Control And Characterization Of Memory Function
Deisseroth; Karl ; et al.
U.S. patent application number 16/112202 was filed with the patent office on 2019-02-14 for control and characterization of memory function. The applicant listed for this patent is The Board of Trustees of the Leland Stanford Junior University. Invention is credited to Karl Deisseroth, Inbal Goshen.
| Application Number | 20190046554 16/112202 |
| Document ID | / |
| Family ID | 46024836 |
| Filed Date | 2019-02-14 |







| United States Patent Application | 20190046554 |
| Kind Code | A1 |
| Deisseroth; Karl ; et al. | February 14, 2019 |
CONTROL AND CHARACTERIZATION OF MEMORY FUNCTION
Abstract
Provided herein are devices and methods for reversibly controlling memory function in living non-human animals. Some variations of methods for affecting memory function comprise temporarily inhibiting neurons of the hippocampus (e.g., neurons of the dorsal CA1 field of the hippocampus) during the acquisition or retrieval of a memory. Alternatively or additionally, methods for reversibly affecting memory function comprise inhibiting neurons of the amygdala (e.g. basolateral amygdala) and/or neurons of the cingulate cortex (e.g., anterior cingulated cortex). Methods for disrupting the formation and recall of memories by inhibiting excitatory neurons expressing light-activated proteins are disclosed herein. One or more methods for reversibly affecting memory function described herein can be used to evaluate the effectiveness of pharmacological agents in treating PTSD and/or various memory disorders.
| Inventors: | Deisseroth; Karl; (Stanford, CA) ; Goshen; Inbal; (Palo Alto, CA) | ||||||||||
| Applicant: |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID: | 46024836 | ||||||||||
| Appl. No.: | 16/112202 | ||||||||||
| Filed: | August 24, 2018 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 13882705 | Jul 25, 2013 | 10086012 | ||
| PCT/US2011/059283 | Nov 4, 2011 | |||
| 16112202 | ||||
| 61540926 | Sep 29, 2011 | |||
| 61410732 | Nov 5, 2010 | |||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | A61P 25/00 20180101; C07K 2319/10 20130101; A61P 25/22 20180101; C07K 2319/04 20130101; A61K 31/7088 20130101; A61K 38/1709 20130101; A61P 25/28 20180101; A01K 67/0275 20130101; A01K 2217/052 20130101 |
| International Class: | A61K 31/7088 20060101 A61K031/7088; A61K 38/17 20060101 A61K038/17; A01K 67/027 20060101 A01K067/027 |
Claims
1. A light-activated protein expressed on the cell membrane of excitatory neurons in the dorsal CA1 field of the hippocampus, the anterior cingulated cortex, or the basolateral amygdala of an animal, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, wherein the illumination of the protein reversibly affects memory function.
2.-3. (canceled)
4. The non-human animal of claim 1, wherein the memory function that is affected is memory retrieval or memory formation.
5. The non-human animal of claim 1, wherein the memory is a fearful memory.
6. The non-human animal of claim 1, wherein the memory is a remote memory.
7. The non-human animal of claim 1, wherein the protein is selected from the group consisting of NpHR, BR, AR, and GtR3.
8. The non-human animal of claim 7, wherein the protein comprises an amino acid sequence at least 95% identical to the sequence shown in SEQ ID NO:3.
9. The non-human animal of claim 8, wherein the protein further comprises an endoplasmic reticulum (ER) export signal and/or a membrane trafficking signal.
10. The non-human animal of claim 8, wherein the amino acid sequence is linked to the ER export signal through a linker.
11. The non-human animal of claim 9, wherein the ER export signal comprises the amino acid sequence FCYENEV.
12. The non-human animal of claim 9, wherein the membrane trafficking signal comprises the amino acid sequence KSRITSEGEYIPLDQIDINV.
13. The non-human animal of claim 7, wherein the protein comprises the amino acid sequence shown in SEQ ID NO:5 or SEQ ID NO:6.
14. A brain tissue slice comprising a brain region selected from the group consisting of the dorsal CA1 field of the hippocampus, the basolateral amygdala, and the anterior cingulated cortex, wherein a light-activated protein is expressed on the cell membrane of excitatory neurons of the brain region, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, wherein the illumination of the protein reversibly affects memory function.
15. A method for reversibly affecting memory retrieval or formation in an individual comprising: administering a polynucleotide encoding a light-activated protein to the dorsal CA1 field of the hippocampus, the anterior cingulated cortex, or the basolateral amygdala in the individual, wherein light-activated protein is expressed on the cell membrane of the excitatory neurons in the dorsal CA1 field of the hippocampus, the anterior cingulated cortex, or the basolateral amygdala, and the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, whereby activating the protein by the light reversibly affects memory retrieval or formation of an event in the individual.
16.-17. (canceled)
18. The method of claim 15, wherein the polynucleotide is a vector.
19. The method of claim 18, wherein the vector is a viral vector selected from the group consisting of an AAV vector, a retroviral vector, an adenoviral vector, an HSV vector, and a lentiviral vector.
20. A method for reversibly affecting memory retrieval or formation comprising: inhibiting depolarization of excitatory neurons in the dorsal CA1 field of the hippocampus, the anterior cingulated cortex, or the basolateral amygdala during memory retrieval or formation of an event in an individual, wherein a light-activated protein is expressed on the cell membrane of the excitatory neurons in the dorsal CA1 field of the hippocampus, the anterior cingulated cortex, or the basolateral amygdala of the individual, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light.
21.-22. (canceled)
23. The method of claim 15, wherein the event is a fearful event.
24. The method of claim 15, wherein the individual is a human.
25. The method of claim 15, wherein the individual is a non-human animal.
26. A method for treating post-traumatic stress disorder in an individual comprising: administering a polynucleotide encoding a light-activated protein to the dorsal CA1 field of the hippocampus, the anterior cingulated cortex, or the basolateral amygdala in the individual, wherein light-activated protein is expressed on the cell membrane of the excitatory neurons in the dorsal CA1 field of the hippocampus, the anterior cingulated cortex, or the basolateral amygdala and the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, whereby activating the protein by the light reversibly affects memory retrieval or formation of an event in the individual.
27. (canceled)
28. The method of claim 26, wherein the polynucleotide is a vector.
29. The method of claim 28, wherein the vector is a viral vector selected from the group consisting of an AAV vector, a retroviral vector, an adenoviral vector, an HSV vector, and a lentiviral vector.
30. A method of screening a pharmacological agent that affects memory retrieval or formation comprising: a) contacting excitatory neurons in the dorsal CA1 field of the hippocampus, the anterior cingulated cortex, or the basolateral amygdala during memory retrieval or formation of an event in a non-human animal with a pharmacological agent, wherein the non-human animal comprises a light-activated protein expressed on the cell membrane of excitatory neurons in the dorsal CA1 field of the hippocampus, the anterior cingulated cortex, or the basolateral amygdala of the animal, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light; b) inhibiting depolarization of the excitatory neurons in the dorsal CA1 field of the hippocampus, the anterior cingulated cortex, or the basolateral amygdala during memory retrieval or formation of an event; and c) determining if the pharmacological agent affects memory retrieval or formation in the presence or absence of the light.
31.-32. (canceled)
33. The method of claim 15, wherein the protein is selected from the group consisting of NpHR, BR, AR, and GtR3.
34. The method of claim 33, wherein the NpHR protein comprises an amino acid sequence at least 95% identical to the sequence shown in SEQ ID NO:3.
35. The method of claim 34 wherein the NpHR protein further comprises an endoplasmic reticulum (ER) export signal and/or a membrane trafficking signal.
36. The method of claim 35, wherein the amino acid sequence at least 95% identical to the sequence shown in SEQ ID NO:3 is linked to the ER export signal and/or the membrane trafficking signal through a linker.
37. The method of claim 35, wherein the ER export signal comprises the amino acid sequence FCYENEV.
38. The method of claim 35, wherein the membrane trafficking signal comprise the amino acid sequence KSRITSEGEYIPLDQIDINV.
39. The method of claim 33, wherein the NpHR protein comprises the amino acid sequence shown in SEQ ID NO:5 or SEQ ID NO:6.
Description
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the priority benefit of U.S. Provisional Patent Application Nos. 61/410,732 filed on Nov. 5, 2010, and 61/540,926, filed on Sep. 29, 2011, the contents of each of which are incorporated herein by reference in their entirety.
BACKGROUND
[0002] The consolidation of remote memories relies on both synaptic consolidation processes on the timescale of minutes to hours, and circuit consolidation over weeks to years (Frankland and Bontempi, 2005; Squire and Bayley, 2007). The process of long-term contextual fear memory consolidation requires early involvement of the hippocampus, followed by the neocortex; in the course of this process, an influence of hippocampus on neocortex may enable the hippocampus to facilitate the long-term cortical storage of memory, rather than stably store the memory itself. Studies have shown that hippocampal lesions impair recent memory one day after training, but the same lesions had no effect on remote memory, several weeks after training (Anagnostaras et al., 1999; Bontempi et al., 1999; Debiec et al., 2002; Frankland et al., 2004; Kim and Fanselow, 1992; Kitamura et al., 2009; Maren et al., 1997; Maviel et al., 2004; Shimizu et al., 2000; Wang et al., 2003; Winocur et al., 2009). Additional studies suggest that both hippocampal and cortical memories are in continuous interplay.
[0003] Previous work on the circuitry of memory has involved physical, pharmacological and genetic lesion studies, which have greatly enhanced our understanding of neural systems but also have suffered from certain well-known challenges; for example, physical lesions are highly effective but lack both cellular and temporal precision, and other methods typically involve tradeoffs between cellular and temporal precision. Elegant genetic interventions can be cell-type specific (McHugh et al., 2007; Nakashiba et al., 2008), but are slow on the timescale of days. Pharmacological lesions enable higher temporal resolution on the timescale of minutes (Kitamura et al., 2009; Wiltgen et al., 2010), but are still slower than neurons and not typically cell-specific. There is a need for developing methods and tools that enable both cell-type precision and temporal control on the millisecond timescale for the study of memory in animals.
[0004] Various psychiatric conditions may arise due to a disorder in the circuitry of memory. For example, amnesia (e.g., non-graded, graded retrograde, focal retrograde amnesia, etc.) involves an inability to retrieve certain memories, while post traumatic stress disorder (PTSD) involves undesired retrieval of fearful memories. PTSD is a common debilitating psychiatric condition in which a single exposure to a traumatic event can lead to years of compromised function due to repeated re-experiencing of the trauma.
[0005] Understanding the neural pathways that underlie undesired memory recall may help aid in the discovery and screening of pharmacological therapies to treat patients with such memory disorders.
[0006] All references cited herein, including patent applications and publications, are incorporated by reference in their entirety.
SUMMARY
[0007] Aspects of the present disclosure relates to control or characterization of memory function in living animals, as described herein. While the present disclosure is not necessarily limited in these contexts, embodiments of the invention may be appreciated through a discussion of examples using these and other contexts.
[0008] Certain embodiments of the present disclosure are directed toward specially-targeted circuits that are associated with memory function. More particular embodiments relate to spatio-temporal control over neural circuitry to identify specific circuit targets associated and corresponding with memory function(s) (e.g., memory formation and/or retrieval).
[0009] Particular embodiments of the present disclosure are directed toward temporally precise inhibition of neural circuits in the hippocampus (such as the neurons of the dorsal CA1 field of the hippocampus), the precision being sufficient to disrupt memory function. It has been discovered that temporal precision of neural inhibition is effective to disrupt remote memory retrieval, whereas prolonged inhibition has no significant effect on remote memory retrieval. Accordingly, aspects of the present disclosure relate to temporal aspects of such inhibition. Alternatively or additionally, methods for reversibly affecting memory function may comprise temporarily inhibiting neurons of the amygdala (e.g. basolateral amygdala) and/or neurons of the cingulate cortex (e.g., anterior cingulated cortex). In certain embodiments, this inhibition is performed using an optogenetic system that involves the expression of light-activated proteins (e.g., opsins) in the cells of the neural circuit. In other embodiments, the inhibition can be performed using direct electrical stimulus. Still other embodiments allow for the use of temporally-precise pharmaceuticals.
[0010] Various embodiments of the present disclosure relate to an optogenetic system or method that correlates temporal control over a neural circuit with measurable metrics. For instance, a particular memory function might be associated with a neurological disorder. The optogenetic system targets a neural circuit within an individual for selective control thereof. The optogenetic system involves monitoring the individual for metrics (e.g., symptoms) associated with the neurological disorder. In this manner the optogenetic system can provide detailed information about the neural circuit, its function and/or the neurological disorder. One or more methods for reversibly affecting memory function may be used to evaluate the effectiveness of pharmacological agents in treating PTSD and/or various memory disorders.
[0011] Provided herein are methods for affecting memory using optogenetic techniques by expressing light-activated proteins in a specific population of neurons involved in memory function, and affecting memory function by activating the protein by light. In some variations, the light-activated proteins may be configured to inhibit depolarization of a neuron in the presence of light having a specific wavelength. In some variations, the light-activated proteins may be configured to promote depolarization of a neuron in the presence of a light having a specific wavelength.
[0012] Provided herein is a non-human animal comprising a light-activated protein expressed on the cell membrane of excitatory neurons in the dorsal CA1 field of the hippocampus of the animal, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, wherein the illumination of the protein reversibly affects memory function. Also provided herein is a non-human animal comprising a light-activated protein expressed on the cell membrane of excitatory neurons in the anterior cingulated cortex of the animal, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, wherein the illumination of the protein reversibly affects memory function. Also provided herein is a non-human animal comprising a light-activated protein expressed on the cell membrane of excitatory neurons in the basolateral amygdala of the animal, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, wherein the illumination of the protein reversibly affects memory function. In some embodiments, the memory function that is affected when the neurons are illuminated may be memory retrieval and/or memory formation. In some embodiments, the memory is a fearful memory and/or a remote memory.
[0013] Also provided herein is a brain tissue slice comprising a brain region selected from the group consisting of the dorsal CA1 field of the hippocampus, the basolateral amygdala, and the anterior cingulated cortex, wherein a light-activated protein is expressed on the cell membrane of excitatory neurons of the brain region, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, wherein the illumination of the protein reversibly affects memory function.
[0014] Also provide herein are methods of reversibly affecting memory retrieval or formation in an individual.
[0015] In some embodiments, the method for reversibly affecting memory retrieval or formation in an individual comprises: administering a polynucleotide encoding a light-activated protein to the dorsal CA1 field of the hippocampus in the individual, wherein light-activated protein is expressed on the cell membrane of the excitatory neurons in the dorsal CA1 field of the hippocampus and the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, whereby activating the protein by the light reversibly affects memory retrieval or formation of an event in the individual. In some embodiments, the method for reversibly affecting memory retrieval or formation comprises: inhibiting depolarization of excitatory neurons in the dorsal CA1 field of the hippocampus during memory retrieval or formation of an event in an individual, wherein a light-activated protein is expressed on the cell membrane of the excitatory neurons in the dorsal CA1 field of the hippocampus of the individual, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light.
[0016] In some embodiments, the method for reversibly affecting memory retrieval or formation in an individual comprises: administering a polynucleotide encoding a light-activated protein to the anterior cingulated cortex in the individual, wherein light-activated protein is expressed on the cell membrane of the excitatory neurons in the anterior cingulated cortex and the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, whereby activating the protein by the light reversibly affects memory retrieval or formation of an event in the individual. In some embodiments, the method for reversibly affecting memory retrieval or formation comprises: inhibiting depolarization of excitatory neurons in the anterior cingulated cortex during memory retrieval or formation of an event in an individual, wherein a light-activated protein is expressed on the cell membrane of the excitatory neurons in the anterior cingulated cortex of the individual, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light.
[0017] In some embodiments, the method for reversibly affecting memory retrieval or formation in an individual comprises: administering a polynucleotide encoding a light-activated protein to the basolateral amygdala in the individual, wherein light-activated protein is expressed on the cell membrane of the excitatory neurons in the basolateral amygdala and the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, whereby activating the protein by the light reversibly affects memory retrieval or formation of an event in the individual. In some embodiments, the method for reversibly affecting memory retrieval or formation comprises: inhibiting depolarization of excitatory neurons in the basolateral amygdala during memory retrieval or formation of an event in an individual, wherein a light-activated protein is expressed on the cell membrane of the excitatory neurons in the basolateral amygdala of the individual, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light.
[0018] Also provided herein are methods for treating post-traumatic stress disorder in an individual. In some embodiments, the method for treating post-traumatic stress disorder in an individual comprises: administering a polynucleotide encoding a light-activated protein to the dorsal CA1 field of the hippocampus in the individual, wherein light-activated protein is expressed on the cell membrane of the excitatory neurons in the dorsal CA1 field of the hippocampus and the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, whereby activating the protein by the light reversibly affects memory retrieval or formation of an event in the individual. In some embodiments, the method for treating post-traumatic stress disorder in an individual comprises: administering a polynucleotide encoding a light-activated protein to the anterior cingulated cortex in the individual, wherein light-activated protein is expressed on the cell membrane of the excitatory neurons in the anterior cingulated cortex and the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, whereby activating the protein by the light reversibly affects memory retrieval or formation of an event in the individual.
[0019] Also provided herein are methods of screening a pharmacological agent that affects memory retrieval or formation comprising: a) contacting excitatory neurons in the dorsal CA1 field of the hippocampus during memory retrieval or formation of an event in a non-human animal with a pharmacological agent, wherein the non-human animal comprises a light-activated protein expressed on the cell membrane of excitatory neurons in the dorsal CA1 field of the hippocampus of the animal, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light; b) inhibiting depolarization of the excitatory neurons in the dorsal CA1 field of the hippocampus during memory retrieval or formation of an event; and c) determining if the pharmacological agent affects memory retrieval or formation in the presence or absence of the light. Also provided herein are methods of screening a pharmacological agent that affects memory retrieval or formation comprising: a) contacting excitatory neurons in the anterior cingulated cortex during memory retrieval or formation of an event in a non-human animal with a pharmacological agent, wherein the non-human animal comprises a light-activated protein expressed on the cell membrane of excitatory neurons in the anterior cingulated cortex of the animal, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light; b) inhibiting depolarization of the excitatory neurons in the anterior cingulated cortex during memory retrieval or formation of an event; and c) determining if the pharmacological agent affects memory retrieval or formation in the presence or absence of the light. Also provided herein are methods of screening a pharmacological agent that affects memory retrieval or formation comprising: a) contacting excitatory neurons in the basolateral amygdala during memory retrieval or formation of an event in a non-human animal with a pharmacological agent, wherein the non-human animal comprises a light-activated protein expressed on the cell membrane of excitatory neurons in the basolateral amygdala of the animal, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light; b) inhibiting depolarization of the excitatory neurons in the basolateral amygdala during memory retrieval or formation of an event; and c) determining if the pharmacological agent affects memory retrieval or formation in the presence or absence of the light.
[0020] The light-activated protein may be responsive to light and configured such that the protein is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light. In some embodiments, the light-activated protein may be selected from the group consisting of NpHR, BR, AR, and GtR3 described herein. In some embodiments, the light-activated protein is a NpHR protein comprising an amino acid sequence at least 95%, at least 96%, at least 97%, at least 98%, at least 99% or 100% identical to the sequence shown in SEQ ID NO:3. In some embodiments, the NpHR protein further comprises an endoplasmic reticulum (ER) export signal and/or a membrane trafficking signal. For example, the NpHR protein comprises an amino acid sequence at least 95% identical to the sequence shown in SEQ ID NO:3 and an endoplasmic reticulum (ER) export signal. In some embodiments, the amino acid sequence at least 95% identical to the sequence shown in SEQ ID NO:3 is linked to the ER export signal through a linker. In some embodiments, the ER export signal comprises the amino acid sequence FXYENE, where X can be any amino acid. In another embodiment, the ER export signal comprises the amino acid sequence VXXSL, where X can be any amino acid. In some embodiments, the ER export signal comprises the amino acid sequence FCYENEV. In some embodiments, the NpHR protein comprises an amino acid sequence at least 95% identical to the sequence shown in SEQ ID NO:3, an ER export signal, and a membrane trafficking signal. In other embodiments, the NpHR protein comprises, from the N-terminus to the C-terminus, the amino acid sequence at least 95% identical to the sequence shown in SEQ ID NO:3, the ER export signal, and the membrane trafficking signal. In other embodiments, the NpHR protein comprises, from the N-terminus to the C-terminus, the amino acid sequence at least 95% identical to the sequence shown in SEQ ID NO:3, the membrane trafficking signal, and the ER export signal. In some embodiments, the membrane trafficking signal is derived from the amino acid sequence of the human inward rectifier potassium channel Kir2.1. In some embodiments, the membrane trafficking signal comprises the amino acid sequence K S R I T S E G E Y I P L D Q I D I N V. In some embodiments, the membrane trafficking signal is linked to the amino acid sequence at least 95% identical to the sequence shown in SEQ ID NO:3 by a linker. In some embodiments, the membrane trafficking signal is linked to the ER export signal through a linker. The linker may comprise any of 5, 10, 20, 30, 40, 50, 75, 100, 125, 150, 175, 200, 225, 250, 275, 300, 400, or 500 amino acids in length. The linker may further comprise a fluorescent protein, for example, but not limited to, a yellow fluorescent protein, a red fluorescent protein, a green fluorescent protein, or a cyan fluorescent protein. In some embodiments, the light-activated protein further comprises an N-terminal signal peptide. In some embodiments, the light-activated protein comprises the amino acid sequence of SEQ ID NO:5. In some embodiments, the light-activated protein comprises the amino acid sequence of SEQ ID NO:6.
