Apparatus and Methods for Preventing or Treating Failure of Hemodialysis Vascular Access and Other Vascular Grafts
Iyer; Sriram S. ; et al.
U.S. patent application number 16/297006 was filed with the patent office on 2019-07-04 for apparatus and methods for preventing or treating failure of hemodialysis vascular access and other vascular grafts. The applicant listed for this patent is Vascular Therapies, Inc.. Invention is credited to Sriram S. Iyer, Nicholas N. Kipshidze, Victor V. Nikolaychik.
| Application Number | 20190201383 16/297006 |
| Document ID | / |
| Family ID | 22996277 |
| Filed Date | 2019-07-04 |









View All Diagrams
| United States Patent Application | 20190201383 |
| Kind Code | A1 |
| Iyer; Sriram S. ; et al. | July 4, 2019 |
Apparatus and Methods for Preventing or Treating Failure of Hemodialysis Vascular Access and Other Vascular Grafts
Abstract
This invention is a prosthetic device generally placed on the outside surface of the vessel or graft which then elutes antiproliferative drugs or agents from a drug-eluting matrix material. Methods of perivascular antiproliferative drug administration also are disclosed.
| Inventors: | Iyer; Sriram S.; (New York, NY) ; Kipshidze; Nicholas N.; (New York, NY) ; Nikolaychik; Victor V.; (Mequon, WI) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 22996277 | ||||||||||
| Appl. No.: | 16/297006 | ||||||||||
| Filed: | March 8, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 14509279 | Oct 8, 2014 | 10272073 | ||
| 16297006 | ||||
| 11931143 | Oct 31, 2007 | 8858982 | ||
| 14509279 | ||||
| 10832048 | Apr 26, 2004 | 7807191 | ||
| 11931143 | ||||
| 10051708 | Jan 16, 2002 | 6726923 | ||
| 10832048 | ||||
| 60262132 | Jan 16, 2001 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61P 7/02 20180101; A61M 39/0247 20130101; A61L 2300/416 20130101; A61K 31/337 20130101; A61K 31/727 20130101; A61L 27/44 20130101; A61M 2202/0468 20130101; A61L 27/225 20130101; A61K 31/122 20130101; A61P 9/00 20180101; A61L 27/20 20130101; A61P 37/06 20180101; A61K 31/573 20130101; A61M 1/16 20130101; A61M 2202/0478 20130101; A61K 31/395 20130101; A61L 2300/252 20130101; A61P 9/10 20180101; A61M 2039/0261 20130101; A61K 9/0024 20130101; A61M 2039/0258 20130101; A61P 31/00 20180101; A61K 31/436 20130101; A61L 2300/204 20130101; A61P 41/00 20180101; A61P 7/08 20180101; A61M 1/3655 20130101; A61P 43/00 20180101; A61L 27/54 20130101; A61L 31/16 20130101 |
| International Class: | A61K 31/436 20060101 A61K031/436; A61M 39/02 20060101 A61M039/02; A61K 31/122 20060101 A61K031/122; A61K 31/337 20060101 A61K031/337; A61K 31/395 20060101 A61K031/395; A61K 31/573 20060101 A61K031/573; A61K 31/727 20060101 A61K031/727; A61L 31/16 20060101 A61L031/16; A61M 1/36 20060101 A61M001/36; A61L 27/20 20060101 A61L027/20; A61L 27/22 20060101 A61L027/22; A61L 27/44 20060101 A61L027/44; A61L 27/54 20060101 A61L027/54; A61K 9/00 20060101 A61K009/00 |
Claims
1. A method of preventing or treating vasculoproliferative disease in vascular structures, which comprises the step of: administering extravascularly and locally an antiproliferative effective amount of an antiproliferative agent to the vascular structure.
2. A method according to claim 1 wherein the agent comprises rapamycin.
3. A method according to claim 1 wherein the antiproliferative agent is administered perivascularly.
4. A method according to claim 1 wherein extravascular, local administration is accomplished by means of an implantable, antiproliferative agent eluting, perivascular vascular sleeve, the sleeve comprising a matrix material imbibed with the agent.
5. A method according to claim 4 wherein the sleeve is substantially circumvascular.
6. A method according to claim 4 wherein the matrix material comprises fibrin.
7. A method according to claim 4 wherein the agent comprises rapamycin and heparin
8. A method according to claim 4 wherein the matrix material comprises collagen.
9. A method according to claim 4 wherein the matrix material comprises chitosan.
10. An implantable, antiproliferative agent-administering perivascular sleeve adapted to be placed in contact with the exterior of a vascular structure comprising: a) A flexible, bioabsorbing, agent-eluting matrix material, the material having a vascular-sized lumen passing substantially through said matrix material, the matrix material having dispersed therein: b) an antiproliferative agent.
Description
CROSS-REFERENCED TO RELATED APPLICATIONS
[0001] This application is a continuation patent application of U.S. patent application Ser. No. 14/509,279 filed Oct. 8, 2014, which is a divisional patent application of U.S. patent application Ser. No. 11/931,143 filed Oct. 31, 2007, now U.S. Pat. No. 8,858,982, which is a continuation patent application of U.S. patent application Ser. No. 10/832,048 filed Apr. 26, 2004, now U.S. Pat. No.7,807,191, which is a continuation patent application of U.S. patent application Ser. No. 10/051,708, filed Jan. 16, 2002, now U.S. Pat. No. 6,726,923, which claims priority to U.S. Provisional Patent Application Ser. No. 60/262,132, filed Jan. 16, 2001, each of which are incorporated herein by reference in their entireties.
STATEMENT REGARDING FEDERALLY SPONSORED RESEARCH OR DEVELOPMENT
[0002] Not Applicable
BACKGROUND OF THE INVENTION
[0003] Failure of hemodialysis vascular access and other vascular grafts becomes evident as compromise of the lumen of the native vessel (vein or artery) or of the prosthetic conduit at o or away from the anastamotic site. Compromise of the lumen manifests as either stenosis or occlusion and is a result of either intraluminal thrombus and/or a vasculoproliferative response. The etiology of graft failures may be related to a variety of physical (e.g., shear stress causing hemodynamic disturbance), chemical and/or biological stimuli as well as infection and foreign body rejection which may explain why fistulae which do not involve a foreign body (in this case, for example, polytetrafluroethylene, PTFE) remain patent for a longer time compared to vascular access grafts that involve interposition of a PTFE graft.
[0004] The present invention relates generally to therapeutic implant, apparatus and methods useful for preventing, suppressing (inhibiting) or treating failure of hemodialysis vascular access and other vascular grafts.
[0005] Vascular access grafts, specifically, hemodialysis access grafts are well known to the art. Approximately 100,000 vascular access procedures are performed yearly in the United States. Hemodialysis vascular access can be constructed in one of several ways: as an arterio-venous fistula (e.g.; Brecisa-Cimino), or as a graft, interposing either prosthetic (e.g., PTFE) or biologic tissue (e.g., vein) between the artery and the vein. Such grafts are usually constructed using a tubular or cylindrical segment of suitably bio-compatible, substantially inert material such as polytetrafluoroethylene (PTFE). In fact, PTFE is the most common material used for prosthetic dialysis access. In one approach, a segment of PTFE is surgically interposed between an artery and a vein in the arm, forearm or thigh. The graft is then available for repeated vascular access for performing hemodialysis.
[0006] Subsequent to placement of the access graft the sutured sites in the artery and the vein undergo healing. Sixty percent of these grafts fail each year, usually because of narrowing (stenosis) at the venous end. Similar lesions develop in PTFE grafts placed in the arterial circulation, where there is a similar tendency for the distal end of the graft to be affected. Dysfunction or failure of veing grafts and/or other graft conduits used in coronary artery bypass graft surgery or in peripheral vascular surgery (e.g., aorta-iliac, femoral-femoral, femoral-popliteal, femoral tibial, etc.) are well known. Development of arterial access graft stenosis is not as rapid as development of access graft stenosis at the venous end. Proliferation and migration of smooth muscle cells resulting in intimal hyperplasia in the vein and the adjacent graft orifice has been described in human dialysis access stenosis. As the stenosis in the graft becomes progressively more severe, the graft becomes dysfunctional and hemodialysis is suboptimal. If the stenosis in the graft is not treated, it eventually leads to occlusion and to graft failure.
[0007] The reasons why the venous ends of access graft have such a marked propensity for narrowing are multifactorial. Features unique to this location include exposure to arterial pressures and arterial flow rates, dissipation of acoustic (vibratory) energy in the vessel wall and surrounding tissue, repeated puncture of the graft, and infusion of processed blood. In addition, the venous end of the graft may be bathed in mitogens released during passage of the blood through the dialysis tubing or during activation of platelets at the site of needle puncture.
[0008] Tissue samples collected from the graft-vein anastomosis site of stenotic PTFE grafts during surgical revision showed significant narrowing of the lumen and were characterized by the (i) presence of smooth muscle cells, (ii) accumulation of extra-cellular matrix, (iii) angiogenesis within the neointima and adventitia, and (iv) presence of an active macrophage cell layer lining the PTFE graft material. A large variety of cytokines and cell growth stimulating factors like platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), and vascular endothelial growth factor (VEGF) were expressed by smooth muscle cells/myofibroblasts within the venous neointima, by macrophages lining both sides of the PTFE graft, and by vessels within the neointima and adventitia. It has been suggested that macrophages, specific cytokines (bFGF, PDGF, and VEGF), and angiogenesis within the neointima and adventitia are likely to contribute to the pathogenesis of venous neointimal hyperplasia (VNH) a manifestation of the vasculoproliferative response in PTFE dialysis grafts.
[0009] Survival of patients with chronic renal failure depends on optimal regular performance of dialysis. If this is not possible (for example as a result of vascular access dysfunction or failure), it leads to rapid clinical deterioration and unless the situation is remedied, these patients will die. Vascular access dysfunction is the most important cause of morbidity and hospitalization in the hemodialysis population in the United States at an estimated cost of approximately one billion US dollars per annum. Venous neointimal hyperplasia characterized by stenosis and subsequent thrombosis accounts for the overwhelming majority of pathology resulting in PTFE dialysis graft failure. Despite the magnitude of the problem and the enormity of the cost, there are currently no effective therapies for the prevention or treatment of venous neointimal hyperplasia in PTFE dialysis grafts. Consequently, interventions aimed at the specific mediators and processes may be successful in reducing the very significant human and economic costs of vascular access dysfunction.
[0010] Once the stenosis has occurred, one of the current methods of treatment involves reduction or obliteration of the narrowing and restoration of blood flow through the graft (permitting the performance of adequate hemodialysis) by means of non-surgical, percutaneous catheter based treatments such as balloon angioplasty. Balloon angioplasty, in one aspect, involves deployment of a balloon catheter at the site of the blockage and inflating the balloon to increase the minimum luminal diameter (MLD) of the vessel by compressing the material causing the restriction against the interior of the vessel wall, thereby dilating the vessel. Depending upon the length and severity of the restriction, the procedure may be repeated several times (by inflating and deflating the balloon). When completed, the balloon catheter is withdrawn from the system.
[0011] Although balloon angioplasty can be used as a "stand alone" procedure, it is frequently accompanied by deployment of what is called a stent. A stent is an expandable scaffolding or support device which is placed within the vasculature to prevent mechanical recoil and reduce the chance of renarrowing (restenosis) at the site of the original restriction. Stents are either "balloon-expandable" or "self-expanding" and when deployed endovascularly, abut against the inner vessel wall. Whether or not a stent is placed, this form of treatment has a high risk of failure i.e., the risk of renarrowing (restenosis) at the treatment site is very high. Unless stenosis within the access graft can be effectively and permanently treated, graft failure tends to follow. In the event of graft failure, the patient has to undergo an endovascular procedure i.e., a non-surgical, catheter-based percutaneous procedure, repeat vascular surgery e.g., thrombectomy to "declot" the graft or to place another vascular access graft or a shunt (as it is sometimes referred to) at a different site, unless the patient receives a kidney transplant. Given the obvious problems of repeat surgery(ies) and the limited availability of transplants, there is a need for a treatment that is both effective and long lasting (durable) in the prevention and treatment of dialysis graft stenosis.
