Methods Of Preventing Or Reducing A Fibrotic Response Using CSF1R Inhibitors
Doloff; Joshua C. ; et al.
U.S. patent application number 16/081654 was filed with the patent office on 2019-03-21 for methods of preventing or reducing a fibrotic response using csf1r inhibitors. This patent application is currently assigned to Massachusetts Institute of Technology. The applicant listed for this patent is Massachusetts Institute of Technology. Invention is credited to Daniel G. Anderson, Joshua C. Doloff, Shady Farah, Robert S. Langer, Arturo J. Vegas, Omid Veiseh.
| Application Number | 20190083495 16/081654 |
| Document ID | / |
| Family ID | 58549290 |
| Filed Date | 2019-03-21 |








View All Diagrams
| United States Patent Application | 20190083495 |
| Kind Code | A1 |
| Doloff; Joshua C. ; et al. | March 21, 2019 |
Methods Of Preventing Or Reducing A Fibrotic Response Using CSF1R Inhibitors
Abstract
Described herein are methods of preventing or reducing fibrosis comprising administering CSF1R inhibitors, coating formulations comprising CSF1R inhibitors, coatings comprising CSF1R inhibitors for implantable medical devices, CSF1R inhibitor coated implantable medical devices, as well as corresponding embodiments comprising additional agents.
| Inventors: | Doloff; Joshua C.; (Quincy, MA) ; Farah; Shady; (Boston, MA) ; Veiseh; Omid; (Cambridge, MA) ; Vegas; Arturo J.; (Cambridge, MA) ; Langer; Robert S.; (Newton, MA) ; Anderson; Daniel G.; (Framingham, MA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Assignee: | Massachusetts Institute of
Technology Cambridge MA |
||||||||||
| Family ID: | 58549290 | ||||||||||
| Appl. No.: | 16/081654 | ||||||||||
| Filed: | April 4, 2017 | ||||||||||
| PCT Filed: | April 4, 2017 | ||||||||||
| PCT NO: | PCT/US17/25991 | ||||||||||
| 371 Date: | August 31, 2018 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 62317831 | Apr 4, 2016 | |||
| 62318208 | Apr 4, 2016 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61K 9/0024 20130101; A61K 31/416 20130101; C07B 2200/13 20130101; A61K 31/519 20130101; C07D 239/49 20130101; A61K 31/5025 20130101; A61K 31/444 20130101; A61K 31/5377 20130101; A61K 31/496 20130101; A61K 31/551 20130101; A61K 31/553 20130101; A61K 31/4709 20130101; A61K 31/404 20130101; A61K 31/4439 20130101; A61P 29/00 20180101 |
| International Class: | A61K 31/519 20060101 A61K031/519; A61K 31/4709 20060101 A61K031/4709; A61K 31/4439 20060101 A61K031/4439; A61K 31/5377 20060101 A61K031/5377; A61K 31/416 20060101 A61K031/416; A61K 31/444 20060101 A61K031/444; A61K 31/496 20060101 A61K031/496; A61K 31/404 20060101 A61K031/404; A61K 31/553 20060101 A61K031/553; A61K 31/551 20060101 A61K031/551; A61K 31/5025 20060101 A61K031/5025 |
Goverment Interests
GOVERNMENT SUPPORT
[0002] This invention was made with Government support under Grant No. R01 DE016516 awarded by the National Institutes of Health and under Contract No. W81XWH-13-1-0215 awarded by the U.S. Army Medical Research and Material Command. The Government has certain rights in the invention.
Claims
1. A method of preventing or reducing a fibrotic response to an implanted synthetic material in a patient, the method comprising administering to the patient an effective amount of a CSF1R inhibitor selected from the group consisting of a compound of structural formula ##STR00073## a compound represented by structural formula ##STR00074## a compound represented by structural formula ##STR00075## a compound represented by structural formula ##STR00076## and AC708, 4-(3,4-Dimethylanilino)-7-(4-(methylsulfonyl)phenyl)quinoline-3-carboxami- de, (4-cyano-N-(2-cyclohexenyl-4-(1-(2-(dimethylamino)acetyl)piperidin-4-y- l)phenyl)-1H-imidazole-2-carboxamide), a compound represented by structural formula ##STR00077## ARRY-382, a compound represented by structural formula ##STR00078## a 2'-aminoanilide, a 3-amido-4-anilinocinniline, an indoline-2-one, a 2-(alpha-methylbenzylamino)-pyrazine, an arylamide, a 3,4,6-substituted 2-quinolone, a pyrido[2,3-d]pyrimidin-5-one, a 3-amido-4-anilinoquinoline, a pyridyl bisamide, a thiazolyl bisamide, a 1,4-disubstituted-pyrrolo-[3,2-c]-pyridine, a substituted diphenylurea, a 5'-pyrimidine-2,4-diamine, a compound represented by structural formula ##STR00079## a compound represented by structural formula ##STR00080## anilinoquinazoline, a compound represented by structural formula ##STR00081## a compound represented by structural formula ##STR00082## DCC-3014, a compound represented by structural formula ##STR00083## a compound represented by structural formula ##STR00084## a compound represented by structural formula ##STR00085## a compound represented by structural formula ##STR00086## a compound represented by structural formula ##STR00087## a compound represented by structural formula ##STR00088## and a compound represented by structural formula ##STR00089##
2. The method of claim 1, wherein the CSF1R inhibitor is GW2580.
3. The method of claim 1, wherein the CSF1R inhibitor is Ki20227.
4. The method of claim 1, wherein the CSF1R inhibitor is 4-(3,4-Dimethylanilino)-7-(4-(methylsulfonyl)phenyl)quinoline-3-carboxami- de.
5. The method of claim 1, wherein the CSF1R inhibitor is (4-cyano-N-(2-cyclohexenyl-4-(1-(2-(dimethylamino)acetyl)piperidin-4-yl)p- henyl)-1H-imidazole-2-carboxamide).
6. The method of claim 1, wherein the foreign body is an ingested foreign body or an inhaled foreign body.
7. The method of claim 1, wherein the patient has an implanted medical device comprising the implanted material.
8. The method of claim 7, wherein the implanted medical device is implanted intraperitoneally, subcutaneously, or intramuscularly in the patient.
9. The method of claim 7, wherein the implanted medical device comprises at least one of a polymer, a ceramic, a hydrogel, a rubber, a metal, and glass.
10. The method of claim 9, wherein the polymer is a polysaccharide.
11. The method of claim 10, wherein the polysaccharide is alginate or chitosan.
12. The method of claim 9, wherein the polymer is polytetrafluoroethylene, polystyrene, polycaprolactone (PCL), or polydimethylsiloxane (PDMS).
13. The method of claim 9, wherein the metal is gold or stainless steel.
Description
RELATED APPLICATION(S)
[0001] This application claims the benefit of U.S. Provisional Application No. 62/317,831, filed Apr. 4, 2016. This application also claims the benefit of U.S. Provisional Application No. 62/318,208 filed Apr. 4, 2016. The entire teachings of the above applications are incorporated herein by reference.
INCORPORATION BY REFERENCE OF MATERIAL IN ASCII TEXT FILE
[0003] This application incorporates by reference the Sequence Listing contained in the following ASCII text file being submitted concurrently herewith:
[0004] a) File name: SEQLIST.txt; created Apr. 4, 2017, 7 KB in size.
BACKGROUND
[0005] Implanted biomedical devices currently reside within tens of millions of patients, both juvenile and adult, in the United States alone, and are involved in millions of new as well as revisionary surgeries every year (Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. The Journal of bone and joint surgery American volume 2007, 89(4): 780-785.; Med I. Medical Devices and the Public's Health: The FDA 510(k) Clearance Process at 35 Years. Medical Devices and the Public's Health: The Fda 510(K) Clearance Process at 35 Years 2011: 1-298.). As such, they comprise a major component of modern biomedicine, both in time and cost, and are essential for many clinical applications ranging from hip/knee replacement Cobelli N, Scharf B, Crisi G M, Hardin J, Santambrogio L. Mediators of the inflammatory response to joint replacement devices. Nature reviews Rheumatology 2011, 7(10): 600-608), tissue repair/reconstruction (Hubbell J A, Langer R. Translating materials design to the clinic. Nature materials 2013, 12(11): 963-966), prosthesis and neural interfacing (Fattahi P, Yang G, Kim G, Abidian M R. A review of organic and inorganic biomaterials for neural interfaces. Advanced materials 2014, 26(12): 1846-1885), controlled drug release (Farra R, Sheppard N F, Jr., McCabe L, Neer R M, Anderson J M, Santini J T, Jr., et al. First-in-human testing of a wirelessly controlled drug delivery microchip. Sci Transl Med 2012, 4(122): 122ra121), electronic pacing (Rosen M R, Robinson R B, Brink P R, Cohen I S. The road to biological pacing. Nat Rev Cardiol 2011, 8(11): 656-666), vital sign monitoring (Nichols S P, Koh A, Storm W L, Shin J H, Schoenfisch M H. Biocompatible materials for continuous glucose monitoring devices. Chemical reviews 2013, 113(4): 2528-2549), intraocular lens replacement (Perez-Cambrodi R J, Pinero D P, Ferrer-Blasco T, Cervino A, Brautaset R. The posterior chamber phakic refractive lens (PRL): a review. Eye 2013, 27(1): 14-21), and cell encapsulation and transplantation (Kearney C J, Mooney D J. Macroscale delivery systems for molecular and cellular payloads. Nature materials 2013, 12(11): 1004-1017.). Unlike particulates, which may be phagocytosed by the immune system and cleared via circulation and excretion (Cobelli N, Scharf B, Crisi G M, Hardin J, Santambrogio L. Mediators of the inflammatory response to joint replacement devices. Nature reviews Rheumatology 2011, 7(10): 600-608), larger non-biodegradable macroscale devices cannot be dislodged and extruded from the body. Instead, the host senses these implants as foreign and mounts an immune-mediated rejection response (Anderson J M, Rodriguez A, Chang D T. Foreign body reaction to biomaterials. Semin Immunol 2008, 20(2): 86-100; Wynn T A, Ramalingam T R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nature medicine 2012, 18(7): 1028-1040.). Immune cell adhesion leads to fibrosis, which encapsulates the implants in layers of scar tissue and extracellular matrix (Wynn T A, Ramalingam T R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nature medicine 2012, 18(7): 1028-1040.). Such sequestration can impair and eventually ruins device function by enzyme, acid, or reactive oxygen species-based degradation. This can also prevent necessary interaction with the surrounding microenvironment, including sensing of biochemical stimuli such as pH, oxygen, blood glucose levels, and/or obstructing nutrient flux where internal device components are of biologic origin (Anderson J M, Rodriguez A, Chang D T. Foreign body reaction to biomaterials. Semin Immunol 2008, 20(2): 86-100; Kenneth Ward W. A Review of the Foreign-body Response to Subcutaneously-implanted Devices: The Role of Macrophages and Cytokines in Biofouling and Fibrosis. J Diabetes Sci Technol Online 2008, 2: 768-777.). Furthermore, fibrotic scar tissue can cause pain and discomfort by displacing or abrading normal primary tissues (Bryers J D, Giachelli C M, Ratner B D. Engineering biomaterials to integrate and heal: the biocompatibility paradigm shifts. Biotechnol Bioeng 2012, 109(8): 1898-1911; Williams D F. On the mechanisms of biocompatibility. Biomaterials 2008, 29(20): 2941-2953.). It would be useful to have better methods to prevent or treat detrimental fibrosis.
SUMMARY OF THE INVENTION
[0006] The present invention relates to methods of treating or preventing fibrosis using a CSF1R inhibitor.
[0007] In one example embodiment, the method is a method of preventing or reducing a fibrotic response to an implanted synthetic material in a patient. The method includes administering to the patient an effective amount of a CSF1R inhibitor selected from the group consisting of a compound of structural formula
##STR00001##
of a compound represented by structural formula
##STR00002##
a compound represented by structural formula
##STR00003##
a compound represented by structural formula
##STR00004##
AC708, 4-(3,4-Dimethylanilino)-7-(4-(methylsulfonyl)phenyl)quinoline-3-ca- rboxamide, (4-cyano-N-(2-cyclohexenyl-4-(1-(2-(dimethylamino)acetyl)piperi- din-4-yl)phenyl)-1H-imidazole-2-carboxamide), a compound represented by structural formula
##STR00005##
ARRY-382, a compound represented by structural formula
##STR00006##
a 2'-aminoanilide, a 3-amido-4-anilinocinniline, an indoline-2-one, a 2-(alpha-methylbenzylamino)-pyrazine, an arylamide, a 3,4,6-substituted 2-quinolone, a pyrido[2,3-d]pyrimidin-5-one, a 3-amido-4-anilinoquinoline, a pyridyl bisamide, a thiazolyl bisamide, a 1,4-disubstituted-pyrrolo-[3,2-c]-pyridine, a substituted diphenylurea, a 5'-pyrimidine-2,4-diamine, a compound represented by structural formula
##STR00007##
a compound represented by structural formula
##STR00008##
anilinoquinazoline, a compound represented by structural formula
##STR00009##
a compound represented by structural formula
##STR00010##
DCC-3014, a compound represented by structural formula
##STR00011##
a compound represented by structural formula
##STR00012##
a compound represented by structural formula
##STR00013##
a compound represented by structural formula
##STR00014##
a compound represented by structural formula
##STR00015##
a compound represented by structural formula
##STR00016##
a compound represented by structural formula
##STR00017##
and combinations thereof.
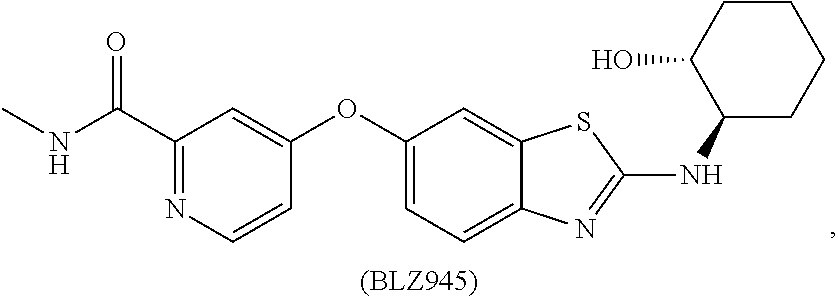
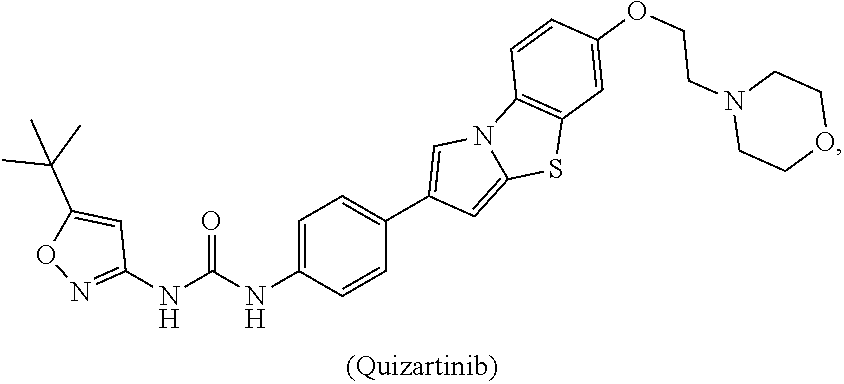
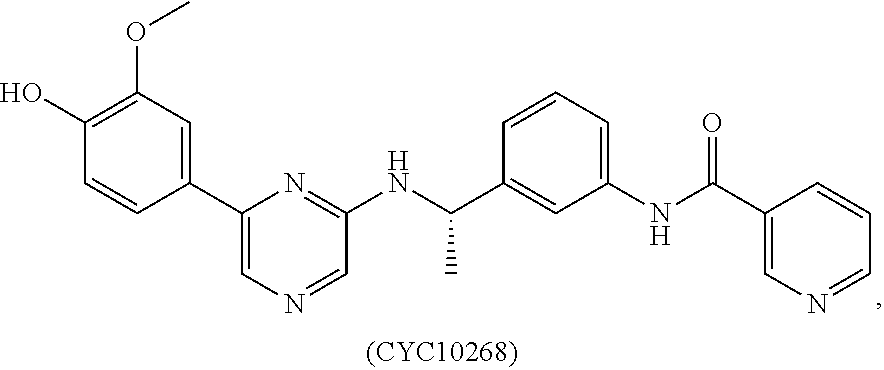
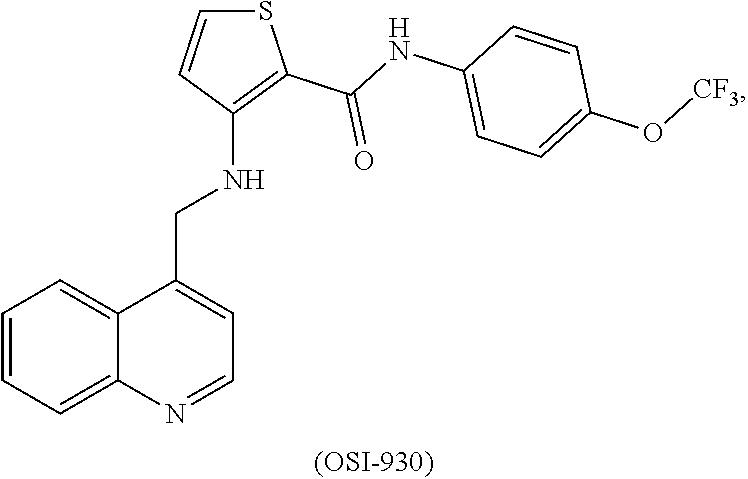
[0008] In some embodiments, the CSF1R inhibitor is at least one CSF1R inhibitor described herein. In some embodiments, for example, the CSF1R inhibitor is one or more CSF1R inhibitors selected from the group consisting of GW2580, Ki20227, 4-(3,4-Dimethylanilino)-7-(4-(methylsulfonyl)phenyl)quinoline-3-carboxami- de, (4-cyano-N-(2-cyclohexenyl-4-(1-(2-(dimethylamino)acetyl)piperidin-4-y- l)phenyl)-1H-imidazole-2-carboxamide), BLZ945, Quizartinib, AC708, Linifanib (a multitargeted receptor tyrosine kinase inhibitor), ARRY-382, Pexidartinib, a 2'-aminoanilide, a 3-amido-4-anilinocinniline, an indoline-2-one, a 2-(alpha-methylbenzylamino)-pyrazine, an arylamide, a 3,4,6-substituted 2-quinolone, a pyrido[2,3-d]pyrimidin-5-one, a 3-amido-4-anilinoquinoline, a pyridyl bisamide, a thiazolyl bisamide, a 1,4-disubstituted-pyrrolo-[3,2-c]-pyridine, a substituted diphenylurea, a 5'-pyrimidine-2,4-diamine, CYC10268, AZ683, anilinoquinazoline, OSI-930, DCC-2618, DCC-3014, JNJ-40346527 (a macrophage colony stimulating factor receptor agonist and CSF-1R inhibitor), Sunitinib, Lestaurtinib, Midostaurin, Tandutinib, Sorafenib, Ponatinib and combinations thereof. In one embodiment, the CSF1R inhibitor is GW2580. In some embodiments, the CSF1R inhibitor is Ki20227. In some embodiments, the CSF1R inhibitor is 4-(3,4-Dimethylanilino)-7-(4-(methylsulfonyl)phenyl)quinoline-3-carbox- amide. In some embodiments, the CSF1R inhibitor is (4-cyano-N-(2-cyclohexenyl-4-(1-(2-(dimethylamino)acetyl)piperidin-4-yl)p- henyl)-1H-imidazole-2-carboxamide). In one embodiment, the CSF1R inhibitor is at least one of GW2580, Ki20227, 4-(3,4-Dimethylanilino)-7-(4-(methylsulfonyl)phenyl)quinoline-3-carboxami- de and (4-cyano-N-(2-cyclohexenyl-4-(1-(2-(dimethylamino)acetyl)piperidin-- 4-yl)phenyl)-1H-imidazole-2-carboxamide).
[0009] In some embodiments, the foreign body is an ingested foreign body or an inhaled foreign body. In some embodiments, the foreign body is a mineral or element.
[0010] In some embodiments, the patient has an implanted medical device comprising the implanted material. In some embodiments, the implanted medical device is implanted, for example, intraperitoneally, subcutaneously, or intramuscularly in the patient. In some embodiments, the implanted medical device comprises, for example, at least one of a polymer, a ceramic, a hydrogel, a rubber, a metal, and glass. In some embodiments, the polymer is a polysaccharide. In some embodiments, the polysaccharide is alginate or chitosan.
[0011] In some embodiments, the polymer is polytetrafluoroethylene, polystyrene, polycaprolactone (PCL), or polydimethylsiloxane (PDMS). In some embodiments, the metal is gold or stainless steel.
BRIEF DESCRIPTION OF THE DRAWINGS
[0012] U.S. Provisional Application No. 62/317,831 ('831 application) contains color drawings which correspond to drawings of the present invention. With regard to indications of color within the instant description of the figures provided herein, reference is made to those corresponding drawings and associated descriptions of (1) the '831 application and (2) Doloff et al., "Colony stimulating factor-1 receptor is a central component of the foreign body response to biomaterial implants in rodents and non-human primates", Nature Materials, Advance Online Publication, published online Mar. 20, 2017 (DOI:10.1038/NMAT4866), both incorporated herein by reference in their entirety.
[0013] The foregoing will be apparent from the following more particular description of example embodiments of the invention, as illustrated in the accompanying drawings in which like reference characters refer to the same parts throughout the different views. The drawings are not necessarily to scale, emphasis instead being placed upon illustrating embodiments of the present invention.
[0014] FIGS. 1A-G depicts that numerous immune populations respond and adhere to implanted biomaterial alginate spheres. SLG20 alginate 500 .mu.m diameter spheres (0.35 ml total implant volume) were implanted into the intraperitoneal space of C57BL/6 mice, where they were retained for 14 days and analyzed for degree of fibrosis upon retrieval. FIG. 1A Dark field phase contrast image and FIG. 1B DAPI immunofluorescence image obtained from retrieved spheres reveal a significant level of cellular overgrowth; scale bar=2000 .mu.m). FIG. 1C: qPCR analysis of innate and adaptive immune population and fibrosis markers present on fibrosed alginate spheres, A, or non-transplant, N, or mock transplant, M, omental and epididymal fat pad tissue after 14 days post-implantation into the IP space of C57BL/6 mice. M.PHI.=macrophages, DCs=Dendritic cells, NKs=Natural Killer cells. Data: mean.+-.SEM, n=5. qPCR statistical analysis: one-way ANOVA with Bonferroni multiple comparison correction **: p<0.001, and ***: p<0.0001; ns=not significantly different. FIG. 1D Confocal staining showing DAPI (cellular nuclei), innate immune macrophage marker CD68 (green), adaptive immune B cells (magenta), alpha smooth muscle actin (.alpha.SMactin, myofibroblasts, red), and overlay making up the fibrosis on 500 .mu.m alginate spheres. FIG. 1E: Confocal staining showing DAPI (cellular nuclei), innate immune neutrophil marker Ly6g/Gr1 (green), alpha smooth muscle actin (.alpha.SMactin, myofibroblasts, red), fluorescent overlay, and brightfield image for the fibrosis on 500 .mu.m alginate spheres. In vivo intravital imaging of adaptive B cell behavior and accumulation at day 14 post-implant for mock transplant (FIG. 1F) or SLG20 sphere implanted (FIG. 1G) C57BL/6-Ccr6 (EGFP) mice. For intravital imaging: N=3 mice per treatment. For all others, N=5 mice per group. Experiments repeated at least 3 times.
[0015] FIG. 2 depicts that the same immune responders adhere to implanted biomaterial alginate spheres in both the intraperitoneal (IP) and subcutaneous (SC) sites. SLG20 alginate 500 .mu.m diameter spheres were implanted into the subcutaneous space of C57BL/6 mice, where they were retained for 14 days and analyzed upon retrieval. qPCR analysis of innate and adaptive immune population and fibrosis markers present in mock transplanted, M, versus fibrosed implanted alginate-embedded subcutaneous tissues, A. M.PHI.=macrophages, DCs=Dendritic cells, NKs=Natural Killer cells. Data: mean.+-.SEM, n=5. qPCR statistical analysis: one-way ANOVA with Bonferroni multiple comparison correction *: p<0.05, **: p<0.001, and ***: p<0.0001; ns=not significantly different. Experiment run at least twice.
[0016] FIGS. 3A-G depicts that the immune response to implanted biomaterial alginate is long lived. Flow analysis, using specific markers for responding host innate immune macrophage (red), neutrophil (blue), and adaptive immune B cells (green) at 1, 4, 7, 14, and 28 days post-implantation among peritoneal exudate (lavage) (FIG. 3A), peripheral fibrosed omental and epididymal fat pads (through which host cells infiltrate and to which material spheres become fibrosed, becoming part of the fibrosis themselves) (FIG. 3B), and directly on fibrosed alginate spheres (FIG. 3C). For sphere-specific FACS, fixation and permeabilization was also carried out to stain for fibrosis-depositing myofibroblasts (.alpha.SMactin, white). NanoString-based analysis for expression of macrophage (FIG. 3D) and neutrophil (FIG. 3E) associated markers analyzed from deposited cell RNA extracts at 14 days post-implant, presented on a base 2 logarithmic scale. (FIG. 3F) qPCR analysis on either peripheral tissue (yellow) or spheres alone (blue), relative to day 1 tissue for all, showing that there is a slightly delayed mobilization of adaptive B cells out of peripheral tissue and onto spheres after 1 week post-implantation. (FIG. 3G) qPCR analysis on either peripheral tissue (yellow) or spheres alone (blue), relative to day 1 for either tissue or spheres, respectively, showing that there is no apparent response (due to high tissue background) across time points for .alpha.SMactin, as opposed to a high dynamic response if spheres alone are examined. Error bars, mean.+-.SEM. For FACS and qPCR analysis N=5 mice per treatment; for NanoString analysis N=4 per treatment. FACS and qPCR experiments were performed twice and NanoString analysis was performed once. FACS time point comparisons were performed by unpaired, two-tailed t-test *: p<0.05, **: p<0.001, and ***: p<0.0001, vs mock or non-implanted controls.
[0017] FIGS. 4A-E depict that kinetic profiling of 30 cytokines in the blood shows no global response to alginate. Multiplexed Luminex kinetic profiling of protein production of 30 inflammatory cytokines in the serum of C57BL/6 mice at 1, 4, 7, 14, and 28 days post-intraperitoneal implantation of 500 .mu.m biomaterial alginate spheres. (FIG. 4A) Fold change numbers of all 30 cytokines at individual points, relative to protein levels of the mock/non-implanted (NT) control serum samples. (FIGS. 4B-E) Individual kinetic plots of the only 4 cytokines (IL-5, IL-6, G-CSF, and KC) that showed any significant responses (red squares in (a)) in the blood of implanted C57BL/6 mice. Responses, however, were transient and gone within 4-7 days post-implantation, suggesting that these increases were instead surgery related. Error bars, mean.+-.SEM. N=5 mice per treatment. Performed at least two times.
[0018] FIG. 5 depicts a photo sequence for retrieval process. 500 .mu.m diameter alginate microspheres are retrieved from the intraperitoneal (IP) space of wildtype C57BL/6 mice following a 2-week implantation. 1-2) Incisions were made first into the skin and then the underlying peritoneum. 3-4) Once the skin and peritoneal wall were successfully resected, the intestines were moved to the side exposing both the IP omental and epididymal fat pads. 5) It has been consistently and reliably observed that alginate spheres are fibrosed to non-collagen-encapsulated adipose tissues (omental, top blue inset square; and epididymal, bottom left and right inset squares) within the IP space (and never to tissues with collagen capsules, such as the liver, kidneys, etc.). Implanted materials suffer immune attack from immune cell responses that extravasate out of these microvessel rich adipose tissues (Med I. Medical Devices and the Public's Health: The FDA 510(k) Clearance Process at 35 Years. Medical Devices and the Public's Health: The Fda 510(K) Clearance Process at 35 Years 2011: 1-298.). 5a & 5b) Large groups of fibrosed alginate microspheres are found around and under both the left and right epididymal fat pads. Black arrows, fibrosed alginate microsphere(s). 6) While numerous microspheres are fibrosed directly to and embedded within adipose tissues, with more adherence over extended implantation times, many individual dirty (fibrosed) microspheres can be flushed out of the IP space following a 2-week implantation (6b). Images, representative across all mice; observed for countless (at least 100-200+) different implantations over the past 3-4 years (as of filing date of the U.S. Provisional Application No. 62/317,831).
[0019] FIGS. 6A-C depict additional kinetic expression profiling for immune-related factors associated with fibrotic cascade. qPCR analysis (Macrophage marker CD68 (FIG. 6A) and Transforming growth factor-beta (TGF.beta.) (FIG. 6B)) on either peripheral tissue (yellow) or alginate spheres alone (blue), relative to day 1 for either tissue or spheres, respectively, showing that there are similar kinetic responses between surrounding fibrosed tissue and embedded, fibrosed 500 .mu.m biomaterial alginate spheres, implanted in the IP space of C57BL/6 mice. Error bars, mean.+-.SEM. N=5 mice per treatment. Experiments were performed twice. qPCR statistical analysis: one-way ANOVA with Bonferroni multiple comparison correction *: p<0.5, **: p<0.001, and ***: p<0.0001, vs non-implanted (NT) controls. MT=mock transplanted, and N/A=not applicable, for spheres alone. (FIG. 6C) Western blotting time course for chemokine CXCL13 expression in mock implant tissue vs. day 1, 4, 7, 14, and 28 tissue and sphere protein samples retrieved from the intraperitoneal space of C57BL/6 mice, showing similar kinetics to Cxcl13 gene expression as shown by Nanostring analysis in FIG. 3D. Run at least twice.
[0020] FIG. 7 depicts knockouts and targeted depletion fibrosis summary. Serial or combined immune perturbations were used across various C57BL/6 strains to determine which cell populations are necessary for immune-mediated fibrosis. After extensive characterization, macrophages, and not neutrophils, are the only cell population required for downstream fibrotic sequestration of implanted alginate spheres. Right column: representative summary responses based on phase contrast images showing fibrosis levels on 500 .mu.m alginate spheres retrieved from wild type C57BL/6 mice (n=5/group), after 14-day intraperitoneal implantations. *, as reported.
[0021] FIGS. 8A-G depict that innate immune macrophage function is required for fibrosis of alginate. SLG20 alginate 500 .mu.m diameter spheres (0.35 ml total implant volume) were implanted into the intraperitoneal space of wild type and various knockout (IghMnull, B cell deficiency; Rag2.sup.null, T and B cell deficiency; and Rag2.sup.null/IL2r.gamma..sup.null, T, B, NK cell deficiency, and M.PHI. and DC dysfunction) C57BL/6 mice, where they were retained for 14 days and analyzed for degree of fibrosis upon retrieval. (FIG. 8A) Dark field phase contrast images showing that fibrosis, as compared to wild type (WT) control, was partially decreased upon removal of adaptive B cells (B KO), increased when T cells were also removed (TB KO), and completely removed with additional M.PHI. dysfunction (Rag2/.gamma. KO). (FIG. 8B) Semi-quantitative western blot analysis of .alpha.-SMact expression in cell overgrowth on microspheres (bands correspond to 5 individual mice), confirming relative changes to the levels of fibrosis observed due to the same immune perturbations in (a). (FIG. 8C) Plot of analyzed band intensities from western blot images shown in f (FIG. 8B). Error bars, mean.+-.SEM. N=5 mice per treatment. All experiments were performed at least three times. (FIG. 8D) q-PCR based expression analysis of fibrotic markers .alpha.-SMactin, Collagen 1a1 (Col1a1), and Collagen 1a2 (Col1a2) directly on retrieved spheres from WT, B KO, and Rag2/.gamma. KO C57BL/6 strains, plotted normalized to relative expression levels on spheres from WT mice. (FIG. 8E) Quantified IVIS live imaging fluorescent ProSense 750 inflammation levels across Mock (saline), WT, B KO, and Rag2/.gamma. KO mice 7 days post-subcutaneous implant. (FIG. 8F) H&E and Masson's Trichrome stained histological sections of excised subcutaneous tissue at 14 days post-implant from various treatments, as noted (FIG. 8F, Scale bar=500 .mu.m). (FIG. 8G) Flow analysis, using specific markers for responding host innate immune macrophage, neutrophil, and adaptive B cells dissociated directly from spheres (normalized to cell counts from 100 .mu.l material) 14 days post-intraperitoneal (i.p.) implantation (note: one WT subcutaneous sample is also included for comparison). qPCR, western blot, IVIS, and FACS statistical analysis: one-way ANOVA with Bonferroni multiple comparison correction *: p<0.05, **: p<0.001, and ***: p<0.0001, vs WT. N=5 mice/group. Experiments repeated at least 2-3 times.
[0022] FIGS. 9A-F depict a complete panel of phase contrast images for all knockouts. Samples, retrieved Ba-crosslinked SLG20 spheres of 500 .mu.m diameter spheres implanted into the intraperitoneal space of wildtype (FIG. 9A) and various immune compromised knockout (IghM.sup.null, B cell deficiency, (FIG. 9B); Rag2.sup.null, T and B cell deficiency, (FIG. 9C); Rag2.sup.null/IL2r.gamma..sup.null, T, B, NK cell deficiency, and M.PHI. and DC dysfunction, (FIG. 9D); T cell knockout (T KO) nude (FIG. 9E) and complement (C3) knockout (FIG. 9F)) C57BL/6 mice. Fibrosis, as compared to wildtype (WT) control, was partially decreased upon losing adaptive B cells (B KO), increased when T cells were also removed (TB KO), and gone completely with additional M.PHI. dysfunction (Rag2/.gamma.). Fibrosis was also not significantly affected by T cell loss alone, nor dependent on C3 complement immune recognition. Images obtained from all spheres retrieved from individual mice (n=5/group). The same material volume of hydrogel spheres was implanted into each mouse in all cases. Experiments were repeated twice.
[0023] FIGS. 10A-C depict additional B cell (IghM.sup.null) knockout model characterization, for reduced B cell response and corresponding reductions in fibrosis. (FIG. 10A) Additional in vivo intravital imaging of adaptive B cell behavior and accumulation at day 14 post-implant for SLG20 sphere implanted C57BL/6-Ccr6 (EGFP) mice (extra images from those shown in FIG. 1G showing B cell responses, extravasation from surrounding IP epididymal fat pads and aggregation between quantum dot (pink) encapsulating SLG20 alginate microspheres. (FIG. 10B) IgM protein levels determined by ELISA for both blood serum (left axis) and IP protein lysates (right axis) taken from tissue and spheres 14 days post-implant from mock (MT) and implanted wildtype (WT) and knockout strains ((IghM.sup.null, B cell knockout, B KO; and Rag2.sup.null/IL2r.gamma..sup.null knockout (Rag2/.gamma. KO), with T, B, NK cell deficiency, and M.PHI. and DC dysfunction). Loss of IgM in both blood and IP lysates taken from B cell deficient strains was confirmed. No IgM increases were detectable in wildtype (WT) responses in the blood but were significantly increased locally in the intraperitoneal space, suggesting that IgM is present not as secreted antibody but as a B cell receptor (BCR) on responding B cells. (FIG. 10C) Confocal staining showing DAPI (cellular nuclei), innate immune macrophage marker CD68 (green), alpha smooth muscle actin (.alpha.SMactin, myofibroblasts, red), B cell marker CD19 (magenta), fluorescent overlay, and brightfield image for the fibrosis on 500 .mu.m alginate spheres in wildtype (WT) C57BL/6 mice. B cell CD19 staining was unsurprisingly lost in B cell knockout (B KO) mice. More so, however, confocal imaging confirmed decreased fibrotic overgrowth due to loss of B cells, as seen by multiple imaging and staining methods (FIGS. 8A-G). Experiments were repeated twice.
