Combination Therapy for Treatment of Cancer
Markland, JR.; Francis S. ; et al.
U.S. patent application number 13/219472 was filed with the patent office on 2011-12-29 for combination therapy for treatment of cancer. This patent application is currently assigned to University of Southern California. Invention is credited to Francis S. Markland, JR., Jacek Pinski, Steven Swenson.
| Application Number | 20110319345 13/219472 |
| Document ID | / |
| Family ID | 38668298 |
| Filed Date | 2011-12-29 |










View All Diagrams
| United States Patent Application | 20110319345 |
| Kind Code | A1 |
| Markland, JR.; Francis S. ; et al. | December 29, 2011 |
Combination Therapy for Treatment of Cancer
Abstract
The invention relates to compositions and methods for treating diseases. In particular aspects, the invention relates to administering a combination of a disintegrin with a microtubule stabilizing agent useful for treatment of cancer.
| Inventors: | Markland, JR.; Francis S.; (Manhattan Beach, CA) ; Swenson; Steven; (Arcadia, CA) ; Pinski; Jacek; (La Canada, CA) |
| Assignee: | University of Southern
California |
| Family ID: | 38668298 |
| Appl. No.: | 13/219472 |
| Filed: | August 26, 2011 |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | ||
|---|---|---|---|---|
| 11742389 | Apr 30, 2007 | 8008256 | ||
| 13219472 | ||||
| 60797030 | May 1, 2006 | |||
| Current U.S. Class: | 514/21.2 ; 514/1.1; 514/21.3 |
| Current CPC Class: | A61P 21/02 20180101; A61P 35/00 20180101; A61K 38/1703 20130101; A61P 13/06 20180101 |
| Class at Publication: | 514/21.2 ; 514/1.1; 514/21.3 |
| International Class: | A61K 38/17 20060101 A61K038/17; A61P 35/00 20060101 A61P035/00 |
Goverment Interests
STATEMENT OF GOVERNMENT SUPPORT
[0002] This invention was made with government support under Contract No. W81XWH-04-1-0817 awarded by the US Army Medical Research and Material Command. The government has certain rights in the invention.
Claims
1.-56. (canceled)
57. A combination comprising a therapeutically effective amount of a disintegrin, characterized by having an integrin binding loop stabilized by disulfide bonds, and a microtubule stabilizing agent.
58.-59. (canceled)
60. The combination of claim 57 wherein said disintegrin is selected from the group consisting of (a) vicrostatin; (b) a contortrostatin monomer; (c) a contortrostatin dimer; and, (d) a contortrostatin precursor or biologically active variant thereof, containing an amino acid sequence selected from the group consisting of: (1) amino acid numbers 419 to 483 of SEQ ID NO: 1; (2) amino acid numbers 191 to 410 of SEQ ID NO: 1; (3) amino acid numbers 1 to 190 of SEQ ID NO: 1; (4) SEQ ID NO: 1; (5) an amino acid sequence at least 90% identical to (1), (2) or (4) as determined by FASTA or BLAST using default opening and gap penalties and a default scoring matrix; or, (6) an amino acid sequence at least 95% identical to (3) as determined by FASTA or BLAST using default opening and gap penalties and a default scoring matrix.
61. The combination of claim 57 wherein said disintegrin comprises a contortrostatin which has an amino acid sequence which is at least 90% percent identical to amino acid numbers 419 to 483 of SEQ ID NO: 1, wherein said disintegrin (i) binds to integrin .alpha.v.beta.5 and (ii) induces .alpha.v.beta.3-mediated tyrosine phosphorylation of CAS and FAK in tumor cells.
62. The combination of claim 57 wherein said disintegrin is a contortrostatin that comprises a monomer having a molecular mass of about 5 to about 7 kDa.
63. The combination of claim 62 wherein said contortrostatin monomer forms a homodimer with another contortrostatin monomer.
64. The combination of claim 57 wherein said disintegrin comprises a constrained Arg-Gly-Asp (RGD) sequence of a peptide loop of about 13 amino acid residues flanked by two Cys residues, wherein the peptide loop is an integrin antagonist which has an amino acid sequence comprising amino acid numbers 457 to 469 of SEQ ID NO: 1.
65. The combination of claim 57 wherein said disintegrin is vicrostatin.
66. The combination of claim 57 wherein said microtubule stabilizing agent is a taxane.
67. The combination of claim 66 wherein said taxane is docetaxel.
68. The combination of claim 66 wherein said taxane is paclitaxel.
69. The combination of claim 57 wherein said taxane has Formula H as follows: ##STR00018## wherein: R.sup.1 and R.sup.2 are independently selected from alkyl, alkenyl, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, or oxy, each of which may be optionally substituted; R.sup.3 and R.sup.4 are independently selected from alkyl, substituted alkyl, hydroxyl, oxy, C(O)H, or OC(O)R.sup.5; and R.sup.5 is alkyl, alkenyl, cycloalkyl, heterocycloalkyl, aryl, or heteroaryl, each of which may be optionally substituted.
70. The combination of claim 66 wherein said taxane has Formula III as follows: ##STR00019## wherein R.sup.10 is selected from alkyl, cycloalkyl, aryl or heteroaryl, each of which may be optionally substituted; and R.sup.11 is selected from hydrogen, alkyl, --C(O)H, --C(O)CH.sub.3, or --C(O)CH.sub.2CH.sub.3.
71. The composition of claim 70 wherein R.sup.10 is --C(CH.sub.3).sub.3 and R.sup.11 is H.
72. The combination of claim 70 wherein R.sup.10 is phenyl and R.sup.11 is acetyl.
73. The combination of claim 57 wherein said microtubule stabilizing agent is a non-taxane.
74. The combination of claim 73 wherein said non-taxane microtubule stabilizing agent has Formula V as follows: ##STR00020## wherein Q is selected from the group consisting of ##STR00021## G is selected from the group consisting of alkyl, substituted alkyl, substituted or unsubstituted aryl, heterocyclo, ##STR00022## W is O or NR.sup.45; X is O or H, H; Y is selected from the group consisting of O; H, OR.sup.46; OR.sup.47, OR.sup.47; NOR.sup.48; H, NOR.sup.49; H, NR.sup.50R.sup.51; H, H; and CHR.sup.52; wherein OR.sup.47 OR.sup.47 can be a cyclic ketal; Z.sup.1 and Z.sup.2 are independently selected from the group consisting of CH.sub.2, O, NR.sup.53, S and SO.sub.2, wherein only one of Z.sup.1 and Z.sup.2 can be a heteroatom; B.sup.1 and B.sup.2 are independently selected from the group consisting of OR.sup.54, OC(O)R.sup.55, and OC(O)NR.sup.56R.sup.57; wherein when B.sup.1 is OH and Y is OH, H, B.sup.1 and Y can form a six-membered ring ketal or acetal; D is selected from the group consisting of NR.sup.58R.sup.59, NR.sup.60COR.sup.61 and saturated heterocycle; R.sup.31, R.sup.32, R.sup.33, R.sup.34, R.sup.35, R.sup.36, R.sup.37, R.sup.48, R.sup.49, R.sup.50, R.sup.51, R.sup.52, R.sup.56 and R.sup.57 are independently selected from H, alkyl, substituted alkyl, or aryl, wherien when R.sup.31 and R.sup.32 are alkyl, they can be joined to form a cycloalkyl; and when R.sup.33 and R.sup.34 are alkyl, they can be joined to form a cycloalkyl; R.sup.39, R.sup.40, R.sup.46 and R.sup.47 are independently selected from H, alkyl, and substituted alkyl; R.sup.38, R.sup.41, R.sup.42, R.sup.58, R.sup.60, R.sup.62 and R.sup.63 are independently selected from the group consisting of H, alkyl, substituted alkyl, aryl, substituted aryl, cycloalkyl, and heterocyclo; R.sup.13, R.sup.14 and R.sup.61 are independently selected from the group consisting of H, alkyl, substituted alkyl, aryl, heteroaryl, cycloalkyl, and heterocyclo; R.sup.54 and R.sup.55 are independently selected from the group consisting of H, alkyl, substituted alkyl, aryl, substituted aryl, and heterocyclo; R.sup.45, R.sup.53 and R.sup.59 are independently selected from the group consisting of H, alkyl, substituted alkyl, aryl, substituted aryl, cycloalkyl, heterocyclo, R.sup.62C(O), R.sup.63SO.sub.2, hydroxy, O-alkyl and O-substituted alkyl.
75. The combination of claim 73 wherein the non-taxane microtubule stabilizing agent has Formula V as follows: ##STR00023## wherein W is O, NH or NR.sup.64; R.sup.35 and R.sup.38 are independently selected from lower alkyl or lower alkenyl; R.sup.64 is selected from H, OH, optionally substituted alkyl, optionally substituted oxy, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; and R.sup.h is selected from cycloalkyl, heterocycloalkyl, aryl or heteroaryl, each of which may be optionally substituted.
76. The combination of claim 75 wherein W is O or NH; R.sup.35 and R.sup.38 are each CH.sub.3, and R.sup.h is selected from the group consisting of thiazole, oxazole or pyridine, each of which is optionally substituted.
77. A pharmaceutical composition comprising the combination of claim 57 in a pharmaceutically acceptable carrier.
Description
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application is a divisional application of U.S. application Ser. No. 11/742,389, which claims the benefit of priority under 35 U.S.C. .sctn.119(e) to U.S. Provisional Application Ser. No. 60/797,030 filed May 1, 2006, which is incorporated by reference herein in its entirety including all figures and tables.
SEQUENCE LISTING
[0003] The instant application contains a Sequence Listing which has been submitted via EFS-Web and is hereby incorporated by reference in its entirety. Said ASCII copy, created on Apr. 29, 2010 is named 07540512.txt, and is 24,000 bytes in size.
FIELD OF THE INVENTION
[0004] The invention relates to compositions and methods for treating diseases. In particular aspects, the invention relates to administering a combination of a disintegrin with a microtubule stabilizing agent useful for treatment of cancer.
BACKGROUND OF THE INVENTION
[0005] The first step of metastasis involves the attachment of cancer cells to tissues around the primary site, i.e., to the extracellular matrix (ECM) via cell surface integrins and other adhesion receptors. Integrin targets of the ECM include fibronectin, fibrinogen, vitronectin, collagen and laminin. Integrins mediate cell-cell and cell-substratum interactions and are involved in bidirectional signaling that links the ECM with cytoskeletal proteins. In the second step, cancer cells secrete digestive enzymes that degrade the surrounding tissues allowing the tumor cells to invade these tissues. Eventually, the tumor cells enter the blood or lymphatic system where they repeat the adhesion and invasion steps at a distant (metastatic) site. At this remote site, tumor cells induce the formation of new blood vessels (a process called neovascularization), in and around the growing tumor. These new blood vessels supply nutrients to the metastatic tumor and allow it to grow. Treatments that block any of these steps should act to inhibit metastasis.
[0006] Integrins are heterodimers composed of alpha and beta submits that are non-covalently associated. Interactions between integrins and ECM proteins have been shown to be mediated via an Arg-Gly-Asp (RGD) sequence present in the matrix proteins. Both the alpha and beta subunits of the integrin are required for fibrinogen binding.
[0007] A well known inhibitor of the integrin-ECM interaction is a disintegrin which represents a family of proteins that include those from venom of snakes of the Crotalidae and Viperidae families have been found to inhibit glycoprotein (GP) IIb/IIIa mediated platelet aggregation. See, e.g., Huang, T. F. et al., J. Biol. Chem. 262:16157 (1987); Gan, Z. R. et al., J. Biol. Chem. 263:19827 (1988); Yasuda, T. et al., J. Am. Coll. Cardiol. 16:714 (1990); Trikha, M. et al., Fibtinolysis 4 (Suppl. 1):105 (1990); Trikha, M. et al., Blood 76 (Suppl. 1):479a (1990); Holahan, M. A. et al., Pharmacology 42:340 (1991); Shebuski, R. J. et al., Circulation 82:169 (1990); Yasuda, T. et al., Circulation 83:1038 (1991). Disintegrins are disulfide rich and, with the exception of barbourin, contain an RGD (Arg-Gly-Asp) sequence that has been implicated in the inhibition of integrin-mediated interactions (Scarborough et al., J. Biol. Chem. 266(20):9359-62 (1991)). Most disintegrins can disrupt different integrin-ECM interactions (e.g., inhibition of .beta.1 integrins (McLane et al. 1998) and .beta.3 integrins such as barbourin are relatively specific and disrupt only .alpha.IIb.beta.3 integrin function (Scarborough et al. (1991)).
[0008] The RGD sequence of disintegrins is located at the tip of a flexible loop, the integrin-binding loop, stabilized by disulfide bonds and protruding from the main body of the polypeptide chain. See, e.g., amino acid residues 457 to 469 of SEQ ID NO: 1. This exposed RGD sequence enables disintegrins to bind to integrins with high affinity. Portions of a disintegrin other than the RGD site may have biological effects on integrins. See, e.g., Connolly, T. M. et al., Circulation 82 (Suppl. III):660 (1990)).
[0009] Disintegrins that are known to disrupt integrin interactions include bitistatin, an 83 amino acid disintegrin isolated from the venom of Bitis arietans; echistatin, a 49 amino acid disintegrin isolated from the venom of Echis cannatus; kistrin, a 68 amino acid disintegrin isolated from the venom of Calloselasma rhodostoma; trigamin, a 72 amino acid disintegrin isolated from the venom of Trimeresurus gramineus, (see U.S. Pat. No. 5,066,592 by Huang et al.); applaggin, isolated from the venom of Agkistrodon piscivorus piscivorus (see e.g., Chao, B. H. et al., Proc. Natl. Acad. Sci. USA 86:8050 (1989); Savage, B. et al., J. Biol. Chem. 265:11766 (1990)); and contortrostatin (CN), isolated from the venom of Agkistrodon contortix contortix (the southern copperhead snake).
[0010] Unlike other monomeric disintegrins, CN is a homodimer with molecular mass (Mr) of 13,505 for the intact molecule and 6,750 for the reduced chains as shown by mass spectrometry (Trikha, Rote, et al., Thrombosis Research 73:39-52 (1994)). CN can be purified from snake venom, as described in Trikha, Rote, et al., Thrombosis Research 73:39-52 (1994).
[0011] CN full-length DNA precursor has been cloned and sequenced (Zhou, Hu et al. (2000)). CN is produced in the snake venom gland as a multidomain precursor of 2027 by having a 1449 by open reading frame encoding a precursor that includes a pro-protein domain (amino acid residues 1 to 190 of SEQ ID NO: 1), a metalloproteinase domain (residues 191 to 410 of SEQ ID NO: 1) and a disintegrin domain (residues 419 to 483 of SEQ ID NO: 1). The CN precursor is proteolytically processed, possibly autocatalytically, to generate mature CN. The CN disintegrin domain encodes 65 amino acids with a molecular weight equal to that of the mature CN subunit. CN displays the classical RGD motif in its integrin-binding loop.
[0012] The CN full-length precursor mRNA sequence can be accessed in the GenBank database using accession number: AF212305. The nucleotide sequence encoding the 65 amino acid disintegrin domain of CN represents the segment from 1339 to 1533 in the mRNA. Plasmids encoding the CN full-length gene have been described (Zhou, Hu et al. (2000)) and are available from the laboratory of Francis S. Markland at University of Southern California (Los Angeles, Calif.). Various recombinant forms of CN are disclosed in U.S. Pat. No. 6,710,030 by Markland.
[0013] CN is cysteine-rich (10 cysteines per monomer), displays no secondary structure and, like other disintegrins, has a complex folding pattern that relies on multiple disulfide bonds (four intrachain and two interchain disulfide bonds) to stabilize its tertiary structure (Zhou, Hu et al. (2000)). The compact structure of CN, achieved by its multiple disulfide bonds, renders it more resistant to proteolytic inactivation as compared to other disintegrins.
[0014] Receptors of CN that have been identified include: integrins .alpha.IIb.beta.3, .alpha.v.beta.3, .alpha.v.beta.5, and .alpha.5.beta.1 (Trikha, De Clerck et al., Cancer Res. 54(18): 4993-98 (1994); Trikha, Rote et al., Thrombosis Res. 73(1): 39-52 (1994); Zhou, Nakada et al., Angiogenesis 3(3): 259-69 (1999); Zhou, Nakada et al., Biochem. Biophys. Res. Commun. 267(1): 350-55 (2000). Interactions between CN and integrins are RGD-dependent. As an anti-cancer agent, CN has effective anti-angiogenic and anti-metastatic properties (Trikha, De Clerck et al. 1994; Trikha, Rote et al. (1994); Schmitmeier et al., Anticancer Res. 20(6B): 4227-33 (2000); Zhou, Hu et al., Biochem. Biophys. 375(2): 278-88 (2000); Markland et al., Haemostasis 31(3-6): 183-91 (2001); Swenson et al., Mol. Cancer. Ther. 3(4): 499-511 (2004)). CN also has the ability to directly engage tumor cells and suppress their growth in a cytostatic manner (Trikha, De Clerck et al. (1994); Trikha, Rote et al. (1994); Schmitmeier et al. (2000)). The antitumoral activity of CN is based on its high affinity interaction with integrins .alpha.5.beta.1, .alpha.v.beta.3 and .alpha.v.beta.5 on both cancer cells and newly growing vascular endothelial cells (Trikha, De Clerck et al. (1994); Zhou, Nakada et al. (1999); Zhou, Nakada et al. (2000); Zhou, Sherwin et al., Breast Cancer Res. Treat. 61(3): 249-60 (2000)). This diverse mechanism of action provides CN with a distinct advantage over many antiangiogenic agents that only block a single angiogenic pathway and/or do not directly target tumor cells.
[0015] The taxanes represent a class of small molecule diterpenoids compounds (i.e., taxoids) that are useful for cancer therapy. Paclitaxel (Taxol.RTM.) and docetaxel (Taxotere.RTM.), are well known taxanes which are efficacious against a range of solid tumors, particularly carcinomas, melanomas, and sarcomas. (See e.g., references cited in Pamela et al., Clin Cancer Res Vol. 8, 846-855 (2002)). Paclitaxel and docetaxel bind to .beta. tubulin and disrupt microtubule assembly/disassembly. Id. Stabilization of microtubules by taxanes causes mitotic arrest and cell death (e.g., apoptosis) reportedly independent of the p53 tumor suppressor. Id. Taxanes induce genes encoding inflammatory mediators such as tumor necrosis factor alpha, interleukins, and enzymes such as NO synthase and COX-2. Id.
