On-demand rapid synthesis of lomustine under continuous flow conditions
Thompson , et al. March 9, 2
U.S. patent number 10,941,111 [Application Number 16/654,103] was granted by the patent office on 2021-03-09 for on-demand rapid synthesis of lomustine under continuous flow conditions. This patent grant is currently assigned to Purdue Research Foundation. The grantee listed for this patent is Purdue Research Foundation. Invention is credited to Robert Graham Cooks, Christina Ramires Ferreira, Zinia Jaman, David H Thompson.











View All Diagrams
| United States Patent | 10,941,111 |
| Thompson , et al. | March 9, 2021 |
On-demand rapid synthesis of lomustine under continuous flow conditions
Abstract
Disclosed herein is a continuous manufacturing process for lomustine that has a short residence time and 63 percent yield. Major advantages of this process are that the total production cost for lomustine is lower, the product is higher quality, and the manufacturing operation is safer for production personnel.
| Inventors: | Thompson; David H (West Lafayette, IN), Cooks; Robert Graham (West Lafayette, IN), Ferreira; Christina Ramires (West Lafayette, IN), Jaman; Zinia (West Lafayette, IN) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Applicant: |
|
||||||||||
| Assignee: | Purdue Research Foundation
(West Lafayette, IN) |
||||||||||
| Family ID: | 1000005409054 | ||||||||||
| Appl. No.: | 16/654,103 | ||||||||||
| Filed: | October 16, 2019 |
Prior Publication Data
| Document Identifier | Publication Date | |
|---|---|---|
| US 20200115330 A1 | Apr 16, 2020 | |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | Issue Date | ||
|---|---|---|---|---|---|
| 62746045 | Oct 16, 2018 | ||||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | C07C 273/1854 (20130101); C07C 273/1818 (20130101); B01J 19/0046 (20130101); C07C 273/1818 (20130101); C07C 275/26 (20130101); C07C 273/1854 (20130101); C07C 275/68 (20130101); C07C 275/68 (20130101); B01J 2219/00058 (20130101); B01J 2219/00033 (20130101); B01J 2219/00279 (20130101) |
| Current International Class: | C07C 273/18 (20060101); B01J 19/00 (20060101); C07C 275/68 (20060101) |
References Cited [Referenced By]
U.S. Patent Documents
| 4182757 | January 1980 | Tsujihara |
| 2012/0142722 | June 2012 | Ziv |
| 2020/0020516 | January 2020 | Cooks |
Other References
|
Johnston ("The Synthesis of Potential Anticancer Agents. XXXVI. N-Nitrosoureas. II. Haloalkyl Derivatives" J. Med. Chem, 1966, 9(6), p. 892-911) (Year: 1966). cited by examiner . Chakkath, T., et al. "Alkylation and Carbamylation Effects of Lomustine and Its Major Metabolites and MGMT Expression in Canine Cells." Veterinary Sciences, (2015), 52-68, 2.2. cited by applicant . Dirikolu, L., et al. "Synthesis of Trans- and Cis-4'-Hydroxylomustine and Development of Validated Analytical Method for Lomustine and Trans- and Cis-4'-Hydroxylomustine in Canine Plasma." Journal of Analytical Toxicology, (2009), 595-603, 33.9, Oxford University Press. cited by applicant . Lown, J. W. et al. "Synthesis of Specifically Nitrogen-15- and Carbon-13-Labeled Antitumor (2-Haloethyl) Nitrosoureas.The Study of Their Conformations in Solution by Nitrogen-15 and Carbon-13 Nuclear Magnetic Resonance Electronic Control in Their Aqueous Decomposition." Journal of Organic Chemistry, (1981), 5309-5321, 46.26, American Chemical Society. cited by applicant . Kaina, B., et al. "MGMT: Key Node in the Battle against Genotoxicity, Carcinogenicity and Apoptosis Induced by Alkylating Agents." DNA Repair, (2007), 1079-1099, 6.8, Elsevier. cited by applicant . Lee, F. Y. F., et al. "Clinical Pharmacokinetics of Oral CCNU (Lomustine)." Cancer Chemotherapy and Pharmacology, (1985), 125-131, 14.2, Springer Nature. cited by applicant . Funaro, J. et al. "A costly rebranding of an old drug comes with a 700% price increase." The Cancer Letter, (2017). cited by applicant . Webb, D. et al. "Continuous Flow Multi-Step Organic Synthesis." Chemical Science, (2010), 675-680, 1.6, The Royal Society of Chemistry. cited by applicant . Wegner, J. et al. "Ten Key Issues in Modern Flow Chemistry." Chemical Communications, (2011), 4583-4592, 47.16, The Royal Society of Chemistry. cited by applicant . Yoshida, J. et al. "Green and Sustainable Chemical Synthesis Using Flow Microreactors." ChemSusChem, (2011), 331-340, 4.3, John Wiley & Sons, Ltd. cited by applicant . Yoshida, J. et al. "Flash Chemistry: Flow Chemistry That Cannot Be Done in Batch." Chemical Communications, (2013), 9896-9904, 49.85, The Royal Society of Chemistry. cited by applicant . Hessel, V., et al. "Novel Process Windows for Enabling, Accelerating, and Uplifting Flow Chemistry." ChemSusChem (2013), 746-789, 6.5, John Wiley & Sons, Ltd. cited by applicant . Jensen, K. F. et al. "Tools for Chemical Synthesis in Microsystems." Lab on a Chip, (2014), 3206-3212, 14.17, The Royal Society of Chemistry. cited by applicant . Gutmann, B. et al. "Continuous-Flow Technology--A Tool for the Safe Manufacturing of Active Pharmaceutical Ingredients." Angewandte Chemie International Edition, (2015), 6688-6728, 54.23, John Wiley & Sons, Ltd. cited by applicant . Cambie, D., et al. "Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment." Chemical Reviews, (2016), 10276-10341, 116.17, American Chemical Society. cited by applicant . Poh, J.S., et al. "Rapid Asymmetric Synthesis of Disubstituted Allenes by Coupling of Flow-Generated Diazo Compounds and Propargylated Amines." Angewandte Chemie International Edition, (2017),1864-1868, 56.7, John Wiley & Sons, Ltd. cited by applicant . Yu, Z., et al. "A Fully Continuous-Flow Process for the Synthesis of p-Cresol: Impurity Analysis and Process Optimization." Organic Process Research & Development, (2017),1644-1652, 21.10, American Chemical Society. cited by applicant . Britton, J. et al. "Multi-Step Continuous-Flow Synthesis." Chemical Society Reviews, (2017),1250-1271, 46.5, The Royal Society of Chemistry. cited by applicant . Berton, M. et al. "On-Demand Synthesis of Organozinc Halides under Continuous Flow Conditions." Nature Protocols, (2018), 324-334, 13.2. Springer Nature. cited by applicant . Hessel, V. "Novel Process Windows--Gate to Maximizing Process Intensification via Flow Chemistry." Chemical Engineering & Technology, (2009), 1655-1681, 32.11, John Wiley & Sons, Ltd. cited by applicant . Zaborenko, N., et al. "Kinetic and Scale-Up Investigations of Epoxide Aminolysis in Microreactors at High Temperatures and Pressures." Organic Process Research & Development, (2011), 131-139, 15.1, American Chemical Society. cited by applicant . Wiles, C. et al. "Recent Advances in Micro Reaction Technology." Chemical Communications, (2011), 6512-6535, 47.23, The Royal Society of Chemistry. cited by applicant . Wegner, J. et al. "Flow Chemistry--A Key Enabling Technology for (Multistep) Organic Synthesis." Advanced Synthesis & Catalysis, (2012), 17-57, 354.1, John Wiley & Sons, Ltd. cited by applicant . Snead, D. R. et al. "End-to-End Continuous Flow Synthesis and Purification of Diphenhydramine Hydrochloride Featuring Atom Economy, in-Line Separation, and Flow of Molten Ammonium Salts." Chemical Science, (2013), 2822-2827, 4.7, The Royal Society of Chemistry. cited by applicant . Len, C. et al. "Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling in Continuous Flow." Catalysts, (2017), 146, 7.5. cited by applicant . Lummiss, J. A. M., et al. "Towards More Efficient, Greener Syntheses through Flow Chemistry." The Chemical Record, (2017), 667-680, 17.7, John Wiley & Sons, Ltd. cited by applicant . Morse, P. D. et al. "Synthesis and Utilization of Nitroalkyne Equivalents in Batch and Continuous Flow." Angewandte Chemie International Edition, (2017), 13999-14002, 56.45, John Wiley & Sons, Ltd. cited by applicant . Gustafsson, T. et al. "Trimethylaluminium Mediated Amide Bond Formation in a Continuous Flow Microreactor as Key to the Synthesis of Rimonabant and Efaproxiral." Chemical Communications, (2008), 1100-1102, 9, The Royal Society of Chemistry. cited by applicant . Hopkin, M. D. et al. "An Expeditious Synthesis of Imatinib and Analogues Utilising Flow Chemistry Methods." Organic & Biomolecular Chemistry, (2013), 1822-1839, 11.11, The Royal Society of Chemistry. cited by applicant . Deadman, B. J., et al. "The Synthesis of Bcr-Abl Inhibiting Anticancer Pharmaceutical Agents Imatinib, Nilotinib and Dasatinib." Organic & Biomolecular Chemistry, (2013), 1766-1800, 11.11, The Royal Society of Chemistry. cited by applicant . Murray, P. R. D., et al. "Continuous Flow-Processing of Organometallic Reagents Using an Advanced Peristaltic Pumping System and the Telescoped Flow Synthesis of (E/Z)-Tamoxifen." Organic Process Research & Development, (2013), 1192-1208, 17.9, American Chemical Society. cited by applicant . Zhang, P. et al. "Continuous Flow Total Synthesis of Rufinamide." Organic Process Research & Development, (2014), 1567-1570, 18.11, American Chemical Society. cited by applicant . Snead, D. R. et al. "A Three-Minute Synthesis and Purification of Ibuprofen: Pushing the Limits of Continuous-Flow Processing." Angewandte Chemie International Edition, (2015), 983-987, 54.3, John Wiley & Sons, Ltd. cited by applicant . Adamo, A., et al. "On-Demand Continuous-Flow Production of Pharmaceuticals in a Compact, Reconfigurable System." Science, (2016), 61-67, 352.6281, The American Association for the Advancement of Science. cited by applicant . Lin, H., et al. "A Rapid Total Synthesis of Ciprofloxacin Hydrochloride in Continuous Flow." Angewandte Chemie International Edition, (2017), 8870-8873, 56.30, John Wiley & Sons, Ltd. cited by applicant . Zhang, P., et al. "Advanced Continuous Flow Platform for On-Demand Pharmaceutical Manufacturing." Chemistry--A European Journal, (2018), 2776-2784, 24.11, John Wiley & Sons, Ltd. cited by applicant . Falcone, C. E., et al. "Reaction Screening and Optimization of Continuous-Flow Atropine Synthesis by Preparative Electrospray Mass Spectrometry." Analyst, (2017), 2836-2845, 142.15, The Royal Society of Chemistry. cited by applicant . Loren, B. P., et al. "Mass Spectrometric Directed System for the Continuous-Flow Synthesis and Purification of Diphenhydramine." Chemical Science, (2017), 4363-4370, 8.6, The Royal Society of Chemistry. cited by applicant . Wleklinski, M., et al. "Can Accelerated Reactions in Droplets Guide Chemistry at Scale?" European Journal of Organic Chemistry, (2016), 5480-5484, 2016.33, John Wiley & Sons, Ltd. cited by applicant . Ewan, H. S., et al. "Multistep Flow Synthesis of Diazepam Guided by Droplet-Accelerated Reaction Screening with Mechanistic Insights from Rapid Mass Spectrometry Analysis." Organic Process Research & Development, (2017), 1566-1570, 21.10, American Chemical Society. cited by applicant . Riva, E., et al. "Synthesis of (+)-Dumetorine and Congeners by Using Flow Chemistry Technologies." Chemistry--A European Journal, (2011), 6221-6226, 17.22, John Wiley & Sons, Ltd. cited by applicant . Ahmed-Omer, B. et al. "Preparation of Fluoxetine by Multiple Flow Processing Steps." Organic & Biomolecular Chemistry, (2011), 3854-3862, 9.10, The Royal Society of Chemistry. cited by applicant . Newton, S., et al. "Accelerating Spirocyclic Polyketide Synthesis Using Flow Chemistry." Angewandte Chemie International Edition, (2014), 4915-4920, 53.19, John Wiley & Sons, Ltd. cited by applicant . Peeva, L., et al. "Continuous Consecutive Reactions with Inter-Reaction Solvent Exchange by Membrane Separation." Angewandte Chemie International Edition, (2016), 13576-13579, 55.43, John Wiley & Sons, Ltd. cited by applicant . Brasholz, M., et al. "A Gram-Scale Batch and Flow Total Synthesis of Perhydrohistrionicotoxin." Chemistry--A European Journal, (2010), 11471-11480, 16.37, John Wiley & Sons, Ltd. cited by applicant . Jensen, K. F. "Silicon-Based Microchemical Systems: Characteristics and Applications." MRS Bulletin, (2006), 101-107, 31.2, Cambridge University Press. cited by applicant . Geyer, K. et al. "Developing Continuous-Flow Microreactors as Tools for Synthetic Chemists." Synlett, (2009), 2382-2391, 15, Georg Thieme Verlag KG. cited by applicant . Kim, H., et al. "Submillisecond Organic Synthesis: Outpacing Fries Rearrangement through Microfluidic Rapid Mixing." Science, (2016), 691-694, 352.6286, The American Association for the Advancement of Science. cited by applicant . Troshin, K. et al. "Snap Deconvolution: An Informatics Approach to High-Throughput Discovery of Catalytic Reactions." Science, (2017), 175-181, 357.6347, The American Association for the Advancement of Science. cited by applicant . Wleklinski, M., et al. "High Throughput Reaction Screening Using Desorption Electrospray Ionization Mass Spectrometry." Chemical Science, (2018), 1647-1653, 9.6, The Royal Society of Chemistry. cited by applicant . Jaman, Z., et al. "High Throughput Experimentation and Continuous Flow Validation of Suzuki-Miyaura Cross-Coupling Reactions." Chemistry--A European Journal, (2018), 9546-9554, 24.38, John Wiley & Sons, Ltd. cited by applicant . Yan, X. et al. "Organic Reactions in Microdroplets: Reaction Acceleration Revealed by Mass Spectrometry." Angewandte Chemie International Edition, (2016), 12960-12972, 55.42, John Wiley & Sons, Ltd. cited by applicant . Ifa, D. R., et al. "Latent Fingerprint Chemical Imaging by Mass Spectrometry." Science, (2008), 805, 321.5890, The American Association for the Advancement of Science. cited by applicant . Freeman, E. S. "The Kinetics of the Thermal Decomposition of Sodium Nitrate and of the Reaction between Sodium Nitrite and Oxygen." Journal of Physical Chemistry, (1956), 1487-1493, 60.11, American Chemical Society. cited by applicant . Fox, J. B. et al. "The Determination of Nitrite: A Critical Review." CRC Critical Reviews in Analytical Chemistry, (1985), 283-313, 15.3, Taylor & Francis. cited by applicant . Johnston, T. P. et al. "Synthesis of Chlorozotocin, the 2-Chloroethyl Analog of the Anticancer Antibiotic Streptozotocin." Journal of Medicinal Chemistry, (1975), 104-106, 18.1, American Chemical Society. cited by applicant . Baracu, I. et al. "Potential Anticancer Agents. XXVIII. Synthesis of Some Cyclohexanone Derived N-Nitrosoureas." Journal fur Praktische Chemie, (1985), 675-681, 327.4, John Wiley & Sons, Ltd. cited by applicant. |
Primary Examiner: Bonaparte; Amy C
Attorney, Agent or Firm: D'Hue Law LLC D'Hue; Cedric A.
Government Interests
GOVERNMENT SUPPORT
This invention was made with government support under CA023168 awarded by National Institutes of Health, and W911NF-16-2-0020 awarded by Army Research Laboratory. The government has certain right in the invention.
Claims
The invention claimed is:
1. A method of producing Lomustine with a continuous flow condition, wherein said Lomustine is prepared using starting materials including cyclohexanamine, 1-chloro-2-isocyanatoethane, triethylamine (TEA), and optionally a solvent via a linear sequence of two chemical reactions performed separately in two telescoped flow reactors, wherein an extractor is coupled with the first telescoping reactor to remove excess starting materials from the first reaction reactor, wherein the second chemical reaction includes the step of nitrosation of an intermediate product, from the first chemical reaction, 1-(2-chloroethyl)-3-cyclohexylurea, using a nitrosation agent, wherein the nitrosation agent is sodium nitrite in formic acid or tert-butyl nitrite.
2. The method of claim 1, further comprising screening the first reaction conditions according to the method of claim 1: ##STR00005## in a continuous flow as a function of temperature, solvent and stoichiometry, wherein the reaction is monitored by thin-layer chromatography (TLC) and mass spectrometry (MS) using a triple quadrupole mass spectrometer operating in positive ion mode to identify an optimum reaction condition for the first reaction.
3. The method of claim 1, comprising sequential)y: mixing cyclohex anamine (1) and 1-chloro-2-isocyanatoethane (2) with triethylamine (TEA) in the first telescoping reactor at 50.degree. C., for about 1 minute to form the first chemical reaction; wherein an intermediate product 1-(2-chloroethyl)-3-cyclohexylurea (3) is formed in the first chemical reaction; coupling the extractor with the first telescoping reactor to remove excess base TEA from the first chemical reaction; wherein the extractor is a liquid-liquid extractor; transferring the intermediate product 1-(2-chloroethyl)-3-cyclohexylurea (3) from the first telescoping reactor to the second telescoping reactor in a continuous flow; and carrying out nitrosation of the intermediate product 1-(2-chloroethyl)-3-cyclohexylurea (3) in the second telescoping reactor at 25.degree. C. for about 8 minutes using tort-butyl nitrite (tBuONO (5) as the nitrosation agent to form the second chemical reaction; wherein the final product Lomustine is formed in the second chemical reaction.
4. The method of claim 1, wherein the first chemical reaction is performed at a temperature within the range of about 25.degree. C. to about 65.degree. C.
5. The method of claim 1, wherein the first chemical reaction is performed at a temperature selected from the group consisting of room temperature, about 25.degree. C., about 50.degree. C., and about 65.degree. C.
6. The method of claim 1, wherein the first chemical reaction is perfomed for a residence time within the range of about 10 seconds to about three minutes.
7. The method of claim 1, wherein the first chemical reaction is. performed for a residence time selected from the group consisting of about 10 seconds, about 30 seconds, about 60 seconds, and about 180 seconds.
8. The method of claim 1, wherein the extractor is coupled with the first telescoping, reactor to remove excess triethylamine (TEA) from the first reaction.
9. The method of claim 8, wherein the extractor is a liquid-liquid extractor.
10. The method of claim 1, wherein the nitrosation is carried out at a temperature of about 0.degree. C. when sodium nitrite in formic acid is used as the nitrosation agent or about 25.degree. C. to about 50.degree. C. when tent-butyl nitrite is used as the nitrosation agent.
11. The method of claim 1, wherein the second chemical reaction is performed in a solvent Selected from the pup consisting of dimethyl sulfoxide (DMSO), toluene, acetonitrile (ACN), dichloromethane (DCM), ethanol (EtOH), and methanol (MeOH).
12. The method of claim 1, wherein the second chemical reaction is performed for a residence time within the range of about 30 seconds to about ten minutes.
13. The method of claim 1, wherein the nitrosation agent is sodium nitrite in formic acid.
14. The method of claim 1, wherein the nitrosation agent is tert-butyl nitrite.
15. The method of claim 13, wherein the second chemical reaction is performed in a solvent selected from the group consisting of ethyl acetate (EtOAc), tetrahydrofuran (THF), dimethyl sulfoxide (DMSO), toluene, acetonitrile (ACN), dichloromethane (DCM), ethanol (DOH), methanol (MeOH), and methanol:water (4:1 v/v).
16. The method of claim 14, wherein the second chemical reaction is performed in a solvent selected from the group consisting of dimethyl sulfoxide (DMSO), toluene, acetonitrile (ACN), dichloromethane (DCM), ethanol (EtOH), methanol (MeOH), and acetonitrile:ethanol (3.7:1 v/v).
17. The method of claim 13, wherein the second chemical reaction is performed at 0.degree. C.
18. The method of claim 1, wherein the two chemical reactions do not include a purification step of a reaction intermediate.
19. The method of claim 1. wherein the first chemical reaction is performed in a solvent selected from the group consisting of ethanol (Et0H). acetonitrile (CCN), Toluene, Ether and tetrahydrofuran (THF).
20. The method of claim 14, wherein the second chemical reaction is performed at a temperature within the range of about 25.degree. C. to about 50.degree. C.
Description
FIELD OF INVENTION
This disclosure provides a novel method of synthesizing lomustine drug in scalable size under continuous flow conditions. Particularly, lomustine is prepared via a linear sequence of two chemical reactions performed separately in two telescoped flow reactors. The process omits isolation and purification of a labile intermediate, providing tremendous advantages of producing active pharmaceutical ingredient at low cost.
BACKGROUND
Lomustine, a widely used anticancer agent, is a highly lipophilic alkylating agent that produces chloroethyl carbonium ions and carbamylating intermediates in vivo..sup.[1] These electrophilic compounds attack the nucleophilic sites on DNA to form alkylated products..sup.[1a] Other anticancer agents such as mitomycin C, streptonigrin, bleomycin, and the anthracyclines require bioactivation to react with their cellular targets, whereas lomustine does not require pre-activation..sup.[2] Unlike alkylating agents that form adducts at the most reactive N.sup.7 position of guanine, chloroethylating compounds like lomustine form adducts at O.sup.6, leading to interstrand DNA cross-linking. If DNA repair does not occur, this crosslinking can cause double strand breaks during DNA replication, eventually leading to cell death via apoptosis..sup.[3]