[0021] It is to be understood that one, some, or all of the properties of the various embodiments described herein may be combined to form other embodiments of the present invention. These and other aspects of the invention will become apparent to one of skill in the art.
BRIEF DESCRIPTION OF THE DRAWINGS
[0022] FIG. 1 depicts one variation of a device or a system that may be used to apply light of selected wavelengths to affect memory function.
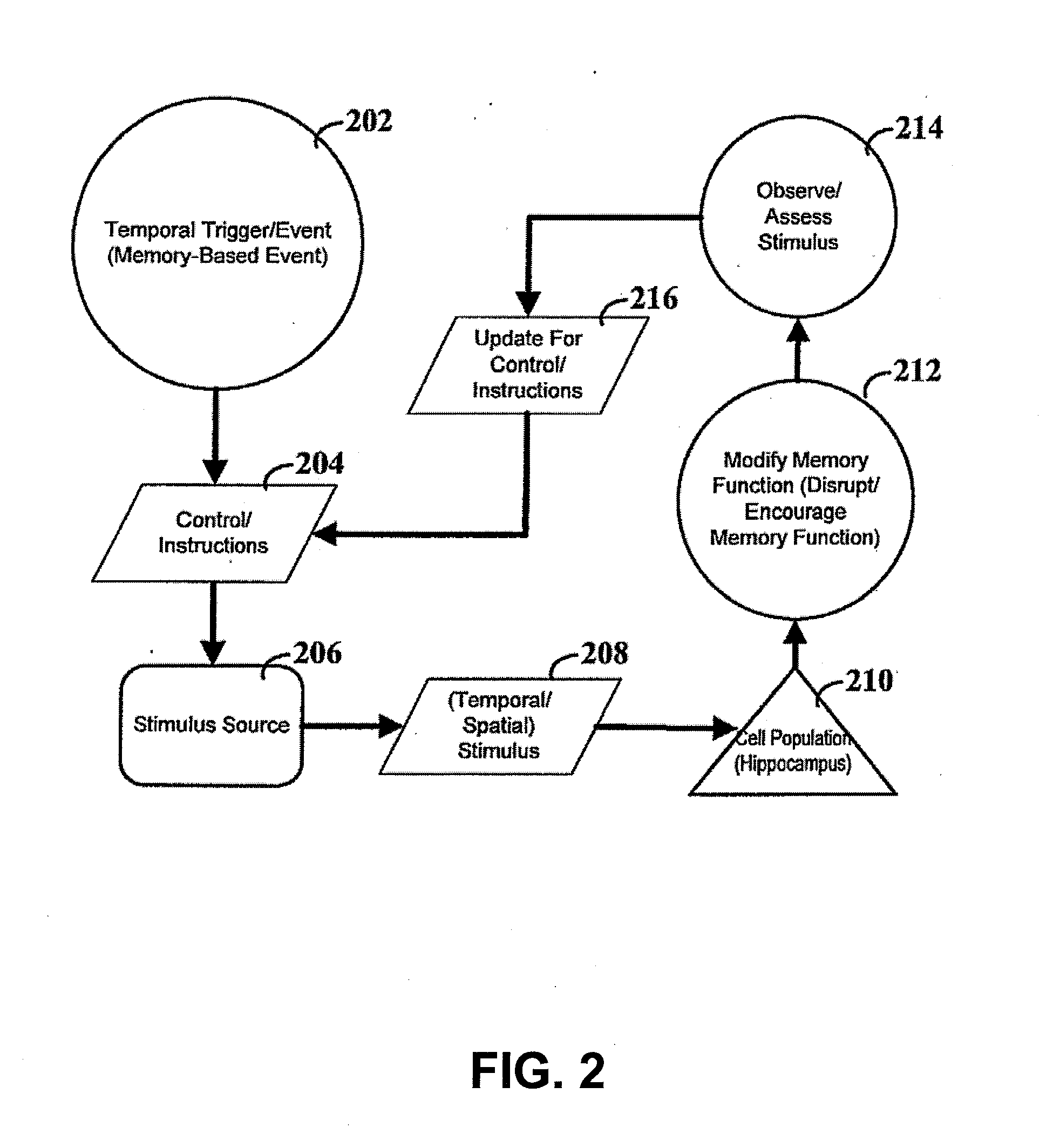
[0023] FIG. 2 depicts a flow diagram for modifying memory function.
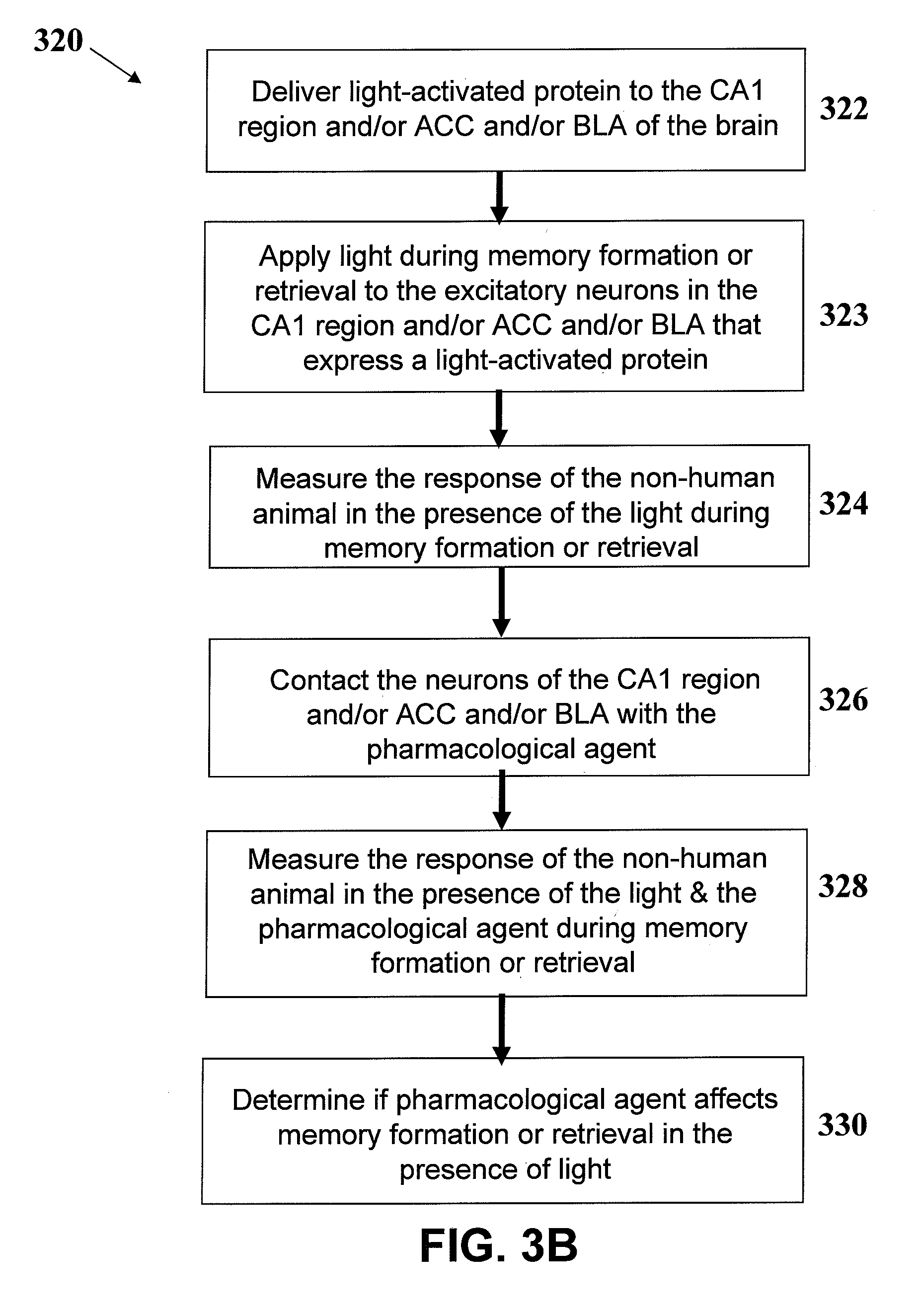
[0024] FIGS. 3A and 3B depict variations of methods for evaluating the effects of a test pharmacological agent on neural circuits that underlie memory function.
[0025] FIGS. 4A-D depicts experimental data showing specific optogenetic inhibition of excitatory neurons in dorsal CA1 reduces neuronal activity. FIG. 4A shows that double lentiviral injection resulted in eNpHR3.1 expression throughout the CA1 only. FIG. 4B shows that eNpHR3.1 is expressed in the neuronal membrane around the soma, as well as in the apical and basal dendrites of CA1 neurons. FIG. 4C depicts data demonstrating that CaMKII.alpha.::eNpHR3.1 was expressed in 94% (458/486 cells, from 3 mice) of CA1 pyramidal neurons, with 100% specificity (all eNpHR3.1-EYFP cells were CaMKII.alpha. positive). FIG. 4D depicts data from iIn-vivo `optrode` light administration and recording performed by inserting an optic fiber coupled to a tungsten electrode to the CA1 in anesthetized mice expressing eNpHR3.1 (left). 561 nm illumination of CA1 neurons in these mice resulted in a reversible, marked reduction in spiking frequency (4.93.+-.1.6 Hz, 1.31.+-.0.15 Hz, and 6.45.+-.2.4 Hz; before, during and after light administration, respectively, in 15 traces from 2 mice, P<0.02), without affecting average spike amplitude (33.55.+-.4.94 .mu.V, 29.20.+-.4.4 .mu.V, and 33.32.+-.5.45 .mu.V; before, during and after light). A representative optrode recording trace, as well as average frequency and amplitude are shown (mean.+-.SEM).
[0026] FIGS. 5A-5I depicts experimental data showing that real time CA1 optogenetic inhibition blocks contextual fear acquisition and retrieval. FIG. 5A shows that bilateral in-vivo light may be administered to CA1 by inserting a double optic fiber through bilateral cannula guide in freely-moving mice. FIG. 5B (top) depicts an experimental sequence where continuous 561 nm illumination was administered during fear-conditioning training, and mice were tested for their memory 24 hr later without light. One day later, mice were re-trained without light, and re-tested without light on the fourth day and with light on the fifth. (bottom) CA1 optogenetic inhibition during fear-conditioning training (Light ON) prevented acquisition in eNpHR3.1 mice (n=5) compared to controls (n=4) (39.+-.5.4 vs. 7.6.+-.4.3% freezing; means.+-.SEM, P<0.005). When re-trained without illumination (Light OFF), the same mice demonstrated intact contextual memory (64.6.+-.6.6 vs. 49.7.+-.11.7% freezing; P>0.5). This contextual fear memory became unavailable for recall upon light administration during testing (light ON) in eNpHR3.1 mice (42.6.+-.10.1 vs. 5.94.+-.4.1% freezing, P<0.01). FIG. 5C shows that CA1 optogenetic inhibition had no effect on either acquisition (left) or recall (right) of the hippocampal-independent auditory-cued fear memory in eNpHR3.1 mice (n=5) compared to controls (n=4). FIG. 5D depicts data showing optogenetic inhibition had no effect on exploration of the context before conditioning in eNpHR3.1 mice (n=5) compared to controls (n=4). CA1 optogenetic inhibition also had no effect on exploration of a novel environment. FIGS. 5E and 5F show that control (n=6) and eNpHR3.1 (n=4) mice explored the field with similar path lengths (564.+-.9 and 618.+-.114 cm, respectively) and similar speeds (3.3.+-.0.1 vs. 3.43.+-.0.6 cm/sec, respectively). FIG. 5G shows that there was no effect on anxiety, as the percent of time that control and eNpHR3.1 mice spent in the center of the open field was similar (23.8.+-.2.76% vs. 20.46.+-.5.97%, P>0.5). Representative exploration traces are presented. FIG. 5H depicts eNpHR3.0 expression in basolateral amygdala (BLA). FIG. 5I shows that light administration to the BLA resulted in impaired contextual (65.5.+-.7.2 vs. 9.6.+-.5.5% freezing; P<0.001) and cued (69.5.+-.9.6 vs. 24.5.+-.13% freezing; P<0.05) memory acquisition in eNpHR3.0 (n=4) mice, compared to controls (n=9).
[0027] FIGS. 6A-6E depicts experimental data showing that CA1 optogenetic inhibition reversibly interferes with remote fear memory recall. FIG. 6A depicts data indicating that CA1 optogenetic inhibition reversibly prevented recall of remote memory that was acquired 28 days earlier, and was never previously evoked (P<0.0001; Control n=14, 69.8.+-.5.3% freezing eNpHR3.1 n=6, 14.+-.6.4% freezing). This recall disruption was reversible, as when the same mice were re-introduced to the conditioning context on the next day with no illumination they demonstrated intact fear responses (52.45.+-.6.0 vs. 45.18.+-.11.5% freezing; P>0.5). FIG. 6B depicts data showing that auditory-cued fear, tested 28 days after conditioning was not affected (Control n=14, 22.3.+-.6.8%, eNpHR3.1 n=6, 11.8.+-.3.5% freezing in the new context; and 72.4.+-.8.4 vs. 58.77.+-.7.9% freezing to the tone; P>0.5).
[0028] FIG. 6C shows that CA1 optogenetic inhibition impaired recall of ultra remote memory that was acquired 63 days earlier, and was never previously evoked (P<0.005; Control n=9, 31.8.+-.3.8% freezing eNpHR3.1 n=6, 11.3.+-.3.6% freezing). FIG. 6D depicts data showing that pharmacological hippocampal inhibition by TTX and CNQX administration one day after conditioning prevented recent fear recall (Saline n=5, 56.86.+-.1.9% freezing; TTX+CNQX n=4, 26.05.+-.10.23% freezing; P<0.05). FIG. 6E shows that TTX and CNQX administration one month after conditioning did not affect remote fear recall (Saline n=8, 93.93.+-.2.54% freezing; TTX+CNQX n=9, 83.8.+-.4.4% freezing; P>0.05).
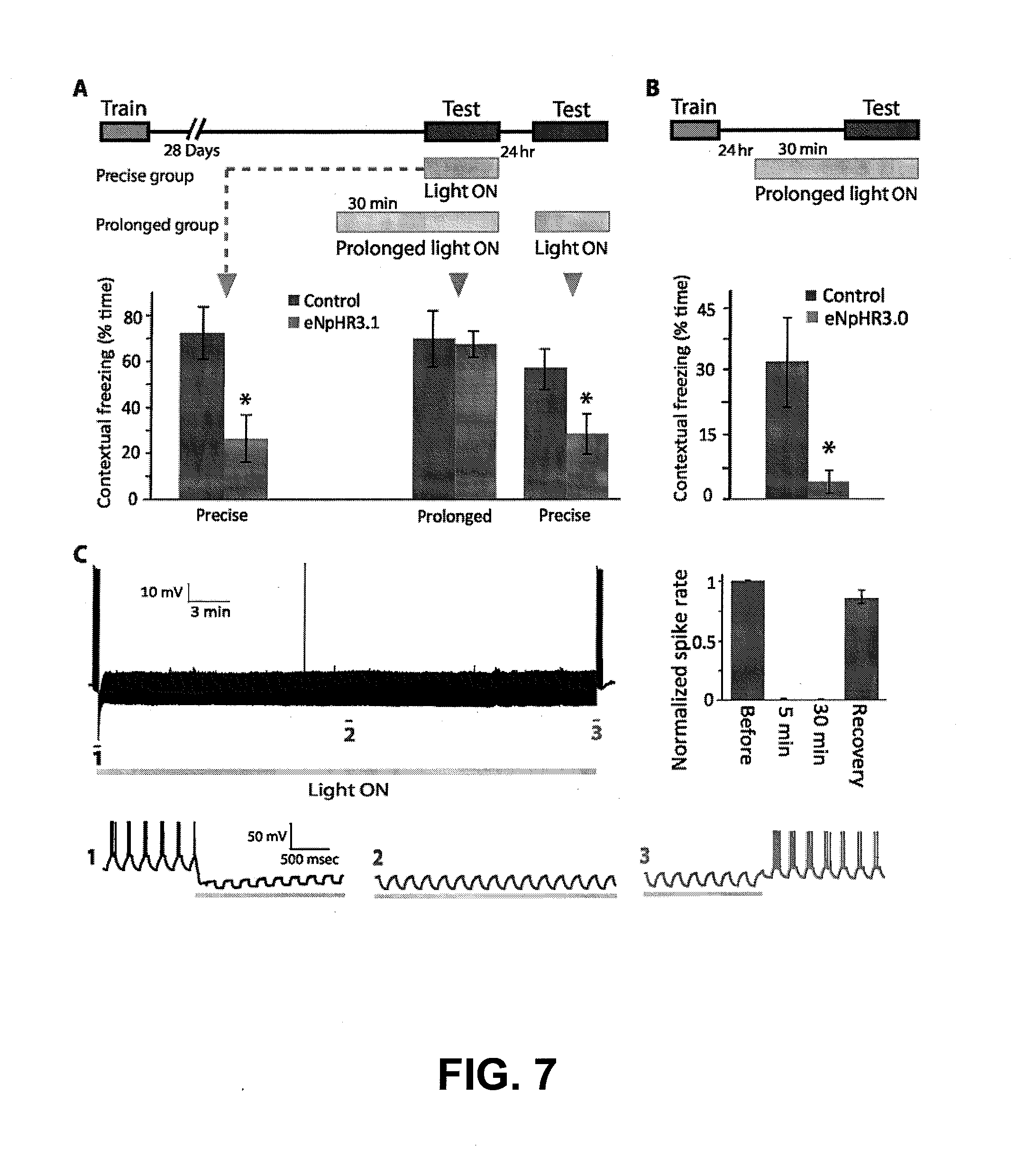
[0029] FIGS. 7A-7C depicts experimental data showing that precise, but not prolonged CA1 optogenetic inhibition blocks remote contextual fear recall. FIG. 7A shows that CA1 optogenetic inhibition prevents remote fear recall of a memory that was acquired 28 days earlier, only when the light was administered precisely during testing (Precise group, Control n=4, 72.65.+-.11.5% freezing, eNpHR3.1 n=8, 26.9.+-.10.4% freezing; P<0.01), but not when the light was ON continuously for 30 min before, as well as during, the test (Prolonged group, middle, Control n=3, 70.13.+-.12.2% freezing, eNpHR3.1 n=4, 67.7.+-.5.6% freezing; P>0.05). When the prolonged group mice were re-tested the next day with light during the test only, their recall was disrupted (Prolonged group, left, 55.5.+-.8.5 vs. 27.6.+-.8.6% freezing; P<0.05). FIG. 7B shows that prolonged light prevents recall of recent memory, 24 hr after conditioning (Control n=7, 32.2.+-.10.6% freezing, eNpHR3.1 n=3, 4.+-.2.6% freezing; P<0.05). FIG. 7C shows that eNpHR3.1 continuously and completely prevented evoked spiking for 30 min, as shown in the recording trace. Detailed traces of sections 1 (inhibition onset) 2 (during continuous inhibition) and 3 (end of inhibition and recovery) are presented on the bottom left. Averaged percent successful evoked spiking before light, during light administration (after 5 min and 30 min of light ON) and recovery after light OFF are presented (bottom right; n=4 mice, 10 cells).
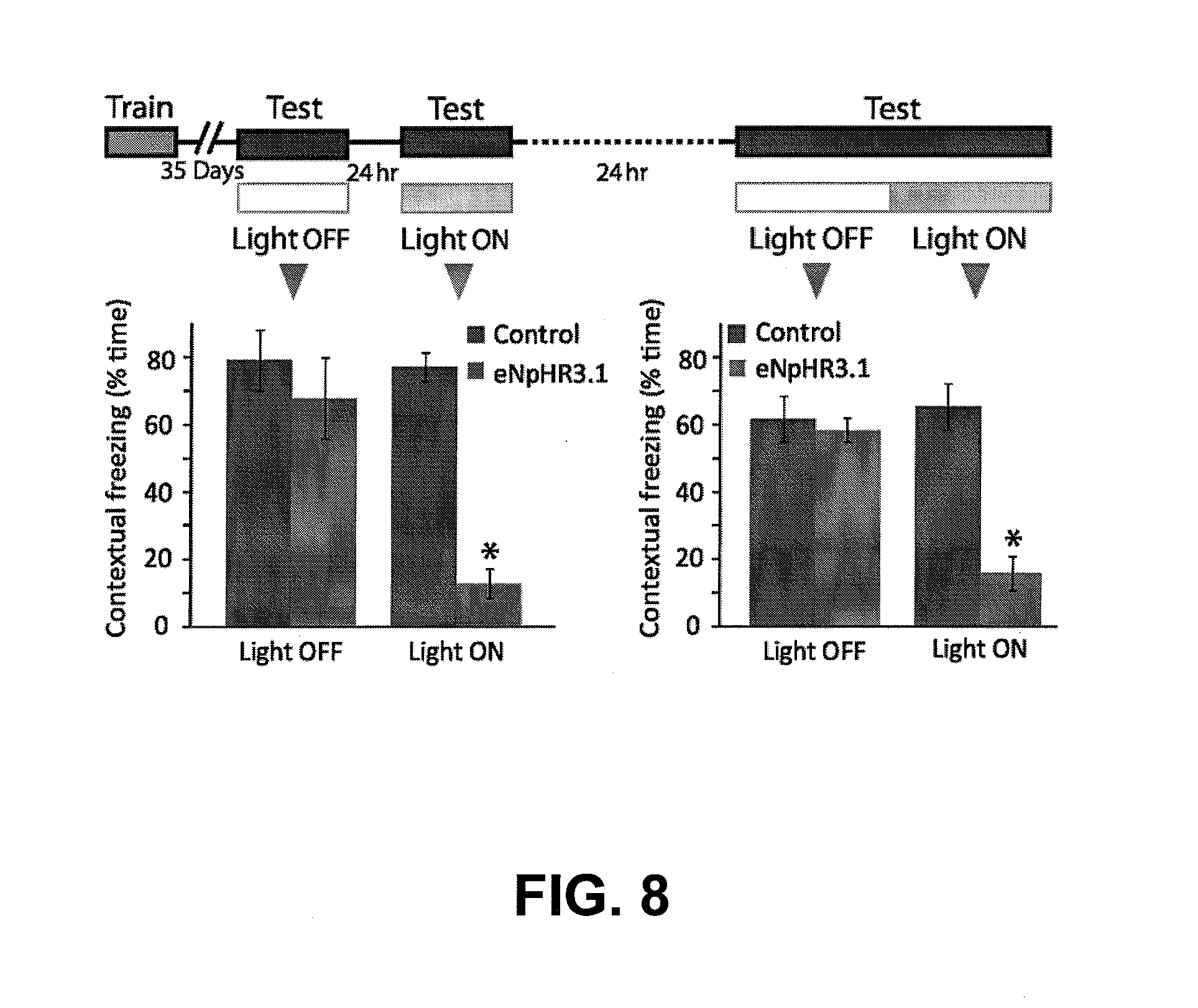
[0030] FIG. 8 depicts experimental data showing that CA1 optogenetic inhibition interferes with ongoing fear recall. Left: Remote fear memory that was acquired 5 weeks before and was efficiently recalled (Control n=8, 79.0.+-.8.9% freezing; eNpHR3.1 n=6, 67.8.+-.12.1% freezing; P>0.5) was no longer available for recall under CA1 optogenetic inhibition (77.2.+-.4.3% vs. 12.8.+-.4.4% freezing; P<0.0001). Right: This recall disruption did not result in memory erasure, as when the same mice were re-introduced to the conditioning context with no illumination they again demonstrated intact fear response (61.5.+-.6.7 vs. 58.3.+-.3.5% freezing; P>0.5). When illumination was introduced again in the middle of the testing trial, after the memory was already recalled, the fear response abruptly ceased (65.2.+-.6.9 vs. 15.9.+-.5.2% freezing; P<0.001).
