Preparation of Treatment Composition and System and Method of Maintaining a Treatment Bath Formed Therefrom
Post; Gordon L. ; et al.
U.S. patent application number 16/324977 was filed with the patent office on 2019-06-13 for preparation of treatment composition and system and method of maintaining a treatment bath formed therefrom. This patent application is currently assigned to PRC-DeSoto International, Inc.. The applicant listed for this patent is PRC-DeSoto International, Inc.. Invention is credited to Michael A. Mayo, Eric L. Morris, Michael J. Pawlik, Gordon L. Post, Edward F. Rakiewicz.
| Application Number | 20190177855 16/324977 |
| Document ID | / |
| Family ID | 59762041 |
| Filed Date | 2019-06-13 |



| United States Patent Application | 20190177855 |
| Kind Code | A1 |
| Post; Gordon L. ; et al. | June 13, 2019 |
Preparation of Treatment Composition and System and Method of Maintaining a Treatment Bath Formed Therefrom
Abstract
Disclosed is a method of making a treatment composition. A lithium cation and carbon dioxide are combined in an aqueous medium to form the treatment composition comprising lithium carbonate in situ. Also disclosed is a system and method for maintaining a treatment bath formed from a treatment composition comprising lithium carbonate. Carbon dioxide and/or a lithium salt are supplied to the bath in an amount sufficient to maintain the pH of the treatment bath at 9.5 to 12.5, lithium in an amount of 5 ppm to 5,500 ppm (calculated as lithium cation) and carbonate in an amount of 15 ppm to 25,000 ppm (calculated as carbonate) based on total weight of the treatment bath. Substrates treated with the composition, system and method also are disclosed.
| Inventors: | Post; Gordon L.; (Pittsburgh, PA) ; Pawlik; Michael J.; (Glenshaw, PA) ; Morris; Eric L.; (Murrieta, CA) ; Rakiewicz; Edward F.; (Gibsonia, PA) ; Mayo; Michael A.; (Pittsburgh, PA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Assignee: | PRC-DeSoto International,
Inc. Sylmar CA |
||||||||||
| Family ID: | 59762041 | ||||||||||
| Appl. No.: | 16/324977 | ||||||||||
| Filed: | August 14, 2017 | ||||||||||
| PCT Filed: | August 14, 2017 | ||||||||||
| PCT NO: | PCT/US2017/046705 | ||||||||||
| 371 Date: | February 12, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 62374188 | Aug 12, 2016 | |||
| 62526382 | Jun 29, 2017 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | C23C 22/66 20130101; C23C 22/86 20130101; C23C 22/83 20130101; C01D 15/00 20130101 |
| International Class: | C23C 22/66 20060101 C23C022/66; C23C 22/83 20060101 C23C022/83; C23C 22/86 20060101 C23C022/86 |
Claims
1. A composition comprising lithium carbonate, wherein the lithium carbonate is formed in situ by reacting a carbon dioxide source and a lithium cation in an aqueous medium.
2. The composition of claim 1, wherein the carbon dioxide source comprises a gas, a solid, or combinations thereof.
3. The composition of claim 1, wherein the lithium cation is present in an amount of 5 ppm to 5500 ppm (calculated as lithium cation) based on total weight of the composition.
4. The composition of claim 1, wherein the pH is 9.5 to 12.5.
5. The composition of claim 1, further comprising a hydroxide source.
6. The composition of claim 1, wherein the carbonate is present in an amount of 15 ppm to 25000 ppm (calculated as carbonate) based on total weight of the treatment composition.
7. A substrate treated with the composition of claim 1.
8. A method of making a treatment composition comprising: combining a lithium cation and a carbon dioxide source in an aqueous medium to form the treatment composition wherein the treatment composition comprises lithium cation in an amount of 5 ppm to 5500 ppm (calculated lithium cation) based on total weight of the treatment composition and carbonate in an amount of 15 ppm to 25000 ppm (calculated as carbonate) based on total weight of the treatment composition.
9. The method of claim 8, wherein the pH of the treatment composition is 9.5 to 12.5.
10. The method of claim 8, further comprising adding an acid other than carbon dioxide to the treatment composition.
11. The method of claim 8, wherein the lithium cation is present as a salt comprising lithium carbonate, lithium hydroxide, or combinations thereof.
12. The method of claim 8, further comprising adding a hydroxide source to the aqueous medium.
13. The method of claim 8, wherein the hydroxide source comprises lithium hydroxide, sodium hydroxide, potassium hydroxide, or combinations thereof.
14. The method of claim 8, wherein the carbon dioxide source comprises a gas, a solid, or combinations thereof.
15. A substrate treated with the method of claim 8.
16. A system for maintaining carbonate levels in a treatment bath containing a treatment composition, comprising: a lithium cation; and/or a carbon dioxide source; and optionally, a hydroxide source.
17. The system of claim 16, wherein the lithium cation comprises lithium carbonate, lithium hydroxide, or combinations thereof.
18. The system of claim 16, wherein the carbon dioxide source comprises a gas, a solid, or combinations thereof.
19. The system of claim 16, wherein the hydroxide source comprises lithium hydroxide, sodium hydroxide, potassium hydroxide, or combinations thereof.
20. A substrate treated with the treatment composition in the bath maintained according to the system of claim 16.
21. A method for maintaining carbonate levels in a treatment bath containing a treatment composition, comprising: supplying at least one of a carbon dioxide source and a lithium source to the bath in an amount sufficient to maintain the pH of the treatment composition at 9.5 to 12.5, lithium in an amount of 5 ppm to 5500 ppm (calculated as lithium cation) based on total weight of the treatment composition, and carbonate in an amount of 15 ppm to 25000 ppm (calculated as carbonate) based on total weight of the treatment composition.
22. The method of claim 21, wherein the carbon dioxide source comprises a gas, a solid, or combinations thereof.
23. The method of claim 21, wherein the lithium source comprises lithium carbonate, lithium hydroxide, or combinations thereof.
24. The method of claim 21, further comprising supplying to the bath a hydroxide source.
25. The method of claim 21, wherein the amount of lithium carbonate in the treatment bath following the supplying is 25 ppm to 30000 ppm (calculated as total compound) based on total weight of the treatment composition.
26. The method of claim 21, further comprising monitoring pH of the treatment bath, amount of carbonate in the treatment bath, amount of lithium in the treatment bath, or combinations thereof.
27. The method of claim 21, further comprising adding an acid other than carbon dioxide to the treatment bath.
28. A substrate treated with the treatment composition in the bath maintained according to the method of claim 21.
Description
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims priority to U.S. Provisional Application No. 62/374,188, filed on Aug. 12, 2016 and entitled "Sealing Composition" and to U.S. Provisional Application No. 62/526,382, filed on Jun. 29, 2017 and entitled "Preparation of Treatment Composition and System and Method of Maintaining a Treatment Bath Formed Therefrom," both of which are incorporated in their entireties herein by reference.
FIELD OF THE INVENTION
[0002] The present invention relates to treatment compositions for the treatment of substrates such as metal substrates, such as to treatment compositions for forming a protective coating on the surface, and also to the preparation of such compositions and systems and methods of maintaining treatment baths formed from such treatment composition.
BACKGROUND OF THE INVENTION
[0003] The use of protective coatings on metal surfaces for improved corrosion resistance and paint adhesion characteristics is well known in the metal finishing arts. Conventional techniques involve treating metal substrates with treatment compositions containing phosphate and chromium for promoting corrosion resistance and adherence of the coating formed by the treatment composition to the substrate surface. The use of such phosphate and/or chromate-containing compositions, however, gives rise to environmental and health concerns. As a result, chromate-free and/or phosphate-free treatment compositions have been developed.
[0004] During a typical treatment process, as a treatment composition is contacted with a substrate, certain ingredients, such as metal ions in the treatment composition, deposit on or bind to the substrate's surface to form a protective layer. As a result, the concentration of those ions in the composition may be diminished during the process, which may adversely affect the coating characteristics and reproducibility among substrates coated successively in the same coating composition. Accordingly, it would be desirable to provide treatment compositions which do not give rise to environmental and health concerns and can be used to form protective coatings having efficient corrosion protection and adhesion characteristics on a substrate surface, and means to avoid or at least alleviate compositional variations upon continued use of such compositions for treating substrates and associated adverse effects on coating characteristics and reproducibility. The present invention therefore aims to provide treatment compositions which are environmentally safe and health benign, can be produced in a cost-efficient manner from readily available resources and yet may form a protective layer imparting efficient corrosion protection and having suitable adhesion on a substrate surface comparable to phosphate and/or chromate-containing compositions.
[0005] Another objective resides in providing a method and a system which enable continued use of treatment baths formed from such compositions for treating substrates yielding coatings of desirable characteristics in a reproducible manner without compositional variations that impact corrosion or adhesion performance.
SUMMARY OF THE INVENTION
[0006] These objectives are solved by the treatment composition and method of making the same and the method and system for maintaining a treatment bath as specified in the appended claims and described in more detail in the following description.
[0007] The treatment compositions described herein generally comprise a carbon dioxide source, a lithium cation, which may be in the form of a lithium salt, and an aqueous medium.
[0008] The treatment composition may comprise lithium carbonate, wherein the lithium carbonate may be formed by reacting carbon dioxide and a lithium cation in situ in an aqueous medium.
[0009] The present invention thus relates to a method of making a treatment composition comprising: combining a lithium cation and carbon dioxide in an aqueous medium to form a treatment composition comprising lithium in an amount of 5 ppm to 5,500 ppm (calculated as lithium cation) based on total weight of the treatment composition and carbonate in an amount of 15 ppm to 25,000 ppm (calculated as carbonate) based on total weight of the treatment composition.
[0010] The present invention relates furthermore to a system for maintaining a treatment bath formed from a treatment composition comprising lithium carbonate, the system comprising: a lithium salt; and/or carbon dioxide; and optionally, a hydroxide source.
[0011] Also part of the present invention is a method for maintaining a treatment bath formed from a treatment composition comprising lithium carbonate, the method comprising: supplying during and/or after treatment of a substrate with the bath at least one of carbon dioxide and a lithium salt to the bath in an amount sufficient to maintain the pH of the treatment bath at 9.5 to 12.5, lithium in an amount of 5 ppm to 5,500 ppm (calculated as lithium cation) based on total weight of the treatment bath, and carbonate in an amount of 15 ppm to 25,000 ppm (calculated as carbonate) based on total weight of the treatment bath.
[0012] The present invention moreover relates to substrates treated with such compositions and maintained treatment baths. The coating characteristics and reproducibility of coatings formed on substrates treated with such compositions and maintained treatment baths are more consistent in successively treated substrates than are coating characteristics and reproducibility of coatings formed on substrates treated with compositions that are not formed or maintained in this manner. Accordingly, the protective coatings formed from compositions and treatment baths maintained according to the present invention are reproducible and exhibit suitable corrosion performance and adhesion on the substrate surface.
BRIEF DESCRIPTION OF THE DRAWINGS
[0013] FIG. 1 shows a flow diagram detailing the sequential steps used to prepare the treatment baths containing the treatment compositions used in Examples D to J.
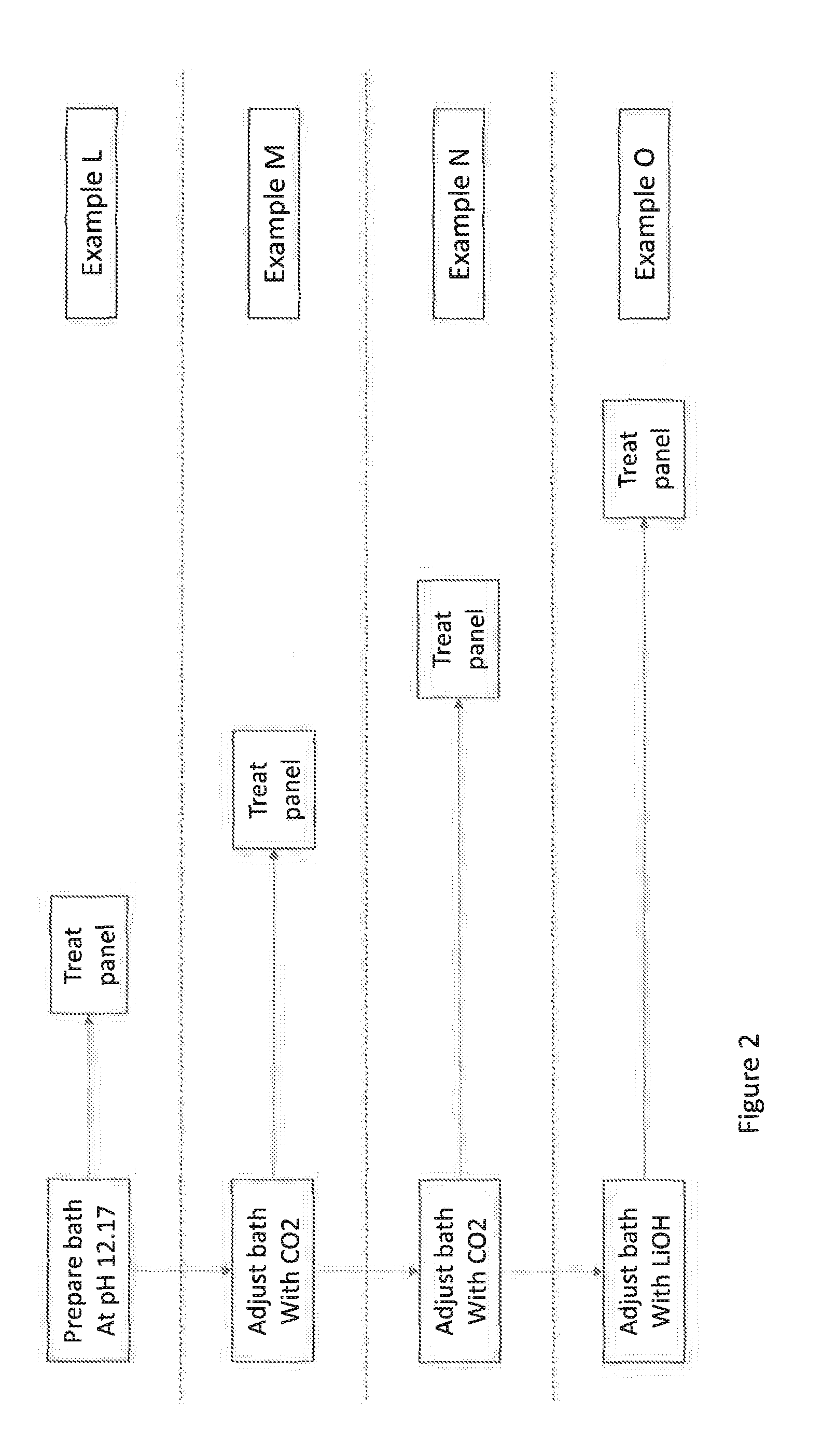
[0014] FIG. 2 shows a flow diagram detailing the sequential steps used to prepare the treatment baths containing the compositions used in Examples L to O.
[0015] FIG. 3 shows a schematic illustrating the thickness of a layer of the treatment composition on a substrate surface.
DETAILED DESCRIPTION OF THE INVENTION
[0016] For purposes of the following detailed description, it is to be understood that the invention may assume various alternative variations and step sequences, except where expressly specified to the contrary. Moreover, other than in any operating examples, or where otherwise indicated, all numbers such as those expressing values, amounts, percentages, ranges, subranges and fractions may be read as if prefaced by the word "about," even if the term does not expressly appear. Accordingly, unless indicated to the contrary, the numerical parameters set forth in the following specification and attached claims are approximations that may vary depending upon the desired properties to be obtained by the present invention. At the very least, and not as an attempt to limit the application of the doctrine of equivalents to the scope of the claims, each numerical parameter should at least be construed in light of the number of reported significant digits and by applying ordinary rounding techniques. Where a closed or open-ended numerical range is described herein, all numbers, values, amounts, percentages, subranges and fractions within or encompassed by the numerical range are to be considered as being specifically included in and belonging to the original disclosure of this application as if these numbers, values, amounts, percentages, subranges and fractions had been explicitly written out in their entirety.