Lomustine, 1-(2-chloroethyl)-3-cyclohexyl-1-nitroso-urea (commercial names: CCNU, CeeNU, Gleostine) is used as an oral antineoplastic agent that is administered every 6 weeks. It was first evaluated in clinical trials in the late 1960s.sup.[4] and approved by the US FDA in 1976.sup.[5] for primary and metastatic brain tumors as well as Hodgkin's lymphoma..sup.[3] Bristol-Myers Squibb originally held the patent for the agent under the brand name CeeNu. In 2014, Next Source Biotechnology LLC (NSB) was approved by the FDA for the rebranding of lomustine under the trade name Gleostine..sup.[5] The average wholesale price for one dose of rebranded Gleostine is $1,645.68, while the generic formulation costs $203.38..sup.[5] The huge price discrepancy (700%) between Gleostine and the generic formulation has created patient access problems, thus motivating our effort to develop a rapid and low cost lomustine synthesis method by continuous flow.
Continuous flow synthesis has been reported as an efficient methodology and has been explored in both industry and academic labs for the last few decades:.sup.[6] Compared to traditional batch synthesis processes, flow reactors provide better control over reaction conditions and selectivity owing to rapid mixing and precise control of reaction parameters such as temperature, stoichiometry, pressure, and residence time. The enhanced heat and mass transfer capabilities also provide safer and greener operational conditions:.sup.[6b, 7] Generally, these aspects of continuous flow synthesis contribute to improved chemical reaction efficiency.sup.[7g, 8] and shorter reaction times, enabling process intensification.sup.[7a], and more facile scale-up, with improved quality and consistency in production. Motivated by these factors, continuous flow synthesis of active pharmaceutical ingredient has recently become more attractive,.sup.[7g, 9] however efficient execution of multistep reactions in a telescoped manner still remains a challenge due to challenges arising from workup conditions.sup.[9f, 10], solvent switches,.sup.[11] and flow rate differences..sup.[11a, 12] Moreover, optimization of continuous flow conditions and analysis require significant investments in time and material..sup.[9f, 13]
SUMMARY OF THE INVENTION
This disclosure provides a method of producing Lomustine with continuous flow condition, the method comprises the steps illustrated in FIG. 6, or steps illustrated in FIG. 20.
This disclosure further provides a method of identifying optimum reaction condition of producing Lomustine. The method comprises screening the reaction condition according to scheme 1 in a continuous flow as a function of temperature, solvent and stoichiometry, wherein the reaction is monitored by TLC and MS using a triple quadrupole mass spectrometer operating in positive ion mode to afford rapid investigation of full mass spectra and product ion distribution for each reaction condition.
This disclosure further provides a system for telescoped Lomustine synthesis. The system comprises sequentially: a first telescoping chamber mixing cyclohexylamine (1) and 1-chloro-2-isocyanatoethane (2) with triethyalamine (TEA) at 50 C for about 1 minute; a Zaiput liquid-liquid extractor to couple with the first telescoping chamber to remove the base TEA from the first reaction chamber; and a second telescoping chamber for nitrosation of the intermediate product 1-(2-chloroethyl) -3-cyclohexylurea (3) at 25 C for about 8 minutes using tBuONO (5) as the nitrosation agent.
These and other features, aspects and advantages of the present invention will become better understood with reference to the following figures, associated descriptions and claims.
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. 1: Microfluidic synthesis of intermediate 3 in a glass reactor chip (SOR 3225). A=1 in THF; B=2 in THF, C=TEA in THF.
FIG. 2: DESI-MS plate maps showing the presence or absence of expected ions at each spot where the nitrosation reaction conditions were tested using different stoichiometries and with commercially available standards. Blue dots indicate the presence of the m/z 169 expected stable fragment for the reaction product (successful reaction), Red dots indicate that the expected m/z for the reaction product was not present at the reaction spot (unsuccessful reaction condition). A: Concentration screening using the lomustine ion (m/z 169) intensity and NaNO.sub.2 as nitrosation reagent; B: Solvent screening using the lomustine ion (m/z 169) intensity and NaNO.sub.2 as nitrosation reagent; C: Solvent screening using the lomustine ion (m/z 169) intensity and tBuONO as nitrosation reagent.
FIG. 3: Microfluidic synthesis of lomustine using NaNO.sub.2/HCO.sub.2H as nitrosation reagent in a Chemtrix 3225 SOR glass reactor chip. A=3 in THF; B=HCO.sub.2H (neat), C=NaNO.sub.2 in MeOH:H.sub.2O (4:1).
FIG. 4: Microfluidic synthesis of lomustine using 5 as nitrosation reagent in a Chemtrix 3223 SOR glass reactor chip. A=3 in ACN:EtOH (3.7:1) ; B=5 in ACN.
FIG. 5: Schematic for the telescoped synthesis of lomustine using NaNO.sub.2/HCO.sub.2H (4) as nitrosation reagent.
FIG. 6: Schematic for the final telescoped synthesis of lomustine using 5 as a nitrosation reagent.
FIG. 7: Total ion chromatogram (TIC) from HPLC-MS/MS and comparison between synthesized lomustine (top, red) and commercially available lomustine (bottom, black).
FIG. 8A and FIG. 8B: Full ESI-MS scan and MS/MS of commercially available 3 and synthesized 3 derived from flow under the reaction conditions of 50.degree. C., 1 min.
FIG. 9: DESI master plate layout using four different concentrations in two solvents.
FIG. 10: DESI master plate layout using eight different solvents.
FIG. 11: DESI master plate layout using only commercially available 3 and lomustine.
FIG. 12: (left) Direct DESI-MS data comparison between the two nitrosation reactions in THF and ACN in different concentration. (right) DESI-MS data of commercially available 3 and lomustine standards. The data was analyzed using BioMAP imaging software.
FIG. 13: (left & center) Direct DESI-MS data comparison between the two nitrosation reactions in different solvents. (right) DESI-MS data of commercially available 3 and lomustine standards. The data was analyzed using BioMAP imaging software.
FIG. 14A to FIG. 14C: Map of the DESI-MS plates showing some of the expected ions where the nitrosation reaction was screened using different stoichiometries as well as commercially available standards. Green dots indicate the presence of the m/z expected for the reaction product (successful reaction), Red dots indicate that the expected m/z for the reaction product was not present at the reaction spot (unsuccessful reaction condition). A: NaNO.sub.2, concentration screening using the lomustine ion (m/z 169) intensity; B: NaNO.sub.2, solvent screening using the lomustine ion (m/z 169) intensity; C: TBN, solvent screening using the lomustine ion (m/z 169) intensity
FIG. 15: Comparison of .sup.1H NMR of lomustine synthesized by continuous flow for different methods of purification.
FIG. 16: Full MS scan and MS/MS of m/z=169 from commercially available lomustine and synthesized lomustine form flow. Reaction conditions were 25.degree. C., 8 min.
FIG. 17: Telescoped two step synthesis of lomustine using sodium nitrite in the second step.
FIG. 18: First step of the telescoped synthesis of lomustine.
FIG. 19: Second step of the telescoped synthesis of lomustine using NaNO.sub.2 as a nitrosation reagent.
FIG. 20: Telescoped lomustine synthesis using TBN as a nitrosation reagent, before reaction initiation.
FIG. 21: Telescoped lomustine synthesis using TBN (protected from light).
FIG. 22: Second step of the telescoped synthesis of lomustine using TBN.
FIG. 23: TLC monitoring during lomustine synthesis in flow
FIG. 24: Comparison of TLC of telescoped lomustine synthesis using different equivalents of base
DETAILED DESCRIPTION
While the concepts of the present disclosure are illustrated and described in detail in the figures and the description herein, results in the figures and their description are to be considered as exemplary and not restrictive in character; it being understood that only the illustrative embodiments are shown and described and that all changes and modifications that come within the spirit of the disclosure are desired to be protected.
Unless defined otherwise, the scientific and technology nomenclatures have the same meaning as commonly understood by a person in the ordinary skill in the art pertaining to this disclosure.
Lomustine, an important agent for treatment of brain tumors and Hodgkin's lymphoma, has been synthesized using continuous flow methodology. Desorption electrospray ionization mass spectrometry (DESI-MS) was used to quickly explore a large number of reaction conditions and guide the efficient translation of optimized conditions to continuous lomustine production at a rate of approximately one dose/h. Using only four inexpensive commercially available starting materials and a total residence time of 9 min, lomustine was prepared via a linear sequence of two chemical reactions performed separately in two telescoped flow reactors. Sequential offline extraction and filtration resulted in 63% overall yield of pure lomustine at a production rate of 110 mg/h. The primary advantage of this approach lies in the rapid manufacture of lomustine with two telescoped steps to avoid isolation and purification of a labile intermediate, thereby decreasing the production cost of this active pharmaceutical ingredient to approximately $5/gram in total material cost.
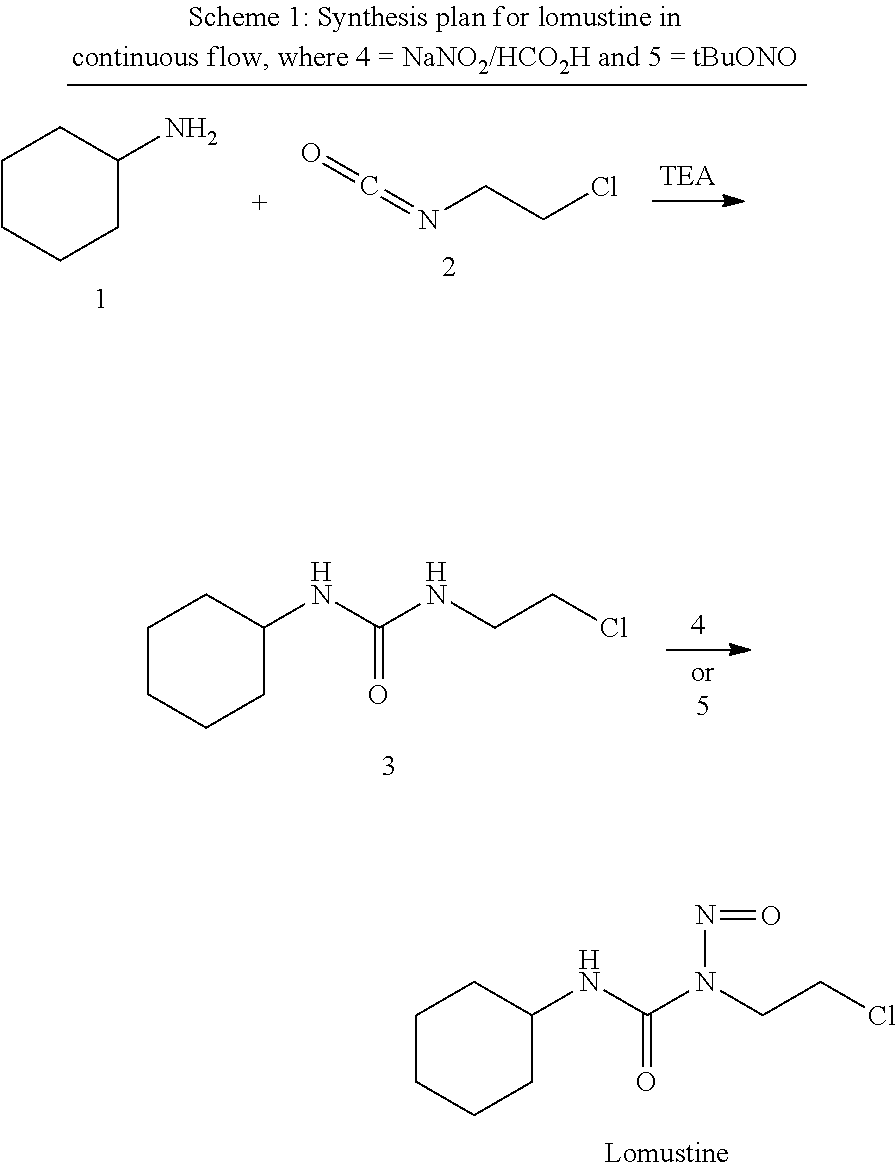
Briefly, herein we disclose robust high throughput reaction screening method using desorption electrospray ionization mass spectrometry (DESI-MS) to guide scalable synthesis in continuous flow reactors at scale..sup.[13e] Herein, we report the continuous flow synthesis of lomustine using an optimization protocol that was guided by DESI-MS. The final method consists of two reactions telescoped without isolation or purification of intermediates. This approach can reduce production costs radically by using a simple reactor set up and inexpensive starting materials (Scheme 1). To the best of our knowledge, this is the first synthesis of lomustine telescoped in continuous flow using an approach that does not interrupt the flow sequence due to intermediate workup requirements.