[0031] FIG. 9A-9H depicts experimental data showing brain-wide mapping of circuit activity controlled by the hippocampus during remote recall. FIG. 9A depicts an experiment where mice were fear-conditioned under light delivery, and brains were collected 90 min after training. FIG. 9B shows brain slices stained for c-Fos and DAPI. Expression of YFP control and eNpHR3.1 are shown. The CA1 region from which these images were taken is marked by a white square in FIG. 9C. FIG. 9C depicts representative images of CA1, ACC and BLA. Anatomy is shown by DAPI nuclear staining, and the margins of the amygdala are marked with a dashed yellow line. White scalebar: 150 .mu.m.
[0032] FIG. 9D shows that CA1 optogenetic inhibition during FC reduced the expression of the neuronal activation marker c-Fos in CA1 (n=2 to 4 mice, 6 to 15 slices per group; P<0.01), but not in the ACC or BLA. In the BLA, activity levels were similarly elevated in both control and eNpHR3.1 mice (p<0.0001). FIG. 9E depicts an experiment where another group of mice was trained, and then re-exposed to the conditioning context 28 days after conditioning. Brains were collected for staining 90 min after testing. FIG. 9F depicts representative CA1, ACC and BLA images following remote memory are shown. White scalebar: 150 .mu.m. FIG. 9G shows that remote recall 28 days following conditioning resulted in a small but significant increase in CA1 c-Fos expression in control mice (P<0.005), and highly increased activity levels in ACC (P<0.0001) and BLA (P<0.0001). Light inhibition during exposure to the context completely blocked CA1 activity (P<0.05), and significantly reduced ACC and BLA activity (P<0.0001 and P<0.0001, respectively), compared to control. FIG. 9H shows global patterns in brain activity between conditioning (day 0) and remote recall (day 28). Activity levels in CA1 significantly decreased in control (P<0.005) mice from day 0 to day 28. Activity levels in ACC significantly increased in both control (P<0.0001) and eNpHR3.1 (P<0.001) mice day 0 to day 28. Activity levels in BLA significantly increased in control (P<0.001) but not in eNHR3.1 mice.
[0033] FIG. 10 depicts experimental data showing that precise and prolonged anterior cingulate cortex (ACC) optogenetic inhibition disrupts remote, but not recent, fear memory recall. FIG. 10A depicts eNpHR3.0 expression in the anterior cingulate cortex (ACC). FIG. 10B depicts an experiment where precise light administration resulted in inhibition of remote (Control n=5, 81.6.+-.4.9% freezing; eNpHR3.0 n=5, 53.8.+-.11% freezing; P<0.05), but not recent (75.9.+-.5.4 vs. 76.+-.2.9% freezing) memory recall. FIG. 10C depicts another experiment where prolonged light in ACC also resulted in inhibition of remote (Control n=3, 78.0.+-.6.2% freezing; eNpHR3.0 n=8, 45.0.+-.5.2% freezing; P<0.05), but not recent (78.5.+-.12.7 vs. 74.3.+-.4.3% freezing) memory recall.
DETAILED DESCRIPTION
[0034] The present disclosure is believed to be useful for modifying memory function on a temporal basis. Specific applications of the present invention facilitate disrupting memory retrieval and/or emotional responses linked to memory retrieval. As many aspects of the example embodiments disclosed herein relate to and significantly build on previous developments in this field, the following discussion summarizes such previous developments to provide a solid understanding of the foundation and underlying teachings from which implementation details and modifications might be drawn. It is in this context that the following discussion is provided and with the teachings in these references incorporated herein by reference. While the present invention is not necessarily limited to such applications, various aspects of the invention may be appreciated through a discussion of various examples using this context.
[0035] It has been discovered that (temporal) disruption of the dorsal CA1 hippocampus circuit is effective to prevent contextual fear memory acquisition. Consistent therewith, a prevailing neural network theory suggests that the process of memory consolidation starts with short-term modifications in the connections between the hippocampus and the cortex, which enable the hippocampus to activate the relevant cortical sites that contribute to the complete memory, rather than to store the memory itself. While these cortical traces are repeatedly co-activated, gradual long-lasting changes in the connections between them occur until eventually these connections are strong enough to support the memory without any hippocampal involvement.
[0036] Surprisingly, it has been discovered that the disruption of the dorsal CA1 hippocampus circuit is effective to block fear-memory recall, even after cortical reorganization is believed to have occurred.
[0037] Consistent with various embodiments of the present disclosure, methods, systems or devices are discussed that relate to controlling neural circuits. Control over the neural circuit can include inhibition or excitation, which can each include coordinated firing, and/or modified susceptibility to external circuit inputs. For instance, inhibition can be accomplished using a light-activated protein, such as an ion channel and/or ionic pump (e.g., NpHR and NpHR variants). Such ion channels move the membrane potential of the neuron away from its threshold voltage to dissuade or inhibit action potentials. In another instance, excitation can be accomplished using a light-activated protein, such as an ion channel (e.g., ChR2 and ChR2 variants). Such ion channels can cause the membrane potential to move toward and/or past the threshold voltage, thereby exciting or encouraging action potentials. Consistent with various embodiments, a light-activated protein can be used to (temporarily) shift the resting potential of a neuron to increase or decrease its susceptibility to external circuit inputs. These various options can also be used in combination.
[0038] The devices and methods provided herein may reversibly affect memory function. For example, the methods described below may be used to control and/or characterize the neural circuitry that underlies long-term and short-term memory, as well as various types of memories, including fearful or stressful memories. The methods may also affect various stages of memory function (e.g., memory acquisition, consolidation, and recall). In some variations for affecting memory function (e.g., such as memory formation and/or retrieval), memory function is affected by applying light to neurons of the dorsal CA1 region of the hippocampus, in the basolateral amygdala (BLA), and/or in the anterior cingulated cortex (ACC) that express light-activated proteins. In the presence of light, these light-activated proteins may inhibit depolarization of the neurons, thereby disturbing the formation and/or retrieval of memories. While the exemplary methods are described in the context of the acquisition and recall of contextual remote and recent fear-based memories, it should be understood that the devices and methods disclosed herein may be used to affect other stages of memory function, as well as other types of memories (e.g., cued memories).
[0039] Various embodiments described herein and shown in the figures may be implemented together and/or in other manners. One or more of the items depicted in the drawings/figures can also be implemented in a more separated or integrated manner, or removed and/or rendered as inoperable in certain cases, as is useful in accordance with particular applications. For example, embodiments involving the treatments for PTSD as discussed herein may be implemented using temporally-controlled drug release. In view of the description herein, those skilled in the art will recognize that many changes may be made thereto without departing from the spirit and scope of the present invention.
[0040] Expressing Light-Activated Proteins in Target Cells
[0041] The activity of a neuron (e.g., neurons involved in memory function) may be affected using a variety of mechanisms. Deterministic methods of affecting neuronal activity may be used to control and/or characterize the neural circuits that underlie various brain functions. For example, neuronal responses may be affected by applying pharmacological agents (e.g., tetrodotoxin (TTX), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), picrotoxin, strychnine, etc.) and/or by electrical stimulation (e.g., electrodes). In some variations, neuronal activity may be affected by activating certain types of proteins on the membrane of the neuron, which may hyperpolarize or depolarize the cell membrane. For example, light-activated proteins that become permeable to certain ions (e.g., cations, anions) in the presence of light with a certain wavelength may be expressed in a neuron. Examples of light-activated proteins may include light-activated ion channels and/or pumps, which are further described below.
[0042] In some variations, microbial opsin genes may be adapted for uses in neuroscience. These opsins allow transduction of light pulse trains into millisecond-timescale membrane potential changes in specific cell types within the intact mammalian brain (e.g., channelrhodopsin (ChR2), Volvox channelrhodopsin (VChR1) and halorhodopsin (NpHR)). ChR2 is a rhodopsin derived from the unicellular green alga Chlamydomonas reinhardtii. The term "rhodopsin" as used herein is a protein that comprises at least two building blocks, an opsin protein, and a covalently bound cofactor, usually retinal (retinaldehyde). The rhodopsin ChR2 is derived from the opsin Channelopsin-2 (Chop2), originally named Chlamyopsin-4 (Cop4) in the Chlamydomonas genome. The temporal properties of one depolarizing channelrhodopsin, ChR2, include fast kinetics of activation and deactivation, affording generation of precisely timed action potential trains. For applications seeking long timescale activation, it has been discovered that the normally fast off-kinetics of the channelrhodopsins can be slowed. For example, certain implementations of channelrhodopsins apply 1 mW/mm.sup.2 light for virtually the entire time in which depolarization is desired, which can be less than desirable.
[0043] Light-activated proteins that generate hyperpolarization or inhibit depolarization of the membrane in response to light with certain wavelength(s) may be expressed in the excitatory neurons (e.g., glutamatergic neurons) of the dorsal CA1 region of the hippocampus (CA1), basolateral amygdala (BLA), and anterior cingulated cortex (ACC) regions. Table 1 below shows various examples of light-activated proteins that may be expressed in the excitatory neurons to inhibit depolarization or hyperpolarize the neurons in the presence of light of a certain wavelength. Further description of these and other light-activated proteins may be found in PCT App. No. PCT/US11/028893, titled "LIGHT SENSITIVE ION PASSING MOLECULES", filed on Mar. 17, 2011, which is incorporated by reference in its entirety. As used herein, "NpHR", "BR", "AR", and "GtR3" include wild type proteins and functional variants (including naturally occurring variants).
TABLE-US-00001 TABLE 1 Light- activated Biological Wavelength proteins Origin Sensitivity Defined Action NpHR Natronomonas 680 nm utility Inhibition pharaonis (with 3.0 series) (hyperpolarization) 589 nm max BR Halobacterium 570 nm max Inhibition helobium (hyperpolarization) AR Acetabulaira 518 nm max Inhibition acetabulum (hyperpolarization) GtR3 Guillardia theta 472 nm max Inhibition (hyperpolarization)
[0044] Embodiments of the present invention include relatively minor amino acid variants of the naturally occurring sequences. In one instance, the variants are greater than about 75% homologous to the protein sequence of the naturally occurring sequences. In other variants, the homology is greater than about 80%. Yet other variants have homology greater than about 85%, greater than 90%, or even as high as about 93% to about 95% or about 98%. Homology in this context means sequence similarity or identity, with identity being preferred. This homology can be determined using standard techniques known in the field of sequence analysis. The compositions of embodiments of the present invention include the protein and nucleic acid sequences provided herein, including variants which are more than about 50% homologous to the provided sequence, more than about 55% homologous to the provided sequence, more than about 60% homologous to the provided sequence, more than about 65% homologous to the provided sequence, more than about 70% homologous to the provided sequence, more than about 75% homologous to the provided sequence, more than about 80% homologous to the provided sequence, more than about 85% homologous to the provided sequence, more than about 90% homologous to the provided sequence, or more than about 95% homologous to the provided sequence.
[0045] Provided herein are non-human animals comprising a light-activated protein expressed on the cell membrane of excitatory neurons in the dorsal CA1 field of the hippocampus, anterior cingulated cortex, and/or basolateral amygdala of the animal, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, wherein the illumination of the protein reversibly affects memory function. In some embodiments, the light-activated protein is selected from the group consisting of NpHR, BR, AR and GtR3 described herein. For example, any of the NpHR proteins described herein may be expressed on the cell membrane of the target neurons.
[0046] Also provided herein are brain tissue slices comprising a brain region selected from the group consisting of the dorsal CA1 field of the hippocampus, the basolateral amygdala, and the anterior cingulated cortex, wherein a light-activated protein is expressed on the cell membrane of excitatory neurons of the brain region, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, wherein the illumination of the protein reversibly affects memory function. In some embodiments, the brain tissue slices are cultured tissue slices taken from the non-human animals described herein. In some embodiments, the light-activated protein is selected from the group consisting of NpHR, BR, AR and GtR3 described herein. For example, any of the NpHR proteins described herein may be expressed on the cell membrane of the target neurons.
[0047] In some embodiments, neurons of the CA1, BLA, and/or ACC regions may express ChR2. Unless otherwise stated, the invention includes a number of similar variants. Examples include, but are not limited to, Chop2, ChR2-310, Chop2-310, and Volvox channelrhodopsin (VChR1). For further details on VChR1, reference can be made to "Red-shifted optogenetic excitation: a tool for fast neural control derived from Volvox carteri," Nat Neurosci. June 2008, 11(6):631-3. Epub 2008 Apr. 23, which is fully incorporated herein by reference. In other implementations, similar modifications can be made to other opsin or light-activated molecules. For instance, modifications/mutations can be made to ChR2 or VChR1 variants. Moreover, the modified variants can be used in combination with light-activated ion pumps.
[0048] As used herein, stimulation of a target cell is generally used to describe modification of properties of the cell. For instance, the stimulus of a target cell may result in a change in the properties of the cell membrane that can lead to the depolarization or polarization of the target cell. In a particular instance, the target cell is a neuron and the stimulus may affect the transmission of impulses by facilitating or inhibiting the generation of impulses (action potentials) by the neuron.
[0049] For further details on light-activated proteins (e.g., opsins), reference can be made to PCT Publ. No. WO 2010/056970, entitled "OPTICALLY-BASED STIMULATION OF TARGET CELLS AND MODIFICATIONS THERETO," to Deisseroth et al., which is fully incorporated herein by reference.
[0050] Embodiments of the present disclosure are directed toward implementation of bistable changes in the excitability of targeted populations. This includes, but is not necessarily limited to, the double-mutant ChR2-C128S/D156A. This double-mutant ChR2-C128S/D156A has been found to be well-tolerated in cultured hippocampal neurons and preserved the essential SFO properties of rapid step-like activation with single brief pulses of blue light, and deactivation with green or yellow light. In particular, the activation spectrum of ChR2-C128S/D156A peaks at 445 nm. A second deactivation peak was found at 390-400 nm, with faster but less complete deactivation by comparison with the 590 nm deactivation peak. Peak photocurrents in cells expressing ChR2-C128S/D156A were found to be robust and comparable to those of ChR2-D156A (231.08.+-.31.19 s.e.m; n=9 cells and 320.96.+-.78.26 s.e.m; n=7 cells, respectively).
[0051] Individual transfected and patch-clamped neurons were next activated with 100 ms pulses of 470 nm light. To ensure over very long recordings that current decay would not be attributable to cell rundown, each cell was deactivated with prolonged 590 nm light pulses at distinct intervals to determine the magnitude of remaining SFO current at each time point. Surprisingly, neurons expressing ChR2-C128S/D156A gave rise to sustained photocurrents that were more stable than those from cells expressing either single mutant alone. Fitting a mono-exponential decay curve to the ratio of Ideactivation/Iactivation over time revealed a spontaneous decay time constant of 29.3 minutes for ChR2-C128S/D156A, indicating that the C128 and D156 mutations act synergistically to delay the decay of the open state of ChR2. Consistent with the required improvement for the anticipated application to complex mammalian behaviors, significant portions of the double-mutant SFO current were still present up to 20 minutes after the single photoactivation pulse.
[0052] Based on these surprisingly slow decay kinetics, the double-mutant gene is referred to as SSFO (for stabilized step-function opsin) gene. SSFO is also used as shorthand for the active protein. Both residues likely are involved in ChR2 channel closure (gating), and both mutations likely stabilize the open state configuration of the channel
[0053] Without being limited by theory, aspects of the present disclosure relate to the discovery that SSFO may be completely blocked in photocycle progression, and may therefore represent the maximal stability possible with photocycle engineering. For instance, in contrast to ChR2 C128X and ChR2-D156A, the SSFO photocycle does not appear to access additional inactive deprotonated side products which likely split off the photocycle at later photocycle stages not reached in this mutant, in turn making the SSFO even more reliable for repeated use in vivo than the parental single mutations.
[0054] Embodiments of the present disclosure are directed toward the sensitivity of the SSFO to light. For instance, channelrhodopsins with slow decay constants effectively act as photon integrators. This can be particularly useful for more-sensitive, less-invasive approaches to optogenetic circuit modulation, still with readily titratable action on the target neuronal population via modulation of light pulse length. It has been discovered that, even at extraordinarily low light intensities (as low as 8 .mu.W mm.sup.-2), hundreds of picoamps of whole-cell photocurrents could be obtained from neurons expressing SSFO, which increased with monoexponential kinetics in response to 470 nm light during the entire time of illumination. Other aspects relate to the use of activation time constants that are linearly correlated with the activation light power on a log-log scale, which is indicative of a power-law relationship and suggesting that the SSFO is a pure integrator, with total photon exposure over time as the only determinant of photocurrent. For instance, it is believed that the number of photons per membrane area required for photocurrents to reach a given sub-maximal activation (time to r) is constant regardless of activation light power.
[0055] Example embodiments of the present disclosure relate to the use of a hybrid ChRI/VChRI chimera, which contains no ChR2 sequence at all and is derived from two opsins genes that do not express well individually, and is herein referred to as C1V1. Embodiments of the present disclosure also relate to improvements of the membrane targeting of VChR1 through the addition of a membrane trafficking signal derived from the K.sub.ir2.1 channel. Confocal images from cultured neurons expressing VChR1-EYFP revealed a large proportion of intracellular protein compared with ChR2; therefore, to improve the membrane targeting of VChR1, we added a membrane trafficking signal derived from the Kir2.1 channel. Membrane targeting of this VChR1-ts-EYFP was slightly enhanced compared with VChR1-EYFP; however, mean photocurrents recorded from cultured hippocampal neurons expressing VChR1-ts-EYFP were only slightly larger than those of VChR1-EYFP. Accordingly, embodiments of the present disclosure relate VChR1 that is modified by exchanging helices with corresponding helices from other ChRs. For example, robust improvement has been discovered in two chimeras where helices 1 and 2 were replaced with the homologous segments from ChR1. It was discovered that whether splice sites were in the intracellular loop between helices 2 and 3 (at ChR1 residue Ala145) or within helix 3 (at ChR1 residue Trp163), the resulting chimeras were both robustly expressed and showed similarly enhanced photocurrent and spectral properties. This result was unexpected as ChR1 is only weakly expressed and poorly integrated into membranes of most mammalian host cells. The resulting hybrid ChR1/VChR1 chimera is herein referred to as C1V1.
[0056] Aspects of the present disclosure relate to the expression of C1V1 in cultured neurons (e.g., hippocampal neurons). Experimental tests have shown a number of surprising and useful results, which are discussed in more detail hereafter. C1V1-EYFP exhibits surprisingly improved average fluorescence compared with VChR1-EYFP. Whole cell photocurrents in neurons expressing C1V1 were much larger than those of VChR1-EYFP and VChR1-ts-EYFP, and ionic selectivity was similar to that of ChR2 and VChR1. The addition of the Kir2.1 trafficking signal between C1V1 and YFP further enhanced photocurrents by an additional 41%. (C1V1-ts-EYFP mean photocurrents were extremely large, nearly tenfold greater than wild type (WT) VChR1). Mean fluorescence levels closely matched the measured photocurrents (mean fluorescence 9.3.+-.1, 19.6.+-.3.4, 19.8.+-.2.8 and 36.3.+-.3.8 for VChR1-EYFP, VChR1-ts-EYFP, C1V1-EYFP and C1V1-ts-EYFP, respectively), suggesting that the increase in photocurrent sizes resulted mainly from the improved expression of these channels in mammalian neurons. Total somatic fluorescence (measured as integrated pixel density) was linearly correlated with photocurrent size in individual recorded/imaged cells across the different constructs (VChR1, VChR1-ts-EYFP, C1V1, C1V1-ts-EYFP). This suggests (without being limited by theory) that the increased photocurrent of C1V1 results from functional expression changes in neurons.