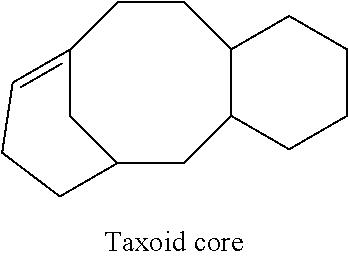
[0016] Taxanes have a common "taxoid" core structure shown below.
##STR00001##
[0017] Taxol.RTM. was first isolated from the bark of the Pacific yew (Taxus brevifolia Nutt.) but is presently derived mainly by semisynthesis from the advanced taxoid 10-deacetylbaccatin III, which can be obtained from bark or needles of the European yew, Taxus baccata. (See e.g., references 15-20 in Jennewein, et al., PNAS, 98(24):13595-13560 (2001); see also Holton, et al., J. Am. Chem. Soc., 116:1597-1601 (1994)).
[0018] A number of modified taxanes or taxoid analogs have been prepared which have a taxane ring bearing modified side chains. These modified taxanes or taxoid analogs inhibit cancer growth while having greater water solubility and stability than naturally occurring Taxol.RTM.. Analogs also include fatty acid conjugates. Exemplary derivatives of Taxol.RTM. are described in U.S. Pat. Nos. 6,638,742; 5,278,324; 5,272,171; 5,254,580; 5,250,683; 5,248,796; and 5,227,400; and U.S. Pub. App. No. 2005/0148657; and the references cited therein, as well as those compounds disclosed in Villalva-Servin, et al., Can. J. Chem., 82: 227-39 (2004); Shen, et al., Chem. Pharm. Bull., 53(7): 808-10 (2005); Ono, et al., Biol. Pharm. Bull., 27(3): 345-51 (2004); Sampath, et al., Mol. Cancer. Ther., 2(9): 873-74 (2003); and Wolff, et al., Clin. Cancer Res., 9(10): 3589-97 (2003).
[0019] The co-administration of taxanes or taxane derivatives with at least one active agent has been reported. Taxotere.RTM. in combination with prednisone has been approved by the US Food and Drug Administration for the treatment of metastatic androgen-independent prostate cancer. Rose et al. reported the administration of the oral taxane BMS-275183 in combination with cetuximab (an anti-epidermal growth factor receptor monoclonal antibody) (Rose, et al., Clin. Cancer Res., 10(21): 7413-17 (2004)). Levy, et al. reported the administration of antimetabolite-taxane combinations (specifically, the administration of gemcitabine and docetaxel) in women with anthracycline pretreated metastatic breast cancer (Levy, et al., Cancer Treat. Rev., 31: S17-22 (2005)).
SUMMARY OF THE INVENTION
[0020] The invention relates to compositions and methods for treating diseases. In particular aspects, the invention relates to administering a combination of a disintegrin with a small molecule cell division inhibitor for treating cancer. In preferred embodiments, the small molecule cell division inhibitor is a microtubule stabilizing agent. The inventors have discovered that administration of disintegrins in combination with a microtubule stabilizing agent is particularly effective in inhibiting cancer and/or preventing metastasis.
[0021] In one aspect, the invention provides a method of treating an individual suffering from cancer, including administering to the individual a therapeutically effective amount of a disintegrin and a microtubule stabilizing agent. In another aspect, the invention provides a method of preventing or inhibiting the growth of metastases in an individual having cancer, the method including administering to the individual an effective amount of a disintegrin and a microtubule stabilizing agent. In yet a further aspect, the invention provides a combination including a therapeutically effective amount of a disintegrin and a microtubule stabilizing agent. In a preferred embodiment, the microtubule stabilizing agent is a taxane.
[0022] In some embodiments, the cancer expresses an integrin; preferably, the integrin is .alpha.v.beta.5. In other embodiments, the cancer is one or more cancers selected from the group consisting of prostate cancer, breast cancer, lung cancer, colon cancer, ovarian cancer, renal cancer, central nervous system (CNS) cancer, and leukemia. In a preferred embodiment, the cancer is prostate cancer.
[0023] As used herein, "disintegrin" refers to a class of cysteine-rich proteins that are potent soluble ligands of integrins and which are involved in regulating many processes such as cell-cell and cell-extracellular matrix adhesion, migration and invasion, cell cycle progression, differentiation and cell type speciation during development of many metazoan organisms, cell death and apoptosis. The tri-peptide motif RGD (Arg-Gly-Asp) is conserved in most monomeric disintegrins and is located at the tip of a flexible loop, the integrin-binding loop, which is stabilized by disulfide bonds and protruding from the main body of the polypeptide chain. All disintegrins purified from snake venoms bind to the fibrinogen receptor, integrin .alpha.IIb.beta.3, the binding of which results in the inhibition of fibrinogen-dependent platelet aggregation. Most disintegrins also bind to integrins .alpha.v.beta.3 (a vitronectin receptor) and .alpha.5.beta.1 (a fibronectin receptor) in an RGD-dependent manner. Also included within the meaning of disintegrins are biologically active variants and fragments thereof, which variants include for example without limitation, fusion proteins which include disintegrins or fragments thereof.
[0024] In preferred embodiments, the disintegrin is a contortrostatin (CN). CN is a disintegrin isolated from Agkistrodon contortrix contortrix (southern copperhead) venom (Trikha, Rote et al. 1994). CN is produced in the snake venom gland as a multidomain precursor of 2027 by having a 1449 by open reading frame encoding the pro-protein, metalloproteinase and disintegrin domains. The precursor is proteolytically processed, possibly autocatalytically, to generate mature CN. The full length CN proprotein is encoded by the nucleotide sequence 85-1536 of the full length mRNA (GenBank AF212305), whereas the disintegrin domain of CN represents 1339-1533 of the mRNA. The CN disintegrin domain, which contains 65 amino acids, is shown below with the RGD sequence underlined.
TABLE-US-00001 (SEQ ID NO: 3) DAPANPCCDAATCKLTTGSQCADGLCCDQCKFMKEGTVCRRARGDDLDD YCNGISAGCPRNPFHA
[0025] Contortrostatin as used herein includes the native homodimer as well as the monomer, precursor or biologically active variant thereof. In some embodiments, the biologically active variant includes an amino acid sequence selected from the group consisting of: (a) amino acid numbers 419 to 483 of SEQ ID NO: 1; (b) amino acid numbers 191 to 410 of SEQ ID NO: 1; (c) amino acid numbers 1 to 190 of SEQ ID NO: 1; (d) SEQ ID NO: 1; (e) an amino acid sequence at least 90% identical to (a), (b) or (d) as determined by FASTA or BLAST using default opening and gap penalties and a default scoring matrix; and (f) an amino acid sequence at least 95% identical to (c) as determined by FASTA or BLAST using default opening and gap penalties and a default scoring matrix.
[0026] In certain embodiments, the disintegrin includes a contortrostatin amino acid sequence which is at least 90% percent identical to amino acid numbers 419 to 483 of SEQ ID NO: 1, wherein the contortrostatin amino acid sequence (i) binds to integrin .alpha.v.beta.5 and (ii) induces .alpha.v.beta.3-mediated tyrosine phosphorylation of CAS and FAK in tumor cells.
[0027] In yet further embodiments, the disintegrin includes a constrained Arg-Gly-Asp (RGD) sequence of a peptide loop of about 13 amino acid residues flanked by two Cys residues, where the peptide loop is an integrin antagonist which has an amino acid sequence comprising amino acid numbers 457 to 469 of SEQ ID NO: 1.
[0028] In other embodiments, the disintegrin is vicrostatin, which is a fusion protein that includes a contortrostatin domain N-terminal to the sequence HKGPAT (SEQ ID NO: 47):
[0029] As used herein, the term "purified" in reference to polypeptides (or proteins) does not require absolute purity. Instead, it represents an indication that the polypeptide(s) of interest is (are) in an environment in which the protein is more abundant (on a mass basis) than the environment from which the protein was initially produced. Purified polypeptides may be obtained by a number of methods including, for example, chromatography, preparative electrophoresis, centrifugation, precipitation, affinity purification, etc. The degree of purity is preferably at least 10%. One or more "substantially purified" polypeptides are at least 50% of the protein content of the environment, more preferably at least 75% of the protein content of the environment, and most preferably at least 95% of the protein content of the environment. Protein content may be determined using a modification of the method of Lowry, et al. (Lowry, Rosebrough et al. 1951), described by Hartree (Hartree 1972), using bovine serum albumin as a protein standard.
[0030] As described herein, cancer therapy is achieved by administering a combination of a disintegrin with an agent that inhibits cell division. Preferably, the cell division inhibitor is a microtubule stabilizing agent.
[0031] As used herein, "microtubule stabilizing agent" refers to any compound which inhibits cell division by binding to .beta. tubulin and thereby disrupting the equilibrium between the free .beta. tubulin and microtubules (See e.g., Pamela et al., Clin Cancer Res Vol. 8, 846-855 (2002)). Stabilization of microtubules by a microtubule stabilizing agents causes mitotic arrest and cell death (e.g., apoptosis). At certain doses, microtubule stabilizing agents may have other effects including induction of genes encoding inflammatory mediators such as tumor necrosis factor alpha, interleukins, and enzymes such as NO synthase and COX-2. Microtubule stabilizing agents are preferably small molecules of 1,500 daltons or less, preferably 1,000 daltons or less. Exemplary microtubule stabilizing agents include, but are not limited to, taxanes and non-taxanes such as epothilones.
[0032] "Taxane" refers to a chemical class of diterpenoids compounds that inhibit cell division. Taxanes as used herein share a common core structure (i.e., a taxoid core) shown below.
##STR00002##
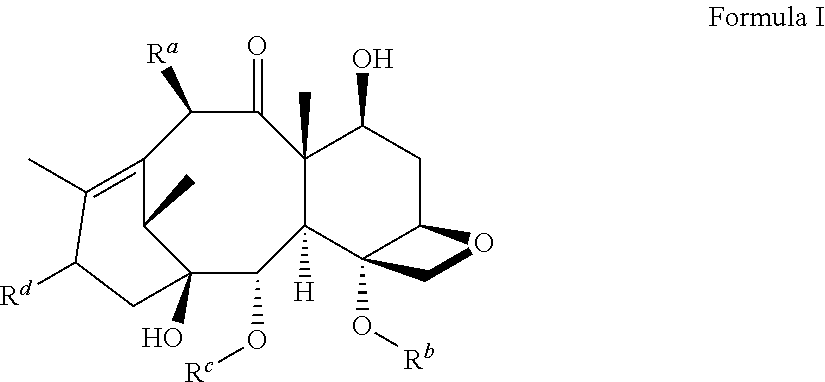
[0033] Taxol.RTM. (paclitaxel), and Taxotere.RTM. (docetaxel) are well known microtubule stabilizing agents of the taxane family. The term "taxane" as used herein also encompasses derivatives of naturally occurring taxanes referred to herein as a "taxane derivative" or "taxoid analog." A preferred taxane is shown in Formula I.
##STR00003##
wherein: [0034] R.sup.a is hydrogen, hydroxyl, alkyl, substituted alkyl, oxy, substituted oxy, cycloalkyl, substituted cycloalkyl, heterocycloalkyl, substituted heterocycloalkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, or --C(O)R.sup.e; [0035] R.sup.b is hydrogen, alkyl, substituted alkyl, or C(O)R.sup.e; [0036] R.sup.c is hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, or C(O)R.sup.e; [0037] R.sup.d is alkyl, alkenyl, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, alkoxy, alkenoxy, or --OC(O)R.sup.e, each of which may be optionally substituted; [0038] R.sup.e is hydrogen, alkyl, alkenyl, amino, cycloalkyl, heterocycloalkyl, aryl, heteroaryl or --(CH.sub.2).sub.nNHC(O)R.sup.f, each of which may be optionally substituted; and [0039] R.sup.f is alkyl, alkenyl, oxy, cycloalkyl, heterocycloalkyl, aryl, or heteroaryl, each of which may be optionally substituted; wherein n is an integer between 1 and 5.
[0040] In particular embodiments, the taxane has the structure shown as Formula II.
##STR00004##
wherein: [0041] R.sup.1 and R.sup.2 are independently selected from alkyl, alkenyl, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, or oxy, each of which may be optionally substituted; [0042] R.sup.3 and R.sup.4 are independently selected from alkyl, substituted alkyl, hydroxyl, oxy, C(O)H, or OC(O)R.sup.5; and [0043] R.sup.5 is alkyl, alkenyl, cycloalkyl, heterocycloalkyl, aryl, or heteroaryl, each of which may be optionally substituted.
[0044] In other embodiments, the taxane has the structure shown as Formula III.
##STR00005##
wherein [0045] R.sup.10 is selected from alkyl, cycloalkyl, aryl or heteroaryl, each of which may be optionally substituted; and [0046] R.sup.11 is selected from hydrogen, alkyl, --C(O)H, --C(O)CH.sub.3, or --C(O)CH.sub.2CH.sub.3.
[0047] In further embodiments of Formula III, R.sup.10 is selected from--C(CH.sub.3).sub.3 or phenyl and R.sup.11 is selected from hydrogen, --C(O)CH.sub.3 or --C(O)CH.sub.2CH.sub.3. In a one embodiment, R.sup.10 is--C(CH.sub.3).sub.3 and R.sup.11 is H. In another embodiment, R.sup.10 is phenyl and R.sup.11 is --C(O)CH.sub.3.
[0048] Also included within the meaning of "taxane" as used herein are rearranged taxoids having the structure shown in Formula IV, which are described, for example, in Choudhary, et al., Chem. Pharm. Bull., 50(11): 1488-90 (2002). Rearranged taxoids useful in the invention are microtubule stabilizing agents.
##STR00006##
wherein [0049] R.sup.21 and R.sup.23 are independently selected from hydrogen, lower alkyl, substituted lower alkyl, acetyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heteroaryl or substituted heteroaryl; [0050] R.sup.22 is selected from hydrogen, hydroxy, lower alkyl, substituted lower alkyl or acetyl; [0051] R.sup.24, R.sup.25 and R.sup.26 are each independently selected from hydrogen, hydroxy, lower alkyl, substituted lower alkyl, oxy, acetyl, cycloalkyl, substituted cycloalkyl, heterocycloalkyl, substituted heterocycloalkyl, aryl, substituted aryl, heteroaryl, or substituted heteroaryl.
[0052] In further embodiments of Formula IV, R.sup.21 is acetyl, R.sup.22 is acetyl, R.sup.23 and R.sup.26 are hydrogen and R.sup.24 and R.sup.25 are hydroxy. In other embodiments, R.sup.21, R.sup.22 and R.sup.24 are each hydrogen, and R.sup.23, R.sup.25 and R.sup.26 are each --C(O)OCH.sub.3.
[0053] In preferred embodiments, the taxanes including those of Formulas I-III are administered in combination with a disintegrin, preferably contortrostatin. In preferred embodiments, the taxanes including Formulas I-III are administered in combination with vicrostatin.
[0054] In another aspect of the present invention, a non-taxane microtubule stabilizing agent having the structure shown in Formula V is administered in combination with a disintegrin:
##STR00007##
wherein [0055] Q is selected from the group consisting of
[0055] ##STR00008## [0056] G is selected from the group consisting of alkyl, substituted alkyl, substituted or unsubstituted aryl, heterocyclo,
[0056] ##STR00009## [0057] W is O or NR.sup.45; [0058] X is O or H, H; [0059] Y is selected from the group consisting of O; H, OR.sup.46; OR.sup.47, OR.sup.47; NOR.sup.48; H, NOR.sup.49; H, NR.sup.50R.sup.51; H, H; and CHR.sup.52; wherein OR.sup.47 OR.sup.47 can be a cyclic ketal; [0060] Z.sup.1 and Z.sup.2 are independently selected from the group consisting of CH.sub.2, O, NR.sup.53, S and SO.sub.2, wherein only one of Z.sup.1 and Z.sup.2 can be a heteroatom; [0061] B.sup.1 and B.sup.2 are independently selected from the group consisting of OR.sup.54, OC(O)R.sup.55, and OC(O)NR.sup.56R.sup.57; wherein when B.sup.1 is OH and Y is OH, H, B.sup.1 and Y can form a six-membered ring ketal or acetal; [0062] D is selected from the group consisting of NR.sup.58R.sup.59, NR.sup.60COR.sup.61 and saturated heterocycle; [0063] R.sup.31, R.sup.32, R.sup.33, R.sup.34, R.sup.35, R.sup.36, R.sup.37, R.sup.48, R.sup.49, R.sup.50, R.sup.51, R.sup.52, R.sup.56 and R.sup.57 are independently selected from H, alkyl, substituted alkyl, or aryl, wherien when R.sup.31 and R.sup.32 are alkyl, they can be joined to form a cycloalkyl; and when R.sup.33 and R.sup.34 are alkyl, they can be joined to form a cycloalkyl; [0064] R.sup.39, R.sup.40, R.sup.46 and R.sup.47 are independently selected from H, alkyl, and substituted alkyl; [0065] R.sup.38, R.sup.41, R.sup.42, R.sup.58, R.sup.60, R.sup.62 and R.sup.63 are independently selected from the group consisting of H, alkyl, substituted alkyl, aryl, substituted aryl, cycloalkyl, and heterocyclo; [0066] R.sup.13, R.sup.14 and R.sup.61 are independently selected from the group consisting of H, alkyl, substituted alkyl, aryl, heteroaryl, cycloalkyl, and heterocyclo; [0067] R.sup.54 and R.sup.55 are independently selected from the group consisting of H, alkyl, substituted alkyl, aryl, substituted aryl, and heterocyclo; [0068] R.sup.45, R.sup.53 and R.sup.59 are independently selected from the group consisting of H, alkyl, substituted alkyl, aryl, substituted aryl, cycloalkyl, heterocyclo, R.sup.62C(O), R.sup.63SO.sub.2, hydroxy, O-alkyl and O-substituted alkyl; [0069] and any salts, solvates or hydrates thereof.