[0017] Notwithstanding that the numerical ranges and parameters setting forth the broad scope of the invention are approximations, the numerical values set forth in the specific examples are reported as precisely as possible. Any numerical value, however, inherently contains certain errors necessarily resulting from the standard variation found in their respective testing measurements.
[0018] As used herein, unless indicated otherwise, a plural term can encompass its singular counterpart and vice versa, unless indicated otherwise. For example, although reference is made herein to "a" lithium salt, "a" hydroxide, and "a" treatment composition, a combination (i.e., a plurality) of these components can be used. In addition, in this application, the use of "or" means "and/or" unless specifically stated otherwise, even though "and/or" may be explicitly used in certain instances.
[0019] As used herein, "including," "containing" and like terms are understood in the context of this application to be synonymous with "comprising" and are therefore open-ended and do not exclude the presence of additional undescribed and/or unrecited elements, materials, ingredients and/or method steps. As used herein, "consisting of" is understood in the context of this application to exclude the presence of any unspecified element, ingredient and/or method step. As used herein, "consisting essentially of" is understood in the context of this application to include the specified elements, materials, ingredients and/or method steps "and those that do not materially affect the basic and novel characteristic(s)" of what is being described.
[0020] As used herein, the terms "on," "onto," "applied on," "applied onto," "formed on," "deposited on," "deposited onto," mean formed, overlaid, deposited, and/or provided on but not necessarily in contact with the surface. For example, a coating layer "formed over" a substrate does not preclude the presence of one or more other intervening coating layers of the same or different composition located between the formed coating layer and the substrate.
[0021] Unless otherwise disclosed herein, the term "substantially free," when used with respect to the absence of a particular material, means that such material, if present at all in a composition, a bath containing the composition, and/or layers formed from and comprising the composition, only is present in a trace amount of 5 ppm or less based on a total weight of the composition, bath and/or layer(s), as the case may be. Unless otherwise disclosed herein, the term "essentially free," when used with respect to the absence of a particular material, means that such material, if present at all in a composition, a bath containing the composition, and/or layers formed from and comprising the composition, only is present in a trace amount of 1 ppm or less based on a total weight of the composition, bath and/or layer(s), as the case may be. Unless otherwise disclosed herein, the term "completely free," when used with respect to the absence of a particular material, means that such material, if present at all in a composition, a bath containing the composition, and/or layers formed from and comprising the composition, is absent from the composition, the bath containing the composition, and/or layers formed from and comprising same (i.e., the composition, bath containing the composition, and/or layers formed from and comprising the composition contain 0 ppm of such material). When a composition, bath containing a composition, and/or a layer(s) formed from and comprising the same is substantially free, essentially free, or completely free of a particular material, this means that such material is excluded therefrom, except that the material may be present as a result of, for example, carry-over from prior treatment baths in the processing line, municipal water sources, substrate(s), and/or dissolution of equipment.
[0022] As used herein, a "salt" refers to an ionic compound made up of metal cations and non-metallic anions and having an overall electrical charge of zero. Salts may be hydrated or anhydrous.
[0023] As used herein, "aqueous composition" refers to a solution or dispersion in a medium that comprises predominantly water. For example, the aqueous medium may comprise water in an amount of more than 50 wt. %, or more than 70 wt. % or more than 80 wt. % or more than 90 wt. % or more than 95 wt. %, based on the total weight of the medium. The aqueous medium may for example consist substantially of water.
[0024] As used herein, the term "oxidizing agent," when used with respect to a component of the sealing composition, refers to a chemical which is capable of oxidizing at least one of: a metal present in the substrate which is contacted by the sealing composition and/or a metal-complexing agent present in the sealing composition. As used herein with respect to "oxidizing agent," the phrase "capable of oxidizing" means capable of removing electrons from an atom or a molecule present in the substrate or the sealing composition, as the case may be, thereby decreasing the number of electrons.
[0025] As used herein, the term "Group IA metal" refers to an element that is in Group IA of the CAS version of the Periodic Table of the Elements as is shown, for example, in the Handbook of Chemistry and Physics, 63.sup.rd edition (1983), corresponding to Group 1 in the actual IUPAC numbering.
[0026] As used herein, the term "Group IA metal compound" refers to compounds that include at least one element that is in Group IA of the CAS version of the Periodic Table of the Elements.
[0027] As used herein, the term "Group IIA metal" refers to an element that is in Group IIA of the CAS version of the Periodic Table of the Elements as is shown, for example, in the Handbook of Chemistry and Physics, 63.sup.rd edition (1983), corresponding to Group 2 in the actual IUPAC numbering.
[0028] As used herein, the term "Group IIA metal compound" refers to compounds that include at least one element that is in Group IIA of the CAS version of the Periodic Table of the Elements.
[0029] As used herein, the term "Group IIIB metal" refers to yttrium and scandium of the CAS version of the Periodic Table of the Elements as is shown, for example, in the Handbook of Chemistry and Physics, 63.sup.rd edition (1983), corresponding to Group 3 in the actual IUPAC numbering. For clarity, "Group IIIB metal" expressly excludes lanthanide series elements.
[0030] As used herein, the term "Group IIIB metal compound" refers to compounds that include at least one element that is in group IIIB of the CAS version of the Periodic Table of the Elements as defined above.
[0031] As used herein, the term "Group IVB metal" refers to an element that is in group IVB of the CAS version of the Periodic Table of the Elements as is shown, for example, in the Handbook of Chemistry and Physics, 63.sup.rd edition (1983), corresponding to Group 4 in the actual IUPAC numbering.
[0032] As used herein, the term "Group IVB metal compound" refers to compounds that include at least one element that is in Group IVB of the CAS version of the Periodic Table of the Elements.
[0033] As used herein, the term "Group VB metal" refers to an element that is in group VB of the CAS version of the Periodic Table of the Elements as is shown, for example, in the Handbook of Chemistry and Physics, 63.sup.rd edition (1983), corresponding to Group 5 in the actual IUPAC numbering.
[0034] As used herein, the term "Group VB metal compound" refers to compounds that include at least one element that is in Group VB of the CAS version of the Periodic Table of the Elements.
[0035] As used herein, the term "Group VIB metal" refers to an element that is in group VIB of the CAS version of the Periodic Table of the Elements as is shown, for example, in the Handbook of Chemistry and Physics, 63.sup.rd edition (1983), corresponding to Group 6 in the actual IUPAC numbering.
[0036] As used herein, the term "Group VIB metal compound" refers to compounds that include at least one element that is in Group VIB of the CAS version of the Periodic Table of the Elements.
[0037] As used herein, the term "Group VIIB metal" refers to an element that is in Group IA of the CAS version of the Periodic Table of the Elements as is shown, for example, in the Handbook of Chemistry and Physics, 63.sup.rd edition (1983), corresponding to Group 7 in the actual IUPAC numbering.
[0038] As used herein, the term "Group VIIB metal compound" refers to compounds that include at least one element that is in Group VIIB of the CAS version of the Periodic Table of the Elements.
[0039] As used herein, the term "Group XII metal" refers to an element that is in Group IA of the CAS version of the Periodic Table of the Elements as is shown, for example, in the Handbook of Chemistry and Physics, 63.sup.rd edition (1983), corresponding to Group 12 in the actual IUPAC numbering.
[0040] As used herein, the term "Group XII metal compound" refers to compounds that include at least one element that is in Group XII of the CAS version of the Periodic Table of the Elements.
[0041] As used herein, the term "lanthanide series elements" refers to elements 57-71 of the CAS version of the Periodic Table of the Elements and includes elemental versions of the lanthanide series elements. According to the present invention, the lanthanide series elements may be those which have both common oxidation states of +3 and +4, referred to hereinafter as +3/+4 oxidation states.
[0042] As used herein, the term "lanthanide compound" refers to compounds that include at least one of elements 57-71 of the CAS version of the Periodic Table of the Elements.
[0043] As used herein, a "sealing composition" refers to a composition, e.g. a solution or dispersion, that affects a substrate surface or a material deposited onto a substrate surface in such a way as to alter the physical and/or chemical properties of the substrate surface (e.g., the composition affords corrosion protection).
[0044] As used herein, a "conversion composition" refers to a composition, e.g., a solution or dispersion, that is capable of reacting with and chemically altering the substrate surface and binding to it to form a film that affords corrosion protection.
[0045] As used herein, a "treatment bath" refers to an aqueous bath formed from an initial treatment composition. The treatment bath may contain components that are byproducts of the process of contacting a substrate with the treatment composition.
[0046] As used herein, "maintaining" a treatment bath formed from a treatment composition refers to keeping certain parameters of the treatment bath including the concentration of certain ingredients and/or the pH in desirable ranges. This can be achieved, as described in more detail below, by the addition of one or more materials from a respective source to the treatment bath on-shift and/or off-shift. As used herein, "on-shift" means that an article to be treated is present in the treatment bath. As used herein, "off-shift" means that an article to be treated by the treatment composition is absent from the treatment bath, but does not mean that the treatment bath is necessarily removed from the process line.
[0047] Pitting corrosion is the localized formation of corrosion by which cavities or holes are produced in a substrate. The term "pit," as used herein, refers to such cavities or holes resulting from pitting corrosion and is characterized by (1) a rounded, elongated or irregular appearance when viewed normal to the test panel surface, (2) a "comet-tail", a line, or a "halo" (i.e., a surface discoloration) emanating from the pitting cavity, and (3) the presence of corrosion byproduct (e.g., white, grayish or black granular, powdery or amorphous material) inside or immediately around the pit. An observed surface cavity or hole must exhibit at least two of the above characteristics to be considered a corrosion pit. Surface cavities or holes that exhibit only one of these characteristics may require additional analysis before being classified as a corrosion pit. Visual inspection using a microscope with 10.times. magnification is used to determine the presence of corrosion byproducts when corrosion byproducts are not visible with the unaided eye.
[0048] Unless otherwise disclosed herein, as used herein, the terms "total composition weight", "total bath weight", "total weight of a composition", "total weight of a treatment bath" or similar terms refer to the total weight of all ingredients being present in the respective composition or bath including any carriers and solvents.
[0049] As mentioned above, according to the present invention, disclosed is a treatment composition comprising lithium carbonate. The lithium carbonate may in particular be formed in situ as set forth above by reacting carbon dioxide and a lithium cation, which may be in the form of a lithium salt, for example, in an aqueous medium. The treatment composition may be a sealing composition, a conversion composition, or the like.
[0050] The treatment composition of the present invention is typically alkaline. According to the present invention, the pH of the treatment composition may be at least 9.5, such as at least 10, such as at least 11, and in some instances, may be no greater than 12.5, such as no greater than 12, such as no greater than 11.5. According to the present invention, the pH of the treatment composition may be 9.5 to 12.5, such as 10 to 12, such as 11 to 11.5. According to the present invention, the pH of the treatment composition may be adjusted through the inclusion of an acidic material, including carbon dioxide, water soluble and/or water dispersible acids, such as nitric acid, sulfuric acid, and/or phosphoric acid. According to the present invention, the pH of the treatment composition may be adjusted through the inclusion of a basic material, including water soluble and/or water dispersible bases including carbonates, such as Group I carbonates, Group II carbonates, hydroxides, such as lithium hydroxide, sodium hydroxide, potassium hydroxide, or ammonium hydroxide, ammonia, and/or amines such as triethylamine, methylethyl amine, or mixtures thereof.
[0051] According to the present invention, the carbon dioxide used to form the treatment composition of the present invention may be a gas, a solid (i.e., dry ice), or a combination thereof.
[0052] According to the present invention, the lithium salt used to form the treatment composition of the present invention may comprise an inorganic lithium salt, an organic lithium salt, or combinations thereof. According to the present invention, the anion and the cation of the lithium salt both may be soluble in water. According to the present invention, the lithium salt may have a solubility constant in water at a temperature of 25.degree. C. (K; 25.degree. C.) of at least 1.times.10.sup.-11, such as least 1.times.10.sup.-4, and in some instances, may be no more than 5.times.10.sup.+2. According to the present invention, the lithium salt may have a solubility constant in water at a temperature of 25.degree. C. (K; 25.degree. C.) of 1.times.10.sup.-11 to 5.times.10.sup.+2, such as 1.times.10.sup.-4 to 5.times.10.sup.+2. As used herein, "solubility constant" means the product of the equilibrium concentrations of the ions in a saturated aqueous solution of the respective lithium salt. Each concentration is raised to the power of the respective coefficient of ion in the balanced equation. The solubility constants for various salts can be found in the Handbook of Chemistry and Physics. Examples of suitable lithium salts are lithium carbonate, lithium hydroxide, lithium phosphate, lithium sulphate, and lithium tetraborate.
[0053] Optionally, the treatment composition also may comprise a hydroxide, such as an alkaline metal hydroxide, an alkaline earth metal hydroxide, or a combination thereof. According to the present invention, the hydroxide may be one or more Group I hydroxide(s), ammonium hydroxide, or mixtures thereof. The hydroxide, if present at all, may be present in any amount, such as in an amount that the pH of the treatment composition remains 9.5 to 12.5. Nonlimiting examples of Group I hydroxides include sodium hydroxide, potassium hydroxide, lithium hydroxide, or mixtures thereof. Accordingly, the hydroxide, if used, may be supplied as the lithium salt component used to form the treatment composition or part thereof, e.g. as lithium hydroxide, optionally in combination with other lithium salts such as lithium carbonate. The treatment composition may however also comprise one or more hydroxide different from lithium salts such as for example sodium hydroxide, potassium hydroxide, or a combination thereof.
[0054] The treatment composition of the present invention generally comprises an aqueous medium as a carrier. The composition may thus be in the form of a solution or dispersion of the lithium salt in the carrier.
[0055] According to the present invention, lithium carbonate is formed by combining carbon dioxide and a lithium cation in the aqueous carrier medium wherein the carbon dioxide and lithium cation are balanced to be present in amounts such that lithium is present in the treatment composition in an amount of 5 ppm to 5,500 ppm (calculated as lithium cation) based on total weight of the treatment composition, carbonate is present in the treatment composition in an amount of 15 ppm to 25,000 ppm (calculated as carbonate) based on total weight of the treatment composition. As set forth above, optionally further one or more pH modifier(s) such as one or more acidic material(s) and/or one or more basic material(s) such as one or more hydroxide is added to the aqueous carrier medium wherein the amounts of such optional pH modifier(s), carbon dioxide and lithium salt may be balanced such that the pH of the treatment composition is 9.5 to 12.5.
[0056] According to the present invention, the treatment composition may further comprise at least one Group IA metal cation other than lithium, a Group VB metal cation, and/or Group VIB metal cation. According to the present invention, the at least one Group IA metal cation other than lithium, a Group VB metal cation, and/or Group VIB metal cation may be in the form of a salt and cation each may be present in the treatment composition in an amount of at least 5 ppm, such as at least 50 ppm, such as at least 150 ppm, such as at least 250 ppm (calculated as metal cation) based on total weight of the treatment composition, and in some instances, may be present in an amount of no more than 5,500 ppm, such as no more than 1,200 ppm, such as no more than 1,000 ppm, such as no more than 500 ppm, (calculated as metal cation) based on total weight of the treatment composition. In some instances, according to the present invention, the lithium metal may be present in the treatment composition in an amount of 5 ppm to 5,500 ppm, such as 50 ppm to 1,000 ppm, (calculated as metal cation) based on total weight of the treatment composition, such as 150 ppm to 500 ppm.
