Detection of lung neoplasia by analysis of methylated DNA
Allawi , et al. A
U.S. patent number 10,385,406 [Application Number 15/471,337] was granted by the patent office on 2019-08-20 for detection of lung neoplasia by analysis of methylated dna. This patent grant is currently assigned to EXACT SCIENCES DEVELOPMENT COMPANY, LLC, Mayo Foundation for Medical Education and Research. The grantee listed for this patent is EXACT SCIENCES DEVELOPMENT COMPANY, LLC, Mayo Foundation for Medical Education and Research. Invention is credited to David A. Ahlquist, Hatim Allawi, Maria Giakoumopoulos, Graham P. Lidgard, Douglas Mahoney, William R. Taylor.












View All Diagrams
| United States Patent | 10,385,406 |
| Allawi , et al. | August 20, 2019 |
Detection of lung neoplasia by analysis of methylated DNA
Abstract
Provided herein is technology for lung neoplasia screening and particularly, but not exclusively, to methods, compositions, and related uses for detecting the presence of lung cancer.
| Inventors: | Allawi; Hatim (Middleton, WI), Lidgard; Graham P. (Middleton, WI), Giakoumopoulos; Maria (Middleton, WI), Ahlquist; David A. (Rochester, MN), Taylor; William R. (Lake City, MN), Mahoney; Douglas (Rochester, MN) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Applicant: |
|
||||||||||
| Assignee: | EXACT SCIENCES DEVELOPMENT COMPANY,
LLC (Madison, WI) Mayo Foundation for Medical Education and Research (Rochesher, MN) |
||||||||||
| Family ID: | 60203203 | ||||||||||
| Appl. No.: | 15/471,337 | ||||||||||
| Filed: | March 28, 2017 |
Prior Publication Data
| Document Identifier | Publication Date | |
|---|---|---|
| US 20170335401 A1 | Nov 23, 2017 | |
Related U.S. Patent Documents
| Application Number | Filing Date | Patent Number | Issue Date | ||
|---|---|---|---|---|---|
| 62462677 | Feb 23, 2017 | ||||
| 62332295 | May 5, 2016 | ||||
| Current U.S. Class: | 1/1 |
| Current CPC Class: | C12Q 1/6806 (20130101); C12Q 1/6886 (20130101); C12Q 2600/16 (20130101); C12Q 2600/154 (20130101) |
| Current International Class: | C12Q 1/6886 (20180101); C12Q 1/6806 (20180101); A61K 31/713 (20060101) |
References Cited [Referenced By]
U.S. Patent Documents
| 4683195 | July 1987 | Mullis et al. |
| 4683202 | July 1987 | Mullis |
| 4965188 | October 1990 | Mullis et al. |
| 5011769 | April 1991 | Duck et al. |
| 5124246 | June 1992 | Urdea et al. |
| 5288609 | February 1994 | Engelhardt et al. |
| 5338671 | August 1994 | Scalice et al. |
| 5403711 | April 1995 | Walder et al. |
| 5409818 | April 1995 | Davey et al. |
| 5494810 | February 1996 | Barany et al. |
| 5508169 | April 1996 | Deugau et al. |
| 5624802 | April 1997 | Urdea et al. |
| 5639611 | June 1997 | Wallace et al. |
| 5660988 | August 1997 | Duck et al. |
| 5710264 | January 1998 | Urdea et al. |
| 5773258 | June 1998 | Birch et al. |
| 5786146 | July 1998 | Herman et al. |
| 5792614 | August 1998 | Western et al. |
| 5846717 | December 1998 | Brow et al. |
| 5849481 | December 1998 | Urdea et al. |
| 5851770 | December 1998 | Babon et al. |
| 5882867 | March 1999 | Ullman et al. |
| 5914230 | June 1999 | Liu et al. |
| 5958692 | September 1999 | Cotton et al. |
| 5965408 | October 1999 | Short |
| 5985557 | November 1999 | Prudent et al. |
| 5994069 | November 1999 | Hall et al. |
| 6001567 | December 1999 | Brow et al. |
| 6013170 | January 2000 | Meade |
| 6063573 | May 2000 | Kayyem |
| 6090543 | July 2000 | Prudent et al. |
| 6110677 | August 2000 | Western et al. |
| 6110684 | August 2000 | Kemper et al. |
| 6121001 | September 2000 | Western et al. |
| 6150097 | November 2000 | Tyagi et al. |
| 6183960 | February 2001 | Lizardi |
| 6210884 | April 2001 | Lizardi |
| 6221583 | April 2001 | Kayyem et al. |
| 6235502 | May 2001 | Weissman et al. |
| 6248229 | June 2001 | Meade |
| 6251594 | June 2001 | Gonzalgo et al. |
| 7037650 | May 2006 | Gonzalgo et al. |
| 7662594 | February 2010 | Kong et al. |
| 8361720 | January 2013 | Oldham-Haltom et al. |
| 8715937 | May 2014 | Zou et al. |
| 8808990 | August 2014 | Lidagrad et al. |
| 8916344 | December 2014 | Zou et al. |
| 9000146 | April 2015 | Bruinsma et al. |
| 9096893 | August 2015 | Allawi et al. |
| 9163278 | October 2015 | Bruinsma et al. |
| 9169511 | October 2015 | Lidagrad et al. |
| 9212392 | December 2015 | Allawi et al. |
| 9315853 | April 2016 | Domanico et al. |
| 9657511 | May 2017 | Lidagrad et al. |
| 2005/0214926 | September 2005 | Zielenski |
| 2007/0202525 | August 2007 | Quake et al. |
| 2011/0160446 | June 2011 | Ritt |
| 2016/0010081 | January 2016 | Allawi et al. |
| 2016/0168643 | June 2016 | Ahlquist |
| 2016/0194721 | July 2016 | Allawi et al. |
| 2017/0121704 | May 2017 | Allawi et al. |
| 2017/0121757 | May 2017 | Lidgard et al. |
| 2018/0143198 | May 2018 | Wen |
| WO 1995/000669 | Jan 1995 | WO | |||
| WO 1995/015373 | Jun 1995 | WO | |||
| WO 1997/046705 | Dec 1997 | WO | |||
| WO 1999/028498 | Jun 1998 | WO | |||
| WO 2002/070755 | Sep 2002 | WO | |||
| WO 2005/023091 | Mar 2005 | WO | |||
| WO 2005/038051 | Apr 2005 | WO | |||
| WO 2012/155072 | Nov 2012 | WO | |||
| WO 2013/116375 | Aug 2013 | WO | |||
| WO 2017/075061 | May 2017 | WO | |||
| WO 2017/192221 | Nov 2017 | WO | |||
Other References
|
Kneip, C. et al., SHOX2 DNA Methylation Is a Biomarker for the Diagnosis of Lung Cancer in Plasma, J. Thoracic Oncol., vol. 6, pp. 1632-1638 (Year: 2011). cited by examiner . Schmidt, B. et al., SHOX2 DNA Methylation is a Biomarker for the Diagnosis of Lung Cancer Based on Bronchial Aspirates, BMC Cancer, vol. 10:600, pp. 1-9 (Year: 2010). cited by examiner . Yamada, H. et al., Fluorometric Identification of 5-Methylcytosine Modification in DNA: Combination of Photosensitized Oxidation and Invasive Cleavage, Bioconjugate Chem., vol. 19, pp. 20-23 (Year: 2008). cited by examiner . Zou, H. et al., Quantification of Methylated Markers with a Multiplex Methylation-Specific Technology, Clin. Chem., vol. 58, pp. 375-383 (Year: 2012). cited by examiner . Antequera et al., High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell. Aug. 10, 1990;62(3):503-14. cited by applicant . Ballabio, et al., Screening for steroid sulfatase (STS) gene deletions by multiplex DNA amplification, Human Genetics, 1990, 84(6): 571-573. cited by applicant . Barnay, Genetic disease detection and DNA amplification using cloned thermostable ligase, Proc. Natl. Acad. Sci USA, 1991, 88:189-93. cited by applicant . Budd et al., Circulating tumor cells versus imaging--predicting overall survival in metastatic breast cancer. Clin Cancer Res. Nov. 1, 2006;12(21):6403-9. cited by applicant . Bustin, Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays, J. Molecular Endocrinology, 2000, 25:169-193. cited by applicant . Carvalho et al., Genome-wide DNA methylation profiling of non-small cell lung carcinomas. Epigenetics Chromatin. Jun. 22, 2012;5(1):9. cited by applicant . Ceska et al., Structure-specific DNA cleavage by 5' nucleases. Trends Biochem Sci. Sep. 1998;23(9):331-6. cited by applicant . Chamberlain et al., Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification, Nucleic Acids Research, 1988, 16(23):11141-11156. cited by applicant . Cohen et al., Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. Jul. 1, 2008;26(19):3213-21. cited by applicant . Cristofanilli et al., Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. Aug. 19, 2004;351(8):781-91. cited by applicant . Don et al., `Touchdown` PCR to circumvent spurious priming during gene amplification, Nucleic Acids Research, 1991, 19(14):4008. cited by applicant . Eads et al., CpG island hypermethylation in human colorectal tumors is not associated with DNA methyltransferase overexpression. Cancer Res. May 15, 1999;59(10):2302-6. cited by applicant . Feil et al., Methylation analysis on individual chromosomes: improved protocol for bisulphite genomic sequencing. Nucleic Acids Res. Feb. 25, 1994;22(4):695-6. cited by applicant . Frommer et al., A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. Mar. 1, 1992;89(5):1827-31. cited by applicant . Gonzalgo et al., Identification and characterization of differentially methylated regions of genomic DNA by methylation-sensitive arbitrarily primed PCR. Cancer Res. Feb. 15, 1997;57(4):594-9. cited by applicant . Gonzalgo et al., Rapid quantitation of methylation differences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE). Nucleic Acids Res. Jun. 15, 1997;25(12):2529-31. cited by applicant . Grafstrom et al., The characteristics of DNA methylation in an in vitro DNA synthesizing system from mouse fibroblasts. Nucleic Acids Res. Apr. 25, 1985;13(8):2827-42. cited by applicant . Grigg et al., Sequencing 5-methylcytosine residues in genomic DNA. Bioessays. Jun. 1994;16(6):431-6. cited by applicant . Grigg, Sequencing 5-methylcytosine residues by the bisulphite method. DNA Seq. 1996;6(4):189-98. cited by applicant . Gu et al., Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat Methods. Feb. 2010;7(2):133-6. cited by applicant . Guilfoyle et al., Ligation-mediated PCR amplification of specific fragments from a class-II restriction endonuclease total digest, Nucleic Acids Research, 1997, 25:1854-1858. cited by applicant . Hall et al., Sensitive detection of DNA polymorphisms by the serial invasive signal amplification reaction, PNAS, 2000, 97:8272. cited by applicant . Hayden et al., Multiplex-Ready PCR: A new method for multiplexed SSR and SNP genotyping, BMC Genomics, 2008, 9:80. cited by applicant . Hayes et al., Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin Cancer Res. Jul. 15, 2006;12(14 Pt 1):4218-24. cited by applicant . Hecker et al., High and low annealing temperatures increase both specificity and yield in touchdown and stepdown PCR, Biotechniques, 1996, 20(3):478-485. cited by applicant . Herman et al., Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996; 93: 9821-9826. cited by applicant . Higuchi et al., A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions, Nucleic Acids Research, 1988, 16(15):7351-7367. cited by applicant . Higuchi et al., Simultaneous amplification and detection of specific DNA sequences, Biotechnology, 1992, 10:413-417. cited by applicant . Higuchi et al.,Kinetic PCR analysis: real-time monitoring of DNA amplification reactions, Biotechnology, 1993, 11:1026-1030. cited by applicant . Kaiser et al., A comparison of eubacterial and archaeal structure-specific 5'-exonucleases. J Biol Chem. Jul. 23, 1999;274(30):21387-94. cited by applicant . Kalinina et al., Nanoliter scale PCR with TaqMan detection, Nucleic Acids Research, 1997, 25:1999-2004. cited by applicant . Kober et al., Methyl-CpG binding column-based identification of nine genes hypermethylated in colorectal cancer. Mol Carcinog. Nov. 2011;50(11):846-56. cited by applicant . Kuppuswamy et al., Single nucleotide primer extension to detect genetic diseases: experimental application to hemophilia B (factor IX) and cystic fibrosis genes. Proc Natl Acad Sci U S A. Feb. 15, 1991;88(4):1143-7. cited by applicant . Liu et al., Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem. 2004;73:589-615. cited by applicant . Lyamichev et al.,Polymorphism identification and quantitative detection of genomic DNA by invasive cleavage of oligonucleotide probes, Nat. Biotech., 1999, 17:292-296. cited by applicant . Martin et al., Genomic sequencing indicates a correlation between DNA hypomethylation in the 5' region of the pS2 gene and its expression in human breast cancer cell lines. Gene. May 19, 1995;157(1-2):261-4. cited by applicant . Meissner et al., Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. Oct. 13, 2005;33(18):5868-77. cited by applicant . Moreno et al., Circulating tumor cells predict survival in patients with metastatic prostate cancer. Urology. Apr. 2005;65(4):713-8. cited by applicant . Nyce et al., Variable effects of DNA-synthesis inhibitors upon DNA methylation in mammalian cells. Nucleic Acids Res. May 27, 1986;14(10):4353-67. cited by applicant . Olek et al., A modified and improved method for bisulphite based cytosine methylation analysis. Nucleic Acids Res. Dec. 15, 1996;24(24):5064-6. cited by applicant . Olek et al., The pre-implantation ontogeny of the H19 methylation imprint. Nat Genet. Nov. 1997;17(3):275-6. cited by applicant . Olivier, The Invader assay for SNP genotyping, Mutat Res. Jun. 3, 2005;573(1-2):103-10. cited by applicant . Orpana, Fluorescence resonance energy transfer (FRET) using ssDNA binding fluorescent dye, Biomol Eng. Apr. 2004;21(2):45-50. cited by applicant . Pantel et al., Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat Rev Cancer. May 2008;8(5):329-40. cited by applicant . Ponomaryova et al., Potentialities of aberrantly methylated circulating DNA for diagnostics and post-treatment follow-up of lung cancer patients. Lung Cancer. Sep. 2013;81(3):397-403. cited by applicant . Ramsahoye et al., Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A. May 9, 2000;97(10):5237-42. cited by applicant . Rein et al., Identifying 5-methylcytosine and related modifications in DNA genomes. Nucleic Acids Res. May 15, 1998;26(10):2255-64. cited by applicant . Roux, Using mismatched primer-template pairs in touchdown PCR, Biotechniques, 1994, 16(5):812-814. cited by applicant . Sadri et al., Rapid analysis of DNA methylation using new restriction enzyme sites created by bisulfite modification. Nucleic Acids Res. Dec. 15, 1996;24(24):5058-9. cited by applicant . Salomon et al., Methylation of mouse DNA in vivo: di- and tripyrimidine sequences containing 5-methylcytosine. Biochim Biophys Acta. Apr. 15, 1970;204(2):340-51. cited by applicant . Schouten et al., Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification, Nucleic Acids Research, 2002, 30(12): e57. cited by applicant . Selvin, Fluorescence resonance energy transfer, 1995, Methods Enzymol. 1995;246:300-34. cited by applicant . Singer-Sam et al., A quantitative Hpall-PCR assay to measure methylation of DNA from a small number of cells. Nucleic Acids Res. Feb. 11, 1990;18(3):687. cited by applicant . Singer-Sam et al., A sensitive, quantitative assay for measurement of allele-specific transcripts differing by a single nucleotide. PCR Methods Appl. Feb. 1992;1(3):160-3. cited by applicant . Stryer, Fluorescence energy transfer as a spectroscopic ruler, Annu Rev Biochem. 1978;47:819-46. cited by applicant . Szabo et al., Allele-specific expression and total expression levels of imprinted genes during early mouse development: implications for imprinting mechanisms. Genes Dev. Dec. 15, 1995;9(24):3097-108. cited by applicant . Toyota et al., Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res. May 15, 1999;59(10):2307-12. cited by applicant . Triglia et al., A procedure for in vitro amplification of DNA segments that lie outside the boundaries of known sequences, Nucleic Acids Res., 1988, 16:8186. cited by applicant . Vogelstein et al., Digital PCR, PNAS, 1999, 96: 9236-41. cited by applicant . Woodcock et al., The majority of methylated deoxycytidines in human DNA are not in the CpG dinucleotide. Biochem Biophys Res Commun. Jun. 15, 1987;145(2):888-94. cited by applicant . Xiong et al., COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. Jun. 15, 1997;25(12):2532-4. cited by applicant . Zeschnigk et al., Imprinted segments in the human genome: different DNA methylation patterns in the Prader-Willi/Angelman syndrome region as determined by the genomic sequencing method. Hum Mol Genet. Mar. 1997;6(3):387-95. cited by applicant . Zou et al., Sensitive quantification of methylated markers with a novel methylation specific technology. Abstract D-144, Clin Chem 2010;56(6)Suppl:A199. cited by applicant . International Search Report and Written Opinion for PCT/US2017/024468, dated Sep. 1, 2017, 17 pages. cited by applicant. |
Primary Examiner: Strzelecka; Teresa E
Attorney, Agent or Firm: Casimir Jones, S.C. Brow; Mary Ann D.
Parent Case Text
The present application claims priority to U.S. Provisional Application Ser. No. 62/332,295, filed May 5, 2016 and U.S. Provisional Application Ser. No. 62/462,677, filed Feb. 23, 2017, each of which is incorporated herein by reference.
Claims
What is claimed is:
1. A method of processing a sample, the method comprising: a) assaying a sample from a subject for an amount of at least one methylation marker selected from the group consisting of BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, and ZNF329; b) assaying said sample for an amount of a reference marker; c) comparing the amount of said at least one methylation marker to the amount of reference marker in said sample to determine a methylation state for said at least one methylation marker in said sample; and optionally d) generating a record reporting the methylation state for said at least one methylation marker in said sample; wherein said sample is a plasma sample obtained from a subject having or suspected of having a lung neoplasm, and wherein said method comprises: A) combining the plasma sample with: i) protease; and ii) a first lysis reagent, said first lysis reagent comprising guanidine thiocyanate; and non-ionic detergent; to form a mixture wherein proteins are digested by said protease; B) to the mixture of step a) adding iii) silica particles, and iv) a second lysis reagent, said second lysis reagent comprising: guanidine thiocyanate; non-ionic detergent; and isopropyl alcohol; under conditions wherein DNA is bound to said silica particles; C) separating silica particles with bound DNA from the mixture of B); D) to the separated silica particles with bound DNA adding a first wash solution, said first wash solution comprising a) guanidine hydrochloride or guanidine thiocyanate, and b) ethyl alcohol; E) separating the silica particles with bound DNA from said first wash solution; F) to the separated silica particles with bound DNA adding a second wash solution, said second wash solution comprising a buffer and ethyl alcohol; G) separating washed silica particles with bound DNA from said second wash solution; and H) eluting DNA from the washed silica particles with bound DNA separated in step G); I) assaying eluted DNA for an amount of at least one methylated methylation marker and for an amount of reference marker in said eluted DNA; and J) comparing the amount of said at least one methylated methylation marker to the amount of reference marker in said DNA to determine a methylation state for said at least one methylation marker in said plasma sample; wherein assaying said eluted DNA comprises analyzing multiple DNA methylation markers using a PCR pre-amplification and a PCR-flap assay by a process comprising: K) combining eluted DNA comprising a plurality of different DNA methylation marker target regions in a first reaction mixture with PCR amplification reagents, wherein said PCR amplification reagents comprise: i) a plurality of different primer pairs for amplifying said plurality of different target regions, if present in said sample, from said eluted DNA; ii) thermostable DNA polymerase; iii) dNTPs; and iv) a buffer comprising Mg.sup.++ L) exposing said first reaction mixture to thermal cycling conditions wherein a plurality of different DNA methylation marker target regions, if present in the sample, are amplified to produce a pre-amplified mixture, and wherein said thermal cycling conditions are limited to a number of thermal cycles that maintain amplification in an exponential range; M) partitioning said pre-amplified mixture into a plurality of PCR-flap assay reaction mixtures, wherein each PCR-flap assay reaction mixture comprises: i) an additional amount of a primer pair selected from said plurality of different primer pairs of step K) i); ii) thermostable DNA polymerase; iii) dNTPs; iv) said buffer comprising Mg.sup.++ v) a flap endonuclease; vi) a flap oligonucleotide; and vi) a hairpin oligonucleotide comprising a region that is complementary to a portion of said flap oligonucleotide; and N) detecting amplification of one or more different DNA methylation marker target regions from said eluted DNA during PCR-flap assay reactions.
2. The method of claim 1, wherein said assaying comprises treating the eluted DNA with a methylation-sensitive restriction enzyme or with a reagent that selectively modifies unmethylated cytosine residues in the eluted DNA.
3. The method of claim 1, wherein said at least one methylation marker comprises a methylation marker selected from the group consisting of SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, EMX1, CYP26C1, SOBP, SUCLG2, SHOX2, NFIX, FLJ45983, HOXA9, B3GALT6, ZNF781, SP9, BARX1, and SKI.
4. The method of claim 1, wherein said at least one methylation marker comprises a group of methylation markers selected from: the group consisting of ZNF781, BARX1, and EMX1; the group consisting of SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI; the group consisting of SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, and EMX1; the group consisting of SHOX2, SOBP, ZNF781, BTACT, CYP26C1, and DLX4; the group consisting of SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI; and the group consisting of ZNF781, BARX1, and EMX1, and further comprising SOBP and/or HOXA9.
5. The method of claim 1, wherein assaying the methylation state of a methylation marker in the sample comprises determining the extent of methylation at a plurality of bases.
6. The method of claim 2, wherein the reagent that selectively modifies unmethylated cytosine residues comprises bisulfite, and wherein said assaying comprises bisulfate converting methylation marker DNA and reference marker DNA.
7. The method of claim 1, wherein said eluted DNA is prepared from a plasma sample of at least one mL, and wherein the volume of said eluted DNA comprising a plurality of different DNA methylation marker target regions in the first reaction mixture is at least 20 to 50% of the total volume of the first reaction mixture.
8. A method of processing a sample, the method comprising: a) assaying a sample from a subject for an amount of at least one methylation marker selected from the group consisting of BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX62, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, and ZNF329; b) assaying said sample for an amount of a reference marker; c) comparing the amount of said at least one methylation marker to the amount of reference marker in said sample to determine a methylation state for said at least one methylation marker in said sample; wherein assaying said sample comprises analyzing multiple DNA methylation markers using a PCR pre-amplification and a PCR-flap assay by a process comprising: I) combining DNA from the sample comprising a plurality of different DNA methylation marker target regions in a first reaction mixture with PCR amplification reagents, wherein said PCR amplification reagents comprise: i) a plurality of different primer pairs for amplifying said plurality of different target regions, if present in the sample; ii) thermostable DNA polymerase; iii) dNTPs; and iv) a buffer comprising Mg.sup.++; II) exposing said first reaction mixture to thermal cycling conditions wherein a plurality of different DNA methylation marker target regions, if present in the sample, are amplified to produce a pre-amplified mixture, and wherein said thermal cycling conditions are limited to a number of thermal cycles that maintain amplification in an exponential range; III) partitioning said pre-amplified mixture into a plurality of PCR-flap assay reaction mixtures, wherein each PCR-flap assay reaction mixture comprises: i) an additional amount of a primer pair selected from said plurality of different primer pairs of step I) i); ii) thermostable DNA polymerase; iii) dNTPs; iv) said buffer comprising Mg.sup.++; v) a flap endonuclease; vi) a flap oligonucleotide; and vi) a hairpin oligonucleotide comprising a region that is complementary to a portion of said flap oligonucleotide; and IV) detecting amplification of one or more different DNA methylation marker target regions from said DNA from said sample during PCR-flap assay reactions.
9. The method of claim 1, wherein said assaying comprises treating the DNA obtained from the sample with a methylation-sensitive restriction enzyme or with a reagent that selectively modifies unmethylated cytosine residues in the obtained DNA.
10. The method of claim 9, wherein the reagent that selectively modifies unmethylated cytosine residues comprises bisulfate.
11. The method of claim 10, wherein the assaying comprises bisulfite-converting methylation marker DNA and reference marker DNA.
12. The method of claim 8, wherein said at least one methylation marker comprises a methylation marker selected from the group consisting of SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, EMX1, CYP26C1, SOBP, SUCLG2, SHOX2, NFIX, FLJ45983, HOXA9, B3GALT6, ZNF781, SP9, BARX1, and SKI.
13. The method of claim 8, wherein said at least one methylation marker comprises a group of methylation markers selected from: the group consisting of ZNF781, BARX1, and EMX1; the group consisting of SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI; the group consisting of SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, and EMX1; the group consisting of SHOX2, SOBP, ZNF781, BTACT, CYP26C1, and DLX4; the group consisting of SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI; and the group consisting of ZNF781, BARX1, and EMX1, and further comprising SOBP and/or HOXA9.
14. The method of claim 8, wherein assaying the methylation state of a methylation marker in the sample comprises determining the extent of methylation at a plurality of bases.
15. The method of claim 8, wherein said DNA from the sample is prepared from a plasma sample of at least one mL, and wherein the volume of said DNA from the sample comprising a plurality of different DNA methylation marker target regions in the first reaction mixture is at least 20 to 50% of the total volume of the first reaction mixture.
Description
FIELD OF THE INVENTION
Provided herein is technology relating to detecting neoplasia and particularly, but not exclusively, to methods, compositions, and related uses for detecting neoplasms such as lung cancer.
BACKGROUND OF THE INVENTION
Lung cancer remains the number one cancer killer in the US, and effective screening approaches are desperately needed. Lung cancer alone accounts for 221,000 deaths annually. DNA methylation profiling has shown unique patterns in DNA promoter regions with cancer and has potential application for detection of lung malignancies. However, optimally discriminant markers and marker panels are needed.
SUMMARY OF THE INVENTION
Provided herein is a collection of methylated methylation markers assayed on tissue that achieves extremely high discrimination for all types of lung cancer while remaining negative in normal lung tissue and benign nodules. Markers selected from the collection can be used alone or in a panel, for example, to characterize blood or bodily fluid, with applications in lung cancer screening and discrimination of malignant from benign nodules. In some embodiments, markers from the panel are used to distinguish one form of lung cancer from another, e.g., for distinguishing the presence of a lung adenocarcinoma or large cell carcinoma from the presence of a lung small cell carcinoma, or for detecting mixed pathology carcinomas. Provided herein is technology for screening markers that provide a high signal-to-noise ratio and a low background level when detected from samples taken from a subject.
Methylation markers and/or panels of markers (e.g., chromosomal region(s)) having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329 were identified in studies by comparing the methylation state of methylation markers from lung cancer samples to the corresponding markers in normal (non-cancerous) samples.
As described herein, the technology provides a number of methylation markers and subsets thereof (e.g., sets of 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 or more markers) with high discrimination for lung cancer and, in some embodiments, with discrimination between lung cancer types. Experiments applied a selection filter to candidate markers to identify markers that provide a high signal to noise ratio and a low background level to provide high specificity and selectivity for purposes of characterizing biological samples, e.g., for cancer screening or diagnosis. For example, as described herein below, analysis of methylation of combination of 8 markers, SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, and EMX1, resulted in 98.5% sensitivity (134/136 cancers) for all of the cancer tissues tested, with 100% specificity. In another embodiment, a panel of 6 markers (SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI) resulted in a sensitivity of 92.2% at 93% specificity, and a panel of 4 markers (ZNF781, BARX1, EMX1, and HOXA9) resulted in an overall sensitivity of 96% and specificity of 94%.
Accordingly, provided herein is technology related to a method of processing a sample obtained from a subject, the method comprising assaying a methylation state of one or more marker genes in the sample. In preferred embodiments, the methylation state of the methylation marker is determined by measuring the amounts of a methylated marker and of a reference marker in the sample, and comparing the amount of the methylated marker to the amount of reference marker in the sample to determine a methylation state for the methylation marker in the sample. While not limiting the invention to any particular application or applications, the method finds use, e.g., in characterizing samples from a subject having or suspected of having lung cancer, when the methylation state of the methylation marker is different than a methylation state of that marker assayed in a subject that does not have a neoplasm. In preferred embodiments, the methylation marker comprises a chromosomal region having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329.
In some embodiments, the technology comprises assaying a plurality of markers, e.g., comprising assaying the methylation states of 2 to 21 markers, preferably 2 to 8 markers, preferably 4 to 6 markers. For example, in some embodiments, the method comprises analysis of the methylation status of two or more markers selected from SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, EMX1, CYP26C1, SOBP, SUCLG2, SHOX2, ZDHHC1, NFIX, FLJ45983, HOXA9, B3GALT6, ZNF781, SP9, BARX1, and SKI. In some preferred embodiments, the method comprises analysis of the methylation status of a set of markers comprising SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, and EMX1. In some embodiments, the method comprises analysis of the methylation status of a set of markers selected from: the group consisting of ZNF781, BARX1, and EMX1; the group consisting of SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI; the group consisting of SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, and EMX1; the group consisting of SHOX2, SOBP, ZNF781, BTACT, CYP26C1, and DLX4; and the group consisting of SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI. In certain embodiments, the at least one methylation marker comprises the group selected from ZNF781, BARX1, and EMX1, and further comprises SOBP and/or HOXA9.
The technology is not limited in the methylation state assessed. In some embodiments assessing the methylation state of the methylation marker in the sample comprises determining the methylation state of one base. In some embodiments, assaying the methylation state of the marker in the sample comprises determining the extent of methylation at a plurality of bases. Moreover, in some embodiments the methylation state of the marker comprises an increased methylation of the marker relative to a normal methylation state of the marker. In some embodiments, the methylation state of the marker comprises a decreased methylation of the marker relative to a normal methylation state of the marker. In some embodiments the methylation state of the marker comprises a different pattern of methylation of the marker relative to a normal methylation state of the marker.
In some embodiments, the technology provides a method of generating a record reporting a lung neoplasm in a subject, the method comprising the steps of:
a) assaying a sample from a subject for an amount of at least one methylated methylation marker gene selected from the group consisting of BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329 in a sample obtained from a subject;
b) assaying said sample for an amount of reference marker in said sample;
c) comparing the amount of said at least one methylated methylation marker to the amount of reference marker in said sample to determine a methylation state for said at least one methylation marker in said sample; and d) generating a record reporting the methylation state for said at least one marker gene in said sample, wherein the methylation state of said methylation marker is indicative of the presence or absence of a lung neoplasm in said subject.
