As used herein, the term "prodrug" means a derivative of a compound described herein (e.g., apilimod) that can hydrolyze, oxidize, or otherwise react under biological conditions (in vitro or in vivo) to provide a compound of the invention. Prodrugs may only become active upon such reaction under biological conditions, or they may have activity in their unreacted forms. Examples of prodrugs contemplated in this invention include, but are not limited to, analogs or derivatives of a compound described herein (e.g., apilimod) that comprise biohydrolyzable moieties such as biohydrolyzable amides, biohydrolyzable esters, biohydrolyzable carbamates, biohydrolyzable carbonates, biohydrolyzable ureides, and biohydrolyzable phosphate analogues. Other examples of prodrugs include derivatives of compounds of any one of the formulae disclosed herein that comprise --NO, --NO.sub.2, --ONO, or --ONO.sub.2 moieties. Prodrugs can typically be prepared using well-known methods, such as those described by Burger's Medicinal Chemistry and Drug Discovery (1995) 172-178, 949-982 (Manfred E. Wolff ed., 5th ed).
[0048] As used herein, the term "solvate" or "pharmaceutically acceptable solvate," is a solvate formed from the association of one or more solvent molecules to one of the compounds disclosed herein (e.g., apilimod). The term solvate includes hydrates (e.g., hemi-hydrate, mono-hydrate, dihydrate, trihydrate, tetrahydrate, and the like).
[0049] As used herein, the term "analog" refers to a chemical compound that is structurally similar to another but differs slightly in composition (as in the replacement of one atom by an atom of a different element or in the presence of a particular functional group, or the replacement of one functional group by another functional group). Thus, an analog is a compound that is similar or comparable in function and appearance, but not in structure or origin to the reference compound. As used herein, the term "derivative" refers to compounds that have a common core structure, and are substituted with various groups as described herein.
Colorectal Cancer
[0050] Colorectal cancer (also known as colon cancer, rectal cancer, or bowel cancer) is the development of cancer in the colon or rectum. Colon cancer is staged according to the TNM staging system. The TNM system is one of the most widely used cancer staging systems and has been adopted by the Union for International Cancer Control (UICC) and the American Joint Committee on Cancer (AJCC). The TNM system is based on the size and/or extent (reach) of the primary tumor (T), the amount of spread to nearby lymph nodes (N), and the presence of metastasis (M) or secondary tumors formed by the spread of cancer cells to other parts of the body. A number is added to each letter to indicate the size and/or extent of the primary tumor and the degree of cancer spread.
[0051] In both cancer of the colon and rectum, chemotherapy may be used in addition to surgery in certain cases. The decision to add chemotherapy in management of colon and rectal cancer depends on the stage of the disease and is more common for later stages, e.g., stages III and IV, compared to early stages (I and II) where surgery alone is the standard treatment. For stage III and IV colon cancer (cancer in which the cancer has spread to the lymph nodes or distant organs), fluorouracil, capecitabine or oxaliplatin chemotherapy is standard treatment.
[0052] If the cancer is widely metastatic or unresectable, treatment is then palliative. Typical chemotherapy medications used may include capecitabine, fluorouracil, irinotecan, and oxaliplatin. Anti-angiogenic drugs such as bevacizumab or epidermal growth factor receptor inhibitors, such as cetuximab and panitumumab, may also be used.
Methods of Treatment
[0053] The present invention provides methods for the treatment of colorectal cancer in a subject in need thereof by administering to the subject a therapeutically effective amount of an apilimod composition of the invention, said composition comprising apilimod, or a pharmaceutically acceptable salt, solvate, clathrate, hydrate, polymorph, prodrug, analog or derivative thereof. In one embodiment, the apilimod composition comprises apilimod free base or apilimod dimesylate. The present invention further provides the use of an apilimod composition for the preparation of a medicament useful for the treatment of colorectal cancer.
[0054] In the context of the methods described herein, the amount of an apilimod composition administered to the subject is a therapeutically effective amount. The term "therapeutically effective amount" refers to an amount sufficient to treat, ameliorate a symptom of, reduce the severity of, or stabilize or cause the regression of colorectal cancer in the subject being treated, or to enhance or improve the therapeutic effect of another therapy, such as vemurafenib.
[0055] In accordance with the methods described herein, a "subject in need of" is a subject having colorectal cancer. In one aspect, the subject is a human patient having malignant colorectal cancer or late-stage colorectal cancer. In this context, "stage" refers to the clinical stage of the cancer. For example, stage 0 to 2 colorectal cancer or stage 3 or stage 4 colorectal cancer. In embodiments, the subject is a human patient having stage 3 or stage 4 colorectal cancer. The subject in need of treatment can also be one that is "non-responsive" or "refractory" to a currently available therapy. For example the subject's cancer may be resistant or refractory to treatment with vemurafenib or one or more of oxaliplatin, regorafenib, irinotecan, 5-fluorouracil. In embodiments, the subject in need of treatment is one having a cancer characterized by the V600E or V600K mutation of human BRAF. In the context of the present disclosure, the terms "non-responsive" and "refractory" refer to the subject's response to therapy as not clinically significant according to the definition for a clinical response in standard medical practice.
Combination Therapy
[0056] The present invention also provides methods comprising combination therapy. As used herein, "combination therapy" or "co-therapy" includes the administration of a therapeutically effective amount of an apilimod composition with at least one additional active agent, as part of a specific treatment regimen intended to provide a beneficial effect from the co-action of the apilimod composition and the additional active agent. "Combination therapy" is not intended to encompass the administration of two or more therapeutic compounds as part of separate monotherapy regimens that incidentally and arbitrarily result in a beneficial effect that was not intended or predicted.
[0057] In one embodiment, the method is a method of treating colorectal cancer using a combination therapy comprising apilimod and a chemotherapy regimen for the treatment of colorectal cancer. In embodiments, the chemotherapy regimen comprises one or more of vemurafenib, oxaliplatin, regorafenib, irinotecan, or 5-fluorouracil.
[0058] The at least one additional active agent may be a therapeutic agent, for example an anti-cancer agent or a cancer chemotherapeutic agent, or a non-therapeutic agent, and combinations thereof. With respect to therapeutic agents, the beneficial effect of the combination includes, but is not limited to, pharmacokinetic or pharmacodynamic co-action resulting from the combination of therapeutically active compounds. With respect to non-therapeutic agents, the beneficial effect of the combination may relate to the mitigation of a toxicity, side effect, or adverse event associated with a therapeutically active agent in the combination.