[0057] Nonlimiting examples of anions suitable for forming a salt with lithium cation, Group IA cations other than lithium, Group VB cations, and/or Group VIB cations include carbonates, hydroxides, nitrates, halogens, sulfates, phosphates and silicates (e.g., orthosilicates and metasilicates) such that the metal salt may comprise a carbonate, an hydroxide, a nitrate, a halide, a sulfate, a phosphate, a silicate (e.g., orthosilicate or metasilicate), a permanganate, a chromate, a vanadate, a molybdate, and/or a perchlorate.
[0058] According to the present invention, the metal salts of the treatment composition (i.e., the salts of lithium, Group IA metals other than lithium, Group VB, and/or Group VIB) each may be present in the treatment composition in an amount of at least 25 ppm, such as at least 150 ppm, such as at least 500 ppm (calculated as total compound) based on total weight of the treatment composition, and in some instances, no more than 30,000 ppm, such as no more than 2,000 ppm, such as no more than 1,500 ppm (calculated as total compound) based on total weight of the treatment composition. According to the present invention, the metal salts each may be present in the treatment composition in an amount of 25 ppm to 30,000 ppm, such as 150 ppm to 2,000 ppm, such as 500 ppm to 1,500 (calculated as total compound) based on total weight of the treatment composition.
[0059] According to the present invention, the sealing composition of the present invention may an include oxidizing agent, such as hydrogen peroxide, persulfates, perchlorates, sparged oxygen, bromates, peroxi-benzoates, ozone, and the like, or combinations thereof. For example, the sealing composition may comprise 0.1 wt % to 15 wt % of an oxidizing agent based on total weight of the sealing composition, such as 2 wt % to 10 wt %, such as 6 wt % to 8 wt %. Alternatively, according to the present invention, the sealing composition may be substantially free, or in some cases, essentially free, or in some cases, completely free, of an oxidizing agent.
[0060] According to the present invention, the treatment composition may exclude chromium or chromium-containing compounds. As used herein, the term "chromium-containing compound" refers to materials that include hexavalent chromium. Non-limiting examples of such materials include chromic acid, chromium trioxide, chromic acid anhydride, dichromate salts, such as ammonium dichromate, sodium dichromate, potassium dichromate, and calcium, barium, magnesium, zinc, cadmium, and strontium dichromate. When a treatment composition and/or a bath, or a coating or a layer formed from the same is substantially free, essentially free, or completely free of chromium, this includes chromium in any form, such as, but not limited to, the hexavalent chromium-containing compounds listed above.
[0061] Thus, optionally, according to the present invention, the present treatment compositions and/or treatment baths and/or coatings or layers formed from the same may be substantially free, may be essentially free, and/or may be completely free of one or more of any of the elements or compounds listed in the preceding paragraph. A treatment composition and/or bath and/or coating or layer formed from the same that is substantially free of chromium or chromium-containing compounds means that chromium or chromium-containing compounds are not intentionally added, but may be present in trace amounts, such as because of impurities or unavoidable contamination from the environment. In other words, the amount of material is so small that it does not affect the properties of the treatment composition and/or bath and/or coating or layer formed from the same; in the case of chromium, this may further include that the element or compounds thereof are not present in the treatment compositions and/or baths and/or coatings or layers formed from the same, in such a level that it causes a burden on the environment. The term "substantially free" may thus for example mean that the treatment compositions and/or baths and/or coating or layers formed from the same contain less than 10 ppm of any or all of the elements or compounds listed in the preceding paragraph, based on total weight of the composition, bath, coating or layer, as the case may be, if any at all. The term "essentially free" means that the treatment compositions and/or baths and/or coatings or layers formed from the same contain less than 1 ppm of any or all of the elements or compounds listed in the preceding paragraph, based on total weight of the composition, bath, coating or layer, as the case may be, if any at all. The term "completely free" means that the treatment compositions and/or baths and/or coatings or layers formed from the same contain less than 1 ppb of any or all of the elements or compounds listed in the preceding paragraph, based on total weight of the composition, bath, coating or layer, as the case may be, if any at all.
[0062] According to the present invention, the present treatment compositions and/or treatment baths and/or coatings or layers formed from the same may, in some instances, exclude phosphate ions or phosphate-containing compounds and/or the formation of sludge, such as aluminum phosphate, iron phosphate, and/or zinc phosphate, formed for example in the case of using a treating agent based on zinc phosphate. As used herein, "phosphate-containing compounds" include compounds containing the element phosphorous such as ortho phosphate, pyrophosphate, metaphosphate, tripolyphosphate, organophosphonates, and the like, and can include, but are not limited to, monovalent, divalent, or trivalent cations such as: sodium, potassium, calcium, zinc, nickel, manganese, aluminum and/or iron. When a treatment composition and/or bath and/or coatings or layers formed from the same is substantially free, essentially free, or completely free of phosphate, this includes phosphate ions or compounds containing phosphate in any form.
[0063] Thus, according to the present invention, the treatment compositions and/or baths and/or coatings or layers formed from the same disclosed herein may be substantially free, or in some cases may be essentially free, or in some cases may be completely free, of one or more of any of the ions or compounds listed in the preceding paragraph. A treatment compositions and/or baths and/or coatings or layers formed from the same that is substantially free of phosphate means that phosphate ions or compounds containing phosphate are not intentionally added, but may be present in trace amounts, such as because of impurities or unavoidable contamination from the environment. In other words, the amount of material is so small that it does not affect the properties of the treatment compositions and/or baths and/or coatings or layers formed from the same; this may further include that phosphate is not present in the treatment compositions and/or baths and/or coatings or layers formed from the same in such a level that they cause a burden on the environment. The term "substantially free" may in particular mean that the treatment compositions and/or baths and/or coatings or layers formed from the same contain less than 5 ppm of any or all of the phosphate anions or compounds listed in the preceding paragraph, based on total weight of the composition, bath, coating or layer, as the case may be, if any at all. The term "essentially free" means that the treatment compositions and/or baths and/or coatings or layers formed from the same contain less than 1 ppm of any or all of the phosphate anions or compounds listed in the preceding paragraph, based on total weight of the composition, bath, coating or layer, as the case may be, if any at all. The term "completely free" means that the treatment compositions and/or baths and/or coatings or layers formed from the same contain less than 1 ppb of any or all of the phosphate anions or compounds listed in the preceding paragraph, based on total weight of the composition, bath, coating or layer, as the case may be, if any at all.
[0064] A According to the present invention, the sealing composition may exclude Group IIA metal cations or Group IIA metal-containing compounds, including but not limited to calcium. Non-limiting examples of such materials include Group IIA metal hydroxides, Group IIA metal nitrates, Group IIA metal halides, Group IIA metal sulfamates, Group IIA metal sulfates, Group IIA carbonates and/or Group IIA metal carboxylates. When a sealing composition and/or a coating or a layer, respectively, formed from the same is substantially free, essentially free, or completely free of a Group IIA metal cation, this includes Group IIA metal cations in any form, such as, but not limited to, the Group IIA metal-containing compounds listed above.
[0065] According to the present invention, the sealing composition may, in some instances, exclude fluoride or fluoride sources. As used herein, "fluoride sources" include monofluorides, bifluorides, fluoride complexes, and mixtures thereof known to generate fluoride ions. When a composition and/or a layer or coating comprising the same is substantially free, essentially free, or completely free of fluoride, this includes fluoride ions or fluoride sources in any form, but does not include unintentional fluoride that may be present in a bath as a result of, for example, carry-over from prior treatment baths in the processing line, municipal water sources (e.g.: fluoride added to water supplies to prevent tooth decay), fluoride from a pretreated substrate, or the like. That is, a bath that is substantially free, essentially free, or completely free of fluoride, may have unintentional fluoride that may be derived from these external sources, even though the composition used to make the bath prior to use on the processing line was substantially free, essentially free, or completely free of fluoride.
[0066] For example, the sealing composition may be substantially free of any fluoride-sources, such as ammonium and alkali metal fluorides, acid fluorides, fluoroboric, fluorosilicic, fluorotitanic, and fluorozirconic acids and their ammonium and alkali metal salts, and other inorganic fluorides, nonexclusive examples of which are: zinc fluoride, zinc aluminum fluoride, titanium fluoride, zirconium fluoride, nickel fluoride, ammonium fluoride, sodium fluoride, potassium fluoride, and hydrofluoric acid, as well as other similar materials known to those skilled in the art.
[0067] Fluoride present in the sealing composition that is not bound to metals ions such as Group IVB metal ions, or hydrogen ion, defined herein as "free fluoride," may be measured as an operational parameter in the sealing composition bath using, for example, an Orion Dual Star Dual Channel Benchtop Meter equipped with a fluoride ion selective electrode ("ISE") available from Thermoscientific, the Symphony.RTM. Fluoride Ion Selective Combination Electrode supplied by VWR International, or similar electrodes. See, e.g., Light and Cappuccino, Determination of fluoride in toothpaste using an ion-selective electrode, J. Chem. Educ., 52:4, 247-250, April 1975. The fluoride ISE may be standardized by immersing the electrode into solutions of known fluoride concentration and recording the reading in millivolts, and then plotting these millivolt readings in a logarithmic graph. The millivolt reading of an unknown sample can then be compared to this calibration graph and the concentration of fluoride determined. Alternatively, the fluoride ISE can be used with a meter that will perform the calibration calculations internally and thus, after calibration, the concentration of the unknown sample can be read directly.
[0068] Fluoride ion is a small negative ion with a high charge density, so in aqueous solution it is frequently complexed with metal ions having a high positive charge density, such as Group IVB metal ions, or with hydrogen ion. Fluoride anions in solution that are ionically or covalently bound to metal cations or hydrogen ion are defined herein as "bound fluoride." The fluoride ions thus complexed are not measurable with the fluoride ISE unless the solution they are present in is mixed with an ionic strength adjustment buffer (e.g.: citrate anion or EDTA) that releases the fluoride ions from such complexes. At that point (all of) the fluoride ions are measurable by the fluoride ISE, and the measurement is known as "total fluoride". Alternatively, the total fluoride can be calculated by comparing the weight of the fluoride supplied in the sealer composition by the total weight of the composition.
[0069] According to the present invention, the treatment composition may, in some instances, be substantially free, or in some instances, essentially free, or in some instances, completely free, of cobalt ions or cobalt-containing compounds. As used herein, "cobalt-containing compounds" include compounds, complexes or salts containing the element cobalt such as, for example, cobalt sulfate, cobalt nitrate, cobalt carbonate and cobalt acetate. When a composition and/or a layer or coating comprising the same is substantially free, essentially free, or completely free of cobalt, this includes cobalt ions or compounds containing cobalt in any form.
[0070] According to the present invention, the treatment composition may, in some instances, be substantially free, or in some instances, essentially free, or in some instances, completely free, of vanadium ions or vanadium-containing compounds. As used herein, "vanadium-containing compounds" include compounds, complexes or salts containing the element vanadium such as, for example, vanadates and decavanadates that include counterions of alkali metal or ammonium cations, including, for example, sodium ammonium decavanadate. When a composition and/or a layer or coating comprising the same is substantially free, essentially free, or completely free of vanadium, this includes vanadium ions or compounds containing vanadium in any form.
[0071] According to the present invention, the treatment composition may optionally further contain an indicator compound, so named because it indicates, for example, the presence of a chemical species, such as a metal ion, the pH of a composition, and the like. An "indicator", "indicator compound", and like terms as used herein refer to a compound that changes color in response to some external stimulus, parameter, or condition, such as the presence of a metal ion, or in response to a specific pH or range of pHs.
[0072] The indicator compound used according to the present invention can be any indicator known in the art that indicates the presence of a species, a particular pH, and the like. For example, a suitable indicator may be one that changes color after forming a metal ion complex with a particular metal ion. The metal ion indicator is generally a highly conjugated organic compound. A "conjugated compound" as used herein, and as will be understood by those skilled in the art, refers to a compound having two double bonds separated by a single bond, for example two carbon-carbon double bonds with a single carbon-carbon bond between them. Any conjugated compound can be used according to the present invention.
[0073] Similarly, the indicator compound can be one in which the color changes upon change of the pH; for example, the compound may be one color at an acidic or neutral pH and change color in an alkaline pH, or vice versa. Such indicators are well known and widely commercially available. An indicator that "changes color upon transition from a first pH to a second pH" (i.e., from a first pH to a second pH that is more or less acidic or alkaline) therefore has a first color (or is colorless) when exposed to a first pH and changes to a second color (or goes from colorless to colored) upon transition to a second pH (i.e., one that is either more or less acidic or alkaline than the first pH). For example, an indicator that "changes color upon transition to a more alkaline pH (or less acidic pH) goes from a first color/colorless to a second color/color when the pH transitions from acidic/neutral to alkaline. For example, an indicator that "changes color upon transition to a more acidic pH (or less alkaline pH) goes from a first color/colorless to a second color/color when the pH transitions from alkaline/neutral to acidic.
[0074] Non-limiting examples of such indicator compounds include methyl orange, xylenol orange, catechol violet, bromophenol blue, green and purple, eriochrome black T, Celestine blue, hematoxylin, calmagite, gallocyanine, and combinations thereof. Optionally, the indicator compound may comprise an organic indicator compound that is a metal ion indicator. Nonlimiting examples of indicator compounds include those found in Table 1. Fluorescent indicators, which will emit light in certain conditions, can also be used according to the present invention, although the use of a fluorescent indicator also may be specifically excluded. That is, alternatively, conjugated compounds that exhibit fluorescence are specifically excluded. As used herein, "fluorescent indicator" and like terms refer to compounds, molecules, pigments, and/or dyes that will fluoresce or otherwise exhibit color upon exposure to ultraviolet or visible light. To "fluoresce" will be understood as emitting light following absorption of shorter wavelength light or other electromagnetic radiation. Examples of such indicators, often referred to as "tags," include acridine, anthraquinone, coumarin, diphenylmethane, diphenylnaphthlymethane, quinoline, stilbene, triphenylmethane, anthracine and/or molecules containing any of these moieties and/or derivatives of any of these such as rhodamines, phenanthridines, oxazines, fluorones, cyanines and/or acridines.
TABLE-US-00001 TABLE 1 Compound Structure CAS Reg. No. Catechol Violet Synonyms: Catecholsulfonphthalein; Pyrocatecholsulfonephthalein; Pyrocatechol Violet ##STR00001## 115-41-3 Xylenol Orange Synonym: 3,3'-Bis[N,N- bis(carboxymethyl) aminomethyl]- o-cresolsulfonephthalein tetrasodium salt ##STR00002## 3618-43-7
[0075] According to the present invention, the conjugated compound useful as indicator may for example comprise catechol violet, as shown in Table 1. Catechol violet (CV) is a sulfone phthalein dye made from condensing two moles of pyrocatechol with one mole of o-sulfobenzoic acid anhydride. It has been found that CV has indicator properties and when incorporated into compositions having metal ions, it forms complexes, making it useful as a complexiometric reagent. As the composition containing the CV chelates metal ions coming from the metal substrate (i.e., those having bi- or higher valence), a generally blue to blue-violet color is observed.
[0076] Xylenol orange, as shown in Table 1 may likewise be employed in the compositions according to the present invention. It has been found that xylenol orange has metal ion (i.e., those having bi- or higher valence) indicator properties and when incorporated into compositions having metal ions, it forms complexes, making it useful as a complexiometric reagent. As the composition containing the xylenol orange chelates metal ions, a solution of xylenol orange turns from red to a generally blue color.
[0077] According to the present invention, the indicator compound may be present in the treatment composition in an amount of at least 0.01 g/1000 g treatment composition, such as at least 0.05 g/1000 g treatment composition, and in some instances, no more than 3 g/1000 g treatment composition, such as no more than 0.3 g/1000 g treatment composition. According to the present invention, the indicator compound may be present in the treatment composition in an amount of 0.01 g/1000 g treatment composition to 3 g/1000 g treatment composition, such as 0.05 g/1000 g treatment composition to 0.3 g/1000 g treatment composition.