[0070] In one embodiment, Y and X are O; W is O or NH; B.sup.1 and B.sup.2 are OH; R.sup.31 and R.sup.32 are H; R.sup.33 R.sup.34 and R.sup.36 are CH.sub.3; Z.sup.1 and Z.sup.2 are CH; and G is --R.sup.gR.sup.h wherein R.sup.g is lower alkyl or lower alkenyl and R.sup.h is an optionally substituted herteroaryl or heterocycle. In one embodiment, the disintegrin is contortrostatin. In another embodiment, the disintegrin is vicrostatin.
[0071] In yet another aspect of the present invention, a non-taxane microtubule stabilizing agent having the structure shown in Formula Va is administered in combination with a disintegrin:
##STR00010##
wherein [0072] Q is selected from the group consisting of
[0072] ##STR00011## [0073] G is selected from the group consisting of alkyl, substituted alkyl, substituted or unsubstituted aryl, heterocyclo,
[0073] ##STR00012## [0074] W is O or NR.sup.45; [0075] X is O or H, H; [0076] Y is selected from the group consisting of 0; H, OR.sup.46; OR.sup.47, OR.sup.47; NOR.sup.48; H, NOR.sup.49; H, NR.sup.50R.sup.51; H, H; and CHR.sup.52; wherein OR.sup.47 OR.sup.47 can be a cyclic ketal; [0077] Z.sup.1 and Z.sup.2 are independently selected from the group consisting of CH.sub.2, O, NR.sup.53, S and SO.sub.2, wherein only one of Z.sup.1 and Z.sup.2 can be a heteroatom; [0078] B.sup.1 and B.sup.2 are independently OC(O)NR.sup.56R.sup.57; [0079] D is selected from the group consisting of NR.sup.58R.sup.59 and saturated heterocycle; [0080] R.sup.31, R.sup.32, R.sup.33, R.sup.34, R.sup.35, R.sup.36, R.sup.37, R.sup.48, R.sup.49, R.sup.50, R.sup.51, R.sup.52, R.sup.56 and R.sup.57 are independently selected from H, alkyl, substituted alkyl, or aryl, wherien when R.sup.31 and R.sup.32 are alkyl, they can be joined to form a cycloalkyl; and when R.sup.33 and R.sup.34 are alkyl, they can be joined to form a cycloalkyl; [0081] R.sup.39, R.sup.40, R.sup.46 and R.sup.47 are independently selected from H, alkyl, and substituted alkyl; [0082] R.sup.38, R.sup.41, R.sup.42, R.sup.58, R.sup.62 and R.sup.63 are independently selected from the group consisting of H, alkyl, substituted alkyl, aryl, substituted aryl, cycloalkyl, and heterocyclo; [0083] R.sup.45, R.sup.53 and R.sup.59 are independently selected from the group consisting of H, alkyl, substituted alkyl, aryl, substituted aryl, cycloalkyl, heterocyclo, R.sup.62C(O), R.sup.63SO.sub.2, hydroxy, O-alkyl and O-substituted alkyl; [0084] and any salts, solvates or hydrates thereof.
[0085] In another aspect of the present invention, a non-taxane microtubule stabilizing agent having the structure shown in Formula VI is administered in combination with a disintegrin.
##STR00013##
[0086] wherein W is O, NH or NR.sup.64; [0087] R.sup.35 and R.sup.38 are independently selected from lower alkyl or lower alkenyl; [0088] R.sup.64 is selected from H, OH, optionally substituted alkyl, optionally substituted oxy, optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, optionally substituted aryl, or optionally substituted heteroaryl; and [0089] R.sup.h is selected from cycloalkyl, heterocycloalkyl, aryl or heteroaryl, each of which may be optionally substituted.
[0090] In one embodiment, W is O or NH; R.sup.35 and R.sup.38 are CH.sub.3, and R.sup.h is selected from a substituted thiazole, oxazole or pyridine. In another embodiment, the disintegrin is contortrostatin. In another embodiment, the disintegrin is vicrostatin.
[0091] "Halo" and "halogen" refer to all halogens, that is, chloro (Cl), fluoro (F), bromo (Br), or iodo (I).
[0092] "Hydroxyl" and "hydroxy" refer to the group OH.
[0093] "Oxy" refers to the group OR, where R can be alkyl, acyl, aryl, heteroaryl, aralkyl, cycloalkyl, or heterocyclyl.
[0094] "Substituted oxy" refers to the group OR, where R can be substituted alkyl, substituted acyl, substituted aryl, substituted heteroaryl, substituted aralkyl, substituted cycloalkyl or substituted heterocyclyl.
[0095] "Alkoxy" refers to the group OR.sup.cc, where R.sup.cc is alkyl, wherein alkyl is as defined herein.
[0096] "Substituted alkoxy" refers to the group OR.sup.dd, where R.sup.dd is an alkyl group as defined herein, substituted with one or more groups or substituents such as halo, hydroxy, oxy, amino, alkylamino, arylamino, aralkylamino, cycloalkylamino, or heterocycloamino.
[0097] "Alkyl" refers to an alkane-derived radical containing from 1 to 20, preferably 1 to 8, more preferably 1-4, yet more preferably 1-2, carbon atoms. Alkyl includes straight chain alkyl, and branched alkyl such as methyl, ethyl, propyl, isopropyl, butyl, t-butyl, and the like, as well as cycloalkyl as defined herein. The alkyl group can be attached at any available point to produce a stable compound.
[0098] "Substituted alkyl" is an alkyl group independently substituted with one or more, e.g., 1, 2, or 3, groups or substituents such as halo, trifluoromethyl, trifluoromethoxy, hydroxy, alkoxy, cycloalkyoxy, heterocylooxy, oxo, alkanoyl, aryloxy, alkanoyloxy, amino, alkylamino, arylamino, aralkylamino, cycloalkylamino, heterocycloamino, disubstituted amines in which the 2 amino substituents are selected from alkyl, aryl or aralkyl, alkanoylamino, aroylamino, aralkanoylamino, substituted alkanoylamino, substituted arylamino, substituted aralkanoylamino, thiol, alkylthio, arylthio, aralkylthio, cycloalkylthio, heterocyclothio, alkylthiono, arylthiono, aralkylthiono, alkylsulfonyl, arylsulfonyl, aralkylsulfonyl, sulfonamido (e.g. SO.sub.2, NH.sub.2), substituted sulfonamido, nitro, cyano, carboxy, carbamyl (e.g. CONH.sub.2), substituted carbamyl (e.g. CONH alkyl, CONH aryl, CONH aralkyl or cases where there are two substituents on the nitrogen selected from alkyl, aryl or aralkyl), alkoxycarbonyl, aryl, substituted aryl, guanidino and heterocyclos, such as, indolyl, imidazolyl, furyl, thienyl, thiazolyl, pyrrolidyl, pyridyl, pyrimidyl and the like. Where noted above where the substituent is further substituted it will be with halogen, alkyl, alkoxy, aryl or aralkyl.
[0099] "Lower alkyl" refers to an alkyl group having 1-6 carbon atoms.
[0100] "Substituted lower alkyl" is a lower alkyl which is substituted with one or more, e.g., 1, 2, or 3, groups or substituents, as defined above, attached at any available point to produce a stable compound.
[0101] "Aryl" means a monocyclic or bicyclic aromatic hydrocarbon group having 6 to 12 carbon atoms in the ring portion, such as phenyl, naphthyl, biphenyl and diphenyl groups.
[0102] "Substituted aryl" refers to an aryl group as defined above independently substituted with one or more, e.g., 1, 2, or 3, groups or substituents such as halo, hydroxy, optionally substituted alkoxy, optionally substituted alkylthio, alkylsulfinyl, alkylsulfonyl, optionally substituted amino, optionally substituted amido, amidino, urea optionally substituted with alkyl, aminosulfonyl optionally N-mono- or N,N-di-substituted with alkyl, alkylsulfonylamino, carboxyl, heterocycle, substituted heterocycle, nitro, cyano, thiol, sulfonylamino, or the like, attached at any available point to produce a stable compound.
[0103] "Aralkyl" refers to an aryl substituted alkyl group, such as benzyl.
[0104] "Cycloalkyl" refers to optionally substituted, saturated cyclic hydrocarbon ring systems, preferably containing 1 to 3 rings and 3 to 10 carbons per ring which may be further fused with an unsaturated C.sub.3-C.sub.7 carbocyclic ring. Exemplary groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclodecyl, cyclododecyl, and adamantyl. Exemplary substituents include one or more alkyl groups as described above, or one or more groups described above as alkyl substituents.
[0105] The terms "heterocycle", "heterocyclic" and "heterocyclo" refer to an optionally substituted, fully saturated or unsaturated, aromatic or nonaromatic cyclic group, for example, which is a 4 to 7 membered monocyclic, 7 to 11 membered bicyclic, or 10 to 15 membered tricyclic ring system, which has at least one heteroatom in at least one carbon atom-containing ring. Each ring of the heterocyclic group containing a heteroatom may have 1, 2 or 3 heteroatoms selected from nitrogen atoms, oxygen atoms and sulfur atoms, where the nitrogen and sulfur heteroatoms may also optionally be oxidized and the nitrogen heteroatoms may also optionally be quaternized. The heterocyclic group may be attached at any heteroatom or carbon atom.
[0106] The term "heteroatoms" includes oxygen, sulfur and nitrogen.
[0107] Exemplary monocyclic heterocyclic groups include pyrrolidinyl, pyrrolyl, indolyl, pyrazolyl, oxetanyl, pyrazolinyl, imidazolyl, imidazolinyl, imidazolidinyl, oxazolyl, oxazolidinyl, isoxazolinyl, isoxazolyl, thiazolyl, thiadiazolyl, thiazolidinyl, isothiazolyl, isothiazolidinyl, furyl, tetrahydrofuryl, thienyl, oxadiazolyl, piperidinyl, piperazinyl, 2-oxopiperazinyl, 2-oxopiperidinyl, 2-oxopyrrolidinyl, 2-oxazepinyl, azepinyl, 4-piperidonyl, pyridyl, N-oxo-pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, tetrahydropyranyl, tetrahydrothiopyranyl, tetrahydrothiopyranyl sulfone, morpholinyl, thiomorpholinyl, thiomorpholinyl sulfoxide, thiomorpholinyl sulfone, 1,3-dioxolane and tetrahydro-1,1-dioxothienyl, dioxanyl, isothiazolidinyl, thietanyl, thiiranyl, triazinyl, and triazolyl, and the like.
[0108] Exemplary bicyclic heterocyclic groups include benzothiazolyl, benzoxazolyl, benzothienyl, quinuclidinyl, quinolinyl, quinolinyl-N-oxide, tetrahydroisoquinolinyl, isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, benzofuryl, chromonyl, coumarinyl, cinnolinyl, quinoxalinyl, indazolyl, pyrrolopyridyl, furopyridinyl (such as furo[2,3-c]pyridinyl, furo[3,1-b]pyridinyl] or furo[2,3-b]pyridinyl), dihydroisoindolyl, dihydroquinazolinyl (such as 3,4-dihydro-4-oxo-quinazolinyl), benzisothiazolyl, benzisoxazolyl, benzodiazinyl, benzofurazanyl, benzothiopyranyl, benzotriazolyl, benzpyrazolyl, dihydrobenzofuryl, dihydrobenzothienyl, dihydrobenzothiopyranyl, dihydrobenzothiopyranyl sulfone, dihydrobenzopyranyl, indolinyl, isochromanyl, isoindolinyl, naphthyridinyl, phthalazinyl, piperonyl, purinyl, pyridopyridyl, quinazolinyl, tetrahydroquinolinyl, thienofuryl, thienopyridyl, thienothienyl, and the like.
[0109] Exemplary substituents include one or more alkyl groups as described above or one or more groups described above as alkyl substituents. Also included are smaller heterocyclos, such as, epoxides and aziridines.
[0110] "Amino" or "amine" denotes the group--NH.sub.2. A "disubstituted amine" denotes--NR.sub.2 where R is lower alkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, acyl, substituted acyl, sulfonyl or substituted sulfonyl.
[0111] "Alkenyl" refers to a straight chain, branched, or cyclic hydrocarbon containing 2-20, preferably 2-17, more preferably 2-10, even more preferably 2-8, most preferably 2-4, carbon atoms, and which contains at least one, preferably 1-3, more preferably 1-2, and most preferably one, carbon to carbon double bond. In the case of a cycloalkyl group, conjugation of more than one carbon to carbon double bond is not such as to confer aromaticity to the ring. Carbon to carbon double bonds may be either contained within a cycloalkyl portion, or within a straight chain or branched portion. Examples of alkenyl groups include, but are not limited to, ethenyl, propenyl, isopropenyl, butenyl, cyclohexenyl, cyclohexenylalkyl, and the like.
[0112] "Substituted alkenyl" is an alkenyl which is independently substituted with one or more, e.g., 1, 2, or 3, groups or substituents such as halo, hydroxy, optionally substituted alkoxy, optionally substituted alkylthio, alkylsulfinyl, alkylsulfonyl, acyloxy, optionally substituted aryl, optionally substituted aryloxy, optionally substituted heteroaryloxy, optionally substituted amino, optionally substituted amido, amidino, urea optionally substituted with alkyl, aryl, heteroaryl or heterocyclyl groups, aminosulfonyl optionally N-mono- or N,N-di-substituted with alkyl, aryl or heteroaryl groups, alkylsulfonylamino, arylsulfonylamino, heteroarylsulfonylamino, alkylcarbonylamino, arylcarbonylamino, heteroarylcarbonylamino, carboxyl, heterocycle, substituted heterocycle, heteroaryl, substituted heteroaryl, nitro, cyano, thiol, sulfonylamino or the like attached at any available point to produce a stable compound.
[0113] "Lower alkenyl" refers to an alkenyl group having 1-6 carbon atoms.
[0114] "Substituted lower alkenyl" is a lower alkenyl which is substituted with 1 or more, e.g., 1, 2, or 3, groups or substitutents such as halo, hydroxy, optionally substituted alkoxy, optionally substituted alkylthio, alkylsulfinyl, alkylsulfonyl, acyloxy, optionally substituted aryl, optionally substituted aryloxy, optionally substituted heteroaryloxy, optionally substituted amino, optionally substituted amido, amidino, urea optionally substituted with alkyl, aryl, heteroaryl or heterocyclyl groups, aminosulfonyl optionally N-mono- or N,N-di-substituted with alkyl, aryl or heteroaryl groups, alkylsulfonylamino, arylsulfonylamino, heteroarylsulfonylamino, alkylcarbonylamino, arylcarbonylamino, heteroarylcarbonylamino, carboxyl, heterocycle, substituted heterocycle, heteroaryl, substituted heteroaryl, nitro, cyano, thiol, sulfonylamino or the like attached at any available point to produce a stable compound.
[0115] In another aspect of the present invention, an individual suffering from cancer is treated by administering an effective amount of a disintegrin in combination with an effective amount of at least one microtubule stabilizing agent. In one embodiment, the disintegrin is selected from contortrostatin or vicrostatin. In another embodiment, an effective amount of a disintegrin in administered in combination with an effective amount of a taxane microtubule stabilizing agent and an effective amount of a non-taxane microtubule stabilizing agent.
[0116] In one aspect of the present invention, the disintegrin is administered before the microtubule stabilizing agent. In another aspect, the disintegrin is administered after the microtubule stabilizing agent. In yet another aspect, the disintegrin and microtubule stabilizing agent are co-administered.
[0117] In accordance with the methods of the invention, the disintegrin and microtubule stabilizing agent may be co-adminstered, or administered separately in any order. Co-administration refers to simultaneouos delivery of two or more drugs. Treatment which combines administration of a disintegrin and a microtubule stabilizing agent, if co-administered, is preferably administered so that both drugs are in the body in active form at the same time.
[0118] In accordance with the methods of the invention, treatment with a disintegrin and microtubule stabilizing agent may be repeated at later times. Multiple treatments are likely to be necessary in most instances. When repeat administrations are used, the disintegrin and the microtubule stabilizing agent need not be administered an equal number of times. In addition, the dose of the disintegrin and the microtubule stabilizing agent may be modified for repeat administrations as medically required.
[0119] As used herein, "treating" refers to the administration of an agent (for example a disintegrin or a microtubule stabilizing agent) to a subject. Although it is preferred that treating a condition such as cancer will result in an improvement of the condition, the term treating as used herein does not indicate, imply, or require that the administration of the agent is successful in reducing or ameliorating symptoms associated with any particular condition. In some individuals, a treatment may result in adverse effects or even worsen a condition which the treatment was intended to improve.
[0120] As used herein, "administration" or "administer" or "administering" refers to dispensing, applying, or tendering an agent (for example a disintegrin or taxane) to a subject. Administration may be performed using any of a number of methods known in the art.
[0121] As used herein, "effective amount" refers to a dose sufficient to provide a concentration high enough to impart a beneficial effect on the recipient thereof. An "effective amount" is that which is determined by conducting clinical trials in accordance with generally accepted or legal guidelines. The specific therapeutically effective dose level for any particular subject will depend upon a variety of factors including the disorder being treated, the severity of the disorder, the activity of the specific compound, the route of administration, the rate of clearance of the compound, the duration of treatment, the drugs used in combination or coincident with the compound, the age, body weight, sex, diet and general health of the subject, and like factors well known in the medical arts and sciences. Various general considerations taken into account in determining the "therapeutically effective amount" are known to those of skill in the art and are described, e.g., in Gilman et al., eds., Goodman And Gilman's: The Pharmacological Bases of Therapeutics, 8th ed., Pergamon Press, 1990; and Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Co., Easton, Pa., 1990.
[0122] As used herein, "composition" refers to a formulation suitable for administration to an intended animal subject for therapeutic purposes that contains at least one pharmaceutically active compound and at least one pharmaceutically acceptable carrier or excipient. The term "pharmaceutically acceptable" indicates that the identified material does not have properties that would cause a reasonably prudent medical practitioner to avoid administration of the material to a patient, taking into consideration the disease or conditions to be treated and the respective route of administration. For example, it is commonly required that such a material be essentially sterile, e.g., for injectibles. Techniques for formulation and administration may be found, for example, in "Remington's Pharmaceutical Sciences," (18th ed., Mack Publishing Co., Easton Pa., 1990).