##STR00001##
Continuous Synthesis of 1-(2-Chloroethyl)-3-cyclohexylurea, 3.
The first step in the synthesis of lomustine (Scheme 1) is a fast reaction at room temperature. This transformation was optimized in continuous flow as a function of temperature, solvent, and stoichiometry to discover the conditions for maximum product yield. The reactions were monitored by TLC and MS using a triple quadrupole mass spectrometer operating in positive ion mode to afford rapid investigation of full mass spectra and product ion distribution for each reaction condition.
TABLE-US-00001 TABLE 1 Reaction conditions evaluated for the synthesis of 3 in flow using a Chemtrix S1 glass system fitted with a 3225 SOR reactor chip. 2 Temperature, Residence Isolated Entry Solvent equivalent .degree. C. time, sec Yield % 1 EtOH 1 50 10 42.8 2 EtOH 1 50 30 0.00 3 EtOH 1 50 60 0.00 4 ACN 1 50 10 56.2 5 ACN 1 50 30 clogged 6 ACN 1 25 30 clogged 7 Toluene 1 50 10 clogged 8 Ether 1 50 10 clogged 9 THF 1 25 10 50.0 10 THF 1 25 30 56.4 11 THF 1 50 10 59.5 12 THF 1 50 30 62.1 13 THF 1 65 10 47.5 14 THF 1 65 30 29.1 15 THF 1.2 50 10 61.2 16 THF 1.2 50 30 64.8 17 THF 1.2 50 60 71.3 18 THF 1.4 50 10 67.1 19 THF 1.4 50 30 82.0 20 THF 1.4 50 60 91.7 21 THF 1.4 50 180 82.8
A cascade method was designed to reveal the best conditions for the first step. Cyclohexylamine, 1, 1-chloro-2-isocyanatoethane, 2, and triethylamine (TEA) solutions were pumped through a Chemtrix 3225 SOR chip with automatic collection of the products in vials via an autosampler (FIG. 1). The individual reaction mixtures were evaporated and the white solid washed with cold Et.sub.2O before drying under vacuum for overnight prior to analysis by TLC, MS, MS/MS, and NMR (.sup.1H and .sup.13C).
Parameters such as residence time (t), reaction temperature (T), 1-chloro-2-isocyanatoethane:cyclohexylamine ratio were investigated systematically under continuous flow conditions. As shown in Table 1, product yield dropped sharply in EtOH at longer residence times (entries 1-3) due to ethanolysis of the 1-chloro-2-isocyanato-ethane starting material. Though the yield of 3 in ACN was significant, its low solubility in this solvent led to significant chip clogging (entries 4-6). Similar clogging problems were found for toluene and Et.sub.2O, even at low concentrations (entries 7-8). The yield increased to 50-62% with longer residence times (entries 9-14) in THF, reaching a maximum when .tau.=30 s at 50.degree. C. and decayed rapidly with increased temperature (entries 13, 14) due to increased product decomposition at elevated temperature. The yield of 3 was also found to increase with proportions of 2 ratio (entries 15-21), whereas longer residence times promoted product decomposition (entry 21). Consequently, a maximum yield of intermediate 3 (92%) was achieved under conditions of .tau.=60 s, T=50.degree. C., and 1.4 equivalents of 1-chloro-2-isocyanatoethane (entry 20).
Synthesis of Lomustine, Guided by DESI-MS High Throughput Experimentation.
We employed DESI-MS to evaluate the impact of solvent, concentration and nitrosation reagent type on the efficiency of lomustine production. DESI-MS was originally applied to the surface analysis of intact samples such as biological tissues for cancer diagnosis or human fingerprints for drug detection,.sup.[14] although more recently, the DESI-MS approach has been used for reaction analysis..sup.[13e] This approach is based on the phenomena of reaction acceleration that occurs within confined volumes such as microdroplets that originate from spray-based MS ionization processes..sup.[14a] MS analysis speeds approaching 10,000 reaction spots/hour can be achieved by this technique..sup.[13e]
As shown in Scheme 1 (2.sup.nd step), two nitrosation methods were investigated. First, the efficiency of sodium nitrite, 4, in formic acid as the nitrosation reagent toward the conversion of 3 to lomustine was evaluated. This conversion was then compared with tert-butyl nitrite (TBN, 5) as the nitrosation reagent.
DESI-MS was used to evaluate the NaNO.sub.2/HCO.sub.2H transformation under different reactant concentrations and solvents. The expected m/z 234 value for lomustine were not observed. Analysis of a commercial lomustine sample yielded a very similar MS, suggesting that loss of NO occurs readily during the ionization process. Triple quadrupole MS with electrospray ionization, as well as ion trap mass spectrometry coupled with DESI, revealed the presence of a stable lomustine ion at m/z 169. This was further confirmed by the fact that the losmustine fragments from this reaction matched with commercially available lomustine. Reactions for all NaNO.sub.2 concentrations in THF:H.sub.2O worked better than for ACN:H.sub.2O (FIG. 2, A). Also, the fragments from 3 were most abundant in the ACN:H.sub.2O reactions compared to reactions in THF:H.sub.2O, suggesting that the conversion of starting material was comparatively sluggish in ACN:H.sub.2O. Next, eight different solvents (EtOH, MeOH, DCM, ACN, toluene, DMSO, THF and EtOAc) were compared for guiding the continuous flow synthesis. All eight solvents produced similar outcomes using 4 as the nitrosation reagent (FIG. 2, B). When 5 was used, lomustine was detected in all solvents except THF and EtOAc (FIG. 2, C).
Nitrosation Reaction Optimization in Continuous Flow.
We utilized the reaction conditions emerging from the DESI-MS high throughput experiments to optimize the flow synthesis and also check whether unsuccessful reactions identified by DESI-MS would also negatively translate under flow conditions. From the DESI-MS experiments, THF was identified as the best solvent for nitrosation using 4. Excess 4 was also used to maximize conversion of 3 to lomustine since the nitrosation reagents are hygroscopic and readily oxidize to nitrate..sup.[15] Overall, the DESI-MS experiments were in excellent agreement with the outcomes of flow reaction conditions in terms of stoichiometry, reagents and solvents.
A series of reactions (Table 2) were performed to maximize the conversion of 3 to lomustine under continuous synthesis conditions (FIG. 3). Initially, we examined the reaction at 0.degree. C. with a residence time of 30 sec using TLC and MS to monitor the reaction progress. Gratifyingly, lomustine was obtained through this very short reaction time, albeit in low conversion (Table 2, entry 1). A systematic evaluation of residence times led to good lomustine yields and starting material conversions, however, longer residence times appeared enhance the decomposition of lomustine (Table 2, entries 2-6). Consequently, we kept the temperature constant at 0.degree. C. to avoid NaNO.sub.2 decomposition of sodium nitrite to the diazonium salt that occurs at higher temperatures..sup.[15b]
Different purification methods were evaluated to isolate pure lomustine. At first, the product was extracted with Et.sub.2O (3 times) to exploit the low solubility of 3 in this solvent (Method 1). Unfortunately, the TLC analysis revealed the presence of 3 in the organic layer as well as in the aqueous layer. Therefore, the combined organic extracts were dried over anhydrous Na.sub.2SO.sub.4 and concentrated in vacuo to produce a yellowish oil that re-dissolved in Et.sub.2O, heated and cooled in an ice bath to precipitate 3 from the mixture. NMR analysis of the filtrate revealed that 22% of 3 remained in the isolated lomustine product. Next, we purified the product by recrystallization from ACN (Method 2). Although we obtained very pure lomustine as identified by NMR analysis, recovery using this approach was low. Finally, we found that hot filtration from petroleum ether removed the insoluble 3 impurity (Method 3). Concentration of the filtrate after drying gave pure lomustine without detectable amount of 3 by NMR. Using this method, we obtained 74% isolated yield of pure lomustine under the conditions of 0.degree. C. reaction and a residence time of 5 min using 3 equivalents of 4.
TABLE-US-00002 TABLE 2 Isolated yields of lomustine under different reaction conditions using 4 as the nitrosation reagent. Temperture = 0.degree. C., solvent = MeOH:H.sub.2O (4:1) Residence Isolated Yield Isolated Yield Isolated Yield Entry time, min (Method 1) % (Method 2) % (Method 3) % 1 0.5 26.8 -- 43.7 2 1 48.8 -- 50.6 3 3 51.6 41.2 63.1 4 5 79.0 54.4 74.5 5 8 59.0 -- 68.7 6 10 50.2 -- 65.3
The reaction was also optimized with respect to residence time, temperature, and solvent using 5 as nitrosation agent (FIG. 4 and Table 3). This optimization process led to the finding that an elevated residence time (8 min), lower temperature (25.degree. C.) and increased 5 ratio (3 equiv) resulted in the most efficient conversion to lomustine (91% isolated yield) after Method 3 purification (Table 3, entry 11).
TABLE-US-00003 TABLE 3 Synthesis of lomustine under at different reaction conditions using 5 as the nitrosation reagent. Isolated Temperature, Residence Lomustine Entry Solvent (3.7:1) (.degree. C.) time, (min) Yield (%) 1 ACN:EtOH 50 0.5 68.3 2 ACN:EtOH 50 1 69.8 3 ACN:EtOH 50 3 60.0 4 ACN:EtOH 50 5 58.8 5 ACN:EtOH 50 8 51.9 6 ACN:EtOH 50 10 49.4 7 ACN:EtOH 25 0.5 48.8 8 ACN:EtOH 25 1 54.1 9 ACN:EtOH 25 3 66.5 10 ACN:EtOH 25 5 79.3 11 ACN:EtOH 25 8 91.2 12 ACN:EtOH 25 10 89.8 13 THF(100%) 25 3 36.5
Telescoped Synthesis of Lomustine
The next step was to adapt the whole two step sequence to a continuous flow setup. We sought to telescope the carbamylation and nitrosation reactions using the Chemtrix reactor chips, however, an extraction needed to be incorporated between the steps to remove the TEA present in the first reaction to avoid competitive consumption of the nitrosation reagent in the second step. We achieved this objective by incorporating a commercially available Zaiput liquid-liquid extractor to remove the base before the nitrosation step and by re-optimizing the synthesis using FEP tubing reactors (FIG. 5). In the beginning, the best reaction conditions were a 1 min residence time at 50.degree. C. for the first step and a 5 min residence time at 0.degree. C. for the second step, yielding 43 mg (51.8% overall yield) of pure lomustine (Table 4, entry 1). Efforts to improve the lomustine yield by changing the residence times of either the first or second steps were unsuccessful in the FEP tubing reactor using 4 as nitrosation reagent (Table 4, entry 1-4).