[0024] FIGS. 11A-D depict additional histology (H&E and Masson's Trichrome staining) panels for subcutaneously implanted wildtype and knockout mice. Mock (saline injected) (FIG. 11A) and SLG20 500 .mu.m diameter alginate sphere implanted wildtype (WT) (FIG. 11B), B cell knockout (B KO) (FIG. 11C), and M.PHI. dysfunctional Rag2.sup.null/IL2r.gamma..sup.null (Rag2/.gamma.) C57BL/6 mice (FIG. 11D). 2.times. and 20.times. magnifications are shown in all cases to show both the scale as well as cellular details of varying levels of fibrosis in each treatment group. Arrows, showing implanted regions surrounding by an outer fibrosis collagen capsule. *, denote individual fibrosed alginate spheres. As shown in FIGS. 8A-G, B cell loss contributes to fibrotic reduction, while M.PHI. dysfunction results in loss of fibrosis (similar to mock), as compared to WT implanted controls.
[0025] FIGS. 12A-C depict additional FACS characterization of mock (left), distant (middle), and implanted (right) subcutaneous sites. Subcutaneous tissue groups, comparing immune population compositions of mock implanted (saline injected) to that from alginate-implanted C57BL/6 mice either at a site distant to (at least 1 cm away) or directly at material delivery (Implanted). Flow analysis, using specific markers for responding host innate immune macrophage (CD68.sup.+CD11b.sup.+) (FIG. 12A), neutrophil (Ly6g/Gr1.sup.+CD11b.sup.+) (FIG. 12B), and adaptive B cells (CD19.sup.+IgM.sup.+) (FIG. 12C) from dissociated subcutaneous tissue (as percent) 14 days post-subcutaneous (s.c.) alginate sphere implantation. Interestingly, macrophage percentage is slightly increased over that observed on intraperitoneally implantated alginate spheres. Distant tissues, taken from the same mice implanted s.c. with 500 .mu.m SLG20 alginate spheres, appear no different than s.c. tissue taken from mock-implanted (saline injected) mouse controls. Experiments were repeated twice.
[0026] FIGS. 13A-F depict that innate immune macrophages, and not neutrophils, are necessary and sufficient for fibrosis of biomaterial alginate spheres. SLG20 alginate 500 .mu.m diameter spheres (0.35 ml total implant volume) were implanted into the intraperitoneal space of different groups of wild type C57BL/6 mice, either treated with vehicle (WT) or targeted depletion agents to eliminate macrophages and/or neutrophils over a 14 day implantation and analyzed for degree of fibrosis upon retrieval. (FIG. 13A) Dark field phase contrast images showing that fibrosis, as compared to wild type (WT) control, was completely eliminated following removal of innate immune macrophages (- M.PHI.) either alone or in combination with neutrophil depletion (- Neutros & M.PHI.). Neutrophil depletion alone (- Neutros) did not alleviate fibrosis, and may have augmented sphere clumping. (FIG. 13B) Flow analysis, using specific markers for responding host innate immune macrophage, neutrophil, and adaptive B cells dissociated directly from spheres (as percent composition) 14 days post-intraperitoneal (i.p.) implantation, illustrating specificity of depletions. Shown to be recruited downstream of macrophages, adaptive B cells also decreased upon macrophage depletions. (FIG. 13C) Flow analysis for host innate immune macrophage, neutrophil, and adaptive B cells dissociated directly from spheres (normalized to cell counts from 100 .mu.l material) 14 days post-intraperitoneal (i.p.) implantation, showing absolute cell presence, and the lack thereof on spheres in mice which received macrophage depletion treatments. NanoString analysis for expression of all known cytokine and cytokine receptors (see FIG. 16A-C) for complete data set, excerpted based on response here) for macrophage-specific factors (by depletion and cell sorting) also inhibited by the CSF1R inhibitor GW2580 (FIG. 13D), for macrophage-specific factors (corroborated by cell sorting) removed by depletion but not affected by CSF1R blockade, suggesting altered macrophage polarization/phenotype and residual function (FIG. 13E), and for macrophage-associated factors (by depletion, but NOT cell sorting) also inhibited by the CSF1R inhibitor GW2580 (FIG. 13F), analyzed from RNA extracts at 14 days post-implant, presented on a base 2 logarithmic scale. Since these factors were removed by macrophage depletion, but not expressed by sorted macrophages (decreased/diluted green expression profile as compared to mock controls), they likely belong to cells that are recruited by macrophages downstream in the fibrotic cascade (ie, CD19, B cell marker and fibrosis protein collagen 1a1, Col1a1). FACS statistical analysis: one-way ANOVA with Bonferroni multiple comparison correction ***: p<0.0001, vs WT. ns=not significantly different. N=5 mice/group. Experiments repeated at least 3 times.
[0027] FIGS. 14A-D depict a complete panel of phase contrast images for targeted, serial innate immune depletions (as shown in FIG. 13). Images of retrieved 500 .mu.m diameter SLG20 spheres following implantation into the intraperitoneal space of wildtype C57BL/6 mice treated with either saline vehicle (Veh) (FIG. 14A), neutrophil depleting Gr1 antibody (- N) (FIG. 14B), macrophage depleting clodrosome (- M.PHI.) (FIG. 14C), and both neutrophil and macrophage depletion agents (- M.PHI. & N) (FIG. 14D). Fibrosis, as compared to vehicle-treated wildtype (WT) controls, was not decreased with neutrophil depletion (--N), increased when T cells were also removed (TB KO), and gone completely with additional M.PHI. dysfunction (Rag2/.gamma.). Images obtained from all spheres retrieved from individual mice (n=5/group). The same material volume of hydrogel spheres was implanted into each mouse in all cases. Experiments were repeated twice.
[0028] FIG. 15A-C depict flow analysis plots, comparing wildtype versus targeted innate immune depletion responses to implantation of alginate. Flow analysis, using specific markers for responding host innate immune macrophage (CD68.sup.+CD11b.sup.+) (FIG. 15A), neutrophil (Ly6g/Gr1.sup.+CD11b.sup.+) (FIG. 15B), and adaptive B cells (CD19.sup.+IgM.sup.+) (FIG. 15C) from cells dissociated from fibrosed or clean tissue/spheres as well as spleens (as percent composition) taken 14 days post-intraperitoneal (i.p.) implantation, from wildtype C57BL/6 mice treated with either saline vehicle or macrophage-depleting clodrosome, corresponding to the fibrosis images shown in FIG. 12A-C. Interestingly, M.PHI. depletion by clodrosome was specific, leaving neutrophil responses intact. B cells were also decreased in M.PHI. depleted mice, suggesting that macrophages are responsible for their recruitment. These results combined with fibrosis images and data from FIG. 8 and FIG. 12A-C suggest that neutrophils alone are not capable of fibrosing biomaterial alginate. Cell population percentages in the spleen (global immune reservoir) were unaffected by i.p. injected clodrosome treatment. Experiments were repeated twice.
[0029] FIG. 16A-C depict flow analysis plots, showing specificity of serial innate immune depletions. Specific markers used are for responding host innate immune macrophage (CD68.sup.+CD11b.sup.+) (FIG. 16A), neutrophil (Ly6g/Gr1.sup.+CD11b.sup.+) (FIG. 16B), and adaptive B cells (CD19.sup.+IgM.sup.+) (FIG. 16C) from cells dissociated from fibrosed or clean tissue/spheres (as percent composition) taken 14 days post-intraperitoneal (i.p.) implantation, from wildtype C57BL/6 mice treated with either saline (Vehicle, left column), neutrophil depleting Gr1 antibody (- N, middle column), or both macrophage and neutrophil depletion agents (- M.PHI. & N, right column), corresponding to the fibrosis images shown in FIG. 12A-C. All depletion agents proved to be population specific. Furthermore, neutrophil depletion neither affected macrophage nor B cell presence on fibrosed spheres, again suggesting their non-importance in a macrophage and B cell driven fibrotic response. Related, B cells were once again decreased in M.PHI. depleted mice. A CD11b.sup.loGr1.sup.lo/-CD68.sup.- population, likely repopulating monocytes, was also apparent in M.PHI. depleted mice. These results combined with fibrosis images and data from FIG. 8 and FIG. 12A-C suggest that neutrophils alone are not capable of fibrosing biomaterial alginate, nor are they required. Experiments were repeated twice.
[0030] FIG. 17 depicts a complete NanoString analysis for identification of inflammation and immune population-specific factors. Expression of all known mouse (host) cytokine and cytokine receptors, corresponding to sorted truncated heatmaps in FIGS. 13D-F for macrophage-specific or associated (downstream) factors, based on removal by depletion and corroborated by cell sorting. Subsets not affected by CSF1R blockade suggest altered macrophage polarization/phenotype and residual function (corroborated by FIG. 17. All samples were analyzed from RNA extracts at 14 days post-implant from each treatment group, presented on a base 2 logarithmic scale. White, within 2 standard deviations of the mean background of the assay.
[0031] FIG. 18A-D depicts elucidation of upstream inflammation induced by implanted biomaterial alginate, and peripheral macrophage function spared by CSF1R inhibition. (FIG. 18A) NanoString analysis heatmap was enriched from expression profiling of all known cytokine and cytokine receptors for all C57BL/6 wildtype (unperturbed and perturbed) and knockout models for mock and alginate material implanted treatment groups (see FIG. 18) for factors not associated with any immune population (not affected upon perturbations or CSF1R blockade), therefore likely associated with an upstream inflammation response. (FIG. 18B) Interestingly, some factors not associated with the removal of individual innate or adaptive immune populations, were found to be decreased or eliminated by CSF1R inhibition. These factors are likely induced inflammation response genes that are being negated by peripheral intact macrophage immune functions (ie., wound healing), implicated by a subset of macrophage-specific factors, removed by macrophage population depletion, but unaffected by CSF1R blockade (FIG. 13E), suggesting altered macrophage polarization/phenotype and residual preserved function. N=4 per treatment group. White boxes indicate values within 2 standard deviations of the background noise of the assay, indicating that they are statistically not detectable. (FIG. 18C) CXCL10 ELISA quantification for protein lysates derived from various wildtype and knockout model fibrosed or clean alginate spheres and tissue and retrieved at 14 days. CXCL10, an immune-mobilizing chemokine presented on the surface during periods of stress/damage, is increased upon B cell removal and even moreso upon macrophage depletion during implantation of C57BL/6 mice with 500 .mu.m alginate spheres, suggesting that these cell populations are not just involved in fibrosis initiation but also inflamed tissue repair. Interestingly, the CSF1R inhibitor GW2580 (GW) resulted in increased reduction of CXCL10 back to wildtype levels, suggesting that residual would healing functions are intact in CSF1R-inhibited monocyte/macrophages. CXCL10 protein levels also matched RNA expression changes in panel (b). (FIG. 18D) VEGF Luminex protein quantification for the same protein lysates used in (c) wildtype (control and perturbed) and knockout models, as in (c). VEGF, important for neovascularization and wound healing, is significantly reduced in both macrophage depletion groups, but returned to normal and not significantly different (ns) than levels observed in fully functional wildtype (WT) immune competent and implanted control mice. N=5/group. Luminex and ELISA run once each.
[0032] FIGS. 19A-G depict that CSF1R-dependent macrophages recruit fibrosis-potentiating adaptive B cells via chemokine CXCL13. CSF1R inhibition prevents the entire immune response to implanted biomaterials. Dark field phase contrast images showing that fibrosis, as compared to wild type (WT) control, was partially eliminated by CXCL13 neutralization, and completely eliminated with continuous CSF1R inhibition (160 mg/kg BW GW2580 s.c.) over a 14 day implant period (FIG. 19A). Fibrosis, was reduced the same extent as that of the B cell knockout (B KO) shown in FIG. 8a. (FIG. 19B) Flow analysis for responding host innate immune macrophage, neutrophil, and adaptive B cells dissociated directly from spheres (normalized to cell counts from 100 .mu.l material) 14 days post-intraperitoneal (i.p.) implant, showing partial loss of cell presence with CXCL13 neutralization, and complete loss in WT mice treated with the CSF1R inhibitor GW2580. (FIG. 19C) Brightfield images showing that fibrosis, as compared to vehicle controls, was completely eliminated by both macrophage depletion (- M.PHI.) and CSF1R inhibition (inh.). (FIG. 19D) Flow analysis for responding host innate immune macrophages and neutrophils dissociated directly from spheres (normalized to cell counts from 100 .mu.l of each material) 14 days post-intraperitoneal (i.p.) implantation, showing the loss of immune adhesion with loss of fibrosis due to either macrophage depletion (- M.PHI.) or CSF1R inhibition (inh.). (FIG. 19E) NanoString analysis for expression of all known cytokine and cytokine receptors (see FIG. 22A-C for complete data set, excerpted here), showing similar unique factors increased across all material (hydrogel alginate, ceramic glass, and polymer polystyrene (PS)) groups. (FIG. 19F) Confocal staining showing DAPI (cellular nuclei), innate immune macrophage marker CD68 (green), adaptive immune B cell marker CD19 (magenta), alpha smooth muscle actin (.alpha.SMactin, myofibroblasts, red), overlay, and brightfield making up the fibrosis on 500 .mu.m alginate spheres, showing that CXCL13 neutralization resulted in loss of B cell recruitment. (FIG. 19G) qPCR based expression analysis of fibrotic marker .alpha.-SMactin directly on retrieved spheres from WT, B KO, and CXCL13 neutralized mice, plotted normalized to relative expression levels on spheres from WT mice. qPCR and FACS statistical analysis: one-way ANOVA with Bonferroni multiple comparison correction ***: p<0.0001, vs Vehicle. N=5 mice/group. Experiments repeated at least 2 times.
[0033] FIG. 20A-B depict CSF1R inhibition prevents fibrosis of IP implanted alginate spheres (cont.). Complete phase contrast fibrosis images for wildtype C57BL/6 mice treated with either saline vehicle (Veh) (FIG. 20A) or CSF1R inhibitor GW2580 (FIG. 20B), corresponding to the images in FIG. 19D. Images obtained from all spheres retrieved from individual mice (n=5/group). The same material volume of hydrogel spheres was implanted into each mouse in all cases. Experiments were repeated twice.
[0034] FIGS. 21A-C depict that macrophage elimination or, minimally, CSF1R inhibition prevents fibrosis of IP implanted 500 .mu.m glass ceramic spheres. Complete brightfield fibrosis images for wildtype C57BL/6 mice treated with either saline vehicle (Veh) (FIG. 21A), macrophage depletion agent clodrosomes (FIG. 21B), or CSF1R inhibitor GW2580 (FIG. 21C), corresponding to the images in FIG. 19F. Images obtained from all spheres retrieved from individual mice (n=4/group). The same material volume of hydrogel spheres was implanted into each mouse in all cases. Experiments were repeated twice.
[0035] FIG. 22A-C depict that macrophage elimination or, minimally, CSF1R inhibition prevents fibrosis of IP implanted 500 .mu.m polystyrene polymer spheres. Complete brightfield fibrosis images for wildtype C57BL/6 mice treated with either saline vehicle (Veh) (FIG. 22A), macrophage depletion agent clodrosomes (FIG. 22B), or CSF1R inhibitor GW2580 (FIG. 22C), corresponding to the images in FIG. 19F. Images obtained from all spheres retrieved from individual mice (n=4/group). The same material volume of hydrogel spheres was implanted into each mouse in all cases. Experiments were repeated twice.
[0036] FIG. 23 depicts a NanoString analysis for complete cytokine signaling common to host responses across multiple material classes. Expression of all known mouse (host) cytokine and cytokine receptors to identify common inflammation and immune signaling across implanted hydrogel alginate, ceramic glass, and polymer polystyrene spheres, all retrieved 14 days after IP implantation into C57BL/6 mice. N=4/group. Presented on a base 2 logarithmic scale. Corresponds to excerpted heat map in FIG. 19G. Green box in Mock column, within 2 standard deviations of the mean background of the assay.
[0037] FIGS. 24A-D depict FACS plots and phase contrast images showing effects of CXCL13 antibody neutralization on B cell recruitment and downstream fibrosis. Flow analysis, using specific markers for responding host adaptive B cells (CD19.sup.+IgM.sup.+) from cells dissociated from fibrosed tissue/spheres (as percent composition) taken 14 days post-intraperitoneal (i.p.) implantation, from wildtype C57BL/6 mice treated with either saline (Vehicle) (FIG. 24A) or CXCL13-neutralizing antibody (- CXCL13) (FIG. 24B). Complete phase contrast fibrosis images for wildtype C57BL/6 mice treated with either saline vehicle (Veh) (FIG. 24C) or CXCL13-neutralizing antibody (- CXCL13) (FIG. 24D), corresponding to the images in FIG. 19. Images obtained from all spheres retrieved from individual mice (n=5/group). The same material volume of hydrogel spheres was implanted into each mouse in all cases. Experiments were repeated twice.
[0038] FIGS. 25A-E. Essential fibrotic cascade players are also increased in non-human primates. 0.5 mm-sized spheres of SLG20 hydrogels were implanted either intraperitoneally or subcutaneously in the dorsal region of cynomolgus macaque monkeys and retrieved by laparoscopy-guided tissue excision (control mock or implanted and sphere-embedded omentum fat tissue, (FIG. 25A)) or biopsy punch after 28 days (Veiseh O, Doloff J C, Ma M, Vegas A J, Tam H H, Bader A R, et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature materials 2015, 14(6): 643-651.). (FIG. 25B) H&E and Masson's Trichrome stained histological sections of excised IP omentum tissue at 28 days for mock and implanted groups, showing clean non-fibrosed fat-laden (Mock) or heavily collagen-deposited and sphere-embedded omental tissue (Implanted). (FIG. 25C) Despite a limited amount of functional antibodies for the cynomolgus species, we performed confocal staining showing DAPI (cellular nuclei), innate immune macrophage marker CD68 (green), and fibrosis-associated activated myofibroblast alpha smooth muscle actin (.alpha.SMactin, myofibroblasts, red), overlaid together, showing cellular infiltration around and fibrosis deposition on an embedded 500 .mu.m alginate sphere (20.times. magnification). White scale bars: both 200 um for each respective image. (FIG. 25D) Flow analysis showing similar host innate immune macrophage (CD68.sup.+CD11b.sup.+, top right quadrants) and neutrophil/myeloid (CD68.sup.-CD11b.sup.+, bottom right quadrants) cells across C57BL/6 mice and cynomolgus macaque monkeys, dissociated directly from fibrosed spheres and adjacent fibrosed omentum tissue (as percent composition) 28 days post-intraperitoneal (i.p.) implantation. While the prominence of CD11b seems to be inverted between macrophages and neutrophils in C57BL/6 mice vs cynomolgus monkeys, population response percentages are similar 28 days post-IP implantation. (FIG. 25E) NanoString analysis for immune markers and cytokines, originally identified in C57BL/6 mice (Note: CD66b is used here as a neutrophil marker, as Ly6g/Gr1 does not exist in NHPs or humans). Significant increases are observed for macrophage markers, as well as CSF1R and CXCL13 in both peritoneal and subcutaneous implant sites, as compared to mock (saline-injected) controls (there was no difference between SC and IP mock controls). N=2 for IP implanted groups; N=4 for subcutaneous (s.c.) treatment groups. These experiments were performed once for s.c. and twice for IP delivery.
[0039] FIGS. 26A-F depict additional primate histology and immunofluorescence panels showing similar foreign body responses. (FIG. 26A) H&E (a) and Masson's Trichrome (FIG. 26B) (b) stained histological sections of excised IP omentum tissue at 28 days for mock and implanted groups, showing clean non-fibrosed fat-laden (Mock) or heavily collagen-deposited and sphere-embedded omental tissue (Implanted). Corresponds to panels in FIG. 25. Magnifications: 10 and 40.times.. Additional confocal images for immunostained sections from implant alginate sphere embedded omental tissue excised at 28 days from cynomolgus monkeys. Shown: DAPI (cellular nuclei), innate immune macrophage marker CD68 (green), and alpha smooth muscle actin (.alpha.SMactin, myofibroblasts, red; also refer to blue arrows)), overlayed together, showing cellular infiltration around and fibrosis deposition on embedded 500 .mu.m alginate spheres. 5.times. (c) (FIG. 26C) and 20.times. (d) (FIG. 26D) magnifications. (FIG. 26E) e) Single red channel images (corresponding to the same images in (d)) showing material-bordering and more distant punctate .alpha.SMactin-staining myofibroblasts (see blue arrows for examples). It should also be noted, to not be confused, that pericytes covering larger circular blood vessels are also positive for .alpha.SMactin. (FIG. 26F) f) Single channels for DAPI (cellular nuclei), colony stimulating factor-1 receptor (CSF1R) (green), and brightfield views showing high CSF1R staining on both fused foreign body giants cells (FBGCs) and individual macrophages around embedded 500 .mu.m alginate spheres. 20.times. magnification. n=2 NHPs/group. These experiments were performed once for SC and twice for IP delivery.
[0040] FIGS. 27A-G depict CSF1R inhibition leaves many macrophage functions intact. (FIG. 27A) a) Skin incisions (all 1.5 cm in length, rulers are visible on left in all images) were made on day 0, and then wound clipped shut for both vehicle and daily GW2580-treated C57BL/6 mice (top left). Wound clips were removed and then replaced each imaging day up until day 7, after which clips were left off completely. By day 7, upon stretching the skin apart, incision sites on GW2580-treated mice were shut and not pulling apart (top right, red inset). By day 14, there appeared to be very little scarring in both vehicle and GW2580-treated groups (bottom middle). (FIG. 27B) b) After both 4 and 14 days, IP immune cells were taken by peritoneal lavage and analyzed by FACS for innate immune macrophage phenotype (CD68 & CD11b staining). As expected, at both time points, the mature tissue-resident macrophage phenotype observed in vehicle-treated mice was shifted (decreased CD68 & CD11b intensities) following daily GW2580 treatment. (FIG. 27C) c) Despite a phenotype shift, overall cell numbers in the peritoneal exudate were unchanged across untreated, vehicle-treated, or GW2580-treated groups. (FIG. 27 D) d) Confirming visibly healing skin incisions, histological assessment (H&E and Masson's Trichrome) show no significant (ns) differences by width and depth measurements (FIG. 27E) (e) in wound resolution and healing potential between vehicle or GW2580 treatment groups, by day 14 post-incision; scale bar: 400 .mu.m. (FIG. 27F) f) Peritoneal exudate macrophages isolated by IP lavage from (n=5) mice in each treatment group were immediately plated and incubated with fluorospheres for 90 minutes to determine phagocytic activity. Again, no significant differences were observed between macrophages isolated from vehicle and GW2580-treated mice. (FIG. 27G) g) Protein lysates were prepared from alginate spheres retrieved 14 days after IP implantation, and incubation with two different reactive oxygen specie (ROS) substrate solutions. Once again, no differences in ROS activity were observed between untreated, vehicle-treated, and GW2580-treated mice. n=5 mice for all assays. Error bars, mean+/-SE. Run 1-2 times, depending on the assay.
DETAILED DESCRIPTION OF THE INVENTION
[0041] A description of example embodiments of the invention follows below; additional description is found in International application Ser. No. ______, entitled "Compositions Of Crystallized Hydrophobic Compounds And Methods Of Making And Using Same" (HBSR Attorney Docket No. 0050.2293-001), filed concurrently with the instant application on Apr. 4, 2017, incorporated herein by reference in its entirety.
[0042] Regardless of the specific stimulus that initiates device rejection, aspects of the biology involved in the ensuing immune response have been characterized. Macrophages have remarkable plasticity, responding to numerous signals (Gordon S. Alternative activation of macrophages. Nat Rev Immunol 2003, 3(1): 23-35), and are a key component of material recognition, actively adhering to the surface of foreign objects (Anderson J M, Rodriguez A, Chang D T. Foreign body reaction to biomaterials. Semin Immunol 2008, 20(2): 86-100; Kenneth Ward W. A Review of the Foreign-body Response to Subcutaneously-implanted Devices: The Role of Macrophages and Cytokines in Biofouling and Fibrosis. . J Diabetes Sci Technol Online 2008, 2: 768-777; Grainger D W. All charged up about implanted biomaterials. Nat Biotechnol 2013, 31(6): 507-509; Sussman E M, Halpin M C, Muster J, Moon R T, Ratner B D. Porous implants modulate healing and induce shifts in local macrophage polarization in the foreign body reaction. Annals of biomedical engineering 2014, 42(7): 1508-1516.). They are increased locally throughout the implant site within days and may persist at the material surface even for the life of the implant (Anderson J M, Rodriguez A, Chang D T. Foreign body reaction to biomaterials. Semin Immunol 2008, 20(2): 86-100; Kenneth Ward W. A Review of the Foreign-body Response to Subcutaneously-implanted Devices: The Role of Macrophages and Cytokines in Biofouling and Fibrosis. J Diabetes Sci Technol Online 2008, 2: 768-777; Sussman E M, Halpin M C, Muster J, Moon R T, Ratner B D. Porous implants modulate healing and induce shifts in local macrophage polarization in the foreign body reaction. Annals of biomedical engineering 2014, 42(7): 1508-1516.). Devices too large to be cleared by phagocytosis instead initiate macrophage fusion into foreign-body giant cells (Anderson J M, Rodriguez A, Chang D T. Foreign body reaction to biomaterials. Semin Immunol 2008, 20(2): 86-100; Kyriakides T R, Foster M J, Keeney G E, Tsai A, Giachelli C M, Clark-Lewis I, et al. The CC chemokine ligand, CCL2/MCP1, participates in macrophage fusion and foreign body giant cell formation. Am J Pathol 2004, 165(6): 2157-2166), which recruit fibroblasts responsible for final fibrous collagen and matrix protein deposition (Anderson J M, Rodriguez A, Chang D T. Foreign body reaction to biomaterials. Semin Immunol 2008, 20(2): 86-100; Kenneth Ward W. A Review of the Foreign-body Response to Subcutaneously-implanted Devices: The Role of Macrophages and Cytokines in Biofouling and Fibrosis. . J Diabetes Sci Technol Online 2008, 2: 768-777; Rodriguez A, Meyerson H, Anderson J M. Quantitative in vivo cytokine analysis at synthetic biomaterial implant sites. Journal of biomedical materials research Part A 2009, 89(1): 152-159.). Ultimately, this fate is the same for many implanted materials of both natural and synthetic origin, including polysaccharides, polymers, ceramics such as silica and alumina, rubber, Teflon, and metals such as gold, stainless steel and titanium (Kenneth Ward W. A Review of the Foreign-body Response to Subcutaneously-implanted Devices: The Role of Macrophages and Cytokines in Biofouling and Fibrosis. J Diabetes Sci Technol Online 2008, 2: 768-777.).
[0043] One natural polysaccharide, alginate, is a multipurpose biomaterial that has been evaluated for use in numerous biomedical applications including biosensors, tissue regeneration, cell encapsulation, and drug delivery (Kearney C J, Mooney D J. Macroscale delivery systems for molecular and cellular payloads. Nature materials 2013, 12(11): 1004-1017; Lee K Y, Mooney D J. Alginate: properties and biomedical applications. Progress in polymer science 2012, 37(1): 106-126.). While non-biodegradable alginate capsules are used to immunoisolate transplanted islets for type 1 diabetes therapy, host immune and fibrosis responses directed to the encapsulating biomaterial results in device failure (de Vos P, Faas M M, Strand B, Calafiore R. Alginate-based microcapsules for immunoisolation of pancreatic islets. Biomaterials 2006, 27(32): 5603-5617; Jacobs-Tulleneers-Thevissen D, Chintinne M, Ling Z, Gillard P, Schoonjans L, Delvaux G, et al. Sustained function of alginate-encapsulated human islet cell implants in the peritoneal cavity of mice leading to a pilot study in a type 1 diabetic patient. Diabetologia 2013, 56(7): 1605-1614; Tuch B E, Keogh G W, Williams L J, Wu W, Foster J L, Vaithilingam V, et al. Safety and viability of microencapsulated human islets transplanted into diabetic humans. Diabetes care 2009, 32(10): 1887-1889; Weir G C. Islet encapsulation: advances and obstacles. Diabetologia 2013, 56(7): 1458-1461.). The combination of biologic and biomaterial complicates deconvoluting host immune responses, but empty alginate microspheres alone elicit rejection (Dang T T, Thai A V, Cohen J, Slosberg J E, Siniakowicz K, Doloff J C, et al. Enhanced function of immuno-isolated islets in diabetes therapy by co-encapsulation with an anti-inflammatory drug. Biomaterials 2013, 34(23): 5792-5801; Robitaille R, Dusseault J, Henley N, Desbiens K, Labrecque N, Halle J P. Inflammatory response to peritoneal implantation of alginate-poly-L-lysine microcapsules. Biomaterials 2005, 26(19): 4119-4127), thus immune attack of biomaterial alginate occurs independently of an encapsulated tissue of foreign origin. This response has been a fundamental barrier to translation of encapsulated islets for several decades (Tuch B E, Keogh G W, Williams L J, Wu W, Foster J L, Vaithilingam V, et al. Safety and viability of microencapsulated human islets transplanted into diabetic humans. Diabetes care 2009, 32(10): 1887-1889; Weir G C. Islet encapsulation: advances and obstacles. Diabetologia 2013, 56(7): 1458-1461.).