[0123] As used herein, "about" means in quantitative terms plus or minus 10%.
[0124] As used herein, "analog" means a compound that resembles another in structure but differs by at least one atom.
[0125] As used herein, "combination" refers to any association between or among two or more items. The combination can be two or more separate items, such as two compositions or two collections. It can be a mixture thereof, such as a single mixture of the two or more items, or any variation thereof.
[0126] As used herein "derivative" is a chemicalsubstance derived from another substance by modification or substitution.
BRIEF DESCRIPTION OF THE FIGURES
[0127] FIG. 1 shows the inhibition of PC-3 xenograft tumor growth of four treatment groups. Mice were treated with either PBS, CN, docetaxel, or CN plus docetaxel. The group that received a combination of CN and docetaxel resulted in the greatest inhibition of tumor growth. Error bars represent SEM. Experimental details are described in Example 7.
[0128] FIG. 2 shows the amino acid sequence of contortrostatin (SEQ ID NO:1).
[0129] FIG. 3 shows the full-length nucleotide sequence of contortrostatin cDNA (SEQ ID NO:2).
DETAILED DESCRIPTION OF THE INVENTION
[0130] The invention relates to compositions and methods for treating cancer. In particular aspects, the invention relates to administering a combination of a disintegrin with a microtubule stabiling agent useful for treatment of cancer. The methods and compositions of the invention are useful for inhibiting the growth of a cancer or inhibiting the emergence or growth of metastases. The invention methods and compositions are particularly suited for inhibiting the appearance or growth of cancer metastatic to the bone such as in cases of breast and prostate cancer.
[0131] Prostate cancer is a major public health issue. With the exception of skin cancer, prostate cancer is the most prevalent cancer in American men and the second leading cause of cancer death. American Cancer Society data indicated 220,900 men diagnosed with and 28,900 deaths from prostate cancer in the United States in 2005. Despite improvements in diagnosis, surgical techniques, and local and systemic adjuvant therapies, most deaths from prostate cancer are still caused by metastases, especially to the bones, that are resistant to conventional therapies. Osteoblastic metastases are common in lethal prostate cancer.
[0132] The therapeutic efficacy of CN has been proven in a subcutaneous human prostate cancer nude mouse model. See Pinski, et al., Proc. Am. Soc. Clin. Oncol. 22: 218 (2003) (abstr 874). In one aspect of the invention, contortrostatin (CN) is combined with a taxane for inhihbiting tumor growth and appearance or growth of metastases.
Preparation of Disintegrin
[0133] Disintegrins may be obtained by purifying them from natural sources such as snake venom using methods well known in the art. For example, the purification of contortrostatin from Agkistrodon contortrix contortrix (Southern copperhead) venom using a four step HPLC procedure is described in U.S. Pat. No. 5,731,288 (Markland, et al.). Also described therein are methods to characterize the purified disintegrin such as SDS-polyacrylamide gel electrophoresis (SDS-PAGE), mass spectrometry, Scatchard analysis of binding to unactivated human platelets to determine the IC.sub.50 of the preparation.
[0134] Disintegrins also may be obtained by synthetic methods or by recombinant expression techniquies. In this regards, U.S. Pat. No. 6,710,030 (Markland et al.) discloses the nucleotide and amino acid sequence of native contortrostatin which results from proteolytic processing of a contortrostatin precursor. The precursor is a multidomain protein that includes pro-protein, metalloproteinase, and disintegrin (mature contortrostatin) domains. U.S. Pat. No. 6,710,030 also describes various biologically active variants and fragments of contortrostatin.
[0135] Methods of expressing distintegrins by recombinant means in prokaryotic organisms is described in international Application Serial No. PCT/US2006/004413 (see also U.S. application Ser. No. 11/351,311), filed Feb. 9, 2006. As described therein, expression of the disintegrin in prokaryotic host cells is achieved by expressing as a genetic fusion a bacterial thioredoxin such as thioredoxin A (TrxA). This is achieved by cloning DNA sequence encoding the disintegrin downstream (i.e., 3') to sequence encoding the thioredoxin. This can be cloned into a suitable expression vector such as pET32a.
[0136] An exemplary thioredoxin is thioredoxin A (TrxA) from E. coli, which is about 109 amino acids in length and is encoded by the trxA gene. The amino acid sequence of E. coli wild type thioredoxin A is shown below with the active site CXXC bolded and underlined.
TABLE-US-00002 (SEQ ID NO: 4) MSDKIIHLTDDSFDTDVLKADGAILVDFWAEWCGPCKMIAPILDEIADE YQGKLTVAKLNIDQNPGTAPKYGIRGIPTLLLFKNGEVAATKVGALSKG QLKEFLDANLA.
[0137] Active site mutants of thioredoxin may be used in place of wild type thioredoxin in the fusion protein. Thus, thioredoxin active-site motif CXXC can be replaced with an active-site motif from another oxido-reductase. For example, active site mutants of wild type thioredoxin A may be used in place of wild type thioredoxin in the fusion construct with the eukaryotic protein. In this regard, thioredoxin A's active site motif CGPC (SEQ ID NO: 48) may be replaced with the active site motif CPYC (SEQ ID NO: 49), taken from another bacterial oxido-reductase, glutaredoxin A (also called glutaredoxin 1). This mutant may be referred to as a glutaredoxin-like thioredoxin. Another thioredoxin active site mutant is the PDI-like thioredoxin, generated by replacing the active site wild type motif CGPC (SEQ ID NO: 48) with the active site motif CGHC (SEQ ID NO: 50), taken from eukaryotic protein disulfide isomerase (PDI).
[0138] Also described in PCT/US2006/004413 is to transform the disintegrin expression vector into prokaryotic host cells that have been are engineered in ways to enhance expression of proteins with large numbers of disulfide bridging such as disintegrins. Host cell engineering includes cytoplasmic expression of a disulfide isomerase (such as DsbC) normally targeted to the periplasmic space in bacteria and/or cytoplasmic expression of a redox catalyst such as the a-domain of the bacterial thiol-disulfide interchange protein DsbD also normally targeted to the periplasmic space. Cytoplasmic localization of DsbC or the a-domain of DsbD can be achieved by expressing the mature protein without a signal sequence. PCT/US2006/004413 also describes active site mutants of DsbC that have increased isomerase activity. This may be achieved by replacing the E. coli wildtype sequence CGYC (SEQ ID NO: 51) with CGFC (SEQ ID NO: 52) or CTFC (SEQ ID NO: 53).
[0139] The sequence of E. coli DsbC is shown below without the signal sequence and with the active site CGYC (SEQ ID NO: 51) underlined and bolded.
TABLE-US-00003 (SEQ ID NO: 5) DDAAIQQTLAKMGIKSSDIQPAPVAGMKTVLTNSGVLYITDDGKHIIQ GPMYDVSGTAPVNVTNKMLLKQLNALEKEMIVYKAPQEKHVITVFTDI TCGYCHKLHEQMADYNALGITVRYLAFPRQGLDSDAEKEMKAIWCAKD KNKAFDDVMAGKSVAPASCDVDIADHYALGVQLGVSGTPAVVLSNGTL VPGYQPPKEMKEFXDEHQKMTSGK
[0140] The DsbD .alpha.-domain represents the first 132 amino acids of mature DsbD from which a cleavable signal sequence of 19 aa is removed. The sequence of the DsbD .alpha.-domain without the leader sequence and with the catalytic site underlined is shown below.
TABLE-US-00004 (SEQ ID NO: 6) GLFDAPGRSQFVPADQAFAFDFQQNQHDLNLTWQIKDGYYLYRKQIRIT PEHAKIADVQLPQGVWHEDEFYGKSEIYRDRLTLPVTINQASAGATLTV TYQGCADAGFCYPPETKTVPLSEVVANNEASQPV
[0141] PCT/US2006/004413 also describes other useful bacterial host cell mutants including a mutant trxB gene and/or a mutant gor gene, rendering the cell deficient in thioredoxin reductase activity and/or glutathione reductase activity. Other host cell mutations include deficiency in one or more proteases such as those encloded by ompT and lon genes. For example, E. coli host cells AD494(DE3)pLysS are deficient in trxB gene as well as ompT and lon. E. coli strain Origami B(DE3)pLysS and Rosetta-gami B(DE3)pLysS are deficient in trxB, gor, ompT and lon gene products. These mutations may be used in combination with any other host cells variations described above.
[0142] Also described in PCT/US2006/004413 is the use of a cleavage site engineered between thioredoxin and the disintegrin to enable isolation of the disintegrin from the fusion protein following expression. Any number of well known cleavage sites may be used for this purpose. A suitable protease cleavage site is the TEV protease cleavage site, which comprises the amino acid sequence ENLYFQG/S (three letter code: Glu-Asn-Leu-Tyr-Phe-Gln-Gly/Ser) (SEQ ID NO: 7). The TEV site may be engineered just upstream of the N-terminus of the disulfide containing disintegrin. A chemical cleavage site also may be used for this purpose. For example, a DP (Asp-Pro) dipeptide sequence can be engineered in a similar location to that of the TEV site in the fusion protein. Formic acid hydrolysis can then be used to cleave the protein at the DP site. The cleavage site is preferably placed between the thioredoxin and the disintegrin (e.g., downstream of the thioredoxin sequence and upstream of the N-terminal end of the disintegrin) in order to obtain the disintegrin free from thioredoxin.
[0143] Recombinantly expressed disintegrin may include functionally useful sequences that are taken or modeled from other proteins of the same structural class. These functional sequences, non-native to the disintegrin, may be located at either terminus of the disintegrin or within the distintegrin as dictated by the effect of the addition on the biological function of the disintegrin. Such functional sequences include the amino acid residues located downstream from the most C-terminal Cys residue in mono- or dimeric disintegrin primary sequences. For example, a biologically active disintegrin domain may include sequence at its C-terminus that directs binding to a particular type of integrin. For example, the CN full length disintegrin precursor or its disintegrin domain may be expressed with the C-terminal extension, HKGPAT (SEQ ID NO: 47) (three letter code: His-Lys-Gly-Pro-Ala-Thr), which represents the C-terminal amino acid sequence of echistatin, a disintegrin which is monomeric in its native state. The addition of the HKGPAT (SEQ ID NO: 47) at the C-terminus of the CN monomer can be used to increase the affinity of the expressed recombinant CN disintegrin domain for .alpha.5.beta.1 integrin. This C-terminal fusion also can facilitate the proper folding of nascent recombinant CN disintegrin domain in the C-terminal half of the molecule where the integrin-binding loop key structural element resides.
[0144] Recombinantly expressed monomeric disintegrin or monomeric disintegrin domain may comprise a C-terminal sequence non-native to the disintegrin or disintegrin domain, such as the C-terminal sequence encoding a functional integrin-binding loop. In one embodiment, integrin binding loop is selected from any loops that bind to integrin .alpha. Ib.beta.3, .alpha. v.beta.3, .alpha.v.beta.5, or .alpha.5.beta.1. In another embodiment, the integrin binding loop C-terminal sequence comprises HKGPAT (SEQ ID NO: 47). In a further embodiment, the integrin binding loop is stabilized by at least one intramolecular disulfide bridge. In yet another embodiment, the monomeric disintegrin or monomeric disintegrin domain is from contortrostatin.
[0145] Pharmaceutical compositions containing homodimeric and monomeric disintegrins should comprise at a minimum an amount of protein effective to achieve the desired effect (i.e., inhibit cancer growth or prevent or inhibit cancer metastasis) and a suitable carrier or excipient. Generally, in these compositions, homodimeric and monomeric disintegrins are present in an amount sufficient to provide about 0.01 mg/kg to about 50 mg/kg per day, preferably about 0.1 mg/kg to about 5.0 mg/kg per day, and most preferably about 0.1 mg/kg to about 0.5 mg/kg per day.
[0146] Homodimeric and monomeric disintegrins may be administered by a variety of heretofore known means suitable for delivery thereof into the blood stream in substantial amounts. Intravenous administration of homodimeric and monomeric disintegrins in a suitable liquid vehicle or excipient is presently contemplated as the preferred route of administration. Homodimeric and monomeric disintegrins are soluble in water, and may therefore be effectively administered in a suitable aqueous solution (e.g., phosphate buffered saline). Alternatively, homodimeric and monomeric disintegrins may be administered orally (in the form of tablets or capsules formulated with a suitable binder or excipient material, or in the form of aqueous or oily suspensions, solutions, emulsions, syrups or elixirs) or as a parenteral suspension. As is well known in the art, adjuvants or excipients such as local anesthetics, preservatives, buffering agents, lubricants, wetting agents, colorants, flavorings, fillers and diluents may suitably be included in any of these formulations.
Preparation of Microtubule Stabilizing Agents
[0147] Microtubule stabilizing agents are combined with a disintegrin in the methods and compositions of the present invention. Taxanes, in particular paclitaxel, docetaxel and derivatives thereof, are preferred microtubule stabilizing agents for use in combination with a disintegrin in the methods and compositions of the invention. Taxanes have a common core structure (i.e., a taxoid core) shown below.
##STR00014##
[0148] The chemical structure of Taxol.RTM. and Taxotere.RTM. are shown below.
##STR00015##
[0149] A number of non-natural taxanes have been prepared which have a taxane ring bearing modified side chains, which may include fatty acids. These modified taxanes or taxoid analogs inhibit cancer growth while having greater water solubility and stability than naturally occurring Taxol.RTM.. Exemplary derivatives of Taxol.RTM. are described in U.S. Pat. Nos. 6,638,742; 5,278,324; 5,272,171; 5,254,580; 5,250,683; 5,248,796; and 5,227,400; and US Pub. App. No. 2005/0148657; and the references cited therein, as well as those compounds disclosed in Villalva-Servin, et al., Can. J. Chem., 82:227-239 (2004); Shen, et al., Chem. Pharm. Bull., 53(7):808-10 (2005); Ono, et al., Biol. Pharm. Bull., 27(3):345-51 (2004); Sampath, et al., Mol. Cancer. Ther., 2(9):873-74 (2003); and Wolff, et al., Clin. Cancer Res., 9(10):3589-97 (2003).
[0150] In addition, non-taxane microtubule stabilizing agents, such as epothilones and derivatives thereof, also may be administered in combination with a disintegrin for the treatment of cancer in the methods and compositions of the present invention. Epothilones A and B (shown below), for example, have been found to exert microtubule stabilizing effects and cytotoxic activity against rapidly proliferating cells, such as tumor cells or other hyperproliferative cellular diseases, with results similar to those observed with Taxol.RTM.. Epothilones have a similar mechanism of action to taxanes despite the structural disimilarity. Epothilones, however, display some superior qualities to taxanes: namely water solubility, production in large quantities from bacteria fermentation, and retention of activity against multi-drug resistant cell lines and tumors. (See Giannakakou, et al., PNAS, 97(6): 2904-09 (2000) and references cited therein).
##STR00016##
[0151] Epothilone derivatives have been previously administered in combination with therapeutic agents. For example, Mani, et al., describes administering the epothilone B derivative BMS-247550 in combination with capecitabine to breast cancer patients resistant to taxane therapy. See, e.g., Mani, et al., Clin. Cancer Res., 10:1289-98 (2004). BMS-247550 has been shown to have anti-tumor activity in paclitaxel-resistant tumor models. Id.
##STR00017##
[0152] Examples of epothilone compounds and derivatives contemplated for use herein are disclosed in U.S. Pat. Nos. 6,294,374; 6,365,749; 6,380,394; 6,380,395; 6,387,927; 6,399,638; 6,441,186; 6,489,314; 6,498,257; 6,518,421; 6,531,497; 6,583,290; 6,589,968; 6,593,115; 6,596,875; 6,605,599; 6,605,726; 6,610,736; 6,624,310; 6,660,758; 6,670,384; 6,686,380; 6,689,802; 6,719,540; 6,727,276; 6,730,803; 6,780,620; 6,800,653; 6,831,090; 6,858,411; 6,867,333; 6,893,859; 6,900,331; 6,906,188; 6,921,650; 6,930,102; 6,930,187; 6,958,401; 6,982,276; 6,982,280; 6,998,256; and 7,008,936; and U.S. Pub. App. Nos. 20020042109; 20020045609; 20020062030; 20020143038; 20020156110; 20020165257; 20020165258; 20020169190; 20020188014; 20020193361; 20030004338; 20030023082; 20030045711; 20030060623; 20030073677; 20030087888; 20030144523; 20030144533; 20030149281; 20030176473; 20030176710; 20030186965; 20030187039; 20030187273; 20030191089; 20030203938; 20030219877; 20030220295; 20030220503; 20040014978; 20040023345; 20040024032; 20040030147; 20040038324; 20040039026; 20040049051; 20040053978; 20040058969; 20040072870; 20040072882; 20040082651; 20040092478; 20040127432; 20040132146; 20040132754; 20040157897; 20040176429; 20040214871; 20040253697; 20040259922; 20050038086; 20050042275; 20050113429; 20050159461; 20050187270; 20050192440; 20050267306; 20050282873; 20060013836; 20060014796; 20060040990; 20060046997; and 20060063815.
[0153] Other non-taxane microtubule stabilizing agents contemplated for use herein include taccalonolides and analogues thereof (see, e.g., U.S. Pat. No. 6,878,699 and U.S. Pub. App. No. 2002/0094991 and 2004/0022869); dictyostatin and analogues thereof (see, e.g., Madiraju et al., Biochem. 44(45) 15053-63 (2005)); laulimalide and analogues thereof (see Mooberry et al., PNAS 101(23) 8803-08 (2004)); and discodermolides and analogues thereof (see Kowalski et al., Mol. Pharm. 52(4) 613-22 (1997)).
[0154] A composition comprising a combination of a disintegrin or fragment thereof and a microtubule stabilizing agent can be administered as a pharmaceutical composition wherein the composition is formulated with a pharmaceutically acceptable carrier as is well known in the art. Techniques for formulation and administration may be found, for example, in "Remington's Pharmaceutical Sciences," (18th ed., Mack Publishing Co., Easton, Pa., 1990). Accordingly, the invention compounds and combination of compounds may be used in the manufacture of a medicament. It is understood that a pharmaceutically acceptable carrier, or a pharmaceutical composition, or any substance suitable for administration to a mammal should be manufactured and stored in accordance with standards of local regulations. For example, many governments have guidelines or rules that regulate various aspects of the manufacture and handling of compositions which are for administration into mammals and/or humans such as sanitation, process validation, equipment and document traceability, and personnel qualification. Preferably, a pharmaceutical composition or a pharmaceutically acceptable carrier is suitable for administration to a human and pharmaceutically complies with GMP (Good Manufacturing Practices) regulations set forth by the United States Food and Drug Administration for such a purpose.