[0057] Various embodiments of the present disclosure relate to opsins or light-activated proteins with fast decay constants. This property can be particularly useful for providing precise control over spiking, e.g., in order to interfere minimally with intrinsic conductances, trigger single spikes per light pulse and/or minimize plateau potentials during light pulse trains. Experimental results suggest that the light-evoked photocurrents recorded in C1V1-ts-EYFP decayed with a time constant similar to that of VChR1. Aspects of the present disclosure are therefore directed toward modifications in the chromophore region to improve photocycle kinetics, reduced inactivation and/or possible further red-shifted absorption.
[0058] One embodiment is directed toward a corresponding ChETA mutation E162T, which experiments suggest provides an accelerated photocycle (e.g., almost 3-fold), (reference can be made to Gunaydin, et al., Ultrafast optogenetic control, Nat Neurosci, 2010, which is fully incorporated herein by reference). Surprisingly, this mutation was shown to shift the action spectrum hypsochromic to 530 nm, whereas analogous mutations in ChR2 or other microbial rhodopsins have caused a red-shift.
[0059] Another embodiment is directed toward a mutation of glutamate-122 to threonine (C1V1-E122T). Experimental tests showed that C1V1-E122T is inactivated only by 26% compared to 46% inactivation of ChR2; in addition, the spectrum was further red-shifted to 546 nm.
[0060] Another embodiment of the present disclosure is directed toward a double mutant of C1V1 including both E122T and E162T mutations. Experimental tests have shown that the inactivation of the current was even lower than in the E122T mutant and the photocycle was faster compared to E162T. This suggests that multiple useful properties of the individual mutations were conserved together in the double mutant.
[0061] Polynucleotides Encoding Light-Activated Proteins
[0062] Light-activated proteins or opsins described herein may be delivered into neurons by methods known in the art, such as by a polynucleotide comprising a sequence encoding the proteins. In some embodiments, the polynucleotide comprises an expression cassette. In some embodiments, the polynucleotide is a vector, such as a viral vector selected from the group consisting of an AAV vector, a retroviral vector, an adenoviral vector, an HSV vector, and a lentiviral vector.
[0063] For example, neurons may be contacted with a vector comprising a nucleic acid sequence encoding a light-activated protein operably linked to a cell specific promoter, wherein said neurons express the light-activated protein on the cell membrane. In some variations, the cell specific promoter is a calcium/calmodulin-dependent protein kinase Ha (CaMKII.alpha.) promoter. In some variations, a nucleic acid sequence encoding light activatable eNpHR3.1 or eNpHR3.0 is operably linked to a CaMKII.alpha. promoter in the vector. In some variations, the light-activated protein is expressed in excitatory glutamatergic neuron in the CA1 region, BLA and/or ACC. Any vectors that may be used for gene delivery may be used. In some variations, a viral vector (such as AAV, adenovirus, lentivirus, a retrovirus) may be used.
[0064] In some embodiments, the vector is a recombinant AAV vector. AAV vectors are DNA viruses of relatively small size that can integrate, in a stable and sitespecific manner, into the genome of the cells that they infect. They are able to infect a wide spectrum of cells without inducing any effects on cellular growth, morphology or differentiation, and they do not appear to be involved in human pathologies. The AAV genome has been cloned, sequenced and characterized. It encompasses approximately 4700 bases and contains an inverted terminal repeat (ITR) region of approximately 145 bases at each end, which serves as an origin of replication for the virus. The remainder of the genome is divided into two essential regions that carry the encapsidation functions: the left-hand part of the genome, that contains the rep gene involved in viral replication and expression of the viral genes; and the right-hand part of the genome, that contains the cap gene encoding the capsid proteins of the virus.
[0065] AAV vectors may be prepared using standard methods in the art. Adeno-associated viruses of any serotype are suitable (see, e.g., Blacklow, pp. 165-174 of "Parvoviruses and Human Disease" J. R. Pattison, ed. (1988); Rose, Comprehensive Virology 3:1, 1974; P. Tattersall "The Evolution of Parvovirus Taxonomy" In Parvoviruses (J R Kerr, S F Cotmore. M E Bloom, R M Linden, C R Parrish, Eds.) p5-14, Hudder Arnold, London, U K (2006); and D E Bowles, J E Rabinowitz, R J Samulski "The Genus Dependovirus" (J R Kerr, S F Cotmore. M E Bloom, R M Linden, C R Parrish, Eds.) p15-23, Hudder Arnold, London, UK (2006), the disclosures of which are hereby incorporated by reference herein in their entireties). Methods for purifying for vectors may be found in, for example, U.S. Pat. Nos. 6,566,118, 6,989,264, and 6,995,006 and WO/1999/011764 titled "Methods for Generating High Titer Helper-free Preparation of Recombinant AAV Vectors", the disclosures of which are herein incorporated by reference in their entirety. Preparation of hybrid vectors is described in, for example, PCT Application No. PCT/US2005/027091, the disclosure of which is herein incorporated by reference in its entirety. The use of vectors derived from the AAVs for transferring genes in vitro and in vivo has been described (See e.g., International Patent Application Publication Nos: 91/18088 and WO 93/09239; U.S. Pat. Nos. 4,797,368, 6,596,535, and 5,139,941; and European Patent No: 0488528, all of which are herein incorporated by reference in their entirety). These publications describe various AAV-derived constructs in which the rep and/or cap genes are deleted and replaced by a gene of interest, and the use of these constructs for transferring the gene of interest in vitro (into cultured cells) or in vivo (directly into an organism). The replication defective recombinant AAVs according to the invention can be prepared by co-transfecting a plasmid containing the nucleic acid sequence of interest flanked by two AAV inverted terminal repeat (ITR) regions, and a plasmid carrying the AAV encapsidation genes (rep and cap genes), into a cell line that is infected with a human helper virus (for example an adenovirus). The AAV recombinants that are produced are then purified by standard techniques.
[0066] In some embodiments, the vector(s) for use in the methods of the invention are encapsidated into a virus particle (e.g. AAV virus particle including, but not limited to, AAV1, AAV2, AAV3, AAV4, AAV5, AAV6, AAV7, AAV8, AAV9, AAV10, AAV11, AAV12, AAV13, AAV14, AAV15, and AAV16). Accordingly, the invention includes a recombinant virus particle (recombinant because it contains a recombinant polynucleotide) comprising any of the vectors described herein. Methods of producing such particles are known in the art and are described in U.S. Pat. No. 6,596,535.
[0067] For the animal cells described herein, it is understood that one or more vectors may be administered to neural cells, heart cells, or stem cells. If more than one vector is used, it is understood that they may be administered at the same or at different times to the animal cells.
[0068] For example, in some variations, C1V1 opsin genes in neurons were carried out by generating lentiviral vectors encoding C1V1-ts-EYFP and various point mutation combinations discussed herein. The opsins were then expressed in cultured hippocampal neurons and recorded whole-cell photocurrents under identical stimulation conditions (2 ms pulses, 542 nm light, 5.5 mW/mm.sup.2). Photocurrents in cells expressing C1V1, C1V1-E162T and C1V1-E122T/E162T were all robust and trended larger than the photocurrents of ChR2-H134R. The experiments also included a comparison of integrated somatic YFP fluorescence and photocurrents from cells expressing C1V1-E122T/E162T and from cells expressing ChR2-H134R. Surprisingly, C1V1-E122T/E162T cells showed stronger photocurrents than ChR2-H134R cells at equivalent fluorescence levels. This suggests that C1V1 could possess a higher unitary conductance compared with ChR2-H134R. The test results suggest that the kinetics of C1V1-E122T were slower than those of C1V1-E122T/E162T and that cells expressing C1V1-E122T responded more strongly to red light (630 nm) than cells expressing the double mutant. This can be particularly useful for generating optogenetic spiking in response to red light.
[0069] Consistent with various embodiments of the present disclosure, inhibitory and/or excitatory neurons residing within the same microcircuit are be targeted with the introduction of various light-activated proteins (e.g., opsins). Experimental tests were performed by separately expressed C1V1-E122T/E162T and ChR2-H134R under the CaMKII.alpha. promoter in cultured hippocampal neurons. Cells expressing C1V1-E122T/E162T spiked in response to 2 ms green light pulses (560 nm) but not to violet light pulses (405 nm). In contrast, cells expressing ChR2-H134R spiked in response to 2 ms 405 nm light pulses, but not to 2 ms 561 nm light pulses.
[0070] Various embodiments of the present disclosure relate to independent activation of two neuronal populations within living brain slices. Experimental tests were performed by CaMKII.alpha.-C1V1-E122T/E162Tts-eYFP and EF1a-DIO-ChR2-H134R-EYFP in mPFC of 20 PV::Cre mice. In non-expressing PYR cells, 405 nm light pulses triggered robust and fast inhibitory postsynaptic currents (IPSCs) due to direct activation of PV cells, while 561 nm light pulses triggered only the expected long-latency polysynaptic IPSCs arising from C1V1-expressing pyramidal cell drive of local inhibitory neurons.
[0071] Light Activation of Proteins Expressed in Neurons
[0072] Any device that is capable of applying light having a wavelength to activate the light-activated proteins expressed in a neuron may be used to depolarize and/or hyperpolarize the neuron. For example, a light-delivery device (100) for activating ion channels and/or ionic pumps to affect the membrane voltage of one or more neurons depicted in FIG. 1 may be used. As shown there, the light-delivery device (100) is configured to provide optical stimulus to a target region of the brain. The light-delivery device (100) may comprise a base (102), a cannula guide (104) that is attached to the base, and one or more optical conduits (106) attached to the base via the cannula guide. The base (102) may comprise one or more light delivery ports (108) that are positioned to deliver light from the optical conduits (106) to targeted tissue regions (101), such as the CA1 region (103). The optical conduits (106) may be optical fibers, where the proximal end of the fiber is attached to an optical light source (not shown), and the distal end is in communication with the light delivery ports (108). The optical light source may be capable of providing continuous light and/or pulsed light, and may be programmable to provide light in pre-determined pulse sequences. The light delivery device (100) may have any number of optical conduits (106) as may be desirable, e.g., 1, 2, 3, 4, 5, 10, 15, 20, etc. The optical conduits (106) may each carry light of the same or different wavelengths. The delivered light may have a wavelength between 450 nm and 600 nm, such as yellow or green light.
[0073] The light delivery device (100) may have any number of light delivery ports (108) as may be desirable, e.g., 1, 2, 3, 4, 5, 10, 15, 20, etc. In some variations, there may be the same number of light delivery ports as optical conduits while in other variations, there may be different number of optical conduits and light delivery ports. For example, there may be a single optical conduit that conveys light to two or more light delivery ports. Alternatively or additionally, a single optical conduit may connect to a single light delivery port. The cannula guide (104) may be configured to help secure and align the optical conduits (106) with the light delivery ports (108). In some embodiments, the light delivery device (100) is configured to deliver bilateral light to the CA1 region (103) to affect the formation and retrieval of memories. Light delivery devices may also comprise one or more measurement electrodes that may be configured for measuring neural activity. For example, measurement electrodes may record changes in the membrane potential (e.g., action potentials) and/or current flow across a membrane of one or more neurons as the neurons respond to a stimulus. In some variations, the measurement electrodes may measure the electrical response of one or more neurons to optical stimulation. Measurement electrodes may be extracellular or intracellular electrodes.
[0074] Methods of Affecting Memory Function
[0075] As described herein, the target tissue regions (101) may include neural tissue with cells that have light-activated proteins designed to modify the membrane voltage of the cells in response to light. In some variations, light-activated proteins may be used to disrupt the formation and/or retrieval of memories by inhibiting the depolarization of the neurons in the CA1, BLA, and ACC regions of the brain. Embodiments of the present disclosure are directed towards disrupting memory acquisition, recall and/or associations between memory and emotional responses, such as fear. In a particular embodiment, function of a neural circuit involved in memory is disrupted by activation of light-activated ion channels (e.g., using NpHR, BR, AR, etc.) and/or pumps (e.g., a proton pump GtR3). In certain implementations, this disruption can be implemented during memory formation. In other implementations, this disruption can be implemented before or during memory retrieval. This can be particularly useful for psychiatric or neurological disorders involving memory recall, such as PTSD. Consistent with certain embodiments, the disruption can be triggered in response to a memory trigger event or other external stimulus that is presented and/or controlled for the disruption. For instance, the disruption can be provided in response to a trigger for a memory to an individual conditioned to respond to the trigger. In another instance, an individual can actively trigger the disruption. For instance, an individual may trigger the disruption when experiencing a memory associated with PTSD. Other embodiments of the present disclosure are directed toward encouraging memory acquisition, recall and/or associations between memory and emotional responses. The methods described herein may be used to ascertain the role of neuron(s) and/or neuronal circuits in memory function, and/or to treat disorders associated with memory impairment.
[0076] In some embodiments, the methods provided herein for reversibly affecting memory retrieval or formation in an individual comprise administering a polynucleotide encoding a light-activated protein to the dorsal CA1 field of the hippocampus, anterior cingulated cortex, or basolateral amygdala in the individual, wherein light-activated protein is expressed on the cell membrane of the excitatory neurons in the dorsal CA1 field of the hippocampus, anterior cingulated cortex, or basolateral amygdala and the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, whereby activating the protein by the light reversibly affects memory retrieval or formation of an event in the individual. In some embodiments, the methods provided herein for reversibly affecting memory retrieval or formation in an individual comprise inhibiting depolarization of excitatory neurons in the dorsal CA1 field of the hippocampus, anterior cingulated cortex, or basolateral amygdala during memory retrieval or formation of an event in an individual, wherein a light-activated protein is expressed on the cell membrane of the excitatory neurons in the dorsal CA1 field of the hippocampus, anterior cingulated cortex, or basolateral amygdala of the individual, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light. In some embodiments, the event is a fearful event.
[0077] Provided herein are methods for treating post-traumatic stress disorder in an individual comprising: administering a polynucleotide encoding a light-activated protein to the dorsal CA1 field of the hippocampus, anterior cingulated cortex, or basolateral amygdala in the individual, wherein light-activated protein is expressed on the cell membrane of the excitatory neurons in the dorsal CA1 field of the hippocampus, anterior cingulated cortex, or basolateral amygdala and the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light, whereby activating the protein by the light reversibly affects memory retrieval or formation of an event in the individual.
[0078] Provided herein are methods for screening a pharmacological agent that affects memory retrieval or formation comprising: a) contacting excitatory neurons in the dorsal CA1 field of the hippocampus, anterior cingulated cortex, or basolateral amygdala during memory retrieval or formation of an event in a non-human animal with a pharmacological agent, wherein the non-human animal comprises a light-activated protein expressed on the cell membrane of excitatory neurons in the dorsal CA1 field of the hippocampus, anterior cingulated cortex, or basolateral amygdala of the animal, wherein the protein is responsive to light and is capable of inhibiting depolarization of the neurons when the neurons are illuminated with the light; b) inhibiting depolarization of the excitatory neurons in the dorsal CA1 field of the hippocampus, anterior cingulated cortex, or basolateral amygdala during memory retrieval or formation of an event; and c) determining if the pharmacological agent affects memory retrieval or formation in the presence or absence of the light.
[0079] As used herein, an "individual" is a mammal including a human. Mammals include, but are not limited to, farm animals, sport animals, pets, primates, mice and rats. Individuals also include companion animals including, but not limited to, dogs and cats. In one aspect, an individual is a human. In another aspect, an individual is a non-human animal. As used herein, "non-human animals" include non-human mammals.
[0080] One example of a method for controlling or modifying memory function consistent with the embodiments of the present disclosure is depicted in FIG. 2. A temporal-trigger event (202) provides a reference point for implementing control over memory function. As discussed herein, the temporal nature of the control can be particularly useful. Although not limited thereto, the memory-trigger event (202) can be linked to a training event. For instance, an individual (e.g., non-human animals, mammals, humans) can be introduced to a stimulus designed to train the individual to respond to a particular stimulus. The memory-trigger event (202) could be the introduction of the particular stimulus to the individual. In another instance, the memory-trigger event could be in response to a response or action of the individual (e.g., an indication that the individual is experiencing a PTSD event). Control instructions (204) determine how stimulus source (206) applies a stimulus (208) to a cell population (210). These control instructions can be determined and applied as a function of a desired target. The desired target can be defined by, for example, one or more of temporal attributes, spatial location and/or cell-type. The stimulus (208) results in the modification of memory function (212). The effect of the stimulus can then be monitored, observed and/or assessed (214). The monitoring can be used to adjust (216) the control instructions (204), thereby fine-tuning the stimulus for the intended result. Various embodiments discussed herein provide further examples that can be used in connection with (or in addition to) such a process for controlling and characterizing the neural circuits that underlie memory function.
[0081] Affecting Memory Retrieval by Inhibiting Neurons of CA1 and ACC
[0082] One variation of a method for disrupting memory retrieval may comprise inhibiting the excitatory neurons of the CA1 region (e.g., by blocking or reducing membrane depolarization, and/or by promoting membrane hyperpolarization). Light-activated ion channels, such as eNpHR3.1 or NpHR3.0, may be expressed on neurons located in the CA1 region of an individual by administering a polynucleotide encoding the channel protein to the region. The eNpHR3.1 or NpHR3.0 ion channel is activated in the presence of yellow light (e.g., having a wavelength of about 591 nm). The individual may be provided with a light-delivery device, such as the light-delivery device (100) described above. The light-delivery device may be positioned on the individual such that yellow light is capable of being delivered to the CA1 neurons. After or during the retrieval of a memory (e.g., any undesired memory such as a fearful or stressful memory), the light-delivery device may be activated to deliver yellow light to the CA1 neurons, thereby inhibiting their depolarization, and disrupting the recall of the memory. Once the memory recall has been sufficiently disrupted, the light-delivery device may be de-activated. Upon de-activation of the light-delivery device, the individual may regain the ability retrieve memories without disruption. This method may be used to disrupt recall of recent memories (e.g., memories of events that occurred less than one day in the past) and recall of remote memories (e.g., memories of events that occurred more than one day in the past, 1 week in the past, 2 weeks in the past, 4 weeks in the past, 8 weeks or more in the past, etc.). In some variations, excitatory neurons of the ACC may express similar light-activated proteins, and may be similarly inhibited to disrupt the retrieval of remote memories.
[0083] Methods for disrupting memory retrieval comprising inhibiting the neurons of the CA1 region may be used in a non-human animal, such as a mouse. For example, mice expressing eNpHR3.1 or NpHR3.0 in the neurons of the CA1 region were trained in a customized FC chamber, where they were introduced into context A and then presented twice with a tone followed by a foot-shock. In a testing session, green light delivered to the eNpHR3.1 or NpHR3.0 CA1 neurons interfered with the ability of the mice to recall the memory (i.e., a fearful or stressful memory), as measured by a reduction in freezing (e.g., contextual freezing). In a separate testing session where the eNpHR3.1 or NpHR3.0 CA1 neurons are not exposed to light, the mice are able to recall the fearful memory formed during the training session, as measured by normal rates of freezing. In some variations, the testing session may occur one day or less after the training session, while in other variations, the testing session may occur four weeks or more after the training session. Applying green light to the eNpHR3.1 CA1 neurons of the mice reversibly inhibits the depolarization of the neurons, thereby disrupting the recall of recent and/or remote contextual fearful memories. Removing the green light from the eNpHR3.1 or NpHR3.0 CA1 neurons restores the ability of the mice to recall recent and/or remote contextual fearful memories.
[0084] Methods for reversibly disrupting the recall or retrieval of remote memories may also be used after the memory has been repeatedly recalled and consolidated. For example, mice having CA1 neurons expressing eNpHR3.1 or NpHR3.0 may be trained as described above. In a testing session five weeks after the training session, the mice were able to recall the memory formed during training, however, when the eNpHR3.1 or NpHR3.0 CA1 neurons were exposed to green light, they were no longer able to recall the memory. Subsequent exposure of the eNpHR3.1 or NpHR3.0 CA1 neurons to green light disrupted retrieval of the fearful memory. In some methods, memory recall may be disrupted by exposing the eNpHR3.1 or NpHR3.0 CA1 neurons to light upon initiation of the memory recall and/or during the memory recall. For example, applying green light to the eNpHR3.1 or NpHR3.0 CA1 neurons at the same time as recall initiation (e.g., at the beginning of the testing session) disrupts recall of the memory. When green light was applied to the eNpHR3.1 or NpHR3.0 CA1 neurons during memory recall (e.g., applying the light some time after the testing session has begun, such as in the middle of the testing session), the mice initially recalled and responded to the fearful memory (by freezing), but then quickly ceased exhibiting the fear response after the light was applied. These methods may be used in an individual with PTSD having CA1 neurons expressing eNpHR3.1, where a light-delivery device may be activated at the same time and/or during the retrieval of a fearful memory in order to reversibly disrupt and/or discontinue recall of that fearful memory. Subsequent de-activation of the light-delivery device may restore the ability of the individual to recall this and other memories.
[0085] Methods for disrupting memory retrieval comprising inhibiting the neurons of the ACC region may be used in a non-human, such as a mouse. For example, mice expressing eNpHR3.1 in the neurons of the ACC may be trained as described above. In a testing session four weeks after the training session, green light delivered to the eNpHR3.1 ACC neurons interfered with the ability of the mice to recall the memory formed during training. Removing the green light from the eNpHR3.1 CA1 neurons restores the ability of the mice to remote fearful memories.