[0044] Overcoming the rejection of implanted biomaterial devices could allow for a range of medical advancements (Harding J L, Reynolds M M. Combating medical device fouling. Trends in biotechnology 2014, 32(3): 140-146; Langer R. Perspectives and challenges in tissue engineering and regenerative medicine. Advanced materials 2009, 21(32-33): 3235-3236.). Current approaches for immune suppression or management of long-term biomedical device implantation often involve broad-spectrum anti-inflammatories (Rhen T, Cidlowski J A. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. New England Journal of Medicine 2005, 353(16): 1711.). The major immunosuppressive agents used as standard care for implantation procedures are rapamycin (sirolimus), tacrolimus, everolimus, cyclosporine, and corticosteroids, as well as azathioprine, mycophenolate mofetil (MMF), mycophenolate sodium (Myfortic), and belatacept for transplantation (Denton M D, Magee C C, Sayegh M H. Immunosuppressive strategies in transplantation. Lancet 1999, 353(9158): 1083-1091; Halloran P F. Immunosuppressive drugs for kidney transplantation. N Engl J Med 2004, 351(26): 2715-2729; Khan W, Muntimadugu E, Jaffe M, Domb A J. Implantable Medical Devices. Focal Controlled Drug Delivery. Springer US, 2014, pp 33-59; Wong W, Venetz J P, Tolkoff-Rubin N, Pascual M. 2005 immunosuppressive strategies in kidney transplantation: which role for the calcineurin inhibitors? Transplantation 2005, 80(3): 289-296.). However, many anti-inflammatory drugs, including rapamycin, FK-506 (tacrolimus), cyclosporine, and numerous glucocorticosteroids, are not specific to individual immune populations, having multiple targets and differential effects in vivo (Rhen T, Cidlowski J A. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. New England Journal of Medicine 2005, 353(16): 1711; Attur M G, Patel R, Thakker G, Vyas P, Levartovsky D, Patel P, et al. Differential anti-inflammatory effects of immunosuppressive drugs: cyclosporin, rapamycin and FK-506 on inducible nitric oxide synthase, nitric oxide, cyclooxygenase-2 and PGE 2 production. Inflammation Research 2000, 49(1): 20-26.). Another anti-oxidant and immunomodulatory agent, curcumin, has also been shown to inhibit numerous immune cell populations (Dang T T, Thai A V, Cohen J, Slosberg J E, Siniakowicz K, Doloff J C, et al. Enhanced function of immuno-isolated islets in diabetes therapy by co-encapsulation with an anti-inflammatory drug. Biomaterials 2013, 34(23): 5792-5801), decrease macrophage and natural killer cell nitric oxide synthesis (Bhaumik S, Jyothi M D, Khar A. Differential modulation of nitric oxide production by curcumin in host macrophages and NK cells. FEBS Lett 2000, 483(1): 78-82), inhibit dendritic cell stimulation and cytokine production (Kim G Y, Kim K H, Lee S H, Yoon M S, Lee H J, Moon D O, et al. Curcumin inhibits immunostimulatory function of dendritic cells: MAPKs and translocation of NF-kappa B as potential targets. J Immunol 2005, 174(12): 8116-8124), and decrease T cell proliferation (Kim W, Fan Y Y, Smith R, Patil B, Jayaprakasha G K, McMurray D N, et al. Dietary curcumin and limonin suppress CD4+ T-cell proliferation and interleukin-2 production in mice. J Nutr 2009, 139(5): 1042-1048), thereby leading to functional decreases in multiple innate and adaptive immune populations. In general, broad-spectrum immune inhibition results in unwanted side effects. As such, improved drug targets and corresponding inhibitory compounds, capable of specific immune population inhibition or modulation, need to be identified. However, to do so, deeper understanding of the host immune-mediated foreign body rejection response must be achieved.
[0045] Foreign body responses are one of the largest impediments to biomedical device success. Up until now, the reasons why they occur are poorly understood. To identify key cell and cytokine targets, we performed in-depth systems analysis of innate and adaptive immune systems. While innate macrophages were indispensable to the fibrotic cascade, contrary to established belief, neutrophils and complement were not. Macrophages, via CXCL13, also led to downstream recruitment of B cells, which further potentiated fibrosis. Previously unimplicated, CSF1R was significantly increased upon implantation of multiple biomaterial classes: ceramic, polymer, and hydrogel. Its inhibition, like macrophage depletion, led to complete loss of fibrosis. CSF1R blockade, however, spared other macrophage function such as wound healing, establishing a more selective method of fibrosis inhibition. Immune cell and cytokine targets were additionally confirmed in non-human primates.
[0046] Here, we sought to further examine the role of innate and adaptive immunity on biomaterial biocompatibility in vivo. First, we focused on interrogating immune and fibrosis responses to implanted alginate hydrogels, then extended this characterization to include other materials as well. It is important to note that the fibrosis of alginate microspheres in rodents has been shown to be strain dependent (King A, Sandler S, Andersson A. The effect of host factors and capsule composition on the cellular overgrowth on implanted alginate capsules. Journal of biomedical materials research 2001, 57(3): 374-383; Manoury B, Caulet-Maugendre S, Guenon I, Lagente V, Boichot E. TIMP-1 is a key factor of fibrogenic response to bleomycin in mouse lung. International journal of immunopathology and pharmacology 2006, 19(3): 471-487.). Implantation of alginate into the intraperitoneal (IP) space of immune compliant BALB/c mice yields little to no fibrosis (King A, Sandler S, Andersson A. The effect of host factors and capsule composition on the cellular overgrowth on implanted alginate capsules. Journal of biomedical materials research 2001, 57(3): 374-383; Manoury B, Caulet-Maugendre S, Guenon I, Lagente V, Boichot E. TIMP-1 is a key factor of fibrogenic response to bleomycin in mouse lung. International journal of immunopathology and pharmacology 2006, 19(3): 471-487), whereas alginate retrieved from C57BL/6 mice, which have more aggressive innate immunity, is densely covered with fibrous overgrowth (King A, Sandler S, Andersson A. The effect of host factors and capsule composition on the cellular overgrowth on implanted alginate capsules. Journal of biomedical materials research 2001, 57(3): 374-383), mimicking the foreign body response observed in humans and non-human primates (NHPs). Therefore, we sought to further elucidate the immunity involved in the foreign body response in various C57BL/6 strains, with additional confirmation in cynomolgus monkeys, across which our recent work has translated successfully (Veiseh O, Doloff J C, Ma M, Vegas A J, Tam H H, Bader A R, et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature materials 2015, 14(6): 643-651; Vegas A J, Veiseh O, Doloff J C, Ma M, Tam H H, Bratlie K, et al. Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nat Biotechnol 2016.).
[0047] In one example embodiment, the method is a method of preventing or reducing a fibrotic response to a foreign body or to an implanted material in a patient comprising administering to the patient an effective amount of a CSF1R inhibitor. In some embodiments, the foreign body and/or the implanted material is of natural origin (e.g., natural). In some embodiments, it is of synthetic origin (e.g., synthetic). In some embodiments, the foreign body and/or the implanted material or device does not comprise biologics, cells and/or tissues.
[0048] In one embodiment, the method is a method of preventing or reducing a fibrotic response to a foreign body or an implanted material in a patient, the method comprising administering to the patient an effective amount of a CSF1R inhibitor selected from the group consisting of GW2580, Ki20227, BLZ945, Quizartinib, AC708, 4-(3,4-Dimethylanilino)-7-(4-(methylsulfonyl)phenyl)quinoline-3-carboxami- de, (4-cyano-N-(2-cyclohexenyl-4-(1-(2-(dimethylamino)acetyl)piperidin-4-y- l)phenyl)-1H-imidazole-2-carboxamide), Linifanib, ARRY-382, Pexidartinib, a 2'-aminoanilide, a 3-amido-4-anilinocinniline, an indoline-2-one, a 2-(alpha-methylbenzylamino)-pyrazine, an arylamide, a 3,4,6-substituted 2-quinolone, a pyrido[2,3-d]pyrimidin-5-one, a 3-amido-4-anilinoquinoline, a pyridyl bisamide, and a thiazolyl bisamide, a 1,4-disubstituted-pyrrolo-[3,2-c]-pyridine, a substituted diphenylurea, a 5'-pyrimidine-2,4-diamine, CYC10268, AZ683, anilinoquinazoline, OSI-930, DCC-2618, DCC-3014, JNJ-40346527, Sunitinib, Lestaurtinib, Midostaurin, Tandutinib, Sorafenib, and Ponatinib.
[0049] The methods described herein pertain to preventing or reducing a fibrotic response to a substance in a subject (e.g., a human, e.g., a patient). In some embodiments, the substance is a foreign body or an implanted material in a patient. Generally, a foreign body refers to a substance that has entered the body, e.g., unintentionally, such as by ingestion (e.g., due to debris in drinking water and/or food) or by inhalation (e.g., due to airborne debris). In some instances, a foreign body cannot be extruded or expelled by the body, either by responding immune cells or through normal circulation and excretion. Such materials, including nano-scale materials, can lead to downstream fibrosis and scar tissue deposition in major organs. Implanted materials generally refer to materials that have been introduced into the body, e.g., intentionally (e.g., by a surgeon or physician).
[0050] In some embodiments, the devices, materials and/or foreign bodies can be organic or inorganic. In some embodiments, they are synthetic.
[0051] The implanted medical devices can be implanted into a patient in a variety of locations. For example, they can be implanted intraperitoneally, subcutaneously, or intramuscularly. The implanted medical device or material (e.g., the implanted synthetic material) can be made from a variety of suitable materials, such as polymers, ceramics, hydrogels, rubbers, metals, and glasses. The implanted synthetic material can comprise a plurality of materials or be made from a combination of materials, such as such as polymers, ceramics, hydrogels, rubbers, metals, and glasses. The polymer can be a natural polymer, which refers to polymers that can be found in nature regardless of the method by which the polymer was actually made. Examples of natural polymers include polysaccharides, such as alginate or chitosan. For example, alginate can be purified from biological sources (e.g., seaweed). Alginate can also be synthetically derived. The polymer can also be a synthetic polymer, which refers to polymer that are not typically found in nature. Examples of synthetic polymers include polytetrafluoroethylene and polystyrene. Other examples of polymers include polycaprolactone (PCL) and polydimethylsiloxane (PDMS).
[0052] In some embodiments, the implanted material is not from a biological source. For example, in some embodiments, the material does not comprise a biologic (e.g., an antibody or fragment thereof), cells, tissues, protein or a combination thereof.
[0053] As used herein, the term "preventing" or "reducing," such as in the context of preventing or reducing a fibrotic response, refers to obtaining desired pharmacological and/or physiological effect. The effect can be prophylactic or therapeutic, which includes achieving, partially or substantially, one or more of the following results: partially or totally reducing the extent of the disease, disorder or syndrome; ameliorating or improving a clinical symptom or indicator associated with the disorder; delaying, inhibiting or decreasing the likelihood of the progression of the disease, disorder or syndrome; or partially or totally delaying, inhibiting or reducing the likelihood of the onset or development of disease, disorder or syndrome.
[0054] "Effective amount" means that amount of active compound agent that elicits the desired biological response in a subject. Such response includes alleviation of the symptoms of the disease or disorder being treated. The effective amount of a compound of the invention in such a therapeutic method is from about 0.01 mg/kg/day to about 1000 mg/kg/day, from about 0.1 mg/kg/day to about 300 mg/kg/day, from about 50 mg/kg/day to about 250 mg/kg/day, from about 100 mg/kg/day to 200 mg/kg/day, or from about 125 mg/kg/day to about 175 mg/kg/day.
[0055] "Pharmaceutically acceptable carrier" means compounds and compositions that are of sufficient purity and quality for use in the formulation of a composition of the invention and that, when appropriately administered to an animal or human, do not produce an adverse reaction.
[0056] In certain embodiments, the compositions of the invention described herein are formulated for therapeutic (e.g., pharmaceutical) use with one or more pharmaceutically-acceptable carriers or excipients. The term "pharmaceutically acceptable carrier" means a non-toxic solvent, dispersant, excipient, adjuvant or other material which is mixed with the active ingredient in order to permit the formation of a pharmaceutical composition, i.e., a dosage form capable of administration to the patient. Generally, pharmaceutically-acceptable carriers or excipients may be present in amounts having no substantial effect on the stability and release rate profiles of the hydrophobic compound(s) in the composition. Suitable excipients/carriers are well known in the art, including those described in Gennaro et al., Remington's Pharmaceutical Sciences (18th ed., Mack Publishing Company, 1990, see especially Part 8: Pharmaceutical Preparations and their Manufacture), which is incorporated herein by reference in its entirety. The compositions of the invention formulated for therapeutic use may be used as is, or may be used as a pharmaceutically acceptable salt thereof. The term "pharmaceutically acceptable salt" means either an acid addition salt or a basic addition salt which is compatible with the treatment of patients/subjects.
[0057] In some embodiments, exemplary inorganic acids which form suitable salts include, but are not limited thereto, hydrochloric, hydrobromic, sulfuric and phosphoric acid and acid metal salts such as sodium monohydrogen orthophosphate and potassium hydrogen sulfate. Illustrative organic acids which form suitable salts include the mono-, di- and tricarboxylic acids. Illustrative of such acids are, for example, acetic, glycolic, lactic, pyruvic, malonic, succinic, glutaric, fumaric, malic, tartaric, citric, ascorbic, maleic, hydroxymaleic, benzoic, hydroxybenzoic, phenylacetic, cinnamic, salicylic, 2-phenoxybenzoic, p-toluenesulfonic acid and other sulfonic acids such as methanesulfonic acid and 2-hydroxyethanesulfonic acid. Either the mono- or di-acid salts can be formed, and such salts can exist in either a hydrated, solvated or substantially anhydrous form. In general, the acid addition salts of these compounds are more soluble in water and various hydrophilic organic solvents, and generally demonstrate higher melting points in comparison to their free base forms. Other non-pharmaceutically acceptable salts e.g. oxalates may be used.
[0058] The compositions of the invention can be in a solid form or liquid form. Typically, they are in dosage unit form, such as tablet, powder, sachet, bead, pellet, osmotic dosage form, etc., but they may as well be in a liquid, cream or aerosol form for use in various applications, i.e., parenteral, oral, buccal, ophthalmic, nasal, dermal, rectal, and pulmonary routes. In one embodiment, the compositions provided in the present invention are encapsulated. Non limiting examples of materials used for encapsulation of the composition of the current invention include materials composed of ceramic, glass, metal, poly lactic-co-glycolic acid (PLGA) co-polymer, polymer (e.g., polystyrene beads) and alginate hydrogels. In a particular embodiment, the compositions provided in the present invention are encapsulated in a biocompatible polymer (e.g., alginate hydrogel).
[0059] The compositions of the present invention can be formulated for different modes of administration, including, but not limited to, parenteral, oral, buccal, ophthalmic, nasal, dermal, rectal, and pulmonary routes. In one embodiment, the compositions are in an oral delivery form, such as a tablet, capsule or osmotic dosage form. In another embodiment, the compositions are in a form suitable for administration by injection. In another embodiment, the compositions are in a form suitable for administration by implantation.
[0060] Compositions and compounds of the invention suitable for parenteral injection include sterile solutions.
[0061] The dosage form containing the composition of the invention contains an effective amount of the active ingredient necessary to provide a therapeutic effect. The composition may contain from about 5,000 mg to about 0.5 mg (preferably, from about 1,000 mg to about 0.5 mg) of a compound of the invention or salt form thereof and may be constituted into any form suitable for the selected mode of administration. The composition may be administered about 1 to about 5 times per day. Daily administration or post-periodic dosing may be employed.
[0062] In some embodiments, the compounds may be administered parenterally via injection. A parenteral formulation may consist of the active ingredient dissolved in or mixed with an appropriate inert liquid carrier. Acceptable liquid carriers usually comprise aqueous solvents and other optional ingredients for aiding solubility or preservation. Such aqueous solvents include sterile water, Ringer's solution, or an isotonic aqueous saline solution. Other optional ingredients include vegetable oils (such as peanut oil, cottonseed oil, and sesame oil), and organic solvents (such as solketal, glycerol, and formyl). A sterile, non-volatile oil may be employed as a solvent or suspending agent. The parenteral formulation is prepared by dissolving or suspending the active ingredient in the liquid carrier whereby the final dosage unit contains from 0.005 to 10% by weight of the active ingredient. Other additives include preservatives, isotonizers, solubilizers, stabilizers, and pain-soothing agents. Injectable suspensions may also be prepared, in which case appropriate liquid carriers, suspending agents and the like may be employed.
[0063] A number of compounds can be administered to a patient in order prevent or reduce a fibrotic response to a foreign body or to an implanted material. In general, the compounds inhibit CSF1R. Examples of compounds include GW2580; Ki20227; 4-(3,4-Dimethylanilino)-7-(4-(methylsulfonyl)phenyl)quinoline-3-carboxami- de; (4-cyano-N-(2-cyclohexenyl-4-(1-(2-(dimethylamino)acetyl)piperidin-4-y- l)phenyl)-1H-imidazole-2-carboxamide).
[0064] The compound GW2580 has the following structure:
##STR00018##
[0065] The compound Ki20227 has the following structure:
##STR00019##
[0066] The compound 4-(3,4-Dimethylanilino)-7-(4-(methylsulfonyl)phenyl)quinoline-3-carboxami- de, also known as cFMS Receptor Inhibitor III, has the following structure:
##STR00020##
[0067] The compound (4-cyano-N-(2-cyclohexenyl-4-(1-(2-(dimethylamino)acetyl)piperidin-4-yl)p- henyl)-1H-imidazole-2-carboxamide), also known as JNJ-28312141, has the following structure:
##STR00021##
[0068] A class of compounds that inhibit CSF1R include 2'-aminoanilides, of which the following compound is an example:
##STR00022##
[0069] wherein NR.sup.1R.sup.2 is selected from the group consisting of: piperidino; morpholino; piperazine; N-ethyl-N-propylamino; N,N-dipropylamino; anilino; azetidino; pyrrolidino; azepino; 2-methylpiperidino; 3-methylpiperidino; 3,5-dimethylpiperidino; 4-methylpiperidino; 4-methylpiperazino; 4-phenylpiperazino; 4-hydroxypiperidino; 4-hydroxymethylpiperidino; 4-(2-hydroxyethyl)piperidino; 3-hydroxypyrrolidino; and 3-hydroxymethylpyrrolidino. (See also Raymond J. Patch et al, Bioorganic & Medicinal Chemistry Letters 17 (2007) 6070-6074.) For example, the compound of the following structural formula:
##STR00023##
[0070] A class of compounds that inhibit CSF1R include 3-amido-4-anilinocinnolines, of which the following compound is an example:
##STR00024##
[0071] wherein R.sup.3 is selected from the group consisting of EtO and MeO; and wherein each R.sup.4 is independently selected from fluorine, chlorine, and methyl. (See also David A. Scott et al. Bioorganic & Medicinal Chemistry Letters 21 (2011) 1382-1384.)
[0072] A class of compounds that inhibit CSF1R include indoline-2-ones (e.g., of structural formula
##STR00025##
such as sunitinib, lestaurtinib, midostaurin, tandutinib, sorafenib, ponatinib, and quizartinib. (See also Li Sun et al, J. Med. Chem. 1998, 41, 2588-2603.) Other indoline-2-ones include compounds having the following structure:
##STR00026##
[0073] wherein R.sup.5 is selected from the group consisting of --H; 1'-CH.sub.3; 4'-CH.sub.3; 6'-F; 5'-Cl; 5'-Br; and wherein R.sup.6 is selected from the group consisting of 4'-N(CH.sub.3).sub.2;
##STR00027##
4'-OH; 4'-OCH.sub.3; 4'-Br; 4'-COOH; 3'- and 5'-C(CH.sub.3).sub.3, 4-OH; 3'- and 5'-CH(CH.sub.3).sub.2, 4-OH; 3'-C(CH.sub.3).sub.3, 4-OCH.sub.3, and 5'-Br; 3'-C(CH.sub.3).sub.3 and 4'-OCH.sub.3; 4'-CH(CH.sub.3).sub.2; 3'- and 5'-CH(CH.sub.3).sub.2 and 4'-OH; 3'-C(CH.sub.3).sub.3 and 4'-OCH.sub.3; 4'-Br; and 3'-C(CH.sub.3).sub.3, 4'-OCH.sub.3, and 5'-Br; and wherein the indicated stereochemistry can be either the E or Z configuration.
[0074] Another indole-2-one compound is a compound having the following structure:
##STR00028##
[0075] Other indoline-2-ones include compounds having the following structure:
##STR00029##
[0076] wherein R.sup.7 is selected from the group consisting of --H; 4'-CH.sub.3; 5'-CH.sub.3; 6'-F; 5'-Cl; and 5'-NO.sub.2; and wherein R.sup.8 is selected from the group consisting of --H; 3'- and 4'-CH.sub.3; 3'-CH.sub.2CH.sub.2COOH and 4'-CH.sub.3; 3'-CH.sub.3 and 4'-CH.sub.2CH.sub.2COOCH.sub.3; 3'-CH.sub.3 and COOCH.sub.2CH.sub.3; 3'- and 5'-CH.sub.3; 3'- and 5' CH.sub.3, 4'-CH.sub.2CH.sub.3; 3'- and 5'-CH.sub.3, 4'-COOCH.sub.2CH.sub.3; 3'-CH.sub.2CH.sub.3, 4'- and 5'-CH.sub.3; 1'-CH.sub.3; and 3'- and 5'-CH.sub.3.
[0077] Other indoline-2-ones include compounds having the following structure:
##STR00030##
[0078] wherein R.sup.9 is selected from the group consisting of --H and 4'-CH.sub.3; and wherein R.sup.10 is selected from the group consisting of 3'-Br; 4'-Br; 5'-SCH.sub.3; and 5'-CH.sub.2CH.sub.3.
[0079] Other indoline-2-ones include compounds having the following structure:
##STR00031##
[0080] wherein R.sup.11 is --H; and wherein R.sup.12 is selected from the group consisting of --H, -5'-CH.sub.3, and 5'-CH.sub.2CH.sub.3.
[0081] Other indoline-2-ones include compounds having the following structure:
##STR00032##
[0082] wherein R.sup.13 is selected from the group consisting of 4'-Cl; and 1'-CH.sub.3 and 4'-Cl.
[0083] A class of compounds that inhibit CSF1R include 2-(.alpha.-methylbenzylamino) pyrazines, of which the following compound is an example:
##STR00033##
(CYC10268). See also Christopher J. Burns et al, Bioorganic & Medicinal Chemistry Letters 19 (2009) 1206-1209).
[0084] Other examples of 2-(.alpha.-methylbenzylamino) pyrazines include compounds having the following structure:
##STR00034##
[0085] wherein R.sup.14 is selected from the group consisting of
##STR00035##
[0086] Other examples of 2-(.alpha.-methylbenzylamino) pyrazines include compounds having the following structure:
##STR00036##
[0087] wherein R.sup.15 is selected from the group consisting of
##STR00037##
and wherein X is N or C.
[0088] A further class of compounds that inhibits CSF1R is arylamides. For example, the compound of structural formula
##STR00038##
(See also Carl R. Illig et al, J. Med. Chem. 2011, 54, 7860-7883 and Hui Huang et al, J. Med. Chem. 2009, 52, 1081-1099)
[0089] A further class of compounds that inhibits CSF1R is 3,4,6-Substituted-2-quinolone, for example, the compound of structural formula
##STR00039##
wherein R is a substituent (see Mark J. Wall et al. Bioorganic & Medicinal Chemistry Letters 18 (2008) 2097-2102).
[0090] A further class of compounds that inhibits CSF1R is 3-amido-4-anilinoquinolines, for example, the compound of structural formula
##STR00040##
wherein R.sup.1 is a substituent.
[0091] Further suitable compounds include cFMS Receptor Inhibitor III--CAS 959861-21-3, represented by structural formula
##STR00041##
Ki20227, represented by structural formula
##STR00042##
AZ683, and OSI-930, represented by structural formula
##STR00043##
See also David A. Scott et al. Bioorganic & Medicinal Chemistry Letters 19 (2009) 697-700.
[0092] A further class of compounds that inhibits CSF1R is the class of pyridyl and thiazolyl bisamides, such as the compound represented by structural formula
##STR00044##
wherein R.sup.1, R.sup.2 and R.sup.3 are independently selected substituents; and the compound represented by structural formula CSF-1R enzyme and cell activity--pyridal bisamides
##STR00045##
wherein R.sup.1 and R.sup.2 are independently selected substituents, and the X stands for a ring atom, such as C or N; see also David A. Scott et al. Bioorganic & Medicinal Chemistry Letters 18 (2008) 4794-4797.)
[0093] Another suitable compound is Linifanib (ABT-869); N-[4-(3-amino-1H-indazol-4-yl)phenyl]-N1-(2-fluoro-5-methylphenyl) urea, represented by structural formula
##STR00046##
See also Daniel H. Albert et al, Mol Cancer Ther 2006; 5(4) 995-1006).
[0094] Another suitable compound is the compound is ARRY-382 (Array BioPharma); see also J. Bendell, et al., EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics.
[0095] Another suitable compound is the compound is Pexidartinib, PLX3397 (Plexxikon), represented by structural formula
##STR00047##
See also Nicholas Butowsk et al, Neuro-Oncology 18(4), 557-564, 2016.
[0096] A further class of compounds that inhibits CSF1R is 7-azaindoles, for example, the compound represented by structural formula
##STR00048##
wherein R and R' stand are independently selected substituents (for example, PLX3397; see also Chao Zhang et al, PNAS, 110, 14, 2013, 5689-5694.)
[0097] A further class of compounds that inhibits CSF1R is 1,4-disubstituted-pyrrolo-[3,2-c]-pyridines, for example, the compound represented by structural formula
##STR00049##
wherein R and R' stand are independently selected substituents; see also Chao Zhang et al, Bioorganic & Medicinal Chemistry Letters 22 (2012) 4362-4367.
[0098] A further class of compounds that inhibits CSF1R is substituted diphenylurea compounds, for example, the compound represented by structural formula
##STR00050##
wherein R and R' are independently selected substituents. For example, Linifanib (ABT-869). See also Jun Guo et al, Mol Cancer Ther 2006; 5(4) 1007-1013.
[0099] A further class of compounds that inhibits CSF1R is 5'-pyrimidine-2,4-diamines, for example, the compound represented by structural formula
##STR00051##
wherein R is a substituent. For example GW2580. See also Vadim Bernard-Gauthier et al, Bioorganic & Medicinal Chemistry Letters 24 (2014) 4784-4790.
[0100] A further class of compounds that inhibits CSF1R is pyrido[2,3-d]pyrimidin-5-ones, for example, the compound represented by structural formula
##STR00052##
wherein R, R' and R'' are independently selected substituents. For example, the compound represented by structural formula
##STR00053##
See also Hui Huang et al, J. Med. Chem. 2009, 52, 1081-1099.
[0101] Another suitable compound is AZ683 (a 3-amidoquinoline), represented by structural formula
##STR00054##
[0102] Another suitable compound is anilinoquinazoline, represented by structural formula
##STR00055##
[0103] In other embodiment of the present inventions a plurality of CSF1R inhibitors are used in combination, for example, two, three, or four of the CSF1R inhibitors described herein. For example, one embodiment of the present invention is a method of preventing or reducing a fibrotic response to an implanted synthetic material in a patient, the method comprising administering to the patient an effective amount of GW2580 and an effective amount of Ki20227.
[0104] In some embodiments (e.g., where fibrosis is a concern, the CSF1R inhibitor compounds and compositions disclosed herein drugs can be merged or integrated into already existing platforms (e.g., implants). Examples of implants and implanted medical devices include, but are not limited to, the following: 1) Breast implants, 2) implanted sensors, for continuous sensing and monitoring of physiological conditions (e.g., subcutaneously embedded continuous glucose monitors, CGMs), 3) implants for nerve/muscle enervation (STIMs, stimulation systems) for preventing muscle and/or nerve atrophy following injury, 4) implants for any pacing/pacemaker, for monitoring/regulating heart rhythm, 5) implants for hip/knee replacement, 6) implants for tissue repair/reconstruction, 7) implants for prosthesis and neural interfacing, 8) implants for controlled drug release, 9) implants for vital sign monitoring, 10) implants for intraocular lens replacement, 11) implants for cell encapsulation and transplantation, and 12) implants for tissue engineering/regeneration.
[0105] In another embodiment of the present invention, the CSF1R inhibitor compounds disclosed herein (for example, GW2580) can be incorporated into compositions for coating an implantable medical device, such as prostheses, artificial valves, vascular grafts, stents, or catheters. Suitable coatings and the general preparation of coated implantable devices are known in the art. The coatings are typically biocompatible polymeric materials such as a hydrogel polymer, polymethyldisiloxane, polycaprolactone, polyethylene glycol, polylactic acid, ethylene vinyl acetate, and mixtures of any of the foregoing. The coatings can optionally be further covered by a suitable topcoat of fluorosilicone, polysaccharides, polyethylene glycol, phospholipids or combinations of any of the foregoing to impart controlled release characteristics in the composition.
[0106] Another embodiment of the present invention is an implantable medical device having a coating comprising a CSF1R inhibitor. In a specific aspect, the CSF1R inhibitor is GW2580. In another specific aspect, the CSF1R inhibitor (for example, CSF1R) and an effective amount of an additional agent (selected from the ones set forth below) are comprised in one or more coatings of the implantable medical device.
[0107] According to another embodiment, the invention provides a method of impregnating an implantable medical device comprising the step of contacting the device with a CSF1R inhibitor disclosed herein or composition of this invention. Implantable drug release devices include, but are not limited to, biodegradable polymer capsules or bullets, non-degradable, diffusible polymer capsules and biodegradable polymer wafers. In a specific aspect, the CSF1R inhibitor is GW2580.
[0108] Another embodiment of the present invention, is a CSF1R inhibitor, for use in preventing or reducing a fibrotic response to an implanted synthetic material in a patient. In a specific aspect, the CSF1R inhibitor is GW2580.
[0109] Yet another embodiment is the use of a CSF1R inhibitor for the manufacture of a medicament for preventing or reducing a fibrotic response to an implanted synthetic material in a patient. In a specific aspect, the CSF1R inhibitor is GW2580.
[0110] In some embodiments, the CSF1R inhibitor can be administered in combination with one or more additional agents, e.g., additional therapeutic agents, either simultaneously or sequentially. In some embodiments, the CSF1R inhibitor can be administered with one or more additional agents in a composition, e.g., a pharmaceutical composition. For example, the additional agent can be administered in an effective amount, using the routes of administration described herein.
[0111] In some embodiments, the additional agent is an anti-inflammatory, e.g., a broad-spectrum anti-inflammatory (e.g., rapamycin/sirolimus, tacrolimus, everolimus, cyclosporine, corticosteroids, other NSAIDS, etc.) for preventing (with one or more CSF1R inhibitors) fibrosis either due to implant or damaged tissues (i.e., due to cirrhosis of the liver, fatty liver disease, torn muscle and wear and tear, following surgery procedures, etc.), as well as, by the other anti-inflammatories, preventing rejection, mediated by other immune cells, of tissue/organ or implant-incorporated biologics (for repairing, supplementing, replacing, or regenerating disease/condition-affected tissues).
[0112] In some embodiments, the additional agent is an immunomodulatory agent (i.e., immune checkpoint inhibitors such as PD 1, PDL1, CLTA4 antibodies or others) for preventing suppressive immune cells (ie., T regulatory suppressor cells) from inhibiting the anti-tumor actions of other immune populations (ie., cytotoxic T, macrophage, or other cells) in addition to using CSF1R inhibitors for reducing fibrosis/scarring following cytotoxic cancer drug treatments, which can also damage normal tissues. CSF1R inhibitors also have the added benefit of influencing macrophage phenotype (moving them from pro-tumor to anti-tumor function/behaviors) for cancer therapy. In some embodiments, the CSF1R agents would also inhibit fibrosis from damage caused by dysfunctional immune cells just prior to or during treatment. In some embodiments, coadministration with other immunodulatory agents can change osteoblast/osteoclast function to prevent and treat osteoporosis (bone loss).