[0155] A combination of a disintegrin and a microtubule stabilizing agent may be formulated as solutions or lyophilized powders for parenteral administration. Powders may be reconstituted by addition of a suitable diluent or other pharmaceutically acceptable carrier prior to use. Liquid formulations may be buffered, isotonic, aqueous solutions. Powders also may be sprayed in dry form. Examples of suitable diluents are normal isotonic saline solution, standard 5% dextrose in water, or buffered sodium or ammonium acetate solution. Such formulations are especially suitable for parenteral administration, but may also be used for oral administration or contained in a metered dose inhaler or nebulizer for insufflation. It may be desirable to add excipients such as polyvinylpyrrolidone, gelatin, hydroxy cellulose, acacia, polyethylene glycol, mannitol, sodium chloride, sodium citrate, and the like.
[0156] Alternately, a combination of a disintegrin and a microtubule stabilizing agent may be prepared for oral administration. Pharmaceutically acceptable solid or liquid carriers may be added to enhance or stabilize the composition, or to facilitate preparation of the vectors. Solid carriers include starch, lactose, calcium sulfate dihydrate, terra alba, magnesium stearate or stearic acid, talc, pectin, acacia, agar or gelatin. Liquid carriers include syrup, peanut oil, olive oil, saline and water. The carrier may also include a sustained release material such as glyceryl monostearate or glyceryl distearate, alone or with a wax. The amount of solid carrier varies but, preferably, will be between about 20 mg to about 1 g per dosage unit. When a liquid carrier is used, the preparation may be in the form of a syrup, elixir, emulsion, or an aqueous or non-aqueous suspension.
[0157] A combination of a disintegrin and a microtubule stabilizing agent may be formulated to include other medically useful drugs or biological agents and/or may be administered in conjunction with the administration of other drugs or biological agents useful for the disease or condition that the invention compounds are directed.
[0158] The dosage to be administered depends to a large extent on the condition and size of the subject being treated as well as the frequency of treatment and the route of administration. Regimens for continuing therapy, including dose and frequency may be guided by the initial response and clinical judgment. For general purposes, the small molecule microtubule stabilizing agent could be administered at about 60-75 mg/m.sup.2 every 3 weeks while the disintegrin dose could be from 0.1 mg/kg to 1 mg/kg for each administration.
[0159] As such, the invention provides a pharmaceutical product, comprising a combination of a disintegrin and a microtubule stabilizing agent, in solution in a physiologically acceptable injectable carrier and suitable for introduction into an individual, a container enclosing the solution, and a notice associated with the container in form prescribed by a governmental agency regulating the manufacture, use, or sale of pharmaceuticals, which notice is reflective of approval by the agency of manufacture, use, or sale of the solution of the combination (or separate individual components) for human administration.
[0160] Disintegrin and/or microtubule stabilizing agents may be delivered by way of liposomes, which may incorporate one or both of these compounds. Liposomal delivery is well known in the art and has been described for delivery of both distintegrins and microtubule stabilizing agents. For example, Swenson et al. Cancer Ther. 2004, 3(4):499-511 describes use of intravenous delivery of contortrostatin in liposomes for therapy of breast cancer. See also, Fujii, Chang et al. Biochemistry 1997, 36(16):4959-68.
[0161] Another embodiment is to administer an expression vector encoding the disintegrin to obtain the disintegrin by recombinant expression in the individual with cancer. An expression vector encoding the disintegrin can be formulated to facilitate transfection delivery to the interior of a cell, and/or to a desired location within a cell. Many such transfection facilitating materials are commercially available, for example Lipofectin, Lipofectamine, Lipofectamine 2000, Optifect, SuperFect. Examples of transfection facilitating materials include, but are not limited to lipids, preferably cationic lipids; inorganic materials such as calcium phosphate, and metal (e.g., gold or tungsten) particles (e.g., "powder" type delivery solutions); peptides, including cationic peptides, targeting peptides for selective delivery to certain cells or intracellular organelles such as the nucleus or nucleolus, and amphipathic peptides, i.e., helix forming or pore forming peptides; basic proteins, such as histones; asialoproteins; viral proteins (e.g., Sendai virus coat protein); pore-forming proteins; and polymers, including dendrimers, star-polymers, "homogenous" poly-amino acids (e.g., poly-lysine, poly-arginine), "heterogeneous" poly-amino acids (e.g., mixtures of lysine & glycine), co-polymers, polyvinylpyrrolidinone (PVP), and polyethylene glycol (PEG). Furthermore, those auxiliary agents of the invention which facilitate and enhance the entry of a polynucleotide into vertebrate cells in vivo, may also be considered "transfection facilitating materials."
[0162] Lipofection facilitated transfection is well known in the art as described, for example, in U.S. Pat. Nos. 6,034,072, 6,040,295 and 6,710,035. Certain embodiments may include lipids as a transfection facilitating material, including cationic lipids (e.g., DOTMA, DMRIE, DOSPA, DC-Chol, GAP-DLRIE), basic lipids (e.g., steryl amine), neutral lipids (e.g., cholesterol), anionic lipids (e.g., phosphatidyl serine), and zwitterionic lipids (e.g., DOPE, DOPC). Preferably, the cationic lipid is mixed with one or more co-lipids. For purposes of definition, the term "co-lipid" refers to any hydrophobic material which may be combined with the cationic lipid component and includes amphipathic lipids, such as phospholipids, and neutral lipids, such as cholesterol. Cationic lipids and co-lipids may be mixed or combined in a number of ways to produce a variety of non-covalently bonded macroscopic structures, including, for example, liposomes, multilamellar vesicles, unilamellar vesicles, micelles, and simple films.
[0163] Viral vectors suitable for delivery in vivo and expression of a disintegrin are well known and include adenoviral vectors, adeno-associated viral vectors, retroviral vectors, herpes simplex viral vectors, and the like. Viral vectors are preferably made replication defective in normal cells. See U.S. Pat. Nos. 6,669,942; 6,566,128; 6,794,188; 6,110,744; and 6,133,029. Suitable adenoviral vectors include those capable of replicating and being packaged when any deficient essential genes are provided in trans. A suitable adenoviral vector desirably contains at least a portion of each terminal repeat required to support the replication of the viral DNA, preferably at least about 90% of the full ITR sequence, and the DNA required to encapsidate the genome into a viral capsid. Many suitable adenoviral vectors have been described in the art. See U.S. Pat. Nos. 6,440,944 and 6,040,174 (replication defective E1 deleted vectors and specialized packaging cell lines). A preferred adenoviral expression vector is one that is replication defective in normal cells.
[0164] Adeno-associated viruses represent a class of small, single-stranded DNA viruses that can insert their genetic material at a specific site on chromosome 19. The preparation and use of adeno-associated viral vectors for gene delivery is described in U.S. Pat. No. 5,658,785.
[0165] Non-viral vectors for gene delivery comprise various types of expression vectors (e.g., plasmids) which are combined with lipids, proteins and other molecules (or combinations of thereof) in order to protect the DNA of the vector during delivery. Fusigenic non-viral particles can be constructed by combining viral fusion proteins with expression vectors as described. Kaneda, Curr. Drug Targets (2003) 4(8):599-602. Reconstituted HVJ (hemagglutinating virus of Japan; Sendai virus)-liposomes can be used to deliver expression vectors or the vectors may be incorporated directly into inactivated HVJ particles without liposomes. See Kaneda, Curr Drug Targets (2003) 4(8):599-602. DMRIE/DOPE lipid mixture are useful a vehicle for non-viral expression vectors. See U.S. Pat. No. 6,147,055. Polycation-DNA complexes also may be used as a non-viral gene delivery vehicle. See Thomas et al., Appl Microbiol Biotechnol (2003) 62(1):27-34.
[0166] Various examples describing genetic delivery and expression of a disintegrin and achieving therapeutic cancer effects have been reported. See, e.g., Soo In Kim et al., Cancer Research 63: 6458-62 (2003).
[0167] The versatility of the invention is illustrated by the following Examples which illustrate preferred embodiments of the invention and are not limiting of the claims or specification in any way.
EXAMPLES
Example 1
Expression of contortrostatin in Origami B strain of E. coli
[0168] The sequence HKGPAT (SEQ ID NO: 47), which represents the C-terminal amino acid sequence of the monomeric disintegrin, echistatin, was included at the C-terminal end of the CN disintegrin domain sequence. This construct is a chimera that combines by the means of genetic engineering the sequences of two snake venom disintegrins with different originis: echistatin (a viperid disintegrin) and contortrostatin (a crotalid disintegrin). For this reason, this disintegrin construct that carries a C-terminal graft is referred to as "Vicrostatin" or "VN." CN disintegrin domain without the HKGPAT (SEQ ID NO: 47) sequence is referred to as "rCN" or "rCN construct." The amino acid sequence of vicrostatin is shown below as SEQ ID NO: 8
TABLE-US-00005 (SEQ ID NO: 8) GDAPANPCCDAATCKLTTGSQCADGLCCDQCKFMKEGTVCRRARGDDLDDYC NGISAGCPRNP HKGPAT.
[0169] The sequence of VN results from its expression as a fusion to thioredoxin and post expression processing as described below.
[0170] Contortrostatin wild-type disintegrin domain or the disintegrin domain with echistatin C-terminal graft was directionally cloned by PCR into the pET32a vector (Novagen, Inc.), downstream of the thioredoxin sequence. The set of restriction enzymes used for cloning was: BglII/NcoI. The oligonucleotide primers employed for cloning were as follows: [0171] CNfor1--forward primer for rCN (disintegrin domain) and VN (disintegrin domain) introducing BglII restriction site
TABLE-US-00006 [0171] (SEQ ID NO: 9) 5'GTTCCAGATCTCGAGAATCTTTACTTCCAAGGAGACGCTCCTGCAAA TCCGTGCTGCGATGCTGCA3'
[0172] CNback1--reverse primer for rCN (disintegrin domain) introducing the NcoI restriction site
TABLE-US-00007 [0172] (SEQ ID NO: 10) 5'GTTATTCGCCATGGCTTAGGCATGGAAGGGATTTCTGGGACAGCCAG CAGA3'
[0173] CNback2--reverse primer for VN (disintegrin domain) introduction the NcoI restriction site
TABLE-US-00008 [0173] (SEQ ID NO: 11) 5'GTTATTCGCCATGGCTTAAGTAGCTGGACCCTTGTGGGGATTTCTGG GACAGCCAGCAGATATGCC3'
[0174] The forward primer introduces a unique TEV protease cleavage site, which makes possible the removal of the thioredoxin fusion partner after purification of the fusion protein by Ni-column chromatography. The TEV protease recognizes with high specificity the canonical ENLYFQG (SEQ ID NO: 54) amino acid sequence engineered between recombinant CN and the thioredoxin fusion partner in this construct and following cleavage leaves a glycine at the N-terminus of rCN and VN. The reverse primer grafts the HKGPAT (SEQ ID NO: 47) segment to the C-terminus of the fusion protein. Thus, two recombinant fusion proteins, designated Trx-rCN and Trx-VN, were generated using the above described cloning strategy.
[0175] The initial cloning was carried out in the DH5.alpha. strain, which is recA.sup.- endA.sup.- and has high transformation efficiency and good plasmid yield. After validating the cloning by sequencing the constructs retrieved from DH5.alpha. transformants, the vector was used to transform the expression host, Origami B(DE3)pLysS, for expression optimization.
[0176] The Origami B/pET32a system produced up to 20 mg/L of recombinant CN (both Trx-rCN and Trx-VN constructs) without optimization. A single colony of transformed Origami B cells was used to inoculate a primary culture containing 10 mL LB broth with carbenicillin (100 .mu.g/mL), tetracycline (12.5 .mu.g/mL), kanamycin (15 .mu.g/mL) and chloramphenicol (34 .mu.g/mL). The culture was grown overnight to high turbidity and was used to inoculate 1 L of fresh LB broth with all 4 antibiotics. The first culture was used to inoculate a larger volume of LB broth plus antibiotics which was grown at 37.degree. C. with shaking at 250 rpm to an OD.sub.600 of 1-2. At this point, 1 mM IPTG was added and the cells further grown for another 3-5 hours at 37.degree. C. with shaking at 250 rpm.
[0177] The cells were harvested and resuspended in 5 mL of cold 20 mM Tris-HCl, pH 7.5, and lysed by sonication. The insoluble cellular debris was removed by centrifugation at 40,000.times.g and the total soluble protein fraction collected. The total soluble protein fractions retrieved from cell lysates and analyzed by SDS-PAGE showed that the fusion proteins (Trx-rCN and Trx-VN) were the prevalent species in this cell fraction.
[0178] The fusion proteins in the total soluble protein fractions were subjected to proteolysis by recombinant TEV protease following the manufacturer's protocol (Invitrogen, Carlsbad, Calif.) so as to cleave rCN or VN from its fusion partner, thioredoxin. Following TEV protease treatment (monitored by SDS-PAGE), the protein lysates were sterilized by passage through a 0.22 .mu.m filter and further passed through a 30 kDa molecular cut-off filter (Millipore, Bedford, Mass.). The recombinant disintegrin species (rCN or VN) contained in the filtrate were further recovered by reverse phase HPLC purification. Alternatively, the fusion proteins containing a His-tag sequence were initially purified by Ni-chelation affinity chromatography using a commercially available His.cndot.Bind resin kit (Novagen, Madison, Wis.). After buffer exchange (removal of imidazole excess), the fusion proteins were subjected to overnight proteolysis at room temperature using TEV protease in the presence of a very small amount of DTT or GSH/GSSG to keep the TEV protease (a cysteine-protease) in a reduced (active) state. When proteolysis was complete (assessed by SDS-PAGE), the recombinant CN species (rCN or VN) were recovered by reverse phase HPLC purification.
[0179] C18-Reverse Phase HPLC was employed to purify recombinant CN constructs following TEV cleavage of the fusion protein. The HPLC column conditions used for rCN and VN were the same as for native CN. HPLC was conducted using a Vydac C18 column (218TP54, Temecula, Calif.) in a solution of 0.1% TFA in water. A ten-minute rinse (at 1 ml/ml) of the column with the loading solution was followed by a linear gradient (0-100%) elution over 50 minutes with a mobile phase containing 80% acetonitrile in 0.1% TFA. Under these conditions, native CN and both forms of recombinant CN elute at 41% acetonitrile. The eluted material analyzed by reducing SDS-PAGE showed that VN as a single band with a molecular weight of .about.8 kDa, slightly larger than native CN, which agrees with the primary structure containing five additional amino acids. The recovered rCN was almost identical in size to native CN.
[0180] HPLC purified rCN and VN were recognized by a polyclonal antisera raised against native CN in both ELISA and Western blotting assays (data not shown).
Example 2
Biological Activity of Recombinant Contortrostatin Constructs
[0181] A. In Vitro Functional Assays
[0182] The recombinant CN products were evaluated for biological activity by a platelet aggregation inhibition assay. According to this assay, CN binding to GPIIb/IIIa (integrin .alpha.IIb.beta.3) in an RGD dependent manner inhibits ADP induced platelet aggregation (Trikha, Rote, et al. 1994). In this assay, potential inhibitors are added to fresh platelet-rich plasma, and after minute, ADP is added to a final concentration of 1 .mu.M to induce aggregation. If inhibitor is present, functional aggregation will not occur. The IC.sub.50 is defined as the concentration at which 50% of the activity is inhibited, and is used as a measure of the potency of an inhibitor.
[0183] It was shown that VN exhibited an IC.sub.50 of 59 nM in the platelet inhibition assay, which is almost identical to that observed for native CN. The rCN conformer however did not inhibit platelet aggregation, even at low .mu.M concentrations which are effective for small, synthetic RGD peptides. Furthermore, the chimeric recombinant disintegrin VN showed similar results in several other in vitro integrin-based functional assays: inhibition of MDA-MB-435 carcinoma cell adhesion to immobilized fibronectin (Fn) and vitronectin (Vn), or inhibition of MDA-MB-435 cell invasion through an artificial basement membrane (Matrigel) in a modified Boyden chamber. For the cell adhesion assay, the pretreatment MDA-MB-435 breast carcinoma cells with various concentrations (0-1000 nM) of either native CN or VN for 30 min inhibited adhesion of MDA-MB-435 cells (100 .mu.l of cells, 10.sup.5 cells/ml) to either immobilized fibronectin (Fn) or vitronectin (Vn). Pretreated cells were allowed to adhere for 1 hr at 25.degree. C., and after non-adherent cells were washed away, the number of adherent cells for each condition was estimated using the MTS cell viability assay. In the cell invasion assay, an invasion chamber consisting of cell culture inserts that fit into 24-well tissue culture plate has been utilized. The inserts contain an 8 .mu.m-pore size polycarbonate membrane, over which a thin layer of ECMatrix.TM. was applied. The ECMatrix.TM. serves as an in vitro reconstituted basement membrane and is a solid gel of ECM proteins prepared from the Engelbreth Holm-Swarm (EHS) mouse tumor. The ECM layer occludes the membranes pores, blocking non-invasive cells to migrate through. The cells were incubated in the presence of various concentrations (10, 100, 1000 nM) of either native CN or Vicrostatin for 30 min at 25.degree. C. and then allowed to migrate in the Boyden chamber for 8 hrs. At the 8 hr time point the cells that invaded through the pores into the lower chamber were measured. The numbers of invaded cells for each condition were approximated by quantitating the retrieved labeled DNA using a fluorescent plate reader. The results were calculated in % invasion, where the untreated control was considered as 100% invasion. In all these in vitro functional assays, only Vicrostatin (VN) showed the same potency and exhibited and IC.sub.50 almost identical to that of native CN. In all in vitro functional assays tested, rCN construct was inactive in the nanomolar range.