[0012] The vast majority of current approaches for reducing or preventing the vasculoproliferative response (believed to be the pathophysiological basis of restenosis), are based on treatment options that originate from within the vascular or graft lumen. One current, novel approach utilizes drug coated or drug impregnated stents which are then deployed within the lumen of the blood vessel. Examples of drugs used to coat stents include Rapamycin commercially available from the Wyeth Ayerst company (Sirolimus.RTM.), and Paclitaxel commercially available from the Bristol-Myers Squibb Company (Taxol.RTM.). In this stent-based approach, Rapamycin or Paclitaxel is gradually eluted from the stent and diffuses into the vessel wall from the intima (the innermost layer of the vessel wall) to the adventitia (the outermost layer of the vessel wall). Studies have shown that Rapamycin and Paclitaxel tend to inhibit smooth muscle cell proliferation.
[0013] Delivery from the perivascular or extravascular space through the arterial or vascular wall utilizing a synthetic matrix material (ethylene-vinyl acetate copolymer, EVA) together with an anticoagulant that also has antiproliferative properties e.g., heparin, has been suggested. There are two disadvantages of this approach: heparin is a soluble substance and rapidly disappears from the vascular wall and, ethylene-vinyl acetate copolymer is not biodegradable potentially raising concerns about long term effects, in vivo.
[0014] If a therapeutic agent is delivered locally using a matrix material-based system, the matrix material should preferably have the following characteristics:
[0015] 1. The matrix material has to permit the loading of adequate quantity of the therapeutic agent.
[0016] 2. The matrix material must elute the therapeutic agent at an appropriate, well defined rate.
[0017] 3. The matrix material should preferably be implantable and biodegradable. Thus, physical removal of the matrix material from recipient's tissue following drug delivery would not be necessary and obviates concerns about the long term effects of the residual matrix.
[0018] 4. Neither matrix material nor its biodegradation products should provoke a significant inflammatory or proliferative tissue response, nor should they alter or interfere with the recipient's natural defense systems or healing.
[0019] 5. The device (comprising the matrix material and the drug) should be flexible enough to mould to the contours of the vasculature and
[0020] 6. The device should be amenable to be fixed in place preventing its migration to an unintended location.
[0021] Polymer matrix materials used for drug delivery within the context of implantable devices can be either natural or synthetic. Examples include but are not limited to polymers composed of chemical substances like polyglycolic acid or polyhydroxybutyrate, EVA or natural polymers like collagen, fibrin or polysaccharides like chitosan. However, not all of these matrix materials are ideal; inappropriate features include poor mechanical characteristics, potential immunogenicity, and cost. In addition, some may produce toxic degradation products and induce inflammatory reactions or a proliferative response.
[0022] A well known biocompatible, biodegradable, resorbable matrix material for drug delivery is collagen. The use of collagen as a material for fabrication of biodegradable medical devices is and has undergone serious scrutiny. U.S. Pat. Nos. 6,323,184, 6,206,931; 4,164,559; 4,409,332; 6,162,247. One current focus involves delivery of pharmaceutical agents including antibiotics and physiologically active proteins and peptides such as growth factors.
[0023] Under scanning electron microscopy, the collagen matrix has a morphology of condensed laminated film with a textured surface and a range of pore sizes. It can be produced in a wide range of effective pore sizes from 0.001 microns to 100 microns or even larger. This internal pore network (porous material) creates a high surface area and serves as a microreservoir for storage and delivery of the therapeutic agent. Several features make collagen an excellent and ideal matrix material for drug delivery. Collagen exhibits a high degree of flexibility and mechanical durability, as well as intrinsic water wettability, semipermeability and consistent flow characteristics. More importantly, collagen, a naturally occurring substance is biodegradable and non-toxic. In addition, collagen has favorable biodegradation characteristics and time to complete degradation or resorption i.e., durability of the collagen matrix for drug delivery can be modified.
[0024] A second protein matrix suitable for drug delivery is fibrin. A fibrin matrix is comprised of cross-linked fibrin units that are a reticular network of thrombin-modified fibrinogen molecules. This matrix is similar to a natural blood clot. In contrast to natural clot, the size of pores in a fibrin matrix can be controlled and varies from 0.001 millimicrons to 0.004 millimicrons, so-called micropores. The differences in pore sizes between collagen and fibrin matrices permit the binding of therapeutic agents with distinct rates of drug release. The ability to control bleeding, to remain firmly fixed in place, and to be naturally biodegradable have all made fibrin a good matrix material for drug delivery and confers fibrin some advantages over synthetic matrices. Most of the early applications of fibrin as a matrix were for delivery of antibiotics and other biologics.
[0025] The fibrin matrices are prepared in a dry granular form. (cf., PCT/EP99/08128). This formulation, manufactured by HyQ Solvelopment, Buhlmhle, Germany, contains D-mannitol, D-Sorbit, fibrinogen-aqueous solution, and a thrombin-organic suspension. The formulation is manufactured by fluid bed granulation. The applications for dry fibrin are manifold: wound closure, promotion of healing, and homeostasis. However, application for drug delivery is limited since such a formulation does not allow for a target-oriented shaping of solid particles around the vessel wall and delivery of exact dosages is difficult. Porosity and capacity of dry fibrin particles are low, physical stability is poor.
[0026] Another group of potentially useful resorbable, natural polymer matrix material is chitosan. Chitosan has proven to be a useful biocompatible aminopolysaccharide and a matrix for controlled release of the agent for local delivery. Chitosan implants cause no systemic and local side effects or immunologic responses, and are suitably biodegradable. Chitosan can be prepared from the degradation of slow chitin (molecular weight 1.times.10.sup.6) using high temperature sodium hydroxide hydrolysis to a molecular weight of 5.times.10.sup.5. The inability to control porosity is a disadvantage of this matrix material.
BRIEF DESCRIPTION OF THE PRESENT INVENTION
[0027] The present invention is unique in at least two respects: 1) Whereas the majority of current methods of preventing suppressing or treating the vasculoproliferative response (smooth muscle cell hyperplasia, restenosis, vascular occlusion) do so from inside the vascular (i.e., vein and/or artery) or graft lumen, the present invention is a method of doing so extravascularly or perivascularly i.e., from outside the vascular or graft lumen and through the vascular wall. 2) All current treatment approaches are relevant only after the narrowing or stenosis has actually taken place. The current invention is, in one aspect, a method of preventing or suppressing vasculoproliferative disease, in contradistinction to curing it.
[0028] In a further embodiment, the present invention is an implantable prosthetic device placed on the outer surface of the vessel or graft which then elutes anti-vasculoproliferative drugs or agents such as Rapamycin, Paclitaxel, Tacrolimus, and other cell cycle inhibitor or similarly-functioning agents. In addition to a resorbable matrix material, e.g., protein, and an antiproliferative agent, this implantable device contains optionally, agents that inhibit collagen accumulation in the tunica media and adventitia of the vascular wall and pharmaceuticals that help reduce calcification of the vascular wall. This invention provides a method of preventing or treating neo intimal hyperplasia (an expression of the vasculoproliferative response) and calcification by extravascular delivery of an effective amount of an antiproliferative agent with low water solubility alone or in combination with adjuvants, and other antiproliferative agents. Rapamycin is a particularly preferred drug with antiproliferative properties for use with the present invention. A mixture of suitable drugs may be used. The Rapamycin diffuses from the outside and through the vessel and/or graft wall to the interior of the vein and/or artery and/or graft. Elution of Rapamycin (and other drugs with antiproliferative effect), into and through the vascular wall from the outside starts soon after the device is implanted and the drug will inhibit smooth muscle cell proliferation within the hemodialysis and other vascular grafts and/or at their anastamotic sites. Thus, in one aspect, the present invention is a method of inhibiting smooth muscle cell proliferation of a vascular access graft or shunt by the gradual elution or timed release of a drug from outside the vascular access site vessel wall to the vessel interior i.e., by extravascular or perivascular delivery.
[0029] In another aspect the present invention is a prosthetic device comprising a cylindrical, antiproliferative-imbibed, protein interior layer and, optionally, an exterior support or skeletal structure or layer. In one embodiment, the imbibed protein layer is collagen and the exterior skeletal support structure is a sheet of PTFE. The antiproliferative drug, in this embodiment, is preferably Rapamycin. Paclitaxel (or Taxol) is another antiproliferative drug or agent well-suited to the embodiment of the invention.
[0030] A third embodiment of the present invention is a method of inhibiting stenosis of hemodialysis access graft comprising the method of placing a prosthetic device (described above) over a graft or vascular structure and/or at the site of anastomosis and anchoring the prosthetic device at the desired site (e.g., by suturing).
[0031] A device of this invention may employ a biocompatible matrix material such as collagen, fibrin or chitosan. An important factor in the selection of a particular matrix material is the porosity of the material and a controllable rate of biodegradation. Use of a matrix material is important because it creates a delivery reservoir and controls the agent delivery kinetics.
[0032] A preferred device of this invention comprises a collagen matrix material imbibed with Rapamycin, which will be placed in position so as to extravascularly deliver the agent.
[0033] In a preferred embodiment, about 120 micrograms/cm.sup.2 of Rapamycin (Range: 50 micrograms to 10 mg/cm.sup.2) is combined with a collagen matrix material sheet with a thickness in the dry state between 0.3 and 2.0 mm sheet which is then implanted or wrapped upon the outside of the vascular or graft wall.
[0034] A further aspect of the present invention is "self fixation" of the device delivering the drug or agent to the outer surface of the vascular or graft wall. The collagen-device could be made more adhesive to the vascular wall if in the final stage collagen is combined with photoreactive groups such as FITS (fluorescein isothiocyanate) or Bengal Rose both from Sigma Chemicals, St Louis, Mo. Stimulation of the device with ultra violet light will activate these photoreactive groups and will increase adhesion. Fibrin sealant and acetylated collagen also have been found to increase adhesion of collagen matrix material to the outside vascular wall.
[0035] Early work showed a relationship between local vessel trauma and expedited calcification. Recently, a study in humans has shown that the matrix Gla-protein (protein .gamma.-carboxylated vitamin K-dependent .gamma.-carboxylase) is constitutively expressed by normal vascular smooth muscle cells and bone cells. High levels of Gla-protein mRNA and non-.gamma.-carboxylated protein were found in atherosclerotic vessel tissues. This .gamma.-carboxylated protein is necessary to prevent or postpone beginning of vascular calcification (Price, P. et al., "Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves," Atheroscler Thromb Vasc Biol, (1998) 18: 1400-1407). These data indicate that calcification caused by injury must be actively inhibited. Introduction of pharmaceuticals preventing calcium accumulation helps to postpone calcification and helps prevent, suppress or treat the vasculoproliferative processes. In one aspect of this invention, local delivery of Vitamin K counteracts the calcification effect associated with vessel injury by timely activation of .gamma.-carboxylase (in this case Gla-protein) and ensures other calcium-binding proteins function properly and do not bind excess of calcium (Hermann, S. M. et al., "Polymorphisms of the human matrix Gla-protein gene (MGP) vascular calcification and myocardial infarction," Arterioscler Thromb Vasc Biol. (2000) 20:2836-2893. A mixture of Vitamin K and other anti-proliferative drugs may be used.
[0036] The acute response, characterized by an inflammatory reaction, is an attempt to limit disturbances in the homeostasis. Hallmarks of this inflammatory reaction include leukocyte accumulation, increased fibrin deposition and release of cytokines. Addition of synthetic glucocorticoids like dexamethasone decreases this inflammatory response and may eventually decrease the vasculoproliferative process. Since the pharmacological mechanisms of action of the antiproliferative agents and synthetic glucocorticoids are different, agents with different "mechanisms of action" may be expected to act synergistically. It may be useful, therefore, to combine two or more of these agents.
[0037] This invention thus provides a method of preventing, suppressing, or treating neointimal hyperplasia by extravascular, (e.g., perivascular) local delivery of an effective amount of an anti-vasculoproliferative agent with low water solubility (e.g., Rapamycin) alone or in combination with other antiproliferative agents and adjuvants.