[0086] Affecting Memory Formation by Inhibiting CA1 Hippocampus
[0087] While inhibiting the depolarization of excitatory neurons in the CA1 region (and in some cases hyperpolarizing these neurons) may interfere with memory retrieval, such inhibition may also disrupt memory formation. One variation of a method for disrupting memory formation may comprise inhibiting the neurons of the CA1 region during the formation of a memory such as a contextual memory. Light-activated ion channels, such as eNpHR3.1, may be expressed on neurons located in the CA1 region of an individual as previously described. The individual may be provided with a light-delivery device, such as the light-delivery device (100) described herein. During the formation of a memory (e.g., a fearful or stressful memory), the light-delivery device may be activated to deliver green light to the CA1 neurons, thereby inhibiting their depolarization and disrupting the formation of the memory. Once the memory formation has been sufficiently disrupted, the light-delivery device may be de-activated. Upon de-activation of the light-delivery device, the individual may regain the ability form memories without disruption.
[0088] Methods for disrupting memory formation comprising inhibiting the neurons of the CA1 region may be used in a non-human animal, such as a mouse. For example, mice expressing eNpHR3.1 in the neurons of the CA1 region were trained in a customized FC chamber, while delivering green light to the eNpHR3.1 CA1 neurons. During the training, the mice were introduced into a first context and then exposed to a tone followed by a foot-shock. In a subsequent testing session without the application of light, the mice exhibited no memory of the training, as measured by a reduction in contextual freezing. The same mice underwent a separate training session where the eNpHR3.1 CA1 neurons were not exposed to light. The mice were then able to recall the memory in a subsequent testing session. In some variations, the testing session may occur one day or less after the training session, while in other variations, the testing session may occur four weeks after the training session. Applying green light to the eNpHR3.1 CA1 neurons of the mice reversibly inhibited the depolarization of the neurons, thereby disrupting the formation of recent and/or remote memories. Removing the green light from the eNpHR3.1 CA1 neurons restored the ability of the mice to form fearful memories.
[0089] Affecting Memory Formation by Inhibiting Basolateral Amygdala
[0090] Some variations of methods for disrupting memory formation may comprise delivering light to neurons expressing eNpHR3.1 in the BLA during memory formation. Light-activated ion channels, such as eNpHR3.1, may be expressed on neurons located in the BLA of an individual. The individual may be provided with a light-delivery device, such as the light-delivery device (100) described above. The light-delivery device may be positioned on the individual such that green light is capable of being delivered to the BLA neurons. After or during the formation of a memory (e.g., a fearful or stressful memory), the light-delivery device may be activated to deliver green light to the BLA neurons, thereby inhibiting their depolarization, and disrupting the formation of the memory. Once the memory formation has been sufficiently disrupted, the light-delivery device may be de-activated. Upon de-activation of the light-delivery device, the individual may regain the ability acquire memories without disruption.
[0091] Methods for disrupting memory acquisition comprising inhibiting the neurons of the BLA region may be used in a non-human animal, such as a mouse. For example, green light may be delivered to mice expressing eNpHR3.1 in the neurons of the BLA during a fear conditioning training session as described above. The mice may then be tested to determine whether they acquired the fearful memory of the training session. Green light delivered to the BLA during the training session may disrupt the ability of the mice to acquire a fearful or stressful memory.
[0092] Screening for Drugs that Repair Memory Formation or Retrieval
[0093] Controlling the neural circuit that underlies memory function may provide a tool for evaluating the effect of pharmacological agents on memory retrieval. For example, inhibiting the neurons expressing eNpHR3.1 of the CA1 region and/or ACC and/or BLA may be used to evaluate the effectiveness of various pharmacological agents for the restoration of memory recall. One example of a method for identifying a pharmacological agent that activates depolarization or excitation of non-human excitatory neurons in the CA1 region and/or ACC and/or BLA is depicted in FIG. 3A. The method (300) may comprise delivering a light-activated protein to the CA1 region and/or ACC and/or BLA of the brain (302) and inhibiting depolarization of excitatory neurons of the CA1 and/or ACC region (303). As described above, inhibiting depolarization may comprise applying light having a selected wavelength (e.g., yellow or green) to eNpHR3.1 ion channels expressed on the neurons of the CA1 and/or ACC region to prevent the generation of action potentials. Other types of light-activated channels may also be expressed to inhibit depolarization of these excitatory cells, such as variants of NpHR, BR, AR, and proton pumps such as GtR3. The effect of the inhibition from activating the eNpHR3.1 ion channels may be electrically measured by using loose-cell or whole-cell patch clamp methods (304). In some variations, the electrical activity of the excitatory cells of the CA1 and/or ACC region may be measured using single electrodes and/or multielectrode arrays. The inhibited neurons of the CA1 and/or ACC region may then be contacted with a test pharmacological agent (306). The electrical activity of the neurons may be similarly measured (308). The electrical measurements of the excitatory neurons of the CA1 region and/or ACC and/or BLA before and after contacting with the test pharmacological agent may be compared to determine if the test agent activates and/or restores the depolarization of the neurons (310). The method (300) may be used repeatedly as desired to screen any number or variety of pharmacological agents.
[0094] One example of a method for identifying a pharmacological agent that may be effective for restoring memory formation or retrieval in a non-human animal is depicted in FIG. 3B. The method (320) may comprise delivering a light-activated protein to the CA1 region and/or ACC and/or BLA of the brain (322) and applying light have a selected wavelength (e.g., yellow or green) to eNpHR3.1 ion channels expressed on the neurons of the CA1 and/or ACC and/or BLA region to prevent the generation of action potentials (323). Other types of light-activated channels may also be expressed to inhibit depolarization of these excitatory cells, such as variants of NpHR, BR, AR, and proton pumps such as GtR3. The response of the non-human animal in the presence of the light during memory formation or retrieval may be measured (324). In some variations, the memory may be formed during a training session where the individual is introduced into context A and exposed to a tone accompanied by a foot-shock, and the response to memory retrieval may be freezing when introduced into the context A and/or when the tone is played. The inhibited neurons of the CA1 and/or ACC region may then be contacted with a test pharmacological agent (326). The response of the non-human animal may be similarly measured (328). The response of the non-human animal before and after contacting with the test pharmacological agent may be evaluated to determine if the test agent affects memory formation or retrieval in the presence of light (330). In some variations, the method (320) may be used during memory formation (e.g., a training session) to evaluate the effect of the pharmacological agent on memory formation. The method (320) may also be used during memory retrieval (e.g., a testing session some time after a training session) to evaluate the effect of the pharmacological agent on memory retrieval. The method (320) may be used repeatedly as desired to screen any number or variety of pharmacological agents.
[0095] Variations on Temporal Precision that can Apply to all Above Methods
[0096] In some variations of the methods described above, inhibition of the neurons expressing the light-activated protein (e.g., eNpHR3.1 or eNpHR3.0) may be applied at a precise point in time. For example, neurons expressing eNpHR3.1 in the CA1 region may illuminated by light during the testing session only. Temporally precise inhibition of neurons expressing eNpHR3.1 may disrupt memory recall. Precisely applying light to neurons expressing eNpHR3.1 in the CA1 region of mice during the testing session may inhibit remote and/or recent fear memory retrieval in an animal. In other variations of the methods described above, inhibition of the neurons expressing eNpHR3.1 may be applied over a prolonged period of time. For example, neurons expressing eNpHR3.1 in the CA1 region may be illuminated by light before the testing session (e.g., 30 minutes or more before the testing session). Prolonged inhibition of the neurons expressing eNpHR3.1 in the CA1 region of the hippocampus may affect the retrieval of memories differently from precise inhibition of the CA1 neurons. For example, prolonged light application (i.e., prolonged inhibition) to CA1 neurons may affect recent contextual fear recall, but may not affect remote contextual memory recall.
[0097] Methods of Treating PTSD
[0098] One or more of the methods described above may be used to treat individuals with PTSD. Aspects of the present disclosure may be used to treat PTSD patients, in which a recurring disturbing memory may be stopped as it appears by reversibly shutting down a remote fearful memory in real-time before and after reconsolidation, or in real-time after it has already been retrieved. In some variations, a method for treating PTSD may comprise administering a viral vector encoding a light-activated protein to an individual. The light-activated protein may be configured to inhibit depolarization of the neuron in the presence of light with a specific wavelength. Examples of such light-activated proteins may include NpHR, BR, AR, and GrR3. As described previously, the viral vector may be delivered to any neuron population or type (e.g., the excitatory neurons of the CA1, ACC, and BLA brain regions). During the recall of an undesired memory (e.g., a fearful or stressful memory), the neuron(s) expressing the light-activated protein may be inhibited from depolarizing, thereby disrupting the retrieval of the undesired memory. In some variations, inhibiting depolarization of the neuron(s) may comprise applying light of the specific wavelength to the neurons expressing the light-activated proteins. Subsequently (e.g., after recall of the undesired memory has been disrupted), the light may be removed. This may restore memory function such that memories may be recalled without disruption. These steps may be repeated as may be desirable in the course of PTSD treatment.
[0099] Consistent with another embodiment of the present disclosure, memories related to drugs of abuse can be inhibited to reduce drug-seeking behavior. Other embodiments are directed toward the ability to instantaneously affect cognition by modulation of different brain areas in order to study the role of specific neuronal populations in memory processes. Inhibition of neurons by certain light-activated proteins and activation by other light-activated proteins may enable a finer temporal, genetic and spatial dissection of the neuronal circuits that underlie various brain function and behaviors.
[0100] Provided herein are methods of disrupting memory recall, the method comprising: inhibiting the function of the dorsal CA1 hippocampus circuit with a temporal precision of the inhibition that is sufficient to disrupt the effects of remote memory retrieval. In some embodiments, the step of inhibiting is responsive to a memory trigger event. In some embodiments, the step of inhibiting includes activating light-responsive opsins expressed in cells of the dorsal CA1 hippocampus circuit. In some embodiments, the step of inhibiting includes applying an electrical pulse through one or more electrodes positioned near the dorsal CA1 hippocampus circuit. In some embodiments, the step of inhibiting includes releasing a drug at a location proximate to the dorsal CA1 hippocampus circuit. In some embodiments, the effects of remote memory retrieval include emotional responses to a remote memory.
[0101] Also provided herein are methods of disrupting memory creation, the method comprising: inhibiting the function of the dorsal CA1 hippocampus circuit with a temporal precision of the inhibition that is sufficient to disrupt remote memory creation. In some embodiments, the step of inhibiting is responsive to a memory trigger event. In some embodiments, the step of inhibiting includes activating light-responsive opsins expressed in cells of the dorsal CA1 hippocampus circuit. In some embodiments, the step of inhibiting includes applying an electrical pulse through one or more electrodes positioned near the dorsal CA1 hippocampus circuit. In some embodiments, the step of inhibiting includes releasing a drug at a location proximate to the dorsal CA1 hippocampus circuit. In some embodiments, the effects of remote memory retrieval include emotional responses to a remote memory.
[0102] Also provided herein are methods of encouraging memory function, the method comprising: exciting the function of the dorsal CA1 hippocampus circuit to promote remote memory creation or remote memory recall.
[0103] Also provided herein are methods for treatment of a neurological disorder associated with remote memory recall, the method comprising: in response to retrieval of the remote memory, inhibiting the function of the dorsal CA1 hippocampus circuit with a temporal precision of the inhibition that is sufficient to disrupt the effects of the retrieval of the remote memory.
EXAMPLES
[0104] Various experiments and examples in accordance with the disclosure herein are provided below.
[0105] In exploring the contribution of defined cell types to remote memory using optogenetic methods (which are orders of magnitude faster in onset and offset than earlier methods), it was found that even many weeks after contextual conditioning (far into the "remote" phase), recall of contextual fear memory was abolished by optogenetic inhibition of excitatory neurons in the CA1 region of the hippocampus--at times when earlier studies had found no detectable influence of hippocampus. No effects of this intervention were observed on locomotion, anxiety, or cued memory formation, and remarkably, remote contextual memory could be instantaneously suppressed by CA1 inhibition even in the midst of a freely-moving behavioral session. The experiments described below confirmed that earlier observations however, as extending optogenetic inhibition of hippocampus to match typical pharmacological timescales converted the remote hippocampus-dependence to remote hippocampus-independence; optogenetic methods also confirmed the remote-timescale importance of anterior cingulate cortex (ACC), and showed that the hippocampus is involved in the recruitment of the ACC for remote recall. These findings have broad implications for the interpretation of drug and lesion data, illuminate puzzling aspects of the clinical hippocampus literature, and uncover a remarkable dynamism in memory retrieval, in which underlying neural circuitry adaptively shifts the default structures involved in memory--normally depending upon the hippocampus even at remote timepoints, but flexibly moving to alternate mechanisms on the timescale of minutes.
[0106] Various types of light-activated proteins may be used to control and characterize the neural circuits that underlie memory function. For example, variants of NpHR may be used to inhibit depolarization and/or hyperpolarize a neuron. The third generation eNpHR has a trafficking signal between the gene and the fluorophore and has shown improved membrane targeting and increased light-induced hyperpolarizations. This third generation eNpHR was used to perturb the neurons in the CA1 region of the hippocampus to determine their role in both recent and remote memory acquisition and recall. A lentiviral vector encoding eNpHR3.1 fused in-frame to enhanced yellow fluorescent protein (eNpHR3.1-EYFP) under control of the calcium/calmodulin-dependent protein kinase IIa (CaMKII.alpha.) promoter, selective for excitatory glutamatergic neurons in hippocampus was used. eNpHR3.1 is a truncated version of eNpHR3.0 with a deletion of the intrinsic N-terminal signal peptide that is similar to eNpHR3.0 in both the photocurrent and the hyperpolarization induced in neurons.
EXPERIMENTAL PROCEDURES
[0107] Subjects.
[0108] C57BL6 mice aged 6 to 8 weeks were obtained from Charles River. Mice were housed four to five per cage in a colony maintained on a reversed 12 hr light/dark cycle and given food and water ad libitum. Experimental protocols were approved by Stanford University IACUC and meet guidelines of the National Institutes of Health guide for the Care and Use of Laboratory Animals.
[0109] Virus Production.
[0110] The CaMKII.alpha.-eNpHR3.1-EYFP lentivirus for in vivo injection was produced as previously described (Gradinaru et al., 2010; Zhang et al., 2007). The adeno-associated virus (AAV) CaMKII.alpha.-eNpHR3.0-EYFP plasmid was constructed by cloning eNpHR3.0-EYFP into an AAV backbone carrying the CaMKII.alpha. promoter using BamHI and EcoRI restriction sites. The recombinant AAV vectors were serotyped with AAV5 coat proteins and packaged by the Vector Core at the University of North Carolina; titers were 2.times.1012 particles/mL. The maps for AAV CaMKII.alpha.::eNpHR3.0 and Lenti CaMKII.alpha.::eNpHR3.1 are available online at www.optogenetics.org.
[0111] Stereotactic Virus Injection, Cannula/Patchcord Implantation, and Light Delivery.
[0112] Mice were anesthetized with isoflurane, the head was placed in a stereotactic apparatus (Kopf Instruments, Tujunga, CA; Leica stereomicroscope). Ophthalmic ointment was applied to prevent eye drying. A midline scalp incision was made and then a small craniotomy was performed and the virus was delivered using a 10 .mu.l syringe and a thin 34 gauge metal needle (World Precision Instruments, Sarasota, Fla.). The injection volume and flow rate (1 .mu.l at 0.1 .mu.l/min) were controlled by an injection pump (WPI). After injection the needle was left in place for 5 additional minutes and then slowly withdrawn. For CA1 optogenetic inhibition, 2 .mu.l of concentrated lentivirus carrying CaMKII.alpha.::eNpHR3.1-EYFP was microinjected into two sites in the CA1 (1 .mu.l/site) of both left and right adult hippocampus. Site one: anteroposterior (AP), -1.5 mm from bregma, mediolateral (ML), .+-.1 mm, dorsoventral (DV) -1.5; site two: AP, -2.5 mm, ML, .+-.2 mm, DV -1.5 mm. A bilateral guide cannula (2.5 mm center to center; PlasticsOne, Roanoke, Va.) was then placed 0.5 mm above CA1 (AP, -1.94 mm, ML, .+-.1.25 mm, DV -1 mm), and secured to the skull using dental cement (C&B metabond, Parkell, Edgwood, N.Y.). The skin was glued back with Vetbond tissue adhesive. The animal was kept on a heating pad until it recovered from anesthetic. Buprenorphine (0.03 mg/kg) was given subcutaneously at the beginning of the surgical procedure to minimize discomfort. To inhibit neuronal activity, green light (561 nm, describe laser etc) was bilaterally delivered through two 300 .mu.m thick optic fibers (Thorlabs, Newton, N.J.) that were inserted through the guide cannulas, with a 0.5 mm projection. Control mice were either uninfected with eNpHR3.1 but still implanted with the cannula delivering light into CA1, or were infected with eNpHR3.1 and implanted, but connected to a dummy fiber that terminated the light delivery at the surface of the brain. Control mice therefore experienced identical visual cues and contextual information as the experimental mice associated with laser light delivery. For basolateral amygdala (BLA) optogenetic inhibition, 1.5 .mu.l of AAV5 CaMKII.alpha.::eNpHR3.0-EYFP was microinjected into both left and right BLA (AP, -1.5 mm, ML, .+-.3.47 mm, DV -5 mm). A patchcord (a metal ferrule, 2.5 mm in diameter with a 200 .mu.m thick, 5 mm long, cleaved bare optic fiber; Doric lenses Inc., Quebec, Canada) was then placed in each BLA (AP, -1.5 mm, ML, .+-.3.47 mm, DV -4.8 mm), and secured to the skull using dental cement. Green light was bilaterally delivered through two 200 .mu.m thick optic fibers (Doric lenses) that were attached to the patchcord using a connecting plastic sleeve. For anterior cingulate cortex (ACC) optogenetic inhibition, 1.0 .mu.l of AAV5 CaMKII.alpha.::eNpHR3.0-EYFP was microinjected into both left and right ACC (AP, .+-.1 mm, ML, .+-.0.35 mm, DV -2.2 mm). A patchcord (Doric lenses Inc.) was then unilaterally placed above one ACC, as close as possible to the midline (AP, +1 mm, ML, .+-.0.2 mm, DV -1.25 mm), and secured to the skull using dental cement. Green light was delivered through a 200 .mu.m thick optic fiber (Doric lenses) attached to the patchcord. For olfactory bulb (OB) optogenetic inhibition, 1.0 .mu.l of AAV5 CaMKII.alpha.::eNpHR3.0-EYFP was microinjected into both left and right OB (AP, +4.5 mm, ML, .+-.0.75 mm, DV -3.25 and -2 mm). A patchcord (Doric lenses Inc.) was then unilaterally placed above one OB, as close as possible to the midline (AP, +4.5 mm, ML, .+-.0.15 mm, DV -1.4 mm), and secured to the skull using dental cement. Green light was delivered through a 200 .mu.m thick optic fiber (Doric lenses) attached to the patchcord.
[0113] Immunohistochemistry.
[0114] To measure the spread and determine the specificity of eNpHR-EYFP expression in CaMKII.alpha. positive neurons, mice were anesthetized with ketamine/xylazine and perfused transcardially with cold PBS followed by 4% paraformaldehyde (PFA) dissolved in phosphate-buffered saline (PBS, pH 7.4). The brains were removed and post-fixed in 4% PFA in PBS for 3 hr at 4.degree. C., and then equilibrated in 30% sucrose in PBS. 40 .mu.m-thick coronal sections were cut on a freezing microtome (Leica) and stored in cryoprotectant (25% glycerol, 30% ethylene glycol, in PBS) at 4.degree. C. until processed for immunohistochemistry. Free-floating sections were washed in PBS and then incubated for 30 min in 0.2% Triton X-100 (Tx100) and 2% normal donkey serum (NDS). Slices were incubated overnight with primary antibody in 2% NDS (Mouse anti-CaMKII.alpha. 1:500, Abcam, Cambridge, Mass.; Rabbit anti GABA 1:500, Millipore, Billerica, Mass.; Rabbit anti c-Fos 1:500, EMD Darmstadt, Germany). Sections were then washed with PBS and incubated for 2 hr at room temperature with secondary antibodies (Donkey anti mouse conjugated to Cy3, donkey anti rabbit conjugated to either Cy3 or Cy5, all 1:1000, Jackson Laboratories, West grove, PA). Slices were then washed, incubated with DAPI (1:50,000) for 20 min, washed again, and mounted on slides with PVA-Dabco (Sigma). Confocal fluorescence images were acquired on a scanning laser microscope using a 5.times. or a 10.times. air objectives, or a 40.times. oil immersion objective. To determine the rate of viral transduction we calculated the percentage of CaMKII.alpha.-immunoreactive neurons per 40.times. field that were also eNpHR-EYFP-positive.
[0115] In Vivo Optrode Recording.
[0116] Simultaneous optical stimulation and electrical recording in the CA1 was carried out as described previously (Gradinaru et al., 2007) using an optrode consisting of an extracellular tungsten electrode (1 M.OMEGA., .about.125 .mu.m) tightly bundled with an optical fiber (200 .mu.m core diameter, 0.2 N.A.), with the tip of the electrode protruding slightly beyond the fiber end (.about.0.4 mm) to ensure illumination of the recorded neurons. Recordings were conducted with the optrode initially placed at the boundary of CA1 (AP, -1.94 mm; ML, 1.4 mm; DV, -1.1) and gradually lowered in 0.1 mm increments. The optical fiber was coupled to a 473 nm solid-state laser diode with .about.20 mW of output from the 200 .mu.m fiber. Single unit recordings were done in mice anesthetized with a ketamine/xylazine mixture (ketamine, 80 mg/kg; xylazine, 15-20 mg/kg) diluted in PBS. Signals were recorded and band-pass filtered at 300 Hz low/5 kHz high using an 1800 Microelectrode AC Amplifier.