[0184] B. Preparation of Recombinant Disintegrin Containing Liposomes
[0185] Endotoxin-free VN containing liposomes (referred to as LVN) and endotoxin-free native CN containing liposomes (referred to as LCN) were prepared utilizing a probe sonication previously described (Fujii, Chang et al. 1997). Briefly, the lipids (disteroylphosphatidylcholine, cholesterol and polyethylene glycol derivatized lipid) were dissolved in a chloroform/methanol solution. Thin lipid films were created by pipetting aliquots of the lipid solution into round bottom glass tubes followed by solvent evaporation at 65.degree. C. under a stream of nitrogen gas. The films were placed under vacuum for at least 24 hours to remove residual organic solvent. Liposomes formed following hydration of the lipid films with native CN or VN dissolved in 10 mM sodium phosphate, 9% sucrose, pH 7.2. The mixture was incubated at 65.degree. C. for 5-10 minutes. Hydration was followed by probe sonication until the suspension was translucent. The resultant suspension contained liposomes entrapping CN/VN and unencapsulated CN/VN. The unencapsulated fractions were removed by ultrafiltration. Following clean-up, the suspension was sterilized by passage through a 0.22 .mu.m filter.
[0186] The concentration of liposome entrapped CN/VN was determined by disruption of the liposomes with chloroform/methanol/water (10:40:50) followed by centrifugation at 14,000.times.g. The supernatant was analyzed for CN/VN concentration using BCA protein assay (Smith et al., Anal. Biochem. 150(1): 76-85 (1985)). The encapsulation efficiency was assessed by BCA protein determination following disruption of the LrCN with a solution of H.sub.2O:methanol:chloroform.
[0187] It was observed that 72% of the recombinant protein VN in the encapsulation solution was entrapped within the liposomes, as compared to 80% with native CN. LVN showed identical stability and size distribution (average particle size 140 nm) as encapsulated native CN.
[0188] C. Tumor Therapy Using Recombinant Disintegrin-Containing Liposomes
[0189] Biological activity of liposome encapsulated CN was evaluated as previously described (Swenson et al. (2004)). Briefly, three groups of five nude mice had MDA-MB-435 human mammary carcinoma cells implanted in the mammary fat pad. Two weeks following implantation, small tumors were palpable and treatment was commenced. Animals were treated with LCN or LVN (105 .mu.g, twice weekly, i.v. administration); a PBS treated control was included. A significant inhibitory effect on tumor growth by LVN was observed. The functional activity of VN was indicated by its in vivo cancer therapeutic effect, which was found to be similar to native CN.
[0190] D. Anti-Angiogenic Activity of Recombinant Disintegrin-Containing Liposomes
[0191] Previous in vivo studies with native CN and encapsulated native CN (LCN) demonstrated a dramatic inhibitory effect on angiogenesis in growing tumors (Zhou, Nakada et al. (1999); Zhou, Sherwin et al. (2000); Markland et al. (2001); Golubkov et al., Angiogenesis 6(3): 213-24 (2003); Swenson et al. (2004)). Consequently, the effect of LVN on tumor angiogenesis in the MDA-MB-435 breast cancer model was examined by histochemical identification of blood vessels with anti-CD31 (anti-PECAM-1) monoclonal antibody. CD31 has been reported to be highly expressed in the angiogenic vasculature with approximately one million copies reported on the surface of endothelial cells (Newman, Ann. N.Y. Acad. Sci. 714: 165-74 (1994)). CD31 also has been reported to be involved with the initial formation and stabilization of cell-cell contacts at lateral junctions of endothelial cells, the maintenance of the vascular permeability barrier, the regulation of cell migration, and the formation of new blood vessels during angiogenesis (Newman et al., Science 247(4947): 1219-22 (1990); Ferrero et al., FEBS Lett. 374(3): 323-26 (1995); DeLisser et al., Am. J. Pathol. 151(3): 671-77 (1997)). These combined properties of CD31 make it an optimal reporter molecule for determinations of angiogenic growth.
[0192] Briefly, tumors from treated and untreated mice from the LCN/LVN efficacy studies in the MDA-MB-435 animal tumor model were fixed in 4% normal buffered formalin and embedded in paraffin blocks as previously described (Shi et al. J. Histochem. Cytochem 39(6): 741-48 (1991)). The paraffin blocks were cut into 5 .mu.m sections and placed on glass slides. Tissue sections underwent deparaffinazation, rehydration, and antigen retrieval as described previously (Pileri, Roncador et al., J. Pathol. 183(1): 116-23 (1997)). Endogenous peroxidase activity was blocked by exposure of the sections to 3% H2O2. Specimens were blocked with normal goat serum (1:20) for 30 minutes, followed by incubation with the primary antibody for 1 hour. Rabbit monoclonal antibody to CD31 (Sigma, St. Louis, Mo.) was used as a primary antibody to detect small vessels. The secondary (detection) goat anti-rabbit antibody conjugated with peroxidase (Zymed, San Francisco, Calif.) was then applied to the samples and incubated for 10 minutes at room temperature followed by removal of unbound antibody by multiple washes with PBS. Detection of the secondary antibody using 3,3'-diaminobenzidine (DAB) as the chromogen, was performed following the manufacturers instructions (Zymed HistoMouse Max). Slides were counterstained with hematoxylin. Quantitation of the stained vessels was performed using "hot spot" analysis (Gasparini et al., Int. J. Cancer 55(5): 739-44 (1993)), with "hot spots" being defined as areas of high vessel density (Weidner et al., J. Natl. Cancer Inst. 84(24): 1875-87 (1992); Swenson et al. (2004)). Areas showing positive staining (100.times. magnification) were quantitated in terms of pixels within a given hot spot using SimplePCI advanced imaging software (C-Imaging Systems, Cranberry Township, Pa.).
[0193] Vessel detection by CD31 in MDA-MB-435 tumor sections indicated differences in positive staining in each of the treatment groups: PBS, intravenous liposomal encapsulated native CN (LCN) and intravenous liposomal encapsulated VN (referred to as LVN). In both the LCN, and LVN treated tumors, there is a statistically significant (p<0.0005) reduction of microvascular density, which corresponds to a 90% reduction in angiogenesis in the LCN group and 92% reduction in the LVN group. The reduction in angiogenesis, as observed by CD31 immunostaining in all treatment groups in the MDA-MB-435 breast cancer xenograft model indicates that LVN is an effective inhibitor of angiogenesis.
[0194] E. Structural Analysis of Recombinant Disintegrin
[0195] The structure of native CN and VN was evaluated by mass spectrometry. MALDI-TOF mass spectrometry was performed using a matrix of .alpha.-cyano-4-hydroxycinnamic acid. Native CN was observed as a dimer while VN was observed as a monomeric peak with Mr of 7143.41.
[0196] Electron spray ionization mass spectrometry was also used to evaluate native CN and VN. A large peak of 13507.0, for CN representing the dimer was observed, and two smaller peaks, probably CN, representing a single amino acid cleavage fragment. A single peak of 7146.0, for VN was observed confirming that it is a monomer.
[0197] Mass spectrometry data showed that VN is a monomeric structure unlike the dimer form of native CN. Because the biological activities measured for CN as described above reside in the C-terminal portion of the molecule, this indicates that VN folded correctly at least in the C-terminal part of the molecule, making the correct disulfide bridge combinations and preserving the integrin binding loop that exists in the native conformer. However, the failure to obtain the native dimer configuration indicates that the N-terminal portion of VN folded in a different manner than native CN, which compromised the ability of the N-terminal cysteines of VN to participate in intermolecular disulfide bond formation. This was confirmed by the detection of at least one free thiol in VN. The first cysteine residue (Cys-7) which pairs in the native state with the seventh cysteine (Cys-30) in CN are the furthest apart of the cysteines that bridge in CN. Difficulty inherent in bridging the C7 and C30 cysteines in CN is a possible explanation for the failure of VN to form dimers.
Example 3
Optimizing Codon Usage
[0198] A potential issue with Origami E. coli strain (FA113) is its lack of codon usage optimization. In many organisms, not all of the 61 tRNA species are used equally. The so-called major codons are those that occur in highly expressed genes, whereas the minor or rare codons tend to be in genes expressed at lower levels; which of the 61 codons are the rare ones depends strongly on the organism.
[0199] Eukaryotic proteins tend to translate inefficiently in E. coli because of mismatched codon use that hampers protein production in heterologous expression systems (Makrides, Microbiol. Rev. 60(3): 512-38 (1996)). The codon usage per organism can be found in codon usage databases well known in the art and available online.
[0200] The following overlapping oligonucleotide primers were generated and used to replace the CGG and ACA codons in the wild type CN gene. [0201] CNCGGfor--CN disintegrin domain forward primer that replaces CGG and the eleventh ACA codons:
TABLE-US-00009 [0201] (SEQ ID NO: 12) 5'ACCGTATGCCGTAGAGCAAGGGGTGATGACCTGGATGATTAC3'
[0202] CNCGGback--CN disintegrin domain reverse primer that replaces CGG and the eleventh ACA codons:
TABLE-US-00010 [0202] (SEQ ID NO: 13) 5'TGCTCTACGGCATACGGTTCCTTCTTTCATAAATTTGCACTG3'
[0203] CNACAfor--CN disintegrin domain forward primer that replaces the eight, ninth and tenth ACA codons:
TABLE-US-00011 [0203] (SEQ ID NO: 14) 5'TGCGATGCTGCAACCTGTAAACTGACCACCGGGTCACAGTGTGCAGA T3'
[0204] CNACAback--CN disintegrin domain reverse primer that replaces the eight, ninth and tenth ACA codons:
TABLE-US-00012 [0204] (SEQ ID NO: 15) 5'CAGTTTACAGGTTGCAGCATCGCAGCACGGATTTGC3'
[0205] CNMACA12for--CN metalloprotease domain forward primer that replaces the first two ACA codons:
TABLE-US-00013 [0205] (SEQ ID NO: 16) 5'TCTGATGGCAGAAAAATTACCACCAACCCTCCGGTTGAG3'
[0206] CNMACA12back--CN metalloprotease domain reverse primer that replaces the first two ACA codons:
TABLE-US-00014 [0206] (SEQ ID NO: 17) 5'AATTTTTCTGCCATCAGAGGAATAATG3'
[0207] CNMACA45for--CN metalloprotease domain forward primer that replaces the fourth and fifth ACA codons:
TABLE-US-00015 [0207] (SEQ ID NO: 18) 5'CATAGTGCAATAAATCTTTGGGTTGCAGTTACTATGGCCCATGAG3'
[0208] CNMACA45back--CN metalloprotease domain reverse primer that replaces the fourth and fifth ACA codons:
TABLE-US-00016 [0208] (SEQ ID NO: 19) 5'ATTTATTGCACTATGATCCTGAACAATTCCGGTAGAAAGCTTCGG3'
Example 4
Engineered Hosts System
[0209] An engineered Rosetta-gami B host with disulfide isomerase activity in the cytoplasm and including auto-regenerating capabilities for its oxido-reductive enzymatic equipment in the same compartment may be used for recombinant CN production in bacteria. The host can be engineered to concomitantly overexpress in its cytoplasm the disulfide containing eukaryotic protein fused to thioredoxin along with .DELTA.ssDsbC and .DELTA.ssDsbD.alpha.. This goal can be achieved using a pair of vectors that can coexist together in the same system. The minimum features of this vector set are: the presence of a strong promoter-like T7lac, that can be used for all three proteins simultaneously, as well as the presence of convenient multiple restriction sites in different MCSs (multiple cloning sites) incorporated in the vectors. Two Novagen vectors (pET32a and pCDFDuet-1) that are compatible with each other by having different replicons, and also compatible with Rosetta-gami expression host, have the aforementioned characteristics and may be used in the system described herein. By employing these two vectors, the scenario of using an integrated system, in which the expression of all three proteins would be simultaneously controlled by a single strong promoter T7lac, is achieved.
[0210] Several wild-type and active site mutated thioredoxin-CN genetic constructs were prepared which express a fusion protein containing thioredoxin at the N-terminus and disintegrin domain (CN), or with the disintegrin domain including echistatin C-terminal graft (VN), or with larger eukaryotic proteins consisting of proprotein, metalloproteinase and disintegrin domains of CN with or without the echistatin C-terminal graft (designated rCN PMD and VN PMD). The broad term "TrxA-disintegrin construct" used below refers to the following constructs prepared as described herein: TrxA-rCN (thioredoxin A fused to CN disintegrin domain), TrxA-VN (thioredoxin A fused to CN disintegrin domain including echistatin C-terminal graft), TrxA-rCN PMD (thioredoxin A fused to a large protein consisting of CN proprotein, metalloproteinase and disintegrin domains), TrxA-VN PMD (thioredoxin A fused to a large protein consisting of CN proprotein, metalloproteinase and disintegrin domains with echistatin C-terminal graft), TrxA (CPYC (SEQ ID NO: 49))-rCN (an active site mutated thioredoxin A with the CPYC (SEQ ID NO: 49) motif fused to CN disintegrin domain), TrxA (CPYC (SEQ ID NO: 49))-VN (an active site mutated thioredoxin A including the CPYC (SEQ ID NO: 49) motif fused to CN disintegrin domain with echistatin C-terminal graft), TrxA (CPYC (SEQ ID NO: 49))-rCN PMD (an active site thioredoxin A including the CPYC (SEQ ID NO: 49) motif fused to a large protein consisting of CN proprotein, metalloproteinase and disintegrin domains), TrxA (CPYC (SEQ ID NO: 49))-VN PMD (an active site thioredoxin A including the CPYC (SEQ ID NO: 49) motif fused to a large protein consisting of CN proprotein, metalloproteinase, and disintegrin domains with echistatin C-terminal graft), TrxA (CGHC (SEQ ID NO: 50))-rCN (an active site mutated thioredoxin A including the CGHC (SEQ ID NO: 50) motif fused to CN disintegrin domain), TrxA (CGHC (SEQ ID NO: 50))-VN (an active site mutated thioredoxin A including the CGHC (SEQ ID NO: 50) motif fused to CN disintegrin domain with echistatin C-terminal graft), TrxA (CGHC (SEQ ID NO: 50))-rCN PMD (an active site mutated thioredoxin A including the CGHC (SEQ ID NO: 50) motif fused to a large protein consisting of CN proprotein, metalloproteinase and disintegrin domains), and TrxA (CGHC (SEQ ID NO: 50))-VN PMD (an active site mutated thioredoxin A including the CGHC (SEQ ID NO: 50) motif fused to a large protein consisting of CN proprotein, metalloproteinase and disintegrin domains with echistatin C-terminal graft).
[0211] To increase the stability of some recombinant eukaryotic protein transcripts in the cytoplasm of the expression host, especially of those large transcripts containing the proprotein, metalloprotease and disintegrin domains (with or without echistatin C-terminal graft), some recombinant constructs were designed to include nucleotide sequences of various length that can normally be found in the 3' non-translatable regions of CN native mRNA, downstream of the stop codon signaling the end of translation found in the CN native transcript. Several disintegrin constructs were cloned with extra non-coding nucleotide regions modeled from CN native mRNA by using CN cDNA as a template (Zhou, Hu et al. 2000). Native CN cDNA is available in Francis S. Markland laboratory at USC.
[0212] The primers that were used to PCR clone downstream of TrxA nucleotide sequence, the CN disintegrin domain sequence with or without the echistatin C-terminal graft or the larger CN sequences consisting of proprotein, metalloprotease, and disintegrin domains with or without echistatin C-terminal graft into the pET32a vector were the following: [0213] CNfor2--forward primer for CN disintegrin domain introducing the NcoI restriction site and the TEV protease cleavage site:
TABLE-US-00017 [0213] (SEQ ID NO: 20) 5'GTTCCCCATGGATGAGAATCTTTACTTCCAAGGAGACGCTCCTGCAA ATCCGTGCTGCGATGCTGCA3'
[0214] CNfor3--forward primer for full-length CN introducing the NcoI restriction site and the TEV protease cleavage site:
TABLE-US-00018 [0214] (SEQ ID NO: 21) 5'GTTCCCCATGGATGAGAATCTTTACTTCCAAGGAATGATCCAGGTTC TCTTGGTGACTCTATGCTTA3'
[0215] CNback3--reverse primer for CN constructs without echistatin C-terminal graft introducing the EcoRI restriction site:
TABLE-US-00019 [0215] (SEQ ID NO: 22) 5'GTTATTCGGAATTCTTAGGCATGGAAGGGATTTCTGGGACAGCCAGCA GA3'
[0216] CNback4--reverse primer for CN constructs with echistatin C-terminal graft introducing the EcoRI restriction site:
TABLE-US-00020 [0216] (SEQ ID NO: 23) 5'GTTATTCGGAATTCTTAAGTAGCTGGACCCTTGTGGGGATTTCTGGG ACAGCCAGCAGATATGCC3'
[0217] The reverse primers used to clone various disintegrin constructs including the non-translatable nucleotide sequences of CN native mRNA into the pET32a vector were the following: [0218] CNback5--reverse primer for generating CN native transcripts introducing the EcoRI restriction site:
TABLE-US-00021 [0218] (SEQ ID NO: 24) 5'GTTATTCGGAATTCATATTACAGAATTTGGATACCATCTGGAAGCTA3'
[0219] CNback6--reverse primer for generating CN native transcripts introducing the EcoRI restriction site:
TABLE-US-00022 [0219] (SEQ ID NO: 25) 5'GTTATTCGGAATTCGAATGAGAATAGTTTGTTTATTGACGGAAGCAG3'
[0220] The oligonucleotide primers that were used to amplify the active-site thioredoxin mutants and clone them into pET32a vector replacing the wild type TrxA nucleotide sequence were the following: [0221] Trxfor--Trx forward external primer introducing the XbaI restriction site and designed for inserting the 5' end of the active site mutants into pET32a vector:
TABLE-US-00023 [0221] 5'CCCCTCTAGAAATAATTTTGTTTAACT3' (SEQ ID NO: 26)
[0222] Trxback--Trx reverse external primer introducing the BglII restriction site and designed for inserting the 3' end of the active site mutants into pET32a vector:
TABLE-US-00024 [0222] 5'TACCCAGATCTGGGCTGTCCATGTGCT3' (SEQ ID NO: 27)
[0223] TrxGrxfor--Trx forward primer that mutates TrxA active site to a glutaredoxin-like one:
TABLE-US-00025 [0223] (SEQ ID NO: 28) 5'TTCTGGGCAGAGTGGTGCCCGTATTGCAAAATGATCGCCCCG3'
[0224] TrxGrxback--Trx reverse primer that mutates TrxA active site to a glutaredoxin-like one:
TABLE-US-00026 [0224] 5'GCACCACTCTGCCCAGAAATC3' (SEQ ID NO:29)
[0225] TrxPDIfor--Trx forward primer that mutates TrxA active site to a PDI-like one:
TABLE-US-00027 [0225] (SEQ ID NO: 30) 5'TTCTGGGCAGAGTGGTGCGGTCATTGCAAAATGATCGCCCCG3'
[0226] TrxPDlback--Trx reverse primer that mutates TrxA active site to a PDI-like one:
TABLE-US-00028 [0226] 5'GCACCACTCTGCCCAGAAATC3' (SEQ ID NO: 31)
[0227] For DsbD cloning, the restriction sites employed were NcoI/EcoRI. This restriction enzyme pair was used because it removed the His tag-sequence from the pCDFDuet-1 vector first multiple cloning site, so .DELTA.ssDsbD .alpha.-domain would be expressed as a non-tagged molecule. For DsbC cloning the NdeI/XhoI restriction enzyme pair was used, so that .DELTA.ssDsbC protein would be expressed un-tagged.