TABLE-US-00004 TABLE 4 Lomustine synthesis yields for telescoped reactions under different eagents, residence time, stoichiometry, and temperature conditions. Isolated Nitrosation TEA Lomustine Entry reagent Step 1 Step 2 Stoichiometry Yield % 1 4 1 min, 50.degree. C. 5 min, 0.degree. C. 1 51.8 2 4 2 min, 50.degree. C. 5 min, 0.degree. C. 1 44.2 3 4 10 min, 50.degree. C. 5 min, 0.degree. C. 1 38.6 4 4 10 min, 50.degree. C. 3 min, 0.degree. C. 1 24.0 5 5 1 min, 50.degree. C. 5 min, 50.degree. C. 1 41.5 6 5 1 min, 50.degree. C. 5 min, 25.degree. C. 1 21.8 7 5 1 min, 50.degree. C. 8 min, 25.degree. C. 1 24.6 8 5 1 min, 50.degree. C. 8 min, 25.degree. C. 0.1 55.5 9 5 1 min, 50.degree. C. 8 min, 25.degree. C. 0.01 63.7
Subsequent telescoping experiments indicated that 5 was a better nitrosation reagent than 4 for the efficient synthesis of lomustine in flow. This is also true when the synthesis of lomustine were performed separately in glass reactor chips (Table 2 and 3). The final optimized conditions were 1 min reaction time at 50.degree. C. for the first step and 8 min reaction time at 25.degree. C. for the second step (FIG. 6). As the small amount of extracted TEA from the first step minimized the TBN activity, lowering the amount of TEA used led to an increased production of lomustine (Table 4, entries 7-9) such that 1% TEA produced 110 mg (63%) of pure isolate lomustine via this reaction setup (Table 4, entry 9). TLC, NMR (.sup.1H and .sup.13C), MS and MS/MS data of lomustine obtained by this telescoped continuous route were a direct match with values measured for commercially available lomustine.
HPLC-MS was used to certify the purity of the synthesized lomustine and compare the total ion chromatogram (TIC) values of this material with a commercial lomustine standard. The TIC profile fully overlapped with the commercial standard without the appearance of any new byproducts (FIG. 7).
We have developed a rapid continuous synthesis of lomustine using DESI-MS to guide the selection of reaction conditions in the second step of the two step overall transformation. The total residence time is 9 minutes to produce pure lomustine in 63% overall isolated yield compared to over 2 hours to generate a lower product yield using batch conditions..sup.[1b, 2, 16] The two synthetic steps were optimized separately in glass chips and then translated to FEP tubing for telescoped scaling of the whole process. Only one in-line workup step was required for the two-step reaction sequence. Mixed solvents were used in the telescoped reaction to avoid clogging due to the low solubility of 3. tButyl nitrite, 5, was found to be a milder and more efficient nitrosation reagent in this process to enable the isolation of pure lomustine via simple extraction, filtration and washing. This synthesis is a faster and greener process that affords a significant reduction in reaction time, lower waste production, and avoidance of any chromatographic steps. Using this method, 110 mg/hour of lomustine can be produced, equivalent to one dose/h for an agent that is administered orally every 6 weeks. Scale up and in-line recrystallization of lomustine are in progress.
Supporting Information
Materials
All chemicals and reagents were purchased from Sigma-Aldrich (St Louis, Mo.) and used without any purification. Intermediate 3 standard was purchased from 1Click Chemistry, Inc. (Kendall Park, N.J.). Lomustine was purchased from ApexBio (Houston, Tex.).
Liquid Handling Robot
Assay plate set up and sample preparation steps for DESI-MS were done using a Biomek i7 (Beckman Coulter, Inc., Indianapolis, Ind.) dual-bridge liquid handling robot. A 384-tip head was employed to enable simultaneous transfer of 384 samples under the same conditions (speed of aspiration and dispensing, height of pipetting at source and destination positions, pattern of pipetting, etc.). An 8-channel head was used to provide more flexibility in terms of volumes transferred, layout of source and destination places, pipetting height, speed, and reaction stoichiometry. The high capacity deck accommodated all labware (robotic tips, plates, reservoirs) needed for assembling one reaction step. All robotic tips were made of chemically resistant polypropylene and disposable. Polypropylene multi-well plates and reservoirs, as well as custom made Teflon reservoirs were used during the experiments for reagent solutions. Methods were developed and validated using the Biomek point-and-click programming tool. Standard pipetting techniques used in this software were modified to optimize accurate transfer of highly volatile liquids.
Mass Spectrometry
Samples were analyzed using a Thermo Fisher TSQ Quantum Access MAX mass spectrometer that was connected with a Dionex Ultimate 3000 Series Pump and WPS-3000 Autosampler (Thermo Fisher Scientific, Waltham, MA). Electrospray ionization (ESI) analysis in full scan mode was used to monitor each reaction in both positive and negative ion modes. These data were recorded using optimized parameters for the ESI source and MS as follows: spraying solvent, ACN; spray voltage +3.5 kV (positive mode) and -4.0 kV (negative mode); capillary temperature, 250.degree. C.; Sheath gas pressure, 10 psi; scan time, 1 s; Q1 peak width (FWHM), 0.70 Th; micro scans, 3. The autosampler settings were as follows: MS acquire time, 2 min; sample injection volume, 10 .mu.L. Thermo Fisher Xcalibur software was used to process the data from the MS spectrometer.
NMR Analysis
.sup.1H-NMR and .sup.13C-NMR were acquired using a Bruker AV-III-500-HD NMR spectrometer (Billerica, Mass., USA). Samples for NMR were prepared by dissolving .about.5 mg of sample in CDCl.sub.3. MestReNova 10.0 software was used to analyze the .sup.1H-NMR and .sup.13C-NMR.
Chemtrix S1 Microfluidics System.
The two step synthesis of lomustine was performed using a Labtrix 51 microfluidic reactor system (Chemtrix, Ltd, Netherlands). The reactor parts are made of PPS (polyphenylsulfide) and perfluoroelastomer to provide excellent chemical resistance. The system has a temperature range for synthesis from -20.degree. C. to +195.degree. C. and pressures of up to 35 bar. The outer diameter of the fluorinated ethylene propylene (FEP) tube is 1/32 inches and the inner diameter is 150 .mu.L. The 3223 and 3225 microreactors are made of glass and were used for all conducted reactions. The staggered orientated microreactor (SOR) chip 3223 (three inlets and one outlet, volume 10 .mu.L) and 3225 (four inlets and one outlet, volume 10 .mu.L) have a channel width of 300 .mu.m and a channel depth of 120 .mu.m. The Labtrix unit is able to independently pump five syringes into the microreactor seated on a Peltier heating and cooling unit. All the gastight glass syringes were bought separately from Hamilton Company (Hamilton, Reno, Nev.). The tubing and fittings connect the syringes with the selected connection port on the microreactor. All operations were controlled using a Chemtrix GUI software installed on a laptop that was connected to the Labtrix S1 casing with a USB cable.
Liquid-Liquid Separator.
All the liquid-liquid separations were performed using a SEP-10 unit (Zaiput Flow Technologies, Cambridge, Mass.). The separator uses a porous hydrophobic PTFE membrane (OB-900) which allows flow of the organic phase through the membrane. The organic phase wets the membrane while the aqueous phase does not. A built-in pressure controller is used to maintain the appropriate pressure differential that is required of the flow for both sides of the membrane.
DESI-MS Analysis
The DESI-MS evaluation was done following the previously published method of Wleklinski et al.sup.[1] except that the density of reaction spots was 1536 spots/plate instead of 6144/plate using reagents that were pipetted into standard polypropylene 384-well plates using a liquid handling robot (Biomek i7; Beckman-Coulter, US). DESI-MS slides were fabricated from porous PTFE sheets (EMD, Millipore Fluoropore, Saint-Gobain) glued onto a glass support (Foxx Life Sciences). The PTFE sheet was cut with scissors and bonded to the glass slides using spray adhesive (Scotch Spray mount). No signs of interference from the glue was observed. The reagents were mixed at 1:1 stoichiometry in various solvents (EtOAc, THF, DMSO, Toluene, ACN, DCM, EtOH and MeOH) and rhodamine B dye was added to some wells of the plate as a fiducial marker. After the reagents were mixed, 50 nL of the reactions were deposited onto a porous PTFE surface at 1,536 spot density using a magnetic pin tool equipped with slotted transfer pins. DESI-MS data was acquired using a linear ion trap mass spectrometer (LTQ XL; Thermo Scientific, San Jose, Calif.) equipped with a commercial DESI-imaging source (DESI 2D source, Prosolia Inc., Indianapolis, Ind.). The instrument was controlled using Xcalibur v. 4.0 software to run worklists for DESI-MS data acquisition. The DESI spray angle was 55.degree. using MeOH as spray solvent, and with an applied voltage of 5 kV. Mass spectra were acquired at the positive ion mode over the m/z range of 50-500. The DESI-MS imaging lateral resolution was 350 .mu.m. This was achieved using stage speed of 4,376 .mu.m/sec and the instrument scan time of 80 ms. For data processing, data were visualized using an in-house software designed.sup.[1] to automatically search for the m/z values of reactants, intermediates, and lomustine fragments to generate a YES/NO visualization output for each spot in the PTFE plate imaged by DESI-MS. Data files also were combined into .img files using Firefly software (Prosolia Inc., Indianapolis, Ind.). Ion images were plotted using BioMAP (Novartis, freeware). The expected m/z values for the lomustine fragments were plotted and visualized using the BioMAP rainbow false color scale where the minimum and maximum ion intensity values were set to the best contrast for each ion.
HPLC-MS Analysis