[0078] According to the present invention, the indicator compound changing color in response to a certain external stimulus provides a benefit when using the treatment composition in that it can serve, for example, as a visual indication that a substrate has been treated with the composition. For example, a treatment composition comprising an indicator that changes color when exposed to a metal ion that is present in the substrate will change color upon complexing with metal ions in that substrate; this allows the user to see that the substrate has been contacted with the composition. Similar benefits can be realized by depositing an alkaline or acid layer on a substrate and contacting the substrate with a composition of the present invention that changes color when exposed to an alkaline or acidic pH.
[0079] Optionally, the treatment composition of the present invention may further comprise a nitrogen-containing heterocyclic compound. The nitrogen-containing heterocyclic compound may include cyclic compounds having 1 nitrogen atom, such as pyrroles, and azole compounds having 2 or more nitrogen atoms, such as pyrazoles, imidazoles, triazoles, tetrazoles and pentazoles, 1 nitrogen atom and 1 oxygen atom, such as oxazoles and isoxazoles, or 1 nitrogen atom and 1 sulfur atom, such as thiazoles and isothiazoles. Nonlimiting examples of suitable azole compounds include 2,5-dimercapto-1,3,4-thiadiazole (CAS:1072-71-5), 1H-benzotriazole (CAS: 95-14-7), 1H-1,2,3-triazole (CAS: 288-36-8), 2-amino-5-mercapto-1,3,4-thiadiazole (CAS: 2349-67-9), also named 5-amino-1,3,4-thiadiazole-2-thiol, and 2-amino-1,3,4-thiadiazole (CAS: 4005-51-0). In some embodiments, for example, the azole compound comprises 2,5-dimercapto-1,3,4-thiadiazole. Additionally, according to the present invention, the nitrogen-containing heterocyclic compound may be in the form of a salt, such as a sodium salt.
[0080] The nitrogen-containing heterocyclic compound may be present in the treatment composition at a concentration of at least 0.0005 g per liter of composition, such as at least 0.0008 g per liter of composition, such as at least 0.002 g per liter of composition, and in some instances, may be present in the treatment composition in an amount of no more than 3 g per liter of composition, such as no more than 0.2 g per liter of composition, such as no more than 0.1 g per liter of composition. According to the present invention, the nitrogen-containing heterocyclic compound may be present in the treatment composition (if at all) at a concentration of 0.0005 g per liter of composition to 3 g per liter of composition, such as 0.0008 g per liter of composition to 0.2 g per liter of composition, such as 0.002 g per liter of composition to 0.1 g per liter of composition.
[0081] As indicated above, the treatment composition of the present invention comprises an aqueous medium as carrier. The aqueous carrier may optionally contain other materials such as at least one organic solvent. Nonlimiting examples of suitable solvents include propylene glycol, ethylene glycol, glycerol, low molecular weight alcohols (i.e., C1-C12 alcohols), and the like. When present, if at all, the organic solvent may be present in the treatment composition in an amount of at least 1 g solvent per liter of treatment composition, such as at least about 2 g solvent per liter of treatment composition, and in some instances, may be present in an amount of no more than 40 g solvent per liter of treatment composition, such as no more than 20 g solvent per liter of treatment composition. According to the present invention, the organic solvent may be present in the treatment composition, if at all, in an amount of 1 g solvent per liter of treatment composition to 40 g solvent per liter of treatment composition, such as 2 g solvent per liter of treatment composition to 20 g solvent per liter of treatment composition.
[0082] As set forth above, the treatment composition of the present invention described above may be prepared by a method that comprises combining a lithium salt and carbon dioxide in an aqueous carrier medium to form the treatment composition comprising lithium in an amount of 5 ppm to 5,500 ppm (calculated as lithium cation) based on total weight of the treatment composition and carbonate in an amount of 15 ppm to 25,000 ppm (calculated as carbonate) based on total weight of the treatment composition. Suitable lithium salts and amounts of lithium in the treatment composition are described above. For example, the lithium salt used in the method of forming the treatment composition can comprise lithium carbonate, lithium hydroxide, or a combination thereof. The method of making a treatment composition of the present invention may furthermore comprise adjusting the pH of the treatment composition to a pH of at least 9.5, such as at least 10, such as at least 11, and in some instances to a pH no greater than 12.5, such as no greater than 12, such as no greater than 11.5. According to the present invention, the treatment composition may thus be adjusted to have a pH of 9.5 to 12.5, such as 10 to 12, such as 11 to 11.5. The pH of the treatment composition may be measured according to any of the methods described below and may be adjusted using, for example, any acid and/or base as is necessary, as described above.
[0083] According to the present invention, the method of making the treatment composition comprises combining carbon dioxide, and the lithium salt in an aqueous medium. According to the present invention, the carbon dioxide may be supplied to the aqueous carrier medium in the form of a gas, a solid, or a combination thereof. As used herein, "supplied," when used with respect to carbon dioxide, refers to introducing carbon dioxide to the composition using a source other than the atmosphere. The carbon dioxide is supplied to the aqueous medium in an amount sufficient to form the treatment composition comprising carbonate (calculated as carbonate) in an amount of at least 15 ppm based on total weight of the treatment composition, such as at least 50 ppm, such as at least 200 ppm, and in some instances, no more than 25,000 ppm based on total weight of the treatment composition, such as no more than 15,000 ppm, such as no more than 2,400 ppm. In some instances, according to the present invention, the carbon dioxide may be combined with water in an amount sufficient to form the treatment composition comprising carbonate (calculated as carbonate) in an amount of 15 ppm to 25,000 ppm based on total weight of the treatment composition, such as 50 ppm to 15,000 ppm, such as 200 ppm to 2,400 ppm.
[0084] As pointed out above, the method of making the treatment composition according to the present invention also may comprise adding a hydroxide, such as Group I hydroxides, ammonium hydroxide, or mixtures thereof. The hydroxide source, if present at all, may be present in any amount, such as in an amount such that the pH of the treatment composition is within the range of 9.5 to 12.5. Nonlimiting examples of Group I hydroxides include sodium hydroxide, potassium hydroxide, lithium hydroxide, or mixtures thereof.
[0085] As mentioned above, according to the present invention, also disclosed is a system and method of maintaining a treatment bath formed from a treatment composition comprising lithium carbonate. The treatment composition may be the treatment composition described above and may be made according to the method described herein above or may be made by any method known to those of skill in the art. In an example, according to the present invention, a "treatment bath" may refer to an aqueous bath formed from an initial treatment composition comprising lithium carbonate, e.g. as described above, upon treatment of one or more substrate(s). As used "maintaining" a treatment bath formed from the treatment composition comprising lithium carbonate (regardless of how the lithium carbonate composition was formed) refers to keeping certain parameters of the treatment bath including the concentration of lithium and carbonate and the pH in desirable ranges such as those indicated above for the treatment composition according to the present invention. This can be achieved, as described in more detail below, by the addition of one or more materials from a respective source to the treatment bath on-shift and/or off-shift.
[0086] According to the present invention, the system or method of maintaining may comprise (i) adding materials to the treatment bath formed from the treatment composition that are different from materials used to formulate the treatment composition and/or (ii) adding materials to the treatment bath formed from the treatment composition that are the same as those materials used to formulate the treatment composition. For example, while the method of maintaining the treatment bath containing the treatment composition may comprise adding carbon dioxide to the treatment bath, the treatment composition may be formulated using a carbonate.
[0087] According to the present invention, the system or method of maintaining may comprise adding materials to the treatment bath containing the treatment composition that are the same as materials used to formulate the treatment composition. For example, the treatment composition may be formulated using carbon dioxide (as described above), and the method of maintaining the treatment bath containing the treatment composition may comprise adding carbon dioxide to the treatment bath.
[0088] The system or method of the present invention is not directed to simply adding more treatment composition to the treatment bath in order to maintain the bath. Rather, as mentioned above, the system and method of the present invention are directed to adding carbon dioxide and/or a lithium salt and/or a hydroxide to the treatment bath in an amount sufficient to maintain the pH of the treatment bath at 9.5 to 12.5, lithium in an amount of 5 ppm to 5,500 ppm (calculated as lithium cation) based on total weight of the treatment bath, and carbonate in an amount of 15 ppm to 25,000 ppm (calculated as carbonate) based on total weight of the treatment bath. The supplying can be carried out on-shift or off-shift.
[0089] As mentioned above, according to the present invention, the system for maintaining a treatment bath formed from a treatment composition comprising lithium carbonate is disclosed. According to the present invention, the system may comprise a lithium salt and/or a carbon dioxide, optionally a hydroxide, or a combination of any of the foregoing. The lithium salt may comprise one or more of any of the lithium salts described above, such as for example lithium carbonate, lithium hydroxide or a combination thereof. The carbon dioxide may comprise carbon dioxide as a gas, a solid, or a combination thereof. The hydroxide may comprise one or more of any of the hydroxides mentioned above such as for example lithium hydroxide, sodium hydroxide, potassium hydroxide or a combination thereof. The lithium salt, carbon dioxide, and/or hydroxide described above may be included in the system individually or in any combination and may be added from their respective sources of the system to the treatment bath formed from the treatment composition to achieve a treatment bath being maintained having a pH and amounts of lithium and carbonate as described above.
[0090] As mentioned above, according to the present invention, also disclosed is a method of maintaining a treatment bath formed from a treatment composition comprising lithium carbonate. According to the present invention, the method comprises supplying during and/or after treatment of a substrate with the bath at least one of carbon dioxide and a lithium salt and, optionally, a hydroxide to the treatment bath in an amount sufficient to maintain the pH of the treatment bath at 9.5 to 12.5, lithium in an amount of 5 ppm to 5,500 ppm (calculated as lithium cation) based on total weight of the treatment bath, and carbonate in an amount of 15 ppm to 25,000 ppm (calculated as carbonate) based on total weight of the treatment bath. The lithium salt, carbon dioxide, and hydroxide described above may be added to the treatment bath formed from the treatment composition to achieve a treatment bath being maintained having a pH and amounts of lithium and carbonate as described above in more detail in the context of the treatment composition according to the present invention. For example, according to the present invention, the method of maintaining may comprise adding carbon dioxide to the treatment bath formed from the treatment composition in such amount that the pH of the treatment bath is maintained below 12.5 and/or adding a hydroxide to the treatment bath in such amount that the pH of the treatment bath is maintained above 9.5. In examples, according to the present invention, the carbon dioxide may be slowly bubbled into the treatment bath or may be added by dropping in dry ice piece by piece. According to the present invention, the pH may be periodically or continually monitored (described below) and/or hydroxide may be added to the treatment bath as discussed above to maintain pH between 9.5 and 12.5.
[0091] According to the present invention, as described above, following the supplying of the carbon dioxide and/or the lithium salt and/or the hydroxide, lithium (calculated as lithium cation) may be present in the treatment composition in an amount of at least 5 ppm, such as at least 50 ppm, such as at least 150 ppm, such as at least 250 ppm, based on total weight of the treatment bath, and in some instances, may be present in an amount of no more than 5,500 ppm, such as no more than 1,200 ppm, such as no more than 1,000 ppm, such as no more than 500 ppm, based on total weight of the treatment bath. In some instances, according to the present invention, following the supplying of the carbon dioxide and/or the lithium salt, lithium (calculated as lithium cation) may be present in the treatment bath in an amount of 5 ppm to 5,500 ppm based on total weight of the treatment bath, such as 50 ppm to 1,200 ppm, such as 150 ppm to 1,000 ppm, such as 250 ppm to 500 ppm.
[0092] According to the present invention, following the supplying of the carbon dioxide and/or the lithium salt and/or the hydroxide, carbonate (calculated as carbonate) may be present in the treatment bath in an amount of at least 15 ppm based on total weight of the treatment bath, such as at least 50 ppm, such as at least 200 ppm, and in some instances, may be present in an amount of no more than 25,000 ppm based on total weight of the treatment bath, such as no more than 15,000 ppm, such as no more than 2,400 ppm. In some instances, according to the present invention, following the supplying of the carbon dioxide and/or the lithium salt and/or the hydroxide, the carbonate (calculated as carbonate) may be present in the treatment bath in an amount of 15 ppm to 25,000 ppm based on total weight of the treatment bath, such as 50 ppm to 15,000 ppm, such as 200 ppm to 2,400 ppm.
[0093] According to the present invention, following the supplying of the carbon dioxide and/or the lithium salt and/or the hydroxide, the treatment bath may have a pH of at least 9.5, such as at least 10, such as at least 11, and in some instances, may have a pH no greater than 12.5, such as no greater than 12, such as no greater than 11.5. According to the present invention, following the supplying of the carbon dioxide and/or the lithium salt and/or the hydroxide, the treatment bath may have a pH of 9.5 to 12.5, such as 10 to 12, such as 11 to 11.5.
[0094] According to the present invention, the method of maintaining a treatment bath may further comprise adjusting a pH of the treatment bath, such as by adding any acid and/or base as is necessary. According to the present invention, the treatment bath may be maintained through the inclusion of an acidic material, including water soluble and/or water dispersible acids, such as nitric acid, sulfuric acid, and/or phosphoric acid. According to the present invention, the pH of the treatment bath may be maintained through the inclusion of a basic material, including water soluble and/or water dispersible bases, such as Group I carbonates, Group II carbonates, hydroxides, such as sodium hydroxide, potassium hydroxide, lithium hydroxide, ammonium hydroxide, ammonia, amines such as triethylamine, methylethyl amine, or mixtures thereof.
[0095] The method of maintaining a treatment bath of the present invention may further comprise monitoring the pH of the treatment bath using a pH meter and probe appropriate for the size of the bath formed from the treatment composition comprising lithium carbonate. An example of a suitable pH meter and probe includes, but is not limited to, the Accumet AB15 (available from Fisher Scientific) and a single junction electrode (Ag/AgCl reference; Fisher Scientific).
[0096] The method of maintaining a treatment bath of the present invention may further comprise monitoring the amount of lithium, carbonate, or lithium carbonate in the treatment bath by any method known to those skilled in the art. For example, according to the present invention, the method of monitoring lithium may comprise, for example, using an optical emission spectrometer or equivalent instrumentation and using a standard sample with a defined concentration of lithium (e.g. a standard of known concentration (such as a 500 ppm Li standard diluted to 5 ppm Li) at a specified wavelength (e.g., 670.784 nm) to calculate the concentration of lithium (metal cation) in the treatment bath. The method of maintaining a treatment bath of the present invention may further comprise monitoring the amount of carbonate in the treatment bath by any method known to those skilled in the art, including for example, using a manual titration or an autotitration method.
[0097] It has been unexpectedly discovered that carbon dioxide and/or a lithium salt and, optionally a hydroxide, may be used to maintain a treatment bath formed from a lithium carbonate containing treatment composition such that the pH and lithium cation concentration, and/or lithium cation concentration and carbonate (anion) concentration may be independently manipulated or adjusted, depending on bath conditions, compared to maintenance of a bath with, for example, lithium carbonate, where pH, lithium concentration, and carbonate concentration are all changed upon addition of lithium carbonate to the bath (i.e., there is no independent control of each such parameter). For example, carbon dioxide and/or a lithium salt and, optionally a hydroxide, may be used to maintain a treatment bath formed from a lithium carbonate containing treatment composition such that the treatment bath has a pH of 9.5 to 12.5, a lithium concentration of 5 ppm to 5,500 ppm (calculated as lithium cation) based on total weight of the treatment bath, and a carbonate concentration of 15 ppm to 25,000 ppm (calculated as carbonate) based on total weight of the treatment bath.