[0228] Wild type DsbC gene carries an EcoRI restriction site. For this reason, the foldase sequences were cloned by PCR in a stepwise manner as following: the .DELTA.ssDsbD .alpha.-domain nucleotide sequence was inserted in one multiple cloning site of pCDFDuet-1 vector in the first cloning step, followed by .DELTA.ssDsbC nucleotide sequence, which was inserted in the other multiple cloning site of the vector in a second cloning step. The only His-tagged proteins expressed in the system described herein were the TrxA-disintegrin fusion constructs, so they can be easily separated from the other two co-overexpressed proteins by employing the Ni-column chromatography purification technique. All TrxA-disintegrin constructs included a TEV protease cleavage-site engineered just upstream of the disulfide containing recombinant protein (eukaryotic protein) nucleotide sequences. All purification steps of TrxA-disintegrin constructs were performed in the identical manner to those described in the section discussing the Origami system. However, some TrxA-disintegrin constructs also carried a formic acid cleavage site (Asp-Pro) instead of a TEV protease cleavage site, also engineered just upstream of the N-terminus of disulfide containing recombinant eukaryotic protein nucleotide sequences. Use of formic acid for hydrolysis reduces costs as compared with other protease cleavage systems such as the TEV proteolysis system.
[0229] The oligonucleotide primers that were used to clone various disintegrin constructs engineered to carry an Asp-Pro formic acid cleavage site just upstream of the N-terminus of various CN constructs (with or without multiple domains or echistatin C-terminal graft) into pET32a vector were the following: [0230] CNfor4--forward primer for CN disintegrin domain introducing the NcoI restriction site and the Asp-Pro cleavage site:
TABLE-US-00029 [0230] (SEQ ID NO: 32) 5'GTTCCCCATGGATGACCCTGCAAATCCGTGCTGCGATGCTGCAA CA3'
[0231] CNfor5--forward primer for full-length CN introducing the NcoI restriction site and the Asp-Pro cleavage site:
TABLE-US-00030 [0231] (SEQ ID NO: 33) 5'GTTCCCCATGGATGACCCTATGATCCAGGTTCTCTTGGTGACTCTAT GCTTA3'
[0232] The oligonucleotide primers that were used to PCR clone the .DELTA.ssDsbC, .DELTA.ssDsbD .alpha.-domain nucleotide sequences as well as their active-site mutants sequences into pCDFDuet-1 vector were the following: [0233] DsbCUP--DsbC forward primer introducing the NdeI restriction site:
TABLE-US-00031 [0233] (SEQ ID NO: 34) 5'GTATTCATATGGATGACGCGGCAATTCAACAAACGTTA3'
[0234] DsbCDN--DsbC reverse primer introducing the XhoI restriction site:
TABLE-US-00032 [0234] (SEQ ID NO: 35) 5'GTTCCCTCGAGTTATTTACCGCTGGTCATTTTTTGGTG3'
[0235] DsbDUP--DsbD forward primer introducing the NcoI restriction site:
TABLE-US-00033 [0235] (SEQ ID NO: 36) 5'GTTATTCGCCATGGGATTATTCGACGCGCCGGGACGTTCA3'
[0236] DsbDDN--DsbD reverse primer introducing the EcoRI restriction site:
TABLE-US-00034 [0236] (SEQ ID NO: 37) 5'GTCTACGAATTCGCTTAAGGCTGTGGCGCTGCGTTGTTGGC3'
[0237] The overlap extension oligonucleotide primers that were used to generate the DsbC active site mutants were the following: [0238] DsbCTFfor--active site mutated DsbC (CTFC) overlap extension forward primer:
TABLE-US-00035 [0238] (SEQ ID NO: 38) 5'TTTACTGATATTACCTGTACCTTCTGCCACAAACTGCATGAG3'
[0239] DsbCGFfor--active site mutated DsbC (CGFC) overlap extension forward primer:
TABLE-US-00036 [0239] (SEQ ID NO: 39) 5'TTTACTGATATTACCTGTGGTTTCTGCCACAAACTGCATGAG3'
[0240] DsbCOEback--active site mutated DsbC overlap extension backward primer:
TABLE-US-00037 [0240] 5'ACAGGTAATATCAGTAAACAC3' (SEQ ID NO: 40)
[0241] The pET32a and pCDFDuet-1 external and internal oligonucleotide primers that were employed for sequencing were the following:
TABLE-US-00038 DuetCDFUP1: 5'GGATCTCGACGCTCTCCCTTA3' (SEQ ID NO: 41) DuetCDFUP2: 5'TTGTACACGGCCGCATAATCG3' (SEQ ID NO: 42) DuetCDFDN1: 5'CGATTATGCGGCCGTGTACAA3' (SEQ ID NO: 43) PETUP1 : 5'GGAATTGTGAGCGGATAACAATTC3' (SEQ ID NO: 44) PETUP2: 5'CGCGGTTCTGGTATGAAAGAAACC3' (SEQ ID NO: 45) PETDN1: 5'GTTATGCTAGTTATTGCTCAGCGG3' (SEQ ID NO: 46)
[0242] The bacterial thiol-disulfide interchange protein DsbD .alpha.-domain and disulfide isomerase DsbC nucleotide sequences were directly amplified by PCR from E. coli K-12 MG1655 strain genomic DNA prepared and purified in the Francis S. Markland laboratory at the University of Southern California, using the afore-mentioned oligonucleotide primers. The CN sequences were amplified by PCR from plasmids and/or mutated first to replace all native codons that were rarely used in bacteria or those for which Rosetta-gami B did not provide support. The CN nucleotide sequence encompassing the proprotein, metalloproteinase and disintegrin domain was mutated by utilizing the site-directed mutagenesis technique, employing the overlap extension oligonucleotide primers in several PCR steps.
[0243] Following PCR amplification of the wild-type full-length CN nucleotide sequence and replacement of optimized codons was completed, and all foldases sequences amplified (with or without active site mutations), these sequences were cloned into pET32a and pCDFDuet-1 vectors in a stepwise manner. The full-length CN nucleotide sequence with replaced codons further served as templates to build the disintegrin constructs including the echistatin C-terminal graft. The wild-type TrxA and the disintegrin nucleotide sequences with or without the echistatin C-terminal graft were directly inserted into the pET32a vector using the BglII/NcoI restriction sites. To build the TrxA-disintegrin constructs with TrxA active site mutants, the TrxA mutants were first separately amplified using the overlap extension primers and then inserted in the pET32a vector to replacing the wild type TrxA sequence using the XbaI/BglII set of restriction enzymes. The pET32a vector including the wild type TrxA nucleotide sequence was used as a template for all the PCR amplification steps necessary to generate TrxA active site mutants. In a further step, after the active site TrxA mutants were inserted into the vector, the disintegrin nucleotide sequences were also inserted in pET32a, by employing the NcoI/EcoRI set of restriction enzymes.
[0244] The following active site Trx A mutants were used in the expression system described herein: glutaredoxin-like TrxA (thioredoxin A with a bacterial glutaredoxin A active site) and PDI-like TrxA (thioredoxin A with a eukaryotic protein disulfide isomerase active site).
[0245] The active site mutated sequences of .DELTA.ssDsbC were directly amplified by PCR from E. coli K-12 MG1655 strain genomic DNA using the overlap extension primers. The following active-site mutants were used in the expression system described herein: .DELTA.ssDsbC (CGFC (SEQ ID NO: 52)), and .DELTA.ssDsbC (CTFC (SEQ ID NO: 53)). The wild-type nucleotide sequences of .DELTA.ssDsbD .alpha.-domain and .DELTA.ssDsbC or the active site mutated sequences of .DELTA.ssDsbC were cloned into separate multiple cloning sites of pCDFDuet-1 vector using two sets of restriction enzymes: NcoI/EcoRI and NdeI/XhoI respectively. The pETDuet-1 and pCDFDuet-1 vector constructs were used to co-transform electrocompetent DH5.alpha. cells that are further amplified in culture. All constructs were then validated by sequencing and the recombinant plasmids further used to co-transform the Rosetta-gami B expression host.
[0246] All growing steps were the same as those previously described for the Origami system, except for the antibody usage. The Rosetta-gami B co-transformants were grown in five antibiotics: carbenicillin (100 .mu.g/mL), spectinomycin (50 .mu.g/mL), tetracycline (12.5 .mu.g/mL), kanamycin (15 .mu.g/mL) and chloramphenicol (34 .mu.g/mL). All processing and purification step of various recombinant proteins were identical to those previously described for the Origami system.
[0247] The production level and the biological activity of recombinant disintegrin variants with different co-overexpressed foldases are initially determined after employing the following expression combinations:
[0248] 1. TrxA-disintegrin+.DELTA.ssDsbC+.DELTA.ssDsbD.alpha.
[0249] 2. TrxA (CPYC (SEQ ID NO: 49))-disintegrin+.DELTA.ssDsbC+.DELTA.ssDsbD.alpha.
[0250] 3. TrxA (CGHC (SEQ ID NO: 50))-disintegrin+.DELTA.ssDsbC+.DELTA.ssDsbD.alpha.
[0251] By comparing the structures and yields of different TrxA-disintegrin fusion proteins, the version that generates the properly folded dimer at the best level of expression is chosen. To further improve the expression level of correctly folded proteins, the best oxidase version, referred to as "best oxidase-disintegrin," is further tested in combination with two mutated variants of DsbC.
[0252] 1. Best oxidase-disintegrin+.DELTA.ssDsbC (CGFC (SEQ ID NO: 52))+.DELTA.ssDsbD.alpha.
[0253] 2. Best oxidase-disintegrin+.DELTA.ssDsbC (CTFC (SEQ ID NO: 53))+.DELTA.ssDsbD.alpha.
[0254] For production of recombinant disintegrins the same steps described in Example 1 were employed. However, the expressed recombinant variants including the pro-protein, metalloproteinase and disintegrin domains may undergo a post-translational autocatalytical proteolysis freeing the C-terminal recombinant disintegrin domains with or without echistatin C-terminal graft, disintegrin domains that can be further purified directly from the total soluble protein fraction retrieved form bacteria by reverse-phase HPLC. This desirable event would obviate the need for Ni-column affinity chromatography purification and TEV-protease proteolysis or formic acid hydrolysis intermediary steps employed in recombinant disintegrin production.
Example 5
Optimizing Expression
[0255] The system can be optimized to achieve better yields by changing several parameters such as the use of: increased concentrations of carbenicillin (a more stable form of penicillin) that is more resistant to .beta.-lactamase degradation for all growing steps; an optimal IPTG concentration (a feature that can be easily achieved in a Tuner derivative host); and, finally, an optimal induction temperature for generating the best yields. The simplest way to achieve a more homogenous culture and prevent the plasmid loss is to use the more stable antibiotic carbenicillin rather than ampicillin. For tighter regulation of basal expression, a phenomenon that also leads to plasmid instability, the culture medium may be supplemented with 1% glucose to repress induction of the lac promoter by lactose, which is present in most rich media (such as LB). Moreover, addition of small quantities of ethanol, of polyols and sucrose, or low molecular weight thiols can be used to significantly boost the expression of soluble proteins in E. coli (e.g., from several milligrams of recombinant protein to tens or even hundreds of milligrams). Also, codon optimization can be used as already discussed. In an effort to optimize the codon usage, the Rosetta-gami B(DE3)pLysS expression host, a strain supplemented with rare tRNAs, may be preferentially employed instead of Origami B(DE3)pLysS.
Example 6
Expression System Combinations
[0256] The following elements may be used in obtaining recombinant disintegrin expression. "TP" refers to eukaryotic protein.
[0257] 1. TrxA-TP or TrxA (CPYC (SEQ ID NO: 49))-TP or TrxA (CGHC (SEQ ID NO: 50)-TP
[0258] 2. Integrin binding (e.g. HKGPAT (SEQ ID NO: 47)) C-terminal sequence for TP
[0259] 3. "Tag" sequence for item no. 1.
[0260] 4. Cleavage site sequence for item no. 1.
[0261] 5. .DELTA.ssDsbC
[0262] 6. .DELTA.ssDsbD.alpha.
[0263] 7. trxB mutant
[0264] 8. gor mutant
[0265] 9. ompT mutant
[0266] 10. lon mutant
[0267] The methods may include any combination of 1-10 above for expressing TP. In another approach, any of the following elements may be combined for expressing a eukaryotic protein that is not fused to thioredoxin.
[0268] 1. Eukaryotic protein unfused to thioredoxin
[0269] 2. Integrin binding (e.g. HKGPAT (SEQ ID NO: 47)) C-terminal sequence for TP
[0270] 3. Tag sequence for item no. 1.
[0271] 4. Cleavage site sequence for item no. 1.
[0272] 5. .DELTA.ssDsbC
[0273] 6. .DELTA.ssDsbD.alpha.
[0274] 7. trxB mutant
[0275] 8. gor mutant
[0276] 9. ompT mutant
[0277] 10. lon mutant
Example 7
Antitumor Activity of CN and Docetaxel
[0278] 100 .mu.l of PC-3 cell suspension (1.times.10.sup.6 cells) (ATCC.org, Manassas, Va.) was mixed with an equal volume, 100 .mu.l of Matrigel basement membrane matrix (BD Biosciences, Bedford, Mass.) and injected subcutaneously into the flank region of athymic male nude mice (Balb/c/nu/nu mice) (Charles River Laboratory, Wilmington, Mass.). Four weeks after inoculation, 32 mice with PC-3 tumors were randomized into four groups with 8 mice in each group. The control group was treated with PBS and released from Alzet osmotic mini-pumps for four weeks. The second group was treated with docetaxel injections intraperitoneally at a dose of 6 mg/kg twice a week for two weeks and then switched to 4 mg/kg twice a week for two weeks. The third group was treated with contortrostatin (CN) at a dose of 60 .mu.g/day which was continually released from subcutaneously implanted Alzet osmotic mini-pumps for four weeks. The fourth group was treated with a combination of docetaxel and CN at doses indicated in groups 2 and 3 for four weeks. Tumor volume was measured one a week and are calculated according to the formula:
length.times.width.sup.2.times.0.52.
[0279] During the experiment, there were no significant differences in body weights between the groups. Both docetaxel and CN individually significantly suppressed growth of PC-3 tumors. After four weeks, the mean tumor volume was significantly reduced in groups receiving CN or docetaxel with tumor volumes at 195.+-.40 mm.sup.3 and 259.+-.38 mm.sup.3, respectively as compared with the control group which had a tumor volume of 378.+-.49 mm.sup.3. The group receiving a combination of both CN and docetaxel was much more effective that either agent individually. The final tumor volume was significantly (p<0.01) reduced to 95.+-.38 mm.sup.3. See FIG. 1.
[0280] Unless otherwise defined, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs.
[0281] The inventions illustratively described herein may suitably be practiced in the absence of any element or elements, limitation or limitations, not specifically disclosed herein. Thus, for example, the terms "comprising," "including," "containing," etc. shall be read expansively and without limitation. Additionally, the terms and expressions employed herein have been used as terms of description and not of limitation, and there is no intention in the use of such terms and expressions of excluding any equivalents of the features shown and described or portions thereof, but it is recognized that various modifications are possible within the scope of the invention claimed.
[0282] Thus, it should be understood that although the present invention has been specifically disclosed by preferred embodiments and optional features, modification, improvement and variation of the inventions embodied therein herein disclosed may be resorted to by those skilled in the art, and that such modifications, improvements and variations are considered to be within the scope of this invention. The materials, methods, and examples provided here are representative of preferred embodiments, are exemplary, and are not intended as limitations on the scope of the invention.
[0283] The invention has been described broadly and generically herein. Each of the narrower species and subgeneric groupings falling within the generic disclosure also form part of the invention. This includes the generic description of the invention with a proviso or negative limitation removing any subject matter from the genus, regardless of whether or not the excised material is specifically recited herein.
[0284] In addition, where features or aspects of the invention are described in terms of Markush groups, those skilled in the art will recognize that the invention is also thereby described in terms of any individual member or subgroup of members of the Markush group.
[0285] All publications, patent applications, patents, and other references mentioned herein are expressly incorporated by reference in their entirety, including all formulas and figures, to the same extent as if each were incorporated by reference individually. In case of conflict, the present specification, including definitions, will control.
[0286] Other embodiments are set forth within the following claims.