[0113] In some embodiments, the additional agent is an anti-angiogenic agent (ie., anti-VEGFR1, 2, or 3 monoclonal antibodies or small molecular tyrosine kinase inhibitors), cytotoxic (chemotherapeutics), or anti-proliferative (ie. Paclitaxel) agents to simultaneously treat cancer while also preventing tissue damage-induced fibrosis (by the CSF1R agents).
[0114] In some embodiments, the additional agent is an antibiotic, e.g., to prevent infection following implant/transplant surgeries, while also blocking fibrosis.
[0115] A further embodiment is a method of preventing or reducing a fibrotic response to an implanted synthetic material in a patient, the method comprising administering to the patient an effective amount of a CSF1R inhibitor and an effective amount of one or more additional agents set forth above. In a specific aspect, the CSF1R inhibitor is GW2580 and the additional agent is an anti-inflammatory agent, an immunomodulatory agent, an anti-angiogenic agent, or an antibiotic. In a specific aspect, the CSF1R inhibitor (e.g., GW2580) and an additional agent are comprised in a coating of an implanted medical device.
[0116] A further embodiment of the present invention is a coating formulation, comprising a CSF1R inhibitor (e.g., GW2580) and optionally one or more additional agents selected from an anti-inflammatory agent, an immunomodulatory agent, an anti-angiogenic agent, and an antibiotic.
[0117] A further embodiment of the present invention is a coating of an implantable medical device, comprising a CSF1R inhibitor (e.g., GW2580) and optionally one or more additional agents selected from an anti-inflammatory agent, an immunomodulatory agent, an anti-angiogenic agent, and an antibiotic.
[0118] In some embodiments of the present invention, a CSF1R inhibitor is a compound of structural formula
##STR00056##
a compound represented by structural formula
##STR00057##
4-(3,4-Dimethylanilino)-7-(4-(methylsulfonyl)phenyl)quinoline-3-carboxami- de, (4-cyano-N-(2-cyclohexenyl-4-(1-(2-(dimethylamino)acetyl)piperidin-4-y- l)phenyl)-1H-imidazole-2-carboxamide), a compound represented by structural formula
##STR00058##
ARRY-382, a compound represented by structural formula
##STR00059##
a 2'-aminoanilide, a 3-amido-4-anilinocinniline, an indoline-2-one, a 2-(alpha-methylbenzylamino)-pyrazine, an arylamide, a 3,4,6-substituted 2-quinolone, a pyrido[2,3-d]pyrimidin-5-one, a 3-amido-4-anilinoquinoline, a pyridyl bisamide, a thiazolyl bisamide, a 1,4-disubstituted-pyrrolo-[3,2-c]-pyridine, a substituted diphenylurea, a 5'-pyrimidine-2,4-diamine, a compound represented by structural formula
##STR00060##
a compound represented by structural formula
##STR00061##
anilinoquinazoline, a compound represented by structural formula
##STR00062##
a compound represented by structural formula
##STR00063##
a compound represented by structural formula
##STR00064##
DCC-3014, a compound represented by structural formula
##STR00065##
a compound represented by structural formula
##STR00066##
a compound represented by structural formula
##STR00067##
a compound represented by structural formula
##STR00068##
a compound represented by structural formula
##STR00069##
a compound represented by structural formula
##STR00070##
a compound represented by structural formula
##STR00071##
a compound represented by structural formula
##STR00072##
or AC708.
EXEMPLIFICATION
Methods and Materials
[0119] In brief, all materials were implanted intraperitoneally or subcutaneously into and retrieved at specified times from C57BL/6 (wild type, knockout, or serially immune depleted/perturbed) mice, or non-human primate cynomolgus macaques in accordance with approved protocols and federal guidelines. Sample processing, staining, FACS, NanoString expression analysis, and imaging were performed as detailed below. Shown are representative images in all cases from n=5 mice per treatment group. Quantified data shown are group mean values.+-.SEM.
[0120] a. Materials/Reagents
[0121] All in vitro reagents were obtained from Life Technologies (Carlsbad, Calif.), unless otherwise noted. Antibodies: Alexa Fluor-conjugated anti-mouse CD68, Ly-6G/Ly-6C (Gr-1), CD11b, CD19, and IgM (described below) were purchased from BioLegend Inc. (San Diego, Calif.). For primate immunostaining, anti-human CD68 Alexa Fluor-conjugated antibody was purchased from Santa Cruz (Dallas, Tex.). The same CD11b (anti-mouse/human) antibody (BioLegend) was used for both primate and mouse staining. Cy3-conjugated anti-mouse alpha smooth muscle actin antibody and glass spheres (acid washed) of medium (500 .mu.m) size were purchased from Sigma Aldrich (St. Louis, Mo.). Polystyrene spheres of medium (400-500 .mu.m) size were purchased from Phosphorex (Hopkinton, Mass.). A sampling of materials used in this study were submitted for endotoxin testing by a commercial vendor (Charles River, Wilmington, Mass.) and the results showed that spheres contained <0.05 EU/ml of endotoxin levels (below detectable limits) (Table 1). Table 1. Negative endotoxin and glucan results for all materials used in this study. As determined by Charles River Labs sample submission, as well as in-house testing, for bacterial pyrogen and endotoxin. Specifically, E. coli and Limulus Amebocyte Lysates were used as positive controls to test for the presence of general endotoxin. BDL=below detectable limits. These negative results have also been corroborated by others in our group, having now been published in multiple studies (Veiseh O, Doloff J C, Ma M, Vegas A J, Tam H H, Bader A R, et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature materials 2015, 14(6): 643-651; Jhunjhunwala S, Aresta-DaSilva S, Tang K, Alvarez D, Webber M J, Tang B C, et al. Neutrophil Responses to Sterile Implant Materials. PloS one 2015, 10(9): e0137550; Vegas A J, Veiseh O, Doloff J C, Ma M, Tam H H, Bratlie K, et al. Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nat Biotechnol 2016).
TABLE-US-00001 Sample Endotoxin Test Glucan Test Saline control <0.05 EU/mL (BDL) <10 .rho.g/ml (BDL) SLG20 alginate <0.05 EU/mL (BDL) <10 .rho.g/ml (BDL) 500 .mu.m spheres SLG20 alginate <0.05 EU/mL (BDL) <10 .rho.g/ml (BDL) solution Glass spheres <0.05 EU/mL (BDL) <10 .rho.g/ml (BDL) Polystyrene <0.05 EU/mL (BDL) <10 .rho.g/ml (BDL) spheres
[0122] b. Fabrication of Alginate Hydrogel Spheres
[0123] Alginate hydrogel spheres were made with an in-house customized electro-jetting system: voltage generator, vertical syringe pump (Harvard Apparatus), and a gelation bath basin. Voltage was coupled to the syringe needle dispensing the alginate and grounded to the gelling bath vessel. Spheres were made with a 1.4% solution of commercially available sterile alginate (PRONOVA SLG20, NovaMatrix, Sandvika, Norway) dissolved in 0.9% saline (pH.apprxeq.7.4, Osmotic pressure.apprxeq.290 mOsm), and crosslinked with 250 mL of sterile BaCl.sub.2 gelling solution (20 mM BaCl.sub.2, 250 mM D-Mannitol, 25 mM HEPES, pH.apprxeq.7.4, Osmotic pressure.apprxeq.290 mOsm) (see Morch Y A, Donati I, Strand B L, Skjak-Braek G. Effect of Ca.sup.2+, Ba.sup.2+, and Sr.sup.2+ on alginate microbeads. Biomacromolecules 2006, 7(5): 1471-1480.)
[0124] Alginate hydrogel 500 .mu.m diameter microspheres were generated with a 25G blunt needle, a voltage of 5 kV and a 200 .mu.l/min flow rate. Immediately after gelation, alginate spheres were washed with HEPES buffer (25 mM HEPES, 1.2 mM MgCl.sub.2.times.6H2O, 4.7 mM KCl, 132 mM NaCl.sub.2, pH.apprxeq.7.4, .apprxeq.290 mOsm) 4 times and stored overnight at 4.degree. C. Immediately prior to implantation, spheres were washed an additional 2 times with 0.9% saline. A sampling of the fabricated hydrogels was submitted for endotoxin testing by a commercial vendor (Charles River, Wilmington, Mass.) and the results showed that SLG20 hydrogels contained <0.05 EU/ml of endotoxin levels (below detectable limits).
[0125] c. Implantation Surgeries
[0126] All protocols were approved by the MIT Committee on Animal Care, and all surgical procedures and post-operative care were supervised by MIT Division of Comparative Medicine veterinary staff. All mice, wild type male immune-competent (non-diabetic), STZ-induced diabetic, as well as knockout C57BL/6 mice, were ordered pathogen-free from Jackson Laboratory (Bar Harbor, Me.) or Taconic (Hudson, N.Y.). The status of all mice was subsequently verified by testing sentinel animals, shown to be negative for at least 12 known mouse pathogens. Implanted mice were anesthetized with 3% isoflurane in oxygen and had their abdomens shaved and sterilized using betadine and isopropanol. C57BL/6-Nude (T KO, B6NU), C57BL/6-Rag2.sup.null (T & B KO, RAGN12), C57BL/6-Rag2.sup.null/Il2r.gamma..sup.null (Rag2/.gamma., 4111) were ordered from Taconic (Hudson, N.Y.), and C57BL/6-IghM.sup.null (BKO, 002288), (C57BL/6) B6.12956-Ccr6tm1(EGFP)Irw/J. (Ccr6, 013061), and C57BL/6-C3 KO (C3 KO, 003641) mice were ordered from Jackson Laboratory, Bar Harbor, Me.). Preoperatively, all mice received 0.05 mg/kg buprenorphine and 0.2 mL of 0.9% saline subcutaneously for pre-surgery analgesia and dehydration prevention. A midline (abdomen) incision (0.5 mm) was made and the peritoneal lining was exposed using blunt dissection. The peritoneal wall was then grasped with forceps and a 0.5-1 mm incision was made along the linea alba. A desired volume of spheres (all materials) were then loaded into a sterile pipette and implanted into the peritoneal cavity. The incision was then closed using 5-0 taper-tipped polydioxanone (PDS II) absorbable sutures, and the skin was closed using a wound clip and VetBond tissue glue. For subcutaneous implantation, .about.200-300 .mu.L of 500 .mu.m SLG20 spheres were injected s.c. following anesthesia with isofluorane. All primate implant samples were derived by excising fibrosed, material-containing omentum and subcutaneous tissues 28 days following implantation of SLG20 500 .mu.m diameter spheres in cynomolgus macaques, as described (see Veiseh O, Doloff J C, Ma M, Vegas A J, Tam H H, Bader A R, et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature materials 2015, 14(6): 643-651).
[0127] For targeted macrophage depletions, clodrosome (200 .mu.L/mouse) (Encapsula Nano Sciences, Nashville, Tenn.) were injected intraperitoneally starting at -3 days (prior to implantation, day 0), and for every 7 days thereafter (so days -3, 4, and 11). To achieve neutrophil depletion, affinity purified anti-Ly6g (Clone 1A8) antibody (250 ug/mouse) (BioLegend, San Diego, Calif.) was administered also starting at day -3, and every 3 days thereafter (except the day of implantation). For the combination therapy, in order to avoid drug-antibody interactions, each agent was administered 4 hours apart from one another. To neutralize secreted CXCL13, anti-CXCL13 antibody (mAB470, R&D Systems) was injected i.p. in sterile 1.times.PBS at a dose of 100 .mu.g/mouse, once every 3 days, starting 3 days prior to implantation as well. For selective macrophage inhibition and polarization, the CSF1R-targeted inhibitor GW2580 (LC Labs, Woburn, Mass.), was dissolved in a 1:1 DMSO/PEG400 solution, and injected daily (starting 1 day prior to implantation) at a dose of 160 mg/kg BW subcutaneously (s.c.), to eliminate concerns of vehicle directly perturbing responding immune cells as well as a recovering implant site.
[0128] d. IVIS Imaging
[0129] C57BL/6 mice (6-8 weeks old) were utilized for this assay. 200-300 .mu.l of alginate spheres were resuspended in saline, and injected subcutaneously into the mouse on both left and right sides of upper back. The mice were shaved to get rid of the hair on the entire back before injection, and fed on sterilized AIN-93G purified rodent diet (TD 94045, Harlan) to minimize the fluorescent background after injection. Six days later, 100 .mu.l (4 nmol) of ProSence 750 FAST (NEV11171, PerkinElmer Inc.) per mouse was injected intravenously via tail vein. At day 7 (24 hours post the ProSense 750 FAST intravenous administration), the mice were scanned by IVIS Spectrum system (Xenogen, Caliper LifeScience). The mice were anesthetized using 3% isofluorane in oxygen and maintained at the same rate throughout the procedure, the new grown hair were removed by Nair hair removal lotion, and then the mice were scanned by the IVIS Spectrum system at the settings of Exposure=7.50, Binning=Medium, FStop=2, Excitation=605-640 nm and Emission=660-760 nm. The images were analyzed with LivingImage Software, and the duplicated ROI of lower back on the same mouse was used as control during the signal quantification.
[0130] e. Retrieval of Cells, Tissues, and Materials
[0131] At desired time points post-implantation or transplantation (with encapsulated islets), as specified in figures, mice were euthanized by CO.sub.2 administration, followed by cervical dislocation. In certain instances, 5 ml of ice cold PBS was first injected in order perform an intraperitoneal lavage to rinse out and collect free-floating intraperitoneal immune cells. An incision was then made using forceps and scissors along the abdomen, and intraperitoneal lavage volumes were pipetted out into fresh 15 ml falcon tubes (each prepared with 5 ml of RPMI cell culture media). Next, a wash bottle tip was inserted into the abdominal cavity. KREBS buffer was then used to wash out all material spheres into petri dishes for collection. After ensuring all the spheres were washed out or manually retrieved (if fibrosed directly to intraperitoneal tissues, in particular epididymal and omental fat pads), they were transferred into 50 mL conical tubes for downstream processing and imaging. In certain instances, after intraperitoneal lavage, portions of fibrosed intraperitoneal tissues and material spheres were also excised for downstream FACS and expression analyses.
[0132] f. Imaging of the Retrieved Material Spheres
[0133] For phase contrast imaging retrieved materials were gently washed using Krebs buffer and transferred into 35 mm petri dishes for phase contrast microscopy using an Evos X1 microscope (Advanced Microscopy Group). For bright-field imaging of retrieved materials, samples were gently washed using Krebs buffer and transferred into 35 mm petri dishes for bright-field imaging using a Leica Stereoscopic microscope.
[0134] g. Confocal Immunofluorescence
[0135] Immunofluorescence imaging was used to determine immune populations attached to spheres. Materials were retrieved from mice and fixed overnight using 4% paraformaldehyde at 4.degree. C. Samples were then washed twice with KREBS buffer, permeabilized for 30 min using a 0.1% Triton X100 solution, and subsequently blocked for 1 hour using a 1% bovine serum albumin (BSA) solution. Next, the spheres were incubated for 1 hour in an immunostaining cocktail solution consisting of DAPI (500 nM), specific marker probes (1:200 dilution) in BSA. After staining, spheres were washed three times with a 0.1% Tween 20 solution and maintained in a 50% glycerol solution. Spheres were then transferred to glass bottom dishes and imaged using an LSM 700 point scanning confocal microscope (Carl Zeiss Microscopy, Jena Germany) equipped with 5 and 10.times. objectives. Obtained images were adjusted linearly for presentation using Photoshop (Adobe Inc. Seattle, Wash.). For antigen-specific immunostaining, non-human primate (cynomolgus macaque) intraperitoneal sphere-embedded omenta were sectioned and stained according to traditional antigen retrieval and immunofluorescent methods, specifically looking at cellular nuclei (DAPI), macrophage marker CD68-AF488 (Santa Cruz, Dallas, Tex.) and Cy3-conjugated anti-mouse alpha smooth muscle actin (fibrosis) (Sigma Aldrich, St. Louis, Mo.).
[0136] h. Histological Processing for H&E and Masson's Trichrome Staining
[0137] Retrieved material-containing tissue (omentum and/or subcutaneous) was fixed overnight using 4% paraformaldehyde at 4.degree. C. After fixation, alginate sphere or retrieved tissue samples were washed using 70% alcohol. The materials were then paraffin embedded, sectioned and stained according to either standard histological (H&E or Masson's Trichrome) or antigen-specific methods (as described above).
[0138] i. Western Blotting
[0139] Protein was extracted directly from materials for western blot analysis. Retrieved materials were prepared by lysis of covering cellular overgrowth in Pierce RIPA buffer (Cat. #89901, Thermo Scientific) with protease inhibitors (Halt Protease inhibitor single-use cocktail, Cat. #78430, Thermo Scientific) on ice, and then lysed by sonication (for 30 seconds on, 30 seconds off, twice at 70% amplitude). Samples were then agitated constantly for 2 hours at 4.degree. C. Lysates were centrifuged for 20 min at 12,000 rpm at 4.degree. C., and protein-containing supernatants were collected in fresh tubes, on ice. In samples from fatty tissue, an excess of fat (a top layer on the supernatant) was removed before supernatant transfer. 20 .mu.g protein (quantified by BCA assay, Pierce BCA protein assay kit, Cat. #23225, Thermo Scientific) for each lane was boiled at 95.degree. C. for 5 min and electrophoresed on SDS-polyacrylamide gels (Any kD 15-well comb mini-gel, BioRad, Cat. #456-9036) and then blotted onto nitrocellulose membranes (BioRad, Cat. #162-0213). Blots were probed with anti-alpha Smooth Muscle actin antibody (1:400 dilution, Rabbit polyclonal to alpha smooth muscle actin; Cat. #ab5694, AbCam) and anti-.beta.-actin antibody (1:4000 dilution, monoclonal anti-.beta.-actin antibody produced in mouse; Cat. #A1978, Sigma Aldrich) as a loading control followed by donkey anti-rabbit (1:15,000 dilution, Cat. #926-32213, Li-Cor) and goat anti-mouse (1:15,000 dilution, Cat. #926-68070, Li-Cor) fluor-conjugated secondary antibodies. Bands were visualized using an Odyssey detector (Li-Cor, Serial No. ODY-2329) at 700 and 800 nm wavelengths. For CXCL13 detection, a rabbit anti-CXCL13/BCA1 polyclonal antibody (Bioss, Woburn, Mass.) was used.
[0140] j. qPCR Analysis
[0141] Total RNA was isolated from tissue (peripheral epididymal and omental adipose tissue alone for mock controls, or fibrosed spheres with adhered adipose tissue and immune overgrowth, if present), liquid nitrogen snap-frozen immediately following excision, using TRIzol (Invitrogen, Carlsbad, Calif.) according to the manufacturer's instructions. In addition, to help ensure complete tissue disruption, we also employed strong mechanical disruption with a Polytron homogenizer. Thus, gene expression signatures shown throughout are proportional and representative of the entire cell population present on and/or around retrieved materials. Before reverse transcription using the High Capacity cDNA Reverse Transcription kit (Cat. #4368814; Applied Biosystems, Foster City, Calif.), all samples were first normalized for comparison by loading the same input 1 .mu.g total RNA in a volume of 20 .mu.l for each sample. cDNA (4.8 .mu.l; 1:20 dilution) in a total volume of 16 .mu.l (including SYBR Green and PCR primers) was amplified by qPCR with the following primers. Primers (see Table 2 below) were designed using Primer Express software (Applied Biosystems, Carlsbad, Calif., USA) and evaluated using LaserGene software (DNAStar, Madison, Wis., USA) to ensure either mouse (host)-specificity. Samples were incubated at 95.degree. C. for 10 min followed by 40 cycles of 95.degree. C. for 15 sec and 60.degree. C. for 1 min in an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). Results were analyzed using the comparative C.sub.T (DDC.sub.T) method as described by the manufacturer. Results were analyzed using the comparative C.sub.T (.DELTA..DELTA.C.sub.T) method and are presented as relative RNA levels, as compared to control cell samples as specified in figure legends after normalization to the .beta.-actin RNA content of each sample. To further ensure proper normalization and sample handling across multiple retrieval time points, RNA for all samples for each harvest condition (ie., ip lavage, spheres with or without adhered cells and fibrosis, and peripheral tissues with infiltration, as described), were quantified, reverse transcribed, and analyzed by qPCR in parallel.
TABLE-US-00002 TABLE 2 Mouse (m)-specific (host) forward and reverse oligonucleotide primer sets used for qPCR analysis of RNA levels. Gene names are also shown in parentheses. Primers (5' to 3'): Gene Sequence Sense & Antisense Mouse Collagen 1 Forward: 1a1 (mCol1a1) 5'-CATGTTCAGCTTTGTGG ACCT-3' 2 Reverse: 5'-GCAGCTGACTTCAGGGA TGT-3' Mouse Collagen 3 Forward: 1a2 (mCol1a2) 5'-GCAGGTTCACCTACTCT GTCCT-3' 4 Reverse: 5'-CTTGCCCCATTCATTTG TCT-3' Mouse Alpha 5 Forward: Smooth Muscle 5'-CGCTTCCGCTGCCCAGA actin (mActa2) GACT-3' 6 Reverse: 5'-TATAGGTGGTTTCGTGG ATGCCCGCT-3' Mouse 7 Forward: Inflammation 5'-CCTGAGTGGCTGTCTTT marker TGAC-3' Transforming 8 Reverse: Growth Factor 5'-ACAAGAGCAGTGAGCGC beta1 (mTGFb1) TGAAT-3' Mouse 9 Forward: Macrophage 5'-GATACAGCAATGCCAAG marker F4/80 CAGT-3' (mEmr1) 10 Reverse: 5'-TTGTGAAGGTAGCATTC ACAAGTGTA-3' Mouse 11 Forward: Macrophage 5'-GCCCGAGTACAGTCTAC marker CD68 CTGG-3' (mCd68) 12 Reverse: 5'-AGAGATGAATTCTGCGC CAT-3' Mouse Myeloid 13 Forward: cell marker 5'-CCAAGAGAATGCAAAAG CD11b GCTTT-3' (mItgam) 14 Reverse: 5'-GGGGGGCTGCAACAACC ACA-3' Mouse 15 Forward: neutrophil 5'-TGCCCCTTCTCTGATGG marker Gr1 ATT-3' (mLy6g) 16 Reverse: 5'-TGCTCTTGACTTTGCTT CTGTGA-3' Mouse B cell 17 Forward: marker CD19 5'-GGAAACCTGACCATCGA (mCd19) GAG-3' 18 Reverse: 5'-TGGGACTATCCATCCAC CAGTT-3' Mouse 19 Forward: Dendritic cell 5'-CCCAGGACCATGTGATG (DC) marker CAT-3' CD74 (mCd74) 20 Reverse: 5'-CTTAAGATGCTTCAGAT TCTCT-3' Mouse 21 Forward: Langerhans 5'-GGACTACAGAACAGCTT Dendritic cell GGAGAATG-3' (DC) marker 22 Reverse: CD207 (Langerin) 5'-TACTTCCAGCCTCGAGC (mCd207) CAC-3' Mouse Natural 23 Forward: killer (NK) cell 5'-GCAACCCCCTGAAACTG marker NKp46 GTA-3' (mNcr1) 24 Reverse: 5'-AAGGTTACCTCAGGCTG TGGATA-3' Mouse 25 Forward: Adaptive helper 5'-GAAGATTCTGGGGCAGC T cell marker ATGGCAAAG-3' CD4 (mCd4) 26 Reverse: 5'-TTTGGAATCAAAACGAT CAA-3' Mouse 27 Forward: Cytotoxic T cell 5'-CTGCGTGGCCCTTCTGC marker CD8a TGTCCT-3' (mCd8a) 28 Reverse: 5'-GGGACATTTGCAAACAC GCT-3' Mouse 29 Forward: regulatory T 5'-GCCTTCAGACGAGACTT suppressor cell GGAA-3' marker FoxP3 30 Reverse: (mFoxP3) 5'-CTGGCCTAGGGTTGGGC ATT-3' Mouse .beta.-actin 31 Forward: (mActB) 5'-GCTTCTTTGCAGCTCCT TCGTT-3' 32 Reverse: 5'-CGGAGCCGTTGTCGACG ACC-3'
[0142] k. Luminex Multiplexed Cytokine and ELISA Analyses
[0143] Cytokine array analysis was performed using the Luminex Bio-Rad Bio-Plex Pro mouse cytokine panel based on Luminex beads (multiplexing both the 23 and 8-cytokine arrays) (Bio-Rad, Hercules, Calif.), according to manufacturer's instructions. 1:4 diluted mouse sera samples were analyzed for the time course, and protein lysates taken from fibrosed tissue and alginate microspheres, loaded at a concentration of 500 .mu.g/mL (200 uL per sample), were run to compare cytokine responses across mock (saline) treated versus wildtype, knockout or serially perturbed/depleted C57BL/6 mice implanted with 500 .mu.m SLG20 alginate spheres. Samples were added to the panel of beads bearing capture antibodies for the analytes of interest, and agitated for 30 minutes. Sample plates were subsequently washed using an automated magnetic plate washer (Bio-Rad), and a biotinylated detection antibody for each of the 32 cytokines was added followed by agitation for 30 minutes. After another wash, streptavidin-PE was added, the plate was agitated for 10 minutes, washed, and resuspended in assay buffer. The plate was then read on a BioPlex-200 plate reader, with absolute concentrations obtained through fitting a standard curve. For Luminex runs, protein lysates were prepared using an NP-40-based lysis buffer, as opposed to the SDS-based RIPA buffer used for western blotting (described above) to maintain native state protein folding. For mouse CXCL10 (IP-10) and IgM analyses, an ELISA from eBioscience (San Diego, Calif.) was used, according to manufacturer's specifications, with protein lysates for CXCL10 and both sera and protein lysates for IgM.
[0144] l. FACS Analysis
[0145] Single-cell suspensions of freshly excised tissues were prepared using a gentleMACS Dissociator (Miltenyi Biotec, Auburn, Calif.) according to the manufacturer's protocol. Single-cell suspensions were prepared in a passive PEB dissociation buffer (1.times.PBS, pH 7.2, 0.5% BSA, and 2 mM EDTA) and suspensions were passed through 70 .mu.m filters (Cat. #22363548, Fisher Scientific, Pittsburgh, Pa.). This process removed the majority of cells adhered to the surface (>90%) (See Supplemental FIG. 17 from Veiseh O, Doloff J C, Ma M, Vegas A J, Tam H H, Bader A R, et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature materials 2015, 14(6): 643-651). All tissue and material sample-derived, single-cell populations were then subjected to red blood cell lysis with 5 ml of 1.times.RBC lysis buffer (Cat. #00-4333, eBioscience, San Diego, Calif., USA) for 5 min at 4.degree. C. The reaction was terminated by the addition of 20 ml of sterile 1.times.PBS. The cells remaining were centrifuged at 300-400g at 4.degree. C. and resuspended in a minimal volume (.about.50 .mu.l) of eBioscience Staining Buffer (cat. #00-4222) for antibody incubation. All samples were then co-stained in the dark for 25 min at 4.degree. C. with two of the fluorescently tagged monoclonal antibodies specific for the cell markers CD68 (1 .mu.l (0.5 .mu.g) per sample; CD68-Alexa647, Clone FA-11, Cat. #11-5931, BioLegend), Ly-6G (Gr-1) (1 .mu.l (0.5 .mu.g) per sample; Ly-6G-Alexa-647, Clone RB6-8C5, Cat. #108418, BioLegend), CD11b (1 .mu.l (0.2 .mu.g) per sample; or CD11b-Alexa-488, Clone M1/70, Cat. #101217, BioLegend). For alpha smooth muscle actin (fibrosis) analysis, additional cell aliquots were also fixed in 1% paraformaldehyde and permeabilized with 0.1% triton X-100 before being stained with Cy3-conjugated anti-mouse .alpha.SM actin antibody (1:100) (Sigma Aldrich, St. Louis, Mo.). Two ml of eBioscience Flow Cytometry Staining Buffer (cat. #00-4222, eBioscience) was then added, and the samples were centrifuged at 400-500g for 5 min at 4.degree. C. Supernatants were removed by aspiration, and this wash step was repeated two more times with staining buffer. Following the third wash, each sample was resuspended in 500 .mu.l of Flow Cytometry Staining Buffer and run through a 40 .mu.m filter (Cat. #22363547, Fisher Scientific) for eventual FACS analysis using a BD FACSCalibur (cat. #342975), BD Biosciences, San Jose, Calif., USA). For proper background and laser intensity settings, unstained, single antibody, and IgG (labeled with either Alexa-488 or Alexa-647, BioLegend) controls were also run.
[0146] m. Intravital Imaging--Ccr6-EGFP Mice
[0147] For intravital imaging, 500 .mu.m SLG20 hydrogel spheres were loaded with Qdot 605 (Life technologies, Grand Island, N.Y.) and surgically implanted into (C57BL/6) B6.129S6-Ccr6tm1(EGFP)Irw/J (Ccr6, 013061, Jackson Laboratory, Bar Harbor, Me.) mice, as described above. After 14 days post implantation, the mice were placed under isoflurane anesthesia throughout and a small incision was made at the site of the original surgery to expose beads. The mice were placed on an inverted microscope and imaged using a 25.times., N.A. 1.05 objective on an Olympus FVB-1000 MP multiphoton microscope at an excitation wavelength of 860 nm. While Ccr6 can show EGFP in adaptive T and B cells as well as innate immune dendritic cells (DCs), only B cells respond in our alginate material model.
[0148] n. NanoString Analysis
[0149] RNAs for mock-implanted (saline) treated controls, or for 500 .mu.m alginate sphere-bearing C57BL/6 mouse strains (n=4/group), including wild type, knockouts, and serially immune depleted wild type mice, were isolated from tissue samples taken at various time points after implantation, as described. For each of the knockout strains, they were normalized to their own (strain-specific) mock (saline) implanted controls, in order to eliminate any artifacts due to shifts in immune homeostasis, since gene knockouts were also present throughout development. Furthermore, to corroborate macrophage-specific changes, macrophages were also sorted from implanted wild type C57BL/6 mice, and also plotted relative to expression levels from the entire mock tissue cell population. Thus, macrophage-specific genes show enriched (red, increased) expression fold changes, and macrophage-independent gene show diluted (green, decreased) expression fold changes. In general, respective RNAs were quantified, normalized to the appropriate loading concentration (100 ng/.mu.l), and then 500 ng of each sample was processed according to NanoString manufacturer protocols for expression analysis via our customized multiplexed total (known) mouse cytokine and cytokine receptor expression panel, used for both immune strain comparisons with alginate, and material class comparisons just the wild type C57BL/6 strain. Another non-human primate custom panel was also used to corroborate gene hits in our cynomolgus macaque intraperitoneal and subcutaneous models. RNA levels (absolute copy numbers) were obtained following nCounter (NanoString Technologies Inc., Seattle, Wash.) quantification, and group samples were analyzed using nSolver analysis software (NanoString Technologies Inc., Seattle, Wash.).
[0150] o. Statistical Analysis
[0151] Data are expressed as mean.+-.SEM, and N=5 mice per time point and per treatment group. These sample sizes were chosen based on previous literature. All animals were included in analyses except in instances of unforeseen sickness or morbidity. Animal cohorts were randomly selected. Investigators were not blind to performed experiments. For qPCR, western blot quantification, IVIS imaging, or FACS, data were analyzed for statistical significance either by unpaired, two-tailed t-test, or one-way ANOVA with Bonferroni multiple comparison correction, unless indicated otherwise, as implemented in GraphPad Prism 5; *: p<0.05, **: p<0.001, and ***: p<0.0001. For Nanostring, data was normalized using the geometric means of the NanoString positive controls and background levels were established using the means of the negative controls. Housekeeping genes Tubb5, Hprt1, Bact, and Cltc were used to normalize between samples. Data was then log-transformed
Example 1: Implanted Biomaterial Alginate Elicits a Multi-Immune Population Host Rejection Response
[0152] Host immune-mediated foreign body rejection of commercially purified biomaterial alginate, implanted as 500 .mu.m spheres, is complex, engaging both innate and adaptive immune cell populations (FIGS. 1A-G). Historically, responses were thought to be a function of contaminating endotoxins (Paredes-Juarez G A, de Haan B J, Faas M M, de Vos P. The role of pathogen-associated molecular patterns in inflammatory responses against alginate based microcapsules. J Control Release 2013, 172(3): 983-992.). However, the clinical grade alginates used here do not contain detectable endotoxins (Veiseh O, Doloff J C, Ma M, Vegas A J, Tam H H, Bader A R, et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature materials 2015, 14(6): 643-651; Vegas A J, Veiseh O, Doloff J C, Ma M, Tam H H, Bratlie K, et al. Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nat Biotechnol 2016; Jhunjhunwala S, Aresta-DaSilva S, Tang K, Alvarez D, Webber M J, Tang B C, et al. Neutrophil Responses to Sterile Implant Materials. PloS one 2015, 10(9): e0137550.). For thoroughness, both the alginate solution, prior to use, as well as spheres post-gelation were again tested and verified to be endotoxin-free (See Table 1 above).