In some embodiments, the sample is assayed for at least two of the markers, and preferably the at least two methylated marker genes are selected from the group consisting of SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, EMX1 CYP26C1, SOBP, SUCLG2, SHOX2, ZDHHC1, NFIX, FLJ45983, HOXA9, B3GALT6, ZNF781, SP9, BARX1, and SKI. In certain preferred embodiments, the method comprises analysis of the methylation status of a set of markers selected from: the group consisting of ZNF781, BARX1, and EMX1; the group consisting of SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI; the group consisting of SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, and EMX1; the group consisting of SHOX2, SOBP, ZNF781, BTACT, CYP26C1, and DLX4; and the group consisting of SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI. In certain embodiments, the at least one methylation marker comprises the group selected from ZNF781, BARX1, and EMX1, and further comprises SOBP and/or HOXA9. In some embodiments, methylation markers are selected such that the methylation status of said one or more markers is indicative of only one of lung adenocarcinoma, large cell carcinoma, squamous cell carcinoma, or small cell carcinoma. In other embodiments, methylation markers are selected such that the methylation status of said one or more markers is indicative of more than one of lung adenocarcinoma, large cell carcinoma, squamous cell carcinoma, and small cell carcinoma. In yet other embodiments, methylation markers are selected such that the methylation status of said one or more markers is indicative of any one of or combination of lung adenocarcinoma, large cell carcinoma, squamous cell carcinoma, small cell carcinoma, generic non-small cell lung cancer, and/or undefined lung carcinoma.
In some embodiments the method used for assaying comprises obtaining a sample comprising DNA from a subject, and treating DNA obtained from the sample with a reagent that selectively modifies unmethylated cytosine residues in the obtained DNA to produce modified residues. In preferred embodiments the reagent comprises a bisulfate reagent.
In some embodiments assaying the methylation state of the methylation marker in the sample comprises determining the methylation state of one base, while in other embodiments the assay comprises determining the extent of methylation at a plurality of bases. In some embodiments the methylation state of the marker comprises an increased or decreased methylation of the marker relative to a normal methylation state of the marker, e.g., as the marker would appear in a non-cancerous sample, while in some embodiments the methylation state of the marker comprises a different pattern of methylation of the marker relative to a normal methylation state of the marker. In preferred embodiments the reference marker is a methylated reference marker.
The technology is not limited to particular sample types. For example, in some embodiments the sample is a tissue sample, a blood sample, a plasma sample, a serum sample, or a sputum sample. In certain preferred embodiments a tissue sample comprises lung tissue. In certain preferred embodiments, the sample comprises DNA isolated from plasma.
The technology is not limited to any particular method of assaying DNA from samples. For example, in some embodiments the assaying comprises using polymerase chain reaction, nucleic acid sequencing, mass spectrometry, methylation specific nuclease, mass-based separation, and/or target capture. In certain preferred embodiments the assaying comprises using a flap endonuclease assay. In particularly preferred embodiments the sample DNA and/or reference marker DNA are bisulfite-converted and the assay for determining the methylation level of the DNA is achieved by a technique comprising the use of methylation-specific PCR, quantitative methylation-specific PCR, methylation-sensitive DNA restriction enzyme analysis, quantitative bisulfite pyrosequencing, flap endonuclease assay (e.g., a QUARTS flap endonuclease assay), and/or bisulfite genomic sequencing PCR.
The technology also provides kits. For example, in some embodiments the technology provides a kit, comprising a) at least one oligonucleotide, wherein at least a portion of the oligonucleotide specifically hybridizes to a marker selected from the group consisting of BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329. In preferred embodiments, the portion of the oligonucleotide that hybridizes to the marker specifically hybridizes to bisulfite-treated DNA comprising the methylation marker. In some embodiments, the kit comprises at least one additional oligonucleotide, wherein at least a portion of the additional oligonucleotide specifically hybridizes to a reference nucleic acid. In some embodiments the kit comprises at least two additional oligonucleotides and, in some embodiments, the kit further comprises a bisulfite reagent.
In certain embodiments at least a portion of the oligonucleotide specifically hybridizes to a least one the marker selected from the group consisting of SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, EMX1, CYP26C1, SOBP, SUCLG2, SHOX2, ZDHHC1, NFIX, FLJ45983, HOXA9, B3GALT6, ZNF781, SP9, BARX1, and SKI. In preferred embodiments, the kit comprises a set of oligonucleotides, each of which hybridizes to one marker in a set of markers, the set of markers selected from: the group consisting of ZNF781, BARX1, and EMX1; the group consisting of SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI the group consisting of SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, and EMX1; the group consisting of SHOX2, SOBP, ZNF781, BTACT, CYP26C1, and DLX4; and the group consisting of SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI. In certain embodiments, the set of methylation markers comprises the group selected from ZNF781, BARX1, and EMX1, and further comprises SOBP and/or HOXA9.
In some embodiments, the at least one oligonucleotide in the kit is selected to hybridize to methylation marker(s) that are indicative of only one of type of lung carcinoma, e.g., lung adenocarcinoma, large cell carcinoma, squamous cell carcinoma, or small cell carcinoma. In other embodiments, the at least one oligonucleotide is selected to hybridize to methylation marker(s) that are indicative of more than one of lung adenocarcinoma, large cell carcinoma, squamous cell carcinoma, and small cell carcinoma. In yet other embodiments, the at least one oligonucleotide is selected to hybridize to methylation marker(s) that are indicative of any one of, or any combination of lung adenocarcinoma, large cell carcinoma, squamous cell carcinoma, small cell carcinoma, and/or undefined lung carcinoma.
In preferred embodiments, oligonucleotide(s) provided in the kit are selected from one or more of a capture oligonucleotide, a pair of nucleic acid primers, a nucleic acid probe, and an invasive oligonucleotide. In preferred embodiments, oligonucleotide(s) specifically hybridize to bisulfite-treated DNA comprising said methylation marker(s).
In some embodiments the kit further comprises a solid support, such a magnetic bead or particle. In preferred embodiments, a solid support comprises one or more capture reagents, e.g., oligonucleotides complementary said one or more markers genes.
The technology also provides compositions. For example, in some embodiments the technology provides a composition comprising a mixture, e.g., a reaction mixture, that comprises a complex of a target nucleic acid selected from the group consisting of BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12a, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329 and an oligonucleotide that specifically hybridizes to the target nucleic acid. In some embodiments, the target nucleic acid is bisulfite-converted target nucleic acid. In preferred embodiments, the mixture comprises a complex of a target nucleic acid selected from the group consisting of SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, EMX1, CYP26C1, SOBP, SUCLG2, SHOX2, ZDHHC1, NFIX, FLJ45983, HOXA9, B3GALT6, ZNF781, SP9, BARX1, and SKI, and an oligonucleotide that specifically hybridizes to the target nucleic acid (whether unconverted or bisulfite-converted). Oligonucleotides in the mixture include but are not limited to one or more of a capture oligonucleotide, a pair of nucleic acid primers, a hybridization probe, a hydrolysis probe, a flap assay probe, and an invasive oligonucleotide.
In some embodiments, the target nucleic acid in the mixture comprises a nucleic acid sequence selected from the group consisting of SEQ ID NOS: 1, 6, 11, 16, 21, 28, 33, 38, 43, 48, 53, 58, 63, 68, 73, 78, 86, 91, 96, 101, 106, 111, 116, 121, 126, 131, 136, 141, 146, 151, 156, 161, 166, 171, 176, 181, 186, 191, 196, 201, 214, 219, 224, 229, 234, 239, 247, 252, 257, 262, 267, 272, 277, 282, 287, 292, 298, 303, 308, 313, 319, 327, 336, 341, 346, 351, 356, 361, 366, 371, 384, and 403.
In some embodiments, the mixture comprises bisulfate-converted target nucleic acid that comprises a nucleic acid sequence selected from the group consisting of SEQ ID NOS: 2, 7, 12, 17, 22, 29, 34, 39, 44, 49, 54, 59, 64, 69, 74, 79, 87, 92, 97, 102, 107, 112, 117, 122, 127, 132, 137, 142, 147, 152, 157, 162, 167, 172, 177, 182, 187, 192, 197, 202, 210, 215, 220, 225, 230, 235, 240, 248, 253, 258, 263, 268, 273, 278, 283, 288, 293, 299, 304, 309, 314, 320, 328, 337, 342, 347, 352, 357, 362, 367, 372, 385, and 404.
In some embodiments, an oligonucleotide in said mixture comprises a reporter molecule, and in preferred embodiments, the reporter molecule comprises a fluorophore. In some embodiments the oligonucleotide comprises a flap sequence. In some embodiments the mixture further comprises one or more of a FRET cassette; a FEN-1 endonuclease and/or a thermostable DNA polymerase, preferably a bacterial DNA polymerase.
Definitions
To facilitate an understanding of the present technology, a number of terms and phrases are defined below. Additional definitions are set forth throughout the detailed description.
Throughout the specification and claims, the following terms take the meanings explicitly associated herein, unless the context clearly dictates otherwise. The phrase "in one embodiment" as used herein does not necessarily refer to the same embodiment, though it may. Furthermore, the phrase "in another embodiment" as used herein does not necessarily refer to a different embodiment, although it may. Thus, as described below, various embodiments of the invention may be readily combined, without departing from the scope or spirit of the invention.
In addition, as used herein, the term "or" is an inclusive "or" operator and is equivalent to the term "and/or" unless the context clearly dictates otherwise. The term "based on" is not exclusive and allows for being based on additional factors not described, unless the context clearly dictates otherwise. In addition, throughout the specification, the meaning of "a", "an", and "the" include plural references. The meaning of "in" includes "in" and "on."
The transitional phrase "consisting essentially of" as used in claims in the present application limits the scope of a claim to the specified materials or steps "and those that do not materially affect the basic and novel characteristic(s)" of the claimed invention, as discussed in In re Herz, 537 E2d 549, 551-52, 190 USPQ 461, 463 (CCPR 1976). For example, a composition "consisting essentially of" recited elements may contain an unrecited contaminant at a level such that, though present, the contaminant does not alter the function of the recited composition as compared to a pure composition, i.e., a composition "consisting of" the recited components.
As used herein, "methylation" refers to cytosine methylation at positions C5 or N4 of cytosine, the N6 position of adenine, or other types of nucleic acid methylation. In vitro amplified DNA is usually unmethylated because typical in vitro DNA amplification methods do not retain the methylation pattern of the amplification template. However, "unmethylated DNA" or "methylated DNA" can also refer to amplified DNA whose original template was unmethylated or methylated, respectively.
Accordingly, as used herein a "methylated nucleotide" or a "methylated nucleotide base" refers to the presence of a methyl moiety on a nucleotide base, where the methyl moiety is not present in a recognized typical nucleotide base. For example, cytosine does not contain a methyl moiety on its pyrimidine ring, but 5-methylcytosine contains a methyl moiety at position 5 of its pyrimidine ring. Therefore, cytosine is not a methylated nucleotide and 5-methylcytosine is a methylated nucleotide. In another example, thymine contains a methyl moiety at position 5 of its pyrimidine ring; however, for purposes herein, thymine is not considered a methylated nucleotide when present in DNA since thymine is a typical nucleotide base of DNA.
As used herein, a "methylated nucleic acid molecule" refers to a nucleic acid molecule that contains one or more methylated nucleotides.
As used herein, a "methylation state", "methylation profile", and "methylation status" of a nucleic acid molecule refers to the presence of absence of one or more methylated nucleotide bases in the nucleic acid molecule. For example, a nucleic acid molecule containing a methylated cytosine is considered methylated (e.g., the methylation state of the nucleic acid molecule is methylated). A nucleic acid molecule that does not contain any methylated nucleotides is considered unmethylated. In some embodiments, a nucleic acid may be characterized as "unmethylated" if it is not methylated at a specific locus (e.g., the locus of a specific single CpG dinucleotide) or specific combination of loci, even if it is methylated at other loci in the same gene or molecule.
The methylation state of a particular nucleic acid sequence (e.g., a gene marker or DNA region as described herein) can indicate the methylation state of every base in the sequence or can indicate the methylation state of a subset of the bases (e.g., of one or more cytosines) within the sequence, or can indicate information regarding regional methylation density within the sequence with or without providing precise information of the locations within the sequence the methylation occurs. As used herein, the terms "marker gene" and "marker" are used interchangeably to refer to DNA that is associated with a condition, e.g., cancer, regardless of whether the marker region is in a coding region of DNA. Markers may include, e.g., regulatory regions, flanking regions, intergenic regions, etc.
The methylation state of a nucleotide locus in a nucleic acid molecule refers to the presence or absence of a methylated nucleotide at a particular locus in the nucleic acid molecule. For example, the methylation state of a cytosine at the 7th nucleotide in a nucleic acid molecule is methylated when the nucleotide present at the 7th nucleotide in the nucleic acid molecule is 5-methylcytosine. Similarly, the methylation state of a cytosine at the 7th nucleotide in a nucleic acid molecule is unmethylated when the nucleotide present at the 7th nucleotide in the nucleic acid molecule is cytosine (and not 5-methylcytosine).
The methylation status can optionally be represented or indicated by a "methylation value" (e.g., representing a methylation frequency, fraction, ratio, percent, etc.) A methylation value can be generated, for example, by quantifying the amount of intact nucleic acid present following restriction digestion with a methylation dependent restriction enzyme or by comparing amplification profiles after bisulfite reaction or by comparing sequences of bisulfite-treated and untreated nucleic acids. Accordingly, a value, e.g., a methylation value, represents the methylation status and can thus be used as a quantitative indicator of methylation status across multiple copies of a locus. This is of particular use when it is desirable to compare the methylation status of a sequence in a sample to a threshold or reference value.
As used herein, "methylation frequency" or "methylation percent (%)" refer to the number of instances in which a molecule or locus is methylated relative to the number of instances the molecule or locus is unmethylated.
As such, the methylation state describes the state of methylation of a nucleic acid (e.g., a genomic sequence). In addition, the methylation state refers to the characteristics of a nucleic acid segment at a particular genomic locus relevant to methylation. Such characteristics include, but are not limited to, whether any of the cytosine (C) residues within this DNA sequence are methylated, the location of methylated C residue(s), the frequency or percentage of methylated C throughout any particular region of a nucleic acid, and allelic differences in methylation due to, e.g., difference in the origin of the alleles. The terms "methylation state", "methylation profile", and "methylation status" also refer to the relative concentration, absolute concentration, or pattern of methylated C or unmethylated C throughout any particular region of a nucleic acid in a biological sample. For example, if the cytosine (C) residue(s) within a nucleic acid sequence are methylated it may be referred to as "hypermethylated" or having "increased methylation", whereas if the cytosine (C) residue(s) within a DNA sequence are not methylated it may be referred to as "hypomethylated" or having "decreased methylation". Likewise, if the cytosine (C) residue(s) within a nucleic acid sequence are methylated as compared to another nucleic acid sequence (e.g., from a different region or from a different individual, etc.) that sequence is considered hypermethylated or having increased methylation compared to the other nucleic acid sequence. Alternatively, if the cytosine (C) residue(s) within a DNA sequence are not methylated as compared to another nucleic acid sequence (e.g., from a different region or from a different individual, etc.) that sequence is considered hypomethylated or having decreased methylation compared to the other nucleic acid sequence. Additionally, the term "methylation pattern" as used herein refers to the collective sites of methylated and unmethylated nucleotides over a region of a nucleic acid. Two nucleic acids may have the same or similar methylation frequency or methylation percent but have different methylation patterns when the number of methylated and unmethylated nucleotides is the same or similar throughout the region but the locations of methylated and unmethylated nucleotides are different. Sequences are said to be "differentially methylated" or as having a "difference in methylation" or having a "different methylation state" when they differ in the extent (e.g., one has increased or decreased methylation relative to the other), frequency, or pattern of methylation. The term "differential methylation" refers to a difference in the level or pattern of nucleic acid methylation in a cancer positive sample as compared with the level or pattern of nucleic acid methylation in a cancer negative sample. It may also refer to the difference in levels or patterns between patients that have recurrence of cancer after surgery versus patients who not have recurrence. Differential methylation and specific levels or patterns of DNA methylation are prognostic and predictive biomarkers, e.g., once the correct cut-off or predictive characteristics have been defined.
Methylation state frequency can be used to describe a population of individuals or a sample from a single individual. For example, a nucleotide locus having a methylation state frequency of 50% is methylated in 50% of instances and unmethylated in 50% of instances. Such a frequency can be used, for example, to describe the degree to which a nucleotide locus or nucleic acid region is methylated in a population of individuals or a collection of nucleic acids. Thus, when methylation in a first population or pool of nucleic acid molecules is different from methylation in a second population or pool of nucleic acid molecules, the methylation state frequency of the first population or pool will be different from the methylation state frequency of the second population or pool. Such a frequency also can be used, for example, to describe the degree to which a nucleotide locus or nucleic acid region is methylated in a single individual. For example, such a frequency can be used to describe the degree to which a group of cells from a tissue sample are methylated or unmethylated at a nucleotide locus or nucleic acid region.
As used herein a "nucleotide locus" refers to the location of a nucleotide in a nucleic acid molecule. A nucleotide locus of a methylated nucleotide refers to the location of a methylated nucleotide in a nucleic acid molecule.
Typically, methylation of human DNA occurs on a dinucleotide sequence including an adjacent guanine and cytosine where the cytosine is located 5' of the guanine (also termed CpG dinucleotide sequences). Most cytosines within the CpG dinucleotides are methylated in the human genome, however some remain unmethylated in specific CpG dinucleotide rich genomic regions, known as CpG islands (see, e.g., Antequera, et al. (1990) Cell 62: 503-514).
As used herein, a "CpG island" refers to a G:C-rich region of genomic DNA containing an increased number of CpG dinucleotides relative to total genomic DNA. A CpG island can be at least 100, 200, or more base pairs in length, where the G:C content of the region is at least 50% and the ratio of observed CpG frequency over expected frequency is 0.6; in some instances, a CpG island can be at least 500 base pairs in length, where the G:C content of the region is at least 55%) and the ratio of observed CpG frequency over expected frequency is 0.65. The observed CpG frequency over expected frequency can be calculated according to the method provided in Gardiner-Garden et al (1987) J. Mol. Biol. 196: 261-281. For example, the observed CpG frequency over expected frequency can be calculated according to the formula R=(A.times.B)/(C.times.D), where R is the ratio of observed CpG frequency over expected frequency, A is the number of CpG dinucleotides in an analyzed sequence, B is the total number of nucleotides in the analyzed sequence, C is the total number of C nucleotides in the analyzed sequence, and D is the total number of G nucleotides in the analyzed sequence. Methylation state is typically determined in CpG islands, e.g., at promoter regions. It will be appreciated though that other sequences in the human genome are prone to DNA methylation such as CpA and CpT (see Ramsahoye (2000) Proc. Natl. Acad. Sci. USA 97: 5237-5242; Salmon and Kaye (1970) Biochim. Biophys. Acta. 204: 340-351; Grafstrom (1985) Nucleic Acids Res. 13: 2827-2842; Nyce (1986) Nucleic Acids Res. 14: 4353-4367; Woodcock (1987) Biochem. Biophys. Res. Commun. 145: 888-894).
As used herein, a "methylation-specific reagent" refers to a reagent that modifies a nucleotide of the nucleic acid molecule as a function of the methylation state of the nucleic acid molecule, or a methylation-specific reagent, refers to a compound or composition or other agent that can change the nucleotide sequence of a nucleic acid molecule in a manner that reflects the methylation state of the nucleic acid molecule. Methods of treating a nucleic acid molecule with such a reagent can include contacting the nucleic acid molecule with the reagent, coupled with additional steps, if desired, to accomplish the desired change of nucleotide sequence. Such methods can be applied in a manner in which unmethylated nucleotides (e.g., each unmethylated cytosine) is modified to a different nucleotide. For example, in some embodiments, such a reagent can deaminate unmethylated cytosine nucleotides to produce deoxy uracil residues. An exemplary reagent is a bisulfite reagent.
The term "bisulfite reagent" refers to a reagent comprising bisulfite, disulfite, hydrogen sulfite, or combinations thereof, useful as disclosed herein to distinguish between methylated and unmethylated CpG dinucleotide sequences. Methods of said treatment are known in the art (e.g., PCT/EP2004/011715 and WO 2013/116375, each of which is incorporated by reference in its entirety). In some embodiments, bisulfite treatment is conducted in the presence of denaturing solvents such as but not limited to n-alkyleneglycol or diethylene glycol dimethyl ether (DME), or in the presence of dioxane or dioxane derivatives. In some embodiments the denaturing solvents are used in concentrations between 1% and 35% (v/v). In some embodiments, the bisulfite reaction is carried out in the presence of scavengers such as but not limited to chromane derivatives, e.g., 6-hydroxy-2,5,7,8,-tetramethylchromane 2-carboxylic acid or trihydroxybenzone acid and derivates thereof, e.g., Gallic acid (see: PCT/EP2004/011715, which is incorporated by reference in its entirety). In certain preferred embodiments, the bisulfite reaction comprises treatment with ammonium hydrogen sulfite, e.g., as described in WO 2013/116375.
A change in the nucleic acid nucleotide sequence by a methylation--specific reagent can also result in a nucleic acid molecule in which each methylated nucleotide is modified to a different nucleotide.
The term "methylation assay" refers to any assay for determining the methylation state of one or more CpG dinucleotide sequences within a sequence of a nucleic acid.
As used herein, the "sensitivity" of a given marker (or set of markers used together) refers to the percentage of samples that report a DNA methylation value above a threshold value that distinguishes between neoplastic and non-neoplastic samples. In some embodiments, a positive is defined as a histology-confirmed neoplasia that reports a DNA methylation value above a threshold value (e.g., the range associated with disease), and a false negative is defined as a histology-confirmed neoplasia that reports a DNA methylation value below the threshold value (e.g., the range associated with no disease). The value of sensitivity, therefore, reflects the probability that a DNA methylation measurement for a given marker obtained from a known diseased sample will be in the range of disease-associated measurements. As defined here, the clinical relevance of the calculated sensitivity value represents an estimation of the probability that a given marker would detect the presence of a clinical condition when applied to a subject with that condition.
As used herein, the "specificity" of a given marker (or set of markers used together) refers to the percentage of non-neoplastic samples that report a DNA methylation value below a threshold value that distinguishes between neoplastic and non-neoplastic samples. In some embodiments, a negative is defined as a histology-confirmed non-neoplastic sample that reports a DNA methylation value below the threshold value (e.g., the range associated with no disease) and a false positive is defined as a histology-confirmed non-neoplastic sample that reports a DNA methylation value above the threshold value (e.g., the range associated with disease). The value of specificity, therefore, reflects the probability that a DNA methylation measurement for a given marker obtained from a known non-neoplastic sample will be in the range of non-disease associated measurements. As defined here, the clinical relevance of the calculated specificity value represents an estimation of the probability that a given marker would detect the absence of a clinical condition when applied to a patient without that condition.
As used herein, a "selected nucleotide" refers to one nucleotide of the four typically occurring nucleotides in a nucleic acid molecule (C, G, T, and A for DNA and C, G, U, and A for RNA), and can include methylated derivatives of the typically occurring nucleotides (e.g., when C is the selected nucleotide, both methylated and unmethylated C are included within the meaning of a selected nucleotide), whereas a methylated selected nucleotide refers specifically to a nucleotide that is typically methylated and an unmethylated selected nucleotides refers specifically to a nucleotide that typically occurs in unmethylated form.
The terms "methylation-specific restriction enzyme" or "methylation-sensitive restriction enzyme" refers to an enzyme that selectively digests a nucleic acid dependent on the methylation state of its recognition site. In the case of a restriction enzyme that specifically cuts if the recognition site is not methylated or is hemi-methylated, the cut will not take place or will take place with a significantly reduced efficiency if the recognition site is methylated. In the case of a restriction enzyme that specifically cuts if the recognition site is methylated, the cut will not take place or will take place with a significantly reduced efficiency if the recognition site is not methylated. Preferred are methylation-specific restriction enzymes, the recognition sequence of which contains a CG dinucleotide (for instance a recognition sequence such as CGCG or CCCGGG). Further preferred for some embodiments are restriction enzymes that do not cut if the cytosine in this dinucleotide is methylated at the carbon atom C5.
The term "primer" refers to an oligonucleotide, whether occurring naturally as, e.g., a nucleic acid fragment from a restriction digest, or produced synthetically, that is capable of acting as a point of initiation of synthesis when placed under conditions in which synthesis of a primer extension product that is complementary to a nucleic acid template strand is induced, (e.g., in the presence of nucleotides and an inducing agent such as a DNA polymerase, and at a suitable temperature and pH). The primer is preferably single stranded for maximum efficiency in amplification, but may alternatively be double stranded. If double stranded, the primer is first treated to separate its strands before being used to prepare extension products. Preferably, the primer is an oligodeoxyribonucleotide. The primer must be sufficiently long to prime the synthesis of extension products in the presence of the inducing agent. The exact lengths of the primers will depend on many factors, including temperature, source of primer, and the use of the method.
The term "probe" refers to an oligonucleotide (e.g., a sequence of nucleotides), whether occurring naturally as in a purified restriction digest or produced synthetically, recombinantly, or by PCR amplification, that is capable of hybridizing to another oligonucleotide of interest. A probe may be single-stranded or double-stranded. Probes are useful in the detection, identification, and isolation of particular gene sequences (e.g., a "capture probe"). It is contemplated that any probe used in the present invention may, in some embodiments, be labeled with any "reporter molecule," so that is detectable in any detection system, including, but not limited to enzyme (e.g., ELISA, as well as enzyme-based histochemical assays), fluorescent, radioactive, and luminescent systems. It is not intended that the present invention be limited to any particular detection system or label.
The term "target," as used herein refers to a nucleic acid sought to be sorted out from other nucleic acids, e.g., by probe binding, amplification, isolation, capture, etc. For example, when used in reference to the polymerase chain reaction, "target" refers to the region of nucleic acid bounded by the primers used for polymerase chain reaction, while when used in an assay in which target DNA is not amplified, e.g., in some embodiments of an invasive cleavage assay, a target comprises the site at which a probe and invasive oligonucleotides (e.g., INVADER oligonucleotide) bind to form an invasive cleavage structure, such that the presence of the target nucleic acid can be detected. A "segment" is defined as a region of nucleic acid within the target sequence.
The term "marker", as used herein, refers to a substance (e.g., a nucleic acid, or a region of a nucleic acid, or a protein) that may be used to distinguish non-normal cells (e.g., cancer cells) from normal cells (non-cancerous cells), e.g., based on presence, absence, or status (e.g., methylation state) of the marker substance. As used herein "normal" methylation of a marker refers to a degree of methylation typically found in normal cells, e.g., in non-cancerous cells.
The term "neoplasm" as used herein refers to any new and abnormal growth of tissue. Thus, a neoplasm can be a premalignant neoplasm or a malignant neoplasm.
The term "neoplasm-specific marker," as used herein, refers to any biological material or element that can be used to indicate the presence of a neoplasm. Examples of biological materials include, without limitation, nucleic acids, polypeptides, carbohydrates, fatty acids, cellular components (e.g., cell membranes and mitochondria), and whole cells. In some instances, markers are particular nucleic acid regions (e.g., genes, intragenic regions, specific loci, etc.). Regions of nucleic acid that are markers may be referred to, e.g., as "marker genes," "marker regions," "marker sequences," "marker loci," etc.
The term "sample" is used in its broadest sense. In one sense it can refer to an animal cell or tissue. In another sense, it refers to a specimen or culture obtained from any source, as well as biological and environmental samples. Biological samples may be obtained from plants or animals (including humans) and encompass fluids, solids, tissues, and gases. Environmental samples include environmental material such as surface matter, soil, water, and industrial samples. These examples are not to be construed as limiting the sample types applicable to the present invention.