Sequence CWU 1
1
541483PRTAgkistrodon contortrix contortrix 1Met Ile Gln Val Leu Leu
Val Thr Leu Cys Leu Ala Ala Phe Pro Tyr1 5 10 15Gln Gly Ser Ser Ile
Ile Leu Glu Ser Gly Asn Val Asn Asp Tyr Glu 20 25 30Val Leu Tyr Pro
Gln Lys Val Thr Ala Leu Pro Lys Gly Ala Val Gln 35 40 45Pro Lys Tyr
Glu Asp Thr Met Gln Tyr Glu Phe Lys Val Asn Gly Glu 50 55 60Pro Val
Val Leu His Leu Glu Lys Asn Lys Gly Leu Phe Ser Lys Asp65 70 75
80Tyr Ser Glu Thr His Tyr Ser Ser Asp Gly Arg Lys Ile Thr Thr Asn
85 90 95Pro Pro Val Glu Asp His Cys Tyr Tyr His Gly Arg Ile Gln Asn
Asp 100 105 110Ala Asp Ser Thr Ala Ser Ile Ser Ala Cys Asn Gly Leu
Lys Gly His 115 120 125Phe Lys Leu Gln Gly Glu Thr Tyr Leu Ile Glu
Pro Leu Lys Leu Ser 130 135 140Asp Ser Glu Ala His Ala Val Tyr Lys
Tyr Glu Asn Val Glu Lys Glu145 150 155 160Asp Glu Ala Pro Lys Met
Cys Gly Val Thr Gln Thr Asn Trp Glu Ser 165 170 175Asp Glu Pro Ile
Lys Lys Ala Ser Gln Leu Asn Leu Thr Pro Glu Gln 180 185 190Gln Gly
Phe Pro Gln Arg Tyr Ile Glu Leu Val Val Val Ala Asp His 195 200
205Arg Met Phe Thr Lys Tyr Asn Gly Asn Leu Asn Thr Ile Arg Ile Trp
210 215 220Val His Glu Leu Val Asn Thr Met Asn Val Phe Tyr Arg Pro
Leu Asn225 230 235 240Ile Arg Val Ser Leu Thr Asp Leu Glu Val Trp
Ser Asp Gln Asp Leu 245 250 255Ile Asn Val Gln Pro Ala Ala Ala Asp
Thr Leu Glu Ala Phe Gly Asp 260 265 270Trp Arg Glu Thr Val Leu Leu
Asn Arg Ile Ser His Asp Asn Ala Gln 275 280 285Leu Leu Thr Ala Ile
Glu Leu Asp Gly Glu Thr Ile Gly Leu Ala Asn 290 295 300Arg Gly Thr
Met Cys Asp Pro Lys Leu Ser Thr Gly Ile Val Gln Asp305 310 315
320His Ser Ala Ile Asn Leu Trp Val Ala Val Thr Met Ala His Glu Met
325 330 335Gly His Asn Leu Gly Ile Ser His Asp Gly Asn Gln Cys His
Cys Asp 340 345 350Ala Asn Ser Cys Ile Met Ser Glu Glu Leu Arg Glu
Gln Leu Ser Phe 355 360 365Glu Phe Ser Asp Cys Ser Gln Asn Gln Tyr
Gln Thr Tyr Leu Thr Asp 370 375 380His Asn Pro Gln Cys Met Leu Asn
Glu Pro Leu Arg Thr Asp Ile Val385 390 395 400Ser Thr Pro Val Ser
Gly Asn Glu Leu Leu Glu Thr Gly Glu Glu Ser 405 410 415Asp Phe Asp
Ala Pro Ala Asn Pro Cys Cys Asp Ala Ala Thr Cys Lys 420 425 430Leu
Thr Thr Gly Ser Gln Cys Ala Asp Gly Leu Cys Cys Asp Gln Cys 435 440
445Lys Phe Met Lys Glu Gly Thr Val Cys Arg Arg Ala Arg Gly Asp Asp
450 455 460Leu Asp Asp Tyr Cys Asn Gly Ile Ser Ala Gly Cys Pro Arg
Asn Pro465 470 475 480Phe His Ala22029DNAAgkistrodon contortrix
contortrix 2gaattcgggg tcaatagagg aagagctcaa gttggcttga aagcaggaag
agattgcctg 60tcttccagcc aaatccagcc gccaaaatga tccaggttct cttggtgact
ctatgcttag 120cagcttttcc ttatcaaggg agctctataa tcctggaatc
tgggaatgtt aatgattatg 180aagtactgta tccacaaaaa gtcactgcat
tgcccaaagg agcagttcag ccaaagtatg 240aagacaccat gcaatatgaa
tttaaagtga atggagagcc agtggtcctt cacctggaaa 300aaaataaagg
acttttttca aaagattaca gcgagactca ttattcctct gatggcagaa
360aaattacaac aaaccctccg gttgaggatc actgctatta tcatggacgc
atccagaatg 420atgctgactc aactgcaagc atcagtgcat gcaacggttt
gaaaggacat ttcaagcttc 480aaggggagac gtaccttatt gaacccttga
agctttccga cagtgaagcc catgcagtct 540acaaatatga aaacgtagaa
aaagaagatg aggcccccaa aatgtgtggg gtaacccaga 600ctaattggga
atcagatgag cccatcaaaa aggcctctca gttaaatctt actcctgaac
660aacaaggatt cccccaaaga tacattgagc ttgttgtagt tgcagatcac
agaatgttca 720cgaaatacaa cggcaattta aatactatta gaatatgggt
acatgaactt gtcaacacta 780tgaatgtgtt ttacagacct ttgaatattc
gtgtctcact gactgaccta gaagtttggt 840cagaccaaga tttgatcaac
gtgcagccag cagcggctga tactttggaa gcatttggag 900actggagaga
gacagtcttg ctgaatcgca taagtcatga taatgctcag ttactcacgg
960ccattgagct tgatggagaa actataggat tggctaacag gggcaccatg
tgcgacccga 1020agctttctac aggaattgtt caggatcata gtgcaataaa
tctttgggtt gcagttacaa 1080tgccccatga gatgggtcat aatctgggta
ttagtcacga tggaaatcag tgtcattgcg 1140atgctaactc atgcattatg
agtgaagaac taagagaaca actttccttt gagttcagcg 1200attgtagtca
gaatcaatat cagacatatc ttactgatca taacccacaa tgcatgctca
1260atgaaccctt gagaacagat attgtttcaa ctccagtttc tggaaatgaa
cttttggaga 1320cgggagaaga aagtgacttt gacgctcctg caaatccgtg
ctgcgatgct gcaacatgta 1380aactgacaac agggtcacag tgtgcagatg
gactgtgttg tgaccagtgc aaatttatga 1440aagaaggaac agtatgccgg
agagcaaggg gtgatgacct ggatgattac tgcaatggca 1500tatctgctgg
ctgtcccaga aatcccttcc atgcctaacc aacaatggag atggaatggt
1560ctgcagcaac aggcagtgtg ttgatctgaa tacagcctaa taatcaacct
ctggcttctc 1620tcagatttga tcatggagat ccttcttcca gaaggtttca
cttccctcaa atccaaagag 1680acccatctgc ctgcatccta ctagtaaatc
acccttagct tccagatggt atccaaattc 1740tgtaatattt cttctccata
tttaatctat ttaccttttg ctgtaacaaa acctttttcc 1800tgtcacaaag
ctccatgggc atgtacagct tatctgctgt caagaaaaaa aatggccatt
1860ttaccgtttg ccagttacaa agcacattta atgcaacaag ttcttccttt
tgagctgatg 1920tattcaaagt caatgcttcc tctcccaaaa tttcatgctg
gcttcccaag atgtagctgc 1980ttccgtcaat aaacaaacta ttctcattca
aaaaaaaaaa cccgaattc 2029365PRTAgkistrodon contortrix contortrix
3Asp Ala Pro Ala Asn Pro Cys Cys Asp Ala Ala Thr Cys Lys Leu Thr1 5
10 15Thr Gly Ser Gln Cys Ala Asp Gly Leu Cys Cys Asp Gln Cys Lys
Phe 20 25 30Met Lys Glu Gly Thr Val Cys Arg Arg Ala Arg Gly Asp Asp
Leu Asp 35 40 45Asp Tyr Cys Asn Gly Ile Ser Ala Gly Cys Pro Arg Asn
Pro Phe His 50 55 60Ala654109PRTEscherichia coli 4Met Ser Asp Lys
Ile Ile His Leu Thr Asp Asp Ser Phe Asp Thr Asp1 5 10 15Val Leu Lys
Ala Asp Gly Ala Ile Leu Val Asp Phe Trp Ala Glu Trp 20 25 30Cys Gly
Pro Cys Lys Met Ile Ala Pro Ile Leu Asp Glu Ile Ala Asp 35 40 45Glu
Tyr Gln Gly Lys Leu Thr Val Ala Lys Leu Asn Ile Asp Gln Asn 50 55
60Pro Gly Thr Ala Pro Lys Tyr Gly Ile Arg Gly Ile Pro Thr Leu Leu65
70 75 80Leu Phe Lys Asn Gly Glu Val Ala Ala Thr Lys Val Gly Ala Leu
Ser 85 90 95Lys Gly Gln Leu Lys Glu Phe Leu Asp Ala Asn Leu Ala 100
1055216PRTEscherichia coliMOD_RES(206)..(206)Any amino acid 5Asp
Asp Ala Ala Ile Gln Gln Thr Leu Ala Lys Met Gly Ile Lys Ser1 5 10
15Ser Asp Ile Gln Pro Ala Pro Val Ala Gly Met Lys Thr Val Leu Thr
20 25 30Asn Ser Gly Val Leu Tyr Ile Thr Asp Asp Gly Lys His Ile Ile
Gln 35 40 45Gly Pro Met Tyr Asp Val Ser Gly Thr Ala Pro Val Asn Val
Thr Asn 50 55 60Lys Met Leu Leu Lys Gln Leu Asn Ala Leu Glu Lys Glu
Met Ile Val65 70 75 80Tyr Lys Ala Pro Gln Glu Lys His Val Ile Thr
Val Phe Thr Asp Ile 85 90 95Thr Cys Gly Tyr Cys His Lys Leu His Glu
Gln Met Ala Asp Tyr Asn 100 105 110Ala Leu Gly Ile Thr Val Arg Tyr
Leu Ala Phe Pro Arg Gln Gly Leu 115 120 125Asp Ser Asp Ala Glu Lys
Glu Met Lys Ala Ile Trp Cys Ala Lys Asp 130 135 140Lys Asn Lys Ala
Phe Asp Asp Val Met Ala Gly Lys Ser Val Ala Pro145 150 155 160Ala
Ser Cys Asp Val Asp Ile Ala Asp His Tyr Ala Leu Gly Val Gln 165 170
175Leu Gly Val Ser Gly Thr Pro Ala Val Val Leu Ser Asn Gly Thr Leu
180 185 190Val Pro Gly Tyr Gln Pro Pro Lys Glu Met Lys Glu Phe Xaa
Asp Glu 195 200 205His Gln Lys Met Thr Ser Gly Lys 210
2156132PRTEscherichia coli 6Gly Leu Phe Asp Ala Pro Gly Arg Ser Gln
Phe Val Pro Ala Asp Gln1 5 10 15Ala Phe Ala Phe Asp Phe Gln Gln Asn
Gln His Asp Leu Asn Leu Thr 20 25 30Trp Gln Ile Lys Asp Gly Tyr Tyr
Leu Tyr Arg Lys Gln Ile Arg Ile 35 40 45Thr Pro Glu His Ala Lys Ile
Ala Asp Val Gln Leu Pro Gln Gly Val 50 55 60Trp His Glu Asp Glu Phe
Tyr Gly Lys Ser Glu Ile Tyr Arg Asp Arg65 70 75 80Leu Thr Leu Pro
Val Thr Ile Asn Gln Ala Ser Ala Gly Ala Thr Leu 85 90 95Thr Val Thr
Tyr Gln Gly Cys Ala Asp Ala Gly Phe Cys Tyr Pro Pro 100 105 110Glu
Thr Lys Thr Val Pro Leu Ser Glu Val Val Ala Asn Asn Glu Ala 115 120
125Ser Gln Pro Val 13077PRTArtificial SequenceDescription of
Artificial Sequence Synthetic peptide 7Glu Asn Leu Tyr Phe Gln Xaa1
5869PRTArtificial SequenceDescription of Artificial Sequence
Synthetic polypeptide 8Gly Asp Ala Pro Ala Asn Pro Cys Cys Asp Ala
Ala Thr Cys Lys Leu1 5 10 15Thr Thr Gly Ser Gln Cys Ala Asp Gly Leu
Cys Cys Asp Gln Cys Lys 20 25 30Phe Met Lys Glu Gly Thr Val Cys Arg
Arg Ala Arg Gly Asp Asp Leu 35 40 45Asp Asp Tyr Cys Asn Gly Ile Ser
Ala Gly Cys Pro Arg Asn Pro His 50 55 60Lys Gly Pro Ala
Thr65966DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 9gttccagatc tcgagaatct ttacttccaa ggagacgctc
ctgcaaatcc gtgctgcgat 60gctgca 661051DNAArtificial
SequenceDescription of Artificial Sequence Synthetic primer
10gttattcgcc atggcttagg catggaaggg atttctggga cagccagcag a
511166DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 11gttattcgcc atggcttaag tagctggacc cttgtgggga
tttctgggac agccagcaga 60tatgcc 661242DNAArtificial
SequenceDescription of Artificial Sequence Synthetic primer
12accgtatgcc gtagagcaag gggtgatgac ctggatgatt ac
421342DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 13tgctctacgg catacggttc cttctttcat aaatttgcac tg
421448DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 14tgcgatgctg caacctgtaa actgaccacc gggtcacagt
gtgcagat 481536DNAArtificial SequenceDescription of Artificial
Sequence Synthetic primer 15cagtttacag gttgcagcat cgcagcacgg atttgc
361639DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 16tctgatggca gaaaaattac caccaaccct ccggttgag
391727DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 17aatttttctg ccatcagagg aataatg
271845DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 18catagtgcaa taaatctttg ggttgcagtt actatggccc
atgag 451945DNAArtificial SequenceDescription of Artificial
Sequence Synthetic primer 19atttattgca ctatgatcct gaacaattcc
ggtagaaagc ttcgg 452067DNAArtificial SequenceDescription of
Artificial Sequence Synthetic primer 20gttccccatg gatgagaatc
tttacttcca aggagacgct cctgcaaatc cgtgctgcga 60tgctgca
672167DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 21gttccccatg gatgagaatc tttacttcca aggaatgatc
caggttctct tggtgactct 60atgctta 672250DNAArtificial
SequenceDescription of Artificial Sequence Synthetic primer
22gttattcgga attcttaggc atggaaggga tttctgggac agccagcaga
502365DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 23gttattcgga attcttaagt agctggaccc ttgtggggat
ttctgggaca gccagcagat 60atgcc 652447DNAArtificial
SequenceDescription of Artificial Sequence Synthetic primer
24gttattcgga attcatatta cagaatttgg ataccatctg gaagcta
472547DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 25gttattcgga attcgaatga gaatagtttg tttattgacg
gaagcag 472627DNAArtificial SequenceDescription of Artificial
Sequence Synthetic primer 26cccctctaga aataattttg tttaact
272727DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 27tacccagatc tgggctgtcc atgtgct
272842DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 28ttctgggcag agtggtgccc gtattgcaaa atgatcgccc cg
422921DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 29gcaccactct gcccagaaat c 213042DNAArtificial
SequenceDescription of Artificial Sequence Synthetic primer
30ttctgggcag agtggtgcgg tcattgcaaa atgatcgccc cg
423121DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 31gcaccactct gcccagaaat c 213246DNAArtificial
SequenceDescription of Artificial Sequence Synthetic primer
32gttccccatg gatgaccctg caaatccgtg ctgcgatgct gcaaca
463352DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 33gttccccatg gatgacccta tgatccaggt tctcttggtg
actctatgct ta 523438DNAArtificial SequenceDescription of Artificial
Sequence Synthetic primer 34gtattcatat ggatgacgcg gcaattcaac
aaacgtta 383538DNAArtificial SequenceDescription of Artificial
Sequence Synthetic primer 35gttccctcga gttatttacc gctggtcatt
ttttggtg 383640DNAArtificial SequenceDescription of Artificial
Sequence Synthetic primer 36gttattcgcc atgggattat tcgacgcgcc
gggacgttca 403741DNAArtificial SequenceDescription of Artificial
Sequence Synthetic primer 37gtctacgaat tcgcttaagg ctgtggcgct
gcgttgttgg c 413842DNAArtificial SequenceDescription of Artificial
Sequence Synthetic primer 38tttactgata ttacctgtac cttctgccac
aaactgcatg ag 423942DNAArtificial SequenceDescription of Artificial
Sequence Synthetic primer 39tttactgata ttacctgtgg tttctgccac
aaactgcatg ag 424021DNAArtificial SequenceDescription of Artificial
Sequence Synthetic primer 40acaggtaata tcagtaaaca c
214121DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 41ggatctcgac gctctccctt a 214221DNAArtificial
SequenceDescription of Artificial Sequence Synthetic primer
42ttgtacacgg ccgcataatc g 214321DNAArtificial SequenceDescription
of Artificial Sequence Synthetic primer 43cgattatgcg gccgtgtaca a
214424DNAArtificial SequenceDescription of Artificial Sequence
Synthetic primer 44ggaattgtga gcggataaca attc 244524DNAArtificial
SequenceDescription of Artificial Sequence Synthetic primer
45cgcggttctg gtatgaaaga aacc 244624DNAArtificial
SequenceDescription of Artificial Sequence Synthetic primer
46gttatgctag ttattgctca gcgg 24476PRTArtificial SequenceDescription
of Artificial Sequence Synthetic peptide 47His Lys Gly Pro Ala Thr1
5484PRTArtificial SequenceDescription of Artificial Sequence
Synthetic peptide 48Cys Gly Pro Cys1494PRTArtificial
SequenceDescription of Artificial Sequence Synthetic peptide 49Cys
Pro Tyr Cys1504PRTArtificial SequenceDescription of Artificial
Sequence Synthetic peptide 50Cys Gly His Cys1514PRTArtificial
SequenceDescription of Artificial Sequence Synthetic peptide 51Cys
Gly Tyr Cys1524PRTArtificial SequenceDescription of Artificial
Sequence Synthetic peptide 52Cys Gly Phe Cys1534PRTArtificial
SequenceDescription of Artificial Sequence Synthetic peptide 53Cys
Thr Phe Cys1547PRTArtificial SequenceDescription of Artificial
Sequence Synthetic peptide 54Glu Asn Leu Tyr Phe Gln Gly1
5











D00001

D00002

D00003

D00004

P00001

S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.