[0059] In one embodiment, the at least one additional agent is a non-therapeutic agent which mitigates one or more side effects of an apilimod composition, the one or more side effects selected from any of nausea, vomiting, headache, dizziness, lightheadedness, drowsiness and stress. In one aspect of this embodiment, the non-therapeutic agent is an antagonist of a serotonin receptor, also known as 5-hydroxytryptamine receptors or 5-HT receptors. In one aspect, the non-therapeutic agent is an antagonist of a 5-HT.sub.3 or 5-HT.sub.1a receptor. In one aspect, the non-therapeutic agent is selected from the group consisting of ondansetron, granisetron, dolasetron and palonosetron. In another aspect, the non-therapeutic agent is selected from the group consisting of pindolol and risperidone.
[0060] In one embodiment, the at least one additional active agent is a therapeutic agent. In one embodiment, the therapeutic agent is an anti-cancer agent. In embodiments, the anti-cancer agent is vemurafenib, oxaliplatin, regorafenib, irinotecan, or 5-fluorouracil, and combinations thereof. In embodiments, the anti-cancer agent is vemurafenib or regoragenib.
[0061] In embodiments, a composition comprising apilimod is administered along with the at least one additional active agent in a single dosage form or in separate dosage forms. In one embodiment, the dosage form is an oral dosage form. In another embodiment, the dosage form is suitable for intravenous administration.
[0062] In one embodiment, the anti-cancer agent is a drug that is approved for use in treating colorectal cancer. Non-limiting examples of such drugs include oxaliplatin, regorafenib, irinotecan, and 5-fluorouracil.
[0063] In embodiments, the anti-cancer agent is selected from vermurafenib, oxaliplatin, regorafenib, irinotecan, and 5-fluorouracil.
[0064] In embodiments, the at least one additional agent is a monoclonal antibody such as, for example, alemtuzumab, bevacizumab, catumaxomab, cetuximab, edrecolomab, gemtuzumab, ofatumumab, panitumumab, rituximab, trastuzumab, eculizumab, efalizumab, muromab-CD3, natalizumab, adalimumab, afelimomab, certolizumab pegol, golimumab, infliximab, basiliximab, canakinumab, daclizumab, mepolizumab, tocilizumab, ustekinumab, ibritumomab tiuxetan, tositumomab, abagovomab, adecatumumab, alemtuzumab, anti-CD30 monoclonal antibody Xmab2513, anti-MET monoclonal antibody MetMab, apolizumab, apomab, arcitumomab, basiliximab, bispecific antibody 2B1, blinatumomab, brentuximab vedotin, capromab pendetide, cixutumumab, claudiximab, conatumumab, dacetuzumab, denosumab, eculizumab, epratuzumab, ertumaxomab, etaracizumab, figitumumab, fresolimumab, galiximab, ganitumab, gemtuzumab ozogamicin, glembatumumab, ibritumomab, inotuzumab ozogamicin, ipilimumab, lexatumumab, lintuzumab, lintuzumab, lucatumumab, mapatumumab, matuzumab, milatuzumab, monoclonal antibody CC49, necitumumab, nimotuzumab, ofatumumab, oregovomab, pertuzumab, ramacurimab, ranibizumab, siplizumab, sonepcizumab, tanezumab, tositumomab, trastuzumab, tremelimumab, tucotuzumab celmoleukin, veltuzumab, visilizumab, volociximab, and zalutumumab.
[0065] In embodiments, the at least one additional inhibitor is a BRAF inhibitor, a MEK inhibitor, PD-1/PDL-1 pathway inhibitor, or a check point inhibitor. In embodiments, the PD-1/PDL-1 pathway inhibitor is selected from pembrolizumab (Keytruda), avelumab, atezolizumab (MPDL3280A), nivolumab (BMS-936558), pidilizumab (MK-3475), MSB0010718C, and MEDI4736.
[0066] In the context of combination therapy, administration of the apilimod may be simultaneous with or sequential to the administration of the one or more additional active agents. In another embodiment, administration of the different components of a combination therapy may be at different frequencies. The one or more additional agents may be administered prior to (e.g., 5 minutes, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks before), concomitantly with, or subsequent to (e.g., 5 minutes, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks after) the administration of a compound of the present invention.
[0067] The one or more additional active agents can be formulated for co-administration with apilimod in a single dosage form, as described in greater detail herein. The one or more additional active agents can be administered separately from the dosage form that comprises the compound of the present invention. When the additional active agent is administered separately from the apilimod composition, it can be by the same or a different route of administration as the apilimod composition.
[0068] Preferably, the administration of apilimod in combination with one or more additional agents provides a synergistic response in the subject being treated. In this context, the term "synergistic" refers to the efficacy of the combination being more effective than the additive effects of either single therapy alone. The synergistic effect of a combination therapy according to the invention can permit the use of lower dosages and/or less frequent administration of at least one agent in the combination compared to its dose and/or frequency outside of the combination. Additional beneficial effects of the combination can be manifested in the avoidance or reduction of adverse or unwanted side effects associated with the use of either therapy in the combination alone (also referred to as monotherapy).
[0069] "Combination therapy" also embraces the administration of the compounds of the present invention in further combination with non-drug therapies (e.g., surgery or radiation treatment). Where the combination therapy further comprises a non-drug treatment, the non-drug treatment may be conducted at any suitable time so long as a beneficial effect from the co-action of the combination of the therapeutic compounds and non-drug treatment is achieved. For example, in appropriate cases, the beneficial effect is still achieved when the non-drug treatment is temporally removed from the administration of the therapeutic compounds, perhaps by days or even weeks.
[0070] In accordance with any of the methods described herein, a therapeutically effective amount of apilimod, e.g., apilimod dimesylate, can range from about 0.001 mg/kg to about 1000 mg/kg, about 0.01 mg/kg to about 100 mg/kg, about 10 mg/kg to about 250 mg/kg, about 0.1 mg/kg to about 15 mg/kg; or any range in which the low end of the range is any amount between 0.001 mg/kg and 900 mg/kg and the upper end of the range is any amount between 0.1 mg/kg and 1000 mg/kg (e.g., 0.005 mg/kg and 200 mg/kg, 0.5 mg/kg and 20 mg/kg). Effective doses will also vary, as recognized by those skilled in the art, depending on the diseases treated, route of administration, excipient usage, and the possibility of co-usage with other therapeutic treatments such as use of other agents. See, e.g., U.S. Pat. No. 7,863,270, incorporated herein by reference.
[0071] In more specific aspects, apilimod, e.g., apilimod dimesylate, is administered at a dosage regimen of 30-1000 mg/day (e.g., 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 125, 150, 175, 200, 225, 250, 275, or 300 mg/day) for at least 1 week (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 36, 48, or more weeks). Preferably, apilimod is administered at a dosage regimen of 100-1000 mg/day for 4 or 16 weeks. Alternatively or subsequently, apilimod is administered at a dosage regimen of 100 mg-300 mg twice a day for 8 weeks, or optionally, for 52 weeks. Alternatively or subsequently, apilimod is administered at a dosage regimen of 50 mg-1000 mg twice a day for 8 weeks, or optionally, for 52 weeks.
