Compositions And Methods For Treating Hcv
Collins; Christine A. ; et al.
U.S. patent application number 16/272599 was filed with the patent office on 2019-06-06 for compositions and methods for treating hcv. The applicant listed for this patent is AbbVie Inc.. Invention is credited to Daniel E. Cohen, Christine A. Collins, Gennadiy Koev, Preethi Krishnan, Tami J. Pilot-Matias.
| Application Number | 20190167684 16/272599 |
| Document ID | / |
| Family ID | 65242294 |
| Filed Date | 2019-06-06 |

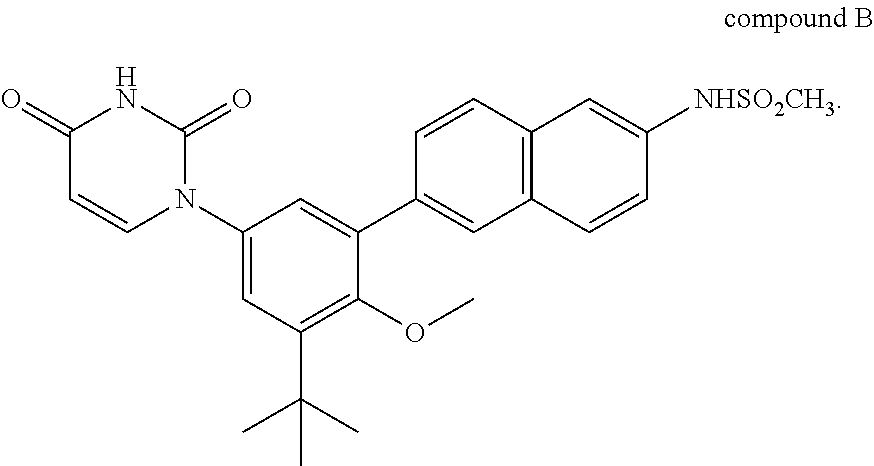
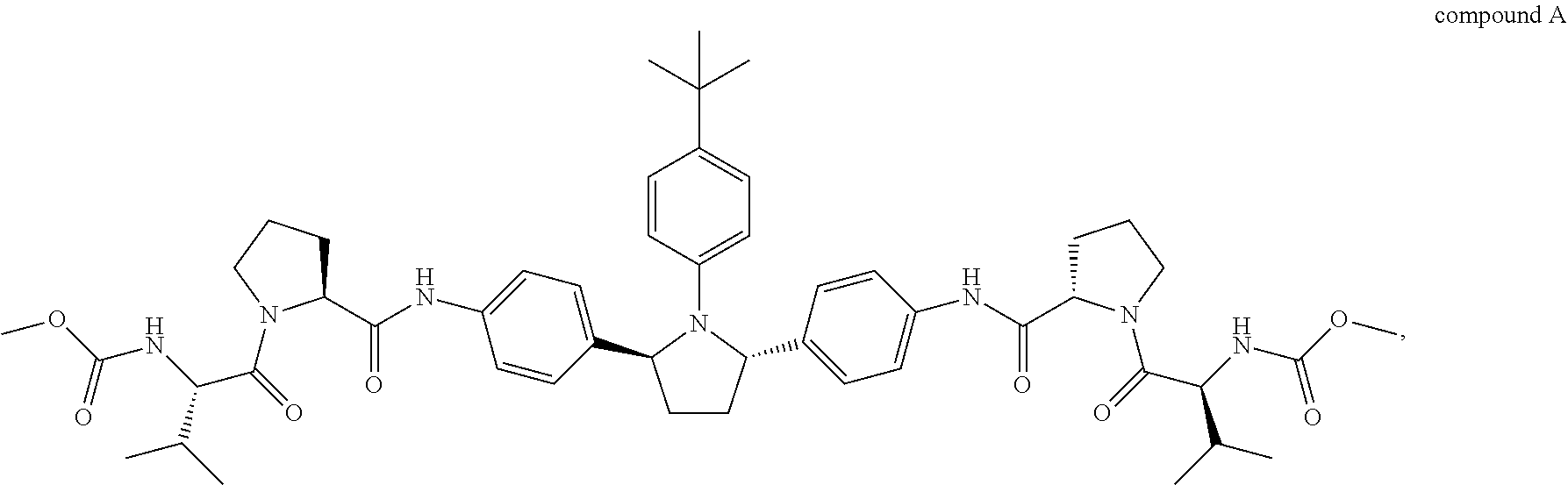
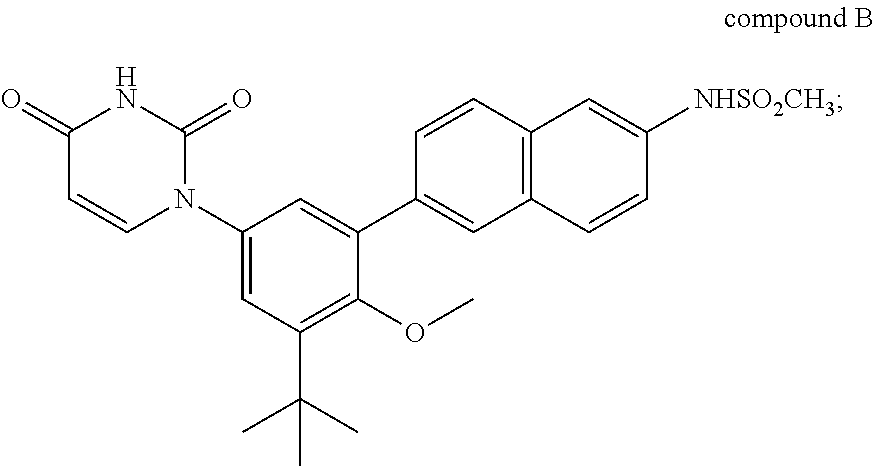

| United States Patent Application | 20190167684 |
| Kind Code | A1 |
| Collins; Christine A. ; et al. | June 6, 2019 |
COMPOSITIONS AND METHODS FOR TREATING HCV
Abstract
This disclosure is directed to pharmaceutical compositions that comprise two or more therapeutic agents that, inter alia, are useful for inhibiting hepatitis C virus (HCV) and methods for inhibiting HCV by co-administering two or more anti-HCV therapeutic agents.