As used herein, the terms "patient" or "subject" refer to organisms to be subject to various tests provided by the technology. The term "subject" includes animals, preferably mammals, including humans. In a preferred embodiment, the subject is a primate. In an even more preferred embodiment, the subject is a human. Further with respect to diagnostic methods, a preferred subject is a vertebrate subject. A preferred vertebrate is warm-blooded; a preferred warm-blooded vertebrate is a mammal. A preferred mammal is most preferably a human. As used herein, the term "subject` includes both human and animal subjects. Thus, veterinary therapeutic uses are provided herein. As such, the present technology provides for the diagnosis of mammals such as humans, as well as those mammals of importance due to being endangered, such as Siberian tigers; of economic importance, such as animals raised on farms for consumption by humans; and/or animals of social importance to humans, such as animals kept as pets or in zoos. Examples of such animals include but are not limited to: carnivores such as cats and dogs; swine, including pigs, hogs, and wild boars; ruminants and/or ungulates such as cattle, oxen, sheep, giraffes, deer, goats, bison, and camels; pinnipeds; and horses. Thus, also provided is the diagnosis and treatment of livestock, including, but not limited to, domesticated swine, ruminants, ungulates, horses (including race horses), and the like. The presently-disclosed subject matter further includes a system for diagnosing a lung cancer in a subject. The system can be provided, for example, as a commercial kit that can be used to screen for a risk of lung cancer or diagnose a lung cancer in a subject from whom a biological sample has been collected. An exemplary system provided in accordance with the present technology includes assessing the methylation state of a marker described herein.
The term "amplifying" or "amplification" in the context of nucleic acids refers to the production of multiple copies of a polynucleotide, or a portion of the polynucleotide, typically starting from a small amount of the polynucleotide (e.g., a single polynucleotide molecule), where the amplification products or amplicons are generally detectable. Amplification of polynucleotides encompasses a variety of chemical and enzymatic processes. The generation of multiple DNA copies from one or a few copies of a target or template DNA molecule during a polymerase chain reaction (PCR) or a ligase chain reaction (LCR; see, e.g., U.S. Pat. No. 5,494,810; herein incorporated by reference in its entirety) are forms of amplification. Additional types of amplification include, but are not limited to, allele-specific PCR (see, e.g., U.S. Pat. No. 5,639,611; herein incorporated by reference in its entirety), assembly PCR (see, e.g., U.S. Pat. No. 5,965,408; herein incorporated by reference in its entirety), helicase-dependent amplification (see, e.g., U.S. Pat. No. 7,662,594; herein incorporated by reference in its entirety), hot-start PCR (see, e.g., U.S. Pat. Nos. 5,773,258 and 5,338,671; each herein incorporated by reference in their entireties), intersequence-specific PCR, inverse PCR (see, e.g., Triglia, et al. (1988) Nucleic Acids Res., 16:8186; herein incorporated by reference in its entirety), ligation-mediated PCR (see, e.g., Guilfoyle, R. et al., Nucleic Acids Research, 25:1854-1858 (1997); U.S. Pat. No. 5,508,169; each of which are herein incorporated by reference in their entireties), methylation-specific PCR (see, e.g., Herman, et al., (1996) PNAS 93(13) 9821-9826; herein incorporated by reference in its entirety), miniprimer PCR, multiplex ligation-dependent probe amplification (see, e.g., Schouten, et al., (2002) Nucleic Acids Research 30(12): e57; herein incorporated by reference in its entirety), multiplex PCR (see, e.g., Chamberlain, et al., (1988) Nucleic Acids Research 16(23) 11141-11156; Ballabio, et al., (1990) Human Genetics 84(6) 571-573; Hayden, et al., (2008) BMC Genetics 9:80; each of which are herein incorporated by reference in their entireties), nested PCR, overlap-extension PCR (see, e.g., Higuchi, et al., (1988) Nucleic Acids Research 16(15) 7351-7367; herein incorporated by reference in its entirety), real time PCR (see, e.g., Higuchi, et al., (1992) Biotechnology 10:413-417; Higuchi, et al., (1993) Biotechnology 11:1026-1030; each of which are herein incorporated by reference in their entireties), reverse transcription PCR (see, e.g., Bustin, S. A. (2000) J. Molecular Endocrinology 25:169-193; herein incorporated by reference in its entirety), solid phase PCR, thermal asymmetric interlaced PCR, and Touchdown PCR (see, e.g., Don, et al., Nucleic Acids Research (1991) 19(14) 4008; Roux, K. (1994) Biotechniques 16(5) 812-814; Hecker, et al., (1996) Biotechniques 20(3) 478-485; each of which are herein incorporated by reference in their entireties). Polynucleotide amplification also can be accomplished using digital PCR (see, e.g., Kalinina, et al., Nucleic Acids Research. 25; 1999-2004, (1997); Vogelstein and Kinzler, Proc Natl Acad Sci USA. 96; 9236-41, (1999); International Patent Publication No. WO05023091A2; U.S. Patent Application Publication No. 20070202525; each of which are incorporated herein by reference in their entireties).
The term "polymerase chain reaction" ("PCR") refers to the method of K. B. Mullis U.S. Pat. Nos. 4,683,195, 4,683,202, and 4,965,188, that describe a method for increasing the concentration of a segment of a target sequence in a mixture of genomic or other DNA or RNA, without cloning or purification. This process for amplifying the target sequence consists of introducing a large excess of two oligonucleotide primers to the DNA mixture containing the desired target sequence, followed by a precise sequence of thermal cycling in the presence of a DNA polymerase. The two primers are complementary to their respective strands of the double stranded target sequence. To effect amplification, the mixture is denatured and the primers then annealed to their complementary sequences within the target molecule. Following annealing, the primers are extended with a polymerase so as to form a new pair of complementary strands. The steps of denaturation, primer annealing, and polymerase extension can be repeated many times (i.e., denaturation, annealing and extension constitute one "cycle"; there can be numerous "cycles") to obtain a high concentration of an amplified segment of the desired target sequence. The length of the amplified segment of the desired target sequence is determined by the relative positions of the primers with respect to each other, and therefore, this length is a controllable parameter. By virtue of the repeating aspect of the process, the method is referred to as the "polymerase chain reaction" ("PCR"). Because the desired amplified segments of the target sequence become the predominant sequences (in terms of concentration) in the mixture, they are said to be "PCR amplified" and are "PCR products" or "amplicons." Those of skill in the art will understand the term "PCR" encompasses many variants of the originally described method using, e.g., real time PCR, nested PCR, reverse transcription PCR (RT-PCR), single primer and arbitrarily primed PCR, etc.
As used herein, the term "nucleic acid detection assay" refers to any method of determining the nucleotide composition of a nucleic acid of interest. Nucleic acid detection assay include but are not limited to, DNA sequencing methods, probe hybridization methods, structure specific cleavage assays (e.g., the INVADER assay, (Hologic, Inc.) and are described, e.g., in U.S. Pat. Nos. 5,846,717, 5,985,557, 5,994,069, 6,001,567, 6,090,543, and 6,872,816; Lyamichev et al., Nat. Biotech., 17:292 (1999), Hall et al., PNAS, USA, 97:8272 (2000), and U.S. Pat. No. 9,096,893, each of which is herein incorporated by reference in its entirety for all purposes); enzyme mismatch cleavage methods (e.g., Variagenics, U.S. Pat. Nos. 6,110,684, 5,958,692, 5,851,770, herein incorporated by reference in their entireties); polymerase chain reaction (PCR), described above; branched hybridization methods (e.g., Chiron, U.S. Pat. Nos. 5,849,481, 5,710,264, 5,124,246, and 5,624,802, herein incorporated by reference in their entireties); rolling circle replication (e.g., U.S. Pat. Nos. 6,210,884, 6,183,960 and 6,235,502, herein incorporated by reference in their entireties); NASBA (e.g., U.S. Pat. No. 5,409,818, herein incorporated by reference in its entirety); molecular beacon technology (e.g., U.S. Pat. No. 6,150,097, herein incorporated by reference in its entirety); E-sensor technology (Motorola, U.S. Pat. Nos. 6,248,229, 6,221,583, 6,013,170, and 6,063,573, herein incorporated by reference in their entireties); cycling probe technology (e.g., U.S. Pat. Nos. 5,403,711, 5,011,769, and 5,660,988, herein incorporated by reference in their entireties); Dade Behring signal amplification methods (e.g., U.S. Pat. Nos. 6,121,001, 6,110,677, 5,914,230, 5,882,867, and 5,792,614, herein incorporated by reference in their entireties); ligase chain reaction (e.g., Baranay Proc. Natl. Acad. Sci USA 88, 189-93 (1991)); and sandwich hybridization methods (e.g., U.S. Pat. No. 5,288,609, herein incorporated by reference in its entirety).
In some embodiments, target nucleic acid is amplified (e.g., by PCR) and amplified nucleic acid is detected simultaneously using an invasive cleavage assay. Assays configured for performing a detection assay (e.g., invasive cleavage assay) in combination with an amplification assay are described in U.S. Pat. No. 9,096,893, incorporated herein by reference in its entirety for all purposes. Additional amplification plus invasive cleavage detection configurations, termed the QUARTS method, are described in, e.g., in U.S. Pat. Nos. 8,361,720; 8,715,937; 8,916,344; and 9,212,392, each of which is incorporated herein by reference for all purposes. The term "invasive cleavage structure" as used herein refers to a cleavage structure comprising i) a target nucleic acid, ii) an upstream nucleic acid (e.g., an invasive or "INVADER" oligonucleotide), and iii) a downstream nucleic acid (e.g., a probe), where the upstream and downstream nucleic acids anneal to contiguous regions of the target nucleic acid, and where an overlap forms between the a 3' portion of the upstream nucleic acid and duplex formed between the downstream nucleic acid and the target nucleic acid. An overlap occurs where one or more bases from the upstream and downstream nucleic acids occupy the same position with respect to a target nucleic acid base, whether or not the overlapping base(s) of the upstream nucleic acid are complementary with the target nucleic acid, and whether or not those bases are natural bases or non-natural bases. In some embodiments, the 3' portion of the upstream nucleic acid that overlaps with the downstream duplex is a non-base chemical moiety such as an aromatic ring structure, e.g., as disclosed, for example, in U.S. Pat. No. 6,090,543, incorporated herein by reference in its entirety. In some embodiments, one or more of the nucleic acids may be attached to each other, e.g., through a covalent linkage such as nucleic acid stem-loop, or through a non-nucleic acid chemical linkage (e.g., a multi-carbon chain). As used herein, the term "flap endonuclease assay" includes "INVADER" invasive cleavage assays and QuARTS assays, as described above.
The term "probe oligonucleotide" or "flap oligonucleotide" when used in reference to flap assay, refers to an oligonucleotide that interacts with a target nucleic acid to form a cleavage structure in the presence of an invasive oligonucleotide.
The term "invasive oligonucleotide" refers to an oligonucleotide that hybridizes to a target nucleic acid at a location adjacent to the region of hybridization between a probe and the target nucleic acid, wherein the 3' end of the invasive oligonucleotide comprises a portion (e.g., a chemical moiety, or one or more nucleotides) that overlaps with the region of hybridization between the probe and target. The 3' terminal nucleotide of the invasive oligonucleotide may or may not base pair a nucleotide in the target. In some embodiments, the invasive oligonucleotide contains sequences at its 3' end that are substantially the same as sequences located at the 5' end of a portion of the probe oligonucleotide that anneals to the target strand.
The term "flap endonuclease" or "FEN," as used herein, refers to a class of nucleolytic enzymes, typically 5' nucleases, that act as structure-specific endonucleases on DNA structures with a duplex containing a single stranded 5' overhang, or flap, on one of the strands that is displaced by another strand of nucleic acid (e.g., such that there are overlapping nucleotides at the junction between the single and double-stranded DNA). FENs catalyze hydrolytic cleavage of the phosphodiester bond at the junction of single and double stranded DNA, releasing the overhang, or the flap. Flap endonucleases are reviewed by Ceska and Savers (Trends Biochem. Sci. 1998 23:331-336) and Liu et al (Annu. Rev. Biochem. 2004 73: 589-615; herein incorporated by reference in its entirety). FENs may be individual enzymes, multi-subunit enzymes, or may exist as an activity of another enzyme or protein complex (e.g., a DNA polymerase).
A flap endonuclease may be thermostable. For example, FEN-1 flap endonuclease from archival thermophiles organisms are typical thermostable. As used herein, the term "FEN-1" refers to a non-polymerase flap endonuclease from a eukaryote or archaeal organism. See, e.g., WO 02/070755, and Kaiser M. W., et al. (1999) J. Biol. Chem., 274:21387, which are incorporated by reference herein in their entireties for all purposes.
As used herein, the term "cleaved flap" refers to a single-stranded oligonucleotide that is a cleavage product of a flap assay.
The term "cassette," when used in reference to a flap cleavage reaction, refers to an oligonucleotide or combination of oligonucleotides configured to generate a detectable signal in response to cleavage of a flap or probe oligonucleotide, e.g., in a primary or first cleavage structure formed in a flap cleavage assay. In preferred embodiments, the cassette hybridizes to a non-target cleavage product produced by cleavage of a flap oligonucleotide to form a second overlapping cleavage structure, such that the cassette can then be cleaved by the same enzyme, e.g., a FEN-1 endonuclease.
In some embodiments, the cassette is a single oligonucleotide comprising a hairpin portion (i.e., a region wherein one portion of the cassette oligonucleotide hybridizes to a second portion of the same oligonucleotide under reaction conditions, to form a duplex). In other embodiments, a cassette comprises at least two oligonucleotides comprising complementary portions that can form a duplex under reaction conditions. In preferred embodiments, the cassette comprises a label, e.g., a fluorophore. In particularly preferred embodiments, a cassette comprises labeled moieties that produce a FRET effect.
As used herein, the term "FRET" refers to fluorescence resonance energy transfer, a process in which moieties (e.g., fluorophores) transfer energy e.g., among themselves, or, from a fluorophore to a non-fluorophore (e.g., a quencher molecule). In some circumstances, FRET involves an excited donor fluorophore transferring energy to a lower-energy acceptor fluorophore via a short-range (e.g., about 10 nm or less) dipole-dipole interaction. In other circumstances, FRET involves a loss of fluorescence energy from a donor and an increase in fluorescence in an acceptor fluorophore. In still other forms of FRET, energy can be exchanged from an excited donor fluorophore to a non-fluorescing molecule (e.g., a "dark" quenching molecule). FRET is known to those of skill in the art and has been described (See, e.g., Stryer et al., 1978, Ann. Rev. Biochem., 47:819; Selvin, 1995, Methods Enzymol., 246:300; Orpana, 2004 Biomol Eng 21, 45-50; Olivier, 2005 Mutant Res 573, 103-110, each of which is incorporated herein by reference in its entirety).
In an exemplary flap detection assay, an invasive oligonucleotide and flap oligonucleotide are hybridized to a target nucleic acid to produce a first complex having an overlap as described above. An unpaired "flap" is included on the 5' end of the flap oligonucleotide. The first complex is a substrate for a flap endonuclease, e.g., a FEN-1 endonuclease, which cleaves the flap oligonucleotide to release the 5' flap portion. In a secondary reaction, the released 5' flap product serves as an invasive oligonucleotide on a FRET cassette to again create the structure recognized by the flap endonuclease, such that the FRET cassette is cleaved. When the fluorophore and the quencher are separated by cleavage of the FRET cassette, a detectable fluorescent signal above background fluorescence is produced.
The term "real time" as used herein in reference to detection of nucleic acid amplification or signal amplification refers to the detection or measurement of the accumulation of products or signal in the reaction while the reaction is in progress, e.g., during incubation or thermal cycling. Such detection or measurement may occur continuously, or it may occur at a plurality of discrete points during the progress of the amplification reaction, or it may be a combination. For example, in a polymerase chain reaction, detection (e.g., of fluorescence) may occur continuously during all or part of thermal cycling, or it may occur transiently, at one or more points during one or more cycles. In some embodiments, real time detection of PCR or QUARTS reactions is accomplished by determining a level of fluorescence at the same point (e.g., a time point in the cycle, or temperature step in the cycle) in each of a plurality of cycles, or in every cycle. Real time detection of amplification may also be referred to as detection "during" the amplification reaction.
As used herein, the term "quantitative amplification data set" refers to the data obtained during quantitative amplification of the target sample, e.g., target DNA. In the case of quantitative PCR or QuARTS assays, the quantitative amplification data set is a collection of fluorescence values obtained at during amplification, e.g., during a plurality of, or all of the thermal cycles. Data for quantitative amplification is not limited to data collected at any particular point in a reaction, and fluorescence may be measured at a discrete point in each cycle or continuously throughout each cycle.
The abbreviations "Ct" and "Cp" as used herein in reference to data collected during real time PCR and PCR+INVADER assays refer to the cycle at which signal (e.g., fluorescent signal) crosses a predetermined threshold value indicative of positive signal. Various methods have been used to calculate the threshold that is used as a determinant of signal verses concentration, and the value is generally expressed as either the "crossing threshold" (Ct) or the "crossing point" (Cp). Either Cp values or Ct values may be used in embodiments of the methods presented herein for analysis of real-time signal for the determination of the percentage of variant and/or non-variant constituents in an assay or sample.
As used herein, the term "kit" refers to any delivery system for delivering materials. In the context of reaction assays, such delivery systems include systems that allow for the storage, transport, or delivery of reaction reagents (e.g., oligonucleotides, enzymes, etc. in the appropriate containers) and/or supporting materials (e.g., buffers, written instructions for performing the assay etc.) from one location to another. For example, kits include one or more enclosures (e.g., boxes) containing the relevant reaction reagents and/or supporting materials. As used herein, the term "fragmented kit" refers to delivery systems comprising two or more separate containers that each contains a subportion of the total kit components. The containers may be delivered to the intended recipient together or separately. For example, a first container may contain an enzyme for use in an assay, while a second container contains oligonucleotides.
The term "system" as used herein refers to a collection of articles for use for a particular purpose. In some embodiments, the articles comprise instructions for use, as information supplied on e.g., an article, on paper, or on recordable media (e.g., DVD, CD, flash drive, etc.). In some embodiments, instructions direct a user to an online location, e.g., a website.
As used herein, the term "information" refers to any collection of facts or data. In reference to information stored or processed using a computer system(s), including but not limited to internets, the term refers to any data stored in any format (e.g., analog, digital, optical, etc.). As used herein, the term "information related to a subject" refers to facts or data pertaining to a subject (e.g., a human, plant, or animal). The term "genomic information" refers to information pertaining to a genome including, but not limited to, nucleic acid sequences, genes, percentage methylation, allele frequencies, RNA expression levels, protein expression, phenotypes correlating to genotypes, etc. "Allele frequency information" refers to facts or data pertaining to allele frequencies, including, but not limited to, allele identities, statistical correlations between the presence of an allele and a characteristic of a subject (e.g., a human subject), the presence or absence of an allele in an individual or population, the percentage likelihood of an allele being present in an individual having one or more particular characteristics, etc.
DESCRIPTION OF THE DRAWINGS
FIG. 1 shows schematic diagrams of marker target regions in unconverted form and bisulfite-converted form. Flap assay primers and probes for detection of bisulfate-converted target DNA are shown.
FIGS. 2-5 provide tables comparing Reduced Representation Bisulfite Sequencing (RRBS) results for selecting markers associated with lung carcinomas as described in Example 2, with each row showing the mean values for the indicated marker region (identified by chromosome and start and stop positions). The ratio of mean methylation for each tissue type (normal (Norm), adenocarcinoma (Ad), large cell carcinoma (LC), small cell carcinoma(SC), squamous cell carcinoma (SQ) and undefined cancer (UND)) is compared to the mean methylation of buffy coat samples from normal subjects (WBC or BC)) is shown for each region, and genes and transcripts identified with each region are indicated.
FIG. 2 provides a table comparing RRBS results for selecting markers associated with lung adenocarcinoma.
FIG. 3 provides a table comparing RRBS results for selecting markers associated with lung large cell carcinoma.
FIG. 4 provides a table comparing RRBS results for selecting markers associated with lung small cell carcinoma.
FIG. 5 provides a table comparing RRBS results for selecting markers associated with lung squamous cell carcinoma.
FIG. 6 provides a table of nucleic acid sequences of assay targets and detection oligonucleotides, with corresponding SEQ ID NOS.
FIG. 7 provides a graph showing a 6-marker logistic fit of data from Example 3, using markers SHOX2, SOBP, ZNF781, BTACT, CYP26C1, and DLX4. The ROC curve analysis shows an area under the curve (AUC) of 0.973.
FIG. 8 provides a graph showing a 6-marker logistic fit of data from Example 3, using markers SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI. The ROC curve analysis shows an area under the curve (AUC) of 0.97982.
DETAILED DESCRIPTION OF THE INVENTION
Provided herein is technology relating to selection of nucleic acid markers for use in assays for detection and quantification of DNA, e.g., methylated DNA, and use of the markers in nucleic acid detection assays. In particular, the technology relates to use of methylation assays to detect lung cancer.
In this detailed description of the various embodiments, for purposes of explanation, numerous specific details are set forth to provide a thorough understanding of the embodiments disclosed. One skilled in the art will appreciate, however, that these various embodiments may be practiced with or without these specific details. In other instances, structures and devices are shown in block diagram form. Furthermore, one skilled in the art can readily appreciate that the specific sequences in which methods are presented and performed are illustrative and it is contemplated that the sequences can be varied and still remain within the spirit and scope of the various embodiments disclosed herein.
In some embodiments, a marker is a region of 100 or fewer bases, the marker is a region of 500 or fewer bases, the marker is a region of 1000 or fewer bases, the marker is a region of 5000 or fewer bases, or, in some embodiments, the marker is one base. In some embodiments the marker is in a high CpG density promoter.
The technology is not limited by sample type. For example, in some embodiments the sample is a stool sample, a tissue sample, sputum, a blood sample (e.g., plasma, serum, whole blood), an excretion, or a urine sample.
Furthermore, the technology is not limited in the method used to determine methylation state. In some embodiments the assaying comprises using methylation specific polymerase chain reaction, nucleic acid sequencing, mass spectrometry, methylation specific nuclease, mass-based separation, or target capture. In some embodiments, the assaying comprises use of a methylation specific oligonucleotide. In some embodiments, the technology uses massively parallel sequencing (e.g., next-generation sequencing) to determine methylation state, e.g., sequencing-by-synthesis, real-time (e.g., single-molecule) sequencing, bead emulsion sequencing, nanopore sequencing, etc.
The technology provides reagents for detecting a differentially methylated region (DMR). In some embodiments, an oligonucleotide is provided, the oligonucleotide comprising a sequence complementary to a chromosomal region having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSFJ, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably to a marker selected from the subset SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, EMX1, CYP26C1, SOBP, SUCLG2, SHOX2, ZDHHC1, NFIX, FLJ45983, HOXA9, B3GALT6, ZNF781, SP9, BARX1, and SKI; or a marker selected from any of the subsets of markers defining the group consisting of ZNF781, BARX1, and EMX1; the group consisting of SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI; the group consisting of SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, and EMX1; the group consisting of SHOX2, SOBP, ZNF781, BTACT, CYP26C1, and DLX4; or the group consisting of SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI.
Kit embodiments are provided, e.g., a kit comprising a bisulfate reagent; and a control nucleic acid comprising a chromosomal region having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably from any of the subsets of markers as recited above, and having a methylation state associated with a subject who does not have a cancer (e.g., lung cancer). In some embodiments, kits comprise a bisulfite reagent and an oligonucleotide as described herein. In some embodiments, kits comprise a bisulfite reagent; and a control nucleic acid comprising a sequence from such a chromosomal region and having a methylation state associated with a subject who has lung cancer.
The technology is related to embodiments of compositions (e.g., reaction mixtures). In some embodiments are provided a composition comprising a nucleic acid comprising a chromosomal region having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably from any of the subsets of markers as recited above, and a bisulfite reagent. Some embodiments provide a composition comprising a nucleic acid comprising a chromosomal region having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably from any of the subsets of markers as recited above, and an oligonucleotide as described herein. Some embodiments provide a composition comprising a nucleic acid comprising a chromosomal region having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably from any of the subsets of markers as recited above, and a methylation-sensitive restriction enzyme. Some embodiments provide a composition comprising a nucleic acid comprising a chromosomal region having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably from any of the subsets of markers as recited above, and a polymerase.
Additional related method embodiments are provided for screening for a neoplasm (e.g., lung carcinoma) in a sample obtained from a subject, e.g., a method comprising determining a methylation state of a marker in the sample comprising a base in a chromosomal region having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably from any of the subsets of markers as recited above, comparing the methylation state of the marker from the subject sample to a methylation state of the marker from a normal control sample from a subject who does not have lung cancer; and determining a confidence interval and/or a p value of the difference in the methylation state of the subject sample and the normal control sample. In some embodiments, the confidence interval is 90%, 95%, 97.5%, 98%, 99%, 99.5%, 99.9% or 99.99% and the p value is 0.1, 0.05, 0.025, 0.02, 0.01, 0.005, 0.001, or 0.0001. Some embodiments of methods provide steps of reacting a nucleic acid comprising a chromosomal region having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably from any of the subsets of markers as recited above, with a bisulfite reagent to produce a bisulfite-reacted nucleic acid; sequencing the bisulfite-reacted nucleic acid to provide a nucleotide sequence of the bisulfite-reacted nucleic acid; comparing the nucleotide sequence of the bisulfite-reacted nucleic acid with a nucleotide sequence of a nucleic acid comprising the chromosomal region from a subject who does not have lung cancer to identify differences in the two sequences; and identifying the subject as having a neoplasm when a difference is present.
Systems for screening for lung cancer in a sample obtained from a subject are provided by the technology. Exemplary embodiments of systems include, e.g., a system for screening for lung cancer in a sample obtained from a subject, the system comprising an analysis component configured to determine the methylation state of a sample, a software component configured to compare the methylation state of the sample with a control sample or a reference sample methylation state recorded in a database, and an alert component configured to alert a user of a cancer-associated methylation state. An alert is determined in some embodiments by a software component that receives the results from multiple assays (e.g., determining the methylation states of multiple markers, e.g., a chromosomal region having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably from any of the subsets of markers as recited above, and calculating a value or result to report based on the multiple results. Some embodiments provide a database of weighted parameters associated with each a chromosomal region having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably from any of the subsets of markers as recited above, provided herein for use in calculating a value or result and/or an alert to report to a user (e.g., such as a physician, nurse, clinician, etc.). In some embodiments all results from multiple assays are reported and in some embodiments one or more results are used to provide a score, value, or result based on a composite of one or more results from multiple assays that is indicative of a lung cancer risk in a subject.
In some embodiments of systems, a sample comprises a nucleic acid comprising a chromosomal region having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably from any of the subsets of markers as recited above. In some embodiments the system further comprises a component for isolating a nucleic acid, a component for collecting a sample such as a component for collecting a stool sample. In some embodiments, the system comprises nucleic acid sequences comprising a chromosomal region having an annotation selected from BARU, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably from any of the subsets of markers as recited above. In some embodiments the database comprises nucleic acid sequences from subjects who do not have lung cancer. Also provided are nucleic acids, e.g., a set of nucleic acids, each nucleic acid having a sequence comprising a chromosomal region having an annotation selected from BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably from any of the subsets of markers as recited above.
Related system embodiments comprise a set of nucleic acids as described and a database of nucleic acid sequences associated with the set of nucleic acids. Some embodiments further comprise a bisulfate reagent. And, some embodiments further comprise a nucleic acid sequencer.
In certain embodiments, methods for characterizing a sample obtained from a human subject are provided, comprising a) obtaining a sample from a human subject; b) assaying a methylation state of one or more markers in the sample, wherein the marker comprises a base in a chromosomal region having an annotation selected from the following groups of markers: BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329, preferably from any of the subsets of markers as recited above; and c) comparing the methylation state of the assayed marker to the methylation state of the marker assayed in a subject that does not have a neoplasm.