[0072] A therapeutically effective amount of the apilimod can be administered once daily, from two to five times daily, up to two times or up to three times daily, or up to eight times daily. In one embodiment, the apilimod is administered thrice daily, twice daily, once daily, fourteen days on (four times daily, thrice daily or twice daily, or once daily) and 7 days off in a 3-week cycle, up to five or seven days on (four times daily, thrice daily or twice daily, or once daily) and 14-16 days off in 3 week cycle, or once every two days, or once a week, or once every 2 weeks, or once every 3 weeks.
[0073] A "subject" as used in the context of the methods described herein is preferably a human subject but may also include other mammals. The mammal can be e.g., any mammal, e.g., a human, primate, vertebrate, bird, mouse, rat, fowl, dog, cat, cow, horse, goat, camel, sheep or a pig. The term "patient" refers to a human subject.
[0074] The present invention also provides a monotherapy for the treatment of colorectal cancer as described herein. As used herein, "monotherapy" refers to the administration of a single active agent (also referred to as the therapeutic agent), e.g., apilimod, and in embodiments, apilimod dimesylate, to a subject in need thereof.
[0075] As used herein, "treatment", "treating" or "treat" describes the management and care of a patient for the purpose of combating the colorectal cancer and includes alleviating one or more symptoms or complications of the cancer, including for example slowing the growth of the cancer, slowing or preventing the occurrence of metastases, or further metastases, and promoting regression of one or more tumors in the subject being treated.
[0076] In embodiments, the administration of a composition as described herein leads to the elimination of a symptom or complication of the cancer being treated, however, elimination is not required. In one embodiment, the severity of the symptom is decreased. In the context of cancer, such symptoms may include clinical markers of severity or progression including the degree to which a tumor secrets growth factors, degrades the extracellular matrix, becomes vascularized, loses adhesion to juxtaposed tissues, or metastasizes, as well as the number of metastases.
[0077] Treating colorectal cancer according to the methods described herein can result in a reduction in size of a tumor. A reduction in size of a tumor may also be referred to as "tumor regression". Preferably, after treatment, tumor size is reduced by 5% or greater relative to its size prior to treatment; more preferably, tumor size is reduced by 10% or greater; more preferably, reduced by 20% or greater; more preferably, reduced by 30% or greater; more preferably, reduced by 40% or greater; even more preferably, reduced by 50% or greater; and most preferably, reduced by greater than 75% or greater. Size of a tumor may be measured by any reproducible means of measurement. The size of a tumor may be measured as a diameter of the tumor.
[0078] Treating colorectal cancer according to the methods described herein can result in a reduction in tumor volume. Preferably, after treatment, tumor volume is reduced by 5% or greater relative to its size prior to treatment; more preferably, tumor volume is reduced by 10% or greater; more preferably, reduced by 20% or greater; more preferably, reduced by 30% or greater; more preferably, reduced by 40% or greater; even more preferably, reduced by 50% or greater; and most preferably, reduced by greater than 75% or greater. Tumor volume may be measured by any reproducible means of measurement.
[0079] Treating colorectal cancer according to the methods described herein can result in a decrease in number of tumors. Preferably, after treatment, tumor number is reduced by 5% or greater relative to number prior to treatment; more preferably, tumor number is reduced by 10% or greater; more preferably, reduced by 20% or greater; more preferably, reduced by 30% or greater; more preferably, reduced by 40% or greater; even more preferably, reduced by 50% or greater; and most preferably, reduced by greater than 75%. Number of tumors may be measured by any reproducible means of measurement. The number of tumors may be measured by counting tumors visible to the naked eye or at a specified magnification. Preferably, the specified magnification is 2.times., 3.times., 4.times., 5.times., 10.times., or 50.times..
[0080] Treating colorectal cancer according to the methods described herein can result in a decrease in number of metastatic lesions in other tissues or organs distant from the primary tumor site. Preferably, after treatment, the number of metastatic lesions is reduced by 5% or greater relative to number prior to treatment; more preferably, the number of metastatic lesions is reduced by 10% or greater; more preferably, reduced by 20% or greater; more preferably, reduced by 30% or greater; more preferably, reduced by 40% or greater; even more preferably, reduced by 50% or greater; and most preferably, reduced by greater than 75%. The number of metastatic lesions may be measured by any reproducible means of measurement. The number of metastatic lesions may be measured by counting metastatic lesions visible to the naked eye or at a specified magnification. Preferably, the specified magnification is 2.times., 3.times., 4.times., 5.times., 10.times., or 50.times..
[0081] Treating colorectal cancer according to the methods described herein can result in an increase in average survival time of a population of treated subjects in comparison to a population receiving carrier alone. Preferably, the average survival time is increased by more than 30 days; more preferably, by more than 60 days; more preferably, by more than 90 days; and most preferably, by more than 120 days. An increase in average survival time of a population may be measured by any reproducible means. An increase in average survival time of a population may be measured, for example, by calculating for a population the average length of survival following initiation of treatment with an active compound. An increase in average survival time of a population may also be measured, for example, by calculating for a population the average length of survival following completion of a first round of treatment with an active compound.
[0082] Treating colorectal cancer according to the methods described herein can result in increase in average survival time of a population of treated subjects in comparison to a population receiving monotherapy with a drug that is not an apilimod composition as described herein. Preferably, the average survival time is increased by more than 30 days; more preferably, by more than 60 days; more preferably, by more than 90 days; and most preferably, by more than 120 days. An increase in average survival time of a population may be measured by any reproducible means. An increase in average survival time of a population may be measured, for example, by calculating for a population the average length of survival following initiation of treatment with an active compound. An increase in average survival time of a population may also be measured, for example, by calculating for a population the average length of survival following completion of a first round of treatment with an active compound.
[0083] Treating colorectal cancer according to the methods described herein can result in a decrease in the mortality rate of a population of treated subjects in comparison to a population receiving carrier alone. Treating colorectal cancer according to the methods described herein can result in a decrease in the mortality rate of a population of treated subjects in comparison to an untreated population. Treating colorectal cancer according to the methods described herein can result in a decrease in the mortality rate of a population of treated subjects in comparison to a population receiving monotherapy with a drug that is not an apilimod composition. Preferably, the mortality rate is decreased by more than 2%; more preferably, by more than 5%; more preferably, by more than 10%; and most preferably, by more than 25%. A decrease in the mortality rate of a population of treated subjects may be measured by any reproducible means. A decrease in the mortality rate of a population may be measured, for example, by calculating for a population the average number of disease-related deaths per unit time following initiation of treatment with an active compound. A decrease in the mortality rate of a population may also be measured, for example, by calculating for a population the average number of disease-related deaths per unit time following completion of a first round of treatment with an active compound.