| Inventors: | Collins; Christine A.; (Skokie, IL) ; Cohen; Daniel E.; (Highland Park, IL) ; Koev; Gennadiy; (Libertyville, IL) ; Krishnan; Preethi; (Gurnee, IL) ; Pilot-Matias; Tami J.; (Green Oaks, IL) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 65242294 | ||||||||||
| Appl. No.: | 16/272599 | ||||||||||
| Filed: | February 11, 2019 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 15398390 | Jan 4, 2017 | 10201541 | ||
| 16272599 | ||||
| 14247975 | Apr 8, 2014 | 10201584 | ||
| 15398390 | ||||
| 13474411 | May 17, 2012 | |||
| 14247975 | ||||
| 61486842 | May 17, 2011 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61K 38/05 20130101; A61K 31/513 20130101; A61K 31/4025 20130101; A61P 31/12 20180101; A61K 31/427 20130101; A01N 37/18 20130101; A61K 45/06 20130101; A61K 38/00 20130101; A61K 38/21 20130101; A61K 31/427 20130101; A61K 2300/00 20130101; A61K 31/513 20130101; A61K 2300/00 20130101; A61K 31/4025 20130101; A61K 2300/00 20130101; A61K 38/21 20130101; A61K 2300/00 20130101 |
| International Class: | A61K 31/513 20060101 A61K031/513; A01N 37/18 20060101 A01N037/18; A61K 45/06 20060101 A61K045/06; A61K 31/4025 20060101 A61K031/4025; A61K 38/05 20060101 A61K038/05; A61K 38/00 20060101 A61K038/00 |
Claims
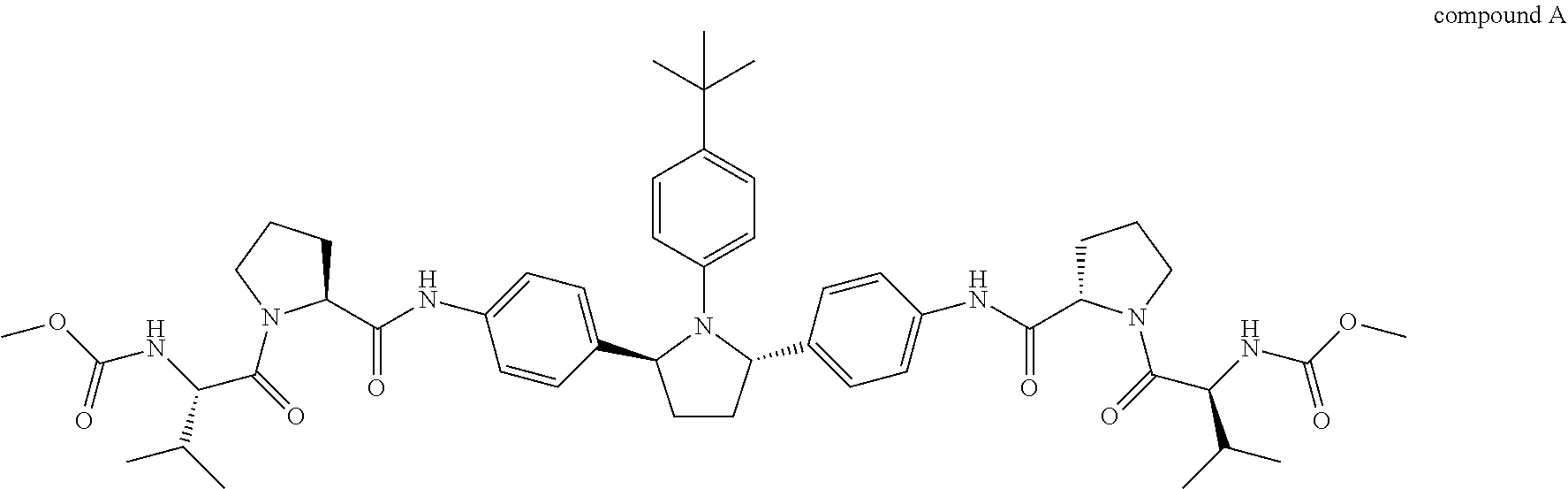
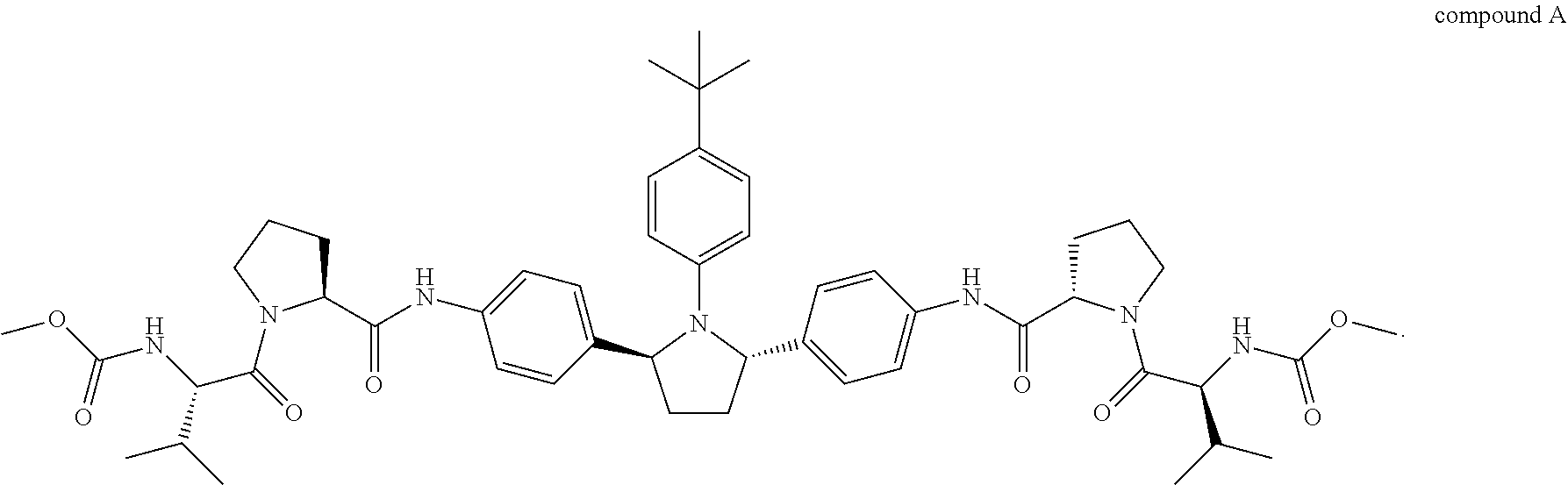
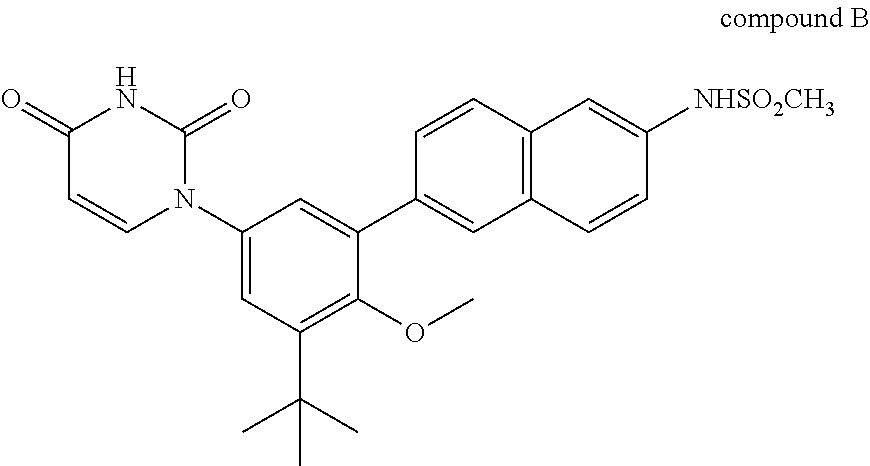
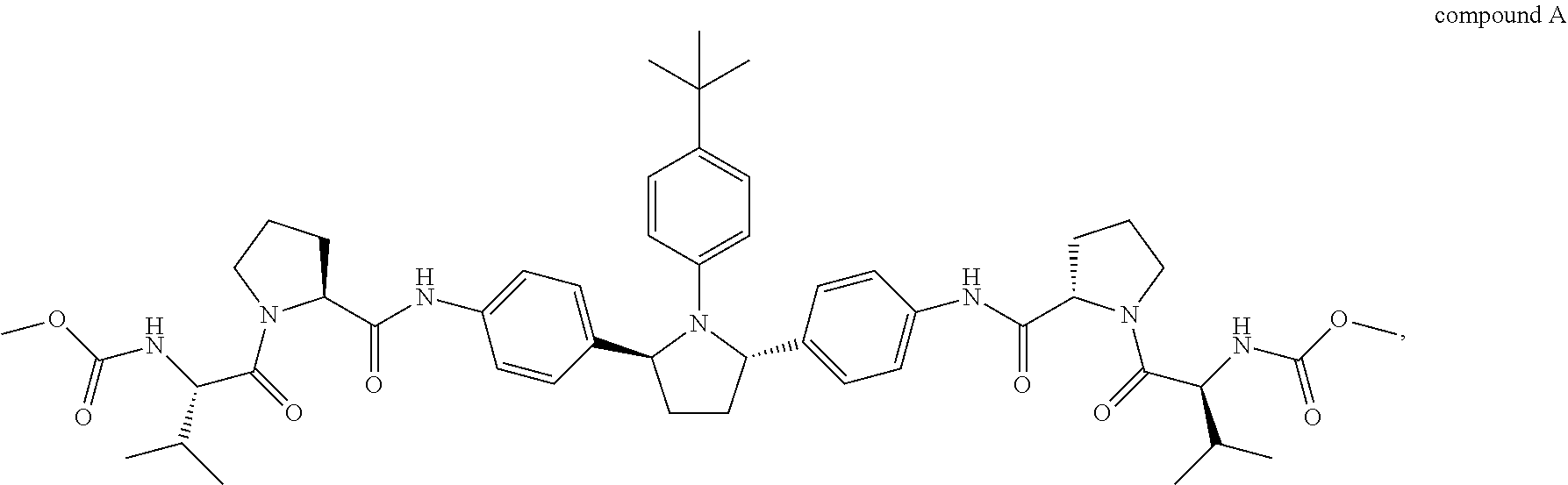
1. A combination or pharmaceutical composition comprising an amount of therapeutic agent A and an amount of therapeutic agent B, wherein therapeutic agent A is compound A or a salt thereof: ##STR00005## and wherein therapeutic agent B is compound B or a salt thereof: ##STR00006##