[0098] As mentioned above, the treatment composition or bath formed therefrom comprises an aqueous medium as a carrier. Accordingly, the composition or bath may be in the form of a solution or dispersion of the lithium salt in the carrier. According to the present invention, the solution or dispersion may be brought into contact with a substrate to be treated with the composition or bath by any of a variety of known techniques, such as dipping or immersion, spraying, intermittent spraying, dipping followed by spraying, spraying followed by dipping, brushing, or roll-coating. According to the invention, the solution or dispersion when applied to the substrate may be at a temperature ranging from 40.degree. F. (5.degree. C.) to about 160.degree. F. (71.degree. C.), such as 60.degree. F. (16.degree. C.) to 110.degree. F. (43.degree. C.). For example, the process of contacting the substrate with the treatment composition or bath may be carried out at ambient or room temperature, such as 23.degree. C., if not indicated otherwise. The contact time is often from 1 second to 2 hours, such as 5 minutes to 60 minutes.
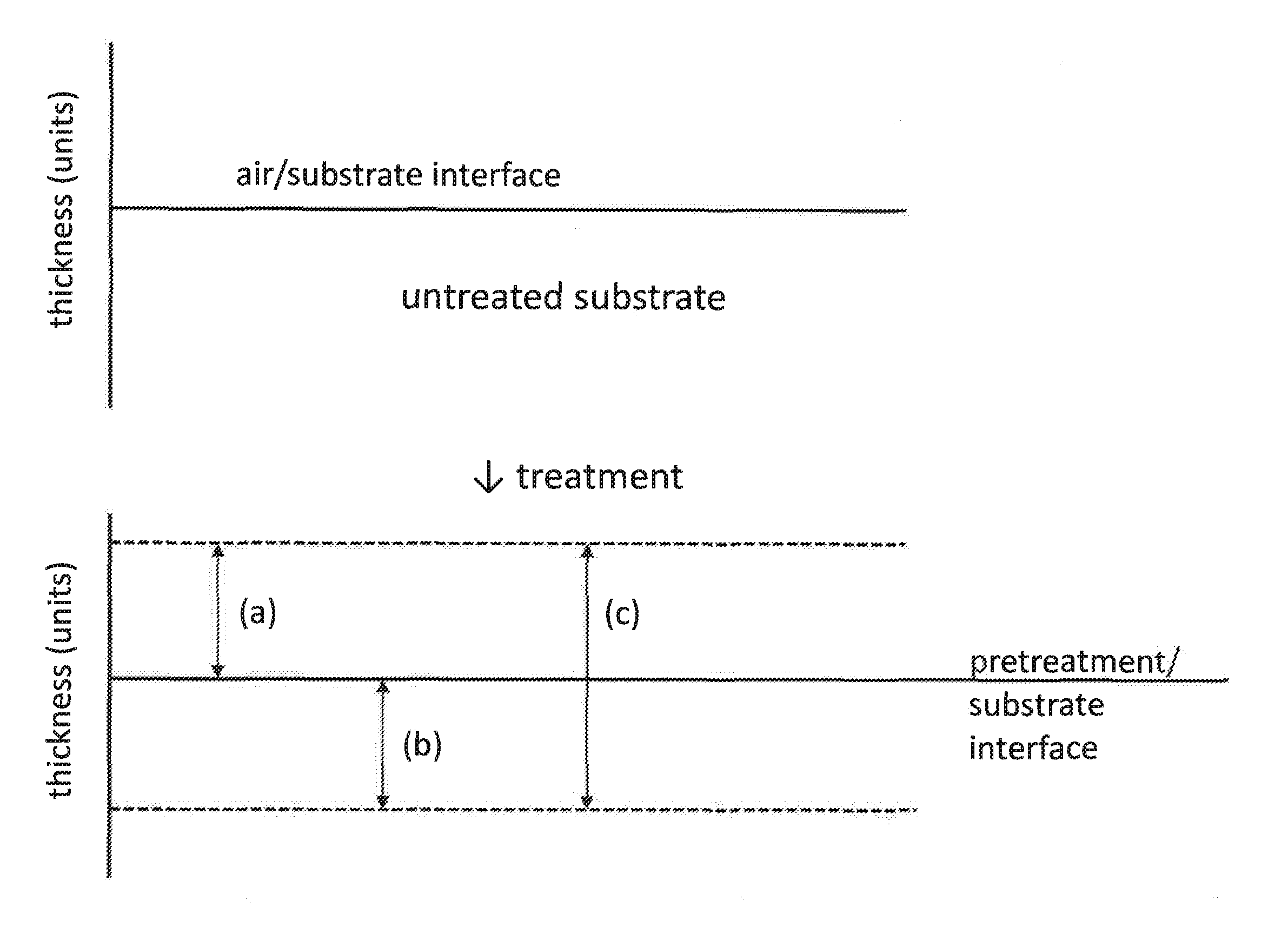
[0099] According to the present invention, the thickness of the layer formed by the treatment composition may for instance be up to 550 nm, such as 5 nm to 550 nm, such as 10 nm to 400 nm, such as 25 nm to 250 nm. Thickness of layer formed from the treatment composition can be determined using a handful of analytical techniques including, but not limited to XPS (x-ray photoelectron spectroscopy) depth profiling or TEM (transmission electron microscopy). As used herein, "thickness," when used with respect to a layer formed by the treatment composition of the present invention, refers to either (a) a layer formed above the original air/substrate interface, (b) a modified layer formed below the pretreatment/substrate interface, or (c) a combination of (a) and (b), as illustrated in FIG. 3. Although modified layer (b) is shown extending to the pretreatment/substrate interface in FIG. 3, an intervening layer may be present between the modified layer (b) and the pretreatment/substrate interface. Likewise, (c), a combination of (a) and (b), is not limited to a continuous layer and may include multiple layers with intervening layers therebetween, and the measurement of the thickness of layer (c) may exclude the intervening layers.
[0100] Suitable substrates that may be used in the present invention include metal substrates, metal alloy substrates, and/or substrates that have been metallized, such as nickel plated plastic. According to the present invention, the metal or metal alloy can comprise or be steel, aluminum, zinc, nickel, and/or magnesium. For example, the steel substrate could be cold rolled steel, hot rolled steel, electrogalvanized steel, and/or hot dipped galvanized steel. Aluminum alloys of the 1XXX, 2XXX, 3XXX, 4XXX, 5XXX, 6XXX, or 7XXX series as well as clad aluminum alloys also may be used as the substrate. Aluminum alloys may comprise 0.01% by weight copper to 10% by weight copper. Aluminum alloys which are treated may also include castings, such as 1XX.X, 2XX.X, 3XX.X, 4XX.X, 5XX.X, 6XX.X, 7XX.X, 8XX.X, or 9XX.X (e.g.: A356.0). Magnesium alloys of the AZ31B, AZ91C, AM60B, or EV31A series also may be used as the substrate. The substrate used in the present invention may also comprise titanium and/or titanium alloys, zinc and/or zinc alloys, and/or nickel and/or nickel alloys. According to the present invention, the substrate may comprise a portion of a vehicle such as a vehicular body (e.g., without limitation, door, body panel, trunk deck lid, roof panel, hood, roof and/or stringers, rivets, landing gear components, and/or skins used on an aircraft) and/or a vehicular frame. As used herein, "vehicle" or variations thereof includes, but is not limited to, civilian, commercial and military aircraft, and/or land vehicles such as cars, motorcycles, and/or trucks.
[0101] According to the present invention, at least a portion of the substrate surface may be cleaned and/or deoxidized and/or otherwise pretreated by any conventional means known in the art of cleaning or pretreating a metal substrate prior to contacting at least a portion of the substrate surface with a treatment composition or bath described above, in order to remove grease, dirt, and/or other extraneous matter. At least a portion of the surface of the substrate may be cleaned by physical and/or chemical means, such as mechanically abrading the surface and/or cleaning/degreasing the surface with commercially available alkaline or acidic cleaning agents that are well known to those skilled in the art. Examples of alkaline cleaners suitable for use in the present invention include Chemkleen.TM. 166tlP, 166 m/c, 177, 490MX, 2010LP, and Surface Prep 1 (SP1), Ultrax 32, Ultrax 97, Ultrax 29 and 92D, each of which are commercially available from PPG Industries, Inc. (Cleveland, Ohio), and any of the DFM Series, RECC 1001, and 88X1002 cleaners commercially available from PRC-DeSoto International, Sylmar, Calif.), and Turco 4215-NCLT and Ridolene (commercially available from Henkel Technologies, Madison Heights, Mich.). Such cleaners are often preceded or followed by a water rinse, such as with tap water, distilled water, or combinations thereof.
[0102] As mentioned above, according to the present invention, at least a portion of the cleaned substrate surface may be deoxidized, mechanically and/or chemically. As used herein, the term "deoxidize" means removal of the oxide layer found on the surface of the substrate in order to promote uniform deposition of a conversion or pretreatment composition as well as to promote the adhesion of the such a composition coating to the substrate surface. Suitable deoxidizers will be familiar to those skilled in the art. A typical mechanical deoxidizer may be uniform roughening of the substrate surface, such as by using a scouring or cleaning pad. Typical chemical deoxidizers include, for example, acid-based deoxidizers such as phosphoric acid, nitric acid, fluoroboric acid, sulfuric acid, chromic acid, hydrofluoric acid, and ammonium bifluoride, or Amchem 7/17 deoxidizers (available from Henkel Technologies, Madison Heights, Mich.), OAKITE DEOXIDIZER LNC (commercially available from Chemetall), TURCO DEOXIDIZER 6 (commercially available from Henkel), or combinations thereof. Often, the chemical deoxidizer comprises a carrier, often an aqueous medium, so that the deoxidizer may be in the form of a solution or dispersion in the carrier, in which case the solution or dispersion may be brought into contact with the substrate by any of a variety of known techniques, such as dipping or immersion, spraying, intermittent spraying, dipping followed by spraying, spraying followed by dipping, brushing, or roll-coating. According to the present invention, the skilled artisan will select a temperature range of the solution or dispersion, when applied to the metal substrate, based on etch rates, for example, at a temperature ranging from 50.degree. F. to 150.degree. F. (10.degree. C. to 66.degree. C.), such as from 70.degree. F. to 130.degree. F. (21.degree. C. to 54.degree. C.), such as from 80.degree. F. to 120.degree. F. (27.degree. C. to 49.degree. C.). The contact time may be from 30 seconds to 20 minutes, such as 1 minute to 15 minutes, such as 90 seconds to 12 minutes, such as 3 minutes to 9 minutes.
[0103] Following the cleaning and/or deoxidizing step(s), the substrate optionally may be rinsed with tap water, deionized water, and/or an aqueous solution of rinsing agents in order to remove any residue. According to the present invention, the wet substrate surface may be pretreated by any method familiar to those skilled in the art of substrate protection, such an anodized or treated with a conversion composition, and/or may be treated one of the treatment compositions described above, or the substrate may be dried prior to treating the substrate surface, such as air dried, for example, by using an air knife, by flashing off the water by brief exposure of the substrate to a high temperature, such as 15.degree. C. to 100.degree. C., such as 20.degree. C. to 90.degree. C., or in a heater assembly using, for example, infrared heat, such as for 10 minutes at 70.degree. C., or by passing the substrate between squeegee rolls. According to the present invention, the conversion composition may comprise, for example, a lanthanide series element, a Group IIIB metal, and/or a Group IVB metal, and may further comprise a Group IIA metal, a Group VB metal, a Group VIB metal, a Group VIIB metal, and/or a Group XII. According to the present invention, the lanthanide series element may, for example, comprise cerium, praseodymium, terbium, or combinations thereof; the Group IIA metal may comprise magnesium; the Group IIIB metal may comprise yttrium, scandium, or combinations thereof; the Group IVB metal may comprise zirconium, titanium, hafnium, or combinations thereof; the Group VB metal may comprise vanadium; the Group VIB metal may comprise trivalent or hexavalent chromium and/or molybdenum; the Group VIIB metal may comprise manganese; and the Group XII metal may comprise zinc.
[0104] According to the present invention, after the substrate is contacted with the treatment composition, a coating composition comprising a film-forming resin may be deposited onto at least a portion of the surface of the substrate that has been contacted with the treatment composition. Any suitable technique may be used to deposit such a coating composition onto the substrate, including, for example, brushing, dipping, flow coating, spraying and the like. In some instances, however, as described in more detail below, such depositing of a coating composition may comprise an electrocoating step wherein an electrodepositable composition is deposited onto a metal substrate by electrodeposition. In certain other instances, as described in more detail below, such depositing of a coating composition comprises a powder coating step. In still other instances, the coating composition may be a liquid coating composition.
[0105] According to the present invention, the coating composition may comprise a thermosetting film-forming resin or a thermoplastic film-forming resin. As used herein, the term "film-forming resin" refers to resins that can form a self-supporting continuous film on at least a horizontal surface of a substrate upon removal of any diluents or carriers present in the composition or upon curing at ambient or elevated temperature. Conventional film-forming resins that may be used include, without limitation, those typically used in automotive OEM coating compositions, automotive refinish coating compositions, industrial coating compositions, architectural coating compositions, coil coating compositions, and aerospace coating compositions, among others. As used herein, the term "thermosetting" refers to resins that "set" irreversibly upon curing or crosslinking, wherein the polymer chains of the polymeric components are joined together by covalent bonds. This property is usually associated with a cross-linking reaction of the composition constituents often induced, for example, by heat or radiation. Curing or crosslinking reactions also may be carried out under ambient conditions. Once cured or crosslinked, a thermosetting resin will not melt upon the application of heat and is insoluble in solvents. As used herein, the term "thermoplastic" refers to resins that comprise polymeric components that are not joined by covalent bonds and thereby can undergo liquid flow upon heating and are soluble in solvents.
[0106] As previously indicated, according to the present invention, an electrodepositable coating composition comprising a water-dispersible, ionic salt group-containing film-forming resin that may be deposited onto the substrate by an electrocoating step wherein the electrodepositable coating composition is deposited onto the metal substrate by electrodeposition. The ionic salt group-containing film-forming polymer may comprise a cationic salt group containing film-forming polymer for use in a cationic electrodepositable coating composition. As used herein, the term "cationic salt group-containing film-forming polymer" refers to polymers that include at least partially neutralized cationic groups, such as sulfonium groups and ammonium groups, that impart a positive charge. The cationic salt group-containing film-forming polymer may comprise active hydrogen functional groups, including, for example, hydroxyl groups, primary or secondary amine groups, and thiol groups. Cationic salt group-containing film-forming polymers that comprise active hydrogen functional groups may be referred to as active hydrogen-containing, cationic salt group-containing film-forming polymers. Examples of polymers that are suitable for use as the cationic salt group-containing film-forming polymer include, but are not limited to, alkyd polymers, acrylics, polyepoxides, polyamides, polyurethanes, polyureas, polyethers, and polyesters, among others. The cationic salt group-containing film-forming polymer may be present in the cationic electrodepositable coating composition in an amount of 40% to 90% by weight, such as 50% to 80% by weight, such as 60% to 75% by weight, based on the total weight of the resin solids of the electrodepositable coating composition. As used herein, the "resin solids" include the ionic salt group-containing film-forming polymer, curing agent, and any additional water-dispersible non-pigmented component(s) present in the electrodepositable coating composition.