[0084] Treating colorectal cancer according to the methods described herein can result in a decrease in tumor growth rate. Preferably, after treatment, tumor growth rate is reduced by at least 5% relative to number prior to treatment; more preferably, tumor growth rate is reduced by at least 10%; more preferably, reduced by at least 20%; more preferably, reduced by at least 30%; more preferably, reduced by at least 40%; more preferably, reduced by at least 50%; even more preferably, reduced by at least 50%; and most preferably, reduced by at least 75%. Tumor growth rate may be measured by any reproducible means of measurement. Tumor growth rate can be measured according to a change in tumor diameter per unit time. In one embodiment, after treatment the tumor growth rate may be about zero and is determined to maintain the same size, e.g., has stopped growing.
[0085] Treating a colorectal cancer according to the methods described herein can result in a decrease in tumor regrowth. Preferably, after treatment, tumor regrowth is less than 5%; more preferably, tumor regrowth is less than 10%; more preferably, less than 20%; more preferably, less than 30%; more preferably, less than 40%; more preferably, less than 50%; even more preferably, less than 50%; and most preferably, less than 75%. Tumor regrowth may be measured by any reproducible means of measurement. Tumor regrowth is measured, for example, by measuring an increase in the diameter of a tumor after a prior tumor shrinkage that followed treatment. A decrease in tumor regrowth is indicated by failure of tumors to reoccur after treatment has stopped.
[0086] As used herein, the term "selectively" means tending to occur at a higher frequency in one population than in another population. The compared populations can be cell populations. Preferably, an apilimod composition as described herein acts selectively on hyper-proliferating cells or abnormally proliferating cells, compared to normal cells. As used herein, a "normal cell" is a cell that cannot be classified as part of a "cell proliferative disorder". A normal cell lacks unregulated or abnormal growth, or both, that can lead to the development of an unwanted condition or disease. Preferably, a normal cell possesses normally functioning cell cycle checkpoint control mechanisms. Preferably, an apilimod composition acts selectively to modulate one molecular target (e.g., a target kinase) but does not significantly modulate another molecular target (e.g., a non-target kinase). The invention also provides a method for selectively inhibiting the activity of an enzyme, such as a kinase. Preferably, an event occurs selectively in population A relative to population B if it occurs greater than two times more frequently in population A as compared to population B. An event occurs selectively if it occurs greater than five times more frequently in population A. An event occurs selectively if it occurs greater than ten times more frequently in population A; more preferably, greater than fifty times; even more preferably, greater than 100 times; and most preferably, greater than 1000 times more frequently in population A as compared to population B. For example, cell death would be said to occur selectively in diseased or hyper-proliferating cells if it occurred greater than twice as frequently in diseased or hyper-proliferating cells as compared to normal cells.
Pharmaceutical Compositions and Formulations
[0087] The present invention provides apilimod compositions that are preferably pharmaceutically acceptable compositions suitable for use in a mammal, preferably a human. In this context, the compositions may further comprise at least one pharmaceutically acceptable excipient or carrier, wherein the amount is effective for the treatment of colorectal cancer.
[0088] In one embodiment, the apilimod composition comprises apilimod free base or apilimod dimesylate.
[0089] In one embodiment, the apilimod composition is combined with at least one additional active agent in a single dosage form. In one embodiment, the composition further comprises an antioxidant.
[0090] In one embodiment, the at least one additional active agent is selected from the group consisting of an alkylating agent, an intercalating agent, a tubulin binding agent, a PD-1/PDL-1 pathway inhibitor, a corticosteroid, and combinations thereof. In embodiments, the at least one additional active agent is a therapeutic agent selected from the group consisting of vemurafenib, ibrutinib, rituximab, doxorubicin, prednisolone, vincristine, velcade, and everolimus, and combinations thereof. In embodiments, the at least one additional active agent is a therapeutic agent selected from the group consisting of vemurafenib, oxaliplatin, regorafenib, irinotecan, and 5-fluorouracil. In embodiments, the at least one additional active agent is a therapeutic agent selected from the group consisting of pembrolizumab (Keytruda), avelumab, atezolizumab (MPDL3280A), nivolumab (BMS-936558), pidilizumab (MK-3475), MSB0010718C, and MEDI4736.
[0091] In embodiments, the at least one additional active agent is a therapeutic agent selected from the group consisting of dacarbazine, temozolomide, Nab-paclitaxel, carmustine, cisplatin, carboplatin, vinblastine, ipilimumab, Interleukin-2 (IL-2, Proleukin), pembrolizumab (Keytruda), dabrafenib (Tafinlar), vemurafenib (Zelboraf), trametinib (Mekinist), dasatinib (Sprycel), imatinib (Gleevec), and nilotinib (Tasigna) and combinations thereof.
[0092] In embodiments, the at least one additional active agent is a non-therapeutic agent selected to ameliorate one or more side effects of the apilimod composition. In one embodiment, the non-therapeutic agent is selected from the group consisting of ondansetron, granisetron, dolasetron and palonosetron. In one embodiment, the non-therapeutic agent is selected from the group consisting of pindolol and risperidone.
[0093] In embodiments, the at least one additional active agent is selected from an inhibitor of BRAF, an inhibitor of the Raf/MEK/ERK pathway, an inhibitor of the mTOR pathway, a PI3K inhibitor, a dual PI3K/mTOR inhibitor, a SRC inhibitor, a VEGF inhibitor, a Janus kinase (JAK) inhibitor, a Raf inhibitor, an Erk inhibitor, a farnesyltransferase inhibitor, a histone deacetylase inhibitor, an anti-mitotic agent, a multi-drug resistance efflux inhibitor, an antibiotic, and a therapeutic antibody. In one embodiment, the at least one additional active agent is selected from a farnesyltransferase inhibitor (e.g., tipifarnib), an anti-mitotic agent (e.g., docetaxel), a histone deacetylase inhibitor (e.g., vorinostat), and a multi-drug resistance efflux inhibitor.
[0094] In one embodiment, the mTOR inhibitor is selected from the group consisting of rapamycin (also referred to as sirolimus), everolimus, temsirolimus, ridaforolimus, umirolimus, zotarolimus, AZD8055, INK128, WYE-132, Torin-1, pyrazolopyrimidine analogs PP242, PP30, PP487, PP121, KU0063794, KU-BMCL-200908069-1, Wyeth-BMCL-200910075-9b, INK-128, XL388, AZD8055, P2281, and P529. See, e.g., Liu et al. Drug Disc. Today Ther. Strateg., 6(2): 47-55 (2009).