[0038] In one aspect, the present invention is a prosthetic device that consists of a resorbable protein matrix combined with a drug, placed on the outer surface of a blood vessel or graft. The device then elutes the drug which inhibits smooth muscle cell proliferation (anti-vasculoproliferative). Examples of such drugs include Rapamycin, Paclitaxel, Tacrolimus, other cell cycle inhibitors or similarly-functioning agents. A mixture of suitable drugs and/or additives may be used. In addition to a resorbable protein matrix and an antiproliferative agent, this implantable device contains optionally, agents that inhibit collagen accumulation in the vascular wall and pharmaceuticals that help reduce calcification of the vascular wall.
[0039] Rapamycin is a particularly preferred drug for use with the present invention. The Rapamycin [or other drug(s)] elutes from the outside and diffuses through the vessel and/or graft wall to the interior of the vein and/or artery and/or graft. Elution of Rapamycin (or a similarly acting drug or a drug having similar properties), into and through the vascular wall from the outside takes place during the healing phase of the anastamotic sites and the drug will prevent suppress/inhibit or treat smooth muscle cell proliferation that accompanies such healing. Thus, in one aspect, the present invention is a method of inhibiting the vasculoproliferative response at the anastamotic ends of a vascular access graft or shunt by the gradual elution or timed release of a drug from outside to the vessel interior i.e., by transvascular delivery using an extravascular source.
[0040] In another aspect the present invention is a prosthetic device comprising a antiproliferative-imbibed, protein interior layer and, optionally, an exterior support or skeletal structure or layer. In one embodiment, the imbibed protein layer is collagen and the exterior skeletal support material structure is a sheet of PTFE. The antiproliferative drug, in that embodiment, is preferably Rapamycin, or other similarly-functioning drugs.
[0041] Another embodiment of the present invention is a method of inhibiting stenosis of hemodialysis access graft comprising the method of placing the prosthetic device (described above) over a graft or vascular structure and/or at the site of anastomosis and anchoring the prosthetic device at the desired site (e.g., by suturing).
BRIEF DESCRIPTION OF FIGURES
[0042] FIGS. 1A, 1B, 2A, and 2B illustrate preferred embodiments of the present invention.
[0043] FIGS. 2A and 2B illustrate another embodiment of the present invention in which an exterior support or skeletal structure are employed.
[0044] FIGS. 3A-3C illustrate a self-interlocking embodiment of this invention.
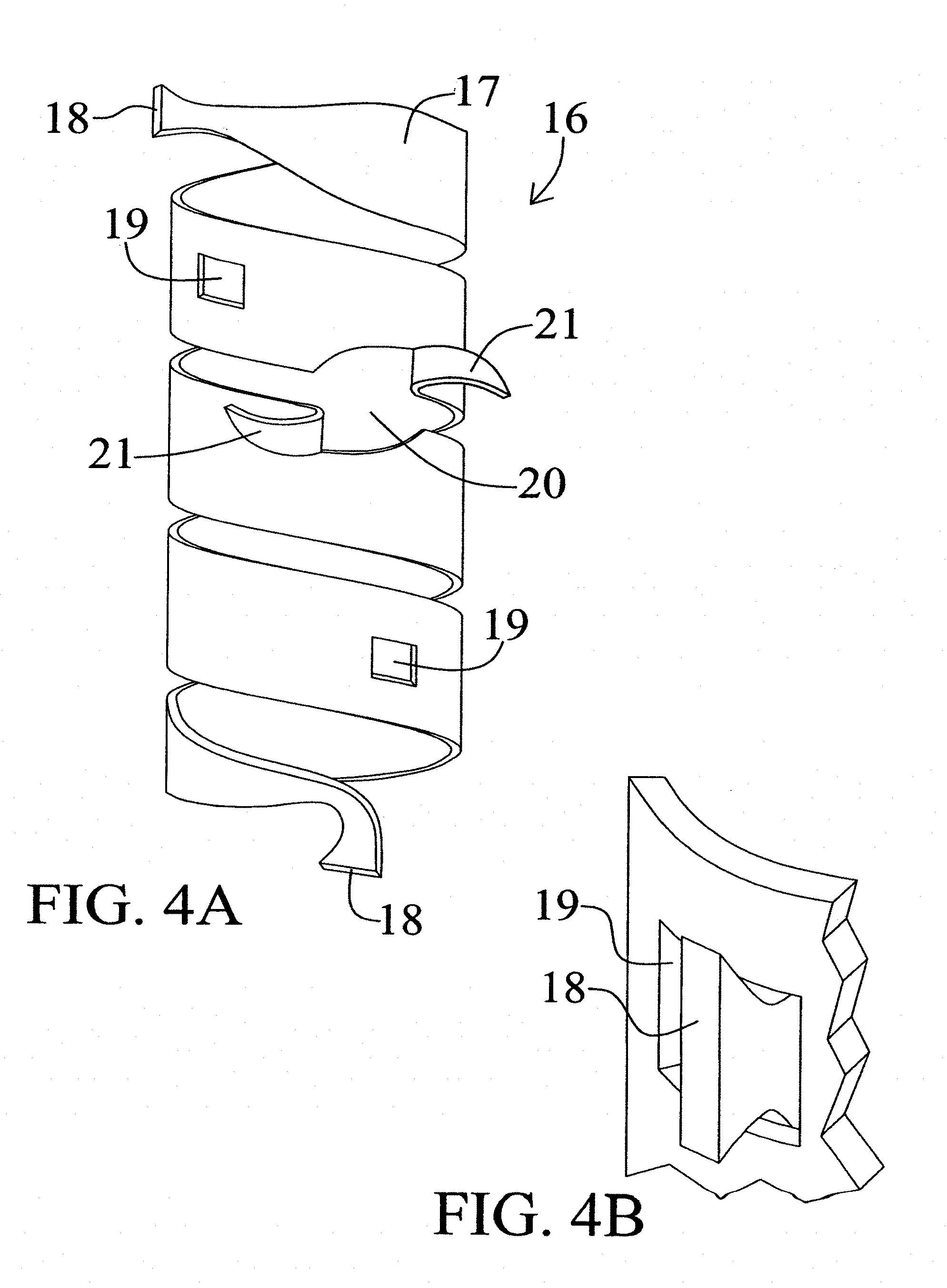
[0045] FIGS. 4A and 4B illustrate a second interlocking embodiment of the present invention.
[0046] FIG. 5 Shows the basic device shown in FIGS. 1A-1B/2A-2B include an exterior wire support or framework, which assists retention of sleeve shape.
[0047] FIGS. 6-13 Illustrate various possible deployments of the drug-eluting sleeve of the present invention in view of various vessel reparative needs.
[0048] FIG. 14 Shows rates of release of collagen saturated with tetracycline and rapamycin. Rapamycin was combined with a collagen matrix material using four different formats. Numbers on y-axis shows concentration of drug in micrograms per ml. Legend: A=Collagen saturated with Tetracycline. B=Collagen Saturated with Rapamycin. C=Rapamycin Dispersed throughout collagen. D=Collagen conjugated with Rapamycin. E=Combination of dispersed and conjugated forms of Rapamycin.
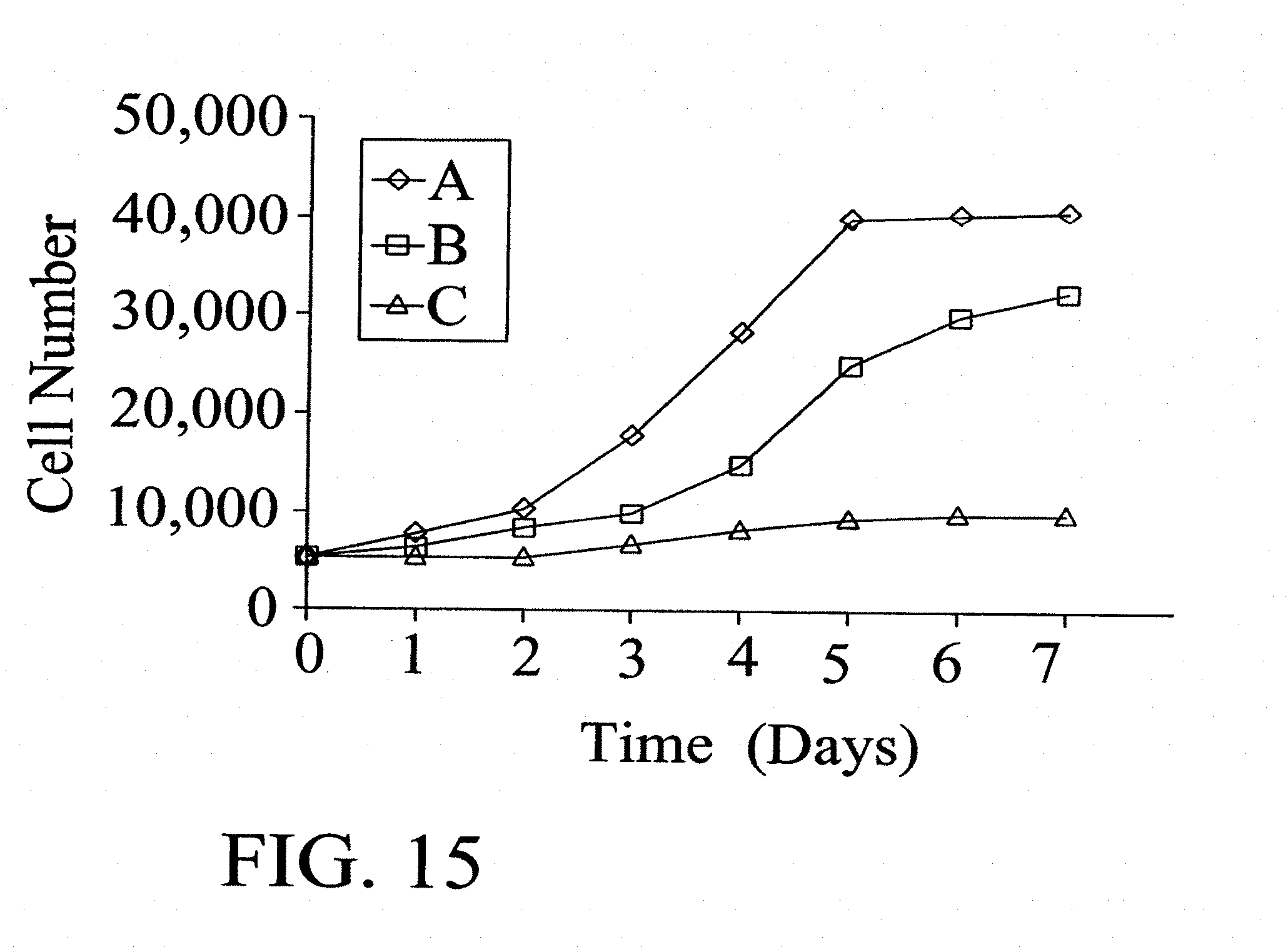
[0049] FIG. 15: Is a comparison of inhibition of growth of Smooth Muscle Cells using collagen matrices combined with different anti-proliferative agents. Numbers on .gamma.-axis denotes cell numbers. Legend: A=Control; B=Collagen+Actinomycin D; and C=Collagen+Rapamycin.
[0050] FIG. 16 Is a comparison of the effect of Rapamycin, Tacrolimus and Paclitaxel (3 doses) on Human Smooth Muscle Cells.