2. The combination or composition of claim 1, wherein the amount of therapeutic agent A is about 25 mg.
3. The combination or composition of claim 1, wherein therapeutic agent B is a sodium salt of compound B.
4. The combination or composition of claim 1, wherein the amount of therapeutic agent B is from about 400 to about 800 mg.
5. The combination or composition of claim 1, wherein the amount of therapeutic agent A and the amount of therapeutic agent B together constitute a therapeutically effective amount.
6. The combination or composition of claim 1, wherein the composition further comprises one or more additional therapeutic agents.
7. The combination or composition of claim 6, wherein the additional therapeutic agent is independently selected from the group consisting of ribavirin, taribavirin, and HCV inhibitor.
8. The combination or composition of claim 6, wherein the one or more additional therapeutic agents include an HCV protease inhibitor.
9. The combination or composition of claim 8, wherein the one or more additional therapeutic agents include a cytochrome P-450 inhibitor.
10. The combination or composition of claim 9, wherein the cytochrome P-450 inhibitor is ritonavir.
11. The combination or composition of claim 6, wherein the amount of therapeutic agent A, the amount of therapeutic agent B ant the amount of the additional therapeutic agent together constitute a therapeutically effective amount.
12. A pharmaceutical composition comprising an amount of therapeutic agent A and an amount of therapeutic agent B, wherein therapeutic agent A is compound A or a salt thereof: ##STR00007## wherein therapeutic agent B is compound B or a salt thereof: ##STR00008## and wherein the ratio of therapeutic agent A to therapeutic agent B is about 1:20 to about 1:2000.
13. The pharmaceutical composition of claim 12, wherein the amount of therapeutic agent B is about 200 mg.
14. The pharmaceutical composition of claim 12, wherein the amount of therapeutic agent A is from about 5 mg to about 200 mg.
15. The pharmaceutical composition of claim 12, wherein the composition further comprises one or more additional therapeutic agents
16. The pharmaceutical composition of claim 15, wherein the one or more additional therapeutic agents include an HCV protease inhibitor.
17. The pharmaceutical composition of claim 16, wherein the one or more additional therapeutic agents include a cytochrome P-450 inhibitor.
18. The pharmaceutical composition of claim 12, further comprising an HCV protease inhibitor and a cytochrome P-450 inhibitor.
19. The pharmaceutical composition of claim 18, wherein the cytochrome P-450 inhibitor is ritonavir.
20. The pharmaceutical composition of claim 12, wherein therapeutic agent B is a sodium salt of compound B.
Description
CROSS-REFERENCE TO RELATED PATENT APPLICATION
[0001] This patent application is a continuation of U.S. patent application Ser. No. 13/474,411 filed May 17, 2012, which claims priority to U.S. Provisional Patent Application No. 61/486,842, filed May 17, 2011. The entire text of these applications is incorporated by reference into this patent application.
TECHNICAL FIELD
[0002] This disclosure is directed to: (a) pharmaceutical compositions that comprise two or more therapeutic agents that, inter alia, are useful for inhibiting hepatitis C virus (HCV); (b) methods for preparing such compositions; and (c) methods of use of such compositions; as well as (d) methods for inhibiting HCV by co-administering two or more anti-HCV therapeutic agents.
BACKGROUND
[0003] Hepatitis C is a blood-borne, infectious, viral disease that is caused by an RNA virus belonging to the Hepacivirus genus in the Flaviviridae family called HCV. The enveloped HCV virion contains a positive stranded RNA genome encoding all known virus-specific proteins in a single, uninterrupted, open reading frame. The open reading frame comprises approximately 9500 nucleotides and encodes a single large polyprotein of about 3000 amino acids. The polyprotein comprises a core protein, envelope proteins E1 and E2, a membrane bound protein p7, and the non-structural proteins NS2, NS3, NS4A, NS4B, NS5A and NS5B.
[0004] At least six different HCV genotypes (with several subtypes within each genotype) are known to date. In North America, HCV genotype 1a predominates, followed by HCV genotypes 1b, 2a, 2b, and 3a. In the United States, HCV genotypes 1, 2, and 3 are the most common, with about 80% of the hepatitis C patients having HCV genotype 1. In Europe, HCV genotype 1b is predominant, followed by HCV genotypes 2a, 2b, 2c, and 3a. HCV genotypes 4 and 5 are found almost exclusively in Africa. As discussed below, the patient's HCV genotype is clinically important in determining the patient's potential response to therapy and the required duration of such therapy.
[0005] An HCV infection can cause liver inflammation (hepatitis) that is often asymptomatic, but ensuring chronic hepatitis can result in cirrhosis of the liver (fibrotic scarring of the liver), liver cancer (hepatocellular carcinoma), and/or liver failure. The World Health Organization estimates that about 170 million persons worldwide are chronically infected with HCV, and from about three to about four million persons are newly infected globally each year. According to the Centers for Disease Control and Prevention, about four million people in the United States are infected with HCV. Co-infection with the human immunodeficiency virus (HIV) is common, and rates of HCV infection among HIV positive populations are higher.
[0006] There is a small chance of clearing the virus spontaneously, but the majority of patients with chronic hepatitis C will not clear the virus without treatment. Indications for treatment typically include proven HCV infection and persistent abnormal liver function tests. These are two treatment regimens that are primarily used to treat hepatitis C: monotherapy (using an interferon agent--either a "conventional" or longer-acting pegylated interferon) and combination therapy (using an interferon agent and ribavirin). Interferon, which is injected into the bloodstream, works by bolstering the immune response to HCV; and ribavirin, which is taken orally, is believed to work by preventing HCV replication. Taken alone, ribavirin does not effectively suppress HCV levels, but an interferon/ribavirin combination is more effective than interferon alone. Typically, hepatitis C is treated with a combination of pegylated interferon alpha and ribavirin for a period of 24 or 48 weeks, depending on the HCV genotype.
[0007] The goal of treatment is sustained viral response--meaning that HCV is not measurable in the blood after therapy is completed. Following treatment with a combination of pegylated interferon alpha and ribavirin, sustained cure rates (sustained viral response) of about 75% occur in people with HCV genotypes 2 and 3 in 24 weeks of treatment, about 50% in those with HCV genotype 1 with 48 weeks of treatment, and about 65% in those with HCV genotype 4 in 48 weeks of treatment.
[0008] Thus, there continues to be a need for new compositions and methods of treatment to prevent the progression of liver damage from hepatitis C. This disclosure provides compositions and methods of treatment that generally address such a need.
SUMMARY
[0009] This disclosure is directed, in part, to the co-administration of an amount of therapeutic agent A with an amount of therapeutic agent B. Therapeutic agent A is compound A or a salt thereof:
##STR00001##
Therapeutic agent B is compound B or a salt thereof:
##STR00002##
[0010] This disclosure is also directed, in part, to combinations or pharmaceutical compositions comprising therapeutic agent A and therapeutic agent B. The combinations or compositions may comprise one or more additional therapeutic agents.