[0153] Cellular adhesion and fibrotic overgrowth of alginate microspheres implanted in the intraperitoneal space of C57/B6 mice after 14 days is apparent as a white plaque by phase contrast imaging (FIG. 1A), with individual cells resolvable by nuclear DAPI staining (FIG. 1B). Specific markers for innate immune cell macrophages (F4/80 and CD68) and neutrophils (Ly6g), adaptive immune B cells (CD19), as well as fibrosis (alpha smooth muscle actin and collagen 1a1) were identified by qPCR, and increased over time on alginate spheres, as compared to non-implanted and mock (saline injected) controls (FIG. 1C and FIG. 2). Immunofluorescent staining and confocal imaging of spheres retrieved 14 days post-intraperitoneal (IP) implantation reveals that all three of these responding immune populations (macrophages: CD68; neutrophils, Ly6g/Gr1; B cells, CD19) reside within the fibrotic plaque directly enveloping implanted alginate spheres (FIGS. 1D-E). In addition, intravital imaging at 14 days post-implant in Ccr6-EGFP transgenic C57BL/6 mice, verified trafficking of adaptive B cells onto implanted quantum-dot labeled (pink) alginate spheres (FIGS. 1F-G).
Example 2: Local Immune Responses to Alginate are Complex and Long-Lived
[0154] To determine whether the host immune response to implanted biomaterial alginate was acute and short-lived or manifested over a longer period of time, we performed FACS on cells obtained by both peritoneal lavage and dissociation directly from the surface of 500 alginate spheres and associated peripheral fibrosed tissue retrieved at a range of time points (1, 4, 7, 14, and 28) days following IP implantation into C57BL/6 mice (FIGS. 3A-C). In general, peritoneal exudate cell number reflected an increase in neutrophils (as % total), B cells decreased and macrophages were unchanged (FIG. 3A). However, these observable responses in the peritoneal exudate do not depict earlier identified cell population increases directly on the implanted material surface (FIGS. 1A-G and FIG. 2).
[0155] Luminex analysis of serum levels of cytokines following implantation showed an acute and transient increase of 4 out of 32 cytokines (FIGS. 4A-E), suggesting an early, perhaps surgery-related, event that goes away within .about.1-3 days post-implantation. This is in stark contrast to the previous mention of longer-term evidence of immunogenicity or host reactivity to implanted alginate spheres. Such long-term responses, however, do not appear to be discernable by looking at global, blood-circulating cytokines, which have likely become too dilute. When FACS was performed on dissociated fibrosed IP tissue (repeatedly observed to be only non-collagen-encapsulated epididymal and omental fat pads) immediately adjacent to and often stuck to the fibrosed capsules (FIG. 5), as well as specifically cells taken directly from the surface of implanted alginate spheres, immune cells increased over a 28-day period, with macrophages the major responding cell population (FIGS. 3B-C). FACS analysis of alginate-associated cells on day 1 show the presence of immature CD68.sup.-Gr1.sup.lo/-CD11b.sup.+ monocytes, known for being early responders patrolling out from the blood (Shi C, Pamer E G. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 2011, 11(11): 762-774.). On days 4 and 7, this immature population disappeared as mature CD68.sup.+CD11b.sup.+ macrophages increased in number (FIG. 3C, purple vs red). Macrophage subtyping carried out on surface-dissociated cells at 1, 4, and 7 days also suggest this change is due to early-stage monocyte recruitment and differentiation (Veiseh O, Doloff J C, Ma M, Vegas A J, Tam H H, Bader A R, et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature materials 2015, 14(6): 643-651.). Later stage (day 7 and beyond) recruitment of both adaptive immune CD19.sup.+IgM.sup.+ B cells and fibrosis-associated alpha smooth muscle actin (.alpha.SMactin)-positive myofibroblasts was observed (FIG. 3C, white). Multiplexed NanoString gene expression analysis showed longer-term (up to 4 weeks) increases in macrophage and neutrophil-associated markers (FIGS. 3D-E). Tissue immediately adjacent to alginate capsules (FIG. 5, epididymal and omental fat pads) as well as cellular material taken directly from the surface of implanted spheres were both analyzed over a 28-day time course to ascertain localization of various macrophage and inflammation markers (FIGS. 6A-B). By contrast, qPCR-determined expression of B cell marker CD19 showed a delayed (day 7 and later) mobilization out of peripheral fibrosed epididymal and omental tissue and onto alginate spheres (FIG. 3F), indicating later adaptive B cell recruitment not just to the site but also directly onto implanted alginate. Lastly, expression analysis of fibrosis marker .alpha.SMactin was masked in peripheral tissue due to very high background in IP fat pads (also the case in subcutaneous tissue). However, we were able to ascertain an appropriate kinetic, with a significant, delayed expansion at and beyond day 7, when fibrotic material was instead taken directly from implanted spheres (FIG. 3G).
Example 3: Adaptive as Well as Innate Immune Cells are Involved in the Downstream Fibrotic Cascade
[0156] To better understand the cellular requirements driving the fibrotic response, we examined fibrosis to implanted 500 .mu.m alginate spheres retrieved after 14 days, in a range of C57BL/6-derived immune mutant rodents. A total of 7 strains were studied, ranging from fully immune competent wild type versus knockout (KO) strains to strains with varying levels of immunodeficiency (FIG. 7). Phase contrast imaging was used to show relative levels of fibrotic overgrowth across mice with varying immune deficiency after 14 days (FIG. 8A and FIGS. 9A-F). Implicating importance in the fibrotic cascade, loss of B cells alone (IghM.sup.null, B KO) resulted in a partial loss of fibrosis (FIG. 8A, and FIGS. 9A-F, and FIGS. 10A-C). Additional T cell loss (Rag2.sup.null, T & B KO) made fibrosis worse and more comparable to wild type (WT) levels (FIG. 8A and FIG. 9E), perhaps due to the loss of the regulatory T cell subset important for suppressing overreaching immune reactions (Wood K J, Bushell A, Hester J. Regulatory immune cells in transplantation. Nat Rev Immunol 2012, 12(6): 417-430.). Ultimately, only with innate immune cell macrophage dysfunction in Rag2.sup.null/Il2r.gamma..sup.null (Rag2/.gamma.KO) was a complete loss of fibrosis observed (FIG. 8A and FIG. 9D). Individual T cell (T KO, nude) and complement C3 knockouts did not result in the loss of fibrosis (FIGS. 9E-F).
[0157] Western blot assays were performed on extracted proteins from retrieved spheres in all groups exhibiting significant decreases in fibrosis (B KO and Rag2/.gamma. KO), as compared to wild type controls, to quantify changes in the fibrosis marker .alpha.SMactin (FIG. 8B-C). Expression results were further corroborated using qPCR analysis of RNA isolated from retrieved spheres, where .alpha.SMactin and Collagens 1a1 and 1a2 showed similar significant decreases across both knockout models, as compared to WT (FIG. 8D).
[0158] In addition to study of spheres retrieved from the peritoneal space, which is therapeutically relevant for cell encapsulation and transplantation (Jacobs-Tulleneers-Thevissen D, Chintinne M, Ling Z, Gillard P, Schoonjans L, Delvaux G, et al. Sustained function of alginate-encapsulated human islet cell implants in the peritoneal cavity of mice leading to a pilot study in a type 1 diabetic patient. Diabetologia 2013, 56(7): 1605-1614; Weir G C. Islet encapsulation: advances and obstacles. Diabetologia 2013, 56(7): 1458-1461; Veiseh O, Doloff J C, Ma M, Vegas A J, Tam H H, Bader A R, et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature materials 2015, 14(6): 643-651), alginate spheres were also implanted into the subcutaneous compartment in the same wild type and knockout strains (FIGS. 8E-F). Immune cell activity at the site of implantation was analyzed using Prosense, a fluorescent indicator of secreted immune inflammation cathepsins (Bratlie K M, Dang T T, Lyle S, Nahrendorf M, Weissleder R, Langer R, et al. Rapid biocompatibility analysis of materials via in vivo fluorescence imaging of mouse models. PloS one 2010, 5(4): e10032.). IVIS imaging at 7 days post-implant showed increasingly significant decreases in alginate-induced inflammation across both knock-out models (FIG. 8E). Following a 1-month implantation, subcutaneous spheres were excised and processed for histology (H&E and Masson's Trichrome). In general, spheres were completely embedded and individually sequestered in WT mice, but only had an outer, thin fibrotic capsule in B KO mice, while showing no significant deposition in macrophage dysfunctional (Rag2/.gamma. KO) mice (FIG. 8F and FIG. 11A-D). FACS analysis of retrieved capsules and surrounding tissue verified that wildtype subcutaneous immune responses are also localized to the implant (FIG. 12A-C) and similar in composition to those in the IP space (FIG. 8G). Lastly, FACS analysis corroborated earlier findings of decreased immunity and fibrosis rejection across both knock out strains, with an .about.60% decrease in macrophage and neutrophil presence on spheres taken from B KO mice, and an essentially complete loss of adherent cells in macrophage dysfunctional (Rag2/.gamma. KO) mice (FIG. 8G). Loss of macrophage presence resulted in complete B cell loss as well, suggesting that macrophages may be required for B cell recruitment to the capsules.
Example 4: Macrophages, not Neutrophils, are Necessary for Fibrosis of Alginate Microcapsules
[0159] Both macrophage and neutrophil populations were present on the surface of alginate spheres even 1 day following alginate sphere implantation into wild type C57BL/6 mice (FIG. 3C). Our observation that microcapsules implanted into the Rag2/.gamma. KO model being fibrosis-free indicates that macrophages are necessary for fibrosis (FIG. 8A-G). However, Rag2/.gamma. KO rodents still have macrophages, although they are reported to be reduced in number and dysfunctional (Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 2002, 100(9): 3175-3182) (FIG. 7). Since the Rag2/.gamma. model also lacks adaptive T and B cells, innate immune natural killer cells, as well as dendritic cell function, other unknown consequences affecting immune signaling and homeostasis may be possible. It has also recently been reported that neutrophils, and then macrophages, are recruited as first and second line responders in an initiated fibrotic cascade (Grainger D W. All charged up about implanted biomaterials. Nat Biotechnol 2013, 31(6): 507-509.). To further clarify the role of macrophages and neutrophils in fibrosis we induced depletion of macrophages, neutrophils, or both together by clodronate liposome (clodrosome) and/or targeted Ly6g (clone 1A8)-antibody depletion, in implanted wild type C57BL/6 mice (FIG. 13A and FIG. 14A-D). Depletion was started 3 days prior to implantation in all cases in order to ensure immune population ablation prior to alginate microcapsule exposure. The extent and specificity of all targeted depletions was verified by FACS (FIGS. 13B-C, FIGS. 15A-C, and FIGS. 16A-C). Interestingly, fibrosis was only eliminated with targeted macrophage depletion, both alone and in combination with neutrophil removal (FIG. 13A and FIGS. 14A-D). Neutrophil depletion caused implanted alginate spheres to clump more aggressively, an effect seen with exacerbated immunity in the case of smaller diameter spheres (Veiseh O, Doloff J C, Ma M, Vegas A J, Tam H H, Bader A R, et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature materials 2015, 14(6): 643-651) (FIG. 13A and FIG. 14A-D). This maybe a consequence of the loss of the Ly6g-granulocyte myeloid derived suppressor cell (MDSC) subset, which like T.sub.regs, prevent excessive immune reactions (Wood K J, Bushell A, Hester J. Regulatory immune cells in transplantation. Nat Rev Immunol 2012, 12(6): 417-430.).
[0160] Lastly, in order to elucidate immune cell signaling, RNA samples derived from all day 14 treatment groups (fibrosed capsules+epididymal/omental fat): mock vs. alginate-implanted wild type, and knockouts or depletions) were analyzed in parallel with a multiplexed NanoString probe set for all known mouse cytokines and cytokine receptors (FIGS. 13D-F and FIG. 17). Numerous genes, induced in alginate-implanted over mock (saline injected), after a 14-day period in wild type C57BL/6 mice (2.sup.nd versus 1.sup.st column), were eliminated upon macrophage depletion, both alone or in combination with neutrophil depletion (3.sup.rd and 5.sup.th columns). Many of these hits were then corroborated by expression enrichment (increased in red) or dilution (either decreasing back to mock levels, black, or below, green) upon sorting and analyzing just macrophages from extracted samples, with comparison to levels in the original mixed tissue population in mock implanted mice (2.sup.nd to last vs. 1.sup.st column). Macrophage-specific gene subsets were identified reflecting macrophage-specific factors, concomitantly lost upon macrophage depletion and increased in expression upon analysis of macrophages isolated and enriched by cell sorting (FIGS. 13D-E, partial). In addition, genes lost upon macrophage depletion, and not enriched, but instead reduced upon macrophage sorting, were identified (FIG. 13E, partial, & FIG. 13F). This subset is associated with cells (ie., B cell marker CD19) and recruitment events downstream of macrophages in the fibrotic cascade. Furthermore, since expression of this second gene category is lost upon macrophage depletion; they are therefore dependent on and only occur downstream of successful macrophage activation and recruitment. Additional genes, not affected by any of the immune population depletions utilized in this study, were also identified, and are likely associated with upstream biomaterial-induced tissue damage and inflammation events prior to immune cell recruitment (FIG. 18A-D).
Example 5: CSF1R Inhibition Prevents Immune and Fibrotic Responses to Implanted Microcapsules
[0161] While elimination of macrophages by clodrosome treatment can prevent fibrosis of hydrogel alginate (FIG. 13A), it is unlikely this approach would be suitable for human use (Diel I J, Bergner R, Grotz K A. Adverse effects of bisphosphonates: current issues. The journal of supportive oncology 2007, 5(10): 475-482.). An ideal anti-fibrotic drug therapy would only modulate and not eliminate this immune population, thus avoiding unnecessary immune suppression and resulting side effects. One macrophage-specific factor identified by our NanoString analysis was the cytokine receptor CSF1R (FIG. 13D). This receptor had been previously reported to play a role in selectively polarizing and modulating macrophage phenotypes in cancer (Pyonteck S M, Akkari L, Schuhmacher A J, Bowman R L, Sevenich L, Quail D F, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 2013, 19(10): 1264-1272.). To test whether inhibition of this receptor could prevent macrophage-dependent biomaterial fibrosis, we examined the potential the inhibitory small molecule GW2580 to inhibit CSF1R (Conway J G, McDonald B, Parham J, Keith B, Rusnak D W, Shaw E, et al. Inhibition of colony-stimulating-factor-1 signaling in vivo with the orally bioavailable cFMS kinase inhibitor GW2580. Proceedings of the National Academy of Sciences of the U.S. Pat. No. 2,005,102(44): 16078-16083) and the fibrotic response to implanted microspheres. Treatment of C57BL/6 mice with GW2580 prevented the fibrotic response to a range of implanted microcapsules, including 500 .mu.m alginate spheres, and 500 .mu.m glass and polystyrene spheres, all implanted for 14 days into the intraperitoneal space (FIGS. 19A-D, FIGS. 20A-B, FIGS. 21A-C, and FIGS. 22A-C). CSF1R blockade by GW2580 was as effective as clodrosome-based macrophage depletion at inhibiting host immune attack to alginate, glass and polystyrene spheres (FIG. 13C vs. FIG. 19B, and FIG. 19D). Uninhibited fibrotic responses against all three materials importantly share the same core immune marker, cytokine and cytokine receptor signaling, including CSF1R (FIG. 19E and FIG. 23). This response was dependent upon the implanted material, and not endotoxin, as analysis did not detect the presence of general pyrogens, endotoxins or glucan (See Table 1 above).
[0162] RNA samples derived from alginate-implanted C57BL/6 mice also treated with the CSF1R small molecule inhibitor GW2580, were also analyzed and their gene expression patterns were integrated into the cytokine genetic profiling array. This was done to determine which of the earlier identified macrophage-specific and (non-associated) inflammation genes would or would not be affected upon CSF1R blockade (FIG. 13D vs. FIG. 13E, and FIG. 18A vs. FIG. 18B). Specifically, we inspected all macrophage-associating genes that were both significantly increased 2 weeks following implantation into the intraperitoneal space (WT vs Mock controls), as well as eliminated or significantly decreased following macrophage depletion. Interestingly, the presence of many macrophage-specific factors not affected by CSF1R blockade (FIG. 13E) suggests possible spared macrophage function, not observed with more blunt macrophage depletion and removal. This theory is supported by the presence of a large non-immune-associated (due to the lack of removal following any of the earlier serial depletions) and, as such, likely upstream inflammation-associated gene family that was decreased back to mock implanted (saline) control levels upon CSF1R inhibition by GW2580 treatment (FIG. 18B). Many of these factors are known stress/inflammation response genes that are decreased or turned off as tissue is repaired due to spared residual macrophage repair function, as a consequence of their polarization upon CSF1R inhibition (Pyonteck S M, Akkari L, Schuhmacher A J, Bowman R L, Sevenich L, Quail D F, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 2013, 19(10): 1264-1272.). Further supporting the hypothesis of preserved macrophage tissue repair function (MacDonald K P, Palmer J S, Cronau S, Seppanen E, Olver S, Raffelt N C, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood 2010, 116(19): 3955-3963), tissue/cell stress chemokine CXCL10 levels were increased significantly in inflamed intaperitoneal epididymal and omental fat pad tissue immediately adjacent to implanted alginate spheres in both macrophage depletion treatment groups, instead of being decreased back to low levels, as normally seen in both non-depleted as well as CSF1R inhibited B6 mice (FIG. 18C). VEGF protein production, directly associated with macrophage repair function (Laskin D L, Sunil V R, Gardner C R, Laskin J D. Macrophages and tissue injury: agents of defense or destruction? Annual review of pharmacology and toxicology 2011, 51: 267-288), was significantly diminished in both macrophage depletion groups, but spared to normal levels with GW2580 treatment (FIG. 18D).
Example 6: Innate Immune Macrophages Recruit Fibrosis-Potentiating Adaptive B Cells Via Chemokine CXCL13
[0163] The chemokine CXCL13 has been shown to be expressed by monocytes/macrophages and responsible for B cell recruitment in a model of lymphoid neogenesis (Carlsen H S, Baekkevold E S, Morton H C, Haraldsen G, Brandtzaeg P. Monocyte-like and mature macrophages produce CXCL13 (B cell-attracting chemokine 1) in inflammatory lesions with lymphoid neogenesis. Blood 2004, 104(10): 3021-3027.). Interestingly, CXCL13 was also identified by profiling the kinetics of host immune adhesion and fibrotic sequestration (FIG. 3D and FIG. 6C), as well as macrophage depletion, sorting and cytokine NanoString characterization (FIG. 13D). CXCL13 was one of numerous immune markers and cytokines that either disappeared or were decreased in expression below background levels following macrophage depletion. Conversely, CXCL13 was also enriched upon sorting the macrophage population away from the bulk heterogeneous host cell infiltrate. To determine whether it plays a role in B cell recruitment in the observed biomaterial-induced fibrotic cascade, neutralizing antibody was administered to alginate-implanted wild type C57BL/6 mice. CXCL13 neutralization resulted in loss of B cell recruitment and reduced fibrosis (FIG. 19A, FIG. 19F, and FIGS. 24A-D), similar to levels observed with B cell loss (FIG. 19G and FIG. 8, B KO). These findings suggest a role for B cells in potentiating fibrosis, perhaps due to their ability to regulate macrophage phenotype and response (Affara N I, Ruffell B, Medler T R, Gunderson A J, Johansson M, Bornstein S, et al. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer cell 2014, 25(6): 809-821.). Interestingly, B cell loss was also observed with both clodrosome macrophage depletion and CSF1R inhibition (FIG. 13B, FIG. 13C, FIG. 13F, and FIG. 19B), once again suggesting that macrophages are responsible for their downstream recruitment. NanoString and western blot analysis for CXCL13 over 1, 4, 7, 14, and 28-day alginate implantation showed initial expression at both the RNA and protein levels by day 4 onward (FIG. 3D and FIG. 6C), correlating with maturation of macrophages from immature monocytes (FIG. 3C), and just preceding apparent day 7 B cell recruitment (FIG. 3B and FIG. 3G). Taken together, these results demonstrate that macrophages are required and responsible for downstream B cell recruitment via the B cell chemoattractant CXCL13, and that, upon arrival, these B cells enhance fibrotic deposition. Also highlighting the translational significance of these findings, CXCL13 was recently shown in human patients to also be a biomarker for idiopathic pulmonary fibrosis (Vuga L J, Tedrow J R, Pandit K V, Tan J, Kass D J, Xue J, et al. C-X-C motif chemokine 13 (CXCL13) is a prognostic biomarker of idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine 2014, 189(8): 966-974.).
Example 7: Immune Factors Identified in Response to Implanted Alginate in C57BL/6 Mice are Also Preserved in Non-Human Primates
[0164] Next, with potential implications for clinical translation, we wanted to study if these findings were also relevant in higher order non-human primates (NHPs). To evaluate this, we implanted 500 .mu.m diameter SLG20 alginate spheres, either intraperitoneally (N=2) or subcutaneously into the dorsal regions (N=4) of cynomolgus monkeys for 28 days. At 28 days post-implantation the 500 .mu.m SLG20 alginate spheres were heavily embedded in host omental or subcutaneous tissues upon minimally invasive laparoscopic surgery or biopsy punch retrieval (FIG. 25A and Ref.: Veiseh O, Doloff J C, Ma M, Vegas A J, Tam H H, Bader A R, et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature materials 2015, 14(6): 643-651.). Excised tissue obtained from the IP implanted 500 .mu.m SLG20 alginate spheres or mock control tissue were examined through histological analysis with H&E and Masson's Trichrome staining (FIG. 25B and FIGS. 26A-B). Extensive embedding and fibrosis buildup (up to 100 .mu.m thick), similar to that seen in C57BL/6 mice, is visible enveloping the implanted spheres. Confocal imaging of sections taken from retrieved 28-day IP omentum tissue confirmed extensive macrophage cellular deposition and fibrosis-associated activated myofibroblast coverage (FIG. 25C and FIGS. 26C-D). Immune responses (macrophage and myeloid/neutrophil) were determined by FACS to be similar between C57BL/6 mice and NHP cynomolgus monkeys (FIG. 25D). Lastly, to confirm that the immune factors identified in the C57BL/6 model are also relevant for both intraperitoneal as well as subcutaneous alginate implants isolated previously (Veiseh O, Doloff J C, Ma M, Vegas A J, Tam H H, Bader A R, et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature materials 2015, 14(6): 643-651) from NHPs, NanoString analysis was performed on RNA derived from excised fibrotic alginate implants. Results showed increased expression of numerous factors common to the C57BL/6 model: cytokine receptors and cytokines, such as CSF1R and CXCL13, and immune markers, such as CD68 and CD66b (in place of Ly6g/Gr1, which does not exist in NHPs and humans) (FIG. 25E). Together, these findings support the use of the C57BL/6 mouse model to recapitulate the foreign body response seen in higher species such as non-human primate cynomolgus monkeys and human patients. This is relevant not only for choosing the right mouse strain for research but also confirming findings in a higher model organism before moving to human patients. Lastly, the fact that both models share similar immunologic responses, not just in content but also kinetics and magnitude, adds additional credence to their use for research validation before translation into the clinic.
Example 8: Macrophage Functional Testing
[0165] For in vivo wound healing: C57BL/6 mice were anesthetized (same as above) and shaved, and skin incisions 1.5 cm in length were made along the midline. Each incision was then wound clipped shut for both vehicle and daily GW2580-treated C57BL/6 mice. Wound clips were removed and then replaced each imaging day up until day 7 (required to maintain them up until at least this time), after which clips were left off completely. Photos were acquired on days 0, 1, 4, 7, 10, and 14 post-incision. After 14 days, mice were sacrificed, and skin samples were prepared for histological assessment (H&E and Masson's Trichrome) of both vehicle and GW2580 treatment groups. Cross-sections through the incision line were generated to assess the level of in vivo healing following a 14-day treatment regimen. For IP innate immune cell counts, FACS, and phagocytosis assays: cells were isolated by peritoneal lavage with 8 mL DPBS, diluted into RPMI, and then subsequently counted, and aliquoted either for staining and FACS analysis (as described above), or for phagocytosis assays. Peritoneal exudate macrophages isolated by IP lavage from (n=5) mice in each treatment group were immediately plated (400,000 cells/well in a 24-well plate) and incubated with fluorospheres for 90 minutes to determine phagocytic activity. 1 .mu.m orange fluorospheres (540/560) (Cat# F13082, Invitrogen, Carlsbad, Calif.) were added to each well in a volume of 500 .mu.L from an initial dilution of 20 .mu.L into 10 mL. After addition, plates were spun down at 1,000 rpm for 1 min, and then placed in an incubator for 90 minutes to allow IP macrophages time to phagocytose the 1 .mu.m diameter fluorospheres. Following the 90-minute incubation, all wells were fixed with 4% paraformaldehyde and stored at 4 C prior to imaging using a fluorescent and brightfield-capable EVOS microscope (Advanced Microscopy Group). For ROS activity assessment: protein lysates were prepared from alginate spheres retrieved 14 days after IP implantation into untreated, vehicle-treated, and daily GW2580-treated C57BL/6 mice. 50 .mu.g of each lysate were aliquoted twice and incubated for 30 min. at 37 C with two different reactive oxygen specie (ROS) substrate solutions: 10 .mu.M APF (Cat# A36003, Invitrogen, Carlsbad, Calif.) and 5 .mu.M CellROX (Cat# C10422, Invitrogen, Carlsbad, Calif.) diluted in PBS. Following appropriate incubation times, samples were read in a black-wall 96-well plate with a Tecan fluorescent-capable Infinite M1000 plate reader. Results of the macrophage functional testing are shown in FIGS. 27A-G.
SUMMARY
[0166] In summary, we have demonstrated that by inhibiting CSF1R, as opposed to more blunt macrophage depletion, can eliminate host immune-mediated recognition and propagation of foreign body rejection responses for a broad spectrum of materials encompassing hydrogels, ceramics, and plastics. We believe these findings have important implications for the integration of a highly macrophage-specific agent for localized delivery at the host-material interface of implanted biomedical devices, to prevent fibrosis and ensure long-term success for a vast range of biomedical device applications. Lastly, similar host rejection responses and immunobiology across C57BL/6 mice and cynomolgus monkeys implicates the clinical importance of these findings.
[0167] The teachings of all patents, published applications and references cited herein are incorporated by reference in their entirety.
[0168] While this invention has been particularly shown and described with references to example embodiments thereof, it will be understood by those skilled in the art that various changes in form and details may be made therein without departing from the scope of the invention encompassed by the appended claims.
Sequence CWU 1
1
32121DNAArtificial Sequenceoligonucleotide primer 1catgttcagc
tttgtggacc t 21220DNAArtificial Sequenceoligonucleotide primer
2gcagctgact tcagggatgt 20322DNAArtificial Sequenceoligonucleotide
primer 3gcaggttcac ctactctgtc ct 22420DNAArtificial
Sequenceoligonucleotide primer 4cttgccccat tcatttgtct
20521DNAArtificial Sequenceoligonucleotide primer 5cgcttccgct
gcccagagac t 21626DNAArtificial Sequenceoligonucleotide primer
6tataggtggt ttcgtggatg cccgct 26721DNAArtificial
Sequenceoligonucleotide primer 7cctgagtggc tgtcttttga c
21822DNAArtificial Sequenceoligonucleotide primer 8acaagagcag
tgagcgctga at 22921DNAArtificial Sequenceoligonucleotide primer
9gatacagcaa tgccaagcag t 211026DNAArtificial
Sequenceoligonucleotide primer 10ttgtgaaggt agcattcaca agtgta
261121DNAArtificial Sequenceoligonucleotide primer 11gcccgagtac
agtctacctg g 211220DNAArtificial Sequenceoligonucleotide primer
12agagatgaat tctgcgccat 201322DNAArtificial Sequenceoligonucleotide
primer 13ccaagagaat gcaaaaggct tt 221420DNAArtificial
Sequenceoligonucleotide primer 14ggggggctgc aacaaccaca
201520DNAArtificial Sequenceoligonucleotide primer 15tgccccttct
ctgatggatt 201623DNAArtificial Sequenceoligonucleotide primer
16tgctcttgac tttgcttctg tga 231720DNAArtificial
Sequenceoligonucleotide primer 17ggaaacctga ccatcgagag
201822DNAArtificial Sequenceoligonucleotide primer 18tgggactatc
catccaccag tt 221920DNAArtificial Sequenceoligonucleotide primer
19cccaggacca tgtgatgcat 202022DNAArtificial Sequenceoligonucleotide
primer 20cttaagatgc ttcagattct ct 222125DNAArtificial
Sequenceoligonucleotide primer 21ggactacaga acagcttgga gaatg
252220DNAArtificial Sequenceoligonucleotide primer 22tacttccagc
ctcgagccac 202320DNAArtificial Sequenceoligonucleotide primer
23gcaaccccct gaaactggta 202423DNAArtificial Sequenceoligonucleotide
primer 24aaggttacct caggctgtgg ata 232526DNAArtificial
Sequenceoligonucleotide primer 25gaagattctg gggcagcatg gcaaag
262620DNAArtificial Sequenceoligonucleotide primer 26tttggaatca
aaacgatcaa 202723DNAArtificial Sequenceoligonucleotide primer
27ctgcgtggcc cttctgctgt cct 232820DNAArtificial
Sequenceoligonucleotide primer 28gggacatttg caaacacgct
202921DNAArtificial Sequenceoligonucleotide primer 29gccttcagac
gagacttgga a 213020DNAArtificial Sequenceoligonucleotide primer
30ctggcctagg gttgggcatt 203122DNAArtificial Sequenceoligonucleotide
primer 31gcttctttgc agctccttcg tt 223220DNAArtificial
Sequenceoligonucleotide primer 32cggagccgtt gtcgacgacc 20
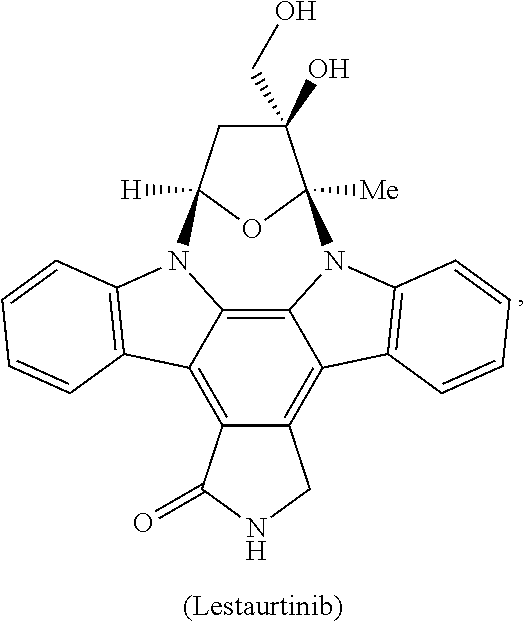