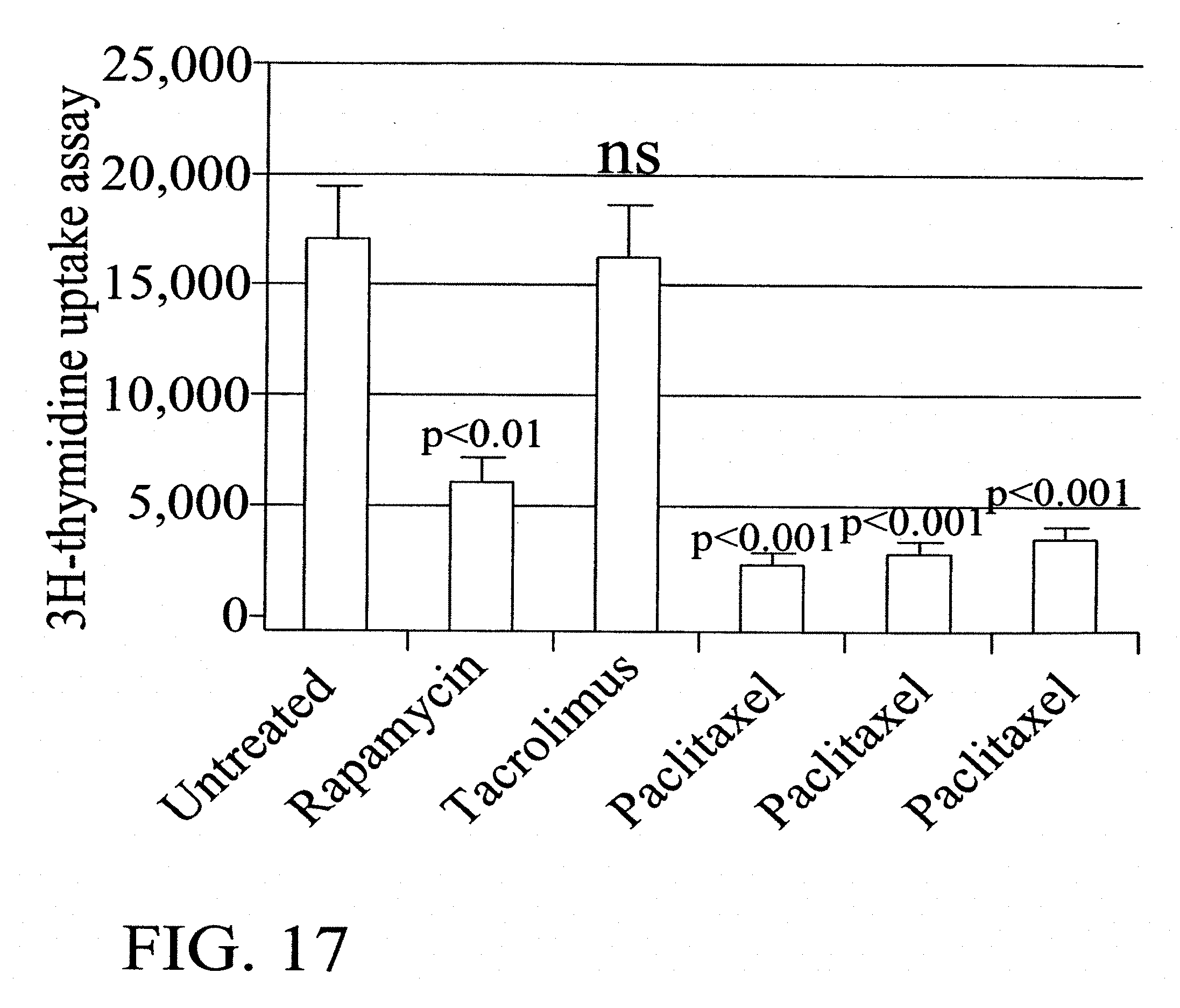
[0051] FIG. 17: Is a comparison of the effect of Rapamycin, Tacrolimus and Paclitaxel (3 doses) on Human Endothelial Cells.
[0052] FIGS. 18A, 18B, 19A, 19B, and 20 illustrate some results obtained using the present invention.
DETAILED DESCRIPTION OF THE INVENTION
[0053] In one aspect the present invention is a prosthetic device adapted for extravascular drug or agent delivery comprising a drug or agent-eluting matrix material combined with a drug(s) that can prevent, suppress or treat vasculoproliferation.
[0054] Matrix Materials: Material for the matrix may be from natural sources or may be synthetically manufactured or may be a combination of the two. A device of this invention may employ a biocompatible, biodegradable resorbable matrix material such as collagen, fibrin or chitosan. A suitably biocompatible, nonbiodegradable matrix may be also be used. Combination of degradable and nonbiodegradable or two or more biodegradable substances (e.g., collagen plus fibrin) or two or more nonbiodegradable substances may be selected for the matrix material. An important factor in the selection of a particular matrix material is the porosity of the material and where applicable, a controllable rate of biodegradation. The characteristics of the matrix material is important because the material creates a delivery depot or reservoir and control the kinetics of agent delivery. The characteristics with respect to thickness, porosity, rate of biodegradation etc. need not be identical throughout the matrix. It is also conceivable that by creating a polymer from the drug (for example, the antiproliferative), the matrix and the drug are one and the same, and, as the polymer degrades it releases the drug.
[0055] Collagen (Type I) is a preferred biocompatible biodegradable resorbable material for the matrix of the drug eluting sleeve of the present invention. The collagen source may be animal or human or may be produced using recombinant DNA techniques. Other types of collagen e.g., types II, III, V, XI singularly or in combination with Type I may be used. Although collagen matrix in the form of a sheet or membrane is the preferred embodiment of this invention, other forms of collagen e.g., gel, fibrilla, sponge, tubular etc., may also be used. As is well known, the rate at which resorption of the collagen occurs can be modified by cross-linking the protein.
[0056] Therapeutic Agents: In order to prevent suppress or treat the smooth muscle proliferative response that predominantly contributes to the neointimal hyperplasia, therapeutic agents that have significant antivasculoproliferative properties will be used in this invention. It is to be understood that as presently informed it is smooth muscle proliferation, which is believed to be primarily responsible for the stenosis and luminal compromise leading to graft failure. The present invention should not be interpreted to require that failure mechanism for its operation. Stated differently, applicants do not wish to be bound by any theory of graft failure, which would tend to narrow the scope of their invention. Examples of drugs with significant anti proliferative effects include but are not limited to Rapamycin, paclitaxel, other taxanes, tacrolimus, actinomycin D, angiopeptin, vassenoids, flavoperidol, hormones such as estrogen, halofuginone, matrix metalloprotienase inhibitors, ribosimes, interferons and antisense compounds. Analogues of the parent compound e.g., those of rapamycin, paclitaxel and tacrolimus may be used. Examples of other therapeutic agents include anti-inflammatory compounds, dexamethasone and other steroids, antiplatelet agents including aspirin, clopidogrel, IIBIIIA antagonists, antithrombins, anticoagulants including unfractionated and fractionated heparin, statins, calcium channel blockers, protease inhibitors, alcohol, botulin and genetic material. Vascular, bone marrow and stem cells may also be used
[0057] These agents can be combined to the matrix singly or in combination. Depending on the therapeutic agent, the agent can be combined with the matrix using physical, chemical and/or biological methods. A combination of techniques can be used. It will also be appreciated that drug concentration need not be (and often will not be) the same throughout the entire matrix.
[0058] It is to be understood that the process of elution of drug from the matrix material (sleeve) to and through the vessel wall is merely illustrative of one possible drug delivery process. For example, a drug may be released by application of a stimulus or a trigger e.g., light, temperature variation, pressure, ultrasound-ionizing energy, electromagnetic or magnetic field. Also, the drug may reside in the matrix as a pro-drug or in an inactive form. Application of the stimulus referred to above triggers conversion to the active form of the drug which is then released. Illustrating this application, it is known that Porphyrins and Psoralens are activated and may be released from a matrix to which they are absorbed or bound, by application of visible or ultraviolet light. Application of light modifies the drug structure causing the association between the drug and the protein reservoir or source to be disrupted. Thus, the drug is released from its matrix or reservoir and elutes to and through the vessel wall and into the vessel lumen in accordance with this invention.
[0059] A device of this invention optionally includes agents that accomplish other objectives e.g., that inhibit collagen accumulation and help reduce calcification of the vascular wall. Early work by Selye and colleagues showed a relationship between local vessel trauma and expedited calcification. Recently, a study in humans has shown that the matrix Gla-protein (protein .gamma.-carboxylated vitamin K-dependent .gamma.-carboxylase) is constitutively expressed by normal vascular smooth muscle cells and bone cells. High levels of Gla-protein mRNA and non-.gamma.-carboxylated protein were found in atherosclerotic vessel tissues. This .gamma.-carboxylated protein is necessary to prevent or postpone beginning of vascular calcification (Price P. et al., "Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves," Atheroscler Thromb. Vasc. Biol. (1998); 18:1400-1407). These data indicate that calcification caused by injury must be actively inhibited. Introduction of pharmaceuticals preventing calcium accumulation helps to postpone calcification and the restenotic processes. In this invention, local delivery of Vitamin K counteracts the calcification effect associated with vessel injury, by timely activation of .gamma.-carboxylase (in this case Gla-protein) and ensures other calcium-binding proteins function properly and do not bind excess of calcium (Hermann S. M. et al., "Polymorphisms of the human matrix Gla-protein gene (MGP) vascular calcification and myocardial infarction," Arterioscler Thromb. Vasc. Biol. (2000); 20: 2836-93). A mixture of Vitamin K along with other anti-proliferative drugs may be used.
[0060] The acute response to any injury, (in this instance, surgical trauma) characterized by an inflammatory reaction, is an attempt to limit disturbances in the homeostasis. Hallmarks of this inflammatory reaction include leukocyte accumulation, increased fibrin deposition and release of cytokines. Addition of synthetic glucocorticoids like dexamethasone decreases this inflammatory response and may eventually decrease the restenotic process. Since the pharmacological mechanisms of action of the antiproliferative agents and synthetic glucocorticoids are different, agents with different "antirestenotic mechanisms" may be expected to act synergistically. It may be useful, therefore, to combine two or more of these agents.
[0061] Numerous other antiproliferative or anti-stenosis drugs and other suitable therapeutics and adjuvants will likely occur to one skilled in the art in light of the present disclosure.
[0062] Method of Making the Sleeve: In view of the above disclosure several potential processes for making the prosthetic device and for its application will occur to one skilled in the art.
[0063] Single or Uni Layer Device: In a preferred embodiment of this invention, the protein matrix is a sheet or membrane of Type I bovine collagen and the drug is Rapamycin. Collagen is a particularly preferred example for the matrix because it has the property of being biodegradable and reabsorbable. The durability of the matrix reflects the time to complete reabsorption of the collagen, the porosity influences the drug binding capacity of the collagen matrix, both of these features can be controlled and varied. As an example, a relatively flat sheet of collagen is impregnated, absorbed, saturated, dispersed or immobilized with Rapamycin. About 120 micrograms/cm.sup.2 (Range: 50 micrograms-2 milligrams/cm.sup.2) of Rapamycin is combined with the collagen matrix material which in the dry form is in the form of a sheet that is 0.3 to 2.0 mm thick. This drug combined collagen sheet (sleeve), modified into a tube (cylinder) or other geometrical shapes, is directly secured to the outside of the native vessel, at the site of graft anastamosis and/or over the vein, artery or graft itself. The device may be secured by sutures or staples. The suture material itself may be combined with an anti vasculoproliferative drug. In this aspect, the chosen antiproliferative agent permeates through the vessel wall the rate of drug elution from the membrane can be varied and can continue until the collagen matrix material is completely resorbed. Tacrolimus, paclitaxel, other taxanes, flavoperidol, antisense, analogues of Paclitaxel, Rapamycin and tacrolimus, and other adjuvants well known to one skilled in the art, may be used.
[0064] Double or Dual or Multi layer Device: In another aspect, the present invention is a dual layered prosthetic device comprising an antiproliferative-imbibed, inner matrix layer and, an external support skeletal structure or layer. In this embodiment, the inner matrix material is a sheet or membrane of type I collagen and the exterior skeletal support material structure is a sheet of PTFE. The antiproliferative drug, in this embodiment, is Rapamycin. The sheet of collagen will be attached to the PTFE sheet using a variety of techniques e.g., physically using sutures, adhesives, staples or the two may be chemically bonded. The two sheath composite would then be rolled to create either a tubular structure or geometrical variations thereof. The composite device or sleeve is then suitably trimmed so that it can be applied over the desired site(s): artery, vein, graft anastomotic site etc., and the free edges of the PTFE sleeve are attached to each other by adhesive, sutures, staples etc. This stabilizes the entire device on the outside of the vascular structure or graft. The drug then permeates through the vascular or prosthetic material wall and while in the wall the drug inhibits smooth cell proliferation, an integral part of the healing response that follows surgical construction of the graft.