[0011] This disclosure is also directed, in part, to methods for treating hepatitis C in a subject in need of such treatment. The methods comprise administering to the subject an amount of therapeutic agent A and an amount of therapeutic agent B. The methods may optionally comprise administering to the subject an amount of one or more additional therapeutic agents.
[0012] This disclosure is also directed, in part, to the use of therapeutic agent A and therapeutic agent B, to prepare a medicament. In embodiments, the medicament is useful for treating hepatitis C.
[0013] This disclosure is also directed, in part, to methods of using therapeutic agent A and therapeutic agent B, for example, to inhibit replication of a ribonucleic acid (RNA) virus (including HCV) or to treat a disease treatable by inhibiting HCV RNA polymerase and/or the NS5A protein of HCV.
[0014] Further benefits of the disclosed embodiments will be apparent to one skilled in the art from reading this disclosure.
BRIEF DESCRIPTION OF THE DRAWINGS
[0015] FIG. 1 is a three-dimensional surface plot illustrating the statistically significant anti-HCV effect for the combination of compound A and compound B in the HCV Genotype 1b (Con1) replicon. FIG. 1 details the mean differences between the observed anti-HCV effect and the calculated additivity of that effect in percent inhibition at various concentrations of compound A and compound B according to the Prichard and Shipman model. The concentrations for each of compound A and compound B are expressed in a log.sub.2 scale.
[0016] FIG. 2 is a two-dimensional contour plot illustrating the statistically significant synergistic, additive or antagonistic anti-HCV effects at various concentrations of the combination of compound A and compound B in the HCV Genotype 1b (Con1) replicon using the Prichard and Shipman model as a reference.
[0017] FIG. 3 is a bar graph illustrating the percentage of replicon colonies surviving exposure to various concentrations of therapeutic agent A, therapeutic agent B and therapeutic agent C in a replicon colony count assay.
DETAILED DESCRIPTION
[0018] This detailed description is intended only to acquaint others skilled in the art with the disclosed embodiments, their principles, and their practical applications so that others skilled in the art may adapt and apply the disclosed embodiments in their numerous forms, as they may be best suited to the requirements of a particular use. This description and its specific examples are intended for purposes of illustration only. This disclosure, therefore, is not limited to the embodiments described in this patent application, and may be variously modified.
[0019] The disclosure is directed, in part, to the co-administration of an amount of therapeutic agent A with an amount of therapeutic agent B. Therapeutic agent A is compound A or a salt thereof.
##STR00003##
Compound A is also known as dimethyl (2S2'S)-2,2'-(4,4'-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bi- s(4,1-phenylene))bis(azanedily)bis(oxomethylene)bis(pyrrolidine-2,1-diyl))- bis(3-methyl-1-oxobutane-2,1-diyl)dicarbamate. Compound A can be prepared as described in, for example, U.S. Publication No. 2010/0317568, which is incorporated herein by reference.
[0020] Therapeutic agent B is compound B or a salt thereof.
##STR00004##
Compound B is also known as N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl-2-methoxyph- enyl)naphthalen-2-yl)methanesulfonamide. As described in, for example, International Publication No. WO2009/039127, therapeutic agent B includes various salts of compound B, such as sodium salts, potassium salts, and choline salts. Therapeutic agent B also includes crystalline forms of compound B and its salts such as solvate, hydrate, and solvent-free crystalline forms of compound B and its salts. Compositions comprising compound B can be prepared as described in, for example, International Publication No. WO2009/039127 which is incorporated herein by reference.
[0021] The total daily dose of the disclosed compounds of their salts (administered in single or divided doses) may typically be from about 0.001 mg/kg to about 200 mg/kg, or from about 0.001 mg/kg to about 30 mg/kg, or from about 0.01 mg/kg to about 10 mg/kg (i.e., mg of the compound or salt per kg body weight).
[0022] Therapeutic agent A may be administered, for example and without limitation, as a free acid or salt. Therapeutic agent A may be administered in any suitable amount such as, for example, in doses of from about 0.1 mg/kg to about 200 mg/kg body weight, or from about 0.25 mg/kg to about 100 mg/kg, or from about 0.3 mg/kg to about 3.0 mg/kg. As non-limiting examples, therapeutic agent A may be administered in a total daily dose amount of from about 5 mg to about 300 mg, or from about 25 mg to about 200 mg, or from about 25 mg to about 50 mg. In embodiments, the total daily dosage amount for therapeutic agent A is about 25 mg. In embodiments, the total daily dosage amount for therapeutic agent A is about 50 mg.
[0023] Therapeutic agent B may be administered as a free acid, salt or particular crystalline form of compound B. In embodiments, therapeutic agent B is administered as a sodium salt of compound B. Therapeutic agent B may be administered in any suitable amount such as, for example, in doses of from about 5 mg/kg to about 30 mg/kg. As non-limiting examples, therapeutic agent B may be administered in a total daily dose amount of from about 300 mg to about 1800 mg, or from about 400 mg to about 1600 mg, or from about 600 mg to about 1800 mg, or from about 800 mg to about 1600 mg. In embodiments, the total daily dosage amount for therapeutic agent B is about 300 mg. In embodiments, the total daily dosage amount for therapeutic agent B is about 400 mg. In embodiments, the total daily dosage amount for therapeutic agent B is about 600 mg. In embodiments, the total daily dosage amount for therapeutic agent B is about 800 mg. In embodiments, the total daily dosage amount for therapeutic agent B is about 1200 mg. In embodiments, the total daily dosage amount for therapeutic agent B is about 1600 mg.
[0024] Therapeutic agent A and therapeutic agent B may also be co-administered with interferon. Interferon may include any suitable form of interferon such as interferon alpha, interferon alpha 2a, interferon alpha 2b such as LOCTERON.RTM., interferon omega, interferon lambda, and albinterferon, such as ZALBIN.RTM. and JOULFERON.RTM. or albinterferon as disclosed in International Publication No. WO2007/021494A2. In embodiments, the interferon is pegylated. Pegylated interferon may include pegylated interferon alpha 2a, such as PEGASYS.RTM., or pegylated interferon alpha 2b, such as PEGINTRON.RTM.; pegylated interferon omega, such as Biomed-510, or pegylated interferon omega as disclosed in U.S. Publication No. 2006/263433; and pegylated interferon lambda, such as PEG-rIL-29, or pegylated interferon lambda as disclosed in International Publication No. WO2007/041713A1.
[0025] Interferon may be administered in accordance with interferon administration well known in the art. For example, interferon may be administered in a total weekly dose amount of from about 0.1 mcg/kg to about 2.5 mcg/kg. In embodiments, alpha-2b pegylated interferon is administered in a total weekly dose of about 0.5 mcg/kg to about 1.5 mcg/kg. Interferon may be administered in a total weekly dose amount of 50 mcg to about 250 mcg. In embodiments, alpha-2a pegylated interferon is administered in a total weekly dose of from about 90 mcg to about 180 mcg.
[0026] LOCTERON.RTM. is an example of an interferon that can be co-administered with the disclosed compositions, compounds and their salts. LOCTERON.RTM. is a controlled-release formulation of interferon alpha-2b interferon that allows the interferon to be administered every two weeks rather than every week. LOCTERON.RTM. may be administered in accordance with LOCTERON.RTM. administration well known in the art. For example, the interferon may be administered at least once every one to two weeks at a dose of from about 250 mcg to about 750 mcg or from about 320 mcg to about 640 mcg as a single or as multiple subcutaneous injections at the same or different doses in each injection. In embodiments, the peginterferon is administered subcutaneously at a dose of 480 mcg every two weeks.
[0027] ZALBIN.RTM. and JOULFERON.RTM. (formerly known as Albuferon.RTM. and ABF-656) are other examples of an interferon that can be co-administered with the disclosed compositions, compounds and their salts. ZALBIN.RTM. and JOULFERON.RTM. are an albumin interferon alpha-2b which is a recombinant fusion protein composed of recombinant human albumin genetically fused at its C-terminus to the N-terminus of recombinant human interferon alfa-2b. ZALBIN.RTM. and JOULFERON.RTM. may be administered in accordance with ZALBIN.RTM. and JOULFERON.RTM. administration well known in the art. For example, the albinterferon may be administered at least once every one to two weeks at a dose of from about 1 to about 2000 mcg as a single or as multiple subcutaneous injections at the same or different doses in each injection. In embodiments, the albinterferon is administered subcutaneously at a dose of from about 7 to about 900 mcg as single or double (14 days apart) injections.