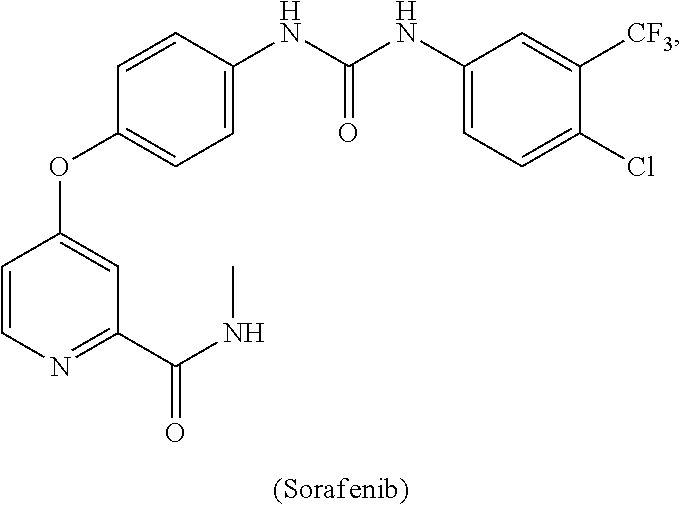






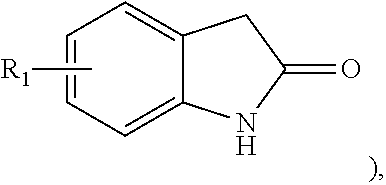


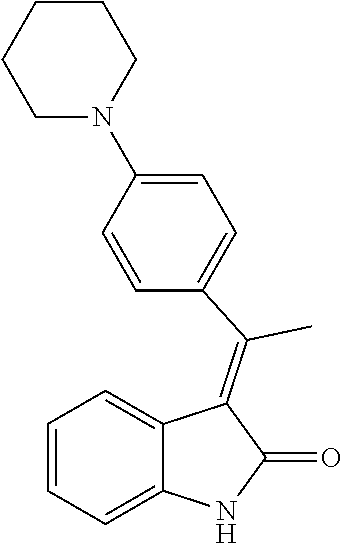
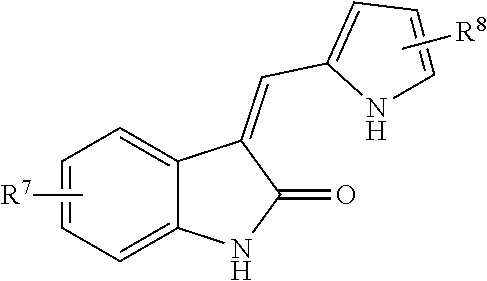






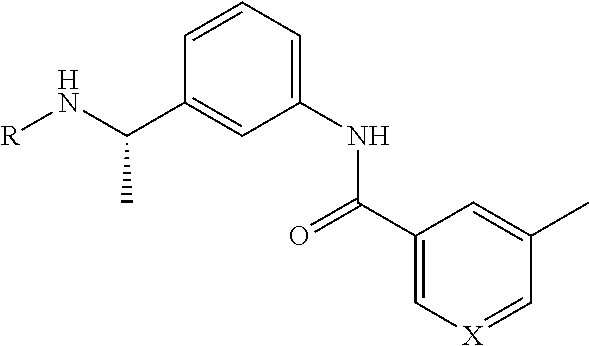
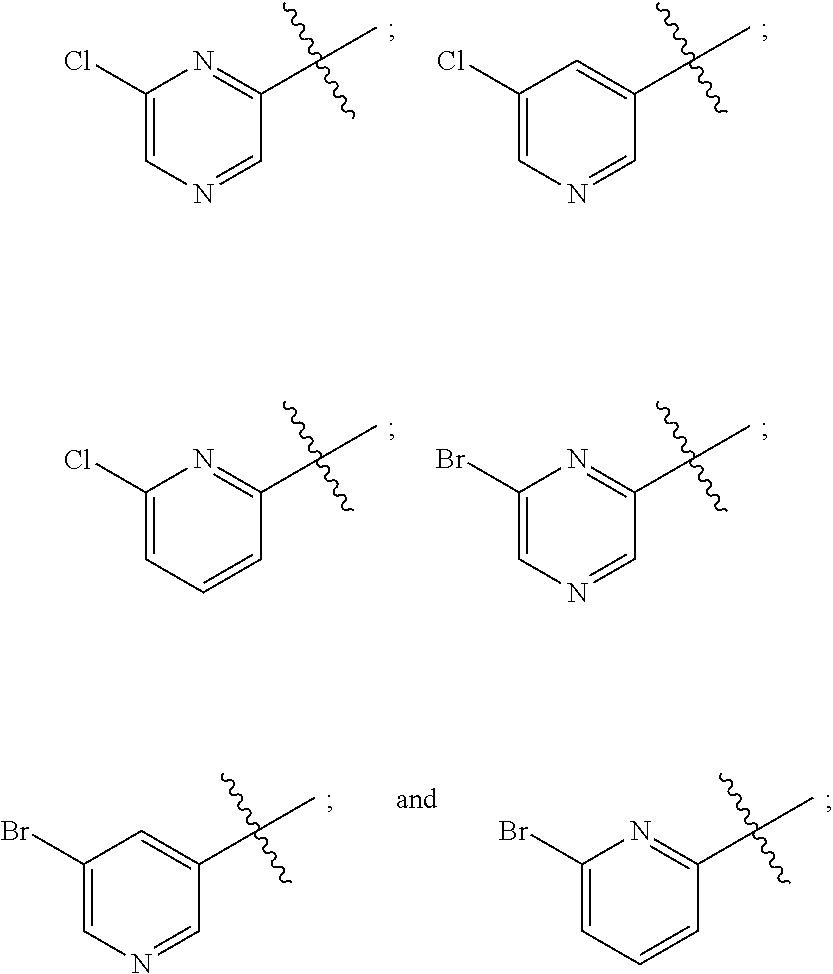