In some embodiments, the technology is related to assessing the presence of and methylation state of one or more of the markers identified herein in a biological sample. These markers comprise one or more differentially methylated regions (DMR) as discussed herein. Methylation state is assessed in embodiments of the technology. As such, the technology provided herein is not restricted in the method by which a gene's methylation state is measured. For example, in some embodiments the methylation state is measured by a genome scanning method. For example, one method involves restriction landmark genomic scanning (Kawai et al. (1994) Mol. Cell. Biol. 14: 7421-7427) and another example involves methylation-sensitive arbitrarily primed PCR (Gonzalgo et al. (1997) Cancer Res. 57: 594-599). In some embodiments, changes in methylation patterns at specific CpG sites are monitored by digestion of genomic DNA with methylation-sensitive restriction enzymes followed by Southern analysis of the regions of interest (digestion-Southern method). In some embodiments, analyzing changes in methylation patterns involves a PCR-based process that involves digestion of genomic DNA with methylation-sensitive restriction enzymes prior to PCR amplification (Singer-Sam et al. (1990) Nucl. Acids Res. 18: 687). In addition, other techniques have been reported that utilize bisulfate treatment of DNA as a starting point for methylation analysis. These include methylation-specific PCR (MSP) (Herman et al. (1992) Proc. Natl. Acad. Sci. USA 93: 9821-9826) and restriction enzyme digestion of PCR products amplified from bisulfite-converted DNA (Sadri and Hornsby (1996) Nucl. Acids Res. 24: 5058-5059; and Xiong and Laird (1997) Nucl. Acids Res. 25: 2532-2534). PCR techniques have been developed for detection of gene mutations (Kuppuswamy et al. (1991) Proc. Natl. Acad. Sci. USA 88: 1143-1147) and quantification of allelic-specific expression (Szabo and Mann (1995) Genes Dev. 9: 3097-3108; and Singer-Sam et al. (1992) PCR Methods Appl. 1: 160-163). Such techniques use internal primers, which anneal to a PCR-generated template and terminate immediately 5' of the single nucleotide to be assayed. Methods using a "quantitative Ms-SNuPE assay" as described in U.S. Pat. No. 7,037,650 are used in some embodiments.
Upon evaluating a methylation state, the methylation state is often expressed as the fraction or percentage of individual strands of DNA that is methylated at a particular site (e.g., at a single nucleotide, at a particular region or locus, at a longer sequence of interest, e.g., up to a .about.100-bp, 200-bp, 500-bp, 1000-bp subsequence of a DNA or longer) relative to the total population of DNA in the sample comprising that particular site. Traditionally, the amount of the unmethylated nucleic acid is determined by PCR using calibrators. Then, a known amount of DNA is bisulfite treated and the resulting methylation-specific sequence is determined using either a real-time PCR or other exponential amplification, e.g., a QuARTS assay (e.g., as provided by U.S. Pat. Nos. 8,361,720; 8,715,937; 8,916,344; and 9,212,392).
For example, in some embodiments methods comprise generating a standard curve for the unmethylated target by using external standards. The standard curve is constructed from at least two points and relates the real-time Ct value for unmethylated DNA to known quantitative standards. Then, a second standard curve for the methylated target is constructed from at least two points and external standards. This second standard curve relates the Ct for methylated DNA to known quantitative standards. Next, the test sample Ct values are determined for the methylated and unmethylated populations and the genomic equivalents of DNA are calculated from the standard curves produced by the first two steps. The percentage of methylation at the site of interest is calculated from the amount of methylated DNAs relative to the total amount of DNAs in the population, e.g., (number of methylated DNAs)/(the number of methylated DNAs+number of unmethylated DNAs).times.100.
Also provided herein are compositions and kits for practicing the methods. For example, in some embodiments, reagents (e.g., primers, probes) specific for one or more markers are provided alone or in sets (e.g., sets of primers pairs for amplifying a plurality of markers). Additional reagents for conducting a detection assay may also be provided (e.g., enzymes, buffers, positive and negative controls for conducting QuARTS, PCR, sequencing, bisulfite, or other assays). In some embodiments, the kits containing one or more reagent necessary, sufficient, or useful for conducting a method are provided. Also provided are reactions mixtures containing the reagents. Further provided are master mix reagent sets containing a plurality of reagents that may be added to each other and/or to a test sample to complete a reaction mixture.
Methods for isolating DNA suitable for these assay technologies are known in the art. In particular, some embodiments comprise isolation of nucleic acids as described in U.S. patent application Ser. No. 13/470,251 ("Isolation of Nucleic Acids"), incorporated herein by reference in its entirety.
Genomic DNA may be isolated by any means, including the use of commercially available kits. Briefly, wherein the DNA of interest is encapsulated by a cellular membrane the biological sample must be disrupted and lysed by enzymatic, chemical or mechanical means. The DNA solution may then be cleared of proteins and other contaminants, e.g., by digestion with proteinase K. The genomic DNA is then recovered from the solution. This may be carried out by means of a variety of methods including salting out, organic extraction, or binding of the DNA to a solid phase support. The choice of method will be affected by several factors including time, expense, and required quantity of DNA. All clinical sample types comprising neoplastic matter or pre-neoplastic matter are suitable for use in the present method, e.g., cell lines, histological slides, biopsies, paraffin-embedded tissue, body fluids, stool, colonic effluent, urine, blood plasma, blood serum, whole blood, isolated blood cells, cells isolated from the blood, and combinations thereof.
The technology is not limited in the methods used to prepare the samples and provide a nucleic acid for testing. For example, in some embodiments, a DNA is isolated from a stool sample or from blood or from a plasma sample using direct gene capture, e.g., as detailed in U.S. Pat. Appl. Ser. No. 61/485,386 or by a related method.
The technology relates to the analysis of any sample that may be associated with lung cancer, or that may be examined to establish the absence of lung cancer. For example, in some embodiments the sample comprises a tissue and/or biological fluid obtained from a patient. In some embodiments, the sample comprises a secretion. In some embodiments, the sample comprises sputum, blood, serum, plasma, gastric secretions, lung tissue samples, lung cells or lung DNA recovered from stool. In some embodiments, the subject is human. Such samples can be obtained by any number of means known in the art, such as will be apparent to the skilled person.
I. Methylation Assays to Detect Lung Cancer
Candidate methylated DNA markers were identified by unbiased whole methylome sequencing of selected lung cancer case and lung control tissues. The top marker candidates were further evaluated in 255 independent patients with 119 controls, of which 37 were from benign nodules, and 136 cases inclusive of all lung cancer subtypes. DNA extracted from patient tissue samples was bisulfite treated and then candidate markers and .beta.-actin (ACTB) as a normalizing gene were assayed by Quantitative Allele-Specific Real-time Target and Signal amplification (QUARTS amplification). QuARTS assay chemistry yields high discrimination for methylated marker selection and screening.
On receiver operator characteristics analyses of individual marker candidates, areas under the curve (AUCs) ranged from 0.512 to 0.941. At 100% specificity, a combined panel of 8 methylation markers (SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX 12.526, HOXB2, and EMX1) yielded a sensitivity of 98.5% across all subtypes of lung cancer. Furthermore, using the 8 markers panel, benign lung nodules yielded no false positives.
II. Methylation Detection Assays and Kits
The markers described herein find use in a variety of methylation detection assays. The most frequently used method for analyzing a nucleic acid for the presence of 5-methylcytosine is based upon the bisulfite method described by Frommer, et al. for the detection of 5-methylcytosines in DNA (Frommer et al. (1992) Proc. Natl. Acad. Sci. USA 89: 1827-31 explicitly incorporated herein by reference in its entirety for all purposes) or variations thereof. The bisulfite method of mapping 5-methylcytosines is based on the observation that cytosine, but not 5-methylcytosine, reacts with hydrogen sulfite ion (also known as bisulfite). The reaction is usually performed according to the following steps: first, cytosine reacts with hydrogen sulfite to form a sulfonated cytosine. Next, spontaneous deamination of the sulfonated reaction intermediate results in a sulfonated uracil. Finally, the sulfonated uracil is desulfonated under alkaline conditions to form uracil. Detection is possible because uracil base pairs with adenine (thus behaving like thymine), whereas 5-methylcytosine base pairs with guanine (thus behaving like cytosine). This makes the discrimination of methylated cytosines from non-methylated cytosines possible by, e.g., bisulfite genomic sequencing (Grigg G, & Clark S, Bioessays (1994) 16: 431-36; Grigg G, DNA Seq. (1996) 6: 189-98),methylation-specific PCR (MSP) as is disclosed, e.g., in U.S. Pat. No. 5,786,146, or using an assay comprising sequence-specific probe cleavage, e.g., a QuARTS flap endonuclease assay (see, e.g., Zou et al. (2010) "Sensitive quantification of methylated markers with a novel methylation specific technology" Clin Chem 56: A199; and in U.S. Pat. Nos. 8,361,720; 8,715,937; 8,916,344; and 9,212,392.
Some conventional technologies are related to methods comprising enclosing the DNA to be analyzed in an agarose matrix, thereby preventing the diffusion and renaturation of the DNA (bisulfite only reacts with single-stranded DNA), and replacing precipitation and purification steps with a fast dialysis (Olek A, et al. (1996) "A modified and improved method for bisulfite based cytosine methylation analysis" Nucleic Acids Res. 24: 5064-6). It is thus possible to analyze individual cells for methylation status, illustrating the utility and sensitivity of the method. An overview of conventional methods for detecting 5-methylcytosine is provided by Rein, T., et al. (1998) Nucleic Acids Res. 26: 2255.
The bisulfite technique typically involves amplifying short, specific fragments of a known nucleic acid subsequent to a bisulfite treatment, then either assaying the product by sequencing (Olek & Walter (1997) Nat. Genet. 17: 275-6) or a primer extension reaction (Gonzalgo & Jones (1997) Nucleic Acids Res. 25: 2529-31; WO 95/00669; U.S. Pat. No. 6,251,594) to analyze individual cytosine positions. Some methods use enzymatic digestion (Xiong & Laird (1997) Nucleic Acids Res. 25: 2532-4). Detection by hybridization has also been described in the art (Olek et al., WO 99/28498). Additionally, use of the bisulfite technique for methylation detection with respect to individual genes has been described (Grigg & Clark (1994) Bioessays 16: 431-6; Zeschnigk et al. (1997) Hum Mol Genet. 6: 387-95; Feil et al. (1994) Nucleic Acids Res. 22: 695; Martin et al. (1995) Gene 157: 261-4; WO 9746705; WO 9515373).
Various methylation assay procedures can be used in conjunction with bisulfite treatment according to the present technology. These assays allow for determination of the methylation state of one or a plurality of CpG dinucleotides (e.g., CpG islands) within a nucleic acid sequence. Such assays involve, among other techniques, sequencing of bisulfite-treated nucleic acid, PCR (for sequence-specific amplification), Southern blot analysis, and use of methylation-sensitive restriction enzymes.
For example, genomic sequencing has been simplified for analysis of methylation patterns and 5-methylcytosine distributions by using bisulfite treatment (Frommer et al. (1992) Proc. Natl. Acad. Sci. USA 89: 1827-1831). Additionally, restriction enzyme digestion of PCR products amplified from bisulfite-converted DNA finds use in assessing methylation state, e.g., as described by Sadri & Hornsby (1997) Nucl. Acids Res. 24: 5058-5059 or as embodied in the method known as COBRA (Combined Bisulfite Restriction Analysis) (Xiong & Laird (1997) Nucleic Acids Res. 25: 2532-2534).
COBRA.TM. analysis is a quantitative methylation assay useful for determining DNA methylation levels at specific loci in small amounts of genomic DNA (Xiong & Laird, Nucleic Acids Res. 25:2532-2534, 1997). Briefly, restriction enzyme digestion is used to reveal methylation-dependent sequence differences in PCR products of sodium bisulfite-treated DNA. Methylation-dependent sequence differences are first introduced into the genomic DNA by standard bisulfite treatment according to the procedure described by Frommer et al. (Proc. Natl. Acad. Sci. USA 89:1827-1831, 1992). PCR amplification of the bisulfite converted DNA is then performed using primers specific for the CpG islands of interest, followed by restriction endonuclease digestion, gel electrophoresis, and detection using specific, labeled hybridization probes. Methylation levels in the original DNA sample are represented by the relative amounts of digested and undigested PCR product in a linearly quantitative fashion across a wide spectrum of DNA methylation levels. In addition, this technique can be reliably applied to DNA obtained from microdissected paraffin-embedded tissue samples.
Typical reagents (e.g., as might be found in a typical COBRA.TM.-based kit) for COBRA.TM. analysis may include, but are not limited to: PCR primers for specific loci (e.g., specific genes, markers, regions of genes, regions of markers, bisulfite treated DNA sequence, CpG island, etc.); restriction enzyme and appropriate buffer; gene-hybridization oligonucleotide; control hybridization oligonucleotide; kinase labeling kit for oligonucleotide probe; and labeled nucleotides. Additionally, bisulfite conversion reagents may include: DNA denaturation buffer; sulfonation buffer; DNA recovery reagents or kits (e.g., precipitation, ultrafiltration, affinity column); desulfonation buffer; and DNA recovery components.
Assays such as "MethyLight.TM." (a fluorescence-based real-time PCR technique) (Eads et al., Cancer Res. 59:2302-2306, 1999), Ms-SNuPE.TM. (Methylation-sensitive Single Nucleotide Primer Extension) reactions (Gonzalgo & Jones, Nucleic Acids Res. 25:2529-2531, 1997), methylation-specific PCR ("MSP"; Herman et al., Proc. Natl. Acad. Sci. USA 93:9821-9826, 1996; U.S. Pat. No. 5,786,146), and methylated CpG island amplification ("MCA"; Toyota et al., Cancer Res. 59:2307-12, 1999) are used alone or in combination with one or more of these methods.
The "HeavyMethyl.TM." assay, technique is a quantitative method for assessing methylation differences based on methylation-specific amplification of bisulfite-treated DNA. Methylation-specific blocking probes ("blockers") covering CpG positions between, or covered by, the amplification primers enable methylation-specific selective amplification of a nucleic acid sample.
The term "HeavyMethyl.TM. MethyLight.TM." assay refers to a HeavyMethyl.TM. MethyLight.TM. assay, which is a variation of the MethyLight.TM. assay, wherein the MethyLight.TM. assay is combined with methylation specific blocking probes covering CpG positions between the amplification primers. The HeavyMethyl.TM. assay may also be used in combination with methylation specific amplification primers.
Typical reagents (e.g., as might be found in a typical MethyLight.TM.-based kit) for HeavyMethyl.TM. analysis may include, but are not limited to: PCR primers for specific loci (e.g., specific genes, markers, regions of genes, regions of markers, bisulfite treated DNA sequence, CpG island, or bisulfite treated DNA sequence or CpG island, etc.); blocking oligonucleotides; optimized PCR buffers and deoxynucleotides; and Taq polymerase.
MSP (methylation-specific PCR) allows for assessing the methylation status of virtually any group of CpG sites within a CpG island, independent of the use of methylation-sensitive restriction enzymes (Herman et al. Proc. Natl. Acad. Sci. USA 93:9821-9826, 1996; U.S. Pat. No. 5,786,146). Briefly, DNA is modified by sodium bisulfite, which converts unmethylated, but not methylated cytosines, to uracil, and the products are subsequently amplified with primers specific for methylated versus unmethylated DNA. MSP requires only small quantities of DNA, is sensitive to 0.1% methylated alleles of a given CpG island locus, and can be performed on DNA extracted from paraffin-embedded samples. Typical reagents (e.g., as might be found in a typical MSP-based kit) for MSP analysis may include, but are not limited to: methylated and unmethylated PCR primers for specific loci (e.g., specific genes, markers, regions of genes, regions of markers, bisulfite treated DNA sequence, CpG island, etc.); optimized PCR buffers and deoxynucleotides, and specific probes.
The MethyLight.TM. assay is a high-throughput quantitative methylation assay that utilizes fluorescence-based real-time PCR (e.g., TaqMan0) that requires no further manipulations after the PCR step (Eads et al., Cancer Res. 59:2302-2306, 1999). Briefly, the MethyLight.TM. process begins with a mixed sample of genomic DNA that is converted, in a sodium bisulfite reaction, to a mixed pool of methylation-dependent sequence differences according to standard procedures (the bisulfite process converts unmethylated cytosine residues to uracil). Fluorescence-based PCR is then performed in a "biased" reaction, e.g., with PCR primers that overlap known CpG dinucleotides. Sequence discrimination occurs both at the level of the amplification process and at the level of the fluorescence detection process.
The MethyLight.TM. assay is used as a quantitative test for methylation patterns in a nucleic acid, e.g., a genomic DNA sample, wherein sequence discrimination occurs at the level of probe hybridization. In a quantitative version, the PCR reaction provides for a methylation specific amplification in the presence of a fluorescent probe that overlaps a particular putative methylation site. An unbiased control for the amount of input DNA is provided by a reaction in which neither the primers, nor the probe, overlie any CpG dinucleotides. Alternatively, a qualitative test for genomic methylation is achieved by probing the biased PCR pool with either control oligonucleotides that do not cover known methylation sites (e.g., a fluorescence-based version of the HeavyMethyl.TM. and MSP techniques) or with oligonucleotides covering potential methylation sites.
The MethyLight.TM. process is used with any suitable probe (e.g. a "TaqMan.RTM." probe, a Lightcycler.RTM. probe, etc.) For example, in some applications double-stranded genomic DNA is treated with sodium bisulfite and subjected to one of two sets of PCR reactions using TaqMan.RTM. probes, e.g., with MSP primers and/or HeavyMethyl blocker oligonucleotides and a TaqMan.RTM. probe. The TaqMan.RTM. probe is dual-labeled with fluorescent "reporter" and "quencher" molecules and is designed to be specific for a relatively high GC content region so that it melts at about a 10.degree. C. higher temperature in the PCR cycle than the forward or reverse primers. This allows the TaqMan.RTM. probe to remain fully hybridized during the PCR annealing/extension step. As the Taq polymerase enzymatically synthesizes a new strand during PCR, it will eventually reach the annealed TaqMan.RTM. probe. The Taq polymerase 5' to 3' endonuclease activity will then displace the TaqMan.RTM. probe by digesting it to release the fluorescent reporter molecule for quantitative detection of its now unquenched signal using a real-time fluorescent detection system.
Typical reagents (e.g., as might be found in a typical MethyLight.TM.-based kit) for MethyLight.TM. analysis may include, but are not limited to: PCR primers for specific loci (e.g., specific genes, markers, regions of genes, regions of markers, bisulfite treated DNA sequence, CpG island, etc.); TaqMan.RTM. or Lightcycler.RTM. probes; optimized PCR buffers and deoxynucleotides; and Taq polymerase.
The QM.TM. (quantitative methylation) assay is an alternative quantitative test for methylation patterns in genomic DNA samples, wherein sequence discrimination occurs at the level of probe hybridization. In this quantitative version, the PCR reaction provides for unbiased amplification in the presence of a fluorescent probe that overlaps a particular putative methylation site. An unbiased control for the amount of input DNA is provided by a reaction in which neither the primers, nor the probe, overlie any CpG dinucleotides. Alternatively, a qualitative test for genomic methylation is achieved by probing the biased PCR pool with either control oligonucleotides that do not cover known methylation sites (a fluorescence-based version of the HeavyMethyl.TM. and MSP techniques) or with oligonucleotides covering potential methylation sites.
The QM.TM. process can be used with any suitable probe, e.g., "TaqMan.RTM." probes, Lightcycler.RTM. probes, in the amplification process. For example, double-stranded genomic DNA is treated with sodium bisulfite and subjected to unbiased primers and the TaqMan.RTM. probe. The TaqMan.RTM. probe is dual-labeled with fluorescent "reporter" and "quencher" molecules, and is designed to be specific for a relatively high GC content region so that it melts out at about a 10.degree. C. higher temperature in the PCR cycle than the forward or reverse primers. This allows the TaqMan.RTM. probe to remain fully hybridized during the PCR annealing/extension step. As the Taq polymerase enzymatically synthesizes a new strand during PCR, it will eventually reach the annealed TaqMan.RTM. probe. The Taq polymerase 5' to 3' endonuclease activity will then displace the TaqMan.RTM. probe by digesting it to release the fluorescent reporter molecule for quantitative detection of its now unquenched signal using a real-time fluorescent detection system. Typical reagents (e.g., as might be found in a typical QM.TM.-based kit) for QM.TM. analysis may include, but are not limited to: PCR primers for specific loci (e.g., specific genes, markers, regions of genes, regions of markers, bisulfite treated DNA sequence, CpG island, etc.); TaqMan.RTM. or Lightcycler.RTM. probes; optimized PCR buffers and deoxynucleotides; and Taq polymerase.
The Ms-SNuPE.TM. technique is a quantitative method for assessing methylation differences at specific CpG sites based on bisulfite treatment of DNA, followed by single-nucleotide primer extension (Gonzalgo & Jones, Nucleic Acids Res. 25:2529-2531, 1997). Briefly, genomic DNA is reacted with sodium bisulfite to convert unmethylated cytosine to uracil while leaving 5-methylcytosine unchanged. Amplification of the desired target sequence is then performed using PCR primers specific for bisulfite-converted DNA, and the resulting product is isolated and used as a template for methylation analysis at the CpG site of interest. Small amounts of DNA can be analyzed (e.g., microdissected pathology sections) and it avoids utilization of restriction enzymes for determining the methylation status at CpG sites.
Typical reagents (e.g., as might be found in a typical Ms-SNuPE.TM.-based kit) for Ms-SNuPE.TM. analysis may include, but are not limited to: PCR primers for specific loci (e.g., specific genes, markers, regions of genes, regions of markers, bisulfite treated DNA sequence, CpG island, etc.); optimized PCR buffers and deoxynucleotides; gel extraction kit; positive control primers; Ms-SNuPE.TM. primers for specific loci; reaction buffer (for the Ms-SNuPE reaction); and labeled nucleotides. Additionally, bisulfite conversion reagents may include: DNA denaturation buffer; sulfonation buffer; DNA recovery reagents or kit (e.g., precipitation, ultrafiltration, affinity column); desulfonation buffer; and DNA recovery components.
Reduced Representation Bisulfite Sequencing (RRBS) begins with bisulfite treatment of nucleic acid to convert all unmethylated cytosines to uracil, followed by restriction enzyme digestion (e.g., by an enzyme that recognizes a site including a CG sequence such as Mspl) and complete sequencing of fragments after coupling to an adapter ligand. The choice of restriction enzyme enriches the fragments for CpG dense regions, reducing the number of redundant sequences that may map to multiple gene positions during analysis. As such, RRBS reduces the complexity of the nucleic acid sample by selecting a subset (e.g., by size selection using preparative gel electrophoresis) of restriction fragments for sequencing. As opposed to whole-genome bisulfite sequencing, every fragment produced by the restriction enzyme digestion contains DNA methylation information for at least one CpG dinucleotide. As such, RRBS enriches the sample for promoters, CpG islands, and other genomic features with a high frequency of restriction enzyme cut sites in these regions and thus provides an assay to assess the methylation state of one or more genomic loci.
A typical protocol for RRBS comprises the steps of digesting a nucleic acid sample with a restriction enzyme such as Mspl, filling in overhangs and A-tailing, ligating adaptors, bisulfite conversion, and PCR. See, e.g., et al. (2005) "Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution" Nat Methods 7: 133-6; Meissner et al. (2005) "Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis" Nucleic Acids Res. 33: 5868-77.
In some embodiments, a quantitative allele-specific real-time target and signal amplification (QUARTS) assay is used to evaluate methylation state. Three reactions sequentially occur in each QuARTS assay, including amplification (reaction 1) and target probe cleavage (reaction 2) in the primary reaction; and FRET cleavage and fluorescent signal generation (reaction 3) in the secondary reaction. When target nucleic acid is amplified with specific primers, a specific detection probe with a flap sequence loosely binds to the amplicon. The presence of the specific invasive oligonucleotide at the target binding site causes a 5' nuclease, e.g., a FEN-1 endonuclease, to release the flap sequence by cutting between the detection probe and the flap sequence. The flap sequence is complementary to a non-hairpin portion of a corresponding FRET cassette. Accordingly, the flap sequence functions as an invasive oligonucleotide on the FRET cassette and effects a cleavage between the FRET cassette fluorophore and a quencher, which produces a fluorescent signal. The cleavage reaction can cut multiple probes per target and thus release multiple fluorophore per flap, providing exponential signal amplification. QuARTS can detect multiple targets in a single reaction well by using FRET cassettes with different dyes. See, e.g., in Zou et al. (2010) "Sensitive quantification of methylated markers with a novel methylation specific technology" Clin Chem 56: A199), and U.S. Pat. Nos. 8,361,720; 8,715,937; 8,916,344; and 9,212,392, each of which is incorporated herein by reference for all purposes.
In some embodiments, the bisulfite-treated DNA is purified prior to the quantification. This may be conducted by any means known in the art, such as but not limited to ultrafiltration, e.g., by means of Microcon.TM. columns (manufactured by Millipore.TM.). The purification is carried out according to a modified manufacturer's protocol (see, e.g., PCT/EP2004/011715, which is incorporated by reference in its entirety). In some embodiments, the bisulfate treated DNA is bound to a solid support, e.g., a magnetic bead, and desulfonation and washing occurs while the DNA is bound to the support. Examples of such embodiments are provided, e.g., in WO 2013/116375 and U.S. Pat. No. 9,315,853. In certain preferred embodiments, support-bound DNA is ready for a methylation assay immediately after desulfonation and washing on the support. In some embodiments, the desulfonated DNA is eluted from the support prior to assay.
In some embodiments, fragments of the treated DNA are amplified using sets of primer oligonucleotides according to the present invention (e.g., see FIG. 1) and an amplification enzyme. The amplification of several DNA segments can be carried out simultaneously in one and the same reaction vessel. Typically, the amplification is carried out using a polymerase chain reaction (PCR).
Methods for isolating DNA suitable for these assay technologies are known in the art. In particular, some embodiments comprise isolation of nucleic acids as described in U.S. Pat. Nos. 9,000,146 and 9,163,278, each incorporated herein by reference in its entirety.
In some embodiments, the markers described herein find use in QUARTS assays performed on stool samples. In some embodiments, methods for producing DNA samples and, in particular, to methods for producing DNA samples that comprise highly purified, low-abundance nucleic acids in a small volume (e.g., less than 100, less than 60 microliters) and that are substantially and/or effectively free of substances that inhibit assays used to test the DNA samples (e.g., PCR, INVADER, QuARTS assays, etc.) are provided. Such DNA samples find use in diagnostic assays that qualitatively detect the presence of, or quantitatively measure the activity, expression, or amount of, a gene, a gene variant (e.g., an allele), or a gene modification (e.g., methylation) present in a sample taken from a patient. For example, some cancers are correlated with the presence of particular mutant alleles or particular methylation states, and thus detecting and/or quantifying such mutant alleles or methylation states has predictive value in the diagnosis and treatment of cancer.
Many valuable genetic markers are present in extremely low amounts in samples and many of the events that produce such markers are rare. Consequently, even sensitive detection methods such as PCR require a large amount of DNA to provide enough of a low-abundance target to meet or supersede the detection threshold of the assay. Moreover, the presence of even low amounts of inhibitory substances compromise the accuracy and precision of these assays directed to detecting such low amounts of a target. Accordingly, provided herein are methods providing the requisite management of volume and concentration to produce such DNA samples.
In some embodiments, the sample comprises blood, serum, plasma, or saliva. In some embodiments, the subject is human. Such samples can be obtained by any number of means known in the art, such as will be apparent to the skilled person. Cell free or substantially cell free samples can be obtained by subjecting the sample to various techniques known to those of skill in the art which include, but are not limited to, centrifugation and filtration. Although it is generally preferred that no invasive techniques are used to obtain the sample, it still may be preferable to obtain samples such as tissue homogenates, tissue sections, and biopsy specimens. The technology is not limited in the methods used to prepare the samples and provide a nucleic acid for testing. For example, in some embodiments, a DNA is isolated from a stool sample or from blood or from a plasma sample using direct gene capture, e.g., as detailed in U.S. Pat. Nos. 8,808,990 and 9,169,511, and in WO 2012/155072, or by a related method.
The analysis of markers can be carried out separately or simultaneously with additional markers within one test sample. For example, several markers can be combined into one test for efficient processing of multiple samples and for potentially providing greater diagnostic and/or prognostic accuracy. In addition, one skilled in the art would recognize the value of testing multiple samples (for example, at successive time points) from the same subject. Such testing of serial samples can allow the identification of changes in marker methylation states over time. Changes in methylation state, as well as the absence of change in methylation state, can provide useful information about the disease status that includes, but is not limited to, identifying the approximate time from onset of the event, the presence and amount of salvageable tissue, the appropriateness of drug therapies, the effectiveness of various therapies, and identification of the subject's outcome, including risk of future events.
The analysis of biomarkers can be carried out in a variety of physical formats. For example, the use of microtiter plates or automation can be used to facilitate the processing of large numbers of test samples. Alternatively, single sample formats could be developed to facilitate immediate treatment and diagnosis in a timely fashion, for example, in ambulatory transport or emergency room settings.