[0028] PEGASYS.RTM. is a further example of an interferon that can be co-administered with the disclosed compositions, compounds and their salts. PEGASYS.RTM. is a pegylated interferon alpha-2a which is a covalent conjugate of recombinant alfa-2a interferon with a single branched bis-monomethoxy polyethylene glycol (PEG) chain. PEGASYS.RTM. may be administered in accordance with PEGASYS.RTM. administration well known in the art. For example, the peginterferon may be administered at least once every one to two weeks at a dose of from about 100 mcg to about 400 mcg as a single or as multiple subcutaneous injections at the same or different doses in each injection. In embodiments, the peginterferon is administered subcutaneously at a dose of about 180 mcg as a single weekly injection.
[0029] PEGINTRON.RTM. is an additional example of an interferon that can be co-administered with the disclosed compositions, compounds and their salts. PEGINTRON.RTM. is a pegylated interferon alpha-2b which is a covalent conjugate of recombinant alpha-2b interferon with monomethoxy polyethylene glycol (PEG). PEGINTRON.RTM. may be administered in accordance with PEGINTRON.RTM. administration well known in the art. For example, the peginterferon may be administered at least once every one to two weeks at a dose of from about 1 mcg/kg to about 3 mcg/kg or from about 40 mcg/m.sup.2 to about 80 mcg/m.sup.2. The peginterferon may be administered at least once every one to two weeks at a dose of from about 25 mcg to about 200 mcg or from about 50 mcg to about 150 mcg as a single or as multiple subcutaneous injections at the same or different doses in each injection. In embodiments, the peginterferon is administered subcutaneously at a dose of about 1.5 mcg/kg as single weekly injection. In embodiments, the peginterferon is administered subcutaneously at a dose of about 60 mcg/m.sup.2as single weekly injection.
[0030] Therapeutic agent A and therapeutic agent B may also be co-administered with ribavirin, or a pro-drug thereof, in the same or separate pharmaceutical compositions. Ribavirin may include any suitable form or formulation of ribavirin. Exemplary formulations of ribavirin include COPEGUS.RTM., REBETOL.RTM. and RIBASPHERE.RTM.. An exemplary pro-drug of ribavirin is taribavirin having the chemical name of 1-.beta.-D-ribofuranosyl-1,2,4-triazole-3-carboxamidine.
[0031] Ribavirin and taribavirin may be administered in accordance with ribavirin and taribavirin administration well known in the art. For example, ribavirin or taribavirin may be administered in a total daily dose of from about 5 mg to about 1500 mg. In embodiments, COPEGUS.RTM. or REBETOL.RTM. is administered in a daily dosage amount of from about 500 mg to about 1500 mg in one dose or in divided doses. In embodiments, COPEGUS.RTM. or REBETOL.RTM. is administered in a daily dosage amount of about 800 mg. In embodiments, REBETOL.RTM. is administered in a daily dosage amount of about 100 mg. In embodiments, COPEGUS.RTM. or REBETOL.RTM. is administered in a daily dosage amount of about 1200 mg. In embodiments, REBETOL.RTM. is administered in a daily dosage amount of about 1400 mg.
[0032] Ribavirin may be co-administered with the interferon, together with therapeutic agent A and therapeutic agent B. In embodiments, ribavirin is administered with pegylated interferon alpha 2a, such as PEGASYS.RTM., together with therapeutic agent A and therapeutic agent B. For example, in embodiments, a daily dose of COPEGUS.RTM. of 800 mg to 1200 mg is administered in combination with a weekly dose of PEGASYS.RTM. of 180 mcg, together with daily administration of therapeutic agent A and therapeutic agent B. In embodiments, ribavirin is administered with pegylated interferon alpha 2b, such as PEGINTRON.RTM., together with therapeutic agent A and therapeutic agent B. For example, in embodiments, a daily dose of REBETOL.RTM. of 800 mg to 1400 mg is administered in combination with a weekly dose of PEGINTRON.RTM. of 1.5 mcg/kg, together with daily administration of therapeutic agent A and therapeutic agent B.
[0033] Therapeutic agent A and therapeutic agent B may be co-administered with interferon and ribavirin or a pro-drug thereof.
[0034] Therapeutic agent A and therapeutic agent B may be co-administered with an HIV inhibitor including an HIV protease inhibitor, with or without a cytochrome P-450 inhibitor (e.g., ritonavir), in the same or separate pharmaceutical compositions.
[0035] The cytochrome P-450 inhibitor may be administered in any suitable amount such as, for example, in dose of from about 0.3 mg/kg to about 2 mg/kg or from about 0.6 mg/kg to about 1.5 mg/kg. As non-limiting examples, the cytochrome P-450 inhibitor may be administered in a total daily dose amount of from about 25 mg to about 300 mg, or from about 50 mg to about 250 mg, or from about 100 mg to about 200 mg. In embodiments, the cytochrome P-450 inhibitor is administered in a total daily dose amount of about 25 mg. In embodiments, the cytochrome P-450 inhibitor is administered in a total daily dose amount of about 50 mg. In embodiments, the cytochrome P-450 inhibitor is administered in a total daily dose amount of about 75 mg. In embodiments, the cytochrome P-450 inhibitor is administered in a total daily dose amount of about 100 mg. In embodiments, the cytochrome P-450 inhibitor is administered in a total daily dose amount of about 125 mg.
[0036] Therapeutic agent A and therapeutic agent B may be co-administered with an HCV protease inhibitor in the same or separate pharmaceutical compositions. HCV protease inhibitors may include, for example, ACH-1625 (Achillion), ACH-2684 (Achillion), AVL-181 (Avila Therapeutics), AVL-192 (Avila Therapeutics), BI 201335 (Boehringer Ingelheim), BMS-791325 (Bristol-Myers Squibb), GS 9256 (Gilead), IDX320 (Idenix), danoprevir or ITMN-191 or R7227 (RO5190591) (Intermune/Roche), TMC435 (Medivir/Tibotec/JnJ), Boceprevir or SCH503034 (Merck), Vaniprevir or MK-7009 (Merck), PHX1766 (Phenomix), Telaprevir or VX-950 (Vertex), VX-985 (Vertex) and VX-500 (Vertex).
[0037] In embodiments, therapeutic agent A and therapeutic agent B are co-administered with interferon and an HCV protease inhibitor. In embodiments, therapeutic agent A and therapeutic agent B are co-administered with ribavirin and an HCV protease inhibitor. In embodiments, therapeutic agent A and therapeutic agent B are co-administered with interferon, ribavirin and an HCV protease inhibitor. In embodiments, therapeutic agent A and therapeutic agent B are co-administered with an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir). In embodiments, therapeutic agent A and therapeutic agent B are co-administered with ribavirin and an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir). In embodiments, therapeutic agent A and therapeutic agent B are co-administered with interferon, ribavirin, and an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir).
[0038] Factors affecting the dosage regimen include the route of administration; the type, age, weight, sex, diet, and condition of the patient; the severity of the pathological condition; pharmacological considerations, such as the activity, efficacy, pharmacokinetic, and toxicology profiles of the particular compound or salt used; whether a drug delivery system is utilized; and the specific drug combination. Thus, the dosage regimen actually employed can vary widely and, therefore, can deviate from the disclosed dosage regimen set forth above.
[0039] In embodiments, the combination or pharmaceutical composition comprises an amount of therapeutic agent A and an amount of therapeutic agent B. The amount of therapeutic agent A and therapeutic agent B may be any suitable amount that provides the desired total periodic dosing amount such as the total daily dosing amount. For example, the amount of therapeutic agent A in the combination or pharmaceutical composition may be any suitable amount such as from about 5 mg to about 200 mg or from about 10 to about 100 mg or from about 25 mg to about 50 mg. In embodiments, the total daily dosage amount for therapeutic agent A is about 25 mg. In embodiments, the total daily dosage amount for therapeutic agent A is about 50 mg.