[0095] In one embodiment, the mTOR inhibitor is trans-4-[4-amino-5-(7-methoxy-1H-indol-2-yeimidazo[5,1-f][1,2,4]triazin-7- -yl]cyclohexane carboxylic acid (also known as OSI-027), and any salts, solvates, hydrates, and other physical forms, crystalline or amorphous, thereof. See US 2007/0112005. OSI-027 can be prepared according to US 2007/0112005, incorporated herein by reference. In one embodiment, the mTOR inhibitor is OXA-01. See e.g., WO 2013152342 A1.
[0096] In one embodiment, the PI3K inhibitor is selected from the group consisting of GS-1101 (Idelalisib), GDC0941 (Pictilisib), LY294002, BKM120 (Buparlisib), PI-103, TGX-221, IC-87114, XL 147, ZSTK474, BYL719, AS-605240, PIK-75, 3-methyladenine, A66, PIK-93, PIK-90, AZD6482, IPI-145 (Duvelisib), TG100-115, AS-252424, PIK294, AS-604850, GSK2636771, BAY 80-6946 (Copanlisib), CH5132799, CAY10505, PIK-293, TG100713, CZC24832 and HS-173.
[0097] In one embodiment, the dual PI3K/mTOR inhibitor is selected from the group consisting of, GDC-094, WAY-001, WYE-354, WAY-600, WYE-687, Wyeth-BMCL-200910075-16b, Wyeth-BMCL-200910096-27, KU0063794 and KUBMCL-200908069-5, NVP-BEZ235, XL-765, PF-04691502, GDC-0980 (Apitolisib), GSK1059615, PF-05212384, BGT226, PKI-402, VS-558 and GSK2126458. See, e.g., Liu et al. Drug Disc. Today Ther. Strateg., 6(2): 47-55 (2009), incorporated herein by reference.
[0098] In one embodiment, the mTOR pathway inhibitor is a polypeptide (e.g., an antibody or fragment thereof) or a nucleic acid (e.g., a double-stranded small interfering RNA, a short hairpin RNA, a micro-RNA, an antisense oligonucleotide, a locked nucleic acid, or an aptamer) that binds to and inhibits the expression level or activity or a protein (or nucleic acid encoding the protein) in the mTOR pathway. For example, the polypeptide or nucleic acid inhibits mTOR Complex 1 (mTORC1), regulatory-associated protein of mTOR (Raptor), mammalian lethal with SEC13 protein 8 (MLST8), proline-rich Akt substrate of 40 kDa (PRAS40), DEP domain-containing mTOR-interacting protein (DEPTOR), mTOR Complex 2 (mTORC2), rapamycin-insensitive companion of mTOR (RICTOR), G protein beta subunit-like (G.beta.L), mammalian stress-activated protein kinase interacting protein 1 (mSIN1), paxillin, RhoA, Ras-related C3 botulinum toxin substrate 1 (Rac1), Cell division control protein 42 homolog (Cdc42), protein kinase C .alpha. (PKC.alpha.), the serine/threonine protein kinase Akt, phosphoinositide 3-kinase (PI3K), p70S6K, Ras, and/or eukaryotic translation initiation factor 4E (eIF4E)-binding proteins (4EBPs), or the nucleic acid encoding one of these proteins.
[0099] In one embodiment, the SRC inhibitor is selected from the group consisting of bosutinib, saracatinib, dasatinib, ponatinib, KX2-391, XL-228, TG100435/TG100855, and DCC2036. See, e.g., Puls et al. Oncologist. 2011 May; 16(5): 566-578. In one embodiment, the SRC inhibitor is a polypeptide (e.g., an antibody or fragment thereof) or nucleic acid (e.g., a double-stranded small interfering RNA, a short hairpin RNA, a micro-RNA, an antisense oligonucleotide, a locked nucleic acid, or an aptamer) that binds to and inhibits the expression level or activity of the SRC protein or a nucleic acid encoding the SRC protein.
[0100] In one embodiment, the VEGF inhibitor is selected from bevacizumab, sunitinib, pazopanib, axitinib, sorafenib, regorafenib, lenvatinib, and motesanib. In one embodiment, the VEGF inhibitor is a polypeptide (e.g., an antibody or fragment thereof) or nucleic acid (e.g., a double-stranded small interfering RNA, a short hairpin RNA, a micro-RNA, an antisense oligonucleotide, a morpholino, a locked nucleic acid, or an aptamer) that binds to and inhibits the expression level or activity of a VEGF protein, a VEGF receptor protein, or a nucleic acid encoding one of these proteins. For example, the VEGF inhibitor is a soluble VEGF receptor (e.g., a soluble VEGF-C/D receptor (sVEGFR-3)).
[0101] In one embodiment, the JAK inhibitor is selected from facitinib, ruxolitinib, baricitinib, CYT387 (CAS number 1056634-68-4), lestaurtinib, pacritinib, and TG101348 (CAS number 936091-26-8). In one embodiment, the JAK inhibitor is a polypeptide (e.g., an antibody or fragment thereof) or nucleic acid (e.g., a double-stranded small interfering RNA, a short hairpin RNA, a micro-RNA, an antisense oligonucleotide, a morpholino, a locked nucleic acid, or an aptamer) that binds to and inhibits the expression level or activity of a JAK (e.g., JAK1, JAK2, JAK3, or TYK2) or a nucleic acid encoding the JAK protein.
[0102] In one embodiment, the Raf inhibitor is selected from PLX4032 (vemurafenib), sorafenib, PLX-4720, GSK2118436 (dabrafenib), GDC-0879, RAF265, AZ 628, NVP-BHG712, SB90885, ZM 336372, GW5074, TAK-632, CEP-32496 and LGX818 (Encorafenib). In one embodiment, the Raf inhibitor is a polypeptide (e.g., an antibody or fragment thereof) or nucleic acid (e.g., a double-stranded small interfering RNA, a short hairpin RNA, a micro-RNA, an antisense oligonucleotide, a morpholino, a locked nucleic acid, or an aptamer) that binds to and inhibits the expression level or activity of a Raf (e.g., A-Raf, B-Raf, C-Raf) or a nucleic acid encoding the Raf protein. In one embodiment, the MEK inhibitor is selected from AZD6244 (Selumetinib), PD0325901, GSK1120212 (Trametinib), U0126-EtOH, PD184352, RDEA119 (Rafametinib), PD98059, BIX 02189, MEK162 (Binimetinib), AS-703026 (Pimasertib), SL-327, BIX02188, AZD8330, TAK-733 and PD318088. In one embodiment, the MEK inhibitor is a polypeptide (e.g., an antibody or fragment thereof) or nucleic acid (e.g., a double-stranded small interfering RNA, a short hairpin RNA, a micro-RNA, an antisense oligonucleotide, a morpholino, a locked nucleic acid, or an aptamer) that binds to and inhibits the expression level or activity of a MEK (e.g., MEK-1, MEK-2) or a nucleic acid encoding the MEK protein.