It is contemplated that embodiments of the technology are provided in the form of a kit. The kits comprise embodiments of the compositions, devices, apparatuses, etc. described herein, and instructions for use of the kit. Such instructions describe appropriate methods for preparing an analyte from a sample, e.g., for collecting a sample and preparing a nucleic acid from the sample. Individual components of the kit are packaged in appropriate containers and packaging (e.g., vials, boxes, blister packs, ampules, jars, bottles, tubes, and the like) and the components are packaged together in an appropriate container (e.g., a box or boxes) for convenient storage, shipping, and/or use by the user of the kit. It is understood that liquid components (e.g., a buffer) may be provided in a lyophilized form to be reconstituted by the user. Kits may include a control or reference for assessing, validating, and/or assuring the performance of the kit. For example, a kit for assaying the amount of a nucleic acid present in a sample may include a control comprising a known concentration of the same or another nucleic acid for comparison and, in some embodiments, a detection reagent (e.g., a primer) specific for the control nucleic acid. The kits are appropriate for use in a clinical setting and, in some embodiments, for use in a user's home. The components of a kit, in some embodiments, provide the functionalities of a system for preparing a nucleic acid solution from a sample. In some embodiments, certain components of the system are provided by the user.
III. Applications
In some embodiments, diagnostic assays identify the presence of a disease or condition in an individual. In some embodiments, the disease is cancer (e.g., lung cancer). In some embodiments, markers whose aberrant methylation is associated with a lung cancer (e.g., one or more markers selected from the markers listed in Table 1, or preferably one or more of BARX1, LOC100129726, SPOCK2, TSC22D4, MAX.chr8.124, RASSF1, ZNF671, ST8SIA1, NKX6_2, FAM59B, DIDO1, MAX_Chr1.110, AGRN, SOBP, MAX_chr10.226, ZMIZ1, MAX_chr8.145, MAX_chr10.225, PRDM14, ANGPT1, MAX.chr16.50, PTGDR_9, ANKRD13B, DOCK2, MAX_chr19.163, ZNF132, MAX chr19.372, HOXA9, TRH, SP9, DMRTA2, ARHGEF4, CYP26C1, ZNF781, PTGDR, GRIN2D, MATK, BCAT1, PRKCB_28, ST8SIA_22, FLJ45983, DLX4, SHOX2, EMX1, HOXB2, MAX.chr12.526, BCL2L11, OPLAH, PARP15, KLHDC7B, SLC12A8, BHLHE23, CAPN2, FGF14, FLJ34208, B3GALT6, BIN2_Z, DNMT3A, FERMT3, NFIX, S1PR4, SKI, SUCLG2, TBX15, ZDHHC1, and ZNF329) are used. In some embodiments, an assay further comprises detection of a reference gene (e.g., .beta.-actin, ZDHHC1, B3GALT6. See, e.g., U.S. patent application Ser. No. 14/966,617, filed Dec. 11, 2015, and U.S. Pat. Appl. No. 62/364,082, filed Jul. 19, 2016, each of which is incorporated herein by reference for all purposes).
In some embodiments, the technology finds application in treating a patient (e.g., a patient with lung cancer, with early stage lung cancer, or who may develop lung cancer), the method comprising determining the methylation state of one or more markers as provided herein and administering a treatment to the patient based on the results of determining the methylation state. The treatment may be administration of a pharmaceutical compound, a vaccine, performing a surgery, imaging the patient, performing another test. Preferably, said use is in a method of clinical screening, a method of prognosis assessment, a method of monitoring the results of therapy, a method to identify patients most likely to respond to a particular therapeutic treatment, a method of imaging a patient or subject, and a method for drug screening and development.
In some embodiments, the technology finds application in methods for diagnosing lung cancer in a subject is provided. The terms "diagnosing" and "diagnosis" as used herein refer to methods by which the skilled artisan can estimate and even determine whether or not a subject is suffering from a given disease or condition or may develop a given disease or condition in the future. The skilled artisan often makes a diagnosis on the basis of one or more diagnostic indicators, such as for example a biomarker, the methylation state of which is indicative of the presence, severity, or absence of the condition.
Along with diagnosis, clinical cancer prognosis relates to determining the aggressiveness of the cancer and the likelihood of tumor recurrence to plan the most effective therapy. If a more accurate prognosis can be made or even a potential risk for developing the cancer can be assessed, appropriate therapy, and in some instances less severe therapy for the patient can be chosen. Assessment (e.g., determining methylation state) of cancer biomarkers is useful to separate subjects with good prognosis and/or low risk of developing cancer who will need no therapy or limited therapy from those more likely to develop cancer or suffer a recurrence of cancer who might benefit from more intensive treatments.
As such, "making a diagnosis" or "diagnosing", as used herein, is further inclusive of making determining a risk of developing cancer or determining a prognosis, which can provide for predicting a clinical outcome (with or without medical treatment), selecting an appropriate treatment (or whether treatment would be effective), or monitoring a current treatment and potentially changing the treatment, based on the measure of the diagnostic biomarkers disclosed herein.
Further, in some embodiments of the technology, multiple determinations of the biomarkers over time can be made to facilitate diagnosis and/or prognosis. A temporal change in the biomarker can be used to predict a clinical outcome, monitor the progression of lung cancer, and/or monitor the efficacy of appropriate therapies directed against the cancer. In such an embodiment for example, one might expect to see a change in the methylation state of one or more biomarkers disclosed herein (and potentially one or more additional biomarker(s), if monitored) in a biological sample over time during the course of an effective therapy.
The technology further finds application in methods for determining whether to initiate or continue prophylaxis or treatment of a cancer in a subject. In some embodiments, the method comprises providing a series of biological samples over a time period from the subject; analyzing the series of biological samples to determine a methylation state of at least one biomarker disclosed herein in each of the biological samples; and comparing any measurable change in the methylation states of one or more of the biomarkers in each of the biological samples. Any changes in the methylation states of biomarkers over the time period can be used to predict risk of developing cancer, predict clinical outcome, determine whether to initiate or continue the prophylaxis or therapy of the cancer, and whether a current therapy is effectively treating the cancer. For example, a first time point can be selected prior to initiation of a treatment and a second time point can be selected at some time after initiation of the treatment. Methylation states can be measured in each of the samples taken from different time points and qualitative and/or quantitative differences noted. A change in the methylation states of the biomarker levels from the different samples can be correlated with risk for developing lung, prognosis, determining treatment efficacy, and/or progression of the cancer in the subject.
In preferred embodiments, the methods and compositions of the invention are for treatment or diagnosis of disease at an early stage, for example, before symptoms of the disease appear. In some embodiments, the methods and compositions of the invention are for treatment or diagnosis of disease at a clinical stage.
As noted above, in some embodiments multiple determinations of one or more diagnostic or prognostic biomarkers can be made, and a temporal change in the marker can be used to determine a diagnosis or prognosis. For example, a diagnostic marker can be determined at an initial time, and again at a second time. In such embodiments, an increase in the marker from the initial time to the second time can be diagnostic of a particular type or severity of cancer, or a given prognosis. Likewise, a decrease in the marker from the initial time to the second time can be indicative of a particular type or severity of cancer, or a given prognosis. Furthermore, the degree of change of one or more markers can be related to the severity of the cancer and future adverse events. The skilled artisan will understand that, while in certain embodiments comparative measurements can be made of the same biomarker at multiple time points, one can also measure a given biomarker at one time point, and a second biomarker at a second time point, and a comparison of these markers can provide diagnostic information.
As used herein, the phrase "determining the prognosis" refers to methods by which the skilled artisan can predict the course or outcome of a condition in a subject. The term "prognosis" does not refer to the ability to predict the course or outcome of a condition with 100% accuracy, or even that a given course or outcome is predictably more or less likely to occur based on the methylation state of a biomarker. Instead, the skilled artisan will understand that the term "prognosis" refers to an increased probability that a certain course or outcome will occur; that is, that a course or outcome is more likely to occur in a subject exhibiting a given condition, when compared to those individuals not exhibiting the condition. For example, in individuals not exhibiting the condition, the chance of a given outcome (e.g., suffering from lung cancer) may be very low.
In some embodiments, a statistical analysis associates a prognostic indicator with a predisposition to an adverse outcome. For example, in some embodiments, a methylation state different from that in a normal control sample obtained from a patient who does not have a cancer can signal that a subject is more likely to suffer from a cancer than subjects with a level that is more similar to the methylation state in the control sample, as determined by a level of statistical significance. Additionally, a change in methylation state from a baseline (e.g., "normal") level can be reflective of subject prognosis, and the degree of change in methylation state can be related to the severity of adverse events. Statistical significance is often determined by comparing two or more populations and determining a confidence interval and/or ap value. See, e.g., Dowdy and Wearden, Statistics for Research, John Wiley & Sons, New York, 1983, incorporated herein by reference in its entirety. Exemplary confidence intervals of the present subject matter are 90%, 95%, 97.5%, 98%, 99%, 99.5%, 99.9% and 99.99%, while exemplary p values are 0.1, 0.05, 0.025, 0.02, 0.01, 0.005, 0.001, and 0.0001.
In other embodiments, a threshold degree of change in the methylation state of a prognostic or diagnostic biomarker disclosed herein can be established, and the degree of change in the methylation state of the biomarker in a biological sample is simply compared to the threshold degree of change in the methylation state. A preferred threshold change in the methylation state for biomarkers provided herein is about 5%, about 10%, about 15%, about 20%, about 25%, about 30%, about 50%, about 75%, about 100%, and about 150%. In yet other embodiments, a "nomogram" can be established, by which a methylation state of a prognostic or diagnostic indicator (biomarker or combination of biomarkers) is directly related to an associated disposition towards a given outcome. The skilled artisan is acquainted with the use of such nomograms to relate two numeric values with the understanding that the uncertainty in this measurement is the same as the uncertainty in the marker concentration because individual sample measurements are referenced, not population averages.
In some embodiments, a control sample is analyzed concurrently with the biological sample, such that the results obtained from the biological sample can be compared to the results obtained from the control sample. Additionally, it is contemplated that standard curves can be provided, with which assay results for the biological sample may be compared. Such standard curves present methylation states of a biomarker as a function of assay units, e.g., fluorescent signal intensity, if a fluorescent label is used. Using samples taken from multiple donors, standard curves can be provided for control methylation states of the one or more biomarkers in normal tissue, as well as for "at-risk" levels of the one or more biomarkers in tissue taken from donors with lung cancer.
The analysis of markers can be carried out separately or simultaneously with additional markers within one test sample. For example, several markers can be combined into one test for efficient processing of a multiple of samples and for potentially providing greater diagnostic and/or prognostic accuracy. In addition, one skilled in the art would recognize the value of testing multiple samples (for example, at successive time points) from the same subject. Such testing of serial samples can allow the identification of changes in marker methylation states over time. Changes in methylation state, as well as the absence of change in methylation state, can provide useful information about the disease status that includes, but is not limited to, identifying the approximate time from onset of the event, the presence and amount of salvageable tissue, the appropriateness of drug therapies, the effectiveness of various therapies, and identification of the subject's outcome, including risk of future events.
The analysis of biomarkers can be carried out in a variety of physical formats. For example, the use of microtiter plates or automation can be used to facilitate the processing of large numbers of test samples. Alternatively, single sample formats could be developed to facilitate immediate treatment and diagnosis in a timely fashion, for example, in ambulatory transport or emergency room settings.
In some embodiments, the subject is diagnosed as having lung cancer if, when compared to a control methylation state, there is a measurable difference in the methylation state of at least one biomarker in the sample. Conversely, when no change in methylation state is identified in the biological sample, the subject can be identified as not having lung cancer, not being at risk for the cancer, or as having a low risk of the cancer. In this regard, subjects having lung cancer or risk thereof can be differentiated from subjects having low to substantially no cancer or risk thereof. Those subjects having a risk of developing lung cancer can be placed on a more intensive and/or regular screening schedule. On the other hand, those subjects having low to substantially no risk may avoid being subjected to screening procedures, until such time as a future screening, for example, a screening conducted in accordance with the present technology, indicates that a risk of lung cancer has appeared in those subjects.
As mentioned above, depending on the embodiment of the method of the present technology, detecting a change in methylation state of the one or more biomarkers can be a qualitative determination or it can be a quantitative determination. As such, the step of diagnosing a subject as having, or at risk of developing, lung cancer indicates that certain threshold measurements are made, e.g., the methylation state of the one or more biomarkers in the biological sample varies from a predetermined control methylation state. In some embodiments of the method, the control methylation state is any detectable methylation state of the biomarker. In other embodiments of the method where a control sample is tested concurrently with the biological sample, the predetermined methylation state is the methylation state in the control sample. In other embodiments of the method, the predetermined methylation state is based upon and/or identified by a standard curve. In other embodiments of the method, the predetermined methylation state is a specifically state or range of state. As such, the predetermined methylation state can be chosen, within acceptable limits that will be apparent to those skilled in the art, based in part on the embodiment of the method being practiced and the desired specificity, etc.
In some embodiments, a sample from a subject having or suspected of having lung cancer is screened using one or more methylation markers and suitable assay methods that provide data that differentiate between different types of lung cancer, e.g., non-small cell (adenocarcinoma, large cell carcinoma, squamous cell carcinoma) and small cell carcinomas. See, e.g., marker ref # AC27 (FIG. 2; PLEC), which is highly methylated (shown as mean methylation compared to mean methylation at that locus in normal buffy coat) in adenocarcinoma and small cell carcinomas, but not in large cell or squamous cell carcinoma; marker ref # AC23 (FIG. 2; ITPRIPL1), which is more highly methylated in adenocarcinoma than in any other sample type; marker ref # LC2 (FIG. 3; DOCK2)), which is more highly methylated in large cell carcinomas than in any other sample type; marker ref # SC221 (FIG. 4; ST8SIA4), which is more highly methylated in small cell carcinomas than in any other sample type; and marker ref # SQ36 (FIG. 5, DOK1), which is more highly methylated in squamous cell carcinoma than in than in any other sample type.
Methylation markers selected as described herein may be used alone or in combination (e.g., in panels) such that analysis of a sample from a subject reveals the presence of a lung neoplasm and also provides sufficient information to distinguish between lung cancer type, e.g., small cell carcinoma vs. non-small cell carcinoma. In preferred embodiments, a marker or combination of markers further provide data sufficient to distinguish between adenomcarcinomas, large cell carcinomas, and squamous cell carcinomas; and/or to characterize carcinomas of undetermined or mixed pathologies. In other embodiments, methylation markers or combinations thereof are selected to provide a positive result (i.e., a result indicating the presence of lung neoplasm) regardless of the type of lung carcinoma present, without differentiating data.
Over recent years, it has become apparent that circulating epithelial cells, representing metastatic tumor cells, can be detected in the blood of many patients with cancer. Molecular profiling of rare cells is important in biological and clinical studies. Applications range from characterization of circulating epithelial cells (CEpCs) in the peripheral blood of cancer patients for disease prognosis and personalized treatment (See e.g., Cristofanilli M, et al. (2004) N Engl J Med 351:781-791; Hayes D F, et al. (2006) Clin Cancer Res 12:4218-4224; Budd G T, et al., (2006) Clin Cancer Res 12:6403-6409; Moreno J G, et al. (2005) Urology 65:713-718; Pantel et al., (2008) Nat Rev 8:329-340; and Cohen S J, et al. (2008) J Clin Oncol 26:3213-3221). Accordingly, embodiments of the present disclosure provide compositions and methods for detecting the presence of metastatic cancer in a subject by identifying the presence of methylated markers in plasma or whole blood.
EXPERIMENTAL EXAMPLES
Example 1
Sample Preparation Methods
Methods for DNA Isolation and QUARTS Assay
The following provides exemplary method for DNA isolation prior to analysis, and an exemplary QUARTS assay, such as may be used in accordance with embodiments of the technology. Application of QUARTS technology to DNA from blood and various tissue samples is described in this example, but the technology is readily applied to other nucleic acid samples, as shown in other examples.
DNA Isolation From Cells and Plasma
For cell lines, genomic DNA may be isolated from cell conditioned media using, for example, the "Maxwell.RTM. RSC ccfDNA Plasma Kit (Promega Corp., Madison, Wis.). Following the kit protocol, 1 mL of cell conditioned media (CCM) is used in place of plasma, and processed according to the kit procedure. The elution volume is 100 .mu.L, of which 70 .mu.L are generally used for bisulfite conversion.
An exemplary procedure for isolating DNA from a 4 mL sample of plasma is as follows: To a 4 mL sample of plasma, 300 .mu.L of Proteinase K (20 mg/mL) is added and mixed. Add 3 .mu.L of 1 .mu.g/.mu.L of Fish DNA to the plasma-proteinase K mixture. Add 2 mL of plasma lysis buffer to plasma. Plasma lysis buffer is: 4.3M guanidine thiocyanate 10% IGEPAL CA-630 (Octylphenoxy poly(ethyleneoxy)ethanol, branched) (5.3 g of IGEPAL CA-630 combined with 45 mL of 4.8 M guanidine thiocyanate) Incubate mixtures at 55.degree. C. for 1 hour with shaking at 500 rpm. Add 3 mL of plasma lysis buffer and mix. Add 200 .mu.L magnetic silica binding beads (16 .mu.g of beads/.mu.L} and mix again. Add 2 mL of 100% isopropanol and mix. Incubate at 30.degree. C. for 30 minutes with shaking at 500 rpm. Place tube(s) on magnet and let the beads collect. Aspirate and discard the supernatant. Add 750 .mu.L GuHC1-EtOH to vessel containing the binding beads and mix. GuHCl-EtOH wash buffer is: 3M GuHCl (guanidine hydrochloride) 57% EtOH (ethyl alcohol) Shake at 400 rpm for 1 minute. Transfer samples to a deep well plate or 2 mL microcentrifuge tubes. Place tubes on magnet and let the beads collect for 10 minutes. Aspirate and discard the supernatant. Add 1000 .mu.L wash buffer (10 mM Tris HC1, 80% EtOH) to the beads, and incubate at 30.degree. C. for 3 minutes with shaking. Place tubes on magnet and let the beads collect. Aspirate and discard the supernatant. Add 500 .mu.L wash buffer to the beads and incubate at 30.degree. C. for 3 minutes with shaking. Place tubes on magnet and let the beads collect. Aspirate and discard the supernatant. Add 250 .mu.L wash buffer and incubate at 30.degree. C. for 3 minutes with shaking. Place tubes on magnet and let the beads collect. Aspirate and discard the remaining buffer. Add 250 .mu.L wash buffer and incubate at 30.degree. C. for 3 minutes with shaking. Place tubes on magnet and let the beads collect. Aspirate and discard the remaining buffer. Dry the beads at 70.degree. C. for 15 minutes, with shaking. Add 125 .mu.L elution buffer (10 mM Tris HC1, pH 8.0, 0.1 mM EDTA) to the beads and incubate at 65.degree. C. for 25 minutes with shaking. Place tubes on magnet and let the beads collect for 10 minutes. Aspirate and transfer the supernatant containing the DNA to a new vessel or tube. Bisulfite Conversion I. Sulfonation of DNA Using Ammonium Hydrogen Sulfite 1. In each tube, combine 64 .mu.L DNA, 7 .mu.L 1 N NaOH, and 9 .mu.L of carrier solution containing 0.2 mg/mL BSA and 0.25 mg/mL of fish DNA. 2. Incubate at 42.degree. C. for 20 minutes. 3. Add 120 .mu.L of 45% ammonium hydrogen sulfite and incubate at 66.degree. for 75 minutes. 4. Incubate at 4.degree. C. for 10 minutes. II. Desulfonation Using Magnetic Beads Materials Magnetic beads (Promega MagneSil Paramagnetic Particles, Promega catalogue number AS1050, 16 .mu.g/.mu.L). Binding buffer: 6.5-7 M guanidine hydrochoride. Post-conversion Wash buffer: 80% ethanol with 10 mM Tris HCl (pH 8.0). Desulfonation buffer: 70% isopropyl alcohol, 0.1 N NaOH was selected for the desulfonation buffer.
Samples are mixed using any appropriate device or technology to mix or incubate samples at the temperatures and mixing speeds essentially as described below. For example, a Thermomixer (Eppendorf) can be used for the mixing or incubation of samples. An exemplary desulfonation is as follows: 1. Mix bead stock thoroughly by vortexing bottle for 1 minute. 2. Aliquot 50 .mu.L of beads into a 2.0 mL tube (e.g., from USA Scientific). 3. Add 750 .mu.L of binding buffer to the beads. 4. Add 150 .mu.L of sulfonated DNA from step I. 5. Mix (e.g., 1000 RPM at 30.degree. C. for 30 minutes). 6. Place tube on the magnet stand and leave in place for 5 minutes. With the tubes on the stand, remove and discard the supernatant. 7. Add 1,000 .mu.L of wash buffer. Mix (e.g., 1000 RPM at 30.degree. C. for 3 minutes). 8. Place tube on the magnet stand and leave in place for 5 minutes. With the tubes on the stand, remove and discard the supernatant. 9. Add 250 .mu.L of wash buffer. Mix (e.g., 1000 RPM at 30.degree. C. for 3 minutes). 10. Place tube on magnetic rack; remove and discard supernatant after 1 minute. 11. Add 200 .mu.L of desulfonation buffer. Mix (e.g., 1000 RPM at 30.degree. C. for 5 minutes). 12. Place tube on magnetic rack; remove and discard supernatant after 1 minute. 13. Add 250 .mu.L of wash buffer. Mix (e.g., 1000 RPM at 30.degree. C. for 3 minutes). 14. Place tube on magnetic rack; remove and discard supernatant after 1 minute. 15. Add 250 .mu.L of wash buffer to the tube. Mix (e.g., 1000 RPM at 30.degree. C. for 3 minutes). 16. Place tube on magnetic rack; remove and discard supernatant after 1 minute. 17. Incubate all tubes at 30.degree. C. with the lid open for 15 minutes. 18. Remove tube from magnetic rack and add 70 .mu.L of elution buffer directly to the beads. 19. Incubate the beads with elution-buffer (e.g., 1000 RPM at 40.degree. C. for 45 minutes). 20. Place tubes on magnetic rack for about one minute; remove and save the supernatant.
The converted DNA is then used in a detection assay, e.g., a pre-amplification and/or flap endonuclease assays, as described below.
See also U.S. Patent Appl. Ser. No. 62/249,097, filed Oct. 30, 2015; U.S. patent application Ser. Nos. 15/335,111 and 15/335,096, both filed Oct. 26, 2016; and International Appl. Ser. No. PCT/US16/58875, filed Oct. 26, 2016, each of which is incorporated herein by reference in its entirety, for all purposes.
QuARTS Assay
The QUARTS technology combines a polymerase-based target DNA amplification process with an invasive cleavage-based signal amplification process. The technology is described, e.g., in U.S. Pat. Nos. 8,361,720; 8,715,937; 8,916,344; and 9,212,392, each of which is incorporated herein by reference. Fluorescence signal generated by the QUARTS reaction is monitored in a fashion similar to real-time PCR and permits quantitation of the amount of a target nucleic acid in a sample.
An exemplary QUARTS reaction typically comprises approximately 400-600 n mol/L (e.g., 500 n mol/L) of each primer and detection probe, approximately 100 n mol/L of the invasive oligonucleotide, approximately 600-700 n mol/L of each FRET cassette (FAM, e.g., as supplied commercially by Hologic, Inc.; HEX, e.g., as supplied commercially by BioSearch Technologies; and Quasar 670, e.g., as supplied commercially by BioSearch Technologies), 6.675 ng/.mu.L FEN-1 endonuclease (e.g., Cleavase.RTM. 2.0, Hologic, Inc.), 1 unit Taq DNA polymerase in a 30 .mu.L reaction volume (e.g., GoTaq.RTM. DNA polymerase, Promega Corp., Madison, Wis.), 10 m mol/L 3-(n-morpholino) propanesulfonic acid (MOPS), 7.5 m mol/L MgCl.sub.2, and 250 .mu.mol/L of each dNTP. Exemplary QUARTS cycling conditions are as shown in the table below. In some applications, analysis of the quantification cycle (C.sub.q) provides a measure of the initial number of target DNA strands (e.g., copy number) in the sample.
TABLE-US-00001 Stage Temp/Time # of Cycles Denaturation 95.degree. C./3' 1 Amplification 1 95.degree. C./20'' 10 67.degree. C./30'' 70.degree. C./30'' Amplification 2 95.degree. C./20'' 37 53.degree. C./1' 70.degree. C./30'' Cooling 40.degree. C./30'' 1
Multiplex Targeted Pre-amplification of Large-Volume Bisulfite-Converted DNA
To pre-amplify most or all of the bisulfite-treated DNA from an input sample, a large volume of the treated DNA may be used in a single, large-volume multiplex amplification reaction. For example, DNA is extracted from a cell lines (e.g., DFCI032 cell line (adenocarcinoma); H1755 cell line (neuroendocrine), using, for example, the Maxwell Promega blood kit # AS1400, as described above. The DNA is bisulfate converted, e.g., as described above.
A pre-amplification is conducted, for example, in a reaction mixture containing 7.5 mM MgCl.sub.2, 10 mM MOPS, 0.3 mM Tris-HC1, pH 8.0, 0.8 mM KC1, 0.1 .mu.g/.mu.L BSA, 0.0001% Tween-20, 0.0001% IGEPAL CA-630, 250 .mu.M each dNTP, oligonucleotide primers, (e.g., for 12 targets, 12 primer pairs/24 primers, in equimolar amounts (including but not limited to the ranges of, e.g., 200-500 nM each primer), or with individual primer concentrations adjusted to balance amplification efficiencies of the different target regions), 0.025 units/.mu.L HotStart GoTaq concentration, and 20 to 50% by volume of bisulfate-treated target DNA (e.g., 10 .mu.L of target DNA into a 50 .mu.L reaction mixture, or 50 .mu.L of target DNA into a 125 .mu.L reaction mixture). Thermal cycling times and temperatures are selected to be appropriate for the volume of the reaction and the amplification vessel. For example, the reactions may be cycled as follows
TABLE-US-00002 Stage Temp/Time #of Cycles Pre-incubation 95.degree. C./5' 1 Amplification 1 95.degree. C./30'' 10 64.degree. C./30'' 72.degree. C./30'' Cooling 4.degree. C./Hold 1
After thermal cycling, aliquots of the pre-amplification reaction (e.g., 10 .mu.L) are diluted to 500 .mu.L in 10 mM Tris, 0.1 mM EDTA, with or without fish DNA. Aliquots of the diluted pre-amplified DNA (e.g., 10 .mu.L) are used in a QUARTS PCR-flap assay, e.g., as described above. See also U.S. Patent Appl. Ser. No. 62/249,097, filed Oct. 30, 2015; application Ser. No. 15/335,096, filed Oct. 26, 2016, and PCT/US16/58875, filed Oct. 26, 2016, each of which is incorporated herein by reference in its entirety for all purposes.
Example 2
Selection and Testing of Methylation Markers
Marker Selection Process:
Reduced Representation Bisulfite Sequencing (RRBS) data was obtained on tissues from 16 adenocarcinoma lung cancer, 11 large cell lung cancer, 14 small cell lung cancer, 24 squamous cell lung cancer, and 18 non-cancer lung as well as RRBS results of buffy coat samples obtained from 26 healthy patients.
After alignment to a bisulfite-converted form of the human genome sequence, average methylation at each CpG island was computed for each sample type (i.e., tissue or buffy coat) and marker regions were selected based on the following criteria: Regions were selected to be 50 base pairs or longer. For QUARTS flap assay designs, regions were selected to have a minimum of 1 methylated CpG under each of: a) the probe region, b) the forward primer binding region, and c) the reverse primer binding region. For the forward and reverse primers, it is preferred that the methylated CpGs are close to the 3'-ends of the primers, but not at the 3'terminal nucleotide. Exemplary flap endonuclease assay oligonucleotides are shown in FIG. 1. Preferably, buffy coat methylation at any CpG in a region of interest is no more than >0.5%. Preferably, cancer tissue methylation in a region of interest is >10%. For assays designed for tissue analysis, normal tissue methylation in a region of interest is preferably <0.5%.
RRBS data for different lung cancer tissue types is shown in FIGS. 2-5. Based on the criteria above, the markers shown in the table below were selected and QuARTS flap assays were designed for them, as shown in FIG. 1.
TABLE-US-00003 TABLE l Marker Name Genomic coordinates AGRN chr1: 968467-968582, strand = + ANGPT1 chr8: 108509559-108509684, strand = - ANKRD13B chr17: 27940470-27940578, strand = + ARHGEF4 chr2: 131792758-131792900, strand = - B3GALT6 chr1: 1163595-1163733, strand = + BARX1 chr9: 96721498-96721597, strand = - BCAT1 chr12: 25055868-25055986, strand = - BCL2L11 chr2: 111876620-111876759, strand = - BHLHE23 chr20: 61638462-61638546, strand = - BIN2 chr12: 51717898-51717971, strand = - BIN2_Z chr12: 51718088-51718165, strand = + CAPN2 chr1: 223936858-223936998, strand = + chr17_737 chr17: 73749814-73749919, strand = - chr5_132 chr5: 132161371-132161482, Strand = + chr7_636 chr7: 104581684-104581817, Strand = - CYP26C1 chr10: 94822396-94822502, strand = + DIDO1 chr20: 61560669-61560753, strand = - DLX4 chr17: 48042426-48042820, strand = - DMRTA2 chr1: 50884390-50884519, strand = - DNMT3A chr2: 25499967-25500072, strand = - DOCK2 chr5: 169064370- 169064454, strand = - EMX1 chr2: 73147685-73147792, strand = + FAM59B chr2: 26407701-26407828, strand = + FERMT3 chr11: 63974820-63974959, strand = + FGF14 chr13: 103046888-103046991, strand = + FLJ34208 chr3: 194208249-194208355, strand = + FLJ45983 chr10: 8097592-8097699, strand = + GRIN2D chr19: 48918160-48918300, strand = - HIST1H2BE chr6: 26184248-26184340, strand = + HOXA9 chr7: 27205002-27205102, strand = - HOXB2 chr17: 46620545-46620639, strand = - KLHDC7B chr22: 50987199-50987256, strand = + LOC100129726 chr2: 43451705-43451810, strand = + MATK chr19: 3786127-3786197, strand = + MAX.chr10.22541891-22541946 chr10: 22541881-22541975, strand = + MAX.chr10.22624430-22624544 chr10: 22624411-22624553, strand = - MAX.chr12.52652268-52652362 chr12: 52652262-52652377, strand = - MAX.chr16.50875223-50875241 chr16: 50875167-50875274, strand = - MAX.chr19.16394489-16394575 chr19: 16394457-16394593, strand = - MAX.chr19.37288426-37288480 range = chr19: 37288396-37288512, strand = - MAX.chr8.124173236-124173370 chr8: 124173231-124173386, strand = - MAX.chr8.145105646-145105653 chr8: 145105572-145105685, strand = - MAX_Chr1.110 chr1: 110627118-110627224 strand = - NFIX chr19: 13207426-13207513, strand = + NKX2-6 chr8: 23564052-23564145, strand = - OPLAH chr8: 145106777-145106865, strand = - PARP15 chr3: 122296692-122296805, strand = + PRDM14 chr8: 70981945-70982039, strand = - PRKAR1B chr7: 644172-644237, strand = + PRKCB_28 chr16: 23847607-23847698, strand = - PTGDR chr14: 52735270-52735400, strand = - PTGDR_9 chr14: 52735221-52735300, strand = + RASSF1 chr3: 50378408-50378550, strand = - SHOX2 chr3: 157821263-157821382, strand = - SHROOM1 chr5: 132161371-132161425, strand = + SIPR4 chr19: 3179921-3180068 strand = - SKI chr1: 2232328-2232423, strand = + SLC12A8 chr3: 124860704-124860791, strand = + SOBP chr6: 107956176-107956234, strand = + SP9 chr2: 175201210-175201341, strand = - SPOCK2 chr10: 73847236-73847324, strand = - ST8SIA1 chr12: 22487518-22487630, strand = + ST8SIA1_22 chr12: 22486873-22487009, strand = - SUCLG2 chr3: 67706477-677065610, strand = - TBX15 Region 1 chr1: 119527066-119527655, strand = + TBX15 Region 2 chr1: 119532813-119532920 strand = - TRH chr3: 129693481-129693580, strand = + TSC22D4 chr7: 100075328-100075445, strand = - ZDHHC1 chr16: 67428559-67428628, strand = - ZMIZ1 chr10: 81002910-81003005, strand = + ZNF132 chr19: 58951403-58951529, strand = - ZNF329 chr19: 58661889-58662028, strand = - ZNF671 chr19: 58238790-58238906, strand = + ZNF781 ch19 : 38183018-38183137, strand = -