[0065] Following placement on the outside of a vessel or prosthetic surface, after a period of time the body absorbs the collagen leaving its exterior support skeleton or structure intact. One skilled in the art will appreciate that the body-resorbable aspect of the protein layer chosen to imbibe the drug, is an optional preferred practice of the present invention. The PTFE not being bioabsorbable, tends to hold the resorbable protein layer in place for a length of time sufficient for the drug to permeate through the vascular or graft or prosthetic material wall. Besides its value in supporting the drug eluting inner membrane or matrix material there are other potential advantages of the external layer. Although the desired effect of the drugs is their ability to inhibit the smooth muscle cell proliferative response, it is this proliferative response that contributes to the formation of a good quality (firm) surgical scar. A weak scar at the site of surgical anastamosis can potentially lead to graft disruption or aneurysm formation. Having an external PTFE skeleton functions as an additional reinforcement layer and prophylactically addresses the treatment for problems related to a weak scar, graft disruption, and/or aneurysm formation. The external PTFE layer serves to keep the drug in close apposition with the outer aspect of the vessel or graft wall and limits its diffusion to the surrounding tissues and skin. It is also within the contemplation of the present invention that the exterior skeletal or support aspect of the prosthetic device could, itself, be biodegradable. Thus, a resorbable external skeletal structure combined with a resorbable internal drug eluting collagen layer, the two layers having the same or different rate of degradability and resorption, would generate a healed vascular or graft structure without the necessity of foreign material remaining after the procedure. One skilled in the art would understand in view of this disclosure that numerous other such materials are likely to be usable in this invention. For example, Dacron.RTM. polyester can also be a suitable material for the external support structure.
[0066] A further object of the present invention is device self-fixation to the outer surface of the vascular wall. The device could be made more adhesive to the vascular wall if in the final stage collagen is combined with photoreactive groups such as FITS (fluorescein isothiocyanate) or Bengal Rose both from Sigma Chemicals, St Louis, Mo., USA. Stimulation of the device with ultra violet light activates the photoreactive groups and will increase adhesion. Fibrin sealant and acetylated collagen have been found to increase adhesion of collagen matrix material to the outside vascular wall.
[0067] Another embodiment of the present invention is a method of inhibiting stenosis of hemodialysis access graft comprising the method of placing the prosthetic device (described above) over a graft or vascular structure and/or at the site of anastomosis and anchoring the prosthetic device at the desired site (e.g., by suturing).
[0068] FIGS. 1A, 1B, 2A, and 2B illustrate preferred embodiments of the present invention 1. In FIG. 1A there is shown a rectangular sheet of a matrix material 2 having disbursed or distributed therein an agent 3 of the present invention (shown by stippling). FIG. 1B illustrates a further embodiment of the invention shown in FIG. 1A in which a hole 4 has been created in the drug-containing matrix material 3,2. It will be understood by one skilled in the art that the diameter of hole 4 will be adjusted to accommodate the outside diameter of any vascular or graft structure passing therethrough. In one embodiment, the diameter of hole 4 is 6 millimeters.
[0069] FIGS. 2A and 2B illustrate a further embodiment to the present invention in which an exterior support or skeletal structure or means 5 is employed. Support 5 is exterior to matrix material sheet 2 when sheet 2 is rolled or coiled into a cylindrical shape. Exterior skeletal means such as polytetrafluoro ethylene (PTFE) and dacron sheets are among the support materials presently contemplated. Many other such exterior skeletal support means will occur to one skilled in this art. As is shown, FIG. 2B illustrates an embodiment to the invention in which a hole 4 (which may vary in diameter) is employed.
[0070] FIGS. 3A, 3B, and 3C illustrate an embodiment of the invention employing an interlocking design in which one edge of the rectangular agent-eluting sheet or matrix material interlocks adjacent the opposite edge. More specifically, FIG. 3A shows a rectangular matrix material 2 having agent 3 (shown in stippling) disposed or disbursed therein. Also shown on the sheet illustrated in FIG. 3A are a series of v-shaped notches 6 located approximately adjacent one edge 7 of the agent-containing matrix material. Cooperating with notches 6 on the opposite edge 8 are a series of projections 9. Projections 9 are arrow-head shaped. However, other combinations of projection 9 and slots 6 certainly are contemplated by this invention. Thus, assembly of a sleeve embodiment of the present invention involves rolling edge 8 toward edge 7 (shown in FIG. 3B) and inserting projections 9 into slots 6. As is shown in FIG. 3C projections 9 have been inserted into slots 6 from the inside of the tubular structure meaning that the points 10 of projections 9 project from the inside to the outside of the structure. As is shown, the following edges 11 of projections 9 cooperate with v-shaped slots 6 to lock the flat structure into a cylindrical vascular-dimensioned sleeve 12. Vascular sleeve 12 further then defines a lumen 14. Lumen 14 is of a vascular dimension such that the interior surface of sleeve 12 would be in contact with the exterior surface of a vascular structure to which sleeve 12 was attached. In this fashion, the drug or agent-eluting, vascular-dimension sleeve is deployed over and around the vascular structure with which this invention is to be used.
[0071] FIGS. 4A and 4B illustrate a second interlocking embodiment of the present invention. In embodiment, a strip-form of the present invention is utilized. Agent-eluting sleeve 16 comprises an elongate drug or agent-eluting matrix material 17 (alone or in conjunction with an external support means, not shown). Created in matrix material 17 are two locks 18 located on opposite ends thereof. Cooperating with lock 18 are windows 19 into which locks 18 are inserted such that sleeve 16 is deployed against and on the exterior of the operant vascular structure. As is shown on FIG. 4B, lock 18 may be inserted into window 19 from the inside toward the outside. In an alternative embodiment lock 18 may be inserted into window 19, from the outside toward the interior of the sleeve structure. Also shown in FIG. 4A is a representative shunt opening 20 including two shunt contact wings or flaps 21.
[0072] FIG. 5 illustrates another embodiment to the present invention in which an external wire support or framework means is employed. External wire framework 20 surrounds a preferred embodiment of the present invention i.e. a PTFE and drug-coated collagen matrix material 22 disposed around vessel 24.
[0073] FIGS. 6-13 illustrate various arterio-venous fistuale. A drug eluting sleeve or matrix material of the present invention 26 is shown to be implanted, wrapped or placed around the various fistulae 32 shown in the several figures. In each of these figures venous structures are designated 28 and arterial structures are designated 30. Arrows 34 illustrate the direction of blood flow.
[0074] FIGS. 10-13 illustrate a further embodiment of this invention in which a graft e.g., a PTFE graft, 36 is used in conjunction with the present invention. As is shown in FIG. 13, graft 36 may itself include a matrix material with a drug or agent 36 (shown in stippling) of this invention.
[0075] A further application of the present sleeve involves utilization of the interior drug-imbibing protein layer as a drug source or drug reservoir. In that application the drug selected may be replenished periodically, e.g., by puncturing the sleeve with a needle and delivering additional drug thereto or creating a reservoir for the drug within the sleeve from which it can be gradually eluted.
EXAMPLES
[0076] The following examples are set forth to illustrate the device and the method of preparing matrices for delivering antiproliferative drug(s) and other therapeutics. The examples are set forth for purpose of illustration and not intended in a limiting sense.
Example 1
Inhibitory Effect of Different Antiproliferative Agents
[0077] Prefabricated collagen matrices were placed in different antiproliferative drug solutions until complete saturation occurred. The antiproliferative drugs were chosen to represent the more active compounds capable of smooth muscle cell and fibroblast inhibition without inhibiting collagenase and elastase enzymes. (Collagenase and elastase enzymatically inhibit collagen accumulation--one cause of restenosis). The collagen matrices were saturated with these compounds at concentration of 25 .mu.g/ml lyophilized, washed with 0.066 M phosphate buffer (pH 7.4) at 37.degree. C. for 24 hours and cut in the shape of a disc with density of compound about 5 .mu.g per cm.sup.2. After washing, sterile discs, 15 mm in diameter were placed in 24-well culture plate and cells at a density of 5000 per cm.sup.2 were seeded. Five days later cell number was measured and enzymatic activity was evaluated in the aliquots of media via chromogenic substrates hydrolysis and spectrophotometry. These data are presented in Table 1.
TABLE-US-00001 TABLE 1 Inhibitory effect of different antiproliferative agents SMC Fibroblast Collagenase Elastase Agent Inhibition % Inhibition % Activity % Activity % Control, plain 0 0 100 100 matrix Paclitaxel 88 .+-. 6 62 .+-. 11 98 .+-. 5 90 .+-. 4 Rapamycin 94 .+-. 5 90 .+-. 12 137 .+-. 8 142 .+-. 5 Cyclosporin A 61 .+-. 7 53 .+-. 7 104 .+-. 5 87 .+-. 7 Tetracycline free 11 .+-. 8 13 .+-. 5 56 .+-. 8 81 .+-. 4 base Methotrexate 32 .+-. 9 28 .+-. 6 23 .+-. 12 14 .+-. 3 Actinomycin D 44 .+-. 11 35 .+-. 8 55 .+-. 9 84 .+-. 11
[0078] In this comparative in vitro test, among tested agents, Paclitaxel and Rapamycin performed similarly.
Example 2
Capacity of Different Types of Matrices to Bind Rapamycin
[0079] In the next in vitro study, the ability of different matrices to bind Rapamycin was tested. A prefabricated (BioMend, Sulzer Calcitek, Inc or Biopatch, Ethicon Inc, containing collagen-alginate) collagen matrix with Rapamycin was prepared as described in Example 1 at initial Rapamycin concentration of 250 .mu.g/ml. Prefabricated chitosan (using technique described in: Almin, C., Chunlin, H., Juliang, B. et al "Antibiotic loaded chitosan bar. In vitro, in vivo study of a possible treatment for osteomyelitis," Clin Orthop pp. 239247 (September 1999) and fibrin matrices (using technique mentioned in example 5) were also placed in of 250 .mu.g/ml of rapamycin in DMSO solution until complete saturation occurred. After solvent evaporation, the matrices combined with drugs were washed with 0.066 M phosphate buffer (pH 7.4) at 37.degree. C. for 24 hours.
[0080] To compare matrix capacity, fluorescent Rapamycin derivate loaded onto 1.88 cm.sup.2 matrix surface of the same thickness was used. After incubation with 0.14 M NaCl solution, the residual rapamycin was extracted with dimethylsulfoxide (DMSO) and yield was measured using fluorescence spectroscopy. These data are presented in Table 2.
TABLE-US-00002 TABLE 2 Matrix Capacity for Rapamycin Matrix Rapamycin capacity (.mu.g per cm.sup.2) Collagen 124.5 .+-. 14.3 Collagen-alginate 131.1 .+-. 12.3 Chitosan 78.7 .+-. 8.9 Fibrin 145.8 .+-. 12.7
[0081] As expected, capacity of protein matrices was found to be higher than the chitosan matrix, usefulness of fibrin or collagen as therapeutic matrix for antiproliferative drug delivery may depend on particular combination or additional components or requirements of longevity of the matrix.