[0103] In one embodiment, the Akt inhibitor is selected from MK-2206, KRX-0401 (perifosine), GSK690693, GDC-0068 (Ipatasertib), AZD5363, CCT128930, A-674563, PHT-427. In one embodiment, the Akt inhibitor is a polypeptide (e.g., an antibody or fragment thereof) or nucleic acid (e.g., a double-stranded small interfering RNA, a short hairpin RNA, a micro-RNA, an antisense oligonucleotide, a morpholino, a locked nucleic acid, or an aptamer) that binds to and inhibits the expression level or activity of a Akt (e.g., Akt-1, Akt-2, Akt-3) or a nucleic acid encoding the Akt protein.
[0104] In one embodiment, the farnesyltransferase inhibitor is selected from LB42708 or tipifarnib. In one embodiment, the farnesyltransferase inhibitor is a polypeptide (e.g., an antibody or fragment thereof) or nucleic acid (e.g., a double-stranded small interfering RNA, a short hairpin RNA, a micro-RNA, an antisense oligonucleotide, a morpholino, a locked nucleic acid, or an aptamer) that binds to and inhibits the expression level or activity of farnesyltransferase or a nucleic acid encoding the farnesyltransferase protein. In one embodiment, the histone modulating inhibitor is selected from anacardic acid, C646, MG149 (histone acetyltransferase), GSK J4 Hcl (histone demethylase), GSK343 (active against EZH2), BIX 01294 (histone methyltransferase), MK0683 (Vorinostat), MS275 (Entinostat), LBH589 (Panobinostat), Trichostatin A, MGCD0103 (Mocetinostat), Tasquinimod, TMP269, Nexturastat A, RG2833, PDX101 (Belinostat).
[0105] In one embodiment, the anti-mitotic agent is selected from Griseofulvin, vinorelbine tartrate, paclitaxel, docetaxel, vincristine, vinblastine, Epothilone A, Epothilone B, ABT-751, CYT997 (Lexibulin), vinflunine tartrate, Fosbretabulin, GSK461364, ON-01910 (Rigosertib), Ro3280, BI2536, NMS-P937, BI 6727 (Volasertib), HMN-214 and MLN0905.
[0106] In one embodiment, the polyether antibiotic is selected from sodium monensin, nigericin, valinomycin, salinomycin.
[0107] A "pharmaceutical composition" is a formulation containing the compounds described herein in a pharmaceutically acceptable form suitable for administration to a subject. As used herein, the phrase "pharmaceutically acceptable" refers to those compounds, materials, compositions, carriers, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
[0108] "Pharmaceutically acceptable excipient" means an excipient that is useful in preparing a pharmaceutical composition that is generally safe, non-toxic and neither biologically nor otherwise undesirable, and includes excipient that is acceptable for veterinary use as well as human pharmaceutical use. Examples of pharmaceutically acceptable excipients include, without limitation, sterile liquids, water, buffered saline, ethanol, polyol (for example, glycerol, propylene glycol, liquid polyethylene glycol and the like), oils, detergents, suspending agents, carbohydrates (e.g., glucose, lactose, sucrose or dextran), antioxidants (e.g., ascorbic acid or glutathione), chelating agents, low molecular weight proteins, or suitable mixtures thereof.
[0109] A pharmaceutical composition can be provided in bulk or in dosage unit form. It is especially advantageous to formulate pharmaceutical compositions in dosage unit form for ease of administration and uniformity of dosage. The term "dosage unit form" as used herein refers to physically discrete units suited as unitary dosages for the subject to be treated; each unit containing a predetermined quantity of active compound calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier. The specification for the dosage unit forms of the invention are dictated by and directly dependent on the unique characteristics of the active compound and the particular therapeutic effect to be achieved. A dosage unit form can be an ampoule, a vial, a suppository, a dragee, a tablet, a capsule, an IV bag, or a single pump on an aerosol inhaler.
[0110] In therapeutic applications, the dosages vary depending on the agent, the age, weight, and clinical condition of the recipient patient, and the experience and judgment of the clinician or practitioner administering the therapy, among other factors affecting the selected dosage. Generally, the dose should be a therapeutically effective amount. Dosages can be provided in mg/kg/day units of measurement (which dose may be adjusted for the patient's weight in kg, body surface area in m.sup.2, and age in years). An effective amount of a pharmaceutical composition is that which provides an objectively identifiable improvement as noted by the clinician or other qualified observer. For example, alleviating a symptom of a disorder, disease or condition. As used herein, the term "dosage effective manner" refers to amount of a pharmaceutical composition to produce the desired biological effect in a subject or cell.
[0111] For example, the dosage unit form can comprise 1 nanogram to 2 milligrams, or 0.1 milligrams to 2 grams; or from 10 milligrams to 1 gram, or from 50 milligrams to 500 milligrams or from 1 microgram to 20 milligrams; or from 1 microgram to 10 milligrams; or from 0.1 milligrams to 2 milligrams.
[0112] The pharmaceutical compositions can take any suitable form (e.g, liquids, aerosols, solutions, inhalants, mists, sprays; or solids, powders, ointments, pastes, creams, lotions, gels, patches and the like) for administration by any desired route (e.g, pulmonary, inhalation, intranasal, oral, buccal, sublingual, parenteral, subcutaneous, intravenous, intramuscular, intraperitoneal, intrapleural, intrathecal, transdermal, transmucosal, rectal, and the like). For example, a pharmaceutical composition of the invention may be in the form of an aqueous solution or powder for aerosol administration by inhalation or insufflation (either through the mouth or the nose), in the form of a tablet or capsule for oral administration; in the form of a sterile aqueous solution or dispersion suitable for administration by either direct injection or by addition to sterile infusion fluids for intravenous infusion; or in the form of a lotion, cream, foam, patch, suspension, solution, or suppository for transdermal or transmucosal administration.