[0117] Measurement of Learning and Memory in the Fear Conditioning Paradigm.
[0118] The fear conditioning apparatus consisted of a square conditioning cage (18.times.18.times.30 cm), with a grid floor wired to a shock generator and a scrambler, surrounded by an acoustic chamber (Coulbourn instruments, PA, USA.). The apparatus was modified to enable light delivery during training and/or testing. To induce fear-conditioning mice were placed in the cage for 120 seconds, and then a pure tone (2.9 kHz) was sound for 20 sec, followed by a 2 sec, foot-shock (0.5 mA for short-term memory, 1 mA for long-term memory). This procedure was then repeated, and 30 sec after the delivery of the second shock mice were returned to their home cage. Fear conditioning was assessed by a continuous measurement of freezing (complete immobility), the dominant behavioral fear response (Fanselow, 2000). Freezing was measured continuously throughout the testing trial by an experienced experimenter blind to the treatment group. To test contextual fear conditioning mice were placed in the original conditioning cage, and freezing was measured for 5 min. To test auditory-cued fear conditioning mice were placed in a different context--a pyramid shaped cage with a smooth floor. As a control for the influence of the novel environment, freezing was measured for 2.5 min in this new cage, and then a 2.9 kHz tone was sound for 2.5 min, during which conditioned freezing was measured. This basic paradigm was applied under variable conditions in the different experiments: In the first experiment (FIG. 5) mice were trained and tested as follows: Day 1--training with continuous 561 nm light administration (light ON). Day 2--contextual and cued tests (2 hr apart) without light administration (light OFF). Day 3--training, light OFF. Day 4--test, light OFF. Day 5--contextual and cued tests, light ON. In the first remote memory experiment (FIG. 6A): Day 1--training, light OFF. Day 29--contextual and cued tests, light ON. Day 30--test light OFF. In a second remote memory experiment (FIG. 6C): Day 1--training, light OFF. Day 64--contextual test, light ON. In a third experiment (FIG. 8): Day 1--training, light OFF. Day 36--test, light OFF. Day 37--test light ON. Day 38--test with 3 min light OFF followed by 3 min light ON.
[0119] In the BLA experiment (FIGS. 5H-I) mice were trained on day 1 with light ON, and tested for contextual and cued fear on day 2 with light OFF. In the ACC (FIG. 10A-B) and OB experiments mice were trained on day 1 with the light OFF, tested on day 2 with the light ON, and then tested on day 29 with light ON. For prolonged light exposure (FIGS. 7A,B, 10C), the optic fibers were passed through the conditioning cage into a regular housing cage with bedding, and light was delivered in this cage for 30 min. The mouse was then placed in the conditioning cage for a five min test, as light delivery continued without interruption. The results of the contextual- and cued-conditioning tests were analyzed by a Student's t-test or 2-way ANOVA, followed by post-hoc tests, as applicable.
[0120] Drug Delivery.
[0121] For the pharmacological experiments (FIGS. 6D-E), mice were implanted with a double cannula above CA1. The cannula, surgical procedure and location were the same as in the light delivery experiments. As described by Kitamura et al. (Kitamura et al., 2009) TTX (Sigma, 20 .mu.M) and CNQX (Tocris Bioscience, Ellisville, Mo.; 3 mM) or saline were infused in a volume of 1 .mu.l through a 28 gauge stainless steel internal cannula (PlasticsOne) that was 0.5 mm longer than the guide cannula. The internal cannula was connected to a micro-syringe pump (Harvard Apparatus, Holliston, Mass.) by a PE20 tube. Solutions were administered at a constant rate of 200 nl/min, and the injection cannula was removed 2 min following the termination of the injection to avoid spillage from the guide cannula.
[0122] Open Field Test.
[0123] The open field test was conducted in an open plastic arena (50 cm long.times.50 cm wide.times.40 cm deep). Mice were individually placed in the center of the chamber and allowed to freely explore for 3 min. Activity in both the central and periphery of the field was measured using an automated video-tracking system (Biobserve, Bonn, Germany). Percentage of time in center is defined as the percent of total time that was spent in the central 35.times.35 cm area of the open field.
[0124] Electrophysiological Measurement of Continuous Inhibition of Evoked Spiking by eNpHR3.1.
[0125] Four mice from the prolonged light exposure experiment were injected as described above, went through behavioral testing, and then sacrificed and sliced for physiology. Coronal slices containing dorsal CA1 were prepared by perfusing ice cold sucrose solution transcardially which contained (in mM): 26 NaHCO.sub.3, 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4H14O7, 0.5 O2H4O2, 11 glucose, and 234 sucrose, and subsequently cutting 300 micron slices in the same ice cold sucrose solution. Electrophysiological recordings were made under the constant perfusion of aCSF, which contained (in mM): 126 NaCl, 26 NaHCO.sub.3, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl, and 10 glucose. All recordings were performed at 32.degree. C. Patch electrodes (tip resistance=2-6 M.OMEGA.) were filled with (in mM): 130 K-gluconate, 10 KCl, 10 Hepes, 10 EGTA, and 2 MgCl (pH adjusted to 7.3 with KOH). Series resistance was usually 10-20 M.OMEGA., and experiments were discontinued if it exceeded 30 M.OMEGA.. The membrane potential was corrected for a measured liquid junction potential of 7 mV. Induction of action potentials was done by injecting current ranging from 200 pA at 10 hz. Light for the activation of eNpHR3.1 was delivered using a X-Cite 120 W halogen light source through a 531.+-.20 nm filter and a 40.times./0.8 NA water objective at 7 mW/mm.sup.2.
[0126] Electrophysiological Comparison Between eNpHR3.1 and eNpHR 3.0 in Cultured Neurons.
[0127] Hippocampal Cultures: Primary cultured hippocampal neurons were prepared from PO Sprague-Dawley rat pups. The CA1 and CA3 regions were isolated, digested with 0.4 mg/mL papain (Worthington, Lakewood, N.J.), and plated onto glass coverslips precoated with 1:30 Matrigel (Beckton Dickinson Labware, Bedford, Mass.) at a density of 65,000/cm.sup.2. Cultures were maintained in a 5% CO2 humid incubator with Neurobasal-A medium (Invitrogen Carlsbad, Calif.) containing 1.25% FBS (Hyclone, Logan, Utah), 4% B-27 supplement (GIBCO, Grand Island, N.Y.), 2 mM Glutamax (GIBCO), and FUDR (2 mg/ml, Sigma).
[0128] Calcium Phosphate Transfection.
[0129] 6-10 div hippocampal neurons were grown at 65,000 cells/well in a 24-well plate. DNA/CaCl2 mix for each well: 1.5-3 .mu.g DNA (QIAGEN endotoxin-free preparation)+1.875 .mu.l 2M CaCl2 (final Ca2+ concentration 250 mM) in 15 .mu.l total H20. To DNA/CaCl2 was added 15 .mu.l of 2.times.HEPES-buffered saline (pH 7.05), and the final volume was mixed well by pipetting. After 20 min at RT, the 30 .mu.l DNA/CaCl2/HBS mixture was dropped into each well (from which the growth medium had been temporarily removed and replaced with 400 .mu.l warm MEM) and transfection allowed to proceed at 37 C for 45-60 min. Each well was then washed with 3.times.1 mL warm MEM and the growth medium replaced. Opsin expression was generally observed within 20-24 hr.
[0130] Electrophysiology.
[0131] Whole-cell patch clamp recordings were performed as previously described (intracellular solution: 129 mM K-gluconate, 10 mM HEPES, 10 mM KCl, 4 mM MgATP, 0.3 mM Na3GTP, titrated to pH 7.2; extracellular Tyrode: 125 mM NaCl, 2 mM KCl, 3 mM CaCl2, 1 mM MgCl2, 30 mM glucose, and 25 mM HEPES, titrated to pH 7.3). For voltage clamp recordings cells were held at -70 mV. Light was delivered from a 300W DG-4 lamp (Sutter Instruments, Novato, Calif.) through a 593/40 nm filter (Semrock, Rochester, N.Y.) and a Leica 40.times./0.8 NA water objective; light power at the sample was 3 mW/mm.sup.2. Whole-cell patch clamp data are from cultured hippocampal neurons either transfected or transduced with lentiviral eNpHR3.0 and eNpHR3.1 and allowed to express for one week. Expression was driven by the human CaMKII.alpha. promoter and visualized by fusion to EYFP.
[0132] Neuronal Activation Imaging by cFos Staining.
[0133] YFP control and eNpHR3.1 mice were trained with light administration during conditioning (without tone presentation, so that only fear of the context would be induced), and sacrificed 90 min later to test for c-Fos levels (described in detail in the immunohistochemistry section above). Two other groups of non-trained control and eNpHR3.1 mice were sacrificed from their home cages. For remote memory, YFP controls and eNpHR3.1 mice were fear-conditioned without light, exposed to the conditioning context with light 28 days later, and sacrificed 90 min afterwards to test for cFos levels. The control groups at this time point were control and eNpHR3.1 mice that were trained, and then sacrificed from their home cages 28 days later without being re-exposed to the conditioning context.
[0134] Results
[0135] Specific Optogenetic Inhibition of Excitatory Neurons in Dorsal CA1 Reduces Neuronal Activity.
[0136] Stereotactic delivery of the CaMKII.alpha.::eNpHR3.1 vector was found to result in CA1-specific expression (FIG. 4A). eNpHR3.1 is a truncated version of eNpHR3.0 with a deletion of the intrinsic N-terminal signal peptide, that has comparable effects on membrane potential. eNpHR3.1 is targeted to the neuronal membrane, and is expressed around the soma, as well as in the apical and basal dendrites of CA1 neurons (FIG. 4B). Within the transfected area, 94% (458/486 cells, from 3 mice) of the CaMKII.alpha. cells expressed eNpHR3.1, and the promoter provided complete specificity; all eNpHR3.1-EYFP cells were also CaMKII.alpha. positive (FIG. 4C). The eNpHR3.1 protein was expressed in CA1, but under these expression conditions not in other hippocampal sub-fields, in the parietal cortex above the injection sites, in thalamus or in habenula. The cannula track (at bregma -1.94) could be seen above the expression sites. The volume of infection covered a substantial fraction of dorsal CA1 (0.875.+-.0.05 mm.sup.3; N=12 mice).
[0137] To verify the physiological effect of eNpHR3.1 on CA1 neuronal activity, `optrode` recordings (simultaneous optical stimulation and electrical recording using an extracellular electrode coupled to a fiber optic cable) of CA1 neurons in anesthetized mice were performed (FIG. 4D left), and the experiments confirmed that continuous 561 nm illumination of excitatory CA1 neurons potently inhibited spiking in vivo (FIG. 4D) in a temporally precise and reversible manner, without affecting spike amplitudes. 561 nm illumination of CA1 neurons in these mice resulted in a reversible, marked reduction in spiking frequency (4.93.+-.1.6 Hz, 1.31.+-.0.15 Hz, and 6.45.+-.2.4 Hz; before, during and after light administration, respectively, in 15 traces from 2 mice, P<0.02), without affecting average spike amplitude (33.55.+-.4.94 .mu.V, 29.20.+-.4.4 .mu.V, and 33.32.+-.5.45 .mu.V; before, during and after light). A representative optrode recording trace, as well as average frequency and amplitude are shown (mean.+-.SEM).
[0138] CA1 Optogenetic Inhibition Blocks Contextual Fear Acquisition and Retrieval.
[0139] The involvement of the hippocampus in contextual fear conditioning is based on physical, pharmacological and genetic lesions to this structure, in which the interval between lesion and testing ranges from tens of minutes to several weeks (Anagnostaras et al., 1999; Kim and Fanselow, 1992; Kitamura et al., 2009; Shimizu et al., 2000; Wiltgen et al., 2010), which could allow for adaptation and compensation within the relevant neural circuitry. To first test if real-time optogenetic inhibition of CA1 could modulate memory formation, bilateral continuous green (561 nm) light via two optical fibers inserted through a double cannula system was delivered targeting dorsal CA1 (FIG. 5A) in freely-moving mice in a customized FC chamber. Light was delivered to all mice, and was accompanied by CA1 inhibition in eNpHR3.1 but not control mice (which were either not infected but implanted with a cannula and received light into CA1, or mice infected and implanted connected to a dummy fiber that did not extend into the brain). During fear-conditioning training, mice were introduced into context A, and then presented twice with a tone followed by a foot-shock, under continuous bilateral 561 nm light delivery, and mice were tested for their memory 24 hr later without light. Fear memory was then assessed the next day in the absence of optical inhibition. Dorsal CA1 optogenetic inhibition during training completely prevented contextual fear acquisition eNpHR3.1 mice (n=5) compared to controls (n=4) (39.+-.5.4 vs. 7.6.+-.4.3% freezing; means.+-.SEM, P<0.005 (FIG. 5B, left). To test whether the effect of optogenetic inhibition was reversible, all mice were then re-trained in the same context without light administration, and tested again on the next day; indeed, eNpHR3.1 mice exhibited intact contextual memory (64.6.+-.6.6 vs. 49.7.+-.11.7% freezing; P>0.5) when no light was administered during training (FIG. 5B, middle).
[0140] Next, whether dorsal CA1 optogenetic inhibition could also interfere with memory recall was tested. To that end the same mice were tested, this time with light delivery during recall, and it was found that the memory that was present the day before became unavailable for recall under illumination (FIG. 5B, right; 42.6.+-.10.1 vs. 5.94.+-.4.1% freezing, P<0.01). These experiments support prior understanding that the hippocampus is required for acquisition and recall of recent contextual fear memory, by directly demonstrating the real-time importance of CA1 excitatory cells in these processes. To verify that these effects were specific to contextual fear memories and not fear acquisition and fear expression mechanisms in general, the same mice were tested in a different context for their memory of the tone; eNpHR3.1 mice (n=5) demonstrated intact auditory-cued fear memory acquisition following CA1 light inhibition during training (FIG. 5C, left), as well as intact cued fear recall with illumination during the test (FIG. 5C, right) as compared to controls (n=4). These findings demonstrate the functional specificity of the optogenetic manipulation in affecting only the hippocampus-dependent task.
[0141] To further validate the optogenetic system, a number of additional control experiments were carried out. Because spatial exploration is critical for contextual fear acquisition (McHugh and Tonegawa, 2007), exploration time within the conditioning chamber during training under light stimulation was measured, and it was found no difference between eNpHR3.1-expressing animals (n=5) and control animals (n=5; FIG. 5D). CA1 optogenetic inhibition also had no effect on exploration of a novel environment. To verify that CA1 optogenetic inhibition did not have an anxiolytic effect, mice were tested for open field exploration during light administration; no differences in path length (FIG. 5E; 564.+-.9 and 618.+-.114 cm, eNpHR3.1 and control respectively), velocity (FIG. 5F; 3.3.+-.0.1 vs. 3.43.+-.0.6 cm/sec, eNpHR3.1 and control respectively), or the percent of time spent in the center of the field (which serves as a sign of anxiety-related behavior) were found between eNpHR3.1-expressing (n=6) and control mice (n=4; FIG. 5G; 23.8.+-.2.76% vs. 20.46.+-.5.97%, P>0.5).
[0142] Finally, mice were bilaterally injected in the basolateral amygdala (BLA; FIG. 5H) instead of hippocampus and it was found that it was possible to optogenetically inhibit both contextual (FIG. 5I; 65.5.+-.7.2 vs. 9.6.+-.5.5% freezing; P<0.001) and auditory-cued FC acquisition (FIG. 5I; 69.5.+-.9.6 vs. 24.5.+-.13% freezing; P<0.05) in eNpHR3.0 (n=4) mice, compared to controls (n=9), as expected from prior findings that acquisition of fear itself and the expression of recent and remote fear depend on the amygdala (Han et al., 2009; Johansen et al., 2010; Killcross et al., 1997; LeDoux, 2000; Lee et al., 2006; Maren and Quirk, 2004). Together this constellation of findings confirm the validity of the real-time, fast, cell type-specific, reversible optogenetic system, and support a wide array of major prior findings in the memory literature by directly demonstrating the real-time role of the hippocampus in acquisition and recall.
[0143] CA1 Optogenetic Inhibition Reversibly Interferes with Remote Fear Memory Recall.
[0144] The role of the hippocampus in remote memory recall was explored. A group of mice with contextual FC as before was trained and the subjects were tested 4 weeks later (FIG. 6A), far into the remote phase when no hippocampus involvement is expected. Surprisingly, it was found that CA1 inhibition during recall completely blocked remote fear memory (P<0.0001; Control n=14, 69.8.+-.5.3% freezing eNpHR3.1 n=6, 14.+-.6.4% freezing). This interference with recall was reversible; when the same mice were re-tested on the next day without illumination, the fear memory was fully expressed as in controls (FIG. 6A; 52.45.+-.6.0 vs. 45.18.+-.11.5% freezing; P>0.5). Moreover, eNpHR3.1 mice demonstrated intact remote auditory-cued fear memory recall with illumination during the cued test (FIG. 6B; Control n=14, 22.3.+-.6.8%, eNpHR3.1 n=6, 11.8.+-.3.5% freezing in the new context; and 72.4.+-.8.4 vs. 58.77.+-.7.9% freezing to the tone; P>0.5), further demonstrating that fear expression mechanisms remained intact. To test if the hippocampus would still be involved in contextual fear recall even at much longer time intervals, another population of mice were trained and this cohort was tested 9 weeks after contextual FC. It was found that CA1 inhibition during recall blocked remote fear memory even after this very long interval and was never previously evoked (FIG. 6C; P<0.005; Control n=9, 31.8.+-.3.8% freezing eNpHR3.1 n=6, 11.3.+-.3.6% freezing).
[0145] These results point to ongoing involvement of the hippocampus in remote contextual fear memories, suggesting that the intact hippocampus is still the default activator of the memory trace. They stand in contrast with prevailing theories based on elegant and pioneering physical, pharmacological or genetic lesions to the hippocampus, in which the interval between lesion and recall-test ranges from tens of minutes to several weeks (Anagnostaras et al., 1999; Kim and Fanselow, 1992; Kitamura et al., 2009; Shimizu et al., 2000; Wiltgen et al., 2010). Indeed, the experiments demonstrated that pharmacological inhibition of hippocampus using TTX and CNQX, as previously reported (Kitamura et al., 2009), disturbed only recent (FIG. 6D; saline n=5, 56.86.+-.1.9% freezing; TTX+CNQX n=4, 26.05.+-.10.23% freezing; P<0.05) but not remote (FIG. 6E; saline n=8, 93.93.+-.2.54% freezing; TTX+CNQX n=9, 83.8.+-.4.4% freezing; P>0.05) fear recall when using the FC protocol, confirming earlier results. Thus, the speed and specificity of optogenetics could instead permit testing the causal role of cells and circuits as they are employed in behaving animals, by not allowing expression of compensatory mechanisms. This hypothesis was next explicitly tested.
[0146] Precise, but not Prolonged CA1 Optogenetic Inhibition Blocks Remote Contextual Fear Recall.
[0147] To test the hypothesis that temporal precision is a critical factor accounting for the discrepancy between the optogenetic and pharmacological findings, the remote optogenetic experiment was repeated with either illumination limited to the duration of the test as before (FIG. 7A "precise"), or with prolonged illumination for 30 min before testing and during the test to mimic a slower intervention and allow time for putative compensatory mechanisms to be engaged (FIG. 7A "prolonged"). Precise optogenetic inhibition significantly inhibited remote memory, whereas prolonged inhibition had no detectable effect on remote memory retrieval (FIG. 7A). Furthermore, when mice from the prolonged group were re-tested on the next day with precise light administration (during the test only), the same mice displayed inhibited fear recall (FIG. 7A right). In other words, CA1 optogenetic inhibition prevents remote fear recall of a memory that was acquired 28 days earlier, only when the light was administered precisely during testing (Precise group, Control n=4, 72.65.+-.11.5% freezing, eNpHR3.1 n=8, 26.9.+-.10.4% freezing; P<0.01), but not when the light was ON continuously for 30 min before, as well as during, the test (Prolonged group, middle, Control n=3, 70.13.+-.12.2% freezing, eNpHR3.1 n=4, 67.7.+-.5.6% freezing; P>0.05). When the prolonged group mice were re-tested the next day with light during the test only, their recall was disrupted (Prolonged group, left, 55.5.+-.8.5 vs. 27.6.+-.8.6% freezing; P<0.05).
[0148] To validate these results, both behavioral and physiological controls were performed. First, it was confirmed that prolonged eNpHR3.1-mediated CA1 inhibition, which had no effect on remote memory, still could block recent memory. To that end, a new group of mice were trained and tested on the next day with prolonged illumination for 30 min before testing and then during the test. It was found that prolonged optogenetic inhibition significantly inhibited recent fear memory recall (FIG. 7B; Control n=7, 32.2.+-.10.6% freezing, eNpHR3.1 n=3, 4.+-.2.6% freezing; P<0.05), similar to the pharmacological effect (FIG. 6D). Second, whole-cell patch clamp recordings (in slices prepared from the prolonged group in FIG. 4A) was performed, which revealed that the ability of eNpHR3.1 to suppress spiking was stable throughout 30 min recording periods, as expected (Gradinaru et al., 2010), and was completely reversible (FIG. 7C). Detailed traces of sections 1 (inhibition onset) 2 (during continuous inhibition) and 3 (end of inhibition and recovery) are presented on the bottom left. Averaged percent successful evoked spiking before light, during light administration (after 5 min and 30 min of light ON) and recovery after light OFF are presented (bottom right; n=4 mice, 10 cells).