[0107] Alternatively, the ionic salt group containing film-forming polymer may comprise an anionic salt group containing film-forming polymer for use in an anionic electrodepositable coating composition. As used herein, the term "anionic salt group containing film-forming polymer" refers to an anionic polymer comprising at least partially neutralized anionic functional groups, such as carboxylic acid and phosphoric acid groups that impart a negative charge. The anionic salt group-containing film-forming polymer may comprise active hydrogen functional groups. Anionic salt group-containing film-forming polymers that comprise active hydrogen functional groups may be referred to as active hydrogen-containing, anionic salt group-containing film-forming polymers. The anionic salt group-containing film-forming polymer may comprise base-solubilized, carboxylic acid group-containing film-forming polymers such as the reaction product or adduct of a drying oil or semi-drying fatty acid ester with a dicarboxylic acid or anhydride; and the reaction product of a fatty acid ester, unsaturated acid or anhydride and any additional unsaturated modifying materials which are further reacted with polyol. Also suitable are the at least partially neutralized interpolymers of hydroxy-alkyl esters of unsaturated carboxylic acids, unsaturated carboxylic acid and at least one other ethylenically unsaturated monomer. Still another suitable anionic electrodepositable resin comprises an alkyd-aminoplast vehicle, i.e., a vehicle containing an alkyd resin and an amine-aldehyde resin. Another suitable anionic electrodepositable resin composition comprises mixed esters of a resinous polyol. Other acid functional polymers may also be used such as phosphatized polyepoxide or phosphatized acrylic polymers. Exemplary phosphatized polyepoxides are disclosed in U.S. Patent Application Publication No. 2009-0045071 at [0004]-[0015] and U.S. patent application Ser. No. 13/232,093 at [0014]-[0040], the cited portions of which being incorporated herein by reference. The anionic salt group-containing film-forming polymer may be present in the anionic electrodepositable coating composition in an amount 50% to 90%, such as 55% to 80%, such as 60% to 75%, based on the total weight of the resin solids of the electrodepositable coating composition.
[0108] The electrodepositable coating composition may further comprise a curing agent. The curing agent may react with the reactive groups, such as active hydrogen groups, of the ionic salt group-containing film-forming polymer to effectuate cure of the coating composition to form a coating. Non-limiting examples of suitable curing agents are at least partially blocked polyisocyanates, aminoplast resins and phenoplast resins, such as phenolformaldehyde condensates including allyl ether derivatives thereof. The curing agent may be present in the cationic electrodepositable coating composition in an amount of 10% to 60% by weight, such as 20% to 50% by weight, such as 25% to 40% by weight, based on the total weight of the resin solids of the electrodepositable coating composition. Alternatively, the curing agent may be present in the anionic electrodepositable coating composition in an amount of 10% to 50% by weight, such as 20% to 45% by weight, such as 25% to 40% by weight, based on the total weight of the resin solids of the electrodepositable coating composition.
[0109] The electrodepositable coating composition may further comprise other optional ingredients, such as a pigment composition and, if desired, various additives such as fillers, plasticizers, anti-oxidants, biocides, UV light absorbers and stabilizers, hindered amine light stabilizers, defoamers, fungicides, dispersing aids, flow control agents, surfactants, wetting agents, or combinations thereof. The electrodepositable coating composition may comprise water and/or one or more organic solvent(s). Water can for example be present in amounts of 40% to 90% by weight, such as 50% to 75% by weight, based on total weight of the electrodepositable coating composition. If used, the organic solvents may typically be present in an amount of less than 10% by weight, such as less than 5% by weight, based on total weight of the electrodepositable coating composition. The electrodepositable coating composition may in particular be provided in the form of an aqueous dispersion. The total solids content of the electrodepositable coating composition may be from 1% to 50% by weight, such as 5% to 40% by weight, such as 5% to 20% by weight, based on the total weight of the electrodepositable coating composition. As used herein, "total solids" refers to the non-volatile content of the electrodepositable coating composition, i.e., materials which will not volatilize when heated to 110.degree. C. for 15 minutes.
[0110] The cationic electrodepositable coating composition may be deposited upon an electrically conductive substrate by placing the composition in contact with an electrically conductive cathode and an electrically conductive anode, with the surface to be coated being the cathode. Alternatively, the anionic electrodepositable coating composition may be deposited upon an electrically conductive substrate by placing the composition in contact with an electrically conductive cathode and an electrically conductive anode, with the surface to be coated being the anode. An adherent film of the electrodepositable coating composition is deposited in a substantially continuous manner on the cathode or anode, respectively, when a sufficient voltage is impressed between the electrodes. The applied voltage may be varied and can be, for example, as low as one volt to as high as several thousand volts, such as between 50 and 500 volts. Current density is usually between 1.0 ampere and 15 amperes per square foot (10.8 to 161.5 amperes per square meter) and tends to decrease quickly during the electrodeposition process, indicating formation of a continuous self-insulating film.
[0111] Once the cationic or anionic electrodepositable coating composition is electrodeposited over at least a portion of the electroconductive substrate, the coated substrate may be heated to a temperature and for a time sufficient to cure the electrodeposited coating on the substrate. For cationic electrodeposition, the coated substrate may be heated to a temperature ranging from 250.degree. F. to 450.degree. F. (121.1.degree. C. to 232.2.degree. C.), such as from 275.degree. F. to 400.degree. F. (135.degree. C. to 204.4.degree. C.), such as from 300.degree. F. to 360.degree. F. (149.degree. C. to 180.degree. C.). For anionic electrodeposition, the coated substrate may be heated to a temperature ranging from 200.degree. F. to 450.degree. F. (93.degree. C. to 232.2.degree. C.), such as from 275.degree. F. to 400.degree. F. (135.degree. C. to 204.4.degree. C.), such as from 300.degree. F. to 360.degree. F. (149.degree. C. to 180.degree. C.), such as 200.degree. F. to 210.2.degree. F. (93.degree. C. to 99.degree. C.). The curing time may be dependent upon the curing temperature as well as other variables, for example, the film thickness of the electrodeposited coating, level and type of catalyst present in the composition and the like. For example, the curing time can range from 10 minutes to 60 minutes, such as 20 to 40 minutes. The thickness of the resultant cured electrodeposited coating may range from 2 to 50 microns.
[0112] Alternatively, as mentioned above, according to the present invention, after the substrate has been contacted the treatment composition, a powder coating composition may then be deposited onto at least a portion of the surface of the substrate that has been contacted with the treatment composition. As used herein, "powder coating composition" refers to a coating composition which is completely free of water and/or solvent. Accordingly, the powder coating composition disclosed herein is not synonymous to waterborne and/or solvent-borne coating compositions known in the art. According to the present invention, the powder coating composition may comprise (a) a film forming polymer having a reactive functional group; and (b) a curing agent that is reactive with the functional group. Examples of powder coating compositions that may be used in the present invention include the polyester-based ENVIROCRON line of powder coating compositions (commercially available from PPG Industries, Inc.) or epoxy-polyester hybrid powder coating compositions. Alternative examples of powder coating compositions that may be used in the present invention include low temperature cure thermosetting powder coating compositions comprising (a) at least one tertiary aminourea compound, at least one tertiary aminourethane compound, or mixtures thereof, and (b) at least one film-forming epoxy-containing resin and/or at least one siloxane-containing resin (such as those described in U.S. Pat. No. 7,470,752, assigned to PPG Industries, Inc. and incorporated herein by reference); curable powder coating compositions generally comprising (a) at least one tertiary aminourea compound, at least one tertiary aminourethane compound, or mixtures thereof, and (b) at least one film-forming epoxy-containing resin and/or at least one siloxane-containing resin (such as those described in U.S. Pat. No. 7,432,333, assigned to PPG Industries, Inc. and incorporated herein by reference); and those ccomprising a solid particulate mixture of a reactive group-containing polymer having a T.sub.g of at least 30.degree. C. (such as those described in U.S. Pat. No. 6,797,387, assigned to PPG Industries, Inc. and incorporated herein by reference).
[0113] After deposition of the powder coating composition, the coating is often heated to cure the deposited composition. The heating or curing operation is often carried out at a temperature in the range of from 150.degree. C. to 200.degree. C., such as from 170.degree. C. to 190.degree. C., for a period of time ranging from 10 to 20 minutes. According to the invention, the thickness of the resultant film is from 50 microns to 125 microns.
[0114] As mentioned above, according to the present invention, the coating composition may be a liquid coating composition. As used herein, "liquid coating composition" refers to a coating composition which contains a portion of water and/or solvent. Accordingly, the liquid coating composition disclosed herein is synonymous to waterborne and/or solventborne coating compositions known in the art. According to the present invention, the liquid coating composition may comprise, for example, (a) a film forming polymer having a reactive functional group; and (b) a curing agent that is reactive with the functional group. In other examples, the liquid coating may contain a film forming polymer that may react with oxygen in the air or coalesce into a film with the evaporation of water and/or solvents. These film forming mechanisms may require or be accelerated by the application of heat or some type of radiation such as Ultraviolet or Infrared. Examples of liquid coating compositions that may be used in the present invention include the SPECTRACRON.RTM. line of solventbased coating compositions, the AQUACRON.RTM. line of waterbased coating compositions, and the RAYCRON.RTM. line of UV cured coatings (all commercially available from PPG Industries, Inc.). Suitable film forming polymers that may be used in the liquid coating composition of the present invention may comprise a (poly)ester, an alkyd, a (poly)urethane, an isocyanurate, a (poly)urea, a (poly)epoxy, an anhydride, an acrylic, a (poly)ether, a (poly)sulfide, a (poly)amine, a (poly)amide, (poly)vinyl chloride, (poly)olefin, (poly)vinylidene fluoride, (poly)siloxane, or combinations thereof.
[0115] According to the present invention, the substrate that has been contacted with the treatment composition described herein may also be contacted with a primer composition and/or a topcoat composition. The primer coat may be, for examples, chromate-based primers and advanced performance topcoats. According to the present invention, the primer coat can be a conventional chromate based primer coat, such as those available from PPG Industries, Inc. (product code 44GN072), or a chrome-free primer such as those available from PPG (DESOPRIME CA7502, DESOPRIME CA7521, Deft 02GN083, Deft 02GN084). Alternately, the primer coat can be a chromate-free primer coat, such as the coating compositions described in U.S. patent application Ser. No. 10/758,973, titled "CORROSION RESISTANT COATINGS CONTAINING CARBON", and U.S. patent application Ser. No. 10/758,972, and Ser. No. 10/758,972, both titled "CORROSION RESISTANT COATINGS", all of which are incorporated herein by reference, and other chrome-free primers that are known in the art, and which can pass the military requirement of MIL-PRF-85582 Class N or MIL-PRF-23377 Class N may also be used with the current invention.
[0116] As mentioned above, the substrate of the present invention also may comprise a topcoat. As used herein, the term "topcoat" refers to a mixture of binder(s) which can be an organic or inorganic based polymer or a blend of polymers, typically at least one pigment, can optionally contain at least one solvent or mixture of solvents, and can optionally contain at least one curing agent. A topcoat is typically the coating layer in a single or multi-layer coating system whose outer surface is exposed to the atmosphere or environment, and its inner surface is in contact with another coating layer or polymeric substrate. Examples of suitable topcoats include those conforming to MIL-PRF-85285D, such as those available from PPG (Deft 03W127A and Deft 03GY292). According to the present invention, the topcoat may be an advanced performance topcoat, such as those available from PPG (Defthane.RTM. ELT.RTM. 99GY001 and 99W009). However, other topcoats and advanced performance topcoats can be used in the present invention as will be understood by those of skill in the art with reference to this disclosure.
[0117] According to the present invention, the metal substrate also may comprise a self-priming topcoat, or an enhanced self-priming topcoat. The term "self-priming topcoat", also referred to as a "direct to substrate" or "direct to metal" coating, refers to a mixture of a binder(s), which can be an organic or inorganic based polymer or blend of polymers, typically at least one pigment, can optionally contain at least one solvent or mixture of solvents, and can optionally contain at least one curing agent. The term "enhanced self-priming topcoat", also referred to as an "enhanced direct to substrate coating" refers to a mixture of functionalized fluorinated binders, such as a fluoroethylene-alkyl vinyl ether in whole or in part with other binder(s), which can be an organic or inorganic based polymer or blend of polymers, typically at least one pigment, can optionally contain at least one solvent or mixture of solvents, and can optionally contain at least one curing agent. Examples of self-priming topcoats include those that conform to TT-P-2756A. Examples of self-priming topcoats include those available from PPG (03W169 and 03GY369), and examples of enhanced self-priming topcoats include Defthane.RTM. ELT.TM./ESPT and product code number 97GY121, available from PPG. However, other self-priming topcoats and enhanced self-priming topcoats can be used in the coating system according to the present invention as will be understood by those of skill in the art with reference to this disclosure.
[0118] According to the present invention, the self-priming topcoat and enhanced self-priming topcoat may be applied directly to the sealed substrate. The self-priming topcoat and enhanced self-priming topcoat can optionally be applied to an organic or inorganic polymeric coating, such as a primer or paint film. The self-priming topcoat layer and enhanced self-priming topcoat is typically the coating layer in a single or multi-layer coating system where the outer surface of the coating is exposed to the atmosphere or environment, and the inner surface of the coating is typically in contact with the substrate or optional polymer coating or primer.
[0119] According to the present invention, the topcoat, self-priming topcoat, and enhanced self-priming topcoat can be applied to the sealed substrate, in either a wet or "not fully cured" condition that dries or cures over time, that is, solvent evaporates and/or there is a chemical reaction. The coatings can dry or cure either naturally or by accelerated means for example, an ultraviolet light cured system to form a film or "cured" paint. The coatings can also be applied in a semi or fully cured state, such as an adhesive.
[0120] In addition, a colorant and, if desired, various additives such as surfactants, wetting agents or catalyst can be included in the coating composition (electrodepositable, powder, or liquid). As used herein, the term "colorant" means any substance that imparts color and/or other opacity and/or other visual effect to the composition. Example colorants include pigments, dyes and tints, such as those used in the paint industry and/or listed in the Dry Color Manufacturers Association (DCMA), as well as special effect compositions. In general, the colorant can be present in the coating composition in any amount sufficient to impart the desired visual and/or color effect. The colorant may comprise from 1 to 65 weight percent, such as from 3 to 40 weight percent or 5 to 35 weight percent, with weight percent based on the total weight of the composition.
Aspects
[0121] In view of the foregoing the present application thus relates in particular, without being limited thereto, to the following Aspects 1 to 24:
[0122] 1. A composition comprising carbon dioxide and a lithium cation, in an aqueous medium.
[0123] 2. The composition according to Aspect 1, wherein the carbon dioxide comprises a gas, a solid, or combinations thereof.
[0124] 3. The composition according to any one of Aspects 1 or 2, wherein the lithium cation is present in an amount of 5 ppm to 5500 ppm (calculated as lithium cation) based on total weight of the treatment composition.
[0125] 4. The composition according to any one of the preceding Aspects, wherein the pH is 9.5 to 12.5.
[0126] 5. The composition according to any one of the preceding Aspects, further comprising a hydroxide.
[0127] 6. The composition according to any one of the preceding Aspects, wherein the carbonate is present in an amount of 15 ppm to 25,000 ppm (calculated as carbonate) based on total weight of the treatment composition.
[0128] 7. A method of making a treatment composition comprising: [0129] combining a lithium cation and carbon dioxide in an aqueous medium to form the treatment composition in situ, wherein the treatment composition comprises comprising lithium in an amount of 5 ppm to 5,500 ppm (calculated as lithium cation) based on total weight of the treatment composition and carbonate in an amount of 15 ppm to 25,000 ppm (calculated as carbonate) based on total weight of the treatment composition.
[0130] 8. The method of making a treatment composition according to Aspect 7, wherein the lithium cation is present as lithium carbonate, lithium hydroxide, or a combination thereof.
[0131] 9. The method of making a treatment composition according to any one of Aspects 7 or 8, wherein the carbon dioxide is supplied to the aqueous medium as a gas, a solid, or a combination thereof.
[0132] 10. The method of making a treatment composition according to any one of Aspects 7 to 9, comprising adding a hydroxide to the aqueous medium.
[0133] 11. The method of making a treatment composition according to Aspect 10, wherein the hydroxide comprises lithium hydroxide, sodium hydroxide, potassium hydroxide, or a combination thereof.
[0134] 12. The method of making a treatment composition according to any one of Aspects 7 to 11, wherein the method comprises adjusting the pH of the treatment composition to from 9.5 to 12.5.