Analyzing Selected Markers for Cross-Reactivity with Buffy Coat. 1) Buffy Coat Screening
Markers from the list above were screened on DNA extracted from buffy coat obtained from 10 mL blood of a healthy patient. DNA was extracted using Promega Maxwell RSC system (Promega Corp., Fitchburg, Wis.) and converted using Zymo EZ DNA Methylation.TM. Kit (Zymo Research, Irvine, Calif.). Using biplexed reaction with bisulfite-converted .beta.-actin DNA ("BTACT"), and using approximately 40,000 strands of target genomic DNA, the samples were tested using a QuARTS flap endonuclease assay as described above, to test for cross reactivity. Doing so, the assays for 3 markers showed significant cross reactivity:
TABLE-US-00004 % Cross Marker reactivity HIST1H2B 72.93% chr7_636 3495.47% chr5_132 0.20%
2) Tissue Screening
264 tissue samples were obtained from various commercial and non-commercial sources (Asuragen, BioServe, ConversantBio, Cureline, Mayo Clinic, M D Anderson, and PrecisionMed), as shown below in Table 2.
TABLE-US-00005 No. of cases Pathology Subtype Details 82 Normal NA 68 smokers, 34 37 Normal benign nodule never smokers, 17 7 NSCLC bronchioalveolar smoking unknown 13 NSCLC large cell 2 NSCLC neuroendocrine 42 NSCLC squamous cell 68 NSCLC adenocarcinomas 4 SCLC small cell 9 NSCLC carcinoid
Tissue sections were examined by a pathologist, who circled histologically distinct lesions to direct the micro-dissection. Total nucleic acid extraction was performed using the Promega Maxwell RSC system. Formalin-fixed, paraffin-embedded (FFPE) slides were scraped and the DNA was extracted using the Maxwell.RTM. RSC DNA FFPE Kit (#AS1450) using the manufacturer's procedure but skipping the RNase treatment step. The same procedure was used for FFPE curls. For frozen punch biopsy samples, a modified procedure using the lysis buffer from the RSC DNA FFPE kit with the Maxwell.RTM. RSC Blood DNA kit (#AS1400) was utilized omitting the RNase step. Samples were eluted in 10 mM Tris, 0.1 mM EDTA, pH 8.5 and 10 uL were used to setup 6 multiplex PCR reactions.
The following multiplex PCR primer mixes were made at 10.times. concentration (10.times.=2 .mu.M each primer): Multiplex PCR reaction 1 consisted of each of the following markers: BARX1,
LOC100129726, SPOCK2, TSC22D4, PARP15, MAX.chr8.145105646-145105653, ST8SIA1_22, ZDHHC1, BIN2_Z, SKI, DNMT3A, BCL2L11, RASSF1, FERMT3, and BTACT. Multiplex PCR reaction 2 consisted of each of the following markers: ZNF671,
ST8SIA1, NKX6-2, SLC12A8, FAM59B, DIDO1, MAX_Chr1.110, AGRN, PRKCB_28, SOBP, and BTACT. Multiplex PCR reaction 3 consisted of each of the following markers: MAX.chr10.22624430-22624544, ZMIZ1, MAX.chr8.145105646-145105653, MAX.chr10.22541891-22541946, PRDM14, ANGPT1, MAX.chr16.50875223-50875241, PTGDR_9, ANKRD13B, DOCK2, and BTACT. Multiplex PCR reaction 4 consisted of each of the following markers: MAX.chr19.16394489-16394575, HOXB2, ZNF132, MAX.chr19.37288426-37288480, MAX.chr12.52652268-52652362, FLJ45983, HOXA9, TRH, SP9, DMRTA2, and BTACT. Multiplex PCR reaction 5 consisted of each of the following markers: EMX1, ARHGEF4, OPLAH, CYP26C1, ZNF781, DLX4, PTGDR, KLHDC7B, GRIN2D, chr17_737, and BTACT. Multiplex PCR reaction 6 consisted of each of the following markers: TBX15,
MATK, SHOX2, BCAT1, SUCLG2, BIN2, PRKAR1B, SHROOM1, S1PR4, NFIX, and BTACT.
Each multiplex PCR reaction was setup to a final concentration of 0.2 .mu.M reaction buffer, 0.2 .mu.M each primer, 0.05 .mu.M Hotstart Go Taq (5U/.mu.L), resulting in 40 .mu.L, of master mix that was combined with 10 .mu.L of DNA template for a final reaction volume of 50 .mu.L.
The thermal profile for the multiplex PCR entailed a pre-incubation stage of 95.degree. for 5 minutes, 10 cycles of amplification at 95.degree. for 30 seconds, 64.degree. for 30 seconds, 72.degree. for 30 seconds, and a cooling stage of 4.degree. that was held until further processing. Once the multiplex PCR was complete, the PCR product was diluted 1:10 using a diluent of 20 ng/.mu.L of fish DNA (e.g., in water or buffer, see U.S. Pat. No. 9,212,392, incorporated herein by reference) and 10 .mu.L of diluted amplified sample were used for each QuARTS assay reaction.
Each QuARTS assay was configured in triplex form, consisting of 2 methylation markers and BTACT as the reference gene. From multiplex PCR product 1, the following 7 triplex QuARTS assays were run: (1) BARX1, LOC100129726, BTACT; (2) SPOCK2, TSC22D4, BTACT; (3) PARP15, MAXchr8145105646-145105653, BTACT; (4) ST8SIA1_22, ZDHHC1, BTACT; (5) BIN2_Z, SKI, BTACT; (6) DNMT3A, BCL2L11, BTACT; (7) RASSF1, FERMT3, and BTACT. From multiplex PCR product 2, the following 5 triplex QuARTS assays were run: (1) ZNF671, ST8SIA1, BTACT; (2) NKX6-2, SLC12A8, BTACT; (3) FAM59B, DIDO1, BTACT; (4) MAX_Chr1110, AGRN, BTACT; (5) PRKCB_28, SOBP, and BTACT. From multiplex PCR product 3, the following 5 triplex QuARTS assays were run: (1) MAXchr1022624430-22624544, ZMIZ1, BTACT; (2) MAXchr8145105646-145105653, MAXchr1022541891-22541946, BTACT; (3) PRDM14, ANGPT1, BTACT; (4) MAXchr1650875223-50875241, PTGDR_9, BTACT; (5) ANKRD13B, DOCK2, and BTACT. From multiplex PCR product 4, the following 5 triplex QuARTS assays were run: (1) MAXchr1916394489-16394575, HOXB2, BTACT; (2) ZNF132, MAXchr1937288426-37288480, BTACT; (3) MAXchr1252652268-52652362, FLJ45983, BTACT; (4) HOXA9, TRH, BTACT; (5) SP9, DMRTA2, and BTACT. From multiplex PCR product 5, the following 5 triplex QuARTS assays were run: (1) EMX1, ARHGEF4, BTACT; (2) OPLAH, CYP26C1, BTACT; (3) ZNF781, DLX4, BTACT; (4) PTGDR, KLHDC7B, BTACT; (5) GRIN2D, chr17_737, and BTACT. From multiplex PCR product 6, the following 5 triplex QuARTS assays were run: (1) TBX15, MATK, BTACT; (2) SHOX2, BCAT1, BTACT; (3) SUCLG2, BIN2, BTACT; (4) PRKAR1B, SHROOM1, BTACT; (5) S1PR4, NFIX, and BTACT. 3) Data Analysis:
For tissue data analysis, markers that were selected based on RRBS criteria with <0.5% methylation in normal tissue and >10% methylation in cancer tissue were included. This resulted in 51 markers for further analysis.
To determine marker sensitivities, the following was performed: 1. % methylation for each marker was computed by dividing strand values obtained for that specific marker by the strand values of ACTB ((.beta.-actin). 2. The maximum % methylation for each marker was determined on normal tissue. This is defined as 100% specificity. 3. The cancer tissue positivity for each marker was determined as the number of cancer tissues that had greater than the maximum normal tissue % methylation for that marker.
The sensitivities for the 51 markers are shown below.
TABLE-US-00006 TABLE 2 Maximum % methylation for Cancer (N = 136) Marker normal # Negative # Positive sensitivity BARX1 1.665 66 70 51% LOC100129726 1.847 109 27 20% SPOCK2 0.261 86 50 37% TSC22D4 0.618 70 66 49% MAX.chr8.124 0.293 45 91 67% RASSF1 1.605 79 57 42% ZNF671 0.441 73 63 46% ST8SIA1 1.56 119 17 13% NKX6_2 15.58 102 34 25% FAM59B 0.433 85 51 38% DIDO1 2.29 93 43 32% MAX_Chr1.110 0.076 85 51 38% AGRN 2.16 66 70 51% SOBP 38.5 110 26 19% MAX_chr10.226 0.7 52 84 62% ZMIZ1 0.025 72 64 47% MAX_chr8.145 5.56 57 79 58% MAX_chr10.225 0.77 72 64 47% PRDM14 0.22 35 101 74% ANGPT1 1.6 99 37 27% MAX.chr16.50 0.27 92 44 32% PTGDR_9 4.62 82 54 40% ANKRD13B 7.03 93 43 32% DOCK2 0.001 71 65 48% MAX_chr19.163 0.61 56 80 59% ZNF132 1.3 83 53 39% MAX chr19.372 0.676 79 57 42% HOXA9 16.7 53 83 61% TRH 2.64 61 75 55% SP9 14.99 75 61 45% DMRTA2 7.9 55 81 60% ARHGEF4 7.41 113 23 17% CYP26C1 39.2 101 35 26% ZNF781 5.28 44 92 68% PTGDR 6.13 76 60 44% GRIN2D 16.1 113 23 17% MATK 0.04 93 43 32% BCAT1 0.64 75 61 45% PRKCB_28 1.68 57 79 58% ST8SIA_22 1.934 55 81 60% FLJ45983 8.34 39 97 71% DLX4 15.1 41 95 70% SHOX2 7.48 32 104 76% EMX1 11.34 34 102 75% HOXB2 0.114 61 75 55% MAX.chr12.526 5.58 34 102 75% BCL2L11 10.7 44 92 68% OPLAH 5.11 29 107 79% PARP15 3.077 42 94 69% KLHDC7B 8.86 38 98 72% SLC12A8 0.883 34 102 75%
Combinations of markers may be used to increase specificity and sensitivity. For example, a combination of the 8 markers SLC12A8, KLHDC7B, PARP15, OPLAH, BCL2L11, MAX.chr12.526, HOXB2, and EMX1 resulted in 98.5% sensitivity (134/136 cancers) for all of the cancer tissues tested, with 100% specificity.
In some embodiments, markers are selected for sensitive and specific detection associated with a particular type of lung cancer tissue, e.g., adenocarcinoma, large cell carcinoma, squamous cell carcinoma, or small cell carcinoma, e.g., by use of markers that show sensitivity and specificity for particular cancer types or combinations of types.
This panel of methylated DNA markers assayed on tissue achieves extremely high discrimination for all types of lung cancer while remaining negative in normal lung tissue and benign nodules. Assays for this panel of markers can be also be applied to blood or bodily fluid-based testing, and finds applications in, e.g., lung cancer screening and discrimination of malignant from benign nodules.
Example 3
Testing a 30-Marker Set on Plasma Samples
From the list of markers in Example 2, 30 markers were selected for use in testing DNA from plasma samples from 295 subjects (64 with lung cancer, 231 normal controls. DNA was extracted from 2 mL of plasma from each subject and treated with bisulfite as described in Example 1. Aliquots of the bisulfite-converted DNA were used in two multiplex QuARTS assays, as described in Example 1. The markers selected for analysis are: 1. BARX1 2. BCL2L11 3. BIN2_Z 4. CYP26C1 5. DLX4 6. DMRTA2 7. DNMT3A 8. EMX1 9. FERMT3 10. FLJ45983 11. HOXA9 12. KLHDC7B 13. MAX.chr10.22624430-22624544 14. MAX.chr12.52652268-52652362 15. MAX.chr8.124173236-124173370 16. MAX.chr8.145105646-145105653 17. NFIX 18. OPLAH 19. PARP15 20. PRKCB 28 21. S1PR4 22. SHOX2 23. SKI 24. SLC12A8 25. SOBP 26. SP9 27. SUCLG2 28. TBX15 29. ZDHHC1 30. ZNF781
The target sequences, bisulfite converted target sequences, and the assay oligonucleotides for these markers were as shown in FIG. 1. The primers and flap oligonucleotides (probes) used for each converted target were as follows:
TABLE-US-00007 TABLE 3 Marker Oligonucleotide Name Component Sequence (5'-3') SEQ ID NO: BARX1 BARX1_FP Forward Primer CGTTAATTTGTTAGATAGAGGGCG 23 BARX1_RP Reverse Primer ACGATCGTCCGAACAACC 24 BARX1_PB_A5 Flap Oligo. CCACGGACGCGCCTACGAAAA/3C6/ 25 SLC12A8 SLC12A8_FP Forward Primer TTAGGAGGGTGGGGTTCG 289 SLC12A8_RP Reverse Primer CTTTCCTCGCAAAACCGC 290 SLC12A8_Pb_A1 Flap Oligo. CCACGGACGGGAGGGCGTAGG/3C6/ 291 PARP15 PARP15_FP Forward Primer GGTTGAGTTTGGGGTTCG 236 PARP15_RP Reverse Primer CGTAACGTAAAATCTCTACGCCC 237 PARP15_Pb_A5 Flap Oligo. CCACGGACGCGCTCGAACTAC/3C6/ 238 MAX.Chr8.124 MAX.Chr8.124_FP Forward Primer GGTTGAGGTTTTCGGGTTTTTAG 203 MAX.Chr8.124_RP Reverse Primer CCTCCCCACGAAATCGC 204 MAX.Chr8.124_Pb_A1 Flap Oligo. CGCCGAGGGCGGGTTTTCGT/3C6/ 205 SHOX2 SHOX2_FP Forward Primer GTTCGAGTTTAGGGGTAGCG 269 SHOX2_RP Reverse Primer CCGCACAAAAAACCGCA 270 SHOX2_Pb_A5 Flap Oligo. CCACGGACGATCCGCAAACGC/3C6/ 271 ZDHHC1 ZDHHC1FP Forward Primer GTCGGGGTCGATAGTTTACG 348 ZDHHC1RP_V3 Reverse Primer ACTCGAACTCACGAAAACG 349 ZDHHC1Probe_v3_A1 Flap Oligo. CGCCGAGGGACGAACGCACG/3C6/ 250 BIN2_Z BIN2_FP_Z Forward Primer GGGTTTATTTTTAGGTAGCGTTCG 50 BIN2_RP_Z Reverse Primer CGAAATTTCGAACAAAAATTAAAACTCGA 51 BIN2_Pb_A5_Z Flap Oligo. CCACGGACGGTTCGAGGTTAG/3C6/ 52 SKI SKI_FP Forward Primer ACGGTTTTTTCGTTATTTTTACGGG 279 SKI_RP Reverse Primer CAACGCCTAAAAACACGACTC 280 SKI_Pb_A1 Flap Oligo. CGCCGAGGGGCGGTTGTTGG/3C6/ 281 DNMT3A DNMT3A_FP Forward Primer GTTACGAATAAAGCGTTGGCG 93 DNMT3A_RP Reverse Primer AACGAAACGTCTTATCGCGA 94 DNMT3A_Pb_A5 Flap Oligo. CCACGGACGGAGTGCGCGTTC/3C6/ 95 BC2L11 BCL2L11_FP Forward Primer CGTAATGTTTCGCGTTTTTCG 35 BCL2L11_RP Reverse Primer ACTTTCTTCTACGTAATTCTTTTCCGA 36 BCL2L11_Pb_A1 Flap Oligo. CGCCGAGGGCGGGGTCGGGC/3C6/ 37 TBX15 TBX15_Reg2_FP Forward Primer AGGAAATTGCGGGTTTTCG 332 TBX15_Reg2_RP Reverse Primer CCAAAAATCGTCGCTAAAAATCAAC 334 TBX15_Reg2_Pb_A5 Flap Oligo. CCACGGACGCGCGCATTCACT/3C6/ 335 FERMT3 FERMT3_FP Forward Primer GTTTTCGGGGATTATATCGATTCG 118 FERMT3_RP Reverse Primer CCCAATAACCCGCAAAATAACC 119 FERMT3_Pb_A1 Flap Oligo. CGCCGAGGCGACTCGACCTC/3C6/ 120 PRKCB_28 PRKCB_28_FP Forward Primer GGAAGGTGTTTTGCGCG 249 PRKCB_28_RP Reverse Primer CTTCTACAACCACTACACCGA 250 PRKCB_28_Pb_A5 Flap Oligo. CCACGGACGGCGCGCGTTTAT/3C6/ 251 SOBP_HM SOBP_HM_FP Forward Primer TTTCGGCGGGTTTCGAG 294 SOBP_HM_RP Reverse Primer CGTACCGTTCACGATAACGT 295 SOBP_HM_Pb_A1 Flap Oligo. CGCCGAGGGGCGGTCGCGGT/3C6/ 296 MAX.chr8.145 MAX.Chr8.145_FP Forward Primer GCGGTATTAGTTAGAGTTTTAGTCG 211 MAX.Chr8.145_RP Reverse Primer ACAACCCTAAACCCTAAATATCGT 212 MAX.Chr8.145_Pb_A5 Flap Oligo. CCACGGACGGACGGCGTTTTT/3C6/ 213 MAX.chr10.226 MAX.Chr10.226_FP Forward Primer GGGAAATTTGTATTTCGTAAAATCG 178 MAX.Chr10.226_RP Reverse Primer ACAACTAACTTATCTACGTAACATCGT 179 MAX_Chr10.226_Pb_A1 Flap Oligo. CGCCGAGGGCGGTTAAGAAA/3C6/ 180 MAX.chr12.52 MAX.Chr12.52_FP Forward Primer TCGTTCGTTTTTGTCGTTATCG 183 MAX.Chr12.52_RP Reverse Primer AACCGAAATACAACTAAAAACGC 184 MAX.Chr12.52PbA1 Flap Oligo. CCACGGACGCGAACCCCGCAA/3C6/ 185 FLI45983 FLI45983_FP Forward Primer GGGCGCGAGTATAGTCG 133 FLI45983_RP Reverse Primer CAACGCGACTAATCCGC 134 FLI45983_Pb_A1 Flap Oligo. CGCCGAGGCCGTCACCTCCA/3C6/ 135 HOXA9 HOXA9_FP Forward Primer TTGGGTAATTATTACGTGGATTCG 148 HOXA9_RP Reverse Primer ACTCATCCGCGACGTC 149 HOXA9_Pb_A5 Flap Oligo. CCACGGACGCGACGCCCAACA/3C6/ 150 EMX1 EMX1_FP Forward Primer GGCGTCGCGTTTTTTAGAGAA 108 EMX1_RP Reverse Primer TTCCTTTTCGTTCGTATAAAATTTCGTT 109 EMX1PbA1 Flap Oligo. CGCCGAGGATCGGGTTTTAG/3C6/ 110 SP9 SP9_FP Forward Primer TAGCGTCGAATGGAAGTTCGA 315 SP9_RP Reverse Primer GCGCGTAAACATAACGCACC 317 SP9_Pb_A5 Flap Oligo. CCACGGACGCCGTACGAATCC/3C6/ 318 DMRTA2 DMRTA2_FP Forward Primer TGGTGTTTACGTTCGGTTTTCGT 88 DMRTA2_RP Reverse Primer CCGCAACAACGACGACC 89 DMRTA2_Pb_A1 Flap Oligo. CGCCGAGGCGAACGATCACG/3C6/ 90 OPLAH FPrimerOPLAH Forward Primer cGTcGcGTTTTTcGGTTATACG 231 RPrimerOPLAH Reverse Primer CGCGAAAACTAAAAAACCGCG 232 ProbeA5OPLAH Flap Oligo. CCACGGACG-GCACCGTAAAAC/3C6/ 233 CYP26C1 CYP26C1_FP Forward Primer TGGTTTTTTGGTTATTTCGGAATCGT 70 CYP26C1_RP Reverse Primer GCGCGTAATCAACGCTAAC 71 CYP26C1_Pb_A1 Flap Oligo. CGCCGAGGCGACGATCTAAC/3C6/ 72 ZNF781 ZNF781F.primer Forward Primer CGTTTTTTGTTTTTCGAGTGCG 373 ZNF781R.primer Reverse Primer TCAATAACTAAACTCACCGCGTC 374 ZNF781probe.A5 Flap Oligo. CCACGGACGGCGGATTTATCG/3C6/ 375 DLX4 DLX4_FP Forward Primer TGAGTGCGTAGTGTTTTCGG 80 DLX4_RP Reverse Primer CTCCTCTACTAAAACGTACGATAAACA 81 DLX4_Pb_A1 Flap Oligo. CGCCGAGGATCGTATAAAAC/3C6/ 82 SUCLG2 SUCLG2_HM_FP Forward Primer TCGTGGGTTTTTAATCGTTTCG 321 SUCLG2_HM_RP Reverse Primer TCACGCCATCTTTACCGC 322 SUCLG2_HM_Pb_A5 Flap Oligo. CCACGGACGCGAAAATCTACA/3C6/ 323 KLHDC7B KLHDC7B_FP Forward Primer AGTTTTCGGGTTTTGGAGTTCGTTA 158 KLHDC7B_RP Reverse Primer CCAAATCCAACCGCCGC 159 KLHDC7B_Pb_A1 Flap Oligo. CGCCGAGGACGGCGGTAGTT/3C6/ 160 S1PR4_HM S1PR4_HM_FP Forward Primer TTATATAGGCGAGGTTGCGT 284 S1PR4_HM_RP Reverse Primer CTTACGTATAAATAATACAACCACCGAATA 285 S1PR4_HM_Pb_A5 Flap Oligo. CCACGGACGACGTACCAAACA/3C6/ 286 NFIX_HM NFIX_HM_FP Forward Primer TGGTTCGGGCGTGACGCG 221 NFIX_HM_RP Reverse Primer TCTAACCCTATTTAACCAACCGA 222 NFIX_HM_Pb_A1 Flap Oligo. CGCCGAGGGCGGTTAAAGTG/3C6/ 223 Reference DNAs Oligonucleotide Name Component Sequence (5'-3') Zebrafish ZF_RASSF1_FP BT Forward TGCGTATGGTGGGCGAG 394 Synthetic Primer (RASSF1) ZF_RASSF1_RP BT Reverse Primer CCTAATTTACACGTCAACCAATCGAA 395 BT converted).dagger. ZF_RASSF1_Pb_A5 BT Flap Oligo. CCACGGACGGCGCGTGCGTTT/3C6/ 397 B3GALT6* B3GALT6_FP_V2 Forward Primer GGTTTATTTTGGTTTTTTGAGTTTTCGG 386 B3GALT6_RP Reverse Primer TCCAACCTACTATATTTACGCGAA 387 B3GALT6_Pb_A1 Flap Oligo. CCACGGACGGCGGATTTAGGG/3C6/ 388 BTACT ACTB_BT_FP65 Forward Primer GTGTTTGTTTTTTGATTAGGTGTTTAAGA 381 ACTB_BT_RP65 Reverse Primer CTTTACACCAACCTCATAACCTTATC 382 ACTBBTPbA3 Flap Oligo. GACGCGGAGATAGTGTTGTGG/3C6/ 383 *The B3GALT6 marker is used as both a cancer methylation marker and as a reference target. See U.S. Patent Application Ser. No. 62/364,082, filed Jul. 19, 2016, which is incorporated herein by reference in its entirety. .dagger.For zebrafish reference DNA see U.S. Patent Application Ser. No. 62/364,049, filed Jul. 19, 2016, which is incorporated herein by reference in its entirety.
The DNA prepared from plasma as described above was amplified in two multiplexed pre-amplification reactions, as described in Example 1. The multiplex pre-amplification reactions comprised reagents to amplify the following marker combinations.
TABLE-US-00008 TABLE 4 Multiplex Mix 1 Multiplex Mix 2 B3GALT6 (reference) B3GALT6 (reference) ZF_RASSF1 (reference) ZF_RASSF1 (reference) BARX1 CYP26C1 BCL2L11 DLX4 BCL2L11 DMRTA2 BIN2_Z EMX1 DNMT3A HOXA9 FERMT3 KLHDC7B PARP15 MAX.chr8.125 PRKCB_28 MAX_chr10.226 SHOX2 NFIX SLC12A8 OPLAH SOBP S1PR4 TBX15_Reg2 SP9 ZDHHC1 SUCLG2 ZNF781
Following pre-amplification, aliquots of the pre-amplified mixtures were diluted 1:10 in 10 mM Tris HC1, 0.1 mM EDTA, then were assayed in triplex QuARTS PCR-flap assays, as described in Example 1. The Group 1 triplex reactions used pre-amplified material from Multiplex Mix 1, and the Group 2 reactions used the pre-amplified material from Multiplex Mix 2. The triplex combinations were as follows:
Group 1:
TABLE-US-00009 ZF_RASSF1-B3GALT6-BTACT (ZBA Triplex) BARX1-SLC12A8-BTACT (BSA2 Triplex) PARP15-MAX.chr8.124-BTACT (PMA Triplex) SHOX2-ZDHHC1-BTACT (SZA2 Triplex) BIN2_Z-SKI-BTACT (BSA Triplex) DNMT3A-BCL2L11-BTACT (DBA Triplex) TBX15-FERMT3-BTACT (TFA Triplex) PRKCB_28-SOBP-BTACT (PSA2 Triplex)
Group 2:
TABLE-US-00010 ZF_RASSF1-B3GALT6-BTACT (ZBA Triplex) MAX.chr8.145-MAX_chr10.226-BTACT (MMA2 Triplex) MAX.chr12.526-FLJ45983-BTACT (MFA Triplex) HOXA9-EMX1-BTACT (HEA Triplex) SP9-DMRTA2-BTACT (SDA Triplex) OPLAH-CYP26C1-BTACT (OCA Triplex) ZNF781-DLX4-BTACT (ZDA Triplex) SUCLG2-KLHDC7B-BTACT (SKA Triplex) S1PR4-NFIX-BTACT (SNA Triplex)
Each triplex acronym uses the first letter of each gene name (for example, the combination of HOXA9-EMX1-BTACT="HEA"). If an acronym is repeated for a different combination of markers or from another experiment, the second grouping having that acronym includes the number 2. The dye reporters used on the FRET cassettes for each member of the triplexes listed above is FAM-HEX-Quasar670, respectively.
Plasmids containing target DNA sequences were used to calibrate the quantitative reactions. For each calibrator plasmid, a series of 10.times. calibrator dilution stocks, having from 10 to 10.sup.6 copies of the target strand per .mu.l in fish DNA diluent (20 ng/mL fish DNA in 10 mM Tris-HCl, 0.1 mM EDTA) were prepared. For triplex reactions, a combined stock having plasmids that contain each of the targets of the triplex were used. A mixture having each plasmid at 1.times.10.sup.5 copies per .mu.L was prepared and used to create a 1:10 dilution series. Strands in unknown samples were back calculated using standard curves generated by plotting Cp vs Log (strands of plasmid).
Using receiver operating characteristic (ROC) curve analysis, the area under the curve (AUC) for each marker was calculated and is shown in the table below, sorted by Upper 95 Pct Coverage Interval.
TABLE-US-00011 TABLE 5 Sensitivity at Marker Name AUC 90% specificity CYP26C1 0.940 80% SOBP 0.929 80% SHOX2 0.905 73% SUCLG2 0.905 64% NFIX 0.895 63% ZDHHC1 0.890 69% BIN2_Z 0.872 59% DLX4 0.856 56% FLJ45983 0.834 67% HOXA9 0.824 53% TBX15 0.813 53% ACTB 0.803 50% S1PR4 0.802 55% SP9 0.782 38% FERMT3 0.773 36% ZNF781 0.769 55% B3GALT6 0.746 39% BTACT 0.742 44% BCL2L11 0.732 39% PARP15 0.673 31% DNMT3A 0.689 20% MAX.chr12.526 0.668 33% MAX.chr10.226 0.671 30% SLC12A8 0.655 19% BARX1 0.663 25% KLHDC7B 0.604 10% OPLAH 0.571 14% MAX.chr8.145 0.572 16% SKI 0.521 14%
The markers worked very well in distinguishing samples from cancer patients from samples from normal subjects (see ROC table, above). Use of the markers in combination improved sensitivity. For example, using a logistic fit of the data and a six-marker fit, ROC curve analysis shows an AUC=0.973.
Using a 6-marker fit, sensitivity of 92.2% is obtained at 93% specificity. The group of 6 markers that together resulted in the best fit was SHOX2, SOBP, ZNF781, BTACT, CYP26C1, and DLX4 (see FIG. 7). Using SHOX2, SOBP, ZNF781, CYP26C1, SUCLG2, and SKI gave an ROC curve with AUC of 0.97982 (see FIG. 8).
Example 4
Archival plasmas from a second independent study group were tested in blinded fashion. Lung cancer cases and controls (apparently healthy smokers) for each group were balanced on age and sex (23 cases, 80 controls). Using multiplex PCR followed by QuARTS (Quantitative Allele-Specific Real-time Target and Signal amplification) assay as described in Example 1, a post-bisulfite quantification of methylated DNA markers on DNA extracted from plasma was performed. Top individual methylated markers from Example 3 were tested in this experiment to identify optimal marker panels for lung cancer detection (2 ml/patient).
Results: 13 high performance methylated DNA markers were tested (CYP26C1, SOBP, SUCLG2, SHOX2, ZDHHC1, NFIX, FLJ45983, HOXA9, B3GALT6, ZNF781, SP9, BARX1, and EMX1). Data were analyzed using two methods: a logistic regression fit and a regression partition tree approach. The logistic fit model identified a 4-marker panel (ZNF781, BARX1, EMX1, and SOBP) with an AUC of 0.96 and an overall sensitivity of 91% and 90% specificity. Analysis of the data using a regression partition tree approach identified 4 markers (ZNF781, BARX1, EMX1, and HOXA9) with AUC of 0.96 and an overall sensitivity of 96% and specificity of 94%. For both approaches, B3GALT6 was used as a standardizing marker of total DNA input. These panels of methylated DNA markers assayed in plasma achieved high sensitivity and specificity for all types of lung cancer.
Example 5
Differentiating Lung Cancers
Using the methods described above, methylation markers are selected that exhibit high performance in detecting methylation associated with specific types of lung cancer.
For a subject suspected of having lung cancer, a sample is collected, e.g., a plasma sample, and DNA is isolated from the sample and treated with bisulfite reagent, e.g., as described in Example 1. The converted DNA is analyzed using a multiplex PCR followed by QuARTS flap endonuclease assay as described in Example 1, configured to provide different identifiable signals for different methylation markers or combinations of methylation markers, thereby providing data sets configured to specifically identify the presence of one or more different types of lung carcinoma in the subject (e.g., adenocarcinoma, large cell carcinoma, squamous cell carcinoma, and/or small cell carcinoma). In preferred embodiments, a report is generated indicating the presence or absence of an assay result indicative of the presence of lung carcinoma and, if present, further indicative of the presence of one or more identified types of lung carcinoma. In some embodiments, samples from a subject are collected over the course of a period of time or a course of treatment, and assay results are compared to monitor changes in the cancer pathology.
Marker and marker panels sensitive to different types of lung cancer find use, e.g., in classifying type(s) of cancer present, identifying mixed pathologies, and/or in monitoring cancer progression over time and/or in response to treatment.
All literature and similar materials cited in this application, including but not limited to, patents, patent applications, articles, books, treatises, and internet web pages are expressly incorporated by reference in their entirety for any purpose. Unless defined otherwise, all technical and scientific terms used herein have the same meaning as is commonly understood by one of ordinary skill in the art to which the various embodiments described herein belongs. When definitions of terms in incorporated references appear to differ from the definitions provided in the present teachings, the definition provided in the present teachings shall control.
Various modifications and variations of the described compositions, methods, and uses of the technology will be apparent to those skilled in the art without departing from the scope and spirit of the technology as described. Although the technology has been described in connection with specific exemplary embodiments, it should be understood that the invention as claimed should not be unduly limited to such specific embodiments. Indeed, various modifications of the described modes for carrying out the invention that are obvious to those skilled in pharmacology, biochemistry, medical science, or related fields are intended to be within the scope of the following claims.
SEQUENCE LISTINGS
1
4041116DNAHomo sapiens 1gttcccggaa cggcctcttg ggggcgttcc agccccacgg acccgcaggg agtccccgcc 60gcaatttgca tggggctcat ttgcatgacc ccgccccgcg cgggagtcgg gggcgc 1162116DNAArtificial sequenceSynthetic DNA 2gttttcggaa cggttttttg ggggcgtttt agttttacgg attcgtaggg agttttcgtc 60gtaatttgta tggggtttat ttgtatgatt tcgtttcgcg cgggagtcgg gggcgt 116323DNAArtificial sequenceSynthetic DNA 3ggcgttttag ttttacggat tcg 23427DNAArtificial sequenceSynthetic DNA 4acaaataaac cccatacaaa ttacgac 27520DNAArtificial sequenceSynthetic DNA 5cgccgaggcg aaaactccct 206126DNAHomo sapiens 6cggattcaac atgggcaatg tgcctacact ttcattcttc cagaacacga tggcaactgt 60cgtgagagta cgacagacca gtacaacaca aacgctctgc agagagatgc tccacacgtg 120gaaccg 1267126DNAArtificial sequenceSynthetic DNA 7cggatttaat atgggtaatg tgtttatatt tttatttttt tagaatacga tggtaattgt 60cgtgagagta cgatagatta gtataatata aacgttttgt agagagatgt tttatacgtg 120gaatcg 126826DNAArtificial sequenceSynthetic DNA 8ttttagaata cgatggtaat tgtcgt 26935DNAArtificial sequenceSynthetic DNA 9acatctctct acaaaacgtt tatattatac taatc 351020DNAArtificial sequenceSynthetic DNA 10cgccgaggct atcgtactct 2011109DNAHomo sapiens 11ggagctacga cgagcagctg cggctggcga tggaactgtc ggcgcaggag caggaggaga 60ggcggcggcg cgcgcgccag gaggaggagg agctggagcg catcctgag 10912109DNAArtificial sequenceSynthetic DNA 12ggagttacga cgagtagttg cggttggcga tggaattgtc ggcgtaggag taggaggaga 60ggcggcggcg cgcgcgttag gaggaggagg agttggagcg tattttgag 1091320DNAArtificial sequenceSynthetic DNA 13agttacgacg agtagttgcg 201418DNAArtificial sequenceSynthetic DNA 14tcctcctact cctacgcc 181521DNAArtificial sequenceSynthetic DNA 15ccacggacgc gacaattcca t 2116143DNAHomo sapiens 16ggtggcaacg gctggagtgc cgtcgcccgc gccactcacc ccggcgcggc gccctgcgcg 60gccgctcagc ggaaggccag caggaagatc agtacgacgt tgatgagaac caggagcgcc 120agcacggcgg agaccaccac gcg 14317143DNAArtificial sequenceSynthetic DNA 17ggtggtaacg gttggagtgt cgtcgttcgc gttatttatt tcggcgcggc gttttgcgcg 60gtcgtttagc ggaaggttag taggaagatt agtacgacgt tgatgagaat taggagcgtt 120agtacggcgg agattattac gcg 1431823DNAArtificial sequenceSynthetic DNA 18cgttcgcgtt atttatttcg gcg 231924DNAArtificial sequenceSynthetic DNA 19gctcctaatt ctcatcaacg tcgt 242020DNAArtificial sequenceSynthetic DNA 20cgccgagggc ggcgttttgc 2021100DNAHomo sapiens 21ggcccggggc cgcctgggcc cctaggggct ggacgtcaac ctgttagata gagggcgtgg 60gaccccccgc aggcggctgc tcggacgacc gcatccggag 10022100DNAArtificial sequenceSynthetic DNA 22ggttcggggt cgtttgggtt tttaggggtt ggacgttaat ttgttagata gagggcgtgg 60gatttttcgt aggcggttgt tcggacgatc gtattcggag 1002324DNAArtificial sequenceSynthetic DNA 23cgttaatttg ttagatagag ggcg 242418DNAArtificial sequenceSynthetic DNA 24acgatcgtcc gaacaacc 182521DNAArtificial sequenceSynthetic DNA 25ccacggacgc gcctacgaaa a 212617DNAArtificial sequenceSynthetic DNA 26tccgaacaac cgcctac 172721DNAArtificial sequenceSynthetic DNA 27ccacggacgc gaaaaatccc a 2128119DNAHomo sapiens 28gcttccagcc gcgcgctccg tgccactgcc gctctctgca gccccgcgtc cccgcagcct 60ccccatggcc agcccgcttc gctccgctgc ggcccttgcc cgccaggtac ctcgaaccc 11929119DNAArtificial sequenceSynthetic DNA 29gtttttagtc gcgcgtttcg tgttattgtc gttttttgta gtttcgcgtt ttcgtagttt 60ttttatggtt agttcgtttc gtttcgttgc ggtttttgtt cgttaggtat ttcgaattt 1193027DNAArtificial sequenceSynthetic DNA 30gtgttattgt cgttttttgt agtttcg 273118DNAArtificial sequenceSynthetic DNA 31cgcaacgaaa cgaaacga 183220DNAArtificial sequenceSynthetic DNA 32cgccgagggc gttttcgtag 2033140DNAHomo sapiens 33gcccgccgca cgccgcaatg ctccgcgctc cccgcggggt cgggcgactc agacagggac 60cggaaaagaa ccacgcagaa gaaagcccta tttcttgtcg tctgttcctg tgcagccttg 120cagcctcgcc gcccccgcgt 14034140DNAArtificial sequenceSynthetic DNA 34gttcgtcgta cgtcgtaatg tttcgcgttt ttcgcggggt cgggcgattt agatagggat 60cggaaaagaa ttacgtagaa gaaagtttta ttttttgtcg tttgtttttg tgtagttttg 120tagtttcgtc gttttcgcgt 1403521DNAArtificial sequenceSynthetic DNA 35cgtaatgttt cgcgtttttc g 213627DNAArtificial sequenceSynthetic DNA 36actttcttct acgtaattct tttccga 273720DNAArtificial sequenceSynthetic DNA 37cgccgagggc ggggtcgggc 203885DNAHomo sapiens 38gccggggagt cgagaagcaa gtactagcgc tccaggaccg cgcgcgccgc cccgcgccgc 60cccgcgccgc ccctcggtcc agagc 853985DNAArtificial sequenceSynthetic DNA 39gtcggggagt cgagaagtaa gtattagcgt tttaggatcg cgcgcgtcgt ttcgcgtcgt 60ttcgcgtcgt ttttcggttt agagt 854023DNAArtificial sequenceSynthetic DNA 40agtattagcg ttttaggatc gcg 234121DNAArtificial sequenceSynthetic DNA 41actctaaacc gaaaaacgac g 214221DNAArtificial sequenceSynthetic DNA 42ccacggacgg cgaaacgacg c 214374DNAHomo sapiens 43gccgggagcc cgcacttcct cctcgggggc ctcagaaaac cacagggcgc ggggccaggg 60cggcggcccc cagg 744474DNAArtificial sequenceSynthetic DNA 44gtcgggagtt cgtatttttt tttcgggggt tttagaaaat tatagggcgc ggggttaggg 60cggcggtttt tagg 744525DNAArtificial sequenceSynthetic DNA 45tcgggagttc gtattttttt ttcgg 254617DNAArtificial sequenceSynthetic DNA 46aaaaccgccg ccctaac 174720DNAArtificial sequenceSynthetic DNA 47cgccgaggcc ccgcgcccta 204878DNAHomo sapiens 48cggggcctac cctcaggcag cgctcgctcg aggccagctt ccgagctcca acccctgccc 60gaaacctcgg cctcactg 784978DNAArtificial sequenceSynthetic DNA 49cggggtttat ttttaggtag cgttcgttcg aggttagttt tcgagtttta atttttgttc 60gaaatttcgg ttttattg 785024DNAArtificial sequenceSynthetic DNA 50gggtttattt ttaggtagcg ttcg 245129DNAArtificial sequenceSynthetic DNA 51cgaaatttcg aacaaaaatt aaaactcga 295221DNAArtificial sequenceSynthetic DNA 52ccacggacgg ttcgaggtta g 2153141DNAHomo sapiens 53tgtcctgaca cgatggccac aggcacagtt tgtggtgatg cccaggggcc cgcgcggccc 60cacggtggtc cagtttacac tcgggccccg cactcctgaa gttccgcgcg ggaggagaag 120ggcgtccctt tcgcagctcg g 14154141DNAArtificial sequenceSynthetic DNA 54tgttttgata cgatggttat aggtatagtt tgtggtgatg tttaggggtt cgcgcggttt 60tacggtggtt tagtttatat tcgggtttcg tatttttgaa gtttcgcgcg ggaggagaag 120ggcgtttttt tcgtagttcg g 1415519DNAArtificial sequenceSynthetic DNA 55tgatgtttag gggttcgcg 195626DNAArtificial sequenceSynthetic DNA 56cgaaacttca aaaatacgaa acccga 265720DNAArtificial sequenceSynthetic DNA 57cgccgagggc ggttttacgg 2058112DNAHomo sapiens 58ccggagcact cgccgctgcg cgccctgaag ccgctggcgg taggcggccc tcgaggccgg 60cgggctgggc ggctcggcag cctgcgccgc ggcctccgcc tcggccgcca gc 11259112DNAArtificial sequenceSynthetic DNA 59tcggagtatt cgtcgttgcg cgttttgaag tcgttggcgg taggcggttt tcgaggtcgg 60cgggttgggc ggttcggtag tttgcgtcgc ggttttcgtt tcggtcgtta gt 1126017DNAArtificial sequenceSynthetic DNA 60gtattcgtcg ttgcgcg 176118DNAArtificial sequenceSynthetic DNA 61cctcgaaaac cgcctacc 186221DNAArtificial sequenceSynthetic DNA 62ccacggacgc gccaacgact t 2163134DNAHomo sapiens 63cgccgtgagt gttatagttc ttaaaggcgg cgtgtccgga gtttcttcct tctggtgggg 60ttcgtggtct cgccggctca ggagtgaagc tgcagatctt cgcggtgagt gttacagctc 120ctaaggcggc gcat 13464134DNAArtificial sequenceSynthetic DNA 64cgtcgtgagt gttatagttt ttaaaggcgg cgtgttcgga gttttttttt tttggtgggg 60ttcgtggttt cgtcggttta ggagtgaagt tgtagatttt cgcggtgagt gttatagttt 120ttaaggcggc gtat 1346517DNAArtificial sequenceSynthetic DNA 65taaaggcggc gtgttcg 176621DNAArtificial sequenceSynthetic DNA 66caacttcact cctaaaccga c 216721DNAArtificial sequenceSynthetic DNA 67ccacggacgc gaaaccacga a 2168107DNAHomo sapiens 68aactggcctt ctggctactc cggaatcgcc aagcagatga ggccagaccg ccgccagcgc 60tgatcacgcg cgctcccaca ggtcctggcg cgcgtgttca gccgcgc 10769107DNAArtificial sequenceSynthetic DNA 69aattggtttt ttggttattt cggaatcgtt aagtagatga ggttagatcg tcgttagcgt 60tgattacgcg cgtttttata ggttttggcg cgcgtgttta gtcgcgt 1077026DNAArtificial sequenceSynthetic DNA 70tggttttttg gttatttcgg aatcgt 267119DNAArtificial sequenceSynthetic DNA 71gcgcgtaatc aacgctaac 197220DNAArtificial sequenceSynthetic DNA 72cgccgaggcg acgatctaac 207385DNAHomo sapiens 73ggagcgggca gaggaggagc ccagcgccga ggcccaggcg cgccccgccc tcgcccctcc 60ccgtgcccct cccccgctgc tcccc 857485DNAArtificial sequenceSynthetic DNA 74ggagcgggta gaggaggagt ttagcgtcga ggtttaggcg cgtttcgttt tcgttttttt 60tcgtgttttt ttttcgttgt ttttt 857519DNAArtificial sequenceSynthetic DNA 75gaggaggagt ttagcgtcg 197624DNAArtificial sequenceSynthetic DNA 76cacgaaaaaa aacgaaaacg aaac 247720DNAArtificial sequenceSynthetic DNA 77cgccgaggcg cgcctaaacc 2078107DNAHomo sapiens 78gcggtctatc acgggcaccc ctaacacttg gtgagtgcgc agtgctctcg gcagtctctg 60ggctccatac gatgcctacc gcacgcccta gcagaggagg tctctgt 10779107DNAArtificial sequenceSynthetic DNA 79gcggtttatt acgggtattt ttaatatttg gtgagtgcgt agtgttttcg gtagtttttg 60ggttttatac gatgtttatc gtacgtttta gtagaggagg tttttgt 1078020DNAArtificial sequenceSynthetic DNA 80tgagtgcgta gtgttttcgg 208127DNAArtificial sequenceSynthetic DNA 81ctcctctact aaaacgtacg ataaaca 278220DNAArtificial sequenceSynthetic DNA 82cgccgaggat cgtataaaac 208321DNAArtificial sequenceSynthetic DNA 83atatttggtg agtgcgtagt g 218426DNAArtificial sequenceSynthetic DNA 84acgtacgata aacatcgtat aaaacc 268520DNAArtificial sequenceSynthetic DNA 85cgccgagggt tttcggtagt 2086121DNAHomo sapiens 86tactccactg ccggcttggt gcccacgctc ggcttccgcc cacccatgga ctacgccttt 60agcgatctca tgcgtgaccg ctcggccgcc gctgctgcgg cggtgcacaa ggagccgacc 120t 12187121DNAArtificial sequenceSynthetic DNA 87tattttattg tcggtttggt gtttacgttc ggttttcgtt tatttatgga ttacgttttt 60agcgatttta tgcgtgatcg ttcggtcgtc gttgttgcgg cggtgtataa ggagtcgatt 120t 1218823DNAArtificial sequenceSynthetic DNA 88tggtgtttac gttcggtttt cgt 238917DNAArtificial sequenceSynthetic DNA 89ccgcaacaac gacgacc 179020DNAArtificial sequenceSynthetic DNA 90cgccgaggcg aacgatcacg 2091106DNAHomo sapiens 91aggccggtca cgaacaaagc gctggcgagt gcgcgcccgc ccacgcgcac aggtgcccgc 60gacaagacgc cccgtccccg cccacgcggc ccccgcgggc tgagcc 10692106DNAArtificial sequenceSynthetic DNA 92aggtcggtta cgaataaagc gttggcgagt gcgcgttcgt ttacgcgtat aggtgttcgc 60gataagacgt ttcgttttcg tttacgcggt tttcgcgggt tgagtt 1069321DNAArtificial sequenceSynthetic DNA 93gttacgaata aagcgttggc g 219420DNAArtificial sequenceSynthetic DNA 94aacgaaacgt cttatcgcga 209521DNAArtificial sequenceSynthetic DNA 95ccacggacgg agtgcgcgtt c 219685DNAHomo sapiens 96gccggccccg cagcatcctc ctgctcgcgg ctctcccgcc acctgtcccg ctccctgccg 60cgccctgggg cccgcaccta cccac 859785DNAArtificial sequenceSynthetic DNA 97gtcggtttcg tagtattttt ttgttcgcgg ttttttcgtt atttgtttcg ttttttgtcg 60cgttttgggg ttcgtattta tttat 859825DNAArtificial sequenceSynthetic DNA 98cggtttcgta gtattttttt gttcg 259917DNAArtificial sequenceSynthetic DNA 99gaaccccaaa acgcgac 1710020DNAArtificial sequenceSynthetic DNA 100cgccgagggc ggttttttcg 20101134DNAHomo sapiens 101cgcctcctgg gctccccccg gagtgggagg gagccgcggt cccgcctccg cgcccgttcc 60ctcccaggcc cctcggccgc cgcgccgagc tttccgcgcg tggacagact gcccggccga 120cggacggacg cagg 134102134DNAArtificial sequenceSynthetic DNA 102cgttttttgg gtttttttcg gagtgggagg gagtcgcggt ttcgttttcg cgttcgtttt 60tttttaggtt tttcggtcgt cgcgtcgagt ttttcgcgcg tggatagatt gttcggtcga 120cggacggacg tagg 13410319DNAArtificial sequenceSynthetic DNA 103gagtcgcggt ttcgttttc 1910419DNAArtificial sequenceSynthetic DNA 104gacgcgacga ccgaaaaac 1910520DNAArtificial sequenceSynthetic DNA 105cgccgaggcg cgttcgtttt 20106108DNAHomo sapiens 106tccggcgccg cgttttctag agaaccgggt ctcagcgatg ctcatttcag ccccgtctta 60atgcaacaaa cgaaacccca cacgaacgaa aaggaacatg tctgcgct 108107107DNAArtificial sequenceSynthetic DNA 107tcggcgtcgc gttttttaga gaatcgggtt ttagcgatgt ttattttagt ttcgttttaa 60tgtaataaac gaaattttat acgaacgaaa aggaatatgt ttgcgtt 10710821DNAArtificial sequenceSynthetic DNA 108ggcgtcgcgt tttttagaga a 2110928DNAArtificial sequenceSynthetic DNA 109ttccttttcg ttcgtataaa atttcgtt 2811021DNAArtificial sequenceSynthetic DNA 110ccacggacga tcgggtttta g 21111128DNAHomo sapiens 111gggcctgctg gccggggacc cgcgcgtcga gcgcctggtg cgcgacagcg cctcctactg 60ccgcgagcgc ttcgaccccg acgagtactc cacggccgtg cgcgaggcgc cagcggagct 120cgccgaag 128112128DNAArtificial sequenceSynthetic DNA 112gggtttgttg gtcggggatt cgcgcgtcga gcgtttggtg cgcgatagcg ttttttattg 60tcgcgagcgt ttcgatttcg acgagtattt tacggtcgtg cgcgaggcgt tagcggagtt 120cgtcgaag 12811323DNAArtificial sequenceSynthetic DNA 113cgatagcgtt ttttattgtc gcg 2311422DNAArtificial sequenceSynthetic DNA 114gcacgaccgt aaaatactcg tc 2211521DNAArtificial sequenceSynthetic DNA 115ccacggacgc gaaatcgaaa c 21116140DNAHomo sapiens 116tagcagcagc cgcagccatg gcggggatga agacagcctc cggggactac atcgactcgt 60catgggagct gcgggtgttt gtgggagagg aggacccaga ggccgagtcg gtcaccctgc 120gggtcactgg ggagtcgcac 140117140DNAArtificial sequenceSynthetic DNA 117tagtagtagt cgtagttatg gcggggatga agatagtttt cggggattat atcgattcgt 60tatgggagtt gcgggtgttt gtgggagagg aggatttaga ggtcgagtcg gttattttgc 120gggttattgg ggagtcgtat