HPLC/MS analysis was performed using an Agilent 6545 UPLC/quadrupole time-of-flight (Q-TOF) mass spectrometer (Palo Alto, Calif.), with an Agilent XDB-C18 column (3.5 .mu.m, 150.times.2.1 mm i.d.) and 5 .mu.L injection volume. A binary mobile phase, consisting of solvent systems A and B was used. A was 0.1% formic acid (v/v) in ddH.sub.2O and B was 0.1% formic acid (v/v) in ACN. Isocratic elution of A:B at 95:5 was used, with a column flow rate of 0.3 mL/min. Following the separation, the column effluent was introduced by positive mode electrospray ionization (ESI) into the mass spectrometer. High mass accuracy spectra were collected between 70-1000 m/z. Mass accuracy was improved by continuously infusing Agilent Reference Mass Correction Solution (G1969-85001). The MS detection conditions were: ESI capillary voltage, 3.5 kV; nebulizer gas pressure, 30 psig; gas temperature, 325.degree. C.; drying gas flow rate, 8.0 L/min; fragmentor voltage, 130 V; skimmer, 45 V; and OCT RF V, 750 V.
Carbamylation of Cyclohexylamine
Experimentation.
A solution of cyclohexylamine (1, 500 mmol, 1 equiv) in THF was loaded into 1 mL Hamilton gas tight glass syringe. Triethylamine (TEA) (500 mmol, 1 equiv) and 1-chloro-2-isocyanatoethane (2, 700 mmol, 1.4 equiv) solutions in THF were individually loaded into another two 1 mL Hamilton gas tight glass syringes. Each solution was simultaneously dispensed into the SOR 3225 reactor to engage the reactants. The syringe containing 2 was protected from light by covering it with aluminum foil. The reactions were run at 25.degree. C., 50.degree. C. and 65.degree. C. at residence times of 10 sec, 30 sec, 60 sec and 180 sec. The reactions were monitored by TLC and ESI-MS. Product 3 was collected after evaporation and washing with cold Et2O. The white solid product was stored in the dark at 4.degree. C. Any clogged chips or tubing of the setup was cleaned using THF and EtOH. The subsequent TLC, ESI-MS, MS/MS and NMR (.sup.1H and .sup.13C) analyses were performed after purification.
##STR00002##
NMR
.sup.1H NMR (500 MHz, CDCl.sub.3, ppm): .delta..sub.H=4.84 (t, J=5.85, 1 H), 4.42 (d, J=7.35 Hz, 1 H), 3.62 (t, J=5.60Hz, 2 H), 3.54 (t, J=5.70Hz, 2 H), 3.51-3.45 (m, 1 H), 1.95-1.92 (m, 2 H), 1.72-1.67 (m, 2 H), 1.62-1.58 (m, 1 H), 1.39-1.30 (m, 2 H), 1.19-1.06 (m, 3 H); .sup.13C NMR (500 MHz, CDCl.sub.3, ppm): .delta..sub.c=157.04, 49.29, 45.25, 42.12, 33.88, 25.57, 24.9
##STR00003##
ESI-MS (m/z): 205/207 (M+H.sup.+), 227/229 (M+Na.sup.+), 408/410 (2M.sup.+).
ESI-MS/MS of m/z 205: 205 (M+H.sup.+), 123 (C.sub.3H.sub.7ClN.sub.2O+H.sup.+), 83 (C.sub.6H.sub.11.sup.+) (80 (C.sub.2H.sub.6ClN.sup.+)
Full ESI-MS scan and MS/MS of commercially available 3 and synthesized 3 derived from flow under the reaction conditions of 50.degree. C., 1 min are shown in FIG. 8A and FIG. 8B.
DESI-MS Screening.
Preparation of Reaction (Master) Plates and Stamping on DESI-MS Slides.
Stock solutions of 3 and nitrosation reagents were made at 4 different concentrations (50, 100, 150, 200 mmol) in THF and ACN. Each reagent solution was also prepared (0.1 M) in eight different solvents ((EtOAc, THF, DMSO, Toluene, ACN, DCM, EtOH and MeOH). At first, 20 .mu.L of 3 solution was dispensed into a 384-well master plate and then the corresponding nitrosation reagents added to the plate in a stoichiometry 1:1 using a Beckman i7 liquid handling robot, resulting in a final volume of 40 .mu.L in each well. Moreover, a master plate was made using only commercially available 3 and lomustine to compare the data. Rhodamine was dissolved in acetonitrile (0.25 mg/mL) and transferred to a reservoir. A pin tool fitted with 50 nL pins was used to transfer solutions from the master plates as well as from the Rhodamine reservoir onto the DESI-MS substrates. The master plate was pinned three times in separate locations with reaction mixtures and Rhodamine once, resulting in 1536 density on the microtiter plate used as the DESI-MS substrate.
DESI-MS Outline is shown in FIGS. 9-11
Nitrosation of 1-(2-chloroethyl)-3-cyclohexylurea, 3.
Experimentation.
A solution of 3 (245 mmol, 1 equiv) in 98% formic acid was loaded into a 1 mL Hamilton gas tight glass syringe. NaNO.sub.2 (735 mmol, 3 equiv) solution in MeOH:H.sub.2O (4:1) was separately loaded into another 1 mL Hamilton gas tight glass syringe and dispensed into the SOR 3225 reactor to engage the reactants. The reactions were run at 0.degree. C. at residence times of 30 sec, 1 min, 3 min, 5 min, 8 min, and 10 min. For nitrosation with 5, a solution of 3 in ACN:EtOH (3.7:1) (200 mmol, 1 equiv) and 5 in ACN (600 mmol, 3 equiv, protected from light by covering the syringe with aluminum foil) were loaded into two separate 1 mL Hamilton gas tight glass syringes and dispensed into the SOR 3223 reactor. All the reactions were monitored at two different temperatures (50.degree. C. and 25.degree. C.) at residence times of 30 sec, 1 min, 3 min, 5 min, 8 min, and 10 min. Reaction progress was monitored by TLC and ESI-MS. The reaction mixtures were extracted by Et.sub.2O, evaporated, and dried over anhydrous Na.sub.2SO.sub.4. The crude oily product was purified by dissolving it in hot petroleum ether, hot filtering the solution, and evaporating the filtrate to dryness in vaccuo to give the yellowish solid lomustine that was stored at -20.degree. C. TLC, ESI-MS, MS/MS, NMR (.sup.1H and .sup.13C), and yield analyses were performed after purification Three purification methods were examined to purify the compounds as described in the main manuscript. The NMR spectra for the different purification methods are shown here for comparison.
ESI-MS:
##STR00004##
ESI-MS (m/z): 169 (C.sub.9H.sub.17N.sub.2O.sup.+), 100 (C.sub.6H.sub.13N+H.sup.+), 87(C.sub.3H.sub.7N.sub.2O.sup.+), 83 (C.sub.6H.sub.11.sup.+)
ESI-MS/MS of m/z 169: 169 (C.sub.9H.sub.17N.sub.2O.sup.+), 100 (C.sub.6H.sub.13N+H.sup.+), 87(C.sub.3H.sub.7N.sub.2O.sup.+)
Telescopped Synthesis of Lomustine
Reactors.
For scale up and telescoping of the two steps, fluorinated ethylene propylene (FEP) tubing was used. The outer diameter of the FEP tube was 1/16 inches and the inner diameter is 0.8 mm. The first reactor volume was 5 .mu.L and the second reactor volume was 100 .mu.L.
Experimentation.
Cyclohexaneamine, 1 (1 M, 1 equiv) and triethylamine (0.01 M, 0.01 equiv) were prepared in DCM separately. Next, the two separate solutions were mixed in a 1:1 (v:v) ratio and loaded into a 5 mL Hamilton gastight syringe. Then, a solution of 1-chloro-2-isocyanatoethane, 2 (0.7 M) was prepared in THF and loaded into a 5 mL Hamilton gastight syringe that was covered with aluminum-tape for light protection since it is light sensitive. The two syringes were connected to a T-connection and outlet of the T-connector was connected to the first tubing reactor using micro-tubes, check valves and other connectors (FIG. 17 and FIG. 18). The setup for producing 3 was assembled and placed in a heated H.sub.2O bath that was maintained at 50.degree. C. The outlet of the tube-reactor was connected to a four-way connector, where two of the outlets of the connectors were connected to a 10 mL Hamilton gastight syringe containing H.sub.2O and a 5 mL Hamilton gastight syringe containing DCM. The four-way connector provide sufficient mixing for the extraction of the triethylamine base in the aqueous phase and leaving 3 in the organic phase. The fourth outlet was connected to the liquid-liquid separator (SEP-10) in which the DCM passes through the membrane carrying with it 3 to the next reaction step. The outlet of the aqueous phase from the separator was connected to a waste vial. For using sodium nitrite as a nitrosation reagent, the outlet of the organic phase was connected to a four-way connector. Sodium nitrite, 4 (1.5 M, 3 equiv) solution in THF and formic acid were loaded into two separate 5 mL Hamilton gastight syringes and connected to the a four-way connector. The outlet of the four-way connector is connected to the second tubing reactor. When using TBN, 5 as a nitrosation reagent, we doubled the concentration of the starting material. The outlet of the organic phase from liquid-liquid extractor was connected to a T-connector, where one outlet was connected to a 5 mL Hamilton gastight syringe containing tert-Butyl nitrite, 5 (5 M) in ACN. The outlet of the T-connector was connected to the second tubing reactor. The second reactor was placed in a H.sub.2O bath with a constant temperature of 25.degree. C. and the outlet of this reactor was connected to a collection vial. The reactions were monitored by TLC and ESI-MS. The purification and analyses were conducted as described above, except that HPLC-MS analysis was performed to evaluate the purity of the product.
TLC and Purification. Reaction progress was monitored by TLC using 1:1 EtOAc:Hexanes as eluent. Lomustine was visualized under shortwave UV light (230 nm), while 3 was observed after staining with ninhydrin solution and heating. The extraction and purification was conducted by taking 500 .mu.L from the collection vial and washing it with 2 mL of H.sub.2O and 2 mL of Et.sub.2O and extracted three times. The combined organic layers were dried using anhydrous NaSO.sub.4. The Et.sub.2O was evaporated and the yellowish oil/solid was dissolved in hot petroleum ether, hot filtered, and the filtrate was removed under vacuum. The resulting solid was recrystallized from petroleum ether.
NMR
.sup.1H NMR (500 MHz, CDCl.sub.3, ppm): .delta..sub.H=6.78 (s, 1H), 4.18 (t, J=7.5, 2 H), 3.92-3.84 (m, 1H), 3.50 (t, J=7.5 Hz, 2 H), 2.07-2.04 (m, 2 H), 1.79-1.75 (m, 2 H), 1.68-1.63 (m, 1 H), 1.45-1.39 (m, 2 H), 1.32-1.24 (m, 3 H); .sup.13C NMR (500 MHz, CDCl.sub.3, ppm): .delta..sub.C=151.78, 49.98, 40.03, 38.89, 33.09, 25.39, 24.76
Proton NMR of lomustine from flow synthesis, or Carbon NMR of lomustine, 6 from flow synthesis are available. Comparison of Proton NMR of lomustine, 6 in flow synthesis with commercially available lomustine, 6, and comparison of carbon NMR of lomustine, 6 in flow synthesis with commercially available lomustine, 6 are available upon request.
HPLC/MS-MS Analysis
HPLC/MS analysis was performed on an Agilent 6545 UPLC/quadrupole time-of-flight (Q-TOF) mass spectrometer (Palo Alto, Calif.), with an Agilent XDB-C18 column (3.5 .mu.m, 150.times.2.1 mm i.d) and 5 uL injection volume. A binary mobile phase consisting of solvent systems A and B were used. A was 0.1% formic acid (v/v) in ddH.sub.2O and B was 0.1% formic acid (v/v) in acetonitrile. Isocratic elution of A:B at 95:5 was used, with a column flow rate of 0.3 mL/min. Following the separation, the column effluent was introduced by positive mode electrospray ionization (ESI) into the mass spectrometer. High mass accuracy spectra was collected between 70-1000 m/z. Mass accuracy was improved by continuously infusing Agilent Reference Mass Correction Solution (G1969-85001). ESI capillary voltage was 3.5 kV, nebulizer gas pressure was 30 psig, gas temperature was 325.degree. C., drying gas flow rate was 8.0 L/min, fragmentor voltage was 130 V, skimmer was 45 V, and OCT RF V was 750 V.