[0149] CA1 Optogenetic Inhibition Interferes with Ongoing Fear Recall.
[0150] Another population of mice were trained and the cohorts were tested 5 weeks after contextual FC with the remote light-on and light-off recall probe order reversed, first verifying persistence of the memory trace (without light during testing, observing similar performance in both eNpHR3.1 and control groups as expected; FIG. 8 left; control n=8, 79.0.+-.8.9% freezing; eNpHR3.1 n=6, 67.8.+-.12.1% freezing; P>0.5). On the next day, the same mice were tested under illumination, and the eNpHR3.1 group failed to recall the contextual memory (FIG. 8 left; 77.2.+-.4.3% vs. 12.8.+-.4.4% freezing; P<0.0001). This effect was in turn fully reversible, as on the next day, when tested without light delivery, eNpHR3.1 mice demonstrated intact contextual memory (FIG. 8 right; 61.5.+-.6.7 vs. 58.3.+-.3.5% freezing; P>0.5). Most importantly, as soon as the light was delivered again to CA1 within this session, after the mice had already recalled the aversive context and expressed fear, the fear response immediately ceased (FIG. 8 right, 65.2.+-.6.9 vs. 15.9.+-.5.2% freezing; P<0.001) in eNpHR3.1 but not control animals.
[0151] Together these data may unify certain disparate findings, at once supporting prior work by revealing that the remote memory trace is not stored only in the hippocampus (since when given enough time to compensate for hippocampal inactivation, the memory trace can still be retrieved by other structures, in line with previous reports), but at the same time revealing the surprising finding that the intact hippocampus may be a default activator of the remote memory trace and actively participates in its maintenance throughout the recall session.
[0152] Brain-Wide Mapping of Circuit Activity Controlled by Hippocampus During Remote Recall.
[0153] Previous studies of the expression of immediate-early gene products (e.g. zif268 and c-Fos), and other global measures of neural activity, have indicated that the transition from recent to remote memory can be accompanied by a decrease in hippocampal activity and an increase in neocortical activity (in ACC and prefrontal cortex; Bontempi et al., 1999; Frankland et al., 2004; Hall et al., 2001; Maviel et al., 2004). To extend this activity mapping approach to the setting of CA1 optogenetic control, eNpHR3.1-mediated inhibition was delivered during training or remote recall, and assessed induction of the immediate early gene product c-Fos across the entire brain. Mice were fear-conditioned under light delivery, and brains were collected 90 min after training (FIG. 9A). Brain slices were stained for c-Fos and DAPI (FIG. 9B). Expression of YFP control and eNpHR3.1 are shown. The CA1 region from which these images were taken is marked by a white square in FIG. 9C. Following training, eNpHR3.1-expressing mice demonstrated markedly reduced c-Fos expression specifically in CA1 compared with trained control animals (FIG. 9C-D; n=2 to 4 mice, 6 to 15 slices per group; P<0.01), but showed BLA activity equivalent to that of trained controls (FIG. 9C-D; p<0.0001) revealing the expected hippocampus-independent engagement of fear circuitry during training. Note that the bars and lines of FIGS. 9D, 9G, and 9H referenced by (900) are data of the "Control-None" group, (902) are data of the "NpHR-None" group, (904) are data of the "Control-Fear" group, and (906) are data of the "NpHR-Fear" group. No significant changes in ACC activity levels were observed at this time point. Representative images of CA1, ACC and BLA are shown. Anatomy is shown by DAPI nuclear staining, and the margins of the amygdala are marked with a dashed line. White scalebar: 150 .mu.m.
[0154] Another group of mice was conditioned, and then re-exposed to the context 28 days after conditioning in the presence or absence of CA1 optogenetic inhibition; as before, the eNpHR3.1-expressing mice demonstrated impaired remote recall. 90 min later the brains were collected and stained for c-Fos (FIG. 9E) to capture putative memory-related brain-wide activity patterns under control of the hippocampus at this remote timepoint. Intriguingly, a small but significant increase in CA1c-Fos was observed in control, but not eNpHR3.1 mice (FIG. 9F-G; P<0.005) following remote recall. Representative CA1, ACC and BLA images following remote memory are shown. White scalebar: 150 .mu.m. This population of CA1 cells appeared to be causally involved in recruiting brain-wide remote memory-related activity, as the increase in ACC activity (P<0.0001) at this remote timepoint observed in control animals was reduced in eNpHR3.1/CA1-inhibited mice (P<0.0001). Even more strikingly, activated cell populations in the BLA (P<0.0001) were observed in control mice (which recognized the context and expressed fear), but not in the CA1-inhibited eNpHR3.1 mice (which were moreover found to be unable to remember the context; FIG. 9F-G; P<0.0001). As depicted in FIG. 9G, remote recall 28 days following conditioning resulted in a small but significant increase in CA1 c-Fos expression in control mice, and highly increased activity levels in ACC and BLA. Light inhibition during exposure to the context completely blocked CA1 activity (P<0.05), and significantly reduced ACC and BLA activity, compared to control.
[0155] Additional observations point to the specificity of this CA1-recruited population at the remote timepoint. eNpHR3.1-expressing mice showed an elevation in prefrontal cortex activity equivalent to that of controls, and no significant changes in parietal cortex activity levels were observed in any of the groups. In contrast, as noted above, activity levels in the ACC were significantly recruited in remote memory only, and to a lesser extent in the setting of eNpHR3.1-mediated CA1 inhibition (FIG. 9H middle), also in agreement with previous reports (Bontempi et al., 1999; Frankland et al., 2004; Hall et al., 2001; Maviel et al., 2004). FIG. 9H depicts global patterns in brain activity between conditioning (day 0) and remote recall (day 28). Activity levels in CA1 significantly decreased in control (P<0.005) mice from day 0 to day 28. Activity levels in ACC significantly increased in both control (P<0.0001) and eNpHR3.1 (P<0.001) mice day 0 to day 28. Activity levels in BLA significantly increased in control (P<0.001) but not in eNHR3.1 mice. Together these data point to a role for this small population of CA1 neurons in organizing the brain-wide activity patterns associated with remote contextual memory.
[0156] Optogenetic Inhibition of ACC Inhibits Remote but not Recent Contextual Memory.
[0157] Since the population of CA1 neurons active during remote contextual memory was found to be causally involved in fully organizing ACC neuronal activity as shown above, and since previous research has implicated the ACC in remote fear memory storage (Bontempi et al., 1999; Fischer et al., 2007; Frankland et al., 2004; Maviel et al., 2004), optogenetic inhibition of memories was explored by targeting ACC directly either one day or one month following contextual FC. FIG. 10A depicts eNpHR3.0 expression in the anterior cingulate cortex (ACC). In full accordance with previous studies (Frankland et al., 2004), optogenetic inhibition of ACC had no effect on recent memory (75.9.+-.5.4 vs. 76.+-.2.9% freezing), but significantly impaired remote memory (FIG. 10B; Control n=5, 81.6.+-.4.9% freezing; eNpHR3.0 n=5, 53.8.+-.11% freezing; P<0.05).
[0158] The same experiment was repeated in a new group of mice, but this time delivered prolonged illumination for 30 min before testing and then during the test. Again it was found that optogenetic inhibition of ACC significantly impaired remote memory (Control n=3, 78.0.+-.6.2% freezing; eNpHR3.0 n=8, 45.0.+-.5.2% freezing; P<0.05), but had no effect on recent memory (FIG. 10C; 78.5.+-.12.7 vs. 74.3.+-.4.3% freezing). In contrast, when another major cortical input region was targeted for control purposes, the olfactory bulbs (OB), and the effect of optogenetic inhibition was tested during both recent and remote fear recall, it was found no effect on recall at either time point This result at once demonstrates that a sudden drop in a major source of synaptic input to cortex does not nonspecifically influence recall, and also points to the specificity of ACC in remote memory (consistent with prior work). Together, these findings support the remote importance of neocortex, and also illustrate that even following cortical reorganization, there exists a default requirement for the hippocampus in recalling remote memory traces.
[0159] Irreversible erasure of remote memories was recently demonstrated in the hippocampus and cortex by PKW administration (Migues et al; Pastalkova et al 2006; Shema et al 2009; Shema et al 2007) and in the amygdala by selective ablation of pre-tagged neurons (Han et al 2009). On the other hand, remote memory traces that were assumed to be lost due to neuronal damage became available for recall following environmental enrichment and chromatin modifications (Fischer et al 2007). Optogenetics, on the other hand, enables reversible recall prevention, without permanent memory erasure. The finding that the hippocampus is still the default activator of contextual fear memory recall may be due to the fact that many place cells (Moser et al 2008) in CA1 remap in response to fear conditioning (Moita et al 2004), and may contribute to a faster recognition of the context. Indeed, hippocampal lesions were repeatedly shown to induce retrograde amnesia for spatial memory (Broadbent et al 2006; Martin et al 2005).
[0160] When remote memories are retrieved they become available for reconsolidation, which renders them susceptible for disruption but may also strengthen the trace (Dudai 2006; Morris et al 2006; Nader and Hardt 2009; Tronson and Taylor 2007; Wang and Morris). The ability to reversibly shut down a remote fearful memory in real-time, before and after reconsolidation, and even in real-time after it had already been retrieved, may open an exciting therapeutic avenue for PTSD patients, in which a recurring disturbing memory may be stopped as it appears, without permanently affecting other memories. Additionally, memories related to drugs of abuse can be inhibited to reduce drug seeking behavior (Everitt et al 2001; Lee et al 2005; Robbins et al 2008). The ability to instantaneously affect cognition by optogenetic modulation of different brain areas may serve as a basis for future studies re-examining the role of specific neuronal populations in memory processes and enable a finer temporal, genetic and spatial dissection of the neuronal circuits that underlie them.
[0161] Although the foregoing invention has been described in some detail by way of illustration and example for purposes of clarity of understanding, the descriptions and examples should not be construed as limiting the scope of the invention.
REFERENCES
[0162] Adamantidis, A. R., Zhang, F., Aravanis, A. M., Deisseroth, K., and de Lecea, L. (2007). Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature 450, 420-424. [0163] Anagnostaras, S. G., Maren, S., and Fanselow, M. S. (1999). Temporally graded retrograde amnesia of contextual fear after hippocampal damage in rats: within-subjects examination. J Neurosci 19, 1106-1114. [0164] Aravanis, A. M., Wang, L. P., Zhang, F., Meltzer, L. A., Mogri, M. Z., Schneider, M. B., and Deisseroth, K. (2007). An optical neural interface: in vivo control of rodent motor cortex with integrated fiberoptic and optogenetic technology. J Neural Eng 4, S143-156. [0165] Bolhuis, J. J., Stewart, C. A., and Forrest, E. M. (1994). Retrograde amnesia and memory reactivation in rats with ibotenate lesions to the hippocampus or subiculum. Q J Exp Psychol B 47, 129-150. [0166] Bontempi, B., Laurent-Demir, C., Destrade, C., and Jaffard, R. (1999). Time-dependent reorganization of brain circuitry underlying long-term memory storage. Nature 400, 671-675. [0167] Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G., and Deisseroth, K. (2005). Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci 8, 1263-1268. [0168] Broadbent, N. J., Squire, L. R., and Clark, R. E. (2006). Reversible hippocampal lesions disrupt water maze performance during both recent and remote memory tests. Learn Mem 13, 187-191. [0169] Cipolotti, L., and Bird, C. M. (2006). Amnesia and the hippocampus. Curr Opin Neurol 19, 593-598. [0170] Debiec, J., LeDoux, J. E., and Nader, K. (2002). Cellular and systems reconsolidation in the hippocampus. Neuron 36, 527-538. [0171] Deisseroth, K., Feng, G., Majewska, A. K., Miesenbock, G., Ting, A., and Schnitzer, M. J. (2006). Next-generation optical technologies for illuminating genetically targeted brain circuits. J Neurosci 26, 10380-10386. [0172] Dudai, Y. (2006). Reconsolidation: the advantage of being refocused. Curr Opin Neurobiol 16, 174-178. [0173] Everitt, B. J., Dickinson, A., and Robbins, T. W. (2001). The neuropsychological basis of addictive behaviour. Brain Res Brain Res Rev 36, 129-138. [0174] Fanselow, M. S. (2000). Contextual fear, gestalt memories, and the hippocampus. Behav Brain Res 110, 73-81. [0175] Fischer, A., Sananbenesi, F., Wang, X., Dobbin, M., and Tsai, L. H. (2007). Recovery of learning and memory is associated with chromatin remodelling. Nature 447, 178-182. [0176] Frankland, P. W., and Bontempi, B. (2005). The organization of recent and remote memories. Nat Rev Neurosci 6, 119-130. [0177] Frankland, P. W., Bontempi, B., Talton, L. E., Kaczmarek, L., and Silva, A. J. (2004). The involvement of the anterior cingulate cortex in remote contextual fear memory. Science 304, 881-883. [0178] Gradinaru, V., Thompson, K. R., Zhang, F., Mogri, M., Kay, K., Schneider, M. B., and Deisseroth, K. (2007). Targeting and readout strategies for fast optical neural control in vitro and in vivo. J Neurosci 27, 14231-14238. [0179] Gradinaru, V., Zhang, F., Ramakrishnan, C., Mattis, J., Prakash, R., Diester, I., Goshen, I., Thompson, K. R., and Deisseroth, K. (2010). Molecular and cellular approaches for diversifying and extending optogenetics. Cell 141, 154-165. [0180] Hall, J., Thomas, K. L., and Everitt, B. J. (2001). Cellular imaging of zif268 expression in the hippocampus and amygdala during contextual and cued fear memory retrieval: selective activation of hippocampal CA1 neurons during the recall of contextual memories. J Neurosci 21, 2186-2193. [0181] Han, J. H., Kushner, S. A., Yiu, A. P., Hsiang, H. L., Buch, T., Waisman, A., Bontempi, B., Neve, R. L., Frankland, P. W., and Josselyn, S. A. (2009). Selective erasure of a fear memory. Science 323, 1492-1496. [0182] Johansen, J. P., Hamanaka, H., Monfils, M. H., Behnia, R., Deisseroth, K., Blair, H. T., and LeDoux, J. E. (2010). Optical activation of lateral amygdala pyramidal cells instructs associative fear learning. Proc Natl Acad Sci USA 107, 12692-12697. [0183] Killcross, S., Robbins, T. W., and Everitt, B. J. (1997). Different types of fear-conditioned behaviour mediated by separate nuclei within amygdala. Nature 388, 377-380. [0184] Kim, J. J., and Fanselow, M. S. (1992). Modality-specific retrograde amnesia of fear. Science 256, 675-677. [0185] Kitamura, T., Saitoh, Y., Takashima, N., Murayama, A., Niibori, Y., Ageta, H., Sekiguchi, M., Sugiyama, H., and Inokuchi, K. (2009). Adult neurogenesis modulates the hippocampus-dependent period of associative fear memory. Cell 139, 814-827. [0186] LeDoux, J. E. (2000). Emotion circuits in the brain. Annu Rev Neurosci 23, 155-184. [0187] Lee, J. L., Di Ciano, P., Thomas, K. L., and Everitt, B. J. (2005). Disrupting reconsolidation of drug memories reduces cocaine-seeking behavior. Neuron 47, 795-801. [0188] Lee, J. L., Milton, A. L., and Everitt, B. J. (2006). Reconsolidation and extinction of conditioned fear: inhibition and potentiation. J Neurosci 26, 10051-10056. [0189] Maren, S. (2001). Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci 24, 897-931. [0190] Maren, S., Aharonov, G., and Fanselow, M. S. (1997). Neurotoxic lesions of the dorsal hippocampus and Pavlovian fear conditioning in rats. Behav Brain Res 88, 261-274. [0191] Maren, S., and Quirk, G. J. (2004). Neuronal signalling of fear memory. Nat Rev Neurosci 5, 844-852. [0192] Martin, S. J., de Hoz, L., and Morris, R. G. (2005). Retrograde amnesia: neither partial nor complete hippocampal lesions in rats result in preferential sparing of remote spatial memory, even after reminding. Neuropsychologia 43, 609-624. [0193] Maviel, T., Durkin, T. P., Menzaghi, F., and Bontempi, B. (2004). Sites of neocortical reorganization critical for remote spatial memory. Science 305, 96-99. [0194] McHugh, T. J., Jones, M. W., Quinn, J. J., Balthasar, N., Coppari, R., Elmquist, J. K., Lowell, B. B., Fanselow, M. S., Wilson, M. A., and Tonegawa, S. (2007). Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science 317, 94-99. [0195] McHugh, T. J., and Tonegawa, S. (2007). Spatial exploration is required for the formation of contextual fear memory. Behav Neurosci 121, 335-339. [0196] Moita, M. A., Rosis, S., Zhou, Y., LeDoux, J. E., and Blair, H. T. (2004). Putting fear in its place: remapping of hippocampal place cells during fear conditioning. J Neurosci 24, 7015-7023. [0197] Morris, R. G., Inglis, J., Ainge, J. A., Olverman, H. J., Tulloch, J., Dudai, Y., and Kelly, P. A. (2006). Memory reconsolidation: sensitivity of spatial memory to inhibition of protein synthesis in dorsal hippocampus during encoding and retrieval. Neuron 50, 479-489. [0198] Moscovitch, M., Nadel, L., Winocur, G., Gilboa, A., and Rosenbaum, R. S. (2006). The cognitive neuroscience of remote episodic, semantic and spatial memory. Curr Opin Neurobiol 16, 179-190. [0199] Moser, E. I., Kropff, E., and Moser, M. B. (2008). Place cells, grid cells, and the brain's spatial representation system. Annu Rev Neurosci 31, 69-89. [0200] Nadel, L., and Moscovitch, M. (1997). Memory consolidation, retrograde amnesia and the hippocampal complex. Curr Opin Neurobiol 7, 217-227. [0201] Nader, K., and Hardt, O. (2009). A single standard for memory: the case for reconsolidation. Nat Rev Neurosci 10, 224-234. [0202] Nakashiba, T., Young, J. Z., McHugh, T. J., Buhl, D. L., and Tonegawa, S. (2008). Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science 319, 1260-1264. [0203] Phelps, E. A., and LeDoux, J. E. (2005). Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron 48, 175-187. [0204] Riedel, G., Micheau, J., Lam, A. G., Roloff, E. L., Martin, S. J., Bridge, H., de Hoz, L., Poeschel, B., McCulloch, J., and Morris, R. G. (1999). Reversible neural inactivation reveals hippocampal participation in several memory processes. Nat Neurosci 2, 898-905. [0205] Robbins, T. W., Ersche, K. D., and Everitt, B. J. (2008). Drug addiction and the memory systems of the brain. Ann N Y Acad Sci 1141, 1-21. [0206] Shimizu, E., Tang, Y. P., Rampon, C., and Tsien, J. Z. (2000). NMDA receptor-dependent synaptic reinforcement as a crucial process for memory consolidation. Science 290, 1170-1174. [0207] Squire, L. R., and Alvarez, P. (1995). Retrograde amnesia and memory consolidation: a neurobiological perspective. Curr Opin Neurobiol 5, 169-177. [0208] Squire, L. R., and Bayley, P. J. (2007). The neuroscience of remote memory. Curr Opin Neurobiol 17, 185-196. [0209] Sutherland, R. J., O'Brien, J., and Lehmann, H. (2008). Absence of systems consolidation of fear memories after dorsal, ventral, or complete hippocampal damage. Hippocampus 18, 710-718. [0210] Sutherland, R. J., Sparks, F. T., and Lehmann, H. (2010). Hippocampus and retrograde amnesia in the rat model: a modest proposal for the situation of systems consolidation. Neuropsychologia 48, 2357-2369. [0211] Tronson, N. C., and Taylor, J. R. (2007). Molecular mechanisms of memory reconsolidation. Nat Rev Neurosci 8, 262-275. [0212] Wang, H., Shimizu, E., Tang, Y. P., Cho, M., Kyin, M., Zuo, W., Robinson, D. A., Alaimo, P. J., Zhang, C., Morimoto, H., et al. (2003). Inducible protein knockout reveals temporal requirement of CaMKII reactivation for memory consolidation in the brain. Proc Natl Acad Sci USA 100, 4287-4292. [0213] Wang, S. H., and Morris, R. G. (2010). Hippocampal-neocortical interactions in memory formation, consolidation, and reconsolidation. Annu Rev Psychol 61, 49-79, C41-44. [0214] Wang, S. H., Teixeira, C. M., Wheeler, A. L., and Frankland, P. W. (2009). The precision of remote context memories does not require the hippocampus. Nat Neurosci 12, 253-255. [0215] Wiltgen, B. J., Zhou, M., Cai, Y., Balaji, J., Karlsson, M. G., Parivash, S. N., Li, W., and Silva, A. J. (2010). The Hippocampus Plays a Selective Role in the Retrieval of Detailed Contextual Memories. Curr Biol 20, 1336-1344. [0216] Winocur, G., Frankland, P. W., Sekeres, M., Fogel, S., and Moscovitch, M. (2009). Changes in context-specificity during memory reconsolidation: selective effects of hippocampal lesions. Learn Mem 16, 722-729. [0217] Winocur, G., Moscovitch, M., and Bontempi, B. (2010). Memory formation and long-term retention in humans and animals: convergence towards a transformation account of hippocampal-neocortical interactions. Neuropsychologia 48, 2339-2356. [0218] Winocur, G., Moscovitch, M., and Sekeres, M. (2007). Memory consolidation or transformation: context manipulation and hippocampal representations of memory. Nat Neurosci 10, 555-557. [0219] Zhang, F., Wang, L. P., Brauner, M., Liewald, J. F., Kay, K., Watzke, N., Wood, P. G., Bamberg, E., Nagel, G., Gottschalk, A., et al. (2007). Multimodal fast optical interrogation of neural circuitry. Nature 446, 633-639.