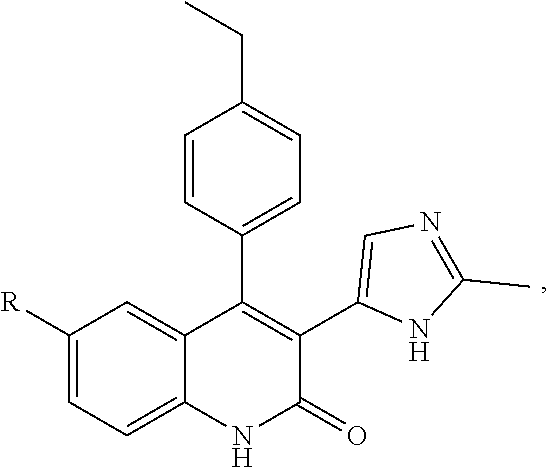


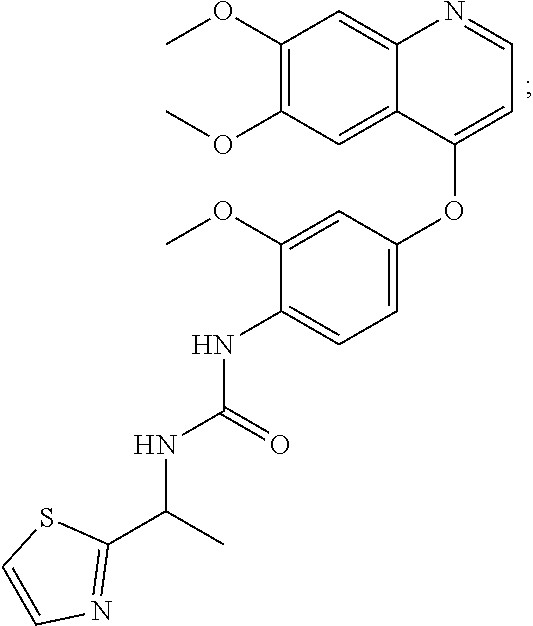
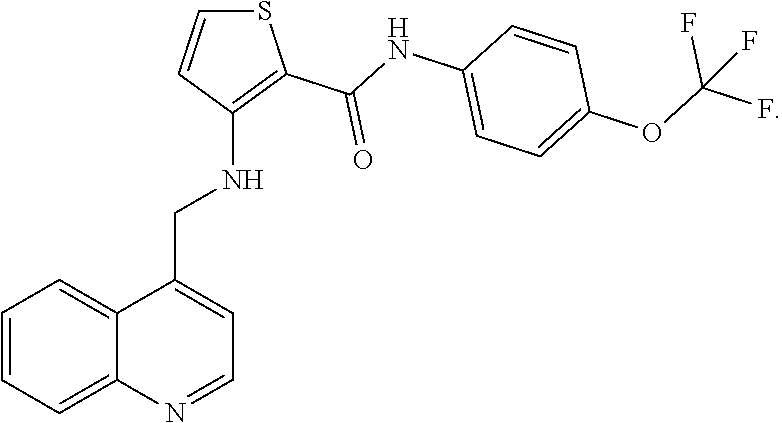





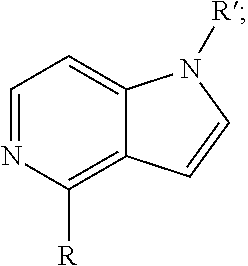


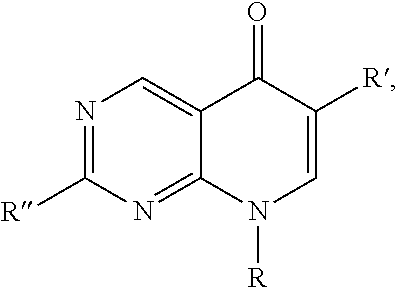

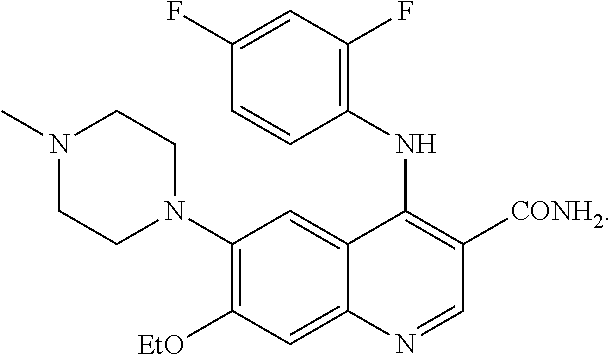
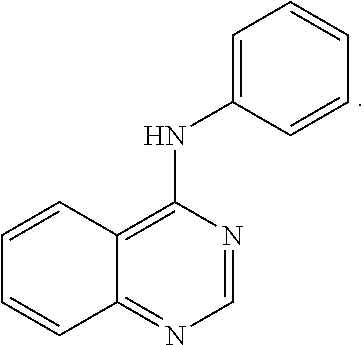
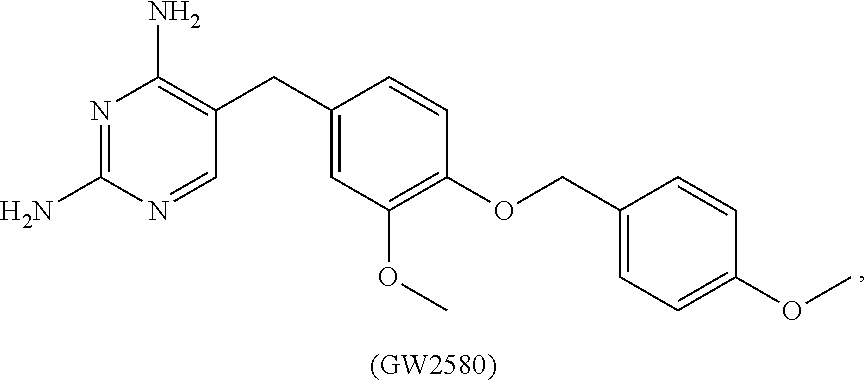

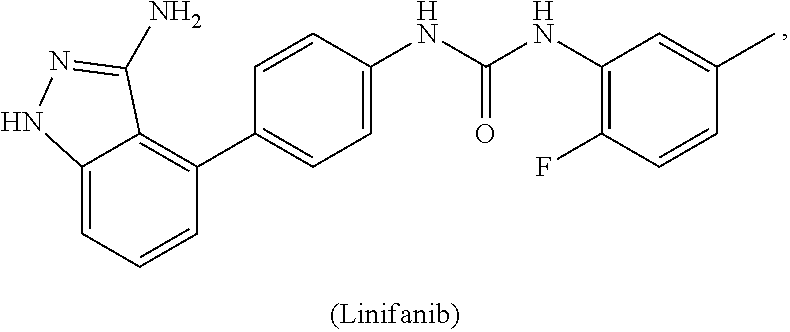
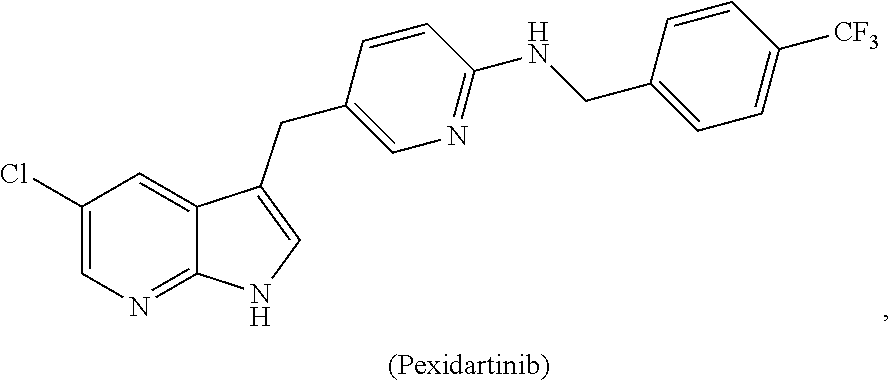
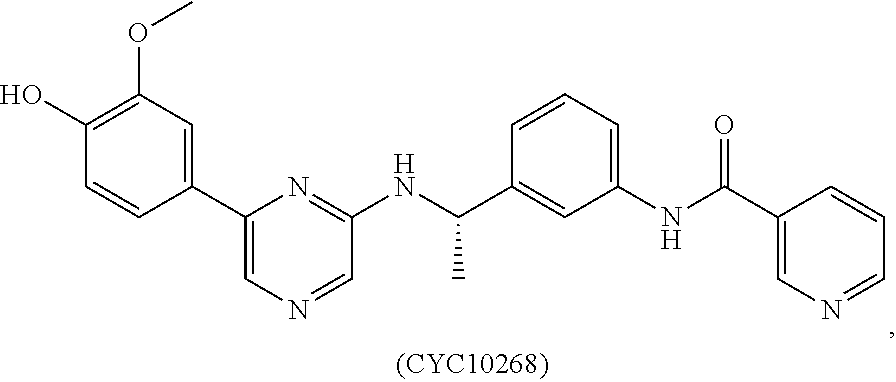
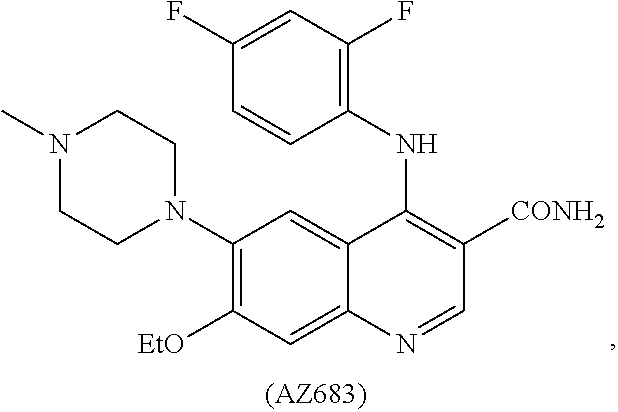
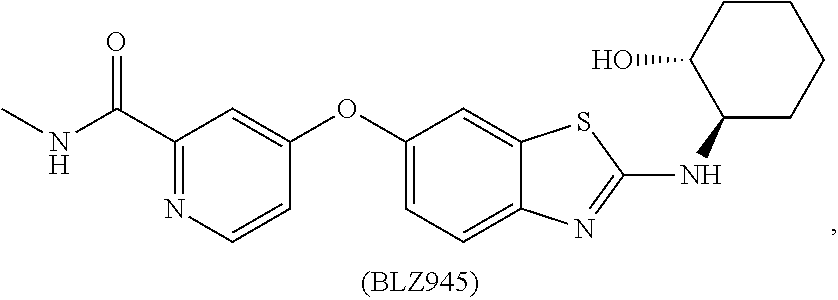






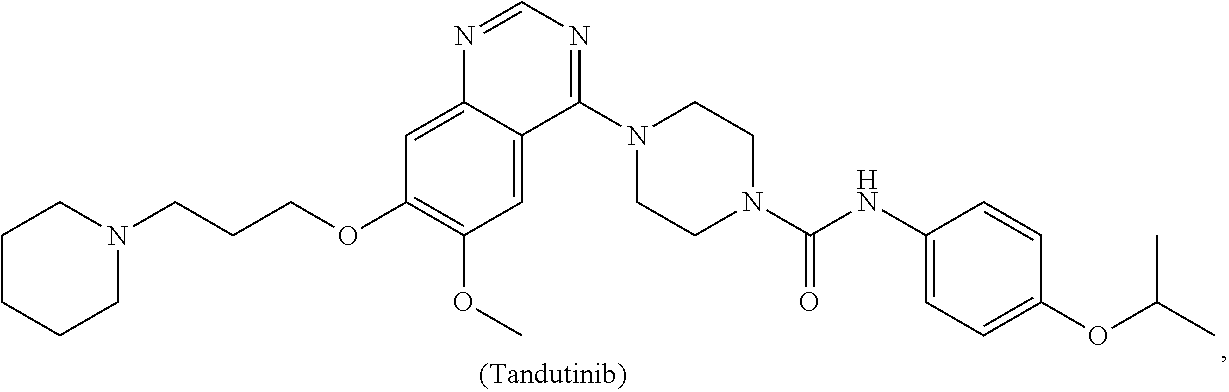
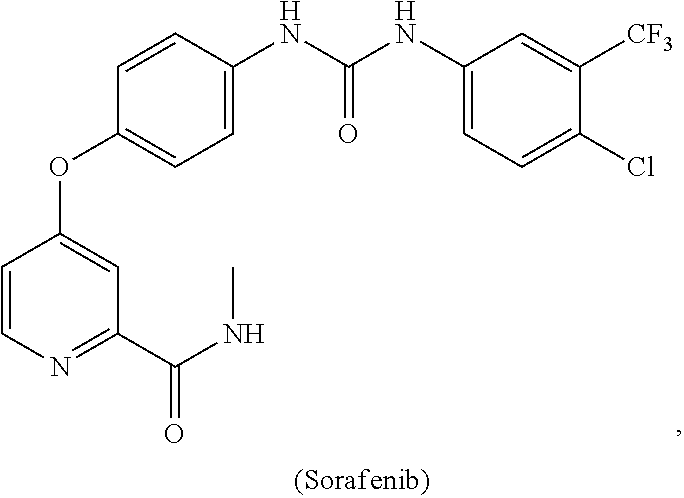




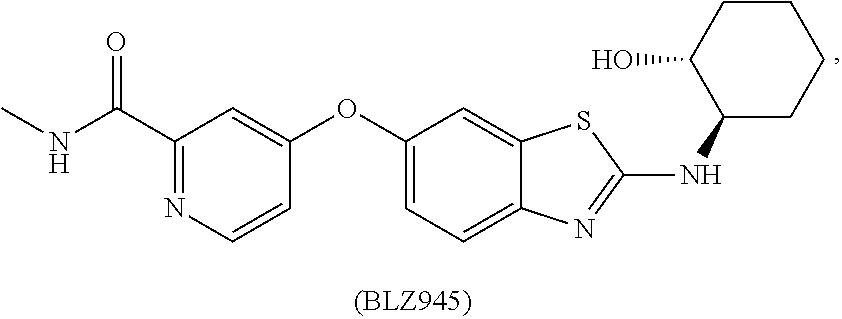

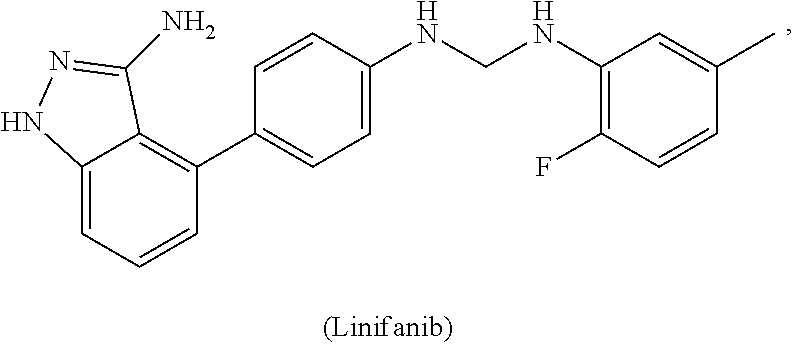
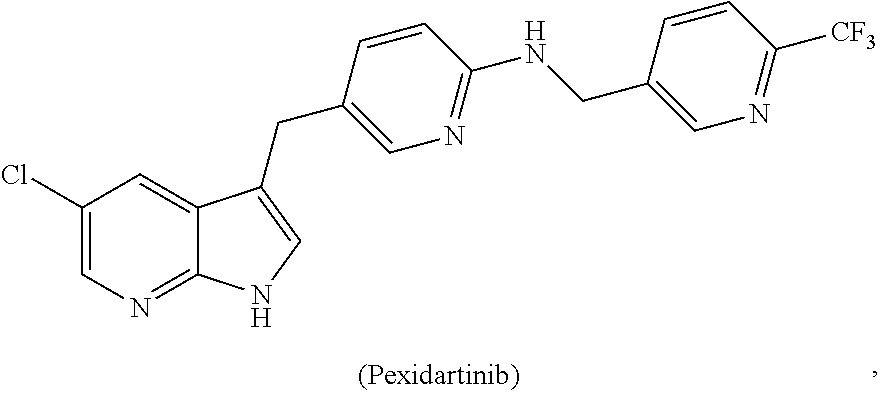
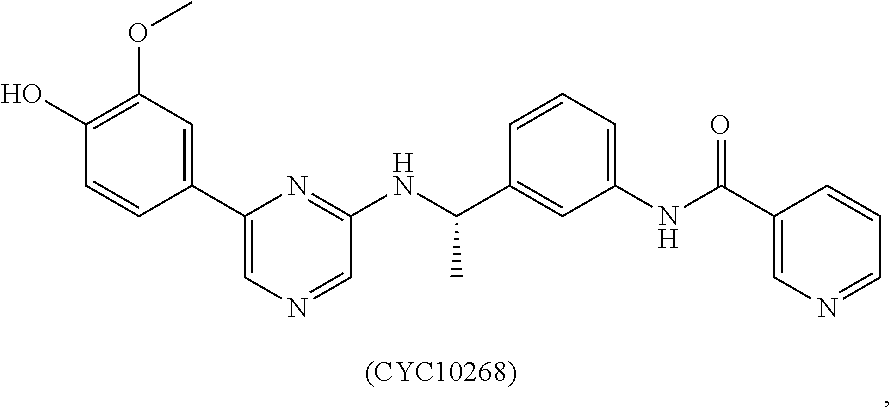


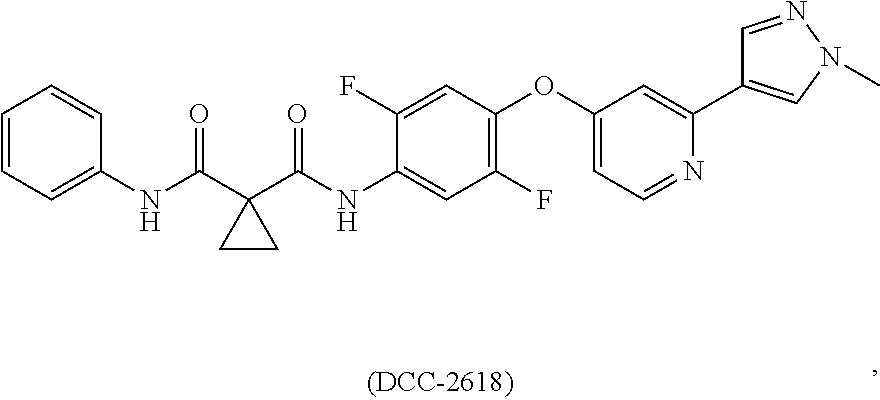
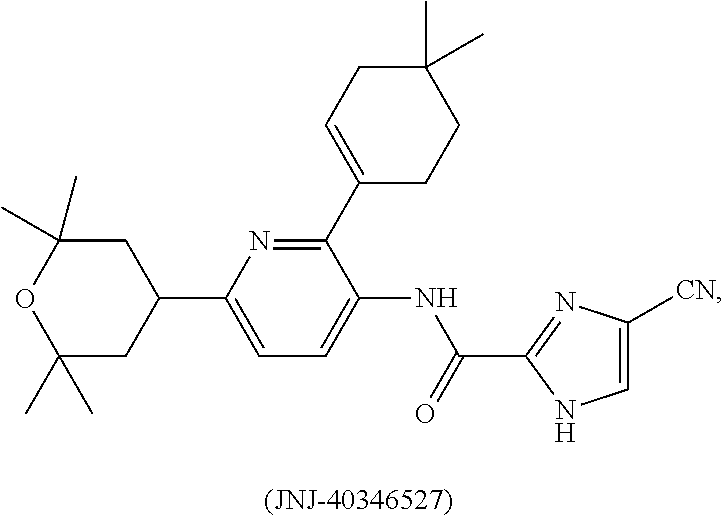