Full MS from HPLC-MS/MS and comparison between synthesized lomustine and commercially available lomustine are available upon request.
Flow Rates:
Flow Rates for 1.sup.st step Reaction in Labtrix S1 system:
Chemtrix reactor chip: 3225, 10 .mu.L, pressure: ambient pressure
TABLE-US-00005 R1 R3 Cyclohexane R2 2-Chloroethyl Residence amine, Triethylamine isocyanate, Time Temperature 1 .mu.L/min .mu.L/min 2 .mu.L/min in min .degree. C. 20 20 20 0.167 50 6.67 6.67 6.67 0.5 50 3.33 3.33 3.33 1 50 1.11 1.11 1.11 3 50 0.67 0.67 0.67 5 50 0.417 0.417 0.417 8 50 0.333 0.333 0.333 10 50
Flow Rates for 2.sup.nd step Reaction using NaNO.sub.2/HCO.sub.2H, 4 in Labtrix S1 system: Chemtrix reactor chip: 3225, 10 .mu.L, pressure: ambient pressure
TABLE-US-00006 R2 R3 Sodium Formic R1 Nitrite Acid Residence Time 3 .mu.L/min .mu.L/min .mu.L/min in min Temperature .degree. C. 6.67 6.67 6.67 0.5 0 3.33 3.33 3.33 1 0 1.11 1.11 1.11 3 0 0.67 0.67 0.67 5 0 0.417 0.417 0.417 8 0 0.333 0.333 0.333 10 0
Flow Rates for 2.sup.nd step Reaction using TBN, 5 in Labtrix S1 system. Chemtrix reactor chip: 3223, 10 .mu.L, pressure: ambient pressure
TABLE-US-00007 R2 R1 tert-Butyl nitrite Residence Time 3 .mu.L/min .mu.L/min in min Temperature .degree. C. 10 10 0.5 50 5 5 1 50 1.67 1.67 3 50 1 1 5 50 0.625 0.625 8 50 0.5 0.5 10 50 10 10 0.5 25 5 5 1 25 1.67 1.67 3 25 1 1 5 25 0.625 0.625 8 25 0.5 0.5 10 25
Telescoped Reaction in Tube:
Nitrosation Reagent: NaNO.sub.2/HCO.sub.2H , 4
TABLE-US-00008 R1 1 + Extraction Triethyl Reactor Extraction step, R3 R3 amine volume, step, H.sub.2O DCM NaNO.sub.2 HCO.sub.2H .mu.L/min R2 2 .mu.L/min cm Step 1 .mu.L/min .mu.L/min .mu.L/min .mu.L/min Step 2 12.56 12.56 Step 1: 5 1 min, 50.24 25.12 25.12 25.12 5 min, Step 2: 50.degree. C. 0.degree. C. 100 12.56 12.56 Step 1: 10 2 min, 50.24 25.12 25.12 25.12 5 min, Step 2: 50.degree. C. 0.degree. C. 100 12.56 12.56 Step 1: 50 10 min, 50.24 25.12 25.12 25.12 5 min, Step 2: 50.degree. C. 0.degree. C. 100 12.56 12.56 Step 1: 50 10 min, 50.24 25.12 50.24 67.02 3 min, Step 2: 50.degree. C. 0.degree. C. 100
Nitrosation Reagent: TBN , 5
Reactor volume: Step 1=5 cm; Step 2=100 cm
TABLE-US-00009 R1 Extraction 1 + Triethyl Extraction step, R3 amine step, H.sub.2O DCM TBN .mu.L/min R2 2 .mu.L/min Step 1 .mu.L/min .mu.L/min .mu.L/min Step 2 12.56 12.56 1 min, 50.degree. C. 50.24 25.12 50.2 5 min, 25/50.degree. C. 12.56 12.56 1 min, 50.degree. C. 50.24 25.12 12.56 8 min, 25.degree. C.
TABLE-US-00010 DESI-MS data of the nitrosation reaction using 4 in different stoichiometries Normalized Normalized Starting Starting Product Intensity Intensity Intensity Normalized Inte- nsity no of Material Material Solvent Stoichiometry m/z (Average) Stdev (max) (Average- ) Stdev (max) spots, n 3 4 Acetonitrile 150 169.2 213.3 191.6 934.1 0.025 0.030 0.166 144 3 4 Acetonitrile 150 234.7 8.0 6.7 45.1 0.001 0.001 0.007 144 3 4 Acetonitrile 150 239.4 1206.9 1349.7 6575.7 0.069 0.066 0.284 144 3 4 Acetonitrile 150 241.0 390.4 412.9 1980.9 0.022 0.020 0.088 144 3 4 Acetonitrile 150 250.4 143.7 184.3 923.0 0.008 0.005 0.024 144 3 4 Acetonitrile 150 251.2 34.7 40.6 205.3 0.002 0.001 0.006 144 3 4 Acetonitrile 150 337.4 831.0 707.6 4214.5 0.043 0.016 0.100 144 3 4 Acetonitrile 150 373.4 195.1 203.4 1017.1 0.010 0.006 0.026 144 3 4 Acetonitrile 100 169.2 428.3 487.1 3538.6 0.021 0.019 0.124 144 3 4 Acetonitrile 100 234.7 19.3 15.1 80.5 0.002 0.002 0.008 144 3 4 Acetonitrile 100 239.4 2066.8 2519.0 12435.6 0.061 0.049 0.220 144 3 4 Acetonitrile 100 241.0 664.8 776.1 3676.1 0.020 0.015 0.070 144 3 4 Acetonitrile 100 250.4 179.0 200.3 1072.1 0.006 0.003 0.016 144 3 4 Acetonitrile 100 251.2 50.4 57.4 348.6 0.002 0.001 0.005 144 3 4 Acetonitrile 100 337.4 1541.9 1537.4 9588.3 0.050 0.018 0.105 144 3 4 Acetonitrile 100 373.4 261.3 307.9 1751.0 0.008 0.004 0.022 144 3 4 Acetonitrile 50 169.2 53.9 58.1 322.0 0.018 0.019 0.105 144 3 4 Acetonitrile 50 234.7 11.7 16.2 177.5 0.004 0.002 0.015 144 3 4 Acetonitrile 50 239.4 382.9 825.0 5452.0 0.045 0.062 0.326 144 3 4 Acetonitrile 50 241.0 124.8 257.2 1692.1 0.016 0.019 0.101 144 3 4 Acetonitrile 50 250.4 21.3 24.1 145.3 0.005 0.002 0.012 144 3 4 Acetonitrile 50 251.2 6.3 6.7 37.3 0.001 0.001 0.004 144 3 4 Acetonitrile 50 337.4 116.2 130.7 681.0 0.020 0.015 0.075 144 3 4 Acetonitrile 50 373.4 21.0 24.5 133.5 0.004 0.003 0.014 144 3 4 Acetonitrile 200 169.2 134.4 122.3 642.8 0.007 0.002 0.012 144 3 4 Acetonitrile 200 234.7 7.7 8.7 57.9 0.001 0.001 0.003 144 3 4 Acetonitrile 200 239.4 2162.9 2123.7 15812.4 0.119 0.056 0.297 144 3 4 Acetonitrile 200 241.0 608.6 518.4 4106.0 0.035 0.017 0.090 144 3 4 Acetonitrile 200 250.4 39.5 26.3 138.1 0.003 0.001 0.006 144 3 4 Acetonitrile 200 251.2 20.5 49.7 382.9 0.001 0.000 0.002 144 3 4 Acetonitrile 200 337.4 485.0 296.1 1620.7 0.028 0.007 0.042 144 3 4 Acetonitrile 200 373.4 73.1 51.3 324.3 0.004 0.001 0.007 144 3 4 THF 200 169.2 1024.5 744.7 6746.3 0.041 0.059 0.443 144 3 4 THF 200 234.7 30.8 36.3 222.7 0.002 0.001 0.006 144 3 4 THF 200 239.4 1067.2 1527.8 7808.4 0.022 0.034 0.174 144 3 4 THF 200 241.0 333.9 480.7 2373.1 0.007 0.010 0.057 144 3 4 THF 200 250.4 749.2 367.0 2331.3 0.011 0.004 0.022 144 3 4 THF 200 251.2 237.1 227.8 1263.0 0.003 0.002 0.009 144 3 4 THF 200 337.4 13765.9 7680.8 50455.2 0.186 0.054 0.343 144 3 4 THF 200 373.4 1096.5 568.4 3314.2 0.016 0.005 0.038 144 3 4 THF 150 169.2 680.3 705.5 3251.7 0.036 0.033 0.202 144 3 4 THF 150 234.7 43.2 44.1 295.0 0.003 0.001 0.009 144 3 4 THF 150 239.4 1106.3 2107.4 12553.7 0.021 0.031 0.143 144 3 4 THF 150 241.0 366.5 601.2 2985.4 0.008 0.009 0.044 144 3 4 THF 150 250.4 349.0 338.2 1672.1 0.007 0.004 0.020 144 3 4 THF 150 251.2 92.1 92.6 353.0 0.002 0.001 0.008 144 3 4 THF 150 337.4 2590.0 3621.7 15618.2 0.044 0.051 0.185 144 3 4 THF 150 373.4 291.1 374.6 1736.1 0.006 0.006 0.024 144 3 4 THF 100 169.2 1152.9 849.2 4316.7 0.053 0.038 0.236 144 3 4 THF 100 234.7 40.4 42.7 243.0 0.002 0.001 0.014 144 3 4 THF 100 239.4 486.4 1259.8 8600.9 0.008 0.019 0.149 144 3 4 THF 100 241.0 166.2 407.2 2879.3 0.003 0.006 0.044 144 3 4 THF 100 250.4 1070.5 451.4 2449.5 0.013 0.004 0.020 144 3 4 THF 100 251.2 260.9 172.7 1492.2 0.003 0.001 0.011 144 3 4 THF 100 337.4 14213.4 9456.4 39713.7 0.157 0.076 0.339 144 3 4 THF 100 373.4 1262.2 675.0 3233.6 0.016 0.006 0.030 144 3 4 THF 50 169.2 708.7 560.9 3726.2 0.060 0.034 0.211 144 3 4 THF 50 234.7 46.1 56.3 341.8 0.003 0.002 0.020 144 3 4 THF 50 239.4 91.6 164.1 938.4 0.003 0.003 0.023 144 3 4 THF 50 241.0 54.0 70.6 369.5 0.003 0.003 0.015 144 3 4 THF 50 250.4 783.5 422.1 2172.0 0.012 0.003 0.018 144 3 4 THF 50 251.2 227.3 310.1 2172.0 0.003 0.002 0.014 144 3 4 THF 50 337.4 10050.7 9141.9 52775.8 0.125 0.074 0.268 144 3 4 THF 50 373.4 911.9 744.6 4270.9 0.012 0.006 0.022 144 Rhodamine 443.3 12213.1 8739.2 70545.6 0.491 0.101 0.690 384 DESI-MS data of the nitrosation reaction using 4 as a nitrosation reagent Normalized Normalized Starting Starting Product Intensity Intensity Intensity Normalized Inten- sity No of Material Material Solvent m/z (Average) Stdev (max) (Average) Stdev (max) - spots, n 3 4 Toluene 169.2 2554.4 5564.3 36332.5 0.406 0.173 0.719 144 3 4 Toluene 205.7 7.2 10.4 63.4 0.003 0.003 0.015 144 3 4 Toluene 234.7 2.8 4.5 24.2 0.001 0.001 0.004 144 3 4 Toluene 239.4 2.7 5.8 38.3 0.000 0.001 0.007 144 3 4 Toluene 241.0 2.5 5.8 38.1 0.000 0.001 0.004 144 3 4 Toluene 250.4 1.4 4.4 43.3 0.000 0.001 0.004 144 3 4 Toluene 251.2 30.9 134.5 1279.6 0.002 0.006 0.039 144 3 4 Toluene 337.4 100.7 267.5 1413.6 0.007 0.018 0.146 144 3 4 Toluene 373.4 440.4 1209.9 8746.2 0.026 0.043 0.215 144 3 4 Acetonitrile 169.2 861.8 2266.6 24796.5 0.471 0.136 0.774 144 3 4 Acetonitrile 205.7 6.4 12.7 84.5 0.003 0.004 0.026 144 3 4 Acetonitrile 234.7 0.8 2.0 15.5 0.000 0.001 0.007 144 3 4 Acetonitrile 239.4 1.1 2.4 17.8 0.000 0.000 0.002 144 3 4 Acetonitrile 241.0 6.9 18.6 151.1 0.001 0.002 0.008 144 3 4 Acetonitrile 250.4 5.3 13.5 106.8 0.001 0.002 0.012 144 3 4 Acetonitrile 251.2 330.2 517.7 2676.7 0.039 0.035 0.213 144 3 4 Acetonitrile 337.4 46.4 73.3 493.7 0.005 0.006 0.033 144 3 4 Acetonitrile 373.4 651.5 1237.0 9488.0 0.065 0.035 0.190 144 3 4 DMSO 169.2 4904.5 8789.6 53558.7 0.355 0.137 0.642 144 3 4 DMSO 205.7 25.7 26.7 151.4 0.002 0.002 0.012 144 3 4 DMSO 234.7 1.8 2.3 13.1 0.000 0.001 0.002 144 3 4 DMSO 239.4 17.1 26.6 228.2 0.001 0.000 0.002 144 3 4 DMSO 241.0 5.2 6.1 26.9 0.000 0.000 0.002 144 3 4 DMSO 250.4 1.6 9.6 110.6 0.000 0.000 0.001 144 3 4 DMSO 251.2 101.2 179.2 1054.9 0.002 0.003 0.021 144 3 4 DMSO 337.4 443.8 791.7 4937.7 0.009 0.011 0.053 144 3 4 DMSO 373.4 2602.8 3506.2 30323.8 0.049 0.033 0.232 144 3 4 THF 169.2 1604.6 1828.4 9837.0 0.343 0.165 0.671 144 3 4 THF 205.7 14.3 16.2 107.3 0.002 0.003 0.014 144 3 4 THF 234.7 9.2 20.8 141.7 0.000 0.001 0.003 144 3 4 THF 239.4 68.5 72.4 381.6 0.002 0.002 0.007 144 3 4 THF 241.0 22.7 22.2 99.5 0.001 0.000 0.004 144 3 4 THF 250.4 4.1 7.3 32.2 0.000 0.000 0.001 144 3 4 THF 251.2 254.1 351.0 1963.4 0.008 0.008 0.040 144 3 4 THF 337.4 122.6 116.9 840.1 0.003 0.002 0.019 144 3 4 THF 373.4 3643.6 3629.0 24476.2 0.079 0.036 0.222 144 3 4 Ethanol 169.2 1939.9 2663.0 23988.5 0.315 0.159 0.644 144 3 4 Ethanol 205.7 18.0 22.0 108.9 0.003 0.003 0.017 144 3 4 Ethanol 234.7 9.7 28.9 313.9 0.001 0.001 0.003 144 3 4 Ethanol 239.4 3.1 3.4 20.9 0.000 0.000 0.002 144 3 4 Ethanol 241.0 4.1 3.4 17.3 0.000 0.000 0.003 144 3 4 Ethanol 250.4 3.5 3.4 17.0 0.000 0.000 0.002 144 3 4 Ethanol 251.2 53.0 37.6 152.7 0.002 0.001 0.006 144 3 4 Ethanol 337.4 263.7 193.3 1112.6 0.009 0.005 0.049 144 3 4 Ethanol 373.4 553.9 508.5 2842.8 0.024 0.010 0.074 144 3 4 DCM 169.2 463.4 643.7 3574.7 0.429 0.135 0.733 144 3 4 DCM 205.7 6.8 10.5 72.0 0.005 0.008 0.056 144 3 4 DCM 234.7 1.8 9.0 77.5 0.000 0.001 0.006 144 3 4 DCM 239.4 1.6 3.8 22.9 0.000 0.001 0.005 144 3 4 DCM 241.0 6.9 17.3 111.6 0.001 0.003 0.027 144 3 4 DCM 250.4 4.4 11.7 91.1 0.001 0.002 0.009 144 3 4 DCM 251.2 68.1 211.5 1683.3 0.009 0.014 0.103 144 3 4 DCM 337.4 32.4 77.4 572.0 0.007 0.013 0.060 144 3 4 DCM 373.4 381.0 586.2 3159.2 0.072 0.062 0.206 144 3 4 Ethyl Acetate 169.2 2710.5 3037.9 15437.7 0.325 0.124 0.586 144 3 4 Ethyl Acetate 205.7 23.4 33.0 173.7 0.005 0.006 0.042 144 3 4 Ethyl Acetate 234.7 7.6 29.1 260.3 0.001 0.002 0.017 144 3 4 Ethyl Acetate 239.4 2.0 3.6 16.7 0.000 0.000 0.002 144 