[0040] The amount of therapeutic agent B in the combination or pharmaceutical composition may be from about 100 mg to about 1800 mg or from about 300 to about 1600 mg or from about 400 mg to about 1200 mg. In embodiments, the amount of therapeutic agent B in a combination or pharmaceutical composition is about 100 mg. In embodiments, the amount of therapeutic agent B in a combination or pharmaceutical composition is about 200 mg. In embodiments, the amount of therapeutic agent B in a combination or pharmaceutical composition is about 300 mg. In embodiments, the amount of therapeutic agent B in a combination or pharmaceutical composition is about 400 mg. In embodiments, the amount of therapeutic agent B in a combination or pharmaceutical composition is about 600 mg. In embodiments, the amount of therapeutic agent B in a combination or pharmaceutical composition is about 800 mg. In embodiments, the amount of therapeutic agent B in a combination or pharmaceutical composition is about 1000 mg. In embodiments, the amount of therapeutic agent B in a combination or pharmaceutical composition is about 1200 mg. In embodiments, the amount of therapeutic agent B in a combination or pharmaceutical composition is about 1600 mg.
[0041] The combinations or pharmaceutical compositions may also comprise other therapeutic agents and combinations thereof, used to treat hepatitis C, such as any suitable amount of ribavirin and pro-drugs thereof, HCV inhibitors such as, for example, HCV helicase inhibitors, HCV polymerase inhibitors, HCV protease inhibitors, HCV NS5A inhibitors, CD81 inhibitors, cyclophilin inhibitors, or internal ribosome entry site (IRES) inhibitors; and HIV inhibitors.
[0042] In embodiments, the combination or pharmaceutical composition comprises an amount of therapeutic agent A and an amount of therapeutic agent B. In embodiments, the combination or pharmaceutical composition comprises an amount of therapeutic agent A, an amount of therapeutic agent B and an amount of HCV protease inhibitor. In embodiments, the combination or pharmaceutical composition comprises an amount of therapeutic agent A, an amount of therapeutic agent B and ribavirin. In embodiments, the combination or pharmaceutical composition comprises an amount of therapeutic agent A, an amount of therapeutic agent B, and an amount of HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir). In embodiments, the combination or pharmaceutical composition comprises an amount of therapeutic agent A, an amount of therapeutic agent B, an amount of HCV protease inhibitor (with our without a cytochrome P-450 inhibitor such as ritonavir), and ribavirin. In embodiments, interferon is co-administered with the above-mentioned combination or pharmaceutical composition.
[0043] Dosage unit compositions may contain such amounts or submultiples thereof to make up the total daily dose. The administration of the therapeutic agent may be repeated a plurality of times. Multiple doses per day may be used to achieve the total daily dose, if desired. For example, a combination or pharmaceutical composition comprising a dose of about 25 mg or 50 mg of therapeutic agent A may be administered at least twice per day to achieve a total daily dosage amount of about 50 mg or 100 mg of therapeutic agent A, respectively. A dose of about 400 mg or 800 mg of therapeutic agent B may be administered at least twice per day to achieve a total daily dosage amount of about 800 mg or 1600 mg of therapeutic agent B, respectively.
[0044] The disclosed compositions may comprise one or more conventional pharmaceutically acceptable carriers, adjuvants, and/or vehicles (together referred to as "excipients"). The disclosed compositions may be prepared in a form for oral administration such as in a solid dosage form. Such solid dosage forms include, for example, capsules, tablets, pills, powders, and granules. In such solid dosage forms, the compounds or salts may be combined with one or more excipients. If administered per os, the compounds or salts may be mixed with, for example, lactose, sucrose, starch powder, cellulose esters of alkanoic acids, cellulose alkyl esters, talc, stearic acid, magnesium stearate, magnesium oxide, sodium and calcium salts of phosphoric and sulfuric acids, gelatin, acacia gum, sodium alginate, polyvinylpyrrolidone, and/or polyvinyl alcohol, and then tableted or encapsulated for convenient administration. Such capsules or tablets may contain a controlled-release formulation, as may be provided in, for example, a dispersion of the compound or its salt in hydroxypropylmethyl cellulose. In the case of capsules, tablets, and pills, the dosage forms also may comprise buffering agents, such as sodium citrate, or magnesium or calcium carbonate or bicarbonate. In addition, tablets and pills may be prepared with enteric coatings or other sustained/delayed/controlled release excipients known in the art. In embodiments, therapeutic agent A may be formulated as described in U.S. Provisional Application No. 61/353,553, filed Jun. 10, 2010, which is incorporated herein by reference.
[0045] One or more of interferon, an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor, such as ritonavir), and ribavirin may be co-administered with therapeutic agent A and therapeutic agent B.
[0046] The disclosed combination(s)/composition(s) may be administered at any suitable frequency such as at least three times daily (e.g., every 8 hours in a 24-hour period), at least two times daily (e.g., every 12 hours in a 24-hour period), at least once daily (e.g., once in a 24-hour period), or at least once weekly (e.g., once in a 7-day period).
[0047] This disclosure is also directed, in part, to methods of using the disclosed combination(s)/compositions(s). The disclosed combination(s)/composition(s) may be used in a method for inhibiting replication of an RNA virus. In embodiments, the method comprises exposing the virus to a disclosed combination(s)/composition(s) and, optionally one or more additional therapeutic agents. The disclosed combination(s)/composition(s) may be administered with one or more of an HCV protease inhibitor (with or without a cytochrome-P-450 inhibitor such as ritonavir), interferon and ribavirin in the same or separate pharmaceutical compositions to inhibit replication of an RNA virus. In embodiments, replication of the RNA virus is inhibited in vitro. In embodiments, replication of the RNA virus is inhibited in vivo. In embodiments, the RNA virus whose replication is being inhibited is a single-stranded, positive sense RNA virus. In embodiments, the RNA virus whose replication is being inhibited is a virus from the Flaviviridae family. In embodiments, the RNA virus whose replication is being inhibited is HCV.
[0048] The disclosed combination(s)/composition(s) may be used in a method for inhibiting HCV RNA polymerase. In embodiments, the method comprises exposing the polymerase to a disclosed combination(s)/composition(s) and, optionally one or more additional therapeutic agents. The disclosed combination(s)/composition(s) may be administered with one or more of an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir), interferon and ribavirin in the same or separate pharmaceutical compositions to inhibit HCV RNA polymerase. In embodiments, HCV RNA polymerase activity is inhibited in vitro. In embodiments, HCV RNA polymerase activity is inhibited in vivo.
[0049] The disclosed combination(s)/composition(s) may be used in a method for inhibiting the HCV non-structural protein 5A (NS5A protein). In embodiments, the method comprises exposing the polymerase to a disclosed combination(s)/composition(s) and, optionally one or more additional therapeutic agents. The disclosed combination(s)/composition(s) may be administered with one or more of an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir), interferon and ribavirin in the same or separate pharmaceutical compositions to inhibit the HCV NS5A protein. In embodiments, the HCV NS5A protein is inhibited in vitro. In embodiments, the HCV NS5A protein is inhibited in vivo.
[0050] The term "inhibiting" means reducing the level of RNA virus replication/HCV polymerase activity either in vitro or in vivo. for example, if a disclosed combination(s)/composition(s) reduces the level of RNA virus replication by at least about 10% compared to the level of RNA virus replication before the virus was exposed to the combination(s)/composition(s), then the combination(s)/composition(s) inhibits RNA virus replication. In some embodiments, the disclosed combination(s)/composition(s) can inhibit RNA virus replication by at least about 20%, at least about 30%, at least about 40%, at least about 50%, at least abut 60%, at least about 70%, at least about 80%, at least about 90%, or at least about 95%.