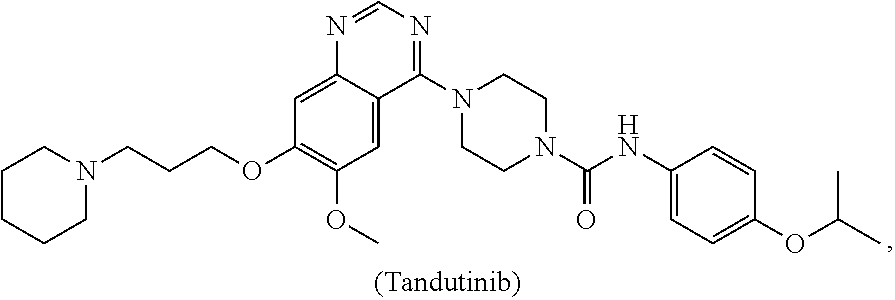

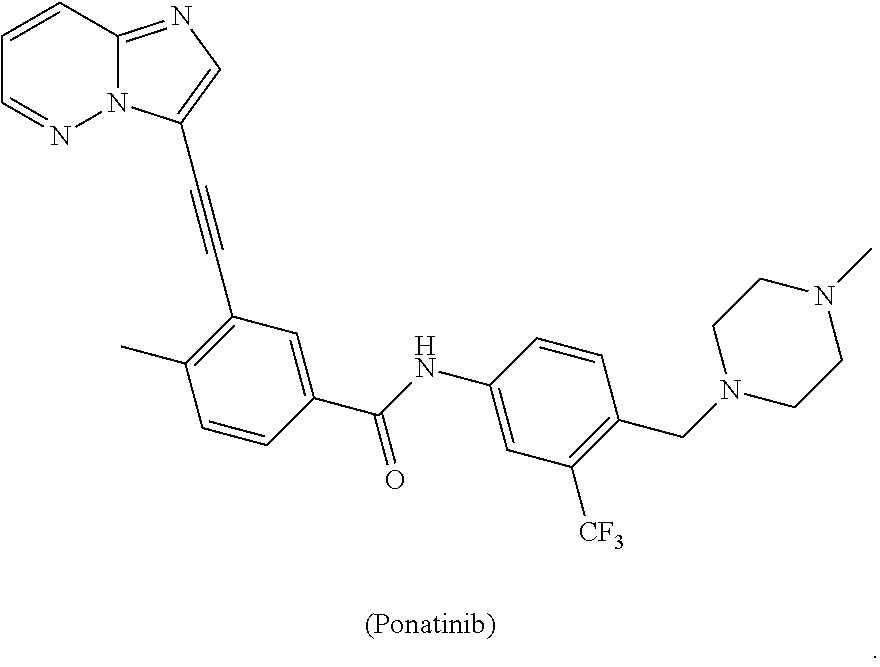
D00000

D00001

D00002

D00003

D00004
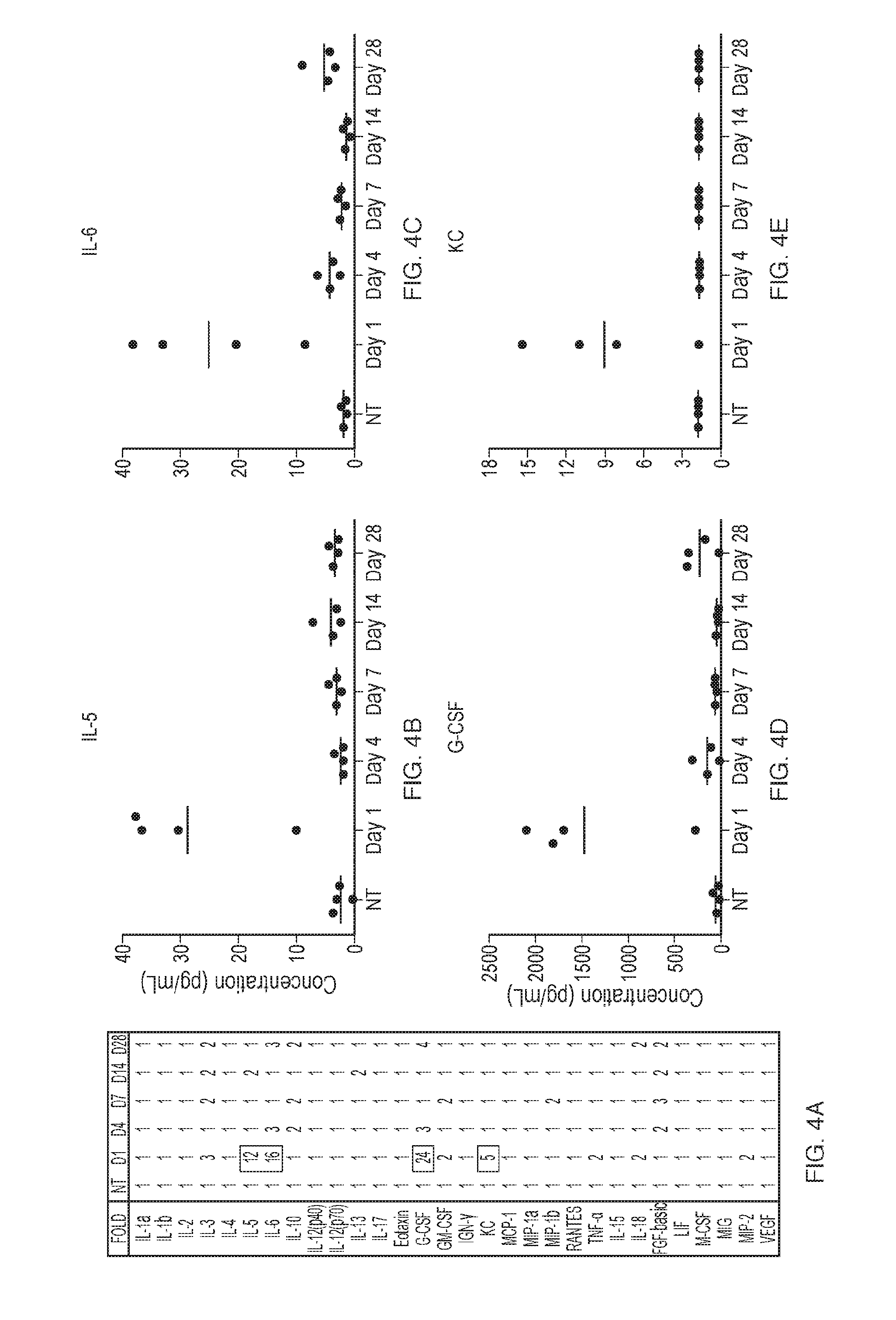
D00005

D00006

D00007

D00008

D00009

D00010
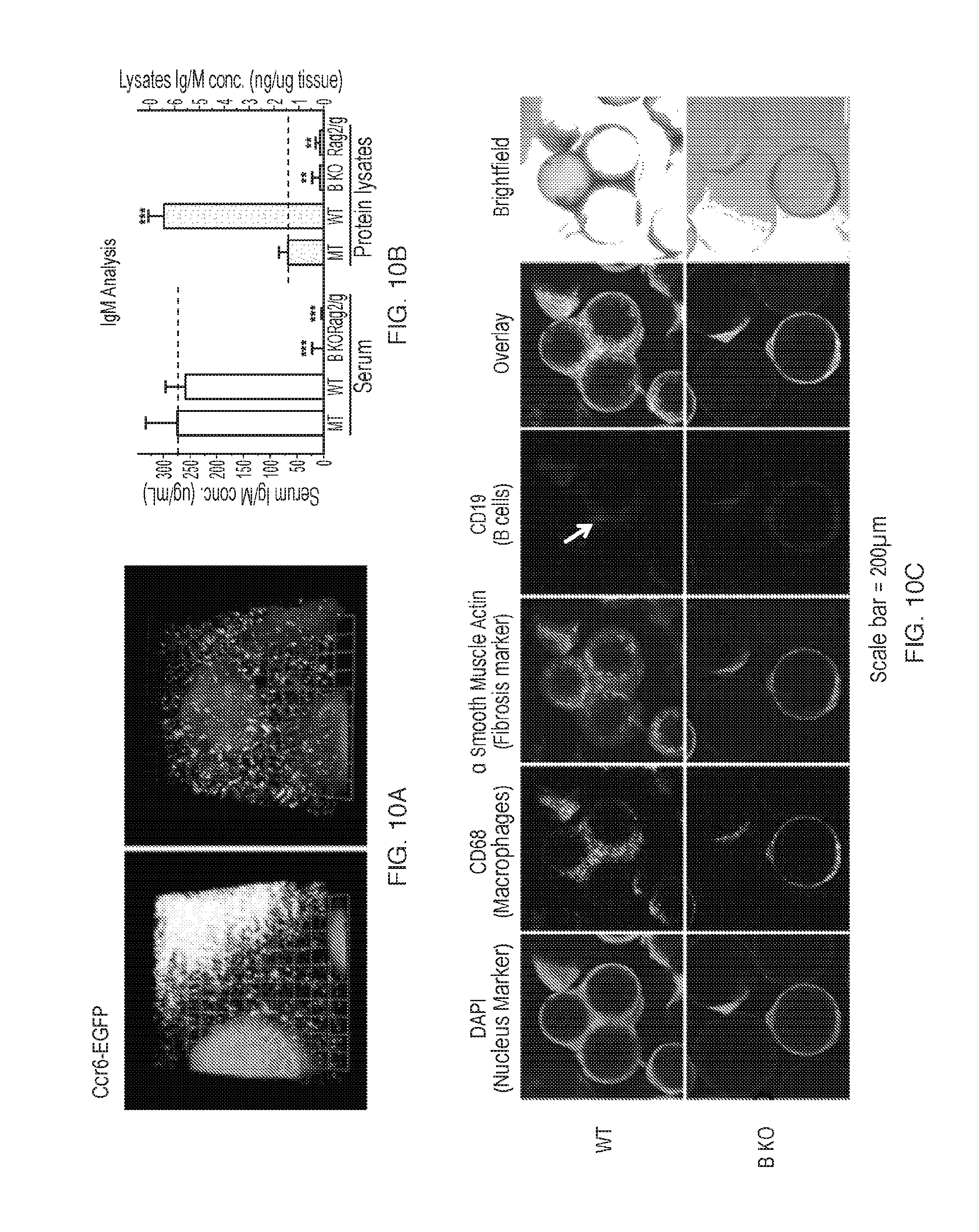
D00011

D00012

D00013
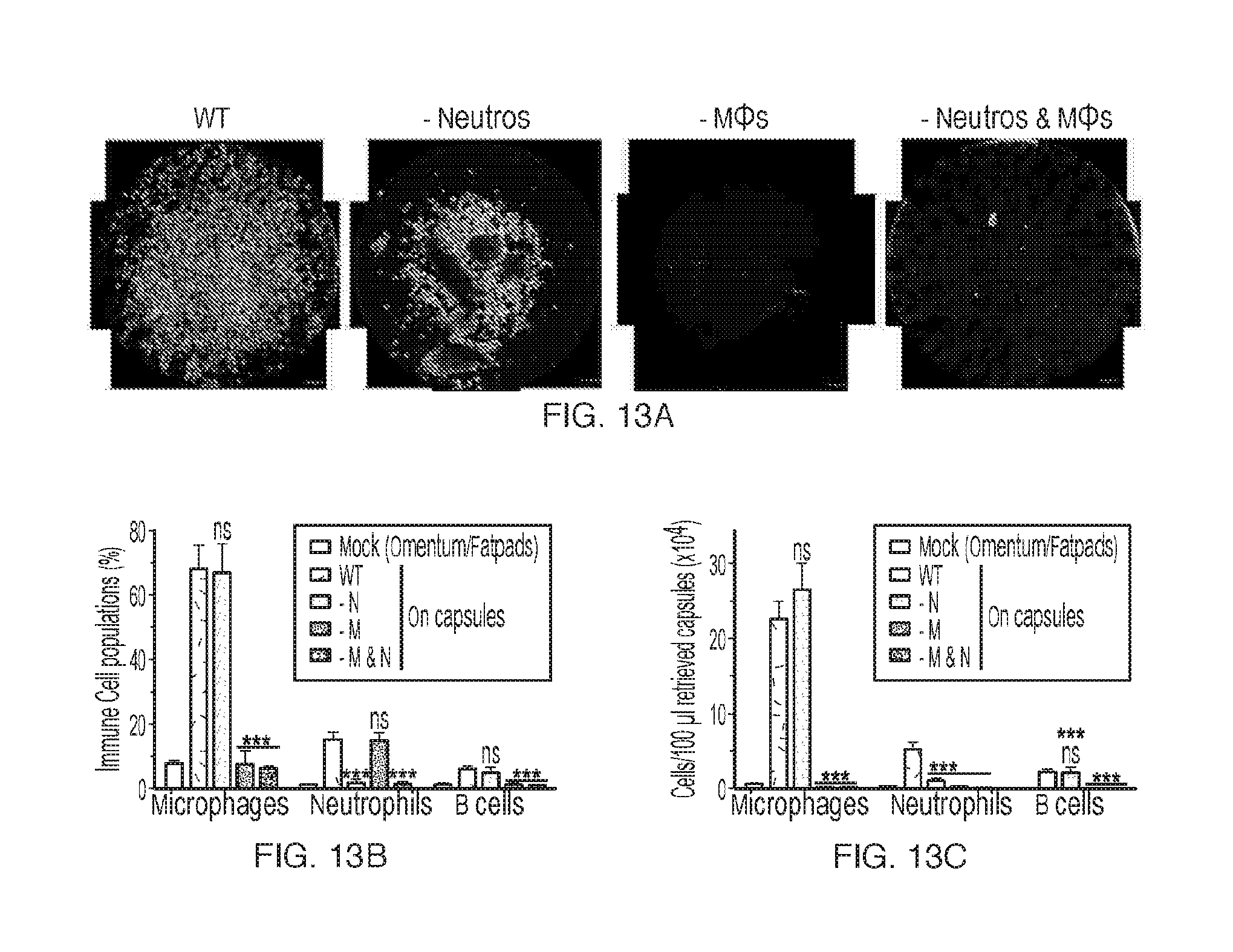
D00014

D00015

D00016
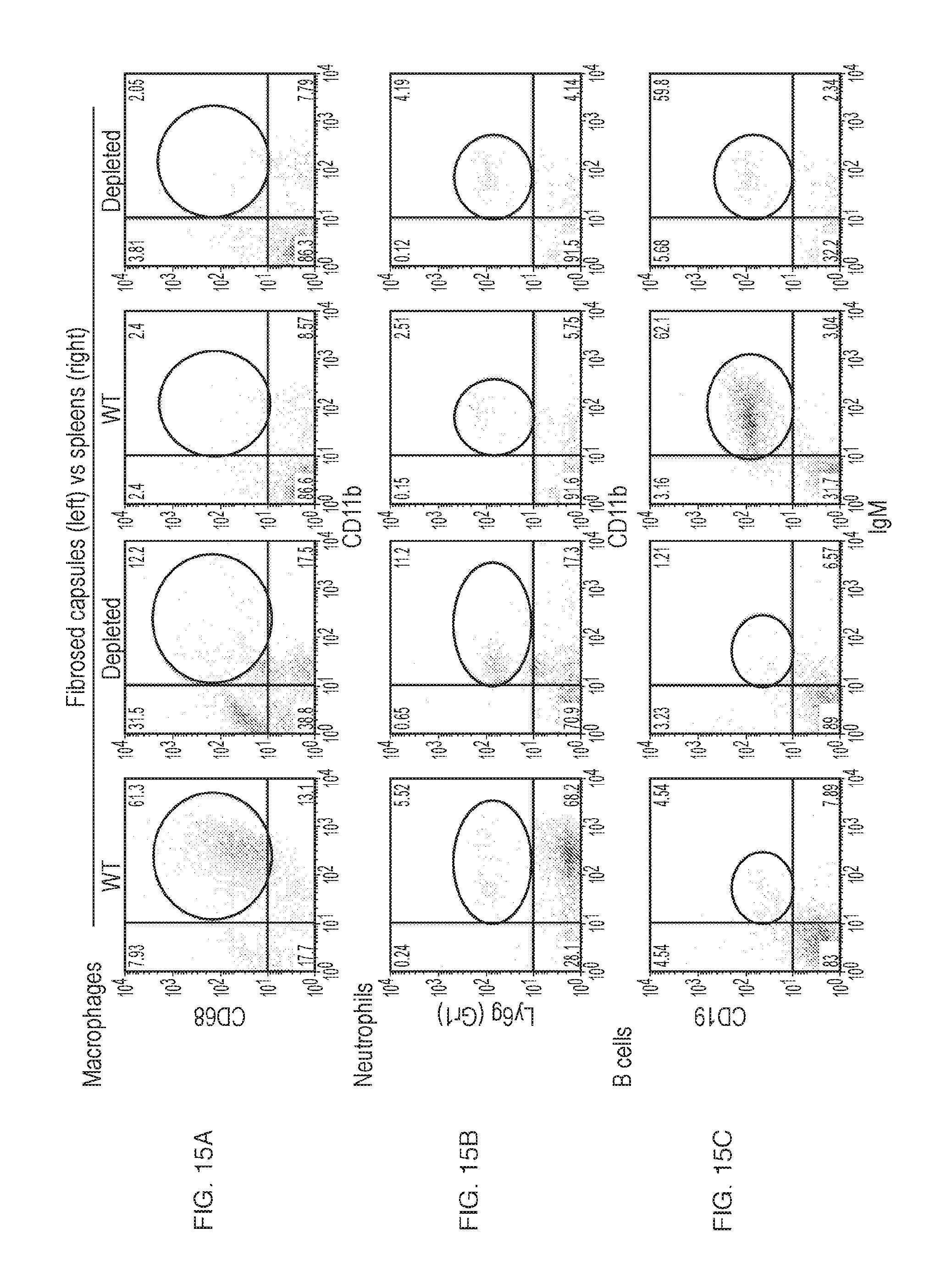
D00017
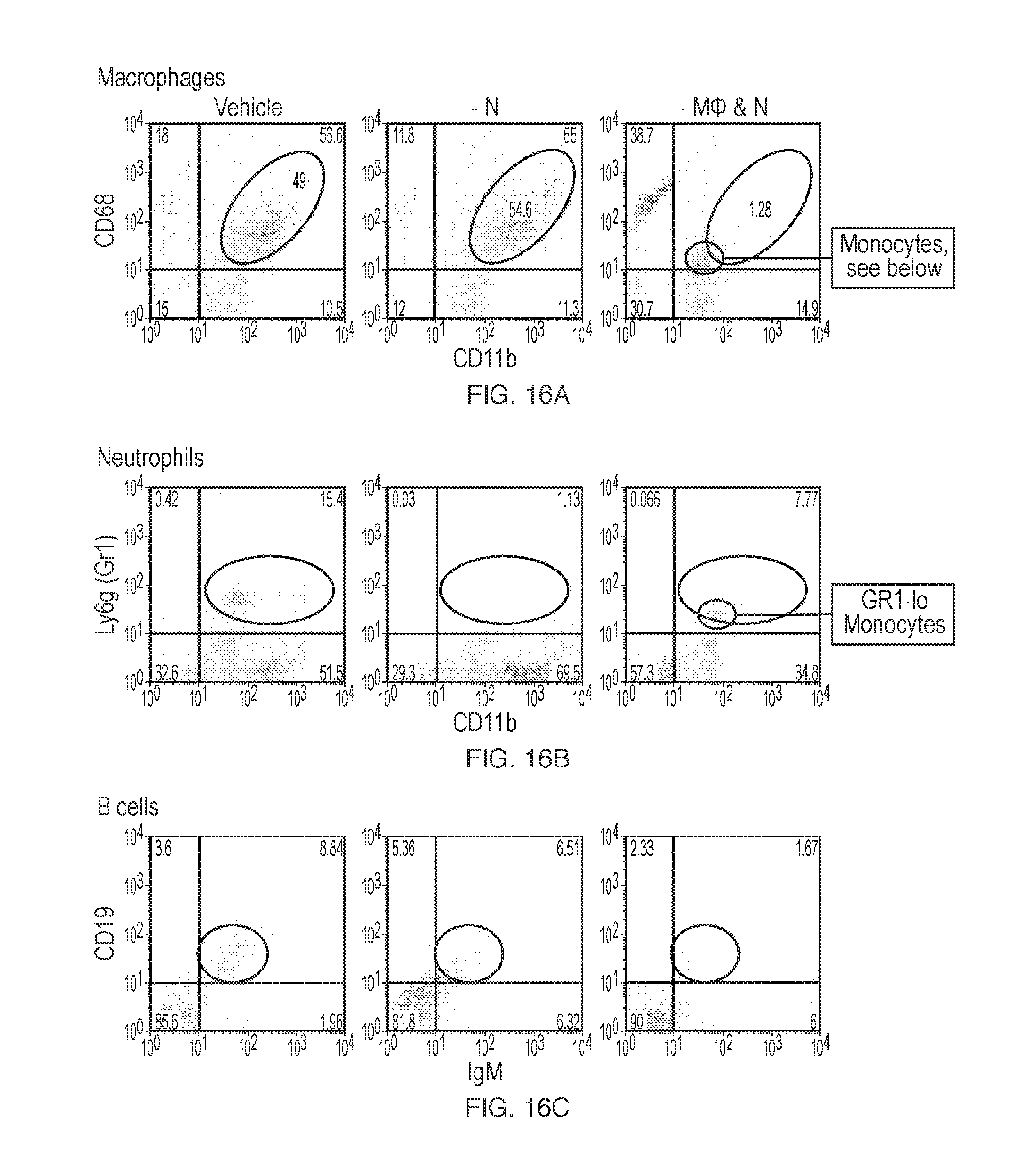
D00018

D00019

D00020

D00021
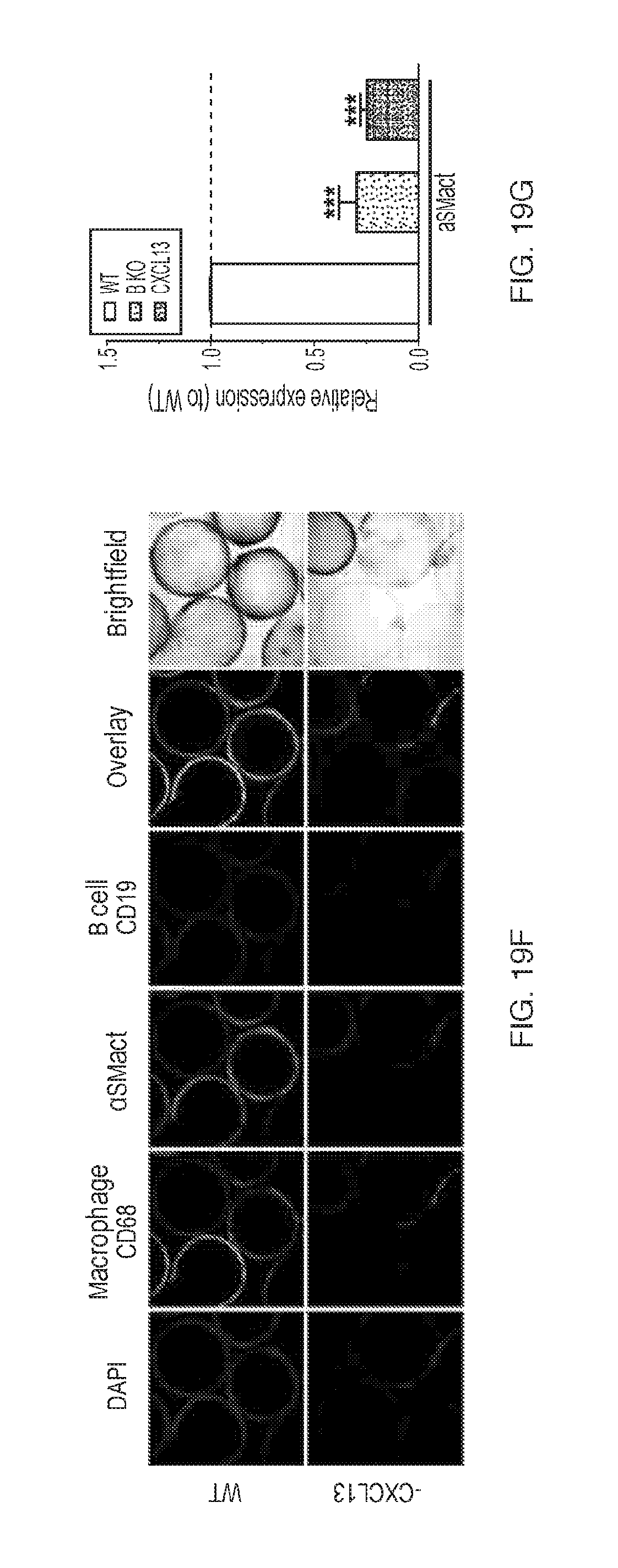
D00022

D00023

D00024

D00025

D00026

D00027

D00028

D00029
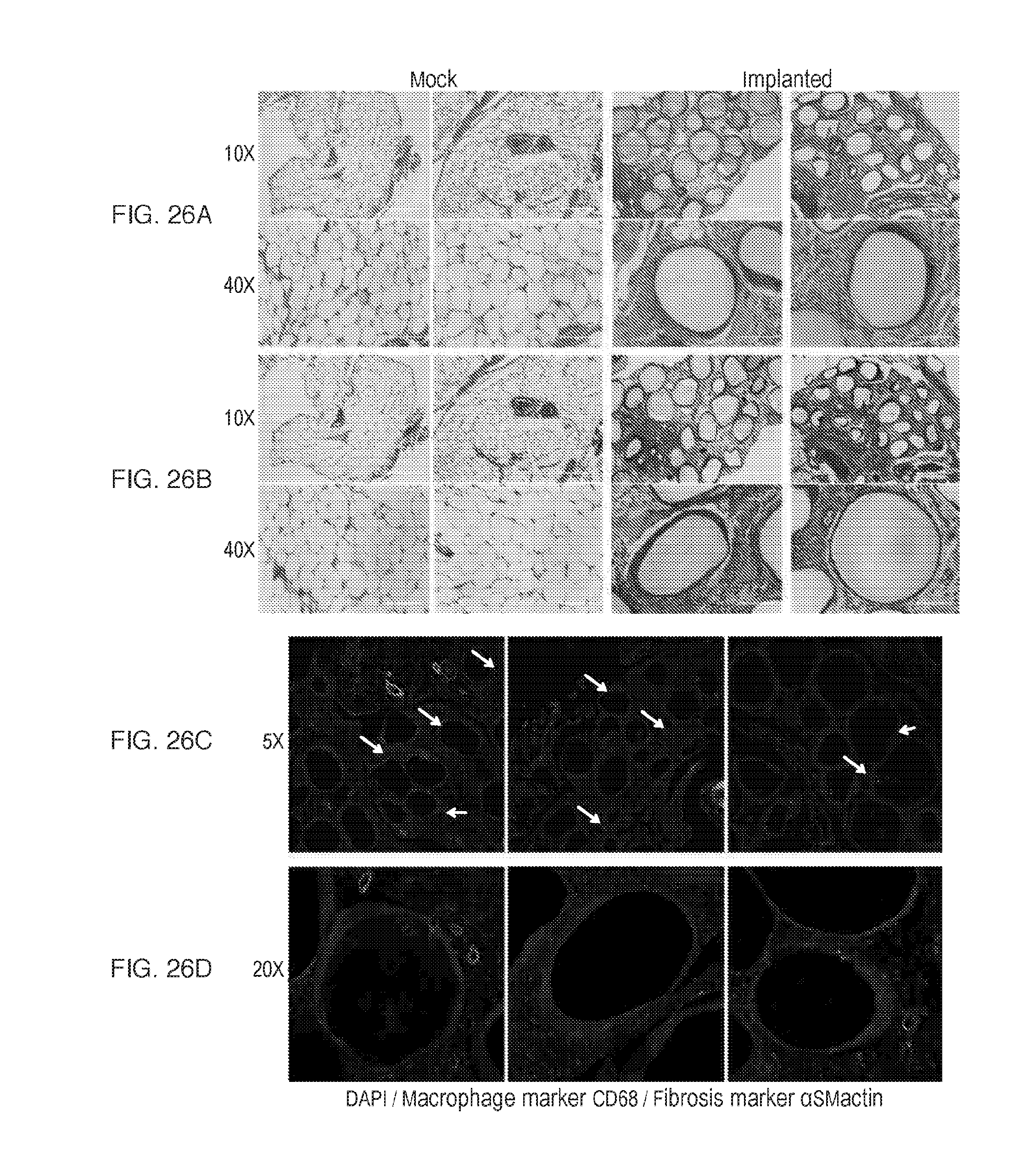
D00030
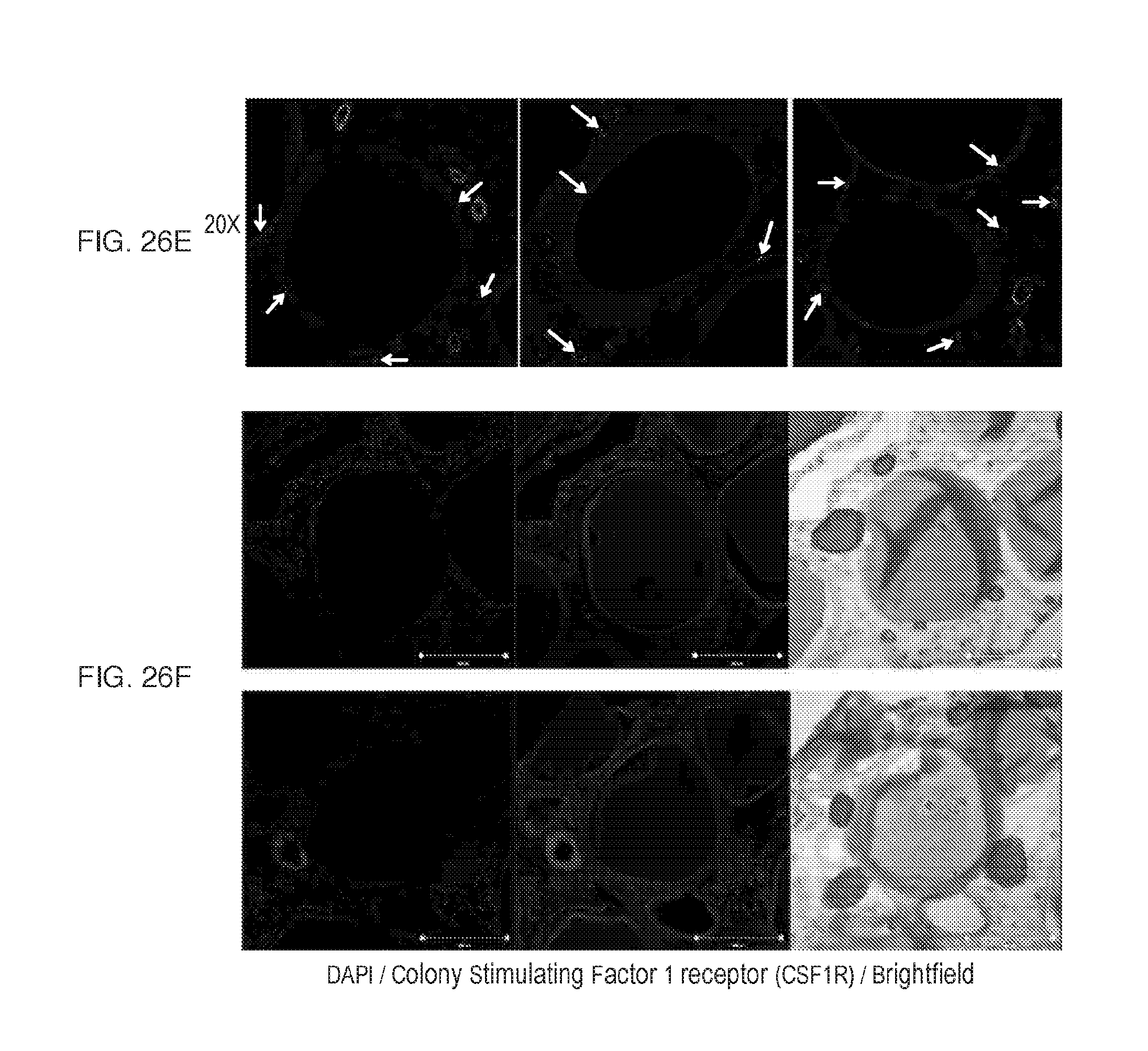
D00031
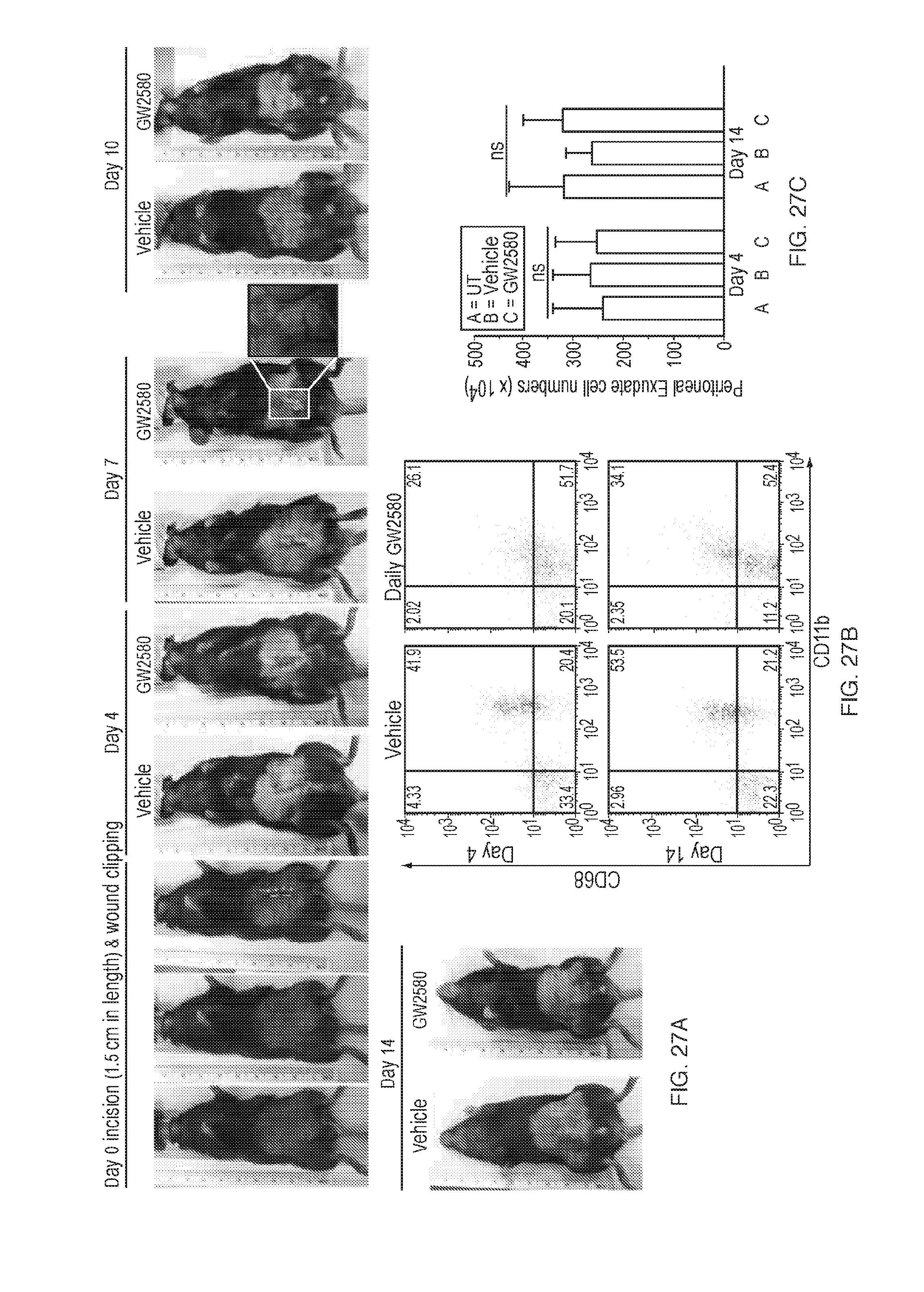
D00032

S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.