[0135] 13. A treatment composition obtained according to the method of any one of Aspects 7 to 12.
[0136] 14. A method for maintaining a treatment bath formed from a treatment composition comprising lithium carbonate, the method comprising: [0137] supplying during and/or after treatment of a substrate with the bath at least one of carbon dioxide and a lithium salt to the bath in an amount sufficient to maintain the pH of the bath at 9.5 to 12.5, lithium in an amount of 5 ppm to 5,500 ppm (calculated as lithium cation) based on total weight of the treatment bath, and carbonate in an amount of 15 ppm to 25,000 ppm (calculated as carbonate) based on total weight of the treatment bath.
[0138] 15. The method for maintaining a treatment bath according to Aspect 14, wherein the lithium salt comprises lithium carbonate, lithium hydroxide, or a combination thereof.
[0139] 16. The method for maintaining a treatment bath according to any one of Aspects 14 or 15, wherein the carbon dioxide is supplied to the bath as a gas, a solid, or a combination thereof.
[0140] 17. The method for maintaining a treatment bath according to any one of Aspects 14 to 16, comprising supplying a hydroxide to the bath.
[0141] 18. The method for maintaining a treatment bath according to Aspect 17, wherein the hydroxide comprises lithium hydroxide, sodium hydroxide, potassium hydroxide, or a combination thereof.
[0142] 19. The method for maintaining a treatment bath according to any one of Aspects 14 to 18, further comprising monitoring pH of the treatment bath, amount of carbonate in the treatment bath, amount of lithium in the treatment bath, or a combination thereof.
[0143] 20. A substrate treated with the treatment composition of any one of Aspects 1 to 6 or 13 or with the treatment bath maintained according to the method of any one of Aspects 14 to 19.
[0144] 21. A system for maintaining a treatment bath formed from a treatment composition comprising lithium carbonate, the system comprising: [0145] a lithium salt source; and/or [0146] a carbon dioxide source; and [0147] optionally, a hydroxide source.
[0148] 22. The system for maintaining a treatment bath according to Aspect 21, wherein the lithium salt source comprises lithium carbonate, lithium hydroxide, or a combination thereof.
[0149] 23. The system for maintaining a treatment bath according to any one of Aspects 21 or 22, wherein the carbon dioxide source comprises carbon dioxide as a gas, a solid, or a combination thereof.
[0150] 24. The system for maintaining a treatment bath according to any one of Aspects 21 to 23, wherein the system includes a hydroxide source comprising lithium hydroxide, sodium hydroxide, potassium hydroxide, or a combination thereof.
[0151] Whereas particular features of the present invention have been described above for purposes of illustration, it will be evident to those skilled in the art that numerous variations of the details of the treatment composition and bath formed therefrom and methods of preparing or maintaining the same disclosed herein may be made without departing from the scope in the appended claims.
[0152] Illustrating the invention are the following examples that are not to be considered as limiting the invention to their details. All parts and percentages in the examples, as well as throughout the specification, are by weight unless otherwise indicated.
EXAMPLES
TABLE-US-00002 [0153] TABLE 2 Materials lithium carbonate, 98% Alfa Aesar lithium hydroxide mono hydrate, 98% min Alfa Aesar carbon dioxide gas Air Gas sodium hydroxide pellets, 98% Alfa Aesar sodium phosphate dodecahydrate, 97% Alfa Aesar polyvinylpyrrolidone (PVP), 8000 m.w. Alfa Aesar Allantoin, 98% Alfa Aesar 2,5-dimercapto-1,3,4-thiadiazole, 98% Acros Organics Carbowet GA100, 100% Air Products cerium nitrate solution (65.37% Ce(NO.sub.3).sub.3.cndot.6H.sub.2O) ProChem Inc. yttrium nitrate solution (72.45% Y(NO.sub.3).sub.3.cndot.6H.sub.2O) ProChem Inc. cerium chloride solution (32.2% as CeO.sub.2*) ProChem Inc. hydrogen peroxide solution (30% H.sub.2O.sub.2) Alfa Aesar *per supplier's certificate of analysis
TABLE-US-00003 TABLE 3 Cleaner/Deoxidizer Composition (Example A) WEIGHT (g) sodium hydroxide pellets, 98% 0.016 sodium phosphate dodecahydrate, 97% 0.063 polyvinylpyrrolidone (PVP), 8000 m.w. 0.002 Allantoin, 98% 0.003 2,5-dimercapto-1,3,4-thiadiazole(DMTD), 98% 0.100 Carbowet GA100 0.410 deionized water 98.7
[0154] The ingredients and their relative amounts used to prepare cleaner/deoxidizer composition Example A are provided in Table 3. Sodium hydroxide and sodium phosphate were completely dissolved in deionized water under mild mechanical agitation using a stir plate (VWR, 7.times.7 CER HOT/STIR). Next, once the sodium hydroxide and sodium phosphate were completely dissolved, the PVP was stirred in until dissolved, and then Allantoin was added and stirred until dissolved, and then the DMTD was added and stirred until dissolved. After the DMTD was completely dissolved, Carbowet GA100 was stirred in under mild mechanical agitation as above.
TABLE-US-00004 TABLE 4 Conversion Composition (Example B) Mass (g) Yttrium Nitrate Solution 12.48 Cerium Nitrate Solution 10.40 Cerium Chloride Solution 0.04 Hydrogen Peroxide Solution 1.04 Deionized Water 1953
[0155] The ingredients used to prepare a solution of conversion coating composition Example B and their amounts are provided in Table 4. Cerium nitrate, yttrium nitrate and cerium chloride solutions were weighed into individual cups. Then using 500 grams of deionized water, the solutions were transferred to a vessel containing 1,000 grams of deionized water under mild agitation. The remaining 453 grams of water was added and the solution was stirred for 10 minutes to ensure uniformity before the hydrogen peroxide was added. The final solution was stirred for a minimum of 30 minutes before use.
TABLE-US-00005 TABLE 5 Sealing Compositions (Examples C-O) LiCO.sub.3 LiOH 5% LiOH Deionized (98% purity) (98% purity) Li Solution Water (g) (g) (moles) CO.sub.2 (g) (g) pH Ex. C 3.06 -- 0.081 -- -- 1996.94 11.52 Ex. D -- 1.99 0.081 -- -- 1998.01 12.69 Ex. E -- -- -- Bubbled -- -- 11.42 Ex. F -- -- -- Bubbled -- -- 10.54 Ex. G -- -- -- Bubbled -- -- 9.47 Ex. H -- -- -- -- 15.3 -- 10.47 Ex. I -- -- -- -- 15.5 -- 11.48 Ex. J -- -- -- -- 23.2 -- 12.47 Ex. K 3.06 -- 0.081 -- -- 1996.94 11.14 Ex. L -- 1.99 0.081 -- -- 1998.01 12.17 Ex. M -- -- -- Bubbled -- -- 11.37 Ex. N -- -- -- Bubbled -- -- 9.50 Ex. O -- -- -- -- 15.54 -- 11.37
[0156] In preparing the sealing compositions, pH for each Example C-J was measured using a pH meter (Accumet AB15, Fisher Scientific) and a single junction electrode (Ag/AgCl reference; Fisher Scientific) and the pH for each Example K-O was measured using a pH meter (Mettler Toledo, Seven2Go, model S2) and a double open junction electrode (Mettler Toledo, Xerolyt.RTM. polymer reference).
[0157] Sealing composition Example C and Example K each were prepared using the ingredients shown in Table 5 by dissolving lithium carbonate into deionized water under mild agitation using the stir plate as described above (VWR, 7.times.7 CER HOT/STIR). Example C had a final pH of 11.52. Example C was used to treat panels in Comparative Example 1 (described below). Example K had a final pH of 11.14. Example K was used to treat panels in Comparative Example 9 (described below).
[0158] Sealing composition Example D and Example L each were prepared using the ingredients shown in Table 5 by dissolving lithium hydroxide into deionized water under mild agitation using the stir plate as described above. Example D had a final pH value of 12.69. Example D was used to treat panels in Comparative Example 2 (described below). Example L had a final pH value of 12.17. Example L was used to treat panels in Comparative Example 10 (described below).
[0159] Following use of the bath containing the composition of Example D to treat panels according to Comparative Example 2, sealing composition Example E was prepared by bubbling carbon dioxide gas into the bath containing the composition of Example D until a final pH value of 11.42 was obtained. See FIG. 1 and Table 5. Example E was used to treat panels in Example 3 (described below).
[0160] Following use of the bath containing the composition of Example E to treat panels according to Example 3, sealing composition Example F was prepared by bubbling additional carbon dioxide gas into the composition of Example E until a final pH value of 10.54 was obtained. See FIG. 1 and Table 5. Example F was used to treat panels in Example 4 (described below).
[0161] Following use of the bath containing the composition of Example F to treat panels according to Example 4, sealing composition Example G was prepared by bubbling additional carbon dioxide gas into the composition of Example F until a final pH value of 9.47 was obtained. See FIG. 1 and Table 5. Example G was used to treat panels in Example 5 (described below).
[0162] Following the use of the bath containing the composition of Example G to treat panels according to Example 5, sealing composition Example H was prepared by adding 5% lithium hydroxide solution into the composition of Example G until a final pH value of 10.47 was obtained. See FIG. 1 and Table 5. Example H was used to treat panels in Example 6 (described below).
[0163] Following the use of the bath containing the composition of Example H according to Example 6, sealing composition Example I was prepared by adding 5% lithium hydroxide solution into the composition of Example H until a final pH value of 11.48 was obtained. See FIG. 1 and Table 5. Example I was used to treat panels in Example 7 (described below).
[0164] Following the use of the bath containing the composition of Example I according to Example 7, sealing composition Example J was prepared by adding 5% lithium hydroxide solution into the composition of Example I until a final pH value of 12.47 was obtained. See FIG. 1 and Table 5. Example J was used to treat panels in Example 8 (described below).
[0165] Following use of the bath containing the composition of Example L to treat panels according to Comparative Example 10, sealing composition Example M was prepared by bubbling carbon dioxide gas into the bath containing the composition of Example L until a final pH value of 11.37 was obtained. See FIG. 2 and Table 5. Example M was used to treat panels in Example 11 (described below).
[0166] Following use of the bath containing the composition of Example M to treat panels according to Example 11, sealing composition Example N was prepared by bubbling additional carbon dioxide gas into the composition of Example M until a final pH value of 9.5 was obtained. See FIG. 2 and Table 5. Example N was used to treat panels in Example 12 (described below).
[0167] Following the use of the bath containing the composition of Example N according to Example 12, sealing composition Example O was prepared by adding 5% lithium hydroxide solution into the composition of Example N until a final pH value of 11.37 was obtained. See FIG. 2 and Table 5. Example O was used to treat panels in Example 13 (described below).
Panel Preparation
Comparative Example 1
[0168] Aluminum 2024T3 bare substrate (obtained from Priority Metals, Orange County, Calif.) measuring 3''.times.5''.times.0.032'' was hand-wiped with methyl ethyl ketone and a disposable cloth and allowed to air dry prior to chemical cleaning. The panel was immersed in the cleaner-deoxidizer composition Example A for 3.5 minutes at ambient temperature with intermittent agitation. The panel was then immersed in two subsequent deionized water rinses for two minutes each, both at ambient temperature with intermittent agitation. After the second rinse, the panel was rinsed with a cascading deionized water rinse for 10 seconds. The panel was then immersed in the conversion composition Example B for 5 minutes at ambient temperature without agitation. Next, the panel was rinsed by immersion in deionized water for 2 minutes at ambient temperature with intermittent agitation followed by a 10 second cascading deionized water rinse. The panel was then immersed in the sealing composition Example C for 2 minutes at ambient temperature with intermittent agitation. The panel was air dried at ambient conditions overnight before corrosion testing as described below.
Comparative Example 2
[0169] Aluminum 2024T3 bare substrate (obtained from Priority Metals, Orange County, Calif.) measuring 3''.times.5''.times.0.032'' was hand-wiped with methyl ethyl ketone and a disposable cloth and allowed to air dry prior to chemical cleaning. The panel was immersed in the cleaner-deoxidizer composition Example A for 3.5 minutes at ambient temperature with intermittent agitation. The panel was then immersed in two subsequent deionized water rinses for two minutes each, both at ambient temperature with intermittent agitation. After the second rinse, the panel was rinsed with a cascading deionized water rinse for 10 seconds. The panel was then immersed in the conversion composition Example B for 5 minutes at ambient temperature and without agitation. Next, the panel was rinsed by immersion in deionized water for 2 minutes at ambient temperature with intermittent agitation followed by a 10 second cascading deionized water rinse. The panel was then immersed in the sealing composition Example D for 2 minutes at ambient temperature with intermittent agitation. The panel was air dried at ambient conditions overnight before corrosion testing as described below.
Example 3
[0170] After panels from Comparative Example 2 were processed through the seal solution of Example D, the pH of the bath was adjusted by bubbling carbon dioxide gas into the bath until the pH was 11.42 (i.e., to form Example E as described above). See FIG. 1.
[0171] Next, additional aluminum 2024T3 bare substrate measuring 3''.times.5''.times.0.032'' (Priority Metals, Orange County, Calif.) was hand-wiped with methyl ethyl ketone and a disposable cloth and allowed to air dry prior to chemical cleaning. The panel was immersed in the cleaner-deoxidizer composition Example A for 3.5 minutes at ambient temperature with intermittent agitation. The panel was then immersed in two subsequent deionized water rinses for two minutes each, both at ambient temperature with intermittent agitation. After the second rinse, the panel was rinsed with a cascading deionized water rinse for 10 seconds. The panel was then immersed in the conversion composition Example B for 5 minutes at ambient temperature and without agitation. Next, the panel was rinsed by immersion in deionized water for 2 minutes at ambient temperature with intermittent agitation followed by a 10 second cascading deionized water rinse. The panel was then immersed in the sealing composition Example E for 2 minutes at ambient temperature with intermittent agitation. The panel was air dried at ambient conditions overnight before corrosion testing as described below.
Example 4
[0172] After panels were processed through the seal solution of Example E, the pH of the bath was adjusted by bubbling carbon dioxide gas into the bath until the pH was 10.54 (i.e., to form Example F as described above). See FIG. 1.
[0173] Next, additional aluminum 2024T3 bare substrate measuring 3''.times.5''.times.0.032'' (Priority Metals, Orange County, Calif.) was hand-wiped with methyl ethyl ketone and a disposable cloth and allowed to air dry prior to chemical cleaning. The panel was immersed in the cleaner-deoxidizer composition Example A for 3.5 minutes at ambient temperature with intermittent agitation. The panel was then immersed in two subsequent deionized water rinses for two minutes each, both at ambient temperature with intermittent agitation. After the second rinse, the panel was rinsed with a cascading deionized water rinse for 10 seconds. The panel was then immersed in the conversion composition Example B for 5 minutes at ambient temperature and without agitation. Next, the panel was rinsed by immersion in deionized water for 2 minutes at ambient temperature with intermittent agitation followed by a 10 second cascading deionized water rinse. The panel was then immersed in the sealing composition Example F for 2 minutes at ambient temperature with intermittent agitation. The panel was air dried at ambient conditions overnight before corrosion testing as described below.
Example 5
[0174] After panels were processed through the seal solution of Example F, the pH of the bath was adjusted by bubbling carbon dioxide gas into the bath until the pH was 9.47 (i.e., to form Example G as described above). See FIG. 1.