[0051] The disclosed combination(s)/compositions(s) may be used in a method for reducing HCV viral load. In embodiments, the method comprises exposing the polymerase to a disclosed combination(s)/composition(s) and, optionally one or more additional therapeutic agents. The disclosed combination(s)/composition(s) may be administered with one or more of an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir), interferon and ribavirin in the same or separate pharmaceutical compositions to reduce HCV viral load. In embodiments, HCV viral load is reduced in vitro. In embodiments, HCV viral load is reduced in vivo. For example, if a disclosed combination(s)/composition(s) reduces the HCV viral load by at least about 10% compared to the HCV viral load before the virus was exposed to the combination(s)/composition(s), then the combination(s)/composition(s) reduces the HCV viral load. In some embodiments, the disclosed combination(s)/composition(s) can reduce viral load by at least about 20%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, or at least about 95%.
[0052] The disclosed combination(s)/composition(s) may be used in a method for treating a disease that can be treated by inhibiting HCV RNA polymerase and/or the HCV NS5A protein. Thus, this disclosure is also directed, in part, to a method for treating hepatitis C in an animal in need of such treatment. These methods comprise administering to the animal a disclosed combination(s)/composition(s) and, optionally one or more additional therapeutic agents. The disclosed combination(s)/composition(s) may be administered with one or more of an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir), interferon and ribavirin in the same or separate pharmaceutical compositions to treat hepatitis C. In some embodiments, a therapeutically effective amount of the disclosed combination(s)/composition(s) is administered to the animal.
[0053] "Treating" means ameliorating, suppressing, eradicating, preventing, reducing the risk of, and/or delaying the onset of the disease being treated. The term "treating" encompasses administration of the disclosed combination(s)/composition(s) to an HCV-negative patient that is a candidate for an organ transplant.
[0054] The methods of treatment are particularly suitable for use with humans, but may be used with other animals, particularly mammals. A "therapeutically-effective amount" or "effective amount" is an amount that will achieve the goal of treating the targeted condition.
[0055] In embodiments, therapeutic agent A is administered in combination with therapeutic agent B to reduce side effects associated with the administration of an interferon and ribavirin, either alone or in combination.
[0056] This disclosure is also directed, in part, to use of the disclosed combination(s)/composition(s), and, optionally one or more additional therapeutic agents in preparation of a medicament for use in one or more of the disclosed methods. The disclosed combination(s)/composition(s) may be combined with one or more of an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir), interferon and ribavirin in the same or separate medicaments for use in one or more of the disclosed methods.
[0057] In embodiments, therapeutic agent A and therapeutic agent B are used in the preparation of a medicament. In embodiments, therapeutic agent A, therapeutic agent B and an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir) are used in the preparation of a medicament. In embodiments, therapeutic agent A, therapeutic agent B and ribavirin are used in the preparation of a medicament. In embodiments, therapeutic agent A, therapeutic agent B, an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir), and ribavirin are used in the preparation of a medicament.
[0058] In embodiments, the disclosed medicaments are for inhibiting replication of an RNA virus.
[0059] In embodiments, the disclosed medicaments are for inhibiting HCV RNA polymerase activity.
[0060] In embodiments, the disclosed medicaments are for inhibiting the HCV NS4A protein.
[0061] In embodiments, the disclosed medicaments are for decreasing HCV viral load in a subject.
[0062] In embodiments, the disclosed medicaments are for treating hepatitis C.
[0063] In embodiments, the disclosed medicaments are for reducing side effects associated with the administration of an interferon and ribavirin, either alone or in combination.
[0064] The disclosed medicaments may be for co-administration with one or more additional therapeutic agents. For example, the medicaments may be for co-administration with one or more of an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir), interferon and ribavirin.
[0065] In embodiments, the disclosed medicaments may be for co-administration with interferon. In embodiments, the disclosed medicaments are for co-administration with one or more of an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir), interferon and ribavirin for inhibiting replication of an RNA virus.
[0066] In embodiments, the disclosed medicaments are for co-administration with one or more of an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir), interferon and ribavirin for inhibiting HCV RNA polymerase activity.
[0067] In embodiments, the disclosed medicaments are for co-administration with one or more of an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir), interferon and ribavirin for inhibiting HCV NS5A protein.
[0068] In embodiments, the disclosed medicaments are for co-administration with one or more of an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir), interferon and ribavirin for decreasing HCV viral load in a subject.
[0069] In embodiments, the disclosed medicaments are for co-administration with one or more of an HCV protease inhibitor (with or without a cytochrome P-450 inhibitor such as ritonavir), interferon and ribavirin for treating hepatitis C.
EXAMPLES
[0070] The following examples are for illustration purposes and do not limit the scope of this disclosure in any way.
Materials.
[0071] The replicon cell line was derived from the human hepatoma cell line Huh7. It was derived from HCV genotype 1b (Con1), and is a bicistronic subgenomic replicon, essentially similar to those described in Science 285(5424):110-3 (1999). The first cistron of the construct contains a firefly luciferase reporter and a neomycin phosphotransferase selectable marker.
Replicon Cell Culture.
[0072] Replicon cells were seeded at a density of 5000 cells per well of a 96-well plate in 100 .mu.l Dulbecco's Modified Eagle Media (DMEM) containing 5% FBS. Replicon cells were maintained in DMEM containing 100 IU/ml penicillin, 100 mg/ml streptomycin (Invitrogen), 200 mg/ml G418 (Invitrogen) and 10% fetal bovine serum (FBS) at 37.degree. C. and 5% CO.sub.2.
Combination Studies.
[0073] The replicon cell culture was used to determine the dose or concentration of therapeutic agent A that produces a synergistic, additive or antagonistic inhibitory effects on HCV replication when combined with therapeutic agent B.
[0074] The compounds were diluted in dimethyl sulfoxide (DMSO) to generate a 200X stock in a series of 6 two-fold dilutions. The dilution series was then further diluted 100-fold in the medium containing 5% FBS.
[0075] The dilutions of each compound were combined in a checkerboard fashion in the cell culture plates. Three experiments with three plates in each experiment were performed. In particular, six concentrations of compound A alone and six concentrations of the sodium salt of compound B alone were assayed in each plate. In addition, 36 combinations of various concentrations of the two compounds were assayed for each plate. The concentrations of compound A and compound B were chosen to ensure that the EC.sub.50s of the compounds were substantially in the middle of the serial dilution range. For compound A, concentrations ranged from 0.0002 nM (1.95.times.10.sup.-4 nM) to 0.0063 nM (6.25.times.10.sup.-3 nM), and for compound B, concentrations ranged from 0.10 nM (0.0977 nM) to 3.13 nM. The cells were incubated in a tissue culture incubator at 37.degree. C. and 5% CO.sub.2 for three days.
[0076] The inhibitor effects of compounds on HCV replication were analyzed by determining the fraction of inhibition of the luciferase signal which was determined by measuring activity of a luciferase reporter gene using a Luciferase Assay System kit (Promega) according to the manufacturer's instructions. Passive Lysis buffer (30 .mu.l, Promega) was added to each well, and the plates were incubated for 15 minutes with rocking to lyse the cells. Luciferin solution (100 .mu.l, Promega) was added to each well and the luciferase activity was measured using a Victor II luminometer (Perkin-Elmer). To determine the EC.sub.50, the luciferase inhibition data were analyzed using GraphPad Prism 4 software.
Combination Analysis.
[0077] Synergy or antagonism from combining therapeutic agent A with therapeutic agent B was quantified for direct comparison of inhibitor effects on HCV replication. The percent inhibition results were analyzed for synergy, additivity and antagonism according to the Bliss independence, Lowe additivity, and Pritchard-Direct models (Pharmacol. Rev. 47(2):331-85 (1995); Antiviral Research 14:181-206 (1990)).