14011824DNAArtificial sequenceSynthetic DNA 118gttttcgggg attatatcga ttcg 2411922DNAArtificial sequenceSynthetic DNA 119cccaataacc cgcaaaataa cc 2212020DNAArtificial sequenceSynthetic DNA 120cgccgaggcg actcgacctc 20121104DNAHomo sapiens 121gtcccagaga cgccctaggg tcagaggtca tctccgtggc aacggaaact tcccgcgcta 60cggcggctcc aacgggccgc ttccgccgca ttgcgtagcg aagc 104122104DNAArtificial sequenceSynthetic DNA 122gttttagaga cgttttaggg ttagaggtta ttttcgtggt aacggaaatt tttcgcgtta 60cggcggtttt aacgggtcgt tttcgtcgta ttgcgtagcg aagt 10412324DNAArtificial sequenceSynthetic DNA 123tttcgtggta acggaaattt ttcg 2412417DNAArtificial sequenceSynthetic DNA 124cgacgaaaac gacccgt 1712520DNAArtificial sequenceSynthetic DNA 125cgccgagggc gttacggcgg 20126107DNAHomo sapiens 126gcgccccggc cgcaggcgga ggacagggag gagcgcacac gagaaagctc ccacgcgccc 60gcgcctcgcc tccgacggga aggcgcctct tccgaccgtc ctggatg 107127107DNAArtificial sequenceSynthetic DNA 127gcgtttcggt cgtaggcgga ggatagggag gagcgtatac gagaaagttt ttacgcgttc 60gcgtttcgtt ttcgacggga aggcgttttt ttcgatcgtt ttggatg 10712825DNAArtificial sequenceSynthetic DNA 128gagcgtatac gagaaagttt ttacg 2512917DNAArtificial sequenceSynthetic DNA 129aacgccttcc cgtcgaa 1713021DNAArtificial sequenceSynthetic DNA 130ccacggacgg cgttcgcgtt t 21131108DNAHomo sapiens 131cgagagggcg cgagcacagc cgaggccatg gaggtgacgg cggaccagcc gcgctgggtg 60agccaccacc accccgccgt gctcaacggg cagcacccgg acacgcac 108132108DNAArtificial sequenceSynthetic DNA 132cgagagggcg cgagtatagt cgaggttatg gaggtgacgg cggattagtc gcgttgggtg 60agttattatt atttcgtcgt gtttaacggg tagtattcgg atacgtat 10813317DNAArtificial sequenceSynthetic DNA 133gggcgcgagt atagtcg 1713417DNAArtificial sequenceSynthetic DNA 134caacgcgact aatccgc 1713520DNAArtificial sequenceSynthetic DNA 135cgccgaggcc gtcacctcca 20136141DNAHomo sapiens 136cgccccctca cctccccgat catgccgttc cagacgccat cgatcttctt tccgtgcttg 60ccattggtga ccaggtagag gtcgtagctg aagccgatgg tatgcgccag ccgcttcaga 120atgtcgatgc agaaaccctt g 141137141DNAArtificial sequenceSynthetic DNA 137cgttttttta ttttttcgat tatgtcgttt tagacgttat cgattttttt ttcgtgtttg 60ttattggtga ttaggtagag gtcgtagttg aagtcgatgg tatgcgttag tcgttttaga 120atgtcgatgt agaaattttt g 14113827DNAArtificial sequenceSynthetic DNA 138tcgattatgt cgttttagac gttatcg 2713928DNAArtificial sequenceSynthetic DNA 139tctacatcga cattctaaaa cgactaac 2814021DNAArtificial sequenceSynthetic DNA 140ccacggacgc gcataccatc g 2114193DNAHomo sapiens 141cggcgaggct tcccgcctgg cgcattacaa caagcgctcg accatcacct ccagggagat 60ccagacggcc gtgcgcctgc tgcttcccgg gga 9314293DNAArtificial sequenceSynthetic DNA 142cggcgaggtt tttcgtttgg cgtattataa taagcgttcg attattattt ttagggagat 60ttagacggtc gtgcgtttgt tgtttttcgg gga 9314323DNAArtificial sequenceSynthetic DNA 143tggcgtatta taataagcgt tcg 2314418DNAArtificial sequenceSynthetic DNA 144aacaacaaac gcacgacc 1814521DNAArtificial sequenceSynthetic DNA 145ccacggacgc gtctaaatct c 21146101DNAHomo sapiens 146gggcgggcca ggcgctgggc acggtgatgg ccaccactgg ggccctgggc aactactacg 60tggactcgtt cctgctgggc gccgacgccg cggatgagct g 101147101DNAArtificial sequenceSynthetic DNA 147gggcgggtta ggcgttgggt acggtgatgg ttattattgg ggttttgggt aattattacg 60tggattcgtt tttgttgggc gtcgacgtcg cggatgagtt g 10114824DNAArtificial sequenceSynthetic DNA 148ttgggtaatt attacgtgga ttcg 2414916DNAArtificial sequenceSynthetic DNA 149actcatccgc gacgtc 1615021DNAArtificial sequenceSynthetic DNA 150ccacggacgc gacgcccaac a 2115195DNAHomo sapiens 151gggccattgc cagaagacgt cttctcgggg cgccaggatt cacctttcct tcccgacctc 60aacttcttcg cggccgactc ctgtctccag ctatc 9515295DNAArtificial sequenceSynthetic DNA 152gggttattgt tagaagacgt tttttcgggg cgttaggatt tatttttttt tttcgatttt 60aattttttcg cggtcgattt ttgtttttag ttatt 9515322DNAArtificial sequenceSynthetic DNA 153gttagaagac gttttttcgg gg 2215420DNAArtificial sequenceSynthetic DNA 154aaaacaaaaa tcgaccgcga 2015520DNAArtificial sequenceSynthetic DNA 155cgccgagggc gttaggattt 20156106DNAHomo sapiens 156ggccccggaa gcccagctcc cgggccctgg agcccgccac ggcggcagcc ctgcggcggc 60ggctggacct gggcagttgc ctggacgtgc tggcctttgc ccagca 106157106DNAArtificial sequenceSynthetic DNA 157ggtttcggaa gtttagtttt cgggttttgg agttcgttac ggcggtagtt ttgcggcggc 60ggttggattt gggtagttgt ttggacgtgt tggtttttgt ttagta 10615825DNAArtificial sequenceSynthetic DNA 158agttttcggg ttttggagtt cgtta 2515917DNAArtificial sequenceSynthetic DNA 159ccaaatccaa ccgccgc 1716020DNAArtificial sequenceSynthetic DNA 160cgccgaggac ggcggtagtt 20161106DNAHomo sapiens 161ggcggcgccg gcggctgcgc ggggggcgcc aggccctgct gctgctgctg ctgctgactg 60cggtagtagg cggcggcggc cacggcggca aagttgtggg tctgga 106162106DNAArtificial sequenceSynthetic DNA 162ggcggcgtcg gcggttgcgc ggggggcgtt aggttttgtt gttgttgttg ttgttgattg 60cggtagtagg cggcggcggt tacggcggta aagttgtggg tttgga 10616319DNAArtificial sequenceSynthetic DNA 163ttgattgcgg tagtaggcg 1916419DNAArtificial sequenceSynthetic DNA 164aacccacaac tttaccgcc 1916520DNAArtificial sequenceSynthetic DNA 165cgccgaggcg taaccgccgc 20166154DNAHomo sapiens 166ggtttccccc caccccggcc tcggggtctc tccacgtctc cccgccgacg tgctcacctg 60ctcagggggc gcccccgagc cgcgccccgc gcccgccccc aggagggcct ccgcgagccg 120gctgcacacc ccgaggcggt cccggctgca caac 154167154DNAArtificial sequenceSynthetic DNA 167ggtttttttt tatttcggtt tcggggtttt tttacgtttt ttcgtcgacg tgtttatttg 60tttagggggc gttttcgagt cgcgtttcgc gttcgttttt aggagggttt tcgcgagtcg 120gttgtatatt tcgaggcggt ttcggttgta taat 15416827DNAArtificial sequenceSynthetic DNA 168gtttcggggt ttttttacgt tttttcg 2716919DNAArtificial sequenceSynthetic DNA 169aaacgcgact cgaaaacgc 1917020DNAArtificial sequenceSynthetic DNA 170cgccgagggt cgacgtgttt 2017195DNAHomo sapiens 171ctccggtttt cgcggttctc agcgatatta ggcgcggcca gtgtctgaaa gctcctcggg 60gttacgtcct ggggcgactg gaggcggctc acgac 9517295DNAArtificial sequenceSynthetic DNA 172tttcggtttt cgcggttttt agcgatatta ggcgcggtta gtgtttgaaa gtttttcggg 60gttacgtttt ggggcgattg gaggcggttt acgat 9517322DNAArtificial sequenceSynthetic DNA 173cggtttttag cgatattagg cg 2217418DNAArtificial sequenceSynthetic DNA 174cccaaaacgt aaccccga 1817520DNAArtificial sequenceSynthetic DNA 175cgccgagggc ggttagtgtt 20176143DNAHomo sapiens 176cgacggccgc ggaggaggaa ggccaggggg aaatttgcat ttcgtaaaac cgcggttaag 60aaatgacgat gccacgtaga caagccagtt gtgacgttca gcacaacgtg ctactgaact 120accgagatcc gccaccaaat ggc 143177143DNAArtificial sequenceSynthetic DNA 177cgacggtcgc ggaggaggaa ggttaggggg aaatttgtat ttcgtaaaat cgcggttaag 60aaatgacgat gttacgtaga taagttagtt gtgacgttta gtataacgtg ttattgaatt 120atcgagattc gttattaaat ggt 14317825DNAArtificial sequenceSynthetic DNA 178gggaaatttg tatttcgtaa aatcg 2517927DNAArtificial sequenceSynthetic DNA 179acaactaact tatctacgta acatcgt 2718021DNAArtificial sequenceSynthetic DNA 180ccacggacgg cggttaagaa a 21181116DNAHomo sapiens 181ggcttggggt ccagccgccc gcccctgccg ccaccgcacc atgtcctgcc tctactcccg 60cctcagcgcc ccctgcgggg tccgcgcctt cagctgcatc tcggcctgcg ggcccc 116182116DNAArtificial sequenceSynthetic DNA 182ggtttggggt ttagtcgttc gtttttgtcg ttatcgtatt atgttttgtt tttattttcg 60ttttagcgtt ttttgcgggg ttcgcgtttt tagttgtatt tcggtttgcg ggtttt 11618322DNAArtificial sequenceSynthetic DNA 183tcgttcgttt ttgtcgttat cg 2218423DNAArtificial sequenceSynthetic DNA 184aaccgaaata caactaaaaa cgc 2318521DNAArtificial sequenceSynthetic DNA 185ccacggacgc gaaccccgca a 21186108DNAHomo sapiens 186ggaaggctgc agcgagagat ttacatattc atccgagctt aaggaagccg cgataatgca 60ggtacagccc gaaacccacg cccccagacc ttatctgcgc gccccgcc 108187108DNAArtificial sequenceSynthetic DNA 187ggaaggttgt agcgagagat ttatatattt attcgagttt aaggaagtcg cgataatgta 60ggtatagttc gaaatttacg tttttagatt ttatttgcgc gtttcgtt 10818819DNAArtificial sequenceSynthetic DNA 188ttcgagttta aggaagtcg 1918923DNAArtificial sequenceSynthetic DNA 189tctaaaaacg taaatttcga act 2319021DNAArtificial sequenceSynthetic DNA 190ccacggacgg cgataatgta g 21191137DNAHomo sapiens 191ggagttattt ttaaccatcg cctcccagaa cattacggag cttcctctct ccaacacgca 60ggaaacccta cttggctgtg cttcctgcta acacgaggcc ctgcgattgc tgagaacaac 120agccccgaga ctgcgcg 137192137DNAArtificial sequenceSynthetic DNA 192ggagttattt ttaattatcg ttttttagaa tattacggag tttttttttt ttaatacgta 60ggaaatttta tttggttgtg ttttttgtta atacgaggtt ttgcgattgt tgagaataat 120agtttcgaga ttgcgcg 13719330DNAArtificial sequenceSynthetic DNA 193tttaattatc gttttttaga atattacgga 3019426DNAArtificial sequenceSynthetic DNA 194actattattc tcaacaatcg caaaac 2619521DNAArtificial sequenceSynthetic DNA 195ccacggacgc ctcgtattaa c 21196117DNAHomo sapiens 196ggcgggcgct tggccaaaca gcccaagact gcggaatcac actcgccact gtgtacctgg 60acgccatctg cagacccagc gcctgcgggg attccggaaa cgggagagcg ggcttcc 117197117DNAArtificial sequenceSynthetic DNA 197ggcgggcgtt tggttaaata gtttaagatt gcggaattat attcgttatt gtgtatttgg 60acgttatttg tagatttagc gtttgcgggg atttcggaaa cgggagagcg ggttttt 11719827DNAArtificial sequenceSynthetic DNA 198agtttaagat tgcggaatta tattcgt 2719917DNAArtificial sequenceSynthetic DNA 199ttccgaaatc cccgcaa 1720020DNAArtificial sequenceSynthetic DNA 200cgccgaggaa cgctaaatct 20201156DNAHomo sapiens 201cgcaggctga ggccctcggg tccccagcgg gtcctcgcca tcagtcactc tctacgggcc 60aggcctgggg gtcacggcct gcaggagcct ccctgcgcgg ccccactccc tcatctgcga 120ccccgtgggg aggcgaccct gaccaccctc gttccg 156202156DNAArtificial sequenceSynthetic DNA 202cgtaggttga ggttttcggg tttttagcgg gttttcgtta ttagttattt tttacgggtt 60aggtttgggg gttacggttt gtaggagttt ttttgcgcgg ttttattttt ttatttgcga 120tttcgtgggg aggcgatttt gattattttc gtttcg 15620323DNAArtificial sequenceSynthetic DNA 203ggttgaggtt ttcgggtttt tag 2320417DNAArtificial sequenceSynthetic DNA 204cctccccacg aaatcgc 1720520DNAArtificial sequenceSynthetic DNA 205cgccgagggc gggttttcgt 2020618DNAArtificial sequenceSynthetic DNA 206aggagttttt ttgcgcgg 1820724DNAArtificial sequenceSynthetic DNA 207acgaaaataa tcaaaatcgc ctcc 2420820DNAArtificial sequenceSynthetic DNA 208cgccgaggcc cacgaaatcg 20209114DNAHomo sapiens 209cgggggaggg cggcatcagc cagagcctca gccgacggcg ctccccaggt ccacttcccg 60ctccgatacc ctccccctaa gcacgatacc cagggcccag ggctgctctt ggcg 114210114DNAArtificial sequenceSynthetic DNA 210cgggggaggg cggtattagt tagagtttta gtcgacggcg ttttttaggt ttatttttcg 60tttcgatatt ttttttttaa gtacgatatt tagggtttag ggttgttttt ggcg 11421125DNAArtificial sequenceSynthetic DNA 211gcggtattag ttagagtttt agtcg 2521224DNAArtificial sequenceSynthetic DNA 212acaaccctaa accctaaata tcgt 2421321DNAArtificial sequenceSynthetic DNA 213ccacggacgg acggcgtttt t 21214107DNAHomo sapiens 214ctccgctccc cgcaggcctg gccgcgcgac gggcacccag cgggttgtta tcaattattc 60aggccccaag ttcacgggca ctgcatccat ttccctcgcg tgcgccc 107215107DNAArtificial sequenceSynthetic DNA 215tttcgttttt cgtaggtttg gtcgcgcgac gggtatttag cgggttgtta ttaattattt 60aggttttaag tttacgggta ttgtatttat ttttttcgcg tgcgttt 10721619DNAArtificial sequenceSynthetic DNA 216tttcgtaggt ttggtcgcg 1921726DNAArtificial sequenceSynthetic DNA 217aacctaaata attaataaca acccgc 2621821DNAArtificial sequenceSynthetic DNA 218ccacggacgg cgacgggtat t 2121988DNAHomo sapiens 219gtgggccggg cgtgacgcgc ggtcaaagtg caatgatttt tcagttcggt tggctaaaca 60gggtcagagc tgagagcgaa gcagaagg 8822088DNAArtificial sequenceSynthetic DNA 220gtgggtcggg cgtgacgcgc ggttaaagtg taatgatttt ttagttcggt tggttaaata 60gggttagagt tgagagcgaa gtagaagg 8822118DNAArtificial sequenceSynthetic DNA 221tggttcgggc gtgacgcg 1822223DNAArtificial sequenceSynthetic DNA 222tctaacccta tttaaccaac cga 2322320DNAArtificial sequenceSynthetic DNA 223cgccgagggc ggttaaagtg 2022494DNAHomo sapiens 224ggacctcctc ggccccgccc catccgcctt cgggatgctg ctgagccccg tcacctccac 60ccccttctcg gtcaaggaca tcctgcgact ggag 9422594DNAArtificial sequenceSynthetic DNA 225ggattttttc ggtttcgttt tattcgtttt cgggatgttg ttgagtttcg ttatttttat 60ttttttttcg gttaaggata ttttgcgatt ggag 9422625DNAArtificial sequenceSynthetic DNA 226gattttttcg gtttcgtttt attcg 2522724DNAArtificial sequenceSynthetic DNA 227caatcgcaaa atatccttaa ccga 2422821DNAArtificial sequenceSynthetic DNA 228ccacggacgg ttttcgggat g 2122989DNAHomo sapiens 229ctgtcagtgc tgaccgagcg ccgcgccttc cggccatacg ggctccacgg tgcgcggttc 60cccagccctc gcggccctcc ccgcccccg 8923089DNAArtificial sequenceSynthetic DNA 230ttgttagtgt tgatcgagcg tcgcgttttt cggttatacg ggttttacgg tgcgcggttt 60tttagttttc gcggtttttt tcgttttcg 8923122DNAArtificial sequenceSynthetic DNA 231cgtcgcgttt ttcggttata cg 2223221DNAArtificial sequenceSynthetic DNA 232cgcgaaaact aaaaaaccgc g 2123321DNAArtificial sequenceSynthetic DNA 233ccacggacgg caccgtaaaa c 21234114DNAHomo sapiens 234cggagtatgg tgaggagcgc gggggacggg tgcgggaagg ggacagcagg gctgagcctg 60gggcccgcaa gacccagcag cccgagcggg cgcagagacc ccacgccacg caca 114235114DNAArtificial sequenceSynthetic DNA 235cggagtatgg tgaggagcgc gggggacggg tgcgggaagg ggatagtagg gttgagtttg 60gggttcgtaa gatttagtag ttcgagcggg cgtagagatt ttacgttacg tata 11423618DNAArtificial sequenceSynthetic DNA 236ggttgagttt ggggttcg 1823723DNAArtificial sequenceSynthetic DNA 237cgtaacgtaa aatctctacg ccc 2323821DNAArtificial sequenceSynthetic DNA 238ccacggacgc gctcgaacta c 2123995DNAHomo sapiens 239ggagagcagc ccgcagaacc
tggccgcgta ctacacgcct ttcccgtcct atggacacta 60cagaaacagc ctggccaccg tggaggaaga cttcc 9524095DNAArtificial sequenceSynthetic DNA 240ggagagtagt tcgtagaatt tggtcgcgta ttatacgttt ttttcgtttt atggatatta 60tagaaatagt ttggttatcg tggaggaaga ttttt 9524123DNAArtificial sequenceSynthetic DNA 241gagtagttcg tagaatttgg tcg 2324231DNAArtificial sequenceSynthetic DNA 242ccacgataac caaactattt ctataatatc c 3124321DNAArtificial sequenceSynthetic DNA 243ccacggacgg cgtattatac g 2124423DNAArtificial sequenceSynthetic DNA 244ggagagtagt tcgtagaatt tgg 2324535DNAArtificial sequenceSynthetic DNA 245ctatttctat aatatccata aaacgaaaaa aacgt 3524621DNAArtificial sequenceSynthetic DNA 246ccacggacgg tcgcgtatta t 2124792DNAHomo sapiens 247gggaaggtgc cctgcgcgcg cgcgctcacc agatgaagtc ggtgcagtgg ctgcagaagg 60tgggctgctt gaagaagcgg gcggtgaatt tg 9224892DNAArtificial sequenceSynthetic DNA 248gggaaggtgt tttgcgcgcg cgcgtttatt agatgaagtc ggtgtagtgg ttgtagaagg 60tgggttgttt gaagaagcgg gcggtgaatt tg 9224917DNAArtificial sequenceSynthetic DNA 249ggaaggtgtt ttgcgcg 1725021DNAArtificial sequenceSynthetic DNA 250cttctacaac cactacaccg a 2125121DNAArtificial sequenceSynthetic DNA 251ccacggacgg cgcgcgttta t 21252107DNAHomo sapiens 252gcctcggggc ccggggactc acaattacgg gcagagaaca catagtgaag agcacggtca 60tcagcgccag cagcaggagg tgatccagct cctccagggg ctgaggg 107253107DNAArtificial sequenceSynthetic DNA 253gtttcggggt tcggggattt ataattacgg gtagagaata tatagtgaag agtacggtta 60ttagcgttag tagtaggagg tgatttagtt tttttagggg ttgaggg 10725424DNAArtificial sequenceSynthetic DNA 254gggttcgggg atttataatt acgg 2425525DNAArtificial sequenceSynthetic DNA 255cctcctacta ctaacgctaa taacc 2525621DNAArtificial sequenceSynthetic DNA 256ccacggacgc gtactcttca c 2125780DNAHomo sapiens 257ggcggctgca gcggcacccg cgctcctgca ccagggactg tgccgagccg cgcgcggacg 60ggagggaagc gtcccctcag 8025880DNAArtificial sequenceSynthetic DNA 258ggcggttgta gcggtattcg cgtttttgta ttagggattg tgtcgagtcg cgcgcggacg 60ggagggaagc gtttttttag 8025918DNAArtificial sequenceSynthetic DNA 259gttgtagcgg tattcgcg 1826018DNAArtificial sequenceSynthetic DNA 260cttctctccc gtccgcgc 1826120DNAArtificial sequenceSynthetic DNA 261cgccgaggcg cgactcgaca 20262143DNAHomo sapiens 262tccagaaaca cgggtatctc cgcgtggtgc tttgcggtcg ccgtcgttgt ggccgtccgg 60ggtggggtgt gaggagggga cgaaggaggg aaggaagggc aaggcggggg gggctctgcg 120agagcgcgcc cagccccgcc ttc 143263143DNAArtificial sequenceSynthetic DNA 263tttagaaata cgggtatttt cgcgtggtgt tttgcggtcg tcgtcgttgt ggtcgttcgg 60ggtggggtgt gaggagggga cgaaggaggg aaggaagggt aaggcggggg gggttttgcg 120agagcgcgtt tagtttcgtt ttt 14326421DNAArtificial sequenceSynthetic DNA 264agaaatacgg gtattttcgc g 2126517DNAArtificial sequenceSynthetic DNA 265ccacaacgac gacgacc 1726621DNAArtificial sequenceSynthetic DNA 266ccacggacgc gcaaaacacc a 21267120DNAHomo sapiens 267cggtcgggca ggcgggacgg agattacctg gctgtccagg ggaccttatg cagggtttgg 60cccgagccca ggggcagcga ggggcgtctg cggatgcggc tccctgtgcg gcacaacacc 120268120DNAArtificial sequenceSynthetic DNA 268cggtcgggta ggcgggacgg agattatttg gttgtttagg ggattttatg tagggtttgg 60ttcgagttta ggggtagcga ggggcgtttg cggatgcggt tttttgtgcg gtataatatt 12026920DNAArtificial sequenceSynthetic DNA 269gttcgagttt aggggtagcg 2027017DNAArtificial sequenceSynthetic DNA 270ccgcacaaaa aaccgca 1727121DNAArtificial sequenceSynthetic DNA 271ccacggacga tccgcaaacg c 2127255DNAHomo sapiens 272ccggagcact cgccgctgcg cgccctgaag ccgctggcgg taggcggccc tcgag 5527355DNAArtificial sequenceSynthetic DNA 273tcggagtatt cgtcgttgcg cgttttgaag tcgttggcgg taggcggttt tcgag 5527418DNAArtificial sequenceSynthetic DNA 274ggagtattcg tcgttgcg 1827517DNAArtificial sequenceSynthetic DNA 275cgaaaaccgc ctaccgc 1727620DNAArtificial sequenceSynthetic DNA 276cgccgagggc gttttgaagt 2027796DNAHomo sapiens 277cccgggccta cggtcctccc gccacctcca cggggcggct gttggggccc caccaggcag 60agccgtgttc tcaggcgttg gctctcatgg aggtgg 9627896DNAArtificial sequenceSynthetic DNA 278ttcgggttta cggttttttc gttattttta cggggcggtt gttggggttt tattaggtag 60agtcgtgttt ttaggcgttg gtttttatgg aggtgg 9627925DNAArtificial sequenceSynthetic DNA 279acggtttttt cgttattttt acggg 2528021DNAArtificial sequenceSynthetic DNA 280caacgcctaa aaacacgact c 2128120DNAArtificial sequenceSynthetic DNA 281cgccgagggg cggttgttgg 20282148DNAHomo sapiens 282gggcctgtcc cgttccctgc tccccataca ggcgaggctg cgtgcacaca gcttcctgta 60ccccaggagg gcctgcctgg cacgcacccg gtggctgcac catccacacg caagactgca 120acttcagatg ctccgcacgc tggagatg 148283148DNAArtificial sequenceSynthetic DNA 283gggtttgttt cgttttttgt tttttatata ggcgaggttg cgtgtatata gttttttgta 60ttttaggagg gtttgtttgg tacgtattcg gtggttgtat tatttatacg taagattgta 120attttagatg tttcgtacgt tggagatg 14828420DNAArtificial sequenceSynthetic DNA 284ttatataggc gaggttgcgt 2028530DNAArtificial sequenceSynthetic DNA 285cttacgtata aataatacaa ccaccgaata 3028621DNAArtificial sequenceSynthetic DNA 286ccacggacga cgtaccaaac a 2128788DNAHomo sapiens 287cggagctagg agggtggggc tcggagggcg caggaagagc ggctctgcga ggaaagggaa 60aggagaggcc gcttctggga agggaccc 8828888DNAArtificial sequenceSynthetic DNA 288cggagttagg agggtggggt tcggagggcg taggaagagc ggttttgcga ggaaagggaa 60aggagaggtc gtttttggga agggattt 8828918DNAArtificial sequenceSynthetic DNA 289ttaggagggt ggggttcg 1829018DNAArtificial sequenceSynthetic DNA 290ctttcctcgc aaaaccgc 1829121DNAArtificial sequenceSynthetic DNA 291ccacggacgg gagggcgtag g 2129259DNAHomo sapiens 292gccccggcgg gccccgaggc ggccgcggcc tgcaacgtca tcgtgaacgg cacgcgcgg 5929359DNAArtificial sequenceSynthetic DNA 293gtttcggcgg gtttcgaggc ggtcgcggtt tgtaacgtta tcgtgaacgg tacgcgcgg 5929417DNAArtificial sequenceSynthetic DNA 294tttcggcggg tttcgag 1729520DNAArtificial sequenceSynthetic DNA 295cgtaccgttc acgataacgt 2029620DNAArtificial sequenceSynthetic DNA 296cgccgagggg cggtcgcggt 2029720DNAArtificial sequenceSynthetic DNA 297cgccgaggtt acaaaccgcg 2029889DNAHomo sapiens 298ctaggcgaga tggtggaagg cgtgtccgta cgggggtggg ctggggtccc cgtgcagaag 60ggcgcgcgag gacccaggct ggttttccc 8929989DNAArtificial sequenceSynthetic DNA 299ttaggcgaga tggtggaagg cgtgttcgta cgggggtggg ttggggtttt cgtgtagaag 60ggcgcgcgag gatttaggtt ggttttttt 8930017DNAArtificial sequenceSynthetic DNA 300cgagatggtg gaaggcg 1730117DNAArtificial sequenceSynthetic DNA 301gcgcccttct acacgaa 1730221DNAArtificial sequenceSynthetic DNA 302ccacggacgg tgttcgtacg g 21303145DNAHomo sapiens 303gcgctgctgc gccgccaggc aaggcgaggg tccgggagaa ggctcggctc cctcctaaac 60atgtggcccg tggcgtcccc ttgtcccctc cgagcgatgc tcctgcgccc ttcgccgcct 120cccgcgctgc tgcgccgcca ggcaa 145304123DNAArtificial sequenceSynthetic DNA 304ggcgagggtt cgggagaagg ttcggttttt ttttaaatat gtggttcgtg gcgttttttt 60gtttttttcg agcgatgttt ttgcgttttt cgtcgttttt cgcgttgttg cgtcgttagg 120taa 12330521DNAArtificial sequenceSynthetic DNA 305aaatatgtgg ttcgtggcgt t 2130619DNAArtificial sequenceSynthetic DNA 306acgcaacaac gcgaaaaac 1930720DNAArtificial sequenceSynthetic DNA 307cgccgaggcg acgaaaaacg 20308137DNAHomo sapiens 308acgagaaaga gatcgtgcag ggggtgctgc aacagggcac ggcgtggagg aggaaccaga 60ccgcggccag agcgttcagg tactcctgcc ctcgcggctc ctcccctcta gcgtcctttc 120ctccccgagt gcagagg 137309137DNAArtificial sequenceSynthetic DNA 309acgagaaaga gatcgtgtag ggggtgttgt aatagggtac ggcgtggagg aggaattaga 60tcgcggttag agcgtttagg tatttttgtt ttcgcggttt ttttttttta gcgttttttt 120tttttcgagt gtagagg 13731021DNAArtificial sequenceSynthetic DNA 310ggggtgttgt aatagggtac g 2131119DNAArtificial sequenceSynthetic DNA 311ctaaacgctc taaccgcga 1931221DNAArtificial sequenceSynthetic DNA 312ccacggacgg gcgtggagga g 21313117DNAHomo sapiens 313cgcgccgttg gtcacctcgc cggccgccag cgtcgaatgg aagcccgact tgtaccagga 60ctcgtacggg tgcgccatgc ccacgcgcgg gtacagcccg tcggctgccg tcgtgtg 117314117DNAArtificial sequenceSynthetic DNA 314cgcgtcgttg gttatttcgt cggtcgttag cgtcgaatgg aagttcgatt tgtattagga 60ttcgtacggg tgcgttatgt ttacgcgcgg gtatagttcg tcggttgtcg tcgtgtg 11731521DNAArtificial sequenceSynthetic DNA 315tagcgtcgaa tggaagttcg a 2131618DNAArtificial sequenceSynthetic DNA 316ggtcgttagc gtcgaatg 1831720DNAArtificial sequenceSynthetic DNA 317gcgcgtaaac ataacgcacc 2031821DNAArtificial sequenceSynthetic DNA 318ccacggacgc cgtacgaatc c 2131985DNAHomo sapiens 319ggttccttcc cgtgggttct taatcgtctc gctgacttcc agaatgaaac tgcagaccct 60cgcggtaaag atggcgtgac cagaa 8532085DNAArtificial sequenceSynthetic DNA 320ggtttttttt cgtgggtttt taatcgtttc gttgattttt agaatgaaat tgtagatttt 60cgcggtaaag atggcgtgat tagaa 8532122DNAArtificial sequenceSynthetic DNA 321tcgtgggttt ttaatcgttt cg 2232218DNAArtificial sequenceSynthetic DNA 322tcacgccatc tttaccgc 1832321DNAArtificial sequenceSynthetic DNA 323ccacggacgc gaaaatctac a 2132426DNAArtificial sequenceSynthetic DNA 324ggtttttttt cgtgggtttt taatcg 2632521DNAArtificial sequenceSynthetic DNA 325ctaatcacgc catctttacc g 2132621DNAArtificial sequenceSynthetic DNA 326ccacggacgg tttcgttgat t 21327115DNAHomo sapiens 327ggagtgagtg cctacaacgc gcaggccgga ctgatccccc gttgctgcag gttggtgccc 60caagctgcgg gtgctcgggc gccaactaaa gccagctctg tccagacgcg gaaag 115328115DNAArtificial sequenceSynthetic DNA 328ggagtgagtg tttataacgc gtaggtcgga ttgatttttc gttgttgtag gttggtgttt 60taagttgcgg gtgttcgggc gttaattaaa gttagttttg tttagacgcg gaaag 11532923DNAArtificial sequenceSynthetic DNA 329cgtaggtcgg attgattttt cgt 2333030DNAArtificial sequenceSynthetic DNA 330tctaaacaaa actaacttta attaacgccc 3033121DNAArtificial sequenceSynthetic DNA 331ccacggacgc gaacacccgc a 2133219DNAArtificial sequenceSynthetic DNA 332aggaaattgc gggttttcg 1933321DNAArtificial sequenceSynthetic DNA 333ggaaggaaat tgcgggtttt c 2133425DNAArtificial sequenceSynthetic DNA 334ccaaaaatcg tcgctaaaaa tcaac 2533521DNAArtificial sequenceSynthetic DNA 335ccacggacgc gcgcattcac t 21336100DNAHomo sapiens 336ggcggccgcg acccctcccc gctgacctca ctcgagccgc cgcctggcgc agatataagc 60ggcggcccat ctgaagaggg ctcggcaggc gcccggggtc 100337100DNAArtificial sequenceSynthetic DNA 337ggcggtcgcg attttttttc gttgatttta ttcgagtcgt cgtttggcgt agatataagc 60ggcggtttat ttgaagaggg ttcggtaggc gttcggggtt 10033823DNAArtificial sequenceSynthetic DNA 338tttcgttgat tttattcgag tcg 2333919DNAArtificial sequenceSynthetic DNA 339tcttcaaata aaccgccgc 1934020DNAArtificial sequenceSynthetic DNA 340cgccgagggt cgtttggcgt 20341118DNAHomo sapiens 341cgggtggtga agctgcccca cggcctggga gagccttatc gccgcggtcg ctggacgtgt 60gtggatgttt atgagcgaga cctggagccc cacagcttcg gcggactcct ggagggaa 118342118DNAArtificial sequenceSynthetic DNA 342cgggtggtga agttgtttta cggtttggga gagttttatc gtcgcggtcg ttggacgtgt 60gtggatgttt atgagcgaga tttggagttt tatagtttcg gcggattttt ggagggaa 11834322DNAArtificial sequenceSynthetic DNA 343gtttgggaga gttttatcgt cg 2234418DNAArtificial sequenceSynthetic DNA 344cctccaaaaa tccgccga 1834520DNAArtificial sequenceSynthetic DNA 345cgccgagggc ggtcgttgga 2034670DNAHomo sapiens 346ggggcggggg ccgacagccc acgctggcgc ggcaggcgcg tgcgcccgcc gttttcgtga 60gcccgagcag 7034770DNAArtificial sequenceSynthetic DNA 347ggggtcgggg tcgatagttt acgttggcgc ggtaggcgcg tgcgttcgtc gttttcgtga 60gttcgagtag 7034820DNAArtificial sequenceSynthetic DNA 348gtcggggtcg atagtttacg 2034919DNAArtificial sequenceSynthetic DNA 349actcgaactc acgaaaacg 1935020DNAArtificial sequenceSynthetic DNA 350cgccgaggga cgaacgcacg 2035196DNAHomo sapiens 351ggagccccca gccccacgcg ggcacacgca gggtgggtgg tcacgcccgc agggtccgcg 60agcgcggcgc agagcgcggg ccgtgggaag tttctc 9635296DNAArtificial sequenceSynthetic DNA 352ggagttttta gttttacgcg ggtatacgta gggtgggtgg ttacgttcgt agggttcgcg 60agcgcggcgt agagcgcggg tcgtgggaag tttttt 9635318DNAArtificial sequenceSynthetic DNA 353gtagggtggg tggttacg 1835417DNAArtificial sequenceSynthetic DNA 354aacttcccac gacccgc 1735520DNAArtificial sequenceSynthetic DNA 355cgccgagggt tcgtagggtt 20356127DNAHomo sapiens 356ggcgccgcca ttgcggtcct cattttgctg ctggtgggtt gggctacagc aggcctctgg 60agccacacca gggcacggga gtgggtgcag ggaccgtcac cgcgccttca cacgcaccat 120agtgccc 127357127DNAArtificial sequenceSynthetic DNA 357ggcgtcgtta ttgcggtttt tattttgttg ttggtgggtt gggttatagt aggtttttgg 60agttatatta gggtacggga gtgggtgtag ggatcgttat cgcgttttta tacgtattat 120agtgttt 12735823DNAArtificial sequenceSynthetic DNA 358tggagttata ttagggtacg gga 2335928DNAArtificial sequenceSynthetic DNA 359acactataat acgtataaaa acgcgata 2836021DNAArtificial sequenceSynthetic DNA 360ccacggacga acgatcccta c 21361140DNAHomo sapiens 361ggcggcgagg ggcgcgtccg cgggtgggtt tcacctgggt ggtgggcatg tcgggcccgc 60tagggcgagg gtctggccag gggcgtagtt ctcctggtgg gtggggacgc tccgtggcga 120ttggggtcac tcctctgagg 140362140DNAArtificial sequenceSynthetic DNA 362ggcggcgagg ggcgcgttcg cgggtgggtt ttatttgggt ggtgggtatg tcgggttcgt 60tagggcgagg gtttggttag gggcgtagtt tttttggtgg
gtggggacgt ttcgtggcga 120ttggggttat ttttttgagg 14036317DNAArtificial sequenceSynthetic DNA 363ggtggtgggt atgtcgg 1736417DNAArtificial sequenceSynthetic DNA 364ccaatcgcca cgaaacg 1736521DNAArtificial sequenceSynthetic DNA 365ccacggacgg gttcgttagg g 21366117DNAHomo sapiens 366ccgtgggcgc ggacagctgc cgggagcggc aggcgtctcg atcggggacg caggcacttc 60cgtccctgca gagcatcaga cgcgtctcgg gacactgggg acaacatctc ctccgcg 117367117DNAArtificial sequenceSynthetic DNA 367tcgtgggcgc ggatagttgt cgggagcggt aggcgtttcg atcggggacg taggtatttt 60cgtttttgta gagtattaga cgcgtttcgg gatattgggg ataatatttt tttcgcg 11736818DNAArtificial sequenceSynthetic DNA 368gttgtcggga gcggtagg 1836921DNAArtificial sequenceSynthetic DNA 369ccaatatccc gaaacgcgtc t 2137021DNAArtificial sequenceSynthetic DNA 370ccacggacgg cgtttcgatc g 21371120DNAHomo sapiens 371aagctgcgcc cggagacgtg ggagcgttct cttgttttcc gagtgcgcgg actcatcggg 60tcacagttta tgcttttatg acgcggtgag tccagccact gattcctaac ggtttagagt 120372120DNAArtificial sequenceSynthetic DNA 372aagttgcgtt cggagacgtg ggagcgtttt tttgtttttc gagtgcgcgg atttatcggg 60ttatagttta tgtttttatg acgcggtgag tttagttatt gatttttaac ggtttagagt 12037323DNAArtificial sequenceSynthetic DNA 373cgtttttttg tttttcgagt gcg 2337423DNAArtificial sequenceSynthetic DNA 374tcaataacta aactcaccgc gtc 2337521DNAArtificial sequenceSynthetic DNA 375ccacggacgg cggatttatc g 21376224DNAHomo sapiens 376ctctgacctg agtctccttt ggaactctgc aggttctatt tgctttttcc cagatgagct 60ctttttctgg tgtttgtctc tctgactagg tgtctaagac agtgttgtgg gtgtaggtac 120taacactggc tcgtgtgaca aggccatgag gctggtgtaa agcggccttg gagtgtgtat 180taagtaggtg cacagtaggt ctgaacagac tccccatccc aaga 224377224DNAArtificial sequenceSynthetic DNA 377ttttgatttg agtttttttt ggaattttgt aggttttatt tgtttttttt tagatgagtt 60ttttttttgg tgtttgtttt tttgattagg tgtttaagat agtgttgtgg gtgtaggtat 120taatattggt ttgtgtgata aggttatgag gttggtgtaa agtggttttg gagtgtgtat 180taagtaggtg tatagtaggt ttgaatagat tttttatttt aaga 22437819DNAArtificial sequenceSynthetic DNA 378ccatgaggct ggtgtaaag 1937924DNAArtificial sequenceSynthetic DNA 379ctactgtgca cctacttaat acac 2438020DNAArtificial sequenceSynthetic DNA 380cgccgagggc ggccttggag 2038130DNAArtificial sequenceSynthetic DNA 381gtgtttgttt ttttgattag gtgtttaaga 3038226DNAArtificial sequenceSynthetic DNA 382ctttacacca acctcataac cttatc 2638321DNAArtificial sequenceSynthetic DNA 383gacgcggaga tagtgttgtg g 21384139DNAHomo sapiens 384ggccacacag gcccactctg gccctctgag cccccggcgg acccagggca ttcaaggagc 60ggctctgggc tgccagcgca ggcctccgcg caaacacagc aggctggaag tggcgctcat 120caccggcacg tcttcccag 139385139DNAArtificial sequenceSynthetic DNA 385ggttatatag gtttattttg gttttttgag ttttcggcgg atttagggta tttaaggagc 60ggttttgggt tgttagcgta ggttttcgcg taaatatagt aggttggaag tggcgtttat 120tatcggtacg tttttttag 13938628DNAArtificial sequenceSynthetic DNA 386ggtttatttt ggttttttga gttttcgg 2838724DNAArtificial sequenceSynthetic DNA 387tccaacctac tatatttacg cgaa 2438821DNAArtificial sequenceSynthetic DNA 388ccacggacgg cggatttagg g 21389171DNAArtificial sequenceSynthetic DNA 389tccacgtggt gcccactctg gacaggtgga gcagagggaa ggtggtggca tggtggggag 60ggtggcctgg aggacccgat tggctgagtg taaaccagga gaggacatga ctttcagccc 120tgcagccaga cacagctgag ctggtgtgac ctgtgtggag agttcatctg g 171390180DNAArtificial sequenceSynthetic DNA 390tttatcgtgg tgtttatttt ggataggtgg agtagaggga aggtggtgcg tatggtgggc 60gagcgcgtgc gtttggagga tttcgattgg ttgacgtgta aattaggacg aggatatgat 120ttttagtttt gtagttagat atagttgagt tggtgtgatt tgtgtggaga gtttatttgg 18039115DNAArtificial sequenceSynthetic DNA 391cgcatggtgg gcgag 1539218DNAArtificial sequenceSynthetic DNA 392acacgtcagc caatcggg 1839320DNAArtificial sequenceSynthetic DNA 393gacgcggagg cgcgtgcgcc 2039417DNAArtificial sequenceSynthetic DNA 394tgcgtatggt gggcgag 1739526DNAArtificial sequenceSynthetic DNA 395cctaatttac acgtcaacca atcgaa 2639621DNAArtificial sequenceSynthetic DNA 396gacgcggagg cgcgtgcgtt t 2139721DNAArtificial sequenceSynthetic DNA 397ccacggacgg cgcgtgcgtt t 2139828DNAArtificial sequenceSynthetic DNA 398agccggtttt ccggctgaga cctcggcg 2839928DNAArtificial sequenceSynthetic DNA 399agccggtttt ccggctgaga cctcggcg 2840029DNAArtificial sequenceSynthetic DNA 400agccggtttt ccggctgaga ctccgcgtc 2940129DNAArtificial sequenceSynthetic DNA 401agccggtttt ccggctgaga cgtccgtgg 2940228DNAArtificial sequenceSynthetic DNA 402agccggtttt ccggctgaga ggacgcgc 28403108DNAHomo sapiens 403ggaaggaaat tgcgggttcc cgtctgcctt gtctccagct tctctgctga agcccggtag 60cagtgaatgc gcgctgactt tcagcgacga ctcctggaag caacgcca 108404108DNAArtificial sequenceSynthetic DNA 404ggaaggaaat tgcgggtttt cgtttgtttt gtttttagtt tttttgttga agttcggtag 60tagtgaatgc gcgttgattt ttagcgacga tttttggaag taacgtta 108
* * * * *
D00001
D00002
D00003
D00004
D00005
D00006
D00007
D00008
D00009
D00010
D00011
D00012
D00013