[0113] A pharmaceutical composition can be in the form of an orally acceptable dosage form including, but not limited to, capsules, tablets, buccal forms, troches, lozenges, and oral liquids in the form of emulsions, aqueous suspensions, dispersions or solutions. Capsules may contain mixtures of a compound of the present invention with inert fillers and/or diluents such as the pharmaceutically acceptable starches (e.g., corn, potato or tapioca starch), sugars, artificial sweetening agents, powdered celluloses, such as crystalline and microcrystalline celluloses, flours, gelatins, gums, etc. In the case of tablets for oral use, carriers which are commonly used include lactose and corn starch. Lubricating agents, such as magnesium stearate, can also be added. For oral administration in a capsule form, useful diluents include lactose and dried corn starch. When aqueous suspensions and/or emulsions are administered orally, the compound of the present invention may be suspended or dissolved in an oily phase is combined with emulsifying and/or suspending agents. If desired, certain sweetening and/or flavoring and/or coloring agents may be added.
[0114] A pharmaceutical composition can be in the form of a tablet. The tablet can comprise a unit dosage of a compound of the present invention together with an inert diluent or carrier such as a sugar or sugar alcohol, for example lactose, sucrose, sorbitol or mannitol. The tablet can further comprise a non-sugar derived diluent such as sodium carbonate, calcium phosphate, calcium carbonate, or a cellulose or derivative thereof such as methyl cellulose, ethyl cellulose, hydroxypropyl methyl cellulose, and starches such as corn starch. The tablet can further comprise binding and granulating agents such as polyvinylpyrrolidone, disintegrants (e.g. swellable crosslinked polymers such as crosslinked carboxymethylcellulose), lubricating agents (e.g. stearates), preservatives (e.g. parabens), antioxidants (e.g. BHT), buffering agents (for example phosphate or citrate buffers), and effervescent agents such as citrate/bicarbonate mixtures.
[0115] The tablet can be a coated tablet. The coating can be a protective film coating (e.g. a wax or varnish) or a coating designed to control the release of the active agent, for example a delayed release (release of the active after a predetermined lag time following ingestion) or release at a particular location in the gastrointestinal tract. The latter can be achieved, for example, using enteric film coatings such as those sold under the brand name Eudragit.RTM..
[0116] Tablet formulations may be made by conventional compression, wet granulation or dry granulation methods and utilize pharmaceutically acceptable diluents, binding agents, lubricants, disintegrants, surface modifying agents (including surfactants), suspending or stabilizing agents, including, but not limited to, magnesium stearate, stearic acid, talc, sodium lauryl sulfate, microcrystalline cellulose, carboxymethylcellulose calcium, polyvinylpyrrolidone, gelatin, alginic acid, acacia gum, xanthan gum, sodium citrate, complex silicates, calcium carbonate, glycine, dextrin, sucrose, sorbitol, dicalcium phosphate, calcium sulfate, lactose, kaolin, mannitol, sodium chloride, talc, dry starches and powdered sugar. Preferred surface modifying agents include nonionic and anionic surface modifying agents. Representative examples of surface modifying agents include, but are not limited to, poloxamer 188, benzalkonium chloride, calcium stearate, cetostearyl alcohol, cetomacrogol emulsifying wax, sorbitan esters, colloidal silicon dioxide, phosphates, sodium dodecylsulfate, magnesium aluminum silicate, and triethanolamine.
[0117] A pharmaceutical composition can be in the form of a hard or soft gelatin capsule. In accordance with this formulation, the compound of the present invention may be in a solid, semi-solid, or liquid form.
[0118] A pharmaceutical composition can be in the form of a sterile aqueous solution or dispersion suitable for parenteral administration. The term parenteral as used herein includes subcutaneous, intracutaneous, intravenous, intramuscular, intra-articular, intraarterial, intrasynovial, intrasternal, intrathecal, intralesional and intracranial injection or infusion techniques.
[0119] A pharmaceutical composition can be in the form of a sterile aqueous solution or dispersion suitable for administration by either direct injection or by addition to sterile infusion fluids for intravenous infusion, and comprises a solvent or dispersion medium containing, water, ethanol, a polyol (e.g., glycerol, propylene glycol and liquid polyethylene glycol), suitable mixtures thereof, or one or more vegetable oils. Solutions or suspensions of the compound of the present invention as a free base or pharmacologically acceptable salt can be prepared in water suitably mixed with a surfactant. Examples of suitable surfactants are given below. Dispersions can also be prepared, for example, in glycerol, liquid polyethylene glycols and mixtures of the same in oils.
[0120] The pharmaceutical compositions for use in the methods of the present invention can further comprise one or more additives in addition to any carrier or diluent (such as lactose or mannitol) that is present in the formulation. The one or more additives can comprise or consist of one or more surfactants. Surfactants typically have one or more long aliphatic chains such as fatty acids which enables them to insert directly into the lipid structures of cells to enhance drug penetration and absorption. An empirical parameter commonly used to characterize the relative hydrophilicity and hydrophobicity of surfactants is the hydrophilic-lipophilic balance ("HLB" value). Surfactants with lower HLB values are more hydrophobic, and have greater solubility in oils, while surfactants with higher HLB values are more hydrophilic, and have greater solubility in aqueous solutions. Thus, hydrophilic surfactants are generally considered to be those compounds having an HLB value greater than about 10, and hydrophobic surfactants are generally those having an HLB value less than about 10. However, these HLB values are merely a guide since for many surfactants, the HLB values can differ by as much as about 8 HLB units, depending upon the empirical method chosen to determine the HLB value.
[0121] Among the surfactants for use in the compositions of the invention are polyethylene glycol (PEG)-fatty acids and PEG-fatty acid mono and diesters, PEG glycerol esters, alcohol-oil transesterification products, polyglyceryl fatty acids, propylene glycol fatty acid esters, sterol and sterol derivatives, polyethylene glycol sorbitan fatty acid esters, polyethylene glycol alkyl ethers, sugar and its derivatives, polyethylene glycol alkyl phenols, polyoxyethylene-polyoxypropylene (POE-POP) block copolymers, sorbitan fatty acid esters, ionic surfactants, fat-soluble vitamins and their salts, water-soluble vitamins and their amphiphilic derivatives, amino acids and their salts, and organic acids and their esters and anhydrides.
[0122] The present invention also provides packaging and kits comprising pharmaceutical compositions for use in the methods of the present invention. The kit can comprise one or more containers selected from the group consisting of a bottle, a vial, an ampoule, a blister pack, and a syringe. The kit can further include one or more of instructions for use in treating and/or preventing a disease, condition or disorder of the present invention, one or more syringes, one or more applicators, or a sterile solution suitable for reconstituting a pharmaceutical composition of the present invention.
[0123] All percentages and ratios used herein, unless otherwise indicated, are by weight. Other features and advantages of the present invention are apparent from the different examples. The provided examples illustrate different components and methodology useful in practicing the present invention. The examples do not limit the claimed invention. Based on the present disclosure the skilled artisan can identify and employ other components and methodology useful for practicing the present invention.