[0175] Next, additional aluminum 2024T3 bare substrate measuring 3''.times.5''.times.0.032'' (Priority Metals, Orange County, Calif.) was hand-wiped with methyl ethyl ketone and a disposable cloth and allowed to air dry prior to chemical cleaning. The panel was immersed in the cleaner-deoxidizer composition Example A for 3.5 minutes at ambient temperature with intermittent agitation. The panel was then immersed in two subsequent deionized water rinses for two minutes each, both at ambient temperature with intermittent agitation. After the second rinse, the panel was rinsed with a cascading deionized water rinse for 10 seconds. The panel was then immersed in the conversion composition Example B for 5 minutes at ambient temperature and without agitation. Next, the panel was rinsed by immersion in deionized water for 2 minutes at ambient temperature with intermittent agitation followed by a 10 second cascading deionized water rinse. The panel was then immersed in the sealing composition Example G for 2 minutes at ambient temperature with intermittent agitation. The panel was air dried at ambient conditions overnight before corrosion testing as described below.
Example 6
[0176] After panels were processed through the seal solution of Example G, the pH of the bath was adjusted using lithium hydroxide solution as described above (i.e., to form Example H as described above). See FIG. 1.
[0177] Next, additional aluminum 2024T3 bare substrate measuring 3''.times.5''.times.0.032'' (Priority Metals, Orange County, Calif.) was hand-wiped with methyl ethyl ketone and a disposable cloth and allowed to air dry prior to chemical cleaning. The panel was immersed in the cleaner-deoxidizer composition Example A for 3.5 minutes at ambient temperature with intermittent agitation. The panel was then immersed in two subsequent deionized water rinses for two minutes each, both at ambient temperature with intermittent agitation. After the second rinse, the panel was rinsed with a cascading deionized water rinse for 10 seconds. The panel was then immersed in the conversion composition Example B for 5 minutes at ambient temperature and without agitation. Next, the panel was rinsed by immersion rinse in deionized water for 2 minutes at ambient temperature with intermittent agitation followed by a 10 second cascading deionized water rinse. The panel was then immersed in the sealing composition Example H for 2 minutes at ambient temperature with intermittent agitation. The panel was air dried at ambient conditions overnight before corrosion testing as described below.
Example 7
[0178] After panels were processed through the seal solution of Example H, the pH of the bath was adjusted using lithium hydroxide solution as described above (i.e., to form Example I as described above). See FIG. 1.
[0179] Next, additional aluminum 2024T3 bare substrate measuring 3''.times.5''.times.0.032'' (Priority Metals, Orange County, Calif.) was hand-wiped with methyl ethyl ketone and a disposable cloth and allowed to air dry prior to chemical cleaning. The panel was immersed in the cleaner-deoxidizer composition Example A for 3.5 minutes at ambient temperature with intermittent agitation. The panel was then immersed in two subsequent deionized water rinses for two minutes each, both at ambient temperature with intermittent agitation. After the second rinse, the panel was rinsed with a cascading deionized water rinse for 10 seconds. The panel was then immersed in the conversion composition Example B for 5 minutes at ambient temperature and without agitation. Next, the panel was rinsed by immersion rinse in deionized water for 2 minutes at ambient temperature with intermittent agitation followed by a 10 second cascading deionized water rinse. The panel was then immersed in the sealing composition Example I for 2 minutes at ambient temperature with intermittent agitation. The panel was air dried at ambient conditions overnight before corrosion testing as described below.
Example 8
[0180] After panels were processed through the seal solution of Example I, the pH of the bath was adjusted using lithium hydroxide solution as described above (i.e., to form Example I as described above). See FIG. 1.
[0181] Next, additional aluminum 2024T3 bare substrate measuring 3''.times.5''.times.0.032'' (Priority Metals, Orange County, Calif.) was hand-wiped with methyl ethyl ketone and a disposable cloth and allowed to air dry prior to chemical cleaning. The panel was immersed in the cleaner-deoxidizer composition Example A for 3.5 minutes at ambient temperature with intermittent agitation. The panel was then immersed in two subsequent deionized water rinses for two minutes each, both at ambient temperature with intermittent agitation. After the second rinse, the panel received a cascading deionized water rinse for 10 seconds. The panel was then immersed in the conversion composition Example B for 5 minutes at ambient temperature and without agitation. Next, the panel was rinsed by immersion in deionized water for 2 minutes at ambient temperature with intermittent agitation followed by a 10 second cascading deionized water rinse. The panel was then immersed in the sealing composition Example J for 2 minutes at ambient temperature with intermittent agitation. The panel was air dried at ambient conditions overnight before corrosion testing as described below.
Comparative Example 9
[0182] Aluminum 2024T3 bare substrate (obtained from Priority Metals, Orange County, Calif.) measuring 3''.times.5''.times.0.032'' was hand-wiped with methyl ethyl ketone and a disposable cloth and allowed to air dry prior to chemical cleaning. The panel was immersed in the cleaner-deoxidizer composition Example A for 3.5 minutes at ambient temperature with intermittent agitation. The panel was then immersed in two subsequent deionized water rinses for two minutes each, both at ambient temperature with intermittent agitation. After the second rinse, the panel was rinsed with a cascading deionized water rinse for 10 seconds. The panel was then immersed in the conversion composition Example B for 5 minutes at ambient temperature without agitation. Next, the panel was rinsed by immersion in deionized water for 2 minutes at ambient temperature with intermittent agitation followed by a 10 second cascading deionized water rinse. The panel was then immersed in the sealing composition Example K for 2 minutes at ambient temperature with intermittent agitation. The panel was air dried at ambient conditions overnight before corrosion testing as described below.
Comparative Example 10
[0183] Aluminum 2024T3 bare substrate (obtained from Priority Metals, Orange County, Calif.) measuring 3''.times.5''.times.0.032'' was hand-wiped with methyl ethyl ketone and a disposable cloth and allowed to air dry prior to chemical cleaning. The panel was immersed in the cleaner-deoxidizer composition Example A for 3.5 minutes at ambient temperature with intermittent agitation. The panel was then immersed in two subsequent deionized water rinses for two minutes each, both at ambient temperature with intermittent agitation. After the second rinse, the panel was rinsed with a cascading deionized water rinse for 10 seconds. The panel was then immersed in the conversion composition Example B for 5 minutes at ambient temperature and without agitation. Next, the panel was rinsed by immersion in deionized water for 2 minutes at ambient temperature with intermittent agitation followed by a 10 second cascading deionized water rinse. The panel was then immersed in the sealing composition Example L for 2 minutes at ambient temperature with intermittent agitation. The panel was air dried at ambient conditions overnight before corrosion testing as described below.
Example 11
[0184] After panels from Comparative Example 10 were processed through the seal solution of Example L, the pH of the bath was adjusted by bubbling carbon dioxide gas into the bath until the pH was 11.37 (i.e., to form Example M as described above). See FIG. 2.
[0185] Next, additional aluminum 2024T3 bare substrate measuring 3''.times.5''.times.0.032'' (Priority Metals, Orange County, Calif.) was hand-wiped with methyl ethyl ketone and a disposable cloth and allowed to air dry prior to chemical cleaning. The panel was immersed in the cleaner-deoxidizer composition Example A for 3.5 minutes at ambient temperature with intermittent agitation. The panel was then immersed in two subsequent deionized water rinses for two minutes each, both at ambient temperature with intermittent agitation. After the second rinse, the panel was rinsed with a cascading deionized water rinse for 10 seconds. The panel was then immersed in the conversion composition Example B for 5 minutes at ambient temperature and without agitation. Next, the panel was rinsed by immersion in deionized water for 2 minutes at ambient temperature with intermittent agitation followed by a 10 second cascading deionized water rinse. The panel was then immersed in the sealing composition Example M for 2 minutes at ambient temperature with intermittent agitation. The panel was air dried at ambient conditions overnight before corrosion testing as described below.
Example 12
[0186] After panels were processed through the seal solution of Example M, the pH of the bath was adjusted by bubbling carbon dioxide gas into the bath until the pH was 9.50 (i.e., to form Example N as described above). See FIG. 2.
[0187] Next, additional aluminum 2024T3 bare substrate measuring 3''.times.5''.times.0.032'' (Priority Metals, Orange County, Calif.) was hand-wiped with methyl ethyl ketone and a disposable cloth and allowed to air dry prior to chemical cleaning. The panel was immersed in the cleaner-deoxidizer composition Example A for 3.5 minutes at ambient temperature with intermittent agitation. The panel was then immersed in two subsequent deionized water rinses for two minutes each, both at ambient temperature with intermittent agitation. After the second rinse, the panel was rinsed with a cascading deionized water rinse for 10 seconds. The panel was then immersed in the conversion composition Example B for 5 minutes at ambient temperature and without agitation. Next, the panel was rinsed by immersion in deionized water for 2 minutes at ambient temperature with intermittent agitation followed by a 10 second cascading deionized water rinse. The panel was then immersed in the sealing composition Example N for 2 minutes at ambient temperature with intermittent agitation. The panel was air dried at ambient conditions overnight before corrosion testing as described below.
Example 13
[0188] After panels were processed through the seal solution of Example N, the pH of the bath was adjusted using lithium hydroxide solution as described above (i.e., to form Example O as described above). See FIG. 2.
[0189] Next, additional aluminum 2024T3 bare substrate measuring 3''.times.5''.times.0.032'' (Priority Metals, Orange County, Calif.) was hand-wiped with methyl ethyl ketone and a disposable cloth and allowed to air dry prior to chemical cleaning. The panel was immersed in the cleaner-deoxidizer composition Example A for 3.5 minutes at ambient temperature with intermittent agitation. The panel was then immersed in two subsequent deionized water rinses for two minutes each, both at ambient temperature with intermittent agitation. After the second rinse, the panel was rinsed with a cascading deionized water rinse for 10 seconds. The panel was then immersed in the conversion composition Example B for 5 minutes at ambient temperature and without agitation. Next, the panel was rinsed by immersion rinse in deionized water for 2 minutes at ambient temperature with intermittent agitation followed by a 10 second cascading deionized water rinse. The panel was then immersed in the sealing composition Example O for 2 minutes at ambient temperature with intermittent agitation. The panel was air dried at ambient conditions overnight before corrosion testing as described below.
Corrosion Testing
[0190] Panels from Examples 1-8 were placed in a 7-day exposure in a neutral salt spray cabinet operated according to ASTM B117. Corrosion performance was evaluated by counting the number of pits visible to the naked eye on the panels following the 7-day exposure. Pits that were pre-existing to testing, on an edge, or resulting from a scratch were excluded from the counts. The corrosion performance data are reported in Table 6.
[0191] Baths from Examples 9-13 were evaluated for lithium carbonate content using an autotitration method (Metrohm 799 GPT Titrino, Software by Tiamo 2.3). % Li.sub.2CO.sub.3 and % CO.sub.3 were calculated using the following formulae:
% Li.sub.2CO.sub.3=[(Volume at EP3-Volume at EP2).times.MW Li.sub.2CO.sub.3.times.HCl conc..times.100]/(SW.times.1000) and
% CO.sub.3=[(Volume at EP3-Volume at EP2).times.MW CO.sub.3.times.HCl conc..times.100]/(SW.times.1000), [0192] where EP2 is the second endpoint and EP3 is the third endpoint. HCl concentration (N) was 0.1012.
[0193] These values were used to calculate the amount of carbonate in baths containing the sealing compositions of Examples 9-13, as reported in Table 6.
TABLE-US-00006 TABLE 6 Corrosion Results and Carbonate Measurements Pits/ Sample HCl HCl Avg Avg Corrosion Weight (mL) (mL) % % CO.sub.3 CO.sub.3 Example pH Sites (g) EP2* EP3** Li.sub.2CO.sub.3 CO.sub.3 (%) (ppm) Example 1 11.52 5 -- -- -- -- -- -- -- (Comparative) Example 2 12.69 100+ -- -- -- -- -- -- -- (Comparative) Example 3 11.42 6 -- -- -- -- -- -- -- Example 4 10.54 10 -- -- -- -- -- -- -- Example 5 9.47 18 -- -- -- -- -- -- -- Example 6 10.47 14 -- -- -- -- -- -- -- Example 7 11.48 3 -- -- -- -- -- -- -- Example 8 12.47 39 -- -- -- -- -- -- -- Example 9 11.14 -- 10.0781 2.0809 4.1378 0.153 0.124 0.125 1248 (Comparative) 10.0877 2.0735 4.1618 0.155 0.126 Example 10 12.17 -- 10.1993 4.0802 4.1572 0.006 0.005 0.005 48 (Comparative) 10.3639 4.1290 4.2158 0.006 0.005 Example 11 11.37 -- 10.1814 2.2263 4.1518 0.141 0.115 0.115 1147 10.3984 2.2655 4.2272 0.141 0.115 Example 12 9.50 -- 10.0817 0.8203 4.0719 0.241 0.196 0.196 1960 10.8563 0.8730 4.3789 0.241 0.196 Example 13 11.37 -- 10.4032 3.6583 7.0788 0.246 0.200 0.200 2002 10.3221 3.5725 6.9857 0.247 0.201
[0194] Comparative Examples 1 and 9 illustrate treatment baths containing a composition made from lithium carbonate. These Examples illustrate the pH of such a treatment bath containing a composition made from lithium carbonate, and Comparative Example 9 also demonstrated the amount of lithium carbonate and carbonate in the treatment bath. The pH of the treatment bath of Comparative Example 1 was 11.52 and there were 5 pits on the treated panel following salt spray exposure. The treatment bath of Comparative Example 9 had a pH of 11.14, and contained 0.154% lithium carbonate and 1248 ppm carbonate.
[0195] Comparative Example 2 and 10 illustrated a treatment bath containing a composition made from lithium hydroxide. Notably, the amount of lithium in Example D (used to make Comparative Example 2) was the same as the amount of lithium in Example C (used to make Comparative Example 1) (0.081 mol lithium). The pH of the treatment bath of Comparative Example 2 was 12.69 and there were more than 100 pits on the treated panel following salt spray exposure. The treatment bath of Comparative Example 10 had a pH of 12.17 and contained only 48 ppm of carbonate. While not wishing to be bound by theory, it is hypothesized that the carbonate present in the treatment bath of Comparative Example 10 is the result of the conversion of CO.sub.2 to CO.sup.3-. These data demonstrate that lithium in the absence of sufficient carbonate does not provide corrosion protection for a metal substrate treated with the treatment bath.
[0196] Example 3 demonstrated that by bubbling CO.sub.2 into the treatment bath, pH can be lowered to a range comparable to Comparative Example 1, while reducing the number of pits on the treated panel to 6. As demonstrated by Example 11, CO.sub.2 also can be used to form lithium carbonate in a bath that had only a trace amount of lithium carbonate prior to addition of CO.sub.2.
[0197] Examples 4, 5, and 12 also demonstrated that bubbling additional quantities of CO.sub.2 into the treatment bath lowers pH, but these Examples show a trend of increasingly more corrosion pits on the treated panels after salt spray exposure as pH of the treatment bath is lowered (i.e., the panels treated in Examples 4 and 5 had 10 and 18 pits at pH 10.54 and 9.47, respectively) compared to Comparative Example 1, which had a pH of 11.52 and 5 pits on the treated panel and compared to Example 3, which had a pH of 11.42 and 6 pits on the treated panel. As demonstrated by Example 12, there was 1960 ppm of carbonate in the treatment bath. These data demonstrate that in a bath that contains lithium carbonate, pH is critical to corrosion performance.
[0198] Examples 6, 7, and 13 demonstrated that the addition of LiOH to increase pH to 10.47 and 11.48, respectively, resulted in improved corrosion performance, as panels treated in Example 6 had 14 pits, while those treated in Example 7 had 3 pits.
[0199] Example 8 demonstrated that raising the pH to 12.5 impaired corrosion performance even though the carbonate level was sufficient, with the treated panel having 39 corrosion sites, an improvement over Comparative Example 2, which did not include any lithium carbonate and which had more than 100 pits and no lithium carbonate added to the bath.
[0200] The Examples demonstrate the interaction of pH, lithium concentration, and carbonate concentration with respect to corrosion resistance.
* * * * *
D00000
D00001
D00002
D00003
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.