3 4 Ethyl Acetate 241.0 4.7 9.6 73.9 0.000 0.000 0.002 144 3 4 Ethyl Acetate 250.4 3.5 7.4 58.3 0.000 0.000 0.002 144 3 4 Ethyl Acetate 251.2 115.4 304.2 2298.6 0.005 0.009 0.058 144 3 4 Ethyl Acetate 337.4 34.3 65.0 512.9 0.002 0.002 0.012 144 3 4 Ethyl Acetate 373.4 2627.7 3123.4 13965.7 0.104 0.081 0.247 144 3 4 Methanol 169.2 1516.5 3046.6 34891.7 0.369 0.172 0.780 144 3 4 Methanol 205.7 15.8 21.8 167.1 0.003 0.004 0.028 144 3 4 Methanol 234.7 1.3 1.6 7.2 0.000 0.001 0.002 144 3 4 Methanol 239.4 1.6 2.1 15.3 0.000 0.000 0.002 144 3 4 Methanol 241.0 2.7 4.0 22.3 0.000 0.000 0.002 144 3 4 Methanol 250.4 1.3 2.3 14.7 0.000 0.000 0.002 144 3 4 Methanol 251.2 19.6 25.3 173.7 0.001 0.001 0.006 144 3 4 Methanol 337.4 288.8 315.4 1590.6 0.013 0.010 0.052 144 3 4 Methanol 373.4 510.8 507.8 2698.7 0.027 0.014 0.072 144 3 443.3 12333.9 13328.7 384.0 0.354 0.083 0.646 384 3 5 Ethyl Acetate 169.2 41.1 47.5 326.1 0.006 0.005 0.028 144 3 5 Ethyl Acetate 205.7 26.2 10.1 59.8 0.004 0.001 0.011 144 3 5 Ethyl Acetate 234.7 46.6 22.4 225.0 0.007 0.002 0.026 144 3 5 Ethyl Acetate 239.4 54.1 121.6 768.5 0.005 0.006 0.027 144 3 5 Ethyl Acetate 241.0 18.6 34.1 215.5 0.002 0.002 0.010 144 3 5 Ethyl Acetate 250.4 243.1 89.4 605.5 0.038 0.008 0.076 144 3 5 Ethyl Acetate 251.2 239.7 89.6 605.5 0.039 0.010 0.112 144 3 5 Ethyl Acetate 337.4 6.5 4.5 38.0 0.001 0.001 0.004 144 3 5 Ethyl Acetate 373.4 19.5 23.7 107.1 0.003 0.004 0.022 144 3 5 Ethanol 169.2 1059.5 1113.0 5710.9 0.047 0.027 0.128 144 3 5 Ethanol 205.7 44.2 47.6 224.7 0.002 0.001 0.006 144 3 5 Ethanol 234.7 22.5 14.2 73.3 0.001 0.000 0.002 144 3 5 Ethanol 239.4 437.1 485.3 2453.7 0.016 0.012 0.091 144 3 5 Ethanol 241.0 155.0 199.6 1496.5 0.005 0.004 0.027 144 3 5 Ethanol 250.4 75.4 57.5 339.3 0.003 0.002 0.009 144 3 5 Ethanol 251.2 33.5 21.6 123.8 0.001 0.000 0.003 144 3 5 Ethanol 337.4 28.4 16.3 85.8 0.001 0.000 0.003 144 3 5 Ethanol 373.4 547.1 727.2 4628.6 0.021 0.014 0.065 144 3 5 THF 169.2 77.4 147.1 824.4 0.008 0.009 0.066 144 3 5 THF 205.7 23.6 18.8 81.5 0.004 0.002 0.009 144 3 5 THF 234.7 55.6 74.9 771.9 0.008 0.004 0.020 144 3 5 THF 239.4 98.5 215.8 1279.6 0.008 0.010 0.046 144 3 5 THF 241.0 31.2 63.7 401.6 0.002 0.003 0.013 144 3 5 THF 250.4 278.7 221.3 1061.8 0.046 0.021 0.088 144 3 5 THF 251.2 285.5 222.9 1061.8 0.048 0.020 0.088 144 3 5 THF 337.4 5.7 5.9 46.3 0.001 0.001 0.007 144 3 5 THF 373.4 23.1 41.3 258.5 0.003 0.004 0.021 144 3 5 DCM 169.2 357.7 408.6 2428.0 0.028 0.018 0.076 144 3 5 DCM 205.7 56.1 22.9 159.1 0.005 0.002 0.011 144 3 5 DCM 234.7 111.7 69.4 561.9 0.010 0.003 0.017 144 3 5 DCM 239.4 255.9 372.5 1692.9 0.019 0.016 0.055 144 3 5 DCM 241.0 76.3 115.2 670.7 0.006 0.005 0.018 144 3 5 DCM 250.4 538.9 212.4 1632.1 0.057 0.017 0.086 144 3 5 DCM 251.2 551.4 202.1 1632.1 0.058 0.015 0.086 144 3 5 DCM 337.4 3.3 3.5 28.0 0.000 0.000 0.002 144 3 5 DCM 373.4 102.8 212.9 2118.2 0.007 0.007 0.038 144 3 5 Toluene 169.2 292.1 271.9 1570.8 0.025 0.016 0.098 144 3 5 Toluene 205.7 25.0 15.0 107.4 0.003 0.001 0.011 144 3 5 Toluene 234.7 37.9 32.5 268.9 0.005 0.001 0.012 144 3 5 Toluene 239.4 179.6 173.3 1095.6 0.012 0.007 0.034 144 3 5 Toluene 241.0 57.9 59.1 394.2 0.004 0.002 0.011 144 3 5 Toluene 250.4 257.8 80.3 515.9 0.040 0.006 0.054 144 3 5 Toluene 251.2 257.5 78.3 463.2 0.040 0.006 0.054 144 3 5 Toluene 337.4 5.5 4.5 24.3 0.001 0.000 0.002 144 3 5 Toluene 373.4 136.8 119.1 632.3 0.012 0.008 0.045 144 3 5 DMSO 169.2 3063.0 4126.1 20263.6 0.192 0.113 0.754 144 3 5 DMSO 205.7 28.5 52.8 347.2 0.003 0.002 0.014 144 3 5 DMSO 234.7 19.5 14.6 117.7 0.004 0.003 0.033 144 3 5 DMSO 239.4 30.2 77.6 745.0 0.002 0.003 0.016 144 3 5 DMSO 241.0 10.1 24.1 233.9 0.001 0.001 0.005 144 3 5 DMSO 250.4 108.8 94.5 872.6 0.020 0.008 0.039 144 3 5 DMSO 251.2 105.8 71.3 325.7 0.022 0.007 0.040 144 3 5 DMSO 337.4 5.5 8.9 57.0 0.001 0.001 0.007 144 3 5 DMSO 373.4 2038.4 3628.2 27327.2 0.095 0.060 0.239 144 3 5 Methanol 169.2 1526.0 1662.0 6465.5 0.070 0.055 0.179 144 3 5 Methanol 205.7 46.2 46.0 249.9 0.002 0.001 0.006 144 3 5 Methanol 234.7 24.3 25.1 113.3 0.001 0.000 0.003 144 3 5 Methanol 239.4 483.9 642.3 3403.1 0.019 0.017 0.090 144 3 5 Methanol 241.0 194.0 382.7 3346.8 0.006 0.006 0.029 144 3 5 Methanol 250.4 71.5 104.6 744.7 0.003 0.002 0.010 144 3 5 Methanol 251.2 43.1 65.4 548.5 0.002 0.001 0.006 144 3 5 Methanol 337.4 26.3 29.5 167.7 0.001 0.001 0.004 144 3 5 Methanol 373.4 1043.9 1687.0 11607.0 0.039 0.035 0.128 144 3 5 Acetonitrile 169.2 3290.0 3100.9 15073.0 0.150 0.056 0.313 144 3 5 Acetonitrile 205.7 33.4 12.5 102.0 0.004 0.001 0.008 144 3 5 Acetonitrile 234.7 57.2 44.7 317.8 0.006 0.002 0.012 144 3 5 Acetonitrile 239.4 187.1 221.0 1440.7 0.008 0.004 0.025 144 3 5 Acetonitrile 241.0 64.9 87.3 669.7 0.003 0.001 0.008 144 3 5 Acetonitrile 250.4 299.6 100.6 629.8 0.038 0.010 0.063 144 3 5 Acetonitrile 251.2 311.5 96.9 629.8 0.040 0.009 0.063 144 3 5 Acetonitrile 337.4 8.2 9.1 50.7 0.000 0.000 0.005 144 3 5 Acetonitrile 373.4 1274.8 1544.7 12113.3 0.049 0.025 0.120 144 3 443.3 40997.0 24891.7 384.0 0.654 0.118 0.807 384
REFERENCES
[1] a) T. Chakkath, S. Lavergne, T. M. Fan, D. Bunick, L. Dirikolu, Vet. Sci. 2015, 2, 52-68; b) L. Dirikolu, T. Chakkath, T. Fan, N. R. Mente, J. Anal. Toxicol. 2009, 33, 595-603. [2] J. W. Lown, S. M. S. Chauhan, J. Org. Chem. 1981, 46, 5309-5321. [3] B. Kaina, M. Christmann, S. Naumann, W. P. Roos, DNA Repair 2007, 6, 1079-1099. [4] F. Y. Lee, P. Workman, J. T. Roberts, N. M. Bleehen, Cancer Chemother. Pharmacol. 1985, 14, 125-131. [5] J. Funaro, H. Friedman, M. Weant, Cancer Lett. 2017, 13-14. [6] a) D. Webb, T. F. Jamison, Chem. Sci. 2010, 1, 675-680; b) J. Wegner, S. Ceylan, A. Kirschning, Chem. Commun. 2011, 47, 4583-4592; c) J. I. Yoshida, H. Kim, A. Nagaki, Chemsuschem 2011, 4, 331-340; d) J.-i. Yoshida, Y. Takahashi, A. Nagaki, Chem. Commun. 2013, 49, 9896-9904; e) V. Hessel, D. Kralisch, N. Kockmann, T. Noel, Q. Wang, ChemSusChem 2013, 6, 746-789; f) K. F. Jensen, B. J. Reizman, S. G. Newman, Lab Chip 2014, 14, 3206-3212; g) B. Gutmann, D. Cantillo, C. O. Kappe, Angew. Chem. Int. Ed. 2015, 54, 6688-6728; h) D. Cambie, C. Bottecchia, N. J. W. Straathof, V. Hessel, T. Noel, Chem. Rev. 2016, 116, 10276-10341; i) J. S. Poh, S. Makai, T. von Keutz, D. N. Tran, C. Battilocchio, P. Pasau, S. V. Ley, Angew. Chem. Int. Ed. 2017, 56, 1864-1868; j) Z. Yu, X. Ye, Q. Xu, X. Xie, H. Dong, W. Su, Org. Process Res. Dev. 2017, 21, 1644-1652; k) J. Britton, C. L. Raston, Chem. Soc. Rev. 2017, 46, 1250-1271;1) M. Berton, L. Huck, J. Alcazar, Nat. Protoc. 2018, 13, 324. [7] a) H. V., Chem. Eng. Technol. 2009, 32, 1655-1681; b) N. Zaborenko, M. W. Bedore, T.
F. Jamison, K. F. Jensen, Org. Process Res. Dev. 2011, 15, 131-139; c) C. Wiles, P. Watts, Chem. Commun. 2011, 47, 6512-6535; d) J. Wegner, S. Ceylan, A. Kirschning, Adv. Synth. Catal. 2012, 354, 17-57; e) D. R. Snead, T. F. Jamison, Chem. Sci. 2013, 4, 2822-2827; f) C. Len, S. Bruniaux, F. Delbecq, V. Parmar, Catalysts 2017, 7, 146; g) L. J. A. M., M. P. D., B. R. L., J. T. F., Chem. Rec. 2017, 17, 667-680. [8] P. D. Morse, T. F. Jamison, Angew. Chem. Int. Ed. 2017, 56, 13999-14002. [9] a) T. Gustafsson, F. Ponten, P. H. Seeberger, Chem. Commun. 2008, 1100-1102; b) M. D. Hopkin, I. R. Baxendale, S. V. Ley,Org. Biomol. Chem. 2013, 11, 1822-1839; c) B. J. Deadman, M. D. Hopkin, I. R. Baxendale, S. V. Ley, Org. Biomol. Chem. 2013, 11, 1766-1800; d) P. R. D. Murray, D. L. Browne, J. C. Pastre, C. Butters, D. Guthrie, S. V. Ley, Org. Process Res. Dev. 2013, 17, 1192-1208; e) P. Zhang, M. G. Russell, T. F. Jamison, Org. Process Res. Dev. 2014, 18, 1567-1570; f) D. R. Snead, T. F. Jamison, Angew. Chem. Int. Ed. 2015, 54, 983-987; g) A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J. C. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong, P. Zhang, Science 2016, 352, 61-67; h) H. Lin, C. Dai, T. F. Jamison, K. F. Jensen, Angew. Chem. Int. Ed. 2017, 56, 8870-8873; i) P. Zhang, N. Weeranoppanant, D. A. Thomas, K. Tahara, T. Stelzer, M. G. Russell, M. O'Mahony, A. S. Myerson, H. Lin, L. P. Kelly, K. F. Jensen, T. F. Jamison, C. Dai, Y. Cui, N. Briggs, R. L. Beingessner, A. Adamo, Chem. Eur. J. 2018, 24, 2776-2784; j) C. E. Falcone, Z. Jaman, M. Wleklinski, A. Koswara, D. H. Thompson, R. G. Cooks, Analyst 2017, 142, 2836-2845; k) B. P. Loren, M. Wleklinski, A. Koswara, K. Yammine, Y. Hu, Z. K. Nagy, D. H. Thompson, R. G. Cooks, Chem. Sci. 2017, 8, 4363-4370; 1) M. Wleklinski, C. E. Falcone, B. P. Loren, Z. Jaman, K. Iyer, H. S. Ewan, S.-H. Hyun, D. H. Thompson, R. G. Cooks, Eur. J. Org. Chem. 2016, 2016, 5480-5484; m) H. S. Ewan, K. Iyer, S.-H. Hyun, M. Wleklinski, R. G. Cooks, D. H. Thompson, Org. Process Res. Dev. 2017, 21, 1566-1570. [10] a) R. Elena, R. Anna, G. Stefania, P. Daniele, M. Marisa, Chem. Eur. J. 2011, 17, 6221-6226; b) B. Ahmed-Omer, A. J. Sanderson, Org. Biomol. Chem. 2011, 9, 3854-3862. [11] a) S. Newton, C. F. Carter, C. M. Pearson, L. D. Alves, H. Lange, P. Thansandote, S. V. Ley, Angew. Chem. Int. Ed. 2014, 53, 4915-4920; b) P. Ludmila, D. S. B. Joao, H. Zsofia, B. Florine, C. Florian, L. Andrew, Angew. Chem. Int. Ed. 2016, 55, 13576-13579. [12] B. Make, M. J. M., S. Simon, R. J. H., H. A. B., Chem. Eur. J. 2010, 16, 11471-11480. [13] a) K. F. Jensen, MRS Bull. 2006, 31, 101-107; b) K. Geyer, T. Gustafsson, P. H. Seeberger, Synlett 2009, 2382-2391; c) H. Kim, K. I. Min, K. Inoue, D. J. Im, D. P. Kim, J. Yoshida, Science 2016, 352, 691-694; d) K. Troshin, J. F. Hartwig, Science 2017, 357, 175-181; e) M. Wleklinski, B. P. Loren, C. R. Ferreira, Z. Jaman, L. Avramova, T. J. P. Sobreira, D. H. Thompson, R. G. Cooks, Chem. Sci. 2018, 9, 1647-1653; f) Z. Jaman, A. Mufti, S. Sah, L. Avramova, D. H. Thompson, Chem. Eur. J. 2018, 24, 9546-9554. [14] a) Y. Xin, B. R. M., C. R. Graham, Angew. Chem. Int. Ed. 2016, 55, 12960-12972; b) D. R. Ifa, N. E. Manicke, A. L. Dill, R. G. Cooks, Science 2008, 321, 805-805. [15] a) E. S. Freeman, J. Phys. Chem. 1956, 60, 1487-1493; b) J. B. Fox, F. B. Suhre, CRC Cr. Rev. Anal. Chem 1985, 15 (3), 283 [16] a) T. P. Johnston, G. S. McCaleb, J. A. Montgomery, J. Med. Chem. 1975, 18, 104-106; b)B. Ileana, N.-D. I., J. Prakt. Chem. 1985, 327, 675-681.
* * * * *
C00001
C00002
C00003
C00004
C00005
D00000
D00001
D00002
D00003
D00004
D00005
D00006
D00007

D00008

D00009

D00010

D00011

D00012

D00013

D00014

D00015

D00016

D00017

D00018

D00019

D00020

D00021

D00022

D00023

D00024

D00025

D00026

D00027

XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.