Example 3
Delivery Systems Using Liposomes
[0082] Liposomes represent a form of drug delivery system, and offer controlled release of biologically active agents. They are used in pharmaceutical formulations especially for water insoluble drugs. Rapamycin is a typical example. Liposomal entrapment has been shown to have considerable effect on the pharmacokinetics and tissue distribution of administered drugs. The formulations tested included nonionic liposomal formulation composed of glyceryl dilaureate (Sigma Chemicals, St Louis, Mo.), cholesterol(Sigma Chemicals, St. Louis, Mo.), and polyoxylene-10-stearyl (Sigma Chemicals, St. Louis, Mo.) either at a weight ratio of 56:12:32 (Formulation 1) or nonionic 40% hydroalcoholic oil-in-water liposomal emulsion containing isopropyl myristate (Sigma Chemicals, St. Louis, Mo.) and mineral oil (Sigma Chemicals, St. Louis, Mo.) (Formulation 2). Rapamycin was entrapped into each formulation at a concentration of 250 .mu.g/ml in dimethylsulfoxide or isopropanol and formed liposomes were applied on surface of prefabricated collagen sheets to create maximal surface density of Rapamycin. Samples were washed with 0.066 M phosphate buffer (pH 7.4) at 37.degree. C. for 24 hours. To compare matrix capacity, liposomes loaded with fluorescent Rapamycin derivate placed onto 1.88 cm.sup.2 disc was used. After incubation with 0.14 M NaCI solution, matrices with remaining Rapamycin were extracted with dimethylsulfoxide (DMSO) and fluorescent yield was measured.
TABLE-US-00003 TABLE 3 Liposomal Delivery System Rapamycin Liposome Type Binding Capacity .mu.g per cm.sup.2 Nonionic cholesterol liposomes 117.4 .+-. 10.9 (Formulation1) Nonionic oil-in-water emulsion 89.6 .+-. 7.5 (Formulation 2) Saturated collagen matrix (DMSO) 124.5 .+-. 14.3 Saturated collagen matrix (isopropanol) 105.6 .+-. 9.7
[0083] Liposomal delivery systems do not have significant advantages over saturated collagen matrix in ability to bind Rapamycin. However the liposomal approach may be useful for other antiproliferative drugs.
Example 4
Preparation of a Laminated Collagen Film
[0084] In order to prepare a textured, surface neutralized, laminated collagen film an isotonic suspension of insoluble fibrillar collagen was obtained. Three liters of chilled collagen suspension at concentration of 5 to 18%, (preferred 12%) was swollen overnight in 0.3-0.6 M acetic acid, (preferred 0.52 M), at 4.degree. C. The swollen suspension was dispersed with 3 liters of crushed ice for 10-20 min, (preferred 12 min.) in a blender and thereafter homogenized for 30 min in an Ultra-Turrax (Alfa, Sweden). The resulting slurry was filtered through a series of filters (Cellector, Bellco, UK) with pore sizes decreasing from 250 .mu.m to 20 .mu.m, mounted in filter holder (Millipore). After degasation at 0.04-0.09 mbar, preferred 0.06 mbar, the slurry was mixed with 2 liters of chilled 0.1-0.05 M NaOH, final pH adjusted to 7.4.+-.0.3. The neutralized suspension can be stored at 4-6.degree. C. only for several hours prior to matrix formation. This neutralized suspension serves as a foundation for preparation of a saturated or dispersed form of a matrix containing rapamycin. The neutralized slurry may be directly cast as a wet film with a thickness of 3 mm on a flat hydrophobic surface at room temperature. A dry film with a thickness of approximately 60-70 .mu.m is formed. Three to five ml of slurry cover an area of 10 cm.sup.2 area. On top of such a surface several layers may be formed. The layers will serve as a basis for preparation of saturated form of anti proliferative agent by immersing the collagen film into solutions of rapamycin, Taxol or combinations thereof. Simultaneous combination of neutralized slurry and rapamycin or other agents in suspension may be used for preparation of film with dispersed form of active ingredients.
[0085] An important factor in the preparation of the matrix material is the porosity of the protein carrier from which the device is to be formed. Porosity may be regulated by drying rate, temperature, and the characteristics of the initial collagen. Porosity is significant because it controls the kinetics of drug release. It is desirable for the matrix to be sufficiently porous to bind small molecules such as rapamycin (Molecular weight 914.2) and durable enough to maintain the shape of device. Samples of collagen matrix with effective pore size of 0.002 to 0.1 microns were tested. Higher binding capacity (to bind rapamycin in saturation experiments) was observed with the matrix having pore size of 0.004 microns. In addition, collagen matrices with bigger pore sizes are fragile. Since the binding capacity of the matrix to the antiproliferative agent is critical for this application, three different concentrations of rapamycin were used to prepare a rapamycin -collagen matrix combination from commercially available collagen prepared at optimal density of pores. The three different concentrations labeled high, medium and low, were 120.+-.5 .mu.g/cm.sup.2, 60.+-.4 .mu.g/cm.sup.2, and 30.+-.3 .mu.g/cm.sup.2, respectively. None of these matrices were fragile or had non-uniform rapamycin distribution. Different densities permit regulating kinetics of drug release.
Example 5
Preparation of an Implantable Fibrin Matrix Device Combined with an Antiproliferative Agent
[0086] In general, to make a device based on a fibrin matrix loaded with an antiproliferative agent, aqueous fibrinogen and thrombin solutions are prepared as described below. Commercial fibrinogen can be acquired from such vendors as Sigma, American Red Cross, or can be prepared from plasma by well-known techniques. Alternatively, fibrinogen prepared by recombinant methods is suitable for use. Commercial active thrombin can be acquired from Sigma or from Johnson and Johnson as thrombin, topical USP, Thrombogen. To make the fibrinogen and thrombin solutions used to prepare the matrix, the necessary components are measured, weighed and dissolved in about 900 ml of deionized water. Tables 4 and 5 disclose preferable compositions used to prepare fibrinogen and thrombin solutions to prefabricate matrix, respectively.
[0087] The glycerol in Table 4 used as a plasticizer. Other plasticizers would also be suitable for the present invention. TRIS buffer is used for pH adjustment. Suitable alternatives for TRIS include HEPES, Tricine and other buffers with a pKa between 6.8 and 8.3. Triton X-100 is a non-ionic detergent and stabilizer and may be substituted by other detergents and stabilizers. Caprylic acid may be substituted by other agents that provide protection from denaturation, for example, alginic acid.
TABLE-US-00004 TABLE 4 Fibrinogen Solution Composition Composition Range Composition Preferred Component g/liter g/liter Fibrinogen 50-120 76 Glycerol 20-80 40.5 TRIS buffer 3-25 12.1 Caprylic Acid 10-35 18.7 Triton X-100 2-8 5.4 Heparin 0.5-6 2.38
TABLE-US-00005 TABLE 5 Thrombin composition Composition range Composition preferred Component (g/liter) (g/liter) Thrombin 5,000-100,000 units 8,000 units Albumin 1-100 50 Factor XIII 1,000-5,000 units 2,500 units CaCl2 50-250 mg/liter 123 mg/liter Troglitazone 3-24 8
[0088] Fibrinogen converted to fibrin is the most critical reagent in the matrix because it controls the material properties of the matrix, such as flexibility, pore size and fiber mass density. These features determine how easily other molecules can diffuse within the matrix and how long the matrix may remain intact before it is resorbed.
[0089] In Table 5, albumin is a stabilizer of thrombin. Thrombin controls the rate of fibrin matrix formation. The presence of Factor XIII is preferred but not necessary. Factor XIII covalently cross-links fibrin, making the matrix more stable. Calcium ions are needed for activation of thrombin. Troglitozone (Sankyo, Japan) is a thiazollidione derivate, which decreases collagen accumulation in the vascular wall. (Yao L, Mizushige K, Murakami K et al. Troglitozone decreases collagen accumulation in prediabetic stage of a type II diabetic rat model. Heart 2000: 84: 209-210
[0090] It is preferable to completely dissolve each component before adding the next component. If necessary, after the last component is dissolved, the pH is adjusted to 7.0-7.4 and the solution volume is adjusted to 1 liter with water. The solutions are then degassed. Both solutions are dispensed by pump through mixture chamber onto a non-stick, preferably hydrophobic, surface to form a film approximately 2 mm thick. The film is then dried for about 3 to 6 hours at temperature in the range of about 20.degree. C. to 60.degree. C., at a pressure of about 30 Torr. Residual moisture of the film is about 10%, preferably less than 3%, of the total wet weight.
[0091] On this surface dry solid Rapamycin is added to create density in the range of 100 to 500 .mu.g per cm.sup.2 of film. A second layer of fibrin matrix is formed on top of this surface such that the drug is sandwiched between the two layers of fibrin.
[0092] In one embodiment of the present invention, one would add (and/or) an antiproliferative/anti restenotic agent like Rapamycin or Taxol, an anti rejection drug like Rapamycin or tacrolimus, an anti-inflammatory drug and/or an antisense oligonucleotide to enhance antirestenotic effects. These solid materials would be added to supplement the fibrin-Rapamycin sandwich complex described above.
Example 6
Method of Cross Linking Chitosan Matrix
[0093] In order to increase binding capacity of a chitosan matrix for antiproliferative drug, cross-linking of fiber is used. Fifty ml of chilled chitosan suspension at concentration from 10% to 25%, (preferred 12%) was gently and slowly mixed with 5 to 25 ml of acrylic acid chloranhydride for 30 min. to acetylate this polymer. After this time period, a solution of rapamycin in DMSO at concentration of 250 .mu.g/ml was added, mixed vigorously, and poured onto the chitosan matrix surface for spontaneous cross-linking and formation of conjugated rapamycin. This approach, because of the microporous structure of the chitozan, allows increasing the binding capacity of the matrix from 15% to 45%.
Example 7
Incorporation of Rapamycin into Collagen Matrix by Dispersion, Immobilization and Immobilization-Dispersion
[0094] Besides the technique of saturation, rapamycin was incorporated into the collagen matrix by three different methods: dispersion, immobilization, and immobilization-dispersion.
[0095] Dispersion technique: an aqueous slurry of water insoluble collagen was prepared using non-crosslinked dry, highly purified, lyophilized calfskin collagen obtained from Elastin Product Co., Inc. (Owensville, Mo.). This collagen and solubilizing buffer are chilled to a temperature of 2-8.degree. C., preferred 4.degree. C. and vigorously mixed to prepare collagen slurry containing 10-21%, (preferred 12%) of collagen protein. Such slurry includes 9% of plasticizer, glycerol 15% o rapamycin in DMSO at concentration of 250 .mu.g/ml and water. The solution had a viscosity of 50,000 cps. Immediately after mixing with rapamycin, 8% glutaraldehyde is added to the slurry (100-350 ml per liter of slurry). The aqueous slurry must be homogenous and degassed, the pH is adjusted to 6.0-7.1. The solution is constantly vigorously mixed and dispersed by pump onto a non-stick surface to form a film approximately 2 mm thick. All procedures are carried out at a temperature of 4.degree. C. The film is then dried for about 3-7 hours at temperatures in the vicinity of 45.degree. C., and a pressure of 15 Torr until its residual moisture is less than about 10% of the total weight. The drug solution application and drying steps are repeated three more times.
[0096] II): Immobilization technique: The same collagen preparation from Elastin Product Co. is used. One volume of 12% collagen slurry is chilled and coupled with rapamycin via esterification of antiproliferative drug. Esterification is carried out with 0.9 M N-hydroxysuccynimide (Pierce Biochemical, Rockford, Ill.) in the presence of 0.9 M N-dicyclohexylocarbodimide (Pierce Biochemical, Rockford, Ill.) at 2-4.degree. C. for 2 days. Conjugates are prepared by titration of active N-hydroxysuccynimide ester of rapamycin in DMSO under the surface of stirred collagen suspension, the pH of the reaction is maintained between 7.0 and 8.5, preferred 7.8. After drying, the films with conjugated rapamycin are washed with 0.15 M NaCl containing 0.02 M sodium bicarbonate at a pH of 7.4. HPLC reveals no free rapamycin in the matrix. Rapamycin ester reacts with amino- or hydroxyl-groups of aminoacid residues forming a covalent linkage with collagen. After such immobilization, Rapamycin is released as a result of in vivo or in vitro degradation-erosion of the matrix. Nakano et al make reference to collagen (SM-10500) degradation and resorption via natural metabolic process in Rhesus monkeys during 6 months Ref: Nakano M, Nakayama Y, Kohda A et al: Acute subcutaneous toxicity of SM-10500 in rats. Kisoto Rinsho (Clinical Report) 1995; 29: 1675-1699]
[0097] In order to study the rate of rapamycin release from the matrix, samples are washed with 0.066 M phosphate buffer (pH 7.4) at 37.degree. C. for 24 hours and cut to give a shape of disc with area of 1.88 cm.sup.2, and placed into 24 well culture plate containing 0.14 M NaCl, 0.05M Tris buffer, 0.5% of albumin, and 0.1 mg/ml collagenase, at pH 7.0. Collagenase is added to increase erosion of collagen matrix and facilitate release of rapamycin. Aliquots are collected at various time intervals from the wells.