EXAMPLES
Example 1: Apilimod Inhibits Proliferation of Diverse Colon Cancer Cell Lines
[0124] The colon cancer cell lines HCT116 (BRAF wild-type) and HT-29 (BRAF mutated, V600E) cells were grown in DMEM (Corning), RKO (BRAF mutated, V600E) were grown in MEM (Corning), and HCT-15 (BRAF wild-type), SW1116 (BRAF wild-type), SW480 (BRAF wild-type) and SW620 (BRAF wild-type) were grown in RPMI-1640 (Corning) and supplemented with 10% FBS (Sigma Aldrich F2442-500ML, Lot 12D370) and Penicillin/Streptomycin (100.times.) (CellGro Ref 30-002). For drug studies, HCT116, HT-29, RKO, HCT-15, SW1116, SW480 and SW620 cells were seeded at a density of 750, 8000, 300, 480, 1600, 2000, and 2000 cells per well, respectively, into 96 well plates in a final volume of 50 .mu.L.
[0125] The apilimod used in these studies was apilimod dimesylate. The term "LAM-002" refers to apilimod dimesylate.
[0126] For single treatment studies, 24 h after seeding, cells were treated with apilimod alone (final concentration 0.5-10000 nM; 3-fold dilutions and a total of 10 dilutions), or with one of vemurafenib (final concentration 56.6-30000 nM; 2-fold dilutions and a total of 10 dilutions), regorafenib, oxaliplatin, 5-fluorouracil or irinotecan (all drugs screened at final concentration 2.5-50000 nM; 3-fold dilutions and a total of 10 dilutions). All drug dilutions were made up as a 2.times. stock and 50 .mu.L added to appropriate wells. Cells were treated for 120 h before viability was assessed using CellTiterGlo.RTM. (Promega) where the relative luminescence of untreated cells was set to 100% viability and each drug concentration expressed as a percentage of untreated cells. EC.sub.50 values were determined using GraphPad Prism (GraphPad Software, Inc). Briefly, raw data was log transformed and then analyzed using nonlinear regression (curve fit) where the data were constrained (bottom=0, top=100).
[0127] For determination of synergy between apilimod and each of vemurafenib, regorafenib, oxaliplatin, 5-fluorouracil or irinotecan, HCT116, HT-29, RKO, HCT-15, SW1116, SW480, and SW620 cells were seeded as outlined above. 24 h later, cells were treated with apilimod dimesylate alone (final concentration 2-250 nM; 2-fold dilutions and a total of 8 dilutions), with vemurafenib alone (final concentration 234-30000 nM; 2-fold dilutions and a total of 8 dilutions), with oxaliplatin, irinotecan, 5-fluorouracil or regorafenib alone (for all, final concentration 195-25000 nM; 2-fold dilutions and a total of 8 dilutions) or the combination of each concentration of apilimod mesylate with each concentration of vemurafenib, oxaliplatin, irinotecan, 5-fluorouracil or regorafenib (8.times.8 matrix). Cells were treated for 120 h before viability was assessed using CellTiterGlo.RTM. (Promega) where the relative luminescence of untreated cells was set to 100% viability and each drug concentration expressed as a percentage of untreated cells.
[0128] Bar graphs show the effect of a single concentration of apilimod, a single concentration of vemurafenib and the effect of the combination of drugs (at the single agent concentrations) on cell viability. The expected value was calculated (fraction of viability for apilimod multiplied by fraction of viability for vemurafenib) and is shown.
[0129] For calculation of synergy, CalcuSyn (version 2.11, Biosoft) was used to determine the combination index (CI) as defined by Chou et al. (Chou T C, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22:27-55). We constrained the analysis to assess CI values which were within a clinically achievable concentration and also where the fraction effect (Fa) was greater than 0.75 (ie a greater than 75% reduction in cell viability with the combination of drugs). CI versus fractional effect graphs are shown (datapoints denoted by `x`, lines show 95% confidence interval), where drug combinations producing CI values >1 are antagonistic, CI=1 are additive and CI<1 are synergistic. In addition, the CI value at the ED50, ED75 and ED90 are shown for apilimod and vemurafenib. The same methodology was applied to cells treated with apilimod in combination with oxaliplatin, irinotecan, regorafenib, and 5-fluorouracil.
[0130] Using this approach, apilimod was found to act synergistically in HCT116 cells with each of oxaliplatin, irinotecan, regorafenib, and 5-fluorouracil. Apilimod also demonstrated synergistic activity with both regorafenib in RKO cells. Apilimod was also found to be synergistic with vemurafenib in HCT116, HT-29, RKO, HCT-15, SW1116, SW480, and SW620 cells. These data demonstrate that apilimod can act both alone and synergistically with other anti-cancer agents against colon cancer cells. In addition, the data show that the combination of apilimod with vemurafenib shows synergistic activity in cells with wild-type BRAF as well as cells harboring mutated BRAF (V600E), which are often resistant to standard therapy.
TABLE-US-00001 TABLE 1 Apilimod and vemurafenib synergy in colorectal cancer cells (5 day assays). Cell Line BRAF status EC50 (nM) HCT116 WT 10 HCT-15 WT 15 SW1116 WT 12 SW480 WT 24 SW620 WT 9 RKO Mutant (V600E) 52 HT-29 Mutant (V600E) 19
Example 2: SNX10 Expression as a Biomarker for LAM-002 Response in Colon Cancer Lines
[0131] SNX10 is a gene involved in intracellular trafficking that was identified in a high-throughput screen conducted to identify genes that confer resistance to LAM-002. Genetic ablation of SNX10 conferred resistance to LAM-002 treatment. This suggested that over-expression of SNX10 would induce sensitivity to LAM-002. To validate this hypothesis, the expression level of SNX10 was correlated with LAM-002 sensitivity in various colon cancer lines.
[0132] Expression profiles were obtained from the public databases CCLE (Barretina, Caponigro, Stransky et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012 Mar. 28; 483(7391):603-7) and COSMIC (Forbes SA1, Beare D2, Gunasekaran P2, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015 January; 43 (Database issue):D805-11). R statistical package (R Development Core Team (2008). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0) was used for the normalization of expression and the correlation analysis.
[0133] To determine LAM-002 sensitivity in colon cancer cell lines, the EC90 was calculated using a 10 point dose response as described above for EC50 determination. The results revealed a correlation between SNX10 expression and LAM-002 sensitivity whereby colon cancer cells expressing higher levels of SNX10 are the most sensitive to LAM-002 (Table 2). These findings support SNX10 expression as a predictive biomarker for LAM-002 sensitivity in colon cancer cells.
* * * * *
D00000
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011

D00012

P00001

P00002

XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.