Sequence CWU 1
1
101223PRTGuillardia theta 1Ala Ser Ser Phe Gly Lys Ala Leu Leu Glu
Phe Val Phe Ile Val Phe 1 5 10 15 Ala Cys Ile Thr Leu Leu Leu Gly
Ile Asn Ala Ala Lys Ser Lys Ala 20 25 30 Ala Ser Arg Val Leu Phe
Pro Ala Thr Phe Val Thr Gly Ile Ala Ser 35 40 45 Ile Ala Tyr Phe
Ser Met Ala Ser Gly Gly Gly Trp Val Ile Ala Pro 50 55 60 Asp Cys
Arg Gln Leu Phe Val Ala Arg Tyr Leu Asp Trp Leu Ile Thr 65 70 75 80
Thr Pro Leu Leu Leu Ile Asp Leu Gly Leu Val Ala Gly Val Ser Arg 85
90 95 Trp Asp Ile Met Ala Leu Cys Leu Ser Asp Val Leu Met Ile Ala
Thr 100 105 110 Gly Ala Phe Gly Ser Leu Thr Val Gly Asn Val Lys Trp
Val Trp Trp 115 120 125 Phe Phe Gly Met Cys Trp Phe Leu His Ile Ile
Phe Ala Leu Gly Lys 130 135 140 Ser Trp Ala Glu Ala Ala Lys Ala Lys
Gly Gly Asp Ser Ala Ser Val 145 150 155 160 Tyr Ser Lys Ile Ala Gly
Ile Thr Val Ile Thr Trp Phe Cys Tyr Pro 165 170 175 Val Val Trp Val
Phe Ala Glu Gly Phe Gly Asn Phe Ser Val Thr Phe 180 185 190 Glu Val
Leu Ile Tyr Gly Val Leu Asp Val Ile Ser Lys Ala Val Phe 195 200 205
Gly Leu Ile Leu Met Ser Gly Ala Ala Thr Gly Tyr Glu Ser Ile 210 215
220 2270PRTArtificial SequenceSynthetic peptide 2Met Asp Tyr Gly
Gly Ala Leu Ser Ala Val Gly Arg Glu Leu Leu Phe 1 5 10 15 Val Thr
Asn Pro Val Val Val Asn Gly Ser Val Leu Val Pro Glu Asp 20 25 30
Gln Cys Tyr Cys Ala Gly Trp Ile Glu Ser Arg Gly Thr Asn Gly Ala 35
40 45 Ser Ser Phe Gly Lys Ala Leu Leu Glu Phe Val Phe Ile Val Phe
Ala 50 55 60 Cys Ile Thr Leu Leu Leu Gly Ile Asn Ala Ala Lys Ser
Lys Ala Ala 65 70 75 80 Ser Arg Val Leu Phe Pro Ala Thr Phe Val Thr
Gly Ile Ala Ser Ile 85 90 95 Ala Tyr Phe Ser Met Ala Ser Gly Gly
Gly Trp Val Ile Ala Pro Asp 100 105 110 Cys Arg Gln Leu Phe Val Ala
Arg Tyr Leu Asp Trp Leu Ile Thr Thr 115 120 125 Pro Leu Leu Leu Ile
Asp Leu Gly Leu Val Ala Gly Val Ser Arg Trp 130 135 140 Asp Ile Met
Ala Leu Cys Leu Ser Asp Val Leu Met Ile Ala Thr Gly 145 150 155 160
Ala Phe Gly Ser Leu Thr Val Gly Asn Val Lys Trp Val Trp Trp Phe 165
170 175 Phe Gly Met Cys Trp Phe Leu His Ile Ile Phe Ala Leu Gly Lys
Ser 180 185 190 Trp Ala Glu Ala Ala Lys Ala Lys Gly Gly Asp Ser Ala
Ser Val Tyr 195 200 205 Ser Lys Ile Ala Gly Ile Thr Val Ile Thr Trp
Phe Cys Tyr Pro Val 210 215 220 Val Trp Val Phe Ala Glu Gly Phe Gly
Asn Phe Ser Val Thr Phe Glu 225 230 235 240 Val Leu Ile Tyr Gly Val
Leu Asp Val Ile Ser Lys Ala Val Phe Gly 245 250 255 Leu Ile Leu Met
Ser Gly Ala Ala Thr Gly Tyr Glu Ser Ile 260 265 270
3273PRTNatronomonas pharaonis 3Val Thr Gln Arg Glu Leu Phe Glu Phe
Val Leu Asn Asp Pro Leu Leu 1 5 10 15 Ala Ser Ser Leu Tyr Ile Asn
Ile Ala Leu Ala Gly Leu Ser Ile Leu 20 25 30 Leu Phe Val Phe Met
Thr Arg Gly Leu Asp Asp Pro Arg Ala Lys Leu 35 40 45 Ile Ala Val
Ser Thr Ile Leu Val Pro Val Val Ser Ile Ala Ser Tyr 50 55 60 Thr
Gly Leu Ala Ser Gly Leu Thr Ile Ser Val Leu Glu Met Pro Ala 65 70
75 80 Gly His Phe Ala Glu Gly Ser Ser Val Met Leu Gly Gly Glu Glu
Val 85 90 95 Asp Gly Val Val Thr Met Trp Gly Arg Tyr Leu Thr Trp
Ala Leu Ser 100 105 110 Thr Pro Met Ile Leu Leu Ala Leu Gly Leu Leu
Ala Gly Ser Asn Ala 115 120 125 Thr Lys Leu Phe Thr Ala Ile Thr Phe
Asp Ile Ala Met Cys Val Thr 130 135 140 Gly Leu Ala Ala Ala Leu Thr
Thr Ser Ser His Leu Met Arg Trp Phe 145 150 155 160 Trp Tyr Ala Ile
Ser Cys Ala Cys Phe Leu Val Val Leu Tyr Ile Leu 165 170 175 Leu Val
Glu Trp Ala Gln Asp Ala Lys Ala Ala Gly Thr Ala Asp Met 180 185 190
Phe Asn Thr Leu Lys Leu Leu Thr Val Val Met Trp Leu Gly Tyr Pro 195
200 205 Ile Val Trp Ala Leu Gly Val Glu Gly Ile Ala Val Leu Pro Val
Gly 210 215 220 Val Thr Ser Trp Gly Tyr Ser Phe Leu Asp Ile Val Ala
Lys Tyr Ile 225 230 235 240 Phe Ala Phe Leu Leu Leu Asn Tyr Leu Thr
Ser Asn Glu Ser Val Val 245 250 255 Ser Gly Ser Ile Leu Asp Val Pro
Ser Ala Ser Gly Thr Pro Ala Asp 260 265 270 Asp 4291PRTArtificial
SequenceSynthetic peptide 4Met Thr Glu Thr Leu Pro Pro Val Thr Glu
Ser Ala Val Ala Leu Gln 1 5 10 15 Ala Glu Val Thr Gln Arg Glu Leu
Phe Glu Phe Val Leu Asn Asp Pro 20 25 30 Leu Leu Ala Ser Ser Leu
Tyr Ile Asn Ile Ala Leu Ala Gly Leu Ser 35 40 45 Ile Leu Leu Phe
Val Phe Met Thr Arg Gly Leu Asp Asp Pro Arg Ala 50 55 60 Lys Leu
Ile Ala Val Ser Thr Ile Leu Val Pro Val Val Ser Ile Ala 65 70 75 80
Ser Tyr Thr Gly Leu Ala Ser Gly Leu Thr Ile Ser Val Leu Glu Met 85
90 95 Pro Ala Gly His Phe Ala Glu Gly Ser Ser Val Met Leu Gly Gly
Glu 100 105 110 Glu Val Asp Gly Val Val Thr Met Trp Gly Arg Tyr Leu
Thr Trp Ala 115 120 125 Leu Ser Thr Pro Met Ile Leu Leu Ala Leu Gly
Leu Leu Ala Gly Ser 130 135 140 Asn Ala Thr Lys Leu Phe Thr Ala Ile
Thr Phe Asp Ile Ala Met Cys 145 150 155 160 Val Thr Gly Leu Ala Ala
Ala Leu Thr Thr Ser Ser His Leu Met Arg 165 170 175 Trp Phe Trp Tyr
Ala Ile Ser Cys Ala Cys Phe Leu Val Val Leu Tyr 180 185 190 Ile Leu
Leu Val Glu Trp Ala Gln Asp Ala Lys Ala Ala Gly Thr Ala 195 200 205
Asp Met Phe Asn Thr Leu Lys Leu Leu Thr Val Val Met Trp Leu Gly 210
215 220 Tyr Pro Ile Val Trp Ala Leu Gly Val Glu Gly Ile Ala Val Leu
Pro 225 230 235 240 Val Gly Val Thr Ser Trp Gly Tyr Ser Phe Leu Asp
Ile Val Ala Lys 245 250 255 Tyr Ile Phe Ala Phe Leu Leu Leu Asn Tyr
Leu Thr Ser Asn Glu Ser 260 265 270 Val Val Ser Gly Ser Ile Leu Asp
Val Pro Ser Ala Ser Gly Thr Pro 275 280 285 Ala Asp Asp 290
5559PRTArtificial SequenceSynthetic peptide 5Met Thr Glu Thr Leu
Pro Pro Val Thr Glu Ser Ala Val Ala Leu Gln 1 5 10 15 Ala Glu Val
Thr Gln Arg Glu Leu Phe Glu Phe Val Leu Asn Asp Pro 20 25 30 Leu
Leu Ala Ser Ser Leu Tyr Ile Asn Ile Ala Leu Ala Gly Leu Ser 35 40
45 Ile Leu Leu Phe Val Phe Met Thr Arg Gly Leu Asp Asp Pro Arg Ala
50 55 60 Lys Leu Ile Ala Val Ser Thr Ile Leu Val Pro Val Val Ser
Ile Ala 65 70 75 80 Ser Tyr Thr Gly Leu Ala Ser Gly Leu Thr Ile Ser
Val Leu Glu Met 85 90 95 Pro Ala Gly His Phe Ala Glu Gly Ser Ser
Val Met Leu Gly Gly Glu 100 105 110 Glu Val Asp Gly Val Val Thr Met
Trp Gly Arg Tyr Leu Thr Trp Ala 115 120 125 Leu Ser Thr Pro Met Ile
Leu Leu Ala Leu Gly Leu Leu Ala Gly Ser 130 135 140 Asn Ala Thr Lys
Leu Phe Thr Ala Ile Thr Phe Asp Ile Ala Met Cys 145 150 155 160 Val
Thr Gly Leu Ala Ala Ala Leu Thr Thr Ser Ser His Leu Met Arg 165 170
175 Trp Phe Trp Tyr Ala Ile Ser Cys Ala Cys Phe Leu Val Val Leu Tyr
180 185 190 Ile Leu Leu Val Glu Trp Ala Gln Asp Ala Lys Ala Ala Gly
Thr Ala 195 200 205 Asp Met Phe Asn Thr Leu Lys Leu Leu Thr Val Val
Met Trp Leu Gly 210 215 220 Tyr Pro Ile Val Trp Ala Leu Gly Val Glu
Gly Ile Ala Val Leu Pro 225 230 235 240 Val Gly Val Thr Ser Trp Gly
Tyr Ser Phe Leu Asp Ile Val Ala Lys 245 250 255 Tyr Ile Phe Ala Phe
Leu Leu Leu Asn Tyr Leu Thr Ser Asn Glu Ser 260 265 270 Val Val Ser
Gly Ser Ile Leu Asp Val Pro Ser Ala Ser Gly Thr Pro 275 280 285 Ala
Asp Asp Ala Ala Ala Lys Ser Arg Ile Thr Ser Glu Gly Glu Tyr 290 295
300 Ile Pro Leu Asp Gln Ile Asp Ile Asn Val Val Ser Lys Gly Glu Glu
305 310 315 320 Leu Phe Thr Gly Val Val Pro Ile Leu Val Glu Leu Asp
Gly Asp Val 325 330 335 Asn Gly His Lys Phe Ser Val Ser Gly Glu Gly
Glu Gly Asp Ala Thr 340 345 350 Tyr Gly Lys Leu Thr Leu Lys Phe Ile
Cys Thr Thr Gly Lys Leu Pro 355 360 365 Val Pro Trp Pro Thr Leu Val
Thr Thr Phe Gly Tyr Gly Leu Gln Cys 370 375 380 Phe Ala Arg Tyr Pro
Asp His Met Lys Gln His Asp Phe Phe Lys Ser 385 390 395 400 Ala Met
Pro Glu Gly Tyr Val Gln Glu Arg Thr Ile Phe Phe Lys Asp 405 410 415
Asp Gly Asn Tyr Lys Thr Arg Ala Glu Val Lys Phe Glu Gly Asp Thr 420
425 430 Leu Val Asn Arg Ile Glu Leu Lys Gly Ile Asp Phe Lys Glu Asp
Gly 435 440 445 Asn Ile Leu Gly His Lys Leu Glu Tyr Asn Tyr Asn Ser
His Asn Val 450 455 460 Tyr Ile Met Ala Asp Lys Gln Lys Asn Gly Ile
Lys Val Asn Phe Lys 465 470 475 480 Ile Arg His Asn Ile Glu Asp Gly
Ser Val Gln Leu Ala Asp His Tyr 485 490 495 Gln Gln Asn Thr Pro Ile
Gly Asp Gly Pro Val Leu Leu Pro Asp Asn 500 505 510 His Tyr Leu Ser
Tyr Gln Ser Ala Leu Ser Lys Asp Pro Asn Glu Lys 515 520 525 Arg Asp
His Met Val Leu Leu Glu Phe Val Thr Ala Ala Gly Ile Thr 530 535 540
Leu Gly Met Asp Glu Leu Tyr Lys Phe Cys Tyr Glu Asn Glu Val 545 550
555 6542PRTArtificial SequenceSynthetic peptide 6Met Val Thr Gln
Arg Glu Leu Phe Glu Phe Val Leu Asn Asp Pro Leu 1 5 10 15 Leu Ala
Ser Ser Leu Tyr Ile Asn Ile Ala Leu Ala Gly Leu Ser Ile 20 25 30
Leu Leu Phe Val Phe Met Thr Arg Gly Leu Asp Asp Pro Arg Ala Lys 35
40 45 Leu Ile Ala Val Ser Thr Ile Leu Val Pro Val Val Ser Ile Ala
Ser 50 55 60 Tyr Thr Gly Leu Ala Ser Gly Leu Thr Ile Ser Val Leu
Glu Met Pro 65 70 75 80 Ala Gly His Phe Ala Glu Gly Ser Ser Val Met
Leu Gly Gly Glu Glu 85 90 95 Val Asp Gly Val Val Thr Met Trp Gly
Arg Tyr Leu Thr Trp Ala Leu 100 105 110 Ser Thr Pro Met Ile Leu Leu
Ala Leu Gly Leu Leu Ala Gly Ser Asn 115 120 125 Ala Thr Lys Leu Phe
Thr Ala Ile Thr Phe Asp Ile Ala Met Cys Val 130 135 140 Thr Gly Leu
Ala Ala Ala Leu Thr Thr Ser Ser His Leu Met Arg Trp 145 150 155 160
Phe Trp Tyr Ala Ile Ser Cys Ala Cys Phe Leu Val Val Leu Tyr Ile 165
170 175 Leu Leu Val Glu Trp Ala Gln Asp Ala Lys Ala Ala Gly Thr Ala
Asp 180 185 190 Met Phe Asn Thr Leu Lys Leu Leu Thr Val Val Met Trp
Leu Gly Tyr 195 200 205 Pro Ile Val Trp Ala Leu Gly Val Glu Gly Ile
Ala Val Leu Pro Val 210 215 220 Gly Val Thr Ser Trp Gly Tyr Ser Phe
Leu Asp Ile Val Ala Lys Tyr 225 230 235 240 Ile Phe Ala Phe Leu Leu
Leu Asn Tyr Leu Thr Ser Asn Glu Ser Val 245 250 255 Val Ser Gly Ser
Ile Leu Asp Val Pro Ser Ala Ser Gly Thr Pro Ala 260 265 270 Asp Asp
Ala Ala Ala Lys Ser Arg Ile Thr Ser Glu Gly Glu Tyr Ile 275 280 285
Pro Leu Asp Gln Ile Asp Ile Asn Val Val Ser Lys Gly Glu Glu Leu 290
295 300 Phe Thr Gly Val Val Pro Ile Leu Val Glu Leu Asp Gly Asp Val
Asn 305 310 315 320 Gly His Lys Phe Ser Val Ser Gly Glu Gly Glu Gly
Asp Ala Thr Tyr 325 330 335 Gly Lys Leu Thr Leu Lys Phe Ile Cys Thr
Thr Gly Lys Leu Pro Val 340 345 350 Pro Trp Pro Thr Leu Val Thr Thr
Phe Gly Tyr Gly Leu Gln Cys Phe 355 360 365 Ala Arg Tyr Pro Asp His
Met Lys Gln His Asp Phe Phe Lys Ser Ala 370 375 380 Met Pro Glu Gly
Tyr Val Gln Glu Arg Thr Ile Phe Phe Lys Asp Asp 385 390 395 400 Gly
Asn Tyr Lys Thr Arg Ala Glu Val Lys Phe Glu Gly Asp Thr Leu 405 410
415 Val Asn Arg Ile Glu Leu Lys Gly Ile Asp Phe Lys Glu Asp Gly Asn
420 425 430 Ile Leu Gly His Lys Leu Glu Tyr Asn Tyr Asn Ser His Asn
Val Tyr 435 440 445 Ile Met Ala Asp Lys Gln Lys Asn Gly Ile Lys Val
Asn Phe Lys Ile 450 455 460 Arg His Asn Ile Glu Asp Gly Ser Val Gln
Leu Ala Asp His Tyr Gln 465 470 475 480 Gln Asn Thr Pro Ile Gly Asp
Gly Pro Val Leu Leu Pro Asp Asn His 485 490 495 Tyr Leu Ser Tyr Gln
Ser Ala Leu Ser Lys Asp Pro Asn Glu Lys Arg 500 505 510 Asp His Met
Val Leu Leu Glu Phe Val Thr Ala Ala Gly Ile Thr Leu 515 520 525 Gly
Met Asp Glu Leu Tyr Lys Phe Cys Tyr Glu Asn Glu Val 530 535 540
7262PRTHalobacterium helobium 7Met Leu Glu Leu Leu Pro Thr Ala Val
Glu Gly Val Ser Gln Ala Gln 1 5 10 15 Ile Thr Gly Arg Pro Glu Trp
Ile Trp Leu Ala Leu Gly Thr Ala Leu 20 25 30 Met Gly Leu Gly Thr
Leu Tyr Phe Leu Val Lys Gly Met Gly Val Ser 35 40 45 Asp Pro Asp
Ala Lys Lys Phe Tyr Ala Ile Thr Thr Leu Val Pro Ala 50 55 60 Ile
Ala Phe Thr Met Tyr Leu Ser Met Leu Leu Gly Tyr Gly Leu Thr 65 70
75 80 Met Val Pro Phe Gly Gly Glu Gln Asn Pro Ile Tyr Trp Ala Arg
Tyr 85 90 95 Ala Asp Trp Leu Phe Thr Thr Pro Leu Leu Leu Leu Asp
Leu Ala Leu 100 105 110 Leu Val
Asp Ala Asp Gln Gly Thr Ile Leu Ala Leu Val Gly Ala Asp 115 120 125
Gly Ile Met Ile Gly Thr Gly Leu Val Gly Ala Leu Thr Lys Val Tyr 130
135 140 Ser Tyr Arg Phe Val Trp Trp Ala Ile Ser Thr Ala Ala Met Leu
Tyr 145 150 155 160 Ile Leu Tyr Val Leu Phe Phe Gly Phe Thr Ser Lys
Ala Glu Ser Met 165 170 175 Arg Pro Glu Val Ala Ser Thr Phe Lys Val
Leu Arg Asn Val Thr Val 180 185 190 Val Leu Trp Ser Ala Tyr Pro Val
Val Trp Leu Ile Gly Ser Glu Gly 195 200 205 Ala Gly Ile Val Pro Leu
Asn Ile Glu Thr Leu Leu Phe Met Val Leu 210 215 220 Asp Val Ser Ala
Lys Val Gly Phe Gly Leu Ile Leu Leu Arg Ser Arg 225 230 235 240 Ala
Ile Phe Gly Glu Ala Glu Ala Pro Glu Pro Ser Ala Gly Asp Gly 245 250
255 Ala Ala Ala Thr Ser Asp 260 86PRTArtificial SequenceSynthetic
peptideVARIANT(2)..(2)xaa = any amino acid 8Phe Xaa Tyr Glu Asn Glu
1 5 97PRTArtificial SequenceSynthetic Peptide 9Phe Cys Tyr Glu Asn
Glu Val 1 5 1020PRTArtificial SequenceSynthetic Peptide 10Lys Ser
Arg Ile Thr Ser Glu Gly Glu Tyr Ile Pro Leu Asp Gln Ile 1 5 10 15
Asp Ile Asn Val 20
References
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.