D00014

D00015

D00016

D00017

D00018

D00019

D00020

D00021

D00022

D00023

D00024

D00025

D00026

D00027

D00028

D00029

D00030

D00031

D00032

D00033

D00034

D00035

D00036

D00037

D00038

D00039

D00040

D00041

D00042

D00043

D00044

D00045

D00046

D00047

D00048

D00049

D00050

D00051

D00052

D00053

D00054

D00055

D00056

D00057

D00058

D00059

D00060

D00061

D00062

D00063

D00064

D00065

D00066

D00067

D00068

D00069

D00070

D00071

D00072

D00073

D00074

D00075

D00076

D00077

D00078
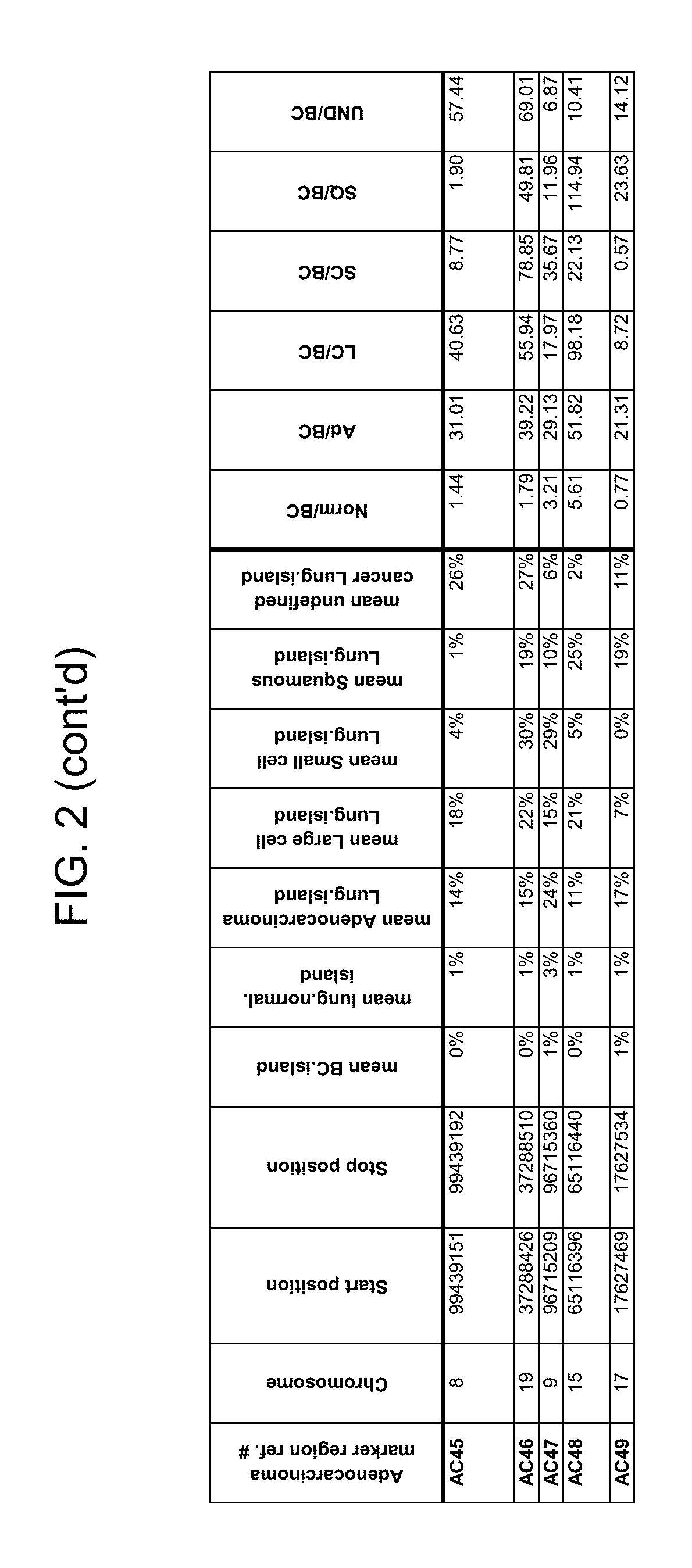
D00079

D00080

D00081
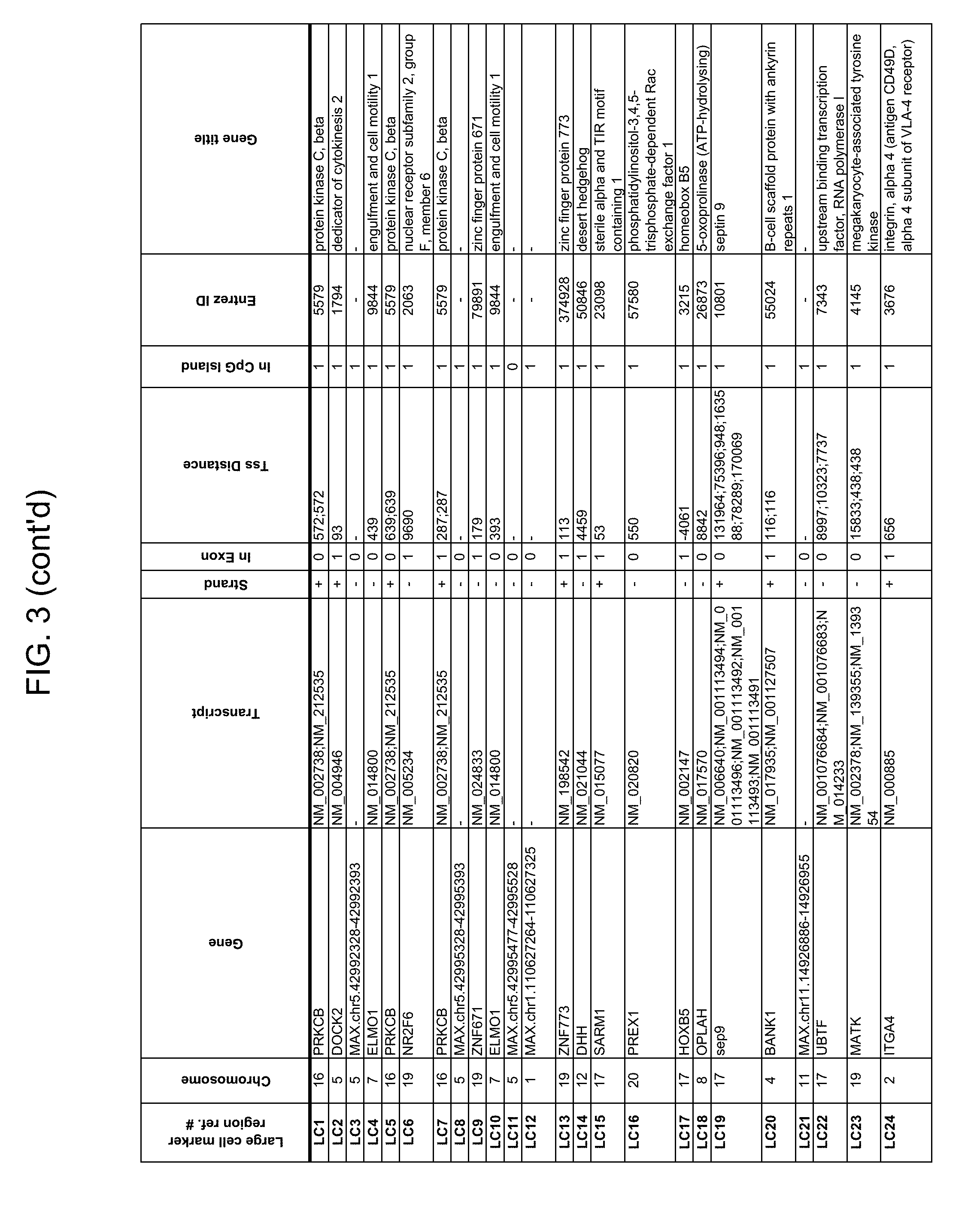
D00082
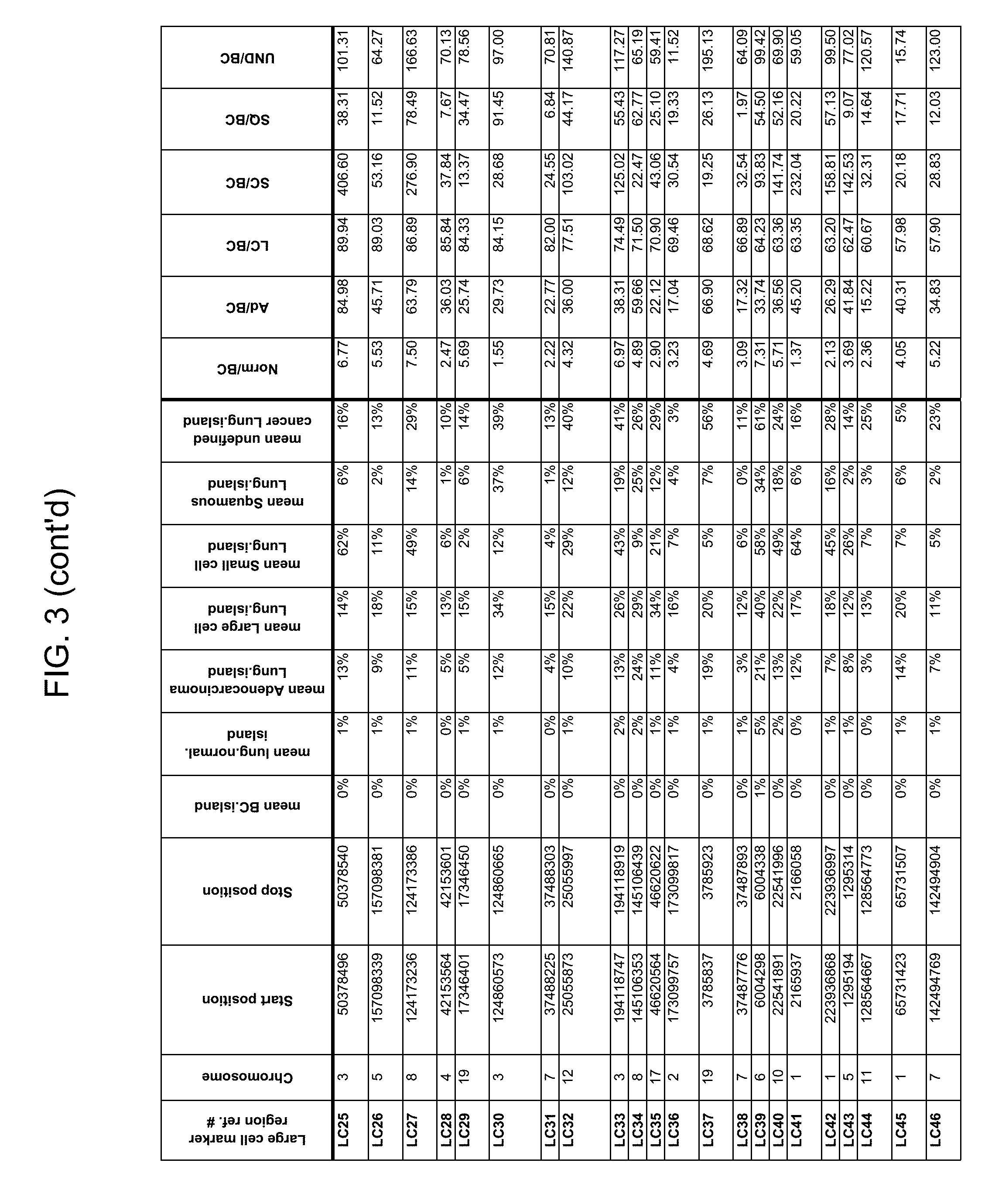
D00083

D00084

D00085

D00086

D00087
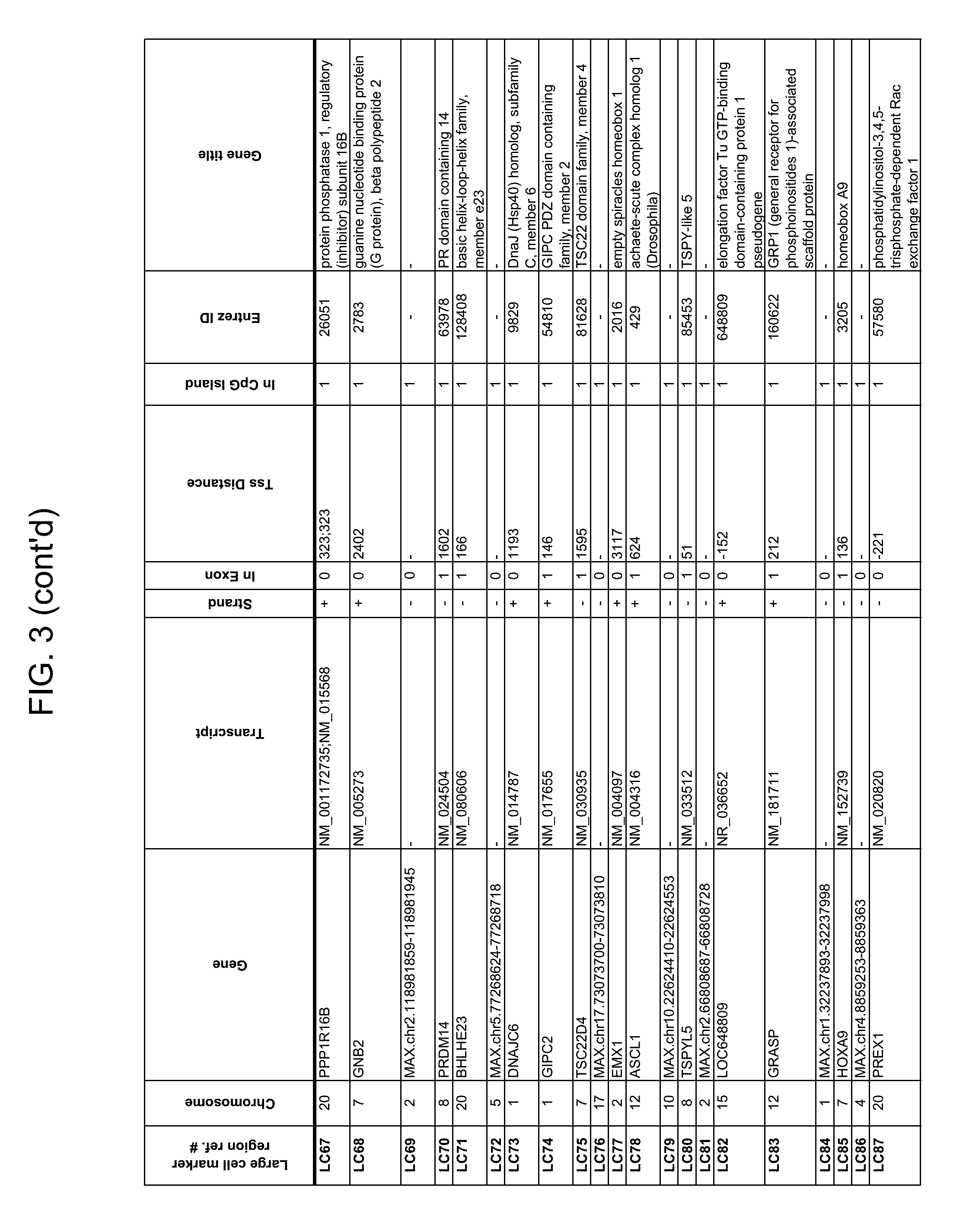
D00088
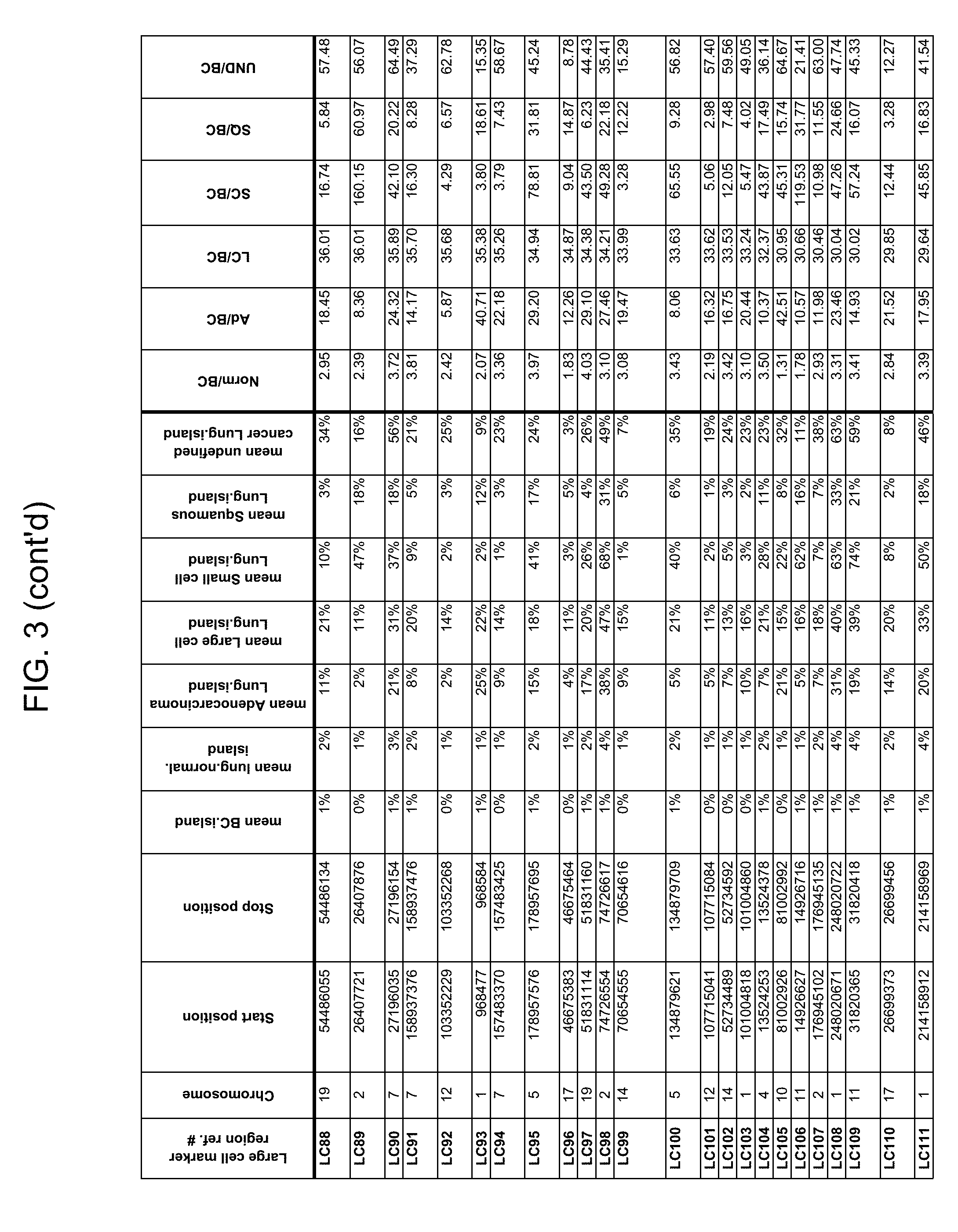
D00089
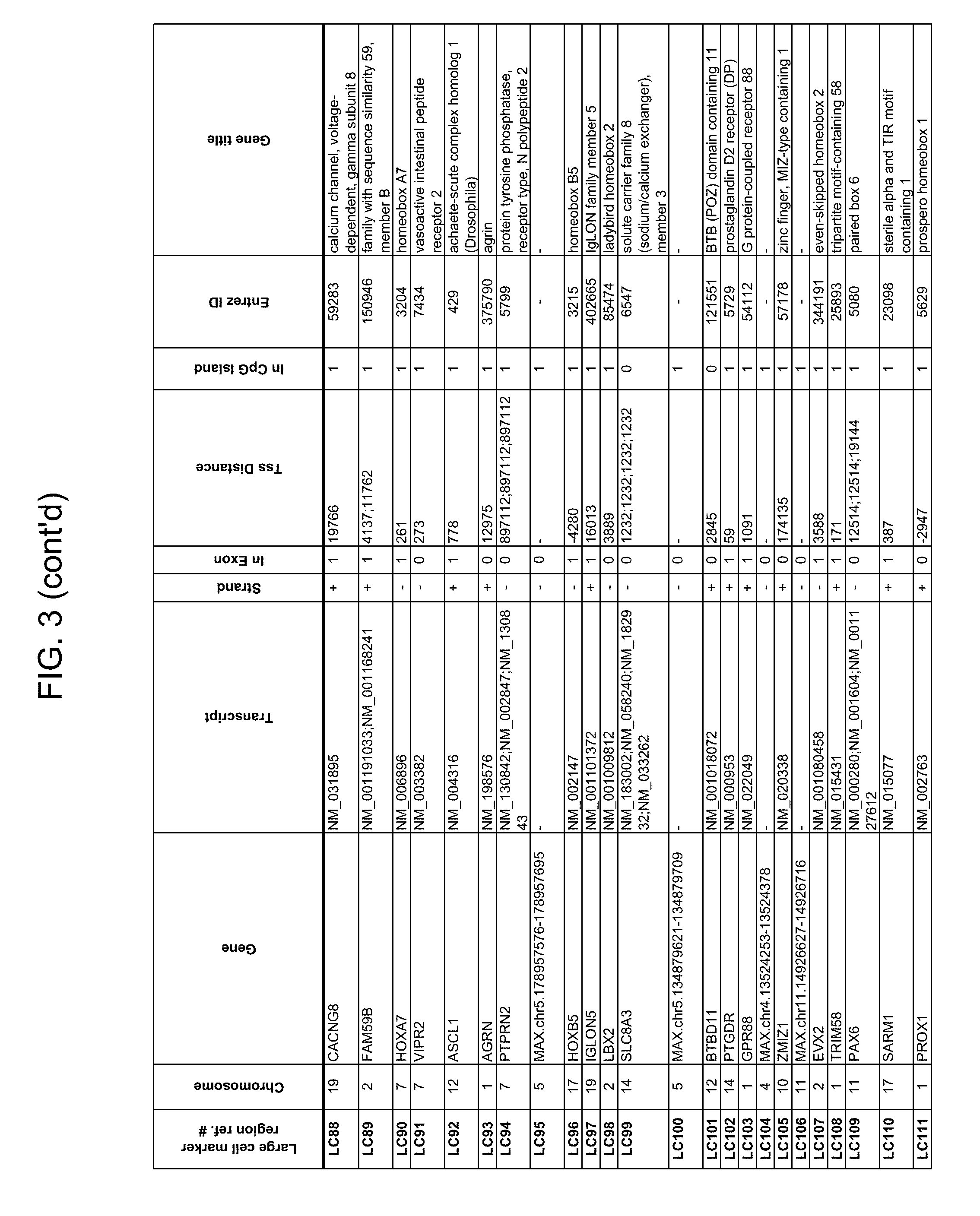
D00090

D00091

D00092

D00093

D00094

D00095

D00096

D00097

D00098

D00099

D00100

D00101
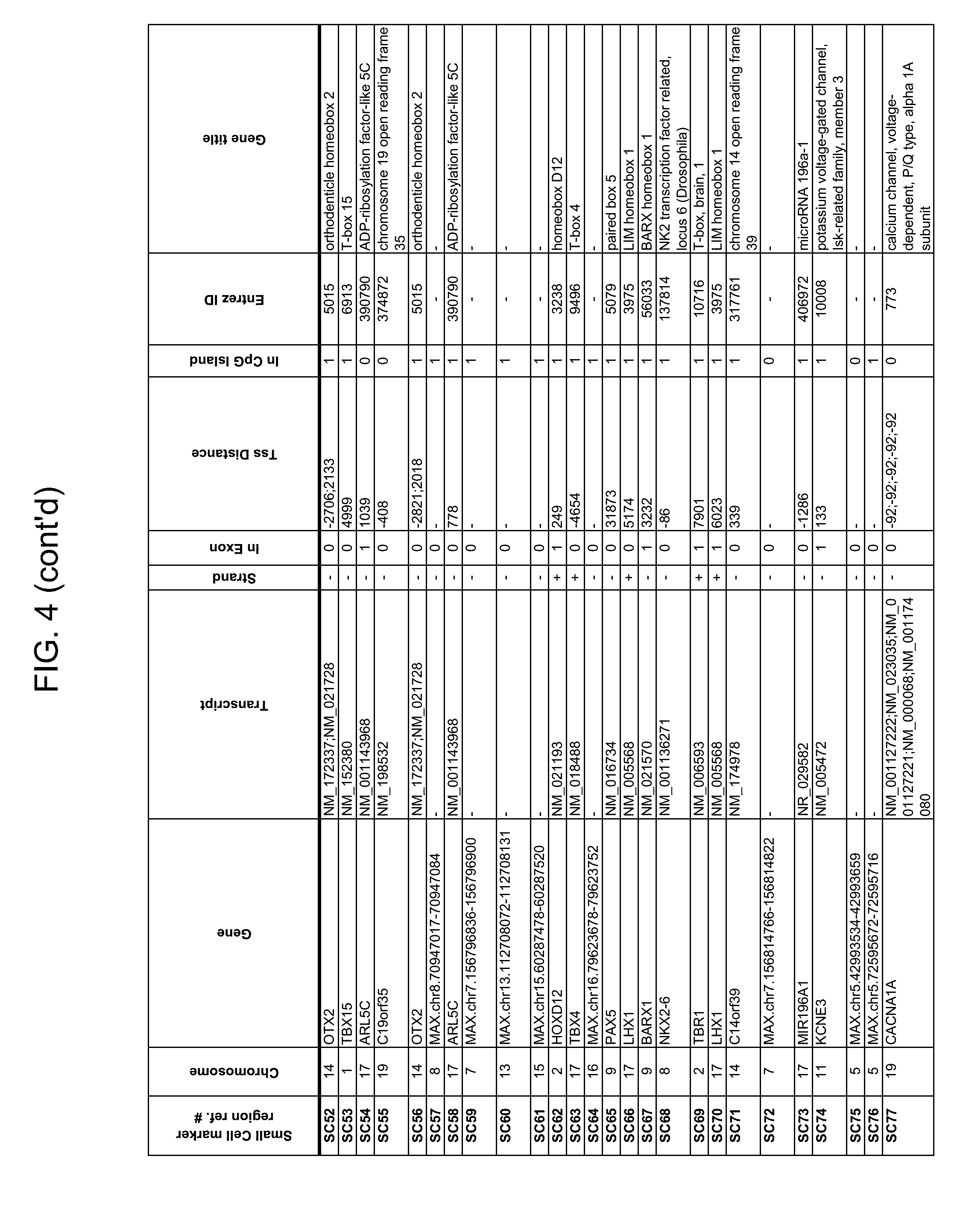
D00102

D00103

D00104
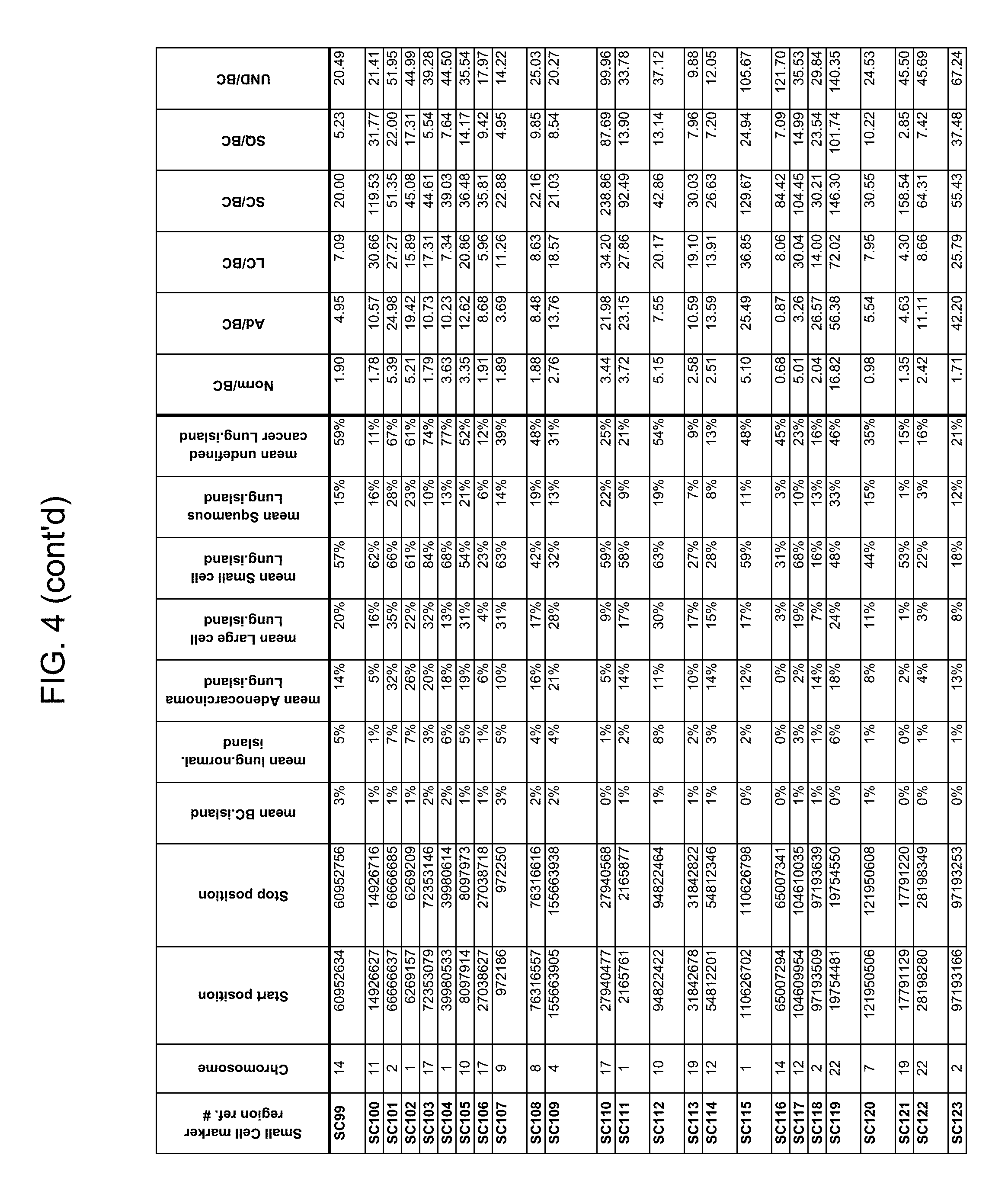
D00105

D00106

D00107

D00108

D00109

D00110

D00111

D00112

D00113

D00114

D00115

D00116

D00117

D00118

D00119

D00120

D00121

D00122

D00123

D00124

D00125

D00126

D00127
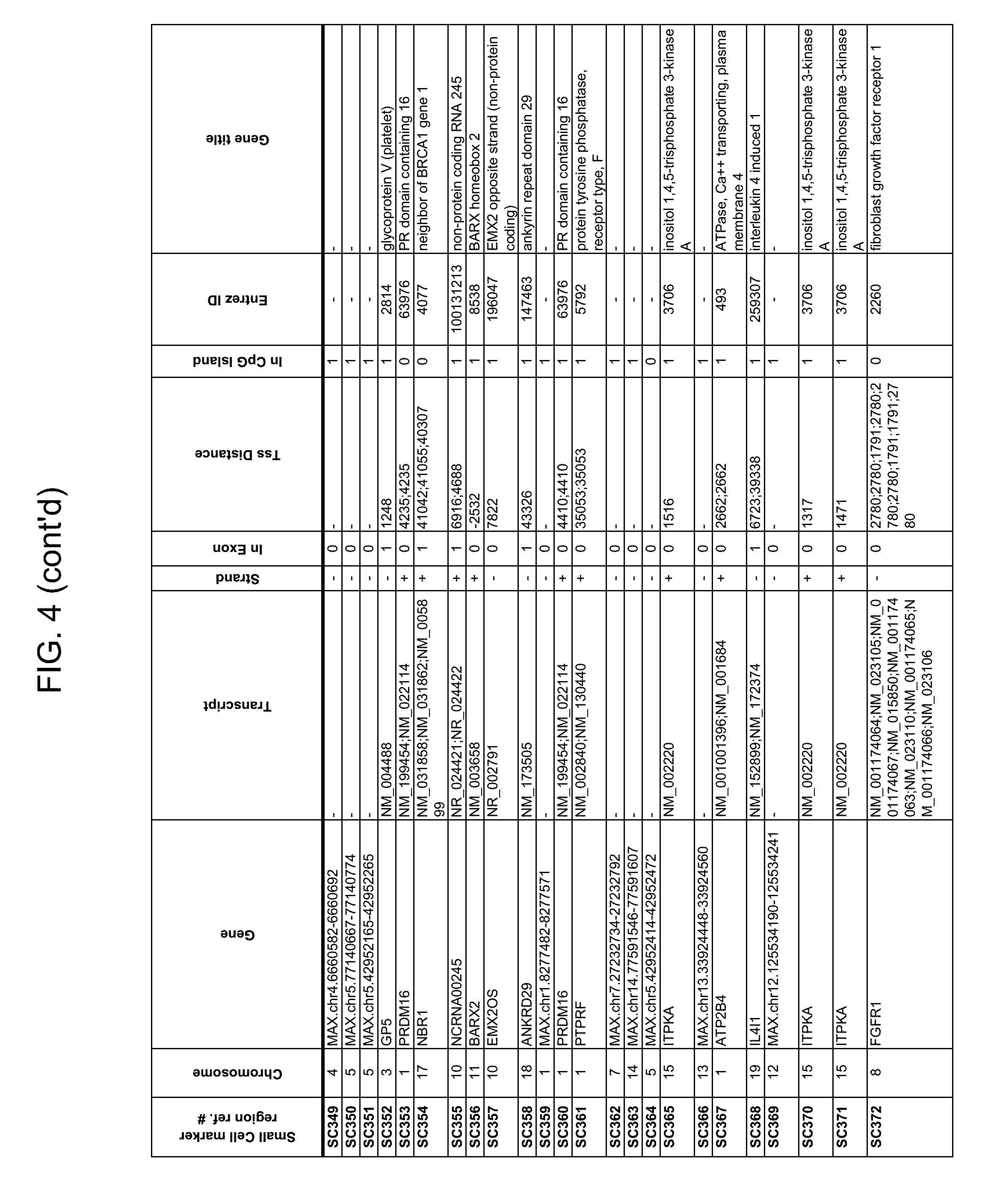
D00128

D00129

D00130

D00131

D00132

D00133

D00134

D00135

D00136

D00137

D00138

D00139

D00140

D00141

D00142

D00143

D00144

D00145

D00146

D00147
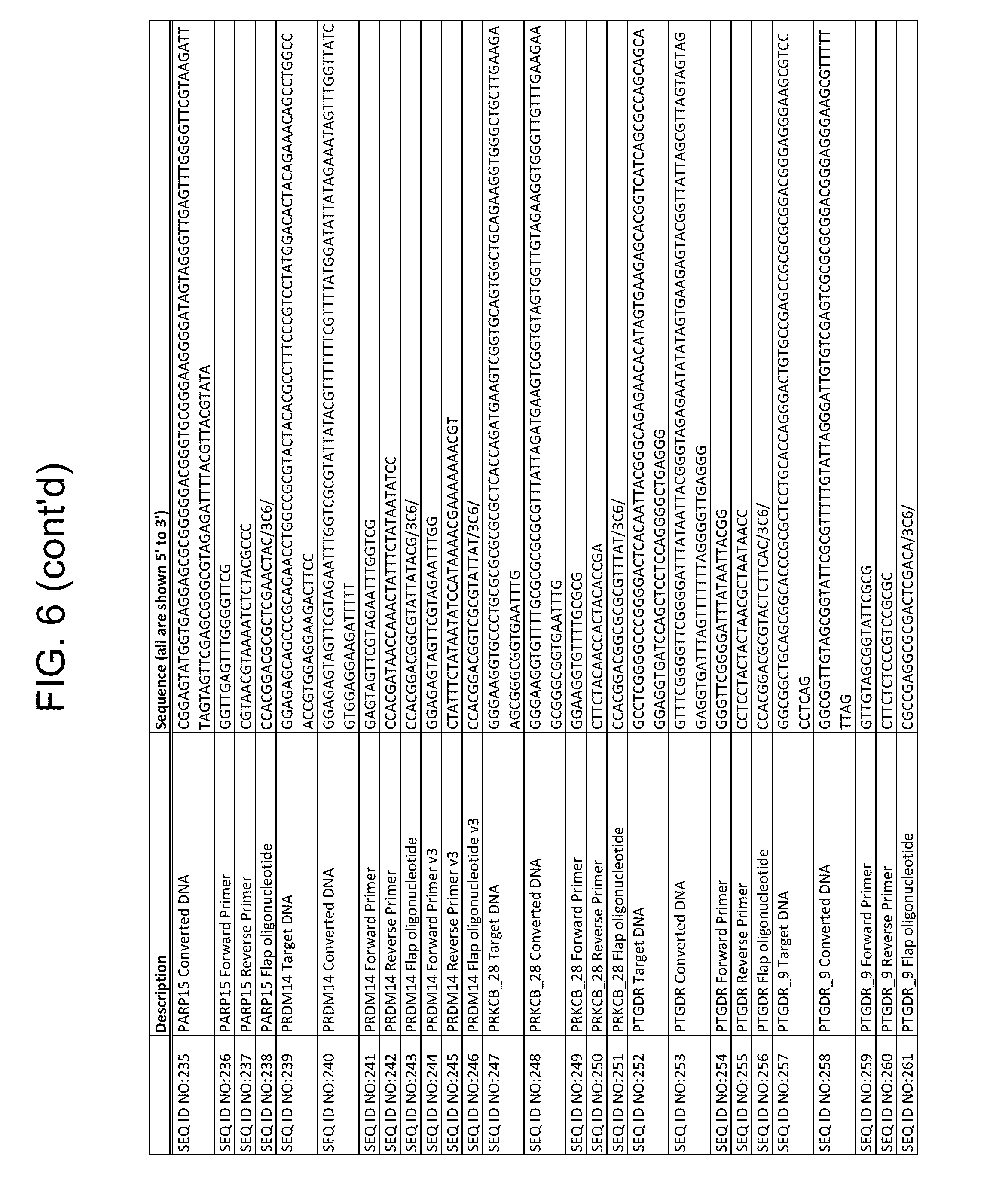
D00148

D00149

D00150

D00151

D00152

D00153

D00154

D00155

S00001
XML
uspto.report is an independent third-party trademark research tool that is not affiliated, endorsed, or sponsored by the United States Patent and Trademark Office (USPTO) or any other governmental organization. The information provided by uspto.report is based on publicly available data at the time of writing and is intended for informational purposes only.
While we strive to provide accurate and up-to-date information, we do not guarantee the accuracy, completeness, reliability, or suitability of the information displayed on this site. The use of this site is at your own risk. Any reliance you place on such information is therefore strictly at your own risk.
All official trademark data, including owner information, should be verified by visiting the official USPTO website at www.uspto.gov. This site is not intended to replace professional legal advice and should not be used as a substitute for consulting with a legal professional who is knowledgeable about trademark law.