[0098] A combination of dispersed and conjugated forms is also prepared. In all these forms, the content of rapamycin is 5.0 .mu.g is per cm.sup.2. The samples are placed in wells and 1 ml of elution media containing serum are added. Aliquots are taken every hour.
[0099] The content of Rapamycin is measured according to the procedure of Ferron et al. (Ferron G M, Conway W D, and Jusko W J. Lipophilic benzamide and anilide derivatives as high-performance liquid chromatography internal standard: application to sirolimus (rapamycin) determination. J Chromatogr B Biomed Sci Appl 1997; December 703: 243-251.) These measurements are made using batch assay and, therefore, represent release rates at 0 ml/min flow rate. The results are tabulated in Table 6 and graphically illustrated in FIG. 14; concentrations of antiproliferative drug are in .mu.g/ml.
[0100] These data show that different forms of drug imbedding and drugs with different solubility have distinct kinetics. In the case of comparatively soluble Tetracycline, after saturation of the collagen matrix with the free base, peak release occurs in a short period of time, whereas for less soluble rapamycin this peak is postponed for several hours. It has been shown in experiments in vitro, that collagen saturated with soluble antibiotics such as gentamicin, cefotaxin, tetracycline or clindamycin delivers these antibiotics at effective concentrations for 4 days. [Wachol-Drewek Z, Pfeifer M, Scholl E. "Comparative investigation of drug delivery of collagen implants saturated in antibiotic solutions and sponge containing gentamicin." (Biomaterials 1996; 17: 1733-1738)].
TABLE-US-00006 TABLE 6 Rate of release of collagen saturated with Tetracycline and Rapamycin. Rapamycin was combined with collagen matrix using four different methods. Combination Collagen Collagen Rapamycin Collagen of Saturated Saturated Dispersed Conjugated Dispersed and Time With With Throughout With Conjugated (Hour) Tetracycline Rapamycin Collagen Rapamycin Forms 1 0.06 0.01 0.01 0 0.01 2 0.4 0.05 0.03 0 0.02 3 0.96 0.09 0.06 0.01 0.07 4 0.54 0.15 0.08 0.02 0.09 5 0.15 0.19 0.12 0.05 0.17 6 0.08 0.28 0.18 0.07 0.26 7 0.02 0.57 0.19 0.11 0.31 8 0.01 0.44 0.29 0.13 0.32 9 0.01 0.24 0.41 0.19 0.34 10 -- 0.20 0.62 0.27 0.41 11 -- 0.19 0.61 0.31 0.78 12 -- 0.18 0.40 0.42 0.76 13 -- 0.15 0.32 0.45 0.79 14 -- 0.02 0.16 0.32 0.45 24 -- 0.11 0.24 0.42 Totally 0 0.003 0.23 0.53 0.39 Dissolved matrix
[0101] In other laboratories is also was shown in vivo, that, collagen saturated with gentamycin at concentration of 3 .mu.g/g and implanted into muscle tissue is capable of delivering antibiotic into blood through day 28. However, concentration was less than optimal. (Mehta S, Humphrey J S, Schenkman D I, et al., "Gentamycin distribution from a collagen carver." J Orthop. Res., 1996; 14: 749-754.). It is theorized that knowing the low concentration of collagenase in perivascular space and the low flow of perivascular fluid (only a few milliliters per day) a matrix material, saturated with rapamycin might produce in vivo delivery kinetics, which will support effective local concentration of antiproliferative drug for a period of several weeks to prevent and combat progress of SMC proliferation. Inhibitory concentrations for SMC would be in the range of 0.001 to 0.005 .mu.g/ml culture media. Such levels are met or exceeded in vitro for 3 weeks. Moreover, Rapamycin dispersed into collagen matrix may exhibit an antiproliferative effect for a month or longer. Finally, conjugated and combined forms may support treatment until complete matrix erosion.
Example 8
Biological Activity of Rapamycin in the Rapamycin-Collagen Matrix
[0102] The most important parameter when assessing the combination of rapamycin and collagen is inhibition of smooth muscle cell (SMC) growth. To evaluate this parameter SMC's at density of 5,000 cells per cm.sup.2 are seeded onto control tissue culture surface and testing matrices (Table 7). Cell growth curves are presented in FIG. 15.
[0103] Actinomycin D is quickly released from the drug matrix and suppresses cell growth for only a short period of time. A change of media removes soluble Actinomycin and after several washes no antibiotic is present in the media or in the matrix. As a result, cells start proliferating as usual. Because of a slow gradual release of rapamycin suppression of cell growth continued throughout the observation period.
TABLE-US-00007 TABLE 7 Comparison of inhibition of growth of smooth muscle cells using collagen matrices saturated with Actinomycin D and Rapamycin Cell number Collagen + Days in Culture Control Axtinomycin D Collagen + Rapamycin 0 5000 5000 5000 1 6430 .+-. 20.4 5230 .+-. 16.8 4800 .+-. 9 2 10240 .+-. 27.1 7350 .+-. 19.5 5040 .+-. 11.2 3 16340 .+-. 30.12 9400 .+-. 13.2 6230 .+-. 13.4 4 27100 .+-. 25.4 14280 .+-. 17.6 7400 .+-. 15.1 5 38450 .+-. 22.6 23540 .+-. 17.8 8000 .+-. 17.8 6 40000 .+-. 20.7 29300 .+-. 19.4 8550 .+-. 13.9 7 40100 .+-. 20.5 32090 .+-. 32.1 8500 .+-. 14.4
Example 9
[0104] Two different types of matrices, collagen and fibrin combined with antiproliferative agents (singly or in combination) along with Vitamin K are added to the cell culture medium in different ratios. Cells are seeded at the same density, on day 5 numbers of viable cells are measured by Alamar blue assay. Data are presented in Table 8.
TABLE-US-00008 TABLE 8 Inhibition of cell growth (%) Collagen Collagen Fibrin Plus plus plus Collagen Rapamycin Rapamycin Fibrin Rapamycin Matrix to Media plus Plus plus plus plus Ratio Rapamycin Taxol Vitamin K Rapamycin Taxol 1:400 5 4 8 3 2 1:200 25 27 34 21 19 1:100 54 50 77 56 55 1:50 73 76 99 79 78 1:25 88 88 99 79 84 1:12.5 95 99 99 98 96 1:6.25 95 99 99 100 98
Example 10
Antiproliferative Effect of Combination of Rapamycin and Heparin Combined to a Collagen Matrix
[0105] Antiproliferative effects of different components combined within a matrix may exhibit a synergy. A combination of dispersed Rapamycin, soluble and immobilized heparin are used. In order to immobilize heparin 5 ml of chilled heparin solution at concentration of 1 mg/ml to 10 mg/ml, (preferred 5 mg/ml) is mixed with 5 to 20 ml, (preferred 11.4 ml) of acrylic acid chloranhydride at the rate of approximately 1 .mu.l per min, (preferred 2.5 .mu.l per min). After addition, mixture is agitated for 30 minutes at a temperature of 4-8.degree. C. The heparinized collagen is extensively washed with sodium phosphate buffered saline at pH 7.4. A colorimetric assay with Eosin A is used to determine the concentration of heparin immobilized on matrix. Using this method between 0.01 mg/cm.sup.2 and 0.1 mg/cm.sup.2 may be covalently linked to the matrix.
[0106] Such a formulation combined with Rapamycin has inhibitory effect on SMC growth in culture if added in the form of suspension into the media at ratio 1:100, whereas individual forms have lesser effects; ratio of 1:25 for heparin alone to 1:65 for dispersed rapamycin. Each of these drugs can inhibit restenosis via different mechanisms, hence it is reasonable to expect synergistic effect when used in combination. Heparin can also be used in matrix saturated form in combination with antiproliferatives.
Example 11
[0107] Sustained local delivery of Dexamethasone in combination with Rapamycin (or other antiproliferative agents) can be used to simultaneously inhibit restenosis as well as inflammatory reactions. Twenty percent (weight/weight) collagen slurry is prepared, to which is added a 2% (weight/weight) suspension of dexamethasone. This mixture is sprayed on to a plastic surface to form the film. The final thickness of the film ranged from 1.92 to 2.14 mm (mean 2 mm). This sheet is flexible and mechanically stable. The kinetics of dexamethasone elution from the c matrix (collagen plus rapamycin) were characterized in an in vitro system. Fifteen mm diameter sheets were placed in the wells and immersed in 2.5 ml of phosphate buffered solution. At time points ranging from 1 to seven days, concentration of dexamethasone in aliquots of elution buffer were measured by spectrophotometry. Chemical stability of the dexamethasone through the sheet formation, drying storage and elution process was confirmed by HPLC. Cumulative in vitro elution of dexamethasone is shown in Table 9.
[0108] More than 50% of the dexamethasone elution occurred within the first three days, with a leveling off of the elution curves after 6 days. Dexamethasone can prevent a severe inflammatory response, which is maximal during this time period and can act synergistically with rapamycin to reduce restenosis. In contrast to a dexamethasone eluting stent, perivascular delivery does not inhibit endothelial cell regeneration and acts directly on fibroblasts and smooth muscle cells.
TABLE-US-00009 TABLE 9 Cumulative in-vitro elution of dexamethasone from a collagen matrix. Eluted Dexamethasone Mass (micrograms) Time (days) 0 0 211 .+-. 23 1 489 .+-. 31 2 605 .+-. 42 3 672 .+-. 38 4 725 .+-. 21 5 733 .+-. 18 6 745 .+-. 13 7
[0109] Combination of macro and micro porosity may increase capacity of the device. Collagen and fibrin matrices were mixed to obtain such a combination. In addition, good mechanical characteristics of collagen improved stability of fibrin. To prepare fibrin-Rapamycin loaded matrix, (Rapamycin density of 150 .mu.g/cm.sup.2) compositions disclosed in Tables 4 and 5 were used. 2. After formation of first dry layer of fibrin, second layer of collagen, rapamycin and heparin was formed as described in example 4 (Rapamycin density of 128 .mu.g/cm.sup.2, heparin density of 5000 U/cm.sup.2). The collagen fibrin sheaths loaded with medicine (thickness 2mm) were formed as tubular structures and externally crosslinked using high concentration of glutaraldehyde (25%) for one minute. After drying, spiral form of sleeve shown in FIG. 4 was prepared. This sleeve was made planar on ten occasions, the spiral shape was restored each time. The Rapamycin capacity of the final sleeve was 143 .mu.g/cm.sup.2. In vitro elution of heparin continues for 7 days.
[0110] Heparin concentration was measured as in example 10, buffer for the dilution was replenished each day. The data are shown in Table 10.
[0111] It is known that effective concentration of heparin to inhibit SMC proliferation is in the range of 100 .mu.g/ml. In this example, heparin can significantly inhibit SMC proliferation for at least 4 days In addition diffusion of heparin form the sleeve can prevent thrombotic events on the inner surface of the shunt and damaged vessel wall for longer periods of time. Besides, concentration of soluble heparin can be increased up to 20,000 units/cm.sup.2 without changing mechanical characteristics of the matrix. Therefore, anti smooth muscle cell proliferation as well as antithrombotic effect can be prolonged.