[0078] An E.sub.max model in the following form is used to fit the data from each single drug for each plate in each experiment using the NLIN procedure of SAS (SAS 9.1, SAS Institute Inc. 2004),
f o = 1 - 1 ( 1 + ( C I ) h ) g , ##EQU00001##
where f.sub.a is the fraction of inhibition, C is the concentration, l is the location of the concentration-response curve's inflection point (point of greatest slope), g is the degree of asymmetry, and h is the shape parameter of the curve. Using the estimated g, l, and h for each single drug for each plate in an experiment, the fraction of inhibition for any concentration combination of the two drugs is predicted by one of two reference models: Loewe additivity and Bliss independence (Pharmacol. Rev. 47(2):331-85 (1995)). A difference between the actual observed fraction of inhibition and the predicted value is calculated for each concentration combination for each plate in each experiment to determine whether the observed combined effect is greater than that predicted by Loewe additivity or Bliss independence. For each concentration combination, the replicates (across all plates and experiments) were used to calculate a mean difference between observed and predicted fraction of inhibition, its standard error and its two-sided 95% confidence interval (CI).
[0079] The Prichard-Shipman method, similar to the E.sub.max methods, is used to calculate the difference between the actual observed fraction of inhibition and the predicted value for each concentration combination for each plate in each experiment to determine whether the observed combined effect is greater than the theoretical additive effect determined directly from the individual dose-response curves in the assays described above (Antiviral Research 14:181-206 (1990)). The calculated theoretical additivity is then compared to the experimental dose-response surface, and subsequently subtracted to reveal any areas of aberrant interaction. The following equation is used to calculate the theoretical additive effects:
Z=X+Y(1-X)=X+Y-XY,
where Z is the total inhibition produced by the combination of drugs X and Y, with X and Y representing the inhibition produced by drugs X and Y alone, respectively.
[0080] A difference between the actual observed fraction of inhibition and the predicted value is calculated for each concentration combination for each plate in each experiment to determine whether the observed combined effect is greater than the theoretical additive effect, Z, calculated from equation (1). The mean difference between the observed and predicted fraction of inhibition, its standard error and its two-sided 95% CI is then calculated for each concentration combination across all plates and experiments.
[0081] Synergy or antagonism for a concentration combination is determined by calculating at each concentration combination the 95% confidence interval (CI) of the mean difference between observed and predicted fraction of inhibition. If the lower bound of 95% CI is larger than zero, then the drug combination is considered to have a synergistic effect; it the upper bound of 95% CI is less than zero, then the drug combination is considered to have an antagonistic effect; otherwise, the effect of the combination is considered to be purely additive, and no significant antagonism or synergy exists at this concentration combination. Small differences of statistical significance caused by very small variance were excluded if the relative mean difference (i.e., the absolute mean difference divided by its corresponding observed mean inhibition) of the synergistic or antagonistic effect is less than about one percent.
Results.
[0082] The results of the replicon assay analysis using the Prichard-Shipman Model are illustrated in Table 1 and FIGS. 1 and 2.
[0083] Table 1 below lists various combinations of concentrations of compound A and compound B. For each combination of concentrations, Table 1 includes the mean difference in the observed and predicted fraction of inhibition, the standard deviation or error of the mean difference, and the upper and lower limits of the 95% confidence interval of the mean difference between observed and predicted fractions of inhibition.
TABLE-US-00001 TABLE 1 Mean difference in Standard Lower 95% Upper 95% Compound A, Compound B, fraction of inhibition: error of mean confidence confidence nM nM Observed-Predicted difference limit limit 0.000195 0.390625 -0.16428 0.054657 -0.29032 -0.03824 0.000391 0.097656 0.17174 0.035757 0.08929 0.25420 0.000781 0.097656 0.16218 0.063815 0.01502 0.30933 0.000781 0.195313 0.13851 0.054433 0.01298 0.26403 0.000781 0.390625 0.09246 0.021657 0.04252 0.14240 0.000781 0.781250 0.08495 0.016567 0.04674 0.12315 0.001563 0.195313 0.07811 0.032064 0.00417 0.15205 0.001563 0.390625 0.05619 0.016132 0.01900 0.09339 0.001563 0.781250 0.05043 0.012437 0.02175 0.07911
[0084] According to Table 1, all but one of the concentration combinations of compound A and compound B listed in the table have statistically significant synergistic effects.
[0085] FIG. 1 illustrates deviations from expected interactions between compound A and compound B are purely additive at concentrations associated with a horizontal plane at 0%. Synergistic interactions between compound A and compound B appear as a peak above the horizontal plane with a height corresponding to the percent above calculated additivity. Antagonistic interactions between compound A and compound B appear as a pit or trough below the horizontal plane with a negative value signifying the percent below the calculated additivity. It is apparent from FIG. 1 that synergistic interactions between compound A and compound B exist at many of the concentration combinations of compounds A and B.
[0086] The contour plot of FIG. 2 displays the region of concentration combinations with a statistically significant synergistic, antagonistic, or additive effect. Synergistic interactions appear as dark grey, additive interactions appear white, and antagonistic interactions appear as light grey. As illustrated in FIG. 2, an additive or synergistic effect exists at most of the concentrations for compound A and compound B. In particular, there is a concentration region showing synergy at the lower dose concentrations of compounds A and B.
[0087] The results presented in Table 1 and FIGS. 1 and 2 demonstrate that the combination of therapeutic agent A and therapeutic agent B achieves additivity or synergy at most concentration combinations of therapeutic agent A and therapeutic agent B. Taken together, these in vitro replicon results suggest that therapeutic agent A should produce a significant antiviral effect in patients when administered in combination with therapeutic agent B in patients infected with HCV.
Colony Counting Assay
[0088] Replicon colonies were exposed to therapeutic agent A, therapeutic agent B, an HCV protease inhibitor (a macrocyclic compound comprising a 9-membered fused bicycle, herein referred to as "therapeutic agent C"), and various combinations of these agents, to quantify the frequency of resistance of these replicon colonies to these agents.
[0089] The stable subgenomic bicistronic replicon cell line derived from HCV genotype 1a (H77; Genbank accession number AF011751) was generated by introducing the constructs into cell lines derived from the human hepatoma cell line Huh-7. The replicon also contains a firefly luciferase reporter and a neomycin phosphotransferase (Neo) selectable marker. The first cistron and the second cistron of the bicistronic replicon construct are separated by the FMDV 2a protease, and the second cistron comprises the HCV NS3-NS5B coding region with addition of adaptive mutations E1202G, K1691R, K2040R and S22041.
[0090] The HCV replicon cell line was maintained in Dulbecco's modified Eagles medium (DMEM; Invitrogen) containing 10% (v/v) fetal bovine serum, 100 IU/ml penicillin, 100 .mu.g/ml streptomycin, and 200 .mu.g/ml G418 (all from Invitrogen). 1a-H77 replicon cells (10.sup.5-10.sup.6) were plated in 150 mm cell culture plates and grown in the presence of G418 (400 .mu.g/ml) and therapeutic agent A, the potassium salt of compound B and/or therapeutic agent C at concentrations that were either 10-fold or 100-fold above the EC.sub.50 value for the HCV genotype 1a replicon cell line. After three weeks of treatment, the majority of replicon cells were cleared of replicon RNA and, therefore, were unable to survive in the G418-containing medium. The cells containing resistant replicon variants survived and formed colonies. These colonies were stained with 1% crystal violet in 10% Protocol SafeFix II reagent (Fisher Scientific) and counted.
[0091] As shown in FIG. 3, the combination of therapeutic agent A and therapeutic agent B, and the combination of therapeutic agent A and therapeutic agent C, at concentrations either 10-fold or 100-fold above their respective EC.sub.50 values, resulted in significantly fewer colonies than therapeutic agent A, therapeutic agent B or therapeutic agent C alone at concentrations 10-fold or 100-fold above their respective EC.sub.50 values.
[0092] All references (patent and non-patent) cited above are incorporated by reference into this patent application. The discussion of those references is intended merely to summarize assertions made by their authors. No admission is made that any reference (or a portion of a reference) is relevant prior art (or prior art at all). Applicants reserve the right to challenge the accuracy and pertinence of the cited references.
* * * * *
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.