TABLE-US-00010 TABLE 10 Elution profile of heparin from a collagen matrix combined with rapamycin and heparin. Time (days) Eluted Heparin Mass (U/ml) 0 0 1 341 2 275 3 188 4 103 5 57 6 24
Examples 13 and 14
Comparison of In Vitro Effect of Rapamycin, Tacrolimus and Paclitaxel on Human Smooth Muscle and Endothelial Cells
[0112] Human smooth muscle cells and endothelial cells (Clonetics, USA) were seeded (100,000 cells) in 24 well plates overnight. Both cell types were grown and maintained in OPTI-MEM (Gibco, Long Island, N.Y.) and 5% fetal bovine serum at 37.degree. C. in a 5% carbon dioxide and 95% atmospheric air. Cells were exposed to a range of concentrations of Rapamycin (10-100 nM), Paclitaxel (0.1-10 mM) and Tacrolimus (10-100 nM). Each cell type was allowed to grow for 24 hours, last four hours in the presence of [.sup.3H]-thymidine. Proliferation of cells was quantified as new DNA synthesis using .sup.3H-thymidine uptake assay. After 72 hours of culture, cells were washed twice with cold phosphate buffered saline (PBS) and 1 ml of methanol was added to the contents of each well, the plates were kept at 4.degree. C. for 60 minutes, cells were then washed once with cold PBS and 500 microliter of 0.2 m NaOH was added to each well and the plates were kept at 4.degree. C. for 30 minutes. The contents of each well were transferred into scintillation vials and liquid scintillation fluid was added to quantify radioactivity using a liquid scintillation counter and results expressed as counts per minute.
[0113] Results are shown in Tables 11 and 12 and corresponding FIGS. 16 and 17 respectively. Rapamycin and Paclitaxel inhibit proliferation of both human smooth muscle and endothelial cells (new DNA synthesis). Tacrolimus appears to preferentially inhibit new DNA synthesis in human smooth muscle cells, sparing endothelial cells. This differential effect may be extremely important and can be beneficially exploited if Tacrolimus were to be used for inhibition of smooth muscle cell proliferation.
TABLE-US-00011 TABLE 11 Comparison of Effect of Rapamycin, Tacrolimus and Paclitaxel (3 doses) on Human Smooth Muscle Cells [.sup.3H] - thymidine uptake Assay Mean (.+-.SD) p Untreated (Control) 17434 (1822) Rapamycin 6498 (245) <0.01 Tacrolimus 11995 (1850) <0.05 Paclitaxel 2421 (206) <0.001 Paclitaxel 2527 (195) <0.001 Paclitaxel 2710 (162) <0.001
TABLE-US-00012 TABLE 12 Comparison of Effect of Rapamycin, Tacrolimus and Paclitaxel (3 doses) on Human Endothelial Cells [.sup.3H] - thymidine uptake Assay Mean (.+-.SD) p Unttreated (Control) 16342 (3039) Rapamycin 5787 (1323) <0.01 Tacrolimus 16073 (3008) ns Paclitaxel 2222 (228) <0.001 Paclitaxel 2648 (248) <0.001 Paclitaxel 3459 (272) <0.001
[0114] Animal Studies
[0115] A proof of principle study was performed using a porcine model. A total of 6 pigs were studied, 2 were used as controls and 4 were treated. A 6 mm PTFE vascular graft was anastomosed between the carotid artery on one side and the contralateral jugular vein, this created an arterio venous (AV) loop that is similar in construction to the human hemodialysis access loop. A collagen sleeve combined with a known dose of Rapamycin (approximately 500 .mu.gm/cm.sup.2) was placed around the distal end of the PTFE vascular graft just proximal to the venous anastomosis in the treated group.
[0116] After 30 days an angiogram was performed to demonstrate vessel and graft patency. The animals were euthanized and the relevant segments dissected. The inhibitory effect of Rapamycin on cell cycle progression, is believed to be via induction of cyclin inhibitors. Hence, expression of p21 will increase in tissues obtained from rapamycin treated animals but not from controls. In other words, the presence of p21 is confirmation that that the observed effect is attributable to Rapamycin. Tissues from treated and untreated animals were obtained, RNA was prepared and reverse transcribed to cDNA, which was amplified for house keeping gene, b-actin and p21 by PCR.
[0117] Results
[0118] Both controls had luminal narrowing caused by severe neo-intimal hyperplasia at the site of venous anastomosis (FIGS. 18A and 19A). All 4 treated animals had significantly higher luminal patency of the vein and the graft, with minimal to absent neo intimal hyperplasia (FIGS. 18B and 19B). Expression of p21 mRNA was observed in venous tissue at the perianastamotic site obtained from rapamycin treated animals (FIG. 20) but not from controls. This demonstrates that the Rapamycin contained in the sleeve matrix was responsible for the reduction/virtual abolition of neo intimal hyperplasia (an expression of the vasculoproliferative response) an effect mediated through rapamycin induced inhibition of cellular proliferation.
* * * * *
D00000
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
D00012

D00013
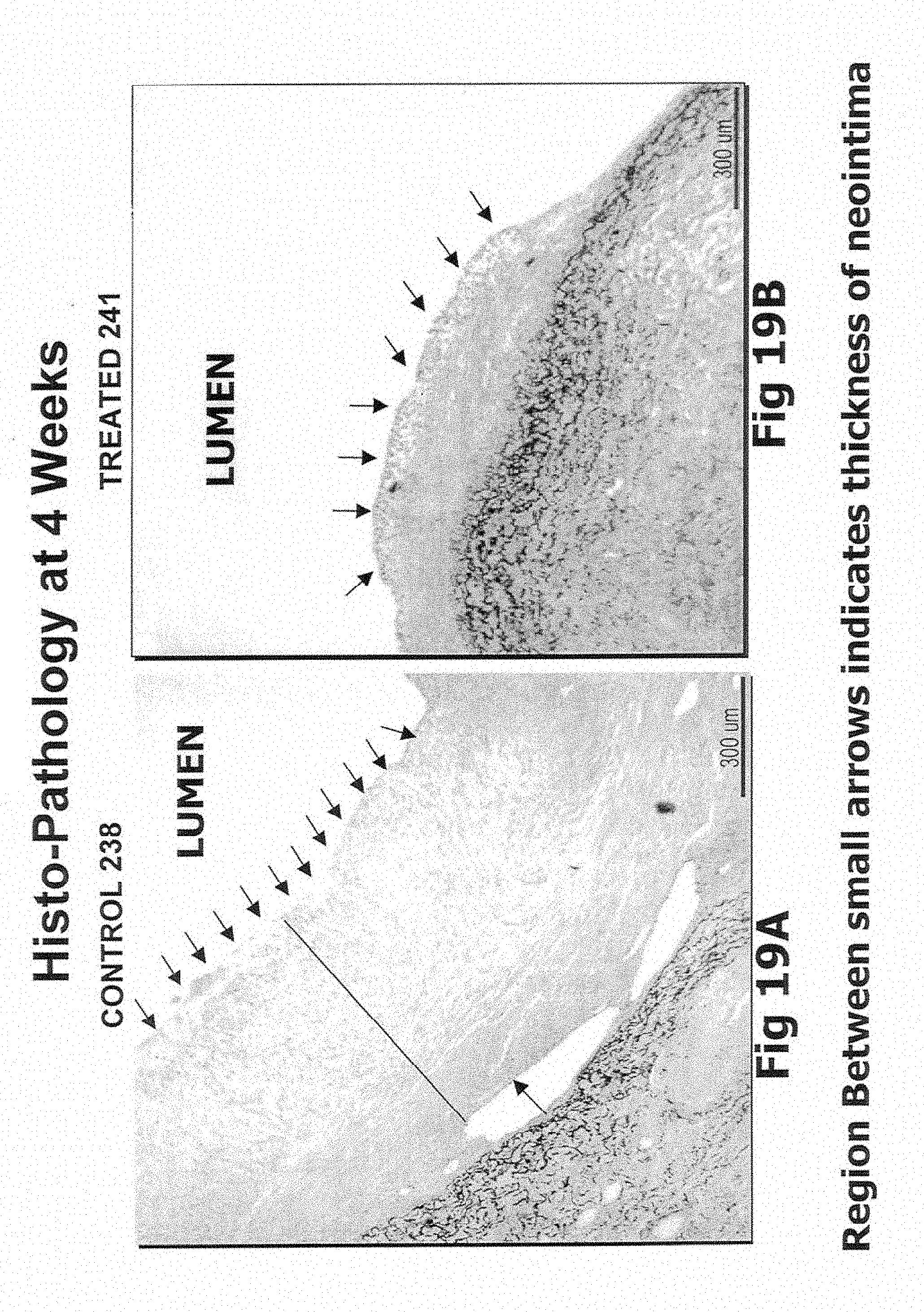
D00014
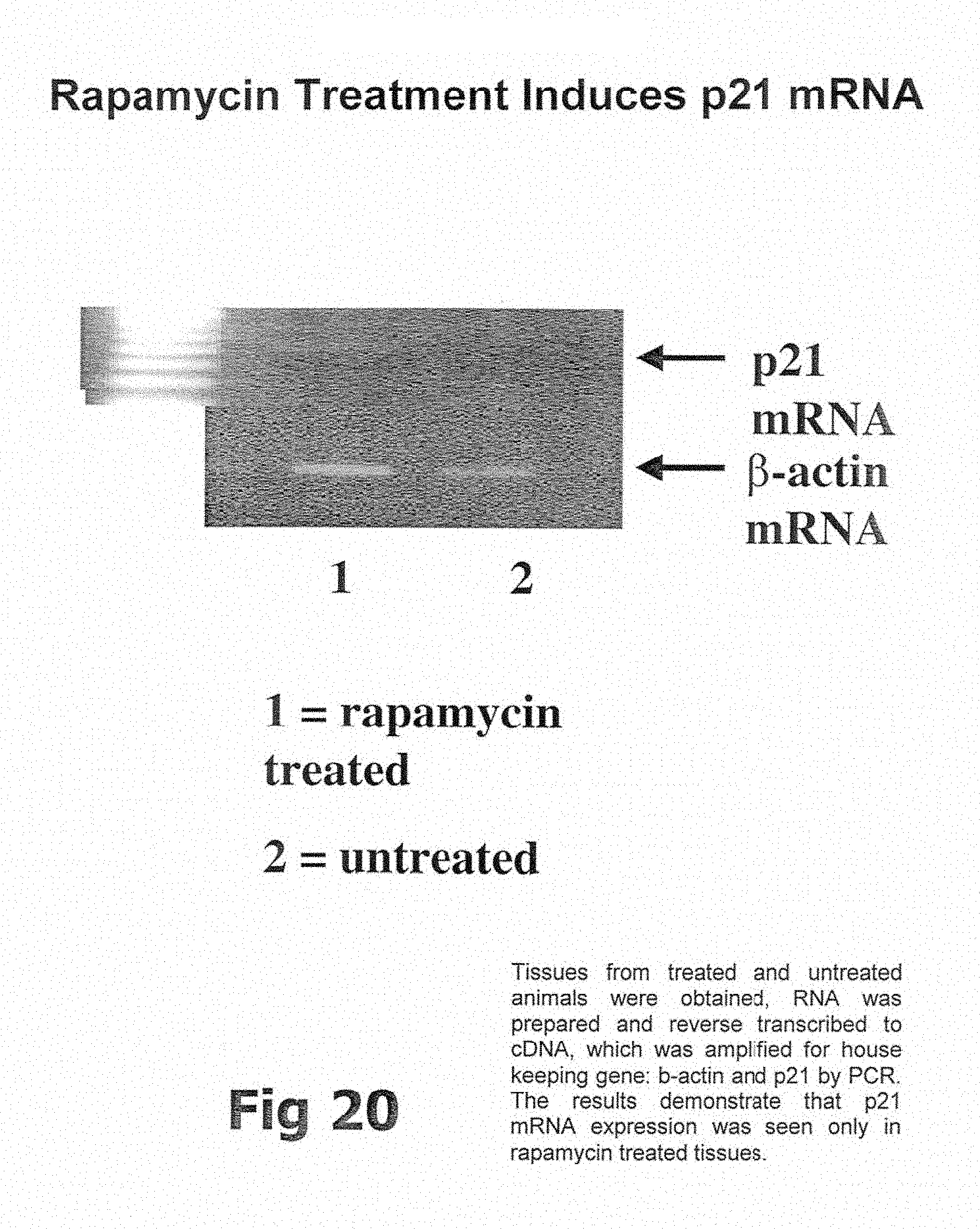
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.